Note: Descriptions are shown in the official language in which they were submitted.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
1
85262fft
Administration form for the oral application of poorly soluble acidic and
amphoteric drugs
s The invention relates to a formulation for the oral administration of acidic
and
amphoteric active substances with pH-dependent solubility characteristics, and
the
salts thereof.
The term "active substance" for the purposes of this invention refers to any
~o pharmacologically effective compound which (as such or in the form of the
pharmaceutically acceptable salts thereof) is a weak acid or behaves
amphoterically
and in the range from pH 1 to pH 11 exhibits pH-dependent solubility
characteristics
(with greater solubility under basic conditions and less solubility under
neutral and/or
acidic conditions). Acidic active substances are preferred. In these active
substances,
~s in fact, the bioavailability may be dependent on the pH in the
gastrointestinal tract
when administered orally. Preferably, active substances in the sense of this
invention
have a relatively high saturation solubility in aqueous solutions at higher pH
levels,
whereas at pH values of about 5 they are virtually insoluble according to the
definition in the European Pharmacopoeia (saturation solubility less than 100
pg /
2o ml).
The oral formulation according to the invention may contain as active
substance for
example telmisartan, meloxicam, DT-TX 30 SE, BIBV 308 SE (terbogrel), AGEE 623
r (repaglinide), gliquidone or glibenclamide or a physiologically acceptable
salt thereof.
2s The physiologically acceptable salts include for example the sodium,
potassium,
calcium and ammonium salt as well as salts with diethanolamine, meglumine,
lysine,
arginine, ethanolamine, piperazine or triethanolamine.
The solubility of a compound may be determined by dispersing an excess of the
3o compound at ambient temperature in the medium in question and shaking it
vigorously for a defined length of time (approx. 1 to 24 h) until equilibrium
is
achieved. After filtration the pH is determined in the clear filtrate and the
CA 02537776 2006-03-03
Case 111563
foreign filing text
2
concentration of the dissolved substance is determined by spectral photometry
or
some other suitable analytical process.
The pH-dependent solubility characteristics of the active substance may mean
that,
s depending on the dose, when administered orally in solid preparations of
conventional composition, the active substance is only totally dissolved when
it
reaches lower parts of the patient's intestines. This then leads to
significantly delayed
and in some cases incomplete absorption, i.e. there is no therapeutic
certainty.
to The effect of the dose of the active substance on its bioavailability can
be
quantitatively described by means of the concept of the (dimensionless) dose
number
(Do). The dose number is defined as:
Do = (Mo I Vo) I Cs ,
Is where
Mo = dose (mg),
Vo = liquid volume present (ml) and
Cs = saturation solubility (mg / ml).
2o According to an assumption which is conventional nowadays the liquid volume
in the
stomach after taking a preparation is about 250 ml. (Lobenberg, R., Amidon,
G.L.:
Modern bioavailability, bioequivalence and biopharmaceutics classification
system.
New scientific approaches to international regulatory standards (Eur. J.
Pharm.
Biopharm. 50 (2000) 3-12).
At dosages which give a dose number of less than 1, no solubility problems
occur.
Only if the critical dose number of 1 is exceeded may there be significant
reductions
in solubility and hence a decreased bioavailability. As a rule the actual
problem area
only begins at doses which give a dose number significantly above 1, as at
least
3o some of the dissolved substance can be constantly eliminated from the
equilibrium by
the absorption process.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
3
The active substances contained in the oral formulation according to the
invention
have a dose number significantly higher than 1, based on the solubility in the
range
from pH 1 to pH 7 for acidic active substances or in the range from pH 3 to 7
for
amphoteric active substances, i.e. for the oral formulation according to the
invention
s both the degree of pH-dependence of the solubility of the active substance
and the
size of the dose of active substance are of interest. The formulation
according to the
invention is particularly suitable for active substances which have poor
solubility
below pH 6 according to the dose number defined above.
to Generally increasing the dose in order to compensate for the reduced
bioavailability
in patients is frequently undesirable because of the waste of active substance
and
the greater burden on the patient and the associated risk of side effects, for
example,
or even totally impossible, on the grounds of drug safety. In any case, with
active
substances which have poor solubility increasing the dose does not necessarily
lead
~s to the expected plasma levels: in the case of DT-TX 30, in a phase I study
only
about 50 % higher plasma levels were obtained by increasing the dose from 100
to
600 mg, and levels equal to those obtained with 600 mg were achieved with 200
mg.
The aim of the invention is to provide a pharmaceutical composition for oral
2o administration of active substances with pH-dependent solubility
characteristics
which guarantees largely pH-independent bioavailability of the active
substance.
Surprisingly, it has now been found that the use of pharmaceutically
acceptable
inorganic or organic bases with a water solubility of more than 1 g / 250 ml
at 20° C,
2s preferably more than 1 g / 160 ml at 25 °C, in solid oral
formulations can ensure
sufficient bioavailability of active substances with pH-dependent solubility
characteristics. Organic bases and mixtures of inorganic and organic bases are
preferred.
~o Pharmaceutically suitable bases for the purposes of this invention are for
example
sodium hydroxide (NaOH), potassium hydroxide (KOH), calcium hydroxide
(Ca(OH)2J, sodium carbonate (Na2C03), ammonia, diethanolamine, meglumine,
lysine, arginine, ethanolamine, piperazine, trometamol and triethanolamine.
NaOH,
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
4
KOH, Ca(OH)2, ammonia, diethanolamine, meglumine, lysine and arginine are
particularly suitable for the purposes of this invention. Most particularly
preferred are
meglumine, lysine, arginine and trometamol. The advantage of the inorganic
bases is
the low molecular weight, which makes it possible to produce formulations with
very
high contents of active substance, while the ratio of base to active substance
must be
selected so that no irritation occurs in the stomach or intestines as a result
of higher
pH levels. If desired, inorganic and organic bases may also be combined so as
to
achieve physiologically acceptable pH values of not more than 12.
~o Numerous active substances display a more or less marked tendency to
hydrolytic
decomposition in the presence of bases and traces of water. In individual
cases there
may even be a direct chemical reaction between the active substance and bases,
e.g. ester formation. When developing a product which remains stable when
stored it
is therefore advantageous to separate the base spatially from the active
substance in
is the formulation. Only after the administration of the formulation does the
base
dissolve and produce a basic microclimate in which the active substance can
dissolve.
A further aim of the invention is to prevent the undesirable interactions
between base
?o and active substance in spite of the use of a base to improve the
solubility.
Multiparticulate formulations in which the individual particles have the
structure
shown in Figure 2 are particularly suitable for the preferred spatial
separation of
active substance and base.
2s
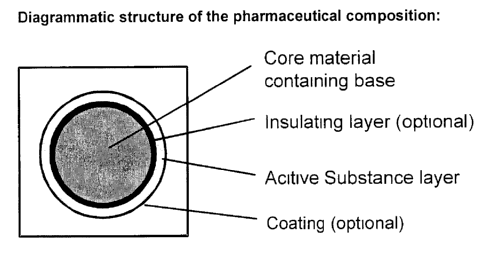
Figure 2 shows the diagrammatic structure of the pharmaceutical composition by
means of a section through a pellet which is suitable for producing the
pharmaceutical composition according to the invention. The roughly spherical
core
portion of this pellet contains or consists of one or more pharmaceutically
acceptable
~o inorganic or organic bases. This is optionally followed by a layer which
separates the
base-core from the layer containing the active substance, the so-called
insulating
layer. The insulating layer in turn, or the core material in the absence of an
insulating
layer, is surrounded by the active substance layer, which is also spherical,
which
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
may itself be surrounded by a coating to increase the abrasion resistance and
shelf
life of the pellets.
One advantage of a formulation in the form of pellets is the possibility of
individual
s dosing, which is important when medicating children, for example.
The core material used is a pharmaceutically acceptable base with a water
solubility
of > 1 g / 250 ml at 20° C, such as e.g. NaOH, KOH, Ca(OH)2, Na2C03,
ammonia,
diethanolamine, meglumine, lysine, arginine, ethanoiamine, piperazine,
trometamol
to or triethanolamine, or mixtures of such bases, to which a small amount of 1
to 10
by weight, preferably 3 to 6 % by weight of a suitable binder is optionally
added. The
use of a binder may be necessary, for example, if the core material is
produced by a
pan build-up process. If the method used is extrusion or spheronisation, other
technological adjuvants such as microcrystalline cellulose will be needed
instead of
Is binders, optionally together with binders for increasing the mechanical
stability. It is
also possible to use pure (100 %) base as the starting material if it can be
obtained in
a sufficiently narrow range of particle sizes: The pharmaceutically acceptable
bases
used are preferably NaOH, KOH, Ca(OH)2, ammonia, diethanolamine, meglumine,
lysine, arginine or trometamol; particularly preferred are meglumine, lysine,
arginine
zo and trometamol or mixtures thereof; meglumine is particularly preferred. As
binder, it
is possible to use gum arabic or a partially or totally synthetic polymer
selected from
among the hydroxypropylcelluloses, hydroxypropylmethylcelluloses,
methylcelluloses, hydroxyethylcelluloses, carboxymethylcelluloses, polyvinyl-
pyrrolidones, the copolymers of N-vinylpyrrolidone and vinyl acetate, or
combinations
~s of these polymers; gum arabic is preferred. The spherical core material
preferably
has an average diameter of 0.4 - 1.5 mm. The content of the pharmaceutically
acceptable inorganic or organic base is usually between 30 and 100 % in the
core
material. This base-containing spherical core material is also referred to as
starter
pellets or starter, for short (e.g. meglumine starter, lysine starter).
To increase the durability of the finished product, in the case of active
substances
which are unstable in the presence of bases, it is advantageous to coat the
core
material before the application of the active substance with an insulating
layer based
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
6
on a water-soluble, pharmaceutically acceptable polymer. Examples of such
water-
soluble polymers include for example gum arabic or a partially or totally
synthetic
polymer selected from among the hydroxypropylcelluloses, hydroxypropyl-
methylcelluloses, methylcelluloses, hydroxyethylcelluloses,
carboxymethylcelluloses,
s polyvinylpyrrolidones, the copolymers of N-vinylpyrrolidone and vinyl
acetate, or
combinations of these polymers. Gum arable or a hydroxypropylmethylcellulose
is
preferably used. If desired, the coating with the water-soluble,
pharmaceutically
acceptable polymer may be carried out with the addition of suitable
plasticisers,
separating agents and pigments, such as for example triethylcitrate,
tributylcitrate,
~o triacetin, polyethyleneglycols (plasticisers), talc, silicic acid
(separating agents),
titanium dioxide or iron oxide pigments (pigments).
The active substance layer contains the active substance as well as binders
and
optionally separating agents. Suitable binders include for example
~s hydroxypropylcellulose, hydroxypropylmethylcellulose, methylcellulose,
hydroxy-
ethylcellulose, carboxymethylcellulose, poiyvinylpyrrolidone, copolymers of N-
vinylpyrrolidone and vinyl acetate or combinations of these polymers.
Preferably,
hydroxypropylcellulose or copolymers of N-vinylpyrrolidone and vinyl acetate
are
used. The addition of separating agents such as e.g. talc, magnesium stearate
or
2o silicic acid serves to prevent the particles from aggregating during the
process. The
active substance content is not more than 60 %, preferably not more than 50 %
of the
pharmaceutical composition.
The optional outermost layer, which serves to reduce any increased abrasion
during
2s packing into capsules and/or to increase the shelf life, consists of
pharmaceutically
conventional film-forming agents, plasticisers and optionally pigments.
Suitable film-
forming agents include for example hydroxypropylcellulose, hydroxypropyl-
methylcellulose, methylcellulose, polymers and copolymers of acrylic and
methacrylic
acid and the esters thereof, or combinations of these polymers. Suitable
plasticisers
~o include inter alia triethylcitrate, tributylcitrate, triacetin or
polyethyleneglycols. The
pigments used may be e.g. titanium dioxide or iron oxide pigments. Preferably,
the
outer coating consists of hydroxypropylmethylcellulose and/or methylcellulose,
optionally with the addition of polyethylenegiycois as plasticisers.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
7
The pellets may be prepared by the method described hereinafter:
The base-containing core material consists either of crystals of the
particular base
s used or, more advantageously, of roughly spherical particles of the desired
size
containing a large amount of base, which can be produced by methods known and
established in pharmaceutical technology. The core material may be produced,
in
particular, by pan methods, on pelleting plates or by
extrusion/spheronisation. Then
the core material thus obtained may be divided into fractions of the desired
diameter
io by screening. Suitable core material has an average diameter of 0.4 to 1.5
mm,
preferably 0.6 to 0.8 mm.
First, the insulating layer is applied to this base-containing core material.
This can be
done by conventional methods, e.g. by applying an aqueous dispersion of the
water-
~s soluble, pharmaceutically acceptable polymer, optionally with the addition
of
plasticisers, separating agents and/or pigments, in a fluidised bed, in
coating pans or
in conventional film coating apparatus. If necessary the product can then be
screened again.
zo Then the active substance is applied from a dispersion containing binder
and
optionally separating agent. The volatile dispersant is removed during and/or
after
the process by drying. Suitable binders in the dispersion may be for example
hydroxypropylcellulose, hydroxypropylmethylcellulose, methylcellulose,
hydroxyethylcellulose, carboxymethylcellulose, polyvinylpyrrolidone,
copolymers of
2s N-vinylpyrrolidone and vinyl acetate or combinations of these polymers.
Preferably,
hydroxypropylcellulose or copolymers of N-vinylpyrrolidone and vinyl acetate
are
used. Suitable separating agents include e.g. talc, magnesium stearate or
silicic acid;
preferably, talc is used. The dispersants may be for example water, ethanol, 2-
propanol, acetone or mixtures of these solvents with one another. The
application of
~o active substance to the core material may be carried out by established
methods
known in pharmaceutical technology, e.g. in coating pans, conventional film
coating
apparatus or by the fluidised bed method. Then a further screening process may
be
carried out.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
8
To reduce any increased abrasion during transfer into capsules or to increase
the
shelf life the system may finally be coated with a coating of a
pharmaceutically
conventional film forming agent, plasticiser and optionally pigment. This may
be
s done by conventional methods as mentioned earlier in the description of the
application of the insulating layer.
When core material with an average diameter of 0.4 - 1.5 mm is used, the
process
described above produces pellets containing active substance, which can then
be
~o packed into hard capsules, for example. To do this, a number of these units
corresponding to the required dosage are packed into hard capsules in a
standard
capsule filling machine. Suitable hard capsules include, for example, hard
gelatine
capsules or hard capsules of hydroxypropylmethylcellulose (HPMC). The
preferred
active substance content of the pharmaceutical composition is not more than 60
%,
~s preferably not more than 50 % .
Unless otherwise stated, percentages specified are always percent by weight.
All the
data on the active substance content relate to the active substance acid (not
to a
specific salt) unless otherwise stated.
The amount of active substance per capsule is preferably such that 1 to 2
capsules a
day are sufficient to produce the desired activity.
For repaglinide, for example, daily doses of 0.2 mg to 5 mg, preferably
capsules
containing 0.5 mg, 1.0 mg or 2.0 mg are suitable.
zs For telmisartan, for example, daily doses of 10 mg to 150 mg, preferably
capsules
containing 20 mg, 40 mg or 80 mg are suitable.
For meloxicam, for example, daily doses of 4 mg to 30 mg, preferably capsules
containing 7.5 mg or 15.0 mg are suitable.
For DT-TX 30 , for example, daily doses of 50 mg to 300 mg, preferably
capsules
~o containing 100 mg or 200 mg are suitable.
For gliquidone, for example, daily doses of 10 mg to 50 mg, preferably
capsules
containing 30 mg are suitable.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
9
For glibenclamide, for example, daily doses of 1.9 mg to 5.0 mg, preferably
capsules
containing 3.5 mg are suitable.
The preferred ratio of base to active substance is approx. 1:1 to approx.
20:1. The
s theoretical lower limit at which the system can still function is 1
equivalent of base per
mol of active substance. The upper limit of approx. 20:1 (base to active
substance) is
determined by the size of the formulation at the desired doses (number of
pellets per
capsule).
~o In quality control, in vitro releases are measured by USP methods. The drug
is
released in a volume of 900 ml and the pH is selected so as to obtain "sink
conditions", i.e. the entire dose of active substance is soluble in these 900
ml. This in
vitro method cannot be predictive of absorption in humans in most cases as a
patient
will generally take the drug with approx. 200 ml of liquid and in a non-acidic
stomach
Is the solubility is often only sufficient for a fraction of the dose. Non-
acidic stomach
occurs at a rate of about 25% of the population in older patients and is often
also
caused by co-medication with H2 biockers or proton pump inhibitors.
Therefore, within the scope of the invention, an empirical test method was
developed
2o which has a better correlation with the in vivo performance in humans. In
this
procedure, a drug preparation which contains the maximum dose used in humans
is
released in a volume of 200 ml (this corresponds to the dose in humans) in
buffer at
a pH with reduced solubility of the active substance in the physiologically
acceptable
range, i.e. between pH 1 - 7. As the absorbability can also be predicted with
some
Zs accuracy using this method, even at non-acidic gastric pH levels, it is
suitable for
optimising drug preparations. In order to identify the most favourable
formulation in
each case from a number of possible recipes, the maximum release and/or the
area
under the curve (AUC) from time 0 to the end of the release may be used as
relevant
characteristics.
This is clear from the example of the comparison of the formulation examples
c1 - c2
(reference to neutral starter) and c3 - c5 (Example according to the invention
with
meglumine starter) (Figure 3 and Table 1). By "neutral starter", neutral core"
and
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
neutral pellet" are meant in each case standard commercial pellets of sucrose
or
microcrystalline cellulose.
Table 1 a shows the in vitro releases and the characteristics of the area
under the
5 release curve (AUC) and the maximum release of Examples c3 - c5 according to
the
invention (active substance: telmisartan) compared with the reference forms of
Examples c1 and c2 with an even lower content of active substance in 0.005 mol
citrate buffer pH 5.0
~o Table 1a: Comparison of the in vitro releases of pellets on base starters
(Examples
c3 - c5) and neutral starters (Examples c1- c2)
Examplecontent in AUC maximum
of vitro
release
after
...
minutes
active 0 4 8 12 16 20 release
substance
c1 2.6 0 0 0 0 0 0 0 0
c2 16.8 0 0 0 0 0 0 0 0
c3 8.9 0 44 50 62 72 69 1046 72
c4 23.2 0 18 14 17 17 14 293 18
c5 29.4 0 9 9 8 8 6 147 9
Other Examples are the comparisons of formulation examples c28 (reference to
~5 neutral starter) with c31, c32 (Example according to the invention with
meglumine
starter) and c33- c35 (Example according to the invention with arginine
starter) as
well as the comparisons of formulation examples c43 and c45 (reference to
neutral
starter) with c46 and c47 (Example according to the invention with meglumine
starter)
and c49 and c49a (Example according to the invention with arginine starter).
Table 1 b shows the in vitro releases and the characteristics of area under
the release
curve (AUC) and maximum release of the Examples c31, c32, c33, c34 and c35
according to the invention (active substance: meloxicam) compared with the
reference form of Example c28 in 0.01 mol phosphate buffer pH 5.0
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
11
Table 1 b: Comparison of the in vitro releases of pellets on base starters
(Examples
c31, c32, c33, c34, c35) and neutral starters (Example c28)
Examplecontent in AUC maximum
of vitro release
active release
after
...
minutes
substance0 5 10 15 20
c28 16.5 0 17 6 3 0 126 17
c31 16.8 0 71 83 87 88 1426 88
c32 26.1 0 58 61 62 52 1034 62
c33 17.1 0 55 57 52 53 951 57
c34 28.6 0 34 37 41 36 652 41
c35 33.3 0 34 36 35 29 596 36
s Table 1 c shows the in vitro releases and the characteristics of area under
the release
curve (AUC) and maximum release of Examples c46, c47, c49 and c49a according
to
the invention (active substance: DT-TX 30) compared with reference forms of
Examples c43 and c45 in 0.025 mol phosphate buffer pH 6.0
~o Table 1c: Comparison of the in vitro releases of pellets on base starters
(Examples
c46, c47, c49 and c49a and neutral starters (Examples c43, c45)
Exa content in AUC maximum
mple of vitro release
active release
after
...
minutes
substance0 4 8 12 16 20
c43 15.8 0 7 6 5 0 0 74 7
c45 31.2 0 7 8 0 0 0 58 8
c46 16.3 0 19 51 55 56 58 845 58
c47 25.7 0 8 35 38 39 39 558 39
c49 26.7 0 19 29 27 27 27 463 29
c49a 19.5 0 30 32 30 35 36 578 36
~s Interpretation of the results:
All the Examples according to the invention are clearly superior to the
reference
formulation as the reference forms do not achieve any measurable releases, or
only
slight ones.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
12
As the content of active substance increases the in vitro release declines, as
the
amount of base is smaller for the same dose of active substance.
All the bases and other excipients are generally suitable, but display
somewhat
s different release characteristics at comparable active substance
concentrations.
Examples c3, c4, c16, c21, c24, c25, c31, c32, c33, c46, c47, c49 and c49a
have
proved particularly suitable for the purposes of the invention.
The invention further relates to base-containing formulations for amphoteric
active
~o substances, such as for example telmisartan.
What is particularly surprising is the superiority of the forms according to
the
invention with amphoteric, i.e. acidically and basically soluble active
substances, if
one compares the in vitro releases of the pellets according to the invention
on base
Is starters with pellets in which the active substance has been applied to
acid-
containing cores. With a similar charge of amphoteric active substance is
present the
release of the base-containing pellets is significantly better than that of
the acid-
containing pellets, as shown by the comparison of the in vitro releases of
Examples
c10, c13, c14 and c15 (for releases see Table 2 and Figure 1).
CA 02537776 2006-03-03
13
Case 1/1563
foreign filing text
Table 2: Comparison of the in vitro releases and the characteristics of area
under the
release curve (AUC) and maximum release of pellets on base starters (Examples
c3
- c5) and acid starters (Examples c13 - c15 contain tartaric acid starter and
Example
c10 contains succinic acid starter)
s
I Examplecontent in AUC maximum
of vitro
release
after
...
minutes
active 0 4 8 12 16 20 release
substance
c3 8.9 0 44 50 62 72 69 1046 72
c4 23.2 0 18 14 17 17 14 293 18
c5 29.4 0 9 9 8 8 6 147 9
c13 3.4 0 16 17 17 17 18 302 18
c14 9.5 0 7 8 8 7 8 134 8
c15 17.4 0 4 4 3 3 3 63 4
c10 19.7 10 10 0 0 0 0 10 0
The data show that Example c3 with approx 9% active substance content achieves
a
maximum release of 72 %, whereas Example c13, also with approx 9% active
substance content achieves a maximum release of only 8 %, i.e. the maximum
~o release with base starter is higher by about a factor of 9, and with regard
to the AUC
by a factor of 8. Example c4 with an active substance content of approx 23%
achieves 18 % release whereas Example c15 with an active substance content of
only 17.4% achieves only 4% release, i.e. the maximum release and also the AUC
value is higher by a factor of 5. Example c5 with an active substance content
of 29
achieves 9 % release and is thus comparable with Example c14 with an active
substance content of only 9.5 %. This means that e.g. for 1 capsule containing
80 mg
of active substance for Example c5 only 235 mg of pellets are needed, which
can
very easily be packed into a size 3 capsule (approx 6*16 mm), whereas for 1
capsule
containing 80 mg of active substance for Example c14 , 762 mg of pellets are
2o required, which can only be fitted into 2 size 1 capsules (approx 8*24 mm),
i.e. the
medication is substantially more difficult for the patient to take. Examples
c15 with
an active substance content of 17 % achieve only 4 % maximum release and
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
14
Example c10 (succinic acid starter with active substance content of 20 %) does
not
release the active substance at aii.
Legend relating to the Figures:
Figure 1 shows the in vitro releases of the Examples c3 - c5 according to the
invention compared with the formulations on tartaric acid starters (Examples
c13 -
c15) and on succinic acid starter (c10) with different contents of active
substance in
0.005 mol citrate buffer pH 5Ø
io
Figure 2 shows the schematic structure of a pharmaceutical composition
according to
the invention in the form of a sectional view of a pellet.
Figure 3 shows the in vitro releases of the Examples c3 - c5 according to the
~5 invention compared with the reference formulations c1 - c2 with different
contents of
active substance in 0.005 mol citrate buffer pH 5Ø
Figure 4 shows the in vitro releases of the Examples c33 - c35 according to
the
invention compared with the reference formulation c28 with different contents
of
2o active substance in 0.01 mol phosphate buffer pH 5Ø
Figure 5 shows the in vitro releases of the Examples c31 and c32 according to
the
invention compared with the reference formulation c28 with different contents
of
active substance in 0.01 mol phosphate buffer pH 5Ø
Figure 6 shows the in vitro releases of the Examples c46 and c47 according to
the
invention compared with the reference formulations c43 and c45 with different
contents of active substance in 0.0025 mol phosphate buffer pH 6Ø
~o Figure 7 shows the in vitro releases of the Examples c49 and c49a according
to the
invention compared with the reference formulations c43 and c45 with different
contents of active substance in 0.0025 mol phosphate buffer pH 6Ø
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
The Examples that follow are intended to illustrate the invention:
Examples:
The preparation of the following Examples usually takes place over 5 steps:
s a: preparation of base-containing core material
b: isolation of the base-containing core material
c: preparation of the active substance layer
d: isolation of the pellets consisting of base-containing core
e: packing into capsules
~o
Step b is absolutely essential if there is any incompatibility between base
and active
substance layer, otherwise this step may be omitted to simplify the production
method. Step d is necessary if the mechanical stability of the active
substance layer
is insufficient to dissolve the active substance completely.
~s
The brand names used in the Examples and not separately characterised refer to
the
following substances:
Koflidon K25 povidone (polyvinylpyrrolidone)
2o Avicel PH101 microcrystalline cellulose
Klucel hydroxypropylcellulose
i.e. the Examples should be read as meaning that povidone, e.g. Kollidon K25
is
used.
2s
a: Examples of the preparation of base-containing core material
Example a1: Preparation of meglumine-containing core material with binder
~o
Composition:
Kollidon K25 3 parts by weight
Avicel PH101 20 parts by weight
meglumine 77 parts by weight
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
16
77 parts by weight of meglumine, 20 parts by weight of Avicel PH 101 and 3
parts by
weight of Kollidon K25 are mixed for 15 minutes in a gyrowheel mixer. Then the
powder mixture is transferred into a twin-screw metering device. This mixture
is
s introduced into a twin-screw extruder of the Werner & Pfleiderer 37/18D type
(or any
other suitable type of extruder) at a speed of about 1 kg/h, together with
water which
is added by means of a ProMinent metering pump. The water is automatically
regulated so as to obtain a nominal torque of approx. 19% in the extruder. The
extrusion is carried out using a die plate with bores 8 mm in diameter.
to The extruded strips are rounded off to form pellets in a WyPro Spharomat,
the
process taking approx. 3 minutes at approx. 850 RPM.
Drying of the pellets at 80°C for approx. 1.5 h in the GPCG1 fluidised
bed dryer.
The core material is fractionated using a tumbler screening machine with
different
Is perforated plates having nominal mesh sizes of 0.71 to 1.25 mm. The
suitable
fractions of materials of between 0.71 and 0.90 or 0.90 and 1.12 mm are used
in
subsequent processes.
Example a2: Preparation of meqlumine-containing core material without binder
2o
Composition:
Avicel PH101 40 parts by weight
megiumine 60 parts by weight
2s
60 parts by weight of meglumine and 40 parts by weight of Avicel PH 101 are
mixed
for 15 minutes in a gyrowheel mixer. Then the powder mixture is transferred
into a
twin-screw metering device. This mixture is introduced into a twin-screw
extruder of
the Werner & Pfleiderer 37/18D type (or any other suitable type of extruder)
at a
~o speed of about 1 kg/h, together with water which is added by means of a
ProMinent
metering pump. The water is automatically regulated so as to obtain a nominal
torque
of approx. 19% in the extruder. The extrusion is carried out using a die plate
with
bores 8 mm in diameter.
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
17
The extruded strips are rounded off to form pellets in a WyPro Spharomat, the
process taking about 3 minutes at approx. 850 RPM.
Drying of the pellets at 80°C for approx. 1.5 h in the GPCG1 fluidised
bed dryer.
s The core material is fractionated using a tumbler screening machine with
different
perforated plates having nominal mesh sizes of 0.71 to 1.25 mm. The suitable
fractions of materials of between 0.71 and 0.90 or 0.90 and 1.12 mm are used
in
subsequent processes.
to Example a3: Preparation of arqinine-containing core material
Composition:
Avicel PH101 40 parts by weight
arginine 60 parts by weight
~s
Prepared analogously to Example a2
Example a4: Preparation of trometamol-containing core material
2o Composition:
Avicel PH101 40 parts by weight
trometamol 60 parts by weight
Prepared analogously to Example a2
2s
Example a5: Preparation bf piperazine-containing core material
Composition:
Avicel PH101 40 parts by weight
~o piperazine 60 parts by weight
Prepared analogously to Example a2
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
18
Example a6: Preparation of sodium hydroxide-containing core material
Composition:
Avicel PH101 30 parts by weight
s sodium hydroxide 70 parts by weight
Prepared by dissolving sodium hydroxide in water, then adding Avicel PH101.
Further processing is carried out analogously to Example a2
~o Example a7: Preparation of potassium hydroxide-containing core material
Composition:
Avicel PH101 40 parts by weight
potassium hydroxide 60 parts by weight
Is
Prepared analogously to Example a6
Example a8: Preparation of calcium hydroxide-containing core material
2o Composition:
Avicet PH101 70 parts by weight
Calcium hydroxide 30 parts by weight
Prepared analogously to Example a6
Zs
Example a9: Preparation of sodium h~rdroxide- and me4lumine-containing core
material
~o Composition:
Avicel PH101 30 parts by weight
sodium hydroxide 20 parts by weight
meglumine 50 parts by weight
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
19
Prepared by dissolving sodium hydroxide in water, then adding Avicel PH101 and
meglumine. Further processing is carried out analogously to Example a2
s b: Example for the isolation of the base-containing core material
Composition:
Base-containing core material 23 parts by weight
Gum arabic 1 part by weight
to Taic 2 parts by weight
1 part by weight of gum arabic is dissolved with stirring in a mixture of 6.7
parts by
weight of 96 % ethanol and 13.5 parts by weight of purified water. Then 2
parts by
weight of talc are dispersed in the solution with stirring.
In a fluidised bed processing plant, 23 parts by weight of base-containing
core
material are sprayed with the gum arabic/talc dispersion at an air entry
temperature
of 35° - 40° C by the under-bed spraying method.
2o The isolated base-containing core material is then dried at 40° C in
the circulating air
dryer for 8 hours.
To remove lumps the dried isolated base-containing core material is screened
through a screen with a nominal mesh size of 1.0 mm. The fraction of material
2s (particle size less than 1 mm) is further processed.
c: Examples of the preparation of the active substance layer
The active substance layer is generally prepared in the same way, but with
variations
3o in the nature and quantity of the active substance, the nature and quantity
of the
binder, the amount of talc and isopropanol or the amount of ethanol. The
preparation
is therefore only described for Example c9, and the particular compositions
for each
active substance are shown in table form.
CA 02537776 2006-03-03
Preparation of Example c9:
Composition:
s isolated meglumine-containing core material 12 parts by weight
hydroxypropylcellulose 2.5 parts by weight
talc 5 parts by weight
active substance (e.g. telmisartan) 10 parts by weight
Case 1/1563
foreign filing text
~o Hydroxypropylcellulose is dissolved In 255 parts by weight of 2-propanol
with stirring
and then the active substance and talc are dispersed in this solution with
stirring.
In a fluidised bed processing plant, 12 parts by weight of meglumine-
containing core
material are sprayed with the dispersion containing the active substance at an
air
~s entry temperature of 20° - 30° C by the under-bed spraying
method.
The pellets containing the active substance are then dried at 35° C in
the circulating
air dryer for 8 hours.
2o To remove lumps the pellets containing the active substance are screened
through a
screen with a nominal mesh size of 1.25 mm. The product fraction (particle
size less
than 1.25 mm) is further processed.
For other Examples of step c see below.
CA 02537776 2006-03-03
21
Case 111563
foreign filing text
d' Example of the isolation of the pellefs containing active substance
Composition:
pellets containing active substance 23 parts by weight
gum arabic 1 part by weight
talc 2 parts by weight
~0 1 part by weight of gum arabic is dissolved with stirring in a mixture of
6.7 parts by
weight of 96 % ethanol and 13.5 parts by weight of purified water. Then 2
parts by
weight of talc are dispersed in the solution with stirring.
In a fluidised bed processing plant, 23 parts by weight of pellets containing
active
~s substance are sprayed with the gum arabic/talc dispersion at an air entry
temperature
of 35° - 40° C by the under-bed spraying method.
The isolated meglumine-containing core material is then dried at 40° C
in the
circulating air dryer for 8 hours.
To remove lumps the dried pellets containing active substance are screened
through
a screen with a nominal mesh size of 1.25 mm. The product fraction (particle
size
less than 1.25 mm) is further processed.
2s e) Packing into capsules
A quantity of pellets containing active substance corresponding to the desired
dosage in each case is packed into hard capsules, e.g. hard gelatine capsules,
of
suitable size using a capsule filling machine.
JO
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
22
Other Examples of compositions for step c
The numbers given below, unless otherwise stated, are parts by weight. In each
case parts by weight are specified which correspond to the active substance
content
determined experimentally, i.e. the spray losses, which may fluctuate somewhat
from
s one batch to the next, were compensated in the calculation in each case so
as to
obtain truly comparable data.
For example, the 10-fold values may be regarded as amounts given in grams,
i.e. for
Example c1, 200.0 g of neutral pellets, 5.4 g of telmisartan, 1.4 g of
povidone K90,
2.7 g of talc and 142.1 g of isopropanoi.
ao The Examples which contain a commercially available neutral core instead of
the
base-containing starter cores according to the invention serve in each case as
reference values for the in vitro testing.
Telmisartan Examples
Is Examples c1 - c2 contain a commercially available neutral core instead of
the base-
containing starter cores according to the invention. These cores serve as
reference
values for the in vitro testing (see Table 1 ). Examples c10 - c15 comprise
acid-
containing cores instead of the base-containing starter cores according to the
invention, so as to demonstrate the unforeseeable advantage of the base
starters
Zo according to the invention over acid starters, which should theoretically
be at least
equally soluble, as telmisartan is also readily soluble in an acidic medium.
Example c1 c2
active substance content2.60 16.80
(wt.%):
neutral pellets 20.00 20.00
telmisartan 0.54 4.76
povidone K 90 0.14 1.19
talc 0.27 2.38
i isopropanol (for preparation14.21 124.36
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
23
Example c3 c4 c5 c6 c7 c8 c9
active substance8.9 23.2 29.4 33.7 10.6 14.5 33.7
content (wt.%):
meglumine starter12.00 12.00 12.00 12.00 12.00 12.00 12.00
t telmisartan 1.26 4.68 7.28 9.88 1.56 2.34 9.88
povidone K 90 0.32 1.17 1.82 2.47 - - -
talc 0.63 2.34 3.64 4.94 0.78 1.17 4.94
Klucel - - - - 0.39 0.59 2.47
isopropanol 32.94 122.27190.19 258.12 40.37 60.55 255.65
(for
preparation
only)
Example c10 c11 c12
active substance content 19.70 20.70 32.10
(wt.%):
succinic acid starter 12.00 12.00 12.00
telmisartan 3.61 3.90 8.81
talc 1.81 1.95 4.41
Klucel 0.90 0.98 2.20
I isopropanol (for preparation93.51 100.91 228.06
only) ~
Example c13 c14 c15
active substance content 3.40 9.50 17.40
(wt.%):
tartaric acid starter 20.00 20.00 20.00
telmisartan 0.73 2.29 4.99
talc 0.36 1.14 2.50
i Klucel 0.18 0.57 1.25
isopropanol (for preparation18.84 59.20 129.17
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
24
Example c16 c17 c18
I
active substance content 19.70 28.50 32.90
(wt.%):
arginine starter 12.00 12.00 12.00
telmisartan 3.60 6.80 9.30
povidone K90 0.90 1.70 3.40
talc 1.80 3.40 4.65
isopropanol (for preparation93.60 176.80 241.80
only)
Example c19 c20 i
active substance content 17.10 26.20
(wt.%):
trometamol starter 12.00 12.00
telmisartan 2.93 5.80
povidone K 90 0.73 1.45
talc 1.47 2.90
isopropanol (for preparation76.18 150.80
only)
Example c21 c22 c23
active substance content 19.80 28.50 32.90
(wt.%):
lysine starter 12.00 12.00 12.00
telmisartan 3.64 6.82 9.29
povidone K90 0.91 1.71 3.32
talc 1.82 3.41 4.65
isopropano! (for preparation94.64 177.32 241.54
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
Example c24 c25
active substance content 18.20 25.80
(vut.%):
sodium hydroxide starter 12.00 12.00
telmisartan 3.20 5.63
povidone K 90 0.80 1.41
talc 1.60 2.82
isopropanol (for preparation83.20 146.38
only)
Example c26 c27
active substance content 26.90 33.90
(wt.%):
potassium hydroxide starter12.00 12.00
telmisartan 6.12 9.98
povidone K 90 1.53 2.50
talc 3.06 4.99
isopropanol (for preparation159.12 259.48
only)
Meloxicam Examples:
Example c28 c29 c30
active substance content 14.80 25.20 33.00
(wt.%):
neutral pellets 12.00 12.00 12.00
meloxicam 2.40 5.42 9.40
povidone K90 0.60 1.36 2.35
talc 1.20 2.71 4.70
isopropanol (for preparation62.40 140.92 244.40
only) ~
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
26
Example c31 c32
active substance content 16.80 26.10
(wt.%):
meglumine starter 12.00 12.00
meloxicam 2.85 5.78
povidone K 90 0.71 1.45
talc 1.43 2.89
isopropanol (for preparation74.10 150.28
only)
Example c33 c34 c35
active substance content 17.10 28.60 33.30
(wt.%):
arginine starter 12.00 12.00 12.00
meloxicam 2.93 6.87 9.58
povidone K90 0.73 1.72 2.40
talc 1.47 3.44 4.79
isopropanol (for preparation76.18 178.62 249.08
only)
Example . c36 c37
active substance content 15.90 25.70
(wt.%):
trometamol starter 12.00 12.00
meloxicam 2.64 5.61
povidone K 90 0.66 1.40
talc 1.32 2.81
isopropanol (for preparation68.64 145.86
only) ~
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
27
Example c38 c39 c40
I
active substance content 16.10 26.50 36.60
(wt.%):
lysine starter 12.00 12.00 12.00
meloxicam 2.70 5.94 11.30
povidone K90 0.68 1.49 2.83
talc 1.35 2.97 5.65
isopropanol (for preparation70.20 154.44 293.80
only)
Example c41 c42
active substance content 28.30 36.60
(wt.%):
calcium hydroxide starter12.00 12.00
meloxicam 6.75 12.21
povidone K 90 1.69 3.05
talc 3.38 6.11
isopropanol (for preparation~ 175.50 317.46
only)
DT-TX 30-Examples
Example c43 c44 c45
active substance content 15.80 22.20 31.20
(wt.%):
neutral pellets 12.00 12.00 12.00
DT-TX 30 2.63 4.35 8.26
povidone K90 0.66 1.09 2.07
talc 1.32 2.18 4.13
isopropanol (for preparation68.38 113.10 214.76
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
28
Example c46 c47 c48
active substance content16.30 25.70 33.60
(wt.%):
meglumine starter 12.00 12.00 12.00
DT-TX 30 2.73 5.60 9.76
povidone K90 0.68 1.40 2.44
talc 1.37 2.80 4.88
isopropanol (for preparation70.98 145.60 253.76
only) ~
Example c49 c49a c50
active substance content26.70 19.50 34.20
(wt.%):
arginine starter 12.00 12.00 12.00
DT-TX 30 6.00 4.00 10.20
povidone K 90 1.50 1.50 2.55
talc 3.00 3.00 5.10
isopropanol (for preparation156.00 156.00 265:20
only)
Example c51 c52 c53
active substance content15.70 26.00 37.60
(wt.%):
lysine starter 12.00 12.00 12.00
DT-TX 30 2.59 5.74 13.20
povidone K90 0.65 1.44 3.30
talc 1.30 2.87 6.60
isopropanol (for preparation67.34 149.24 343.20
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
29
Gliguidone Examples
Example c54 c55 c56
active substance content14.80 21.70 31.90
(wt.%):
neutral pellets 12.00 12.00 12.00
gliquidone 2.40 4.21 8.67
povidone K90 0.60 1.05 2.17
talc 1.20 2.11 4.34
isopropanol (for preparation62.40 109.46 225.42
only) I
Example c57 c58 c59
active substance content15.60 22.30 31.20
(wt.%):
meglumine starter 12.00 12.00 12.00
gliquidone 2.57 4.38 8.26
povidone K90 0.64 1.10 2.07
talc 1.29 2.19 4.13
isopropanol (for preparation66.82 113.88 214.76
only)
Example c60 c61
active substance content 17.80 27.10
(wt. %):
arginine starter 12.00 12.00
giiquidone 3.10 6.20
povidone K 90 0.78 1.55
talc 1.55 3.10
isopropanol (for preparation 80.60 161.20
only)
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
Example c62 c63 c64
active substance content 14.90 25.00 32.60
(wt.%):
lysine starter 12.00 12.00 12.00
gliquidone 2.41 5.32 9.12
povidone K90 0.60 1.33 2.28
talc 1.21 2.66 4.56
isopropanol (for preparation62.66 138.32 237.12
only)
Repaglinide Examples
Example c65 c66 c67
active substance content 4.30 11.00 16.50
(wt.%):
neutral pellets 12.00 12.00 12.00
repaglinide 0.56 1.63 2.78
povidone K90 0.14 0.41 0.70
talc 0.28 0.82 1.39
isopropanol (for preparation14.56 42.38 72.28
only)
Example c68 c69 c70
active substance content 3.40 11.40 13.50
(wt.%):
meglumine starter 12.00 12.00 12.00
repaglinide 0.44 1.70 2.12
povidone K90 0.11 0.43 0.53
I talc 0.22 0.85 1.06
isopropanol (for preparation11.44 44.20 55.12
only) ~
CA 02537776 2006-03-03
Case 1/1563
foreign filing text
31
'~ Example c71 c72
active substance content 5.10 8.00
(wt.%):
arginine starter 12.00 12.00
repaglinide 0.67 1.11
povidone K 90 0.17 0.28
talc 0.34 0.56
isopropanol (for preparation17.42 28.86
only)
Example c73 c74 c75
I
I~active substance content5.00 8.60 15.00
(wt.%):
lysine starter 12.00 12.00 12.00
repaglinide 0.66 1.21 2.44
povidone K90 0.17 0.30 0.61
talc 0.33 0.61 1.22
isopropanol (for preparation17.16 31.46 63.44
only)