Note: Descriptions are shown in the official language in which they were submitted.
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
AMINO ACID PRODRUGS
SCOPE OF THE INVENTION
The invention relates to amino acid derivatives of pharmaceutical compounds,
methods
of treating particular ailments, which are ameliorated by the administration
of these
drugs and pharmaceutical compositions containing these drugs.
The current invention involves improving many physicochemical,
biopharmaceutical,
and clinical efficacy of various drugs using amino acids as covalently bonded
carriers for
these drugs.
The development of chemical compounds for the treatment of disorders, maladies
and
diseases has become increasingly difficult and costly. The probability of
success for such
development is often discouragingly low. Further, the time for development can
approach or exceed ten years, leaving large numbers of patients without remedy
for an
extended period of time.
Even in cases in which effective pharmaceutical compounds have been developed,
there
are often disadvantages associated with their administration. These
disadvantages can
include aesthetic, and pharmacokinetic bafflers affecting the effectiveness of
some
existing pharmaceutical compounds, For example, unpleasant taste or smell of a
pharmaceutical compound or composition can be a significant barrier to patient
compliance with respect to the administration regimen. The undesirable
solubility
characteristics of a pharmaceutical compound can lead to difficulty in
formulation of a
homogeneous composition. Other disadvantages associated with known
pharmaceutical
compounds include: poor absorption of orally administered formulations; poor
bioavailability of the pharmaceutical compounds in oral formulations; lack of
dose
proportionality; low stability of pharmaceutical compounds in vitro and in
vivo; poor
penetration of the blood/brain barrier; excessive first-pass metabolism of
pharmaceutical
compounds as they pass through the liver; excessive enterohepatic
recirculation; low
absorption rates; ineffective compound release at the site of action;
excessive irritation,
1
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
for example, gastro-intestinal irritability and/or ulceration; painful
injection of
parenterally administered pharmaceutical compounds and compositions;
excessively
high dosages required for some pharmaceutical compounds and compositions, and
other
undesirable characteristics. Some pharmaceutical compounds are processed by
the body
to produce toxic by-products with harmful effects.
The art is continually seeking new chemical compounds for the treatment of a
wide
variety of disorders, with improved properties to overcome the disadvantages
of known
pharmaceutical compounds mentioned above.
The present invention has overcome many problems associated with currently
marketed
drugs by making prodrugs. The concept of prodrugs is well known, and there are
a
number of examples of such prodrugs enumerated in the literature and there are
a
number of prodrugs available in the market, including such diverse groups as
statin
drugs, ACE inhibitors, antiviral drugs such as Acyclovir and the like.
The present invention, however, uses amino acids as the moiety to make the
prodrugs.
The prodrugs of the present invention have a number of advantages. For
example, when
amino acid prodrugs are administered by a number of routes such as oral, Iv,
rectal or
other such methods, these prodrugs are converted into active drug molecules. A
significant advantage of the amino acid prodrug is that it is non-toxic, and
hence either
assimilated into the body or safely excreted. This is quite unlike the
characteristics
exhibited by a number of prodrugs available in the market, where the promoiety
itself is
toxic, as is the case with statin drugs, Enalapril, Benazapril and the like
group of ace
inhibitors, and a number of antibiotics such as pivoxil, isopropyl, Axetil,
Cilexetil and
the like groups, which are highly toxic, thereby reducing the therapeutic
index of the
active drug.
On the other hand, the amino acid prodrugs of the present invention also
impart a
number of advantages as shown herein below.
2
11\work\1652\17690\SPEC\17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
SUMMARY OF THE INVENTION
The present invention is directed to a pharmaceutically active prodrug, having
amino
acid covalently bonded to a pharmaceutical compound to form said acid prodrug,
which
is administered in this form to the subject, such as a mammal.
The amino acid is an ideal model to be used as a prodrug, because it is
capable of
forming various types of linkages between itself and the drug. By definition,
an amino
acid has at least two functionalities thereon, an amino group (NH2) and a
carboxy group
(COOH). For example, the a- amino acids have the well known structure
H2N¨T_co0H
R0
wherever R0 is the side group or chain of the amino acid. The H 2N ¨C¨ COOH
as defined herein, is the main chain of the amino acid. Thus, for example,
beside the
amino group and the carboxyl group on the main chain, the side chain may have
functional groups thereon. It is the functional group on the amino acid moiety
that
permits the covalent linkage to occur between the amino acid and the drug.
The drug or medicament useful in the present invention contains functional
groups
thereon that permit the drug to react with and form a covalent bond with the
amino acid.
Examples of functional groups present on the drugs include NH2, OH, COOH or
acid
derivatives thereof, such as esters, amides and the like.
The mode of attachment between the pharmaceutical compound and the amino acid
can
be via:
1) An ester bond (-00-0-) arising from condensation of a carboxylic acid and
an
alcohol or phenolic hydroxyl group, or through transesterification, for
example:
3
H:\work\ 1652\17690 \SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
a) Where the pharmaceutical compound has an aliphatic or aromatic
hydroxyl group an ester bond can be formed with the backbone carboxylic acid
group of
the amino group under esterification conditions; or
b) Where the pharmaceutical compound has an aliphatic or aromatic
hydroxyl group and the amino acid has a side chain carboxylic acid group, an
ester bond
can be formed therebetween under esterification conditions; or
c) Where the pharmaceutical compound has a carboxylic acid group
and the amino acid has a side chain aliphatic or aromatic hydroxyl group, an
ester bond
can be formed therebetween under esterification condition; or
d) Where the pharmaceutical compound has an ester group with a
substituted or unsubstituted acyloxy (e.g., alkoxy- or arylalkoxy-, aryloxy
carbonyl)
substituent (compound-O-CO-substituent) and the amino acid has a backbone
carboxylic
acid group, an ester bond can be formed therebetween through
transesterification; or
e) Where the pharmaceutical compound has an ester group
with a
substituted or unsubstituted acyloxy (e.g., alkoxy- or arylalkoxy-, aryloxy
carbonyl)
substituent (compound-O-CO-substituent) and the amino acid has a side chain
carboxylic acid group, an ester bond can be formed therebetween through
transesterification; or
Where the pharmaceutical compound has an ester group with a
substituted or unsubstituted alkoxy- or arylalkoxy- or aryloxy carbonyl
substituent
(compound-00-0-Substituent) and the amino acid has a side chain aliphatic or
aromatic
hydroxyl group, an ester bond can form therebetween though
transesterification; or
The alcohol and carboxylic acid moieties may be on the same
molecule such they can form an internal ester. For example, certain compounds
like
Gabapentin can form an internal ester under ester forming conditions, is also
included
with the scope of the present invention.
2) An amide bond (-CO-NH -) arising from condensation of a carboxylic acid
and an amine, for example:
4
FI:\work\1652\17690 \SPEC\ 17690.spec without claimsli.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
a) Where the pharmaceutical compound has an amino group and the
amino acid has a backbone carboxylic acid group, an amide can be formed under
amide
forming conditions; or
b) Where the pharmaceutical compound has an amino group and the
amino acid has a side chain carboxylic acid group, an amide bond can form
therebetween under amide forming conditions; or
c) Where the pharmaceutical compound has a carboxylic acid group
and the amino acid has a backbone amino group, an amide bond can form
therebetween
under amide forming conditions; or
d) Where the
pharmaceutical compound has a carboxylic acid group
and the amino acid has a side chain amino group, an amide bond can be formed
therebetween under amide forming conditions.
Thus, the present invention is directed to the prodrugs thus formed. As shown
hereinbelow the prodrug thus formed has advantages not realized relative to
the drug
without the amino acid attached thereto. For example, it can improve
bioavailability,
efficacy, be less toxic, exhibit greater solubility in water and/or improve
the ability of
the drug to pass into the cell membrane or through blood brain barrier,
exhibit less side
effects, such as gastro-intestinal irritability, enhanced therapeutic index
and the like.
Thus, the present invention is directed to a method of improving the
therapeutic quality
of a drug wherein the improvement in the therapeutic quality is selected from
the group
consisting of improved efficacy, enhanced therapeutic index, increased
solubility in the
mammal's internal fluid, improved passage through the cell membrane, improved
passage through the blood brain barrier, decreased side effects, such as
significantly
decreased irritation and/or ulcerations, less toxicity, enhanced absorption
ratio and the
like relative to the corresponding drug administered to the patient in the non-
prodrug
form, said method comprising reacting the drug with an amino acid to form a
covalent
bond therebetween and administering the product thereof (hereinafter
"prodrug") to a
patient. The prodrugs of the present invention have at least one improved
quality. In
5
FlAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
fact, they exhibit preferably at least two of the improved qualities cited
hereinabove.
Other advantages of the prodrug include the wide availability of the amino
acids and the
ease in which the reactions take place. The reaction to form the amide is
generally
efficient and yield are very high, presumably above about 70% and more
preferably
above about 80% and most preferably above about 90%.
BRIEF DESCRIPTION OF THE DRAWINGS
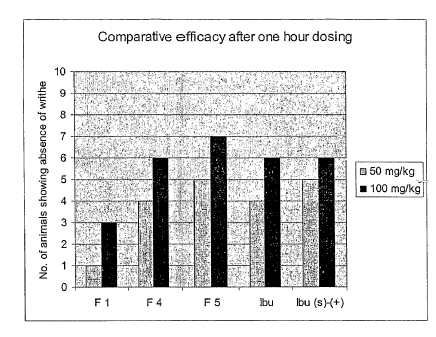
Figure 1 graphically compares the efficacy of L- serine ester of (+) Ibuprofen
(F1), L-
threonine ester ft-) Ibuprofen (F2) and L-hydroxyproline ester of ( )
Ibuprofen (F3), (+)
Ibuprofen (i.e., the racemic mixture) and Ibuprofen (S)(+), after one hour
dosing, based
on the antagonizing property of Acetylcholine induced writhe in Albino mice.
Figure 2 graphically compares the efficacy of L- serine ester of ( )
Ibuprofen, (F1), L-
threonine ester of, (I) Ibuprofen (F2), L-hydroxyproline ester of ( )
Ibuprofen (F3), +
Ibuprofen and S(+) Ibuprofen after 3 hour dosing, based on the antagonizing
property of
Acetylcholine induced writhes in albino mice.
Figure 3 depicts graphically the dose response relationship to mean clotting
time (MCT)
in minutes for the L-serine ester of acetylsalicylic acid (Formulation 1).
Figure 4 depicts graphically the dose response relationship to mean clotting
time (MCT)
minutes for the L-hydroxyproline ester of acetylsalicylic acid (Formulation
2).
Figure 5 depicts the dose response relationship to mean clotting time (MCT) in
minutes
for the L-threonine ester of acetylsalicylic acid (Formulation 3)
Figure 6 depicts the dose response relationship to mean clotting time (MCT) in
minutes
for control (acetylsalicylic acid).
6
H:\work\1652 \17690 \SPEC\ 17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Figure 7 graphically compares the relative efficacy of L-serine (ester of
acetyl salicyclic
acid (F.1), L-threonine ester of acetyl salicylic acid (F.2), L-hydroxyproline
ester of
acetylsalicylic acid (F.3), and acetylsalicylic acid (PC) as a function of
mean clotting
time in minutes.
DETAILED DESCRIPTION OF THE PRESENT INVENTION
As used here, the term "drug", "medicament ", and "pharmaceutical" are being
used
interchangeably and refer to the active compound that is administered to the
patient
without attachment of the amino acid thereto. Moreover, as used herein, the
drug
contains a functional group thereon capable of reacting with the amino acid,
such as
NH2, OH, COOH or acylating derivatives thereof (e.g., ester, anhydride, amide,
and the
like) and the like. When the drug is linked to an amino acid, the term "amino
acid
prodrug" or "prodrug of the present invention" or synonym thereto is utilized.
Among the amino acids useful as promoieties (i.e., reacting with the drugs)
are those
containing the free amino and/or carboxylic acid groups of all conventional
amino acids.
Of those, some preferred embodiments include those amino acids having
relatively high
solubility in aqueous media, for example, in deionized water at unbuffered
aqueous
solution at 25 C, of at least 100 g/L, and more preferably, at least 250 g/L,
and even
more preferably at least 500 g/L. For example, glycine and proline have
solubilities in_
deionized water at 25 C of approximately 250 g/L and 1620 g/L, respectively.
Other amino acids useful as promoieties are those containing basic amino side
chains,
such as lysine. For example, lysine has solubility in deionized water at 25 C
of
approximately 700 g/L.
Among other amino acids useful as promoieties are those containing hydroxyl
side
chains, such as hydroxyproline, serine, and threonine. For example, threonine,
hydroxyproline and serine have solubilities in deionized water at 25 C of
approximately
100 g/L, 369 g/L and 420 g/L, respectively.
7
H:\work\1652\17690\SPEC\17690.spec without claimsil.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Other preferred embodiments include those amino acids with relatively low
solubility in
aqueous media, for example, in deionized water at 25 C of at most 10 g/L, or
for
example, at most 2 g/L, or for example at most 0.6 g/L. For example, the
solubility of
tyrosine in deionized water at 25 C is approximately 0.5 g/L. Such prodrugs
could be
used to produce formulations with extended release characteristics, due to the
limited
solubility of the prodrugs.
Among other amino acids useful as promoieties are those containing carboxylic
acid side
chains, such as glutamic acid and aspartic acid. Other amino acids useful as
promoieties
are the non-essential amino acids, and the non-naturally occurring amino
acids.
The following reaction schemes depict the reactions discussed hereinabove with
respect
to reaction of hydroxyl, carboxyl and amine containing drugs with various
amino acids.
In the schemes below, R is the drug less the functional OH, COOH or NH2 group
¨ C ¨ R0
whichever is present, and R1 is
wherein Ro is the side chain of the amino acid listed hereinbelow:
Reaction Scheme A: Where the hydroxyl group of the drug is reacted with the
carboxyl
group of an amino acid to from the ester prodrug
R-OH + HOOC-R1- NH2 9 R-0-(C=0)-Ri- NH2
Drug Amino Acid Amino acid Ester Prodrug
Reaction Scheme B: Where the carboxyl group of the drug is reacted with the
hydroxyl
group of a hydroxy amino acid wherein the hydroxy group is on the side chain
to form
the ester prodrug.
8
H:\work\1652\17690\SPEC\17690.spec without clairnsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
R-COOH + HO-R1-N112 4 R-(C=0)-0-R1-N/12
CLH COOH
Drug Hydroxy Amino Acid Amino Acid Ester Prodrug
Reaction Scheme C: Where the amine group of the drug is reacted with the
carboxyl
group of the amino acid to from the amide prodrug
R- NH2 + HOOC-Ri- NH2 --> R-NH(C=0)-Ri- NH2
Drug Amino Acid Amino Acid Amide Prodrug
Reaction Scheme D: Where the carboxyl group of the drug is reacted with the
carboxyl
group of the amino acid to form the anhydride prodrug.
R-COOH + HOOC-R1- NH2 4 H-(C=0)-0-(C=0)-Hi- NH2
Drug Amino Acid Amino Acid Anhydride Prodrug
Reaction Scheme E: Where the amine group of the drug is reacted with the amine
group
of the amino acid to form the azo prodrug derivative.
R- NH2 + NH2-121- COOH R-N=N-R1-COOH
Drug Amino Acid Amino Acid Azo Prodrug
Reaction Scheme F: Where the carboxyl group of the drug is reacted with amine
group
of the amino acid to form the amide prodrug.
R-COOH + NH2-R1- COOH 3 R-(C=0)NH-Ri- COOH
Drug Amino Acid Amino Acid Amide Prodrug
In the above schemes A-F, the preferred amino acids used are shown
hereinbelow:
9
H:\work\1652\17690\SPEC\17690.spec without clairnsadoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
_o 9 . P . 9
. pi . lp . 9 0-4T Nz.,'-' o*--i ti4.--;+
o--r.='-------- .t14-.;+ o'------ 4 ,..,..+ o--j-....---õ Liz+
glycine (gly) sarcosine (sar) GAM alanine (ala) vallne
(val) isoleucine (ile) leucine (leu)
. 9 . 9
9
9 - ...i tli + ..... + o--3 NZ+
o-) 4;
o----' . '
, 1 +
HO
. :
N'T: HO'"--------- " ---. 0
HO \ 0 --:.õ 0
HO
serine (ser) homoserine threonine (thr) carnitine choline
aspartate (asp) glutamate (glu)
9 o
0---' 14.7 01-i kl,'
o 9
o---J kl+i
97- 1 i +
/ +
O.-- N
0 I
N
o
"--)4 --1( !. \ - N.
---k
I + / I + 1
...N 4' _ N N
omithine i µ lysine (lys) I = calanine / \ canavanine arginine
(arg)
9 . P . 9 9
'+
+
o"-:-"=--N :,- (3.-J 4 .;
= , - N
ti I' - N N =-
=-= N_.::- 0
1,1
N...1.,1
!I 0
praline (pro) asparagine (asn) glutamine (gin) histidine (hls)
carnosine
0 0
-
9
+ o
j
. . = 1 +
: 1 + 1 + o-- N.: ry-' N.:- -. ;
0--1
o---' N.: 0-- NZ ...
00'
N '.. ::-.N N OH
N =-
/4W HO 4 P
phenylalanine (phe) tyrosine (tyr) tryptophan (trp) kyurenine
kyurenio acid hypaphorine
. 9 9 o
. - P o
. 9 ..1 .l. + o---; 4; : , +=:
---J --- 4.,.. ,-.-' r'z
,
:Tx 4
:f
s' + -
N'
o 4; o
-i
HS / /
I
HS ---------Nc S S 0 = S
4
HO %
HS s cystine s 2 djenkolic acid /S
cysteine (cys) homowsteine cysteamIne :!: N , .. 0
methionine (met) taurine
+l
O +'
ci
As used herein the term "amino acid" refers to an organic compound having
therein a
carboxyl group (COOH) and an amino group (NH2) or salts thereof. In solution,
these
two terminal groups ionize to form a double ionized, through overall neutral
entity
identified as zwitterions. The amine donates an electron to the carboxyl group
and the
ionic ends are stabilized in aqueous solution by polar water molecules.
1-1:\work\1652\17690\SPEC\17690.spec without claimsIl.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
It is the side groups that distinguish the amino acids from each other. Some
amino acids,
such as lysine, have amino groups on the side chain; other amino acids have
side chains
containing hydroxy groups, such as threonine, serine, hydroxyproline, and
tyrosine;
some amino acids have carboxy groups on the side chain, such as glutamic acid
or
aspartic acid. These functional groups on the side chain also can form a
covalent bond
with the drug, such as esters, amides, and the like. When these side groups
become
involved in these linkages, such as hydroxy group, the bond may be depicted as
OAA,
wherein AA is an amino acid residue having a side chain with a hydroxy group,
but
without the hydroxy group. Thus, AA by this definition, refers to the amino
acid
without the hydroxy side group since it took part in the reaction in forming
the ester.
Moreover, when an ester is formed between the hydroxy group of the amino acid
and the
OH group of the drug, the hydroxy group on the carboxy group forms a byproduct
with
the hydrogen of the hydroxy group, thus, the resulting product does not have
the OH
group on the carboxy group, but just the acyl moiety. When the bond is
depicted as
C(=0)-NHAA, this means that the amino acid forms as an amide bond between the
carboxy group on the drug and the amino group of the amino acid. However, as
written,
since the NH from the amide bond comes from the amino acid, AA is the amino
acid
without the amino group.
The preferred amino acids are the naturally occurring amino acids. It is more
preferred
that the amino acids are the cc-amino acids. It is also preferred that the
amino acids are
in the L-configuration. The preferred amino acids include the twenty essential
amino
acids. The preferred amino acids are Lysine (Lys), Leucine (Leu), Isoleucine
(Ile),
Glycine (Gly), Aspartic Acid (Asp), Glutamic Acid (Glu), Methionine (Met),
Alanine
(Ala), Valine (Val), Proline (Pro), Histidine (His), Tyrosine (Tyr), Serine
(Ser),
Norleucine (Nor), Arginine (Arg), Phenylalanine (Phe), Tryptophan (Trp),
Hydroxyproline (Hyp), Homoserine (Hsr), Carnitine (Car), Ornithine (Ort),
Canavanine
(Cav), Asparagine (Asn), Glutamine (Gln), Carnosine (Can), Taurine (Tau),
djenkolic
= Acid (Djk), 7-aminobutyric Acid (GABA), Cysteine (Cys) Cystine (Dcy),
Sarcosine
, 11
H: \work\1652 \17690 \SPEC \17690.spec without clahnsILdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
(Sar), Threonine (Thr) and the like. The even more preferred amino acids are
the twenty
essential amino acids, Lys, Leu, Ile, Gly, Asp, Glu, Met, Ala, Val, Pro, His,
Tyr, Thr,
Arg, Phe, Trp, Gln, Asn, Cys and Ser.
The prodrugs are prepared from a drug having a group thereon which can react
with the
amino acid.
The preferred drugs that are reacted with amino acids in accordance with
various
schemes are as follows:
Reaction Schemes
Drugs A BCDE F
Cyclosporins YES
Lopinavir YES YES YES
Ritonavir YES YES YES
Cefdinir YES YES YES YES YES
Zileuton YES YES YES
Nelfinavir YES YES YES
Flavoxate YES YES YES
Candesarten YES YES YES YES YES
Propofol YES
Nisoldipine YES YES YES YES YES
Amlodipine YES YES YES YES YES
Ciprofloxacin YES YES YES YES
Ofloxacin YES YES YES YES
Fosinopril YES YES YES
Enalapril YES YES YES
Ramipril YES YES YES
Benazepril YES YES YES
Moexipril YES YES YES
Trandolapril YES YES YES
12
H:\work\1652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Cromolyn YES YES YES YES
Amoxicillin YES YES YES YES YES YES
Cefuroxime YES YES YES YES YES YES
Ceftazimide YES YES YES YES YES YES
Cefpodoxime YES YES YES YES YES YES
Atovaquone YES
Gancyclovir YES YES YES
Pencielovir YES YES YES
Fameiclovir YES YES YES
Acyclovir YES YES YES
Niacin YES YES YES
Bexarotene YES YES YES
Propoxyphene YES
Salsalate YES YES YES YES
Acetaminophen YES
Ibuprofen YES YES YES
Lovastatin YES YES YES YES
Simavastatin YES YES YES YES
Atorvastatin YES YES YES YES
Pravastatin YES YES YES YES
Fluvastatin YES YES YES YES
Nadolol YES
Valsartan YES YES YES
Methylphenidate YES YES YES YES
Sulfa Drugs YES YES
Sulfasalazine YES
Methylprednisolone YES
Medroxyprogesterone YES
Estramustine YES
Miglitol YES
13
H:\work\1652\17690\SPEC\17690.spec without clairnsIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Mefloquine YES YES
Capacitabine YES
Danazol YES
Eprosartan YES YES YES
Divalproex YES YES YES
Fenofibrate YES YES YES
Gabapentin* YES YES YES YES YES
Omeprazole YES
Lansoprazole YES
Megestrol YES
Metformin YES
Tazorotene YES YES YES
Sumitriptan YES
Naratriptan YES
Zolmitriptan YES
Aspirin YES YES YES
Olmesartan YES YES YES
Sirolimus YES
Tacrolimus YES
Clopidogyel YES YES YES
Amphotericin B YES YES YES YES
Tenofovir YES
Unoprostone YES YES YES
Fulvestrant YES
Cefditoren YES YES YES
Efavirenz YES
Eplerenone YES YES YES
Treprostinil YES YES YES YES
Adefovir YES
14
li:\work\1652\17690\SPEC\17690.spec without claims11.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The prodrug of the present invention contains amino groups and as such are
basic in
nature. They are capable of forming a wide variety of pharmaceutically
acceptable salts
with various inorganic and organic acids. These acids that may be used to
prepare
pharmaceutically acceptable acid addition salts of such basic compounds are
those that
form non-toxic acid addition salts, i.e., salts containing pharmaceutically
acceptable
anions, such as the hydrochloride, hydrobromide, hydroiodide, nitride,
sulfate, bisulfate,
phosphate, formate, acetate, citrate, tartate, lactate, and the like.
As indicated herein, in one embodiment, the present invention is directed to a
prodrug
wherein the prodrug comprises a drug, e.g., cyclosporine and an amino acid
esterified to
the MeBmt (x-y=CH=CH) or dihydro MeBmt moiety, (x-y=CH2CH2). The amino acid
is attached to the cyclosporine and to the other other drugs by a covalent
bond.
The compounds of the present invention are prepared by art recognized
techniques. For
examples, if the drug contains an OH group, said as cyclosporin, then an amino
acid or
an acylating derivatives thereof, such as the acid halide, e.g., amino acid
fluoride, amino
acid chloride, or an amino acid alkyl ester wherein alkyl group contains 1-6
carbon
atoms is reacted with the carboxy group of the drug, e.g., cyclosporine under
esterification condition. Preferably, the reaction is conducted in the
presence of an acid,
such as hydrochloric acid, hydrobromic acid, p-toluenesulfonic acid and the
like.
Alternatively, as described hereinabove, if the drug has an amino group
thereon, then the
amino acid may be reacted with the drug under amide forming conditions to form
an
amide as the covalent bond. Or if the drug has a carboxy group or acylating
derivative
thereon, it may be reacted with the amino group of the amino acid to form an
amide
under amide forming conditions to form an amide bond between the amino acid
and the
drug. Additionally if the drug has a carboxy group therein, the hydroxy group
of the
side chain of the amino acid may be reacted with the carboxy group or
acylating
derivative, therein under esteiification conditions to form the ester linkage
between the
amino acid and the drug, as described hereinabove.
15
1-1:\work\1652\17690\SPEC \17690.spec without clainisiLdoc
CA 02534342 2011-12-06
If the amino acid has a group thereon which is reactive under the reaction
conditions it is
protected by a protecting group known in the art. After the completion of the
reaction,
the protecting group is removed. Examples of protecting groups that could be
used are
described in the book entitled, "Protective Group in Organic Synthesis" by
Theodora W.
Greene, John Wiley & Sons, 19g1.
For example, if amino acids with carboxylic groups in their side chains, for
example,
aspartic acid and glutamic acid, are used in the aforementioned synthesis,
these will
generally require protection of the side chain carboxylic acid. Suitable
protecting groups
can be esters, such as cyclohexyl esters, t-butyl esters, benzyl esters, allyl
esters, 9-
fluorophenyl-methyl groups or adamantyl groups, such as 1-or 2-adamantyl which
can
be protected after the esterfication reaction is completed using techniques
known to one
of ordinary skill in the art.
If amino acids with hydroxyl groups in their side chains, for example, serine,
threonine,
hydroxyproline, and the like and amino acids with phenolic groups in their
side chains,
for example, tyrosine, and the like are used in the aforementioned
esterification,
reaction, they will desirably require protection of the chain hydroxyl or
phenolic group.
Suitable for protecting groups for the hydroxyl side chain groups can be
ethers, such as
benzyl ether or t-butyl ether. Removal of the benzyl ether can be effected by
liquid
hydrogen fluoride, while the t-butyl ether can be removed by treatment with
trifluoroacetic acid. Suitable protecting groups for phenolic side chain
groups can be
ethers, as above, including benzyl or t-butyl ether or 2,6-dichlorobenzyl, 2-
bromobenzyloxycarbonyl, 2,4-dintrophenyl and the like.
Moreover, the products can be purified to be made substantially pure by
techniques
known to one of ordinary skill in the art, such as by chromatography, e.g.,
HPLC,
crystallization and the like. By substantially "pure" it is meant that the
product contains
no more than about 10% impurity therein.
16
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The prodrugs can be made into pharmaceutical compositions including prodrugs
of, or
pharmaceutical acceptable salts, pharmaceutical acceptable solvates, esters,
enantiomers,
diastereomers, N-Oxides, polymorphs, thereof, as described herein, along with
a
pharmaceutical acceptable carrier, and optionally but desirably
pharmaceutically
acceptable excipients using techniques known to one of ordinary skill in the
art.
The prodrugs utilized in the present method are used in therapeutically
effective
amounts.
The physician will determine the dosage of the prodrugs of the present
invention which
will be most suitable and it will vary with the form of administration and the
particular
compound chosen, and furthermore, it will vary depending upon various factors,
including but not limited to the patient under treatment and the age of the
patient, the
severity of the condition being treated and the like and the identify of the
prodrug
administered. He will generally wish to initiate treatment with small dosages
substantially less than the optimum dose of the compound and increase the
dosage by
small increments until the optimum effect under the circumstances is reached.
It will
generally be found that when the composition is administered orally, larger
quantities of
the active agent will be required to produce the same effect as a smaller
quantity given
parenterally. The compounds are useful in the same manner as the corresponding
drug
in the non-prolong form and the dosage level is of the same order of magnitude
as is
generally employed with these other therapeutic agents. When given
parenterally, the
compounds are administered generally in dosages of, for example, about 0.001
to about
10,000 mg/kg/day, also depending upon the host and the severity of the
condition being
treated and the compound utilized.
In a preferred embodiment, the compounds utilized are orally administered in
amounts
ranging from about 0.01 mg to about 1000 mg per kilogram of body weight per
day,
depending upon the particular mammalian host or the disease to be treated,
more
preferably from about 0.1 to about 500 mg/kg body weight per day. This dosage
17
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
regimen may be adjusted by the physician to provide the optimum therapeutic
response.
For example, several divided doses may be administered daily or the dose may
be
proportionally reduced as indicated by the exigencies of the therapeutic
situation.
The prodrug may be administered in any convenient manner, such as by oral,
intravenous, intramuscular or subcutaneous routes.
The prodrug may be orally administered, for example, with an inert diluent or
with an
assimilable edible carrier, or it may be enclosed in hard or soft shell
gelatin capsules, or
it may be compressed into tablets, or it may be incorporated directly into the
food of the
diet. For oral therapeutic administration, the prodrug may be incorporated
with
excipients and used in the form of ingestible tablets, buccal tablets,
troches, capsules,
elixirs, suspensions, syrups, wafers, and the like. Such compositions and
preparations
should contain at least 1% of the prodrug. The percentage of the compositions
and
preparations may, of course, be varied and may conveniently be between about 5
to
about 80% of the weight of the unit. The amount of the prodrug used in such
therapeutic
compositions is such that a suitable dosage will be obtained. Preferred
compositions or
preparations according to the present invention contain between about 200 mg
and about
4000 mg of prodrug. The tablets, troches, pills, capsules and the like may
also contain
the following: A binder such as gum tragacanth, acacia, corn starch or
gelatin; excipients
such as dicalcium phosphate; a disintegrating agent such as corn starch,
potato starch,
alginic acid and the like; a lubricant such as magnesium stearate; and a
sweetening agent
such as sucrose, lactose or saccharin may be added or a flavoring agent such
as
peppermint, oil of wintergreen, or cherry flavoring. When the dosage unit form
is a
capsule, it may contain, in addition to materials of the above type, a liquid
carrier.
Various other materials may be present as coatings or otherwise modify the
physical
form of the dosage unit. For instance, tablets, pills, or capsules may be
coated with
shellac, sugar or both. A syrup or elixir may contain the active compound,
sucrose as a
sweetening agent, methyl and propylparabens as preservatives, a dye and
flavoring such
18
H:\work\1652\17690\SPEC\17690.spec without claimsli.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
as cherry or orange flavor. Of course, any material used in preparing any
dosage unit
form should be pharmaceutically pure and substantially non-toxic in the
amounts
employed. In addition, the active compound may be incorporated into sustained-
release
preparations and formulations. For example, sustained release dosage forms are
contemplated wherein the active ingredient is bound to an ion exchange resin
which,
optionally, can be coated with a diffusion barrier coating to modify the
release properties
of the resin or wherein the prodrug of the present invention is associated
with a sustained
release polymer known in the art, such as hydroxypropylmethylcellulose and the
like.
The prodrug may also be administered parenterally or intraperitoneally. It is
especially
advantageous to formulate parenteral compositions in dosage unit form for ease
of
administration and uniformity of dosage. Dispersions can also be prepared in
glycerol,
liquid polyethylene glycols, e.g., PEG 100, PEG 200, PEG 300, PEG 400, and the
like,
and mixtures thereof and in oils. Under ordinary conditions of storage and
use, these
preparations contain a preservative to prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous
solutions
(where water soluble) or dispersions and sterile powders for the
extemporaneous
preparation of sterile injectable solutions or dispersions. In all cases the
form is usually
sterile and must be fluid to the extent that syringability exists. It must be
stable under
the conditions of manufacture and storage and usually must be preserved
against the
contaminating action of microorganisms such as bacteria and fungi. The carrier
can be a
solvent or dispersion medium containing, for example, water, ethanol, polyol
(for
example, glycerol, propylene glycol, and one or more liquid polyethylene
glycol, e.g. as
disclosed herein and the like), suitable mixtures thereof, and vegetable oils.
The proper
fluidity can be maintained, for example, by the use of a coating such as
lecithin, by the
maintenance of the required particle size in the case of dispersions and by
the use of
surfactants. The prevention of the action of microorganisms can be brought
about by
various antibacterial and antifungal agents, for example, parabens,
chlorobutanol,
phenol, sorbic acid, thimerosal, and the like. In many cases, it will be
preferable to
19
FlAwork\1652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
include isotonic agents, for example, sugars or sodium chloride. Prolonged
absorption
of the injectable compositions can be brought about by the use in the
compositions of
agents delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating the prodrug in the
required
amount in the appropriate solvent with various of the other ingredients
enumerated
above, as required, followed by filtered sterilization. Generally, dispersions
are prepared
by incorporating the various sterilized active ingredient into a sterile
vehicle which
contains the basic dispersion medium and the required other ingredients from
those
enumerated above. In the case of sterile powders, the above solutions are
vacuum dried
or freeze-dried, as necessary.
The prodrug can also be applied topically, as e.g., through a patch using
techniques
known to one of ordinary skill in the art. The prodrug can be administered
buccally by
preparing a suitable formulation of the prodrug of the present invention and
utilizing
procedures well known to those skilled in the art. These formulations are
prepared with
suitable non-toxic pharmaceutically acceptable ingredients. These ingredients
are
known to those skilled in the preparation of buccal dosage forms. Some of
these
ingredients can be found in Remington's Pharmaceutical Sciences, 17th edition,
1985, a
standard reference in the field. The choice of suitable carriers is highly
dependent upon
the exact nature of the buccal dosage form desired, e.g., tablets, lozenges,
gels, patches
and the like. All of these buccal dosage forms are contemplated to be within
the scope
of the present invention and they are formulated in a conventional manner.
The formulation of the pharmaceutical compositions may be prepared using
conventional methods using one or more physiologically and/or pharmaceutically
acceptable carriers or excipients. Thus, the compounds and their
pharmaceutically
acceptable salts and solvates may be formulated for administration by
inhalation or
insuffiation (either through the mouth or the nose) or oral, buccal,
parenteral, or rectal
administration. For oral administration, the pharmaceutical compositions may
take the
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
form of, for example, tablets or capsules prepared by conventional means with
pharmaceutically acceptable excipients such as binding agents (for example,
pregelatinized maize starch, polyvinylpyrrolidone, or hydroxypropylmethyl
cellulose);
fillers (for example, lactose, microcrystalline cellulose or calcium hydrogen
phosphate);
lubricants (for example, magnesium stearate, talc, or silica); disintegrants
(for example,
potato starch, or sodium starch glycolate); or wetting agents (for example,
sodium lauryl
sulfate). The tablets may be coated by methods well known in the art.
Liquid preparations for oral administration may take the form of, for example,
solutions,
syrups, or suspensions, or they may be presented as a dry product for
constitution with
water or other suitable vehicles before use. Such liquid preparations may be
prepared by
conventional means with pharmaceutically acceptable additives, such as
suspending
agents (for example, sorbitol syrup, corn syrup, cellulose derivatives or
hydrogenated
edible oils and fats); emulsifying agents (for example, lecithin or acacia);
non-aqueous
vehicles (for example, almond oil, oily esters, ethyl alcohol or fractionated
vegetable
oils); and preservatives (for example, methyl or propyl p-hydroxybenzoates or
sorbic
acid). The preparations may also contain buffer salts, flavoring, coloring and
sweetening
agents as appropriate. Preparations for oral administration may be suitably
formulated to
give controlled release of the active prodrug.
The prodrug of the present invention may be formulated for parenteral
administration by
injection, for example, by bolus injection or continuous infusion.
Formulations for
injection may be presented in unit dosage form, for example, in ampoules, or
in multi-
dose containers, with an added preservative. The compositions may take such
forms as
suspension, solutions or emulsions in oily or aqueous vehicles, and may
contain
formulatory agents such as suspending, stabilizing and/or dispersing agents.
Alternatively, the prodrug may be in the powder form for constitution with a
suitable
vehicle, for example, sterile pyrogen-free water, before use.
21
H:\work\1652\17690\SPEC\17690.spec without claimadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The prodrugs of the present invention may also be formulated in rectal
compositions
such as suppositories or retention enemas, for example, containing
conventional
suppository bases such as cocoa butter or other glycerides.
In addition to the formulations described previously, the prodrug of the
present invention
may also be formulated as a depot preparation. Such long acting formulations
may be
administered by implantation (for example, subcutaneously or intramuscularly)
or by
intramuscular injection. Thus, for example, the prodrugs may be formulated
with
suitable polymeric or hydrophobic materials (for example, as an emulsion in an
acceptable oil) or ion exchange resins, or as sparingly soluble derivatives,
for example,
as a sparingly soluble salt.
The pharmaceutical compositions containing the prodrugs of the present
invention may,
if desired, be presented in a pack or dispenser device which may contain one
or more
unit dosage forms containing the active ingredients. The pack may for example
comprise metal or plastic foil, such as blister pack. The pack or dispenser
device may be
accompanied by instructions for administration.
In tablet form, it is desirable to include a lubricant which facilitates the
process of
manufacturing the dosage units; lubricants may also optimize erosion rate and
drug flux.
If a lubricant is present, it will be present on the order of 0.01 wt. % to
about 2 wt. %,
preferably about 0.01 wt. % to 0.5 wt, %, of the dosage unit. Suitable
lubricants include,
but are not limited to, magnesium stearate, calcium stearate, stearic acid,
sodium
stearylfumarate, talc, hydrogenated vegetable oils and polyethylene glycol. As
will be
appreciated by those skilled in the art, however, modulating the particle size
of the
components in the dosage unit and/or the density of the unit can provide a
similar effect-
-i.e., improved manufacturability and optimization of erosion rate and drug
flux--without
addition of a lubricant.
22
H:\work\1652 \17690 \SPEC \I7690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Other components may also optionally be incorporated into the dosage unit.
Such
additional optional components include, for example, one or more
disintegrants,
diluents, binders, enhancers, or the like. Examples of disintegrants that may
be used
include, but are not limited to, crosslinked polyvinylpyrrolidones, such as
crospovidone
(e.g., Polyplasdone XL, which may be obtained from GAF), cross-linked
carboxylic
methylcelluloses, such as croscanrnelose (e.g., Ac-di-sol , which may be
obtained from
FMC), alginic acid, and sodium carboxymethyl starches (e.g., Explotab , which
may be
obtained from Edward Medell Co., Inc.), agar bentonite and alginic acid.
Suitable
diluents are those which are generally useful in pharmaceutical formulations
prepared
using compression techniques, e.g., dicalcium phosphate dihydrate (e.g., Di-
Tab ,
which may be obtained from Stauffer), sugars that have been processed by
crystallization with dextrin (e.g., co-crystallized sucrose and dextrin such
as Di-Pak ,
which may be obtained from Amstar), calcium phosphate, cellulose, kaolin,
mannitol,
sodium chloride, dry starch, powdered sugar and the like. Binders, if used,
are those that
enhance adhesion. Examples of such binders include, but are not limited to,
starch,
gelatin and sugars such as sucrose, dextrose, molasses, and lactose.
Permeation
enhancers may also be present in the novel dosage units in order to increase
the rate at
which the active agents pass through the buccal mucosa. Examples of permeation
enhancers include, but are not limited to, dimethylsulfoxide ("DMSO"),
dimethyl
formamide ("DMF"), N,N-dimethylacetamide ("DMA"), decylmethylsulfoxide
("CioMSO"), polyethylene glycol monolaurate ("PEGML"), glycerol monolaurate,
lecithin, the 1-substituted azacycloheptan-2-ones, particularly 1-n-
dodecylcyclazacycloheptan-2-one (available under the trademark Azone® from
Nelson Research & Development Co., Irvine, Calif.), lower alkanols (e.g.,
ethanol),
SEPA (available from Macrochem Co., Lexington, Mass.), cholic acid,
taurocholic
acid, bile salt type enhancers, and surfactants such as Tergitol , Nonoxyno1-9
and
TWEEN-800.
Flavorings may be optionally included in the various pharmaceutical
formulations. Any
suitable flavoring may be used, e.g., mannitol, lactose or artificial
sweeteners such as
23
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
aspartame. Coloring agents may be added, although again, such agents are not
required.
Examples of coloring agents include any of the water soluble FD&C dyes,
mixtures of
the same, or their corresponding lakes.
In addition, if desired, the present dosage units may be formulated with one
or more
preservatives or bacteriostatic agents, e.g., methyl hydroxybenzoate, propyl
hydroxybenzoate, chlorocresol, benzalkonium chloride, or the like.
As used herein, "pharmaceutically acceptable carrier" includes any and all
solvents,
dispersion media, coatings, antibacterial and antifungal agents, isotonic and
absorption
delaying agents for pharmaceutical active substances well known in the art.
Except
insofar as any conventional media or agent is incompatible with the prodrug,
their use in
the therapeutic compositions is contemplated. Supplementary active ingredients
can also
be incorporated into the compositions.
Dosage unit form as used herein refers to physically discrete units suited as
unitary
dosages for the subjects to be treated; each unit containing a predetermined
quantity of
prodrug calculated to produce the desired therapeutic effect in association
with the
required pharmaceutical carrier.
The prodrug is compounded for convenient and effective administration in
effective
amounts with a suitable pharmaceutically acceptable carrier in dosage unit
form as
hereinbefore described. A unit dosage, for example, contains the principal
active
compound in amounts ranging from about 10 mg e.g. in humans, or as low as 1 mg
(for
small animals) to about 2000 mg. If placed in solution, the concentration of
the prodrug
preferably ranges from about 10 mg/mL to about 250 mg/mL. In the case of
compositions containing supplementary active ingredients, the dosages are
determined
by reference to the usual dose and mariner of administration of the said
ingredients. In
the case of buccal administration, the prodrugs are preferably in the buccal
unit dosage
= form present in an amount ranging from about 10 to about 50 mg.
24
H:\wor1\1652\17690\SPEC\17690.spec without clairnsEdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The prodrugs of the present invention are effective in treating disease or
conditions in
which the corresponding drug (without the amino acid prodrug of the present
invention)
normally are used.
As used herein the term "treating" refers to reversing, alleviating or
inhibiting the
progress of a disease, disorder or condition, or one or more symptoms of such
disease,
disorder or condition, to which such term applies. As used herein, "treating"
may also
refer to decreasing the probability or incidence of the occurrence of a
disease, disorder or
condition in a mammal as compared to an untreated control population, or as
compared
to the same mammal prior to treatment. For example, as used herein, "treating"
may
refer to preventing a disease, disorder or condition, and may include delaying
or
preventing the onset of a disease, disorder or condition, or delaying or
preventing the
symptoms associated with a disease, disorder or condition. As used herein,
"treating"
may also refer to reducing the severity of a disease, disorder or condition or
symptoms
associated with such disease, disorder or condition prior to a mammal's
affliction with
the disease, disorder or condition. Such prevention or reduction of the
severity of a
disease, disorder or condition prior to affliction relates to the
administration of the
composition of the present invention, as described herein, to a subject that
is not at the
time of administration afflicted with the disease, disorder or condition. As
used herein
"treating" may also refer to preventing the recurrence of a disease, disorder
or condition
or of one or more symptoms associated with such disease, disorder or
condition. The
terms "treatment" and "therapeutically," as used herein, refer to the act of
treating, as
"treating" is defined above.
As used herein the term "patient" or "subject" refers to a warm blooded
animal, and
preferably mammals, such as, for example, cats, dogs, horses, cows, pigs,
mice, rats and
primates, including humans. The preferred patient is humans.
H:\work\1652\17690\SPEC\17690.speo without clainasILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The prodrugs of the present invention exhibit the same utility as the
corresponding drug
without the amino acid linkage. The prodrug exhibits an enhanced therapeutic
quality.
That is, they exhibit at least one and more preferably at least two enhanced
therapeutic
qualities relative to the drug which has not been transformed to the prodrug
of the
present invention prior to administration. These include, but are not limited
to
a. Improved taste, smell
b. Desired octanol/water partition coefficient (i.e., solubility in water/fat)
The various amino acids have different solubility in aqueous solutions. By
selecting a
particular amino acid, the octanol water partition coefficient can be
affected. For
example, many drugs in the following list are highly hydrophobic. The amino
acids are
highly hydrophilic. For example, assume propofol is the drug and lysine is the
amino
acid. Propofol is completely insoluble in water, while lysine is soluble to
the extent of
700 mg/ml. When these two diverse molecules are esterified via an ester bond,
the
resulting lysine ester of propfol has a solubility in water in excess of 250
mg/ml.
On the other hand, cromolyn sodium is highly water soluble. For all practical
purpose, it
is not absorbed when administered orally. By affecting its water solubility
one could
improve absorption. In this case, one would look for conditions opposite to
that of
propofol, i.e., the goal is to decrease water solubility. By choosing
apporoprieate low
water soluble amino acids, such as tyrosine, one can achieve proper
hydrophilic/lipophilic balance.
c. Improved stability in-vitro and in-vivo
d. Enhanced penetration of blood-brain barrier
e. Elimination of first-pass effect in liver, i.e., the drug not metabolized
in liver and
therefore more drug in system circulation
f. Reduction of entero-hepatic recirculation (this improves bio-
availability)
g. Painless injections with parenteral formulations
h. Improved bio-availability
i. Improved changes in the rate of absorption (increase vs lack thereof)
26
H:\work\1652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
j. Reduced side effects
k. Dose proportionality
A dose proportionality claim requires that when the drug is administered in
escalating
doses, proportionally escalating amounts of active drug is delivered into the
blood
stream. This is measured by determining the area under the plasma
concentration vs.
time curve obtained after administering a drug via any route other than IV
route and
measuring the same in plasma/blood. A simple mathematical procedure is as
follows:
For example, a drug is administered at e.g., 3 different doses, 10, 100 and
1000 mg,
orally to a patient, the area under the plasma concentration time curve (AUC)
is
measured. Then each total AUC is divided by the dose, and the result should be
the same
for all three doses. If it is the case, then there is dose proportionality.
Lack of dose
proportionality indicates any one or more of the
pharmacokinetic/pharmacological
mechanisms are saturable, including absorption, metabolism or the number of
receptor
sites available for pharmacological response.
For example in the above study, assume the AUC values of 100, 1000 and 10,000
are
obtained, in this case the dose proportionality is inappropriate. When there
is lack of
dose proportionality, there is either more or less amount of drug in the
plasma,
depending upon which mechanism is saturable. The following are the
possibilities:
Saturable Absorption. If this is the case, as the dose is increased,
proportionally less and
less of the drug is absorbed, hence overall AUC will decrease as the dose is
increased.
Saturable metabolism of elimination. If thus is the case, then more and more
of the drug
will be circulating in the blood, and the AUC will increase with increasing
dose.
Saturable pharmacological receptor sites: In this case, since all the receptor
sites will
eventually be occupied by the drug, any additional drug will not increase the
response.
Thus, increasing dose will not result in increasing response.
27
H:\work\ 1652 \I7690 \SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Dose proportionality is an excellent response profile, since one can predict
accurately the
pharmacological response and curative power at all doses. Thus dose
proportionality is
a desirable quality for any drug. Furthermore, achievement of dose
proportionality is
also dependent upon the formulation, and fed/fasted differences.
1. Selective hydrolysis of the prodrug at site of action
m. Controlled release properties
n. Targeted drug delivery
o. Reduction in toxicity, hence, improved therapeutic ratio
p. Reduced dose
q. Alteration of metabolic pathway to deliver more drug at the site of
action
r. Increased solubility in aqueous solution
s. Enhanced efficacy
Thus, various dosage forms available with amino acid pro-drugs and they are
prepared
by conventional methods:
i. Oral liquid dosage (Controlled release and immediate release
liquids
containing sugar and sugar free, dye and dye free, alcohol and alcohol free
formulations, including chewable tablets)
ii. Oral solid dosage (Controlled release and immediate release tablets,
capsules
and caplets
Intravenous (Injections, both ready to use and lyophilized powders)
iv. Intramuscular (Injections, both ready to use and lyophilized powders)
v. Subcutaneous (Injections, both ready to use and lyophilized powders)
vi. Transdermal (Mainly patches)
vii. Nasal (Sprays, formulations for nebulizer treatments)
viii. Topical (Creams, ointments)
ix. Rectal (Creams, ointments and suppositories)
x. Vaginal (Creams, ointments and pessaries)
xi. Ocular (Drops and ointments)
28
FlAwork\ 1652 \ 17690 \SPEC\ 17690.spec without claiinsILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
xii. Buccal (Chewable and now chewable tables)
Many drugs discussed herein, especially in the table hereinbelow are
characteristically
highly hydrophobic and readily precipitate in the presence of even very minor
amounts
of water, e.g., on contact with the body (e.g., stomach fluids). It is
accordingly
extremely difficult to provide e.g., oral formulations which are acceptable to
the patient
in terms of form and taste, which are stable on storage and which can be
administered on
a regular basis to provide suitable and controlling patient dosing.
Proposed liquid formulations, e.g., for oral administration of a number of
drugs shown
herein in the table have heretofore been based primarily on the use of ethanol
and oils or
similar excipient as carrier media. Thus, the commercially available drink-
solutions of a
number of drugs employ ethanol and olive oil or corn oil as carrier medium in
conjunction with solvent systems comprising e.g., ethanol and LABRIFIL and
equivalent excipient as carrier media. For example, the commercially available
Cyclosporin drink solution employs ethanol and olive oil or corn oil as
carrier medium
in conjunctions with a Labroid as a surfactant. See e.g., U.S. Patent NO.
4,388,307. Use
of the drink solution and similar composition as proposed in the art is
however
accompanied by a variety of difficulties.
Further, the palatability of the known oil based system has proved
problematic. The
taste of the known drink-solution of several drugs is, in particular,
unpleasant.
Admixture with an appropriate flavored drink, for example, chocolate drink
preparation,
at high dilution immediately prior to ingestion has generally been practiced
in order to
make regular therapy at all acceptable. Adoption of oil-based systems has also
required
the use of high ethanol concentrations which is itself inherently undesirable,
in particular
where administration to children is foreseen. In addition, evaporation of the
ethanol,
e.g., from capsules (adopted in large part, to meet problems of palatability,
as discussed
or other forms (e.g., when opened)) results in the development of a drug
precipitate.
Where such compositions are presented in, for example, soft gelatin
encapsulated form,
29
HAwork\1652\176901SPEC\17690.spec without claiinsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
this particular difficulty necessitates packaging of the encapsulated product
in an air-
tight component, for example, an air-tight blister or aluminum-foil blister
package. This
in turn renders the product both bulky and more expensive to produce. The
storage
characteristics of the aforesaid formulations are, in addition, far from
ideal.
Bioavailability levels achieved using existing oral dosage system for a number
of drugs
described herein are also low and exhibit wide variation between individuals,
individual
patient types and even for single individuals at different times during the
course of
therapy. Reports in the literature indicates that currently available therapy
employing
the commercially available drug drink solution provides an average absolute
bioavailability of approximately 10-30% only, with the marked variation
between
individual groups, e.g., between liver (relatively low bioavailability) and
bone-marrow
(relatively high bioavailability) transplant recipients. Reported variation in
bioavailability between subjects has varied from one or a few percent for some
patients,
to as much as 90% or more for others. And as already noted, marked change in
bioavailability for individuals with time is frequently observed. Thus, there
is a need for
a more uniform and high bioavailability of a number drugs shown herein in
patients.
Use of such dosage forms is also characterized by extreme variation in
required patient
dosing. To achieve effective therapy, drug blood or blood serum levels have to
be
maintained within a specified range. This required range can in turn, vary,
depending on
the particular condition being treated, e.g., whether therapy is to prevent
one or more
pharmacological actions of a specific drug and when alternative therapy is
employed
concomitantly with principal therapy. Because of the wide variations in
bioavailability
levels achieved with conventional dosage forms, daily dosages needed to
achieve
required blood serum levels will also vary considerably from individual to
individual
and even for a single individual. For this reason it may be necessary to
monitor
blood/blood-serum levels of patients receiving drug therapy at regular and
frequent
intervals. Monitoring of blood/blood-serum levels has to be carried out on a
regular
H:\work\1652\17690\SPEC\17690.spec without claimsIl.doe
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
basis. This is inevitably time consuming and inconvenient and adds
substantially to the
overall cost of therapy.
It is also the case that blood/blood serum levels of a number of drugs
described herein
achieved using available dosage systems exhibit extreme variation between peak
and
trough levels. That is for each patient, effective drug levels in the blood
vary widely
between administrations of individual dosages.
There is also a need for providing a number of drugs described herein,
especially the
beta-lactum antibiotics, Cyclosporin, cephalosporins, steroids, quinolone
antibiotics and
Cyclosporin, in a water-soluble form for injection. It is well known that
Cremaphore LID
(CreL) used in current formulations of a number of drugs described hereinbelow
is a
polyoxyethylated derivative of castor oil and is a toxic vehicle. There have
been a
number of incidences of anaphylaxis due to the castor oil component. At
present there is
no formulation that would allow many of these drugs to be in aqueous solution
at the
concentrations needed due to poor water solubility of the drag.
Beyond all these very evident practical difficulties lies the occurrence of
undesirable
side reactions already alluded to, observed employing available oral dosage
forms.
Several proposals to meet these various problems have been suggested in the
art,
including both solid and liquid oral dosage forms. An overriding difficulty
which has
however remained is the inherent insolubility of the several of the drugs
shown in the
table hereinbelow in aqueous media, hence preventing the use of a dosage form
which
can contain the drugs in sufficiently high concentration to permit convenient
use and yet
meet the required criteria in terms of bioavailability, e.g. enabling
effective absorption
from the stomach or gut lumen and achievement of consistent and appropriately
high
blood/blood-serum levels.
31
H:\wor1\I652\l7690\SPEC\17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The particular difficulties encountered in relation to oral dosing with these
drags have
inevitably led to restrictions in the use of specific drug therapy for the
treatment of
relatively less severe or endangering disease conditions. For example, taking
Cyclosporin as a test drug, a particular area of difficulty in this respect
has been the
adoption of Cyclosporin therapy in the treatment of autoimmune diseases and
other
conditions affecting the skin, for example for the treatment of atopic
dermatitis and
psoriasis and, as also widely proposed in the art, for hair growth
stimulation, e.g. in the
treatment of alopecia due to ageing or disease.
Thus while oral Cyclosporin therapy has shown that the drug is of considerable
potential
benefit to patients suffering e.g. from psoriasis, the risk of side-reaction
following oral
therapy has prevented common use. Various proposals have been made in the art
for
application of Cyclosporins, e.g. Cyclosporin, in topical form and a number of
topical
delivery systems have been described. Attempts at topical application have
however
failed to provide any demonstrably effective therapy.
However, the present invention overcomes the problems described hereinabove.
More
specifically, the prodrug of the present invention significantly enhances its
solubility in
aqueous solutions relative to the non-prodrug form of the pharmaceutical,
thereby
avoiding the need to utilize a carrier, such as ethanol or castor oil when
administered as a
solution. Moreover, the prodrugs of these drugs, in accordance with the
present
invention, do not exhibit the side effects of the prior art formulations.
Further, it has
been found that when many of the drugs in the table hereinbelow is
administered in its
prodrug form in accordance with the present invention, there is enhanced oral
absorption, thereby enhancing significantly its bioavailability and its
efficacy.
The preferred drugs used in combination with the amino acids are forming
prodrugs are
listed hereinbelow in the following table and the benefits found are as listed
in the
penultimate column of the table. In the table, the key is as follows:
a) Improved taste smell
32
EI:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2011-12-06
b) Desired Octanol/water partition coefficient (i.e. solubility in water)
c) Improved stability in vitro and in vivo
d) Penetration of blood-brain barrier
e) Elimination of first pass effect in liver
f) Reduction of enterohepatic recirculation
g) Painless injections with parenteral formulations
h) Improved bioavailability
i) Increased rate of absorption
j) Reduced side effects
k) Dose proportionability
1) Selective hydrolysis of the prodrug at site of actions
m) Controlled release properties
n) Targeted drug delivery
o) Reduction in toxicity, hence improved therapeutic ratio
p) Reduced dose
q) Alteration of metabolic pathway to deliver more drug at site of action.
Moreover, the table indicates the utility of the prodrug. The utility of the
prodrug is the
same as the corresponding drug (without the amino acid moiety attached). The
utiluity
is described in the literature such as the Physicians Desk Reference, 2004
edition.
33
Amino Acid Applicable Preferred Most Amino Acids that can Preferred
Most Improvem Utility
Prodrugs of Dose Dose Preferred be reacted with the
Amino Acids Preferred ents with
Range Range Dose Range drug to form the Amino
prodrugs 0
n.)
ster/arnide/azo/anhydri AC-ids
utility =
o
de prodrugs
immuno un
. 6 .
c:
Cyclosporin All doses expressed as drug base
Lys, Leu, Ile, Gly, Lys, Leu, Ile, Gly, Lys, Pro, b,e, f,
g, h, prophylaxis of organ un
--4
Preferred Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, & Gly and k, 1, o, and
rejection, e.g., kidney, un
Forms 5-1000 mg 20-250 25-100 mg Val, Pro, His, Tyr, Ser, Val, Pro,
His, Tyr, dipeptides p liver and heart allogenic
Oral Tab/Cap 1-25 mg 10 mg/ml Nor, Arg, Phe, Trp,
Thr, Arg, Phe, Trp, of Lys- transplants, treatment of
Oral Liquid mg/ml 5-15 50 mg/5m1 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and Gly, Pro- rheumatoid arthritis and
IV Injections 10-250 mg mg/ml Cav, Asn, Gln, Can,
Ser, Hyp, Sar or Gly, Gly- psoriosis
per 5 nil 25-100 Tau, Djk, GABA, Cys, dipeptides of
Gly
mg per 5 Dcy, Thr, and Sar or combination of
any
ml dipeptide of two amino acids
n
combination of any especially AA-Gly,
0
two amino acids where Gly is a
iv
in
especially AA-Gly, spacer attached to
co
a,
where Gly is a spacer cyclosporin and
u.)
a,
attached to AA is the above-
N)
cyclosporin and AA is cited amino acids.
iv
0
the above-cited amino
0
0,
1
acids.
0
H
Lopinavir All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Lys, Pro, b, h, j, k, treatment of
HIV 1
u.)
Preferred Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Gly, & and o infections, e.g., AIDS 0
Forms 0.1-1 g 200-800 400-500 mg Val,
Pro, His, Tyr, Ser, Val, Pro, His, Tyr, Ala
Oral Tab/Cap m mg 400 mg/5m1 Nor, Arg, Phe, Trp, Thr, Arg, Phe,
Trp,
Oral Liquid 0.1-1 gm/5 0.2-0.8 Hyp, Hsr, Car,
Ort, Gln, Asn, Cys and
nil g/5 ml _ Cav, Asn, Gln, Can, Ser, Hyp, Sar
Tau, Djk, GABA, Cys,
Dcy, Thr, and Sar
Iv
n
c 4
k . ,
=
=
. 6 .
7: -:- 5
k . ,
. 6 .
34
H:\work\1652\17690\SPEC\17690.speo without claiinsILdoc vD
1¨,
Cefdinir All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Ser, a, b, e, f, h, antibiotic treatment of
diseases caused.
Preferred Asp, Glu, Met, Ala, ______________________ Asp, Glu, Met, Ala,
Hyp, i, o, and p by Haemophilus influenzae, includinP
Forms 0.1-1gm 0.2-0.5 200-400 mg Val,
Pro, His, Tyr, Ser, Val, Pro, His, Tyr, Tyr, & B-lactamase producing
strains, e.g., in
Oral Tab/Cap 0.1-1 gm 0.2- Nor, Arg, Phe, Trp,
Thr, Arg, Phe, Trp, Thr Haemophilus parainfluenzae (includid
0
Oral Liquid gm/5m1 0.2- 0.4grn/.5m1 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and f3 - 1 ac t o s am a s e producing
strains) and\ n.)
o
IV Infusions 0.01-1 0.5gm/5m 50-150 Cav, Asn, Gln, Can,
Ser, Hyp, Sar moraxella catarihalis (including 13-
un
gm/100 1 mg/100 ml Tau, Djk, GABA, Cys,
lactamase producing strains), and 01 t:
ml 20-500 Dcy, Thr, and Sar
streptococcus pyogenes; such as
mg/100
pneumonia, bronchitis and sinusitis,
ml
pharyngitis and tonsillitis
-..,
'-'
..,,.
B z =
Zileuton All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Gly, Lys, b, h, i, j, k, treatment of asthma
..----t-- I
Preferred Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Sar, Ala, o, p ,...-
Forms Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, ________________ Pro
..----
.:---:
Nor, Arg, Phe, Trp, Thr, Arg, Phe, Trp,
Oral Tab/Cap 200-1200 200-800 300-400 mg Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and F :
r=
n
Oral Liquid nag mg 200400 Cav, Asn, Gin, Can,
Ser, Hyp, Sar
200-1200 200-800 mg/5m1 Tau, Djk, GABA, Cys,
0
iv
mg/5ra1 mg/5m1 Dcy, Thr, and Sar
in
u.)
a,
Nelfutavir All Doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Gly, Lys, b,
h, i, j, k, treatment of HIV, infected patients, u.)
a,
Preferred Asp, Glu, Met, Ala, ______________________ Asp, Glu, Met, Ala,
Sar, Ala, o, p e.g., AIDS "
forms 0.05-1 gm 0.1-0.5 0.2-0.4 gm
Val, Pro, His, Tyr, Ser, Val, Pro,
His, Tyr, Pro iv
0
Oral Tab/Cap 10-250 gm 40-100 mg/gm Nor, Arg, Phe, Tip,
Thr, Arg, Phe, Trp, 0
c7,
1
Oral Powder mg/gm 20-200 40-100 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and 0
H
IV 10-250 mg/gm mg/100m1 Cav, Asn, Gln, Can,
Ser, Hyp, Sar 1
u.)
Formulation mg/100 20-200 Tau, Djk, GABA, Cys,
0
ml mg/100m Dcy, Thr, and Sar
1
1-5
oo
n
1-i
cp
tµ.)
o
o
.6.
k . ,
. 6 .
, . z
=
35
.
HAwork\1652\17690\SPEC\17690.spec without claimsli.doc
Flavoxate All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly,
Hyp, Ser, Tyr, b, h, i, j, k, treatment of urinary spasms
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Thr 1, o,& p
Oral Tab/Cap 10-1000 mg 20-500 mg 50-250 mg His, Tyr, Ser, Nor, Arg, Val,
Pro, His, Tyr, 73
P-8
Oral Liquid 10-1000 20-500 50-250 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Tip, 4 ''' o
mg/5m1 mg/5m1 mg/5m1 Car, Ort, Cav, Asn, Gin, Asn,
Cys and
. o
Gln, Can, Tau, Djk, Ser, Hyp, Sar
.,.
GABA, Cys, Dcy, Thr,
C t:
and Sar
Lil c3;
Candesarten All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Tyr, b, c, e, f,
treatment of hypertension
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, GM,
Met, Ala, & Thr h, I, .i, k, I,
-r-
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, o, p, q -.
1-100 mg 2-75 mg 4-50
.,
,
Oral Liquid Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Tip, ...-
1-100 2-75 4-50
¨
mg/5m1 mg/5m1 mg/5m1 Car, Ort, Cav, Asn, Gln, Asn,
Cys and
Gln, Can, Tau, Djk, Ser, Hyp, Sar
...
,.
GABA, Cys, Dcy, Thr,
......i_z .--._
_
, ..
and Sar
....-...
n
Propofol All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Sar, b, c, d, g,
provides central nervous i ---'
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Pro, Ala, & h, j, k, 1, system anethesia
o
iv
IV Infusions 5-15 raginil His, Tyr, Ser, Nor, Arg, Val,
Pro, His, Tyr, Val m, n, o, p, in
1-25 mg/ml 2.0-20
u.)
mg/
Phe, Trp, Hyp, Hsr, Thr, Arg, Phe, Trp,
q u.)
Car, Ort, Cav, Asn, Gln, Asn, Cys and
a,
iv
Gln, Can, Tau, Djk, Ser, Hyp, Sar
iv
GABA, Cys, Dcy, Thr,
0
0
c7,
and Sar
1
- -
- - o
Nisoldipine All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Ser, b, e, h, i,
j, calcium channel blocker, H
I
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Hyp o treatment of hypertension u.)
o
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr,
Oral Liquid 2-100 mg 2.5-75 mg 5-50 mg Phe, Tip, Hyp, Hsr,
Thr, Arg, Phe, Tip,
2-100 2.5-75 5-50 Car, Ort, Cav, Asn, Gln, Asn, Cys
and
mg/5m1 mg/5m1 mg/5m1 Gln, Can, Tau, Djk, Ser, Hyp,
Sar
GABA, Cys, Dcy, Thr,
and Sar
.o
n
,-i
cp
t.,
=
=
.6.
-a-,
t.,
.6.
=
36
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
¨
Amlodipine All Doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Gly, Lys, Ser, b, e, h, i, j, calcium channel
blocker,
Preferred forms IV Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, & Hyp o treatment of hypertensiodr,
Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr,
4
Oral Tab/Cap 0.1 ¨ 20 mg 1-10 mg 2.5-5 mg Nor, Arg, Phe, Trp,
Thr, Arg, Phe, Trp, o
0
Oral Liquid 0.1-20 1-10 2.5-5 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and n.)
o
mg/5m1 mg/5m1 mg/5m1 Cav, Asn, Gln, Can, Ser, Hyp,
Sar
un
Tau, Djk, GABA, Cys,
Dcy, Thr, and Sar
c:
.....¨...
un
Ciprofloxacin All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Hyp, Ser, a, b, c, g,
Antibiotic; inhibits variou un
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly, & h, i, j, k, o, bacteria, e.g.,
pseudomonds,
Oral Tab/Cap 0.1-1.5 gm 0.1-1.0 g 0.2-0.8 gm
Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, LysP aeruginosa,
staphy1ococcu5 i l
Oral Liquid 0.05- m 0.12-1 g/5m1 Nor, Arg, Phe, Tip, Thr, Arg,
Phe, Trp,.,
aureus or proteus mirabilisr
IV Bulk (Sterile) igm/5nil 0.08-1 5-15 mg/m1 Hyp,
Hsr, Car, Ort, Gln, Asn, Cys and treatment of comeal ulcersi,,
2-25 mg/m1 gm/5m1 Cav, Asn, Gln, Can,
Ser, Hyp, Sar conjunctivitis, acute otitis
3-20 mg/ml Tau, Djk, GABA, Cys,
extema,
Dcy, Thr, and Sar
!-----?-
n_
Ramipril All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Hyp, Ser, j, o treatment of hypertension
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly, & 0
iv
Oral Tab/Cap Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, ______________ Lys
in
0.1-20 mg 0.5-12 mg 1-10 mg
u.,
Oral Liquid Nor, Arg, Phe, Trp,
Thr, Arg, Phe, Trp, u.)
0.1-20 0.5-12 1-10 mg/5m1
a,
mg/5m1 mg/5m1 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and I.)
Cav, Asn, Gln, Can, Ser, Hyp, Sar
I.)
0
Tau, Djk, GABA, Cys,
0
c7,
Dcy, Thr, and Sar
1
0
H
Trandolapril All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Ser, Hyp, j, o treatment of
hypertension 1
u.)
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly & 0
Oral Tab/Cap Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, ______________ Lys
0.1-10 mg 0.5-7.5 mg 1-4 mg 0.1-10 0.5-7.5 1-4 mg/5m1
Oral Liquid Nor, Arg, Phe, Trp, Thr, Arg,
Phe, Trp,
Hyp, Hsr, Car, Ort, Gln, Asn, Cys and
mg/5m1 mg/5m1
Cav, Asn, Gln, Can, Ser, Hyp, Sar
Tau, Djk, GABA, Cys,
Dcy, Thr, and Sar
1 Iv
n
c 4
k . ,
=
=
. 6 .
k . ,
. 6 .
37
o
o
HAwork\1652\17690\SPEC\17690.spec without claimsII.doo
1¨,
Fosinopril All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Ser, Hyp, j, o treatment of hypertension
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly, & ,..,...
Oral Tab/Cap 1-100 mg 2-75 mg
5_50 mg Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, Lys ri
Oral Liquid 1-100 2-75 mg/5m1 5-50 Nor, Arg, Phe, Trp,
Thr, Arg, Phe, Trp, o
0
mg/5m1 mg/5m1 Hyp, Hsr, Car, Ort, Gln, Asn,
Cys and
o
Cav, Asn, Gln, Can, Ser, Hyp, Sar
un
Tau, Djk, GABA, Cys,
Dcy, Thr, and Sar
o
::---, un
Enalapril All Doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Ser, Hyp, j, o treatment of
hypertension-Q. --4
un
Preferred forms 0.5-100 mg 1-50 mg 2-25 mg Asp, Glu,
Met, Ala, Asp, Glu, Met, Ala, Thr, Gly & .--'
Oral Tab/Cap 0.5-100 1-50 mg/5m1 2-25
Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, Lys ..,
Oral Liquid mg/5m1 monil Nor, Arg, Phe, Trp, Thr, Arg, Phe,
Trp,
...,..,...
Hyp, Hsr, Car, Ort, Gln, Asn, Cys and
Cav, Asn, Gln, Can, Ser, Hyp, Sar
Tau, Djk, Djk, GABA, Cys,
-a......5
Dcy, Thr, and Sar
F: :
.---,----
Benazepril All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Hyp, Ser, j, o treatment of
hypertension n
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly, & 0
Oral Tab/Cap 1-100 mg 2-75 mg 2.5-50
Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, Lys iv
in
Oral Liquid 1-100 2-75 mg/5m1 mg Nor, Arg, Phe, Trp,
Ur, Arg, Phe, Trp, u.)
a,
u.)
mg/5m1 2.5-50 Hyp, Hsr, Car, Ort,
Gln, Asn, Cys and a,
iv
ing/5mi, Cav, Asn, Gln, Can,
Ser, Hyp, Sar iv
Tau, Djk, GABA, Cys,
0
0
Dcy, Thr, and Sar
c7,
1
Perindopril All doses expressed as drug base Lys, Leu, Ile, Gly,
Lys, Leu, Ile, Gly, Hyp, Ser, j, o treatment of
hypertension 0
H
Preferred Forms Asp, Glu, Met, Ala,
Asp, Glu, Met, Ala, Thr, Gly, & 1
u.)
0
Oral Tab/Cap Val, Pro, His, Tyr, Ser, Val, Pro, His, Tyr, ______________ Lys
0.1-20 mg 0.5-15 mg 1-10 mg
Oral Liquid Nor, Arg, Phe, Trp, Thr, Arg,
Phe, Trp,
0.1-20 0.5-15 mg/5m1 1-10
mg/5m1 mg/5m1 Hyp, Hsr, Car, Ort, Gln, Asn,
Cys and
Cav, Asn, Gln, Can, Ser, Hyp, Sar
Tau, Djk, GABA, Cys,
Dcy, Thr, and Sar
1
.0
n
,-i
cp
t..,
=
=
.6.
t..,
.6.
38
=
HAwork\1652\17690\SPEC\17690.spec without claimsII.doc 1¨,
Moexipril All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, j, o
treatment of hypertension
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Gly & Lys
1:f
Oral Tab/Cap His Tyr Ser, Nor Arg Val, Pro, His, Tyr,
________________________________________________________ n
1-30 mg 2-20 mg 5-15 mg ' " "
= f
Oral Liquid Phe Tr Hyp Hsr,
, p, , , Thr, Arg, Phe,
Trp, 0
E
1-30 mg/5m1 2-20 mg/5m1 5-15
',
mg/5m1
Car, Ort, Cav, Asn, Gln, Asn, Cys and
%
Gln, Can, Tau, Djk, Ser, Hyp, Sar
un
-:-..
GABA, Cys, Dcy, Thr,
,....: :,. .6.
and Sar
o
Cromolyn All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, & b, c, h, i,
j, inhibits release of histiffane --4
un
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Pro k, 1, n, o, and leukotrienes fromPT-a. st
Oral Tab/Cap 10-200 mg
20-100 mg 20-50 mg His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
I), q,
cell; treatment of
--- Ai
Oral Liquid 10-200 20-100 20-50 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, mastocytosis, asthma w
mg/5m1 mg/5m1 mg/5m1 Car, Ort, Cav, Asn,
Gln, Asn, Cys and .---.
,.-_-
,..
Gln, Can, Tau, Djk, Ser, Hyp, Sar
,-..
GABA, Cys, Dcy, Thr,
f't
.-....,
and Sar
n
Amoxicillin All Doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, a, b, c, h,
antibiotic effective against
Preferred forms ________________________ Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Gly & Lys i, j, k, 1, o, 13-lactamase
negative strains 0
Oral Tab/Cap* 0.1-1.5 gm 0.2-1.2
0.25-1 gm His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, p
causing infections of ear, N)
in
Oral Liquid 0.1-1.5 gm 0.25-1 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, nose, throat, e.g., u.)
a,
gm/5m1 0.2-1.2 gm/5m1 Car, Ort, Cav, Asn,
Gln, Asn, Cys and streptococcus, u.)
a,
Oral Powder 0.1-0.75 gm gm/5m1 0.125-0.5 Gln, Can, Tau, Djk,
Ser, Hyp, Sar staphylococcus or H iv
(*also chewable) 0.1-0.6 gm GABA, Cys, Dcy, Thr,
influenzae;treatment of iv
0
0
gm and Sar
infections of genitourinary c7,
1
tact due to E. coil, P.
0
H
1
mirabilis, E. faecalis,
u.)
0
infections of skin due to
streptococcus,
staphylococcus or E. coli,
infections of lower
respiratory tract due to
streptoccus, staphyloccus.
H. influenzae, and
Iv
n
gonorrhea
1-3
Cefuroxime All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Thr, b, c, e, f,
antibiotic; treatment of
cp
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Lys h, i, j, k, o,
pharyngitis/tonsillitis caused n.)
o
=
.6.
7:-:-..,
t.,
.6.
39
=
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
1¨,
Oral Tab/Cap 10-1000 mg 50-750 100-600 His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, p by streptococcus, acuto7,
Oral Liquid 10-1000 mg mg Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, bacterialotitis media cats' ed
mg/5m1 50-750 100-600 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and by streptococcus, H J.
mg/5m1 mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar influenzae, moraxella 0
Dcy, Thr, and Sar
catarihalis or streptocabus,
urinary tract infectionsC
caused by E coli or ILI
Klebsiella pneumonia,
uri
gonorrhea, skin infection3
cause by staphylococcukor
straptococcuso
o
o
T
0
HAworld1652\17690 \SPEC\17690.spec without claimsTI.doc
Ceftazidime All doses expressed as drug base Lys, L,eu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Thr, a, b, c, g,
antibiotic, treatmtent
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Lys h, i, j, k, 1, lower respiratory
tractn
Powder for IV His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, o, p, q infections, including
0.1-5 gm 0.25-4 gm 0.5-2 gm
0
Oral Tab/Cap Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, pneumonia caused by
0.1-1 gm 0.25-1 gm 0.5-1 gm
t=.)
Oral Liquid 0.1-2.5 0.25-2 0.5-1 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and pseudomonas, H.
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
influenzae, Klebsiella,
gm/5m1 gm/5m1 gm/5m1
-:-
Dcy, Thr, and Sar
Enterbacter, E. coli, proteus
mirabilis, streptococcu
staphylococcus; skin anT1
skin structure infectionAl
caused by peudomonue,
=
aeruginosa,
coli, Proteus enterbact&-','
staphylococcus,
streptococcus, urinary it-act
infections casued by
pseudomonas aeruginosa,
enterbacter, proteus,
0
Klebsiella, E. coli; bone and
joint infections caused by
pseudomonas, eruginosa,
Klebsiella, Enterbacter, or
0
staphylococcus;
0
gynecologic infections
0
including endometritis,
pelvic cellulits and
0
infections of the female
genital tract caused by E.
coli, intra-abdominal
infections and central
nervous system infections,
including meningitis
tµ.)
-:-
41
HAwork\1652\17690\SPEC\17690.spec without clafinsII.doc
Cefpodoxime All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, a, b, c, g, h, i,
antibiotic especial%
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly & Lys j, k, 1, o, p, q against
streptococcils,
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, H. influenzae, s
10-500 mg 25-350 mg 50-250
Oral Liquid 10-500 25-350 mg Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, moraxella catarrhalig; 0
mg/5m1 mg/5m1 50-250
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
treatment of acutelaiis n.)
o
mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar media, pharyngitii7 un
1.17,
-:-..
Dcy, Thr, and Sar
tonsillitis, pneumcma,
.6.
bronchitis, gonorrtid,
o
un
r-
and rectal infectioin
--4
un
in women
'---.
Atovaquone All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Sar, a, b, h, i, j, k,
treatment of malarrej A
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ala, Pro & Ser o, p caused by
plasmodPmli
Oral Tab/Cap 50-1000 mg 100-500 200-300 His, Tyr,
Ser, Nor, Arg, Val, Pro, His, Tyr, parasite ¨
¨ .
E-_-:
Oral Liquid above/5 ml mg mg Phe, Trp, Hyp,
Hsr, Car, Thr, Arg, Phe, Trp, ri
For Pediatric Use 10-150 above/5 ml above/5 Ort, Cav, Asn,
Gln, Can, Gln, Asn, Cys and
P
mg/5m1 25-100 ml Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
n
mg/5m1 50-75 Dcy, Thr, and Sar
mg/5m1
0
iv
_
Acyclovir All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Sar, Hyp b, c, h, i,
j, k, treatment of human in
u.)
Preferred forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Pro & Ser o, p cytomegalovirus a,
u.)
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, (HCMV) a,
iv
Oral Liquid 50-1000 mg 100-750 ml 150-
500 Phe, Tip, Hyp, Hsr, Car, Thr, Arg, Phe, Tip, iv
50-1000 100-750 mg Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and 0
0
mg/5m1 mg/5m1 150-500 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar c7,
1
0
mg/5m1 Dcy, Thr, and Sar
H
I
CA
0
IV
n
,-i
cp
t.,
=
=
.6.
7:-:-..,
t.,
.6.
42
o
o
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
1¨,
Gancyclovir All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Thr,
b, c, e, f, h, i, j, treatment of human '
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Lys k, o,
pcytomegat , . :,-...
o virus V'
Oral Tab/Cap 0.1-1 gm 0.2-0.8 gm 0.2-0.6 His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, (HCMV) 0
Oral Liquid 0.1-1gm/5m1 0.2- gm Phe, Tip, Hyp,
Hsr, Car, Thr, Arg, Phe, Trp, ..J
E
IV Infusions 10-200 mg/ml 0.8mg/5m1 0.2- Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and \ O
tµ.)
25-100 0.6mg/5 Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
C =
o
mg/ml ml Dcy, Tin, and Sar
fit u.
=
30-60
o
mg/ml
'f-- -4
Penciclovir All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leta, Ile, Gly, Hyp, Ser, Thr,
, b, c, e, f, h, i, j, treatment of humair
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Lys k, o, p.
cytomegaloviru' s
si.
Powder for IV His, Tyr, Ser, Nor, Arg, ___________________ Val, Pro, His,
Tyr, (HCMV) ...¨..-
10-1000mg/m1 25-750 50-500
_=
Topical Cream 0.1_5% mg/m1 mg/ml Phe, Trp, Hyp, Hsr, Car, Thr,
Arg, Phe, Trp,
¨
Oral Cap/Tab Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
10-500 mg 0.25-3% 0.5-2.5%
20-300 mg 25-250 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar 0
Dcy, Thr, and Sar
1 :
IF--
mg
.
.
Niacin ER All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Tin, a, b, h, i,
j, 1, lipid management n
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Tyr, Gly &
Lys m, n, o, p, q 0
iv
Oral Tab/Cap His Tyr Ser, Nor, Arg, ______________________ Val, Pro, His,
Tyr, in
0.2-2 gm 0.25-1.5 0.5-1 gm '
" u.)
Oral Liquid Phe Trp,, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, a,
0.2-2 gm/5m1 gm
0.5-1u.)
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
a,
0.25-1 grn/5m1
iv
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
gm/5m1iv
Dcy, Thr, and Sar
0
_
0
_
c7,
1
Bexarotene - All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Re, Gly, Ser, Hyp, Thr, b, c, h, i,
j, k, 1, treatment of skin 0
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Lys o, p conditions, especially
H
1
u.)
Oral Tab/Cap 10-500 mg 25-250 mg 50-100 His, Tyr,
Ser, Nor, Arg, Val, Pro, His, Tyr, those requiring 0
Oral Liquid above/5m1 above/5m mg Phe, Tip, Hyp,
Hsr, Car, Thr, Arg, Phe, Trp, activation of
retinoid
Topical Gel 0.1-5% 0.25-2.5%1 above/5 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and X receptors
ml Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
0.5- Dcy, Thr, and Sar
1.5%
4
.0
n
,-i
cp
.
t.,
=
=
.6.
41
=
,.,
.6.
HAwork\1652\17690\SPEC\17690.spec without claimadoc
g
-
Propoxyphene All Doses expressed as drug base Lys, Leu, Ile, Gly,
Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, a, b, c, h, i, j,
treatment of pain
Preferred forms Glu, Met, Ala, Val,
Pro, Asp, Glu, Met, Ala, Gly, & Lys k, 1, o, 0 p
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
_ In o
Oral Liquid 20-400 mg 25-250 mg 30-150 Phe, Tip,
Hyp, Hsr, Car, Thr, Arg, Phe, Trp, n.)
20-400 25-250 mg Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and 7 =
o
mg/5m1 mg/5m1 30-150 Tau, Djk,
GABA, Cys, Ser, Hyp, Sar -...
=
un
7:-:-..,
mg/51n1 Dcy, Thr, and Sar
Salsalate All doses expressed as drug base Lys, Leu, Ile, Gly,
Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Thr, Gly, b, c, h, i,
j, treatment of
un
Preferred Forms Glu, Met, Ala, Val,
Pro, Asp, Glu, Met, Ala, & Lys . k, o, p inflammatory 7.----
, ...----
Oral Tab/Cap 0.2-2 gm 0.25-1.5 gm 0.3-1 His, Tyr, Ser, Nor,
Arg, Val, Pro, His, Tyr, conditions
...,
Oral Liquid 0.2-2 gin/5m1 0.25-1.5 gm Phe, Trp, Hyp,
Hsr, Car, Thr, Arg, Phe, Tip, '
gm/5m1 0.3-1 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
,--
gmi5mi Tau, Djk, GABA, Cys, Ser, Hyp, Sar -,---
--
Dcy, Thr, and Sar
4,fi
_ ._
Acetaminophen All doses expressed as drug base Lys, Leu, Ile, Gly,
Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Sar, Gly, a, b, c, e, h, treatment of
pain=eir
Preferred Forms Glu, Met, Ala, Val,
Pro, Asp, Glu, Met, Ala, & Lys i, j, k, o, p fever
r- n
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, 0
20-1000 mg 50-800 mg 100-600
iv
Oral Liquid Phe T
, rp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp, in
20-1000 50-800 mg
u.)
, ln, ,
Gln, Asn, Cys and a,
mg/5m1 mg/5m1 100-600 Ort, Cav, Asn G
Can u.)
mg/5m1
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
a,
N)
Dcy, Thr, and Sar
iv
0
_
_
_
- 0
Ibuprofen All doses expressed as drug base Lys, Leu, Ile, Gly,
Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Tyr, a, b, h,
i, j, 1, treatment of pain, fever c7,
I
Preferred Forms Glu, Met, Ala, Val,
Pro, Asp, Glu, Met, Ala, Gly & Lys m, n, o, p, q or inflammation
0
H
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, 1
20-1000 mg 50-800 mg 100-600
u.)
Oral Liquid 20-1000 50-800 mg Phe, Trp, Hyp, Hsr,
Car, Thr, Arg, Phe, Trp, 0
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
mg/5m1 mg/5m1 100-600
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
mg/5m1
Dcy, Thr, and Sar
,
_______________________________________________________________________________
____________________________________ -
1-10
n
,-i
c,
,.,
=
=
.6.
,.,
. .6.
,.,
44 =
11:\work\1652\17690\SPEC\17690.spec without claimsII.doe
Lovastatin A11 doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Gly, b, c,
e, f, lowers choloesterol
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Lys h, i, j, k, concentration; II
Oral Tab/Cap 1-100 mg 2-80 mg 5-50 mg
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, 1, o, p
inhibits HMG-CoAn
Oral Liquid 1-100 mg/5m1 2-80 5-50 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, reductase....-4
,.: 0
mg/5m1 mg/5m1 Car, Ort, Cav, Asn,
Gln, Asn, Cys and -...
, n.)
=
Gln, Can, Tau, Djk, Ser, Hyp, Sar
un
GABA, Cys, Dcy, Thr,
.6.
and Sar
.= - .., un
Simavastatin All Doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Gly, b, c,
e, f, lowers choloesterol7-
, --
Preferred forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Lys h, i, j, k, concentration;
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, 1, o, p inhibits HMG-CoA
.._...
Oral Liquid 1-200 mg 2-150 mg 2.5-100 mg Phe,
T¨, Hyp, Hsr, Thr, Arg, Phe, Trp, reductase -
,
1-200 mg/5m1 2-150 2.5-100 Car, Ort, Cav, Asn,
Gln, Asn, Cys and ..--.----
mg/5m1 mg/5m1 Gln, Can, Tau, Djk,
Ser, Hyp, Sar .:.-
....:._:-.
GABA, Cys, Dcy, Thr,
and Sar
_
n
Atorvastatin All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Gly, b, c,
e, f, lowers choloesterol
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Lys h, i, j, k, concentration; 0
iv
Oral Tab/Cap 1-250 mg 2-125 mg 5-100 mg
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, 1, o, p
inhibits HMG-CoA in
u.)
Oral Liquid 1-250 mg/5m1 2-125 5-100 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, reductase a,
u.)
mg/5m1 mg/51n1 Car, Ort, Cav, Asn,
Gln, Asn, Cys and a,
iv
Gln, Can, Tau, Djk, Ser, Hyp, Sar
iv
GABA, Cys, Dcy, Thr,
0
0
c7,
and Sar
1
0
Pravastatin All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Gly, b, c,
e, f, lowers choloesterol H
I
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Lys h, i, j, k, concentration; u.)
0
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, 1, o, p inhibits HMG-CoA
1-250 mg 2-125 mg 5-75 mg
Oral Liquid Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, reductase
1-250 mg/5m1 2-125 5-75
Car, Ort, Cav, Asn, Gln, Asn, Cys and
mg/5m1 mg/5m1
Gln, Can, Tau, Djk, Ser, Hyp, Sar
GABA, Cys, Dcy, Thr,
and Sar
4
.0
n
,-i
c 4
t.,
=
=
.6.
t.,
45
.6.
=
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
1¨,
Fluvastatin All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Tyr, b, c,
e, f, lowers choloesterol
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Gly & Lys h, i, j, k, concentration; 'Irt
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, 1, o, p inhibits HMG-CoAn
1-250 mg 2-125 mg 5-75 mg
O
Oral Liquid 1-250 mg/5m1 2-125 Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, reductase _k:
5-75
' -'1 t=.)
mg/Sml mg/5m1
Car, Ort, Cav, Asn, Gln, Asn, Cys and
%, o
Gln, Can, Tau, Djk, Ser, Hyp, Sar
f"'W u.
7:-:-..,
GABA, Cys, Dcy, Thr,
rw.-..
and Sar
un
_
:7-'=--: ---1
Nadolol All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Sar, Ser, b, h,
i, j, treatment of angina' un
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Pro k, 1, o, p pectoris and ..----
,---
...
Oral Tab/Cap 1-250 mg 5-225 mg 10-200
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
hypertension; p- ".--- 4.
Oral Liquid 1-250 mg/5m1 5-225 mg Phe, Trp, Hyp, Hsr,
Thr, Arg, Phe, Trp, adrenergic receptorri li
mg/5m1 10-200 Car, Ort, Cav, Asn,
Gln, Asn, Cys and antagonist -:.,--..---
momi Gln, Can, Tau, Djk, Ser, Hyp,
Sar
.,_...
GABA, Cys, Dcy, Thr,
.----,
and Sar
n
Valsartan All Doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Ser, Thr, Lys, b, f,
i, j, treating hypertension,
Preferred forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Gly & Sar k, 1, o, p angiotension II 0
iv
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, antagonist in
u.)
Oral Liquid 10-500 mg 25-250 mg 50-200 Phe, Trp, Hyp,
Hsr, Thr, Arg, Phe, Tip, a,
u.)
10-500 25-250 mg Car, Ort, Cav, Asn,
Gln, Asn, Cys and a,
iv
mg/5m1 mg/5m1 50-200 Gln, Can, Tau, Djk,
Ser, Hyp, Sar iv
0
mg/5m1 GABA, Cys, Dcy, Thr,
0
c7,
d Sanar
1
_
0
Methyl All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Hyp, Sar, a, b,
c, h, treatment of attention H
1
phenidate Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, & Ser j, k, 1, o, deficit disorders and u.)
0
Preferred Forms His, Tyr, Ser, Nor, Arg, Val, Pro,
His, Tyr, P narcolepsy
Oral Tab/Cap 1_50 mg 2-40 mg 2.5-25 mg Phe, Trp, Hyp,
Hsr, Thr, Arg, Phe, Trp,
Oral Liquid 1-50 mg/5m1 2-40 2.5-25 mg/5m1
Car, Ort, Cav, Asn, Gln, Asn, Cys and
mg/5m1 Gln, Can, Tau, Djk, Ser, Hyp,
Sar
GABA, Cys, Dcy, Thr,
and Sar
4 ,
n
,-i
cp
t.,
=
=
.6.
t.,
.6.
46
o
o
HAwork\1652\17690\SPEC\17690.spec without claimsll.doc
1¨,
Trovafloxacin All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Ser, Pro, a, b, e, h, j,
antibiotic; inhib0
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Hyp & Thr k, o, p bacteria such as E.
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, coli, pseudomorfal
10-500 mg 50-300 80-250 mg
'
Phe, Trp, Hyp, Hsr, Car, Thr, Arg, Phe,
Trp, aeruginosa, H.
Oral Liquid
==1
10-500 mg 80-250
mg/5m1 50-300 mg/5ml Ort, Cav, Asia, Gln, Can,
Gln, Asn, Cys and influenzae,
Tau, Pik, GABA, Cys, Ser, Hyp, Sar
streptococous, a: 0
mg/inl S
n.)
Dcy, Thr, and Sar
Klebsiella, Li fl o
o
staphylococcus,in
u,
Ci5
mycoplasma
c:
pneu.moniae, ,..7...
un
--.1
peptostreptococcA,
un
prevotella; treatiiiint
of pneumonia, t a
postsurgical
infections;
graecolgic and
pelvic infections,
infections,
such as
n
endomyometritis,
parametritis, septic
0
iv
abortions,and post-
in
u.)
partum infections;
a,
u.)
skin infections, e.g.,
a,
iv
diabetic foot
iv
infections
0
0
5-AS* All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Glu, Gly, Tyr & Lys b, c, i,
j, 1, treatment of c7,
1
0
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, m, n, o, p, tuberculosis H
1
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, q u.)
1-200 mg 5-150 10-125 mg
0
ar,
Phe, Trp, Hyp, Hsr, C, Thr, Arg, Phe,
Trp,
Oral Liquid
1-200 mg/5m1 mg 10-125
(*5-Arnino- Ort, Cav, Asti, Gln, Can, Gln,
Asn, Cys and
5-150 mg/Sinl
Salicylic acid) Tau, Djk, GABA, Cys, Ser, Hyp, Sar
mg/5m1
Dcy, Thr, and Sar
_______________________________________________________________________________
__________________________________ I
IV
n
,-i
cp
,.,
=
=
.6.
47
t..,
.6.
HAwork\1652 \I7690 \SPEC \I7690.spec without claimsII.doc
g
Methyl All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Pro, b, c, g, j, 1,
treatment of 177
prednisolone Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar & Ser m, n, o, p, inflammation ,r3
Preferred Forms His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, q especially from ---4 o
IM Injection 2-200 mg/ml 5-150 10-100 Phe, Trp, Hyp,
Hsr, Car, Thr, Arg, Phe, Tip, infections, tissud-,õ n.)
o
mg/ml mg/ml Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and damage, allergyptd a
.--
Topical Cream 0.001-5% Tau, Djk, GABA,
Cys, Ser, Hyp, Sar auto-immune 7:-:-..,
0.01- 0.1-2% Dcy, Thr, and Sar
disease .
un
,-,¨; --4
2.5%,....-_-õ
un
_
¨
._
Medroxy All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Pro, b, c, g, j, 1,
providing ,
Progesterone Glu, Met, Ala, Val, Pro, Asp,
Glu, Met, Ala, Sar & Ser
m, n, o, p,
contraception
Preferred forms ________________________ His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, q ¨ A
IM Injections Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Tip, 4- Ill
1 mg ¨ 4 10 mg ¨ 40 rag ¨ 1 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
gm/ml 2 gm/m1 gm/ml Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
,
Dcy, Thr, and Sar
ILL
_
n
Estramustine All doses expressed as drug base Lys, Lau, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Pro, Ala, b, c, h, i, j,
treatment of cancer
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar & Val k, 1, o, p especially
treatment 0
iv
Oral Tab/Cap 10-500 mg 25-250 50-200 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, of metastatic or in
u.)
Oral Liquid 10-500 mg 50-200 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Tip, progressive a,
u.)
mg/5ral 25-250 mg/5m1 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and carcinoma of a,
iv
rug/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar prostate iv
0
Dcy, Thr, and Sar0
'
Miglitol All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Sar,
Pro, & b, c, i, j, n, treatment of type II 0
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ser q diabetes H
1
Oral Tab/Cap 1-250 mg 2-150 _______ 10-125 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, u.)
0
Oral Liquid Phe, Tip, Hyp, Hsr, Car, Thr,
Arg, Phe, Tip,
1-250 mg/5m1 mg 10-125
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
2-150 mg/Sral
mg/5m1 Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
Dcy, 'Thr, and Sar
,
_______________________________________________________________________________
___________________________________
Iv
n
,-i
t.,
=
=
.6.
7:-:-..,
t.,
.6.
48
o
H:\work\1652\17690\SPEC\17690.spec without clairnsadoc
Mefloquine All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Sar, Pro, a, b, c, h, i,
treatment of malgia
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Val, Ala .i, 1c 1, 0, I), qj
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, n
10-500 mg 100-400 150-300 mg
q , o
Oral Liquid Phe Tr p, Hsr, C
, p, Hyp, , Car, , g,
, p, ..--,..E
10-500 mg 150-300 Thr, Arg, Phe,
Tip,
mg/5m1
, , , , , , , y
mg/5m1 25-400 mg/5m1 Ort Cav, Asn Gln Can
Gln Asn Cs and un
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
..----- ,
mg/5m1 m..-.. o
Dcy, Thr, and Sar
Danazol All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Hyp, Pro, Ala, Val, a, b, c, e, f,
treatment of
-_-.
Preferred forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ser & Thr g, h, i, j, k, endometriosis
atitt-
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, 1, n, o, p, q flbrostatic breast\
Oral Liquid 2-500 mg 10-350 mg 25-250 mg Phe, Tip, Hyp,
Hsr, Car, Thr, Arg, Phe, Tip, disease .---i
:-........4
2-500 10-350 25-250 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
mg/5m1 mg/5m1 mg/5m1 Tau, Djk, GABA, Cys, Ser, Hyp,
Sar
..-4-,-
Dcy, Thr, and Sar
Eprosartan All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Mr, Lys, b, c, h,
i, j, ACE inhibitors,p_, n
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly & Val k, 1, o, p treatment of
Oral Tab/Cap 0.1-1 gm 200-800 300-750 mg His,
Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, hypertension o
iv
Oral Liquid 0.1-1 mg 300-750 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Tip, in
co
gm/5m1 200-800 mg/5m1 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and a,
u.)
a,
mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar iv
Dcy, Thr, and Sar
iv
_______________________________________________________________________________
_________________________________________ o
Divalproex Na All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, a, b, c,
f, h, treatment of epilepsy 0
c7,
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Val i, j, k, 1, o, '
0
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, p H
1
50-800 mg 75-750 mg 100-600 mg
u.)
Oral Liquid 50-800 75-750 100-60 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, 0
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
mg/5m1 mg/5m1 mg/Sml
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
Dcy, Thr, and Sar
Iv
n
c 4
k . ,
=
=
. 6 .
-a 5
k . ,
. 6 .
49 ,. z
=
HAwork\1652\17690\SPEC\17690.spec without claimsII.doe
1-"
Fenotibrate All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, b, c, h, i, j,
treatment of
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Ala k, 1, o, p, q hypercholestemii
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, i.--
10-800 mg 20-750 mg 100-600 mg
o
Oral Liquid Phe Tr H Hsr, Car,
, p, Hyp, , ,
Thr, Arg, Phe, Trp, i
=
10-800 20-750 100-600
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
,
mg/5m1 mg/5m1 mg/5m1
7. =
un
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
r..
Dcy, Thr, and Sar
.,..-...
c:
un
Gabapentin All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Cyclic Deriv. & Tyr b, c, d,
e, f, treatment of un
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, h, i, j, k, 1, convulsions
Oral Tab/Cap 10_800 mg 25-750 mg 50-500 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, n, 0, 13, p
Oral Liquid 10-800 25-750 50-500 Phe, Trp, Hyp, Hsr, Car, Thr,
Arg, Phe, Trp,
mg/5m1 mg/5m1 mg/5m1 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
Dcy, Thr, and Sar
,
Lansoprazole All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Pro, Sar, b, e,
f, h, i, suppression of daitric n
Preferred forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ser & Val j, k, 1, o, p acid secretion
by 0
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, inhibition of (H+, K+) iv
Oral Liquid 1-60 mg 2-50 mg 10-40 mg Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, ATP-ase enzyme in
co
1-6- 2-50 mg/5m1 1040 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and system at the secretory a,
u.)
a,
mg/5m1 mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar surface of the gastric K)
Dcy, Thr, and Sar
parietal cell; treatment "
0
of gastric hyperacidity
0
c7,
_
1
Omeprazole All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys,
Gly, Val, Pro & b, e, f, h, i, suppression of gastric 0
H
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar j, k, 1, o, p acid secretion
by 1
u.)
Oral Tab/Cap 1-200 mg 2-100 mg 5-60 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, inhibition of (H+, K+) 0
Oral Liquid 1-200 2-100 5-60 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, ATP-ase enzyme
mg/5m1 mg/5m1 mg/5m1 Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and system at the secretory
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
surface of the gastric
. Dcy, Thr, and Sar
parietal cell, treatment
of gastric hyperacidity
Iv
n
c 4
k . ,
=
=
. 6 .
k . ,
. 6 .
o
,-,
H:\work\1652 \17690 \SPEC \17690.spec without claimsil.doc
Megestrol All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Sar, Pro, b, c, h, i, j,
treatment of anorexia;
,....-.
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ser & Ala k, 1, n, o, p improving appetite
in
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, .
2-100 mg 4-80 mg 20-60 mg
anorexic and pents
a't'r
Oral Liquid 2-100 4-80 mg/5m1 20-60 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, suffering from ADS O
mg/5m1 mg/5m1
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
o
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
C =
un
Dcy, Thr, and Sar
c:
Metformin All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Asp, Glu, Lys &
Azo o, p4 .7.
treatment of
--4
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, dimer hyperglycemia; Qs un
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, insulin to improirre.
0.2 ¨ 3 gm 0.25-1.5 gm 0.5-1 gm
Oral Liquid 0.2-1 0,25-1.5 0.5-1 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, transport of gluaie, 1
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
into cells ,--_
gm/5m1 mg/5m1 gm/5m1
1=
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
pri
Dcy, Thr, and Sar
----,
. .
,-..---:
Tazarotene All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser,
Hyp, Thr, Lys, & B, c, h, I, j, treatment of psoliasis n
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly k, 1, o, p and acne especially
Topical Gel 0.01-0.3% 0.02-0.25% 0.025-
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
those caused by 0
iv
0.125% Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, pathogenic in
u.)
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
microorganisms, allergy a,
u.)
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
and inflammation a,
iv
Dcy, Thr, and Sar
iv
0
Sumatriptan All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Ala, Pro, b, c,
d, g, h, 5 HT subtype receptor 0
c7,
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar & Val i, j, k, 1, n, agonist,
treatment of 1
0
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, o, p, q migraine headaches
H
1
Oral Liquid 5-250 mg 10-200 mg 20-125 mg Phe,
Trp, Hyp, Hsr, Car, Thr, Arg, Phe, Trp, u.)
0
5-250 10-200 20-125 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
IM Injections mg/5m1 mg/5m1 mg/5m1 Tau, Djk, GABA,
Cys, Ser, Hyp, Sar
1-36 mg/ml 2-24 mg/ml 4-20 Dcy, Thr, and Sar
mg/ml
Iv
n
c 4
k . ,
=
=
. 6 .
k . ,
51
.6.
H:\work\1652\17690\SPEC\17690.spec without clainasII.doc
o
1¨,
,--
Naratriptan All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly Lys, Gly, Sar, Val, b, h, i, j, k,
5 HT subtype _
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Alf,, Ala, & Pro 1, o, p receptor agonise
Oral Tab/Cap 0.1-10 mg 0.25-5 mg 0.5-4 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, treatment of i 4
Oral Liquid 0.1-10 0.25-5 0.5-4 Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, migraine headades 0
n.)
mg/5m1 mg/5m1 me5mi Ort, Cav, Asn, Gln, Can, Gln, Asn,
Cys and
o
-:-
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
C un
7: 5
- Dcy, Thr, and Sar
_
_
_
Zolmitriptan All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Sar, Val, b, h, i,
j, k, 5 HT subtype ri un
.....,:. -4
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ala, & Pro 1, o, p receptor .----
un
---....-z-4
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, agortist;treatmerikof
0.1-12 mg 0.5-10 mg 1-7.5 mg
Oral Liquid Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, migraine headaclie's 4
1-12 0.5-10 1-7.5
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
m1
mg/5m1 mg/5m1 mg/5
.
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
.".:¨...
Dcy, Thr, and Sar
.-....,
El
Aspirin All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, a, b, c, e, f,
antipyretic, antilu.
n
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Ala g, h, j, k, I, inflammatory,
Oral Tab/Cap His, Tyr, Ser, Nor, Arg, ____________________ Val, Pro, His,
Tyr, m, n, o, p, q analgesic, 0
10-1000 20-800 mg 25-600
iv
Oral Liquid Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, thrombolytic; in
mg 20-800 mg
u.)
Ort, Cav, Asn, G C Gn, Cys and
treatment of a,
10-1000 mg/ml 25-600 ln, an,
ln, As u.)
. Tau, Djk, GABA, Cys,
Ser, Hyp, Sar hyperthermia, a,
mg/ml mg/ml
"
Dcy, Thr, and Sar
myocardial iv
infarction and
0
0
thrombolysis
c7,
_
.
1
Olmesartan All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, b, h,
i, j,k, 1, ACE inhibitor, 0
H
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, GIu, Met, Ala, Gly, & Ala o, p treatment of 1
u.)
0
Oral Tab/Cap 1-100 mg 2-80 mg 4-50 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, hypertension
Oral Liquid 1-100 2-80 4-50 mg/5m1 Pile, Tip, Hyp, Hsr, Car, Thr,
Arg, Phe, Tip,
mg/5m1 mg/5m1 Ort, Cav, Asn, Gln, Can, Gln, Asn,
Cys and
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
Dcy, Thr, and Sar
Iv
n
k . ,
=
=
. 6 .
7: -:- 5
k . ,
. 6 .
52 o
o
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
1¨,
Sirolimns All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, b, h, i, j,k, 1,
immunosuppregant
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Gly, & Ala o, p in surgical hump
Oral Tab/Cap His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, patients with õ.4.
Oral Liquid 0.1-20 mg 0.5-10 mg 1-8 mg Phe, Trp, Hyp,
Hsr, Car, Thr, Arg, Phe, Trp, transplants;
0.1-20 0.5-10 1-8 mg/5m1 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and
antibiotic; treating
n.)
IM Injections mg/51111 mg/5m1 Tau, Djk, GABA,
Cys, Ser, Hyp, Sar -,,-,..
vitiligo psoriassit-E
o
un
7: -:- 5
Dcy, Thr, and Sar
acne
c:
Tacrolimus All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Ala, Thr, b, c, g,
h, i, immunosuppresRint un
--4
,. un
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar & Pro j, k, 1, o, p in surgical
human¨
Oral Tab/Cap 0.1-20 mg 0.2-15 mg 0.25-10 mg
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
patients with ...".-";
Oral Liquid above/5m1 above/5m1 above/5m1
Phe, Tip, Hyp, Hsr, Car, Thr, Arg, Phe, Trp,
transplants; l'-..
¨ A
IV Infusions 1-20 mg/ml 2-15 2.5-8 mg/ml Ort, Cav, Asn, Gln, Can,
Gln, Asn, Cys and antibiotic; treating, 13
mg/ml Tau, Djk, GABA, Cys,
Ser, Hyp, Sar vitiligo psoriass
Dcy, Thr, and Sar
acne ...f9
õ:,..i,
Pimeerolimus All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Ala,
Thr, = b, c, g, h, i, immunosuppre4int
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Sar & Pro j, k, 1, o, p in surgical
human n
Oral Tab/Cap 0.1-20 mg 0.2-15 mg 0.25-10 mg
His, Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr,
patients with 0
Oral Liquid above/5m1 above/5m1 above/5m1
Phe, Trp, Hyp, Hsr, Car, Thr, Arg, Phe, Trp,
transplants; "
in
Ointment Cream 0.01 ¨ 10% 0.1-5% 0.5-2% Ort, Cav, Asn,
Gln, Can, Gln, Asn, Cys and antibiotic treating u.)
a,
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
vitiligo psoriass, u.)
a,
Dcy, Thr, and Sar
acne "
_
Clopidogrel All doses expressed as drug base 20-125 mg 25-
100 mg Ser, Hyp, Thr, Lys, b, c, h, i, j, treatment of
"
0
Preferred Forms 20-125 mg/5m1 25-100 mg/5m1
Ala, & Gly k, 1, m, o, p, myocardial 0
c7,
1
Oral Tab/Cap
q infections 0
10-250 mg 20-125 25-100 mg
H
Oral Liquid
(Ai
10-250 mg 25-100
0
mg/5m1 20-125 mg/5m1
mg/5m1
,
Iv
n
c 4
k . ,
=
=
. 6 .
7: -:- 5
k . ,
. 6 .
53
o
o
HAwork\1652\17690\SPEC\17690.spec without claimsILdoc 1¨,
Aruphoteriein All doses expressed as drug base
Lys, Leu, Ile, Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, b,
c, g, i, j, treatment of futtps,
B Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ala, & Gly 1, m, n, o, p, expecially
those=:.
Preferred Forms His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, q acting on cell ir,k
0.5-20 1-15 2-10
IV Infusion
mg/kg/day mg/kg/day mg/kg/day Phe, Trp, Hyp, Hsr, Car, Thr,
Arg, Phe, Trp, membrane claangim
0
Ort, Cav, Asn, Gin, Can, Gba, Asn, Cys and
its permeability '''-i.
0.01-10% 0.1-5% 0.5-2%
n.)
Topical Cream Tau, Djk, GABA, Cys,
Ser, Hyp, Sar d.---.
o
Dcy, Thr, and Sar
---.
o
_
.
_
Tenofovir All doses expressed as drug base - Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Ala, Pro, b, c, h, i, j,
inhibitor of HP
e.n
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Ser & Sar k, 1, o, p virus, treatment,*
---.)
cm
Oral Tab/Cap 10-900 mg 50-750 100-500 mg His,
Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, AIDS infections.::,,:
Oral Liquid 10-900 mg 100-500 Phe, Trp, Hyp, Hsr, Car, Thr,
Arg, Phe, Trp,f..ig 4
,
mg/5m1 50-750 mg/5m1 Ort, Cav, Asn, Gin, Can, Gin, Asn,
Cys and r
-a-
mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar 4
Dcy, Tim-, and Sar
n
-
. -
Unoprostone All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, lie, Gly, Ser, Hyp, Thr, Tyr, b, c, h, i, j,
treatment of ''
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Pro, & Lys k, 1, n, o, p, glaucoma, n
Ocular Drops - His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, q especially caused by
0.01-1% 0.05-0.5% 0.01-0.25% Phe, Trp, Hyp, Hsr, Car,
Thr, Arg, Phe, Trp, age; lowers 0
iv
Ort, Cav, Asn, Gin, Can, Gln, Asn, Cys and
intraocular pressure (.71
w
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
A.
w
Dcy, Thr, and Sar
A.
n)
_
_
_
Fulvestrant All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Pro, Ala, B, c,
g, j, 1, treating cancer, iv
0
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Val & Sar o,p especially breast o
0,
IM Injection 2-1250 10-1000 ' 20-500 His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, cancer 1
0
mg/5m1 mg/5m1 mg/5m1 Phe, Trp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp, '-
Ort, Cav, Asn, Gin, Can, Gin, Asn, Cys and
w
0
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
Dcy, Thr, and Sar
_
1-e
n
1 7..3.
c A
w
c ,
4-.
---,
0
54
t..,
4-.
HAwork\ 1652 \17690\SPEC117690.spec without claims11.cloc
vz
1¨k
Cefditoren All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Gly, b, c, h, i, j,
antibiotics,
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Lys & Ala k, 1, o, p especially inhibTig
Oral Tab/Cap
His, Tyr, Ser, Nor, Arg, Val, Pro, His,
Tyr, H. influenzae;
20-500 mg 100-400 150-300 mg
n
Oral Liquid Phe Trp H
, , Hyp, Hsr, Car, Thr, Arg, ,
,
20-500 mg 150-300 Phe
Trp Haemophilus pas-
0
mg/5m1 100-400 mg/5m1
Ort, Cav, Asn, Gln, Can, Gln, Asn, Cys and
influenzae,
n.)
mg/5m1
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
streptococcus,
o
Dcy, Thr, and Sar
Maraxella
. 6 .
catarrhalis;
un
treatment of 2
,
un
bronchitis,
,,,,,
pharyngitis,
tonsillitis, skin
infections
47-
_
Efavirenz All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Pro, Ala, b, c, h, i, j,
inhibitor of HIVa
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Sar, & Val k, 1, o, p specific, non- r-g
Oral Tab/Cap
His, Tyr, Ser, Nor, Arg, Val, Pro, His,
Tyr, nucleoside, reveXse
0.2-1.2 gm 300-800 400-750
Oral Liquid Phe, Trp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp, transcirptase; n
0.2-1.2 mg mg
gm/5m1 300-800 400-750 Ort, Cav, Asn, Gln, Can, Gln,
Asn, Cys and treatment of AIDS
0
nag/5m1 mg/5m1
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
infections iv
in
Dcy, Thr, and Sar
u.)
a,
Eplerenone All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Ser, Hyp, Thr, Lys, b, c,
h, i, j, treatment of a,
iv
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Gly & Val k, 1, o, p hypertension, blocks iv
Oral Tab/Cap 10-250 mg 15-200 ¨20-150 mg His,
Tyr, Ser, Nor, Arg, Val, Pro, His, Tyr, binding of 0
0
Oral Liquid 10-250 mg 20-150 Phe, Trp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp, aldosterone to c7,
1
mg/5m1 15-200 mg/5m1 Ort, Cav, Asn, Gln, Can, Gln, Asn,
Cys and mineralo-corticoid 0
H
1
mg/5m1 Tau, Djk, GABA, Cys,
Ser, Hyp, Sar receptors u.)
0
Dcy, Thr, and Sar
_
Iv
n
_
c 4
=
=
. 6 .
7: -:- 5
55
t..,
.6.
HAwork\1652\17690\SPEC\17690.spec without clahnsII.doo
o
1¨,
Treprostinil All Doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Gly, Lys, Val, Hyp, b, c, g, h, i,
Inhibits platelet,..
Preferred Forms Glu, Met, Ala, Val, Pro, Asp, Glu,
Met, Ala, Thr & Ser j, k, 1, o, p aggregation and:
SC infusion His, Tyr, Ser, Nor, Arg,
Val, Pro, His, Tyr, vasodilation of
0.1-100 0.2-50 0.5-20 Phe, Trp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp,
systemic and 7'9
C)
Oral Tab/Cap mg/ml mg/ml mg/ml Ort, Cav, Asn, Gln, Can, Gln, Asn,
Cys and pulmonary vasCillar
10-1000 mg 20-800 mg 25-500 mg Tau, Djk, GABA, Cys, Ser, Hyp, Sar
bed, treatment aZ
C-5
Dcy, Thr, and Sar
cardiovascular M
related conditioe
Adefovir All doses expressed as drug base Lys, Leu, Ile,
Gly, Asp, Lys, Leu, Ile, Gly, Lys, Gly, Val, Ser, b, c, h, i, j,
HIV reverse
Preferred Forms Glu, Met, Ala, Val, Pro,
Asp, Glu, Met, Ala, Hyp, & Pro k, 1, 0, P. transcriptase
Oral Tab/Cap 1-100 mg 2-50 mg 5-20 mg His, Tyr, Ser,
Nor, Arg, Val, Pro, His, Tyr, inhibitors; treatment.
.
-
Oral Liquid 1-100 2-50 5-20 Phe, Trp, Hyp, Hsr, Car, Thr, Arg,
Phe, Trp, of HIV mfection,
mg/5m1 mg/5m1 moini Ort, Cav, Asn, Gln, Can, Gln, Asn,
Cys and and AIDS
Tau, Djk, GABA, Cys, Ser, Hyp, Sar
Dcy, Thr, and Saro
.1õ,ff=-=
o
o
c7,
o
o
56
11:\work\1652\17690\SPEC\17690.spec without clairnsli.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The following non-limiting examples further illustrate the invention:
Synthesis of Various Amino Acid Derivatives of Selected Drugs
I. Propofol Derivatives
Propofol (2,6-diisopropylphenol) is a low molecular weight phenol which widely
used as
a central nervous system anesthetic, and posses sedative and hypnotic
activities. It is
administered intravenously in the induction and maintenance of anesthesia
and/or
sedation in mammals. The major advantages of Propofol are that it can induce
anesthesia
rapidly, minimal side effects and upon withdrawal, the patient recovers
quickly without
prolonged sedation.
PROPOFOL
01-13. OH CH3
H3C
CHIA
Propofol has been shown to have a large number of therapeutic applications,
which are
quite varying and somewhat surprising. For example, it has been shown to be an
effective antioxidant, anti-emetic, anti-pruritic, anti-epileptic, anti-
inflammatory, and
even seems to possess anti-cancer properties.
Mechanism of Action:
The mechanism of action of Propofol has been extensively studied. Its central
nervous
system anesthetic activity has been shown to be related its high affinity for
a specific
subclass of GABA receptors (Collins G.G.S., 1988, Br. J. Pharmacology. 542,
225-232).
However, there are a number of different receptors in the brain which are
substrates for
propofol, hence its varied activities.
57
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Propofol also has significant biological effect as an antioxidant. Because of
this
generalized activity of propofol, it is theoretically useful in the treatment
of a number of
inflammatory processes where oxidation is an important factor. For example,
cyclooxygenase mediated prostaglandin synthesis results in inflammation. By
inhibiting
oxidation in the respiratory tract, one could use propofol in the treatment of
acid
aspiration, adult/infant respiratory distress syndrome, airway obstructive
diseases,
asthma, cancer and a number of other similar pathological conditions.
Since oxidative tissue damage is a very common occurrence, it has been
suggested that
propofol could be useful in the treatment of Parkinson's disease, Alzheimer
disease,
Friedrich's disease, Huntington's disease, multiple sclerosis, amyotrophic
lateral
sclerosis, spinal chord injuries, and various other neurodegenerative
diseases.
Propofol is currently available in the US market as an intravenous emulsion
marketed by
Astra Zenaca under the brand name Diprivane. It is one the most widely used
short
acting central nervous system anesthetics in the market. The concentration of
propofol is
10 mg/mL in non-pyrogenic sterile emulsion and the formula contains soybean
oil,
glycerol, egg lecithin, disodium edetate and sodium hydroxide.
A significant disadvantage of Propofol is that it is completely insoluble in
water. Even at
very low concentrations of 10 mg/mL, the drug precipitates out of an aqueous
solution in
room temperature. Therefore, manufacturers of this formulation use heroic
methods to
emulsify this product in water using extraordinarily complex and toxic
emulsifying
agents. For example, manufacturers of the IV formulations use egg lecithin,
Cremaphor
Le, castor oil, and other similar emulsifiers.
However, use of such emulsifiers is associated with number of problems. It is
well now
that various types of Cremaphor Le emulsifiers can precipitate allergic
reactions. Egg
lecithin and castor oil have been shown to produce anaphylactic shock in some
patients.
Furthermore, maintenance of stability of propofol in these emulsions is short
lived and
58
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
more expensive. Moreover, the presence of egg lecithin and castor oil make the
emulsion
prone to microbial growth. It may be possible to dissolve propofol in water by
complexing it with cyclodextrin, but cyclodextrin has not been approved by the
FDA for
use in intravenous therapy.
Heretofore, no one has made a safe prodrug of propofol. The British patents
1,102,011,
and 1,160,468 and US Patent 3,389,138 describe the various phenol esters of
amino
acids, wherein the propofol is attached to a number of side chains which when
released
in the body produce toxic effects.
US Patent 6,451,854 describe a number of substituted alpha amino acetic acid
esters of
propofol, wherein propofol and the side chain were substituted with a number
of
different chemical groups. All the N,N-disubstituted glycine esters of
propofol have not
shown to be non-toxic and there many of the compounds described are derivative
of
propofol. Thus when released in the body after the cleavage of ester by the
enzymes,
many of the active drugs released are not propofol, and hence they do not
possesses any
toxicity data and are entirely new molecules with unknown therapeutic efficacy
in man.
In yet another published paper on the water soluble salts of amino acid esters
of
anesthetic agent propofol, (Int. J. Pharmaceutics, 175[4 195-204, 1998)
authors have
synthesized a number of water soluble derivatives of propofol. However, when
these
prodrugs are cleaved by esterase enzymes, substituted non-natural amino acids
with
unknown toxicity profile are released in the body.
Until now there has been no pharmaceutical preparation has been available in
the market
that can deliver propofol without harmful side effects. The present invention
however,
has produced a number of water soluble, non-toxic derivatives of propofol
which are
suitable for delivering propofol in the body without any harmful side effects
and without
the needs for toxic and expensive additives, solubilizers and emulsifiers.
59
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Accordingly, in one aspect, the present invention is directed to a class of
prodrugs of
Propofol. The prodrug consists of the carboxyl group of an amino acid
esterified to the
free hydroxyl group present on the propofol molecules.
=
More specifically, one aspect of the present invention is directed to, the
compounds of
the formulae
AA PROPOFOL
CH3 0 CH3
H3C
111 CH3
or pharmaceutically acceptable salts thereof; wherein AA is an amino acid, in
which the
carboxyl group of AA is reacted with the hydroxyl group of the Propofol.
In antoher aspect, the present invention is also directed to a pharmaceutical
composition
comprising a therapeutically effective amount of the various Propofol prodrugs
above
and a pharmaceutical carrier therefor.
In another embodiment, the present invention is directed to a method of
treating a patient
in need of propofol therapy, which method comprises administering to said
patient an
effective amount of the Propofol.
In a further embodiment, the present invention is directed to a method of
enhancing the
solubility of propofol in an aqueous solution comprising reacting the hydroxyl
functionality of the Propofol and isolating the products thereof.
In a still further embodiment, the present invention is directed to a method
of
substantially and in a therapeutically efficacious manner, reducing or
eliminating the
potential toxic side effects of current formulations containing toxic
exepients when
HAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
administered to a patient which comprises reacting the hydroxyl functionality
of the
propofol molecule with carboxyl function of selected amino acids to form an
ester
covalent bond respectively and isolating the product thereof and administering
said
product to the patient.
The current invention shows that when unsubstituted naturally occurring amino
acids are
esterified to propofol, the resulting prodrugs are highly water soluble, (>200
mg/L in
water), release non-toxic amino acids upon cleavage in the body and require
none of the
toxic emulsifier, additives and other exepients.
Furthermore, it has been shown that the current invention also produced drugs,
while
they are prodrugs of propofol of the present invention are highly effective
central
nervous system anesthetics. Thus the current amino acid prodrugs are effective
central
nervous system anesthetics, with or without releasing the active parent drug.
The amino acid esters of the present invention are at least 10 times more
soluble that
propofol in water in room temperature. Especially the glycine, proline and
lysine esters
of propofol are soluble at the range of more than 100 mg/ml, and in case of
lysine it is
greater than 250 mg/mL.
While the prodrugs of the present invention are not expected to possess any
antioxidant
activity due to blockage of the phenolic group responsible for such; however
the present
inventor has found that the prodrugs of propofol are effective anesthetics
with or without
releasing propofol. The propofol prodrugs described release the propofol when
administered in vivo and the resulting drug maintains its pharmacological and
anti-
oxidant properties.
The prodrug of propofol of the present invention clearly provides a number of
advantages over propofol, for example, all of the side chains cleaved from
these
prodrugs are naturally occurring essential amino acids and hence are non-
toxic. This
61
11:\work\ 1652 \ 17690 \ SPEC\ 17690.spec without claimsn.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
results in high therapeutic index. Secondly all the prodrugs are readily
cleaved in the
body to release propofol. Furthermore, due their high water solubility, they
can be easily
administered by either forming an in-situ solution just before IV
administration using
lyophilized sterile powder or providing the drug in solution in prefilled
syringe or bottles
for infusion. The aminoacid esters are more stable than propofol since OH
group in
propofol is blocked to oxidation. Thus the propofol prodrugs of the present
invention are
more effective then propofol itself without the toxicity and other
pharmaceutical
problems associated with current marketed formulations.
The prodrugs of propofol of the present invention possess anti-inflammatory,
anti-
oxidant, anti-cancer, anti-convulsive, anti-emetic and anti-pruritic
properties.
These prodrugs of propofol of the present invention are effective in treating
diseases or
conditions in which Propofol normally are used. The prodrugs disclosed herein
are
transformed within the body to release the active compound and enhances the
therapeutic benefits of the Propofol by reducing or eliminating
biopharmaceutical and
pharmacokenetic barriers associated with each of them. However it should be
noted that
these prodrugs themselves will have sufficient activity without releasing any
active drug
in the mammals. Since the prodrugs are more soluble in water then Propofol, it
does not
need to be associated with a carrier vehicle, such as alcohol or castor oil
which may be
toxic or produce unwanted side reactions. Moreover, oral formulations
containing the
prodrugs of Propofol are absorbed into the blood and are quite effective.
Thus, the prodrug of the present invention enhances the therapeutic benefits
by removing
biopharmaceutical and pharmacokenetic barriers of existing drugs.
Furthermore, the prodrugs are easily synthesized in high yields using reagents
which are
readily and commercially available.
62
H:\work\1652\17690\SPEC\17690.speo without clainisIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Overview:
The procedure for the synthesis of the glycine, L-proline, and L-lysine esters
of Propofol
is depicted hereinbelow. However, these are exemplary and any amino acid
produrugs
thereof can be prepared using the following methodology. The complete
procedure and
analytical data is given in the Experimental Section. In general, as shown in
the
following scheme Propofol (10 g) was coupled with the N-Boc protected amino
acid (1
equivalent) with 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide, hydrochloride
(EDC)
in the presence of a catalytic amount of 4-(N,N-dimethyamino)-pyridine (DMAP).
The
EDC was removed by extraction with water. After drying over sodium sulfate,
filtration,
and concentration the crude protected amino acid ester of Propofol was
purified by flash
chromatography to generate the protected esters in 50-60% yield. The
protecting groups
were then removed by stirring the protected esters in diethyl ether saturated
with
hydrochloric acid (gas) at room temperature. Yields for the deprotection step
were
generally 60-95%. After filtration and drying the hydrochloride salts of the
glycine and
L-proline esters of Propofol did not require additional purification. The
hydrochloride
salt of the L-lysine-Propofol ester was crystallized once from ethanol to
remove a trace
of mono-protected L-lysine-Propofol ester.
Synthetic Sequence:
63
HAwork\1652\17690\SPEC\17690.spec without claimsEdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
1. SPI0010
Cl-
0 H
=OH + HO 0 a) y b) 0 y
o)-LN+1-1
8
0
Propofol Boc-glycine SPI0010
2. SPI0011
O O
\\P-8 CI¨
b)
t-0 a) 0
OH
0101 YL612.-H
HO))1 sõN o =
SPI0011
Propofol Boc-L-proline
3. SPI0013
0 a)40 0 01,\"---0 b) o CI-
OH 0 õ1\1744
101
HONI-1 9 ( 0,1L(NH 0 H
/---\ A ><
ANOci-
SPI0013
Propofol Boc-L-lysine H HH
SCHEME
Synthesis of the glycine, L-proline, and L-lysine esters of Propofol: a) EDC,
DMAP, CH2C12; b) HC1 (g), Et20.
Experimental Section:
The synthesis of SPI0010, SPI0011 and SPI0013 were conducted in batches.
Generally
a small-scale experiment was performed first followed by a larger batch.
Reagents
mentioned in the experimental section were purchased at the highest obtainable
purity
from Aldrich, Acros, or Bachem, except for solvents, which were purchased from
either
Fisher Scientific or Mallinlcrodt.
64
H:\work\1652\17690\SPEC\17690.spec without clafinsILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
1) SPI0010
Propofol (9.98 g, 55.97 mmole) was dissolved in dichloromethane (200 mL) at
room
temperature, under an argon atmosphere. N-t-Butyloxocarbonyl-glycine (11.2 g,
63.91
mmole) was added along with 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide,
hydrochloride (EDC, 11.1g, 57.9 mmole) and 4-(N,N-dimethylamino)-pyridine
(DMAP,
1.5 g, 12.27 mmole). After stirring for 21 hours under an argon atmosphere at
room
temperature, water (200 mL) was added and the layers were separated. The
dichloromethane layer was washed again with water (200 mL) and dried for 1
hour over
sodium sulfate (5 g). After filtration and concentration under reduced
pressure, the
remaining oil was purified by flash chromatography on silica gel (250 g),
eluting with
hexanes/ethyl acetate (10:1). The procedure generated the protected N-BOC
protected
glycine ester of Propofol as a white solid (11.34g, 60% yield).
tert-Butoxycarbonylamino-acetic acid 2,6-diisopropylphenyl ester:
111 NMR (300 MHz, CDC13): 6 = 7.25-7.13 (m, 3H), 5.18 (br s, 1H), 4.22 (d, 2H,
J= 5.7
Hz), 2.89 (m, 2H), 1.46 (s, 9H), 1.18 (d, 12H, Jr= 6.9 Hz).
13C NMR (75 MHz, CDC13): = 169.35, 155.75, 145.22, 140.35, 126.90, 124.14,
80.32,
42.66, 28.54, 27.79, 23.57.
The Propofol-Boc-gycine ester (11.28 g, 33.6 mmole) was dissolved in anhydrous
diethyl ether (200 mL) at room temperature. Hydrochloric acid (gas) was passed
through the solution for 45 minutes while stirring. The mixture was allowed to
stir at
rooin temperature for 48 hours under an argon atmosphere. After 48 hours
hexanes (200
mL) were added and the precipitate was filtered. The white solid was dried
under high
FlAwork\1652\17690\SPEC\17690.spec without clahnsil.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
vacuum for 5 hours at 88 C. The experiment produced SPI0010 (8.73 g, 95%
yield,
purity 99.9% by HPLC) as a white solid.
CI
H
\N H
0
SPI0010
Amino-acetic acid 2,6-diisopropyl-phenyl ester, hydrochloride:
1HNMR (300 MHz, CDC13): 6 = 8.77 (br s, 3H), 7.20-7.08 (m, 3H), 4.14 (m, 2H),
2.87
(m, 2H), 1.11 (d, 12H, J= 7 Hz).
13C NMR (75 MHz, CDC13): 6 =166.42, 144.84, 140.42, 127.10, 124.06, 40.47,
27.61,
23.55.
CHN analysis:
cafe.: C 61.87, H 8.16, N 5.15; found: C 61.14, H 8.20, N 5.14.
2) SPI0011
Propofol (10.03 g, 56.23 mmole) was dissolved in dichloromethane (100 mL) at
room
temperature, under an argon atmosphere. N-t-Butyloxocarbonyl-L-proline (14.04
g,
65.22 mmole) was added along with 1-(3-dimethylaminopropy1)-3-
ethylcarbodiimide,
hydrochloride (EDC, 11.95 g, 62.33 mmole) and 4-(N,N-dimethylamino)-pyridine
(DMAP, 1.1 g, 9.0 mmole). After stirring for 3 hours under an argon atmosphere
at
room temperature, water (100 mL) was added and the layers were separated. The
dichloromethane layer was washed again with water (100 mL) and dried for 1
hour over
sodium sulfate (5 g). After filtration and concentration under reduced
pressure, the
remaining oil was purified by flash chromatography on silica gel (250 g),
eluting with
hexanes/ethyl acetate (10:1). The procedure generated the protected N-BOC
protected
66
HAwork\1652\17690\SPEC\17690.spec without clairnadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
L-proline ester of Propofol as a clear oil (11.34g, 66% yield) that solidified
on standing
in the freezer.
0 ==
Pynolidine-1,2-dicarboxylic acid 1-tert-butyl ester 2-(2,6-diisopropyl-phenyl)
ester:
NMR (300 MHz, CDC13): 8 = 7.31-7.20 (m, 3H), 4.73 (m, 1H), 3.70-3.50 (m, 2H),
3.20-2.94 (m, 2H), 2.46-2.20 (m, 2H), 2.20-2.0 (m, 2H), 1.55 (m, 9H), 1.25 (m,
12H).
13C NMR (75 MHz, CDC13): ö = 171.87, 171.01, 154.34, 153.93, 145.35 145.23,
140.06,
140.21, 126.69, 126.53, 123.95, 80.28, 79.89, 59.14, 46.67, 46.42, 31.10,
30.17, 28.61,
28.56, 28.56, 27.44, 27.18, 23.47.
The Propofol-Boc-L-proline ester (13.95 g, 37.14 mmole) was dissolved in
anhydrous
diethyl ether (100 mL) at room temperature. Hydrochloric acid (gas) was passed
through the solution for 60 minutes while stirring. The mixture was allowed to
stir at
room temperature for 22 hours under an argon atmosphere. After 22 hours
hexanes (50
mL) were added and the precipitate was filtered. The white solid was dried
under high
vacuum for 5 hours at 88 C. The experiment produced SPI0011 (9.1 g, 81%
yield,
purity 99.1% by HPLC) as a white solid.
CI-
O 0
oç
SPI0011
Pyrrolidine-2(S)-carboxylic acid 2,6-diisopropyl-phenyl ester, hydrochloride:
67
11:\work\1652\17690\SPEC\17690.spec without clahnsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
11-INMR (300 MHz, CDC13): 5 = 10.15 (br s, 2H), 7.27-7.14 (m, 3H), 4.78 (t,
1H, J= 7.8
Hz), 3.56 (m, 2H), 2.85 (m, 2H), 2.64 (m, 1H), 2.40 (m, 1H), 2.20 (m, 1H),
2.05 (m,
1H), 1.18 (m, 12H).
13C NMR (75 MHz, CDC13): 5 = 168.30, 144.23, 139.74, 126.98, 123.96, 51.58,
38.21,
29.32, 26.64, 26.18, 23.71, 23.02, 21.67.
CHN analysis:
calc.: C 65.48, H 8.40, N 4.49; found: C 65.50, H 8.43, N 4.50.
3) SPI0013
The dicyclohexylamine salt of di-N-boc-L-lysine (23.62 g, 0.0447 mole) was
added to
diethyl ether (200 mL) and potassium hydrogen sulfate (9.14 g) in water (200
mL) that
was cooled in an ice/water bath. After strining for 20 minutes, the layers
were
separated. The ether layer was extracted three times with cold water (100 mL).
The
ether layer was then dried over sodium sulfate (15 g) for one hour, filtered,
and
concentrated under reduced pressure. The procedure generated the free acid of
N,N'- di-
boc-L-lysine (15.5 g, 100% recovery).
Propofol (8.0 g, 45 mmole) was dissolved in dichloromethane (100 mL) at room
temperature, under an argon atmosphere. N, N'-di-t-Butyloxocarbonyl-L-lysine
(15.5 g,
44.7 mmole) was added along with 1-(3-dimethylaminopropy1)-3-
ethylcarbodiimide,
hydrochloride (EDC, 8.62 g, 45 mmole) and 4-(N,N-dimethylamino)-pyridine
(DMAP,
0.55 g, 4.5 mmole). After stirring for 3 hours under an argon atmosphere at
room
temperature, water (100 mL) was added and the layers were separated. The
dichloromethane layer was washed again with water (100 mL) and dried for 1
hour over
sodium sulfate (5 g). After filtration and concentration under reduced
pressure, the
remaining oil was purified by flash chromatography on silica gel (250 g),
eluting with
hexanes/ethyl acetate (9:1). The procedure generated the protected N-BOC
protected L-
lysine ester of Propofol as a white foam (12.42 g, 55% yield).
68
FlAwork\1652\17690\SPEC\17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
J 1;i
O
o r'NÄ(X
2(S),6-Bis-t-butoxycarbonylarnino-hexanoic acid 2,6-diisopropyl-phenyl ester:
NMR (300 MHz, CDC13): S = 7.28-7.15 (m, 3H), 5.22 (d, 1H, J= 8.4 Hz), 4.70 (m,
1H), 4.59 (m, 1H), 3.17 (m, 211), 2.93 (m, 2H), 2.09 (m, 1H), 1.86 (m, 1H),
1.67-1.54
(m, 411), 1.48 (s, 9H), 1.46 (s, 9H), 1.20 (m, 12H).
13C NMR (75 MHz, CDC13): 5 = 171.82, 156.10, 155.65, 145.25, 140.30, 126.80,
124.03, 80.14, 79.28, 53.76, 40.29, 32.09, 28.66, 28.54, 27.48, 23.91, 23.10.
The Propofol-di-Boc-L-lysine ester (12.34 g, 24.37 mmole) was dissolved in
anhydrous
diethyl ether (250 mL) at room temperature. Hydrochloric acid (gas) was passed
through the solution for 60 minutes while stirring and cooling in an ice/water
bath. The
mixture was allowed to stir at room temperature for 48 hours under an argon
atmosphere. After 48 hours the precipitate was filtered and crystallized from
ethanol
(100 mL). The white solid was dried under high vacuum for 4 hours at 90 C.
The
experiment produced SPI0013 (5.5 g, 60% yield, purity 98.6% by HPLC) as a
white
solid.
H CI
0)0 \ H
sµr\L¨H
\--_\ CI¨
SP10013
,N¨H
H I
2(S),6-Diamino-hexanoic acid 2,6-diisopropyl-phenyl ester, dihydrochloride:
69
\work\1652 \17690 \SPEC \17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
IIINMR (300 MHz, CDC13): = 9.05 (br s, 3H), 8.35 (br s, 3H), 7.26-7.13 (m,
3H),
4.43 (t, 1H, .1= 6 Hz), 3.0-2.6 (m, 4H), 2.09 (m, 2H), 1.80-1.50 (m, 4H), 1.10
(d, 12H, J=
7 Hz).
13C NMR (75 MHz, CDC13): 8 = 168.30, 144.23, 139.74, 126.98, 123.96, 51.58,
38.21,
29.32, 26.64, 23.71, 23.02, 21.67.
CHN analysis:
calc.: C 56.99, H 8.50, N 7.38; found: C 56.48, H 8.56, N 7.30.
II. PRO DRUGS OF NON-STEROIDAL ANTI-INFLAMMATORY DRUGS
(NSAIDs)
The NSAIDs comprise a class of structurally distinctive, carboxylic acid
moiety attached
to a planar aromatic functionality, Examples include: acetyl salicyclic acid,
salicyclic
acid, diflunisal, ibuprofen, fenoprofen, carprofen, flurbiprofen, ketoprofen,
naproxen,
sulindac, indomethacin, etodolac, tolmetin, ketorolac, diclofenac, and
meclofenamate.
The NSADIs posess anti-inflammatory, analgesic, antipyretic and anti-clotting
activity.
Examples of the chemical structures of this uniques class of compounds showing
wide
variety of pharmacological activities are shown below.
HAwor1\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0 all: F
cH3 0
0OH 00 101 . ,
411,
OH OH
OH ,CH3
Salicylic Acid 8 011 cil3 c.,
Ibuprofen
Acetylsalicylic Acid Diflunisal
CH3 0H CH3 0 CH3 0
P
Oli 01 ,11 _ 011 OH *a
= 1.11 illin
Fenoprofen Carprofen
Flurbiprofen
o ca 3 o CH3 0 0
i illt \
oso
OH OS OH "Ilki N 0 is .
c . . c.,.Ø
Ketoprofen Naproxen Etodolac
ct o 0
o
F Cal 01.1 ato OH
OH
0111111 40 \- =OH
_CHs
11
/
ofe0
Cfil....
0 CH.3
Sulindac Indomethacin Tolmetin
Ketorolac
NSAIDs are widely used for the treatment of acute and chronic pain, management
of
edema, tissue damage resulting from inflammatory joint diseases and also,
effective anti-
clotting agents in the treatment of myocardial infraction. A number of the
agents also
possess antipyretic activity in addition to analgesic and anti-inflammatory
action, thus
useful in reducing fever.
Some drugs in the above group have also been prescribed for Rheumatoid
Arthritis,
Osteoarthritis, acute gout, ankolysing spondylitis, and dysmenorrhea.
71
H: \ work\1652 \ 17690 \ SPEC \ 17690.spe,c without clainisll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Mechanism of Action:
The major mechanism by which the NSAIDs produce their therapeutic effect is
via
inhibition of prostaglandin synthesis. Specifically NSAIDs inhibit
cyclooxygenases,
such as COX-1 and COX-2 enzymes, where these two enzymes are responsible for
synthesis of prostaglandins. While COX-1 enzyme is important for the
regulation of
platelet aggregation, regulation of blood flow in kidney and stomach, and
regulation of
gastric acid secretion, COX-2 enzyme plays an important role in the pain and
inflammatory processes. NSAIDs significantly increase clotting time and can be
used for
prophylaxis of thromboembolism and myocardial infarction.
All NSAlDs are relatively medium to strong organic acids with pKa's in the 3-6
range.
Most of them are carboxylic acid derivatives. Acidic group is essential for
COX
inhibitory activity and in physiological pH, all the NSAIDs are ionized. All
of them have
quite varying hydrophilic lipophilic balance, and these are functions of their
aryl,
aromatic and aliphatic side chains and other heterocyclic variations in their
structures.
Most of the NSAIDs are highly bound to plasma proteins and often competitively
replace other drugs which have similar affinity for plasma proteins. Hence
concomitant
administration of NSAIDs with other therapeutic class must be carefully
evaluated to
prevent drug interactions. Most of the drugs, due to acidic carboxyl group are
metabolized by the manunals via conjugation. The major pathway of metabolic
clearance of a number of NSAIDs is glucuronidation followed by renal
elimination.
Use of acetylsalicylic acid (aspirin) in the prophylaxis of coronary heart
diseases is now
well known, and this drug has proved to be a lifesaver for a number of
patients with
myocardial infarction. Several additional uses have already been documents for
aspirin,
for example, it was recently reported in the medical journal Lancet (Vol 349,
p 1641)
that aspirin reduces the risk of stroke in patients with early warning signs
of transient
ischemic heart attacks. Pre-eclampsia and fetal growth retardation, both
caused by
blockages of the blood vessels of the placenta, are two of the commonest
complications
of pregnancy ¨ there are millions of cases of pre-eclampsia in the world each
year. In a
72
FlAwork\1652\17690\SPEC\17690.spec without claims11.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
trial involving more than 9000 women in 16 countries, a daily dose of 60mg
aspirin
reduced the risk of pre-eclampsia by 13 per cent. (Aspirin Foundation
website). Aspirin
has also been shown to be effective in some studies to prevent colon cancer,
lung cancer
and pancreatic cancer in post-menopausal women. Since aspirin can improve
blood flow,
its usefulness in the treatment of diabetes certain forms of dementia such as
Alzheimer's
disease are becoming increasingly clear.
Because of their unique pharmaceutical potential, the NSAIDs have attracted
considerable attention in the press. The primary area of clinical
investigation for above
drugs has been as non-steroidal anti-inflammatory agents, in particular in
relation to their
application to patients suffering from pain, arthritis, (Rheumatoid and Osteo)
other
inflammatory reactions, fever and for the prophylaxis of coronary heart
diseases. These
drugs are also used in the treatment of migraine headache, menstrual
syndromes, back
pain and gout.
Despite the very major contribution which NSAIDs have made, difficulties have
been
encountered in providing more effective and convenient means of administration
(e.g.,
galenic formulations, for example, oral dosage form, which are both convenient
and for
the patient as well as providing appropriate bioavailability and allowing
dosaging at an
appropriate and controlled dosage rate) as well as the reported occurrence of
undesirable
side reactions; in particular severe gastric and duodenal ulcers, mucosal
erythema, and
edema, erosions, perforations, blood in stool, ulcerative colitis have been
obvious serious
impediments to their wider use or application. The dual injury theory involves
NSAID-
mediated direct damage, followed by a systemic effect in which prostaglandin
synthesis
is inhibited. Topical injury may also occur as a result of the biliary
excretion of active
hepatic metabolites and subsequent duodenogastric reflux. (Arthritis and
Rheumatism
1995; 38(1):5-18) The effects are additive; either topical or systemic
mechanisms alone
are sufficient to produce gastro duodenal mucosal damage.
73
H:\work\1652 \17690 \SPEC \17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Moreover, the above mentioned NSAIDs are characteristically highly hydrophobic
and
readily precipitate in the presence of even very minor amounts of water, e.g.,
on contact
with the body (e.g., stomach fluids). It is accordingly extremely difficult to
provide e.g.,
oral formulations which are acceptable to the patient in terms of form and
taste, which
are stable on storage and which can be administered on a regular basis to
provide
suitable and controlling patient dosaging.
Proposed liquid formulations, e.g., for oral administration of NSAIDs, have
heretofore
been based primarily on the use of natural gums, like Xanthan, cellulose,
citric acid, and
lime flavor etc. See e.g., U.S. Patent NO. 5,780,046. Commercially available
NSAIDs
drink-solution employs incompatible orange color and berry flavor, citric
acid, Xanthan
Gum, polysorbate 80, pregelatinized starch, glycerin, sodium benzoate, and
additional
artificial colors and flavors. Use of the drink solution and similar
composition as
proposed in the art is however accompanied by a variety of difficulties.
Further, the palatability of the known oil based system has proved
problematic. The
taste of the known drink-solution is, in particular, unpleasant. Admixture
with an
appropriate flavored drink, for example, chocolate drink preparation, at high
dilution
immediately prior to ingestion has generally been practiced in order to make
regular
therapy at all acceptable. Adoption of oil based systems has also required the
use of
high ethanol concentrations to itself inherently undesirable, in particular
where
administration to children is forseen. In addition, evaporation of the
ethanol, e.g., from
capsules (adopted in large part, to meet problems of palatability, as
discussed or other
forms (e.g., when opened) results in the development of a NSAID precipitate.
Where
such compositions are presented in, for example, soft gelatin encapsulated
form; this
particular difficulty necessitates packaging of the encapsulated product in an
air-tight
component, for example, an air-tight blister or aluminum-foil blister package.
This in
turn renders the product both bulky and more expensive to produce. The storage
characteristics of the aforesaid formulations are, in addition, far from
ideal.
74
flAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Gastric irritability of the NSAIDs has been a topic of great concern to the
practicing
physicians and as well as patients. Acute uses of aspirin, fenoprofen,
flurbiprofen,
indomethacin, ketorolac, meclofenamate, mefanamic acid, and piroxicam produce
serious GI side effects. Even Ibuprofen is shown to cause severe gastric
lesions upon
long term use. Gastrointestinal toxicity is the most frequently encountered
side effect
associated with NSAIDs and presents considerable concern. Approximately one
half of
all hospital admissions for a bleeding ulcer are attributed to the use of
NSAIDs, aspirin,
or the two taken in combination during the week prior to admission. (Faulkner
G,
Prichard P, Somerville K, et al. Aspirin and bleeding peptic ulcers in the
elderly. Br Med
J. 1988; 297:1311-1313). A survey of Tennessee Medicaid patients who were
hospitalized with GI complications showed that patients who used NSAIDs had
approximately a fourfold greater risk for developing GI hemorrhage or peptic
ulcer
disease than patients not taking NSAIDs. (Griffin MR, Piper JM, Daugherty JR,
et al.
Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer
disease in
elderly persons. Ann Intern Med. 1991; 114:257-263). Serious GI events,
according to
the FDA, occur in as many as 2% to 4% of patients per year who are taking
continuous
NSAID therapy for rheumatoid arthritis. The relative risk of gastric ulcer
(4.725),
duodenal ulcer (1.1 to 1.6), bleeding (3.8), perforation, and death are all
increased by
NSAID use when such patients are compared to those who do not take these
products. In
1989, patients with rheumatoid arthritis had approximately 20,000
hospitalizations per
year with an estimated cost of $10,000 per stay. (Fries JF, Miller SR, Spitz
PW, et al.
Toward an epidemiology of gastropathy associated with nonsteroidal anti-
inflammatory
drug use. J Gastroenterology. 1989; 96:647-655).
There is also a need for providing some of the NSAIDs in a water soluble form
for
injection. It is well known that high concentrations of alcohol and
tromethamine used to
form a salt in the current formulations of Ketorolac are toxic. At present
there is no
formulation that would allow the NSAIDs to be in aqueous solution at the
concentrations
needed due to poor water solubility of the drug.
flAwork\1652\17690\SPEC\17690.spec without clahnsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Beyond all these very evident practical difficulties lies the occurrence of
undesirable
side reactions already alluded to, observed employing available oral dosage
forms.
Several proposals to meet these various problems have been suggested in the
art,
including both solid and liquid oral dosage forms. An overriding difficulty
which has
however remained is the inherent insolubility of the NSAIDs in aqueous media,
hence
preventing the use of a dosage form which can contain NSAIDs in sufficiently
high
concentration to permit convenient use and yet meet the required criteria in
ten-ns of
bioavailability, e.g. enabling effective resorption from the stomach or gut
lumen and
achievement of consistent and appropriately high blood/blood-serum levels.
The present prodrugs of NSAIDs overcome the problems described hereinabove.
More
specifically, an embodiment of the present invention is directed to a prodrug
of NSAID
which significantly enhances its solubility in aqueous solutions, thereby
avoiding the
need to utilize a carrier, such as ethanol or castor oil when administered as
a solution.
Moreover, the prodrugs of NSAID, in accordance with the present invention, do
not
exhibit the side effects of the prior art formulations. Further, the prodrugs
of the present
invention are almost completely devoid of gastric irritability upon oral
administration,
thereby enhancing significantly the therapeutic index of the prodrugs tested
and their
efficacy.
Accordingly, in one aspect, the present invention is directed to a prodrug of
NSAIDs.
The preferred prodrugs of the NSAIDs have the formula
76
II: \work\ 1652\17690 \SPEC\ 17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
0 F
a Y 41 Y Mr 0 cE3 0
CH3 * i
OH
Salicylic Acid 6 - OH cH3
Ibuprofen
Acetylsalicylic Acid Diflunisal
cs3 0 a CH3 0 CH3 0
Y /Y P ailh Olt
01110 Y
Fenoprofen Carprofen Flurbiprofen
0 cH3. 0 CE3 0 0
i \
40 . Y 40* Y IS N
I o
c s H Y
CH3
CHICH:f
Ketoprofen Naproxen Etodolac
0 o o
o
F Y .0130 0* alit, Y cal
1,11 \ Y -- ii-c213 ccril% Y
I
,c2)iir0 N
0
411 0
Cr23,,
4, Cl
0 Cfis
Sulindac Indomethacin Tolmetin Ketorolac
or pharmaceutically acceptable salts thereof; wherein Y is either NH-AA or 0-
AA and
AA is an amino acid, in which either an amine group or the hydroxyl group of
AA is
reacted with the carboxylic acid group of the NSAIDs.
The present invention is also directed to a pharmaceutical composition
comprising a
therapeutically effective amount of the various NSAIDs above and a
pharmaceutical
carrier therefor.
77
11:\work\1652\17690\SPEC\17690.spec without clainisILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
In another embodiment, the present invention is directed to a method of
treating a patient
in need of NSAID therapy, which method comprises administering to said patient
an
effective amount of the NSAIDs.
In a further embodiment, the present invention is directed to a method of
enhancing the
solubility of NSAID in an aqueous solution comprising reacting the carboxyl
functionality of each of the NSAIDs and isolating the products thereof.
In a still further embodiment, the present invention is directed to a method
of
substantially and in a therapeutically efficacious manner, reducing or
eliminating the
gastric mucosal damage of NSAIDs when administered to a patient which
comprises
reacting the carboxyl functionality of each of the NSAID molecule with either
amine or
hydroxyl function of selected amino acids to form either an amide or ester
covalent bond
respectively and isolating the product thereof and administering said product
to the
patient.
A. Synthesis of Ibuprofen Amino Acid Derivaties
Overview:
The procedure for the synthesis of the L-serine, L-threonine, and L-
hydroxyproline
esters of Ibuprofen is outlined in Synthetic Sequence section. The complete
procedure
and analytical data is given in the Experimental Section. Again, these
synthetic
schemes are exemplary. The scheme is applicable for other amino acids in the
preparation of the NSAID prodrugs of the present invention. In general, ( )-
Ibuprofen
(4-10 g, in batches) was coupled with the N-benzyloxy/benzyl ester protected
amino
acids (1 equivalent) with 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide,
hydrochloride (EDC, 1 equivalent) in the presence of a catalytic amount of 4-
(N,N-
dimethyamino)-pyridine (DMAP). Once the reactions were complete, any excess
EDC
was removed by extraction with water, DMAP was removed by extraction with
dilute
acid, and Ibuprofen was removed by extraction with sodium bicarbonate. After
drying
over sodium sulfate, filtration, and concentration the crude protected amino
acid esters
78
1-1:\work\1652\17690\SPEC\17690.spec without claiinsIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
of ( )-Ibuprofen were either used directly or purified by flash chromatography
on silica
gel to generate the protected esters in good yield (85-95%). The column
chrornatogaphy
was generally not necessary if a slight excess of Ibuprofen and coupling agent
were
used, and a thorough extraction procedure was conducted. The protecting graups
were
removed by hydrogenation (25-35 psi H2) in the presence of 10% palladium on
carbon
and hydrochloric acid. Yields for the deprotection step ranged from 70-90%.
After
filtration and drying the hydrochloride salts of the serine and threonine
esters of ( )-
Ibuprofen were purified by crystallization. The hydrochloride salt of the L-
hydroxyproline-lbuprofen ester was a gel that would not solidify/crystallize.
In this case
the hydrogenation was repeated without the use of acid and the neutral
compound was
purified.
Because the Ibuprofen started as a mixture of enantiomers, the final products
were
delivered as a mixture of diastereomers except for the threonine ester. In the
case of the
threonine ester of Ibuprofen, washing with water, acetone or acetonitrile
could readily
separate the final diastereomeric salts. The insoluble isomer (SPI0016A) was
determined to be the active isomer by comparison with an authentic standard
prepared
from S-(+)-Ibuprofen. The serine and hydroxyproline esters of ( )-Ibuproferi
could not
be readily separated in this fashion.
79
HAwork\1652\17690\SPEC\17690.spec without claimsIl.doc
CA 02534342 2006-01-30
PCT/US2004/024901
WO 2005/046575
Synthetic Sequence:
1. SPI0015
0 o a) 0
b)
1101 OH õ..---rkObz ----*" = 0 401 0:
il
HO
+ HAls-cbz \¨clObz OH
H,N1,--cbz
14 I H CI
-
I-I
( )-Ibuprofen Z-Ser-OBz SPI001501 SPI0015
2. SPI0016A and SPI0016B
0 o 0 o
%
0
0õ.\ ____________________________________________ CILObz 0.3---
(ILOH
a)
, b)
110 OH :3--rit'Obz ---÷. ,..N-- =. ,N,
H cuz H I H
HO
* li H
,NI-.. b
H c z CI-
( )-Ibuprofen Z-Thr-OBz SPI001601 SPI0016
c)/
o 0 0
..)..._11
OH OH
0
0 ,N H I H
Ark H. +
I H H
VW H
CI-Cl 41 SPI0016A SPI0016B
3. SPI0017
o 0
0 OH HO,,,,Cf 0
'Obz --""a) o d)
=410 0
04,0)...0bz
+
`cbz
`cbz H
( )-Ibuprofen Z-Hyp-OBz SPI001701
SPI0017
Synthesis of the L-serine, L-threonine, and L-hydroxyproline esters of
( )-Ibuprofen: a) EDC, DMAP, CH2C12; b) HC1, 10% Pd/C, Et0H c) acetone,
d) 10% Pd/C, Et0H.
flAwork\1652\17690\SPEC\17690.spec without claiinsIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Experimental Section:
The synthesis of SPI0015, SPI0016 and SPI0017 were conducted in two or three
batches. Reagents mentioned in the experimental section were purchased at the
highest
obtainable purity from Sigma-Aldrich, Acros, or Bachem, except for solvents,
which
were purchased from either Fisher Scientific or Mallinkrodt.
1) Preparation of ( )-Ibuprofen-L-serine ester, hydrochloride (SPI0015).
( )-Ibuprofen (5.04 g, 24.4 mmole), N-carbobenzyloxy-L-serine benzyl ester
(8.11 g,
24.6 mmole), 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide, hydrochloride
(EDC,
4.87g, 25.4 mmole), and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.40 g, 3.27
mmole)
were dissolved in dichloromethane (150 mL) at room temperature, under an argon
atmosphere. After stirring for 22 hours under an argon atmosphere at room
temperature, water (100 mL) was added and the layers were separated. The
dichloromethane layer was washed again with water (100 mL) and dried for 1
hour over
sodium sulfate (5 g). After filtration and concentration under reduced
pressure, the
remaining oil was purified by flash chromatography on silica gel (250 g),
eluting with
hexanes/ethyl acetate (3:1). The procedure generated the protected L-serine-(
)-
Ibuprofen ester (SPI001501) as a colorless solid (11.4 g, 90% yield).
= o
Obz
HNCbZ
SPI001501
2(S)-Benzyloxycarbonylamino-342(R,S)-(4-isobutyl-pheny1)-propionyloxy]-
propionic
acid benzyl ester:
1H NMR (300 MHz, CDC13): 5 = 7.40-7.20 (m, 10H), 7.14-7.01 (m, 4H), 5.50 (d,
V2H,
J= 8.4 Hz), 5.29 (d, V2H, J= 8.4 Hz), 5.11-5.02 (m, 2.5H), 4.90 (d, 1/2H, ,T=
12 Hz), 4.62
81
HAwor1\1652\17690\SPEC\17690.speo without clainasII.doc
CA 02534342 2013-04-18
,
WO 2005/046575
PCT/US2004/024901
(m, 1H), 4.49-4.43 (m, 1H), 4.36-4.32 (m, 1H), 3.59 (m, 1H), 2.39-2.35 (m,
2H), 1.78
(m, 1H), 1.42-1.39 (m, 3H), 0.85 (d, 6H, J= 6.6 Hz).
=
13C NMR (75 MHz, CDC13): 8 = 174.05, 169.19, 169.07, 155.68, 140.73, 137.20,
136.12, 135.05, 134.91, 129.44, 128.67, 128.65, 128.60, 128.41, 128.33,
128.30, 128.19,
127.19, 127.16, 67.75, 67.32, 64.51, 64.32, 53.71, 45.16, 45.02, 30.35, 22.60,
18.27.
The protected Ibuprofen-L-serine ester (22.50 g, 43.4 mmole) was dissolved in
ethanol
(200 mL) at room temperature and added to a Parr bottle that contained 10%
palladium
on carbon (3.86 g, 50% wet) under a nitrogen atmosphere. Hydrochloric acid (10
mL
37% HC1 in 30 mL water) was added and the nitrogen atmosphere was replaced
with
hydrogen gas (25 psi). After 4 hours of shaking, the palladium catalyst was
removed by
filtration through celite. The ethanol/water was removed under reduced
pressure. The
remaining white solids were washed with water (25 mL), acetone (20 mL) and
dried
under high vacuum (4 hours at 88 C). The experiment produced ( )-Ibuprofen-L-
sarine
ester, hydrochloride SPI0015 (11.3 g, 80% yield) as a colorless solid.
O
\¨rit0H
H H ¨
H CI
SPI0015
2(S)-Amino-342(R,S)-(4-isobutylpheny1)-propionyloxy)-propionic acid,
hydrochloride;
((R,S)-Ibuprofen-L-Serine ester, hydrochloride):
NMR (300 MHz, DMS0): 8 = 8.92 (br s, 3H), 7.22 (t, 2H, J= 7.5 Hz), 7.10 (d,
2H,
J= 7.5 Hz), 4.56 (in, 1H), 4.37-4.20 (m, 211), 3.83 (q, 1H, J= 6.9 Hz), 2.41
(d, 2H, J= 6.9
Hz), 1.80 (m, 1H), 1.41 (d, 3H, J= 6.9 Hz), 0.85 (d, 611, J= 6.9 Hz).
*Trade-mark
82
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
13C NMR (75 MHz, DMS0): 6 = 173.36, 173.32, 168.08, 168.04, 139.70, 128.96,
129.92, 127.20, 127.05, 62.47, 51.59, 51.49, 44.28, 44.00, 43.90, 29.68,
22.28, 18.70,
18.42.
HPLC analysis:
99.13% purity; rt = 3.133 min; Luna C18 5u column (sn 167917-13); 4.6x250 mm;
254
nm; 50% ACN/50% TFA buffer (0.1%); 35 C; 20 ul inj.; lml/min; 1 mg/mL sample
size; sample dissolved in mobile phase.
CHN analysis:
cale.: C 58.27, H 7.33, N 4.25; found: C 58.44, H 7.46, N 4.25.
Melting point: 169.5 - 170.5 C
2a) Preparation and Separation of ( )-Ibuprofen-L-threonine ester,
hydrochloride
(SPI0016A and SPI0016B).
( )-Ibuprofen (4.15 g, 20.11 mmole), N-carbobenzyloxy-L-threonine benzyl ester
(6.90
g, 20.11 mmole), 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide, hydrochloride
(EDC,
3.95 g, 20.6 mmole), and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.25 g, 2.0
mmole)
were dissolved in dichloromethane (50 mL) at room temperature, under an argon
atmosphere. After stirring for 19 hours, the dichloromethane layer was washed
with
water (50 mL), 5% hydrochloric acid (2x25 mL), water (25 mL), saturated sodium
bicarbonate (2x25 mL), and water (50 mL). After drying for one hour over
sodium
sulfate (5 g), filtration, and concentration under reduced pressure, the
remaining oil was
used without further purification. The procedure generated the protected L-
threonine-
( )-Ibuprofen ester (SPI001601) as a light yellow oil (10.2 g, 95.3% yield),
which
solidified on standing.
83
H:\work\1652\17690\SPEC\17690.spec without clahnsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
0 0
H
N
SPI001601 lp
2(S)-Benzyloxycarbonylamino-342(R,S)-(4-isobutyl-pheny1)-propionyloxykbutyric
acid benzyl ester:
1IINMR (300 MHz, CDC13): = 7.40-7.15 (m, 10H), 7.14-7.01 (m, 4H), 5.48-5.25
(m,
2H), 5.11-5.01 (m, 3H), 4.90 (d, 1/2H, J= 12 Hz), 4.68 (d, 1/2H, J= 12 Hz),
4.48 (m, 1H),
3.60-3.48 (m, 1H), 2.39(m,.2H), 1.79 (m, 1H), 1.42-1.35 (m, 3H), 1.27 (d, 1.5
H, J= 6.6
Hz), 1.17 (d, 1.5 H, J= 6.6 Hz), 0.85 (m, 6 H).
13C NMR (75 MHz, CDC13): 8 = 173.32, 169.70, 169.30, 156.55, 140.75, 137.38,
137.22, 136.14, 135.07, 134.99, 129.45, 129.41, 128.65, 128.39, 128.22,
127.21, 127.14,
70.97, 70.70, 67.81, 67.66, 67.53, 57.83, 45.19, 30.39, 22.61, 18.57, 18.30,
17.18, 16.87.
The protected Ibuprofen-L-threonine ester (10.15 g, 19.0 mmole) was dissolved
in warm
ethanol (150 mL) and added to a Parr bottle that contained 10% palladium on
carbon
(3.4 g, 50% wet) under a nitrogen atmosphere. Hydrochloric acid (6 mL 37% HC1
in 20
mL water) was added and the nitrogen atmosphere was replaced with hydrogen gas
(30
psi). After 3 hours of shaking, the palladium catalyst was removed by
filtration through
celite (30 g). The ethanol/water was removed under reduced pressure. The
experiment
produced ( )-Ibuprofen-L-threonine ester, hydrochloride (SPI0016A and
SPI0016B, 6.4
g, 97% crude yield) as a colorless solid. The crude mixture of diastereomers
was stirred
in acetone (200 mL) for 2 hours at room temperature under an argon atmosphere.
After
2 hours the solids (2.84 g, SPI0016A) were filtered. The filtrate (SPI0016B,
3.0 g) was
concentrated under reduced pressure.
84
HAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
1.) Purification of SPI0016A (active isomer):
After 3 batches of the S-Ibuprofen-L-threonine ester (SPI0016A) had been
completed,
the batches were combined (8.78 g total) and crystallized three times from
DIUF water
(100 mL). Each time a small amount of zwitterion was generated. In order to
regenerate
the salt, the solid generated (from each crystallization) was dissolved in 1%
hydrochloric
acid in ethanol (3 mL 37% hydrochloric acid in 100 mL ethanol). The ethanol
solution
was then concentrated under reduced pressure at room temperature. After the
third
crystallization and regeneration procedure, the salt (5.6 g) was stirred in
acetonitrile (100
mL) for 44 hours at room temperature, under an argon atmosphere. The salt was
then
filtered and dried under high vacuum at 50-55 until the weight was constant
(5.5 g).
0
N, OH
HI H
Cl-
SPI0016A
2(S)-Amino-3(R)42(S)-(4-isobutyl-phenyl)-propionyloxy]-butyric acid;
(S-Ibuprofen-L-threonine ester, hydrochloride, active isomer):
1H NMR (300 MHz, DMS0): 8 = 8.76 (br s, 3H), 7.19 (d, 211, J= 8.1 Hz), 7.11
(d, 2H,
J= 8.1 Hz), 5.28 (dq, 1H, J= 6.3, 3.6 Hz), 4.14 (q, 1H, J= 3.6 Hz), 3.80 (q,
1H, J= 7.2
Hz), 2.41 (d, 2H, J= 7.2 Hz), 1.80 (m, 1H), 1.37 (d, 3H, J= 7.2 Hz), 1.21 (d,
3H, J= 6.3
Hz), 0.85 (d, 6H, J= 6.6 Hz).
13C NMR (75 MHz, DMS0): 8 = 172.66, 168.24, 139.68, 137.24, 128.95, 126.97,
67.98,
55.35, 44.23, 43.83, 29.66, 22.24, 18.52, 16.47.
CHN analysis:
calc.: C 59.38, H 7.62, N 4.07; found: C 59.17, H 7.63, N 4.04.
FlAwork\1652 \I7690 \SPEC \17690,spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
HPLC analysis:
98.28% purity; r.t.= 6.951 min.; 60% TFA (0.1%)/40% acetonitaile; 1 mL/min;
37.5 C;
Luna C18, 3u column (SN 167917-13), 4.6x250 mm; 22 ul injection.
Optical rotation: + 24.5
Melting Point: 189-190 C
2) Purification of SPI0016B (inactive isomer):
After 3 batches of the R-Ibuprofen-L-threonine ester (SPI0016B) had been
completed,
the batches were combined (9.02 g total) and crystallized from DIUF water (50
mL). A
small amount of zwitterion was generated during the crystallization. In order
to
regenerate the salt, the solid generated was dissolved in 1% hydrochloric acid
in ethanol
(3 mL 37% hydrochloric acid in 100 mL ethanol). The ethanol solution was then
concentrated under reduced pressure at room temperature. The remaining salt
(5.93 g)
was crystallized three times from hot toluene (100 mL) with the addition of a
small
amount on acetone (1 mL). The salt was then filtered and dried under high
vacuum at
room temperature until the weight was constant (5.1 g).
0
110 OH:10
N+N, OH
"
CI
SPI0016B
2(S)-Amino-3(R)42(R)-(4-isobutyl-phenyl)-propionyloxyl-butyric acid;
(R-Ibuprofen-L-threonine ester, hydrochloride, inactive isomer):
86
FlAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
1H NMR (300 MHz, DMS0): 8 = 8.82 (br s, 3H), 7.23 (d, 2H, J= 7.8 Hz), 7.10 (d,
2H,
J= 7.8 Hz), 5.27 (m, 1H), 4.18 (m, 1H), 3.80 (q, 1H, J= 7.2 Hz), 2.41 (d, 2H,
J= 7.2 Hz),
1.81 (m, 1H), 1.41 (d, 3H, J= 6.9 Hz), 1.34 (d, 3H, J= 6.3 Hz), 0.85 (d, 6H,
J= 6.3 Hz).
13C NMR (75 MHz, DMS0): 6 = 72.56, 168.08, 139.64, 136.98, 128.84, 127.14,
68.8,
55.29, 44.28, 29.69, 22.28, 18.24, 16.41.
CHN analysis:
calc.: C 59.38, H 7.62, N 4.07; found: C 59.30, H 7.60, N 4.05.
HPLC analysis:
98.43% purity; r.t.= 6.19 min.; 60% TFA (0.1%)/40% acetonitrile; 1 mL/min;
37.5 C;
Luna C18, 3u column (SN 167917-13), 4.6x250 mm; 22 ul injection.
Optical Rotation: + 10.4
Melting Point: 176-177 C
2b) Preparation of the S-(+)-Ibuprofen-L-threonine ester, hydrochloride
standard
(SPI0016S).
S-(+)-Ibuprofen (2.0 g, 9.69 mmole), N-carbobenzyloxy-L-threonine benzyl ester
(3.25
g, 9.91 mmole), 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide, hydrochloride
(EDC,
1.90 g, 9.91 mmole), and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.12 g, 1.0
mmole)
were dissolved in dichloromethane (25 mL) at room temperature, under an argon
atmosphere. After stirring for 4 hours, the dichloromethane layer was washed
with
water (25 mL), 5% hydrochloric acid (25 mL), saturated sodium bicarbonate
(2x25 mL),
and water (25 mL). After drying for one hour over sodium sulfate (5 g),
filtration, and
concentration under reduced pressure, the remaining oil was used without
further
purification. The procedure generated the protected S-(+)-Ibuprofen-L-
threonine ester
(SPI001601S) as a light yellow oil (5.01 g, 98 % yield), which solidified on
standing.
87
H:\work\1652\17690\SPEC\17690.spec without claimadoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
1.s
0
HD¨e
N 0
d--C)
SPI001601S
2(S)-Benzyloxycarbonylamino-342(R,S)-(4-isobutyl-pheny1)-propionyloxy]-butyric
acid benzyl ester:
1H NMR (300 MHz, CDC13): 6 = 7.35-7.23 (m, 10H), 7.10 (d, 2H, J= 7.8 Hz), 7.05
(d,
2H, J= 7.8 Hz), 5.48-5.25 (m, 2H), 5.17-5.01 (m, 4H), 4.50 (dd, 1H, J= 9.6,
1.8 Hz),
3.50 (q,11-1, J= 7.2 Hz), 2.40 (d, 2H, J= 7.2 Hz), 1.80 (m, 1H), 1.37 (d, 3H,
J= 7.2 Hz),
1.17 (d, 3 H, J= 6.3 Hz), 0.86 (d, 6 H, J= 6.6 Hz).
13C NMR (75 MHz, CDC13): 8 = 173.29, 169.69, 156.51, 140.68, 137.21, 136.08,
135.06, 129.40, 128.70, 128.66, 128.57, 128.38, 128.24, 127.14, 70.70, 67.80,
67.53,
57.87, 45.19, 45.11, 30.39, 22.61, 18.57, 16.87.
The protected S-(+)-Ibuprofen-L-threonine ester (5.0 g, 9.40 mmole) was
dissolved in
warm ethanol (100 mL) and added to a Parr bottle that contained 10% palladium
on
carbon (1.0 g, 50% wet) under a nitrogen atmosphere. Hydrochloric acid (1 mL
37%
HC1 in 10 mL water) was added and the nitrogen atmosphere was replaced with
hydrogen gas (32 psi). After 2 hours of shaking, the palladium catalyst was
removed by
filtration through celite (30 g). The ethanol/water was removed under reduced
pressure.
The experiment produced S-(+)-Ibuprofen-L-threonine ester, hydrochloride
(SPI0016S,
2.8 g, 85% crude yield) as a colorless solid. The salt was stirred in acetone
(50 mL) for
3 hours at room temperature under an argon atmosphere. After 3 hours the
solids (2.24
g, 69% yield) were filtered and dried under high vacuum at room temperature,
until the
weight was constant.
88
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
s 0
= H,
1\1, OH
H , H
CI
SPI0016S
2(S)-Amino-3(R)42(S)-(4-isobutyl-phenyl)-propionyloxy]-butyric acid;
(S-Ibuprofen-L-threonine ester, hydrochloride, active isomer):
1H NMR (300 MHz, DMS0): 6 = 8.76 (br s, 3H), 7.19 (d, 2H, J= 8.1 Hz), 7.11 (d,
2H,
J= 8.1 Hz), 5.28 (dq, 1H, J= 6.3, 3.6 Hz), 4.14 (q, 1H, J= 3.6 Hz), 3.80 (q,
1H, J= 7.2
Hz), 2.41 (d, 211, J= 7.2 Hz), 1.80 (m, 111), 1.37 (d, 3H, J= 7.2 Hz), 1.21
(d, 3H, J= 6.3
Hz), 0.85 (d, 6H, J= 6.6 Hz).
13C NMR (75 MHz, DMS0): 8 = 172.66, 168.24, 139.68, 137.24, 128.95, 126.97,
67.98,
55.35, 44.23, 43.83, 29.66, 22.24, 18.52, 16.47.
HPLC analysis:
98.28% purity; r.t.= 6.951 min.; 60% TFA (0.1%)/40% acetonitrile; 1 mL/min;
37.5 C;
Luna C18, 3u column (SN 167917-13), 4.6x250 mm; 22 ul injection.
Optical rotation: + 26.5
Melting Point: 189-190 C
3) Preparation of the ( )-Ibuprofen-L-hydroxyproline ester (SPI0017).
( )-Ibuprofen (5.10 g, 24.7 mmole), N-carbobenzyloxy-L-hydroxyproline benzyl
ester
(8.80 g, 24.7 mmole), 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide,
hydrochloride
(EDC, 5.10g, 26.0 mmole), and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.30 g,
2.40
mmole) were dissolved in dichloromethane (100 mL) at room temperature, under
an
89
H:\work\1652\17690\SPEC\17690.spec without clairnadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
argon atmosphere. After stirring for 24 hours under an argon atmosphere at
room
temperature, water (100 mL) was added and the layers were separated. The
dichloromethane layer was washed again with water (100 mL), 5% sodium
bicarbonate
(2x50 mL) and dried for 1 hour over sodium sulfate (5 g). After filtration and
concentration under reduced pressure, the remaining oil was used without
further
purification. The procedure generated the protected ( )-Ibuprofen-L-
hydroxyproline
ester (SPI001701) as a light yellow oil (11.5 g, 85% yield).
0
o
0 IP
0
SPI001701
4(R)42-(4-Isobutyl-pheny1)-propionyloxyl-pyrrolidine-2(S)-carboxylic acid;
((R,S)-Ibuprofen-L-hydroxyproline ester):
1H NMR (300 MHz, CDC13): 5 = 7.33-7.02 (m, 14H), 5.25-4.95 (m, 5H), 4.51-4.19
(m,
1H), 3.75-3.50 (m, 3H), 2.40 (d, 2H, J= 6.9 Hz), 2.15 (m, 1H), 1.81 (m, 1H),
1.44 (d,
3H, J= 7.0 Hz), 0.87 (d, 6H, J= 6.6 Hz).
13C NMR (75 MHz, CDC13): 5 = 173.99, 171.93, 171.72, 154.68, 154.15, 140.70,
137.23, 137.04, 136.23, 135.44, 135.23, 129.41, 128.59, 128.47, 128.35,
128.19, 128.08,
127.89, 127.02, 72.86, 72.16, 67.40, 67.18, 67.09, 58.12, 57.83, 52.66, 52.49,
52.13,
45.15, 36.63, 35.67, 32.07, 30.33, 29.23, 22.90, 22.58, 18.36.
The protected Ibuprofen-L-hydroxyproline ester (11.40 g, 43.4 mmole) was
dissolved in
ethanol (150 mL) at room temperature and added to a Parr bottle that contained
10%
palladium on carbon (2.73 g, 50% wet) under a nitrogen atmosphere. The
nitrogen
atmosphere was replaced with hydrogen gas (34 psi). After 5 hours of shaking,
the
= palladium catalyst was removed by filtration through celite. The ethanol was
removed
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
under reduced pressure. The remaining white solids (6.60 g) were washed with
DIUF
water (50 mL), diethyl ether (50 mL) and dried under high vacuum until the
weight was
constant. The experiment produced ( )-Ibuprofen-L-hydroxyproline ester SPI0017
(5.64 g, 84% yield) as a colorless solid.
o
SPI0017
4(R)42-(4-Isobutyl-pheny1)-propionyloxy]-pyrrolidine-2(S)-carboxylic acid;
((R,S)-Ibuprofen-L-hydroxyproline ester):
IHNMR (300 MHz, CDC13): 5 = 7.22 (d, 2H, J= 7.2 Hz), 7.09 (d, 2H, J= 7.2 Hz),
5.27
(m, 1H), 4.40 (t, 0.5H, J= 7 Hz), 4.24 (t, 0.5 H, J= 9 Hz), 3.75 (m, 1H),
3.61(m, 1H),
3.28 (d, 0.5H, J= 13 Hz), 3.15 (d, 0.5H, J= 13 Hz), 2.42-2.10 (m, 4H), 1.78
(m, 1H),
1.40 (br t, 3H, J= 6 Hz), 0.82 (d, 6H, J= 6 Hz). (mixture of diastereomers)
13C NMR (75 MHz, CDC13): 5 = 173.28, 173.23, 168.98, 139.88, 137.33, 137.23,
129.12, 127.26, 127.17, 72.58, 57.60, 57.50, 50.24, 50.12, 44.34, 44.15,
34.31, 34.16,
29.77, 22.34, 18.43, 18.23. (mixture of diastereomers)
HPLC analysis:
100% purity; r.t.= 5.35, 5.22 min.; 55% TFA (0.1%), 45% ACN; 1 mL/min; 32.3 C,
Luna C18, serial # 188255-37; 20 ul inj..
CHN analysis:
calc.: C 67.69, H 7.89, N 4.39; found: C 67.47, H 7.87, N 4.30.
Melting Point: 198-199 C
91
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Efficacy (anti nociceptive potential) of Synthesis of the L-serine, L-
threonine, and
L-hydroxyproline esters of ( )-Ibuprofen by employing acetylcholine induced
abdominal constriction method in male albino mice:
The present study was conducted to evaluate the efficacy of L-serine, L-
threonine, and
L-hydroxyproline esters of ( )-Ibuprofen taking into account the antagonizing
property
on acetylcholine induced writhe as an index in albino mice. Ibuprofen (racemic
mixture)
and ibuprofen (S)-(+) served as reference controls.
Different new formulations of ibuprofen and reference controls viz., ibuprofen
(racemic
mixture) and ibuprofen (S)-(+) were administered by gavage to male albino mice
(Swiss
strain), using 5% (v/v) Tween 80 in milli Q water as the vehicle. The study
was
conducted at two dose levels viz. 50mg and 100mg/kg body weight along with a
vehicle
control group. At each dose level 10 animals were used. All the doses were
expressed as
ibuprofen molar equivalents. The doses used as well as the molar equivalents
are
presented below.
92
1-1:\work\1652\17690\SPEC\17690.spec without claim.sII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 2: Formulation: Molar Equivalent:
Formulation Molar equivalent
0.833 units are equivalent to 1 unit of
S-(+)-Ibuprofen-L-threonine ester
Ibuprofen
1.6 units are equivalent to 1 unit of
( )-Ibuprofen-L-serine ester
Ibuprofen
1.55 units are equivalent to 1 unit of
( )-Ibuprofen-L-hydroxproline ester
Ibuprofen
Table 3: Test Item: Group: Dose(mg/kg): Equivalent wt. Of the test item:
Equivalent
Dose (mg per kg)
weight of the
Test Item Group [in terms of
Test item
Ibuprofen]
[mg/kg]
Vehicle control 0.0
Vehicle
Group
S-(+)-Ibuprofen-L- Test Group 1 50.0 41.65
threonine ester Test Group 2 100.0 83.30
( )-Ibuprofen-L- Test Group 3 50.0 80.0
serine ester Test Group 4 100.0 160.0
(Ibuprofen S)
( )-Ibuprofen-L- Test Group 5 50.0 77.5
hydroxyproline ester Test Group 6 100.0 155.0
Ibuprofen (racemic Test Group 7 50.0 50.0
mixture) Test Group 8 100.0 100.0
Ibuprofen +
Test Group 9 50.0 25.0
S
Test Group 10 100.0 50.0
The efficacy in terms of antagonizing effect on acetylcholine induced single
writhe at
two dose levels ¨ 50.0 and 100.0 mg/kg for the three formulations and
reference controls
are presented below.
93
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 4: Test Item: Group: Dose (mg/kg): Number of animals showing absence of
single
writhe (out of 10)
Number of animals
showing absence of single
Dose (mg
writhe (number of animals
per kg)
Test Item Group per dose = 10)
[in terms of
One hour Three hours
Ibuprofen]
after after dosing
dosing
Vehicle Vehicle control 0.0 0 0
S-(+)-
Low dose 50.0 1 0
Ibuprofen-L-
threonine
High dose 100.0 3 0
ester
Low dose 50.0 4 2
( )-
Ibuprofen-L-
High dose 100.0 6 4
serine ester
Low dose 50.0 5 4
( )-
Ibuprofen-L-
hydroxyprolin High dose 100.0 7 7
e ester
Low dose 50.0 4 2
Ibuprofen
(racemic
High dose 100.0 6 6
mixture)
Low dose 50.0 5 1
Ibuprofen S +
High dose 100.0 6 6
Statistical analysis employing Chi ¨ square test procedure did not show any
statistically
significant difference among the formulations in comparison to reference
control, while
comparing the number of animals not showing writhe in each groups, as the
respective
"p" was found to be greater than 0.05, the level of significance.
From clinical observation based on the number of animals not showing writhes
due to
administration of acetylcholine, ( )-Ibuprofen-L-hydroxyproline ester was
found to be
94
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
more effective in antagonizing the acetylcholine induced writhe when compared
to other
formulations and Ibuprofen (racemic) and Ibuprofen (S)-(+).
Table 5: Summary of Efficacy of L-serine, L-threonine, and L-hydroxyproline
esters of ( )-Ibuprofen, Ibuprofen (racemic mixture) and Ibuprofen (S)-(+) -
Based
on Antagonizing Property of Acetylcholine Induced Writhe in Albino Mice
Number of animals showing
Dose absence of single writhe (number
(mg/kg) Test Item of animals per dose = 10)
[in terms of One hour after Three hours
Ibuprofen] dosing after dosing
Vehicle control 0 0
S-(+)-Ibuprofen-L-
1 0
threonine ester
( )-Ibuprofen-L-
4 2
serine ester
50mg/kg ( )-Ibuprofen-L-
hydroxyproline 5 4
' ester
Ibuprofen
4 2
(racemic mixture)
Ibuprofen (S)-(+) 5 1
Table 6:
Vehicle control 0 0
S-(+)-Ibuprofen-
3 0
L-threonine ester
( )-Ibuprofen-L-
6 4
seine ester
100mg/kg ( )-Ibuprofen-L-
hydroxyproline 7 7
ester
Ibuprofen
6 6
(racemic mixture)
Ibuprofen (S)-(+) 6 6
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The data were subjected to statistical analysis employing Chi ¨ square test
procedure for
evaluating the efficacy of the new formulations in comparison to the reference
controls.
The test did not show any statistically significant difference among the
formulations in
comparison to reference control, while comparing the number of animals not
showing
writhe in each groups, as the respective "p" was found to be greater than
0.05, the level
of significance.
The data is also summarized in FIGS. 1 and 2. From clinical observations and
bar
diagram for comparative efficacy (FIGS. 1 and 2) based on the number of
animals not
showing writhes due to administration of acetylcholine, ( )-Ibuprofen-L-
hydroxyproline
ester was found to be more effective in antagonizing the acetylcholine induced
writhe
when compared to other formulations and Ibuprofen (racemic) and Ibuprofen (S)-
(+).
CONCLUSION
The present study was conducted to evaluate the relative efficacy of new
formulations of
ibuprofen. For this the antagonizing property of new formulations on
acetylcholine
writhes was taken as an index to determine the relative efficacy of the
formulations.
Ibuprofen (racemic mixture and ibuprofen (S)-(+) served as reference controls.
The
study was conducted at two dose levels (50.0 and 100.0 mg/kg) along with a
vehicle
control group.
The efficacy in terms of antagonizing effect of acetylcholine induced single
writhe at
two dose levels ¨ 50.0 and 100.0 mg/kg for the three formulations and
reference controls
are presented below.
30
96
Mwork\1652\17690\SPEC\17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Table 7: Test Item: Group: Dose (mg/kg): No. of animals showing absence of
single
writhe (out of 10)
Number of animals showing
absence of single writhe
Dose (mg per
(number of animals per dose =
kg)
Test Item Group 10)
[in terms of
Three hours
Ibuprofen] One hour after
after dosing
dosing
Vehicle
Vehicle 0.0 0 0
control
S-(+)-Ibuprofen- Low dose 50.0 1 0
L-threonine
ester High dose 100.0 3 0
Low dose 50.0 4 2
( )-Ibuprofen-L-
serine ester High dose 100.0 6 4
Low dose 50.0 5 4
( )-Ibuprofen-L-
hydroxyproline
High dose 100.0 7 7
ester
Low dose 50.0 4
Ibuprofen
(racemic
High dose 100.0 6 6
mixture)
Low dose 50.0 5 1
Ibuprofen (S)-
(-0 High dose 100.0 6 6
Statistical analysis employing Chi ¨ square test procedure did not show any
statistically
significant difference among the formulations in comparison to reference
control, while
comparing the number of animals not showing writhe in each groups, as the
respective
"p" was found to be greater than 0.05, the level of significance.
However from clinical observation based on the number of animals not showing
writhes
due to administration of acetylcholine ( )-Ibuprofen-L-hydroxyproline ester
was found
97
H:\work\1652 \17690 \SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
to be more effective in antagonizing the acetylcholine induced writhe when
compared to
other formulations and Ibuprofen (racemic) and Ibuprofen (S)-(+).
Gastric mucosal irritation potential of L-serine, L-threonine, and L-
hydroxyproline
esters of ( )-Ibuprofen in fasted male albino rats
SUMMARY
The present study was conducted to determine the relative potential of new
formulations
of ibuprofen (L-serine, L-threonine, and L-hydroxyproline esters of ( )-
Ibuprofen) to
cause gastric mucosal irritation/lesions in fasted male albino rats. Ibuprofen
(racemic
mixture) and Ibuprofen(S)-(+) served as reference controls.
Different new formulations of ibuprofen and ibuprofen (racemic mixture) and
ibuprofen(S)-(+) were administered by gavage to fasted male albino rats
(Wistar strain),
using 5% solution of Tween 80 in milli Q water as the vehicle. The study was
conducted
at two dose levels viz. 200mg and 300mg/kg body weight along with a vehicle
control
group. At each dose level 5 animals were used. All the doses were expressed as
ibuprofen (racemic mixture) molar equivalents. The doses used as well as the
molar
equivalents were presented below.
Table 8: Formulation: Molar Equivalent
Formulation Molar equivalent
0.833 units are equivalent to 1 unit of
S-(+)-Ibuprofen-L-threonine ester
Ibuprofen
1.60 units are equivalent to 1 unit of
( )-Ibuprofen-L-serine ester
Ibuprofen
1.55 units are equivalent to 1 unit of
( )-Ibuprofen-L-hydroxyproline ester
Ibuprofen
98
H:\work\1652 \I7690 \SPEC \I7690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
The various groups used are tabulated hereinbelow:
Table 9: Test item: group: Dose (mg/kg) Equivalent wt.
Equivalent
Dose (mg per kg) weight of
Test item Group [in terms of the
Ibuprofen] Test
item
[mg/kg]
Vehicle control 0.0
Vehicle
Group
S-(+)-Ibuprofen-L- Test Group 1 200.0 0.0
threonine ester Test Group 2 300.0 166.6
( )-Ibuprofen-L-serine Test Group 1 200.0 249.9
ester Test Group 2 300.0 320.0
( )-Ibuprofen-L- Test Group 1 200.0 480.0
_
hydroxyproline ester Test Group 2 300.0 310.0
Ibuprofen (racemic Test Group 1 200.0 465.0
mixture) Test Group 2 300.0 300.0
_
Test Group 1 200.0 100.0
Ibuprofen (S)- (+)
Test Group 2 300.0 150.0
The rats were fasted for a period of 18 to 22 hours before dosing. The test
item was
administered as a single dose by gavage. Three hours after drug
administration, the
animals were killed humanely by CO2 gas inhalation. The stomach was dissected
out and
observed for
= the quantity of mucous exudate,
= degree of hyperemia and thickening of stomach wall,
= hemorrhagic spots (focal or diffuse), nature of hemorrhages (petechial or
ecchymotic) along with the size and
= perforations or any other lesions
99
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The observations on gastric mucosal irritation of animals of various groups
were
summarized below
Table 10: Test item: Group: Dose (mg/kg): Observation
Doe mg/kg
Test Item Group ( as per Observation
ibuprofen)
None of the animals
Vehicle control showed any evidence
Vehicle control 0.0
Group of gastric mucosal
irritation
None of the dosed
animals showed any
Test Group 1 200.0
evidence of gastric
S-(+)-Ibuprofen- mucosal irritation
L-threonine ester None of the dosed
animals showed any
Test Group 2 300.0
evidence of gastric
mucosal irritation.
None of the dosed
animals showed any
Test Group 1 200.0
evidence of gastric
( )-Ibuprofen-L- mucosal irritation.
setine ester None of the dosed
animals showed any
Test Group 2 300.0
evidence of gastric
mucosal irritation
None of the dosed
animals showed any
Test Group 1 200.0
evidence of gastric
( )-Ibuprofen-L-
mucosal irritation
hydroxyproline
None of the dosed
ester
animals showed any
Test Group 2 300.0
evidence of gastric
mucosal irritation
Gastric mucosal
irritation was
Ibuprofen
(racemic mixture) Test Group 1 200.0 observed in one
animal out of 5
animals dosed.
100
H:\wor1\1652\17690\SPEC\17690.spec without claimsndoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Gastric mucosal
irritation was
Test Group 2 300.0 observed
in two
animals out of 5
animals dosed.
Gastric mucosal
irritation was
Test Group 1 200.0
observed in all the 5
animals dosed.
Ibuprofen (S)
Gastric mucosal
irritation was
Test Group 2 300.0 observed in three
animals out of 5
animals dosed.
The results of the present study showed that none of the formulations of
ibuprofen had
caused any evidence of irritation of gastric mucosa in fasted male albino rats
of male sex
at the two dose levels tested (200mg and 300mg/kg body weight). In contrast
both
ibuprofen (racemic mixture) and ibuprofen (S) ¨ (+) had caused irritation of
gastric
mucosa at the two dose levels tested. Further ibuprofen(S ) - (+) was found to
be more
gastric mucosal irritant than ibuprofen (racemic mixture).
Overview Ketoprofen S(+) Threonine Ester Synthesis:
The procedure for the synthesis of the L-threonine esters of Ketoprofen is
outlined in
Synthetic Sequence section. This synthesis is exemplary and is equally
applicable for
the other amino acids. The complete procedure and analytical data is given in
the
Experimental Section. In general, ( )-Ketoprofen (5 g) was coupled with N-boc-
L-
threonine t-butyl ester' (1 equivalent) with 1-(3-dimethylaminopropy1)-3-
ethylcarbodiimide, hydrochloride (EDC, 1 equivalent) in the presence of a
catalytic
amount of 4-(N,N-dimethyamino)-pyridine (DMAP). Once the reaction was
complete,
any excess EDC was removed by extraction with water, DMAP was removed by
extraction with dilute acid, and Ketoprofen was removed by extraction with
sodium
bicarbonate. After drying over sodium sulfate, filtration, and concentration
the crude
101
H:\work\1652\17690\SPEC\17690.spec without claimsiLdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
protected L-threonine-( )-Ketoprofen was purified by flash chromatography on
silica gel
to generate the protected L-threonine ester in good yield (98%). The
protecting groups
were removed by treatment with 2M hydrochloric acid in diethyl ether to cleave
the boc
group, followed by treatment with trifluoroacetic acid to remove the t-butyl
ester. After
drying, the mixture of L-threonine-R,S( )-Ketoprofen esters was separated by
crystallization from acetonitrile. The hydrochloride salt of the L-threonine-
S(+)-
Ketoprofen ester preferentially precipitated from acetonitrile. A sample of an
optically
pure standard was prepared starting with S(+)-ketoprofen for comparison. After
drying
and analysis, a sample of L-threonine-S(+)-Ketoprofen ester, hydrochloride
(1.75 g) was
1 0 separated from the mixture.
Synthetic Sequence:
SPI0018A
O
o a)
I. 0. 0
HO 1,1¨boc 10 0 /
õ0
H-14'
%boo
( )-Ketoprofen Boc-Thr-O-tBu SPI001801
b)
c)
0
0 0
4. = 10 0 /
10 110 0 0 d)
10 10 0
0 0
Fri 0, 0,
WI 0, H H H H
H H
SPI0018A SPI0018A SPI0018B
1 5 Synthesis of the L-threonine esters of ( )-Ketoprofen: a) EDC, DMAP,
CH2C12; b)
FIC1 (2M); c) TFA; d) ACN (crystallization).
Experimental Section:
The synthesis of SPI0018A was conducted in a single batch. Reagents mentioned
in the
experimental section were purchased at the highest obtainable purity from
Sigma-
1 02
H:\work\1652\17690\SPEC\17690.spec without clahnsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Aldrich, Acros, or Bachem, except for solvents, which were purchased from
either
Fisher Scientific or Mallinkrodt.
Preparation and Separation of S(+)-Ketoprofen-L-threonine ester, hydrochloride
(SPI0018A).
( )-Ketoprofen (5.32 g, 20.92 mmol), N-t-butylcarbonyl-L-threonine t-butyl
ester (Boc-
Thr-OtBu, 5.17 g, 18.72 mmol, (prepared in accordance with the literature), 1-
(3-
dimethylaminopropy1)-3-ethylcarbodiimide, hydrochloride (EDC, 4.0 g, 20.9
mmol),
and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.22 g) were dissolved in
dichloromethane (50 mL) at room temperature, under an argon atmosphere. After
stirring for 5 hours, the dichloromethane layer was washed with water (50 mL),
5%
hydrochloric acid (2x25 mL), water (25 mL), saturated sodium bicarbonate (2x25
mL),
and water (50 mL). After drying for one hour over sodium sulfate (5 g),
filtration, and
concentration under reduced pressure, the remaining oil (10.3 g) was purified
by column
chromatography on silica gel (150 g), eluting with hexanes/ethyl acetate
(2:1). After
combining the product containing fractions, concentration and drying under
high
vacuum, the procedure generated the protected L-threonine-( )-Ketoprofen ester
(SPI001801) as a clear oil (9.42 g, 98% yield).
µboc
SPI001801
20 342(R,S)-(3-Benzoyl-pheny1)-propionyloxy]-2(S)-tert-butoxycarbonylamino-
butyric
acid tert-butyl ester: (mix of diastereomers)
IFINMR (300 MHz, CDC13): 5 = 7.83-7.42 (m, 9H), 5.43 (dd, 1H, J= 13.2, 6.9
Hz), 5.10
(dd, 1H, Jr= 20.7, 9.3), 4.29 (t,11-1, J= 11.7 Hz), 3.75 (q, 1H, J= 7.2 Hz),
1.50-1.42 (m,
25 19.5H), 1.30-1.18 (m, 4.5H).
103
11:\wor1\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
13C NMR (75 MHz, CDC13): 8 =. 196.18, 172.62, 172.55, 168.85, 168.58, 155.81,
140.33, 140.23, 137.86, 137.39, 132.46, 132.42, 131.54, 131.38, 130.00,
129.31, 129.13,
129.02, 128.54, 128.27, 82.50, 82.37, 80.05, 71.38, 71.22, 57.59, 57.52,
45.46, 45.31,
28.40, 27.98, 27.84, 18.54, 18.48, 17.19, 16.84.
The protected (R,S)-Ketoprofen-L-threonine ester (9.42 g, 18.41 mmol) was
dissolved in
dichloromethane (25 mL) under an argon atmosphere, at room temperature.
Anhydrous
hydrochloric acid in diethyl ether (2M, 25 mL) was added to the solution and
the
mixture was allowed to stir for 17 hours at room temperature. The mixture was
concentrated under reduced pressure. The remaining foam (8.2 g) was dissolved
in a
mixture of dichloromethane (10 mL) and trifluoroacetic acid (20 mL). After
stirring at
room temperature for 6.5 hours the solution was concentrated under reduced
pressure.
Toluene (25 mL) was added to the remaining oil and the mixture was
concentrated a
second time. A mixture of ethanol (20 mL) and anhydrous hydrochloric acid in
diethyl
ether (2M, 20 mL) was added and the solution was concentrated a third time.
After
drying under high vacuum for 2 hours at room temperature, the experiment
produced
( )-Ketoprofen-L-threonine ester, hydrochloride (mix of diastereomers, 7.11 g,
98%
crude yield) as an off-white solid. The crude mixture of diastereomers (7.0 g)
was
crystallized 3 times from acetoniffile (200 mL). After the third
crystallization, the
remaining white solid was dried under high vacuum at 50 C until the weight
was
constant (4 hours). The experiment produced L-threonine-S(+)-Ketoprofen ester,
hydrochloride SPI0018A (2.2 g, 30% yield from SPI001801).
0
o
O \>/<
H_N+ _OH
H Cl SPI0018A
2(S)-Amino-3(R)-[2(S)-(3-benzoyl-phenyl)-propionyloxy]-butyric acid,
hydrochloride
(L-threonine-S(+)-Ketoprofen ester, hydrochloride):
104
H:\work\1652 \17690 \SPEC \17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
1H NMR (300 MHz, DMS0): 8 = 14.08 (br s, 1H), 8.72 (br s, 3H), 7.74-7.51 (m,
9H),
5.29 (t, 1H, J= 4.5 Hz), 4.16 (m, 1H), 3.97 (q, 1H, J= 6.3 Hz), 1.42 (d, 3H,
J= 6.9 Hz),
1.23 (d, 3H, J= 6.3 Hz).
13C NMR (75 MHz, DMS0): 6= 195.34, 172.26, 168.21, 140.42, 137.05, 136.74,
132.66, 131.66, 129.48, 128.73, 128.49, 128.30, 68.23, 55.31, 44.00, 18.44,
16.45.
CHN analysis:
calc.: C 61.30, ILI 5.66, N 3.57; found: C 61.02, H 5.58, N 3.58.
HPLC analysis:
98.28% purity; r.t.= 25.14min.; 55% DTUF water (0.1% TFA)/45% methanol; 1
mL/min;
36.4 C; Luna Cl 8, 5u column (serial # 211739-42), 4.6x250 mm; 20 ul
injection.
Optical rotation: + 27.0 (20 C, 174.4 mg/10 mL ethanol, 589 nm); Melting
Point:
166-167 C
Preparation of the S-(+)-Ketoprofen-L-threonine ester, hydrochloride standard.
(+)-Ketoprofen (1.87 g, 7.74 mmol), N-t-butylcarbonyl-L-threonine t-butyl
ester (Boc-
Thr-OtBu, 2.25 g, 8.14 mmol, prepared in accordance with the literature
method), 1-(3-
dimethylaminopropy1)-3-ethylcarbodihnide, hydrochloride (EDC, 1.65 g, 8.60
mmol),
and 4-(N,N-dimethylamino)-pyridine (DMAP, 0.1 g) were dissolved in
dichloromethane
(25 mL) at room temperature, under an argon atmosphere. After stirring for 4
hours, the
dichloromethane layer was washed with water (25 mL). After drying for one hour
over
sodium sulfate (5 g), filtration, and concentration under reduced pressure,
the remaining
oil was used without purification. The procedure generated the protected L-
threonine-
(+)-Ketoprofen ester as a clear oil (4.01 g, ¨100% yield).
105
1-Bwork\1652\17690\SPEC\17690.spec without clahnsIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
of
Si 0
boc
1H NMR (300 MHz, CDC13): 8 = 7.81-7.42 (m, 9H), 5.43 (m, 1H), 5.10 (d, 1H,
.1.= 9.3),
4.29 (d, 1H, J= 9.6 Hz), 3.75 (q, 1H, J= 7.2 Hz), 1.50-1.42 (m, 21H), 1.18 (d,
3H, .1.= 6.3
Hz).
13C NMR (75 MHz, CDC13): 8 = 196.4, 172.79, 168.99, 155.94, 140.44, 137.99,
137.51,
132.59, 131.50, 130.13, 129.31, 129.25, 129.15, 128.66, 128.40, 82.68, 80.24,
71.37,
57.71, 45.43, 28.53, 28.10, 18.99, 16.96.
The protected (S)-Ketoprofen-L-threonine ester (3.92 g, 7.66 mmol) was
dissolved in
anhydrous hydrochloric acid in diethyl ether (2M, 50 mL) and stirred for 17
hours at
room temperature. The mixture was concentrated under reduced pressure. The
remaining foam (3.4 g) was dissolved in a mixture of dichloromethane (20 mL)
and
trifluoroacetic acid (20 mL). After stirring at room temperature for 6.5 hours
the
solution was concentrated under reduced pressure. Toluene (25 mL) was added to
the
remaining oil and the mixture was concentrated a second time. A mixture of
ethanol (20
mL) and anhydrous hydrochloric acid in diethyl ether (2M, 20 mL) was added and
the
solution was concentrated a third time. After drying under high vacuum for 2
hours at
room temperature, the experiment produced S(+)-Ketoprofen-L-threonine ester,
hydrochloride (3.05g crude) as an off-white solid. The crude material was
stirred with
acetone (50 mL) for 2 hours at room temperature under an argon atmosphere. The
remaining white solid was filtered and dried under high vacuum at 50 C until
the weight
was constant (4 hours). The experiment produced L-threonine-S(+)-Ketoprofen
ester,
hydrochloride (2.04 g, 67 % yield).
106
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
NMR (300 MHz, DMS0): 5 = 14.08 (br s, 111), 8.72 (br s, 3H), 7.74-7.51 (m,
9H),
5.29 (t, 1H, J= 4.5 Hz), 4.16 (m, 1H), 3.97 (q, 1H, J= 6.3 Hz), 1.42 (d, 3H,
J= 6.9 Hz),
1.23 (d, 3H, J= 6.3 Hz).
13C NMR (75 MHz, DMS0): 5 = 195.34, 172.26, 168.21, 140.42, 137.05, 136.74,
132.66, 131.66, 129.48, 128.73, 128.49, 128.30, 68.23, 55.31, 44.00, 18.44,
16.45.
HPLC analysis:
99.43% purity; r.t.= 25.14min.; 55% DIUF water (0.1% TFA)/45% methanol; 1
mL/min;
36.4 C; Luna C18, 5u column (serial # 211739-42), 4.6x250 mm; 20 ul injection.
Optical rotation: + 27.1 (20 C, 177.8 mg/10 mL ethanol, 589 nm); Melting
Point:
166-167 C
C. Amino Acid Derivaties of Aspirin
Overview:
The procedure for the synthesis of the L-serine, L-threonine, and L-
hydroxyproline
esters of acetylsalicylic acid is outlined in Synthetic Sequence section and
is exemplary
for other amino acids. The complete procedure and analytical data is given in
the
Experimental Section. In general, acetylsalicyloyl chloride (10 g-25 g, in
batches) was
coupled with the N-benzyloxy/benzyl ester protected amino acids in the
presence of
pyridine. Once the reactions were complete (24 to 48 hours at room
temperature), the
mixture was poured into ice-cold 2N hydrochloric acid. The dichloromethane
fraction
was then washed with sodium bicarbonate, water and brine. After drying over
sodium
sulfate, filtration, and concentration the crude protected amino acid esters
of
acetylsalicylic acid were purified by flash chromatography on silica gel. The
procedure
generated the protected amino acid esters of acetylsalicylic acid in yields
ranging from
68% to 95%. The protecting groups were removed by hydrogenation (20 psi H2) in
the
presence of 10% palladium on carbon. The amino acid esters of acetylsalicylic
acid
were extracted away from the palladium catalyst with water, concentrated, and
dried.
107
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The final compounds were washed with solvent (water, dioxane, acetonitile,
and/or
dichloromethane) until pure and dried under high vacuum until a constant
weight was
achieved.
Synthetic Sequence:
1. SPIB00102
0 o o o o
o a) 40 0,.)-µ
,...... OBzl H'
b)
0 OH
1 ____________________ cit. ¨i.- 0 0-)----OH
HO., OBzl H" H'
CIDZ
0 N,H
0 + H __ N¨cbz 0
0 0
0\
Acetylsalicylic acid Z-Thr-OBzI SPIB0010201 SPIB00102
2. SPIB00101
O o o o o
o
0 OH a) HO LOBz1 0 r.,,,¨(1L0Bz1 b) ('OH---- ---.-
''' N¨ --0-
H... cb z 0 00- ,N,
O H H
bz
H c
0\ --N¨= 0
0---ec
0
Acetylsalicylic acid Z-Ser-OBzI SPIB0010101 SPIB00101
3. SPIB00103
O o o
40 OH + HOii., OBzi a) 04 b) 40
OBz1 0
''''CrYLOH
'''CrL ----..
0 N
\ cbz 0
0 \ cbz
0 0
o=
H
Acetylsalicylic acid Z-Hyp-OBzI SPIB0010301 SPIB00103
Synthesis of the L-serine, L-threonine, and L-hydroxyproline esters of
acetylsalicylic acid: a) pyridine, CH2C12; b) 10% Pd/C, Et0H, Et0Ac.
Experimental Section:
The synthesis of SPIB00101, SPII300102 and SPIB00103 was conducted in one or
two
batches. Reagents mentioned in the experimental section were purchased at the
highest
108
1-1:\work\1652\17690\SPEC\17690.spec without clairnslidoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
obtainable purity from Lancaster, Sigma-Aldrich, Acros, or Bachem, except for
solvents,
which were purchased from either Fisher Scientific or Mallinkrodt.
1) SPIB00102: 2-0-Acetylsalicylic acid (2S, 3R)-(-)-threonine ester
A mixture of N-carbobenzyloxy-L-threonine benzyl ester (Z-Thr-OBz1, 21.77 g,
63.40
mmole) and pyridine (25 mL) in anhydrous dichloromethane (500 mL) was cooled
in an
ice bath while under a nitrogen atmosphere. Acetylsalicyloyl chloride (17.63
g, 88.76
mmole) was added and the mixture was allowed to warm to room temperature and
stir
overnight. After 24 hours, the mixture was poured into ice-cold 2N
hydrochloric acid
(400 mL). After mixing, the layers were separated and the dichloromethane
fraction was
washed with water (500 mL), saturated sodium bicarbonate solution (500 mL),
water
(500 mL), brine (500 mL) and dried over sodium sulfate (25 g). After
filtration,
concentration under reduced pressure, and drying under high vacuum, the
remaining
yellow oil (35. 43 g) was purified by flash chromatography on silica gel (300
g, 0.035-
0.070 mm, 6 nn pore diameter), eluting with hexanes/ethyl acetate (3:1). After
concentration of the product containing fractions under reduced pressure and
drying
under high vacuum until the weight was constant, the experiment produced the
protected
acetylsalicylic-L-threonine ester SPIB0010201 (28.1 g, 88% yield) as a
colorless oil.
0 0
2
0 H \O
0\ 0 =
SPIB0010201
1H NMR (300 MHz, CDC13): 5 = 7.74 (1H, d, .1= 7.5 Hz), 7.51 (1H, dt, J= 7.5,
1.5 Hz),
7.34-7.17 (11H, m), 7.06 (1H, d, .1= 7.2 Hz), 5.62 (2H, m), 5.13 (4H, m), 4.65
(1H, dd,
.1= 9.6, 2.4 Hz), 2.29 (3H, s), 1.38 (3H, d, J.= 6.6 Hz).
109
H: \work\ 1652 \ 17690 \SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
13C NMR (75 MHz, CDC13): 8 = 169.35, 169.22, 162.73, 156.26, 150.41, 135.79,
134.67, 133.77, 131.24, 128.35, 128.24, 128.08, 127.95, 125.78, 123.51,
122.61, 71.22,
67.72, 67.26, 57.64, 20.98, 16.88.
The protected acetylsalicylic-L-threonine ester SEIB0010201 (14.50 g, 28.68
mmole)
was dissolved in ethanol (100 mL) and ethyl acetate (100 mL) at room
temperature and
added to a Parr bottle that contained 10% palladium on carbon (3.0 g, 50% wet)
under a
nitrogen atmosphere. The nitrogen atmosphere was replaced with hydrogen gas
(20 psi).
After 20 hours of shaking, the palladium catalyst was removed by filtration
through
celite. The remaining solids (palladiurn/celite and product) were washed with
water
(600x4 mL) until the product was removed. The ethanol and water fractions were
concentrated under reduced pressure at room temperature. The remaining solids
were
washed with water (20 mL) and dioxane (20 mL) for 48 hours. After filtration,
the
remaining white solid was dried at room temperature under high vacuum until
the
product weight was constant (16 hours). The experiment produced
acetylsaficylic-L-
threonine ester, SPIB00102 (4.40 g, 55% yield) as a white solid.
0 0 H
0) (C3'''
-N,
H H
0
0\
SPIB00102
1H NMR (300 MHz, D20-DC1): = 8.00 (1H, dd, J= 7.8, 1.5 Hz), 7.74 (111, dt, J=
7.8,
1.5 Hz), 7.47 (1H, dt, J= 7.8, 1.5 Hz), 7.27 (1H, dd, J= 7.8, 1.5 Hz), 5.76
(1H, dq, J= 6.9,
3.0 Hz), 4.49 (1H, d, J= 3.0 Hz), 2.39 (3H, s), 1.55 (3H, d, J= 6.9 Hz).
13C NMR (75 MHz, D20-DC1): 8 = 173.03, 168.84, 163.97, 149.56, 135.32, 131.26,
126.85, 123.48, 121.49, 69.16, 56.36, 20.45, 15.86.
110
HAwork\1652 \17690 \SPEC \17690.spec without claints11.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
HPLC analysis:
98.7% purity; rt= 6.233 min; Luna C18 5u column (sn 167917-13); 4.6x250 mm;
254
nm; 35% Me0H/65% TFA (0.1%) pH= 1.95; 35 C; 20 ul inj.; lml/min; sample
dissolved in mobile phase with 1 drop phosphoric acid.
CHN analysis:
calc.: C 55.51, H 5.38, N 4.98; found: C 55.37, H 5.40, N 5.03.
Melting point: 153.5 C (dec.)
2) SPIB00101: 2-0-Acetylsalicylic acid (2S)-(+)-serine ester
A mixture of N-carbobenzyloxy-L-serine benzyl ester (Z-Ser-OBz1, 23.17 g,
70.34 mmole) and pyridine (30 mL) in anhydrous dichloromethane (500 mL) was
cooled
in an ice bath while under a nitrogen atmosphere. Acetylsalicyloyl chloride
(21.07 g,
106.1 mmole) was added and the mixture was allowed to warm to room temperature
and
stir over two days. After 48 hours, the mixture was poured into ice-cold 2N
hydrochloric acid (400 mL). After mixing, the layers were separated and the
dichloromethane fraction was washed water (500 mL), saturated sodium
bicarbonate
solution (500 mL), water (500 mL), brine (500 mL) and dried over sodium
sulfate (25
g). After filtration, concentration under reduced pressure, and drying under
high
vacuum, the remaining brown solid (47.19 g) was purified by flash
chromatography on
silica gel (200 g, 0.035-0.070 min, 6 nm pore diameter), eluting with
hexanes/ethyl
acetate (3:1). After concentration of the product containing fractions under
reduced
pressure and drying under high vacuum until the weight was constant, the
experiment
produced the protected acetylsalicylic-L-serine ester SPIB0010101 (32.97 g,
95% yield)
as a white solid.
111
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
1.1 0 __________ /NI
H \C)
0
0\ 0 =
SPIB0010101
1H NMR (300 MHz, CDC13): ö = 7.74 (1H, d, J= 7.8 Hz), 7.55 (1H, dt, J= 7.8,
1.5 Hz),
7.33-7.21 (11H, m), 7.08 (1H, d, J= 7.5 Hz), 5.68 (1H, d, J= 8.4 Hz), 5.20
(2H, s), 5.12
(2H, s), 4.77 (1H, m), 4.66 (1H, dd, J= 11.4, 3.3 Hz), 4.57 (1H, dd, ./.=
11.4, 3.3 Hz),
2.30 (3H, s).
13C NMR (75 MHz, CDC13): 8 = 169.45, 169.09, 163.68, 163.35, 155.57, 150.77,
135.87, 134.75, 134.07, 131.44, 128.50, 128.43, 128.27, 128.14, 128.04,
125.92, 123.71,
122.18, 67.83, 67.27, 64.63, 53.55, 21.03.
The protected acetylsalicylic-L-serine ester SPEB0010101 (21.0 g, 42.7 mmole)
was
dissolved in ethanol (100 mL) and ethyl acetate (100 mL) at room temperature
and
added to a Parr bottle that contained 10% palladium on carbon (4.20 g, 50%
wet) under a
nitrogen atmosphere. The nitrogen atmosphere was replaced with hydrogen gas
(20 psi).
After 5 hours additional 10% palladium catalyst (4.26 g) was added and the
hydrogen
atmospere was returned (20 psi). After an additional 20 hours of shaking at
room
temperature, the palladium catalyst was removed by filtration through celite.
The
remaining solids (palladium/celite and product) were washed with water (1500x2
mL)
until the product was removed. The ethanol and water fractions were
concentrated under
reduced pressure at room temperature. The remaining solid (7.17 g) was
dissolved in
DIUF water (4.3 L), filtered through celite to remove insoluble material, and
concentrated under high vacuum at room temperature. The white solid was then
washed
with 1,4-dioxane (100 mL) and DIUF water (50 mL) overnight. After 24 hours the
solid
was filtered and dried under high vacuum until the weight was constant (24
hours).
112
1-1:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The experiment produced the acetylsalicylic-L-serine ester SPIB00101 (6.17 g,
54%
yield) as a white solid.
o 0
C)
-
H'N H
0
0
SPIB001 01
11-1 NMR (300 MHz, D20-DC1): 8 = 8.05 (1H, dd, J= 7.8, 1.5 Hz), 7.75 (1H, dt,
J 7.8,
1.5 Hz), 7.47 (1H, dt, J= 7.8, 0.9 Hz), 7.27 (1H, dd, J= 7.8, 0.9 Hz), 4.87
(1H, dd, J=
12.6, 4.2 Hz), 4.79 (1H, dd, J= 12.6, 3.0 Hz), 4.62 (1H, dd, J= 4.2, 3.0 Hz),
2.39 (3H, s).
13C NMR (75 MHz, D20-DC1): 5 = 173.01, 168.58, 164.54, 149.72, 135.39, 131.59,
126.87, 123.62, 121.15, 62.38, 52.05, 20.44.
HPLC analysis:
98.1% purity; r.t.= 5.839 min.; 65% TFA (0.1%)/35% methanol; 1 mL/min; 35 C;
Luna
C18, 3u column (SN 184225-37), 4.6x250 min; 22 ul injection; DAD1B, Sig = 240,
4
Ref= 550,100.
CHN analysis:
calc.: C 53.93, H 4.90, N 5.24; found: C 54.02, H 5.00, N 5.23.
Melting point: 147.0 C (dec.)
3) SPIB00103: 2-0-Acetylsalicylic acid (2S, 4R)-4-hydroxyproline ester
A mixture of N-carbobenzyloxy-L-hydroxyproline benzyl ester (Z-Ser-OBz1,
21.5 g, 60.5 mmole)1 and pyridine (25 mL) in anhydrous dichloromethane (500
mL) was
cooled in an ice bath while under a nitrogen atmosphere. Acetylsalicyloyl
chloride (13.2
113
1-1:\work\1652\17690\SPEC\17690.spec without clahnslicloc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
g, 66.6 mmole) was added and the mixture was allowed to warm to room
temperature
and stir overnight. After 24 hours, additional acetylsalicyloyl chloride (5.0
g, 25.2
mmole) was added and the mixture was allowed to stir overnight. After 48
hours, the
mixture was poured into ice-cold 1N hydrochloric acid (500 mL). After mixing,
the
layers were separated and the dichloroMethane fraction was washed with water
(500
mL), saturated sodium bicarbonate solution (500 mL), water (500 mL), brine
(500 mL)
and dried over sodium sulfate (25 g). After filtration, concentration under
reduced
pressure, and drying under high vacuum, the remaining yellow oil (40.7 g) was
purified
by flash chromatography on silica gel (460 g, 0.035-0.070 mm, 6 nm pore
diameter),
eluting with heptane/ethyl acetate (3:1). After concentration of the product
containing
fractions under reduced pressure and drying under high vacuum until the weight
was
constant, the experiment produced the protected acetylsalicylic-L-
hydroxyproline ester
SPIB0010301 (21.31 g, 68% yield) as a colorless oil.
o 0 0
1.1
ìr o
c)\
0
SPIB0010301
1H NMR (300 MHz, CDC13): 8 = 7.92 (1H, d, J= 7.8 Hz), 7.56 (1H, t, J= 7.8 Hz),
7.34-
7.21 (10H, m), 7.09 (1H, d, J= 7.8 Hz), 5.48 (1H, s), 5.21 (2H, m), 5.03 (2H,
d, J=15
Hz), 4.57 (1H, m), 3.85 (2H, m), 2.53 (1H, m), 2.28 (4H, m).
13C NMR (75 MHz, CDC13): 6= 171.72, 171.49, 169.25, 163.47, 163.30, 154.52,
153.93, 150.54, 136.05, 135.94, 135.21, 135.00, 134.17, 134.12, 128.43,
128.32, 128.28,
128.20, 128.05, 127.98, 127.94, 127.79, 125.89, 123.70, 122.46, 122.38, 73.24,
72.59,
67.33, 67.11, 66.97, 58.02, 57.69, 52.47, 52.15, 36.74, 35.65, 20.90.
114
1-1:\work\1652\17690\SPEC\17690.spec without clairnslI.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The protected acetylsalicylic-L-hydroxyproline ester SPI110010301 (10.6 g,
20.5
mmole) was dissolved in ethanol (75 mL) and ethyl acetate (75 mL) at room
temperature
and added to a Parr bottle that contained 10% palladium on carbon (3.0 g, 50%
wet)
under a nitrogen atmosphere. The nitrogen atmosphere was replaced with
hydrogen gas
(20 psi). After 17 hours of shaking at room temperature, the reaction mixture
was
washed with water (500 mL) for two hours. The organic layer (top) was removed
via
pipette and the aqueous layer was filtered through celite. The water fraction
was
concentrated under reduced pressure at room temperature. The remaining solid
(6.71 g)
was then washed with anhydrous dichloromethane (35 mL) overnight. After 24
hours
the solid was filtered and dried under high vacuum until the weight was
constant (24
hours). The experiment produced acetylsalicylic-L-hydroxyproline ester,
SPIB00301
(2.87 g, 47.7 % yield) as a white solid.
0 0
=
d\--OH
o
SPIB00103
1H NMR (300 MHz, D20-DC1): 8 = 8.09 (1H, d, J= 7.5 Hz), 7.75 (1H, t, J= 7.5
Hz),
7.48 (1H, t, J= 7.5 Hz), 7.28 (1H, d, J= 7.5 Hz), 5.69 (1H, m), 4.76 (1H, t,
J=7.5 Hz),
3.86 (1H, dd, J= 13.5, 3.9 Hz), 3.74 (1H, d, J= 13.5 Hz), 2.81 (1H, dd, J=
15.0, 7.5 Hz),
2.60 (1H, m), 2.40 (3H, s).
13C NMR (75 MHz, D20-DC1): 8 = 173.13, 170.25, 164.31, 149.65, 135.36, 131.54,
126.87, 123.54, 121.37, 73.86, 58.34, 50.95, 34.38, 20.48.
HPLC analysis:
98.3% purity; r.t.= 7.201 min.; 65% TFA (0.1%)/35% methanol; 1 mL/min; 35 C;
Luna
C18, 3u column (SN 184225-37), 4.6x250 mm; 22 ul injection; DAD1B, Sig= 240, 4
Ref= 550,100.
115
FlAwork\1652117690 \SPEC \ 17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
CHN analysis:
calc.: C 57.34, H 5.16, N 4.78; found: C 57.09, H 5.23, N 4.91.
Melting point: 162 C (dec.)
Gastric Mucosa irritation potential of the L-serine, L-threonine, and L-
Hydroxyproline esters of acetylsalicylic acid compared to acetylsalicylic
acid:-
The present study was conducted to determine the relative potential of new
formulations
of aspirin (L-serine, L-threonine, and L-Hydroxyproline esters of
acetylsalicylic
acid) to cause gastric mucosal irritation/lesions in fasted male albino rats.
Aspirin served
as reference control.
Different new formulations of aspirin and aspirin were administered by gavage
to fasted
male albino rats (Wistar strain), using 0.5% (w/v) Carboxymethylcellulose
(CMC) in
Phosphate Buffer (pH 2.6) solution as the vehicle. The study was conducted at
two dose
levels viz. 100mg and 200mg/kg body weight along with a vehicle control group.
At
each dose level 5 animals were used. All the doses were expressed as aspirin
molar
equivalents. The doses used as well as the molar equivalents were presented
below.
Table 11:Formulation: Molar equivalent
Formulation Molar equivalent
L-serine ester of acetylsalicylic 1.483 units are equivalent to 1 unit of
aspirin
acid
L-Hydroxyproline ester of
1.628 units are equivalent to 1 unit of aspirin
acetylsalicylic acid
L-threonine ester of
1.561 units are equivalent to 1 unit of aspirin.
acetylsalicylic acid
116
11:\work\1652\17690\SPEC\17690.spec without claimslidoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 12: Test Item: Group: Dose (mg per kg) [in terms of acetylsalicylic
acid] :
Equivalent weight of the Test item [mg]
Dose (mg per kg)
Equivalent
[in terms of
Test Item
Groupweight of the
acetylsalicylic
Test item [mg]
acid]
Vehicle control 0.0
Vehicle control
Group
L-serine ester of Test Group 1 100.0 148.3
acetylsalicylic Test Group 2 200.0 296.6
acid
L-Hydroxyproline Test Group 1 100.0 162.8
ester of Test Group 2 200.0 325.6
acetylsalicylic
acid
L-threonine, ester Test Group 1 100.0 156.1
of acetylsalicylic Test Group 2 200.0 312.2
acid
Reference control Test Group 1 100.0 100.0
acetylsalicylic Test Group 2 200.0 200.0
acid
The rats were fasted for a period of 18 to 22 hours before dosing. The test
item was
administered as a single dose by gavage. Three hours after drug
administration, the
animals were killed humanely by CO2 gas inhalation. The stomach was dissected
out and
observed for
= the quantity of mucous exudate,
= degree of hyperemia and thickening of stomach wall,
= hemorrhagic spots (focal or diffuse), nature of hemorrhages (petechial or
ecchymotic) along with the size and
= perforations
The observations on gastric mucosal irritation of animals of various groups
were
summarized below
117
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 13: Test Item: Group: Dose mg/kg (as acetylsalicylic acid): Observation
Dose mg/kg
(as
Test Item GroupObservation
acetylsalicylic
acid
None of the animals showed
Vehicle
Vehicle control 0.0 any evidence of gastric
control Group
mucosal irritation
None of the dosed animals
Test Group 1 100.0 showed any evidence of
L-serine ester of gastric mucosal irritation
acetylsalicylic acid None of the dosed animals
Test Group 2 200.0 showed any evidence of
gastric mucosal irritation.
None of the dosed animals
Test Group 1 100.0 showed any evidence of
L-Hydroxyprolin ester gastric mucosal irritation.
of acetylsalicylic acid None of the dosed animals
Test Group 2 200.0 showed any evidence of
gastric mucosal irritation
None of the dosed animals
Test Group 1 100.0 showed any evidence of
L-threonine, ester of gastric mucosal irritation
acetylsalicylic acid None of the dosed animals
Test Group 2 200.0 showed any evidence of
gastric mucosal irritation
None of the dosed animals
Test Group 1 100.0 showed any evidence of
Reference control gastric mucosal irritation
(acetylsalicylic acid) All the 5 animals dosed,
Test Group 2 200.0 showed evidence of gastric
mucosal irritation.
In conclusion it was observed that none of the L-serine, L-threonine, and L-
Hydroxyproline esters of acetylsalicylic acid induced any evidence of
irritation of gastric
mucosa at the two doses tested viz., 100 and 200mg/kg body weight In contrast,
aspirin
(acetylsalicylic acid) has caused irritation of the gastric mucosal in all the
fasted male
albino rats at the dose level of 200mg/kg. However at the dose level of
100mg/kg aspirin
failed to cause any evidence of gastric mucosal irritation in the male rats.
118
FlAwor1\1652\17690\SPEC\17690.spee without clahnsthdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Further none of the animals of different test groups showed any clinical
symptoms of
toxicity through out the observation period of three hours.
Efficacy of L-serine, L-threonine, and L-Hydroxyproline esters of
acetylsalicylic
acid compared to acetylsalicylic acid on Clotting Time in rats estimated one
hour
after dosing
Observations of blood clotting time
The data on the mean clotting time (MCT) of the animals of low, intermediate
and high
dose groups of different formulations, vehicle control and positive control
groups
estimated one hour after dosing were presented below (Table 14):
Table 14: Summary of Mean Clotting Time ( S.D.) in Minutes - L-serine, L-
threonine,
and L-Hydroxyproline esters of acetylsalicylic acid and Aspirin (Positive
control): Low
dose: Intermediate dose: High dose
Low Dose Intermediate Dose , High Dose
Vehicle control 4.9 1.10
L-serine ester of 5.7 1.34 6.8 1.48 6.9 1.37
acetylsalicylic acid
L-Hydroxyprolin ester 6.1 1.10 5.7 0.82 7.5 1.18
of acetylsalicylic acid _
L-threonine, ester of 5.2 1.14 5.6 0.84 7.4 0.97
acetylsalicylic acid
Positive control 6.2 1.40 8.1 1.97 9.8 1.32
(acetylsalicylic acid)
FIG. 3-6 depict the group mean data of animals regarding the dose relationship
+ mean
clotting time in minutes for the L-series ester of aspirin and for the
control.
The statistical analysis showed a significant improvement at 5% significance
level in the
efficacy for the high dose and mid dose when compared to the vehicle control
group
(Figure 7).
119
FlAwork\1652\176901SPEC\17690.spec without clairnsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
FIG. 4 shows the group mean data of animals. It provides the dose response
relationship
to mean clotting time (MCT) in minutes with respect to L-hydroxyproline ester
of
asperin. The statistical analysis of FIG. 4 showed a significant improvement
at 5%
significance level in the efficacy for the high dose and low dose when
compared to the
vehicle control group (Figure 6)
FIG. 5 depicts the dose response relationship to mean clotting time (MCT) in
minutes of
L-threonine ester of acetylsalicylic acid. The statistical analysis showed a
significant
improvement at 5% significance level in the efficacy for the high dose when
compared
to the vehicle control.
FIG. 6 depicts the dose response relationship to mean clotting time for
acetylsalicylic
acid. The statistical analysis showed a significant improvement at 5%
significance level
in the efficacy for the intermediate and high dose when compared to the
vehicle control.
The dose response effect were statistically significant and clearly evident
(Figure 7).
CONCLUSION
The present study was conducted to evaluate the efficacy of new formulations
of aspirin
using blood clotting time as an index in albino rats. Aspirin served as
positive control.
The study was conducted at three dose levels with the new formulations and
positive
control along with a vehicle control group.
Doses
The doses for the main study were selected based on the dose range finding
experiments
with acetylsalicylic acid. All the doses were expressed as aspirin molar
equivalents. The
doses used for the main experiment for different formulations and positive
control were
same and presented below.
120
flAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 15: Test Item: Low Dose (mg/kg): Intermediate dose 9mg/kg): High dose
(mg/kg)
Low Dose Intermediate High Dose
Test Item
(mg/kg) Dose (mg/kg) (mg/kg)
L-serine ester of
1.0 4.0 10.0
acetylsalicylic acid
L-Hydroxyprolin ester of
1.0 4.0 10.0
acetylsalicylic acid
L-threonine, ester of
1.0 4.0 10.0
acetylsalicylic acid
Aspirin (Positive control) 1.0 4.0 10.0
Efficacy (Blood clotting time)
The efficacy in terms of time required for the blood clotting time at
different dose levels
- low, intermediate and high dose for different formulations and
acetylsalicylic acid are
presented below.
Table 16: Low dose: Intermediate dose: High dose
Low Dose Intermediate Dose High Dose
Vehicle control 4.9 1.10
L-serine ester of 5.7 1.34 6.8 1.48 6.9 1.37
acetylsalicylic acid
L-Hydroxyprolin ester 6.1 1.10 5.7 0.82 7.5 1.18
of acetylsalicylic acid
L-threonine, ester of 5.2 1.14 5.6 0.84 7.4 0.97
acetylsalicylic acid
Positive control 6.2 1.40 8.1 1.97 9.8
1.32
L-serine, L-threonine, and L-Hydroxyproline esters of acetylsalicylic acid are
as
significant as acetylsalicylic acid with respect to clotting time observed
after one hour
but are far superior in terms of the absence of gastric irritation at all
levels compared to
acetylsalicylic acid.
Efficacy of L-serine, L-threonine, and L-Hydroxyproline esters of
acetylsalicylic
acid compared to acetylsalicylic acid on Clotting Time in rats estimated two
hours
after dosing
121
H:\work\1652\17690\SPEC\17690.spec without claimsILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The present study was conducted to evaluate the efficacy of L-serine, L-
threonine, and
L-Hydroxyproline esters of acetylsalicylic acid compared to acetylsalicylic
acid using
blood clotting time, estimated 2 hours ( 10 minutes) after dosing, as an
index in albino
rats. Aspirin served as positive control. Male albino rats were exposed to
aspirin and to 3
new formulations of aspirin at one dose level of 20mg/kg body weight. No
vehicle
control group was used. The doses were expressed as aspirin molar equivalents.
The
doses used for the main experiment for different formulations and positive
control was
presented below.
Table 17: Test Item: Dose in terms of Acetylsalicylic acid 9mg/kg)
Dose in terms of
Test Item
Acetylsalicylic acid (mg/kg)
L-serine ester of acetylsalicylic acid 20.0
L-Hydroxyproline ester of
20.0
acetylsalicylic acid
L-threonine ester of acetylsalicylic acid 20.0
Aspirin (Positive control) 20.0
Efficacy (Blood clotting time)
The efficacy in terms of time required for the blood clotting time at the dose
level of 20
mg/kg body weight for different formulations and aspirin (positive control)
are presented
below.
Observations of Blood clotting time
The data on the mean clotting time (MCT) of the animals, estimated 2 hours (
10
minutes) after dosing, at the dose level of 20mg/kg body weight for different
formulations, vehicle control and positive control are presented below
122
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Table 18: Summary of Mean Clotting Time ( S.D.) in Minutes of L-serine, L-
threonine,
and L-Hydroxyproline esters of acetylsalicylic acid compared to
acetylsalicylic acid
(Positive control)
Dose
(20mg/kg)
L-serine ester of 3.8 0.92
acetylsalicylic acid
L-Hydroxyproline 4.2 1.32
ester of
acetylsalicylic acid
L-threonine ester of 5.3 1.06
acetylsalicylic acid
Positive control 5.4 1.17
(acetylsalicylic acid)
L-serine, L-threonine, and L-Hydroxyproline esters of acetylsalicylic acid
were found to
be effective on clotting time.
In conclusion, it was observed that based on the time required for the blood
to clot
(clotting time), when estimated 2 hours after dosing, the amino acid prodrugs
were
efficacous. However, the L-threonine ester of acetylsalicyclic acid was found
to have
relatively better efficacy than the other two formulations.
As shown by FIG. 7 the statistical analysis showed that L-threonine, and L-
Hydroxyproline esters of acetylsalicylic acid are as effective as
acetylsalicylic acid there
is no significant difference at 5% significance level for L-Hydroxyproline
ester of
acetylsalicylic acid and L-threonine ester of acetylsalicylic with respect to
positive
control for the mean blood clotting time observed after two hours. However,
combined
with the gastric irritation potential, the L-serine, L-threonine, and L-
Hydroxyproline
esters of acetylsalicylic acid are far superior.
123
11:\work\1652\17690\SPEC\17690.speo without claintsadoc
CA 02534342 2011-12-06
There are a number of screening tests to determine the utility of the prodrugs
created
according to the disclosed methods. These include both in vitro and in vivo
screening
methods.
The in vitro methods include acid/base hydrolysis of the prodrugs, hydrolysis
in pig
pancreas hydrolysis in rat intestinal fluid, hydrolysis in human gastric
fluid, hydrolysis
in human intestinal fluid, and hydrolysis in human blood plasma. These assays
are
described in Simmons, DM, Chandran, VR and Portmann, GA, Danazol Amino Acid
Prodrugs: In Vitro and In Situ Biopharmaceutical Evaluation, Drug Development
and
Industrial Pharmacy, Vol 21, Issue 6, Page 687, 1995.
The compounds of the present invention are effective in treating diseases or
conditions
in which NSAIDs normally are used. The prodrugs disclosed herein are
transformed
within the body to release the active compound and enhances the therapeutic
benefits of
the NSAIDs by reducing or eliminating biopharmaceutical and pharmacokenetic
barriers
associated with each of them. However it should be noted that these prodrugs
themselves
will have sufficient activity without releasing any active drug in the
mammals. Since the
prodrugs is more soluble in water then Ibuprofen or other NSAIDs, it does not
need to be
associated with a carrier vehicle, such as alcohol or castor oil which may be
toxic or
produce unwanted side reactions. Moreover, oral formulations containing the
NSAID
prodrugs are absorbed into the blood and are quite effective.
Thus, the prodrug of the present invention enhances the therapeutic benefits
by removing
biopharmaceutical and pharmacokenetic barriers of existing drugs.
Furthermore, the prodrugs are easily synthesized in high yields using reagents
which are
readily and commercially available.
124
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
IV. Proline Derivative of Acetaminophen
Overview:
The procedure for the synthesis of the L-proline ester of acetaminophen is
outlined in
Synthetic Sequence section. The synthesis is exemplary. The complete procedure
and
analytical data is given in the Experimental Section. Acetaminophen (10 g) was
coupled with Boc-L-proline with EDC in the presence of a catalytic amount of
DMAP.
Once the reaction was complete (3 hours at room temperature), the solution was
washed
with water. After drying over sodium sulfate, filtration, and concentration
the crude
protected amino acid ester of acetaminophen was purified by flash
chromatography on
silica gel. The procedure generated the protected L-proline ester of
acetaminophen in
72%. The protecting group was removed by dissolving the ester in
dichloromethane and
passing hydrogen chloride through the solution at room temperature. After
filtration, the
final salt was stirred in tetrahydrofuran until pure. The yield for the
deprotection step
was 91.4% after filtration and drying under high vacuum at 90 C for 4 hours.
Synthetic Sequence:
125
1-1:\work\1652 \17690\SPEC \17690.spec without clainisIi.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/oUS200,4/,00N24901
0 0
= OH a)
HO
H,
0
Acetaminophen boc-L-proline SP1001401
0 ,0
b)
0
H,N
Cl- H
C)
SPI0014
Synthesis of the L-proline ester of acetaminophen: a) EDC, DMAP, CH2C12; b)
HC1
(g), CH2C12.
Experimental Section:
The synthesis of SPI0014 was conducted in one batch. Reagents mentioned in the
experimental section were purchased at the highest obtainable purity from
Lancaster,
Sigma-Aldrich, or Acros, except for solvents, which were purchased from either
Fisher
Scientific or Mallinkrodt.
SPI0014: Pyrrolidine-2(S)-carboxylic acid 4-acetylamino-phenyl ester,
hydrochloride
A mixture of Boc-L-proline (14.39 g, 68.80 mmole), acetaminophen (10.02 g,
66.28
mmole), EDC (12.9 g, 67.29 mmole) and DMAP (1.10 g, 9.0 mmole) in anhydrous
dichloromethane (100 mL) was stirred for 3 hours at room temperature under an
argon
126
H:\work\1652 \17690 \SPEC \17690.spec without claimsn.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
atmosphere. After 3 hours, water (120 mL) was added. After mixing for 5
minutes, the
layers were separated and the dichloromethane fraction was washed with water
(120
mL) and dried over sodium sulfate (5 g). After filtration, concentration under
reduced
pressure, and drying under high vacuum, the remaining oil (24.10 g) was
purified by
flash chromatography on silica gel (100 g, 0.035-0.070 mm, 6 nm pore
diameter),
eluting with hexanes/ethyl acetate (1:2). After concentration of the product
containing
fractions under reduced pressure and drying at high vacuum until the weight
was
constant, the experiment produced the protected acetaminophen-L-proline ester
SPI001401 (16.71 g, 72:30: yield) as a white solid (foam).
o
0 ,i
H,N 1.1
SPI001401
1H NMR (300 MHz, CDC13): 5 = 8.83 (1/2 H, s), 8.70 (1/2 H, s), 7.58 (1/2 H, d,
J= 7.5
Hz), 7.46 (1/2 H, d, J= 7.5 Hz), 6.96 (2 H, m), 4.47 (1 H, m), 3.59-3.45 (2H,
m), 2.36 (1
H, m), 2.17-1.90 (6 H, m), 1.46 (9 H, m).
13C NMR (75 MHz, CDC13): 5 = 171.91, 171.75, 169.02, 154.44, 153.78, 146.36,
146.21, 121.44, 121.23, 120.82, 80.41, 80.17, 59.16, 46.78, 46.55, 31.06,
30.11, 28.50,
24.57, 24.28, 23.78.
The protected acetaminophen-L-proline ester SPI001401 (16.60 g, 47.64 mmole)
was
dissolved in dichloromethane (400 mL) and hydrogen chloride gas was passed
through
the solution for 2 hours at room temperature. The remaining solids were
allowed to
settle (for 1 hour). The dichloromethane was carefully decanted away from the
white
precipitate. Tetrahydrofuran (200 mL) was added to the precipitate and the
mixture
stirred for 2 hours under an argon atmosphere. After filtration, the remaining
white solid
127
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
was dried under high vacuum at 90 C until the product weight was constant (4
hours).
The experiment produced acetaminophen-L-proline ester, hydrochloride SPI0014
(12.4
g, 91.4% yield) as a white solid.
1HNMR (300 MHz, CDCL3-DMS0): 8 = 10.41 (1H, br s), 10.26 (1H, s), 9.55 (1H, br
s), 7.70 (2H, d, J= 9 Hz), 7.12 (2H, d, J= 9 Hz), 4.66 (t, 1H, J= 8.4 Hz),
3.33 (2H, m),
2.43 (1H, m), 2.28 (1H, m), 2.08 (s, 3H), 2.04 (2H, m).
13C NMR (75 MHz, CDCL3-DMS0): 8 = 168.08, 167.25, 144.55, 137.40, 121.12,
119.64, 58.53, 45.33, 27.74, 23.86, 23.08.
HPLC analysis:
99.45% purity; rt= 5.733 min; Luna C18 5u column (sn 167917-13); 4.6x250 mm;
254
nm; 15% Me0H/85% hexane sulfonate buffer (110mMol, pH= 6); 35 C; 20 ul inj.;
lml/min; 5 mg/mL sample size.
CHN analysis:
calc.: C 54.84, H 6.02, N 9.84; found: C 54.66, H 5.98, N 9.65.
Melting point: 221-222 C
V. Amino Acid Derivative of Cyclosporine A
The macrocyclic immunosuppresants comprise a class of structurally
distinctive, cyclic,
poly, N-methylated undecaptides, and similar semi-synthetic macrolide
structures
commonly possessing pharmacological, in particular immunosuppressive, anti-
inflammatory and/or anti-parasitic activity. The first of the cyclosporine to
be isolated
was the naturally occurring fungal metabolite Ciclosporin or Cyclosporine also
known as
cyclosporine A, which has the formula:
128
H:\work\1652\17690\SPEC\17690.spec without clainisadoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
MeBmt-aAbu-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal
r1 2 3 4 5 6 7 8 9 10 11
wherein MeBmt represents N-methyl-(4R)-4-but-2E-en-1-y1-4-methyl-(L) threonyl
residue of the formula
CH3
CH2
HO (R) ,CH
CH (R) CH3
¨ N ¨CH-- CO
(S)
CH3
in which ¨x-y- is CH=CH ¨(trans). Other similar products include, sirolimus
(b),
tacrolimus (c), and pimecrolimus (d), having the following structures:
129
\wor1\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
(b)
4scrOH
H
"4
0i/, H
a
N CHs
0 0 0
0
OH
H3C
= H3C /
H
OH
H3C
CH3, 0
HC0
H300 1 4..3
õ
I 'CH3
'bi^i3 Sirolimus
(c)
H3C0 si
cii3
1 i OH
H3C .
0 i
--CH
2
--
N 0
0
0 OH H3C
H3C g
,
i
(SCH3
00H3 Tacrolimus
130
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
(d)
CI
=
1-1300
01-f3
OH
1-130
0
6
0
0
0 OH H3C
1-13C q
ba-13
OCH3 Pimecrolimus
The class comprised by the cyclosporines is thus now very large indeed and
includes, for
example, [Thr]2-, [Va1]2-, [Nva]2- and [Nva]2-[Nva]5-Cielosporin (also known
as
cyclosporines C, D, G and M respectively), [Dihydrop-MeBmt]l-[Val]2-
ciclosporin (also
known as dihydro-cyclosporine D), [(D)Ser]8-Ciclosporin,
[(D)MeVall11-Ciclosporin (also known as cyclosporine H), [MeAlar-Ciclosporin,
[(D)Pre-Ciclosporin and so on.
In accordance with conventional nomenclature for cyclosporines, these are
defined
throughout the present specification and claims by reference to the structure
of
cyclosporine (i.e., Cyclosporine A). This is done by first indicating the
amino acid
residues present which differ from those present ion Ciclosporin (e.g.,
"[(D)Pro]3" to
indicate that the cyclosporine in question has a ¨(D)Pro- rather than ¨Sar-
residue at the
3-position) and then applying the term Cyclosporine to characterize remaining
residues
which are identical to those present in Cyclosporine A.
131
H:\work\1652\17690\SPEC\17690.spec without clahnsltdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
As used herein, the term "cyclosporines" refers to the various types of
cyclosporines, in
which x-y in the MeBmt residue has a cis or trans CH=CH or in which x-y
therein is
also included in those derivatives in which one or more of those amino acids
in positions
2-11 of Cyclosporine A is replaced by a different amino acid. It is preferred;
however,
that not more than two of the amino acids are replaced in the formula of
cyclosporine A
and more preferentially not more than one of the amino acids is replaced by an
amino
acid.
In addition, amino acid residues referred to by abbreviation, e.g., -Ala-, -
MeVal- and -
aAbu-, are, in accordance with conventional practice, to be understood as
having the
(L)-configuration unless otherwise indicated, e.g. as in the case of "-(D)Ala-
". Residue
abbreviations preceded by "Me" as in the case of "-MeLeu-", represent a-N-
methylated
residues. Individual residues of the cyclosporine molecule are numbered, as in
the art,
clockwise and starting with the residue ¨MeBmt-, dihydro-MeBmt- etc...in
position 1.
The same numerical sequence is employed throughout the present specification
and
claims.
Because of their unique pharmaceutical potential, the macrocyclic
immunosuppressants
have attracted considerable attention in the press. The term "macrocyclic
immuno-
suppressants" includes various natural and semi-synthetic derivatives of
cyclosporine,
and other macrolides such as sirolimus, tacrolimus and pimecrolimus. The
primary area
of clinical investigation for above drugs has been as immunosuppressive
agents, in
particular in relation to its application to recipients of organ transplants,
e.g., heart, lung,
combined heart-lung, liver, kidney, pancreatic, bone-marrow, skin and corneal
transplants, and in particular allogenic organ transplants. These drugs are
also used in
the treatment of psoriasis, atompic dermatitis, rheumatoid arthritis and
nephritic
syndrome.
Macrocyclic immunosuppressants are also useful for treating various autoimmune
diseases and inflammatory conditions and especially inflammatory conditions
with an
132
flAwork\1652 \17690 \SPEC\ 17690.spec without clairasEdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
aetiology, including an autoirnmune component, such as arthritis (for example,
rheumatoid arthritis, arthritis chronica progredient and arthritis deformons)
and
rheumatic diseases. Specific autoinunune diseases for which cyclosporine
therapy has
been proposed or applied include, autoirrunune hematological disorder
(including, e.g.,
hemolytic anemia, aplastic anemia, pure red cell anemia, and idiopathic
thrombocytopaenia), systemic lupus erythematosus, polychondritis, sclerodoma,
Wegener granulamatosis, dermatomyositis, chronic active hepatitis, myasthenia
gravis,
psoriasis, Steven-Johnson syndrome, idiopathic sprue, autoimmune inflammatory
bowel
disease, including, e.g., ulcerative colitis and Crohn's disease), endocrine
opthalmopathy Graves disease, sarcoidosis, multiple sclerosis, primary
billiary cirrhosis,
juvenile diabetes (diabetes mellitus type I), uvetis (anterior and posterior),
keratoconjunctivitis sicca and vernal keratoconjunctivitis, interstial lung
fibrosis,
psoriatic arthritis, atopic dermatitis and glomerulonephritis (with and
without nephrotic
syndrome, e.g., including idiopathic nephritic syndrome or minimal change
nephropathy).
Furthermore, macrocyclic immunosuppressants also have applicability as an anti-
parasitic, in particular anti-protozoal agent, and are suggested to be useful
for treating
malaria, coccidiomycosis and schistomsomiasis. More recently, they have been
taught
to be useful as an agent for reversing or abrogating anti-neoplastic agent
resistance
contumors, and the like.
Despite the very major contribution which macrocyclic immunosuppressants have
made,
difficulties have been encountered in providing more effective and convenient
means of
administration (e.g., galenic formulations, for example, oral dosage form,
which are both
convenient and for the patient as well as providing appropriate
bioavailability and
allowing dosaging at an appropriate and controlled dosage rate) as well as the
reported
occurrence of undesirable side reactions; in particular nephrotoxic reactions
have been
obvious serious impediments to its wider use or application.
133
HAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Moreover, the above mentioned macrocyclic immunosuppressants are
characteristically
highly hydrophobic and readily precipitate in the presence of even very minor
amounts
of water, e.g., on contact with the body (e.g., stomach fluids). It is
accordingly
extremely difficult to provide e.g., oral formulations which are acceptable to
the patient
in terms of form and taste, which are stable on storage and which can be
administered on
a regular basis to provide suitable and controlling patient dosaging.
Proposed liquid formulations, e.g., for oral administration of macrocyclic
immunosuppressants, have heretofore been based primarily on the use of ethanol
and
oils or similar excipients as carrier media. Thus, the commercially available
macrocyclic immunosupressant drink-solution employs ethanol and olive oil or
corn-oil
as carrier medium in conjunction with solvent systems comprising e.g., ethanol
and
LABR1FIL and equivalent excipients as carrier media. Thus, the commercially
available
macrocyclic immunosupressant drink solution employs ethanol and olive oil or
corn-oil
as carrier medium in conjunctions with a Labrffil as a surfactant. $ee e.g.,
U.S. Patent
NO. 4,388,307. Use of the drink solution and similar composition as proposed
in the art
is however accompanied by a variety of difficulties.
Further, the palatability of the known oil based system has proved
problematic. The
taste of the known drink-solution is, in particular, unpleasant. Admixture
with an
appropriate flavored drink, for example, chocolate drink preparation, at high
dilution
immediately prior to ingestion has generally been practiced in order to make
regular
therapy at all acceptable. Adoption of oil based systems has also required the
use of
high ethanol concentrations to itself inherently undesirable, in particular
where
administration to children is forseen. In addition, evaporation of the
ethanol, e.g., from
capsules (adopted in large part, to meet problems of palatability, as
discussed or other
forms (e.g., when opened) results in the development of a macrocyclic
immunosupressant precipitate. When such compositions are presented in, for
example,
soft gelatin encapsulated form an additional problem arises. This particular
difficulty
necessitates packaging of the encapsulated product in an air-tight component,
for
134
FlAwork\1652 \ 17690 \SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
example, an air-tight blister or aluminum-foil blister package. This in turn
renders the
product both bulky and more expensive to produce. The storage characteristics
of the
aforesaid formulations are, in addition, far from ideal.
Bioavailability levels achieved using existing oral macrocyclic
immunosupressant
dosage system are also low and exhibit wide variation between individuals,
individual
patient types and even for single individuals at different times during the
course of
therapy. Reports in the literature indicates that currently available therapy
employing
the commercially available macrocyclic immunosupressant drink solution
provides an
average absolute bioavailability of approximately 30% only, with the marked
variation
between individual groups, e.g., between liver (relatively low
bioavailability) and bone-
marrow (relatively high bioavailability) transplant recipients. Reported
variation in
bioavailability between subjects has varied from one or a few percent for some
patients,
to as much as 90% or more for others. And as already noted, marked change in
bioavailability for individuals with time is frequently observed. Thus, there
is a need for
a more uniform and high bioavailability of macrocyclic immunosupressant in
patients.
Use of such dosage forms is also characterized by extreme variation in
required patient
dosaging. To achieve effective immunosuppressive therapy, blood or blood serum
levels
compounds of the cyclosporin have to be maintained within a specified range.
This
required range can in turn, vary, depending on the particular condition being
treated,
e.g., whether therapy is to prevent transplant rejection or for the control of
an
autoimmune disease, or condition and on whether or not alternative
immunosuppressive
therapy is employed concomitantly with any of the immunosuppressants of the
formula
a-d therapy. Because of the wide variations in bioavailability levels achieved
with
conventional dosage forms, daily dosages needed to achieve required blood
serum levels
will also vary considerably from individual to individual and even for a
single
individual. For this reason it is necessary to monitor blood/blood-serum
levels of
patients receiving macrocyclic immunosuppressant therapy at regular and
frequent
intervals. Monitoring of blood/blood-serum levels, which is generally
performed by
135
H:\work\1652 \ 17690 \SPEC\ 17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
RIA or equivalent immunoassay technique, e.g. employing monoclonal antibody
based
technology, has to be carried out on a regular basis. This is inevitably time
consuming
and inconvenient and adds substantially to the overall cost of therapy.
It is also the case that blood/blood serum macrocyclic immunosuppressant
levels
achieved using available dosage systems exhibit extreme variation between peak
and
trough levels. That is for each patient, effective macrocyclic
immunosuppressant levels
in the blood vary widely between administrations of individual dosages.
There is also a need for providing macrocyclic immunosuppressant in a water
soluble
form for injection. It is well known that Cremephore L used in a current
formulations of
macrocyclic immunosuppressants is a polyoxyethylated derivative of castor oil
and is a
toxic vehicle. There have been a number of incidences of anaphylaxis due to
the castor
oil component. At present there is no formulation that would allow the
macrocyclic
immunosuppressants to be in aqueous solution at the concentrations needed due
to poor
water solubility of the drug.
Beyond all these very evident practical difficulties lies the occurrence of
undesirable
side reactions already alluded to, observed employing available oral dosage
forms.
Several proposals to meet these various problems have been suggested in the
art,
including both solid and liquid oral dosage forms. An overriding difficulty
which has
however remained is the inherent insolubility of the macrocyclic
immunosuppressants in
aqueous media, hence preventing the use of a dosage form which can contain
macrocyclic immunosuppressants in sufficiently high concentration to permit
convenient
use and yet meet the required criteria in terms of bioavailability, e.g.
enabling effective
resorption from the stomach or gut lumen and achievement of consistent and
appropriately high blood/blood-serum levels.
136
FlAwork\1652\17690\SPEC\17690.spec without claims11.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The particular difficulties encountered in relation to oral dosaging with
macrocyclic
immunosuppressants have inevitably led to restrictions in the use of
macrocyclic
immunosuppressant therapy for the treatment of relatively less severe or
endangering
disease conditions. A particular area of difficulty in this respect has been
the adoption of
macrocyclic immunosuppressant therapy in the treatment of autoimmune diseases
and
other conditions affecting the skin, for example for the treatment of atopic
dermatitis and
psoriasis and, as also widely proposed in the art, for hair growth
stimulation, e.g. in the
treatment of alopecia due to ageing or disease.
Thus while oral macrocyclic immunosuppressant therapy has shown that the drug
is of
considerable potential benefit to patients suffering e.g. from psoriasis, the
risk of side-
reaction following oral therapy has prevented common use. Various proposals
have
been made in the art for application of macrocyclic immunosuppressants, e.g.
cyclosporine, in topical form and a number of topical delivery systems have
been
described. Attempts at topical application have however failed to provide any
demonstrably effective therapy.
However, the present invention overcomes the problems described hereinabove.
More
specifically, an embodiment of the present invention is a prodrug of
macrocyclic
immunosuppressant which significantly enhances its solubility in aqueous
solutions,
thereby avoiding the need to utilize a carrier, such as ethanol or castor oil
when
administered as a solution. Moreover, the prodrugs of macrocyclic
immunosuppressant,
in accordance with the present invention, do not exhibit the side effects of
the prior art
formulations. Further, the inventor has found that the macrocyclic
immunosuppressant
prodrugs of the present invention enhance its absorption when administered in
the
prodrug form to a patient, thereby enhancing significantly its bioavailability
and its
efficacy.
Accordingly, in one aspect, the present invention is directed to a prodrug of
macrocyclic
immunosuppressants. The prodrug consists of an amino acid esterified to the
free
137
FlAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
hydroxy group present on the side chain of cyclospoiine, sirolimus, tacrolimus
and either
one of the hydroxyl groups of the pimecrolimus molecule.
For example, an aspect of the present invention is directed to, the compounds
of the
formulas
(a)
CH3
X
CH2
AA0 CH
HC CH3
________________________ N¨CH¨CO¨CYCLO _________________
CH3
138
1{:\work\1652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
(b)
0
.s." AA or GLYAA
CH .-"-`0CH3
CH3
0 0 0 SIROLIMUS
0
OH
H3C
H H3C
OH
H3C
CH3 õ 0
H3C0 co
õ,CH3
''CF13
(c)
AA-GLY or AA ¨ O. TACROL1MUS
1-11,,C0 cm3
' OH
H30
0
Cpuipc..6CH
0
H30
0
OH H3C
H3C
oCH3
OCH3
139
1-1:\work\ 1652 \17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
(d)
Cl PIMECROLIMUS
H,co 9H,
0 ¨AA or GLY,AA
H3C
0
CH3
H3C
0
0 OH H3C
H3C q
'0013
OCH3
or pharmaceutically acceptable salts thereof;
wherein CYCLO represents the residues at positions 2-11 of the cyclosporine
molecule;
x-y is CH=CH or CH2CH2 and AA is an amino acid or a dipeptide of the formula
GLY-
AA. In the latter case, GLY is glycine and AA is any a-amino acid. In the
dipeptide
structure, an AA is attached to the drug via OH group using glycine as the
spacer.
Glycine is esterified to cyclospoiine and then glycine is bonded to any AA via
amide
linkage using amino group of glycine and carboxylic acid group of AA.
The present invention is also directed to a pharmaceutical composition
comprising a
therapeutically effective amount of the compounds of the Formulas a-d above
and a
pharmaceutical carrier therefor.
In another embodiment, the present invention is directed to a method of
treating a patient
in need of macrocyclic immunosuppressant therapy, which method comprises
administering to said patient an effective amount of the compounds of Formulas
a-d.
140
H:\work\1652\17690\SPEC\17690.spec without claimsILdoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
In a further embodiment, the present invention is directed to a method of
enhancing the
solubility of macrocyclic immunosuppressant in an aqueous solution comprising
reacting
the hydroxy functionality in the MeBmt moiety at position 1 of the
cyclosporine
molecule as well as the specified hydroxyl functions in formulas b-d with an
amino acid
or acylating derivative thereof under ester forming conditions or by using a
simple
amino acid or a dipeptide structure wherein the AA is attached to drug using
glycine as
the spacer and isolating and isolating the product thereof.
In a still further embodiment, the present invention is directed to a method
of enhancing
the bioavailability of macrocyclic immunosuppressants when administered to a
patient
which comprises reacting the hydroxy functionality in the MeBmt moiety in
position of
the cyclospotine molecule with an amino acid or acylating derivative under
ester
forming conditions and as well as the specified hydroxyl functions in formulas
b-d with
an amino acid or acylating derivative thereof under ester forming conditions
or by using
a simple amino acid or a dipeptide structure wherein the AA is attached to
drug using
glycine as the spacer and isolating the product thereof and administering said
product to
the patient.
Overview:
The procedure for the synthesis of the N-(L-proline)-glycine and N-(L-lysine)-
glycine
esters of Cyclosporine A is outlined in Synthetic Sequence section. These
examples are
exemplary of the synthetic scheme using amino acids. The complete procedure
and
analytical data is given in the Experimental Section. Cyclosporine A (15 g)
was
coupled with chloroacetic anhydride (4 equivalent) in anhydrous pyridine. The
experiment produced the chloroacetate ester of Cyclosporine A (SPI001201, 14
g, 88%
yield) in good yield. The chloroacetate ester (10.1 g) was then treated with
sodium azide
in DMF to generate the azidoacetate ester of Cyclosporine A (SPI001202, 9.9 g,
97%
yield). The azidoacetate (9.8 g) was then reduced with tin chloride (9 g) to
prepare the
glycine ester of Cyclosporine A (8.54 g, 89% yield). The glycine ester of
Cyclosporine
A (SPI001203) was then coupled with a two-fold excess of either boc-L-proline
or Boc-
141
H: \work\1652\17690 \SPEC \17690.spec without clahnsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
L-lysine using EDC as the coupling agent. After purification by column
chromatography, the boc protecting groups were removed from the dipeptide
esters of
Cyclospotine A at low temperature (5 C) by treatment with 2M hydrochloric
acid in
diethyl ether. The L-lysine-glycine ester salt of Cyclosporine A did not
require
additional purification and was dried. The L-proline-glycine ester salt of
Cyclosporine
A required purification. The salt was converted to the free-base with sodium
bicarbonate and purified by filtration through silica gel (eluting with
acetone). The salt
was then formed at low temperature with dilute anhydrous hydrochloric acid and
dried.
142
11:\work\1652\17690\SPEC\17690.spec without claims1I.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Synthetic Sequence:
N,
NCI N /
ji 0:
r 1
0 I 0 i 3
....0
N 0 N¨ a N 0 N¨
O + c====1-0-JUI --3.- o 0 \ 0
H.N...<1\....r
0 \ N
.../ 0 H
.44)N.1 14_,NN-1.),\
.... o Nx.. ;
SPI001201
Cyclosporin A
1 b
....õ/ o (3, N3L i
......, 0õ,
r i c
0E-
g7 . c-Nro 0 . . . \ro
'0 N¨ N 0 N¨
H 0
0
0 \ .NICk"--( //. 0 H
\N=1),\1CCC--
441/ 0 H O H N-.11---5,\
oNy.
oK,'N-y
SPI001203 SPI001202
1 d,f
cwsLro
H CI- H H HN,..31,
H-.N
H = \--"NoriC4H 0 Cr-
ci- HT1N-H ....v.., 04,
C /
N 0 N¨
......\____µ".\A.i).-1011 0 Nrror N ..'c, 0
"N o 0
N =-= N-
4/
YJY 141H43Y1'4N -\
H.
__fr/0 0
0 \tsi____ci\..,r
_
---CONN
SPI0023
SPI0022
Synthesis of the N-(L-proline)-glycine and N-(L-Lysine)-glycine esters of
Cyclosporine A: a) pyridine; b) NaN3, DMF; c) SnC12, methanol; d) boc-L-
lysine,
EDC; e) boc-L-proline, EDC; f) HC1, Et20.
143
H: \work\1652\17690 \SPEC \17690.spe,c without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Experimental Section:
The synthesis of SPI0022 and SPI0023 was conducted in batches. Generally a
small-
scale experiment was performed first followed by a larger batch. Reagents
mentioned in
the experimental section were purchased at the highest obtainable purity from
Aldrich,
Acros, or Bachem, except for solvents, which were purchased from either Fisher
Scientific or Mallincicrodt. The Cyclosporine A (USP grade) used in these
procedures
was provided by Signature Pharmaceuticals, Inc.
1) SPI001201
ci,_ jok
/
01, 0 ro
0
Cyclosporine A (15.01 g, 0.0124 moles) was dissolved in anhydrous pyridine (35
mL) at
room temperature, under an argon atmosphere. The solution was cooled to 5 C
in an
ice/water batch and chloroacetic anhydride (9.10 g, 0.053 moles) was added.
After
stirring for 10 minutes, the ice bath was removed and the solution was allowed
to stir
under an argon atmosphere at room temperature for 17 hours. After 17 hours,
diethyl
ether (200 mL) was added. The ether was washed with water (2x100 mL) and dried
for
1 hour over sodium sulfate (10 g). After filtration and concentration under
reduced
pressure, the remaining yellow foam was dried under high vacuum (1 hour at
room
temperature) and purified by flash chromatography on silica gel (200 g),
eluting with
heptane/acetone (2:1). After combining and concentrating the product
containing
fractions, the remaining light yellow foam (14.8 g) was purified a final time
by
crystallization from hot diethyl ether (140 mL). After cooling (-10 C, 2
hours),
filtration, and drying under high vacuum, the procedure generated the
chloroacetate ester
of Cyclosporine A SPI001201 as a white solid (14.0 g, 88.3% yield).
144
H:\work\ 1652 \ 17690 \ SPEC\ 17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Cyclosporine A chloroacetate ester:
1H NMR (300 MHz, CDC13):
5 = 8.50 (d, 1H, J= 9.6 Hz), 7.95 (d, 1H, J= 6.6 Hz), 7.46 (d, 1H, J= 9.0 Hz),
7.40 (d, 1H,
J= 7.8 Hz), 5.35-4.52 (m, 15 H), 4.37 (t, 1H, J= 7.2 Hz), 4.12 (d, 1H, J= 14.7
Hz), 3.89
(d, 1H, J= 14.7 Hz), 3.45-3.0 (m, 15 H), 2.8-2.5 (m, 6H), 2.5-1.5 (m, 16H),
1.5-0.7 (m,
53 H).
13C NMR (75 MHz, CDC13):
ö= 173.78, 173.37, 172.86, 172.61, 171.28, 171.18, 170.91, 170.79, 168.78,
167.64,
167.18, 128.77, 126.68, 75.46, 65.95, 58.89, 57.47, 55.80, 55.31, 54.86,
54.34, 50.19,
48.91, 48.35, 48.02, 44.80, 40.96, 39.44, 37.07, 35.93, 33.85, 33.25, 32.40,
31.74, 31.50,
30.38, 30.12, 29.82, 29.53, 25.13, 24.92, 24.78, 24.40, 23.99, 23.75, 22.85,
21.94, 21.41,
21.25, 20.84, 19.85, 18.79, 18.32, 17.89, 17.82, 15.46, 15.24, 10.08.
2) SPI001202
0
0
0 \
The chloroacetate ester of Cyclosporine A SPI001201 (10.10 g, 7.89 mmole) was
dissolved in anhydrous N,N-dimethlformamide (30 mL) at room temperature.
Sodium
azide (2.15 g, 33.0 mmole) was added. The mixture was allowed to stir at room
temperature for 24 hours in the dark, under an argon atmosphere. After 24
hours, diethyl
ether (150 mL) was added and the precipitate was filtered. The ether was
washed with
water (2x100 mL), dried over sodium sulfate (15 g) for 30 minutes, filtered,
and
145
H:\work\1652\17690\SPEC\17690.spec without claimsli.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
concentrated under reduced pressure. The remaining white solid was dried under
high
vacuum for 1 hour at room temperature. The experiment produced the
azidoacetate ester
of Cyclosporine A SPI001202 (9.90 g, 97% yield) as a white solid, which was
used
without further purification.
Cyclosporine A azidoacetate ester:
1HNMR (300 MHz, CDC13):
8 = 8.48 (d, 1H, J= 9.3 Hz), 7.95 (d, 1H, J= 6.9 Hz), 7.45 (d, 111, J= 9.0
Hz), 7.39 (d, 1H,
J= 7.8 Hz), 5.5-4.5 (m, 15 H), 4.31 (t, 1H, J= 6.6 Hz), 4.04 (d, 1H, J= 17.3
Hz), 3.53 (d,
1H, J= 17.3 Hz), 3.45-3.0 (m, 15 H), 2.8-2.5 (m, 6H), 2.5-1.5 (m, 16H), 1.5-
0.7 (m, 53
H).
13C NMR (75 MHz, CDC13):
8 = 173.76, 173.32, 172.82, 172.53, 171.13, 170.89, 170.76, 170.69, 169.70,
168.20,
167.49, 128.63, 126.61, 74.96, 58.91, 57.39, 55.56, 55.21, 54.80, 54.23,
50.14, 48.99,
48.23, 48.24, 47.93, 44.71, 40.89, 39.33, 39.22, 37.02, 35.83, 33.81, 32.96,
32.31, 31.67,
31.42, 30.31, 30.09, 29.76, 29.47, 25.08, 24.92, 24.84, 24.67, 24.51, 24.40,
23.94, 23.82,
23.71, 21.85, 21.33, 21.25, 20.82, 19.79, 18.71, 18.25, 17.92, 17.81, 15.17,
10.03.
3) SPI001203
NE1,,.... A
/
tsil.--0 14 ¨
H
0 =
µ,
oLy1:1;4õitisN
The azidoacetate ester of Cyclosporine A SPI001202 (9.80 g, 7.62 mmole) was
dissolved in methanol (250 mL) at room temperature. Water (40 mL) was added
146
H:\work\1652\17690\SPEC\17690.spec without clainislI.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
followed by tin (II) chloride (5 g, 26.3 nunole). The solution was allowed to
stir for 1
hour at room temperature when an additional quantity of tin (II) chloride (4
g, 21.0
rnmole) was added. The solution was allowed to stir for an additional 2 hours
at room
temperature. Water (200 mL) containing ammonium hydroxide (40 mL, 29%) was
added. After filtration, the solution was concentrated (to 200 mL) under
reduced
pressure. The remaining aqueous solution was extracted with ethyl acetate
(2x200 mL).
The ethyl acetate fractions were combined, dried over sodium sulfate (20 g),
filtered and
concentrated under reduced pressure. The remaining clear foam was purified by
filtration through silica gel (150 g), eluting with dichloromethane/methanol
(20:1). The
procedure generated the glycine ester of Cyclosporine A as a clear, solid foam
(8.54g,
89% yield).
Glycine ester of Cyclosporine A:
1H NMR (300 MHz, CDC13):
= 8.60 (d, 1H, J= 9.6 Hz), 8.06 (d, 1H, J= 6.9 Hz), 7.53 (d, 1H, J= 8.4 Hz),
7.51 (d, 1H,
J= 6.6 Hz), 5.7-4.52 (m, 15 H), 4.41 (t, 1H, J= 6.9 Hz), 3.5-3.0 (m, 17 H),
2.82-2.5 (m,
8H), 2.5-1.5 (m, 16H), 1.5-0.7 (m, 53 H).
13C NMR (75 MHz, CDC13):
8 = 174.10, 173.67, 173.23, 172.72, 172.55, 171.18, 171.10, 170.73, 170.61,
169.68,
167.77, 128.82, 126.42, 73.83, 58.57, 57.32, 55.99, 55.20, 54.74, 54.31,
50.08, 48.82,
48.28, 47.90, 44.70, 43.81, 40.74, 39.33, 39.24, 37.02, 35.84, 33.72, 33.07,
32.39, 31.72,
31.41, 30.25, 29.98, 29.74, 29.51, 25.05, 24.81, 24.73, 24.54, 24.31, 23.91,
23.78, 23.68,
21.86, 21.33, 21.25, 20.68, 19.76, 18.74, 18.24, 17.94, 17.79, 15.18, 10.03.
4) SPI0022
147
FI:\wor1\1652\176901SPEC\17690.spec without claitnsil.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
CI¨
H¨.N.
0
0õ,
0 N,H-Hcr, Nrc,
N N-
0
o'YjLrN
The glycine ester of Cyclosporine A (SPI001203, 2.0 g, 1.59 mmole) was
dissolved in
anhydrous dichloromethane (25 mL) with boc-L-lysine (1.31 g, 3.78 mmole) and
EDC
(0.75 g, 3.9 mmole), under an argon atmosphere at room temperature. The boc-L-
lysine
was prepared from the dicyclohexylamine salt (2.0 g in 50 mL ether) by
extraction with
cold potassium hydrogen sulfate solution (1 g in 50 mL water) followed by cold
water
(2x50 mL). The ether containing the boc-L-lysine was dried over sodium sulfate
(5 g),
filtered, concentrated and dried under high vacuum for one hour at room
temperature. A
few crystals of DMAP were added to the mixture of EDC, boc-L-lysine, and the
glycine
ester of Cyclosporine A and the solution was allowed to stir for 4 hours at
room
temperature. The dichloromethane solution was extracted with DIUF water (50
mL),
5% sodium bicarbonate solution (50 mL), and with DIUF water (50 mL). After
drying
over sodium sulfate (10 g), the dichloromethane solution was filtered and
concentrated
under reduced pressure. The remaining white foam (3.01 g) was purified by
flash
column chromatography on silica gel (50 g), eluting with heptane/acetone
(2:1). The
product containing fractions were combined, concentrated under reduced
pressure, and
dried under high vacuum. The purified protected intermediate (2.34 g white
solid,
92.8% yield) was placed in a flask under an argon atmosphere, which was cooled
in an
ice-water bath. Cold anhydrous 2 M hydrochloric acid in diethyl ether (20 mL)
was
added and the solution stirred for 8 hours (at 5 C). The mixture was slowly
allowed to
warm to room temperature overnight. After stirring for a total of 20 hours,
the flask was
cooled again in an ice-water bath for 30 minutes. The product was filtered and
dried
under high vacuum for 1 hour at room temperature and then at 50 C for 4
hours. The
148
FlAwork11652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
experiment produced Cyclosporine A N-(L-lysine)-glycine ester, dihydrochloride
trihydrate (SPI0022, 1.59 g, 73.9% yield) as a white solid.
11-1NMR (300 MHz, CDC13, NMR data is for the free base):
5 = 8.58 (d, 111, J= 9.3 Hz), 8.04 (d, 1H, J= 6 Hz), 7.80 (d, 1H, J= 6 Hz),
7.49 (d, 2H, J=
8.4 Hz), 5.70-4.6 (m, 17 H), 4.41 (m, 1H), 4.28 (dd, 1H, J= 17, 7.2 Hz), 3.67
(d, 1H, J=
17 Hz), 3.46 (s 3H), 3.4-2.8 (m, 16 H), 2.8-2.5 (m, 8H), 2.5-1.35 (m, 24H),
1.5-0.7 (m,
50H).
13C NMR (75 MHz, CDC13, NMR data is for the free base):
5 = 175.23, 173.77, 173.34, 172.75, 172.63, 171.34, 171.22, 170.94, 170.84,
170.91,
169.89, 169.70, 128.74, 126.67, 74.41, 58.82, 57.43, 55.91, 55.21, 54.81,
54.42, 50.17,
48.89, 48.31, 47.98, 44.78, 41.92, 40.82, 40.69, 39.44, 39.32, 27.19, 35.91,
34.88, 33.71,
, 33.25, 33.12, 32.44, 31.83, 31.50, 30.38, 30.06, 29.81, 29.55, 25.14, 24.90,
24.52, 24.43,
24.00, 23.76, 21.93, 21.42, 21.29, 20.81, 19.84, 18.82, 18.32, 17.96, 17.86,
15.21, 10.10.
= CHN analysis:
Calculated for C7011128a2N14015-3H20: C 55.50, H 8.92, and N 12.74; found: C
58.28,
H 8.98, and N 13.16.
HPLC analysis:
99.60% purity; r.t.= 14.763 min.; 80% acetonitrile/20% Tris base in DIUF
water; 1
mL/min; 60C; Synergi Hydro RP, 4u column (serial # 163383-7), 4.6x250 mm; 20
ul;
UV= 210 nm.
Melting point: 196.0-198 C (uncorrected)
149
H:\work\1652 \I7690 \SPEC \ 17690.spec without claintsILdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
5) SPI0023
fo
fr `ii HN.....ik I
N
\
H
.....<...t
N" H H. 1,1=3')\-10.
0 E
The glycine ester of Cyclosporine A (SPI001203, 7.50 g, 5.95 mmole) was
dissolved in
anhydrous dichloromethane (50 mL) with boc-L-proline (2.56 g, 11.90 mmole) and
EDC
(2.28 g, 11.9 mmole), under an argon atmosphere at room temperature. A few
crystals
of DMAP were added to the mixture of EDC, boc-L-proline, and the glycin.e
ester of
Cyclosporine A and the solution was allowed to stir for 3 hours at room
temperature.
The dichloromethane solution was extracted with DIUF water (50 mL), 5% sodium
bicarbonate solution (2x50 mL), and with DIUF water (50 mL). After drying over
sodium sulfate (10 g), the dichloromethane was filtered and concentrated under
reduced
pressure. The remaining white foam (9.50 g) was purified by flash column
chromatography on silica gel (150 g), eluting with heptane/acetone (2:1
followed by
1:1). The product containing fractions were combined, concentrated under
reduced
pressure, and dried under high vacuum (7.94 g white solid, 91.7% yield) for 10
minutes
at room temperature.
The purified protected intermediate (6.46 g) was placed in a flask under an
argon
atmosphere, which was cooled in an ice-water bath. Cold anhydrous 2 M
hydrochloric
acid in diethyl ether (150 mL) was added and the solution stirred for 8 hours
(at 5 C).
The mixture was slowly allowed to warm to room temperature overnight. After
stirring
for a total of 20 hours, the flask was cooled again in an ice-water bath for
30 minutes.
The product was filtered and dried under high vacuum for 30 minutes at room
temperature. The Cyclosporine A N-(L-proline)-glycine ester, hydrochloride
(5.17 g,
84.6% yield, and 90% purity by HPLC) was converted to the free base by
dissolving the
1 50
H:\work\1652\17690\SPEC\17690.spec without claimsadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
salt in DIUF water (25 mL) that contained sodium bicarbonate (1 g). The free
base was
extracted with dichloromethane (3x25 mL), which was dried over sodium sulfate
(5 g),
filtered and concentrated. The remaining off-white solid (5 g) was purified by
filtration
through silica gel (100 g), eluting with acetone. The product containing
fractions were
combined, concentrated under reduced pressure, and dried under high vacuum for
30
minutes at room temperature. The hydrochloride salt was regenerated by
dissolving the
free base (3.8 g) in diethyl ether (25 mL) and adding it to telydrous 2M
hydrochloric
acid (5 mL) in heptarie (50 mL), while cooling in an ice-water bath. After 20
minutes at
5 C, the white solid was filtered and dried under high vacuum for 6 hours at
room
temperature. The experiment produced Cyclosporine A N-(L-proline)-glycine
ester,
hydrochloride (SPI0023, 3.8 g) as a white solid.
1H NMR (300 MHz, CDC13):
8 = 14.20 (br s, 2H), 8.62 (d, 1H, J= 10 Hz), 8.06 (d, 1H, J= 6.9 Hz), 7.61
(d, 1H, J= 8.1
Hz), 7.48 (d, 1H, J= 9 Hz), 5.70-5.50 (m, 3H), 5.40-4.60 (m, 12H), 4.37 (m,
1H), 4.20
(d, 1H, J= 18 Hz), 3.97 (d, 1H, J= 18 Hz), 3.70 (m, 1H), 3.45 (s, 3H), 3.23-
3.08 (m,
12H), 2.66 (s, 3H), 2.60 (s, 3H), 2.50-1.80 (m, 15H), 1.78-1.20 (m, 15H), 1.15-
0.66 (m,
46H).
13C NMR (75 MHz, CDC13):
= 174.15, 173.49, 172.67, 172.59, 171.86, 171.20, 171.13, 171.02, 170.83,
169.68,
168.77, 167.55, 128.30, 127.10, 80.09, 75.58, 62.65, 59.35, 57.36, 55.53,
55.30, 54.78,
54.35, 53.60, 50.25, 50.09, 48.92, 48.18, 48.12, 44.62, 40.59, 40.02, 39.43,
39.30, 37.13,
35.88, 33.74, 33.07, 32.19, 32.01, 31.86, 31.50, 31.43, 30.43, 29.93, 29.72,
29.30, 29.16,
27.56, 26.04, 25.00, 24.86, 24.74, 24.39, 20.96, 19.81, 18.71, 18.26, 18.09,
17.85, 17.79,
15.09, 14.30, 10.00.
CHN analysis:
Calculated for C691-1122C1N13014: C 59.48, H 8.83, and N 13.07; found: C
59.84, H 9.02,
and N 12.65.
151
11:\work\ 1652\17690 \SPEC\17690.spec without claimsn.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
HPLC analysis:
99.59% purity; r.t.= 10.613 min.; 85% acetonitaile/15% Tris base in DIUF
water; 1.2
mL/min; 60C; Synergi Hydro RP, 4u column (serial # 163383-7), 4.6x250 mm; 20
ul;
UV= 210 nm.
Melting point: 197.0-199 C (uncorrected)
These prodrugs of cyclosporin of the present invention are effective in
treating diseases
or conditions in which macrocyclic immunosuppressants normally are used. These
prodrugs are transformed within the body to release the active compound and
enhances
the therapeutic benefits of the macrocyclic immunosuppressants by reducing or
eliminating biopharmaceutical and pharmacokenetic barriers associated with
each of
them. However it should be noted that these prodrugs themselves will have
sufficient
activity without releasing any active drug in the mammals. Since the prodrugs
are more
soluble in water then cyclosporine or other macrocyclic immunosuppressants, it
does not
need to be associated with a carrier vehicle, such as alcohol or castor oil
which may be
toxic or produce unwanted side reactions. Moreover, oral formulations
containing the
prodrugs of the prodrugs are absorbed into the blood and are quite effective.
Thus, the prodrug of cyclosporin of the present invention enhances the
therapeutic
benefits by removing biopharmaceutical and pharmacokenetic barriers of
existing drugs.
Furthermore, the prodrugs are easily synthesized in high yields using reagents
which are
readily and commercially available.
VI. Valproic Acid Esters
Valproic acid (2-Propylpentanoic acid) is low molecular weight carboxylic acid
derivative which is widely used as an anti-convulsive agent, useful in the
treatment of
epilepsy and also possess vasodilatation activity in the brain to relieve
migraine
152
H:\work\1652\17690\SPEC\17690.spec without claimsIl.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
headaches. It is administered orally to control epileptic episodes in humans
and also
alleviate severe pain associated with migraine headaches.
113C
OH
o
VALPROIC ACID
Valproic acid has been shown to have a large number of therapeutic
applications, which
are quite varying and somewhat surprising. For example, in addition to its
efficacy in the
treatment of epilepsy and migraine headaches, it has been shown to be
effective in the
treatment of certain psychiatric illnesses, such as bipolar disorder, mood
stabilization,
control of aggression, impulsivity in personality disorder, agitation in
dementia, and has
also been of use as adjunct therapy in the treatment of post traumatic stress
disorder
(PTSD).
Mechanism of Action:
In spite of being used in the treatment of epilepsy for a number of years, the
exact
mechanism of action of Valproic acid is still unknown. It has been postulated
that it
exerts its action by increasing concentration of gamma-amino butyric acid
(GABA) in
the brain. Gamma-amino butyric acid is a neurotransmitter, a chemical that
nerves use to
communicate with one another.
Valproate is the drug of choice in myoclonic epilepsy, with or without
generalized tonic-
clonic seizures, including juvenile myoclonic epilepsy of Janz that begins in
adolescence
or early adulthood. Photosensitive myoclonus is usually easily controlled.
Valproate also
is effective in the treatment of benign myoclonic epilepsy, postanoxic
myoclonus, and,
with clonazepam, in severe progressive myoclonic epilepsy that is
characterized by
tonic-clonic seizures as well. It also may be preferred in certain stimulus-
sensitive
(reflex, startle) epilepsies.
153
11:\work\1652\17690\SPEC\17690.spee without clainisII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Although Valproate may be effective for infantile spasms; it is relatively
contraindicated
in children whose spasms are due to hyperglycinemia or other underlying
metabolic
(mitochondrial) abnormalities. In general, atonic and akinetic seizures in
patients with
Lennox-Gastaut syndrome are difficult to control, but Valproate is the drug of
choice for
treatment of mixed seizure types. Since this drug has been useful in some
patients who
are refractory to all other antiepileptic drugs, it may warrant a trial in
nearly all
nonresponsive patients regardless of seizure type.
In spite of it usefulness, hepatotoxicity may be fatal, but is idiosyncratic
and not
preventable by routinely monitoring liver enzymes. Hepatotoxicity occurs in
very young
children, most often those on multiple anticonvulsants. Valproate-induced
cytopenias
may be dose-related and warrant monitoring of complete blood counts during
therapy.
Encephalopathy with hyperammonemia without liver function test abnormalities
may
occur. Pregnant women in first month are at risk for neural tube defects.
Valproic acid is a low molecular weight liquid with characteristic odor. Taken
orally it
has unpleasant taste and can severely irritate mouth and throat. In order to
convert
Valproic acid into a solid dosage form convenient for oral administration, a
number of
derivatives with covalent and ionic bond with the carboxylic acid have been
made. A
simple sodium salt of Valproic acid, resulting in Valproate sodium is
available as a solid.
However a stable coordination complex, know as Divalproex sodium was formed by
partial neutralization of two molecules of Valproic acid with one atom of
sodium. This
product is the most widely available Valproic acid hemi salt marketed by
Abbott
Laboratories in the USA under the brand name Depakote . Depakote is also
available
in extended release formulation for oral administration.
A significant disadvantage of Valproic acid is that it in liquid form it is
difficult to
administer. Furthermore, administration of Valproic acid in different forms
does not
uniformly produce desired bioavailability. For example, the overall
bioavailability of
Valproate from Valproic acid, its sodium salt, Divalproex , and their extended
release
154
11:\work\1652\176901SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
formulations are not quite interchangeable. Since continuous monitoring of
plasma
profile of Valproic acid is essential, any change in plasma concentration due
to changes
in the formulation adversely affect overall therapeutic outcome.
In order to improve the therapeutic effectiveness, uniform blood profile,
develop
pharmaceutically elegant formulation and reduce first pass metabolism, present
invention discusses prodrugs of Valproic acid which overcome some of the
difficulties
stated above.
Until now there has been no pharmaceutical preparation has been available in
the market
that can deliver Valproic acid with out harmful side effects. The present
inventior.
however, has produced a number of water soluble, non-toxic derivatives of
Valproic acid
which are suitable for delivering Valproic acid consistently in the body
without any
harmful side effects and without the needs for expensive additives, and
exepients.
Accordingly, in one aspect, the present invention is directed to a class of
prodrugs of
Valproic acid. The prodrug consists of the hydroxyl group of an amino acid
esterified to
the free carboxyl group present on the Valproic acid molecules. In another
embodiment,
the amine group of the amino acid is reacted with COOH group to form an amide
linkage.
More specifically, an embodiment of the present invention is directed to, the
compounds
of the formula
HC
VALPROIC ACID
155
1-1:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
or pharmaceutically acceptable salts thereof; wherein R is either NH-AA or 0-
AA and
AA is an amino acid, in which either an amine group or the hydroxyl group is
reacted_
with the carboxylic acid group of Valproic Acid.
The present invention is also directed to a pharmaceutical composition
comprising a
therapeutically effective amount of the various Valproic acid prodrugs above
and a
pharmaceutical carrier therefor.
In another embodiment, the present invention is directed to a method of
treating a patient
in need of Valproic acid therapy, which method comprises administering to said
patient
an effective amount of the Valproic acid.
In a further embodiment, the present invention is directed to a method of
converting
liquid Valproic acid into a solid powder by reacting the carboxyl
functionality of the
Valproic acid with either amine or hydroxyl functionality of an amino acid and
isolating
the products thereof.
In a still further embodiment, the present invention is directed to a method
of
substantially and in a therapeutically efficacious manner, reducing or
eliminating the
potential first pass metabolism thereby improving the consistent therapeutic
effect by
administering to a patient a prodrug which comprises reacting the COOH
functionality
of the Valproic acid molecule with either NH2 or OH functionality of selected
amino
acids to form an ester or amide covalent bond respectively and isolating the
product
thereof and administering said product to the patient.
It has been found that when unsubstituted naturally occurring amino acids are
esterified
to Valproic acid, the resulting prodrugs are pharmaceutically elegant free
flowing
powders, and are rapidly absorbed into the body and release non-toxic amino
acids upon
cleavage in the body and require none of the emulsifiers, additives and other
exepients.
156
H:\work\1652\17690\SPEC\17690.spec without clainisadoc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Furthermore, it has been shown +11it the current invention also produced
drugs, while
they are prodrugs of Valproic acid; they were highly effective anti-epileptics
and were
exhibiting such effect intact. Thus the current amino acid prodrugs are
effective anti-
epileptics and useful in the treatment of a number of psychiatric illnesses
and exhibit
such potential with or without releasing the active parent drug.
The Valproic acid prodrug bulk density is much higher than the corresponding
sodium
salts, and they are suitable for compacting large weight tablets and capsules.
Furthermore, their do not exhibit bitter taste and unusual odor of the
Valproic acid.
While the prodrugs my invention are not supposed to possess any acidic
activity due to
blockage of the carboxylic acid group responsible for such, however it has
been shown
that the prodrugs are effective anti-epileptics with or without releasing
Valproic acid.
However, all of the Valproic acid prodrugs described are released in vivo the
active drug
with all its pharmacological and psychoactive properties.
The prodrug of Valproic acid clearly provides a number of advantages over
Valproic
acid, for example, all of the side chains cleaved from these prodrugs are
naturally
occurring essential amino acids hence are non-toxic. This results in high
therapeutic
index. Secondly all the prodrugs are readily cleaved in the body to release
Valproic acid.
Furthermore, due their high water solubility, they can be easily administered
by either
forming an in-situ solution just before IV administration using lyophilized
sterile powder
or providing the drug in solution in prefilled syringe or bottles for
infusion. The
aminoacid esters are more stable than Valproic acid since COOH group in
Valproic acid
is blocked to reaction with bases. Thus the Valproic acid prodrugs invented
here are
more effective then Valproic acid itself without the toxicity and other
pharmaceutical
problems associated with current marketed formulations.
The procedure for the synthesis of the L-serine, L-threonine, and L-
hydroxyproline
30= esters of valproic acid (2-propylpentanoic acid) is outlined in
Synthetic Sequence
157
FlAwork\1652\17690\SPEC\17690.speo without claiinsIl.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
section and is exemplary for alai preparation of the various prodrugs of the
present
invention. The complete procedure and analytical data is given in the
Experimental
Section. In general, valproic acid (2-8 g, in batches) was coupled with the N-
benzyloxy/benzyl ester protected amino acids using EDC in the presence of a
catalytic
amount of DMAP. Once the reactions were complete (20 hours at room
temperature),
the mixture was extracted with DIUF water, dried over sodium sulfate, and
concentrated
under reduced pressure. The crude material was either used directly for the
deprotection
step or purified by column chromatography. The procedure generated the
protected
amino acid esters of valproic acid in yields ranging from 72% to 92%. The
protecting
groups were removed by hydrogenation (30 psi H2) in the presence of 10%
palladium on
carbon. The amino acid esters of valproic acid were extracted away from the
palladium
catalyst with ethanol, concentrated, and dried. The final salts were formed by
acidification with hydrochloric acid. The crude salts (yields ranging from 57%
to 92%)
were then purified by the methods described in the Experimental Section.
158
H:\wor1\1652\17690\SPEC\17690.spec without claiins11.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Synthetic Sequence:
1. SPIC001
O o
o
4. HO.---CLOBz1 a" b) 1L'O 0
-4 OH ,.. r IYICE3zI YCH
z c)
H c.
,N
H 'cbz H I H
CI
H
Valproic acid Z-Ser-OBzI SPIC00101 SPIC001
2. SPIC002
O 0 li
0
+ r r .crioszi cyLoBzi b)
N
\cbz a) )((:)
N
0
k.
0
0,tcr-koH
N
H
Valproic acid Z-Hyp-OBzI SPIC00201 SPIC002
3. SPIC003
1 0
a)
ecm + HO;"¨jOM ....:bz ----"-
H c II, 1 0
0:0Bz1 b)
N
Hõcbz c)
(
H I H -
H Cl
Valproic acid 2-Thr-OBzI SPIC00301 SPIC003
Synthesis of the L-serine, L-threonine, and L-hydroxyproline esters of
valproicacid: a) EDC, DMAP, CH2C12; b) H2, 10% Pd/C, Et0H, Et0Ac; c) HC1.
,
159
H:\work\1652\17690\SPEC\17690.spec without claiinsILdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Experimental Section:
The synthesis of SPIC001, SPIC002 and SPIC003 was conducted in one or two
batches. Reagents mentioned in the experimental section were purchased at the
highest
obtainable purity from Lancaster, Sigma-Aldrich, Acros, or Bachem, except for
solvents,
which were purchased from either Fisher Scientific or Mallinkrodt.
1) SPIC001: 2-Propylpentanoic acid 2(S)-amino-2-carboxy-ethyl ester,
hydrochloride
(L-Serine-valproic acid ester, hydrochloride)
A mixture of 2-propylpentanoic acid (valproic acid, 6.48 g, 44.93 mmole), N-
carbobenzyloxy-L-serine benzyl ester (Z-Ser-OBz1, 14.80 g, 44.93 mmole), EDC
(8.61
g, 44.91mmole), and DMAP (549 mg, 4.49 mmole) in anhydrous dichloromethane (50
mL) was stirred under an argon atmosphere at room temperature for 20 hours.
After 20
hours, the dichloromethane was washed with water (3x50 mL), dried over
magnesium
sulfate (5 g), filtered and concentrated under reduced pressure. The remaining
colorless
oil (20.87 g) was purified by column chromatography on silica gel (150 g,
0.035-0.070
mm, 6 nm pore diameter), eluting with hexanes/ethyl acetate (3:1). After
concentration
of the product containing fractions under reduced pressure and drying under
high
vacuum until the weight was constant, the experiment produced the protected L-
serine-
valproate ester SPIC00101 (18.9 g, 92% yield) as a colorless oil.
0
o
YL0
HNsr0
SPIC00101 0 A ik
160
H:\work\1652\17690\SPEC\17690.spec without clairnsILdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
IHNMR (300 MHz, DMS0): 8 = 7.96 (1H, d, J= 8.1 Hz), 7.35 (10H, m), 5.14 (2H,
s),
5.05 (2H, s), 4.51 (1H, m), 4.29 (2H, m), 2.29 (1H, m), 1.50-1.25 (4H, m),
1.25-1.10
(4H, m), 0.80 (6H, t, J= 6.6 Hz).
13C NMR (75 MHz, DMS0): 8 = 174.88, 169.15, 155.85, 136.58, 135.45, 128.26,
128.18, 127.47, 127.71, 127.57, 66.32, 65.66, 62.47, 53.09, 44.20, 33.86,
33.79, 19.95,
13.85.
The protected L-serine-valproate ester SPIC00101 (18.9 g, 41.48 mmole) was
dissolved
in ethanol (60 mL) and ethyl acetate (60 mL) at room temperature and added to
a Parr
bottle (500 mL) that contained 10% palladium on carbon (3.0 g, 50% wet) under
a
nitrogen atmosphere. The nitrogen atmosphere was replaced with hydrogen gas
(30 psi).
After 4 hours of shaking, additional palladium catalyst (1.0 g) in
ethanol\ethyl acetate
(1:1, 100 mL) was added and the reaction mixture shook overnight under
hydrogen gas
(30 psi) at room temperature. After 24 hours the catalyst was removed by
filtration
through a thin layer of activated carbon. The ethanol and ethyl acetate were
concentrated
under reduced pressure at room temperature. After drying under high vacuum,
the
remaining solids were acidified with hydrochloric acid in diethyl ether (2M,
24.6 mL).
The mixture was stored in a refrigerator for two hours before filtration and
washing with
additional cold diethyl ether (10 mL). After filtration, the remaining white
solid was
dried at room temperature under high vacuum until the product weight was
constant (24
hours). The experiment produced L-serine-valproic acid ester, hydrochloride
SPIC001
(6.34 g, 57% yield) as a white solid.
0
0
N
I 1-1 Cl
SPIC001
161
FlAwork\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
1HNMR (300 MHz, DMS0): 8 = 8.73 (br s, 3H), 4.47 (dd, 111, J= 12.9, 4.5 Hz),
4.31
(dd, 2H, J= 12.9, 3.6 Hz), 2.36 (m, 1H), 1.50 (m, 2H), 1.39 (m, 2H), 1.20 (m,
4H), 0.84
(t, 6H, J= 7 Hz).
13C NMR (75 MHz, DMS0): 8 = 174.67, 168.19, 61.84, 51.16, 44.12, 33.76, 33.58,
20.07, 19.92, 13.97, 13.89.
HPLC analysis:
98.49% purity; rt= 4.767 min; Luna C18 5u column (sn 167917-13); 4.6x250 mm;
254
nm; 33% ACN/66% DIUF water; 35 C; 20 ul inj.; lml/min; 20 mg/mL sample size;
sample dissolved in mobile phase.
CHN analysis:
calc.: C 49.34, H 8.28, N 5.23; found: C 49.22, H 8.35, N 5.24.
Melting point: 159-160 C
2) SPIC002: 4(R)-(2-Propyl-pentanoyloxy)-pyrrolidine-2(S)-carboxylic acid
(L-Hydroxyproline-valproic acid ester)
A mixture of 2-propylpentanoic acid (valproic acid, 4.32 g, 30 mmole), N-
carbobenzyloxy-L-hydroxyproline benzyl ester (Z-Hyp-OBz1, 10.66 g, 30 mmole)',
EDC (5.74 g, 30 mmole), and DMAP (366 mg, 3 mmole) in anhydrous
dichloromethane
(30 mL) was stirred under an argon atmosphere at room temperature for 20
hours. After
20 hours, the dichloromethane was washed with water (3x30 mL), dried over
magnesium sulfate (5 g), filtered and concentrated under reduced pressure. The
remaining colorless oil SPIC00201 (11.95 g, 24.7 mmole, 82.4% yield) was used
without purification.
162
11:\work\1652\17690\SPEC\17690.spec without claimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
0
0 ,,,CYLO
0 *I
SPIC00201
1H NMR (300 MHz, CDC13): = 7.29 (10H, m), 5.28-5.00 (5H, m), 4.55 (1/2H, t, J--
= 8
Hz), 4.46 (1/2H, t, J= 8 Hz), 3.80-3.60 (2H, m), 2.43-2.16 (3H, m), 1.60-1.45
(2H, m),
1.40-1.32 (2H, m), 1.28-1.20 (4H, m), 0.86 (6H, m).
13C NMR (75 MHz, DMS0): 8 = 174.74, 171.40, 171.05, 153.79, 153.31, 136.34,
136.20, 135.57, 135.38, 128.24, 128.13, 127.95, 127.87, 127.67, 127.52,
127.28, 127.10,
72.29, 71.53, 66.34, 66.10, 57.66, 57.19, 52.27, 51.89, 44.13, 40.33, 35.78,
34.79, 34.04,
33.92, 33.35, 20.00, 19.91, 13.79, 13.73.
The protected L-hydroxyproline-valproate ester SPIC00201 (17.24 g, 35.79
mmole) was
dissolved in ethanol (50 mL) and ethyl acetate (100 mL) at room temperature
and added
to a Parr bottle (500 mL) that contained 10% palladium on carbon (3.5 g, 50%
wet)
under a nitrogen atmosphere. The nitrogen atmosphere was replaced with
hydrogen gas
(30 psi). After 15 hours of shaking, the catalyst was removed by filtration
through a thin
layer of celite and activated carbon. The ethanol and ethyl acetate mixture
was
concentrated under reduced pressure at room temperature. After drying
overnight under
high vacuum at room temperature, the experiment produced L-hydroxyproline-
valproic
acid ester SPIC002 (9.2 g, 99.8% yield) as a white solid. In order to remove
trace
impurities, the zwitterion was purified by reverse-phase column chromatography
(50 g
ODS silica gel) in two batches. The zwitterion was placed on the column in
DIUF water
and eluted with mixture of DIUF water/methanol (2:1, 1:1, 1:2, 100% methanol).
163
11:\work\1652\17690\SPEC\17690,spec without claitnsEdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
The product containing fractions were combined, concentrated under reduced
pressure at
20 C (or less), and dried under high vacuum at room temperature until the
weight was
constant (24 hours, 6.4 g white solid recovered).
0 +.0)LOH
Ns
SPIC002
1H NMR (300 MHz, CDC13): = 12.40 (br s, 1H), 8.32 (br s, 1H), 5.28 (m, 1H),
4.11 (t,
1H, Jr= 7.2 Hz), 3.59 (m, 1H), 3.34 (br d, 1H, Jr= 10.5 Hz), 2.50-2.22 (m,
3H), 1.62-1.50
(m, 2H), 1.50-1.32 (m, 2H), 1.32-1.19 (m, 4H), 0.88 (t, 6H, Jr= 7.2 Hz).
13C NMR (75 MHz, CDC13): 8 = 175.99, 173.35, 71.83, 59.56, 49.77, 45.08,
36.19,
34.51, 20.87, 14.31.
HPLC analysis:
99.20% purity; r.t.= 7.228 min.; 70% DIUF water/30% acetonitrile; 1 mL/min;
36.8C;
Luna C18, 5u column (serial # 167917-13), 4.6x250 mm; 22 ul injection; sample
dissolved in mobile phase.
CHN analysis:
calc.: C 60.68, H 9.01, N 5.44; found: C 60.58, H 9.12, N 5.48.
Melting point: 179.0-180.0 C
3) SPIC003: 2-Propyl-pentanoic acid 2(S)-amino-2-carboxy-1(R)-methy1-ethy1
ester,
hydrochloride
164
FlAwork\1652\17690\SPEC\17690.spec without claimsII.doe
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
(L-Threonine-valproic acid ester, hydrochloride)
A mixture of 2-propylpentanoic acid (valproic acid, 4.32 g, 30 mmole), N-
carbobenzyloxy-L-threonine benzyl ester (Z-Thr-OBz1, 10.30 g, 30 mmole), EDC
(5.74
g, 30 mmole), and DMAP (366 mg, 3.0 mmole) in anhydrous dichloromethane (30
mL)
was stirred under an argon atmosphere at room temperature for 20 hours. After
20
hours, the dichloromethane was washed with water (3x30 mL), dried over
magnesium
sulfate (5 g), filtered and concentrated under reduced pressure. The remaining
colorless
oil (13.44 g) was purified by column chromatography on silica gel (100 g,
0.035-0.070
mm, 6 nm pore diameter), eluting with hexanes/ethyl acetate (4:1).
After concentration of the product containing fractions under reduced pressure
and
drying under high vacuum until the weight was constant, the experiment
produced the
protected L-threonine-valproate ester SPIC00301 (12.65 g, 89.8% yield) as a
colorless
oil.
0 t: 0
-)L0).Y0
Ny
O
SPIC00301
111 NMR (300 MHz, CDC13): 8 = 7.40-7.05 (11H, m), 5.45 (1H, m), 5.17-5.02 (4H,
m),
4.53 (1H, d, J= 9.6 Hz), 2.24 (1H, m), 1.58-1.40 (2H, m), 1.40-1.15 (9H, m),
0.86 (6H,
m).
13C NMR (75 MHz, DMS0): ô = 174.24, 169.29, 156.48, 136.61, 135.34, 128.26,
128.20, 127.74, 127.67, 127.58, 69.04, 66.33, 65.78, 57.62, 44.50, 33.89,
33.80, 20.03,
19.91, 16.40, 13.87.
165
H:\work\1652\17690\SPEC\17690.spec without clairnsII.doc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
The protected L-threonine-valproate ester SPIC00301 (12.65 g, 26.9 mmole) was
dissolved in ethanol (50 mL) and ethyl acetate (50 mL) at room temperature and
added
to a Parr bottle (500 mL) that contained 10% palladium on carbon (2.53 g, 50%
wet)
under a nitrogen atmosphere. The nitrogen atmosphere was replaced with
hydrogen gas
(30 psi). After 20 hours the catalyst was removed by filtration through a thin
layer of
activated carbon, washing with ethanol (25 mL). The ethanol and ethyl acetate
were
concentrated under reduced pressure at room temperature. After drying under
high
vacuum, the remaining solids (6.13 g) were acidified with hydrochloric acid
(3.1 mL
conc.) in DIUF water (50 mL). The solution was filtered a second time through
activated carbon and dried overnight in a freeze-dryer. The experiment
produced L-
threonine-valproic acid ester, hydrochloride SPIC003 (6.52 g, 86.0 % yield) as
a white
solid.
The combined batches of the L-threonine-valproic acid ester, hydrochloride
SPIC003
(8.8 g) were purified by crystallization form acetonitrile. After the salt was
dissolved in
hot acetonitrile (225 mL), the material was treated activated acrbon,
filtered, and placed
in a 5 C refrigerator overnight. The white solids were filtered after 18
hours, washed
with cold acetonitrile (10 mL), and dried under high vacuum at room
temperature until
the product weight was constant (24 hours). The process recovered L-threonine-
valproic
acid ester, hydrochloride SPIC003 (6.82 g, 77.5 % recovery) as a white solid.
166
1-Bwork\1652\17690\SPEC\17690.spec without claimsILdoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
0 0
OOH
H I H ¨
H CI
SPIC003
111NMR (300 MHz, DMS0): 8 = 8.71 (br s, 3H), 5.28 (m, 1H), 4.16 (d, 1H, J=
2.7Hz),
2.33 (m, 1H), 1.56-1.40 (m, 2 H), 1.37-1.27 (m, 5H), 1.21-1.13 (m, 4H), 0.84
(t, 6H, J=
6.6 Hz).
13C NMR (75 MHz, DMS0): 8 = 173.97, 168.19, 67.69, 55.42, 44.43, 33.95, 33.78,
20.07, 19.95, 16.54, 13.94.
HPLC analysis:
98.88% purity; r.t.= 4.864 min.; 70% DIUF water/30% acetonitrile; 1 mL/min;
40C;
Luna C18, 5u column (serial # 211739-42), 4.6x250 mm; 20 ul injection; sample
dissolved in mobile phase.
CHN analysis:
calc.: C 51.15, H 8.59, N 4.97; found: C 51.29, H 8.59, N 4.98.
Melting point: 144 C
Solubility of the above esters wee determined in water at room temperature by
dissolving excess of each of the drug and allowing them to settle for a few
hours. The
resulting solutions were centrifuged at 150Orpm for 3 min and the supernatant
liquid was
analyzed. It was shown that these esters possess solubility in water in excess
of 50
mg/mL.
167
11:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2011-12-06
There are a number of screening tests to determine the utility of the prodrugs
created
according to the disclosed methods. These include both in vitro and in vivo
screening
methods.
The in vitro methods include acid/base hydrolysis of the prodrugs, hydrolysis
in pig
pancreas, hydrolysis in rat intesetinal fluid, hydrolysis in human gastric
fluid, hydrolysis
in human intestinal fluid, and hydrolysis in human blood plasma. These assays
are
described in Simmons, DM, Chandran, VR and Portmann, GA, Danazol, Amino Acid
Prodrugs: In Vitro and In Situ Biopharmaceutical Evaluation, Drug Development
and
Industrial Pharmacy, Vol. 21, Issue 6, Page 687, 1995,.
Prodrugs of Valproic acid of the present invention are effective in treating
diseases or
condidiotns in which Valproic acid normally are used. The prodrugs disclosed
herein
are transformed within the body to release the active compond and enhances the
therapeutic benefits of the Valproic acid by reducing or eliminating
biopharmaceutical
and pharmacokinetic barriers associated with each of them. However it should
be noted
that these prodrugs themselves will have sufficient activity without releasing
any active
drug in the mammals.
Thus, the prodrug of the present invention enhances the therapeutic benefits
by removing
biopharmaceutical and pharmacokenetic barriers of existing drugs.
Furthermore, the prodrugs are easily synthesized in high yields using reagents
which are readily and commercially available.
VII WATER SOLUBLE PRODRUGS OF FIBRIC ACID DERIVATIVES
Fibric acid derivatives are useful anti-hyperlipidemic drugs useful in the
treatment of
hyperlipidemia in mammals where the symptoms are elevated triglycerides, low
HDL
(High density lipoproteins or "good" cholesterol, and elevated cholesterol.
Fibric Acid
derivatives are also useful in reducing LDL (Low density lipoproteins, or
"bad"
168
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
cholesterol). The general structure of the filmic acid analogs is represented
below, where
X is various mixed aliphatic and aromatic functionalities. Specific
derivatives included
in this formula are clofibric acid, fenofibric acid, ciprfibrate and
gemfibrozil and the
like.
X xl,L,
OH
H3C CH3
FIBRIC ACID ANALOGS
Typical examples of the chemical moiety X in the above structure are shown
below.
0
=Ciprofibric Acid
a a
Clofibric Acid
Çl
cl 0,
411
Fenofibric Acid
o
ei,3
Gemfibrozil
H3c
169
IT: \work\ 1652 \17690 \SPEC\17690.spec without claimsIl.doc
CA 02534342 2011-12-06
Fibric acid analogs shown in the structure above have been shown to have a
large
number of therapeutic applications, which are quite varying and somewhat
surprising.
Broadly, these derivatives are useful in the treatment dyslipidemia and
dyslipoproteinemia. Dyslipidemia and dyslipoproteinemia are herein defined to
include
the group selected from hypercholesterolemia, abnormal and elevated levels of
cholesterol, abnormal and elevated levels of LDL cholesterol, abnormal and
elevated
levels of total cholesterol, abnormal and elevated levels of plasma
cholesterol, abnormal
and elevated levels of triglycerides, hypertrigylceridaemia, abnormal levels
of
lipoproteins, abnormal and elevated levels of low density lipoproteins (LDLs),
abnormal
and elevated levels of very low density lipoproteins, abnormal and elevated
levels of
very low intermediate density lipoproteins, abnormal levels of high density
lipoproteins,
hyperlipidemia, hyperchylomicronemia, abnormal levels of chylomicrons, related
disorders, and combinations thereof such as those described in The ILIB Lipid
Handbook for Clinical Practice, Blood Lipids and Coronary Heart Disease,
Second
Edition, A. M. Gotto et al, International Lipid Information Bureau, New York,
N.Y.,
2000,.
Mechanism of Action:
The mechanism of action of Fibric acid derivatives seen in clinical practice
have been
explained in-vivo in transgenic mice and in vitro in human hepatocyte cultures
by the
activation of peroxisome proliferator activated receptor alpha (PPAR-alpha).
Through
this mechanism, Fibric acid derivatives increase lipolysis and elimination of
triglyceride-
rich particles from plasma by activating lipoprotein lipase and reducing
production of
apoprotein C-III (an inhibitor of lipoprotein lipase activity).
The resulting fall in ttiglycerides produces an alteration in the size and
composition of
LDL from small, dense particles (which are thought to be atherogenic due their
susceptibility to oxidation), to large buoyant particles. These larger
particles have greater
affinity for cholesterol receptors and are catabolized rapidly. Activation of
PPAR-alpha
also induces an increase in the synthesis of apoproteins A-I, A-II, and HDL
cholesterol.
170
CA 02534342 2011-12-06
Fibric Acid derivatives are also useful in the treatment of gout, as they
reduce serum uric
acid levels in hyperurecemic patients.
Hyperlipidemia types include type I, type IIa, type Ilb, type III, type IV,
and type V.
These types can be characterized according to the levels relative to normal of
lipids
(cholesterol and triglycerides) and lipoproteins described above. Different
classifications
are derived from Drug Facts and Comparisons, 52nd Edition (1998) page 1066 .
Many of the fibric acid derivatives when administered orally do not have
sufficient
bioavilability and absorption are variable, erratic and depended upon food. In
fact
absolute bioavialbility of many of the fibric acid derivatives is not possible
since the
prodrugs of fibric acids currently marketed as insoluble in water, hence a
parenteral
formuation is difficult or not available. Furthermore, since these drugs
usually
administered as esters, they are in fact prodrugs. These prodrugs have to be
metabolized
in the body to release active drug, which are the filmic acids. However, due
to the ester
formation of these drugs, they are quite insoluble in water, hence are
difficult to
formulate, and are not easily broken down in the body to release active drugs.
Many of the Fibric acid derivatives are low to medium molecular weight solids
with
characteristic odor. Taken orally it has unpleasant taste and can severely
irritate mouth
and throat. Taken with food provides more blood concentration compared to
fasting.
This fed/fast difference in bioavailability is more pronounced when Filmic
acid
derivatives are compared against their corresponding prodrug derivatives.
Overall
bioavailability has been reported anywhere between 40-60 and quite variable
among
patients.
One of the significant problems associated with currently marketed fibric acid
derivatives being that when these prodrugs are cleaved in the body, they
release the
prodrug moiety, which themselves are highly toxic. For example, in the case of
171
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
fenofibrate and gemfibrozil isopropyl alcohol is released as the esterase
enzyme cleave
the pro-moiety from the fenofibric acid. It is well know isopropanol is highly
toxic when
released into any of the mammalian tissues.
In order to improve the therapeutic effectiveness, uniform blood profile,
develop
pharmaceutically elegant formulation and improve the solubility of the drug in
water,
present invention discusses alternative prodrugs of Fibric acid derivatives
which
overcome many of the difficulties stated above.
Accordingly, in one aspect, the present invention is directed to alternate
class of
prodrugs of Fibric acid derivatives. The prodrug consists of the hydroxyl
group of an
amino acid esterffied to the free carboxyl group present on the Fibric acid
derivatives
molecules. In another embodiment, the amine group of the amino acid is reacted
with
COOH of the filmic acids to form an amide linkage.
More specifically, in one aspect of the present invention is directed to, the
compounds of
the formulas
H><
tise
FIBRIC ACID ANALOGS
where x is as defined hereinabove
or pharmaceutically acceptable salts thereof; wherein R is either NH-AA or 0-
AA and
AA is an amino acid, in which either an amine group or the hydroxyl group is
reacted
with the carboxylic acid group of Fibric acid derivatives.
172
H:\work\1652\17690\SPEC\17690.spec without claimslicloc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
The present invention is also directed in an embodiment to a pharmaceutical
composition comprising a therapeutically effective amount of the various
Fibric acid
derivatives prodrugs above and a pharmaceutical carrier therefor.
In another embodiment, the present invention is directed to a method of
treating a patient
in need of Fibric acid derivatives therapy, which method comprises
administering to said
patient an effective amount of the Fibric acid derivatives.
In a further embodiment, the present invention is directed to a method of
converting
liquid Fibric acid derivatives into a solid powder by reacting the carboxyl
functionality
of the Fibric acid derivatives with either amine or hydroxyl functionality of
an amino
acid and isolating the products thereof.
In a still further embodiment, the present invention is directed to a method
of
substantially and in a therapeutically efficacious manner, make the
derivatives absorbed
easily upon oral administration thereby improving the consistent therapeutic
effect by
administering to a patient a prodrug which comprises reacting the COOH
functionality
of the Fibric acid derivatives molecule with either NH2 or OH functionality of
selected
amino acids to form an ester or amide covalent bond respectively and isolating
the
product thereof and administering said product to the patient.
It was determined that when unsubstituted naturally occurring amino acids are
esterified
to Fibric acid derivatives, the resulting prodrugs are pharmaceutically
elegant free
flowing powders, and are rapidly absorbed into the body and release non-toxic
amino
acids upon cleavage in the body and require none of the emulsifiers, additives
and other
exepients.
Furthermore, it has been found that the current invention also produced drugs,
while they
are prodrugs of Fibric acid derivatives; they were highly effective anti-
hyperlipidemics
and were exhibiting such effect intact. Thus the current amino acid prodrugs
are
173
H:\work\1652\17690\SPEC\17690.spec without claims11.cloc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
effective anti-hyperlipidemics and useful in the treatment of a number of high
cholesterol related illnesses and exhibit such potential with or without
releasing the
active parent drug.
While the prodrugs of fibric acidn of the present invention are not expected
to possess
any acidic activity due to blockage of the carboxylic acid group responsible
for such,
however it has been shown that the prodrugs of fibric acid are effective anti-
hyperlipidemics with or without releasing Fibric acid derivatives. However,
all of the
Fibric acid derivatives prodrugs described are released in vivo the active
drug with all its
pharmacological and cholesterol lowering properties.
The present invention clearly provides a number of advantages over Fibric acid
derivatives, for example, all of the side chains cleaved from these prodrugs
are naturally
occurring essential amino acids hence are non-toxic. This results in high
therapeutic
index. Secondly all the prodrugs are readily cleaved in the body to release
Fibric acid
derivatives. Furthermore, due their high water solubility, they can be easily
administered
by either forming an in-situ solution just before IV administration using
lyophilized
sterile powder or providing the drug in solution in prefilled syringe or
bottles for
infusion. The aminoacid esters are more stable than Fibric acid derivatives
since COOH
group in Fibric acid derivatives is blocked to reaction with bases. Thus the
Fibric acid
derivatives prodrugs described here are more effective then Fibric acid
derivatives itself
without the toxicity and other pharmaceutical problems associated with current
marketed
formulations.
The prodrugs of this invention are anti-hyperlipidemic drugs useful in the
treatment of
hyperlipidemia in mammals where the symptoms are elevated triglycerides, low
HDL
(High density lipoproteins or "good" cholesterol, and elevated cholesterol.
Fibric Acid
derivatives are also useful in reducing LDL (Low density lipoproteins, or
"bad"
cholesterol).
174
H:\work\1652\17690\SPEC\17690.spec without clahnsil.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
Typical examples of synthesis of L-threonine, L-hydroxyproline and L-serine
esters of
Fibric acid derivatives are shown in the synthetic processes outlined below.
These
procedures are applicable to all other compounds of the Fibric acid
derivatives class as
well.
Synthesis of Fibric acid derivatives Prodrugs
The procedure for the synthesis of the L-serine, L-threonine, and L-
hydroxyproline
esters of fenofibric acid is outlined in Synthetic Sequence section and is
exemplary.
The complete procedure and analytical data is given in the Experimental
Section. In
general, fenofibric acid (100 g batches) was prepared from 4-chloro-4'-
hydroxybezophenone in accordane with the procedures in the literature.
Fenofibric acid
was coupled with the t-butyl esters of N-Boc protected amino acid (L-serine, L-
threonine, and L-hydroxyproline) using EDC as the coupling agents and a
catalytic
amount of DMAP. The protecting groups were removed at low temperature (5 C, 3-
6
days) with a mixture of hydrochloric acid in acetic acid (1M) with
dichloromethane.
The amino acid ester salts of fenofibric acid were purified by crystallization
from ethyl
acetate, and dried under high vacuum.
Synthetic Sequence:
175
H:\work\1652\17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
D
0 to + A + CHID. + Na0N
a OH
V
0
ci 1><TrOH
110 0
Fenotibric Acid o
\Nõ,
[ b
,ti 0 0
ci Itill 110 oXi -,----iir -.),_ CI 110 itill oxõ...e.
N
o I µ
o
SP1130D20101 H A )4, 0
'14 0 SPI130020301
)Cr-
C 0I 11 I 1110 xe0 ' 0
1 d - II ri µ-l<
o i
SPI90020201 d
0
o 0
H CI-
H,4,_H i d
0.
01 AO ISO y....r. µ on 01
0 ni 0 CI- o .1.1
o H
H..4..H a-
SP1800201 ur ir Off SPIB00203
CI
o i O
SPIBIDDX2
Synthesis of the L-serine, L-threonine, and L-hydroxyproline esters of
fenofibric acid: a) Boc-Ser-OtBu, EDC, DMAP, CH2C12; b) Boc-Thr-OtBu, EDC,
DMAP, CH2C12; c) Boc-Hyp-OtBu, EDC, DMAP, CH2C12; d) HC1, AcOH, CH2C12.
,
176
H:\work\1652\17690\SPEC\17690.spec without clairnsII.doe
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Experimental Section:
The synthesis of SPIB00201, SPIB00202 and SPIB00203 was conducted in one or
two
batches. Reagents mentioned in the experimental section were purchased at the
highest
obtainable purity from Lancaster, Sigma-Aldrich, Acros, or Bachem, except for
solvents,
which were purchased from either Fisher Scientific or Mallinkrodt.
1) Synthesis of fenofibric acid:
C1OOOfOH
0
A mixture of 4-chloro-4'-hydroxybezophenone (116 g, 0.500 mole) and sodium
hydroxide (120 g, 3.00 mole) in acetone (1 L) was heated to reflux for 2
hours. The
heating was stopped and the heating source was removed. A mixture of
chloroform (179
g, 1.50 mole) in acetone (300 mL) was added drop-wise. The reaction mixture
was
stirred overnight without heating. The mixture was heated to reflux for 8
hours and then
allowed to cool to room temperature. The precipitate was removed by filtration
and
washed with acetone (100 mL). The filtrate was concentrated under reduced
pressure to
give a brown oil. Water (200 mL) was added to the brown oil and was acidified
(to
pH=1) with 1N hydrochloric acid. The precipitate, which formed was filtered
and dried
under high vacuum. The remaining yellow solid (268 g) was recrystallized from
toluene
in 4 batches (400 mL toluene each). After filtration and drying under high
vacuum, the
experiment produced fenofibric acid (116 g, 73% yield) as a light yellow
solid.
NMR (300 MHz, DMSO-d6): 6 = 13.22 (1H, s, br), 7.72 (4H, d, J= 8.4 Hz), 7.61
(2H,
d, J= 7.8 Hz), 6.93 (2H, d, J= 7.8 Hz), 1.60 (6H, s).
I3C NMR (75 MHz, DMSO-d6): 8 = 192.96, 174.18, 159.35, 136.84, 136.12, 131.67,
131.02, 129.12, 128.43, 116.91, 78.87, 25.13.
177
H:\work\1652 \17690 \SPEC \17690.spec without clairnsli.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
2) SPIB00201: L-serine-fenofibric acid ester
To a mixture of fenofibric acid (11.6 g, 36.3 mmol), N-carbobenzyloxy-L-serine
t-butyl
ester (Boc-Ser-OtBu, 8.62 g, 33.0 mmol), EDC (7.59 g, 39.6 mmol), and DMAP
(484
mg, 3.96 mmol) cooled in an ice-water bath was added anhydrous dichloromethane
(150
mL) dropwise. After the addition was complete, the ice bath was removed and
the
reaction mixture was stirred under an argon atmosphere at room temperature for
20
hours. After 20 hours, the additional dichloromethane (200 mL) was added and
the
solution was washed with water (2x200 mL) and brine (200 mL). After drying
over
sodium sulfate and filtration, the solution was concentrated under reduced
pressure. The
remaining yellow oil (21.2 g) was purified by column chromatography on silica
gel (400
g, 0.035-0.070 mm, 6 nm pore diameter), eluting with heptane/ethyl acetate
(3:1). After
concentration of the product-containing fractions under reduced pressure and
drying
under high vacuum until the weight was constant, the experiment produced the
protected
L-serine-fenofibric acid ester SPIB0020101 (16.2 g, 87% yield) as a light
yellow oil.
o o _
10 =
o
1HNMR (300 MHz, CDC13): = 7.75 (2H, d, J= 9.0 Hz), 7.72 (211, d, J= 9.0 Hz),
7.45
(2H, d, J= 8.7 Hz), 6.86 (2H, d, ./.= 8.7 Hz), 5.04 (1H, d, J= 6.9 Hz), 4.55-
4.42 (3H, m),
1.66 (3H, s), 1.65 (3H, s), 1.43 (9H, s), 1.39 (9H, s).
13C NMR (75 MHz, CDC13): 8 = 193.92, 172.99, 168.07, 159.24, 154.87, 138.24,
136.19, 131.94, 131.06, 130.40, 128.41, 117.26, 82.88, 80.13, 79.24, 65.44,
53.44,
28.27, 27.92, 25.70, 25.30.
To a stirred solution of the protected L-serine-fenofibric acid ester
SPEB0020101 (16.2
g, 28.8 mmol) in anhydrous dichloromethane (100 mL) cooled to 5 C, under an
argon
atmosphere was added a solution of hydrogen chloride in acetic acid (400 mL,
1M, 400
178
H:\work\1652 \17690 \SPEC \17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
minol) drop-wise. The reaction mixture stirred for 3 days at 5 C. After three
days the
mixture was concentrated under reduced pressure and dried under high vacuum to
remove acetic acid. To the remaining light yellow oil (24.7 g) was added ethyl
acetate
(100 mL). The solution was concentrated and dried a second time. To the
remaining
light yellow oil (17.0 g) was added ethyl acetate (65 mL). The mixture was
heated to
reflux for 5 minutes and cooled to room temperature. The precipitate was
removed by
filtration and dried under high vacuum overnight at room temperature, then at
43 C for
one hour. The experiment produced the L-serine-fenofibric acid ester,
hydrochloride
SPLB00201 (7.66 g, 60% yield) as a white solid.
Cl
110 c>rOjirF1 1.-H OH
CI
0 0
NMR (300 MHz, DMSO-d6): 8 = 14.12 (1H, s, br), 8.77 (3H, s, br), 7.72 (411,
m),
7.62 (2H, d, J= 8.4 Hz), 6.92 (2H, d, J= 9.0 Hz), 4.62 (1H, dd, .,T= 12.0, 4.2
Hz), 4.50
(1H, dd, J= 12.0, 2.4 Hz), 4.41 (1H, m), 1.64 (3H, s), 1.63 (3H, s).
13C NMR (75 MHz, DMSO-d6): 8 = 193.06, 171.70, 168.06, 158.72, 136.93, 136.06,
131 .73, 131.09, 129.62, 128.49, 117.64, 79.02, 62.99, 51.11, 25.04, 24.94.
HPL,C analysis:
100% purity; r.t.= 4.361 min.; 55% TFA (0.1%), 45% ACN; 1 mL/min; 32.3 C, Luna
C18, serial # 167917-13; 20 ul inj., NB275-49.
CHN analysis:
cale.: C 54.31, H 4.79, N 3.17; found: C 54.37, H 4.78, N 3.12.
Melting point: 151 C (dec.)
179
H:\work\1652\17690\SPEC\17690.spec without claimsIl.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
3) SPI1300202: L-threonine-fenofibric acid ester
To a mixture of fenofibric acid (25.5 g, 79.9 mmol), N-carbobenzyloxy-L-
threonine t-
butyl ester (Boc-Thr-OtBu, 20.0 g, 72.6 mmol, prepared by the literature
method), EDC
(16.7 g, 87.1 mmol), and DMAP (1.06 g, 8.71 mmol) cooled in an ice-water bath
was
added anhydrous dichloromethane (200 mL), dropwise. After the addition was
complete, the ice bath was removed and the reaction mixture was stirred under
an argon
atmosphere at room temperature for 20 hours. After 20 hours, additional EDC
(1.39 g,
7.26 mmol) was added and the experiment was allowed to stir over the weekend
at room
temperature under an argon atmosphere. After 4 days, additional
dichloromethane (300
mL,) was added and the solution was washed with water (300 mL) and brine (300
mL).
After drying over sodium sulfate and filtration, the solution was concentrated
under
reduced pressure. The remaining yellow oil (53.5 g) was purified by column
chromatography on silica gel (500 g, 0.035-0.070 mm, 6 nm pore diameter),
eluting with
heptane/ethyl acetate (3:1). After concentration of the product-containing
fractions
under reduced pressure and drying under high vacuum until the weight was
constant, the
experiment produced the protected L-threonine-fenofibric acid ester
SPIB0020201 (34.1
g, 82% yield) as a white foam.
H,o31--ok
CI 1. 0
0
0 I ti
1H INIMR (300 MHz, CDC13): 6 = 7.74 (211, d, J= 8.4 Hz), 7.72 (2H, d, J= 8.4
Hz), 7.45
(2R, d, J= 8.4 Hz), 6.87 (2H, d, J= 8.4 Hz), 5.47 (1H, m), 4.98 (1H, d, .1-=
9.9 Hz), 4,31
(1R, d, J= 9.9 Hz), 1.65 (3H, s), 1.64 (3H, s), 1.45 (9H, s), 1.42 (9H, s),
1.22 (3H, d,
6.3 Hz).
13C NMR (75 MHz, CDC13): 8 = 193.94, 172.14, 168.70, 159.26, 155.62, 138.28,
136.18, 131.90, 131.08, 130.37, 128.43, 117.40, 82.70, 80.17, 79.38, 72.02,
57.46,
28.30, 27.99, 26.44, 24.79, 16.90.
180
1-1:\wor111652\17690\SPEC\17690.spec without clahnsil.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
To a stirred solution of the protected L-threonine-fenofibric acid ester
SPD30020201
(34.1 g, 59.2 mtnol) in anhydrous dichloromethane (100 mL) cooled to 5 C,
under an
argon atmosphere was added a solution of hydrogen chloride in acetic acid (600
mL,
1M, 600 mmol) drop-wise. The reaction mixture was kept for 6 days at 5 C. The
mixture was concentrated under reduced pressure and dried under high vacuum to
remove acetic acid. To the remaining white solid (45.8 g) was added ethyl
acetate (500
mL). The mixture was heated to reflux for 10 minutes and cooled to room
temperature.
The precipitate was removed by filtration and dried under high vacuum
overnight at
room temperature. The experiment produced the L-threonine-fenofibric acid
ester,
hydrochloride SPD300202 (26.3 g, 97% yield) as a white solid.
Cl-
CI 110 401HI H
-
CX/r H
0 8
1H NMR (300 MHz, DMSO-d6): 8 = 14.10 (1H, s, br), 8.84 (3H, s, br), 7.73 (4H,
m),
7.63 (2H, d, J= 8.1 Hz), 6.89 (2H, d, J= 8.7 Hz), 5.44 (1H, m), 4.31 (1H, s),
1.64 (3H, s),
1.62 (3H, s), 1.38 (3H, d, J= 6.3 Hz).
13C NMR (75 MHz, DMSO-d6): 8 = 193.04, 171.00, 168.13, 158.76, 136.90, 136.08,
131.70, 131.06, 129.49, 128.48, 117.41, 78.99, 69.40, 55.21, 25.59, 24.22,
16.06.
HPLC analysis:
98.59% purity; r.t.= 4.687 min.; 55% TFA (0.1%), 45% ACN; 1 mL/min; 32.3 C,
Luna
C18, serial # 167917-13; 20 ul inj., NB275-49, DAD1 B, Sig--210.4,
Ref=550,100.
CHN analysis:
calc.: C 55.27, H 5.08, N 3.07; found: C 54.98, H 5.13, N 3.03.
Melting point: 160.5 C (dec.)
181
H:\work\1652\17690\SPEC\17690.spec without olaimsll.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
4) SP1B00203: L-hydroxyproline-fenofibric acid ester
To a mixture of fenodbric acid (24.9 g, 78.1 mmol), N-carbobenzyloxy-L-
hydroxyproline t-butyl ester (Boc-Hyp-OtBu, 20.4 g, 71.0 mmole, prepared in
accordance with the procedure in the literature), EDC (16.3 g, 85.2 mmol), and
DMAP
(1.04 g, 8.52 rnmol) cooled in an ice-water bath was added anhydrous
dichloromethane
(200 mL) dropwise. After the addition was complete, the ice bath was removed
and the
reaction mixture was stirred under an argon atmosphere at room temperature for
20
hours. After 20 hours, additional EDC (1.63 g, 8.52 nnnol) was added and the
experiment was allowed to stir over the weekend at room temperature under an
argon
atmosphere. After 4 days the solution was washed with water (200 mL) and brine
(200
mL). After drying over sodium sulfate and filtration, the solution was
concentrated
under reduced pressure. The remaining yellow oil (49.4 g) was purified by
column
chromatography on silica gel (500 g, 0.035-0.070 mm, 6 nm pore diameter),
eluting with
heptane/ethyl acetate (2:1). After concentration of the product containing
fractions
under reduced pressure and drying under high vacuum until the weight was
constant, the
experiment produced the protected L-hydroxyproline-fenofibric acid ester
SPIB0020301
(26.4 g, 63% yield) as a colorless oil.
ci
0 \O
0
SPIB0020301
i\
NMR (300 MHz, CDC13): S = 7.76 (2H, d, J= 8.1 Hz), 7.73 (2H, d, J= 8.1 Hz),
7.46
(2H, d, J= 8.1 Hz), 6..84 (2H, d, J= 8.1 Hz), 5.32 (1H, m), 4.13 (0.38H, t,
Je= 7.8 Hz),
4.00 (0.62H, t, J= 7.8 Hz), 3.67 (1.62H, m), 3.46 (0.38H, d, J=12.6 Hz), 2.29
(1H, m),
2.15 (1H, m), 1.68 (3H, s), 1.66 (3H, s), 1.44-1.38 (18H, m).
182
FlAwork\1652\17690\SPEC\17690.spec without clainisII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
13C NMR (75 MHz, CDC13): 8 = 193.88, 172.98, 171.14, 159.25, 153.48, 138.23,
136.16, 131.99, 131.08, 130.36, 128.44, 117.03, 116.91, 81.48, 80.32, 80.20,
79.19,
74.03, 73.26, 58.23, 51.88, 51.58, 36.33, 35.31, 31.92, 28.29, 28.00, 25.89,
24.95.
To a stirred solution of the protected L-hydroxyproline-fenofibric acid ester
SPIB0020301 (26.0 g, 44.2 mmol) in anhydrous dichloromethane (100 mL) cooled
to 5
C, under an argon atmosphere was added a solution of hydrogen chloride in
acetic acid
(450 mL, 1M, 450 mmol) drop-wise. The reaction mixture stirred for 4 days at 5
C.
After four days the mixture was concentrated under reduced pressure and dried
under
high vacuum to remove acetic acid. To the remaining yellow oil (31.5 g) was
added
ethyl acetate (200 mL). The mixture was sonicated and then concentrated under
reduced
pressure and dried under high vacuum. To the remaining white solid (23.2 g)
was added
ethyl acetate (300 mL). The ethyl acetate mixture was heated to reflux for 10
minutes
and cooled to room temperature. The precipitate was removed by filtration and
dried
under high vacuum overnight at room temperature. The experiment produced the L-
hydroxyproline-fenoftbric acid ester, hydrochloride SPII300203 (15.8 g, 76%
yield) as a
white solid.
o
ci 40 40 Xiroõ,,Qc_H OH
0
1H NMR (300 MHz, DMSO-d6): 8 = 14.07 (1H, s, br), 10.75 (1H, s, br), 9.40 (1H,
s, br),
7.71 (4H, d, J= 8.1 Hz), 7.60 (2H, d, J= 8.1 Hz), 6.96 (2H, d, J= 8.1 Hz),
5.42 (1H, m),
4.24 (1H, t, J= 9.0 Hz), 3.61 (1H, dd, J= 13.2, 4.2 Hz), 3.28 (1H, d, J= 13.2
Hz), 2.35
(2H, m), 1.66 (3H, s), 1.64 (3H, s).
13C NMR (75 MHz, DMSO-d6): 8 = 193.00, 171.52, 169.14, 158.81, 136.87, 136.09,
131.81, 131.05, 129.48, 128.46, 117.28, 78.99, 73.79, 57.54, 50.23, 34.13,
25.69, 24.49.
HPLC analysis:
183
H:\work\1652\17690\SPEC\17690.spec without claimadoc
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
100% purity; r.t.= 8.369 min.; 60% DRJF water (0.1% TFA)/40% acetonitrile; 1
mL/min; 36.4 C; Luna Cl 8, 5u column (serial # 191070-3), 4.6x250 mm; 20 ul
injection; DAD1 A, Sig =- 210.4, Ref= 550,100.
HPLC-MS (ESI): calculated: M+ = 431; found M+H= 432.3
Melting point: 187.5 C (dec.)
Solubility of the above esters were determined in water at room temperature by
dissolving excess of each of the drug and let them settle for few hours. The
resulting
solutions were centrifuged at 150Orpm for 3 min and the supernatant liquid was
analyzed. It was shown that these esters possess solubility in water in excess
of 50
mg/mL.
EXPERIMENTAL
Rats were checked for time zero tiiglyceride level in blood. Then the rats
were set on
high sugar diet, such as 30% surcorse in water for 1 week. Then at the end of
1 week,
rats were tested for triglycerides, and were put on normal diet. From day 7-14
the rats
were administered either test or control drug. Triglycerides were again tested
on the 14th
day in rat blood.
In the Fenofibrate (control) vs L-Serine Ester of Fenofibric acid (test drug),
3 rats each
for each of the drug and control at equivalent doses of 50, 100 and 200 mg/kg
were
tested.
The results are shown below.
184
H: \work\ 1652 \17690\SPEC\17690.spec without claimsII.doc
CA 02534342 2006-01-30
WO 2005/046575 PCT/US2004/024901
SUMMARY ¨ DOSE RANGE FINDING STUDY ¨ HYPOLIPIDEMIC
PROPERTY ¨ FENOFLBRATE AND ITS FORMULATION
Test Substance: L-Serine Ester of Fenofibric Acid
Vehicle: 1% Tween 80 in milli Q -water
Dose.
Test Item Animal No. Triglycerides (mg/di)
Mg/kg)
Day zero Day 7 Day 14
1 81 168 121
Vehicle 0 2 88 171 222
3 114 133 162
4 95 157 101
50 5 92 228 76
6 80 150 73
Reference control 7 110 204 62
Fenofibrate 100 8 115 195 69
9 96 167 93
144 90 48
200 11 56 106 51
12 58 125 38
--
13 88 148 86
50 14 94 145 86
100 127 73
L-Serine Ester of 16 109 46
Fenofibric Acid 100 17 129 100 69
18 71 183 47
19 74 240 83
200 20 81 158 61
21 42 77 46
From the above results, it can be concluded the highly water soluble serine
ester was
effectively performed.
10 There are a number of screening tests to determine the utility of the
prodrugs created
according tio the disclosed methods. These include both in vitro and in vivo
Iscreening
methods.
185
H:\work\1652 \17690 \SPEC \17690.spec without claimsll.doc
CA 02534342 2011-12-06
The in vitro methods include acid/base hydrolysis of the prodrugs, hydrolysis
in pig
pancreas hydrolysis in rat intestinal fluid, hydrolysis in human gastric
fluid, hydrolysis
in human intestinal fluid, and hydrolysis in human blood plasma. These assays
are
dscribed in Simmons, DM, Chandran, VR and Portmann, GA, Danazol Amino Acid
Prodrugs: In Vitro and In Situ Biopharmaaceutical Evaluation, Drug Development
and
Industrial Pharmacy, Vol 21, Issue 6, Page 687, 1995.
The prodrugs of Fibric Acid of the present invention are effective in treating
diseases or
conditions in which Fibric acid derivatives normally are used. The prodrugs
disclosed
herein are transformed within the body to release the active compound and
enhances the
therapeutic benefits of the Fibric acid derivatices by reducing or eliminating
biopharmaceutical and pharmacoldnetic barriers associated with each of them.
However
it should be noted that these prodrugs themselves will have sufficient
activity without
releasing any active drug in the mammals.
Thus, the prodrug of the present invention enhances the therapeutic benefits
by removing
biopharmaceutical and pharmacokenetic barriers of existing drugs.
Furtheremore, the prodrugs are easily synthesized in high yields using
reagents which
are readily and commercially available.
The prodrugs of Fibric acid of the present invention are effective in treating
diseases or
conditions in which Fibric acid derivatives normally are used. The prodrugs
disclosed
herein are transformed within the body to release the active compound and
enhances the
therapeutic benefits of the Fibric acid derivatives by reducing or eliminating
biopharmaceutical and pharmacokenetic barriers associated with each of them.
However
it should be noted that these prodrugs themselves will have sufficient
activity without
releasing any active drug in the mammals.
186
CA 02534342 2006-01-30
WO 2005/046575
PCT/US2004/024901
Thus, the prodrug of fibric acid of the present invention enhances the
therapeutic
benefits by removing biopharmaceutical and pharmacokenetic barriers of
existing drugs.
Furthermore, the prodrugs are easily synthesized in high yields using reagents
which are
readily and commercially available.
In the formula hereinabove and in the claims it is to be understood that the
AA has the
following definition in the following contents
0
I I
1)
¨ C ¨NH ¨AA ¨
AA in this definition refers to the amino acid residue without an amino group
either on
the main chain or the side chain.
¨ ¨ 0 ¨AA ¨
0
2)
AA in this definition is an amino acid residue less the hydroxy group on the
side chain.
NH ¨ ¨ AA
3) 0
AA refers to an amino acid group without the carboxy group, either on the main
chain or
side group.
187
H:\work\1652\17690\SPEC\17690.spec without claimsadoc
CA 02534342 2013-04-18
WO 2005/046575 PCT/US2004/024901
4) OAA- This is a ester bond between the hydroxy group of the drug and the
carboxy
group of the amino acid either on the main chain or side chain. Thus, as
written OAA is
H
C NH2
Ro
wherein Ro is the side chain amino acid as defined hereinabove.
Alternatively, it may refer to an ester bond between the carboxy group of the
drug and
the hydroxy group on the side chain of those amino acids which have a hydroxy
group
thereon such as threonine, serine, hydroxyprofine, tyrosine and the like. The
hydroxy
group forms part of the ester linkage which is depicted hereinabove with O.
Thus, as
written, the AA refers to an amino acid with a hydroxy group on the side
chain, but as
depicted OAAõ the AA is without the hydroxy group since the oxygen atom is
depicted
in the formula.
The scope of the claims shot'. ld not be limited by the preferred embodiments
set forth in
the examples, but should be given the broadest interpretation consistent with
the
description as a whole.
=
188