Note: Descriptions are shown in the official language in which they were submitted.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
FUSED LACTAM COMPOUNDS
1
Technical Field
This invention relates to novel fused lactam compounds. These compounds
are useful as antagonists, of NMDA (N-methyl-D-aspartate) NR2B receptor, and
are
thus useful for the treatment of pain, stroke, traumatic brain injury,
Parkinson's
disease, Alzheimer's disease, depression, anxiety, migraine, or the like in
mammalian,
especially humans. The present invention also relates to a pharmaceutical
composition comprising the above compounds.
Background Art
Glutamate plays dual role in the central nervous system (CNS) as essential
amino acid and the principal excitatory neurotransmitters. There are at least
four
classes of receptors, specifically N-methyl-aspartate (NMDA), 2-amino-3-
(methyl-3-
hydroxyisoxazol-4-yl)propionic acid, (AMPA), kainate and metabotropic. There
is
considerable preclinical evidence that hyperalgesia and allodynia following
peripheral
tissue or nerve injury is not only due to an increase in the sensitivity of
primary
afferent nociceptors at the site of injury but also depends on NMDA receptor-
mediated central changes in synaptic excitability. In humans, NMDA receptor
antagonists have also been found to decrease both pain perception and
sensitization.
Also, overactivation of NMDA receptor is a key event for triggering neuronal
cell
death under pathological conditions of acute and chronic forms of
neurodegeneration.
However, while NMDA receptor inhibition has therapeutic utility in the
treatment of
pain and neurodegenerative diseases, there are significant liabilities to many
available
NMDA receptor antagonists that can cause potentially serious side effects.
NMDA
subunits are differentially distributed in the CNS. Especially, NR2B is
believed to
be restricted to the forebrain and laminas I and II of the dosal horn. The
more
discrete distribution of NR2B subunit in the CNS may support a reduced side-
effect
profile of agents that act selectively at this site.
For example, NMDA NR2B selective antagonists may have clinical utility
for the treatment of neuropathic and other pain conditions in human with a
reduced
side-effect profile than existing NMDA antagonists (S. Royce, et al.,
Neuropharmacology, 38, pp.611-623 (1999)).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
2
International Publication Number WO 91/17156 and W094/10166 discloses
a variety of 3,4-dihydroquinolin-2(II~-one compounds. Especially, a compound
represented by the following formula is disclosed in WO 94110166:
However, the known compounds have potential to prolong the QT-interval
due to their potent inhibitory activity at HERG (human ether-a-go-go related
gene)
potassium channel. QT prolongation is known to have a potential liability to
produce fatal cardiac arrhythmias of Torsades de Pointes (TdP) . The ability
to
prolong the cardiac action potential duration was identified as being due to
an action
at the HERG potassium channel. For example, drugs withdrawn from the market
due to QT prolongation, such as Cisapride and Terfenadine, are known to be
potent
HERG potassium channel Mocker (Expert Opinion of Pharmacotherapy.; 2, pp947-
973, 2000). Therefore, it would be desirable if there were provided a novel
NMDA
NR2B selective antagonist with analgesic activity by systemic administration
and
with reduced inhibitory activity at HERG potassium channel.
Brief Disclosure of the Invention
It has now surprisingly been found that fused lactam compounds of present
invention are NMDA NR2B selective antagonists with analgesic activity by
systemic
administration and with reduced inhibitory activity at HERG channel.
Inhibitory
activity at HERG channel was estimated from affinity for HERD type potassium
channel was investigated by checking [3H]dofetilide binding, which can predict
inhibitory activity at HERG channel (Eur. J. Pharmacol., 430, pp147-148,
2001).
Selected compounds with low [3H]dofetilide binding activity were evaluated in
I~RG
assay to check activity at HERG channel. The compounds of the present
invention
show a reduced QT prolongation by removing a methyl group from the carbon atom
adjacent to nitrogen atom on piperidine ring of the formula (I).
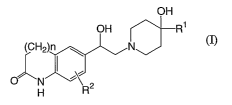
The present invention provides a compound of the following formula (I):
Compound A
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
3
OH
OH R1
(CH2)n ~ N
O N ~~ a
H R
(n
wherein
R1 represents an aryl group having from 6 to 10 ring carbon atoms or a
heteroaryl
group having from 5 to 10 ring atoms which consists of from 1 to 4 heteroatoms
independently selected from the group consisting of sulfur atoms, oxygen atoms
and
nitrogen atoms;
R2 represents a hydrogen atom, a halogen atom, or an alkyl group having from 1
to 6
carbon atoms; and
n represents 0, 1 or 2;
said heteroaryl group is unsubstituted or substituted and said aryl is
substituted by at
least one substituent selected from the group consisting of substituents oc;
said substituents oc are selected from the group consisting of halogen atoms,
alkyl
groups having from 1 to 6 carbon atoms, alkoxy groups having from 1 to 6
carbon
atoms, alkoxyalkyl groups having from 1 to 6 carbon atoms, alkylthio groups
having
from 1 to 6 carbon atoms, mono- or di-alkylamino groups having from 1 to 6
carbon
atoms in said alkyl groups or two adjacent a groups are optionally joined
together to
form an alkylene chain having 3 or 4 carbon atoms in which one or two (non-
adjacent) carbon atoms are optionally replaced by oxygen atoms;
with the proviso that said aryl group is substituted by at least one
alkoxyalkyl group
having from 1 to 6 carbon atoms
or a pharmaceutically acceptable ester of such compound,
or a pharmaceutically acceptable salt thereof.
The fused lactam compounds of this invention have an antagonistic action
towards NMDA NR2B receptor subtype selectively and are thus useful in
therapeutics,
particularly for the treatment of stroke or brain injury, chronic
neurodegenerative
disease such as Parkinson's disease, Alzheimer's disease, Huntington's disease
or
amyotrophic lateral sclerosis (ALS), epilepsy, convulsive disorder, pain,
anxiety,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
4
human immunodeficiency virus (HIV) related neuronal injury, migraine,
depression,
schizophrenia, tumor, post-anesthesia cognitive decline (PACD), glaucoma,
tinnitus,
tradive dyskinesia, allergic encephalomyelitis, opioid tolerance, drug abuse,
alcohol
abuse, Irritable bowel syndrome (IBS), or the like in mammalian, especially
humans.
The compounds of the present invention are useful for the general
treatment of pain, particularly neuropathic pain. Physiological pain is an
important
protective mechanism designed to warn of danger from potentially injurious
stimuli
from the external environment. The system operates through a specific set of
primary sensory neurons and is exclusively activated by noxious stimuli via
peripheral
transducing mechanisms (Millan 1999 Prog. Neurobio. 57: 1-164 for an
integrative
Review). These sensory fibres are known as nociceptors and are characterized
by
small diameter axons with slow conduction velocities. Nociceptors encode the
intensity, duration and quality of noxious stimulus and by virtue of their
topographically organized projection to the spinal cord, the location of the
stimulus.
The nociceptors are found on nociceptive nerve fibres of which there are two
main
types, A-delta fibres (myelinated) and C fibres (non-myelinated). The activity
generated by nociceptor input is transferred after complex processing in the
dorsal
horn, either directly or via brain stem relay nuclei to the ventrobasal
thalamus and
then on to the cortex, where the sensation of pain is generated.
Intense acute pain and chronic pain may involve the same pathways driven
by pathophysiological processes and as such cease to provide a protective
mechanism
and instead contribute to debilitating symptoms associated with a wide range
of
disease states. Pain is a feature of many trauma and disease states. When a
substantial injury, via disease or trauma, to body tissue occurs the
characteristics of
nociceptor activation are altered. There is sensitisation in the periphery,
locally
around the injury and centrally where the nociceptors terminate. This leads to
hypersensitivity at the site of damage and in nearby normal tissue. In acute
pain
these mechanisms can be useful and allow for the repair processes to take
place and
the hypersensitivity returns to normal once the injury has healed. However, in
many
chronic pain states, the hypersensitivity far outlasts the healing process and
is
normally due to nervous system injury. This injury often leads to
maladaptation of
the afferent fibres (Woolf & Salter 2000 Science 288: 1765-1768). Clinical
pain is
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
present when discomfort and abnormal sensitivity feature among the patient's
symptoms. Patients tend to be quite heterogeneous and may present with various
pain symptoms. There are a number of typical pain subtypes: 1) spontaneous
pain
which may be dull, burning, or stabbing; 2) pain responses to noxious stimuli
are
5 exaggerated (hyperalgesia); 3) pain is produced by normally innocuous
stimuli
(allodynia) (Meyer et al., 1994 Textbook of Pain 13-44). Although patients
with
back pain, arthritis pain, CNS trauma, or neuropathic pain may have similar
symptoms, the underlying mechanisms are different and, therefore, may require
different treatment strategies. Therefore pain can be divided into a number of
different areas because of differing pathophysiology, these include
nociceptive,
inflammatory, neuropathic pain etc. It should be noted that some types of pain
have
multiple aetiologies and thus can be classified in more than one area, e.g.
Back pain,
Cancer pain have both nociceptive and neuropathic components.
Nociceptive pain is induced by tissue injury or by intense stimuli with the
potential to cause injury. Pain afferents are activated by transduction of
stimuli by
nociceptors at the site of injury and sensitise the spinal cord at the level
of their
termination. This is then relayed up the spinal tracts to the brain where pain
is
perceived (Meyer et al., 1994 Textbook of Pain 13-44). The activation of
nociceptors activates two types of afferent nerve fibres. Myelinated A-delta
fibres
transmitted rapidly and are responsible for the sharp and stabbing pain
sensations,
whilst unmyelinated C fibres transmit at a slower rate and convey the dull or
aching
pain. Moderate to severe acute nociceptive pain is a prominent feature of, but
is not
limited to pain from strains/sprains, post-operative pain (pain following any
type of
surgical procedure), posttraumatic pain, burns, myocardial infarction, acute
pancreatitis, and renal colic. Also cancer related acute pain syndromes
commonly due
to therapeutic interactions such as chemotherapy toxicity, immunotherapy,
hormonal
therapy and radiotherapy. Moderate to severe acute nociceptive pain is a
prominent
feature of, but is not limited to, cancer pain which may be tumour related
pain, (e.g.
bone pain, headache and facial pain, viscera pain) or associated with cancer
therapy
(e.g. postchemotherapy syndromes, chronic postsurgical pain syndromes, post
radiation syndromes), back pain which may be due to herniated or ruptured
intervertabral discs or abnormalities of the lumber facet joints, sacroiliac
joints,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
6
paraspinal muscles or the posterior longitudinal ligament.
Neuropathic pain is defined as pain initiated or caused by a primary lesion
or dysfunction in the nervous system (IASP definition). Nerve damage can be
caused by trauma and disease and thus the term 'neuropathic pain' encompasses
many
disorders with diverse aetiologies. These include but are not limited to,
Diabetic
neuropathy, Post herpetic neuralgia, Back pain, Cancer neuropathy, HIV
neuropathy,
Phantom limb pain, Carpal Tunnel Syndrome, chronic alcoholism, hypothyroidism,
trigeminal neuralgia, uremia, or vitamin deficiencies. Neuropathic pain is
pathological
as it has no protective role. It is often present well after the original
cause has
dissipated, commonly lasting for years, significantly decreasing a patients
quality of
life (Woolf and Mannion 1999 Lancet 353: 1959-1964). The symptoms of
neuropathic pain are difficult to treat, as they are often heterogeneous even
between
patients with the same disease (Woolf & Decosterd 1999 Pain Supp. 6: S 141-S
147;
Woolf and Mannion 1999 Lancet 353: 1959-1964). They include spontaneous pain,
which can be continuous, or paroxysmal and abnormal evoked pain, such as
hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia
(sensitivity to
a normally innocuous stimulus).
The inflammatory process is a complex series of biochemical and cellular
events activated in response to tissue injury or the presence of foreign
substances,
which result in swelling and pain (Levine and Taiwo 1994: Textbook of Pain 45-
56).
Arthritic pain makes up the majority of the inflammatory pain population.
Rheumatoid disease is one of the commonest chronic inflammatory conditions in
developed countries and rheumatoid arthritis is a common cause of disability.
The
exact aetiology of RA is unknown, but current hypotheses suggest that both
genetic
and microbiological factors may be important (Grennan & Jayson 1994 Textbook
of
Pain 397-407). It has been estimated that almost 16 million Americans have
symptomatic osteoarthritis (OA) or degenerative joint disease, most of whom
are over
60 years of age, and this is expected to increase to 40 million as the age of
the
population increases, making this a public health problem of enormous
magnitude
(Houge & Mersfelder 2002 Ann Pharmacother. 36: 679-686; McCarthy et al., 1994
Textbook of Pain 387-395). Most patients with OA seek medical attention
because of
pain. Arthritis has a significant impact on psychosocial and physical function
and is
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
7
known to be the leading cause of disability in later life. Other types of
inflammatory
pain include but are not limited to inflammatory bowel diseases (IBD),
Other types of pain include but are not limited to;
- Musculo-skeletal disorders including but not limited to myalgia,
fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-
articular
rheumatism, dystrophinopathy, Glycogenolysis, polymyositis, pyomyositis.
- Central pain or 'thalamic pain' as defined by pain caused by lesion or
dysfunction of the nervous system including but not limited to central post-
stroke pain,
multiple sclerosis, spinal cord injury, Parkinson's disease and epilepsy.
- Heart and vascular pain including but not limited to angina, myocardical
infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma,
scleredoma, skeletal muscle ischemia.
- Visceral pain, and gastrointestinal disorders. The viscera encompasses the
organs of the abdominal cavity. These organs include the sex organs, spleen
and
part of the digestive system. Pain associated with the viscera can be divided
into
digestive visceral pain and non-digestive visceral pain. Commonly encountered
gastrointestinal (GI) disorders include the functional bowel disorders (FBD)
and the
inflammatory bowel diseases (IBD). These GI disorders include a wide range of
disease states that are currently only moderately controlled, including - for
FBD,
gastro-esophageal reflux, dyspepsia, the irritable bowel syndrome (IBS) and
functional abdominal pain syndrome (FAPS), and - for IBD, Crohn's disease,
ileitis,
and ulcerative colitis, which all regularly produce visceral pain. Other types
of
visceral pain include the pain associated with dysmenorrhea, pelvic pain,
cystitis and
pancreatitis.
- Head pain including but not limited to migraine, migraine with aura,
migraine without aura cluster headache, tension-type headache.
- Orofacial pain including but not limited to dental pain,
temporomandibular myofascial pain.
The present invention provides a pharmaceutical composition for the
treatment of disease conditions caused by overactivation of NMDA NR2B
receptor,
in a mammalian subject, which comprises administering to said subject a
therapeutically effective amount of a compound of formula (I).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
8
Further, the present invention also provides a composition which comprises a
therapeutically effective amount of the cycloalkylene amide compound of
formula (1)
or its pharmaceutically acceptable salt together with a pharmaceutically
acceptable
carrier. Among them, the composition is preferably for the treatment of
disease
defined above.
Also, the present invention provides for the use of a compound of formula (>),
or a pharmaceutically acceptable ester of such compound, or a pharmaceutically
acceptable salt thereof, as a medicament.
Also, the present invention provides a method for the treatment of disease
conditions defined above, which comprises administering to said subject a
therapeutically effective amount of a compound of formula (I).
Further, the present invention provides a method for the treatment of
disease conditions defined above in a mammal, preferably human, which
comprises
administering to said subject a therapeutically effective amount of a compound
of
formula (n.
Yet further, the present invention provides the use of a therapeutically
effective amount of a compound of formula (n in the manufacture of a
medicament
for the treatment of the disease conditions defined above.
Detailed Description of the Invention
As used herein, the term "halogen" means fluoro, chloro, bromo and iodo,
preferably fluoro or chloro.
As used herein, the term "alkyl" means straight or branched chain saturated
radicals, including, but not limited to methyl, ethyl, n-propyl, isopropyl, n-
butyl, iso-
butyl, secondary-butyl, tertiary-butyl.
As used herein, the term "alkoxy" means alkyl-O-, including, but not limited
to methoxy, ethoxy, rz-propoxy, isopropoxy, ra-butoxy, iso-butoxy, secondary-
butoxy,
tertiary-butoxy.
As used herein, the term "alkoxyalkyl" means alkyl-O-alkyl, including, but
not limited to methoxymethyl, methoxyethyl, methoxypropyl, methoxybutyl,
ethoxymethyl, ethoxyethyl, ethoxypropyl, ethoxbutyl, ra-propoxymethyl, h-
propoxyethyl, n-propoxypropyl, n-propoxybutyl, isopropoxymethyl,
isopropoxyethyl,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
9
isopropoxypropyl, isopropoxybutyl, n-butoxymethyl, ~-butoxyethyl, n-
butoxypropyl,
iso-butoxymethyl, iso-butoxyethyl, iso-butoxypropyl, iso-butoxybutyl,
secorzdary
butoxymethyl, secondary-butoxyethyl, secondary-butoxypropyl, seco~zdary
butoxybutyl, tertiary-butoxymethyl, tertiary-butoxyethyl, tertiary-
butoxypropyl or
tertiary-butoxybutyl.
As used herein, the term "alkylene", as used herein, means a saturated
hydrocarbon (straight chain or branched) wherein a hydrogen atom is removed
from
each of the terminal carbons such as methylene, ethylene, methylethylene,
propylene,
butylene, pentylene, hexylene and the like.
As used herein, the term "aryl a monocyclic or bicyclic aromatic carbocyclic
ring of 6 to 10 carbon atoms, including, but not limited to, phenyl or
naphtyl,
preferably phenyl.
The term "heteroaryl" means a 5- to 10-membered monocyclic or bicyclic
aromatic heterocyclic ring which consists of from 1 to 4 heteroatoms
independently
selected from the group consisting of sulfur atoms, oxygen atoms and nitrogen
atoms
including, but not limited to, pyrazolyl, furyl, thienyl, oxazolyl,
tetrazolyl, thiazolyl,
imidazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyrrolyl, thiophenyl,
pyrazinyl,
pyridazinyl, isooxazolyl, isothiazolyl, triazolyl, furazanyl, quinolyl,
isoquinolyl,
tetrahydroquinolyl, tetrahydroisoquinolyl, chromanyl or isochromanyl group,
and the
like.
The term "ordinary protecting group" means a protecting group, which can
be cleaved by a chemical method such as hydrogenolysis, hydrolysis,
electrolysis or
photolysis.
The term "esters " means a protecting group which can be cleaved in vivo by
a biological method such as hydrolysis and forms a free acid or salt thereof.
Whether
a compound is such a derivative or not can be determined by administering it
by
intravenous injection to an experimental animal, such as a rat or mouse, and
then
studying the body fluids of the animal to determine whether or not the
compound or a
pharmaceutically acceptable salt thereof can be detected.
Preferred examples of groups for an ester of a hydroxy group include: lower
aliphatic alkanoyl groups, for example: alkanoyl groups, such as the formyl,
acetyl,
propionyl, butyryl, isobutyryl, pentanoyl, pivaloyl, valeryl, isovaleryl,
octanoyl,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
nonanoyl, decanoyl, 3-methylnonanoyl, 8-methylnonanoyl, 3-ethyloctanoyl, 3,7-
dimethyloctanoyl, undecanoyl, dodecanoyl, tridecanoyl, tetradecanoyl,
pentadecanoyl,
hexadecanoyl, 1-methylpentadecanoyl, 14-methylpentadecanoyl, 13,13-
dimethyltetradecanoyl, heptadecanoyl, 15-methylhexadecanoyl, octadecanoyl, 1-
5 methylheptadecanoyl, nonadecanoyl, icosanoyl and henicosanoyl groups;
halogenated
alkylcarbonyl groups, such as the chloroacetyl, dichloroacetyl,
trichloroacetyl, and
trifluoroacetyl groups; alkoxyalkylcarbonyl groups, such as the methoxyacetyl
group;
and unsaturated alkylcarbonyl groups, such as the acryloyl, propioloyl,
methacryloyl,
crotonoyl, isocrotonoyl and (E)-2-methyl- 2-butenoyl groups; more preferably,
the
10 lower aliphatic alkanoyl groups having from 1 to 6 carbon atoms; aromatic
alkanoyl
groups, for example: arylcarbonyl groups, such as the benzoyl, a -naphthoyl
and ~3 -
naphthoyl groups; halogenated arylcarbonyl groups, such as the 2-bromobenzoyl
and
4-chlorobenzoyol groups; lower alkylated arylcarbonyl groups, such as the 2,
4,6-
trimethylbenzoyl and 4-toluoyl groups; lower alkoxylated arylcarbonyl groups,
such
as the 4-anisoyl group; nitrated arylcarbonyl groups, such as the 4-
nitrobenzoyl and 2-
nitrobenzoyl groups; lower alkoxycarbonylated arylcarbonyl groups, such as the
2-
(methoxycarbonyl)benzoyl group; and arylated arylcarbonyl groups, such as the
4-
phenylbenzoyl group; alkoxycarbonyl groups, for example: lower alkoxycarbonyl
groups, such as the methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl,
butoxycarbonyl, sec-butoxycarbonyl, t-butoxycarbonyl and isobutoxycarbonyl
groups;
and halogen- or tri(lower alkyl)silyl-substituted lower alkoxycarbonyl groups,
such as
the 2,2,2-trichloroethoxycarbonyl and 2-trimethylsilylethoxycarbonyl groups;
tetrahydropyranyl or tetrahydrothiopyranyl groups, such as: tetrahydropyran- 2-
yl, 3-
bromotetrahydropyran-2-yl, 4-methoxytetrahydropyran-4-yl, tetrahydrothiopyran-
2-yl,
and 4-methoxytetrahydrothiopyran-4-yl groups; tetrahydrofuranyl or
tetrahydrothiofuranyl groups, such as: tetrahydrofuran-2-yl and
tetrahydrothiofuran-
2-yl groups; silyl groups, for example: tri(lower alkyl)silyl groups, such as
the
trimethylsilyl, triethylsilyl, isopropyldimethylsilyl, t-butyldimethylsilyl,
methyldiisopropylsilyl, methyldi-t-butylsilyl and triisopropylsilyl groups;
and
tri(lower alkyl)silyl groups substituted by 1 or 2 aryl groups, such as the
diphenylmethylsilyl, diphenylbutylsilyl, diphenylisopropylsilyl and
phenyldiisopropylsilyl groups; alkoxymethyl groups, for example: lower
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
11
alkoxymethyl groups, such as the methoxymethyl, 1,1-dimethyl-1-methoxymethyl,
ethoxymethyl, propoxymethyl, isopropoxymethyl, butoxymethyl and t-butoxymethyl
groups; lower alkoxylated lower alkoxymethyl groups, such as the 2-
methoxyethoxymethyl group; and halo(lower alkoxy)methyl groups, such as the
2,2,2-
trichloroethoxymethyl and bis(2-chloroethoxy)methyl groups; substituted ethyl
groups,
for example: lower alkoxylated ethyl groups, such as the 1-ethoxyethyl and 1-
(isopropoxy)ethyl groups; and halogenated ethyl groups, such as the 2,2,2-
trichloroethyl group; aralkyl groups, for example: lower alkyl groups
substituted by
from 1 to 3 aryl groups, such as the benzyl, a -naphthylmethyl, ~3 -
naphthylmethyl,
diphenylmethyl, triphenylmethyl, a - naphthyldiphenylmethyl and 9-
anthrylmethyl
groups; and lower alkyl groups substituted by from 1 to 3 substituted aryl
groups,
where one or more of the aryl groups is substituted by one or more lower
alkyl, lower
alkoxy, nitro, halogen or cyano substituents, such as the 4-methylbenzyl,
2,4,6-
trimethylbenzyl, 3,4,5-trimethylbenzyl, 4-methoxybenzyl, 4-
s15 methoxyphenyldiphenylmethyl, 2-nitrobenzyl, 4-nitrobenzyl, 4-chlorobenzyl,
4-
bromobenzyl and 4-cyanobenzyl groups; alkenyloxycarbonyl groups: such as the
vinyloxycarbonyl and aryloxycarbonyl groups; and aralkyloxycarbonyl groups in
which the aryl ring may be substituted by 1 or 2 lower alkoxy or nitro groups:
such as
the benzyloxycarbonyl, 4-methoxybenzyloxycarbonyl, 3,4-
dimethoxybenzyloxycarbonyl, 2-nitrobenzyloxycarbonyl and 4-
nitrobenzyloxycarbonyl groups.
The term "treating", as used herein, refers to reversing, alleviating,
inhibiting
the progress of, or preventing the disorder or condition to which such term
applies, or
one or more symptoms of such disorder or condition. The term "treatment" as
used
herein refers to the act of treating, as "treating" is defined immediately
above.
A preferred compound of formula (n of this invention is that wherein
represents
a phenyl group being substituted by at least one alkoxyalkyl groups having
from 1 to
6 carbon atoms or a heteroaryl group having from 5 to 10 ring atoms which
consists
of from 1 to 2 heteroatoms independently selected from the group consisting of
sulfur
atoms, oxygen atoms and nitrogen atoms; said heteroaryl groups having from 5
to 10
atoms are unsubstituted or are substituted by at least one substituent
selected from the
group consisting of substituents oc; said substituents oc; are selected from
the group
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
12
consisting of halogen atoms, alkyl groups having from 1 to 6 carbon atoms,
alkoxy
groups having from 1 to 6 carbon atoms, alkoxyalkyl groups having from 1 to 6
carbon atoms, alkylthio groups having from 1 to 6 carbon atoms or mono- or di-
alkylamino groups having from 1 to 6 carbon atoms in said alkyl groups.
. More preferably R1 represents a phenyl group being substituted by at least
one
alkoxyalkyl groups having from 1 to 6 carbon atoms, a thiazolyl group, an
isothiazolyl
group, an oxazolyl group, an isoxazolyl group, a pyrrolyl group, a pyridyl
group, a
pyrimidine group, a quinolyl group, an isoquinolyl group, a tetrahydroquinolyl
group,
a tetrahydroisoquinolyl group, a chromanyl group or an isochromanyl group.
said thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, pyrrolyl, pyridyl,
pyrimidine,
quinolyl, isoquinolyl, tetrahydroquinolyl, tetrahydroisoquinolyl, chromanyl or
isochromanyl groups are unsubstituted or are substituted by at least one
substituent
selected from the group consisting of substituents a; said substituents oc;
are selected
from the group consisting of halogen atoms, alkyl groups having from 1 to 6
carbon
atoms, alkoxy groups having from 1 to 6 carbon atoms, alkoxyalkyl groups
having
from 1 to 6 carbon atoms, alkylthio groups having from 1 to 6 carbon atoms or
mono-
or di-alkylamino groups having from 1 to 6 carbon atoms in said alkyl groups.
Most preferably R1 represents a phenyl group being substituted by at least one
alkoxyalkyl groups having from 1 to 6 carbon atoms, a thiazolyl group, a
pyridyl
group, or an isochromanyl group. When Rl is phenyl, it is preferably
substituted by
one alkoxyalkyl group having from 1 to 6 carbon atoms, preferably para. No
substitution or one or two substituents are preferred when Rl is heteroaryl.
When Rl
is monocyclic heteroaryl, one or two substituents are preferred.
A preferred compound of formula (~ of this invention is that wherein R2
represents
a hydrogen atom or a halogen atom. More preferably, R2 represents a hydrogen
atom or a fluorine atom.
A preferred compound of formula (n of this invention is that wherein n
represents 1 or 2.
A preferred individual compound of this invention is selected from
6-{ 2-[4-(6-chloro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
13
3,4-dihydroquinolin-2(lI~-one;
5-fluoro-6-{ 2-[4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-3,4-dihydroquinolin-2( lI~-one;
6-{ 2-[4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yl]-1-hydroxyethyl }-
3,4-
dihydroquinolin-2(lI~-one;
6-(2-{ 4-[5-(dimethylamino)-6-fluoropyridin-2-yl]-4-hydroxypiperidin-1-y1 }-1-
hydroxyethyl)-3,4-dihydroquinolin-2( lI~-one;
6-{2-[4-(3,4-Dihydro-1H isochromen-7-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-5-fluoro-3,4-dihydroquinolin-2(lI~-one;
5-fluoro-6-{ 1-hydroxy-2.-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-
yl]ethyl}-
3,4-dihydroquinolin-2( 11~-one;
6-{ 1-hydroxy-2-[4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yl]ethyl}-3,4-
dihydroquinolin-2( 11~-one;
6- { 1-Hydoxy-2 [4-methyl-4-(3-methylisothiazole-5-yl)-1-yl] ethyl } -3,4-
dihydroquinolin-2(1H)-one;
6-{ 1-hydroxy-2-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yl]ethyl}-3,4-
dihydroquinolin-2( l I~-one;
6-{ 1-hydroxy-2-[4-hydroxy-4-(4-methylpyridin-2.-yl)piperidin-1-yl]ethyl}-3,4-
dihydroquinolin-2(ll~-one;
6-{2-[4-(3,4-Dihydro-1H isochromen-7-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-3,4-dihydroquinolin-2( lI~-one;
6-( 1-Hydroxy-2- { 4-hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-yl }
ethyl)-3,4-
dihydroquinolin-2( l I~-one;
7-{ 1-hydroxy-2-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yl]ethyl}-
1,3,4,5-
tetrahydro-2H 1-benzazepin-2-one;
. 6-{ 2-[4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yl]-1-hydroxyethyl }-
3,4-
dihydroquinolin-2( l I~-one;
5-fluoro-6-(1-hydroxy-2-{4-hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-
yl}ethyl)-3,4-dihydroquinolin-2(1F~-one; and
6-{2-[4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl}-
3,4-dihydroquinolin-2( 11~-one;
or a pharmaceutically acceptable salt thereof.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
14
A further preferred individual compound of this invention is selected from
6-{ 1-hydroxy-2-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yl]ethyl}-3,4-
dihydroquinolin-2( 1I~-one;
6-{ 1-hydroxy-2-[4-hydroxy-4-(4-methylpyridin-2-yl)piperidin-1-yl]ethyl }-3,4-
dihydroquinolin-2(lI~-one;
6-{2-[4-(3,4-Dihydro-1H isochromen-7-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-3,4-dihydroquinolin-2( 1F~-one;
6-( 1-Hydroxy-2-{ 4-hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-yl } ethyl)-
3,4-
dihydroquinolin-2( 1I~-one;
7-{ 1-hydroxy-2-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yl]ethyl}-
1,3,4,5-
tetrahydro-2H 1-benzazepin-2-one;
6-{ 2-[4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yl]-1-hydroxyethyl }-3,4-
dihydroquinolin-2( 1~-one;
5-fluoro-6-(1-hydroxy-2-{4-hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-
y1 } ethyl)-3,4-dihydroquinolin-2( lI~-one; and
6-{ 2-[4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yl]-1-
hydroxyethyl }-
3,4-dihydroquinolin-2( 1F~-one;
or a pharmaceutically acceptable salt thereof.
General Synthesis
The compounds of the present invention may be prepared by a variety of
processes well known for the preparation of compounds of this type, for
example as
shown in the following reaction Schemes. Unless otherwise indicated Rl, RZ and
R3
in the reaction Schemes and discussion that follow are defined as above. The
term
"protecting group", as used hereinafter, means a hydroxy or anuno protecting
group
which is selected from typical hydroxy or amino protecting groups described in
Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John
Wiley &
Sons, 1991);
The following reaction Schemes illustrate the preparation of compounds of
formula (1].
Scheme 1:
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
This illustrates the preparation of compounds of formula (~.
R1 -Y
(1-1)
Step 1 A
R1 -~ off
PG1 N~ _ (1 2) R1 deprotection
PGy N~ .N
(1-3) Step 1 B Step 1 C H
(1_4) (1_5)
O
(~ z) ~ X
O N I ~ 2 OH OH
R O R1 reduction OH N R1
H (1-6) (CH2) ~ N~ (CH2)
,
Step 1 D o H R2 (1-~) Step 1 E O H R2
(I)
In the above formula, X represents a leaving group. Example of suitable
leaving groups include: halogen atoms, such as chlorine, bromine and iodine;
sulfonic
5 esters such as Tf0 (triflates), Ms0 (mesylates), Ts0 (tosylates); and the
like. Y
represents a hydrogen atom, a halogen atom such as, fluorine, chlorine,
bromine or
iodine; .L represents metal such as lithium, or MgY. PG1 represents a
protecting
group. The term "protecting group", as used herein, means a hydroxy or amino
protecting group which is selected from typical hydroxy or amino protecting
groups
10 described in Protective Groups in Organic Synthesis edited by T. W. Greene
et al.
(John Wiley & Sons, 1991). Typical hydroxy or amino protecting groups include
benzyl, CZH50(C=O)-, CH3(C=O)-, t-butyldimethylsilyl (TBS), t-
butyldiphenylsilyl,
benzyloxycarbonyl represented as Z and t-buthoxycarbonyl represented as t-Boc
or
Boc.
15 Step 1A
In this Step, the organometallic compound of formula (1-2) can be prepared
by reaction of a halide compound of formula (1-1). This reaction may be
carried out
in the presence of an organometallic reagent or a metal. Examples of suitable
organometallic reagents include; alkyllithiums such as n-butyllithium, sec-
butyllithium and tent-butyllithium; aryllithiums such as phenyllithium and
lithium
naphtilide. Examples of suitable metal include magnesium. . Preferred reaction
inert solvents include, for example, hydrocarbons, such as hexane; ethers,
such as
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
16
diethyl ether, diisopropyl ether, dimethoxyethane (DME) tetrahydrofuran (THF)
and
dioxane; or mixtures thereof. Reaction temperatures are generally in the range
of
100 to 50 °C, preferably in the range of from -100 °C. to room
temperature.
Reaction times are, in general, from 1 minute to a day, preferably from 1 hour
to 10
hours.
Step 1B
In this Step, an alcohol compound of formula (1-4) can be prepared by the
nucleophilic addition of a ketone compound of formula (1-3) with the
organometallic
compound of formula (1-2). The reaction may be carried out in the presence of
a
solvent. Examples of suitable solvents include for example, hydrocarbons, such
as
hexane; ethers, such as diethyl ether, diisopropyl ether, dimethoxyethane
(DME)
tetrahydrofuran (THF) and dioxane; or mixtures thereof. Reaction temperatures
are
generally in the range of -100 to 50 °C, preferably in the range of
from -100 °C. to
room temperature. Reaction times are, in general, from 1 minute to a day,
preferably
from 1 hour to 10 hours.
Step 1C
In this Step, the desired compound of formula (1-5) may be prepared by the
deprotection of the compound of formula 1-4, prepared as described in Step 1B,
according to known procedures such as those described in Protective Groups in
Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1991).
In the case of Bn or Z protection, the removal of the protecting groups may
be carried out under, for example, known hydrogenolysis conditions in the
presence
of a metal catalyst under hydrogen atmosphere or in the presence of hydrogen
sources
such as formic acid or ammonium formate in a reaction inert solvent. If
desired, the
reaction is carried out under acidic conditions, for example, in the presence
of
hydrochloric acid or acetic acid. A preferred metal catalyst is selected from,
for
example, palladium-carbon, palladiumhydroxide-carbon, platinumoxide, platinum-
carbon, ruthenium-carbon, rhodium-aluminumoxide, tris[triphenyphosphine]
rhodiumchlrodie. Example of suitable reaction inert aqueous or non-aqueous
organic solvents include: alcohols, such as methanol, ethanol; ethers, such as
tetrahydrofuran or dioxane; acetone; dimethylformamide; halogenated
hydrocarbons,
such as dichloromethane, dichloroethane or chloroform; and acetic acid or
mixtures
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
17
thereof. The reaction may be carried out at a temperature in the range from of
20 °C
to 100 °C, preferably in the range of 20°C to 60°C.
Reaction times are, in general,
from 10 minutes to 48 hours, preferably 30 minutes to 24 hours. This reaction
may
be carried out under hydrogen atmosphere at a pressure ranging from 1 to 100
atom,
preferably from 1 to 10 atom.
Step 1D
In this Step, the desired beta-carbonyl piperidne compound of formula 1-7
may be prepared by the coupling of a halide compound of formula 1-6 with the
piperidine compound of formula 1-5 in an inert solvent, e.g. aliphatic
hydrocarbons,
such as hexane, heptane and petroleum ether; aromatic hydrocarbons, such as
benzene,
toluene, xylene and nitrobenzene; halogenated hydrocarbons, such as methylene
chloride, chloroform, carbon tetrachloride and dichloroethane; ethers, such as
diethyl
ether, diisopropyl ether, tetrahydrofuran and dioxane; alcohols, such as
methanol,
ethanol, propanol, isopropanol and butanol; and dimethylformamide (DMF),
dimethylsulfoxide (DMSO), 1,3-dimethyl-2-imidazolidinone(DMn or acetonitrile.
This reaction may be carried out in the presence of a base, e.g. an alkali or
alkaline
earth metal hydroxide, alkoxide, carbonate, or hydride, such as sodium
hydroxide,
potassium hydroxide, sodium methoxide, sodium ethoxide, potassium tent-
butoxide,
sodium carbonate, potassium carbonate, cesium carbonate, sodium hydride or
potassium hydride, or an amine such as triethylamine, tributylamine,
diisopropylethylamine, pyridine or dimethylaminopyridine. This reaction may be
carried out in the presence of a suitable additive, e.g.
tetrakis(triphenylphosphine)-
palladium, bis(triphenylphosphine)palladium(II) chloride, copper(0), copper(
acetate,
copper(n bromide, copper(1] chloride, copper(n iodide, copper( oxide, copper(n
trifluoromethanesulfonate, copper(In acetate, copper(In bromide, copper(In
chloride,
copper(I~ iodide, copper(1~ oxide, 1,10-phenanthroline, dibenzanthracene(DBA)
or
copper(II] trifluoromethanesulfonate. The reaction may be carried out at a
temperature in the range from of 0 °C to 100 °C, preferably in
the range of 20°C to
100°C. Reaction times are, in general, from 5 minutes to 48 hours,
preferably 30
minutes to 24 hours.
Step 1E
In this Step, the desired compound of formula (~ can be prepared by the
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
18
reduction of the ketone compound of formula (1-7) with a reducing agent, e.g.
NaBH4,
LiAlH4, LiBH4, or ZnBH4 in an inert solvent, e.g. methanol, ethanol, diglyme,
or
mixtures thereof. The reaction may be carried out at a temperature in the
range from
of 0 °C to 100 °C, preferably in the range of 20°C to
80°C. Reaction times are, in
general, from 5 minutes to 48 hours, preferably 30 minutes to 24 hours.
Scheme 2
OR3
~I 4
O HN~OR OR3 4 OR3
(CH2) w X ~(2-1 ) (CH2j \ O NOR reduction OH N~OR4
I I (CH2)
O~N ~R2 Step 2A O~N I ~ z Step 2B
H (1-6) H R (2-2) H R~
Ri -Y
(1-1 )
Step 2D
OH
OH ~O R1 -L OH ~Ri
deprotection ~ H2) I ~ ,N J (~ (~ 2j I \ , ~N
Step 2C ~ H /R2 (2-4) Step 2E O H R2
(I)
In the above formula, R3 and R4 represents an alkyl group or R3 and R4 may
be joined together to form an ethylene or a propylene group; said ethylene or
propylene group are optionally substituted by hydroxy groups.
Step 2A
In this Step, a desired beta-carbonyl piperidne compound of formula 2-2 may
be prepared by the coupling of a halide compound of formula 1-6 with an ketal
piperidine compound of formula 2-1. This reaction is essentially the same as
and
may be carried out in the same manner as and using the same reagents and
reaction
conditions as Step 1D in Scheme 1.
Step 2B
In this Step, an alcohol compound of formula (2-3) can be prepared by the
reduction of the ketone compound of formula (2-2) with a reducing agent. This
reaction is essentially the same as and may be carried out in the same manner
as and
using the same reagents and reaction conditions as Step 1E in Scheme 1.
Step 2C
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
19
In this Step, a piperidone compound of formula (2-4) can be prepared by the
deprotection of the ketal compound of formula (2-3) in the presence or the
absence of
a catalyst in a reaction-inert solvent.
The hydrolysis reaction may be carried out in an aqueous or non-aqueous
organic
solvent. Examples of suitable solvents include: alcohols, such as methanol or
ethanol; ethers, such as tetrahydrofuran or dioxane; acetone;
dimethylformamide;
halogenated hydrocarbons, such as dichloromethane, dichloroethane or
chloroform;
acids, such as acetic acid, hydrogen chloride, hydrogen bromide and sulfuric
acid.
Example of suitable catalysts include: hydrogen halides, such as hydrogen
chloride
and hydrogen bromide; sulfonic acids, such as p-toluenesulfonic acid and
benzenesulfonic acid; ammonium salts, such as pyridium p-toluenesulfonate and
ammonium chloride; and carboxylic acid, such as acetic acid and
trifluoroacetic acid.
This reaction can be carried out at temperature of 0 ~C to 200 ~C, preferably
from
about 20 ~C to 120 ~C for 5 minutes to 48 hours, preferably 30 minutes to 24
hours.
Step 2D
In this Step, the organometallic compound of formula (1-2) can be prepared
by reaction of a halide compound of formula (1-1) in the same manner as and
using
the same reagents and reaction conditions as Step 1A in Scheme 1.
Step 2E
In this Step, the desired compound of formula (~ can be prepared by the
nucleophilic addition of the ketone compound of formula (2-4) with the
organometallic compound of formula (1-2). This reaction is essentially the
same as
and may be carried out in the same manner as and using the same reagents and
reaction conditions as Step 1B in Scheme 1.
Scheme 3
Y'
~1 -H 2 ~ ~1 -Y
3-1 Step 3A 3-2
In the above formula, Y' represents a halogen atom such as chlorine, bromine
or iodine.
Step 3A
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
The halogenated inetermediate compound 3-2 may be generally prepared by
halogenation with a halogenating reagent in a reaction-inert solvent. Examples
of
suitable solvents include: such as aqueous or non-aqueous organic solvents
such as
tetrahydrofuran, dioxane, acetone, dimethylformamide, acetonitrile;
halogenated
5 hydrocarbons, such as dichloromethane, dichloroethane or chloroform; and
acetic acid.
Suitable halogenating reagents include, for example, bromine, chlorine,
iodine, N-
chlorosuccimide, N-bromosuccimide, 1,3-dibromo-5,5-dimethylhydantoin,
bis(dimethylacetamide)hydrogen tribromide, tetrabutylammonium tribromide,
bromodimethylsulfonium bromide, hydrogen bromide-hydrogen peroxide,
10 nitrodibromoacetonitrile or copper(II] bronude. The reaction can take place
over a
wide range of temperatures, and the precise reaction temperature is not
critical to the
invention. The preferred reaction temperature will depend upon such factors as
the
nature of the solvent, and the starting material or reagent used. However, in
general,
we find it convenient to carry out the reaction at a temperature of from 0 ~C
to 200 ~C,
15 more preferably from 20 ~C to 120 ~C. The time required for the reaction
may also
vary widely, depending on many factors, notably the reaction temperature and
the
nature of the reagents and solvent employed. However, provided that the
reaction is
effected under the preferred conditions outlined above, a period of 5 minutes
to
4~hours, more preferably 30 minutes to 24 hours, will usually suffice.
Scheme 4
R5OOC (CH2)n \ HO2C~.(CH2)n \
R500C~ ~ ,y ~ .y
O2N R2 O2N R2
(4-1 ) Step 4A (4-2)
O
~~ O
(CH2)n \ X " X (CH2)n \
i
O N 2 O N ~~ 2
Step 4B H 4-3 Step 4C H R
( )
(1-6)
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
21
In the above formula, R5 represents an alkyl group.
Step 4A
In this Step, a desired beta-carboxylic acid compound of formula 4-2 may be
prepared by the decarboxylation of a di-ester compound of formula 4-1.
Hydrolytic
decarboxylation can be carried out in a reaction inert solvent. Example of
suitable
solvents include: alcohols, such as methanol and ethanol; ethers
tetrahydrofuran and
dioxane; dimethylformamide. The solvents contain an aqueous alkaline solution
such as sodium hydroxide, potassium hydroxide and potassium carbonate. This
reaction can be carried out at a temperature in the range from 0 °C to
100 °C,
preferably from about 20 °C to 80 °C for 5 minutes to 48 hours,
preferably 30 minutes
to 24 hours. Then the reaction mixture can be acidified with an acid. Example
of
suitable acids include: hydrogen halide, such as hydrogen chloride and
hydrogen
bromide; sulfonic acids, such as p-toluenesulfonic acid and benzenesulfonic
acid;
ammonium salts, such as pyridium p-toluenesulfonate and ammonium chloride;
carboxylic acids, such as acetic acid and trifluoroacetic acid. This reaction
can be
carried out at a temperature in the range from 0 °C to 200 °C,
preferably from about
°C to 120 °C for 5 minutes to 48 hours, preferably 30 minutes to
24 hours.
Step 4B
In this Step, a cyclized compound of formula (4-3) can be prepared by the
20 cyclization of the carboxylic acid compound of formula (4-2) with a
reducing agent in
an inert solvent, e.g. methanol, ethanol, ethyl acetate, THF or mixtures
thereof. The
reduction may be carried out under known hydrogenation conditions in the
presence
of a metal catalyst, e.g. nickel catalysts such as Raney nickel, palladium
catalysts such
as Pd(OH)2-C, platinum catalysts such as Pt02, or ruthenium catalysts such as
RuCl2
(Ph3P)3 under hydrogen atmosphere or in the presence of hydrogen sources such
as
hydrazine or formic acid. If desired, the reaction is carried out under acidic
conditions, e.g. in the presence of hydrochloric acid or acetic acid. The
reduction
may also be carried out in the presence of a suitable reducing agent, e.g.
LiAlH4,
LiBH4, Fe, Sn or Zn, in a reaction inert solvent, e.g. methanol, ethanol,
diglyme,
benzene, toluene, xylene, o-dichlorobenzene, dichloromethane, dichloroethane,
tetrahydrofuran, dioxane, or mixtures thereof; or without solvent. If desired,
when a
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
22
reducing reagent is Fe, Sn or Zn, the reaction is carried out under acidic
conditions in
the presence of water.
Step 4C
In this Step, a beta-halocarbonyl compound of formula (1-6) can be prepared
by the acylation of the compound of formula (4-3) under the Friedel-Crafts
reaction
condition. This reaction may be carried out in an inert solvent. Examples of
suitable solvents include: halogenated hydrocarbons, such as dichloromethane,
dichloroethane or chloroform; aromatic hydrocarbons, such as nitrobenzene and
chlorobenzene. Example of suitable catalysts include: aluminum halide, such as
aluminum chloride and aluminum ch bromide. This reaction can be carried out at
temperature of -50 ~C to 200 ~C, preferably from about -10 ~C to 150 ~C for 5
minutes
to 48 hours, preferably 30 minutes to 24 hours.
in the same manner as and using the same reagents and reaction conditions as
Step 1B
in Scheme 1.
The starting materials in the aforementioned general syntheses may be
commercially available or obtained by conventional methods known to those
skilled
in the art.
In the above Schemes from 1 to 4, examples of suitable solvents include a
mixture of any two or more of those solvents described in each Step.
The compounds of formula (1], and the intermediates above-mentioned
preparation methods can be isolated and purified by conventional procedures,
such as
recrystallization or chromatographic purification.
The optically active compounds of this invention can be prepared by several
methods. For example, the optically active compounds of this invention may be
obtained by chromatographic separation, enzymatic resolution or fractional
crystallization from the final compounds.
Method for assessing biological activities:
NR2B binding Assay
The activity of the bicyclic amide compounds of the present invention, as
NR2B antagonists, is determined by their ability to inhibit the binding of
NR2B
subunit at its receptor sites employing radioactive ligands.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
23
The NR2B antagonist activity of the bicyclic amide compounds is evaluated
by using the standard assay procedure described in, for example, J.
Pharmacol., 331,
pp117-126, 1997. This method essentially involves determining the
concentration of
the individual compound required to reduce the amount of radiolabelled NR2B
ligands by 50% at their receptor sites, thereby affording characteristic ICSO
values for
each compound tested. More specifically, the assay is carried out as follows.
Membranes were prepared by homogenization of forebrain of male CD rats
weighing between 170 190 g by using glass-Teflon homogenizer in 0.32 M sucrose
at
4°C. The crude nuclear pellet was removed by centrifugation at 1000xg
for 10 min,
and the supernatant centrifuged at 17000xg for 25 min. The resulting pellet
was
resuspended in 5 mM Tris acetate pH 7.4 at 4°C for 10 min to lyse
cellular particles
and again centrifuged at 17000xg. The resulting pellet (P2 membrane) was
washed
twice in Tris acetate, resuspended at 5.5 mg proteinlml and stored at -
20°C until use.
All the manipulation was done on ice, and stock solution and equipment were
kept on
ice at all time.
For the saturation assay, receptor saturation was determined by incubating
[3H]-CP-98,113 and 50 ~,g protein of P2 membrane for 60 minutes at room
temperature in a final 100 ~ul of incubation buffer (50 mM Tris HCI, pH7.4).
Total
and non-specific bindings (in the presence of 10 ~,M of unlabeled CP-98,113)
were
determined in a range of [3H]-CP-98113 concentrations (0.625 nM to 60nM). [3H]-
CP-98,113 is as follows:
T
OH
OH
N
T
CH3 3
H~ (wherein T is tritio ( H)).
For the competition assay, test compounds were incubated in duplicate with
5 nM [3H]-CP-98,113 and 50 ~g protein of P2 membrane for 60 minutes at room
temperature in a final 100 ~,l of 50 mM Tris HCl buffer (pH7.4). Nonspecific
binding was determined by 10 ~,M of unlabeled CP-98,113 (25 ~,1). The
saturation
derived IUD gained in saturation assay was used for all Ki calculations.
All incubations were terminated by rapid vacuum filtration over 0.2%
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
24
polyethyleneimine soaked Whatman GFB glass fibre filter paper using a SKATRON
cell harvester followed by three washes with ice-cold filtration buffer (5 mM
Tris HCI,
pH 7.4.). Receptor-bound radioactivity was quantified by liquid scintillation
counting
using Packard LS counter. Competition assays were performed by counting Wallac
GFB filters on Betaplate scintillation counter (Wallac).
All compounds prepared in the working examples as described below were
tested by this method, and they showed Ki values from 4 nM to 30 nM with
respect to
inhibition of binding at the NR2B receptor.
Human NR2B cell functional assay
HEK293 cells stably expressing human NRlb/2B receptor were used for cell
functional assay. Cells were grown in 75-cm2 culture flasks, using Dulbecco's
modified Eagle's medium (DMEM, high glucose) supplemented with 10% fetal
bovine, 52 ~,glml Zeocin, 530 ~,g/ml Geneticin, 100 unitslml penicillin and
100 ~.g/ml
,15 ' streptomycin. Cells were maintained in a humidified atmosphere in 5% C02
at 37°C,
and 50-60% confluent cells were harvested by 0.05% trypsin containing 0.53 mM
EDTA. The day before the experiment, expression of NRlb/2B receptor was
induced by 5 p.M ponasteron A in DMEM (40 ml) in the presence of 400 pM
ketamine to prevent excitotoxicity. The induction was performed for 19-24
hours,
using 50-60% confluent cells.
Cells were washed with 10 ml of Ca2+-free I~rebs-Ringer Hepes buffer
(KRH) containing 400 p.M ketamine, and the loading of 5 p,M fura-2
acetoxymethyl
ester was made for 2hrs at room temperature in the presence of 400 p.M
ketamine in
Ca2+-free KRH (10 ml). Subsequently, cells were collected in 50 ml tube by
pipetting manipulation and centrifuged at . 850 rpm for 2 min. Supernatant was
removed, and cells were washed with 10 ml of Ca2+-free KRH buffer, followed by
centrifugation again. This manipulation was repeated 4 times to remove
ketamine,
glutamate and glycine. Cells were re-suspended in Ca2+-free KRH buffer, and 50
~,1 of
cell suspension was added to each well of 96-well plates at a density of
100,000
cells/well, followed by adding test compounds dissolved in 50 p,1 of Ca2+-free
KRH.
After pre-incubation for 30 min, agonists (final 100 p,M glutamic acid and 10
~,M
glycine) dissolved in 25 ~,l of KRH containing 9 mM Ca2+ (final 1.8 mM) were
added.
Fura-2 fluorescence (excitation wavelengths: 340 nm and 380 nm; emission
wavelengths 510-520 nm) was monitored with a fluorescence imaging system,
FDSS6000. The 0 fluorescence ratio F340/F380 (i.e., the fluorescence ratio
immediately post-agonist - the basal fluorescence ratio; calculated as AUC)
was used
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
for evaluation of drug effects on agonists-induced changes in intracellular
Ca2+. The
basal fluorescence ratio was determined in the presence of 10 ~,M MK-801.
rat haloneridol-induced catalepsy assay:
5 Fasted male CD rats were used (7-8 weeks old). Test compound or
vehicle was given subcutaneously then haloperidol 0.5 mg/kg s.c.. Sixty
minutes
after haloperidol-injection, the duration of catalepsy was quantified by
placing the
animals forepaws on an elevated bar and determining the latency to remove both
forepaws from the bar. The cutoff latency was 60 seconds. Experimenter was
10 blind to treatments during testing.
Human dofetilide binding
Human HERG transfected HEK293S cells were prepared and grown in-house. The
collected cells were suspended in 50 mM Tris-HCl (pH 7.4 at 4°C) and
homogenized
15 using a hand held Polytron PT 1200 disruptor set at full power for 20 sec
on ice. The
homogenates were centrifuged at 48,000 x g at 4 °C for 20 min. The
pellets were then
resuspended, homogenized, and centrifuged once more in the same manner. The
final
pellets were resuspended in an appropriate volume of 50 mM Tris-HCI, 10 mM
KCI,
1 mM MgCl2 (pH 7.4 at 4°C), homogenized, aliquoted and stored at -
80°C until use.
20 An aliquot of membrane fractions was used for protein concentration
determination
using BCA protein assay kit (PIERCE) and ARVOsx plate reader (Wallac).
Binding assays were conducted in a total volume of 200 ~,1 in 96-well plates.
Twenty
~,l of test compounds were incubated with 20 ~,1 of [3H]-dofetilide (Amersham,
final 5
nM) and 160 ~,1 of membrane homogenate (25 ~g protein) for 60 minutes at room
25 temperature. Nonspecific binding was determined by 10 ~M dofetilide at the
final
concentration. Incubation was terminated by rapid vacuum filtration over 0.5%
presoaked GF/B Betaplate filter using Skatron cell harvester with 50 mM Tris-
HCl,
10 mM KCI, 1 mM MgCl2, pH 7.4 at 4°C. The filters were dried, put into
sample
bags and filled with Betaplate Scint. Radioactivity bound to filter was
counted with
Wallac Betaplate counter.
All compounds prepared in the working examples as described below showed
a TI (TI is a value of { Dofetilide Binding Ki [~,M ]/ NR2B Binding Ki
[nM]x1000}
value in the range of 900-3900, whereas a structurally similar comparative
compound
A showed a TI value of 490.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
26
IrrFR~ assay
HEK 293 cells which stably express the HERG potassium channel were used
for electrophysiological study. The methodology for stable transfection of
this
channel in HEK cells can be found elsewhere (Z.Zhou et al., 1998, Biophysical
journal, 74, pp230-241). Before the day of experimentation, the cells were
harvested
from culture flasks and plated onto glass coverslips in a standard MEM medium
with
10% FCS. The plated cells were stored in an incubator at 37°C
maintained in an
atmosphere of 95%02/5%C02. Cells were studied between 15-28hrs after harvest.
HERD currents were studied using standard patch clamp techniques in the
whole-cell mode. During the experiment the cells were superfused with a
standard
external solution of the following composition (mM); NaCI, 130; KCI, 4; CaCl2,
2;
MgCh, 1; Glucose, 10; HEPES, 5; pH 7.4 with NaOH. Whole-cell recordings was
made using a patch clamp amplifier and patch pipettes which have a resistance
of 1-
3MOhm when filled with the standard internal solution of the following
composition
(mM); KCI, 130; MgATP, 5; MgCl2, 1.0; HEPES, 10; EGTA 5, pH 7.2 with KOH.
Only those cells with access resistances below 15MS2 and seal resistances
>1GS2 was
accepted for further experimentation. Series resistance compensation was
applied
up to a maximum of 80%. No leak subtraction was done. However, acceptable
access resistance depended on the size of the recorded currents and the level
of series
resistance compensation that can safely be used. Following the achievement of
whole cell configuration and sufficient for cell dialysis with pipette
solution (>5min),
a standard voltage protocol was applied to the cell to evoke membrane
currents. The
voltage protocol is as follows. The membrane was depolarized from a holding
potential of -80mV to +20mV for 1000ms. This was followed by a descending
voltage ramp (rate 0.5mV msec 1) back to the holding potential. The voltage
protocol was applied to a cell continuously throughout the experiment every 4
seconds (0.25Hz). The amplitude of the peak current elicited around -40mV
during
the ramp was measured. Once stable evoked current responses were obtained in
the
external solution, vehicle (0.5% DMSO in the standard external solution) was
applied
for 10-20 min by a peristalic pump. Provided there were minimal changes in the
amplitude of the evoked current response in the vehicle control condition, the
test
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
27
compound of either 0.3, l, 3, 10~.M was applied for a 10 min period. The 10
min
period included the time which supplying solution was passing through the tube
from
solution reservoir to the recording chamber via the pump. Exposing time of
cells to
the compound solution was more than 5min after the drug concentration in the
chamber well reached the attempting concentration. There reversibility.
Finally,
the cells was exposed to high dose of dofetilide (S~t.M), a specific II~r
blocker, to
evaluate the insensitive endogenous current.
All experiments were performed at room temperature (23 ~ 1 °C).
Evoked
membrane currents were recorded on-line on a computer, filtered at 500-lKHz
(Bessel -3dB) and sampled at 1-2KHz using the patch clamp amplifier and a
specific
data analyzing software. Peak current amplitude, which occurred at around
~I.OmV,
was measured off line on the computer.
The arithmetic mean of the ten values of amplitude was calculated under
control
conditions and in the presence of drug. Percent decrease of IN in each
experiment
was obtained by the normalized current value using the following formula: IN =
(1
h/I~ )x100, where ID is the mean current value in the presence of drug and I~
is the
mean current value under control conditions. Separate experiments were
performed
for each drug concentration or time-matched control, and arithmetic mean in
each
experiment is defined as the result of the study.
Mice PSL Method
Surgery of partial sciatic nerve ligation (PSL) was made according to Seltzer
et al.
(Pain 43, 1990, 205-218). Von Fray hair test was applied slowly to the plantar
surface of the hind operated paw until the hairs bent. Each hair was tested 10
times
in ascending order of force to different loci of the paw with one to two
second
intervals between each application. Once a withdrawal response was
established, the
paw was re-tested with the same hair. The lowest amount of force required to
elicit
a response was recorded as the paw-withdrawal threshold, measured in grams.
Chronic Contriction Injury Model (CCI Model):
Male Sprague-Dawley rats (270-300 g; B.W., Charles River, Tsukuba, Japan) were
used.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
28
The chronic constriction injury (CCI) operation was performed according to the
method described by Bennett and Xie 1~. Briefly, animals were anesthetized
with
sodium pentobarbital (64.8 mglkg, i.p.) and the left common sciatic nerve was
exposed at the level of the middle of the thigh by blunt dissection through
biceps
femoris. Proximal to the sciatic's trifurcation was freed of adhering tissue
and 4
ligatures (4-0 silk) were tided loosely around it with about 1 mm space. Sham
operation was performed as same as CCI surgery except for sciatic nerve
ligation.
Two weeks after surgery, mechanical allodynia was evaluated by application of
von
Frey hairs (VFHs) to the plantar surface of the hind paw. The lowest amount of
force of VFH required to elicit a response was recorded as paw withdrawal
threshold
(PWT). VFH test was performed at 0.5, 1 and 2 hr post-dosing. Experimental
data
were analyzed using I~ruskal-Wallis test followed by Dunn's test for multiple
comparisons or Mann-Whitney U-test for paired comparison.
1~ Bennett, G.J. and Xie, Y.K. Pairz, 33:87-107,1988
Pharmaceutically acceptable salts of the compounds of formula (1) include the
acid addition and base salts (including disalts) thereof.
Suitable acid addition salts are formed from acids which form -non-toxic
salts.
Examples include the acetate, aspartate, benzoate, besylate,
bicarbonate/carbonate,
bisulphate, camsylate, citrate, edisylate, esylate, fumarate, gluceptate,
gluconate,
glucuronate, hibenzate, hydrochloride/chloride, hydrobromidelbromide,
hydroiodide/iodide, hydrogen phosphate, isethionate, D- and L-lactate, malate,
maleate, malonate, mesylate, methylsulphate, 2-napsylate, nicotinate, nitrate,
orotate,
palmoate, phosphate, saccharate, stearate, succinate sulphate, D- and L-
tartrate, and
tosylate salts. Suitable base salts are formed from bases which form non-toxic
salts.
Examples include the aluminium, arginine, benzathine, calcium, choline,
diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine,
potassium,
sodium, tromethamine and zinc salts.
For a review on suitable salts, see Stahl and Wermuth, Handbook of
Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH, Weinheim,
Germany (2002).
A pharmaceutically acceptable salt of a compound of formula (I) may be readily
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
29
prepared by mixing together solutions of the compound of formula (1) and the
desired
acid or base, as appropriate. The salt may precipitate from solution and be
collected
by filtration or may be recovered by evaporation of the solvent.
Pharmaceutically acceptable solvates in accordance with the invention include
hydrates and solvates wherein the solvent of crystallization may be
isotopically
substituted, e.g. D20, d6-acetone, d6-DMSO.
Also within the scope of the invention are clathrates, drug-host inclusion
complexes
wherein, in contrast to the aforementioned solvates, the drug and host are
present in
non-stoichiometric amounts. For a review of such complexes, see J Pharm Sci,
64 (8),
1269-1288 by Haleblian (August 1975).
Hereinafter all references to compounds of formula (I) include references to
salts
thereof and to solvates and clathrates of compounds of formula (1J and salts
thereof.
The invention includes all polymorphs of the compounds of formula (I) as
hereinbefore defined.
Also within the scope of the invention are so-called "prodrugs" of the
compounds of
formula (I). Thus certain derivatives of compounds of formula (I) which have
little or
no pharmacological activity themselves can, when metabolised upon
administration
into or onto the body, give rise to compounds of formula (I) having the
desired
activity. Such derivatives are referred to as "prodrugs".
Prodrugs in accordance with the invention can, for example, be produced by
replacing
appropriate functionalities present in the compounds of formula (1J with
certain
moieties known to those skilled in the art as "pro-moieties" as described, for
example,
in "Design of Prodrugs" by H Bundgaard (Elsevier, 1985).
Finally, certain compounds of formula (1J may themselves act as prodrugs of
other
compounds of formula (I).
Compounds of formula (I) containing one or more asymmetric carbon atoms can
exist
as two or more optical isomers. Where a compound of formula (1) contains an
alkenyl
or alkenylene group, geometric cisltrares (or Z/E) isomers are possible, and
where the
compound contains, for example, a keto or oxime group, tautomeric isomerism
('tautomerism') may occur. It follows that a single compound may exhibit more
than
one type of isomerism.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
Included within the scope of the present invention are all optical isomers,
geometric
isomers and tautomeric forms of the compounds of formula (I), including
compounds
exhibiting more than one type of isomerism, and mixtures of one or more
thereof.
Cisltrans isomers may be separated by conventional techniques well known to
those
5 skilled in the art, for example, fractional crystallization and
chromatography.
Conventional techniques for the preparationlisolation of individual
stereoisomers
include the conversion of a suitable optically pure precursor, resolution of
the
racemate (or the racemate of a salt or derivative) using, for example, chiral
HPLC, or
fractional crystallization of diastereoisomeric salts formed by reaction of
the racemate
10 with a suitable optically active acid or base, for example, tartaric acid.
The present invention also includes all pharmaceutically acceptable isotopic
variations of a compound of formula (I). An isotopic variation is defined as
one in
which at least one atom is replaced by an atom having the same atomic number,
but
an atomic mass different from the atomic mass usually found in nature.
15 Examples of isotopes suitable for inclusion in the compounds of the
invention include
isotopes of hydrogen, such as 2H and 3H, carbon, such as 13C and 14C,
nitrogen, such
as 15N, oxygen, such as 1'O and 180, phosphorus, such as 32P, sulphur, such as
355,
fluorine, such as 18F, and chlorine, such as 36C1.
Substitution of the compounds of the invention with isotopes such as
deuterium, i.e.
20 2H, may afford certain therapeutic advantages resulting from greater
metabolic
stability, for example, increased in vavo half life or reduced dosage
requirements, and
hence may be preferred in some circumstances.
Certain isotopic variations of the compounds of formula (I), for example,
those
25 incorporating a radioactive isotope, are useful in drug andlor substrate
tissue
distribution studies. The radioactive isotopes tritium, i.e. 3H, and carbon-
14, i.e. 14C,
are particularly useful for this purpose in view of their ease of
incorporation and ready
means of detection.
Isotopic variations of the compounds of formula (I) can generally be prepared
by
30 conventional techniques known to those skilled in the art or by processes
analogous to
those described in the accompanying Examples and Preparations using
appropriate
isotopic variations of suitable reagents.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
31
The compounds of formula (n may be freeze-dried, spray-dried, or evaporatively
dried to provide a solid plug, powder, or film of crystalline or amorphous
material.
Microwave or radio frequency drying may be used for this purpose.
The compounds of the invention may be administered alone or in combination
with
other drugs and will generally be administered as a formulation in association
with
one or more pharmaceutically acceptable excipients. The term "excipient" is
used
herein to describe any ingredient other than the compound of the invention.
The
choice of excipient will to a large extent depend on the particular mode of
administration.
The compounds of the invention may be administered in combination,
separately, simultaneously or sequentially, with one or more other
pharmacologically active agents. Suitable agents, particularly for the
treatment of
pain, include:
(i) opioid analgesics, e.g. morphine, heroin, hydromorphone, oxymorphone,
levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine,
dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene,
nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine and
pentazocine;
(ii) nonsteroidal antiinflammatory drugs (NSA~s), e.g. aspirin, diclofenac,
diflusinal, etodolac, fenbufen, fenoprofen, flufenisal,
flurbiprofen,ibuprofen,
indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid,
nabumetone, naproxen, oxaprozin, phenylbutazone, piroxicam, sulindac,
tolmetin, zomepirac, and their pharmaceutically acceptable salts;
(iii) barbiturate sedatives, e.g. amobarbital, aprobarbital, butabarbital,
butabital,
mephobarbital, metharbital, methohexital, pentobarbital, phenobartital,
secobarbital, talbutal, theamylal, thiopental and their pharmaceutically
acceptable salts;
(iv) benzodiazepines having a sedative action, e.g. chlordiazepoxide,
clorazepate,
diazepam, flurazepam, lorazepam, oxazepam, temazepam, triazolam and their
pharmaceutically acceptable salts,
(v) Hl antagonists having a sedative action, e.g. diphenhydramine, pyrilamine,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
32
promethazine, chlorpheniramine, chlorcyclizine and their pharmaceutically
acceptable salts;
(vi) miscellaneous sedatives such as glutethimide, meprobamate, methaqualone,
dichloralphenazone and their pharmaceutically acceptable salts;
(vii) skeletal muscle relaxants, e.g. baclofen, carisoprodol, chlorzoxazone,
cyclobenzaprine, methocarbamol, orphrenadine and their pharmaceutically
acceptable salts,
(viii) alpha-2-delta ligands, e.g. gabapentin and pregabalin;
(ix) alpha-adrenergic active compounds, e.g. doxazosin, tamsulosin, clonidine
and
4-amino-6,7-dimethoxy-2-(5-methanesulfonamido-1,2,3,4-
tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
(x) tricyclic antidepressants, e.g. desipramine, imipramine, amytriptiline and
nortriptiline;
(xi) anticonvulsants; e.g. carbamazepine and valproate;
(xii) serotonin reuptake inhibitors, e.g. fluoxetine, paroxetine, citalopram
and
sertraline;
(xiii) mixed serotonin-noradrenaline reuptake inhibitors, e.g. milnacipran,
venlafaxine and duloxetine;
(xiv) noradrenaline reuptake inhibitors , e.g. reboxetine;
(xv) Tachykinin (NK) antagonists, particularly Nk-3, NK-2 and NK-1
antagonists,
e.g. (ocR,9R)-7-[3,5-bis(trifluoromethyl)benzyl] -8,9,10,11-tetrahydro-9-
methyl-5-(4-methylphenyl)-7H-[1,4]diazocino[2,1-g] [1,7]naphthridine-6-13-
dione (TAK-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-
bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]methyl]-
1,2-dihydro-3H-1,2,4-triazol-3-one (MK-869), lanepitant, dapitant and 3-
[[2-methoxy-5-(trifluoromethoxy)phenyl]methylamino]-2-phenyl-piperidine
(2S,3S)
(xvi) Muscarinic antagonists, e.g oxybutin, tolterodine, propiverine, tropsium
chloride and darifenacin;
(xvii) COX-2 inhibitors, e.g. celecoxib, rofecoxib and valdecoxib;
(xviii) Non-selective COX inhibitors (preferably with GI protection), e.g.
nitroflurbiprofen (HCT-1026);
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
33
(xix) coal-tar analgesics, in particular, paracetamol;
(xx) neuroleptics, such as droperidol;
(xxi) Vanilloid receptor agonists, e.g. resinferatoxin;
(xxii) Beta-adrenergic compounds such as propranolol;
(xxiii) Local anaesthetics, such as mexiletine;
(xxiv) Corticosteriods, such as dexamethasone
(xxv) serotonin receptor agonists and antagonists;
(xxvi) cholinergic (nicotinic) analgesics; and
(xxvii) miscellaneous analgesic agents, such as Tramadol~.
Thus, the invention further provides a combination comprising a compound of
the invention or a pharmaceutically acceptable salt, solvate or pro-drug
thereof, and a
compound or class of compounds selected from the group (i)-(xxvii), above.
There
is also provided a pharmaceutical composition comprising such a combination,
together with a pharmaceutically acceptable excipient, diluent or carrier,
particularly
for the treatment of a disease for which an alpha-2-delta ligand is
implicated.
Combinations of the compounds of the present invention and other
therapeutic agents may be administered separately, sequentially or
simultaneously.
Thus, the present invention extends to a kit comprising a compound of the
invention,
one or more other therapeutic agents, such as those listed above, and a
suitable
container.
The compounds of the present invention may be formulated by any convenient
means using well-known carriers and excipients. Thus, the present invention
also
provides a pharmaceutical composition comprising a compound of the invention
or a
pharmaceutically acceptable ester or a pharmaceutically acceptable salt
thereof with
one or more pharmaceutically acceptable carriers.
ORAL ADMINISTRATION
The compounds of the invention may be administered orally. Oral administration
may
involve swallowing, so that the compound enters the gastrointestinal tract, or
buccal
or sublingual administration may be employed by which the compound enters the
blood stream directly from the mouth.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
34
Formulations suitable for oral administration include solid formulations such
as
tablets, capsules containing particulates, liquids, or powders, lozenges
(including
liquid-filled), chews, multi- and nano-particulates, gels, films (including
muco-
adhesive), ovules, sprays and liquid formulations.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such
formulations may be employed as fillers in soft or hard capsules and typically
comprise a carrier, for example, water, ethanol, propylene glycol,
methylcellulose, or
a suitable oil, and one or more emulsifying agents and/or suspending agents.
Liquid
formulations may also be prepared by the reconstitution of a solid, for
example, from
a sachet.
The compounds of the invention may also be used in fast-dissolving, fast-
disintegrating dosage forms such as those described in Expert Opinion in
Therapeutic
Patents, 11 (6), 981-986 by Liang and Chen (2001).
The composition of a typical tablet in accordance with the invention may
comprise:
Ingredient 7o w/w
Compound of formula (I) 10.00*
Microcrystalline cellulose 64.12
Lactose 21.38
Croscarmellose sodium 3.00
Magnesium stearate 1.50
Quantity adjusted in accordance with drug activity.
A typical tablet may be prepared using standard processes known to a
formulation
chemist, for example, by direct compression, granulation (dry, wet, or
melt), melt congealing, or extrusion. The tablet formulation may comprise one
or
more layers and may be coated or uncoated.
Examples of excipients suitable for oral administration include carriers, for
example,
cellulose, calcium carbonate, dibasic calcium phosphate, mannitol and
sodium citrate, granulation binders, for example, polyvinylpyrrolidine,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
hydroxypropylcellulose, hydroxypropylmethylcellulose and gelatin,
disintegrants, for
example, sodium starch glycolate and silicates, lubricating agents, for
example,
magnesium stearate and stearic acid, wetting agents, for example, sodium
lauryl
sulphate, preservatives, anti-oxidants, flavours and colourants.
5 Solid formulations for oral administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled dual-, targeted and programmed release. Details of suitable
modified
release technologies such as high energy dispersions, osmotic and coated
particles are
to be found in Verma et al, Pharmaceutical Technology On-line, 25(2), 1-14
(2001).
10 Other modified release formulations are described in US Patent No.
6,106,864.
PARENTERAL ADMINISTRATION
The compounds of the invention may also be administered directly into the
blood
stream, into muscle, or into an internal organ. Suitable means for parenteral
15 administration include intravenous, intraarterial, intraperitoneal,
intrathecal,
intraventricular, intraurethral, intrasternal, intracranial, intramuscular and
subcutaneous. Suitable devices for parenteral administration include needle
(including
microneedle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain
excipients
20 such as salts, carbohydrates and buffering agents (preferably to a pH of
from 3 to 9),
but, for some applications, they may be more suitably formulated as a sterile
non-
aqueous solution or as a dried form to be used in conjunction with a suitable
vehicle
such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for
example, by
25 lyophilisation, may readily be accomplished using standard pharmaceutical
techniques
well known to those skilled in the art.
The solubility of compounds of formula (I) used in the preparation of
parenteral
solutions may be increased by suitable processing, for example, the use of
high energy
spray-dried dispersions (see WO 01/47495) and/or by the use of appropriate
30 formulation techniques, such as the use of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate
andlor
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
36
controlled dual-, targeted and programmed release.
TOPICAL ADMINISTRATION
The compounds of the invention may also be administered topically to the skin
or
mucosa, either dermally or transdermally. Typical formulations for this
purpose
include gels, hydrogels, lotions, solutions, creams, ointments, dusting
powders,
dressings, foams, films, skin patches, wafers, implants, sponges, fibres,
bandages and
microemulsions. Liposomes may also be used. Typical carriers include alcohol,
water,
mineral oil, liquid petrolatum, white petrolatum, glycerin and propylene
glycol.
Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88
(10),
955-958 by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by iontophoresis,
electroporation, phonophoresis, sonophoresis and needle-free or microneedle
injection.
Formulations for topical administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled dual-, targeted and programmed release. Thus compounds of the
invention may be formulated in a more solid form for administration as an
implanted
depot providing long-term release of the active compound.
II~IHALEDl11~1TRANASAL ADMINISTRATION
The compounds of the invention can also be administered intranasally or by
inhalation, typically in the form of a dry powder (either alone, as a mixture,
for
example, in a dry blend with lactose, or as a mixed component particle, for
example,
mixed with phospholipids) from a dry powder inhaler or as an aerosol spray
from a
pressurised container, pump, spray, atomiser (preferably an atomiser using
electrohydrodynamics to produce a fine mist), or nebuliser, with or without
the use of
a suitable propellant, such as dichlorofluoromethane.
The pressurised container, pump, spray, atomizer, or nebuliser contains a
solution or
suspension of the active compound comprising, for example, ethanol
(optionally,
aqueous ethanol) or a suitable alternative agent for dispersing, solubilising,
or
extending release of the active, the propellants) as solvent and an optional
surfactant,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
37
such as sorbitan trioleate or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is
micronised to a size suitable for delivery by inhalation (typically less than
5 microns).
This may be achieved by any appropriate comminuting method, such as spiral jet
milling, fluid bed jet milling, supercritical fluid processing to foam
nanoparticles, high
pressure homogenisation, or spray drying.
A suitable solution formulation for use in an atomiser using
electrohydrodynamics to
produce a fine mist may contain from 1 ~,g to l Omg of the compound of the
invention
per actuation and the actuation volume may vary from 1 ~.l to 100,1. A typical
formulation may comprise a compound of formula (1J, propylene glycol, sterile
water,
ethanol and sodium chloride. Alternative solvents which may be used instead of
propylene glycol include glycerol and polyethylene glycol.
Capsules, blisters and cartridges (made, for example, from gelatin or HPMC)
for use
in an inhaler or insufflator may be formulated to contain a powder mix of the
compound of the invention, a suitable powder base such as lactose or starch
and a
performance modifier such as l-leucine, mannitol, or magnesium stearate.
In the case of dry powder inhalers and aerosols, the dosage unit is determined
by
means of a valve which delivers a metered amount. Units in accordance with the
invention are typically arranged to administer a metered dose or "pufp'.
Formulations for inhaled/intranasal administration may be formulated to be
immediate and/or modified release. Modified release formulations include
delayed-,
sustained-, pulsed-, controlled dual-, targeted and programmed release.
RECTAL/INTRAVAGINAL ADMINISTRATION
The compounds of the invention may be administered rectally or vaginally, for
example, in the form of a suppository, pessary, or enema. Cocoa butter is a
traditional
suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be
immediate
and/or modified release. Modified release formulations include delayed-,
sustained-,
pulsed-, controlled dual-, targeted and programmed release.
CA 02541162 2006-04-03
WO 2005/035523 PCTlIB2004/003125
38
OCULAR/ANDIAL ADMINISTRATION
The compounds of the invention may also be administered directly to the eye or
ear,
typically in the form of drops of a micronised suspension or solution in
isotonic, pH-
adjusted, sterile saline. Other formulations suitable for ocular and andial
administration include ointments, biodegradable (e.g. absorbable gel sponges,
collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and
particulate or vesicular systems, such as niosomes or liposomes. A polymer
such as
crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a
cellulosic
polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or
methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum,
may be
incorporated together with a preservative, such as benzalkonium chloride. Such
formulations may also be delivered by iontophoresis.
Formulations for ocular/andial administration may be formulated to be
immediate
and/or modified release. Modified release formulations include delayed-,
sustained-,
pulsed-, controlled dual-, targeted, or programmed release.
ENABLING TECHNOLOGIES
The compounds of the invention may be combined with soluble macromolecular
entities such as cyclodextrin or polyethylene glycol-containing polymers to
improve
their solubility, dissolution rate, taste-masking, bioavailability and/or
stability.
Drug-cyclodextrin complexes, for example, are found to be generally useful for
most
dosage forms and administration routes. Both inclusion and non-inclusion
complexes
may be used. As an alternative to direct complexation with the drug, the
cyclodextrin
may be used as an auxiliary additive, i.e. as a carrier, diluent, or
solubiliser. Most
commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins,
examples of which may be found in International Patent Applications Nos. WO
91/11172, WO 94/02518 and WO 98/55148.
DOSAGE
The compounds of the invention can be administered via either the oral,
parenteral or
topical routes to mammals. In general, these compounds are most desirably
administered to humans in doses ranging from 0.1 mg to 3000 mg, preferably
from 1
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
39
mg to 500 mg, which may be administered in a single dose or in divided doses
throughout the day, although variations will necessarily occur depending upon
the
weight and condition of the subject being treated, the disease state being
treated and
the particular route of administration chosen.
These dosages are based on an average human subject having a weight of about
65 to
70kg. The physician will readily be able to determine doses for subjects whose
weight
falls outside this range, such as infants and the elderly.
For example, a dosage level that is in the range of from 0.01 mg to 10 mg per
kg of
body weight per day is most desirably employed for treatment of pain
associated with
inflammation.
Examples
The invention is illustrated in the following non-limiting examples in which,
unless stated otherwise: all operations were carried out at room or ambient
temperature, that is, in the range of 18-25 °C; evaporation of solvent
was carried out
using a rotary evaporator under reduced pressure with a bath temperature of up
to 60
°C; reactions were monitored by thin layer chromatography (tlc) and
reaction times
are given for illustration only; melting points (m.p.) given are uncorrected
(polymorphism may result in different melting points); the structure and
purity of all
isolated compounds were assured by at least one of the following techniques:
tlc
(Merck silica gel 60 FZS4 precoated TLC plates or Merck NH2 FZSøs precoated
HPTLC
plates), mass spectrometry, nuclear magnetic resonance (NMR), infrared red
absorption spectra (1R) or microanalysis. Yields are given for illustrative
purposes
only. Flash column chromatography was carried out using Merck silica gel 60
(230-
400 mesh ASTM) or Fuji Silysia Chromatorex° DU3050 (Amino Type, 3050
p.m).
Low-resolution mass spectral data (E1) were obtained on an Automass 120 (JEOL)
mass spectrometer. Low-resolution mass spectral data (ESI) were obtained on a
Quattro II (Micromass) mass spectrometer. Melting point was obtained using
Seiko
Instruments Inc. Exstar 6000. NMR data was determined at 270 MHz (JEOL JNM-
LA 270 spectrometer) or 300 MHz (JEOL JNM-LA300) using deuterated chloroform
(99.8% D) or dimethylsulfoxide (99.9% D) as solvent unless indicated
otherwise,
relative to tetramethylsilane (TMS) as internal standard in parts per million
(ppm);
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
conventional abbreviations used are: s = singlet, d = doublet, t = triplet, q
= quartet, m
= multiplet, br. = broad, etc. IR spectra were measured by a Shimazu infrared
spectrometer (IR-470). Optical rotations were measured using a JASCO DIP-370
Digital Polarimeter (Japan Spectroscopic CO, Ltd.).
5 Chemical symbols have their usual meanings; b.p. (boiling point), m.p.
(melting point), 1 (liter(s)), mL (milliliter(s)), g (gram(s)),
mg(milligram(s)), mol
(moles), mmol (millimoles), eq. (equivalent(s)).
10 Example 1
6-f ~4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yllacetyl)-3,4-
dihydroguinolin-2(1I~-one
A. tent-butyl 4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidine-1-carboxylate
To a stirred solution of 5-bromo-2-ethoxypyridine (Yakugaku Zasshi, 1952, 72,
15 381)(5.02 g, 24.8 mmol) in diethyl ether (90 mL) was added dropwise n-
butyllithium
(1.56 M, 15.9 mL, 24.8 mmol) at -78 °C under nitrogen and the mixture
was stirred
for 50 minutes at -78 °C. To the mixture, a solution of tent-butyl 4-
oxopiperidine-1-
carboxylate (4.49 g, 22.5 mmol) in diethyl ether (10 mL) was added at -78
°C. The
mixture was stirred at -78 °C for 2 hours and at room temperature for 3
hours. The
20 mixture was treated with H20 and extracted with ethyl acetate. The combined
organic
layer was dried and evaporated. The residue was purified by chromatography on
silica
gel, eluting with triethylamine / ethyl acetate / hexane (0.05:1:2 v/v/v), to
afford the
titled compound as a yellow oil (3.50 g, 48 %).
1H NMR (300MHz, CDCl3) 8 = 8.23 (dd, J = 2.6, 0.7 Hz, 1H), 7.68 (dd, J = 8.8,
2.6
25 Hz, 1H), 6.71 (dd, J= 8.8, 0.7 Hz, 1H), 4.34 (q, J= 7.1 Hz, 2H), 4.00
(br.s, 2H), 3.16-
3.30 (m, 2H), 2.02-1.86 (m, 2H), 1.80-1.70 (m, 2H), 1.48 (s, 9H), 1.39 (t, J =
7.1 Hz,
3H) ppm.
MS (El]; M+=322
B. 4-(6-ethoxypyridin-3-yl)piperidin-4-of dihydrochloride
30 Hydrogen chloride (10 mL, 40 mmol), 4.0 M solution in ethyl acetate, was
added to a solution of tert-butyl 4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidine-
1-
carboxylate (3.50 g, 10.9 mmol) in ethyl acetate (40 mL). The mixture was
stiiTed at
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
41
50 °C for 1 hour. The precipitate was collected by filtration to afford
the title
compound as a yellow solid (3.09 g, 96 %).
1H NMR (300MHz, DMSO) ~ = 9.34 (br.s, 1H), 9.21 (br.s, 1H), 8.23 (d, J = 2.6
Hz,
1H), 7.89 (dd, J = 8.8, 2.6 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 6.36 (br.s,
2H), 4.34 (q,
J = 7.0 Hz, 2H), 3.30-1.75 (m, 8H), 1.33 (t, J = 7.0 Hz, 3H) ppm.
MS (E~; M+=295
C. 6-~ ~4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yllacetyl~-3,4-
dihydropuinolin-2(1Fn-one
To a stirred solution of 6-(chloroacetyl)-3,4-dihydroquinolin-2(1F~-one
(1.03 g, 4.62 mmol) in ethanol (20 mL) were added 4-(6-ethoxypyridin-3
yl)piperidin-4-ol, dihydrochloride (1.50 g, 5.08 mmol) and triethylamine (3.22
mL,
23.1 mmol) at room temperature under nitrogen and the mixture was stirred
under
reflux for 4 hours. The mixture was treated with H20 and the precipitate was
colleted
by filtration to afford the titled compound as a yellow solid (1.09 g, 58 %).
1H NMR (300MHz, DMSO-d6) 8 = 10.43 (br.s, 1H), 8.22 (d, J = 2.4 Hz, 1H), 7.86
(d,
J = 8.4 Hz, 1H), 7.85 (s, 1H), 7.76 (dd, J = 8.6, 2.4 Hz, 1H), 6.93 (d, J =
8.6 Hz, 1H),
6.72 (d, J= 8.4 Hz, 1H), 4.89 (s, 1H), 4.27 (q, J= 7.0 Hz, 2H), 3.77 (s, 2H),
3.00-2.48
(m, 8H), 2.00-1.58 (m, 4H), 1.30 (t, J = 7.0 Hz, 3H) ppm.
MS (ESn; (M+H)+ = 410.15, (M-H)- =4.08.21
m.p.213.1°C
IR (KBr); 3489, 1678 cm 1
Example 2
A. 6-~2- f 4-(6-ethoxyuyridin-3-yl)-4-hydroxypiperidin-1-yll-1-hydroxyethyl~-
3,4-
dihydropuinolin-2(lI~-one
The mixture of 6-{ [4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yl]acetyl}-
3,4-
dihydroquinolin-2(1H)-one (1.49 g, 3.64 mmol) and sodium borohydride (206 mg,
5.46 mmol) in ethanol (30 mL) was stirred at room temperature for 17 h. After
ethanol was removed, the residue was treated with HZO and extracted with
dichloromethane. The combined organic layer was dried and evaporated. The
residue
was purified by chromatography on silica gel, eluting with methanol /
dichloromethane (1:7 vlv), to afford the titled compound as a white solid
(1.06 g,
71 %).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
42
1H NMR (270MHz, DMSO-d6) 8 = 10.02 (s, 1H), 8.21 (d, J = 2.4 Hz, 1H), 7.76
(dd, J
= 8.7, 2.4 Hz, 1H), 7.15 (s, 1H), 7.10 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1
Hz, 1H),
6.72 (d, J = 8.7 Hz, 1H), 4.84 (s, 1H), 4.81 (d, J = 3.0 Hz, 1H), 4.68-4.60
(m, 1H),
4.27 (q, J = 7.1 Hz, 2H), 2.96-2.34 (m, 10H), 2.00-1.90 (m, 2H), 1.68-1.54 (m,
2H),
1.30 (t, J = 7.1 Hz, 3H) ppm.
B. 6-~2-f 4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-1-yll-1-hydroxyethyll-
3,4-
dihydroauinolin-~(lI~-one hydrochloride
Hydrogen chloride (0.456 mL" 1.83 mmol), 4.0 M solution in ethyl acetate,
was added to a suspension of 6-{2-[4-(6-ethoxypyridin-3-yl)-4-hydroxypiperidin-
1
yl]-1-hydroxyethyl}-3,4-dihydroquinolin-2(11-one (751 mg, 1.83 mmol) in methyl
alcohol (20 mL). The mixture was stirred for 30 minutes at room temperature
and
filtered. The filtrate was evaporated and the residue was recrystallized from
methanol/
2-propanol to afford the titled compound as a white solid (727 mg, 89 %).
1H NMR (300MHz, DMSO-d6) 8 = 10.20 (s, 1H), 10.14 (s, 1H), 8.25 (s, 1H), 7.76
(d,
J = 8.1 Hz, 1H), 7.25 (s, 1H), 7.21 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 8.1 Hz,
1H), 6.80
(d, J = 8.4 Hz, 1H), 6.17 (br.s, 1H), 5.61 (br.s, 1H), 5.13 (br.s, 1H), 4.29
(q, J = 7.1
Hz, 2H), 3.57 (s, 2H), 3.50-3.-2.42 (m, 10H), 1.95-1.80 (m, 2H), 1.31 (t, J =
7.1 Hz,
3H) ppm.
MS (ESn; (M+H)+ = 412.14, (M-H)- =410.23
m.p. 225.1 °C
1R (KBr); 3294, 3182, 1674 cm 1
Example 3
6-f 2-~4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl)-3,4-dihydroauinolin-2(lI~-one hydrochloride
A.6-bromo-2-fluoropyridin-3-of
To a stirred solution of 2-fluoropyridin-3-of (J. Labelled Coumpound.
Radiophann., 1998, 41, 451)(3.81 g, 33.7 mmol) and sodium acetate (2.76 g,
33.7
mmol) in acetic acid (30 mL) was added bromine (1.74 mL, 33.7 mmol) at 0
°C, and
the mixture was stirred at room temperature for 3.5 hours. The mixture was
poured
onto ice-aq.sodium hydroxide and extracted with ethyl acetate. The combined
organic
layer was dried and evaporated to afford the titled compound as a yellow solid
(4.67 g,
72 %).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
43
1H NMR (270MHz, DMSO-d6) b = 7.29 (d, J = 8.2 Hz, 1H), 7.28 (s, 1H), 7.23 (d,
J =
8.2 Hz, 1H) ppm.
MS (E1]; M+=191, 193
B. 6-bromo-2-fluoro-3-methoxypyridine
To a stirred solution of 6-bromo-2-fluoropyridin-3-of (4.67 g, 24.3 mmol) and
sodium methoxide (1.38 g, 25.5 mmol) in N,N dimethylformamide (50 mL) was
added methyl iodide (1.59 mL, 25.5 mmol) at 0 °C, and the mixture was
stirred at
room temperature for 12 hours. The mixture was treated with HZO and extracted
with
ethyl acetate. The combined organic layer was dried and evaporated. The
residue was
purified by chromatography on silica gel, eluting with ethyl acetate / hexane
(1:5 v/v),
to afford the titled compound as a yellow oil (2.43 g, 49 %).
1H NMR (270MHz, CDCl3) 8 = 7.32-7.26 (m, 1H), 7.22-7.15 (m, 1H), 3.90 (s, 3H)
ppm.
MS (El]; M+=205, 207
C. tent-butyl 4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidine-1-
carboxylate
The title compound was prepared according to the procedure described in
Example 1 from 6-bromo-2-fluoro-3-methoxypyridine: 948 mg (49°Io) as a
colorless
oil.
1H NMR (300MHz, CDCl3) ~ = 7.31 (dd, J=9.9, 8.3Hz, 1H), 7.18 (dd, J=8.3, 0.9
Hz,
1H), 4.15-3.95 (m, 2H), 3.91 (s, 3H), 3.32-3.15 (m, 2H), 2.02-1.80 (m, 2H),
1.70-1.53
(m, 2H), 1.48 (s, 9H) ppm.
D. 4-(6-fluoro-5-methoxypyridin-2-yl)piperidin-4-of dihydrochloride
The title compound was prepared according to the procedure described in
Example 1 from tert-butyl 4-(6-fluoro-5-methoxypyridin-2-yl)-4-
hydroxypiperidine-
1-carboxylate: 623 mg (72%) as a white solid.
1H NMR (300MHz, DMSO-d6) 8 = 9.24-9.12 (m, 1H), 8.78 (br.s, 1H), 7.69 (dd,
J=10.6, 8.2 Hz, 1H), 7.53 (d, J=8.2 Hz, 1H), 5.74 (br.s, 1H), 3.88 (s, 3H),
3.20-3.10
(m, 4H), 2.30-2.16 (m, 2H), 1.80-1.66 (m, 2H) ppm.
MS (ESn; (M+H)+ = 227.01
E. 6-~ f 4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yl~ acetyl ~-
3,4-
dihydroauinolin-2(1Fn-one
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
44
To a stirred solution of 6-(chloroacetyl)-3,4-dihydroquinolin-2(lI~-one (1.02
g, 4.57 mmol) in N,N dimethylformamide (20 mL) were added 4-(6-fluoro-5-
methoxypyridin-2-yl)piperidin-4-of dihydrochloride (1.50 g, 5.03 mmol) and
triethylamine (1.91 mL, 13.7 mmol) at room temperature under nitrogen and the
mixture was stirred at room temperature for 4 hours. The mixture was treated
with
H20 and the precipitate was colleted by filtration to afford the titled
compound as a
yellow solid (1.34 g, 71 %).
1H NMR (270MHz, DMSO-d6) 8 = 10.41 (s, 1H), 7.92-7.82 (m, 2H), 7.62 (dd, J =
10.4, 8.2 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 5.02
(s, 1H),
3.85 (s, 3H), 3.72 (s, 2H), 3.00-2.90 (m, 2H), 2.72-2.63 (m, 2H), 2.56-2.44
(m, 4H),
2.10-1.96 (m, 2H), 1.52-1.44 (m, 2H) ppm.
MS (ES)); (M+H)+ = 414.12, (M-H)- = 412.19
m.p. 231.1°C
IR (KBr); 3541, 1674 cm 1
F.6-~2-f4-(6-fluoro-S-methoxypyridin-2-yl)-4-hydroxypiperidin-1-y11-1-
hydroxyethyl~-3,4-dihydroauinolin-2(lI~-one
The title compound was prepared according to the procedure described in
Example 2 from 6-{ [4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-
yl]acetyl}-3,4-dihydroquinolin-2(11-one: 1.07 g (76%) as a yellow solid.
1H NMR (300MHz, DMSO-d6) S = 10.02 (s, 1H), 7.63 (dd, J = 10.4, 8.3 Hz, 1H),
7.49 (d, J = 8.3 Hz, 1H), 7.15 (s, 1H), 7.10 (d, J = 8.0 Hz, 1H), 6.79 (d, J =
8.0 Hz,
1H), 5.00 (s, 1H), 4.81 (br.s, 1H), 4.66-4.58 (m, 1H), 3.86 (s, 3H), 2.90-2.84
(m, 2H),
2.76-2.32 (m, 8H), 2.15-1.92 (m, 2H), 1.54-1.45 (m, 2H) ppm.
G. 6-~2-f 4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl~-3,4-dihydroauinolin-2(1I~-one hydrochloride
By the procedures of Example 2, 6-{2-[4-(6-fluoro-5-methoxypyridin-2-yl)-4-
hydroxypiperidin-1-yl]-1-hydroxyethyl}-3,4-dihydroquinolin-2(11-one was
converted to the title compound obtained as a white solid in 83% (110 mg)
after
recrystallization from 2-propanol.
1H NMR (270MHz, DMSO-d6) 8 = 10.12 (s, 1H), 9.66 (s, 1H), 7.73-7.65 (m, 1H),
7.54 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.20 (d, J = 8.1 Hz, 1H), 6.86 (d, J =
8.1 Hz,
1H), 6.12 (s, 1H), 5.72 (s, 1H), 5.05 (br.s, 1H), 3.88 (s, 3H), 3.56-2.42 (m,
12H),
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
1.89-1.70 (m, 2H), ppm.
MS (ESZ]; (M+H)+ = 416.12, (M-H)- = 414.21
IR (KBr); 3274, 1670 cm 1
Example 4
5 6-~1-hydroxy-2-f4-hydroxy-4-(6-propoxypyridin-3-yl)niperidin-1-yllethyl)-3,4-
dihydroauinolin-2(1H~-one hydrochloride
A. 5-bromo-2-uropoxypyridine
To a stirred solution of sodium (591 mg, 24.6 mmol) in 2-propanol (20 mL)
was added a solution of 5-bromo-2-nitropyridine (5 g, 24.6 mmol) in 2-propanol
(10
10 mL) at room temperature and the mixture was stirred under reflux for 2.5
hours. After
all solvents were removed, the residue was diluted with dichloromethane and
H20,
and extracted with dichloromethane. The combined organic layer was dried and
evaporated. The residue was purified by chromatography on silica gel, eluting
with
ethyl acetate / hexane (1:10, v/v), to afford the titled compound as a
colorless oil (3.84
15 g, 72 %).
1H NMR (270MHz, CDC13) 8 = 8.17 (d, J=2.6 Hz, 1H), 7.63 (dd, J=8.7, 2.6 Hz,
1H),
6.64 (d, J=8.7 Hz, 1H), 4.21 (t, J=6.6 Hz, 2H), 1.84-1.70 (m, 2H), 1.01 (t,
J=7.4 Hz,
3H) ppm.
MS (En; M + = 215, 217
20 B. tent-butyl 4-hydroxy-4-(6-propoxypyridin-3-yl)piperidine-1-carboxylate
The title compound was prepared according to the procedure described in
Example 1 from 5-bromo-2-propoxypyridine: 2.57 g (75%) as yellow oil.
1H NMR (270MHz, CDCl3) 8 = 8.23 (d, J=2.6 Hz, 1H), 7.68 (dd, J=8.7, 2.6 Hz,
1H),
6.72 (d, J=8.7Hz, 1H), 4.24 (t, J=6.8 Hz, 2H), 4.00 (br.s, 2H), 3.29-3.19 (m,
2H),
25 2.04-1.70 (m, 6H), 1.48 (s, 9H), 1.02 (t, J=7.4 Hz, 3H) ppm.
MS (E~; M+=336
C. 4-(fi-propoxypyridin-3-yl)piperidin-4-of dihydrochloride
The title compound was prepared according to the procedure described in
Example 1 from ter-t-butyl 4-hydroxy-4-(6-propoxypyridin-3-yl)piperidine-1
30 carboxylate: 2.5 g (quant.) as a yellow solid.
1H NMR (300MHz, DMSO) 8 = 9.35-9.00 (m, 1H), 8.22 (d, J = 2.4 Hz, 1H), 7.94-
7.80 (m, 1H), 7.02-6.88 (m, 1H), 4.60-4.00 (m, 4H), 3.22-3.10 (m, 4H), 2.32-
2.15 (m,
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB20041003125
46
2H), 1.83-1.76 (m, 2H), 1.77-1.66 (m, 2H), 0.96 (t, J = 6.8 Hz, 3H) ppm.
MS (EI); M+=236
D. 6-~C4-hydroxy-4-(6-propoxypyridin-3-Ylluiperidin-1-yllacetyl~-3,4-
dihydrocruinolin-2(lF,n-one
The title compound was prepared according to the procedure described in
Example 1 from 4-(6-propoxypyridin-3-yl)piperidin-4-ol: 584 mg (69%) as a
yellow
solid.
1H NMR (270MHz, DMSO-d6) 8 = 10.42 (s, 1H), 8.21 (d, J = 2.5 Hz, 1H), 7.88-
7.84
(m, 2H), 7.76 (dd, J = 8.6, 2.5 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 6.73 (d, J
= 8.6 Hz,
1H), 4.88 (s, 1H), 4.18 (t, J = 6.6 Hz, 2H), 3.77 (s, 2H), 3.02-2.90 (m, 2H),
2.69-2.47
(m, 8H), 1.95-1.85 (m, 2H), 1.75-1.58 (m, 2H), 0.95 (t, J= 7.3 Hz, 3H) ppm.
MS (ESI); (M+H)+ = 424.18, (M-H)T =422.24
m.p. 185.9°C
IR (KBr); 3431, 3192, 1665 cm 1
E.6-d1-hydroxy-2-[4-hydroxy-4-(6-propoxyuyridin-3-~l)piperidin-1-yllethyl~-
3,4-dihydroauinolin-2(1F~-one
The title compound was prepared according to the procedure described in
Example 2 6-{[4-hydroxy-4-(6-propoxypyridin-3-yl)piperidin-1-yl]acetyl}-3,4-
dihydroquinolin-2(lI~-one: 408 mg (74%) as a yellow solid.
1H NMR (300MHz, DMSO-d6) 8 = 10.03 (s, 1H), 8.21 (d, J = 2.3 Hz, 1H), 7.76
(dd, J
= 8.6, 2.3 Hz, 1H), 7.15 (s, 1H), 7.11 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0
Hz, 1H),
6.74 (d, J = 8.6 Hz, 1H), 4.90-4.70 (m, 2H), 4.63 (br.s, 1H), 4.18 (t, J = 6.8
Hz, 2H),
3.75-2.40 (m, 10H), 1.96-1.89 (m, 2H), 1.68-1.54 (m, 4H), 0.95 (t, J = 7.3 Hz,
3H)
ppm.
MS (ESIJ; (M+H)+ = 426.21, (M-H)- =424.25
F. 6-~1-hydroxy-2-f 4-hydroxy-4-(6-propoxypyridin-3-yl)piperidin-1-yllethyl}-
3,4-dihydroauinolin-2(11-one hydrochloride
By the procedures of Example 2 6-{ 1-hydroxy-2-[4-hydroxy-4-(6
propoxypyridin-3-yl)piperidin-1-yl]ethyl}-3,4-dihydroquinolin-2(1,F~-one was
converted to the title compound obtained as a white solid in 93% (408 mg)
after
recrystallization from 2-propanol.
1H NMR (270MHz, DMSO-d6) 8 = 10.12 (s, 1H), 9.82 (s, 1H), 8.23 (s, 1H), 7.76
(d, J
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB20041003125
47
= 8.1 Hz, 1 H), 7.24 (s, 1 H), 7.20 (d, J = 8.7 Hz, 1 H), 6.86 (d, J = 8.1 Hz,
1 H), 6.81 (d,
J = 8.7 Hz, 1 H), 6.16 (s, 1 H), 5.53 (s, 1 H), 5.07 (br. s, 1 H), 4.20 (t, J
= 6.8 Hz, 2H),
3.57-2.42 (m, 12H), 1.92-1.66 (m, 4H), 0.96 (t, J = 7.2 Hz, 3H) ppm.
MS (ESI); (M+H)+ = 426.19, (M-H)- =424.25
m.p.218.5°C
IR (KBr); 3240, 3136, 1678 cm 1
Example 5
A. 6-bromo-2-chloropyridin-3-of
The title compound was prepared according to the procedure described in
Example 3 from 2-chloropyridin-3-ol: 7.6 g (quant.) as a yellow solid.
1H NMR (270MHz, CDCl3) b = 7.35 (d, J = 8.3 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H)
ppm.
MS (En; M+=207, 209
B. 6-bromo-2-chloro-3-methoxypyridine
The title compound was prepared according to the procedure described in
Example 3 from 6-bromo-2-chloropyridin-3-ol: 4.37 g (54%) as a white solid.
1H NMR (300MHz, CDC13) b = 7.38 (d, J--8.4 Hz, 1H), 7.11 (d, J--8.4 Hz, 1H),
3.92
(s, 3H) ppm.
C. tart-butyl 4-(6-chloro-5-methoxypyridin-2-yl)-4-hydroxypiperidine-1-
carboxylate
The title compound was prepared according to the procedure described in
Example 1 from 6-bromo-2-chloro-3-methoxypyridine: 1.30 g (46%) as a yellow
solid.
1H NMR (270MHz, CDCl3) ~ = 7.28-7.21 (m, 2H), 4.19-4.03 (m, 2H), 3.93 (s, 3H),
3.30-3.20 (m, 2H), 2.00-1.86 (m, 2H), 1.72-1.60 (m, 2H), 1.48 (s, 9H) ppm.
MS (E17; M~ =342
D. 4-(6-chloro-5-methoxypyridin-2-yl)piperidin-4-of dihydrochloride
The title compound was prepared according to the procedure described in
Example 1 from tent-butyl 4-(6-chloro-5-methoxypyridin-2-yl)-4-
hydroxypiperidine
1-carboxylate: 963 mg (81 %) as a yellow solid.
1H NMR (270MHz, DMSO-d6) 8 = 9.32 (br.s, 1H), 8.78 (br.s, 1H), 7.67-7.60 (m,
2H),
3.90 (s, 3H), 4.20-3.10 (m, 6H), 2.33-2.20 (m, 2H), 1.80-1.70 (m, 2H) ppm.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
48
MS (En; M +-18 = 224
E. 6-f f 4-(6-chloro-5-methoxynyridin-2-yl)-4-hydroxypiperidin-1-yllacetyl}-
3,4-
dihydroauinolin-2(1~-one
The title compound was prepared according to the procedure described in
Example 3 from 4-(6-chloro-5-methoxypyridin-2-yl)piperidin-4-ol: 671 mg (78%)
as
a yellow solid.
1H NMR (300MHz, DMSO-d6) 8 = 10.43 (s, 1H), 7.89-7.83 (m, 2H), 7.62-7.54 (m,
2H), 6.93 (d, J = 8.0 Hz, 1H), 5.07 (s, 1H), 3.87 (s, 3H), 3.74 (s, 2H), 2.98-
2.92 (m,
2H), 2.74-2.64 (m, 2H), 2.56-2.42 (m, 4H), 2.14-2.00 (m, 2H), 1.53-1.46 (m,
2H)
ppm.
MS (ESn; (M+H)+ = 430.13, (M-H)' = 428.20
m.p. 215.9°C
IR (KBr); 3508, 3195, 1678 cm 1
F. 6-f 2-f 4-(6-chloro-5-methoxypyridin-2-yl)-4-hydroxypineridin-1-yll-1-
hydroxyethyl~-3,4-dihydroauinolin-2(1F~-one
The title compound was prepared according to the procedure described in
Example 2 from 6-{ [4-(6-chloro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-
yl]acetyl}-3,4-dihydroquinolin-2(lf~-one: 357 mg (57%) as ayellow solid.
1H NMR (270MHz, DMSO-d6) 8 = 10.02 (s, 1H), 7.63-7.54 (m, 2H), 7.15 (s, 1H),
7.10 (d, J = 8.1 Hz, 1 H), 6.79 (d, J = 8.1 Hz, 1 H), 5.02 (s, 1 H), 4. 82 (s,
1 H), 4.67-4.58
(m, 1H), 3.87 (s, 3H), 2.90-2.68 (m, 4H), 2.54-2.34 (m, 6H), 2.18-2.00 (m,
2H), 1.54-
1.46 (m, 2H) ppm.
G. 6-(2-f 4-(6-chloro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl}-3,4-dihydroguinolin-2(1F~1-one hydrochloride
By the procedures of Example 2, 6-{2-[4-(6-chloro-5-methoxypyridin-2-yl)-4-
hydroxypiperidin-1-yl]-1-hydroxyethyl}-3,4-dihydroquinolin-2(lI-~-one was
converted to the title compound obtained as a white solid in 100% (284 mg)
after
recrystallization from methanol-2-propanol.
1H NMR (270MHz, DMSO-d6) S = 10.09 (s, 1H), 7.66-7.58 (m, 2H), 7.21 (s, 1H),
7.17 (d, J = 8.1 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 5.49 (s, 1H), 4.90 (s,
1H), 3.89 (s,
3H), 3.33-2.30 (m, 12H), 1.78-1.50 (m, 2H) ppm.
MS (ESA; (M+H)+ = 432.14, (M-H)- = 430.22
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
49
1R (KBr); 3206, 1686 cni I
Example 6
6-;1-hydroxy-2-f4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yllethyl)-3,4-
dihydroauinolin-2(~-one hydrochloride
A. tent-butyl 4-hydroxy-4-(5-methoxypyridin-2-yl)piperidine-1-carboxylate
The title compound was prepared according to the procedure described in
Example 1 from 2-bromo-5-methoxypyridine (J. Chem. Soc. Perkirc Trans. 1,
1988,
3085): 647 mg (49%) as yellow oil.
1H NMR (300MHz, CDCl3) 8 = 8.21 (t, J=1.8 Hz, 1H), 7.25 (d, J=1.8 Hz, 2H),
4.97
(s, 1H), 4.20-1.56 (m, 8H), 3.87 (s, 3H), 1.49 (s, 9H) ppm.
MS (E~; M+ = 308
E. 4-(5-methoxypyridin-2-yl)piperidin-4-of
The title compound was prepared according to the procedure described in
Example 1 from tart-butyl 4-hydroxy-4-(5-methoxypyridin-2-yl)piperidine-1
carboxylate: 1.53 g (87%) as a yellow solid.
MS (E~; M + = 208
C. 6-~~4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yllacetyl)-3,4-
dihydropuinolin-2(1IT)-one
The title compound was prepared according to the procedure described in
Example 3 from 4-(5-methoxypyridin-2-yl)piperidin-4-ol: 729 mg (92%) as a
yellow
solid.
1H NMR (300MHz, DMSO-d6) 8 = 10.43 (s, 1H), 8.20 (d, J = 2.9 Hz, 1H), 7.90-
7.83
(m, 2H), 7.58 (d, J = 8.8 Hz, 1H), 7.36 (dd, J = 8.8, 2.9 Hz, 1H), 6.93 (d, J
= 8.1 Hz,
1H), 4.96 (s, 1H), 3.81 (s, 3H), 3.73 (s, 2H), 2.98-2.90 (m, 2H), 2.75-2.55
(m, 2H),
2.54-2.45 (m, 4H), 2.17-2.09 (m, 2H), 1.52-1.43 (m, 2H) ppm.
MS (ESA; (M+H)+ = 396.14, (M-H)- =394.21
m.p. 218.8°C
1R (I~Br); 3327, 3188, 1681 cm 1
D. 6-~1-hydroxy-2-f4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yllethyl)-
3,4-dihydroauinolin-2(1F~-one
The title compound was prepared according to the procedure described in
Example 2 from 6-{[4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yl]acetyl}-
3,4-
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
dihydroquinolin-2(11-one: 162 mg (24%) as a yellow solid.
1H NMR (270MHz, DMSO-d6) S = 10.02 (s, 1H), 8.20 (d, J = 3.0 Hz, 1H), 7.58 (d,
J
= 8.8 Hz, 1H), 7.36 (dd, J = 8.8, 3.0 Hz, 1H), 7.15 (s, 1H), 7.10 (d, J = 7.9
Hz, 1H),
6.79 (d, J= 7.9 Hz, 1H), 4.92 (s, 1H), 4.79 (br.s, 1H), 4.67-4.56 (m, 1H),
3.81 (s, 3H),
5 2.90-2.66 (m, 4H), 2.60-2.35 (m, 6H), 2.24-2.05 (m, 2H), 1.52-1.44 (m, 2H)
ppm.
E. 6-~1-hydroxy-2-f 4-hydroxy-4-(5-methoxypyridin-2-yl)piperidin-1-yllethyl)-
3,4-dihydroaluinolin-2(1I~-one hydrochloride
By the procedures of Example 2, 6-{ 1-hydroxy-2-[4-hydroxy-4-(5
methoxypyridin-2-yl)piperidin-1-yl]ethyl}-3,4-dihydroquinolin-2(11-one was
10 converted to the title compound obtained as a white solid in 72% (103 mg)
after
recrystallization from 2-propanol.
1H NMR (270MHz, DMSO-d6) 8 = 10.12 (s, 1H), 9.82 (s, 1H), 8.25 (d, J = 2.6 Hz,
1 H), 7.61 (d, J = 8.7 Hz, 1 H), 7.43 (dd, J = 8.7, 2.6 Hz, 1 H), 7.24 (s, 1
H), 7.20 (d, J =
7.9 Hz, 1H), 6.86 (d, J= 7.9 Hz, 1H), 6.13 (s, 1H), 5.61 (s, 1H), 5.07 (br.s,
1H), 3.83
15 (s, 3H), 3.65-2.40 (m, 12H), 1.90-1.70 (m, 2H) ppm.
MS (ESA; (M+H)+ = 398.15
m.p. 236.5°C
1R (KBr); 3188, 1686 cm 1
Example 7
20 6-~1-hydroxy-2-f4-hydroxy-4-(4-methylpyridin-2-yl)piperidin-1-yllethyl)-3,4-
dihydroauinolin-2(1Fn-one hydrochloride
A. tart-butyl 4-hydroxy-4-(4-methylpyridin-2-yl)piperidine-1-carboxylate
The title compound was prepared according to the procedure described in
Example l from 2-bromo-4-methylpyridine: 1.72 g (53%) as a yellow oil.
25 1H NMR (270MHz, CDCl3) b = 8.38 (d, J = 5.1 Hz, 1H), 7.13 (s, 1H), 7.04 (d,
J = 5.1
Hz, 1H), 5.32 (s, 1H), 4.19-4.03 (m, 2H), 3.37-3.17 (m, 2H), 2.38 (s, 3H),
2.00-1.85
(m, 2H), 1.64-1.50 (m, 2H), 1.48 (s, 9H) ppm.
MS (El); M+ =292
B. 4-(4-methylpyridin-2-yl)piperidin-4-of dihydrochloride
30 The title compound was prepared according to the procedure described in
Example 1 from tart-butyl 4-hydroxy-4-(4-methylpyridin-2-yl)piperidine-1-
carboxylate: 1.26 g (81%) as a yellow solid.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
51
1H NMR (270MHz, DMSO-d6) ~ = 9.34 (br.s, 2H), 8.62 (d, J=5.8 Hz, 1H), 7.83
(br.s,
1H), 7.70 (d, J=5.8 Hz, 1H), 3.30-2.34 (m, 6H), 2.58 (s, 3H), 2.00-1.88 (m,
2H) ppm.
C. 6-f [4-hydroxy-4-(4-methvlpyridin-2-vl)piperidin-1-vllacetvl~-3.4-
dihydroauinolin-2(lI~-one
The title compound was prepared according to the procedure described in
Example 3 from 4-(4-methylpyridin-2-yl)piperidin-4-ol: 501 mg (66%) as a
yellow
solid.
1H NMR (270MHz, DMSO-d6) b = 10.42 (s, 1H), 8.34 (d, J = 4.9 Hz, 1H), 7.88 (d,
J
= 8.2 Hz , 1H), 7.85 (s, 1H), 7.49 (s, 1H), 7.05 (d, J= 4.9 Hz, 1H), 6.93 (d,
J= 8.2 Hz,
1H), 5.01 (s, 1H), 3.73 (s, 2H), 3.00-2.46 (m, 8H), 2.32 (s, 3H), 2.24-2.10
(m, 2H),
1.52-1.40 (m, 2H) ppm.
MS (ESn; (M+H)+ = 380.17, (M-H)- =378.24
IR (KBr); 3234, 1682 cm 1
D. 6-~1-hydroxy-2-f4-hydroxy-4-(4-methylpyridin-2-yl)piperidin-1-yllethyl)-3,4-
dihydroauinolin-2(1I3~-one
The title compound was prepared according to the procedure described in
Example 2 from 6-{[4-hydroxy-4-(4-methylpyridin-2-yl)piperidin-1-yl]acetyl}-
3,4-
dihydroquinolin-2(lI~-one: 134 mg (28%) as a yellow solid.
1H NMR (270MHz, DMSO-d6) b = 10.02(s, 1H), 8.37-8.32 (m, 1H), 7.49 (s, 1H),
7.18-7.04 (m, 3H), 6.79 (d, J--8.1 Hz, 1H), 4.98 (s, 1H), 4.80 (br.s, 1H),
4.63 (br.s,
1H), 2.90-2.70 (m, 4H), 2.54-2.06 (m, 8H), 2.32 (s, 3H), 1.50-1.40 (m, 2H)
ppm.
E. 6-f 1-hydroxy-2-f4-hydroxy-4-(4-methylpyridin-2-yl)piperidin-1-yllethyl)-
3,4-
dihydropuinolin-2(1I3~-one hydrochloride
By the procedures of Example 2, 6-{1-hydroxy-2-[4-hydroxy-4-(4
methylpyridin-2-yl)piperidin-1-yl]ethyl}-3,4-dihydroquinolin-2(1F~-one was
converted to the title compound obtained as a white solid in 71% (104 mg)
after
recrystallization from 2-propanol.
1H NMR (270MHz, DMSO-d6) 8 = 10.13 (s, 1H), 9.83 (br.s, 1H), 8.40 (d, J = 4.9
Hz,
1H), 7.52 (s, 1H), 7.24 (s, 1H), 7.21 (d, J= 8.2 Hz, 1H), 7.14 (d, J= 4.9 Hz,
1H), 6.86
(d, J = 8.2 Hz, 1H), 6.13 (s, 1H), 5.67 (s, 1H), 5.08 (br.s, 1H), 3.65-2.42
(m, 12H),
2.35 (s, 3H), 1.86-1.70 (m, 2H) ppm.
MS (ESn; (M+H)+ = 382.19
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
52
m.p. 251.7°C
IR (KBr); 3242, 1661 cm 1
Example 8
6-(2-f 4-(5-(dimethylamino)-6-fluoropyridin-2-yll-4-hydroxypiperidin-1-yl~-1-
hydroxyethyl)-3,4-dihydroauinolin-2(lli~-one hydrochloride
A. 6-bromo-2-fluoronyridin-3-amine
The title compound was prepared according to the procedure described in
Example 3 from 2-fluoropyridin-3-amine (Heterocycles, 1986, 24, 3213): 1.68 g
(76%) as a red solid.
1H NMR (270MHz, CDCl3) b = 7.15 (d, J=7.9 Hz, 1H), 7.00 (dd, J=10.2, 7.9 Hz,
1H),
3.82 (br.s, 2H) ppm.
MS (E1); M+=190, 192
B. 6-bromo-2-fluoro-N, N-dimethylpyridin-3-amine
A mixture of 6-bromo-2-fluoropyridin-3-amine (800 mg, 4.19 mmol) and
formaldehyde (35% in H20, 2 mL) in formic acid (2 mL) was heated under reflux
for
5 hours. The mixture was treated with aq. sodium hydroxide and extracted with
ethyl
acetate. The combined organic layer was dried and evaporated. The residue was
purified by chromatography on silica gel, eluting with ethyl acetate / hexane
(1:10
v/v), to afford the titled compound as a colorless oil (770 mg, 84 %).
1H NMR (300MHz, CDCl3) 8 = 7.22 (dd, J=8.3, 1.1 Hz, 1H), 7.03 (dd, J=10.3, 8.3
Hz,
1H), 2.88 (s, 3H), 2.88 (s, 3H) ppm.
MS (El]; M+ =218, 220
C. tent-butyl 4-(5-(dimethylamino)-6-fluoropyridin-2-yll-4-hydroxypiperidine-1-
carboxylate
The title compound was prepared according to the procedure described in
Example 1 from 6-bromo-2-fluoro-N,N dimethylpyridin-3-amine: 718 mg (61 %) as
a
yellow solid.
1H NMR (270MHz, CDCl3) 8 = 7.19 (dd, J=10.0, 7.9 Hz, 1H), 7.09 (dd, J=7.9, 2.1
Hz, 1H), 4.15-4.00 (m, 2H), 3.40-3.07 (m, 2H), 2.89 (s, 3H), 2.88 (s, 3H),
2.00-1.86
(m, 2H), 1.70-1.59 (m, 2H), 1.48 (s, 9H) ppm.
MS (El); M+=339
D. 4-(5-(dimethylamino)-6-fluoropyridin-2-yllpiperidin-4-of dihydrochloride
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
53
The title compound was prepared according to the procedure described in
Example 1 from tert-butyl 4-[5-(dimethylamino)-6-fluoropyridin-2-yl]-4-
hydroxypiperidine-1-carboxylate: 643 mg (87%) as a yellow solid.
1H NMR (270MHz, DMSO-d6) b = 9.10 (br.s, 1H), 8.72 (br.s, 1H), 7.50-7.40 (m,
1H),
3.25-1.69 (m, 14H) ppm.
MS (E~; M+=239
E. 6-(f 4-[5-(dimethylamino)-6-fluoropyridin-2-yll-4-hydroxypiperidin-1-
yl~acetyl)-3,4-dihydroguinolin-2(1I~-one
The title compound was prepared according to the procedure described in
Example 3 from 4-[5-(dimethylamino)-6-fluoropyridin-2-yl]piperidin-4-ol: 239
mg,
56%) as a yellow solid.
1H NMR (270MHz, DMSO-d6) b = 10.42 (s, 1H), 7.90-7.82 (m, 2H), 7.44-7.32, (m,
2H), 6.93 (d, J = 8.2 Hz, 1H), 4.95 (s, 1H), 3.72 (s, 2H), 3.02-2.45 (m, 8H),
2.78 (s,
6H), 2.10-2.06 (m, 2H), 1.54-1.43 (m, 2H) ppm.
MS (ESn; (M+H)+ = 427.16, (M-H)- = 425.10
F. 6-(2-~4-~5-(dimethylamino)-6-fluoropyridin-2-yll-4-hydroxypiperidin-1-yl)-1-
hydroxyethyl)-3,4-dihydroguinolin-2(lI~-one
The title compound was prepared according to the procedure described in
Example 2 from 6-({4-[5-(dimethylamino)-6-fluoropyridin-2-yl]-4-
hydroxypiperidin-
1-yl}acetyl)-3,4-dihydroquinolin-2(lI~-one: 120 mg, 50%) as a yellow solid.
1H NMR (270MHz, DMSO-d6) 8 = 10.02 (s, 1H), 7.44-7.32 (m, 2H), 7.15 (s, 1H),
7.10 (d, J= 8.2 Hz, 1H), 6.78 (d, J= 8.2 Hz, 1H), 4.92 (s, 1H), 4.81 (s, 1H),
4.63 (br.s,
1H), 2.90-2.40 (m, 10H), 2.78 (s, 6H), 2.10-2.00 (m, 2H), 1.58-1.45 (m, 2H)
ppm.
MS (ESA; (M+H)+ = 429.22, (M-H)- = 427.19
G. 6-(2-~4-[5-(dimethylamino)-6-fluoropyridin-2-yll-4-hydroxypiperidin-1-yl~-1-
hydroxyethyl)-3,4-dihydroguinolin-2(lI~-one hydrochloride
By the procedures of Example 2, 6-(2-{4-[5-(dimethylamino)-6-fluoropyridin-
2-yl]-4-hydroxypiperidin-1-yl}-1-hydroxyethyl)-3,4-dihydroquinolin-2(1I~-one
was
converted to the title compound obtained as a white solid in 95% (114 mg)
after
recrystallization from 2-propanol.
1H NMR (270MHz, DMSO-d6) 8 = 10.09 (s, 1H), 9.58 (s, 1H), 7.51-7.36 (m, 2H),
7.25-7.15 (m, 2H), 6.84 (d, J = 8.0 Hz, 1H), 4.96 (br.s, 1H), 3.73-1.57 (m,
14H), 2.79
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
54
(s, 6H) ppm.
MS (ESn; (M+H)+ = 428.99, (M-H)- = 426.93
IR (KBr); 3223, 1691 cm 1
Example 9
A. 4-[4-(Methoxymethyl)phenyllpiperidin-4-of
To a stirred solution of 1-bromo-4-(methoxymethyl)benzene (J.Med. Chem. 1998,
~l ,
940 - 951.) (3.4 g, 20 mmol) in tetrahydrofuran (60 mL), ~c-buthyllithium
(1.56 M in
hexane, 13.5 mL, 21 mmol) was added at - 78 °C and the mixture was
stirred for 1 h.
Then to the mixture, a solution of ethyl 4-oxopiperidine-1-carboxylate in
tetrahydrofuran was added at - 78 °C and the mixture was stirred at
room temperature
for 16 h. The mixture was treated with sat. aq. Ammonium chloride and was
extracted
with CH2C12. The extract was dried over MgS04. After evaporation, the crude
was
dissolved with ethyl alcohol (10 mL). To the solution, KOH (5.6 g, 100 mmol)
was
added and the mixture was refluxed for 2 h. The mixture was diluted with water
and
extracted with CH2C12. The extract was dried over MgS04. 4-[4-
(Methoxymethyl)phenyl]piperidin-4-of (450 mg) was afforded by amino type gel
(Fuji
Silysia ; DU3050) column chromatography (CH2Cl2-MeOH 8:1).
1H NMR (270 MHz, CDC13) S = 7.50 (d, J = 8.1 Hz, 2H), 7.34 (d, J = 8.1 Hz,
2H),
4.45 (s, 2H), 3.40 (s, 3H), 3.19-2.97 (m, 4H), 2.10-1.96 (m, 2H), 1.78-1.70
(m, 2H)
ppm.
B. 6-(1-Hydroxy-2-~4-hydroxy-4-[4-(methoxymethyl)phenyllpiperidin-1-
yl}ethyl)-3,4-dihydroauinolin-2(115n-one hydrochloride
A mixture of 6-(chloroacetyl)-3,4-dihydroquinolin-2(lI~-one (450 mg, 2.0
mmol),
4-[4-(methoxymethyl)phenyl]piperidin-4-of (450 mg, 2.0 mmol) and triethylamine
(0.56 mL, 4.0 mmol) in N.N-dimethyl formamide (4 mL) was stirred at room
temperature for 24 h. Then to a suspension of sodium borohydride (227 mg, 6.0
mmol) in ethyl alcohol (20 mL) , the reaction mixture was added at room
temperature
and the mixture was stirred for 16 h. The mixture was diluted with ethyl
acetate and
was washed with water. The organic layer was dried over MgS04. After
evaporation,
6-( 1-hydroxy-2-{ 4-hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-yl } ethyl)-
3,4-
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
dihydroquinolin-2(1F~-one (155 mg) was afforded by silica-gel column
chromatography (CH2C12-MeOH 8:1). To a suspension of 6-(1-hydroxy-2-{4-
hydroxy-4-[4-(methoxymethyl)phenyl]piperidin-1-yl } ethyl)-3,4-dihydroquinolin-
2(lI~-one (155 mg, 0.38 mmol) in ethyl Alcohol (3 mL) , 4N hydogenchloride in
5 ethyl acetate (0.1 mL, 0.4 mmol) was added. 6-(1-Hydroxy-2-{4-hydroxy-4-[4
(methoxymethyl)phenyl]piperidin-1-yl } ethyl)-3,4-dihydroquinolin-2(ll~-one
hydrochloride (112 mg) was crystallized from ethyl alcohol.
1H NMR (300 MHz, DMSO-d6) 8 = 10.22-10.10 (br, 2H), 7.60-7.18 (m, 6H), 6.87
(d,
J = 8.1 Hz, 1H), 6.18 (br, 1H), 5.52 (br, 1H), 5.17-5.10 (m, 1H), 4.40 (s,
2H), 3.65
10 3.15 (m, 9H), 2.88 (t, J = 7.9, 2H), 2.56-2.37 (m, 2H), 1.87-1.74 (m, 2H)
ppm.
MS (ESn; (M+H)+ (411.14), (M-H)- (409.19)
IR (F~Br) 3404, 1674, 1508, 1375, 1238, 1203 cm 1
m.p. 208.4 °C
15 Example 10
tent-butyl 4-hydroxy-4-(6-methoxypyridin-3-yl)piperidine-1-carboxylate
A solution of 5-bromo-2-methoxypyridine (36g / 193mmo1) in diethyl ether
(200m1) was added dropwise to a solution of n-butyl lithium in hexane (1.59M /
121m1) and diethyl ether (500m1) at -78°C. After addition was
completed, the mixture
20 was stirred at -78 °C for 30min and to the mixture was added a
solution of tart-butyl
4-oxopiperidine-1-carboxylate-in diethyl ether (300m1) at -78°C. The
mixture was
allowed to warm to room temperature and stirred overnight. To the mixture was
added water (400m1) and the organic layer was extracted with diethyl ether
(500m1).
The combined organic layer was washed with brine and dried over NaS04 and
25 concentrated. The residue was purified by column chromatography on silica
gel (ethyl
acetate / hexane = 1/2) to afford the titled compound (16.5g / 42%) as oil.
4-(6-methoxypyridin-3-yl)piperidin-4-of hydrochloride
To a stirred solution of tart-butyl 4-hydroxy-4-(6-methoxypyridin-3
30 yl)piperidine-1-carboxylate (15g / 49mmo1) in ethyl acetate (300m1) was
added 4N
hydrochloride in ethyl acetate (45m1 / 150mmol) and the resulting suspension
was
stirred at 50°C for 2h. To the suspension was added additional 4N
hydrogen chloride
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
56
in ethyl acetate (27.5m1 / 75mmo1) and stirred at 50°C for 3h. After
cooling, the
precipitate was collected and dried in vacuo for 1h to afford the titled
compound as a
white solid (12.7g / 93%).
1H NMR (300 MHz, DMSO-d6) S = 9.25-8.90 (br, 2H), 8.24 (dd, J = 0.5, 2.6Hz,
1H),
7.79 (dd, J = 2.6, 8.6Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H), 6.00-5.40 (br, 1H),
3.86 (s,
3H), 3.30-3.00 (m, 4H), 2.30-2.10 (m, 2H), 1.86-1.76 (m, 2H) ppm.
6-~f4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yllacetyl~-3,4-
dihydroauinolin-2(1FP-one
To a stirred solution of 6-(Chloroacetyl)-3,4-dihydroquinolin-2(lI~-one (WO
9302052)(544mg, 2.42 mmol) in DMF (15m1) were added 4-(6-methoxypyridin-3
yl)piperidin-4-of hydrochloride (680mg, 2.42mmol) and triethylamine (3.6m1, 26
mmol) at room temperature under nitrogen and the mixture was stirred for 1h at
room
temperature. The reaction mixture was added water and the resulting solid was
collected to afford the titled compound (650mg / 68%).
1H NMR (300MHz, DMSO-d6) ~ =10.4 (s, 1H), 8.24 (d, J = 2.4 Hz, 1H), 7.90-7.80
(m, 2H), 7.78 (dd, J = 2.4, 8.4 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 6.75 (d, J
= 8.4 Hz,
1H, 4.90 (s, 1H), 3.82 (s, 3H), 3.77 (s, 2H), 2.95 (t, J = 8.1 Hz, 2H), 2.75-
2.65 (m,
2H), 2.65-2.45 (m, 4H), 2.00-1.85 (m, 2H), 1.70-1.55 (m, 2H) ppm
MS (ESI); (M+H)+ (427.25), (M-H)- (425.32)
6-~1-hydroxy-2-~4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yllethyl~-3,4-
dihydroauinolin-2(lI~-one
The mixture of 6-{ [4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-
yl]acetyl}-3,4-dihydroquinolin-2(11-one (560mg, 1.4 mmol) and sodium
borohydride (60mg, 1.56mmol) in methanol (15m1) was stirred at room
temperature
for 18h and 60°C for 1h. The resulting white precipitate was collected
(366mg / 65%).
1H NMR (300MHz, DMSO-d6) ~ =10.0 (s, 1H), 8.24 (d, J = 2.6 Hz, 1H), 7.76 (dd,
J
= 2.6, 8.8 Hz, 1H), 7.15 (s, 1H), 7.10 (d, J = 9.3 Hz, 1H), 6.78 (d, J = 7.9
Hz, 1H),
6.77 (d, J = 8.1 Hz, 1H) , 4.95-4.75 (br, 1H), 4.70-4.55 (m, 1H), 3.82 (s,
3H), 2.86 (t,
J = 7.9 Hz, 2H), 2.80-2.65 (m, 2H), 2.65-2.35 (m, 6H), 2.00-1.85 (m, 2H), 1.65-
1.55
(m, 2H) ppm
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
57
6-f 1-hydroxy-2-f4-hydroxy-4-(fi-methoxypyridin-3-yl)piperidin-1-yllethyl~-3,4-
dihydroguinolin-2(11~-one hydrochloride
To a suspension of 6-{ 1-hydroxy-2- [4-hydroxy-4-(6-methoxypyridin-3
yl)piperidin-1-yl]ethyl}-3,4-dihydroquinolin-2(lI~-one (538m g, 1.36 mmol) in
methanol (14 ml) was added hydrogen chloride 4.0 M solution in ethyl acetate
(0.339mL, 1.36 mmol) and the mixture was concentrated. The residue was
crystallized from IPA to afford the titled compound as a white solid (430mg /
73%).
1H NMR (270MHz, DMSO-d6) ~ 9.9 (s, 1H), 8.27 (d, J--2.6Hz, 1H), 7.78 (dd, .1--
2.6,
8.9 Hz, 1H), 7.23 (s, 1H), 7.20 (d, J= 8.1 Hz, 1H), 6.86 (d, J= 8.1 Hz, 1H),
6.79 (d, J
= 8.6 Hz, 1H) , 5.40-5.20 (br, 1H), 5.20-4.90 (br, 1H), 3.86 (s, 3H), 3.60-
2.80 (m, 8H),
2.60-2.20 (m, 4H), 2.00-1.70 (m, 2H) ppm
MS (ESIJ; (M+H)+ (398.18), (M-H)- (396.25)
IR (KBr) 3308, 3173, 2949, 2823, 1653, 1599, 1493, 1389, 1292, 1223, 1200,
1134,
1051, 1029, 914, 829 cm 1
Example 11
Diethyl 2-(2-fluoro-6-nitrobenzyl)malonate
To a suspension of sodium hydride (5.2g, 130mmo1) in N.N-
dimethylformamide / tetrahydrofuran (110m1/45m1) was added Diethyl malonate
(19m1, 125mmol) dropwise and stirred for 30min at room temperature. To the
suspension was added 2-(bromomethyl)-1-fluoro-3-nitrobenzene (29g, 124mmol) in
DMF/THF (40m1/30m1) and refluxed for 3h. After cooling, the mixture was
quenched
with brine and extracted with ethyl acetate. The extract was dried over sodium
sulfate
and concentrated in vacuo to afford the titled compound as brown oil (48g).
This
crude product was used in the next step without further purification.
1H NMR (CDC13) ~ 7.77 (d, J = 8.lHz, 1H), 7.50-7.24 (m, 2H), 4.18 (q, J = 7.1
Hz,
4H), 3.76 (t, J = 7.7 Hz, 1H), 3.55 (dd, J = 7.7, 1.6 Hz, 2H), 1.23 (t, J = 7.
lHz, 6H)
3-(2-fluoro-6-nitrophenyl)propan0ic acid
The mixture of Diethyl 2-(2-fluoro-6-nitrobenzyl) malonate (crude 48g) and
6N hydrogen chloride aq. (100m1) in acetic acid (100m1) was refluxed for 7h.
After
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
58
evaporating solvent, the resulting solid was collected and triturated with
water to
afford 3-(2-fluoro-6-nitrophenyl) propanoic acid and Ethyl 3-(2-fluoro-6-
nitrophenyl )propanoate mixture. The mixture (18g) and 2N NaOH aq. (450m1) in
ethanol (450m1) were refluxed for lh.After cooling, the mixture was acidified
with
2N hydrogen chloride aq. and extracted with dichloromethane. The extract was
concentrated in vacuo to afford the titled compound (15g) as a brown solid.
This
crude product was used in the next step without further purification.
1H NMR (d-DMSO) ~ 7.82 (d, J = 7.9Hz, 1H), 7.66-7.50 (m, 2H), 3.03 (t, J = 7.5
Hz,
2H), 2.54 (t, J = 7.5 Hz, 2H)
5-fluoro-3, 4-dihydro-2(lFi~-auinolinone
The suspension of 3-(2-fluoro-6-nitrophenyl) propanoic acid( 6.0g, 28mmol)
and 10°Io Pd-C (300mg) was hydrogenated under HZ(4atm) for 3h. After
filtration, the
filtrate was concentrated in vacuo to afford the titled compound (4.6g) as a
slightly
brown solid. This crude product was used in the next step without further
purification.
1H NMR (d-DMSO) ~S 10.2 (s, 1H), 7.16 (dd, J = 14.5, 8.2 Hz, 1H), 6.77 (d, J =
8.4
Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 2.87 (t, J = 7.5 Hz, 2H), 2.47 (t, J = 7.3
Hz, 2H)
6-(bromoacetyl)-5-fluoro-3, 4-dihydro-2(1I~-auinolinone
To a stirring suspension of aluminum chloride (8.0g, 60mmo1) in 1,2-
dichloroethane (16m1) was added bromoacetyl bromide (4.2m1, 48mmo1) at
0°C.
After 30min, to a suspension was added 5-fluoro-3, 4-dihydro-2 (lI~-
quinolinone
(4.0g, 24mmo1) portionwise, then the mixture was allowed to warm to
50°C and
stirred for 4h. After cooling, the solvent was evaporated in vacuo and the
residue was
quenched with ice water and extracted with ethyl acetate. The extract was
dried over
sodium sulfate and concentrated in vacuo. The residue was purified by column
chromatography (hexane / ethyl acetate = 1 / 1) to afford the titled compound
(2g) as a
yellow solid.
'H NMR (d-DMSO) ~ 10.7(s, 1H), 7.75 (t, J = 8.3 Hz, 1H), 7.49 (t, J = 8.4 Hz,
1H),
4.74 (d, J = 2.4 Hz, 1H), 2.95 (t, J = 7.9 Hz, 2H), 2.54 (t, J = 8.1 Hz, 2H)
5-fluoro-6-~ f 4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yllacetyl~-3,4-
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
59
dihydroauinolin-2(11-one
To a cooling solution of 6-(bromoacetyl)-5-fluoro-3, 4-dihydro-2 (lI~-
quinolinone (1g, 3.5 mmol) and triethylamine (4.5 ml, 32 mmol) in DMF (70 ml)
at
0°C was added a solution of 4-(3-methoxyphenyl)-4-piperidinol (1.2g,
4.2 mmol) in
DMF (15m1) dropwise and stirred at the same temperature for 3h. The reaction
mixture was added ice water and the resulting precipitate was collected to
afford
product the titled compound as a brown solid (943mg / 65%).
1H NMR (300 MHz, DMSO-d6) ~ 10.6 (s, 1H), 8.23 (d, J--2.7Hz, 1H), 7.70 (dd,
J=2.6, 8.6Hz, 1H), 7.69 (t, J--8.3Hz, 1H), 6.76 (t, J=8.6Hz, 2H), 4.89 (s,
1H), 3.82 (s,
3H), 3.71 (d, J--2.2Hz, 2H), 2.93 (t, J = 7.5 Hz, 2H,), 2.75-2.50 (m, 4H),
2.00-1.80 (m,
2H), 1.65-1.55 (m, 2H) ppm
5-fluoro-6-~1-hydroxy-2-[4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-
yllethyl}-3,4-dihydroguinolin-2(ll~-one
The mixture of 5-fluoro-6-{ [4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-
1-yl]acetyl}-3,4-dihydroquinolin-2(11-one (943mg, 2.3mmo1) and sodium
borohydride (95mg, 2.5mmol) in ethanol (23m1) was stirred at room temperature
overnight. The resulting white precipitate was collected (586mg / 61 %).
1H NMR (270 MHz, DMSO-d6) ~ 10.2 (s, 1H), 8.23 (d, J--2.OHz, 1H), 7.70 (dd,
J--2.3, 8.6Hz, 1H), 7.26 (t, J--8.4Hz, 1H), 6.75 (d, J--8.4Hz, 1H), 6.67 (d, J-
-8.2Hz,
1H), 5.20-4.75 (br, 2H), 3.82 (s, 3H), 2.87(t, J = 7.6 Hz, 2H,), 2.80-2.35 (m,
6H),
2.00-1.80 (m, 2H), 1.65-1.55 (m, 2H) ppm
5-fluoro-6-f 1-hydroxy-2-f4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-
yllethyl}-3,4-dihydroguinolin-2(1I~-one hydrochloride
To a suspension of 5-fluoro-6-{ 1-hydroxy-2-[4-hydroxy-4-(6-
methoxypyridin-3-yl)piperidin-1-yl]ethyl}-3,4-dihydroquinolin-2(1F~-one (586m
g,
1.41 mmol) in methanol (14 ml) was added 4N hydrogen chloride 4.0 M solution
in
ethyl acetate (0.355mL, 1.42 mmol) and the mixture was concentrated. The
residue
was crystallized from Il'A to afford the titled compound as a white solid
(500mg /
79%).
1H NMR (300 MHz, DMSO-d6) ~ 10.3 (s, 1H), 10.1-9.80 (br, 1H), 8.26 (s, 1H),
7.77
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
(d, J--7.OHz, 1H), 7.32 (t, J--7.9Hz, 1H), 6.83 (d, J--8.4Hz, 1H), 6.75 (d, J--
8.2Hz, 1H),
6.40-6.15 (br, 1H), 5.65-5.50 (br, 1H), 5.40-5.20 (br, 1H), 3.84 (s, 3H), 3.70-
3.00 (m,
6H), 2.90(t, J= 7.5 Hz, 2H,), 2.54-2.44 (m, 2H), 1.96-1.76 (m, 2H) ppm
MS (ESI]; (M+H)+ (416.10), (M-H)- (414.17)
5 IR (KBr) 3333, 2955, 2748, 2345, 1670, 1630, 1604, 1489, 1377, 1286, 1223,
1069,
1022, 960, 831 cm 1
m.p. 204.1 °C
Example 12
10 . 7-(bromoacetyl)-1,3,4,5-tetrahydro-2H-1-benzazepin-2-one
N.N-Dimethylformamide(3ml) was added dropwise to aluminum
chloride(22g / 168mmo1). The suspension was stirred at 45 °C and added
bromoacetyl
bromide (4.2m1 / 48m1) and 1,3,4,5-tetrahydro-2H 1-benzazepin-2-one
(Terahedoron.,
49, 1867 (1993)).3.8g / 24mmo1). The suspension was warmed to 78°C
under stirring
15 for 3h, poured onto ice and the resulting precipitate was collected to
afford the titled
compound (7g). The crude product was used in the next step without further
purification.
7-~f4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-yl~acetyl)-1,3,4,5-
20 tetrahydro-2FI-1-benzazepin-2-one
To a stirred solution of 7-(bromoacetyl)-1,3,4,5-tetrahydro-2H 1-benzazepin-
2-one (500mg, 1.78 mmol) in DMF (6m1) were added 4-(6-methoxypyridin-3-yl)
piperidin-4-of hydrochloride (400mg, 1.42mmol) and triethylamine (1.2m1, 8.9
mmol)
at room temperature and the mixture was stirred for 2days at room temperature.
To
25 the reaction mixture was added sat.NaHCO3aq. and extracted with ethyl
acetate. The
combined organic layer was washed with brine and dried over NaS04 and
concentrated to afford the titled compound as a yellow solid (126mg / 22%).
1H NMR (300MHz, DMSO-d6) ~ =9.85 (s, 1H), 8.24 (d, J = 2.4 Hz, 1H), 7.98-7.86
(m, 2H), 7.78 (dd, J = 2.6, 8.6 Hz, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.75 (d, J
= 8.6 Hz,
30 1H), 4.91 (s, 1H), 3.82 (s, 3H), 2.84-2.48 (m, 4H), 2.30-2.04 (m, 6H), 2.00-
1.86 (m,
2H), 1.70-1.56 (m, 2H) ppm
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
61
7-f 1-hydroxy-2-~4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-vllethvll-
1,3,4,5-tetrahydro-2H-1-benzazepin-2-one
The mixture of 7-( [4-hydroxy-4-(6-methoxypyridin-3-yl)piperidin-1-
yl]acetyl}-1,3,4,5-tetrahydro-2H 1-benzazepin-2-one (126mg, 0.31 mmol) alld
sodium borohydride (l3mg, 0.34mmo1) in ethanol (3m1) was stirred at room
temperature for 2days. To the reaction mixture was added water and extracted
with
dichloromethane and methanol. The combined organic layer was washed with brine
and dried over NaS04 and concentrated. The residue was purified by column
chromatography on silica gel (dichloromethane / methanol = 5/1) to afford the
solid
(70mg). This solid was crystallized from methanol-ethyl acetate-hexane to
afford the
titled compound as a white solid (40mg / 30%).
1H NMR (300MHz, DMSO-d6) ~ =9.47 (s, 1H), 8.24 (d, J = 2.4 Hz, 1H), 7.78 (dd,
J
= 2.6, 8.6 Hz, 1H), 7.23 (s, 1H), 7.20 (d, J = 8.1 Hz, 1H), 6.90 (d, J = 8.1
Hz, 1H),
6.76 (d, J = 8.6 Hz, 1H), 4.95-4.85 (m, 2H), 4.75-4.65 (br, 1H), 3.82 (s, 3H),
2.80
2.35 (m, 8H), 2.20-1.85 (m, 6H), 1.70-1.50 (m, 2H) ppm
MS (ESI); (M+H)+ (412.13), (M-H)- (410.23)
IR (I~Br) 3246, 2943, 2827, 1662, 1606, 1496, 1373, 1288, 1120, 1028, 831crri
1
m.p. 187.7 °C
Example 13
5-fluoro-6-(d4-hydroxy-4-f4-(methoxymethyl)phenyllpiperidin-1-yl)acetyl)-3,4-
dihydroguinolin-2(1I3~-one
To a cooling solution of 6-(bromoacetyl)-5-fluoro-3, 4-dihydro-2 (lI~
quinolinone (425mg, 1.5 mmol) and triethylamine (0.63 ml, 4.5 mmol) in DMF (10
ml) at 0°C was added a solution of 4-[4-(methoxymethyl)phenyl]piperidin-
4-of
(492mg, 2.23 mmol) in DMF (5m1) dropwise. The reaction mixture was added ice
water and extracted with ethyl acetate. The combined organic layer was washed
with
brine and dried over NaS04 and concentrated to afford the titled compound as a
brown solid (600mg). The crude product (600mg) included impurities but used in
the
next step without further purification.
5-fluoro-6-(1-hydroxy-2-;4-hydroxy-4-(4-(methoxymethyl)phenyll piperidin-1-
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
62
yl)ethyl)-3,4-dihydroauinolin-2(lI~-one
The mixture of 5-fluoro-6- ({4-hydroxy-4- [4-(methoxymethyl)
phenyl]piperidin-1-yl}acetyl)-3,4-dihydroquinolin-2(11-one (600mg) and sodium
borohydride (114mg, 3.Ommol) in ethanol was stirred for 2days at room
temperature.
To the reaction mixture was added water and extracted with dichloromethane.
The
combined organic layer was washed with brine and dried over NaS04 and
concentrated. The residue was purified by column chromatography on silica gel
(dichloromethane / methanol = 10/1) to afford the titled compound (123mg / 20%
2steps).
1H NMR (300MHz, DMSO-d6) ~ =10.2 (s, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.30-7.20
(m, 3H), 6.68 (d, J = 8.3 Hz, 1H), 5.10-4.90 (m, 2H), 4.78 (s, 1H), 4.37 (s,
2H), 3.27
(s, 3H), 2.87 (t, J = 7.7 Hz, 2H), 2.85-2.65 (m, ZH), 2.65-2.35 (m, 6H), 2.05-
1.85 (m,
2H), 1.65-1.50 (m, 2H) ppm
5-fluoro-6-(1-hydroxv-2-f 4-hvdroxv-4-f4-(methoxvmethvl)nhenvllnineridin-1-
yl)ethyl)-3,4-dihydropuinolin-2(1I~-one hydrochloride
To a suspension of 5-fluoro-6- (1-hydroxy-2-{4-hydroxy-4-[4-
(methoxymethyl) phenyl] piperidin-1-yl}ethyl)-3,4-dihydroquinolin-2(1~-one
(123m
g, 0.30 mmol) in methanol (3 ml) was added 4N hydrogen chloride 4.0 M solution
in
ethyl acetate (0.075mL, 0.30 mmol) and the mixture was concentrated. The
residue
was crystallized from IPA to afford the titled compound as a white solid
(100mg /
75%).
1H NMR (300MHz, DMSO-d6) ~S =10.3 (s, 1H), 10.3-10.0 (br, 1H), 7.47 (d, J=
8.3Hz,
2H), 7.38-7.26 (m, 3H), 6.75 (d, J = 8.3 Hz, 2H), 6.27(d, J = 4.2 Hz, 1H),
5.49 (s, 1H),
5.49-5.30 (m, 1H), 4.40 (s, 2H), 3.70-3.00 (m, 9H), 2.90 (t, J = 7.7 Hz, 2H),
2.60-
2.25(m, 4H), 1.90-1.70 (m, 2H) ppm
MS (ESI); (M+H)+ (429.17), (M-H)- (427.25)
IR (KBr) 3331, 3198, 2936, 2668, 1684, 1633, 1604, 1380, 1098, 1064, 1037,
815crri
i
m.p. 238.3 °C
Example 14
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
63
5-fluoro-6-f ~4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydrox piperidin-1-
yllacetyl)-
3,4-dihydroauinolin-2(1I~-one
To a cooling solution of 6-(bromoacetyl)-5-fluoro-3, 4-dihydro-2 (lI~-
quinolinone (912mg, 3.2 mmol) and triethylamine (2.3 ml, 16 mmol) in DMF (20
ml)
at 0°C was added a solution of 4-(6-fluoro-5-methoxypyridin-2-
yl)pipcridin-4-of in
DMF (12m1) drop wise and stirred for 1.5h at the same temperature. The
reaction
mixture was added ice water (100m1) and the resulting precipitate was
collected to
afford the titled compound as a yellow solid (794mg / 57%).
1H NMR (300 MHz, DMSO-d6) ~S 10.6 (s, 1H), 7.69 (t, ,1--8.3Hz, 1H), 7.62 (dd,
J--8.2,10.6Hz, 1H), 7.49 (d, J--8.3Hz, 1H), 6.78 (d, J--8.4Hz, 1H), 5.04 (s,
1H), 3.85
(s, 3H), 3.67 (d, J--2.lHz, 2H), 2.94 (t, J = 7.5 Hz, 2H,), 2.70-2.60 (m, 2H),
2.60-
2.46(m, 4H), 2.10-1.94 (m, 2H), 1.53-1.40 (m, 2H) ppm
MS (ESn; (M+H)+ (432.13), (M-H)- (430.20)
5-fluoro-6-f 2-~4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl)-3,4-dihydropuinolin-2(1Fn-one
The mixture of 5-fluoro-6-{ [4-(6-fluoro-5-methoxypyridin-2-yl)-4-
hydroxypiperidin-1-yl]acetyl}-3,4-dihydroquinolin-2(11-one (750mg, 1.73mmo1)
and sodium borohydride (69mg, 1.82mmo1) in ethanol (18m1) was stirred at room
temperature for 15h. The resulting white precipitate was collected to afford
the titled
compound (506mg / 68%).
1H NMR (300 MHz, DMSO-d6) ~ 10.2 (s, 1H), 7.62 (dd, J--8.4,10.6Hz, 1H), 7.49
(d,
J=7.9Hz, 1H), 7.26 (t, J=7.9Hz, 1H), 6.68 (d, J=8.3Hz, 1H), 5.08-4.98 (m, 2H),
3.85
(s, 3H), 2.87 (t, J = 7.3 Hz, 2H), 2.77-2.60 (br, 2H), 2.58-2.36 (m, 6H), 2.12-
1.96(m,
2H), 1.54-1.40 (m, 2H) ppm
5-fluoro-6-f 2-~4-(6-fluoro-5-methoxypyridin-2-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl)-3,4-dihydroauinolin-2(lI~-one hydrochloride
To a suspension of 5-fluoro-6-{2-[4-(6-fluoro-5-methoxypyridin-2-yl)-4-
hydroxypiperidin-1-yl]-1-hydroxyethyl}-3,4-dihydroquinolin-2(11-one (506mg,
1.17
mmol) in methanol (10 ml) was added 4N hydrogen chloride 4.0 M solution in
ethyl
acetate (0.292mL, 1.17 mmol) and the mixture was concentrated. The residue was
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
64
crystallized from methanol-ethylacetate-2-propanol to afford the titled
compound as a
white solid (360mg / 66%).
1H NMR (300 MHz, DMSO-d6) ~ 10.3 (s, 1H), 10.1-9.8 (br, 1H), 7.69 (dd,
J--8.3,10.5Hz, 1H), 7.54 (d, J--7.9Hz, 1H), 7.31 (t, J--7.9Hz, 1H), 6.75 (d, J-
-8.2Hz,
1H), 6.35-6.15 (br, 1H), 5.85-5.60 (br, 1H), 5.45-5.20 (br, 1H), 3.88 (s, 3H),
3.70
3.45 (br, 2H), 3.40-3.10 (br, 4H), 2.87 (t, J = 8.3 Hz, 2H), 2.55-2.25 (m,
4H), 1.90-
1.65 (m, 2H) ppm
MS (ES)]; (M+H)+ (434.14), (M-H)- (432.23)
IR (KBr) 3325, 3196, 3082, 2908, 2651, 2570, 1693, 1630, 1601, 1483, 1379,
1313,
1134, 1034, 993, 827 cm 1
m.p. 251.7 °C
Example 15
Ethyl 4-(3,4-dihydro-lII-isochromen-7-yl)-4-hydroxypiperidine-1-carboxylate
To a solution of 7-bromoisochroman (WO 9305772 A1) (5.3 g, 25 mmol) in
THF (35 ml) was added 1.5 M solution of n-butyllithium in hexane (17 ml, 26
mmol)
dropwise at -78 °C. The mixture was stirred at -78 °C for 1 h.
To this mixture
was added a solution of N carbethoxy-4-piperidone (4.3 g, 25 mmol) in THF (35
ml)
at -78 °C. The mixture was stirred at -78 °C for an additional 1
h and warmed to
room temperature. Water (30 ml) was carefully added to the mixture and the
organic
layer was separated. The aqueous layer was extracted with ethyl acetate (30 ml
x2).
The combined organic layers were washed with saturated aqueous sodium chloride
solution, dried over potassium carbonate, filtered and concentrated under
reduced
pressure to give 7.8 g of a white powder. The powder was washed with 2-
propanol to
give the titled compound as a white powder (5.7 g, 75%).
1H NMR (270 MHz, CDC13) ~ = 7.36-7.08 (m, 3H), 4.77 (s, 2H), 4.31-3.93 (m,
4H),
4.16 (q, J = 7.1 Hz, 2H), 3.44-3.20 (m, 2H), 2.93-2.80 (m, 2H), 2.09-1.91 (m,
2H),
1.81-1.67 (m, 2H), 1.28 (t, J = 7.1 Hz, 3H) ppm.
4-(3,4-Dihydro-1FI-isochromen-7-yl)piperidin-4-of
To a suspension of ethyl 4-(3,4-dihydro-1H isochromen-7-yl)-4-
hydroxypiperidine-1-carboxylate (5.7 g, 19 mmol) in ethanol (6.3 ml) was added
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
potassium hydroxide (5.3 g, 94 mmol). The mixture was stirred under reflux for
2 h.
The mixture was concentrated under reduced pressure. The residue was diluted
with
dichloromethane (20 ml) and the resulting suspension was washed with water (20
ml).
The organic layer was separated and the aqueous layer was extracted with
5 dichloromethane (20 ml x 2). The combined organic layers were washed with
brine,
dried over potassium carbonate, filtered and concentrated under reduced
pressure to
give the titled compound as a pale yellow color solid (3.9 g, 89%).
1H NMR (270 MHz, CDC13) b = 7.38-7.23 (m, 1H), 7.20-7.02 (m, 2H), 4.77 (s,
2H),
4.03-3.90 (m, 2H), 3.20-3.02 (m, 2H), 3.00-2.77 (m, 4H), 2.12-1.90 (m, 2H),
1.87-
10 1.58 (m, 2H) ppm.
6-~2-~4-(3,4-Dihydro-1H-isochromen-7-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl)-3,4-dihydroauinolin-2(1F~-one mesylate
To a stirred solution of 6-(Chloroacetyl)-3,4-dihydroquinolin-2(IF~-one (WO
15 9302052)(0.61 g, 2.7 mmol) in DMF (2.7 ml) were added 4-(3,4-dihydro-1H
isochromen-7-yl)piperidin-4-of (0.64 g, 2.7 mmol) and. triethylamine (0.55 g,
5.5
mmol) at room temperature under nitrogen and the mixture was stirred at room
temperature overnight. The reaction mixture was poured into water (40 ml) and
the
precipitate was collected by filtration then dissolved into methanol (40 ml).
The
20 filtrate was extracted with dichloromethane (40 ml x1). The organic layer
was
separated and combined with the methanol solution of the precipitation. The
combined organic solutions were dried over potassium carbonate, filtered and
concentrated under reduced pressure to give 1.4 g of brown color oil. To the
suspension of the crude oil in ethanol (14 ml) was added a suspension of
sodium
25 borohydride (0.10 mg, 2.7 mmol) in ethanol ( 14 ml) at room temperature.
The
mixture was stirred at room temperature overnight. The volatile materials were
removed under reduced pressure to give a residue, which was diluted with
dichloromethane (20 ml). The solution was washed with saturated aqueous
ammonium chloride solution, dried over potassium carbonate, filtered and
30 concentrated under reduced pressure. Purification by flash column
chromatography
(silica gel, eluent dichloromethane:methanol = 7:1) to give the titled
compound as a
pale yellow color powder (0.25 g, 22% for 2steps).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
66
1H NMR (270 MHz, DMSO-d6) 8 = 10.02 (br, 1H), 7.29-7.23 (m, 1H), 7.18-7.04 (m,
4H), 6.79 (d, J = 8.1 Hz, 1H), 4.83-4.58 (m, 5H), 3.68 (t, J = 5.7 Hz, 2H),
2.91-2.65
(m, 6H), 2.63-2.39 (m, 6H), 2.05-1.83 (m, 2H), 1.64-1.47 (m, 2H) ppm.
To a suspension of Example 15 (0.25 g, 0.59 mmol) in isopropanol (5.9 ml)
was added methanesulfonic acid (57 mg, 0.59 mmol) at room temperature. The
mixture was turned into a clear solution upon heating (60 °C). The
solution was
filtered and cooled to room temperature. A white color solid (0.23g,
76°Io) was
formed after a night.
1H NMR (300 MHz, DMSO-d6) 8 = 10.13 (br, 1H), 9.19 (br, 1H), 7.30-7.09 (m,
5H),
6.90-6.83 (m, 1H), 6.19 (br, 1H), 5.41 (br, 1H), 5.07-4.97 (m, 1H), 4.69 (s,
2H), 3.92-
3.84 (m, 2H), 3.70-3.55 (m, 2H), 3.54-3.16 (m, 4H), 2.94-2.84 (m, 2H), 2.80-
2.71 (m,
2H), 2.50-2.17 (m, 4H), 2.31 (s, 3H), 1.88-1.68 (m, 2H) ppm.
MS (ESI]; M+H~ = 423.06, M-H+ = 421.03
IR (KBr); 3244, 2847, 1688, 1151 cm i
m.p. 229 °C
Example 16
6-f 2-~4-(3,4-Dihydro-1H-isochromen-7-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl~-5-fluoro-3,4-dihydropuinolin-2(lFi~-one Hydrochloride
To a stirred solution of 4-(3,4-dihydro-1H isochromen-7-yl)piperidin-4-of
(0.29 g, 1.2 mmol) and triethylamine (0.25 g, 2.5 mmol) in DMF (6.0 ml) was
added a
solution of 6-(bromoacetyl)-5-fluoro-3, 4-dihydro-2 (lI~-quinolinone (0.35 g,
1.2
mmol) in DMF (6.0 ml) at 0 °C under nitrogen and the mixture was
stirred at 0 °C for
2 h. The reaction mixture was poured into water (100 ml) and the precipitate
was
collected by filtration. The solid was washed with water (10 ml x 2),
isopropanol (15
ml x 2) and dried to give 0.34 g of a crude product as a pale orange color
powder.
To a suspension of the crude product in ethanol (8 ml) was added sodium
borohydride
(0.030 mg, 0.79 mmol) at 0 °C and the mixture was stirred at room
temperature for 2
h. The reaction mixture was poured into water (50 ml) and the precipitate was
collected by filtration. The precipitation was washed with isopropanol (15 ml
x 2) and
dried to give the titled compound as a white powder (0.26 g, 49% for 2steps).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
67
1H NMR (300 MHz, DMSO-d6) 8 = 10.21 (br, 1H), 7.29-7.22 (m, 2H), 7.12 (s, 1H),
7.06 (d, J, = 8.2 Hz, 1H), 6.67 (d, J = 8.2 Hz, 1H), 5.02-4.98 (m, 1H), 4.97-
4.90 (m,
1H), 4.69 (s, 1H), 4.66 (s, 2H), 3.86 (t, J = 5.7 Hz, 2H), 2.91-2.83 (m, 2H),
2.77-2.66
(m, 4H), 2.60-2.40 (m, 6H), 1.97-1.80 (m, 2H), 1.57-1.48 (m, 2H) ppm.
To a suspension of Example 16 (0.26 mg, 0.59 mmol) in 2-propanol (6.0 ml)
was added hydrogen chloride 4.0 M solution in ethyl acetate (0.15 mL, 0.59
mmol) at
room temperature. 70 ml of methanol was added to the mixture to dissolve the
precipitate. The solution was filtered and concentrated to give a white powder
(0.23
g, 82%).
1H NMR (300 MHz, DMSO-d6) ~ = 10.34 (br, 1H), 10.09 (br, 1H), 7.40-7.21 (m,
2H),
7.18-7.10 (m, 2H), 6.79-6.72 (m, 1H), 6.27 (br, 1H), 5.48-5.31(m, 2H), 4.69
(s, 2H),
3.92-3.83 (m, 2H), 3.66-3.49 (m, 2H), 3.46-3.20 (m, 4H), 2.94-2.85 (m, 2H),
2.80-
2.72 (m, 2H), 2.52-2.30 (m, 4H), 1.83-1.70 (m, 2H) ppm.
MS (ESn; M+H+ = 441.03, M-H+ = 439.00
IR (KBr); 3300, 3178, 2939, 2671, 1661, 1383 cm 1
m.p. 264 °C
Example 17
6-{2-[4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yl]-1-hydroxyethyl}-
3,4-
dihydroquinolin-2(ll~-one hydrochloride
A. ethyl4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidine-1-carboxylate
To a stirred solution of 2-ethoxy-1,3-thiazole (J. Chem. Soc. Perkin Trczns2.,
1589 (1986))(1.02 g, 7.90 mmol) in tetrahydrofuran (15 ml) was added drop wise
n
butyllithium (1.55 M, 5.10 ml) at 0 °C under nitrogen and the mixture
was stirred
for 10 minutes at 0 °C. To the mixture, a solution of 1-carbethoxy-4-
piperidone
(1.13 g, 6.58 mmol) in tetrahydrofuran (3 ml) was added and the mixture was
stirred
for 2 hours at 0 °C and 2 hours at room temperature. The mixture was
treated with
H20 and extracted with ethyl acetate. The combined organic layer was dried and
evaporated. The residue was purified by chromatography on silica gel, eluting
with
methyl alcohol / dichloromethane (1:20 v/v), to afford the titled compound as
a
yellow oil (1.36 g, 69 %).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
68
1H NMR (270 MHz, CDC13) b = 6.95 (s, 1H), 4.42 (q, J = 7.08 Hz, 2H), 4.14 (q,
J =
7.08 Hz, 2H), 4.05-3.84 (m, 2H), 3.45-3.25 (m, 2H), 2.10- 1.75 (m, 5H), 1.42
(t, J =
7.08 Hz, 3H), 1.27 (t, J = 7.08 Hz, 3H) ppm.
B. 4-(2-ethoxy-1,3-thiazol-5-yl)piperidin-4-of
A mixture of ethyl 4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidine-1-
carboxylate (1.36 g, 4.53 mmol) and potassium hydroxide (1.27 g, 22.7 mmol) in
ethyl alcohol (3 ml) was heated under reflux for 7 hours. The mixture was
evaporated in vacuo, and the residue was treated with dichloromethane (200 ml)
and
filtered. The filtrate was concentrated in vacuo to afford the titled compound
as a
yellow solid (0.81 g, 78 %).
1H NMR (270 MHz, CDC13) ~ = 6.96 (s, 1H), 4.42 (q, J = 7.08 Hz, 2H), 3.12-2.97
(m,
2H), 2.95-2.83 (m, 2H), 2.06- 1.80 (m, 4H), 1.42 (t, J = 7.08 Hz, 3H) ppm.
C. 6-df4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yllacetyl~-3,4-
dihydroguinolin-2(1F~-one
A mixture of 6-(chloroacetyl)-3,4-dihydroquinolin-2(lI~-one (J. Med. Chem.,
35,
620 (1992)), 4-(2-ethoxy-1,3-thiazol-5-yl)piperidin-4-of (0.81 g, 3.55 mmol)
and
triethylamine (1.0 ml, 7.11 mmol) in dimethylformamide (3.3 ml) was stirred
for 1
day at room temperature. The mixture was treated with HZO and the precipitate
was
colleted by filtration to afford the titled compound as a yellow solid (0.49
g, 50 %).
1H NMR (300 MHz, CDC13) S = 10.42 (s, 1H), 7.89-7.78 (m, 2H), 6.97 (s, 1H),
6.92
(d, J = 8.80 Hz, 1 H), 4.34 (q, J = 7.15 Hz, 2H), 3.74 (s, 2H), 3.39-2.42 (m,
7H), 3.00-
2.86 (m, 2H), 1.96-1.68 (m, 4H), 1.33 (t, J = 7.15 Hz, 3H) ppm.
MS (ESn; M+H+ = 416.10, M-H- = 414.18
D. 6-~2-~4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl~-3,4-dihydroguinolin-2(1f~-one
To a stirred solution of sodium borohydride (0.074 g, 1.95 mmol) in ethanol
(8.5
ml) was added suspension of 6-{ [4-(2-ethoxy-1,3-thiazol-5-yl)-4-
hydroxypiperidin-1-
yl]acetyl}-3,4-dihydroquinolin-2(1F~-one (0.41 g, 0.98 mmol) in ethanol (8.5
ml) at
0 °C and the mixture was stirred at room temperature overnight. The
precipitate was
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
69
collected by filtration to afford the title compound as a white solid (0.26 g,
64 %).
1H NMR (300 MHz, DMSO-d6) b = 10.02 (s, 1H), 7.13 (s, 1H), 7.17-7.04 (m, 1H),
6.95 (s, 1H), 6.78 (d, J= 8.07 Hz, 1 H), 5.34 (brs, 1H), 4.81 (brs, 1H), 4.66-
4.54 (m,
1H), 4.35 (q, J= 6.97 Hz, 2H), 2.91-2.79 (m, 2H), 2.71-2.29 (m, 8H), 1.97-1.81
(m,
4H), 1.33 (t, J = 6.97 Hz, 3H) ppm.
MS (ESA; M+H+ = 418.12, M-H- = 416.21
E. 6-f 2-~4-(2-ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yll-1-
hydroxyethyl)-3,4-dihydroe~uinolin-2(lIn-one hydrochloride
Hydrogen chloride (0.17 ml, 0.69 mmol), 4.0 M solution in ethyl acetate, was
added to a suspension of 6-{2-[4-(2-ethoxy-1,3-thiazol-5-yl)-4-
hydroxypiperidin-1-
yl]-1-hydroxyethyl}-3,4-dihydroquinolin-2(lI~-one (0.26 g, 0.63 mmol) in
methyl
alcohol (3 ml). The mixture was stirred for 5 hours at room temperature and
filtered.
The filtrate was evaporated and the residue was recrystallized from 2-propanol
to
afford the titled compound as a wr~ti~ 6o~,t8 (0.22 y, 77 %).
1H NMP (300 MHO, OM~O-86) S = 10.12 (s, 1H), 10.20-9.67 (m, 1H), 7.23 (s, 1H),
7.28-7.13 (m, 1H), 7.03 (s, 1H), 6.85 (d, J = 8.07 Hz, 1H), 6.16 (brs, 1H),
6.01 (brs,
1H), 5.14-4.96 (m, 1H), 4.38 (q, J = 6.97 Hz, 2H), 3.70-2.19 (m, 10H), 2.95-
2.81 (m,
2H), 2.10-1.88 (m, 2H), 1.34 (t, J = 6.97 Hz, 3H) ppm.
MS (ES)]; M+H+ = 418.11, M-H- = 416.20
IR (I~Br) 3275, 2739, 1655, 1601, 1545, 1514, 1472, 1379, 1286, 1248, 1169,
1061,
1026, 988, 955, 866, 826, 789 cm 1.
Example 18
6-f 1-Hydoxy-2f4-methyl-4-(3-methylisothiazole-5-yl)-1-yllethyl~-3,4-
dihydroauinolin-2(1H)-one
A. ethyl4-hydroxy-4-(3-methylisothiazol-5-yl)piueridine-1-carboxylate
The title compound is prepared from 3-methylisothiazole (J. Chem. Soc., 2032
(1963)) (1.9g) instead of 2-ethoxy thiazole according to the method described
in
Example 17 as oil (0.57g).
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
1H NMR (270 MHz, CDC13) 8 = 6.84 (s, 1H), 4.15 (q, J = 7 Hz, 2H), 4.03 (br,
2H),
3.36-3.20 (m, 2H), 2.90 (s, 1H), 2.45 (s, 3H), 1.94 (br, 4H), 1.27 (t, J =
7Hz, 3H) ppm.
B. 4-(3-methylisothiazol-5-yl)niperidine-4-of
5 The title compound is prepared from ethyl 4-hydroxy-4-(3-methylisothiazol-5-
yl)piperidine-1-carboxylate (0.63g) to the method described in Example 17 as a
solid
(0.36g).
1H NMR (270 MHz, CDC13) & = 6.86 (s, 1H), 3.12-2.91 (m, 4H), 2.47 (s, 3H),
2.07-
10 1.80 (m, 5H) ppm.
C. 6-~ f 4-hydroxy-4-(3-methylisothiazol-5-yl)piperidin-1-yllacetyl~-3,4-
dihydroauinolin-Z(lI~-one
The title compound is prepared from 4-(3-methylisothiazol-5-yl)piperidine-4-of
15 (0.36 g) instead of 4-(2-ethoxy-1,3-thiazol-5-yl)piperidin-4-of according
to the
method described in Example 17 as a solid (0.57g).
1H NMR (300 MHz, DMSO-d6) 8 = 10.43 (s, 1H), 7.87-7.83 (rn, 2H), 7.04 (s, 1H),
6.93 (d, J= 9 Hz, 1H), 5.73 (br, 1H), 3.80 (br, 2H), 2.95 (dd, J-- 8, 7 Hz,
2H), 2.8-2.6
20 (br, 2H), 2.6-2.45 (m, 4H), 2.36 (s, 3H), 2.00-1.85 (m, 2H), 1.82-1.72 (br,
2H) ppm.
D. 6-f 1-hydroxy-2-f 4-hydroxy-4-(3-methylisothiazol-5-yl)Piperidin-1-
yllethyl)-
3,4-dihydroauinolin-2(lI~-one
The title compound is prepared from 6-{ [4-hydroxy-4-(3-methylisothiazol-5-
25 yl)piperidin-1-yl]acetyl}-3,4-dihydroquinolin-2(11-one (0.57g) instead of
26-{[4-(2-
ethoxy-1,3-thiazol-5-yl)-4-hydroxypiperidin-1-yl] acetyl }-3,4-dihydroquinolin-
2(1F~-
one according to the method described in Example 17 as an amorphous. (0.35g).
1H NMR (300 MHz, DMSO-d6) 8 = 10.01 (s, 1H), 7.13-7.16 (m, 2H), 7.00-6.99 (m,
1H), 6.77 (d, J= 8 Hz, 1H), 5.65 (s, 1H), 4.82 (d, J-- 3Hz, 1H), 4.65-4.56 (m,
1H),
30 2.87-2.79 (m, 2H), 2.74-2.65 (m, 2H), 2.53-2.33 (m, 6H), 2.34 (s, 3H), 1.97-
1.83 (m,
2H), 1.79-1.70 (m, 2H) ppm.
CA 02541162 2006-04-03
WO 2005/035523 PCT/IB2004/003125
71
E. 6-dl-hydroxy-2-~4-hydroxy-4-(3-methylisothiazol-5-yl)piperidin-1-yllethyl~-
3,4-dihydroguinolin-2(1F~-one hydrochloride
Hydrogen chloride 4.0 M solution in ethyl acetate (0.23 mL) was added to a
solution of 6-{ 1-hydroxy-2-[4-hydroxy-4-(3-methylisothiazol-5-yl)piperidin-1-
yl]ethyl}-3,4-dihydroquinolin-2(lI~-one (0.35 g) in methyl alcohol (3 ml)-
tetrahydrofuran (6m1). The mixture was stirred for 0.5 hours at room
temperature
and concentrated in vacuo. The precipitate was washed with isopropyl alcohol,
the
titled compound as white amorphous. (0.21g).
1H NMR (300 MHz, DMSO-d6) b = 10.08 (s, 1H), 7.20-7.12 (m, 2H), 7.01 (br, 1H),
6.82 (d, J = 8 Hz, 1H), 4.93 (br, 1H), 3.35-3.27 (2H), 2.88-2.83 (m, 2H), 2.50-
2.35 (m,
10H), 2.37 (s, 3H), 2.00-1.85 (br, 2H) ppm.
Anal.Calcd.for.C~oH26N303SC1:0.3H20: C, 56.87; H, 6.25; N, 9.77. Found: C,
55.91;
H, 6.27; N, 9.83.
MS (ESA; M+H+ = 387.96, M-H- = 385.91