Note: Descriptions are shown in the official language in which they were submitted.
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
TITLE: Epitope protection assay and method for detecting protein
conformations
FIELD OF THE INVENTION
The invention relates to an epitope protection assay for use in diagnosis,
prognosis and therapeutic intervention in diseases, for example, diseases
involving polypeptide aggregation such as prion infections.
BACKGROUND OF THE INVENTION
Protein Misfolding and Aggregation
Proteins can fold into complex and close-packed structures. Folding is not
only crucial for biological activity but failure of proteins to fold properly
or
remain folded can give rise to disease (Dobson CM, Methods (2004) 34:4-14).
Misfolding can in some cases cause protein aggregation which can further give
rise to discrete deposits extracellularly (e.g., plaques) or intracellularly
(e.g.,
inclusions in the cytosol or nucleus).
Neurogenerative diseases such as Alzheimer's disease (AD), Parkinson's
disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS)
and prion diseases are characterized by neural deposits of misfolded
aggregated protein. Type II diabetes and cancer have also been linked to
protein misfolding and it is likely that there are yet to be identified
diseases that
result from errors in protein folding and that in some cases lead to
consequences such as aggregation. The nature of the misfolding and any
aggregation in such diseases is typically not well characterized.
Prion Diseases
Prion diseases have become a major health concern since the outbreak of
BSE or "Mad Cow Disease" (reviewed above, 40, 41). BSE was first
discovered in the United Kingdom but has now spread to many other countries
in Europe and Japan. In the UK alone there has been close to 180,000 cases
of BSE, which resulted in the destruction of cattle and possible infection of
an
estimated 3-5 million head. The total cost estimated to the UK was in excess
of $2.5 billion. BSE is believed to be transmitted among cattle through feed
1
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
that contains prions rendered from infected cattle, and it is thought to be
transmitted to humans through eating beef or other cattle products from
infected animals.
Emerciinq Prion Diseases
The prion diseases are a group of rapidly progressive and untreatable
neurodegenerative syndromes, neuropathologically characterized by
spongiform change, neuronal cell loss, gliosis, and brain accumulation of
abnormal amyloid polypeptide. Human prion diseases include classical
Creutzfeldt-Jakob disease (CJD), which has sporadic, iatrogenic, and familial
forms. Since 1996, a "new variant" of CJD (vCJD) has been identified in the
United Kingdom, France, the Republic of Ireland, Hong Kong, Italy, the United
States, and Canada (40,41). Variant CJD is capable of killing individuals as
young as age 14 with unknown incubation period. There is little doubt that
vCJD is a human form of bovine spongiform encephalopathy (BSE)(42). The
primary epidemic from consumption of contaminated cattle tissue has affected
over 130 individuals as of this filing.
The spectre of vCJD "secondary epidemics" through blood, blood products,
surgery, dentistry, vaccines, and cosmetics is of great concern(40,41).
Detection of blood prion infectivity in experimental BSE/vCJD infections of
mice and sheep (40) suggests a special risk exists for the transmission of
vCJD through blood and blood products. The recent reports of vCJD in two
recipients of a donor who developed the disease is also troubling (52, 53).
Canada and the United States have recently expanded vCJD blood donor
deferrals to all countries in Western Europe.
Although sheep scrapie has been known for centuries, the most important
animal prion disease at present is BSE. More than 173,000 cattle, primarily
from Britain, have developed symptomatic BSE, and as many as 3 million have
entered the food supply undetected. BSE is now being increasingly reported
in cattle which were "born after the ban" in 1996 of food supplementation with
meat and bone meal, suggesting that alternate routes may exist to keep the
epidemic from being readily extinguished. Another troubling issue is the
2
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
possible transmission of BSE to sheep, which may expose additional human
populations to the BSE/vCJD prion strain. Recent reports show that prions can
replicate in certain muscle groups of sheep, experimental animals and humans
(54-57),indicating a potential risk in tissues previously considered safe for
human consumption.
Chronic wasting disease (CVVD) of captive and wild cervids (deer and elk)
represents another newly emergent animal prion disease in North America,
whose impact on human health is yet unknown. It is apparent that newly-
recognized prion diseases pose a threat to the safety of foods, blood
products,
and medical-surgical treatments.
Prions: Atypical Pathogens
Newly emergent prion diseases, and the polypeptide-only nature of prions,
have created serious medical, veterinary, and economic challenges worldwide.
To date, the only commercialised tests for prion infection have been based on
post-mortem brain samples. No biochemical test exists to detect prions in the
blood of infected animals, despite detection by experimental transmission
studies. The development of sensitive and specific diagnostic tests for prion
infection is a challenging task, in part due to the unusual nature of the
prion
infectious agent. The infectious agents that transmit the prion diseases
differ
from other pathogens in that no nucleic acid component has been detected in
infectious materials (41). According to the prion theory developed by Nobel
Laureate Dr. Stanley Prusiner, infectivity resides in PrPs c, a misfolded
conformational isoform of the near-ubiquitous normal cellular prion
polypeptide
PrPc. PrPsc is indeed the most prominent (or perhaps sole) macromolecule in
preparations of prion infectivity, and minimally appears to be a reliable
surrogate for prion infection. PrPsc is partially resistant to protease
digestion,
poorly soluble, and exists in an aggregated state, in contrast to the protease
sensitive, soluble, monomeric isoform PrPc (29, 31, 43-46).
PrPsc is derived from its normal cellular isoform (PrPc), which is rich in a-
helical structure, by a posttranslational process involving a conformational
3
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
transition. While the primary structure of PrPc is identical to that of PrPsc,
secondary and tertiary structural changes are responsible for the distinct
physicochemical properties of the two isoforms.
One of the difficulties in assessing the safety of food or blood products from
potentially infected humans with prions is the lack of an accurate diagnostic
test for blood or other accessible biosamples. Currently, there are no
diagnostic tests that can be applied for screening live animals, humans, blood
or blood products at an early stage. This also provides a further problem in
organ transplantation, adding unknown risk to organ recipients. Therefore, as
a preventative measure, countries such as the UK no longer source plasma
from its inhabitants. The risk of spreading prion diseases has affected other
countries as well. For example, the United States and Canada do not accept
blood donations from individuals who have resided in the UK or France for
more than 3-6 months.
Currently, the diagnosis of vCJD can only be confirmed following pathological
examination of the brain at autopsy or biopsy. Some complimentary strategies
in early CJD detection include electroencephalograms (EEG), magnetic
resonance imaging (MRI) scans, and cerebrospinal fluid (CSF) tests, which
may be useful "surrogate" or "proxy" markers. The absence of a "direct test"
for prion infection stands in stark contrast to conventional infectious
agents,
such as viruses and bacteria.
Some tests that are in the process of being commercialized are based on
surrogate markers of infection which are "once removed" from actual infectious
prions.
PrP protease resistance is the basis of most commercially available diagnostic
tests for prion disease. In the current methodologies, a sample of brain is
removed and digested with proteases that can eliminate PrPc, but leave a
protease-resistant core of PrPsc. The protease-resistant fragment of PrPsc is
then detected by immunoblotting (as in the Prionics test) or by capture ELISA
(as in the BioRad and Enfer tests, and in a new test from Prionics). However,
4
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
digestion with proteases is cumbersome and variable, leading to false
negatives and positives. Moreover, there are some prion strains which are
reported to contain PrPsc which is infectious and aggregated, but which is not
protease resistant. Protease-sensitive PrPsc also predominates early in
infection and in cross-species transmission of disease (31).
Detection of protease-resistant PrP fragments is also the basis of a urine
diagnostic test (47) which is being commercially developed by Prionics.
However, detection of protease-resistant PrP in urine is subject to the same
limitations as the post-mortem brain test, and has the additional disadvantage
of requiring precipitation from large volumes of urine, and poor sensitivity
(for
example, only detecting PrPsc in late stages of the disease, not pre-
symptomatically).
Other Neurodegenerative Diseases
Neurodegenerative diseases, such as Alzheimer's disease (AD), Huntington's
disease, amyotrophic lateral sclerosis (ALS) and Parkinson's disease/Lewy
body dementia (PD, LBD) also pose major challenges to our aging population
and health care system (reviewed in 1). An estimated 364,000 Canadians
over 65 are currently diagnosed with AD or a related dementia
(http://www.alzheimerca/). With increased life expectancy, the incidence of
neurodegenerative disease is expected to grow. By 2025, AD will affect as
many as a million Canadians, and by 2050, this number will double.
Sporadic AD, ALS, and PD/LBD are all associated with neural accumulation of
pathological multimers of misfolded polypeptides (these could potentially be
fibrils, protofilaments, and amorphous aggregates), including the amyloid-beta
(Abeta) fragment of the amyloid precursor protein (APP) in AD; superoxide
dismutase-1 (SOD1) in ALS, and alpha-synuclein in PD and LBD (1).
Additionally familial amyloidotic polyneuropathy (FAP) results from the
aggregation of transthyretin to form amyloid deposits. As with prion diseases,
mutations in genes encoding these polypeptides are associated with
autosomal dominant familial forms of AD, ALS, and PD.
CA 02536305 2006-02-20
WO 2005/019828 PCT/CA2004/001503
Alzheimer's Disease
AD is a common dementing (disordered memory and cognition)
neurodegenerative disease associated with brain accumulation of extracellular
plaques composed predominantly of the Abeta (1-40), Abeta (1-42) and Abeta
(1-43) peptides, all of which are proteolytic products of APP (reviewed in 4).
In
addition, neurofibrillary tangles, composed principally of abnormally
phosphorylated tau protein (a neuronal microtubule-associated protein),
accumulate intracellularly in dying neurons (4). Familial forms of AD can be
caused by mutations in the APP gene, or in the presenilin 1 or 2 genes
(www.websiteformutations.com), the protein products of which are implicated
in the processing of APP to Abeta. Apolipoprotein E allelic variants also
influence the age at onset of both sporadic and familial forms of AD (reviewed
.
in 5). Abeta has been detected in the blood and CSF of AD patients and in
normal controls (6). Abeta is also present in vascular and plaque arnyloid
filaments in trisomy 21 (Down's syndrome), hereditary cerebral hemorrhage
with amyloidosis (HCHWA)-Dutch type, and normal brain aging (Mori, H et al.
JBC (1992) 267: 17082-86). Tau and phosphorylated tau have been detected
in the cerebral spinal fluid (CSF) of AD patients and patients with other
neurological diseases (7; reviewed in 8).
Amyotrophic Lateral Sclerosis
ALS is a fatal neuromuscular disease, with an incidence of 1 in 1000 adults,
presenting as progressive weakness, muscle atrophy, and spasticity, which is
due to degeneration of ¨500,000 "lower motor neurons" in the spinal cord and
brainstem, and innumerable "upper motor neurons" in the brain cortex. An
important clue to the etiology of ALS came with the finding that about 20% of
familial ALS (fALS) cases are due to mutations in superoxide dismutase-1
(SOD1) (10,11), a free radical defense enzyme. Over 100 fALS SOD1
missense, nonsense, and intronic splice-disrupting mutations have been
catalogued to date (12; www.alsod.orq). Transgenic mice expressing mutant
human SOD1 (mtHuSOD1) develop a motor neuron syndrome with clinical and
pathological similarities to human ALS (13, 14), whereas mice expressing wild-
type human SOD1 (wtHuSOD1) do not develop disease (13). SOD1-containing
cytoplasmic inclusions can be detected in many diseased motor neurons from
6
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
familial and sporadic ALS patients (15), and in most transgenic mouse (16, 17)
and tissue culture (18) models of the disease.
Parkinson's and Lewy Body Disease
PD is a neurodegenerative movement disorder second only to AD in
prevalence (-350 per 100,000 population; 1). It is clinically characterized by
rigidity, slowness of movement, and tremor (reviewed in 21). Most cases of
Parkinson's disease are sporadic, but both sporadic and familial forms of the
disease are characterized by intracellular Lewy bodies in dying neurons of the
substantia nigra, a population of midbrain neurons (-60,000) that are
selectively decimated in PD. Lewy bodies are predominantly composed of
alpha-synuclein (22). Mutations in the gene encoding alpha-synuclein have
been found in patients with familial Parkinson's disease (reviewed in 23;
www.parkinsonsmutation.com). Another gene associated with autosomal
recessive PD is parkin, which is involved in alpha-synuclein degradation (22,
23). Diffuse cortical Lewy bodies composed of alpha-synuclein are observed in
Lewy body disease (LBD), a dementing syndrome associated with
parkinsonian tone changes, hallucinations, and rapid symptom fluctuation (24).
LBD may be the second most common form of neurodegenerative dementia
after AD, accounting for 20 to 30 percent of cases among persons over the
age of 60 years (1, 24).
Huntington's Disease and Related Diseases
HD is a progressive neurodegenerative disorder characterized by expansion of
polyglutamine encoding CAG repeats in the N-terminus of the huntingtin
protein (reviewed in 48). Polyglutamine stretches of > 36 cause disease and
longer repeats cause earlier onset (49, 50).
Other polyglutamine diseases such as dentate-rubral and pallido-luysian
atrophy (DRPLA) and some forms of sino-cerebellar ataxia (SCA) also have
intracellular inclusions that roughly correlate to regions of neuronal death.
Interruptions in the expanded polyglutamine repeat in the SCA-1 gene product
result in the absence of disease (51),
7
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Neurodegenerative diseases, such as Alzheimer's disease (AD), amyotrophic
lateral sclerosis (ALS) and Parkinson's disease/Lewy body disease (PD, LBD)
pose major challenges to our aging population and health care system. No
specific biochemical test exists for neurodegenerative diseases as a group
(1,2). Since neurodegenerative diseases are regarded as "diagnoses of
exclusion," very broad investigation is required to achieve "clinically
probable"
diagnosis for these progressive, incurable, and usually fatal conditions.
Expensive surrogate testing, such as neuroimaging, is utilized to increase
diagnostic probability (2). The availability of specific, sensitive, and
inexpensive biochemical tests for this devastating group of diseases could
potentially conserve financial resources for over-burdened health care
systems. Moreover, secure diagnosis of these diseases at an earlier
symptomatic stage increases the window for enhanced treatment efficacy at a
time at which the disease pathophysiology is generally more responsive to
treatment (3).
Effective, efficient and inexpensive diagnostic and screening strategies for
antemortem diagnosis of human neurodegenerative diseases are urgently
needed, given the aging population and continued financial pressure on the
health care system.
Diabetes
Protein aggregation is also observed in patients with type II diabetes.
Increased expression of the adipocyte-derived peptide, resistin, has been
observed in diabetes type II patients (Youn BS et al. J Clin Endocrinol Metab.
(2004);89:150-6) and studies suggest that elevated resistin levels may play a
role in obesity and insulin resistance. Additionally, islet amyloid
polypeptide
(also known as annylin) deposition is pathogenically associated with type 2
diabetes. These deposits contain islet amyloid polypeptide, a unique
amyloidogenic peptide and are associated with beta cell death. Recent studies
suggest that the species responsible for islet amyloid-induced beta-cell death
are formed early in islet amyloid formation, when islet amyloid polypeptide
accumulation begins (Hull RL et al. J Clin Endocrinol Metab. (2004) 89:3629-
43). A diagnostic test that can identify pathogenic islet amyloid polypeptide
8
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
would be very useful for detecting type 2 diabetes in its early stages, when
dietary and therapeutic interventions are most effective.
Cancer
Many forms of cancer are also considered to be protein conformation diseases
(Ishimaru D. et al. Biochemistry (2003) 42:9022-7). A subset of
neuroblastomas, carcinomas and myelomas show an abnormal accumulation
of tumor suppressor p53 protein aggregates (Butler JS et at. Biochemistry
(2003) 42: 2396-403; Ishimaru D. et al. Biochemistry (2003) 42:9022-7). This
accumulation could contribute to the loss of p53 function in some cancerous
cells (Ishimaru D. et al. Biochemistry (2003) 42:9022-7). Assays able to
detect
accumulated p53 could provide a diagnostically useful detection system and
could enhance therapeutic intervention by individualizing therapeutic
intervention.
SUMMARY OF THE INVENTION
The inventors have recently developed the epitope protection assay (EPA), a
novel method that yields sensitive and specific antemortem detection of
disease proteins in blood and other accessible tissues and fluids. The
invention shows the role of aggregation in diseases, such as prion disease,
and provides an assay that overcomes problems in the prior art. In prion
diseases, the normal cellular monomeric prion polypeptide PrPc undergoes
refolding to an abnormal, aggregated isoform, generically designated PrPsc.
Diseases such as AD, PD, LBD, ALS and HD are also characterized by
misfolded and/or aggregated conformations of cellular proteins. This property
is exploited by the methods of the invention to provide sensitive and specific
diagnostic tests for these and other diseases.
According to the invention, the methods are useful where a target epitope is
accessible in either one of a non-wildtype protein (i.e. disease protein) or a
wild
type protein and inaccessible in the other. Inaccessibility is often due to
aggregation making the target epitope inaccessible.
9
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
The invention includes a method of detecting whether a candidate polypeptide
including a target epitope is a disease (disorder) polypeptide or a wild type
polypeptide, comprising:
- contacting the candidate polypeptide with a blocking agent; and
- determining whether the target epitope is inacessible or accessible to
chemical modification by the blocking agent.
The accessibility or inaccessibility of the target epitope is indicative of
whether
the candidate polypeptide is a disease (disorder) polypeptide or a wild type
polypeptide because in one of the disease (disorder) protein and the wild type
protein, the target epitope is accessible. In the other polypeptide the target
epitope is inaccessible.
In one embodiment, the invention provides a method of detecting prion
diseases, for example, determining whether a candidate polypeptide including
a target epitope is in a wildtype conformation or in a non-wildtype
conformation
in which it is aggregated, comprising:
- reacting a sample of polypeptide (the sample typically contains PrPsc
and/or PrF)c, and in many cases an abundance of one or the other) with
a chemical modifying agent, typically an agent which chemically reacts
with proteins such as peroxynitrite, which modifies accessible epitopes
(target epitopes) so that they cannot bind to a detection agent;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with detection agents, such as an antibody against a target
epitope, to determine whether the polypeptide (such as prior to
disaggregation and/or denaturing) included inaccessible target epitopes.
PrPc is rendered "invisible" in the assay, because epitopes on the monomeric
molecules are blocked to antibody recognition by the chemical modifying
agent, whereas molecules of PrPsc are "protected" from chemical modification
by virtue of being sequestered within aggregates or otherwise unavailable for
reacting. Alternatively, epitopes on the nnultimeric molecules are blocked to
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
antibody recognition by'the chemical modifying agent, whereas molecules of
PrPc are "protected" from chemical modification by virtue of a difference in
accessible epitopes.
In another embodiment, the Alzheimer's disease detection method comprises:
- reacting a sample of polypeptide (the sample typically contains all or
part of diseased amyloid precursor polypeptide or A beta or tau and/or
the corresponding wild type polypeptide, and in many cases an
abundance of one or the other) with a chemical modifying agent,
typically an agent which chemically reacts with proteins such as
peroxynitrite, which modifies exposed epitopes so that they cannot bind
to a detection agent;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with detection agents, such as an antibody against a target
epitope to determine whether the polypeptide prior to disaggregation
and/or denaturing, included inaccessible target epitopes.
In further embodiments, the invention provides disease detection methods for
other diseases characterized by differentially accessible target epitopes in
disease and wildtype conformations, for example, resulting from misfolded
and/or aggregated proteins such as Parkinson's disease (PD), Lewy Body
disease (LBD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS),
diabetes, and cancer. These methods similarly include steps such as reacting
a sample of polypeptide (eg. a disease polypeptide described herein) with a
chemical modifying agent, which modifies exposed epitopes so that they
cannot bind to a detection agent; then disaggregating and/or denaturing the
polypeptide in the sample; and probing with detection agents, such as an
antibody against a target epitope to determine whether the polypeptide prior
to
disaggregation and/or denaturing, included inaccessible target epitopes.
These steps are similarly adapted for other purposes, such as screening blood
and blood products, and other uses described herein.
11
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
The method of the invention has many advantages over existing technology.
As noted above, the invention is optionally referred to as "EPA", which in the
case of prion protein disease detection is a simple, efficient method for
detecting aggregated disease proteins such as PrPse, the pathogenic molecule
which is thought to constitute the infectious particle in prion diseases and
polypeptide, associated with AD.
The invention is useful in high-throughput robotic-capable platforms. For
example, EPA is not dependent on PrP protease resistance, the basis of most
commercially available diagnostic tests for prion disease. Epitope protection
technology does not require a protease digestion step, which makes it more
sensitive to early infection. Certainly, the absence of a protease digestion
step
permits EPA to be more amenable to high-throughput robotic platforms.
In addition, the methods of the invention can be used to detect any protein
that
exists in two or more conformations, where one or more target epitopes are
concealed in at least one conformation.
Accordingly, the invention relates to a detection method comprising:
- reacting a sample of polypeptide with a chemical modifying agent,
typically an agent which chemically reacts with proteins, which is
defined to modify exposed epitopes so that they cannot bind to
detection agents;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with detection agents, such as antibodies against a target
epitope to determine whether the polypeptide prior to disaggregation
and/or denaturing, included target epitopes inaccessible to the chemical
modifying agent.
The result indicates whether the polypeptide includes inaccessible epitopes,
which is indicative of the type of polypeptide that is present (i.e. wild type
protein or non-wild type protein).
12
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
In one embodiment, the invention includes a method of detecting whether a
candidate polypeptide including a target epitope is in a wildtype conformation
or a non-wildtype conformation (in one embodiment, in the non-wildtype
conformation, the candidate polypeptide aggregates with aggregated
polypeptide), comprising:
- contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitopes, wherein in one of the non-wildtype
conformation or the wildtype conformation, the target epitope is
accessible and reacts with the blocking agent, and wherein in the other
conformation, the target epitope is inaccessible and does not react with
the blocking agent. Unreacted blocking agent is removed from contact
with the polypeptide, for example, by allowing time for blocking agent to
be consumed or degraded or by actively removing it by physical or
chemical processes as described below.;
-modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope; and
-contacting the polypeptide with a detection agent that binds selectively
to target epitope that was converted from inaccessible target epitope to
accessible target epitope, wherein binding between detection agent and
converted target epitope indicates that prior to conversion the
candidate polypeptide was in a conformation in which the target epitope
was inaccessible and wherein lack of binding between the detection
agent and the target epitope indicates that the polypeptide was in a
conformation in which the target epitope was accessible, thereby
indicating whether the polypeptide was in a wildtype conformation or a
non-wildtype conformation.
The invention also includes a method of detecting whether a candidate
polypeptide including a target epitope is in a wildtype conformation or a non-
wildtype conformation, comprising:
13
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
,
- contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitope, wherein in the wildtype conformation, the
target epitope is accessible and reacts with the blocking agent, and
wherein in the non-wildtype conformation, the target epitope is
inaccessible and does not react with the blocking agent. Unreacted
blocking agent is removed from contact with the polypeptide, for
example, by allowing time for blocking agent to be consumed or
degraded or by actively removing it by physical or chemical processes
as described below.;
- modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope; and
- contacting the polypeptide with a detection agent that binds selectively
to the target epitope that was converted from inaccessible target epitope
to accessible target epitope, wherein binding between detection agent
and converted target epitope indicates that the candidate polypeptide
was in a non-wildtype conformation and wherein lack of binding
between the detection agent and the target epitope indicates that the
polypeptide was in a wildtype conformation.
The invention also includes a method of detecting whether a candidate
polypeptide including a target epitope is in a wildtype conformation or a non-
wildtype conformation, comprising:
- contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitope, wherein in the non-wildtype conformation, the
target epitope is accessible and reacts with the blocking agent, and
wherein in the wildtype conformation, the target epitope is inaccessible
and does not react with the blocking agent. Unreacted blocking agent is
removed from contact with the polypeptide, for example, by allowing
time for blocking agent to be consumed or degraded or by actively
removing it by physical or chemical processes as described below.;
14
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
- modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope; and
- contacting the polypeptide with a detection agent that binds selectively
to target epitope that was converted from inaccessible target epitope to
accessible target epitope, wherein binding between detection agent and
converted target epitope indicates that the candidate polypeptide was in
a wildtype conformation and wherein lack of binding between the
detection agent and the target epitope indicates that the polypeptide
was in a non-wildtype conformation.
The invention also includes a method of detecting whether a candidate
polypeptide including target epitope which has been reacted with a blocking
agent, is in a wildtype conformation or a non-wildtype conformation,
comprising:
- modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope;
- contacting the polypeptide with a detection agent that binds selectively
to the target epitope that was converted from inaccessible target epitope
to accessible target epitope, wherein binding between detection agent
and converted target epitope indicates that the candidate polypeptide
was in a non-wildtype conformation and wherein lack of binding
between the detection agent and the target epitope indicates that the
polypeptide was in a wild type conformation.
In the methods of the invention, the epitope is in many cases inaccessible in
the misfolded or non-wild type conformation because i) the differential
misfolding of the polypeptide compared to the wild type folded polypeptide
prevents or reduces reaction between the blocking agent and the target
epitope, ii) the polypeptide in the misfolded conformation aggregates with
itself
or other polypeptides in the misfolded conformation to prevent or reduce
reaction between the protecting/blocking agent and the target epitope, and/or
iii) post translational modifications of the polypeptide prevent or reduce
reactions between the blocking agent and the target epitope.
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
In one example, the candidate polypeptide comprises prion protein, the wild
type folded conformation comprises the conformation of wild type folded prion
protein and the misfolded conformation comprises the conformation of PrPsc.
Alternatively, the wild type folded protein comprises the conformation of APP
or its cleavage product amyloid beta, and the misfolded conformation
comprises the conformation of Alzheimer's disease APP or its cleavage
product amyloid beta.
In another example, the candidate polypeptide comprises, SOD1,
alpha-synuclein, islet amyloid polypeptide, resistin or p53 protein. The
methods and kits'of the invention described in the application are useful, for
example, for application to a non-wildtype polypeptide having a conformation
comprising multiple copies of a polypeptide aggregated together through
interactions of beta-sheet-rich areas of the polypeptide. In one embodiment
the
polypeptide is polypeptide that is aggregated in prion protein aggregates. In
another embodiment, the polypeptide is polypeptide that is aggregated in
amyloid plaques.
The invention also includes i) polypeptide of the invention modified by
reaction
with a blocking agent listed herein and ii) the polypeptide modified by
reaction
with a detecting agent. The invention also includes compositions and kits of
the invention including these modified polypeptide.
The blocking agent is optionally peroxynitrite, hydrogen peroxide, diethyl
pyrocarbonate (DEPC), 4-hydroxynonenal (4HNE) an epoxide such as
conduritol-B-epoxide and 1,2-epoxy-3-(p-nitrophenoxy)propane, methylene or
diazirine and related compounds. In the methods, the polypeptide is optionally
modified by denaturing the polypeptide, for example with heat, detergent
and/or chaotropic agents. The polypeptide is optionally modified by treatment
with a disaggregation agent to disaggregate the polypeptide from other
polypeptides of the same type, and from other molecules, wherein the
disaggregation agent is optionally selected from at least one of the group
consisting of chaotropic agent (including guanidine salts, urea or thiourea),
detergent and heat.
16
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
It is readily apparent to a skilled person that the method steps of the
invention
recited here involving removing the blocking agent typically involve
physically,
chemically or otherwise removing the blocking agent away from the candidate
polypeptide to prevent further reaction. Removal optionally involves allowing
a
sufficient time to pass so that the blocking agent is removed from the
candidate polypeptide by being consumed or degraded (for example, such that
the blocking agent becomes inert or oxidized). Removal optionally involves
adding a compound to react with any excess blocking agent to inactivate it.
Removal also optionally involves physical filtering of the blocking agent by
conventional filtration techniques or centrifugation to separate the candidate
polypeptide and blocking agent, or physical binding to a substrate useful for
removing the blocking agent, such as by binding of blocking agent or
candidate polypeptide to an immobilized substrate in a column.
Removing means preventing further reactions by the blocking agent by, for
example, physically or chemically inactivating the blocking agent, taking the
blocking agent out of contact with the sample including the candidate
polypeptide or allowing a sufficient amount of time to pass for the blocking
agent to be consumed or degraded.
The detection agent optionally comprises an antibody directed against a prion
polypeptide epitope, an amyloid beta epitope, an alpha-synuclein epitope or a
SOD1 epitope. The antibody optionally comprises all or part of the anti-prion
antibodies 6H4 and 3F4, and the anti-amyloid beta antibodies 6E10 and 408.
The methods of the invention are preferably used with mammals, such as
humans. In addition, the methods of the invention are preferably used with
mammals, such as livestock. In addition the methods of the invention are used
with food items, cosmetic items, dental and surgical instruments vacuums and
pharmaceutical products.
The invention also includes a method of testing a sample from an animal,
using methods of the invention described herein, to determine if the animal
has
a disease characterized by the presence of candidate polypeptide in a non-
17
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
wildtype conformation in the sample, wherein the candidate polypeptide
includes a target epitope. Such diseases are described herein. In one
embodiment, the method comprises; determining whether the candidate
polypeptide is in i) a wildtype conformation or ii) a non-wildtype
conformation,
by a method comprising the steps of:
contacting the sample with a blocking agent that selectively blocks
accessible target epitope in the candidate polypeptide, wherein in the
wildtype conformation, the target epitope is accessible and reacts with
the blocking agent, and wherein in the non-wildtype conformation, the
target epitope is inaccessible because the candidate polypeptide is
aggregated with the aggregated polypeptide and the target epitope
cannot react with the blocking agent;
contacting the sample with a conversion agent to modify the candidate
polypeptide to convert any inaccessible target epitope in the sample to
accessible target epitope;
contacting the polypeptide with a detection agent that binds selectively
to target epitope that was converted from inaccessible target epitope to
accessible target epitope, wherein binding between detection agent and
converted target epitope indicates that the candidate polypeptide was in
a non-wildtype conformation and the animal has a disease and wherein
lack of binding between the detection agent and the target epitope
indicates that the polypeptide was in a wild type conformation.
The invention also includes a method of the invention described herein for
screening, for example, by testing a sample, such as blood or blood products
and other samples, to determine if the sample comprises a candidate
polypeptide in a non-wildtype conformation wherein the candidate polypeptide
includes a target epitope. In one embodiment, the method comprises;
determining whether the candidate polypeptide is in i) a wildtype conformation
or ii) a non-wildtype conformation, by a method comprising the steps of:
18
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
contacting the sample with a blocking agent that selectively blocks
accessible target epitope in the candidate polypeptide, wherein in the
wildtype conformation, the target epitope is accessible and reacts with
the blocking agent, and wherein in the non-wildtype conformation, the
target epitope is inaccessible and cannot react with the blocking agent;
contacting the sample with a conversion agent to modify the candidate
polypeptide to convert any inaccessible target epitope in the sample to
accessible target epitope;
contacting the polypeptide with a detection agent that binds selectively to
target epitope that was converted from inaccessible target epitope to
accessible target epitope, wherein binding between detection agent and
converted target epitope indicates that the candidate polypeptide was in a non-
wildtype conformation and the sample comprises a candidate polypeptide in a
non-wildtype conformation and wherein lack of binding between the detection
agent and the target epitope indicates that the polypeptide was in a wild type
conformation.
In another embodiment, the invention relates to a method of detecting whether
a candidate polypeptide including a target epitope is in i) a wildtype
conformation or ii) a non-wildtype conformation (for example, wherein the
polypeptide aggregates in the non-wildtype conformation), comprising:
contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitope, wherein in the wildtype conformation, the
target epitope is accessible and reacts with the blocking agent, and
wherein in the non-wildtype conformation, the target epitope is
inaccessible (for example, because the candidate polypeptide is
aggregated) and the target epitope cannot react with the blocking agent;
modifying the candidate polypeptide to convert inaccessible target
epitope to accessible target epitope; and
19
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
contacting the polypeptide with a detection agent that binds selectively
to the target epitope that was converted from inaccessible target epitope
to accessible target epitope, wherein binding between detection agent
and converted target epitope indicates that the candidate polypeptide
was in a non-wildtype conformation and wherein lack of binding
between the detection agent and the target epitope indicates that the
polypeptide was in a wild type conformation. One also removes
unreacted blocking agent from contact with the polypeptide, for
example, by allowing it to be consumed or degraded or removing it from
the reaction by physical or chemical processes.
The candidate polypeptide optionally comprises prion protein, the wild type
conformation comprises the conformation of wild type prion protein and the
non-wildtype conformation comprises the conformation of PrPsc. The
candidate polypeptide optionally comprises beta-amyloid polypeptide, tau
protein or APP protein, SOD1, alpha-synuclein, huntingtin protein, p53 or
islet
amyloid polypeptide or resistin. The blocking agent is optionally selected
from
the group consisting of peroxynitrite, hydrogen peroxide, methylene
compounds, succinic anhydride, epoxides, diethyl pyrocarbonate, 4-
hydroxynonenal (4HNE) and diazirine. The polypeptide is optionally modified
by denaturing the polypeptide. The polypeptide is also optionally denatured by
heat and/or detergent and/or chaotropic agents. The polypeptide is optionally
modified by treatment with a disaggregation agent to disaggregate the
polypeptide from the aggregated polypeptides. The disaggregation agent is
optionally selected from at least one of the group consisting of chaotropic
agents , detergent and heat. The detergent optionally comprises SDS. The
detection agent optionally comprises an aptamer or an antibody, for example,
directed against a prion polypeptide epitope. The antibody optionally
comprises 6H4 or 3F4. The aptamer or antibody is optionally directed against
an amyloid beta epitope. The antibody optionally comprises 6E10 or 4G8.
The non-wildtype conformation is in certain embodiments indicative of a
disease caused by protein aggregation, such as prion disease (eg. BSE or
CJD), Alzheimer's disease, Parkinson's disease or Lewy body disease,
Huntington's disease, amyotrophic lateral sclerosis, cancer or diabetes.
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Optionally, prior to contacting the blocking agent with the candidate
polypeptide, the candidate polypeptide is in a sample that is pretreated by
one
or more of the following methods: adsorption, precipitation, or
centrifugation.
Optionally, prior to contacting the blocking agent with the candidate
polypeptide, the target epitope is mapped (ie., the epitope is identified, for
example, as described below under the "Target Epitopes" section). Optionally,
the polypeptide is in a postmortem or antemortem sample selected from the
group of: CSF, serum, blood, urine, biopsy sample or brain tissue. Another
aspect of the invention relates to a kit for detecting whether a candidate
polypeptide including a target epitope is in i) a wildtype conformation or ii)
a
non-wildtype conformation, comprising a detecting agent that recognizes the
target epitope and instructions for at least one of i) mapping a target
epitope, ii)
contacting a candidate polypeptide with a blocking agent, and iii) contacting
a
candidate polypeptide with a detecting agent. The kit is useful to implement
method of the invention described herein. The detecting agent optionally
comprises an aptamer or an antibody. The antibody optionally comprises 6H4,
3F4, 6E10 or 4G8, optionally immobilized to a solid support. The kit
optionally
further comprises buffers and reagents, for example, for ELISA, such as
sandwhich ELISA, fluorescent ELISA. The kit optionally further comprises a
blocking agent. The kit optionally further comprises a denaturing agent
selected from at least one of the group of detergents and chaotropic agents.
The kit optionally further comprises a polypeptide standard. The kit
optionally
comprises a recombinant disease protein or a recombinant protein that mimics
a disease protein. In another embodiment, the invention relates to method of
detecting whether a candidate polypeptide that has been contacted with a
blocking agent is i) a wildtype conformation or ii) a non-wildtype
conformation,
wherein the candidate polypeptide comprises at least one target epitope and,
following contact with the blocking agent and removal of the blocking agent,
the candidate polypeptide has been modified to convert any inaccessible
target epitope to accessible target epitope, the method comprising: contacting
the polypeptide with a detection agent that binds selectively to the target
epitope that was converted from inaccessible target epitope to accessible
target epitope, wherein binding between detection agent and converted target
epitope indicates that the candidate polypeptide was in a non-wildtype
21
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
conformation (for example, an aggregated conformation) and wherein lack of
binding between the detection agent and the target epitope indicates that the
polypeptide was in a wild type conformation. Diseases, blocking agents, target
epitopes, detecting agents and other aspects described herein are also useful
in this method. The diseases, blocking agents, target epitopes, detecting
agents and other aspects described herein are also readily adapted for the
methods described in preceding paragraphs, such as methods for testing a
sample from an animal (such as a human, livestock etc.) to determine if the
animal has a disease or screening a sample.
The reverse situation to the methods described in some of the aforementioned
paragraphs is also usefully detected, for example, where the wildtype
conformation includes an inaccessible epitope and the non-wild type
conformation has an accessible epitope. This situation is also readily adapted
to methods described herein, such as diagnosing disease or screening
samples.
Other features and advantages of the present invention will become apparent
from the following detailed description. It should be understood, however,
that
the detailed description and the specific examples while indicating preferred
embodiments of the invention are given by way of illustration only, since
various changes and modifications within the spirit and scope of the invention
will become apparent to those skilled in the art from this detailed
description.
BRIEF DESCRIPTION OF THE DRAWINGS
Embodiments of the invention are described in relation to the drawings in
which:
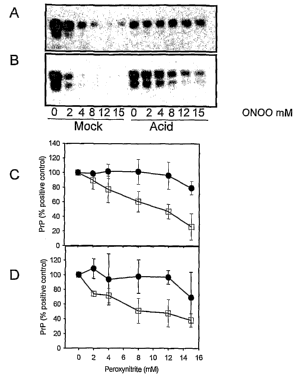
Fig. 1. Brain PrP aggregated in vitro by acid treatment is protected from
modification by peroxynitrite
Mock or acid treated human brain homogenate was treated with increasing
concentrations of peroxynitrite (ON00) and then subjected to immunoblotting
with 3F4 (panel A ) or 6H4 (panel B). Effect of peroxynitrite on the 3F4 (C)
and
6H4 (D) epitope in mock (D) and acid treated ( = ) brain homogenate.
lmmunoblot films were scanned and band intensities determined by Unscanit
22
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
software. The results are the combined relative intensities of 3 separate
experiments.
Fig. 2. PrP in scrapie infected hamster brain is protected from
modification by peroxynitrite
(A) Effect of peroxynitrite treatment on the 6H4 epitope in scrapie infected
hamster brain. (B) The blot in (A) was scanned and relative band intensities
determined using Unscanit software. ( = ) Scrapie infected hamster brain. (0)
Normal hamster brain.
Fig. 3 Protection from peroxynitrite induced modification is due to
aggregation in acid treated brain
(A) Effect of peroxynitrite on the immunoprecipitation (IP) of PrP in mock and
acid treated brain homogenate. Brain homogenate was treated with 10 mM
peroxynitrite followed by incubation for 2 h at RT with (+) or without (-) 2.5
M
guanidine hydrochloride (Gu). The resulting samples were immunoprecipitated
with 6H4 or 3F4. More PrP is precipitated in the acid treated sample following
treatment with peroxynitrite + Gu whereas in the mock sample, Gu has no
effect. This suggests that Gu is able to break up aggregated PrP in the acid
sample that is protected from destruction by peroxynitrite. (B) Effect of
peroxynitrite on PrP in mock and acid treated brain homogenate as measured
by ELISA. Brain homogenate was treated with increasing concentrations of
peroxynitrite followed by 2.5 M Gu. Following a 10-fold dilution, the samples
were analyzed by sandwich ELISA with 6H4 as the capture Ab and 3F4 as the
detection Ab. Similar to the immunoblot and IP data, the results show that
misfolded PrP is protected from destruction by peroxynitrite treatment, due to
aggregation.
Fig. 4 Detection of aggregated amyloid beta (Abeta) using EPA
The 6E10 epitope in the Abeta region of APP is less accessible to
peroxynitrite
modification in Alzheimer's disease brain compared to normal brain (panel A),
and in brain homogenates that have been treated at low pH to induce protein
aggregation (panel B). Abeta 1-42 peptide aggregated in vitro shows
prominent epitope protection of the 6E10 epitope to peroxynitrite
modification,
in comparison with soluble non-aggregated Abeta 1-42 (panel C). Normal and
aggregated Abeta 1-42 treated with increasing concentrations of peroxynitrite
23
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
and them immunoblotted with 6E10 antibody also shows prominent epitope
protection (panel D).
Fig. 5 Detection of epitopes modifiable by DEPC in SOD1
(A) Western blot showing soluble SOD1 treated with increasing concentrations
of DEPC and immunoblotted with sheep anti-SOD1. (B) Graphical
representation of decreasing antibody binding to SOD1 with increasing DEPC-
concentration.
Fig. 6 Detection of aggregated alpha-synuclein using EPA
(A) Western blot showing effect of increasing concentrations of DEPC on
antibody binding to soluble and insoluble alpha-synuclein. (B) Graphical
representation showing the extent of antibody binding to normal (¨) and
insoluble (n) alpha-synuclein.
DETAILED DESCRIPTION OF THE INVENTION
The current invention provides a useful method for the detection of a disease
related polypeptide counterpart of a normal cellular polypeptide which forms
aggregates or otherwise leads to the obscuration of one or more epitopes that
are not obscured in the normal or wild type polypeptide. The invention
recognizes the importance of aggregation in the pathology of diseases such as
prion disease. The invention also takes advantage of this aggregation effect
and provides an assay that overcomes problems with prior art detection
assays. In one embodiment, the method of the invention is applied to the
detection of PrPsc in plasma, serum, urine or other biological sample. The
methods of the invention are further useful for detecting any polypeptide that
exists in two or more conformations, where one or more target epitopes are
inaccessible in at least one conformation. In one embodiment the invention
includes a method of detecting whether a candidate polypeptide including a
target epitope is in a wild type or non-wild type conformation.
"Epitope" refers to a portion of a sequence of contiguous or non-contiguous
amino acids (antigen) which is recognized by and bound by a detection agent
such as an antibody. Preferably, the epitope is a linear epitope on a
polypeptide which typically includes 3 to 10 or 6 to 10 or more contiguous
\
24
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
amino acids that are recognized and bound by a detection agent. A
conformational epitope includes non-contiguous amino acids. Sometimes
conformational epitopes can re-establish themselves after denaturation by
partial refolding on, e.g, an immunoblot membrane. The detection agent such
as an antibody recognizes the 3-dimensional structure. When a protein
molecule is folded into a three dimensional structure the amino acids forming
the epitope are positioned in a manner that permits the detection agent to
recognize and bind to the amino acids. In an unfolded (denatured) protein only
the linear epitope is recognized and bound by the detection agent. Since the
protein is unfolded prior to contact with the detection agent, the
inaccessible
epitope will typically be a linear epitope.
"Blocking agent" refers to an agent that reduces epitope reactivity, for
example by binding to the epitope or by modifying and destroying epitope
reactivity, for example on an amino acid side group within a linear epitope,
so
that the epitope is prevented from binding to detection agent (usually but not
always an antibody). An example of a blocking agent is peroxynitrite. Other
examples would include methylene, hydrogen peroxide, diethyl pyrocarbonate,
4-hydroxynonenal (4HNE) epoxides such as conduritol-B-epoxide and 1,2-
epoxy-3-(p-nitrophenoxy)propane and diazirine. Chemical modifying agents
that saturate accessible amino acids critical for epitope recognition in
native
conditions are most useful in the applications of epitope protection
technology.
Additionally the blocking agent may phosphorylate, glycosylate or otherwise
modify a target-epitope. The blocking agent may also include peptides,
antibodies or antibody fragments that bind to the epitope. The blocking agent
should efficiently modify accessible amino acids (e.g. modify at least: 50%,
75%, 90%, 95% or 99% of accessible amino acids).
"Accessible epitope" is target epitope that is available to react with
blocking
agent in methods of the invention. For example, epitope that is available to
react with blocking agent is accessible epitope. After reacting with blocking
agent, the accessible epitope is prevented from binding to detection agent
(after this reacting step, the reacted epitope may be referred to as the
blocked
epitope).
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
"Antibody" is intended to include whole antibodies and fragments thereof
which also specifically react with one or more protein epitopes, such as
disease protein epitopes. Antibodies can be fragmented using conventional
techniques and the fragments screened for utility in the same manner as
described below. For example, F(ab')2 fragments can be generated by
treating antibody with pepsin. The resulting F(ab')2 fragment can be treated
to
reduce disulfide bridges to produce Fab' fragments.
"Aptamer" means a macromolecule such as a peptide, RNA or DNA
molecule that is able to specifically interact with a protein or peptide
target.
"Inaccessible epitope" means that target epitope modification by the chemical
blocking agent is prevented or significantly reduced (e.g. reduced by at
least:
50%, 75%, 90%, or 95%), for example, by differential misfolding relative to
the
wild type polypeptide, by aggregation of misfolded polypeptide or by post-
translational modifications of the polypeptide. In some cases, inaccessible
epitope is converted to accessible epitope by removing the hindrance (e.g.
misfolding or aggregation) that prevents or significantly reduces target
epitope
modification by the blocking agent. The inaccessible epitope that is converted
to accessible epitope may also be called "revealed epitope".
"Detection agent" refers to an agent that binds to epitope and which may be
detected, such as antibody specific for prion polypeptide epitopes that can be
used to probe the sample containing the polypeptide. The detection agent is
used after the polypeptide is unfolded such that the detection preferentially
binds the unblocked, unmodified epitopes.
"Disease protein or disease polypeptide" refers to a polypeptide associated
with a disease or disorder state where the modular or higher order
conformation of the polypeptide differs from the wild type or non-disease
conformation and includes mutants, variants and polymorphic versions thereof.
A disease protein or disease polypeptide can also be referred to as non-wild
type conformation protein or polypeptide. The modular conformation refers to
26
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
conformational changes in the three dimensional structure of a single protein
molecule. The higher order conformation refers to conformational changes in
the three dimensional structure of many protein molecules aggregated
together. The aggregation can consist of one or more different proteins and
can be associated with non-protein molecules. The wildtype and non-wildtype
candidate polypeptides including disease proteins or polypeptides also include
recombinant proteins, such as cellularly expressed (i.e. bacteria, using
baculovirus sytems etc.) and in vitro translated polypeptides
"Wildtype folded conformation" refers to the wild type, folded conformation
of protein in a non-disease or non-disorder state.
"Misfolded conformation" refers to the folded conformation of polypeptide in
a disease or disorder state where the conformation differs from the wild type
conformation. The difference in conformation is as a result of differential
folding. The differential folding may cause protein aggregation.
"Wildtype conformation" refers to the conformation of polypeptide in its usual
or normal state or in a reference or desired state and can include polypeptide
in a non-disease or disorder state.
"Non-wildtype conformation" refers to a conformation of polypeptide that
differs from the conformation of the wild type polypeptide and can include a
conformation of polypeptide in a disease or disorder, where the conformation
differs from the wild type conformation. The difference in conformation may be
as a result of differential folding, polypeptide aggregation or differential
post-
translational modification compared to the wild type polypeptide. In the case
of
polypeptide aggregation, the aggregation may prevent accessibility of the
epitope rather than the changed conformation.
Neurodegenerative diseases, such as Alzheimer's disease (AD), amyotrophic
lateral sclerosis (ALS) and Parkinson's disease/Lewy body dementia (PD,
LBD) pose major challenges to the aging population and health care system.
No specific biochemical test exists for neurodegenerative diseases as a group
27
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
(1,2). Sporadic AD, ALS, and PD/LBD are all associated with neural
accumulation of pathological multimers of nnisfolded polypeptides (such as
fibrils, protofilaments, and amorphous aggregates), including the Abeta
fragment of the amyloid precursor protein (APP) in AD; superoxide dismutase-
1 (SOD1) in ALS, and alpha-synuclein in PD and LBD (1). As with prion
diseases, mutations in genes encoding these aggregation-prone polypeptides
are associated with autosomal dominant familial forms of AD, ALS, and PD.
The detection of disease-associated misfolded polypeptide aggregates
enables specific and sensitive antemortem diagnostic tests for
neurodegenerative diseases.
To this end the inventors have invented the "epitope protection assay" (EPA),
an innovative technology for detection of aggregated polypeptides in tissues
and accessible biological fluids, such as blood.and CSF, which serve as
"sinks" for the aggregates released from dying neurons. The method optionally
consists of:
- reacting a sample with a chemical modifying agent;
- disaggregating and denaturing the treated polypeptides;
- probing the sample with detection agents such as antibodies against
specific epitopes blocked by the chemical modifier; and
- detection of agent-bound polypeptides (e.g., by ELISA).
Normal soluble polypeptides in the sample are rendered "invisible" in the
assay, because accessible epitopes are not detected by a detecting agent (eg.
blocked to antibody recognition), whereas a proportion of polypeptides in
aggregates are "protected" from chemical modification by virtue of their
interior
sequestration, and are still available to be detected by a detecting agent
(eg.
bind antibody) after disaggregation.
The methods of the invention are useful to diagnose diseases characterized by
polypeptide misfolding and/or aggregation such as in the diseases mentioned
above or for diseases or disorders characterized by polypeptides with
28
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
otherwise differentially accessible target epitopes in disease and wildtype
protein conformations.
The present inventors have also found that treatment of recombinant mouse
prion polypeptide (rmPrP) at low pH in the presence of low concentrations of
denaturants causes the polypeptide to acquire increased beta-sheet content,
reminiscent of the misfolded disease-associated prion polypeptide isoform,
PrP. This conversion of rmPrP is associated with increased solvent
accessibility of tyrosine side chains4. The inventors have found that
treatment
of normal brain homogenate with acid and denaturants causes PrP to become
detergent insoluble (29). In order to probe the surface accessibility of
tyrosines
and other residues in normal and misfolded PrPc, normal and acid-misfolded
human brain tissue was treated with the chemical nitrating compound
peroxynitrite. Peroxynitrite treatment of brain tissue caused a reduction in
the
binding of the anti-PrP antibodies 3F4 and 6H4 as measured by
innmunoblotting, immunoprecipitation and ELISA.
Peroxynitrite-induced
epitope blocking was more pronounced on normal brain PrP than on misfolded
PrP, showing a protective effect of aggregation. Similar findings were
observed in normal and scrapie-infected hamster brain, in which 3F4 and 6H4
epitopes of scrapie brain PrP were partially protected from peroxynitrite-
induced modification. lmmunoprecipitation of peroxynitrite-treated brain with
anti-nitrotyrosine antibodies suggests that either PrP is nitrated on tyrosine
residues or another polypeptide in proximity to PrP is nitrated and
coimmunoprecipitates PrP.
Accordingly, the invention includes a method of determining polypeptide
aggregation, including but not limited to PrPs, comprising:
= reacting a sample with a chemical modifying agent where such agent
could be, but is not limited to, peroxynitrite
= disaggregating and/or denaturing the chemically modified sample with
heat, detergent, or chaotropic agents; and
= probing with antibodies specific for prion polypeptide epitopes.
29
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
The inventors have further shown that the methods of the invention are useful
for detecting Alzheimer disease proteins.
In one embodiment, the Alzheimer's disease detection method comprises:
- reacting a sample of polypeptide (the sample typically contains all or
part of a disease protein or polypeptide such as amyloid precursor
polypeptide or amyloid beta or tau and/or the corresponding wild type
polypeptide, and in many cases an abundance of one or the other) with
a chemical modifying agent, typically a blocking agent such as
peroxynitrite, which modifies exposed epitopes so that they cannot bind
to a detection agent;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with detection agents, such as an antibody against a target
epitope to determine whether the polypeptide prior to disaggregation
and/or denaturing, included inaccessible target epitopes.
Abeta containing vascular or plaque filaments are also associated with
conditions such as trisomy 21 (Down's syndrome), hereditary cerebral
hemorrhage with amyloidosis (HCHWA)-Dutch type, and normal brain aging
(Mori, H et al. JBC (1992) 267: 17082-86). Accordingly, in one embodiment
detection of Abeta disease protein is prognostic for diseases HCHWA-Dutch
type or normal brain aging.
In addition, the methods of the invention can be combined with other
diagnostic methods such as magnetic resonance imaging (MRI) or computed
tomography (CT) scans to confirm diagnosis.
The methods of the invention are useful to detect protein or polypeptide
including target that exists in two or more conformations, where one or more
target epitopes are concealed in at least one conformation.
Accordingly, the invention relates to a detection method comprising:
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
- reacting polypeptide with a chemical modifying agent, typically a
blocking agent , which is defined to modify exposed epitopes so that
they cannot bind to detection agents;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with detection agents, such as antibodies against target
epitope to determine whether the polypeptide prior to disaggregation
and/or denaturing, included target epitopes inaccessible to the chemical
modifying agent.
The result indicates whether the polypeptide includes inaccessible epitopes,
which is indicative of the type of polypeptide that is present (i.e. wild type
or
non-wild type protein).
The invention also includes a method of detecting whether a candidate
polypeptide including a target epitope that has been reacted with a blocking
agent, is in a wildtype conformation or a non-wildtype conformation,
comprising:
- modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope; and
- contacting the polypeptide with a detection agent that binds selectively
to the target epitope that was converted from inaccessible target epitope
to accessible target epitope, wherein binding between detection agent
and converted target epitope indicates that the candidate polypeptide
was in a non-wildtype conformation and wherein lack of binding
between the detection agent and the target epitope indicates that the
polypeptide was in a wild type conformation.
In another application, the invention also includes a method of detecting
intrinsically modified polypeptide, wherein the modification protects target
epitope from reacting with the detecting agent, comprising:
31
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
- contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitope, wherein in one of the non-wildtype
conformation or the wildtype conformation, the target epitope is
accessible and reacts with the blocking agent, and wherein in the other
conformation, the target epitope is inaccessible and does not react with
the blocking agent;
- reacting the sample with an agent that removes the intrinsic
modification from the intrinsically modified polypeptide target epitope;
- disaggregating and/or denaturing the polypeptide in the sample; and
- probing with a detection agent, such as antibodies against the target
epitope, to determine whether the candidate polypeptide is an
intrinsically modified polypeptide.
Chemical Modifying Agents
The chemical modifying agent of the invention comprises any chemical
(including a biological agent) that modifies target epitope residues such that
the epitope is rendered invisible by the methods of the invention (ie. not
detected by the detecting agent or detection is reduced). For example,
peroxynitrite preferentially modifies tyrosine, serine, methionine, histidine
and
tryptophan as well as cysteine and other amino acids (25, 26). DEPC
preferentially modifies histidines (37), and succinic anhydride preferentially
modifies residues comprising amines. Epoxides, including conduritol-B-
epoxide and 1,2-epoxy-3-(p-nitrophenoxy) propane) are a reactive group used
widely for "suicide inhibition" of carboxyl group side chains, such as the
catalytic residues of aspartyl proteases (19, 20). Hydrogen peroxide and
methylene are also useful. The chemicals may modify the target epitope by
oxidizing, nitrating, reducing, or otherwise modifying the epitope. In
addition,
the epitope may be modified by a chemical modifying agent that is a
phosphate group (by phosphorylation), or a gylcosyl group (by gylcosylation),
and/or other chemical group that obscures the target epitope.
Accordingly, in one embodiment the chemical modifying agent is chosen from
32
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
the group peroxynitrite, DEPC, hydrogen peroxide, succinic anhydride,
methylene and epoxides (conduritol-B-epoxide and 1,2-epoxy-3-)p-
nitrophenoxy)propane and/or related variants thereof.
After reacting with candidate polypeptides, the chemical modifying agent is
removed. It is readily apparent to a skilled person that the method steps of
the
invention recited here involving removing the blocking agent typically involve
physically, chemically or otherwise removing the blocking agent away from the
candidate polypeptide to prevent further reaction. Removal optionally involves
allowing a sufficient time to pass so that the blocking agent is removed from
the candidate polypeptide by being consumed or degraded (for example, such
that the blocking agent becomes inert or oxidized). Removal optionally
involves adding a compound to react with any excess blocking agent to
inactivate it. Removal also optionally involves physical filtering of the
blocking
agent by conventional filtration techniques or centrifugation to separate the
candidate polypeptide and blocking agent, or physical binding to a substrate
useful for removing the blocking agent, such as by binding of blocking agent
or
candidate polypeptide to an immobilized substrate in a column.
Removing means preventing further reactions by the blocking agent by, for
example, physically or chemically inactivating the blocking agent, taking the
blocking agent out of contact with the sample including the candidate
polypeptide or allowing a sufficient amount of time to pass for the blocking
agent to be consumed or degraded.
Chemical modification of a target epitope leads to obscuration of an epitope
to
antibody recognition. In one embodiment treatment with a blocking agent such
as peroxynitrite leads to destruction of epitopes on monomeric proteins but
not
epitopes on aggregated proteins such as non-wild type polypeptides or
disease proteins.
Pretreatment
The methods of the invention also contemplate pretreatment of the sample to
enhance EPA detection. For example if decreased detection of aggregated
33
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
proteins such as prions in blood or urine is observed, pre-clearing strategies
are readily employed to enhance detection with detergents, precipitating
agents, and adsorbents such as those typically used in commercial ELISA
assays which are known to one skilled in the art. Polypeptide samples may
also be pretreated with agents such as detergents or guanidine or heat.
Finally samples maybe concentrated or precleared by methods such as
centrifugation. Accordingly, in one embodiment the samples are pretreated
before employing a method of the invention.
Detecting Misfolded or Aggregated Proteins and Polypeptides
The inventors have found a method that detects polypeptides that have target
epitopes that are accessible to detection in one conformation and inaccessible
in another by modification of inaccessible epitopes by a modifying agent. The
inventors have identified several epitopes that are useful as target epitopes
in
the methods of the invention. Other target epitopes are identified as
described
below.
Target Epitopes
Target epitopes are identified for polypeptides that exist in two or more
conformations wherein epitopes that can be detected by detecting agents such
as antibodies, aptamers or peptides, are accessible in one conformation and
inaccessible in the other conformation. Where an epitope is found to be
blocked from detection by a blocking agent in one conformation of the
polypeptide, the epitope is a target epitope. To identify target epitopes, a
detection agent such as an antibody is chosen. If the detection agent is an
antibody it is preferably a monoclonal antibody although polyclonal antibodies
are also usable. The epitope, which can be a linear or non-linear epitope, and
which is specifically recognized by the antibody, is optionally a known
epitope.
A candidate chemical modifying agent such as peroxynitrate is chosen. If the
epitope recognized by the detection agent is known, the candidate chemical
modifying agent is preferably chosen based on its ability to modify amino acid
residues in the target epitope. For example peroxynitrite preferentially
34
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
modifies tyrosine and histidine residues with some modification of cysteine
and
other amino acids. Peroxynitrite is optionally chosen as chemical modifying
agent if tyrosines and/or histidines are present in the target epitope.
Aliquots
of a sample comprising wild type polypeptide and aliquots of a sample
comprising non-wildtype polypeptide are reacted with increasing
concentrations of the chosen chemical modifying agent. Each sample
comprises one or more of recombinant polypeptide, cell extracts or tissue
samples known to express the polypeptide in either the wild type or non-wild
type conformation. Preferably samples of polypeptide have similar
concentrations of polypeptide. The non-wildtype conformation polypeptide
sample is alternatively obtained by treating a polypeptide in wild type
conformation with an agent, such as acid, that induces conversion to a non-
wildtype conformation.
Each sample of polypeptide is denatured and/or disaggregated to convert any
inaccessible putative target epitope to accessible target epitope. Each sample
of polypeptide is then contacted with the chosen detection agent. Detection is
performed using techniques known in the art such as ELISA, and Western
blotting. The amount of signal generated by the detection agent for sample
comprising polypeptide in a wildtype conformation treated with protection
agent
and for sample comprising polypeptide in a non-wildtype conformation treated
with protection agent are compared. A difference in detection at one or more
concentrations of chemical modifying agent indicates that the epitope is
protected in one conformation and further indicates that the epitope is a
target
epitope. A difference over a range of chemical modifying agent concentrations
indicates that the target epitope is useful for EPA. The process is repeated
with
different blocking agents and/or detecting agents and target epitopes are
identified. One typically standardizes and titrates the blocking agent and to
performs experiments using a "universal" chemical modifier such as
methylene24'25, which optionally yields more uniform and complete protection
of
the target epitope.
Accordingly in one example, a method of identifying a target epitope in a
polypeptide that has two or more conformations wherein the target epitope is
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
accessible to detection in one conformation and inaccessible in another
conformation comprises:
- reacting a sample comprising polypeptide in a wild type
conformation and a sample comprising polypeptide in a non-wild
type conformation typically with one or more concentrations of a
chemical modifying agent;
- denaturing and/or disaggregating each sample to convert any
inaccessible target epitope to accessible target epitope;
- contacting the samples with a detection agent; and
- comparing the signal generated by the detection agent for
samples comprising polypeptide in a wildtype conformation
treated with chemical modifying agent and for samples
comprising polypeptide in a non-wildtype conformation treated
with chemical modifying agent wherein a difference in detection
between sample comprising wildtype polypeptide and sample
comprising non-wild type sample indicates that the epitope is
protected in one conformation and further indicates that the
epitope is a target epitope.
Conditions for EPA
Titration experiments with peroxynitrite, hydrogen peroxide and methylene
(based on UV light photolysis of the precursor diazirine) or other modifying
agents, are useful to improve conditions for epitope protection.
Samples known to contain polypeptides in two or more conformations,
including disease proteins, are optionally reacted using immunoblotting and
ELISA. In each case, samples are prepared and optionally mixed with
increasing concentrations of the modifying agent and processed, for example,
by immunoblotting, ELISA and/or time resolved fluorescence. This defines the
type and concentration of chemical agent allowing the maximal distinction
between monomeric and aggregated proteins including disease proteins.
Additional informative experiments can involve using recombinant disease
(non-wild type) protein (such as acid treated prion proteins or mutant p53
36
CA 02536305 2006-02-20
WO 2005/019828 PCT/CA2004/001503
proteins and other protein listed in this application) and normal (wild type)
protein in place of samples containing disease proteins.
In some cases, disease proteins may have different properties for chemical
modification than do recombinant disease proteins, and disease proteins in
one sample type (such as brain tissue) may display different chemical
modification properties than disease proteins circulating in blood, or
detectable
in urine. One of skill in the art shall readily identify the optimal
conditions for
endogenous prions using known techniques.
The EPA achieves superior commercial utility by detecting disease proteins in
biological tissues and fluids for which no present technology exists. Some
disease proteins are in very low abundance. For example prions are in very
low abundance (10-100 prions/mL by bioassay), and protease-resistant PrP in
urine is only intermittently/sporadically detectable by precipitation of large
fluid
volumes. Also, any prospective blood test must contend with high
concentrations of wildtype protein (i.e. for PrPc (typically 106-fold more
than
PrPsc)) and "blocking" by heterologous plasma proteins. Using the optimized
chemical modification regimen and the DELFIA-TRF system, the sensitivity
thresholds for EPA in blood and urine are optionally determined using:
1. Animal and human plasma and urine "spiked" with a titration of
disease protein;
2. Plasma and urine from model disease animals expressing a disease
protein.
Biological fluids clinically accessible by non-invasive routes provide a
substrate -
for a practical antemortem test for diagnosis and screening of diseases
involving aggregated disease proteins in humans and animals. The methods
of this invention are also useful in post-mortem testing. One of skill in the
art
readily determines whether EPA with "disease protein spike" titration in
normal
blood and urine reveals similar DELFIA-TRF signals to the same disease
protein titration in buffer, showing that the EPA is not affected by "blocking
factors" in these biological fluids. Preferential "blocking" of disease
protein by
37
CA 02536305 2007-08-02
heterologous proteins may actually enhance epitope protection to chemical
modifying agents. If decreased detection of disease protein in blood or urine
is
observed, pre-clearing strategies are readily employed to enhance disease
protein detection, for example, with detergents, precipitating agents, and
adsorbents typically used in commercial ELISA assays which are known to
one skilled in the art.
In one embodiment, the methods of the invention involve the detection of
target epitopes in misfolded or aggregated polypeptides. In another
embodiment, the invention provides a method for improving or optimizing the
detection of polypeptides that can exist in 2 or more conformations. In
another
embodiment the detection of misfolded and/or aggregated polypeptides is
indicative of disease (disease proteins). In another embodiment, polypeptides
are detected using detection agents such as antibodies, aptamers or peptides
that specifically bind to epitopes of polypeptides that can be chemically
modified. In another embodiment these polypeptides are disease proteins.
Antibodies to candidate protein epitopes are commercially available antibodies
or are readily prepared by a person skilled in the art and include antibody
fragments, and single chain antibodies. All the aforementioned methods are
readily implemented using steps described in this application.
Antibodies
The invention contemplates the use of known antibodies as the binding agent
including biotin-3F4 and 3F4 and 6H4 which recognize prion disease proteins.
3F4 reacts against the MKHV (SEQ ID NO: 1) epitope and 6H4 reacts against
the DYEDRYYRE (SEQ ID NO: 2) epitope. Additionally, 6E10 which
recognizes Abeta, reacts against the EFRHDS (SEQ ID NO: 3) epitope
(residues 3-8).
Other antibodies and the epitopes recognized (if known) which are optionally
used with the methods of the invention are listed in the table below.
Table. Antibodies useful to detect disease proteins
38
CA 02536305 2006-02-20
WO 2005/019828 PCT/CA2004/001503
Protein Antibody Mono/Poly Epitope Company
4G8 Monoclonal Within aa18-22 of human Abeta Signet
6E10 Monoclonal Within aa3-8 of human Abeta Signet
ab2539 Polyclonal NA Abcam
Abeta QED
Abeta-NT Polyclonal NA Bioscience
DE2B4 Monoclonal Within aa1-17 of human Abeta Acris
antibodies
NBA-104E Monoclonal Within aa1-16 of human Abeta Stressgen
4D6 Monoclonal Unknown Acris
antibodies
ab6162 Polyclonal NA Abcam
LB509 Monoclonal Unknown Zymed
Alpha- Syn-1 Monoclonal Within aa91-99 of human a-syn BD
Biosciences
synuclein Within aa87-110 of human a-
Syn-204 Monoclonal syn Lab Vision
Within aa121-125 of human a-
Syn-211 Monoclonal syn Lab Vision
Mouse Anti-Tau-
1' Monoclonal Within aa95-108 of human tau Biomeda
Mouse Anti-Tau-
T 2 Monoclonal Unknown Stressgen
au
T14 Monoclonal Within aa141-178 of human tau Zymed
T46 Monoclonal Within aa404-441 of human tau Zymed
Tau-2 Monoclonal Unknown Acris
antibodies.
Tau-5 (ab3931) Monoclonal Unknown Abcam
Mouse SOD1 Monoclonal Unknown Sigma-aldrich
SOWRabbit SOD1 Polyclonal Unknown Stressgen
Rat SOD1 Polyclonal Unknown Stressgen
Sheep SOD1 Polyclonal Unknown OxisResearch
Antibodies to disease protein epitopes are prepared using techniques known in
the art. For example, by using a peptide of a disease protein including a
putative target epitope, polyclonal antisera or monoclonal antibodies are made
=
using standard methods. A mammal, (e.g., a mouse, hamster, or rabbit) can
be immunized with an immunogenic form of the peptide which elicits an
antibody response in the mammal. Techniques for conferring immunogenicity
on a peptide include conjugation to carriers or other techniques well known in
the art. For example, the protein or peptide is administered in the presence
of
adjuvant. The progress of immunization can be monitored by detection of
antibody titers in plasma or serum. Standard ELISA or other...immunoassay
procedures are optionally used with the immunogen as antigen to assess the
39
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
levels of antibodies. Following immunization, antisera can be obtained and, if
desired, polyclonal antibodies isolated from the sera.
To produce monoclonal antibodies, antibody producing cells (lymphocytes) are
optionally harvested from an immunized animal and fused with myeloma cells
by standard somatic cell fusion procedures thus immortalizing these cells and
yielding hybridoma cells. Such techniques are well known in the art, (e.g.,
the
hybridoma technique originally developed by Kohler and Milstein (Nature 256,
495-497 (1975)) as well as other techniques such as the human B-cell
hybridoma technique (Kozbor et al., lmmunol. Today 4, 72 (1983)), the EBV-
hybridoma technique to produce human monoclonal antibodies (Cole et al.
Monoclonal Antibodies in Cancer Therapy (1985) Allen R. Bliss, Inc., pages
77-96), and screening of combinatorial antibody libraries (Huse et al.,
Science
246, 1275 (1989)). Hybridoma cells can be screened immunochemically for
production of antibodies specifically reactive with the peptide and the
monoclonal antibodies can be isolated.
Chimeric antibody derivatives, i.e., antibody molecules that combine a non-
human animal variable region and a human constant region are also
contemplated within the scope of the invention. Chimeric antibody molecules
include, for example, the antigen binding domain from an antibody of a mouse,
rat, or other species, with human constant regions. Conventional methods
used to make chimeric antibodies containing the immunoglobulin variable
region which recognizes disease protein epitopes of the invention (See, for
example, Morrison et al., Proc. Natl Acad. Sci. U.S.A. 81,6851 (1985);
Takeda et al., Nature 314, 452 (1985), Cabilly et al., U.S. Patent No.
4,816,567; Boss et al., U.S. Patent No. 4,816,397; Tanaguchi et al.,
European Patent Publication EP171496; European Patent Publication
0173494, United Kingdom patent GB 2177096B).
Specific antibodies, or antibody fragments, such as, but not limited to,
single-
chain Fv monoclonal antibodies reactive against disease protein epitopes are
readily generated by screening expression libraries encoding immunoglobulin
genes, or portions thereof, expressed in bacteria with peptides produced from
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
the nucleic acid molecules of disease proteins. For example, complete Fab
fragments, VH regions and FV regions are expressed in bacteria using phage
expression libraries (See for example Ward et al., Nature 341, 544-546:
(1989); Huse et al., Science 246, 1275-1281 (1989); and McCafferty et al.
Nature 348, 552-554 (1990)). Alternatively, a SCID-hu mouse, for example the
model developed by Genpharm, is used to produce antibodies or fragments
thereof.
Antibodies specifically reactive with disease protein epitopes, or
derivatives,
such as enzyme conjugates or labeled derivatives, are useful to detect disease
protein epitopes in various samples (e.g. biological materials). They are
useful
as diagnostic or prognostic reagents and are readily used to detect
abnormalities in the level of protein expression, or abnormalities in the
structure, and/or temporal, tissue, cellular, or subcellular location of
disease
protein epitopes. In vitro immunoassays are also useful to assess or monitor
the efficacy of particular therapies. The antibodies of the invention may also
be used in vitro to determine the level of expression of a gene of a
polypeptide
that exists in two or more conformations such as a disease protein in cells
genetically engineered to produce the disease protein.
The antibodies are useful in any known immunoassays which rely on the
binding interaction between an antigenic determinant of the disease protein
epitopes and the antibodies. Examples
of such assays are
radioimmunoassays, enzyme immunoassays (e.g. ELISA including Sandwich
ELISA), immunofluorescence, immunoprecipitation, latex agglutination,
hemagglutination, and histochemical tests. The antibodies are useful to detect
and quantify the disease protein in a sample in order to determine its role
and
to diagnose the disease caused by the disease protein.
In particular, the antibodies of the invention are useful in
immunohistochennical
analyses, for example, at the cellular and subcellular level, to detect a
disease
protein, to localize it to particular cells and tissues, and to specific
subcellular
locations, and to quantitate the level of expression.
41
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Cytochemical techniques known in the art for localizing antigens using light
and electron microscopy to detect polypeptides such as disease proteins.
Generally, an antibody of the invention is optionally labeled with a
detectable
substance and the recognized polypeptide is localised in tissues and cells
based upon the presence of the detectable substance. Examples of detectable
substances include, but are not limited to, the following: radioisotopes
(e.g., 3H,
14C, 35s, 1251, 131.,i),
fluorescent labels (e.g., FITC, rhodamine, lanthanide
phosphors), luminescent labels such as luminol; enzymatic labels (e.g.,
horseradish peroxidase, beta-galactosidase, luciferase, alkaline phosphatase,
acetylcholinesterase), biotinyl groups (which can be detected by marked avid
in
e.g., streptavidin containing a fluorescent marker or enzymatic activity that
can
be detected by optical or colorimetric methods), predetermined polypeptide
epitopes recognized by a secondary reporter (e.g., leucine zipper pair
sequences, binding sites for secondary antibodies, metal binding domains,
epitope tags). In some embodiments, labels are attached via spacer arms of
various lengths to reduce potential steric hindrance. Antibodies may also be
coupled to electron dense substances, such as ferritin or colloidal gold,
which
are readily visualized by electron microscopy.
The antibody or sample may be immobilized on a carrier or solid support which
is capable of immobilizing cells, antibodies etc. For example, the carrier or
support may be nitrocellulose, or glass, polyacrylamides, gabbros, and
magnetite. The support material may have any possible configuration
including spherical (e.g. bead), cylindrical (e.g. inside surface of a test
tube or
well, or the external surface of a rod), or flat (e.g. sheet, test strip).
Indirect
methods may also be employed in which the primary antigen-antibody reaction
is amplified by the introduction of a second antibody, having specificity for
the
antibody reactive against disease protein epitopes. By way of example, if the
antibody having specificity against a polypeptide epitope such as a disease
protein epitope is a rabbit IgG antibody, the second antibody may be goat anti-
rabbit gamma-globulin labeled with a detectable substance as described
herein.
42
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Where a radioactive label is used as a detectable substance, disease proteins
may be localized by autoradiography. The results of autoradiography may be
quantitated by determining the density of particles in the autoradiographs by
various optical methods, or by counting the grains.
Aptamers
Aptamers are also useful in the methods of the invention to detect
polypeptides
such as disease proteins. Aptamers are macromolecules that can recognize
targets such as proteins with high specificity and sensitivity.
Nucleic acid aptamers are small molecules isolated from combinatorial
libraries
by a procedure named systemic evolution of ligands by exponential enrichment
(SELEX) (reviewed in Cerchia L et al, FEBS Letters 528 (2002) 12-12). Using
this technology aptamers that bind proteins with high target specificity and
selectivity can be identified. The affinities can be comparable to antibody
antigen interactions. Discrimination between native and denatured protein has
been shown (Bianchini et al. Immunol Methods (2001) 252:191-97) making
aptamers useful detection agents for the methods of the invention.
Peptide aptamers, also known as paptamers, thioredoxin-insert proteins or
pertubagens are artificial proteins where an inserted peptide is expressed on
a
solvent exposed surface of a structurally stable protein which functions as a
scaffold (Crawford M. et al. Brief Funct Genomic Proteomic. 2003 Apr;2:72-9).
Peptide aptamers can function similarly to antibodies and have dissociation
constants that are comparable to, and sometimes better than, antibodies.
They can be used to probe immobilized proteins on nitrocellulose (Crawford M.
et al. Brief Funct Genomic Proteomic. 2003 Apr;2:72-9). Peptide aptamers
have been shown to exhibit different affinities for small changes such as
single
amino acid differences making them useful for the detection of polypeptides
that exist in two or more conformations such as disease proteins that exhibit
different folding or aggregation conformations.
Accordingly, in one embodiment of the invention, nucleic acid and/or peptide
aptamers are used with the methods of the invention to distinguish between
wild-type and disease conformation proteins. In one embodiment the disease
43
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
protein is a prion protein. In another embodiment the disease protein is
amyloid-beta. In another embodiment the disease protein is tau protein. In
another embodiment the disease protein is alpha-synuclein. In another
embodiment the disease protein is SOD-1.
Denaturing and Disaggregation
In the methods, the polypeptide is optionally modified by denaturing the
polypeptide, for example with heat, detergent and/or chaotropic agents. The
polypeptide is optionally modified by treatment with a disaggregation agent to
disaggregate the polypeptide from other polypeptides of the same type, and
from other molecules, wherein the disaggregation agent is optionally selected
from at least one of the group consisting of chaotropic agents, detergent and
heat. Chaotropic agents can include but are not limited to such as guanidine
salts, urea, and thiourea.
The inventors have shown that treating proteins with guanidine hydrochloride
increases the amount of protected protein detectable.
Combining disaggregation methods can result in optimised disaggregation.
For example boiling samples in sodium dodecyl sulfate (SDS; also known as
sodium lauryl sulfate) loading buffer can increase solubilization of
polypeptides
such as disease proteins, increasing the epitopes available for interacting
with
the detecting agent. For example, boiling samples in SDS loading buffer
results in enhanced solubilization, and allows detection of protected epitopes
by sandwich ELISA. The sandwich ELISA assay system is able to identify
aggregated disease protein in tissue homogenate samples if the samples are
boiled in SDS loading buffer after peroxynitrite treatment. At peroxynitrite
concentrations greater than 8 mM, there is 2.5-3x as much PrP detected in the
acid treated sample as compared to the mock treated sample. Accordingly, in
one embodiment the sample is boiled in SDS loading after treatment with
modifying agent and before detection with a detecting agent such as an
antibody.
Time resolved Fluorescence (TRF) two point ELISA and Dissociation
44
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Enhanced Lanthanide Fluorolmmunoassay (DELFIA)
As previously mentioned ELISA techniques can be employed by the methods
of the invention. Time resolved flouresence two-point ELISA employing
Dissociation Enhanced Lanthanide Fluorolmmunassay (DELFIA) technology is
1000 fold more sensitive than conventional ELISA techniques and can be used
with the methods of the invention to detect polypeptides aggregated in vitro,
in
neural tissue of transgenic mouse models of neurodegeneration, and in human
AD, ALS, PD and LBD patient brain samples.
The DELFIA assay uses a chelated lanthanide-labeled tracer, such as
europium (Eu) and time-resolved fluorescence (TRF) to measure output signal
(33). The benefit of lanthanide chelates is that their fluorescence is intense
and lasts up to 200,000 times longer than conventional fluorophores, allowing
signal capture after non-specific interfering fluorescence has faded
(particularly
critical for biological samples, which may possess considerable intrinsic
fluorescence, the emission of which is comparatively short-lived). DELFIA-
based systems can measure as little as 100 fmol/well of Eu (33).
In one embodiment of the invention, a chemical modifying agent and antibody
are employed in a sensitive capture-detection "sandwich" 96-well plate DELFIA
TRF system in the detection of aggregated disease specific proteins described
herein, such as Abeta, tau, SOD1, huntingtin alpha-synuclein, islet amyloid
polypeptide, resistin and p53.
A two-point EPA increases the specificity for detection of proteins
sequestered
in aggregates of a clinical sample. In one embodiment, two or more chemically
modifiable epitopes are present in each test polypeptide, which would increase
the specificity of diagnostic tests employing this technology (e.g., use in
two-
point ELISA). In one embodiment, the chemically modifiable epitopes are
modified by the same chemical. In another embodiment, the epitopes are
modified by one of two or more different chemicals. The modified epitopes
may be recognized by the same antibody or they may be recognized by two or
more different antibodies. For clinical and commercial use, EPA must be
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
sensitive and specific for polypeptides aggregated in vitro and in vivo. With
optimal antibodies and chemical modifying regimens, and the DELFIA-TRF
system EPA can detect 105- 106 molecules of soluble polypeptides This may
correspond to a single polypeptide aggregate, if these aggregates are of
similar size to prion protein aggregates in disease (35, 36).
Accordingly in one embodiment, the DELFIA-TRF system EPA can be used to
identify disease proteins that are in very low abundance, as low as a single
polypeptide aggregate.
Diagnostic and screening applications
Effective, efficient and inexpensive diagnostic and screening strategies for
antemortem diagnosis of human neurodegenerative diseases are urgently
needed, given the aging population and continued financial pressure on the
health care system. EPA will achieve clinical utility by detecting polypeptide
aggregates in relevant and accessible biological tissues and fluids, for which
no present technology exists. In one embodiment the methods of the invention
are used to diagnose individuals who have a disease protein related disease.
In one embodiment, the invention is used to diagnose individuals who have a
neurodegenerative disease. In another embodiment, the invention is used to
diagnose individuals who have a neurodegenerative disease selected from the
group comprising prion related diseases, AD, HD, ALS and PD. In a further
embodiment, the methods of the invention are used post-mortem to determine
if the individual had a disease protein related disease.
The methods of the invention are used to detect whether a human has a
disease protein related disease. In another embodiment, the methods are
used to detect if a non-human animal has a disease protein related disease. In
a further embodiment the non-human animal is one of the group comprising
cattle, sheep and cervids. In another embodiment, the methods of the
invention are used to detect if livestock has a disease protein related
disease.
In one embodiment the methods of the invention are used to detect disease
proteins in biological specimens. The biological specimens may comprise
46
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
biological fluids, such as CSF, serum, blood, tears, peritoneal exudates, or
urine, or tissue samples such as biopsies or brain tissue. The samples in one
embodiment are antemortem samples. In another embodiment they are
postmortem samples.
The methods of the invention are useful to quantify detection of soluble form
of
disease related proteins such as Abeta, tau, SOD1 huntingtin,alpha-synuclein,
islet amyloid polypeptide, resistin and p53 protein.
In another embodiment, EPA is used to determine the sensitivity and
specificity
of aggregate detection in homogenates from CRND8 (human mutant APP)
mouse brain and CSF (34) and G93A human mutant SOD1 transgenic mice
(13).
In another embodiment the invention is used to determine the sensitivity and
specificity of aggregate detection in homogenates from normal (treated and
untreated at low pH) and diseased frozen human brain (AD, ALS, PD, LBD).
The methods of the invention are used in one embodiment to ensure
preparations derived from mammalian blood or tissues or involving processes
where mammalian blood or tissues come into contact with preparations, are
free of disease proteins. In one embodiment the preparation is a
pharmaceutical product. In another embodiment the preparation is a vaccine.
In a further embodiment, the preparation is a cosmetic. In one embodiment,
the preparations are tested for prion proteins. In another embodiment, the
preparations are tested for amyloid-beta. In another embodiment, the
preparations are tested for tau protein. In another embodiment, the
preparations are tested for alpha-synuclein. In a further embodiment, the
preparations are tested for SOD-1.
In another embodiment, the methods of the invention are used to screen
blood, and blood products (eg. blood fractions such as blood plasma or
compounds isolated or manufactured from blood) used for transfusions or
other medical procedures for disease proteins. In another embodiment, the
invention is used to screen organ transplants for disease proteins. In one
47
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
embodiment, the preparations are screened for prion proteins. In another
embodiment, the preparations are screened for amyloid-beta. In one
embodiment, the preparations are screened for tau protein. In another one
embodiment, the preparations are screened for alpha-synuclein. In a further
embodiment, the preparations are screened for SOD-1.
The invention is also useful for ensuring that food sources are free of
disease
proteins. In another embodiment the methods of the invention are used to test
edible products derived from mammals such as meats and meat products; and
dairy products. Foods potentially contaminated with neural tissue (such as
"mechanically separated meat," and meat cuts containing dorsal root ganglia
or other neural tissue) are particularly important to screen for prion
contamination.
Instruments that are used for invasive procedures may also be a source of
transmitting disease. In one embodiment instruments used for medical and
surgical procedures are tested for the presence of disease proteins using
methods of the invention. In another embodiment instruments used for dental
hygiene are tested for the presence of disease proteins.
In a further embodiment, the invention provides methods to ensure that
decontamination methods for removing disease proteins and disease protein
containing tissues, have been successful. In one embodiment the methods of
the invention are used to assess decontamination procedures in a meat
processing plant. In another embodiment the methods of the invention are
used to assess decontamination in a food processing plant. In another
embodiment instruments used for surgery or dentistry are tested for the
presence of disease proteins.
Prognostic Applications
Prion protein conversion, Alzheimer's disease related polypeptide or other
disease/disorder polypeptide may be periodically monitored in a subject over
time (e.g. at a first time and a second time at least a week or at least a
month
48
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
after the first time) to identify, for example, increased or decreased levels
of
PrPc or increased or decreased levels of PrPsc in the subject. The methods of
the invention are also useful to measure a subject's level of PrPc or PrPsc to
determine the subject's response to drug therapy. Decreasing levels of prion
protein in the subject over time indicate a positive response to drug therapy.
The same methods are used with other disease or disorder protein.
Since many neurological diseases are associated with aggregated proteins,
similar methods are useful for these diseases and their aggregated proteins,
including, but not limited to: amyotrophic lateral sclerosis (superoxide
dismutase
1), Alzheimer's disease (amyloid beta), Parkinson's disease (alpha synuclein),
Huntington's disease (huntingtin), cancer (p53), diabetes (eg. islet amyloid
polypeptide and resistin) and other diseases involving abnormal protein
folding,
aggregation or post-translational modification. Such a test is useful in the
spinal
fluid and other bodily fluids in addition to peripheral blood. In Alzheimer' s
disease, the aggregation status of the amyloid beta peptide is optionally
monitored by determining the accessibility of two epitopes detected by the
monoclonal antibodies 6E10 and 4G8, in addition to other amyloid beta
epitopes, using the methods described in this application, for example, with
an
anti-6E10 or anti-4G8 antibody (detection agent) known in the art.
Identifying Prion Conversion Inhibitors
Since the invention is useful for detecting differences between polypeptides,
the invention further includes an assay for evaluating whether a candidate
compound is capable of inhibiting or stabilizing prion conversion or formation
of
other disease or disorder polypeptides, such as amyloid beta, tau and APP in
Alzheimer's disease, SOD1 in amyotrophic lateral sclerosis, alpha-synuclein in
Parkinson's and Lewy body disease, huntingtin in Huntington's disease islet
amyloid polypeptide and resistin in diabetes and p53 in cancer. The invention
also includes compounds for inhibiting or stabilizing prion conversion (or
conversion of other disease or disorder polypeptides) identified by the
methods
described in the application. Decreased protein conversion to an intermediate
prion protein substrate or PrPsc (or other disease or disorder polypeptides
shows that the candidate compound is useful for treating prion disease.
49
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
The assays of the invention are useful to screen candidate compounds to
determine if they inhibit PrPsc formation (or formation of other disease or
disorder polypeptides from wild type protein). Protein may be contacted with a
candidate compound in vivo or in vitro and then used in the methods of the
invention to determine if wild type protein has been converted to PrPsc or if
PrPsc has been converted to wild type protein. Similar methods are used with
respect to other disease or disorder polypeptides. Recombinant proteins are
useful for identifying aggregation inhibitors.
Therefore, the invention also provides methods for identifying substances that
inhibit conversion to PrPsc (e.g. prion protein conversion from wild type
protein
or intermediate to PrPsc) comprising the steps of:
- reacting a polypeptide and a candidate substance, and
- determining whether the protein has been converted to PrPsc using
themethods of the invention.
Similar methods are optionally performed to identify compounds which stabilize
the wild-type prion state, or bind to PrPsc and block conversion of
recruitable
PrP isoforms.
The invention also provides methods for identifying substances that inhibit
conversion to disease or disorder polypeptides (e.g. conversion from wild type
protein to the amyloid beta, tau or APP protein in Alzheimer's disease and
other proteins and diseases described in this application) comprising the
steps
of:
- reacting a polypeptide and a candidate substance, and
- determining whether the protein has been converted to the amyloid
betaor APP protein in Alzheimer's disease using the methods of
the invention.
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Another aspect of the invention provides a method of identifying substances
which reverse PrPsc formation comprising the steps of:
- reacting a polypeptide and a candidate substance, and
- determining whether the PrPsc has been converted to wild type protein
using the methods of the invention.
Another aspect of the invention provides a method of identifying substances
which reverse amyloid beta or APP protein in Alzheimer's disease formation
comprising the steps of:
- reacting a polypeptide and a candidate substance; and
- determining whether the amyloid beta or APP protein in Alzheimer's
disease has been converted to wild type protein using the methods
of the invention.
The same methods are used with other polypeptides associated with diseases
and disorders described in this application.
Biological samples and commercially available libraries may be tested for
substances such as proteins or small organic molecules that bind to a protein.
Inhibitors are preferably directed towards specific domains of disease
proteins
such as prion protein. To achieve specificity, inhibitors should target the
unique sequences and or conformational features of the disease protein.
Protein Conformation Detection
The invention includes a method of detecting whether a candidate polypeptide
including a target epitope is a non-wild type conformation polypeptide or a
wild
type conformation polypeptide, comprising:
- contacting the candidate polypeptide with a blocking agent; and
- determining whether the target epitope is inaccessible or accessible to
chemical modification by the blocking agent.
51
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
The accessibility or inaccessibility of the target epitope is indicative of
whether
the candidate polypeptide is non-wild type conformation polypeptide or a wild
type conformation polypeptide because in one of the non-wild type protein and
the wild type protein, the target epitope is accessible. In the other
polypeptide,
the target epitope is inaccessible.
In one embodiment, the invention includes a method of detecting whether a
candidate polypeptide including a target epitope is in a wildtype conformation
or a non-wildtype conformation, comprising:
- contacting the polypeptide with a blocking agent that selectively blocks
accessible target epitope, wherein in one of the non-wildtype
conformation or the wildtype conformation, the target epitope is
accessible and reacts with the blocking agent, and wherein in the other
conformation, the target epitope is inaccessible and does not react with
the blocking agent;
- removing unreacted blocking agent from contact with the polypeptide
(eg. by allowing blocking agent to be consumed or degraded in the
sample comprising the candidate polypeptide or by physical or chemical
removal processes);
- modifying the candidate polypeptide to convert any inaccessible target
epitope to accessible target epitope; and
- contacting the polypeptide with a detection agent that binds selectively
to target epitope that was converted from inaccessible target epitope to
accessible target epitope, wherein binding between detection agent and
converted target epitope indicates that prior to conversion the
candidate polypeptide was in a conformation in which the target epitope
was inaccessible and wherein lack of binding between the detection
agent and the target epitope indicates that the polypeptide was in a
conformation in which the target epitope was inaccessible, thereby
indicating whether the polypeptide was in a wildtype conformation or a
non-wildtype conformation.
52
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
A polypeptide may have more than two conformations. For example a
polypeptide may exist in a wild-type conformation, in a benign misfolded,
aggregated or otherwise non-wildtype conformation not associated with
disease, and a disease associated conformation (i.e. aggregated in higher
order structures). The methods of the invention can be applied to distinguish
each of these states through the use of one or more chemical modifying
agents and/or one or more detecting agents such as antibodies.
Detection of Intrinsically Modified Polypeptides
The invention also provides a method of detecting polypeptides that exist in
two or more conformations wherein the target epitopes in one of the
conformations is modified by an intrinsic mechanism. The intrinsic mechanism
can include intracellular and/or post-translational modification of a
polypeptide
such as phosphorylation and/or glycosylation or a modification resulting from
an additive used in a process. The intrinsic modification blocks a target
epitope
obscuring it from detection with a detection agent. The sample of polypeptide
is reacted with a blocking agent that reacts with available target epitope in
polypeptide that is not intrinsically modified. The intrinsic modification is
then
removed. For example if the intrinsic modification is phosphorylation, the
polypeptide is treated with a phosphatase which removes the phosphorylation
and converts the inaccessible target epitope in the previously intrinsically
modified polypeptide, to accessible epitope. The polypeptide is then detected
with a detecting agent such as an antibody.
Accordingly in one embodiment, the invention provides a method of detecting
intrinsically modified target epitopes in a polypeptide having two or more
conformations comprising;
- contacting the polypeptide with a blocking agent that selectively
blocks accessible target epitope, wherein in one of the non-
wildtype conformation or the wildtype conformation, the target
epitope is accessible and reacts with the blocking agent, and
53
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
wherein in the other conformation, the target epitope is
inaccessible and does not react with the blocking agent;
- reacting the sample with an agent that removes the intrinsic
modification from the intrinsically modified polypeptide target
epitope;
- disaggregating and/or denaturing the polypeptide in the sample;
and
- probing with a detection agent, such as antibodies against the
target epitope, to determine whether the candidate polypeptideis
an intrinsically modified polypeptide.
In one application, the methods of the invention can be used to detect whether
polypeptides present in food items have been chemically modified by
manufacturing processes. For example dairy products can be tested for the
presence of formaldehyde, which is used as a bacteriostatic agent.
Formaldehyde formylates gamma(2) casein (Pizzano R. et al J. Agric Food
Chem (2004) 52:649-54) obscuring modified epitopes from subsequent
detection by the detecting agent.
Kits
The methods described herein are optionally performed by utilizing pre-
packaged diagnostic kits comprising the necessary reagents to perform any of
the methods of the invention. For example, the kits typically include at least
one specific nucleic acid, peptide orantibody described herein, which are
conveniently used, e.g., in clinical settings, to screen and diagnose patients
and to screen and identify those individuals expressing a disease conformation
protein. Kit antibodies can comprise whole antibody, antibody fragments,
single chain antibody, monoclonal antibody and/or polyclonal antibody. The
kits optionally also include at least one chemical agent for modifying
epitopes
recognized by an antibody or aptamer. The kit is optionally based on ELISA
technology such as sandwich ELISA and DELFIA and may employ detergents,
precipitation agents (such as phosphotungstic acid) and adsorbents typically
used in ELISA technology and known to one skilled in the art. The kit will
also
54
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
include detailed instructions for carrying out the methods of the invention.
Recombinant protein are useful for standards in kits.
All such assays could be adapted and optimised to a simple high-throughput
platform.
The following non-limiting examples are illustrative of the present invention.
EXAMPLES
Example 1
Peroxynitrite reacts differently with PrP in normal and acid treated or
scrapie brain homogenate
When brain homogenate is incubated at pH 3.5 in the presence of guanidine,
PrP becomes detergent insoluble and is more susceptible to misfolding to a
PK-resistant isoform in the presence of PrP sc (29). This acid treated PrP is
a
'model prion' which is partially misfolded and/or aggregated resembling
characteristics of PrP. When mock (0) and acid treated ( = ) brain
homogenate is incubated with increasing concentrations of peroxynitrite and
then subjected to immunoblotting, there is less PrP recognized by both 3F4
(Figure 1A and C) and 6H4 (Figure 1B and D) in mock treated brain
homogenate than in acid treated brain homogenate. The PrP in the acid
treated brain homogenate is protected from modification by peroxynitrite.
Example 2
PrP in scrapie infected hamster brain is protected from modification by
peroxynitrite
The epitope protection phenomenon for 'model prions' as observed in example
1 was also observed for authentic disease-nnisfolded prion protein in scrapie
infected hamster (Ha) brain (Figure 2A and B). As with model prions, the 3F4
and 6H4 epitopes of PrP in Has c brain homogenate are protected from
modification by peroxynitrite. It is clear that 'model prions' and HaPrpsc
share
characteristics that provide protection from chemical modification by
peroxynitrite, such as differential misfolding or aggregation.
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Example 3
Aggregation is responsible for the reduction in peroxynitrite-induced
epitope modification of misfolded PrP
To show that epitope protection of acid treated and scrapie brain was due to
aggregation, samples were treated with peroxynitrite and then incubated with
or without guanidine before immunoprecipitation. Treatment of the samples
with guanidine dissociates aggregates of PrP (43-45) that protect the
polypeptide from modification by peroxynitrite. Incubation of mock treated
brain with 2.5 M guanidine after peroxynitrite treatment did not show an
increase in 3F4 and 6H4 epitopes as revealed by immunoprecipitation (Figure
3A lanes 1-4). However, when peroxynitrite-treated acid brain homogenate
was incubated with guanidine, there was an increase in PrP that could be
detected by immunoprecipitation with 3F4 and 6H4 immunobeads (Figure 3A
lanes 5-8). This shows that guanidine is able to dissociate aggregates of acid
treated brain homogenate and release PrP that is protected from modification
by peroxynitrite. Other means of solubilizing PrP aggregates were used and
boiling samples in SDS loading buffer resulted in the greatest observed
solubilization to date.
Example 4
Optimization of EPA parameters
Titration experiments with peroxynitrite, hydrogen peroxide and methylene
(based on UV light photolysis of the precursor diazirine) or other modifying
agents, identify the optimal conditions for epitope protection in:
1. Normal hamster and human brain "model prions", using immunoblotting and
conventional fluorescence ELISA.
2. Infectious prions from hamster and human brain, using immunoblotting
analysis and time-resolved fluorescence
In each case, brain homogenates are prepared and mixed with increasing
concentrations of the modifying agent and processed as described
(immunoblotting, and time resolved fluorescence). This defines the type and
concentration of chemical agent allowing the maximal distinction between
monomeric and aggregated prion proteins. Additional informative control
56
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
experiments include using recombinant hamster PrPc in buffer and in PrP -/-
knockout mouse brain, and by mouse normal and scrapie-infected brain
(murine PrP is 6H4+ and 3F4-).
In some cases, infectious prions may have different properties for chemical
modification than do "model prions," and brain prions may display different
chemical modification properties than do endogenous prions circulating in
blood, or PrPs' detectable in urine of infected animals. One of skill in the
art
shall readily identify the optimal conditions for authentic endogenous prions
using known techniques.
Example 5
EPA adapted to a fluorescent ELISA system
The epitope protection assay for aggregated PrP was adapted to a fluorescent
sandwich ELISA system using 6H4 as the capture antibody and 3F4 as the
detection antibody (Figure 3B). The sandwich ELISA assay system is able to
identify aggregated PrP in acid treated brain homogenate but only if the
samples are boiled in SDS loading buffer after peroxynitrite treatment. At
peroxynitrite concentrations greater than 8 mM, there is 2.5-3x as much PrP
detected in the acid treated sample as compared to the mock treated sample.
Example 6
Detection of a single brain prion
A single brain prion has been estimated to comprise 105-106 molecules of
PrP. Detection of 108-109 molecules of recombinant PrP using conventional
fluorescence ELISA has been accomplished. The assay used is about 1000-
fold more sensitive for single-prion detection ¨ the necessary sensitivity is
provided by the Dissociation enhanced lanthanide fluoroimmunoassay
(DELFIA). DELFIA uses a chelated lanthanide-labeled tracer, such as
europium (Eu) and time-resolved fluorescence (TRF) to measure output signal.
The benefit of lanthanide chelates is that their fluorescence duration is
200,000
times longer than conventional fluorophors, allowing signal capture after non-
specific interfering fluorescence has faded (particularly critical for
biological
samples, which may possess considerable non-specific fluorescence).
57
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
DELFIA-based systems can measure as little as 100 fmol/well of Eu which is
>1000 times more sensitive than conventional ELISA assays, which detects
single prions by EPA. The optimal TRF 96-well plate reader for the DELFIA
system is manufactured by Wallac-Victor (Perkin-Elmer), and is used to
automate sample analysis.
Using an optimal chemical modifier and optimal conditions a sensitive capture
96-well plate assay for detection of hamster and human prions, using the
DELFIA TRF system is provided. This is used to:
1. Characterize, optimize and quantify detection of recombinant prion
protein by TRF.
2. Determine the sensitivity of the DELFIA-TRF for hamster and human
brain prions.
Example 7
Detection of prion proteins in biological fluids
The EPA achieves commercial utility by detecting PrPsc in biological tissues
and fluids for which no present technology exists. Blood prions are in very
low
abundance (10-100 prions/mL by bioassay, and protease-resistant PrP in urine
is only intermittently/sporadically detectable by precipitation of large fluid
volumes. Also, any prospective blood test must contend with high
concentrations of PrPc (106-fold more than PrPs) and "blocking" by
heterologous plasma proteins. Using the optimized chemical modification
regimen and the DELFIA-TRF system, the sensitivity thresholds for EPA in
blood and urine are determined using:
1. Hamster and human plasma and urine "spiked" with a titration of
263K hamster prions;
2. Plasma and urine from Syrian hamsters "endogenously" infected with
263K prion disease
Biological fluids clinically accessible by non-invasive routes provide a
substrate
for a practical antemortenn test for diagnosis and screening of prion
infection in
58
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
humans and animals. The methods of this invention may also be used in post-
mortem testing. One of skill in the art readily determines whether EPA with
"prion spike" titration in normal blood and urine reveals similar DELFIA-TRF
signals to the same prion titration in buffer, showing that the EPA is not
affected by "blocking factors" in these biological fluids.
Interestingly,
preferential "blocking" of PrPsc by heterologous proteins may actually enhance
epitope protection to chemical modifying agents. If decreased detection of
prions in blood or urine is observed, pre-clearing strategies are readily
employed to enhance PrPsc detection with detergents, precipitating agents,
and adsorbents typically used in commercial ELISA assays which are known to
one skilled in the art.
Human and bovine plasma and urine and other bodily fluids are tested using
optimized EPA conditions and compared to samples from human variant CJD
and BSE, respectively. Although the monoclonal antibody 6H4 recognizes PrP
from all relevant species, other antibodies (commercially available) are used
for the DELFIA TRF system for cattle, sheep, and cervids, which lack the 3F4
epitope. Other antibodies and epitopes useful in methods described in this
application will be readily apparent to those of skill in the art.
Example 8
Detection of aggregated amyloid beta (Abeta) using EPA
Amyloid beta peptide (Abeta) is a normal cleavage product of the proteolytic
processing of amyloid precursor protein (APP). Abeta accumulates in discrete
plaques in affected regions of Alzheimer's disease brain, and triggers
neuronal
death and gliosis observed in this disease. Plaque Abeta is aggregated and
rich in beta-sheet structure, in contrast to the Abeta region of APP expressed
by normal cells. Using epitope protection technology, it was demonstrated that
the 6E10 epitope in the Abeta region of APP is less accessible to
peroxynitrite
modification in Alzheimer's disease brain compared to normal brain (Figure 4
panel A). Similarly, the 6E10 epitope is partially protected in brain
homogenates that have been treated at low pH to induce protein aggregation
(Figure 4 panel B). Abeta 1-42 peptide aggregated by overnight incubation at
1 mg/ml in water shows prominent epitope protection of the 6E10 epitope to
59
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
peroxynitrite modification, in comparison with soluble non-aggregated Abeta 1-
42 (Figure 4 panel C). A further example of this phenomenon is presented in
Figure 4, panel D which shows normal (0) and aggregated (0) Abeta 1-42
treated with increasing concentrations of peroxynitrite, subjected to
immunoblotting with 6E10 antibody.
The 6E10 epitope of APP is also unavailable to peroxynitrite modification in
AD
brain homogenates and in normal brain homogenates aggregated by low pH,
but normal untreated brain does not show this protection (Figure 4 panel A and
B), showing molecular interaction in vivo of APP with an Abeta-domain
blocking molecule (perhaps Abeta itself; ref. 9).
The sensitive and specific EPA detection of aggregated Abeta in biological
fluids (such as blood and spinal fluid), or protection of Abeta epitopes in
APP. in
cells and tissues, provides an antemortem diagnostic test for Alzheimer's
disease. The methods of the invention described in this application are used
for this diagnostic test.
Detection of aggregated Tau protein by EPA
Dying neurons release intracellular proteins such as tau into the CSF (39) and
likely ultimately blood. Tau studies are directly performed on brain specimens
including Alzheimer patient samples and control brain.
Example 9
Detection of aggregated superoxide dismutase 1 (SOD1) by EPA
SOD1-containing cytoplasmic inclusions are detected in many diseased motor
neurons from familial and sporadic ALS patients (15), and in most transgenic
mouse (16, 17) and tissue culture models (18) of the disease. Human SOD1
can be aggregated in vitro. Further SOD1 is modifiable by succinic anhydride
and DEPC. This property can be exploited by EPA technology to discriminate
between aggregated and unaggregated SOD1 protein.
Purified SOD1 from human erythrocytes (Sigma) was aggregated in a metal-
catalyzed oxidation reaction. Soluble SOD1 was treated with varying
concentrations of DEPC, denatured with heat, and immunoblotted with anti-
CA 02536305 2007-08-02
SOD1 antibodies. Western blotting shows that increasing concentrations of
DEPC are associated with decreases in antibody binding in soluble SOD1
(Figure 5) showing that sites are available for modification by a blocking
agent.
Antibodies against SOD1 selected a priori for utility in EPA are used to
distinguish disease specific aggregated SOD1 from wildtype SOD1. Relevant
factors include:
1) selecting an epitope on the molecular surface of the native dimer to
be accessible to chemical modification in the native soluble state;
2) identifying a linear epitope to optimize detection in the denatured
state on immunoblots and ELISA;
3) immunogenicity;
4) uniqueness to SOD1; and
5) presence of acidic amino acids (Glu and Asp) that are readily
modified by epoxides.
The five SOD1 sequences that meet these criteria are: 22QKESNG27 (SEQ ID
NO: 4); 51EDNTAGCTSA60 (SEQ ID NO: 5); 74PKDEERHV81 (SEQ ID
NO:6); 89ADKDG93 (SEQ ID NO: 7); and 127GKGGNEQSTK136 (SEQ ID
NO: 8) (in bold: solvated side chains).
Additionally the electrostatic loop sequence and zinc binding loop of human
SOD1 are surface-accessible sequences and are involved in aggregate
formation (Elan, J. et al. Nature Structural Biology (2003) 10:461-67).
These sequences are:
Electrostatic loop of human SOD1: Asp Leu Gly Lys Gly Gly Asn Glu Glu Ser
Thr Lys Thr Gly Asn Ala Gly Ser (SEQ ID NO: 9)
Zinc-binding loop of human SOD1: Asn Pro Leu Ser Arg Lys His Gly Gly Pro
Lys Asp Glu Glu (SEQ ID NO: 10)
61
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Example 10
Detection of aggregated alpha-synuclein by EPA
Most cases of Parkinson's disease are sporadic, but both sporadic and familial
forms of the disease are characterized by intracellular Lewy bodies in dying
neurons of the substantia nigra, a population of midbrain neurons (-60,000)
that are selectively decimated in PD. Lewy bodies are predominantly
composed of alpha-synuclein (22). Mutations in the gene encoding alpha-
synuclein have been found in patients with familial Parkinson's disease
(reviewed in 23). Another gene associated with autosomal recessive PD is
parkin, which is involved in alpha- synuclein degradation (22, 23). Diffuse
cortical Lewy bodies composed of alpha-synuclein are observed in Lewy body
disease (LBD), a dementing syndrome associated with Parkinsonian tone
changes, hallucinations, and rapid symptom fluctuation (24).
The Syn-1 epitope is optionally blocked by chemical modification of
recombinant alpha-synuclein with DEPC (histidine reactive), and alpha-
synuclein aggregated in vitro is partially protected from DEPC epitope
blocking
(Figure 6).
Aggregated alpha-synuclein in vitro is protected from modification by DEPC
whereas normal protein is not. Three mg/mL mutant A53T alpha-synuclein
was incubated at 37 C for three days for aggregation. The aggregation
reaction was applied to ultracentrifugation. Normal protein prior to
aggregation
(containing soluble alpha-synuclein) and the pellet resuspension from the
ultracentrifugation (containing insoluble alpha-synuclein) were treated with
varying concentrations of DEPC, denatured with heat, and blotted with Syn-1
antibody from BD Biosciences. Figure 6A shows that increasing
concentrations of DEPC are associated with a gradual decrease in antibody
binding in normal alpha-synuclein. Insoluble alpha-synuclein shows little
change in antibody binding with increasing concentrations of DEPC until the
DEPC concentrations reach 1mM. A graphical representation of these findings
is presented in Figure 6B. The extent of antibody binding to DEPC-treated
normal alpha-synuclein (¨) decreases gradually overall but more rapidly at
62
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
higher concentrations of DEPC. Insoluble alpha-synuclein (n), on the other
hand, shows little change in the extent of antibody binding. The last data
point
at 0.01M DEPC for insoluble alpha-synuclein increases due to the darkening of
the film.
Example 11
Detection of aggregated proteins in CSF
Extracellularly deposited Abeta has been quantified in CSF and blood of
patients with AD and normal controls (6-8). Intracellular neuronal proteins,
such as alpha-synuclein, have been detected in CSF and blood (37, 38).
Dying neurons release intracellular proteins 14-3-3, neuron-specific enolase,
tau, and alpha-synuclein into the CSF (39), and likely ultimately blood. A
proportion of released protein in disease is in an aggregated form. EPA
technology is applied to determine the proportion of polypeptide aggregates in
CSF samples from patients with AD, ALS, PD, and LBD. Signal is measured
for polypeptides disaggregated before and after chemical treatment,
representing "total" and "protected" epitopes, respectively, to determine the
proportion of polypeptide in the aggregated state. Using the optimized mAbs
and chemical modification regimens, and the DELFIA-TRF system, EPA
sensitivity is determined in:
1. Normal CSF "spiked" with polypeptides aggregated in vitro.
2. CSF from patients with AD, ALS, PD, and LBD.
The proportion of aggregated polypeptides in a CSF sample, is determined
even if it constitutes only 105-106 molecules. Detergents, precipitating
agents
(such as phosphotungstic acid), and adsorbents typically used in commercial
ELISA assays to enrich for relevant species are optionally employed.
Biological fluids clinically accessible by non-invasive routes provides an
ideal
substrate for a practical antemortem test for diagnosis and screening of
neurodegenerative diseases.
Materials and Methods
63
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
Materials
Recombinant hamster PrP (rhaPrP) and 6H4 was from Prionics. Recombinant
human PrP (rhuPrP) was from Roboscreen. Biotin-3F4 and 3F4 were from
Signet. 3F4 reacts against MKHV and 6H4 reacts against DYEDRYYRE. 6E10
anti-Abeta (from Signet) reacts against EFRHDS (residues 3-8).
Other antibodies and the epitopes recognized if known, are provided in table 1
above.
Preparation of Acid-misfolded PrP and APP.
Acid misfolded PrP was used as "model prions" in this study and was prepared
as in (29). Briefly, 100 RI of 10% brain homogenate was mixed with an equal
volume of 3.0 M GdnHCI (final concentration 1.5 M) in PBS at pH 7.4 or pH 3.5
adjusted with 1 N HCI, followed by rotation at room temperature. After 5 h
incubation, samples were methanol precipitated with 5 volumes of ice-cold
methanol and pellets were resuspended in 100 I of lysis buffer. The samples
treated at pH 7.4 were designated as mock-treated samples.
Peroxynitrite treatment of Brain Homogenates
An aliquot (18p1) of normal or misfolded/diseased brain homogenate was
vortexed while 2 pl of peroxynitrite in 100mM NaOH/60 mM H202 was added to
give a final peroxynitrite concentration of 0-15 mM. After vortexing for a
further
15 s, the samples were subjected to Western blotting, immunoprecipitation or
sandwich ELISA.
DEPC treatment of Erythrocyte SOD1
Purified SOD1 from human erythrocytes (Sigma) is aggregated in a metal-
catalyzed oxidation reaction consisting of 40 M SOD1, 4mM ascorbic acid,
and 0.2mM CuCl2 in 10mM Iris-acetate buffer (pH7) at 37 C for three days.
Ultracentrifuged supernatant (containing soluble SOD1) and the pellet
resuspension (containing insoluble SOD1) are treated with varying
64
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
concentrations of DEPC (100 pM to 0.1 M), denatured with heat, and
immunoblotted with anti-SOD1.
Western Blotting
Samples were boiled in SDS loading buffer (62 mM Tris (pH 6.8), 10%
glycerol, 2% SDS, 5% beta-mercaptoethanol and 0.01% bronnphenol blue) for
min. and separated on 12% Tris-Glycine polyacrylamide gels followed by
transfer to Hybond-P. PrP was detected using 3F4 (1:50000) 6H4 (1:10000) or
6E10 (1:1000) as the primary antibodies and HRP-conjugated goat anti-mouse
(1:10000) as the secondary antibody followed by exposure to ECL-Plus and
visualization by exposure to Kodak X-OMAT film. Band intensities were
quantitated using UnScan-IT software.
Immunoprecipitation
Samples were incubated with 50 pl of Ab-conjugated (100pg/m1) Dynal M-280
magnetic beads in a final volume of 1 ml binding buffer (3 % NP-40; 3%
Tween-20) for 3 h at room temperature with rotation. Beads were washed in
wash buffer (2% NP-40; 2% Tween-20) x3 and boiled in 30 pl SDS loading
buffer without beta-mercaptoethanol for 5 min. Supernatants were analyzed
by Western blotting as described above.
Sandwich ELISA
The capture antibody (6H4; 1:5000 in 50 mM bicarbonate binding buffer, pH
9.6) was bound to an opaque 96-well plate (Nunc Maxisorp) by overnight
incubation at 4 C. After blocking with 1% BSA in 0.05% TBST for 2 h, plates
were washed 3x in TBST and incubated overnight at 4 C with standard
concentrations of rhuPrP or rHaPrP along with unknown brain homogenates.
Plates were washed 3x and incubated with the detecting antibody biotin-3F4
(1:5000) at RT for 1h. After washing 3x, avidin-HRP (1:5000) was added and
incubated for 30 min. at RT. Following a final wash step (x3) the plate was
developed with Quantablu fluorescent substrate for 10-90 min at RT and
fluorescent intensities determined with an excitation of 325nm and emission of
420 nm.
CA 02536305 2011-01-19
While the present invention has been described with reference to what are
presently considered to be the preferred examples, it is to be understood that
the invention is not limited to the disclosed examples.
66
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
REFERENCES
1. Prusiner SB. Shattuck lecture--neurodegenerative diseases and prions.
N Engl J Med. 344:1516-26, 2001.
2. CaseIli RJ. Current issues in the diagnosis and management of dementia.
'Semin Neurol. 23:231-40, 2003.
3. Cashman NR. Do the benefits of currently available treatments justify early
diagnosis and announcement? Arguments for. Neurology. 53(Suppl 5):S50-2,
1999.
4. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy.
Physiol Rev. 81:741-66, 2001.
5. Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol
connection.
Nat Neurosci. 6:345-51, 2003.
6. Mehta PD, Pirttila T, Mehta SP. Plasma and cerebrospinal fluid levels of
amyloid beta proteins 1-40 and 1-42 in Alzheimer disease.Arch Neurol.
57:100-5, 2000.
7. Clark CM, Xie S, Chittams J et al. Cerebrospinal fluid tau and beta-
amyloid:
how well do these biomarkers reflect autopsy-confirmed dementia diagnoses?
Arch Neurol. 60:1696-702, 2003.
8. Green AJ. Cerebrospinal fluid brain-derived proteins in the diagnosis of
Alzheimer's disease and Creutzfeldt-Jakob disease. Neuropathol Appl
Neurobiol. 28:427-40, 2002.
9. Lorenzo A, Yuan M, Zhang Z, et al. Amyloid beta interacts with the amyloid
precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat
Neurosci. 3:460-4, 2000.
10. Rosen DR, Siddique T, Patterson D. et al. Mutations in Cu/Zn superoxide
dismutase gene are associated with familial amyotrophic lateral sclerosis.
Nature. 362:59-62, 1993.
11. Deng HX, Hentati A, Tainer JA et al. Amyotrophic lateral sclerosis and
structural defects in Cu,Zn superoxide dismutase. Science. 20;261:1047-51,
1993.
12. Anderson, PM in Brown RH, Meininger V, Swash eds. Amyotrophic Lateral
Sclerosis. London: Martin Dunitz. 2000.
13. Gurney,M.E., Pu,H., Chiu,A.Y., et al. Motor neuron degeneration in mice
that express a human Cu,Zn superoxide dismutase mutation. Science
264:1772-1775, 1994.
67
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
14. Ripps,M.E., Huntley,G.W., Hof,P.R., et al. Transgenic mice expressing an
altered murine superoxide dismutase gene provide an animal model of
amyotrophic lateral sclerosis. Proc.NatI.Acad.Sci.U.S.A 92: 689-693, 1995.
15. Kato,S., Takikawa,M., Nakashima,K., et al. New consensus research on
neuropathological aspects of familial amyotrophic lateral sclerosis with
superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1
in neurons and astrocytes. Amyotroph.Lateral.Scler.Other Motor Neuron
Disord. 1:163-184, 2000.
16. Bruijn,L.I., Becher,M.W., Lee,M.K. ALS-linked SOD1 mutant G85R
mediates damage to astrocytes and promotes rapidly progressive disease with
SOD1-containing inclusions. Neuron 18: 327-338, 1997.
17. Bruijn,L.I., Houseweart,M.K., Kato,S., Aggregation and motor neuron
toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1.
Science 281: 1851-1854, 1998.
18. Durham,H.D., Roy,J., Dong,L., and Figlewicz,D.A. Aggregation of mutant
Cu/Zn superoxide dismutase proteins in a culture model of ALS.
J.Neuropathol.Exp.Neurol. 56:523-530, 1997.
19 Li YK, Chir J, Chen FY. Catalytic mechanism of a family 3 beta-glucosidase
and mutagenesis study on residue Asp-247. Biochem J 355(Pt 3):835-40,
2001
20 Rose RB, Rose JR, Salto R, Craik CS, Stroud RM. Structure of the
protease from simian immunodeficiency virus: complex with an irreversible
nonpeptide inhibitor. Biochemistry 32:12498-507, 1993.
21. Olanow CW. The scientific basis for the current treatment of Parkinson's
disease. Annu Rev Med 55:41-60, 2004.
22. lwatsubo T. Aggregation of alpha-synuclein in the pathogenesis of
Parkinson's disease. J Neurol 250 Suppl 3:11111-4, 2003.
23. Eriksen JL, Dawson TM, Dickson DW, Petrucelli L Caught in the ac: alpha-
synuclein is the culprit in Parkinson's disease. Neuron 40:453-6, 2003.
24. McKeith I, Mintzer J, Aarsland D et al. Dementia with Lewy bodies. Lancet
Neurol 3:19-28, 2004.
25 Alvarez B, Ferrer-Sueta G, Freeman BA, Radi R. Kinetics of peroxynitrite
reaction with amino acids and human serum albumin. J Biol Chem 274:842-8,
1999.
26. Alvarez B, Radi R Peroxynitrite reactivity with amino acids and proteins.
Amino Acids 25:295-311, 2003.
68
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
27. Sokol PP, Holohan PD, Ross CR. Arginyl and histidyl groups are essential
for organic anion exchange in renal brush-border membrane vesicles. J Biol
Chem 263:7118-23, 1988.
28. Zou W-Q, Yang D-S, Fraser PE, Cashman NR, Chakrabartty A. All-or-none
fibrillogenesis of a prion peptide. Europ J Biochem 268:4885-4891, 2001.
29. Zou W-Q, Cashman NR. Acidic pH and detergents enhance in vitro
conversion of human brain PrPC to a PrPSc-like form. J Biol Chem 277:43942-
43947 2002.
30. Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi X-F, Crow J,
Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding
and aggregation of superoxide dismutase and its implications for amyotrophic
lateral sclerosis. J Biol Chem 277:47551-62002, 2002.
31. Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou W-
Q, Estey LA., Kondejewski LH, Francoeur GP, Papadopoulos M, Haghighat A,
Spatz SJ, ToneIli Q, Ledebur HC, Chakrabartty A, Cashman NR. A PrPSc -
specific immunological epitope. Nature Medicine 9:893-9, 2003.
32. Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR,
Chakrabartty A. Monomeric Cu/Zn superoxide dismutase is a common
misfolding intermediate in the oxidation models of sporadic and familial ALS.
J
Biol Chem e-pub Jan 2004.
33. MacGregor,I., Hope,J., Barnard,G. Application of a time-resolved
fluoroimmunoassay for the analysis of normal prion protein in human blood
and its components. Vox Sang 77:88-96, 1999.
34. Chishti MA, Yang DS, Janus C et al. Early-onset amyloid deposition and
cognitive deficits in transgenic mice expressing a double mutant form of
amyloid precursor protein 695. J Biol Chem 276:21562-70, 2001.
35. Bolton D.C., McKinley M.P., and Prusiner S.B. Identification of a protein
that purifies with the scrapie prion. Science 218:1309-1311, 1982.
36. Beekes M., Baldauf E., and Diringer H. Sequential appearance and
accumulation of pathognomic markers in the central nervous system of
hamsters orally infected with scrapie J.Gen. Virol 77:1925-1934, 1996.
37. Borghi R, Marchese R, Negro A, et al. Full length alpha-synuclein is
present in cerebrospinal fluid from Parkinson's disease and normal subjects.
Neurosci Lett. 287:65-7, 2000.
38. El-Agnaf OM, Salem SA, Paleologou KE, et al. Alpha-synuclein implicated
in Parkinson's disease is present in extracellular biological fluids,
including
human plasma. FASEB J 17:1945-7, 2003.
69
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
39. Verbeek MM, De Jong D, Kremer HP. Brain-specific proteins in
cerebrospinal fluid for the diagnosis of neurodegenerative diseases. Ann Olin
Biochem 40(Pt 1):25-40, 2003.
40. Coulthart, M. B. and Cashman, N. R. (2001) CMAJ. 165, 51-58
41.Prusiner, S. B. (1998) Proc.NatI.Acad.Sci.U.S.A 95, 13363-13383
42 Will, R. G., lronside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K.,
Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., and Smith, P. G.
(1996) Lancet 347, 921-925.
43. Kocisko, D. A., Lansbury, P. T., Jr., and Caughey, B. (1996) Biochemistry
35, 13434-13442
44, Barnard, G., Helmick, B., Madden, S., Gilbourne, C., and Patel, R. (2000)
Luminescence. 15, 357-362
45. Meyer, R. K., Oesch, B., Fatzer, R., Zurbriggen, A., and Vandevelde, M.
(1999) J.Virol. 73, 9386-9392
46. Kang, S. C., Li, R., Wang, C., Pan, T., Liu, T., Rubenstein, R., Barnard,
G., Wong, B. S., and Sy, M. S. (2003) J.Pathol. 199, 534-541.
47. Shaked GM, Shaked Y, Kariv-lnbal Z, Halimi M, Avraham I, Gabizon R.
(2001) J Biol Chem. 276, 31479-82.
48. Ross CA et at. Nature Medicine, (2004) S10-17.)
49. Davies SW et at Cell 90, 537-548 (1997).
50. Scherzinger E et al. Proc. Natl. Acad. Sci. USA 96, 4604-9, (1999).
51. Sen S et at Protein Sci. (2003) 12:953-962.
52. Llewelyn CA etal. Lancet (2004) 363:417-421
53. Peden AH et a/. Lancet (2004) 364:527-529
CA 02536305 2006-02-20
WO 2005/019828
PCT/CA2004/001503
54. Andreoletti 0 etal. Nat. Med. (2004) 6:591-593
55. Thomzig A etal. J Clin Invest. (2004) 10:1465-72.
56 Glatzel M etal. N Engl J Med. (2003) 349:1812-20
57 Bosque PJ etal. Proc Natl Acad Sci U S A. (2002) 99:3812-7
71
CA 02536305 2007-08-02
1/3
SEQUENCE LISTING
<110> Amorfix Life Sciences Ltd.
<120> EPITOPE PROTECTION ASSAY AND METHOD FOR DETECTING PROTEIN
CONFORMATIONS
<130> 15289-12
<140> CA 2,536,305
<141> 2004-08-20
<150> US 60/496,381
<151> 2003-08-20
<150> CA 2,437,675
<151> 2003-08-20
<150> US 60/497,362
<151> 2003-08-21
<150> CA 2,437,999
<151> 2003-08-21
<160> 10
<170> PatentIn version 3.3
<210> 1
<211> 4
<212> PRT
<213> Artificial sequence
<220>
<223> epitope
<400> 1
Met Lys His Val
1
<210> 2
<211> 9
<212> PRT
<213> Artificial sequence
<220>
<223> epitope
<400> 2
Asp Tyr Glu Asp Arg Tyr Tyr Arg Glu
1 5
<210> 3
<211> 6
<212> PRT
<213> Artificial sequence
CA 02536305 2007-08-02
2/3
<220>
<223> epitope
<400> 3
Glu Phe Arg His Asp Ser
1 5
<210> 4
<211> 6
<212> PRT
<213> Homo sapiens
<400> 4
Gin Lys Glu Ser Asn Gly
1 5
<210> 5
<211> 10
<212> PRT
<213> Homo sapiens
<400> 5
Glu Asp Asn Thr Ala Gly Cys Thr Ser Ala
1 5 10
<210> 6
<211> 8
<212> PRT
<213> Homo sapiens
<400> 6
Pro Lys Asp Glu Glu Arg His Val
1 5
<210> 7
<211> 5
<212> PRT
<213> Homo sapiens
<400> 7
Ala Asp Lys Asp Gly
1 5
<210> 8
<211> 10
<212> PRT
<213> Homo sapiens
<400> 8
CA 02536305 2007-08-02
3/3
Gly Lys Gly Gly Asn Glu Gin Ser Thr Lys
1 5 10
<210> 9
<211> 18
<212> PRT
<213> Homo sapiens
<400> 9
Asp Leu Gly Lys Gly Gly Asn Glu Glu Ser Thr Lys Thr Gly Asn Ala
1 5 10 15
Gly Ser
<210> 10
<211> 14
<212> PRT
<213> Homo sapiens
<400> 10
Asn Pro Leu Ser Arg Lys His Gly Gly Pro Lys Asp Glu Glu
1 5 10