Note: Descriptions are shown in the official language in which they were submitted.
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
Resolution of a-(Phenoxy)phenylacetic Acid Derivatives
FIELD OF THE INVENTION
[0001] The present invention relates to an enantioselective resolution process
for the
separation of a-(phenoxy)phenylacetic acids from its enantiomeric mixture.
BACKGROUND OF THE INVENTION
[0002] Esters and amides derivatives of a-(phenoxy)phenylacetic acids, such as
halofenate,
are chiral compounds and are useful in ameliorating a variety of physiological
conditions,
including conditions associated with blood lipid deposition, e.g., Type II
diabetes and
hyperlipidema. See, for example, U.S. Patent Nos. 3,517,050 and 6,262,118. a-
(phenoxy)phenylacetic acids contain a single chiral center at an
asymmetrically substituted
carbon atom alpha to the carbonyl carbon atom, and therefore exist in two
enantiomeric
forms.
[0003] Cytochrome P450 2C9 is an enzyme known to play a significant role in
the
metabolism of specific drugs. It is known to one skilled in the art that
changes in drug
metabolism mediated by inhibition of cytochrome P450 enzymes has a high
potential to
precipitate significant adverse effects in patients. It is also known that a
racemic a-
(phenoxy)phenylacetic acid, e.g., halofenic acid, inhibits cytochrome P450
2C9. See, for
example, U.S. patent No. 6,262,118. Thus, administration of a racemic a-
(phenoxy)phenyl-
acetic acid, such as halofenic acid or its derivatives, can lead to a variety
of drug interaction
problems with other drugs, including anticoagulants, anti-inflammatory agents
and other
drugs that are metabolized by this enzyme. It has been found that the (-)-
enantiomer of
halofenic acid is about twenty-fold less active in its ability to inhibit
cytochrome P450 2C9
compared to the (+)-enantiomer. Id. Thus, it is desirable to administer the (-
)-enantiomer of
halofenic acid or its derivatives which is substantially free of the (+)-
enantiomer to reduce the
possibility of drug interactions.
[0004] Therefore, there is a need for an efficient process for producing a
product enriched
in a desired enantiomer of a a-(phenoxy)phenylacidic acid, e.g., (-)-halofenic
acid.
SUMMARY OF THE INVENTION
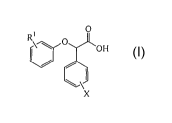
[0005] One aspect of the present invention provides a method for producing an
enantiomerically enriched a-(phenoxy)phenylacetic acid compound of the
formula:
1
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
0
RI II
OH
I
wherein
RI is alkyl or haloalkyl, and
X is halide;
from an enantiomeric mixture of the a-(phenoxy)phenylacetic acid compound
comprising a
first and a second enantiomers. In one particular embodiment, the enantiomeric
mixture is a
racemic mixture.
[0006] Methods of the present invention includes:
(a) producing a solution comprising a solid enantiomerically
enriched
acid-base salt of the first enantiomer by contacting the enantiomeric mixture
of the a-
(phenoxy)phenylacetic acid compound with less than 0.5 molar equivalents of an
enantiomerically enriched chiral amine compound under conditions sufficient to
produce the
ratio of the amount of free first enantiomer to the amount of the free second
enantiomer in the
solution is about 1 to 3; and
(b) separating the solid acid-base salt of the first enantiomer from the
solution at a temperature where the concentration of an acid-base salt of the
second
enantiomer of the a-(phenoxy)phenylacetic acid compound is near or below its
saturation
point.
[0007] At least a portion of the second enantiomer can be converted to the
first enantiomer,
e.g., racemized, by contacting the second enantiomer with a base. The
resulting enatiomeric
mixture can then be recycled and subjected to a similar enantiomeric
enrichment process to
increase the yield of the first enantiomer acid-base salt.
[0008] In one particular embodiment, the chiral amine compound is of the
formula:
OR2 OR3
Ar R4
NR5R6
wherein
each of R2 and R3 is independently hydrogen or alkyl; or R2 and R3 together
with atoms to which they are attached to form a heterocyclic ring moiety;
R4 is hydrogen or alkyl;
2
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
each of R5 and R6 is independently hydrogen or alkyl, or one of R5 or R6 is an
amine protecting group; and
Ar is aryl.
BRIEF DESCRIPTION OF THE DRAWINGS
[0009] Figure 1 is a graph showing the solubility profiles of (-)- and (+)-
CPTA/CAF D-
Base salts in 2-propanol.
[0010] Figure 2 shows results of a process for resolving a racemic mixture of
CPTA using
CAF D-Base under a variety of crystallization conditions.
[0011] Figure 3 is a graph showing the solubility of(-)- and (+)-CPTAJCAF D-
Base salts
in pure isopropanol and a solution comprising a mixture of isopropanol and
CPTA (11%).
[0012] Figure 4 is a graph showing the composition of a mixture with a various
amount of
each components.
[0013] Figure 5 is a graph showing a (-/+)-salt saturation profile for
crystallization and
heating.
[0014] Figure 6 is a table showing comparison of the model prediction to
experimental
results for entry 4 of Figure 2.
[0015] Figure 7 is a graph showing the amount of (+)-salt formation as a
function of the
amount of CAF D-Base added.
[0016] Figure 8 is a graphic representation of experimental data for the
resolution shown in
entry 11 of Figure 2.
[0017] Figure 9 shows the actual and calculated amount of CPTA in mother
liquor and a
graphic comparison of a calculated percentage of (+)-CPTA salt with the
experimental data.
[0018] Figure 10A shows tables showing experimental data and a solubility
model
calculation for Figure 7 (i.e., entry 13 of Figure 2).
[0019] Figure 10B is a table showing experimental data and a solubility model
calculation
for entry 4 of Figure 2 at 28.3 C.
[0020] Figure 11 is a graph showing solubility of racemic CPTA at various
temperatures in
1,2-dichloroethane.
3
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0021] Figure 12 is a graph showing solubility of racemic CPTA at various
temperatures in
heptane.
[0022] Figure 13 is a table of results in Example 24 showing yield of CPTA
resolution
using CAF D-Base under variety of crystallization conditions.
[0023] Figure 14 shows a cooling profiles for the resolution crystallization
of various
entries in Figure 2.
[0024] Figure 15 is a table showing the amount of (-)-halofenate yield from (-
)-CPTA salt
in Example 26.
[0025] Figure 16 is a graph showing solubility of racemic CPTA sodium salt at
various
temperatures in water.
[0026] Figure 17 is a graph showing CPTA racemization profile at various pH
during
hydrolysis of (-)-halofenate.
[0027] Figure 18 is a table showing the results of CAF D-Base recovery at
various pH as
described in Example 30.
=
[0028] Figure 19 is experimental results of solubility determination of
racemic CPTA in
1,2-dichloroethane and heptane as determined in Example 33.
[0029] Figure 20 is experimental results of solubility determination of
racemic CPTA
sodium salt in water as determined in Example 41.
[0030] Figure 21 is experimental results of basic hydrolysis of (+)-halofenate
as determined
in Example 42.
DETAILED DESCRIPTION
I. Definitions
[0031] "Alkyl" refers to straight or branched aliphatic hydrocarbons chain
groups of one to
ten carbon atoms, preferably one to six carbon atoms, and more preferably one
to four carbon
atoms. Exemplary alkyl groups include, but are not limited to, methyl, ethyl,
n-propyl,
2-propyl, tert-butyl, pentyl, and the like.
[0032] "Aryl" refers to a monovalent monocyclic or bicyclic aromatic
hydrocarbon moiety
of 6 to 10 carbon ring atoms. Unless stated or indicated otherwise, an aryl
group can be
substituted with one or more substituents, preferably one, two, or three
substituents, and more
4
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
preferably one or two substituents selected from alkyl, haloalkyl, nitro, and
halo. More
specifically the term aryl includes, but is not limited to, phenyl, 1-
naphthyl, and 2-naphthyl,
and the like, each of which is optionally substituted with one or more
substituent(s) discussed
above.
[0033] "CAF D base" refers to chloramphenicol D base, i.e., D-threo+)-2-amino-
1-
(nitropheny1)-1,3-propanediol.
[0034] "Chiral" or "chiral center" refers to a carbon atom having four
different substituents.
However, the ultimate criterion of chirality is non-superimposability of
mirror images.
[0035] The terms "CPTA" and "halofenic acid" are used interchangeably herein
and refer
to (4-chlorophenyl)(3-trifluoromethylphenoxy)acetic acid.
[0036] "Enantiomeric mixture" means a chiral compound having a mixture of
enantiomers,
including a racemic mixture. Preferably, enantiomeric mixture refers to a
chiral compound
having a substantially equal amounts of each enantiomers. More preferably,
enantiomeric
mixture refers to a racemic mixture where each enantiomer is present in an
equal amount.
[0037] "Enantiomerically enriched" refers to a composition where one
enantiomer is
present in a higher amount than prior to being subjected to a separation
process.
[0038] "Enantiomeric excess" or "%cc" refers to the amount of difference
between the first
enantiomer and the second enantiomer. Enantiomeric excess is defined by the
equation: %ee
= (% of the first enantiomer) - (% of the second enantiomer). Thus, if a
composition
comprises 98% of the first enantiomer and 2% of the second enantiomer, the
enantiomeric
excess of the first enantiomer is 98%-2% or 96%.
[0039] The terms "halide" and "halo" are used interchangeably herein and refer
to halogen,
which includes F, Cl, Br, and I, as well as pseudohalides, such as ¨CN and
¨SCN.
[0040] "Haloalkyl" refers to alkyl group as defined herein in which one or
more hydrogen
atoms have been replaced with halogens, including perhaloalkyls, such as
trifluoromethyl.
[0041] "Halo fenate" refers to 2-acetamidoethyl 4-chlorophenyl-(3-
trifluoromethyl-
phenoxy)acetate (i.e., 4-chloro-a-(3-(trifluoromethyl)phenoxy)benzeneacetic
acid, 2-
(acetylamino)ethyl ester or (4-chlorophenyl)(3-trifluoromethylphenoxy)acetic
acid), 2-
,
(acetylamino)ethyl ester).
5
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0042] "Heteroalkyl" means a branched or unbranched acyclic saturated alkyl
moiety
containing one or more heteroatoms or one or more heteroatom-containing
substituents,
where the heteroatom is 0, N, or S. Exemplary heteroatom-containing
substituents include
=0, -0Ra, -C(=0)Ra, -NRaRb, -N(Ra)C(=0)Rb, -C(=0)NRaRb and -S(0)nRa (where n
is an
integer from 0 to 2). Each of Ra and RI) is independently hydrogen, alkyl,
haloalkyl, aryl, or
aralkyl. Representative examples of heteroalkyl include, for example, N-acetyl
2-aminoethyl
(i.e., ¨CH2CH2NHC(=0)CH3).
[0043] The terms "heterocyclyl" and "heterocyclic ring" are used
interchangeably and refer
to a non-aromatic cyclic moiety of 3 to 8 ring atoms in which one, two, or
three ring atoms
are heteroatoms selected from N, 0, or S(0)õ (where n is an integer from 0 to
2), the
remaining ring atoms being C, where one or two C atoms may optionally be
replaced by a
carbonyl group. Unless stated or indicated otherwise, the heterocyclyl ring
can be optionally
substituted independently with one, two, or three substituents selected from
halogen, alkyl,
aryl, hydroxy, amino, or alkoxy. More specifically the term heterocyclyl
includes, but is not
limited to, 1,3-dioxane and its derivatives, and the like.
[0044] "Leaving group" has the meaning conventionally associated with it in
synthetic
organic chemistry, i.e., an atom or a group capable of being displaced by a
nucleophile and
includes halo (such as chloro, bromo, and iodo), alkanesulfonyloxy,
arenesulfonyloxy,
alkylcarbonyloxy (e.g., acetoxy), arylcarbonyloxy, mesyloxy, tosyloxy,
trifluoromethanesulfonyloxy, aryloxy (e.g., 2,4-dinitrophenoxy), methoxy, N,0-
dimethylhydroxylamino, and the like.
[0045] The term "metal" includes Group I, II, and transition metals as well as
main grouop
metals, such as B and Si.
[0046] "Optical purity" refers to the amount of a particular enantiomer
present in the
composition. For example, if a composition comprises 98% of the first
enantiomer and 2%
of the second enantiomer, the optical purity of the first enantiomer is 98%.
[0047] Unless otherwise stated, the term "phenyl" refers to an optionally
substituted phenyl
group. Suitable phenyl substituents are same as those described in the
definition of "aryl."
Similarly, the term "phenoxy" refers to a moiety of the formula ¨0Ara, wherein
Ara is phenyl
as defined herein. Thus, the term "a-(phenoxy)phenylacetic acid" refers to
acetic acid that is
substituted on the 2-position with an optionally substituted phenyl and
optionally substituted
phenoxy moieties.
6
CA 02529774 2012-02-03
[00481 "Protecting group" refers to a moiety that when attached to a reactive
group in a
molecule masks, reduces or prevents that reactivity. Examples of protecting
groups can be
found in T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis,
3"I edition,
John Wiley & Sons, New York, 1999, and Harrison and Harrison et al.,
Compendium of
Synthetic Organic Methods, Vols. 1-8 (John Wiley and Sons, 1971-1996).
Representative hydroxy protecting groups
include acyl groups, benzyl and trityl ethers, tetrahydropyranyl ethers,
trialkylsily1 ethers and
ally1 ethers. Representative amino protecting groups include, formyl, acetyl,
trifluoroacetyl,
benzyl, benzyloxycarbonyl (CBZ), tert-butoxycarbonyl (Boe), trimethyl silyl
(TMS), 2-
trimethylsilyl-ethanesulfonyl (SES), trityl and substituted trityl groups,
allyloxycarbonyl, 9-
fluorenylmethyloxycarbonyl (FMOC), nitro-veratryloxycarbonyl (NVOC), and the
like.
[00491 The term "rate" when referring to a formation of a salt refers to
kinetic and/or
thermodynamic rates.
[00501 As used herein, the term "treating", "contacting" or "reacting" refers
to adding or
mixing two or more reagents under appropriate conditions to produce the
indicated and/or the
desired product. It should be appreciated that the reaction which produces the
indicated
and/or the desired product may not necessarily result directly from the
combination of two
reagents which were initially added, i.e., there may be one or more
intermediates which are
produced in the mixture which ultimately leads to the formation of the
indicated and/or the
desired product.
[0051] As used herein, the terms "those defined above" and "those defined
herein" when
referring to a variable incorporates by reference the broad definition of the
variable as well as
preferred, more preferred and most preferred definitions, if any.
[00521 Many organic compounds exist in optically active forms, i.e., they have
the ability
to rotate the plane of plane-polarized light. In describing an optically
active compound, the
prefixes R and S are used to denote the absolute configuration of the molecule
about its chiral
center(s). The prefixes "d" and "1" or (+) and (-) are employed to designate
the sign of
rotation of plane-polarized light by the compound, with (-) or (1) meaning
that the compound
is "levorotatory" and with (+) or (d) is meaning that the compound is
"dextrorotatory". There
is no correlation between nomenclature for the absolute stereochemistry and
for the rotation
of an enantiomer. For a given chemical structure, these compounds, called
"stereoisomers,"
are identical except that they are mirror images of one another. A specific
stereoisomer can
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
also be referred to as an "enantiomer," and a mixture of such isomers is often
called an
"enantiomeric" or "racemic" mixture. See, e.g., Streitwiesser, A. & Heathcock,
C. H.,
INTRODUCTION TO ORGANIC CHEMISTRY, 2nd Edition, Chapter 7 (MacMillan
Publishing Co., U.S.A. 1981).
[0053] The terms "substantially free of its (+)-stereoisomer," "substantially
free of its (+)-
enantiomer," are used interchangeably herein and mean that the compositions
contain a
substantially greater proportion of the (-)-isomer in relation to the (+)-
isomer. In a preferred
embodiment, the term "substantially free of its (+) stereoisomer" means that
the composition
is at least 90% by weight of the (-)-isomer and 10% by weight or less of the
(+)-isomer. In a
more preferred embodiment, the term "substantially free of its (+)-
stereoisomer" means that
the composition contains at least 99% by weight of the (-)-isomer and 1% by
weight or less
of the (+)-isomer. In the most preferred embodiment, the term "substantially
free of its (+)-
stereoisomer" means that the composition contains greater than 99% by weight
of the (-)-
isomer. These percentages are based upon the total amount of isomers in the
composition.
II. Introduction
[0054] While chiral synthesis has made an extensive progress in recent years,
resolution of
racemates still remains the method of choice in industrial process for
preparation of optically
active, i.e., chiral, compounds. Typically, a chiral compound is synthesized
in a racemic
4µ. form and the final product is resolved to yield an enantiomerically
enriched compound.
[0055] This process of resolving the final product is particularly useful in a
large scale
preparation of pharmaceutically active chiral compounds. Although enantiomers
of a chiral
compound have exact same chemical bonds, the spatial orientation of atoms in
enantiomers is
different. Thus, one enantiomer of a chiral drug often exerts desired activity
with a
significantly less side-effect(s) than the other enantiomer. While such
relationship between
chirality of an optically active drug and its side-effect(s) has been known
for sometime, many
chiral drugs are still administered in a racemic form.
[0056] Diastereomeric crystallization is widely used on industrial scale. The
theoretical
once-through yield of a resolution via diastereomer crystallization is 50
percent. Typically,
however, more than one re-crystallization process is necessary in order to
produce a
composition that is of a sufficient optical purity.
[0057] The present invention provides a method for enantiomerically enriching
an
enantiomeric mixture, preferably a racemic mixture, of a-(phenoxy)phenylacetic
acid
8
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
compound, e.g., halofenic acid. Preferably, methods of the present invention
provides a solid
acid-base salt of the (-)-enantiomer of a-(phenoxy)phenylacetic acid compound.
In this
manner, the (-)-enantiomer can be readily separated from the solution.
[0058] The carboxylic acid group of the enantiomerically enriched a-
(phenoxy)phenylacetic acid can then be activated by a carboxylic acid
activation group to
produce an activated a-(phenoxy)phenylacetic acid, which can be reacted with
an alcohol, an
amine, a thiol, or other nucleophilic compounds to produce an enantiomerically
enriched a-
(phenoxy)phenylacetic acid esters, amides, thioesters, or other derivatives,
respectively.
Thus, enantiomerically enriched a-(phenoxy)phenylacetic acid compounds
produced using
methods of the present invention are useful in producing a-
(phenoxy)phenylacetic acid
derivatives such as those disclosed in U.S. Patent No. 3,517,050. In
particular, methods of
the present invention are useful in producing (-)-halofenate.
III. Enantioselective Crystallization
[0059] As noted above, most enantioselective crystallization processes require
more than
one re-crystallization process in order to produce a composition that is of a
sufficient optical
purity. However, present inventors have found that under certain conditions
disclosed herein,
a-(phenoxy)phenylacetic acid compound of a sufficient optical purity can be
produced by a
single crystallization process. Thus, in one aspect, methods of the present
invention are
based on the surprising and unexpected discovery by the present inventors that
an
enantiomeric mixture of a a-(phenoxy)phenylacetic acid compound can be
enantiomerically
enriched using a chiral amine compound. In particular, methods of the present
invention
provide a desired enantiomer of the a-(phenoxy)phenylacetic acid compound in
optical purity
of at least about 90%, preferably at least about 95%, more preferably at least
about 97%, and
most preferably at least about 98%.
[0060] In one embodiment, methods of the present invention provide
enantiomeric
enrichment of an enantiomeric mixture, preferably a racemic mixture, of a a-
(phenoxy)phenylacetic acid compound of the formula:
RA'
, OH
X
9
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
wherein RI is alkyl or haloalkyl, and X is halide. The process generally
involves forming a
solid enantiomerically enriched acid-base salt of the a-(phenoxy)phenylacetic
acid compound
uing a chiral amine compound.
[0061] In particular, methods of the present invention are directed to the
resolution of a-
(phenoxy)phenylacetate acid, e.g., halofenic acid (where RI is CF3 and X is
Cl), of the
formula:
Ri * 0
OH
X
II
wherein RI is alkyl or haloalkyl, and X is halide.
[0062] In one particular embodiment, methods of the present invention are
directed to the
resolution of a-(phenoxy)phenylacetate acid of Formula I or, preferably of
Formula II, where
X is chloro.
[0063] Yet in another embodiment, methods of the present invention are
directed to the
resolution of a-(phenoxy)phenylacetic acid of Formula I or, preferably,
Formula II, where R1
,15 is haloalkyl, preferably trifluoromethyl.
[0064] In one particular embodiment, a-(phenoxy)phenylacetic acid is
crystallized using a
chiral base. A wide variety of chiral bases can be used, including those
disclosed in the
Examples section below. Preferably, the chiral base used results in a solid
acid-base salt of
the (-)-enantiomer of a-(phenoxy)phenylacetic acid. In this manner, the (-)-
enantiomer is
readily separated from the solution, for example, by filtration. In one
particular embodiment,
the chiral base is an amine compound of the formula:
OR2 OR3
ArR4
NR5R6
III
wherein each of R2 and R3 is independently hydrogen, alkyl or a hydroxy
protecting group; or
R2 and R3 together with atoms to which they are attached to form a
heterocyclic ring moiety;
R4 is hydrogen or alkyl; each of R5 and R6 is independently hydrogen or alkyl,
or one of R5 or
R6 is an amine protecting group; and Ar is aryl.
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0065] In one particular embodiment, R2 and R3 together along with oxygen
atoms to
which they are attached to form 1,3-dioxane, a substituted 1,3-dioxane (e.g.,
dialkyl
substistuted 1,3-dioxane, such as 5,5-dimethy1-1,3-dioxane), or a derivative
thereof.
[0066] In another embodiment, R2 and R3 are hydrogen.
[0067] Yet in another embodiment, R4 is hydrogen.
[0068] In still another embodiment, Ar is a substituted aryl. A particularly
preferred Ar
moiety is optionally substituted phenyl. An especially preferred Ar moiety is
4-nitrophenyl.
[0069] Still further, combinations of the preferred groups described above
will form other
preferred embodiments. For example, one particularly preferred chiral base is
an amine
compound of Formula III above, wherein R2, R3, R4, R5 and R6 are hydrogen; and
Ar is 4-
nitrophenyl. And a particularly preferred a-(phenoxy)phenylacetic acid
compound is of
Formula II above, wherein RI is trifluoromethyl and X is chloro. In this
manner, a wide
variety of preferred chiral bases and a-(phenoxy)phenylacetic acid compounds
are embodied
within the present invention.
[0070] The present inventors have found that the amount of chiral base used in
crystallization of the a-(phenoxy)phenylacetic acid has a significant effect
on the optical
purity of the enantiomeric enrichment. For example, when a chiral amine
compound of the
formula:
oR2 oR3
Ar)Y( R4
NH2
(wherein R2, R3, R4 and Ar are those defined herein) is used in
crystallization of the a-
(phenoxy)phenylacetic acid compound, higher %ee obtained by using the chiral
amine
compound in an amount less than 0.5 molar equivalent, preferably about 0.48
molar
equivalent or less, more preferably about 0.47 molar equivalent or less, and
most preferably
about 0.45 molar equivalent or less. It should be recognized that the chiral
amine compound
itself should be of a sufficient enantiomeric purity in order to yield a
highly enantiomerically
enriched a-(phenoxy)phenylacetic acid derivatives.
[0071] The crystallization is typically conducted in a solvent that allows a
different
solubility of salts that are formed between two enantiomers of the a-
(phenoxy)phenylacetic
acid and the chiral amine. In this manner, one of the diastereomeric salt
precipitates out of
11
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
the solution preferentially. Suitable crystallization solvents include protic
solvents, such as
alcohols. A particularly preferred crystallization solvent is isopropyl
alcohol.
[0072] The yield of enantiomerically enriched a-(phenoxy)phenylacetic acid
also depends
on, among others, the amount of crystallization solvent used. For example, if
a large quantity
of crystallization solvent is used, the mixture becomes too dilute and the
solid formation is
reduced. If the amount of crystallization solvent used is too small, the
solution will be
supersaturated with the undesired diastereomeric salt which may lead to
crystallization of the
undesired diastereomeric salt, thereby reducing the optical purity of a
desired enantiomer.
Thus, when isopropanol is used as the crystallization solvent, the amount of
crystallization
solvent used is preferably from about 2 grams to about 6 grams per one gram of
the a-
(phenoxy)phenylacetic acid compound, more preferably from about 3 grams to
about 5
grams, still more preferably from about 3.5 grams to about 4.5 grams, and most
preferably
about 4 grams.
[00731 In one embodiment, the crystallization process involves heating the
crystallization
solution mixture to a temperature above the nucleation temperature of both
enantiomers to
dissolve substantially all of both enantiomers. For example, the
crystallization solution is
heated to a temperature in the range of from about 60 C to the boiling point
of the solution,
preferably from about 70 C to about 80 C. More preferably, the
crystallization solution is
heated to about 75 C. The solution can be heated prior to and/or after the
chiral amine
compound is added. Heating is carried out until the solid materials are
substantially
completely dissolved, which typically ranges from about 0.5 to about 16 hours,
preferably
from about 1 to about 8 hours.
[00741 The Crystallization solution is then cooled until it is at or below the
nucleation
=
temperature of the first diastereomeric salt, e.g., salt of (-)-enantiomer of
the a-(phenoxy)-
phenylacetic acid, but preferably above the nucleation temperature of the
second
diastereomeric salt, e.g., salt of (+)-enantiomer of the a-
(phenoxy)phenylacetic acid. This
allows formation of a solid acid-base salt of the first enantiomer with the
chiral amine
compound. Without being bound by any theory, it is believed that the use of a
chiral amine
compound results in formation of an acid-base salt with one of the enantiomer
at a
significantly faster rate than formation of an acid-base salt of the other
enantiomer. This rate
may be due to kinetic and/or thermodynamic rate difference between the two
enantiomers.
As with a typical compound, the solubility profile of the a-
(phenoxy)phenylacetic acid
12
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
compound of the present invention has a higher solubility at a higher
temperature. Therefore,
by cooling the crystallization solution to just above the nucleation
temperature of the second
diastereomeric salt affords a higher recovery yield of the solid first
diastereomeric salt.
[0075] After the slurry is formed, the crystallization solution can be further
cooled until the
temperature of the solution is near or above the saturation point of the
second diastereomeric
salt. This prevents formation of a diastereomeric solid acid-base salt from
the second
enantiomer while increasing the formation of the diastereomeric solid acid-
base salt of the
first enantiomer.
[0076] The rate of cooling the crystallization solution may affect the optical
purity of the
solid acid-base salt that is formed. For example, if the crystallization
solution is cooled too
fast, the undesirable enantiomer may get trapped within the lattice of the
solid acid-base salt
of the desired enantiomer. However, a too slow cooling rate increases the
production time
and cost. Therefore, the crystallization solution should be cooled at a rate
which minimizes
the loss of optical impurity but at a rate sufficient to be economical.
Typically, the
crystallization solution cooling rate is from about 0.05 C/min to about 1
C/min, preferably
from about 0.1 C/min to about 0.7 C/min, and more preferably from about 0.25
C/min to
about 0.4 C. The crystallization solution is then maintained at above the
saturation point of
the solid acid-base salt of the second, i.e., undesired, enantiomer.
Typically, the
crystallization solution is maintained at this temperature for about 1 to
about 72 hours,
preferably from about 2 to about 48 hours, and more preferably from about 3 to
about 30
hours.
[0077] As expected, using a small amount of chiral amine compound allows
selective
formation of the solid acid-base salt of the first enantiomer. However, the
resulting yield will
correspondingly be small. Theoretically, the amount of yield of the desired
enantiomer from
a racemic mixture is 50%. Thus, if 0.5 molar equivalent of the chiral amine
compound is
used, the theoretical yield is 50% of the total a-(phenoxy)phenylacetic acid
(or 100% of the
desired enantiomer). In order to be economically desirable, methods of the
present invention
provide at least about 50% yield of the desired enantiomer, preferably at
least about 60%,
more preferably at least about 70%, and most preferably at least about 75%.
Assuming 100%
selectivity, these yields.correspond to adding about 0.25, 0.30, 0.35 and
0.375 molar
equivalent of the chiral amine compound, which represent a minimum amount of
the chiral
amine compound that need to be added to the crystallization solution.
13
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0078] It is believed that the tendency for the second enantiomer to form a
solid acid-base
salt with the chiral amine compound is one of the major causes for variability
of conventional
crystallization processes. Thus, by determining the supersaturation point of
the second, i.e.,
undesired, enantiomer, one can minimize or prevent unpredictability of a solid
acid-base
formation of the second enantiomer. Supersaturation points can be readily
determined by one
skilled in the art, e.g., by a solubility experiment.
[0079] It should be noted that while methods of the present invention are
discussed in
reference to the enrichment of (-)-enantiomer that is present in the racemic
mixtures, methods
of the present invention are also applicable for enriching the (+)-enantiomer.
The method of
the present invention essentially provides a solid precipitate enriched in the
(-)-enantiomer
and a liquid filtrate, i.e., mother liquor, enriched in the (+)- enantiomer.
Liberation of the
desired (-)-enantiomer and recovery of the chiral amine compound from the
precipitated salt
can be readily accomplished by acidification of the salt with, for example, a
dilute mineral
acid or any other inorganic or organic acid conventionally known to hydrolyze
salts of this
nature. While this procedure leaves the filtrate as an undesired by-product,
the filtrate can be
further treated with acid or, preferably, base to convert the (+)-enantiomer
enriched filtrate to
the racemic mixture. For example, the (+)-enantiomer can be racemized using
aqueous
sodium hydroxide solution. This racemic mixture can then be reused, i.e.,
recycled. In
addition, the chiral amine compound can also be recovered from the above
described
conversion step and recycled. Thus, the process of the present invention lends
itself readily
to a recycling-type of procedure.
IV. Synthesis of racemic a-(phenoxy)phenylacetic acid
[0080] One method of producing a racemic mixture of a-(phenoxy)phenylacetic
acid of
Formula I is shown in Scheme I below.
Br
CO2H 1. SOC12
ROH
2 Br
X¨ I
1
2 RI
0 0
3
O
OH R
Hydrolysis ii+ Base
4
1
Scheme I
14
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0081] Thus, conversion of phenylacetic acid 1 to an activated carboxylic acid
derivative,
e.g, acid chloride, followed by a-bromination gave a-bromophenylacetyl
chloride (not
shown). The acid chloride was then converted to ester 2, where R is typically
alkyl.
Preferably, alcohol ROH, which is used to convert the acid chloride to ester
2, is the same
alcohol that is used as a solvent in a subsequent reaction. In this manner,
the number of
different solvent types is minimized. In addition, by using the same ROH as
the solvent in
the subsequent reaction, the amount of by-product, e.g., by trans-
esterification, formation is
minimized. For example, isopropyl ester 2, i.e., where R is isopropyl, is
particularly
advantages as the subsequent reaction is conveniently carried out in
isopropanol solvent. A
displacement reaction of ester 2 with a phenol compound 3 in the presence of a
base, such as
a hydroxide (e.g., potassium hydroxide), gave a a-(phenoxy)phenylacetic acid
ester 4.
Hydrolysis of a-(phenoxy)phenylacetic acid ester 4 afforded a-
(phenoxy)phenylacetic acid I.
[0082] In this manner, (4-chloropheny1)-(3-trifluoromethylphenoxy)-acetic
acid, i.e.,
CPTA, can be prepared in five steps without intermediate isolation in about
85% yield
following crystallization from heptane.
V. Utility of enantiomerically enriched a-(phenoxy)phenylacetic acid
[0083] Enantiomerically enriched a-(phenoxy)phenylacetic acid compounds are
useful
intermediates in preparing a variety of phan-naceutically active compounds,
including a-
(phenoxy)phenylacetic acid compounds disclosed in U.S. Patent No. 3,517,050.
Thus, anther
aspect of the present invention provides a method for enantioselectively
producing a a-
(phenoxy)phenylacetate compound of the formula:
oa7
x
IV
from a racemic mixture of a a-(phenoxy)phenylacetic acid compound Formula I,
wherein RI
is alkyl or haloalkyl, X is halide and R7 is heteroalkyl, preferably N-acetyl
2-aminoethyl (i.e.,
a moiety of the formula ¨CH2CH2NHC(=0)CH3). The method involves resolving the
racemic mixture of the a-(phenoxy)phenylacetic acid compound of Formula I as
described
above and producing an enantiomerically enriched activated a-
(phenoxy)phenylacetic acid
by reacting the enantiomerically enriched a-(phenoxy)phenylacetic acid with a
carboxylic
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
acid activating reagent. Suitable carboxylic acid activating reagents include
thionyl halides
(e.g., thionyl chloride), anhydrides, thioester generating reagents, and other
carboxylic acid
activating reagents known to one skilled in the art.
[00841 The activated a-(phenoxy)phenylacetic acid is than reacted with a
compound of the
formula (R7--0)M, e.g., N-acetyl ethanolamine derivative, to produce
enantiomerically
enriched a-(phenoxy)phenylacetate compound of Formula III, where R7 is as
defined above,
M is hydrogen or a metal, e.g., Na, K, Li, Ca, Mg, Cs, etc. and the
superscript w is the
oxidation state of M. The present inventors have discovered that the reaction
between the
activated acid and the compound of formula (R7-0)wM can be carried out without
any
significant racemization.
[00851 Additional objects, advantages, and novel features of this invention
will become
apparent to those skilled in the art upon examination of the following
examples thereof,
which are not intended to be limiting.
EXAMPLES
Reagents and Experimental Setup
[00861 Unless otherwise stated, reagents and solvents were purchased from
Aldrich
Chemical or Fisher Scientific. N-Acetylethanolamine was also obtained from
Lancaster
Synthesis. The racemic CPTA, i.e., halofenic acid was prepared according to
the procedures
disclosed in U.S Patent Nos. 3,517,050 and 6,262,118 all of which are
incorporated herein by
reference in their entirety. (1R,2R)-(-)-2-Amino-1-(4-nitropheny1)-1,3-
propandiol (i.e., CAF
D-Base) was obtained from TCI Americas.
[00871 Operations were conducted under a positive nitrogen atmosphere. A
Camile
process control computer attached to a recirculating heating and cooling
system was used to
regulate jacket temperatures in the jacketed straight-walled bottom-drain
glass reactors.
Unless otherwise indicated, solvents were removed using a Buchi rotary
evaporator at 15 to
25 ton with a bath temperature of up to 40 C. Solid samples were dried in a
vacuum oven at
40 C, 15 to 25 ton. A Cenco HYVAC vacuum pump was used to supply vacuum of
less
than 1 ton for vacuum distillations. Water levels were determined by Karl
Fisher analysis
using a Metrohm 756 KF Coulometer and HYDRANAL Coulomat AG reagent. Melting
points were determined using a Mettler Toledo FP62 melting point apparatus. pH
was
measured using a calibrated Orion Model 290A pH meter. Proton and 13C NMR
spectra were
recorded on a Braker Avance 300 MHz spectrometer.
16
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0088] Chiral HPLC analysis was carried out at X=240 mn by injecting 10 tit of
sample
dissolved in mobile phase onto a (R,R)'WHELK-0 1.5 jun 250 x 4.6 mm column
(Regis
Technologies) and eluting with a 1.0 mL/min flow of 95/5/0.4 (v/v/v) hexanes/2-
propanol/acetic acid. For solid samples of the CPTA/CAF D-Base diastereomeric
salt, the
solid was added to aqueous hydrochloric acid and the CPTA was extracted into
methylene
chloride; after removing the solvent from the methylene chloride layer, the
residue was
dissolved in mobile phase for analysis.
[0089] Achiral HPLC analysis was carried out at X=220 nm by injecting 5 pL of
sample
dissolved in mobile phase onto a Phenomenex LUNA 5 pm C18(2) 250 x 4.6 mm
column at
25 C. A 1.5 mL/min flow of the gradient starting at 66 vol% water/34 vol%
acetonitrile/0.1
vol% trifluoroacetic acid and increasing linearly to 26 vol% water/74 vol%
acetonitrile/0.1
vol% trifluoroacetic acid at 20 minutes was used.
[0090] For analysis of acidic solutions of esters, such as halofenate,
acetonitrile was used as
the injection solvent. When determined, product concentrations for CPTA and
halofenate
were evaluated by HPLC assay using the external standard method and the
achiral analysis
procedure at sample concentrations of less than 2.5 mg/mL.
Example 1
[0091] Previous resolution of CPTA has been reported in U.S. Patent No.
3,517,050, in
which cinchonidine was used as the chiral base, and the (+)-enantiomer of CPTA
precipitated
as the diastereomeric salt. One major drawback to this procedure was that the
desired (-)-
enantiomer remained in the mother liquor, making separation of a pure (-)-
enantiomer
fraction difficult.
[0092] This example shows the results of resolving a racemic mixture of CPTA
using a =
variety of different chiral bases to obtain a solid enantiomerically enriched
(-)-isomer. Unlike
the previous method, methods of the present invention allow the solid
enantiomerically
enriched (-)-CPTA to be readily isolated from the solution.
[0093] Racemic CPTA was prepared by the potassium hydroxide hydrolysis of
racemic
halofenate. For chiral base screening, equal molar mixtures of CPTA and the
chiral base
were mixed in ethanol, methanol and acetone in glass vials, and the solutions
were allowed to
stand undisturbed. After holding overnight at ambient temperature, the samples
that
remained in solution were placed in a refrigerator at 5 C. After holding
overnight in the
17
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
refrigerator, a small amount of water was added to the samples that remained a
solution in
ethanol. After four days at ambient temperature, the aqueous ethanol solutions
were placed
back in the refrigerator. All of the samples remained in the refrigerator, and
were
periodically checked for precipitate formation over the course of a month. A
list of the bases
and solvent conditions examined, and temperatures at which crystalline salts
were found is
shown in Table 1.
Table 1. Bases Examined for CPTA Resolution.
Solvent System
Base Et0H Et0H (aq) Acetone
Me0H
S )-Methylbenzylamine
Quinine C (22 C) C (22 C) C
(22 C)
Quinidine
L-Tyrosine Hydrazide C (22 C)
L-Leucine Methyl Ester Hydrochloride*
1-2-Amino-1-butanol
Brucine
(S)-(+)-2-Pyrrolidine-methanol
(S)-(4-)-2-Amino-3-methy1-1-butanol
(S)-(+)-2-Amino-1-propanol
(S)-(+2-Amino-3-phenyl-1-propanol
(I S,2S)-(+)-Pseudoephedrine
(1S,2S)-(+)-2-Amino-l-phenyl-1,3-propanediol E
(1 S,2S)-(+)-2-Amino-1-(4-nitropheny1)-1,3-propandiol C (5 C)
(1R,2S)-(-)-Norephecirine
(1R,2S)-(-)-Ephedrine
(1R,2R)-(+2-Amino-1-(4-nitropheny1)-1,3-propandiol C (22 C)
(+)-Cinchonone
(-)-Cinchonidine C (22 C)
(-)-Strychnine
E ¨ Evaluated
C ¨ Crystallized at (Temperature)
* - With 1 mol/mol of Aqueous Sodium Hydroxide
18
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0094] Four chiral bases, quinine, L-tyrosine hydrazide, (-)-cinchonidine, and
both
enantiomers of 2-amino-1-(4-nitropheny1)-1,3-propandiol, were found to give
crystalline salts
from racemic CPTA. For samples that crystallized, the solid was isolated by
filtration, and
both the solid phase and mother liquor were analyzed by chiral HPLC to
determine the
enantiomeric composition of both streams. The results from the screen are
shown in Table 2.
Three of the bases shown in Table 2 gave the (+)-enantiomer enrichment in the
solid phase.
Table 2. Results from Chiral Base Screen.
Solid
Mother Liquor % Yield
Base % (+)
(-) % (+) % (-) Calculated
Solvent Temp C
L-Tyrosine Hydrazide Acetone 22 86.6 13.4 40.7 59.3
20.3
(-)-Cinchonidine Ethanol 22
66.8 33.2 12.0 88.0 69.3
(1S,2S)-(+)-2-Amino-1- (4- Ethanol 22 93.2 6.8 28.5 71.5
33.2
nitropheny1)-1,3-propandiol
Quinine Ethanol 22 39.9 60.1 60.1 39.9 50.1
Acetone 22
28.2 71.8 58.9 41.1 28.9
Acetone* 5 23.0 77.0 83.5 16.5 55.4
= Methanol 22 25.8 74.2 53.0 47.0 10.9
2-Propanol 30 43.2 56.8 64.3 35.7 67.6
2-Propanol** 30 40.4 59.6 78.8 21.1 75.0
2-Propanol* 21 42.3 57.7 59.1 40.9 53.9
* - More Dilute
** - Slower Cooling Profile
[0095] Included in Table 2 is the percent yield of solid calculated from the
isomeric ratio in
the solid and mother liquor streams. The equation used is shown below. The
maximum
theoretical yield with 100% isomeric purity is 50%. Yields over 50% indicate
inclusion of
the other isomer.
Equation to calculate yield from isomer ratios.
Set: a = area % Component 1 in starting material; b = area % Component 2 in
starting
material; x = area % Component 1 in isolated; y = area % Component 2 in
isolated; w = area
% Component 1 in mother liquor; z = area % Component 2 in mother liquor; E = g
material
isolated; F = g material in mother liquor.
And: a + b = 100%; E + F = 1
Then: xE + wF = a; yE + zF = b
Solving: xE + w(1-E) = a; yE + Z(1-E) = b
E = isolated yield = (a-w)/(x-w) = (b-z)/(y-z)
19
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 2
[0096] This example shows the results of resolving CPTA with CAF D base in
ethanol and
2-propanol.
[0097] The results for ethanol and 2-propanol are summarized in Table 3 below.
For this
evaluation, the slurries were sampled at various points in the cooling
profile, and the
enantiomeric composition of both the solid and solution phases determined.
From this
information, the %ee of the solid phase and expected weight percent yield
(maximum 50%
yield with 100 %ee), calculated from the isomer ratio, were determined.
Included in Table 3
is the yield of(-)-CPTA, which is derived from the weight percent yield and
the (-)-CPTA
content of the solid phase (maximum 100% yield with 100 %ee).
[0098] In this particular study, the best results in ethanol used 1 mole of
CAF D Base per
mole of CPTA. Approximately 72% yield of the (-)-CPTA CAF D Base salt was
calculated
from the chiral composition of both phases, with an 87.6 %ee of the (-)-CPTA
salt in the
solid phase. Use of one molar equivalent of CAF D Base in 2-propanol at a
similar
concentration gave a lower resolution. Higher enantiomeric enrichment was
achieved when
0.55 mole of CAF D Base per mole of CPTA was used. Under these conditions,
approximately 76-79% yield of the (-)-CPTA CAF D Base salt was calculated from
the phase
compositions, with an 87-90 %ee of (-)-CPTA in the solid phase. Calculated
weight percent
yields, which do not take into account physical losses, were 41 to 42%; actual
weighed
isolated yields were 37 to 39%.
Table 3. Resolution of CPTA with CAF D Base.
wt% mole/mole T C Solid M.L. %Yield %Yield
CPTA base %(+) %(-) %(+) %(-)
%ee 50% Max (-)-CPTA
Ethanol
13.68 1.02 11 10.2 89.8
75.8 24.2 79.7 39.4 68.9
0 8.9 91.1 76.4 23.6 82.3 39.1 69.9
-9 6.2 93.8 78.0 22.0 87.6 39.0 72.4
14.09 0.50 18 6.6 93.4 55.5
44.5 86.7 11.2 20.7
-5 10.3 89.7 59.7 40.3 79.3 19.7 35.0
2-Propanol
15.72 1.01 12 45.9 54.1 66.9
33.1 8.2 80.4 85.8
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
-8 46.6 53.4 68.0 32.0 6.7 84.2 87.1
16.6 0.50 36 8.3 91.7 69.6 30.4 83.5
32.0 58.6
22 10.2 89.8 73.9 26.1 79.6 37.5 62.3
2 8.0 92.0 74.9 25.1 84.0 37.2 68.5
16.7 0.55 49 26.3 73.7 64.5 35.5 47.4
38.0 56.1
50 7.5 92.5 63.3 36.7 85.0 -
23,8 44.0
20 6.7 93.3 79.7 20.3 86.6 40.7 75.6
16.7 0.55 50 8.8 91.2 64.2 35.8 82.3
25.6 46.6
35 9.1 90.9 69.0 31.0 81.7 31.7 57.6
6 5.2 94.8 75.0 25.0 89.6 35.8 67.9
5 5.7 94.3 81.9 18.1 88.6 41.8 78.9
1 5.3 94.7 82.2 17.8 89.5 41.9 78.8
18.34 0.54 6 6.3 93.7 82i 17.9 87.3 42.4
79.2
[0099] Recrystallization of the CPTA CAF D Base salt from 2-propanol increased
the
optical purity from approximately 87 %ee to 98 %ee with 87% mass recovery, or
93%
recovery based on the (-)-CPTA content of the feed (Table 4).
Table 4. Recrystallization of (-)-CPTA CAF D Base from 2-Propanol.
%ee M.L. wt% % Yield
%ee Feed wt% Salt Isolated %(l) %(-)
Yield (-)-CPTA
86.6 13.0 97.7 48.8 51.2 87.8 93.3
87.3 12.9 98.0 45.0 55.0 87.8 92.8
[0100] Overall, an approximately 35% yield out of a maximum 50% of the (-)-
CPTA CAF
D Base salt, with an optical purity of approximately 98 %ee, was obtained from
racemic
CPTA.
[0101] Crystallization of optically enriched enantiomers often increases the
chiral purity.
Following removal of the resolving agent, crystallization of (-)-CPTA from
methylcyclohexanone will also increase the optical purity to some degree. In
one
experiment, crystallization of (+)-CPTA increased the optical purity from 99.1
to 100 %ee;
the mother liquor was 95 %ee.
21
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 3
[0102] This example illustrates the solubility profiles of CAF D Base salt of
(+)- and (-)-
isomers of CPTA in 2-propanol.
[0103] To aid in optimization of the CPTA resolution using CAF D Base, the
solubility
profiles of both of the diastereomeric salts in 2-propanol were determined.
The results are
shown in Figure 1. The (+)-CPTA CAF D Base salt was prepared using
cinchonidine-
resolved (+)-CPTA. As Figure 1 shows, the desired (-)-CPTA diastereomer is
approximately
three times less soluble than the (+)-CPTA form. Equations describing the
solubilities
included in the figure were calculated by least squares analysis (R2>0.99).
The data point for
the (-)-CPTA salt at 82 C was not included in determining the equation, but
closely fits the
calculated solubility.
[0104] Racemization of the undesired CPTA enantiomer could be recycled back
into the
process. Thus, it was found that heating an enantiomerically enriched
undesired isomer of
CPTA in 1 N aqueous sodium hydroxide at reflux resulted in racemization in
less than one
hour. No other by-products were detected by HPLC analysis of the isolated
CPTA.
Example 4
[0105] This example illustrates a method for obtaining (+)-CPTA.
[0106] A 2-L round-bottom flask with an overhead stirrer was charged with 33.0
g of crude
(+)-CPTA - chinconidine salt, 610 mL of ethanol, and 125 mL of methanol. The
slurry was
heated to reflux to give a solution, then cooled. A very thick slurry formed
at 42 C. The
slurry was heated to 68 C to give a light slurry, then allowed to cool to
ambient temperature.
The mixture was filtered at 26 C and rinsed with 150 mL of ethanol to give,
after drying
under vacuum at 40 C, 23.48 g of (+)-CPTA ¨ chinconidine salt. The
recrystallization
procedure was repeated with 600 mL of ethanol and 120 mL methanol to give
18.23 g of (+)-
CPTA ¨ chinconidine salt (55% recovery from two crystallizations). No (-)-CPTA
was
detected by chiral chromatography, although the degree of separation did not
allow for an
assessment of low levels (the halofenate chiral analysis conditions were also
used at that time
for the CPTA analysis).
[0107] A 3.61 g sample of the purified salt was mixed with 50 mL of water and
50 mL of
toluene, and 2.9 g of sulfuric acid was added. The organic phase was washed
with 30 mL of
water, then evaporated to a residue. The residue was crystallized from 20 mL
of cyclohexane
22
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
to give 1.22 g of (+)-CPTA. Alternatively, 6.3 g of the (+)-CPTA ¨
chinconidine salt (10.2
mmol) was mixed with 56 g of diethyl ether and 29 g of water, and acidified to
a pH of 1.9
with drops of sulfuric acid. The organic phase was washed with 25 mL of water,
dried
(magnesium sulfate), filtered, and evaporated to.a residue. The residue was
stirred with 22
mL of methylcyclohexane at ambient temperature to form a slurry. The slurry
was warmed
to 40 C, then cooled in an ice bath and the solid isolated by filtration to
give, after drying at
40 C under vacuum, 2.62 g (7.92 mmol, 78% yield) of (+)-CPTA.
Example 5
[0108] This example illustrates a method for synthesizing (+)-halofenate from
(+)-CPTA.
[01091 A 25-mL round-bottom flask was charged with 0.91 g of (+)-CPTA and 2.6
g of
thionyl chloride, and the mixture heated to reflux to give a solution.
Conversion to acid
chloride was monitored by quenching a sample with methanol and analyzing the
product with
HPLC. To the acid chloride solution was added 4.8 g of diethyl ether, and this
solution was
added to 2.0 g of N-acetylethanolamine in 12 mL of N,N-dimethylformamide (DMF)
with
0.37 g of pyridine chilled in an ice bath. The resulting solution was added to
25 mL of water
and 30 mL of diethyl ether. The organic phase was separated, washed with 25 ml
of water,
dried (MgSO4), and filtered to give, after removal of the solvent, 0.92 g of
an oil. HPLC
analysis showed 45 area% of halofenate and 50 area% of CPTA. Chiral HPLC
analysis
indicated that the halofenate was 99.78 %ee of the (+)-enantiomer.
Example 6
[0110] This example illustrates a method for preparing racemic CPTA.
[0111] A 2-L round-bottom flask with an overhead stirrer was charged with
102.7 g of
halofenate, 500 mL of water, and 16.3 g of 2-propanol. The slurry was stirred,
and 32.3 g of
aqueous 45% potassium hydroxide was added. After heating to reflux for 1 hour,
the solution
was cooled to ambient temperature and charged with 380 mL of hexanes. The pH
was
adjusted from 12.5 to 2 with 24.57 g of 37% hydrochloric acid. The three phase
mixture was
heated to 60 C to give two phases. The lower aqueous phase was removed and
extracted
with 50 mL of hexanes. The combined organic layers were heated to distill at
atmospheric
pressure to remove 100 mL of cloudy distillate. The solution was cooled to 30
C and seeded
with CPTA. A slurry formed. The slurry was cooled in an ice bath and the solid
isolated by
filtration to afford 64.0 g (78.4% yield) of racemic CPTA, i.e., (4-
chlorophenyl)(3-trifluoro-
methylphenoxy)acetic acid.
23
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 7
[0112] This example shows representative results of chiral resolution
screening in ethanol
using a variety of chiral bases.
[0113] A sample of 1.16 g (3.51 mmol) of CPTA was dissolved in 6.98 g of
ethanol to give
filtration, and also analyzed. Some of the results are shown in Table 2 (see
Example 1
above). The remaining vials were placed in a refrigerator at 5 C. After one
day, 7E give a
precipitate. The sample was analyzed as previously described. The remaining
vials were
charged with 50111, of water, and held at ambient temperature for three days
before placing in
15 the refrigerator. No additional precipitates were noted after one month.
Table 5. Base Screening in Ethanol.
wt CPTA Wt
Et0H Water
Base Solution (g) Base (g) Added
Added
7A S-(-)-Methylbenzylamine 0.8836 0.4620 0 g
0.05 g
7B 1-2-Amino-l-butanol 0.8198 0.0314 0
0.05
7C (1R,2S)-(-)-Norephedrine 0.5273 0.0342 0.2007 0.05
7D (1S,2S)-(+)-Pseudoephedrine 0.7295
0.0515 0.1459 0.05
7E (1S,2S)-( )-2-Amino-1-(4-nitropheny1)-1,3- 0.5580 0.0510 0.1228 0
propanediol
7F (1,2S)-(+)-2-Amino-l-phenyl-1,3-propanediol 0.5640 0.0405 0.1287 0.05
7G (-)-Cinchonidine 0.3484 0.4390 0.3637 0
7H (+)-Cinchonine 0.6409 0.0796 0.2103 0.05
71 Quinine 0.5391 0.0750 0.1735 0
7J (-)-Strychnine 0.5812 0.0828 0.2295 0.05
7K Brucine 0.7566 0.1287 0
0.05
7L (S)-(+)-2-pyrrolidine-methanol
0.8681 0.0383 0 0.05
24
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 8
[01141 This example shows representative results of chiral resolution
screening in acetone
using a variety of chiral bases.
[0115] A sample of 1.67 g of CPTA was dissolved in 7.57 g of HPLC grade
acetone to give
a solution. Glass vials were individually charged with the amounts of each
base listed in
Table 6, and the amount of the CPTA solution calculated to give a 1 to 1 molar
ratio of acid
to base was added. In some cases, a small amount of acetone was added and the
mixture was
warmed to about 40 C to give a solution. Additionally, 0.300 mL of 1 N sodium
hydroxide
was added to vial 16M. The vials were allowed to stand overnight at ambient
temperature.
Vial 16D formed a precipitate, and was analyzed as described above. Some of
the results are
summarized in Table 2 (see Example 1). The remaining vials were placed in the
refrigerator.
Vial 16 N formed a precipitate, and was analyzed. Vial 16G formed a very light
precipitate.
After one week, vial 16L was found to contain a precipitate. The sample was
analyzed as
previously indicated. No additional precipitates were noted.
Table 6. Base Screening in Acetone.
wt CPTA Wt
Acetone
Base
Solution (g) Base (g) Added (g)
16A (1R,2S)-(-)-Norephedrine 0.8568
0.0704
16B (1S,2S)-(+)-2-Amino-1-pheny1-1,3-propanediol 0.1824
0.0168
16C S-(+Methylbenzylamine 0.8948 0.0592
16D Quinine 0.1968 0.0347
0.85
16E (S)-(+)--2-pyrrolidine-methanol 0.8181 0.0452
16F Brucine 0.2163 0.0463
16G (+)-Cinchonine 0.3987 0.0630
1611 (1S,2S)-(+)-Pseudoephedrine 1.0835
0.0974
161 (-)-Strychnine 0.1462 0.0265
0.25
163 Quinidine 0.3753 0.0663
16K 1-2-Amino-1-butanol 0.7248 0.0353
16L L-Tyrosine Hydrazide 0.4508 0.0472
0.39
16M L-Leucine Methyl Ester Hydrochloride 0.5585 0.0544
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
16N Quinine 0.4712 0.0829
2.00
; 160 (+)-Cinchonine 0.3363 0.0539
0.30
Example 9 =
[0116] This example shows representative results of chiral resolution
screening in methanol
using a variety of chiral bases.
[0117] A sample of 2.00 g of CPTA was dissolved in 8.03 g of HPLC grade
methanol to
give a solution. Glass vials were individually charged with the amounts of
each base listed in
Table 7, and the amount of the CPTA solution calculated to give a Ito 1 molar
ratio of acid
to base was added. Additionally, 0.300 mL of 1 N sodium hydroxide was added to
vial 27J.
The vials were allowed to stand overnight at ambient temperature. Vial 27B
solidified, and
an additional 300 ,L of methanol was added before the sample was analyzed as
described
above. The remaining vials were placed in the refrigerator. No additional
precipitates were
noted after one month.
Table 7. Base Screening in Methanol.
Base wt CPTA solution (g) wt
base (g)
27A (1R,2S)-(-)-Ephedrine 0.4896 g 0.0478 g
27B Quinine 0.1420
0.0282
1 27C (+)-Cinchonine 0.1822
0.0324
27D 1-2-Amino-l-butanol 1.0012
0.0539
27E S-(-)-Methylbenzylamine 0.7892
0.0576
27F (1S,2S)-(+)-Pseudoephedrine 0.7600
0.0749
27G Brucine 0.1891
0.0436
2711 Quinidine 0.5845
0.1144
271 (1S,2S)-(+)-2-Amino-l-pheny1-1,3-propandiol 0.3032
0.0299
27J L-Leucine Methyl Ester Hydrochloride 0.5033
0.0545
27K (S)-(+)-2-Pyrrolidine-methanol 0.7133
0.0434
1 ____________________________________________________________________________
27L (1R,2S)-(-)-Norephedrine 1.1788
0.1070
27M (-)-Strychnine 0.4525
0.0905
27N (S)-(+)-2-Amino-3-methyl-1-butanol 0.1478
0.0092
26
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
270 (S)-(+)-2-Amino-l-propanol 0.9268
0.0417
_
27P (S)-(-)-2-mino-3-pheny1-1-propanol 0.3406
0.0307
Example 10
[0118] This example shows the result of resolving CPTA with quinine.
[0119] A 150-mL jacketed bottom-drain flask was charged with 2.70 g (8.17
mmol) of
CPTA, 2.65 g (8.17 mmol) of quinine, and 50 mL of 2-propanol. The mixture was
heated to
70 C to give a solution, then cooled to 30 C at a rate of 0.2 C/min and
held for 2 hours to
give a slurry. Chiral HPLC analysis of a sample showed 42.88 and 56.47 area%
of (+) and (-
)-CPTA, respectively, in the solid phase, and 61.54 and 34.19 area% of (+) and
(-)-CPTA,
respectively, in the solution. The slurry was heated to 60 C, then cooled to
30 C at a rate of
0.04 C/min and held overnight to give a slurry. Chiral HPLC analysis showed
29.94 and
44.19 area% of (+) and (-)-CPTA, respectively, in the solid phase, and 77.54
and 20.88 area%
of (+) and (-)-CPTA, respectively, in the solution. The slurry was diluted
with 50 mL of 2-
propanol and heated to 57 C to give a solution, then cooled to 30 C at a
rate of 0.2 C/min.
A slurry started to form after 1 hour at 30 C. The mixture was stirred for 2
days at ambient
temperature, then the solid was isolated by filtration and rinsed with 2-
propanol to give, after
drying under vacuum, 2.89 g (54% yield by mass) of the quinine salt of CPTA.
Chiral HPLC
analysis found 42.25 and 57.75 area% of (+) and (-)-CPTA, respectively, in the
solid phase
and 56.56 and 39.20 area% of (+) and (-)-CPTA, respectively, in the mother
liquor. The
results are also included in Table 2 (see Example 1).
Example 11
[0120] This example shows the result of resolving CPTA with GAF D base.
[0121] A 150-mL bottom-drain flask was charged with 19.54 g of CPTA, 6.82 g of
CAF D
Base (i.e., D-threo-(-)-2-amino-1-(nitropheny1)-1,3-propandiol), and 80.2 g of
2-propanol.
The mixture was warmed to 70 C to give a solution, then cooled to a jacket
temperature of 5
C at a rate of 0.1 C/min. The mixture was hazy at 62 C. After holding at 6
C for 9 hours,
the solid was isolated by filtration, rinsed with 5 mL of 2-propanol, and
dried at 40 C under
vacuum to give 12.03 g (37.4 wt% yield) of(-)-CPTA CAF D Base salt. Chiral
HPLC
analysis of the solid found 6.34 area% of (+)-CPTA and 93.46 area% of(-)-CPTA;
the
mother liquor contained 81.41 area% of (+)-CPTA and 17.76 area% of(-)-CPTA.
27
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
Example 12
[01221 This example shows the result of recrystallizing (-)-CPTA CAF D Base
salt.
[01231 A 150-mL bottom-drain flask was charged with 8.00 g of the (-)-CPTA CAF
D
Base salt (from Example 11 above) and 54.2 g of 2-propanol. The mixture was
heated to
reflux to give a solution, then cool to a jacket temperature of 20 C at a
rate of 0.1 C/min
and held at an internal temperature of 22 C for 6 hours. The solid was
isolated by filtration,
rinsed with 2-propanol, and dried at 40 C under vacuum to give 6.93 g (86.6
wt% recovery)
of(-)-CPTA CAF D Base salt (m.p. 184-185 C). The solid contained 0.995 area%
of (+)-
CPTA and 99.01 area% of(-)-CPTA; the mother liquor contained 44.53 area% of
(+)-CPTA
and 54.47 area% of(-)-CPTA. The reactor was cleaned out with acetone. The
acetone was
evaporated to a residue of 0.27 g (3.4 wt%).
Example 13
[0124] This example illustrates a method for preparing (+)-CPTA CAF D Base
salt.
[0125] A 1-L flask was charged with 10.94 g (17.5 mmol) of the (+)-CPTA
cinchonidine
salt, 200 mL of water, and 100 mL of methylene chloride. The pH was adjusted
to 1.9 by the
addition of 1.8 g of sulfuric acid. The organic layer was washed three times
with 100-mL
portions of dilute aqueous sulfuric acid, dried (magnesium sulfate), filtered,
and evaporated
to a residue of 5.79 g. The residue was dissolved in 22.2 g of 2-propanol, and
3.5 g of CAF
D Base was added. The resulting slurry was heated to reflux to give a
solution, then cooled
to ambient temperature and the slurry stirred for three hours. After cooling
in an ice bath, the
solid was isolated by vacuum filtration, rinsed with 5 mL of 2-propanol, and
dried under
vacuum at 40 C to give 7.39 g (80% yield) of (+)-CPTA CAF D Base salt (m.p.
172-173
C).
Example 14
[0126] This example shows solubility of diastereomeric CPTA-CAF D base salts
in 2-
propanol.
[0127] Samples of(-)-CPTA CAF D Base and (+)-CPTA CAF D Base (>98 %ee) were
added to 2-propanol in the amounts shown in Table 8, and mixed using an
ultrasonic bath.
All samples remained slurries. The slurries were held overnight at the
temperature listed,
then samples of the supernates were removed and analyzed by quantitative HPLC
analysis to
determine the CPTA concentration. The results are shown in the table, and in
Figure 1.
28
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Additionally, 8.00 g of (-)-CPTA CAF D Base salt required 54.2 g of 2-propanol
for solution
at 82 C (14.7 wt%). This data point was included in Figure 1, but not
included in the
solubility equation.
Table 8. Solubility in 2-Propanol.
Wt Salt (g) Wt 2-propanol (g) T C Wt% in
solution
(-)-CPTA CAF D Base Salt
0.31 1.17 45.3 2.35
0.23 2.48 7.8 0.376
0.21 1.31 19.4 0.688
(+)-CPTA CAF D Base Salt
0.25 1.84 20.0 1.85
0.27 1.83 45.8 6.03
0.17 2.09 8.5 1.32
Example 15
[0128] This example illustrates a method for racemizing enantiomerically
enriched CPTA.
[0129] A 50-mL round bottom flask was charged with 0.31 g of (-)-CPTA (68.7%
ee) and
9.4 g of 1N sodium hydroxide. The solution was heated to reflux for one hour,
then cool to
ambient temperature and acidified with 1 g of 37% hydrochloric acid. The CPTA
was
extracted into methylene chloride, and the solvent was evaporated to an oil of
0.46 g. HPLC
analysis found 99.4 area% of CPTA, and chiral HPLC analysis found a 50/50
mixture of the
CPTA enantiomers.
Example 16
[0130] This example illustrates a process for resolving a racemic mixture of
CPTA using
CAF D-Base under a variety of crystallization conditions.
[0131] The general crystallization procedure was to charge CPTA, CAF D-Base,
and 2-
propanol at room temperature and heat to a solution at about 75 C. The
solution was cooled
to about 60 C and held until nucleation occurred. Several batches were seeded
with (-)-Salt
(i.e., salt of(-)-CPTA and CAF D-Base) to induce nucleation. After the slurry
had developed
over about an hour, the vessel was cooled to the isolation temperature. The
first 5 entries in
Figure 2 used a slow cooling rate of about 0.05-0.10 C / minute to reach the
isolation
29
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
temperature. The other experiments used a faster cooling rate of 0.25-0.40 C
/ minute. A
fiber optic probe is inserted directly into the crystallizer to determine the
slurry density.
[0132] The amount of CAF D-Base added and the solute concentration are some of
the
important variables which give rise to the final batch composition. The
tendency for the (+)-
Salt (i.e., salt of (+)-CPTA and CAF D-Base) to remain supersaturated for
varying amounts
of time is believed to be a major cause for variability in some experiments.
This is
demonstrated in entry 5 in Figure 2, whereby the slurry was held for 8 hours
at 13 C and
produced high purity crystal (99.7% (-)-Salt). Three hours later, an increase
in the signal of
the fiber optic probe indicated the likely nucleation of the (+)-Salt. After
another 27 hours,
the slurry was isolated and the crystal product contained a (- / +)-CPTA ratio
of 83.3 / 16.7%.
Analysis of the crystal product by HPLC gives the ratio of(-)-CPTA and (+)-
CPTA. Since
the free CPTA in solution is undersaturated the crystal analysis therefore
gives the
diasteriomeric salt ratio. Mother liquors contain both dissolved salt and free
CPTA. Analysis
by HPLC reports the combined amount of each enantiomer as CPTA. Similarly,
entry 6 of
Figure 2 shows that the slurry was held for 20 hours at 1 C and produced high
purity salt
(>98% (-)-CPTA). After heating to 17 C, the (+)-Salt nucleated and gave
poorer quality
product [(- / +)-CPTA = 81.2 / 18.8%].
[0133] In other trials, nucleation of the (+)-Salt occurred more quickly, as
in entries 2, 8,
and 10 of Figure 2. A crystallization is desirable for which isolation could
be done near,
preferably just above, the saturation temperature of the (+)-Salt.
[0134] At a loading of 3.9 g of 2-propanol per gram of CPTA and with 0.45
equivalent of
CAF D-Base, an isolation at room temperature appears to be very near the
saturation level (or
within the metastable zone) of the (+)-Salt. Entry 12 in Figure 2 started with
0.43 equivalents
of base, and the crystal product at 21 C remained pure (>99% (-)-Salt), even
after seeding
with (+)-Salt. After adding more CAF D-Base to give 0.45 equivalents, the
slurry was held
for 14 hours, and then for 6 more hours after seeding with (+)-Salt. The
crystal product
analyzed at 98.7% (-)-CPTA ratio. Increasing the total base to 0.47 equivalent
gave crystal
product which slowly increased in (+)-Salt composition to (- / +)-CPTA = 92.3
/ 7.7%.
[0135] Entry 11 of Figure 2 (3.9 g of 2-propanol per gram of CPTA, 0.45 eq.
base)
maintained high purity of the (-)-Salt (99.1%) after 14 hours, but upon
addition of more base
to 0.48 eq., the resulting ratio of the product was (- +)-Salt = 89.2 / 10.8%.
Entry 9 of
Figure 2 (at 0.45 eq. base) maintained 99.5% (-)-Salt purity after 16 hours at
22 C.
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Calculated yields of (-)-CPTA from the three batches under these conditions
were 70.7-
71.6%. Calculated yields are derived from a forced mass balance from the
racemic CPTA
feed, by knowing the crystal and mother liquor composition of (-)-CPTA and (+)-
CPTA.
[0136] These loadings of about 0.45 equivalent of CAF D-Base and about 4 g of
2-
propanol per gram of CPTA provide a high purity (-)-Salt ( >98.5%) product,
which can be
used without a further recrystallization.
Example 17
[0137] This example provides a model to describe the
resolution/crystallization of CPTA
salt.
[0138] The concentration of free CPTA depends on the amount of base charged
and the
solvent loading. For example, a resolution of CPTA by charging 4.0 grams of 2-
propanol and
0.50 equivalent of CAF D-Base, results in formation of the salt in 2-propanol
which contains
11% free CPTA. This solvent possesses greater solubility for both the (-)-Salt
and the (+)-
Salt, and was determined as shown in Figure 3. Figure 3 also includes the
solubility data in
pure 2-propanol, expressed in gram of component per gram of 2-propanol. As
Figure 3
shows the curves for the respective salts are of similar shape.
[0139] By other combinations of the loading of CPTA, CAF D-Base, and 2-
propanol, a ,
system resulting in 11.0% free CPTA in 2-propanol can also be attained, as
shown in Figure
4. As Figure 4 shows, the loading for various experiments in Figure 2 did not
usually fall
exactly on this line. However, the (-)-Salt and the (+)-Salt solubility can be
estimated as
follows: a loading which gives a point above the "11.0% free CPTA" line is
more dilute (i.e.,
<11.0% free CPTA in 2-propanol), and exhibits a lower solubility than the
"11.0%" line.
Conversely, points below the "11.0%" line result in solvent containing > 11.0%
free CPTA,
and the salt solubility is greater than determined in Figure 3. To estimate
component
solubility, a constant multiplier factor, k, was used. The modified solubility
equations for the
(-)-Salt and the (+)-Salt are therefore S() = 0.01421ke 0.02613T and S(+) =
0.02868ke002771T.
[0140] Even with a good estimation of the (-)-Salt and the (+)-Salt solubility
by adjusting k,
one can still not describe the crystallization, for the other unknown is the
ratio of (-)-Salt and
(+)-Salt which is formed upon addition of the resolving agent base. One of the
more detailed
experiments is shown in Figure 5 (see also Figure 2). This experiment used
0.75 equivalent
of base and when sampled at 21.5 C, gave the product with (- / +)-Salt ratio
of 66.4/33.6%.
31
____________________________________ CA 02529774 2009-06-11
____________________________________
By heating the slurry and continuing to take samples, the saturation line for
both (-)-Salt and
the (+)-Salt in the solvent can be followed.
[01411 To match the solubility model to the actual data, a regression
technique was used,
whereby the solubility factor k and the feed ratio of(-)-Salt and (+)-Salt
were Manipulated to
give an answer (i.e., crystal composition, mother liquor composition, and
crystal yield) which
was consistent with the observed data. By selecting k= 0.68 and a feed ratio
for 0.75
equivalent of salt at 58.1% (-)-Salt / 41.9% (+)-Salt (i.e., 0.436 eq. of(-)-
Salt and 0.314 eq. of
(+)-Salt were formed upon addition of CAF D-Base), a good agreement was
obtained. Figure
6 shows the comparison. The solubility model allows calculation of the
complete mass
balance for the isolation: the amount of (-)-Salt and (+)-Salt in the crystal,
the amount of(-)-
Salt and (+)-Salt in the mother liquor, and also the amount of (-)-free CPTA
and (+)-free
CPTA in the mother liquor. One procedure for quantifying (- / +)-Salt and (- /
+)-free CPTA
in mother liquor by an extractive work-up, using solubility differences, is
provide in Example
19 below.
[0142] The regression technique with the solubility model was applied to other
experiments
which fed differing amounts of resolving agent. Using a combination of the
solubility factor
k and the composition of the salt as feed (i.e., the ratio of (-)-Salt and (+)-
Salt which was
formed upon the addition of base), the model tended to a unique solution which
fit the
experimental results. From these, the graph in Figure 7 was constructed. This
result shows
that as more resolving agent is added (above the extrapolated minimum-point of
0.34 eq.), an
increasing amount of (+)-Salt is formed. Without being bound by any theory, in
some
embodiments, it is believed that if less than 0.34 equivalent is added, the
CAF D:-Base will
coordinate substantially only with (-)-CPTA, forming almost exclusively (-)-
Salt.
Additionally, by aid of the curve in Figure 7, the amount of(-)-CPTA and (+)-
CPTA (free
acid) can be calculated. Between 0.35-0.75 equivalent of base charged, the %
ratio of {(-)-
CPTA / total CPTA free acid) is around 25% (23.3-27.1%), The "selectivity" for
the ratio of
(- / +)-Salt that is formed thus is dependent on the amount of free (-)-CPTA
that remains (in
solution), which comes to an endpoint of about (-)-CPTA / (+)-CPTA = 1 / 3. It
is believed
that once the (-)-CPTA concentration is depleted by addition of about 0.34 eq.
of base to a (- /
+)-CPTA ratio of 1 / 3, continued addition of base forms the (- / +)-Salt at a
ratio of 3 / 1 (to
keep free (- / +)-CPTA at a constant 1 /3 ratio in solution).
32
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 18
[0143] This example illustrates resolution of a racemic mixture of CPTA.
[0144] A 200-mL vessel was charged with 17.0 g of CPTA (51.4 mmol), 4.91 g of
CAF D-
Base (23.1 mmol, 0.450 eq.), and 85 mL of 2-propanol. The mixture was heated
to a solution
at 78 C, and then cooled at 0.5 C / min to 54 C. About 1/2 hour later, the
solution was
seeded with (-)-Salt to induce nucleation. After holding at 54 C for about 1-
1/2 hours, the
slurry was cooled to 22 C at 0.25 C / minute. After holding for 14 hours at
22 C, a small
sample (-5 mL) was taken and separated on a 15-mL, medium-fritted funnel. The
mother
liquor was weighted and saved, and the solid was washed with 2 mL of 2-
propanol. The
wash was weighed and saved, and suction was continued to dry the crystal.
Analysis by the
standardized HPLC system allowed calculation of weight% (-)-CPTA and (+)-CPTA
in each
stream. A mass balance around this sample (total accountability of CPTA in the
crystal,
mother liquor, and wash was 0.85 g) gave a 31.9% isolated yield of crystal
product from the
total CPTA. Crystal purity was 99.1 / 0.9% = (- / +)-CPTA ratio by weight.
Figure 8 shows
the analytical and mass balance results in the rectangular boxes. The
calculated yield (from
CPTA) based on feed/mother liquor/crystal composition is given inside the
circles.
Abbreviations in Figure 8 are as follows: R.A. = resolving agent, x or xtal =
crystal, ML =
mother liquor, Yld = yield.
[0145] The vessel was seeded several times with crystal containing (+)-Salt,
and about 2
hours later, 0.31 g of CAF D-Base (1.46 mmol, ¨0.03 eq.) was added. The vessel
was
sampled two times (see Figure 8) before the final isolation on a 60-mL medium-
fitted
funnel. The mother liquor was clear, pale yellow-gold, 59.1 g. The solid was
washed with
19.2 g of 2-propanol, with recovery of 18.8 g of wash solution. The washed
solid (10.07 g)
was further dried by suction on the funnel for an hour to 8.36 g (15.4 mmol
salt). Analysis of
all streams from the final isolation accounted for 13.45 g (40.67 mmol) of
CPTA. The final
crystal product ratio was (- /+)-CPTA = 89.2 / 10.8%, for an isolated yield of
(-)-CPTA =
33.8 /a (from CPTA). The calculated yield of(-)-CPTA, based upon the feed,
mother liquor,
and crystal composition, was 35.0%.
Example 19
[0146] This example illustrates an extractive work-up process to quantify (-
/+)-Salt and (-
/+)-CPTA in Mother Liquor.
33
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[01471 A mixture of the (-/+)-Salt, 80/20, was only sparingly soluble in
methylene chloride
at about 0.016%, while racemic CPTA was considerably more soluble at a little
less than
3.4%. The final mother liquor from separation of entry 4 of Figure 2 at 55.3 C
(see Figures
2 and 5) was analyzed by evaporating 0.1286 g to a glassy residue of 0.0242 g.
The residue
was dissolved in 5 mL of methylene chloride, seeded with (-/+)-Salt = 80/20,
and allowed to
stand overnight. The bulk of the supernatant liquid was removed, 3 mL of
methylene
chloride were added, and the bulk of the liquid was removed and combined with
the first
extract. The methylene chloride extract was evaporated to give a glassy solid,
0.0074 g, and
then analyzed by HPLC. The remaining thick slurry was evaporated to 0.0162 g
and
analyzed by HPLC. Results from the extractive work-up procedure are generally
similar to
the composition predicted by the solubility model, as shown in Figure 9.
Example 20
[0148] This example shows solubility of(-)- and (+)-CPTA=CAF D-Base salts in
alcohols
containing CPTA.
[0149] "Solvent" was prepared by dissolving 2.40 g of racemic CPTA in 19.42 g
of 2-
propanol (Fisher HPLC Grade) or 4.90 g of racemic CPTA in 31.4 g of ethanol.
The
respective concentrations of CPTA in solution were 11.0% and 13.5%. Solubility
of the (-)-
CPTA = CAF D-Base Salt (i.e., (-)-Salt) or (+)-CPTA = CAF D-Base Salt (i.e.,
(+)-Salt) was
determined by a gravimetric method. At a given temperature, a portion of the
supernatant
liquid from a saturated solution was remove to a vial of known weight. The
solution weight
was determined, and the volatile solvent was evaporated with a purge of
nitrogen. The solid
was further dried to constant weight in a vacuum oven at about 50 C/1 mm Hg.
The vial
was re-weighed to determine the loss of volatile solvent and weight of solid
remaining. From
this, the amount of dissolved CPTA from the "solvent" could be calculated.
Subtracting the
weight of total solid from the CPTA gave the weight of soluble salt in the
solvent. Data are
shown in Figures 10A and 10B.
Example 21
[0150] This example illustrates a method for preparing enantiomerically
enriched (-)-
halofenate.
[0151] CPTA was prepared in five steps, as discussed above, without
intermediate isolation
in about 85% yield following crystallization from heptane. Resolution gave an
average of
32% yield (max 50%) of >98% optically pure (-)-CPTA diastereomeric salt. After
removing
34
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
the resolving agent, the (-)-CPTA was esterified to give (-)-halofenate in
about 55% yield
using thionyl chloride and N-acetylethanolamine. By hydrolyzing the mother
liquor residue
with aqueous sodium hydroxide, (-)-CPTA can be recovered from the final
product mother
liquor and cycled back through the process. The resolving agent was isolated
from water in
about 90% recovery by a pH adjustment. Recovery and racemization of the (+)-
CPTA using
aqueous sodium hydroxide gave about 90% recovery. Overall, the first pass
yield from 4-
chlorophenylacetic acid was 15-17%. The entire eight-step process used three
organic
solvents, and three solid isolation steps.
Example 22
[0152] This example illustrates a method for preparing CPTA.
Br Br
CO2H COR Br2 COR 0
SOCl2
CI CI CI CI 0
1 R=C1 2 R=CI
CF3
1401
0 CF NaOH f-,, 1.1 ^
KOH OH
3
401
0 2-Propanol
0
01
CI OH CI
4
[0153] The synthetic route to CPTA is outlined above. Following bromination of
the acid
chloride 1 in 1,2-dichloroethane to give 2 2-propanol was added to give the
isopropyl ester 3.
The displacement reaction with a,a,a-trifluoro-m-cresol was accomplished using
potassium
hydroxide in 2-propanol. Following a water quench and wash and removal of the
1,2-
dichloroethane, the liquid 3 was added to a solution of a,a,a-trifluoro-m-
cresol and potassium
hydroxide in 2-propanol to give 4. The 2-propanol solvent was removed, and the
hydrolysis
to CPTA was completed by heating with aqueous sodium hydroxide.
[0154] The sodium salt of CPTA can be isolated as a solid by simply cooling
the reaction
mixture. Better isolated yields were obtained, however, by isolation of the
carboxylic acid.
For isolation, the basic aqueous CPTA reaction mixture was acidified with
hydrochloric acid,
and the CPTA was extracted into 1,2-dichloroethane. Solvent exchange of the
separated
organic phase from 1,2-dichloroethane to heptane afforded CPTA as a white
solid in
approximately 85% yield from 4-chlorophenylacetic acid.
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 23
[0155] This example shows solubility of CPTA in 1,2-dichloroethane and
heptane.
[0156] The solubility of racemic CPTA in 1,2-dichloroethane and heptane are
shown in
Figures 11 and 12, respectively. Included in the Figures are the equations for
the least-
squares fit of the data.
[0157] Based on the solubility profile of Figure 11, a concentration of
approximately 25
wt% CPTA in 1,2-dichloroethane at a temperature of approximately 35 C was
chosen for the
CPTA extraction conditions.
[0158] CPTA crystallization from heptane was exothermic. Seeding of a solution
of
approximately 170 g of CPTA in 500 mL of heptane at 46 C resulted in a
temperature
increase to 54 C as the crystallization progressed. Crystallization increased
the CPTA purity
as determined by HPLC analysis from 93-95 to >99 area %. HPLC assay of a
crystallization
mother liquor, which contained 15 area % of CPTA, found less than 3% yield
loss to the
mother liquor. As the purity was improved by crystallization, isolated yields
were high, and
the loss to the mother liquor was minor.
Example 24
[0159] This example shows yield of CPTA resolution under variety of
crystallization
conditions.
[0160] Results of CPTA resolution using CAF D-Base under various
crystallization
conditions are shown in Figure 13. Final chiral purity for each preparation,
obtained after
zero, one, or two recrystallizations, is in bold type. The molar ratio of the
CAF D-Base was
varied from 0.5 to 0.56. The amount of 2-propanol solvent listed for the
crystallizations and
reerystallizations are both based on the initial charge of racemic CPTA.
Chiral HPLC results
for both the isolated solids and mother liquors are normalized to 100%. The
calculated yield
and overall yield are calculated from the ratio of the (+)-enantiomer and (-)-
enantiomer forms
in the isolated solids and mother liquors. The actual percent yield in the
last column is of
weighed, dried material, and is based on a maximum yield of 50%.
[0161] Overall yields of the diastereomeric salt at >98% optical purity ranged
from 28 to
35%, and averaged 32%. In one case, using the lowest ratio of resolving agent,
this was
obtained without recrystallization (experiment 2 in Figure 13). The chiral
purity of the first
isolated solid ranged from 73% to 98%. A single recrystallization was
generally sufficient to
36
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
obtain the desired optical purity. A high overall yield was obtained when the
mother liquor
reached a 20/80 ratio of(-)-CPTA to (+)-CPTA.
[0162] Figure 14 shows the cooling profiles for the resolution
crystallizations listed in
order of decreasing yield of(-)-CPTA. Experiment number in Figure 14
corresponds to the
experiment number in Figure 13. The isolated yield of (-)-CPTA was determined
using the
calculated yield of Figure 13 and the percent of (-)-CPTA in the isolated
material. In general,
longer hold times at low temperatures led to an increase in yield.
[0163] Use of 0.45 molar equivalents of CAF D-Base consistently gave 35-37%
yield of
material that was >98% optically pure without the need for recrystallization.
Example 25
[0164] This example shows a method for separating (-)-CPTA from the CAF D-
Base.
[0165] To separate (-)-CPTA from the CAF D-Base, the diastereomeric salt was
mixed
with 1,2-dichloroethane, and aqueous hydrochloric acid was added to give a pH
in the
aqueous phase of less than about 2. The aqueous phase containing the
hydrochloride salt of
the CAF D-Base was separated. After a water wash of the organic phase, the
bulk of the 1,2-
dichloroethane was removed by distillation to remove residual water. Complete
solvent
removal gave an oil.
Example 26
[0166] This example shows a method for esterifying (-)-CPTA without any
significant
racemization.
0
,,c F3C OH 0
Soc12 HO
101 0
C
CI I
[0167] (-)-CPTA was reacted with thionyl chloride in 1,2-dichloroethane at
reflux to yield a
corresponding acid chloride. Reaction progress can be monitored by HPLC
analysis. A
small amount of distillate was removed to remove excess thionyl chloride. The
mixture was
cooled, and a large excess of vacuum distilled N-acetylethanolamine was added.
Stirring at
ambient temperature gave (-)- halofenate.
37
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[01681 The esterification reaction mixture was quenched by adding the reaction
mixture to
an aqueous potassium carbonate solution. (-)- Halofenate was isolated by
solvent exchange
and crystallization from the 6:1 heptane:2-propanol. Results are summarized in
Figure 15.
[01691 First crop isolated yields ranged from 47 to 59% and averaged 55%. This
isolated
yield represents a reaction yield of 75 to 80% for this step. A second crop
afforded a higher
overall yield; however, the product quality was poorer with the second crop
material.
[0170] Molar accountability of the CPTA loaded, found as isolated halofenate,
and
halofenate and CPTA in the mother liquor, ranged from 90 to 99%.
Example 27
[01711 This example shows a method for recovering and recycling (+)-CPTA.
[01721 Heating CPTA in aqueous base caused racemization. The remaining CPTA
from
the resolution step in Example 25 was approximately 47 %ee of the (+)-
enantiomer, which
also contains residual CAF 0-Base.
[0173] To recover and racemize the (+)-CPTA, the 2-propanol solvent was
removed and
replaced with 1,2-dichloroethane. Washing with water at a pH below about 2
removed the
CAF-D-Base for subsequent recovery. Aqueous sodium hydroxide was added, and
the
aqueous solution heated to reflux. The 1,2-dichloroethane was either removed
by distillation
prior to the addition of the basic solution, or by a phase separation
following addition of the
basic solution. An 89% yield of racemic CPTA was isolated from heptane after
heating an
aqueous solution for four hours with 1.4 molar equivalents of sodium
hydroxide. Isolation of
CPTA as a crystallized intermediate provided a more consistent quality feed
for the
resolution step.
[01741 The solubility of the sodium salt of racemic CPTA in water, determined
and
expressed as the acid form, is shown in Figure 16. Addition of the isolated
sodium salt to
water gave a pH of about 9.5, and the solubility profile shown in the upper
solubility curve.
Addition of a small amount of sodium hydroxide to give a pH of about 12.6
decreased the
aqueous solubility to that shown on the lower curve.
Example 28
101751 This example shows a method for producing CPTA from (+)-halofenate.
38
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
[0176] Addition of from 1 to 3 molar equivalents of sodium hydroxide to about
10 wt% of
87 %ee (+)-halofenate in water and warming to 50 to 60 C resulted in a
substantially
complete hydrolysis to CPTA. Partial racemization to give approximately 70 %ee
(+)-CPTA
occurred (Time = 0 of Figure 17). The solution was heated to reflux, and the
enantiomeric
ratio monitored over time. With 3 molar equivalents of base, almost complete
racemization
(<3 %ee by the chiral HPLC analysis method) occurred in less than 2 hours at
reflex. The
pH dropped from 12.8 to 12.6 over the course of the racemization. A slightly
longer reaction
time was required with 2 molar equivalents (pH 12.6 to 11.6). With 1 molar
equivalent,
racemization stopped at approximately 60 to 70 %ee, with a final pH of 9.4.
[0177] Use of 0.5 molar equivalents of sodium hydroxide left approximately 40%
of the
halofenate unhydrolyzed after 2 hours at 60 C; heating to reflux overnight
left approximately
1 % halofenate at a final pH of 4.8. This did not significantly minimize
racemization. The
amount of CPTA produced was 72.6 %ee of the (+)-enantiomer.
Example 29
[0178] This example illustrates a method for recovering (-)-CPTA from (-)-
halofenate
crystallization mother liquor.
[0179] As noted previously and shown in Figure 15, the (-)-halofenate
crystallization
mother liquor contains a large amount of (-)-halofenate and (-)-CPTA. By
hydrolysis of the
(-)-halofenate, additional (-)-CPTA can be generated as feed for the
resolution step.
[01801 Hydrolysis of a (-)-halofenate crystallization mother liquor (88.3 %ee
of(-)-
halofenate) at 50 C and a final pH of 12.7 rapidly gave 65.8 %ee (-)-CPTA.
The (-)-CPTA
was recovered as the CAF D-Base diastereomeric salt (96.4 %ee) by addition of
CAF D-Base
to a 2-propanol solution. From the amount of diastereomeric salt initially
loaded, 55 mol%
was obtained as (-)-halofenate, 28% was recovered as the (-)-CPTA CAF D-Base
salt, and
14 mol% remained as CPTA in the mother liquor.
Example 30
[0181] This example illustrates a method for recovering CAF D-Base.
[0182] The CAF D-Base is found in the acidic phase from separation of(-)-CPTA
from the
diastereomeric salt, and from the acidic wash step of the CPTA recovery from
the resolution
mother liquors. Basification with aqueous sodium hydroxide to a pH greater
than about 12
resulted in precipitation with good recovery in a form that was easily
filtered. Results are
39
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
shown in Figure 18. Recovery from the diastereomeric salt was generally
greater than 90%;
recovery from the resolution mother liquor was lower. Concentrations in the
aqueous
solution ranged from about 5 to 20%.
[0183] The enantiomeric purity of the CAF D-Base can be determined by careful
analysis
of the melting point by DSC (D. Pitre, M. Nebuloni, and V. Ferri; Arch. Pharm.
(Weinheim)
324, 525 (1991)). As the conglomerate of the (+)- and (-)-forms, e.g.,
racemate, melts more
than 20 C lower than the pure enantiomer, melting point was found to be a
sensitive method
for assessing enantiomeric purity. However, measurement of the enantiomeric
purity of two
of the samples by chromatographic separation of a derivative showed no loss of
chiral purity.
The enantiomeric purity of the recovered CAF D-Base, near the detection limit
of the HPLC
analysis method, was indistinguishable from the source material.
Example 31
[0184] This example illustrates another method for preparing racemic CPTA.
[0185] A 500-mL round-bottom flask in a heating mantel and fitted with an
overhead stirrer
and condenser was charged with 73.28 g (0.430 mol) of 4-chlorophenylacetic
acid, 70 ml, of
1,2- dichloroethane, and 41 mL (0.56 mol) of thionyl chloride. The mixture was
warmed at
50 to 55 C for 19 h. The reaction mixture was analyzed by HPLC analysis. To
the solution
of acid chloride was added 29 mL (0.57 mol) of bromine, and the solution was
warmed at 70
to 75 C for 20 h. The resulting a-bromo product was cooled in an ice bath and
100 mL (1.31
mol) of 2-propanol was added dropwise. The maximum temperature reached was 17
C.
After cooling to 4 C, the reaction mixture was added to water. The solution
was warmed to
ambient temperature, and the aqueous layer was removed. The organic phase was
washed
with 37 mL of water. The separated 1,2- dichloroethane solution was evaporated
to give
134.1 g of an oil.
[0186] A 1-L round-bottom flask with an overhead stirrer was charged with 34.0
g (0.515
mol) of 85% potassium hydroxide and 370 mL of 2-propanol. The mixture was
warmed to
41 C using a water bath to dissolve much of the solid. The mixture was cooled
in an ice
bath, and 73.8 g (0.455 mol) of a,a,a-trifluoro-m-cresol was added dropwise.
The maximum
temperature reached was 13 C. The solution was cooled to 5 C before the
dropwise
addition of 134.1 g of the oil obtained above. The material was rinsed in with
18 g of 2-
propanol. The slurry was evaporated to a residue, then charged with 250 mL of
water and
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
42.8 g (0.535 mol) of 50% aqueous sodium hydroxide. The mixture was heated to
reflux for
1 h.
[0187] After cooling to ambient temperature, the mixture was diluted with 250
mL of 1,2-
_
dichloroethane, and the pH was decreased to 0.3 by the dropwise addition of 71
g (0.72 mol)
of 37% hydrochloric acid. After a phase separation, the solvent was removed
from the 1,2-
dichloroethane phase to give 202.2 g of residue. The residue was treated with
131 g of
heptane, and evaporated to a residue of 164 g. The process was repeated with
97 g of
heptane, giving 160 g of an oil. The residual oil was stirred at ambient
temperature with 257
g of heptane to give a slurry, which was chilled in an ice bath before
isolation of the solid by
filtration. The filter cake was washed with 49 g of heptane, then dried under
a vacuum to
give 125.58 g (0.380 mol, 88% yield) of CPTA.
Example 32
[0188] This example illustrates a method for preparing a racemic mixture of
compound 4 of
Example 22.
[0189] A 50-mL round-bottom flask equipped with a magnetic stirrer and reflux
condenser
was charged with 2.10 g (6.35 mmol) of racemic CPTA, 21 g of 2-propanol, and
0.50 g (4.2
mmol) of thionyl chloride. HPLC analysis after 90 minutes at reflux indicated
84.2 area% of
7 and 12.7 area% of CPTA. An additional 1.0 g (8.4 mmol) of thionyl chloride
was added to
give less than 1 area% of CPTA. The solution was cooled to ambient temperature
and treated
with 1.0 g (12 mmol) of solid sodium bicarbonate. The solvent was evaporated,
and the
residue dissolved in 25 mL of toluene. After washing with water (2 x 10 mL),
the solvent
was evaporated to a residue of 2.31 g (6.2 mmol, 98% yield) of compound 4 of
Example 22
(95.8 area% of 7, 2.4 area% of toluene).
Example 33
[0190] This example illustrates a method for determining solubility of racemic
CPTA.
[0191] A 100 mL water jacketed resin pot with a magnetic stirrer was connected
to a
recirculating water bath and charged with 9.44 g of racemic CPTA and 16.78 g
of 1,2-
dichloroethane. The bath temperature was warmed to 35 C, and the slurry was
stirred for
one hour. The agitator was shut off, and the solid was allowed to settle for
30 min. A 0.1360
g sample of the supernate was removed and diluted to 25.00 mL with
acetonitrile, and the
solution was assayed by HPLC analysis. Results for this and a series of other
measurements
are shown in Figures 11 and 19. For analysis at about 2 C, a 0.54 g-sample of
CPTA in 1.92
41
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
g of 1,2-dichloroethane was stored in a refrigerator overnight before analysis
of the supernate
by HPLC analysis. The solubility of CPTA in heptane, included in Figure 19
shown in
Figure 12, was determined in a similar fashion.
Example 34
[0192] This example illustrates a method for resolving a racemic mixture of
CPTA.
[0193] A 1-L bottom-drain reactor was charged with 48.2 g (146 mmol) of CPTA,
16.4 g
(77.3 mmol) of (1R,2R)-(-)-2-amino-1-(4-nitropheny1)-1,3-propanediol (CAF D-
Base), and
193 g of 2-propanol. The slurry was heated to 70 C to give a solution, then
cooled to 60 C
and held for 1 h. The resulting slurry was cooled at 0.25 C/min to a jacket
temperature of 2
C and held for 14 h; the internal temperature was 4 C. The solid was isolated
by vacuum
filtration and rinsed with 27 g of 2-propanol. The mother liquor and wash
solution was
sampled for HPLC analysis, and the results are shown in Figure 13. The 50.48-g
wetcake
was reloaded to the 1-L reactor with 193 g of 2-propanol, and the slurry
warmed to a gentle
reflux with a jacket temperature of 85 C to give a solution. The solution
was, sampled for
HPLC analysis; the results are listed in Figure 13. A slurry formed upon
cooling to 65 C.
After warming to 68 C for 30 min, the slurry was cooled to 40 C at 0.25
C/min, then to 18
C at 0.4 C/min, then to 2 C at 1 C/min. (In other preparations, linear
cooling rates
recorded in Figure 14 were used.) The solid was isolated by vacuum filtration,
rinsed with
4., 18 g of 2-propanol, and dried under vacuum to give 27.29 g (50.4 mmol,
34.5% yield) of(-)-
CPTA / CAF D-Base. HPLC analysis results for the isolated solid and mother
liquor and
wash are included in Figure 13.
Example 35
[0194] This example illustrates preparation and resolution of racemic CPTA
from
halofenate.
[0195] A 1-L round-bottom flask with an overhead stirrer was charged with
129.75 g
(0.312 mol) of racemic halofenate, 325 g of water, and 32.6 g (0.408 mol) of
50% aqueous
sodium hydroxide. The slurry was heated to 60 C for 1 hour to give a
solution, then cooled.
At a temperature of 40 C, 328.5 g of 1,2-dichloroethane and 44 g (0.45 mol)
of 37%
hydrochloric acid were added, and the two-phase mixture was cooled to 29 C.
The pH of the
aqueous phase was 0.85. The organic phase was separated and washed with 250 mL
of
water, then evaporated to a residue of 118.2 g. 2-Propanol (149 g) was added,
and
evaporated to a residue of 131.2 g. The residue, containing theoretically
103.2 g of racemic
42
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
CPTA based on the amount of halofenate loaded, was charged to a 1-L bottom-
drain reactor
with 33.10 g (0.1556 mol) of CAF D-Base and 400 g of 2-propanol. The mixture
was
warmed to 67 C to give a light slurry, then cooled to 1 C at 0.075 C/min.
The mixture was
chilled to -7 C, and the solid isolated by vacuum filtration and washed with
60 mL of 2-
propanol. HPLC analysis results of the isolated solid and the 492.8-g mother
liquor and wash
solution are shown in Figure 13 (experiment 9). The 92.74-g wetcake was
reloaded to the 1-
L reactor along with 477 g of 2-propanol, and the mixture heated to 75 C to
give a solution.
The solution was cooled to 5 C at 0.5 C/min, and the crystallized solid
isolated by vacuum
filtration, rinsed with 60 mL of 2-propanol, and dried to give 51.81 g (0.0956
mol, 31%
yield) of the (-)-CPTA CAF D-Base diastereomeric salt. HPLC analysis results
for the
isolated solid and 529.9 g of mother liquor and wash solution are included in
Figure 13.
Example 36
[0196] This example illustrates a method for racemizing (+)-CPTA and
recovering racemic
CPTA.
[0197] The resolution and recrystallization mother liquors from the resolution
of 103.2 g of
CPTA described in Example 35 above, containing 71.6 g (0.217 mol) of CPTA (44
%ee of
the (+)-enantiomer) based on the yield and purity of the isolated
diastereomeric salt, was
evaporated to a residue of 108.7 g. The residue was treated with 176 g of 1,2-
dichloroethane,
35.2 g of water, and 6.8 g of 37% hydrochloric acid. The organic phase was
removed and
evaporated to a residue to 79.0 g. Water (80 g) was added, and the solvent
evaporated to a
residue of 78.1 g. The residue was treated with 141.9 g of water and 24.6 g
(0.308 mol) of
50% aqueous sodium hydroxide, and the solution was heated to reflux for 4
hours to give a
racemate by chiral HPLC analysis. The solution was cooled and treated with 140
ml, of 1,2-
dichloroethane and 32.0 g (0.325 mol) of 37% hydrochloric acid. The organic
phase was
removed and evaporated to a residue of 80.1 g, which was treated with 250 mL
of heptane in
a 40 C water bath to give a slurry. The solid was isolated by vacuum
filtration and dried to
give 63.83 g (0.193 mol, 89% yield) of racemic CPTA. Resolution of a sample
gave results
consistent with those of fresh CPTA (entry 10 of Figure 13).
Example 37
[0198] This example illustrates a method for isolating (-)-CPTA from the
diastereomeric
salt.
43
CA 02529774 2005-12-16
WO 2004/112774
PCT/US2004/019616
[0199] A 500-mL flask with a magnetic stirrer was charged with 40.0 g (73.7
mmol) of(-)-
CPTA / CAF D-Base, 100 g of 1,2-dichloroethane, 40 g of water and 7.6 g (77
mmol) of 37%
hydrochloric acid. After complete dissolution of the solid, the lower organic
phase was
removed and washed with 10 mL of water. The pH of the combined aqueous phase
was 0.9.
HPLC assay of 128.2 g of the organic phase found 24.32 g (73.6 mmol, 99.8% of
theory) of
(-)-CPTA as a solution in 1,2-dichloroethane.
Example 38
[0200] This example illustrates a vacuum purification of N-acetylethanolamine.
[0201] A 50-mL round-bottom flask equipped with a magnetic stirrer, heating
mantel and a
short path distillation head was charged with 29.09 g of N-acetylethanolamine
and placed
under a vacuum of approximately 0.8 torr. Bubbles formed as the liquid was
heated,
although no condensate was collected. Distillate was collected at a head
temperature of
approximately 130 C to afford 26.71 g (92% recovery) of N-acetylethanolamine
as a clear
liquid.
Example 39
[0202] This example illustrates a method for producing (-)-halofenate.
[0203] A 500-mL round-bottom flask with a magnetic stirrer was charged with
35.5 g (65.4
mmol) of the (-)-CPTA / CAF D-Base diastereomeric salt (99.4 %ee), 89.0 g of
1,2-
.,
dichloroethane, and 35.5 mL of water. To the slurry was added 6.7 g (68 mmol)
of 37%
hydrochloric acid, and the mixture was stirred at ambient temperature to give
two clear
phases. The lower organic phase was removed and washed with 7.0 g of water.
The organic
phase was evaporated to a residue of 26.13 g, then dissolved in 55.6 g of 1,2-
dichloroethane
and placed in a 250-mL round-bottom flask in a heating mantel with a magnetic
stirrer and
fitted with a reflux / distillation head. HPLC assay of the solution found
22.06 g (66.7 mmol,
102% of theory) of CPTA. To the solution was added 7.5 mL (100 mmol) of
thionyl
chloride, and the solution was heated to reflux for 2 hours. Heating was
continued to collect
6.1 g of distillate. The solution was cooled to ambient temperature, then
chilled in an ice bath
for the addition of 25.85 g (251 mmol) of distilled N-acetylethanolamine (KF
analysis 1176
and 1288 ppm water). The temperature rose to about 26 C after the addition.
The solution
was added slowly with stirring to 9.90 g (71.6 mmol) of potassium carbonate in
36 g of water
chilled in an ice bath. The maximum temperature reached was 15 C. The
reaction mixture
was rinsed in with 5 mL of 1,2-dichloroethane. The lower organic phase was
removed and
44
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
washed with 37 mL of water. The solution was evaporated to give an oil (32.84
g). The oil
was treated with 54 g of heptane, and the solvent was removed to give 31.56 g
of a solid
residue. To the solid was added 76 g of heptane, and the solvent was removed
to give 29.19
of a solid residue. The solid was dissolved in 28 mL of 2-propanol at 40 C,
then diluted with
an additional 28 mL of 20 propanol and 334 mL of heptane. Cooling to ambient
temperature
gave a thin slurry. A thick slurry formed upon cooling in an ice bath. After
stirring for 2
hours, the solid was isolated by vacuum filtration, rinsed with 29 g of
heptane, and dried to
give 14.21 g (34.2 mmol, 52.3% yield) of(-)-halofenate. No (+)-halofenate was
detected by
chiral HPLC analysis (>99.8% ee).
[0204] HPLC assay of 294.1 g of the mother liquor and wash found 11.2 g of
halofenate
and 1.26 g of CPTA. The solvent was evaporated, and 12.47 g of the residue was
dissolved
in 14 mL of 2-propanol. Addition of 84 mL of heptane gave a slurry after
stirring overnight
at ambient temperature. The slurry was chilled in an ice bath and the solid
was collected,
rinsed with 9 g of heptane, and dried to give 5.64 g (13.6 mmol, 20.7% yield,
89.9%
halofenate and 3.9% CPTA by HPLC analysis, 99.6 %ee) of(-)-halofenate. HPLC
assay of
81.74 g of the mother liquor and wash found 3.66 g (8.8 mmol, 13.5%) of
halofenate and
0.93 g (2.8 mmol, 4.8%) of CPTA.
Example 40
[0205] This example illustrates a method for isolating racemic CPTA sodium
salt.
[0206] The mother liquors from a resolution crystallization and
recrystallization containing
in theory 63.9 g (0.193 mol) of CPTA based on the resolution recovery was
evaporated to a
residue of 91 g. The residue was dissolved in 146 g of 1,2-dichloroethane and
treated with
28.6 g of water and 6.3 g of 37% hydrochloric acid at 40 C. The 219 g organic
phase was
evaporated to a residue of 71.86 g. To the residue was added 120 g of water
and 21.5 g
(0.269 mol) of 50% sodium hydroxide. The solution was heated to reflux, then
allowed to
cool to ambient temperature to give a thick slurry. The solid that formed upon
cooling was
isolated by vacuum filtration, rinsed with 25 mL of water, then dried to give
31.78 g (0.0901
mol, 46.7% recovery) of the sodium salt of CPTA. Chiral HPLC analysis found
that the
material was racemic. HPLC assay of the 188.6 g mother liquor and wash found
28.3 g
(0.0856 mol, 44.4%) of CPTA.
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
Example 41
[0207] This example illustrates a method for determining the solubility of
racemic CPTA
sodium salt.
[0208] A 100-mL water jacketed resin pot with a magnetic stirrer was connected
to a
recirculating water bath and charged with 3.48 g of the racemic CPTA sodium
salt and 20.0 g
of water. The bath temperature was warmed to 35 C, and the slurry was stirred
for one hour.
The agitator was shut off, and the solid was allowed to settle for 30 min. The
pH was 9.4. A
0.3036 g-sample of the supernate was removed and diluted to 25.00 mL with
acetonitrile, and
the solution was assayed by HPLC analysis. Analysis was repeated at 47 C and
at 19 C.
An additional 3.01 g of CPTA sodium salt was added to maintain a slurry at the
higher
temperature, and 25 g of water was added to give a thinner slurry at the lower
temperature.
The pH was increased to 12.7 at ambient temperature by the addition of 50%
aqueous sodium
hydroxide, and the analysis was continued at 13.5, 25, 34, and 42 C. Results
are shown in
Figures 16 and 20.
Example 42
[0209] This example illustrates hydrolysis and racemization of (+)-halofenate.
[0210] A 250-mL round-bottom flask equipped with a magnetic stirrer and
heating mantel
was charged with 7.28 g (17.5 mmol) of (+)-halofenate (86.9 %ee), 72.2 g of
water, and 4.21
g (52.6 mmol) of 50% aqueous sodium hydroxide. The slurry was heated to 50 to
60 C. The
pH of the resulting solution was 12.8. Chiral HPLC analysis showed 80.4% of
(+)-CPTA and
10.5% of(-)-CPTA. The solution was heated to reflux for 90 minutes. Chiral
HPLC analysis
showed 49.6% of (+)-CPTA and 47.0% of(-)-CPTA. The pH was 12.6. After cooling
to
ambient temperature, approximately 50 mL of 1,2-dichloroethane was added, and
the pH was
adjusted to 0.8 by the addition of 7.3 g (74 mmol) of 37% hydrochloric acid.
The organic
phase was evaporated to a residue of 6.0 g. The residue was treated with 25 mL
of heptane,
warmed to dissolve the oil, and then cooled in an ice bath. The solid was
collected by
vacuum filtration and dried to give 5.10 g (15.4 mmol, 88% yield) of racemic
CPTA. Data
for this and two similar hydrolyses are shown in Figures 17 and 21.
[0211] Similarly, heating 6.75 g (16.3 mmol) of (+)-halofenate with 0.65 g
(8.1 mmol) of
50% aqueous sodium hydroxide in 67.5 g of water for 2 hours at 60 C gave
37.5% of
halofenate and 54.2% of CPTA. Heating to reflux overnight gave 92.1% of CPTA
and 1.1%
46
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
of halofenate, with a final pH of 4.8. Chiral HPLC analysis found an 80.3/12.8
ratio of (+)/(-
)-CPTA.
Example 43
[02121 This example illustrates preparation of (-)-halofenate with recovery of
the (-)-
CPTA/CAF D-Base diastereomeric salt from the (-)-halofenate crystallization
mother liquors.
[02131 A 1-L round bottom flask with magnetic stirring was charged with 50.0 g
(92.3
mmol) of the (-)-CPTA /CAF D-Base diastereomeric salt (97.1 %ee), 124 g of 1,2-
dichloroethane, 50 mL of water, and 9.6 g (98 mmol) of 37% hydrochloric acid.
The organic
phase was separated and washed with 50 mL of water, then placed in a 250-mL
round-bottom
flask in a heating mantel with a magnetic stirrer. A reflux / distillation
head was attached,
and the solution was heated to remove 35.4 g of distillate by distillation.
After cooling to 40
C, the solution was diluted with 25 mL of 1,2-dichloroethane, and 11 mL (150
mmol) of
thionyl chloride was added. After heating at reflux for 2 hours and removing
22.6 g of
distillate, the solution was cooled in an ice bath for the dropwise addition
of 38.6 g (374
mmol) of distilled N-acetylethanolamine. The reaction temperature rose from 7
to 18 C
during the addition. After stirring overnight at ambient temperature, the
solution was added
with stirring to 12.7 g of potassium carbonate in 51 mL of water chilled in an
ice bath. The
organic phase was removed and washed with 51 g of water. The organic phase
(85.2% of
halofenate and 6.1% CPTA by HPLC analysis) was evaporated to an oil of 44.3 g,
treated
with 133 g of heptane, then evaporated to a solid of 43.3 g. The solid residue
was dissolved
in 61.5 g of 2-propanol and charged to the 1-L bottom-drain reactor along with
320 g of
heptane, warmed to 50 C, and cooled at 3 C/min to 20 C, then at 1 C/min to
-3 C. The
solution became hazy at 27 C, and a thick slurry formed at 15 C. The solid
was isolated by
vacuum filtration, washed with 40 mL of heptane containing 5 mL of 2-propanol,
and dried
to give 21.01 g (50.6 mmol. 55% yield, 98.93% by HPLC) of (-)-halofenate (99.9
%ee). The
395.7 g mother liquor and wash solution, containing 14.65 g (35.3 mmol) of
halofenate (88.3
%ee) and 1.78 g (5.4 mmol) of CPTA by HPLC assay, was evaporated to a residue
of 21.57
g. The residue was heated to 50 C with 100 mL of water and 5.0 g (63 mmol) of
50%
aqueous sodium hydroxide to give a solution. HPLC analysis after about 10
minutes found
83.6% of CPTA and 0.3% of halofenate. The solution was cooled, diluted with 50
mL of 1,2-
dichloroethane, and the pH decreased from 12.7 to 1.6 with 7.3 g (74 mmol) of
37%
hydrochloric acid. After washing with 30 mL of water, the 72.9 g organic
phase, containing
11.32 g (34.2 mmol) of CPTA by HPLC assay, was evaporated to a residue,
treated with 36 g
47
CA 02529774 2005-12-16
WO 2004/112774 PCT/US2004/019616
of heptane, then evaporated to a residue of 14.9 g. The oily residue was
dissolved in 38 g of
heptane with heating. Cooling gave an oil. The solvent was removed and the
residual oil
dissolved in 34.8 g of methylcyclohexane. An oil formed with cooling. The
solvent was
removed and replaced with 45.6 g of 2-propanol. Chiral HPLC analysis found
65.8 %ee of(-
)-CPTA (a (+)/(-)-ratio of 16.9/81.6). To the solution at ambient temperature
was added 6.50
g (30.6 mmol) of CAF D-Base. A thick slurry rapidly formed. The slurry was
warmed to 40
C with stirring, then cooled in an ice bath and the solid isolated by a vacuum
filtration,
washed with 7 g of 2-propanol, and dried to give 13.91 g (25.7 mmol) of (-)-
CPTA/CAF D-
Base diastereomeric salt, which corresponds to 28% recovery of the 50.0 g of
salt initially
loaded. The (+)/(-)-CPTA ratio was 1.77/97.86. HPLC assay of the 45.34-g
mother liquor
and wash solution found 4.34 g (13.1 mmol) of CPTA, which corresponds to 14
mol% of the
50.0 g of salt initially loaded.
Example 44
[0214] This example illustrates a process for recovering CAF D-Base from
CPTA/CAF D-
Base salt.
[02151 A 1-L round-bottom flask with a magnetic stirrer was charged with 80.16
g (0.148
mol) of the (-)-CPTA/CAF D-Base salt, 237 g of 1,2-dichloroethane, and 80 mL
of water. To
the slurry was added 15.2 g (0.154 mol) of 37% hydrochloric acid, giving two
clear phases.
The pH of the aqueous layer was 1.2. The lower organic layer was removed and
washed with
16 mL of water. The combined aqueous phase (140.7 g) was treated with 12.9 g
(0.161 mol)
of 50% aqueous sodium hydroxide to reach a pH of 12.1. The resulting slurry
was filtered
and the solid was rinsed with 25 mL of water and dried to give 30.79 g (0.145
mol, 98%
recovery) of CAF D-Base (mp 160.4-161.0 C).
Example 45
[0216] This example illustrates a process for recovering CAF D-Base from the
resolution
mother liquor.
[0217] A 60.0 g sample of racemic CPTA was resolved with 20.88 g of CAF D-Base
in
240 g of 2-propanol as described above to give a 74.7 g wetcake. The wetcake
was
recrystallized in 218 g of 2-propanol to give 32.35 g (32.8% yield) of (-)-
CPTA/CAF D-Base
salt. The mother liquor and wash solutions from the crystallization and
recrystallization,
theoretically containing 40.32 g of CPTA and 8.23 g (38.8 mmol) of CAF D-Base
from the
amount of salt obtained, was evaporated to a residue of 72.9 g. The residue
was dissolved in
48
CA 02529774 2012-11-05
-
265 g of 1,2-dichloroethane, 50 mL of water, and 4.0 g (40.6 mmol) of 37%
hydrochloric
acid. The aqueous layer was separated, and the pH was increased from 0.6 to
12.3 by the
addition of 3.88 g (48.5 mmol) of 50% aqueous sodium hydroxide. The resulting
slurry was
filtered and the solid was collected and rinsed with water to give 7.12 g
(33.6 mmol, 87%
recovery) of CAF D-Base (mp 162.4-163.0 C).
49