Note: Descriptions are shown in the official language in which they were submitted.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
PHENYL CARBOXAMIDE AND SULFONAMIDE DERIVATIVES FOR USE AS 11-BETA-
HYDROXYSTEROID
DEHYDROGENASE
FIELD OF INVENTION
The present invention relates to a compound. In particular the present
invention
provides compounds capable of inhibiting 11 [3-hydroxysteroid dehydrogenase
(11 (3-
HSD).
INTRODUCTION
The role of glucocorticoids
Glucocorticoids are synthesised in the adrenal cortex from cholesterol. The
principle
glucocorticoid in the human body is cortisol, this hormone is synthesised and
secreted in
response to the adrenocortictrophic hormone (ACTH) from the pituitary gland in
a
circadian, episodic manner, but the secretion of this hormone can also be
stimulated by
stress, exercise and infection. Cortisol circulates mainly bound to
transcortin (cortisol
binding protein) or albumin and only a small fraction is free (5-10%) for
biological
processes [1 ].
Cortisol has a wide range of physiological effects, including regulation of
carbohydrate,
protein and lipid metabolism, regulation of normal growth and development,
influence on
cognitive function, resistance to stress and mineralocorticoid activity.
Cortisol works in
the opposite direction compared to insulin meaning a stimulation of hepatic
gluconeogenesis, inhibition of peripheral glucose uptake and increased blood
glucose
concentration. Glucocorticoids are also essential in the regulation of the
immune
response. When circulating at higher concentrations glucocorticoids are
immunosuppressive and are used pharmacologically as anti-inflammatory agents.
Glucocorticoids like other steroid hormones are lipophilic and penetrate the
cell
membrane freely. Cortisol binds, primarily, to the intracellular
glucocorticoid receptor
(GR) that then acts as a transcription factor to induce the expression of
glucocorticoid
responsive genes, and as a result of that protein synthesis.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
2
The role of the 11 [3-HSD enzyme
The conversion of cortisol (F) to its inactive metabolite cortisone (E) by 11
[3-HSD was
first described in the 1950's, however it was not until later that the
biological importance
for this conversion was suggested [2]. In 1983 Krozowski et al. showed that
the
mineralocorticoid receptor (MR) has epual binding affinities for
glucocorticoids and
mineralocorticoids [3]. Because the circulating concentration of cortisol is a
100 times
higher than that of aldosterone and during times of stress or high activity
even more, it
was not clear how the MR remained mineralocorticoid specific and was not
constantly
occupied by glucocorticoids. Earlier Ulick et al. [4] had described the
hypertensive
condition known as, "apparent mineralocorticoid excess" (AME), and observed
that
whilst secretion of aldosterone from the adrenals was in fact low the
peripheral
metabolism of cortisol was disrupted. These discoveries lead to the suggestion
of a
protective role for the enzymes. By converting cortisol to cortisone in
mineralocorticoid
dependent tissues 11 [i-HSD enzymes protects the MR from occupation by
glucocorticoids and allows it to be mineralcorticoid specific. Aldosterone
itself is
protected from the enzyme by the presence of an aldehyde group at the C-18
position.
Congenital defects in the 11 [3-HSD enzyme results in over occupation of the
MR by
cortisol and hypertensive and hypokalemic symptoms seen in AME.
Localisation of the 11 ~-HSD showed that the enzyme and its activity is highly
present in
the MR dependent tissues, kidney and parotid. However in tissues where the MR
is not
mineralocorticoid specific and is normally occupied by glucocorticoids, 11 [i-
HSD is not
present in these tissues, for example in the heart and hippocampus [5]. This
research
also showed that inhibition of 11 [i-HSD caused a loss of the aldosterone
specificity of
the MR in these mineralocorticoid dependent tissues.
It has been shown that two iso-enzymes of 11 (3-HSD exist. Both are members of
the
short chain alcohol dehydrogenase (SCAD) superfamily which have been widely
conserved throughout evolution. 11 [3-HSD type 2 acts as a dehydrogenase to
convert
the secondary alcohol group at the C-11 position of cortisol to a secondary
ketone, so
producing the less active metabolite cortisone. 11 (3-HSD type 1 is thought to
act mainly
in vivo as a reductase, that is in the opposite direction to type 2 [6] [see
below]. 11 [i-
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
3
HSD type 1 and type 2 have only a 30% amino acid homology.
Ig4tt .,'.tt_Otd
CC1I'riSOL C01'~tSOI~~
C"- rU C== U
II° ..»UEi ~~. ~ ~~~ ~~~r~ ~ ...C.If
11l3 1~51~ 'TVt~81 f
U U
11 (3-HSD enzyme activity
The intracellular activity of cortisol is dependent on the concentration of
glucocorticoids
and can be modified and independently controlled without involving the overall
secretion
and synthesis of the hormone.
The role of 11 [3-HSD Type 1
The direction of 11 (3-HSD type 1 reaction in vivo is generally accepted to be
opposite to
the dehydrogenation of type 2. In vivo homozygous mice with a disrupted type 1
gene
are unable to convert cortisone to cortisol, giving further evidence for the
reductive
activity of the enzyme [7]. 11 ~-HSD type 1 is expressed in many key
glucocorticoid
regulated tissues like the liver, pituitary, gonad, brain, adipose and
adrenals ,however,
the function of the enzyme in many of these tissues is poorly understood [8].
The concentration of cortisone in the body is higher than that of cortisol ,
cortisone also
binds poorly to binding globulins, making cortisone many times more
biologically
available. Although cortisol is secreted by the adrenal cortex, there is a
growing amount
of evidence that the intracellular conversion of E to F may be an important
mechanism in
regulating the action of glucocorticoids [9].
It may be that 11 ~-HSD type 1 allows certain tissues to convert cortisone to
cortisol to
increase local glucocorticoid activity and potentiate adaptive response and
counteracting
the type 2 activity that could result in a fall in active glucocorticoids
[10]. Potentiation of
the stress response would be especially important in the brain and high levels
of 11 ~i-
HSD type 1 are found around the hippocampus, further proving the role of the
enzyme.
11 [3-HSD type 1 also seems to play an important role in hepatocyte maturation
[8].
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
4
Another emerging role of the 11 a-HSD type 1 enzyme is in the detoxification
process of
many non-steroidal carbonyl compounds, reduction of the carbonyl group of many
toxic
compounds is a common way to increase solubility and therefore increase their
excretion. The 11 ~-HSD type1 enzyme has recently been shown to be active in
lung
tissue [11]. Type 1 activity is not seen until after birth, therefore mothers
who smoke
during pregnancy expose their children to the harmful effects of tobacco
before the child
is able to metabolically detoxify this compound.
The role of 11 [3-HSD Type 2
As already stated earlier the 11 [3-HSD type 2 converts cortisol to cortisone,
thus
protecting the MR in many key regulatory tissues of the body. The importance
of
protecting the MR from occupation by glucocorticoids is seen in patients with
AME or
liquorice intoxification. Defects or inactivity of the type 2 enzyme results
in hypertensive
syndromes and research has shown that patients with an hypertensive syndrome
have
an increased urinary excretion ratio of cortisol : cortisone. This along with
a reported
increase in the half life of radiolabelled cortisol suggests a reduction of 11
(3-HSD type 2
activity [12].
Rationale for the development of 11 (3-HSD inhibitors
As said earlier cortisol opposes the action of insulin meaning a stimulation
of hepatic
gluconeogenesis, inhibition of peripheral glucose uptake and increased blood
glucose
concentration. The effects of cortisol appear to be enhanced in patients
suffering from
glucose intolerance or diabetes mellitus. Inhibition of the enzyme 11 [i-HSD
type 1
would increase glucose uptake and inhibit hepatic gluconeogenesis, giving a
reduction in
circulatory glucose levels. The development of a potent 11 ~i-HSD type 1
inhibitor could
therefore have considerable therapeutic potential for conditions associated
with elevated
blood glucose levels.
An excess in glucocorticoids can result in neuronal dysfunctions and also
impair
cognitive functions. A specific 11 [i-HSD type 1 inhibitor might be of some
importance
by reducing neuronal dysfunctions and the loss of cognitive functions
associated with
ageing, by blocking the conversion of cortisone to cortisol.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
Glucocorticoids also have an important role in regulating part of the immune
response
[13]. Glucocorticoids can suppress the production of cytokines and regulate
the receptor
levels. They are also involved in determining whether T-helper (Th)
lymphocytes
5 progress into either Th1 or Th2 phenotype. These two different types of Th
cells secrete
a different profile of cytokines, Th2 is predominant in a glucocorticoid
environment. By
inhibiting 11 (3-HSD type 1, Th1 cytokine response would be favoured. It is
also possible
to inhibit 11 [3-HSD type 2 , thus by inhibiting the inactivation of cortisol,
it may be
possible to potentiate the anti-inflammatory effects of glucocorticoids.
Aspects of the invention are defined in the appended claims.
SUMMARY ASPECTS OF THE PRESENT INVENTION
In one aspect the present invention provides a compound having Formula I
R~-Z-R~ Formula I
wherein R~ is an optionally substituted phenyl ring; R~ is or comprises an
optionally
substituted aromatic ring; and Z is -X-Y-L- or -Y-X-L- wherein either X is
selected from -
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-; L is an optional linker; and R3, R4 and R5 are each
independently
selected' from H and hydrocarbyl; and wherein when R~ comprises the following
structural moiety
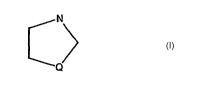
N
Q
wherein Q is an atom selected from the group consisting of S, O, N and C; the
compound is selected from compounds of the formulae R~-C(=O)-NR3-L-R2; R~
S(=O)(=O)-C(R4)(Rs)-L-R~; R~-S-C(Ra)(Rs)-L-R2; R~-NR3-S(=O)(=O)-L-R2; R~-NR3_
C(=O)-L-R2; R~-C(R4)(R5)-S(=O)(=O)-L-R2; and R~-C(Ra)(Rs)-S-L-R~.
In one aspect the present invention provides a pharmaceutical composition
comprising
(i) a compound having Formula I
R~-Z-R2 Formula I
wherein R~ is an optionally substituted phenyl ring; R2 is or comprises an
optionally
substituted aromatic ring; and Z is -X-Y-L- or -Y-X-L- wherein either X is
selected from -
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
6
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-; L is an optional linker; and R3, R4 and R5 'are each
independently
selected from H and hydrocarbyl; and wherein when R~ comprises the following
structural moiety
N
Q
wherein Q is an atom selected from the group consisting of S, O, N and C; the
compound is selected from compounds of the formulae R~-C(=O)-NR3-L-R2; R~-
S(=O)(=O)-C(R4)(R5)-L-Ra; R~-S-C(R4)(Rs)-L-R2; R~-NR3-S(=O)(=O)-L-R2; R~-NR3_
C(=O)-L-R2; R~-C(Ra)(Rs)-S(=O)(=O)-L-R2; and R~-C(R4)(R5)-S-L-R2.
(ii) optionally admixed with a pharmaceutically acceptable carrier, diluent,
excipient or
adjuvant.
In one aspect the present invention provides a compound for use in medicine
wherein
the compound has Formula I
R~-Z-R2 Formula I
wherein R~ is an optionally substituted phenyl ring; R~ is or comprises an
optionally
substituted aromatic ring; and Z, is -X-Y-L- or -Y-X-L- wherein either X is
selected from -
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-; L is an optional linker; and R3, R4 and R5 are each
independently
selected from H and hydrocarbyl; and wherein when R2 comprises the following
structural moiety
N
Qe
wherein Q is an atom selected from the group consisting of S, O, N and C; the
compound is selected from compounds of the formulae R~-C(=O)-NR3-L-R~; R~
S(=O)(=O)-C(Ra)(Rs)-L-R2; R~-S-C(Ra)(Rs)-L-Ra; R~-NR3-S(=O)(=O)-L-R2; R~-NR3_
C(=O)-L-R2; R~-C(R4)(R5)-S(=O)(=O)-L-R2; and R~-C(R4)(R5)-S-L-R~.
In one aspect the present invention provides a use of a compound in the
manufacture of
a medicament for use in the therapy of a condition or disease associated with
11 (3-HSD,
wherein the compound has Formula I
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
7
R~-Z-R2 Formula I
wherein R~ is an optionally substituted phenyl ring; RZ is or comprises an
optionally
substituted aromatic ring; and Z is -X-Y-L- or -Y-X-L- wherein either X is
selected from -
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-; L is an optional linker; and R3, R4 and R5 are each
independently
selected from H and hydrocarbyl; and wherein when R2 comprises the following
structural moiety
N
Qs
wherein Q is an atom selected from the group consisting of S, O, N and C; the
compound is selected from compounds of the formulae R~-C(=O)-NR3-L-R~; R~-
S(=O)(=O)-C(R4)(R5)-L-R~; R~-S-C(R4)(R5)-L-R~; R~-NR3-S(=O)(=O)-L-R~; R~-NR3-
C(=O)-L-R2; R~-C(R4)(R5)-S(=~)(=O)-L-R2; and R~-C(Ra)(Rs)-S-L-R2.
SOME ADVANTAGES
One key advantage of the present invention is that the compounds of the
present
invention can act as 11 ~i-HSD inhibitors. The compounds may inhibit the
interconversion
of inactive 11-keto steroids with their active hydroxy equivalents. Thus
present invention
provides methods by which the conversion of the inactive to the active form
may be
controlled, and to useful therapeutic effects which may be obtained as a
result of such
control. More specifically, but not exclusively, the invention is concerned
with
interconversion between cortisone and cortisol in humans.
Another advantage of the compounds of the present invention is that they may
be potent
11 (3-HSD inhibitors in vivo.
Some of the compounds of the present invention are also advantageous in that
they may
be orally active.
The present invention may provide for a medicament for one or more of (i)
regulation of
carbohydrate metabolism, (ii) regulation of protein metabolism, (iii)
regulation of lipid
metabolism, (iv) regulation of normal growth and/or development, (v) influence
on
cognitive function, (vi) resistance to stress and mineralocorticoid activity.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
g
Some of the compounds of the present invention may also be useful for
inhibiting
hepatic gluconeogenesis. The present invention may also provide a medicament
to
relieve the effects of endogenous glucocorticoids in diabetes mellitus,
obesity (including
centripetal obesity), neuronal loss and/or the cognitive impairment of old
age. Thus, in a
further aspect, the invention provides the use of an inhibitor of 11 (3-HSD in
the
manufacture of a medicament for producing one or more therapeutic effects in a
patient
to whom the medicament is administered, said therapeutic effects selected from
inhibition of hepatic gluconeogenesis, an increase in insulin sensitivity in
adipose tissue
and muscle, and the prevention of or reduction in neuronal loss/cognitive
impairment
due to glucocorticoid-potentiated neurotoxicity or neural dysfunction or
damage.
From an alternative point of view, the invention provides a method of
treatment of a
human or animal patient suffering from a condition selected from the group
consisting of:
hepatic insulin resistance, adipose tissue insulin resistance, muscle insulin
resistance,
neuronal loss or dysfunction due to glucocorticoid potentiated neurotoxicity,
and any
combination of the aforementioned conditions, the method comprising the step
of
administering to said patient a medicament comprising a pharmaceutically
active amount
of a compound in accordance with the present invention.
Some of the compounds of the present invention may be useful for the treatment
of
cancer, such as breast cancer, as well as (or in the alternative) non-
malignant
conditions, such as the prevention of auto-immune diseases, particularly when
pharmaceuticals may need to be administered from an early age.
DETAILED ASPECTS OF THE PRESENT INVENTION
As previously mentioned, in one aspect the present invention provides a
compound
having Formula I defined above.
As previously, mentioned, in one aspect the present invention provides a
pharmaceutical
composition comprising
(i) a compound having Formula I defined above
(ii) optionally admixed with a pharmaceutically acceptable carrier, diluent,
excipient or
adjuvant.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
9
As previously mentioned, in one aspect the present invention provides a
compound
having Formula I defined above, for use in medicine.
As previously mentioned, in one aspect the present invention provides a use of
a
compound having Formula I defined above in the manufacture of a medicament for
use
in the therapy of a condition or disease associated with 11 ~3-HSD.
In one aspect the present invention provides a use of a compound having
Formula I
defined above in the manufacture of a medicament for use in the therapy of a
condition
or disease associated with adverse 11 ~i-HSD levels.
In one aspect the present invention provides a use of a compound having
Formula I
defined above in the manufacture of a pharmaceutical for modulating 11 ~i-HSD
activity.
In one aspect the present invention provides a use of a compound having
Formula I
defined above in the manufacture of a pharmaceutical for inhibiting 11 (3-HSD
activity.
In one aspect the present invention provides a method comprising (a)
performing a 11 (3-
HSD assay with one or more candidate compounds having Formula I defined above;
(b)
determining whether one or more of said candidate compounds is/are capable of
modulating 11 ~i-HSD activity; and (c) selecting one or more of said candidate
compounds that is/are capable of modulating 11 (3-HSD activity.
In one aspect the present invention provides a method comprising (a)
performing a 11~i-
HSD assay with one or more candidate compounds having Formula I defined above;
(b)
determining whether one or more of said candidate compounds is/are capable of
inhibiting 11 (3-HSD activity; and (c) selecting one or more of said candidate
compounds
that is/are capable of inhibiting 11 (3-HSD activity.
In one aspect the present invention provides
~ a compound identified by the above method,
~ the use of the said compound in medicine,
~ a pharmaceutical composition comprising the said compound, optionally
admixed with
a pharmaceutically acceptable carrier, diluent, excipient or adjuvant,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
~ use of the said compound in the manufacture of a medicament for use in the
therapy
of a condition or disease associated with 11 ~i-HSD, and
~ use of the said compound in the manufacture of a medicament for use in the
therapy
of a condition or disease associated with adverse 11 ~i-HSD levels.
5
For ease of reference, these and further aspects of the present invention are
now
discussed under appropriate section headings. However, the teachings under
each
section are not necessarily limited to each particular section.
10 PREFERABLE ASPECTS
Compound
As previously mentioned, in one aspect the present invention provides a
compound
having Formula I
R~-Z-R~ Formula I
wherein R~ is an optionally substituted phenyl ring; R~ is or comprises an
optionally
substituted aromatic ring; and Z is -X-Y-L- or -Y-X-L- wherein either X is
selected from -
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-; L is an optional linker; and R3, R4 and R5 are each
independently
selected from H and hydrocarbyl; and wherein when R~ comprises the following
structural moiety
C>
wherein Q is an atom selected from the group consisting of S, O, N and C; the
compound is selected from compounds of the formulae R~-C(=O)-NR3-L-R2; R~
S(=O)(=O)-C(Ra)(Rs)-L-Rz; R~-S-C(R4)(R5)-L-R2; R~-NR3-S(=O)(=O)-L-R~; R~-NR3_
C(=O)-L-R2; R~-C(R4)(R5)-S(=O)(=O)-L-R~; and R~-C(R4)(R5)-S-L-R2.
It will be readily appreciated that references to the structural moiety
N
Q/
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
11
wherein Q is an atom selected from the group consisting of S, O, N and C,
include the
following structural moieties
N N N N
S O N C/
The structural moiety may be saturated or may include unsaturation, such as
one or
more double bonds.
Thus, references to the structural moiety
N
~s
include structural moieties such as
N N N
Q Q Q
The structural moiety may be substituted. The structural moiety may be part of
a
polycyclic system such as
N , for example / N
Q
R~ and R2
R~ is an optionally substituted phenyl ring and R~ is or comprises an
optionally
substituted aromatic ring. R~ and R~ are referred to collectively as the ring
systems.
R~ is an optionally substituted phenyl ring.
R~ may be substituted or unsubstituted. Preferably R~ is substituted.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
12
R~ may be substituted with one or more hydrocarbyl substituents. Preferably
the
substituents are selected from hydrocarbon groups, oxyhydrocarbon groups,
halogens,
amines and amides. More preferably the substituents are selected from aromatic
hydrocarbon groups, alkyl groups, oxyalkyl groups, halogens, amines and
amides, such
as from aromatic hydrocarbon groups, alkyl groups, oxyalkyl groups and
halogens.
In a highly preferred aspect the substituents are selected from phenyl groups,
C~_5alkyl
groups, oxy-C~_5-alkyl groups, chloro, bromo and iodo. More preferably the
substituents
are selected from phenyl, methyl, ethyl, propyl, O-methyl, O-ethyl, O-propyl
and chloro.
Preferably R~ is selected from the following:
i~ i~ i
o~ o~ o/
\ \ ~ o~
CI CI CI
CI
CI
CI CI NHS
H
NHZ ~ N
O
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
13
Preferably R~ is or comprises a group selected from the following wherein ----
indicates
the point of attachment to Z.
N
HN O
O~ O~
~~
CI
CI
CI CI CI
NHZ
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
14
CI NHZ
H
N
O
/ \ / I ,
More preferably R~ is selected from the following:
of a
R2 is or comprises an optionally substituted aromatic ring. Preferably the
optionally
substituted aromatic ring is a five or six membered ring. In one aspect
preferably R~ is
an optionally substituted five membered aromatic ring. In another aspect
preferably R2 is
an optionally substituted six membered aromatic ring.
In one preferred aspect, the optionally substituted aromatic ring is a
heterocyclic ring.
Preferably R~ is an optionally substituted five or six membered aromatic
heterocyclic
ring.
Preferably the optionally substituted aromatic ring is a heterocyclic ring
comprising a
carbon and a hetero atom selected from O, S and N. More preferably the
optionally
substituted aromatic ring is a heterocyclic ring comprising a carbon and a
hetero atom
selected from O and N.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
1S
Preferably R2 is or comprises:
\\ \ %
\ /
v ~% I w o
Preferably R~ is
s
Preferably Ra is or comprises a group selected from the following wherein ----
indicates
the point of attachment to Z.
~J
In one preferred aspect, R2 is an optionally substituted five membered
heterocyclic
aromatic ring. In this aspect preferably R2 is or comprises:
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
16
In this aspect more preferably R~ is or comprises:
n_
In one preferred aspect the optionally substituted aromatic ring is a
carbocyclic ring.
R2 may be substituted or unsubstituted. Preferably R2 is substituted, more
preferably Ra
is a substituted aromatic carbocyclic ring.
In one aspect the substituents are selected from hydrocarbon groups,
oxyhydrocarbon
groups, halogens, amines and amides. More preferably the substituents are
selected
from aromatic hydrocarbon groups, alkyl groups, oxyalkyl groups, halogens,
amines and
amides, such as from aromatic hydrocarbon groups, alkyl groups, oxyalkyl
groups and
halogens.
In one aspect R2 is substituted with two or more substituents. In a preferred
aspect Rz is
substituted with two or more substituents and the two or more substituents
together form
a ring which is fused to the carbocyclic ring of Rz.
Preferably the carbocyclic ring is a five or six membered aromatic carbocyclic
ring. More
preferably the carbocyclic ring is a phenyl ring.
In a preferred aspect R2 is a substituted phenyl ring. Preferably R~ is
substituted with
two or more substituents and the two or more substituents together form a ring
which is
fused to the phenyl ring of R2.
In this aspect preferably R2 comprises the following structure:
i
wherein A represents a heterocyclic ring, preferably a five or six membered
heterocyclic
ring.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
17
Thus, in one aspect preferably R2 comprises the following structure:
I A
wherein A represents a five membered heterocyclic ring. In this aspect
preferably R~ is
or comprises a group selected from the following:
/ s / o \ \
\ \ / N
H
\ S~N
/ ~ / ° \ \
\ \ / N
~O ~O
\ \ \ S
\N \N
/ N~ / S /
H
HN
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
18
~ iN I ~ iN
More preferably R~ is or comprises a group selected from the following:
I i ~ i
v
O \N
~ /
In another preferred aspect R~ comprises the following structure:
=i~~
wherein A represents a six membered heterocyclic ring. In this aspect
preferably R~ is
or comprises a group selected from the following:
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
19
More preferably R~ is or comprises a group selected from the following:
N\ ~ N\ ~ N\
N ~ N/
In one preferred aspect R2 is or comprises a group selected from the
following:
S ~ O ~ N
/ ~ / ~ \
\ \
N\ ~ N\ ~ N\
N/ / N/
s
O \ O
\ \ \ ~N
~ \N ~ /
0 0
0
In another preferred aspect R~ is or comprises a group selected from the
following:
s ~ o ~ N
/ ~ / ~ \
\ \
N\ \ N\ \ N\
N/ / N/
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
/ \ o I / i.- I / /v
Preferably Ra is or comprises a group selected from the following wherein ----
indicates
the point of attachment to Z.
/ s / o \ N
.\ , -\ / s
\ N~ \ N\ \ N\
- / N~ - ~ N - / /
O \ O
\ ~ \N
\ \ N . / /
- / o o /
0
N
____ O
N _
\O
5 Preferably in this aspect, R~ is a substituted phenyl and Z is -S(=O)(=O)NH-
or -
NHS(=O)(=O) -.
The compound of the present invention may have substituents other than those
of the
ring systems show herein. Furthermore the ring systems herein are given as
general
10 formulae and should be interpreted as such. The absence of any specifically
shown
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
21
substituents on a given ring member indicates that the ring member may
substituted with
any moiety of which H is only one example. Each ring system may contain one or
more
degrees of unsaturation, for example is some aspects one or more rings of a
ring
system is aromatic. Each ring system may be carbocyclic or may contain one or
more
hetero atoms.
The compound of the invention, in particular the ring systems of the compound
of the
invention may contain substituents other than those show herein. By way of
example,
these other substituents may be one or more of: one or more halo groups, one
or more
O groups, one or more hydroxy groups, one or more amino groups, one or more
sulphur
containing group(s), one or more hydrocarbyl groups) - such as an
oxyhydrocarbyl
group.
In general terms the ring systems of the present compounds may contain a
variety of non-
interfering substituents. In particular, the ring systems may contain one or
more hydroxy,
alkyl especially lower (C~-C6) alkyl, e.g. methyl, ethyl, n-propyl, isopropyl,
n-butyl, sec-
butyl, tent-butyl, n-pentyl and other pentyl isomers, and n-hexyl and other
hexyl isomers,
alkoxy especially lower (C,-C6) alkoxy, e.g. methoxy, ethoxy, propoxy etc.,
alkinyl, e.g.
ethinyl, or halogen, e.g. fluoro substituents.
X. Y and L
As previously mentioned, Z in Formula I is -X-Y-L- or -Y-X-L-; wherein either
X is
selected from -S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -
S(=O)(=O)- and -S-, and Y is -C(R4)(R5)-; L is an optional linker; and R3, R4
and R5 are
each independently selected from H and hydrocarbyl.
In one preferred aspect, X is -C(=O)-. In this aspect preferably Z is selected
from -
C(=O)-NR3-, -C(=O)-NR3-L-, -NR3-C(=O)-, and -NR3-C(=O)-L-.
In one preferred aspect, X is -S(=O)(=O)-. In this aspect preferably Z is
selected from -
S(=O)(=O)-NR3-, -S(=O)(=O)-NR3-L-, -NR3-S(=O)(=O)-, -NR3-S(=O)(=O)-L-, -
S(=O)(=O)-C(R4)(Rs)-~ -S(=O)(=O)-C(Ra)(Rs)-L-~ -C(Ra)(Rs)-S(=O)(=O)- and -
C(R4)(Rs)-
S(=O)(=O)-L-.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
22
In one preferred aspect, X is -S-. In this aspect preferably Z is selected
from -S-
C(Ra)(Rs)-, -S-C(R4)(Rs)-L-, -C(Ra)(Rs)-S- and -C(R4)(Rs)-S-L-.
In one preferred aspect, Y is -NR3-. In this aspect preferably Z is selected
from -C(=O)
NR3-, -C(=O)-NR3-L-, -NR3-C(=O)-, -NR3-C(=O)-L-, -S(=O)(=O)-NR3-, -S(=O)(=O)-
NR3_
L-, -NR3-S(=O)(=O)- and -NR3-S(=O)(=O)-L-.
In another preferred aspect, Y is -C(R4)(R5)-. In this aspect preferably Z is
selected from
-S(-0)(-O)-C(R4)(R5)-r -S(=O)(=O)-C(R4)(Rs)-L-~ -C(Ra)(Rs)-S(=O)(=O)- and -
C(R4)(Rs)
S(=O)(=O)-L-, -S-C(R4)(R5)-, -S-C(R4)(R5)-L-, -C(R4)(R5)-S- and -C(R4)(R5)-S-L-
.
In one preferred aspect, X is -S(=O)(=O)- and Y is -NR3-. In this aspect
preferably Z is -
S(=O)(=O)NR3-or - NR3S(=O)(=O) -, such as -S(=O)(=O)NH-or - NHS(=O)(=O) -
In one preferred aspect, X is -S(=O)(=O)- and Y is -C(R4)(R5)-. In this aspect
preferably
Z is -S(=O)(=O)C(R4)(R5)- or -C(R4)(R5)S(=O)(=O)-, such as -S(=O)(=O)CH2- or -
CH~S(=O)(=O)-
In one preferred aspect, X is -S- and Y is -C(R4)(R5)-. In this aspect
preferably Z is -
SC(R4)(R5)- or -C(R4)(R5)S-, such as -SCH2- or -CHaS-
L is an optional linker. In one aspect L is present. L may be any suitable
group, for
example L may a hydrocarbyl group or a hetero atom, in particular L may be a
hydrocarbon group such a C~_~o alkyl group. In one aspect, L is selected from -
C(=O)-, -
S(=O)(=O)-, -S-, -NR3-, -[C(R4)(R5)]n , -C6H4- and combinations thereof,
wherein R3, R4
and R5 are each independently selected from H and hydrocarbyl and wherein n is
an
integer from 1 to 10, preferably from 1 to 5, more preferably 1 or 2.. In one
preferred
aspect, L is selected from -C(=O)-, -S(=O)(=O)-, -S-, -NR3-, -C(R4)(R5)-, -
C6H4- and
combinations thereof, wherein R3, R4 and R5 are each independently selected
from H
and hydrocarbyl. In these aspects X-Y is preferably S(=O)(=O)NR3- or -
NR3S(=O)(=O)
-, such as -S(=O)(=O)NH-or - NHS(=O)(=O) -.
In a preferred aspect, L is selected from -NR3-C(=O)-C(R4)(R5)-S- and -
S(=O)(=O)-NR3
NR3-C(=O)-. In these aspects X-Y is preferably S(=O)(=O)NR3- or - NR3S(=O)(=O)-
,
such as -S(=O)(=O)NH-or - NHS(=O)(=O) -.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
23
In a highly preferred aspect, L is selected from -NH-C(=O)-CH2-S- and -
S(=O)(=O)-NH-
NH-C(=O)-. In these aspects X-Y is preferably S(=O)(=O)NR3- or - NR3S(=O)(=O)-
, such
as -S(=O)(=O)NH-or -NHS(=O)(=O)-.
R3,,_R4 and R5
As previously mentioned, Z in Formula I is -X-Y-L- or -Y-X-L-; wherein either
X is
selected from -S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -
S(=O)(=O)- and -S-, and Y is -C(R4)(R5)-; L is an optional linker; and R3, R4
and R5 are
each independently selected from H and hydrocarbyl.
The term "hydrocarbyl group" as used herein means a group comprising at least
C and
H and may optionally comprise one or more other suitable substituents.
Examples of
such substituents may include halo, alkoxy, nitro, an alkyl group, a cyclic
group etc. In
addition to the possibility of the substituents being a cyclic group, a
combination of
substituents may form a cyclic group. If the hydrocarbyl group comprises more
than one
C then those carbons need not necessarily be linked to each other. For
example, at
least two of the carbons may be linked via a suitable element or group. Thus,
the
hydrocarbyl group may contain hetero atoms. Suitable hetero atoms will be
apparent to
those skilled in the art and include, for instance, sulphur, nitrogen and
oxygen. A non-
limiting example of a hydrocarbyl group is an acyl group.
A typical hydrocarbyl group is a hydrocarbon group. Here the term
"hydrocarbon"
means any one of an alkyl group, an alkenyl group, an alkynyl group, which
groups may
be linear, branched or cyclic, or an aryl group. The term hydrocarbon also
includes
those groups but wherein they have been opfiionally substituted. If the
hydrocarbon is a
branched structure having substituent(s) thereon, then the substitution may be
on either
the hydrocarbon backbone or on the branch; alternatively the substitutions may
be on
the hydrocarbon backbone and on the branch.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from optionally substituted alkyl group, optionally
substituted
haloalkyl group, aryl group, alkylaryl group, alkylarylakyl group, and an
alkene group.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
24
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from C~-Coo alkyl group, such as C~-C6 alkyl group, and
C~-C3
alkyl group. Typical alkyl groups include C~ alkyl, C~ alkyl, C3 alkyl, C4
alkyl, C5 alkyl, C~
alkyl, and C$ alkyl.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from C~-Coo haloalkyl group, C~-C6 haloalkyl group, C~-
C3
haloalkyl group, C~-Coo bromoalkyl group, C~-C6 bromoalkyl group, and C~-C3
bromoalkyl
group. Typical haloalkyl groups include C~ haloalkyl, C~ haloalkyl, C3
haloalkyl, C4
haloalkyl, C5 haloalkyl, C~ haloalkyl, C$ haloalkyl, C~ bromoalkyl, C~
bromoalkyl, C3
bromoalkyl, C4 bromoalkyl, C5 bromoalkyl, C~ bromoalkyl, and C$ bromoalkyl.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from aryl groups, alkylaryl groups, alkylarylakyl
groups, -(CH2)~-
~o-aryl, -(CHz)~-~o-Ph, (CHz)~-~o-Ph-C~_~o alkyl, -(CH2)~-5-Ph, (CH~)1-5-Ph-
C~_5 alkyl, -(CH2)1-
3-Ph, (CH2)~_3-Ph-C~_3 alkyl, -CHI-Ph, and -CH2-Ph-C(CH3)3. The aryl groups
may
contain a hetero atom. Thus the aryl group or one or more of the aryl groups
may be
carbocyclic or more may heterocyclic. Typical hetero atoms include O, N and S,
in
particular N.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from -(CH~)~-~o-cycloalkyl, -(CH~)~_~o-C3_~ocycloalkyl,
-(CH2)~-~-C3_
~cycloalkyl, -(CH2)~_5-C3-5cycloalkyl, -(CH2)~_3-C3_5cycloalkyl, and -CH2-
C3cycloalkyl.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from alkene groups. Typical alkene groups include C~-
Coo
alkene group, C~-C6 alkene group, C~-C3 alkene group, such as C~, C2, C3, C4,
C5, C6, or
C~ alkene group. In a preferred aspect the alkene group contains 1, 2 or 3 C=C
bonds.
In a preferred aspect the alkene group contains 1 C=C bond. In some preferred
aspect
at least one C=C bond or the only C=C bond is to the terminal C of the alkene
chain,
that is the bond is at the distal end of the chain to the ring system.
In some aspects of the present invention, one or more hydrocarbyl groups is
independently selected from oxyhydrocarbyl groups.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
The term "oxyhydrocarbyl" group as used herein means a group comprising at
least C, H
and O and may optionally comprise one or more other suitable substituents.
Examples
of such substituents may include halo-, alkoxy-, nitro-, an alkyl group, a
cyclic group etc.
In addition to the possibility of the substituents being a cyclic group, a
combination of
5 substituents may form a cyclic group. If the oxyhydrocarbyl group comprises
more than
one C then those carbons need not necessarily be linked to each other. For
example, at
least two of the carbons may be linked via a suitable element or group. Thus,
the
oxyhydrocarbyl group may contain hetero atoms. Suitable hetero atoms will be
apparent
to those skilled in the art and include, for instance, sulphur and nitrogen.
In one embodiment of the present invention, the oxyhydrocarbyl group is a
oxyhydrocarbon group.
Here the term "oxyhydrocarbon" means any one of an alkoxy group, an oxyalkenyl
group, an oxyalkynyl group, which groups may be linear, branched or cyclic, or
an
oxyaryl group. The term oxyhydrocarbon also includes those groups but wherein
they
have been optionally substituted. If the oxyhydrocarbon is a branched
structure having
substituent(s) thereon, then the substitution may be on either the hydrocarbon
backbone
or on the branch; alternatively the substitutions may be on the hydrocarbon
backbone
and on the branch.
Typically, the oxyhydrocarbyl group is of the formula C~_60 (such as a C~_30).
In a preferred aspect, R3 is selected from H and hydrocarbon groups.
Preferably, R3 is
selected from H and alkyl groups, preferably from H and C~_~oalkyl groups,
preferably
from H and C~_5alkyl groups. In a highly preferred aspect, R3 is H.
In a preferred aspect, R3 may be equivalent to -L-R2 wherein L and R2 are
independently
selected from the possibilities defined herein. In this aspect the present
compound
contains two groups of the formula -L-R~ wherein each L and R~ are selected
independently of each other. However in one aspect, each of L and R2 may be
the same
as the other of L and R~ present in the compound. Examples of compounds
meeting
such requirements are shown below.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
26
In a preferred aspect, R4 and R5 are independently selected from H and
hydrocarbon
groups. Preferably R4 and R5 are independently selected from H and alkyl
groups,
preferably independently selected from H and C~_~oalkyl groups, preferably
independently
selected from H and C~_5alkyl groups. In a preferred aspect at least one of R4
and R5 is
H. In a highly preferred aspect, R4 and RS are both H.
FURTHER ASPECTS
For some applications, preferably the compounds have a reversible action.
For some applications, preferably the compounds have an irreversible action.
In one embodiment, the compounds of the present invention are useful for the
treatment
of breast cancer.
The compounds of the present invention may be in the form of a salt.
The present invention also covers novel intermediates that are useful to
prepare the
compounds of the present invention. For example, the present invention covers
novel
alcohol precursors for the compounds. The present invention also encompasses a
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
27
process comprising precursors for the synthesis of the compounds of the
present
invention.
Steroid Dehydrogenase
11 (3 Steroid dehydrogenase may be referred to as "11 f3-HSD" or "HD" for
short
In some aspects of the invention 11 ~i-HSD is preferably 11 ~i-HSD Type 1.
In some aspects of the invention 11 ~i-HSD is preferably 11 ~i-HSD Type 2.
Steroid Dehydrogenase Inhibition
It is believed that some disease conditions associated with HD activity are
due to
conversion of a inactive, cortisone to an active, cortisol. In disease
conditions
associated with HD activity, it would be desirable to inhibit HD activity.
Here, the term "inhibit" includes reduce and/or eliminate and/or mask and/or
prevent the
detrimental action of HD.
HD Inhibitor
In accordance with the present invention, the compound of the present
invention is
capable of acting as an HD inhibitor.
Here, the term "inhibitor" as used herein with respect to the compound of the
present
invention means a compound that can inhibit HD activity - such as reduce
and/or
eliminate and/or mask and/or prevent the detrimental action of HD. The HD
inhibitor
may act as an antagonist.
The ability of compounds to inhibit steroid dehydrogenase activity can be
assessed
using the suitable biological assay presented in the Examples section.
It is to be noted that the compound of the present invention may have other
beneficial
properties in addition to or in the alternative to its ability to inhibit HD
activity.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
28
Therapy
The compounds .of the present invention may be used as therapeutic agents -
i.e. in
therapy applications.
The term "therapy" includes curative effects, alleviation effects, and
prophylactic effects.
The therapy may be on humans or animals, preferably female animals.
Pharmaceutical Compositions
In one aspect, the present invention provides a pharmaceutical composition,
which
comprises a compound according to the present invention and optionally a
pharmaceutically acceptable carrier, diluent or excipient (including
combinations
the reof).
The pharmaceutical compositions may be for human or animal usage in human and
veterinary medicine and will typically comprise any one or more of a
pharmaceutically
acceptable diluent, carrier, or excipient. Acceptable carriers or diluents for
therapeutic
use are well known in the pharmaceutical art, and are described, for example,
in
Rernington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit.
1985). The choice of pharmaceutical carrier, excipient or diluent can be
selected with
reg and to the intended route of administration and standard pharmaceutical
practice.
The pharmaceutical compositions may comprise as - or in addition to - the
carrier,
excipient or diluent any suitable binder(s), lubricant(s), suspending
agent(s), coating
agent(s), solubilising agent(s).
Preservatives, stabilisers, dyes and even flavouring agents may be provided in
the
pharmaceutical composition. Examples of preservatives include sodium benzoate,
sorbic acid and esters of p-hydroxybenzoic acid. Antioxidants and suspending
agents
may be also used.
There may be different composition/formulation requirements dependent on the
different
delivery systems. By way of example, the pharmaceutical composition of the
present
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
29
invention may be formulated to be delivered using a mini-pump or by a mucosal
route,
for example, as a nasal spray or aerosol for inhalation or ingestable
solution, or
parenterally in which the composition is formulated by an injectable form, for
delivery, by,
for example, an.intravenous, intramuscular or subcutaneous route.
Alternatively, the
formulation may be designed to be delivered by both routes.
Where the agent is to be delivered mucosally through the gastrointestinal
mucosa, it
should be able to remain stable during transit though the gastrointestinal
tract; for
example, it should be resistant to proteolytic degradation, stable at acid pH
and resistant
to the detergent effects of bile.
Where appropriate, the pharmaceutical compositions can be administered by
inhalation,
in the form of a suppository or pessary, topically in the form of a lotion,
solution, cream,
ointment or dusting powder, by use of a skin patch, orally in the form of
tablets
containing excipients such as starch or lactose, or in capsules or ovules
either alone or
in admixture with excipients, or in the form of elixirs, solutions or
suspensions containing
flavouring or colouring agents, or they can be injected parenterally, for
example
intravenously, intramuscularly or subcutaneously. For parenteral
administration, the
compositions may be best used in the form of a sterile aqueous solution which
may
contain other substances, for example enough salts or monosaccharides to make
the
solution isotonic with blood. For buccal or sublingual administration the
compositions
may be administered in the form of tablets or lozenges which can be formulated
in a
conventional manner.
Combination Pharmaceutical
The compound of the present invention may be used in combination with one or
more
other active agents, such as one or more other pharmaceutically active agents.
By way of example, the compounds of the present invention may be used in
combination
with other 11 (3-HSD inhibitors and/or other inhibitors such as an aromatase
inhibitor (such
as for example, 4hydroxyandrostenedione (4-OHA)), and/or a steroid sulphatase
inhibitors such as EMATE and/or steroids - such as the naturally occurring
sterneurosteroids dehydroepiandrosterone sulfate (DHEAS) and pregnenolone
sulfate
(PS) and/or other structurally similar organic compounds.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
In addition, or in the alternative, the compound of the present invention may
be used in
combination with a biological response modifier.
5 The term biological response modifier ("BRM") includes cytokines, immune
modulators,
growth factors, haematopoiesis regulating factors, colony stimulating factors,
chemotactic, haemolytic and thrombolytic factors, cell surface receptors,
ligands,
leukocyte adhesion molecules, monoclonal antibodies, preventative and
therapeutic
vaccines, hormones, extracellular matrix components, fibronectin, etc. For
some
10 applications, preferably, the biological response modifier is a cytokine.
Examples of
cytokines include: interleukins (IL) - such as IL-1, IL-2, IL-3, IL-4, IL-5,
IL-6, IL-7, IL-8, IL-
9, IL-10, IL-11, IL-12, IL-19; Tumour Necrosis Factor (TNF) - such.as TNF-a;
Interferon
alpha, beta and gamma; TGF-(3. For some applications, preferably the cytokine
is
tumour necrosis factor (TNF). For some applications, the TNF may be any type
of TNF -
15 such as TNF-a, TNF-(3, including derivatives or mixtures thereof. More
preferably the
cytokine is TNF-a. Teachings on TNF may be found in the art - such as WO-A-
98/08870
and WO-A-98/13348.
Administration
Typically, a physician will determine the actual dosage which will be most
suitable for an
individual subject and it will vary with the age, weight and response of the
particular
patient. The dosages below are exemplary of the average case. There can, of
course,
be individual instances where higher or lower dosage ranges are merited.
The compositions of the present invention may be administered by direct
injection. The
composition may be formulated for parenteral, mucosal, intramuscular,
intravenous,
subcutaneous, intraocular or transdermal administration. Depending upon the
need, the
agent may be administered at a dose of from 0.01 to 30 mg/kg body weight, such
as
from 0.1 to 10 mg/kg, more preferably from 0.1 to 1 mglkg body weight.
By way of further example, the agents of the present invention may be
administered in
accordance with a regimen of 1 to 4 times per day, preferably once or twice
per day.
The specific dose level and frequency of dosage for any particular patient may
be varied
and will depend upon a variety of factors including the activity of the
specific compound
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
31
employed, the metabolic stability and length of action of that compound, the
age, body
weight, general health, sex, diet, mode and time of administration, rate of
excretion, drug
combination, the severity of the particular condition, and the host undergoing
therapy.
Aside from the typical modes of delivery - indicated above - the term
"administered"
also includes delivery by techniques such as lipid mediated transfection,
liposomes,
immunoliposomes, lipofectin, cationic facial amphiphiles (CFAs) and
combinations
thereof. The routes for such delivery mechanisms include but are not limited
to
mucosal, nasal, oral, parenteral, gastrointestinal, topical, or sublingual
routes.
The term "administered" includes but is not limited to delivery by a mucosal
route, for
example, as a nasal spray or aerosol for inhalation or as an ingestable
solution; a
parenteral route where delivery is by an injectable form, such as, for
example, an
intravenous, intramuscular or subcutaneous route.
Thus, for pharmaceutical administration, the compounds of the present
invention can be
formulated in any suitable manner utilising conventional pharmaceutical
formulating
techniques and pharmaceutical carriers, adjuvants, excipients, diluents etc.
and usually
for parenteral administration. Approximate effective dose rates may be in the
range
from 1 to 1000 mg/day, such as from 10 to 900 mg/day or even from 100 to 800
mg/day
depending on the individual activities of the compounds in question and for a
patient of
average (70Kg) bodyweight. More usual dosage rates for the preferred and more
active
compounds will be in the range 200 to 800 mg/day, more preferably, 200 to 500
mg/day,
most preferably from 200 to 250 mg/day. They may be given in single dose
regimes,
split dose regimes and/or in multiple dose regimes lasting over several days.
For oral
administration they may be formulated in tablets, capsules, solution or
suspension
containing from 100 to 500 mg of compound per unit dose. Alternatively and
preferably
the compounds will be formulated for parenteral administration in a suitable
parenterally
admin istrable carrier and providing single daily dosage rates in the range
200 to 800 mg,
preferably 200 to 500, more preferably 200 to 250 mg. Such effective daily
doses will,
however, vary depending on inherent activity of the active ingredient and on
the
bodyvveight of the patient, such variations being within the skill and
judgement of the
physician.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
32
Cell Cycling
The compounds of the present invention may be useful in the method of
treatment of a
cell cycling disorder.
As discussed in "Molecular Cell Biology" 3rd Ed. Lodish et al. pages 177-181
different
eukaryotic cells can grow and divide at quite different rates. Yeast cells,
for example,
can divide every 120 min., and the first divisions of fertilised eggs in the
embryonic cells
of sea urchins and insects take only 1530 min. because one large pre-existing
cell is
subdivided. However, most growing plant and animal cells take 10-20 hours to
double in
number, and some duplicate at a much slower rate. Many cells in adults, such
as nerve
cells and striated muscle cells, do not divide at all; others, like the
fibroblasts that assist
in healing wounds, grow on demand but are otherwise quiescent.
Still, every eukaryotic cell that divides must be ready to donate equal
genetic material to
two daughter cells. DNA synthesis in eukaryotes does not occur throughout the
cell
division cycle but is restricted to a part of it before cell division.
The relationship between eukaryotic DNA synthesis and cell division has been
thoroughly analysed in cultures of mammalian cells that were all capable of
growth and
division. In contrast to bacteria, it was found, eukaryotic cells spend only a
part of their
time in DNA synthesis, and it is completed hours before cell division
(mitosis). Thus a
gap of time occurs after DNA synthesis and before cell division; another gap
was found
to occur after division and before the next round of DNA synthesis. This
analysis led to
the conclusion that the eukaryotic cell cycle consists of an M (mitotic)
phase, a G~ phase
(the first gap), the S (DNA synthesis) phase, a G2 phase (the second gap), and
back to
M. The phases between mitoses (G~, S, and G~) are known collectively as the
interphase.
Many nondividing cells in tissues (for example, all quiescent fibroblasts)
suspend the
cycle after mitosis and just prior to DNA synthesis; such "resting" cells are
said to have
exited from the cell cycle and to be in the Go state.
It is possible to identify cells when they are in one of the three interphase
stages of the
cell cycle, by using a fluorescence-activated cell sorter (FACS) to measure
their relative
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
33
DNA content: a cell that is in G~ (before DNA synthesis) has a defined amount
x of DNA;
during S (DNA replication), it has between x and 2x; and when in G2 (or M), it
has 2x of
DNA.
The stages of mitosis and cytokinesis in an animal cell are as follows
(a) Interphase. The G2 stage of interphase immediately precedes the beginning
of
mitosis. Chromosomal DNA has been replicated and bound to protein during the S
phase, but chromosomes are not yet seen as distinct structures. The nucleolus
is the
only nuclear substructure that is visible under light microscope. In a diploid
cell before
DNA replication there are two morphologic chromosomes of each type, and the
cell is
said to be 2n. In G2, after DNA replication, the cell is 4n. There are four
copies of each
chromosomal DNA. Since the sister chromosomes have not yet separated from each
other, they are called sister chromatids.
b) Early prophase. Centrioles, each with a newly formed daughter centriole,
begin
moving toward opposite poles of the cell; the chromosomes can be seen as long
threads. The nuclear membrane begins to disaggregate into small vesicles.
(c) Middle and late prophase. Chromosome condensation is completed; each
visible
chromosome structure is composed of two chromatids held together at their
centromeres. Each chromatid contains one of the two newly replicated daughter
DNA
molecules. The microtubular spindle begins to radiate from the regions just
adjacent to
the centrioles, which are moving closer to their poles. Some spindle fibres
reach from
pole to pole; most go to chromatids and attach at kinetochores.
(d) Metaphase. The chromosomes move toward the equator of the cell, where they
become aligned in the equatorial plane. The sister chromatids have not yet
separated.
(e) Anaphase. The two sister chromatids separate into independent chromosomes.
Each contains a centromere that is linked by a spindle fibre to one pole, to
which it
moves. Thus one copy of each chromosome is donated to each daughter cell.
Simultaneously, the cell elongates, as do the pole-to-pole spindles.
Cytokinesis begins
as the cleavage furrow starts to form.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
34
(f) Telophase. New membranes form around the daughter nuclei; the
chromosomes uncoil and become less distinct, the nucleolus becomes visible
again, and
the nuclear membrane forms around each daughter nucleus. Cytokinesis is nearly
complete, and the spindle disappears as the microtubules and other fibres
depolymerise.
Throughout mitosis the "daughter" centriole at each pole grows until it is
full-length. At
telophase the duplication of each of the original centrioles is completed, and
new
daughter centrioles will be generated during the next interphase.
(g) Interphase. Upon the completion of cytokinesis, the cell enters the G~
phase of
the cell cycle and proceeds again around the cycle.
It will be appreciated that cell cycling is an extremely important cell
process. Deviations
from normal cell cycling can result in a number of medical disorders.
Increased and/or
unrestricted cell cycling may result in cancer. Reduced cell cycling may
result in
degenerative conditions. Use of the compound of the present invention may
provide a
means to treat such disorders and conditions.
Thus, the compound of the present invention may be suitable for use in the
treatment of
cell cycling disorders such as cancers, including hormone dependent and
hormone
independent cancers.
In addition, the compound of the present invention may be suitable for the
treatment of
cancers such as breast cancer, ovarian cancer, endometrial cancer, sarcomas,
melanomas, prostate cancer, pancreatic cancer etc. and other solid tumours.
For some applications, cell cycling is inhibited and/or prevented and/or
arrested,
preferably wherein cell cycling is prevented and/or arrested. In one aspect
cell cycling
may be inhibited and/or prevented and/or arrested in the G2/M phase. In one
aspect cell
cycling may be irreversibly prevented and/or inhibited and/or arrested,
preferably wherein
cell cycling is irreversibly prevented and/or arrested.
By the term "irreversibly prevented and/or inhibited and/or arrested" it is
meant after
application of a compound of the present invention, on removal of the compound
the
effects of the compound, namely prevention and/or inhibition andlor arrest of
cell cycling,
are still observable. More particularly by the term "irreversibly prevented
and/or inhibited
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
and/or arrested" it is meant that when assayed in accordance with the cell
cycling assay
protocol presented herein, cells treated with a compound of interest show less
growth after
Stage 2 of the protocol I than control cells. Details on this protocol are
presented below.
5 Thus, the present invention provides compounds which: cause inhibition of
growth of
oestrogen receptor positive (ER+) and ER negative (ER-) breast cancer cells in
vitro by
preventing and/or inhibiting and/or arresting cell cycling; and/or cause
regression of
nitroso-methyl urea (NMU)-induced mammary tumours in intact animals (i.e. not
ovariectomised), and/or prevent and/or inhibit and/or arrest cell cycling in
cancer cells;
10 and/or act in vivo by preventing and/or inhibiting and/or arresting cell
cycling and/or act as
a cell cycling agonist.
CELL CYCLING ASSAY
(PROTOCOL 2)
15 Procedure
Stage 1
MCF-7 breast cancer cells are seeded into multi-well culture plates at a
density of 105
cells/well. Cells were allowed to attach and grown until about 30% confluent
when they
20 are treated as follows:
Control - no treatment
Compound of Interest (COI) 20p,M
25 Cells are grown for 6 days in growth medium containing the COI with changes
of
medium/COI every 3 days. At the end of this period cell numbers were counted
using a
Coulter cell counter.
Stage 2
After treatment of cells for a 6-day period with the COI cells are re-seeded
at a density of
104 cells/well. No further treatments are added. Cells are allowed to continue
to grow
for a further 6 days in the presence of growth medium. At the end of this
period cell
numbers are again counted.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
36
Cancer
As indicated, the compounds of the present invention may be useful in the
treatment of a
cell cycling disorder. A particular cell cycling disorder is cancer.
Cancer remains a major cause of mortality in most Western countries. Cancer
therapies
developed so far have included blocking the action or synthesis of hormones to
inhibit
the growth of hormone-dependent tumours. However, more aggressive chemotherapy
is currently employed for the treatment of hormone-independent tumours.
Hence, the development of a pharmaceutical for anti-cancer treatment of
hormone
dependent and/or hormone independent tumours, yet lacking some or all of the
side-
effects associated with chemotherapy, would represent a major therapeutic
advance.
We believe that the compound of the present invention provides a means for the
treatment of cancers and, especially, breast cancer.
In addition or in the alternative the compound of the present invention may be
useful in
the blocking the growth of cancers including leukaemias and solid tumours such
as
breast, endometrium, prostate, ovary and pancreatic tumours.
Other Therapies
As previously mentioned, in one aspect the present invention provides use of a
compound as described herein in the manufacture of a medicament for use in the
therapy of a condition or disease associated with 11(3-HSD.
Conditions and diseases associated with 11 ~i-HSD have been reviewed in
Walker, E. A,;
Stewart, P. M.; Trends in Endocrinology and Metabolism, 2003, 14 (7), 334-339.
In a preferred aspect, the condition or disease is selected from the group
consisting of:
~ metabolic disorders, such as diabetes and obesity
~ cardiovascular disorders, such as hypertension
~ glaucoma
~ inflammatory disorders, such as arthritis or asthma
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
37
~ immune disorders
~ bone disorders, such as osteoporosis
~ cancer
~ intra-uterine growth retardation
~ apparent mineralocorticoid excess syndrome (AME)
~ polycystic ovary syndrome (PCOS)
~ hirsutism
~ acne
~ oligo- or amenorrhea
~ adrenal cortical adenoma and carcinoma
~ Cushing's syndrome
~ pituitary tumours
~ invasive carcinomas
~ breast cancer; and
~ endometrial cancer.
It is also to be understood that the compound/composition of the present
invention may
have other important medical implications.
For example, the compound or composition of the present invention may be
useful in the
treatment of the disorders listed in WO-A-99/52890 - viz:
In addition, or in the alternative, the compound or composition of the present
invention
may be useful in the treatment of the disorders listed in WO-A-98/05635. For
ease of
reference, part of that list is now provided: diabetes including Type II
diabetes, obesity,
cancer, inflammation or inflammatory disease, dermatological disorders, fever,
cardiovascular effects, haemorrhage, coagulation and acute phase response,
cachexia,
anorexia, acute infection, HIV infection, shock states, graft-versus-host
reactions,
autoimmune disease, reperfusion injury, meningitis, migraine and aspirin-
dependent
anti-thrombosis; tumour growth, invasion and spread, angiogenesis, metastases,
malignant, ascites and malignant pleural effusion; cerebral ischaemia,
ischaemic heart
disease, osteoarthritis, rheumatoid arthritis, osteoporosis, asthma, multiple
sclerosis,
neurodegeneration, Alzheimer's disease, atherosclerosis, stroke, vasculitis,
Crohn's
disease and ulcerative colitis; periodontitis, gingivitis; psoriasis, atopic
dermatitis, chronic
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
38
ulcers, epidermolysis bullosa; corneal ulceration, retinopathy and surgical
wound
healing; rhinitis, allergic conjunctivitis, eczema, anaphylaxis; restenosis,
congestive heart
failure, endometriosis, atherosclerosis or endosclerosis.
In addition, or in the alternative, the compound or composition of the present
invention
may be useful in the treatment of disorders listed in WO-A-98/07859. For ease
of
reference, part of that list is now provided: cytokine and cell
proliferation/differentiation
activity; immunosuppressant or immunostimulant activity (e.g. for treating
immune
deficiency, including infection with human immune deficiency virus; regulation
of
lymphocyte growth; treating cancer and many autoimmune diseases, and to
prevent
transplant rejection or induce tumour immunity); regulation of haematopoiesis,
e.g.
treatment of myeloid or lymphoid diseases; promoting growth of bone,
cartilage, tendon,
ligament and nerve tissue, e.g. for healing wounds, treatment of burns, ulcers
and
periodontal disease and neurodegeneration; inhibition or activation of
follicle-stimulating
hormone (modulation of fertility); chemotactic/chemokinetic activity (e.g. for
mobilising
specific cell types to sites of injury or infection); haemostatic and
thrombolytic activity
(e.g. for treating haemophilia and stroke); antiinflammatory activity (for
treating e.g.
septic shock or Crohn's disease); as antimicrobials; modulators of e.g.
metabolism or
behaviour; as analgesics; treating specific deficiency disorders; in treatment
of e.g.
psoriasis, in human or veterinary medicine.
In addition, or in the alternative, the composition of the present invention
may be useful
in the treatment of disorders listed in WO-A-98/09985. For ease of reference,
part of
that list is now provided: macrophage inhibitory and/or T cell inhibitory
activity and thus,
anti-inflammatory activity; anti-immune activity, i.e. inhibitory effects
against a cellular
and/or humoral immune response, including a response not associated with
inflammation; inhibit the ability of macrophages and T cells to adhere to
extracellular
matrix components and fibronectin, as well as up-regulated fas receptor
expression in T
cells; inhibit unwanted immune reaction and inflammation including arthritis,
including
rheumatoid arthritis, inflammation associated with hypersensitivity, allergic
reactions,
asthma, systemic lupus erythematosus, collagen diseases and other autoimmune
diseases, inflammation associated with atherosclerosis, arteriosclerosis,
atherosclerotic
heart disease, reperfusion injury, cardiac arrest, myocardial infarction,
vascular
inflammatory disorders, respiratory distress syndrome or other cardiopulmonary
diseases, inflammation associated with peptic ulcer, ulcerative colitis and
other diseases
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
39
of the gastrointestinal tract, hepatic fibrosis, liver cirrhosis or other
hepatic diseases,
thyroiditis or other glandular diseases, glomerulonephritis or other renal and
urologic
diseases, otitis or other oto-rhino-laryngological diseases, dermatitis or
other dermal
diseases, periodontal diseases or other dental diseases, orchitis or epididimo-
orchitis,
infertility, orchidal trauma or other immune-related testicular diseases,
placental
dysfunction, placental insufficiency, habitual abortion, eclampsia, pre-
eclampsia and
other immune and/or inflammatory-related gynaecological diseases, posterior
uveitis,
intermediate uveitis, anterior uveitis, conjunctivitis, chorioretinitis,
uveoretinitis, optic
neuritis, intraocular inflammation, e.g. retinitis or cystoid macular oedema,
sympathetic
ophthalmia, scleritis, retinitis pigmentosa, immune and inflammatory
components of
degenerative fondus disease, inflammatory components of ocular trauma, ocular
inflammation caused by infection, proliferative vitreo-retinopathies, acute
ischaemic optic
neuropathy, excessive scarring, e.g. following glaucoma filtration operation,
immune
and/or inflammation reaction against ocular implants and other immune and
inflammatory-related ophthalmic diseases, inflammation associated with
autoimmune
diseases or conditions or disorders where, both in the central nervous system
(CNS) or
in any other organ, immune and/or inflammation suppression would be
beneficial,
Parkinson's disease, complication and/or side effects from treatment of
Parkinson's
disease, AIDS-related dementia complex HIV-related encephalopathy, Devic's
disease,
Sydenham chorea, Alzheimer's disease and other degenerative diseases,
conditions or
disorders of the CNS, inflammatory components of stokes, post-polio syndrome,
immune and inflammatory components of psychiatric disorders, myelitis,
encephalitis,
subacute sclerosing pan-encephalitis, encephalomyelitis, acute neuropathy,
subacute
neuropathy, chronic neuropathy, Guillaim-Barre syndrome, Sydenham chora,
myasthenia gravis, pseudo-tumour cerebri, Down's Syndrome, Huntington's
disease,
amyotrophic lateral sclerosis, inflammatory components of CNS compression or
CNS
trauma or infections of the CNS, inflammatory components of muscular atrophies
and
dystrophies, and immune and inflammatory related diseases, conditions or
disorders of
the central and peripheral nervous systems, post-traumatic inflammation,
septic shock,
infectious diseases, inflammatory complications or side effects of surgery,
bone marrow
transplantation or other transplantation complications and/or side effects,
inflammatory
and/or immune complications and side effects of gene therapy, e.g. due to
infection with
a viral carrier, or inflammation associated with AIDS, to suppress or inhibit
a humoral
and/or cellular immune response, to treat or ameliorate monocyte or leukocyte
proliferative diseases, e.g. leukaemia, by reducing the amount of monocytes or
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
lymphocytes, for the prevention and/or treatment of graft rejection in cases
of
transplantation of natural or artificial cells, tissue and organs such as
cornea, bone
marrow, organs, lenses, pacemakers, natural or artificial skin tissue.
5 Summary
In summation, the present invention provides compounds for use as steroid
dehydrogenase inhibitors, and pharmaceutical compositions for the same.
10 BROAD ASPECTS
In one broad aspect the present invention provides a compound having Formula I
R~-Z-R2 Formula I
wherein Z is -X-Y-L- or -Y-X-L-; R~ is an aromatic ring; R2 is or comprises a
ring; X is
selected from -C(=O)-, -S(=O)(=O)- and -S-; Y is selected from -NR3- and -
C(R4)(R5)-; L
15 is an optional linker; R3, R4 and R5 are each independently selected from H
and
hydrocarbyl.
In one broad aspect the present invention provides a pharmaceutical
composition
comprising
20 (i) a compound having Formula I
R~-Z-R~ Formula I
wherein Z is -X-Y-L- or -Y-X-L-; R~ is an aromatic ring; R~ is or comprises a
ring; X is
selected from -C(=O)-, -S(=O)(=O)- and -S-; Y is selected from -NR3- and -
C(R4)(R5)-; L
is an optional linker; R3, R4 and R5 are each independently selected from H
and
hydrocarbyl;
25 (ii) optionally admixed with a pharmaceutically acceptable carrier,
diluent, excipient or
adjuvant.
In one broad aspect the present invention provides a compound for use in
medicine
wherein the compound has Formula I
R~-Z-R~ Formula I
30 wherein Z is -X-Y-L- or -Y-X-L-; R~ is an aromatic ring; R~ is or comprises
a ring; X is
selected from -C(=O)-, -S(=O)(=O)- and -S-; Y is selected from -NR3- and -
C(R4)(R5)-; L
is an optional linker; R3, R4 and R5 are each independently selected from H
and
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
41
hydrocarbyl.
In one broad aspect the present invention provides a use of a compound in the
manufacture of a medicament for use in the therapy of a condition or disease
associated
with 11 ~i-HSD, wherein the compound has Formula I
R~-Z-R~ Formula I
wherein Z is -X-Y-L- or -Y-X-L-; R~ is an aromatic ring; R~ is or comprises a
ring; X is
selected from -C(=O)-, -S(=O)(=O)- and -S-; Y is selected from -NR3- and -
C(R4)(R5)-; L
is an optional linker; R3, R4 and R5 are each independently selected from H
and
hydrocarbyl.
In these broad aspects, R~ is an aromatic ring. Preferably R~ is a five or six
membered
aromatic ring, more preferably a six mernbered aromatic ring. Preferably, R~
is a
carbocyclic ring. In a highly preferred aspect, R~ is an optionally
substituted phenyl ring.
In these broad aspects, R~ is or comprises a ring, preferably an aromatic
ring, more
preferably an optionally substituted aromatic ring
In one broad aspect R2 comprises the following structure:
A
wherein A represents a heterocyclic ring, preferably a five or six membered
heterocyclic
ring.
In these broad aspects X is selected from -C(=O)-, -S(=O)(=O)- and -S-; and Y
is
selected from -NR3- and -C(R4)(R5)-. In a preferred aspect either X is
selected from -
S(=O)(=O)- and -C(=O)-, and Y is -NR3-; or X is selected from -S(=O)(=O)- and -
S-, and
Y is -C(R4)(R5)-.
BRIEF DESCRIPTION OF THE FIGURES
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
42
The present invention will be described in further detail by way of example
only with
reference to the accompanying figures in which:-
Figure 1 is a graph showing extraction efficiencies obtained with four
extraction
methods.
Figure 2 is a graph showing a comparison of 11 j3-HSD1 activity in rat and
human hepatic
microsomes.
Figure 3 is a series of graphs showing the effect of incubation time on human
microsomal 11 ~i-HSD1 activity
Figure 4 is a series of graphs showing the effect of microsomal protein
concentration on
human microsomal 11(3-HSD1 activity.
Figure 5 is a graph showing the substrate (cortisone) saturation curve for
human hepatic
microsomal 11 (3 HSD1.
Figure 6 is a Lineweaver-Burke plot.
Figure 7 is a graph showing the ICSO determination for Glycyrrhetinic acid.
Figure 8 is a graph showing the ICSO determination for Carbenoxolone.
Figures 9(A), 9(B) and 9(C) are graphs showing the 11 [3-HSD1 activity
measured by
Immunoassay. Figure 9(A) shows the effect of protein; Figure 9(B) shows the
effect of
cortisone; and Figure 9(C) shows the effect of Tween-80.
Figure 10 is a graph showing the performance of the cortisol immunoassay:
various
experimental designs.
Figure 11 is a graph showing the effect of increasing microsomal protein on
measurement of 11 (3 HSD1 activity detected by Assay Designs Immunoassay.
Figure 12 is a graph showing the detection of 11 j3 HSD1 activity by RIA using
the
Immunotech anti-cortisol antibody.
Figure 13 is a graph showing the effect of lowering the Immunotech antibody
concentration on the signal to noise (microsorne group compared to GA blank
group).
Figure 14 is a graph showing the Immunotech antibody saturation curve for
detection of
11 [3 HSD1 activity by RIA.
Figure 15 is a graph showing the linearity of human hepatic microsomal 11 a
HSD1
activity detected by RIA.
Figure 16 is a graph showing the effect of Tween 80 on detection of human
hepatic
microsomal 11 ~i HSD1 activity by RIA.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
43
Figure 17 is a graph showing the effect of buffer systems on detection of
human hepatic
microsomal 11 ~i HDS1 activity by RIA.
Figure 18 is a graph showing the linearity of human hepatic microsomal 11 ~3
HSD1
activity with incubation time detected by RIA.
Figure 19 is a graph showing the substrate saturation curve for human hepatic
microsomal 11 ~i HDS1 activity detected by RIA.
Figure 20 is a Lineweaver-Burke plot
Figure 21 is an ICSO curve for inhibition of human hepatic rnicrosomal 11 [3
HSD1 activity
by Glycyrrhetinic acid.
Figure 22 is an ICSO curve for inhibition of human hepatic nnicrosomal 11 (3
HSD1 activity
by Glycyrrhetinic acid in the presence of 350 nM cortisone.
Figure 23 is an ICSO curve for inhibition of human hepatic nnicrosomal 11 (3
HSD1 activity
by Carbenoxolone in the presence of 350 nM cortisone.
The present invention will now be described in further detail in the following
examples.
EXAMPLES
The present invention will now be described only by way of example.
Biological Assays
Standard Operating Procedure for the 11 (3-Hydroxysteroid Dehydrogenase Type 1
cortisol Radioimmunoassay (11 (3 HSD1 Cortisol RIA).
Reagents
Cortisone, Cortisol (Hydrocortisone), NADPH, Glucose-6-phosphate,
Glycyrrhetinic acid
(GA), Dextran coated charcoal (C6197) and DMSO were obtained from Sigma
Aldrich,
Carbenoxolone was obtained from ICN Biomedicals, Product 215493001, 3H-
cortisone
was obtained from American Radiolabelled Compounds Inc, Product ART-743, 3H-
cortisol was obtained from NEN, Product NET 396, '4C-cortisol was obtained
from NEN,
Product NEC 163, human hepatic microsomes were obtained from XenoTech, product
H0610 / Lot 0210078, rat hepatic microsomes were obtained from XenoTech, SPA
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
44
beads were obtained from Amersham, Product RPNQ0017, the Immunoassay kit was
obtained from Assay Designs, Product 900-071, the Immunologicals Direct anti-
cortisol
antibody was Product OBT 0646, the Sigma anti-cortisol antibody was Product
C8409
and the Immunotech antibody was supplied by Beckman, Product IMBULK3 6D6.
Buffer Solutions
Buffer 1, from Barf et al., (2002) [14]: 30 mM Tris-HCL, pH 7.2, containing 1
mM EDTA
Buffer 2, from the Sterix protocol: PBS (pH 7.4) containing 0.25M sucrose
Buffer 3, from the Sigma RIA protocol: 50 mM Tris-HCL, pH 8, containing 0.1 M
NaCI
and 0.1 % gelatin
Stop solution, from Barf et al., (2002) [14]: 1 mM Glycyrrhetinic acid in 100
% DMSO
Enzyme assays were carried out in the presence of 181 ~M NADPH, 1 mM Glucose-6-
Phosphate and cortisone concentrations indicated for each experiment.
Enzyme Assay Buffer
30 mM Tris-HCL, pH 7.2 containing 1 mM EDTA
Antibody Binding Buffer
50 mM Tris-HCL, pH 8, containing 0.1 M NaCI and 0.1 % gelatin
Compound Preparation
Prepare 10 mM stock solutions in 100% DMSO at 100 times the required assay
concentration. Dilute into assay buffer 1 in 25. Also dilute neat DMSO 1 in 25
into
assay buffer for controls.
Substrate Preaaration
Prepare a solution of cortisone in ethanol 600 times the required assay
concentration
(175 nM). Dilute this 1 in 50 into assay buffer.
Prepare NADPH as a 1.8 mg/ml solution in assay buffer.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
Prepare G-6-P as a 3.65 mg/ml solution in assay buffer.
Mix these 3 solutions 1:1:1 to make a solution of sufficient volume for 25 p.1
additions to
each sample. Add 0.5 ~Ci tritiated cortisone per 25 ~I and mix the solution
well.
5 Microsome Preparation
Dilute stock 20 mg/ml solution 1 in 100 with assay buffer.
Antibody Preparation
Dilute stock antibody solution to 17 p.g/ml in antibody binding buffer.
Dextran Coated Charcoal Preparation
Make a 20 mg/ml solution in antibody binding buffer and chill on ice.
Enzyme Assay
To a u-bottom polypropylene 96 well plate add:
25 ~,I compound dilution or diluted DMSO to controls, NSB's and blanks
10 w1 1 mM GA in DMSO (enzyme stop solution) to blanks
~I substrate mixture to all samples
50 p1 diluted microsomes to all samples
Incubate plate for 30 min at 37°C shaking
25 Add 10 ~,I enzyme stop solution to all wells except the blanks
Add 100 p,1 antibody solution to all wells except the NSB's, add antibody
binding buffer to
these wells
Incubate at 37°C for 1 h
Chill plate on ice for 15 min
Add 50 p,1 / well charcoal solution and mix with an 8-channel pipette (4 - 5
aspirations)
Chill the plate on ice
Centrifuge at 4°C, 2000 x g for 15 min
Transfer 100 p,1 supernatant into an Optiplate, also add 25 1u.1 substrate
mixture to 2
empty wells to indicate counting efficiency
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
46
Add 200 p,1 Microscint-40 to all wells and count on a Topcount
Radioimmunoassay
The 11 ~i HSD'I enzyme assay was carried out following the standard operating
procedure described above in u-bottom polypropylene 96 well plates or 1.5 ml
Eppendorf
tubes as indicated for each experiment. Subsequent to stopping the enzyme
reaction,
100 p,1 antibody prepared in buffer 3 unless otherwise indicated was added to
test
samples and 100 p1 buffer 3 was added to the NSB samples. The samples were
incubated for 1 hour at 37°C and the chilled on ice for 15 mins.
Dextran coated charcoal
(50 p.1 / sample) prepared to the indicated concentration in buffer 3 was
added and the
samples were mixed (vortex for tubes and aspiration 5 times with an 8-channel
pipette
for 96 well plates) and chilled for a further 10 min. The samples were
centrifuged at
2000 x g for 15 min at 4°C to pellet the charcoal. Aliquots of the
supernatant (100 p,1)
were transferred to an Optiplate and counted on the Topcount in 150-200 p1
Microscint
40. In some experiments, aliquots of supernatant were transferred to
scintillation vials
and counted on the Tricarb LSC in 5 ml Ultima Gold scintillant.
Inhibition Data
STX No. Structure % inhibition of Human 11 ~i HSD1
10 M t ical sd ~ 5% N=2
990 73.3
N O
\/ \
0
472 71.4
~N
/S
N O
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
47
STX No. Structure % inhibition of Human 11(3 HSD1
M t ical sd ~ 5% N=2
956 64.8
o
HN
N
1015 64.6
ci
/o
HN
1033 62.6
N
H~ ~/
\
0
980 61.2
0
/ ci
O O
955 60.8
0
N N
ci
O \ N
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
48
STX No. Structure % inhibition of Human 11 (3 HSD1
1 O M ical sd ~ 5% N=2
972 59.2
ci
o~so
HN
958 58.1
/o
o/ \
HN ~ N
N
988 56
\ \
N
N~
\ Hr \\
957 55.5
0
N N
'N
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
49
STX No. Structure % inhibition of Human 11 (3 HSD1
M ical sd ~ 5% N=2
577 54.6
o~
\ jw
0
N- \H
979 54.1
0
0
-N
CI
0
471 53.7
~N a
N O
H
555 53.0
~ ~N
NN
557 51.3
0
H
HH-
O r
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
STX No. Structure % inhibition~of Human 11 (3 HSD1
10 M fi ical sd ~ 5% N=2
971 49.6
~i
°e
HN
O
/O
646 48.5
~OH
H
O=S=O
\~NH
Is
576 47.7
°
II_
II \' °/~\
° ~ \
\ .NH
556 \\~~66 46.2
,,o
~~'',~//H
HN
O-
0
~~ / .S \O
NH
O~
1031 45.6
ci
s
s
ci
N
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
51
STX No. Structure % inhibition of Human 11 ~i HSD1
M t ical sd ~ 5% N=2
520 43.5
H
O N
\
\S
N
CI \ CI
823 42.9
w
HN
O
CI O~~ N~
O
CI
936 42.3
/~o
HN ~ N
645 42.0
N~
/ O
HN
~o
0
NH CI
983 41.9
O H
N
c1 o ~~ ~ \
i H/
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
52
STX No. Structure % inhibition of Human 11(3 HSD1
__ 10 M ical sd ~ 5% N=2
702 37.6
N
N-H
0
\O
644 37.1
N/ , _O
HN
O=S=O
\\ ~NH
CI ~ CI
937 36.6
~N ~ NH
O
CI
O
CI
608 36.1
H2N
/ ~/ w ~o
~N
N
H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
53
STX No. Structure % inhibition of Human 11 (3 HSD1
M ical sd ~ 5% N=2
653 36.4
'OH
H JJ/N
O=S=O
\\~NH
\~
ci ~ ci
647 34.5
\ /
H
~O
/ \
\ II HH
973 33.1
CI
\ i
°s \
H ~ ~ o
O
~O
654 32.1
NHp
CI
O=S=C
IH
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
54
STX No. Structure % inhibition of Human 11 (3 HSD1-
_ _ 10 M t ical sd ~ 5% N=2
652 31.8
a
w
H
558 31.2
N
HN~N
O=S=O
NH
/o
92g 31.0
io
o
HN
~N
HN'
\/~'O
1038 28.7
o ~ o
oy w
\ N
H
CI
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
STX No. Structure % inhibition of Human 11 (3 HSD1
10 M t ical sd ~ 5% N=2
920 27.5
N~
NH
0
CI
0
CI
649 26.3
'OH
H JJ/N
O=S=O
~S/NH
O
O
919 25.4
/o
0
HN ~ 0
~N
876 23.6
~N
HN
CI O
O
CI
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
56
STX No. Structure % inhibition of Human 11 (3 HSD1
M ical sd ~ 5% N=2
825 23.2
\~
HN
O
O\ \ N/
O
CI
606 22.1
HN
O
O\
/ ~o
~NH
O
N
935 20.7
O H
/ \
/ \
Chemistry Experimental Section and Compound Examples
5 General. All chemicals were either purchased from the Aldrich Chemical Co.
(Gillingham, UK), Lancaster Synthesis (Morecambe, UK) or ACROS Organics
(Loughborough, UK). All organic solvents of A.R. grade were supplied by Fisher
Scientific (Loughborough, UK).
Thin layer chromatography (TLC) was performed on precoated plates (Merck TLC
10 aluminium sheets silica gel 60 F2s4, Art. No. 5554). Compounds were
visualised by either
viewing under UV light or treating with an ethanolic solution of
phosphomolybdic acid
(PMA) followed by heating. Flash chromatography was carried out using Sorbsil
C60
silica gel or Isolute~ pre-packed Flash Si columns from Argonaut Technologies.
Parallel
synthesis was performed on either Radleys Carousel reaction stations or
Radleys
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
57
GreenHouse parallel synthesisers. Solvent removal from parallel syntheses was
performed on a GeneVac DD4 evaporation system. NMR spectra were recorded with
a
JEOL GX-270 or Varian-Mercury-400 spectrometer, and chemical shifts are
reported in
parts per million (ppm, i5relative to tetramethylsilane (TMS) as an internal
standard.
Mass spectra were recorded at the Mass Spectrometry Service Centre, University
of
Bath. FAB-MS were carried out using m-nitrobenzyl alcohol (NBA) as the matrix.
High
performance liquid chromatography (HPLC) analysis was performed with a Waters
Delta
600 liquid chromatograph with a Waters 996 photodiode Array Detector using a
Waters
Radialpack C18, 8x100 mm column. Melting points (Mp) were measured with a
Reichert-Jung ThermoGalen Kofler block or a Sanyo Gallenkamp melting point
apparatus and are uncorrected.
Synthesis of Benzofuran and Benzo[b]thiophene Derivatives
S O- ~ / O
c1 I ~ ,s~ ~ ~ / o c1 I / ,s~
N N v
O H O H
STX 971 STX 972
O O- ~ / S
c1 I ~ ,s~ ~ ~ / o c1 I ~ ,s~ ~ ~ /
N N v
O H O H
STX 973 STX 1015
o O ~ / S
,O
CI / oS,N ~ l / CI OS,N ~
H H
STX 1038 STX 1049
Synthesis of 5-(3-chloro-2-methyl-benzenesulfonylamino)
-benzofblthiophene-2-carboxylic acid methyl ester, STX 971 (KRB01096):
s o-
i
c1 I / ,s N ~ ~ ~ o
O H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
5~
5-amino-benzo[b]thiophene-2-carboxylic acid methyl ester (KRB01094): To a
solution of
5-nitro-benzo[b]thiophene-2-carboxylic acid methyl ester [15] (130 mg, 0.548
mmol) in
methanol (30 mL) was added 5% palladium on carbon (20 mg) and the mixture was
stirred under 1 atm Ha for 2 h. The mixture was filtered through celite and
the filtrate
evaporated. The residue was passed through a silica column to afford 5-amino-
benzo(b]thiophene-2-carboxylic acid methyl ester as a pale yellow solid (94
mg, 83%),
single spot at Rf 0.75 (95:5 dichloromethane:methanol). 'H NMR (CDCI3): 5 7.86
(1H,
s), 7.61 (1 H, d, J=8.7 Hz), 7.11 (1 H, d, J=2.2 Hz), 6.88 (1 H, dd, J=8.7,
2.5 Hz), 3.91 (3H,
s), 3.76 (2H, s, N-H~) [16].
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (46 mg, 0.20 mmol)
in
dichloromethane (2 mL) was added pyridine (40 pL, 0.48 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-benzo[b]thiophene-2-
carboxylic acid
methyl ester (40 mg, 0.19 mmol) was added. The resulting mixture was stirred
for 2 h at
room temperature, then saturated NaHC03 solution (6 mL) was added and the
mixture
was extracted into ethyl acetate (12 mL). The organic phase was washed with
brine,
dried (Na~S04), filtered and evaporated to give a residue that was purified
using flash
chromatography to afford a pale yellow solid (65 mg, 85%), single spot at Rf
0.52 (2:1
hexane:ethyl acetate). mp 173.6-174.2°C, HPLC purity 99% (tR 2.13 min
in 10% water-
acetonitrile). 'H NMR (CDCI3): b 7.91 (1 H, s), 7.88 (1 H, d, J=7.9 Hz), 7.70
(1 H, d, J=8.7
Hz), 7.55-7.52 (2H, m), 7.18 (1 H, t, J=8.0 Hz), 7.12 (1 H, dd, J=8.7, 2.2
Hz), 6.98 (1 H, s,
N-I-~, 3.92 (3H, s), 2.73 (3H, s). LCMS: 394.12 (M-). FAB-MS (MH+,
C~7H~4CINO4S2):
calcd 395.0053, found 395.0045.
Synthesis of 3-chloro-2-methyl-N-(3-methyl-benzofuran-5-
-benzenesulfonamide. STX 972 (KRB01097):
O
i
,o
ci oS,N ~
H
5-amino-3-methyl-benzofuran (KRB01095): To a solution of 3-methyl-5-nitro-
benzofuran
[17] (135 mg, 0.762 mmol) in methanol (30 mL) was added 5% palladium on carbon
(30
mg) and the mixture was stirred under 1 atm Ha for 8 h. The mixture was
filtered
through celite and the filtrate evaporated. The residue was passed through a
silica plug
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
59
to afford 5-amino-3-methyl-benzofuran as a pale pink oil (85 mg, 76%), single
spot at Rf
0.28 (2:1 hexane:ethyl acetate). ' H NMR (CDCI3): b 7.31 (1 H, s), 7.22 (1 H,
d, J=8.7 Hz),
6.78 (1 H, d, J=2.5 Hz), 6.66 (1 H, dd, J=8.7, 2.5 Hz), 3.60 (2H, s, N-H~),
2.16 (3H, s).
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (128 mg, 0.571
mmol) in
dichloromethane (3 mL) was added pyridine (110 pL, 1.36 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 5-amino-3-methyl-benzofuran (80
mg, 0.54
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (8 mL) was added and the mixture was extracted into
ethyl
acetate (15 mL). The organic phase was washed with brine, dried (Na2SO4),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
yellow solid (159 mg, 87%), single spot at Rf 0.65 (2:1 hexane:ethyl acetate).
mp 119.5-
120.1°C, HPLC purity 99% (tR 2.22 min in 10% water-acetonitrile). 'H
NMR (CDC13): S
7.82 (1 H, d, J=7.9 Hz), 7.54 (1 H, d, J=7.9 Hz), 7.38 (1 H, s), 7.28-7.22
(2H, m), 7.15 (1 H,
t, J=8.0 Hz), 6.83 (1 H, dd, J=8.7, 2.2 Hz), 6.56 (1 H, s, N-l~, 2.72 (3H, s),
2.15 (3H, s).
LCMS: 334.07 (M-). FAB-MS (MH+, C~sH14CINO3S): calcd 335.0383, found 335.0383.
Synthesis of 5-(3-chloro-2-methyl-benzenesulfonylamino)-benzofuran-2-
carboxylic acid
methyl ester. STX 973 (KRB01100):
0 0-
,o
ci ' os_N w I o
H
5-vitro-benzofuran-2-carboxylic acid methyl ester (KRB01098): To a suspension
of NaH
(258 mg, 60% dispersion in mineral oil, 6.46 mmol) in anhydrous DMF (10 mL)
was
added methyl glycolate (420 pL, 5.39 mmol) dropwise. After 10 min, a solution
of 2-
chloro-5-nitrobenzaldehyde (1.00 g, 5.39 mmol) in anhydrous DMF (3 mL) was
added
and the resulting solution was stirred at room temperature for 1 h followed by
100°C for
5 h. After cooling, the mixture was poured into 1 N HCI (50 mL) and the
resulting
precipitate filtered and washed with water. The solid was purified by flash
chromatography (4:1 hexane:ethyl acetate) and then recrystallized from
hexane/ethyl
acetate to yield 5-vitro-benzofuran-2-carboxylic acid methyl ester as yellow
needles (450
mg, 37%). mp 168.0-168.4°C. 'H NMR (CDC13): S 8.65 (1 H, d, J=2.2 Hz),
8.37 (1 H, dd,
J=9.0, 2.2 Hz), 7.70 (1 H, d, J=9.1 Hz), 7.64 (1 H, s), 4.01 (3H, s).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
5-amino-benzofuran-2-carboxylic acid methyl ester (KRB01099): To a solution of
5-
nitrobenzofuran-2-carboxylic acid methyl ester (100 mg, 0.452 mmol) in
methanol (20
mL) was added 5% palladium on carbon (20 mg) and the mixture was stirred under
1
5 atm H2 for 4h. The mixture was filtered through celite and the filtrate
evaporated. The
residue was passed through a silica plug to afford 5-amino-benzofuran-2-
carboxylic acid
methyl ester as an orange solid (87 mg, 100%), single spot at Rf 0.34 (1:1
hexane:ethyl
acetate). mp 112.4-113.0°C. ' H NMR (CDC13): b 7.36 (1 H, s), 7.36 (1
H, d, J=8.2 Hz),
6.89 (1 H, d, J=2.2 Hz), 6.83 (1 H, dd, J=8.2, 2.2 Hz), 3.95 (3H, s), 3.66
(2H, s, N-H2).
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (99 mg, 0.44 mmol)
in
dichloromethane (3 mL) was added pyridine (85 pL, 1.05 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-benzofuran-2-carboxylic
acid methyl
ester (80 mg, 0.42 mmol) was added. The resulting mixture was stirred for 2 h
at room
temperature, then saturated NaHC03 solution (10 mL) was added and the mixture
was
extracted into ethyl acetate (15 mL). The organic phase was washed with brine,
dried
(Na~S04), filtered and evaporated to give a residue that was purified using
flash
chromatography to afford an off-white solid (122 mg, 77%), single spot at Rf
0.47 (2:1
hexane:ethyl acetate). mp 139.4-140.1°C , HPLC purity 99+% (tR 1.90 min
in 10%
water-acetonitrile). 'H NMR (CDCI3): b 7.83 (1 H, d, J=7.9 Hz), 7.55 (1 H, d,
J=7.9 Hz),
7.44 (1 H, d, J=9.0 Hz), 7.42 (1 H, s), 7.39 (1 H, d, J=2.2 Hz), 7.17 (1 H, t,
J=8.0 Hz), 7.05
(1 H, dd, J=8.7, 2.2 Hz), 6.65 (1 H, s, N-H), 3.95 (3H, s), 2.72 (3H, s).
LCMS: 378.16.
FAB-MS (MH+, C~7H~4CINO5S): calcd 379.0281, found 379.0281.
Synthesis of 3-chloro-2-methyl-N-(3-methyl-benzo~blthiophen-5-yl)-
benzenesulfonamide.
STX 1015 (KRB01108):
a S
i
~ ,o I /
CI OS,N
H
3-methyl-benzo[b[thiophen-5-ylamine (KRB01106): To a solution of 3-methyl-5-
nitro-
benzo[b]thiophene [18] (95 mg, 0.492 mmol) in methanol (20 mL) was added 5%
palladium on carbon (20 mg) and the mixture was stirred under 1 atm H2 for
8.5h. The
mixture was filtered through celite and the filtrate evaporated. The residue
was passed
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
61
through a silica plug to afford 3-methyl-benzo[b]thiophen-5-ylamine as a dark
orange
solid '(31 mg, 39%), single spot at Rf 0.31 (4:1 hexane:ethyl acetate). 'H NMR
(CDCI3):
S 7.60 (1 H, d, J=8.4 Hz), 7.02 (1 H, s), 7.00 (1 H, d, J=2.2 Hz), 6.79 (1 H,
dd, J=8.4, 2.2
Hz), 2.37 (3H, s).
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (43 mg, 0.19 mmol)
in
dichloromethane (2 mL) was added pyridine (40 pL, 0.46 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 3-methyl benzo[b]thiophen-5-
ylamine (30 mg,
0.18 mmol) was added. The resulting mixture was stirred for 2 h at room
temperature,
then saturated NaHC03 solution (10 mL) was added and the mixture was extracted
into
ethyl acetate (15 mL). The organic phase was washed with brine, dried
(Na2S04),
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford a pale brown solid (30 mg, 46%), single spot at Rf 0.39 (3:1
hexane:ethyl acetate).
mp 152.6-153.0°C , HPLC purity 99% (tR 2.42 min in 10% water-
acetonitrile). 'H NMR
(CDCI3): b 7.90 (1 H, d, J=7.9 Hz), 7.65 (1 H, d, J=8.7 Hz), 7.53 (1 H, d,
J=7.9 Hz), 7.37
(1 H, d, J=2.0 Hz), 7.16 (1 H, t, J=7.9 Hz), 7.07 (1 H, s), 7.02 (1 H, dd,
J=8.7, 2.0 Hz), 2.74
(3H, s), 2.32 (3H,. s). LCMS: 350.09. FAB-MS (MH+, C16H14CINO~S~): calcd
351.0155,
found 351.0155
~nthesis of 3-chloro-2-methyl-N-(2-methyl-benzofuran-5-yl)-benzenesulfonamide,
STX
1038 (KRB01114):
O
CI ~ OS.N W
H
5-amino-2-methylbenzofuran (ICRB01112): To a solution of 2-methyl-5-vitro-
benzofuran
[19] (125 mg, 0.706 mmol) in methanol (20 mL) was added 5% palladium on carbon
(20
mg) and the mixture was stirred under 1 atm H~ for 4h. The mixture was
filtered through
celite and the filtrate evaporated. The residue was passed through a silica
plug to afford
5-amino-2-methyl benzofuran as a red-brown oil (78 mg, 75%), single spot at Rf
0.45
(1:1 hexane:ethyl acetate). 'H NMR (CDC13): S 7.17 (1 H, d, J=8.7 Hz), 6.75 (1
H, d,
J=2.5 Hz), 6.57 (1 H, dd, J=8.7, 2.5 Hz), 6.20 (1 H, s), 3.53 (2H, s, NHS),
2.39 (3H, s).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
62
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (112 mg, 0.499
mmol) in
dichloromethane (3 mL) was added pyridine (95 pL, 1.2 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-2-methyl benzofuran (70
mg, 0.48
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (15 mL). The organic phase was washed with brine, dried (Na2S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale brown solid (120 mg, 75%), single spot at Rf 0.51 (3:1 hexane:ethyl
acetate). mp
123.2-123.6°C , HPLC purity 98% (tR 3.14 min in 10% water-
acetonitrile). 'H NMR
(CDCI3): 5 7.81 (1 H, d, J=7.7 Hz), 7.52 (1 H, d, J=7.7 Hz), 7.21 (1 H, d,
J=8.7 Hz), 7.16-
7.11 (2H, m), 6.78 (1 H, dd, J=8.7, 2.2 Hz), 6.48 (1 H, s, N-f~, 6.27 (1 H,
s), 2.70 (3H, s),
2.40 (3H, s). LCMS: 334.13. FAB-MS (MH+, C~6H14CINO3S): calcd 335.0383, found
335.0381
~nthesis of N benzofblthiophen-5-yl-3-chloro-2-methyl-
benzenesulfonamide. STX 1049 (KRB01121):
i ,o
CI OS.N \
H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (47 mg, 0.21 mmol)
in
dichloromethane (2 mL) was added pyridine (40 pL, 0.50 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-benzo[b]thiophene [20]
(30 mg, 0.20
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (15 mL). The organic phase was washed with brine, dried (Na~S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
dark brown solid (50 mg, 74%), single spot at Rf 0.58 (3:1 hexane:ethyl
acetate). mp
108.5-109.0°C , HPLC purity 99% (tR 2.20 min in 10% water-
acetonitrile). 'H NMR
(CDCI3): S 7.87 (1 H, d, J=7.9 Hz), 7.70 (1 H, d, J=8.7 Hz), 7.54-7.51 (2H,
m), 7.44 (1 H, d,
J=5.4 Hz), 7.21 (1 H, d, J=5.4 Hz), 7.16 (1 H, t, J=7.9 Hz), 6.98 (1 H, dd,
J=8.7, 2.2 Hz),
6.73 (1 H, s, N-I~, 2.74 (3H, s). LCMS: 336.07. FAB-MS (MH+, C~5H~2CIN02S2):
calcd
336.9998, found 337.0004.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
63
Synthesis of Quinoline, Quinazoline, and Quinoxaline Derivatives
R~ R~
z
z
R I ~ O s N~ R I ~ O / N
R3 / ,S, ~ I / R3 / ,S. ~ I
N ~ N
Ra. O H Ra. O H
STX 932: R~=R2=H, R3=CI, R4=Me STX 935: R~=R2=H, R3=CI, R4=Me
STX 933: R~=R3=R4=H, R2=n-propyl STX 936: R~=R3=R4=H, R2=n-propyl
STX 934: R~=R4=CI, R2=R3=H STX 937: R~=R4=CI, R2=R3=H
R~ R~
R2 I ~ O / N ~ R2 I ~ O / N
R3 / ,S, ~ I ~ N Rs / ,
N ' N N
R4 O H Ra. O H
STX 943: R~=R2=H, R3=CI, R4=Me STX 953: R~=R2=H, R3=CI, R4=Me
STX 944: R~=R3=R4=H, R~=n-propyl STX 954: R~=R3=R4=H, R2=n-propyl
R~ R~
2
2
R I ~ O / N~ R I ~ O / N
R3 / S. ~ I ~ R3 / ,S,
'' N N ' N N
R4 O H R4 O H
STX 955: R~=R2=H, R3=CI, R4=Me STX 957: R~=R2=H, R3=CI, R4=Me
STX 956: R~=R3=R4=H, RZ=n-propyl STX 958: R~=R3=R4=H, R2=n-propyl
Synthesis of 3-chloro-2-methyl-N-auinolin-6-yl-benzenesulfonamide, STX 932
(KRB01058):
/ N~
CI ,S, N w
O H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (164 mg, 0.728
mmol) in
dichloromethane (4 mL) was added pyridine (140 pL, 1.74 mmol) and the mixture
was
stirred under NZ for 5 min, after which time 6-aminoquinoline (100 mg, 0.694
mmol) was
added. The resulting mixture was stirred for 2 h at room temperature, then
saturated
NaHC03 solution (10 mL) was added and the mixture was extracted into ethyl
acetate
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
64
(20 mL). The organic phase was washed with brine, dried (Na~S04), filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale pink solid (48 mg, 21 %), single spot at Rf 0.71 (ethyl acetate). mp
228.0-228.5°C,
HPLC purity 99+% (tR 2.43 min in 10% water-acetonitrile). 'H NMR (CDCI3): i5
8.83 (1 H,
dd, J=4.2, 1.7 Hz), 7.98 (3H, m), 7.54 (1 H, d, J=7.9 Hz), 7.48 (1 H, d, J=2.7
Hz), 7.39-
7.32 (2H, m), 7.20 (1 H, t, J=7.9 Hz), 7.05 (1 H, s, N-I-n, 2.75 (3H, s).
LCMS: 331.16 (M-).
FAB-MS (MH+, ClsH~3CIN20~S): calcd 333.0464, found 333.0461.
~nthesis of 4-propyl-N-auinolin-6-yl-benzenesulfonamide STX 933 (KRB010590
N~
~O
,S. N ~
O H
To a solution of 4n-propylbenzenesulphonyl chloride (159 mg, 0.728 mmol) in
dichloromethane (4 mL) was added pyridine (140 pL, 1.74 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 6-aminoquinoline (100 mg, 0.694
mmol) was
added. The resulting mixture was stirred for 2 h at room temperature, then
saturated
NaHC03 solution (10 mL) was added and the mixture was extracted into ethyl
acetate
(20 mL). The organic phase was washed with brine, dried (Na2S04), filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
white solid (210 mg, 93%), single spot at Rf 0.71 (ethyl acetate). mp 180.1-
180.7°C,
HPLC purity 99+% (tR 2.37 min in 10% water-acetonitrile). 'H NMR (CDC13): b
8.83 (1 H,
dd, J=4.2, 1.7 Hz), 8.05 (1 H, d, J=8.4 Hz), 7.97 (1 H, d, J=9.2 Hz), 7.70
(2H, d, J=8.2
Hz), 7.57 (1 H, d, J=2.5 Hz), 7.40-7.34 (2H, m), 7.22 (2H, m), 6.85 (1 H, s, N-
I-~, 2.57
(2H, t, J=7.2 Hz), 1.57 (2H, sextet, J=7.2 Hz), 0.87 (3H, t, J=7.3 Hz). LCMS:
325.23 (M
). FAB-MS (MH+, C~BH~$N~O~S): calcd 327.1167, found 327.1167.
Synthesis of 2,5-dichloro-N-auinolin-6-yl-benzenesulfonamide STX 934
(KRB010600
CI
Nw
~O
,S. ~ I /
CI O N
H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
To a solution of 2,5-dichlorobenzenesulphonyl chloride (179 mg, 0.728 mmol) in
dichloromethane (4 mL) was added pyridine (140 pL, 1.74 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 6-aminoquinoline (100 mg, 0.694
mmol) was
added. The resulting mixture was stirred for 2 h at room temperature, then
saturated
5 NaHC03 solution (10 mL) was added and the mixture was extracted into ethyl
acetate
(20 mL). The organic phase was washed with brine, dried (Na~S04), filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
white solid (168 mg, 69%), single spot at Rf 0.74 (ethyl acetate). mp 213.4-
213.8°C,
HPLC purity 99+% (tR 2.28 min in 10% water-acetonitrile). 'H NMR (CDCI3): 5
8.85 (1H,
10 dd, J=4.2, 1.5 Hz), 8.09 (1 H, d, J=8.4 Hz), 8.03-8.00 (2H, m), 7.62 (1 H,
d, J=2.5 Hz),
7.56 (1 H, s, N-I-n, 7.47-7.38 (4H, m). LCMS: 351.10 (M-). FAB-MS (MH+,
C~SH~oChN20~S): calcd 352.9918, found 352.9922.
~nthesis of 3-chloro-2-methyl-N-(2-methyl-auinolin-6-
15 -benzenesulfonamide, STX 935 (KRB01061 ):
o Nw
CI OS,N ~ /
H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (149 mg, 0.664
mmol) in
20 dichloromethane (4 mL) was added pyridine (130 pL, 1.58 mmol) and the
mixture was
stirred under N2 for 5 min, after which time 6-amino-2-methylquinoline (100
mg, 0.632
mmol) was added. The resulting mixture was stirred for 90 min at room
temperature,
then saturated NaHC03 solution (10 mL) was added and the mixture was extracted
into
ethyl acetate (20 mL). The organic phase was washed with brine, dried
(Na~S04),
25 filtered and evaporated to give a residue that was purified using flash
chromatography to
afford an off-white solid (148 mg, 68%), single spot at Rf 0.64 (ethyl
acetate). mp 178.1
178.4°C, HPLC purity 99+% (tR 2.27 min in 10% water-acetonitrile). 'H
NMR (CDCI3): S
7.93-7.89 (3H, m), 7.53 (1 H, d, J=8.2 Hz), 7.44 (1 H, d, J=2.5 Hz), 7.31-7.24
(2H, m),
7.18 (1 H, t, J=8.1 Hz), 2.73 (3H, s), 2.69 (3H, s). LCMS: 345.17 (M-). FAB-MS
(MH+,
30 C~~H~SCINzOZS): calcd 347.0621, found 347.0622.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
66
~nthesis of N-(2-methyl-auinolin-6-yl)-4-propel-benzenesulfonamide, STX 936
(KRB01062):
/ N~
a
~S, N w a
H
To a solution of 4n-propylbenzenesulphonyl chloride (145 mg, 0.664 mmol) in
dichloromethane (4 mL) was added pyridine (130 pL, 1.58 mmol) and the mixture
was
stirred under Nz for 5 min, after which time 6-amino-2-methylquinoline (100
mg, 0.632
mmol) was added. The resulting mixture was stirred for 1 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (NazS04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford an
off-white solid (186 mg, 86%), single spot at Rf 0.70 (ethyl acetate). mp
173.7-174.0°C,
HPLC purity 99+% (tR 2.32 min in 10% water-acetonitrile). 'H NMR (CDCI3): b
7.94 (1 H,
d, J=8.4 Hz), 7.89 (1 H, d, J=9.2 Hz), 7.67 (2H, d, J=8.4 Hz), 7.54 (1 H, d,
J=2.5 Hz), 7.31
(1 H, dd, J=8.9, 2.5 Hz), 7.25 (1 H, d, J=8.4 Hz), 7.19 (2H, d, J=8.4 Hz),
7.07 (1 H, s, N-
I~, 2.70 (3H, s), 2.56 (2H, t, J=7.5 Hz), 1.58 (2H, sextet, J=7.4 Hz), 0.87
(3H, t, J=7.4
Hz). LCMS: 339.24 (M-). FAB-MS (MH+, C,9HzoNzOzS): calcd 341.1323, found
341.1324.
Synthesis of 2.5-dichloro-N-(2-methyl-auinolin-6-yl)-
benzenesulfonamide. STX 937 (KRB01063):
CI
N~
~S. N ~
CI ~ H
To a solution of 2,5-dichlorobenzenesulphonyl chloride (163 mg, 0.664 mmol) in
dichloromethane (4 mL) was added pyridine (130 pL, 1.58 mmol) and the mixture
was
stirred under Nz for 5 min, after which time 6-amino-2-methylquinoline (100
mg, 0.632
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (NazS04),
filtered and
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
67
evaporated to give a residue that was purified using flash chromatography to
afford an
off-white solid (120 mg, 52%), single spot at Rf 0.68 (ethyl acetate), mp
124.6-125.0°C,
HPLC purity 99+% (tR 2.22 min in 10% water-acetonitrile). 'H NMR (CDCI3): b
7.98 (1 H,
d, J=2.2 Hz), 7.94 (1 H, d, J=8.4 Hz), 7.88 (1 H, d, J=8.9 Hz), 7.57 (1 H, d,
J=2.2 Hz),
7.45-7.35 (3H, m), 7.27-7.24 (2H, m), 2.68 (3H, s). LCMS: 365.10 (M-). FAB-MS
(MH+,
C~6H~~CI2N~OZS): calcd 367.0075, found 367.0074.
Synthesis of 3-chloro-2-methyl-N-auinazolin-6-yl-benzenesulfonamide, STX 943
(KRB01068):
N\
~ \1O
CI ~S.N w I ~ N
O H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (163 mg, 0.723
mmol) in
dichloromethane (4 mL) was added pyridine (140 pL, 1.72 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 6-aminoquinazoline [21] (100 mg,
0.689
mmol) was added. The resulting mixture was stirred for 4 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (Na~S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale yellow solid (55 mg, 24%), single spot at Rf 0.56 (ethyl acetate). mp
>270°C
(appeared to decompose at about 150°C), HPLC purity 98% (tR 2.06 min in
10% water-
acetonitrile). 'H NMR (CDCI3): 5 9.28 (1 H, s), 9.25 (1 H, s), 7.98 (1 H, d,
J=4.4 Hz), 7.95
(1 H, d, J=5.2 Hz), 7.59-7.53 (3H, m), 7.25-7.20 (1 H, m), 2.76 (3H, s). LCMS:
332.11 (M-
). FAB-MS (MH+, C~5H~2CIN3O2S): calcd 334.0417, found 334.0420.
Synthesis of 4-propel-N-auinazolin-6-yl-benzenesulfonamide. STX 944
(KRB01069):
N\
I , ~ \1O
~S. ~ o N
O N
H
To a solution of 4n-propylbenzenesulphonyl chloride (151 mg, 0.723 mmol) in
dichloromethane (4 mL) was added pyridine (140 pL, 1.72 mmol) and the mixture
was
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
68
stirred under N2 for 5 min, after which time 6-aminoquinazoline (100 mg, 0.689
mmol)
was added. The resulting mixture was stirred for 4 h at room temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (Na2S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale yellow solid (47 mg, 21 %), single spot at Rf 0.57 (ethyl acetate). mp
202.1-202.6°C,
HPLC purity 96% (tR 2.14 min in 10% water-acetonitrile). 'H NMR (CDCI3): 8
9.30 (1H,
s), 9.24 (1 H, s), 7.99-7.92 (2H, m), 7.77 (2H, d, J=8.4 Hz), 7.68-7.63 (2H,
m), 7.23 (1 H,
d, J=7.7 Hz), 2.57 (2H, t, J=7.6 Hz), 1.57 (2H, sextet, J=7.6 Hz), 0.87 (3H,
t, J=7.4 Hz).
LCMS: 326.24 (M-). FAB-MS (MH+, C~~H~~N30~S): calcd 328.1119, found 328.1118.
Synthesis of 3-chloro-N (2,3-dimethyl-auinoxalin-6-yl)-2-methyl-
benzenesulfonamide, STX 953 (KRB01074):
N
CI I s ,S N \ I N-
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (109 mg, 0.485
mmol) in
dichloromethane (3 mL) was added pyridine (90 pL, 1.2 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 6-amino-2,3-dimethylquinoxaline
[22] (80 mg,
0.46 mmol) was added. The resulting mixture was stirred for 6 h at room
temperature,
then saturated NaHC03 solution (8 mL) was added and the mixture was extracted
into
ethyl acetate (15 mL). The organic phase was washed with brine, dried
(Na~S04),
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford an off-white solid (152 mg, 91 %), single spot at Rf 0.70 (ethyl
acetate). mp 225.3-
225.7°C, HPLC purity 99+% (tR 2.29 min in 10% water-acetonitrile). 'H
NMR (CDCI3): b
7.99 (1 H, d, J=7.9 Hz), 7.85 (1 H, d, J=9.2 Hz), 7.54-7.52 (2H, m), 7.40 (1
H, dd, J=9.1,
2.6 Hz), 7.21 (1 H, t, J=7.9 Hz), 6.95 (1 H, s, N-f-n, 2.75 (3H, s), 2.67 (6H,
s). LCMS:
360.24 (M-). FAB-MS (MH+, C~~H~6CIN30~S): calcd 362.0730, found 362.0732.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
69
Synthesis of N-(2,3-dimethyl-ctuinoxalin-6-yl)-4-propyl-
benzenesulfonamide. STX 954 (KRB01075):
~ I N
,o
iSe N~ I~j
O H
To a solution of 4n-propylbenzenesulphonyl chloride (106 mg, 0.485 mmol) in
dichloromethane (3 mL) was added pyridine (90 pL, 1.2 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 6-amino-2,3-dimethylquinoxaline
(80 mg,
0.46 mmol) was added. The resulting mixture was stirred for 6 h at room
temperature,
then saturated NaHC03 solution (8 mL) was added and the mixture was extracted
into
ethyl acetate (15 mL). The organic phase was washed with brine, dried
(Na~S04),
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford an off-white solid (147 mg, 90%), single spot at Rf 0.69 (ethyl
acetate). mp 198.4-
198.8°C, HPLC purity 99+% (tR 2.39 min in 10% water-acetonitrile). 'H
NMR (CDC13): 5
7.86 (1 H, d, J=8.9 Hz), 7.72 (2H, d, J=8.4 Hz), 7.56 (1 H, d, J=2.5 Hz), 7.48
(1 H, dd,
J=9.2, 2.5 Hz), 7.20 (2H, d, J=7.9 Hz), 6.82 (1 H, s, N-I~, 2.67 (6H, s), 2.56
(2H, t, J=7.5
Hz), 1.57 (2H, sextet, J=7.4 Hz), 0.87 (3H, t, J=7.4 Hz). LCMS: 354.31 (M-).
FAB-MS
(MH+, C~gH2~N3O2S): calcd 356.1432, found 356.1433.
Synthesis of 3-chloro-N-(2-methyl-auinoxalin-6-yl)-2-methyl-
benzenesulfonamide, STX 955 (KRB01079):
N
,O
CI ~ ,S~N~N
O H
6-amino-2-methylquinoxaline (KRB01078): To a solution of 2-methyl-6-
nitroquinoxaline
[23] (500 mg, 2.64 mmol) in methanol (22 mL) was added 10% palladium on carbon
(50
mg) and the mixture was stirred under 1 atm H~ for 4 h. The mixture was
filtered
through celite and the filtrate evaporated. The residue was passed through a
silica plug
and evaporated to afford 6-amino-2-methylquinoxaline as a yellow solid (376
mg, 89%),
single spot at Rf 0.33 (ethyl acetate). mp 163-164°C ([23] 164-
165°C). ~H NMR
(CDCI3): b 8.57 (1 H, s), 7.78 (1 H, d, J=8.4 Hz), 7.17-7.12 (2H, m), 4.11
(2H, s -NHS),
2.67 (3H, s).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (119 mg, 0.528
mmol) in
dichloromethane (3 mL) was added pyridine (100 pL, 1.26 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 6-amino-2-methylquinoxaline (80
mg, 0.50
5 mmol) was added. The resulting mixture was stirred for 5 h at room
temperature, then
saturated NaHC03 solution (8 mL) was added and the mixture was extracted into
ethyl
acetate (15 mL). The organic phase was washed with brine, dried (Na2S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale pink solid (161 mg, 92%), single spot at Rf 0.67 (ethyl acetate). mp
158.7-159.3°C,
10 HPLC purity 99+% (tR 2.23 min in 10% water-acetonitrile). 'H NMR (CDCI3): b
8.66 (1 H,
s), 8.02 (1 H, d, J=7.7 Hz), 7.89 (1 H, d, J=8.9 Hz), 7.61 (1 H, d, J=2.5 Hz),
7.55 (1 H, d,
J=8.2 Hz), 7.46 (1 H, dd, J=8.9, 2.5 Hz), 7.23 (1 H, t, J=7.9 Hz), 7.08 (1 H,
s, N-f~, 2.76
(3H, s), 2.71 (3H, s). LCMS: 346.11 (M-). FAB-MS (MH+, C~sH14CIN3O2S): calcd
348.0573, found 348.0589.
Synthesis of N-(2-methyl-auinoxalin-6-yl)-4-propel-benzenesulfonamide. STX 956
(KRB01080):
,1N
S N \ N
O H
To a solution of 4n-propylbenzenesulphonyl chloride (115 mg, 0.528 mmol) in
dichloromethane (3 mL) was added pyridine (100 pL, 1.26 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 6-amino-2-methylquinoxaline (80
mg, 0.50
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (8 mL) was added and the mixture was extracted into
ethyl
acetate (15 mL). The organic phase was washed with brine, dried (Na2S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
white solid (168 mg, 95%), single spot at Rf 0.72 (ethyl acetate). mp 135.6-
136.2°C,
HPLC purity 99+% (tR 2.23 min in 10% water-acetonitrile). ~H NMR (CDC13): b
8.66 (1H,
s), 7.89 (1 H, d, J=9.2 Hz), 7.77 (2H, d, J=8.4 Hz), 7.67 (1 H, d, J=2.5 Hz),
7.54 (1 H, dd,
J=8.9, 2.5 Hz), 7.22 (2H, d, J=8.4 Hz), 2.71 (3H, s), 2.56 (2H, t, J=7.7 Hz),
1.58 (2H,
sextet, J=7.7 Hz), 0.87 (3H, t, J=7.4 Hz). LCMS: 340.25 (M-). FAB-MS (MH+,
C~gH~gN3O~S): calcd 342.1276, found 342.1290.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
71
~nthesis of 3-chloro-2-methyl-N-auinoxalin-6-yl-benzenesulfonamide, STX 957:
N~
CI I ~ OS N w I N
H
6-aminoquinoxaline (KRB01083): To a solution of 6-nitroquinoxaline [24] (500
mg, 2.86
mmol) in methanol (20 mL) was added 10% palladium on carbon (50 mg) and the
mixture was stirred under 1 atm HZ for 4 h. The mixture was filtered through
celite and
the filtrate evaporated. The residue was passed through a silica plug and
evaporated to
afford 6-aminoquinoxaline as a yellow solid (342 mg, 82%), single spot at Rf
0.32 (ethyl
acetate). ' H NMR (CDC13): 8 8.65 (1 H, d, J= 1.7 Hz), 8.55 (1 H, d, J=1.7
Hz), 7.87 (1 H,
d, J=S.9 Hz), 7.18 (1 H, dd, J=8.9, 2.5 Hz), 7.13 (1 H, d, J=2.5 Hz), 4.20
(2H, br.s, -NHS)
[25].
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (147 mg, 0.651
mmol) in
dichloromethane (4 mL) was added pyridine (125 pL, 1.55 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 6-aminoquinoxaline (90 mg, 0.62
mmol) was
added. The resulting mixture was stirred for 5 h at room temperature, then
saturated
NaHC03 solution (10 mL) was added and the mixture was extracted into ethyl
acetate
(20 mL). The organic phase was washed with brine, dried (Na~S04), filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
pale yellow solid (180 mg, 87%), single spot at Rf 0.60 (ethyl acetate). mp
166.5-
166.9°C, HPLC purity 99+% (tR 2.15 min in 10% water-acetonitrile). 'H
NMR (CDC13): 8
8.77 (1 H, d, J=1.7 Hz), 8.74 (1 H, d, J=1.7 Hz), 8.06 (1 H, d, J=8.2 Hz),
8.00 (1 H, d, J=9.2
Hz), 7.66 (1 H, d, J=2.5 Hz), 7.56 (1 H, d, J=7.9 Hz), 7.50 (1 H, dd, J=9.1,
2.6 Hz), 7.28-
7.22 (1 H, obscured under CHC13), 2.76 (3H, s). LCMS: 332.24 (M-). FAB-MS
(MH+,
C~SH~~CIN302S): calcd 334.0417, found 334.0433.
Synthesis of 4-propel-N-auinoxalin-6-yl-benzenesulfonamide, STX 958
(KRB01085):
N\
\Jll
~S.N~N
O H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
72
To a solution of 4n-propylbenzenesulphonyl chloride (142 mg, 0.651 mmol) in
dichloromethane (4 mL) was added pyridine (125 pL, 1.55 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 6-aminoquinoxaline (90 mg, 0.62
mmol) was
added. The resulting mixture was stirred for 4 h at room temperature, then
saturated
NaHC03 solution (10 mL) was added and the mixture was extracted into ethyl
acetate
(20 mL). The organic phase was washed with brine, dried (Na2SO4), filtered and
evaporated to give a residue that was purified using flash chromatography to
afford an
off-white solid (190 mg, 94%), single spot at Rf 0.66 (ethyl acetate). mp
198.4-198.9°C,
HPLC purity 99+% (tR 2.22 min in 10% water-acetonitrile). 'H NMR (CDCI3): b
8.77 (1H,
d, J=1.7 Hz), 8.73 (1 H, d, J=1.7 Hz), 8.00 (1 H, d, J=8.9 Hz), 7.80 (2H, d,
J=8.2 Hz), 7.71
(1 H, d, J=2.5 Hz), 7.59 (1 H, dd, J=8.9, 2.5 Hz), 7.23 (2H, obscured under
CHCI3), 2.57
(2H, t, J=7.6 Hz), 1.58 (2H, sextet, J=7.5 Hz), 0.87 (3H, t, J=7.4 Hz). LCMS:
326.24 (M-
). FAB-MS (MH+, C~7H17N3~2S): calcd 328.1119, found 328.1136.
Synthesis of benzisothiazole derivatives
R1
R2
S
,O ~ i N
R3 ~ ,S. N
R4 O H N H
O
STX 929: R1=R2=H, R3=CI, R4=Me
STX 930: R1=R3=R4=H, R~=n-propyl
STX 931: R1=R~=CI, R2=R3=H
Synthesis of N-acetyl-N-(5-amino-benzofdlisothiazol-3-yl)-acetamide
(KRB01050):
a S N AczO _ ~ I S.N Fe, HOAc ~ I S
p2N I ~ DMF O~N \ ~ O HZN ~ ~ N O
NHS
O O
N acetyl-N-(5-nitro-benzo[djisothiazol-3-yl)-acetamide (KRB01049): To a
solution of 3-
amino-5-nitrobenzisothiazole (1.00g, 5.12 mmol) in DMF (20 mL) was added
acetic
anhydride (1 mL) and triethylamine (1 mL) and the resulting solution was
stirred for 4 h.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
73
The yellow precipitate was filtered, washed quickly with cold ethyl acetate,
and dried in
vacuo (1.139g, 80%). 'H NMR (CDCI3): S 9.20 (1 H, d, J=2.5 Hz), 8.87 (1 H, d,
J=9.4 Hz),
8.55 (1 H, dd, J=9.4, 2.5 Hz), 2.69 (3H, s), 2.63 (3H, s).
N-acetyl-N-(5-amino-benzo[d]isothiazol-3-yl)-acetamide: To a suspension of N
acetyl-N-
(5-nitro-benzo[d]isothiazol-3-yl)-acetamide (1.133g, 4.06 mmol) in a 1:1
acetic acid:ethyl
acetate solution (70 mL) at 50°C was added iron powder (680 mg, 12.2
mmol). The
resulting mixture was stirred at 50°C for 5 h, then cooled, poured into
saturated sodium
bicarbonate and extracted into ethyl acetate. The organic layers were
combined,
washed with water and then brine, dried (MgS04), filtered, and evaporated. The
residue
was purified using flash chromatography on silica to afford the desired amine
as an
orange solid (383 mg, 38%), single spot at Rf 0.30 (1:1 dichloromethane:ethyl
acetate).
mp 213.0-214.2°C. 'H NMR (CDCI3): 5 8.56 (1H, d, J=9.2 Hz), 7.48 (d,
J=2.7 Hz), 7.14
(1 H, dd, J=9.2, 2.7 Hz), 2.62 (3H, s), 2.56 (3H, s).
Synthesis of N f5-(3-chloro-2-methyl-benzenesulfonylamino)-
benzofdlisothiazol-3-yll-acetamide, STX 929 (KRB01055):
s~
i
,o ~ ~N
CI ~ O ,N s
H NH
O
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (95 mg, 0.42 mmol)
in
dichloromethane (3 mL) was added pyridine (80 pL, 1.0 mmol) and the mixture
was
stirred under N2 for 5 minutes, at which time N-acetyl-N-(5-amino-
benzo[d]isothiazol-3-
yl)-acetamide (100 mg, 0.401 mmol) was added. The resulting mixture was
stirred for 4
h at room temperature. 5% NaHC03 solution (8 mL) was added and the mixture was
extracted into ethyl acetate (15 mL). The organic phase was washed with brine,
dried
(Na2S04), filtered and evaporated to give a residue that was purified using
flash
chromatography to afford a yellow solid (82 mg, 47%), single spot at Rf 0.71
(1:1
hexane:ethyl acetate). This solid was stirred in a solution of 1:1 THF:6N HCI
(6 mL) for
16 h. The solution was then made slightly basic by the addition of NaaC03, and
extracted into ethyl acetate (15 mL). The organic layer was dried (MgS04),
filtered, and
evaporated. The residue was purified using flash chromatography to afford a
dark
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
74
brown solid (32 mg, 44%), single spot at Rf 0.84 (ethyl acetate). mp 142-146
°C, HPLC
purity 96% (tR 2.34 min in 10% water-acetonitrile). 'H NMR (d6 DMSO): b 12.32
(1H, s),
10.61 (1 H, s), 7.93-7.88 (2H, m), 7.68 (1 H, d, J=8.2 Hz), 7.47 (1 H, d,
J=9.4 Hz), 7.34
(1 H, t, J=8.1 Hz), 7.13 (1 H, dd, J=9.4, 2.2 Hz), 2.68 (3H, s), 2.34 (3H, s).
LCMS: 394.18
(M-). FAB-MS (MH+, C~gH14CIN3O3S2): calcd 396.0243, found 396.0238.
Synthesis of N-f5-(4-propel-benzenesulphonylamino)-benzofdlisothiazol-3-yll-
acetamide
STX 930 (KRB01056):
~ so ~ I sN
N
H NH
To a solution of 4n-propylbenzenesulphonyl chloride (92 mg, 0.42 mmol) in
dichloromethane (3 mL) was added pyridine (80 pL, 1.0 mmol) and the mixture
was
stirred under N~ for 5 minutes, at which time N-acetyl-N-(5-amino-
benzo[d]isothiazol-3-
yl)-acetamide (100 mg, 0.401 mmol) was added. The resulting mixture was
stirred for 3
h at room temperature. 5% NaHC03 solution (8 mL) was added and the mixture was
extracted into ethyl acetate (15 mL). The organic phase was washed with brine,
dried
(Na2S04), filtered and evaporated to give a residue that was purified using
flash
chromatography to afford a yellow solid (115 mg, 66%), single spot at Rf 0.66
(1:1
hexane:ethyl acetate). This solid was stirred in a solution of 1:1 THF:6N HCI
(6 mL) for
6 h. The solution was then made slightly basic by the addition of Na~C03, and
extracted
into ethyl acetate (15 mL). The organic layer was dried (MgS04), filtered, and
evaporated. The residue was purified using flash chromatography to afford a
pale
brown solid (88 mg, 91 %), single spot at Rf 0.71 (ethyl acetate). mp 254.3-
255.0°C,
HPLC purity 99+% (tR 2.32 min in 10% water-acetonitrile). 'H NMR (d6 DMSO): b
12.35
(1 H, s), 10.23 (1 H, s), 7.94 (1 H, d, J=2.0 Hz), 7.70 (2H, d, J=8.2 Hz),
7.43 (1 H, d, J=9.4
Hz), 7.33 (2H, d, J=8.2 Hz), 7.08 (1 H, dd, J=9.4, 2.0 Hz), 2.50 (2H, t, J=7.4
Hz), 2.35
(3H, s), 1.53 (2H, sextet, J=7.4 Hz), 0.82 (3H, t, J=7.3 Hz). LCMS: 388.25 (M-
). FAB-
MS (MH+, C~gH~gN3O3S~): calcd 390.0946, found 390.0941.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
Synthesis of N-f5-(2,5-dichloro-benzenesulphonylamino)-
benzo~dlisothiazol-3-yll-acetamide, STX 931 (KRB01057):
CI
s
,~ ~ oN
,S.N
CI ~ H NH
O
5
To a solution of 2,5-dichlorobenzenesulphonyl chloride (103 mg, 0.421 mmol) in
dichloromethane (3. mL) was added pyridine (80 pL, 1.0 mmol) and the mixture
was
stirred under N2 for 5 minutes, at which time N-acetyl-N-(5-amino-
benzo[djisothiazol-3-
yl)-acetamide (100 mg, 0.401 mmol) was added. The resulting mixture was
stirred for 6
10 h at room temperature. 5% NaHC03 solution (8 mL) was added and the mixture
was
extracted into ethyl acetate (15 mL). The organic phase was washed with brine,
dried
(NaZS04), filtered and evaporated to give a residue that was purified using
flash
chromatography to afford a yellow solid (114 mg, 62%), single spot at Rf 0.74
(1:1
hexane:ethyl acetate). This solid was stirred in a solution of 1:1 THF:6N HCI
(6 mL) for
15 5 h. The solution was then made slightly basic by the addition of Na~C03,
and extracted
into ethyl acetate (15 mL). The organic layer was dried (MgS04), filtered, and
evaporated. The residue was purified using flash chromatography to afford a
pale
brown solid (28 mg, 29%), single spot at Rf 0.64 (ethyl acetate). mp 169-
172°C, HPLC
purity 99+% (tR 2.85 min in 10% water-acetonitrile). 'H NMR (d6 DMSO): S 12.34
(1H,
20 s), 10.84 (1 H, s), 8.12 (1 H, d, J=1.7 Hz), 7.89 (1 H, d, J=1.7 Hz), 7.68
(2H, m), 7.49 (1 H,
d, J=9.4 Hz), 7.18 (1 H, dd, J=9.4, 2.0 Hz), 2.35 (3H, s). LCMS: 414.11 (M-).
FAB-MS
(MH+, C~SH~~CI2N303S~): calcd 415.9697, found 415.9697.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
76
Synthesis of Benzisoxazole Derivatives
R~ R~
R2 ~ ~ O ~ O R2 ~ ~ O ~ O
R3 s S. W I ~ N R3 / ~S. W I s N
N v ~ ~ N
R4 O H Rq. O H
STX 874: R~=R2=H, R3=CI, R4=Me STX 918: R~=R2=H, R3=CI, R4=Me
STX 875: R~=R3=R4=H, R2=n-propyl STX 919: R~=R3=R4=H, R2=n-propyl
STX 876: R~=R4=CI, R2=R3=H STX 920: R~=Rq=CI, R2=R3=H
Synthesis of 3-chloro-2-methyl-N-(3-methyl-benzofdlisoxazol-5-yl)-
benzenesulfonamide, STX 874 (KRB01027):
CI I ~ S~ ~ ~ O N
O N ~
H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (128 mg, 0.567
mmol) in
dichloromethane (3 mL) was added pyridine (110 pL, 1.35 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-3-methyl-1,2-
benzisoxazole [26] (80
mg, 0.54 mmol) was added. The resulting mixture was stirred for 4 h at room
temperature, then saturated NaHC03 solution (8 mL) was added and the mixture
was
extracted into ethyl acetate (15 mL). The organic phase was washed with brine,
dried
(Na2S04), filtered and evaporated to give a residue that was purified using
flash
chromatography to afford a white solid (88 mg, 48%), single spot at Rf 0.58
(1:1
hexane:ethyl acetate). mp 162.8-163.2°C, HPLC purity 99+% (tR 2.26 min
in 10% water-
acetonitrile). 'H NMR (CDC13): 8 7.80 (1 H, dd, J=7.9, 1.3 Hz), 7.55 (1 H, dd,
J=8.1, 1.1
Hz), 7.38 (2H, m), 7.17 (1 H, t, J=8.1 Hz), 7.10 (1 H, dd, J=8.8, 2.2 Hz),
6.72 (1 H, s, N-l~,
2.72 (3H, s), 2.50 (3H, s). LCMS: 320.00 (M-CH3). FAB-MS (MH+, C~5H~3CIN~O3S):
calcd 337.0413, found 337.0422.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
77
Synthesis of N (3-methyl-benzo~dlisozazol-5-yl)-4-prop rLl-
benzenesulfonamide, STX 875 (KRB01028):
O
I ,N
O .N
H
To a solution of 4n-propylbenzenesulphonyl chloride (124 mg, 0.567 mmol) in
dichloromethane (3 mL) was added pyridine (110 pL, 1.35 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-3-methyl-1,2-
benzisoxazole (80 mg,
0.54 mmol) was added. The resulting mixture was stirred for 3 h at room
temperature,
then saturated NaHC03 solution (8 mL) was added and the mixture was extracted
into
ethyl acetate (15 mL). The organic phase was washed with brine, dried
(Na2SO4),
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford a pale pink solid (150 mg, 84%), single spot at Rf 0.62 (1:1
hexane:ethyl acetate).
mp 119.5-120.0°C, HPLC purity 99+% (tR 2.29 min in 10% water-
acetonitrile). ~H NMR
(CDCI3): b 7.57 (2H, d, J=8.4 Hz), 7.40 (1 H, d, J=2.2 Hz), 7.37 (1 H, d,
J=8.8 Hz), 7.20
(2H, d, J=8.4 Hz), 7.10 (1 H, dd, J=8.8, 2.2 Hz), 6.71 (1 H, s, N-I~, 2.59
(2H, t, J=7.5 Hz),
2.51 (3H, s), 1.59 (2H, sextet, J= 7.5 Hz), 0.88 (3H, t, J=7.5 Hz). LCMS:
314.07 (M-
CH3). FAB-MS (MH+, C~~H~$N~03S): calcd 331.1116, found 331.1117.
Synthesis of 2,5-dichloro-N-(3-methyl-benzofdlisozazol-5-yl)-
benzenesulfonamide, STX 876 (KRB01030):
CI
a
~ ,o
;s,
CI 0 H
To a solution of 2, 5-dichlorobenzenesulphonyl chloride (105 mg, 0.428 mmol)
in
dichloromethane (3 mL) was added pyridine (100 pL, 1.02 mmol) and the mixture
was
stirred under N2 for 5 min, after which time 5-amino-3-methyl-1,2-
benzisoxazole (60 mg,
0.41 mmol) was added. The resulting mixture was stirred for 2 h at room
temperature,
then saturated NaHC03 solution (8 mL) was added and the mixture was extracted
into
ethyl acetate (15 mL). The organic phase was washed with brine, dried
(Na2S04),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
7~
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford a yellow solid (100 mg, 68%), single spot at Rf 0.62 (1:1 hexane:ethyl
acetate).
mp 229.4-230.0°C, HPLC purity 94% (tR 2.18 min in 10% water-
acetonitrile). 'H NMR
(CDCI3): b 7.89 (1 H, d, J=2.2 Hz), 7.43 (4H, m), 7.22 (1 H, m), 7.09 (1 H, s,
N-f~, 2.54
(3H, s). LCMS: 340.06 (M-CH3). FAB-MS (MH+, C~4H~pChN2O3S): calcd 356.9867,
found 356.9860.
Synthesis of N benzofdlisoxazol-5-yl-3-chloro-2-methyl-
benzenesulfonamide, STX 918 (KRB01046):
I o
CI I ~ SO ~ ~ N
O N
H
To a solution of 3-chloro-2-methylbenzenesulphonyl chloride (176 mg, 0.783
mmol) in
dichloromethane (4 mL) was added pyridine (150 pL, 1.86 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-1,2-benzisoxazole [27]
(100 mg,
0.746 mmol) was added. The resulting mixture was stirred for 2 h at room
temperature,
then saturated NaHC03 solution (10 mL) was added and the mixture was extracted
into
ethyl acetate (20 mL). The organic phase was washed with brine, dried
(Na~S04),
filtered and evaporated to give a residue that was purified using flash
chromatography to
afford a white solid (178 mg, 74%), single spot at Rf 0.69 (1:1 hexane:ethyl
acetate). mp
111.9-112.4°C, HPLC purity 97% (tR 2.44 min in 10% water-acetonitrile).
'H NMR
(CDCI3): 5 8.62 (1 H, d, J=1.0 Hz), 7.81 (1 H, dd, J=7.9, 1.2 Hz), 7.55 (1 H,
dd, J=7.9, 1.0
Hz), 7.45 (2H, m), 7.17 (2H, m), 6.77 (1 H, s, N-I~, 2.72 (3H, s). LCMS:
321.01 (M-).
FAB-MS (MH+, C~4H~~CIN203S): calcd 323.0257, found 323.0271.
Synthesis of N-benzo~dlisoxazol-5-yl-4-propel-benzenesulfonamide. STX 919
(KRB01047):
O
,o ~ I ~ N
,s, ~~r
O N
H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
79
To a solution of 4n-propylbenzenesulphonyl chloride (171 mg, 0.783 mmol) in
dichloromethane (4 mL) was added pyridine (150 pL, 1.86 mmol) and the mixture
was
stirred under N~ for 5 min, after which time 5-amino-1,2-benzisoxazole (100
mg, 0.746
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (Na~S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
white solid (170 mg, 72%), single spot at Rf 0.68 (1:1 hexane:ethyl acetate).
mp 130.0-
130.6°C, HPLC purity 99+% (tR 2.44 min in 10% water-acetonitrile). 'H
NMR (CDCI3): 5
8.62 (1 H, d, J=1.0 Hz), 7.60 (2H, d, J=8.4 Hz), 7.48 (2H, m), 7.19 (3H, m),
6.86 (1 H, s,
N-I~, 2.58 (2H, t, J=7.5 Hz), 1.58 (2H, sextet, J=7.4 Hz), 0.88 (3H, t, J=7.4
Hz). LCMS:
315.14 (M-). FAB-MS (MH+, C,6H~6NZO3S): calcd 317.0960, found 317.0962.
Synthesis of N-benzofdlisoxazol-5-yl-2,5-dichloro-benzenesulfonamide. STX 920
(KRB01048):
CI
O
i ,
i ,~ ~ ~ N
~S. N
CI ~ H
To a solution of 2,5-dichlorobenzenesulphonyl chloride (192 mg, 0.783 mmol) in
dichloromethane (4 mL) was added pyridine (150 pL, 1.86 mmol) and the mixture
was
stirred under NZ for 5 min, after which time 5-amino-1,2-benzisoxazole (100
mg, 0.746
mmol) was added. The resulting mixture was stirred for 2 h at room
temperature, then
saturated NaHC03 solution (10 mL) was added and the mixture was extracted into
ethyl
acetate (20 mL). The organic phase was washed with brine, dried (Na~S04),
filtered and
evaporated to give a residue that was purified using flash chromatography to
afford a
white solid (174 mg, 68%), single spot at Rf 0.68 (1:1 hexane:ethyl acetate).
mp 172.9-
173.6°C, HPLC purity 99+% (tR 2.41 min in 10% water-acetonitrile). 'H
NMR (CDCI3): b
8.64 (1 H, s), 7.89 (1 H, d, J=2.2 Hz), 7.56-7.28 (5H, m). LCMS: 341.07 (M-).
FAB-MS
(MH+, C~3HgCI2N~O3S): calcd 342.9711 , found 342.9710.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
Synthesis of Arysulfonamide, Arylthiol Ether and Arylsulfone Derivatives
O H O
O '' ' N i
CI S' I \O
CI ~S~H \ / ~ I / O
STX828 STX885
O S~N ~ O O ~S~N ~ O O
~N
c1 , o c1
STX979 STX980
O H CI
O \S/N \ O I ~ S
S
\ O
C. , CI
STX990 STX1031
CI O O H
'S ~ ~ CI ~S~ N
~O ~ / S O
O
CI STX1032 STX1033
General method for arylsulphonamide formation:
S To a solution arylsulphonyl chloride (1.1 eq.) in DCM were added pyridine
(2.2 eq.) and
catalytic amount of DMAP, followed by the corresponding amine (1 eq.). The
reaction
mixture was stirred at rt under nitrogen for 4-16 h, then partitioned between
ethyl acetate
and 5% sodium bicarbonate after TLC showed the completion of the reaction. The
organic layer was washed with brine, dried over sodium sulphate, and
concentrated in
10 vacuo to give crude product as solid or thick syrup. The compound was then
purified by
flash chromatography (Methanol-DCM gradient elution) to give desired
arylsulphonamide
as crystalline solid. Yield ranges from 40-90%.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
81
3-Chloro-2-methyl-N-(4-morpholin-4-yl-phenyl)-benzenesulfonamide (STX828,
XDS01161 )
Off-white crystalline solid. TLC single spot at fZf 0.70 (70% ethyl
acetate/hexane); HPLC
purity > 99 % (tR 1.8 min in 4 % water-methanol); 'H NMR (400 MHz, DMSO): 5
10.1
(1 H, s, NH), 7.77 (1 H, dd, J = 8.2, 1.2 Hz, ArH ), 7.70 (1 H, dd, J=8.2, 1.2
Hz, ArH), 7.34
(1 H, t, J = 8.2 Hz, ArH), 6.78-6.92 (4H, m, ArH), 3.67 (4H, t, J = 5.1 Hz,
N(CH~)~), 2.99
(4H, t, J = 5.1 Hz, O(CH2)2), 2.60 (3H, s, CH3); APCI-MS 365 (M-H+); FAB-HRMS
calcd
for C~~HaoCIN203S (MH+) 367.0883, found 367.0854
3-Chloro-2-methyl-N-(3-oxo-1.3-dihydro-isobenzofuran-5-yl)-benzenesulfonamide
~STX885, XDS01179)
White crystalline solid. TLC single spot at Rf 0.60 (20% ethyl
acetate/hexane); HPLC
purity > 99 % (tR 2.2 min in 10 % water-methanol); 'H NMR (270 MHz, DMSO): 5
11.0
(1 H, s, NH), 7.84 (1 H, d, J = 7.9 Hz, ArH), 7.66 (1 H, d, J = 7.9 Hz, ArH),
7.49 (1 H, d, J =
8.5 Hz, ArH), 7.31-7.46 (3H, m, ArH), 5.22 (2H, s, CHI), 2.57 (3H, s, CH3);
APCI-MS 338
(M+H+); FAB-HRMS calcd for C~5H~3CINO4S (MH+) 338.0254, found 338.0270
3-Chloro-2-methyl-N-(2-methyl-1,3-dioxo-2.3-dihydro-1H-isoindol-5-yl)-
benzenesulfonamide (STX979, XDS01180)
Yello~ni crystalline solid. TLC single spot at Rf 0.82 (20% ethyl
acetate/hexane); HPLC
purity > 99 % (tR 2.1 min in 10 % water-methanol); 'H NMR (270 MHz, DMSO): 5
11.5
(1 H, s, NH), 7.98 (1 H, d, J = 7.9 Hz, ArH), 7.76 (1 H, d, J = 8.3 Hz, ArH),
7.73 (1 H, d, J =
8.3 Hz, ArH), 7.45 (1 H, t, J = 8.3 Hz, ArH ), 7.41 (1 H, s, ArH), 7.39 (1 H,
d, J = 7.9 Hz,
ArH ),2.94 (3H, s, NCH3), 2.64 (3H, s, CH3); APCI-MS 363 (M-H+); FAB-HRMS
calcd for
~16H14CINZOqS (MH+) 365.0363, found 365.0375
3-Chloro-2-methyl-N-(4-methyl-2-oxo-2H-chrornen-7-yl)-benzenesulfonamide
(STX980,
XDS02018)
White crystalline solid. TLC single spot at Rf 0.78 (6% methanol/DCM); HPLC
purity >
99 % (tR 2.1 min in 10 % water-methanol); 'H NMR (270 MHz, DMSO): S 11.3 (1H,
s,
NH), 8.03 (1 H, d, J = 7.9 Hz, ArH), 7.76 (1 H, d, J = 7.9 Hz, ArH), 7.65 (1
H, d, J = 8.6 Hz,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
82
ArH), 7.46 (1 H, t, J = 7.9 Hz, ArH), 7.07 (1 H, dd, J=8.6, 1.8 Hz, ArH), 6.98
(1 H, d, J =
1.5 Hz, ArH), 6.24 (1 H, s, 3-H), 2.69 (3H, s, CH3), 2.33 (3H, s, CH3); APCI-
MS 362 (M-
H+); FAB-HRMS calcd for C~~H~5CIN04S (MH+) 364.0410, found 364.0414
3-Chloro-N-(2,3-dihydro-benzof1,41dioxin-6-yl)-2-methyl-benzenesulfonamide
(STX990.
XDS02039)
White crystalline solid. TLC single spot at Rf 0.78 (30% ethyl acetate/DCM);
HPLC
purity 94 % (tR 2.3 min in 10 % water-methanol); ' H NMR (270 MHz, DMSO): 5
10.3 (1 H,
s, NH), 7.82 (1 H, d, J = 7.9 Hz, ArH), 7.72 (1 H, dd, J = 7.9, 1.0 Hz, ArH),
7.39 (1 H, t, J =
7.9 Hz, ArH ), 6.72 (1 H, d, J = 8.4 Hz, ArH ), 6.49-6.55 (2H, m, ArH), 4.15
(4H, s,
(CH~)2), 2.62 (3H, s, CH3); APCI-MS 338 (M-H+); FAB-HRMS calcd for
C~5H~5CINO4S
(MH+) 340.0410, found 340.0387
3-Chloro-2-methyl-N-(9-oxo-9H-fluoren-3-yl)-benzenesulfonamide (STX1033,
XDS02074)
Yellow solid. TLC single spot at Rf 0.55 (8% ethyl acetate/DCM); HPLC purity >
99 % (tR
2.5 min in 20 % water-methanol); 'H NMR (270 MHz, DMSO): b 10.3 (1 H, s, NH),
7.82
(1 H, d, J = 7.9 Hz, ArH), 7.72 (1 H, dd, J = 7.9, 1.0 Hz, ArH), 7.39 (1 H, t,
J = 7.9 Hz, ArH
), 6.72 (1 H, d, J = 8.4 Hz, ArH ), 6.49-6.55 (2H, m, ArH), 4.15 (4H, s,
(CH~)z), 2.62 (3H,
s, CH3); APCI-MS 382 (M-H+); FAB-HRMS calcd for C2pH15CINO3S (MH+) 384.0461,
found 384.0457.
5-(2,5-Dichloro-phenylsulfanylmethyl)-2-methylbenzothiazole (STX1031.
XDS02072)
To a solution of 2,5-dichlorobenzothiol (179 mg, 1.0 mmol) in absolute ethanol
(3 mL)
were added 5-(bromomethyl)-2-methylbenzothiazole (182 mg, 0.75 mmol) and
triethylamine (0.15 mL). After stirred at rt for 3h, the mixture was
partitioned between
ethyl acetate and 1 % KOH solution. The organic phase was washed with brine,
dried
over sodium sulphate and concentrated in vacuo to give a residue that was
subjected to
flash chromatography. Colorless needles (220 mg, 86%) were obtained. TLC
single spot
at Rf 0.55 (5% ethyl acetate/DCM); HPLC purity > 99% (tR 6.8 min in 20% water-
methanol); 'H NMR (270 MHz, DMSO): 5 8.00 (1 H, d, J = 8.1 Hz, ArH), 7.97 (1
H, s,
ArH), 7.53 (1 H, d, J = 2.5 Hz, ArH ), 7.46-7.49 (2H, m, ArH ), 7.24 (1 H, dd,
J = 8.1, 2.5
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
83
Hz, ArH), 4.54 (2H, s, CHI), 2.79 (3H, s, CH3); APCI-MS 338 (M+); FAB-HRMS
calcd for
C15H12CI2NS2 (MH+) 339.9788, found 339.9779.
5-(2,5-Dichloro-benzenesulfonylmethyl)-2-methylbenzothiazole (STX1032,
XDS02075)
To a solution of 5-(2,5-Dichloro-phenylsulfanylmethyl)-2-methylbenzothiazole
(STX1031,
125 mg, 0.367 mmol) in DCM (5 mL) was added 3-cholroperoxybenzoic acid (370
mg,
2.14 mmol). The mixture was stirred at rt for 2h, then partitioned between DCM
and
saturated sodium carbonate solution. The organic phase was washed with brine,
dried
over sodium sulphate and concentrated to give a residue that was purified with
flash
chromatography. White crystals (55 mg, 40%) were obtained after
recrystallization from
DCM. TLC single spot at Rf 0.20 (5% ethyl acetate/DCM); HPLC purity 97% (tR
3.0 min
in 20% water-methanol); 'H NMR (270 MHz, DMSO): 5 7.99 (1 H, d, J = 8.2 Hz,
ArH),
7.82-7.83 (2H, m, ArH), 7.77 (1 H, s, ArH), 7.69 (1 H, d, J = 2.5 Hz, ArH ),
7.23 (1 H, dd, J
= 8.2, 1.5 Hz, ArH), 5.07 (2H, s, CHI), 2.79 (3H, s, CH3); APCI-MS 370 (M+);
FAB-HRMS
calcd for C15H12CI2N02S2 (MH+) 371.9686, found 371.9691.
Synthesis of N-2-Hydroxyethyl Arysulfonamides and Arysulfonic Acid Esters
'O O ~S'-O O
\ HN ~ ~ S=O I \ HN ~ ~ S=O
w ~ w
O HN~ O HN~O
OH ,
STX646 XDS01110 O=S=O
STX647 XDS01110B
s0
O~ .O O O ,.O O
H N ~ ~ DSO~ /O I ~ H N ~ ~ DSO~
W
HN~ O HN~
CI OH STX649 XDS01112 OH
STX648 XDS01111
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
84
O ~ %O O
\ H N ~ ~ DSO~ I H N ~ ~ S=O
w ~ w
O HN~O O a W HN~
STX650 XDS01112B O~S-O ST~C651 XDS01117 OH
O
/O
O ~S%O O
HN ~ ~ S=O I \ HN ~ ~ S=O
O HN~O CI CI HN
i OOH
O=S=O ST7C653 XDS01118
STX 652 XDS01117B
,O
General Method for coupling 4-amino-N-(2-hydroxyethyl)benzenesulfonamide with
arylsulphonyl chloride (STX646-STX653)
To a solution arylsulphonyl chloride (1.1 eq.) in DCM were added pyridine (10
eq.) and
4-amino-N-(2-hydroxy-ethyl)-benzenesulfonamide (1 eq.). The reaction mixture
was
stirred at rt under nitrogen for 20 h, then partitioned bet~rveen DCM and 1 %
HCI solution.
The organic layer was washed with brine, dried over sodium sulphate, and
concentrated
in vaeuo to give crude oily product, which was seperated by flash
chromatography (ethyl
acetate-DCM gradient elution) to give arylsulphonamide (STX646, 648, 649, 651,
653)
and arylsulphonamide arylsulphonic acid ester (STX647, 650, 652) in 10 : 1
ratio.
N-f4-(2-Hydroxyethylsulfamoyl)phenyll-4'-methoxybenzenesulfonamide (STX646.
XDS01110)
White foam . TLC single spot at Rf 0.21 (50% DCM-ethyl acetate); HPLC purity >
99%
(tR 2.0 min in 10% water-methanol);'H NMR (400 MHz, DMSO): 5 10.8 (1H, s, NH),
7.76
(2H, m, ArH), 7.64 (2H, d, J = 9 Hz, ArH), 7.46 (1 H, t, J = 5.9 Hz, NH), 7.24
(2H, d, J = 9
Hz, ArH), 7.08 (2H, m, ArH), 4.66 (1 H, t, J = 5.5 Hz, OH), 3.80 (3H, s,
OCH3), 3.30 (2H,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
m, OCH2), 2.72 (2H, m, NCH); FAB-MS 387 (MH+); FAB-HRMS calcd for
C15H1gN20gS2 (MH+) 387.0685, found 387.0685
4-Methoxy-benzenesulfonic acid 2-f4-(4-methoxybenzenesulfonylamino)-
5 benzenesulfonylaminol ethyl ester (STX647 XDS01110B)
White foam TLC single spot at Rf 0.62 (50% DCM-ethyl acetate); HPLC purity >
99% (tR
1.6 min methanol); ~H NMR (400 MHz, DMSO): 5 '10.8 (1 H, s, NH), 7.82 (1 H, t,
J = 5.9
Hz, NH), 7.75-7.81 (4H, m, ArH), 7.59 (2H, d, J = 9 Hz, ArH), 7.22-7.25 (2H,
2d, J = 9
10 Hz, ArH), 7.17 (2H, m, ArH), 7.08 (2H, m, ArH), 3.88 (2H, m, OCH~), 3.87
(3H, s, OCH3),
3.79 (3H, s, OCH3), 2.90 (2H, q, J = 5.9 Hz, NCH2); FAB-MS 557 (MH+); FAB-HRMS
calcd for C22H25N209S3 (MH+) 557.0722, found 557.0717.
N-f4-(2-Hydroxyethylsulfamoyl)phenyll-2'-methyl-3'-chlorobenzenesulfonamide
15 (STX648, XDS01111 )
White foam. TLC single spot at Rf 0.28 (50% DCM-ethyl acetate); HPLC purity >
99%
(tR 1.7 min in methanol); 'H NMR (400 MHz, DMSO): 5 11.2 (1 H, s, NH), 7.96 (1
H, dd, J
= 8.2, 1.1 Hz, ArH), 7.75 (1 H, dd, J = 8.2, 1.1 Hz, ArH), 7.65 (2H, m, ArH),
7.46 (1 H, t, J
20 = 8.2 Hz, ArH ), 7.44 (1 H, t, J = 6.4 Hz, NH), 7.22 (2H, m, ArH ), 4.66 (1
H, t, J = 5.8 Hz,
OH), 3.32 (2H, m, OCH~), 2.71 (2H, m, NCHa), 2.65 (3H, s, CH3); APCI-MS 405
(MH+);
FAB-HRMS calcd for C15H18CIN205S2 (MH+) 405.0345, found 405.0334
N-f4-(2-Hydroxyethylsulfamoyl)phenyll-3' 4'-dimethoxy Benzene
25 Sulfonamide (STX649, STX01112)
White foam. TLC single spot at Rf 0.17 (50% DCM-ethyl acetate); HPLC purity >
99
(tR 1.6 min in methanol); 'H NMR (400 MHz, DMSO): ~ 10.7 (1 H, s, NH), 7.64
(2H, d, J =
8.6 Hz, ArH), 7.46 (1 H, t, J = 6.4 Hz, NH), 7.39 (1 H, dd, J = 8.5, 2.3 Hz,
ArH), 7.28 (1 H,
30 d, J = 2.3 Hz, ArH), 7.26 (2H, d, J = 8.6 Hz, ArH), 7.08 (1 H, d, J = 8.5
Hz, ArH ), 4.66
(1 H, t, J = 5.4 Hz, OH), 3.79 (3H, s, OCH3), 3.77 (3H, s, OCH3), 3.33 (2H, m,
OCH~),
2.71 (2H, m, NCH); FAB-MS 417 (MH+); FAB-HRMS calcd for C1gH21N2O7S2 (MH+)
417.0790, found 417.0783.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
86
3,4-Dimethoxybenzenesulfonic acid 2-f4-(3,4-dimethoxybenzenesulfonylamino)-
benzenesulfonylaminolethyl ester (STX650, XDS01112B)
White foam TLC single spot at Rf 0.50 (50% DCM-ethyl acetate); HPLC purity >
99
(tR 1.6 min in methanol); 'H NMR (400 MHz, DMSO): 5 10.7 (1 H, s, NH), 7.80(1
H, t, J =
5.9 Hz, NH), ,7.60 (2H, d, J = 8.9 Hz, ArH), 7.38-7.45 (2H, m ArH), 7.23-7.29
(4H, m
ArH), 7.17 (2H, d, J = 8.6 Hz, ArH), 7.07 (2H, d, J = 8.6 Hz, ArH), 3.92 (2H,
t, J = 5.1 Hz,
OCH2), 3.87 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.76 (3H,
s, OCH3),
2.90 (2H, q, J = 5.9 Hz, NCH); FAB-MS 617 (M-H+); FAB-HRMS calcd for
C24H2gN2011 S3 (MH+) 617.0933, found 617.0926
N-f4-(2-Hydroxyethylsulfamoyl)phenyll-2',3'.6'-trimethyl-4'-methoxy-
benzenesulfonamide
(STX651, XDS01117)
White solid. TLC single spot at Rf0.28 (60% DCM-ethyl acetate); HPLC purity >
99 % (tR
1.6 min in methanol); 'H NMR (400 MHz, DMSO): 5 10.8 (1H, s, NH), 7.60 (2H, d,
J =
8.9 Hz, ArH), 7.41 (1 H, t, J = 5.8 Hz, NH), 7.07 (2H, d, J = 8.9 Hz, ArH),
6.83 (1 H, s,
ArH), 4.65 (1 H, broad, OH), 3.81 (3H, s, OCH3), 3.33 (2H, m, OCH2), 2.70 (2H,
m,
NCH), 2.65 (3H, s CH3), 2.53 (3H, s CH3), 2.05 (3H, s CH3); FAB-MS 429 (MH+);
FAB
HRMS calcd for C18H25N20gS2 (MH+) 429.1154, found 429.1143.
4-Methoxy-2,3,6-trimethylbenzenesulfonic acid 2-f4-(4-methoxy-2.3,6-trimethyl-
benzenesulfonylamino)benzenesulfonylaminol ethyl ester (STX652, XDS01117B)
White foam TLC single spot at Rf 0.80 (60% DCM-ethyl acetate); HPLC purity >
99
(tR 1.7 min in methanol); 'H NMR (400 MHz, DMSO): b 10.8 (1 H, s, NH), 7.72(1
H, t, J =
5.9 Hz, NH), 7.56 (2H, d, J = 9 Hz, ArH), 7.03 (2H, d, J = 9 Hz, ArH), 6.89 (1
H, s, ArH),
6.84 (1 H, s, ArH), 3.86 (3H, s, OCH3), 3.80 (5H, m, OCH3, and OCH2), 2.89
(2H, m,
NCH), 2.65 (3H, s CH3), 2.54 (3H, s CH3), 2.52 (3H, s CH3), 2.42 (3H, s CH3),
2.08 (3H,
s CH3), 2.03 (3H, s CH3); FAB-MS 641 (MH+); FAB-HRMS calcd for C28H37N20gS3
(MH+) 641.1661, found 641.1642
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
87
N-f4-(2-Hydroxy-ethylsulfamoyl)-phenyll-2',4'-dichlorobenzenesulfonamide
(STX653,
XDS01118)
White crystalline solid. TLC single spot at Rf 0.36 (60% DCM-ethyl acetate);
HPLC purity
> 99 % (tR 1.9 min in 4 % water-methanol);'H NMR (400 MHz, DMSO): b 11.3 (1H,
s,
NH), 8.11 (1 H, d, J = 8.2 Hz, ArH), 7.88 (1 H, d, J = 2.3 Hz, ArH), 7.62-7.68
(3H, m, ArH),
7.47 (1 H, t, J = 5.8 Hz, NH), 7.23 (2H, d, J = 8.6 Hz, ArH), 4.66 (1 H, t, J
= 5.9 Hz, OH),
3.33 (2H, m, OCH2), 2.71 (2H, m, NCH2); FAB-MS 424.9 (MH+); FAB-HRMS calcd for
C14H15CI2N205S2 (MH+) 424.9799, found 424.9800
Synthesis of N-(5-Methylisoxazol-3-yl)-arysulphonamides
CI
w O N-O I W O N_O
I \ O N-O / ~S. I ~ CI ~S I /'
N N
OS.N I / O H O H
CI H
STX 824 XDS0156B STX825 XDS01157
STX823 XDS01155B
O
O ~ OSLO N,O .~ CI / \ ,O, \ ~S\'O N~O
~HN~ HN S-O ~HN~
N ~ HN ~N'O HEN '' \/
H
STX606 XDS01096 STX645 XDS01109 STX608 XDS01099
CI O ~ ~SeO c ~s~o 0
H2N ~ ~ ~S-O \ ~ ~ FiN ~ ~ S=O \ ~ / HN ~ ~ DSO~
HN [~ ~ HN ,N O HN N O
O _
STX642 XDS01106
STX638 XDS01105
STX654 XDS01119
OSLO O OSLO O
I j HN ~ ~ DSO~ I j HN ~ ~ DSO~
HN ~N CI CI HN N,
CI O O
STX644 XDS01108
STX643 XDS01107
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
88
O ~O 0 ~~ %O 0
\ \HN ~ ~ S=O ~O I \ HN ~ ~ S=O
w i w i
O HN N O HN N
CI ' 'O CI ' 'p
STX728 XDS01120 STX729 XDS01121
General Method for coupling 3-amino-5-methylisooxazole with arylsulphonyl
chloride
(STX606, STX645, STX823-825)
To a solution arylsulphonyl chloride (1.1 eq.) in DCM were added pyridine (1
.2 eq.) and
3-amino-5-methylisooxazole (1 eq.). The reaction mixture was stirred at rt
under
nitrogen for 24 h, then partitioned between ethyl acetate and 5% sodium
bicarbonate
solution. The organic layer was washed with 1 % HCI solution and brine, dried
over
sodium sulphate, and concentrated in vacuo to give crude product that was
purified by
flash chromatography (ethyl acetate-DCM gradient elution) to give
arylsulphonamide as
white or off-white crystalline solid (Yield 50-80%).
2,5-Dichloro-N-(5-methylisoxazol-3-yl)benzenesulphonamide (STX823 XDS01155B)
White crystalline solid. TLC single spot at Rf 0.45 (10% ethyl acetate-DCM);
HPLC purity
as rotational isomers > 99% (tR 2.2 min in 15% water-methanol); 'H NMR (270
MHz,
DMSO-d6): b 12.0 (1 H, s, NH), 8.01 (1 H, d, J = 2 Hz, ArH), 7.78 (1 H, dd, J
= 8.0, 2.0 Hz,
ArH), 7.71 (1 H, d, J = 8.0 Hz, ArH), 6.04 (1 H, s, ArH), 2.27 (3H, s, CH3);
APCI-MS 306
(M+); FAB-HRMS calcd for CIpHgC12N203S (MH+) 306.9711, found 306.9718
N-(5-Methylisoxazol-3-yl)-4-propylbenzenesulphonamide (STX824 XDS01156B)
White crystalline solid. TLC single spot at Rf 0.65 (10% ethyl acetate-DCM);
HPLC purity
> 99% (tR 2.3 min in 10% water-methanol); 'H NMR (270 MHz, DMSO-d6): ~ 11.3 (1
H,
s, NH), 7.72 (2H, d, J = 8.1 Hz, ArH), 7.38 (2H, d, J = 8.1 Hz, ArH), 6.10 ('1
H, s, ArH),
2.58 (2H, t, J = 8.0 Hz, CH2), 2.25 (3H, s, CH3), 1.53 (2H, t, J = 7.9 Hz,
CH2), 0.83 (3H, t,
J = 7.9 Hz, CH3); APCI-MS 281 (MH+); FAB-HRMS calcd for C13H17N2~3S (MH+)
281.0960, found 281.0970
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
89
3-Chloro-2-methyl-N-(5-methylisoxazol-3-yl)benzenesulphonamide (STX825
XDS01157)
White crystalline solid. TLC single spot at Rf 0.50 (10% ethyl acetate-DCM); H
PLC purity
as rotational isomers > 99% (tR 2.4 min in 15% water-methanol); 'H NMR (270
MHz,
DMSO-d6): 5 11.8 (1 H, s, NH), 7.91 (1 H, d, J = 8.1 Hz, ArH), 7.76 (1 H, d, J
= 8.1 Hz,
ArH), 7.43 (1 H, t, J = 8.1 Hz, ArH), 6.01 (1 H, s, ArH), 2.61 (3H, s, CH3),
2_ 26 (3H, s,
CH3); FAB-MS 286 (M+); FAB-HRMS calcd for C11 H12CIN2O3S (MH+) 287.0257, found
287.0258
N-(5-methylisoxazol-3-yl)-4-acetamidobenzenesulphonamide (STX606, XDSO'1096)
White solid. Mp 220-222°C (lit [28], 225-228°C); TLC single spot
at Rf 0.62 (10% ethyl
acetate-DCM); HPLC purity 99% (tR 1.7 min in methanol); 'H NMR (270 MHz, DMSO-
d6): S 11.3 (1 H, s, NH), 10.4 (1 H, s, AcNH), 7.76 (4H, s, ArH), 6.12 (1 H,
s, ArH), 2.29
(3H, s, CH3), 2.07 (3H, s, COCH3); FAB-MS 296 (MH+); FAB-HRMS calcd for
C12H14N304S (MH+) 296.0705, found 296.0701
N-(5-methylisoxazol-3-yl)-3-chloro-4-acetamidobenzenesulphonamide (STX645,
XDS011091
White solid. TLC single spot at Rf 0.52 (6% methanol-DCM); HPLC purity as
rotational
isomers > 99% (tR 1.7 min in 4% water-methanol);'H NMR (400 MHz, DMSO-d6): S
11.5
(1 H, s, NH), 9.78 (1 H, s, AcNH), 8.10 (1 H, d, J = 8.2 Hz, ArH), 7.90 (1 H,
s, ArH), 7.77
(1 H, d, J = 8.2 Hz, ArH), 6.15 (1 H, s, ArH), 2.31 (3H, s, CH3), 2.16 (3H, s,
COCH3); FAB-
MS 330 (MH+); FAB-HRMS calcd for C12H13CIN304S (MH+) 330.0315, found 330.0321
4-Amino-N-(5-methyl-isoxazol-3-yl)-benzenesulphonamide (STX608, XDS01099)
The solution of N-(5-methylisoxazol-3-yl)-4-acetamidobenzenesulphonamid (3.4
g, 11.5
mmol) in 10% NaOH solution (15 mL) was stirred at 80°C for 1 h, cooled
to rt and
neutralized to pH 6 with acetic acid. The precipitate was washed with water,
dried in
vacuo to yield off-white solid (2,8 g, 96%). Mp167-169°C (lit [29], 168-
171 °C); TLC
single spot at Rf 0.39 (6% methanol-DCM); HPLC purity > 99% (tR 1.6 min in 4%
water-
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
methanol); 'H NMR (400 MHz, CD30D): S 7.54 (2H, m, ArH), 6.63 (2H, m, ArH),
6.08
(1 H, s, ArH), 2.30 (3H, s, CH3); FAB-MS 254 (MH+); FAB-HRMS calcd for
C10H12N303S (MH+) 254.0599, found 254.0605.
5 4-Amino-3-chloro-N-(5-methylisoxazol-3-yl)benzenesulphonamide (STX654.
XDS01119)
The compound was prepared as described above. White solid (370 mg,
94°Oo) was
obtained. TLC single spot at Rf 0.58 (30% ethyl acetate-DCM); HPLC purity >
99% (tR
1.7 min in methanol); 'H NMR (400 MHz, DMSO-d6): b 11.1 (1 H. s, NH), 7.60 (1
H, d, J =
10 2.3 Hz, ArH), 7.45 (1 H, dd, .J = 8.5, 2.3 Hz, ArH),6.82 (1 H, d, J = 8.5
Hz, ArH), ), 6.23
(2H, s, NHS), 6.10 (1 H, s, ArH), 2.29 (3H, s, CH3); FAB-MS 288 (MH+); FAB-
HRMS calcd
for ClpHIpCIN3O3S (MH+) 288.0210, found 288.0213.
General Method for coupling 4-Amino-N-(5-methyl-isoxazol-3-yl)-
benzenesulphonamide
15 or 4-Amino-3-chloro-N-(5-methylisoxazol-3-yl)benzenesulphonamide with
arylsulphonyl
chloride (STX638, 642-644. 728, 729)
To a solution arylsulphonyl chloride (1.1 eq.) in DCM were added pyridine (1.3
eq.) and
catalytic amount of DMAP, followed by the amine (1 eq.). The reaction mixture
was
20 stirred at rt or 40°C under nitrogen for 24-48 h, then partitioned
between DCM and water
after TLC showed completion of the reaction. The organic layer was washed
v~ith 3%
HCI solution and brine, dried over magnesium sulphate, and concentrated in
vacuo to
give crude product that was purified by recrystallization from ethyl acetate-
DCM or by
flash chromatography (ethyl acetate-DCM gradient elution) to give
arylsulphonamide as
25 white or off-white crystalline solid (Yield 40-80%).
N-f4-(5-Meth~~l-isoxazol-3-ylsulfamoyl)phenyll-3',4'-dimethoxy-
benzenesulphonamide
(STX638, XDS01105)
30 White crystalline solid. TLC single spot at Rf 0.30 (6% methanol-DCM); HPLC
purity as
rotational isomers > 99% (tR 1.7 min in 4% water-methanol); ~H NMR (400 MHz,
DMSO
d6): ~ 11.3 (1 H, s, NH), 10.8 (1 H, s, NH), 7.71 (2H, d, J = 8.2 Hz, ArH),
7.41 (1 H, m,
ArH), 7.38 (3H, m, ArH), 7.07 (1 H, d, J = 8.6 Hz, ArH), 6.08 (1 H, s, ArH),
3.79 (3H, s,
OCH3), 3.74 (3H, s, OCH3), 2.28 (3H, s, CH3); FAB-MS 454 (MH+); FAB-HRMS calcd
for
35 C18H2pN3O7S2 (MH+) 454.0743, found 454.0746.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
91
N-f4-(5-Methyl-isoxazol-3-ylsulfamoyl)-phenyll-2',3',6'-trimethyl-4'-methoxy-
benzenesulfonamide (STX642, XDS01106)
White crystalline solid. TLC single spot at Rf 0.60 (10% methanol-DCM); HPLC
purity as
rotational isomers > 99% (tR 1.8 min in 4% water-methanol);'H NMR (400 MHz,
DMSO
d6): S 11.3 (1 H, s, NH), 10.9 (1 H, s, NH), 7.67 (2H, d, J = 9.0 Hz, ArH),
7.04 (2H, d, J =
9.0 Hz, ArH), 6.83 (1 H, s, ArH), 6.06 (1 H, s, ArH), 3.81 (3H, s, OCH3), 2.64
(3H, s, CH3),
2.27 (3H, s, CH3), 2.03 (3H, s, CH3); FAB-MS 466 (MH+); FAB-HRMS calcd for
C2pH24N3OgS2 (MH+) 466.1107, found 466.1109.
N-f4-(5-Methyl-isoxazol-3-ylsulfamoyl)-phenyll-2'-methyl-3'-
chlorobenzenesulfonamide
(STX643, XDS01107)
White crystalline solid. TLC single spot at Rf 0.56 (10% methanol-DCM); HPLC
purity as
rotational isomers > 99% (tR 2.0 min in 4% water-methanol); 'H NMR (400 MHz,
DMSO-
d6): 5 11.3 (2H, s, NH), 7.97 (1 H, d, J = 8.2 Hz, ArH), 7.75 (1 H, d, J = 8.2
Hz, ArH), 7.71
(2H, d, J = 8.6 Hz, ArH), 7.43 (1 H, t, J = 8.2 Hz, ArH), 7.21 (2H, m, ArH),
6.08 (1 H, s,
ArH), 2.62 (3H, s, CH3), 2.28 (3H, s, CH3); FAB-MS 442 (MH+); FAB-HRMS calcd
forC17H17CIN305S2 (MH+) 442.0298, found 442.0297.
N-f4-(5-Methyl-isoxazol-3-ylsulfamoyl)phenyll-2',4'-dichlorobenzenesulfonamide
(STX644, XDS01108)
White crystalline solid. TLC single spot at Rf 0.60 (10% methanol-DCM); HPLC
purity as
rotational isomers > 99% (tR 2.2 min in 10% water-methanol); 'H NMR (400 MHz,
DMSO-d6): ~ 11.5 (1 H, s, NH), 11.3 (1 H, s, NH), 8.12 (1 H, d, J = 8.5 Hz,
ArH), 7.88 (1 H,
s, ArH), 7.72 (2H, d, J = 8.6 Hz, ArH), 7.65 (1 H, d, J = 8.5 Hz, ArH), 7.23
(2H, d, J = 8.6
Hz, ArH), 6.08 (1 H, s, ArH), 2.29 (3H, s, CH3); FAB-MS 462 (MH+); FAB-HRMS
calcd for
C1gH14C12N305S2 (MH+) 461.9752, found 461.9756.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
92
3-Chloro-4-(4-methoxybenzenesulfonylamino)-N-(5-methylisoxazol-3-yl)-
benzenesulfonamide (STX728. XDS01120)
Off-white solid. TLC single spot at Rf 0.64 (30% ethyl acetate-DCM); HPLC
purity > 99%
(tR 1.8 min in 10% water-methanol); 'H NMR (400 MHz, DMSO-d6): S 11.5 (1 H, s,
NH),
10.4 (1 H, s, NH), 7.80 (1 H, d, J = 2.0 Hz, ArH), 7.70-7.76 (3H, m, ArH),
7.52 (1 H, d, J =
8.0 Hz, ArH), 7.08 (2H, d, J = 8.0 Hz, ArH), 6.13 (1 H, s, ArH), 3.82 (3H, s,
OCH3), 2.30
(3H, s, CH3); FAB-MS 458 (MH+); FAB-HRMS calcd for C17H17CIN30gS2 (MH+)
458.0247, found 458.0245.
3-Chloro-4-(3.4-dimethoxybenzenesulfonylamino)-N-(5-methylisoxazol-3-yl)-
benzenesulfonamide (STX729, XDS01121)
White crystalline solid. TLC single spot at Rf 0.49 (25% ethyl acetate-DCM);
HPLC
purity as rotational isomers > 99% (tR 1.8 min in methanol); ~H NMR (270 MHz,
DMSO-
d6): b 11.5 (1 H, s, NH), 10.3 (1 H, s, NH), 7.77 (1 H, d, J = 2.1 Hz, ArH),
7.69 (1 H, dd, J =
8.2, 2.1 Hz ArH), 7.48 (1 H, d, J = 8.2 Hz, ArH), 7.36 (1 H, dd, J = 8.6, 2.2
Hz, ArH), 7.23
(1 H, d, J = 2.2 Hz, ArH), 7.05 (1 H, d, J = 8.6 Hz, ArH), 6.07 (1 H, s, ArH),
3.77 (3H, s,
OCH3), 3.67 (3H, s, OCH3), 2.25 (3H, s, CH3); FAB-MS 488 (MH+); FAB-HRMS calcd
for
C18H1gCIN3O7S2 (MH+) 488.0353, found 488.0360.
Synthesis of pyrazolyl arysulphonamides and benzopyrazole arysulphonamides
CI CI
N
ii0
N-N ~ / ~~ N-N
o S ~ ~S~N ~ ~ ~S~N~
~ H ~ H
STX769, XDS01152 STX880, XDS01174 STX881, XDS01175C
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
93
CI CI CI
N_N ~ \ _N I ~ O \ _N
~ ~O I I / ~O N v / ~~ N v
S, ~ S, S,
O
O=S=O O=S=O
O O
STX884, XDS01178B
CI ~ CI
STX882, XDS01075B STX883, XDS01178A
CI I \ S~ I \ ~O NH2 O
~N CI ,S~
O N ~ NH2 O N
N ~ O
O STX 830, XDS01162B
O
STX829, XDS01162A
O H O H O H
CI ~ ~S\ N I ~ ~'N O 'S~N I ~ ~'N O ~S~N I ~ ~ N
O
',O ~ \ N ~ \ N
O%S CI ~ H CI
STX983, XDS02021 f STX988, XDS02037
CI
STX826, XDS01159
General Method for synthesis of an/Isulphonamide and N-arylsulphonyl
arylsulphonamide (STX769, STX829-830, STX880-884)
To a solution arylsulphonyl chloride (1.05 eq.) in DCM were added pyridine
(2.1 eq.) and
the amine (1 eq.). The reaction mixture was stirred at rt or 40°C under
nitrogen for 4-14
h, then partitioned between ethyl acetate and 5% sodium bicarbonate solution
after TLC
showed completion of the reaction. The organic layer was washed with brine,
dried over
sodium sulphate, and concentrated in vacuo to give crude product that was
separated by
flash chromatography (ethyl acetate-DCM gradient elution) to give
arylsulphonamide and
N-arylsulphonyl arylsulphonamide as white or off-white solid (Yield 30-80%).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
94
N-(2-N-ethyl-2H-pyrazol-3-yl)-N-(4-n-propylphenylsulphonyl)-4-n-
propylbenzenesulfonamide (STX769, XDS01152)
White powder. TLC single spot at Rf 0.71 (10% ethyl acetate-DCM); HPLC purity
99% (tR
4.1 min in 10% water-methanol); 'H NMR (400 MHz, DMSO-d6): 5 7.62-7.66 (4H, m,
ArH), 7.56 (1 H, d, J = 2.3 Hz, ArH), 7.47-7.50 (4H, m, ArH), 5.93 (1 H, d, J
= 2.2 Hz,
ArH), 3.66 (2H, q, J = 7.0 Hz, NCH), 2.68 (4H, t J = 7.8 Hz, CH2) 1.63(4H, m,
2 x CH2),
1.18 (3H, t, J = 7.0 Hz, CH3), 0.90 (6H, t, J = 7.6 Hz, 2 x CH3); FAB-MS 476
(MH+); FAB-
HRMS calcd for C23H3pN3O4S2 (MH+) 476.1678, found 476.1682
N-(2,5-dimethyl-2H-prrazol-3-yl)-3-Chloro-2-methylbenzenesulfonamide (STX880.
XDS01174)
White crystalline solid. TLC single spot at Rf 0.76 (10% methanol-DCM); HPLC
purity as
rotational isomers 96% (tR 2.0 min in 20% water-methanol); 'H NMR (270 MHz,
DMSO-
d6): b 7.75 (2H, d, J = 8.2 Hz, ArH), 7.37 (1 H, d, J = 8.1 Hz, ArH), 5.49 (1
H, s, ArH),
3.48 (3H, s, CH3), 2.61 (3H, s, CH3), 1.98 (3H, s, CH3); APCI-MS 300 (MH+);
FAB-HRMS
calcd for C12H15CIN302S (MH+) 300.0573, found 300.0572
N-(1-Ethyl-1 H-pyrazol-3~r1)-3-Chloro-2-methylbenzenesulfonamide (STX881.
XDS01175C)
White solid. TLC single spot at Rf 0.80 (10% methanol-DCM); HPLC purity as
rotational
isomers 95% (tR 2.0 min in 20% water-methanol);'H NMR (270 MHz, DMSO-d6): 5
10.5
(1 H, s, NH), 7.71 (1 H, d, J = 8.1 Hz, ArH), 7.68 (1 H, d, J = 8.1 Hz, ArH),
7.32 (1 H, t, J =
8.1 Hz, ArH), 7.24 (1 H, broad s, ArH), 5.57 (1 H, d, J = 1.8 Hz, ArH), 3.88
(2H, q, J = 7.3
Hz, CH2), 2.57 (3H, s, CH3), 1.10 (3H, t, J = 7.3 Hz, CH3); APCI-MS 300 (MH+);
FAB-
HRMS calcd for C12H15CIN302S (MH+) 300.0573, found 300.0583
N-(1-Ethyl-1 H-pyrazol-3-yl)-N-(3-chloro-2-methyl~henylsulphonyl)-3-chloro-2-
methylbenzenesulfonamide (STX882, XDS01175B)
Colorless oil. TLC single spot at Rf 0.82 (20% ethyl acetate-DCM); HPLC purity
98% (tR
4.5 min in 20% water-methanol); 'H NMR (270 MHz, CDCI3): 5 7.95 (2H, d, J =
8.1 Hz,
ArH), 7.70 (2H, d, J = 8.1 Hz, ArH), 7.56 (1 H, d, J = 2.1 Hz, ArH), 7.32 (2H,
t, J = 8.1 Hz,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
ArH), 6.21 (1 H, d, J = 1.8 Hz, ArH), 3.71 (2H, q, J = 7.3 Hz, CHI), 2.47 (6H,
s, 2 x CH3),
1.28 (3H, t, J = 7.3 Hz, CH3); APCI-MS 488 (MH+); FAB-HRMS calcd for
C1 gH2pC12N304S2 (MH*) 488.0272, found 488.0263
5 5-(N-3-Chloro-2-methylphenylsulphonyl-f3-Chloro-2-methyl-
benzenesulfonylaminol)-1-
methyl-1 H-pyrazole-4-carboxylic acid ethyl ester (STX883, XDS01178A)
White crystalline solid. TLC single spot at Rf 0.81 (20% ethyl acetate-DCM);
HPLC purity
93% (tR 2.2 min in 15% water-methanol); 'H NMR (270 MHz, CDCI3): b 8.09 (2H,
dd, J =
10 8.1, 1.5 Hz, ArH), 7.95 (1 H, s, ArH), 7.63 (2H, dd, J = 8.1, 1.3 Hz, ArH),
7.30 (2H, t, J =
8.1 Hz, ArH), 3.88 (2H, q, J = 7.0 Hz, CHI), 3.54 (3H, s, CH3), 2.29 (6H, s, 2
x CH3), 0.96
(3H, t, J = 7.3 Hz, CH3); FAB-MS 546 (MH+); FAB-HRMS calcd for C21
H22C12N30gS2
(MH+) 546.0327, found 546.0320
15 5-(3-Chloro-2-methyl-benzenesulfonylamino)-1-methyl-1 H-pyrazole-4-
carboxylic acid
ethyl ester (STX884. XDS01178B)
Off-white solid. TLC single spot at Rf 0.49 (20% ethyl acetate-DCM); HPLC
purity 91
(tR 2.0 min in 20% water-methanol); 'H NMR (270 MHz, CDCI3): 5 7.60 (1 H, s,
ArH), 7.58
20 (1 H, dd, J = 7.9, 2.0 Hz, ArH), 7.50 (1 H, dd, J = 8.0, 2.0 Hz, ArH), 7.09
(1 H, t, J = 8.1
Hz, ArH), 3.91 (3H, s, CH3), 3.90 (2H, q, J = 7.0 Hz, CHZ), 2.50 (3H, s, CH3),
1.05 (3H, t,
J = 7.0 Hz, CH3); APCI-MS 358 (MH+); FAB-HRMS calcd for C14H17CIN304S (MH+)
358.0628, found 358.0640
25 3-Amino-1-(3-chloro-2-methylbenzenesulfonyl)-1 H-pyrazole-4-carboxylic acid
ethyl ester
(STX829, XDS01162A)
White crystalline solid. TLC single spot at Rf 0.25 (DCM); HPLC purity > 99%
(tR 2.0 min
in 4% water-methanol); ~H NMR (270 MHz, DMSO-d6): b 8.62 (1 H, s, ArH), 7.95
(1 H, d,
30 J = 8.2 Hz, ArH), 7.86 (1 H, d, J = 8.2 Hz, ArH), 7.48 (1 H, t, J = 8.2 Hz,
ArH), 5.95 (2H, s,
NH2), 4.17 (2H, q, J = 7.0 Hz, CHI), 2.49 (3H, s, CH3), 1.21 (3H, t, J = 7.0
Hz, CH3);
APCI-MS 344 (MH+); FAB-HRMS calcd for C13H15CIN3O4S (MH+) 344.0472, found
344.0477
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
96
5-Amino-1-(3-chloro-2-methyl-benzenesulfonyl)-1 H-pyrazole-4-carboxylic acid
ethyl ester
(STX830, XDS01162B)
Off-white crystalline solid. TLC single spot at Rf 0.30 (DCM); HPLC purity 91
% (tR 2.0
min in 4% water-methanol); 'H NMR (270 MHz, DMSO-d6): 5 7.97 (1 H, d, J = 8.0
Hz,
ArH), 7.87 (1 H, d, J = 8.0 Hz, ArH), 7.77 (1 H, s, ArH), 7.50 (1 H, t, J =
8.0 Hz, ArH), 7.03
(2H, s, NH2), 4.14 (2H, q, J = 6.9 Hz, CH2), 2.50 (3H, s, CH3), 1.18 (3H, t, J
= 6.9 Hz,
CH3); FAB-MS 344 (MH+); FAB-HRMS calcd for C13H15CIN3O4S (MH+) 344.0472,
found 344.0472
N-f 1-(3-Chloro-2-methylbenzenesulfonyl)-1 H-indazol-5-yll-3-chloro-2-
methylbenzenesulfonamide (STX826, XDS01159)
Off-white foam. TLC single spot at Rf 0.65 (10% ethyl acetate-DCM); HPLC
purity 96%
(tR 2.2 min in 15% water-methanol); 'H NMR (270 MHz, DMSO-d6): b 10.7 (1 H, s,
NH),
9.01 (1 H, s, 3-H), 8.05 (1 H, d, J = 8.1 Hz, ArH), 7.84 (1 H, d, J = 8.0 Hz,
ArH), 7.80 (1 H,
d, J = 8.0 Hz, ArH), 7.62 (1 H, d, J = 8.0 Hz, ArH), 7.51 (1 H, d, J = 8.0 Hz,
ArH), 7.49
(1 H, t, J = 8.0 Hz, ArH), 7.29 (1 H, t, J = 8.0 Hz, ArH), 7.24 (1 H, d, J =
1.1 Hz, ArH),. 7.11
(1 H, dd, J = 8.0, 1.2 Hz, ArH), 2.36 (6H, s, 2 x CH3); APCI-MS 510 (MH+); FAB-
HRMS
calcd for C21 H18C12N304S2 (MH+) 510.0116, found 510.0106
3-Chloro-N-(1 H-indazol-5-yl)-2-methyl-benzenesulfonamide (STX983, XDS02021 B)
Off-white solid. TLC single spot at Rf 0.46 (6% methanol-DCM); HPLC purity >
99% (tR
2.0 min in 10% water-methanol); 'H NMR (270 MHz, DMSO-d6): b 10.4 (1H, s, NH),
7.99 (1 H, s, 3-H), 7.79 (1 H, d, J = 7.9 Hz, ArH), 7.68 (1 H, d, J = 7.9 Hz,
ArH), 7.43 (1 H,
d, J = 8.9 Hz, ArH), 7.41 (1 H, s, ArH), 7.32 (1 H, t, J = 8.0 Hz, ArH), 7.08
(1 H, d, J = 8.9
Hz, ArH),. 2.64 (3H, s, CH3); APCI-MS 322 (MH+); FAB-HRMS calcd for
C14H13CIN3O2S (MH+) 322.0417, found 322.0417
3-Chloro-2-methyl-N-(1-methyl-1 H-indazol-5-yl)-benzenesulfonamide (STX988,
XDS02037)
Off-white solid. TLC single spot at Rf 0.46 (7% methanol-DCM); HPLC purity 96%
(tR
2.2 min in 10% water-methanol); 'H NMR (270 MHz, DMSO-d6): b 10.4 (1 H, s,
NH),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
97
7.95 (1 H, s, 3-H), 7.78 (1 H, d, J = 7.9 Hz, ArH), 7.68 (1 H, d, J = 7.9 Hz,
ArH), 7.52 (1 H,
d, J = 9.0 Hz, ArH), 7.40 (1 H, J = 2.0 Hz, ArH), 7.31 (1 H, t, J = 7.9 Hz,
ArH), 7.12 (1 H,
dd, J = 9.0, 2.0 Hz, ArH),. 3.97 (3H, s, NCH3), 2.65 (3H, s, CH3); APCI-MS 336
(MH+);
FAB-HRMS calcd for C15H15CIN302S (MH+) 336.0573, found 336.0575
Synthesis of benzamides and arysulphonamides
w H ci I w I
N ~ H i N ~ N
I / N ~ N o I ~ s
STX471, DGS03074B STX472, DGS03076A
STX520, DGS03080A
N O~
o ~ s0 I
W S~N ~ \ S~ '
N
I~ "~N~ ~I / H
pS O
STX576, DGSO3os4A STX577, DGS03096A
\ OS N \ O O N O~
I '' ~~ I
I / H N / \
N
STX702, DGS03132A I / H
STX770, XDS01153
N-(2-Methylbenzothiazol-5-yl)-4-propylbenzamide (STX471, DGS030748)
To a solution of 5-amino-2-methylbenzothiazole (150 mg, 0.91 mmol) in THF (1
mL) was
added triethylamine (5 mL). After stirring at rt for 15 min, 4-propylbenzoyl
chloride(200
mg, 1.09 mmol) was added. The mixture was kept stirring at rt for 1 h,
extracted with
ethyl acetate. The organic phase was washed with brine, dried over sodium
sulphate
and concentrated in vacuo to yield a white solid that was recrystallized from
ethyl acetate
to give white needles (216 mg, 76%). mp 148-149°C; TLC single spot at
Rf 0.56 (60%
ethyl acetate-hexane); HPLC purity 99% (tR 3.1 min in 10% water-methanol); 'H
NMR
(400 MHz, DMSO-d6): b 10.3 (1 H, s, NH), 8.41 (1 H, d, J = 2.0 Hz, ArH), 7.88-
7.96 (3H,
m, ArH), 7.50 (1 H, dd, J = 9.0, 2.3 Hz, ArH), 7.33-7.36 (1 H, m, ArH), 2.78
(3H, s, CH3),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
98
2.63 (2H, t J = 7.4 Hz, CHI) 1.62(2H, m, CH2), 0.98 (3H, t, J = 7.4 Hz, CH3);
FAB-MS
311 (MH+); FAB-HRMS calcd for C18H1gN2OS (MH+) 311.1218, found 311.1227.
3,5-Dichloro-N-(2-methylbenzothiazol-5-yl)-benzamide (STX520. DGS03080A)
STX520 was synthesized as described for STX471. White crystalline solid (166
mg,
54%) was obtained. mp 194°C; TLC single spot at Rf 0.76 (60% ethyl
acetate-hexane);
HPLC purity 97% (tR 1.6 min in 4% water-methanol); 'H NMR (400 MHz, DMSO-d6):
~
10.6 (1 H, s, NH), 8.40 (1 H, d, J = 2.0 Hz, ArH), 7.99-8.02 (3H, m, ArH),
7.89-7.90 (1 H,
m, ArH), 7.74 (1 H, dd, J = 8.9, 2.3 Hz, ArH), 2.80 (3H, s, CH3); FAB-MS 337
(MH+);
FAB-HRMS calcd for C15H11CI2N2OS (MH+) 336.9969, found 336.9972.
N-Methyl-N-(2-methylbenzothiazol-5-yl)-4-propylbenzamide (STX472, DGS03076A)
To a solution of N-(2-methylbenzothiazol-5-yl)-4-propylbenzamide (130 mg, 0.4
mmol) in
DMF ( 5 mL) was added sodium hydride (17 mg, 0.44 mmol), followed by methyl
iodide
(85 mg, 059 mmol). The mixture was stirred at rt overnight, partitioned
between water
and ethyl acetate. The organic phase was washed with brine, dried over sodium
sulphate and concentrated to give a residue that wad purified with flash
chromatography
(ethyl acetate-hexane gradient elution). A thick syrup (41 mg, 26% was
obtained. 'H
NMR (400 MHz, CDC13): S 7.61-7.79 (3H, m, ArH), 7.22-7.26 (2H, m, ArH), 6.92-
7.02
(2H, m, ArH), 3.54 (3H, s, CH3). 2.81 (3H, s, CH3), 2.45 (2H, t J = 7.4 Hz,
CH2) 1.52(2H,
m, CHI), 0.83 (3H, t, J = 7.4 Hz, CH3); FAB-MS 325 (MH+); FAB-HRMS calcd for
C1gH21N20S (MH+) 325.1375, found 325.1992.
General Method for synthesis of arylsulphonamide (, STX576-577, . STX702.
STX770)
To a solution of amine (1 eq.) in DMF was added Et3N (5 eq.), followed by
corresponding
sulphonyl chloride (1.2 eq.). The reaction mixture was stirred at rt under N~
overnight,
poured into water after TLC showed completion of the reaction, and extracted
with ethyl
acetate, dried (MgS04), concentrated under reduced pressure to give the
desired
sulphonamide as crystalline solid or as a thick syrup. The crude compound was
then
purified by flash chromatography using EtOAc/hexane (3:2) or CH2CI2/EtOAc
(4:1) as
eluent to give crystalline solid. Yield 20-80%.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
99
4-Propel-N-f 1-(4-propylbenzenesulfonyl)-pi~eridin-4-ylmethyll-
benzenesulfonamide
(STX576, DGS03094A)
White solid. mp 158-159°C; TLC single spot at Rf 0.59 (20% ethyl
acetate-DCM); HPLC
purity 99% (tR 2.0 min in 4% water-methanol); 'H NMR (400 MHz, DMSO-d6): 5
7.60
7.66 (4H, m, ArH), 7.55 (1 H, broad, NH), 7.35-7.45 (4H, m, ArH), 3.55-3.58
(2H, dd, J =
7.2, 2.3 Hz, CH2), 2.56-2.67 (6H, m, 3 x CHZ), 2.04-2.10 (2H, m, CH2), 1.54-
1.65 (6H, m,
3 x CHI), 1.23 (1 H, m, CH), 1.02-1.11 (2H, m, CHI), 0.85-0.89 (6H, m, 2 x
CH3); FAB
MS 479 (MH+).
N-(6-Methoxypyridin-3-yl)-4-propylbenzenesulfonamide (STX577, DGS03096A)
White solid. mp 93-94°C; TLC single spot at Rf 0.54 (20% ethyl acetate-
DCM); HPLC
purity 99% (tR 2.0 min in 4% water-methanol); 'H NMR (400 MHz, DMSO-d6): b
10.0
(1 H, s, NH), 7.77 (1 H, d, J = 2.7 Hz, ArH), 7.56-7.59 (2H, m, ArH), 7.35-
7.38 (3H, m,
ArH), 6.72 (1 H, m, ArH), 3.75 (3H, s, OCH3), 2.59 (2H, t, J = 7.4 Hz, CH2),
1.61 (2H, m,
CHI), 0.85 (3H, t, J = 7.4 Hz, CH3); FAB-MS 307 (MH+); FAB-HRMS calcd for
C15H19N203S (MH+) 307.1048, found 307.1061.
4-Propel-N-(pyridin-2-yl-methyl)-benzenesulfonamide (STX702, DGS03132A)
White crystalline solid. mp 109°C; TLC single spot at Rf 0.50 (20%
ethyl acetate-DCM);
HPLC purity > 99% (tR 1.8 min in 4% water-methanol);'H NMR (400 MHz, DMSO-d6):
5
8.41 (1 H, m, ArH), 8.19 (1 H, s, NH), 7.66-7.72 (3H, m, ArH), 7.31-7.37 (3H,
m, ArH),
7.22 (1 H, m, ArH), 4.06 (2H, s, CHI), 2.61 (2H, t, J = 7.0 Hz, CH2), 1.60
(2H, m, CH2),
0.88 (3H, t, J = 7.4 Hz, CH3); FAB-MS 291 (MH+); FAB-HRMS calcd for C15H1
gN202S
(MH+) 291.1167, found 291.1164.
N-(2,6-Dimethoxypyridin-3-yl)-4-propylbenzenesulfonamide (STX770, XDS01153)
Off-white crystalline solid. TLC single spot at Rf 0.37 (30% ethyl acetate-
hexane); HPLC
purity 98% (tR 2.7 min in 10% water-methanol);'H NMR (400 MHz, DMSO-d6): b
9.39
(1 H, s, NH), 7.48-7.54 (2H, m, ArH), 7.42 (1 H, d, J = 8.8 Hz, ArH), 7.33
(2H, d, J = 8.6
Hz, ArH), 6.30 (1 H, d, J = 8.8 Hz, ArH), 3.76(3H, s, OCH3), 3.45(3H, s,
OCH3), 2.60 (2H,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
100
t, J = 7.8 Hz, CHZ), 1.55 (2H, sextet, J = 7.8 Hz, CH2), 0.88 (3H, t, J = 7.8
Hz, CH3);
FAB-MS 291 (MH+); FAB-HRMS calcd for C~gH~~N2O4S (MH+) 337.1222, found
337.1222.
Synthesis of N-Benzimidazole Aryl Amide Derivatives
O I ~ N~- O ~ N O ~ N
N i N i~ i
H \ i N i N s N i N
I ~ \ ~ I H ~ I H
I
STX1129, JWS01109 STX 1130, JWS01111 ' STX1131, JWS01113
/ / /
I ~ ~ ~ I ~ N~-- ~ I
/ I H N / I I N / I I N
CI \ CI I ~ \ CI ~ CI
STX1132, JWS01116 STX1134, XDS02113
STX1133,XDS02112
General method for benzamide formation:
To a solution of substituted benzoyl chloride (1.2 eq.) in THF-triethylamine
(1 : 4) was
added the corresponding amine (1 eq.). The reaction miXture was stirred at rt
under
nitrogen for 16 h, partitioned between DCM and 5% sodium bicarbonate after TLC
showed completion of the reaction. The organic layer was washed with brine,
dried over
sodium sulphate, and concentrated in vacuo to give crude product as solid or
thick
syrup. The compound was then purified by flash chromatography (Methanol-DCM
gradient elution) to give desired N-benzimidazole aryl amide as crystalline
solid. Yield
ranges from 60-80%.
Biphenyl-4-carboxylic acid (1.2-dimethyl-3H-benzoimidazol-6-yl)-amide
(STX1129,
JWS01109)
Off-white solid. Mp 270-270.3°C; TLC single spot at Rf 0.52 (10%
methanol/DCM);
HPLC purity 98% (tR 2.2 min in 20 % water-methanol); 'H NMR (400 MHz, DMSO): b
10.3 (1 H, s, NH), 8.05-8.10 (3H, m, ArH), 7.82 (2H, d, J = 8.2 Hz, ArH), 7.75
(2H, dd, J =
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
101
8.1, 1.2 Hz, ArH), 7.38-7.52 (5H, m, ArH), 3.70 (3H, s, NCH3), 2.52 (3H, s,
CH3); FAB-
MS 342 (MH+); FAB-HRMS calcd for C22H20N30 (MH+) 342.1606, found 342.1615
Biphenyl-4-carboxylic acid (1,2-dimethyl-3H-benzoimidazol-5-yl)-amide
(STX1130,
JWS01112)
Off-white solid. Mp 272-274°C; TLC single spot at Rf 0.52 (10%
methanol/DCM); HPLC
purity 98% (tR 2.2 min in 20% water-methanol); 'H NMR (400 MHz, DMSO): 5 10.2
(1 H,
s, NH), 8.06 (2H, d, J = 8.2 Hz, ArH), 7.95 (1 H, d, J = 2.0 Hz, ArH), 7.82
(2H, d, J = 8.2
Hz, ArH), 7.74 (2H, dd, J = 8.2, 1.9 Hz, ArH), 7.48-7.56 (5H, m, ArH), 3.72
(3H, s,
NCH3), 2.52 (3H, s, CH3); FAB-MS 342 (MH+); FAB-HRMS calcd for C22H20N30 (MH+)
342.1606, found 342.1611
N-(1,2-Dimethyl-1H-benzoimidazol-5-yl)-4-propylbenzamide (STX1131, JWS01113)
Off-white solid. Mp 214-218°C; TLC single spot at Rf 0.43 (10%
methanol/DCM); HPLC
purity > 99% (tR 2.2 min in 20% water-methanol); 'H NMR (400 MHz, DMSO): S
10.1
(1 H, s, NH), 7.95 (1 H, d, J = 1.5 Hz, ArH), 7.87 (2H, d, J = 8.2 Hz, ArH),
7.51 (1 H, dd, J
= 8.5, 2.0 Hz, ArH), 7.39 (1 H, d, J = 8.2 Hz, ArH), 7.32 (2H, d, J = 8.5 Hz,
ArH), 3.70
(3H, s, NCH3), 2.63 (2H, t, J = 7.1 Hz, CH2), 2.50 (3H, s, CH3), 1.62 (2H,
sextet, J = 7.2
Hz, CH2), 0.91 (3H, t, J = 7.3 Hz, CHZ); FAB-MS 308 (MH+); FAB-HRMS calcd for
C1gH22N30 (MH+) 308.1763, found 308.1778
2,4-Dichloro-N-(1,2-dimethyl-1H-benzoimidazol-5-yl)-benzamide (STX1132,
JWS01116)
light yellow solid. Mp 275-277°C; TLC single spot at Rf 0.43 (10%
methanol/DCM);
HPLC purity > 99% (tR 2.0 min in 10% water-methanol); 'H NMR (400 MHz, DMSO):
5
10.4 (1 H, s, NH), 7.92 (1 H, d, J = 2.0 Hz, ArH), 7.74 (1 H, d, J = 1.9 Hz,
ArH), 7.62 (1 H,
d, J = 8.2 Hz, ArH), 7.53 (1 H, dd, J = 8.2, 2.0 Hz, ArH), 7.39-7.42 (2H, m,
ArH), 3.70
(3H, s, NCH3), 2.50 (3H, s, CH3); FAB-MS 334 (MH+); FAB-HRMS calcd for
C16H14C12N30 (MH+) 334.0514, found 334.0517
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
102
Biphenyl-4-carboxylic acid (1,2-dimethyl-3H-benzoimidazol-5-yl)-N-methyl-amide
(STX1133, XDS02112)
White crystals. Mp 213-214.5°C; TLC single spot at Rf 0.79 (10%
methanol/DCM);
HPLC purity 99% (tR 2.2 min in 20% water-methanol); 'H NMR (270 MHz, DMSO): 8
7.58 (2H, m, ArH), 7.30-7.50 (9H, m, ArH), 7.07 (1 H, dd, J = 8.1, 1.9 Hz,
ArH), 3.65 (3H,
s, NCH3), 3.29 (3H, s, NCH3), 2.45 (3H, s, CH3); APCI-MS 356 (MH+)
2,4-Dichloro-N-(1,2-dimethyl-1H-benzoimidazol-5-yl)-N-methyl-benzamide
(STX1134,
XDS02113)
White crystals. Mp 245-247°C; TLC single spot at Rf 0.70 (10%
methanol/DCM); HPLC
purity > 99% (tR 2.2 min in 10% water-methanol); 'H NMR (270 MHz, DMSO): 5
7.44
(1 H, d, J = 2.0 Hz, ArH), 7.41 (1 H, broad, VI/"2 = 1.7 Hz, ArH), 7.39 (1 H,
d, J = 8.2 Hz,
ArH), 7.35 (1 H, d, J = 8.5 Hz, ArH), 7.22 (1 H, dd, J = 8.2, 1.9 Hz, ArH),
7.09 (1 H, dd, J =
8.5, 1.9 Hz, ArH), 3.64 (3H, s, NCH3), 3.39 (3H, s, NCH3), 2.45 (3H, s, CH3);
APCI-MS
348 (MH+)
The compounds shown in the following table were synthesised in the manner
described.
STX COMPOUND STRUCTURE
CODE CODE
ci
N
1264 XDS03019 0 \ N
\ //
/~ ~ N ~ N
0 H
O
1317 XDS03047B II-NH
O
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
103
STX COMPOUND STRUCTURE
CODE CODE
CI
0
1318 XDS03048 ~ -NH
O
CI
CI
O
1319 XDS03049
II-NH ~S
O
0
NH
1320 XDS03050
0
s
0
0
1321 XDS03051
// ~H i
0
s
0
s
1327 XDS03061 A ~ I ~-N
II
0
s
0
1328 XDS03061 B ~ ~ ~ -NH _
S
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
104
STX COMPOUND STRUCTURE
CODE CODE
ci
\
o \
s
1329 XDS03062A ~ ~ I ~-N
s
c1
0
1330 XDS03062B ~ ~ ~ ~-NH
\ S
CI
O
S
1331 XDS03063A ~ -N
\ s
cr
0
-NH
1332 XDS03063B
0
s
c~
! -N \ S
1333 XDS03064A
~s
0
-NH
1334 XDS03064B ~ ~s
0
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
105
STX COMPOUND STRUCTURE
CODE CODE
o H
\\ sN
1335 XDS03065 \ \\
s
O H
N
1336 XDS03066 I \ \\
/ /
ci
s
O H
\\,N
1337 XDS03067
s
ci
O H
\\ rN
1338 XDS03068 /
a
s
ci
/ ~s
i
1339 XDS03070 ~ ~ s
ci
0
ci \\ /
1340 XDS03071 B ~ \~ ~ s
a
~ci
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
106
STX COMPOUND STRUCTURE
CODE CODE
o / s
CI
w
\ N
1355 CCM01002 / H
\ c1
c1
o / s
1356 CCM01003 / N ~ N
H
\ CI
CI
1357 CCM01004
H
N N
o \
s
s
CI O
i N \
1358 CCM01006 ( H
\ c1
c1
/o \ o \ s
1363 CCM01008
/ N / N
H
CI
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
1~7
STX COMPOUND STRUCTURE
CODE CODE
c1
H
\ N \ N
1364 CCM01009
\ ~ ° ~ s
c1
H
1365 CCM01010 \° ~ N \ \
s
/o
\ c1
H
1366 CCM01011 / N \
/ s
\ o \ s
1367 CCM01012
\O ~ N ~ N
H
CI
CI
H
N N
1376 CCM01013 ~ ~ \
0
c1 / / s
\ ~ \ °
1377 CCM01015 ~ N s
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
10~
STX COMPOUND STRUCTURE
CODE CODE
ci
HN
1378 CCM01016
0
ci
o s
1379 CCM01017 \ ~N
H
\O
O
1380 CCM01018 HN g
O
O S
1381 CCM01020
\ ~N
H
CI
1382 CCM01021
ci ~ /
o s~
c1 c1
H
N N
1396 CCM01022
0
ci
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
109
STX COMPOUND STRUCTURE
CODE CODE
c1
1397 CCM01023 0
ci
0
0
1398 CCM01024 ~ ~ HN
0
1399 CCM01025 ~ ~ o
HN
CI
O
\ O
1400 CCM01026 ~ HN
0
0
1401 CCM01027 HN
0
0
1402 CCM01028 ~ o
HN
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
110
ST~C COMPOUND STRUCTURE
CO DE CODE
O
s
1405 XDS03101 ~ ~N
H I
O
CI g
\ N
1406 XDS03102 I H I
CI
s
1407 XDS03103
HN
O
S
1408 XDS03104 ~ ~N
H I
\O
O
O S
1409 XDS03105 ~ ~ ~N
H I
O
-N H
1414 XDS03111
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
111
STX COMPOUND STRUCTURE
CODE CODE
c1
0
1415 XDS03112 ~ \ ~ NH g
O
CI
O
II-NH S
1416 XDS03113
\ /
0
w
1417 XDS03114
0
c1
~N
1418 XDS03115
a
w
1419 XDS03116 / H
1430 CCM01029
HN
O
0
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
112
STX COMPOUND STRUCTURE.
CODE CODE
c1 ci
/ o
1431 CCM01031 ~ / \
\ /
O N
H
0
O /
1432 CCM01032
/° / N \ /
H
CI
O
1433 CCMO1034 c1
o N
H
O
o / ~ \
1434 CCM01036
\ N \
H
O
° ~ \
1435 CCM01037
\ \N
H
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
113
STX COMPOUND STRUCTURE
CODE CODE
ci
H
N N
1445 CCM01038
ci
ci
H
1446 CCM01029 c~ ~ s~N ~ N
s
HN
1461 CCM01040A /
0
0
ci
HN
1462 CCM01041 ~ ~ ~ ~ o
W
0
N
1463 CCM01042
o
HN
1464 CCM01043 ~ \ \ / o
'o
ci
H
1465 CCM01044 ~ N
ci o
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
114
STX COMPOUND STRUCTURE
CODE CODE
w
H
N
1468 CCM01047
0
H
\O
1469 CCM01048
o
1470 CCM01049 ~ ~ ~ ~ HN
0
0
Cl -
H
1472 CCM01050 ~, ~ N
0
1473 CCM01051 ~ ~ HN
O
'V
N
1474 CCM01052
0
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
115
STX COMPOUND
STRUCTURE
CODE CODE
H
~O ~ N
1475 CCM01052
0
Synthesis of N-(2-thiopheneethyl)-benzamide derivatives
CI O
o I H
I H W N S
w N I s CI ~ N I ~ O
O ~..~ O
STX1377, CCM01015 STX1378, CCM01016 STX1379, CCM01017
CI
~O \ I N S \ I N S W I N
O I ~ O I / I /
CI O
STX1380, CCM01018 STX1381, CCM01020 STX1382, CCM01021
General method for synthesis of N-(2-thiopheneethyl)-benzamide
derivatives (STX1377-'1382):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the acyl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers are washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers are then washed with brine, dried (MgS04),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
116
Biphenyl-4-carbox lic acid 2-thiophen-2-yl-ethyl)-amide (STX1377, CCMO1015)
Reaction of 2-thiopheneethylamine (SO ~L, 0.42 mmol) in THF (2.5 mL) with 4-
biphenylcarbonyl chloride (120 mg, 0.55 mmol) in presence of triethylamine (78
~L, 0.55
mmol) according to method A gave biphenyl-4-carboxylic acid (2-thiophen-2-yl-
ethyl)-
amide (70 mg, 0.23 mmol, 55% yield) as a grey powder after crystallisation in
hexane/EtOAc.
Rf : 0.4 (EtOAc/DCM 5/95) ; M.p.:164-166 °C ; 1H NMR (270MHz, CDCl3) 8H
3.16 (t,
2H, J = 6.6 Hz, CH2Ar), 3.71-3.77 (m, 2H, CH2NH), 6.27 (bs, 1H, NH), 6.88 (bs,
1H,
Hue.), 6.95-6.98 (m, 1H, HA,.), 7.17 (d, 1H, J = 4.7 Hz, HA,.thiophene), 7.36-
7.47 (m, 3H,
Hue), 7.57-7.64 (m, 4H, Hue), 7.77 (d, 2H, J = 8.1 Hz, HA,.); 13C NMR (400MHz,
CDC13)
SC 30.0 (CH2), 41.4 (CH2), 124.2 (CHI.), 125.6 (CHI), 127.2 (2*CH~), 127.3
(CHI),
127.4 (CHI.), 128.0 (CHI.), 129.0 (CHI.), 133.2 (Cq), 140.0 (Cq), 141.3 (C9),
144.3 (Cq),
167.7 (C=O); LC/MS (AP-) n2/z 305.8 (M-H); tR = 2.3 min (99.6%); HRMS (FAB+)
Calculated for C19H1~NOS 307.1031; Found 307.1016
3,5-Dichloro-N (2-thiophen-2-yl-ethyl)-benzamide (STX1378, CCM01016)
Reaction of 2-thiopheneethylamine (50 ~L, 0.42 mmol) in THF (2.5 mL) with 3,5
dichlorobenzoyl chloride (120 mg, 0.57 mmol) in presence of triethylamine (78
~L, 0.55
mmol) according to method A gave 3,5-dichloro-N (2-tluophen-2-yl-ethyl)-
benzamide
(118 mg, 0.39 mmol, 92°J° yield) as a white powder after
purification by chromatography
on silica gel (eluent : EtOAc/hexane 2/8).
Rf : 0.3 (EtOAc/hexane 2/8); M.p.:100-102 °C; 1H NMR (270MHz, CDC13) 8H
3.13 (t,
2H, J = 6.4 Hz, CH2-Ar), 3.69 (dt, 2H, J = 6.4, 6.4 Hz, CH2NH), 6.24 (bs, 1H,
NH), 6.85
(dd, 1 H, J = 1.0, 3 .5 Hz, H~.thiophene), 6.96 (dd, 1 H, J = 3.5, 5.2 Hz,
H~.thiophene), 7.18
(dd, 1H, J = 1.0, 5.2 Hz, H~.thiophene), 7.45 (t, 1H, J = 2.0 Hz, HA,.), 7.56
(d, 2H, J = 2.0
Hz, HA,.); LC/MS (AP-) m/z 297.8 (M-H); tR = 2.5 min (99.3%); HRMS (FAB+)
Calculated for C13H1iC12NOS 298.9938; Found 298.9934
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
117
4-Methoxy-N (2-thiophen-2 yl-ether)-benzamide (STX1379, CCM01017)
Reaction of 2-thiopheneethylamine (50 ~,L, 0.42 mmol) in THF (2.5 mL) with p-
anisoyl
chloride (74 ~,L, 0.57 mmol) in presence of triethylamine (78 ~,L, 0.55 mmol)
according
to method A gave 4-methoxy-N (2-thiophen-2-yl-ethyl)-benzamid (105 mg, 0.40
mmol,
95% yield) as a white powder after purification by chromatography on silica
gel (eluent
EtOAc/hexane 5/95 to 20/80).
Rf : 0.4 (EtOAc/DCM 1/9) ; M.p.: 109-111 °C ; 1H NMR (270MHz, CDCl3) 8H
3.13 (t,
2H, J = 6.4 Hz, CH2Ar), 3.69 (dt, 2H, J = 6.4, 6.4 Hz, CH2NH), 3.82 (s, 3H,
OCH3), 6.19
(bs, 1H, NH), 6.85-6.90 (m, 3H, Hue.), 6.94 (dd, 1H, J = 3.5, 5.4 Hz,
H~.thiophene), 7.16
(dd, 1H, J = 1.2, 5.4 Hz, HA,.thiophene), 7.65-7.70 (m, 2H, H,~.); LC/MS (AP-)
m/z 259.9
(M-H); tR = 2.0 min (95.4%); HRMS (FAB+) Calculated for Cl4HisNOaS 261.0824;
Found 261.0826
3-Methox~2-thiophen-2yl-ethyl)-benzamide (STX1380, CCM01018)
Reaction of 2-thiopheneethylamine (50 ~L, 0.42 mmol) in THF (2.5 mL) with m-
anisoyl
chloride (74 ~,L, 0.57 mmol) in presence of triethylamine (78 ~,L, 0.55 mmol)
according
to method A gave 3-methoxy-N (2-thiophen-2-yl-ethyl)-benzamide (100 mg, 0.38
mmol,
90% yield) as a yellow wax after purification on silica gel (eluent :
EtOAc/hexane 5/95 to
20/80).
Rf : 0.3 (EtOAc/DCM 5/95); 1H NMR (270MHz, CDCl3) 8H 3.13 (t, 2H, J = 6.4 Hz,
CH2Ar), 3.70 (dt, 2H, J = 6.4, 6.4 Hz, CHaNH), 3.81 (s, 3H, OCH3), 6.27 (bs,
1H, NH),
6.85 (dd, 1 H, J = 1.2, 3.4 Hz, H~thiophene), 6.94 (dd, 1 H, J = 3 .5, 5.2 Hz,
HA,.thiophene),
7.00 (ddd, 1 H, J =1.3, 2.8, 8.2 Hz, HA,.), 7.16 (dd, 1 H, J = 1.2, 5.2. Hz,
HA,.thiophene),
7.19 (ddd, 1H, J = 1.3, 1.3, 7.7 Hz, Hue.), 7.24-7.32 (m, 2H, H~,.); LC/MS (AP-
) m/z 259.9
(M-H), tR = 2.0 min (99.5%); HRMS (FAB+) Calculated for C14H1sN02S 261.0824;
Found 261.0827
4-Propyl-N (2-thiophen-2-~hyl)-benzamide (STX1381, CCM01020)
Reaction of 2-thiopheneethylamine (50 ~L, 0.42 mmol) in THF (2.5 mL) with 4
propylbenzoyl chloride (92 p,L, 0.55 mmol) in presence of triethylamine (78
~.L, 0.55
mmol) according to method A gave 4-propyl-N (2-thiophen-2-yl-ethyl)-benzamide
(100
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
118
mg, 0.36 mmol, 85% yield) as a off white powder after purification on silica
gel (eluent
EtOAc/hexane 5/95 to 20/80).
Rf: 0.4 (EtOAc/DCM 5/95) ; M.p.: 97-99 °C ; 1H NMR (270MHz, CDCl3) ~H
0.93 (t, 3H,
J = 7.4 Hz, CH3), 1.55-1.69 (m, 2H, CH2-GH3), 2.60 (t, 2H, J = 7.4 Hz, CH2Ar),
3.13 (t,
2H, J = 6.4 Hz, CH2thiophene), 3.70 (dt, 2H, J = 6.4, 6.4 Hz, CH2NH), 6.22
(bs, 1H, NH),
6.85 (d, 1H, J = 3.4 Hz, H~.thiophene), 6.94 (dd, 1H, J = 3.5, 5.0 Hz,
HA,.thiophene), 7.16
(dd, 1 H, J =1.0, 5.2. Hz, H~.thiophene), 7.20 (d, 2H, J = 8.1 Hz, HA,.), 7.62
(d, 2H, J = 8.1
Hz, Hue.); LC/MS (AP-) m/z 271.8 (M-H); tR = 2.4 min (95.8%); HRMS (FAB+)
Calculated for C16Hi9NOS 273.1187; Found 273.1176
2 5-Dichloro-N (2-thiophen-2-yl-ethy_l~-benzamide (STX1382 CCM01021)
Reaction of 2,5-dichlorobenzoic acid (164 mg, 0.85 mmol) in thionyl chloride
(2 mL)
then with 2-thiopheneethylamine (50 pL, 0.42 mmol) in presence of
triethylamine (0.5
mL) in THF (3 mL) according to method B gave 2,5-dichloro-N (2-thiophen-2-yl-
ethyl)-
benzamide (83 mg, 0.28 mmol, 66% yield) as a off white powder after
purification on
silica gel (eluent : EtOAc/hexane 5/95 to 20/80).
Rf: 0.2 (EtOAc/hexane 2/8); M.p.: 99-100 °C; 1H NMR (270MHz, CDCl3) 8H
3.15 (t, 2H,
J = 6.6 Hz, CH2thiophene), 3.73 (dt, 2H, J = 6.6, 6.6 Hz, CH2NH), 6.33 (bs,
1H, NH),
6.86 (d, 1 H, J = 3.4 Hz, H~.thiophene), 6.94 (dd, 1 H, J = 3.5, 5.1 Hz,
H~.thiophene), 7.15
(dd, 1 H, J = 1.2, 5.1. Hz, HE".thiophene), 7.28 (d, 2H, J = 8.1 Hz, HA,.), 7.
S 8-7.59 (m, 1 H,
Hue); 13C NMR (400MHz, CDCl3) 8e 29.7 (CHZ-thiophene), 41.6 (CH2NH), 124.2
(CHA,.), 125.7 (CHA,.), 127.2 (CHI.), 128.9 (Cq), 130.2 (CHI), 131.3 (CHI.),
131.5
(CH,~,.), 133.3 (Cq), 136.2 (Cg), 140.9 (Cq), 165.1(C=0); LC/MS (AP-) fnlz
298.0 (M
H); tR = 2.2 min (99.9%); HRMS (FAB+) Calculated for C13H11C12NOS 298.9938;
Found
298.9933
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
119
Synthesis of N-(5-indanone)-benzamide derivatives
CI o I CI
H o 0
H
\ I N \ \ I N \ \ I N \
CI
CI O I o ~ I o O I o
O p O
STX1397, CCM01023 STX1398, CCM0101624 STX1399, CCM01025
O
o H
\ I N \ \ I N \ ~O \ I N \
O I o O .I o O I o
O O
STX1400, CCM01026 STX1401, CCM01027 STX1402, CCM01028
General method for synthesis of N-(5-indanone)-benzamide derivatives
(STX1397-1402):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the acyl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers are washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers are then washed with brine, dried (MgS04),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
2,5-Dichloro-N (1-oxo-indan-5-yl)-benzamide (STX1397 CCM01023)
Reaction of 2,5-dichlorobenzoic acid (287 mg, 1.50 mmol) in thionyl chloride
(3.5 mL)
then with 5-amino-indan-1-one (74mg, 0.50 rillnol) in presence of
triethylamine (0.5 mL)
in THF (6 mL) according to method B gave 2,5-dichloro-N (1-oxo-indan-S-yl)-
benzamide
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
120
(33 nzg, 0. 09 ~rz~raol, 35% yield) as a green powder after purification by
chromatography on
silica gel (eluent : DCM).
Rf: 0.4 (EtOAc/DCM 1/9) ; M.p.: 218-221 °C ; 1H NMR (270MHz, DMSO-d6)
bH 2.58
2.62 (m, 2H, CH2), 3.06-3.10 (m, 2H, CH2), 7.57-7.60 (m, 4H, Hue.), 7.77-7.78
(m, 1H,
HA,.), 8.01 (s, 1 H, H,e,,.), 10.94 (s, 1 H, NH); LC/MS (AP-) m/z 317.7 (M-H);
tR = 2.1 min
(98.0%)
Biphenyl-4-carboxylic acid (1-oxo-indan-5 ,~)-amide~SXT1398, CCM01024)
Reaction of 5-amino-indan-1-one (73 mg, 0.50 mmol) in THF (6 mL) with 4
biphenylcarbonyl chloride (140 mg, 0.65 mmol) in presence of triethylamine (90
~L, 0.65
mmol) according to method A gave biphenyl-4-carboxylic acid (1-oxo-indan-5-yl)-
amide
(100 mg, 0.30 mmol, 60% yield) as a brown powder after purification by washing
the
crude product with ethyl acetate.
Rf: 0.4 (EtOAc/DCM 1/9) ; M.p.: 217-219 °C ; 1H NMR (270MHz, DMSO-d6)
SH 2.58-
2.62 (m, 2H, CHZ), 3.07-3.11 (m, 2H, CH2), 7.41-7.45 (m, 1H, H,~.), 7.48-7.54
(m, 1H,
H,er), 7.63 (d, 1H, J = 8.4 Hz, Hue), 7.75-7.80 (m, 3H, Hue.), 7.84-7.87 (m,
2H, HA,.), 8.05-
8.08 (m, 2H, Hue), 8.12 (bs, 1H, H~,.), 10.62 (s, 1H, NH); LC/MS (AP-) m/z
325.9 (M-H);
tR = 2.3 min (97.4%); HRMS (FAB+) Calculated for C22H1~N02 327.1259; Found
327.1266
3~5-Dichloro-N (1-oxo-indan-5-Xl)-benzamide (STX1399, CCM01025)
Reaction of 5-amino-indan-1-one (73 mg, 0.50 mmol) in THF (6 mL) with 3,5-
dichlorobenzoyl chloride (136 mg, 0.65 mmol) in presence of triethylamine (90
~L, 0.65
mmol) according to method A gave biphenyl-4-carboxylic acid (1-oxo-indan-5-yl)-
amide
(115 mg, 0.36 mmol, 72% yield) as a brown powder after purification by washing
the
crude product with ethyl acetate.
Rf: 0.4 (EtOAc/DGM 1/9) ; M.p.: 261-263 °C ; 1H NMR (270MHz, DMSO-d6)
8H 2.58-
2.62 (m, 2H, CH2), 3.06-3.10 (m, 2H, CH2), 7.63 (d, 1H, J = 8.4 Hz, CHA,.-C-
CO), 7.72
(dd, 1H, J = 1.7, 8.4 Hz, N-C-CHA,.-CHI.), 7.88 (t, 1H, J = 2.0 Hz, CCl-CH,er-
CCl), 7.97
(d, 2H, J = 2Hz, Hue.), 8.05 (d, 1H, J = 1.7 Hz, CH,y,.-CH,~,,.-C-CO), 10.70
(s, 1H, NH); 13C
NMR (400MHz, DMSO-d6) 80 26.0 (CH2), 36.5 (CHI), 117.5 (CHI.), 120.0 (CHI),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
121
124.2 (CHI), 127.1 (CH~,I), 131.7 (CH,~,.), 132.9 (Cq), 134.8 (Cq), 138.2
(Cq), 144.9 (Cq),
157.1 (Cg), 163.8 (C=O), 205.4 (C=O); LC/MS (AP-) m/z 318.0 (M-H); tR = 2.5
min
(99.9%); HRMS (FAB+) Calculated for C1gH11C12NO2 319.0167; Found 319.0156
N ~1-Oxo-indan-5-yl~-4-propyl-benzamide (STX1400, CCM010261
Reaction of 5-amino-indan-1-one (75 mg, 0.50 mmol) in THF (6 mL) with 4
propylbenzoyl chloride (108 ~L, 0.65 mmol) in presence of triethylamine (90
~,L, 0.65
mmol) according to method A gave N (1-oxo-indan-5-yl)-4-propyl-benzamide (94
mg,
0.32 mmol, 64% yield) as a white powder after purification on silica gel
(eluent
EtOAc/DCM 0/10 to 1/9).
Rf: 0.4 (EtOAc/DCM 1/9) ; M.p.: 181-184 °C ; 1H NMR (270MHz, DMSO-d6)
8H 0.88
(t, 3H, J = 7.2 Hz, CH3), 1.56-1.65 (m, 2H, CH2-CH3), 2.57-2.65 (m, 4H,
2*CH2), 3.08
(bt, 2H, J = 5.4 Hz, CH2CO), 7.35 (d, 2H, J = 8.1 Hz, HA,.), 7.60 (d, 1H, J =
8.4 Hz, CH~.-
C-CO), 7.74 (d, 1H, J = 8.4 Hz, N-C-CHI.-CHI-), 7.87 (d, 2H, J = 8.1 Hz,
CHI.), 8.09 (d,
1H, J = 8.1 Hz, CHI-CH,~,.-C-CO), 10.50 (s, 1H, NH); LC/MS (AP-) nz/~, 291.9
(M-H);
tR = 2.4 min (99.1 %)
4-Methoxy-N (1-oxo-indan-5-~)-benzamide (STX1401, CCM01027)
Reaction of 5-amino-indan-1-one (73 mg, 0.50 mmol) in THF (6 xnL) with ~a-
anisoyl
chloride (88 ~L, 0.65 mmol) in presence of triethylamine (90 ~L, 0.65 mmol)
according
to method A gave 4-methoxy-N (1-oxo-indan-5-yl)-benzamide (107 mg, 0.38 mmol,
76%
yield) as a yellow powder after purification by chromatography on silica gel
(eluent
EtOAc/DCM 0/10 to 2/8).
Rf: 0.2 (EtOAc/DCM 1/9) ; M.p.: 237-238 °C ; 1H NMR (270MHz, DMSO-d6)
bH 2.57
2.62 (m, 2H, CH2), 3.08 (bt, 2H, J = 5.5 Hz, CH~,CO), 3.83 (s, 3H, OCH3), 7.07
(d, 2H, J
= 8.9 Hz, HA,.), 7.60 (d, 1H, J = 8.4 Hz, CHA,.-C-CO), 7.74 (d, 1H, J = 1.6
Hz, N-C-CH~.
CH,~,.), 7.96 (d, 2H, J = 8.9 Hz, CHI), 8.08 (d, 1H, J = 1.6 Hz, CHI.-CHI-C-
CO), 10.42
(s, 1H, NH); 13C NMR (400MHz, DMSO-d6) ~0 26.0 (CH2), 36.5 (CHz), 56.0 (OCH3),
114.2 (CH,~,.), 117.1 (CHI.), 119.8 (CHI.), 124.1 (CHI.), 127.0 (Cq), 130.3
(CHA,.), 132.3
(Cq), 145.8 (Cq), 157.1 (Cq), 162.7 (Cq), 165.9 (Cq), 205.3 (C=O); LC/MS (AP-)
~ralz 279.9
(M-H); tR = 2.0 min (99.9%)
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
122
3-Methox~(1-oxo-indan-5-~)-benzamide (STX1402, CCM01028)
Reaction of 5-amino-indan-1-one (73 mg, 0.50 mmol) in THF (6 mL) with m-
anisoyl
chloride (90 p,L, 0.65 mmol) in presence of triethylamine (90 ~,L, 0.65 mmol)
according
to method A gave 3-methoxy-N (1-oxo-indan-5-yl)-benzamide (73 mg, 0.26 mmol,
52%
yield) as a yellow powder after purification by chromatography on silica gel
(eluent
EtOAc/DCM 0/10 to 1/9).
Rf: 0.2 (EtOAc/DCM 1/9) ; M.p.: 203-204 °C ; 1H NMR (270MHz, DMSO-d6)
8H 2.59-
2.63 (m, 2H, CH2), 3.09 (bt, 2H, J = 5.7 Hz, CHZCO), 3.84 ~s, 3H, OCH3), 7.16-
7.21 (m,
1 H, H,~), 7.44-7.49 (m, 2H, H,9,.), 7.52-7.56 (m, 1 H, Hue.), 7. 62 (d, 1 H,
J = 8.4 Hz, CH~-
C-CO), 7.76 (dd, 1H, J = 1.8, 8.4 Hz, N-C-CHI-CHI), 8.09 (d, 1H, J = 1.8 Hz,
CH~.-
CHA,.-C-CO), 10.55 (s, 1H, NH); 13C NMR (400MHz, DMSO-d6) b~ 26.0 (CH2), 36.5
(CHZ), 55.9 (OCH3), 113.6 (CHI.), 117.3 (CH,~.), 118.1 ~CH~), 119.9 (CHI),
120.5
(CHI.), 124.2 (CH,~,.), 130.2 (CHI.), 132.5 (Cq), 136.5 (Cq), 145.4 (Cq),
157.1 (Cq), 159.7
(Cq), 166.3 (C=O), 205.3 (C=O); LC/MS (AP-) f~zlz 280.0 (M-H); tR = 2.0 min
(99.7%
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
123
Synthesis of Thiophene or Benzothiophene Arysulfonamide Derivatives
0 0
s. s c1 ~ s, s I
I a O H I / I / O H I / s
CI
CI
STX1317, XDS03047B STX1318, XDS03048 STX1319, XDS03049
O S
I ~ S. S O
~ H I / a I I W I S/ O~ ~ N~S
a ~ ~ O
Ie
I v ~~S\
STX1320, XDS03050 STX1321, XDS03051 STX1327, XDS03061A
S
O
ii
O\ N ~ S CI ~ S.N
I a O H~ CI ~ S~ I O H I
~ O ~CI S
-CI
STX1328, XDS03061B STX1329, XDS03062A STX1330, XDS03062B
S
0
CI OS'N ~ S CI I \ O H I \ \ ~S\ N ~ S
'o a S I
a I
a
STX1331, XDS03063A STX1332, XDS03063B STX1333, XDS03064A
O
ii
S.N O I \ O I \
I I a O H I S I O H S CI I j O H S
a CI
5TX1334, XDS03064B STX1335, XDS03065 STX1336, XDS03066
o I \ ~ I \ o
CI ~ S.N S I ~ ~ H S ~ S.N S
I o H ~ _e I ~ O H I / \
a I
s
STX1337, XDS03067 STX1338, XDS03068 STX1414, XDS03111
O
ii
O S.N S
CI ~ S. S I a O H I
I a O H I / \
CI I a
STX1415,XDS03112 STX1416,XDS03113
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
124
General method for synthesis of thiophene or benzothiophene
arylsulphonamide derivatives (STX1317-1321, STX1327-1338, STX1414-
1416):
To a solution of arylsulphonyl chloride (1.1 eq.) in DCM was added pyridine
(2.2
eq.), followed by the corresponding amine (1 eq.). The reaction mixture was
stirred at ambient temperature under nitrogen for 3-6 h. After TLC showed
completion of the reaction, the mixture was partitioned between ethyl acetate
and
5% sodium bicarbonate solution. The organic layer was washed with brine, dried
over sodium sulfate, and concentrated in vacuo to give the crude product as
solid
or thick syrup. The compound was then purified by flash chromatography (Ethyl
acetate-hexane gradient elution) to give desired arylsulphonamide as
crystalline
solid or amorphous solid. Yield ranged from 35-65%.
4-Propyl-N-thiophen-2-ylmethyl-benzenesulfonamide (STX1317,
XDS03047B)
White solid. TLC single spot at Rf 0.25 (20% ethyl acetatelhexane); HPLC
purity
> 99 % (tR 3.1 min in 30% water-acetonitrile); ~H NMR (270 MHz, DMSO): b 8.20
(1 H, broad, NH), 7.70 (2H, d, J = 8.4 Hz, ArH), 7.37-7.40 (3H, m, ArH), 6.89
(2H,
m, ArH), 4.15 (2H, s, NCH2), 2.65 (2H, t, J = 7.4 Hz, CH2), 1.61 (2H, sextet,
J =
7.4 Hz, CH2), 0.89 (3H, t, J = 7.4 Hz, CH3); FAB-MS 296 (MH+); FAB-HRMS
calcd for C~4H~$NO~S~ (MH+) 296.0779, found 296.0776.
2,5-Dichloro-N-thiophen-2-ylmethyl-benzenesulfonamide (STX1318,
XDS03048)
White crystalline solid. TLC single spot at Rf 0.29 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.8 min in 30% water-acetonitrile); ~ H NMR (270 MHz,
DMSO): S 8.73 (1 H, s, NH), 7.80 (1 H, d, J = 2.5 Hz, ArH), 7.60-7.69 (2H, m,
ArH), 7.33 (1 H, dd, J = 5.0, 1.5 Hz, ArH), 6.82-6.87 (2H, m, ArH), 4.36 (2H,
s,
NCH2); FAB-MS 322 (MH+); FAB-HRMS calcd for C~~H10ChNO2S2 (MH+)
321.9530, found 321.9426.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
125
3-Chloro-2-methyl-N-thiophen-2-ylmethyl-benzenesulfonamide (STX1319,
XDS03049)
White crystalline solid. TLC single spot at Rf 0.28 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 3.1 min in 30% water-acetonitrile); ~ H NMR (270 MHz,
DMSO): 5 8.58 (1 H, s, NH), 7.82 (1 H, d, J = 7.9 Hz, ArH), 7.70 (1 H, d, J =
7.9 Hz,
ArH), 7.35-7.40 (2H, m, ArH), 6.84-6.87 (2H, m, ArH), 4.26 (2H, s, NCH2), 2.57
(3H, s, CH3); FAB-MS 302 (MH+); FAB-HRMS calcd forC~2H~3CINO~S2 (MH+)
302.0076, found 301.9988.
Biphenyl-4-sulfonic acid (thiophen-2-ylmethyl)-amide (ST~1320, XDS03050)
White solid. TLC single spot at Rf 0.29 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 2.5 min in 30% water-acetonitrile); ~H NMR (270 MHz, DMSO): b 8.32
(1 H, s, NH), 7.87 (4H, s, ArH), 7.73-7.76 (2H, m, ArH), 7.39-7.55 (4H, m,
ArH),
6.91 (2H, m, ArH), 4.21 (2H, s, NCH2); FAB-MS 330 (MH+); FAB-HRMS calcd for
C~~H~6NOZS2 (MH+) 330.0622, found 330.0601.
4-Phenoxy-N-thiophen-2-ylmethyl-benzenesulfonamide (STX1321 XDS03051
White solid. TLC single spot at Rf 0.29 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 2.4 min in 20% water-acetonitrile); ~H NMR (270 MHz, DMSO): 5 8.22
(1 H, s, NH), 7.75-7.80 (2H, m, ArH), 7.39-7.51 (3H, m, ArH), 7.07-7.28 (5H,
m,
ArH), 6.90-6.92 (2H, m, ArH), 4.18 (2H, s, NCH2); FAB-MS 346 (MH+); FAB-
HRMS calcd for C~7H~~NO3S2 (MH+) 346.0572, found 346.0574.
4-Propyl-N,N-bis-thiophen-3-ylmethyl-benzenesulfonamide (STX1327
XDS03061 A)
White solid. TLC single spot at Rf 0.56 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 4.5 min in 20% water-acetonitrile); ~H NMR (270 MHz, CDC13): 5 7.72
(2H, dt, J = 8.4, 2.0 Hz, ArH), .7.29 (2H, dt, J = 8.5, 1.9 Hz, ArH), 7.17
(2H, dd, J
= 5.0, 3.0 Hz, ArH), 6.90 (2H, dd, J = 3.0, 1.5 Hz, ArH), 6.74 (2H, dd, J =
5.0, 1.5
Hz, ArH), 4.31 (4H, s, 2 x NCH2), 2.66 (2H, t, J = 7.1 Hz, CH2), 1.67 (2H,
sextet, J
= 7.5 Hz, CH2), 0.95 (3H, t, J = 7.3 Hz, CH3); APCI-MS 392 (MH+); FAB-HRMS
calcd for C~gH2~NO2S3 (MH+) 392.0813, found 392.0805.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
126
4-Propyl-N-thiophen-3-ylmethyl-benzenesulfonamide (STX1328,
XDS03061 B)
White solid. TLC single spot at Rf 0.29 (20% ethyl acetate/hexane); HPLC
purity
92 % (tR 2.6 min in 20% water-acetonitrile); ~H NMR (270 MHz, CDC13): ~ 7.75
(2H, dt, J = 8.5, 2.0 Hz, ArH), 7.29 (2H, dt, J = 8.4, 1.8 Hz, ArH), 7.20 (1
H, dd, J
= 5.2, 3.2 Hz, ArH), 7.03 (1 H, dd, J = 3.1, 1.1 Hz, ArH), 6.86 (1 H, dd, J =
5.1, 1.1
Hz, ArH), 4.73 (1 H, t, J = 6.1 Hz, NH), 4.15 (2H, d, J = 5.9 Hz, NCH2), 2.65
(2H, t,
J = 7.9 Hz CH2), 1.67 (2H, sextet, J = 8.0 Hz, CH2), 0.94 (3H, t, J = 7.3 Hz,
CH3);
APCI-MS 294 (M-H+); FAB-HRMS calcd for C~4H~gNO2S~ (MH+) 296.0779, found
296.0784.
2,5-Dichloro-N,N-bis-thiophen-3-ylmethyl-benzenesulfonamide (STX1329,
XDS03062A)
White crystalline solid. TLC single spot at Rf 0.50 (20% ethyl
acetate/hexane);
HPLC purity > 98 % (tR 4.0 min in20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): ~ 8.02 (1 H, m, ArH), 7.42-7.45 (2H, m, ArH), 7.23 (2H, dd, J = 5.0,
3.0
Hz, ArH), 7.01 (2H, dd, J = 3.0, 1.0 Hz, ArH), 6.82 (2H, dd, J = 5.0, 1.3 Hz,
ArH),
4.41 (4H, s, 2 x NCH2); APCI-MS 418 (MH+); FAB-HRMS calcd for
2O C~6H~4CI2N02S3 (MH+) 417.9564, found 417.9510.
2,5-Dichloro-N-thiophen-3-ylmethyl-benzenesulfonamide (STX1330,
XDS03062B)
White crystalline solid. TLC single spot at Rf 0.25 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.4 min in20% water-acetonitrile); 'H NMR (270 MHz,
CDC13): ~ 7.99 (1 H, d, J = 2.0 Hz, ArH), 7.36-7.45 (2H, m, ArH), 7.17 (1 H,
dd, J =
5.2, 3.2 Hz, ArH), 7.06 (1 H, dd, J = 3.0, 1.0 Hz, ArH), 6.86 (1 H, dd, J =
5.0, 1.3
Hz, ArH), 5.25 (1 H, t, J = 6.1 Hz, NH), 4.19 (2H, d, J = 6.2 Hz, NCH2); APCI-
MS
320 (M-H+); FAB-HRMS calcd for C~~H~oChN02S2 (MH+) 321.9530, found
321.9473.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
127
3-Chloro-2-methyl-N,N-bis-thiophen-3-ylmethyl-benzenesulfonamide
(STX1331, XDS03063A)
White solid. TLC single spot at Rf 0.55 (20% ethyl acetate/hexane); HPLC
purity
> 98 % (tR 6.8 min in 30% water-acetonitrile); ~ H NMR (270 MHz, CDCI3): ~
7.90
(1 H, dd, J = 7.8, 1.2 Hz, ArH), 7.60 (1 H, dd, J = 7.8, 1.1 Hz, ArH), 7.20-
7.27 (3H,
m, ArH), 7.00 (2H, dd, J = 3.0, 1.0 Hz, ArH), 6.76 (2H, dd, J = 5.0, 1.3 Hz,
ArH),
4.32 (4H, s, 2 x NCH2), 2.61 (3H, s, CH3); APCI-MS 398 (MH+); FAB-HRMS calcd
for C~7H~~CINOZS3 (MH+) 398.0110, found 398.0098.
3-Chloro-2-methyl-N-thiophen-3-ylmethyl-benzenesulfonamide (STX1332,
XDS03063B)
White solid. TLC single spot at Rf 0.30 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 2.5 min in 20% water-acetonitrile); ~H NMR (270 MHz, CDC13): ~ 7.91
(1 H, dd, J = 7.9, 1.0 Hz, ArH), 7.57 (1 H, dd, J = 7.8, 1.0 Hz, ArH), 7.20-
7.27 (2H ,
m, ArH), 7.02 (1 H, m, ArH), 6.84 (2H, dd, J = 5.1, 1.3 Hz, ArH), 4.79 (1 H,
t, J =
5.9 Hz, NH), 4.16 (2H, d, J = 5.9 Hz, NCH2); 2.62 (3H, s, CH3); APCI-MS 302
(MH+); FAB-HRMS calcd for C~~H~3CINO~S~ (MH+) 302.0076, found 302.0056.
Biphenyl-4-sulfonic acid bis-thiophen-3-ylmethyl-amide (STX1333,
XDS03064A)
White solid. TLC single spot at Rf 0.45 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 4.3 min in 20% water-acetonitrile); ~H NMR (270 MHz, CDC13): 5 7.86
(2H, dt, J = 8.6, 1.8 Hz, ArH), 7.70 (2H, dt, J = 8.5, 1.8 Hz, ArH), 7.59-7.63
(2H ,
m, ArH), 7.39-7.52 (3H, m, ArH), 7.19 (2H, dd, J = 5.3, 3.3 Hz, ArH), 6.95 (2H
,
dd, J = 3.2, 1.2 Hz, ArH), 6.80 (2H, dd, J = 5.3, 1.4 Hz, ArH), 4.36 (4H, s, 2
x
NCH2); APCI-MS 426 (MH+); FAB-HRMS calcd for C22H2oN02S3 (MH+) 426.0656,
found 426.0628.
Biphenyl-4-sulfonic acid (thiophen-3-ylmethyl)-amide (STX1334,
XDS03064B)
White solid. TLC single spot at Rf 0.45 (20% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 3.1 min in 30% water-acetonitrile); ~H NMR (270 MHz, CDC13): b
7.9'1
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
128
(2H, dt, J = 8.5, 1.7 Hz, ArH), 7.71 (2H, dt, J = 8.8, 1.8 Hz, ArH), 7.57-7.62
(2H,
m, ArH), 7.42-7.52 (3H, m, ArH), 7.22 (1 H, dd, J = 5.2, 3.2 Hz, ArH), 7.08 (1
H, m,
ArH), 6.90 (1 H, dd, J = 5.2, 1.2 Hz, ArH), 4.61 (1 H, t, J = 6.4 Hz, NH),
4.22 (2H,
d, J = 6.5 Hz, NCH2); APCI-MS 330 (MH+); FAB-HRMS calcd for C~7H~gNO2S2
(MH+) 330.0622, found 330.0627.
4-Propyl-N-(2-thiophen-2-yl-ethyl)-benzenesulfonamide (STX1335,
XDS03065)
White solid. TLC single spot at Rf 0.40 (25% ethyl acetate/hexane); HPLC
purity
> 99 % (tR 3.5 min in 30% water-acetonitrile); ~H NMR (270 MHz, CDC13): ~ 7.72
(2H, dt, J = 8.2, 1.1 Hz, ArH), 7.28 (2H, m, ArH), 7.11 (1 H, dd, J = 5.2, 1.2
Hz,
ArH), 6.88 (1 H, dd, J = 5.2, 3.2 Hz, ArH), 6.73 (1 H, dd, J = 3.0, 1.0 Hz,
ArH), 4.78
(1 H, t, J = 6.2 Hz, NH), 3.21 (2H, q, J = 6.4 Hz, CH2) 2.95 (2H, t, J = 6.8
Hz,
CH2), 2.64 (2H, t, J = 6.5 Hz, CHI), 1.65 (2H, sextet, J = 6.3 Hz, CH2), 0.90
(3H, t,
J = 6.5 Hz, CH3); APCI-MS 308 (M-H+)
2,5-Dichloro-N-(2-thiophen-2-yl-ethyl)-benzenesulfonamide (STX1336,
XDS03066)
Off-white crystalline solid. TLC single spot at Rf 0.37 (25% ethyl
acetate/hexane); HPLC purity > 99 % (tR 3.3 min 30% water-acetonitrile); ~H
NMR (270 MHz, CDC13): 5 8.06 (1 H, d, J = 2.5 Hz, ArH), 7.39-7.49 (2H, m,
ArH),
7.16 (1 H, dd, J = 5.2, 1.2 Hz, ArH), 6.91 (1 H, dd, J = 5.2, 3.2 Hz, ArH),
6.79 (1 H,
dd, J = 3.2, 1.1 Hz, ArH), 5.06 (1 H, t, J = 5.8 Hz, NH), 3.24 (2H, q, J = 5.9
Hz,
CH2) 3.01 (2H, t, J = 5.8 Hz, CH2); APCI-MS 334 (M-H+).
3-Chloro-2-methyl-N-(2-thiophen-2-yl-ethyl)-benzenesulfonamide (STX1337,
XDS03067)
White crystalline solid. TLC single spot at Rf 0.34 (25% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 3.3 min in 30% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.90 (1 H, dd, J = 7.9, 1.2 Hz, ArH), 7.57 (1 H, dd, J = 7.8, 1.2
Hz, ArH),
7.25 (1 H, t, J = 7.7 Hz, ArH), 7.16 (1 H, dd, J = 5.2, 1.3 Hz, ArH), 6.92 (1
H, dd, J
= 5.2, 3.3 Hz, ArH), 6.73 (1 H, dd, J = 3.2, 1.2 Hz, ArH), 4.54 (1 H, t, J =
6.5 Hz,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
129
NH), 3.24 (2H, q, J = 6.4 Hz, CH2), 2.98 (2H, t, J = 6.4 Hz, CH2), 2.50 (3H,
s,
CH3); APCI-MS 314 (M-H+).
Biphenyl-4-sulfonic acid (2-thiophen-2-yl-ethyl)-amide (STX1338, XDS03068)
Off-white crystalline solid. TLC single spot at Rf 0.29 (25% ethyl
acetate/hexane); HPLC purity > 99 % (tR 2.7 min in 30% water-acetonitrile); ~H
NMR (270 MHz, CDC13): 5 7.87 (2H, dt, J = 7.9, 1.2 Hz, ArH), 7.70 (2H, dt, J =
8.1, 1.2 Hz, ArH), 7.58-7.62 (2H, m, ArH), 7.41-7.50 (3H, m, ArH), 7.15 (1H,
dd, J
= 5.1, 1.2 Hz, ArH), 6.91 (1 H, dd, J = 5.2, 3.2 Hz, ArH), 6.77 (1 H, dd, J =
3.0, 1.0
Hz, ArH), 4.50 (1 H, t, J = 6.5 Hz, NH), 3.29 (2H, q, J = 6.5 Hz, CH2), 3.00
(2H, t, J
= 6.5 Hz, CH2); APCI-MS 342 (M-H+)
N-Benzo[b]thiophen-2-ylmethyl-4-propyl-benzenesulfonamide (STX1414,
XDS03111 )
Off-white solid. TLC single spot at Rf 0.47 (25% ethyl acetate/hexane); HPLC
purity > 99 % (tR 2.4 min in 20% water-acetonitrile); ~H NMR (270 MHz, CDC13):
b
7.76 (2H, dt, J = 7.9, 1.7 Hz, ArH), 7.61-7.72 (2H, m, ArH), 7.24-7.32 (4H, m,
ArH), 7.05 (1 H, d, J = 1.2 Hz, ArH), 4.73 (1 H, t, J = 6.7 Hz, NH), 4.43 (2H,
d, J =
6.5 Hz, NCH2), 2.61 (2H, t, J = 7.6 Hz, CH2), 1.62 (2H, sextet, J = 7.4 Hz,
CH2),
0.92 (3H, t, J = 7.3 Hz, CH3); APCI-MS 344 (M-H+).
N-Benzo[b]thiophen-2-ylmethyl-2,5-dichloro-benzenesulfonamide (STX1415,
XDS03112)
Off-white solid. TLC single spot at Rf 0.46 (20% ethyl acetate/hexane); HPLC
purity > 99 % (tR 2.4 min in 30% water-acetonitrile); ~H NMR (270 MHz, CDC13):
5
7.98 (1 H, dd, J = 1.5, 0.5 Hz, ArH), 7.61-7.69 (2H, m, ArH), 7.26-7.30 (4H,
m,
ArH), 7.05 (1 H, d, J = 0.8 Hz, ArH), 5.41 (1 H, t, J = 5.9 Hz, NH), 4.47 (2H,
d, J =
5.7 Hz, NCH2); APCI-MS 370 (M-H+).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
130
Biphenyl-4-sulfonic acid (benzo[b~thiophen-2-ylmethyl)-amide (STX1415,
XDS030113)
Off-white solid. TLC single spot at Rt 0.58 (30% ethyl acetate/hexane); HPLC
purity > 99 % (tR 2.4 min in 30% water-acetonitrile); 1H NMR (270 MHz, CDC13):
5
7.92 (2H, dt, J = 7.9, 1.7 Hz, ArH), 7.61-7.72 (4H, m, ArH), 7.40-7.55 (5H, m,
ArH), 7.24-7.30 (2H, m, ArH), 7.08 (1 H, d, J = 1.2 Hz, ArH), 4.81 (1 H, t, J
= 5.2
Hz, NH), 4.48 (2H, d, J = 5.4 Hz, NCH2); APCI-MS 378 (M-H+).
Synthesis of 3-(2,5-Dichloro-benzenesulfonylmethyl)-thiophene
ci S ,,S ci S ,,S
CI I ~ SH + CI I ~ Et3N ~ ~ ~-- ~~ mCPBA
CI ~ CH30H ' DCM °O
CI CI
STX1339, XDS03070 STX1340, XDS03071 B
3-(2,5-Dichloro-phenylsulfanylmethyl)-thiophene (STX1339, XDS03070):
To a solution of 2,5-dichlorobenzenethiol (420 mg, 2.35 mmol) in methanol (8
mL) was added triethylamine (0.4 mL), followed by 3-chloromethyl-thiophene
(266 mg, 2.00 mmol). The reaction mixture was stirred at ambient temperature
under nitrogen for 4 hours, and then partitioned between ethyl acetate and
water.
The organic layer was washed with 1 N HCI, 5% sodium carbonate solution and
brine, dried over sodium sulphate, and concentrated in vacuo to give the crude
product. The compound was purified by flash chromatography (Ethyl acetate-
hexane gradient elution). White crystalline solid (440 mg, yield 80%) was
obtained. TLC single spot at Rt 0.49 (8% ethyl acetate/hexane); HPLC purity >
99 % (tR 7.1 min in 30% water-acetonitrile); 1H NMR (270 MHz, CDC13): S 7.27-
7.30 (2H, m, ArH), 7.16-7.19 (2H, m, ArH), 7.04-7.09 (2H, m, ArH), 4.16 (2H,
s,
CH2); FAB-MS 274 (M+); FAB-HRMS calcd for C11H$C12S2 (M+) 273.9444, found
273.9439.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
131
3-(2,5-Dichloro-benzenesulfonylmethyl)-thiophene (STX1340, XDS03071 B):
To a cold solution (-5°C) of 3-(2,5-Dichloro-phenylsulfanylmethyl)-
thiophene (360
mg, 1.31 mmol) in DCM (10 mL) was added 3-chloroperoxy benzoic acid (800
mg, 57-86% pure). The reaction mixture was stirred at -5 - 0°C for 6
hours, and
then partitioned between DCM and 5% sodium carbonate solution. The organic
layer was washed with brine, dried over sodium sulphate, and concentrated in
vacuo to give a yellow residue which was purified by flash chromatography
(Ethyl
acetate-hexane gradient elution). White crystalline solid (295 mg, 73%) was
obtained. TLC single spot at Rf 0.52 (20% ethyl acetate/hexane); HPLC purity >
99 % (tR 3.0 min in 30% water-acetonitrile); 1H NMR (270 MHz, CDC13): b 7.68
(1 H, t, J = 1.3 Hz, ArH), 7.46 (2H, d, J = 1.2 Hz, ArH), 7.23 (1 H, m, ArH),
7.17
(1 H, m, ArH), 6.96 (1 H, dd, J = 5.0, 1.2 Hz, ArH), 4.70 (2H, s, CH2); FAB-MS
307
(MH+); FAB-HRMS calcd for C11H9C12O2S2 (MH+) 306.9421, found 306.9397
Synthesis of Thiophene or Benzothiophene Benzamide Derivatives
0
0 o s
N
I H 1S/ CI I ~ H 1S/ I I
CI
STX1405,XDS03101 STX1406,XDS03102 STX1407,XDS03103
O p O
/ H ~S/ /O I / H IS/ I , H ~S/ \
O
STX1408,XDS03104 STX1409,XDS03105 STX1417,XDS03114
O O
CI S
I i H ~ / \ w I s H ~S/ \
O
CI
STX1418, XDS03115 STX1419, XDS03116
General method for synthesis of thiophene or benzothiophene benzamide
derivatives (STX1405-1409, STX1417-1419):
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
132
To a solution of substituted benzoyl chloride (1.1 eq.) in DCM was added
triethylamine (2.2 eq.), followed by the corresponding amine (1 eq.). The
reaction mixture was stirred at ambient temperature under nitrogen overnight.
PS-Trisamine (0.2 eq.) was added and the mixture was stirred for another 3
hours at ambient temperature, filtered. The solution was concentrated in vacuo
to give crude product as solid or thick syrup. The compound was then purified
by
flash chromatography (Ethyl acetate-hexane gradient elution) to give desired
benzamide as crystalline solid. Yield ranged from 70-96%.
4-Propyl-N-thiophen-2-ylmethyl-benzamide (STX1405, XDS03101)
White crystalline solid. TLC single spot at Rf 0.49 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.3 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.68 (2H, m, ArH), 7.20-7.25 (3H, m, ArH), 7.02 (1 H, dd, J = 3.0,
1.3
Hz, ArH), 6.96 (1 H, dd, J = 5.2, 3.2 Hz, ArH), 6.37 (1 H, broad, NH), 4.80
(2H, d, J
= 5.4 Hz, CH2), 2.61 (2H, t, J = 7.7 Hz, CH2), 1.61 (2H, sextet, J = 7.4 Hz,
CH2),
0.92 (3H, t, J = 7.3 Hz, CH3); APCI-MS 258 (M-H+)
3,5-Dichloro-N-thiophen-2-ylmethyl-benzamide (STX1406, XDS03102)
White crystalline solid. TLC single spot at Rf 0.45 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.4 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.62 (2H, d, J = 2.0 Hz, ArH), 7.46 (1 H, t, J = 1.9 Hz, ArH), 7.25
(1 H,
dd, J = 5.0, 1.2 Hz, ArH), 7.03 (1 H, dd, J = 3.6, 0.9 Hz, ArH), 6.96 (1 H,
dd, J =
5.0, 3.5 Hz, ArH), 6.38 (1 H, broad, NH), 4.77 (2H, d, J = 5.7 Hz, NCH2); APCI-
MS
284 (M-H+).
Biphenyl-4-carboxylic acid (thiophen-2-ylmethyl)-amide (STX1407,
XDS03103)
White crystalline solid. TLC single spot at Rf 0.52 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.2 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): b 7.85 (2H, dt, J = 8.9, 2.0 Hz, ArH), 7.57-7.66 (4H, m, ArH), 7.35-
7.48
(3H, m, ArH), 7.25 (1 H, dd, J = 5.1, 1.3 Hz, ArH), 7.06 (1 H, dd, J = 3.5,
1.0 Hz,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
133
ArH), 6.97 (1 H, dd, J = 5.0, 3.5 Hz, ArH), 6.46 (1 H, broad, NH), 4.84 (2H,
d, J =
5.7 Hz, NCH2); APCI-MS 294 (MH+).
4-Methoxy-N-thiophen-2-ylmethyl-benzamide (STX1408, XDS03104)
White crystalline solid. TLC single spot at Rf 0.35 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 1.9 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): b 7.74 (2H, dt, J = 9.0, 2.3 Hz, ArH), 7.23 (1 H, dd, J = 5.2, 1.2 Hz,
ArH),
7.02 (1 H, dd, J = 3.5, 1.0 Hz, ArH), 6.96 (1 H, dd, J = 5.1, 3.6 Hz, ArH),
6.31 (1 H,
broad, NH), 4.79 (2H, d, J = 5.7 Hz, CH2), 3.83 (3H, s, OCH3); APCI-MS 246 (M-
H+).
3-Methoxy-N-thiophen-2-ylmethyl-benzamide (STX1409, XDS03105)
White crystalline solid. TLC single spot at Rf 0.35 (20% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.0 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDCI3): 5 7.38 (2H, dd, J = 2.3, 1.7 Hz, ArH), 7.24-7.32 (3H, m, ArH), 7.01-
7.05
(2H, m, ArH), 6.97 (1 H, dd, J = 5.2, 3.4 Hz, ArH), 6.39 (1 H, broad, NH),
4.82 (2H,
d, J = 5.5 Hz, CH2), 3.84 (3H, s, OCH3); APCI-MS 246 (M-H+).
N-Benzo[b]thiophen-2-ylmethyl-4-propyl-benzamide (STX1417, XDS03114)
White crystalline solid. TLC single spot at Rf 0.49 (30% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.5 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.77 (1 H, m, ArH), 7.69-7.73 (3H, m, ArH), 7.28-7.36 (2H, m, ArH),
7.21-7.25 (3H, m, ArH), 6.48 (1 H, broad, NH), 4.88 (2H, dd, J = 5.6, 1.0 Hz,
CH2),
2.61 (2H, t, J = 7.2 Hz, CH2), 1.63 (2H, sextet, J = 7.5 Hz, CH2), 0.92 (3H,
t, J =
7.4 Hz, CH3); APCI-MS 308 (M-H+).
N-Benzo[b]thiophen-2-ylmethyl-3,5-dichloro-benzamide (STX1418,
XDS03115)
White crystalline solid. TLC single spot at Rf 0.45 (20% ethyl
acetate/hexane);
HPLC purity > 98 % (tR 2.7 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.76-7.80 (1 H, m, ArH), 7.69-7.73 (1 H, m, ArH), 7.65 (2H, d, J =
1.7
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
134
Hz, ArH), 7.48 (1 H, t, J = 1.9 Hz, ArH), 7.30-7.35 (2H, m, ArH), 7.25 (1 H,
m,
ArH), 6.45 (1 H, broad, NH), 4.87 (2H, d, J = 5.6 Hz, CH2); APCI-MS 334 (M-
H+).
N-Benzo[b]thiophen-2-ylmethyl-4-methoxy-benzamide (STX1419, XDS03116)
White crystalline solid. TLC single spot at Rf 0.52 (40% ethyl
acetate/hexane);
HPLC purity > 99 % (tR 2.1 min in 20% water-acetonitrile); ~H NMR (270 MHz,
CDC13): 5 7.69-7.79 (4H, m, ArH), 7.24-7.33 (3H, m, ArH), 6.92 (2H, dt, J =
8.8,
2.2 Hz, ArH), 6.43 (1 H, broad, NH), 4.88 (2H, dd, J = 5.6, 1.0 Hz, CH2), 3.84
(3H,
s, OCH3); APCI-MS 296 (M-H+).
Synthesis of N-(4-benzophenone)-benzamide derivatives
c1
s
H
W / CI ~ I N w /
o I ~ w I o I / w I
O O
STX1430, CCM01029 STX1431, CCM01031
CI
H
N ~ I N ~ /
W /
O I / W I CI O I , w I
O O
STX1432, CCM01032 ST?C1433, CCM01034
i I H i0 / I H
W N ~ , o N w
o I / ~ I o I / ~_ I
0 0
STX1434, CCM01036 STX1435, CCM01037
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
135
General method for synthesis of N-(4-benzophenone)-benzamide
derivatives (STX1430-1435):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the acyl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers axe washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers axe then washed with brine, dried (MgS04),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
Biphenyl-4-carboxylic acid (4-benzo T~l-phen~)-amide (STX1430, CCM01029)
Reaction of 4-aminobenzophenone (102 mg, 0.52 mmol) in THF (6 mL) with 4
biphenylcarbonyl chloride (143 mg, 0.65 mmol) in presence of triethylamine (90
p,L, 0.65
mmol) according to method A gave biphenyl-4-carboxylic acid (4-benzoyl-phenyl)-
amide
(125 mg, 0.33 mmol, 63% yield) as a yellow powder after purification by
crystallisation in
DCM/EtOAc.
Rf: 0.2 (DCM); M.p.: 212-213 °C; 1H NMR (270MHz, DMSO-dB) ~H 7.40-7.45
(m, 1H,
Hue), 7.49 (bs, 1H, Hue), 7.52 (bs, 1H, HA,.), 7.54-7.59 (m, 2H, H,~,.), 7.64-
7.68 (m, 1H,
HA,.), 7.72 (d, 1H, J = 1.54 Hz, HA,.), 7.74-7.76 (m, 2H, Hue.), 7.77-7.79 (m,
2H, H,~,.), 7.81
(s, 1H, Hue.), 7.87 (d, 2H, J = 8.4 Hz, HP,L), 8.02 (d, 2H, J = 8.4 Hz, Hue.),
8.09 (d, 2H, J =
8.4 Hz, H~.),10.70 (s, 1H, NH); LC/MS (AP-) m/z 376.0 (M-H); tR = 2.7 min
(99.9%)
N (4-Benzoyl-phen~)-3,5-dichloro-benzamide (STX1431, CCM01031)
Reaction of 4-aminobenzophenone (100 mg, 0.51 mmol) in THF (6 mL) with 3,5
dicchlorobenzoyl chloride (136 mg, 0.65 mmol) in presence of triethylamine (90
~L, 0.65
mmol) according to method A gave N (4-benzoyl-phenyl)-3,5-dichloro-benzamide
(160
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
136
mg, 0.33 mmol, 63% yield) as a white powder after purification by
crystallisation in
DCM.
Rf : 0.3 (DGM); M.p.: 219-220 °C; 1H NMR (270MHz, DMSO-d6) 8H 7.52-7.58
(m, 2H,
Hue.), 7.63-7.67 (m, 1 H, H,~,.), 7.69-7.71 (m, 1 H, HA,.), 7.72-7.73 (m, 1 H,
HA,.), 7.79 (d, 2H,
J = 8.6 Hz, HA,.), 7.89 (t, 1H, J = 2.0 Hz, CCl-CHI.-CCl), 7.94 (d, 2H, J =
8.6 Hz, Hue.),
7.99 (d, 2H, J = 2.0 Hz, HA,.), 10.74 (s, 1H, NH); 13C NMR (400MHz, DMSO-d6)
80
120.1 (CHA,.), 127.2 (CHA,.), 129.0 (CHI,,.), 129.9 (CHI.), 131.5 (CHA,.),
131.7 (CHA,.),
132.7 (Cq), 132.9 (CHI.), 134.8 (Cg), 137.9 (Cq), 138.2 (Cq), 143.3 (Ca),
163.7 (C=O),
195.2 (C=O); LC/MS (AP-) fzZ/z 367.3 (M-H); tR = 3.0 min (99.9%)
N (4-Benzoyl-phenyl)-3-methoxv-benzamide (STX1432, CCM01032)
Reaction of 4-aminobenzophenone (100 mg, 0.51 mmol) in THF (6 mL) with m-
anisoyl
chloride (136 mg, 0.65 mmol) in presence of triethylamine (90 ~L, 0.65 mmol)
according
to method A gave N (4-benzoyl-phenyl)-3-methoxy-benzamide (160 mg, 0.48 mmol,
94%
yield) as a white powder after purification by chromatography on silica gel
(eluent
EtOAc/DCM 0/100 to 5/95.
Rf: 0.4 (EtOAc/DCM 5/95) ; M.p.: 139-141 °C ; 1H NMR (270MHz, CDCl3) 8H
3.76 (s,
3H, OCH3), 7.00 (ddd, 1H, J = 1.4, 2.8, 8.0 Hz, H,~), 7.28 (t, 1H, J = 7.7 Hz,
Hue.), 7.33
7.38 (m, 2H, Hue), 7.41 (dt, 2H, J = 1.5, 7.7 Hz, Hue.), 7.48-7.52 (m, 1H,
Hue.), 7.67-7.70
(m, 2H, Hue.), 7.72-7.77 (m, 4H, Hue:), 8.29 (s, 1H, NH); 13C NMR (400MHz,
DMSO-d6)
8c 55.5 (OCH3), 112.6 (CHA,.), 118.4 (CHA,.), 118.9 (CH,~,.), 119.3 (CHA,.),
128.3 (CHI.),
129.9 (CHA,.), 129.9 (CHI), 131.7 (CHI.), 132.4 (CHI,,.), 133.2 (Cq), 136.0
(Cq), 137.8
(Cq), 142.1 (Cq), 160.0 (Cq), 166.0 (C=O), 195.9 (C=O); LC/MS (AP-) m/z 330.0
(M-H);
tR = 2.2 min (99.9%)
N (4-Benzoyl-phenyl)-2,5-dichloro-benzamide (STX1433, CCM01034)
Reaction of 2,5-dichlorobenzoic acid (300 mg, 1.50 mmol) in thionyl chloride
(3.5 mL)
then with 4-aminobenzophenone (100 mg, 0.51 mmol) in presence of triethylamine
(90
~L) in THF (6 mL) according to method B gave N (4-benzoyl-phenyl)-2,5-dichloro-
benzamide (160 mg, 0.43 mmol, 84% yield) as a off white powder after
purification by
chromatography on silica gel (eluent : EtOAc/exane 1/9 to 3/7).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
137
Rf: 0.3 (DCM); M.p.: 150-153 °C; 1H NMR (270MHz, DMSO-d6) 8H 7.51-7.55
(m, 1H,
Hue), 7.57-7.58 (m, 1H, Hue), 7.59-7.61 (m, 2H, H,~,.), 7.62-7.66 (m, 1H,
Hue.), 7.68-7.70
(m, 1H, Hue.), 7.72-7.76 (m, 1H, H,~.), 7.77-7.80 (m, 2H, H~,.), 7.85-7.88 (m,
3H, HA,.),
10.96 (s, 1H, NH); LC/MS (AP-) m/z 367.8 (M-H), tR = 2.3 min (98.4%)
N (4-Benzo~pheny~l -4-prowl-benzamide (STX1434, CCM01036)
Reaction of 4-aminobenzophenone (99 mg, 0.51 mmol) in THF (6 mL) with 4-
propylbenzoyl chloride (100 ~L, 0.65 mmol) in presence of triethylamine (90
~.L, 0.65
mmol) according to method A gave N (4-benzoyl-phenyl)-4-propyl-benzamide (136
mg,
0.40 mmol, 78% yield) as a white powder after purification by chromatography
on silica
gel (eluent EtOAc/DCM 0/100 to 5/95).
Rf: 0.3 (DCM); M.p.: 119-122 °C; 1H NMR (270MHz, CDCl3) 8H 0.94 (t, 1H,
J = 7.2 Hz,
CH3), 1.59-1.72 (m, 2H, CH2-CH3), 2.65 (t, 2H, J =7.7 Hz, CHI-Ar), 7.27-7.30
(m, 2H,
Hue), 7.44-7.50 (m, 2H, Hue), 7.54-7.60 (m, 1H, Hue.), 7.74-7.76 (m, 2H, Hue),
7.76-7.79
(m, 3H, Hue), 7.80-7.87 (m, 3H, Hue), 8.02 (s, 1H, NH); 13C NMR (400MHz,
CDCl3) ~c
13.8 (CH3), 24.3 (CH2), 37.9 (CH2), 119.1 (CHI.), 127.2 (CHA,.), 128.3
(CHA,.), 129.0
(CHA,.), 129.9 (CHA,.), 131.7 (CHI), 131.8 (Cq), 132.3 (CHI.), 133.1 (Cq),
137.9 (Cq),
142.1 (Cq), 147.7 (G9), 165.9 (C=O), 195.7 (C=O); LC/MS (AP-) nZ/z 341.5 (M-
H); tR =
2.8 min (99.1 %)
N (4-Benzo 1-~en~)-4-methoxy-benzamide (STX1435, CCM01037)
Reaction of 4-aminobenzophenone (98 mg, 0.51 mmol) in THF (6 mL) with p-
anisoyl
chloride (90 ~L, 0.65 mmol) in presence of triethylamine (90 ~.L, 0.65 nunol)
according
to method A gave N (4-benzoyl-phenyl)-4-methoxy-benzamide (165 mg, 0.50 mmol,
98%
yield) as a white powder after purification by washing the crude product with
ethyl acetate
and hexane.
Rf: 0.4 (EtOAc/DCM 5/95) ; M.p.: 171-174 °C ; 1H NMR (270MHz, DMSO-d6)
8H 3.84
(s, 3H, OCH3), 7.08 (d, 2H, J = 8.6 Hz, HA,.), 7.53-7.58 (m, 2H, HA,.), 7.64-
7.66 (m, 1H,
Hue.), 7.71-7.79 (m, 4H, Hue.), 7.97-8.01 (m, 4H, HA,.), 10.45 (s, 1H, NH);
13C NMR
(400MHz, , DMSO-d6) 8c 56.0 (OCH3), 114.2 (CH,~,.), 119.8 (CHA,.), 127.0 (Cq),
129.0
(CHA,.), 129.9 (CHI,,.), 130.3 (CHA,.), 131.5 (CHI,.), 131.9 (Cq), 132.8
(CHI.), 138.1 (Cq),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
138
144.2 (Cq), 162.7 (Cq), 165.8 (C=O), 195.1 (C=O); LC/MS (AP-) rnlz 330.0 (M-
H); tR =
3.6 min (97.2%)
Synthesis of N-benzothiazole benzamide, acetamide and sulfonamide
derivatives
CI
°.
c1 I H \ c1
1i \ N \ N / I H
CI \ N I \ ~ ° \ N
/ / O
STX1355, CCM01002 STX1356, STX1357, CCM01004
CCM01003
CI
/ I CI CI H CI \ N \ N
CI \ N I ~ ~ \° ~ N ~ \ / /
° I/
STX1358, CCM01006 STX1363, STX1364, CCM01009
CCM01008
H CI / / CI H CI
N
/° \ N \ ~ \ I N I \ \ /° I \ N I \
/ ° I / S~ O / / ° / S
STX1365, CCM01010 STX1366, STX1367, CCM01012
CCM01011
CI CI CI
H H
N I \ N I \ N I \ N
c1 / ° / s~ °, s O /
STX1376, CCM01013 STX1396,
CCM01022
General method for synthesis of N-benzothiazole benzamide, acetamide
and sulfonamide derivatives (STX1355-1358, STX1363-1367, STX1376,
STX1396):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the acyl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers are washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
139
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers are then washed with brine, dried (MgS04),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
3,5-Dichloro-N-(4-chloro-2-methyl-benzothiazol-5-yl)-benzamide (STX1355,
CCM01002)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 3,5-dichlorobenzoyl chloride (122 mg, 0.56 mmol) in presence of
triethylamine
(0.5 mL) according to method A gave 3,5-Dichloro-N-(4-chloro-2-methyl-
benzothiazol-5-
yl)-benzamide (66 mg, 0.15 mmol, 60% yield) as a off white powder after
crystallisation
in hexane/DCM.
Rf 0.3 (DCM); M.p. (°C) 252-253; 1H NMR (270MHz, DMSO-d6) 2.85 (s,
3H, CH3),
7.54 (d, 1H, J = 8.7 Hz, HA,.benzothiazole), 7.92-7.94 (m, 1H, HA,.), 8.03-
8.04 (m, 2H,
HA,.), 8.06 (d, 1H, J = 8.7 Hz, H~.benzothiazole), 10.55 (s, 1H, NH); 13C NMR
(400MHz,
DMSO-d6) 20.4 (CH3), 120.8 (CH,~.benzothiazole), 123.2 (Cq), 125.6
(CH~benzothiazole), 127.1 (CHI), 131.8 (GHQ), 133.2 (Cq), 134.8 (Cq), 135.0
(Cq),
137.6 (Cq), 150.8 (Cq), 163.5 (Cq), 170.4 (C=O); LC/MS (AP-) m/z 368.7 (M-H);
tR=3.0
min (99.9%); HRMS (FAB+) Calculated for C15H9C13N20S 369.9501; Found 369.9504
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-4-propyl-benzamide (STX1356,
CCM01003)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 4-propylbenzoyl chloride (90 ~,L, 0.56 mmol) in presence of
triethylamine (0.5
mL) according to method A gave N (4-Chloro-2-methyl-benzothiazol-5-yl)-4-
propyl-
benzamide (44 mg, 0.13 nnnol, 52% yield) as a orange powder after
crystallisation in
hexane and methanol.
Rf 0.3 (DCM); M.p. 184 °C; 1H NMR (270MHz, DMSO-d6) 0.91 (t, 3H, J =
7.3 Hz,
CH2-CH3), 1.59-1.68 (m, 2H, CHZ-CH3), 2.65 (t, 2H, J = 7.6 Hz, CH2-Ar), 2.85
(s, 3H,
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
140
CH3), 7.36 (d, 2H, J = 8.2 Hz, Hue.), 7.58 (d, 1H, J = 8.6 Hz,
H~.benzothiazole), 7.95 (m,
2H, J = 8.2 Hz, HAr), 8.03 (d, 1H, J = 8.6 Hz, H~.benzothiazole), 10.18 (s,
1H, NH); 13C
NMR (400MHz, DMSO-d6) 14.1 (CHZ-CH3), 20.4 (CH3), 24.4 (CHZ-CH3), 37.5 (Ar-
CH2), 120.6 (CH~.benzothiazole), 125.5 (CH~.benzothiazole), 128.3 (CHI.),
128.9
(CHA,), 131.9 (Cq), 133.9 (Cq), 134.2 (Cq), 146.9 (Cq), 150.8 (Cq), 165.9
(Cq), 170.1
(C=O); LC/MS (AP-) m/z 343.0 (M-H); tR=2.9 min (98.8%); HRMS (FAB+) Calculated
for CIgHI~C1N20S 344.0750; Found 344.0744; IR (v, cm 1) (CC14) 3429w, 2962w,
2933w, 1692s (CO), 1611w, 1598, 1561w, 1520s, 1505s, 1430w, 1405, 1308, 1253,
1176w, 1122w, 1097w
Biphenyl-4-carboxylic acid (4-chloro-2-methyl-benzothiazol-5-yl)-amide
(STX1357, CCM01004
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 4-biphenylcarbonyl chloride (50 ~L, 0.30 mmol) in presence of
triethylamine
(42 ~L, 0.30 mmol) according to method A gave biphenyl-4-carboxylic acid (4-
chloro-2-
methyl-benzothiazol-5-yl)-amide (79 mg, 0.21 rmnol, 84% yield) as a pale pink
powder
after crystallisation in hexane/DCM.
Rf 0.3 (DCM); M.p. 210-211 °C; 1H NMR (270MHz, DMSO-d6) 2.86 (s, 3H,
CH3), 7.42-
7.47 (m, 1H, Hue.), 7.49-7.52 (m, 2H, Hue.), 7.61 (d, 1H, J = 8.4 Hz,
H~.benzothiazole),
7.77 (d, 2H, J = 7.4 Hz, HA,.), 7.88 (d, 2H, J = 8.4 Hz, Hue), 8.04 (d, 1H, J
= 8.4 Hz,
H~benzothiazole), 8.13 (d, 2H, J = 8.4 Hz, Hue), 10.33 (s, 1H, NH); 13C NMR
(400MHz,
DMSO-db) 20.4 (CH3), 120.7 (CH,~.benzothiazole), 123.1 (Cq), 125.6
(CH,~,.benzothiazole), 127.2 (CHA,.), 127.4 (CH,~,.), 128.7 (CHA,.), 129.0
(CHI.), 129.6
(CHA,.), 133.1 (Cq), 133.8 (Cq), 134.3 (Ca), 139.5 (C9), 143.9 (Cq), 150.8
(Cq), 165.7 (Cq),
170.2 (C=O); LC/MS (AP+) m/z 379.02 (M+H); tR=2.8 min (99.4%); HRMS (FAB+)
Calculated for C2lHisC1N20S 378.0594; Found 378.0590
2,5-Dichloro-N-(4-chloro-2-methyl-benzothiazol-5-yl)-benzamide (STX1358,
CCM01006)
Reaction of 2,5-dichlorobenzoic acid (145 mg, 0.76 mmol) in thionyl chloride
(5 mL)
then with 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in
presence of
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
141
triethylamine (0.5 mL) in THF (2 mL) according to method B gave 2,5-dichloro-N
(4-
chloro-2-methyl-benzothiazol-5-yl)-benzamide (50 mg, 0.13 mmol, 52% yield) as
a white
powder after purification by chromatography on silica gel (eluent : DCM).
Rf 0.35 (DCM); M.p. 175 °C; 1H NMR (270MHz, DMSO-d6) 2.85 (s, 3H,
CH3), 7.61
7.62 (m, 2H, H,e,,.), 7.66 (d, 1H, J = 8.6 Hz, H,~.benzothiazole), 7.76 (m,
1H, HA,.), 8.05 (d,
1H, J = 8.6 Hz, H~.benzothiazole), 10.52 (s, 1H, NH); LC/MS (AP+) m/z 370.8
(M+H);
tR=2.4 min (99.5%) HRMS (FAB+) Calculated for C15H9C13NZOS 369.9501; Found
369.9503
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-2-(4-methoxy-phenyl)-acetamide
(STX1363, CCM01008)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 4-methoxyphenylacetyl chloride (46 ~L, 0.30 mmol) in presence of
triethylamine (42 ~,L, 0.30 mmol) according to method A gave N (4-chloro-2-
methyl-
benzothiazol-5-yl)-2-(4-methoxy-phenyl)-acetamide (50 mg, 0.14 mmol, 56%
yield) as a
white powder after crystallisation in hexane/DCM.
Rf 0.4 (EtOAc/DCM 1/9); M.p. 173-174 °C; 1H NMR (270MHz, CDC13) 2.83
(s, 3H,
CH3), 3.76 (s, 2H, CH2), 3.82 (s, 3H, OCH3), 6.95 (d, 2H, J = 8.6 Hz, Hue.),
7.30 (d, 2H, J
= 8.6 Hz, Hue), 7.65 (d, 1H, J = 8.9 Hz, H~.benzothiazole), 7.80 (s, 1H, NH),
8.40 (d, 1H,
J = 8.9 Hz, H~.benzotluazole); LC/MS (AP+) m/z 347.0 (M+H); tR=2.1 min
(99.2%);
HRMS (FAB+) Calculated for C1~H15C1N202S 346.0543; Found 346.0542
Biphenyl-4-carboxylic acid (4-chloro-2-methyl-benzothiazol-5-yl)-amide
(STX1364, CCM01009)
Reaction of 4-biphenylacetic acid (160 mg, 0.76 mmol) in thionyl chloride (1
mL) then
with 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in presence
of
triethylamine (0.5 mL) in THF (2 mL) according to method B gave biphenyl-4-
carboxylic
acid (4-chloro-2-methyl-benzothiazol-5-yl)-amide (60 mg, 0.15 mmol, 60% yield)
as a
pink powder after purification by chromatography on silica gel (eluent : DCM).
Rf: 0.2 (DCM); M.p.:168-169 °C; 1H NMR (270MHz, CDC13) 8H 2.83 (s, 3H,
CH3), 3.87
(s, 2H, CH2), 7.35-7.38 (m, 1H, H,e,,.), 7.41-7.48 (m, 4H, Hue.), 7.57-7.61
(m, 2H, H,~,.),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
142
7.64-7.69 (m, 3H, Hue.), 7.82 (s, 1H, NH), 8.42 (d, 1H, J = 9.1 Hz,
H~.benzothiazole);
LC/MS (AP+) ~n/z 393.3; , tR = 3.2 min (99.8%); HRMS (FAB+) Calculated for
C22H1~C1N~OS 392.0750; Found 392.0742
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-3-methoxy-benzamide (STX1365,
CCM01010)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with nz-anisoyl chloride (41 ~L, 0.30 mmol) in presence of triethylamine
(42 ~L,
0.30 mmol) according to method A gave N (4-chloro-2-methyl-benzothiazol-5-yl)-
3-
methoxy-benzamide (70 mg, 0.21 mmol, 84% yield) as a orange powder after
crystallisation in hexane/DCM.
Rf : 0.4 (EtOAc/DCM 5/95); M.p.:177-179 °C; 1H NMR (270MHz, DMSO-d6)
8H 2.83
(s, 3H, CH3), 3.82 (s, 3H, OCH~), 7.17 (ddd, 1H, J = 1.3, 2.7, 8.1 Hz, Hue.),
7.44 (dd, 1H,
J = 7.7, 8.1 Hz, Hue.), 7.54-7.55 (m, 1H, H,~.), 7.55 (d, 1H, J = 8.4 Hz,
H,~.benzothiazole),
7.56-7.61 (m, 1H, Hue.), 8.02 (d, 1H, J = 8.4 Hz, H~.benzothiazole), 10.20 (s,
1H, NH);
LC/MS (AP+) m/z 333.2; . tR = 2.6 min, (99.7%); HRMS (FAB+) Calculated for
CisH13C1N202S 332.0386; Found 332:0383
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-4-methoxy-benzamide (STX1366,
CCM01011 )
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) withp-anisoyl chloride (41 ~L, 0.30 mmol) in presence of triethylamine (42
~,L, 0.30
mmol) according to method A gave N (4-chloro-2-methyl-benzothiazol-5-yl)-4-
methoxy
benzamide (65 mg, 0.19 mmol, 76% yield) as a white powder after
crystallisation in
hexane/DCM.
Rf : 0.4 (EtOAc/DCM 1/9); M.p.:182 °C 1H NMR (270MHz, DMSO-d6) ~H 2.83
(s, 3H,
CH3), 3.83 (s, 3H, OCH3), 7.06 (d, 2H, J = 8.8 Hz, Hue), 7.55 (d, 1H, J = 8.5
Hz,
H,~,.benzothiazole), 7.99 (d, 1H, J = 8.5 Hz, HA,.benzothiazole), 8.00 (d, 2H,
J = 8.8 Hz,
H,~,.), 10.06 (s, 1H, NH); LC/MS (AP+) m/z 333.2; tR = 2.6 min (99.9%); HRMS:
(FAB+)
Calculated for C16H13C1N202S 332.0386; Found 332.0383
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
143
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-2-(3-methoxy-phenyl)-acetamide
(STX1367, CCM01012)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 3-methoxyphenylacetyl chloride (47 ~L, 0.30 mmol) in presence of
triethylamine (42 ~.L, 0.30 mmol) according to method A gave N (4-chloro-2-
methyl-
benzothiazol-5-yl)-2-(3-methoxy-phenyl)-acetamide (40 mg, 0.11 mmol, 48%
yield) as a
white powder after crystallisation in hexane/DCM.
Rf: 0.4 (EtOAc/DCM 1/9); M.p.:147-148 °C; 1H NMR (270MHz, DMSO-d6) 8H
2.82 (s,
3H, CH3), 3.72 (s, 2H, CHZ), 3.75 (s, 3H, OCH3), 6.80-6.84 (m, 1H, Hue), 6.93-
6.96(m,
2H, Hue.), 7.25 (dd, 1H, J = 8.2, 8.2 Hz, HA,.), 7.65 (d, 1H, J = 8.6 Hz,
H,~benzothiazole),
7.95 (d, 1H, J = 8.6 Hz, H~.benzothiazole), 9.90 (s, 1H, NH); LC/MS (AP+) m/z
347.2
(M+H); tR = 2.5 min, (99.3%); HRMS (FAB+) Calculated for C1~H15C1N202S
346.0543;
Found 346.0541
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-2-(4-chloro-phenyl)-acetamide
(STX1376, CCM01013)
Reaction of 4-chloro-2-methyl-benzothiazol-5-ylamine (50 mg, 0.25 mmol) in THF
(1.5
mL) with 4-chlorophenylacetyl chloride (65 mg, 0.30 mmol) in presence of
triethylamine
(42 ~,L, 0.30 mmol) according to method A gave N (4-chloro-2-methyl-
benzothiazol-5-
yl)-2-(4-chloro-phenyl)-acetamide (65 mg, 0.19 xmnol, 80% yield) as a white
powder after
crystallisation in hexane/DCM.
Rf: 0.5 (EtOAc/DCM 1/9); M.p.:227-229 °C; 1H NMR (270MHz, DMSO-db) bH
2.82 (s,
3H, CH3), 3.76 (s, 2H, CH2), 7.38 (s, 4H, HA,.), 7.63 (d, 1H, J = 8.5 Hz,
HA,.benzothiazole), 7.95 (d, 1H, J = 8.5 Hz, H,~,.benzothiazole), 9.96 (s, 1H,
NH); LC/MS
(AP-) fnlz 350.8 (M-H); tR = 2.5 min, (96.5%) HRMS (FAB+) Calculated for
CisHi2C12N~OS 350.0047; Found 350.0048
N-(4-Chloro-2-methyl-benzothiazol-5-yl)-2-(2,4-dichloro-phenyl)-acetamide
(STX1396, CCM01022)
Reaction of 2,4-dichlorophenylacetic acid (163 mg, 0.79 mmol) in thionyl
chloride (2 mL)
then with 4-chloro-2-methyl-benzothiazol-5-ylamine (52mg, 0.26 mmol) in
presence of
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
144
triethylamine (0.5 mL) in THF (3 mL) according to method B gave N (4-chloro-2-
methyl-
benzothiazol-5-yl)-2-(2,4-dichloro-phenyl)-acetamide (33 mg, 0.09 mmol, 35%
yield) as a
white powder after crystallisation in hexane/DCM.
Rf: 0.4 (EtOAc/DCM 1/9); M.p.:257-258 °C; 1H NMR (270MHz, DMSO-d6) 8H
2.81 (s,
3H, CH3), 3.93 (s, 2H, CH2), 7.38-7.42 (m, 1H, HA,.), 7.47-7.52 (m, 1H, H~,.),
7.59-7.60
(m, 1 H, HA,.), 7.62 (d, 1 H, J = 8.7 Hz, H~.benzothiazole), 7.95 (d, 1 H, J =
8.7 Hz,
HA,.benzothiazole), 10.05 (bs, 1H, NH); LC/MS (AP-) m/~; 382.8 (M-H); tR = 2.6
min
(95.1%); HRMS (FAB+) Calculated for C16H11C13N2OS 383.9658; Found 383.9653
Synthesis of N (5-tetralone)-benzamide derivatives
c1 ~ I c1
H ~ ~ H
N ~ ~ I N ~ CI \ l N
CI O I s O ~ , O ~ r
O p O
STX1465, CCM01044 STX1470, CCM01049 STX142, CCM01050
H /O / I H ~ I H
~N ~ w N ~ w0 w N
Ion ~ s o ~ i o ~ s
0 0 0
STX1473, CCM01051 STX1474, CCM01052 STX1475, CCM01053
General method for synthesis of N-(5-tetralone)-benzamide derivatives
(STX1465 and STX1470-1475):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the acyl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers are washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
145
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers are then washed with brine, dried (MgS04),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
2,5-Dichloro-N-(5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-benzamide
(CCM01044, STX1465)
Reaction of 2,5-dichlorobenzoic acid (300 mg, 1.50 mmol) in thionyl chloride
(3.5 mL)
then with 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in
presence of
triethylamine (90 ~,L, 0.65 mmol) in THF (6 mL) according to method B gave 2,5-
dichloro-N (5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-benzamide (150 mg, 0.48
mmol,
76% yield) as a grey powder after purification by crystallisation in DCM.
1H NMR (270 MHz, DMSO-d6) 8H 1.92-1.97 (2H, m, CH2), 2.46-2.50 (2H, m CH2),
2.84
(2H, t, J= 5.7 Hz, CH2), 7.48-7.52 (3H, m, Hue), 7.67 (2H, bs, Hue.), 7.78
(1H, d, J= 8.6
Hz, Hue), 10.78 (1H, s, NH) ; LC/MS (AP-) t,.= 2.2min (93.5%), m/z 315.2 (M-
H).
Biphenyl-4-carboxylic acid (5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-amide
(CCM01049, STX1470)
Reaction of 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in THF
(6
mL,) with 4-biphenylcarbonyl chloride (143 mg, 0.65 mmol) in presence of
triethylamine
(90 ~L, 0.65 mmol) according to method A gave biphenyl-4-carboxylic acid (5-
oxo-
5,6,7,8-tetrahydro-naphthalen-2-yl)-amide (150 mg, 0.43 mmol, 86% yield) as a
white
powder after purification by flash chromatography on silica gel (EtOAc/DCM 1/9
to 2/8).
1H NMR (270 MHz, DMSO-d6) 8H 1.92-1.98 (2H, m, CH2), 2.44-2.51 (2H, m, CH2),
2.85
(2H, t, J= 5.9 Hz, CH2), 7.34-7.45 (3H, m, HA,.), 7.65-7.70 (3H, m, Hue.),
7.75-7.80 (4H,
m, HA,.), 7.98 (2H, d, J= 8.6 Hz, H,9,.), 10.48 (1H, s, NH).
3,5-Dichloro-N-(5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-benzamide
(CCM01050, STX1472)
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
146
Reaction of 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in THF
(6
mL) with 3,5-dichlorobenzoyl chloride (136 mg, 0.65 mmol) in presence of
triethylamine
(90 ~.L, 0.65 mmol) according to method A gave 3,5-dichloro-N (5-oxo-5,6,7,8
tetrahydro-naphthalen-2-yl)-benzamide (140 mg, 0.42 mmol, 84% yield) as a
brown
powder after washing by DCM.
Mp 226-228 °C ; 1H NMR (270 MHz, DMSO-d~) 8H 1.92-1.97 (2H. m, CH2),
2.49 (2H, t,
J= 6.0 Hz, CHa), 2.85 (2H, t, J = 6.0 Hz, CH2), 7.62 (1H, dd, J = 2.1;8.4 Hz,
H,9,.tetralone), 7.71 (1H, d, J= 1.7 Hz, Hue), 7.79 (1H, d, J= 8.4 Hz,
H~tetralone), 7.81
(1H, d, J= 2.1 Hz, HA,.tetralone), 7.90 (2H, d, J= 1.7 Hz, Hue.), 8.08 (2H, d,
J = 8.4 Hz,
H,~.), 10.61 (1H, s, NH) ; 13C NMR (50 MHz, DMSO-d6) SC 23.4 (CH2), 29.8
(CH2), 38.9
(CH2), 118.7 (CHA,.), 119.6 (CH~,L), 127.1 (CH,~.), 128.0 (CHI), 128.6 (Cq),
131.7 (Cq),
134.8 (CH,~.), 13 8.2 (Cq), 143.5 (C~), 146.2 (C9),163.6 (CO), 196.8 (CO).
N-(5-Oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-4-propyl-benzamide
(CCM01051, STX1473)
Reaction of 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in THF
(6
mL) with 4-propylbenzoyl chloride (100 ~,L, 0.65 mmol) in presence of
triethylamine (90
~L, 0.65 mmol) according to method A gave N (5-oxo-5,6,7,8-tetrahydro-
naphthalen-2-
yl)-4-propyl-benzamide (140 mg, 0.45 mmol, 90% yield) as a white powder after
purification by flash chromatography on silica gel (eluent : EtOAc/hexane 2/8
to 4/6).
Mp 168-170 °C ; 1H NMR (270 MHz, DMSO-d6) bH 0.89 (3H, t, J = 7.4 Hz,
CH3), 1.54-
1.68 (2H, m, CH2propyl), 1.97-2.07 (2H. m, CH2tetralone), 2.55 (2H, t, J= 6.5
Hz, CH2),
2.63 (2H, t, J= 7.4 Hz, CH2), 2.91 (2H, t, J = 6.5 Hz, CH2), 7.35 (2H, d, J=
8.4 Hz, HA,.),
7.70 (1H, dd, J= 1.7;8.7 Hz, H~tetralone), 7.83-7.89 (4H, m, Hue.), 10.41 (1H,
s, NH) ;
13C NMR (50 MHz, DMSO-d6) 8~ 14.1 (CH3), 23.4 (CH2tetralone), 24.4 (CH2), 29.8
(CH2tetralone), 37.5 (CH2), 38.9 (CH2tetralone), 118.5 (CHI.), 119.3 (CH~,L),
127.9
(CH,~,.), 128.2 (C4), 128.3 (CH,~,.), 128.9 (CH~,.),132.5 (Cq), 144.2 (Cq),
146.2 (Cq), 147.0
(Cq),166.4 (CO), 196.8 (CO)
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
147
4-Methoxy-N-(5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-benzamide
(CCM01052, STX1474)
Reaction of 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in THF
(6
mL) withp-anisoyl chloride (90 mg, 0.65 mmol) in presence of triethylamine (90
~,L, 0.65
mmol) according to method A gave 4-methoxy-N (5-oxo-5,6,7,8-tetrahydro-
naphthalen-2
yl)-benzamide (135 mg, 0.46 mmol, 92% yield) as a white powder after
purification by
flash chromatography on silica gel (eluent : EtOAc/DCM 0/10 to 2/8).
Mp 163-166 °C ; 1H NMR (270 MHz, DMSO-d6) 8H 1.99-2.06 (2H. m, CHZ),
2.54 (2H, t,
J= 6.0 Hz, CH2), 2.90 (2H, t, J= 5.8 Hz, CH2), 3.82 (3H, s, OCH3), 7.06 (2H,
d, J= 8.7
Hz, HA,.), 7.70 (1H, dd, J= 2.0;8.7 Hz, H~tetralone), 7.81-7.84 (2H, m, H~,.),
7.95 (2H, d,
J= 8.7 Hz, H,~,.), 10.32 (1H, s, NH).
3-Methoxy-N-(5-oxo-5,6,7,8-tetrahydro-naphthalen-2-yl)-benzamide
(CCM01053, STX1475)
Reaction of 6-Amino-3,4-dihydro-2H naphthalen-1-one (80 mg, 0.50 mmol) in THF
(6
mL) with m-anisoyl chloride (90 ~,L, 0.65 mmol) in presence of triethylamine
(90 ~,L,
0.65 mmol) according to method A gave 3-methoxy-N (5-oxo-5,6,7,8-tetrahydro-
naphthalen-2-yl)-benzamide (135 mg, 0.46 mmol, 92% yield) as a white powder
after
purification by flash chromatography on silica gel (eluent : EtOAc/DCM 0/10 to
2/8).
Mp 142-143 °C ; 1H NMR (270 MHz, DMSO-d6) bH 2.01-2.09 (2H. m, CH2),
2.57 (2H, t,
J= 6.3 Hz, CH2), 2.93 (2H, t, J = 6.0 Hz, CH2), 3.84 (3H, s, OCH3), 7.18 (1H,
ddd,
J= 1.0;2.5;7.9 Hz, HA,.), 7.43-7.56 (3H, m, HAr), 7.73 (1H, dd, J= 2.0;8.7 Hz,
HA,.tetralone), 7.83 (1H, d, J = 1.0 Hz, HA,.tetralone), 7.86 (1H, d, J = 8.7
Hz,
H~.tetralone), 10.32 (1H, s, NH).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
148
Synthesis of N (5-acetophenone)-benzamide derivatives
i CI
I ~ H ~ I H ~ ~ N
N w CI ~ N
I ~ o I
O
O
O O
STX1461, CCM01040 STX1462, CCM01041 STX1463, CCM01042
CI
~O
H
N ~ ~O ~ I N w
CI O I / O I ~ O
I O O
O
STX1464, CCM01043 STX1468, CCM01047 STX1469, CCM01048
General method for synthesis of N-(5-acetophenone)-benzamide derivatives
(STX1461-1464 and STX1468-1469):
Method A : to a stirred solution of the amine (n mmol) in THF are added
triethylamine
(1.2n mmol) and the aryl chloride (1.2n mmol) at room temperature. After
completion,
ethyl acetate and water are added. The aqueous layer is extracted by ethyl
acetate. The
combined organic layers are washed with brine, dried (MgS04), filtered and
evaporated
under reduce pressure. The crude product is then purified to give the amide.
Method B : A solution of the acid (3n mmol) in thionyl chloride is refluxed 3
hours.
Thionyl chloride is then removed under reduced pressure. The crude product is
diluted in
dry THF and added to a solution of the amine (n mmol) and triethylamine in
THF. After
completion, ethyl acetate and water are added . The aqueous layer is extracted
by EtOAc.
The combined organic layers are then washed with brine, dried (MgSO4),
filtered and
evaporated under reduce pressure. The crude product is purified to give the
amide.
Biphenyl-4-carboxylic acid (4-acetyl-phenyl)-amide (CCM01040, STX1461 )
Reaction of 4-aminoacetophenone (75 mg, 0.55 mmol) in THF (6 mL) with
4-biphenylcarbonyl chloride (147 mgy 0.68 mmol) in presence of triethylamine
(90 ~L,
0.65 minol) according to method A gave biphenyl-4-carboxylic acid (4-acetyl-
phenyl)-
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
149
amide (90 mg, 0.28 mmol, 50% yield) as a white powder after washing by water
and ethyl
acetate.
Mp 283-285 °C; 1H NMR (270 MHz, DMSO-d6) ~H 2.55 (3H, s, CH3), 7.42-
7.45 (1H, m,
Hue), 7.48-7.54 (2H, m, HA,.), 7.75-7.78 (2H, m, H,~,.), 7.86 (2H, d, J= 8.4
Hz, Hue), 7.97
(4H, s, Hue.), 8.08 (2H, d, J = 8.4 Hz, HA,.), 10.61 ( 1 H, s, NH); LG/MS (AP-
) tr = 2.3 min
(99.2%), m/z 314.3 (M-H)
N-(4-Acetyl-phenyl)-3,5-dichloro-benzamide (CCM01041, STX1462)
Reaction of 4-aininoacetophenone (76 mg, 0.56 mmol) in THF (6 mL) with 3,5
dichlorobenzoyl chloride (145 mg, 0.69 mmol) in presence of triethylamine (90
~L, 0.65
mmol) according to method A gave N (4-acetyl-phenyl)-3,5-dichloro-benzamide
(145 mg,
0.47 mmol, 84% yield) as a white powder after washing by dichloromethane.
Mp 199-200°C ; R~: 0.3 (Ethyl acetate/hexane, 3:7) ; 1H NMR (270 MHz,
DMSO-d6) 8H
2.55 (3H, s, CH3), 7.86-7.92 (3H, m, HA,.), 7.97-8.00 (4H, m, Hue.), 10.69
(1H, s, NH); 13C
NMR (50 MHz, DMSO-d6) 8C 27.2 (CH3), 120.3 (CHI.), 127.3 (CHA,.), 130.0
(CHI.),
131.9 (Cq), 133.1 (Cq), 135.0 (CHI.), 138.4 (Cq), 143.7 (Cq), 163.8 (CO),
197.3 (CO);
LC/MS (AP-) t,.= 2.Smin (99.5%), m/z 306.1 (M-H).
N-(4-Acetyl-phenyl)-4-propyl-benzamide (CCM01042, STX1463)
Reaction of 4-aminoacetophenone (76 mg, 0.56 mmol) in THF (6 mL) with 4-
propylbenzoyl chloride (100 ~,L, 0.65 mmol) in presence of triethylamine (90
~,L, 0.65
mmol) according to method A gave N (4-acetyl-phenyl)-4-propyl-benzamide (125
mg,
0.44 mmol, 78% yield) as a white powder after washing by dichloromethane and
hexane.
Mp 174-176°C ; Rf 0.2 (Ethyl acetate/hexane, 4:8) ; 1H NMR (270 MHz,
DMSO-d6) 8H
0.89 (3H, t, J= 7.2 Hz, CH3), 1.55-1.59 (2H, m, CHI-CH3), 2.54 (3H, s, CH3),
2.63 (2H,
t, J = 7.2 Hz, ArCH2), 7.3 5 (2H, d, J = 8.3 Hz, HA,.), 7.89 (2H, d, J = 8.3
Hz, HA,.), 7.94
7.98 (4H,m, Hue), 10.47 (1H, s, NH); 13C NMR (50 MHz, DMSO-d6) 80 14.3 (CH3),
24.6
(CH2), 27.1 (CH3CO), 37.7 (CH2), 120.0 (CHA,.), 128.5 (CHI.), 129.1 (CHI.),
130.0
(CHp,,.), 132.5 (Cq), 132.7 (Cg), 144.4 (Cq), 147.2 (Cq), 166.6 (CO), 197.3
(CO); LC/MS
(AP-) t,.= 2.4min (99.1%), m/z 280.2 (M-H).
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
150
N-(4-Acetyl-phenyl)-2,5-dichloro-benzamide (CCM01043, STX1464)
Reaction of 2,5-dichlorobenzoic acid (300 mg, 1.50 mmol) in thionyl chloride
(3.5 mL)
then with 4-aminoacetophenone (85 mg, 0.63 mmol) in presence of triethylamine
(90 ~,L,
0.65 mmol) in THF (6 mh) according to method B gave N (4-Acetyl-phenyl)-2,5-
dichloro-benzamide (150 mg, 0.48 rilmol, 76% yield) as a orange powder after
purification by crystallisation in DCM.
Rf 0.35 (Ethyl acetate/hexane, 4:6) ; 1H NMR (270 MHz, DMSO-d6) 8H 2.49 (3H,
s,
CH3), 7.55-7.56 (2H, m, Hue.), 7.73 (1H, dd, J= 1.0; 2.0 Hz, Hue.), 7.75-7.78
(2H, m, Hue),
7.91-7.94 (2H, m, Hue), 10.86 (1H, s, NH).
N-(4-Acetyl-phenyl)-4-methoxy-benzamide (CCM01047, STX1468)
Reaction of 4-aminoacetophenone (74 mg, 0.55 mmol) in THF (6 mL) with p-
anisoyl
chloride (90 mg, 0.65 mmol) in presence of triethylamine (90 ~L, 0.65 rnmol)
according
to method A gave N (4-acetyl-phenyl)-4-methoxy-benzamide (110 mg, 0.41 mmol,
75%
yield) as a white powder after washing by dichloromethane.
Mp 222-223°C ; Rf 0.35 (Ethyl acetate/DCM, 1:9) ; 1H NMR (270 MHz, DMSO-
d6) ~H
2.53 (3H, s, CH3), 3.83 (3H, s, OCH3), 7.05-7.08 (2H, m, Hue), 7.93-7.98 (6H,
m, Hue),
10.38 (1H, s, NH) ; 13C NMR (50 MHz, DMSO-d~) ~c 27.1 (CH3C0), 56.1 (CH3),
114.4
(CHI), 120.0 (CHI), 127.2 (Cq), 130.0 (CHA,.), 130.5 (CHA,.), 132.4 (Cq),
144.5 (Cq),
162.8 (Cq), 166.0 (CO), 197.3 (CO).
N-(4-Acetyl-phenyl)-3-methoxy-benzamide (CCM01048, STX1469)
Reaction of 4-aminoacetophenone (72 mg, 0.53 mmol) in THF (6 mL) with m-
anisoyl
chloride (90 ~L, 0.65 mmol) in presence of triethylamine (90 ~,L, 0.65 mmol)
according
to method A gave N (4-acetyl-phenyl)-3-methoxy-benzamide (110 mg, 0.41 mmol,
77%
yield) as a white powder after washing by dichloromethane.
Mp 152-154°C ; R~: 0.35 (Ethyl acetate/DCM, 1:9) ; 1H NMR (270 MHz,
DMSO-d6) 8H
2.49 (3H, s, CH3), 3.78 (3H, s, OCH3), 7.12 (1H, ddd, J =1.0;2.7;8.1 Hz,
HP,,.), 7.37-7.44
(2H, m, HA,.), 7.49 (1H, ddd, J= 1.0;1.5;7.4 Hz, Hue.), 7.85 7.94 (4H, m,
Hue), 10.48 (1H,
s, NH) ; 13C NMR (50 MHz, DMSO-d6) 8~ 27.4 (CH3CO), 56.2 (OCH3), 113.9
(CHA,.),
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
151
118.5 (CHI), 120.4 (CH,er), 120.9 (CHI), 130.2 (CHI), 130.5 (CHP,,.), 132.9
(Cq), 136.8
(Cq), 144.4 (Cq), 160.1 (Cq), 166.6 (CO), 197.5 (CO).
DETAILED DESCRIPTION OF FIGURES
Separation of Cortisone and Cortisol
Several solvent systems claiming to separate cortisone from cortisol are
detailed in the
literature[30, 31 ] _ Before running an assay, 10 mg l ml solutions of
cortisone and cortisol
were prepared in IMS and 50 p,1 aliquots were spotted separately onto a silica
gel TLC
plate 3 cm from the bottom edge and 2.5 cm apart. The plate was run in a TLC
tank in
200 ml of CH~C12 : IMS 92 : 8 ~/" [30] until the solvent front reached the top
of the plate.
The plate was air dried and sprayed with 0.1 % Rhodamine B in IMS to visualise
the
spots. The Table below describes the separation obtained.
Table . Separation of cortisone from cortisol by TLC
Steroid Distance run from Solvent front migration
origin /
(cm) steroid migration
(cm)
Cortisone 7.5 2.3
Cortisol 4.5 3.8
The separation vvas considered adequate for use in an enzyme assay.
Figure 1 (Extraction efficiencies obtained with four extraction methods)
The literature details several methods of extracting cortisol from aqueous
solution[30,
31]. In order to select a method for use, a'4C-labelled cortisol was obtained
from NEN.
A stock was prepared in PBS containing 4000 DPM in 50 p.1 with cold cortisol
(1 p,g)
added as a carrier. The final ethanol concentration was 0.4 %. Aliquots of
this solution
were added to g lass tubes (100 ~.I) and the following extractions were
carried out: 1. 1
ml CH~CI2, vortex and pass through phase separating filter paper (Whatman,
IPS) 2. 1
ml ethyl acetate, vortex and pass through phase separating filter paper 3. 1
ml CH~CI2
and 200 ~.I 0.05 % CaCl2, vortex, centrifuge (500 X g for 5 min) and remove
upper
aqueous phase 4. 1 ml ethyl acetate and 200 ~,I 0.05 % CaCl2, vortex,
centrifuge (500 X
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
152
g for 5 min) and collect upper organic phase. The organic phases were dried
and the
residues were taken up in 100 g,1 IMS. An aliquot of this (50 ~I) was spotted
onto a TLC
plate and run as before. Following visualisation with Rhodamine B, the spots
were
scraped into scintillation vials and counted on a liquid scintillation counter
(TriCarb) in 5
ml Ultima gold scintillant. Extraction efficiencies were calculated and are
given in Figure
1.
Assay of Human and Rat Hepatic Microsomal 11 (3-HSD1 Activity using a TLC
Separation of Substrate from Product
Figure 2 (Comparison of 11 ~i-HSD1 activity in rat and human hepatic
microsomes)
This experiment was carried out to compare the enzyme activity in hepatic
microsomes
from human and rat and to assess minimum microsomal protein concentrations
necessary for reasonable measurement of enzyme activity. The assay was carried
out
in Buffer 2 and the cortisone concentration used was 2 ~M containing 0.5 ~Ci
per
incubation 3H-cortisone. Rat and human hepatic microsomes were tested at
concentrations ranging from 400 ~g to 50 wg microsomal protein per incubation
in a final
incubation volume of 100 w1 in glass tubes. Buffer was substituted for
microsomal
protein for blanks. Samples were incubated for 1 h in a shaking water bath at
37°C and
the assay was stopped by the addition of 1 ml ethyl acetate. To correct for
recovery, 50
wl'4C-cortisol (approximately 4000 DPM per tube) was added to the samples
followed by
200 g,1 0.05 % CaCl2. The samples were vortexed and centrifuged as detailed in
2.1.
The upper organic phase was removed into clean tubes and dried down. The
residue
was taken up in 100 p.1 IMS and 50 ~.I aliquots were spotted onto TLC plates
which were
run as described in 2.1. DPM were measured on a TriCarb liquid scintillation
counter
using a dual label programme. Recovery was determined from the DPM obtained in
50
w1 ~4C-cortisol solution which was counted with the samples. The results are
given in
Figure 2.
It had been expected that there would be higher activity in the rat microsomes
but this
was not the case. Using 50 wg microsomal protein per well the activities of
the rat and
human enzymes were quite similar, rat microsomal activity was 0.7 pmol/mg/min
and
human was 0.5 pmol/mg/min. Good activity was detected in the human microsomes
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
153
although this was not particularly related to microsomal protein
concentration. It was
suspected that the concentration range examined was too high so in the next
experiment
a lower range was tested. Also, the dependence of apparent enzyme activity
with
incubation time was looked at. A linear relationship between DPM and
incubation time
would indicate that increases over blank were due to enzyme activity and not
due to an
artefact.
Figure 3 (Effect of incubation time on human microsomal 11 (3-HSD1 activity)
and Fiqure
4 (Effect of microsomal protein concentration on human microsomal 11 (3-HSD1
activity)
In the next test, the same assay method was followed except that only human
hepatic
microsomes were examined and the concentration range of these was from 3.7 ~,g
per
sample to 100 p,g per sample. The samples were incubated for 60 min, 45 min,
30 min
or 15 min in a shaking water bath at 37°C and were stopped, extracted
and the
substrate and product were separated as detailed above. Figure 3 and Figure 4
illustrate the results:
Conclusion (Fig. 3):
~ There is linearity of enzyme activity with incubation times up to 30 min
with all
microsomal protein concentrations tested
Conclusion (Fig. 4):
~ Enzyme activity is linear with microsomal protein concentrations below 30
p.g per
sample
Figure 5 (Substrate (cortisone) saturation curve for human hepatic microsomal
1113
HSD11
Substrate requirement was examined using the classical assay. The DPM in each
group
was kept constant (0.5 ~,Ci l sample) and the cold cortisone was varied from 2
wM down
to 43.8 nM. The assay was carried out with 10 pg microsomal protein per sample
and
the incubation time was 30 min at 37°C. The buffer used for this assay
was Buffer 1
Figure 5 shows the data obtained.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
154
Figure 6 (Lineweaver-Burke Plot)
A double reciprocal plot of these data (Lineweaver-Burke) gives an apparent Km
for
cortisone of 660 nM, but it should be noted that it is unlikely that initial
enzyme activity
rates were measured at the lower [cortisone] over a 30 mins incubation. Figure
6 shows
the Lineweaver-Burke plot obtained:
Figure 7 (ICSO determination for Glycyrrhetinic acid) and Figure 8 (ICSO
determination for
Carbenoxolone)
In order to reproduce the inhibition data given in [14], it was decided to use
the cortisone
concentration quoted in the reference (175 nM) to examine compound activity,
even
though Figure 5 and Figure 6 suggest that this concentration is not saturating
with 10
~.g microsomal protein per 30 mins incubation at 37°C. The following
experiment was
carried out with a lower microsomal protein concentration (5 p,g) in the same
buffer
conditions as in the last experiment (Buffer 1) over a 30 mins incubation at
37°C in the
presence of 175 nM cortisone (0.5 wCi / sample). Glycyrrhetinic acid and
carbenoxolone
were examined at concentrations from 3 wM to 0.012p,M (DMSO concentration 1
throughout) and the data are shown in Figure 7 and Figure 8.
The reported ICSO for carbenoxolone is 330 nM [14], which is approximately
three times
less active than observed in the above experiment. It appears that these assay
conditions support good enzyme activity which should be measurable in a 96
well plate
method.
Cortisol Immunoassay
An enzyme immunoassay kit was obtained from Assay Designs, Inc. The antibody
provided in the kit is a mouse monoclonal reported to cross react 100% with
cortisol (the
enzyme product) and <0.1 % with cortisone (the enzyme substrate). The kit is
designed
for the analysis of cortisol levels in saliva, urine, serum and plasma and
also in tissue
culture media, not for microsomal enzyme activity.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
155
Figure 9 (1113-HSD1 activity measured by Immunoassay)
The methodology for the enzyme assay used with the kit was based on the paper
by
Barf et al. [14J Human hepatic microsomes were incubated in Buffer 1 at
concentrations
ranging from 25 ~.g protein per point to 200 ~g protein per point in the
presence of
cortisone ranging from 44 nM to 700 nM for 1 h. Also, these groups were tested
in the
presence and absence of 0.9 % Tween 80 since this detergent may improve the
activity
of enzymes involved in steroid metabolism. The basis of the assay is one of
competition
between the sample cortisol binding and the detector-cortisol binding. The
assay
detected the cortisol in the standard curve (313 pg / ml to 10,000 pg / ml) as
expected
but the signal obtained from the enzyme assay samples decreased with
increasing
microsomal protein concentration, suggesting that the presence of microsomes
interfered with the immunoassay. Figures 9(A), 9(B) and 9(C) shows some of the
data
obtained.
Figure 9(A) shows the effect of protein. Data taken from 700 ~,M cortisone
group tested
in the presence of Tween-80
Figure 9(B) shows the effect of cortisone. Data taken from the 25 wg
microsomal
protein group tested in the presence of Tween-80
Figure 9(C) shows the effect of Tween-80. Data taken from the 25 ~g microsomal
protein group tested in the presence of 700 wM cortisone
Conclusions:
~ Microsomal protein may interfere with the immunoassay (Figure 9(A))
~ Addition of exogenous cortisone had no effect on levels of cortisol detected
in the
enzyme assay samples (Figure 9(B))
~ Inclusion of detergent in the enzyme assay buffer had only a slight effect
(Figure
9(C))
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
156
Figure 10 (Performance of the Cortisol Immunoassay: various experimental
designs)
An 11 (3-HSD1 assay was carried out with 24 p,g microsomal protein / sample
and 2 p.M
cortisone substrate in buffer 2 Enzyme activity was also measured in samples
following
the addition of steroid displacement reagent (kit component) which releases
cortisol from
cortisol binding protein, if present in the sample. The assay detected the
cortisol in the
standard curve (313 pg / ml to 10,000 pg / ml). Figure 10 shows the absorbance
at 405
nm obtained for the different groups:
Conclusions:
~ The lowest and highest concentrations of the cortisol standard have been
included in
Figure 10 as 313 pg/ml and 1000 pg/ml together with the NSB absorbance to show
the dynamic range obtained in the assay.
~ Absorbance obtained in the presence of reaction mixture taken from samples
incubated with microsomal protein ("Enzyme") are lower than those in the
presence
of reaction mixture not containing microsomal protein ("No enzyme") indicating
increases in levels of cortisol.
~ In the presence of the kit steroid displacement reagent ("DR") these two
reaction
mixtures show the same pattern but the signal is depressed.
~ Glycyrrhetinic acid ("GA") in the presence of the top concentration of
cortisol
standard has no effect on the ability of the kit to measure cortisol
concentrations.
Figure 11 (Effect of increasinct microsomal protein on measurement of 11 (3
HSD1 activity
detected by Assay Designs Immunoassay)
When the enzyme assay data vvas calculated as enzyme activity using the pg/ml
cortisol
indicated by the standard curve, the blank value was 47 pg/ml / min incubation
and the
enzyme activity was 119 pg/ml / min, a signal to noise of 2.5. Although the
signal to
noise obtained is rather poor, these data demonstrate that the antibody can
bind the
cortisoI:AP conjugate and that this can be displaced by cortisol. An
experiment was
carried out to examine the effect of slightly increasing the microsomal
protein
concentration in an attempt to improve the signal to noise obtained.
Microsomal protein
was tested from 100 p,g / incubation down to 5 p.g / incubation using 2 pM
cortisone in
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
157
buffer 2. All other conditions were identical to those detailed above. The
results are
shown in Figure 11.
Conclusions:
~ Decreasing microsomal protein from 10 p,g / incubation to 5 p.g / incubation
results in
a corresponding decrease in enzyme activity.
~ Increasing microsomal protein above 10 p.g / incubation results in a
quenching of
signal which may be due to the colour of the microsomes.
~ The dynamic range of this assay cannot be improved by increasing the
microsomal
protein concentration.
Figure 12 (Detection of 11 a HSD1 activity by RIA using the Immunotech anti-
cortisol
antibod
The next experiment was carried out to assess the Immunotech antibody. The
enzyme
assay was carried out in Buffer 2. The substrate (cortisone) concentration of
175 nM
was chosen from the SPA method described by Barf et al. [14] with 0.5 p,Ci l
well 3H-
cortisone. The enzyme assay was carried out in a polypropylene plate in a
final
incubation volume of 100 ~I containing 10 p,g / well human hepatic microsomal
protein.
Blanks had either buffer substituted for microsomal protein or had 10 w1 stop
solution
added prior to the microsomes. The assay was incubated at 37°C for 30
mins and the
reaction was terminated by the addition of the stop solution to all remaining
wells. The
Immunotech antibody was diluted in Buffer 3 to give 25 wg / 100 p1 down to
6.25 ~g / 100
~,I. The antibody (100 p,1) was added to test wells, 100 p1 Buffer 3 was added
to the
antibody blank wells. The remainder of the procedures followed the 96 well
plate RIA
protocol exactly. Results demonstrating 11 (3 HSD1 activity using the
Immunotech are
shown in Figure 12.
Conclusions:
~ Good enzyme activity was detected with this antibody.
~ The signal to noise with 12.5 and 6.1 ~,g antibody per well was similar
hence it may
be possible to reduce the antibody concentration.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
IS$
Figure 13 (Effect of lowering the Immunotech antibody concentration on the
signal to
noise (microsome Group compared to GA blank group)) and Figure 14 (Immunotech
antibody saturation curve for detection of 11 (3 HSD1 activity by RIA)
The antibody titre was examined in the next test, investigating concentrations
per well
from 6.7 ~g down to 0.67 fig. The usual 11 (3 HSD1 assay was carried out
except that
the microsomal protein concentration was doubled to 20 ~,g / well in order to
get the best
signal to noise. The cortisone concentration was 175 nM and the enzyme assay
buffer
was Buffer 2. Each antibody concentration was tested against a "no enzyme"
blank
(buffer substituted for microsomes) and a "GA blank" (10 w1 stop solution
added prior to
microsomes) and a control group. The RIA vvas carried out exactly as indicated
in the
methods for assay in 96 wells. These results are shown in Figure 13 and Figure
14.
Conclusion:
~ The Immunotech antibody performed very well.
~ The saturation curve indicates that there is no difference in the detection
of enzyme
activity above 1.68 wg / well.
~ The antibody will be used at 1.7 ~g / well.
~ The signal to noise ratio (microsome CPM / microsome + GA blank CPM) with
this
concentration of antibody was 6 fold.
Figure 15 (Linearity of human hepatic microsornal 11 f3 HSD1 activity detected
by RIA)
Linearity of enzyme activity with human hepatic microsomal protein
concentration using
RIA detection was examined in the next test. The usual 11 (3 HSD1 assay was
carried
out except that the microsomal protein concentration was varied from 40 ~g /
well down
to 1 ~,g / well. The cortisone concentration v~ras 175 nM and the enzyme assay
buffer
was Buffer 2. 11 (3 HSD1 activity was linear with protein up to concentrations
of 20 ~,g /
well confirming the results obtained with the classical enzyme assay (Figure
4). Data
from these experiments are shown in Figure 15.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
159
Fiaure 16 (Effect of Tween 80 on detection of human hepatic microsomal 11 (3
HSD1
activity by RIA)
The effect of including Tween 80 in the enzyme assay buffer was also
investigated. This
assay was carried out in parallel with the assay above and under the same
conditions
except that the enzyme assay buffer (Buffer 2) contained 0.05 % Tween 80.
Microsomal
protein was tested at four concentrations. Tween 80 was found to increase the
blank
CPM, reducing the signal to noise of the assay. The data in Figure 16 are
taken from
the group tested with 10 ~g / well microsomal protein, but the same effect was
seen with
all protein concentrations examined.
Conclusion:
~ Including 0.05 % Tween 80 in the enzyme assay buffer increases the CPM
obtained
in the blanks thereby reducing the signal to noise to 3 fold (compared to 5
fold in the
absence of Tween 80).
Figure 17 (Effect of buffer systems on detection of human hepatic microsomal
11 (3
HDS1 activity by RIA)
To simplify the protocol such that both enzyme assay and RIA stages are
carried out in
the same buffer, both phases were carried out in either enzyme assay buffer
(buffer 2)
or buffer 3 (RIA buffer). The microsomal protein concentration used was 10 g,g
/ well
and the cortisone concentration was 175 nM. Performing both enzyme assay and
RIA in
enzyme assay buffer gave similar data to the two buffers system but performing
both
enzyme assay and RIA in Buffer 3 appeared to improve the data slightly. These
results
are highlighted in Figure 17.
Figure 18 (Linearity of human hepatic microsomal 1113 HSD1 activity with
incubation time
detected by RIA)
Linearity of enzyme activity with incubation time was investigated. The enzyme
assay
was carried out exactly as indicated in the methods section in buffer 3 with
10 p,g / well
microsomal protein and with 175 nM cortisone. The reaction was stopped at the
times
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
160
indicated in Figure 18 by the addition of 10 ~,I stop solution. The RIA was
carried out
exactly as indicated in the methods section. Figure 18 illustrates these
results.
Conclusion:
~ 30 min incubation is within the linear range of enzyme activity with 10 ~g /
well
microsomal protein and 175 nM substrate.
It is possible that 175 nM substrate is low. The apparent Km observed in the
classical
11 [3 HSD1 assay was 660 nM (Figure 5 and Figure 6), although these assays are
end-
point measurement, hence it is not certain that initial rates were measured in
the low
substrate groups with a 30 min incubation time. However, there are published
Km
values which suggest that the actual Km for cortisone in human hepatic
microsomal 11 (3
HSD1 assays is in the micromolar range [31, 32]. Even though 175 nM substrate
is well
below the apparent Km, it may not be possible to increase the concentration
significantly
for two reasons:
(i) If the compounds are competitive with cortisone, the measured inhibiton
will fall if
the substrate is increased above the concentration used in Reference [14].
(ii) Increasing the substrate will reduce the specific activity of the label,
reducing the
CPM and the sensitivity of the assay - this could be overcome by adding higher
concentrations of 3H-cortisone
Figure 19 (Substrate saturation curve for human hepatic microsomal 11 (3 HDS1
activity
detected by RIA)
The substrate saturation effects were examined in the next assay. The enzyme
assay
was carried out exactly as indicated in the methods section in buffer 3 with
10 ~g / well
microsomal protein and with [cold cortisone] as indicated. 3H-cortisone was
0.5 p,Ci /
sample throughout. The reaction was stopped after 30 min by the addition of 10
w1 stop
solution. The RIA was carried out exactly as indicated in the methods section.
Results
are shown in Figure 19.
Inspection of the data shown in Figure 19 shows that 10 ~.g microsomal protein
is not
saturated with 175 nM cortisone over an incubation period of 30 mins. The
apparent Km
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
161
(700 nM), determined from the Lineweaver-Burke plot of these data shown in
Figure 20
is very similar to that determined in the classical 11[3 HSD1 assay (Figure 6,
apparent
Km 660 nM).
Figure 20 (Lineweaver-Burke plot)
Lowering the microsomal protein concentration or the incubation time to fit
well within the
linear range would partly overcome the problem, but both adjustments would
decrease
the assay sensitivity. All of the tests carried out so far suggest that even
if increasing
the microsomal protein from 10 p.g / sample to 20 ~g / sample does not result
in a
doubling of enzyme activity, decreasing it from 10 ~.g / sample to 5 pg l
sample does
result in a twofold decrease in enzyme activity. Since the purpose of the
assay is to
monitor inhibitory effects of compounds it is probably a better course of
action to leave
the assay parameters as they are.
Figure 21 (ICSO curve for inhibition of human hepatic microsomal 11 (3 HSD1
activity by
Gl~yrrhetinic acid)
In order to assess the quality of compound inhibition data obtained in this
assay format,
an ICSO for Glycyrrhetinic acid was determined in the next test. A 10 mM stock
solution
of Glycyrrhetinic acid was prepared in 100 % DMSO and was further diluted in
100
DMSO to 0.3 mM. This solution was serially diluted in 100 % DMSO 1 in 3 to
obtain the
test range and each solution was diluted in assay buffer (Buffer 3) 1 in 25.
These
solutions were diluted into the final enzyme reaction 1 in 4 to give assay
concentrations
from 3 wM down to 0.012 pM in a final [DMSO] of 1 %. Controls, NSB (no
antibody) and
GA blanks (addition of 10 p,1 stop solution prior to the addition of
microsomes) were
included with and without the addition of 1 % DMSO. Human hepatic microsomal
protein
was tested at 10 wg / well and the substrate concentration (cortisone) was 175
nM, 0.5
wCi / well. All other procedures were as indicated in the methods section. GA
inhibition
data are shown in Figure 21.
The assay control and blank CPM are given in the Table below.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
162
Table. Control and blank CPM obtained in the Glycyrrhetinic acid IC5°
assay showing
effect of 1 % DMSO and signal to noise ratio obtained.
Group 1 % DMSO No DMSO
NSB 670 661
GA blank 640 660
Control 3515 2583
Signal to noise 5 fold 4 fold
Conclusion:
~ Glycyrrhetinic acid gives a concentration-related inhibition of the enzyme
with
reasonable fit values (r2 = 0.962) and Hillslope (Figure 21).
~ In the classical enzyme assay (Figure 7) an ICSO of 40 nM was determined for
this
inhibitor, which is similar to the data given in Figure 21.
~ Glycyrrhetinic acid inhibition of human hepatic microsomal 11 [3 HSD1 using
dehydro-
dexamethasone as the substrate has been reported to have an ICSO of 30 nM [32]
~ However, the compound appears more active than suggested by Barf et al.,
(2002)
[14]
~ Inclusion of DMSO at 1 % in the enzyme assay does not affect NSB or blank
values
but may slightly increase enzyme activity and the signal to noise ratio
~ These data suggest that RIA detection of 11 (3 HSD1 activity can generate
acceptable
compound inhibition data.
Figure 22 (ICSO curve for inhibition of human hepatic microsomal 11 f3 HSD1
activity by
Glycyrrhetinic acid in the presence of 350 nM cortisone) and Ficlure 23 (ICSO
curve for
inhibition of human hepatic microsomal 11~i HSD1 activity by Carbenoxolone in
the
luresence of 350 nM cortisone)
In the next experiment, Glycyrrhetinic acid and its hemisuccinate ester,
carbenoxolone,
were tested for ICSO determination.. In view of the higher than expected
inhibitory activity
obtained with Glycyrrhetinic acid (Figure 22) in the last test and the
apparent non-
saturation obtained with 175 nM cortisone (Figure 19), the substrate
concentration was
increased to 350 nM (0.5 p,Ci / well). In addition, the final DMSO
concentration was
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
163
reduced to 0.3 %. The assay was performed as indicated in the Methods in RIA
buffer.
Results with some of these compounds are shown in Figure 22 and Figure 23 .
Conclusion (Fig. 22):
~ Increasing the substrate concentration and decreasing the DMSO concentration
has
little effect on Glycyrrhetinic acid inhibitory activity.
Conclusion (Fig. 23):
~ Even in the presence of a higher concentration of cortisone, carbenoxolone
is about
5 times more active than is suggested Barf et al. [14]. This could be due to
an effect
of the buffer system used here (Buffer 3) since it differs from that Barf et
al., [14]
(Buffer 1). This compound was less active (ICSO of 119 nM) in the classical
enzyme
assay, which used the buffer described by Bart et al. [14] (Figure 8).
All publications mentioned in the above specification are herein incorporated
by
reference. Various modifications and variations of the described methods and
system of
the invention will be apparent to those skilled in the art without departing
from the scope
and spirit of the invention. Although the invention has been described in
connection with
specific preferred embodiments, it should be understood that the invention as
claimed
should not be unduly limited to such specific embodiments. Indeed, various
modifications of the described modes for carrying out the invention which are
obvious to
those skilled in chemistry or related fields are intended to be within the
scope of the
following claims.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
164
REFERENCES
1. Hammond, GH ( 1990) : Molecular properties of corticosteroid binding
globulin and
sex-steroid binding proteins. Endocr. Rev. 11, 65- 79.
2. Gomez-Sanchez EP,Gomex-Sanchez CE (1997): First there was one, then two
..why
not more 11 (3-Hydroxysteroid Dehydrogenases? Endocrinology vol. 138, 12.
3. Krozowski ~S, Funder JW (1983): Renal mineralocorticosterone receptors and
hippocampal corticosterone binding species have identical intrinsic steroid
specificity .
Proc. Natl. Sci. USA 80: 6056-60
4. Ulick S, Levine LS, Gunczler P, ~anconato G, Rarnirez LC, Rauh W, Rosler A,
Bradlow HL, Mew MI (1979): A syndrome of apparent mineralocorticoid excess
associated with defects in the peripheral metabolism of cortisol. J. Clin.
Endo. And
Metab. 49: 757-64.
5. Edwards CRW, Stewart PM, Burt D, Brett L, Mclntyre MA, Sutanto WS, Kloet
ER,
Monder C (1998): Localisation of 11 (3-HSD-tissue specific protector of the
mineralocorticoid receptor. Lancet 2: 986-989.
6. Moore CCD, Melloh SH, Murai I, Siiteri PK, Miller WL (1993): Structure and
function
of the hepatic form of 11 (3-HSD in the squirrel monkey, an animal model of
glucocorticoid resistance. Endocrinology 133: 368-375.
7. Kotelevtsev YV, larnieson PM, Best R, Stewart F, Edwards CRW, Seckl JR,
Mullins II
( 1996): Inactivation of 11 (3-HSD type 1 by gene targeting in mice.
Endocrinology Res.
22: 791-792.
8. Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM
(1998):
Irnmunohistochemicallocalisation of type 1 11 (3-HSD in human tissues. I.
Clin. Endoc.
Metab. 83: 1325-35.
9. Stewart PM, Sheppard MC (1992): Novel aspects ofhorrnone action:
intracellular
ligand supply and its control by a series of tissue specific enzymes.
Molecular and
Cellular Endocrinology 83: C13-C18.
10. Seckl JR, Chapman KE (1997): The 11 (3-HSD system, a determinant of
glucocorticoid and mineralocorticoid action. Medical and physiological
aspects.
European I. Biochem. 249: 361-364.
11. Maser E (1998): 11 (3-HSD responsible for carbonyl reduction of the
tobacco-
specific nitrosoamine in mouse lung microsomes. Cancer Res. 58: 2996-3003.
12. Walker BR, Stewart PM, Shackleton C H L, Padfield PL, Edwards CRW (1993):
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
165
Deficient inactivation of cortisol by 11 ~3-HSD in essential hypertension.
Clin. Endocr.
38: 221-227.
13. Daynes RA, Araneo BA (1998) : Contrasting effects of glucocorticoids on
the
capacity of T -cells to produce the growth factors interleukin-2 and
interleukin-4. Eur. J.
I m mu nol. 19: 2319-2324.
14. Barf, T. et al., (2002), Arylsulfonamidothiazoles as a new class of
potential
antidiabetic drugs. Discovery of potent and selective inhibitors of the 11 (3-
Hydroxysteroid
Dehydrogenase Type 1. J. Med. Chem., 45, 3813-3815.
15. Matassa, Victor G. et. al. J. Med. Chem.; 33(9); 1990; 2621.
16. This compound is synthesized in the literature and the NMR spectrum is
reported,
however the spectrum obtained here differs from that in the literature_
Baraldi, Pier
Giovanni et. al.; Bioorg. & Med. Chem. Lett.; 10; 2002, 1611.
17. Horaguchi, Takaaki; Matsuda, Shinichi; Tanemura, Kiyoshi; Suzuki, Tsuneo.
J.
Heterocyclic Chem.; 24; 1987; 965.
18. Ple, Patrick A., Marnett, Lawrence J.; J. Heterocyclic Chem.; 25; 1988;
1271.
19. Rao, U. and Balasubramanian, K. K.; Tetrahedron Lett.; 24; 1983; 5023.
20. Bordwell, F.G. and Stange, Hugo; J. Amer. Chem. Soc.; 77; 1955; 5939.
21. Elderfield, Robert C.; Williamson, Thurmond A.; Gensler, Walter J.;
Kremer,
Chester B.; J. Org. Chem; 12; 1947; 405.
22. For 6-nitro-2,3-dimethylquinoxaline see: Barluenga, Jose; Aznar, Fernando;
Liz,
Ramon; Cabal, Maria-Paz; Synthesis; 3; 1985; 313., then for 6-amino-2,3-
dimethylquinoxaline: Salon, Jozef; Milata, Viktor; Pronayova, Nadezda; Lesko,
Jan;
Collect. Czech. Chem. Commun.; 66; 11; 2001; 1691.
23. Klicnar, J.; and Kosek, F.; Collect. Czech. Chem. Commun.; 30; 1965; 3102.
24. Gloster, Daniel F.; Cincotta, Louis; Foley, James W.; J. Heterocyclic
Chem.; 36;
1999; 25.
25. The same reduction was carried out using SnCh by: Case et al.; J. Amer.
Chem.
Soc.; 81; 1959; 6297.
26. Modified procedure from similar compound described in US Patent 6,355,796
(Example 20)
27. Hollfelder, F.; Kirby, A. J.; Tawfik, D. S.; Kikuchi, K.; Hilvert, D.; J_
Amer. Chem.
Soc.; 122 (6); 2000; 1022-1029
28. Fujimoto, M.; Okabe, K.; Chem.Pharm.Bull.; 10; 1962; 572-575.
CA 02540843 2006-03-30
WO 2005/042513 PCT/GB2004/004498
166
29. Kawamura, T.; Yagi, N.; Sugawara, H.; Yamahata, K.; Takada, M.;
Chem.Pharm.Bull.; 28; 1; 1980; 268-276.
30. Stewart, P.M. and Mason, J.I., (1995), Cortisol to cortisone:
Glucocorticoid to
mineralocortcoid. Steriods, 60,143-146.
31. Escher, G. et al., (1995), Furosemide inhibits 11 (3-Hydroxysteroid
Dehydrogenase in
vitro and in vivo. Endocrinology, 136, 1759-1765.
32. Hult, M. et. al., (1998), Selective inhibition of human type 1 11 (3-
hydroxysteroid
dehydrogenase by synthetic steroids and xenobiotics. FEBS Letters, 441, 25-28.
33. Diederich S, Grossmann C, Hanke B, Quinkler M, Herrrnann M, Bahr V,
Oelkers W
(2000): In the search for specific inhibitors ofhuman 11 (3-HSD:
chenodeoxycholic acid
selectively inhibits 11 (3-HSD type 1. Europ. J. Endocrin. 142: 200-207.