Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST ~.E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional vohxmes please contact the Canadian Patent Oi~ice.
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Complete Mitochondrial Genome Sequences as a Diagnostic Tool for the Health
Sciences
For the purposes of the United States, this application is a continuation-in-
part of
10/732,374 filed December 11, 2003, which is a continuation-in-part of
application
PCT/CA02/00848 filed June 10, 2002, claiming priority to 60/297,340 filed June
1 l, 2001.
For all other countries, this application claims priority to 10/732,374 filed
December 11,
2003. All earlier applications are herein incorporated by reference.
Technical Field of the Invention
This invention is related to the field of mitochondrial genomics. In
particular it is
related to mutations in the mitochondria) genome and their utility as an
indicator of the
genesis of disease, for example detecting the presence of pre-neoplasia,
neoplasia and
progression towards potential malignancy even before common clinical symptoms
are
evident.
Background of the Invention
The current mega-trend in the biological sciences is the human genome project,
and
commercial exploitation of the data. However, there is an exceptional
limitation to the use
2o and implementation of this information as the data is not specific at the
level of the
individual. Incredibly the data is from only a few individuals, hardly
representative of the
variation present in human populations, rendering the data useful in general
applications
only. The staggering complexity of the human genome makes application on an
individual
basis impractical. To sequence completely one human nuclear genome the U.S.
Department of Energy and the National Institute of Health have invested 2.5
billion dollars
since 1988 (http://www.ornl.gov/hgmis/project/budget.html).
Mitochondria) Genome
The mitochondria) genome is a compact yet critical sequence of nucleic acid.
The
3o mitochondria) genome codes for enzyme subunits necessary for cellular
respiration.
Mitochondria) DNA, or "mtDNA", is a minuscule genome of nucleic acid at 16,569
base
pairs (bp) Anderson et al., 1981; Andrews et al., 1999) in contrast to the
immense nuclear
genome of 3.3 billion bp. Its genetic complement is astronomically smaller
than that of its
nuclear cell mate (0.0005%). However, individual cells carry anywhere from 103
to 104
1
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
mitochondria depending on specific cellular function (Singh and Modica-
Napolitano
2002). Communication or chemical signalling, routinely occur between the
nuclear and
mitochondria) genomes (Sherratt et al., 1997). Moreover, specific nuclear
components are
responsible for maintenance and integrity of mitochondria) sequence (Croteau
et al., 1999).
When these nuclear areas are rendered non-functional by nuclear rearrangements
indicative
of potential disease, then mutations begin to appear in mtDNA sequences. In
addition,
specific mitochondria may be identified for intracellular destruction by
deletions prompted
by somatic mutations in the mitochondria) genome. This theoretical mechanism
may serve
as an indication of impending disease as well. About 3,000 genes are required
to make a
1o mitochondrion, with only thirty-seven of these coded by the mitochondria)
genome,
indicating heavy mitochondria) dependence on nuclear loci (Naviaux, 1997).
All mitochondria) DNA (mtDNA) genomes in a given individual are identical
given
the clonal expansion of mitochondria within the ovum, once fertilization has
occurred. The
essential role of mtDNA is the generation of the cellular fuel, adenosine
triphosphate
(ATP), which fires cellular metabolism. Significantly, the mitochondria)
genome is
dependent on seventy nuclear encoded proteins to accomplish the oxidation and
reduction
reactions necessary to this vital function, in addition to the thirteen
polypeptides supplied
by the mitochondria) genome (Leonard and Shapira, 1997). Different tissues and
organs
depend on oxidative phosphorylation to a varied extent. Diseases related to
defective
oxidative phosphorylation (OXPHOS) appear to be closely linked to mtDNA
mutations
(Byrne, 1992). Consequently as O~iPHOS diminishes due to increased severity of
mtDNA
mutations, organ specific energetic thresholds are exceeded which give rise to
a variety of
clinical phenotypes. Moreover, mutations in the mitochondria) genome are
associated with
a variety of chronic, degenerative diseases (Gattermann et al. 1995). It is
well known that
aging and specific types of pathology can alter, or mutate mtDNA compromising
the
energy production capacity of the cell. This often results in over-expression
of defective
mitochondria, and/or the cell supplementing the lack of ATP by becoming more
glycolytic
(Carew and Huang, 2002); therefore, changes or mutations, in the mitochondria)
genome
can be used as markers for disease genesis and/or disease progression, when
monitored at
successive intervals.
2
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Recently, Fliss et al. (2000) found, in primary tumors from lung and bladder
cancer,
a high frequency of mtDNA mutations which were predominantly homoplasmic in
nature,
indicating that the mutant mtDNA was dominant in the malignant cells. Point
mutations
and deletions would appear to be the non-programmed but unavoidable side
effect of
oxygen free radical damage to the membrane and genome of mitochondria (Miquel
et al.
1992). This theory is plausible because not only is the mitochondria) genome
lacking
protective histones, but also is vulnerable to oxidative damage being found
near the oxygen
generating inner mitochondria) membrane. Moreover, as mtDNA has a compact
genome
and lacks introns, deleterious events are thus likely to affect a coding
sequence resulting in
to a biochemical dysfunction. This dysfunction will further increase cellular
oxidative stress
which will lead to nuclear as well as mtDNA damage, thereby increasing the
potential for a
cell to enter into the cancer process (Penta et al., 2001). In this respect,
research indicates
that with increasing age there is an increase in mtDNA damage (Cortopassi &
Wang 1995)
and a subsequent decline in respiratory function (Miquel et al. 1992) leading
to eventual
cell death.
MtDNA as a Diagnostic Tool
MtDNA sequence dynamics are important diagnostic tools. Mutations in mtDNA
are often preliminary indicators of developing disease, often associated with
nuclear
2o mutations, and act as biomarkers specifically related to disease, such as
but not limited to:
tissue damage and cancer from smoking and exposure to second hand tobacco
smoke (Lee
et al., 1998; Wei, 1998); longevity, based on accumulation of mitochondria)
genome
mutations beginning around 20 years of age and increasing thereafter (von
Wurmb, 1998);
metastatic disease caused by mutation or exposure to carcinogens, mutagens,
ultraviolet
radiation (Birch-Machin, 2000); osteoarthritis; cardiovascular, Alzheimer,
Parkinson
disease (Shoffner et al., 1993; Sherratt et al., 1997;Zhang et al, 1998); age
associated
hearing loss (Seidman et al., 1997); optic nerve degeneration and cardiac
dysrhythmia
(Brown et al., 1997; Wallace et al., 1988); chronic progressive external
exophthalmoplegia
(Taniike et al., 1992); atherosclerosis (Bogliolo et al., 1999); papillary
thyroid carcinomas
3o and thyroid tumours ('Yeh et al., 2000); as well as others (e.g. Naviaux,
1997; Chinnery and
Turnbull, 1999;).
3
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Specifically, these alterations include point mutations (transitions,
transversions),
deletions (one base to thousands of bases), inversions, duplications, (one
base to thousands
of bases), recombinations and insertions (one base to thousands of bases). In
addition,
specific base pair alterations, deletions, or combinations of are associated
with early onset
of prostate, skin, and lung cancer, as well as aging (e.g. Polyak et al.,
1998), premature
aging, exposure to carcinogens (Lee et al., 1998), etc.
Since mtDNA is passed to offspring exclusively through the ovum, it is
imperative
to understand mitochondria) sequences through this means of inheritance. The
sequence of
to mtDNA varies widely between maternal lineages (Ward et al., 1991), hence
mutations
associated with disease must be clearly understood in comparison to this
variation. For
example, a specific T to C transition noted in the sequence of several
individuals,
associated with a specific cancer, could in reality be natural variation in a
maternal lineage
widespread in a given particular geographical area or associated with
ethnicity. For
example, Native North Americans express an unusually high frequency of adult
onset
diabetes. In addition, all North American Natives are genetically
characterized by five
basic maternal lineages designated A, B, C, D, and X (Schurr et al., 1990;
Stone and
Stoneking, 1993; Smith et al., 1999). Lineage A is distinguished by a simple
point
mutation resulting in a Hae III site at by 663 in the mitochondria) genome,
yet there is no
2o causative relationship between this mutation and the adult onset of
diabetes. In addition,
even within lineage clusters there is sequence variation.
Outside of the specific markers associated with a particular lineage there is
more
intrapopulation variation than interpopulation sequence variation (Easton et
al., 1996;
Ward et al., 1991, 1993;) This divergence must be understood for optimal
identification of
disease associated mutations, hence a maternal line study approach (Parsons et
al., 1997),
mimicking the strengths of a longitudinal design (i.e. subject tracking over a
substantial
period of time), must be used to identify mutations directly associated with
disease, as
opposed to mutations without disease association. Moreover, particular
substances, such as
3o second hand tobacco smoke, low levels of asbestos, lead, all known mutagens
and at low
levels in many environments, may be the cause of specific point mutations, but
not
necessarily a disease specific marker_ Hence, a substantial mtDNA sequence
database is a
4
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
clear prerequisite to accurate forecasting of potential disease as a natural
process, or
through exposure to causative agents. Furthermore, the entire molecule must be
sequenced
for its full information content. The entire suite of point mutations
(transitions,
transversions), deletions (one base to thousands of bases), inversions,
duplications, (one
base to thousands of bases), recombinations and insertions (one base to
thousands of
bases) must be characterized as a whole over the entire mitochondria) genome.
This
ensures that all possible information available in the mitochondria) genome is
captured.
Although the genome of cytoplasrnic mitochondria (16,569bp) has been sequenced
at an
individual level, like its nuclear counterpart, the mitochondria) genome has
not been
to sequenced at a population level for use as a diagnostic tool.
Recently mitochondria have been implicated in the carcinogenic process because
of
their role in apoptosis and other aspects of tumour biology (Green & Reed,
1998, Penta et
al., 2001), in particular somatic mutations of mtDNA (mtDNA) have been
observed in a
number of human tumours (Habano et al. 1998; Polyak et al. 1998; Tamura et al.
1999;
Fliss, et al. 2000). These latter findings were made more interesting by the
claims that the
particular mtDNA mutations appeared to be homoplasmic (Habano et al. 1998;
Polyak et
a1.1998; Fliss, et al. 2000). Additionally researchers have found that
ultraviolet radiation
(UV) is important in the development and pathogenesis of non-melanoma skin
cancer
(NMSC) (Weinstock 1998; Rees, 1998) and UV induces mtDNA damage in human skin
(Birch-Machin, 2000a).
Moreover, through time, mitochondria) sequence loses integrity. For example,
the
4977bp deletion increases in frequency with age (Fahn et al., 1996). Beginning
at age 20,
this deletion begins to occur in small numbers of mitochondria. By age 80, a
substantial
number of molecules have been deleted. This deletion characterizes the normal
aging
process, and as such serves as a biomarker for this process. Quantification of
this aging
process may allow medical or other interventions to slow the process.
3o This application of mitochondria) genomics to medicine has been overlooked
because mtDNA has been used primarily as a tool in population genetics and
more recently
in forensics; however, it is becoming increasingly evident that the
information content of
5
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
mtDNA has substantial application in the field of medical diagnostics.
Moreover,
sequencing the entire complement of mtDNA was a laborious task before the
recent advent
of high capacity, high-throughput robotic DNA sequencing systems. In addition,
population geneticists were able to gather significant data from two highly
variable areas in
the control region; however, these small regions represent a small portion of
the overall
genome, less than 10%, meaning that 90% of the discriminating power of the
data is left
unused! Significantly, many disease associated alterations are outside of the
control
region. The character of the entire genome should be considered to include all
sequence
information for accurate and highly discriminating diagnostics.
Non-Melanoma Skin Cancer
Human non-melanoma skin cancer (NMSC) is the commonest cancer in many
Caucasian populations (Weinstock, 1998; Rees, 1998). The majority of these
tumours are
basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). BCCs are locally
invasive and can cause significant morbidity but rarely metastasis. SCCs show
significant
metastatic potential and the occurrence of multiple NMSCs in patients with
immunosuppression causes significant management problems (Rees, 1998). While
there
are no clinically identified pre-malignant lesions for BCC, some SCCs are
thought to arise
from precursor lesions, namely actinic keratoses (AKs) or areas of Bowen's
disease (in situ
2o carcinoma)(Rees, 1998).
SCCs show loss of heterozygosity affecting several chromosomes which suggests
the involvement of several tumour suppressor genes in their development.
Interestingly, in
AKs, an equal or greater degree of genetic loss is observed in these precursor
lesions
compared to SCCs (Rehman et al. 1994; Rehman et al. 1996). This is important
for the
proposed invention because it suggests that other mechanisms, in addition to
inactivation
of tumour suppressor genes, axe likely to be involved in the development of
SCCs.
A role for mitochondria in tumourigenesis was originally hypothesised when
3o tumour cells were found to have an impaired respiratory system and high
glycolytic activity
(Shay & Werbin, 1987). Recent findings elucidating the role of mitochondria in
apoptosis
(Green & Reed, 1998) together with the high incidence of homoplasmic mtDNA
mutations
6
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
in colon cancer (Habano et al. 1998; Polyak et al. 1998, reviewed in Penta et
al., 2001),
primary tumours of the bladder, neck and lung (Fliss et al. 2000), and gastric
tumours
(Tamara et al. 1999), further support this hypothesis. Furthermore, it has
been proposed
that these mitochondria) mutations may affect the levels of reactive oxygen
species (ROS)
which have been shown to be highly mitogenic (Polyak et al. 1998; Li et al.
1997).
Previous studies by the inventors and others have shown that mutations in
mtDNA
and the associated mitochondria) dysfunction is an important contributor to
human
degenerative diseases (Birch-Machin et al. 1993; Chinnery et al. 1999; Birch-
Machin et al.
2000b). This is because the mitochondria) genome is particularly susceptible
to mutations
due to the high amounts of ROS produced in this organelle coupled with the
lack of
protective histones and a low rate of mtDNA repair (Pascucci et al. 1997;
Sawyer & van
Houten; LeDoux et a1.1999) compared to the nucleus. lndeed, the mutation rate
for mtDNA
is around ten times higher than that of nuclear DNA (Wallace,l994). Most of
the mtDNA
mutations identified in the recent human tumour studies have indicated
possible exposure
to ROS derived mutagens. This is important for the investigation of mtDNA
mutations in
NMSC because there is recent evidence for the direct involvement of W induced
ROS in
the generation of mtDNA deletions in human skin cells (Berneburg et al. 1999,
Lowes et
al., 2002). In addition, the major determinant of NMSC in individuals without
protective
2o pigmentation or genetic predisposition is UV (Weinstock, 1998). The
putative precursor
lesions of SCCs are also found predominantly on constant sun-exposed sites.
This is
important because work by the Birch-Machin laboratory has shown distinct
differences
between the incidence of mitochondria) DNA damage in skin taken from different
sun
exposed body sites. The vast majority of the damage is found on constant sun-
exposed
sites (Krishanan et al., 2002).
One of the inventors was the first to quantitatively show that UV exposure
induces
mtDNA damage (Birch- Machin et al. 1998). MtDNA as a molecular maxker was used
to
study the relation between chronological aging and photo aging in human skin.
A 3-primer
quantitative PCR method was used to study the changes in the ratio of the 4977
bp-deleted
to wild type mtDNA in relation to sun exposure and chronological age of human
skin.
There was a significant increase in the incidence of high levels (i.e. >1 %)
of the 4977bp-
7
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
deleted mtDNA in sun-exposed (27%, [27/100]) compared with sun-protected sites
(1.1%
[1/90]) (Fishers exact test, P<0.0001). Deletions or mutations of mtDNA may
therefore be
useful as a marker of cumulative ultraviolet radiation exposure.
Furthermore, a study using a South-Western Blot approach involving monoclonal
antibodies against thymine dimers, provided direct evidence for the presence
of UV
induced damage in purified mtDNA (Ray et al. 1998). s'
Quantification of a single deletion alone, however, may not provide a reliable
UV
bio-marker because it represents one of many possible deletions or
combinations and other
to associated mutations (Birch-Machin, 2000). Recent work from the inventors'
research
group has used a long extension PCR (LX-PCR) technique to amplify the entire
mitochondria) genome in order to determine the whole deletion spectrum of
mtDNA
secondary to UV exposure (Ray et al. 2000). Long PCR analysis of 71 split skin
samples,
where the epidermis is separated from the underlying dermis, was performed in
relation to
sun exposure. There was a significant increase in the number of deletions with
increasing
UV exposure in the epidermis (Kruskal-Wallis test, p=0.0015). The findings in
the
epidermis are not confounded by any age-dependent increases in mtDNA deletions
also
detected by the long PCR technique. The large spectrum of identified deletions
highlights
the ubiquitous nature and the high mutational load of mtDNA associated with UV
2o exposure. Compared to the detection of single deletions using competitive
PCR, the study
shows that long PCR is a sensitive technique and may therefore provide a more
comprehensive, although not quantitative, index of overall mtDNA damage in
skin. The
studies by one of the inventors described above clearly show that mtDNA is a
significant
target of UV and this together with the role of mitochondria) in skin disease
has been
recently reviewed (Birch-Machin, 2000).
The pigmentation of human hair and skin which is the major co-variant of LTV
sensitivity and human skin cancer has been investigated. These investigations
have centred
on the association of variants of the melanocortin 1 -receptor gene and sun-
sensitivity of
3o individuals and populations (Smith et al. 1998; Healy et al. 1999; Flanagan
et al. 2000;
Healy et al. 2000; Harding et al. 2000; Flanagan et al., 2002) relating to
skin cancer
s
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
susceptibility. However, these studies have not addressed population-level
variation in
mtDNA sequences in association with particular skin types and/or hair colour.
One of the questions which remains largely unanswered by the recent studies of
mtDNA mutations in humor tumours is the incidence of deletions of the
mitochondrial
genome in relationship to these tumours. This is an important question to
answer because a
preliminary study of a single patient in human skin has shown differences in
the incidence
of the common mtDNA deletion between several tumours (AKs and SCCs) and normal
skin (Pang et al. 1994). As well, the inventors' own preliminary data shows an
increased
number of mtDNA deletions in tumours compared to normal skin. Finally, Birch-
Machin
to and others have shown that the incidence of mtDNA deletions, as well as
duplications,
increases with increasing UV exposure (Berneburg et al. 1999; Birch-Machin et
al.
1998;Ray et al. 1998; Ray et al. 1999; Ray et al. 2000), Lindsey et al., 2001;
Birch-Machin
et al., 2001; Lowes et al., 2002, Krishnan et al., 2002).
Apart from the questions relating to tumour progression other vital questions
remain largely unanswered by the recent studies of mtDNA in human tumours
(Habano et
al. 1998; Fiiss et al. 2000). Firstly, due to technical limitations, it is not
clear whether the
mtDNA mutations are truly homoplasmic, as varying levels of heteroplasmy may
indicate
important disease transitions as well (Habano et al. 1998; Polyal~ et al.
1998; Fliss, et al.
2000); secondly, apart from one study (Tamura et al. 1999) the incidence of
mtDNA
deletions and their role as potential biomarkers for NMSC was not
investigated.
Researchers have looked at the common deletion and ignored the rest of the 100
or so
deletions. As well, investigators have been focused on identification of
mutations, rather
than their quantification. It is important to assess accurately in a
quantitative manner the
incidence of deletions because of the threshold effect of mtDNA damage on ATP
production and consequently cell function. In addition, deletions are
difficult to
characterize. Long PCR is typically used which produces a ladder of deletions
which then
have to be characterized.
3o Current diagnosis of NMSC is pathological evaluation of excised tissue.
Accordingly, there is a need for an early marker of UV-induc ed DNA damage
which
9
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
predisposes an individual to NMSC. There is also a need for a genetic-based
diagnostic
tool which allows for early detection and is diagnostically accurate.
Prostate Cancer
Prostate cancer is a frequently diagnosed solid tumour that most likely
originates in
the prostate epithelium (Huang et al. 1999). In 1997, nearly 10 million
American men
were screened for prostate specific antigen (PSA), the presence of which
suggests prostate
cancer (Woodwell, 1999). Indeed, this indicates an even higher number of men
screened
by an initial digital rectal exam (DRE). In the same year, 31 million men had
a DRE
to (Woodwell, 1999). Moreover, the annual number of newly diagnosed cases of
prostate
cancer in the United States is estimated at 179,000 (Landis et al., 1999). It
is the second
most commonly diagnosed cancer and second leading cause of cancer mortality in
Canadian men. In 1997 prostate cancer accounted for 19,800 of newly diagnosed
cancers in
Canadian men (28%) (National Cancer Institute of Canada). It is estimated that
30% to
40% of all men over the age of forty-nine (49) have some cancerous prostate
cells, yet only
20% to 25% of these men have a clinically significant form of prostate cancer
(SpringNet -
CE Connection, Internet, www.springnet.com/ce/j803a.htm). Prostate cancer
exhibits a
wide variety of histological behaviour involving both erogenous and exogenous
factors, i.e.
socio-economic situations, diet, geography, hormonal imbalance, family history
and
2o genetic constitution (Konishi et al. 1997; Hayward et al. 1998).
From a risk standpoint familial and lzer°editary prostate cancers are
not considered
synonymous terms. Familial cancers refer to the incidences within a family,
but are not
inherited. This form accounts for up to 25% of prostate cancers (Walsh &
Partin, 1997).
Hereditary refers to a subtype of prostate cancer with a Mendelian inheritance
of a
predisposing genes) and accounts for approximately 9% of reported cases. A
positive
family history of prostate cancer for this disease suggests that these
predisposing genes)
play an important role in prostate cancer development and progression.
Recently,
susceptibility genes on chromosomes 1 and X have been identified as
predisposing men to
3o prostate cancer, providing greater insight into the etiology of hereditary
cancer (Berthon et
a]. 1998; Xu et al. 1998).
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Prostate cancer prognosis mainly depends on the tumour stage and grade at
diagnosis.
Only localized prostate cancer can be cured by radical treatment. Standard
detection still
relies on digital rectal examination, PSA testing and histopathologic
examination of
prostatic biopsied tissues. Biopsy of a mass is used to confirm malignancy, it
is not an
early detection technique. Unfortunately, some early tumours are impossible to
identify
during rectal exams. PSA tests have a specificity of 60 to 70% and a
sensitivity of 70 to
80% (personal communication, Dr. Sunil Gulavita, Northwestern Ontario Cancer
Centre).
A newer technique which refines diagnosis for tumours of common histologic
grade is
ploidy-DNA analysis employing flow cytometry (Shankey et al. 1995); however,
this
to technique measures chromosomal changes that are only apparent in later
stages of cancer
development and is not sufficiently sensitive for the detection of minor
alterations in DNA
structure or chromosomal inversions, or reciprocal trans-locations in early
cancers. The
invention focuses on early detection since prognosis is heavily dependent on
the stage of
disease at diagnosis.
Our understanding of genetic abnormalities in prostate cancers is scanty.
Research
into prostate cancer has focussed on the development of knowledge in the
following areas:
1) proto-oncogenes (Buttyan et al. 1987); 2) tumour suppressor genes (p53,
p73, I~AIl and
MMACI/PTEN; Dong et al. 1995; Cairns et al. 1997) and 3) telomere/telomerase
activity
2o in metastasis. Up-regulation of telomerase and amplification of telomeric
DNA in prostate
cells may provide effective markers for diagnosis. Moreover, telomeres may
serve as a site
for therapy (Ozen et al. 1998). A number of groups have provided evidence for
a "prostate
cancer gene" in the short arm of chromosome 1 (Berthon et al. 1998). M~re work
is
needed to identify the specific locus within this region. It has been
suggested that this
marker is only one of several possible genes predisposing men to familial
prostate cancer.
Other studies have shown possible marker loci on the X chromosome (Xu et al.
1998). If
some prostate cancers are polygenic, then mtDNA becomes an important
diagnostic tool
since it may be difficult to identify and understand the interplay between all
associated
nuclear genes in such cases.
Certainly, a key issue in prostate cancer research is to identify molecular
markers
that can effectively determine and distinguish tumour progression. Molecular
markers may
11
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
be able to discriminate between those cases of prostate neoplasmy which will
proceed
rapidly to metastatic disease and those with little chance of resulting in
tumour
development. Comparison of molecular markers or mutations can determine
whether the
tumor pathway is latent or aggressive. Up to the present research has focused
primarily on
the secrets hidden within the nuclear genome; however, the much smaller mtDNA
genome
seems to act as a barometer for events in the nucleus and as such provides a
means for the
early detection of human prostate cancer (Zeviani et al. 1990). Importantly,
in this respect,
mitochondria have been implicated in the carcinogenic process because of their
role in
apoptosis and other aspects of tumour biology (Green & Reed 1998). In
particular, somatic
to mutations of mtDNA have been observed in a number of human tumours (Polyax
et al.
1998, Tamura et al. 1999, Fliss et al. 2000). However, previous studies have
been
exclusively cross-sectional as they have not considered the clonal nature of
mtDNA in
maternal lines. These limited cross-sectional studies merely show the mutation
at one time
point. This may or may not give an accurate link between a mutation and the
corresponding disease state. Cross-sectional studies employing a maternal line
have the
advantage of tracking a mutation in mtDNA over time and thus mimic the
strength of a
longitudinal design. Mutations which are common population variants, as
opposed to
mutations associated with disease, can both be identified.
2o Aging
Aging consists of an accumulation of changes with time both at the molecular
and
cellular levels; however, the specific molecular mechanisms underlying the
aging process
remain to be elucidated. In an attempt to explain the aging process,
mitochondrial
genomes in older subjects are compared to the genomes of younger subjects from
the same
maternal lineage. One deletion associated with aging is known as the common
deletion, or
4977-by deletion. Aging research has been limited to this common deletion and
polymorphisms in the control region. Fox a clear understanding of these
mutations, the
entire genome must be analyzed. Other deletions are seen in Table 1 adapted
from Wei,
1992.
Table 1
Deletions References
size (bn)
12
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Deletions References
size (b )
4977 Cortopassi and Arnheim, 1990;
Ikebe et al., 1990; Linnane
et al., 1990;
Corral-Debrinski et al., 1991;
Yen et al.,
1991;
Torii et al., 1992; Zhang et
al., 1992
7436 Corral-Debrinski et al., 1991;
Hattori et al., 1991
Hsieh and Wei, 1992
3610 I~atayama et al., 1991
6063 Hsieh and Wei, 1992
Yen et al., 1992
5827 Zhang et al., 1992
6335 Zhang et al., 1992
7635 Zhang et al., 1992
7737 Zhang et al., 1992
7856 Zhang et al., 1992
8041 Zhang et al., 1992
8044 Zhang et al., 1992
5756 Zhang et al., 1992
Oxygen free radicals, a normal by product of ATP production, are a probable
cause of this
deletion, which increases in frequency with age. Existing literature
demonstrates a strong
association between mtDNA (mtDNA) mutations, chronological age, and the
overall aging
process in postmitotic tissues such as muscle and brain; however, comparative
maternal
line studies are needed to discriminate between aging associated mutational
events and
those mutations without an aging association.
In recent years a variety of chronic degenerative diseases have been shown to
result
to from mutations in mtDNA (Gatterman et al. 1995). Diseases related to
defective OXPHOS
appear to be closely linked to mtDNA mutations (Byrne, 1992). Furthermore, it
has been
shown that these myopathies are often associated with the common deletion of
4977-by of
the mitochondrial genome (Liu et al. 1997). This large deletion has also been
found, at
heteroplasmic levels, in various tissues of normal aging persons and is
consistent with the
13
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Mitochondria) Theory of Aging (Harman, 1981). This is manifest through an
increase in
the deletion frequency (Cortopassi & Wang, 1995) and a subsequent decline in
respiratory
function (Miquel et al. 1992) resulting in eventual cell death in old age. The
early
detection of a predisposition to a disease or disorder presents the best
opportunity for
medical intervention, as early genetic diagnosis may improve the prognosis for
a patient.
Previous studies employing a cross-sectional design have established an
association
or cause and effect relationship between mtDNA mutations, deletions, and/or
combinations
of such and aging; however, in order to obtain accurate data the age specific
deletion andlor
to mutation rate must be determined concisely. Attributing mutations to the
aging process as
opposed to a particular disease at the population level is vital. This
information is
imperative to an understanding of how mtDNA damage accrues over time.
Moreover, the
consequences of these particular mutations, their frequencies, and
associations in the
temporal aspects of aging must be known in order to forecast and eventually
slow aging at
the molecular level. Researchers have not yet determined this rate, which
requires
evaluation of population data through maternal lines. Accordingly, there is a
need for a
biomarker which tracks the aging process.
Accordingly, there is a need for a simple, straightforward system of
monitoring the
mitochondria) genome for mutations which indicate early stage cancer, aging or
other
human diseases with a DNA component. There is also a need for a simple
diagnostic
system for non-melanoma skin cancer, prostate cancer, lung cancer and aging
linked to
defects in the mitochondria) genome. There is a need for a diagnostic system
which
differentiates between mutations in mtDNA which cause disease, and those which
simply
represent variation within and between populations.
Summary of the Invention
An object of the present invention is to provide a simple, straightforward
system for
monitoring the mitochondria) genome for early transitions associated with
cancer, aging,
3o and other human diseases with a DNA component.
14
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
In an embodiment of the present invention a small biological sample which
includes tissue or fluid samples such as urine, prostate fluid, skin cells, or
saliva is taken
from an individual. These samples are examined, using any suitable method
including
histological examination, to identify cells demonstrating disease morphology.
Using any
suitable method, including without limitation; laser capture, identified cells
demonstrating
disease morphology are recovered from the sample and the mtDNA therefrom is
sequenced, followed by comparison to a database of known mitochondria)
sequences
associated with both health and disease.
In a preferred embodiment, the entire mitochondria) genome is sequenced at a
population level to determine the variation of mtDNA sequences associated with
disease.
In an additional embodiment, the presence of mutation progression may signal
the
beginning and continuing development of disease. Mutation load may also
indicate
progression or disease state.
In a preferred embodiment, mtDNA sequences from prostate massage fluid are
compared to a mtDNA sequence database of normal, transitory, and metastatic
mtDNA
sequences clearly associated with prostate cancer. This comparative data set
is based on
2o studies of maternal lines, and other normal maternal line variation present
in the population
stored in a maternal line database affording a lucid picture of mtDNA
mutations clearly
associated with disease, as opposed to variation present in mitochondria)
lineages existing
in the general population.
There may be specific maternal lineages which indicate a predisposition to
disease.
In another embodiment, mtDNA sequences from suspected non-melanoma skin
cancers are compared to a mtDNA sequence database of normal and mtDNA
sequences
clearly associated with non-melanoma skin cancer.
According to an aspect of the present invention, there is provided a method of
detecting in a subj ect containing mtDNA the genesis or progression of disease
comprising
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
obtaining a biological sample from the subject, extractiilg DNA from the
biological
sample, and detecting the presence of mutations in the mtDNA. The step of
detecting the
presence of mutations is chosen from sequencing the mtDNA, amplifying the
mtDNA by
PCR, South-Western blotting, denaturing HPLC, hybridization to microarrays,
gene chips
or biochips, molecular marker analysis or combinations thereof. Further, the
mtDNA of
the biological sample is compared to a database, the database containing data
of mutations
associated with the mtDNA sequences of non-disease and disease associated
mitochondria)
genomes.
to According to an aspect of the present invention, there is provided a method
of
detecting in a human subject the presence of a disease comprising obtaining a
biological
sample from the human subject, extracting DNA from the biological sample,
detecting
mutations in the mitochondria) DNA of the biological sample, and comparing the
mitochondria) DNA sequence of the biological sample to a database, the
database
containing data of mutations associated with the mitochondria) DNA sequences
of non-
disease and disease associated mitochondria) genomes Mutation rates of
mitochondria
DNA associated .with a specific disease may be an important indicator of
disease
development and prognosis. This may allow specific identification of disease
stage,
improving disease definition resulting in better disease intervention and
specific therapy
2o application.
In yet another embodiment, increasing the sensitivity for heteroplasmy
detection
increases the early identification capacity of the test.
In addition, the invention may be used to monitor the progression of disease
by
watching important sites targeted by metastasis.
According to another aspect of the present invention, there is provided a
method of
determining a predisposition to a disease or disorder indicated by mutations
in a
3o mitochondria) DNA sequence comprising: obtaining a biological sample from
the human
subject, extracting DNA from the biological sample, detecting mutations in the
mitochondria) DNA of the biological sample, and comparing the mitochondria)
DNA
16
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
sequence of the biological sample to a database, the database containing data
of mutations
associated with the mitochondria) DNA sequences of individuals who are
predisposed to
the disease or disorder, and individuals who are not predisposed to the
disease or disorder.
In a preferred embodiment, a DNA microarray is used in determining the
sequence
of the mitochondria) DNA. Other technologies can also be used. For example,
direct
sequencing of a subset, or the complete human genome, SNaP shotTM, SNP
detection, real
time PCR or other methods as is standard in the art.
to According to a further aspect of the present invention, there is provided a
method
for assessing the status of the aging process of a human subject comprising
obtaining a
biological sample from the human subject, extracting DNA from the biological
sample,
detecting mutations in the mitochondria) DNA of the biological sample, and
comparing the
mitochondria) DNA sequence of the biological sample to a database, the
database
containing data of mutations of TDNA associated with aging.
The step of detecting the presence of mutations in the mtDNA can be selected
from:
sequencing the mtDNA, amplifying mtDNA by PCR, Southern, Northern, Western,
South-
Western blot hybridizations, denaturing HPLC, hybridization to microarrays,
biochips or
gene chips, molecular marker analysis, biosensors, melting temperature
profiling or a
2o combination of any of the above.
According to yet another aspect of the present invention, there is provided a
database containing a plurality of human mitochondria) DNA sequences, the
mitochondria)
DNA sequences selected from the group of normal control sequences associated
with non-
disease states, sequences associated with the presence of disease or sequences
indicative of
the predisposition to disease.
According to yet another aspect of the present invention, there is provided a
kit for
diagnosis of a disease comprising a disposable chip, microarray, means for
holding the
3o disposable chip, means for extraction of mitochondria) DNA and means for
access to a
database of mitochondria) DNA sequences.
17
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
According to yet another aspect of the present invention there is provided a
method
of diagnosing a disease in a patient comprising hybridizing a nucleic acid
sample obtained
from mitochondria) DNA to an array comprising a solid substrate and a
plurality of nucleic
acid members, wherein each member is indicative of the presence of a disease,
wherein
each nucleic acid member has a unique position and is stably associated with
the solid
substrate, and wherein hybridization of said nucleic acid sample to one or
more nucleic
acid members comprising said array is indicative of the presence of prostate
cancer.
According to yet another aspect of the present invention there is provided a
kit for
to determining predisposition to a disease comprising a disposable chip,
microarray, means
for holding the disposable chip, means for extraction of DNA and means for
access to a
database of mitochondria) DNA sequences.
According to another aspect of the present invention, there is provided a
method of
determining a predisposition to or developing symptoms of a disease or
disorder indicated
by mutations in a mitochondria) DNA sequence comprising obtaining a biological
sample
from the human subject, extracting mitochondria) DNA from the biological
sample,
sequencing the mitochondria) DNA of the biological sample, and comparing the
mitochondria) DNA sequence of the biological sample to a database, the
database
2o containing population-level data of mutations associated with the mtDNA
sequences of
non-disease and disease associated mitochondria) genomes.
According to yet another aspect of the present invention, there is provided a
method
of diagnosing non-melanoma skin cancer in a patient comprising: hybridizing a
nucleic
acid sample obtained from mitochondria) DNA to an array comprising a solid
substrate and
a plurality of nucleic acid members, wherein each member is indicative of non-
melanoma
cancer, wherein each nucleic acid member has a unique position and is stably
associated
with the solid substrate, and wherein hybridization of said nucleic acid
sample to one or
more nucleic acid members comprising said array is indicative of the presence
of non-
melanoma skin cancer. Alternatively, non-specific mutations may reach a
threshold effect
beyond which cancer develops. In a similar manner, prostate cancer can also be
diagnosed.
18
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
According to another aspect of the present invention, there is provided a
method of
detecting heteroplasmy in a subject containing mtDNA comprising obtaining a
biological
sample from the subject; extracting DNA from the biological sample; and
performing
denaturing HPLC on the sample.
According to another aspect of the present invention, there is provided a
method of
detecting mutations associated with disease in a subj ect containing mtDNA
comprising:
obtaining a biological sample from the subject, extracting DNA from the
biological
sample, detecting the presence of mutations in the mtDNA, and comparing the
mtDNA of
1o the biological sample to a database, the database containing data of common
population
variants in non-disease and disease associated mitochondrial genomes.
Another aspect of the invention is to provide a use of any one or any
combination
of the mutations, substitutions, deletions or insertions listed on Table 4 or
SEQ m NOs:
102 to 138 to detect a predisposition to a disease or disorder, early
detection of a disease or
a disorder, genesis of a disease or a disorder, presence of a disease or a
disorder,
progression of a disease or a disorder, or aging in a subject having mtDNA.
Another aspect of the invention is to provide a method of detecting mutations
2o associated with a disease, a disorder or aging in a subject having mtDNA,
comprising:
providing a biological sample from the subject, wherein the biological sample
is chosen
from non-involved tissue, distant benign tissue, adjacent benign tissue,
atypical tissue,
histologically abnormal tissue, and diseased tissue; extracting DNA from the
biological
sample; detecting the presence of mutations in the mtDNA; and determining
whether the
mutations are associated with normal interpopulation or intrapopulation
variations, or
whether the mutations are associated with the disease, the disorder or aging.
Optionally,
the method further comprises at least one of determining total mutation load
in the mtDNA
of the biological sample; and determining the identity of the mutation in the
mtDNA of the
biological sample. The step of determining whether the mutations are
associated with
3o normal interpopulation or intrapopulation variations, or whether the
mutations are
associated with the disease, the disorder or aging may comprise at least one
of: comparing
the mtDNA of the biological sample to a database, the database containing data
of
19
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
interpopulation and intrapopulation variations, and mutations associated with
the disease,
the disorder or aging; and determining total mutation load of the biological
sample.
The diagnosis may be chosen from predisposition to a disease or a disorder,
early
detection of a disease or a disorder, genesis of a disease or a disorder,
presence of a disease
or a disorder, and progression of a disease or a disorder. The step of
detecting the presence
of mutations may be chosen from: sequencing the mtDNA; amplifying mtDNA by
PCR;
Southern, Northern, Western and South-Western blot hybridizations; denaturing
HPLC;
hybridization to microarrays, gene chips or biochips; molecular marker
analysis; and a
to combination of any of the above.
The disease may be non-melanoma skin cancer or prostate cancer. The database
contains at least a statistically significant number of mitochondria) DNA
sequences, the
mitochondria) DNA sequences having been obtained from a maternal line, a non-
maternal
line, or both.
Another aspect of the invention is to provide an array comprising a plurality
of
nucleic acid members, and a solid substrate, wherein the nucleic acid members
are
associated with the mutations listed on Table 4 or SEQ m Nos: 102 to 138 and
are
indicative of the presence or predisposition of a disease, a disorder or
aging, or used to
determine a prohibiting index by quantifying the proportion of base pair
deletions and
mutations associated with a disease, a disorder or aging, and is chosen from
mitochondria)
DNA, RNA transcribed from mitochondria) DNA, and cDNA, wherein each nucleic
acid
member has a unique position on said array and is stably associated with the
solid
substrate. For example, the members may be associated with prostate cancer or
any other
disease or disorder.
Another aspect of the invention is to provide a kit for diagnosing,
determining a
predisposition, or early detection of a disease comprising a disposable chip,
the array
3o described above, means for holding the disposable chip, means for
extraction of
mitochondria) DNA and means for access to a database of mitochondria) DNA
sequences.
For example, the member may be associated with prostate cancer.
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Another aspect of the invention is to provide a database containing a
plurality of
human mitochondria) DNA sequences, the mitochondria) DNA sequences are chosen
from
normal control sequences associated with non-disease states, sequences
associated with
interpopulation variations, sequences associated with intrapopulation
variations, and
sequences associated with the mutations on Table 4 or SEQ m Nos: 102 to 138.
Another aspect of the invention, is a method of monitoring a person for the
presence of pre-neoplasia, neoplasia or progression of neoplasia toward
potential
to malignancy, in a biological sample, comprising (a) providing a biological
sample from the
subject, (b) extracting DNA from the biological sample, (c) detecting the
presence of
mutations in the mtDNA, (d) determining whether the mutations are associated
with
normal interpopulation or intrapopulation variations, or whether the mutations
are
associated with pre-neoplasia, neoplasia or progression of neoplasia toward
potential
malignancy, and (e) repeating steps (a) through (d). The step of determining
whether the
mutations are associated with pre-neoplasia, neoplasia or progression of
neoplasia may be
done by comparing the mutations with DNA from non-involved tissue or bodily
fluid from
the subject, or by comparing the mutations with mitochondria) DNA from a
maternal
relative. The progression of neoplasia may comprise monitoring the person at
successive
2o time periods for an increase in mutations or an increase in mutated
mitochondria) genomes.
Another aspect of the invention, is a method of determining whether pre-
neoplasia,
neoplasia, or malignancy is latent or aggressive in its growth pattern, in a
biological
sample, comprising (a) providing a biological sample from the subject, (b)
extracting DNA
from the biological sample, (c) detecting the presence of mutations in the
mtDNA, (d)
determining whether the mutations are associated with normal interpopulation
or
intrapopulation variations, or whether the mutations are associated with pre-
neoplasia,
neoplasia or progression of neoplasia toward potential malignancy, and (e)
repeating steps
(a) through (d). ). The step of determining whether the mutations are
associated with pre-
3o neoplasia, neoplasia or progression of neoplasia may be done by comparing
the mutations
with DNA from non-involved tissue or bodily fluid from the subject, or by
comparing the
mutations with mitochondria) DNA from a maternal relative. The determination
of
21
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
whether the malignancy is latent or aggressive may comprise monitoring the
person at
successive time periods for an increase in mutations or an increase in mutated
mitochondria) genomes.
Although specific mutation sites may indicate a disease state, a disorder or
aging,
the total mutation load is also important in determining the genesis, presence
and
progression of a disease, a disorder or aging. Accordingly, mutation load can
be used to
diagnose a disease, a disorder or aging.
to The biological sample for the methods of the present invention may be taken
from a
tissue that is chosen from benign tissue, normal tissue, atypical tissue and
histologically/pathologically abnormal tissue. Other clinical methods can also
identify
abnormal tissue. In addition, the sample may be taken from any bodily fluid,
for example,
blood, urine, prostate massage fluid, etc.
The step of deterniining whether the mutations are associated with normal
interpopulation or intrapopulation variations, or whether the mutations are
associated with
pre-neoplasia, neoplasia, progression of neoplasia toward potential
malignancy, or
malignancy comprises comparing the mtDNA of the biological sample to a
database of
2o sequences associated with pre-neoplasia, neoplasia, progression of
neoplasia toward
potential malignancy, malignancy, inter and intra population variations and
normal
sequences. Optionally, one can determine total mutation load of the biological
sample.
Optionally, the step of determining whether the mutations are associated with
pre-
neoplasia, neoplasia or progression of neoplasia may be done by comparing the
mutations
mitochondria) DNA from non-involved tissue or bodily fluid from the subject,
or by
comparing the mutations with mitochondria) DNA from a maternal relative. The
entire
mitochondria) genome, or a subset of the genome, can be monitored for
mutations which
are then compared to the database to aid in the detection of pre-neoplasia
,and/or neoplasia,
progression towards malignancy and malignancy.
Another aspect of the invention is to provide oligonucleotide primers chosen
from
SEQ m NO. 19 to 101. In still another embodiment of the invention, an
oligonucleotide
22
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
primer is provided wluch comprises a sequence of 10, 12, 14, 16, 18, 20, 22,
24, 26, or 30
contiguous nucleotides comprised of sequences from human mtDNA.
The methods, arrays and kits may comprise the mutations listed in Table 4 or
SEQ
s m Nos: 102 to 138.
Another aspect of the invention is provide a use of a primer to amplify a
nucleic
acid molecule comprising at least one mutation listed in Table 4 or SEQ ~ Nos:
102 to
138. The primer may be selected from SEQ m Nos: 19 to 101.
l0
Another aspect of the invention is to provide a method of detecting at least
one
mutation listed in Table 4 or SEQ ID Ns: 102 to 138 in a nucleic acid
molecule,
comprising: amplifying the nucleic acid molecule with a primer associated with
the
mutation; and detecting the mutation. The primer may be selected from SEQ ID
Nos: 19 to
15 101.
Brief Description of the Figure
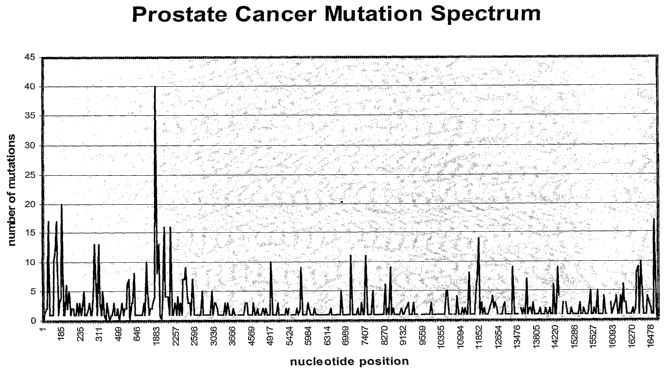
Figure 1 is a histogram showing the number of mutations at nucleotide position
in
2o mitochondria) DNA from patients with prostate cancer.
Brief Description of the Tables
Table 1 is a summary of mutations associated with aging.
25 Table la is a principal component analysis of mutations in mtDNA of seven
protein coding
regions in control, distant benign, adjacent benign and malignant tissue.
Table lb is a neural network analysis of mutations in mtDNA of seven protein
coding
regions in control, distant benign, adjacent benign and malignant tissue.
Table 2 is a summary of the mean number of deletions is epidermal tumours and
adjacent
30 normal tissues.
Table 3 is summary of the standard method of DHPLC.
23
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Table 4 is a summary of mitochondria) mutations (including D-loop) from
prostate needle
biopsies and complete genome mutations from malignant, adjacent and distant
benign
prostate glands from patients with prostate cancer.
Table 5 is a list of primers used for complete mitochondria) genome
amplification for
formalin fixed and normal tissues from blood.
Detailed Description of the Invention
The method of the present invention can be used to diagnose diseases linked to
mtDNA. The method of the present invention provides for analysis of the
mitochondria)
to genome of an individual from a biological sample, for example by
amplification of the
mitochondria) genome, sequencing a portion of the mitochondria) genome,
preferably the
entire mitochondria) genome of the individual using any known means.
Denaturing high
performance liquid chromatography (DHPLC) may also be used to rapidly screen
many
samples. DHPLC can focus on hotspots of mutations. DHPLC is more sensitive
than
automated sequencing in terms of detecting mutations, and can even detect 2%
heteroplasmy, compared with 20-25% for ordinary sequencing. Methods for
detecting
lower levels of heteroplasmy (<2%) may also be developed.
As used herein, the "presence" of a mutation in mtDNA includes heteroplasmic
2o mutations and, therefore, it is contemplated that there may be additionally
the presence of
some normal mtDNA in a sample in which the mutated DNA is present.
As used herein, "actinic kerotoses" means proposed precursor epidermal lesion
of a
squamous cell carcinoma.
As used herein, "aging" refers to an accumulation of changes with time, both
at the
molecular and cellular levels.
As used herein, "alleles" means one of several alternative forms of a given
DNA
3o sequence occupying a specific place on a chromosome.
24
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
As used herein, "attaching" or "spotting" refers to a process of depositing a
nucleic
acid onto a solid substrate to form a nucleic acid array such that the nucleic
acid is
irreversibly bound to the solid substrate via covalent bonds, hydrogen bonds
or ionic
interactions.
As used herein, "atypical" or "abnormal" means cellular appearance which is
not
normal, but also does not appear to be malignant.
As used herein, "basal cell carcinoma" means a type of cancer of skin cells.
to
As used herein, "benign" means of no danger to health; not recurrent or
progressive; not malignant.
As used herein, "Bowen's disease" means in situ epidermal carcinoma.
As used herein, "diagnostic" or "diagnosing" means using the presence or
absence
of a mutation or combination of mutations as a factor in disease diagnosis or
management.
The detection of the mutations) can be a step in the disease state diagnosis.
2o As used herein, "disease" includes a disorder or other abnormal physical
state.
As used herein, "disease associated mitochondiral genomes" means genomes
containing mutations indicative or otherwise associated with a particular
disease.
As used herein, "database" means an electronic storage system (computer based
using standard industry software) which will have the capacity to store and
provide
retrievable information that will enable researchers to rapidly determine the
structure of the
nucleotide sequences. The database will also store descriptive information
about those
individuals who provide the biological samples. This descriptive information
will include
3o health status and other pertinent indices which may be correlated to the
biological sample.
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
As used herein, "deletions" means removal of a region of DNA from a contiguous
sequence of nucleic acids, where once a deletion has occurred, the gap is
repaired by
rejoining of the ends. Deletions can range in size from one base to thousands
of bases or
larger.
As used herein, "duplications" means when a specific sequence of DNA is copied
and inserted behind or forward of the original copy one or more times or
elsewhere in the
genome.
As used herein, "heteroplasmy" is defined by the ratio of mutant: to wild
type mtDNA molecules, where 100% mutant mtDNA is termed "homoplasmic".
Heteroplasmic mutations are those mutations which occur in some, but not all
of the copies
of the mitochondria) genome.
As used herein, "homoplasmy" means all mitochondria) sequences are identical.
As used herein, "hyper-mutation" means accelerated mutation rate which cannot
be
explained by normal cellular processes or standard evolutionary principles.
As used herein, "inversions" refers to when a length of DNA is excised and
reinserted in reverse orientation.
As used herein, "maternal inheritance" means mitochondria which are inherited
through the cytoplasm of the ovum.
As used herein, "maternal line" refers to the clonal sequence of mitochondria)
DNA
as passed down through successive generations from the mother.
As used herein, "mitochondria" means a eukaryotic cytoplasmic organelle that
generates ATP for cellular processes.
26
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
As used herein, "mutation" encompasses any change in a DNA sequence from the
wild type sequence, including without limitation point mutations, transitions,
insertions,
transversions, translocations, deletions, inversions, duplications,
recombinations or
combinations thereof.
As used herein, "mutation load" refers to an increase in mutations in mtDNA
which
eventually leads to compromised function of the involved gene or the entire
genome.
As used herein, "neoplasia" means a pathological process which may result in
transformation to malignant status.
As used herein, "non-involved tissue" means tissue from a part of the body
which is
not associated with the disease in question.
As used herein, "normal tissue" means tissue with no visible manifestations of
disease as determined by lustology.
As defined herein, a "nucleic acid array" refers to a plurality of unique
nucleic acids
attached to one surface of a solid support at a density exceeding 20 different
nucleic
2o acids/cmz wherein each of the nucleic acids is attached to the surface of
the solid support in
a non-identical preselected region. In one embodiment, the nucleic acid
attached to the
surface of the solid support is DNA. In a preferred embodiment, the nucleic
acid attached
to the surface of the solid support is cDNA. In another preferred embodiment,
the nucleic
acid attached to the surface of the solid support is cDNA synthesized by
polymerase chain
reaction (PCR). Preferably, a nucleic acid array according to the invention,
comprises
nucleic acids of at least 150 nucleotides in length. Preferably, a nucleic
acid array
comprises nucleic acids of less than 6,000 nucleotides in length. More
preferably, a
nucleic acid array comprises nucleic acids of less than 500 nucleotides in
length. In one
embodiment, the array comprises at least 500 different nucleic acids attached
to one surface
3o of the solid support. In another embodiment, the array comprises at least
10 different
nucleic acids attached to one surface of the solid support. In yet another
embodiment, the
array comprises at least 10,000 different nucleic acids attached to one
surface of the solid
27
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
support. The teen "nucleic acid", as used herein, is interchangeable with the
term
"polynucleotide".
As used herein, a "nucleic acid taxget" or "a target nucleic acid" is defined
as a
nucleic acid capable of binding to a nucleic acid member of complementary
sequence
through one or more types of chemical bonds, usually through complementary
base pairing,
usually through hydrogen bond formation. As used herein, a nucleic acid target
may
include natural (i. e., A, G, C, or T) or modified bases (7-deazaguanosine,
inosine, etc.). In
addition, the bases in nucleic acid probe may be joined by a linkage other
than a
to phosphodiester bond, so long as it does not interfere with hybridization.
Thus, nucleic acid
targets may be peptide nucleic acids in which the constituent bases are joined
by peptide
bonds rather than phosphodiester linkages. Preferably, the nucleic acid
targets are derived
from human tissue or fluid extracts. More preferably, the nucleic acid targets
are single- or
double-stranded DNA, RNA, or DNA-RNA hybrids synthesized from human tissue of
fluid extracts.
As used herein, "nucleus" means the most conspicuous organelle in the
eucaryotic
cell, contains all of the chromasomal DNA.
2o As used herein, "PSA Test" means prostate-specific antigen test; an antigen
found
in blood that may be indicative of cancer of the prostate.
As used herein, "point mutation" means the change of a single nucleotide in
DNA.
As used herein, "polymorphism" means sequence variation in a population of
alleles or mtDNA genomes.
As used herein, "precursor lesions" means a DNA mutation, or combinations
thereof, indicating potential disease association.
As used herein, "predisposed to a disease" or a "predisposition to a disease"
means
that individuals are at higher risk for developing the disease or disorder or
are at higher risk
28
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
for early onset of the disease or disorder than the average individual, due to
the presence or
absence of mutations which are associated with the disease or disorder.
As used herein, "pre-neoplasia" means indications at the cellular or DNA level
that
a cell may be on the threshold of becoming neoplastic.
As used herein, "preselected region", "predefined region", or "unique
position"
refers to a localized area on a substrate which is, was, or is intended to be
used for the
deposit of a nucleic acid and is otherwise referred to herein in the
alternative as a "selected
to region" or simply a "region." The preselected region may have any
convenient shape, e.g.,
circular, rectangular, elliptical, wedge-shaped, etc. In some embodiments, a
preselected
region is smaller than about 1 cma, more preferably less than 1 mm2, still
more preferably
less than 0.5 mm2, and in some embodiments about 0.125 to 0.5 mm~'.
As used herein, "somatic mutation" means a change in DNA sequence after
fertilization.
As used herein, "solid substrate" or "solid support" refers to a material
having a
rigid or semi-rigid surface. The terms "substrate" and "support" are used
interchangeable
2o herein with the terms "solid substrate" and "solid support". The solid
support may be
biological, non-biological, orgauc, inorganic, or a combination of any of
these, existing as
particles, strands, precipitates, gels, sheets, tubing, spheres, containers,
capillaries, pads,
slices, films, plates, slides, etc. Often, the substrate is a silicon or glass
surface,
(poly)tetrafluoroethylene, (poly)vinylidendifluoride, polystyrene,
polycaxbonate, a charged
membrane, such as nylon 66 or nitrocellulose, or combinations thereof. In a
preferred
embodiment, the solid support is glass. Preferably, at least one surface of
the substrate will
be substantially flat. Preferably, the surface of the solid support will
contain reactive
groups, including, but not limited to, carboxyl, amino, hydroxyl, thiol, or
the like. In one
embodiment, the surface is optically transparent.
As used herein, "squamous cell carcinoma" means a type of cancer of skin
cells.
29
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
As used herein, "stably associated" refers to a nucleic acid that is
irreversibly bound
to a solid substrate to form an array via covalent bonds, hydrogen bonds or
ionic
interactions such that the nucleic acid retains its unique preselected
position relative to all
other nucleic acids that are stably associated with an array, or to all other
preselected
regions on the solid substrate under conditions wherein an array is analyzed
(i.e.,
hybridization and scanning).
A "statistically significant" number of mitochondrial DNA sequences is
determined
by or through the use of standard chi-square statistical algorithms using or
determining
to observed versus expected scores.
As used herein, "subtle mutation" means low level of mutation at the threshold
of
detection.
As used herein, "transitions" means substitution of like nitrogenous bases,
pyrimidine to pyrimidine, purine to purine. A mutation in which one pyrimidine
is
substituted by the other, or in which one purine is substituted by the other.
As used herein, "transversions" means substitution of unlike nitrogenous
bases,
purine to pyrimidine, pyrimidine to purine. A mutation in which a purine is
substituted or
2o replaced by a pyrimidine or vice versa.
MtDNA and diagnosis of specific diseases
In an embodiment of the present invention, methods are provided for monitoring
aging and diagnosing specific diseases such as prostate cancer and non-
melanoma slcin
cancer through comparisons of mtDNA sequences. Diagnosing diseases such as
prostate
cancer with mtDNA, rather than nuclear DNA has several advantages. Firstly,
mtDNA, a
less complex genome, is easily understood at an individual and population
level, hence a
large mtDNA database with normal and disease associated genomes renders
individual
diagnosis extremely accurate. Accordingly, variation, in relationship to
disease, is
3o understood. Secondly, mtDNA has a 10-fold higher mutation rate than nuclear
DNA
(Wallace 1992). Nuclear rearrangements, suggestive of preliminary disease, are
rapidly
communicated to mitochondria, where they appear as somatic mutations. Thirdly,
mtDNA
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
has a maternal inheritance pattern, and is essentially clonal in that all
mitochondria begin
with the same mtDNA sequence, hence variation from this clonal condition is
easily
detected. Additionally, mtDNA does not show convincing evidence of
recombination, thus
any alterations in sequence are a somatic event. Any one mitochondrion
harboring a
mutations) is in a sense 'recessive' as a consequence of there being many
mitochondrial
genomes (2-10 copies) per mitochondrion, and many mitochondria per cell (500-
2,000).
Moreover, mitochondrial genomes can tolerate very high levels (up to 90%) of
mitochondria with damaged genomes. This happens through complementation by the
remaining wild type mtDNA (Chomyn et al. 1992). However, mutated genomes have
a
to replicative advantage over wild type genomes because they are usually
smaller (Hayashi et
al. 1991), hence there is clonal expansion of mutated mtDNA (Brierley et al.
1998),
suggesting that unlike nuclear genes, there is little or no selection against
cells harboring
mtDNA mutations. Because of this elevated mutation rate, mutations and/or
deletions that
appear in mtDNA are maintained through the life span of the cell and may serve
as a record
of exposures to various mutagens. The integrity of mtDNA is maintained by
nuclear repair
mechanisms, and a defect at these loci has been suggested to result in an
autosomal
dominant disorder associated with multiple mitochondrial deletions (Zeviau et
al. 1990).
Consequently, mtDNA may function as an early warning sentinel of early nuclear
events
related to a variety of cancers or other diseases. Finally, the mitochondrial
genome can be
sequenced and monitored for mutations on an individual basis.
The methods and products of the present invention detect both heteroplasmic as
well as
homoplasmic mutations. In fact, heteroplasmic mutations may be key to the
detection of
the early genesis of disease, disorder or aging. In addition, although
specific mutation sites
may indicate a particular disease state, disorder or aging process, the total
mutation load is
also important in determining the genesis, presence and progression of a
disease, a disorder
or aging.
The present invention allows for the ability to examine benign or normal
tissue or bodily
3o fluids to determine the genesis of disease, disorder or aging. For example,
the present
invention allows for the ability to examine benign tissue or bodily fluids for
the presence of
pre-neoplasia, neoplasia, progression toward malignancy and malignancy.
31
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The mitochondria) mutations detected by the methods of the invention are
compared to
inter and intrapopulation variations in mitochondria) DNA, and may include
comparison
with mitochondria) DNA from non-involved tissue from the subject, or with
mitochondria)
DNA from a maternal relative. It is not necessary to analyze the entire
mitochondria)
genome. For example, it is not necessary to sequence the entire mitochondria)
genome,
only a select portion of it. Accordingly, a sample of mitochondria) DNA can
provide a
diagnosis.
to D~osis of Non-Melanoma Skin Cancer
In a preferred embodiment of the invention, a system for early diagnosis of
mtDNA
changes in non-melanoma skin cancer (NMSC) and their precursor lesions
indicative of
solid tumour development is provided. The particular changes, such as the
common
deletion and associated mutations, and the incidence of as yet uncharacterised
deletions in
mtDNA serve as reliable bio-markers of potential skin cancer. The mutation
fingerprint of
the entire mtDNA genome in human NMSC and its precursor lesions is determined.
Thus
mtDNA changes are established as an early bio-marker of human skin cancer and
its
precursor lesions. Denaturing HPLC can then be used to assess low levels of
heteroplasmy
at the sequences of interest. This approach can also provide an insight into
the
2o development of early changes in other human tumours.
Diagnosis of prostate cancer
In another embodiment of the invention, a system for diagnosis of prostate
cancer is
provided. Age related accumulation of mtDNA defects might predispose an
individual to
the appearance of certain clinical disorders such as prostate cancer which is
prevalent in
middle age and older men. In a preferred embodiment, routine prostate cancer
screening
takes place through mitochondria) genome sequencing from prostate massage
fluid. The
presence of epithelial cells transformed into cancer cells, can be determined
through
amplification of mtDNA from prostate massage fluid, eclipsing current
diagnostic
3o techniques such as digital rectal examination and PSA. Recently Fliss et
al. (2000)
identified mutated mtDNA in urine samples of patients with bladder cancer.
Similar
findings in prostate massage fluid provide a non-invasive early detection
method for
32
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
prostate cancer. Different types of prostate cancer can be diagnosed, as well
as
differentiating between aggressive, fast growing cells in younger patients in
contrast to
prostate cancer as a whole.
Early detection and monitoring of prostate cancer pro erg scion
The system and method of the present invention may be used to detect cancer,
and
in particular prostate cancer, at an early stage, and before any histological
abnormalities.
For example, the system and method of the present invention may be used to
detect pre-
1o neoplasia in prostate tissue. The system can be used to detect the genesis
and progression
of prostate cancer. Mutations, including both subtle and hyper-mutation (Chen
et al. 2002;
Chen et al. 2003) in mitochondria) DNA from human prostate tissue, or fluid
associated
with the prostate (for example prostate massage fluid or urine), can be tested
for the
presence of neoplasia, and retested at intervals to follow cancer
transformation, diagnose
malignancy, or confirm continued benign status.
These mutations axe determined by comparison to mitochondria extracted from
non-involved tissue such as, but not limited to: blood, urine, hair and buccal
swabs. This
direct comparison eliminates polymorphisms, maternal background or normal
haplotype
variation unassociated with disease. The mutations can also be compared to
mitochondria)
2o sequences associated with inter and intrapopulation variations. One or more
mutations
from fluid or tissue of the organ or body system in question, indicates
possible disease
genesis. The person is then monitored, at successive intervals, for an
increase in mutations
at other sites, and/or an increase in the number of mutated mitochondria)
genomes,
indicating disease progression. Benign tissue from the prostate cannot always
be
considered non-involved. In fact, as can be seen in Example 9, below, what
appears to be
benign tissue may contain mitochondria) mutations associated with pre-
neoplasia,
neoplasia, progression toward malignancy or malignancy. In addition, mutation
load rather
than specific mutations may be instrumental in determining disease and
progression of
disease. The system and method of the present invention detects heteroplasmic
as well as
3o homoplasmic mutations.
The prostate gland is monitored for mutations in the mitochondria) genome
through
prostate massage fluid (PMF) taken during an initial digital rectal
examination (DRE) of
33
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
the prostate. Cells within the PMF are concentrated, smeared on a slide and
stained with
PSA immunoperoxidase for identification of prostate epithelial cells. These
prostate cells
are selectively recovered through laser capture micro-dissection. The
mitochondria) DNA
from these cells is analyzed and compared to mitochondria) DNA from non-
involved
tissue, and to sequences of inter and intrapopulation variations. For example,
the DNA
analysis can comprise sequencing of the mtDNA. Total DNA is extracted from
these cells
and mitochondria) specific primers, designed for use with biopsy material
treated with
formalin (Table 5), are used to amplify the entire mtDNA genome with
overlapping
amplicons. These PCR products are then sequenced by methods well known to
those in the
1o art, including DNA resequencing arrays. Sequencing results axe screened for
heteroplasmies and mutations and compared to a database of known mtDNA
mutations
associated with malignant and benign prostate tissues. Based on these
comparisons a
designation is returned as to the condition of the prostate in regards to, but
not limited to:
benign (no mutations); pre-neoplasia or neoplasia (low level of mutations); or
malignancy
(high level of mutations). In the situation of benign, pre-neoplasia and
neoplasia, the
prostate can be monitored for progression through regular PMF screenings as
described.
Alternatively, biopsy material which has been diagnosed as benign, atypical,
abnormal can undergo similar testing by either laser capture micro-dissection
of the biopsy,
or the tissue can be scrapped off the slides, followed by DNA extraction,
amplification,
2o sequencing and database comparison.
As an alternative to sequencing, and comparison to a database, micro-array
technology could be used to identify a specific pattern of mutations, or
mutation load based
on any number, or combination of the mutations listed in Table 4, through the
construction
of oligonucleotides, or a specific set of oligonucleotides.
Disease progression can be monitored by comparing mtDNA mutations at
successive intervals to a database of mutations in mitochondria) genomes
associated with
pre-neoplasia, neoplasia and prostate cancer, including calculation of total
mutation load.
Prostate biopsy tissue can be tested for pre-neoplasia, neoplasia and/or
malignant
progression in cells described clinically as benign, normal, atypical or
abnormal by
common histological/pathological, or other clinical methods.
Assessment of mutations associated with aging
34
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The system and method of the present invention may be used to assess aging,
based
on the increasing frequency of mutations such as the "common deletion" of 4977-
by and
other mutations of the mitochondrial genome (Liu et al. 1997). This
information, in
conjunction with health survey data, allows crucial statistical discrimination
between
separate causes resulting in the same mutation/deletion. Fortunately mtDNA is
inherited
exclusively through the ovum and is essentially clonal in nature (Van De
Graaff & Fox,
1995). This permits carefully controlled studies of mutations/deletions within
maternal
lines through several generations to determine a reliable age related deletion
frequency.
This information may be used to develop treatment methods which slow the aging
process.
to
Collection of samples
Biological samples can be collected by any known means, whether for the
purpose of
constructing a mtDNA sequence database, or performing a diagnostic test on an
individual.
Samples destined for database generation include, but are not limited to:
tumour banks,
maternal lineage studies involving affected and unaffected individuals from
the same
maternal lineage, as well as maternal lineage studies from groups or
populations with high
frequencies of specific disease such as, but not limited to: skin and prostate
cancer,
assessment of health status and aging. For example, FTA~ Gene Cards~ may be
used to
collect and archive biological samples. Suitable samples include any tissue or
body fluid
2o derived from mesothelium, epithelium, or endothelium. Such tissues and
fluids include,
but are not limited to blood, sputum, buccal cells, saliva, prostate massage
fluid, sweat,
bone, hair, lymph tissue, cervical smears, breast aspirate, fecal matter,
ejaculate, menstrual
flow and biopsy tissue. Preferably, approximately 100 ~.l of blood, 100 p,g to
25 mg of
solid tissue is sampled. In the case of suspected skin cancer, skin cells or
tissue, (from
normal, NMSC and precursor lesions) is taken from skin biopsy or a routine
suction
blistering technique. Where a disease is suspected, primary care physicians,
oncologists or
other practitioners, may extract both normal and suspected disease tissue from
the patient.
For samples of tumours such as prostate or skin, replicate cross-sections (5
3o microns) of micro-dissected paraffin embedded tissues are de-paraffinized
prior to one
slide being stained with hematoxylin and eosin (HE), with the replicate
stained with methyl
green (MG), as is standard in the art. HE stains are graded by a pathologist
for normal,
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
precursor, and applicable grades of tumour progression. Replicate MG slides
are used for
laser capture, according to manufacturers recommendations (Arcturus) of graded
cells.
Extraction of mtDNA
Extraction of DNA may take place using any method known in the art, followed
by
sequencing of the mitochondrial genome, as described in Current Protocols in
Molecular
Biology.
Analyzing mtDNA
to
The step of detecting the presence of mutations in the mtDNA can be selected
from any
technique as is known to those skilled in the art. For example, analyzing
mtDNA can
comprise sequencing the mtDNA, amplifying mtDNA by PCR, Southern, Northern,
Western South-Western blot hybridizations, denaturing HPLC, hybridization to
microarrays, biochips or gene chips, molecular marker analysis, biosensors,
melting
temperature profiling or a combination of any of the above. In addition,
statistical
techniques such as Inductive Rule Extraction, Neural Networking and Wave
Analysis can
be used.
2o Sequencing of MtDNA
PCR
Polynucleotide sequences of the invention can be amplified by the polymerase
chain reaction (PCR). PCR methods are well-known to those skilled in the art.
PCR
requires the presence of a nucleic acid to be amplified, two single stranded
oligonucleotide
primers flanking the sequence to be amplified, a DNA polymerase,
deoxyribonucleoside
triphosphates, a buffer and salts. The method of PCR is well known in the art.
PCR is
performed as described in Mullis and Faloona, 1987, Methods Enzymol., 155:
335, herein
incorporated by reference.
3o In general, PCR is performed using template DNA (at least lfg; more
usefully, 1-
1000 ng) and at least 25 pmol of oligonucleotide primers. A typical reaction
mixture
includes: 2~,1 of DNA, 25 pmol of oligonucleotide primer, 2.5 ,ul of lOX PCR
buffer 1
36
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
(Perkin-Elmer, Foster City, CA), 0.4 ~,l of 1.25 ,uM dNTP, 0.15 ,ul (or 2.5
units) of Taq
DNA polymerase (Perkin Elmer, Foster City, CA) and deionized water to a total
volume of
25 ~,1. Mineral oil is overlaid and the PCR is performed using a programmable
thermal
cycler.
The length and temperature of each step of a PCR cycle, as well as the number
of
cycles, are adjusted according to the stringency requirements in effect.
Annealing
temperature and timing are determined both by the efficiency with which a
primer is
expected to anneal to a template and the degree of mismatch that is to be
tolerated. The
to ability to optimize the stringency of primer annealing conditions is well
within the
knowledge of one of moderate skill in the art. An annealing temperature of
between 30°C
and 72°C is used. In general, initial denaturation of the template
molecules normally
occurs at between 92°C and 99°C for 4 minutes, followed by 20-40
cycles consisting of
denaturation (94-99°C for 15 seconds to 1 minute), annealing
(temperature determined as
discussed above; 1-2 minutes), and extension (72°C for 1 minute). The
final extension step
is generally carried out for 4 minutes at 72°C, and may be followed by
an indefinite (0-24
hour) step at 4°C.
DNA Sequencing
Any known means to sequence the mitochondrial genome may be used. Preferably,
mtDNA is amplified by PCR prior to sequencing. PCR products can be sequenced
directly
or cloned into a vector which is then placed into a bacterial host. Examples
of DNA
sequencing methods are found in Bromley, R. L. Jr. and Smith, L.M., 1991,
Rapid DNA
sequencing by horizontal ultrathin gel electrophoresis, Nucleic Acids Res.
19:4121-4126
and Luckey, J.A., et al, 1993, High speed DNA sequencing by capillary gel
electrophoresis,
Methods Enzymol. 218: 154-172. The combined use of PCR and sequencing of mtDNA
is
described in Hopgood, R., et al, 1992, Strategies for automated sequencing of
human
mtDNA directly from PCR products, Biotechniques 13:82-92 and Tanaka, M. et al,
1996,
Automated sequencing of mtDNA, Methods Enzymol. 264: 407-421
Deletion Analysis and Detection
37
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
A preferable approach is the long extension PCR (LX-PCR) technique using the
Expand Long Template PCR system (Boehringer Mannheim). Using the LX-PCR
technique, which has been established and validated in the Birch-Machin
laboratory (Ray
et al. 2000), there is the opportunity to rapidly screen for the whole
spectrum of mtDNA
deletions as opposed to the incidence of a single deletion.
A semi-quantitative PCR method (Corral-Debrinski et al 1991) can be used to
estimate the proportion of the mtDNA49~~ deletion in the total mtDNA.
In addition, Southern Blot and probing technology labeled with isotopes or any
other technique as is standard in the art may be used for deletion detection
as well.
to
Sequehczhg of PCR p~oductr
Any known means may be used to sequence the PCR products. Preferably, the
entire DNA sequence is characterized by di-deoxy sequencing using ABI Big Dye
TerminatorTM technology and a series of 72 overlapping primers each for heavy
and light
strands. Sequencing occurs on one, several, or a combination of ABI platforms
such as the
310, 3100, or 3700. Sequencing reactions are performed according to
manufacturer's
recommendation.
Mutational analysis of the mitochondria) genome using denaturing high
performance
liquid chromatography (DHPLC)
Prior to sequencing of the mitochrondrial genome and identification of
mutational
hotspots, DHPLC can be used to rapidly screen mutations in many samples. This
technique provides greater sensitivity in identification of low levels of
heteroplasmy. It
cannot detect homoplasmic changes but will complement traditional sequencing.
Apart
from the homoplasmic mutations recently identified in tumours, the vast
majority of
reported mtDNA mutations are heteroplasmic (Chinnery et al. 1999). These
heteroplasmic
mtDNA changes result in the formation of heteroduplexes after PCR
amplification of the
mtDNA. Rapid screening for heteroplasmic mtDNA mutations is determined using
the
relatively new technique of denaturing high performance liquid chromatography
(DHPLC)
(Oefner & Underhill, 1998). This technique has recently been used to rapidly
screen and
identify whole mtDNA genomes for heteroplasmic point mutations down to levels
<5%
(Van den Bosch et al. 2000).
38
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The DHPLC may be performed on the WAVETM DNA Fragment Analysis System
(Transgenomic, Omaha, USA) which provides a fully automated screening
procedure. The
same technology can be used to screen for mtDNA heteroplasmic mutations.
Preferably,
the entire mtDNA genome is amplified by PCR in 13 overlapping fragments using
two
different PCR conditions as described by van den Bosch et al. (2000). The 1-2
kb PCR
products are digested into fragments of 90-600bp and resolved at their optimal
melting
temperature. Mutations are represented as two peaks and mutations with low
percentages,
such as <2% heteroplasmy as a 'shoulder' in the peal.
l0
DNA sequencing can also take place using a microarray, as is known in the art
(Chee et
al. 1996).
Data Analysis
Once sequenced, normal and disease associated mtDNA sequences are archived for
comparison in a database. Resequencing devices, micro-array technology,
integrated
microfluidic amplification and analysis systems, high-speed, high-throughput,
mutation
detection, and other methods may all be used with the methods of the present
invention.
2o Data obtained from the sequencing of the individual mitochondrial genome is
compared to population level data. The data is obtained through obtaining
samples and
sequencing mtDNA as described above. Preferably, the database contains
information
from maternal line studies. The population level data is maintained in a
database. Any
suitable database can be used.
Preferably, a multidimensional evaluation research database of clinical and
biological data is used, which provides the bio-informatics infrastructure
necessary for the
collection, processing and dissemination of information amassed by the
laboratories
involved in this venture. The database is a centralized electronic system
which links
networks resulting in a dynamic and powerful resource.
39
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The database may be accessed through any known means, and preferably through a
secure Internet pathway. Preferably, the database is developed using an e-
commerce
algorithm, built on a server and deployed using an application server which
supports a high
volume of concurrent users through optimized performance and scalability
features. A
separate "web" server can provide the foundation of the web-site architecture
since it can
serve as the central point through which all content, applications, and
transactions must
flow before reaching users.
Data mining algorithms known in the art are used to discover patterns,
clusters and
to models from data (SAS 2000). Moreover, intelligent algorithms and methods
will be
developed for: occurrence of mutation and mutation rates, patterns of
mutations for disease
detection, information retrieval, and other complex sequence analysis
software.
Nucleic Acid Members and Probes
The invention provides for nucleic acid members and probes that bind
specifically
to a target nucleic acid sequence. The target nucleic acid sequence is a
nucleic acid or a
region of a nucleic acid that is to be detected, as indicative of disease such
as prostate
cancer, non-melanoma skin cancer and the like. The target nucleic acid
sequences to be
2o analyzed using a microarray of the invention are preferably derived from
human tissue or
fluid samples. The invention provides for target nucleic acid sequences
comprising RNA or
nucleic acid corresponding to RNA, (i.e., cDNA), or DNA. Nucleic acid members
are
stably associated with a solid support to comprise an array according to the
invention. The
nucleic acid members may be single or double stranded, and may be a PCR
fragment
amplified from cDNA.
The invention also provides for polynucleotide sequences comprising a probe.
As
used herein, the term "probe" refers to an oligonucleotide which forms a
duplex structure
with a sequence in the target nucleic acid, due to complementarity of at least
one sequence
3o in the probe with a sequence in the target region. The probe may be
labeled, according to
methods known in the art. A probe according to the invention may be single or
double
stranded.
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Diagnostic devices
The invention includes diagnostic devices such as biochips, gene chips or
microarrays used to diagnose specific diseases or identify specific mutations.
All
sequenced mitochondrial genomes are assessed to create a consenus structure of
the base
pair arrangement and are assigned a prohibiting index for proportion of base
pair deletions
and mutations associated with a particular disease or disorder. The diagnostic
arrangement
is then used to create biochips, gene chips, or microarrays.
to Once sequences associated with particular diseases, disease states or
disorders are
identified, hybridization of mtDNA to an array of oligonucleotides can be used
to identify
particular mutations. Any known method of hybridization may be used.
Preferably, an
array is used, which has oligonucleotide probes matching the wild type or
mutated region,
and a control probe. Commercially available arrays such as microarrays or gene
chips are
suitable. These arrays contain thousands of matched and control pairs of
probes on a slide
or microchip, and are capable of sequencing the entire genome very quickly.
Review
articles describing the use of microarrays in genome and DNA sequence analysis
is
available at www.gene-chips.com.
Micf~oa~~ay
Polynucleotide arrays provide a high throughput technique that can assay a
large
number of polynucleotides in a sample comprising one or more target nucleic
acid
sequences. The arrays of the invention are useful for gene expression
analysis, diagnosis of
disease and prognosis of disease (e.g., monitoring a patient's response to
therapy, drug
screening, and the lilee).
Any combination of the polynucleotide sequences of mtDNA indicative of
disease,
aging, or other health related mutations are used for the construction of a
microarray.
3o The target nucleic acid samples to be analyzed using a microarray are
derived from
any human tissue or fluid which contains adequate amounts of mtDNA, as
previously
described, preferably prostate massage fluid, solid tumours, blood, or urine.
The target
41
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
nucleic acid samples are contacted with polynucleotide members under
hybridization
conditions sufficient to produce a hybridization pattern of complementary
nucleic acid
members/target complexes.
CofzstructiofZ of a microarray
The microarray comprises a plurality of unique polynucleotides attached to one
surface of a solid support, wherein each of the polynucleotides is attached to
the surface of
the solid support in a non-identical preselected region. Each associated
sample on the array
comprises a polynucleotide composition, of known identity, usually of known
sequence, as
to described in greater detail below. Any conceivable substrate may be
employed in the
invention.
The array is constructed using any known means. The nucleic acid members may
be produced using established techniques such as polymerase chain reaction
(PCR) and
reverse transcription (RT). These methods are similar to those currently known
in the art
(see e.g. PCR Strategies, Michael A. W nis (Editor), et al. (1995) and PCR:
Introduction to
Biotechniques Series, C. R. Newton, A. Graham (1997)). Amplified
polynucleotides are
purified by methods well known in the art (e.g., column purification). A
polynucleotide is
considered pure when it has been isolated so as to be substantially free of
primers and
2o incomplete products produced during the synthesis of the desired
polynucleotide.
Preferably, a purified polynucleotide will also be substantially free of
contaminants which
may hinder or otherwise mask the binding activity of the molecule.
In the arrays of the invention, the polynucleotide compositions are stably
associated
with the surface of a solid support, wherein the support may be a flexible or
rigid solid
support.
Any solid support to which a nucleic acid member may be attached may be used
in
the invention. Examples of suitable solid support materials include, but are
not limited to,
silicates such as glass and silica gel, cellulose and nitrocellulose papers,
nylon, polystyrene,
polymethacrylate, latex, rubber, and fluorocarbon resins such as TEFLONTM
42
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The solid support material may be used in a wide variety of shapes including,
but
not limited to slides and beads. Slides provide several functional advantages
and thus are a
preferred form of solid support. Due to their flat surface, probe and
hybridization reagents
are minimized using glass slides. Slides also enable the targeted application
of reagents, are
easy to keep at a constant temperature, are easy to wash and facilitate the
direct
visualization of RNA and/or DNA immobilized on the solid support. Removal of
RNA
and/or DNA immobilized on the solid support is also facilitated using slides.
The particular material selected as the solid support is not essential to the
invention,
1o as long as it provides the described function. Normally, those who make or
use the
invention will select the best commercially available material based upon the
economics of
cost and availability, the expected application requirements of the final
product, and the
demands of the overall manufacturing process.
Numerous methods are used for attachment of the nucleic acid members of the
invention to the substrate (a process referred as spotting). For example,
polynucleotides
are attached using the techniques of, for example U.S. Pat. No. 5,807,522,
which is
incorporated herein by reference for teaching methods of polymer attachment.
Alternatively, spotting is carried out using contact printing technology.
2o The amount of polynucleotide present in each composition will be sufficient
to
provide for adequate hybridization and detection of target polynucleotide
sequences during
the assay in which the array is employed. Generally, the amount of each
nucleic acid
member stably associated with the solid support of the array is at least about
0.1 ng,
preferably at least about 0.5 ng and more preferably at least about 1 ng,
where the amount
may be as high as 1000 ng or higher, but will usually not exceed about 20 ng.
Where the
nucleic acid member is "spotted" onto the solid support in a spot comprising
an overall
circular dimension, the diameter of the "spot" will generally range from about
10 to 5,000
p,m, usually from about 20 to 2,000 pm and more usually from about 50 to 1000
pm.
3o Control polynucleotides may be spotted on the array and used as target
expression
control polynucleotides and mismatch control nucleotides to monitor non-
specific binding
or cross-hybridization to a polynucleotide in the sample other than the target
to which the
43
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
probe is directed. Mismatch probes thus indicate whether a hybridization is
specific or not.
For example, if the target is present the perfectly matched probes should be
consistently
brighter than the mismatched probes. In addition, if all central mismatches
are present, the
mismatch probes are used to detect a mutation.
Tafget p~epaf~atio~
The targets for the microarrays, are derived from human fluid or tissue
samples. It
may be desirable to amplify the target nucleic acid sample prior to
hybridization. One of
skill in the art will appreciate that whatever amplification method is used,
if a quantitative
to result is desired, care must be taken to use a method that maintains or
controls for the
relative frequencies of the amplified polynucleotides. Methods of
"quantitative"
amplification are well known to those of skill in the art. For example,
quantitative PCR
involves simultaneously co-amplifying a known quantity of a control sequence
using the
same primers. This provides an internal standard that may be used to calibrate
the PCR
reaction. The high density array may then include probes specific to the
internal standard
for quantification of the amplified polynucleotide. Detailed protocols for
quantitative PCR
are provided in PCR Protocols, A Guide to Methods and Applications, Innis et
al.,
Academic Press, Inc. N.Y., (1990). Other suitable amplification methods
include, but are
not limited to polymerase chain reaction (PCR) (Innis, et al., PCR Protocols.
A guide to
2o Methods and Application. Academic Press, Inc. San Diego, (1990)), ligase
chain reaction
(LCR) (see Wu and Wallace, Genomics, 4: 560 (1989), Landegren, et al.,
Science, 241:
1077 (1988) and Barnnger, et al., Gene, 89: 117 (1990), transcription
amplification (Kwoh,
et al., Proc. Natl. Acad. Sci. USA, 86: 1173 (1989)), and self sustained
sequence
replication (Guatelli, et al., Proc. Nat. Acad. Sci. USA, 87: 1874 (1990)).
The invention provides for labeled target or labeled probe. Any analytically
detectable marker that is attached to or incorporated into a molecule may be
used in the
invention. An analytically detectable marker refers to any molecule, moiety or
atom which
is analytically detected and quantified. Detectable labels suitable for use in
the present
3o invention include any composition detectable by spectroscopic,
photochemical,
biochemical, immunochemical, electrical, optical or chemical means. Useful
labels in the
present invention include biotin for staining with labeled streptavidin
conjugate, magnetic
44
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
beads (e.g., DynabeadsTM), fluorescent dyes (e.g., fluorescein, texas red,
rhodamine, green
fluorescent protein, and the like), radiolabels (e.g., 3H, l2sh 355, 14C, or
3aP), enzymes (e.g.,
horseradish peroxidase, alkaline phosphatase and others commonly used in an
ELISA), and
colorimetric labels such as colloidal gold or colored glass or plastic (e.g.,
polystyrene,
polypropylene, latex, etc.) beads. Patents teaching the use of such labels
include U.S. Pat.
Nos. 3,817,837; 3,850,752; 3,939,350; 3,996,345; 4,277,437; 4,275,149; and
4,366,241.
Means of detecting such labels are well known to those of skill in the art.
Thus, for
example, radiolabels may be detected using photographic film or scintillation
counters,
to fluorescent markers may be detected using a photodetector to detect emitted
light.
Enzymatic labels are typically detected by providing the enzyme with a
substrate and
detecting the reaction product produced by the action of the enzyme on the
substrate, and
colorimetric labels are detected by simply visualizing the colored label.
The labels may be incorporated by any of a number of means well known to those
of skill in the art. However, in a preferred embodiment, the label is
simultaneously
incorporated during the amplification step in the preparation of the sample
polynucleotides.
Thus, for example, polymerase chain reaction (PCR) with labeled primers or
labeled
nucleotides will provide a labeled amplification product. In a preferred
embodiment,
2o transcription amplification, as described above, using a labeled nucleotide
(e.g. fluorescein-
labeled UTP and/or CTP) incorporates a label into the transcribed
polynucleotides.
Alternatively, a label may be added directly to the original polynucleotide
sample (e.g.,
mRNA, polyA mRNA, cDNA, etc.) or to the amplification product after the
amplification
is completed. Means of attaching labels to polynucleotides are well known to
those of skill
in the art and include, for example nick translation or end-labeling (e.g.
with a labeled
RNA) by kinasing of the polynucleotide and subsequent attachment (ligation) of
a
polynucleotide linker joining the sample polynucleotide to a label (e.g., a
fluorophore).
In a preferred embodiment, the target will include one or more control
molecules
3o which hybridize to control probes on the microarray to normalize signals
generated from
the microarray. Labeled normalization targets are polynucleotide sequences
that are
perfectly complementary to control oligonucleotides that are spotted onto the
microarray as
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
described above. The signals obtained from the normalization controls after
hybridization
provide a control for variations in hybridization conditions, label intensity,
"reading"
efficiency and other factors that may cause the signal of a perfect
hybridization to vary
between arrays.
Hybrielization cotzelitioyas
Polynucleotide hybridization involves providing a denatured probe or target
nucleic
acid member and target polynucleotide under conditions where the probe or
target nucleic
acid member and its complementary target can form stable hybrid duplexes
through
to complementary base pairing. The polynucleotides that do not form hybrid
duplexes are
then washed away leaving the hybridized polynucleotides to be detected,
typically through
detection of an attached detectable label. It is generally recognized that
polynucleotides are
denatured by increasing the temperature or.decreasing the salt concentration
of the buffer
containing the polynucleotides. Under low stringency conditions (e.g., low
temperature
and/or high salt) hybrid duplexes (e.g., DNA:DNA, RNA:RNA, RNA:DNA, cDNA:RNA
and cDNA:DNA ) will form even where the annealed sequences are not perfectly
complementary. Thus specificity of hybridization is reduced at lower
stringency.
Conversely, at higher stringency (e.g., higher temperature or lower salt)
successful
hybridization requires fewer mismatches. Methods of optimizing hybridization
conditions
2o are well known to those of skill in the art (see, e.g., Laboratory
Techniques in Biochemistry
and Molecular Biology, Vol. 24: Hybridization With Polynucleotide Probes, P.
Tijssen, ed.
Elsevier, N.Y., (1993)).
Following hybridization, non-hybridized labeled or unlabeled polynucleotide is
removed from the support surface, conveniently by washing, thereby generating
a pattern
of hybridized target polynucleotide on the substrate surface. A variety of
wash solutions
are known to those of skill in the art and may be used. The resultant
hybridization patterns
of labeled, hybridized oligonucleotides and/or polynucleotides may be
visualized or
detected in a variety of ways, with the particular manner of detection being
chosen based
3o on the particular label of the test polynucleotide, where representative
detection means
include scintillation counting, autoradiography, fluorescence measurement,
calorimetric
measurement, light emission measurement and the like.
46
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Image Acquisition and Data Analysis
Following hybridization and any washing steps) and/or subsequent treatments,
as
described above, the resultant hybridization pattern is detected. In detecting
or visualizing
the hybridization pattern, the intensity or signal value of the label will be
not only be
detected but quantified, by which is meant that the signal from each spot of
the
hybridization will be measured and compared to a unit value corresponding to
the signal
emitted by a lcnown number of end labeled target polynucleotides to obtain a
count or
absolute value of the copy number of each end-labeled target that is
hybridized to a
particular spot on the array in the hybridization pattern.
' to
Methods for analyzing the data collected from hybridization to arrays are well
known in the art. For example, where detection of hybridization involves a
fluorescent
label, data analysis can include the steps of determining fluorescent
intensity as a function
of substrate position from the data collected, removing outliers, i.e., data
deviating from a
predetermined statistical distribution, and calculating the relative binding
affinity of the test
polynucleotides from the remaining data. The resulting data is displayed as an
image with
the intensity in each region varying according to the binding affinity between
associated
oligonucleotides and/or polynucleotides and the test polynucleotides.
2o Following detection or visualization, the hybridization pattern is used to
determine
quantitative information about the genetic profile of the labeled target
polynucleotide
sample that was contacted with the array to generate the hybridization
pattern, as well as
the physiological source from which the labeled target polynucleotide sample
was derived.
By genetic profile is meant information regarding the types of polynucleotides
present in
the sample, e.g. in terms of the types of genes to which they are
complementary, as well as
the copy number of each particular polynucleotide in the sample.
Diagnostic or Prognostic Tests
The invention provides for diagnostic tests for detecting diseases. The
invention
also provides for prognostic tests for monitoring a patient's response to
therapy.
According to the method of the invention, the presence of disease or the
patient's response
to therapy is detected by obtaining a fluid or tissue sample from a patient. A
sample
comprising nucleic acid is prepared from the fluid or tissue sample. The
nucleic acid
47
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
extracted from the sample is hybridized to an array comprising a solid
substrate and a
plurality of nucleic acid members, wherein each member is indicative of the
presence of
disease or a predisposition to a disease or disorder. According to this
diagnostic test,
hybridization of the sample comprising nucleic acid to one or more nucleic
acid members
on the array is indicative of disease, a predisposition to a disease or
disorder, or in the case
of a prognostic test, indicative of a patient's response to therapy.
Kits
1o Kits containing reagents and instructions to carry out the methods of the
present invention
are provided. For example, the kit may comprise reagents and instructions for
detecting
mitochondria) mutations, heteroplasmies, homoplasmies in tissue specific
samples and
tissue associated fluids. The kits may also comprise one or more primers which
hybridize
to the mitochondria) genome for making a primer extension product. Kits may
also include
a disposable chip, means for holding the disposable chip, means for extraction
of mtDNA
and means for access to a database of mtDNA sequences.
Other utilities for the present invention, such as that described above and in
the
following examples, will be readily apparent to those skilled in the art.
The present invention is more particularly described in the following examples
which are intended as illustrative only since numerous modifications and
variations will be
apparent to those skilled in the art.
Example 1: Prostate Tumours
Following acquisition of prostate fluid or surgery to remove prostate tumours,
biopsy slides are prepared to identify transforming or cancerous cells. Laser
Capture
Microdissection (LCM) microscopy is used to isolate cells that are either
normal, benign,
or malignant from the tissue section. Procurement of diseased cells of
interest, such as
3o precancerous cells or invading groups of cancer cells is possible from
among the
surrounding heterogeneous cells.
48
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Total DNA extraction from each of these cells was purified according to a
modification of the protocol outlined by Arcturus Engineering Inc. DNA was
extracted
from cells with a 501 volume of 1 mg/ml proteinase K (PK), in IOmM Tris pH
8.0,
O.lmM EDTA pH 8.0, and 0.1% Tween 20, at 42°C overnight. Following
incubation
overnight at 42°C the tubes were removed from the incubation oven. The
samples were
microcentrifuged for 5 min at 6400 rpm(2000 x g). The CapSureTM was removed
from the
tube and discarded. The tube was incubated at 95°C for 10 minutes (PK
is inactivated) and
then cooled to room temperature. 5-50,1 of the sample was used for PCR
amplification.
to Following purification, individual samples are amplified, by LX-PCR using
the
appropriate primers for hypervariable region 1 (HV1), hypervariable region 2
(HV2) and
the entire 12S region. These PCR products are then sequenced using high
throughput
methods as is well known in the art.
Alternatively, full length mitochondria) genomes may be amplified using the
primers in Table 5. Specific capture and amplification of DNA derived from
malignant
tumour cells of any Geason Grade, cells from an adjacent benign gland and
cells from a
"distant" benign gland may be amplified. Other prostate tissues which could
and are
amplified includes: prostatic intraepithelial neoplasia (PIN), benign
prostatic hyperplasia
2o (BPH), hyperplasia of various types, stroma, and cells with undetermined
changes. This
work was done on prostate tissues from 31 individuals electing to have a
prostatectomy
because of a prostate cancer diagnosis. Three tissue types were captured:
malignant,
adjacent benign and distant benign from each individual. Blood from each
patient was
used as a positive, non-diseased tissue control. Amplification and sequencing
of these
samples resulted in the novel mutations seen in Table 4. The mutations of
Table 4 are also
provided in SEQ ID No: 102 which lists the substitutions, SEQ ID NOs: 103 to
109 which
lists the deletions, and SEQ ID No: 110 to 138 which lists the insertions.
Polymorphisms
and mutation positions were determined by comparison to the Revised Cambridge
Reference Sequence (2001), however the historical numbering has been
maintained such
3o that the deletion at position 3106 is denoted as a gap and the rare
polymorphism 750A has
been retained. A subset of this data (7 protein coding regions) was then
subjected to
49
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
principal component analysis, as is standard in the art, with the following
results as shown
in Table 1 a:
Table la
blood distant adjacent malignantunknown
benign benign
blood 100.00% 0.00% 0.00% 0.00% 0.00%
distant 16.13% 35.48% 9.68% 29.03% 16.13%
benign
adjacent12.9% 12.90% 45:16% 3.22% 25.81%
benign
malignant3.22% 0.00% 0.00% 96.78% 0.00%
I I I I I I I
The results demonstrate a clear pattern of malignant transformation. Normal
tissue
(blood) and malignant tissue display high clustering frequencies (1.00 and
0.967).
Interestingly, adjacent and distant benign, both of which appear normal in a
histological
and pathological sense, show levels of transformation with over 50% of the
samples falling
outside the distant benign and adjacent benign intercepts. Moreover, the same
data was
analyzed by a neural network, as is standard in the art, with the following
results as shown
in Table 1b:
Table lb
Blood distant adjacent malignant
benign benign
blood 100.00% 0.00% 0.00% 0.00%
distant 6.45% 0.00% 0.00% 93.55%
benign
adjacent 19.35% 0.00% 0.00% 77.14%
benign
malignant 3.22% 0.00% 0.00% 96.78%
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
This table shows that, in the presence of tumour, all prostate tissue is
considered
malignant at the molecular level, even though anatomical appearance of the
tissue may be
"normal."
Example 2: Duplications in the non-coding region of mtDNA from sun-exposed
skin
DNA was extracted from tissue samples as described in Example 1, with the use
of
DNeasyTM kit supplied by Qiagen. A "back to back" primer methodology was used
to
investigate the incidence of tandem duplications in the non-coding region
(NCR) in
relation to sun-exposure. 32 age-matched, split human skin samples, from sun-
exposed
(n=24) and sun-protected body sites (n=10) were investigated.
The following duplication primers from Brockington et al 1993 and Lee et al
1994
were used:
C L336 AAC ACA TCT CTG CCA AAC CC 20 mer SEQ m NO: 1
D H335 TAA GTG CTG TGG CCA GAA GC 20 mer SEQ m NO: 2
E L467 CCC ATA CTA CTA ATC TCA TC 20 mer SEQ m NO: 3
F H466 AGT GGG AGG GGA AAA TAA TG 20 mer SEQ m NO: 4
Primers pairs C/D and E/F are 'back to back' at the site of two separate sets
of
2o direct repeats in the non-coding region. As a result they only generate a
product if a
duplication is present at these points. Products generated are 260 by and/or
less common
200bp variant. Modified PCR conditions are: 100ng total cellular DNA, 200~M
dNTPs, 2.5
U HotStarTaq polymerase and PCR buffer (Qiagen, Uk), 25 pmoles of primers: one
cycle of
94°C for 4 minutes, 36 cycles of 94 °C x 1 minute, 55°C x
1 minute, 72°C x 1 minute and
one cycle of 72°C x 7 minutes.
An increased incidence of duplications with increasing sun-exposure was
observed,
with duplications identified in 10/24 but 0/10 samples from sun-exposed and
sun-protected
skin respectively (Fisher's exact test, p=0.01 5) (Birch-Machin and Krishnan
2001). The
3o sizes of the most frequent duplications were 200 and 260 base pairs.
Interestingly these
same samples also contained high levels (>l %) of the 4977bp common mtDNA
deletion as
determined by an established quantitative 3-primer PCR assay described in
Example 6.
51
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Example 3: Mutation fingerprint of mtDNA in human NMSC and its precursor
lesions
DNA was extracted from human skin tissue samples as described in Example l,
with the use of DNeasyTM by Qiagen Using specific primers, mtDNA is amplified
by PCR
and following DNA sample preparation (Qiagen), mutations are identified by
automated
sequencing (PE Applied Biosystems) using BigDyeTM Terminator Cycle sequencing.
This
methodology is described in Healy et al. 2000; Harding et al. 2000. The entire
16,569bp
human mitochondria) genome is sequenced using established PCR primer pairs,
which are
1o known not to amplify pseudogenes, or other nuclear loci. Any putative DNA
changes are
confirmed by comparison to the revised "Cambridge" human mtDNA reference
(Andrews
et al. 1999). The sequences obtained from the tumour mtDNA are first compared
for
known polymorphisms (Andrews et al. 1999; MITOMAP) and then compared with the
mtDNA sequence from the normal perilesional skin to identify genuine somatic
mutations.
DHPLC is performed on the WAVETM DNA Fragment Analysis System
(Transgenomic, Omaha, USA) which provides a fully automated screening
procedure. The
same technology is used to screen for heteroplasmic mutations in the skin
tumour mtDNA.
2o Using the back to back primer methodology described in Example 2, the
pattern of
DNA length mutations (i.e. tandem duplications) in the hypervariable segments
of the non-
coding region (NCR) are rapidly screened.
Example 4: deletion spectrum of the entire mitochondria) genome in human NMSC
and its precursor lesions
MtDNA damage in squamous cell carcinomas (SCCS), Basal cell carcinomas
(BCCS) and putative precursor lesions such as Bowen's disease and actinic
keratoses (As)
was compared to adjacent perilesional skin taken from different sun-exposed
body sites. A
long-extension PCR technique (LX-PCR) (Ray et al. 1998) was used to amplify
the entire
3o mitochondria) genome in order to determine the whole deletion spectrum of
mtDNA. A
myriad of specific deletions have been observed to occur in the mitochondria)
genome.
52
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Not all deletions will correlate with non-melanoma skin cancer; however, for
an accurate
diagnostic method, those deletions that are associated with the disease must
be known.
DNA is extracted by use of a commercial kit (Qiangen) according to the
manufacturer's recommendations. The entire mitochondria) genome is amplified
in two
separate reactions using the Expand TM Long Template PCR SystemTM (Boehringer
Manheim, Switzerland). The PCR primers used are those described by Kleinle et
al.
(1997) covering the following regions of the Cambridge sequence (Andrews et
al. 1999):
DIA(nucleotides (nt) 336-363), DIB (nt 282-255), OLA (nt 5756-5781), and OLB
(nt 5745-
5781). These large products eliminate amplification of nuclear pseudogenes.
The
sequences of the primers are as follows:
DIAF: (336-363) 5' AACACATCTCTGCCAAACCCCAAAAACA 3' SEQ m NO: 5
OLBR: (5745-5721) 5' CCGGCGGCGGGAGAAGTAGATTGAA 3' SEQ m NO: 6
OLAF: (5756-5781) 5' GGGAGAAGCCCCGGCAGGTTTGAAGC 3' SEQ m NO: 7
DIBR: (282-255) 5' ATGATGTCTGTGTGGAAAGTGGCTGTGC 3' SEQ m NO: 8
Amplifications are performed in 50 microlitre reactions containing 16 pmol of
each
primer, SOO~,mol dNTPs, 10 x PCR buffer with 22.SmM MgCl2 and detergents(kit),
0.75 ~,1
of enzyme (3.5 x 103 units/ml) and 50-200ng of total DNA. One reaction
generates
11,095bp segments of the genome, while another results in 5,409bp lengths
(e.g. Kleinle et
al, 1997). The PCR amplification conditions consists of a denaturing stage at
93°C for 1
min 30s, followed by 10 cycles of 93°C for 30s, 60°C for 30s and
68°C for 12 min,
followed by a further 20 cycles of the same profile with an additional Ss
added to the
elongation time every cycle. There is a final cycle of 93°C for 30s,
60°C for 30s and an
elongation time of 68°C for 26 minutes. To ensure reproducibility, a
known amount of
DNA is separated on a 1 % agarose gel and only samples which have at least the
same
amount of DNA are included in the analysis.
A greater mean number of deletions is found with increasing UV exposure in the
3o tumour samples, as shown in Table 2.
53
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Mean number of deletionsMean number of deletions
UV exposure in adjacent normal
in epidermal tumour
epidermis
Constant (n=5) 1.0 3.6
Intermittent (n=2) 0 1.5
Sun-protected (n=2) 0 0
Table 2. Comparison of the mean number of deletions observed in the LX-PCR of
mtDNA
between normal and tumour skin taken from different UV-exposed body sites.
Example 5: Aging and MtDNA
Using temporal maternal line comparisons (i.e. great-grandchild through great-
grand parents), the entire sequence of mtDNA extracted from a given tissue is
rapidly, and
accurately sequenced, in order to definitively state the arrangement of
nucleotide base pairs
for that specific molecule and possible changes through time. These
characterizations are
to compared to health status, aging indicators and between specific maternal
lines, within
larger populations. This combined information allows crucial statistical
discrimination
between separate causes resulting in the same mutation/deletion and
establishes that the
mtDNA sequences, used as a bio-maxker, has the required index of specificity
and
sensitivity in order to establish its validity. In addition, the proportions
of base pair
deletions and mutations are compared for consistency in various tissues across
the 4
maternal generations. Recent methodological developments have permitted
detection of
base pair deletions implicated in aging in blood samples (Bassam et al. 1991)
and have
raised the possibility that blood samples may be used to study mtDNA in lieu
of skeletal
muscle (von Wurmb et al. 1998). After establishing the efficacy of employing
leukocytes
in lieu of muscle tissue, as representative of mtDNA deletions and /or
mutations, the next
step measures only mtDNA in leukocytes. MtDNA deletions/mutations are then
determined as previously described.
Skeletal muscle or leukocytes are obtained from a patient. DNA is extracted as
set
out in Example 1. The following primers were used:
54
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
12ST1: (1257-1279) 5' TATACCGCCATCTTCAGCAAAC3' SEQ m NO: 9
12ST2: (1433-1411) 5' TACTGCTAAATCCACCTTCGAC 3' SEQ ll~ NO: 10
D1F: 5' CCTTACACTATTCCTCATCACC 3' SEQ m NO: 11
D1R: 5' TGTGGTCTTTGGAGTAGAAACC 3' SEQ m NO: 12
Amplifications were performed in 50 microlitre reactions containing 2.0 ~,mol
of
each primer, 250~.mol dNTPs, 10 x PCR buffer(Thennopol Reaction Buffer),
bovine
serum albumin, 0.5units Deep vent polymerase and 50-200ng of total DNA. The
PCR
amplification conditions consists of a denaturing stage at 95°C for 5
min (hot start),
1o followed by 30 cycles of 94°C for 30s, 60°C for 60s and
72°C for 30s with a final
extension at 72°C for 10 min. Gel electrophoresis was performed on a 2%
agarose gel at
125 volts for 60 min, stained with ethidium bromide, and visualized under W
light. To
ensure reproducibility, a known amount of DNA was separated on a 2% agarose
gel and
only samples which have the same amount of DNA were included in the analysis.
Example 6: Quantitative detection of the 4977bp common mtDNA deletion by 3-
primer
PCR
Where appropriate the incidence of the common deletion is determined in a
quantitative manner by a 3-primer PCR method which detects levels greater than
1-5% or a
2o dilution PCR method which detects levels less than 1 % down to 10-4%. (See
Example 7)
Samples are obtained and DNA extracted as described in Example 1. To
simultaneously
detect and quantify the ratios of both deleted and wild type (wt) mtDNAs in
the DNA
samples, a 3-primer PCR procedure is used (as described in Birch-Machin et al
1998).
Primers A, and C correspond to heavy strand positions 13720-13705 and 9028-
9008
respectively (Anderson et al., 1981); primer B corresponds to light strand
positions 8273-
8289. Primer C maps to a mtDNA region within the common deletion, whereas
primers A
and B flank the deleted region. Therefore primers B and C only amplify wt-
mtDNAs and
primers A and B only amplify deleted mtDNAs (the distance between the two
primers in the
absence of the deletion, approximately S.Skb, is too long to be amplified
under our PCR
3o conditions as described below).
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Using three primers allowed the simultaneous detection of two bands, the
larger one
(755bp) corresponding to the wt-mtDNA, and the smaller one (470bp)
corresponding to
deleted mtDNA harbouring the 'common deletion'. The PCR reaction mixture (25.1
total
volume) contained 100ng total cellular DNA, 200~M dNTPs, lOmM Tris-HCl (pH
8.8),
SOmM KCI, l.SmM Mg Clz , 0.1% Triton X-100, 2.SU Taq~ DNA polymerase (BioTaq,
BiolineUK Limited, London), 25 pmoles of primers A and B, 6.25 pmoles of
primer C and
3~Ci of [oc-3~ P]-dATP. The PCR conditions were 25 cycles of 94°C at 1
minute, 55°C at 1
minute, 72°C at 2 minutes including a final extension of 15 minutes at
72°C. These PCR
products were then electrophoresed through a 6% nondenaturing polyacrylamide
gel and the
radioactive PCR fragments were quantified by phophorimage analysis using the
ImageQuantTM software (Molecular Dynamics, Chesham UK).
Example 7: Serial Dilution PCR method to quantitatively detect low levels
(<1%) of
the common mtDNA deletion
A semi-quantitative PCR method (Corral-Debrinski et al 1991) is used to
estimate
the proportion of the common deletion in the total mtDNA extracted from the
tissue/cell
samples. Biological samples are obtained and DNA extracted as described in
Example 1.
The DNA sample is initially linearised using the restriction enzyme Bam HI
(lp,l enzyme
and 1 ~.l of commercially supplied buffer) at 37°C for 90 minutes.
Serial dilutions are
performed in two-fold steps (for total mtDNA there was an initial 10-fold
dilution) and
2o PCR performed for each dilution (1~,1) using the following primers:
Primers fog total mtDNA
L3108 (nt3108-3127)
H3717 (nt3717-3701)
Primef°s foY Common Deletion
L8282 (nt8282-8305)
H13851(nt13851-13832)
3o The reaction conditions are as follows:
One cycle 94°C for 2 minutes, 34 cycles of 94°C for 45
seconds, 51°C for 30
seconds (total mtDNA), 56°C for 30 seconds (common deletion),
72°C for 1 minute and
56
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
one final cycle of 72°C for 8 minutes. All PCR reactions are carried
out in the following
mixture (501): Sample DNA 1 ~.1, 0.6~M forward primer, 0.6~,M reverse primer,
0.2mM
dNTP's, 5~,1 GeneAmp~ 10x PCR Buffer, (Perkin Elmer), 0.2,1 Amplitaq~ DNA
polymerase (Perkin Elmer), 35.751 sterile autoclaved double distilled water.
Following electrophoresis the PCR productes are visualised on a UV
transilluminator (TMW-20, Flowgen Ltd., Lichfield, UI~) and a digital image of
the gel
obtained using image acquisition apparatus (Alpha Imager 2000, Alpha Innotech
Corporation, supplied by Flowgen Ltd., Lichfield, UK). The associated image
analysis
to software (Alpha Ease v3.3, Alpha Innotech Corp.) allows the calculation of
the integrated
optical density (IOD) for each PCR product in a dilution series. The band
where an IOD
value of zero is obtained for both total mtDNA and deleted mtDNA and the
corresponding
dilution values are used to calculate the percentage of common deletion in the
sample thus:
15%common deletion = total mtDNA dilution factor ~IOD Zero) x 100
common deletion dilution factor ~IOD Zero)
Example 8: Denaturing high performance liquid chromatography (DHPLC )
Samples are obtained and DNA extracted as in Example 1. PCR in 13 overlapping
20 fragments using two different PCR conditions as described by van den Bosch
et al. (2000).
The following three mtDNA specific primer pairs for PCR:
i. Oligo Sequence
Mt3118F CCCTGTACGAAAGGACAAGAG SEQ ID NO: 13
25 Mt3334R TGAGGAGTAGGAGGTTGG SEQ ID NO: 14
Mt8207F CCCATCGTCCTAGAATTAATTCC SEQ ID NO: 15
Mt8400R ATGGTGGGCCATACGGTAG SEQ 117 NO: 16
3o Mt14427F CCCATGCCTCAGGATACTCCTC SEQ ID NO: 17
Mt14997R GCGTGAAGGTAGCGGATG SEQ ID NO: 18
57
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
The 1-2 kb PCR products are digested into fragments of 90-600bp and resolved
at
their optimal melting temperature. Mutations are represented as two peaks and
mutations
with low percentages, such as <2% heteroplasmy as a "shoulder" in the peak.
DHPLC is performed with a mobile phase consisting of two eluents (pH 7.0).
Buffer A contains triethylammonium acetate (TEAR), which interacts with both
the
negatively charged phosphate groups on the DNA as well as the surface of the
column.
Buffer B contains TEAR with 25% of the denaturing agent acetonitrile.
Fragments were
eluted with a linear acetonitrile gradient at a constant flow rate. Increasing
the
1o concentration of acetonitrile will denature the fragments. Table 3 below is
an example of a
standard method for DHPLC of a PCR reaction generated using the WAVEMAKER
software (Transgenomics) according to manufacturer's instructions.
Table 3: Standard Method for DHPLC
Step Time %A (buffer) %B (buffer) Ml/min (flow
rate)
Loading 0.0 52 48 0.90
Start Gradient0.1 47 53
Stop Gradient4.1 39 61
Start Clean 4.2 0 100
Stop Clean 4.7 0 100
Start Equilibrate4.8 52 48
Stop Equilibrate6.8 52 48
The temperatures for successful resolution of the various heteroduplexes are
detailed
below and can simply be substituted into the relevant places in Table 2:
Fragment Melting temp (°C) Gradient of %Buffer B
Mt3118F 59 51-59
2o Mt8207F 58 50-58
Mt14427F 56 60-68
58
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Example 9
An extensive survey of mtDNA D-loop sequences from 49 prostate needle biopsy
patients
(46 diagnosed with malignancy) demonstrated mtDNA mutations in all prostatic
tissues
inclusive of benign prostatic hyperplasia (BPH), available Gleason grades and
stroma as
compared with the mitochondria) DNA of the patients blood. Moreover, an
expanded
study of mitochondria) genomes from 31 prostatectomy patients demonstrates
equivocable
hyper-mutation (Chen et al. 2002; Chen et al. 2003) loads in matched malignant
glands,
adjacent benign glands (nearby the malignant glands), and distal benign glands
(located in
tissue free of malignant pathology removed from any malignant pathology) as
shown in
Table 4. The mutations of Table 4 are also provided in SEQ ll~ No: 102 which
lists the
substitutions, SEQ m NOs: 103 to 109 which lists the deletions, and SEQ m No:
110 to
13S which lists the insertions. Polymorphisms and mutation positions were
determined by
comparison to the Revised Cambridge Reference Sequence (2001), however the
historical
numbering has been maintained such that the deletion at position 3106 is
denoted as a gap
and the rare polymorphism 750A has been retained. The numbering of the bases
is based
on the revised Cambridge Reference Sequence having a total of 16569 base
positions. A
histogram showing the number of mutations per location of the mitochondria)
genome is
shown in Figure 1. As can be seen in Figure 1, the mutations were found
throughout the
2o mtDNA genome and in all diseased prostates. However, certain "hot spots"
were also
apparent, for example in the D-loop region and the 16s region. These data sets
imply that
the designation of malignant or benign tissue, as made by a qualified
pathologist using
routine histological methods and grading standards, does not identify early
disease
progression. This strongly suggests that malignant transformation begins at
the cellular
level before the morphological characteristics of a cell are altered.
Importantly, the
mutation patterns are completely inconsistent for matched prostate tissue from
an
individual patient, or in comparison to another patient, perhaps indicating
possible tissue
sites where clonal expansion of malignant cells may occur. Moreover, separate
needle
biopsies with the same Gleason score, from the same individual alinost always
demonstrate
3o alternative mtDNA mutation patterns. This indicates that total mutation
load rather than
specific mutation sites may be more representative of the disease and
progression of the
disease.
59
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Since this data was gathered from individuals with known prostate cancer, and
in
the prostatectomy group with known advanced staging, it is likely that
histologically
benign tissue has undergone some intracellular transformations) associated
with neoplasia
and possible progression towards malignancy. Benign tissue harboring mtDNA
mutations
serve as a "biosensor" which can be monitored for increasing mutations
indicative of the
rate of disease progression. This rate may also indicate tumor aggression.
Moreover, the
effectiveness of a specific therapy could also be monitored based on the
change in this
mutational pattern.
This technique may be used as a confirmatory test for benign needle biopsies.
to Currently when a patient has a needle biopsy performed on the prostate and
the tissue looks
histologically benign he is sent home and is usually scheduled for follow-up
needle
biopsies in six months. Use of the above method would examine the already
taken needle
biopsy tissue and either confirm that the tissue is benign on the molecular
level as well, or
find evidence that there is in fact a malignancy in the prostate that was
geographically
missed by the needle biopsy technique, or that the tissue is pre-neoplastic or
neoplastic at
the molecular level. This could potentially save a lot of people from
undergoing multiple
surgeries or allow for early preventative treatment.
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Table 4
Base Mutations: Observed mutations of homoplasmic to homoplasmic,
homoplasmic to heteroplasmic, and heteroplasmic to homoplasmic
* The first nucleotide represents the normal nucleotide, followed by the
mutated nucleotide;
separated by an '=':
* IiVhen no blood is present, it is denoted as an "X':
** Historical numbering wherein deletion at BP 3706 is denoted as a gap and
the rare polymorphism
750A has been retained
BP BP
BP eliminatin BP ' elirninatin
Historicalg positionHomo-Homo- Hetero-Historicalg'positionHomo-Homo-Hetero-
Numbering*gap at Numbering*'
* 3106 Homo HeteroHomo * ' gap Homo HeteroHomo
at
3106
10 10 T-C T-T/C 200 200 A-G A-A/G
31 31 C-C/T 204 204 T-C T-T/C
C-C/T
41 41 C-T C-C/T 205 205 A-A/G
55 55 C-T 207 207 A-A/G
57 57 A-T G-A G-G/A
61 61 C-C/T 208 208 T-T/C
64 64 C-T 214 214 A-A/G
72 72 C-T C-C/T 217 217 C-T
T-C T-T/C 222 222 C-T
73 73 A-G A-A/G 225 225 A-G A-A/G
81.1 81.1 INS 226 226 C-T C-C/T
T
93 93 G-G/A 228 228 A-G A-A/G
94 94 A-A/G G-G/A
104 104 C-C/T 229 229 G-T
113 113 C-C/T 234 234 A-A/G
119 119 C-C/T 235 235 A-G
128 128 C-T G-G/A
146 146 C-T C-C/T 239 239 T-C
T-C T-T/C 247 247 G-G/A
DEL
150 150 C-T C-C/T 248 248 A
T-C T-T/C 262 262 C-C/T
152 152 C-T C-C/T 263 263 G-G/T
T-C T-T/C A-G A-A/G
153 153 A-G A-A/G 264 264 T-C
G-A G-G/A 277 277 C-C/T
170 170 C-C/T 280 280 C-C/T
182 182 C-T C-C/T 295 295 T-C T-T/C
185 185 G-A G-G/A 297 297 G-A G-G/A
A-A/G 303.1 303.1 INS
C
G-G/T 303.2 303.2 INS
C
188 188 A-G 305 305 C-C/T
G-A G-G/A 309 309 T-T/C
189 189 A-G A-A/G C-C/T
DEL
G-A G-G/A 309 309 C
192 192 T-C 309.1 309.1 INS
C
194 194 T-C T-T/C 309.2 309.2 INS
C
C-T 309.3 309.3 INS
C
DEL
195 195 C-T C-C/T 310 310 C T-T/CT/C-T
T-TlC DEL C-C/T
T
196 196 T-C 311 311 C-C/T
198 198 C-C/T 311.1 311.1 INS
C
199 199 T-T/C 312 312 C-C/T
C-C/T 313 313 C-C/T
200 200 G-A G-G/A
61
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
BP -
BP eliminatin BP eliminatin
Historicalg positionHomo-Homo- Hetero-Historicalg positionHomo-Homo-Hetero-
Numbering*gap at Numbering*gap at
* 3106 Homo HeteroHomo * 3106 Homo HeteroHomo
315.1 315.1 INS 1719 1719 A-A/GA/G-A
C
315.2 315.2 INS 1761 1761 A/T-A
C
323 323 G-G/A 1766 1766 C/T-T
325 325 C-T C-C/T 1811 1811 G-A G-GIA
329 329 G-T 1842 1842 A/G-A
394 394 C-C/T 1883 1883 A/G-G
INS
416.1 416.1 G/A 1888 1888 G-A G-GIAA/G-G
419 419 A-A/G A/G-A
456 456 C-T 2005 2005 C/T-C
T-T/C 2056 2056 A/G-G
462 462 T-C 2068.1 2068.1 INS
A
465 465 T-T/C 2075 2075 T/G-T
468 468 C-C/T 2257 2257 C/T-C
477 477 C-T 2258 2258 A/G-A
481 481 C-T C-C/T 2259 2259 T-C
482 482 C-T 2261 2261 C-C/T
489 489 C-T C-C/T 2280 2280 C/T-C
497 497 T-C T-T/C 2351 2351 C/T-T
499 499 A-G 2352 2352 C/T-T
501 501 C-C/T 2357 2357 C/T-C
505 505 C-C/T 2359 2359 C/T-C
506 506 C/T-C2389 2389 C/T-C
508 508 G-A 2596 2596 G-G/A
513 513 A-G 2627 2627 G-G/A
513 513 DEL 2657 2657 C-T
A
514.1 514.1 INS 2683 2683 C-C/T
C
515 515 A-A/G 2689 2689 C-C/T
515 515 D 2706 2706 A-G A-A/G
EL
A
515.1 515.1 INS 2761 2761 C-T
A
517 517 T-T/A 2857 2857 C-C/T
523 523 A-A/G 2885 2885 C-C/T
523 523 D 2927 2927 C-T
EL
A
523.1 523.1 INS 2948 2948 C-T
C
523.2 523.2 INS 2952 2952 C/T-T
A
523.3 523.3 INS 3010 3010 A-G A-A/G
C
523.4 523.4 INS
A
533 533 A-G A-A\G 3013 3013 G-G/A
536 536 C-C/T 3036 3036 A/G-G
567.1 567.1 INS 3040 3040 A/G-G
C
568.1 568.1 INS 3046 3046 C/T-C
C
568.2 568.2 INS
C
709 709 A-A/G 3308 3307 T-T/C
G-GlA 3338 3337 C/T-T
785 785 C-C/G 3349 3348 A/G-A
857 857 G-C 3394 3393 C-T
909 909 G-G/A 3398 3397 T-C
1189 1 189 C-C/T 3469.1 3468.1 INS
T
1247 1247 G-G/A 3480 3479 G-A
1431 1431 G-G/A 3499 3498 A/G-A
1693 1693 C-T 3507 3506 C-A
1709 1709 A/G-G3589 3588 C-C/T
1719 1719 A/G-G3594 3593 C-C/T
62
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
BP BP
BP eliminatinHomo BP eliminatinHomo
Historicalg position Homo- Hetero-Historicalg position Homo- Hetero-
Numbering*gap at - Numbering*gap -
* 3106 Homo HeteroHomo * at Homo HeteroHomo
3106
3657 3656 C-T 5663 5662 C-C/T
3666 3665 G-A 5677 5676 C-T
3688 3687 G-G/A 5882 5881 C-C/T
3693 3692 G-G/A 5897 5896 C-C/T
3744.1 3743.1 INS 5984 5983 A-G A-A/G
T
3908 3907 C-T 5985 5984 A-A/G
3966 3965 C-C/T 5999 5998 C-T
3969 3968 C-T 6009 6008 C-C/T
3992 3991 C-T 6028 6027 A/G-G
4017 4016 C-T 6037 6036 G-GlA
4185 4184 C-T 6041 6040 C-C/T
4216 4215 T-C T-T/C 6047 6046 G-A
4217 4216 A-A/G 6059 6058 C-C/T
4239 4238 C-T 6147 6146 C-T
4418 4417 C-C/T 6219 6218 C-C/T
4561 4560 C-T 6221 6220 C-C/T
4569 4568 G-G/A 6224 6223 C-C/T
4580 4579 A-A/G 6307 6306 A-A/G
4591 4590 T-T/C 6314 6313 C-C/T
4646 4645 C-T 6382 6381 G-A
4655 4654 A-AlG 6548 6547 C-C/T
4703 4702 C-C/T 6553 6552 C-C/T
4716 4715 C-C/T 6557 6556 C-C/T
4722 4721 A-A/G 6579 6578 G-A
4733 4732 C-C/T 6643 6642 T-T/C
4735 4734 C-C/T 6667 6666 C-C/T
4787 4786 G-A G-G/A 6686 6685 T-C
4826 4825 C-C/T 6691 6690 G-A
4864 4863 C-C/T 6776 6775 C-T C-C/T
4892 4891 C-C/T T-C T-T/C
4917 4916 A-G A-A/G 6827 6826 T-T/C
G-G/A 6912 6911 G-G/A
4951 4950 C-C/T 6917.1 6916.1 INS
T
5036 5035 A-A/G 6953 6952 G-A
5046 5045 G-G/A 6989 6988 A-A/G
5102 5101 A-A/G 7007 7006 C-T
5147 5146 G-A G-G/A 7013 7012 G-GlA
A-A/G 7028 7027 C-T C-C/T
5174 5173 C-C/T T-T/C
5198 5197 G-G/A 7055 7054 A-A/G
5213 5212 C-C/T 7059 7058 G-G/A
5300 5299 C-C/T 7146 7145 A-A/G
5312 5311 C-C/T 7159 7158 T-T/C
5371 5370 C-C/T 7184 7183 G-A G-G/A
5424 5423 C-C/T 7256 7255 C-C/T
5440 5439 C-C/T 7309 7308 T-T/C
5456 5455 C-C/T 7389 7388 T-T/C
5593 5592 T-T/C 7406.1 7405.1 INS
C
5633 5632 T-T/C 7407 7406 T-C T-T/C
5650 5649 G-G/A 7412 7411 C/T/
C/T
5655 5654 T-C/T 7476 7475 T-T/C
5656 5655 A-G A-A/G 7521 7520 G-G/A
63
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
BP BP
BP eliminatin BP eliminatin
Historicalg positionHomo-Horno-Hetero-Historicalg positionHomo-Homo-Hetero-
Numbering*gap Numberi~g*gap at
* at Homo Het:eroHomo * 3106 Homo HeteroHomo
3106
7756 7755 C-C/T 10295 10294 G-G/A
7763 7762 G-G/A 10345 10344 T-T/C
7768 7767 G-A A-A/G 10355 10354 C-C/T
7815 7814 C-C/T 10439 10438 C-C/T
7867 7866 C-C/T 10455 10454 G-G/A
7897 7896 G-A 10463 10462 T-C T-TIC
8027 8026 G-A
8065 8064 G/A-G 10550 10549 G-G/A
8117 8116 C-C/T 10679 10678 G-A
8133 8132 C-C/T 10685 10684 A-A/G
8248 8247 A-A/G 10688 10687 G-G/A
8270 8269 C-T C-C/T 10754 10753 A-C
8426.1 8425.1 INS 10810 10809 T-C T-T/C
G
8468 8467 C-C/T 10819 10818 G-G/A
8616 8615 A-A/G 10873 10872 C-C/T
8655 8654 C-C/T T-C T-TIC
8697 8696 G-A G-GlA 10882 10881 C-T
8701 8700 A-A/G 10885 10884 C-T
8718 8717 A-G 10944 10943 C-C/T
8818 8817 T-T/C 10956 10955 X-C/T
8893 8892 A-T 10972 10971 G-A G-G/A
8903 8902 C-C/T 10975 10974 C-C/T
9055 9054 A-G 10978 10977 A-A/G
9093 9092 G-G/A 9667 9666 G-A
9132 9131 A-G 9696 9695 C-C/T
9163 9162 A-G A-A/G 9698 9697 C-T C-C/T
9313 9312 A-A/G 9716 9715 C-T
A-A/C 9767 9766 C-T
9327 9326 A-A/G 9778 9777 G-G/A
9352 9351 C-C/T 9899 9898 C-T
9405 9404 T-T/C 10143 10142 A-G
9413 9412 T-TlC 10295 10294 G-G/A
9419 9418 C-C/T 10345 10344 T-T/C
9445 9444 G-G/A 10355 10354 C-C/T
9477 9476 G-A G-G/A 10439 10438 C-C/T
9502 9501 G-A 10455 10454 G-G/A
9540 9539 C-C/T 10463 10462 T-C T-TlC
9548 9547 A-G
9554 9553 G-G/A 10550 10549 G-GlA
9559 9558 C-C/T 10679 10678 G-A
9564 9563 G-G/A 10685 10684 A-A/G
9574 9573 C-C/T 10688 10687 G-G/A
6-
9591 9590 G/C 10754 10753 A-C
9628 9627 G-G/A 10810 10809 T-C T-T/C
9667 9666 G-A 10819 10818 G-G/A
9696 9695 C-C/T 10873 10872 C-C/T
9698 9697 C-T C-C/T T-C T-T/C
9716 9715 C-T 10882 10881 C-T
9767 9766 C-T 10885 10884 C-T
9778 9777 G-G/A 10944 10943 C-C/T
9899 9898 C-T 10956 10955 7C-C/T
10143 10142 A-G 10972 10971 G-A G-G/A
64
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
BP BP
BP eliminatin BP eliminatin
Historicalg positionHomo-Homo-Hetero-Historicalg positionHomo-Homo-Hetero-
Numbering*gap at Numbering*gap at
* 3106 Homo HeteroHomo * 3106 Homo HeteroHomo
10975 10974 C-C/T 13398 13397 A-A/G
10978 10977 A-A/G 13431 13430 C-C/T
11001 11000 A-A/G 13436 13435 C-C/T
11013 11012 X-C/T 13468 13467 C-C/T
11024 11023 T-T/C 13476 13475 A-A/G
C-C/T 13484 13483 T-T/C
11069 11068 A-A/G 13487 13486 C-C/T
11084 11083 A-A/G 13506 13505 C-C/T
11113 11112 T-C T-T/C 13530 13529 C-C/T
11177 11176 C-C/T 13536 13535 C-C/T
11180 11179 G-G/T 13563 13562 A-A/G
11195 11194 G-G/A 13573 13572 C-C/T
11217 11216 C-C/T 13579 13578 G-A
11251 11250 A-G A-AIG 13609 13608 C-C/T
G-A G-G/A T-T/C
11299 11298 C-T C-C/T 13617 13616 C-T C-C/T
11332 11331 T-T/C T-C T-T/C
11337 11336 G-A/G 13634 13633 G-G/A
11351 11350 G-A/G 13650 13649 C-C/T
11356 11355 T-T/C 13621 13620 T/C-C
11377 11376 A-A/G 13631 13630 C-C/T
11420 11419 G-G/A 13637 13636 G-GlA
11647 11646 C-T 13638 13637 A-A/G
11719 11718 G-A G-G/A 13651 13650 A-A/G
11812 11811 A-G A-A/G 13655 13654 T-T/C
11852 11851 G-G/A 13674 13673 C-T
DEL
13674 13673 C
11857 11856 C-C/T 13680 13679 T-TIC
11864 11863 C-T C-C/T 13687 13686 C-C/T
11881 11880 C-C/T 13707 13706 G-G/A
11907 11906 T-T/C 13708 13707 A-A/G
11914 11913 G-G/A G-G/A
12012 12011 C-C/T 13711 13710 G-GlA
12013 12012 A/G-A 13712 13711 C-C/T
12308 12307 G-G/A 13725 13724 C-C/T
12372 12371 G-G/A 13731 13730 A-A/G
A-A/G 13734 13733 C-C/T
12492 12491 T-T/A 13743 13742 T-T/C
12624 12623 C-T 13748 13747 A-A/G
12633 12632 A-C A-A/C 13759 13758 G-G/A
12654 12653 G-G/A A-A/G
12810 12809 G-G/A 13766 13765 C-C/T
12959 12958 C-C/T 13788 13787 C-C/T
13079 13078 A/G-A 13789 13788 T-T/C
13089 13088 T-A 13805 13804 C-C/T
13105 13104 A-A/G 13841 13840 T-T/C
13111 13110 T-C 13880 13879 C-C/A
13212 13211 C-C/T 13888 13887 X-T X-C/T
13281 13280 T-T/C 13911 13910 G-G/A
13294 13293 A-A/G 13933 13932 A-A/G
13359 13358 G-G/A 14025 14024 C-CIT
13368 13367 G-A G-G/A 14044 14043 C-C/T
A-G A-A/G 14135 14134 T-T/A
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
gp BP
BP eliminatin BP eliminatin
Historicalg positionHomo-Homo-Hetero-Historicalg positionHomo- Homo-Hetero-
Numbering*gap at Numbering*gap
* 3106 Homo HeteroHomo * at Homo HeteroHomo
3106
14139 14138 G-G/A 15889 15888 T-T/C
14167 14166 T-T/C 15904 15903 T-C T-T/C
14178 14177 T-T/C C-A/C
14182 14181 T-C T-TlC 15907 15906 G-G/A
14203 14202 A-A/G 15927 15926 A-G
14220 14219 X-G X-G/A 15928 15927 A-G A-A/G
14233 14232 A-G A-A/G G-A G-G/A
14281 14280 T-C T-T/C 15998 15997 A/T-A
C-C/T 15999.1 15998.1INS
T
14899 14898 G-G/A 16048 16047 G-G/A
14903 14902 G-A X-A/G 16051 16050 A-A/G
14918 14917 G-G/A G-G/A
15043 15042 G-G/A 16063 16062 C-C/T
15115 15114 T-T/C 16067 16066 C-C/T
15162 15161 C-C/T 16069 16068 C-C/T
15218 15217 G-A G-G/A 16093 16092 X-C T-T/C
15244 15243 G-G/A C-T C-C/T
15265 15264 C-C/T 16095 16094 C-T C-C/T
15286 15285 C-C/T 16111 16110 T-TlC
15301 15300 A-A/G 16126 16125 T-C T-T/C
15302 15301 C-C/T C-C/T
15307 15306 C-C/T 16129 16128 G-A G-G/A
15323 15322 A-G A-A/G 16134 16133 C-C/T
G-G/A 16148 16147 C-C/T
15324 15323 C-C/T 16153 16152 A-A/G
15343 15342 X-C X-C/T 16163 16162 G-G/A
15355 15354 X-A X-A/G 16172 16171 C-C/T
15379 15378 C-C/T 16184 16183 C-T C-C/T
15384 15383 X-C X-C/T T-C
15429 15428 A-A/G 16186 16185 T-T/C
15452 15451 C-A C-C/A 16189 16188 T-C T-T/C
A-C A-A/C C-T C-C/T
15523 15522 C-C/T 16190 16189 T-C
15525 15524 G-G/A 16192 16191 C-C/T
15526 15525 C-C/T T-C T-T/C
15527 15526 C-C/T 16209 16208 C-C/T
15557 15556 G-GlA T-T/C
15587 15586 C-C/T 16223 16222 T-T/C
15607 15606 A-G A-A/G C-T
G-A G-G/A 16224 16223 C-T C-C/T
15670 15669 C-C/T T-T/C
15693 15692 C-T C-C/T 16225 16224 C-T
15698 15697 C-C/T 16235 16234 A-A/G
15704 15703 C-C/A 16239 16238 C-T C-C/T
15708 15707 G-G/T X-C
15762 15761 G-G/A 16247 16246 G-G/A
15812 15811 A-A/G 16256 16255 T-T/C
15826.1 15825.1 INS C-C/T
G
15834 15833 T-T/C 16270 16269 C-T C-C/T
15865 15864 A-A/G 16292 16291 C-C/T
G_
15884 15883 C-G C/G 16294 16293 C-T C-C/T
66
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
BP BP
BP eliminatin BP eliminatin
Historicalg positionHomo-Homo-Hetero-Historicalg positionHomo-Homo-Hetero-
Numbering*gap at Numbering*gap at
* 3106 Homo HeteroHomo * 3106 Homo HeteroHomo
A-A/G 16296 16295 C-T C-C/T
16270 16269 T-C T-T/C 16544 16543 T-T/C
C-C/T
16278 16277 T-C
16280 16279 A-A/GA/G-A
16290 16289 C-C/T
T-T/C
16291 16290 C-C/1'
16292 16291 T-T/C
16293 16292 A-A/G
16294 16293 T-T/C
C-T C-C/T
16295 16294 C-C/T
16296 16295 C-T C-C/T
T-T/C
16298 16297 C-T C-C/T
16303 16302 G-G/T
16304 16303 T-T/C
C-T C-C/T
16311 16310 C-T C-C!T
T-C T-T/C
16319 16318 G-A G-G/A
T-T/C
A-A/G
16320 16319 T-T/C.
16325 16324 C-C/T
16344 16343 C-G
16356 16355 C-C/T
16342 16341 C-T C-C/T
16352 16351 T-T/C
16353 16352 C-C/T
16354 16353 T-T/C
16355 16354 T-TlC
16359.1 16358.1 INS
G
16360 16359 C-C/T
16362 16361 C-C/T
T-T/C
16370 16369 G-G/A
16389 16388 G-G/A
16390 16389 X-G X-G/A
16398 16397 A-G G/A-G
16399 16398 G-A G/A-A
16429 16428 X-T/C
16465 16464 C-T C-C/T
T-C T-T/C
16475 16474 T-T/C
16514 16513 C-C/T
16519 16518 T-C T-T/C
C-T C-C/T
16526 16525 A-G
G-G/A
16527 16526 C-C/T
16537 16536 T-C T-T/C
67
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Table 5
D-Loo xed tissue and blood for needle
~ Primers bio sies
used
for
formalin
fi
SE(~ Length (#
ID Primer bases 5'-3'
NO:
19 15971f 20 TTAACTCCACCATTAGCACC
20 15f 20 CACCCTATTAACCACTCACG
21 16211f 22 CAGCAATCAACCCTCAACTATC
22 16410r 19 AGGATGGTGGTCAAGGGAC
23 389r 20 CCTAACACCAGCCTAACCAG
24 420r 18 GTGCATACCGCCAAAAGA
25 711 r 21 AACGGGGATGCTTGCATGTGT
Formalin
Fixed
Tissue
Primers
used
for
31 rostatectomies
SEQ ID Length (#
NO: Primer bases 5'-3'
26 649f 21 TAGGTTTGGTCCTAGCCTTTC
27 1051f 26 ACAATAGCTAAGACCCAAACTGGGAT
28 1247r 22 CAAGAGGTGGTGAGGTTGATCG
29 8959r 22 CGATAATAACTAGTATGGGGAT
30 8814f 22 CCAACTATCTATAAACCTAGCC
31 9247f 19 GCCCATGACCCCTAACAGG
32 9868r 21 CGGATGAAGCAGATAGTGAGG
33 9711f 22 CTGGGTCTCTATTTTACCCTCC
34 10663f 18 TCTTTGCCGCCTGCGAAG
35 10766r 22 TTAGCATTGGAGTAGGTTTAGG
36 11813r 26 GTAGAGTTTGAAGTCCTTGAGAGAGG
37 11629f 23 AATCAGCCACATAGCCCTCGTAG
38 12709r 28 GGAAGATGAGTAGATATTTGAAGAACTG
39 12528f 22 GAACTGACACTGAGCCACAACC
40 13516r 23 GGTCTTTGGAGTAGAAACCTGTG
41 13239f 23 CGTAGCCTTCTCCACTTCAAGTC
42 15351 23 TCGTGCAAGAATAGGAGGTGGAG
r
43 15144f 25 TCCCGTGAGGCCAAATATCATTCTG
44 6145r 24 CAGTTGCCAAAGCCTCCGATTATG
45 5867f 25 CAATGCTTCACTCAGCCATTTTACC
46 13957r 22 CTAGATAGGGGATTGTGCGGTG
47 13838f 23 CCCTAGACCTCAACTACCTAACC
48 15026r 21 GGCAGATAAAGAATATTGAGG
49 14937f 22 CATCAATCGCCCACATCACTCG
50 1938f 24 AGAGCACACCCGTCTATGTAGCAA
51 2084r 26 TACAAGGGGATTTAGAGGGTTCTGTG
52 2973f 24 TAGGGTTTACGACCTCGATGTTGG
53 3101 24 TAGAAACCGACCTGGATTACTCCG
r
54 3728f 23 CATATGAAGTCACCCTAGCCATC
55 3893r 23 GTTCGGTTGGTCTCTGCTAGTGT
56 4888f 27 CAATCATATACCAAATCTCTCCCTCAC
57 5035r 25 CATCCTATGTGGGTAATTGAGGAGT
58 5981f 23 TGGAGTCCTAGGCACAGCTCTAA
59 6154r 24 GGAACTAGTCAGTTGCCAAAGCCT
60 6911f 24 TGCAGTGCTCTGAGCCCTAGGATT
61 7082r 26 GAAGCCTCCTATGATGGCAAATACAG
62 7829f 25 CGCATCCTTTACATAACAGACGAGG
63 8029r 24 GGCTTCAATCGGGAGTACTACTCG
68
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Blood
Primers
rostatectom
SEQ ID Length (#
NO: Primer bases 5'-3'
64 16485f 24 GAACTGTATCCGACATCTGGTTCC
65 919r 22 TTGGGTTAATCGTGTGACCGCG
66 1644r 26 CTCCTAAGTGTAAGTTGGGTGCTTTG
67 615f 24 ATGTTTAGACGGGCTCACATCACC
68 1488f 24 CGTCACCCTCCTCAAGTATACTTC
69 2612r 28 GGAACAAGTGATTATGCTACCTTTGCAC
70 2417f 23 CACTGTCAACCCAACACAGGCAT
71 3641 23 GCTAGGCTAGAGGTGGCTAGAAT
r
72 3230f 23 GTTAAGATGGCAGAGCCCGGTAA
73 4417r 26 TTTAGCTGACCTTACTTTAGGATGGG
74 4337f 24 ATGAGAATCGAACCCATCCCTGAG
75 5551 24 GGCTTTGAAGGCTCTTGGTCTGTA
r
76 6418f 23 AACCCCCTGCCATAACCCAATAC
77 7554r 33 CTTTGACAAAGTTATGAAATGGTTTTTCTAATA
78 7400f 22 CCCACCCTACCACACATTCGAA
79 8441 26 GTTGGGTGATGAGGAATAGTGTAAGG
r
80 8346f 26 CAACACCTCTTTACAGTGAAATGCCC
81 9413r 24 GCCTTGGTATGTGCTTTCTCGTGT
82 10285r 21 GGTAGGGGTAAAAGGAGGGCA
83 9273f 21 TCAGCCCTCCTAATGACCTCC
84 10198f 19 CCCGCGTCCCTTTCTCCAT
85 11408r 25 GGAGTCATAAGTGGAGTCCGTAAAG
86 11210f 24 TTCTACACCCTAGTAGGCTCCCTT
87 12231 26 GTTAGCAGTTCTTGTGAGCTTTCTCG
r
88 12096f 22 TCCTATCCCTCAACCCCGACAT
89 13098r 26 CAACTATAGTGCTTGAGTGGAGTAGG
90 12881f 26 CATCCTCGCCTTAGCATGATTTATCC
91 13851 24 GTTGAGGTCTAGGGCTGTTAGAAG
r
92 14738f 24 AGAACACCAATGACCCCAATACGC
93 15731 28 CTAGGAGTCAATAAAGTGATTGGCTTAG
r
94 15347f 23 CACGAAACGGGATCAAACAACCC
95 16000r 24 CTTAGCTTTGGGTGCTAATGGTGG
96 5544f 21 CACGCTACTCCTACCTATCTC
97 6482r 20 GACTGCTGTGATTAGGACGG
98 13354f 23 TTTATGTGCTCCGGGTCCATCAT
99 14458r 22 GATGGCTATTGAGGAGTATCCT
100 14399f 21 ACACTCACCAAGACCTCAACC
101 15593r 23 ATCGGAGAATTGTGTAGGCGAAT
69
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Refe~ehces
Anderson S, et al., Nature 290:457-464, 1981
Andrews RM, et al., Nature Genetics 23(2):147, 1999
Barringer et al., Gene, 89:117 1990
Bassam BJ, Caetano-Anolles PM, Gresshoff PM., Anal. Biochem. 196: 80-83, 1991
Berneburg M, et al., J. Biol. Chem. 274(22):15345-15349, 1999
Berthon P, Valeri A, Cohen-Akeninc A, Drelon E, Paiss T, Wohr G, Latil A et
al., Am. J.
Hum. Genet., 62: 1416-1424,1998
Birch-Machin MA, et al., Methods in Toxicology, Volume 2, 51-69,1993
Birch-Machin MA, et al., J.Invest.Der~rzatol., 110:149-152 1998
Birch-Machin MA, Online Conference Report (Sunburnt DNA), International
Congress of
Biochemistry and Molecular Biology, New Scientist, 2000(a)
Birch-Machin MA, Taylor RW, Cochran B, Ackrell BAC, Tumbull DM. Anjz Neurol
48:
330-335, 2000(b)
Birch-Machin MA, Clin. Exp. Dermatol,. 25(2), 141-146, 2000 (c)
Birch-Machin MA and Krishnan K. Mitochondrion, 1, p45 (2001).
Birch-Machin MA, Lindsey J. Lusher M and Krishnan K. Mitochondrion, 1 Suppl.
1, S30
(2001).
Bogliolo, M, et al., Mutagenesis, 14: 77-82, 1999
Brierley EJ, Johnson MA, Lightowlers RN, James O, Turnbull DM., Ann Neurol
43(2):217-223, 1998
Broclcington, et al., Nature Genet 4:67-71, 1993
Brown, M.D., et al., Am J. Hump Genet, 60: 381-387, 1997
Brumley, R. L. Jr. and Smith, L.M., 1991, Rapid DNA sequencing by horizontal
ultrathin
gel electrophoresis, Nucleic Acids Res. 19:4121-4126 Nucleic Acids Res. 19:
4121-4126
Buttyan R, Sawczuk IS, Benson MC, Siegal JD, Olsson CA., Prostate 11:327-
337,1987
Byrne E., Curr Opin Reumatol 4(6):784-793, 1992
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J et
al., Cancer
Res 57:4997-5000, 1997
Carew J.S. and Huang P. Molecular Cancer http://www_molecular-cancer.com
(2002)
Chee, M. et al Science 274; 610-614, 1996
Chen J.Z. et al. Cafzcey~ Research (62): 6470-6474 (2002).
Chen J.Z. et al. Carcihogenesis (Vol 24) No. 9 1481-1487 (2003)
Chinnery PF, Howel N, Turnbull DM. J.Med. Genet. ; 3 6: 425-436, 1999
Chinnery PF and Turnbull DM., Lancet 354 (supplement 1): 17-21, 1999
Chinnery PF and Turnbull DM., Lancet 354 (supplement 1): 17-21, 2000
Chollat-Traquet, C, Tobacco or health: a WHO programme., Eur J Cancer, 28(2-
3): 311-
315, 1992
Chomyn A, et al., Proc. Natl. Acad. Sci. USA 89(10):4221-4225, 1992
Cohen D, Barton G, The cost to society of smoking cessation., Thorax. 53( 2):
S38-42,
1998
Corral-Debrinski et al., Mutat Res, 275: 169-180, 1991
Cortopassi G. A. and Arnheim, H. Detection of a specific mitochondiral DNA
deletion in
tissues of older humans, Nucleic Acids Tes. 18, 6927-6933 1990
Cortopassi G, Wang E., Biochim Biophys Acta 1271(1):171-176,1995
Croteau DL, Stierum RH, Bohr VA, Mutat Res 434(3):137-148, 1999
Cut~ren.t Protocols ih Molecular Biology
Davis RM, Boyd GM, Schoenborn CA, "Common courtesy" and the elimination of
passive
smoking. Results of the 1987 National Health Interview Survey. JAMA 263(16):
2208-
10, 1990
Dong JT, Isaacs WB, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaaca JT,
Barrett
JC., Science 268:884-886, 1995
Driezen P, Brown KS., Seaxchable database of questionnaire items from
populations
surveys of tobacco use in Canada: A summary report to the Ontaxio Tobacco
Research Unit
(Toronto, Ontario) 1999
71
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Easton RD, Mernwether AD, Crews DE, and Ferrell RE., Am. J. Hum. Genet. 59:202-
212, 1996
Fahn H, Wang L, Hseith R, Chang S, I~ao S, Huang M, and Wei Y. American.
Journal of
Respiratory Critical Care Medicine, 154:1141-1145, 1996
Fahn HJ, Wang LS, Kao SH, Chang SC, Huang MH, ~VVei YH., Am. J. Respir. Cell.
Mol.
Biol., 19(6): 901-9, 1998
Finegold D., Mitochondrial Disease- Primary Care Physican's Guide. Psy-Ed.
Corp DB/A
Exceptional Parents Guide: 12, 1997
Flanagan N, Birch-Machin MA, Rees JL., HunZ Mol Genet 9 (17):2531-2537,2000
Flanagan N, Ray AJ, Todd C, Birch-Machin MA and Rees JL. J Invest. Dermatol
(2001)
117 (5) 1314-1317
Fliss MS, et al. Science 287: 2017-2019, 2000
Gattermann, N, Berneburg, M, Heinisch, J, Aul, C, Schneider, W., Leukemia
9(10): 1704-
10, 1995
Green R, Reed JC., Science 281 (5381):1309-1312, 1998
Guatelli, et al., Proe. Nat. Acad. Sci. U.S.A. 87: 1874 1990
Gulavita, Sunil Dr. Northwestern Ontario Cancer Centre - Personal
Communication
Habano S, Nakamura, Sugai T., Oncogene 17 (15):1931-1937, 1998
Handing RM, et al., Am. J. Hum. Genet. 66, 1351-1361, 2000
Harman, D., Proc Nati Acad Sci USA 78(11): 7124-8, 1981
Hattori et al, Age-dependant increase in deleted mitochondrial DNA in the
human heart:
possible contributory factor to presbycardia, AM. Heart J., 121, 1735-1742,
1991
Hayashi J, Ohta S, Kikuchi A, Talcemitsu M, Goto Y, Nonalca 1., Proc Natl Acad
Sci U.SA
88 (23):10614-10618, 1991
Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka L, Proc Natl Acad
Sci USA
88 (23):10614-10618, 1991
Hayward SW, Grossfeld GD, Tlsty TD, Cunha GR., Int J Oncol 13:35-47, 1998
Healy E, Birch-Machin MA, Rees JL. Chapter 1 1. The Human Melanocortin 1
Receptor
Gene. In the Melanocortin Receptoys(Cone RD (ed)). Hurnana Press Inc. New
Jersey,
USA, 1999
72
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Healy E, Birch-Machin MA, Rees Jl., Lancet 355, 1072-1073, 2000
Hearst N, Hulley SB. Using secondary data, In Designing clinical research: an
epidemiological approach. Ed. Hulley, S. and Cummings, S., Baltimore: Williams
&
Wilkins, pages 53-62, 1988
Hopgood, R., et al, 1992, Strategies for automated sequencing of human mtDNA
directly
from PCR products, Biotechniques 13:82-92
Hsieh, RH and Wei, YH, Age-dependent multiple deletions in human muscle
mitochondrial DNA, in preparation 1992
Huang GM, Ng WL, Farkas J, He L, Liang HA, Gordon D, Hood R., Genomics
59(2):178-
86,1999
Http://www.ornl.gov/hgmis/proj ect/budget.html)
Ikebe et al., Increase of deleted mitochondrial DNA in the striatum in
Parkinson's disease
and senescence, Biochem. Biophys. Res. ComnZUn. 170, 1044-1048, 1990
Innis et al PCR Protocols, A Guide to Methods and Application, Academic Press
Inc. San
Diego 1990
Kaiserman MJ, Chronic Dis Can 18(1): 13-9, 1997
Kalra J, Chaudhary AK, Prasad K,. Int. J. Exp. Pathol. 72(1): 1-7, 1991
Katayama et al., Deleted mitochondrial DNA in the skeletal muscle of aged
individuals,
Biochem. Int., 25, 47-56 1991
Kleinle S, et al., Human Genet. 290: 457-465, 1997
Konishi N, Cho M, Yamamoto K, Hiasa Y. Pathol. Int. 47:735-747,1997
Krishnan K and Birch-Machin MA. British Journal of Dermatology (2002), 146,723
Kwoh et al. Proc. Natl. Acad. Sci. U.S.A., 86: 1173 1989
Landis SH, Murray T, Bolden S, Wingo PA. Cancer J. Clin. 49:8-31
Landis, SH, et al., CA Cancer J.. Clin. 49: 8-31, 1999
Landegren et al. Science, 241: 1077 1988
LeDoux SP, et al. Mutat Res 434(3):149-159, 1999
Lee HC, et al. FEBS Letters 354:79-83,1994
73
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Lee HC, Lu CY, Fahn HJ, Wei YHu. Federation of European Biochemical Societies,
441: X92-296,1998
Lee HC, et al. Arclz. Bioclaem. Biophys. 362(2): 309-16, 1999
Leonard ~ Shapira 1997
Lindsey J, Lusher M, Krishnan KJ and Birch-Machin MA., British Journal of
Dermatology
(2001), 144,655
Li Y, et al., In: Oxygen Radicals and the Disease Process, Amsterdam, The
Netherlands:
Harwood Academic Publishers, 237-277, 1997
Linnane et al., 1990
Liu CS, I~ao SH, Wei YH. El2VEron. Mol. Mutagen 30(1): 47-55, 1997
Lowes S, Krishnan K, Lindsey J, Lusher M and Brich-Machin MA. British Journal
of
Dermatology (2002, 146,736
Luckey, J.A., et al, 1993, High speed DNA sequencing by capillary gel
electrophoresis,
Methods Ertzyntol. 218: 154-172
McCormack, Douglas. Website: http://cormactech.con~ldna, 2001
Meibner C, von Wurmb N, Oehmichen M., Int. J. Legal Med. 110: 288-291, 1997
Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G., Science 286:
774-
779,1999
Miquel J, de Juan E, Sevila I. EXS 62:47-57,1992
MITOMAP: A human mt genome database (www_gen.emoiy.edu/mitomap.html)
Mullis and Faloona Methods Enzymol 155, 335 1987
Nachman MW, Brown WM, Stoneking M, Aquardo CF., Genetics 142:53-963,1996
National Cancer Institute of Canada, Canadian cancer statistics 2000.,
National Cancer
Institute of Canada., Toronto, Ont. 2000
Naviaux, RK., Mitochondrial Disease- Primary Care Physican's Guide. Psy-Ed.
Corp
DBIA Exceptional Parents Guide: 3-10, 1997
Newton, CR and Graham, A., IntroductiorZ toBiotecniques Series 1997
Oefner PJ, LTnderhill PA., Current protocols in human genetics 19, 7.10.1-12,
1998
74
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Ozen M, et al, Prostate 36:264-271,1998
Pang et al, Arch. Bioclzenz.Biophys. 312: 534-538, 1994
Parsons TJ, et al., Nature Geraet. 15 (4):363-368, 1997
Pascucci B, et al., J.Mol.Biol. 273 (2):417-427, 1997
Penta JS, Johnson FM, Wachsman JT, Copeland WC., Mut. Res. 488, 119-133, 2001
Polyak Y, et al., Nature Genet. 20 (3):291-293, 1998
Ray AJ, Rees JL, Birch-Machin MA., J. Invest.Dernzatol. 110:692, 1998
Ray AJ, Rees JL, Birch-Machin MA., Brit.J.Derrnatol.140:788, 1999
Ray AJ, Pickersgill L, Turner R, Nikaido O, Rees JL, Birch-Machin MA., J.
Irzvest.
Dermatol 115(4):674-679, 2000
Rees JL, Skin cancer. In: The Genetic Basis of Human Cancer, eds Vogelstein B,
Kinzler
K. New York: McGraw-Hill, pp527-536, 1998
Rehman I, Quinn AJ, Healy E, Rees JL. Lancet 344: 788-789, 1994
Rehman I, Takata M, Wu YY, Rees JL. Oncogene 12: 2483-2490, 1996
SAS Enterprise Mirzirzg Users Guide, SAS Inc., 2000
Sawyer E, Van Houten B., Mutatiorz Res: 434(3):161-176, 1999
Schurr TG, Ballinger SW, Gan Y, Hodge JA, Mernwether DA, Lawrence DN, Knowler
WC, Weiss KM, and Wallace DC., Am. J. Hum. Genet. 46:613-623, 1990
Seidman, M.D. et al., Arch. Otolaryngol Head Neck Surg., 123: 1039-1045, 1997
Shankey TV, Jin JK, Dougherty S, Flanigan RC, Graham S, Pyle JM., Cytometry
21:30-
39,1995
Shay JW, Werbin H., Mutat. Res:186: 149, 1987
Sherrat EJ, Thomas AW, Alcolado JC., Clin. Sci. 92:225-235,1997
Shoffiier JM, Brown MD, Torroni A, Lott MT, Cabell MF, Mirra SS, Beal MF, Yang
C,
Gearing M, Salvo R, Watts RL, Juncos JL, Hansen LA, Crain BJ, Fayad M,
Reckford CL,
and Wallace DC., Genomics 17: 171-184, 1993
Singh K.K. and Modica-Napolitano J.S. Expert reviews in molecular medicine.
htt~://www.ermm.cbcu.ac.uk (2002)
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Smith DG, Malhi RS, Eshleman J, Lorenz JG and Kaestle FA., Am. J. Huna. Genet.
110:271-284, 1999
Smith R, Birch-Machin MA, Rees JL. J. Invest. Dernaatol. 111: 101 -104, 1998
SpringNet - CE Connection: Screening, Diagnosis: Improving Primary Care
Outcomes.
Website: http~//www springnet.con~/ce/j803a.htm
Stone AC and Stoneking M. Amer. J., Phys. Anthro. 92(4):463-471, 1993
Tamura S, et al. Eur..LCancer ~AJ 35 (2):316-319, 1999
Tanaka, M. et al, 1996, Automated sequencing of mtDNA, Methods Enzymol. 264:
407-
421
Taniike, M. et al., BioChem BioPhys Res Comun, 186: 47-53, 1992
Taylor RW, Birch-Machin MA, Bartlett K, Turnbull DM., JBiol Chem, 269, 3523-
3528
1994
Tijssen, P. (ed) Laboratory Techniques in Biochemistry and Molecular Biology,
Vol. 24:
Hybridization with Polynucleotide Probes, Elsevier, N.Y., 1993
Tori et al., Ageing-associated deletions of human diaphragmatic mitochondria)
DNA, AM.
J. Respir. Cel l Mol. Biol. in press 1992
Van De Graff, KM, Fox, SI. Concepts of Human Anatomy and Physiology., Dubuque:
WM. C. Brown Publishers, 1995
Van den Bosch BJC, et al., Nucleic Acids Res. 28: 89, 2000
von Wurmb, N, Oehmichen, M, Meissner, C., Mutat Res. 422:247-254, 1998
Wallace DC., Annu Rev Bioclaem, 61: 1175-1212,1992
Wallace DC. Proc. Natl. Acad. Sci. USA 91: X739-X746, 1994
Wald and Wallace, D.C., Mitochondria) Diseases in man and Mouse. Science,
5(283):
1482-1497, 1999
Wald, NJ, Hackshaw,AK, Cigarette Smoking: an epidemiological overview. Br Med
Eull.
52(1): 3-11, 1996
Wallace et al., Mitochondiral DNA MUtatio Assoicated with Leber's Hereditary
Optic
Neuropathy, Science, 1427-1429
Walsh PC, Partin AW. Caracer 80:1871-1874,1997
76
CA 02550135 2006-06-12
WO 2005/056573 PCT/CA2004/002124
Ward RH, Frazier BL, Dew-Jager K, Paabo S., Proc. Natl. Acad. Sci. USA 88:8720-
8724,
1991
Ward 1993
Wei YH, Pang C, You B, Lee H. Ann.NYAcad.Sci. 786:82-101, 1996
Wei YH. Proceedings of the Nat. Sci. Council of the Republic of China April
22(2):5567,
1998
Weinstock MA: In: JJ Stern RS, MacKie RM and Weinstock MA, Grob (eds)
Epidemiology, Blackwell (UK). pp121-128, 1998
Woodwell DA. National Ambulatory Medical Care Survey: 1997 Summary. Advance
data
from vital and health statistics; no. 305. Hyattsville, Maryland: National
Center for Health
Statistics. 1999
Wu & Wallace Genonaics, 4:560, 1989
Xu J, et al., Nature Genet 20: 175-179,1998
Yamaguchi KT, et al., Free Radical Res. Commun. 16(3):167-74, 1992
Yeh, J.J., et al., Oncogerze Journal, 19: 2060-2066, 2000
Yen et al., Ageing-associated Skb deletion in human liver mitochondrial DNA,
Biochem.,
Biophys., Res. Commun., 178, 124-131 1991
Yen et al., Age-dependent 6 kb deletion in human liver mitochondirial DNA,
Biochem. Ifat.
26, 457-468 1992
Zeviani M, et al. Am. J. Hum. Genet. 47:904-914,1990
Zhang et al., Multiple mitochondiral DNA deletions in an elderly human
individual, FEBS
Lett, 297, 34-38 1992
Zhang, C., et al., BioChem. BioPhys. Res. Comun., 195: 1104-1110, 1993
77
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPRI~:ND PLUS D'UN TOME.
CECI EST L,E TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional valumes please contact the Canadian Patent Office.