Note: Descriptions are shown in the official language in which they were submitted.
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
IDENTIFICATION OF KINASE INHIBITORS
Background of the Invention
Field of the Invention
The growth and differentiation of eukaryotic cells is regulated by 'a complex
web of
signal transduction pathways. Precise regulation of these pathways allows
cells to respond to
extracellular stimuli such as hormones, neurotransnutters, and stress as they
proliferate and
differentiate into specific tissues. Protein phosphorylation, a regulatory
mechanism common to
all eukaryotic cells, plays a central role in this signal transduction web.
First discovered as a
regulatory mechanism nearly fifty years ago, protein phosphorylation is very
likely the most
important mechanism for regulation of signal transduction in mammalian cells.
It is therefore
not surprising that protein kinases, enzymes that catalyze the transfer of the
y-phosphatase group
of ATP to the oxygen atom of the hydroxyl group of serine, threonine or
tyrosine residues in
peptides and polypeptides, comprise one of the largest protein superfamilies.
Indeed, with the
complete sequencing of the human genome, it has been asserted that there are
exactly 508 genes
encoding human protein kinases, including 58 receptor tyrosine kinases and 32
nonreceptor
tyrosine kinases.
I~inases and cancer
Cancer consists of a variety of diseases characterized by abnormal cell
growth. Cancer is
caused by both internal and external factors that cause mutations in the
genetic material of the
cells. Cancer causing genetic mutations can be grouped into two categories,
those that act in a
positive manner to increase the rate of cell growth, and those that act in a
negative manner by
removing natural barriers to cell growth and differentiation. Mutated genes
that increase the rate
of cell growth and differentiation are called oncogenes, while those that
remove natural barriers
to growth are called tumor suppressor genes.
The first oncogene identified encoded the Src tyrosine kinase. . Src is a key
regulator of
signal transduction in many different cell types. Present inside nearly all
human cells in an
inactive state,, Src is poised to respond to extracellular signals from a
variety of sources. Once
triggered by a stimulus, Src is transformed into an active state in which it
phosphorylates
tyrosine residues on a number of effector proteins. While the tyrosine kinase
activity of Src is
tightly regulated in normal cells, mutations can occur which transform the
enzyme into a
-1-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
constitutively active state. It was one such mutation, identified over 25
years ago, that gave Src
the dubious honor of being known as the first oncogene. There are now about 30
tumor
suppressor genes and over 100 oncogenes known, about 20% of which encode
tyrosine kinases.
The disregulation of such central regulators of cell growth and
differentiation has disastrous
consequences for the cell.
Kinase inhibitors
Protein kinases play a crucial role not only in signal transduction but also
in cellular
proliferation, differentiation, and various regulatory mechanisms. The casual
role that many
protein kinases play in oncogenesis has made them exciting targets for the
development of novel
anti-cancer chemotherapies. The conserved and extremely well characterized
nature of the ATP
binding pocket has made it the most common, and most successful, target for
kinase inhibition.
Thus, libraries containing ATP (and purine) mimetics have been generated and
screened against
large panels of kinases in the hope of finding those rare pharmacophores that
can outcompete
ATP, thereby blocking kinase activity.
However, this approach has at least two major shortcomings. First, these
inhibitors must
compete directly with ATP for their binding site. ATP, which is used by
thousands of cellular
enzymes, is present in cells in very high concentration. Therefore, kinase
inhibitors that act in a
strictly ATP competitive manner must bind to their target kinase with
extremely high affinity.
Second, the high structural conservation surrounding the ATP binding pocket
(also known as the
purine binding pocket) makes it difficult to design inhibitors that show
specificity for one kinase
over another. Given these two criticisms, it is perhaps not surprising that
after twenty years of
research there are only twelve small molecule tyrosine kinase inhibitors in
clinical trials. All of
these inhibitors compete directly with ATP for the ATP binding pocket, all
bind this pocket
extremely tightly, and all show varying degrees of specificity for their
target kinase.
A possible exception is the small molecule kinase inhibitors, GleevecTM
(Novartis), a
phenylamino-pyrimidine derivative, which binds the purine pocket of Abl
tyrosine kinase._ This
compound shows unique properties that suggest that its mode of action is
somewhat unusual.
While this compound was approximately a thousand fold less potent than most
kinase inhibitor
clinical candidates when assayed in biochemical assays, it did not lose as
much potency as most
inhibitors did when tested in cells, suggesting that it is not competing
directly with cellular ATP
for binding to Abl. Co-crystallization studies have shown that GleevecTM does
indeed occupy
the purine pocket of the Abl lcinase, but it does so only when the kinase is
in an inactive
-2-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
confornlation, with the amino-ternlinal and carboxy-terminal lobes twisted
such that the catalytic
triad of lysine and two aspartic acids is not properly .aligned to accept ATP
or to catalyze the
phosphate transfer reaction. Therefore, GleevecTM makes use of the proven
small molecule
druggability of the purine pocket without directly competing against ATP,
which binds to the
inactive conformation with much lower affinity.
It would be desirable to develop protein kinase inhibitors that do not compete
directly
with ATP for binding to the active conformation of the ATP binding pocket of
the target protein
kinase. It would be further desirable to design fast, reliable, high-
throughput screening assays
for identifying such inhibitors. It would also be desirable to lock the ATP
binding pocket of
protein kinases in an inactive conformation in order to facilitate the design
of such screening
assays and the identification of protein kinase inhibitors with unique
properties, such as
increased specificity.
Sumrizary of the Invention
In one aspect; the invention concerns a method for identifying a ligand
binding to an
inactive conformation of a target protein kinase, comprising
(a) contacting the inactive conformation of the target protein kinase, which
contains or is modified to contain a reactive group at or near a binding site
of interest, with one
or more ligand candidates capable of covalently bonding to the reactive group
thereby forming a
kinase-ligand conjugate; and
(b) detecting the formation of the kinase-ligand conjugate and identifying the
ligand present in the kinase-ligand conjugate.
The kinase and the ligand candidate preferably form a disulfide bond to yield
a kinase-
ligand conjugate. In this embodiment, the kinase and the ligand candidates)
can be contacted in
the presence of a reducing agent, such as 2-mercaptoethanol or cysteamine.
The ligand candidates may be small molecules, and may be less than 1500
daltons,
preferably less than 1000 daltons, more preferably less than 750 daltons, even
more preferably
less than 500 daltons, most preferably less than 250 daltons in size.
In another aspect, the invention concerns a method for identifying a ligand
that binds to
the inactive conformation of a target protein kinase, comprising
(a) obtaining the inactive conformation of the target protein kinase
comprising an
-SH group, masked -SH group, or activated -SH group;
-3-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
(b) combining the inactive conformation of the target protein kinase with one
or
more.ligand candidates wherein said ligand candidates each comprises a
disulfide bond;
(c) forming a kinase-ligand conjugate wherein at least one ligand candidate
binds
to the inactive conformation of the target protein kinase under disulfide
exchange conditions, and
(d) detecting the formation of the kinase-ligand conjugate and identifying the
ligand present in the conjugate.
In another aspect, the invention concerns a method for identifying a ligand
that binds to
the inactive confornlation of a target protein kinase, comprising
(a) obtaining the inactive conformation of the target protein kinase
comprising an
-SH group, masked -SH group, or activated -SH group;
(b) combining in a mixture the inactive conformation of the target protein
kinase
with a plurality of ligand candidates that are each capable of forming a
disulfide bond with the
-SH group, masked -SH group, or activated -SH group thereby forming at least
one kinase-ligand
conjugate;
(c) analyzing the mixture by mass spectrometry; and
(d) detecting the most abundant kinase-ligand conjugate that is formed and
identifying the ligand thereon.
In yet another aspect, the invention concerns a method for identifying ligands
binding to
an inactive conformation of a target protein kinase, comprising
(a) contacting the inactive conformation of the protein kinase having a first
and a
second binding site of interest and containing or modified to contain a
nucleophile at or near the
first site of interest with a plurality of ligand candidates, the candidates
having a functional group
reactive with the nucleophile, under conditions such that a reversible
covalent bond is formed
between the nucleophile and a candidate that has affinity for the first site
of interest, to form a
kinase-first ligand complex;
(b) identifying the first ligand from the complex of (a);
(c) designing a derivative of the first ligand identified in (b) to provide a
small
molecule extender (SME) having a first functional group reactive with the
nucleophile on the
kinase and a second functional group reactive with a second ligand having
affinity for the
binding second site of interest;
(d) contacting the SME with the kinase to form a lcinase-SME complex, and
-4-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
(e) contacting the kinase-SME complex with a plurality of second ligand
candidates, the candidates having a functional group reactive with the SME in
said kinase-SME
complex, wherein a candidate that has affinity for the second binding site of
interest on the
kinase forms a reversible covalent bond with said kinase-SME complex, whereby
a second
ligand is identified.
In a still further aspect, the invention concerns a method for identifying
ligands binding
to an inactive conformation of a target protein kinase, comprising
(a) screening a library of ligand candidates with a kinase-ligand conjugate
formed
by the covalent bonding of the inactive conformation of a kinase comprising a
first reactive
functionality with a compound that comprises (1) a second reactive
functionality and (2) a
chemically reactive group, wherein the second reactive functionality of the
compound reacts
with the first reactive functionality of the inactive conformation of the
target protein kinase to
form a first covalent bond such that the kinase-ligand conjugate contains a
free chemically
reactive group, under conditions wherein at least one member of the library
forms a second
covalent bond with the kinase-ligand conjugate; and
(b) identifying a further ligand that binds covalently to the chemically
reactive
group of the kinase-ligand conjugate.
Brief Description of the Drawings
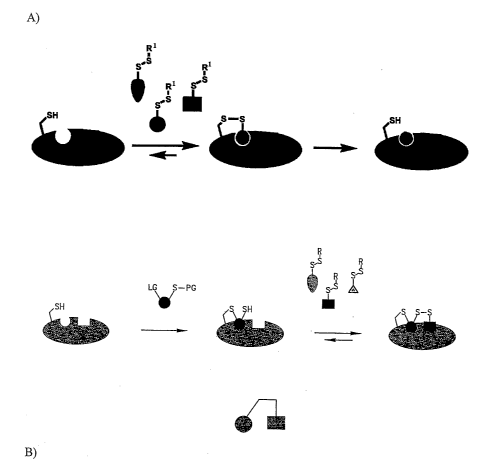
Figure 1A is a schematic illustration of one embodiment of Tethering. A thiol-
containing
protein is reacted with a plurality of ligand candidates. A ligand candidate
that possesses an
inherent binding affinity for the target is identified and a ligand is made
comprising the identified
binding determinant (represented by the circle) that does not include the
disulfide moiety.
Figure 1B is a schematic representation of one embodiment of Tethering where
an
extender comprising a first and second functionality is used. As shown, a
target that includes a
thiol is contacted with an extender comprising a first functionality -LG that
is capable of forming
a covalent bond with the reactive thiol and a second functionality second
functionality -SPG that
is capable of forming a disulfide bond. A target-extender covalent complex is
formed which is
then contacted with a plurality of ligand candidates. The extender provides
one binding
determinant (circle) and the ligand candidate provides the second binding
determinant (square)
and the resulting binding determinants are linked together to form a conjugate
compound.
-5-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Figure 2 illustrates the mass spectrometer profile of purified EGFRl kinase
domain.
Figure 2A is purified EGFR1 in the active conformation. Figure 2B is purified
EGFRl in the
inactive conformation. Figures 2C-E) are purified EGFRl in the inactive
conformation
following incubation with C) cystamine, D) a quinazoline extender, and E) the
quinazoline
extender and cystamine.
Figure 3 is a. schematic depicting the progression from the design and
synthesis of a
purine pocket extender, through a library screen, and ending with a soluble
MEKl inhibitor. The
portion of the molecule that binds to the adaptive binding pocket is indicated
by a circle. The
MEKl construct used in each of these successive steps, either the S 150C
screening mutant or
wild type, are indicated on the left.
Figure 4 is a specificity profile of three inhibitors that were derived from
Tethering that
inhibit MEK1 with ICSO s of 80 nM, 50 nM, and 10 nM respectively. ATP
concentrations were
varied such that the assays were run at or near the Km for ATP for the various
kinases: l OmM
ATP (IKKb, MEK1, MKK4); lSrriM ATP (Aurora-A, CaMKII, CSK, FGFR3, dap-70);
45mM
ATP (GDK2lcyclinA, c-RAF, JNKlal, PI~Ca, Yes); 50mM ATP (MEK1 inactive
conformation); 90mM ATP (SAPK2a); 155mM ATP (MAPK2, PKBa); and 200mM ATP
(cSRC, IR).
Detailed Description of the Preferred Embodiment
A. Definitions
Unless defined otherwise, technical and scientific terms used herein have the
same
meaning as commonly understood by one of ordinary skill in the art to which
this invention
belongs. Singleton et al., Dictionary of Microbiology and Molecular Biolo~y
2nd ed , J. Wiley
& Sons (New York, NY 1994), and Constituents of Si~nalin~ Pathways and their
Chemistry,
New Science Press Ltd. 2002, provide one skilled in the art with a general
guide to many of the
teens used in the present application.
The term "protein kinase" is used to refer to an enzyme that catalyzes the
transfer of the
y-phosphoryl group of ATP (ATP-Mg2+ complex) to the oxygen atom of the
hydroxyl group of
serine, threonine or tyrosine residues in peptides and polypeptides
(substrates).
The term "tyrosine kinase" is used to refer to an enzyme that catalyzes the
transfer of the
y-phosphoryl group from an ATP-Mg2+ complex to the oxygen atom of the hydroxyl
group of
tyrosine residues in another protein (substrate).
-6-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
The term "serine-threonine kinase" is used to refer to an enzyme that
catalyzes the
transfer of the y-phosphoryl group from an ATP-Mg2+ complex to the oxygen atom
of the
hydroxyl group of serine/threonine residues in another protein (substrate).
The teen "dual specificity kinase" is used to refer to kinases that have the
unusual ability
S to phosphorylate both tyrosine and serine/threonine residues of targeted
protein substrates, and
typically function at pivotal positions in signal transduction pathways.
The term "phosphoryl donor" refers to an ATP-Mg2+ complex, where the divalent
Mg2+
ion helps orient the nucleotide and shields' the negative charges on its (3-
and y phosphoryl
groups, reducing electrostatic repulsion of attacking nucleophiles.
The terns "phosphoacceptor" is used to refer to an atom with a free electron
pair that
serves as the nucleophile to attack ATP-Mg2+ (e.g., the oxygen atom of the
deprotonated
hydroxyl groups of the side chains of Ser, Thr, or Tyr residues in a protein).
For example, in the
substrates of tyrosine kinases, the phosphoacceptor is the oxygen atom of the
deprotonated
hydroxyl group of the side chain of a tyrosine (Tyr) residue.
The term "activation loop" is used to a highly variable segment in protein
kinases,
situated between the DFG motif and the APE motif that contains the sites of
activating
phosphorylation in nearly all protein kinases.
The terms "catalytic loop" and "catalytic domain" are used interchangeably and
refer to
residues in conserved protein kinase motif VIb, which contains an invariant
aspartic acid (Asp)
residue that serves as the catalytic base in phosphotransfer and a nearly
invariant arginine (Arg)
residue, that makes electrostatic contact with phosphorylated residues in the
activation loop,
leading to the catalytically active state of the kinase.
The term "APE motif' is used to refer to the residues in conserved protein
kinase motif
VIII, which contains an invariant glutamic acid (Glu) residue that caps a
small helix and an
invariant proline (Pro) residue that terminates the same helix.
The term "DFG motif' is used to refer to the residues in conserved protein
kinase motif
VII, which contains an invariant aspartic acid (Asp) residue that helps mold
the active site by
forming hydrogen-bonds with the invariant lysine (Lys) in motif II and an
invariant asparagine
(Asn) residue in motif VIb, thus helping stabilize the conformation of the
catalytic loop.
The term "inactive conformation," as used herein, refers to a catalytically
inactive state of
the protein. For example, a protein kinase is in an inactive conformation when
the activation
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
loop is not phosphorylated. A kinase is said to be locked in an inactive
conformation when the
kinase assumes the inactive conformation and does not phosphorylate its
intended substrate.
An "inactivation site" on a protein kinase as used herein is any site on the
kinase that,
when occupied by a ligand, adversely affects the formation of the active
conformation or
otherwise impairs the kinase's ability to phosphorylate its intended
substrate. Alternatively, an
inactivation site when referring to an amino acid residue on the kinase is a
residue that is directly
or indirectly involved in the phosphorylation of the activation loop, and/or
in the presentation or
transfer of the y-phosphoryl group of ATP (ATP-Mg2+ complex) to the substrate
of the protein
kinase, and/or in any other interaction between the protein kinase and its
substrate.
A kinase inhibitor binds "preferentially" to an inactive conformation of a
target kiriase, if
its binding affinity to the inactive conformation is at least two fold of its
binding affinity to the
active conformation.
A "ligand" as defined herein is an entity which has an intrinsic binding
affinity for the
taxget. The ligand can be a molecule, or a portion of a molecule which binds
tile target. The
15. ligands are typically small organic molecules which have an intrinsic
binding affinity for the
target molecule, but may also be other sequence-specific binding molecules,
such as peptides (D
L- or a mixture of D- and L-), peptidomimetics, complex carbohydrates or other
oligomers of
individual units or monomers which bind specifically to the target. The term
also includes
various derivatives and modifications that are introduced in order to enhance
binding to the
target. Ligands that inhibit a biological activity of a target molecule are
called "inhibitors" of the
target.
A "ligand candidate" is a compound that has a moiety that is capable of
forming a
covalent bond with a reactive group on a target kinase or with a reactive
group on a target-
kinase-SME covalent complex. A ligand candidate becomes a ligand of a target
once it is
determined that it has an intrinisc binding affinity for the target.
The term "inhibitor" is used in the broadest sense and includes any ligand
that partially or
fully bloclcs, inhibits or neutralizes a biological activity exhibited by a
target protein kinase. In a
similar manner, the term "agonist" is used in the broadest sense and includes
any ligand that
mimics a biological activity exhibited by a target protein kinase.
A "binding site of interest" on a target protein kinase as used herein is a
site to which a
specific ligand binds. Typically, the molecular interactions between the
higand and the binding
site of interest on the target are non-covalent, and include hydrogen bonds,
van der Waals
_g_
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
interactions and electrostatic interactions. On target protein kinases, the
binding site of interest
broadly includes the amino acid residues involved in binding of the target to
a molecule with
which it forms a natural complex iTZ vivo or in vitf~o.
"Small molecules" are usually less than about 10 kDa molecular weight, and
include but
are not limited to synthetic organic or inorganic compounds, peptides,
(poly)nucleotides,
(oligo)saccharides and the like. Small molecules specifically include small
non-polymeric (i.e.
not peptide or polypeptide) organic and inorganic molecules. Many
pharmaceutical companies
have extensive libraries of such molecules, which can be conveniently screened
by using the
extended tethering approach of the present invention. Preferred small
molecules have molecular
weights of less than about 1000 Da, more preferably about 500 Da, and most
preferably about
250 Da.
The phrase "Small Molecule Extender" (SME) as used herein refers to a small
organic
molecule having a molecular weight of from about 75 to about 1,500 daltons and
having a first
functional group reactive with a nucleophile or electrophile on a protein
kinase target and a ,
second functional group reactive with a ligand ca~zdidate or members of a
library of ligand
candidates. Preferably, the first functional group on one end of the SME is
reactive with a
nucleophile on a protein kinase (capable of forming an irreversible or
reversible covalent bond
with such nucleophile), and the reactive group at the other end of the SME is
a free or protected
thiol or a group that is a precursor of a free or protected thiol.
The phrase "reversible covalent bond" as used herein refers to a covalent bond
which can
be broken, preferably under conditions that do not denature the target.
Examples include,
without limitation, disulfides, Schiff bases, thioesters, and the like.
The ternz "reactive group" with reference to a ligand is used to describe a
chemical group
or moiety providing a site at which a covalent bond with the ligand candidates
(e.g. members of
a library or small organic compounds) may be formed. Thus, the reactive group
is chosen such
that it is capable of forniing a covalent bond with members of the library
against which it is
screened.
The phrases "modified to contain" and "modified to possess" are used
interchangeably,
and refer to making a mutant, variant or derivative of the target, or the
reactive nucleophile or
electrophile, including but not limited to chemical modifications. For
example, in a protein one
can substitute an amino acid residue having a side chain containing a
nucleophile or electrophile
_9_
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
for a wild-type residue. Another example is the conversion of the tluol group
of a cysteine
residue to an amine group.
The term "reactive nucleophile" as used herein refers to a nucleoplule that is
capable of
forming a covalent bond with a compatible functional group on another molecule
under
conditions that do not denature or damage the target. The most relevant
nucleophiles are thiols,
alcohols, and amines. Similarly, the term "reactive electroplule" as used
herein refers to an
electrophile that is capable of forming a covalent bond with a compatible
functional group on
another molecule, preferably under conditions that do not denature or
otherwise damage the
target. The most relevant electrophiles are imines, carbonyls, epoxides,
aziridines, sulfonates,
and hemiacetals.
A "first .binding site of interest" on a target protein kinase refers to a
site that can be
contacted by at least a portion of the SME when it is covalently bound to the
reactive nucleophile
or electrophile. The first binding site of interest may, but does not have to
possess the ability to
form a bond with the SME.
The phrases "group reactive with the nucleophile," "nucleophile reactive
group," "group
reactive with an electrophilc," and "electrophile reactive group," as used
herein, refer to a
functional group, e.g. on the SME, that can form a covalent bond with the
nucleophile/electrophile on the target protein kinase under conditions that do
not denature or
otherwise damage the target.
The term "protected thiol" as used herein refers to a thiol that has been
reacted with a
group or molecule to form a covalent bond that renders it less reactive and
which may be
deprotected to regenerate a free tluol.
The phrase "adjusting the conditions" as used herein refers to subjecting a
target protein
kinase, such as a tyrosine kinase, to any individual, combination or~series of
reaction conditions
or reagents necessary to cause a covalent bond to form between the ligand and
the target, such as
a nucleophile and the group reactive with the nucleophile on the SME, or to
break a covalent
bond already formed.
The term "covalent complex" as used .herein refers to the combination of the
SME and
the target, e.g. target protein kinase which is both covalently bonded through
the
nucleophile/electrophile on the target with the group reactive with the
nucleophile/electrophile
on the SME, and non-covalently bonded through a portion of the small molecule
extender and
the first binding site of interest on the target.
-10-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
The phrase "exchangeable disulfide linking group" as used herein refers to the
library of
molecules screened with the covalent complex displaying the thiol-containing
small molecule
extender, where each member of the library contains a disulfide group that can
react with the
thiol or protected thiol displayed on the covalent complex to form a new
disulfide bond when the
reaction conditions are adjusted to favor such thiol exchange.
The phase "highest affinity for the second binding site of interest" as used
herein refers to
the molecule having the greater thermodynamic stability toward the second site
of interest on the
target protein kinase that is preferentially selected from the library of
disulfide-containing library
members.
"Functional variants" of a molecule herein are variants having an activity in
conunon
with the reference molecule.
"Active" or "activity" means a qualitative biological andlor immunological
property.
The term amino acid "alteration" includes amino acid substitutions, deletions,
and/or
insertions.
B. Detailed Description
In one aspect, the present invention provides a method for locking a protein
kinase in an
inactive conformation. In another aspect, the invention concerns the
identification of inhibitors
that preferentially bind to the inactive conformation of a target protein
kinase.
Protein Kinases
Protein l~inases are enzymes that catalyze the transfer of the y-phosphoryl
group of ATP
(ATP-Mg2+ complex) to the oxygen atom of the hydroxyl group of serine,
threonine or tyrosine
residues in peptides and polypeptides (substrates). Protein kinases play a
crucial role in signal
transduction, cellular proliferation, differentiation, and various regulatory
mechanisms. About
3% of the total coding sequences within the human genome encode protein
lcinases.
While there are many different subfamilies within the broad grouping of
protein kinases,
they all share a common feature; they all act as ATP phosphotransferases. It
is, therefore, not
surprising that protein kinases share a very high degree of structural
similarity in the region
where the ATP is bound, the ATP binding pocket (which is also known as the
purine binding
pocket). Structural analysis of many protein kinases shows that the catalytic
domain, responsible
for the phosphotransfer activity, is very highly conserved. This domain is
comprised of two
lobes that are connected by a flexible hinge region. The amino-terminal lobe
is comprised of a
-11-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
single alpha helix and five beta sheets, while the carboxy-terminal lobe is
comprised of a four
alpha helix bundle and a flexible loop called the activation loop. The ATP
binding pocket is
formed at the interface between these two lobes. There are several highly
conserved residues,
including an invariant catalytic triad consisting of a single lysine and two
aspartic acids. The
lysine of this catalytic triad is responsible for properly positioning the y-
phosphate of ATP with
the hydroxyl group of the residue in the substrate to which it is transferred
(phosphoacceptor
residue), while the first aspartic acid acts as a general base catalyst in the
phosphotransfer
reaction. Strikingly, these three crucial residues span the two lobes of the
catalytic domain.
Furthermore, the two aspartic acid,residues within the catalytic triad are
separated from each
other by a second flexible region called the activation loop. To allow the
phosphotransfer
reaction, the structure of a substrate must conform to the geometric
constraints, surface
electrostatics, and other features of the active site of the corresponding
protein kinase. In turn,
substrate binding can induce structural changes in a kinase that stimulate its
catalytic activity. In
particular, for enzyme - substrate interactions, residues within the
activation loop and the
catalytic loop need to be made available to make contacts with side chains in
a substrate.
Outside the conserved motifs crucial for catalytic activity (such as the ATP
binding site), there
are sequence differences in both loops that are critical for substrate
recognition.
Structural States of Kinases and Regulation of Kinase Activity
Proper regulation of protein kinase activity in a cell is critical, and
kinases in a resting
cell generally exist in an inactive conformation. In this inactive
conformation, the catalytic triad
may be oriented in a manner that will not catalyze phosphate transfer, the
substrate binding cleft
may be occluded by the flexible activation loop, or both. Relative to the
active conformation, the
amino- and carboxy-terminal lobes in the inactive conformation may be opened
up with resultant
widening the active site cleft, twisted with resultant tortioning of the
active site cleft, or both.
Only when cells are confronted with specific stimuli do these kinases
transition to a catalytically
active conformation. Transition to the active conformation almost invariably
involves
phosphorylation of a residue in the activation loop, and subsequent formation
of a salt bridge
with a conserved argininc immediately adjacent to the catalytic aspartic acid.
The resultant
rearrangement of the activation loop, stabilized by this newly formed salt
bridge, stabilizes a
catalytically active confornlation characterized by: proper amino- and carboxy-
terminal domain
orientation, proper orientation of the y-phosphate of ATP to allow for
phosphoryl transfer,
-12-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
opening of the substrate binding site, and a favorable electrostatic
environment for the aspartic
acid mediated base catalysis.
While a common function dictates that the structure at the catalytic center is
highly
conserved among kinases in the active confornlation, this is not the case with
kinases in the
inactive conformation. In fact, structural studies of the active and inactive
forms of kinases
reveal that kinases that have highly conserved active site architectures when
in the active
conformation show considerable structural diversity in the same region when
they are in the
inactive conformation. This is particularly true of a region immediately
adjacent to the ATP
binding site that has been ternzed the adaptive binding region. For example,
Gleevec binds to the
ATP binding pocket of the Abl kinase but only when it is in the inactive form.
More
importantly, the bulk of Gleevec binds to the adaptive binding pocket that is
only revealed when
Abl kinase is in the inactive form. Thus, specifically targeting the inactive
form of the kinase
provides a path for mitigating many of the difficulties in developing kinase
inhibitors as drugs.
An important protein kinase target for drug development is the Tyr kinase
EGFRl
(Ullrich et al., Nature 309:418-425 (1984); SwissProt accession code P00533).
EGFRl, a
validated target for chemotherapeutics, is a cell surface receptor that
contains an extracellular
ligand binding domain and an intracellular tyrosine kinase domain. It is a key
regulator of cell
growth, survival, proliferation, and differentiation in epithelial cells. The
binding of a number of
ligands activates EGFR1, including EGF, TGF-a,, amphiregulin, (3-cellulin, and
epiregulin.
Ligand binding leads to receptor dimerization, autophosphorylation at a number
of tyrosine
residues including Tyr845 in the activation loop, and subsequent recruitment
pf substrate
proteins and stabilization of the active conformation of the kinase domain.
EGFRl, -in this
activated state, phosphorylates a variety of downstream targets to propagate
the extracellular
stimulus of ligand binding to the eventual transcriptional upregulation of a
variety of growth
regulatory genes and resultant cell proliferation. In normal cells, EGFRI
regulates cell growth in
a tightly controlled manner. However, overexpression of EGFRl has been
observed in a large
number of tumor types, including breast, bladder, colon, lung, squamous cell
head and neck,
ovarian, and pancreatic cancers. A clear role for EGFRI upregulation in the
initiation and
progression of a variety of cancers has lead to an intense seaxch for
therapeutics that inhibit
signal transduction via EGFRl .
Another important protein kinase target for drug development is the dual
specificity
kinase MEKl (Seger et al., J. Biol. Chern. 267: 25628-31 (1992); Swiss Prot
accession code
-13-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Q02750). It is the central kinase in the mitogen activated Ras -~ Raf -~ MEK -
> ERK signal
transduction cascase (also referred to as the MEK -~ ERK pathway). Conditional
activation of
this pathway transmits mitogenic and cell survival signals from a number of
growth factors and
receptors, including EGFR, VEGFR, PDGFR and FGFR. Overexpression or
consitutive
activation of these same growth factors and receptors in tumors correlates
with a poor prognosis
in cancer patients.
Further validation of MEKl as a general cancer therapeutic target comes from
the
development of two specific MEKl inhibitors. The first, PD98059, is a
specific, albeit relatively
insoluble, MEK1 inhibitor. Though not a therapeutic candidate, this compound
has been used in
over 2,500 publications validating the Ras-~Raf-~MEK-~ERK pathway as a
critical pathway in
transformed cells, and confirming that inhibition of this pathway is
sufficient to reverse the
transformed phenotype of cells that have upregulated this pathway (e.g., cells
transformed with
an activated Ras mutant). The second, PD184352 (also known as CI-1040), is a
specific MEKl
inhibitor currently in Phase II trials for use as a therapeutic in a variety
of solid tumors.
Preclinical and Phase I clinical data have clearly demonstrated that the
MEK~ERK pathway can
be inlubited in vivo, that inhibition of this pathway does not cause general
toxicity, and that
inhibition of this pathway correlates with tumor regression in multiple mouse
xenograft cancer
models.
In addition, the MEK~ERK pathway generally confers resistance to apoptosis.
Thus, it
is believed that cancers with increased MEK~ERK signaling will be more
resistant to
chemotherapy-induced apoptosis, and inhibition of MEKl activity will increase
the sensitivity of
these cancers to traditional chemotherapeutics. In studies in acute and
chronic myelogeneous
leulcemic cell lines, the MEK1 inhibitors PD98059 and PD184352 induced
apoptosis i11 tumor
cell lines in a manner that directly correlated with the level of ERK
activation. As predicted,
these MEK1 inhibitors acted synergistically with a variety of chemotherapeutic
cytotoxins,
including ara-C, cisplatin, and paclitaxel.
Another important family of protein kinases is the Src family. First of all,
the Src family
kinases are well validated casual agents' in a variety of cancers. Second, no
current small
molecule therapeutics effectively targets Src kinases in humans. Finally, Src
family kinases are
the best structurally characterized of all tyrosine kinases.
A representative member of this family, the Tyr kinase Lck (Perlmutter et al.,
J. Cell.
Biochefn. 38:117-126 (1988); Swiss Prot acession code Pb6239), is a cytosolic
tyrosine kinase,
-14-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
which is expressed primarily in T-cells where it is centrally involved in
transducing a signal from
the T-cell receptor (TCR). Lck is fond associated when the inner plasma
membrane where it
phosphorylates the CD3 and zeta chains of the TCR in response to antigenic
stimulation,
initiating a cascade of signal transduction events that eventually result in a
clonal proliferation of
the stimulated T-cell. Thus, Lck is well known as a therapeutic target for
immunological
disorders, such as graft versus host disease. However, Lck is also validated
cancer therapeutic
target. In humans, some neuroblastomas and non-Hodgkin's lymphomas show
chromosomal
abnormalities and translocations in the region of the Lck gene. In at least
one case that has been
molecularly characterized, the "derivative I chromosome" translocation focuses
the
transcriptional regulatory region of the beta T-cell receptor gene with the
coding sequence of
Lck, resulting in increased levels of Lck kinase in patients with T-cell acute
lymphoblastic
leukemia, much like the Philadelplua Chromosome translocation which
upregulates Abl
expression causing CML.
In addition to their value as therapeutic targets, Src family kinases are
extremely well
characterized structurally. Crystal coordinates are' publicly available for
three family members,
hematopoietic cell kinase (Hck), Src, and Lclc, covering both the active and
the inactive
conformational. Furthermore, Lck is known to express well in baculovirus and
to crystallize
readily.
Other illustrative examples of kinase targets include but are not limited to:
Ser/Thr kinase AKTl (Jones et al., PNAS 88: 4171-4175 (1991); Swiss Prot
accession
code P31749);
Ser/Thr kinase AKT2 (Jones et al., Cell Regul. 2(12): 1001-1009 (1991); Swiss
Prot
accession code P31751);
Ser/Thr kinase AKT3 (Brodbeck et al., J. Biol. ClZerrz. 274(14): 9133-9136
(1999); Swiss
Prot acession code Q9Y243);
Tyr kinase BLK (Islam et al., J. Inanautzol. 154(3): 1265-1272 (1995);Swiss
Prot acession
code P51451);
Tyr kinase BTK (Vetrie et al., NatuJ°e 361: 226-233 (1993); Swiss Prot
accession code
Q06187);
Ser/Thr kinase CDKl (Lee et al., Nature 327: 31-35 (1987); Swiss Prot
accession code
P06493);
-15-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Ser/Thr kinase CDK2 (Elledge et al., EMBO J. 10(9): 2653-2659 (1991); Swiss
Prot
accession code P24941 );
Ser/Thr kinase CDK3 (Meyerson et al., EMBO J. 11(8): 2909-2917 (1992); Swiss
Prot
accession code Q00526);
Ser/Thr kinase CDK4 (Hanks et al., PNAS 84: 388-392 (1987); Swiss Prot
accession
code P11802);
Ser/Thr kinase CDKS (Meyerson et al., EMBO J. 11(8): 2909-2917 (1992); Swiss
Prot
accession code Q00535);
Ser/Tlm kinase CDK6 (Meyerson et al., EMBO J. 11(8): 2909-2917 (1992); Swiss
Prot
accession code Q00534);
Ser/Thr kinase CDK7 (Tassan et al, J. Cell Biol. 127(2): 467-478 (1994); Swiss
Prot
accession code P50613);
Ser/Thr kinase CDK8 (Tassan et al., PNAS 92(19): 8871-8875 (1995); Swiss Prot
accession code P49336);
Ser/Thr kinase CDK9 (Gram et al., PNAS 91: 3834-3838 (1994); Swiss Prot
accession
code P50750);
Tyr kinase CSK (Brauninger et al., Oncogene 8(5): 1365-1369 (1993); Swiss Prot
accession code P41240);
Tyr kinase ERB2 (Semba et al., PNAS 82: 6497-6501 (1985); Swiss Prot accession
code
P04626);
Tyr kinase ERB4 (Plowman et al., PNAS 90(5): 1746-1750 (1993); Swiss Prot
accession
code Q15303);
Ser/Thr kinase ERKl (Charest et al., Mol. Cell. Biol. 13(8): 4679-4690 (1993);
Swiss
Prot accession code P27361);
Ser/Thr kinase ERK2 (Owaki et al., Biochem. Bioplzys. Res. Corszmuu. 182(3):
1416-1422
(1992); Swiss Prot accession code P28482);
Ser/Thr lcinase ERK3 (Zhu et al., Mol. Cell. Biol. 14(12): 8202-8211 (1994);
Swiss Prot
accession code Q16659);
Ser/Thr kinase ERK4 (Gonzalez et al., FEBS Lett. 304: 170-178 (1992); Swiss
Prot
accession code P31152);
Ser/Thr lcinase ERKS (Zhou et al., J. Biol. Chefn. 270(21): 12665-12669
(1995); Swiss
Prot accession code Q 13164);
-16-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Ser/Thr kinase ERK6 (Lechner et al., PNAS 93(9): 4355-4359 (1996); Swiss Prot
accession code P53778);
Tyr kinase FAKl (Whitney et al., DNA Cell Biol. 12(9): 823-830 (1993); Swiss
Prot
accession code Q05397); .
Tyr kinase FGFRl (Isacchi et al., Nucleic Acids Res. 18(7): 1906 (1990); Swiss
Prot
accession code P11362);
Tyr kinase FGFR2 (Houssaint et al., PNAS 87(20): 8180-8184 (1990); Swiss Prot
accession code P21802);
Tyr kinase FGFR3 (Keegan et al., PNAS 88(4): 1095-1099 (1991); Swiss Prot
accession
code P22607);
Tyr kinase FGFR4 (Partanen et al., EMBO J. 10(6): 1347-1354 (1991); Swiss Prot
accession code P22455);
Tyr kinase FYN (Semba et al., PNAS 83: 5459-5463 (1986); Swiss Prot accession
code
P06241 );
Tyr kinase HCK (Quintrell et al., Mol. Cell. Biol. 7(6): 2267-2275 (1987);
Swiss Prot
accession code P08631);
Ser/Thr kinase IKK-a (Regnier et al., Cell 90(2): 373-383 (1997); Swiss Prot
accession
code O 15111 );
Ser/Thr kinase IKK-b (Woronicz et al., Science 278: 866-869 (1997); Swiss Prot
accession code 014920);
Ser/Thr kinase IKK-a (Nagase et al., DNA Res. 2(4): 167-174 (1995); Swiss Prot
accession code Q14164);
Tyx kinase JAKl (Wilks et al., Mol. Cell. Biol. 11: 2057-2065 (1991); Swiss
Prot
accession code P23458);
Tyr kinase JAK2 (Saltzman et al., BioclzeyfZ. Bioph,~s. Res. Cof~znzuh.
246(3): 627-633
(1998); Swiss Prot accession code 060674);
Tyr kinase JAK3 (Kawamura et al., PNAS 91: 6374-6378 (1994); Swiss Prot
accession
code P52333);
Ser/Thr kinase JNK1 (Derijard et al., Cell 76: 1025-1037 (1994); Swiss Prot
accession
code P45983);
Ser/Tlir kinase JNK2 (Sluss et al., Mol. Cell. Biol. 14: 8376-8384 (1994);
Swiss Prot
accession code P45984);
-17-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Ser/Thr kinase JNK3 (Mohit et al., Neu~~on 14(1): 67-78 (1995); Swiss Prot
accession
code P53779);
Tyr kinase LCK (Perhnutter et al., J. Cell. Biochem. 38(2): 117-126 (1988);
Swiss Prot
accession code P06239);
Tyr kinase LYN (Yamanashi et al., Mol. Cell. Biol. 7(1): 237-243 (1987); Swiss
Prot
accession code P07948);
Ser/Thr kinase MAPK (Lee et al., Nature 372: 739-746 (1994); Swiss Prot
accession
code Q16539);
Ser/Thr kinase NIK (Malinin et al., Nature 385: 540-544 (1997); Swiss Prot
accession
code Q99558);
Ser/Thr kinase PAKl (Ottilie et al., EMBO J. 14(23): 5908-5919 (1995); Swiss
Prot
accession code P50527);
Ser/Thr kinase PAK2 (Swiss Prot accession code Q13177);
Ser/Thr kinase PAK3 (Allen et al., Nat. Genet. 20(1): 25-30 (1998); Swiss Prot
accession
code 075914);
Ser/Thr kinase PAK4 (Abo et al., EMBO J. 17(22): 6527-6540 (1998); Swiss Prot
accession code 096013);
Ser/Thr kinase PAKS (Swiss Prot accession code Q9P286);
Tyr kinase PDGFR-a (Matsui et al, Science 243: 800-804 (1989); Swiss Prot
accession
code P 16234);
Tyr kinase PDGFR-b (Gronwald et al., PNAS 85(10): 3435-3439 (1988); Swiss Prot
accession code P09619);
Ser/Thr kinase PIM1 (Reeves et al., Gene 90(2): 303-307 (1990); Swiss Prot
accession
code P11309);
Ser/Thr kinase A-Raf (Beck et al., Nucleic Acids Res. 15(2): 595-609 (1987);
Swiss Prot
accession code P10398);
Ser/Thr kinase B-Raf (Sithanandam et al., Oncogene 5: 1775-1780 (1990); Swiss
Prot
accession code P15056);
Ser/Thr kinase C-Raf (Bonner et al, Nucleic Acids Res. 14(2): 1009-1015
(1986); Swiss
Prot accession code P04049);
Tyr kinase SRC (Swiss Prot accession code P12931);
-18-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Tyr kinase SRC2 (c-FGR) (Katamine et al., Mol. Cell. Biol. 8(1): 259-266
(1988); Swiss
Prot accession code P09769);
Tyr kinase STK1 (FLT3) (Small et al., PNAS 91: 459-463 (1994); Swiss Prot
accession
code P36888);
Tyr kinase SYK (Yagi et al., Biochem. Biophys. Res. Comsnun. 200(1): 28-34
(1994);
Swiss Prot accession code P43405);
Tyr kinase TEC (Sato et al., Leukerrzia 8(10): 1663-1672 (1994); Swiss Prot
accession
code P42680);
Ser/Thr kinase TFGRl (Franzen et al., Cell 75(4): 681-692 (1993); Swiss Prot
accession
code P36897);
Ser/Thr kinase TGFR2 (Lin et al., Cell 68(4): 775-785 (1992); Swiss Prot
accession code
P37173);
Tyr kinase TIE1 (Partanen et al., Mol. Cell. Biol. 12(4): 1698-1707 ,(1992);
Swiss Prot
accession code P35590);
Tyr kinase TIE2 (Ziegler et al., Oncogeyze 8(3): 663-670 (1993); Swiss Prot
accession
code Q02763);
Tyr kinase VEGFRI (Yamane et al., Oncogerze 9(9): 2683-2690 (1994); Swiss Prot
accession code P53767);
Tyr kinase VEGFR2 (Swiss Prot accession code P35968);
Tyr kinase VEGFR3 (Galland et al., Oncogene 8(5): 1233-1240 (1993); Swiss Prot
accession code P35916);
Tyr kinase YES (Sukegawa et al., Mol. Cell. Biol. 7: 41-47 (1987); Swiss Prot
accession
code P07947); and,
Tyr kinase ZAP-70 (Chan et al., Cell 71: 649-662 (1992); Swiss Prot accession
code
P43043).
Identification of Protein Kinase Inhibitors Preferential Binding to the
Inactive Conformation
In an important aspect, the present invention provides methods for identifying
protein
kinase inhibitors that specifically target kinases in the inactive
conformation. There are at least
three principle reasons of screening for such inhibitors: (1) the majority of
kinases in a cell exist
in this conformation; (2) relative to the active conformation, kinases in the
inactive conformation
exhibit greater structural diversity; and (3) opening and tortioning of the
active site region in this
-19-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
conformation often results in a decreased affinity for ATP, the primary
intracellular competitor
for small molecule kinase inhibitors.
Traditional high throughput screening techniques detect phosphoryl transfer to
a substrate
molecule by an activated kinase. As such, these assays primarily detect
inhibitors that bind to
the active conformation of kinases and make the identification of inhibitors
targeting the inactive
conformation very unlikely. In contrast, the present invention provides an
efficient, high-
throughput method to identify kinase inhibitors that bind preferentially to
the inactive
conformation of protein kinases. This method includes the step of locking the
protein kinase in
its inactive conformation, and using Tethering to identify inhibitors
specifically targeting the
inactive kinase conformation.
a. Locking kinases in an inactive conformation
In order to identify kinase inhibitors preferentially binding to the inactive
conformation
of the target kinase, according to the invention a target protein kinase is
locked in a catalytically
inactive conformation by introducing one or more amino acid alterations at an
inactivating site
such that the lcinase cannot exert its kinase activity, in most cases because
the alteration inhibits
the phosphorylation of the activation loop. The alteration may target any site
participating
(directly or indirectly) in the fornlation of a catalytically active state of
the kinase. For example,
the alteration may take place at or near amino acid residues participating in
the phosphorylation
of the activation loop, and/or in the presentation or transfer of the y-
phosphoryl group of ATP to
the substrate of the protein lcinase, and/or in any other interaction between
the protein kinase and
its substrate. Alterations within or in the vicinity of the catalytic loop,
e.g. the ATP binding site
including the catalytic triad, the substrate binding channel, a cofactor
binding site, if any,
residues involved in hydrogen bond/acceptor interactions, and/or docking of
the substrate on the
tyrosine kinase are particularly preferred.
For purposes of shorthand designation of the protein kinase variants described
herein, it
is noted that numbers refer to the position of the altered amino acid residue
along the amino acid
sequences of respective wild-type protein kinases. Amino acid identification
uses the single-
letter alphabet of amino acids, as follows:
Asp D Aspartic acid
Ile I Isoleucine
-20-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Thr T Threonine
Leu L Leucine
Ser S Serine
Tyr Y Tyrosine
Glu E Glutamic acid
Phe F Phenylalanine
Pro P Proline
His H Histidine
Gly G Glycine
Lys K Lysine
Ala A Alanine
Arg R Arginine
Cys G Cysteine
Trp W Tryptophan
Val V Valine
Gln Q Glutamine
Met M Methionine
Asn N Asparagine
The designation for a substitution variant herein consists of a letter
followed by a number
followed by a letter. The first (leftmost) letter designates the amino acid in
the wild-type protein
kinase. The number refers to the amino acid position where the amino acid
.substitution is being
made, and the second (right-hand) letter designates the amino acid that is
used to replace the
wild-type amino acid at that position. The designation for an insertion
variant consists of the
letter i followed by a number designating the position of the residue in wild-
type protein kinase
before wluch the insertion starts, followed by one or more capital letters
indicating, inclusively,
the insertion to be made. The designation for a deletion variant consists of
the letter d followed
by the number of the start position of the deletion to the number of the end
position of the
deletion, with the positions being based on the wild-type protein kinase.
Multiple alterations are
separated by a cormna in the notation for ease of reading them.
In one embodiment, the kinase is locked in an inactive conformation by
mutating one or
more residues selected from the group consisting of the invariant aspartic
acid in the catalytic
-21-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
loop; the arginine in the catalytic loop; the invariant aspartic acid in the
DFG motif; and the
invariant lysine in motif II. In preferred embodiments, one or more of these
residues are
substituted by an alanine residue.
Illustrative examples of kinase mutants where the invariant aspartic acid
residue in the
S catalytic loop is mutated to X (wherein X denotes any amino acid residue
other than aspartic
acid) include any combination of the following:
D274X AKTl; D275X AKT2; D271X AKT3; D359X BLK; D521X BTK; D128X
CDKl ; D 127X CDK2; D 127X CDK3; D 140X CDK4; D 126X CDKS; D 145X CDK6; D 13
7X
CDK7; D151X CDKB; D149X CDK9; D314X CSK; D837X EGFRl; D845X ERB2; D843X
ERB4; D166X ERKl; D149X ERK2; D152X ERK3; D149X ERK4; D181X ERKS; D153X
ERK6; D546X FAKl; ID623X FGFRl; .D626X FGFR2; D617X FGFR3; D612X FGFR4;
D389X FYN; D381X HCK; D144X IKK-a; D145X IKK-b; D135X IKK-e; D991X JAKl;
D976X JAK2; D949X JAK3; D151X JNKl; D151X JNK2; D189X JNK3; D363X Lck; D366X
LYN; D150X MAPK; D190X MEK1; DS15X NIK; D389X PAKl; D368X PAK2; D387X
PAK3; D440X PAK4; D568X PAKS; D818X PDGFR-a; D826X PDGFR-b; D167X PIMl;
D429X A-Raf; D575X B-Raf; D468X C-Raf; D388X SRC; D382X SRC2; D811X STKl;
D494X SYK; D489X TEC; D333X TGFRl; D379X TGFR2; D979X TIE1; D964X TIE2;
D1022X VEGFR1; D1028X VEGFR2; D1037X VEGFR3; D386X YES; D461X ZAP-70.
Illustrative examples of kinase mutants where the arginine residue in the
catalytic loop is
mutated to X (wherein X denotes any amino acid residue other than arginine)
include any
combination of the following:
R273X AKTl; R274X AKT2; R270X AKT3; R358X BLK; R520X BTK; R127X
CDKl; R126X CDK2; R126X CDK3; R139X CDK4; R125X CDKS; R144X CDK6; R136X
CDK7; R150X CDK8; R148X CDK9; R313X CSK; R836X EGFRl; R844X ERB2; R842X
ERB4; R165X ERKl; R148X ERK2; R151X ERK3; R148X ERK4; R180X ERKS; R152X
ERK6; R545X FAKE R622X FGFRl; R625X FGFR2; R616X FGFR3; R611X FGFR4; R388X
FYN; R380X HCK; R143X IKK-a; R144X IKK-b; R134X IKK-e; R990X JAKl; R975X JAK2;
R948X JAK3; R150X JNKl; R150X JNK2; R188X JNK3; R362X Lck; R365X LYN; R149X
MAPK; R189X MEKl; R514X NIK; R388X PAKl; R367X PAK2; R386X PAK3; R439X
PAK4; R567X PAKS; R817X PDGFR-a; R825X PDGFR-b; R166X PMl; R428X A-Raf;
R574X B-Raf; R467X C-Raf; R387X SRC; R381X SRC2; R810X STKl; R493X SYK; R488X
-22-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
TEC; R322X TGFRl; R378X TGFR2; R978X TIE1; R963X TIE2; 81021 VEGFRl; R1027X
VEGFR2; R1036X VEGFR3; R395X YES; R460X ZAP-70.
Illustrative examples of kinase mutants where the invariant aspartic acid in
the DFG
motif is mutated to X (wherein X denotes any amino acid residue other than
aspartic acid)
include any combination of the following:
D292X AKT1; D293X AKT2; D289X AKT3; D377X BLK; D539X BTK; D146X
CDKl; D145X CDK2; D145X CDK3; D158X CDK4; D144X CDKS; D163X CDK6; D155X
GDK7; D173X CDK8; D167X CDK9; D332X CSK; D855X EGFR1; D863X ERB2; D861X
ERB4; D184X ERKl; D167X ERK2; D171X ERK3; D168X ERK4; D199X ERKS; D171X
ERK6; D564X FAK1; D641X FGFRI; D644X FGFR2; D635X FGFR3; D630X FGFR4;
D407X FYN; D399X HCK; D165X IKK-a; D166X IKK-b; D157X IKK-e; D1009X JAKl;
D994X JAK2; D967X JAK3; D169X JNKl; D169X JNK2; D207X JNK3; D381X Lck; D384X
LYN; D168X MAPK; D208X MEK1; D534X NIK; D407X PAKl; D386X PAK2; D405X
PAK3; D458X PAK4; D586X PAKS; D836X PDGFR-a; D844X PDGFR-b; D186X PIMI;
D447X A-Raf; D593X B-Raf; D486X C-Raf; D406X SRC; D400X SRC2; D829X STKl;
D512X SYK; D507X TEC; D351X TGFRl; D397X TGFR2; D997X TIE1; D982X TIE2;
D1040X VEGFRl; D1046X VEGFR2; D1055X VEGFR3; D414X YES; D479X ZAP-70.
Illustrative examples of kinase mutants where the invariant lysine in motif II
is mutated
to X (wherein X denotes any amino acid residue other than lysine) include:
K179X AKT1; K181X AKT2; K1~77X AKT3; K268X BLK.; K430X BTK; K33X CDKI;
K33X CDK2; K33X CDK3; K35X CDK4; K33X CDKS; K43X CDK6; K41X CDK7; K52X
CDKB; K48X CDK9; K222X CSK; K745X EGFR1; K753X ERB2; K751X ERB4; K71X
ERKl; K54X ERK2; K49X ERK3; K49X ERK4; K83X ERKS; K56X ERK6; K454X FAKI;
K514X FGFRl; K517X FGFR2; K508X FGFR3; K503X FGFR4; K298X FYN; K290X HCK;
K44X IKK-a; K44X IKK-b; K38X.IKK-e; K896X JAKl; K882X JAK2; K855X JAK3; K55X
JNKl; K55X JNK2; K93X JNK3; K272X Lck; K274X LYN; K53X MAPK; K97X MEK1;
K429X NIK; K299X PAK1; K228X PAK2; K297X PAK3; K350X PAK4; K478X PAKS;
K627X PDGFR-a; K634X PDGFR-b; K67X PIMl; K336X A-Raf; K482X B-Raf; K375X C-
Raf; L297X SRC; K291X SRC2; K644X STKl; K402X SYK; K398X TEC; K232X TGFRI;
K277X TGFR2; K870X TIE1; K855X TIE2; K862X VEGFR1; K868X VEGFR2; K879X
VEGFR3; K305X YES; K369 ZAP-70.
-23-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
It will be appreciated that two or more of the foregoing or similar mutations
can be
combined to produce inactive kinase variants. Protein kinase variants
comprising two or more of
the above-listed mutations in any combination, including double, triple and
quadruple mutants
having mutations other than inactivating mutations described above, are
specifically within the
scope herein.
Those skilled in the art are well aware of various recombinant, chemical,
synthesis and/or
other techniques that can be routinely employed to modify a protein kinase of
interest such that it
possesses a desired number of free thiol groups that are available for
covalent binding to a ligand
candidate comprising a free thiol group. Such techniques include, for example,
site-directed
mutagenesis of the nucleic acid sequence encoding the target protein kinase.
Particularly
preferred is site-directed mutagenesis using polymerase chain reaction (PCR)
amplification (see,
for example, U.S. Pat. No. 4,683,195 issued 28 July 1987; and Current
Protocols In Molecular
Biolo~y, Chapter 15 (Ausubel et al., ed., 1991). Other site-directed
mutagenesis techniques are
also well known in the art and are described, for example, in the following
publications: Ausubel
et al., supt~a, Chapter 8; Molecular Cloning: A Laboratory Manual., 2nd
edition (Sambrook et al.,
1989); Zoller et al., Methods Enzymol. 100:468-500 (1983); Zoller & Smith, DNA
3:479-488
(1984); Zoller et al., Nuel. Acids Res., 10:6487 (1987); Brake et al., PNAS
81:4642-4646 (1984);
Botstein et al., Science 229:1193 (1985); I~unkel et al., Methods EnzynTOl.
154:367-82 (1987),
Adelma~i et al., DNA 2:183 (1983); and Carter et al., Nucl. Acids Res.,
13:4331 (1986). Cassette
mutagenesis (Wells et al., Gene, 34:315 [1985]), and restriction selection
mutagenesis (Wells et
al., Philos. Ti°ans. R. Soc. London SerA, 317:415 [1986]) may also be
used.
Amino acid sequence variants with more than one amino acid substitution may be
generated in one of several ways. If the amino acids are located close
together in the polypeptide
chain, they may be mutated simultaneously, using one oligonucleotide that
codes for all of the
desired amino acid substitutions. If, however, the anuno acids are located
some distance from
one another (e.g. separated by more than ten amino acids), it is more
difficult to generate a single
oligonucleotide that encodes all of the desired changes. Instead, one of two
alternative methods
may be employed. In the first method, a separate oligonucleotide is generated
for each amino
acid to be substituted. The oligonucleotides axe then annealed to the single-
stranded template
DNA simultaneously, and the second strand of DNA that is synthesized from the
template will
encode all of the desired amino acid substitutions. The alternative method
involves two or more
rounds of mutagenesis to produce the desired mutant.
-24-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
The nucleic acid encoding the desired kinase mutant is then inserted into a
replicable
expression vector for further cloning or expression. Expression and cloning
vectors are well
known in the art and contain a nucleic acid sequence that enables the vector
to replicate in one or
more selected host cells. The selection of an appropriate vector will depend
on 1) whether it is to
be used for DNA amplification or for DNA expression, 2) the size of the DNA to
be inserted into
the vector, and 3) the host cell to be transformed with the vector. Each
vector contains various
components depending on its function (amplification of DNA or expression of
DNA) and the
host cell for which it is compatible. The vector components generally include,
but are not
limited to, one or more of the following: a signal sequence, an origin of
replication, one or more
marker genes, an enhancer element, a promoter, and a transcription termination
sequence.
Suitable expression vectors, for use in combination with a variety of host
cells, are well known
in the art and are commercially available.
The protein kinase mutants can be produced in prokaryotic or eukaryotic host
cells,
including bacterial hosts, such as E. eoli, eukaryotic microbes, such as
filamentous fungi or
yeast, and host cells derived from multicellular organisms. Examples of
invertebrate cells
include insect cells such as Drosophila S2 and Spodoptera Sft?, as well as
plant cells, such as cell
cultures of cotton, corn, potato, soybean, petunia, tomato, and tobacco.
Numerous baculoviral
strains and corresponding permissive insect host cells, e.g. cells from
Spodopte~°a fi°ugipef-da,
Aedes aegypti, Aedes albopietus, Drosophila naelaf~~gaster, and Bombyx moy~i
have been
identified. A variety of viral strains for transfection of insect host cells
are publicly available,
including for example variants of Autog~apha California NPV and Bonzbyx f~aot-
i NPV strains.
Further host cells include vertebrate cells. Examples of suitable mammalin
host cell lines
include, without limitation, human embryonic kidney cell line 293, Chinese
hamster ovary
(CHO) cells, etc.
Host cells are transformed with the expression or cloning vectors encoding the
desired
protein kinase mutants, and cultured in conventional nutrient media modified
as appropriate for
inducing promoters, selecting transformants, or amplifying the genes encoding
the desired
sequences.
b. Tethering
According to the present invention, the protein kinases locked in inactive
conformation
are used to screen for inhibitors preferentially binding to the inactive
conformation by using
Tethering. This approach differs significantly from the conventional drug
discovery route that is
-25-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
based on the synthesis of large organic compound libraries, and subsequent
screening, usually
for inhibitory activity against the target protein kinase. In Tethering,
small, drug-like fragments
(monophores) containing or modified to contain a moiety capable of forming a
disulfide bond
are tested for binding activity to the desired kinase. These monophores are
then used to
synthesize conjugates of fragments that bind in non-overlapping sites to
generate molecules that
no longer require the , disulfide for binding. The linking or merging of
multiple fragments
effectively results in the combination of individual binding energies, plus a
favorable entropic
term due to the high local concentration of the second fragment once the first
fragment is bound,
yielding dissociation constants at levels similar to a typical medicinal
chemistry starting point.
In quantitative terms, this means that two fragments, each having ~mM
dissociation constants
(I~a) can be combined to form a molecule having a ~p,M Kd. This "screen then
link" strategy is
much more efficient than the traditional approach, allowing a much larger
survey of chemical
diversity space than is achievable by screening even the largest compound
libraries.
In a preferred embodiment, molecules binding to the target protein kinase
locked in an
inactive conformation are identified using Tethering recently reported by
Erlanson et al., PNAS
97:9367-9372 (2000). This strategy is suitable for rapid and reliable
identification of small
soluble drug fragments that bind with low affinity to a specifically targeted
site on a protein or
other macromolecule, using an intermediary disulfide linker and is illustrated
in Figure 1A.
According to a preferred embodiment of this approach, a library of disulfide-
containing
molecules is allowed to react with a cysteine-containing target protein under
partially reducing
conditions that promote rapid thiol exchange. If a molecule has even weak
affinity for the target
protein, the disulfide bond linking the molecule to the target protein will be
entropically
stabilized. The disulfide-bonded fragments can then be identified by a variety
of methods,
including mass spectrometry (MS), and their affinity improved by traditional
approaches upon
removal of the disulfide tether. See also PCT Publication Nos. WO 00/00823 and
WO
03/046200, the entire disclosures of which are hereby expressly incorporated
by reference.
Briefly, according to preferred embodiments, a disulfide bond is fornled
between the
target protein kinase molecule locked in inactive configuration and a ligand
candidate to yield a
target protein-ligand conjugate, and the ligand present in the conjugate is
identified. Optionally,
the target protein is contacted with a ligand candidate (preferably a library
of ligand candidates)
in the presence of a reducing agent, such as 2-mercaptoethanol, or cysteamine.
Most of the
library members will have little or no intrinsic affinity for the target
molecule, and thus by mass
-26-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
action the equilibrium will lie toward the unbowed target molecule. However,
if a library
member does show intrinsic affinity for the target molecule, the equilibrium
will shift toward the
target molecule, having attached to it the library member with a disulfide
containing linker. If a
plurality of library members have intrinsic affinty for the target molecule,
than the library
member having the greatest affinity for the target molecule will form the most
abundant target
molecule-ligand conjugate.
The target contains, or is modified to contain, free or protected thiol
groups, preferably
not more than about 5 thiol groups, more preferably not more than about 2
thiol groups, more
preferably not more than one free thiol group. The target protein kinase of
interest may be
initially obtained or selected such that it already possesses the desired
number of thiol groups, or
may be modified to possess the desired number of thiol groups.
As noted above, in certain embodiments the kinase of interest possesses at
least one
naturally occurring cysteine that is amenable to Tethering. Illustrative
examples of kinases that
include naturally occurring cysteines that are amenable to Tethering include:
CDKS (C53);
ERKl (C183); ERK2 (C166); ERK3 (C28); FGFRI (C488); FGFR2 (C491); FGFR3
(C482);
FGFR4 (C477); MEKI (C207); NIK (C533); PDGFR-a (C835); PDGFR-b (C843); SRC
(C279);
SRC2 (C273); STK1 (C828); TGFR2 (C396); VEGFR1 (C1039); VEGFR2 (C1045); VEGFR3
(C 1054); YES (C287); ZAP-70 (C346).
In other embodiments, one or more amino acids are mutated into a cysteine. In
general,
cysteine mutants are made using the following guidelines.
Broadly, the "binding site of interest" on a particular target, such as a
target protein
kinase, is defined by the residues that are involved in binding of the target
to a molecule with
which it forms a natural complex in vivo or ifs vitro. If the target is a
peptide, polypeptide, or
protein, the site of interest is defined by the amino acid residues that
participate in binding to
(usually by non-covalent association) to a ligand of the target.
When the target biological molecule is an enzyme, the binding site of interest
can include
amino acids that make contact with, or lie within, about 4 angstroms of a
bound substrate,
inhibitor, activator, cofactor or allosteric modulator of the enzyme. For
protein kinases, the
binding site of interest includes the substrate-binding channel and flee ATP
binding site.
The target protein kinases either contain, or are modified to contain, a
reactive residue at
or near a binding site of interest. Preferably, the target kinases contain or
are modified to contain
a thiol-containing amino acid residue at or near a binding site of interest.
In this case, after a
-27-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
protein kinase is selected, the binding site of interest is calculated. Once
the binding site of
interest is known, a process of determining which amino acid residue within,
or near, the binding
site of interest to modify is undertaken. For example, one preferred
modification results in
substituting a cysteine residue for another amino acid residue located near
the binding site of
interest.
The choice of which residue within, or near, the binding site of interest to
modify is
determined based on the following selection criteria. First, a three
dimensional description of the
target protein kinase is obtained from one of several well-known sources. For
example, the
tertiary structure of many protein kinases has been determined through x-ray
crystallography
experiments. These x-ray structures are available from a wide variety of
sources, such as the
Protein Databank (PDB) which can be found on the Internet at
http://www.rcsb.org. Tertiary
structures can also be found in the Protein Structure Database (PSdb) which is
located at the
Pittsburg Supercomputer Center at http://www.psc.com.
In addition, the tertiary structure of many proteins, and protein complexes,
including
protein kinases, has been determined through computer-based modeling
approaches. Thus,
models of protein three-dimensional conformations are now widely available.
Once the three dimensional structure of the target protein kinase is known, or
modeled
based on homology to a known structure, a measurement is made based on a
structural model of
the wild-type, or a variant form locked in an inactive configuration, from any
atom of an amino
acid within the site of interest across the surface of the protein for a
distance of approximately 10
angstroms. . Since the goal is to identify protein kinase inhibitors that
preferentially bind to an
inactive conformation of the target protein kinase, preferably the sites) of
interest is/are
identified base upon a structural model of the protein kinase locked in an
inactive conformation.
The binding sites (pockets) presented by such inactive conformations axe often
significantly
different from the binding sites (pockets) present on the wild-type structure.
Variants of the
inactive protein kinases, which have been modified to contain the desired
reactive groups (e.g.
thiol groups, or thiol-containing residues) are based on the identification of
one or more wild-
type amino acids) on the surface of the target protein kinase that fall within
that approximate 10-
angstrom radius from the binding site of interest (which may have been first
revealed as a result
of the alteration resulting the stabilization of an inactive conformation).
For the purposes of this
measurement, any amino acid having at least one atom falling within the about
10 angstrom
-28-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
radius from any atom of an amino acid within the binding site of interest is a
potential residue to
be modified to a thiol containing residue.
Preferred residues for modification are those that are solvent-accessible.
Solvent
accessibility may be calculated from structural models using standard numeric
(Lee, B. &
Richards, F. M. .I. Mol. Biol 55:379-400 (1971); Shrake, A. & Rupley, J. A. J.
Mol. Biol. 79:351-
371 (1973)) or analytical (Connolly, M. L. Science 221:709-713 (1983);
Richmond, T. J. J. Mol.
Biol. 178:63-89 (1984)) methods. For example, a potential cysteine variant is
considered
solvent-accessible if the combined surface area of the carbon-beta (CB), or
sulfur-gay~zn2a (SG) is
greater than 21 t~2 when calculated by the method of Lee and Richards (Lee, B.
~& Richards, F.
M. J. Mol. Biol 55:379-400 (1971)). This value represents approximately 33% of
the theoretical
surface area accessible to a cysteine side-chain as described by Creamer et
al. (Creamer, T. P: et
al. Biochemistry 34:16245-16250 (1995)).
It is also preferred that the residue to be mutated to cysteine, or another
thiol-containing
amino acid residue for tethering purposes, not participate in hydrogen-bonding
with backbone
atoms or, that at most, it interacts with the backbone through only one
hydrogen bond. Wild
type residues where the side-chain participates in multiple (>1) hydrogen
bonds with other side-
chains are also less preferred. Variants for which all standard rotamers (chil
angle of-60°, 60°,
or 180°) can introduce unfavorable steric contacts with the N, GA, C,
O, or CB atoms of any
other residue are also less preferred. Unfavorable contacts are defined as
interatomic distances
that are less than 80% of the sum of the van der Waals radii of the
participating atoms.
Additionally, residues found on convex "ridge" regions adjacent to concave
surfaces are
more preferred while those within concave regions are less preferred cysteine
residues to be
modified. Convexity and concavity can be calculated based on surface vectors
(Duncan, B. S. &
Olson, A. J. Biopolyrners 33:219-229 (1993)) or by determining the
accessibility of water probes
placed along the molecular surface (Nicholls, A. et al. Pi°oteitzs
11:281-296 (1991); Brady, G. P.,
Jr. & Stouten, P. F. J. Comput. Aided Mol. Des. 14:383-401 (2000)). Residues
possessing a
backbone conformation that is nominally forbidden for L-amino acids
(Ramachandran, G. N. et
al. J. Mol. Biol. 7:95-99 (1963); Ramachandran, G. N. & Sasisekharahn, V. Adv.
Prot. Clzem.
23:283-437 (1968)) are less preferred targets for modification to a cysteine.
Forbidden
conformations commonly feature a positive value of the phi angle.
Other preferred variants are those which, when mutated to cysteine and linked
via a
disulfide bond to an alkyl tether, would possess a conformation that directs
the atoms of that
-29-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
tether towards the binding site of interest. Two general procedures can be
used to identify these
preferred variants. In the first procedure, a search is~ made of unique
structures (Hobohm, U. et
al. Protein Science 1:409-417 (1992)) in the Protein Databank (Berman, H. M.
et al. Nucleic
Acids Researelz 28 235-242 (2000)) to identify structural fragments containing
a disulfide-
bonded cysteine at position j in which the backbone atoms of residues j-l, j,
and j+1 of the
fragment can be superimposed on the backbone atoms of residues i-1, i, and i+1
of the target
molecule with an RMSD of less than 0.75 A2. If fragments are identified that
place the CB atom
of the residue disulfide-bonded to the cysteine at position j closer to any
atom of the site of
interest than the CB atom of residue i (when mutated to cysteine), position i
is considered
preferred. In an alternative procedure, the residue at position i is
computationally "mutated" to a
cysteine and capped with an S-Methyl group via a disulfide bond.
In still other embodiments, in addition to mutating a naturally- occurring non-
cysteine
residue to a cysteine at a site of interest, one or more naturally occurring
cysteines outside of the
site of interest can be mutated to a non-cysteine residue (such as alanine or
serine) to prevent
unwanted labeling. In particular, those naturally occurring cysteines outside
of the site of
interest and are reactive to cystamine are candidates for being "scrubbed"
(mutated to a non-
cysteine residue).
Further details of identifying binding sites) of interest for tethering
purposes on the
protein kinase targets of the invention are provided in PCT publication WO
03/014308 and co-
pending application Serial No. 10/214,419, filed on August 5, 2002, which
claims priority from
provisional patent application Serial No. 60!310,725, filed on August 7, 2001,
the entire
disclosures of which are hereby expressly incorporated by reference.
Illustrative examples of kinase mutants where a non-native cysteine is
introduced into at
one or more sites of interest are described below.
For the AKTl kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L156C AKT1; K158C AKTl; T160C AKTl; F161C AKTl;
K194C
AKTI; E198C AKT1; M227C AKT1; E278C AKTl; T291C AKT1; K297C AKT1.
For the AKT2 lcinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: K158C AKT2; K160C AKT2; T162C AKT2; F163C AKT2;
H196C
AKT2; E200C AKT2; M229C AKT2; E279C AKT2; T292C AKT2; K298C AKT2.
-3 0-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the AKT3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L154C AKT3; K156C AKT3; T158C AKT3; F159C AKT3;
H192C
AKT3; E196C AKT3; M225C AKT3; E274C AKT3; T288C AKT3; K294C AKT3.
For the BLK kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L246C BLK; S248C BLK; Q151C BLK; F251C BLK; A279C
BLK;
E283C BLK; T311C BLK; A363C BLK; A376C BLK; R382C BLK.
For the BTK kinase, the following cysteine mutants are illustrative examples
of mutants
that re used for Tethering: L408C BTK; T410C BTK; Q313C BTK; F413C BTK; E441C
BTK;
E445C BTK; T474C BTK; R525C BTK; S538C BTK; R544C BTK.
For the CDKl kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: IIOC CDK1; E12C CDKl; T14C CDKl; YlSG CDK1; S53C
CDKl; E57C CDK1; F80C CDKl; Q432C CDK1; A145C CDKl; R151C CDK1.
For the CDK2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: IIOC CDK2; E12C CDK2; T14C CDK2; .YlSC CDK2; S53C
CDK2; E57C CDK2; F80C CDK2; Q431C CDK2; A144C CDK2; R150C CDK2.
For the CDK3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: IIOC CDK3; E12C CDK3; T14C -CDK3;J Y15C CDK3;
S53G
CDK3; E57C CDK3; F80C CDK3; Q431C CDK3; A144C CDK3; R150C CDK3.
For the CDK4 kinase, the following cysteine mutants are illustrative examples
of mutants
that axe used for Tethering: I12C CDK4; V14C CDK4; A16C CDK4; Y17C CDK4; RSSC
CDK4; L59C CDK4; F93C CDK4; E153G CDK4; A157C CDK4; R163C CDK4.
For the CDKS kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: IIOC CDKS; E12C CDKS; T14C CDKS; YlSC CDKS; E57C
CDKS; F80C CDKS; Q430C CDKS; A143C CDKS; R149C CDKS.
For the GDK6 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I19C CDK6; E21C CDK6; A23C CDK6; Y24C CDK6; A63C
CDK6; H67C CDK6; F98C CDK6; Q449C CDK6; A162C CDK6; R168C CDK6.
For the CDK7 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L18C CDK7; E20C CDK7; Q22C CDK7; F23C CDK7; R61C
CDK7; L65G CDK7; F91C CDK7; N141C CDK7; A154C CDK7; K161C CDK7.
-31-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the CDK8 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: V27C CDKB; R29C CDK8; T31C CDKB; Y32C CDKB; R65C
CDK8; L69C CDKB; F97C CDK8; A155C CDKB; A172C CDKB; H178C CDKB.
For the CDK9 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I25C CDK9; Q27C CDK9; T29C CDK9; F30C CDK9; R65C
CDK9; I69C CDK9; F103C CDK9; A153C CDK9; A166C CDK9; R172C CDK9.
For the CSK kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I201C CSK; K203C CSK; E205C CSK; F206C CSK; A232C
CSK;
E236C CSK; T266C CSK; R318C CSK; S331C CSK; K337C CSK.
For the EGFRl kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: L718C EGFRl; S720C EGFRl; A722C EGFRl;
F723C
EGFR1; E758C EGFRl; E762C EGFRl; T790C EGFRl; R841C EGFRl; T854C EGFRl;
K860C EGFRl.
For the ERB2 (also referred to as ErbB2) kinase, the following cysteine
mutants are
illustrative examples of mutants that are used for Tethering: L726C ERB2;
S728C ERB2;
A730C ERB2; F731C ERB2; E766C ERB2; E770C ERB2; T798C ERB2; R849C ERB2; T862C
ERB2; R868C ERB2.
For the ERB4 (also referred to as ErbB4) kinase, the following cysteine
mutants are
illustrative examples of mutants that are used for Tethering: L724C ERB4;
S726C ERB4;
A728C ERB4; F729C ERB4; E764C ERB4; E768C ERB4; T796C ERB4; R847C ERB4; T860C
ERB4; R864C ERB4.
For the ERK1 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I48C ERKI; ESOC ERKI; A52C ERKl; Y53C ERKI; R84C
ERKl;
E88C ERKl; Q122C ERKl; S170C ERKl; R189C ERKl.
For the ERK2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I31C ERK2; E33C ERK2; A35C ERK2; Y36C ERK2; R67C
ERK2;
E71C ERK2; Q105C ERK2; S153C ERK2; R172C ERK2.
For the ERK3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L26G ERK3; G30C ERK3; N31C ERK3; H61C ERK3; E65C
ERK3; Q108C ERK3; A156C ERK3; G170C ERK3; R176C ERK3.
-32-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the ERK4 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L26C ERK4; F28C ERK4; V30C ERK4; N31C ERK4; H61C
ERK4;
E65C ERK4; Q105C ERK4; A153C ERK4; G167C ERK4; R173C ERK4.
For the ERKS kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I60C ERKS; N62C ERKS; A64C ERKS; Y65C ERKS; R97C
ERKS;
ElOlC ERKS; L136C ERKS; S185C ERKS; G198C ERKS; R204C ERKS.
For the ERK6 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: V33C ERK6; S35C ERK6; A37C ERK6; Y38C ERK6; R70C
ERK6; E74C ERK6; M109C ERK6; G157C ERK6; L170C ERK6; R176C ERK6.
For the FAKI kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I428C FAK1; E430C FAK2; Q333C FAKl; F433C FAKl;
K467C
FAK1; E471C FAKl; M499C FAKl; RSSOC FAKI; G563C FAKl; R569C FAK1.
For the FGFRl kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: L484C FGFRI; E486C FGFRl; F489C FGFRl;
L528C
FGFRl; M532C FGFR1; V561C FGFR1; R627C FGFRl; A640C FGFRl; R646C FGFRl.
For the FGFR2 kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: L487C FGFR2; E489C FGFR2; F492C FGFR2;
L531C
FGFR2; M535C FGFR2; V564C FGFR2; R630C FGFR2; A643C FGFR2; R649C FGFR2.
For the FGFR3 kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: L478C FGFR3; E480C FGFR3; F483C FGFR3;
L522C
FGFR3; M526C FGFR3; VSSSC FGFR3; R621G FGFR3; A634G FGFR3; R640C FGFR3.
For the FGFR4 kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: L473C FGFR4;, E475G FGFR4; F478C FGFR4;
L517C
FGFR4; M521C FGFR4; VSSOC FGFR4; R616G FGFR4; A629C FGFR4; R635C FGFR4.
For the FYN kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L276C FYN; N278C FYN; Q181C FYN; F281C FYN; S309C
FYN;
E313C FYN; T341C FYN; A393C FYN; A406C FYN; R412C FYN.
For the HCK kinase, the following cysteine mutaazts are illustrative examples
of mutants
that are used for Tethering: L268C HCK; A274C HCK; Q173G HCK; F273C HCK; A301C
HCK; E305C HCK; T333C HCK; A385C HCK; A398C HCK; R404C HCK.
-33-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the IKK-a kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L21C IKK-a; T23C IKK-a; G25C IKK-a; F26C IKK-a;
R57C IKK-
a; E61C IKK-a; M95C IKK-a; E148C IKK-a; I164C IKK-a; K170C IKK-a.
For the IKK-b kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L21C IKK-b; T23C IKK-b; G25C IKK-b; F26C IKK-b;
R57C IKK-
b; E61C IKK-b; M96C IKK-b; E149C IKK-b; I165C IKK-b; K171C IKK-b:
For the IKK-a kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L15C IKK-e; Q17C IKK-e; A19C IKK-e; T20C LKK-e;
VS1C IKK-
e; ESSC IKK-e; M86C IKK-e; G139C IKK-e; T156C IKK-e; R163C IKK-e.
For the JAKI kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L870C JAKl; E872C JAKl; H874C JAKl; F875C JAKI;
D909C
JAKl; E913C JAKl; M944C JAK1; R995C JAKl; G1008C JAK1; K1014C JAKl.
For the JAK2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L855C JAK2; L857C JAK2; N859C JAK2; F860C JAK2;
D894C
JAK2; E898C JAK2; M929C JAK2; R980C JAK2; G993C JAK2; K999C JAK2.
For the JAK3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L828C JAK3; K830C JAK3; N832C JAK3; F833C JAK3;
D867C
JAK3; E871C JAK3; M902C JAK3; R953C JAK3; A966C JAK3; K972C JAK3.
For the JNKl kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I32C JNK1; S34G JNK1; A36C JNK1; Q37C JNKl; R69C
JNKl;
E73C JNKl; M108C JNK1; S155C JNKl; L168C JNK1; R174C JNK1.
For the JNK2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I32C JNK2; S34C JNK2; A36C JNK2; Q37C JNK2; R69C
JNK2;
E73C JNK2; M108C JNK2; S155C JNK2; L168C JNK2; R174C JNK2.
For the JNK3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I70C JNK3; S72C JNK3; A74C JNK3; Q75C JNK3; R107C
JNK3;
E111C JNK3; M146C JNK3; S193C JNK3; L206C JNK3; R212C JNK3.
For the Lclc ldnase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L250C Lck; A252C Lck; Q155C Lch; F255C Lck; A283G
Lck;
E287C Lck; T315C Lck; A367C Lclc; A380C Lck; R386C Lck.
-34-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the LYN kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L252C LYN; A254C LYN; Q157C LYN; F257C LYN; A285C
LYN; E289C LYN; T318C LYN; A370C LYN; A383C LYN; D389C LYN.
For the MAPK kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: V30C MAPK; S32C MAPK; A34C MAPK; Y35C
MAPK;
R67C MAPK; E71C MAPK; T106C MAPK; S154C MAPK; L167C MAPK; R173C MAPK.
For the NIK kinase, the following cysteine mutants are illustrative examples
of mutants
that are used- for Tethering: L406C NIK; R408C NIK; S410C NIK; F411C NIK;
F436C NIK;
E439C NIK; M469C NIK; D519C NIK; V540C NIK.
For the PAKl kinase, the following cysteine.mutants are illustrative examples
of mutants
that are used for tethering: I276C PAKl; Q179C PAKI; A280C PAKl; S281C PAKl;
N314C
PAK1; V318C PAK1; M344C PAKl; D393C PAKl; T406C PAK1; A412C PAKl.
For the PAK2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I255C PAK2; Q158C PAK2; A259C PAK2; S260C PAK2;
N293C
PAK2; V297C PAK2; M323C PAK2; D372C PAK2; T385C PAK2; A391C PAK2.
For the PAK3 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I274C PAK3; Q177C PAK3; A278C PAK3; S279C PAK3;
N312C
PAK3; V316C PAK3; M342C PAK3; D391G PAK3; T404C PAK3; A410C PAK3.
For the PAK4 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I327C PAK4; E329C PAK4; S331C PAK4; R332C PAK4;
N365C
PAK4; I369C PAK4; M395C PAK4; D444C PAK4; S457C PAK4; A463C PAK4.
For the PAKS kinase, the following cysteine mutants are illustrative examples
of mutants
that.are used for Tethering: I455C PAKS; E457C PAKS; S459C PAKS; T460C PAKS;
N492C
PAKS; I496C PAKS; M523C PAKS; D572C PAKS; D585C PAKS; A591C PAKS.
For the PDGFR-a kinase, the following cysteine mutants are illustrative
examples of
mutants that are used for Tethering: L599C PDGFR-a; S601C PDGFR-a; A603C PDGFR-
a;
F604C PDGFR-a; L641C PDGFR-a; L645C PDGFR-a; T674C PDGR-a; R822C PDGFR-a;
R841C PDGFR-a.
For the PDGFR-b kinase, the following cysteine mutants are illustrative
examples of
mutants that are used for Tethering: L606C PDGFR-b; S608C PDGFR-b; A700C PDGFR-
b;
F701G PDGFR-b; L648C PDGFR-b; L652C PDGFR-b; T681C PDGFR-b; R830C PDGFR-b;
R849C PDGFR-b.
-35-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the PIMI kinase, the following cysteine mutants axe illustrative examples
of mutants
that are used for Tethering: L44C PIM1; S46G PIMI; G48C PIMI; F49C PIMl; M87C
PIM1;
L91C PIM1; E121C PIM1; E171C PIMl; E171C PIM1; I185C PIM1; A192C PIMI.
For the A-Raf kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for tethering: I316C A-Raf; T318C A-Raf; S320C A-Raf; F321C A-
Raf; A350C A-
Raf; E354C A-Raf; T382C A-Raf; N433C A-Raf; G446C A-Raf; T452C A-Raf.
For the B-Raf kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I462C B-Raf; S464C B-Raf; S466C B-Raf; F467C B-
Raf; A496C B-
Raf; ESOOC B-Raf; T528C B-Raf; N579C B-Raf; G592C B-Raf; T598C B-Raf.
For the C-Raf kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I355C C-Raf; S357C C-Raf; S359C C-Raf; F-3606 C-
Raf; A389C
C-Raf; E393C C-Raf; T421C C-Raf; N472C C-Raf; G485C C-Raf; T491C C-Raf.
For the SRC kinase, the following cysteine mutants are illustrative examples
of mutants
that axe used for Tethering: L275C SRC; Q178C SRC; F280C SRC; A308C SRC; E402C
SRC;
T340C SRC; A392C SRC; A405C SRC; R411C SRC.
For the SRC2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L269C SRC2; T271C SRC2; F274G SRC2; A302C SRC2;
E306C
SRC2; T334C SRC2; A386C SRC2; A399C SRC2; R405C SRC2.
For the STKI kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L616C STK1; S618C STK1; A620C STKl; F621C STKl;
L658C
STK1;,L662C STKl; F691C STKl; R815C STKl, R834C STKl.
For the SYK kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L377C SYK; S379C SYK; N381C SYK; F382C SYK; E416C
SYK;
E420C SYK; M448C SYK; R498C SYK; SS11C SYK; K518C SYK.
For the TEC kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L376C TEC; S378C TEC; L380C TEC; F381C TEC; D409C
TEC;
E413C TEC; T442C TEC; R493C TEC; S506C TEC; R513C TEC.
For the TGFRl kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: I211C TGFRl; K213C TGFRl; R215C TGFRI;
F216C
TGFRl; F243C TGFRl; E247C TGFR1; S280C TGFRl; K337C TGFRI; A350C TGFRI;
V357C TGFRI.
-36-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
For the TGFR2 kinase, the following cysteine mutants are illustrative examples
of
mutants that are used for Tethering: V250C TGFR2; K252C TGFR2; R254C TGFR2;
F255C
TGFR2; K288C TGFR2; D292C TGFR2; T325C TGFR2; S383C TGFR2; L403C TGFR2.
For the TIEl kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I845C TIE1; E847C TIE1; N849C TIEl; F850C TIE1;
F884C TIEl;
L888C TIEl; I917C TIEl; R983C TIE1; A996C TIE 1; R1002C TIEl.
For the TIE2 kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: I830C TIE2; E832C TIE2; N834C TIE2; F835C TIE2;
F869C TIE2;
L873C TIE2; I902C TIE2; R968C TIE2; A981C TIE2; R987C TIE2.
For the VEGFRI kinase, the following cysteine mutants are illustrative
examples of
mutants that are used for Tethering: L834C VEGFRI; R836C VEGFRl; A838C VEGFRl;
F839C VEGFRl; L876C VEGFRl; L880C VEGFR1; V910C VEGFRl; R1026C VEGFRl;
R1045C VEGFRl.
For the VEGFR2 kinase, the following cysteine mutants are illustrative
examples of
mutants that are used for Tethering: L840C VEGFR2; R842C VEGFR2; A844C VEGFR2;
F845C VEGFR2; L882C VEGFR2; L886C VEGFR2; V916C VEGFR2; R1032C VEGFR2;
R1051C VEGFR2.
For the VEGFR3 kinase, the following cysteine muta~its are illustrative
examples of
mutants that are used for Tethering: L851C VEGFR3; Y853C VEGFR3; A855C VEGFR3;
F856C VEGFR3; L893C VEGFR3; L987C VEGFR3; V927C VEGFR3; R1041C VEGFR3;
R1060C VEGFR3.
For the YES kinase, the following cysteine mutants are illustrative examples
of mutants
that are used for Tethering: L283C YES; Q286C YES; C287C YES; F288C YES; A316C
YES;
E320C YES; T348C YES; A400C YES; A413C YES; R419C YES.
For the ZAP-70 kinase, the following cysteine mutants are illustrative
examples of
mutants that are used for Tethering: L344C ZAP-70; N348C ZAP-70; F349C ZAP-70;
E382C
ZAP-70; E386C ZAP-70; M414C ZAP-70; R465C ZAP-70; S478C ZAP-70; and K485C ZAP-
70.
Although this approach is typically exemplified with reference to a protein
kinase target
havW g a thiol functionality to screen a disulfide-containing library, other
chemistries are also
available and are readily used.
-37-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
c. Tethering with Extenders
Tethering with extenders is a variation of Tethering described above that uses
a Small
Molecule Extender (SME) to form a target kinase-SME covalent complex. The SME
has a first
reactive functionality that is capable of forming a reversible or irreversible
covalent bond with
the target kinase and a second reactive functionality that is capable of
forming a reversible or
irreversible covalent bond with a ligand candidate. Thus, the SME forms a
first covalent bond
with the target kinase thereby forming a target kinase-SME covalent complex.
In certain .
embodiments, the SME also includes a binding element that has an affinity for
the SME binding
site or a first site of interest. The second reactive functionality on the SME
on the target kinase-
SME covalent complex is used in Tethering to identify ligands that have an
affinity for a site on
the kinase that is adjacent to the SME binding site. This adjacent site is
referred to as the second
site of interest.
In certain embodiments, the first reactive. functionality on a SME forms a
irreversible
covalent bond through the nucleophile or electrophile, preferably nucleophile,
on the protein
kinase target, thereby forming an irreversible protein kinase-SME complex.
Preferred
nucleophiles on the target protein kinase suitable for forming an irreversible
kinase-SME
complex include -SH, -OH, -NH2 and -COOH usually arising from side chains of
Cys, Ser or
Thr, Lys and Asp or Glu respectively. , Protein kinases may be modified (e.g.
mutants or
derivatives) to contain these nucleophiles or may contain them naturally. For
example, BLK,
BTK, EGFRl, ERB2, ERB4, ERK1, ERK2, FGFRl, FGFR2, FGFR3, FGFR4, etc. are
examples
of kinases containing suitable naturally occurring cysteine thiol
nucleophiles.
In other embodiments, the second reactive functionality is a group capable of
forming a
disulfide bond. Illustrative examples of such a group include a free thiol (-
SH) , protected thiol
(-SR' where R' is a thiol protecting group), and a disulfide (-SSR" where R"
is a substituted or
unsubstituted aliphatic or substituted or unsubstituted aryl).
The SME may, but does not have to, include a portion that has binding affinity
(i.e. is
capable of bonding to) a first site of interest on the target kinase. Even if
the SME does not
include such portion, it must be of appropriate length and flexibility to
ensure that the ligand
candidates have free access to the second site of interest on the target.
Figure 1B is a schematic illustration of one embodiment of Tethering with
extenders. As
shown, a target that includes a thiol is contacted with an extender comprising
a first functionality
-3 8-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
-LG that is capable of forming a covalent bond with the reactive thiol and a
second functionality
second functionality -SPG that ~is capable of forming a disulfide bond. The
extender binds to the
first site of interest and forms a target-extender covalent complex which is
then contacted with a
plurality of ligand candidates to identify a ligand for a second site of
interest. The extender
provides one binding determinant (circle) and the ligand candidate provides
the second binding
determinant (square) and the resulting binding determinants are linked
together to form a
conjugate compound.
As illustrated in Figure 1B, in certain embodiments, the SME includes a
binding element
that has affinity for the SME binding site. Thus, compounds having lcnown
affinity for kinases
can be modified to be SME's by adding the first reactive functionality (-LG in
Figure 1 B) and the
second reactive functionality (-SPG in Figure 1 B).
Suitable first reactive functionalities include groups that axe capable of
undergoing SN2-
like or Michael-type addition and thus forming an irreversible covalent bond
with the target
kinase. Examples of SME's having such groups are further described below. For
the purposes of
illustration, the SME's are shown schematically where ~ optionally includes a
binding element
for the intended SME binding site and -SR' is the second reactive
functionality that is capable of
forming a disulfide bond.
a,-halo acids: F, Cl and Br substituted a, to a COOH, P03H2 or P(OR)02H acid
that is
part of the SME can form a thioether with the thiol of the target kinase.
Illustrative examples of
generic a-halo acids are shown below.
~SR' r--SR' ~R'
X COOH X p03H2 ~p(OR)OZH
wliere X is the halogen, R is C1-C20 unsubstituted aliphatic, Cl-C20
substituted aliphatic,
unsubstituted aryl or substituted aryl, and R' is H, SCH3, S(CHZ)"A, where A
is OH, COOH,
S03H, CONH2 or NH2 and n is 1 to 5, preferably n is 2 to 4.
Fluorophosph(on)ates: These are Saxin-like compounds which react readily with
both SH
and OH nucleophiles. Illustrative examples of general fluorophosph(on)ates are
shown below.
-3 9-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
5R' SR' SR' SR'
OH F OH OH O OR
P=O F p-O O~p~O APO~
F F F F
where R and R' are as defined above.
Epoxides, aziridines and thiiranes: SME's containing these reactive functional
groups
are capable of undergoing SN2 ring opening reactions with -SH, -OH and -COOH
nucleophiles.
Preferred examples of the latter are aspartyl proteases like [3-secretase
(BASE). Preferred
generic examples of epoxides, aziridines and thiiranes are shown below.
0 0
'RS 'RS
O
NR NR
'RS 'RS
O
i O~R" i OR"
N N
'RS 'RS
O
5 S
'RS 'RS
O
Here, R' is as defined above, R is usually H or lower alkyl and R" is lower
alkyl, lower alkoxy,
OH, NHZ or SR'. In the case of thiiranes the group SR' is optionally present
because upon
nucleophilic attack and ring opening a free thiol is produced which may be
used in the
subsequent extended tethering reaction.
-40-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Halo-methyl ketones/amides: These compounds have the fornz -(C=O)-CH2-X. Where
X may be a large number of good leaving groups like halogens, NZ, O-R (where R
may be
substituted or unsubstituted heteroaryl, aryl, alkyl, -(P=O)Ar2, -N-O-(C=O)
aryl/alkyl, -(C=O)
aryl/alkyl/alkylaryl and the like), S-Aryl, S-heteroaryl and vinyl sulfones.
'RS
X
O
Fluromethylketones are simple examples of this class of activated ketones
which result in
the formation of a thioether when reacted with a thiol containing protein.
Other well known
examples include acyloxymethyl ketones like benzoyloxymethyl ketone,
aminomethyl ketones
like phenylmethylaminomethyl ketone and sulfonylaminomethyl ketones. These and
other types
. of suitable compounds are reviewed in J. Med. Chem. 43(18) p 3351-71,
September 7, 2000.
Electrophilic aromatic systems: Examples of these include 7-halo-2,1,3-
benzoxadiazoles
and ortholpara nitro substituted halobenzenes.
SR'
N02 SR' p N'
\N /
/ X
X
1$
Compounds of this type fornz arylalkylthioethers with protein kinases
containing a thiol.
Other suitable SN2 like reactions suitable for formation of covalent bonds
with protein
kinase nucleophiles include formation of a Schiff base between an aldehyde and
the amine group
of lysine of enzymes like DNA repair proteins followed by reduction with for
example
NaCNBH4.
5R' SR'
NaCNBHy
+ TBM NHz ~ ~N TBM
O
Michael-type additions: Compounds of the form -RC=CR-Q, or -C=C-Q where Q is
C(=O)H, C(=O)R, COOR, C(=O)NH2, C(=O)NHR, CN, N02, SOR, S02R, where each R is
independently substituted or unsubstituted alkyl, aryl, hydrogen, halogen or
another Q can form
-41-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Michael adducts with SR (where R is H, glutathione or S-lower alkyl
substituted with NH2 or
OH), OH and NHZ on the target protein kinase.
Boronic acids: These compounds can be used where the reactive nucleophile on
the
target kinase is a hydroxyl. For example serine, theonine, or tyrosine on a
target kinase can be
labeled to form kinase-SME complexes for use in the present invention. The
formation of such a
kinase-SME complex is shown below.
SR' SR'
~T -OH
HO~B~OH QT ---~B~OH
where R' is as defined above.
In other embodiments, the first site of interest is the ATP binding pocket. In
these
embodiments, known compounds that target the ATP binding pocket of kinases can
be modified
to be an SME by adding first and second reactive functionalities. Illustrative
examples of such
SME's include those that contain purine or purine mimetics such as the
following:
2
H~N~R R6
~ E ~ 6
N' \N N' \N R
/ R~ R~ ~ / R1
P
R5 R3 R3 R5 ~ R3
R4 > > R4 > >
H caz
R~
R3
where R1, R2, R3, R4, R5, and R6 are each independently selected from the
group consisting of
hydrogen, C1-CS alkyl, C1-CS alkylamine, and aryl provided that at least one R
group on the SME
is a Michael acceptor or -(C=O)CH~X where X is a halogen, and another R group
is selected
-42-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
from -(CHZ)n SR'; -C(=O)-(CH2)n SR; -O-(CH2)n SR;-(CH2)n-SR; and a thiol
protecting group
where R' is hydrogen or sulfide and n is 1-5, preferably 2-4. In certain
embodiments, R' is
-S(CH2)nNH2, -S(CH2)"OH or -S(CH2)"COOH where n is 1-5, preferably 2-4.
Illustrative
examples of Michael acceptors include
H / H H
O , O I , and ~
Illustrative examples of suitable SME's containg quinazolines include:
/Ph , H an
N
2
N \ ~ N R
and
N v ~Rz R
where Rl is -NHC(=O)CH2C1, -NHC(=O)CH=CHZ or -NHC(=O)CCH and RZ is
-(CH2)I"SSCH2CH2NH2 where m is 1-3.
Figure 2 illustrates the use of a quinazoline extender to form a kinase-
extender covalent
complex. Figure 2A is the mass spectrometer profile of purified EGFRl kinase
domain in the
active conformation. Figure 2B is purified EGFRI kinase domain in the inactive
conformation.
Figures 2C-E) are purified EGFRI in the inactive conformation following
incubation with C)
cystamine, D) the quinazoline extender shown, and E) the quinazoline extender
and cystamine.
Alternatively, Tethering can be used to identify novel ligands that bind to
the ATP-
binding pocket. For example, Tethering off the naturally occurring cysteine at
the bottom of the
ATP binding pocket 11 EGFRl (C797), identified the expected purine and purine
mimetic
containing ligands along with several novel scaffolds. Representative ligand
candidates with
novel scaffolds include:
-43-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
H
H5 N NON HS N ~ H CI Hgr''~ . N ..N
l
CHI
l
~--~ ~,CH3
0
NS~N N~ Hg N ..-' ~ H5'~-~ N iN
H3C Cti3 H3
-- ~H3
CI OH
H H H H
HS'~~N ~ NON HS~'rN ~ ~ HS~N~S
'' ~ '~N P \ c~
rs
r. HaC
CH3
Illustrative examples of SME's that can be made using such scaffolds by adding
first and second
functionalities include but are not limited to:
H HN~S~S~NHZ H HN~S.S~NHZ
~N~ ~ J ~N~ , J
N H N
HN~S~S~NH~ HN~S'S~NH2
O ~ ~N
~H ~N~
N N
H
H ~ S~S'~NHZ
and ~N
0 ~ / N
N
H
As described above, certain kinases already possess a naturally occurring
cysteine within
the ATP binding pocket that can be used to identify ligands that bind to this
site. In addition to -
C797 of EGFRl, other examples of kinases that include a naturally occurring
cysteine within the
ATP binding pocket include: BLK (C318); BTK (G481); ERB2 (C805); ERB4 (C803);
JAK3
(C909); TEC (C449). In addition to being used to identify novel ligands that
bind to the ATP-
binding pocket, these cysteines are also good candidates for Tethering with
extenders. If SME's
containing purine or purine mimetics are used (so that the SME binds to the
ATP binding site),
-44-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
the resulting kinase-SME complex can be used to identify ligands to the
adaptive binding region
adjacent to the ATP binding site.
For kinases that do not have a naturally occurring cysteine at the ATP binding
pocket, the
following are illustrative examples of mutants where a cysteine is introduced
at the appropriate
location: E234C AKT1; E236C ALT2; E232C AKT3; D86C CDKl; D86C CDK2; D86C
CDK3; D99C CDK4; D86C CDKS; D104C CDK6; D97C CDK7; D103C CDKB;. D108C
CDK9; S273C CSK; D128C ERKl; D111C ERK2; D114C ERK3; D111C ERK4; D142G
ERKS; ~D115C ERK6; E506C FAKl; N568C FGFR1; N571C FGFR2; N562C FGFR3; N557C
FGFR4; D348C FYN; S340C HCK; D102C IKK-a; D103C IKK-b; S93C IKK-e; S951C JAK1;
S936C JAK2; N114C JNK1; N114G JNK2; N152G JNK3; S322C LCK; S325C LYN; D112C
MAPK; S150C MEKl; S476C NIK; S351C PAKl; S330C PAK2; S349C PAK3; A402C PAK4;
A530C PAKS; D861C PDGFR-a; D688C PDGFR-b; D128C PIM1; S389C A-Raf; S535C B
Raf; S428C C-Raf; S347C SRC; S341G SRC2; D698C STK1; P455C SYK; S287C TGFRl;
N332C TGFR2; N924C TIE1; N909C TIE2; N917C VEGFRI; N923C VEGFR2; N934C
VEGFR3; S355C YES; P421C ZAP-70.
Although Tethering with extenders has been primarily described with target
kinases
having reactive thiols and extenders having a group capable of forming a
covalent bond with the
thiol, other chemistries can be used. For example, the amino group of lysines
are alternative
nucleophiles on the target kinases. The following extender is an exemplary
extender that is
capable of forming a covalent bond with a lysine '
HN~S~S~
~N I ~ N
O N NJ
/ ~.,~OH
~/0
O OH
Because the ATP binding pocket includes a conserved lysine, the 5'-(p-
fluorosulfonylbenzoyl)adenosine-based extender can be used with any kinase
without the need
for making a cysteine mutation in this site. The precursor for installing a
masked thiol onto the
adenosine-containing compound is made by reacting reacting commercially
available N Boc-
cysteamine with commercially available methanethiosulfonic acid S-methyl
ester, followed by
deprotection of the Boc group to generate the hydrochloride salt. The
resulting compomld is
reacted with commercially available 6-chloroadenosine. The installation of the
electrophile as
-45-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
described in J. Biol. Chem. 250: 8140-8147 (1975) and Bioehey~zistry 16: 1333-
1342 (1977).
Once this extender is reacted with the lysine in the ATP-binding site, the
masked thiol can be
reduced to the free thiol by a reducing agent such as 2-mercaptoethanol. The
resulting kinase-
extender complex can then be used in Tethering as described above.
While it is usually preferred that the attachment of the SME does not denature
the target,
the kinase-SME complex may also be formed under denaturing conditions,
followed by refolding
the complex by methods known in the art. Moreover, the SME and the covalent
bond should not
substantially alter the three-dimensional structure of the target protein
kinase, so that the ligands
will recognize and bind to a binding site of interest on the target with
useful site specificity.
Finally, the SME should be substantially unreactive with other sites on the
target under the
reaction and assay conditions.
d. Library of Sulfh ~~dryl-Containing Fra inents
The assembly of a collection of drug-like fragments or "monophores" that
display a
masked sulfhydryl group is used in certain embodiments of Tethering. The
sulfhydryl is
installed such that the fragment can participate in a disulfide exchange
reaction with the cysteine
residue on a kinase target. The monophores fragments are also broadly
representative of
recognized and unique drug-like pharmacophores and fragments thereof. At a
minimum,
candidate fragments satisfy two primary criteria. First, they contain a
functional group that will
permit the installation of a disulfide linker. Suitable functional groups
include a free amine,
carboxylate, sulfonyl chloride, isocyanate, aldehyde, ketone, etc. Second,
they are chosen such
that the, combination of two such entities results in a product with drug-like
physical properties,
including molecular weight (approximately 500 Da or less) and hydrophobicity
(clog P between
-l and 5).
Chemistries for making the sulfhydryl-containing fragments as well as
practicing the
other aspect of the present invention such as forming a reversible or
irreversible covalent bond
between reactive groups on a protein kinase, malting SME's, and compound
advancement, are
well known in the art, and are described in basic textbooks, such as, e.g.
March, Advafzced
Or~gafzic Chemistry, John Wiley 8r Sons, New York, 4t'' edition, 1992.
Reductive aminations
between aldehydes and ketones and amines are described, for example, in March
et al., supra, at
pp. 898-900; alternative methods for preparing amines at page 1276; reactions
between
aldehydes and ketones and hydrazide derivatives to give hydrazones and
hydrazone derivatives
-46-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
such as semicarbazones at pp. 904-906; amide bond formation at p. 1275;
formation of areas at
p. 1299; formation of tluocarbamates at p. 892; formation of carbamates at p.
1280; formation of
sulfonamides at p. 1296; formation of thioethers at p. 1297; formation of
disulfides at p. 1284;
formation of ethers at p. 1285; formation of esters at p. 1281; additions to
epoxides at p. 368;
additions to aziridines at p. 368; formation of acetals and ketals at p. 1269;
formation of
carbonates at p. 392; formation of enamines at p. 1264; metathesis of alkenes
at pp. 1146-1148
(see also Grubbs et al., Acc. Chew. Res. 28:446-453 [1995]); transition metal-
catalyzed
couplings of aryl halides and sulfonates with alkanes and acetylenes, e.g.
Heck reactions, at pp.
717-178; the reaction of aryl halides and sulfonates with organometallic
reagents, such as
organoboron, reagents, at p. 662 (see also Miyaura et al., Chem. Rev. 95:2457
[1995]); organotin,
and organozinc reagents, formation of oxazolidines (Ede et al., Tet~~ahednon
Letts. 28:7119-7122
[1997]); formation of thiazolidines (Patek et al., Tetrahedron Letts. 36:2227-
2230 [1995]);
amines linked through amidine groups by coupling amines through imidoesters
(Davies et al.,
Cafzadian J. BiochenZ. 50:416-422 [1972]), and the like. In particular,
disulfide-containing small
molecule libraries may be made from commercially available carboxylic acids
and protected
cysteamine (e.g. mono-BOC-cysteamine) by adapting the method of Parlow et a1,
Mol. Diversity
1:266-269 (1995), and can be screened for binding to polypeptides that
contain, or have been
modified to contain, reactive cysteines. All of the references cited in this
section are hereby
expressly incorporated by reference.
The monophores library can be derived from commercially available compounds
that
satisfy the above criteria. However, many motifs common in biologically active
compounds are
rare or absent in commercial sources of chemicals. Therefore, the fragment
collection is
preferably supplemented by synthesizing monophores fragments that help fill
these gaps. A
typical library can contain 10,000 or more compounds.
e. Detection and identification of li~ands bound to a target
The ligands bound to a target (or to a target-SME complex) can be readily
detected and
identified by mass spectroscopy (MS). MS detects molecules based on mass-to-
charge ratio
(m/z) and thus can resolve molecules based on their sizes (reviewed in Yates,
Treads Genet. 16:
5-8 [2000]). A mass spectrometer first converts molecules into gas-phase ions,
then individual
ions are separated on the basis of m/z ratios and are finally detected. A mass
analyzer, which is
an integral part of a mass spectrometer, uses a physical property (e.g.
electric or magnetic fields,
-47-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
or time-of flight [TOF]) to separate ions of a particular m/z value that then
strikes the ion
detector. Mass spectrometers are capable of generating data quickly and thus
have a great
potential for high-throughput analysis. MS offers a very versatile tool that
can be used for drug
discovery. Mass spectroscopy may be employed either alone or in combination
with other
means for detection or identifying the organic compound ligand bound to the
target. Techniques
employing mass spectroscopy are well known in the art and have been employed
for a variety of
applications (see, e.g., Fitzgerald and Siuzdak, Chemistfy & Biology 3: 707-
715 [1996]; Chu, et
al., J. Arr. Chena. Soc. 118: 7827-7835 [1996]; Siudzak, Pt-oc. Natl. Acad.
Sci. USA 91: 11290
11297 [1994]; Burlingame et al., Anal. Chem. 68: 5998-6518 [1996]; Wu et al.,
Chemistry &
Biology 4: 653-657 [1997]; and Loo et al., Am. Reports Med. Chem. 31: 319-325
[1996]).
However, the scope of the instant invention is not limited to the use of MS.
In fact, any
other suitable technique for the detection of the adduct formed between the
protein kinase target
molecule and the library member can be used. For example, one may employ
various
chromatographic techniques such as liquid chromatography, thin layer
chromatography a.nd likes
for separation of the components of the reaction mixture so as to enhance the
ability to identify
the covalently bound organic molecule. Such chromatographic techniques may be
employed in
combination with mass spectroscopy or separate from mass spectroscopy. One may
optionally
couple a labeled probe (fluorescently, radioactively, or otherwise) to the
liberated organic
compound so as to facilitate its identification using any of the above
techniques. In yet another
embodiment, the formation of the new bonds liberates a labeled probe, which
can then be
monitored. Other techniques that may find use for identifying the organic
compound bound to
the target molecule include, for example, nuclear magnetic resonance (NMR),
capillary
electrophoresis, X-ray crystallography, and the like, all of which will be
well known to those
skilled in the art.
f. Identification of I~inase Inhibitors from Tethering
As described above, in certain embodiments, pools containing compounds that
covalently
modify the kinase or the kinase-extender covalent complex are identified by
mass spectrometry
(MS) analysis. From the deconvoluted MS profile, the molecular weight of the
bound compound
can be precisely calculated, and thus its identity in the pool deterniined.
The discrete compound
is then tested alone to determine if it can covalently modify the kinase or
the kinase-extender
complex. Each screen is likely to identify multiple hits. Hits are prioritized
according to their
-48-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
relative binding affinities and according to their relative preference for the
inactive enzyme
conformation. Relative enzyme binding affinities, expressed as a BMEso, are
then determined
using a BME titration curve to determine the concentration that allows 50%
modification while
using a constant concentration of compound. From this one can easily rank the
compounds,
based upon their binding affinities.
Upon compilation of the confirmed monophores hits, additional valuable
information can
be gained from analyzing the structure-activity relationship (SAR) between hit
compounds and
their relatives in the monophores ,library. For example, if several hit
molecules for a particular
kinase or kinase-extender pair fall into a closely related family, one may
then go back to the
monophores library and find structurally similar compounds that were not
selected in the initial
screen. These relatives are re-screened as discrete compounds to verify their
activity (or
inactivity), followed by rank ordering of the entire family in terms of
affinity for both the active
and inactive enzyme conformations. From this dataset, one can identify
features critical to
activity, and potential sites of modification the alteration of which is
expected to improve
1 S affinity.
In parallel with the SAR studies, the covalently attached compounds or
extender-
compounds are co-crystallized with the target kinase domain. Alternatively,
the compounds or
the extender-compound complexes lacking the Michael acceptor are synthesized
and either
soaked into crystals of the relevant kinase or co-crystallized with the
relevant kinase. X-ray data
are collected and programmed by using commercially available equipments and
softwares.
The identified ligands can be advanced into lead compounds by any number of
methods
known in the art. In certain embodiments, compound libraries are made based
upon the
identified fragments. In other embodiments, traditional medicinal chemistry
approaches axe
used.
In particular, when Tethering with extenders are used, the binding determinant
from the
extender can be merged with the identified fragment to make a conjugate
compound that is
equivalent or better than a lead compound derived from traditional high-
throughput screening.
Figure 3 illustrates one example of such a conjugate compound in which
subsequent
optimization led to a nanomolar kinase inhibitor. As shown, a cysteine mutant
of MEKl
(S150C) was made that placed a thiol at the bottom of the ATP binding pocket.
A pyrimidine
extender that had been previously been identified as a fragment that had
binding affinity for ATP
binding pocket of kinases was used to form a MEK1-extender covalent complex.
This complex
-49-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
was then used to identify a fragment that binds to the adaptive binding site
that was then merged
with the binding deternlinant from the pyrimidine extender to yield a 33 ~M
MEKl inhibitor
(compound 1). Acylation of the amine resulted in a 170 nM MEKl inhibitor
(compound 2).
Other potent inhibitors that resulted from simple modification of compound 1
include the
following compounds which inhibits MEKl with ICSO's of 80 nM, 50 nM, 30 nM,
and 10 nM
respectively.
HN''''~~"~.,.~.~ ! w ." ~ ~ =.1 I r
~H~N.sI I
3 4
H ~i'"'w.~~~~'
N H
~H~~,,~
H 5
H
Notably, the resulting submicromolar MEK1 inhibitors all preferentially
inhibit the inactive
form. No inhibition of the active form of MEKl was observed at concentrations
of compounds
2-6 at 10 p.M (and ATP concentrations of SO wM). In addition, these compounds
also showed
remarkable specificity for compounds that have yet to be optimized. For
example, neither
compounds 1 nor 2 inhibit Raf kinase at concentrations that inhibit MEKl
completely. In
addition, when the most potent of the above compounds (3, 4 and 6) were tested
in a panel of
kinases, as shown in Figure 4, these compounds were also very specific for the
inactive
conformation of MEKl. Only RAF showed any significant inhibition. However,
because RAF
is a kinase that is immediately upstream from MEKl, inhibition of RAF may also
be
therapeutically desirable.
Further details of the invention are illustrated in the following non-limiting
examples.
Example 1
Construction and Expression of EGFRl and Lck variants
Wild-type human Lck and wild-type human EGFR1 were cloned by RT-PCR from
poly(A)+ enriched mRNA from Jurkat cells and A431 cells, respectively. Jurkat
cells were
grown in suspension in 30 mL of medium containing 10% fetal bovine serum
(FBS), at a
concentration of 8.4 x 107 cells/mL. Approximately 40% of the Jurkat cells
were put into an
-50-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Eppendorf tube and pelleted. Adherent A431 cells were grown in DMEM containing
4 mM
glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, and 10% FBS. A T75
monolayer was
trypsinized and resuspended in 1X phosphate buffered saline. 28 mL of A431
cells at 5.6 x 106
cells/mL were pelleted into a second Eppendorf tube.
The RNA was isolated from each type of cells as follows. The pelleted cells
were lysed
with 1 mL Tri Reagent, microfuged atlSK rpm for 10 min at 4 °C, and the
supernatant was
transferred into a new tube. Chloroform (400 p.L) was added to the tube, which
was vortexed,
allowed to stay at room temperature for 5 min, and then microfuged for 15 min
at 4 °C. The
aqueous phase was transferred to a new tube, to which a half volume of 2-
propanol was added.
The tube was vortexed, allowed to stay at room temperature for 5 min, and then
microfuged for
10 min at 4 °C. The resulting RNA pellet was resuspended in 200 wL
deionized water.
Poly(A)+ mRNA was purified using an Oligotex purification kit (QIAgen), and
stored at -20 °C.
The first strand of cDNA was obtained by reverse transcription from the
poly(A)+
mRNA as follows. Oligonucleotides corresponding to SEQ ID NO:1 and SEQ ID N0:2
were
used as a reverse transcriptase primer for Lck and EGFR, respectively.
AGGGCCTCTCAAGGCCTCCTC SEQ ID N0:1
AGTTGGAGTCTGTAGGACTTGGC SEQ ID N0:2
Reactions containing 4 qL reverse transcriptase primer, 5 p,L poly(A)+ mRNA,
and 13
p.L deionized water were annealed by heating to 70 °C for 10 min, and
then chilled on ice. Two
microliters of reverse transcriptase (Powerscript) were added to a mixture of
8 ~,L SX first strand
buffer, 4 p,L dNTPs, and 4 pL DTT (100 mM).
The reverse transcription reactions containing the first strand of the cDNA
were each
next used in a polymerase chain reaction. Oligonucleotides SEQ ID N0:3 and SEQ
ID N0:4
were used as 5' and 3' PCR primers for Lck, respectively, and oligonucleotides
SEQ ID NO:S
and SEQ ID N0:6 were used as 5' and 3' PCR primers for EGFR, respectively.
CTAGGATATCCTCGAGCAAGCCGTGGTGGGAGGACGAG SEQ ID N0:3
CTAGGATATCAAGCTTTTCAGTCCTCCAGCACACTGCGCAG SEQ ID N0:4
CTAGGATATCCTCGAGCGCTCCCAACCAAGCTCTCTTGAG SEQ ID N0:5
CTAGGATATCAAGCTTTTCATTTGGAGAATTCGATGATCAACTCAC SEQ ID N0:6
One microliter of the first strand cDNA reaction was added to 1 p,L of PCR
primers, 1 p.L
25 mM dNTPs, 5 p,L lOX PFU buffer, 0.5 ~,L DNA polymerase (2:1 KlenTaq:Pfu-
turbo
(vol/vol)), and 41.4 pL deionized water. The resulting PCR reactions were
heated to 94 °C for 4
min, and then cycled 35 times as follows: 94 °C for 30 sec, 57
°G for 30 sec, and 72 °C for 1 min
-51-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
4S sec. FW ally the reactions were allowed to remain at 72 °C for 8
min, and then held at 4 °C
until the following cloning step.
The resulting duplex cDNA was cloned into an RSETB expression vector as
follows.
PCR products were purified on a QIAgen miniprep column, digested in a 80 ~L
volume with
XhoI and HindIII in 1X Buffer2/BSA (New England Biolabs) for 2 hr at 37
°C. Five
micrograms of pRSETB were also digested by XhoI and HindIII in the same
manner. The
resulting digestion products were also purified on a QIAgen miniprep column,
and then used in
ligation reactions. All ligation reactions (Boehringer Rapid Ligation kit)
contained 1 ~,L of
purified vector, and 1-2 ~,L of insert, and were performed according to the
manufacturer's
instructions. The ligated reactions (2 g,L) were transformed into ToplOF'
cells, and 1/10 of the
transformed cells were spread onto LB/amp plates (.100 g.g/mL). Resulting
colonies were
screened for insert by PCR, using the same reaction conditions described
above, and forward and
reverse primers SEQ ID N0:7 and SEQ ID N0:8, respectively; positive clones
were verified by
sequencing.
IS GACCACAACGGTTTCCCTCTAG SEQ ID N0:7
GTTATTGCTCAGCGGTGGCAGC . SEQ ID N0:8
The resulting wild-type Lck pRSETB construct, expressing residues 231-496 of
SEQ ID
N0:9 was altered to express a mutant Lck construct having 5323 mutated to
cysteine. The
mutation was designed to allow covalent attachment of a small molecule
extender by introducing
a cysteine residue into the target kinase in a position analogous to EGFRl
0797 of SEQ ID
NO:10. The resulting EGFRl pRSETB construct encoded residues 698-970 of SEQ ID
NO:1Ø
MGCGCSSHPE DDWMENIDVC ENCHYPIVPL DGKGTLLIRN GSEVRDPLVT SEQ ID N0:9
YEGSNPPASP LQDNLVIALH SYEPSHDGDL GFEKGEQLRI LEQSGEWWKA
QSLTTGQEGF IPFNFVAKAN SLEPEPWFFK NLSRKDAERQ LLAPGNTHGS
2S FLIRESESTA GSFSLSVRDF DQNQGEWKH YKIRNLDNGG FYISPRITFP
GLHELVRHYT NASDGLCTRL SRPCQTQKPQ KPWWEDEWEV PRETLKLVER
LGAGQFGEVW MGYYNGHTKV AVKSLKQGSM SPDAFLAEAN LMKQLQHQRL
VRLYAWTQE PIYIITEYME NGSLVDFLKT PSGIKLTINK LLDMAAQIAE
GMAFIEERNY IHRDLRAANI LVSDTLSCKI ADFGLARLIE DNEYTAREGA
3O KFPIKWTAPE AINYGTFTIK SDVWSFGILL TEIVTHGRIP YPGMTNPEVI
QNLERGYRMV RPDNCPEELY QLMRLCWKER PEDRPTFDYL RSVLEDFFTA
TEGQYQPQP
MRPSGTAGAA LLALLAALCP ASRALEEKKV CQGTSNKLTQ LGTFEDHFLS SEQ ID N0:10
3S LQRMFNNCEV VLGNLEITYV QRNYDLSFLK TIQEVAGYVL IALNTVERIP
LENLQIIRGN MYYENSYALA VLSNYDANKT GLKELPMRNL QEILHGAVRF
SNNPALCNVE SIQWRDIVSS DFLSNMSMDF QNHLGSCQKC DPSCPNGSCW
-S2-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
GAGEENCQKLTKIICAQQCSGRCRGKSPSDCCHNQCAAGCTGPRESDCLV
CRKFRDEATCKDTCPPLMLYNPTTYQMDVNPEGKYSFGATCVKKCPRNYV
VTDHGSCVRACGADSYEMEEDGVRKCKKCEGPCRKVCNGIGIGEFKDSLS
INATNIKHFKNCTSISGDLHILPVAFRGDSFTHTPPLDPQELDILKTVKE
S ITGFLLIQAWPENRTDLHAFENLEIIRGRTKQHGQFSLAVVSLNITSLGL
RSLKEISDGDVIISGNKNLCYANTINWKKLFGTSGQKTKIISNRGENSCK
ATGQVCHALCSPEGCWGPEPRDCVSCRNVSRGRECVDKCNLLEGEPREFV
ENSECIQCHPECLPQAMNITCTGRGPDNCIQCAHYIDGPHCVKTCPAGVM
GENNTLVWKYADAGHVCHLCHPNCTYGCTGPGLEGCPTNGPKIPSIATGM
1O VGALLLLLW ALGIGLFMRRRHIVRKRTLRRLLQERELVEPLTPSGEAPN
QALLRILKETEFKKIKVLGSGAFGTVYKGLWIPEGEKVKIPVAIKELREA
TSPKANKEILDEAYVMASVDNPHVCRLLGICLTSTVQLITQLMPFGCLLD
YVREHKDNIGSQYLLNWCVQIAKGMNYLEDRRLVHRDLAARNVLVKTPQH
VKITDFGLAKLLGAEEKEYHAEGGKVPIKWMALESILHRIYTHQSDVWSY
IS GVTVWELMTFGSKPYDGIPASEISSILEKGERLPQPPICTIDVYMIMVKC
WMIDADSRPKFRELIIEFSKMARDPQRYLVIQGDERMHLPSPTDSNFYRA
LMDEEDMDDVVDADEYLIPQQGFFSSPSTSRTPLLSSLSATSNNSTVACI
DRNGLQSCPIKEDSFLQRYSSDPTGALTEDSIDDTFLPVPEYINQSVPKR
PAGSVQNPVYHNQPLNPAPSRDPHYQDPHSTAVGNPEYLNTVQPTCVNST
2O FDSPAHWAQKGSHQISLDNPDYQQDFFPKEAKPNGIFKGSTAENAEYLRV
APQSSEFIGA
Mutagenesis of the Lck pRSETB construct was performed by PCR using sense and
antisense oligonucleotide of SEQ ID NO:1 l and SEQ ID NO:12, respectively.
CATGGAGAATGGGTGTCTAGTGGATTTTC SEQ ID N0:11
2S GAAAATCCACTAGACACCCATTCTCCATG SEQ ID N0:12
The mutagenesis PCR reactions contained 1 ~.L (approximately 300 ng) of
methylated
pRSETB plasmid isolated from bacteria transformed by pRSETB encoding wild-type
Lck
(residues 231-496), 2.S p.L each of sense and antisense primers (10 ~M stock
concentration), 1
~,L dNTPs (12.5 mM stock concentration), S ~L Pfu-turbo buffer, 37 ~.L
deionized water, and 1
30 ~,L Pfu-turbo polymerase. PCR reactions were heated to 9S °C for 1
min, and then cycled three
times through 9S °C for 30 sec, SS °C for 1 min, and 68
°C for 3 min. Finally the reactions were
allowed to remain at 68 °C for 10 min. DpnI (1 mL) was used to digest
the methylated parent
plasmid, and 1 ~,L of the digest containing the in vitro synthesized,
unmethylated, intact linear
PCR product, including the introduced mutation, was transformed into ToplOF'
cells, where the
3S PCR product was ligated to produce the pRSETB plasmid encoding mutant Lck.
For expression in insect cells, the pRSETB constructs expressing wild-type
EGFRI
residues 698-970 and the Lck mutant were subcloned into a pFastbacHTa vector
(GIBCO-BRL),
such that the resulting encoded proteins contained a (His)6 tag on their N-
termini. S' PCR
-S3-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
primers SEQ ID N0:13 and SEQ ID N0:14 were used to amplify Lck and EGFRI from
the
corresponding pRSETB constructs, respectively; each contain an NcoI site.
CTAGGATATCCCATGGGCAAGCCGTGGTGGGAGGACGAG SEQ ID N0:13
CTAGGATATCCCATGGCTCCCAACCAAGCTCTCTTGAG SEQ ID N0:14
S An EGFR1 construct having a (His)6 tag on its C-terminus instead of its N-
terminus was
constructed to assist in Ni-NTA binding of the expressed protein.
Furtherniore, as the first
EGFRl construct (residues 698-970 of SEQ ID NO:10) was not active, a longer
version was
produced. Two cloning steps were performed. Firstly, the region spanning BamHI-
HindIII was
removed from the polylinker of the plasmid pFastBacl, and replaced with an
annealed
oligonucleotide duplex of SEQ ID NO:15 and SEQ ID N0:16 encoding for a (His)6
tag. This
replacement into the pFastBacl vector obliterates the BamHI and HindIII sites,
and introduces
an NcoI site and a new HindIII site to produce the plasmid pFastBaclC-termHis.
GATCCTCCGAAACCATGGCTCGAGGCGGCCGCAAGCTTGATATCCCAACGACCGAAAACCTGTATTTTCAGGGCCAT
CACCATCACCATCACTAGC SEQ ID N0:15
IS
AGCTGCTAGTGATGGTGATGGTGATGGCCCTGAAAATACAGGTTTTCGGTCGTTGGGATATCAAGCTTGCGGCCGCC
TCGAGCCATGGTTTCGGAG SEQ ID N0:16
Secondly, a longer version of EGFR1 encoding residues 670-988 of SEQ ID N0:10
was
subcloned from the EGFRl FastBacHTa construct described above by amplification
of the
EGFRl coding sequence using primers to extend the coding sequence at the N-
and C-termini.
The primers used for this purpose correspond to SEQ ID N0:17 and SEQ 117
N0:18. The
resulting DNA was inserted into the pFastBaclC-termHis plasmid so as to
replace the NcoI-
HindIII segment. The resulting construct encoded for residues 670-988 of SEQ
ID NO:10, and
contained a (His)6 tag only on the C-terminus.
GGTACCCATGGGAAGGCGCCACATCGTTCGGAAGCGCACGCTGCGGAGGCTGCTGCAGGAGAGGGAGCTTGTGGAGC
2S CTCTTACA . SEQ ID N0:17
GGATCAAGCTTTTCAATGCATTCTTTCATCCCCCTGAATGACAAGGTAGCGCTGGGGGTCTCGGGCCATTTTGGA
SEQ ID N0:18
FastBac plasmids described above encoding the kinases of interest were
transformed into
DHlOBac cells to construct recombinant bacmids by transposition. Specifically,
approximately
S00 ng pFastBac in 1 wL was added to 49 wL l~ I~CM (10 mM Tris-HCl pH 7.7, 120
mM ICI,
?0 mM NaCl, 0.1% Triton X-100) and SO p.L PEG/DMSO competent cells. The cells
were
allowed to sit for 1S min at 4 °C, and then for 10 min at room
temperature. SOC (900 p,L) was
then added to the cells, which were shaken 4 hr at 37 °C. Two hundred
microliters of the cell
mixture was plated onto LB-agar plates containing 50 wg/mL kanamycin, 7 p.g/mL
gentamycin,
-54-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
g,g/mL tetracyclin, 100 ~g/mL Bluo-Gal, and 40 p,g/mL IPTG. Plates were grown
for 2 d at
37 °C, after which white colonies were picked for sequence
verification.
The resulting recombinant bacmids expressing the Lck 5323 mutant and EGFR1
residues
670-988 were transfected into StrJ cells for preparation of recombinant
baculovirus. Transfection
5 of the bacmids, and harvest and storage of the recombinant baculovirus were
performed
according to the manufacturer's instructions (GibcoBRL).
Expression c~v~d Purification
Recombinant baculovirus was used to express the EGFRl and mutated Lck
constructs in
10 High Five insect cells. High Five insect cells (Invitrogen) were grown to 2
X 106 cells/mL, and
then 50 mL of High Five cells were infected with 0.5 mL virus. Standard time
and temperature
of induction variation was used to optimize expression conditions. Typically,
the infected High
Five cells were grown for 2 d at 27 °C.
Insect cells were pelleted and washed with 5 mL PBS; cells were then lysed
with 1 mL
mammalian protein extraction reagent (MPER) solution (23.5 mL MPER (Pierce),
1.5 mL SM
NaCI, 35 p,L 14.3 M (3-mercaptoethanol, 250 mL protease inhibitor-EDTA),
rocking end over
end for 20 min at 4 °C. Lysate was placed in a tube and spun for 15 min
at 4 °C (15K, Sorval
SS-34 rotor). The aqueous layer between the pellet and the lipid layer was
transferred to a fresh
tube, and sufficient 50°J° glycerol was added to a final
concentration of 10% glycerol; the
solution was stored at -20 °C.
N-terminally (His)6-tagged Lck S323C mutant protein was purified from cell
lysates
using standard protein chromatography techniques. Specifically, the Lck S323C
was purified on
a 6 mL Ni-NTA-agarose column. The column was rinsed in deionized water at 2
mL/ min, and
then equilibrated in binding buffer (pH 8.0) containing 50 mM NaH2P04, 0.5 M
NaCI, and 5
mM (3-mercaptoethanol. 50 mL of lysate was added to 250 mL of the binding
buffer and loaded
onto the column at 4 mL/min. The column was washed at 2 mL/min first with
binding buffer
containing no imidazole, and then with binding buffer containing 10 mM
imidazole. Finally, the
protein was eluted at 2 mL/min with binding buffer containing 200 mM
imidazole.
C-ternlinally (His)6-tagged EGFRl (residues 670-988) was expressed and
purified on Ni-
NTA-agarose as described above for Lck mutant. However, for EGFR, it was
necessary to
follow the purification on the Ni-NTA column by purification on an S-sepharose
column (ion-
exchange). A 5 mL SP Sepharose FF (cation exchange) column was equilibrated
with buffer
-55-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
containing 20 mM Tris pH 7.5, 10 mM NaCI and 5 mM [3-mercaptoethanol. Three
milliliters
EGFRl diluted into ~7 mL of a solution of 20 mM Tris pH 7.5, 10 mM NaCl and 5
mM (3-
mercaptoethanol was loaded onto the column at 2 mL/min. Next, a gradient from
0-100% Buffer
B (20 mM Tris pH 7.5, 1.0 M NaCI, 5 mM (i-mercaptoethanol) was run in 45 min,
and the eluate
collected in fractions. The EGFR1 elutes at about 50% buffer B, corresponding
to 0.5 M NaCI.
Example 2
Construction and Expression of MEKl variants
The amino acid sequence of MEKl is shown here as SEQ ID N0:19.
1O MPKKKPTPIQ LNPAPDGSAV NGTSSAETNL EALQKKLEEL ELDEQQRKRL SEQ ID N0:19
EAFLTQKQKV GELKDDDFEK ISELGAGNGG VVFKVSHKPS GLVMARKLIH
LEIKPAIRNQ IIRELQVLHE CNSPYIVGFY GAFYSDGEIS ICMEHMDGGS
LDQVLKKAGR IPEQILGKVS IAVIKGLTYL REKHKIMHRD VKPSNTLVNS
RGEIKLCDFG VSGQLIDSMA NSFVGTRSYM SPERLQGTHY SVQSDIWSMG
IS LSLVEMAVGR YPIPPPDAKE LELMFGCQVE GDAAETPPRP RTPGRPLSSY
GMDSRPPMAI FELLDYIVNE PPPKLPSGVF SLEFQDFVNK CLIKNPAERA
DLKQLMVHAF IKRSDAEEVD FAGWLCSTIG LNQPSTPTHA AGV
The entire coding sequence of MEKl was subcloned into the expression plasmid
pGEX-
6P-1 (Invitrogen) using 5' and 3' PCR primers (SEQ ID N0:20 and SEQ ID N0:21,
20 respectively), along with a cormmercially available MEKl cDNA (Mekl cDNA in
pUSEamp,
Upstate #21-106) as a PCR template.
5'-BamHl CGCGCGGATCCATGCCCAAGAAGAAGCCGACGCCCATCCAGC SEQ ID NO:20
3'-Xhol CGTAGCTCGAGTCAGGTACCGGCAGCGTGGGTTGGTGTGCTGGG SEQ m N0:21
The resulting amplified MEKl DNA was subcloned into pGEX-6P-1 at Ban~HI and
XlaoI. The resulting plasmid, pMekl-001, encodes a GST-MEKl fusion protein in
which the
MEKI portion contains a 14 a~.nino acid insertion between residues M1 and P2
of the GenBank
25 reported sequence, as well as three single amino acid substitutions from
the GenBank reported
sequence: M274L G392S, and V393T, numbering relative to SEQ ID NO:1. Following
cleavage
of this fusion protein with Precission protease (Amersham Biosciences), the
liberated MEKI
protein contains an additional five non-native amino (GPLGS) acids at the
amino terminus.
30 MEIiI CoT'ZStI°ucts
The surface accessibility of native cysteines was assessed by mass
spectrometry,
according to their reactivity with cystine in the presence of 0-16 mM (3-
mercaptoethanol. Of the
-56-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
six naturally occurring cysteines, C207, C277, and C341 were determined to be
reactive
cysteines and were "scrubbed". In addition, a cysteine was introduced in a
location (S 150C)
analogous to that of C797 of EGFRl. All mutations have been introduced using
long-range PCR
with a pair of complementary oligonucleotides containing the desired mutation.
The oligos for making the constructs were:
C121S-s GGTGCTGCATGAGTCCAACTCCCCGTACATAG SEQID
NO:22
C142S-s GCGAGATCAGCATCTCCATGGAGCACATGGATG SEQID
N0:23
S150C-s CATGGATGGTGGGTGCTTGGATCAAGTGCTG SEQID
N0:24
C207S-s GGGAGATCAAACTCTCCGATTTTGGGGTCAG SEQID
N0:25
C207A-s GGGAGATCAAACTCGCCGATTTTGGGGTCAG SEQID
N0:26
S218D,S222D-sCGGGCAGCTAATTGACGACATGGCCAACGACTTCGTGGGAACAAGGSEQID
N0:27
S218D,S222D-sCGGGCAGCTAATTGACGACATGGCCAACGACTTCGTGGGAACAAGGSEQID
N0:28
C277S-s GAGCTGCTGTTTGGATCCCAGGTGGAAGGAG SEQID
N0:29
C341S-s GGATTTTGTGAATAAGTCCTTAATAAAGAACCCTG SEQID
N0:30
C341M-s GGATTTTGTGAATAAGATGTTAATAAAGAACCCTG SEQID
N0:31
C376S-s GACTTCGCAGGCTGGCTCTCCTCCACCATTGGGCTTAACCSEQID
N0:32
Expression of Recoyfabinant MEK1 afzd Mutants
A frozen glycerol stock of E cali (Rosetta DE3 competent cells from Novagen)
containing the desired pGEX-MEK1 construct is used to inoculate 50 mL 2xYT
media
containing 150 p.g/mL ampicillin and 30 p.g/mL chloramphenicol; the resulting
culture is grown
overnight at 37 °C. A portion of the overnight culture (10-15 mL) is
then used to inoculate 1.5 L
2xYT media containing 150 p.g/mL ampicillin and 30 ~.g/mL chloramphenicol, and
the culture is
grown at 37 °C until OD6oo ~ 0.7-1Ø At this point, the cultures are
chilled at 4 °C for 30-60
min; after chilling, IPTG is added to 0.2 mM, and cultures are incubated
overnight at room
temperature with shaking at 225 rpm (20-22 °C).
Cells are harvested by centrifugation at 5000 rpm, media is discarded, and the
pellet is
resuspended in 50 mL freshly made lysis buffer (lx phosphate buffered saline
(PBS), 400 mM
KCI, 1 M urea, 1 tablet Complete Protease Inhibitor Cocktail, 1% (v/v)
aprotinin, DNase I (100
units/mL)). Cells are kept cold during the resuspension procedure, and
immediately after the
cells are resuspended, phenylmethyl sulfonyl fluoride (PMSF) is added to a
final concentration
of 2 mM. Cells are lysed by passing through a micro-fluidizer four separate
times. Lysate is
kept on ice, and inunediately spun at 16,000 rpm at 4 °C for 30 min.
While the lysate is
spinning, a glutathione agarose column is equilibrated with Wash Buffer #1 (lx
PBS, 400 mM
KCI, 1 M urea). Supernatant is removed from the spun lysate, and immediately
loaded onto the
.v _57_
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
equilibrated column at 2-3 mL/min. The column is washed first with Wash Buffer
#1 until the
OD280 drops to a baseline absorbance level, and then with Wash Buffer #2 (lx
PBS, 400 mM
KCl) for several minutes to remove the urea. The bound GST-MEKI fusion protein
is eluted
with Elution Buffer (20 mM HEPES pH 8.4, 100 mM KCI, 10 mM glutathione, 1 mM
DTT).
The column can be regenerated by stripping with 6 M guanidine-HCl and washing
with DI water
after stripping. Next, GST is cleaved off the fusion protein by addition 60
p,L Prescission
Protease (Amersham Biosciences); the digest reaction is transferred into
10,000-14,000 mwco
dialysis tubing and dialyzed against 4 L of 20 mM HEPES pH 7.4, 150 mM NaCI, 1
mM DTT
overnight at 4 °C.
Subsequently the 'digest reaction is removed from the dialysis tubing, and spm
at 16,000
rpm at 4 °C for 30 min. While the digest reaction is spinning, a
glutathione agarose column is
washed with Wash Buffer #3 (20 mM -HEPES pH 7.4, 150 mM NaCI, 1 rnM DTT). The
supernatant is loaded onto the equilibrated column at 1-3 mL/min, and then the
colurm is
washed with Wash Buffer #3 until the OD28o drops to baseline. Flow-through is
collected until
baseline is reached. The flowthrough is then mixed 1:1 with Dilution buffer
(20 mM HEPES pH
8.4, 1 mM DTT), to malce a solution that is 20 mM HEPES pH 8.0, 75 mM NaCI. A
Q-
Sepharose column connected in series with a prepacked 5 mL glutathione agarose
column is
equilibrated with Low Salt Buffer #1 (20 mM HEPES pH 8.0, 75 mM NaCI, 1 mM
DTT). The
diluted flowthrough is loaded onto the equilibrated Q-Sepharose column at 1-3
mL/min, and the
resulting flowthrough is collected. After the entire sample is loaded, the
column is washed with
Low Salt buffer #1 (20 mM HEPES pH 8.0, 75 mM NaCI, 1 mM DTT), and the
flowthrough
containing MEKl is collected until the ODZBO reaches baseline. Bound protein
(GST and
impurities) is eluted by washing the column with High Salt buffer #1 (20 mM
HEPES pH 8.0,
750 mM NaCI, 1 mM DTT), and collected for analysis.
The flowthrough containing MEKl is then mixed with saturated ammonium sulfate
solution (3.9 M), to a final concentration of 1.2 M ammonium 'sulfate. The
resulting solution is
then loaded at 2-3 mL/min onto an HIC phenyl-Sepharose column that has been
equilibrated
with High Salt Buffer #2 (20 mM HEPES pH 7.4, 1.2 M ammonium sulfate). After
loading, the
column is washed with High Salt Buffer #2 until the ODZBO drops to baseline. A
linear gradient
is run from 20 mM HEPES pH 7.4, 1.2 M ammonium sulfate to 20 mM HEPES pH 7.4
with no
ammonium sulfate over 30 min, and 4 mL fractions are collected. The fractions
are run on a gel
to determine which fractions to pool. The pooled fractions are then dialyzed
overnight at 4 °C
-58-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
against 4 L of 20 mM HEPES pH 7.4, 150 mM NaCI, in the absence of DTT.
Finally, the pooled
fractions are dialyzed again against 2 L of 20 mM HEPES pH 7.4, 150 mM NaCI
for 2-4 hr. The
dialyzed protein is quantitated, divided into aliquots and stored frozen at -
80 °C. One
absorbance unit at 280 nm is equivalent to a concentration of 1.86 mglmL, and
1 wg of MEKl is
equivalent to 22.8 pmol, as MEKl has a MW of 43,832.
Example 3
Activi , Assay
MEKI ELISA Assa,~
Phosphorylation of ERK2 by MEKl is measured for two reaction formats. The
first
reaction format is a Raf-~MEKl-~ERK2 cascade where constitutively active
truncated Rafl,
inactive MEKl, inactive biotinylated ERK2, and dephosphorylated MBP (Myelin
Basic Protein)
are present. The second reaction format uses activated MEK1, biotinylated
ERK2, and
dephosphorylated MBP in the absence of Raf. Results can be compared to
determine whether a
compound preferentially inhibits the inactive conformation of MEKl over the
active
conformation of MEK1.
Both reaction formats are run in the presence and absence of compounds, and
use ELISA
as a readout of the extent of phosphorylation of the biotinylated ERK2. For
either format, where
the activity of a potential inhibitor is unknown, generally two sets of
experiments are run. In the
first set, three final concentrations of compound are used, e.g., 50 pM, 10
p.M and 2 p,M. In the
second set, nine concentrations of the compound with 2 fold dilutions are used
to determine the
ICSo for the compound; the concentrations of the compound used depend on the
activity observed
in the three-point experiment. Typical stock concentrations of a moderately
active compound in
a 9-point experiment are 1 mM, 0.5 mM, 0.25 mM, 0.125 mM, 62.5 p,M, 31.2 p,M,
15.6 p.M, 7.8
p.M and 3.9 ~M. The corresponding final concentrations of compound in the
phosphorylation
reaction are 20 p.M, 10 pM, 5 p,M, 2.5 q,M, 1.25 ~M, 0.625 pM, 0.312 p,M,
0.156 p.M, and 0.078
~M. For less active compounds, the most concentrated final concentration of
compound would
be 200 pM, and for more active compounds, the most concentrated final
concentration of
compound would be 2 p.M. Biotinylation of ERK and preparation of ELISA capture
plates axe
described below, followed by conditions for the two reaction formats, and
details on post- .
reaction processing.
-59-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Inactive ERK2 (Cell Signaling #6082) is biotinylated as follows. Twenty-five
microliters of lOX PBS and 200 p.L of 50 mM carbonate buffer pH 9.0 are added
to 250 ~.L of
ERK2 at 2 mg/mL; the resulting solution is kept on ice for 10 min. Next, sulfo-
NHS-LC-LC-
biotin (Pierce) is freshly dissolved in solution to a final concentration of 2
mglmL, and 10 ~,L of
the biotin solution is added immediately to the ERK2 solution. The resulting
reaction is
incubated at room temperature for 1 hr, after which 100 p,L of 3 M
ethanolamine is added to
quench the reaction. Five hundred microliters of the quenched reaction are
loaded onto a Naps
column, discarding the flowthrough. The remaining 85 p,L of the quenched
reaction are then
loaded onto the same column, while collecting the flowthrough, followed by 715
pL Tris-
buffered saline (1X TBS: 10 mM Tris pH 7.5, 150 mM NaCI), while continuing to
collect the
flowthrough. Recovery of biotinylated, inactive ERK2 from the Naps column can
be monitored
by Bradford assay (Bio-Rad Protein Assay Dye Reagent #500-0006) according to
manufacturer's
instructions. Biotin-ERK2 is stored at-20 °C in 1X TBS containing 10%
glycerol.
Avidin-coated capture plates are prepared by adding 100 ~.L of NeutrAvidin
(Pierce
#31000) in PBS at 0.040 mg/mL to each well of 96- well polystyrene plates
(NIJNC brand
maxisorp, VWR #442404). After addition of the NeutrAvidin, the plates are
covered and
allowed to sit at room temperature for 2-4 hr, or overnight at 4 °C.
NeutrAvidin is then
aspirated, and 150 p,L of BLOCK solution (0.05 g/mL BSA, 1X TBS, 0.1% Tween-
20) is added
to each well. The plates are allowed to sit at room temperature for 0.5-2 hr,
until the
phosphorylation reactions are ready to be transferred to the capture plate.
Phosphorylation Cascade Reactions Using Inactive ~IEKI
Typical phosphorylation reactions are performed in either eppendorf tubes or
96-well
plates with conical bottoms (Costar #3363). . The phosphorylation reactions in
this reaction
format contain in a 50 ~.L total volume the following components: 20 ~,g/mL
MBP (Upstate
#13-110), 150 nM biotin-ERK2, 0.7 nM Rafl (residues 306-648, N-terminally GST-
tagged,
Upstate # 14-352), 10 nM MEK1, 4.5 mM MgCl2, 100 ~,M NaOV03, 30 mM Tris HCl
(pH 7.5),
120 mM NaCI, 6 mM DTT, 0.0067% Triton X-100 (vol/vol) and 50 p,M ATP; all
concentrations
are final. Forty-five microliters of all reagents except the ATP, MgCl2 and
NaOV03 are added to
1 pL of stock concentrations of compound in DMSO; thus the phosphorylation
reactions using
inactive MEK1 contain a final amount of DMSO that is 2% by volume. Addition of
5 p,L of a
-60-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
solution of ATP, MgCl2, and NaOV03, each 10 fold higher in concentration than
their respective
final concentrations, starts the phosphorylation reaction. Reactions are
allowed to proceed 30
min at room temperature with gentle shaking.
Phosphosylatiofz Reactions Ilsihg Active MEKI
Typical phosphorylation reactions are performed in either eppendorf tubes or
96-well
plates with conical bottoms (Costar #3363). The phosphorylation reactions in
this fornlat
contain in a 50 p,L total volume the following components: 20 ~.g/mL MBP
(Upstate #13-110),
150 nM biotin-ERK2, 1 nM active MEKl (Upstate #14-429), 4.5 mM MgCh, 100 p,M
NaOV03,
30 mM Tris HCl (pH 7.5), 120 mM NaCI, 6 mM DTT, 0.0067% Triton X-100 (vol/vol)
and 50
~.M ATP; all concentrations are final. The concentration of active MEKl used
is lower than the
concentration of inactive MEKl in the format above, in order to keep readout
in the linear range.
Forty-five microliters of all reagents except the ATP, MgCI~ and NaOV03 are
added to 1 p,L of
stock concentrations of compound in DMSO; thus the phosphorylation reactions
using active
MEK1 also contain a final amount of DMSO that is 2% by volume. Addition of 5
p,L of a
solution of ATP, MgGl2, and NaOV03, each 10 fold higher in concentration than
their respective
final concentrations, starts the phosphorylation reaction. Reactions are
allowed to proceed 30
min at room temperature with gentle shaking.
Post-~eactiorz Ti°eatf~2eszt
Post-reaction treatment is the same for both reaction formats. After reaction,
the solution
phase phosphate-transfer reactions are stopped by addition of 75 p.L stop
buffer containing 0.4 M
EDTA pH 7.5, 1% BSA, 1X TBS and 0.1% Tween-20. At this point, the BLOCK is
removed
from the prepared avidin-coated capture plates, and a 100 pL portion of each
stopped reaction is
transferred to a well of the plate. Bibtinylated ERK2 is captured on the
surface of the avidin-
coated polystyrene plate by incubation of the plate at room temperature with
gentle shaking for
1-2 hr. Subsequently; the reaction mixture is aspirated and the plate is
incubated with a primary
polyclonal antibody (Cell Signaling #9101) that recognizes the activation loop
of ERK2
phosphorylated on T202 and Y204, the antibody diluted 1000 fold in a solution
containing final
concentrations of 1% BSA, 1X TBS and 0.1% Tween-20 by volume. The capture
plate is
incubated with the primaxy antibody solution at room temperature with gentle
shaking for 2-3 hr
prior to aspiration and addition of 100 ~,L of the secondary antibody, which
is horseradish
-61-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
peroxidase (HRP)-conjugated Goat anti Rabbit IgG, (Zymed #62-6120) that has
been diluted
1000 fold in 1% BSA, 1X TBS, 0.1% Tween 20. The secondary antibody is
incubated with the
plate at room temperature for 1-2 hr with gentle shaking, the solution is
aspirated, and the wells
are then washed gently 3 times with 1X PBS with 0.05% Tween-20. The amount of
phosphorylated ERK2 present on the capture plate is quantitated using the
ImmunoPure TMB
substrate kit (Pierce #34021). After the PBS is aspirated, 100 p.L of a
freshly made TMB/H2O2
solution at room temperature containing equal volumes of peroxidase substrate
solution (TMB,
#1854050) and H202 solution (#1854060) is added to the wells, and the plate is
incubated at
room temperature with gentle shaking for 5-20 min. Color development is
stopped by adding
100 p.L of 2.5 M H2S04 to each well of the capture plate and shaking gently
for 1-2 min.
Absorbance of the substrate is measured at 450 nm.
EGFRl arid Lck ELISA Assay
The ELISA assay for EGFRl and Lck are generally similar to that described
above for
MEKl except that biotinylated E4Y substrate is used instead of ERK2. Typical
EGFRI or Lck
kinase assays contain 0.75% BSA, 30 mM Tris pH 7.5, 30 mM MgCl2, 18 mM MnCl2,
45 ~,M
Na2V03, 0.5 mM DTT, 100 pM EGFR or Lck kinase, 30 ~g/ml biotinylated E4Y, and
60 ~M
ATP. Bound substrate/reaction product was reacted with HRP-conjugated anti-
phospho-tyrosine
antibody instead of sequentially with anti-phospho-p44/42 ERKl/2 antibody and
HRP-
conjugated anti-rabbit antibody for the MEKl assays.
MEKI I~TTestef f~ Assay
The MEK1 ELISA does not distinguish between Raf inhibition and MEKl
inhibition.
Therefore, a Western assay was established for independently monitoring Raf
activity. This
assay has a ten fold lower throughput (8-10 compounds per week) than the
ELISA, but it allows
for independent analyses of both MEKl and Raf inhibition in the same assay.
Briefly, assays are
carried out as described for the ELISA format with the exception that ERK2 is
used in place of
biotinylated ERK2 and reactions are ternzinated with the addition of SDS-PAGE
gel loading
buffer. Following SDS-PAGE electrophoresis and transfer to PVDF, transfer
membranes are
incubated overnight with primary antibody in either TBST with 5% BSA and anti
MEKl, anti
phospho MEK1, or anti ERK (Cell Signaling #9122, #9121, and #9102
respectively) or TBST
with 5% nonfat dry milk and anti-phospho ERK (Cell Signaling #9101). All
transfer membranes
-62-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
are then incubated for two hours in TBST with 5% nonfat dry mills and HRf-
conjugated anti-
Rabbit antibody (ZyMed #62-6120) and HRP activity quantified using ECL plus
(Amersham
#RPN2132).
Example 4
Tethering
EGFRl
Tethering was performed on the inactive confornlation of EGFRl (not
phosphorylated on
Y745) using Cys797 as the reactive thiol. The disulfide containing monophore
library was
screened in pools of 10. Using 2 q,M EGFRl, 500 ~M library pool, and 600 ~M
BME, 252
compounds gave >50% conjugation to C97. These 252 compounds were re-tested as
isolated
compounds using 2 q,M EGFRl, 50 ~.M discrete compound, and 600 p.M BME. In
this manner,
214 (85%) screening hits were confirmed. The identified ligands showed clear
preference for
some chemical classes (axomatic 5 and 6 carbon ring systems and aromatic 5,6
carbon
heterocycles, separated from the thiol by a single methylene linker) while
other chemical classes
were not selected (aliphatic chains, aliphatic 5 carbon rings, and aliphatic 6
carbon rings,
separated from the thiol by a 2 or 3 methylene ,linker). Not surprisingly,
these ligands showed
clear enrichment for a number of purine-like compounds, including pyrazines,
pyridines,
quinolines, quinoxalines, pyrazoles, thiazoles, and other substituted
benzenes.
MEKI
Tethering was performed on the inactive conformation (not phosphorylated on
either
5223 or 5227) of a MEI~1 a mutant containing a cysteine at 5150 (ari amino
acid corresponding
to C797 of EGFRl). A partial library screen showed not only a very similar hit
rate to that seen
with the EGFR (1.4% with greater than 50% conjugation), but also a strong
structural similaxity
to the EGFR purine pocket screening hits. Consequently, instead of repeating
the entire library
screen, the hits from EGFRI were tested individually against MEI~l.
Example 5
This example describes the synthesis of
HN~S~S~NH~
~N~ ~ J
N N
H
-63-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
which as prepared according to Scheme 1 and the procedure below.
SCHEME 1
H
CI CI HZN~S~S~N'Boc HN~S~S~N'Bac H2N~NH2 HN~S~S~N'Boc
N 1
Et3N, EtOH '~~' CI NJ 7 H2N~N~~ g
H
1. ~N~ HCI
CI ~ HN~S~S~NHz
O DCM, 0°C
N
OH
2. Intermediate 8 ~N~N NJ 9
H
DIEA, DCM, 0°C
3. TFA, DCM
To a solution of 4,6-dichloropyrimidine (0.500 g, 3.358 mmol) in ethanol (8.3
mL) was
added [2-(2-Amino-ethyldisulfanyl)-ethyl]-carbamic acid tert-butyl ester
(0.706 g, 2.798 mmol)
and triethylamine (0.417 mL, 2.994 mmol). The reaction mixture was refluxed
overnight under
N2 (12 h). The solvent was evaporated and the crude reaction mixture was
purified by silica gel
chromatography (50% ethyl acetate ("EtOAc") in hexanes) to provide
intermediate 7 (0.662 g) as
a white solid in 54% yield. 1H NMR (400 MHz, CHLOROFORM-D) ppm 1.47 (s, 9 H)
2.78 (d,
J 13.99 Hz, 2 H) 2.94 (d, J 10.94 Hz, 2 H) 3.48 (m, 2 H) 3.76 (s, 2 H) 5.07
(s, 1 H) 6.56 (s, 1
H) 8.37 (s, 1 H). LCMS M+1=365.
Intermediate 7 (0.200 g, 0.548 mmol) in neat ethylene diamine (5 mL) was
refluxed
under N2 overnight. The reaction mixture was diluted with EtOAc and
partitioned with saturated
NaHC03. The aqueous layer was extracted with EtOAc (3x). The combined organic
layers were
rinsed with saturated NaCI, dried over Na2S04, filtered and the solvent was
evaporated, to
provide intermediate 8 (0.145 g) as a light yellow solid in 68% yield.
Intermediate 8 was used
without purification in the next step. 'H NMR (400 MHz, MeOD) ppm 1.42 (s, 9
H) 2.79 (m, 3
H) 2.87 (t, J--6.61 Hz, 3 H) 3.31 (d, J--8.90 Hz, 3 H) 3.54 (t, .I--6.36 Hz, 3
H) 5.49 (s, 1 H) 7.92
(s, 1 H). LCMS M+1=389.
To a solution of acrylic acid (0.012 mL, 0.172 mmol) in 1 mL dichloromethane
("DCM")
at 0 °C was added (chloromethylene) dimethyl ammonium chloride. The
reaction mixture was
stirred for 1 hr at 0 °C under N2. This solution was then added
dropwise to a stirred solution of
intermediate 8 (0.067 g, 0.172 mmol) and N, N-diisopropylethylamine (0.061
rnL, 0.343 mmol)
in 1 mL DCM at 0 °C. After stirring for 1 hr under N2, the reaction
mixture was diluted with
DCM, rinsed with 1 M Na2C03, dried over Na2S04, filtered and concentrated down
to a yellow
-64-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
residue. The crude product was deprotected with 1:l TFA/DCM (2 mL) where TFA
is
trifluoroacetic acid. The mixture was stirred for 30 min and the solvent
evaporated. The residue
was purified using reverse phase prep. HPLC to provide the titled compound 9.
1H NMR (400
MHz, MeOD) ppm 2.78 (m, 3 H) 3.11 (m, 3 H) 3.27 (s, 3 H) 3.50 (m, 3 H) 5.48
(m, 1 H) 5.58 (s,
1 H) 6.03 (d, J--5.60 Hz, 2 H) 7.94 (s, 1 H). LCMS M+1=343.
Example 6
This example describes the synthesis of
H HN~S'S~NHz
~N~ . J
N N
H
which as prepared according to Scheme 2 and the procedure below.
SCHEME 2
CI I ~ CI HzNMS.S~H.BOC HNMS'S~H'Boc HZN~NH, H N ~ ~S.S~NH=
N
NvN Et3N, EtOH J 1° N J 11
CI N H
1. \ ~0
v 'CI H HNMS'S~NH,
_DIEA. DCM. 0°C N
I
2. TFA, DCM ~N~N NJ 12
H
To a solution of 4,6-dichloropyrimidine (1.200 g, 8.059 mmol) in ethanol (20
mL) was
added [2-(3-Amino-propyldisulfanyl)-ethyl]-carbamic acid tef°t-butyl
ester (2.147 g, 8.059
mmol) and triethylamine (1.211 mL, 8.623 mmol). The reaction mixture was
refluxed overnight
under NZ (12 hr). The solvent was concentrated under reduced pressure and the
crude reaction
mixture was purified by silica gel chromatography (50% EtOAc in hexanes) to
provide
intermediate 10 (1.978 g) as a clear oil in 78% yield. 1H NMR (400 MHz, MeOD)
ppm 1.33 (s, 9
H) 1.89 (m, 1 H) 2.67 (t, J--6.87 Hz, 4 H) 3.23 (m, 3 H) 3.40 (s, 2 H) 6.40
(s, 1 H) 8.12 (s, 1 H).
LCMS M+1=379.
Intermediate 10 (1.000 g, 2.639 mmol) in neat ethylene diamine (10 mL) was
refluxed
under N2 overnight. The reaction mixture was diluted with EtOAc and
partitioned with saturated
NaHCO3. The aqueous layer was extracted with EtOAc (3x). The combined organic
layers were
rinsed with saturated NaCI, dried over Na2S04, filtered and concentrated under
reduced pressure
to provide internzediate 11 (0.605 g) as a white solid in 57% yield.
Intermediate 11 was used
-65-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
without purification iil the next step. 1H NMR (400 MHz, MeOD) ppm 1.32 (s, 9
H) 1.87 (m, 2
H) 2.69 (m, 4 H) 3.20 (ni, 8 H) S.3S (s, 1 H) 7.79 (s, 1 H). LCMS M+1=403.
To a solution of intermediate 11 (0.100 g, 0.248 mmol) in 1 mL DCM at 0
°C was added
acryloyl chloride (20.2 ,uL, 0.248 mmol) and N, N-diisopropylethylamine (86.5
,uL, 0.497
S mmol). The resulting dark brown solution was stirred at 0 °C for 30
min. The reaction was
diluted with DCM, rinsed with 1 M NaZCO3, dried over NaiS04, filtered and
concentrated to a
yellow solid. The crude product was deprotected with 1:1 TFA/DCM (2 mL). The
mixture was
stirred for 30 min and the solvent evaporated. The residue was purified using
reverse phase prep
HPLC to provide the titled compound 12. 1H NMR (400 MHz, MeOD) ppm 1.94 (m, 2
H) 2.72
(t, J--7.12 Hz, 2 H) 2.83 (t, J--6.61 Hz, 2 H) 3.18 (m, 4 H) 3.35 (s, 4 H)
S.S7 (t, J--5.60 Hz, 1 H)
5.62 (s, 1 H) 6.11 (d, J 6.10 Hz, 2 H) 8.01 (s, 1 H). LCMS M+1=357.
Example 7
This example describes the synthesis of
H HN~S'S~NH2
N\ \ N
l' J
IS H N
which as prepared according to Scheme 3 and the procedure below.
SCHEME 3
I ' Boc
CI~CI HzN~S~S~N.Boc HN'~S~S~N~Boc ~o~p~p~ Boc.N~S.S~N.Boc
TNvN / N ~N
Et3N, EtOH CI ~N CI~N I
,4
.
NH3 . Boc.N~S.S~N~Boc 'I. HN~S~S~NHz
CI
\~°'I ~J
HZN NJ 15 2. TFA, DCM - H \N 16
4,6-Dichloropyrimidine (2.0 g, 13.42 mmol), [2-(2-Amino-ethyldisulfanyl)-
ethyl~-
carbamic acid tert-butyl ester (14.77 mL, 1.0 M in DCM, 14.77 mmol), and
triethylamine (9.35
mL, 67.10 mmol), were dissolved in 70 mL ethanol and heated to 8S °C
for 16 h. The reaction
was cooled to ambient temperature, the solvent evaporated, and the residue
slurried in ethyl
ether. The mixture was filtered, the filtrate concentrated and purified by
flash chromatography
2S (20% EtOAc in hexanes) yielding compound 13 (2.86 g, 7.8 mmol, S8% yield) .
1H NMR (400
-66-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
MHz, CDCl3): 8 1.47 (s, 9 H), 2.78 (m, 2 H), 2.95 (m, 2 H), 3.48 (m, 2 H),
3.75 (m, 2 H), 5.14
(m, 1 H), 6.56 (s, 1 H), 8.37 (s, 1 H). ESI-MS T~~/z: 365 (M + H)+.
Compound 13 (1.40 g, 3.84 mmol), N,N-dimethylaminopyridine ("DMAP") ( 0.047 g,
0.384 mmol), and di-tert-butyl-dicarbonate (5.02 g, 23.02 mmol) were dissolved
in 85 mL dry
tetrahydrofuran ("THF"). The solution was refluxed for 6 h, the solvent
evaporated, and the
residue purified by flash chromatography (20% EtOAc in hexanes) yielding
compound 14 (1.74
g, 3.74 mmol, 97% yield). 1H NMR (400 MHz, CDC13): b 1.52 (s, 18 H), 1.60 (s,
9 H), 2.93 (m,
4 H), 3.92 (m, 2 H), 4.36 (m, 2 H), 8.14 (s, 1 H), 8.67 (s, 1 H). ESI-MS m/z:
566 (M + H)+.
Compound 14 (1.74 g, 3.08 mmol) was dissolved in 100 mL, 7 N NH3 in methanol
and
the solution stirred in a sealed glass bomb at 90 °G for 6 days. The
reaction was cooled to
ambient temperature, the solvent evaporated, and the residue slurried in ethyl
ether. The mixture
was filtered the filtrate concentrated and purified by flash chromatography
(50% EtOAc in
hexanes) yielding compound 15 (0.626 g, 1.15 mmol, 37% yield). 1H NMR (400
MHz, CDC13):
8 1.51 (s, 18 H), 1.56 (s, 9 H), 2.91 (m, 4 H), 3.91 (m, 2 H), 4.28 (m, 2 H),
4.87 (m, 2 H), 7.15 (s,
1 H), 8.34 (s, 1 H). ESI-MS mlz: 546 (M + H)+.
Compound 15 (0.626 g, 1.15 mmol) was dissolved in 12 mL dry DCM under N2, N,N-
diisopropylethylamine ("DIEA") (0.401 mL, 2.3 mmol) was added and the solution
chilled to 0
°C on an ice bath. After stirring on ice for 20 min, acryloyl chloride
(0.104 mL, 1.15 mmol) was
added arid the reaction was allowed to stir for an additional 30 min. The
volatiles were
evaporated and the residue slurried in ethyl ether. The mixture was filtered,
the filtrate
concentrated and redissolved in 10 mL dry DCM. TFA (10 mL) was added and the
solution
stirred at ambient temp for 30 min. The solvent was removed under reduced
pressure and the
crude residue purified by reverse-phase preparatory HPLC to afford compound 16
(0.076 g,
0.254 nunol, 22%). 1H NMR (400 MHz, CD30D): S 2.85 (m, 4 H), 3.18 (m, 2 H),
3.69 (m, 2
H), 5.82 (m, 1 H), 6.35 (m, 2 H), 6.65 (s, 1 H), 8.27 (s, 1 H). ESI-MS m/z:
300 (M + H)+.
Example 8
This example describes the synthesis of
HN~S'S~NH2
O ~ N
w ~J
~N N
H
-67-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
wluch as prepared according to . the procedure of Example 7 except
substituting [2-(3-Amino-
propyldisulfanyl)-ethyl]-carbamic acid tert-butyl ester for [2-(2-Amino-
ethyldisulfanyl)-ethyl]-
carbamic acid tert-butyl ester.1H NMR (400 MHz, CD30D): S 1.91 (m, 2 H), 2.67
(m, 2 H), 2.79
(m, 2 H), 3.14 (m, 2 H), 3.48 (m, 2 H), 5.81 (m, 1 H), 6.31 (m, 2 H), 6.63 (s,
1 H), 8.24 (s, 1 H).
ESI-MS m/z: 314 (M + H)+.
Example 9
This example describes Tethering with extenders on the inactive conformation
of MEKl.
A cysteine mutant of MEKl S150C that also included the following mutations
C207A, C277S,
C376S was used for the following labelling procedure. A frozen aliquot of
MEI~l (20 mM
HEPES pH 7.4 150 mM NaCI) was thawed, and DTT was added to a final
concentration of 2
mM. An extender, stored at a concentration, of 100 mM in DMSO, was added to
the protein so
that the final concentration of extender was 1 mM. Subsequently, protein,
reductant, and
extender were incubated at 4 °C overnight, such that greater than 80%
of protein was labelled
with extender, as detected by mass spectrometry. The samples were injected
onto an HP1100
HPLC and chromatographed on a Protein MicroTrap (Micrhom Bioresources, Inc. #
004/25109/03) attached to a hybrid quadrupole-TOF QSTAR Pulsar i mass
spectrometer (PE
Sciex' Instruments). The QSTAR was outfitted with a MicrolonSpray ESI source,
and was
operated in the positive ion mode, scanning the range of 800-1400 n~/z.
After labelling, the protein-extender covalent complex was dialyzed against 7
L dialysis
buffer (20 mM HEPES pH 7.4, 150 mM NaCI) overnight at 4 °C to remove
unreacted extender
and reductant. After checking for protein labelling again by QSTAR, the
protein-extender
conjugate was split into 1.1 mL working aliquots at 2 p.M, frozen on dry
ice/ethanol, and stored
at -80 °C. Depending upon the reactivity of the protein cysteine(s) and
extender being used,
different reaction conditions, e.g., type of reductant, concentration of
reductant, reaction time,
etc., can be used.
A working aliquot of the MEKI-extender covalent complex .was thawed and placed
on
ice. A library of compounds to be screened was distributed across the wells of
a 96-well plate,
with each well containing a pool of 10 disulfide-containing compounds. The
compounds were
pooled so that each compound in the pool has a unique molecular weight, thus
enabling
deconvolution of the various protein-extender-compound conjugates by mass
spectrometry. The
library pools, stored at stock concentrations of 12.5 ~M/pool in DMSO at 4
°C, were thawed at
-68-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
room temperature for at least 30 min prior to screening. To an assay plate the
following reagents
were added in order: 0.86 p,L of each library pool, 1 pL of 13.5 mM (3-
mercaptoethanol, and 25
~.~,L of protein-extender conjugate. Thus the final screening conditions were
400 pM library
pools, 500 p.M (3-mercaptoethanol, and 2 ~.M protein-extender covalent
complex. The reactions
were incubated at room temperature on a shaker for 1-2 hr. After reaction,
samples were run on
a. QSTAR mass spectrometer as described above for the labelling step, in order
to determine
which of the library compounds reacted with the protein-extender covalent
complex.
Exam 1p a 10
This example describes the synthesis of the following compound
H
N
HN .
O I J 00
~N N
H
which was prepared according to Scheme 4 and the procedure below.
SCHEME4
CI CI O 0 . CI ~ HN~O~
O
( \N NH3 ~N O ~ I %N ~NHzHCI O I %N 0
J ~l J
Cp HZN NJ N N N N
H DIEA H
17 18 19
H
HZN ( ~ HN~N I
Li0 'H w0 / 4 0 I w N 0 0
EDC, HOST, DIEA i "N NJ
H
3
a) 4,6-Dichloropyrimidine (20.85 g, 139 mmol) and 7 N Ammonia in methanol (200
mL)
were heated to 85 C in a sealed glass bomb for 16 h. The reaction was cooled
to ambient
temperature, the solvent evaporated, and the residue recrystalized from water
yielding compound
17 (12.07 g, 93.17 mmol, 67% yield) . 1H NMR (400 MHz, DMSO-D6): 8 6.43 (s, 1
H), 7.22
(s, 2 H), 8.18 (s, 1 H). ESI-MS m/z: 130 (M + H)+.
b) Compound 17 (3.30 g, 25.47 mmol) was mixed with acetic anhydride (50 mL)
and
r~fluxed for 5 h, the solvent evaporated, and the residue co-evaporated with
toluene twice,
yielding compound 18 (4.31 g, 25.06 mmol, 99% yield). 1H NMR (400 MHz, DMSO-
D6): 8
2 .12 (s, 3 H), 8.06 (s, 1 H), 8.71 (s, 1 H), 11.21 (s, 1 H). ESI-MS m/z: 172
(M + H)+.
-69=
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
c) Compound 18 (3.03 g, 17.66 mmol), methyl 6-aminohexanoate hydrochloride
(4.50 g,
24.22'mmol), and DIEA (30.76 mL, 177 mmol) were combined with n-butanol (88
mL) and
refluxed orx a heating mantle for 3 h. The reaction was cooled to ambient
temperature, the
solvent evaporated, and the residue purified by flash chromatography (80%
EtOAc in hexanes)
yielding compound 19 (2.63 g, 9.38 mmol, 53% yield). 1H NMR (400 MHz, DMSO-
D6): ~
1.28 (m, 2 H), 1.49 (m, 4 H), 2.04 (s, 3 H), 2.27 (m, 2 H), 3.21 (s, 1 H),
3.36 (s, 1 H), 3.56 (s, 3
H), 7.13 (s, 1 H), 7.36 (s, 1 H), 8.12 (s, 1 H), 10.24 (s, 1 H). ESI-MS nZ/z:
281 (M + H)+.
d) Compound 19 (1.84 g, 6.57 mmol) was suspended in ~-dioxane (16 mL). Lithium
hydroxide (0.157g, 6.57 mmol) in water (16 mL) was added and the reaction
stirred at ambient
temperature for 16 h. 1N HCL (6.57 mL) was added and the solution stirred for
1 h at which
point the solvent was evaporated to yield crude free acid which was taken on
without further
purification (2.03 g, 6.57 mmol, 99% yield). ESI-MS m/z: 267 (M + H)+. This
free acid (0.216
g, 0.507 mmol) was mixed with 1-ethyl-(3-dimethylaminopropyl)carbodiimide
hydrochloride
(EDC, 0.107 g, 0.558 mmol), hydroxybenzotriazole hydrate (HOBT, 0.085 g, 0.558
mmol), and
5-ter°t-butyl-o-aniside (0.100 g, 0.558 mmol). DMF (3 mL) was added,
followed by DIEA
(0.486 mL, 2.79 mmol) and the reaction stirred at ambient temperature for 16
h. The reaction
was diluted with acetonitrile (3 mL) and the crude mixture purified by reverse-
phase preparatory
HPLC to afford compound 3(0.082 g, 0.151 mmol, 27%). 1H NMR (400 MHz; GD30D):
~ 1.07
(s, 9 H), 1.27 (m, 2 H), 1.51 (m, 4 H), 1.99 (s, 3 H), 2.24 (m, 2 H), 3.10 (m,
2 H), 3.63 (s, 3 H),
2.0 6.40 (s, 1 H), 6.69 (m, 1 H), 6.91 (m, 1 H), 7.79 (s, 1 H), 8.12 (s, 1 H)
ESI-MS ~a/z: 456 (M +
H)+.
Example 11
This example describes the synthesis of the following compound
which was prepared according to the procedure below.
Boc-7-aminoheptanoic acid (4.0 g, 16.31 rmnol) was dissolved in benzene (60
mL) and
methanol (20 mL) was added. (Trimethylsilyl)diazomethane (2.0 M in hexanes)
(16.31 mL,
32.61 mmol) was added and the solution stirred at ambient temperature for 30
min at which point
-70-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
the solvent was removed. The crude residue was then dissolved in 4.0 M HCL in
dioxane (42
mL) and stirred for 2 h at ambient temperature at which point the solvent was
removed, yielding
7-amino-heptanoic acid methyl ester (3.04 g, 15.53 xmnol, 95%). ESI-MS m/z:
196 (M + H)+
The titled compound was prepared according to Example lOc-d except starting
with 7-
~5 amino-heptanoic acid methyl ester instead of methyl 6-aminohexanoate
hydrochloride. 1H NMR
(400 MHz, CD30D): 8 1.32 (m, 4 H), 1.57 (m, 4 H), 2.04 (s, 3 H), 2.91 (m, 2
H), 3.15 (m, 3 H),
6.43 (s, 1 H), 6.91 (m, 1 H), 7.23 (m, 2 H), 8.19 (s, 1 H). ESI-MS m/z: 428 (M
+ H)+.
Example 12
This example describes the synthesis of
O
HN I ~ H
0
N N
H
which was prepared according to Scheme 5 and the protocol below.
SCHEME 5
0
BocHN I ~ NHz HN ~ N
H / I HCI I ~I 0 \N I / H O I /
OH EDC, HOBT, DIEA / N~NHBoc DIEA ~ I
O~ O O~ 0 H NJ
1 5 20 ~ 4
5-Tent-butyl-2-methoxybenzoic acid (0.161 g, 0.773 mmol) was mixed with EDC
(0.153
g, 0.797 mmol), HOBT (0.106 g, 0.785 mmol), dissolved in 5 ml dry DMF, and 1-
(N-Boc-
aminomethyl)-3-(aminometliyl)benzene (0.222 g, 0.939 mmol) was added, along
with DIEA (0.4
ml, 2.3 mmol). The reaction was allowed to stir at ambient temperature for 22
hours, at which
, point it was flooded with 50 rnl EtOAc, rinsed with 2 x 25 ml 1 M sodium
hydrogen sulfate, 2 x
ml saturated sodium bicarbonate, 25 ml brine, dried over sodium sulfate, and
evaporated to
dryness to yield product 20 which was used without further purification. ESI-
MS m/z: 449 (M +
Na)+. ' .
Compound 20 (0.086 g, 0.202 mmol) was dissolved in 4 M HCl in dioxane (5 ml)
and
25 allowed to stir for 30 minutes. The solvent was removed under reduced
pressure and then
evaporated twice from DCM. Compound 18 (0.035 mg, 0.203 mmol) was then added,
along
with DIEA (0.12 ml,, 0.689 mmol) and 2 ml n-butanol. The reaction was then
heated to 100 C for
-71-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
22 hours, at which point the reaction was flooded with EtOAc (40 ml), rinsed
with 3 x 20 ml 1 M
sodium hydrogen sulfate, 20 ml brine, dried over sodium sulfate, and
evaporated to dryness. The
residue was then purified by reverse-phase preparatory HPLC to afford compound
4 (0.010 g,
0.017 mmol, 9%). 1H NMR (400 MHz, CD30D): ~ ppm 1.31 (m, 9 H) 2.18 (m, 3 H)
3.92 (m, 3
H) 4.60 (m, 2 H) 4.71 (m, 2 H) 6.48 (m, 1 H) 7.08 (m, 1 H) 7.24 (m, 1 H) 7.34
(m, 3 H) 7.55 (m,
1 H) 7.97 (m, 1 H) 8.35 (m, 1 H). ESI-MS m/z: 462 (M + H)+.
Example 13
This example describes the synthesis of
H
N
HN' v v O
O I ~~ N
~N NJ
which was made according to the protocol below.
Compound 6 was prepared following the procedure of Example 12, but starting
with [2-
(4-amino-phenyl)-ethyl]-carbamic acid tent-butyl ester instead of 1-(N-Boc-
aminomethyl)-3-
(aminomethyl)benzene. The final product 6 was purified first by reverse-phase
preparatory
HPLC and then by silica gel chromatography, eluting first with 50:50
DCM:EtOAc, then 25:75
DCM:EtOAc, and finally eluting with pure EtOAc. 1H NMR (400 MHz, CD30D): b ppm
1.34
(m, 9 H) 2.17 (m, 3 H) 2.91 (m, 2 H) 3.65 (m, 2 H) 4.00 (m, 3 H) 6.67 (m, 1 H)
7.11 (m, 1 H)
7.24 (m, 2 H) 7.58 (m, 3 H) 7.99 (m, 1 H) 8.27 (m, 1 H). ESI-MS mlz: 462 (M +
H)+.
Example 14
This example describes the synthesis of
w1
HN ~ I O O~
//N I ~ N
\N NJ
H
-72-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
wluch was prepared according to the protocol below.
The titled was prepared following the procedure of Example 13, but the final
coupling
was performed with 6-chloropurine instead of compound 18. 1H NMR (400 MHz,
CD30D): 8
ppm 1.33 (m, 9 H) 3.05 (m, 2 H) 3.93 (m, 2 H) 4.01 (m, 3 H) 7.12 (m, 1 H) 7.29
(m, 2 H) 7.59
(m, 3 H) 7.99 (m, 1 H) 8.30 (m, 1 H) 8.43 (m, 1 H). ESI-MS nZ/z: 445 (M + H)+.
Exam 1p a 15
This example describes the synthesis of the following compounds
H
N W n
=
1:
22
HN~ n=2:
n I 23
~ n=3:
~ 24
O
N n=6:
O 25
O
~N N n=7:
26
H
which were prepared according to Example 10 except for the following changes.
Compound 22 was made using amino-acetic acid methyl ester hydrochloride
instead of
methyl 6-aminohexanoate hydrochloride. ESI-MS m/z: 372 (M + H)+.
Compound 23 was made using 3-amino-propionic acid methyl ester hydrochloride
instead of methyl 6-aminohexanoate hydrochloride. ESI-MS n2/z: 386 (M + H)+.
Compound 24 was made using 4-amino-butyric acid methyl ester hydrochloride
instead
of methyl 6-aminohexanoate hydrochloride. ESI-MS m/z: 400 (M + H)+.
Compound 25 was made using 7-amino-heptanoic acid methyl ester hydrochloride
instead of methyl 6-aminohexanoate hydrochloride. ESI-MS m/z: 442 (M + H)+.
Compound 26 was made using 8-amino-octanoic acid methyl ester hydrochloride
instead
of methyl 6-aminohexanoate hydrochloride. ESI-MS mlz: 456 (M + H)+.
Example 16
This example describes the synthesis of the following compounds
H S
HN~N I
C ~I ~N O HN-N ~ n=5: 27
n = 7: 28
~N~J
H
which were prepared according to Example 11 except for the following changes
Compound 27 was made using 6-aminohexanoate hydrochloride was used instead of
7-
aminoheptanoic acid methyl ester. ESI-MS rnlz: 414 (M + H)+.
-73-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Compound 28 was made using 8-amino-octanoic acid methyl ester hydrochloride
was
used in place of 7-aminoheptanoic acid methyl ester. ESI-MS nalz: 442 (M +
H)+.
Exam 1p a 17
This example describes the synthesis of
H
HN N
O ~N O
~N NJ
H
which was prepared according to Example 10 with 3-tent-butyl-phenylamine
instead of 5-tert-
butyl-o-aniside. ESI-MS m/z: 398 (M + g)+.
Example 18
This example describes the synthesis of
H F F
HN N I ~ F
o /
N N
H
which was prepared according to Example 10 with 3-trifluoromethyl-phenylamine
instead of 5-
~ert-butyl-o-aniside. ESI-MS nZlz: 410 (M + H)+.
Example 19
This example describes the synthesis of
H F F
HN N I ~ F
O I ~N O
~N NJ ,O
H
which was prepared according to Example 10 with, with 3-methoxy-5-
trifluoromethyl-
phenylamine instead of 5-test-butyl-o-aniside. ESI-MS m/z: 440 (M + H)+.
-74-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Example 20
This example describes the synthesis of
H F F
HN N I ~ F
O ~N ° F
~N NJ
H
which was prepared according to Example 1 with 2-fluoro-S-trifluoromethyl-
phenylamine
substituted for 5-tent-butyl-o-aniside. ESI-MS nz/z: 428 (M + H)+.
Exam 1p a 21
This example describes the synthesis of
H F F
HN N I ~ F
o ~ J °ci
~H N
which was prepared according to Example 1 with 2-chloro-5-trifluoromethyl-
phenylamine
substituted for 5-tef-t-butyl-o-aniside. ESI-MS m/z: 444 (M + H)+.
Example 22
This example describes the synthesis of
which was prepared according to Example 12 with 5-tent-butyl-o-aniside and 4-
(2-teYt-
butoxycarbonylamino-ethyl)-benzoic acid replacing 5-tent-butyl-2-
methoxybenzoic acid and 1-
(N-Boc-aminomethyl)-3-(aminomethyl)benzene. ESI-MS nZ/z: 462 (M + H)+.
-75-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Example 23
This example describes the synthesis of
HN I ~ H
~N / N \
N I NJ O O I / _
H
which was prepared according to Example 12 with 5-tet°t-butyl-o-aniside
and 4-(te~t-
butoxycarbonylamino-methyl)-benzoic acid replacing 5-tent-butyl-2-
methoxybenzoic acid and 1-
(N-Boc-aminomethyl)-3-(aminomethyl)benzene. ESI-MS m/z: 448 (M + H)+,
Exam lie 24
This example describes the synthesis of
HN I ~ H / I
wN / N \
N NJ O O~
' H
which was prepared according to Example 12 with (4-aminomethyl-benzyl)-
carbamic acid ter~t-
butyl ester replacing 1-(N-Boc-aminomethyl)-3-(aminomethyl)benzene. ESI-MS
m/z: 462 (M +
H)+.
The examples described above are set forth solely to assist in the
understanding of the
invention, and are not intended to limit the scope of the invention in any
way.
One skilled in the art will readily appreciate that the present invention is
well adapted to
carry out the objects and obtain the ends and advantages mentioned, as well as
those inherent
therein. The methods and procedures described herein are presently
representative of preferred
embodiments and are exemplary and are not intended as limitations on the scope
of the
invention. Changes therein and other uses will occur to those skilled in the
art which are
encompassed within the spirit of. the invention.
It will be readily apparent to one skilled in the art that varying
substitutions and
modifications may be made to the invention disclosed herein without departing
from the scope
and spirit of the invention.
All patents and publications mentioned in the specification are indicative of
the levels of
those skilled in the art to which the invention pertains. All patents and
publications are herein
-76-
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
incorporated by reference to the same extent as if each individual publication
was specifically
and individually indicated to be incorporated by reference.
The invention illustratively described herein suitably may be practiced in the
absence of
any element or elements, limitation or limitations which is not specifically
disclosed herein. The
terms and expressions which have been employed are used as terms of
description and not of
limitation, and there is no intention that in the use of such terms and
expressions indicates the
exclusion of equivalents of the features shown and described or portions
thereof. It is recognized
that various modifications are possible within the scope of the invention.
Thus, it should be
understood that although the present invention has been specifically disclosed
by preferred
embodiments and optional features, modification and variation of the concepts
herein disclosed
may be resorted to by those skilled in the art, and that such modifications
and variations are
considered to be falling within the scope of the invention, which is limited
only by the following
claims.
_77_
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
SEQUENCE LISTING
<110> SUNESIS PHARMACEUTICALS, INC.
Prescott, john C.
Braisted, Andrew
<120> Identification of oinase Inhibitors
<130> 39750-0006PC
<160> 32
<170> FastSEQ for Windows version 4.0
<210>
1
<211>
21
<212>
DNA
<213> sapien
Homo
<400>
1
agggcctctcaaggcctcctc 21
<210>
2
<211>
23
<212>
DNA
<213> sapien
Homo
<400>
2
agttggagtctgtaggacttggc 23
<210>
3
<211>
38
<212>
DNA
<213> sapien
Homo
<400>
3
ctaggatatcctcgagcaagccgtggtgggaggacgag 38
<210>
4
<211>
41
<212>
DNA
<213> sapien
Homo
<400>
4
ctaggatatcaagcttttcagtcctccagcacactgcgca g 41
<210>
<211>
40
<212>
DNA
<213> sapien
Homo
<400>
5
ctaggatatcctcgagcgctcccaaccaagctctcttgag 40
<210>
6
<211>
46
<212>
DNA
<213> sapien
Homo
<400>
6
ctaggatatcaagcttttcatttggagaattcgatgatca actcac 46
<210>
7
<211>
22
<212>
DNA
Page 1
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
<213> Homo Sapien
<400> 7
gaccacaacg gtttccctct ag 22
<210> 8
<211> 22
<212> DNA
<213> Homo Sapien
<400> 8
gttattgctc agcggtggca gc 22
<210> 9
<211> 509
<212> PRT
<213> Homo Sapien
<400> 9
Met Gly Cys Gly Cys Ser Ser His Pro Glu Asp Asp Trp Met Glu Asn
1 5 10 15
Ile Asp Val Cys Glu Asn Cys His Tyr Pro Ile Val Pro Leu Asp Gly
20 25 30
Lys Gly Thr Leu Leu Ile Arg Asn Gly Ser Glu Val Arg Asp Pro Leu
35 40 45
Val Thr Tyr Glu Gly Ser Asn Pro Pro Ala Ser Pro Leu Gln Asp Asn
50 55 60
Leu Val Ile Ala Leu His Ser Tyr Glu Pro Ser His Asp Gly Asp Leu
65 70 75 g0
Gly Phe Glu Lys Gly Glu Gln Leu Arg Ile Leu Glu Gln Ser Gly Glu
85 90 95
Trp Trp Lys Ala Gln Ser Leu Thr Thr Gly Gln Glu Gly Phe Ile Pro
100 105 110
Phe Asn Phe Val Ala Lys Ala Asn Ser Leu Glu Pro Glu Pro Trp Phe
115 120 125
Phe Lys Asn Leu Ser Arg Lys Asp Ala Glu Arg Gln Leu Leu Ala Pro
130 135 140
Gly Asn Thr His Gly Ser Phe Leu Ile Arg Glu Ser Glu Ser Thr Ala
145 150 155 160
Gly Ser Phe Ser Leu Ser Val Arg Asp Phe Asp Gln Asn Gln Gly Glu
165 170 175
Val Val Lys His Tyr Lys Ile Arg Asn Leu Asp Asn Gly Gly Phe Tyr
180 185 190
Ile Ser Pro Arg Ile Thr Phe Pro Gly Leu His Glu Leu Val Arg His
195 200 205
Tyr Thr Asn Ala Ser Asp Gly Leu Cys Thr Arg Leu Ser Arg Pro Cys
210 215 220
Gln Thr Gln Lys Pro Gln Lys Pro Trp Trp Glu Asp Glu Trp Glu Val
225 230 235 240
Pro Arg Glu Thr Leu Lys Leu Val Glu Arg Leu Gly Ala Gly Gln Phe
245 250 255
Gly Glu Val Trp Met Gly Tyr Tyr Asn Gly His Thr Lys Val Ala Val
260 265 270
Lys Ser Leu Lys Gln Gly Ser Met Ser Pro Asp Ala Phe Leu Ala Glu
275 280 285
Ala Asn Leu Met Lys Gln Leu Gln His Gln Arg Leu Val Arg Leu Tyr
290 295 300
Ala Val Val Thr Gln Glu Pro Ile Tyr Ile Ile Thr Glu Tyr Met Glu
305 310 315 320
Asn Gly Ser Leu Val Asp Phe Leu Lys Thr Pro Ser Gly Ile Lys Leu
325 330 335
Thr Ile Asn Lys Leu Leu Asp Met Ala Ala Gln Ile Ala Glu Gly Met
340 345 350
Ala Phe Ile Glu Glu Arg Asn Tyr Ile His Arg Asp Leu Arg Ala Ala
355 360 365
Asn Ile Leu Val Ser Asp Thr Leu Ser Cys Lys Ile Ala Asp Phe Gly
370 375 380
Page 2
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Leu Ala Arg Leu Ile Glu Asp Asn Glu Tyr Thr Ala Arg Glu Gly Ala
385 390 395 400
Lys Phe Pro Ile Lys Trp Thr Ala Pro Glu Ala Ile Asn Tyr Gly Th r
405 410 415
Phe Thr Ile Lys Ser Asp Val Trp Ser Phe Gly Ile Leu Leu Thr G lu
420 425 430
Ile Val Thr His Gly Arg Ile Pro Tyr Pro Gly Met Thr Asn Pro G lu
435 440 445
Val Ile Gln Asn Leu Glu Arg Gly Tyr Arg Met Val Arg Pro Asp A sn
450 455 460
Cys Pro Glu Glu Leu Tyr Gln Leu Met Arg Leu Cys Trp Lys Glu A rg
465 470 475 480
Pro Glu Asp Arg Pro Thr Phe Asp Tyr Leu Arg Ser Val Leu Glu As p
485 490 495
Phe Phe Thr Ala Thr Glu Gly Gln Tyr Gln Pro Gln Pro
500 505
<210> 10
<211> 1210
<212> PRT
<213> Homo Sapien
<400> 10
Met Arg Pro Ser Gly Thr Ala Gly Ala Ala Leu Leu Ala Leu Leu Al a
1 5 10 15
Ala Leu Cys Pro Ala Ser Arg Ala Leu Glu Glu Lys Lys Val Cys Gln
20 25 30
Gly Thr Ser Asn Lys Leu Thr Gln Leu Gly Thr Phe Glu Asp His Ph a
35 40 45
Leu Ser Leu Gln Arg Met Phe Asn Asn Cys Glu Val Val Leu Gly Asn
50 55 60
Leu Glu Ile Thr Tyr Val Gln Arg Asn Tyr Asp Leu Ser Phe Leu Lys
65 70 75 g0
Thr Ile Gln Glu Val Ala Gly Tyr Val Leu Ile Ala Leu Asn Thr Val
85 90 95
Glu Arg Ile Pro Leu Glu Asn Leu Gln Ile Ile Arg Gly Asn Met Ty r
100 105 110
Tyr Glu Asn Ser Tyr Ala Leu Ala Val Leu Ser Asn Tyr Asp Ala Asn
115 120 125
Lys Thr Gly Leu Lys Glu Leu Pro Met Arg Asn Leu Gln Glu Ile Leu
130 135 140
His Gly Ala Val Arg Phe Ser Asn Asn Pro Ala Leu Cys Asn Val Gl a
145 150 15 5 160
Ser Ile Gln Trp Arg Asp Ile Val Ser Ser Asp Phe Leu Ser Asn Me t
165 170 175
Ser Met Asp Phe Gln Asn His Leu Gly Ser Cys Gln Lys Cys Asp Pro
180 185 190
Ser Cys Pro Asn Gly Ser Cys Trp Gly Ala Gly Glu Glu Asn Cys Gln
195 200 205
Lys Leu Thr Lys Ile Ile Cys Ala Gln Gln Cys Ser Gly Arg Cys Arg
210 215 220
Gly Lys Ser Pro Ser Asp Cys Cys His Asn Gln Cys Ala Ala Gly Cys
225 230 235 240
Thr Gly Pro Arg Glu Ser Asp Cys Leu Val Cys Arg Lys Phe Arg As p
245 250 255
Glu Ala Thr Cys Lys Asp Thr Cys Pro Pro Leu Met Leu Tyr Asn Pr~o
260 265 270
Thr Thr Tyr Gln Met Asp Val Asn Pro Glu Gly Lys Tyr Ser Phe Gly
275 280 285
Ala Thr Cys Val Lys Lys Cys Pro Arg Asn Tyr Val Val Thr Asp His
290 295 300
Gly Ser Cys Val Arg Ala Cys Gly Ala Asp Ser Tyr Glu Met Glu Glu
305 310 315 32 0
Asp Gly Val Arg Lys Cys Lys Lys Cys Glu Gly Pro Cys Arg Lys Val
325 330 335
Cys Asn Gly Ile Gly Ile Gly Glu Phe Lys Asp Ser Leu Ser Ile As n
Page 3
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
340 345 350
Ala Thr Asn Ile Lys His Phe Lys Asn Cys Thr Ser Ile Ser Gly Asp
355 360 365
Leu His Ile Leu Pro Val Ala Phe Arg Gly Asp Ser Phe Thr His Thr
370 375 380
Pro Pro Leu Asp Pro Gln Glu Leu Asp Ile Leu Lys Thr Val Lys Glu
385 390 395 400
Ile Thr Gly Phe Leu Leu Ile Gln Ala Trp Pro Glu Asn Arg Thr Asp
405 410 415
Leu His Ala Phe Glu Asn Leu Glu Ile Ile Arg Gly Arg Thr Lys Gln
420 425 430
His Gly Gln Phe Ser Leu Ala Val Val Ser Leu Asn Ile Thr Ser Leu
435 440 445
Gly Leu Arg Ser Leu Lys Glu Ile Ser Asp Gly Asp Val Ile Ile Ser
450 455 460
Gly Asn Lys Asn Leu Cys Tyr Ala Asn Thr Ile Asn Trp Lys Lys Leu
465 470 475 480
Phe Gly Thr Ser Gly Gln Lys Thr Lys Ile Ile Ser Asn Arg Gly Glu
485 490 495
Asn Ser Cys Lys Ala Thr Gly Gln Val Cys His Ala Leu Cys Ser Pro
500 505 510
Glu Gly Cys Trp Gly Pro Glu Pro Arg Asp Cys Val Ser Cys Arg Asn
515 520 525
Val Ser Arg Gly Arg Glu Cys Val Asp Lys Cys Asn Leu Leu Glu Gly
530 535 540
Glu Pro Arg Glu Phe Val Glu Asn Ser Glu Cys Ile Gln Cys His Pro
545 550 555 560
Glu Cys Leu Pro Gln Ala Met Asn Ile Thr Cys Thr Gly Arg Gly Pro
565 570 575
Asp Asn Cys Ile Gln Cys Ala His Tyr Ile Asp Gly Pro His Cys Val
580 585 590
Lys Thr Cys Pro Ala Gly Val Met Gly Glu Asn Asn Thr Leu Val Trp
595 600 605
Lys Tyr Ala Asp Ala Gly His Val Cys His Leu Cys His Pro Asn Cys
610 615 620
Thr Tyr Gly Cys Thr Gly Pro Gly Leu Glu Gly Cys Pro Thr Asn Gly
625 630 635 640
Pro Lys Ile Pro Ser Ile Ala Thr Gly Met Val Gly Ala Leu Leu Leu
645 650 655
Leu Leu Val Val Ala Leu Gly Ile Gly Leu Phe Met Arg Arg Arg His
660 665 670
Ile Val Arg Lys Arg Thr Leu Arg Arg Leu Leu Gln Glu Arg Glu Leu
675 680 685
Val Glu Pro Leu Thr Pro Ser Gly Glu Ala Pro Asn Gln Ala Leu Leu
690 695 700
Arg Ile Leu Lys Glu Thr Glu Phe Lys Lys Ile Lys Val Leu Gly Ser
705 710 715 720
Gly Ala Phe Gly Thr Val Tyr Lys Gly Leu Trp Ile Pro Glu Gly Glu
725 730 735
Lys Val Lys Ile Pro Val Ala Ile Lys Glu Leu Arg Glu Ala Thr Ser
740 745 750
Pro Lys Ala Asn Lys Glu Ile Leu Asp Glu Ala Tyr Val Met Ala Ser
755 760 765
Val Asp Asn Pro His Val Cys Arg Leu Leu Gly Ile Cys Leu Thr Ser
770 775 780
Thr Val Gln Leu Ile Thr Gln Leu Met Pro Phe Gly Cys Leu Leu Asp
785 790 795 800
Tyr Val Arg Glu His Lys Asp Asn Ile Gly Ser Gln Tyr Leu Leu Asn
805 810 815
Trp Cys Val Gln Ile Ala Lys Gly Met Asn Tyr Leu Glu Asp Arg Arg
820 825 830
Leu Val His Arg Asp Leu Ala Ala Arg Asn Val Leu Val Lys Thr Pro
835 840 845
Gln His Val Lys Ile Thr Asp Phe Gly Leu Ala Lys Leu Leu Gly Ala
850 855 860
Glu Glu Lys Glu Tyr His Ala Glu Gly Gly Lys Val Pro Ile Lys Trp
865 870 875 880
Page 4
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Met Ala Leu Glu Ser Ile Leu His Arg Ile Tyr Thr His Gln Ser Asp
885 890 895
Val Trp Ser Tyr Gly Val Thr Val Trp Glu Leu Met Thr Phe Gly Ser
900 905 910
Lys Pro Tyr Asp Gly Ile Pro Ala Ser Glu Ile Ser S er Ile Leu Glu
915 920 925
Lys Gly Glu Arg Leu Pro Gln Pro Pro Ile Cys Thr =1e Asp Val Tyr
930 935 940
Met Ile Met Val Lys Cys Trp Met Ile Asp Ala Asp 5 er Arg Pro Lys
945 950 955 960
Phe Arg Glu Leu Ile Ile Glu Phe Ser Lys Met Ala Arg Asp Pro Gln
965 970 975
Arg Tyr Leu Val Ile Gln Gly Asp Glu Arg Met His Leu Pro Ser Pro
980 985 990
Thr Asp Ser Asn Phe Tyr Arg Ala Leu Met Asp Glu Glu Asp Met Asp
995 1000 1005
Asp Val Val Asp Ala Asp Glu Tyr Leu Ile Pro Gln Gln Gly Phe Phe
1010 1015 1020
Ser Ser Pro Ser Thr Ser Arg Thr Pro Leu Leu Ser 5 er Leu Ser Ala
1025 1030 1035 1040
Thr Ser Asn Asn Ser Thr Val Ala Cys Ile Asp Arg Asn Gly Leu Gln
1045 1050 1055
Ser Cys Pro Ile Lys Glu Asp Ser Phe Leu Gln Arg Tyr Ser Ser Asp
1060 1065 1070
Pro Thr Gly Ala Leu Thr Glu Asp Ser Ile Asp Asp Thr Phe Leu Pro
1075 1080 1085
Val Pro Glu Tyr Ile Asn Gln Ser Val Pro Lys Arg Pro Ala Gly Ser
1090 1095 1100
Val Gln Asn Pro Val Tyr His Asn Gln Pro Leu Asn Pro Ala Pro Ser
1105 1110 1115 1120
Arg Asp Pro His Tyr Gln Asp Pro His Ser Thr Ala dal Gly Asn Pro
1125 1130 1135
Glu Tyr Leu Asn Thr Val Gln Pro Thr Cys Val Asn 5 er Thr Phe Asp
1140 1145 1150
Ser Pro Ala His Trp Ala Gln Lys Gly Ser His Gln =1e Ser Leu Asp
1155 1160 1165
Asn Pro Asp Tyr Gln Gln Asp Phe Phe Pro Lys Glu Ala Lys Pro Asn
1170 1175 1180
Gly Ile Phe Lys Gly Ser Thr Ala Glu Asn Ala Glu Tyr Leu Arg Val
1185 1190 1195 1200
Ala Pro Gln Ser Ser Glu Phe Ile Gly Ala
1205 1210
<210> 11
<211> 29
<212> DNA
<213> Homo 5apien
<400> 11
catggagaat gggtgtctag tggattttc 2g
<210> 12
<211> 29
<212> DNA
<213> Homo Sapien
<400> 12
gaaaatccac tagacaccca ttctccatg 2g
<210> 13
<211> 39
<212> DNA
<213> Homo Sapien
<400> 13
ctaggatatc ccatgggcaa gccgtggtgg gaggacgag 39
Page 5
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
<Z10> 14
<211> 38
<212> DNA
<213> Homo Sapien
<400> 14
ctaggatatc ccatggctcc caaccaagct ctcttgag 38
<210> 15
<211> 96
<212> DNA
<213> Homo Sapien
<400> 15
gatcctccga aaccatggct cgaggcggcc g caagcttga tatcccaacg accgaaaacc 60
tgtattttca gggccatcac catcaccatc actagc g6
<210> 16
<211> 96
<212> DNA
<213> Homo Sapien
<400> 16
agctgctagt gatggtgatg gtgatggccc t gaaaataca ggttttcggt cgttgggata 60
tcaagcttgc ggccgcctcg agccatggtt t cggag g6
<210> 17
<211> 85
<212> DNA
<213> Homo Sapien
<400> 17
ggtacccatg ggaaggcgcc acatcgttcg g aagcgcacg ctgcggaggc tgctgcagga 60
gagggagctt gtggagcctc ttaca g5
<Z10> 18
<211> 75
<212> DNA
<213> Homo Sapien
<400> 18
ggatcaagct tttcaatgca ttctttcatc cccctgaatg acaaggtagc gctgggggtc 60
tcgggccatt ttgga
<210> 19
<211> 393
<212> PRT
<213> Homo sapien
<400> 19
Met Pro Lys Lys Lys Pro Thr Pro Ile Gln Leu Asn Pro Ala Pro Asp
1 5 10 15
Gly Ser Ala Val Asn Gly Thr Ser Se r Ala Glu Thr Asn Leu Glu Ala
20 25 30
Leu Gln Lys Lys Leu Glu Glu Leu Glu Leu Asp Glu Gln Gln Arg Lys
35 40 45
Arg Leu Glu Ala Phe Leu Thr Gln Lys Gln Lys Val Gly Glu Leu Lys
50 55 60
Asp Asp Asp Phe Glu Lys Ile Ser Glu Leu Gly Ala Gly Asn Gly Gly
65 70 75 80
Val Val Phe Lys Val Ser His Lys Pro Ser Gly Leu Val Met Ala Arg
90 95
Lys Leu Ile His Leu Glu Ile Lys Pro Ala Ile Arg Asn Gln Ile Ile
100 10 5 110
Arg Glu Leu Gln Val Leu His Glu Cys Asn Ser Pro Tyr Ile Val Gly
115 120 125
Page 6
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
Phe Tyr Gly Ala Phe Tyr Ser Asp Gly Glu Ile Ser Ile Cys Met Glu
130 135 140
His Met Asp Gly Gly Ser Leu Asp Gln Val Leu Lys Lys Ala Gly Arg
145 150 155 160
Ile Pro Glu Gln Ile Leu Gly Ly s Val Ser Ile Ala Val Ile Lys Gly
165 170 175
Leu Thr Tyr Leu Arg Glu Lys His Lys Ile Met His Arg Asp Val Lys
180 185 190
Pro Ser Asn Ile Leu Val Asn Se r Arg Gly Glu Ile Lys Leu Cys Asp
195 200 205
Phe Gly Val Ser Gly Gln Leu Ile Asp Ser Met Ala Asn Ser Phe Val
210 215 220
Gly Thr Arg Ser Tyr Met Ser Pro Glu Arg Leu Gln Gly Thr His Tyr
225 230 235 240
Ser Val Gln Ser Asp Ile Trp Se r Met Gly Leu Ser Leu Val Glu Met
245 250 255
Ala Val Gly Arg Tyr Pro Ile Pro Pro Pro Asp Ala Lys Glu Leu Glu
260 265 270
Leu Met Phe Gly Cys Gln Val Glu Gly Asp Ala Ala Glu Thr Pro Pro
275 280 285
Arg Pro Arg Thr Pro Gly Arg Pro Leu Ser Ser Tyr Gly Met Asp Ser
290 295 300
Arg Pro Pro Met Ala Ile Phe Glu Leu Leu Asp Tyr Ile Val Asn Glu
305 310 315 320
Pro Pro Pro Lys Leu Pro Ser Gly Val Phe Ser Leu Glu Phe Gln Asp
325 330 335
Phe Val Asn Lys Cys Leu Ile Lys Asn Pro Ala Glu Arg Ala Asp Leu
340 345 350
Lys Gln Leu Met Val His Ala Phe Ile Lys Arg Ser Asp Ala Glu Glu
355 360 365
Val Asp Phe Ala Gly Trp Leu Cys Ser Thr Ile Gly Leu Asn Gln Pro
370 375 380
Ser Thr Pro Thr His Ala Ala Gly Val
385 390
<Z10>
20
<211>
42
<212>
DNA
<213> Sapien
Homo
<400>
20
cgcgcggatccatgcccaagaagaagccg cgcccatcca gc 42
a
<210>
21
<211>
44
<212>
DNA
<213> Sapien
Homo
<400>
21
cgtagctcgagtcaggtaccggcagcgtg gttggtgtgc tggg 44
g
<210>
22
<211>
44
<212>
DNA
<213> Sapien
Homo
<400>
22
cgtagctcgagtcaggtaccggcagcgtg gttggtgtgc tggg 44
g
<210>
23
<211>
33
<212>
DNA
<213> Sapien
Homo
<400>
23
gcgagatcagcatctccatggagcacatg atg 33
g
Page 7
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
<210>
24
<211>
31
<212>
DNA
<213> sapien
Homo
<400>
24
catggatggtgggtgcttggatcaagtgctg 31
<210>
25
<211>
31
<212>
DNA
<213> sapien
Homo
<400>
25
gggagatcaaactctc tttggggtcag
cgat 31
<210>
26
<211>
31
<212>
DNA
<213> Sapien
Homo
<400>
26
gggagatcaaactcgccgattttggggtcag 31
<210>
27
<211>
46
<212>
DNA
<213> Sapien
Homo
<400>
27
cgggcagctaattgacg tggccaacgacttcgtggga acaagg 46
aca
<210>
28
<211>
46
<212>
DNA
<213> sapien
Homo
<400>
28
cgggcagctaattgacgacatggccaacgacttcgtggga acaagg 46
<210>
29
<211>
31
<212>
DNA
<213> Sapien
Homo
<400>
29
gagctgctgtttggat ggtggaaggag 31
ccca
<210>
30
<211>
35
<212>
DNA
<213> sapien
Homo
<400>
30
ggattttgtgaataag-tccttaataaagaaccctg 35
<210>
31
<211>
35
<212>
DNA
<213> sapien
Homo
<400>
31
ggattttgtgaataagatgttaataaagaaccctg 35
<210>
32
<211>
40
Page 8
CA 02539064 2006-03-14
WO 2005/034840 PCT/US2003/029870
<212> DNA
<213> Homo Sapien
<400> 32
gacttcgcag gctggctctc ctccaccatt gggcttaacc 40
Page 9