Note: Descriptions are shown in the official language in which they were submitted.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
HYDRONOPOL DERIVATIVES AS AGONISTS ON HUMAN ORL1 RECEPTORS
The present invention relates to a group of hydronopol derivatives which are
agonists on human ORL1 (nociceptin) receptors. The invention also relates to
the
preparation of these compounds, to pharmaceutical compositions containing a
pharmacologically active amount of at least one of these novel hydronopol
derivatives as an active ingredient, as well as to the use of these
pharmaceutical
compositions for the treatment of disorders in which ORL1 re ceptors are
involved .
The 'Opioid Receptor-Like 1' (ORL1 ) receptor was identified from a human cDNA
library. It was established that this "orphan receptor' has a close homology
with p.-,
x- and 8-opioid receptors (Mollereau et al., FEBS Lett., 341, 33-38, 1994;
Bunzow et
al., FEBS Lett., 347, 284-288, 1994). Despite its close sequential and
structural
resemblance with opioid receptors, classical opioid receptor ligands do not
interact
with ORL1 receptors. In 1995 a 17-amino acid neuropeptide was purified from
brain
extracts, and subsequently shown to be the natural ligand of the G protein -
coupled
ORL1 receptor (Reinscheid et al., Science, 270, 792-794, 1995; Meunier et al.,
Nature, 377, 532-535, 1995). This peptide was named orphanin FQ or nociceptin
and it does not bind to the three traditional opioid receptors. These findings
triggered substantial research into the functional role of, and novel ligands
for, the
ORL1 receptor. That resulted in several hundreds of publications, including
several
reviews (see e.g. Grond et al., Anaesthesist, 51, 996-1005, 2002), and dozens
of
patent applications, describing both peptide and non-peptide ligands, varying
in
potency and selectivity (ORL-1 versus p-opiate). As p-opiate receptors are
widely
distributed throughout the body, a lack of selectivity might lead to a range
of
undesired opiate-Pike side-effects e.g. sedation, respiratory depression,
constipation,
tolerance and dependence ( Druc~News Persaect, 14, 335, 2001 ).
1,3,8-triazaspiro[4,5]decan-4-one derivatives are described in JP-A-20001
169476, published on June 20, 2000; in WO 01!07050 A1, published on February
1,
2001 and in US 2003/0109539 A1, published on June 12, 2003. However, in none
of the applications cited above u-opiate receptors are mentioned. In EP 0 997
464
A1, published on May 3, 2000, 1,3,8-triazaspiro[4,5]decanone compounds as ORL-
1 receptor agonists are said to possess selective affinity for ORL1-receptors,
but
actual information on N-opiate receptor affinity was limited to the statement
that:
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
2
"particularly preferred compounds demonstrated higher affinity for ORL -1
receptors
than for mu-receptors (i.e, IC~ for ORL1-receptors/IC~ for mu-receptors were
less
than 1.0". More specific is US 2001/0041711, published on November 15, 2001.
This patent application describes triazospiro compounds having nociceptin
receptor
affinity. The compounds were also tested on p-, ~c-, and S-opiate receptors
but with
a few exceptions only, found to be more potent on p-opiate receptors than on
ORL-
1 receptors. The exceptions were ORL-1 selective by less than a factor 2. Thus
the
closest prior ark does not teach how to design potent ORL -1 ligands with an
unambiguous selectivity over p-opiate receptors, that is a selectivity of at
least a
factor 10, let alone such compounds which also have a good bioavailability.
Finally,
hydroxy alkyl substituted 1,3,8-triazaspiroj4,5]decan-4-one derivatives useful
for the
treatment of ORL-1 receptor mediated disorders were published on March 18,
2004
in WO 2004/022558, filed on September 5, 2003.
Surprisingly, it has now been found that in a series of hydronopol
derivatives, a
group of compounds was shown to have a very high affinity for human ORL1
receptors. Moreover, these compounds show a good selectivity for ORL1
receptors
relative to p-opiate receptors, and are readily available after oral
administration .
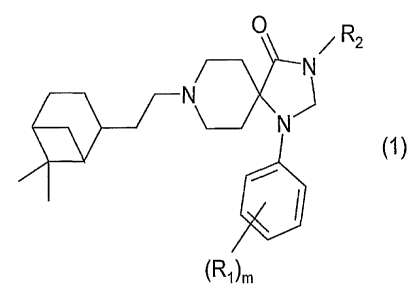
The invention relates to compounds of the general formula (1)
0 R
~ z
N
J
N
(1 )
I
~ROm
wherein:
R~ represents hydrogen, halogen, CF3, alkyl(1-6C), cycloalkyl(3-6C), phenyl,
amino, alkyl(1-3C)amino, dialkyl(1-3C)amino, hydroxy, hydroxyalkyl(1-3C),
(1-3C)alkoxy, OCF3, carboxyl, aminocarbonyl or (1-3C)alkylsulphonyl,
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
3
m is an integer from 1 - 4, with the proviso that when m is 2, 3 or 4, the R~
substituents may be either the same or different,
Rz represents hydrogen, optionally substituted alkyl(1-6C), cycloalkyl(3-6C),
-CHaOH, -CHZOCH3, carboxyl, acetyl , optionally substituted benzyl or a
group Q of the following structure (2):
R3
I
..N
Q - ~~ ~n 'R4 ~2~
wherein:
[]~ symbolizes -(CH2)~ wherein n is an integer from 0 -7,
R3 represents hydrogen or alkyl(1-3C),
R4 represents hydrogen , optionally substituted alkyl(1-6C), a saturated,
w~" unsaturated or partially saturated mono-, di- or tricyclic optionally
substituted
ring, or an alkyl(1-3C) group substituted with a saturated, unsaturated or
partially saturated optionally substituted five or six-membered ring which
optionally contains one or more heteroatoms, or
(R3 + R4) together with the nitrogen atom to which they are bonded,
represent a saturated, unsaturated or partially saturated mono-, di-or
tricyclic optionally substituted ring,
and pharmacologically acceptable salts and prodrugs thereof.
In the description of the substituents the abbreviation 'Ci.~-alkyl' means
'methyl,
ethyl, n-propyl or isopropyl'. 'Optionally substituted' means that a group may
or
may not be further substituted by one or more groups se lected from alkyl,
alkenyl,
alkynyl, aryl, fluoro, chloro, bromo, hydroxyl, alkyloxy, alkenyloxy, aryloxy,
acyloxy,
amino, alkylamino, dialkylamino, arylamino, thio, alkylthio, arylthio, cyano,
oxo, nitro,
acyl, amido, alkylamido, dialkylamido, carboxyl, o r two optional substituents
may
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
4
together with the carbon atoms to which they are attached form a 5 - or 6-
membered
aromatic or non-aromatic ring containing 0, 1 or 2 heteroatoms selected from
nitrogen, oxygen or sulphur. Within the context of the explanation of
'optionally
substituted', 'alkyl' means C,~-alkyl, 'alkenyf means C~.~-alkenyl, 'alkynyf
means
C~_3-alkynyl, 'acyl' means C~_3-acyl and 'aryl' means furyl, thienyl,
pyrrolyl, oxazolyl,
thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, pyr idyl,
pyridazinyl,
pyrimidinyl, pyrazinyl, 1;3,5-triazynyl, phenyl, indazolyl, indolyl,
indolizinyl, isoindolyl,
benzi[b]furanyl, benzo[b]thiophenyl, benz-imidazolyl, benzthiazolyl, purinyl,
quinolynyl, isochinolyl, chinolyl, phtalazinyl, quinazolinyl, qui noxalinyl,
1,8-
naphthyridinyl, pteridinyl, naphthyl or azulenyl, preferably phenyl, pyridyl
or
naphthyl. Optional substituents may themselves bear additional optional
substituents. Preferred optional substituents include C,_3 alkyl such as for
example
methyl, ethyl, and trifluoromethyl, fluoro, chloro, bromo, hydroxyl, C,~
alkyloxy such
as for example methoxy, ethoxy and trifluoromethoxy, and amino. 'Heteroatom'
means an atom such as N, 0 or S. ' Five- or six-membered rings' are for
example:
furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, isoxazole,
isothiazole, 1,2,3-oxadiazole, 1,2,3-triazole, 1,3,4-thiadiazole, pyridine,
pyridazine,
pyrimidine or pyrazine rings.
To the invention belong all compounds having formula (1)',~,racemates,
mixtures of
diastereomers and the individual stereoisomers. Thus compounds in which the
substituents on potentially asymmetrical carbon atoms are in either the R -
configuration or the S-configuration belong to the invention.
Prodrugs are therapeutic agents which are inactive per se but are transformed
into
one or more active metabolites. Prodrugs are bioreversible derivatives of drug
molecules used to overcome some barriers to the utility of the parent drug
molecule.
These barriers include, but are not limited to, solubility, permeability,
stability,
presystemic metabolism and targeting limitations (Medicinal Chemistry:
Principles
and Practice, 1994, ISBN 0-85186-494-5, Ed.: F. D. King, p. 215; J. Stella,
"Prodrugs as therapeutics", Exaert Opin. Ther. Patents, 14(3), 277-280, 2004;
P.
Ettmayer et al., "Lessons learned from marketed and investigational prodrugs
",
J.Med.Chem., 47, 2393-2404, 2004). Pro-drugs, i.e. compounds which when
administered to humans by any known route, are metabolised to compounds having
formula (1), belong to the invention. In particular this relates to compounds
with
primary or secondary amino or hydroxy groups. Such compounds can be reacted
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
with organic acids to yield compounds having formula (1 ) wherein an
additional
group is present which is easily removed after administration, for instance,
but not
limited to amidine, enamine, a Mannich base, a hydroxyl-methylene derivative,
an
0-(acyloxymethylene carbamate) derivative, carbamate, ester, amide or
enaminone.
5
The invention particularly re lates to compounds having formula (1 ) wherein
R~ represents hydrogen, halogen, CF3, alkyl(1-6C), hydroxy, (1-3C)alkoxy or
OCF3,
m =1, and all other symbols have the meanings as given above.
More particular, the invention relates to compounds having fo rmula (1 )
wherein:
R~ represents hydrogen, halogen, CF3, alkyl(1-6C), hydroxy, (1-3C)alkoxy or
OCF3,
m = 1, R~ represents a group Q having general formula (2), and all other
symbols
have the meanings as given above.
Even more particular, the invention relates to compounds having formula (1)
wherein: R~ represents hydrogen, halogen, CF3, alkyl(1-6C), hydroxy, (1-
3C)alkoxy
or OCF3, m = 1, R2 represents a group Q having general formula (2), R3
represents
a methyl group, R4 represents an alkyl(1-3C) group substituted with a
saturated,
optionally substituted six-membered ring which optionally contains one or more
heteroatoms, and [ ]~ has the meanings as given above.
The most preferred compounds of the invention are those having formula (1 )
wherein: R~ represents hydrogen, halogen, CF3, alkyl(1-6C), hydroxy, (1-
3C)alkoxy
or OCF3, m = 1, Rz represents a group Q having general formula (2), R3
represents
a methyl group, R4 represents a methylene group substituted with an optionally
substituted piperidine ring, and [ ]~ has the meanings as given above.
Pharmaceutically acceptable salts may be obtained using standard procedures
well
known in the art, for example by mixing a compound of the present invention
with a
suitable acid, for instance an inorganic acid such as hydrochloric acid, or
with an
organic acid.
The compounds of the invention of the general formula (1 ), as well as the
salts
thereof, have ORL1 agonistic activity. They are useful in the treatment of
disorders
in which ORL1 receptors are involved, or which can be treated via manipulation
of
those receptors especially, but not limited to, acute and chronic pain
conditions ,
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
6
metabolic disorders like anorexia nervosa and bulimia nervosa, obesity ;
gastro-
intestinal disorders in particular irritable bowel syndrome, inflammatory
bowel
disease (Crohn's disease and ulcerative colitis ), diarrhoea, constipation,
visceral
pain, urinary tract inflammation, renal disorders characterized by imbalances
of
water retention/excretion or salt excretion; cardiovascular disorders such as
myocardial infarction, arrhythmias, hypertension, thrombosis, anaemia,
arteriosclerosis, angina pectoris, opthalmological disorders like glaucoma,
respiratory disorders including chronic obstructive pulmonary disease,
bronchitis
and cystic fibrosis; diseases of the immune system, and viral infections.
The in vitro and in vivo ORL1 receptor agonistic properties of the compounds
of the
invention were determined using the method s outlined below.
Affinity for human ORL1 receptors
Affinity of the compounds for human ORL1 receptors was determined using the in
vitro receptor binding assay described by Ardati et al., Mol. Pharmacol., 51,
816,
1997. Briefly, membrane preparations were obtained from CHO (Chinese Hamster
Ovary)-cells in which the human ORL1 receptor was stably expressed. Membranes
were incubated with [3H]-nociceptin in the absence or presence of test-
compounds
in different concentrations, diluted in a suitable buffer. Non specific
binding was
defined as binding remaining in the presence of 10~ M nociceptin. Separation
of
bound radioactivity from free was done by filtration through Packard GFIB
glass
fiber filters with several washings with ice-cold buffer using a Packard cell
harvester.
Bound radioactivity was measured with a scintillation counter (Topcount,
Packard)
using a liquid scintillation cocktail (Microscint 0, Packard). Measured
radioactivity
was plotted against the concentration of the displacing test compound and
displacement curves were calculated by four-parameter logistic regression,
resulting
in ICSO values, i.e. that concentration of displacing compound by which 50% of
the
radioligand is displaced. Affinity pKi values were calculated by correcting
the ICS
values for radioligand concentration and its affinity for the hums n ORL1
receptor
according to the Cheng-Prusoff equation:
pK,= -log (ICS / (1+ S/Kd) )
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
7
in which the ICso is as described above, S is the concentration [3H]-
nociceptin used
in the assay expressed in molll (typically 0.2 nM), and K d is the equilibrium
dissociation constant of [3H]-nociceptin for human ORL1 receptors (0.4 nM).
The compounds of the invention have a high affinity for ORL1 receptors in the
binding assay described above. This property makes them useful in the
treatment of
disorders in which ORL1 receptors are involved, or that can be treated via
manipulation of these receptors.
Affinity for p.-opiate receptors
Affinity of the compounds for p,-opiate receptors was determined using the in
vitro
receptor binding assay described by Wang et al., FEBS Letters, 338, 217, 1994.
Briefly, membrane preparations were obtained from CHO-cells in which the human
p,-opiate receptor was stably expressed, and were incubated with the p.-opiate
specific ligand [3H]-DAMGO (D-Alai, N-Me-Phe4, glycinol5-Enkephalin) in the
absence or presence of test-compounds in different concentrations, diluted in
a
suitable buffer. Non specific binding was defined as binding remaining in the
presence of 10-6 M naloxone. Separation of bound radioactivity from free was
done
as described above, and the affinity of the compounds was calculated in a
similar
way.
The compounds of the invention have a low affinity for tC-opiate receptors in
the
binding assay described above. Thus they are unlikely to evoke the unwanted
side
effects known to occur with opiates like morphine.
In vitro ORL1 receptor agonism
Activation of the G protein-coupled ORL1 receptor inhibits adenylate cyclase
activity
and reduces the intracellular concentration of the second mess enger cAMP.
Using
an assay as described by Jenck et al., Proc. Natl. Acad. Sci USA, 97, 4938-
4943,
2000, the activity of the compounds on ORL1 receptors was measured. They were
demonstrated to be potent agonists with pEC ~-values matching their pK;
values.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
8
Castor oil induced diarrhoea in conscious mice
The compounds of the invention were shown to be able to reduce the cast or oil
induced diarrhea in mice, as was the peptide nociceptin after subcutaneous
administration. Since a peripherally administered peptide does not penetrate
the
blood brain barrier, it is indicated that the ORL1 mediated reduction in
diarrhea is
peripherally mediated.
Animals used: male NMRI mice were used for this model in the model of castor
oil -
induced diarrhoea. In all experiments, a group consisted of 10 to 12 animals .
Experimental procedures: on the day of the experiment, the mice received
either
compound or vehicle (bi-weekly intervals). Castor oil (8 ml/kg body weight)
was
administered orally 30 min later and the animals were placed individually in
cages
with free access to water. The feces were collected after a period of 5 h.
During this
time the quality of the feces was determined every 20 min by visual
inspection. This
diarrhoea score ranged from 0 = no output, 1 = normal output, 2 = slight
diarrh oea,
3 = moderate diarrhoea to 4 = severe diarrhoea. Thus, this score reflects the
onset
and intensity of diarrhoea. In these experiments the mean diarrhoea score and
the
dry weight of the feces were determined.
Data anal,~sis: the effects of the compounds are given as relative numbers
(percent
of control values). The original data registered in the experiments were
compared to
controls (without compound) in the same animals by paired two sided t -tests
or to a
control group by a non paired t-test. Values of p<0.05 were taken as
statistically
significant.
Colon transit in conscious rats
The compounds of the invention were shown to not influence the normal colon
transit in rats. This was also the case for the peptide Nociceptin after
subcutaneous
administration. Since a peripherally administered peptide does not penetrate
the
blood brain barrier, it is indicated that peripheral ORL1 receptor activation
does not
impair the normal gastrointestinal transit. In contrast peripheral p-opiate
receptor
activation is able to strongly impair the transit in this model. Thus, this
test shows
the selectivity of the compounds of the invention for the ORL1 receptor.
Animals used: for the experiments male Sprague Dawley rats were used. In all
experiments, a group consisted of 10 to 12 animals.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
9
Experimental procedures: prior to the experiments the rats were equipped with
a
chronic titanium cecal fistula under general anesthesia. The animals were
allowed to
recover from surgery and were trained to a feeding regimen of free access to
the
chow during 3 h per day. On the day of the experiments after the feeding
period, a
marker substance (2 ml of a suspension containing 80 % barium sulfate) was
injected via the fistula into the caecum and the animals received either
compound or
vehicle. Subsequently they were placed in metabolic cages and fecal pellets
were
collected at hourly intervals for a 21 h period using an automated collection
system.
During this time the animals had free access to water. The ba rium sulfate
content in
the feces was analyzed radiographically and the feces were weighed. The
function
of the marker content in feces versus time and amount of feces enabled the
analysis of the mean retention time of barium sulfate, i.e. the colon transi t
time. The
mean retention time of the BaS04 containing pellets and the total fecal output
were
determined.
Data analysis: the effects of the compounds are given as relative numbers
(percent
of control values). The original data registered in the experimen is were
compared to
controls (without compound) in the same animals by paired two sided t -tests.
Values
of p<0.05 were taken as statistically significant. In the colon transit model,
the
control data represent the mean of two control experiments (before an d after
compound, weekly intervals).
Acetic acid induced visceral hypersensitivity in conscious rats
The compounds of the invention were shown to be able to reduce the visceral
hypersensitivity in rats, as was the peptide Nociceptin after subcutaneous
administration. Since a peripherally administered peptide does not penetrate
the
blood brain barrier, it is indicated that the ORL1 mediated reduction in
visceral
hypersensitivity is peripherally mediated.
Animals used: adult female Sprague Dawley rats, body weight: in the range of
200 -
250 g. A group consists of 5 to 10 animals.
Experimental Procedure: animals were fasted for 24 hours prior to the
experiments
with free access to water. Acetic acid (0,6 °l°, 1.5 ml) was
injected into the colon (10
cm proximal to the anus). After 50 minutes a rubber balloon of 5 cm length (6-
7 ml
volume) was inserted rectally into the distal colon and secured by taping the
attached tubing to the rat's tail. Colorectal distension was performed by
setting the
balloon pressure to 100 mbar for 10 minutes. During this time the number of
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
abdominal constrictions were monitored by visual inspection. The experiments
were
continued only in animals which responded to the colorectal distension with
more
than 10 abdominal constrictions. These animals received a single dose of
substance or vehicle and colorectal distension protocol was repeated at 30,
60, 90
5 and 120 minutes after administration.
Data analysis: results are given as mean ~ SD. The number of abdominal
constrictions at 30, 60, 90 and 120 minutes after administration of substance
or
vehicle as well as the mean values (30-120 min) was compared to prevalues by
paired two sided t-tests. Relative numbers of abdominal constrictions
(°lo of
10 prevalues) at 30, 60, 90 and 120 minutes and the re lative mean values (30-
120 min)
were compared between substance and vehicle by unpaired two sided t-tests.
Values of p<0.05 were taken as statistically significant.
In vivo ORL1 receptor agonism: lack of CNS-penetration
Most of the compounds of the invention were shown to be devoid of activity in
the
Adult stress-induced ultrasonic vocalisation (AUV) procedure as described by
Van
der Poel et al., Psychopharmacoloay. 97, 147-148, 1989. This demonstrates that
the compounds do not penetrate the blood-brain-barrier. The peptide nociceptin
is
also active in this assay, but in order to demonstrate its effect, it needs to
be
administered directly into the brain (by intracerebroventricular injection).
EXAMPLES OF SYNTHESES OF INTERMEDIATES AND END PRODUCTS
(-)-traps-2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (5)
HO~ ~ Br~ ~ NC~
2 3
HO'~
4 5
Myrtanyl bromide (2)
Triphenyl phosphine (116 g, 0.44 mol) was dissolved in acetonitrile (1 I) and
cooled
in an ice bath under Nz atmosphere. Bromine (22.5 ml, 0.44 mol) was added drop
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
11
wise. The temperature of the exothermic reaction was maintained below
10°C. After
complete addition the ice bath was removed and (-) trans-myrtanol (1) (2,686
g,
0.44 mol) dissolved in acetonitrile (250 m I) was added slowly. After complete
addition the light yellow solution was refluxed for 3 h using a Dean-Stark
equipment.
During the reaction, the solvent in the water trap was removed 20 times (ca.
200 m I
of solvents in total). GC analysis revealed complete conversion of the
starting
material. The mixture was evaporated to dryness. The crude mixture was
purified
over silica column (eluent: dichloromethane/diethyl ether 111, v/v). This
resulted in
87.8 g of bromide 2 (91 %) as light yellow oil.
Myrtanyl cyanide (3)
Myrtanyl bromide (2) (87.8 g, 0.41 mol) was dissolved in di methylformamide (1
I ).
Sodium cyanide (40 g, 0.81 mol) was added and the mixture was stirred at
reflux for
5 h. GC analysis revealed complete conversion. The mixture was diluted with
water
(3 I) and extracted with fert.-butyl methyl ether (TBME, 3x 1.5 I). The
organic layer
was washed with brine, dried over Na2S04 and concentrated to dryness. The
crude
mixture was purified over silica column (eluent: heptane/dichloromethane, 1/1,
v/v)
to give 52.4 g (80°l°) of cyanide (3) as colorless liquid.
Ethyl ester (4)
Ethanol (500 ml) was cooled in an ice bath. Sulfuric acid (190 ml) was added
drop
wise. Cyanide (3) (52.4 g, 0.32 mol) dissolved in ethanol (100 ml) was added
and
the mixture was stirred at reflux overnight. GC analysis revealed complete
addition.
The mixture was cooled and water (1.5 I ) was added. The mixture was extracted
with TBME (3x 1.5 1). The organic layer was washed with NaHC03 (sat. 1 I),
dried
over NazS04 and concentrated. Yield: 54.2 g of ester 5 (80%) as near co
lorless
liquid. Crude (4) was used in the next reaction without purification.
(-)-trans-2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (5)
To a suspension of lithium alumin um hydride (20 g, 0,52 mol) in
tetrahydrofuran (1 I )
was added ester (4) (54.2 g, 0.26 mol) dissolved in tetrahydrofuran (500 ml).
After
complete addition the mixture was refluxed for 1 h. GC analysis revealed
complete
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
12
conversion of starting material. The mixture was cooled in an ice bath and HCI
(1M,
1 I) was added carefully. After complete addition, the mixture was diluted
with water
(1 I) and extracted with TBME (3x 1.5 I ). The organic layer was washed with
brine,
dried over Na2S0~ and concentrated to dryness. The crude mixture was purified
by
Kugelrohr distillation (b.p. 85°C, 3.10-a mbar). Yield: 35.9 g of
compound 1 (65%) as
colorless oil.
(+)-trans-2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (10)
HO ,. ~ MsO~,,.~~ ~ NC~,,,,o
6
0
9 10
Myrtanyl mesylate (7)
18.1 g (0.12 mol) of (+)-trans-myrtanol (6) was added to a solution of 18.5 m
I mesyl
chloride (2 eq., 0.24 mol, 27.5 g) and 49 m I pyridine (5 eq., 0.60 mol, 47.5
g) in 400
ml DCM. The reaction mixture was stirred overnight at room temperature. Water
was added and the reaction mixture was stirred for 1h. The organic layer was
extracted and the water layer was extracted two more times. The combined
organic
layers were washed (saturated NaHC03, water, brine), dried (Na2S04) and
evaporated in vacuo to give 25,9 g (91 %) of mesylate (7) as a colourless oil.
Myrtanyl cyanide (8)
Myrtanyl mesylate (7) (25,9 g, 0.11 mol) was dissolved in DMSO (250 m I).
Potassium cyanide (4 eq, 29.2 g, 0.45 mo I) was added and the mixture was
stirred
at 70°C for 2 days. GC analysis revealed complete conversion. The
mixture was
diluted with water (750 ml) and extracted with TBME (3x 300 ml). The organic
layer
was washed with brine, dried over Na2S04 and concentrated to dryness to give
17,7
g (quantitative yield) of cyanide (8) as a colourless oil.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
13
Ethyl ester (9)
Ethanol (200 ml) was cooled in an ice bath. Sulphuric acid (80 ml) was added
dropwise. Cyanide (8) (17.7 g, 0.11 mol) dissolved in ethanol (40 m I) was
added
and the mixture was stirred at reflux overnight. GC analysis revealed complete
addition. The mixture was cooled and water (1 I) was added. The mixture was
extracted with TBME (3x 500 ml). The organic layer was washed with NaHC03
(sat.,
500 ml), dried over Na2S04 and concentrated. Yield: 20.4 g of ester (9) (88%)
as a
yellow oil. Crude (9) was used in the next reaction without purification.
(+)-trans-Dihydronopol (10)
To a suspension of lithium aluminium hydride (7.4 g, 0.19 mol) in
tetrahydrofuran
(350 ml) was added ester (9) (20.1 g, 0.09 mol) dissolved in tetrahydrofuran
(200
ml). After complete addition the mixture was refluxed for 2 h. The mi xture
was
cooled in an ice bath and HCI (1 M, 1 I ) was added carefully. After complete
addition,
the mixture was diluted with water (300 m I) and extracted with TBME (3x 500
ml).
The organic layer was washed with brine, dried over Na 2S04 and concentrated
to
dryness. The crude mixture was purified by Kugelrohr distillation (b.p. 85
°C, 8.10-2
mbar). Yield: 9.2 g of compound (10) (61 %) as colourless oil.
(-)-cis-2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (11)
The synthesis of the cis analog with (-)-a-pinene as starting material is
described in
J. Amer. Chem. Soc. 68, 638, 1946 and US patents 2,427,343, 2,427,344
and 2.427.345(+)-cis-2-(6,6-dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (18)
H0~
12 13 ~ ~1 ~4
MsO~ ' NC~
15 16 17
H0~
18
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
14
(+)-~-pinene (13)
In dried glassware, potassium f butyl oxide (KOt-Bu, 49.4 g; 0.44 mol) was
added to
n-BuLi (176 ml; 2.5 M in hexane). The suspension was cooled to -78°C.
The (+)-a-
pinene (12) (50 g; 0.37 mol) was added drop wise. The reaction mixture was
allowed to warm to room temperature and was stirred for 45 h. The reaction
mixture
was cooled to -78°C and B(OMe)3 (137 ml; 1.20 mol) was added drop wise.
The
reaction mixture was allowed to warm to room temperature (exothermic!). 10%
HCI
(aqueous, 250 ml) was added drop wise and the reaction mixture was stirred for
1 h.
The layers were separated and the water layer was extracted with heptane (2 x
200
ml). The combined organic layers were dried over NazS04 and evaporated to
dryness to give 36.7 g of yellow oil. The raw product was purified usin g
Kugelrohr
distillation (8-12 mbar; 50-60°C) to give 36.6 g (0.27 mol, yield =
73%, 88% pure) of
(+)-~3-pinene (13) as a colourless oil.
Myrtanol (14)
(+)-~-pinene (13) (36.6 g; 0.27 m01) was dissolved in THF (100 m I) and cooled
down
to 0°C. BH3~DMS in THF (2 M; 47.3 mi) was added drop wise. The reaction
mixture
was stirred for 0.5 h. Ethanol (90 ml) was added. 1 M NaOH (aq) (95 ml) was
added.
The reaction mixture was cooled to 0°C. 33 ml 30% H~OZ was added
drop wise
while the temperature was not allowed to rise above 35°C. The reaction
mixture was
refluxed for 1 h and poured into water (1 I). The solution was extracted with
TBME.
The combined organic layers were washed with water and brine, dried over Na
~SOa
and evaporated to dryness. Remaining a pinene was distilled off using
Kugelrohr
distillation (8-12 mbar; 50-60°C), giving 38.6 g (0.25 mol, yield =
93%) of (+)-cis-
myrtanol (14) as a colourless oil.
Myrtanyl mesylate (15)
15.0 g (0.10 mol) of (+)-cis-myrtanol (14) was added to a solution of 15 m I
mesyl
chloride (2 eq., 0.20 mol) and 40 m I pyridine (5 eq., 0.50 mol) in 300 m I
DCM. The
reaction mixture was stirred overnight at room temperature. Water was added
and
the reaction mixture was stirred for 1 h. The organic layer was extracted and
the
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
water layer was extracted two more times. The combined organic layers were
washed (saturated NaHC03, water, brine), dried (Na2S04) and evaporated in
vacuo
to give 21.6 g (yield = 93 %) of mesylate (15) as a colourless oil.
5 Myri;anyi cyanide (16)
Myrtanyl mesylate (15) (21.6 g, 0.093 mol) was dissolved in DMSO (230 m I).
Potassium cyanide (4 eq, 24.2 g, 0.37 mol) was added and the mixture was
stirred
at 70°C for 8 days. GC analysis revealed complete conversion. The
mixture was
10 diluted with water and extracted with heptane. The organic Layer was washed
with
brine, dried over Na2S04 and concentrated to dryness to give 15.8 g (quant.)
of
cyanide (16) as a colourless oil.
Ethyl ester (17)
Ethanol (150 mL) was cooled in an ice bath. Sul phuric acid (60 ml) was added
drop
wise. Cyanide (16) (16 g) dissolved in ethanol (30 m I) was added and the
mixture
was stirred at reflux overnight. GC analysis revealed complete conversion. The
mixture was cooled and water (1 I ) was added. The mixture was extracted with
TBME (3x~~ 500 ml). The organic layer was washed with saturated NaHCO 3
(aqueous, 500 ml), dried over Na2S04 and evaporated to dryness. Yield: 20.6 g
of
ester (17) (quant.) as a yellow oil. Crude (17) was used in the next reaction
without
purification.
(+)-cis-Dihydronopol (18)
To a suspension of lithium aluminium hydride (8.3 g, 0.22 mol) in
tetrahydrofuran
(400 ml) was added ester (17) (23.6 g, 0.11 mol) dissolved in tetrahydrofuran
(200
ml). After complete addition the mixture was refluxed for 2 h. The mixture was
cooled in an ice bath and HCI (1 M, 1 I) was added carefully. After complete
addition, the mixture was diluted with water (300 m I) and extracted with TBME
(3x
500 ml). The organic layer was washed with brine, dried over Na~S04 and
concentrated to dryness, giving a yellow oil (13.4 g). The crude mixture was
purified
by Kugelrohr distillation (b.p. 85°C, 8x10-2 mbar). Yield: 8.7 g (51
mmol; y = 47%) of
compound (18) as colourless oil.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
16
1-Mesyl-2-(6,6-dimethyl-bicyclo[3.1.1]hept-2-yl)-ethanol (20) (for all stereo
isomers of dihydronopol)
HO Ms0
1g 20
To a suspension of 67g (0.4 mol) (-)-cis-2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-
yl)-
ethanoi (19) in 300 ml CHzCIz at 0°C was added 139 ml (1 mol) of
triethylamine. To
this mixture 55.2g (0.48 mol) of mesyl chloride in 100 ml dichloromethane was
added drop wise. After 5h at room temperature the reaction was completed and
300
ml 1 N aqueous HCI solution was added. After separation the aqueous layer was
washed with dichloromethane twice and the combined organi c layers were washed
with water, dried over magnesium sulphate and concentrated in vacuum yielding
91.6 g (0.37 mol 91%) of a crude orange oily product. This raw material was
used
for the next step without further purification.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-(3-methylamino-propyl)-1-
phenyl-1,3,8 triaza-spiro[4.5]decan-4-one (24)
0
NH Mso'
N
21 / \
~Br
CI
~Br
CI
CI~Br H O~NHZ
3
~~~Br
CI
variation of reagent gives comparable high yields
all shown reagents have been synthesized using this protocol
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
17
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-one (22) jexample nr. 7 in fables below]
The spiro compound (21 ) (310 g; 1.34 mol) and (di)hydroponol mesylate (20)
(371
g; 1.51 mol) were dissolved in methyl ethyl ketone (MEK, 15 t). Potassium
carbonate
(735 g; 5.33 mol) and sodium iodide (226 g; 1.51 mol) were added and the
mixture
was refluxed overnight. After cooling the reaction mixture the solvent was
evaporated. The residue was taken up in CH zCl2 (5 I) and shaken with water (4
I ).
The layers were separated, the organic layer dried on Na 2S04, and the solvent
evaporated. The remaining solid was washed with Et20 (3 I) and filtered off.
The
filtrate was evaporated and washed with Et20 (300 ml). The solid was filtered
oft.
(466.3 g; 1.22 mol; 91 %).
3-(3-Chloro-propyl)-8-[2-(6,6-dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-
phenyl-
1,3,8-triaza-spiro[4.5]decan-4~one (23)
THF (1500 ml) was cooled in an icelwater bath. The spiro compound (22) (150.8
g;
0.40 mol) and potassium tent-butoxide (49 g; 0.44 mol) were added and the
resulting
mixture was stirred at 0°C for 30 minutes. The mixture became clear. 1-
bromo-3-
chloropropane (43 ml; 0.44 mol) in THF (150 ml) was added drop wise to the
solution at 0°C. After complete addition the cooling was removed and
the solution
was stirred at 50°C for 4 hours. After cooling the mixture was poured
into sat urated
KHS04 (aq ueous, 1000 ml) and diluted with EtOAc (500 ml). The layers were
separated and the aqueous layer extracted with EtOAc (3x 750 m I). The
combined
organic layers were washed with water and brine (1x 500 m I each). After
drying on
Na2S04 the solvent was evaporated to yield a yellow oil (205.6 g; 0.45 mol;
quantitative yield).
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-(3-methylamino-propyl)-1-
phenyl-1,3,8-triaza-spiro[4.5]decan-4-one (24j (example nr. 73 in tables
below)
The crude spiro compound (23) (162.8 g; 0.36 mol) was dissolved in
methylamine/
EtOH solution (Fluka, 8M; 1154 m I; 9.23 mol). Sodium iodide (2.16 g; 0.014
mol)
was added and the solution was stirred at 70 °C under N2-atmosphere for
3 days.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
18
After cooling the reaction mixture was diluted with water and EtOAc (500 m I
each).
The aqueous layer was extracted with EtOAc (3x 800 m I). The organic layer was
washed with brine (500 m I). After drying on Na~S04 the solvent was evaporated
to
yield a yellow oil. This oil was purified by column chromatography (Si02;
CHZCh /
MeOH 90:10; containing 1 % 7N NH ~/MeOH) to yield 30 g of (24) with a purity
of
93% (according to HPLC/MS) and 85 g of (24) with a purity of 96% (115 g; 0.25
mol;
70%).
Variation of the substitution pattern of the phenyl ring in the spiro core 8-
[2-
{6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-one (22)
Ms0 NHZ
~N~H 20 N + I W
H0~
i
HO O R
26 27 a - f
TMSCN
R R
d
1 / N
N
N . ,.H\~ 28a-f
H 29a - f N ./~vJC
HzN 0
R
a = 3-F
' 30 b = 3-OMe
I 30 c = 3-CI
30 d = 3-CF3
30 a = 4-F
30 f = 4-OMe
(In fhe scheme above TMSCN = trimethylsilylcyanide)
1-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-piperidin-4-one (26):
A mixture of 61.1 g (0.40 mol) of piperidone hydrate hydrochloride (25), 112.8
g
20 (0.46 mol) of dihydronopol mesylate (20), 69.0 g (0.46 mol) of Nal, 273 g
(1.97 mol)
of KZC03 and 4.3 I of MEK was refluxed overnight. The mixture was cooled to
room
temperature and concentrated in vacuo. The residue was dissolved in
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
19
dichloromethane (1.5 I) and water (1.5 I) and the layers were separated. The
organic
layer was washed with water (1 I) and , dried over NazS04. The layer was
concentrated in vacuo to g ive 113 g of crude product, which was purified by
column
chromatography (Si02, heptane:EtOAc, 6:1 X1:1) to yield 77.7 g (0.31 mol, 78
%) of
compound (26) as an orange oil.
1-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-4-(3-fluoro-phenylamino)-
piperidine-4-carbonitrile (28a):
A solution of 20.0 g (80.2 mmol) of (26) and 8.4 ml (87 mmol) of 3-
fluoroaniline
(27a) in 65 ml of acetic acid was cooled with a cold water bath. 10.7 m I
(80.2 mmol)
of trimethylsilylcyanide were added dropwise over a period of 10 min.
maintaining
the temperature below 40°C. The mixture was stirred for 2 h at room
temperature
and poured into a mixture of aqueous ammonia (80 ml) and ice (80 g). The pH
was
adjusted to 10 with concentrated NH3. The mixture was extracted with
chloroform
(3x 200 ml). The combined organic layers were dried over Na ~S04 and
concentrated
in vacuo to give 40.0 g of crude product, which was purified by column
chromatography (SiOz, heptane:EtOAc, 1:1) to give 28.7 g (77.7 mmol, 97%) of
{28a). It is also possible to use crude product in the next step without
purification.
1-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-4-(3-fluoro-phenyfamino)-
piperidine-4-carboxylic acid amide (29a):
A mixture of 28.7 g (78 mmol) (28a), 135 ml of formic acid and 135 ml of
acetic
anhydride was stirred at room temperature for 1 day. The reaction was
monitored by
'H-NMR and MS. After completed reaction the reaction mixture was poured into
ice -
water (800 ml). The pH was adjusted to 10 by the addition of 33% NaOH (aq).
The
aqueous layer was extracted with DCM (3x 1 I). The combined organic layers
were
dried over Na2S04 and concentrated in vacuo. The residue was dissolved in 550
m I
tert.-butylalcohol, 45 ml water and 45 ml concentrated aqueous ammonia. The 90
ml
of 35 % hydrogen peroxide were added drop wise at room temperature. The
mixture
was stirred overnight. The reaction was monitored by TLC. 900 m I of water
were
added and the mixture was extracted with DCM (3x500 m I). The combined organic
layers were dried over NazS04 and concentrated in vacuo to give 29.3 g (76
mmol,
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
98 %) of (29a) as a yellow solid, which was used in the next step without
purification.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-(3-fluoro-phenyl)-1,3,8-
triaza-
5 spiro[4.5]decan-4-one (30a): [example nr. 9 in tables below]
A solution of 29.3 g (76 mmol) of (29a) in 400 ml formamide was heated for 2 h
at
200°G. The solution turned from yellow to black. The reaction was
monitored by 'H-
NMR. After completed reaction the mixture was cooled to room temperatu re and
10 poured into ice-water (800 g). The mixture was extracted with DCM (6x 1 I).
The
combined organic layers were dried over ~da2S04 and concentrated in vacuo. The
residue was dissolved in 1.2 I of methanol and 4.3 g (114 mmol) of
sodiumborohydride were added portion wise. The mixture was stirred for 1 h at
room temperature and another hour at 60°C. The reaction mixture was
cooled to
15 room temperature and quenched with 25 m I of water. The solvent was
evaporated
in vacuo. The residue was dissolved in 750 m I of ammonia and extracted with
DCM
(7x 1.5 I). The combined organic Payers were dried over NazS04 and
concentrated in
vacuo to give 24.8 g of crude product, which was purified by column
chromatography (SiOz, heptane:EtOAc, 1:1-~ 1:3). Trituration with Et20 of the
20 eluated product yielded 3.44 g (8.6 mmol, 11.3 °l° from
compound (26)) of
compound (30a) as a white solid.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]kept-2-yl)-ethyl]-1-(3-methoxy-phenyl)-1,3,8-
triaza-spiro[4.5]decan-4-one (30b): [example nr. 8 in tables below]
The sequence was repeated starting from 68.0 g (0.27 mol) of (26); Compound
(30b) was purified by column chromatography and trituration with diethylether
to
yield 13.9 g (34 mmol, 12 % yield from (26)) as an ofif-white solid.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-(3-chloro-phenyl)-1,3,8-
triaza-
spiro[4.5]decan-4-one (30c): [example nr. 7 in tables below]
The sequence was repeated starting from 67.4 g (0.27 mol) of (26); Compound
(30c) was purified by column chromatography and trituration with diethylether
to
yield 7.42 g (17.8 mmol, 6.6°/° yield from (26)) as an off'-
white solid.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
21
8-[2-(6,6-Dimethyl-bicyclo(3.1.1]hept-2-yl)-ethyl]-1-(3-trifluoromethyl-
phenyl)-
1,3,8-triaza-spiro[4.5]decan-4-one (30d): (example nr. 70 in tables below]
For compound (30d) the same sequence was performed, but instead of the desired
product, compound (29d) was isolated. Therefore the sequence was partially
repeated. The compound was formylated with formic acid and acetic anhydride,
heated in formamide and finally reduced with sodiumborohydride. The crude
product was purified by column chromatography (Si0 ~, EtOAc) and subsequently
trituration with diethylether to yield 7.39 g (6.6% overall yield from (26))
as a white
solid.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-(4-fluoro-phenyl)-1,3,8-
triaza-
spiro(4.5]decan-4-one (30e): [example nr. 5 in tables below]
The sequence was repeated starting from 45.0 g (0.18 mol) of (26); Compound
(30e) was purified by column chromatography and tri turation with diethylether
t o
yield 9.82 g (24.5 mmol, 13.6% yield from (26)) as a grey solid.
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-1-(4-methoxy-phenyl)-1,3,8-
triaza-spiro[4.5]decan-4-one (30f): [example nr. 6 in tables below]
The sequence was repeated starting from 45.0 g (0.18 mol) of (26); Compound
(30f) was purified by column chromatography and tri turation with diethylether
to
yield 8.94 g (21.7 mmol, 12.1% yield from (26)) as a white solid.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
22
,,o
,N
O 31
0
0+ I_
KOH,CH3CN
rf
O
O
EtOH, rf
conc. HCI
THF, rf
1-Oxa-6-aza-spiro[2.5]octane-6-carboxylic acid tert-butyl ester (32):
To a solution of 44.9 g (0.225 mol) of 4-oxo-piperidine-1-carboxylic acid tent-
butyl
ester (31) in 500 ml acetonitrile was added successively 59.5 g (0.27 mol)
trimethyl
sulphoxonium iodide and 18.9 g (0.338 mol) fine crushed potassium hydroxide.
The
reaction mixture was vigorously stirred for two days under a nitrogen
atmosphere at
room temperature. After complete conversion the solvent was evaporated in
vacuo
and the residue was taken up in dichloromethane. The organic layer was washed
with aqueous citric acid solution (6x), dried over sodium sulphate and
concentrated
in vacuo. This crude product was used without further purification for the
next step
(43.5 g, 0.204 mol, 90.6% yield).
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
23
4-([(3-(8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-4-oxo-1-phenyl-
1,3,8-
triaza-spiro[4.5]dec-3-yl} propyl)-methyl-amino]-methyl}-4-hydroxy-piperidine-
1-
carboxylic acid tert-butyl ester (33): [example nr. 35 in fables below]
2.6 g (5.74 mmol) of compound (24) was placed in a flask and diluted with 20
ml
ethanol. To this solution was added 1.88 g (8.81 mmol) of the epoxide (32) and
the
mixture was heated to reflux until TLC showed complete conversion. For work-up
the solvent was evaporated and the residue was taken up with ethyl acetate.
Afte r
washing with aqueous potassium carbonate solution drying over sodium sulphate
and concentration in vacuo the raw material was purified via flash column
chromatography yielding a light yellow viscous oil (3.51 g, 5.15 mmol, 89.8%
yield).
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]~-f 3-[(4-hydroxy-piperidin-
4-
ylmethylj-methyl-amino]-propyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
(34): [example nr. 37 in tables below]
The Boc-derivative (33) (2.79 g, 4.19 mmol) was dissolved in tetrahydrofuran a
(25
ml). To this solution was added 2 ml of concentrated aqueous HCI solution and
the
resulting mixture was stirred at room temperature over night. After TLC
analysis
indicated complete conversion fhe solvent was removed under reduced pressure
and the resulting residue was dissolved in ethyl acetate. Washing with
potassium
carbonate solution, drying of the organic layer with sodium sulphate and
concentration in vacuo provided the pure title compound as a Ii ght yellow
viscous oil
(2.37 g, 3.8 mmol, 90.7% yield).
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
24
0 0 0 off / \ o o / \
~\-N~ --, ~--N~ --
0 EtOH, rf ~ O
35 ~ 36
1
/H
37
conc. NCI
THF, rt
4-[(Benzyl-methyl-amino)-methyl]-4-hydroxy-piperidine-1-carboxylic acid tert-
butyl ester (35)
Epoxide (32) (46 g, 216 mmol) was dissolved in dioxane (30 0 ml). Benzyl
methylamine (75 ml, 583 mmol) was added and the mixture was stirred at reflux
for
90 h. TLC analysis revealed complete conversion. The mixture was evaporated to
dryness. The excess of benzyl methylamine was removed by evaporation in vacuo
(0.05 mbar, 80°C). Yield: 69.3 g of aminoalcohol (35)
(96°l°) as an orange oil.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
4-[(Benzyl-methyl-amino)-methyl]-4-methoxy-piperidine-1-carboxylic acid tert-
butyl ester (36)
Alcohol (35) (69.3 g, 200 mmol) was dissolved in dimethyl formamide (500 ml).
NaH
5 (9.2 g 230 mmol), washed with pentane, was added in 5 portions over 30 min.
After
complete addition the mixture was stirred at ambient temperature fo r 45 min.
Methyl
iodide (14.8 ml, 240 mmol) was added over 1.5 min. The mixture was stirred for
1.5
h at ambient temperature. TLC analysis revealed ca. 80-90°l°
conversion. Extra
NaH, 0.8 g, 20 mmol) and methyl iodide (1.2 ml , 20 mmol) was added and the
10 mixture was stirred for another 2 h at ambient temperature. The excess of
NaH was
destroyed with water (100 ml) and the mixture was further diluted with water
(3.5 I).
The mixture was extracted with ethyl acetate (2x 1 I, 500 ml). The organic
layer was
washed with brine (1 I), dried over Na2S04 and concentrated to dryness. The
traces
of dimethyl formamide were removed by evaporation in vacuo (0.4 mbar,
80°C).
15 The remaining mixture was purified over silica (eluent: heptanelethyl
acetate, 4/1 to
3l1 vlv). Yield: 55.2 g of amine (36) (80%) as light yellow oil.
4-Methoxy-4-methylaminomethyl-piperidine-1-carboxylic acid tert-butyl ester
(37)
Amine (36) (55 g, 158 mmol) was dissolved in ethyl acetate (500 ml). Pd-C
(10°l°,
wet, 5 g) was added and the mixture was stirred for 23 h under hydrogen
atmosphere (1 bar). TLC analysis revealed incomplete conversion. Extra Pd -C
(2.5
g) was added and the mixture was stirred under HZ (1 bar) for 110 h. NMR
analysis
revealed complete conversion. The mixture was filtrated over Celite, the
Celite crop
was washed with ethyl acetate and the filtrate was evaporated to dryness. The
residue was purified by distillation (bulb to bulb, 0.04 mbar, 130°C)
giving 35 g of
compound (37) (86°l°) as colorless oil.
The synthesis of 8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{3-[(4-
methoxy-piperidin-4-ylmethyl)-methyl-amino]-propyl}-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-one 39 [example nr. 45 in tables below] starting with
chlorine
(23) and amine (36) was performed like described below (general methods).
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
26
LIBRARY DESIGN with 8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2 yl)-ethyl]-3-(2-
methylamino-ethyl)-1-phenyl 1,3,8-triaza spiro[4.5]decan-4-one as starting
material
Amide Library, method I:
The N-methyl amine (40) (1.832 g, 4.24 mmol) was dissolved in 170 m( dichloro-
methane. This core solution was used to produce amides with various acid
chloride
solutions in the following manner: 2 ml of the core solution (0.05 mmol of
(40)) was
treated with polymer bound morpholine (0.162 mmol). After stirring for 20 min.
at
room temperature a solution of the corresponding acid chloride (0.06 mmol) in
2 ml
dichloromethane was added and stirring was continue d for 1 day at room
temperature. The reaction was monitored by TLC analysis. To get rid of
remaining
acid chloride and N-methyl amine derivative polymer bound trisamine and
isocyanate reagents (both are used as scavengers) were added, respectively.
Again
stirring was continued at room temperature over night before the polymers were
removed by filtration. The filtrates were concentrated under reduced pressure.
Using this protocol 69 compounds have been synthesized. The affinity of each
synthesized amide to the human ORL1 receptor was measured in the in vifro
binding assay.
Amide Library, method II:
To 200 p1 of the stock solutions of the cores (0.25M in THF) was added 200 p1
of
the stock solutions of the acid chlorides (0.25M in THF), followed by 50 p1 of
the
triethylamine solution (1.0M in THF). After shaking overnight (17 hrs) at
30°C the
solvent was evaporated and the crude products were taken up in DMSO for
analysis. (Remark: insoluble reagents were added by hand). Using this protocol
26
compounds have been synthesized. The affinity of each synthesized amide to the
human ORL1 receptor was measured in the in vitro receptor binding assay.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
27
H
~N
Epoxide Opening Library:
The N-methyl amine (40) (1.316 g, 3.0 mmol) was dissolved in 120 ml isopropyl
alcohol. This core solution was used to produce amino alcohols with various
epoxide solutions in the following manner: to 2 ml of the core solution ( 0.05
mmol
of (40)) was added a solution of the corresponding epoxide (0.075 mmol) in 2
ml
isopropyl alcohol. This mixture was heated to 80°C for 2 days. TLC
analysis was
used to monitor the reactions. For work-up polymer bound trisamine and
isocyanate
reagents (both are used as scavengers) were added, respectively. Again
stirring
was continued at room temperature for two days before the polymers were
removed
by simple filtration. The filtrates were concentrated under reduced pressure.
Using
this protocol 27 compounds have been synthesized. The affinity of each
-: synthesized aminoalcohol to the human ORL1 receptor was measured in the in,
vitro
receptor binding assay.
R-N=0
Urea Library:
To 200 p1 of the stock solutions of the cores (0.25M in THF) was added 200 p1
of
the stock solutions of the isocyanides (0.25M) in THF. The vials were cap ped
and
after shaking overnight (17 hrs) at 30 °C the solvent was evaporated
and the crude
products were taken up in DMSO for analysis. Using this protocol 71 compounds
have been synthesized.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
28
o, ,o
R~S~CI
Sulfonamide Library:
Stock solutions prepared of the cores (0.25M) in THF and of the
sulfonylchlorides
(0.25M) in THF. To 200 p1 of the core solution was added 200 p1 of the
sulfonylchlorides, followed by 50 p1 of a 1.0M DIPEA solution in THF. Vials
were
capped and heated at 30° C for 16 hours. Products were purified by
means of cat-
ion exchange Solid Phase Extraction. Solvent was evaporated and the crude
products were taken up in DMSO for analysis. Using this protocol 69 compounds
have been synthesized.
R~X
Alkylation Library:
Stock solutions were prepared of the cores (0.25M) in DMF and of the halides
(0.25M ) in DMF. To 200 p1 core solution was added 200 tll of the halide
solution
including 1 equivalent of KI, followed by 50 III of a diisopropylethylamine
solution
(1.0 M). Vials were capped and heated for 17 hours. Specific modifications:
alpha
halo ketones at 30°C; the others at 60°C. Products were purified
by means of cat
ion exchange Solid Phase Extraction. The solvent was evaporated and the crude
products were taken up in DMSO for analysis. Using this protocol 61 compounds
have been synthesized.
0 N~ 0
f ~~ C~~O~R
N 40
R
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
29
Carbamate Library:
To a solution of cores (200 pL, 0.25M) in tetrahydrofuran was added a solution
of
diisopropylethylamine (50 pL, 2M) in THF followed b y a stock solution of
chloroformate (200 pL, 0.25M) in THF. The vials were capped and shaken for 24h
at
30 degrees. Products were purified by means of cat-ion exchange Solid Phase
Extraction. The solvent was evaporated and the crude products were taken up in
DMSO for analysis. Using this protocol 21 compounds have been synthesized .
0
step 1
R2
NH Step 2
R1~
R
tert.-Urea Library:
Procedure:
This procedure was followed for the reaction of the carbamoylchloride and 2 x
75
secondary amines. All vials and flasks have to been dried at 100°C
under vacuum.
All the solvents have to been dried (mol sieves for CHZCIz and KZC03 for
CH3CN).
Step 1
9.2 mmol of the core was dissolved in 92 ml of CH 2C12 (mol. sieve 4A) = 0.1 M
solution.
To this solution was added 5.68 ml of DIPEA (3.5 eq.). The mixture was cooled
to 0 °C
(ice-bath) and a solution of 2.728 g (4.6 mmol) of triphosgene in 36.8 ml of
CH ZCIz was
added at once. The ice-bath was removed and the mixture was stirred for 30
minutes.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
The reaction was monitored by TLC and LC-MS. The reaction mixture was
concentrated
under reduced pressure for 1 hr at 40 °C and 20 mbar. The crude product
was dissolved
in 36.8 ml of CH3CN (dried on K~C03) and 1.92 ml of DIPEA was added, obtaining
a
0.25M solution of carbamoylchloride (B).
5
Step 2
To 200 p I (0.25M) of secondary amines in CH3CN was added 200 p I (0.25M)
carbamoylchloride (B) in CH3CN, followed by 1 equivalent of siisopropylamine.
The vials
10 were capped and shaken for 17 hours at 30 °C. Reaction mixtures were
concentrated,
dissolved in EtOAc and washed with 5% NaHC03-solution. The solvent was
evaporated
and the crude products were taken up in DMSO for analysis. Using this protocol
49
compounds have been synthesized.
15 SYNTHESES OF INDIVIDUAL COMPOUNDS
Synthons used for the preparation of the described examples:
0 0
'I N 'I N
HN~~~~ ~N~~~
~N CI ~N
22 23
O 0
N N
/~N /N f N ~u~~
~N 24 N 40
b
Alkylation reactions with compounds (24) 1 (40): general procedure:
The methyl amine compound was dissolved in THF and 1.1 equivalent of
diisopropyl
ethyl amine was added. To this mixture was added the appropr fate alkyiation
reagent (1 equivalent) and the solution was heated to reflux and was monitored
by
TLC. After complete conversion the solution was concentrated and the residue
was
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
31
taken up with aqueous sodium carbonate solution. The aqueous layer was
extracted
several times with dichloromethane. The combined organic layers were dried
over
sodium sulphate, concentrated and the raw product was further purified via
column
chromatography (Si02, ethyl acetate or CHZCIZlMeOH as eluents).
3-(3-[(2,4-Difluoro-benzyl)-methyl-amino]-propyl)-8-[2-(6,6-dimethyl-
bicyclo[3.1.1]hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
yield: 52% [example nr. 23 in tables below]
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-[3-(methyl-pyridin-4-
ylmethyl-
amino)-propyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
yield: 32% (example nr. 25 in tables below]
8-[2-(6,6-Dimethyl-bicyclo[3.1.1 ]hept-2-yl)-ethyl]-3-[3-(methyl-pyridin-3-
ylmethyl-
amino)-propyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one, yield: 15%
[example nr. 26 in tables below]
variation:
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-[3-(methyl-pyridin-2-yl-
amino)-
' propyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
1.7 g of the methyl amine compound 24 was dissolved in 4 ml of 2 -
Fluoropyridin
and refluxed at 150°C. After complete conversion the reaction mixture
was poured
into water and the aqueous layer was extracted with ethyl acetate for several
times.
The combined organic layers were dried over sodium sulphate, concentrated in
vacuo and purified via column chromatography (S102, ethyl acetate)
yield: 60% [example nr. 83 in tables below]
Epoxide opening reactions with compound (24) 1 (40):
0
R N~
f - ~ --~ H~N~ ~~
R N
general procedure:
The methylamine compound was dissolved in EtOHIH 20 (v/v = 10/1, 2 mmollml).
After addition of the epoxide (1.5 eq) the mixture was heated to reflux and
the
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
32
reaction was monitored by TLC. After complete conversion the solution was
concentrated and the residue was taken up with aqueous potassium carbonate
solution. The aqueous layer was extracted several times with ethyl acetate.
The
combined organic layers were dried over sodium sulphate, concentrated and the
raw product was further purified via column chromatography (SiOz, CH2Ch/MeOH
as
eluents).
3-{3-[(2,3-Dihydroxy-propyl)-methyl-amino]-propyl)-8-[2-(6,6-dimethyl-
bicyclo[3.1.1]
hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one yield: 31%
[example nr.
92 in tables below]
3-[2-[(2,3-Dihydroxy-propyl)-methyl-amino]-ethyl}-8-[2-(6,6-dimethyl-
bicyclo[3.1.1]
hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one yield:46%
[example nr.
178 in tables below]
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{3-[(2-hydroxy-
cyclohexyl)-methyl-
amino]-propyl~-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one (example nr. 29 in
tables
below] yield: 80% (remark: potassium carbonate as additional base (2.5 eq) was
used in the synthesis)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1 ]hept-2-yl)-ethyl]-3-{3-[(2-hydroxy-3-
morpholin-4-yl-
propyl)-methyl-amino]-propyl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
[example
nr. 93 in tables below] yield: 65%
Substitution reactions with compound (23):
Generally, substitutions were performed in an aprotic polar solvent (e.g.
acetonitril e,
dimethylsulphoxide or N-dimethyl formamide) in the following manner:
The starting material was dissolved in the appropriate solvent. 0.1 eq of
sodium
3o iodine and 2 eq of a base (e.g. potassium carbonate or diisopropyl ethyl
amine)
were placed into the reaction flask before the corresponding amine (2 to 4 eq)
was
added to this solution. The reaction mixture was heated and monitored by TLC
analysis. After standard aqueous work-up procedures the residues were further
purified via column chromatography (SiOz, CH2CIZlMeOH as eluents).
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
33
Preparation of optically pure starting materials according to literature
procedures [J. Prakt. Chem. 329, 235 (1987)]
2-Methylamino-cyclohexanol
Cyclohexene oxide (147 g, 1.5 mol) was dissolved in a ethanolic 8 M
methylamine
solution (750 ml) and stirred at 40°C for 16 h. The reaction mixture
was
concentrated in vacuo to give racemic 2-methylaminocyclohexanol as a slightly
brown oil (195 g, 100°l°). According to GC-analysis this product
was 99+% pure and
used without further purification.
(1R,2R)-2-methylamino-cyclohexanoh(R)-mandelic acid salt and (1S, 2S)-2-
methylamino cyclohexanoh(S)-mandelic acid
Racemic 2-methylamino-cyclohexanol (195 g, max 1.5 mol) and (R)-(-)-mandelic
acid (228 g, 1.5 mol) were added to 2-propanol (1.2 f) and heafed to reflux
temperature. The solution was allowed to cool slowly to room temperature and
was
stirred overnight. The formed precipitate was collected by filtration, wa shed
with 2-
butanone and dried on air giving a white solid (160 g, enantiomeric excess
(e.e.) _
92°l°). The motherliquor was concentrated giving a brown oil
(275 g) which solidified
on standing. The white solid was heated for 10 min at reflux temperature in 2-
butanone (1.7 I). The mixture was allowed to cool to room temperature under
stirring
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
34
and stirred at room temperature for 16 h ours. The precipitate was collected
by
filtration and dried on air giving ( 1R,2R)-2-methylamino cyclohexanol~(R)-
mandelic
acid salt (151.5 g, 538 mmol, 36%) as a white solid with an e.e. of 99%.
The first motherliquor (275 g, 0.98 mol) was added to a solution of NaOH (200
g, 5
mol) in water (800 ml) and brine (800 ml). Dichloromethane (400 ml) was added
and
the layers were separated after stirring for 15 min. The aqueous layer was
again
extracted with dichloromethane (3 x 400 ml). The combined dichloromethane
layers
were dried (NazS04) and concentrated to give a brown oil (118.5 g, 93.5%
yield).
This oil was Kugelrohr distilled (p=0.3 mbar, T=70-80°C) to give
(1S, 2S)-2
methylamino cyclohexanol (105 g, 813 mmol, 83% yield) with an e.e. of 66%.
This
enriched material was dissolved in 2-propanol (700 mL). (S)-(+)-mandelic acid
(124
g, 815 mmol) was added and the mixture was heated to reflux temperature. The
resulting solution was allowed to cool slowly to room temperature and stirred
for 16
h at that temperature. The formed precipitate was collected by filtration,
washed
with 2-butanone and dried in air to give a white solid (172 g). This first
salt was
heated to reflux for 15 min in 2-butanone (2.0 I). The mixture (no clear
solution was
formed) was allowed to cool slowly to room temperature and stirred 16 h. The
precipitate was collected by filtration and dried to give ( 1S, 2S)-2-
methylamino
cyclohexanol~(S)-mandelic acid salt (160 g, 569 mmol, 38%) as a white solid
with an
e.e. >99%.
(1 R,2R)-(-)-2-Methylamino-cyclohexanol [For configurationlrotation
relationship
see J. Prakt. Chem. 329, 235 (1987), and Tetrahedron Asymm., 10, 4619 (1999) ]
NaOH (108 g, 2.69 mol) was dissolved in water (350 m I). Brine (400 ml) was
added
and cooled to room temperature. Dichloromethane (300 ml) and (1R, 2R)-2-
methylamino cyclohexanol~(R)-mandelic acid salt (151.5 g, 538 mmol) were added
and the mixture was stirred vigorously for 10 min. The layers were separated
and
the aqueous layer was again ext racted with dichloromethane (3 x 200 m I).
(separation of the layers is a time consuming process). The combined
- dichloromethane layers were dried (Na 2S04) and concentrated to give an oil
(67.7 g,
97% yield). This oil was combined with two other batches of 2 .3 g and 6.4 g
(both
with e.e.=99+%) and purified by Kugelrohr distillation (p=0.5 mbar, T=80-
90°C)
giving a colorless oil (72.0 g, 92%) with 94% purity according to GC. This oil
was
again Kugelrohr distilled (p=0.3 mbar) giving two product containing
fractions:
Fraction 1; T=40-60°C : 11.6 g with 89% purity according to GC,
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
Fraction 2; T=60-65°C : 60.1 g 610302-1 as a colorless oil with 99%
purity and an
e.e. of 99.5%. (a]s~o=-51.5 (c=0.14, methanol).
(1S,2S)-(+)-2 -Methylamino-cyclohexanol [For configurationlrotation
relationship
5 see J. Prakt. Chem. 329, 235 (1987), and Tetrahedron Asymm., 10, 4679 (7999)
]
NaOH (108 g, 2.69 mol) was dissolved in water (400 m I). Brine (400 ml) was
added
and cooled to room temperature. Chloroform (300 ml) and (7S, 2S)-2-methylamino
cyclohexanol~(S)-mandelic acid salt (160 g, 569 mmol) were added and the
mixture
10 was stirred vigorously for 5 min. The layers were separated and the aqueous
layer
was again extracted with chloroform (3 x 350 m I). The combined chloroform
layers
were dried (Na2S04) and concentrated to give an oil (68 g, 93% yield). This
oil was
purified by Kugelrohr distillation (p=0.1 mbar) giving two product containing
fractions:
15 Fraction 1; T=40-55°C : 17.5 g as a colorless oil with 99% purity
according to GC,
Fraction 2; T=55-60°C : 48.8 g as a colorless oil with 99.9% purity
according to GC.
Both fractions were combined giving 66.2 g (512 mmol, 90°l°) as
a colorless oil with
an e.e. of 99.9% [a]6,o= +53.6 (c=0.14, methanol).
20 8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{3R-[(2R-hydroxy-
cyclohexyl)-methyl-amino]-propyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
jexampie nr. 30 in fables below]
2.5 g of the bicyclic chioro-compound (23) was dissolved in 5 ml of
acetonitrile. To
25 this solution was successively added 1.5 g (2 eq) of potassium carbonate,
90 mg
(0.1 eq) sodium iodine, 780 mg of 1 R,2R)-(-)- 2 -Methylamino-cyclohexanol and
finally 3 drops of HzO. This mixture was heated to reflux for 8 hours. For
work up the
reaction mixture was diluted with ethyl acetate and washed first with aqueous
citric
acid and afterwards with aqueous sodium hydrogen carbonate solution. The
organic
30 layer was dried over sodium sulphate, concentrated and the residue was
further
purified via column chromatography (SiOa, CNzChI MeOH as eluent) yielding
1.75g
(58%) of a light yellow oil.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
36
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{3S-[(2S-hydroxy-
cyclohexyl)-methyl-amino]-propyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
[example nr. 31 in tables below]
The compound was prepared according to the protocol given above for the
stereoisomer. Yield: 48°l° as a light yellow oil.
Boronic-acid-Mannich-reactions:
OH
i
Ar~B~OH
OH OH
~0
l~! ''n
OH
n=u-a
The following experimental protocol was used to synthesize several compounds
in
the same manner starting with compound (40) and compound (24), respectively
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{3-[(1-furan-2-yl-2,3,4,5-
-~ tetrahydroxy-pentyl)-methyl-amino]-propyl}-1-phenyl-1,3,8-triaza-spiro[4.5]
decan-4-one [example nr. 52 in tables below]
0
~N
N
\N~ ~N
HO
HO OH
HO
A solution of the amine (24) (2.66 g, 5.876 mmol) and 2-furanyl boronic acid
(920
mg, 8.22 mmol) in 30 ml EtOH and 0.5 ml H z0 is heated to 40°C under
vigorous
stirring. To this solution was added 1.06 g, (7.05 mmol) of D-(+)-xylose in
small
portions within 15 min at 40°C.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
37
After 2.5 hours the reaction mixture was poured into aqueous NaHC03 solution
and
the aqueous layer was extracted 3 times with dichloromethane. The combined
organic layers were concentrated and taken up in ethyl acetate. This organic
layer
was washed with aqueous citric acid (10%) several times. The combined aqueous
layers were then neutralized with aqueous NaHC03 solution and the neutral
aqueous layer was extracted with CHzCl2. The combined organic extracts were
dried
over sodium sulphate and concentrated in vacuo to provide a crude yellow oil
which
was purified by column chromatography (SiO~, ethyl acetatelmethanol: 20/1 to
10/1). The purification yielded the title compound as an amorphous white solid
(1.40
g, 2.14 mmol, 37°l°).
Alternative work-up procedure: After complete conversion (TLC) the reaction
mixture was cooled to room temperature, 1 ml of trifluoroacetic acid was added
and
the remaining solution was stirred for 10 min at room temperature.
Concentration in
vacuo was followed by further purification of the raw product via column
chromatography.
Using the same procedure as described above, the following ex amples were
synthesized:
8-[2-(6,6-Dimethyl-bicyclo[3.1.1 ]hept-2-yl)-ethyl]-3-{3-[methyl-(2,3,4,5-
tetrahydroxy-
1-thiophen 2-yl-pentyl)-amino]-propyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-
one
jexample nr. 59 in tables below] yield: 80% (D-xylose and 2-thiophenyl boronic
acid
as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-[methyl-(2,3,4,5-
tetrahydroxy-
1-thiophen-2-yl-pentyl)-amino]-ethyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4.-
one
jexample nr. 58 in tables below] yield: 13% (D-xylose and 2-thiophenyl boronic
acid
as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-[(1-furan-2-yl-
2,3,4,5,6-
pentahydroxy-hexyl)-methyl-amino]-ethyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-
4-
one jexample nr. 55 in tables below] yield: 75% (D-glucose and 2-furanyl
boronic
acid as starting materials)
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
38
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-[(1-furan-2-yl-2,3,4,5-
tetrahydroxy-pentyl)-methyl-amino]-ethyl}-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-
one [example nr. 53 in tables below] yield: 42% (D-xylose and 2-furanyl
boronic acid
as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-[(1-furan-2-yl-2,3,4,5-
tetrahydroxy-pentyl)-methyl-amino]-ethyl}-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-
one [example nr. 52 in tables below] yield: 60% (L-xylose and 2-furanyl
boronic
acid as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-[(1-furan-2-yl-2,3-
dihydroxy-
propyl)-methyl-amino]-ethyl}-1-phenyl-1,3,8-triaza-spiro[4.5]deoan-4-one
[example
nr. 49 in tables below] yield: 66% (D,L-glycerin aldehyde and 2-furanyl
boronic acid
as starting materials)
3-{2-[(2,3-Dihydroxy-1-thiophen-3-yl-propyl)-methyl-amino]-ethyl}-8-[2-(6,6-
dimethyl-
bicyclo[3.1.1]hept-2-yl)-ethyl]-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
[example nr. 47 in tables below] yield: 39% (D,L-glycerin aldehyde and 3-
thiophenyl boronic acid as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-(2-[methyl-(2,3,4,5-
tetrahydroxy
1-thiophen-3-yl-pentyl)-amino]-ethyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4.-
one
[example nr. 48 in tables below] yield: 28% (L-arabinose and 3-thiophenyl
boronic
acid as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1]hept-2-yl)-ethyl]-3-{2-(methyl-(2,3,4,5-
tetrahydroxy
1-thiophen-3-yl-pentyl)-amino]-ethyl}-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-
one
[example nr. 55 in tables below] yield: 21 % (L-xylose and 3-thiophenyl
boronic
acid as starting materials)
8-[2-(6,6-Dimethyl-bicyclo[3.1.1 ]hept-2-yl)-ethyl]-3-{2-[methyl-(2,3,4,5,6-
pentahydroxy-1-thiophen-3-yl-hexyl)-amino]-ethyl}-1-phenyl-1,3,8-triaza-
spiro[4.5]decan-4-one [example nr. 54 in tables below] yield: 16% (D-glucose
and
3-thiophenyl boronic acid as starting materials)
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
39
The invention is further illustrated by mean s of the following specific
examples (only
intended to further illustrate the invention in more detail, and therefore are
not
deemed to restrict the scope of the invention in any way) listed in the table
below
and represented by the general formula a (1 ) and (2):
R~
(1 )
R3
.N
(R1)m Q ~~~~ \R4 (2)
Where in the fable below under the heading "stereochemistry" the name of a
compound is
given (e.g_ ( )-cis-hydronopol or D-xylose), that means that the compound in
question was
used in the final reaction step.
Ex R~ mRz nR3 RQ stereochemistry
1 H 1H - - (-)cis-hydronopol
2 H 1H - - (-)traps-hydronopol
3 H 1H - - +)cis-hydronopol
4 H 1H - - (+ traps-hydronopol
5 4-F 1H - -
6 4-OCH31H - -
7 3-CI 1H - -
8 3-OCHa1H - -
9 3-F 1H - -
10 3-CF31H - -
11 H 1(1)* - -
12 H 1Q 1_ CH3
H
13 H 1Q 2H CH3
14 H 1Q 3H CH3
H 1Q 4H CH3
16 H 1Q 5H CH3
17 H 1Q 1CH3 Benzyl
18 H 1Q 1CH3 2-Morpholin-4-yl-ethyl
19 H 1Q 2H 3,4-Methylendioxybenzyl
H 1Q 2H 4-Sulfamoyl-benzyl
21 H 1Q 2CH3 CH3 (- -cis
22 H 1Q 2CH3 CH3 - -traps
23 H 1Q 2CH3 2,4-Difluorbenzyl
24 H 1Q 2CH3 1-Methyl-pyridinium-2yl
H 1Q 2CH3 Pyridin-4-yl-methyl
26 H 1Q 2CH3 Pyridin-3-yl-methyl
27 H 1Q 2CH(CH3)Z3,4-(Dimethoxy-phenyl)-ethyl
28 H 1Q 1CH3 2-OH-cyclohexyl Racemic
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
ExR~ m RZn R3 R4 stereochemistry
29H 1 Q 2 CH3 2-OH-cyclohexyl racemic
30H 1 Q 2 CH3 2-OH-cyclohexyl trans-diastereomer-
1
31H 1 Q 2 CH3 2-OH-cyclohexyl trans-diastereomer-
2
32H 1 Q 3 CH3 2-OH-cyclohexyl racemic
33H 1 Q 4 CH3 2-Ohi-cyclohexyl racemic
34H 1 Q 5 CH3 2-OH-cyclohexyl racemic
35H 1 Q 2 CH3 4-Hydroxy-(1-tert.butoxypiperidin)-4-
ylmethyl
36H 1 O 1 CH3 4-Hydroxy-piperidin-4-ylmethyl
37H 1 Q 2 CH3 4-Hydroxy-piperidin-4-ylmethyl
38H 1 O 3 CH3 4-Hydroxy-piperidin-4-ylmethyl
39H 1 Q 4 CH3 4-Hydroxy-piperidin-4-ylmethyl
40H 1 Q 2 CH3 4-Hydroxy-(1-naphthalen-2-ylmethyl)-
piperidine-4-ylmethyl
41H 1 Q 2 CH3 4-Hydroxy-1-isopropyl-
piperidine-4-
ylmethyl
42H 1 Q 2 CH3 4-Hydroxy-(1-(3-methoxybenzyl)-
piperidine-4- lmeth
I
43H 1 Q 2 CH3 (4-Hydroxy-4-methyl-piperidin-1-yl)-
acetic acid eth I ester
44H 1 Q 2 CH3 (4-Hydroxy-4-methyl-piperidin-1-yl)-
acetic acid
45H 1 Q 2 CH3 4-Methoxy-piperidin-4-ylmethyl
46H 1 Q 2 CH3 4-Methyl-4-hydroxy-piperidine-1-
carboxamidine
47H 1 Q 2 CH3 1-Acetyl-4-hydroxy-piperidin-4-ylmethyl
48H 1 ~ 2 CH3 (1-Dimethylcarbamoyl-4-hydroxy-
Q
i eridin-4- I
49H 1 Q 1 CH3 2,3-Dihydroxy-1-thiophen-3-yl-propylracemic
'
50H 1 Q 1 CH3 2,3,4,5-Tetrahy pr L-arabinose
ox
y
-i -thiophen-3-yl-
e
t
y
51H 1 Q 1 CH3 1-Furan-2-yl-2,3-dihydroxy-propylracemic
52H 1 Q 2 CH3 1-Furan-2-yl-2,3,4,5-tetrahydroxy-pentylD-xylose
53H 1 Q 1 CH3 1-Furan-2-yl-2,3,4,5-tetrahydroxy-pentyfD-xylose
54H 1 Q 1 CH3 1-Furan-2-yl-2,3,4,5-tetrahydroxy-pentylL-xylose
55N 1 O 1 CH3 1-Furan-2-yl-2,3~e,5 p_glucose
i6-pentahydroxy-
56H 1 Q 1 CH3 2,3,4,5,6-Pentahydroxy-1-thiophen-3-yl-p_glucose
hexyl
57H 1 Q 1 CH3 (2,3,4,5-tetrahypron~'1-thiophen-3-yl-L-xylose
58H 1 Q 1 CH3 (2,3,4,5-tetrahypr p-xylose
o
l thiophen-2-yl-
e
ntyl
59H 1 Q 2 - o . p-xylose
CH3._y
_ l-thiophen-2-yl-
(2,3,4,5-tetrahydr
e
n
l
60H 1 Q 2 3-[3 -(4-hydroxy-4-phenyl-piperidin-1-ylracemic
61H 1 Q 1 3-(2-[4-(4-Chloro-3-trifluoromethyl-phenyl)-4-
h
drox
-
i
eridin
62H 1 Q 2 4-Phenyl-3,6-dihydro-2H-pyridin-1-yl)
63H 1 Q 2 4-(3-Chloro-phenyl)-piperazin-1-yl
64H 1 Q 2 4-(Phenyl)-piperazin-1-yl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
41
ExR~ m RZn R3 stereochemistry
Rq
65H 1 Q 2 4-(3-Fluorophenyl)-piperazin-1-yl
66H 1 Q 2 3-Hydroxymethyl-piperidin-1-yl
67H 1 Q 2 4-carboxylic
acid
amide
piperidine-1yl
68H 1 {2)* -
69H 1 (3)* -
_
70H 1 (4)* -
71H 1 Q 1 CH3 3-(4-Ethyl-piperaon-i racemic
-yl)-2-hydroxy-
72H 1 Q 1 CH3 4-((4-Chloro-phenyl)-piperazin-1-yl)-2-racemic
h dro -pro I
73H 1 Q 1 CH3 2-Hydroxy-3- racemic
4- ridin-2- I-piperazin-1-
I -prop I
74H 1 Q 1 CH3 4-(Benzyl-piperaoP-i racemic
-yl)-2-hydroxy-
75H 1 Q 1 CHs 3-(4-Phenyl-pipepraopyl1-yl)-2-hydroxy-mcemic
76H 1 Q 1 CH3 2-Hydroxy-3-(4-ispporPylyl-piperazin-1-yl)-racemic
77H 1 Q 2 CHs 3-(4-Ethyl-piperpaopylracemic
-yl)-2-hydroxy-
78H 1 Q 2 CH3 4-((4-Chloro-phenyl)-piperazin-1-yl)-2-racemic
h drox - ro I
79H 1 Q 2 CH3 2-Hydroxy-3- racemic
4- ridin-2- I- i erazin-1-
I - rop I
80H 1 Q 2 CH3 4-(Benzyl-pipe p op-i racemic
-yl)-2-hydroxy-
81H 1 Q 2 CH3 3-{4-Phenyl-piperaozinl1-yl)-2-hydroxy-racemic
82H 1 Q 2 CH3 2-Hydroxy-3-(4-ispporPpyl-piperazin-1-yl)-racemic
83H 1 Q 2 CH3 Pyridin-2yl
84H 1 Q 2 H Benzyl
85H 1 (5)* - -
-
86H 1 (6)* _ -
87H 1 (7)* - -
88H 1 Q 9 CH3 2-Cyclohexyl-2-hydroxy-acetyl
89H 1 Q 1 CH3 2-Benzyl-2-hydroxy-acetyl
90H 1 Q 1 CH3 3,4,5-Trimethoxybenzoyl
91H 1 Q 2 CH3 2,3,4,5,6-Pentahydroxy-hexyl(+)-D-Glucosamine
92H 1 Q 2 CH3 2,3-Dihydroxy-propyl racemic
93H 1 Q 2 CH3 2-Hydroxy-3-morpholin-4-yl-propylracemic
94H 1 Q 2 CH3 2-Hydroxy-3-isopropoxy-propyl
95H 1 Q 1 CH3 2-Methoxy-acetyl
96H 1 Q 1 CH3 Benzo[1,3]dioxole-5-carboxyl
97H 1 Q 1 CH3 3,5-Bis-trifluoromethyl-benzoyl
98H 1 Q 1 CH3 Benzoyl
99H 1 Q 1 CH3 2-Bromobenzoyl
100H 1 Q 1 CH3 2,3,4,5,6-Pentafluorobenzoyl
101H 1 Q 1 CH3 2,4-Dichlorobenzoyl
102H 1 Q 1 CH3 2-Methoxybenzoyl
103H 1 Q 1 CH3 2-Trifluoromethyl-benzoyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
42
ExR~ m RZn R3 R4 stereochemistry
104H 1 Q 1 CH3 2-Methylbenzoyl
105H 1 Q 1 CH3 3-Fluorobenzoyl
106H 1 Q 1 CHs 3-Chlorobenzoyl
107H 1 0 1 CH3 3,4-Dichlorobenzoyl
108H 1 Q 1 CH3 3-Methoxybenzoyl
109H 1 Q 1 CH3 4-Fluorobenzoyl
110H 1 Q 1 CH3 4-Chlorobenzoyl
111H 1 Q 1 CH3 4-Methoxybenzoyl
112H 1 Q 1 CH3 4-Hexyloxybenzoyl
113H 1 Q 1 CH3 4-Trifluoromethyl-benzoyl
114H 1 Q 1 CH3 4-tert.-Butylbenzoyl
115H 1 Q 1 CH3 4-Methylbenzoyl
116H 1 Q 1 CH3 N-oxalamic acid methyl
ester
117H 1 Q 1 CH3 2-Acetoxy-2-methyl-propionyl
118H 1 Q 1 CH3 2,2-Dimethylpropionyl
119H 1 Q 1 CH3 2-Acetoxy-2-phenylacetylracemic
120H 1 Q 1 CH3 2-Phenoxyacetyl
121H 1 0 1 CH3 2-Phenylacetyl
122H 1 Q 1 CH3 2,8-Dimethoxybenzoyl
123H 1 Q 1 CH3 3,5-Dichlorobenzoyi
124H 1 Q 1 CH3 2,6-Difluorobenzoyl
125H 1 Q 1 CH3 2,6-Dichlorobenzoyl
126H 1 Q 1 CH3 3-Methylbenzoyl
127H 1 0 1 CH3 2-Ethyl-hexanoyl racemic
128H 1 Q 1 CH3 Cyclobutanecarboxyl
-,
129H 1 Q 1 CH3 3-Nitrobenzoyl
130H 1 Q 1 CH3 3-Cyanobenzoyl
131H 1 Q 1 CH3 (3-Methoxy-phenyl)-acetyl
132H 1 Q 1 CH3 2-Ethylsulfanyl-pyridine-3-carboxyl
133H 1 Q 1 CH3 3,5-Difluorobenzoyl
134H 1 Q 1 CH3 3,4-Difluorobenzoyl
135H 1 Q 1 CH3 2,4-Difluorobenzoyl
136H 1 Q 1 CH3 3-Methyl-but-2-enoyl
137H 1 Q 1 CH3 3,3-Dimethylbutyryl
138H 1 Q 1 CH3 Propionyl
139H 1 Q 1 CH3 2-Benzylacetyl
140H 1 Q 1 CH3 2,2,2-Trichloroacetyl
141H 1 Q 1 CH3 2,2-Dichloroacetyl
142H 1 Q 1 CH3 2-Phenyl-cyclopropanecarboxyl
143H 1 Q 1 CH3 3-Cyclopentylpropionyl
144H 1 Q 1 CH3 Cyclohexylcarboxyl
145H 1 Q 1 CH3 Furan-2yl-carboxyl
146H 1 Q 1 CH3 Thiophen-2yl-carboxyl
147H 1 Q 1 CH3 9-Oxo-9H-fluorene-4-carboxyl
148H 1 Q 1 CH3 2-Benzyloxyacetyl
149H 1 Q 1 CH3 2-Acetoxyacetyl
150H 1 Q 1 CH3 Pyridin-4yl-carboxyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
43
ExR~ m R2n R3 R4 stereochemistry
151H 1 Q 1 CH3 2,2-Diphenylacetyl
152H 1 Q 1 CH3 3-Oxo-propionic acid
methyl ester
153H 1 Q 1 CH3 2-Chlorobutyryl racemic
154H 1 Q 1 CH3 6-Chlorohexanoyl
155H 1 Q 1 CH3 2-[2-(4-Chloro-phenyl)-cyclopentyl]-
ace I
156H 1 Q 1 CH3 2-Phenoxybutyryl racemic
157H 1 Q 1 CH3 Benzo[b]thiophen-2-yl-carboxyl
158N 1 Q 1 CH3 2-(Trifiuormefhoxy)-benzoyl
159N 1 Q 1 CH3 (5-Methyl-2-phenyl-2H-[1,2,3]triazol-4-
yl)-carboxyl
160H 1 Q 1 CH3 2,6-Dichloropyridin-4yi-carboxyl
161H 1 Q 1 CH3 2-(Propylsulfanyl)pyridin-3yl-carboxyl
162H 1 Q 1 CH3 2,3-Dichloropyridin-5yl-carboxyl
163H 1 Q 1 CH3 3-(2-Chloro-6-fluoro-phenyl)-5-meth
I-isoxazole-4-carbox
l
164H 1 Q 1 CH3 2,4,5-Trifluorobenzoyl
165H 1 Q 1 CH3 3,3,3-Trifluoro-2-methoxy-2-phenyl-
ropion
166H 1 Q 1 CH3 2-p-Tolylsulfanyl-pyridin-3-yl-carboxyl
167H 1 Q 1 CH3 2-(4-Chloro-phenoxy)-pyridin-3-yl-
carbox 1
168H 1 Q 1 CH3 2-Chlor-3-methoxy-thiophen-4yl-carboxyl
169H 1 Q 1 CH3 1-Phenyl-5-tritluoromethyl-1
H-pyrazole-
4-carbo I
170H 1 Q 1 CH3 Adamantane-1-carboxyl
171H 1 Q 1 CH3 3-(3-Trifiluoromethyl-phenyl)-propenoylE-isomer
172H 1 Q 1 CH3 2-tert-Butyl-5-methyl-2H-pyrazole-3-
carbox I
173H 1 Q 1 CH3 5-tert-Butyl-2-methyl-2H-pyrazole-3-
carbox I
174H 1 Q 1 CH3 2-Chloro-6-methoxy-pyridin-4yl-carboxyl
175H 1 Q 1 CH3 (2-p-Chlorophenoxy)-2-methyl-propionyl
176H 1 Q 1 CH3 4R,7,7-Trimethyl-3-oxo-2-oxa-bicyclo
2.2.1 he Lane-1-carbo
I
177H 1 Q 1 CH3 3-Phenyl-2S-(toluene-4-sulfonylamino)-
ropionyl
178H 1 Q 1 CH3 2,3-Dihydroxy-propyl racemic
179H 1 Q 1 CH3 2-Hydroxy-2-phenyl-ethylracemic
980H 1 Q 1 CH3 2-hydroxy-propyl racemic
181H 1 Q 1 CH3 (3-Fluoro-2-hydroxy-propyl)racemic
182H 1 Q 1 CH3 4-(Chloro-phenoxy)-2-hydroxy-propylracemic
183H 1 Q 1 CH3 3-(4-Methoxy-phenoxy)-2-hydroxy-propylracemic
184H 1 Q 1 CH3 3-(4-tert.-Butyl-phoeniracemic
xy)-2-hydroxy-
185H 1 Q 1 CH3 3-(iso-Propoxy)-2-hydroxy-propylracemic
186H 1 Q 1 CH3 3-(2-Ethyl-hexyloxy)-2-hydroxy-propylracemic
187H 1 Q 1 CH3 3-Allyloxy-2-hydroxy-propylracemic
188H 1 Q 1 CH3 3-Butoxy-2-hydroxy-propylracemic
189H 1 Q 1 CH3 2-Hydroxy-but-3-enyl racemic
190H 1 Q 1 CH3 2-Hydroxy-but-4-yl racemic
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
44
ExR~ m RZn R3 R4 stereochemistry
191H 1 Q 1 CH3 2-Hydroxy-oct-7-en-1ylracemic
192H 1 Q 1 CH3 2-Hydroxy-oct-1yl racemic
193H 1 Q 1 CH3 3-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-2-racemic
h drox - ro I
194H 1 Q 1 CH3 3-tert.-Butoxy-2-hydroxy-propylracemic
195H 1 Q 1 CH3 2-Hydroxy-hex-5-enyl racemic
196H 1 Q 1 CH3 2R-Hydroxy-3S-(4-methoxy-phenyl)-3yl-
propionic acid meth
I ester
197H 1 Q 1 CH3 3-(Furan-2-ylmethoxy)-2-hydroxy-propylracemic
198H 1 Q 1 CH3 1-Trifluoromethyl-ethan-2yl-1olracemic
199H 1 Q 1 CH3 2-Hydroxy-3-(1,1,2~o2-ptracemic
itrafluoro-ethoxy)-
200H 1 Q 1 CH3 2-Hydroxy-3-morpholin-4-yl-propylracemic
201H 1 Q 1 CH3 2-Hydroxy-dec-9-en-1ylracemic
202H 1 Q 1 CH3 2-Hydroxy-3-phenyl-propylracemic
203H 1 Q 1 CH3 2-Hydroxy-2-methyl-but-3-enylracemic
204H 1 Q 1 CH3 2-Hydroxy-2-methyl-3yl-propionicracemic
acid
methyl ester
205H 1 Q 1 CH3 ~3-[4-(4-Chloro-benzyl)-piperracemic
azin-1- I -2-h dro
- rop I
206H 1 Q 1 CH3 2-Hydroxy-hexyl racemic
207H 1 Q 1 CH3 3-Hydroxy-3-phenyl-2yl-propionicracemic
acid ethyl ester
208H 1 Q 1 CH3 2(R),3-Dihydroxy-propylchiral
209H 1 Q 1 CH3 2-Hydroxy-dodecyl racemic
210H 1 0 1 CH3 2-Hydroxy-tetradecyl racemic
211H 1 Q 1 CH3 2-Hydroxy-3-methoxy-propylracemic
212H 1 Q 1 CHs 2-Hydroxy-hexadecyl racemic
213H 1 Q 1 CH3 2-Hydroxy-octadecyl racemic
214H 1 Q 1 CH3 2-Hydroxy-cyclopentyl racemic
3-Hydroxy-3-(4-methoxy-phenyl)-2yl-
215H 1 Q 1 CH3 propionic acid methyl racemic
ester
216H 1 Q 1 CH3 2-Hydroxy-4-vinyl-cyclohexylracemic
217H 1 Q 1 CH3 2(S)-Hydroxy-1,2-diphenyl-ethyl
218H 1 Q 1 CH3 Biphenyl-4-ylmethyl
219H 1 Q 1 CH3 Naphthalen-2-ylmethyl
220H 1 Q 1 CH3 3-Phenoxy-benzyl
221H 1 Q 1 CH3 Biphenyl-2-ylmethyl
222H 1 Q 1 CH3 Naphthalen-1-ylmethyl
223H 1 Q 1 CH3 (1H-Indol-3-yl)-ethyl
224H 1 Q 1 CH3 Pyridin-2-ylmethyl
225H 1 Q 1 CH3 4-Trifluoromethyl-benzyl
226H 1 Q 1 CH3 Pyridin-3-ylmethyl
227H 1 Q 1 CH3 Cyclopropylmethyl
228H 1 Q 1 CH3 6-Chloro-benzo[1,3]dioxol-5-ylmethyl
229H 1 Q 1 CH3 4-Trifluoromethoxy-benzyl
230H 1 Q 1 CH3 3-Oxo-3-phenyl-propyl
231H 1 Q 1 CH3 2-Cyclohexyl-ethyl
232H 1 Q 1 CH3 4-tert-Butyl-benzyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
ExR~ m RZn R3 Rq stereochemistry
233H 1 Q 1 CH3 2-Phenoxy-ethyl
234H 1 Q 1 CH3 4-Cyanobenzyl
235H 1 Q 1 CH3 3,5-Dimethyl-isoxazol-4-ylmethyl
236H 1 0 1 CH3 2-Benzenesulfonyl-ethyl
237H 1 Q 1 CH3 Phenethyl
238H 1 0 1 CH3 2,4-Dioxo-1,4-dihydro-2H-quinazolin-3-
yl-ethyl
239H 1 Q 1 CH3 3-Fluorobenzyl
240H 1 Q 1 CH3 4-Benzyloxybenzyl
241H 1 Q 1 CH3 4-Chlorobenzyl
242H 1 Q 1 CH3 3,4-Dibenzyloxybenzyl
243H 1 Q 1 CH3 3-Trifluoromethoxybenzyl
244H 1 Q 1 CH3 Pentyl
245H 1 0 1 CH3 3-Phenyl-propyl
246H 1 Q 1 CH3 Propionamide-3yl
247H 1 Q 1 CH3 2,6-Dioxo-1,2,3,6-tetrahydro-pyrimidin-4-
I
248H 1 Q 1 CH3 3-Benzyloxy-propyl
249H 1 Q 1 CH3 5-Chloro-thiophen-2-ylmethyl
250H 1 Q 1 CH3 propionic acid methyl
ester-3yl
251H 1 Q 1 CH3 3,5-Dimethylbenzyl
252H 1 0 1 CH3 Cyanomethyl
253H 1 Q 1 CH3 2-Fluorobenzyl
254H 1 Q 1 CH3 3-Trifluoromethylbenzyl
255H 1 Q 1 CH3 2-Cyanobenzyl
256H 1 Q-1 CH3 3-Methyl-butyl
257H 1 Q 1 CH3 2-Hydroxy-ethyl
258H 1 Q 1 CH3 3-Chlorobenzyl
259H 1 Q 1 CH3 Anthracen-9-ylmethyl
260H 1 Q 1 CH3 2-Methylbenzyl
261H 1 Q 1 CH3 4-Bromobenzyl
262H 1 Q 1 CH3 4-Methylbenzyl
263H 1 Q 1 CH3 3-Cyanobenzyl
264H 1 Q 1 CH3 2-Oxo-2-phenyl-ethyl
265H 1 Q 1 CH3 Acetamide-2yl
266H 1 Q 1 CH3 2-(2,5-Dimethoxyphenyl)-2-oxo-ethyl
267H 1 Q 1 CH3 2-Adamantan-1-yl-2-oxo-ethyl
268H 1 Q 1 CH3 2-(3,4-Dihydro-2H-benzo[b][1,4]dioxepin-
7- I -2-oxo-eth I
269H 1 Q 1 CH3 2-Oxo-1,2-diphenyl-ethylracemic
270H 1 Q 1 CH3 Isobutyl
271H 1 Q 1 CH3 4-Styryl-benzyl E-isomer
272H 1 Q 1 CH3 3-Phenoxypropyl
273H 1 Q 1 CH3 4-Fluorobenzyl
274H 1 Q 1 CH3 3-Methoxybenzyl
275H 1 Q 1 CH3 Pyridin-4-ylmethyl
276H 1 Q 1 CH3 2-Methoxybenzyl
277H 1 Q 1 CH3 N-(4-Phenoxy-phenyl)-formamidyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
46
ExR~ m RZn R3 R4 stereochemistry
~
278H 1 Q 1 CH3 N-Benzyl-formamidyl
279H 1 Q 1 CH3 N-Biphenyl-4-yl-formamidyl
280H 1 Q 1 CH3 N-Biphenyl-2-yl-formamidyl
281H 1 Q 1 CH3 N-(2-Methoxyphenyl)-formamidyl
282H 1 Q 1 CH3 N-(4-Fluorophenyl)-formamidyl
283H 1 Q 1 CH3 N-(4-Cyanophenyl)-formamidyl
284H 1 Q 1 CH3 N-(Adamantan-1-yl)-formamidyl
285H 1 Q 1 CH3 N-(2-Fluorophenyl)-formamidyl
286H 1 Q 1 CH3 N-(4-Methoxyphenyl)-formamidyl
287H 1 Q 1 CH3 N-(3-Cyanophenyl)-formamidyl
288H 1 Q 1 CH3 3-(Formylamino)-benzoic
acid ethyl
ester
289H 1 Q 1 CH3 N-(2-Phenethyl)-formamidyl
290H 1 Q 1 CH3 N-(1-Naphthalen-1-yl-ethyl)-formamidylracemic
291H 1 Q 1 CH3 N-(2,6-Dichlorophenyl)-formamidyl
292H 1 Q 1 CH3 N-(3-Chlorophenyl)-formamidyl
293H 1 Q 1 CH3 N-(4-Chiorophenyl)-formamidyl
294H 1 Q 1 CH3 N-(tert.-Butyl)-formamidyl
295H 1 Q 1 CH3 N-(1-Phenethyl)-formamidylR-isomer
296H 1 Q 1 CH3 N-Butyl-formamidyl
297H 1 Q 1 CH3 N-(3,4,5-Trimethoxyphenyl)-formamidyl
298H 1 Q 1 CH3 N-(2,4-Dimethoxyphenyl)-formamidyl
299H 1 Q 1 CH3 N-Benzoyl-formamidyl
300H 1 Q 1 CH3 N-(iso-Propyl)-formamidyl
301H 1 Q 1 CH3 N-(4-Nitrophenyl)-formamidyl
302H 1 Q 1 CH3 N-(2-Methylsulfanyl-phenyl)-formamidyl
303H 1 Q 1 CH3 (Formylamino)-acetic
acid ethyl ester
304H 1 Q 1 CH3 N-(4-Bromophenyl)-formamidyl
305H 1 Q 1 CH3 N-(4-Butylphenyl)-formamidyl
306H 1 Q 1 CH3 2-Formylamino-3-phenyl-propionicS-isomer
acid
methyl ester
307H 1 Q 1 CH3 N-(2,5-Dimethoxyphenyl)-formamidyl
308N 1 Q 1 CN3 N-(2-Methylphenyl)-formamidyl
309H 1 Q 1 CH3 N-(2,6-Dimethylphenyl)-formamidyl
310H 1 Q 1 CH3 N-(3,4-Dichlorophenyl)-formamidyl
311H 1 Q 1 CH3 4-(Formylamino)-benzoic
acid ethyl
ester
312H 1 Q 1 CH3 N-(3-Nitrophenyl)-formamidyl
313H 1 Q 1 CH3 N-(3,5-Di(trifiuoromethyl)-phenyl)-
formamidyl
314H 1 Q 1 CH3 N-(2,4,6-Trimethylphenyl)-formamidyl
315H 1 Q 1 CH3 4-(Formylamino)-benzoic
acid butyl
ester
316H 1 Q 1 CH3 3-(Formylamino)-propionic
acid ethyl
ester
317H 1 Q 1 CHs N-(1,1,3,3-Tetramethyl-butyl)-formamidyl
318H 1 Q 1 CH3 N-(2,4-Difluorophenyl)-formamidyl
319H 1 Q 1 CH3 N-(2,4-Dichlorophenyl)-formamidyl
320H 1 Q 1 CH3 N-(3-Methylphenyl)-formamidyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
47
ExR~ m R2n R3 , R4 stereochemistry
321H 1 Q 1 CH3 N-Allyl-formamidyl
322H 1 Q 1 CH3 2-Formylamino-acetic
acid butyl ester
323H 1 Q 1 CH 2-Formylamino-3-phenyl-propionicracemic
acid
3 eth I ester
324H 1 Q 1 'CHs N-(2-Trifluoromethoxyphenyl)-
formamidyl
325H 1 Q 1 CH3 N-Pentyl-formamidyl
2-Formylamino-3-methyl-butyric
326H 1 Q 1 CH3 acid S-isomer
meth I ester
327H 1 Q 1 CH3 N-(2-Bromophenyl)-formamidyl
328H 1 Q 1 CH3 N-(2-Methoxyphenyl)-formamidyl
329H 1 Q 1 CH3 2-(Formylamino)-benzoic
acid ethyl
ester
330H 1 Q 1 CH3 N-(4-Ethylphenyl)-formamidyl
331H 1 Q 1 CH3 N-(2,3-Dichlorophenyl)-formamidyl
332H 1 Q 1 CH3 N-(2,5-Dichlorophenyl)-formamidyl
333H 1 Q 1 CH3 N-(3-Bromophenyl)-formamidyl
334H 1 Q 1 CH3 N-(2,6-Di-(iso-propyl)-phenyl)-
formamidyl
335H 1 Q 1 CH3 N-Formyl-carbamic acid
ethyl ester
336H 1 Q 1 CH3 N-(2,4-Dimethylphenyl)-formamidyl
337H 1 Q 1 CH3 N-(5-Chloro-2-methoxyphenyl)-
formamidyl
338H 1 Q 1 CH3 N-(4-Chloro-2-trifluoromethyl-phenyl)-
formamidyl
339H 1 Q 1 CH3 N-(4-Chloro-3-trifluoromethyl-phenyl)-
formamid I
340H 1 Q 1 CH3 N-(4-Ethoxyphenyi)-formamidyl
341H 1 Q 1 CH3 N-(4-Chloro-2-nitro-phenyl)-formamidyl
342H 1 Q 1 CH3 N-(2,6-Diethylphenyl)-formamidyl
343H 1 Q 1 CH3 N-(6-Chloro-2-methyl-phenyl)-
formamid I
344H 1 Q 1 CH3 N-(4-Bromo-2,6-dimethyl-phenyl)-
formamid I
345H 1 Q 1 CHs 6-Formylamino-hexanoic
acid ethyl ester
346H 1 Q 1 CH3 2-Formylamino-propionicracemic
acid ethyl ester
347H 1 Q 1 CH3 N-(2,5-Dinitophenyl)-formamidyl
348H 1 Q 1 CH3 4-Benzo[1,3]dioxol-5-ylmethyl-
piperazine-1-carboxyl
349H 1 Q 1 CH3 4-(2-Oxo-2,3-dihydro-benzoimidazol-1-
yl)-piperidine-1-carboxyl
350H 1 Q 1 CH3 3,4-Dihydro-1 H-isoquinoline-2-carboxyl
351H 1 Q 1 CH3 2,5-Dihydro-pyrrole-1-carboxyl
352H 1 Q 1 CH3 4-Phenyl-piperazine-1-carboxyl
353H 1 Q 1 CH3 Morpholine-4-carboxyl
354H 1 Q 1 CH3 4-Pyridin-2-yl-piperazine-1-carboxyl
355H 1 Q 1 CH3 N-Methyl-N-(2-pyridin-2-yl-ethyl)-
formamid I
356H 1 Q 1 CH3 Pyrrolidine-1-carboxyl
357H 1 Q 1 CH3 1-Formyl-pyrrolidine-2-carboxylicS-isomer
acid
benzyl ester rrolidine
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
48
ExR~ m RZn R3 R4 stereochemistry
358H 1 Q 1 CH3 4-(4-Fluoro-phenyl)-piperazine-1-
carboxyl
359H 1 Q 1 CH3 4-(2-Methoxy-phenyl)-piperazine-1-
carboxyl
360H 1 Q 1 CH3 4-(4-Chloro-phenyl)-4-hydroxy-
i eridine-1-carbox
I
361H 1 Q 1 CH3 4-(4-Trifluoromethy!-phenyl)-piperazine-
1-carbo I
362H 1 Q 1 CH3 4-(4-Chloro-benzyl)-piperazine-1-
carbox I
363H 1 Q 1 CH3 Thiazolidine-3-carboxyl
364H 1 Q 1 CH3 4-[2-(2-Hydroxy-ethoxy)-ethyl]-
piperazine-1-carboxyl
365H 1 Q 1 CH3 N,N-Diethylformamidyl
366H 1 Q 1 CH3 1,4-Dioxa-8-aza-spiro[4.5]decane-8-
carbox I
367H 1 Q 1 CH3 1-Formyl-piperidine-4-carboxylic
acid
eth I ester
368H 1 Q 1 CN3 1,3,4,9-Tetrahydro-beta-carboline-2-
carbox I
369H 1 Q 1 CH3 4-Hydroxy-4-phenyl-piperidine-1-
carboxyl
370H 1 Q 1 CH3 N-Methyl-N-(naphthaien-1-ylmethyl)-
formamidyl
371H 1 Q 1 CHs 4-(4-Methoxy-phenyl)-piperazine-1-
carboxyl
372H 1 Q 1 CH3 2,3,5,6-Tetrahydro-[1,2']bipyrazinyl-4-
carbox I
373H 1 Q 1 CH3 1-Formyl-piperidine-3-carboxylic
acid
amide
374H 1 Q 1 CH3 N-Benzyl-N-phenethyl-formamidyl
375H 1 Q 1 CH3 N,N-Bis-(2-methoxy-ethyl)-formamidyl
376H 1 Q 1 CH3 4-(3-Trifluoromethyl-phenyl)-piperazine-
1-carbo I
377H 1 Q 1 CH3 3-Hydroxy-pyrrolidine-1-carboxylracemic
378H 1 Q 1 CH3 2-Methoxymethyl-pyrrolidine-1-carboxylS-isomer
379H 1 Q 1 CH3 4-Oxo-1-phenyl-1,3,8-triaza-
spiro[4.5]decane-8-carboxyl
380H 1 Q 1 CH3 4-(2-Fluoro-phenyl)-piperazine-1-
carbox I
381H 1 Q 1 CH3 4-Pyridin-4-yl-piperazine-1-carboxyl
382H 1 Q 1 CH3 4-Hydroxy-piperidine-1-carboxylracemic
383H 1 Q 1 CH3 N-Ethyl-N-(2-hydroxy-ethyl)-formamidyl
384H 1 Q 1 CH3 3-Hydroxy-piperidine-1-carboxylracemic
385H 1 Q 1 CH3 N-Methyl-N-propyl-formamidyl
386H 1 Q 1 CH3 2-(Formyl-methyl-amino)-benzoic
acid
meth I ester
387H 1 Q 1 CH3 N-(2-Dimethylamino-ethyl)-N-methyl-
formamid I
388H 1 Q 1 CH3 N-Methyl-N-phenethyl-formamidyl
389H 1 Q 1 CH3 N-Allyl-N-methyl-formamidyl
390H 1 Q 1 CH3 3,6-Dihydro-2H-pyridine-1-carboxyl
391H 1 Q 1 CH3 Pyrrolidine-1-carboxyl-2-carboxylicR-isomer
acid
amide
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
49
ExR~ m R2n R3, R4 stereochemistry
392H 1 Q 1 CH3 4-(2-Methoxy-phenyl)-piperazine-1-
carboxyl
393H 1 Q 1 CH3 N-Methyl-N-ethyl-formamidyl
394H 1 Q 1 CH3 4-Cyclohexyl-piperazine-1-carboxyl
395H 1 Q 1 CH3 N,N-Dimethyl-formamidyl
396H 1 Q 1 CH3 4-Pyrrolidin-1-yl-piperidine-1-carboxyl
397H 1 Q 1 CHs N,N-biphenyl-formamidyl
398H 1 Q 1 CH3 N-Methyl-N-phenyl-formamidyl
399H 1 Q 1 CH3 Formic acid phenyl
ester
400H 1 Q 1 CH3 Formic acid isobutyl
ester
401H 1 Q 1 CH3 Formic acid methyl
ester
402H 1 Q 1 CH3 Formic acid allyl ester
403H 1 Q 1 CH3 Formic acid (4-methoxyphenyl)
ester
404H 1 Q 1 CH3 Formic acid (2-methoxyethyl)
ester
405H 1 Q 1 CH3 Formic acid (2-ethylhexyl)racemic
ester
406H 1 Q 1 CH3 Formic acid propyl
ester
407H 1 Q 1 CH3 Formic acid (4-fluorophenyl)
ester
408H 1 Q 1 CH3 Formic acid (4-chlorophenyl)
ester
409H 1 Q 1 CH3 Formic acid (4-nitrobenzyl)
ester
410H 1 Q 1 CH3 Formic acid 2-isopropyl-5-methyl-( )
cyclohexyl ester - -Menthol
411H 1 Q 1 CH3 Formic acid (4-methylphenyl)
ester
412H 1 Q 1 CH3 Formic acid butyl ester
413H 1 Q 1 CHa Formic acid but-3-enyl
ester
414H 1 Q 1 CH3 Formic acid ethyl ester
415H 1 Q 1 CH3 Formic acid prop-2-ynyl
ester
416H 1 Q 1 CH3 Formic acid 2,2,2-trichloro-1,1-dimethyl-
ethyl ester
417H 1 Q 1 CH3 Formic acid (2-nitrophenyl)
ester
418H 1 Q 1 CH3 Formic acid 2,2,2-trichloro-ethyl
ester
419H 1 Q 1 CH3 Formic acid 2-isopropyl-5-methyl-(+)-Menthol
cyclohexyl ester
420H 1 Q 1 CH3 Naphthalene-1-sulfonyl
421H 1 Q 1 CH3 Thiophene-2-sulfonyl
422H 1 Q 1 CH3 Quinoline-8-sulfonyl
423H 1 Q 1 CH3 Biphenyl-4-sulfonyl
424H 1 Q 1 CH3 Naphthalene-2-sulfonyl
425H 1 Q 1 CH3 Benzenesulfonyl
426H 1 Q 1 CH3 4-Fluoro-benzenesulfonyl
427H 1 Q 1 CH3 4-iso-Propyl-benzenesulfonyl
428H 1 Q 1 CH3 4-Methanesulfonyl-benzenesulfonyl
429H 1 Q 1 CH3 4-Methoxy-benzenesulfonyl
430H 1 Q 1 CH3 2-Fluoro-benzenesulfonyl
431H 1 Q 1 CH3 3,4-Dimethoxy-benzenesulfonyl
432H 1 Q 1 CH3 3-Trifluoromethyl-benzenesulfonyl
433H 1 Q 1 CH3 2-Cyano-benzenesulfonyl
434H 1 Q 1 CH3 4-tert-Butyl-benzenesulfonyl
435H 1 Q 1 CH3 5-Dimethylamino-naphthalene-1-sulfonyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
ExR~ m Rzn R3 Rq stereochemistry
436H 1 Q 1 CH3 4-Chloro-benzenesulfonyl
437H 1 Q 1 CH3 4-Acetylamino-benzenesulfonyl
438H 1 Q 1 CH3 5-Chloro-thiophene-2-sulfonyl
439H 1 Q 1 CH3 4-Trifluoromethyl-benzenesulfonyl
440H 1 Q 1 CH3 Benzo[1,2,5]thiadiazoie-4-sulfonyl
441H 1 Q 1 CH3 2-Acetylamino-4-methyl-thiazole-5-
sulfonyl
442H 1 Q 1 CH3 4-Benzenesulfonyl-thiophene-2-sulfonyl
443H 1 Q 1 CH3 2,4,6-Trimethyl-benzenesulfonyl
444H 1 Q 1 CH3 2-Phenyl-ethenesulfonyl
445H 1 Q 1 CH3 2,5-Dimethoxy-benzenesulfonyl
446H 1 Q 1 CH3 3,4-Dichloro-benzenesulfonyl
447H 1 Q 1 CH3 2,4-Difluoro-benzenesulfonyl
448H 1 Q 1 GH3 4-Methyl-benzenesulfonyl
449H 1 Q 1 CHs 2-Chloro-benzenesulfonyl
450H 1 Q 1 CH3 3-Chloro-benzenesulfonyl
451H 1 Q 1 CH3 2,6-Dichloro-benzenesulfonyl
452H 1 Q 1 CH3 2,5-Dichloro-benzenesulfonyl
453H 1 Q 1 CH3 3-Nitro-benzenesulfonyl
454H 1 Q 1 CH3 Methanesulfonyl
455H 1 Q 1 CH3 4-Trifluoromethoxy-benzenesulfonyl
456H 1 Q 1 CH3 3-Methyl-benzenesulfonyl
457H 1 Q 1 CH3 4-Nitro-benzenesulfonyl
458H 1 Q 1 CH3 4-Propyl-benzenesulfonyl
459H 1 Q 1 CH3 2-Trifluoromethyl-benzenesulfonyl
460H 1 Q 1 CH3 4-Bromo-benzenesulfonyl"
461H 1 Q 1 CH3 5-Benzenesulfonyl-thiophene-2-sulfonyl
462H 1 Q 1 CH3 2-Methanesulfonyl-benzenesulfonyl
463H 1 Q 1 CH3 2-Bromo-benzenesulfonyl
464H 1 Q 1 CH3 4-Sulfamoyl-benzoic
acid
465H 1 Q 1 CH3 2-Nitro-benzenesulfonyl
466H 1 Q 1 CH3 3,5-Dichloro-benzenesulfonyl
467H 1 Q 1 CH3 4,5-Dichloro-thiophene-2-sulfonyl
468H 1 Q 1 CH3 3-Bromo-benzenesulfonyl
469H 1 Q 1 CH3 4-Butoxy-benzenesulfonyl
470H 1 Q 1 CH3 4-Methyl-benzenesulfonyl
471H 1 Q 1 CH3 2,4-Dichloro-thiophene-2-sulfonyl
472H 1 Q 1 CN3 4-(1,1-Dimethyl-propyl-benzenesulfonyl
473H 1 Q 1 CH3 2-Methyl-5-nitro-benzenesulfonyl
474H 1 Q 1 CH3 3,5-Bis-trifluoromethyl-benzenesulfonyl
475H 1 Q 1 CH3 4-Ethyl-benzenesulfonyl
476H 1 Q 1 CH3 2,5-Dichloro-thiophene-3-sulfonyl
477H 1 Q 1 CH3 5-Bromo-2-methoxy-benzenesulfonyl
478H 1 Q 1 CH3 2-Chloro-4-fluoro-benzenesulfonyl
479H 1 Q 1 CH3 5-Fluoro-2-methyl-benzenesulfonyl
480H 1 Q 1 CH3 4-(4-Dimethylamino-phenylazo)
-
benzenesulfon I
481H 1 Q 1 CH3 2,4-Dinitro-benzenesulfonyl
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
51
ExR~ m RZn R3 R4 stereochemistry
482H 1 Q 1 CH3 4,5-Dibromo-thiophene-2-sulfonyl
483H 1 Q 1 CH3 4-Bromo-2,5-dichloro-thiophene-3-
sulfon I
484H 1 Q 1 CH3 2,3-Dichloro-benzenesulfonyl
485H 1 Q 1 CH3 2,4,6-Trichloro-benzenesulfonyl
486H 1 Q 1 CH3 2-Chloro-6-methyl-benzenesulfonyl
487H 1 Q 1 CH3 2,4,6-Triisopropyl-benzenesulfonyl
488H 1 Q 1 CH3 5-Chloro-3-methyl-benzo[b]thiophene-2-
sulfonyl
(1 RZ = (2-methoxy-ethoxymethyl)
)*
(2)* R~ = 4-Hydroxy-piperidin-4-ylmethyl
(3)* RZ = 1-Benzyl-piperidin-4-ylmethyl
RZ = Piperidin-4-ylmethyl
(4)*
(5)* R2 = 4-benzyl-morpholin-2-ylmethyl
(6)* R~ = acetyl
(7)* R~ = 3-aminobenzyl
Analytical data of the examples given in the table above, are given in the
table
below. Details of the analytical methods listed in th is table, viz. "BASIS",
"STANDARD", "CURVE 4", "AMAP 2" and "AMAP 3" are explained below.
Ex Mol Formula Mo1 Melting(MH+) Ret. Method
Wgt pt. Time
1 C24 H35 N3 381.560- 382 7.630 BASIS
0
2 C24 H35 N3 381.560187-189C-
0
3 C24 H35 N3 381.560180.9C - - -
0
4 C24 H35 N3 381.560187.9C - - -
0
5 C24 H34 F N3 399.551189-191C- - -
0
6 C25 H37 N3 411.586156-157C- - -
02
7 C24 H34 CI 416.006144-145C- - -
1 N3 0
8 C25 H37 N3 411.586152-153C- - -
02
9 C24 H34 F N3 399.551198-199C- - -
O
10 C25 H34 F3 449.558174-176C- - -
N3 0
11 C28 H43 N3 469.666- 470 3.990 CURVE
03 4
12 C27 H42 N4 438.656 439 6.500 BASIS
0
13 C28 H44 N4 452.683 453 3.340 CURVE
0 4
14 C29 H46 N4 466.709 467 6.870 BASIS
0
C30 H48 N4 480.736 481 7.030 BASIS
0
16 C31 H50 N4 494.763 495 7.130 BASIS
0
17 C34 H48 N4 528.780 529 1.195 AMAP
0 2
18 C33 H53 N5 551.815 552 1.135 AMAP
02 2
19 C35 H48 N4 572.789 573 3.580 CURVE
03 4
C34 H49 N5 607.859 608 3.430 STANDARD
03 S
21 C29 H46 N4 466.709 467 6.730 BASIS
0
22 C29 H46 N4 466.709 467 3.310 CURVE
O 4
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
52
ExMoi Formula Moi Melting(MH+) Ret. Method
Wgt pt. Time
23C35 H48 F2 578.787 579 9.530 BASIS
N4 0
24C34 H50 N5 544.803 545 3.450 CURVE
0 4
25C34 H49 N5 543.795 544 3.940 CURVE
O 4
26C34 H49 N5 543.795 545 3.770 STANDARD
O
27C40 H60 N4 644.939 645 3.980 CURVE
03 4
28C35 H54 N4 578.837 579 5.350 STANDARD
03
29C34 H54 N4 550.827 551 7.000 BASIS
02
30C34 H54 N4 550.827 551 7.000 BASIS
02
31C34 H54 N4 550.827 551 3.340 CURVE
02 4
32C36 H58 N4 564.853 565 3.350 CURVE
02 4
33C35 H56 N4 578.889 579 3.390 CURVE
02 4
34C37 H60 N4 592.907 593 3.460 CURVE
02 4
35C39 H63 N5 665.958 666 3.690 CURVE
04 4
36C33 H53 N5 551.815 552 4.700 CURVE
02 4
37C34 H55 N5 565.841 566 3.090 CURVE
02 4
38C35 H57 N5 579.868 580 3.140 CURVE
02 4
39C36 H59 N5 593.895 594 3.100 CURVE
02 4
40C45 H63 N5 706.026 706 3.810 CURVE
02 4
41C37 H61 N5 607.922 608 4.460 STANDARD
02
42C42 H63 N5 685.992 686 3.540 CURVE
03 4
43C38 H61 N5 651.931 652 3.480 CURVE
04 4
44C36 H57 N5 623.877 624 3.150 CURVE
04 4
45C35 H57 N5 579.868 580 4.480 STANDARD
02
46C35 H57 N7 607.882 608 3.190 CURVE
02 4
47C36 H57 N5 607.878 608 3.630 CURVE
03 4
48C37...:H60 636.920 637 3.330 CURVE.4.:
N6 03
49C34 H50 N4 594.860 595 8.230 BASIS
03 S
50C36 H54 N4 654.912 655 3.690 CURVE
05 S 4
59C34 H50 N4 578.793 579 8.200 8ASI5
04
52C37 H56 N4 652.871 653 7.870 BASIS
06
53C36 H54 N4 638.845 639 7.870 BASIS
06
54C36 H54 N4 638.845 639 7.830 BASIS
06
55C37 H56 N4 66$.870 669 3.530 CURVE
07 4
56C37 H56 N4 684.937 685 2.720 CURVE
06 S 4
57C36 H54 N4 654.912 655 2.740 CURVE
05 S 4
58C36 H54 N4 654.912 655 3.690 CURVE
05 S 4
59C37 H56 N4 668.938 669 7.370 BASIS
05 S
60C38 H54 N4 598.871 599 7.370 BASIS
02
61C38 H50 CI1 687.286 687 4.600 CURVE
F3 N4 02 4
62C38 H52 N4 580.856 581 8.700 BASIS
O
63C37 H52 CI1 618.305 618 4.820 CURVE
N5 0 4
64C37 H53 N5 583.860 584 4.340 CURVE
O 4
65C37 H52 F1 601.850 602 4.360 CURVE
N5 0 4
66C33 H52 N4 536.800 537 3.260 CURVE
02 4
67C33 H51 N5 549.799 550 3.270 CURVE
02 4
68C30 H46 N4 494.719 495 4.460 STANDARD
02
69C37 H52 N4 568.845 569 5.210 STANDARD
0
70C30 H46 N4 478.720 479 4.58 STANDARD
0
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
53
ExMol Formula Mol Melting(MH+) Ret. Method
Wgt pt. Time
71C36 H60 N6 608.910 609 3.200 CURVE
02 4
72C40 H59 CI1 691.399 691 4.350 CURVE
N6 02 4
73C39 H59 N7 657.942 658 3.630 CURVE
02 4
74C41 H62 N6 670.981 671 3.560 CURVE
02 4
75C40 H60 N6 656.954 657 3.930 CURVE
02 4
76C37 H62 N6 622.937 623 3.180 CURVE
02 4
77C37 H62 N6 622.937 623 3.010 CURVE
02 4
78C41 H61 CI1 705.426 705 3.800 CURVE
N6 02 4
79C40 H61 N7 671.969 ' 672 3.400 CURVE
02 4
80C42 H64 N6 685.008 686 3.300 CURVE
02 4
81C41 H62 N6 670.981 671 3.590 CURVE
02 4
82C38 H64 N6 636.964 638 3.050 CURVE
02 4
83C33 H47 N5 529.768 530 4.370 CURVE
O 4
84C34 H48 N4 528.780 529 3.560 CURVE
0 4
85C36 H50 N4 570.817~ 571 4.470 CURVE
02 4
86C26 H37 N3 439.596 440 4.750 STANDARD
03
87C31 H42 N4 486.700 487 5.900 CURVE
0 4
88C35 H54 N4 578.837 579 4.280 CURVE
03 4
89C35 H48 N4 572.789 573 3.970 CURVE
03 4
90C37 H52 N4 632.841 633 5.760 STANDARD
05
91C34 H56 N4 616.838 617 4.500 STANDARD
06
92C31 H50 N4 526.761 528 3.340 STANDARD
03
93C35 H57 N5 595.867 596 3.240 CURVE
03 4
94C34 H56 N4 568.841 569 3.430 CURVE
03 4
95C30 H46 N4 510.723 , x 5.360 STANDARD
03 ,
511
96C35 H46 N4 586.772 587 5.840 CURVE
04 4
97C36 H44 F6 678.762 679 7.060 STANDARD
N4 02
98C34 H46 N4 542.768 543 6.000 STANDARD
02
99C34 H45 Br1 621.664 621 6.290 STANDARD
N4 02
100C34 H41 F5 632.718 633 6.500 STANDARD
N4 02
101C34 H44 CI2 611.658 611 6.610 STANDARD
N4 02
102C35 H48 N4 572.794 573 5.960 STANDARD
03
103C35 H45 F3 610.765 611 6.400 STANDARD
N4 02
104C35 H48 N4 556.795 557 6.140 STANDARD
02
105C34 H45 F1 560.758 561 6.100 STANDARD
N4 02
106C34 H45 CI1 577.213 577 6.330 STANDARD
N4 02
107C34 H44 CI2 611.658 611 6.690 STANDARD
N4 02
108C35 H48 N4 572.794 573 6.070 STANDARD
03
109C35 H48 N4 572.794 573 6.020 STANDARD
03
110C34 H45 F1 560.758 561 6.350 STANDARD
N4 02
111C34 H45 CI1 577.213 577 5.980 STANDARD
N4 02
112C35 H48 N4 572.794 573 7.680 STANDARD
03
113C40 H58 N4 642.929 643 6.490 STANDARD
03
114C35 H45 F3 610.765 611 6.890 STANDARD
N4 02
115C38 H54 N4 598.876 599 6.210 STANDARD
02
116C35 H48 N4 556.795 557 5.750 STANDARD
02
117C30 H44 N4 524.706 525 5.950 STANDARD
04
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
54
ExMol Formula Mol Melting(MH+) Ret. ~ Method
Wgt pt. Time
118C33 H50 N4 566.787 567 6.150 STANDARD
04
119C32 H50 N4 522.778 523 6.090 STANDARD
02
120C37 H50 N4 614.831 615 6.080 STANDARD
04
121C35 H48 N4 556.795 557 6.090 STANDARD
02
122C34 H45 CI2 626.669 626 1.606 AMAP
N5 02 2
123C31 H47 N5 521.745 522 1.403 AMAP
02 2
124C34 H53 N5 595.824 596 1.460 AMAP
04 2
125C39 H55 N5 657.894 658 1.536 AMAP
04 2
126C35 H46 F3 641.774 642 1.568 AMAP
N5 03 2
127C33 H53 N5 551.815 552 1.494 AMAP
02 2
128C34 H53 N5 595.824 596 1.451 AMAP
04 2
129C34 H46 Br 636.674 636 1.536 AMAP
N5 02 2
130C36 H51 N5 601.831 602 1.553 AMAP
03 2
131C37 H51 N5 629.841 630 1.616 AMAP
04 2
132C36 H51 N5 585.832 586 1.556 AMAP
02 2
133C34 H45 CI2 626.669 626 1.583 AMAP
N5 02 2
134C34 H45 CI2 626.669 626 1.597 AMAP
N5 02 2
135C34 H46 Br 636.674 636 1.552 AMAP
N5 02 2
136C32 H48 N4 520.762 521 5.840 STANDARD
02
137C33 H52 N4 536.805 537 6.310 STANDARD
02
138C30 H46 N4 494.724 495 5.630 STANDARD
02
139C36 H50 N4 570.822 571 6.330 STANDARD
02
140C29 H41 CI3 584.032 583 6.540 STANDARD
N4 02
141C29 H42 CI2 549.587 549 5.980 STANDARD
N4 02
142C37 H50 N4 582.833 583 6.380 STANDARD
Q2
143C35 H54 N4 562.843 563 6.700 STANDARD
02
144C34 H52 N4 548.816 549 6.310 STANDARD
02
145C32 H44 N4 532.729 533 5.710 STANDARD
03
146C33 H46 N4 562.817 563 6.020 STANDARD
02 S1
147C41 H48 N4 644.860 645 6.420 STANDARD
03
148C36 H50 N4 586.821 587 6.150 STANDARD
03
149C31 H46 N4 538.733 539 5.510 STANDARD
04
150C33 H45 N5 543.756 544 5.340 STANDARD
02
151C41 H52 N4 632.893 633 6.760 STANDARD
02
152C31 H46 N4 538.733 539 5.540 STANDARD
04
153C43 H64 N4 543.196 543 6.140 STANDARD
02
154C31 H47 CI1 543.196 543 6.270 STANDARD
N4 02
155C33 H51 CI1 571.250 571 7.540 STANDARD
N4 02
156C39 H53 CI1 645.332 645 6.440 STANDARD
N4 02
157C37 H52 N4 600.848 601 6.510 STANDARD
03
158C36 H46 N4 598.850 599 6.460 STANDARD
02 S1
159C35 H45 F3 626.764 627 6.630 STANDARD
N4 03
160C37 H49 N7 623.846 624 6.410 STANDARD
02
161C33 H43 CI2 612.646 612 6.460 STANDARD
N5 02
162C36 H51 N5 617.897 618 6.380 STANDARD
02 S1
163C33 H43 CI2 612.646 612 6.440 STANDARD
N5 02
164C38 H47 CI1 676.277 676 6.230 STANDARD
F1 N5 03
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
ExMol Formula Mol Melting (MH+) Ret. Method
Wgt pt. Time
165C34 H43 F3 N4 596.738 597 7.000 STANDARD
02
166C37 H49 F3 N4 654.818 655 6.480 STANDARD
03
167C40 H51 N5 02 665.941 666 6.390 STANDARD
S1
168C39 H48 CI1 670.298 670 6.370 STANDARD
N5 03
169C33 H45 CI1 613.261 613 6.490 STANDARD
N4 03 S1
170C38 H47 F3 N6 676.828 677 7.210 STANDARD
02
171C38 H56 N4 02 600.892 601 6.560 STANDARD
172C37 H47 F3 N4 636.803 637 6.180 STANDARD
02
173C36 H54 N6 02 602.868 603 6.300 STANDARD
174C36 H54 N6 02 602.868 603 6.400 STANDARD
175C34 H46 CI1 608.227 608 7.270 STANDARD
N5 03
176C37 H51 CI1 635.293 635 6.300 STANDARD
N4 03
177C37 H54 N4 04 618.863 619 6.730 STANDARD
178C30 H48 N4 03 512.734 513 3.280 CURVE
4
179C35 H50 N4 02 558.806 559 4.140 CURVE
4
180C30 H48 N4 02 496.735 497 3.450 CURVE
4
181C30 H47 F N4 514.725 515 3.710 CURVE
02 4
182C36 H51 CI N4 623.277 623 4.410 CURVE
03 4
183C37 H54 N4 04 618.858 619 4.090 CURVE
4
184C40 H60 N4 03 644.939 645 4.980 CURVE
4
185C33 H54 N4 03 554.815 555 3.810 CURVE
4
186C38 H64 N4 03 624.949 626 5.530 CURVE
4
187C33 H52 N4 03 552.799 553 3.800 CURVE
4
188C34 H56 N4 03 568.841 569 4.110 CURVE
4
189C31 H48 N4 02 508.746 509 3.730 CURVE
-~: 4
190C31 H50 N4 02 510.762 511 3.570 CURVE
4
191C35 H56 N4 02 564.853 565 4.390 CURVE
4
192C35 H58 N4 02 566.869 567 4.740 CURVE
4
193C38 H51 N5 04 641.852 642 3.980 CURVE
4
194C34 H56 N4 03 568.841 569 3.930 CURVE
4
195C33 H52 N4 02 536.800 537 3.960 CURVE
4
196C38 H54 N4 05 646.868 647 4.180 CURVE
4
197C35 H52 N4 04 592.820 593 3.890 CURVE
4
198C30 H45 F3 N4 550.706 551 4.150 CURVE
02 4
199C32 H48 F4 N4 612.748 613 4.150 CURVE
03 4
200C34 H55 N5 03 581.841 582 3.400 CURVE
4
201C37 H60 N4 02 592.907 593 5.090 CURVE
4
202C36 H52 N4 02 572.833 573 4.060 CURVE
4
203C32 H50 N4 02 522.773 523 4.010 CURVE
4
204C32 H50 N4 04 554.771 555 4.020 CURVE
4
205C41 H61 CI N6 705.425 706 1.344 AMAP
02 2
206C33 H54 N4 02 538.815 539 1.414 AMAP
2
207C38 H54 N4 04 630.868 631 1.487 AMAP
2
208C30 H48 N4 03 512.734 513 1.282 AMAP
2
209C39 H66 N4 02 622.976 623 1.713 AMAP
2
210C41 H70 N4 02 651.030 652 1.768 AMAP
2
211C31 H50 N4 03 526.761 527 1.318 AMAP
2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
56
ExMol Formula Mol Melting (MH+) Ret. Method
Wgt pt. Time
212C43 H74 N4 02 679.083 680 1.850 AMAP
2
213C45 H78 N4 02 707.137 708 1.921 AMAP
2
214C32 H50 N4 02 522.773 523 1.327 AMAP
2
215C38 H54 N4 05 646.867 647 1.414 AMAP
2
216C35 H54 N4 02 562.837 563 1.374 AMAP
2
217C41 H54 N4 02 634.903 635 1.483 AMAP
2
218C40 H52 N4 O 604.878 605 1.315 AMAP
2
219C38 H50 N4 0 578.840 579 1.251 AMAP
2
220C40 H52 N4 02 620.877 621 1.307 AMAP
2
221C40 H52 N4 O 604.878 605 1.332 AMAP
2
222C38 H50 N4 0 578.840 579 1.296 AMAP
2
223C37 H51 N5 0 581.844 582 1.216 AMAP
2
224C33 H47 N5 0 529.768 530 1.171 AMAP
2
225C35 H47 F3 N4 596.777 597 1.310 AMAP
O 2
226C33 H47 N5 0 529.768 530 1.136 AMAP
2
227C31 H48 N4 O 492.747 493 1.153 AMAP
2
228C35 H47 CI1 607.234 607 1.273 AMAP
N4 03 2
229C35 H47 F3 N4 612.776 613 1.298 AMAP
02 2
230C36 H50 N4 02 570.817 571 1.212 AMAP
2
231C35 H56 N4 0 548.854 549 1.255 AMAP
2
232C38 H56 N4 0 584.887 585 1.315 AMAP
2
233C35 H50 N4 02 558.806 559 1.217 AMAP
2
234C35 H47 N5 0 553.790 554 1.245 AMAP
2
235C33 H49 N5 02 547.783 548 1.187 AMAP
2
236C35 H50 N4 03 606.871 607 1.305 AMAP
S .. 2
237C35 H50 N4 0 542.807 543 1.212 AMAP
2
238C37 H50 N6 03 626.841 627 1.209 AMAP
2
239C34 H47 F N4 546.770 547 1.214 AMAP
0 2
240C41 H5,4 N4 634.904 635 1.307 AMAP
02 2
241C34 H47 CI1 563.225 563 1.251 AMAP
N4 O 2
242C48 H60 N4 03 741.027 741 1.369 AMAP
2
243C35 H47 F3 N4 612.776 613 1.321 AMAP
02 2
244C32 H52 N4 0 508.790 509 1.192 AMAP
2
245C36 H52 N4 0 556.834 557 1.228 AMAP
2
246C30 H47 N5 02 509.734 510 1.099 AMAP
2
247C32 H46 N6 03 562.754 563 1.273 AMAP
2
248C37 H54 N4 02 586.860 587 1.228 AMAP
2
249C32 H45 CI1 569.253 569 1.339 AMAP
N4 O S 2
250C31 H48 N4 03 524.745 525 1.144 AMAP
2
251C36 H52 N4 0 556.834 557 1.237 AMAP
2
252C29 H43 N5 0 477.693 478 1.392 AMAP
2
253C34 H47 F N4 546.770 547 1.221 AMAP
0 2
254C35 H47 F3 N4 596.777 597 1.301 AMAP
O 2
255C35 H47 N5 0 553.790 554 1.377 AMAP
2
256C32 H52 N4 0 508.790 509 1.197 AMAP
2
257C29 H46 N4 02 482.708 483 1.103 AMAP
2
258C34 H47 CI1 563.225 563 1.268 AMAP
N4 O 2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
57
ExMol Formula Mol Melting(MH+) Ret. Method
Wgt pt. Time
259C42 H52 N4 628.900 629 1.430 AMAP
0 2
260C35 H50 N4 542.807 543 1.229 AMAP
0 2
261C34 H47 Br 607.676 607 1.258 AMAP
N4 0 2
262C35 H50 N4 542.807 543 1.207 AMAP
0 2
263C35 H47 N5 553.790 554 1.244 AMAP
0 2
264C35 H48 N4 556.790 557 1.226 AMAP
02 2
265C29 H45 N5 495.707 496 1.106 AMAP
02 2
266C37 H52 N4 616.842 617 1.235 AMAP
04 2
267C39 H58 N4 614.913 615 1.298 AMAP
02 2
268C38 H52 N4 628.853 629 1.231 AMAP
04 2
269C41 H52 N4 632.888 633 1.435 AMAP
02 2
270C31 H50 N4 494.763 495 1.164 AMAP
0 2
271C42 H54 N4 630.916 631 1.337 AMAP
0 2
272C36 H52 N4 572.833 573 1.227 AMAP
02 2
273C34 H47 F N4 546.770 547 1.199 AMAP
0 2
274C35 H50 N4 558.806 559 1.209 AMAP
02 2
275C33 H47 N5 529.768 530 1.167 AMAP
0 2
276C35 H50 N4 558.806 559 1.202 AMAP
02 2
277C40 H51 N5 649.875 650 1.579 AMAP
03 2
278C35 H49 N5 571.805 572 1.441 AMAP
02 2
279C40 H51 N5 633.876 634 1.594 AMAP
02 2
280C40 H51 N5 633.876 634 1.595 AMAP
02 2
281C35 H49 N5 587.804 588 1.503 AMAP
03 2
282C34 H46 F N5 575.768 576 1.479 AMAP
02 2
283C35 H46 N6 582.788 583 .:1.434AMAP
02 2
284C38 H57 N5 615.901 616 1.599 AMAP
02 2
285C34 H46 F N5 575.768 576 1.465 AMAP
02 2
286C35 H49 N5 587.804 588 1.449 AMAP
03 2
287C35 H46 N6 582.788 583 1.456 AMAP
02 2
288C37 H51 N5 629.841 630 1.516 AMAP
04 2
289C36 H51 N5 585.832 586 1.484 AMAP
02 2
290C40 H53 N5 635.892 636 1.564 AMAP
02 2
291C34 H45 CI2 626.669 626 1.465 AMAP
N5 02 2
292C34 H46 CI1 592.223 592 1.524 AMAP
N5 02 2
293C34 H46 CI1 592.223 592 1.522 AMAP
N5 02 2
294C32 H51 N5 537.788 538 1.467 AMAP
02 2
295C36 H51 N5 585.832 586 1.490 AMAP
02 2
296C32 H51 N5 537.788 538 1.451 AMAP
02 2
297C37 H53 N5 647.856 648 1.417 AMAP
05 2
298C36 N51 N5 617.830 618 1.482 AMAP
04 2
299C35 H47 N5 585.788 586 1.429 AMAP
03 2
300C31 H49 N5 523.761 524 1.390 AMAP
02 2
301C34 H46 N6 602.775 603 1.464 AMAP
04 2
302C35 H49 N5 603.871 604 1.532 AMAP
02 S 2
303C32 H49 N5 567.770 568 1.392 AMAP
04 2
304C34 H46 Br 636.674 636 1.535 AMAP
N5 02 2
305C38 H55 N5 613.885 614 1.650 AMAP
02 2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
58
ExMol Formula Mol Melting(MH+) Ret. Method
Wgt pt. Time
306C38 H53 N5 643.868 644 1.498 AMAP
04 2
307C36 H51 N5 617.830 618 1.510 AMAP
04 2
308C35 H49 N5 571.805 572 1.459 AMAP
02 2
309C36 H51 N5 585.832 586 1.472 AMAP
02 2
310C34 H45 CI2 626.669 626 1.578 AMAP
N5 02 2
311C37 H51 N5 629.841 630 1.501 AMAP
04 2
312C34 H46 N6 602.775 603 1.489 AMAP
04 2
313C36 H45 F6 693.773 694 1.660 AMAP
N5 02 2
314C37 H53 N5 599.859 600 1.509 AMAP
02 2
315C39 H55 N5 657.894 658 1.591 AMAP
04 2
316C33 H51 N5 581.797 582 1.395 AMAP
04 2
317C36 N59 N5 593.895 594 1.631 AMAP
02 2
318C34 H45 F2 593.758 594 1.465 AMAP
N5 02 2
319C34 H45 CI2 626.669 626 1.606 AMAP
N5 02 2
320C35 H49 N5 571.805 572 1.497 AMAP
02 2
321C31 H47 N5 521.745 522 1.403 AMAP
02 2
322C34 H53 N5 595.824 596 1.460 AMAP
04 2
323C39 H55 N5 657.894 658 1.536 AMAP
04 2
324C35 H46 F3 641.774 642 1.568 AMAP
N5 03 2
325C33 H53 N5 551.815 552 1.494 AMAP
02 2
326C34 H53 N5 595.824 596 1.451 AMAP
04 2
327C34 H46 Br 636.674 636 1.536 AMAP
N5 02 2
328C36 H51 N5 601.831 602 1.553 AMAP
03 2
329C37 H51 N5 629.841 630 1.616 AMAP
04 2 .
330C36 H51 N5 ,585.832 586 1.556 AMAP
02 2
331C34 H45 CI2 626.669 626 1.583 AMAP
N5 02 2
332C34 H45 CI2 626.669 626 1.597 AMAP
N5 02 2
333C34 H46 Br 636.674 636 1.552 AMAP
N5 02 2
334C40 H59 N5 641.939 642 1.601 AMAP
02 2
335C31 H47 N5 553.743 554 1.363 AMAP
04 2
336C36 .H51 N5 585.832 586 1.509 AMAP
02 2
337C35 H48 CI1 622.249 622 1.551 AMAP
N5 03 2
338C35 H45 CI1 660.221 660 1.614 AMAP
F3 N5 02 2
339C35 H45 CI1 660.221 660 1.648 AMAP
F3 N5 02 2
340C36 H51 N5 601.831 602 1.475 AMAP
03 2
341C34 H45 CI1 637.221 637 1.596 AMAP
N6 04 2
342C38 H55 N5 613.885 614 1.539 AMAP
02 2
343C35 H48 CI1 606.250 606 1.481 AMAP
N5 02 2
344C36 H50 Br 664.728 664 1.543 AMAP
N5 02 2
345C36 H57 N5 623.877 624 1.470 AMAP
04 2
346C33 H51 N5 581.797 582 1.421 AMAP
04 2
347C34 H45 N7 647.773 648 1.575 AMAP
06 2
348C40 H56 N6 684.920 685 1.169 AMAP
04 2
349C40 H55 N7 681.920 682 1.348 AMAP
03 2
350C37 H51 N5 597.843 598 1.476 AMAP
02 2
351C32 H47 N5 533.756 534 1.372 AMAP
02 2
352C38 H54 N6 626.885 627 1.456 AMAP
02 2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
59
ExMol Formula Mol Melting(MH+) Ret. Method
Wgt pt. Time
353C32 H49 N5 551.771 552 1.320 AMAP
03 2
354C37 H53 N7 627.873 628 1.229 AMAP
02 2
355C36 H52 N6 600.847 601 1.220 AMAP
02 2
356C32 H49 N5 535.772 536 1.357 AMAP
02 2
357C40 H55 N5 669.906 670 1.497 AMAP
04 2
358C38 H53 F N6 644.875 645 1.480 AMAP
02 2
359C39 H56 N6 656.910 657 1.439 AMAP
03 2
360C39 H54 CI1 676.341 676 1.431 AMAP
N 5 03 2
361C39 H53 F3 694.882 695 1.563 AMAP
N6 02 2
362C39 H55 CI1 675.357 676 1.236 AMAP
N6 02 2
363C31 H47 N5 553.811 554 1.374 AMAP
02 S 2
364.C36 H58 N6 638.892 639 1.104 AMAP
04 2
365C32 H51 N5 537.788 538 1.414 AMAP
02 2
366C35 H53 N5 607.835 608 1.386 AMAP
04 2
367C36 H55 N5 621.862 622 1.409 AMAP
04 2
368C39 H52 N6 636.880 637 1.466 AMAP
02 2
369C39 H55 N5 641.896 642 1.388 AMAP
03 2
370C40 H53 N5 635.892 636 1.525 AMAP
02 2
371C39 H58 N6 656.910 657 1.423 AMAP
03 2
372C36 H52 N8 628.861 629 1.332 AMAP
02 2
373C34 H52 N6 592.824 593 1.263 AMAP
03 2
374C43 H57 N5 675.956 676 1.616 AMAP
02 2
375C34 H55 N5 597.839 598 1.355 AMAP
04 2
376C39 H53 F3 694.882 695 1.557 AMAP
N6 02 2
377C32 H49 N5 551.771 552 1.271.AMAP
03 2
378C34 H53 N5 579.825 580 1.368 AMAP
03 2
379C41 H57 N7 695.947 695 1.398 AMAP
03 2
380C38 H53 F N6 644.875 645 1.486 AMAP
02 2
381C37 H53 N7 627.873 628 1.145 AMAP
02 2
382C33 H51 N5 565.798 566 1.258 AMAP
03 2
383C32 H51 N5 553.787 554 1.301 AMAP
03 2
384C33 H51 N5 565.798 566 1.272 AMAP
03 2
385C32 H51 N5 537.788 538 1.410 AMAP
02 2
386C37 H51 N5 629.841 630 1.452 AMAP
04 2
387C33 H54 N6 566.830 566 1.122 AMAP
02 2
388C37 H53 N5 599.859 600 1.484 AMAP
02 2
389C32 H49 N5 535.772 536 1.397 AMAP
02 2
390C33 H49 N5 547.783 548 1.393 AMAP
02 2
391C33 H50 N6 578.797 579 1.256 AMAP
03 2
392C40 H58 N6 670.937 671 1.488 AMAP
03 2
393C31 H49 N5 523.761 524 1.378 AMAP
02 2
394C38 H60 N6 632.932 633 1.163 AMAP
02 2
395C30 H47 N5 509.734 510 1.331 AMAP
02 2
396C37 H58 N6 618.905 619 1.123 AMAP
02 2
397C34 H45 F2 593.758 594 1.465 AMAP
N5 02 2
398C35 H49 N5 571.805 572 1.497 AMAP
02 2
399C34 H46 N4 558.762~ 559 1.985 AMAP
03 ~ I 3
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
ExMol Formula Mol Melting(MH+) Ret. Method
Wgt pt. Time
400C32 H50 N4 538.772 539 2.010 AMAP
03 3
401C29 H44 N4 496.692 497 1.914 AMAP
03 3
402C31 H46 N4 522.729 523 1.950 AMAP
03 3
403C35 H48 N4 588.788 589 1.976 AMAP
04 3
404C31 H48 N4 540.744 541 1.885 AMAP
04 3
405C36 H58 N4 594.879 595 2.206 AMAP
03 3
406C31 H48 N4 524.745 525 1.966 AMAP
03 3
407C34 H45 F N4 576.753 577 2.014 AMAP
03 3
408C34 H45 CI 593.208 593 2.036 AMAP
N4 03 3
409C35 H47 N5 617.786 618 1.981 AMAP
05 3
410C38 H60 N4 620.917 621 2.249 AMAP
03 3
411C35 H48 N4 572.789 573 2.040 AMAP
03 3
412C32 H50 N4 538.772 539 2.002 AMAP
03 3
413C32 H48 N4 536.756 537 1.995 AMAP
03 3
414C30 H46 N4 510.718 511 1.918 AMAP
03 3
415C31 H44 N4 520.714 521 1.919 AMAP
03 3
416C32 H47 CI3 642.107 641 2.137 AMAP
N4 03 3
417C34 H45 N5 603.760 604 2.024 AMAP
05 3
418C30 H43 CI3 614.054 613 2.043 AMAP
N4 03 3
419C38 H60 N4 620.917 621 2.280 AMAP
03 3
420C37 H48 N4 628.877 629 1.519 AMAP
03 S 2
421C31 H44 N4 584.846 585 1.417 AMAP
03 S2 2
422C36 H47 N5 629.865 630 1.428 AMAP
03 S 2
423C39 H50 N4 654.915 655 1.576 AMAP
03 S 2
424C37 H48 N4 628.877 629 1.524 AMAP
03 S 2
425C33 H46 N4 578.817 579 1.427 AMAP
03 S 2
426C33 H45 F N4 596.807 597 1.439 AMAP
03 S 2
427C36 H52 N4 620.898 621 1.571 AMAP
03 S 2
428C34 H48 N4 656.908 657 1.354 AMAP
05 S2 2
429C34 H48 N4 608.843 609 1.409 AMAP
04 S 2
430C33 H45 F N4 596.807 597 1.426 AMAP
03 S 2
431C35 H50 N4 638.869 639 1.375 AMAP
05 S 2
432C34 H45 F3 646.815 647 1.489 AMAP
N4 03 S 2
433C34 H45 N5 603.828 604 1.411 AMAP
03 S 2
434C37 H54 N4 634.925 635 1.593 AMAP
03 S 2
435C39 H53 N5 671.946 672 1.575 AMAP
03 S 2
436C33 H45 CI1 613.263 613 1.502 AMAP
N4 03 S 2
437C35 H49 N5 635.869 636 1.327 AMAP
04 S 2
438C31 H43 CI1 619.291 619 1.504 AMAP
N4 03 S2 2
439C34 H45 F3 646.815 647 1.494 AMAP
N4 03 S 2
440C33 H44 N6 636.882 637 1.433 AMAP
03 S2 2
441C33 H48 N6 656.912 657 1.332 AMAP
04 S2 2
442C37 H48 N4 725.007 725 1.467 AMAP
05 S3 2
443C36 H52 N4 620.898 621 1.545 AMAP
03 S 2
444C35 H48 N4 604.855 605 1.479 AMAP
03 S 2
445C35 H50 N4 638.869 639 1.419 AMAP
05 S 2
446C33 H44 CI2 647.708 647 1.563 AMAP
N4 03 S 2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
61
ExMol Formula Mol Melting (MH+) Ret. Method
Wgt pt. Time
447C33 H44 F2 N4 614.798 615 1.454 AMAP
03 S 2
448C34 H48 N4 03 592.844 593 1.465 AMAP
S 2
449C33 H45 CI1 613.263 613 1.474 AMAP
N4 03 S 2
450C33 H45 CI1 613.263 613 1.474 AMAP
N4 03 S 2
451C33 H44 CI2 647.708 647 1.480 AMAP
N4 03 S 2
452C33 H44 CI2 647.708 647 1.541 AMAP
N4 03 S 2
453C33 H45 N5 05 623.815 624 1.418 AMAP
S 2
454.C28 H44 N4 03 516.747 517 1.280 AMAP
S 2
455C34 H45 F3 N4 662.813 663 1.530 AMAP
04 S 2
456C34 H48 N4 03 592.844 593 1.465 AMAP
S 2
457C33 H45 N5 05 623.815 624 1.434 AMAP
S 2
458C36 H52 N4 03 620.898 621 1.584 AMAP
S 2
459C34 H45 F3 N4 646.815 647 1.499 AMAP
03 S 2
460C33 H45 Br N4 657.714 657 1.524 AMAP
03 S 2
461C37 H48 N4 05 725.007 725 1.498 AMAP
S3 2
462C34 H48 N4 05 656.908 657 1.372 AMAP
S2 2
463C33 H45 Br N4 657.714 657 1.488 AMAP
03 S 2
464C34 H46 N4 05 622.826 623 1.361 AMAP
S 2
465C33 H45 N5 05 623.815 624 1.424 AMAP
S 2
466C33 H44 CI2 647.708 647 1.596 AMAP
N4 03 S 2
467C31 H42 CI2 653.736 653 1.607 AMAP
N4 03 S2 2
468C33 H45 Br N4 657.714 657 1.527 AMAP
03 S 2
469C37 H54 N4 04 650.924 651 1.594 AMAP
S 2
470C33 H45 I N4 704.709 705 1.540 AMAP
03 S 2
471C33 H44,CI2 647.708 647 1.559 AMAP
N4 03 S 2
472C38 H56 N4 03 648.951 649 1.679 AMAP
S 2
473C34 H47 N5 05 637.841 638 1.500 AMAP
S 2
474C35 H44 F6 N4 714.812 715 1.630 AMAP
03 S 2
475C35 H50 N4 03 606.871 607 1.541 AMAP
S 2
476C31 H42 CI2 653.736 653 1.577 AMAP
N4 03 S2 2
477C34 H47 Br N4 687.739 687 1.531 AMAP
04 S 2
478C33 H44 CI1 631.253 631 1.490 AMAP
F N4 03 S 2
479C34 H47 F N4 610.834 611 1.498 AMAP
03 S 2
480C41 H55 N7 03 725.997 726 1.682 AMAP
S 2
481C33 H44 N6 07 668.812 669 1.475 AMAP
S 2
482C31 H42 Br2 742.638 741 1.605 AMAP
N4 03 S2 2
483S21 H41 Br CI2 732.632 731 1.622 AMAP
N4 03 2
484C33 H44 CI2 647.708 647 1.552 AMAP
N4 03 S 2
485C33 H43 CI3 682.153 681 1.587 AMAP
N4 03 S 2
486C34 H47 CI1 627.289 627 1.524 AMAP
N4 03 S 2
487C42 H64 N4 03 705.059 705 1.819 AMAP
S 2
488C36 H47 CI1 683.377 683 1.654 AMAP
N4 03 S2 2
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
62
ANALYTICAL METHODS (GC-MS)
BASIS METHOD
API 100 95: MassChrom
Solvents:
A% 95% NH40Ac buffer +
5% acetonitrile
B% 100% acetonitrile
Flow Ramp 5.00
Flow (mllmin) 1.000
Stop Time (mins) 20.00
Min Pressure (Psi) 0
15Max Pressure (Psi) 6100
LC-200 Quad Pump (Version
1.08)
Column: XTerra (2.5 pm, 4.5
x 50 mm)
Column Temperature (C) 20
20Column Temperature Limit 20
(C)
Gradient Timetable: 0.00 = linear
= isocratic, 1.00
Step Time(min) Dura. (min.)B% Flow(mllmin) Grad.
A%
25
0 -0.10 0.10 100 0 1.000 0.00
1 0.00 10.00 5 95 1.000 1.00
2 10.00 2.00 5 95 1.000 0.00
3 12.00 0.50 100 0 1.000"' 1.00
304 12.50 2.50 100 0 1.000 0.00
Number of Channels: 2
Sampling Rate: 0 points per channel
per second
Voltage Range: 0 till
0.1 volt
35Polarity: UNIPOLAR
Channel A: (A) UV 225
nm
Channel B: (B) ELS Sedex
75 (Temp. 37C)
STANDARD METHOD
Waters Alliance 2790 LC
Mobile Phase
Solvents:
C% 95% NH40Ac buffer+5%
ACN (pH = _+5)
p% 100% acetonitrile (ACN)
Flow Ramp 5.00
Flow (ml/min) 1.000
Stop Time (mins) 11.00
Min Pressure (Bar) 0
Max Pressure (Bar) 300
Degasser OnStroke Length
Auto
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
63
Waters Alliance 2790 LC Column
Column Position Column 1 Equilibration Time (mins) 0.00
Column Temperature (°C) 20
Column Temperature Limit (°C) 20
Waters Alliance 2790 LC Rapid Equilibration
System Path OffSystem Flow (ml/min) 0.00
System Time (mins) 0.00
Re-equilibration Time (mins) 0.00
Pre column volume (p1) 0.00
Waters Alliance 2790 I/0
Switch 1: no change; switch 2: no change; switch 3: no change; switch 4: no
change
Analog Output Setting: Flow Rate
Waters Alliance 2790 LC Gradient Timetable
The gradient Timetable contains 6 entries which are:
Time(min) C% D% Flow(ml/min)Curve
0.00 100.0 0.0 1.000 1
1.00 100.0 0.0 1.000 6
7.00 0.0 100.0 1.000 6
8.00 0.0 100.0 1.000 6
8.50 100.0 0.0 1.000 6
11.00 100.0 0.0 1.000 6
Start Wavelength (nm). 225.00
End Wavelength (nm) 260.00
Resolution (nm) 1.2
Sampling Rate (spectra/second)1.000
Filter Response 1
Exposure Time(ms) Automatic 656
Interpolate
YesAcquisition stop time 10.75
(mins)
Waters996 PDA Analog Channel
1
ELS PL ELS 1000 (Temp. 80C)
CURVE 4 METHOD
Waters Alliance 2790 LC Mobile Phase
Solvents
C% 95% NH40Ac buffer+5% ACN (pH = _+5)
p% 100% acetonitrile
Flow Ramp 5.00
Flow (ml/min) 1.000
Stop Time (mins) 11.00
Min Pressure (Bar) 0
Max Pressure (Bar) 320
Degasser OnStroke LengthAuto
Waters Alliance 2790 LC Column
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
64
Column Position Column 1 Equilibration Time (mins) 0.00
Column Temperature (°C) 20
Column Temperature Limit (°C) 20
Waters Alliance 2790 LC Rapid Equilibration
System Path OffSystem Flow (mllmin) 0.00
System Time (mins) 0.00
Re-equilibration Time (mins) 0.00
Pre column volume (N1) 0.00
Waters Alliance 2790 I/0
Switch 1 No Change5witch 2 No ChangeSwitch 3 No ChangeSwitch 4 No
ChangeAnalog Output Setting Flow Rate
Waters Alliance 2790 LC Gradient Timetable
The gradient Timetable contains 6 entries which are:
Time(min) C% D% Flow Curve
0:00 100.0 0.0 1.0001
1.00 100.0 0.0 1.000 4
7.00 0.0 100.0 1.000 4
8.00 0.0 100.0 1.000 6
9.00 100.0 0.0 1.000 6
11.00 100.0 0.0 1.0006
Start Wavelength (nm) 205.00
End Wavelength (nm) 350.00
Resolution (nm) 1.2
Sampling Rate (spectrals)1.000
Filter Response 1
Exposure Time(ms) Automaticlnterpolate
656
YesAcquisition stop 10.75
time (mins)
Waters996 PDA Analog
Channel 1
ELS PL ELS 1000 (Temp.
80C)
AMAP 2 METHOD
The LC-MS system consists
of 2 Perkin Elmer series
200 micro pumps. The
pumps
are connected to mixer. The mixer is
each other by a 50 connected to the
NI tee
Gilson 215 auto sampler.
The LC method is
step total time flow A(%)B(l)
(pl/min)
0 0 2300 95 5
1 1.6 2300 0 100
2 1.8 2300 0 100
3 1 2300 95 5
4 2.2 2300 95 5
A=100% Water with 0.025% HCOOH and 10mmol NHdHC00 pH=~ 3
B=100% ACN with 0.025% HCOOH
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
The auto sampler has a 2 p1 injection loop. The auto sampler is connected to a
Phenomenex Luna C18(2) 30*4.6 mm column with 3 um particles. The column is
thermo stated in a Perkin Elmer series 200 column oven at 40 °C. The
column is
connected to an Applied biosystems ABI 785 UV m eter with a 2.7 NI flowcell.
The
wavelength is set to 254 nm. The UV meter is connected to a Sciex API 150EX
mass spectrometer. The mass spectrometer has the following parameters:
Scanrange: 150-900
Amu
Polarity: positive
10 Scan mode: profile
Resolution Q1: UNIT
Step size: 0.10 amu
Time per scan: 0.500
sec
Nebulizer (NEB): 10
15 Curtain gas 10
(CUR):
Ion Source (IS): 5200 volt
Temperature (TEM):325C
Deflector (DF): 30 volt
Focussing potential225 volt
(FP):
20 Entrance potential10 volt
(EP):
The light scattering detector is connected to the Sciex API 150. The light
scattering
detector is a Sedere Sedex 55 operating at 50 °C and 3 bar NZ pressure.
The complete system is controlled by a Dell optiplex GX400 compu ter operating
25 under Windows NT.
AMAP 3 METHOD
30 Identical with the AMAP 2 Method, except for the LC method, the latter
being:
step total time flow (pllmin) A(%) B(%
0 0 2300 95 5
1 1.8 2300 0 100
35 2.5 2300 0 100
2
3 2.7 2300 95 5
4 3.0 2300 95 5
40 EXAMPLES OF FORMULATION OF COMPOUND AS USED IN ANIMAL STUDIES
For oral (p.o.) administration the desired quantity (up to 20pmol) of the
solid
Example 1 was added to 1 ml of tot % (w/v) methyl hydroxyethyl cellulose and
0.1
(wlv) poloxamer in water. The compound was suspenaea ay vortexmg Tor n a
45 minutes.
For subcutaneous (s.c.) administration the desired quantity (up to 15pmol) of
the
solid Example 1 was dissolved or suspended in 1 ml saline solution.
CA 02547633 2006-05-29
WO 2005/058890 PCT/EP2004/053373
66
Pharmacological data
af fini in vitro I n vivo ism
a onism a on
transit diarrhoeahypersensitivity
ORL1 p,-opiateCAMP assay Mean mean max inhibition
retentionscore
time
Ex. pK; pK; pECSO % control%control% of control
1 9.3 7.7 10.2 49 s.c.
12 8.8 7.1 8.1 110 s.c.
13 8.9 7.4 8.7 105 s.c.59 s.c.
14 9.2 7.2 9.9 111 s.c.64 s.c.
15 8.5 7.5
16 9.1 7.4
17 8.6 7.0
21 8.1 7.4
22 8.2 7.4
28 9.2 8.0 106 (s.c.)78 (s.c.)35 (s.c.)
94 .o. 58 .o.
29 8.2 7.4
34 8.3 7.6
37 8.8 7.7 10.6 34 s.c.
39 9.4 7.2
42 8.2 7.4
45 8.5 8.1
51 9.1 7.9 102 (s.c.) 45 (s.c.)
105 .o. 44 .o.
58 8.1 7.4
59 8.1 7.4
61 9.2 7.7
68 7.9 7.6
70 8.1 7.6
83 8.6 7.8
91 8.1 7.4
214 8.0 7.3
219 8.2 6.7
223 7.9 7.1
230 8.0 7.0
233 7.6 6.8
238 7.9 7.0
275 8.1 7.4
277 8.1 7.0
279 7.7 6.8
285 7.9 7.1
292 8.2 7.1
293 7.9 7.0
298 8.1 7.1
313 7.8 6.9
315 8.0 6.8
327 7.9 7.1
328 7.7 6.9
346 8.0 6.9