Note: Descriptions are shown in the official language in which they were submitted.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
4-(2-Phenyloxyphenyl)-piperidine or -1,2,3,6-tetrahydropyridine derivatives as
serotonin reuptake inhibitors
The present invention relates to novel compounds which arc serotonin reuptake
inhibitors and as such effective in the treatment of for example depression
and
anxiety.
l~aekground of the invention
to Selective serotonin reuptal~e inhibitors (hereinafter referred to as SSRIs)
have become
first choice therapeutics in the treatment of depression, certain forms of
anxiety and
social phobias, because they are effective, well tolerated a~zd have a
favourable safety
profile compared to the classic tricyclic antidepressants.
15 However, clinical studies on depression indicate that non-response to SSRIs
is
substantial, up to 30%. Another, often neglected, factor in antidepressant
treatment is
compliance, which has a rather profound effect on the patient's motivation to
continue
pharmacother apy.
2o First of all, there is the delay in therapeutic effect of SSRIs. Sometimes
symptoms
even worsen during the first weeps of treatment. Secondly, sexual dysfunction
is a
side effect common to all SSRIs. Without addressing these problems, real
progress in
the pharmacotherapy of depression and anxiety disorders is not lilcely to
happen.
25 The combined effect of serotonin reuptake inhibition and norepinephrine
uptalce
iWibition on depression is explored in clinical studies of compounds such as
Duloxetine (along, Duloxetine (LY-248686): an inhibitor of serotonin and
noradrenaline uptalce and an antidepressant drug candidate Expey~t Opihiora
ofZ
Irzvestigutioraal Dr°ugs, 1998, 7, 10, 1691-1699) and Venlafaxine (Khan-
A et al,
3o Venlafaxine in depressed outpatients 1'syelz.~p7icza°macolog~
Bulletif~, 1991, ~'7, 14.1-
144).
The present invention provides novel compounds which posses the combined
effect of
serotonin reuptake inhibition and norepinephrine uptake inhibition for the
treatment of
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
affective disorders, such as depression, anxiety disorders including general
anxiety
disorder, social anxiety disorder, post traumatic stress disorder, obsessive
compulsive
disorder, panic disorder, panic attacl~s, specific phobias, social phobia and
agoraphobia.
~urramaxy 0f the i~tventi~n
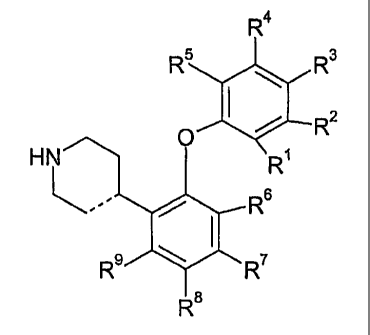
The present invention provides compounds of the general formula I
R4
R5 R3
/ R2
HN R~
.. / . R6
R9'~ R7
R$
to
wherein the dotted line, R1, R2, R3, R4, R5, R~, R', R8, and R~ are as defined
below.
The invention provides a compound according to the above for use as a
medicament.
15 The invention provides a pharmaceutical composition comprising a compound
according to the above or a pharmaceutically acceptable acid addition salt
thereof and
at least one pharmaceutically acceptable carrier or diluent.
The invention provides the use of a compound according to the above or a
20 pharmaceutically acceptable acid addition salt thereof for the preparation
of a
medicament for the treatment of affective disorders, such as depression,
anxiety
disorders including general anxiety disorder, social anxiety disorder, post
traumatic
stress disorder, obsessive compulsive disorder, panic disorder, panic
attacl~s, specific
phobias, social phobia and agoraphobia.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
3
The invention provides a method for the treatment of an affective disorders,
such as
depression, anxiety disorders including general anxiety disorder, social
anxiety
disorder, post traumatic stress disorder, obsessive compulsive disorder, panic
disorder,
panic attacks, specific phobias, social phobia and agoraphobia in a living
animal body,
including a human, comprising administering a therapeutically effective amount
of a
compound according to the above or a pharmaceutically acceptable acid addition
salt
thereof.
~efiniti0ii 0f ~ub~tituent~
to
Halogen means fluoro, chloro, bromo or iodo.
The expression C1_~-allc(enlyn)yl means a C1_~-alkyl, CZ_~-alkenyl or a CZ_G-
allcynyl
group.
The term C 1 _~ alkyl refer s to a branched or unbranched allcyl group having
from one to
six carbon atoms inclusive, including but not limited to methyl, ethyl, 1-
propyl, 2-
propyl, 1-butyl, 2-butyl, 2-methyl-2-propyl and 2-methyl-1-propyl.
Similarly, C2_~ alkenyl and C2_~ alkynyl, respectively, designate such groups
having
from two to six carbon atoms, including one double bond and one triple bond
respectively, including but not limited to ethenyl, propenyl, butenyl,
ethynyl, propynyl
and butynyl.
The terms C1_~-alk(euyn)yloxy, C1_~ allc(en/yn)ylsulfanyl, hydroxy-C1_~-
alk(eWyn)yl,
halo-C1_~-allc(en/yn)yl, cyano-C1_~-alk(en/yn)yl, NRZRW-C1_~-allc(enyn)yl,
C1_~-
allc(enlyn)yloxy-C1_~-allc(en/yn)yl and halo-C1_~-allc(en/yn)yloxy designate
such
groups in which the C1_~-allc(en/yn)yl are as defined above. Halo means
halogen.
NR~RW-C1_G-alk(euyn)yl designate the group
R~
RW N-G~-6a~k(en/yn)yl
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
4
The term C3_8 cycloall~yl designates a monocyclic or bicyclic carbocycle
having three
to eight C-atoms, including but not limited to cyclopropyl, cyclopentyl,
cyclohexyl,
etc.
The term C3_8 cycloal~eriyl designates a monocyclic or bicyclic carbocycle
having
three to eight C-atoms and including ogle double bond.
I11 the te1111 C3_$-cycloallc(en)yl-C1_G-allc(en/y11)yl, C3_8-cycloall~(en)yl
alld Ci-G-
all~(en/yn)yl are as defined above.
to
The term 3-7-membered ring optionally containing one ful-ther heteroatom, such
as N,
O, or S, as used herein refers to ring systems such as 1-morpholinyl, 1-
piperidinyl, 1-
azepinyl, 1-piperazinyl, 1-homopiperazinyl, 1-imidazolyl, 1-pyrrolidinyl, 1-
azetidinyl,
1-pyrrolyl or pyrazolyl, all of which may be further substituted with a group
selected
from a C1_G-all~(en/yn)yl, hydroxy, hydroxy-C1_G-allc(en/yn)yl, C1_G-
allc(en/yn)yloxy-
C 1 _G-all~(en/yn)yl .
Description of the invention
2o The present invention relates to 4-(2-phenyloxyphenyl)-piperidine or -
1,2,3,6-
tetrahydropyridine derivatives which are serotonin reuptal~e inhibitors and as
such
effective in the treatment of for example depression and anxiety. In
particular the
piperidines are also good norepinephrine uptal~e inhibitors.
Accordingly the present invention relates to a compound represented by the
general
formula I
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
R4
R5 R3
R2
H~
R
R6
R7
RV
when ein
the dotted line ---- indicates a single bond or a double bond;
RI, R2, R3, R4, RS are independently selected from hydrogen, halogen, cyano,
C1-~-
allc(en/yn)yl, C1_~-alk(en/yn)yloxy, C1_~-alk(euyn)ylsulfanyl, hydroxy,
hydroxy-C1_~-
alk(en/yn)yl, halo-C1_~-alk(en/yn)yl, halo-C1_~-allc(en/yn)yloxy, or NR"Ry
wherein R"
and Ry are independently selected from hydrogen, C1_~-alk(eWyn)yl, cyano-C1_~-
to allc(en/yn)yl, C3_g-cycloalk(en)yl, C3_$-cycloallc(en)yl-C1_~-alk(enlyn)yl,
or NRZRW-C1_
~-alk(enJyn)yl, wherein RZ and RW are independently selected from hydrogen,
C1_~-
alk(en/yn)yl, C3_$-cycloalk(en)yl, or C3_g-cycloalk(en)yl-C1_~-alk(enyn)yl; or
R" and
RY together with the nitrogen to which they are attached form a 3-7-membered
ring
which optionally contains one further heteroatom; or
RZ and R3 together form a heterocycle fused to the phenyl ring selected from
~o
~o ; and R1, R4, R5 are as defined above;
R~, R', R8, R~ are independently selected from hydrogen, halogen, C1_~-
allc(euyn)yl,
2o C1_~-allc(euyn)yloxy, CI_~-alk(eW~m)ylsulfanyl, hydroxy, hydroxy-C1_~-
alk(euyn)yl,
halo-C1_~-allc(en/yn)yl, halo-C1_~-allc(enyn)yloxy, or NRXRy wherein R" and RY
axe
independently selected from hydrogen, C1_~-alk(en/yn)yl, cyano-C1_~-
allc(en/yn)yl, C3_
8-cycloalk(en)yl, C3_8-cycloall~(en)yl-C1_~-allc(en/yn)yl, or NR~R'~'-C1_~-
allc(en/yn)yl,
wherein RZ and RW are independently selected from hydrogen, C1_~-alk(en/yn)yl,
C3_8-
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
cycloalk(en)yl, or C3_8-cycloallc(en)yl-Cl_~-alk(en/yn)yl; or R" and Ry
together with
the nitrogen to which they are attached form a 3-7-membered ring which
optionally
contains one further heteroatom;
provided that at least one of Rl, R2, R3, R4, R5, R~, R~, R8, and R~ is
different from
hydrogen;
or a salt thereof.
In one embodiment of the compound of formula I, Rl is selected from hydrogen,
halogen, cyano, C1_~-alk(en/yn)yl, C1_~-alk(en/yn)yloxy, C1_~-
alk(en/yn)ylsulfanyl,
to halo-CI_~-alk(en/yn)yl, or NR"Ry wherein RX and RY are independently
selected from
hydrogen, C1_~-alk(en/yn)yl, cyano-C1_~-allc(en/yn)yl, C3_8-cycloallc(en)yl,
C3_8-
cycloalk(en)yl-C1_~-allc(en/yn)yl, or NRZRW-C1_~-alk(en/yn)yl, wherein RZ and
RW are
independently selected from hydrogen, C1_~-alk(en/yn)yl, C3_8-cycloallc(en)yl,
or C3_8-
cycloallc(en)yl-C1_~-alk(en/yn)yl, provided that if one of R" and RY is NRZRW-
C1_~-
15 alk(en/yn)yl then the other is selected from hydrogen, C1_~-allc(en/yn)yl,
cyano-C1_~-
alk(en/yi)yl, C3_8-cycloalk(en)yl, or C3_g-cycloalk(en)yl-C1_~-alk(en/yn)yl;
or R" and
RY together with the nitrogen to which they are attached form a 3-7-membered
ring
which optionally contains one further heteroatom. In a further embodiment of
the
compound of formula I Rl is selected from hydrogen, halogen, cyano, C1_~-
2o alk(eWyn)yl, C1_~-alk(en/yn)yloxy, C1_~-alk(en/yn)ylsulfanyl, halo-C1_~-
alk(euyn)yl.
In a further embodiment Rl is NR"Ry wherein R" and RY are independently
selected
from hydrogen, C1_~-allc(en/yn)yl, cyano-C1_~-allc(en/yn)yl, C3_8-
cycloalk(en)yl, C3_8-
cycloalk(en)yl-C1_~-alk(en/yn)yl, such as hydrogen, cyanomethyl, C1_~-
alk(en/yn)yl. In
a fiuther embodiment Rl is NR"RY wherein R" is NRZRW-C1_~-allc(en/yn)yl,
wherein RZ
25 and RW are independently selected from hydrogen, C1_~-allc(en/yn)yl, C3_8-
cycloalk(en)yl, or C3_8-cycloalk(en)yl-C1_~-allc(euyn)yl, and RY is selected
from
hydrogen, C1_~-allc(en/yn)yl, cyano-C1_~-alk(en/yn)yl, C3_8-cycloallc(en)yl,
or C3_g-
cycloalk(en)yl-C1_~-allc(enyn)yl. In a further embodiment Rl is NR"Ry wherein
R"
and RY together with the nitrogen to which they are attached form a 3-7-
membered
30 ring which optionally contains one further hetcroatom, such as 1-
morpholinyl, 1-
piperidinyl, 1-azepinyl, 1-piperazinyl, 1-homopiperazinyl, 1-imidazolyl, 1-
pyrrolidinyl, 1-azetidinyl, 1-pyrrolyl or pyrazolyl, optionally substituted
with one or
more selected from a C1_~-allc(en/yn)yl, hydroxy, hydroxy-C1_~-allc(euyn)yl,
C1_~-
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
alk(en/yn)yloxy-C1_~-alk(euyn)yl, e.g. one or two selected from hydroxy,
hydroxy-C1_
~-all~yl, C1_~-alkyloxy-C1_~-alkyl, C1_~-allcyl, in particular one or two
selected from
hydroxy, methoxy-methyl, methyl. Typically, Rl is selected from hydrogen;
halogen;
cyano; C~_~-alkyl; C1_~-alhyloxy; C1_~-alkylsulfanyl; halo-C1_~-alkyl; NR"RY
wherein
R~ and Ry are independently selected from hydrogen, C1_~-allcyl, cyanomethyl;
NR"Ry
wherein Ry is selected from hydrogen, or C1_~-alkyl, and R~ is NR~R"'-C1-~-
allc(el~/yn)yl wherein RZ and RW are independently selected from hydrogen, or
C1_~-
alkyl; 1-morpholinyl, 1-piperidinyl, 1-azepinyl, 1-piperazinyl, 1-
homopiperazinyl, 1-
imidazolyl, 1-pyrrolidinyl, 1-azetidinyl, 1-pyrrolyl or pyrazolyl, optionally
substituted
with one or two selected from hydroxy, hydroxy-Cl_~-allcyl, C1_~-allcyloxy-
Cl_~-alkyl,
C1_~-alkyl, in particular one or two selected from hydroxy, methoxy-methyl,
methyl.
To further illustrate without limiting the invention an embodiment of Rl is
hydrogen;
another embodiment of Rl is C1_~-all~yl, such as methyl; a further embodiment
of Rl is
halogen, such as fluoro, or chloro.
In a further embodiment of the compound of formula I, R2 is selected from
hydrogen,
halogen, cyano, C1_~-allc(el~/yn)yl, C1_~-alk(en/yn)yloxy, C1_~-
alk(en/yn)ylsulfanyl,
halo-C1_~-alk(en/yn)yl. Typically, RZ is selected from hydrogen, halogen,
cyano, C1_~-
allcyl, C1_~-allcyloxy, C1_~-alkylsulfanyl, halo-C1_~-alkyl. To ful-ther
illustrate without
limiting the invention an embodiment of RZ is hydrogen; another embodiment of
RZ is
C1_~-alkoxy, such as methoxy; another embodiment of R2 is halo-C1_~-alkyl,
such as
trifluoromethyl; another embodiment of RZ is C1_~-alkyl, such as methyl;
another
embodiment of R2 is halogen, such as chloro.
In a further embodiment of the compound of formula I, R3 is selected from
hydrogen,
halogen, cyano, C1_~-alk(en/yn)yl, C1_~-alk(en/yn)yloxy, C1_~-
alk(enlyn)ylsulfanyl,
halo-C1_~-alk(en/yn)yl. Typically, R3 is selected from hydrogen, halogen,
cyano, C1_~-
allcyl, C1_~-allcyloxy, C1_~-allcylsulfanyl, halo-C1_~-alkyl. To fiu-ther
illustrate without
limiting the invention an embodiment of 8315 hydrogen; another embodiment of
R3 is
3o C1_~-alkyl, such as methyl; a further embodiment of 8315 C1_~-allcoxy, such
as
methoxy; a further embodiment of R3 is halogen, such as chloro, or fluoro;
another
embodiment of R3 is halo-C1_~-alkyl, such as trifluoromethyl.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
In a further embodiment of the compound of formula I, R2 and R3 together form
a
heterocycle fused to the phenyl ring selected from
~o
_~
In a further embodiment of the compound of formula I, R4 is selected from
hydrogen,
halogen, cyano, C1_~-allc(euyn)yl, C1_~-allc(en/yn)yloxy, C1_~-
alk(en/yn)ylsulfanyl,
halo-C1_~-alk(en/yn)yl. Typically, R4 is selected from hydrogen, halogen,
cyano, C1_~-
alkyl, C1_~-alkyloxy, C1_~-alkylsulfanyl, halo-C1_~-allcyl. To further
illustrate without
limiting the invention an embodiment of R4 is hydrogen; another embodiment of
R4 is
to C1_~-allcoxy, such as methoxy; another embodiment of R4 is halo-C1_~-alkyl,
such as
trifluoromethyl; another embodiment of R4 is C1_~-alkyl, such as methyl;
another
embodiment of R4 is halogen, such as chloro.
In a further embodiment of the compound of formula I, RS is selected from
hydrogen,
15 halogen, cyano, C1_~-alk(en/yn)yl, C1_~-alk(euyn)yloxy, C1_~-
alk(en/yn)ylsulfanyl,
halo-C1_~-alk(en/yn)yl, or NR"RY wherein R" and Ry are independently selected
from
hydrogen, C1_~-alk(en/yn)yl, cyano-C1_~-alk(en/yn)yl, C3_8-cycloalk(en)yl,
C3_8-
cycloalk(en)yl-C1_~-alk(en/~m)yl, or NRZRW-C1_G-alk(en/yn)yl, wherein RZ and
RW are
independently selected from hydrogen, C1_~-alk(en/yn)yl, C3_$-cycloall~(en)yl,
or C~_8-
2o cycloallc(en)yl-C1_~-alk(en/yn)yl, provided that if one of R" and RY is
NRZRW-C1_~-
alk(eWyn)yl then the other is selected from hydrogen, C1_~-alk(euyn)yl, cyano-
C1_~-
allc(euyn)yl, C3_g-cycloallc(en)yl, or C3_8-cycloallc(en)yl-C1_~-alk(en/yn)yl;
or R" and
Ry together with the nitrogen to which they are attached form a 3-7-membered
ring
which optionally contains one further heteroatom. In a further embodiment of
the
25 compound of formula I R5 is selected from hydrogen, halogen, cyano, C1_~-
alk(eWyn)yl, C1_~-alk(en/yn)yloxy, C1_~-alk(en/yn)ylsulfanyl, halo-C1_~-
alk(en/yn)yl.
In a further embodiment RS is NR"R'' wherein R" and RY are independently
selected
from hydrogen, C1_~-allc(en/yn)yl, cyano-C1_~-alk(enyn)yl, C3_$-
cycloallc(en)yl, C3_8-
cycloallc(en)yl-C1_~-alk(en/yn)yl, such as hydrogen, cyanomethyl, C1_~-
all~(en/yl)yl. In
3o a further embodiment RS is NRAR'' wherein R~ is NR~R'~'-C1_~-allc(en/yn)yl,
wherein R
and RW are independently selected from hydrogen, C1_~-allc(euyn)yl, C3_8-
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
cycloalk(en)yl, or C3_8-cycloall~(en)yl-C1_~-alk(en/yn)yl, and Ry is selected
from
hydrogen, C1_~-all~(en/yn)yl, cyano-C1_~-alk(en/yn)yl, C3_8-cycloalk(en)yl, or
C3_8-
cycloalk(en)yl-C1_~-alk(en/yn)yl. In a further embodiment RS is NR"Ry wherein
R"
and RY together with the nitrogen to which they are attached form a 3-7-
membered
ring which optionally contains one further heteroatom, such as 1-morpholinyl,
1-
piperidinyl, 1-azepinyl, 1-piperazinyl, 1-homopiperazinyl, 1-imidazolyl, 1-
pyrrolidinyl, 1-azetidinyl, 1-pyrrolyl or pyrazolyl, optionally substituted
with one or
more selected from a C1_~-allc(en/yn)yl, hydroxy, hydroxy-C1_~-allc(en/yn)yl,
C1_~-
alh(en/yn)yloxy-C1_~-allc(euyn)yl, e.g. one or two selected from hydroxy,
hydroxy-Cl_
to ~-alkyl, C~_~-allcyloxy-C1_~-alkyl, C1_~-alkyl, in pauticular one or two
selected from
hydroxy, methoxy-methyl, methyl. Typically, RS is selected from hydrogen;
halogen;
cyano; C1_~-allcyl; C1_~-alkyloxy; C1_~-alkylsulfanyl; halo-C1_~-alkyl; NR"R''
wherein
R" and Ry are independently selected from hydrogen, C1_~-alkyl, cyanomethyl;
NR"RY
wherein R'' is selected from hydrogen, or C1_~-alkyl, and R" is NRZRW-C1_~-
alk(eWyn)yl wherein RZ and RW are independently selected from hydrogen, or
C1_~-
alkyl; 1-morpholinyl, 1-piperidinyl, 1-azepinyl, 1-piperazinyl, 1-
homopiperazinyl, 1-
imidazolyl, 1-pyrrolidinyl, 1-azetidinyl, 1-pyrrolyl or pyrazolyl, optionally
substituted
with one or two selected from hydroxy, hydroxy-C1_~-alkyl, C1_~-alkyloxy-C1_~-
alkyl,
Cl_~-alkyl, in particular one or two selected from hydroxy, methoxy-methyl,
methyl.
2o To further illustrate without limiting the invention an embodiment of RS is
hydrogen;
another embodiment of RS is C1_~-alkyl, such as methyl; a further embodiment
of RS is
halogen, such as chloro, or fluoro.
In a further embodiment of the compound of formula I R~ is selected from
hydrogen,
halogen, C~_~-allc(euyn)yl, halo-C1_~-allc(euyn)yl. Typically, R~ is selected
from
hydrogen, halogen, C1_~-alkyl, halo-C1_~-allcyl. To further illustrate without
limiting
the invention an embodiment of R~ is hydrogen; another embodiment of R~ is
halogen, such as fluoro.
~0 In a further embodiment of the compound of formula I, R' is selected from
hydrogen,
halogen, C1_~-alk(en/yn)yl, halo-C1_~-alk(enlyn)yl. Typically, R~ is selected
from
hydrogen, halogen, C1_~-alkyl, halo-C1_~-alkyl. To further illustrate without
limiting
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
the invention an embodiment of R' is hydrogen; another embodiment of R~ is
halogen, such as fluoro.
In a further embodiment of the compound of formula I, R$ is selected from
hydrogen,
halogen, C1_~-alk(en/yn)yl, halo-C1_~-alk(en/yn)yl, or IVR~Ry wherein R" and
Ry are
independently selected from hydrogen, C1_~-alk(en/yn)yl, cyano-Cl_~-
allc(en/yn)yl, C3_
8-cycloalk(en)yl, C3_8-cycloalk(en)yl-C1_~-all~(en/yn)yl, or NR~R~''-C1_~-
alk(enlyn)yl,
wherein RZ and RW are independently selected from hydrogen, C1_~-alk(en/yn)yl,
C3_$-
cycloalk(en)yl, or C3_8-cycloalk(en)yl-C1_~-alk(en/yn)yl, provided that if one
of R" and
to Ry is NRzRW-C1_~-alk(en/yn)yl then the other is selected from hydrogen,
Cl_~-
alk(en/yn)yl, cyano-C1_~-alk(en/yn)yl, C3_8-cycloallc(en)yl, or C3_$-
cycloalk(en)yl-C1_~-
allc(en/yn)yl; or R" and RY together with the nitrogen to which they are
attached form
a 3-7-membered ring which optionally contains one further heteroatom. In a
further
embodiment of the compound of formula I R8 is selected from hydrogen, halogen,
C1_
~-alk(en/yn)yl, halo-C1_~-allc(en/yn)yl. In a further embodiment R8 is NR"RY
wherein
R" and Ry are independently selected from hydrogen, C1_~-alk(en/yn)yl, cyano-
C1_~-
allc(en/yn)yl, C3_8-cycloalk(en)yl, C3_8-cycloallc(en)yl-C1_~-alk(euyn)yl,
such as
hydrogen, cyanomethyl, C1_~-alk(enlyn)yl. In a further embodiment R8 is NR"R''
wherein R" is NRZRW-C1_~-alk(en/yn)yl, wherein RZ and RW are independently
selected
2o from hydrogen, C1_~-allc(en/yn)yl, C3_8-cycloalk(en)yl, or C3_8-
cycloallc(en)yl-C1_~-
allc(eWyn)yl, and RY is selected from hydrogen, C1_~-allc(euyn)yl, cyano-C1_~-
alk(en/yn)yl, C3_8-cycloalk(en)yl, or C3_8-cycloalk(en)yl-C1_~-alk(en/yn)yl.
In a further
embodiment R$ is NR"Ry wherein R" and Ry together with the nitrogen to which
they
are attached form a 3-7-membered ring which optionally contains one further
heteroatom, such as 1-morpholinyl, 1-piperidinyl, 1-azepinyl, 1-piperazinyl, 1-
homopiperazinyl, 1-imidazolyl, 1-pynolidinyl, 1-azetidinyl, 1-pyrrolyl or
pyrazolyl,
optionally substiW ted with one or more selected from a C1_~-allc(enlyn)yl,
hydroxy,
hydroxy-C1_~-allc(enyn)yl, C1_~-allc(enlyn)yloxy-C1_~-allc(enyn)yl, e.g. one
or two
selected from hydroxy, hydroxy-C1_~-alkyl, C1_~-allcyloxy-C1_~-allcyl, C1_~-
allcyl, in
3o particular one or two selected from hydroxy, methoxy-methyl, methyl.
Typically, R$
is selected from hydrogen; halogen; cyano; C1_G-allcyl; C1_~-alkyloxy; CI_~-
alkylsulfanyl; halo-Cl_~-alkyl; NR~RY wherein RX and Ry are independently
selected
from hydrogen, C1_~-allcyl, cyanomethyl; NR"Ry wherein Ry is selected from
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
11
hydrogen, or C1_6-all~yl, and RX is NRZRW-C1_~-alk(en/yn)yl wherein RZ and RW
are
independently selected from hydrogen, or C1_~-alkyl; 1-morpholinyl, 1-
piperidinyl, 1-
azepinyl, 1-piperazinyl, 1-homopiperazinyl, 1-imida~olyl, 1-pyrrolidinyl, 1-
azetidinyl,
1-pyrrolyl or pyrazolyl, optionally substituted with one or two selected from
hydroxy,
hydro~y-C1_~-alkyl, C1_~-alkyloxy-C1_~-alkyl, C1_~-all~yl, in particular one
or tv,~o
selected from hydroxy, methoxy-methyl, methyl. To further illustrate without
limiting
the invention an embodiment of R8 is hydrogen; another embodiment of R$ is
halogen, such as fluoro, or bromo; a further embodiment of R8 is C1_~-alkyl,
such as
methyl; a further embodiment of R8 is halo-C1_~-allcyl, such as CF3.
to
In a further embodiment of the compound of formula I, R~ is selected from
hydrogen,
halogen, C1_~-allc(euyn)yl, halo-C1_~-alk(en/yn)yl. Typically, R~ is selected
from
hydrogen, halogen, Cl_~-alkyl, halo-C1_~-alkyl. To further illustrate without
limiting
the invention an embodiment of R~ is hydrogen.
In a further embodiment of the compound of formula I, the dotted line ----
indicates a
single bond.
In a further embodiment of the compound of formula I, the dotted line ----
indicates a
double bond.
Typically, the compound of formula I has at least one substituent in the
phenyl
ring(s), selected from any one of Rl-R~, which is different from hydrogen,
such as 1,
2, 3, or 4 substituents in the phenyl ring(s), selected from any one of Rl-R9,
which
is/are different fr0111 hydrogen, and the remaining substituents are hydrogen.
Thus, in
a further embodiment 1 substituent selected from any one of Rl-R~, which is
different
from hydrogen, is present in either of the two phenyl rings, such as 1
substituent
selected from Rl-R5, or the substituent is selected from R~-R~. In a further
embodiment 2 substituents selected from Rl-R~, which are different from
hydrogen,
3o are present in either of the two phenyl rings, such as 1 substituent
selected from Rl-
R5, and the other selected from R~-R~, or both substituents are selected from
Rl-R5; in
this respect R2 and R3 may be talcen together to form the heterocycle as
defined above.
In a further embodiment 3 substituents selected from Rl-R9, which are
different from
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
12
hydrogen, are present in either of the two phenyl rings, such as 2
substituents selected
from Rl-R5, and the last substituent is selected from R~-R~. In each
embodiment, as
mentioned the remaining substituents are hydrogen. To illustrate this further
without
limiting the invention, some typical embodiments are outlined hereafter.
Thus, in a further embodiment of the compound of formula I one substituent is
present
which is R2 as defined above, except hydrogen. In a further embodiment of the
compound of formula I one substituent is present which is R3 as defined above,
except
hydrogen. In a further embodiment of the compound of formula I two
substituents are
to present being R3 and R8, wherein R3 and R$ are as defined above, except
hydrogen. In
a further embodiment of the compound of formula I two substituents are present
being
R3 and RG, wherein R~ and R~ are as defined above, except hydrogen. In a
further
embodiment of the compound of formula I two substituents are present being R3
and
R~, wherein R3 and R' are as defined above, except hydrogen. In a further
15 embodiment of the compound of formula I two substituents are present being
Rl and
R3, wherein Rl and R3 are as defined above, except hydrogen. In a further
embodiment of the compound of formula I two substituents are present being R2
and
R3, wherein R2 and R3 are as defined above, except hydxogen, in this respect
R2 and
R3 may be tal~en together to form the heterocycle as defined above. In a
further
2o embodiment of the compound of formula I three substituents are present
being Rl, R3
and R8, wherein Rl, R3 and R8 are as defined above, except hydrogen. In each
embodiment, as mentioned above the remaining substituents are hydrogen.
In a further embodiment of the compound of formula I, said compound is
selected
25 from
4-~2-(2, 4-Di>7zethylplzetzoxy)phe>zylJ-1, 2, 3, 6-tetrahyd>~opyt~idine,
4-~2-(4-Chlor~oplzet2oxy)phertylJ-1, 2, 3, 6-tetz"a7zyd>"opy>~idi>ze,
4-~2-(4-Fluoz~o-2-metlzylphe~~.~y)pherzylJ-1, ~, 3, 6-tetf-
alzyd>"~pyt°idihe,
4-~2-(~-Fluot°~plze>zoxy)phenylJ-1, ~, 3, 6-tett~ahydropyt~idi>ze,
30 ~-~2-(4-Methylphenoxy)plz~ttylJ-1,x,3,6-tett~altydt°~pyriditte,
4-~2-(~-Meth~xyplz etz~xy)plzetzylJ-1, 2, 3, 6-tett~ahydropyr~idizte,
4-~~',-(2, ~-DinzethylplTez~oxy)plzeraylJpiperidin.e,
4-~~-(4-ClaloYOplzenoxy)phenylJpipez~idi>?e,
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
13
4- j2-(4-Fluoy-o-2-methylphehoxy)phenylJpipe~idihe,
4-j2-(4-FIuoYOpheh.oxy)phe~2ylJpipef°idiyae,
4- j2-(4-Metlaylphenoxy)p7zeraylJpiperidih.e,
4-j2-(4-Chlof°o-2- methyl plaenoxy) plaefZylJ piper~idine,
-l-j?-(2-Clzloro-4-fraeth3rl phenoxy) plaeazylJwiper°idine,
4-j2-(2,4-Dichlo~~o phef2oxy) phenyl) piper°idiyae,
4-j2-(penzo jbJthiophen-5 yloxy) phehylJ pipe~idine,
4-j2-(~enzzojl,3Jdioxol-5-yloxy) pheh>>lJ pipes°idine,
4-j2-(4-Metlaoxy-2-methyl ~ahenoxy) plaeraylJ ~aipef°idine,
4-j2-(3,4-Diclalo~°o pherzoxy) phei~ylJ pipe~~idiyae,
4-j2-(3,4-Diuaetlzyl plaehoxy) plaerrylJ piper°idiy~e,
4-j2-(2,3,4,5-Tetramethyl phef2oxy) phefZylJ pipey~idihe,
4-j2-(4-Trifluo~omethyl phenoxy) phenyl) pipe~idihe,
4-j2-(4-Metl2oxy plae~zoxy) phenyl) piperidine,
4-j2-(2-Chloro-4-methoxy phehoxy) plaenylJ pipe~~idine,
4-j2-(3,4-Dimetlaoxy phenoxy) phe~zylJ piperidi~2e,
4-j2-(4-Clalo~°o-3-tf°ifluo~°omethyl phenoxy) phenyl)
pipe~~idine,
or
a pharmaceutically acceptable salt thereof. Each of these compounds is
considered a
2o specific embodiment and may be subject to individual claims.
As mentioned above, most of the tested compounds posses the combined effect of
serotonin reuptalce inhibition and norepinephrine uptal~e inhibition, however,
a few
compounds selected from
4-j2-(2,4-Dinaethylplaef2oxy)phefzylJ-1,2,3,6-tetralaydy~opy~idih.e,
4- j2-(4-Chloy~oplaenoxy)plaenylJ-1, 2, 3, 6-tet~~alaydy~opyridine,
4-j2-(4-Fluof~o-2-nzethylplze~2oxy)phenylJ-1,2, 3, 6-
tety°alaydropy~idiue,
4- j2-(4-Fluoroplnenoxy)phenylJ-1, 2, 3, 6-tetralaydf~opyYidine,
4- j2-(4-Methylplae~2oxy)phenylJ-1, 2, 3, 6-
tetJ°alaydr~opy°idih.e,
4-j2-(4-Metlno~.yplneraoxy)ph.erzylJ-1,2,3,x-tetrealaydYOpyr-idiyae,
4- j2-(3, 4-Dinaethyl phenoxy) plaef2ylJ piperidine,
4-j2-(2,3,4,5-Tetrarn.etlayl phert.oxy) plaenylJ piperidih.e,
4-j2-(3,4-Di~aet7aoxy plZefaoxy) phetaylJ pipet~idirae,
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
14
did show serotonin reuptal~e inhibition, but did not show norepinephrine
uptal~e
iWibition in the test herein.
The present invention also comprises salts of the present compounds,
typically,
pharmaceutically acceptable salts. Such salts include pharmaceutical
acceptable acid
addition salts, pharmaceutically acceptable metal salts, ammonium and
allcylated
ammonium salts. Acid addition salts include salts of inorganic acids as well
as organic
acids.
to
Representative examples of suitable inorganic acids include hydrochloric,
hydrobromic, hydroiodic, phosphoric, sulfiuic, sulfamic, nitric acids and the
life.
Representative examples of suitable organic acids include formic, acetic,
trichloroacetic, trifluoroacetic, propionic, benzoic, cimzamic, citric,
fumaric, glycolic,
itaconic, lactic, methanesulfonic, malefic, malic, malonic, mandelic, oxalic,
picric,
pyruvic, salicylic, succinic, methane sulfonic, ethanesulfonic, tartaric,
ascorbic,
pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic,
aspartic,
stearic, pahnitic, EDTA, glycolic, p-aminobenzoic, glutamic, benzenesulfonic,
p-
toluenesulfonic acids, theophylline acetic acids, as well as the 8-
halotheophyllines, for
2o example 8-bromotheophylline and the life.
Examples of metal salts include lithium, sodium, potassium, magnesium salts
and the
like.
Examples of ammonium and allcylated ammonium salts include ammonium, methyl-,
dimethyl-, trimethyl-, ethyl-, hydroxyethyl-, diethyl-, n-butyl-, sec-butyl-,
tert-butyl-,
tetramethylammonium salts and the life.
Further, the compounds of this invention may exist in unsolvated as well as in
3o solvated forms with pharmaceutically acceptable solvents such as Water,
ethanol arid
the life. In general, the solvated forms are considered equivalent to the
unsolvated
forms for the purposes of this invention.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
The compounds of the present invention may have one or more asymmetric centres
and it is intended that any optical isomers (i.e. enantiomers or
diastereomers), as
separated, pure or partially purified optical isomers and any mixtures thereof
111Cltldlllg racelnic mixtures are included within the scope of the invention.
5
Racelnic forms can be resolved into the optical antipodes by lgrlown methods,
for
example, by separation of diastereomeric salts thereof with an optically
active acid,
and liberating the optically active amine compound by treatment with a base.
Another
method for resolving racemates into the optical antipodes is based upon
to chromatography on an optically active matrix. Racemic compounds of the
present
invention can also be resolved into their optical antipodes, e.g. by
fractional
crystallization of d- or 1- (tartrates, mandelates or camphorsulphonate)
salts. The
compounds of the present invention may also be resolved by the formation of
diastereomeric derivatives.
Additional methods for the resolution of optical isomers, l~rlown to those
slcilled in the
art, may be used. Such methods include those discussed by J. Jaques, A. Collet
and S.
Wilen in "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New
Yorl~ (1981).
Optically active compounds can also be prepared from optically active starting
materials, or by stereoselective synthesis.
Furthermore, when a double bond or a fully or partially saturated ring system
is
present in the molecule geometric isomers may be formed. It is intended that
any
geometric isomers, as separated, pure or partially purified geometric isomers
or
mixtures thereof are included within the scope of the invention. Lil~ewise,
molecules
having a bond with restricted rotation may form geometric isomers. These are
also
intended to be included within the scope of the present invention.
Furthermore, some of the compounds of the present invention may exist in
different
tautomeric forms and it is intended that any tautomeric forms that the
compounds are
able to form are included within the scope of the present invention.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
16
The invention also encompasses prodrugs of the present compounds, which on
administration undergo chemical conversion by metabolic processes before
becoming
pharmacologically active substances. In general, such prodrugs will be
functional
derivatives of the compounds of the general formula (I), which are readily
convertible
in vivo into the required compomzd of the formula (I). Conventional procedures
for
the selection and preparation of suitable prodrug derivatives are described,
for
example, in "Design of Prodnzgs", ed. H. Bundgaard, Elsevier, 1985.
The invention also encompasses active metabolites of the present compounds.
to
As mentioned above, the compounds of formula I are serotonin reuptal~e
inhibitors,
and accordingly may be applicable for the treatment, including prevention, of
affective disorders, such as depression, anxiety disorders including general
anxiety
disorder and panic disorder and obsessive compulsive disorder.
Accordingly, in a further aspect the invention relates to a compound of
formula I for
use as a medicament.
The present invention also relates to a pharmaceutical composition comprising
a
2o compound of formula I and a pharmaceutically acceptable carrier or diluent.
The
composition may comprise any one of the embodiments of formula I described
above.
In an embodiment of the pharmaceutical composition, the compound of formula I
is
present in an amount of from about 0.001 to about 100 mg/lcg body weight per
day.
The present invention also relates to use of a compound of formula I for the
preparation of a medicament for the treatment of a disease or disorder,
wherein a
serotonin reuptalce inhibitor is beneficial. The medicament may comprise any
one of
the embodiments of formula I described above.
In particular, the present invention also relates to use of a compound of
formula I for
the preparation of a medicament for the treatment of affective disorders.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
17
In a further embodiment the present invention also relates to use of a
compound of
formula I for the preparation of a medicament for the treatment of depression.
In a further embodiment, the present invention also relates to use of a
compound of
foumula I for the preparation of a medicament for the treatment of anxiety
disorders.
In a further embodiment, the present invention also relates to use of a
compound of
formula I for the preparation of a medicament for the treatment of general
anxiety
disorder.
to
In a further embodiment, the present invention also relates to use of a
compound of
formula I for the preparation of a medicament for the treatment of social
anxiety
disorder.
15 In a further embodiment, the present invention also relates to use of a
compound of
fornula I for the preparation of a medicament for the treatment of post
traumatic
stress disorder.
In a further embodiment, the present invention also relates to use of a
compound of
2o fornula I for the preparation of a medicament for the treatment of
obsessive
compulsive disorder.
In a further embodiment, the present invention also relates to use of a
compound of
fornula I for the preparation of a medicament for the treatment of panic
disorder.
In a further embodiment, the present invention also relates to use of a
compound of
formula I for the preparation of a medicament for the treatment of panic
attaclcs.
In a further embodiment, the present invention also relates to use of a
compound of
3o fornula I for the preparation of a medicament for the treatment of specific
phobias.
In a further embodiment, the present invention also relates to use of a
compound of
fornula I for the preparation of a medicament for the treatment of social
phobia.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
18
In a further embodiment, the present invention also relates to use of a
compound of
formula I for the preparation of a medicament for the treatment of
agoraphobia.
A further aspect of the invention relates to a method for the treatment of a
disease or
disorder selected from the group consisting of an affective disorder, such as
depression, anxiety disorders including general anxiety disorder, social
anxiety
disorder, post traumatic stress disorder, obsessive compulsive disorder, panic
disorder,
panic attacl~s, specific phobias, social phobia and agoraphobia in a living
animal body,
including a human, comprising administering to a subj ect in need thereof a
to therapeutically effective amount of a compound of formula I.
In a further aspect, the present invention relates to a method of preparing a
compound
of formula I, comprising
a) Deprotection or cleavage fiom a polymer support of a compound with formula
II
R4
R5 R3
O \ ~ 2
~ o R
R'O- _N R1
.. \ Rs
9
R / R~
R$
II
wherein the dotted line, Rl-R~ are as previously described, and R is a tent-
butyl,
methyl, ethyl, allyl or benzyl group or R OCO is a solid supported carbamate
group;
or
b) Dehydrating and optionally simultaneously deprotecting a compound of
formula III
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
19
R4
R5 Rs
\ R2
R"\~~ OH ~ R~
R6
R9 / R7
R8
III
wherein Ri-R~ are as previously described, and R" is either a hydrogen atom or
a
carbamate group R~OCO wherein R is a tef°t-butyl, methyl, ethyl, allyl
or benzyl
group or R OCO is a solid supported carbamate group; or
c) Reduction of the double bond in a compound of formula IV
R4
R5 R3
/
\ R2
HN~ ~ R~
/ \ R6
9
R / R'
R$
IV
wherein R1-R~ are as previously described.
to
Pharmaceutical compositions
The compounds of the invention may be administered alone or in combination
with
pharmaceutically acceptable carriers or excipients, in either single or
multiple doses.
The pharmaceutical compositions according to the invention may be formulated
with
pharmaceutically acceptable carriers or diluents as well as any other lalown
adjuvants
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
and excipients in accordance with conventional techniques such as those
disclosed in
Remington: The Science and Practice of Pharmacy, 19 Edition, Gennaro, Ed.,
Mach
Publishing Co., Easton, PA, 1995.
The pharmaceutical compositions may be specifically formulated for
administration
by any suitable route such as the oral, rectal, nasal, pulmonary, topical
(including
buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal
and
parenteral (including subcutaneous, intramuscular, intrathecal, intravenous
and
intradermal) route, the oral route being preferred. It will be appreciated
that the
to prefeiTed route will depend on the general condition and age of the subject
to be
treated, the nature of the condition to be treated and the active ingredient
chosen.
Pharmaceutical compositions for oral administration include solid dosage forms
such
as capsules, tablets, dragees, pills, lozenges, powders and granules. Where
15 appropriate, they can be prepared with coatings such as enteric coatings or
they can be
formulated so as to provide controlled release of the active ingredient such
as
sustained or prolonged release according to methods well lmown in the art.
Liquid dosage forms for oral administration include solutions, emulsions,
2o suspensions, syrups and elixirs.
Phamnaceutical compositions for parenteral administration include sterile
aqueous and
nonaqueous injectable solutions, dispersions, suspensions or emulsions as well
as
sterile powders to be reconstituted in sterile injectable solutions or
dispersions prior to
use. Depot injectable formulations are also contemplated as being within the
scope of
the present invention.
Other suitable administration forms include suppositories, sprays, ointments,
cremes,
gels, inhalants, dermal patches, implants etc.
A typical oral dosage is in the range of from about 0.001 to about 100 mg/lcg
body
weight per day, preferably from about 0.01 to about 50 mg/lcg body weight per
day,
and more prefeiTed fiom about 0.05 to about 10 mg/lcg body weight per day
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
21
administered in one or more dosages such as 1 to 3 dosages. The exact dosage
will
depend upon the frequency and mode of administration, the sex, age, weight and
general condition of the subject treated, the nature and severity of the
condition
treated and any concomitant diseases to be treated and other factors evident
to those
skilled in the art.
The formulations may conveniently be presented in unit dosage form by methods
lmown to those spilled in the art. A typical unit dosage form for oral
administration
one or more times per day such as 1 to 3 times per day may contain from 0.01
to
l0 about 1000 mg, preferably from about 0.05 to about 500 mg, and more
preferred from
about 0.5 mg to about 200 mg.
For parenteral routes such as intravenous, intrathecal, intramuscular and
similar
administration, typically doses are in the order of about half the dose
employed for
15 oral administration.
The compounds of this invention are generally utilized as the free substance
or as a
pharmaceutically acceptable salt thereof. One example is an acid addition salt
of a
compound having the utility of a free base. When a compound of the formula (I)
2o contains a free base such salts are prepared in a conventional manner by
treating a
solution or suspension of a free base of the formula (1) with a chemical
equivalent of a
pharmaceutically acceptable acid. Representative examples are mentioned above.
For parenteral administration, solutions of the novel compounds of the formula
(I) in
25 sterile aqueous solution, aqueous propylene glycol, aqueous vitamin E or
sesame or
peanut oil may be employed. Such aqueous solutions should be suitably buffered
if
necessary and the liquid diluent first rendered isotonic with sufficient
saline or
glucose. The aqueous solutions are particularly suitable for intravenous,
intramuscular, subcutaneous and intraperitoneal administration. The sterile
aqueous
30 media employed are all readily available by standard teclmliques known to
those
slcilled in the art.
Suitable pharmaceutical carriers include inert solid diluents or fillers,
sterile aqueous
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
22
solution and various organic solvents. Examples of solid carriers are lactose,
terra
alba, sucrose, cyclodextrin, talc, gelatine, agar, pectin, acacia, magnesium
stearate,
stearic acid and lower all~yl ethers of cellulose. Examples of liquid carriers
are syrup,
peanut oil, olive oil, phospho lipids, fatty acids, fatty acid amines,
polyoxyethylenc
and water. similarly, the carrier or diluent may include any sustained release
material
l~nown in the art, such as glyccryl monostearate or glyceryl distearatc, alone
or mixed
with a wax. The pharmaceutical compositions formed by combining the novel
compounds of the formula (I) and the pharmaceutical acceptable carriers arc
then
readily administered in a variety of dosage forms suitable for the disclosed
routes of
1o administration. The formulations may conveniently be presented in unit
dosage form
by methods l~nown in the art of pharmacy.
Formulations of the present invention suitable for oral administration may be
presented as discrete units such as capsules or tablets, each containing a
predetermined amount of the active ingredient, and which may include a
suitable
excipient. Furthermore, the orally available formulations may be in the form
of a
powder or granules, a solution or suspension in an aqueous or non-aqueous
liquid, or
an oil-in-water or water-in-oil liquid emulsion.
2o If a solid carrier is used for oral administration, the preparation may be
a tablet, placed
in a hard gelatine capsule in powder or pellet fore or it can be in the forn
of a troche
or lozenge.
The amount of solid carrier will vary widely but will usually be from about 25
mg to
about 1 g.
If a liquid carrier is used, the preparation may be in the form of a syrup,
emulsion, soft
gelatine capsule or sterile injectable liquid such as an aqueous or non-
aqueous liquid
suspension or solution.
The compounds of the invention are prepared by the following general methods,
or as
described in the experimental section of this patent:
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
23
a) Deprotection or cleavage from a polymer support of a compound with formula
II
R4
R5 R3
R2
R~~ N
R
.. \ R6
9
R / R7
-, 8
II
wherein R1-R~ are as previously described, and R~ is a tef°t-butyl,
methyl, ethyl, allyl
or benzyl group or ROCO is a solid supported carbamate group, such as the Wang
resin-based carbasnate linlcer;
b) Dehydrating and optionally simultaneously deprotecting a compound of
formula
III
R4
R5 R3
\ R2
R,yN OH o R~
R6
Rg / R~
R$
io
wherein Rl-R~ are as previously described, and R" is either a hydrogen atom or
a
carbamate group R OCO wherein R~ is a tee°t-butyl, methyl, ethyl, allyl
or benzyl
group or R~OCO is a solid suppouted carbamate group, such as the Wang resin-
based
carbamate linlcer;
is
c) Reduction of the double bond in a compound of formula IV
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
24
R4
R5 Rs
\ R2
Hi~~ ~ R~
R6
R7
R8
IV
wherein R1-R~ are as previously described.
The deprotection according to method a) was performed by standard techniques,
knowledgeable to those skilled in the art and detailed in the textboolc Pf-
otective
GT°oups in Organic Synthesis T.W.Greene and P.G.M. Wuts, Wiley
Interscience,
(1991) ISBN 0471623016. The cleavage from a polymer support, such as from the
Wang resin-based carbamate linker, according to method a) may be performed
according to literature known procedures (Zaragoza Tetrah.edf°ora Lett.
1995, 36, 8677-
8678 and Conti et al. Tetnahed~on Lett. 1997, 38, 2915-2918).
Starting materials of formula II can be prepared by removal of the hydroxy
group of
compounds of formula III by a number of methods lmown to the chemist skilled
in the
art, e.g. by the use of triethylsilane in trifluoro acidic acid and boron
trifluoride diethyl
etherate (see Encyclopaedia of Reagefats foy° Organic Synthesis, vol 7,
Paquette, ed.;
John Wiley & Sons, Chichester, 1995, 5122-5123). Starting materials of formula
II,
which are piperidines, may be prepared by reduction of the double bond of the
corresponding tetrahydropyridines by standard hydrogenation procedures, such
as e.g.
catalytic hydrogenation at low pressure (< 3 atm.) in a Parr apparatus.
The dehydration reaction and optional simultaneous deprotection of a compound
of
fOr111ll1a III according to method b) was performed in a similar manner as
described in
Pahner et al J. Mcd. Glaern. 1997, 40, 1982-1989.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
Starting materials of formula III were prepared from the corresponding
properly
substituted 1-bromo-2-phenoxybenzenes of formula VI (wherein Rl-R~ are as
previously described, and G is a bromine or iodine atom) by metal-halogen
exchange
followed by addition of an appropriate electrophile of the formula V (wherein
R' is as
previously described) in a similar manner as described in Pahner et al. .~
Ihfed. C7ze~az.
1997, 40, 1982-1989.
R~
~5 ~3
W~ ~ \ ~2
O
G G
V
VI
The properly substituted 1-bromo-2-phenoxybenzenes were prepared by reaction
of
properly substituted phenols (the sodium salt of the phenols were prepared in
situ by
to the use of sodium hydride) with properly substituted 1-bromo-2-
fluorobenzenes in
dimethyl formamide (DMF) at elevated temperature. The diaryl ethers may also
be
prepared by various modifications of this method (see e.g. Schmittlinger et al
J.Org. Claena. 1993, 58, 3229-3230; Beugelmans et al Tetralzedf°ora
Lett. 1994, 35,
5649-5652; Sawyer et al J.Org.Claem. 1998, 63, 6338-6343), under Ullmann
15 conditions or via arylation of phenols with arylboronic acids (Evans et al
Tetrahedron
Lett 1998, 39, 2937-2940). Phenols and 1-bromo-2-fluorobenzenes are
commercially
available.
The reduction of the double bond according to method c) is generally performed
by
2o catalytic hydrogenation at low pressure (< 3 atm.) in a Parr apparatus.
Starting
materials of formula IV may be prepared from compounds of formula II by
deprotection as described above.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
26
Examples
Analytical LC-MS data were obtained on a PE Sciex API 150EX instrument
equipped
with IonSpray source and Shimadzu LC-8A/SLC-l0A LC system. Column: 30 X 4.6
mm Waters Symmmetry C18 column with 3.5 Nm particle size; Solventsystem: A =
water/trifluoroacetic acid (100:0.05) and B =
water/acetolutrile/trifluoroacetic acid
(5:95:0.03); Method: Linear gradient elution with 90% A to 100% B in 4 min and
with a flow rate of 2 111L/mlll. Purity was determined by integration of the
LTA (254
lnn) and ELSD trace. The retention times (RT) are expressed in minutes.
to Preparative LC-MS-purification was performed on the same instrument.
Column: 50
X 20 mln YMC ~DS-A with 5 ~.m particle size; Method: Linear gradient elution
with
80% A to 100% B in 7 min and with a flow rate of 22.7 mLlmin. Fraction
collection
was performed by split-flow MS detection.
Reactions carried out under , microwave conditions were performed in a
SmithSynthesizer from Personal Chemistry operating at 2450 MHz.
Preparation of intermediates
Preparation of substituted 2-bromo-(substituted-phenoxy)benzenes
1-B~omo-2-(2,4-clifr~ethyl plaeuox~)-benzene
A solution 2,4-dimethylphenol (2.4 g) in dry dimethyl formamide (DMF) (10 mL)
was added drop wise to a mixture of sodium hydride (1.0 g, 60% in mineral oil)
and
dry DMF (25 mL), and the resulting mixture was stirred at 100 °C for 30
min. To this
mixture was further added 1-bromo-2-fluorobenzene (3.5 g) in dry DMF (5 mL),
and
the resulting mixture was stirred at 150 °C for 6 hours. The mixture
was subsequently
poured onto an ice/water mixture, and the aqueous phase was extracted with
diethyl
ether. The combined organic phase was washed with 2N sodium hydroxide, dried
(MgS04), filtered and concentrated iya vacuo. The residue was purified by
flash
chromatography on silica gel (eluent: heptane) to give the cl-ude product (1.2
g, 70%
pure). The chide product was used in the next step without further
purification.
The following compounds were prepared in a similar manner:
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
27
1-BY'oTno-2-(4-chloa~opTzenoxy)-benzene
1-By~omo-2-(4 flzzoz~o-2-znetlzylphenoxy)-benzene
1-Bz-omo-2-(4 fluoy~ophenoxy)-beazzezze
1-Br-omo-2-(4-nzethylplzenoxy)-benzene
1-Bz-omo-2-(~-meth oxyplz.enoxy)-benzene
Preparation of alkyl 4-[B-(substituted-plieno~y)-substituted-phenyl]-4-
hydro~.ypiperidine-1-carbo~ylate~
tent-Butyl 4-~2-(2, 4-Dimethylplzenoxy)plaenylJ-4-hydf-oxv piperidine-1-
caf~boxylate
A solution of 1-bromo-2-(2,4-dimethyl-phenoxy)-benzene (0.7 g) in diy
tetrahydrofuran (THF) (3 mL) was added to a solution of nBuLi (1.6 M in
hexane, 2
mL) in dry THF (15 mL) at -78 °C. The resulting mixture was stirred at -
78 °C for 1
hour and subsequently added a solution of teat-butyl 4-oxo-piperidine-1-
carboxylate
(1.0 g) in dry THF (3 mL). The mixture was stirred for 16 hour at room
temperature,
and then poured onto a saturated solution of ammonium chloride. The aqueous
phase
was extracted with diethyl ether, and the combined organic phase was washed
with
water and brine, and subsequently dried (MgS04), filtered and concentrated in
vacuo.
2o The residue was purified by flash chromatography on silica gel (eluent:
heptane/ethyl
acetate 4:1) to give the crude product (0.55 g). The crude product was used in
the next
step without further purification.
The following compounds were prepared in a similar manner:
Ethyl 4-~2-(2, 4-Dinzethylplzenoxy)plzeaaylJ-4-lzyd~oxy pipes°idine-1-
cay~boxylate
Ethyl 4-~2-(4-Clzlorophenoxy)phenylJ-4-hydt~oxy piper-idine-1-ca~boxylate
tent-Butyl 4-~2-(4-Fluoa°o-2-methylphenoxy)ph.enylJ-4-hydy~oxy
pipe~idine-1-
caYboxylate
Ethyl 4-~2-(4-Flzzo~o-~-nzetlzylplzenoxy)plzenylJ-4-hydroxy pipey-idine-1-
car~boxylate
Et7zyl 4-~2-(~1-Fluoroplzenoxy)plTeraylJ-4-hydrroxy piperidine-1-cay~boxylccte
tea°t-Butyl 4-~2-(4-Metlzylplzeraoxy)phenylJ-4-lzydroxy pipezridirae-1-
carboxylate
Ethyl ~-~2-(4-Metlzylplzefzoxy)plzenylJ-4-Izydnoxy piperidine-1-cccr-boxylate
test-Butyl 4-~2-(4-Methoxyp7zenoxy)plaenylJ-4-lzyda°oxy piper~idine-1-
caa°boxylate
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
28
Preparation of ethyl 4-(2-methoxy-phenyl)-piperidine-1-carboxylate
To 26 mmol 4-(2-methoxyphenyl)-piperidine (Maybridge) in 100 ml chy
dichloromethane were added 28.6 mmol triethylamine and 78 mmol
ethylchlorofonnate at 0 °C. The solution was stirred at room
temperature overnight,
washed twice with 0.5 M HCl (125 ml) then dried over MgSO4 and evaporated. The
product was sufficiently pure to be used in the following steps.
Preparation of ethyl 4-(2-hydroxy-phenyl)-piperidine-1-carboxylate
To 24 mmol ethyl 4-(2-methoxy-phenyl)-piperidine-1-carboxylate in 150 ml dry
to dichloromethane were added 48 mmol BBr3 at 0 °C. The solution was
stirred at room
temperature overnight, washed twice with 0.5 M HCl (125 mL) then dried over
MgS04 and evaporated. The product was sufficiently pure to be used in the
following
steps.
15 Compounds of the invention:
Preparation of 4-[2-(substituted-phenoxy)-substituted-phenyl]-1,2,3,6-
tetrahydropyridines
20 1 a, 4-~2-(2, 4-Dim.etlaylplaefaoxy)plaehyl -1, 2, 3, 6-
teti°ahydf°opyridifze
A mixture of test-butyl 4-[2-(2,4-dimethylphenoxy)phenyl]-4-hydroxy-piperidine-
1-
carboxylate (0.5 g) and a mixture of acidic acid and conc. hydrochloride acid
(3:1)
was boiled under reflux for 16 hours. The mixture was cooled, poured into
allcaline
25 water and extracted with ethyl acetate. The combined organic phase was
dried
(MgS04), filtered and concentrated ih vacuo. The residue was purified by flash
chromatography on silica gel (eluent: ethyl acetate/methanol/triethylamine
8:2:1) to
give the target compound (11 mg, 3%). LC/MS (m/z) 280 (MH+); RT = 2.16; purity
(W, ELSD): 85%, 97%.
The following compounds were prepared in a similar mariner:
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
29
lb, 4-j2-(4-Chlof~ophenoxy)pheyaylJ-1,2,3,6-tetf~ahyd~~opy~idiyae
From ethyl 4-[2-(4-chlorophenoxy)phenyl]-4-hydroxy-piperidine-1-carboxylate.
LC/MS (m/z) 286 (MH+); RT = 2.10; purity (UV, ELSD): 85%, 95%; yield: 33 mg
(6°/~).
lc, 4-j2-(~-Flzco~o-2-rnetlaylphenoxy)plze~aylJ-1,2,3,6-tetr~ahyda~opy~idirte
From ter°t-butyl 4-[2-(4-fluoro-2-methylphenoxy)phenyl]-4-hydroxy-
piperidine-1-
carboxylate. LC/MS (m/z) 284 (MH+); RT = 2.08; purity (UV, ELSD): 97%, 99%;
yield: 100 mg (21 %).
to
ld, 4-j2-(4-Fluo~ophefaoxy)plaenylJ-1,2,3,6-tet~alzyd~opyridiyae
From ethyl 4-[2-(4-fluorophenoxy)phenyl]-4-hydroxy-piperidine-1-carboxylate.
LC/MS (m/z) 270 (MH+); RT = 1.93; purity (UV, ELSD): 87%, 97%; yield: 45 mg
( 11 %).
1 e, 4- j2-(4-Methylphenoxy)phehylJ-1, 2, 3, 6-tet~~ahyd~opy~~idih.e
From test-butyl 4-[2-(4-methylphenoxy)phenyl]-4-hydroxy-piperidine-1-
carboxylate.
LC/MS (m/z) 266 (MH+); RT = 2.04; purity (LJV, ELSD): 98%, 99%; yield: 250 mg
(24%).
lf, 4-j2-(4-Metlaoxyp7xenoxy)pheraylJ-1,2,3,6-tetr~ahyd~opyf~idine
From test-butyl 4-[2-(4-methoxyphenoxy)phenyl]-4-hydroxy-piperidine-1-
carboxylate. LC/MS (m/z) 282 (MH+); RT = 1.95; purity (LTV, ELSD): 79%, 99%;
yield: 14.7 mg (19%).
Preparation of 4-[2-(substituted-phenoxy)-substituted-phenyl)piperidines
Method A
2a, 4-j2-(2,4-l~imetlzylpheraoxy)plaeraylJpipef~idiyi.e
A mixture of ethyl 4-[2-(2,4-dimethylphenoxy)phenyl]-4-hydroxy-piperidine-1-
3o carboxylate (0.6 g), dichloromethane (25 mL), triethylsilane (1 mL),
trifluoro acidic
acid (0.1 mL) and boron trifluoride diethyl etherate (0.2 mL) was stirred at
room
temperature for 16 hours. The resulting mixture was poured onto all~aline
water and
subsequently extracted with ethyl acetate. The combined organic phase was
dried
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
(MgS04), filtered and concentrated in vacuo (0.4 g). The residue was dissolved
in a
mixture of conc. hydrochloric acid and acidic acid (1:3) (25 mL) and boiled
under
reflux for 16 hours. The mixture was poured onto all~aline water and
subsequently
extracted with ethyl acetate. The combined organic phase was dried (MgSO4),
filtered
5 and concentrated iaa vacuo. The residue was purified by flash chromatography
on
silica gel (eluent: ethyl acetate/methanol/triethylamine 8:2:2) to give the
target
compound (10.6 mg, 3%). LC/MS (m/z) 282 (MH+); RT = 2.22; purity (UV, ELSD):
G7%, 83%.
to The following compounds were prepared in a similar manner:
2b, 4-~2-(4-Chlo~ophenoxy)phenylJpiperidihe
From ethyl 4-[2-(4-chlorophenoxy)phenyl]-4-hydroxy-piperidine-1-carboxylate.
LC/MS (m/z) 288 (MH+); RT = 2.1; purity (UV, ELSD): 96%, 97%; yield: 41 mg
is (7%).
2c, 4-~2-(4-Fluo~o-2-methylpheyaoxy)plaenylJpipe~~idis~e
From ethyl 4-[2-(4-fluoro-2-methylphenoxy)phenyl]-4-hydroxy-piperidine-1-
carboxylate. LC/MS (m/z) 286 (MH+); RT = 2.1; purity (UV, ELSD): 89%, 99%;
2o yield: 51 mg (8%).
2d, 4-~2-(4-FluoYOphenoxy)plaerayl jpiperidihe
From ethyl 4-[2-(4-fluorophenoxy)phenyl]-4-hydroxy-piperidine-1-carboxylate.
LC/MS (m/z) 272 (MH+); RT = 1.97; purity (UV, ELSD): 91 %, 99%; yield: 7 mg
25 (5%).
2e, 4-~2-(4-Metlaylplaeyaoxy)phenyl piperidih.e
From ethyl 4-[2-(4-methylphenoxy)phenyl]-4-hydroxy-piperidine-1-carboxylate.
LC/MS (m/z) 268 (MH+); RT = 2.12; purity (UV, ELSD): 88%, 93%; yield: 8 mg
3o (1%).
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
31
Method B
Ethyl 4-(2-Hydroxy-phenyl)-piperidine-1-carboxylate (0.1 mmol ) was combined
in
0.5 mL 1-methyl-pyrrolidin-2-one with 0.12 mmol of an appropriate aryl bromide
or
iodide. CuI catalyst (0.037 rrunol) was added and the vial sealed before it
was heated
for 1 hour in a microwave oven at 220 °C. The solvent was removed from
the
samples, and a solution of I~~H in water (3.7 mmol), dioxane and ethanol
(99,9%)
were added, and the mixture was heated at 130 °C for 1 hour in the
microwave oven.
The samples were then added water and solid IVaCI and then subsequently
extracted
to with ethyl acetate. The organic phase was evaporated and the crude product
purified
by preparative LC-MS. The isolated products were subjected to SCX columns and
the
flee amines submitted for testing as DMSO solutions. The following compounds
were
prepared by this method and measured molecular mass, measured HPLC-retention
time (RT, min) and LTV- and ELSD-purities (%) is described in Table 1.
3a, 4-[2-(4-Chloro-2- methyl-phenoxy)-phenyl]-piperidine
3b, 4-[2-(3-Chloro-2- methyl-phenoxy)-phenyl]-piperidine
3c, 4-[2-(2-Chloro-4-methyl-phenoxy)-phenyl]-piperidine
3d, 4-[2-(2,4-Dichloro-phenoxy)-phenyl]-piperidine
3e, 4-[2-(Benzo[1,3]dioxol-5-yloxy)-phenyl]-piperidine
3f, 4-[2-(4-Methoxy-2-methyl-phenoxy)-phenyl]-piperidine
3g, 4-[2-(3,4-Dichloro-phenoxy)-phenyl]-piperidine
3h, 4-[2-(3,4-Dimethyl-phenoxy)-phenyl]-piperidine
3i, 4-[2-(2,3,4,5-Tetramethyl-phenoxy)-phenyl]-piperidine
3j, 4-[2-(4-Trifluoromethyl-phenoxy)-phenyl]-piperidine
3k, 4-[2-(4-Methoxy-phenoxy)-phenyl]-piperidine
31, 4-[2-(2-Chloro-4-methoxy-phenoxy)-phenyl]-piperidine
3m, 4-[2-(3,4-Dimethoxy-phenoxy)-phenyl]-piperidine
3n, 4-[2-(4-Chloro-3-trifluoromethyl-phenoxy)-phenyl]-piperidine
Table 1. Measured molecular mass, measured HPLC-retention time (RT, min) and
UV- and ELSD-purities (%).
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
32
UV-purity ELSDpurity
compound M+H RT min.
(%) (%)
3a 2,1 88.6% 99.1%
3b 302,1 2,19 92,43 92,56
3c 302,1 2,18 88,64 94,63
3d 321,9 2,23 87,08 92,55
3e 297,9 1,90 92,96 89,84.
3f 298,3 2,02 97,69 96,69
3~ 322,1 2,21 97,55 85,38
3h 282,2 2,18 96,25 91,84
3i 310,2 2,42 83,51 95,12
3j 321,9 2,19 96,8 99,05
31c 284,3 1,92 99,14 97,5
31 318,1 2,07 73,27 85,01
3m 314,1 1,78 97,94 99,05
3n 356,2 2,34 98,68 88,11
Measurements of [3H]-5-HT uptake into rat cortical synaptosomes.
Whole brains from male Wistar rats (125-225 g), excluding cerebellum, are
homogenized in 0.32 M sucrose supplemented with 1mM nialamid with a
glass/teflon
homogenizes. The homogenate is centrifuged at 600 x g for 10 min at 4
°C. The pellet is
discarded and the supernatant is centrifuged at 20.000 x g for 55 min. The
final pellet is
homogenized (20 sec) in this assay buffer (0.5 mg original tissue/well). Test
compounds
(or buffer) and 10 mM [3H]-5-HT are added to 96 well plates and shalcen
briefly.
to Composition of assay buffer: 123 mM NaCI, 4.82 mM KCI, 0.973 mM CaCl2, 1.12
mM
MgS04, 12.66 mM Na2HP04, 2.97 mM NaH2P04, 0.162 mM EDTA, 10 mM glucose
and 1 mM ascorbic acid. Buffer is oxygenated with 95% Oz/5% COZ for 10 min at
37 °C
and pH is adjusted 7.4. The incubation is started by adding tissue to a final
assay volume
of 0.2 mL. After 15 min incubation with radioligand at 37 °C, samples
are filtered
is directly on Unifilter GF/C glass fiber filters (soaked for 1 hour in 0.1%
polyethylenimine) under vacuum and immediately washed with 3 x 0.2 ml assay
buffer.
Non-specific uptalce is determined using citalopram (10 ~.M final
concentration).
Citalopram is included as reference in all experiments as dose-response curve.
CA 02521030 2005-09-30
WO 2004/087155 PCT/DK2004/000241
33
Measurements of [3H]noradrenaline uptake into rat cortical synaptosomes.
Fresh cortex from male Wistar rats (125-225 g) are homogenized in 0.4M sucrose
with a glass/teflon homogenizer. The homogenate is centrifuged at 600 x g for
10 min
at 4 °C. The pellet is discarded and the supernatant is centrifuged at
20.000 x g for 55
min. The final pellet is homogenized (20 sec) in this assay buffer (6 mg
original
tissuchnL = 4 mg/well). Test compounds (or buffer) and 10 nM [3H]-
noradrenaline
are added to deep 96 well plates and shapen briefly. Composition of assay
buffer: 123
mM NaCI, 4.82 mM KCI, 0.973 mM CaCl2, 1.12 mM MgS~4, 12.66 mM Na2HP~4,
2.97 mM NaH2P~4, 0.162 mM EDTA, 10 mM glucose and 1 mM ascorbic acid.
to )3uffer is oxygenated with 95% 02/5% C02 for 10 min at 37 °C and pH
is adjusted 7.4.
The incubation is started by adding tissue to a final assay volume of 1 ml.
After 15
min incubation with radioligand at 37 °C, samples are filtered directly
on Unifilter
GF/C glass fiber filters (soaped for 1 hour in 0.1 % polyethylenimine) under
vacuum
and immediately washed with 3 x 1 mL assay buffer. Non-specific uptape is
15 determined using talsupram (10 ~.M final concentration). Duloxetine is
included as
reference in all experiments as dose-response curve.
Results of the experiments showed that the tested compounds of the invention
inhibit
the norepinephrine and serotonine reuptalce with ICso below 200 nM.