Note: Descriptions are shown in the official language in which they were submitted.
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
SAMPLE PRESENTATION DEVICE WITH DIFFERING WETTABILITY
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates to sample presentation devices useful in
performing
analytical measurements. In addition, the present invention relates to the
fabrication and
use of sample presentation devices.
Background
Most scientific fields that involve some kind of chemical and biological
analysis of
a sample require researchers to be able to identify and measure compounds or
analytes
found in aqueous solutions (e.g., the measurement of proteins in blood plasma
or the
measurement of pesticides in runoff from streams). Here, analytes generally
refer to
component(s) of a liquid sample that are of interest to an investigator.
Typically, fluid
samples containing analytes are presented to an analytical measurement
instrument by
means of a container (e.g., test tube, multiwell plate, or cuvette) or other
presentation
device (e.g., slide or biochip). Because of the overriding interest in
measuring a large
number of samples quickly (so called "high throughput" measurement of
samples), much
attention has been paid to developing standardized containers and devices that
can be used
in connection with automated analytical instruments. For example, in the drug
discovery
field, researchers interested in screening drug candidates frequently screen
thousands or
even millions of possible drug candidates using various analytical techniques
(e.g.,
fluorescence polarization detection), many of which use standard 384 well
plates to contain
the sample solutions containing the drug candidates. As such, sample
presentation devices
constitute a critically important component of a researcher's analytical
equipment in a wide
range of scientific fields, ranging from genomics and proteomics, drug
development,
clinical diagnostics, and analysis of environmental or biological toxins or
agents (e.g.,
assessing environmental contamination and screening for possible agents used
in
bioterrorism).
In genomics and proteomics, for example, the focus is on the identification
and
study of DNA/RNA and proteins/peptides, respectively. These fields
collectively refer to
the systemic study of chemical and biological moieties in living organisms,
their
interactions, and the analytical techniques required to discern them.
Understanding
-1-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
complex living systems, rather than individual cell components, is a major
focus of current
biological and biomedical research in both fields. Specifically, a principal
aim of
genomics is to sequence and generate large databases of the gene content of
entire
organisms. Genomes have been compiled for bacteria, yeast, nematodes,
drosophila, and,
most recently, humans. Similarly, proteomics is the study of all proteins
expressed at a
specific time in the cell, a principal aim of which is to obtain partial
protein amino acid
sequences that can be used with database matching tools to identify an entire
protein, as
opposed to completely sequencing a protein. The identification of proteins
allows for the
study of protein expression (important to identify proteins that are
differentially expressed
under different conditions and biomarkers for disease states) as well as
mapping protein
interactions (which helps develop a picture of the cell architecture).
Understanding the
role of proteins is critical to our understanding of living systems, as
proteins are the main
component of biomatter and perform virtually all critical biological
functions, from
regulating reactions, to transport of oxygen, to providing cellular and
extracellular
structure. As with genomics, the burgeoning field of proteomics has resulted',
in the
generation of information about the proteome of humans and other organisms,
and, while
this information is still incomplete, much of this information is and will be
stored in
databases. It is expected that much of our future understanding of living
systems will be
extracted from these genomic and proteomic databases.
In the field of clinical diagnostics, researchers focus on the identification
and
measurement of a wide range of analytes. The analytes of interest may be the
actual drug
candidates, such as in the example of bioavailability studies conducted in the
course of
clinical trials that reveal the extent to which a drug candidate is present
throughout the
organism. Alternatively, the analyte of interest may reflect a physiological
response to a
drug candidate, such as in the case of measuring the presence or absence of
phosphorylated
reaction products of kinase enzyme reactions. Because kinase enzymes are
important in
the growth and reproduction of cells, a high level of kinase activity is
observed in patients
suffering from diseases in which growth is abnormal (e.g., cancers). Drugs
that result in a
reduction of kinase activity are thus possible anti-cancer therapeutics, and
analytical
methods of detecting the efficacy of such drug candidates often focus on
measuring the
presence or absence of analytes in the form of kinase enzyme reaction
products. These and
other kinds of direct and indirect measurements of analytes of importance in
clinical
-2-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
diagnostics and drug development depend on the existence of analytical
techniques and
sample presentation devices that facilitate their measurement.
The importance of sample presentation devices is by no means limited to the
biomedical context. For example, researchers interested in determining the
extent of
environmental contamination (or remediation) need to be able to screen
environmental
samples of all kinds, including water, air, and soil samples. Many of the
analytical
techniques used to analyze such sample involve analysis of liquid samples, as
is the case of
water quality studies or in the case of soil samples that have been extracted
by diluting in
organic and/or inorganic solvents so as to remove various components. Sample
presentation devices that can present liquid samples for analysis are
therefore an important
tool in accomplishing these kinds of analytical measurements.
In the post-September 11 world, governments are confronted with the need for
platforms and analytical techniques to facilitate the detection of chemical
and biological
agents in both military and civil scenarios. Challenges for biowarfare
detection include
sample collection and distinguishing between innocuous versus toxic organisms.
The
current battlefield technique for bioagents utilizes pyrolysis to convert
biological
compounds to small molecules that can be more easily detected by mass
spectometry
(MS). Development of techniques that rely upon protein or peptide biomarkers
is
anticipated, however, because it would be more specific than currently known
methods,
and could be used to determine potential exposure to warfare agents in
combination with
breath tests, urinalysis, or blood drawing techniques. Stand-alone biosensors
as alerting
devices are also of great interest for use on the battlefield as well as in
public places. All
of these methods present challenges in sample collection, pre-treatment, and
presentation
of samples to detectors.
A wide variety of analytical techniques have been developed to identify and
measure compounds of interest in liquid samples, such as DNA, RNA, proteins,
and
peptides in blood sera, environmental toxins and agents in environmental
samples. While
each of these analytical techniques is useful in its own way, each is at least
partially
dependent upon the type of sample presentation device that is employed. Thus,
limitations
inherent to such devices may adversely affect the measurement of compounds of
interest
using these analytical techniques.
-3-
CA 02531972 2010-11-22
78899-7
Moreover, many analytical techniques focused on identifying, isolating or
measuring analytes in liquid samples require that the sample undergo separate
pre-
processing steps - i.e., processing of the sample before it is exposed to a
particular
analytical technique to determine the presence and amounts of analytes of
interest. For
example, many protein cell extraction techniques yield complex protein
mixtures and
incorporate detergents and salts that interfere with mass spectral analyses
that must be
removed prior to analysis of the proteins. Current methods of fractionation
and
purification are trine consuming. Other purification methods, such as liquid
chromatography and gel electrophoresis used to purify proteins, routinely
involve sample
recovery of volumes greater than 10 L, necessitating additional concentration
prior to
analysis by various protein detection techniques (e.g., MALDI-MS). The demands
of
currently known analytical techniques - and the sample presentation devices
used in
connection with them underscore the importance of sample purification, sample
preparation, automated data acquisition, and automated data analyses.
For example, the most common and preferred type of mass spectrometry used in
the field of proteomics is matrix assisted laser desorption ionization mass
spectrometry
(MALDI-MS). MALDI-MS is a variation of standard laser desorption time-of-
flight mass
spectrometry wherein proteins of relatively high molecular mass are deposited
on a surface
in the presence of a very large molar excess of an acidic, UV absorbing
chemical matrix
(for example, nicotinic acid). This technique allows for desorption of these
high molecular
weight labile macromolecules in the intact state. Mass spectrometry has become
an
important analytical tool in proteomic efforts because it provides mass
accuracy, sensitive
detection, and rapid analysis of minute quantities of samples at moderate
cost.
However, MALDI-MS suffers from various drawbacks, particularly problems
associated with sample preparation. Collectively, present day 1\4ALDI-MS
sample
supports suffer from a severe sample volume limitation in that they are
incompatible with
sample volumes in excess of 2 L. Volumes of up to 2 gL are routinely utilized
and afford
dried-droplets having a diameter of from 1 nun to 2 nun. (Karas, M. and
Hillenkamp, F.
Anal. Chem. 1988, 60, 2299-2301). Because the laser
irradiates only a small portion of the dried-droplet (from 0.015 mm2 to 0.030
nmTrz) during
single-site data acquisition, there is no guarantee that all proteins in a
sample will be
detected. In addition, the sample volume (up to 2 L) is significantly smaller
than the
volume in which samples are routinely recovered after purification
necessitating their
-4-
CA 02531972 2010-11-22
78899-7
further concentration prior to MALDI-MS; for example, peptide and protein
samples
purified by liquid chromatographic and electrophoretic methods are routinely
recovered in
volumes greater than 10 L. As a result, such samples must be further
concentrated prior
to MAI.,DI-MS. Many samples also contain detergents and salts that interfere
with mass
spectral analyses, necessitating their removal prior to MALDI-MS.
Another drawback associated with MALDI-MS is lack of sample homogeneity.
Even volumes as small as 2 L can prove problematic owing to sample
heterogeneity when
the dried-droplet approach to sample application is utilized. Sample volumes
in the range
0.5-2.0 L are routinely utilized and dried, which afford dried-droplets
having a diameter
of from 1 mm to 2 mm. (Karas, M. and Hillenkamp, F. Anal. Chenz. 1988, 60,
2299-2301).
Consequently, only a minute portion of the dried-
droplet (from 0.015 mm2 to 0.030 nun2) is irradiated by the laser during
single-site data
acquisition. Unfortunately, even small volumes of 0.5-2.0 hL are known to
result in
sample heterogeneity (the heterogeneous deposition of analytes), which gives
rise to
significant variations in peak presence, intensity, resolution and mass
accuracy when
focusing the laser on different regions of the dried-droplet (Strupat, K.;
tiaras, M.;
Hillenkamp, F. 157t'1. J. Mass Spectroln. Ion Processes 1991, 111, 89-102;
Cohen, S. L. and
Chart, B. T. Anal. Chem. 1996, 68, 31-37; and Amado, F. M. L.; Domingues, P.;
Santana-
Marques, M. G.; Ferrer-Correia, A. J.; Tourer, K. B. Rapid Coznmun_ Mass
Spectrotn_
1997, 11, 1347-1352). These
phenomena render necessary the critical inspection of the mass spectral data
as well as the
accumulation of a large number of single-site spectra per sample. Therefore,
only a few
hundred samples can be analyzed per day per instrument, and automatic data
acquisition is
often precluded.
It has been demonstrated that the problem of sample heterogeneity can be
minimized as the spot diameter falls to the order of the laser diameter. In
that case, a large
portion of the sample can be irradiated simultaneously, improving sensitivity
and
reproducibility (Little, D. P.; Cornish, T. J.; ODonnell, M. J.; Braun, A.;
Cotter, R. J.;
Koster, H. Proc. Natl. Acad. Sci. U.S.A. 1997, 69, 4540-4546; and Gobom, J.;
Nordhoff,
E.; Mirgorodskaya, E.; Ekman, R.; Roepstorff, P. J. Mass Spectroln. 1999, 34,
105-116).
The sample supports described in United States Patent
No. 6,287,872 are further described (Schuerenberg, M.; Lubbert, C.; Eickhoff,
H.; Kalkum,
M.; Lehrach, H; Nordhoff, E. Anal. Chem. 2000, 72, 3436-3442),
-5-
CA 02531972 2010-11-22
78899-7
wherein it is shown that confining the deposition of analytes to a small spot
diameter not only reduces problems associated with sample heterogeneity, but
also results
in a significant increase in sensitivity of detection. The drawback is that to
obtain this
desired spot size, sample volumes have to be reduced to less than 2 L.
To overcome these sample volume and impurity problems, researchers have
employed sample supports designed or mini-columns used to pre-process samples.
An
example of such a sample support is commercially available as the AnchorChipTM
from
Bruker Daltonics GmbH. The AnchorChipTM products improve MALDI-MS sensitivity
by
concentrating the sample in a precisely-defined location, and specifically
involve a thin
layer of nonwettable hydrophobic material that carries an array of wettable
hydrophilic
spots. A principal limitation associated with the use of the AnchorChipTM is
the
requirement that the volume of liquid sample applied to each anchor be limited
to from
0.50 L to 3.0 L (No. 1 of Eleven General Rules for Sample Preparation on
AnchorChipTM Targets, see AnchorChipTM Technolog),, Reivsion 1.6, Bruker
Daltonics
GmbH, November 2000, incorporated herein by reference); the examples provided
by the
manufacturer in the product's literature further limit the liquid sample drop
volume to
either 0.5 EL or 1.0 EL. Another limitation is that both analytes and
contaminants (salts,
detergents) often get concentrated in the laser-irradiating region. Therefore,
samples must
first be desalted and/or concentrated on a ZipTip`LO or similar mini-column
sample
preparation device prior to application onto mass spectrometer sample
supports, as
described above. (ZipTips , made by Millipore Corp., are micro-columns for
sample
concentration and desalting prepared by packing small pipette tips with
reverse phase
chromatographic media. (Rusconi, F.; Schmitter, J.-M.; Rossier, J.; le Maire,
M. Anal.
Client. 1998, 70, 3046-3052)). However, the use of
home-made micro-columns or commercially available ZipTips" is time consuming,
adds
considerable cost, has proven difficult to automate and often affords only
moderate
recoveries of sample material. Therefore, AnchorChipsTM suffer many of the
same
limitations associated with other present day MALDI-MS sample supports.
An alternative technique to MALDI-MS has been developed for protein profiling
of
serum samples. This technique is called surface enhanced laser desorption
ionization mass
spectrometry (SELDI-MS), and it has produced results with respect to the
discovery of
biomarkers for ovarian cancer and for differentiation of prostate cancer and
benign prostate
hyperplasia. During SELDI-MS, analytes are first selectively retained on a
sample support
-6-
CA 02531972 2010-11-22
78899-7
having a functionalized surface that acts as an affinity capture device. The
retained
analytes are then ionized by laser desorption at the point of capture to
enable their
detection without the need to effect their recovery from the retentive surface
as is required
for other hyphenated Liquid chromatography-mass spectrometry approaches. SELDI-
MS is
described in United States Patent Nos. 5,719,060; 5,894,063; 6,020,208;
6,027,942;
6,124,137; 6,225,047; and 6,579,719. Despite the
restuls recently reported, the SELDI-MS approach is often problematic in
practice as
surfaces which are optimum with respect to retention of biological analytes
can exhibit less
than optimum performance with respect to analyte presentaion during laser
desportion
ionization.
Still other techidques used to isolate and purify analytes, such as proteins,
have
been used. For example, fractionation and purification approaches for
biological samples
via the time consuming techniques of 2D gel electrophoresis and multi
dimensional liquid
chromatography are well known, as are quicker, low sensitivity techniques such
as
consumable columns or pipette tips with chromatography beds. Gel
electrophoresis, which
serves to separate protein mixtures, can be either one or two dimensional. In
1D gel
.electrophoresis, also known as SDS-PAGE (sodium dodecyl sulfate-
polyacrylamide gel
electrophoresis), protein mixtures are separated by their molecular weight
only. In 2D gel
electrophoresis, also known as 2D-PAGE, mixtures are separated by their
isoelectric point
followed by their molecular weight. One disadvantage of the technique is that
the method
has poor resolution, i.e., each resolved spot might contain more than one
protein. Another
disadvantage is that the dyes used to see the separation do not stain all of
the proteins.
Liquid chromatography (LC) is known as "high performance liquid
chromatography"
(HPLC) or "multi-dimensional liquid chromatography," if more than one
chromatographic
column is used. The advantage of LC in general is the availability of diverse
column
chemistries.. In contrast to gel electrophoresis, which cannot efficiently
separate the
smaller peptides, LC can be used to separate peptide mixtures from enzymatic
digests.
Solid phase extraction (SPE) provides a fast way of purification and it is
used in many
areas, from organic synthesis to environmental sample collection. It is faster
than liquid-
liquid extraction or HPLC, it consumes less solvent and can be used to extract
analytes
from gas or liquid samples. The technique of SPE is offered in a variety of
devices, such
as pipette tips, columns, membranes, and 384-well plates, to mention a few.
-7-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
In drug discovery, still other sample presentation devices have been developed
for
use in known analytical methods. For example, ADMET (Absorption Distribution
Metabolism Excretion Toxicology) studies using the Empore card
(http://www.3m.cgm/empore), a C18 RP (reverse phase) sorbent embedded in a
membrane, are touted as capable of reducing the number of steps in sample
purification
and the potential for archiving and concentrating because the loaded samples
are kept dry.
Sample purification requires three steps: loading of samples on to the card,
transferring the
card to the eluter, and eluting 100% of the sample directly into a mass
spectrometer. The
Empore card could be used to load peptide digest samples on a MS if the
elution volumes
are kept as low as possible, otherwise low concentration peptides are below
the limit of
detection.
Therefore, a need exists for sample presentation devices that can be used in
connection with various analytical methods to detect with high sensitivity
biological and
chemical moieties. Moreover, there is a need for sample presentation devices
that are
compatible with the sample volumes routinely recovered from liquid
chromatographic and
electrophoretic separations and other kinds of separation/purification
techniques, that direct
a liquid sample containing analytes to a confined area so as to minimize the
problems
associated with sample heterogeneity, that result in an increase in
sensitivity of detection.
The availability of such sample presentation devices :would enable automated
sample
processing, such as, for example,- on the life science industry's standard
multi-well plate
processors and liquid handling robots. More importantly, they also enable the
direct
collection and subsequent MALDI-MS analysis of chromatographic eluates.
Furthermore,
these capabilities would collectively enhance the throughput of the detection
and
measurement of biological and chemical moieties using the various analytical
techniques
known to those of skill in the art. These and other benefits of the present
invention are
described in more detail below.
SUMMARY OF THE INVENTION
The sample presentation devices of the present invention provide attractive
alternatives to known sample presentation devices used in various analytical
methods used
for the identification of chemical and biological entities. In addition, the
present invention
provides methods of making the sample presentation devices as well as methods
of using
them to perform a wide range of analytical measurements of analytes contained
in liquid
-8-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
samples. The unique properties of the sample presentation devices of the
present invention
address many of the shortcomings (described above) associated with known
analytical
techniques and the sample presentation devices or containers used in
connection with
them.
In fields such as genomics, proteomics, drug discovery, clinical diagnostics,
biosensors, and detection of environmental toxins and agents, mass
spectrometry is a
technique used to identify chemical and biological moieties, wherein often
only very small
quantities of the samples are available, and wherein rapid throughput of large
numbers of
samples is desirable. Other analyte detection methods, such as fluorescence
polarization,
immunofluorescence spectroscopy, gel chromatography, ion exchange
chromatography,
affinity chromatography, can also be used for high throughput detection of
biological and
chemical moieties, and can thus also be used in combination with the sample
presentation
devices of the present invention.
The sample presentation devices of the present invention provide attractive
alternatives to known sample presentation devices used in various analytical
methods. For
example, the present invention allows for analytes to be selectively retained
and
concentrated on the surface of the biochip in volumes up to 100 L. In
addition, because
analytes are detected from a portion of the sample presentation device that is
designed to
be substantially non-binding or binding resistant, they may be detected at
high sensitivity
as compared to direct detection on the surface of a biochip-based affinity
capture device, or
other sample presentation devices in which the surfaces of the devices have
significant
affinity for the analytes.
The present invention further minimizes the potential losses associated with
the
transfer of analytes from one surface to another because the present sample
presentation
devices, in a preferred embodiment, require only a single liquid manipulation.
This,
coupled with the analyte-resistant properties of the sample presentation
device surfaces,
results in a reduction in the loss of the analytes of interest as is the case
in known methods.
In contrast to SELDI-MS, the present invention does not involve desorption of
bound
analytes from the point of capture by an affinity capture device, but rather
uses sample
presentation devices wherein the desorption of analytes from a surface having
no
appreciable affinity or binding of the analytes to that surface.
CA 02531972 2010-11-22
78899-7
In addition, the liquid samples can be manipulated and moved on the
surfaces of the sample presentation devices of the present invention in a
controlled fashion. This allows for the samples to be concentrated to an
analysis
zone where there is no substantial binding of analyte to the surface of the
sample
presentation device. Moreover, this allows the analyte-containing samples to
be
moved to different zones on the surfaces, each zone having different
properties
with respect to an analyte, which allows for purification, isolation and/or
modification of the analytes prior to detection. In addition, the present
invention
involves sample presentation devices in which the properties of various
portions of
the surfaces may change in response to various chemical or physical stimuli
(e.g.,
heat, UV radiation), such that the properties of such surfaces with respect to
analytes can be manipulated during sample handling. Such changes in surface
properties may be designed to be reversible or non-reversible.
According to another aspect of the present invention, there is
provided a sample presentation device for detecting analytes in a sample
comprising a substrate having a surface, wherein the surface is comprised of a
plurality of zones of differing wettability comprising at least a boundary
zone and
an analysis zone, and wherein the boundary zone is a non-wettable zone with
respect to the sample to be analysed and wherein the analysis zone from which
analytes in the sample are detected is the zone that is most wettable and has
the
lowest contact angle with respect to the sample in comparison to the other
zones
in contact with the sample and wherein the analysis zone is substantially
analyte
binding resistant in that it does not exhibit appreciable affinity or binding
to the
analyte to be analysed above the limit of detection of the analysis method
employed.
These and other features of the sample presentation devices of the
present invention are described in more detail below. The present invention
comprises sample presentation devices, methods of making sample presentation
devices, and methods of using sample presentation devices.
-10-
CA 02531972 2010-11-22
78899-7
Sample Presentation Devices
The present invention relates to sample presentation devices that
are useful in performing analytical measurements. In one embodiment, the
present invention involves sample presentation devices having surfaces with
one
or more zones of differing wettability with respect to various samples to be
analyzed. These zones of differing wettability result in zones of differing
abilities
to retain, concentrate, and move analytes in liquid samples. These zones may
be
of various shapes and sizes, and may be continuous or discontinuous with
respect
to each other.
The sample presentation devices of the present invention may be
comprised of distinct zones, one of which is optimal with respect to the
retention of
a liquid sample. The sample presentation devices of the present invention may
further comprise distinct zones of wettabilty, one of which is optimal with
respect
to high sensitivity detection of analytes.
The sample presentation devices of the present invention may
comprise two-dimensional or three-dimensional surfaces, each of which having
two or more zones of differing wettability.
- 10a -
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
The sample presentation devices of the present invention comprise a substrate,
which can be made from a variety of materials, including but not limited to,
for example,
glasses, , semiconductors, metals, polymers (e.g., plastics), and other
hydroxylated
materials, e.g., Si02 on silicon, A1203 on aluminum, etc. Preferably, the
substrate is a
metal, such as gold, or semiconductor, such as silicon.
The sample presentation devices of the present invention further comprise a
substrate that has been surface-modified by methods known to those of ordinary
skill in the
art in order to create various zones on the surface of the substrate, which
zones have
differing properties with respect to wettability. Such surface modifications
include but are
not limited to the addition of self-assembled monolayers (SAMs), polymers
(linear and
branched), and Langmuir-Blodgett assemblies to the substrate. Using SAMs as an
example, when added to the substrate, the SAMs create a surface of the sample
presentation devices to which liquid samples may be exposed. Depending on the
composition of the particular SAMs used, the'. surfaces of the sample
presentation devices
of the present invention may have different properties in terms of
wettability, and in terms
of affinity (or lack thereof) for analytes in liquid samples. The SAMs may be
added to the
sample presentation devices of the present invention in a manner that creates
distinct zones
whose properties reflect the SAMs used in a particular zone. Other surface
modification
techniques known to those of skill in the art are also included in the present
invention.
With respect to the kinds of zones that the surfaces of the sample
presentation
devices may include, they are characterized primarily by virtue of their
differing
wettability with respect to the sample to be analyzed, which in turn results
in zones that
have differing abilities to retain or bind analytes in liquid samples. These
zones are
broadly termed "boundary zones," "liquid retention zones," and "analysis
zones." The
present invention only requires the presence of two types of zones, although
inclusion of
more than two types of zones is also contemplated. The present invention may
also
include more than one zone of each kind - e.g., the sample presentation
devices may
comprise multiple liquid retention zones, each of which may have different
properties with
respect to a liquid sample and/or the analytes contained therein.
A first type of zone is termed a "boundary zone" and involves a substantially
non-
wettable zone with respect to the sample to be analyzed. The boundary zone is
the zone
with the highest contact angle with respect to the sample in comparison to the
other zones.
-11-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
A second type of zone, termed the "liquid retention zone," is relatively more
wettable in comparison to the boundary zone with respect to the sample to be
analyzed
(and is relatively less wettable than the analysis zone, described below). The
liquid
retention zone has a contact angle relatively lower than the contact angle of
the boundary
zone (and a contact angle relatively higher than the contact angle of the
analysis zone,
described below). The liquid retention zone can also have equal or lower
contact angle
than the analysis zone initially, but because of chemical or physical stimuli,
the liquid
retention zone may assume a higher contact, angle than the analysis zone prior
to the
chemical or physical stimuli, which results in the liquid sample being
directed to one zone
preferentially over another.
The liquid retention zone can be of two subtypes. In one subtype, the liquid
retention zone is designed to operate for liquid sample retention purposes,
while being
substantially analyte binding resistant. In a second subtype, the liquid
retention zone is
designed to retain a liquid sample, but also to substantially bind analytes
within a liquid
sample, and can thus be termed a "capture zone" in that it captures the
analytes. This
second subtype may also include a surface that is substantially analyte
binding but that
becomes substantially non-binding upon being subjected to chemical or physical
stimuli,
such as, for example, UV radiation, electricity, or heat.
A third type of zone is termed the "analysis zone" and is the zone that is the
most
wettable (and has the lowest contact angle) with respect to the sample in
comparison to the
other zones. The analysis zone is designed to, be analyte binding resistant.
The analysis
zone may be optimized in terms of size, shape, and surface properties to
enhance the
sensitivity of the analysis of the desired analytes.
The liquid capacity of the sample presentation'devices of the present
invention is
dependent on the sizes of the zones. For a 3 mm diameter circular zone, the
liquid capacity
can be up to about 100 l. The sample presentation devices can contain this
amount of
liquid sample without the need for physical boundaries, reservoirs, or wells.
The various
zones can be precisely positioned in order to facilitate or be compatible with
high
throughput automation on various analytical instruments, such as, for example,
mass
spectrometry instruments.
In another embodiment of the sample presentation devices of the present
invention,
the sample presentation devices can be termed "target chips," and abbreviated
Tn, where
-12-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
"n" is a numerical designation referring to the number of distinct zones on
the surface of
the sample presentation device, where "n" can be any number from 2 to
infinity. Thus, for
example, a T2 target chip has two zones, a T3 target chip has three zones,
etc. The present
invention contemplates sample presentation devices containing many more than 2
or 3
zones and is not limited in any way to a specific number of zones. As the
number of zones
increases, the overall effect approaches a gradient. Target chips are sample
presentation
devices comprised of one or more zones that are designed to be resistant to
analyte
binding.
With respect to a T2 target chip, for example, the sample presentation device
comprises two zones - i.e., a boundary zone and an analysis zone. The surfaces
of the
zone that contacts the liquid sample are designed to be analyte binding
resistant - i.e., the
analysis zone is analyte binding resistant. The surfaces of the zone that
contacts the liquid
sample effectively confine the analytes during the drying step before
analysis.
With respect to a T3 target chip, the sample presentation device comprises
three
zones - i.e., a boundary zone, a liquid retention zone, and an analysis zone.
The surfaces
of the zones that contact the liquid sample are designed to be analyte binding
resistant -
i.e., the liquid retention zone and the analysis zone are analyte binding
resistant. The
surfaces of the zones that contact the liquid sample effectively concentrate
the analytes to
the analysis zone during the drying step.
The sample presentation devices of the , present invention may thus comprise
distinct zones, each of which exhibits a minimum of adsorption with respect to
analytes.
In another embodiment of the sample presentation devices of the present
invention,
the sample presentation devices can be termed "capture chips" or
"capture/concentrate
chips," and abbreviated Xn, where "n" is a numerical designation referring to
the number
of zones on the surface of the sample presentation device, where "n" can be
any number
from 2 to infinity. Thus, for example, an X2 capture chip has two zones, an X3
capture
chip has three zones, etc. The present invention contemplates sample
presentation devices
containing many more than 2 or 3 zones and is not limited in any way to a
specific number
of zones. As the number of zones increases , the overall effect approaches a
gradient.
Capture chips and capture/concentrate chips are sample presentation devices
comprised of
one or more zones that are designed to bind analytes.
-13-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
With respect to an X2 capture chip, for example, the sample presentation
device
comprises two zones - i.e., a boundary zone and a capture zone. The surfaces
of the zones
that contact the liquid sample are designed to capture the analytes - i.e.,
the capture zone
binds the analytes - based on the chemical or biological properties of the
surfaces of the
capture zone. The surfaces of the zones that contact the liquid sample
effectively confine
the analytes during the drying step before analysis.
With respect to an X3 capture/concentrate chip, the sample presentation device
comprises three zones - i.e., a boundary zone, a capture zone, and an analysis
zone. The
boundary zone is designed to be substantially non-wettable. The capture zone
is designed
to capture and bind analytes. The analysis zone is designed to be analyte
binding resistant.
Analytes are transferred between the capture and analysis zones, which is done
prior to
analysis by one of the various known analytical detection methods. The surface
of the
analysis zone that contains the liquid sample effectively confines the
analytes during the
drying step before analysis. The transfer of the liquid sample from the
capture zone to the
analysis zone may be accomplished by virtue of the properties of the surface
of the capture
zone - i.e., if the capture zone has a lower degree of wettability than the
analysis zone, the
liquid sample will move from the capture zone to the analysis zone without
physical
intervention. Alternatively, the capture zone may be designed such that its
properties may
be changed in response to chemical or physical stimuli (e.g., heat, UV
radiation), causing
the capture zone to have a lower degree of wettability than the analysis zone,
and thus
causing the liquid sample to move from the capture zone to the analysis zone.
In yet another embodiment of the sample presentation devices of the present
invention, the sample presentation devices can be combinations of the above-
described
target and capture chips. In this embodiment, the sample presentation devices
are
comprised of surfaces having different functionality. These kinds of sample
presentation
devices may involve the transfer of a liquid sample from one zone to another
by
mechanical means (e.g., via pipetting)or otherwise (e.g., via the differences
in wettability
between zones). As an example, a "capture-transfer-concentrate chip,"
abbreviated X2-
transfer-T3, is a sample presentation device comprised of both an X2 chip
comprised of
two zones (i.e., a boundary zone and a capture zone), as well as a T3 chip
comprised of
three zones (i.e., boundary zone, liquid retention zone, and analysis zone). A
transfer
(mechanical or otherwise) of the analyte occurs between the capture zone of
the X2 chip
and the liquid retention zone of the T3 chip. In addition, the embodiments of
the sample
-14-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
presentation devices that involve combinations of capture zones and liquid
retention zones
may further be used in a combinatorial manner to isolate, concentrate, purify,
and modify
analytes in liquid samples prior to their detection. So, for example, a liquid
sample may be
placed onto a T2 chip such that the analytes in the sample are confined in the
analysis
zone. That sample may then be transferred to an X3 chip that contains a
boundary zone, a
capture zone, and an analysis zone. In this example, the capture zone may be
designed to
bind (and thus remove) lipid moieties from the liquid sample, such that when
the sample is
applied to the X3 chip, it moves from the boundary zone to the capture zone
(which has a
higher degree of wettability), the lipid moieties in the sample bind to the
surface of the
capture zone, and the remaining sample moves to the analysis zone (because it
has the
highest degree of wettability). In this example, the liquid sample is confined
on the T2
chip, and then the lipids are moved on the X3 chip, such that the final sample
that is
analyzed from the analysis zone is concentrated and purified of lipids.
Because the capture
zones can be designed to bind a multitude of different analytes, and because
various
combinations of any of these zones may be used, sample presentation devices
having a vast
range of purification, concentration, isolation, and modification capabilities
(vis-a-vis one
or more analytes) can be created.
The mechanism of transfer of liquid samples from one sample presentation
device
to another may vary. Using the above example, the concentrated sample from T2
may be
removed mechanically (e.g., by pipetting) and placed on a separate X3 sample
presentation
device. Alternatively, the T2 and X3 sample presentation devices may be
connected by a
zone, the wettability of which may be,changed in response to chemical or
physical stimuli
(e.g., UV radiation), such that the concentrated sample in the analysis zone
of the T2
sample presentation device is transferred to the capture zone of the X3 device
when the
exposure of a zone between them to UV radiation results in a wettability that
is higher than
the analysis zone of the T2 device but lower than that of the capture zone of
the X3 device,
such that the sample moves from T2 to X3. Again, with a vast number of
surfaces (having
different wettability and analyte binding properties) and configurations
thereof, sample
presentation devices having a vast range of purification, concentration,
isolation, and
modification capabilities (vis-a-vis one or more analytes) can be created.
The sample presentation devices of the present invention further provide zones
of
different wettability having different shapes or patterns. For example, in one
embodiment,
a sample presentation device may have zones in the form of concentric circles,
with the
-15-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
center zone being the analysis zone, surrounded by the liquid retention zone,
surrounded
by the boundary zone. Because the zones can be created using a various photo-
patterning
techniques, and because known photo-patterning techniques provide for
tremendous
variation in the resulting patterns, there is a vast range of possible shapes,
patterns, and
configurations of the various zones. Moreover, the various properties of the
different
zones of wettability allow for the creation of sample presentation devices
capable of
directing analytes to single or multiple specified or pre-determined locations
on the
surfaces (e.g., addressable sites, lanes, or fields).
The sample presentation devices of the present invention are suitable for the
handling of both biological and non-biological liquid samples. They are also
suitable for
application in a wide range of analyte detection methods, for example,
including but not
limited to, mass spectrometry, various chromatographic methods,
immunofluorescence
spectroscopy, and other known analytical methods of detecting and measuring
analytes in
liquid samples.
Each of the above-described variations is designed to allow for maximum
flexibility in design and use of sample presentation devices having enhanced
capability to
present analytes for detection and analysis over known methods. Thus, the
sample
presentation devices of the present invention have the capability of directing
analytes to an
analysis zone designed to enhance high sensitivity detection of analytes. The
sample
presentation devices of the present invention thus afford improved deposition
of analytes.
Fabrication of Sample Presentation Devices
Still other embodiments of the invention include methods for creating or
fabricating
the sample presentation devices described above.
In an embodiment in which the surfaces are " comprised of self-assembled
monolayers (SAMs) which form distinct zones depending on differences between
the
SAMs used, the sample presentation devices of the present invention may
comprise
various SAM zones that are created by known photo-patterning techniques.
Accordingly,
the present invention further includes methods of creating sample presentation
devices
comprised of SAMs using, as one preferred method, photo-patterning techniques.
The surface of the substrate of the sample presentation device of the present
invention is typically modified or patterned by methods known to those of
skill in the art.
As an example, the substrate's surface can be modified or patterned by means
of applying
-16-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
self-assembled monolayers (SAMs), which modify the surface of the substrate of
the
sample presentation device and whose exposed surfaces may impart particular
chemistries
to the substrate. Selection of various SAMs, including 1 , 2 , 30, or 4
compositions, for a
particular substrate provides the surface of the substrate with unique surface
characteristics
and properties. In particular, application of multiple SAMs results in the
patterning of the
substrate so that it contains a plurality of zones, each zone having different
surface
characteristics and properties. Methods of patterning the SAMs are known in
the art, and
include UV photo-patterning, photolithographic patterning, microstamping.
electron-beam
patterning, and reactive-ion etching.
The zones that are created on the surface of the substrate can be in any
shape, with
circular shapes being preferred. In addition, the zones can be either
continuous or
discontinuous with respect to other zones - i.e., the zones can all be
contiguous with each
other or one or more zones can be discontiguous with one or more other zones.
The zones
that are created on the surface of the substrate of the sample presentation
devices
preferably have a plurality of zones of differing wettability with respect to
the sample to be
analyzed.
As another embodiment of the invention, methods of fabricating a sample
presentation devices that are capable of precisely positioning analytes so as
to facilitate
automated data acquisition are provided.
Uses and Applications o Sample Presentation Devices
In another embodiment, the sample presentation devices of the present
invention
find many uses in combination with various analytical techniques and
procedures. Thus,
the present invention includes methods for using the aforementioned sample
presentation
devices. More specifically, present invention includes methods of using the
sample
presentation devices of the present invention to identify the presence of
analytes in a
sample, and to analyze a plurality of samples, either on a sample presentation
device or on
a plurality of sample presentation devices.
Virtually any analytical method that permits the detection, identification, or
measurement of analytes in a liquid sample can be used in combination with the
sample
presentation devices of the present invention. Examples of such analytical
methods
include but are not limited to, MALDI-MS or electrospray ionization MS. The
sample
presentation devices are particularly well suited to us in combination with
high throughput
-17-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
analytical measurement techniques, such as, for example, for use in MALDI-MS
in which
the sample presentation device analysis zones are configured in such fashion
as to promote
high throughput data acquisition.
The sample presentation devices of the present invention may also be used to
manipulate liquid samples, and the analytes contained therein. Based on the
differing
wettability properties and capture properties that the surfaces of the sample
presentation
devices may be designed to have, the sample presentation devices may be
designed to
manipulate, concentrate, position, store, transfer (with and without
mechanical
intervention), recover (with or without mechanical intervention), analyze,
modify or
process (via use of analyte modifying reagents on the sample presentation
devices), or
fractionate liquid samples or the analytes contained therein. Moreover,
because the sample
presentation devices of the present invention may be designed to accomplish
any,of these
functions in response to chemical or physical stimuli (e.g., heat, W
radiation, pressure,
electromagnetic radiation), the sample presentation devices of the present
invention may
accomplish these functions reversibly or irreversibly, and may further perform
various
combinations of these functions in response to external forces.
Any liquid sample (and analytes) can be used in connection with the sample
presentation devices of the present invention. For example, the, present
invention can be
used to analyze fractions recovered from liquid chromatography. The present
invention
can be used to analyze enzymatic digests prepared from either protein spots
excised from
2D gel electrophoresis or from fractions collected from affinity
chromatography (i.e.,
ICAT (Isotope-Coded Affinity Tags)). The present invention can also be used to
analyze
samples recovered from biosensors. The present invention can also be used for
1:1 sample
transfer with standard multi-well format robotics and assays. Indeed, the
sample
presentation devices of the present invention can be used to handle and
manipulate liquid
samples obtained from virtually any source, whether such samples are the
result of
laboratory experiment (such as the enzymatic digest and biosensor sample
examples
identified above), obtained from the environment (such as a water quality
sample from a
river), or obtained directly from living organisms (such as a human urine
sample).
The present invention can also be used for storage of samples for archival
purposes
or for further analysis. In other words, the detection and analysis of the
analytes contained
-18-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
in liquid samples need not occur immediately following transfer of the liquid
sample to the
analysis zone.
Thus, various embodiments of the present invention provide for sample
presentation devices that serve a variety of liquid-handling functions,
including but not
limited to sample/analyte handling, as well as liquid deposition, retention,
transfer, locating
and re-locating, and storage.
Features and Advantages
In addition to the many features and advantages of the present invention
described
in the summary of the invention section above, additional features and
advantages include
at least the following:
Analytical methods to detect analytes present in a liquid sample, such as
MALDI-
MS, can be performed from a single surface that is substantially analyte non-
binding,
resulting in increased sensitivity of analysis, increased reproducibility of
results, and
comparable results from different capture zones.
With respect to sample liquid handling, increased sample volumes - up to about
100 l for a 3 mm diameter zone - can be analyzed, surfaces can be patterned
having SBS
(Society for Biomolecular Screening) standard well formats (i.e., 96/384/1536
well
formats), and thus are able to be interfaced with common robotics and other
high
throughput analytical methods.
Increased throughput for the various analytical methods (e.g., MALDI-MS) can
be
achieved, in that zones are precision placed for high throughput data
acquisition. With
respect to MALDI-MS, the analysis zone is of optimal size (i.e., less than 2
mm2, and
preferably less than 1 mm). The sample/matrix has improved crystallization,
leading to
improved ionization consistency within the analysis zone. The smaller analysis
zone as
compared to dried spot analysis results in less area to interrogate, resulting
in high
throughput of analysis.
The sample presentation devices of the present invention enable analysis of
diluted
samples by means of the concentration of analyte in the analysis zone.
Separation of analytes in a liquid sample is possible without the need for
multiple
separation steps, such as with binding analytes to an ion exchange
chromatography column
and then having to isolate the analytes from the column in a subsequent wash
step. Indeed,
-19-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
by using SAMs with different surface chemistries designed to bind to different
analytes,
highly specific isolation and purification of particular analytes is possible.
A wide array of liquid samples and analytes can be handled by the sample
presentation devices of the present invention, which avoid the shortcomings of
known
presentation devices and analytical methods described above. While the sample
presentation devices of the present invention are particularly well suited to
use in the
proteomics field and laser desorption ionization mass spectroscopy, as is
described in
detail below the utility of the claimed devices is not in any way, limited to
only that field.
BRIEF DESCRIPTION OF THE FIGURES
The above and other objects of the present invention will become apparent
from'
consideration of the detailed description presented in connection with the
accompanying
drawings in which:
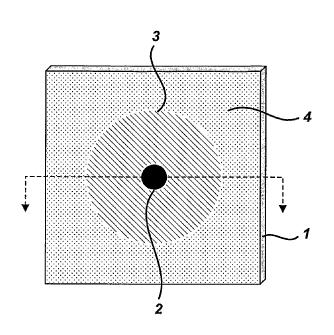
FIG. la depicts a sample presentation device of the present invention, wherein
the
central analysis zone and the surrounding liquid retention zone are concentric
with respect
to one another, and wherein the liquid retention zone is surrounded by a
boundary zone.
FIG. lb depicts a cross-sectional view of the sample presentation device
depicted in FIG.
I a.
FIG. 2 depicts the surface of a sample presentation device of the present
invention,
wherein the surface is further comprised of 16 pairs of analysis zones and
liquid retention
zones, wherein the analysis zones and liquid retention, zones are concentric
with respect to
one another, and wherein pairs of analysis zones and liquid retention zones
are surrounded
by a common boundary zone. In this instance, the sample presentation device is
organized
on geometries corresponding to standard 96-well plate.
FIG. 3 depicts the surface of a sample presentation device of the present
invention,
wherein a portion of the analysis zone and liquid retention zone are
contiguous with
respect to one another, wherein those portions of the analysis and liquid
retention zones
that are not contiguous with respect to one another are surrounded by a common
boundary
zone, and wherein the surface area of the analysis zone is smaller than the
surface of the
liquid retention zone.
FIG. 4a depicts the surface of a sample presentation device of the present
invention, wherein the shape of the analysis zone has been designed to
facilitate the
-20-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
automated acquisition of mass spectral data. FIG. 4b depicts an enlargement of
the analysis
zone indicating 36 regions which measure approximately 100 m2, and which
correspond
to the individual regions that may be sampled by the laser during mass
spectrometry.
FIG. 5 depicts the surface of a sample presentation device of the present
invention,
wherein the surface is further comprised of 96 pairs of analysis zones and
liquid retention
zones, wherein the analysis zones and liquid retention zones are concentric
with respect to
one another, and wherein pairs of analysis zones and liquid retention zones
are surrounded
by a common boundary zone. In this instance, the sample presentation device is
organized
on geometries corresponding to a standard 96-well plate. The liquid retention
zone is
elongated to maximize liquid-holding capacity and minimize the distance
between adjacent
zones. A serpentine pattern is overlaid on the first two rows of the sample
presentation
device to indicate the path described by deposition of a liquid stream of
chromatographic
eluate during automated fraction collection.
FIGS. 6a through 6h illustrate the steps involved in fabrication of a sample
presentation device of the present invention, when alkylthiols on gold are
utilized for
surface modification and UV-photopatterning is exploited for surface
patterning.
FIGS. 7a through 71 illustrate the steps involved in fabrication of a sample
presentation device of the present invention, when alkylthiols on gold are
utilized for
surface modification and photolithography is exploited for surface patterning.
FIGS. 8a through 81 illustrate the steps involved in fabrication of a sample
presentation device of the present invention, when alkylsilanes on silicon are
utilized for
surface modification and photolithography is exploited for surface patterning.
FIGS. 9a through 9f depict various stages during the process whereby a large
volume of aqueous sample deposited on the surface of a sample presentation
device of the
present invention dries within the area corresponding. to the analysis zone.
FIGS. 10a through 10d depict the surface and drop drying characteristics
associated
with a sample presentation device having a liquid retention zone and no
analysis zone.
FIGS. l0e through 10h depict the surface and drop drying characteristics
associated with a
sample presentation device having an analysis zone and no liquid retention
zone.
FIGS. lla through llh depict images recorded on a video contact angle
apparatus
during the drying of a drop on the surface of a sample presentation device of
the present
-21-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
invention, wherein the analysis zone measures 0.6 mm diameter and the liquid
retention
zone measures 1.5 mm diameter.
FIG. 12 is a graph that summarizes the contact angle, drop width and drop
height
associated with the images depicted in FIGS. 11a through 11h.
FIG. 13 is a photograph of a sample presentation device of the present
invention
with liquid volumes of from 5 L to 70 L deposited thereupon.
FIG. 14a is a photograph of a sample presentation device of the present
invention
taken immediately after liquid drops of from 5 L to 40 L were deposited
thereupon.
Each of the liquid drops contained an equivalent amount of alpha-cyan-4-
hydroxycinnamic acid (HCCA). FIG. 14b is a photograph of the HCCA having been
concentrated and directed to the analysis zone due to sample drying on the
sample
presentation device depicted in FIG. 14a. A visual reference to the concentric
zones is
superimposed above the dried HCCA.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Definitions
Unless defined otherwise, all technical and scientific terms used herein have
the
meaning commonly understood by a person skilled in the art to which this
invention
belongs. As used herein, the following terms have the meanings ascribed to
them unless
specified otherwise.
"Analyte(s)" refers to component(s) of a sample which is desirably detected.
The
term can refer to a single component or to multiple components in the sample.
"Sample(s)" refers to any material derived from a biological or non-biological
sources which is presented on the surface of a sample presentation device. The
samples
may be applied to the sample presentation devices in their original, untreated
form and/or
after treatments, including but not limited to modification, fractionation,
extraction, and
concentration. The samples of the present invention can be liquid or non-
liquid samples.
"Substrate" refers to a material that is capable of presenting or supporting a
surface.
"Surface" refers to the exterior or upper boundary of a body or a substrate .
"Substantially non-binding" or "binding resistant" or "analyte binding
resistant"
refers to the property of certain surfaces used in connection with the sample
presentation
-22-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
devices of the present invention that do not exhibit appreciable affinity or
binding of an
analyte to a surface. While some binding may occur, these surfaces are
specifically
designed to minimize binding to levels below the limit of detection of the
analysis method
employed.
"Surface tension" refers to a property of liquids in which a liquid drop
deposited
on a surface tends to contract to the smallest possible contact area because
of unequal
molecular cohesive forces near the surface.
"Wettability" refers to the degree to which a solid surface is wetted by a
liquid
sample. Unless otherwise specified, liquid samples are aqueous in nature.
"Contact angle" refers to the angle between the plane of the solid surface and
the
tangential line to the liquid drop boundary originating at the point of three
phase contact
(solid/liquid/vapor).
"Matrix" refers to materials used in mass spectroscopy techniques, such as
MALDI-MS or SELDI-MS, for absorbing the energy of the laser and transferring
that
energy to analyte molecules, enabling ionization of labile macromolecules. In
SELDI-MS,
the matrix is referred to as "EAM" or "energy absorbing molecule." Reagents
frequently
used as matrices for the detection of biological analytes include but are not
limited to
trans-3,5-dimethoxy-4-hydroxycinnamic acid (sinapinic acid, SA), a-cyano-4-
hydroxycinnamic acid (HCCA) and 2,5-dihydroxybenzoic acid (DHBA). Other
suitable
matrices are known to those skilled in this art.
"SAM" refers to self-assembled monolayers. SAMs are molecular assemblies that
are formed spontaneously by the immersion of an appropriate substrate into a
solution of
an active surfactant in an organic solvent.
Description of the Sample Presentation Devices of the Invention
The following description of the sample presentation devices of the present
invention provides a more detailed understanding than set forth above in the
summary of
the invention. However, the sample presentation devices of the present
invention are
further described by reference to the figures, the methods of fabricating the
sample
presentation devices of the present invention, and the uses and applications
of the sample
presentation devices of the present invention, each of which is described in
detail below.
-23-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
As noted above, the sample presentation devices of the present invention
provide
attractive alternatives to known sample presentation devices used in various
analytical
methods used for the identification of chemical and biological entities. In
addition, the
present invention provides methods of making the sample presentation devices
as well as
methods of using them to perform a wide range of analytical measurements of
analytes
contained in liquid samples. The unique properties of the sample presentation
devices of
the present invention address many of the shortcomings (described in the
background
section above) associated with known analytical techniques and the sample
presentation
devices or containers used in connection with them.
More specifically, the sample presentation devices of the present invention
provide
attractive alternatives to known sample presentation devices used in a wide
range of
analytical methods. They have additional benefits, such as, for example,
allowing for
analytes in a liquid sample to be selectively retained and concentrated on the
surface of the
biochip in volumes up to 100 L. In addition, because analytes are detected
from a portion
of the sample presentation device that is designed to be substantially non-
binding or
binding resistant, they are detected at high sensitivity as compared to direct
detection on
the surface of a biochip-based affinity capture device, or other sample
presentation devices
in which the surfaces of the devices have significant affinity for the
analytes.
The present invention further minimizes the potential losses associated with
the
transfer of analytes from one surface to another because the present sample
presentation
devices, in a preferred embodiment, require only a single liquid manipulation.
This,
coupled with the analyte-resistant properties of the sample presentation
device surfaces,
results in a reduction in the loss of the analytes of interest.
In addition, because the analytes are not bound to affinity capture devices as
in, for
example, SELDI-MS biochips, the liquid samples can be manipulated and moved on
the
surfaces of the sample presentation devices of the present invention in a
controlled fashion.
This allows for the samples to be concentrated to an analysis zone where there
is no
substantial binding of analyte to the surface of the sample presentation
device. Moreover,
this allows the analyte-containing samples to be moved to different zones on
the surfaces,
each zone having different properties with respect to an analyte, which allows
for
purification, isolation and/or modification of the analytes prior to
detection.
-24-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
The present invention involves sample presentation devices in which the
properties
of various portions of the surfaces may change in response to various chemical
or physical
stimuli (e.g., heat, UV radiation), such that the properties of such surfaces
with respect to
analytes can be manipulated during sample handling. Such changes in surface
properties
may be designed to be reversible or non-reversible.
The present invention thus relates to sample presentation devices that are
useful in
performing analytical measurements. In one embodiment, the present invention
involves
sample presentation devices having surfaces with one or more zones of
differing
wettability with respect to various samples to be analyzed. These zones of
differing
wettability result in zones of differing abilities to retain, concentrate, and
move analytes in
liquid samples. These zones may be of various shapes and sizes, and may be
continuous or
discontinuous with respect to each other. The sample presentation devices of
the present
invention may comprise two-dimensional or three-dimensional surfaces, each of
which
having two or more zones of differing wettability.
The sample presentation devices of the present invention comprise a substrate,
which can be made from a variety of materials, including but not limited to,
for example,
glasses, silicates, semiconductors, , metals, polymers (e.g., plastics), and
other
hydroxylated materials, e.g., Si02 on silicon, A1203 on aluminum, etc.
Preferably, the
substrate is a metal, such as gold, or a semiconductor, such as silicon. The
sample
presentation devices of the present invention further comprise a substrate
that has been
surface-modified by methods known to those of ordinary skill in the art in
order to create
various zones on the surface of the substrate, which zones have differing
properties with
respect to wettability. Such surface modifications include but are not limited
to the
addition of self-assembled monolayers (SAMs), polymers (linear and branched),
and
Langmuir-Blodgett assemblies to the substrate. Using SAMs as an example, when
added
to the substrate, the SAMs create a surface of the sample presentation devices
to which
liquid samples may be exposed. Depending on the composition of the particular
SAMs
used, the surfaces of the sample presentation devices of the present invention
may have
different properties in terms of wettability, and in terms of affinity (or
lack thereof) for
analytes in liquid samples. The SAMs may be added to the sample presentation
devices of
the present invention in a manner that creates distinct zones whose properties
reflect the
SAMs used in a particular zone. Other surface modification techniques known to
those of
skill in the art are also included in the present invention.
-25-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
The sample presentation devices of the present invention are comprised of
distinct
zones, one of which is optimal with respect to the retention of a liquid
sample. The sample
presentation devices of the present invention may further comprise distinct
zones of
wettabilty, one of which is optimal with respect to high sensitivity detection
of analytes.
With respect to the kinds of zones that the surfaces of the sample
presentation
devices may include, they are characterized primarily by virtue of their
differing
wettability with respect to the sample to be analyzed, which in turn results
in zones that
have differing abilities to retain or bind analytes in liquid samples. These
zones are
broadly termed "boundary zones," "liquid retention zones," and "analysis
zones." The
present invention only requires the presence of two types of zones, although
inclusion of
more than two types of zones is also contemplated. The present invention may
also
include more than one zone of each kind - e.g., the sample presentation
devices may
comprise multiple liquid retention zones, each of which may have different
properties with
respect to a liquid sample and/or the analytes contained therein. The various
zones can be
precisely positioned in order to facilitate or be compatible with high
throughput automation
on various analytical instruments, such as, for example, mass spectrometry
instruments.
The "boundary zone" involves a substantially non-wettable zone with respect to
the
sample to be analyzed. The boundary zone is the zone with the highest contact
angle with
respect to the sample in comparison to the other zones.
The "liquid retention zone" is relatively more wettable in comparison to the
boundary zone with respect to the sample to be analyzed (and is relatively
less wettable
than the analysis zone, described below). The liquid retention zone has a
contact angle
relatively lower than the contact angle of the boundary zone (and a contact
angle relatively
higher than the contact angle of the analysis zone, described below). The
liquid retention
zone can also have equal or lower contact angle than the analysis zone
initially, but
because of chemical or physical stimuli, the liquid retention zone may assume
a higher
contact angle than the analysis zone prior to the chemical or physical
stimuli, which results
in the liquid sample being directed to one zone preferentially over another.
Moreover, the
liquid retention zone can be of two subtypes. In one subtype, the liquid
retention zone is
designed to operate for liquid sample retention purposes, while being
substantially analyte
binding resistant. In a second subtype, the liquid retention zone is designed
to retain a
liquid sample, but also to substantially bind analytes within a liquid sample,
and can thus
-26-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
be termed a "capture zone" in that it captures the analytes. This second
subtype may also
include a surface that is substantially analyte binding but that becomes
substantially non-
binding upon being subjected to chemical or physical stimuli, such as, for
example, W
radiation, electricity, or heat.
The "analysis zone" is the zone that is the most wettable (and has the lowest
contact angle) with respect to the sample in comparison to the other zones.
The analysis
zone is designed to be analyte binding resistant. The analysis zone may be
optimized in
terms of size, shape, and surface properties to enhance the sensitivity of the
analysis of the
desired analytes.
Among other benefits, the sample presentation devices of the present invention
are
able to retain and handle liquid sample volumes that are larger than other
biochips used in
sample handling, due to the differences in wettability between zones. While
the liquid
capacity of the sample presentation devices of the present invention is
dependent on the
sizes of the zones; for a 3 mm diameter circular zone, the liquid capacity can
be up to about
100 L, and at least up to about 70 L. The sample presentation devices can
contain this
amount of liquid sample without the need for physical boundaries, reservoirs,
or wells.
In another embodiment of the sample presentation devices of the present
invention,
the sample presentation devices can be termed "target 'chips," and abbreviated
Tn, where
"n" is a numerical designation referring to the number' of distinct zones on
the surface of
the sample presentation device, where "n" can be any number from 2 to
infinity. Thus, for
example, a T2 target chip has two zones, a T3 target chip has three zones,
etc. The present
invention contemplates sample presentation devices containing many more than 2
or 3
zones and is not limited in any way to a specific number of zones. As the
number of zones
increases , the overall effect approaches a gradient. Target chips are sample
presentation
devices comprised of one or more zones that are designed to be resistant to
analyte
binding. With respect to a T2 target chip, for example, the sample
presentation device
comprises two zones - i.e., a boundary zone and an analysis zone. The surfaces
of the
zone that contacts the liquid sample are designed to be analyte binding
resistant - i.e., the
analysis zone is analyte binding resistant. The surfaces of the zone that
contacts the liquid
sample effectively confine the analytes during the drying step before
analysis. With
respect to a T3 target chip, the sample presentation device comprises three
zones - i.e., a
boundary zone, a liquid retention zone, and an analysis zone. The surfaces of
the zones
-27-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
that contact the liquid sample are designed to be analyte binding resistant -
i.e., the liquid
retention zone and the analysis zone are analyte binding resistant. The
surfaces of the
zones that contact the liquid sample effectively concentrate the analytes to
the analysis
zone during the drying step. The sample presentation devices of the present
invention may
thus comprise distinct zones, each of which exhibits a minimum of adsorption
with respect
to analytes.
In another embodiment of the sample presentation devices of the present
invention,
the sample presentation devices can be termed "capture chips" or
"capture/concentrate
chips," and abbreviated Xn where "n" is a numerical designation referring to
the number of
zones on the surface of the sample presentation device, where "n' 'can be any
number from
2 to infinity. Thus, for example, an X2 target chip has two zones, an X3
target chip has
three zones, etc. The present invention contemplates sample presentation
devices
containing many more than 2 or 3 zones and is not limited in any way to a
specific number
of zones. As the number of zones increases , the overall effect approaches a
gradient.
Capture chips and capture/concentrate chips are sample presentation devices
comprised of
one or more zones that are designed to bind analytes. The moieties responsible
for
capturing analytes typically comprise specific surface modifications that are
designed as
the distinguishing feature of the capture zone. These surface modifications
may comprise
biological and chemical moieties that bind analytes specifically (such as
monoclonal
antibodies) or non-specifically (such as charged groups that bind on the basis
of
electrostatic attraction) or any combination of such attractive forces. In
addition to the
ability to capture an analyte of interest, these surface modifications may
also retain the
analytes in a liquid sample to permit subsequent modification. So, for
example, a sample
presentation device of the present invention that comprises a capture zone in
which the
surface modification is a monoclonal antibody may bind a complimentary antigen
from a
liquid sample and retain that antigen while the rest of the liquid sample
moves to another
part of the surface of the device, through either physical transfer or
differences in
wettability. The retained antigen may be modified via the addition of other
compounds to
the capture zone of the sample presentation device (e.g., the addition of an
enzyme that
cleaves off a part of the antigen). The modified antigen can then be
transferred to another
portion of the sample presentation device for further handling, or removed
from the device
for analysis by known techniques.
-28-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
With respect to an X2 capture chip, for example, the sample presentation
device
comprises two zones - i.e., a boundary zone and a capture zone. The surfaces
of the zones
that contact the liquid sample are designed to capture the analytes - i.e.,
the capture zone
binds the analytes - based on the chemical or biological properties of the
surfaces of the
capture zone. The surfaces of the zones that contact the liquid sample
effectively confine
the analytes during the drying step before analysis. With respect to an X3
capture/concentrate chip, the sample presentation device comprises three zones
- i.e., a
boundary zone, a capture zone, and an analysis zone. The boundary zone is
designed to be
substantially non-wettable. The capture zone is designed to capture and bind
analytes.
The analysis zone is designed to be analyte binding resistant. Analytes are
transferred
-between the capture and analysis zones, which is done prior to analysis by
one of the
various known analytical detection methods. The surface of the analysis zone
that contains
the liquid sample effectively confines the analytes during the drying step
before analysis.
The transfer of the liquid sample from the capture zone to the analysis zone
may be
accomplished by the properties of the surface of the capture zone - i.e., if
the capture zone
has a lower degree of wettability than the analysis zone, the liquid sample
will move from
the capture zone to the analysis zone without physical intervention.
Alternatively, the
capture zone may be designed such that its properties may be changed in
response to
chemical or physical stimuli (e.g:, heat, UV radiation), causing the capture
zone to have a
lower degree of wettability than the analysis zone, and thus causing the
liquid sample to
move from the capture zone to the analysis zone.
In another embodiment of the sample presentation devices of the present
invention,
the sample presentation devices can be combinations of the above-described
target and
capture chips. In this embodiment, the sample presentation devices are
comprised of
surfaces having different functionality. These kinds of sample presentation
devices may
involve the transfer of a liquid sample from one zone to Another by mechanical
means
(e.g., via pipetting) or otherwise (e.g., via the differences in wettability
between zones).
As an example, a "capture-transfer-concentrate chip," abbreviated X2-transfer-
T3, is a
sample presentation device comprised of both an X2 chip comprised of two zones
(i.e., a
boundary zone and a capture zone), as well as a T3 chip comprised of three
zones (i.e.,
boundary zone, liquid retention zone, and analysis zone). A transfer
(mechanical or
otherwise) of the analyte occurs between the capture zone of the X2 chip and
the liquid
retention zone of the T3 chip.
-29-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
These sample presentation devices may involve more than one "capture zone,"
such
that the surfaces may exhibit binding affinity to one or more analytes. The
ability to bind
analytes seriatim as a liquid sample is moved from one zone to another on the
surface of
the sample presentation devices is a feature of the present invention that
facilitates the
analysis of many different fractions of a liquid sample without the need to
physically
separate them using mechanical intervention. Instead, the different
wettability properties
of the sample presentation devices of the present invention may direct liquid
samples to
different zones of the devices, in the process leaving behind analytes that
bind to different
capture zones, and thereby sequentially process a liquid sample.
More specifically, the embodiments of the sample presentation devices that
involve
combinations of capture zones and liquid retention zones may further be used
in a
combinatorial manner to isolate, concentrate, purify, and modify analytes in
liquid samples
prior to their detection. So, for example, a liquid sample may be placed onto
a T2 chip
such that the analytes in the sample are confined in the analysis zone. That
sample may
then be transferred to an X3 chip that contains a boundary zone, a capture
zone, and an
analysis zone. In this example, the capture zone may be designed to bind (and
thus
remove) lipid moieties from the liquid sample, such that when the sample is
applied to the
X3 chip, it moves from the boundary zone to the capture zone (which has a
higher degree
of wettability), the lipid moieties in the sample bind to the surface of the
capture zone, and
the remaining sample moves to the analysis zone (because it has the highest
degree of
wettability). In this example, the liquid sample is confined on the T2 chip,
and then the
lipids are moved on the X3 chip, such that the final sample that is analyzed
from the
analysis zone is concentrated and purified of lipids. Because the capture
zones can be
designed to bind a multitude of different analytes, and because various
combinations of
any of these zones may be used, sample presentation devices having a vast
range of
purification, concentration, isolation, and modification capabilities (vis-a-
vis one or more
analytes) can be created.
The mechanism of transfer of liquid samples from one sample presentation
device
to another may vary. Using the above example, the concentrated sample from T2
may be
removed mechanically (e.g., by pipetting) and placed on a separate X3 sample
presentation device. Alternatively, the T2 and X3 sample presentation devices
may be
connected by a zone, the wettability of which may be changed in response to
chemical or
physical stimuli (e.g., UV radiation), such that the concentrated sample in
the analysis zone
-30-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
of the T2 sample presentation device is transferred to the capture zone of the
X3 device
when the exposure of a zone between them to UV radiation results in a
wettability that is
higher than the analysis zone of the T2 device but lower than that of the
capture zone of the
X3 device, such that the sample moves from T2 to X3. Again, with a vast number
of
surfaces (having different wettability and analyte binding properties) and
configurations
thereof, sample presentation devices having a vast range of purification,
concentration,
isolation, and modification capabilities (vis-a-vis one or more analytes) can
be created.
The sample presentation devices of the present invention - in each of the
embodiments described above - may further provide zones of different
wettability having
different shapes or patterns (a few examples of which are depicted in the
Figures). For
example, in one embodiment, a sample presentation device may have zones in the
form of
concentric circles, with the center zone being the analysis zone, surrounded
by the liquid
retention zone, surrounded by the boundary zone. Because the zones can be
created using
a various photo-patterning techniques, and because known photo-patterning
techniques
provide for tremendous variation in the resulting patterns, there is a vast
range of possible
shapes, patterns, and configurations of the various zones that can be designed
by those of
skill in the art. Moreover, the various properties of the different zones of
wettability allow
for the creation of sample presentation devices capable of directing analytes
to single or
.multiple specified or pre-determined locations on the surfaces (e.g.,
addressable sites,
lanes, or fields). Addressable in this context simply means that the pre-
determined site,
lane or field can be specified by an automated processing device that works in
concert with
the sample presentation devices of the present invention such that liquid
samples or
analytes retained at those specified locations can be processed by an
analytical device to
measure the analytes of interest. In addition, liquid samples or analytes
present at these
pre-determined locations may be removed from the sample presentation devices
for
subsequent handling or manipulation (e.g., modification, purification,
concentration, etc.)
by another sample presentation device.
The sample presentation devices of the present invention are suitable for the
handling of both biological and non-biological liquid samples. They are also
suitable for
application in a wide range of analyte detection methods, for example,
including but not
limited to, mass spectrometry, various chromatographic methods,
immunofluorescence
spectroscopy, and other known analytical methods of detecting and measuring
analytes in
liquid samples.
-31-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
Each of the above-described variations is designed to allow for maximum
flexibility in design and use of sample presentation devices having enhanced
capability to
present analytes for detection and analysis over known methods. Thus, the
sample
presentation devices of the present invention have the capability of directing
analytes to an
analysis zone designed to enhance high sensitivity detection of analytes. The
sample
presentation devices of the present invention thus afford improved deposition
of analytes.
The sample presentation devices of the present invention may further comprise
devices capable of receiving and retaining liquid samples in volumes up to
about 100 L,
and at least up to about 70 L. The sample presentation devices of the present
invention
may also be utilized as sample positioning devices that directs the deposition
of analytes to
a surface area measuring less than about 2 millimeter squared (2 mm) and
preferably less
than about lmm2. Directing the deposition of analytes to a surface area
measuring less
than about 1 mm2 may facilitate the improved deposition of analytes with a
concomitant
increase in both ease of automated data acquisition and sensitivity of
detection.
Consequently, the sample presentation device of the present invention provides
a surface
that exhibits substantial utility both with respect to liquid-holding capacity
and controlled
deposition of analytes. In preferred embodiments, this combination of
attributes affords an
increase in sensitivity of detection of from about 4-fold to greater than
about 100-fold as
compared to known sample supports.
In one embodiment, the sample presentation device of the present invention is
comprised of a substrate, wherein the surface of the substrate is further
comprised of three
contiguous zones organized in a concentric arrangement, wherein the central
analysis zone
is surrounded by a liquid retention zone, and wherein the liquid retention
zone is
surrounded by a boundary zone. Alternatively, the sample presentation device
of the
present invention may be comprised of a substrate, wherein the surface is
further
comprised of three contiguous zones organized in an adjacent arrangement,
wherein some
portion of the analysis zone and some portion of the liquid retention zone are
contiguous
with respect to one another, and wherein those portions of the analysis and
liquid retention
zones that are not contiguous with respect to one another are surrounded by a
common
boundary zone.
In an embodiment of the sample presentation devices of the present invention,
the
surface of the analysis zone has a contact angle of preferably less than about
40 , more
-32-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
preferably less than about 30 , and most preferably less than about 20 . The
surface of the
analysis zone preferably exhibits minimum affinity or binding with respect to
analytes.
The surface of the liquid retention zone has a contact angle preferably in the
range of about
40 to about 95 , more preferably in the range of about 60 to about 95 , most
preferably in
the range of about 80 to about 95 , and further preferably exhibits minimum
affinity or
binding with respect to analytes. The surface of the boundary zone has a
contact angle of
preferably greater than about 95 , more preferably greater than about 105 ,
most preferably
greater than about 115 , and further preferably exhibits a minimum of
wettability with
respect to liquid samples.
In another embodiment of the sample presentation devices of the present
invention,
the contact angle of the analysis zone is at least about 10 , preferably at
least about 20 ,
more preferably at least about 30 , and most preferably at least about 40
lower then the
contact angle of the liquid retention zone, wherein the contact angle of the
liquid retention
zone is preferably at least about 10 , more preferably at least about 15 , and
most
preferably at least about 20 lower than the contact angle of the boundary
zone. In an
embodiment of the sample presentation devices of the present invention, the
surface area of
the liquid retention zone is preferably at least about 4-fold greater, more
preferably at least
about 10-fold greater, and most preferably at least about 50-fold greater than
the surface
area of the analysis zone, and the surface area of the analysis zone is
preferably less than
about 1 mm2, is more preferably in the range of from about 0.2 mm2 to about
0.8 mm2 , and
is most preferably in the range of from about 0.4 mm2 to about 0.6 mm2.
The sample presentation devices of the present invention may be further
comprised
of a substrate, wherein the surface of the substrate may be further comprised
of, but not
limited to, from 1 to 1536 pairs of analysis zones and liquid retention zones,
wherein pairs
of analysis zones and liquid retention zones are arranged as either concentric
or adjacent
pairs, and wherein pairs of analysis and liquid retention zones are surrounded
by a
common boundary zone. The sample presentation devices comprised of multiple
pairs of
analysis zones and liquid retention zones is preferably configured in a manner
analogous to
the standard 96-well, 384-well and 1536-well plates so as to be compatible
with
standardized multi-well plate processors and laboratory liquid handling
robots.
-33-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
Description of the Figures
The descriptions that follow are merely, exemplary, supplement the disclosure
of
the invention set forth elsewhere, and do not limit the scope of the
invention.
With reference to FIGS. la and 1b, the sample presentation device of the
present
invention is illustrated, showing a substrate 1, wherein the surface of the
substrate is
further comprised of three contiguous zones organized in a concentric
arrangement,
wherein the central analysis zone 2 is surrounded by a liquid retention zone
3, and wherein
the liquid retention zone 3 is surrounded by a boundary zone 4. The surface of
the analysis
zone 2 exhibits a contact angle of preferably less than about 40 , more
preferably less than
about 30 , and most preferably less than about 20 , and further preferably
exhibits a
minimal binding with respect to analytes. The surface of the liquid retention
zone 3
exhibits a contact angle preferably in the range 'of about 40 to about 95 ,
more preferably
in the range of about 60 to about 95 , most preferably in the range of about
80 to about
95 , and further preferably exhibits minimal binding with respect to analytes.
The surface
of the boundary zone 4 exhibits a contact angle of preferably greater than
about 95 , more
preferably greater than about 105 , most preferably greater than about 115 ,
and further
preferably exhibits a minimum of wettability with respect to liquid samples.
With further reference to FIGS. la and 1b, a preferred embodiment of the
sample
presentation device of the present invention is one wherein the contact angle
of the analysis
zone 2 is preferably at least about 10 , more preferably at least about 20 ,
more preferably
at least about 30 , and most preferably at least about 40 lower than the
contact angle of the
liquid retention zone 3, wherein the contact angle of the liquid retention
zone 3 is
preferably at least about 10 , more preferably at least about 15 , and most
preferably at
least about 20 lower than the contact angle of the boundary zone 4, wherein
the surface
area of the liquid retention zone 3 is preferably at least about 4-fold
greater, more
preferably at least about 10-fold greater, and most preferably at least about
50-fold greater
than the surface area of the analysis zone 2, and wherein the surface area of
the analysis
zone 2 is preferably less than about 2 mm2, is more preferably in the range of
from about
0.2 mm2 to about 1.8 mm2 , and is most preferably in the range of from about
0.4 mm2 to
about 1.6 mm2.
With reference to FIG. 2, the sample presentation device of the present
invention is
comprised of a substrate 5 wherein the surface is further comprised of 16
concentric pairs
-34-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
of analysis zones 6 and liquid retention zones 7, all of which are surrounded
by a common
boundary zone 8. In this instance, pairs, of target and liquid retention zones
are arrayed on 9
mm centers that would allow six of these devices to be combined into the
format
corresponding to a standard 96-well plate.
With further reference to FIG. 2, a preferred embodiment of the sample
presentation device of the present invention is one wherein the contact angle
of the analysis
zone 6 is preferably at least about 10 , more preferably at least about 20 ,
more preferably
at least about 30 , and most preferably at least about 40 lower then the
contact angle of the
liquid retention zone 7, wherein the contact angle of the liquid retention
zone 7 is
preferably at least about 10 , more preferably at least about 150, and most
preferably at
least about 20 lower than the contact angle of the boundary zone 8, wherein
the surface
area of the liquid retention zone 7 is preferably at least about 4-fold
greater, more
preferably at least about 10-fold greater, and most preferably at least about
50-fold greater
than the surface area of the analysis zone 6, and wherein the surface area of
the analysis
zone 6 is preferably less than about 2 mm2, is more preferably in the range of
from about
0.2 mm2 to about 1.8 mm2, and is most preferably in the range of from about
0.4 mm2 to
about 1.6 mm2.
It is important to note that neither the analysis zone nor the liquid
retention zone
must be round in shape as illustrated in FIG. la. Both the analysis zone and
the liquid
retention zone may assume a variety of shapes as may be required to optimize
performance
of the sample presentation device with respect to a particular application.
Additionally, it
is important to note that neither the analysis zone nor the liquid retention
zone must be
concentric with one another as illustrated in FIGS. la and 2. Both the
analysis zone and
the liquid retention zone may be positioned accordingly as may be required to
optimize
performance of the sample presentation device with respect to a particular
application.
With reference to FIG. 3, the sample presentation device of the present
invention is
comprised of a substrate 9 having a surface further comprised of three
contiguous zones
organized in an adjacent arrangement, wherein some portion of the analysis
zone 10 and
some portion of the liquid retention zone 11 are contiguous with respect to
one another,
wherein those portions of the analysis zone and liquid retention zone that are
not
contiguous with respect to one another are surrounded by a common boundary
zone 12.
The surface of the analysis zone 10 exhibits a contact angle of preferably
less than about
-35-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
40 , more preferably less than about 30 , and most preferably less than about
20 , and
further preferably exhibiting minimal binding with respect to analytes. The
surface of the
liquid retention zone 11 exhibits a contact angle preferably in the range of
about 40 to
about 95 , more preferably in the range of about 60 to about 95 , most
preferably in the
range of about 80 to about 95 , and further preferably exhibiting minimal
binding with
respect to analytes. The surface of the boundary zone 12 exhibits a contact
angle of
preferably greater than about 95 , more preferably greater than about 105 ,
most preferably
greater than about 1150, and further preferably exhibiting a minimum of
wettability with
respect to liquid samples.
With further reference to FIG. 3, a preferred embodiment of the sample
presentation device of the present invention is one wherein the contact angle
of the analysis
zone 10 is preferably at least about! 10 , more preferably at least about 20 ,
more preferably
at least about 30 , and most preferably at least about 40 lower then the
contact angle of the
liquid retention zone 11, wherein the contact angle of the liquid retention
zone 11 is
preferably at least about 10 , more preferably at least about 15 , and most
preferably at
least about 20 lower than the contact angle of the boundary zone 12, wherein
the surface
area of the liquid retention zone 11 is preferably at least about 4-fold
greater, more
preferably at least about 10-fold greater, and most preferably at least about
50-fold greater
than the surface area of the analysis zone 10, and wherein the surface area of
the analysis
zone 10 is preferably less than about 1 mm2, is more preferably in the range
of from about
0.2 mm2 to about 0.8 mrn2 , and is most preferably in the range of from about
0.4 mm2 to
about 0.6 mm2.
It is important to note that neither the analysis zone nor the liquid
retention zone
must be round in shape as illustrated in FIGS. la, 2 and 3. Both the analysis
zone and the
liquid retention zone may assume a variety of shapes as may be required to
optimize
performance of the sample presentation device with respect to a particular
application.
With reference to FIG. 4a, the sample presentation device of the present
invention
is comprised of a substrate 13 having a surface further comprised of three
contiguous zones
organized in a concentric arrangement, wherein the central analysis zone 14 is
surrounded
by a liquid retention zone 15, and wherein the liquid retention zone 15 is
surrounded by a
boundary zone 16. With reference to FIG. 4b, the shape of the analysis zone 14
(a square)
-36-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
may facilitate automated acquisition of mass spectral data, in that it
corresponds in size to a
raster of 36 regions.
With reference to FIG. 5, the sample presentation device of the present
invention is
comprised of a substrate 17 comprised of 96 pairs of analysis zones 18 and
liquid retention
zones 19, all of which are surrounded by a common boundary zone 20. In this
instance,
the concentric pairs of zones are arrayed on 9 mm centers that correspond to a
standard 96-
well plate. The liquid retention zone 19 was been elongated to maximize liquid-
holding
capacity and minimize the distance between adjacent zones in each row. A
serpentine
pattern is overlaid on the first two rows of the sample presentation device to
indicate the
path described by the deposition of a liquid stream of chromatographic eluate
during
automated fraction collection.
With further reference to FIG. 5, a preferred embodiment of the sample
presentation device of the present invention is one wherein the contact angle
of the analysis
zone 18 is preferably at least about 100, more preferably at least about 20 ,
more preferably
at least about 30 , and most preferably at least about 40 lower then the
contact angle of the
liquid retention zone 19, wherein the contact angle of the liquid retention
zone 19 is
preferably at least about 10 , more preferably at least about 15 , and most
preferably at
least about 20 lower than the contact angle of the boundary zone 20, wherein
the surface
area of the liquid retention zone 19 is preferably at least about 4-fold
greater, more
preferably at least about 10-fold greater, and most preferably at least about
50-fold greater
than the surface area of the analysis zone 18, and wherein the surface area of
the analysis
zone 18 is preferably less than about 2 mm2, is more preferably in the range
of from about
0.2 mm2 to about 1.8 mm2, and is most preferably in the range of from about
0.4 mm2 to
about 1.6 mm2.
Fabrication o Sample Presentation Devices
Still other embodiments of the invention include methods for creating or
fabricating
the sample presentation devices described above. For example, in an embodiment
in
which the surfaces are comprised of one or more self-assembled monolayers
(SAMs)
which form distinct zones depending on differences between the SAMs used, the
sample
presentation devices of the present invention may comprise various SAM zones
that are
created by known photo-patterning techniques. Accordingly, the present
invention further
-37-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
includes methods of creating sample presentation devices comprised of SAMs
using, as
one preferred method, photo-patterning techniques.
More generally, the surface of the substrate of the sample presentation device
of the
present invention is typically modified or patterned by methods known to those
of skill in
the art. As an example, the substrate's surface can be modified or patterned
by means of
applying one or more self-assembled monolayers (SAMs), which modify the
surface of the
substrate of the sample presentation device and whose exposed surfaces may
impart
particular chemistries to the substrate. Selection of various SAMs, including
1 , 2 , 3 , or
4 compositions, for a particular substrate provides the surface of the
substrate with unique
surface characteristics and properties. In particular, application of multiple
SAMs results
in the patterning of the substrate so that it contains a plurality of zones,
each zone having
different surface characteristics and properties. Methods of patterning the
SAMs are
known in the art, and include UV photo-patterning, photolithographic
patterning,
microstamping. electron-beam patterning, and reactive-ion etching.
The zones that are created on the surface of the substrate can be in any
shape, with
circular shapes being preferred. In addition, the zones can be either
continuous or
discontinuous with respect to other zones - i.e., the zones can all be
contiguous with each
other or one or more zones can be discontiguous with one or more other zones.
The zones
that are created on the surface of the substrate of the sample presentation
devices
preferably have a plurality of zones of differing wettability with respect to
the sample to be
analyzed.
As another embodiment of the invention, methods of fabricating a sample
presentation device that is capable of precisely positioning analytes so as to
facilitate
automated data acquisition are provided.
More specifically, approaches to surface patterning, selection of suitable
substrates,
preparation of self-assembled monolayers as well as other approaches to
surface
modification are described below. These descriptions are merely exemplary and
do not
limit the scope of the invention.
The surface of the sample presentation device of the present invention is
patterned
by one of several approaches which preferably include, but are not limited to:
(1) UV-
Photopatterning of self-assembled monolayers (SAMs) prepared from alkylthiols
on a
coinage metal surface; (2) Photolithographic patterning of SAMs prepared from
alkylthiols
-38-
CA 02531972 2010-11-22
78899-7
on a coinage metal surface; (3) Microstamping of SAMs prepared from
alkyltluols on a
coinage metal surface; and (4) Photolithographic patterning of SAMs prepared
from
alkylsilanes on either a silicon or glass surface; (5) Electron-beam
patterning, and (6)
Reactive-ion etching. Preferably, the patterning of the sample presentation
device surface
is achieved either by application of the UV-photopatterning process described
in United
States Patent No. 5,514,501, or by the microstamping process described in
United States
Patent No. 5,512,13 1. Alternatively,
the patterning of the sample presentation device surface may be achieved by
photolithographic patterning processes described in the literature and
understood by those
skilled in the art.
With reference to FIGS. 6a through 6h, the step-wise process for UV-
photopatterning of SAMs comprised of alkylthiols on gold is depicted.
Initially, a suitable
substrate 21 such as a silicon wafer (750 Inn) is appropriately cleaned by a
combination of
wet process and argon plasma etching. An adhesion layer (25-50 run) of either
chromium
or titanium and tungsten (9:1) is first applied to the surface of the wafer
followed by a thin
film (100-1000 am) of gold 22. Metal deposition is accomplished by a
sputtering (vapor
deposition) process that has been calibrated with respect to metal deposition
(thickness) per
unit time. The sputtering process may be undertaken with intact wafers or with
individual
pieces diced from a wafer.
With reference to FIG. 6b, the first monolayer 23 is assembled on the gold
surface
by incubation of the substrate in a solution containing from 0.05 to 5 mM
alkylthiol in
ethanol for a period of from I to 24 hours. The surface-modified substrate is
then washed
with ethanol to remove excess alkylthiol and dried under a stream of nitrogen.
The first
monolayer 23 is prepared from an alkylthiol which affords a surface that
exhibits a contact
angle of greater than about 100 and further exhibits a minimum of wettability
with respect
to liquid samples.
With reference to FIG. 6c, the surface-modified substrate is photo-patterned
by
exposure to an ultraviolet light source through a first mask 24 in the
presence of oxygen so
as to oxidize monomers residing within the exposed zone thereby generating
monomer
sulfonates that exhibit low affinity with respect to the gold surface. The
opening in the
mask 25 results in the creation of features of size and shape corresponding to
the liquid
retention zone.
-39-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
With respect to FIGS. 6d and 6e, subsequent washing of the gold surface
removes
monomer sulfonates and affords an unmodified region of gold 26. The second
monolayer
27 is assembled on the gold surface by incubation of the substrate in a
solution containing
from 0.05 to 5 mM alkylthiol in ethanol for a period of from 1 to 24 hours.
The surface-
modified substrate is then washed with ethanol to remove excess alkylthiol and
dried under
a stream of nitrogen. The second monolayer 27 is prepared from an alkylthiol
that affords
a surface that exhibits a contact angle in the range of about 40 to about 95
and further
affords a surface that exhibits minimal binding with respect to analytes.
With respect to FIG. 6f, the patterned substrate is further photo-patterned by
exposure to an ultraviolet light source through a second mask 28 in the
presence of oxygen
so as to oxidize monomers residing within the exposed zone thereby generating
monomer
sulfonates that exhibit low affinity with respect to the gold surface. The
opening in the
mask 29 results in the creation of features of size and shape corresponding to
the analysis
zone.
With respect to FIGS. 6g and 6h, subsequent washing of the gold surface
removes
monomer sulfonates and affords an unmodified region of gold 30. The third
monolayer 31
is assembled on the gold surface by incubation of the substrate in a solution
containing
from about 0'.05 to about 5 mM alkylthiol in ethanol for a period of from 1 to
24 hours.
The surface-modified substrate is then washed with ethanol to remove excess
alkylthiol
and dried under a stream of nitrogen. The third monolayer 31 is prepared from
analkylthiol that affords a surface that exhibits a contact angle of less than
about 40 and
further exhibits minimal binding with respect to analytes.
In this manner, the step-wise process for UV-photopatterning of self-assembled
monolayers prepared from alkylthiols on gold is exploited to prepare the
sample
presentation device of the present invention. The above-described process of
UV-
photopatterning of self-assembled monolayers prepared from alkylthiols on gold
is
exemplary and the invention is not limited to only the process described.
With reference to FIGS. 7a through 7h, the step-wise process for
photolithographic
patterning of SAMs comprised of alkylthiols on gold is depicted. A suitable
substrate 32
such as a silicon wafer is appropriately cleaned and an adhesion layer and a
thin film of
gold 33 (100-1000 nm) is sputtered thereupon.
-40-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
With reference to FIG. 7b, the first monolayer 34 is assembled on the gold
surface
by incubation of the substrate in a solution containing from 0.05 to 5 mM
alkylthiol in~
ethanol for a period of from 1 to 24 hours. The surface-modified substrate is
then washed
with ethanol to remove excess alkylthiol and dried under a stream of nitrogen.
The first
monolayer 34 is prepared from an alkylthiol that affords a surface that
exhibits a contact
angle of less than 40 and further exhibits minimal binding with respect to
analytes.
With reference to FIG. 7c, the surface-modified substrate is coated with a
photoresist 35 prior to lithography. The resist may be of a negative tone or
positive tone.
A negative resist results in decreased solubility in the exposed regions of
the resist, thus
giving a negative image relative to the mask. A positive resist results in
increased
solubility of the resist in the exposed regions, thus giving a ,positive image
relative to the
mask. The use of a positive resist is depicted. The resist may be applied
through a'dip-
type of process, but is preferable applied using a spin-coater. The
manufacturers'
recommendations with respect to resist thickness and curing time are used as
guidelines.
With reference to FIG. 7d, the surface-modified substrate is photo-patterned
by
exposure to an ultraviolet light source as required for use in conjunction
with the particular
resist employed. The photomask 36 may be prepared from a number of commonly
employed materials which include, but are not limited to, chromium-on-quartz,
Mylar,
acetate, and metallic stencils. The opening in the mask 37 results in the
creation of
features of size and shape corresponding to the analysis zone.
With respect to FIG. 7e, the substrate is initially treated with a commercial'
solution
specific to the resist employed that dissolves the exposed areas of resist
while those regions
not exposed 38 to the ultraviolet light source remain relatively insoluble.
After removal of
exposed resist, an oxygen plasma or UV/ozone treatment may be employed to
oxidize
alkylthiol monomers within the exposed zone thereby generating monomer
sulfonates that'
exhibit low affinity with respect to the gold surface. Subsequent washing of
the gold
surface removes monomer sulfonates and affords an unmodified region of gold
39.
With reference to FIG. 7f, the second monolayer 40 is assembled on the gold
surface by incubation of the substrate in a solution containing from 0.05 to 5
mM alkylthiol
in ethanol for a period of from 1 to 24 hours. The substrate is then washed
with ethanol to
remove excess alkylthiol and dried under a stream of nitrogen. The second
monolayer 40
is prepared from an alkylthiol that affords a surface that exhibits a contact
angle in the
-41-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
range 40 to 95 and further affords a surface that exhibits minimal binding
with respect to
analytes.
With respect to FIGS. 7g and 7h, the remaining photoresist 38 is removed by
further washing the substrate with one of several organic solvents known to
dissolve
unexposed resist (e.g. acetone, 1-methyl-2-pyrrolidinone, etc.) and the
patterned substrate
now comprised of two distinctive zones is coated with fresh photoresist 41
prior to
lithography as described above.
With respect to FIGS. 7i and 7j, the patterned substrate is photo-patterned by
exposure to an ultraviolet light source through a second photomask 42 as
described above.
The opening in the mask 43 results in the creation of features of size and
shape,
corresponding to the liquid retention zone. The substrate is initially treated
with a
commercial solution specific to the resist employed that dissolves the exposed
areas of
resist while those regions not exposed 44 to the ultraviolet light source
remain relatively
insoluble. After removal of exposed resist, an oxygen plasma or UV/ozone
treatment is
employed to oxidize alkylthiol monomers residing within the exposed zone
thereby
generating monomer sulfonates that exhibit low affinitywith respect to the
gold surface.
Subsequent washing of the gold surface removes monomer sulfonates and affords
an
unmodified region of gold 45.
With reference to FIGS. 7k and 71, the third monolayer 46 is assembled on the
gold
surface by incubation of the substrate in a solution containing from 0.05 to 5
mM alkylthiol
in ethanol for a period of from 1 to 24 hours. The substrate is then washed
with ethanol to
remove excess alkylthiol and dried under a stream of nitrogen. The third
monolayer 46 is
prepared from an alkylthiol which affords a surface that exhibits a contact
angle of greater
than 100 and further exhibits a minimum of wettability with respect to liquid
samples.
Finally, the remaining photoresist 44 is removed by further washing the
substrate with one
of several organic solvents known to dissolve unexposed resist to afford a
patterned
surface comprised of three distinctive zones.
In this manner, the step-wise process for photolithographic patterning of SAMs
comprised of alkylthiols on gold is exploited to prepare the sample
presentation device of
the present invention. It should be noted that the sequence of patterning
depicted (analysis
zone, followed by liquid retention zone, followed by boundary zone) was
selected
arbitrarily and that the reverse sequence (boundary zone, followed by liquid
retention zone,
-42-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
followed by analysis zone) would also prove as suitable as the sequence
illustrated. The
above-described process of photolithographic patterning of self-assembled
monolayers
prepared from alkylthiols on gold is exemplary and the invention is not
limited to only the
process described.
Numerous alkylthiol monomers are suitable for use in preparation of the sample
presentation device of the present invention. The synthesis of alkylthiol
monomers, their
assembly into monolayers, and their classification with respect to the surface
tension of the
assembled surfaces has been described (Laibinis, P. E.; Palmer, B. J.; Lee, S.-
W.; Jennings;
G. K. (1998) "The Synthesis of Organothiols and Their Assembly into Monolayers
on
Gold" in Thin Films, Vol. 24 (Ulman, A., ed.) pp. 1-41, Academic Press, San
Diego, CA),
incorporated herein by reference.
The aforementioned review article has classified terminal moieties associated
with
alkylthiol SAMs with respect to the surface energy of the assembled surfaces.
Moieties ,
which afford highly wettable surfaces and are thus suitable for the
preparation of analysis
zone monomers include, but are not limited to:' CO2H, B(OH)2, P03H2, CONH2 and
OR
Each of the aforementioned moieties is reported to afford a surface exhibiting
a contact
angle of less than about 40 . Generally speaking, moieties that afford highly
wettable
surfaces are comprised of hydrogen bond acceptors, hydrogen bond donors, and
combinations thereof. Terminal moieties which afford surfaces of intermediate
wettability
and are thus suitable for the preparation of liquid retention zone monomers
include, but are
not limited to: CN (60 , 10), O2CCH3 (63 , 11), CO2CH3 (67 , 10), NHCOCH3 (68
, 11),
SCOCH3 (70 , 11), OCH3 (74 , 11), CONHCH3 (76 , 11), NHCOCF3 (77 , 11) and
CO2CH2CH3 (89 , 10). The contact angle associated with the assembled surface
and the
corresponding alkyl chain length is shown in parenthesis. Generally speaking,
moieties
which afford intermediately wettable surfaces tend to be comprised of
functionalities that
participate in dipole-dipole interactions. Terminal moieties which afford
minimally
wettable surfaces and are thus suitable for the preparation of boundary zone
monomers
include, but are not limited to: O(CH2)2CH3 (104 , 11), O(CH2)3CH3 (113 , 16),
NHCO(CF2)7CF3 (114.5 , 2), O(CH2)4CH3 (115 , 16), O(CH2)5CH3 (115 , 16),
OCH2CF2CF3 (118 , 11), and (CF2)5CF3 (118 , 2). The contact angle associated
with the
assembled surface and the corresponding alkyl chain length is shown in
parenthesis.
Generally speaking, moieties which afford minimally wettable surfaces tend to
be
comprised of hydrophobic and oleophobic functionalities.
-43-
CA 02531972 2010-11-22
78899-7
Preferably, both the target and liquid retention zones of the sample
presentation
device of the present invention are prepared from monomers that confer protein
resistance
upon the assembled surface. A number of SAMs prepared from alkylthiols on gold
have
been specifically characterized with respect to the adsorption of proteins.
The most protein
resistant of the surfaces thus far reported are those derived from monomers
which present
oligo(ethylene oxide) (OCH2CH2) units. The utility of these surfaces was first
described
by Prime and Whitesides (Prime, K. L. and Whitesides, G. M. J. Arr. Chem.
Soc., 1993,
11J, 10714-21). A survey of structure-property relationships of surfaces that
resist protein
adsorption has appeared (Ostuni, E.; Chapman, R. G.; Holmlin, R. E.; Takayama,
S.;
Whitesides, G. M. Langniuir, 2001, 17, 5605-5620). Recently, a number of
zwitterionic
SAMs have been shown to exhibit good resistance to protein adsorption
(Holmlin, R. E.;
Chen, X.; Chapman, R. G.; Takayama, S.; Whitesides, G. M., Langmuir, 2001, 17,
2841-50) and are therefore potentially useful as analysis zones owing to their
combination
of highly wettable surfaces and good resistance to protein adsorption.
In preferred embodiments, the analysis zone of the sample presentation device
of
the present invention is prepared from monomers of the General Formula I:
HS(CH2)ii-
(OCH2CH2)mOH, wherein m is from 3 to 7. Monomers of this general formula
afford
surfaces that exhibit contact angles in the range of about 30 to about 38 .
Although these
surfaces do not exhibit the lowest possible contact angles, they are
preferably utilized
owing to their superior performance with respect to minimizing the binding of
proteins.
Furthermore, analysis zone monomers of General Formula I are preferably
utilized in
conjunction with liquid retention zone monomers that afford surfaces which
exhibit contact
angles greater than about 60 .
Similarly and preferably,' the liquid retention zone of the sample
presentation
device of the present invention is prepared from monomers of the General
Formula II:
HS(CH2)1 I -(OCH2CH2)mR, wherein in =,3 to 7, and wherein group R is a
terminal moiety
which influences surface tension and wettability. Preferably but not
exclusively, group R
is selected from one of OCH3, OCH2CN, CO2CH3, CONHCH3, and CO2CH2CH3 moieties.
Each of the aforementioned terminal moieties affords a surface that exhibits a
contact angle
in the range of about 62 to about 89 .
-44-
CA 02531972 2010-11-22
78899-7
Alternatively and preferably, the liquid retention zone of the sample
presentation
device of the present invention may be prepared from a monomer of the formula
HS(CH2)110CH2C6H5. The terminal benzyl moiety (CH2C6H5) exhibits particular
utility
with respect to samples dissolved in organic solvents and affords a surface
that exhibits a
contact angle of about 90 .
In preferred embodiments, the boundary zone of the sample presentation device
of
the present invention is prepared from a monomer which confers a minimum of
wettability
with respect to liquid samples wherein the analytes are dissolved in aqueous
buffers,
organic solvents and mixtures thereof. Monomers presenting terminally
perfluorinated
moieties have been shown to have particular utility in this regard (Maud, C.;
Calas, P.;
Blancou, H.; Commeyras, A. J. Fluorine Chem., 2000, 104, 173-183).
A preferred embodiment of the present invention is one wherein the analysis
zone
is prepared from a monomer of the formula HS(CH2)1I(OCH2CH2)30H, wherein the
liquid
retention zone is prepared from a monomer of the formula
HS(CH2)11(OCH2CH2)30CH3,
and wherein the boundary zone is prepared from a monomer of the formula
HS(CH2)11OCH2CH2(CF2)5CF3. This combination of monomers affords a surface
wherein
the contact angle of the analysis zone, liquid retention zone, and boundary
zone are about.
38 , 62 and 117 , respectively.
Another preferred embodiment of the present invention is one wherein the
analysis
zone is prepared from a monomer of the formula HS(CH2)1I(OCH2CH2)30H, wherein
the
liquid retention zone is prepared from a monomer of the formula
HS(CH2)110CH2C6H5,'
and wherein the boundary zone is prepared from a monomer of the formula
HS(CH2)11OCH2CH2(CF2)5CF3. This combination of monomers affords a surface
wherein
the contact angle of the analysis zone, liquid retention zone, and boundary
zone are about
38 , 91 and 117 , respectively.
Mixed (binary) self-assembled monolayers prepared from two alkylthiol monomers
have been exploited to precisely control surface contact angle and
wettability. (Semal, S.;
Bauthier, C.; Voue, M.; Vanden Eynde, J. J.; Gouttebaron, R.; De Coninck, J.
J. Phys.
Chem. B, 2000, 104, 6225-6232). Contact angles have
been adjusted over a range of greater than 40 by mixing monomers utilized to
prepare
highly wettable and intermediately wettable surfaces. Preferably, binary SAMs
are
-45-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
exploited to prepare either the analysis zone or the liquid retention zone.
Alternatively,
ternary and quaternary self-assembled monolayers may be exploited to prepare
either the
analysis zone or the liquid retention zone. Ternary and quaternary SAMs are
prepared
from binary mixtures of either substituted alkylthiols and hetero-substituted
asymmetric
alkyl disulfides (i.e., HS(CH2)11R1 and R2(CH2)11S-S(CH2)11R3) or two hetero-
substituted
asymmetric alkyl disulfides (i.e., R1(CH2)11S-S(CH2)11R2 and R3(CH2)11S-
S(CH2)11R4),
respectively.
With reference to FIGS. 8a through 81, the step-wise process for
photolithographic
patterning of SAMs comprised of alkylsilanes on silicon is depicted.
Modification of
silicon and glass by reaction with either alkyl dimethylchlorosilanes, alkyl
dimethylalkoxysilanes, alkyl trihalosilanes, or alkyl trialkoxysilanes is
described in the
literature and is understood by those skilled in the art.
With reference to FIG. 8a, a suitable substrate 47 such as a silicon wafer,
glass
wafer, or metallic substrate with silicon dioxide deposed thereupon is
appropriately
activated for covalent attachment to an alkylsilane by a process involving
removal of
surface contaminants followed by oxidation of the surface to generate silanol
(Si-OH)
moieties. Preferably, the substrate is briefly treated with oxygen plasma,
washed with an
oxidizing solution (Piranha Solution), and then again treated with oxygen
plasma to afford
an activated surface 48 that presents an average silanol density approaching
4.9
Si-OH/nm2.
With reference to FIG. 8b, following surface activation the first alkylsilane
monolayer 49 is assembled on the silicon surface. Silanization may be
performed neat, by
solution deposition, or by vapor deposition. The first alkylsilane monolayer
49 is
preferably prepared from an alkylsilane which affords a surface that exhibits
a contact
angle of greater than 100 and further exhibits a minimum of wettability with
respect to
liquid samples.
With reference to FIG. 8c, the silanized substrate is coated with a
photoresist 50
prior to lithography. The resist may be of either a negative tone or positive
tone. A
negative resist results in decreased solubility in exposed regions of the
resist, thus giving a
negative image relative to the mask. A positive resist results in increased
solubility in the
exposed regions of the resist, thus giving a positive image relative to the
mask. The use of
a positive resist is depicted throughout FIG. 6. The resist may be applied
through a dip-
-46-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
type of process, but is preferable applied using a spin-coater. The
manufacturers'
recommendations with respect to resist thickness and curing times should be
used as
guidelines.
With reference to FIG. 8d, the substrate is photo-patterned by exposure to an
ultraviolet light source as required for use in conjunction with the
particular resist
employed. The photomask 51 may be prepared from a number of commonly employed
materials which include, but are not limited to, chromium-on-quartz, Mylar,
acetate, and
metallic stencils. The opening in the mask 52 results in the creation of
features of size and
shape corresponding to the liquid retention zone.
With respect to FIGS. 8e and 8f, the substrate is' initially treated with a
commercial
solution specific to the resist employed that dissolves the exposed areas of
resist while
those regions not exposed 53 to the ultraviolet light source remain relatively
insoluble.
After removal of exposed resist, an oxygen plasma treatment is employed to
activate the
surface 54 in preparation for further silanization. The second alkylsilane
monolayer 55 is
assembled on the activated silicon surface. Silanization may be performed
neat, by
solution deposition, or by vapor deposition. The second alkylsilane monolayer
55 is
prepared from an alkylsilane that affords a surface that exhibits a contact
angle in the range
of about 40 to about 95 and further affords a surface that exhibits minimal
binding with
respect to analytes.
With respect of FIGS. 8g and 8h, the remaining photoresist 53 is removed by
further washing the substrate with one of several organic solvents known to
dissolve
unexposed resist (e.g., acetone, 1-methyl-2-pyrrolidinone, etc.) The patterned
substrate
comprised of two distinctive zones is coated with a photoresist 56 prior to
lithography as
described above.
With respect to FIGS. 8i and 8j, the patterned substrate is further photo-
patterned
by exposure to an ultraviolet light source through a photomask 57 as described
above. The
opening in the mask 58 results in the creation of features of size and shape
corresponding
to the analysis zone. The substrate is then treated with a commercial solution
specific to
the resist employed that dissolves the exposed areas of resist while those
regions not
exposed 59 to the ultraviolet light source remain relatively insoluble. After
removal of
exposed resist, an oxygen plasma treatment is employed to activate the surface
60 in
preparation for further silanization.
-47-
CA 02531972 2010-11-22
78899-7
With reference to FIGS. 8k and 81, the third monolayer 61 is assembled on the
activated silicon surface. Silanization may be performed neat, by solution
deposition, or
by vapor deposition. The third alkylsilane monolayer 61 is prepared from an
alkylsilane
that affords a surface that exhibits a contact angle of less than about 400
and further affords
a surface that exhibits minimal binding with respect to analytes. Finally, the
remaining
photoresist 59 is removed by further washing the substrate with one of several
organic
solvents known to dissolve unexposed resist to afford a patterned surface
comprised of
three distinctive zones.
In this manner, the step-wise process for photolithographic patterning of SAMs
prepared from alkylsilanes on silicon is exploited to prepare the sample
presentation device
of the present invention. It should be noted that the sequence of patterning
depicted
(boundary zone, followed by liquid retention zone, followed by analysis zone)
was selected
arbitrarily, and that the reverse sequence (analysis zone, followed by liquid
retention zone,
followed by boundary zone) would also prove as suitable as the sequence
illustrated. The
above-described process of photolithographic patterning of self-assembled
monolayers
prepared from alkylsilanes on silicon is exemplary and the invention is not
limited to only
the process described.
Numerous alkylsilanes are suitable for use in preparation of sample
presentation
device of the present invention. Alkylsilanes are mostly commercially
available and their
synthesis and use in surface modification is understood. (Shriver-Lake, L. C.
(1998)
"Silane-modified surfaces for biomaterial immobilization" Immobilized
Bioinolecules in
- Analysis: A Practical Approach (Cass, T. and Ligler, F. S., eds.) Chapter 1,
Oxford
University Press, Oxford, UK).
Utilizing an approach that differs somewhat from that outlined above,
activated
silicon surfaces may be first derivatized with an appropriate alkylsilane
having a
nucleophilic moiety that is further functionalized by appending a terminal
moiety that
confers the required wettability. Alternatively, when alkylsilanes with
suitable terminal
moieties are available, the surface may be modified in a single step. Terminal
moieties
suitable for use in the preparation of the sample presentation device of the
present
invention include, but are not limited to, those described above.
In preferred embodiments, the analysis zone of the sample presentation device
of
the present invention is initially prepared from 3-
aminopropyltrimnethoxysilane, and then
-48-
CA 02531972 2010-11-22
78899-7
further functionalized to afford an immobilized silane of General Formula III:
(XO)3Si-
CH2CH2CH2NH000H2(OCH2CII2)õOH, wherein X is linkage to either the silicon
surface
or an adjacent immobilized silane, and wherein n is from 4 to 8. Monomers of
General
Formula III afford surfaces that exhibit contact angles in the range from
about 30 to about
40 . Although these surfaces do not exhibit the lowest possible contact
angles, they are
preferably utilized owing to their superior performance with respect to
minimizing the
binding of proteins. Furthermore, analysis zone monomers of General Formula
III are
preferably utilized in conjunction with liquid retention zone monomers that
afford surfaces
which exhibit contact angles greater than 60 .
Similarly and preferably, the liquid retention zone of the sample presentation
device of the present invention is initially prepared from 3-
aminopropyltrimethoxysilane,
and then further functionalized to afford an immobilized silane of General
Formula IV:
(XO)3SiCH2CH2CH2NHCOCH2(OCH2CH2)1R', wherein X is linkage to either the
substrate or an adjacent monomer, wherein n is from 4 to 8, and wherein group
R' is a
ternunal moiety which influences surface tension and wettability. Preferably
but not
exclusively, group R' is selected from one of Cl-I3, CH2CN, CH-2CO2CH3,
CH2CONIICH3,
and CH2CO2CH2CH3 moieties. Each of the afore-mentioned terminal moieties
affords a
surface that exhibits contact angles in the range of about 60 to about 90 .
In preferred embodiments, the boundary zone of the sample presentation device
of
the present invention is prepared in a single step from an alkylsilane which
confers a
minimum of wettability with respect to aqueous samples of General Formula V:
(CH3)2(X')SiCH2CH2-(CF2)2CF3, wherein X' is a surface reactive moiety.
A variety of alternative surface-modification chemistries and surface
patterning
approaches may be exploited to prepare the sample presentation devices of the
present
invention. Polymeric compositions of matter have recently attracted interest
with respect
to the patterning of protein resistant surfaces. Patterned surfaces initially
prepared from
either alkylthiol or alkylsilane SAMs have been further functionalized by
either grafting
polymeric compositions to the surface or growing polymeric compositions from
the
surface (e.g., Husemann, M.; Mecerreyes, D.; Hawker, J. L.; Hedrick, R. S.;
Abbott, N. L.
Angew. Chem. Int. Ed. 1999, 38, 647-649; Shah, R. R.; Merreceyes, D.;
Husemann, M.;
Rees, I.; Abbott, N. L.; Hawker, C. J.; Hedrick, J. L. Macromolecules 2000,
33, 597-605;
and Hyun, J. and Chilkoti, A. Macromolecules 2001, 34, 5644-5652).
-49-
CA 02531972 2010-11-22
78899-7
Recently, the first report of surface patterning by adsorption of block
copolymers appeared
(Deng, T.; Ha, Y.-H.; Cheng, J, Y.; Ross, C. A.; Thomas, E. L. Langnauir,
2002, 18,
6719-6722). Polymeric thin films grafted to SAMs have been shown to resist the
adsorption of proteins to an extent comparable to, or better than, SAMs that
present
tri(ethyleneglycol) groups (Chapman, R. G.; Ostuni, E.; Liang, M. N.;
Meluleni, G.; Kim,
E.; Yan, L.; Pier, G.; Warren, H. S.; Whitesides, G. M. Langmnuir 2001, 17,
1225-1233).
It is understood that even the least wettable surfaces may nevertheless retain
certain
moieties from liquid samples, even if in only a non-specific manner. Such
surfaces in fact
may contribute to the advantages of the sample presentation devices of the
present
invention by, for example, enhancing their ability to concentrate analytes by
removal of
those moieties that are not targets for subsequent analysis. This may be
particularly useful
in the context of retention of non-biological moieties that might interfere
with the analysis
of analytes. However, the surfaces of the sample presentation devices are not
limited to
only this example, but rather may comprise surfaces that bind moieties in
regions other
than the analysis zone that may be handled or processed separately from the
analyte-
containing sample. Indeed, any moiety that may be analyzed by analytical
biochemical
methods may be retained, stored, transported, and subsequently analyzed using
the sample
presentation devices of the invention. The present invention therefore allows
that some
retention of moieties in zones other than that having the highest degree of
wettability is
possible, and that subsequent analysis of those moieties may be desirable.
Substantial
amounts of the analytes of interest, however, are not typically retained in
zones other than
those with the highest degree of wettability. Therefore, in the context of the
example of
analyte analysis by laser desorption spectroscopy, the target analytes
retained in the zone
of highest wettability are not desorbed from a bound state to the surface of
the sample
presentation device.
Uses and Applications of Sample Presentation Devices
The descriptions of various uses and applications of the sample presentation
devices of the present invention that follow are merely exemplary and do not
limit the
scope of the invention.
The sample presentation devices of the present invention find many uses in
combination with various analytical techniques and procedures. Thus, the
present
-50-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
invention includes methods for using the aforementioned sample presentation
devices.
More specifically, present invention includes methods of using the sample
presentation
devices of the present invention to identify the presence of analytes in a
sample, and to
analyze a plurality of samples, either on a sample presentation device or on a
plurality of
sample presentation devices.
Virtually any analytical method that permits the detection, identification, or
measurement of analytes in a liquid sample can be used in combination with the
sample
presentation devices of the present invention. Examples of such analytical
methods
include but are not limited to MALDI-MS or electrospray ionization MS. The
sample
presentation devices are particularly well suited to us in combination with
high throughput
analytical measurement techniques, such as, for example, for use in MALDI-MS
in which
the sample presentation device analysis zones are configured in such fashion
as to promote
high throughput data acquisition.
the sample presentation devices of the present invention may also be used to
manipulate liquid samples, and the analytes 'contained therein. Based on the
differing
wettability properties and capture properties that the surfaces of the sample
presentation
devices may be designed to have, the sample presentation devices may be
designed to
manipulate, concentrate, position, store, transfer (with and without
mechanical
intervention), recover (with or without mechanical intervention), analyze,
modify or
process (via use of analyte modifying reagents on the sample presentation
devices), or
fractionate liquid samples or the analytes contained therein. Moreover,
because the sample
presentation devices of the present invention may be designed to accomplish
any of these
functions in response to chemical or physical stimuli (e.g., heat, UV
radiation, pressure,
electromagnetic radiation), the sample presentation devices of the present
invention may
accomplish these functions reversibly or irreversibly, and may further perform
various
combinations of these functions in response to external forces.
Virtually any liquid sample (and analytes) can be used in connection with the
sample presentation devices of the present invention. For example, the present
invention
can be used to analyze fractions recovered from liquid chromatography. The
present
invention can be used to analyze enzymatic digests prepared from either
protein spots
excised from 2D gel electrophoresis or from fractions collected from affinity
chromatography (i.e. ICAT). The present invention can also be used to analyze
samples
-51-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
recovered from surface plasmon resonance biosensors. The present invention can
also be
used for 1:1 sample transfer with standard multi-well format robotics and
assays. Indeed,
the sample presentation devices of the present invention can be used to handle
and
manipulate liquid samples obtained from virtually any source, whether such
samples are
the result of laboratory experiment (such as the enzymatic digest and surface
plasmon
resonance biosensor sample examples identified above), obtained from the
environment
(such as a water quality sample from a river), or obtained directly from
living organisms
(such as a human urine sample).
The present invention can also be used for storage of samples for archival
purposes
or for further analysis. In other words, the detection and analysis of the
analytes contained
in liquid samples need not occur immediately following transfer of the liquid
sample to the
analysis zone.
Thus, various embodiments 'of the present invention provide for sample
presentation devices that serve a variety of liquid-handling functions,
including but not
limited to sample/analyte handling, as well as liquid deposition, retention,
transfer, locating
and re-locating, and storage. Some examples of these various uses of the
sample
presentation devices of the present invention are provided.
With reference to Figs 9a through 9f, various steps in the process of sample
drying
are illustrated. A cross-sectional view of the sample presentation device of
the present
invention shows the surface deposited on the substrate 62 comprised of three
distinctive
zones, wherein the central analysis zone 63 is surrounded by the liquid
retention zone 64,
and wherein the liquid retention zone 64 is further surrounded by the boundary
zone 65.
With reference to FIG. 9b, depositing a liquid sample drop 66 on the surface
of the
sample presentation device initially results in simultaneous confinement of
the sample drop
volume to the surface of the analysis zone 63 and the liquid retention zone
64. Sample
drop confinement results from the surface tension associated with the limited
wettability of
the boundary region 65. Upon deposition, the contact angle of the sample drop
is
approximately equal to that of a drop residing exclusively on the liquid
retention zone.
With reference to FIGS. 9c through 9e, as the sample drop dries owing to
evaporation, both the radius and the contact angle of the drop recede until
the radius of the
drop corresponds to that of the analysis zone.
-52-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
With reference to FIG. 9f, when the radius of the sample drop 67 and that of
the
analysis zone 63 correspond, the contact angle of the sample drop is found to
be
approximately equal to that of a drop residing on the analysis zone. As the
sample drop
continues to dry owing to evaporation, the radius of the sample drop does not
further
recede, but remains constant as analytes are deposited as a thin film on the
surface of the
analysis zone. In this manner, aqueous samples of variable volume of up to
about 100 L,
deposited on the surface of the sample presentation device, afford upon drying
a thin film
of analytes confined within an area corresponding to the analysis zone.
For example, the sample presentation device of the present invention with a
liquid
retention zone having a 3.0 mm diameter (about 7.069 mm2 surface area) and a
analysis
zone having a 0.5 mm diameter (about 0.196 mm2 surface area), confines the
deposition of
analytes to a analysis zone surface area of about 36-fold smaller than the
surface area of
the liquid retention zone, with an about 36-fold concomitant increase in
average surface
analyte concentration. Consequently, in principal the sample drop drying
process described
above would potentially afford an about 36-fold increase in sensitivity.
With reference to FIGS. 10a through 10d, in the absence of the analysis zone
(only
the liquid retention zone 68 and the boundary zone 69 are present) the sample
drop 70 dries
without a significant reduction in radius resulting in deposition of analytes
over much of
the surface of the liquid retention zone 71. With reference to FIGS. 10e
through 10h, in
the absence of the liquid retention zone (only the analysis zone 72 and the
boundary zone
73 are present) the volume of the sample drop 74 is limited by the liquid-
holding capacity
of the analysis zone 72. The sample drop 74 dries without a significant
reduction in radius
resulting in deposition of analytes over much of the surface of the analysis
zone 75.
A significant increase in the sensitivity of detection results from the
process
described in FIGS. 9b through 9f. This phenomenon is best understood with
reference to
FIGS. 9a through 9d as well as FIGS. 10a through 10d. In the absence of the
analysis zone
(see FIG. 10a), the average analyte surface concentration per unit area in the
liquid
retention zone depicted in Fig 10a, 68 is equal to the total analyte
concentration divided by
the surface area. In the presence of the analysis zone depicted in FIG. 9a,
however, the
deposition of analyte is confined to the analysis zone wherein the average
analyte surface
concentration per unit area is equal to the total analyte concentration
divided by the surface
area of the analysis zone. Therefore, the presence of the analysis zone, 63,
depicted in
-53-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
Figure 9a, affords an increase in average surface concentration of analyte
which is equal to
the ratio of the surface area of the liquid retention zone, 68, depicted in
FIG. 10a, to the
surface area of the analysis zone, 63, depicted in FIG. 9a. Since the surface
area of the
analysis zone is significantly smaller than the surface area of the liquid
retention zone,
confining analyte deposition to the surface area of the analysis zone results
in a significant
increase in the average surface concentration of analyte presented to the mass
spectrometer
with a concomitant increase in sensitivity of detection.
For example, the sample presentation device of the present invention with a
liquid
retention zone having a 3.0 mm diameter (about 7.069 mm2 surface area) and a
analysis
zone having a 0.5 mm diameter (about 0.196 mm surface area), confines the
deposition of
analytes to a analysis zone surface area of about 36-fold smaller than the
surface area of
the liquid retention zone, with an about 36-fold concomitant increase in
average surface
analyte concentration. Consequently, in principal the sample drop drying
process
described above would potentially afford an about 36-fold increase in
sensitivity.
Analyte-confining properties of the analysis zone, which afford an increase in
sensitivity of detection, are demonstrated in the video contact angle images
shown in
FIGS. lla through llh. With reference to FIG. lla, the sample presentation
device of the
present invention was prepared with a liquid retention zone measuring about
1.6 mm OD
and an analysis zone measuring about 0.7 mm OD. To facilitate the observation
of the
focusing effect, the analysis zone was placed off-center. A drop of water was
applied to
the surface of the biochip and was observed to rapidly confine itself to the
surface area
corresponding to the liquid retention zone and the analysis zone. The initial
left-side and
right-side contact angles were recorded and were both found to be 57.1 , a
value which
corresponds to that exhibited by a surface prepared from exclusively the
liquid retention
zone monomer. As the drop dried owing to evaporation (see FIGS. llb through
11h), both
the observed radius and contact angles receded until the radius of the drop
corresponded to
that of the analysis zone. Furthermore, as the drop dried it was observed that
the center of
the drop moved to the right so as to allow the drop to center itself over the
analysis zone.
The left-side and right-side contact angles recorded in FIG. llh were both
found to be
35.4 , a value which corresponds to that exhibited by a surface prepared
exclusively from
the analysis zone monomer. The drop height, width and contact angle data
recorded in
conjunction with the acquisition of the images depicted in FIGS. lla through
llh is
summarized graphically in FIG. 12.
-54-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
The extraordinary liquid-holding capacity of the liquid retention zone is
demonstrated in FIG. 13. A photograph of a 16-site sample presentation device
of the
present invention shows the retention of sample drop volumes in the range 5 L
to 70 L.
The only factor that appears to significantly limit the sample drop volume is
the relative
proximity of the adjacent pairs of analysis and liquid retention zones.
Analyte-confining properties of the analysis zone are further demonstrated in
FIGS.
14a and 14b. The first photograph (FIG. 14a) is of a 16-site sample
presentation device of
the present invention with sample drop volumes in the range 5 L to 40 L
deposed on the
surface of 8 of the 16 sites. Each of the sample drops contained an equivalent
amount of a
soluble dye. The second photograph (FIG. 12b) is of the same sample
presentation device
after allowing the sample drops to dry. The dye is now deposed on the surface
of the
biochip in proximity to the analysis zone. The relative size of the analysis
zone and the
liquid retention zone is superimposed upon the biochip for comparison
purposes. In this
,instance, an excessive amount of dye was required to afford visible material
resulting in
the absence of tightly-focused analyte spots.
The sample presentation device of the present invention may be exploited to
facilitate high sensitivity mass spectrometric detection of chemical and
biological analytes
selected from, but not limited to: biological macromolecules such as peptides,
proteins,
enzymes, enzymes substrates, enzyme substrate analogs, enzyme inhibitors,
polynucleotides, , oligonucleotides, nucleic acids, carbohydrates,
oligosaccharides, poly-
saccharides, avidin, streptavidin, lectins, pepstatin, protease inhibitors,
protein A,
agglutinin, heparin, protein G, concanavalin; fragments of biological
macromolecules set
forth above, such as nucleic acid fragments, peptide fragments, and protein
fragments;
complexes of biological macromolecules set forth above, such as nucleic acid
complexes,
protein-DNA complexes, gene transcription complex, gene translation complex,
membrane, liposomes, membrane receptors, receptor ligand complexes, signaling
pathway
complexes, enzyme-substrate, enzyme inhibitors, peptide complexes, protein
complexes,
carbohydrate complexes, and polysaccharide complexes; and small biological
molecules
such as amino acids, nucleotides, nucleosides, sugars, steroids, lipids, metal
ions, drugs,
hormones, amides, amines, carboxylic acids, vitamins and coenzymes, alcohols,
aldehydes,
ketones, fatty acids, porphyrins, carotenoids, plant growth regulators,
phosphate esters and
nucleoside diphosphosugars, synthetic small molecules such as pharmaceutically
or
therapeutically effective agents, monomers, peptide analogs, steroid analogs,
inhibitors,
-55-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
mutagens, carcinogens, antimitotic drugs, antibiotics, ionophores,
antimetabolites, amino
acid analogs, antibacterial agents, transport inhibitors, surface-active
agents (surfactants),
amine-containing combinatorial libraries, dyes, toxins, biotin, biotinylated
compounds,
DNA, RNA, lysine, acetylglucosamine, procion red, glutathione, adenosine
monophosphate, mitochondrial and chloroplast function inhibitors, electron
donors,
carriers and acceptors, synthetic substrates and analogs for proteases,
substrates and
analogs for phosphatases, substrates and analogs for esterases and lipases and
protein
modification reagents.. Moreover, analytes that may be handled by the sample
presentation devices of the present inventions may be non-biological, and
include but are
not limited to, synthetic polymers, such as oligomers, and copolymers such as
polyalkylenes, polyamides, poly(meth)acrylates, polysulfones, polystyrenes,
polyethers,
polyvinyl ethers, polyvinyl esters, polycarbonates, polyvinyl halides,
polysiloxanes, and
copolymers of any two or more of the above, as well as oather non-biological
analystes
such as pesticides.
Analytes may be dissolved in aqueous buffers, organic solvents or mixtures
thereof.
Buffers are preferably selected from those prepared from volatile constituents
including,
but not limited to: ammonium acetate, ammonium bicarbonate, ammonium
carbonate,
ammonium citrate, triethylammonium acetate and triethylammonium carbonate,
triethyl-
ammonium formate, trimethylammonium acetate, trimethylammonium carbonate and
trimethylammonium formate. Aqueous samples containing high concentrations of
non-
volatile detergents (>0.1%) should be desalted prior to analysis as the
presence of detergent
may counteract and analyte-confining properties of the analysis zone. Organic
solvents are
preferably selected from those know to be miscible in aqueous buffers and to
promote the
solubility of biological analytes including, but not limited to: acetic acid,
acetone,
acetonitrile, ethanol, N,N-dimethylformamide (DMF), N,N-dimethylsulfoxide
(DMSO),
formic acid, heptafluorobutyric acid, methanol, N-methylpyrolidone (NMP),
2,2,2-
trifluoroethanol and trifluoroacetic acid.
The sample presentation device may be heated during the sample drying process
(either on the surface of a heating block, under an infrared lamp or under a
stream of hot
air) to facilitate the evaporation of high-boiling organic solvent or simply
to reduce the
time required for sample drying.
-56-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
Laser desorption time-of-flight mass spectrometry - a preferred analytical
method
to measure analytes using the sample presentation devices of the present
invention requires
a material (matrix) to be applied to the surface of the sample presentation
device to absorb
energy and thereby assist the ionization of analytes. Reagents frequently used
as matrices
for detection of biological analytes include trans-3,5-dimethoxy-4-
hydroxycinnamic acid
(sinapinic acid, SA), a-cyano-4-hydroxycinnamic acid (HCCA) and 2,5-
dihydroxybenzoic
acid (DHBA). Owing to the limited solubility. of the aforementioned matrices
in water,
stock solutions of these reagents often contain 50% to 100% organic solvent.
When
utilized in conjunction with the sample presentation devices of the present
invention, stock
solutions containing matrix are added to aqueous samples prior to applying the
sample to.
the surface of the sample presentation device. Alternatively, stock solutions
containing
matrix may be applied to the surface of the sample presentation device after
sample
deposition and, drying. In this instance, stock solutions containing a high
percentage of
organic solvent are preferably utilized to minimize dissolving of the analytes
deposited on
the surface of the analysis zone into the stock solution.
Numerous applications exist for using the sample presentation devices of the
present invention. Examples of the types of samples that could be used in the
present
invention include, but are not limited to, samples that are to be analyzed
directly without
any processing done before analysis, as well as samples that are to be
analyzed indirectly,
in that the samples are to be analyzed after some sort of processing has
occurred.
Examples of the types of samples that could be used in the present invention
that
fall into the category of samples that are to be analyzed directly without any
processing
done before analysis include, but are not limited to, biofluids; tissue and
cell extracts and
fractions; cells, bacteria, viruses; culture medium; environmental fluids;
environmental air
sampling; environmental media extracts (soil extracts, solid waste extracts,
elution from
wipes, elution from air filters);- forensic samples; and libraries
(combinatorial chemistry,
oligonucleotides, peptides, sugars, lipids, cells and components; chromosomes,
and viruses
and other large protein and nucleoprotein assemblies).
Examples of types of samples that could be used in the present invention that
fall
into the category of samples that are to be analyzed indirectly, i.e., after
some sort of
processing has occurred to the samples include, but are not limited to, liquid
chromatography (LC) output; gas chromatography (GC) output; elution from gels;
digested
-57-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
samples from LC output or gel elutions; mass spectrometry output; elutions
from surface
plasmon resonance (SPR) or other biosensors; desalting column output; solid-
phase
extraction output; liquid phase fractionated environmental samples;
derivatized samples
with respect to any of the above; and other chemical or physical processes and
any
combinations thereof.
The sample presentation device of the present invention further facilitates
the mass
spectrometric analysis of biological analytes recovered from fractionation
schemes that
exploit either column liquid chromatography or electrophoresis. In particular,
utility
results from the combination of the liquid-holding capacity of the device
(which enables
direct collection of chromatographic fractions, ' samples purified by
electrophoresis,
samples recovered from sample presentation devices and samples recovered from
biosensors without prior sample volume reduction) and the precise positioning
of the
sample and increased sensitivity of detection (which enables automated data
acquisition).
The liquid-holding capacity afforded by the sample presentation device of the
present
invention enables direct collection of fractions recovered from, but not
limited to, the
following techniques: affinity chromatography, hydrophobic interaction
chromatography,
ion exchange chromatography, immobilized metal ion affinity chromatography and
size
exclusion chromatography, as well as fractions recovered from orthogonal
separations
involving sequential utilization of two or more of the chromatographic
approaches
enumerated. Furthermore, the availability of the sample presentation device in
standard'
96-well, 384-well and 1536-well formats enables biochip-based sample
collection and
processing on multi-well plate processing devices and laboratory liquid
handling robots.
Consequently, the sample presentation device may be exploited to enable high-
throughput
mass spectrometric platforms as are needed to support the emergence of
proteomics and
other important fields of chemistry and biotechnology.
Contemporary protein identification often involves enzymatic digestion of
proteins
purified either by column liquid chromatography or excised from 2-dimensional
electrophoreses gels. Protein digests generally require desalting on reverse
phase liquid
chromatography (RPLC) or solid-phase extraction (SPE) prior to mass
spectrometry. The
sample presentation devices of the current invention are suitable for direct
collection and
subsequent analysis of protein digests desalted by high performance RPLC or
SPE.
-58-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
As a specific example, surface plasmon resonance (SPR) biosensors exploit
immobilized proteins to study protein-protein and other biological
interactions.
Unfortunately, a large volume of eluant is required to recover an analyte from
a biosensor
and the concentration of analyte in the sample is too low for optimum mass
spectrometry.
The sample presentation device of the present invention is suitable for direct
collection of
analytes recovered from biosensor systems; it may be configured to a standard
96-well
format so as to be compatible with sample collection devices already
integrated into
biosensor systems and can be exploited to enable automated sample collection
for mass
spectrometric analysis, and can concentrate liquid samples of large volumes.
The liquid-holding limitations associated with known mass spectrometer sample
presentation devices have prompted the development of various micro-column
liquid
chromatography approaches involving, the use of small pipette tips packed with
minute
quantities of chromatographic media (e.g., ZipTips ). Micro-column approaches
enable
the desalting of protein digests with a concomitant reduction in sample volume
reported to
be sufficient to enable the sample to be applied directly to prior art mass
spectrometer
devices for retaining samples. The sample presentation devices of the present
invention
are suitable for direct collection and subsequent analysis of protein digests
desalted by
micro-column RPLC.
In general, the sample presentation devices of the present invention can be
used to
accomplish the following with respect to the above-described samples:
concentrating;
diluting; locating; transporting; storing; presenting for analysis;
fractionating; washing; and
post-application processing (including digesting, derivatizing, and eluting).
It should be
understood that this list is not exhaustive and merely provides examples in
general terms as
to the various applications the sample presentation devices of the present
invention can be
used.
Once the samples have been applied to the sample presentation devices of the
present invention, and the samples have undergone any of the above-identified
operations
with respect to movement of liquid samples thereon, the following applications
can be
performed either on the sample presentation device itself or after removal
from the device:
MALDI-MS; other mass spectrometry techniques; surface plasmon resonance (SPR);
fluorescence; atomic force microscopy (AFM); optical spectroscopy; bio- and
chemiluminescence; x-ray photoelectron spectroscopy; ellipsometry;
electrochemical
-59-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
detection; phosphorescence; and UV, visible and IR spectroscopies. It should
be
appreciated that this is only a partial list of such applications. It should
also be understood,
that any of the above analyses may be combined and/or serialized, and that
where
appropriate, these analyses may be performed directly or indirectly upon the
analyte(s).
Numerous fields of use are contemplated as being applicable to the sample
presentation device of the present invention and include, but are not confined
to, such
fields as genomics, proteomics, pharmacogenomics, physiomics, toxiomics,
metabonomics, drug discovery/drug development/clinical trial monitoring,
toxicology,
diagnostics, environmental, biosensors, and biological and chemical
weapons/bioterrorism.
A few specific examples of the applications of the sample presentation devices
are
described below. The descriptions that follow are merely exemplary and do not
limit the
scope of the invention.
Genomics: The application of mass spectrometry to genotypic and phenotypic
problems has an essential prerequisite of desalting the nucleic acid
analyte(s) prior to
ionization. Traditionally this desalting is performed before the sample is
placed on a
MALDI source. In one embodiment, the sample presentation device in an X3
format can
accomplish the desalting simultaneously with concentrating the nucleic acid
analyte(s).
This embodiment is comprised of a reverse phase capture zone and an analyte
binding
resistant analysis zone. Another embodiment may be comprised of an X4, wherein
two
capture zones and a single analysis zone would be employed. In a concentric
arrangement,
the outer , capture zone would specifically bind polynucleotide analytes
through
complementary hybridization with immobilized capture probes; the inner capture
zone
would perform a desalting function as described above, and the analysis zone
presents the
analyte for detection. In both of these embodiments, the performance of
desalting and
presentation for analysis on the same chip increases throughput, minimizes
sample loss,
and decreases cost.
Drug Discovery/Development/Clinical Trial Monitoring: Many drugs are effective
on only a portion of the population. An example of this phenomenon is the drug
Herceptin, which is useful for only about 30% of breast cancer patients. In
the case of
Herceptin, the genetic and protein basis of the sensitivity was integral to
the design of the
drug, but in most cases the population cannot be divided into likely
responders and non-
responders prior to expensive and lengthy clinical trials. One of the
principal challenges of
-60-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
interpreting such clinical trial results is to understand the biological
and/or chemical basis
for response and non-response. That knowledge can then be used both for
targeting of
populations and for further refinement of the drug itself.
One approach to this problem is to obtain profiles (e.g., protein,
carbohydrate,
lipid) from the patients before, during, and after treatment, and to correlate
these profiles
with treatment outcome. Several embodiments of the present device can be
applied to such
studies. Samples (e.g., blood, urine, tissue) obtained from the patients can
be subjected to
one or more of the pre-processing methods enumerated in the section described
above,
such as multi-dimensional liquid chromatography, and the fractionated
materials produced
by that method applied to the device for concentration and presentation for
mass
spectrometric analysis. Alternatively, samples subjected to minimal processing
can be
applied to one or more of the present devices with capture zones of known
specificity. The
analytes are then transferred either to capture zones of complementary
specificity before
transfer to analysis zones, or directly to analysis zones. In this manner,
surfaces with
different specificities can be used both in series and in parallel in an
automated manner,
with the fractionated analytes presented on identical analysis zones for mass
spectrometry.
Mass spectrometry provides both profiles (the full mass spectrum) and the
opportunity to unambiguously identify specific molecular entities of interest.
The mass
spectra can then be collected into a database, and multifactorial analysis
tools applied to
correlate the profiles with patient response. In this way one can discover
patterns within
the profiles and/or specific molecular entities that enable: prediction of
response to
therapy; monitoring of response to therapy; and identification of molecular
entities that
affect response to therapy, thus allowing increasingly sophisticated drug
design.
This area of scientific inquiry, like the others described herein, is
dependent in
large measure on the ability to measure analytes in liquid solution. The
sample
presentation devices of the present invention, and their uses described
herein, represent an
important tool that can be used to conduct further study.
Environmental: Analyzing environmental samples for the presence of
contaminants is a worldwide effort. Among the particular problems faced by
such studies
are the low concentrations of analytes and the diversity of samples that must
be studied, as
contaminants may be present in gaseous, liquid, and solid materials. In
general, such
analyses involve collection, extraction, derivatization, fractionation, and
detection steps.
-61-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
The present devices may be applied in a number of ways to the analysis of
environmental samples. These examples are representative, but by no means
complete.
Devices with capture zones can be used for direct collection of analytes from
gaseous or
liquid media. For example, capture of hydrophobic pesticide residues from
aqueous
solutions by a hydrophobic surface may replace liquid/liquid extractions,
which can be
time-consuming and generate hazardous waste. The collected material can then
be
transferred directly to analysis zones, fractionated by serial or parallel
transfer to capture
zones of complementary specificity prior to transfer to analysis zones, or
transferred from
the device to enable analysis by one or more of the techniques enumerated in
the sections
described above. Mass spectrometry is generally used for identification of
pesticide
residues, but other techniques such as immunoassay may be applied. The present
devices
can also be used as previously described to present and/or fractionate
materials resulting
from any of the steps of environmental analysis listed above. The present
devices can be
used as a platform to derivatize analytes and present them for analysis in
altered form. For
example, silyl- and/or acetyl- moieties may be added to pesticides immobilized
on the
device to enable unambiguous identification of molecular structure.
Biological and Chemical Weapons/Bioterrorism: The United States government is
confronted with the need for platforms and analytical techniques to facilitate
the detection
of chemical and biological agents in both military and civil scenarios.
Challenges for
biowarfare detection include sample collection and distinguishing between
innocuous
versus toxic organisms. The current battlefield technique for bio agents
utilizes pyrolysis
to convert biological compounds to small, easily detectable molecules by MS. A
technique
relying on peptide biomarkers is largely anticipated, since it would be more
specific than
current methods. Tests on individuals to determine potential exposure to
warfare agents
should involve breath tests or blood drawing techniques. Stand-alone
biosensors as alerting
devices are also of great interest for use in public places or in the
battlefield. All these
methods present challenges in sample collection, pre-treatment, and
presentation of
samples to detectors by robotics or other remote means. Techniques that can
store,
manipulate, concentrate or purify samples or those that can be coupled to
aerosol impactors
currently used have the potential of attracting the interest of defense
agencies. The present
devices can be applied to biowarfare/bioterror detection in a manner similar
to that
described for environmental samples. In addition, devices with custom capture
zones can
be designed to collect microorganisms of interest from environmental or
biofluid samples,
-62-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
allow processing of the cells (or viruses) to release key markers, and present
those markers
for detection.
Examples
The following examples provide additional detail about the composition,
manufacture, and use of the sample presentation devices. of the present
invention, but are
exemplary only and do not in any way limit the scope of the present invention.
Example I
Preparation of 11 -(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyloxy)undec- 1 -
ene (1)
F FF FF F
F F F F F F
An amber shell. vial (40 mL) was charged with 3.0 mL of 1 H,1 H,2H,2H-
perfluorooctanol (13.7 mmol) and to this was added 1.4 mL of 50% aqueous
potassium
hydroxide (13.7 mmol). The solution was warmed to 80 C, stirred for 30
minutes and 3.3
mL of 11 -bromoundec- 1 -ene (1.5 mmol) added. The reaction was maintained at
80 C for
52 hours until TLC analysis (hexane) showed the starting material was
consumed. The
product was allowed to cool to room temperature, added to 100 mL ethyl acetate
and
extracted with water (2 x 50 mL) and brine (1 x 50 mL). The ethyl acetate
extract was
dried over magnesium sulfate, filtered and the solvent evaporated in vacuo to
afford an oily
residue. The residue was purified on a silica gel flash column (50 x 300 mm,
0% ethyl
acetate/hexane followed by 10% ethyl acetate/hexane). Fractions containing the
desired
product were combined and the solvent evaporated to afford 4.52 g (64%) of 1
as a
colorless oil. 1H NMR (400 MHz, CDC13): 0 5.80 (m, 1H), 4.95 (m, 2H), 3.69 (t,
J = 6.8
Hz, 2H), 3.43 (t, J = 6.8 Hz, 2H), 2.39 (m, 2H), 2.03 (m, 2H), 1.55 (m, 2H),
1.36 (m, 2H),
1.27 (broad m, l OH).
Example II
Preparation of Thioacetic Acid S-[11-(3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyloxy)undecyl] Ester (2)
0 F F F F FVF
F
F F F F F F
-63-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
A dry round bottom flask (100 mL) was charged with 1.0 g of 1 (1.9 mmol) under
argon and 10 mL of dry methanol added. To the resulting solution was added 426
CL of
thiolacetic acid (6.0 mmol) followed by 52 mg of 2,2'-azobis(2-
methylpropionamidine)
dihydrochloride (0.2 mmol). The reaction was shrouded in a foil tent and
exposed to light
from a low pressure mercury lamp. After 4 hours, TLC analysis (5% ethyl
acetate/hexane)
revealed that the starting material had been consumed. The solvent was
evaporated in
vacuo to give an oily residue. The residue was purified on a silica gel flash
column (40 x
300 mm, 0% ethyl acetate/hexane followed by 5% ethyl acetate/hexane).
Fractions
containing the desired product were combined and the solvent evaporated to
afford 856 mg
(76%) of 2 as a colorless oil. 1H NMR (400 MHz, CDC13): 8 3.69 (t, J= 6.8 Hz,
2H), 3.43
(t, J= 6.8 Hz, 2H), 2.39 (m, 2H), 2.31 (s, 3H),1.55 (m, 2H), 1.33 (m, 2H),
1.25 (broad in,
10H).
Example III
Preparation of 11-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyloxy)undecane-l-
thiol (3)
F F F F F F
HS Q~^\~~~~ F
F F F F F F
An amber shell vial (20 mL) was fitted with a Teflon-lined silicon septum,
charged
with 850 mg of 2 (1.1 mmol) and 5 mL of 3N methanolic hydrogen chloride (15
mmol)
added. The resulting solution was warmed to 40 C for 4 hours. The solvent was
removed
to afford 782 mg (98%) of 3 as a colorless oil. 1H NMR (400 MHz, CDC13): 8
3.69 (t, J
6.8 Hz, 2H), 3.43 (t, J = 6.6 Hz, 2H), 2.51 (dd, J = 7.3, 7.6 Hz, 2H), 2.39
(m, 2H), 1'.58
(m, 4H), 1.32 (t, J= 8.0 Hz, 1H), 1.25 (broad in, 12H).
Example IV
Preparation of 11-{2-[2-(2-Methoxyethoxy)ethoxy]ethoxy}undec-l-ene (4)
A round bottom flask (200 mL) was charged with 27.4 mL of triethyleneglycol
monomethyl ether (171 mmol) and 9.1 mL of 50% aqueous sodium hydroxide (114
mmol)
added. The pale yellow solution was warmed to 80 C, stirred for 30 minutes
and 26.6 mL
-64-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
of 11-bromoundec-1-ene (114 mmol) was added dropwise. The reaction was
maintained at
80 C for 7.5 hours until TLC analysis (100% ethyl acetate) showed the
starting material to
be consumed. The product was cooled to room temperature, diluted into 50 mL of
water
and extracted with hexanes (3 x 50 mL). The hexanes extracts were combined,
dried over
magnesium sulfate, filtered and the solvent evaporated in vacuo to afford 20 g
(56%) of 4
as a clear, colorless oil. 1H NMR (400 MHz, CDC13): 8 5.81 (m, 1H), 4.96 (in,
2H), 3.68-
3.56 (m, 12H), 3.44 (t, J= 6.8 Hz, 2H), 3.38 (s, 3H), 2.04 (m, 2H), 1.57 (m,
2H), 1.36 (m,
2H), 1.27 (broad s, l OH).
Example V
Preparation of Thioacetic acid S-(1 1 - {2- [2-(2-methoxyethoxy)ethoxy]
ethoxy} undecyl)
ester (5)
O
A dry round bottom flask (200 mL) was charged with 5.0 g of 4 (15.8 mmol)
under
argon and 10 mL of dry methanol was added. To this was added 3.6 mL of
thiolacetic acid
(50 mmol) followed by 434 mg of 2,2'-azobis(2-methylpropionamidine)
dihydrochloride
(1.6 mmol). The reaction was shrouded in a foil tent and exposed to light from
a low
pressure mercury lamp. After 15.5 hours, TLC analysis (ethyl acetate/hexane,
1:3) revealed
the starting material had been consumed. The solvent was evaporated in vacuo
to give a
residue with a strong sulfur-like odor. The residue was purified on a silica
gel flash column
(40 x 300 mm, 30% ethyl acetate/hexane, and 50% ethyl acetate/hexane).
Fractions
containing the desired product were combined and the solvent was evaporated to
afford
5.83 g (94%) of 5 as a colorless oil. 1H NMR (400 MHz, CDC13): 8 3.67-3.54 (m,
12H),
3.44 (t, J= 7.2 Hz, 2H), 3.38 (s, 3H), 2.86 (t, J= 7.2 Hz, 2H), 2.32 (s, 3H),
1.57 (m, 4H),
1.36-1.26 (broad in, 14H).
Example VI
Preparation of 11-{2-[2-(2-Methoxyethoxy)ethoxy]ethoxy}undecane-l-thiol (6)
HS O~~O~~O~iO~
An amber shell vial (20 mL) fitted with a Teflon-lined silicon septum was
charged
with 5.0 g of 5 (12.7 mmol) and 7 mL of 3N methanolic hydrogen chloride (21
mmol) was
-65-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
added. The solution was warmed to 40 C for 6 hours. The solvent was then
evaporated in
vacuo to afford 4.40 g (98%) of 6 as a colorless waxy gel. 1H NMR (400 MHz,
CDC13): 8
3.67-3.54 (m, 12H), 3.44 (t, J = 6.8 Hz, 2H), 3.37 (s, 3H), 2.51 (dd, J = 7.3,
8.0 Hz, 2H),
1.57 (m, 4H), 1.32 (t, J= 7.6 Hz, IH), 1.26 (broad m, 14H).
Example VII
Preparation of 2-[2-(2-Undec-10-enyloxyethoxy)ethoxy]ethanol (7)
~ Oi~O~~Oi~OH
A round bottom flask (250 mL) was charged with 67.0 mL of triethyleneglycol
(0.5
mol) and 8.0 mL of 50% aqueous sodium hydroxide (8 mL, 0.1 mol) was added. The
solution was warmed to 100 C, stirred for 30 minutes and 22.0 mL of 11 -
bromoundec- 1
ene (0.1 mol) were added dropwise to give a dark yellow solution which
produced a
precipitate of sodium bromide. The reaction was maintained at 100 C for 2.5
hours until
TLC analysis (methanol/ethyl acetate/hexane, 1:1:8) revealed that the starting
material to
be consumed. The reaction was cooled to room temperature, diluted into 300 mL
of water
and extracted with hexanes (3 x 100 mL). The organic extracts were combined,
washed
with brine (50 mL), dried over magnesium sulfate and filtered. The solvent
evaporated in
vacuo to give an oily residue. The residue was purified on a silica gel flash
column (50 x
400 mm, methanol/ethyl acetate/hexane 5:5:90). Fractions containing the
desired product
were combined and the solvent was evaporated to give 20.8 g (69%) of 7 as a
clear oil. 1H
NMR (400 MHz, CDC13): 8 5.78 (m, 1H), 4.93 (m, 2H), 3.72-3.55 (m, 12H), 3.42
(t, J =
7.2 Hz, 2H), 2.64 (t, J = 5.6 Hz, 1H), 2.01 (m, 2H), 1.54 (m, 2H), 1.34 (m,
2H), 1.25
(broad s, 10H).
Example VIII
Preparation of Thioacetic acid S-(11-{2-[2-(2-
hydroxyethoxy)ethoxy]ethoxy}undecyl)
ester (8)
O
~S O~,O~~Oi~OH
A dry round bottom flask (100 mL) was charged with 2.0 g of 7 (6.6 mmol) under
argon and 10 mL of dry methanol added. To this was added 2.85 mL of
thiolacetic acid (40
mmol) followed by 271 mg of 2,2'-azobis(2-methylpropionamidine)
dihydrochloride (1.0
-66-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
mmol). The reaction was shrouded in a foil tent and exposed to with light from
a low
pressure mercury lamp. After 6 hours, TLC analysis (methanol/ethyl
acetate/hexane,
1:1:8) revealed that the starting material had been consumed. The solvent was
evaporated
in vacuo to give yellow oil. The oil was purified on a silica gel flash column
(50 x 300
mm, methanol/ethyl acetate/hexane, 1:1:8). Fractions containing the desired
product were
combined and the solvent was evaporated to afford 2.44 g (98%) of 8 as a light
yellow oil.
1H NMR (400 MHz, CDC13): 8 3.71-3.54 (m, 12H), 3.42 (t, J = 6.6 Hz, 2H), 2.83
(t, J =
7.2 Hz, 2H), 2.66 (broad s, 1H), 2.29 (s, 3H), 1.52 (m, 4H), 1.36-1.23 (broad
in, 14H).
Example IX
Preparation of 2- {2-[2-( 11 -Mer~aptoundecyloxy)ethoxy]ethoxy} ethanol (9)
HS
An amber shell vial (20 mL) was fitted with a Teflon-lined silicon septum,
charged
with 2.40 g of 8 (6.4 mmol) and 5.0 mL of 3N methanolic hydrogen chloride (15
mmol)
added. The resulting solution was warmed to 40 C for 4 hours. The solvent was
then
evaporated in vacuo to afford 2.05 g (95%) of 9 as a colorless waxy gel. 1H
NMR (400
MHz, CDC13): 8 3.72-3.55 (m, 12H), 3.43 (t, J= 6.8 Hz, 2H), 2.71 (broad s,
1H), 2.50 (dd,
J= 7.6, 7.4 Hz, 2H), 1.62-1.52 (m, 4H), 1.31 (t, J= 7.6 Hz, 1H),'1.26 (broad
in, 14H).
Example X
Preparation of Undec-l0-enyl-oxymethylbenzene (10)
A dry round bottom flask (100 mL) was charged with 5.0 g of undec-10-en-l-ol
(29.4 mmol) under argon and 25 mL of dry N,N-dimethylformamide was added. The
resulting solution was cooled to 0 C and 2.16 g of 60% sodium hydride in
mineral oil (45
mmol) was added in one portion. The frothing mixture was stirred under argon
at 0 C for
minutes. To the chilled, stirred solution was added dropwise 7.7 g of
bromomethylbenzene (45 mmol) in 5mL of dry N,N-dimethylformamide and the
reaction
was allowed to warm to room temperature while stirring for 3 hours. The
reaction was
quenched by the slow addition of lOOmL of ethyl acetate, extracted with 1N
hydrochloric
30 acid (2 x 50 mL) and brine (1 x 50ml). The organic layer was dried over
magnesium
sulfate, filtered and the solvent evaporated to give an oily residue (9.5 g).
The residue was
-67-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
purified on a silica gel flash column (50 x 300 mm, 94:5:1
hexane/toluene/ethyl acetate)
and the fractions containing the desired product were combined. Finally, the
solvent was
evaporated in vacuo to afford 7.lg (93%) of 10 as a colorless oil. 'HNMR (400
MHz,
CDC13): 5 7.32 (d, 4H), 7.28 (m, 1H), 5.81 (m, 1H), 4.95 (m, 2H), 4.49 (s,
2H), 3.46 (t,
2H), 2.03 (in, 2H), 1.61 (m, 2H), 1.35 (broad in, 4H), 1.24 (broad s, 1OH).
Example XI
Preparation of Thioacetic Acid S-(11-Benzyloxyundecyl)ester (11)
O
A jacketed photo-reaction vessel (250 mL) was first charged with 5.0 g of 10
(19.2mmol) and 0.520 g of 2,2'-azobis(2-methylpropionamidine) dihydrochloride
(1.92mmol). The vessel was sealed, evacuated and back-flushed with argon
(several
cycles). While under argon, 60 mL of anhydrous methanol and 0.520 g of
thioacetic acid
(92 mmol) were injected into the reaction vessel and the contents of the
vessel were stirred.
The vessel was again evacuated and back-flushed with argon (several cycles).
The UV
lamp was activated and the mixture irradiated under argon with constant
stirring for 3
hours. The reaction was continually cooled (water jacket) and the temperature
maintained
below 38 C during the photo-reaction process. The reaction vessel was allowed
to cool to
room temperature and the solvent was evaporated to give pale yellow oil (10.8
g). The oil
was purified on a silica gel flash column (50 x 300 mm,' 98:2 hexane/ethyl
acetate) and the
fractions containing the desired product were combined. Finally, the solvent
was removed
in vacuo to afford 5.Og (77%) of 11 as a colorless oil. 1H NMR (400 MHz,
CDC13): 8 7.32
(d, 4H), 7.28 (m, 1H), 4.49 (s, 2H), 3.46 (t, 2H), 2.86(t, 2H), 2.31(s, 3H),
1.50-1.66 (in,
4H), 1.20-1.40 (broad in, 14H).
Example XII
Preparation of 11-Benzyloxyundecane-l-thiol (12)
Hs o ~ ~
An amber shell vial (40 mL) was fitted with a Teflon-lined silicon septum,
charged
with 3.04 g of 11 (9.03mmol) followed by 2mL of dichloromethane, lmL of
hexane, and
12 mL of 4.9 N ethanolic hydrogen chloride. The resulting solution was warmed
to 40 C
-68-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
for 4.5 hours. The solvent was then evaporated in vacuo to afford a colorless
oily residue
(2.8 g). The residue was purified on a silica gel flash column (25 x 450 mm,
9:1
hexane/chloroform) and the fractions containing the desired product were then
combined.
The solvent was evaporated in vacuo to give 2.5 g (94%) of 12 as a colorless
oil. 1H NMR
(400 MHz, CDC13): 8 7.32 (d, 4H), 7.28 (m, 1H), 4.49 (s, 2H), 3.46 (t, 2H),
2.51(q, 2H),
1.55-1.65 (m, 4H), 1.20-1.40 (broad in, broad t, 15H).
Example XIII
Preparation of Self-Assembled Monolayers on Gold-Coated Silicon Substrates
Silicon wafers (200 mm, P-type, Prime Grade Silicon 100) were diced to
individual
substrates and cleaned to afford a surface having fewer than 10 particles
(0.16 m to 3000
m) per substrate. Metal deposition was carried out in a CPA 9900 sputtering
system with
a base pressure of 5 x 10-7 mm. In the sputtering chamber, the substrates were
cleaned and
etched by argon plasma and an adhesive layer of titanium and tungsten (1:9)
was sputtered
at a rate of 5 A/s to a thickness of 250 A. Gold was then sputtered at a rate
of 20 A/s up to a
thickness of 1000 A. Substrates were cooled under an argon flow prior to
removal.
Prior to monolayer assembly, gold-coated substrates were cleaned by treatment
with argon plasma at 200 W for 300 s. The substrates were rinsed with ethanol
and then
'transferred to a 0.1 mM solution of 3 (11-(3,3,4,4,5,5,6,6,7,7,8,8,8-
tridecafluoro-
octyloxy)undecane-1-thiol) in ethanol and incubated at room temperature for a
period
; ranging from 1 to 24 hours. Finally, surface-modified substrates were
removed from the
assembly bath, spin washed at 1000 rpm with ethanol and dried under a stream
of nitrogen.
The advancing contact angles of water drops (0.5 L) applied to the surface-
modified
substrates were in the range 114 to 120 . Surface-modified substrates were
stored in fitted
plastic containers with transparent amber UV resistant covers.
Example XIV
Preparation of Patterned Sample Presentation Devices
Twenty-four (24) surface-modified substrates were prepared as described above,
mounted in a custom alignment jig and covered with a pin-registered etched
stainless steel
shadow mask (0.002 inch) having features corresponding in size and shape to
the liquid
retention zone. The jig was placed on the moving belt of an air-cooled
ultraviolet curing
system fitted with a low-pressure mercury light source rated at 120 W/cm2 and
passed
under the light source 45 to 75 times over the course of one hour. Following
UV exposure,
-69-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
the substrates were removed from the jig, spin washed at 1000 rpm with ethanol
and dried
under a stream of nitrogen. The exposed substrates were placed in a 0.1 mM
solution of 6
(11-{2-[2-(2-methoxyethoxy)ethoxy]ethoxy} undecane-l-thiol) in ethanol and
incubated at
room temperature for a period ranging from 1 to 24 hours. Patterned surface-
modified
substrates were removed from the assembly bath, spin washed at 2400 rpm with
ethanol
and dried under a stream of nitrogen. The advancing contact angles of water
drops applied
to the liquid retention zone were in the range 60 to 65 , and when applied to
the boundary
zone were in the range 110 to 119 .
Patterned surface-modified substrates were mounted in a custom alignment jig
and
covered with a second pin-registered etched stainless steel shadow mask having
features
corresponding in size and shape to the analysis zone. The jig was placed on
the moving
belt of the ultraviolet curing system and passed under the light source 45 to
75 times over
the course of one hour. Following UV,exposure, the substrates were removed
from the jig,
spin washed at 1000 rpm with ethanol and dried under a stream of nitrogen. The
exposed
substrates were placed in a 0.1 mM solution of 9 (2-{2-[2-(11-
mercaptoundecyloxy)ethoxy]ethoxy}ethanol) in ethanol and incubated at room
temperature for 1-24 hours. Finally, twice-patterned surface-modified
substrates were
removed from the assembly bath, spin washed at 1000 rpm with ethanol and dried
under a
stream of nitrogen. The advancing contact angles of water drops applied to the
analysis
zone were less than 47 . Twice-patterned surface-modified substrates were
stored in fitted
plastic containers with amber transparent UV resistant covers.
While various embodiments of the present invention have been described above,
it
should be understood that they have been presented by way of example only, and
not
limitation. In particular, the physical arrangement of the analysis zone,
liquid retention
zone, and boundary zone is not limited by the examples described above. Thus,
the breadth
and scope of the present invention should not be limited by any of the above-
described
exemplary embodiments.
Example XV
Sample Containment and Positioning
Analyte-confining properties of the analysis zone, which afford an increase in
sensitivity of detection, are demonstrated in the video contact angle images
shown in
FIGS. lla through llh. With reference to FIG. lla, the sample presentation
device of the
-70-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
present invention was prepared with a liquid retention zone measuring about
1.6 mm OD
and an analysis zone measuring about 0.7 mm OD. To facilitate the observation
of the
focusing effect, the analysis zone was placed off-center. A drop of water was
applied to the
surface of the biochip and was observed to rapidly confine itself to the
surface area
corresponding to the liquid retention zone and the analysis zone. The initial
left-side and
right-side contact angles were recorded and were both found to be 57.1 , a
value which
corresponds to that exhibited by a surface prepared from exclusively the
liquid retention
zone monomer. As the drop dried owing to evaporation (see FIGS. 11b through
11h), both
the observed radius and contact angles receded until the radius of the drop
corresponded to
that of the analysis zone. Furthermore, as the drop dried it was observed that
the center of
the drop moved to the right so as to allow the drop to center itself over the
analysis zone.
The left-side and right-side contact angles recorded in FIG. 11h were both
found to be
35.4 , a value which corresponds to that exhibited by a surface prepared
exclusively from
the analysis zone monomer. The drop height, width and contact angle data
recorded in
conjunction with the acquisition of the images depicted in FIGS. 11a through
11h is
summarized graphically in FIG. 12.
Example XVI
Liquid-Holding Capacity of Patterned Sample Presentation Devices
The extraordinary liquid-holding capacity of the liquid retention zone is
demonstrated in FIG. 13. A photograph of a 16-site sample presentation device
of the
present invention shows the retention of sample drop volumes in the range 5
.tL to 70 L.
The only factor that appears to significantly limit the sample drop volume is
the relative
.proximity of the adjacent pairs of target and liquid retention zones.
Example XVII
Analyte Directing and Concentration
Analyte-confining properties of the analysis zone are further demonstrated in
FIGS.
14a and 14b. The first photograph (FIG. 14a) is of a 16-site sample
presentation device of
the present invention with sample drop volumes in the range 5 L to 40 L
deposed on the
surface of 8 of the 16 sites. Each of the liquid drops contained an equivalent
amount of
HCCA. FIG. 14b is a photograph of the HCCA having been concentrated and
directed to
the analysis zone due to sample drying on the sample presentation device
depicted in FIG.
-71-
CA 02531972 2006-01-10
WO 2005/016530 PCT/US2003/021786
14a. The relative size of the analysis zone and the liquid retention zone is
superimposed
above the HCCA for comparison purposes.
While various embodiments of the present invention have been described above,
it
should be understood that they have been presented by way of example only, and
are not
limitations. In particular, the physical arrangement of the analysis zone,
liquid retention
zone, and boundary zone is not limited by the examples described above. Thus,
the
breadth and scope of the present invention should not be limited by any of the
above-
described exemplary embodiments.
-72-