Note: Descriptions are shown in the official language in which they were submitted.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-1-
METHODS OF PREPARING COMPOUNDS USEFUL AS PROTEASE INHIBITORS
This application claims priority to United States Provisional Application No.
60/527,477,
filed December 4, 2003, which is hereby incorporated by reference.
Backoround of the Invention
The present invention relates to methods of preparing, and intermediate
compounds
useful in the preparation of, inhibitors of the human immunodeficiency virus
(HIV) protease.
Acquired Immune Deficiency Syndrome (AIDS) causes a gradual breakdown of the
body's immune system as well as progressive deterioration of the central and
peripheral nervous
systems. Since its initial recognition in the early 1980's, AIDS has spread
rapidly and has now
reached epidemic proportions within a relatively limited segment of the
population. Intensive
research has led to the discovery of the responsible agent, human T-
lymphotropic retrovirus III
(HTLV-Ill), now more commonly referred to as HIV.
HIV is a member of the class of viruses known as retroviruses and is the
etiologic agent
of AIDS. The retroviral genome is composed of RNA which is converted to DNA by
reverse
transcription. This retroviral DNA is then stably integrated into a host
cell's chromosome and,
employing the replicative processes of the host cells, produces new retroviral
particles and
advances the infection to other cells. HIV appears to have a particular
affinity for the human T-4
lymphocyte cell which plays a vital role in the body's immune system. HIV
infection of these white
blood cells depletes this white cell population. Eventually, the immune system
is rendered
inoperative and ineffective against various opportunistic diseases such as,
among others,
pneumocystic carini pneumonia, Kaposi's sarcoma, and cancer of the lymph
system.
Although the exact mechanism of the formation and working of the HIV virus is
not
understood, identification of the virus has led to some progress in
controlling the disease. For
example, the drug azidothymidine (AZT) has been found effective for inhibiting
the reverse
transcription of the retroviral genome of the HIV virus, thus giving a measure
of control, though
not a cure, for patients afflicted with AIDS. The search continues for drugs
that can cure or at
least provide an improved measure of control of the deadly HIV virus and thus
the treatment of
AIDS and related diseases.
Retroviral replication routinely features post-translational processing of
polyproteins. This
processing is accomplished by virally encoded HIV protease enzyme. This yields
mature
polypeptides thst will subsequently aid in the formation and function of
infectious virus. If this
molecular processing is stifled, then the normal production of HIV is
terminated. Therefore,
inhibitors of HIV protease may function as anti-HIV viral agents.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
_2_
HIV protease is one of the translated products from the HIV structural protein
pol 25
gene. This retroviral protease specifically cleaves other structural
polypeptides at discrete sites
to release these newly activated structural proteins and enzymes, thereby
rendering the virion
replication-competent. As such, inhibition of the HIV protease by potent
compounds may prevent
proviral integration of infected T-lymphocytes during the early phase of the
HIV-1 life cycle, as
well as inhibit viral proteolytic processing during its late stage.
Additionally, the protease inhibitors
may have the advantages of being more readily available, longer lived in
virus, and less toxic
than currently available drugs, possibly due to their specificity for the
retroviral protease.
Methods for preparing compounds useful as HIV protease inhibitors have been
described
in, e.g., U.S. Patent No. 5,962,640; U.S. Patent No. 5,932,550; U.S. Patent
No. 6,222,043; U.S.
Patent No. 5,644,028; WO 02/100844, Australian Patent No. 705193; Canadian
Patent
Application No. 2,179,935; European Patent Application No. 0 751 145; Japanese
Patent
Application No. 100867489; Y. Hayahsi, et al., J. Org. Chem., 66 5537-5544
(2001); K.
Yoshimura, et al., Proc. Nat!. Acad. Sci. USA, 96, 8675-8680 (1999); and, T.
Mimoto, et al., J.
Med. Chem., 42, 1789-1802 (1999). Thus, methods of preparing compounds useful
as protease
inhibitors have previously been known. However, these methods were linear and
thus inefficient.
The improved methods of the invention provide for convergent synthetic routes
having maximized
efficiency.
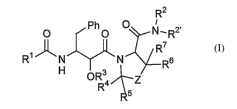
Summary of the Invention
The present invention relates to methods of preparing compounds of formula
(I), or a salt
or solvate thereof:
R2
i
O O N,R2,
R~
HN N V Rs
~ OR3 j~Z
R~ ~O R4
s (I)
wherein:
R' is phenyl optionally substituted by at least one substituent independently
chosen from C~-C6 alkyl, hydroxyl, CRCs alkylcarbonyloxy, C6-C~o
arylcarbonyloxy, and
heteroarylcarbonyloxy;
R2 is C2-C6 alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-(CR4R5)nRs;
n is an integer from 0 to 5;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-3-
R2~ is H or C~-C4 alkyl;
Z is S, O, SO, SO2, CH2, or CFH;
R3 is hydrogen or a hydroxyl protecting group;
each R4, R5, R6 and R' are independently selected from H and C~-C6 alkyl; and
RB is C6-Coo aryl optionally substituted at least one substituent selected
from
C~-Cs alkyl, hydroxyl, and halogen;
comprising:
reacting a compound of formula (II), wherein Y~ is hydroxyl or a leaving group
and R' is
as described for formula (I), with a compound of formula (III), or a salt or
solvate thereof.
R2
,2~
HN~ ~ ~Y~ +
R~~O OR3
(II) (III)
The present invention further comprises deprotecting the compound of formula
(I) when
R3 is a hydroxyl protecting group to afford a compound of formula (I) wherein
R3 is hydrogen.
The present invention also provides intermediate compounds that are useful for
the
preparation of compounds of formula (I).
The following describe further embodiments of the present invention.
In another aspect of the present invention are provided methods for preparing
compounds of formula (I),
R2
O O N , R2,
R~
HN N ~Rs
~ OR3 Z
R~~O R4
s (I)
wherein:
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
R' is phenyl optionally substituted by at least one substituent independently
chosen from C~-Cs alkyl, hydroxyl, C~C6 alkylcarbonyloxy, Cs-Coo
arylcarbonyloxy, and
heteroarylcarbonyloxy;
RZ is C2-Cs alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-(CHz)nR'$~
n is an integer from 0-5;
R2~.is H or C~-C4 alkyl;
Z is S, O, SO, S02, CH2, or CFH;
R3 is hydrogen or a hydroxyl protecting group;
R4, R5, R6 and R' are independently selected from H and C~-C6 alkyl; and
R8 is C6-Coo aryl optionally substituted at least one substituent selected
from
C~-C6 alkyl, hydroxyl, and halogen;
comprising:
reacting a compound of formula (II), wherein Y' is hydroxyl or a leaving
group, with a
compound of formula (III), or a salt or solvate thereof,
R2
I O N_.R2
O R7
HN Y1 + HN ~R6
1~ OR3 R
R O R
(II) (III)
In another aspect of the present invention are provided methods for the
preparation of
compounds of formula (I), comprising:
(i) reacting a compound of formula (IV), wherein Y' is hydroxy or-OP1, wherein
P'
is a suitable protecting group, and R3 is hydrogen, C~-C4 alkyl, or a suitable
hydroxyl protecting
group, with a compound of formula (V), wherein Y2 is a leaving group, to
afford a compound of
formula (II);
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-5-
O O O
R~~Y2 --~ R~~N
H
OR3
(l~ M (I I)
(ii) reacting the compound of formula (II) with a compound of formula (III),
or a salt
or solvate thereof, to afford a compound of formula (I); and
R2
HN' ~ ~Y~ +
R~~O OR3 R4
(II) (III)
(iii) optionally deprotecting those compounds of formula (I) wherein R3 is a
hydroxyl
protecting group, to afford a compound of formula (I) wherein R3 is hydrogen.
In another aspect of the present invention are provided any of the methods
described
herein of preparing the compounds of the formula (I) wherein in the compound
of (II) Y~ is
hydroxyl.
In still another aspect of the present invention are provided any of the
methods described .
herein for the preparation of compounds of formula (I), wherein:
R~ is phenyl optionally substituted by at least one substituent independently
chosen from
C~-C6 alkyl, hydroxyl, C~C6 alkylcarbonyloxy, C6-Coo arylcarbonyloxy, and
heteroarylcarbonyloxy;
RZ is C2-C6 alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-(CHZ)~RB;
n is 0, 1, 2, or 3;
R2~ is H;
Z is S, O, CH2, or CFH;
R3 is hydrogen or a hydroxyl protecting group;
. R4 and R5 are hydrogen;
R6 and R' are C~-C6 alkyl; and
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-6-
R8 is C6-Coo optionally substituted at least one substituent selected from C~-
C6 alkyl,
hydroxyl, and halogen.
Yet another aspect of the present invention provides any of the methods
described herein
for the preparation of compounds of formula (I), wherein:
R' is phenyl optionally substituted by at least one substituent independently
chosen from
C~-Cs alkyl, hydroxyl, C~C6 alkylcarbonyloxy, C6-Coo arylcarbonyloxy, and
heteroarylcarbonyloxy;
R2 is C2-C6 alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-(CH~)nRB~
n is 0, 1, 2, or 3;
R2~ is H;
ZisS;
R3 is hydrogen;
R4 and R5 are hydrogen;
Rs and R~ are methyl; and
Re is phenyl optionally substituted at least one substituent selected from C~-
C6 alkyl,
hydroxyl, and halogen.
In yet another aspect of the present invention provides any of the methods
described
herein for the preparation of compounds of formula (I), wherein:
R~ is phenyl optionally substituted by at least one substituent independently
chosen from
C~-C6 alkyl, hydroxyl, C~C6 alkylcarbonyloxy, Cs-Coo arylcarbonyloxy, and
heteroarylcarbonyloxy;
Rz is C2-Cs alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-(CH2)nRB~
n is 0, 1, 2, or 3;
R2~ is H;
ZisS;
R3 is a hydroxyl protecting group;
R4 and RS are hydrogen;
Rs and R' are methyl; and
Re is phenyl optionally substituted at least one substituent selected from C~-
C6 alkyl,
hydroxyl, and halogen.
Another aspect of the present invention provides any of the methods described
herein for
the preparation of compounds of formula (I), wherein:
R' is phenyl optionally substituted by at least one substituent independently
chosen from
methyl, hydroxyl, and methylcarbonyloxy;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
_7_
RZ is CZ-C6 alkenyl, C~-C6 alkyl optionally substituted with at least one
halogen, or
-CH2R8;
R2~ is H;
ZisS;
R3 is hydrogen or a hydroxyl protecting group;
R4 and R5 are hydrogen;
R6 and R' are methyl; and
R$ is phenyl substituted with at least one methyl.
The present invention also provides any of the methods described herein for
the
preparation of compounds of formula (I), wherein:
R~ is phenyl optionally substituted by at least one substituent independently
chosen from
methyl, hydroxyl, and methylcarbonyloxy;
R2 is Cz-C6 alkenyl;
R2~ is H;
ZisS;
R3 is hydrogen or a hydroxyl protecting group;
R4 and RS are hydrogen; and
Rs and R' are methyl.
Also provided in the present invention are any of the methods described herein
for the
preparation of compounds of formula (I), wherein:
R~ is phenyl substituted by methyl and hydroxyl;
RZ is allyl;
R2~ is H;
ZisS;
R3 is hydrogen or methylcarbonyl;
R4 and R5 are hydrogen; and
R6 and R' are methyl.
The present invention also provides any of the methods described herein for
the
preparation of compounds of formula (I), wherein:
R' is phenyl substituted with methyl and methylcarbonyloxy;
Rz is allyl;
R2~ is H;
ZisS;
R3 is methylcarbonyl;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-g-
R4 and Rsare each H; and
R6 and R' are methyl.
Also provided in the present invention are any of the methods described herein
for the
preparation of compounds of formula (I), wherein the compound of formula (I)
is:
O
CH3 O O NH
HO ~ N N CHa
H OH L.S CH3
Still another aspect of the present invention provides methods for the
preparation of
compounds of formula (I-A),
O
CH3 O O NH
Ac0 ~ '/ , CH
'N N s
H OAc ~S CH3 (I-A)
comprising:
reacting a compound of formula (II-A) with a compound of formula (III-A), or a
salt or
solvate thereof.
O
CH3 O O N
Ac0 + '~CH
~N OH HN ~ a
H OAc ~S CH3
(II-A) (I I I-A)
In still another aspect of the present invention are provided methods for the
preparation
of preparing compounds of formula (I-A),
\ /
O
CH3 O O NH
Ac0 ~ '/' CH
'N N s
/ H OAc ~S CH3
(I-A)
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
_g_
comprising:
(i) reacting a compound of formula (IV-A) with a compound of formula (V-A),
Ph0 CHI O CH3 O Ph0
HEN OH Ac0 ~ CI Ac0 ~ N OH
OH + I i ~ I ~ H OH
(IV-A) (V-A)
(I I-B)
to afford a compound of formula (II-B);
(ii) treating the compound of formula (II-B) with an acetylating agent to
afford a
compound of formula (II-A); and
CH3 O Ph0
Ac0 I ~ H OH
OAc
(I I-A)
(iii) reacting the compound of formula (II-A) with a compound of formula (III-
A).
\ / o
CH3 O O ~NH
Ac0 /'~ CH
N OH + HN ~ s
I i H OAc L-g CH3
(I I-A) (I I I-A)
In yet another aspect of the present invention are provided methods for the
preparation of
compounds of formula (I-B),
\ /
O
CH3 O ~ NH
HO ~ '/ , CH
'N N s
H OH ~S CH3 (1-B)
comprising:
(i) reacting a compound of formula (II-A) with a compound of formula (III-A),
or a
salt or solvate thereof,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-10-
o ~ ~ / o
CH3 O O ~NH CH3 O O ~NH
Ac0 ~ N OH + HN/ CH3 --.~ Ac0 ~ N N/ CHa
/ H OAc ~S CH3 I / H OAc LS CH3
(II-A) (I II-A) (I-A)
to afford a compound of formula (I-A); and
(ii) deprotecting the compound of formula (I-A).
Another aspect of the present invention provides a method of preparing a
compound of
formula (I-B),
O
CH3 0 0 NH
HO ~CH
N N ''' 3
I i H OH ~S CH3
(1-B)
comprising:
(i) reacting a compound of formula (IV-A) with a compound of formula (V-A),
Ph0 CH3 O CH3 O Ph0
HZN OH Ac0 ~ Ci Ac0 ~ N OH
OH + I / I / H OH
(lV-A) tV-A) (li-B)
to afford a compound of formula (II-B);
(ii) treating the compound of formula (II-B) with an acetylating agent to
afford a
compound of formula (II-A); and
CH3 O Ph0
Ac0 ~ N OH
H OAc
(I I-A)
(iii) reacting the compound of formula (II-A) with a compound of formula (III-
A),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-11-
o ~ ~ ~ o
CH3 O O ~NH CH3 O O ~NH
Ac0 ~ N OH + HN/ CH3 Ac0 ~ N N CH3
/ H OAc ~S CH3 I / H OAc L CHs
(II-A) (I II-A) (I-A)
to afford a compound of formula (I-A); and
(iv) deprotecting the compound of formula (I-A).
In another aspect of the present invention are provided any of the methods
described
herein for the preparation of compounds of formula (I), wherein:
R' is phenyl optionally substituted by at least one substituent independently
chosen from
methyl, hydroxyl, and methylcarbonyloxy;
RZ is-CHZRB;
R2~ is H;
Z is S;
R3 is hydrogen or a hydroxyl protecting group;
R4 and R5 are hydrogen;
R6 and R' are methyl; and
Re is phenyl substituted with at least one methyl.
In yet another aspect of the present invention are provided any of the methods
described
herein for preparing compounds of formula (I), wherein:
R~ is phenyl substituted by methyl and methylcarbonyloxy;
R~ is-CH2Rs;
R2~ is H;
Z is S;
R3 is methylcarbonyl;
R4 and R5 are hydrogen;
R6 and R' are methyl; and
R$ is phenyl substituted with at least one methyl.
In still a further aspect of the present invention are provided any of the
methods
described herein for the preparation of compounds of formula (I), wherein:
R' is phenyl substituted by methyl and hydroxyl;
RZ is-CHZRB;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-12-
R2~ is H;
ZisS;
R3 is hydrogen;
R4 and R5 are hydrogen;
Rs and R' are methyl; and
R$ is phenyl substituted with at least one methyl.
Also provided in the present invention are any of the methods described herein
for the
preparation of compounds of formula (I), wherein the compound of formula (I)
is:
H3C
O
CHs O O NH
HO ~ N N CHs
H OH ~S CHs
Yet another aspect of the present invention provides methods for the
preparation of
compounds of formula (I-C),
H3C
0
CHs O O NH
Ac0 ~ ~CH
'N N s
/ H OAc ~S CHs (I-C)
comprising:
reacting a compound of formula (II-A) with a compound of formula (III-B), or a
salt or
solvate thereof.
H3C
O
CHs O 0 ~NH
Ac0 /'~ CH
~N OH + HN ~ a
H OAc LS CHs
(II-A) (III-B)
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-13-
In still another aspect of the present invention are provided methods for the
preparation
of preparing compounds of formula (I-C),
H3C
O
CH3 O O NH
Ac0 ~ '/S CH
'N N s
I / H OAc ~S CH3 (I-C)
comprising:
(i) reacting a compound of formula (IV-A) with a compound of formula (V-A),
\ /
O CH3 O CH3 O O
Ac0 ~ CI Ac0 ~ N OH
HZN OH
OH + I ~ I ~ H OH
(11_B)
(IV-A) (V-A)
to afford a compound of formula (II-B);
(ii) treating the compound of formula (II-B) with an acetylating agent to
afford a
compound of formula (11-A); and
CH3 O PhO
Ac0 ~ N OH
H OAc
(I I-A)
(iii) reacting the compound of formula (II-A) with a compound of formula (III-
B),
H3C
\ l o
\ /
CH3 0 0 ~NH
Ac0 ~'~ CH
N OH + HN ~ s
H OAc ~-S CHs
(I I-A) (I I I-B)
In yet another aspect of the present invention are provided methods for the
preparation of
compounds of formula (I-D),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-14-
H3C
O
CHs O O NH
HO ~ '/, CH
'N N a
H OH ~S CHs
(I-D)
comprising:
(i) reacting a compound of formula (II-A) with a compound of formula (III-B),
or a
salt or solvate thereof,
H3C H3C
O ~ \ / O
CH3 O 0 y-NH ~ ~ CH3 O O ~NH
Ac0 ~ N OH + HN/ CH3 ---~ Ac0 ~ N N CHs
H OAc ~S CH3 I / H OAc LS CH3
(II-A) (III-B) (1-C)
to afford a compound of formula (I-C); and
(ii) deprotecting the compound of formula (I-C).
Another aspect of the present invention provides a method of preparing a
compound of
formula (I-D),
HsC
O
CHs O O ~NH
HO ~ ~/,, CH
'N N a
H OH ~S CHs
(I-D)
comprising:
(i) reacting a compound of formula (IV-A) with a compound of formula (V-A),
O CH3 O CH3 O O
H2N OH Ac0 ~ CI Ac0 ~ N OH
OH + I / I ~ H OH
(IV-A) (V-A)
(I I-B)
to afford a compound of formula (II-B);
(ii) treating the compound of formula (II-B) with an acetylating agent to
afford a
compound of formula (II-A); and
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-15-
CH3 O Ph0
Ac0 I ~ H OH
OAc
(I I-A)
(iii) reacting the compound of formula (II-A) with a compound of formula (III-
B),
H3C H3C
O ~ ~ ~ O
CH3 O O ~NH ~ ~ CH3 O O ~NH
Ac0 ~ N OH + HN/ CH3 Ac0 ~ N N ~ CH3
H OAc ~S CH3 I ~ H OAc LS CH3
(I I-A) (III-B) (I-C)
to afford a compound of formula (I-C); and
(iv) deprotecting the compound of formula (I-C) to afford the compound of
formula
(I-D).
Another aspect of the present invention features compounds of formulae (I-A),
(I-B), (II-
A), (III-A), (III-B), (I-C), and (I-D):
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-16-
\ / \ / O
CH3 O O ~NH CH3 O O ~NH
Ac0 N N ''°' 3 HO ~ '~ CH
w CH I ~H N ~ s
/ H OAc ~S CH3 / OH ~g CHs
(1_A) (1_B)
CH3
Ac0 H
(1_C) (1_D)
(I I-A)
H3C
C ~ C
~NH ~NH
HN/°< CH3 HN~°' CHa
CH3 ~S CH3
(III-A)
(III-B)
all of which are intermediates useful in the preparation of compounds of
formula (I).
Another aspect of the present invention provides for the preparation of
compounds of
formula (II-A),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-17-
CH3 O O
Ac0 ~ N OH
I / H OAc
(II-A)
comprising treating a compound of formula (11-B) with an acetylating agent. In
another aspect of
the present invention, said acetylating agent is chosen from acetic anhydride
and acetyl chloride.
CH3 O O
AcO ~ N OH
H OH
(11_B)
DETAILED DESCRIPTION OF THE INVENTION
0
In accordance with a convention used in the art, ~ is used in structural
formulas
herein to depict the bond that is the point of attachment of the moiety or
substituent to the core or
backbone structure. When the phrase, "substituted with at least one
substituent" is used herein, it
is meant to indicate that the group in question may be substituted by at least
one of the
substituents chosen. The number of substituents a group in the compounds of
the invention may
have depends on the number of positions available for substitution. For
example, an aryl ring in
the compounds of the invention may contain from 1 to 5 additional
substituents, depending on the
degree of substitution present on the ring. The maximum number of substituents
that a group in
the compounds of the invention may have can be determined by those of ordinary
skill in the art.
The term "reacting," as used herein, refers to a chemical process or processes
in which
two or more reactants are allowed to come into contact with each other to
effect a chemical
change or transformation. For example, when reactant A and reactant B are
allowed to come into
contact with each other to afford a new chemical compounds) C, A is said to
have "reacted" with
B to produce C.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-18-
The term "protecting," as used herein, refers to a process in which a
functional group in a
chemical compound is selectively masked by a non-reactive functional group in
order to allow a
selective reactions) to occur elsewhere on said chemical compound. Such non-
reactive
functional groups are herein termed "protecting groups." For example, the term
"hydroxyl
protecting group," as used herein refers to those groups that are capable of
selectively masking
the reactivity of a hydroxyl (-OH) group. The term "suitable protecting
group," as used herein
refers to those protecting groups that are useful in the preparation of the
compounds of the
present invention. Such groups are generally able to be selectively introduced
and removed
using mild reaction conditions that do not interfere with other portions of
the subject compounds.
Protecting groups that are suitable for use in the processes and methods of
the present invention
are known to those of ordinary skill in the art. The chemical properties of
such protecting groups,
methods for their introduction and their removal can be found, for example, in
T. Greene and P.
Wuts, Protective Groups in Organic Synthesis (3'd ed.), John Wiley & Sons, NY
(1999). The
terms "deprotecting," "deprotected," or "deprotect," as used herein, are meant
to refer to the
process of removing a protecting group from a compound.
The term"leaving group," as used herein refers to a chemical functional group
that
generally allows a nucleophilic substitution reaction to take place at the
atom to which it is
attached. For example, in acid chlorides of the formula CI-C(O)R, wherein R is
alkyl, aryl, or
heterocyclic, the -CI group is generally referred to as a leaving group
because it allows
nucleophilic substitution reactions to take place at the carbonyl carbon.
Suitable leaving groups
are known to those of ordinary skill in the art and can include halides,
aromatic heterocycles,
cyano, amino groups (generally under acidic conditions), ammonium groups,
alkoxide groups,
carbonate groups, formates, and hydroxy groups that have been activated by
reaction with
compounds such as carbodiimides. For example, suitable leaving groups can
include, but are not
limited to, chloride, bromide, iodide, cyano, imidazole, and hydroxy groups
that have been
allowed to react with a carbodiimide such as dicyclohexylcarbodiimide
(optionally in the presence
of an additive such as hydroxybenzotriazole) or a carbodiimide derivative.
The term "acetylating agent," as used herein refers to chemical compounds that
are
useful for the introduction of an acetyl group, -C(O)CH3, onto a hydroxyl
group in the compounds
of the invention. The symbol "Ac-," as used in chemical structures herein, is
meant to represent
an acyl group in the compounds of the invention. Useful acetylating agents
include, but are not
limited to, acetic anhydride, acetyl chloride, acetyl bromide, and acetyl
iodide. In addition, such
acetylating agents can be prepared in situ by reaction of an appropriate
combination of
compounds, such as the reaction of acetyl chloride with sodium iodide in
acetone to afford an
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-19-
intermediate acetyl iodide agent. The term "acetic anhydride," as used herein
is meant to
represent a compound with the chemical formula CH3C(O)OC(O)CH3. In addition,
the term
"-OAc," as used in the chemical structures herein, represents the group -
OC(O)CH3.
As used herein, the term "aliphatic" represents a saturated or unsaturated,
straight- or
branched-chain hydrocarbon, containing 1 to 10 carbon atoms which may be
unsubstituted or
substituted by one or more of the substituents described below. The term
"aliphatic" is intended
to encompass alkyl, alkenyl and alkynyl groups.
As used herein, the terms "C~_6 alkyl" and "C~-C6 alkyl," which may be used
interchangeably throughout, represents a straight- or branched-chain saturated
hydrocarbon,
containing 1 to 6 carbon atoms which may be unsubstituted or substituted by
one or more of the
substituents described below. Similarly, the terms "C~~ alkyl" and "C~-C4
alkyl," which may be
used interchangeably throughout, represents a straight- or branched-chain
saturated
hydrocarbon, containing from 1 to 4 carbon atoms which may be unsubstituted or
substituted by
one or more of the substituents described below. Exemplary alkyl substituents
include, but are
not limited to methyl (Me), ethyl (Et), propyl, isopropyl, butyl, isobutyl, t-
butyl, and the like.
The terms "C2_6 alkenyl" and "CZ-C6 alkenyl," which may be used
interchangeably
throughout, represent a straight- or branched-chain hydrocarbon, containing
one or more carbon-
carbon double bonds and having 2 to 6 carbon atoms which may be unsubstituted
or substituted
by one or more of the substituents described below. Exemplary alkenyl
substituents include, but
are not limited to ethenyl, propenyl, butenyl, allyl, pentenyl and the like.
The terms "C~~4 aryl" and "C6-C~4 aryl", which may be used interchangeably
throughout,
and as used herein, mean a group derived from an aromatic hydrocarbon
containing from 6 to 14
carbon atoms. Examples of such groups include, but are not limited to, phenyl
or naphthyl. The
terms "Ph" and "phenyl," as used herein, mean a ~-CsHS group. The term
"benzyl," as used
herein, means a -CH2CsH5 group. The term "phenyl," as used herein refers to a
fully unsaturated
6-membered carbocyclic group. The symbol "Ph," as used in the chemical
structures herein, is
meant to represent a phenyl or C6H5- group.
The term "heteroaryl," as used herein refers to a group comprising an aromatic
monovalent monocyclic, bicyclic, or tricyclic group, containing 5 to 18 ring
atoms, including 1 to 5
heteroatoms selected from nitrogen, oxygen and sulfur, which may be
unsubstituted or
substituted by one or more of the substituents described below. As used
herein, the term
"heteroaryl" is also intended to encompass the N-oxide derivative (or N-oxide
derivatives, if the
heteroaryl group contains more than one nitrogen such that more than one N-
oxide derivative
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-20-
may be formed) of the nitrogen-containing heteroaryl groups described herein.
Illustrative
examples of heteroaryl groups include, but are not limited to, thienyl,
pyrrolyl, imidazolyl,
pyrazolyl, furyl, isothiazolyl, furazanyl, isoxazolyl, thiazolyl, pyridyl,
pyrazinyl, pyrimidinyl,
pyridazinyl, triazinyl, benzo[b]thienyl, naphtho[2,3-b]thianthrenyl,
isobenzofuranyl, chromenyl,
xanthenyl, phenoxathienyl, indolizinyl, isoindolyl, indolyl, indazolyl,
purinyl, isoquinolyl, quinolyl,
phthalazinyl, naphthyridinyl, quinoxyalinyl, quinzolinyl, benzothiazolyl,
benzimidazolyl,
tetrahydroquinolinyl, cinnolinyl, pteridinyl, carbazolyl, beta-carbolinyl,
phenanthridinyl, acridinyl,
perimidinyl, phenanthrolinyl, phenazinyl, isothiazolyl, phenothiazinyl, and
phenoxazinyl.
Illustrative examples of N-oxide derivatives of heteroaryl groups include, but
are not limited to,
pyridyl N-oxide, pyrazinyl N-oxide, pyrimidinyl N-oxide, pyridazinyl N-oxide,
triazinyl N-oxide,
isoquinolyl N-oxide, and quinolyl N-oxide. Further examples of heteroaryl
groups include the
following moieties:
N / \ N
/\ /\ ~ /\ /\
N~ ~ ~\ ~ ~ N ~ %
R , S , N , ~ , R , S , S ,
N / ~ N /N ~ N~\N
/ \ I I I I I N
R , O ' N ~ N N N N
N~N N~N
~\ ' N ~ N
~N NON , R , ~ S , R ,
'
\ ~ ~ I \N
~N
\ O , \ N/ , \ , \ /N , \ ,
R
w
\ i ~ I N\ ~ / \
I /N \ / N ~ ,
, N , R . S
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-21-
N /N ~ ( N
\ ~ . \ I ~ \ / I . \NJ ~ ~ N
0 0 0 0
0 0
\N ~ \N ~ I N\
I /~~ \ N ~ ~ I /N~ \ N
O ~ a O
O O
N/ I \ \N N/ I ~ \N
N/ \ \N~O
N/ / ' \ N
/
o and N , wherein
R is H, alkyl, hydroxyl or represents a compound according to Formula f.
The terms "halogen" and "halo" represent chloro, fluoro, bromo or iodo
substituents.
The terms "C~_s alkylcarbonyloxy" and "C~-Cs alkylcarbonyloxy," which may be
used
interchangeably throughout, and as used herein, refers to groups of the
formula -OC(O)R,
wherein R is an alkyl group comprising from 1 to 6 carbon atoms.
The terms "C~~o arylcarbonyloxy" and "C6-Coo arylcarbonyloxy," which may be
used
interchangeably throughout, and as used herein, refers to a group of the
formula -OC(O)R,
wherein R is an aryl group comprising from 6 to 10 carbons, as defined above.
The term "heteroarylcarbonyloxy," as used herein, refers to a group of the
formula
-OC(O)R, wherein R is a heteroaromatic group as defined above.
If an inventive compound or an intermediate in the present invention is a
base, a desired
salt may be prepared by any suitable method known in the art, including
treatment of the free
base with an inorganic acid, such as hydrochloric acid, hydrobromic acid,
sulfuric acid, nitric acid,
phosphoric acid, and the like, or with an organic acid, such as acetic acid,
malefic acid, succinic
acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid,
glycolic acid, salicylic
acid, pyranosidyl acid, such as glucuronic acid or galacturonic acid, alpha-
hydroxy acid, such as
citric acid or tartaric acid, amino acid, such as aspartic acid or glutamic
acid, aromatic acid, such
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-22-
as benzoic acid or cinnamic acid, sulfonic acid, such as p-toluenesulfonic
acid or ethanesulfonic
acid, or the like.
If an inventive compound or an intermediate in the present inventon is an
acid, a desired
salt may be prepared by any suitable method known to the art, including
treatment of the free
acid with an inorganic or organic base, such as an amine (primary, secondary,
or tertiary); an
alkali metal or alkaline earth metal hydroxide; or the like. Illustrative
examples of suitable salts
include organic salts derived from amino acids such as glycine and arginine;
ammonia; primary,
secondary, and tertiary amines; and cyclic amines, such as piperidine,
morpholine, and
piperazine; as well as inorganic salts derived from sodium, calcium,
potassium, magnesium,
manganese, iron, copper, zinc, aluminum, and lithium.
The compounds of the present invention contain at least one chiral center and
may exist
as single stereoisomers (e.g., single enantiomers or single diastereomers),
any mixture of
stereoisomers (e.g., any mixture of enantiomers or diastereomers) or racemic
mixtures thereof. It
is specifically contemplated that, unless otherwise indicated, all
stereoisomers, mixtures and
racemates of the present compounds are encompassed within the scope of the
present invention.
Compounds identified herein as single stereoisomers are meant to describe
compounds that are
present in a form that contains at least from at least about 90% to at least
about 99% of a single
stereoisomer of each chiral center present in the compounds. Where the
stereochemistry of the
chiral carbons present in the chemical structures illustrated herein are not
specified, it iso
specifically contemplated fihat all possible stereoisomers are encompassed
therein. The
compounds of the present invention may be prepared and used in
stereoisomerically pure form or
substantially stereoisomerically pure form. As used herein, the term
"stereoisomeric" purity refers
to the "enantiomeric" purity and/or "diastereomeric" purity of a compound. The
term
"stereoisomerically pure form," as used herein, is meant to encompass those
compounds that
contain from at least about 95% to at least about 99%, and all values in
between, of a single
stereoisomer. The term "substantially enantiomerically pure," as used herein
is meant to
encompass those compounds that contain from at least about 90% to at least
about 95%, and all
values in between, of a single stereoisomer. The term "diastereomerically
pure," as used herein,
is meant to encompass those compounds that contain from at least about 95% to
at least about
99%, and all values in between, of a single diastereoisomer. The term
"substantially
diastereomerically pure," as used herein, is meant to encompass those
compounds that contain
from at least about 90% to at least about 95%, and all values in between, of a
single
diastereoisomer. The terms "racemic" or "racemic mixture," as used herein,
refer to a mixture
containing equal amounts of stereoisomeric compounds of opposite
configuration. For example,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-23-
a racemic mixture of a compound containing one stereoisomeric center would
comprise equal
amount of that compound in which the stereoisomeric center is of the (S)- and
(R)-configurations.
The term "enantiomerically enriched," as used herein, is meant to refer to
those compositions
wherein one stereoisomer of a compound is present in a greater amount than the
opposite
stereoisomer. Similarly, the term "diastereomerically enriched," as used
herein, refers to those
compositions wherein one diastereomer of compound is present in amount greater
than other
diastereomer(s). The compounds of the present invention may be obtained in
stereoisomerically
pure (i.e., enantiomerically and/or diastereomerically pure) or substantially
stereoisomerically
pure (i.e., substantially enantiomerically and/or diastereomerically pure)
form. Such compounds
may be obtained synthetically, according to the procedures described herein
using
stereoisomerically pure or substantially stereoisomerically pure materials.
Alternatively, these
compounds may be obtained by resolution/separation of mixtures of
stereoisomers, including
racemic and diastereomeric mixtures, using procedures known to those of
ordinary skill in the art.
Exemplary methods that may be useful for the resolution/separation of
stereoisomeric mixtures
include derivitation with stereochemically pure reagents to form
diastereomeric mixtures,
chromatographic separation of diastereomeric mixtures, chromatographic
separation of
enantiomeric mixtures using chiral stationary phases, enzymatic resolution of
covalent
derivatives, and crystallization/re-crystallization. Other useful methods may
be found in
Enantiomers. Racemates. and Resolutions, J. Jacques et al., 1981, John Wiley
and Sons, New
York, NY, the disclosure of which is incorporated herein by reference.
Preferred stereoisomers of
the compounds of this invention are described herein.
If the substituents themselves are not compatible with the synthetic methods
of this
invention, the substituent may be protected with a suitable protecting group
that is stable to the
reaction conditions used in these methods. The protecting group may be removed
at a suitable
point in the reaction sequence of the method to provide a desired intermediate
or target
compound. Suitable protecting groups and the methods for protecting and de-
protecting different
substituents using such suitable protecting groups are well known to those
skilled in the art;
examples of which may be found in T. Greene and P. Wuts, Protective Groups in
Organic
Synthesis (3'~ ed.), John Wiley & Sons, New York (1999), which is incorporated
herein by
reference in its entirety. In some instances, a substituent may be
specifically selected to be
reactive under the reaction conditions used in the methods of this invention.
Under these
circumstances, the reaction conditions convert the selected substituent into
another substituent
that is either useful in an intermediate compound in the methods of this
invention or is a desired
substituent in a target compound.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-24-
In the compounds of this invention, Rz and R2~, independently or taken
together, may be a
suitable nitrogen protecting group. As indicated above, suitable nitrogen
protecting groups are
known to those of ordinary skill in the art and any nitrogen protecting group
that is useful in the
methods of preparing the compounds of this invention or may be useful in the
HIV protease
inhibitory compounds of this invention may be used. Exemplary nitrogen
protecting groups
include alkyl, substituted alkyl, carbamate, urea, amide, imide, enamine,
sulfenyl, sulfonyl, vitro,
nitroso, oxide, phosphinyl, phosphoryl, silyl, organometallic, borinic acid
and boronic acid groups.
Examples of each of these groups, methods for protecting nitrogen moieties
using these groups
and methods for removing these groups from nitrogen moieties are disclosed in
T. Greene and P.
Wuts, supra. Preferably, when R2 and/or R2~ are independently suitable
nitrogen protecting
groups, suitable R2 and R2~ substituents include, but are not limited to,
carbamate protecting
groups such as alkyloxycarbonyl (e.g., Boc: t-butyloxycarbonyl) and
aryloxycarbonyl (e.g., Cbz:
benzyloxycarbonyl, or FMOC: fluorene-9-methyloxycarbonyl), alkyloxycarbonyls
(e.g.,
methyloxycarbonyl), alkyl or arylcarbonyl, substituted alkyl, especially
arylalkyl (e.g., trityl
(triphenylmethyl), benzyl and substituted benzyl), and the like. When RZ and
R2~ taken together
are a suitable nitrogen protecting group, suitable RZ/R2~ substituents include
phthalimido and a
stabase (1,2-bis (dialkylsilyl)) ethylene).
The following processes illustrate the preparation of HIV protease inhibitors
according to
methods of the present invention. These compounds, prepared by the methods of
the present
invention, are potent inhibitors of HIV protease and thus are useful in the
prevention and
treatment of acquired immunodeficiency syndrome (AIDS) and AIDS related
complex ("ARC").
Unless otherwise indicated, variables according to the following processes are
as defined
above.
Starting materials, the synthesis of which are not specifically described
herein or provided
with reference to published references, are either commercially available or
can be prepared
using methods known to those of ordinary skill in the art. Certain synthetic
modifications may be
done according to methods familiar to those of ordinary skill in the art.
Compounds of formula (I),wherein R' is phenyl substituted by at least one
hydroxyl
group, and Z, R~, R2~, R3, R4, R5, Rs, R', are as hereinbefore defined, may be
prepared from
compounds of formula I wherein R' is phenyl substituted by at least one group
selected from C~_s
alkylcarbonyloxy, C~.~o arylcarbonyloxy, and heteroarylcarbonyloxy. The C~_6
alkylcarbonyloxy,
C~~o arylcarbonyloxy, and heteroarylcarbonyloxy groups may be cleaved under
conditions that
directly provide the desired hydroxyl substituted compounds of the invention.
In general, the C~_s
alkylcarbonyloxy, C~~o arylcarbonyloxy, and heteroarylcarbonyloxy groups may
be cleaved under
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-25-
basic conditions, in a solvent that will not intertere with the desired
transformation, and at a
temperature that is compatible With the other reaction parameters, all of
which are known to those
of skill in the art. For example, appropriate bases include, but are not
limited to, sodium
bicarbonate, potassium bicarbonate, sodium carbonate, potassium carbonate,
sodium hydroxide,
potassium hydroxide, a sodium alkoxide such as sodium methoxide or sodium
ethoxide, a
potassium alkoxide such as potassium methoxide or potassium ethoxide, or a
base formed in situ
using an appropriate combination of reagents, such as a combination of a
trialkyl or aryl amine in
combination with an alkanol such as methanol. Or such a transformation may be
accomplished
using an acid that is known to those of skill in the art to be appropriate to
cleave such a group
without intertering with the desired transformation. Such acids include, but
are not limited to,
hydrogen halides such as hydrochloric acid or hydroiodic acid, an alkyl
sulfonic acid such as
methanesulfonic acid, an aryl sulfonic acid such as benzenesulfonic acid,
nitric acid, sulfuric acid,
perchloric acid, or chloric acid. Furthermore, appropriate solvents include
those that are known
to those of skill in the art to be compatible with the reaction conditions and
include alkyl esters
and aryl esters, alkyl, heterocyclic, and aryl ethers, hydrocarbons, alkyl and
aryl alcohols, alkyl
and aryl halogenated compounds, alkyl or aryl nitrites, alkyl and aryl
ketones, and non-erotic
heterocyclic solvents. For example, suitable solvents include, but are not
limited to, ethyl acetate,
isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone,
dimethoxyethane,
diisopropyl ether, chlorobenzene, dimethyl formamide, dimethyl acetamide,
propionitrile,
butyronitrile, t-amyl alcohol, acetic acid, diethyl ether, methyl-t-butyl
ether, diphenyl ether,
methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane,
heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-
butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Finally, these
transformations may be conducted at temperatures from -20 °C to 100
°C, depending on the
specific reactants and solvents and is within the skill of one of ordinary
skill in the art. Further
suitable reaction conditions may be found in Greene et al., Protective Groups
in Organic
Synthesis; John Wiley & Sons, New York, (1999).
Compounds of formulas (I) and (X), wherein R3 is hydrogen and ~, R', R2, R2~,
R4, R5, R6,
R', R8, and R9 are as hereinbefore defined, may be prepared from compounds of
formulas (I) and
(X) wherein R3 is a hydroxyl protecting group. The choice of a suitable
hydroxy protecting group
is within the knowledge of one of ordinary skill in the art. Suitable hydroxyl
protecting groups that
are useful in the present invention include, but are not limited to, alkyl or
aryl esters, alkyl silanes,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-26-
aryl silanes or alkylaryl silanes, alkyl or aryl carbonates, benzyl groups,
substituted benzyl
groups, ethers, or substituted ethers. The various hydroxy protecting groups
can be suitably
cleaved utilizing a number of reaction conditions known to those of ordinary
skill in the art. The
particular conditions used will depend on the particular protecting group as
well as the other
functional groups contained in the subject compound. Choice of suitable
conditions is within the
knowledge of those of ordinary skill in the art.
For example, if the hydroxy protecting group is an alkyl or aryl ester,
cleavage of the
protecting group may be accomplished using a suitable base, such as a
carbonate, a
bicarbonate, a hydroxide, an alkoxide, or a base formed in situ from an
appropriate combination
of agents. Furthermore, such reactions may be performed in a solvent that is
compatible with the
reaction conditions and will not interfere with the desired transformation.
For example, suitable
solvents may include alkyl esters, alkylaryl esters, aryl esters, alkyl
ethers, aryl ethers, alkylaryl
esters, cyclic ethers, hydrocarbons, alcohols, halogenated solvents, alkyl
nitrites, aryl nitrites,
alkyl ketones, aryl ketones, alkylaryl ketones, or non-erotic heterocyclic
compounds. For
example, suitable solvents include, but are not limited to, ethyl acetate,
isobutyl acetate, isopropyl
acetate, n-butyl acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl
ether,
chlorobenzene, dimethyl formamide, dimethyl acetamide, propionitrile,
butyronitrile, t-amyl
alcohol, acetic acid, diethyl ether, methyl-t-butyl ether, diphenyl ether,
methylphenyl ether,
tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, pentane, hexane,
heptane, methanol,
ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-butanol,
dichloromethane, chloroform,
1,2-dichloroethane, acetonitrile, benzonitrile, acetone, 2-butanone, benzene,
toluene, anisole,
xylenes, and pyridine, or any mixture of the above solvents. Additionally,
water may be used as a
co-solvent in this transformation if necessary. Finally, such reactions may be
performed at an
appropriate temperature from -20 °C to 100 °C, depending on the
specific reactants used. The
choice of a suitable temperature is within the skill of one of ordinary skill
in the art. Further
suitable reaction conditions may be found in Greene et al., Protective Groups
in Oraanic
Synthesis, John Wiley & Sons, New York, (1999).
Additionally, if R3 is an alkyl silane, aryl silane or alkylaryl silane, such
groups may be
cleaved under conditions known to those of ordinary skill in the art. For
example, such silane
protecting groups may be cleaved by exposure. of the subject compound to a
source of fluoride
ions, such as the use of an organic fluoride salt such as a tetraalkylammonium
fluoride salt, or an
inorganic fluoride salt. Suitable fluoride ion sources include, but are not
limited to,
tetramethylammonium fluoride, tetraethylammonium fluoride, tetrapropylammonium
fluoride,
tetrabutylammonium fluoride, sodium fluoride, and potassium fluoride.
Alternatively, such silane
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-27-
protecting groups may be cleaved under acidic conditions using organic or
mineral acids, with or
without the use of a buffering agent. For example, suitable acids include, but
are not limited to,
hydrofluoric acid, hydrochloric acid, sulfuric acid, nitric acid, acetic acid,
citric acid, and
methanesulfonic acid. Such silane protecting groups may also be cleaved using
appropriate
Lewis acids. For example, suitable Lewis acids include, but are not limited
to, dimethylbromo
borane, triphenylmethyl tetrafluoroborate, and certain Pd (II) salts. Such
silane protecting groups
can also be cleaved under basic conditions that employ appropriate organic or
inorganic basic
compounds. For example, such basic compounds include, but are not limited to,
sodium
carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate,
sodium hydroxide,
and potassium hydroxide. The cleavage of a silane protecting group may be
conducted in an
appropriate solvent that is compatible with the specific reaction conditions
chosen and will not
interfere with the desired transformation. Among such suitable solvents are,
for example, alkyl
esters, alkylaryl esters, aryl esters, alkyl ethers, aryl ethers, alkylaryl
esters, cyclic ethers,
hydrocarbons, alcohols, halogenated solvents, alkyl nitrites, aryl nitrites,
alkyl ketones, aryl
ketones, alkylaryl ketones, or non-erotic heterocyclic compounds. For example,
suitable solvents
include, but are not limited to, ethyl acetate, isobutyl acetate, isopropyl
acetate, n-butyl acetate,
methyl isobutyl ketone, dimethoxyethane, diisopropyl ether, chlorobenzene,
dimethyl formamide,
dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol, acetic acid,
diethyl ether, methyl-t-
butyl ether, diphenyl ether, methylphenyl ether, tetrahydrofuran, 2-
methyltetrahydrofuran, 1, 4-
dioxane, pentane, hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol,
t-butanol, n-
butanol, 2-butanol, dichloromethane, chloroform, 1,2-dichloroethane,
acetonitrile, benzonitrile,
acetone, 2-butanone, benzene, toluene, anisole, xylenes, and pyridine, or any
mixture of the
above solvents. Additionally, water may be used as a co-solvent in this
transformation if
necessary. Finally, such reactions may be performed at an appropriate
temperature from -20 °C
to 100 °C, depending on the specific reactants used. The choice of a
suitable temperature is
within the skill of one of ordinary skill in the art. Further suitable
reaction conditions may be found
in Greene et al., Protective Groups in Or anic Synthesis, John Wiley & Sons,
New York, (1999).
When R3 is a benzyl or substituted benzyl ether, cleavage of the protecting
group may be
accomplished by treating the subject compound with hydrogen in the presence of
a suitable
catalyst, oxidation with suitable compounds, exposure to light of particular
wavelengths,
electrolysis, treatment with erotic acids, or treatment wifih Lewis acids. The
choice of particular
reagents to effect such a transformation will depend on the specific subject
compound used and
is within the skill of one of ordinary skill in the art. For example, such
benzyl or substituted benzyl
ethers may be cleaved using hydrogen gas in the presence of an appropriate
catalyst. Suitable
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-28-
catalysts include, but are not limited to, 5% palladium on carbon, 10%
palladium on carbon, 5%
platinum on carbon, or 10% platinum on carbon. The choice of a particular
catalyst and the
amounts of catalyst, the amount of hydrogen gas, and the hydrogen gas pressure
used to effect
the desired transformation will depend upon the specific subject compound and
the particular
reaction conditions utilized. Such choices are within the skill of one of
ordinary skill in the art.
Furthermore, such benzyl and substituted benzyl ethers may be cleaved under
oxidative
conditions in which a suitable amount of an oxidizer is used. Such suitable
oxidizers include, but
are not limited to, dichlorodicyanoquinone (DDQ), ceric ammonium nitrate
(CAN), ruthenium
oxide in combination with sodium periodate, iron (III) chloride, or ozone.
Additionally, such ethers
may be cleaved using an appropriate Lewis acid. Such suitable Lewis acids
include, but are not
limited to, dimethylbromo borane, triphenylmethyl tetrafluoroborate, sodium
iodide in combination
with trifluoroborane-etherate, trichloroborane, or tin (IV) chloride. The
cleavage of a benzyl or
substituted benzyl ether protecting group may be conducted in an appropriate
solvent that is
compatible with the specific reaction conditions chosen and will not inten'ere
with the desired
transformation. Among such suitable solvents are, for example, alkyl esters,
alkylaryl esters, aryl
esters, alkyl ethers, aryl ethers, alkylaryl esters, cyclic ethers,
hydrocarbons, alcohols,
halogenated solvents, alkyl nitrites, aryl nitrites, alkyl ketones, aryl
ketones, alkylaryl ketones, or
non-protic heterocyclic compounds. For example, suitable solvents include, but
are not limited to,
ethyl acetate, isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl
isobutyl ketone,
dimethoxyethane, diisopropyl ether, chlorobenzene, dimethyl formamide,
dimethyl acetamide,
propionitrile, butyronitrile, t-amyl alcohol, acetic acid, diethyl ether,
methyl-t-butyl ether, diphenyl
ether, methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-
dioxane, pentane,
hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-
butanol, 2-butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Finally, such
reactions may be performed at an appropriate temperature from -20 °C to
100 °C, depending on
the specific reactants used. The choice of a suitable temperature is within
the skill of one of
ordinary skill in the art. Further°suitable reaction conditions may be
found in Greene et al.,
Protective Grouas in Organic Synthesis, John Wiley 8~ Sons, New York, (1999).
When R3 is a methyl ether, cleavage of the protecting group may be
accomplished by
treating the subject compound with organic or inorganic acids or Lewis acids.
The choice of a
particular reagent will depend upon the type of methyl ether present as well
as the other reaction
conditions. The choice of a suitable reagent for cleaving a methyl ether is
within the skill of one of
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
_29_
ordinary skill in the art. Examples of suitable reagents include, but are not
limited to, hydrochloric
acid, sulfuric acid, nitric acid, para-tol.uenesulfonic acid, or Lewis acids
such as boron trifluoride
etherate. These reactions may be conducted in solvents that are compatible
with the specific
reaction conditions chosen and will not interfere with the desired
transformation. Among such
suitable solvents are, for example, alkyl esters, alkylaryl esters, aryl
esters, alkyl ethers, aryl
ethers, alkylaryl esters, cyclic ethers, hydrocarbons, alcohols, halogenated
solvents, alkyl nitrites,
aryl nitrites, alkyl ketones, aryl ketones, alkylaryl ketones, or non-protic
heterocyclic compounds.
For example, suitable solvents include, but are not limited to, ethyl acetate,
isobutyl acetate,
isopropyl acetate,, n-butyl acetate, methyl isobutyl ketone, dimethoxyethane,
diisopropyl ether,
chlorobenzene, dimethyl formamide, dimethyl acetamide, propionitrile,
butyronitrile, t-amyl
alcohol, acetic acid, diethyl ether, methyl-t-butyl ether, Biphenyl ether,
methylphenyl ether,
tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, pentane, hexane,
heptane, methanol,
ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-butanol,
dichloromethane, chloroform,
1,2-dichloroethane, acetonitrile, benzonitrile, acetone, 2-butanone, benzene,
toluene, anisole,
xylenes, and pyridine, or any mixture of the above solvents. Additionally,
water may be used as a
co-solvent in this transformation if necessary. Finally, such reactions may be
pen'ormed at an
appropriate temperature from -20 °C to 100 °C, depending on the
specific reactants used. The
choice of a suitable temperature is within the skill of one of ordinary skill
in the art. Further
suitable reaction conditions may be found in Greene et al., Protective Groups
in Organic
S nt~, John Wiley & Sons, New York, (1999).
When R3 is a carbonate, cleavage of the protecting group may be accomplished
by
treating the subject compound with suitable basic compounds Such suitable
basic compounds
may include, but are not limited to, sodium carbonate, sodium bicarbonate,
potassium carbonate,
potassium bicarbonate, sodium hydroxide, or potassium hydroxide. The choice of
a particular
reagent will depend upon the type of carbonate present as well as the other
reaction conditions.
These reactions may be conducted in solvents that are compatible with the
specific reaction
conditions chosen and will not intertere with the desired transformation.
Among such suitable
solvents are, for example, alkyl esters, alkylaryl esters, aryl esters, alkyl
ethers, aryl ethers,
alkylaryl esters, cyclic ethers, hydrocarbons, alcohols, halogenated solvents,
alkyl nitrites, aryl
nitrites, alkyl ketones, aryl ketones, alkylaryl ketones, or non-protic
heterocyclic compounds. For
example, suitable solvents include, but are not limited to, ethyl acetate,
isobutyl acetate, isopropyl
acetate, n-butyl acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl
ether,
chlorobenzene, dimethyl formamide, dimethyl acetamide, propionitrile,
butyronitrile, t-amyl
alcohol, acetic acid, diethyl ether, methyl-t-butyl ether, Biphenyl ether,
methylphenyl ether,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-30-
tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, pentane, hexane,
heptane, methanol,
ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-butanol,
dichloromethane, chloroform,
1,2-dichloroethane, acetonitrile, benzonitrile, acetone, 2-butanone, benzene,
toluene, anisole,
xylenes, and pyridine, or any mixture of the above solvents. Additionally,
water may be used as a
co-solvent in this transformation if necessary. Finally, such reactions may be
performed at an
appropriate temperature from -20 °C to 100 °C, depending on the
specific reactants used. The
choice of a suitable temperature is within the skill of one of ordinary skill
in the art. Further
suitable reaction conditions may be found in Greene et al., Protective Groups
in Oraanic
~nthesis; John Wiley & Sons, New York, (1999).
Furthermore, compounds of formula (1) wherein R~ is phenyl substituted by at
least one
hydroxy group, and R3 is hydrogen, may be prepared from compounds of formula
(I) wherein R~
is phenyl optionally substituted by at least one substituent independently
chosen from C~_s
alkylcarbonyloxy, Cs_~o arylcarbonyloxy, and heteroarylcarbonyloxy; and R3 is
a hydroxyl
protecting group. In these compounds, the R~ C~_s alkylcarbonyloxy, Cs_~o
arylcarbonyloxy, and
heteroarylcarbonyloxy group and the R3 hydroxyl protecting group may be
removed using
reactions conditions in which both groups are removed concomitantly or they
may be removed in
step-wise fashion. For example, when R' is phenyl substituted by
alkylcarbonyloxy and R3 is an
alkyl ester, both groups may be cleaved by reacting the subject compound with
a base in an
appropriate solvent and at an appropriate temperature. The choice of a
suitable base, solvent,
and temperature will depend on the particular subject compound and the
particular protecting
groups being utilized. These choices are within the skill of one of ordinary
skill in the art. For
example, in compound (1), wherein R~ is phenyl substituted with
methylcarbonyloxy and methyl
and R3 is acetoxy, the methylcarbonyl and acetoxy protecting groups were
cleaved concomitantly
upon reacting compound (1) with potassium hydroxide in a mixture of methanol
and acetonitrile to
afford the desired compound, as shown below.
\ / \ /
CH3 O O O CH3 O O O
H3C~O~N N~N~ ~ HO ~ N N~N
O \ I / H OAc C j~ H MeOH I / H OH C jC H
S S
(~) CH3CN
Alternatively, in compounds of formula (I) wherein R~ is phenyl substituted by
at least one
group selectEd from C~_s alkylcarbonyloxy, Cs_~o arylcarbonyloxy, and
heteroarylcarbonyloxy, and
R3 is a hydroxyl protecting group, the C~_s alkylcarbonyloxy, Cs_~o
arylcarbonyloxy, and
heteroarylcarbonyloxy group and the R3 hydroxyl protecting group may be
cleaved in a stepwise
manner to afford a compound of formula (I) wherein R~ is phenyl substituted by
hydroxy and R3 is
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-31-
hydrogen. The choice of the R3 hydroxyl protecting group and the conditions to
affect its
cleavage will depend upon the specific subject compound chosen and is within
the knowledge of
one of ordinary skill in the art. For example, in the compounds of formula (I)
wherein R' is phenyl
substituted by C~_s alkylcarbonyloxy and R3 is a silane protecting group, the
R3 silane protecting
group may be cleaved first by treatment of the subject compound with a
fluoride source such as
tetrabutylammonium fluoride in acetonitrile at room temperature, followed by
cleavage of the C~_s
alkylcarbonyloxy group in R' by treatment with a base such as potassium
hydroxide in a mixture
of methanol and acetonitrile at room temperature.
Compounds of formula (I) wherein Z, R~, R2, R2~, R3, R4, R5, Rs, and R', are
as
hereinbefore defined may be prepared by reacting a compound of formula (II),
wherein Y' is a
leaving group and R' and R3 are as hereinbefore defined,
HN'~~Y~
R~ ~O OR3
(II)
with a compound of formula (III),
R~
O N_R2,
R~
HN ~Rs
R4 Z
5
(III)
or a salt or solvate thereof.
The present invention specifically contemplates that the compounds of formula
(I) may be
prepared by reacting compounds of formula (III) with compounds of formula
(II), wherein R3 is
hydrogen, an optionally substituted C~~ alkyl group, or a suitable protecting
group, such as a C~_s
alkylcarbonyl, Cs_~o arylcarbonyl, or heteroarylcarbonyl group. For example,
as shown below,
compound (2), wherein R3 is methylcarboxy, was treated with thionyl chloride
in a mixture of
pyridine and acetonitrile and was then allowed to react with compound (3) to
afford the desired
compound (4), as shown below.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-32-
CH O Ph0 O CH3 O Ph0 O
SOCIa Ac0
Ac0 I ~ N OH + HCI ~ H/N , H~ --. I \ H~N ~ H
/\~H OAc \S%C pyridine ~ OAc C
S
CH3CN
(2) (3) (4)
Alternatively, as shown below, compound (5), wherein R3 is hydrogen, was
allowed to
react with compound (3) to afford the desired product, compound (6).
CH3 O ~Ph0II ~O DIC CH3 O rPhOII O
Ac0 I ~ H~OH + HN~H~ ~ Ac0 ~ N~N N
OH ~ i~ HOBt~H20 I / H OH C ' H
S
2-Me-THF
(5) (3) (6)
Whether R3 in the compounds of formula (II) is hydrogen, an optionally
substituted C~~
alkyl group, or a suitable protecting group is dependent on the specific
product compounds
desired and/or the specific reaction conditions used. Such choices are within
the knowledge of
one of ordinary skill in the art.
For example, as shown below, compound (5) was allowed to react with acetic
anhydride
in ethyl acetate and methanesulfonic acid at about 70 °C to afford
compound (2).
\ / \ /
CH3 O O 1. Ac~O, CH3SO3H CH3 O O
EtOAc
Ac0 ~ N OH Ac0
'H OH
H OH 2. Crystallize / OAc
~5~ (2)
Compounds of formula (II), wherein Y' is hydroxy and R~ and R3 are as
hereinbefore
defined, can be prepared by reaction of compounds of formula (IV), wherein Y~
and R3 are as
hereinbefore defined, with compounds of formula (V), wherein R' is as
hereinbefore defined and
YZ is hydroxy or a suitable leaving group, as shown below.
O
Y1 + R~~Y2 ~ Y~
OR" R~~O OR3
(IV) (V) (II)
In general, these reactions may be performed in a solvent that does not
interfere with the
reaction, for example alkyl or aryl ethers, alkyl or aryl esters, aromatic and
aliphatic
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-33-
hydrocarbons, non-competitive alcohols, halogenated solvents, alkyl or aryl
nitrites, alkyl or aryl
ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For example,
suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, diphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be performed at
temperatures
from -20 °C to 100 °C, depending on the specific reactants,
solvents, and other optional additives
used. Such reactions may also be promoted by the addition of optional
additives. Examples of
such additives include, but are not limited to, hydroxybenzotriazole (HOBt),
hydroxyazabenzotriazole (HOAt), N-hydroxysuccinimide (HOSu), N-hydroxy-5-
norbornene-endo-
2,3-dicarboximide (HONB), and 4-dimethylaminopyridine (DMAP). Whether these
additives are
necessary depends on the identity of the reactants, the solvent, and the
temperature. Such
choices are within the knowledge of one of ordinary skill in the art.
In general, the leaving group YZ in the compounds of formula (V) should be
such that it
provides sufficient reactivity with the amine in the compounds of formula
(IV). Compounds of
formula (V) that contain such suitable leaving groups may be prepared,
isolated andlor purified,
and subsequently reacted with the compounds of formula (IV). Alternatively,
compounds of
formula (V) with suitable leaving groups may be prepared and further reacted
without isolation or
further purification with the compounds of formula (IV) to afford compounds of
formula (11).
Among suitable leaving groups in the compounds of formula (V) are halides,
aromatic
heterocycles, sulfonic acid esters, phosphoric acid esters, anhydrides, or
groups derived from the
reaction of compounds of formula (V) wherein Y2 is hydroxy with reagents such
as carbodiimides
or carbodiimide species. Examples of suitable leaving groups include, but are
not limited to,
chloride, iodide, imidazole, -OC(O)alkyl, -OC(O)aryl, -OC(O)Oalkyl, -
OC(O)Oaryl, -OS(02)alkyl,
-OS(OZ)aryl, -OPO(Oaryl)2, OPO(Oalkyl)2, and those derived from the reaction
of the compounds
of formula (V) wherein Y2 is -OH with carbodiimides. Other suitable leaving
groups are known to
those of ordinary skill in the art and may be found, for example, in Humphrey,
J.M.; Chamberlin,
A.R. Chem. Rev., 1997, 97, 2243; Comprehensive Oraanic S nthesis; Trost, B.
M., Ed.;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-34-
Pergamon: New York, (1991); Vol. 6, pp 301-434; and Comprehensive Oraanic
Transformations;
Larock, R. C.; VCH: New York, (1989), Chapter 9.
Compounds of formula (V) where in Y2 is a halogen can be prepared from
compounds of
formula (V) wherein YZ is hydroxy by reaction with a suitable agent. For
example, the compounds
of formula (V) wherein Y2 is chloro may be prepared from compounds of formula
(V) wherein Y2 is
hydroxy by reaction with agents such as thionyl chloride or oxalyl chloride.
These reactions may
be performed in the presence of a suitable base such as sodium carbonate,
sodium bicarbonate,
potassium carbonate, potassium bicarbonate, sodium hydroxide, potassium
hydroxide, a
trialkylamine, triethylamine for example, or a heteroaromatic base, pyridine
for example. The
resulting compounds may be isolated and then further reacted with the
compounds of formula
(IV) or they may be formed in situ and reacted with the compounds of formula
(IV) without
isolation or further purification. These reactions may be performed in a
solvent that does not
intertere with the desired transformation. Among suitable solvents are alkyl
or aryl ethers, alkyl or
aryl esters, aromatic and aliphatic hydrocarbons, halogenated solvents, alkyl
or aryl nitrites, alkyl
or aryl ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For
example, suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile; butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, diphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be performed at
temperatures
from -20 °C to 100 °C. The specific reaction conditions chosen
will depend on the specific
subject compound and reagents chosen. Such choices are within the knowledge of
one of
ordinary skill in the art. For example, as shown below, compound (7) was
allowed to react with
compound (8) in a mixture of tetrahydrofuran and water, in the presence of
triethylamine, at room
temperature to afford the desired compound (5).
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-35-
CHs O -
Ac0 ~ CI
O ~ CH3 O O
i (~)
H2N OH Ac0 I ~ H OH
OH NEt3, THF, H2O / OH
(~) (5)
Compounds of formula (IV), wherein Y~ is hydroxy and R3 is as defined above,
are either
commercially available or can be prepared by methods known to those of skill
in the art.
Y~
(IV)
For example, the compounds of formula (IV) can be prepared as shown in the
scheme
below. In general, an N-protected amino acid derivative is reduced to an
aldehyde using
reducing agents that are suitable for such a transformation. For example,
suitable reducing
agents are dialkyl aluminum hydride agents, such as diisobutyl aluminum
hydride for example.
Another method of preparing the compounds of formula (IV) is to reduce an
appropriate
carboxylic acid to an alcohol with a suitable reducing agent such as LiAIH4 or
BH3 or NaBH4for
example, followed by oxidation of the alcohol to the corresponding aldehyde
with PCC, under
Swern conditions or using pyr~SO~/DMSO/NEt3for example Another method of
preparing the
compounds of formula (IV) is to reduce an appropriate carboxylic acid
derivative, such as a
Weinreb amide or an acyl imidazole, using a suitable reducing agent such as
LiAIH4 or diisobutyl
aluminum hydride for example. Alternatively, the compounds of formula (IV) can
be prepared by
the preparation of an appropriate aldehyde by reduction of the corresponding
acid chloride. Next,
a compound is added to the aldehyde that is the equivalent of adding a
carboxylate COZ anion.
For example, cyanide can be added to the aldehyde to afford a cyanohydrin that
can then be
hydrolyzed under either acidic or basic conditions to afford the desired
compound, (d).
Alternatively, nitromethane may be added to the aldehyde under basic
conditions to afford an
intermediate that is then converted into the desired compound. These compounds
can be
prepared according to the following procedures. In those compounds where Y3 is
-CN, R.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-36-
Pedrosa et al., Tetrahedron Asymm. 2001, 12, 347. For those compounds in which
Y3 is -
CH2N02, M. Shibasaki et al., Tetrahedron Lett. 1994, 35, 6123.
w1 w1 w1 w1
o
3
PgHN OH PgHN H PgHN Y PgHN OH
O O OH OH
a b c d
Y3=-CN or-CHZNOZ
Pg= protecting group
Compounds of formula (V), wherein YZ is hydroxy and R~ is as hereinbefore
defined, are
either commercially available or can be prepared by methods known to those of
skill in the art.
For example, such compounds can be prepared from the corresponding alcohols by
oxidation
with suitable reagents. Such oxidation agents include, but are not limited to,
KMn04, pyridinium
dichromate (PDC), H2Cr20~ (Jones' reagent), and 2,2,6,6-tetramethylpiperidinyl-
2-oxyl
(TEMPO)/NaClOz.
Compounds of formula (III), wherein Z is S, O, SO, SO~, CH2, or CFH, and R~,
R2~, R4, R5,
Rs, and R' are as hereinbefore defined, are either commercially available or
can be prepared
according to methods known to those of skill in the art. For example, see
Mimoto, T. et al. J.
Med. Chem. 1999, 42, 1789; EP 0751145; U.S. Pat. Nos. 5,644,028, 5,932,550,
5,962,640,
5,932,550, and 6,222,043, H. Hayashi et al., J. Med. Chem. 1999, 42, 1789; and
PCT Publication
No. WO 01/05230 A1, which are hereby incorporated by reference.
Alternatively, the compounds of formula (I), wherein R' is phenyl optionally
substituted by
at least one substituent independently chosen from C~.o alkyl, hydroxyl, C~_6
alkylcarbonyloxy, C6_
~o arylcarbonyloxy, and heteroarylcarbonyloxy, and Z, Rz, R2~, R3, R4, R5, Rs,
and R' are as
hereinbefore defined, may be prepared by reaction of compounds of formula
(VI),
R2
I
N~R2'
~ R'
H2N
R4 i
R5 (VI)
wherein Z, R2, R2~, R3, R4, R5, R6, and R' are as hereinbefore defined with
compounds of formula
(V), wherein R~ and YZ are as hereinbefore defined.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-37-
In general, these reactions may be pertormed in a solvent that does not
intertere with the
reaction, for example alkyl or aryl ethers, alkyl or aryl esters, aromatic and
aliphatic
hydrocarbons, non-competitive alcohols, halogenated solvents, alkyl or aryl
nitrites, alkyl or aryl
ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For example,
suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, diphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be pertormed at
temperatures
from -20 °C to 100 °C, depending on the specific reactants,
solvents, and other optional additives
used. Such reactions may also be promoted by the addition of optional
additives. Examples of
such additives include, but are not limited to, hydroxybenzotriazole (HOBt),
hydroxyazabenzotriazole (HOAt), N-hydroxysuccinimide (HOSu), N-hydroxy-5-
norbornene-endo-
2,3-dicarboximide (HONB), and 4-dimethylaminopyridine (DMAP).Whether these
additives are
necessary depends on the identity of the reactants, the solvent, and the
temperature. Such
choices are within the knowledge of one of ordinary skill in the art.
In general, the leaving group Y2 in the compounds of formula (V) should be
such that it
provides sufficient reactivity with the amino group in the compounds of
formula (VI). Compounds
of formula (V) that contain such suitable leaving groups may be prepared,
isolated and/or purified,
and subsequently reacted with the compounds of formula (VI). Alternatively,
compounds of
formula (V) with suitable leaving groups may be prepared and further reacted
without isolation or
further purification with the compounds of formula (VI) to afford compounds of
formula (I). Among
suitable leaving groups in the compounds of formula (V) are halides, aromatic
heterocycles,
sulfonic acid esters, phosphoric acid esters, anhydrides, or groups derived
from the reaction of
compounds of formula (V) wherein Y2 is hydroxy with reagents such as
carbodiimides or
carbodiimide species. Examples of suitable leaving groups include, but are not
limited to,
chloride, iodide, imidazole, -OC(O)alkyl, -OC(O)aryl, -OC(O)Oalkyl, -
OC(O)Oaryl, -OS(OZ)alkyl,
-OS(02)aryl, -OPO(Oaryl)Z, OPO(Oalkyl)2, and those derived from the reaction
of the compounds
of formula (V), wherein Y2 is -OH, with carbodiimides.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-38-
Compounds of formula (V) where in Y2 is a halogen can be prepared from
compounds of
formula (V) wherein Y2 is hydroxy by reaction with a suitable agent. For
example, the compounds
of formula (V) wherein Y2 is chloro may be prepared from compounds of formula
(V) wherein Y2 is
hydroxy by reaction with agents such as thionyl chloride or oxalyl chloride.
These reactions may
be pertormed in the presence of a suitable base such as sodium carbonate,
sodium bicarbonate,
potassium carbonate, potassium bicarbonate, sodium hydroxide, potassium
hydroxide, a
trialkylamine, triethylamine for example, or a heteroaromatic base, pyridine
for example. The
resulting compounds may be isolated and then further reacted with the
compounds of formula
(VI) or they may be formed in situ and reacted with the compounds of formula
(VI) without
isolation or further purification. These reactions may be pertormed in a
solvent that does not
interfere with the desired transformation. Among suitable solvents are alkyl
or aryl ethers, alkyl or
aryl esters, aromatic and aliphatic hydrocarbons, halogenated solvents, alkyl
or aryl nitrites, alkyl
or aryl ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For
example, suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, diphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be performed at
temperatures
from -20 °C to 100 °C. The specific reaction conditions chosen
will depend on the specific
subject compound and reagents chosen. Such choices are within the knowledge of
one of
ordinary skill in the art.
Compounds of formula (VI),
R2
N~R2'
R~
Rs
Z
(VI)
wherein Z, R2, R~~, R3, R4, R5, R6, and R' are as hereinbefore defined, may be
prepared from
reaction of compounds of formula (VII),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-39-
Y4
OR° (VII)
wherein Pg~ is a suitable nitrogen protecting group, Y4 is hydroxy or a
suitable leaving group, and
R3 is as hereinbefore defined, with a compound of formula (III), wherein Z,
R2, R2~, R4, R5, R6, and
R' are as hereinbefore defined, or a salt or solvate thereof.
A suitable protecting group Pg~ in the compounds of formula (VII) is one that
is stable to
subsequent reaction conditions in which the compounds of formula (VII) are
allowed to react with
the compounds of formula (III). Furthermore, such a protecting group should be
chosen such that
it can be removed after the compounds of formula (VII) have been allowed to
react with the
compounds of formula (III) to afford an intermediate compound that is
subsequently deprotected
to afford a compound of formula (VI). Suitable protecting groups include, but
are not limited to,
carbamates such as t-butyloxycarbonyl and benzyloxycarbonyl, imides such as
phthaloyl, or
suitable benzyl groups. Such protecting groups can be introduced into the
compounds of formula
(VII) and subsequently removed to provide compounds of formula (VI) according
to methods
known to those of ordinary skill in the art and as found in, for example,
Greene et al., Protective
Groups in Oraanic S nthesis; John Wiley & Sons: New York, (1999).
In general, the leaving group Y4 in the compounds of formula (VII) should be
such that it
provides sufficient reactivity with the amino group in the compounds of
formula (III). Compounds
of formula (VII) that contain such suitable leaving groups may be prepared,
isolated and/or
purified, and subsequently reacted with the compounds of formula (III).
Alternatively, compounds
of formula (VII) with suitable leaving groups may be prepared and further
reacted without isolation
or further purification with the compounds of formula (III) to afford
compounds of formula (VI).
Among suitable leaving groups in the compounds of formula (VII) are halides,
aromatic
heterocycles, sulfonic acid esters, phosphoric acid esters, anhydrides, or
groups derived from the
reaction of compounds of formula (VII) wherein Y4 is hydroxy with reagents
such as
carbodiimides or carbodiimide species. Examples of suitable leaving groups
include, but are not
limited to, chloride, iodide, imidazole, -OC(O)alkyl, -OC(O)aryl, -
OC(O)Oalkyl, -OC(O)Oaryl,
-OS(OZ)alkyl, -OS(02)aryl, -OPO(Oaryl)2, -OPO(Oalkyl)2, and those derived from
the reaction of
the compounds of formula (VII), wherein Y4 is -OH, with carbodiimides.
Compounds of formula (VII) where in Y4 is a halogen can be prepared from
compounds
of formula (VII) wherein Y4 is hydroxy by reaction with a suitable agent. For
example, the
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-40-
compounds of formula (VII) wherein Y4 is chloro may be prepared from compounds
of formula
(VII) wherein Y4 is hydroxy by reaction with agents such as thionyl chloride
or oxalyl chloride.
These reactions may be pertormed in the presence of a suitable base such as
sodium carbonate,
sodium bicarbonate, potassium carbonate, potassium bicarbonate, sodium
hydroxide, potassium
hydroxide, a trialkylamine, triethylamine for example, or a heteroaromatic
base, pyridine for
example. The resulting compounds may be isolated and then further reacted with
the
compounds of formula (III) or they may be formed in situ and reacted with the
compounds of
formula (III) without isolation or further purification. These reactions may
be performed in a
solvent that does not intertere with the desired transformation. Among
suitable solvents are alkyl
or aryl ethers, alkyl or aryl esters, aromatic and aliphatic hydrocarbons,
halogenated solvents,
alkyl or aryl nitrites, alkyl or aryl ketones, aromatic hydrocarbons, or
heteroaromatic
hydrocarbons. For example, suitable solvents include, but are not limited to,
ethyl acetate,
isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone,
dimethoxyethane,
diisopropyl ether, chlorobenzene, dimethyl formamide, dimethyl acetamide,
propionitrile,
butyronitrile, t-amyl alcohol, acetic acid, diethyl ether, methyl-t-butyl
ether, diphenyl ether,
methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane,
heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-
butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be performed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Compounds of formula (VII) where in Y4 is an aromatic heterocycle can be
prepared from
compounds of formula (VII) wherein Y4 is hydroxy by reaction with a suitable
agent such as
carbonyl diimidazole. These compounds may be isolated and then further reacted
with the
compounds of formula (III) or they may be formed in situ and reacted with the
compounds of
formula (III) without isolation or further purification. These reactions may
be pertormed in a
solvent that does not interfere with the desired transformation. Among
suitable solvents are alkyl
or aryl ethers, alkyl or aryl esters, aromatic and aliphatic hydrocarbons,
halogenated solvents,
alkyl or aryl nitrites, alkyl or aryl ketones, aromatic hydrocarbons, or
heteroaromatic
hydrocarbons. For example, suitable solvents include, but are not limited to,
ethyl acetate,
isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone,
dimethoxyethane,
diisopropyl ether, chlorobenzene, dimethyl formamide, dimethyl acetamide,
propionitrile,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-41-
butyronitrile, t-amyl alcohol, acetic acid, diethyl ether, methyl-t-butyl
ether, diphenyl ether,
methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane,
heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-
butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be performed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the skill of one of ordinary skill in the art.
Compounds of formula (VII) wherein Y4 is -OC(O)alkyl or-OC(O)aryl may be
prepared
from compounds of formula (VII) wherein Y4 is hydroxy by reaction with
suitable reagents such
aryl halides, acyl imidazoles, or carboxylic acid under dehydrating
conditions. Suitable reagents
may include, but are not limited to, pivaloyl chloride, acetyl chloride,
acetyl iodide formed in situ
from acetyl chloride and sodium iodide, acetyl imidazole, or acetic acid under
dehydrating
conditions. These reactions may be pertormed in the presence of a suitable
base such as
sodium carbonate, sodium bicarbonate, potassium carbonate, potassium
bicarbonate, sodium
hydroxide, potassium hydroxide, a trialkylamine, triethylamine for example, or
a heteroaromatic
base, pyridine for example. The resulting compounds may be isolated and then
further reacted
with the compounds of formula (III) or they may be formed in situ and reacted
with the
compounds of formula (III) without isolation or further purification. These
reactions may be
performed in a solvent that does not interfere with the desired
transformation. Among suitable
solvents are alkyl or aryl ethers, alkyl or aryl esters, aromatic and
aliphatic hydrocarbons,
halogenated solvents, alkyl or aryl nitrites, alkyl or aryl ketones, aromatic
hydrocarbons, or
heteroaromatic hydrocarbons. For example, suitable solvents include, but are
not limited to, ethyl
acetate, isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl
ketone,
dimethoxyethane, diisopropyl ether, chlorobenzene, dimethyl formamide,
dimethyl acetamide,
propionitrile, butyronitrile, t-amyl alcohol, acetic acid, diethyl ether,
methyl-t-butyl ether, diphenyl
ether, methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-
dioxane, pentane,
hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-
butanol, 2-butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be perFormed at temperatures from -20 °C to 100
°C. The specific reaction
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-42-
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Compounds of formula (VII) wherein Y4 is -OC(O)Oalkyl, -OC(O)Oaryl can be
prepared
from compounds of formula (VII) wherein Y4 is hydroxy by reaction with a
suitable agents such as
chloroformates of the formula CI-C(O)Oalkyl or CI-C(O)Oaryl. These reactions
may be pertormed
in the presence of a suitable base such as sodium carbonate, sodium
bicarbonate, potassium
carbonate, potassium bicarbonate, sodium hydroxide, potassium hydroxide, a
trialkylamine,
triethylamine for example, or a heteroaromatic base, pyridine for example. The
resulting
compounds may be isolated and then further reacted with the compounds of
formula (III) or they
may be formed in situ and reacted with the compounds of formula (III) without
isolation or further
purification. These reactions may be pertormed in a solvent that does not
intertere with the
desired transformation. Among suitable solvents are alkyl or aryl ethers,
alkyl or aryl esters,
aromatic and aliphatic hydrocarbons, halogenated solvents, alkyl or aryl
nitrites, alkyl or aryl
ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For example,
suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, Biphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be performed at
temperatures
from -20 °C to 100 °C. The specific reaction conditions chosen
will depend on the specific
subject compound and reagents chosen. Such choices are within the knowledge of
one of
ordinary skill in the art.
Compounds of formula (VII) wherein Y4 is -OS(02)alkyl or-OS(02)aryl can be
prepared
from compounds of formula (VII) wherein Y4 is hydroxy by reaction with a
suitable agent such as
an alkyl or aryl sulfonyl chloride. These reactions may be performed in the
presence of a suitable
base such as sodium carbonate, sodium bicarbonate, potassium carbonate,
potassium
bicarbonate, sodium hydroxide, potassium hydroxide, a trialkylamine,
triethylamine for example,
or a heteroarornatic base, pyridine for example. The resulting compounds may
be isolated and
then further reacted with the compounds of formula (III) or they may be formed
in situ and reacted
with the compounds of formula (III) without isolation or further purification.
These reactions may:
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-43-
be performed in a solvent that does not interfere with the desired
transformation. Among suitable
solvents are alkyl or aryl ethers, alkyl or aryl esters, aromatic and
aliphatic hydrocarbons,
halogenated solvents, alkyl or aryl nitrites, alkyl or aryl ketones, aromatic
hydrocarbons, or
heteroaromatic hydrocarbons. For example, suitable solvents include, but are
not limited to, ethyl
acetate, isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl
ketone,
dimethoxyethane, diisopropyl ether, chlorobenzene, dimethyl formamide,
dimethyl acetamide,
propionitrile, butyronitrile, t-amyl alcohol, acetic acid, diethyl ether,
methyl-t-butyl ether, diphenyl
ether, methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-
dioxane, pentane,
hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-
butanol, 2-butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be pertormed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Alternatively, compounds of formula (VI) may be prepared by reaction of
compounds of
formula (VII), wherein Y4 is -OH, with compounds of formula (III) under
dehydrating conditions
using agents such as carbodiimides or carbodiimide derived species Such
suitable agents
include, but are not limited to, dicyclohexylcarbodiimide,
diisopropylcarbodiimide, 1-[3-
(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC), 2-chloro-4,6-
dimethoxy-1,3,5-
triazine (CDMT), cyanuric chloride, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-
methylmorpholinium
chloride, O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate (HATU),
carbonyldiimidazole (CDI), benzotriazole-1-yl-oxy-tris-(dimethylamino)-
phosphoniumhexafluorophosphate (BOP), 2-ethoxy-1-ethoxycarbonyl-1,2-
dihydroquinoline
(EEDQ), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate (HBTU), 2-
(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrefluoroborate (TBTU),
and 3-
(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4.(3H)-one (DEPBT). These reactions
may be
performed in the presence of optional additives. Suitable additives include,
but are not limited to,
hydroxybenzotriazole (HOBt), hydroxyazabenzotriazole (HOAt), N-
hydroxysuccinimide (HOSu),
N-hydroxy-5-norbornene-endo-2,3-dicarboximide (HONB), and 4-
dimethylaminopyridine (DMAP).
Whether these additives are necessary depends on the identity of the
reactants, the solvent, and
the temperature. Such choices are within the knowledge of one of ordinary
skill in the art.
Alternatively, the compounds of formula (I) may be prepared by reaction of a
compound
of formula (VIII),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-44-
\ . O O Y5
HN N R~
R1 ~O OR3 ~Z R6
R4 R5
(VIII)
wherein Y5 is hydroxy or a suitable leaving group, and Z, R~, R3, R4, R5, R6,
and Rare as
hereinbefore defined, with a compound of formula (IX),
R2
~oNH
R2,/
(IX)
wherein R2 and R~~ are hereinbefore defined, or a salt or solvate thereof.
In general, the leaving group Y$ in the compounds of formula (VIII) should be
such that it
provides sufficient reactivity with the amino group in the compounds of
formula (IX). Compounds
of formula (VIII) that contain such suitable leaving groups may be prepared,
isolated and/or
purified, and subsequently reacted with the compounds of formula (IX).
Alternatively, compounds
of formula (VIII) with suitable leaving groups may be prepared and further
reacted without
isolation or further purification with the compounds of formula (IX) to afford
compounds of formula
(I). Among suitable leaving groups in the compounds of formula (VIII) are
halides, aromatic
heterocycles, sulfonic acid esters, anhydrides, or groups derived from the
reaction of compounds
of formula (VIII) wherein YS is hydroxy with reagents such as carbodiimides or
carbodiimide
species. Examples of suitable leaving groups include, but are not limited to,
chloride, iodide,
imidazole, -OC(O)alkyl, -OC(O)aryl, -OC(O)Oalkyl, -OC(O)Oaryl, -OS(02)alkyl, -
OS(02)aryl,
-OPO(Oalkyl)2, -OPO(Oaryl)2, and those derived from the reaction of the
compounds of formula
(VIII), wherein YS is -OH, with carbodiimides.
Compounds of formula (VIII) where in Y$ is a halogen can be prepared from
compounds
of formula (VIII) wherein Y5 is hydroxy by reaction with a suitable agent. For
example, the
compounds of formula (VIII) wherein YS is chloro may be prepared from
compounds of formula
(VIII) wherein Y5 is hydroxy by reaction with agents such as thionyl chloride
or oxalyl chloride.
These reactions may be pertormed in the presence of a suitable base such as
sodium carbonate,
sodium bicarbonate, potassium carbonate, potassium bicarbonate, sodium
hydroxide, potassium
hydroxide, a trialkylamine, triethylamine for example, or a heteroaromatic
base, pyridine for
example. The resulting compounds may be isolated and then further reacted with
the
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-45-
compounds of formula (IX) or they may be formed in situ and reacted with the
compounds of
formula (IX) without isolation or further purification. These reactions may be
pertormed in a
solvent that does not intertere with the desired transformation. Among
suitable solvents are alkyl
or aryl ethers, alkyl or aryl esters, aromatic and aliphatic hydrocarbons,
halogenated solvents,
alkyl or aryl nitrites, alkyl or aryl ketones, aromatic hydrocarbons, or
heteroaromatic
hydrocarbons. For example, suitable solvents include, but are not limited to,
ethyl acetate,
isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone,
dimethoxyethane,
diisopropyl ether, chlorobenzene, dimethyl formamide, dimethyl acetamide,
propionitrile,
butyronitrile, t-amyl alcohol, acetic acid, diethyl ether, methyl-t-butyl
ether, diphenyl ether,
methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane,
heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-
butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be performed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Compounds of formula (VIII) where in YS is an aromatic heterocycle can be
prepared
from compounds of formula (VIII) wherein YS is hydroxy by reaction with a
suitable agent such as
carbonyl diimidazole. These compounds may be isolated and then further reacted
with the
compounds of formula (IX) or they may be formed in situ and reacted with the
compounds of
formula (IX) without isolation or further purification. These reactions may be
performed in a
solvent that does not interfere with the desired transformation. Among
suitable solvents are alkyl
or aryl ethers, alkyl or aryl esters, aromatic and aliphatic hydrocarbons,
halogenated solvents,
alkyl or aryl nitrites, alkyl or aryl ketones, aromatic hydrocarbons, or
heteroaromatic
hydrocarbons. For example, suitable solvents include, but are not limited to,
ethyl acetate,
isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone,
dimethoxyethane,
diisopropyl ether, chlorobenzene, dimethyl formamide, dimethyl acetamide,
propionitrile,
butyronitrile, t-amyl alcohol, acetic acid, diethyl ether, methyl-t-butyl
ether, diphenyl ether,
methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane,
heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-butanol, 2-
butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-4.6-
such reactions may be performed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Compounds of formula (VIII) wherein Y5 is -OC(O)alkyl or-OC(O)aryl may be
prepared
from compounds of formula (VIII) wherein Y5 is hydroxy by reaction with
suitable reagents such
acyl halides, acyl imidazoles, or carboxylic acid under dehydrating
conditions. Suitable reagents
may include, but are not limited to, pivaloyl chloride, acetyl chloride,
acetyl iodide formed in situ
from acetyl chloride and sodium iodide, acetyl imidazole, or acetic acid under
dehydrating
conditions. These reactions may be performed in the presence of a suitable
base such as
sodium carbonate, sodium bicarbonate, potassium carbonate, potassium
bicarbonate, sodium
hydroxide, potassium hydroxide, a trialkylamine, triethylamine for example, or
a heteroaromatic
base, pyridine for example. The resulting compounds may be isolated and then
further reacted
with the compounds of formula (IX) or they may be formed in situ and reacted
with the
compounds of formula (IX) without isolation or further purification. These
reactions may be
performed in a solvent that does not interfere with the desired
transformation. Among suitable
solvents are alkyl or aryl ethers, alkyl or aryl esters, aromatic and
aliphatic hydrocarbons,
halogenated solvents, alkyl or aryl nitrites, alkyl or aryl ketones, aromatic
hydrocarbons, or
heteroaromatic hydrocarbons. For example, suitable solvents include, but are
not limited to, ethyl
acetate, isobutyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl
ketone,
dimethoxyethane, diisopropyl ether, chlorobenzene, dimethyl formamide,
dimethyl acetamide,
propionitrile, butyronitrile, t-amyl alcohol, acetic acid, diethyl ether,
methyl-t-butyl ether, diphenyl
ether, methylphenyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-
dioxane, pentane,
hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol, t-butanol, n-
butanol, 2-butanol,
dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile, benzonitrile,
acetone, 2-butanone,
benzene, toluene, anisole, xylenes, and pyridine, or any mixture of the above
solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be performed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Compounds of formula (VIII) wherein Y5 is -OC(O)Oalkyl, -OC(O)Oaryl can be
prepared
from compounds of formula (VIII) wherein YS is hydroxy by reaction with a
suitable agents such
as chloroformates of the formula CI-C(O)Oalkyl or CI-C(O)Oaryl. These
reactions may be
pertormed in the presence of a suitable base such as sodium carbonate, sodium
bicarbonate,
potassium carbonate, potassium bicarbonate, sodium hydroxide, potassium
hydroxide, a
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-4.7-
trialkylamine, triethylamine for example, or a heteroaromatic base, pyridine
for example. The
resulting compounds may be isolated and then further reacted with the
compounds of formula
(IX) or they may be formed in situ and reacted with the compounds of formula
(IX) without
isolation or further purification. These reactions may be pertormed in a
solvent that does not
interfere with the desired transformation. Among suitable solvents are alkyl
or aryl ethers, alkyl or
aryl esters, aromatic and aliphatic hydrocarbons, halogenated solvents, alkyl
or aryl nitrites, alkyl
or aryl ketones, aromatic hydrocarbons, or heteroaromatic hydrocarbons. For
example, suitable
solvents include, but are not limited to, ethyl acetate, isobutyl acetate,
isopropyl acetate, n-butyl
acetate, methyl isobutyl ketone, dimethoxyethane, diisopropyl ether,
chlorobenzene, dimethyl
formamide, dimethyl acetamide, propionitrile, butyronitrile, t-amyl alcohol,
acetic acid, diethyl
ether, methyl-t-butyl ether, diphenyl ether, methylphenyl ether,
tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane, pentane, hexane, heptane, methanol,
ethanol, 1-propanol, 2-
propanol, t-butanol, n-butanol, 2-butanol, dichloromethane, chloroform, 1,2-
dichloroethane,
acetonitrile, benzonitrile, acetone, 2-butanone, benzene, toluene, anisole,
xylenes, and pyridine,
or any mixture of the above solvents. Additionally, water may be used as a co-
solvent in this
transformation if necessary. Furthermore, such reactions may be performed at
temperatures
from -20 °C to 100 °C. The specific reaction conditions chosen
will depend on the specific
subject compound and reagents chosen. Such choices are within the knowledge of
one of
ordinary skill in the art.
Compounds of formula (VIII) wherein Y5 is -OS(O~)alkyl or-OS(02)aryl can be
prepared
from compounds of formula (VIII) wherein Y5 is hydroxy by reaction with a
suitable agent such as
an alkyl or aryl sulfonyl chloride. These reactions may be performed in the
presence of a suitable
base such as sodium carbonate, sodium bicarbonate, potassium carbonate,
potassium
bicarbonate, sodium hydroxide, potassium hydroxide, a trialkylamine,
triethylamine for example,
or a heteroaromatic base, pyridine for example. The resulting compounds may be
isolated and
then further reacted with the compounds of formula (IX) or they may be formed
in situ and
reacted with the compounds of formula (IX) without isolation or further
purification. These
reactions may be performed in a solvent that does not intertere with the
desired transformation.
Among suitable solvents are alkyl or aryl ethers, alkyl or aryl esters,
aromatic and aliphatic
hydrocarbons, halogenated solvents, alkyl or aryl nitrites, alkyl or aryl
ketones, aromatic
hydrocarbons, or heteroaromatic hydrocarbons. For example, suitable solvents
include, but are
not limited to, ethyl acetate, isobutyl acetate, isopropyl acetate, n-butyl
acetate, methyl isobutyl
ketone, dimethoxyethane, diisopropyl ether, chlorobenzene, dimethyl formamide,
dimethyl
acetamide, propionitrile, butyronitrile, t-amyl alcohol, acetic acid, diethyl
ether, methyl-t-butyl
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-48-
ether, diphenyl ether, methylphenyl ether, tetrahydrofuran, 2-
methyltetrahydrofuran, 1,4-dioxane,
pentane, hexane, heptane, methanol, ethanol, 1-propanol, 2-propanol, t-
butanol, n-butanol, 2-
butanol, dichloromethane, chloroform, 1,2-dichloroethane, acetonitrile,
benzonitrile, acetone, 2-
butanone, benzene, toluene, anisole, xylenes, and pyridine, or any mixture of
the above solvents.
Additionally, water may be used as a co-solvent in this transformation if
necessary. Furthermore,
such reactions may be pen'ormed at temperatures from -20 °C to 100
°C. The specific reaction
conditions chosen will depend on the specific subject compound and reagents
chosen. Such
choices are within the knowledge of one of ordinary skill in the art.
Alternatively, compounds of formula (I) may be prepared by reaction of
compounds of
formula (VIII), wherein Y5 is -OH, with compounds of formula (IX) under
dehydrating conditions
using agents such as carbodiimides or carbodiimide derived species Such
suitable agents
include, but are not limited to, dicyclohexylcarbodiimide,
diisopropylcarbodiimide, 1-[3-
(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC), 2-chloro-4.,6-
dimethoxy-1,3,5-
triazine (CDMT), cyanuric chloride, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-
methylmorpholinium
chloride, O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate (HATU),
carbonyldiimidazole (CDI), benzotriazole-1-yl-oxy-tris-(dimethylamino)-
phosphoniumhexafluorophosphate (BOP), 2-ethoxy-1-ethoxycarbonyl-1,2-
dihydroquinoline
(EEDQ), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate (HBTU), 2-
(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrefluoroborate (TBTU),
and 3-
(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4.(3H)-one (DEPBT). These reactions
may be
performed in the presence of optional additives. Suitable additives include,
but are not limited to,
hydroxybenzotriazole (HOBt), hydroxyazabenzotriazole (HOAt), N-
hydroxysuccinimide (HOSu),
N-hydroxy-5-norbornene-endo-2,3-dicarboximide (HONB), and 4-
dimethylaminopyridine (DMAP).
Whether these additives are necessary depends on the identity of the
reactants, the solvent, and
the temperature. Such choices are within the knowledge of one of ordinary
skill in the art.
Compounds of formula (IX) are either commercially available or can be prepared
by
methods described herein or methods known to those of ordinary skill in the
art.
The examples and preparations provided below further illustrate and exemplify
the methods
of the present invention. It is to be understood that the scope of the present
invention is not limited
in any way by the scope of the following examples. In the following examples
compounds with
single or multiple stereoisomeric centers, unless otherwise noted, are at
least 95%
stereochemically pure.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-4.9-
EXAMPLES
In the examples described below, unless otherwise indicated, all temperatures
in the
following description are in degrees Celsius (°C) and all parts and
percentages are by weight,
unless indicated otherwise.
Various starting materials and other reagents were purchased from commercial
suppliers,
such as Aldrich Chemical Company or Lancaster Synthesis Ltd., and used without
further
purification, unless otherwise indicated.
The reactions set forth below were pertormed under a positive pressure of
nitrogen,
argon or with a drying tube, at ambient temperature (unless otherwise stated),
in anhydrous
solvents. Analytical thin-layer chromatography was performed on glass-backed
silica gel 60°F
254 plates (Analtech (0.25 mm)) and eluted with the appropriate solvent ratios
(v/v). The
reactions were assayed by high-pressure liquid chromotagraphy (HPLC) or thin-
layer
chromatography (TLC) and terminated as judged by the consumption of starting
material. The
TLC plates were visualized by UV, phosphomolybdic acid stain, or iodine stain.
~H-NMR spectra were recorded on a Bruker instrument operating at 300 MHz and
~3C~NMR spectra were recorded at 75 MHz. NMR spectra are obtained as DMSO-ds
or CDC13
solutions (reported in ppm), using chloroform as the reference standard (7.25
ppm and 77.00
ppm) or DMSO-ds ((2.50 ppm and 39.52 ppm)). Other NMR solvents were used as
needed.
When peak multiplicities are reported, the following abbreviations are used: s
= singlet, d =
doublet, t = triplet, m = multiplet, br = broadened, dd = doublet of doublets,
dt = doublet of triplets.
Coupling constants, when given, are reported in Hertz.
Infrared spectra were recorded on a Perkin-Elmer FT-IR Spectrometer as neat
oils, as
KBr pellets, or as CDCI3 solutions, and when reported are in wave numbers
(crri'). The mass
spectra were obtained using LC/MS or APCI. All melting points are uncorrected.
All final products had greater than 95% purity (by HPLC at wavelengths of
220nm and
254nm).
In the following examples and preparations, "Et" means ethyl,"Ac" means
acetyl, "Me"
means methyl, "Ph" means phenyl, (Ph0)zPOCI means chlorodiphenylphosphate,
"NCI" means
hydrochloric acid, "EtOAc" means ethyl acetate, "Na2C03' means sodium
carbonate, "NaOH"
means sodium hydroxide, "NaCI" means sodium chloride, "NEt3" means
triethylamine , "THF"
means tetrahydrofuran, "DIC" means diisopropylcarbodiimide, "HOBt" means
hydroxy
benzotriazole, "H20" means water, "NaHC03" means sodium hydrogen carbonate,
"K2C03'
means potassium carbonate, "MeOH" means methanol, "i-PrOAc" means isopropyl
acetate,
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-50-
"MgS04' means magnesium sulfate, "DMSO" means dimethylsulfoxide, "AcCI" means
acetyl
chloride, "CH2CI2" means methylene chloride, "MTBE" means methyl t-butyl
ether, "DMF" means
dimethyl formamide, "SOCI2' means thionyl chloride, "H3P04' means phosphoric
acid,
"CH3S03H" means methanesulfonic acid, " Ac20" means acetic anhydride, "CH3CN"
means
acetonitrile, and "ICON" means potassium hydroxide.
Example 1: Preparation of (4R)-4-allylcarbamoyl-5,5-dimethyl-thiazolidine-3-
carboxylic acid
tert-butyl ester:
OII O / OII O
~O~/N~OH H2N ~O~N~N~
(Ph0)~POCI CS jC H
NEt3
EtOAc
(4R)-5,5-Dimethyl-thiazolidine-3,4-dicarboxylic acid 3-tert-butyl ester (which
can be
prepared according to the methods of Ikunaka, M. et al., Tetrahedron Asymm.
2002, 13, 1201;
Mimoto, T. et al., J. Med. Chem. 1999, 42, 1789; and Mimoto, T. et al.,
European Patent
Application 0574135A1 (1993), 250 g; 0.957 mol) was added to an argon-purged 5-
L flask and
was dissolved in EtOAc (1.25 L). The solution was cooled to 2 °C and
(Ph0)2POCI (208 mL; 1.00
mol) was then added in one portion. NEt3 (280 mL; 2.01 mol) was added dropwise
via addition
funnel and the resulting suspension was then stirred at 0 °C. Seven
minutes later, allylamine
(75.4 mL; 1.00 mol) was added dropwise. The ice bath was removed and the
suspension was
allowed to warm to room temperature. One-half hour later, 1 N NCI (750 mL;
0.750 mol) was
added. The mixture was transferred to a 4-L separatory funnel using EtOAc (50
mL) for rinsing.
The layers were separated. The organic fraction was washed with 7.2% aqueous
Na2C03 (2 x
1.25 L), and was then transferred to a 3-L distillation flask and was diluted
with EtOAc (400 mL).
The solution was dried azeotropically and concentrated to a volume of 800 mL
by distillation of
EtOAc at one atmosphere. After cooling to 25 °C, the resulting clear
yellowish EtOAc solution of
(4R)-4-allylcarbamoyl-5,5-dimethyl-thiazolidine-3-carboxylic acid tert-butyl
ester was carried on
directly into the next step. An aliquot was removed and concentrated to give
(4R)-4.-
allylcarbamoyl-5,5-dimethyl-thiazolidine-3-carboxylic acid tert-butyl ester as
a white crystalline
solid: mp = 94 - 98 °C, ' H NMR (300 MHz, CDCI3) b 6.12 (br s, 1 H),
5.88 (app ddt, J = 10.2,
17.1, 5.6 Hz, 1 H), 5.28 (app dq, J = 17.1, 1.5 Hz, 1 H), 5.18 (app dd, J =
1.2, 10.2 Hz, 1 H), 4.68
(s, 2H), 4.14 (br s, 1 H), 3.95 (br t, J = 5.4 Hz, 2H), 1.62 (s, 3H), 1.49 (s,
9H), 1.46 (s, 3H); '3C
NMR (75 MHz, CDCI3) i5 170.0, 154.0, 134.4, 116.9, 82.0, 73.3, 54.0, 48.7,
42.0, 30.6, 28.6, 24.6;
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-51-
MS (CI) m/z 301.1599 (301.1586 calcd for C~4H~SNZO3S, M + H+); elemental
analysis calcd for
Cq4H24N2O3S: C, 55.97; H, 8.05; N, 9.32; found: C, 56.11; H, 8.01; N, 9.11.
Example 2: Preparation of (4R)-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide:
O O
~O /N~N~ CH3S03H HN~N
~S~ H EtOAc CS jC H
Methanesulfonic acid (155 mL; 2.39 mol) was added dropwise to the EtOAc
solution of
(4R)-4-allylcarbamoyl-5,5-dimethyl-thiazolidine-3-carboxylic acid tert-butyl
ester in a 3-L flask.
After stirring at room temperature overnight, the solution was cooled to 7
°C and HZO (400 mL)
was poured in. The mixture was transferred to a 4-L separatory funnel [using
H20 (30 mL) for
rinsing] and the layers were separated. The organic fraction was extracted
with H20 (190 mL).
The combined HZO extracts were transferred to a 5-L flask and were cooled to 8
°C. The pH was
adjusted from 0.4 to 9.3 using 3 N NaOH 01.05 L). 2-Methyltetrahydrofuran
(1.55 L) was poured
in, followed by the addition of NaCI (150 g). The ice bath was removed and the
mixture was
allowed to warm to room temperature. The pH was readjusted to 9.0 using 3 N
NaOH (~1 mL).
The mixture was transferred to a 4-L separatory funnel, using 2-
methyltetrahydrofuran (50 mL) for
rinsing, and the layers were separated. The aqueous phase was extracted with
2-methyltetrahydrofuran (950 mL). The organic extracts were vacuum-filtered
through Celite
directly into a 5-L distillation flask, using 2-methyltetrahydrofuran (200 mL)
for rinsing. The
solution was dried azeotropically and concentrated to a volume of 1.2 L by
distillation of
2-methyltetrahydrofuran at one atmosphere. A measured aliquot was concentrated
and weighed,
which showed that 161 g of (4R)-5,5-Dimethyl-thiazolidine-4-carboxylic acid
allylamide was
present in solution [84% from (4R)-5,5-dimethyl-thiazolidine-3,4-dicarboxylic
acid 3-tert-butyl
ester]. This solution was then carried on directly into the next step. The
concentrated aliquot
from above yielded (4R)-5,5-Dimethyl-thiazolidine-4-carboxylic acid allylamide
as a crystalline
solid: mp = 45 - 47 °C, 'H NMR (300 MHz, CDCI3) S 6.73 (br s, 1H), 5.87
(app ddt, J = 10.2,
17.1, 5.7 Hz, 1 H), 5.17 - 5.27 (m, 2H), 4.27 (AB q, JAB = 9.7 Hz, w = 22.5
Hz, 2H), 2.94 (app tt, J
= 1.5, 5.8 Hz, 2H), 3.51 (s, 1 H), 1.74 (s, 3H), 1.38 (s, 3H); ~3C NMR (75
MHz, CDCI3) b 169.7,
134.4, 116.9, 74.8, 57.2, 51.6, 41.9, 29.1, 27.3; MS (CI) m/z 201.1063
(201.1062 calcd for
C9H~~N20S, M + H+); elemental analysis calcd for C9H~6NZOS: C, 53.97; H, 8.05;
N, 13.99; found:
C, 53.93; H, 8.09; N, 14.07.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-52-
Example 3: Preparation of (2S,3S)-3-(3-acetoxy-2-methyl-benzoylamino)-2-
hydroxy-4-
phenyl-butyric acid:
0
Ph0 Ac0 I ~ O~ O Ph0
Ac0
H2N~OH ~ N~OH
OH NEt3, THF, H20 ~ / H OH
(2S,3S)-3-Amino-2-hydroxy-4-phenyl-butyric acid (which can be prepared
according to
the method of Pedrosa et al., Tetrahedron Asymm. 2001, 12, 347; M. Shibasaki
et al.,
Tetrahedron Lest. 1994, 35, 6123; and Ikunaka, M. et al. Tetrahedron Asymm.
2002, 13, 1201;
185 g; 948 mmol) was added to a 5-L flask and was suspended in THF (695 mL).
H20 (695 mL)
was poured in, followed by NEt3 (277 mL; 1990 mmol). After stirring for 45
min, the solution was
cooled to 6 °C. A solution of acetic acid 3-chlorocarbonyl-2-methyl-
phenyl ester (201 g; 948
mmol) in THF (350 mL) was then added dropwise. One-half hour later, the pH was
adjusted from
8.7 to 2.5 with 6 N HCI 0170 mL). Solid NaCI (46 g) was added, the ice bath
was then removed
and the mixture was stirred vigorously while warming to room temperature. The
mixture was
transferred to 4-L separatory funnel, using 1:1 THF/H20 (50 mL) for the
transfer, and the lower
aqueous phase was then removed. The organic fraction was transferred to a 5-L
distillation flask,
and was then diluted with fresh THF (2.5 L). The solution was azeotropically
dried and
concentrated to a volume of 1.3 L by distillation of THF at one atmosphere. To
complete the
azeotropic drying, fresh THF (2.0 L) was added and the solution was
concentrated to 1.85 L by
distillation at one atmosphere and was then held at 55 °C. n-Heptane
(230 mL) was added
dropwise via addition funnel and the solution was then immediately seeded.
After crystallization
had initiated, additional n-heptane (95 mL) was added dropwise. The resulting
crystal slurry was
stirred vigorously for 7 min. Additional n-heptane (1.52 L) was then added as
a slow stream. The
crystal slurry was then allowed to cool to room temperature slowly and stir
overnight. The
suspension was vacuum-filtered and the filter cake was then washed with 1:1
THF/n-heptane
(700 mL). After drying in a vacuum oven at 45 - 50 °C, 324 g (92%) of
(2S,3S)-3-(3-acetoxy-2-
methyl-benzoylamino)-2-hydroxy-4-phenyl-butyric acid was obtained as a
crystalline solid
contaminated with ~7 mol % Et3N~HCI: mp = 189 - 191 °C, 'H NMR (300
MHz, DMSO-d6) 5
12.65 (br s, 1 H), 3.80 (d, J = 9.7 Hz, 1 H), 7.16 - 7.30 (m, 6H), 7.07 (dd, J
= 1.1, 8.0 Hz, 1 H), 7.00
(dd, J = 1.1, 7.5 Hz), 4.40 - 4.52 (m, 1 H), 4.09 (d, J = 6.0 Hz, 1 H), 2.92
(app dd, J = 2.9, 13.9 Hz,
1 H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1 H), 2.29 (s, 3H), 1.80 (s, 3H); '3C
NMR (75 MHz, DMSO-
ds) 8 174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5, 128.3, 127.9, 126.5,
126.3, 124.8, 123.3,
73.2, 53.5, 35.4, 20.8, 12.6; MS (CI) m/z 372.1464 (372.1447 calcd for
C2oH22N06, M + H+);
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-53-
elemental analysis calcd for C2oHz~N06 ~ 0.07 Et3N~HCI: C, 64.34; H, 5.86; N,
3.95; CI, 0.70;
found: C, 64.27; H, 5.79; N, 3.96; CI; 0.86.
Example 4: Preparation of acetic acid 3-~(1S,2S)-3-[(4R)-4-allylcarbamoyl-5,5-
dimethyl-
thiazolidin-3-yl]-1-benzyl-2-hydroxy-3-oxo-propylcarbamoyl}-2-methyl-phenyl
ester:
O ~Ph0II DIC O Ph0 ~ ~/
Ac0 ~ N~OH + HN , N~ ~ Ac0 ~ N~N , N
I / H OH ~S/C H HOBt~H20 I / H OH 'S j< H
2-Me-THF
(2S,3S)-3-(3-Acetoxy-2-methyl-benzoylamino)-2-hydroxy-4-phenyl-butyric acid
(271 g;
731 mmol) was added to a 5-L flask containing a solution of (4R)-5,5-Dimethyl-
thiazolidine-4-
carboxylic acid allylamide (161 g; 804 mmol) in 2-methyltetrahydrofuran (1.20
L total solution),
while using 2-methyltetrahydrofuran (500 mL) for rinsing. HOBt~H~O (32.6 g;
241 mmol) was
added, using 2-methyltetrahydrofuran (50 mL) for rinsing. The white suspension
was allowed to
stir at room temperature for 10 min. Diisopropylcarbodiimide (119 mL; 760
mmol) was added in
three portions (40 mL + 40 mL + 39 mL) at 30 min intervals. One hour after the
final DIC addition,
Celite (100 g) was added and the suspension was allowed to stir at room
temperature for 3 h.
The mixture was vacuum-filtered, while 2-methyltetrahydrofuran (400 mL) was
used to rinse over
the solids and wash the resulting filter cake. The filtrate was transferred to
4-L separatory funnel,
using 2-methyltetrahydrofuran (50 mL) for rinsing. The solution was washed
with 1 N HCI (1.25
L), and then with an aqueous solution of NaHC03 (27 g), NaCI (134 g) and H20
(1.25 L). The
resulting organic phase was transferred to a 3-L distillation flask and the
solution was then
reduced to a volume of 1.12 L by distillation of 2-methyltetrahydrofuran at
one atmosphere. The
solution was then diluted with 2-methyltetrahydrofuran (230 mL) to bring the
total volume to 1.35
L. After cooling the solution to 23 °C, the solution of crude acetic
acid 3-((1 S,2S)-3-[(4R)-4-
allylcarbamoyl-5,5-dimethyl-thiazolidin-3-yl]-1-benzyl-2-hydroxy-3-oxo-
propylcarbamoyl)-2-
methyl-phenyl ester on directly into the next step.
Example 5: Preparation of (4R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide:
O rPh0II ~ fC CO O Ph0
Ac0 ~ N~N - N~ z a HO \ N~N - N
I / H OOH CSC H MeOH, 2-Me-THF I / H' YOH 'CSC H
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-54-
MeOH (330 mL) and IC2C03 (66.9 g; 484 mmol) were sequentially added to a
2-methyltetrahydrofuran solution of crude acetic acid 3-((1S,2S)-3-[(4R)-4-
allylcarbamoyl-5,5-
dimethyl-thiazolidin-3-yl]-1-benzyl-2-hydroxy-3-oxo-propylcarbamoyl}-2-methyl-
phenyl ester
(theoretical amount: 405 g; 731 mmol) in a 3-L flask at room temperature. Two
and a half hours
later, additional K2C03 (20 g; 144 mmol) was added. Three hours later the
reaction mixture was
vacuum-filtered on a pad of Celite, using 4:1 2-methyltetrahydrofuran/MeOH
(330 mL) for rinsing
over the solids and washing the filter cake. The filtrate was transferred to a
6-L separatory
funnel, using 4:1 2-methyltetrahydrofuran/MeOH (80 mL) for rinsing. The
solution was diluted
with i-PrOAc (1.66 L) and was then washed with a solution of NaCI (83.0 g) in
H20 (1.60 L). The
organic fraction was washed with 0.5 N HCI (1.66 L) and then with a saturated
aqueous NaCI
solution (400 mL). The resulting organic fraction was transferred to a 4-L
Erlenmeyer flask and
MgS04 (120 g) was added. After stirring for 10 min, the mixture was vacuum-
filtered directly into
a 5-L distillation flask, using 2:1 i-PrOAc/2-methyltetrahydrofuran (600 mL)
for rinsing the
separatory funnel and Erlenmeyer flask and washing the MgS04. The 2-
methyltetrahydrofuran
was displaced by distillation at one atmosphere with the simultaneous addition
of i-PrOAc in five
portions (a total of 3.60 L was used), while maintaining a minimum pot volume
of 2.50 L. The
resulting crystallizing mixture was cooled to 75 °C and was held at
this temperature for 30 min.
The suspension was then allowed to slowly cool to room temperature overnight.
The suspension
was vacuum-filtered, using i-PrOAc (600 mL) for transferring and washing the
crystals. After
drying in a vacuum oven at 40 °C, 204 g (54% from (2S,3S)-3-(3-Acetoxy-
2-methyl-
benzoylamino)-2-hydroxy-4-phenyl-butyric acid) of crystalline (4R)-3-[(2S,3S)-
2-Hydroxy-3-(3-
hydroxy-2-methyl-benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-
carboxylic acid
allylamide was obtained. This material was recrystallized as described below.
Example 6: Recrystallization of (4R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-
methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide:
O (Ph0II O
HO ~ N~N~N
H OH CSC H
(4R)-3-[(2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl-
butyryl]-5,5
dimethyl-thiazolidine-4-carboxylic acid allylamide (193 g, 378 mmol) was added
to a 5-L flask and
was then suspended in EtOAc (1.28 L). After heating the suspension to 76
°C, MeOH (68 mL)
was added and the internal temperature was then reduced to 70 °C. n-
Heptane (810 mL) was
added dropwise to the solution, while maintaining the internal temperature at
70 °C. After the n-
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-55-
heptane addition was complete, the resulting crystal suspension was held at 70
°C for 30 min,
and was then allowed to slowly cool to room temperature overnight. The
suspension was
vacuum-filtered, using 1.6:1 EtOAc/n-heptane (500 mL) to transfer and wash the
crystals. The
crystals were then dried in a vacuum oven at 45 °C to give 162 g (84%
recovery) of purified (4R)-
3-[(2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl-butyryl]-5,5-
dimethyl-
thiazolidine-4.-carboxylic acid allylamide as a white crystalline solid: mp =
173 - 175 °C,'H NMR
(300 MHz, DMSO-ds) displayed a 10:1 mixture of rotamers, major rotamer
resonances 8 9.35 (s,
1 H), 8.04 - 8.15 (m, 2H), 7.13 - 7.38 (m, 5H), 6.96 (t, J = 7.7 Hz, 1 H),
6.79 (d, J = 7.2 Hz, 1 H),
6.55 (d, J = 7.5 Hz, 1 H), 5.71 - 5.87 (m, 1 H), 5.45 (br d, J = 6.2 Hz, 1 H),
4.98 - 5.27 (m, 4H), 4.38
- 4.52 (m, 3H), 3.58 - 3.86 (m, 2H), 2.68 - 2.90 (m, 2H), 1.84 (s, 3H), 1.52
(s, 3H), 1.37 (s, 3H)
[characteristic minor rotamer resonances 8 9.36 (s), 8.21 (d, J = 10.5 Hz),
7.82 (5, J = 5.8 Hz),
4.89 (s), 4.78 (AB q, JAB = 9.8 Hz, w = 27.1 Hz), 4.17 - 4.24 (m), 2.93 - 3.01
(m), 1.87 (s), 1.41
(s)]; ~3C NMR (75 MHz, DMSO-ds) displayed a 10:1 mixture of rotamers, major
rotamer
resonances i5 170.4, 169.5, 168.2, 155.7, 139.6, 139.4, 135.5, 135.4, 129.9,
128.2, 126.2, 126.1,
121.9, 117.8, 115.6, 72.4, 72.1, 53.1, 51.4, 48.2, 41.3, 34.2, 30.5, 25.0,
12.6 [characteristic minor
rotamer resonances 8 171.4, 169.7, 168.6, 139.0, 129.5, 128.4, 70.6, 54.2,
49.1, 41.5, 31.4,
24.8]; MS (CI) m/z 512.2224 (512.2219 calcd for C27H34N3O5S, M + H*),
elemental analysis calcd
for C27H33N3~5S~ C, 63.38; H, 6.50; N, 8.22; found: C, 63.19; H, 6.52; N,
8.10.
Example 7: Preparation of (R)-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide;
hydrochloride:
0 0 0
IIJ~ HzN~ II ~ J~
~O~/N~OH ~O~/N~N~ HCI HCI ~ HN~N
(Ph0)ZPOCI ~S~ H EtOAc CS jC H
NEt3
EtOAc
A solution of (R)-5,5-Dimethyl-thiazolidine-3,4-dicarboxylic acid 3-tent-butyl
ester (105 kg,
402 mol) and ethyl acetate (690 L) was treated with diphenylchlorophosphate
(113 kg, 422 mol)
and was then cooled to 0 °C. NEt3 (85.5 kg, 844 mol) was added while
maintaining the
temperature at 5 °C, and the mixture was then held at this temperature
for 2 h. The mixture was
cooled to 0 °C, and allylamine (24.1 kg, 422 mol) was then added while
maintaining the
temperature at 5 °C. The mixture was warmed to 20 °C and was
then quenched with 10 wt.
aqueous HCI (310 L). After separation of the layers, the organic fraction was
washed with 8.6 wt.
aqueous Na2C03 (710 L). After separation of the layers, the aqueous fraction
was extracted
with ethyl acetate (315 L). The combined ethyl acetate extracts containing AG-
074278 were
dried by azeotropic distillation at one atmosphere, while maintaining a
minimum pot volume of
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-56-
approximately 315 L. The resulting suspension of (R)-4-Allylcarbamoyl-5,5-
dimethyl-thiazolidine-
3-carboxylic acid tent-butyl ester was cooled to 5 °C. A 13 wt. %
solution of anhydrous HCI (36.8
kg, 1008 mol) in ethyl acetate (263 L) was cooled to 5 °C and was then
added to the (R)-4.-
Allylcarbamoyl-5,5-dimethyl-thiazolidine-3-carboxylic acid tert-butyl ester
suspension while
maintaining the temperature at 15 °C. The resulting suspension was held
at 20 °C for 19 h, and
was then cooled and held at 5 °C for 2 h. The suspension was then
filtered, using cold ethyl
acetate for rinsing. The wet cake was dried under vacuum at 45 °C to
give 90.5 kg (95.2 %) of
(R)-5,5-Dimethyl-thiazolidine-4-carboxylic acid allylamide hydrochloride as a
white solid: ~H NMR
(300 MHz, DMSO-ds) 8 8.94 (app t, J = 5.5 Hz, 1 H), 5.82 (ddt, J = 10.4, 17.2,
5.2 Hz, 1 H), 5.19 -
5.25 (m, 1 H), 5.10 - 5.14 (m, 1 H), 4.38 (AB q, JAB = 9.8 Hz, w = 14.5 Hz,
2H), 4.08 (s, 1 H), 3.72 -
3.91 (m, 2H), 1.58 (s, 3H), 1.32 (s, 3H); 13C NMR (75 MHz, DMSO-ds) b 161.7,
132.2, 114.0,
67.9, 51.4, 43.5, 39.3, 25.3, 24.3; MS (CI) m/z 201.1070 (201.1062 calcd for
C9H1~N20S, M +
H+); elemental analysis calcd for C9H~~CINzOS: C, 45.65; H, 7.24; N, 11.83;
CI, 14.97; found: C,
45.41; H, 7.33; N, 11.69; CI, 15.22.
Example 8: Preparation of (2S,3S)-2-acetoxy-3-(3-acetoxy-2-methyl-
benzoylamino)-4-
phenyl-butyric acid:
0
Ac0
Ph0 ~ CI O PhO 1. Ac20, CH3S03H 0 Ph0
I ~ Ac0 ~ N OH EtOAc Ac0
H2N OH ~ N~OH
H I H
OH NEt3, THF, H20 / OH 2. Crystallize / OAc
A mixture of (2S,3S)-3-Amino-2-hydroxy-4-phenyl-butyric acid (110 kg, 563
mol), NaCI
(195 kg), and THF (413 L) was charged with NEt3 (120 kg, 1183 mol) and H20
(414 L) at ambient
temperature. The zesulting mixture was cooled to 0 °C. Acetic acid 3-
chlorocarbonyl-2-methyl-
phenyl ester (120 kg, 563 mol) was added to a separate reactor and was then
dissolved in THF
(185 L). The resulting solution of acetic acid 3-chlorocarbonyl-2-methyl-
phenyl ester was cooled
to 10 °C, and was then added to the (2S,3S)-3-amino-2-hydroxy-4-phenyl-
butyric acid mixture
while maintaining the temperature <10 °C during addition. The resulting
biphasic mixture was
agitated at 5 °C for 1 h, and was then adjusted to pH 2.5-3.0 with
concentrated HCI (62 kg). The
mixture was then warmed to 25 °C, and the layers were separated. The
resulting THF fraction,
containing (2S,3S)-3-(3-acetoxy-2-methyl-benzoylamino)-2-hydroxy-4-phenyl-
butyric acid, was
partially concentrated by distillation at one atmosphere. THF was then
replaced with ethyl
acetate by distillation at one atmosphere, while maintaining a minimum pot
volume of 1500 L.
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-57-
The resulting solution was cooled to 25 °C, and was then charged with
acetic anhydride (74.8 kg,
733 mol) and methanesulfonic acid (10.8 kg, 112 mol). The mixture was heated
at 70 °C for
approximately 3 h. The mixture was cooled to 25 °C, and was then
quenched with HBO (1320 L)
while maintaining the temperature at 20 °C. After removal of the
aqueous layer, the organic
fraction was charged with ethyl acetate (658 L) and H20 (563 L). After
agitation, the aqueous
phase was removed. The organic fraction was washed twice with 13 wt. % aqueous
NaCI (2 x
650 L). The organic fraction was partially concentrated and dried by vacuum
distillation (70-140
mm Hg) to a volume of approximately 1500 L. The resulting solution was heated
to 40 °C, and
was then charged with n-heptane (1042 L) while maintaining the temperature at
40 °C. The
solution was seeded with (2S,3S)-2-acetoxy-3-(3-acetoxy-2-methyl-benzoylamino)-
4.-phenyl-
butyric acid (0.1 kg), and additional n-heptane (437 L) was then added slowly.
The crystallizing
mixture was maintained at 40 °C for 1 h. Additional n-heptane (175 L)
was added while
maintaining the temperature at 40 °C. The crystalline suspension was
cooled and held at 25 °C
for 1 h, then at 0 °C for 2 h. The suspension was filtered, using n-
heptane for rinsing. The wet
cake was dried under vacuum at 55 °C to give 174 kg (74.5%) of (2S,3S)-
2-acetoxy-3-(3-acetoxy-
2-methyl-benzoylamino)-4-phenyl-butyric acid as a white solid: m.p. = 152 -
154 °C; 'H NMR
(300 MHz, CDCI3) i5 7.21 - 7.35 (m, 5H), 7.13 (app t, J = 7.9 Hz, 1 H), 7.01
(app d, J = 8.1 Hz,
1 H), 6.94 (app d, J = 7.2 Hz, 1 H), 5.99 (d, J = 9.0 Hz, 1 H), 5.33 (d, J =
4.1 Hz, 1 H), 4.96 - 5.07
(m, 1 H), 3.07 (dd, J = 5.5, 14.6 Hz, 1 H), 2.90 (dd, J = 10.0, 14.5 Hz, 1 H),
2.30 (s, 3H), 2.18 (s,
3H), 1.96 (s, 3H); ~3C NMR (125 MHz, CDCI3) 5 170.4, 170.2, 169.6, 169.5,
149.5, 137.81, 136.5,
129.2, 128.6, 128.4, 127.0, 126.6, 124.5, 123.7, 73.1, 50.9, 35.9, 20.6, 20.5,
12.4; elemental
analysis calcd for C22H23N0~: C, 63.92; H, 5.61; N, 3.39; found: C, 64.22; H,
5.68; N, 3.33; MS
(CI) m/z 414.1572 (414.1553 calcd for C2pH24NO~, M + H+).
Example 9: Preparation of (4R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide:
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-58-
o Pho ° ~ soclz o Pho 0
Ac0 ~ N~OH + HCI ~ HN~N~ --~ Ac0 ~ N~N~N
I / H OAc ~S/~ H pyridine ~ / H OAc C jC H
CH3CN S
1. KOH
MeOH, CH3CN
2. Crystallize
O ~Ph0~~ O
HO ~ N~N~N
H OOH ~S~ H
A solution of (2S,3S)-2-acetoxy-3-(3-acetoxy-2-methyl-benzoylamino)-4.-phenyl-
butyric
acid (140 kg, 339 mol), CH3CN (560 L), and pyridine (64.3 kg, 813 mol) was
cooled to 15 °C.
SOCI2 (44.3 kg, 373 mol) was charged while maintaining the temperature at 15
°C. The mixture
was held at 15 °C for 1 h. A separate reactor was charged with (R)-5,5-
dimethyl-thiazolidine-4-
carboxylic acid allylamide hydrochloride (96.6 kg, 408 mol), CH3CN (254 L),
and pyridine (29.5
kg, 373 mol), and was then cooled to 15 °C. The (2S,3S)-2-acetoxy-3-(3-
acetoxy-2-methyl-
benzoylamino)-4-phenyl-butyric acid chloride solution was added to the (R)-5,5-
dimethyl-
thiazolidine-4-carboxylic acid allylamide solution, while maintaining the
temperature at 15 °C.
The mixture was held at 15 °C for 6 h. A separate reactor was charged
with KOH (167 kg, 2709
mol) and methanol (280 L) using a 0 °C cooling jacket. The resulting
KOH/methanol solution was
cooled to 5 °C. The crude acetic acid 3-{(1S,2S)-2-acetoxy-3-[(R)-4-
allylcarbamoyl-5,5-dimethyl
thiazolidin-3-yl]-1-benzyl-3-oxo-propylcarbamoyl}-2-methyl-phenyl ester
mixture was added to the
KOH/methanol solution while maintaining the temperature at 10 °C. After
addition was complete,
the mixture was held at 25 °C for 3 h. The mixture was charged with HZO
(840 L) and ethyl
acetate (840 L), and was then followed by acidification to pH 5-6.5 with
concentrated HCI (85 kg)
while maintaining the temperature at 20 °C. The resulting layers were
separated. The organic
fraction was sequentially washed with 6.8 wt. % aqueous NaHC03 (770 L), an
aqueous HCI/NaCI
solution (H20: 875 L; conc. NCI: 207 kg; NaCI: 56 kg), 8.5 wt. % aqueous
NaHC03 (322 L), and
then with 3.8 wt. % aqueous NaCI (728 L). The resulting organic fraction was
partially
concentrated by distillation at one atmosphere. The solvent was exchanged with
ethyl acetate by
continuing distillation and maintaining the pot temperature at z70 °C.
Ethyl acetate was added
such that the pot volume remained at approximately 840 L. The solution was
then cooled to 20
°C and held at this temperature until crystallization was observed. n-
Heptane (280 L) was added
and the suspension was agitated at 15 °C for 4 h. The crystals were,
using cold 2.4:1 (v/v) ethyl
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-59-
acetate/n-heptane for rinsing. The wet cake was dried under vacuum at 45
°C to provide crude
(R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl-butyryl]-
5,5-dimethyl-
thiazolidine-4-carboxylic acid allylamide. Decolorization and
recrystallization was conducted as
follows: A mixture of crude (R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-
phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid allylamide, ADP
carbon (21 kg),
Supercel (3 kg), and ethyl acetate (780 L) was heated to 70 °C. CH30H
(40 L) was added to the
mixture. The mixture was filtered, and the resulting clear filtrate was heated
to reflux at one
atmosphere to begin distillation. CH30H was displaced as follows: ethyl
acetate (388 L) was
charged while maintaining the pot volume at approximately 840 L and at 70
°C. The solution was
slowly charged with n-heptane (316 L), while maintaining a temperature of 70
°C. The mixture
was then cooled to 20 °C and was held at this temperature for 4 h. The
crystals were filtered,
using cold 2.1:1 (v/v) ethyl acetate/n-heptane for rinsing. The wet cake was
dried under vacuum
at 45 °C to give 103 kg (59.6°l°) of (4R)-3-[(2S,3S)-2-
hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid
allylamide as a white
crystalline solid: mp = 173 -175 °C, ~H NMR (300 MHz, DMSO-ds)
displayed a 10:1 mixture of
rotamers, major rotamer resonances ~ 9.35 (s, 1 H), 8.04 - 8.15 (m, 2H), 7.13 -
7.38 (m, 5H), 6.96
(t, J = 7.7 Hz, 1 H), 6.79 (d, J = 7.2 Hz, 1 H), 6.55 (d, J = 7.5 Hz, 1 H),
5.71 - 5.87 (m, 1 H), 5.45 (br
d, J = 6.2 Hz, 1 H), 4.98 - 5.27 (m, 4H), 4.38 - 4.52 (m, 3H), 3.58 - 3.86 (m,
2H), 2.68 - 2.90 (m,
2H), 1.84 (s, 3H), 1.52 (s, 3H), 1.37 (s, 3H) [characteristic minor rotamer
resonances S 9.36 (s),
8.21 (d, J = 10.5 Hz), 7.82 (5, J = 5.8 Hz), 4.89 (s), 4.78 (AB q, JAB = 9.8
Hz, w = 27.1 Hz), 4.17 -
4.24 (m), 2.93 - 3.01 (m), 1.87 (s), 1.41 (s)]; ~3C NMR (75 MHz, DMSO-d6)
displayed a 10:1
mixture of rotamers, major rotamer resonances S 170.4, 169.5, 168.2, 155.7,
139.6, 139.4, 135.5,
135.4, 129.9, 128.2, 126.2, 126.1, 121.9, 117.8, 115.6, 72.4, 72.1, 53.1,
51.4, 48.2, 41.3, 34.2,
30.5, 25.0, 12.6 [characteristic minor rotamer resonances i5 171.4, 169.7,
168.6, 139.0, 129.5,
128.4, 70.6, 54.2, 49.1, 41.5, 31.4, 24.8]; MS (CI) m/z 512.2224 (512.2219
calcd for
3O C27H34N3O5S, M + H+), elemental analysis calcd for Ca7H33N3O5S: C, 63.38;
H, 6.50; N, 8.22;
found: C, 63.19; H, 6.52; N, 8.10.
Example 10: Preparation of (2S,3S)-3-Amino-2-hydroxy-4-phenyl-butyric acid;
hydrochloride:
O ph0 Ph0
II HCI (g)
~O~N OH ~ HCI~H2N OH
H OH CH2CI2 OH
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-60-
HCI gas (51 g, 1.4 mol) was bubbled into a suspension of (2S,3S)-3-tert-
butoxycarbonylamino-2-hydroxy-4-phenyl-butyric acid (163 g, 551 mmol) and
CHZCI2 (2.0 L) at 0
°C. The resulting off-white suspension was allowed to warm to ambient
temperature and stir
overnight. 'H NMR analysis of a concentrated aliquot showed approximately 95%
conversion to
product. The suspension was cooled to 0 °C, and additional HCI gas (46
g, 1.3 mol) was bubbled
into the suspension. After warming to ambient temperature, the suspension was
stirred
overnight. The suspension was vacuum-filtered, the solid was rinsed with
CHzCIZ (200 mL), and
the solid was then dried in a vacuum oven at 45 °C for 24 h to give 129
g (100%) of (2S,3S)-3-
amino-2-hydroxy-4-phenyl-butyric acid; hydrochloride as a white solid: 'H NMR
(300 MHz,
DMSO-ds) ~ 13.05 (br s, 1 H), 8.25 (br s, 3H), 7.22-7.34 (m, 5H), 4.41 (d, J =
2.6 Hz, 1 H), 3.66 (br
s, 1 H), 2.84 (AB portion of ABX, J,~ = 11.0 Hz, JBx = 2.8 Hz, Ov = 19.6 Hz,
2H); ~3C NMR (75
MHz, DMSO-ds) 8 172.4, 136.6, 129.8, 128.7, 127.1, 69.6, 55.0, 33.6; MS (CI)
m/z 196.0979
(196.0974 calcd for C~pH14N03, M - CI ). .
Example 11: Preparation of (2S,3S)-3-(3-Acetoxy-2-methyl-benzoylamino)-2-
hydroxy-4-
phenyl-butyric acid:
O
Ph0 Ac0 I ~ CI O ~PhO
AcO
HCI~H2N OH ~ ~N OH
OH NEt3, THF, H20 I / H OH
NEt3 (186 mL, 1.34 mol) was added to a suspension of (2S,3S)-3-amino-2-hydroxy-
4-
phenyl-butyric acid; hydrochloride (100 g, 432 mmol), H20 (320 mL), and
tetrahydrofuran (320
mL). The suspension was cooled to 4 °C and a solution of acetic acid 3-
chlorocarbonyl-2-methyl-
phenyl ester (93.6 g, 440 mmol) and THF (160 mL) was added dropwise. The
resulting solution
was warmed to ambient temperature and stir for 1 h. The solution was cooled to
10 °C and the
pH was adjusted to 2.0 using 6 N HCI (87 mL). NaCI (25 g) and tetrahydrofuran
(200 mL) were
added, and the mixture was warmed to ambient temperature. The phases were
separated and
the tetrahydrofuran fraction was dried over MgS04 and filtered. The filtrate
was concentrated to a
volume of 330 mL using a rotary evaporator, and was then diluted with
tetrahydrofuran (230 mL).
n-Heptane (1.2 L) was added slowly and the resulting white suspension of solid
was stirred at
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-61-
ambient temperature overnight. The suspension was vacuum-filtered, the solid
was rinsed with
n-heptane (2 x 500 mL), and the solid was dried in a vacuum oven at 45
°C for 24 h to give 150 g
(93.6%) of (2S,3S)-3-(3-acetoxy-2-methyl-benzoylamino)-2-hydroxy-4-phenyl-
butyric acid as a
white solid that was contaminated with ~7.7 mol % Et3N~HCI: mp = 189 - 191
°C,'H NMR (300
MHz, DMSO-ds) i5 12.65 (br s, 1 H), 3.80 (d, J = 9.7 Hz, 1 H), 7.16 - 7.30 (m,
6H), 7.07 (dd, J =
1.1, 8.0 Hz, 1 H), 7.00 (dd, J = 1.1, 7.5 Hz), 4.40 - 4.52 (m, 1 H), 4.09 (d,
J = 6.0 Hz, 1 H), 2.92
(app dd, J = 2.9, 13.9 Hz, 1 H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1 H), 2.29
(s, 3H), 1.80 (s, 3H); ~3C
NMR (75 MHz, DMSO-ds) 15174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5,
128.3, 127.9, 126.5,
126.3, 124.8, 123.3, 73.2, 53.5, 35.4, 20.8, 12.6; MS (CI) m/z 372.1464
(372.1447 calcd for
CzoHzzNOs~ M + H+).
Example 12: Preparation of (4R)-3-[(2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid 2-
methyl-
benzylamide:
O Ph0 O O
DCC, HOBt Ac0
Ac0 ~ N~OH + HN N ~ ~ N N . N
H OH L %C H I 1 EtOAc ' I ~ H OH ~ %C H
S
S
NaOMe/MeOH
O Ph0 0
HO ~ N~N~N
I / H OH C ~ H I
A solution of dicyclohexylcarbodiimide (3.05 g, 14.8 mmol) was added dropwise
to a
suspension of (2S,3S)-3-(3-acetoxy-2-methyl-benzoylamino)-2-hydroxy-4-phenyl-
butyric acid
(5.00 g, 13.5 mmol), (4R)-5,5-dimethyl-thiazolidine-4-carboxylic acid 2-methyl-
benzylamide (3.74
g, 14.1 mmol), HOBt~Hz0 (1.82 g, 13.5 mmol), and ethyl acetate (100 mL) at
ambient
temperature. After stirring at ambient temperature overnight, the suspension
was vacuum
filtered. The filtrate was sequentially washed with 5% aqueous NazC03 (50 mL),
1 N HCI (50
mL), and half saturated aqueous NaCI (50 mL). After drying over NazS04, the
ethyl acetate
solution of acetic acid 3-{(1S,2S)-1-benzyl-3-[(4R)-5,5-dimethyl-4.-(2-methyl-
benzylcarbamoyl)-
thiazolidin-3-yl]-2-hydroxy-3-oxo-propylcarbamoyl}-2-methyl-phenyl ester was
concentrated to a
volume of approximately 15 mL using a rotary evaporator. Methanol (11 mL) was
added, and the
solution was then cooled to 0 °C. NaOMe (3.1 mL of a 25 wt. % solution
in methanol, 13.5 mmol)
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-62-
was added dropwise, and the resulting mixture was stirred at 0 °C for 1
h. Ethyl acetate (108 mL)
was added, and 0.15 N HCI (108 mL) was then added slowly. The mixture was
warmed to
ambient temperature and the layers were separated. The organic fraction was
washed with 2.5%
aqueous Na2C03 (30 mL) and then with a solution of NaCI (6.6 g) and 0.1 N HCI
(30 mL). The
resulting organic fraction was dried over Na2SO4, filtered, and then
concentrated to a volume of
approximately 21 mL using a rotary evaporator. Ethyl acetate (15 mL) was
added, followed by
the slow addition of n-heptane (75 mL). The resulting suspension was stirred
overnight, and was
then vacuum-filtered. The solid was rinsed with n-heptane (2 x 25 mL), and was
then dried in a
vacuum oven at 45 °C for 24 h to give 7.41 g (95.4%) of (4R)-3-[(2S,3S)-
2-hydroxy-3-(3-hydroxy-
2-methyl-benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-
carboxylic acid 2-methyl-
benzylamide: 'H NMR (300 MHz, DMSO-ds) displayed a ~7:1 mixture of rotamers,
major rotamer
resonances b 9.37 (s, 1 H), 8.32 (t, J = 5.6 Hz, 1 H), 8.14 (d, J = 8.3 Hz, 1
H), 7.10-7.34 (m, 9H),
6.95 (t, J = 7.7 Hz, 1 H), 6.78 (d, J = 7.7 Hz, 1 H), 6.56 (d, J = 7.1 Hz, 1
H), 5.46 (br s, 1 H), 5.08
(ABq, JAB = 9.1 Hz, 2H), 4.38-4.50 (m, 3H), 4.11 (dd, J = 4.7, 15.1 Hz, 1 H),
2.85 (app dd, J = 2.8,
13.6 Hz, 1 H), 2.73 (app dd, J = 10.5, 13.5 Hz, 1 H), 2.27 (s, 3H), 1.84 (s,
3H), 1.50 (s, 3H), 1.36
(s, 3H) [characteristic minor rotamer resonances b 8.19 (d, J = 8.5 Hz), 8.07
(t, J = 5.7 Hz), 6.49
(d, J = 7.5 Hz), 4.93 (s), 4.80 (ABq, JAB = 9.7 Hz), 1.82 (s), 1.40 (s)]; ~3C
NMR (75 MHz, DMSO-
ds) displayed a ~7:1 mixture of rotamers, major rotamer resonances 5 170.5,
169.5, 168.3, 155.7,
139.7, 139.4, 137.1, 136.0, 130.2, 129.9, 128.3, 128.2, 127.2, 126.2, 126.1,
126.0, 121.8, 117.8,
115.6, 72.4, 71.9, 53.2, 51.5, 48.1, 40.8, 34.2, 30.6, 25.0, 19.1, 12.6
[characteristic minor rotamer
resonances 8 171.4, 169.6, 168.8, 139.0, 137.0, 135.8, 129.5, 128.4, 71.9,
54.3, 31.5, 24.8]; MS
(CI) m/z 576.2552 (576.2532 calcd for C32H38N3O5S, M + H+).
Example 13: Preparation of (2S,3S)-3-Amino-2-hydroxy-4-phenyl-butyric acid
ethyl ester;
hydrochloride:
O ph0 Ph0
II SOC12
~O~N OH ~ HCI~H2N OEt
H OH EtOH OH
SOCI2 (49.4 mL, 677 mmol) was added dropwise to absolute ethanol (500 mL),
which
had been cooled to 2 °C with an ice bath. After stirring the resulting
solution for 0.5 h, (2S,3S)-3-
tent-butoxycarbonylamino-2-hydroxy-4-phenyl-butyric acid (50.0 g, 169 mmol)
was added as a
solid. After stirring the resulting suspension for 20 min, the ice bath was
removed and the mixture
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-63-
was warmed to ambient temperature. Two hours later, the flask was submerged in
an oil bath
and the yellowish solution was heated at reflux overnight. The flask was then
equipped with a
distillation head, the oil bath temperature was increased to 85-90°C,
and 375 mL of distillate
(ethanol) was collected (b.p. = 76 - 82 °C, 1 atm) and was discarded.
The yellowish solution
remaining in the boiling pot was allowed to cool to 35 °C. Methyl t-
butyl ether (400 mL) was
slowly added, followed by the addition of n-heptane (100 mL). The resulting
suspension was
allowed to stir overnight and was then vacuum-filtered. The solid was rinsed
with n-heptane (3 x
150 mL) and was then dried in a vacuum oven at 45 °C overnight to give
40.2 g (91.3%) of
(2S,3S)-3-amino-2-hydroxy-4-phenyl-butyric acid ethyl ester; hydrochloride as
an off white solid:
mp = 129.5-131.5 °C;'H NMR (300 MHz, DMSO-ds) s 8.46 (br s, 3H), 7.19-
7.33 (m, 5H), 6.37 (d,
J = 5.2 Hz, 1 H), 4.48 (dd, J = 2.4, 5.0 Hz, 1 H), 3.68-3.82 (m, 2H), 3.56
(app dq, J = 10.7, 7.1 Hz,
1 H), 2.82-2.95 (m, 2H), 1.00 (t, J = 7.1 Hz, 3H);'H NMR (300 MHz, DSO) 8 7.19-
7.33 (m, 5H),
4.51 (d, J = 2.8 Hz, 1 H), 4.06 (dt, J = 2.8, 7.5 Hz, 1 H), 3.85 (app dq, J =
10.6, 7.2 Hz, 1 H), 3.68
(app dq, J = 10.6, 7.2 Hz, 1H), 2.83-2.97 (m, 2H), 1.07 (t, J = 7.1 Hz,
3H);'3C NMR (75 MHz,
DMSO-ds) ~ 170.7, 136.5, 129.9, 128.5, 127.1, 69.0, 60.8, 54.8, 33.3, 14.0; MS
(CI) m/z
224.1297 (224.1287calcd for C~zH~gN03, M - CI-).
Example 14: Preparation of (2S,3S)-3-(3-Acetoxy-2-methyl-benzoylamino)-2-
hydroxy-4-
phenyl-butyric acid ethyl ester:
O
Ph0 Ac0 I ~ CI O Ph0
HCI~H2N OEt Ac0 ~ N OEt
OH NEt3, CH2CI2 I / H OH
NEt3 (63.0 mL, 450 mmol) was added to an ambient temperature suspension of
(2S,3S)-
3-amino-2-hydroxy-4.-phenyl-butyric acid ethyl ester; hydrochloride (38.9 g,
150 mmol) and
CH2CI2 (800 mL), and the resulting solution was cooled to 1 °C. A
solution of acetic acid 3-
chlorocarbonyl-2-methyl-phenyl ester (35.0 g, 165 mmol) and CHzCl2 (150 mL)
was slowly added,
and the resulting white suspension was then allowed to warm to ambient
temperature and stir
overnight. 0.5 N HCI (400 mL) was added and the resulting layers were
separated. The organic
fraction was washed with H20 (400 mL), and then with one-quarter saturated
aqueous NaHC03.
Methanol (40 mL) was added to the organic fraction, which was then dried over
MgS04 and
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-64-
filtered. The filtrate was concentrated to a solid with a rotary evaporator,
and was further dried in
a vacuum oven at 45 °C overnight to give 60.0 g (101 %) of (2S,3S)-3-(3-
acetoxy-2-methyl-
benzoylamino)-2-hydroxy-4-phenyl-butyric acid ethyl ester as an off white
solid: ' H NMR (300
MHz, CDCI3) 8 6.93-7.25 (m, 8H), 6.02 (d, J = 9.0 Hz, 1 H), 4.78-4.87 (m, 1
H), 4.37 (d, J = 3.0 Hz,
1 H), 4.13 (app dq, J = 10.7, 7.2 Hz, 1 H), 4.04 (app dq, J = 10.7, 7.1 Hz, 1
H), 2.79 (d, J = 7.5 Hz,
2H), 2.24 (s, 3H), 1.95 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H); ~3C NMR (75 MHz,
DMSO-ds) b 172.7,
169.3, 168.1, 149.6, 139.5, 139.2, 129.5, 128.3, 128.0, 126.6, 126.4, 124.8,
123.4, 73.4, 60.7,
53.5, 35.5, 20.8, 14.5, 12.6; MS (CI) m/z 400.1751 (400.1760 calcd for
C2~H26N06, M + H+);
elemental analysis calcd for C22H~5NO6: C, 66.15; H, 6.31; N, 3.51; found: C,
65.90; H, 6.28; N,
3.39.
Example 15: Preparation of (2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-
phenyl-butyric acid:
O Ph0 O PhO
NaOH (aq) HO I ~ H OH
Ac0 ' ~ N OEt
'H OH THF / OH
NaOH (108 mL of a 3 N aqueous solution, 324 mmol) was added to a suspension of
(2S,3S)-3-(3-acetoxy-2-methyl-benzoylamino)-2-hydroxy-4-phenyl-butyric acid
ethyl ester (58.9 g,
147 mmol) and tetrahydrofuran (300 mL) at ambient temperature. The resulting
warm biphasic
solution was stirred at ambient temperature overnight. The flask was equipped
with a distillation
head and was submerged in an oil bath. A total of 340 mL distillate was
collected at one
atmosphere with the oil bath temperature range of 75-125 °C. The
resulting clear yellow solution
remaining in the boiling pot was diluted with HBO (100 mL), and was then
cooled to 0 °C. 6 N HCI
(60 mL) was slowly added, followed by ethyl acetate (250 mL), and the
resulting mixture was
warmed to ambient temperature with vigorous stirring. The resulting layers
were separated. The
organic fraction was washed with one-third saturated aqueous NaCI, and was
then dried over
MgS04, filtered, and was then concentrated to approximately 220 mL using a
rotary evaporator.
The resulting solution was allowed to stir at ambient temperature overnight.
The resulting
suspension of solid was vacuum-filtered, and the solid was rinsed with n-
heptane (2 x 200 mL).
After drying in a vacuum oven at 45 °C for 48 h, 45.1 g (92.8%) of
(2S,3S)-2-hydroxy-3-(3-
hydroxy-2-methyl-benzoylamino)-4-phenyl-butyric acid was obtained as a white
solid: ~H NMR
(300 MHz, DMSO-d6) 8 12.6 (br s, 1 H), 9.35 (s, 1 H), 8.05 (d, J = 9.0 Hz, 1
H), 7.16-7.30 (m, 5H),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-65-
6.95 (t, J = 7.8 Hz, 1 H), 6.77 (d, J = 7.3 Hz, 1 H), 6.53 (d, J = 6.7 Hz, 1
H), 5.63 (br s, 1 H), 4.38-
4.49 (m, 1 H), 4.07 (d, J = 5.9 Hz, 1 H), 2.87 (app dd, J = 3.0, 13.8 Hz, 1
H), 2.74 (app dd, J = 11.2,
13.9 Hz, 1 H), 1.80 (s, 3H); ~3C NMR (75 MHz, DMSO-ds) 8 174.2, 168.9, 155.4,
139.3, 139.2,
129.3, 128.1, 126.0, 125.8, 121.5, 117.6, 115.2, 73.0, 53.1, 35.1, 12.3; MS
(CI) m/z 330.1348
(330.1341 calcd for C~eH~oN05, M + H+).
Example 16: Preparation of (4R)-3-[(2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid 2-
methyl-
benzylamide:
ph Ph
HO O ~O DCC, HOBt O 0
H~OH + H/NJ 'H ~ \ HO I ~ H~N H
OH ~S%~ ~ TNF
A solution of dicyclohexylcarbodiimide (3.29 g, 15.9 mmol) and tetrahydrofuran
(15 mL)
was slowly added to a solution of (2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-
phenyl-butyric acid (5.00 g, 15.2 mmol), (4R)-5,5-dimethyl-thiazolidine-4-
carboxylic acid 2-methyl-
benzylamide (4.21 g, 15.9 mmol) (which can be prepared according to the
procedure found in H.
Hayashi, et al. J. Med. Chem. 1999, 42, 1789; R. Kato et al., United States
Patent No. 5,932,550;
and J. R. Tata et al., PCT Publication No. WO 01/05230 A1) HOBt~H20 (2.05 g,
15.2 mmol), and
tetrahydrofuran (50 mL) at ambient temperature. The resulting suspension was
stirred at ambient
temperature overnight. Ethyl acetate (35 mL) was added and the suspension was
then vacuum-
filtered, using ethyl acetate (20 mL) for rinsing. The filtrate was
sequentially washed with 5%
aqueous Na2C03 (50 mL), 0.5 N HCI (50 mL), and one-quarter saturated aqueous
NaCI (50 mL).
The resulting organic fraction was then dried over MgS04, filtered, and then
concentrated to a
volume of 45 mL using a rotary evaporator. The solution was allowed to stir at
ambient
temperature overnight. The resulting suspension was vacuum-filtered, and the
solid was
discarded. The filtrate was concentrated with a rotary evaporator to give
crude (4R)-3-[(2S,3S)-2-
hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-
thiazolidine-4-
carboxylic acid 2-methyl-benzylamide as a yellowish solid. This solid was
dissolved in isopropyl
acetate (62 mL) and the crystallizing mixture was stirred at ambient
temperature overnight. The
suspension was vacuum-filtered. The solid was rinsed with isopropyl acetate (2
x 20 mL), and
was then dried in a vacuum oven at 45 °C for 24 h to give 5.60 g (64.1
%) of (4R)-3-[(2S,3S)-2-
hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-
thiazolidine-4-
carboxylic acid 2-methyl-benzylamide as a white solid: 'H NMR (300 MHz, DMSO-
ds) displayed
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-66-
a ~7:1 mixture of rotamers, major rotamer resonances b 9.37 (s, 1 H), 8.32 (t,
J = 5.6 Hz, 1 H),
8.14 (d, J = 8.3 Hz, 1 H), 7.10-7.34 (m, 9H), 6.95 (t, J = 7.7 Hz, 1 H), 6.78
(d, J = 7.7 Hz, 1 H), 6.56
(d, J = 7.1 Hz, 1 H), 5.46 (br s, 1 H), 5.08 (ABq, JAB = 9.1 Hz, 2H), 4.38-
4.50 (m, 3H), 4.11 (dd, J =
4.7, 15.1 Hz, 1 H), 2.85 (app dd, J = 2.8, 13.6 Hz, 1 H), 2.73 (app dd, J =
10.5, 13.5 Hz, 1 H), 2.27
(s, 3H), 1.84 (s, 3H), 1.50 (s, 3H), 1.36 (s, 3H) [characteristic minor
rotamer resonances b 8.19
(d, J = 8.5 Hz), 8.07 (t, J = 5.7 Hz), 6.49 (d, J = 7.5 Hz), 4.93 (s), 4.80
(ABq, JAB = 9.7 Hz), 1.82
(s), 1.40 (s)];'3C NMR (75 MHz, DMSO-ds) displayed a ~7:1 mixture of rotamers,
major rotamer
resonances 8 170.5, 169.5, 168.3, 155.7, 139.7, 139.4, 137.1, 136.0, 130.2,
129.9, 128.3, 128.2,
127.2, 126.2, 126.1, 126.0, 121.8, 117.8, 115.6, 72.4, 71.9, 53.2, 51.5, 48.1,
40.8, 34.2, 30.6,
25.0, 19.1, 12.6 [characteristic minor rotamer resonances 8171.4, 169.6,
168.8, 139.0, 137.0,
135.8, 129.5, 128.4, 71.9, 54.3, 31.5, 24.8]; MS (CI) m/z 576.2552 (576.2532
calcd for
C32H38N3~SS~ M + H+).
Example 17: Preparation of (4R)-3-[(2S,3S)-2-Hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4-phenyl-butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid 2-
methyl-
benzylamide:
O ~Ph0~~ ~ O Ph0 O
Ac0 ~ N~H + HN-! 'N' Y \ ~. SOCIZ HO~N~N~N~
I / H ~OAc '' 'SjC H I // 2. KOH, MeOH 'I //' H OH CS%C H I~/
CHaCN
A solution of (2S,3S)-2-acetoxy-3-(3-acetoxy-2-methyl-benzoylamino)-4-phenyl-
butyric
acid (140 kg, 339 mol), CH3CN (560 L), and pyridine (64.3 kg, 813 mol) is
cooled to 15 °C. SOCIZ
(44.3 kg, 373 mol) is charged while maintaining the temperature at 15
°C. The mixture is held at
15 °C for 1 h. A separate reactor is charged with (4R)-5,5-dimethyl-
thiazolidine-4-carboxylic acid
2-methyl-benzylamide (89.7 kg, 339 mol), CH3CN (254 L), and pyridine (29.5 kg,
373 mol), and is
then cooled to 15 °C. The (2S,3S)-2-acetoxy-3-(3-acetoxy-2-methyl-
benzoylamino)-4-phenyl-
butyric acid chloride solution is added to the (4R)-5,5-dimethyl-thiazolidine-
4-carboxylic acid 2-
methyl-benzylamide solution, while maintaining the temperature at 15
°C. The mixture is held at
15 °C for 6 h. A separate reactor is charged with KOH (167 kg, 2709
mol) and methanol (280 L)
using a 0 °C cooling jacket. The resulting KOH/methanol solution is
cooled to 5 °C. The crude
acetic acid ester mixture is added to the KOH/methanol solution while
maintaining the
temperature at 10 °C. After addition is complete, the mixture is held
at 25 °C for 3 h. The mixture
is charged with H20 (840 L) and ethyl acetate (840 L), and is then followed by
acidification to pH
5-6.5 with concentrated HCI (85 kg) while maintaining the temperature at 20
°C. The resulting
layers are separated. The organic fraction is sequentially washed with 6.8 wt.
% aqueous
NaHC03 (770 L), an aqueous HCI/NaCI solution (H20: 875 L; conc. HCI: 207 kg;
NaCI: 56 kg),
CA 02549290 2006-06-02
WO 2005/054214 PCT/IB2004/003823
-67-
8.5 wt. % aqueous NaHC03 (322 L), and then with 3.8 wt. % aqueous NaCI (728
L). The
resulting organic fraction is partially concentrated by distillation at one
atmosphere. The solvent
is exchanged with ethyl acetate by continuing distillation and maintaining the
pot temperature at
z70 °C. Ethyl acetate is added such that the pot volume remained at
approximately 840 L. The
solution is then cooled to 20 °C and held at this temperature until
crystallization is observed. n-
Heptane (280 L) is added and the suspension is agitated at 15 °C for 4
h. The crystals are rinsed
using cold 2.4:1' (v/v) ethyl acetate/n-heptane for rinsing. The wet cake is
dried under vacuum at
45 °C to provide (4R)-3-[(2S,3S)-2-hydroxy-3-(3-hydroxy-2-methyl-
benzoylamino)-4.-phenyl-
butyryl]-5,5-dimethyl-thiazolidine-4-carboxylic acid 2-methyl-benzylamide.