Note: Descriptions are shown in the official language in which they were submitted.
CA 02550627 2006-06-20
-1_
t AS ORIGINALLY FILED
Method for the production of substituted 2-(phenoxymethyl)benzoic acids
The invention relates to a process for the preparation of substituted 2-
(phenoxymethyl)-
benzoic acids of the general formula (I). The present invention further
relates to novel
intermediates which are formed in the process according to the invention for
the
~o preparation of the compounds of the general formula (I).
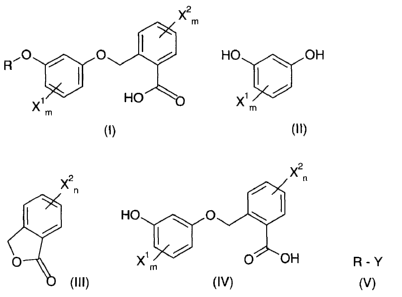
X2
m
O
X, HO
m
Compounds of the general formula (I) are suitable as medicaments, for example
for
lowering blood lipids and for the treatment of diabetes, because they have a
large
pharmacological effect as agonist or antagonist of the peroxisome proliferator-
activated
receptor (PPAR).
A large number of PPAR agonists and antagonists has been disclosed in WO
00/64876,
including the compounds of the general formula (I).
The process described in WO 00/64876 for the preparation of compounds of the
general
formula (I) has, however, some disadvantages, also in relation to an
industrial preparation
of these compounds of pharmacological interest. In this preparation method,
resorcinol is
reacted with 2-bromomethylbenzoic esters, after which the intermediate
containing a free
OH group is reacted with an alkylating reagent in the presence of the base
potassium
carbonate, and finally compounds of the formula I are obtained by hydrolysis
of the ester.
The particular problem in this process is the precursor 2-bromomethylbenzoic
ester,
because this substance is thermally unstable and decomposes even at room
temperature
3o with cyclization and elimination of methyl bromide to give the
corresponding phthalide,
and is in addition highly lachrymatory and potentially carcinogenic and
moreover can be
CA 02550627 2006-06-20
a -2-
prepared pure only by means of chromatography. A further disadvantage is that
N-bromosuccinimide and the explosive dibenzoyl peroxide are employed in the
process
described in WO 00/64876 for the preparation of the precursor (2-
bromomethylbenzoic
ester). An alternative possibility for preparing the precursor is also by
photochemical
bromination as described in J. Org. Chem. 1985, 50, 3355 - 3359. There are
always
problems with such bromination reactions for the preparation of the precursor
if the
appropriate benzoic ester has alkyl groups in addition to that in position 2,
because these
can likewise be brominated. For example, some compounds of pharmacological
interest of
the formula (1) have a 6-methylbenzoic acid fragment, so that industrial
preparation of such
~o compounds by the aforementioned process would also be associated with
distinctly higher
costs as a result of the additional purification steps owing to the
nonselective bromination.
Alternatives to the process described in WO 00/64876 for the preparation of
2-carboxybenzyl aryl ethers have been disclosed. Thus, all the alternatives
are based on the
~5 procc~;s described in J. Chem. Soc. 1964, 4074 - 4075, in which phenol is
reacted with the
a;~t>ropr. ~: phthali~le in tr~~ ;:;e of the base sodium hydroxide at
temperatures above
170°C. 'I hus, US ~,~~ j, ~.~~ relates to a process for the preparation
of E-oxime ethers of
phenylglyoxylic esters, where 2-carboxybenzyl aryl ethers are formed as
intermediate. The
a; ~.. .. gent may in this case be phenyl which is meta-substituted by C1-C4-
alkoxy. The
2o intermeu . , t. ' ~n the nr~~ ~-ice of a base at temperatures between
50°C and 250°C
in the melt. Correspoi~~:. ~ ' . ~ , in DE-A 2 208 893, the preparation of
tricyclic a-oxy
carboxyl aid derivatives starts from the precursor guaiacol, with addition of
a methoxide
solution being followed firstly by removing the solvent methanol by
distillation and, after
addition of the phthalide, heating the reaction mixture at 180 to
190°C.
It is not absolutely necessary to prepare 2-carboxybenzyl aryl ethers in the
melt; on the
contrary, higher-boiling solvents can also be employed where appropriate. DE-A
27 49 957
describes a process for the preparation of quinolizidylidene derivatives of
xanthenes,
thioxanthenes and dibenzoxepines in which 4-methylphenol is reacted with
phthalide using
3o sodium hydride as base and dimethylformamide as solvent under reflux.
Analogously,
JP-A 07002733 describes the preparation of 2-carboxybenzyl aryl ethers using
alcoholates
as base and higher boiling solvents. The aryl fragment may, inter alia, also
be a phenyl
which is substituted in the meta position by lower alkoxy.
It is common to all the processes described above for the preparation of 2-
carboxybenzyl
aryl ethers that the reaction of the alcohol with the phthalide must take
place at high
temperatures (at least 100°C) because, otherwise, only little or no
reaction takes place. An
CA 02550627 2006-06-20
_3_
additional finding is that in no case is resorcinol or a derivative thereof
with two free
hydroxyl groups employed. Where the phenols used have additional alkoxy
substituents
(not only in the meta position), these are unsubstituted lower alkoxy
substituents. The
PPAR agonists and antagonists of the general formula (I) described in WO
00/64876 are,
however, compounds which have as radical R preferably arylalkyl or
heteroarylalkyl
substituents which in turn may be substituted one or more times. The radicals
R thus
predominantly have a moderate to large molecular weight. A process analogous
to, for
example, JP-A 07002733 cannot, however, be employed to prepare compounds of
formula I because the ether linkage between R and the phenyl fragment of the
alcohol is
o unstable because of the high temperatures necessary for the reaction of
alcohol and
phthalide. A higher temperature and a higher molecular weight of the
corresponding
substituent R mean a smaller yield of compounds of the general formula (I).
Accordingly, the object on which the invention is based is to provide a
process for the
~ 5 preparation of PPAR agonists and antagonists of the general formula (I),
which process
does not have the disadvantages of those disclosed in the prior art.
The object is achieved by a process for the preparation of a compound of the
general
formula (I), wherein
a) a compound (II) is reacted in the presence of a base B 1 with a compound
(III) and
X2n
X~ /
HO \ OH ( HO O \
/ ~ /
Xjm O X1m / O OH
(II) (III) (IV)
CA 02550627 2006-06-20
-4-
b) the compound (LV) formed as intermediate in step a) is reacted in the
presence of a
base B2 with a compound (V) to give the compound of the general formula (I)
X2
m
R-Y ~O / O I /
R
(V)
X1m HO O
in which:
R is selected from the group consisting of:
to unsubstituted or at least monosubsti.tuted C,-C"~-alkyl, heterocyclyl, aryl-
(C,-C,o-alkyl)-,
hcteroaryl-(C,-C,~-alkyl)- and heterocyclyl-(C,-C,o-alkyl)-,
where the substituents are selected from halogen, C,-C~-alkyl, -O-aryl, oxo,
C,-C6-alkoxy,
-C(O)O-(C,-C6-alkyl), C2-C6-alkenyl, C2-C6-alkynyl, -C(O)-(C,-C6-alkyl), -
C(O)NH2,
-C(O)NH(C,-C6-alkyl), -C(O)N(C,-C6-alkyl)2, -S-(C,-C6-alkyl), -S02NH2, -SOZ-
(Cl-C6-
alkyl), -NHZ, -N(C,-C~-alkyl)2, -NH(C~-C6-alkyl), -NOZ, -CN, trifluoromethyl,
trifluoromethoxy, aryl, heterocyclyl and heteroaryi,
and aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted
by C,-C3-
alkyl, Ct-C~-alkoxy, halogen or trifluoromethyl;
X~ is halogen, C,-C6-alkyl, C,-C6-alkoxy, trifiuoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl may in turn be at least
monosubstituted by C,-C~-
alkyl, C,-C3-alkoxy, halogen or trifiuoromethyl;
XZ is halogen, C,-C6-alkyl, C,-C6-alkoxy, trifluoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl in turn may be at least
monosubstituted by CI-C3-
3o alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
CA 02550627 2006-06-20
_ -5-
heteroaryl is a 5- to 14-membered, aromatic, mono-, bi- or tricyclic
heterocycle which
contains one or more heteroatoms selected from N, O and S;
aryl is a 6- to 14-rnembered, aromatic mono-, bi- or tricyclic system;
heterocyclyl is a 5- to 14-membered, nonaromatic, mono-, bi- or tricyclic
heterocycle
which contains one or more heteroatoms selected from N, O and S;
mis0, 1,2,3or4;
nis0, 1,2,3or4;
I c~ Y is a leaving group and
B2 is an alkali metal hydroxide, alkaline earth metal hydroxide, alkali metal
alcoholate,
alkaline earth metal alcoholate, alkali metal hydride, alkaline earth metal
hydride, silazide,
alkali metal amide or alkaline earth metal amide.
I _5
The process according to the invention has the advantages compared with those
of the state
of the art that uniform products of compounds of the general formula (I) can
be prepared in
high yields and/or with few intermediate stages, which is economically
worthwhile in
particular in relation to industrial utilization. Compared with the processes
described in
2o W O ~J64876, on the one hand the use of carcinogenic and unstable
intermediate
compounds is dispensed with and, in addition, the free benzoic acid can be
employed in the
alkylation reaction in step b) owing to suitable choice of the base. This
saves two synthetic
steps because it is necessary in the processes described in WO OOJ64876 for
the free
benzoic acid first to be protected in the form of a suitable ester and to be
deprotected again
25 in the final reaction stage. The advantage of the essential reversal of
reaction steps a) and
b) compared with the processes like those described for example in JP-A
07002733
(coupling of meta-lower alkoxyphenols with phthalide) is that it is possible
in this way to
prepare compounds of the formula (I) for the very first time or in
economically worth-
while yields, because the alkylation reaction carried out in step b) of the
process according
3o to the invention can be carried out at distinctly lower temperatures than
the ether linkage
performed in step a).
CA 02550627 2006-06-20
_(-
The process according to the invention can be used to prepare compounds of the
general
formula (1)
X2
m
R~O / O /
X, HO ~O
m
in which:
R is selected from the group consisting of:
o unsubstituted or at least monosubstituted C,-C,o-alkyl, heterocyclyl, aryl-
(C,-C,o-alkyl)-,
heteroaryl-(C,-C"~-alkyl)- and heterocyclyl-(C,-C,o-alkyl)-,
where the substituents are selected from halogen, C,-C6-alkyl, -O-aryl, oxo,
C,-C6-alkoxy,
C2-C6-alkenyl, CZ-C6-alkynyl, -C(O)-(C,-C6-alkyl), -C(O)NH2, -C(O)NH(C,-C6-
alkyl), -
t5 C(O)N(C,-C~-alkyl)2, -S-(C,-C6-alkyl), -S02NH2, -S02-(C,-C6-alkyl), -
C(O)O(C,-C6-
alkyl), -NH2, -N(C,-C6-alkyl)2, -NH(C,-C6-alkyl), -N02, -CN, trifluoromethyl,
trifluoromethoxy, aryl, heterocyclyl and heteroaryl,
and aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted
by CI-C3-
20 alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
X~ is halogen, C,-C6-alkyl, C1-C6-alkoxy, trifluoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl may in turn be at least
monosubstituted by C1-C3-
25 alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
XZ is halogen, C1-C6-alkyl, C,-C6-alkoxy, trifluoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl in turn may be at least
monosubstituted by C,-C3-
3o alkyl, C1-C3-alkoxy, halogen or trifluoromethyl;
CA 02550627 2006-06-20
-
heteroaryl is a 5- to 14-membered, aromatic, mono-, bi- or tricyclic
heterocycle which
contains one or more heteroatoms selected from N, O and S;
aryl is a 6- to 14-mcmbered, aromatic mono-, bi- or tricyclic system;
heterocyclyl is a 5- to 14-membered, nonaromatic, mono-, bi- or tricyclic
heterocycle
which contains one or more heteroatoms selected from N, O and S;
mis0, 1,2,3or4;
o nis0, 1,2,3or4.
Where groups, fragments, radicals or substituents such as, for example, aryl,
heteroaryl,
alkyl, alkoxy etc. are present more than once in the compounds of the formula
(I), they all
have, inclc~pendently of one another, the meanings listed above and may thus
in each
~5 (i;tdi~ ~w~ual) case have either an identical or a mutually independent
meaning. The
following statements apply to (for example) aryl and every other radical
irrespective of
whether it is referred to as aryl group, substituent, fragment or radical. For
example, in a
di(L,-Cc,-alkyl)amino group, the two alkyl substituents may be either
identical or different
(for example 2 x ethyl or 1 x propyl and 1 x hexyl).
Where a substituent, for example aryl, in the above definitions of compounds
of formula
(I) may be unsubstituted or at least monosubstituted by a group of further
substituents, for
example C~-C6-alkyl, Ci-C6-alkoxy, halogen etc., the selection from the series
of further
substituents in the cases where aryl is polysubstituted takes place
independently of one
another. Thus, for example when aryl is disubstituted, all combinations of the
further
substituents are included. Aryl may thus be, for example, disubstituted by
ethyl, aryl may
in each case be monosubstituted by methyl and ethoxy, aryl may in each case be
monosubstituted by ethyl and fluorine, aryl may be disubstituted by methoxy,
etc.
Alkyl radicals may be either linear or branched, acyclic or cyclic. This also
applies when
they are part of another group such as, for example, alkoxy groups (C,-Coo-
alkyl-O-),
alkoxycarbonyl groups or amino groups or if they are substituted.
Examples of alkyl groups are: methyl, ethyl, propyl, butyl, pentyl, hexyl,
heptyl, octyl,
nonyl or decyl. Included therein are the n-isomers of these radicals and
isopropyl, isobutyl,
isopentyl, sec-butyl, tert-butyl, neopentyl, 3,3-dimethylbutyl etc. Unless
described
otherwise, the term alkyl additionally includes alkyl radicals which are
unsubstituted or
CA 02550627 2006-06-20
optionally substituted by one or more further radicals, for example l, 2, 3 or
4 identical or
different radicals such as, for example, aryl, heteroaryl, alkoxy or halogen.
It is moreover
possible for the additional substituents to occur in any position of the alkyl
radical. The
term alkyl also includes cycloalkyl and cycloalkylalkyl (alkyl which in turn
is substituted
by cycloalkyl), where cycloalkyl has at least 3 carbon atoms. Examples of
cycloalkyl
radicals are: cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl,
cyclooctyl,
cyclononyl and cyclodecyl. Polycyclic ring systems are also possible where
appropriate,
such as decalinyl, norbornanyl, bornanyl or adamantanyl. The cycloalkyl
radicals may be
unsubstituted or optionally substituted by one or more further radicals as
listed above by
way of example for the alkyl radicals.
Examples of alkenyl and alkynyl groups are: vinyl, 1-propenyl, 2-propenyl
(allyl),
2-butenyl, 2-methyl-2-propenyl, 3-methyl-2-butenyl, ethynyl, 2-propynyl
(propargyl),
2-butynyl or 3-butynyl. The term alkenyl here expressly includes cycloalkenyl
radicals and
~ 5 cycloalkenylalkyl radicals (alkyl substituted by cycloalkenyl) containing
at least three
carbon atoms. Examples of cycloalkenyl are: cyclopentenyl, cyclohexenyl,
cycloheptenyl
and cyclooctenyl.
The alkenyl radicals may have one to three conjugated or unconjugated double
bonds (i.e.
2o also alk-dienyl and alk-trienyl radicals), preferably one double bond in a
linear or branched
chain, and the same applies to the triple bonds for alkynyl radicals. The
alkenyl and
alkynyl radicals may be unsubstituted or optionally substituted by one or more
further
radicals as listed above by way of example for the alkyl radicals.
25 Unless stated otherwise, the aforementioned aryl, heteroaryl and
heterocyclyl radicals may
be both unsubstituted and have one or more, for example 1, 2, 3 or 4, further
of the
aforementioned substituents in any position. For example, the substituent in
monosubstituted phenyl radicals may be in position 2, 3 or 4, and the
substituents in
disubstituted phenyl radicals may be in the 2,3 position, 2,4 position, 2,5
position, 2,6
3o position, 3,4 position or 3,5 position. The substituents in trisubstituted
phenyl radicals may
be in the 2,3,4 position, 2,3,5 position, 2,3,6 position, 2,4,5 position,
2,4,6 position or
3,4,5 position. The substituents in tetrasubstituted phenyl radicals may be in
the
2,3,4,5 position, the 2,3,4,6 position or in the 2,3,5,6 position.
35 The definitions mentioned above and hereinafter relating to monovalent
radicals apply
equally to divalent radicals such as phenylene, naphthylene or heteroarylene.
These
divalent radicals (fragments) may be linked with the adjacent groups via any
ring carbon
CA 02550627 2006-06-20
-9-
atom. In the case of phenylene radicals, this may be in the 1,2 position
(ortho-phenylene),
1,3 position (meta-phenylene) or 1,4 position (para-phenylene). In the case of
a 5-
membered aromatic system containing a heteroatom, such as, for example,
thiophene or
furan, the two free bonds may be in the 2,3 position, 2,4 position, 2,5
position or 3,4
position. A divalent radical derived from a 6-membered aromatic system having
one
heteroatom, such as, for example, pyridine, may be a 2,3, 2,4, 2,5, 2,6, 3,4
or 3,5
pyridinediyl radical. In the case of unsymmetrical divalent radicals, the
present invention
also includes all positional isomers, i.e. in the case for example of a 2,3-
pyridinediyl
radical the compound in which one adjacent group is in position 2 and the
other adjacent
to group is in position 3 is just as much included as the compound in which
one adjacent
group is in position 3 and the other adjacent group is in position 2.
Unless stated otherwise, heteroaryl radicals, heteroarylene radicals,
heterocyclyl radicals
and heterocyclylene radicals, and rings formed by two groups bonded to
nitrogen, are
preferably derived from completely saturated, partially or wholly unsaturated
heterocycles
(i.e. heterocycloalkanes, heterocycloalkenes, heteroaromatic compounds), which
contain l,
2, 3 ur 4 heteroatoms which may be both different and identical. They are
preferably
derived from heterocycles which contain 1, 2 or 3, particularly preferably 1
or 2,
heteroatoms which may be identical or different. Unless stated otherwise, the
heterocycles
are mono- or polycyclic, for example monocyclic, bicyclic or tricyclic. They
are preferably
monocyclic or bicyclic. Preference is given to 5-membered, 6-membered and 7-
membered
rings, particularly preferably 5-membered and 6-membered rings. In the case of
polycyclic
heterocycles having 2 or more heteroatoms, these may all be in the same ring
or be
distributed over a plurality of rings.
Radicals referred to as heteroaryl in the present invention are those derived
from
monocyclic, bicyclic or tricyclic aromatic heterocycles. Examples of
heteroaryl are:
pyrrolyl, furanyl (furyl), thiophenyl (thienyl), imidazolyl, pyrazolyl, 1,2,3-
triazolyl, 1,2,4-
triazolyl, 1,3-oxazolyl (oxazolyl), 1,2-oxazolyl (isoxazolyl), oxadiazolyl,
1,3-thiazolyl
(thiazolyl), 1,2-thiazolyl (isothiazolyl), tetrazolyl, pyridinyl (pyridyl)
pyridazinyl,
pyrimidinyl, pyrazinyl, 1,2,3-triazinyl, 1,2,4-triazinyl, 1,3,5-triazinyl,
1,2,4,5-tetrazinyl,
indazolyl, indolyl, benzothiophenyl, benzofuranyl, benzothiazolyl,
benzimidazolyl,
benzodioxolyl, acridinyl, quinolinyl, isoquinolinyl, quinazolinyl,
quinoxalinyl,
phthalazinyl, thienothiophenyl, 1,8-naphthyridinyl, other naphthyridinyls,
pteridinyl or
phenothiazinyl. Where the systems are not monocyclic, also included for each
of the
aforementioned heteroaryls for each additional ring is the saturated form
(perhydro form)
or the partially unsaturated form (for example the dihydro form or tetrahydro
form) or the
CA 02550627 2006-06-20
- ID-
maximally unsaturated (nonaromatic) form where the respective forms are known
and
stable. The term heteroaryl thus includes in the present invention for example
bicyclic
radicals in which both the two rings are aromatic and bicyclic radicals in
which only one
ring is aromatic. Such examples of heteroaryl are: 3H-indolinyl, 2(1H)-
quinolinonyl, 4-
oxo-1,4-dihydroquinolinyl, 2H-I-oxoisoquinolyl, 1,2-dihydroquinolinyl,
(2H)quinolinyl N-
oxide, 3,4-dihydroquinolinyl, 1,2-dihydroisoquinolinyl, 3,4-
dihydroisoquinolinyl,
chromonyl, 3,4-dihydroisoquinoxalinyl, 4-(3H)quinazolinonyl, 4H-chromenyl, 4-
chromanonyl, oxindolyl, 1,2,3,4-tetrahydroisoquinolinyl, 1,2,3,4-
tetrahydroquinolinyl,
1 H-2,3-dihydroisoindolyl, 2,3-dihydrobenzo[f]isoindolyl, 1,2,3,4-
o tetrahydrobenzo[g]isoquinolinyl, chromanyl, isochromanonyl, 2,3-
dihydrochromonyl,
1,4-benzodioxanyl, 1,2,3,4-tetrahydroquinoxalinyl, 5,6-dihydroquinolyl, 5,6-
dihydro-
isoquinolyl, 5,6-dihydroquinoxalinyl, 5,6-dihydroquinazolinyl, 4,5-dihydro-1H-
benzimidazolyl, 4,5-dihydrobenzoxazolyl, 1,4-naphthoquinolyl, 5,6,7,8-
tetrahydroquinolinyl, 5,6,7,8-tetrahydroisoquinolyl, 5,6,7,8-
tetrahydroquinoxalinyl,
~5 5,6,7,8-tetrahydroquinazolyl, 4,5,6,7-tetrahydro-1H-benzimidazolyl, 4,5,6,7-
tetra-
hydrobenzoxazolyl, 1H-4-oxa-1,5-diazanaphthalen-2-onyl, 1,3-dihydroimidizolo-
[4,5]-
pyridin-2-onyl, 2,3-dihydro-1,4-dinaphthoquinonyl, 2,3-dihydro-1H-pyrrol[3,4-
b]quinolinyl, 1,2,3,4-tetrahydrobenzo[b][1,7]naphthyridinyl, 1,2,3,4-
tetrahydrobenz[b]
[1,6]naphthyridinyl, 1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indolyl, 1,2,3,4-
tetrahydro-9H-
2o pyrido[4,3-b]indolyl, 2,3-dihydro-1H-pyrrolo[3,4-b]indolyl, 1H-2,3,4,5-
tetra-
hydroazepino[3,4-b]indolyl, 1H-2,3,4,5-tetrahydroazepino[4,3-b]indolyl, 1H-
2,3,4,5-
tetrahydroazepino[4,5-b]indolyl, 5,6,7,8-tetrahydro[1,7]napthyridinyl, 1,2,3,4-
tetra-
hydro[2,7]naphthyridyl, 2,3-dihydro[1,4]dioxino[2,3-b]pyridyl, 2,3-
dihydro[1,4]dioxino-
[2,3-b]pryidyl, 3,4-dihydro-2H-1-oxa[4,6]diazanaphthalenyl, 4,5,6,7-tetrahydro-
3H-
25 imidazo[4,5-c]pyridyl, 6,7-dihydro[5,8]diazanaphthalenyl, 1,2,3,4-
tetrahydro[ 1,5]napthyr-
idinyl, 1,2,3,4-tetrahydro[1,6]napthyridinyl, 1,2,3,4-
tetrahydro[1,7]napthyridinyl, 1,2,3,4-
tetrahydro[1,8]napthyridinyl or 1,2,3,4-tetrahydro[2,6]napthyridinyl.
Radicals referred to as heterocyclyl in the present invention are those
derived from
3o monocyclic, bicyclic or tricyclic nonaromatic heterocycles. Nonaromatic
heterocycles
mean hereinafter in particular heterocycloalkanes (completely saturated
heterocycles) and
heterocycloalkenes (partially unsaturated heterocycles). In the case of the
heterocycloalkenes, also included are compounds having two or more double
bonds which
may also where appropriate be conjugated together. Examples of heterocyclyl
are:
35 pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, pyrazolidinyl,
isothiazolidinyl,
thiazolidinyl, isoxazolidinyl, oxazolidinyl, tetrahydrofuranyl,
tetrahydrothiophenyl 1,3-
dioxolanyl, 1,4-dioxinyl, pyranyl, thiopyranyl, 1,4-dioxinyl, 1,2-oxazinyl,
1,3-oxazinyl,
CA 02550627 2006-06-20
1,4-oxazinyl, 1,2-thiazinyl, 1,3-thiazinyl, 1,4-thiazinyl, azepinyl, 1,2-
diazepinyl, 1,3-
diazepinyl, 1,4-diazepinyl, 1,3-oxazepinyl, 1,3-thiazepinyl, 2-oxo-azepanyl,
morpholinyl,
thiomorpholinyl, 1,2,3,4-tetrahydropyridinyl, 1,2-dihydropyridinyl, 1,4-
dihydropyridinyl,
1,2,3,6-tetrahydropyridinyl, 4(3H)-pyrimidonyl, 1,4,5,6-tetrahydropyrimidinyl,
2-
pyrrolinyl, 3-pyrrolinyl, 2-imidazolinyl, 2-pyrazolinyl, 3,4-dihydro-2H-
pyranyl,
dihydrofuranyl, 7-oxabicyclo[2.2.1 ]heptenyl, dihydrothiophenyl and
dihydrothiopyranyl.
The degree of saturation of heterocyclic groups is indicated in the definition
in each case.
Substituents derived from these heterocycles may be linked via any suitable
carbon atom,
1 o and be provided with further substituents. Radicals derived from nitrogen-
containing
heterocycles may have a hydrogen atom or another substituent on the
appropriate nitrogen
atom. Examples include pyrrole, imidazole, pyrrolidine, morpholine, piperazine
residues
etc. These nitrogen-containing heterocyclic radicals may also be bonded via
the ring
nitrogen atom, especially when the relevant heterocyclic radical is bonded to
a carbon
atom. For example, a thienyl radical may be in the form of 2-thienyl or 3-
thienyl, and a
piperidinyl radical in the form of 1-piperidinyl (piperidino), 2-piperidinyl,
3-piperidinyl or
4-piperidinyl. Suitable nitrogen-containing heterocycles may also be in the
form of N-
oxides or of quarternary salts which have a counter ion which is derived from
a
physiologically acceptable acid. For example, pyridyl radicals may be in the
form of
2o pyridine N-oxides. Suitable sulfur-containing heterocycles may also be in
the form of S-
oxide or 5,S-dioxide.
Radicals referred to as aryl in the present invention are those derived from
monocyclic,
bicyclic or tricyclie aromatic systems which contain no ring heteroatoms.
Where the
systems are not monocyclic, the term aryl includes for each additional ring
also the
saturated form (perhydro form) or the partially unsaturated form (for example
the dihydro
form or tetrahydro form) or the maximally unsaturated (nonaromatic) form where
the
respective forms are known and stable. The term aryl also includes in the
present invention
for example bicyclic radicals in which both the two rings are aromatic and
bicyclic radicals
in which only one ring is aromatic. Examples of aryl are: phenyl, naphthyl,
anthracyl,
indanyl, 1,2-dihydronaphthyl, 1,4-dihydronaphthyl, indenyl, 1,4-
naphthoquinonyl or
1,2,3,4-tetrahydronaphthyl.
Arylalkyl means that an alkyl radical is substituted in turn by an aryl
radical.
Heteroarylalkyl means that an alkyl radical is substituted in turn by a
heteroaryl radical.
Heterocyclylalkyl means that an alkyl radical is substituted in turn by a
heterocyclyl
CA 02550627 2006-06-20
- 12-
radical. For the definitions and possible substitutions of alkyl, heteroaryl,
heterocyclyl and
aryl, reference is made to the definitions above.
Halogen is fluorine, chlorine, bromine or iodine, with preference for
fluorine, chlorine or
.5 bromine, and particular preference for fluorine or chlorine.
Preferred compounds of the general formula (I) which can be prepared by the
process
according to the invention are defined as follows:
o R is selected from the group consisting of:
unsubstituted or at least monosubstituted C,-Clo-alkyl, heterocyclyl, aryl-(C~-
Clo-alkyl)-,
heteroaryl-(Ci-C,«-alkyl)- and heterocyclyl-(C~-CIO-alkyl)-,
S where the substituents are selected from halogen, C~-C6-alkyl, -O-aryl, oxo,
Cl-C6-alkoxy,
-C(O)O-(C,-C~-alkyl), CZ-C6-alkenyl, CZ-C6-alkynyl, -C(O)-(C,-C6-alkyl), -
C(O)NH2,
-C(O)NH(Cl-C6-alkyl), -C(O)N(C1-C6-alkyl)2, -S-(C~-C6-alkyl), -S02NH2, -SO2-
(C~-C6-
al~; , -NHS, -N(C,-C6-alkyl)2, -NH(C~-C6-alkyl), -NO2, -CN, trifluoromethyl,
trifluoromethoxy, aryl, heterocyclyl and heteroaryl,
and aryl, heterocyclyl and heteroaryl in turn may be at least monosubstituted
by C~-C3-
alkyl, C,-C~-alkoxy, halogen or trifluoromethyl;
X' is halogen, C,-C6-alkyl, C1-C6-alkoxy or trifluoromethoxy;
XZ is halogen, C~-C6-alkyl, C,-C6-alkoxy or trifluoromethoxy;
heteroaryl is a 5- to 14-membered, aromatic, mono-, bi- or tricyclic
heterocycle which
contains one or more heteroatoms selected from N, O and S;
aryl is a 6- to 14-membered, aromatic mono-, bi- or tricyclic system;
heterocyclyl is a 5- to 14-membered, nonaromatic, mono-, bi- or tricyclic
heterocycle
which contains one or more heteroatoms selected from N, O and S;
m is 0, f or 2;
nis0, 1 or2.
CA 02550627 2006-06-20
- 13-
More preferred compounds of the general formula (I) are defined as follows:
R is selected from the group consisting of:
unsubstituted or at least monosubstituted aryl-(C,-C6-alkyl)- and heteroaryl-
(C,-C6-alkyl)-,
where the substituents are selected from halogen, C~-C4-alkyl, -O-aryl, oxo,
C,-C4-alkoxy,
aryl, heterocyclyl and heteroaryl,
and aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted
by C~-C3-
alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
X' is halogen, C,-C6-alkyl, C,-C6-alkoxy or trifluoromethoxy;
~5 X'' ist halogen, C,-C6-alkyl, C,-C~-alkoxy or trifluoromethoxy;
heteroaryl is a 5- to 14-membered, aromatic, mono-, bi- or tricyclic
heterocycle which
contains one or more heteroatoms selected from N, O and S;
2o aryl is a 6- to 14-membered, aromatic mono-, bi- or tricyclic system;
heterocyclyl is a 5- bis 14-membered, nonaromatic, mono-, bi- or tricyclic
heterocycle
which contains one or more heteroatoms selected from N, O and S;
25 m is 0, 1 or 2;
n is 0, 1 or 2.
Even more preferred compounds of the general formula (I) are defined as
follows:
3o R is selected from the group consisting of:
unsubstituted or at least monosubstituted aryl-(C1-C6-alkyl)- and heteroaryl-
(C1-C6-alkyl)-,
where the substituents are sleeted from halogen, C~-C4-alkyl, -O-aryl, oxo, C1-
C4-alkoxy,
35 aryl, heterocyclyl and heteroaryl,
CA 02550627 2006-06-20
- 14-
and aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted
by Ci-C3-
alkyl, C,-C~-alkoxy, halogen or trifluoromethyl;
X~ is halogen, C,-C~,-alkyl, C,-C6-alkoxy or trifluoromethoxy;
l0
X'' is halogen, C,-C6-alkyl, C~-C~-alkoxy or trifluoromethoxy;
heteroaryl is a 5- to 10-membered, aromatisc, mono- or bicyclic heterocycle
which
contains one or more heteroatoms selected from N, O and S;
aryl is phenyl, naphthyl, indanyl, dihydronaphthyl, tetrahydronaphthyl or
indenyl;
heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl, tetrahydrofuranyl or
morpholinyl;
I S m i s 0, l or 2;
nis0, 1 or2;
Particularly preferred compounds of the general formula (I) are defined as
follows:
2o R is unsubstituted or at least monosubstituted benzyl or heteroarylmethyl,
where the
substituents are selected from fluorine, chlorine, C~-C4-alkyl, -O-phenyl, C~-
C4-alkoxy,
phenyl and heteroaryl,
and phenyl and heteroaryl may in turn be at least monosubstituted by Cl-C3-
alkyl, C1-C3-
25 alkoxy, chlorine or fluorine;
X~ is C1-C3-alkyl or bromine;
XZ is C,-C3-alkyl, fluorine or chlorine;
heteroaryl is oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl,
pyridinyl,
pyridazinyl, pyrimidinyl, pyrazinyl, indolyl, benzimidazolyl, quinolinyl,
isoquinolinyl,
quinazolinyl or quinoxalinyl;
mis0orl;
nis0, 1 or2.
CA 02550627 2006-06-20
- l5-
Very particularly preferred compounds of the general formula (I) are defined
as follows:
R is unsubstituted or at least monosubstituted benzyl or heteroarylmethyl,
where the
substituents are selected from fluorine, chlorine, C~-Cd-alkyl, phenyl and
heteroaryl,
s
and phenyl and heteroaryl may in turn be at least monosubstituted by C,-C~-
alkyl, chlorine
or fluorine;
X'' is C,-C~-alkyl;
to
heteroaryl is oxazolyl or isoxazolyl;
misO;
nISOOrI.
IS
Step a) of the process according to the invention:
In step a) of the process according to the invention, a compound (II) is
reacted in the
presence of a base B I with a compound (III) to form a compound (IV).
X2 X2n
n
HO .~ OH ~ HO ~ O
' / '
v
Xm O Xm _
(II)
(III) (IV)
The compounds of the formulae (II) and (III) have the corresponding
definitions of the
general formula (I). Compounds suitable as compound (II), which are referred
to
hereinafter as resorcinol derivatives, are only those having two hydroxyl
groups in meta
position relative to one another. The corresponding hydroquinone and catechol
derivatives
cannot, however, be used in the process according to the invention because
they react to
only a small extent or not all with the compounds of the formula (III), which
are referred to
hereinafter as phthalides.
Bases suitable as bases B I are in principle all those familiar to the skilled
worker.
Preferably suitable are alkali metal and alkaline earth metal alcoholates such
as sodium
CA 02550627 2006-06-20
- 16-
methoxide, potassium methoxide and potassium test-butoxide. Equally suitable
are
silazides such as potassium hexamethyldisilazide and lithium
hexamethyldisilazide. The
compounds of the formula (1V) can likewise be prepared using alkali metal
hydroxides
such as sodium hydroxide and potassium hydroxide, although the water resulting
from the
deprotonation likewise reacts with the phthalide employed, with ring opening
which is
irreversible under the reaction conditions, and thus sequesters the precursor
from the
desired reaction, resulting in losses of yield. Alkali metal alcoholates are
particularly
preferred as base B 1, and sodium methoxide, potassium methoxide and potassium
tert-
butoxide are very particularly preferred.
~o
Suitable solvents are solvents or mixtures thereof having a boiling point of >
100°C, such
as, for example, toluene, o-, m-, p-xylene, glyme, dimethylformamide, N-methyl-
1-
pyrrolidone and N,N-dimethylacetamide. N-Methyl-1-pyrrolidone and N,N-
dimethylacetamide are particularly preferred.
The amount of base B 1 to be employed should be equimolar in relation to the
amount of
resorcinol derivative (II) employed, with preference for the exactly equimolar
amount.
Excesses of base B 1 should be avoided because, otherwise, both free hydroxyl
groups of
the compound (II) are alkylated.
Less than equimola.r amounts of base B 1 should likewise be avoided because
this leads to
reduced conversion, likewise having an adverse effect on the yield. The amount
of base B 1
and resorcinol derivative (II) employed must be at least equimolar in relation
to the
phthalide (III) employed. The reaction rate can be increased by simultaneous
use of larger
2S amounts of resorcinol derivative (II) and of base B 1. A one- to six-molar
excess of base B 1
and resorcinol derivative (II), based on the amount of phthalide (III)
employed, is
preferably used.
Phthalide (III), resorincol derivative (II), base B 1 and solvent are mixed at
room
3o temperature, the sequence of addition being immaterial and having no affect
on the
conversion in the reaction and the yield of isolated product. Accordingly, it
is unnecessary,
as described in some of the preceding documents, to carry out the
deprotonation of the
resorcinol derivative (II) with the base B 1 used, possibly also to remove the
corresponding
acid of the base B 1 employed, and subsequently to add the phthalide (III) and
to heat.
CA 02550627 2006-06-20
_ I '7 _
In general, the reaction is carried out after mixing the reactants and solvent
at a
temperature in the range from 80 up to 200°C, preferably between 110
and L50°C. The
reaction time depends on the phthalide (III) employed and its steric demands.
No special conditions in relation to pressure are necessary, and it is
expedient to operate
under atmospheric pressure.
After completion of the reaction, the reaction mixture is diluted with water.
The water is
preferably added at temperatures of > 60°C because solidification of
the reaction mixture
0 may not be preventable, depending on the concentration thereof. The
resulting solution is
acidified to liberate the compounds (IV), preferably with an inorganic acid
such as
hydrochloric acid or sulfuric acid. Methods known to the skilled worker are
used for
further working up.
~ 5 The intermediates of the formula (IV), which have not previously been
disclosed in the
literature, can be obtained in high yields and excellent purities by the
process according to
the invention. On use of N-methyl-1-pyrrolidone and N,N-dimethylacetamide it
is possible
initially to obtained solvates of the compounds of the formula (IV) with
varying contents
of said solvents. Suitable processes, e.g. recrystallization or heating with
water, can be
2o used to reduce or entirely remove the contents of said solvents in the
isolated product.
Removal of the solvate envelope is unnecessary for further use of the
intermediates (IV) in
step b) of the process according to the invention.
The process according to the invention in step a) and/or step b) can be
carried out both
25 batchwise and continuously. In a continuous procedure, the reaction
partners are passed for
example through a pipe reactor or cascades of stirred vessels.
Step b) of the process according to the invention:
3o In step b), the compound (IV) formed as intermediate in step a) of the
process according to
the invention is reacted in the presence of a base B2 with a compound R-Y (V)
to give the
compound of the general formula (I).
The compound (IV) formed as intermediate in step a), and the compound (V) have
the
35 corresponding definitions of the general formula (I). A suitable leaving
group Y in the
compound (V) is any leaving group known to the skilled worker. Preferred for Y
are
CA 02550627 2006-06-20
- 18-
chlorine, bromine, iodine, mesylates or tosylates, particularly preferably
chorine, bromine
or iodine.
Suitable bases B2 are alkali metal and alkaline earth metal hydroxides such as
sodium and
potassium hydroxides, alkali metal and alkaline earth metal alcoholates such
as sodium
methoxide, potassium methoxide and potassium tort-butoxide, alkali metal and
alkaline
earth metal hydrides such as sodium and calcium hydrides. Likewise suitable
are silazides
such as potassium hexamethyldisilazide and lithium hexamethyldisilazide, and
generally
alkali metal and alkaline earth metal amides; alkali metal alcoholates are
preferred, and
o sodium alcoholates are particularly preferred. On use of the aforementioned
bases B2 it is
possible in step b) of the process according to the invention to achieve a
selective
alkylation of the hydroxyl group of the compound (IV) formed as intermediate
in step a).
Additional alkylation of the unprotected carboxyl group of the compound (IV)
takes place
to only a small extent or not at all.
~5
Bases which have proved unsuitable are carbonate bases such as, for example,
potassium
carbonate. Owing to the inadequate basicity, even with large excesses only the
carboxyl
group is deprotonated, leading to selective, but undesired, alkylation of the
acid function of
the compounds of the formula I. This selective alkylation of the acid function
is observed
2o at lower temperatures, in particular at temperatures of less than or equal
to 50°C;
presumably because of the simultaneous presence of a hydroxyl group and of a
carboxyl
group, the basicity of carbonate bases is inadequate at low temperatures.
Carbonate bases
such as potassium carbonate can also be used if the free carboxyl group is
protected with a
suitable protective group, for example an ester as described in WO 00/64876,
before step
25 b) of the process according to the invention is carried out, and is
deprotected again
following step b). Such an additional protection/deprotection of the free
carboxyl group is
also encompassed by the process according to the invention.
Suitable solvents are all solvents or mixtures thereof which are unable to
react with the
3o bases employed:
i) aprotic polar solvents such as acetone and carboxamides, preferably N-
methyl-1-
pyrrolidinone, N,N-dimethylacetamide and dimethylformamide,
35 ii) erotic polar solvents such as, for example, alcohols such as methanol,
ethanol and
tert-butanol.
CA 02550627 2006-06-20
Alcohols and carboxamides are preferred. Carboxamides are particularly
preferred.
The amount of alkylating reagent R-Y of the formula (V) employed is in the
range from
I .0 to 1.5 mole equivalents based on the compound (IV) employed, preferably
between 1.0
and 1.25 mole equivalents. It is also possible where appropriate to employ
more than
1.5 mole equivalents of R-Y. The amount of base B2 employed is at least twice
the molar
amount of compound (IV) employed, with preference for exactly twice the molar
amount
of base B2 in relation to compound (IV).
o The reaction is carried out at temperatures in the range from 20 up to
60°C, preferably
from 20 up to 50°C, particularly preferably between 20 and 25°C,
depending on the
solvent used. The temperatures required by carboxamides as solvents are
generally lower
than by alcohols as solvents. Step b) can also be carried out at temperatures
above 60°C
where appropriate.
l5
The compound (IV) is introduced into the solvent and then the base B 1 is
added. The
mixture is stirred for at least five minutes until both hydroxyl groups have
been completely
deprotonated. The alkylating reagent R-Y of the formula (V) is then added.
2o No special conditions in relation to pressure are necessary; it is
expedient to operate under
atmospheric pressure.
After completion of the reaction, the reaction mixture is diluted for example
with a
solution of sodium chloride, potassium chloride or sodium bicarbonate. The
resulting
25 solution is extracted for example with ethyl acetate, whereupon the organic
constituents in
the reaction solution are transferred into the organic phase. The ethyl
acetate phase is then
extracted with water. The compounds of the general formula (I) are liberated
by acidifying
the aqueous phase, for example, preferably with an inorganic acid such as
hydrochloric
acid or sulfuric acid. Methods known to the skilled worker are used for
further working up.
3o The compounds of the formula (I) according to the invention are obtained in
high yields
and good purities.
The purity of the isolated compounds of the general formula (I) can be
increased where
appropriate by subsequent crystallization.
In a further embodiment of the present compounds it is possible to follow step
b) by
preparing by methods known to the skilled worker - where defined for X1, X2 or
R - from
CA 02550627 2006-06-20
-20-
(-C(O)O-(C,-C~-alkyl)), (-S-(C,-C~-alkyl)) and/or (C,-C~-alkoxy) substituents
by
elimination of alkyl the corresponding compounds of the general formula (I)
substituted by
carboxyl, -SH and/or -OH. These additional substituents may be present
independently of
one another one or more times. The free carboxyl, (-SH) and/or (-OH)
substituents are
preferably obtained by addition of acid. These additional substituents can,
where
appropriate, also be obtained from substituents other than ester, thioalkoxy
or alkoxy
substituents.
The starting materials, solvents, bases etc. used in the process according to
the invention
are all purchasable or can be prepared by methods known to the skilled worker.
A further aspect of the present invention are the compounds of the formula
(IV) which can
be obtained as intermediates in step a) of the process according to the
invention.
X2
n
HO ,~ O
Xim O OH
is (lV)
in which:
X' is halogen, C,-C6-alkyl, C,-C6-alkoxy, trifluoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl may in turn be at least
monosubstituted by C1-C3-
alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
Xz is halogen, C,-C6-alkyl, C,-C6-alkoxy, trifluoromethyl, aryl, heterocyclyl
or heteroaryl,
where aryl, heterocyclyl and heteroaryl may in turn be at least
monosubstituted by C,-C3-
alkyl, C,-C3-alkoxy, halogen or trifluoromethyl;
heteroaryl is a 5- to 14-membered, aromatic, mono-, bi- or tricyclic
heterocycle which
contains one or more heteroatoms selected from N, O and S;
aryl is a 6- to 14-membered, aromatic mono-, bi- or tricyclic system;
CA 02550627 2006-06-20
-21-
heterocyclyl is a 5- to 14-membered, nonaromatic, mono-, bi- or tricyclic
heterocycle
which contains one or more heteroatoms selected from N, O and S;
mis0, 1,2,3or4;
n is (l, I , 2, 3 or 4;
wherein 2-(3-hydroxy-4-methoxyphenoxymethyl)benzoic acid is excluded.
A compound being similar to those of the general formula (IV) is already
known. US
2003/0072842 A and A. Bassoli et al., Quant. Struct.-Act. Relat., 20(2001), p.
3-20
disclose the single compound 2(3-hydroxy-4-methoxyphenoxymethyl)benzoic acid.
This
compound is used as a sweetener, a connection to PPAR-agonists or -antagonists
is not
disclosed. Compounds as such, explicitly disclosed in US 2003/0072842 A or by
A.
Bassoli et al., are not a subject of the present invention.
IS
Preferred compounds of the formula (IV) have the following definition:
X' is halogen, C~-C6-alkyl, C1-C6-alkoxy or trifluoromethyl;
2o X2 is halogen, C~-C6-alkyl, C~-C6-alkoxy or trifluoromethyl;
mis0, 1,2,3or4;
nis0, 1,2,3or4;
25 with the proviso that X~=C,-C6-alkoxy is not in para-position to the ether-
fragment.
More preferred compounds of the formula (IV) have the following definition:
3o
X' is halogen, C,-C6-alkyl, C1-C6-alkoxy or trifluoromethyl;
X2 is halogen, C,-C6-alkyl, C~-C6-alkoxy or trifluoromethyl;
mis0orl;
n is 0, 1 or 2;
with the proviso that X'=C~-C6-alkoxy is not in para-position to the ether-
fragment.
CA 02550627 2006-06-20
-22-
Even more preferred compounds of the formula (IV) have the following
definition:
X~ is halogen, C~-C~-alkyl or trifluoromethyl;
XZ is halogen, C,-C~-alkyl, C,-C~-alkoxy or trifluoromethyl;
mis0or 1;
nis0, lor2.
to Particularly preferred compounds of the formula (IV) have the following
definition:
X~ is C,-C~-alkyl or bromine;
XZ is C,-C~-alkyl, fluorine or chlorine;
IS mis0or l;
n is 0, 1 or 2.
Very particularly preferred compounds of the formula (IV) have the following
definition:
2o Xz is C,-C3-alkyl;
mis0;
nis0orl.
25 As described above, the compounds of the formula (IV) can be prepared by
step a) of the
process according to the invention. They are suitable as starting compounds
for
synthesizing PPAR agonists and antagonists of general formula (I) by step b)
of the
process according to the invention.
3o The following examples are intended to illustrate the process without,
however, limiting it.
CA 02550627 2006-06-20
-23-
1. Preparation of the compounds of the formula (IV)
Example 1.1: 2-( 3-H_ydroxyphenox~methyl)-6-methylbenzoic acid and the
solvates thereof
with N-methyl~yrrolidone
2-(3-H~dro~phenox~methyl)-6-methylbenzoic acid (monosolyate with N-methyl-
py-rrolidone)
7-Methylphthalide (30.1 g, 203 mmol), resorcinol (45.3 g, 407 mmol) and
potassium tert-
to butoxide (47.6 g, 424 mmol) are suspended in 300 ml of N-methylpyrrolidone
(NMP) and
stirred at 140°C for 21 h. After this time, a second amount of
potassium tert-butoxide (2.4
g, 21.4 mmol) is added and stirring is continued at 140°C for 2.5 h.
The reaction mixture is
cooled, 800 ml of water are added, and the mixture is acidified to pH = 2 at
20°C with
concentrated hydrochloric acid. After stirring at 3 to 5°C for 2 hours,
the pale beige solid is
t5 filtered off with suction, washed with water and then dried. The isolated
product contains
2-(3-hydroxyphenoxymethyl)-6-methylbenzoic acid and N-methylpyrrolidone as
solvate
molecule in a ratio of 1:1. Yield 77.7 %, melting point 98 to 99°C.
~ H NMR (400 MHz, DMSO-d6)b
a) 2-(3-H~,droxyphenoxymethyl)-6-methylbenzoic acid
13.3 (br s, I H, COOH), 9.4 (br s 1 H, OH), 7.34 (d, 1 H) 7.33 (s, 1 H), 7.25
(m, 1 H), 7.04 (t,
I H), 6.40 - 6.37 (2H), 5.04 (s, 2H, CH2), 2.69 (s, 3H, CH3)
b) N-Methylpyrrolidone
3.30 (t, 2H), 2.233 (s, 3H), 2.18 (t, 2H), 1.90 (m, 2H)
2-(3-Hydroxyphenoxymethyl)-6-methylbenzaic acid (hemisolvate with N-
methylpyrrol-
idone
53.5 g ( 150 mmol) of 2-(3-hydroxyphenoxymethyl)-6-methylbenzoic acid * NMP
are
dissolved in 250 ml of toluene with heating and cooled at 10 K/h to room
temperature.
This is followed by stirring at 3-5°C for one hour. The crystals are
filtered off with suction
and washed with toluene. The hemisolvate of 2-(3-hydroxyphenoxymethyl)-6-
methyl-
benzoic acid * 0.5 NMP is obtained in a yield of 82%; melting point 112 to
113°C.
CA 02550627 2006-06-20
-24-
2-(3-Hydrox~nhenoxymethyl)-6-methylbenzoic acid (nonsolvate)
5.0 g ( 14 mmol) of 2-(3-hydroxyphenoxymethyl)-6-methylbenzoic acid * NMP are
heated
under reflux in 50 rnl of water for two hours. After cooling, the resulting
solid is altered
off with suction and washed with water. 2-(3-Hydroxyphenoxymethyl)-6-
methylbenzoic
acid is obtained in a yield of 86.4%; melting point 154 to 155°C.
Example 1.2: 2-(3-H droxyphenox~yl)benzoic acid
Phthalide ( 15.0 g, 112 mmol), resorcinol (23.6 g, 214 mmol) and potassium
tert-butoxide
(47.6 g, 220 mmol) are suspended in 150 ml of N-methylpyrrolidone (NMP) and
stirred at
140°C for 5 h. The reaction mixture is cooled, 400 ml of water are
added, and the mixture
is acidified to pH = 4 at 20°C with concentrated hydrochloric acid. The
solution is
is extracted once with 200 ml of ethyl acetate and four times with 100 ml of
ethyl acetate.
The combined ethyl acetate phases are dried over magnesium sulfate and
evaporated to
dryness. A pale brown oil is obtained and crystallizes at 0°C. The
crystals comprise a
mixture of 2-(3-hydroxyphenoxymethyl)benzoic acid and the dialkylated
resorcinol. This
mixture is heated with 10 times the amount of ethyl acetate to dissolve the 2-
(3-hydroxy-
2o phenoxymethyl)benzoic acid, the dialkylated product remaining as solid. The
ethyl acetate
phase is evaporated to dryness, whereupon 2-(3-hydroxyphenoxymethyl)benzoic
acid
remains as pale beige crystals.
~H NMR (400 MHz, DMSO-d6)b
13.1 (br s, 1H, COOH), 9.4 (br s 1H, OH), 7.92 (q, 1H), 7.64 - 7.57 (2H), 7.43
(dt, 1H),
7.06 (m, 1H), 6.41- 6.34 (3H), 5.34 (s, 2H, CH2)
b) Correspondingly overalkylated~roduct of the formula (IV)
13.1 (br s 2H, COOH), 7.94 (d, 2H), 7.66 - 7.58 (4H), 7.45 (d, 2H), 7.21 (t,
1H), 6.62 -
6.58 (2H), 5.44 (s, 4H, CHZ)
CA 02550627 2006-06-20
-25-
Example 1.3: 2-(3-Hydroxyphenoxymethyl)-6-methyl-benzoic acid and the solvate
thereof
with N,N-dimethylacetamide
2-(3-Hydroxynhenox~methyl)-6-methyl-benzoic acid (solvate with N,N-
dimethylacetamide
S in the ratio 5:2)
The procedure takes place in analogy to Example 1.1, but N,N-dimethylacetamide
(DMAA) is employed in place of N-methylpyrrolidone (NMP). The product is
purified by
recrystallization from toluene. Both the crude product and the recrystallized
product have a
2-(3-hydroxyphenoxymethyl)-6-methyl-benzoic acid : DMAA ratio of 5:2. The
yield is
60.0%, melting point 112 - 114°C.
2. Preparation of compounds of the general formula (I)
~5 Example 2.1: 2-43-f2-(4-Fluorophenyl)oxazol-4-ylmethoxylphenoxymethyl~-6-
methyl-
benzoic acid
a) in N-methylpyrrolidone as solvent
20 18.6 g of 30% strength solution of sodium methoxide in methanol ( 104 mmol)
are added to
15.9 g (51.6 mmol) of 2-(3-hydroxyphenoxymethyl)-6-methyl-benzoic acid
(hemisolvate
with N-methylpyrrolidone) in 70 ml of N-methylpyrrolidone while stirring at
room
temperature. After five minutes, 10.0 g (47.3 mmol) of 4-chloromethyl-2-(4-
fluorophenyl)oxazole are added, and the mixture is stirred at room temperature
for 23 h
25 and finally heated at 50°C for one hour. 500 ml of 19% strength
sodium chloride solution
are added to the reaction solution, and the resulting mixture is extracted
twice with 225 ml
of ethyl acetate. The combined ethyl acetate phases are extracted three times
with 225 ml
of water, and the combined aqueous phases are acidified to pH = 3 with 2 M
hydrochloric
acid. The resulting crystals are filtered off with suction and washed with
water. The
30 product is purified further by crystallization from isopropanol. 2-{3-[2-(4-
Fluorophenyl)oxazol-4-ylmethoxy]phenoxymethyl}-6-methylbenzoic acid is
obtained as
colorless crystals in a yield of 58.3%, melting point 168 - 173°C.
~H NMR (400 MHz, DMSO-d6)8
CA 02550627 2006-06-20
-26-
13.2 (br s, 1H, COOH), 8.29 (s, IH, oxazole H), 8.04 (m, 2H, fluorophenyl H),
7.39 (m,
2H, fluorophenyl H), 7.35 - 7.31 (m, 2H), 7.25 (m, 1H), 7.20 (m, 1H), 6.67 -
6.65 (m, 2H),
6.58 (dd, 1 H), 5.11 (s, 2H, CHZ), 5.03 (s, 2H, CHZ), 2.33 (s, 3H, CH3)
b) in methanol as solvent
The reaction is carried out as described under a). The reaction temperature is
50°C over the
entire period of 24 hours. 2-{ 3-[2-phenyloxazol-4-ylmethoxy]phenoxymethyl }-6-
methylbenzoic acid is obtained as colorless crystals in a yield of 53.0%.
0
Example 2.2: 6-MethXl-2-f 3-(2-phenyloxazol-4-ylmethoxy~phenox~methyllbenzoic
acid
The procedure takes place in analogy to Example 2.1, but 4-chloromethyl-2-
phenyloxazole
~ 5 is employed in place of 4-chloromethyl-2-(4-fluorophenyl)oxazole. 2-{ 3-[2-
(4-
Fluorophenyl)oxazol-4-ylmethoxy]phenoxymethyl }-6-methylbenzoic acid is
obtained as
colorless crystals in a yield of 45.0%.
'H NMR (400 MHz, DMSO-d6)8
13.2 (br s, 1H, COOH), 8.29 (s, 1H, oxazole H), 8.02 - 8.00 (m, 2H), 7.56 -
7.52 (m, 3H),
7.37 - 7.19 (4H), 6.69 - 6.59 (3H), 5.13 (s, 2H, CH2), 5.05 (s, 2H, CHZ), 2.35
(s, 3H, CH3)
Example 2.3: 2-(3-Benzyloxyphenoxymethyl)-6-methylbenzoic acid
The procedure takes place in analogy to Example 2.1, but benzyl bromide is
employed in
place of 4-chloromethyl-2-(4-fluorophenyl)oxazole. 2-(3-
Benzyloxyphenoxymethyl)-6-
methylbenzoic acid is obtained as colorless crystals in a yield of 47.3%.
'H NMR (400 MHz, DMSO-d6)8
13.2 (br s, 1H, COOH), 7.45 - 7.30 (7H), 7.25 (m, 1H), 7.18 (t, IH), 6.63 -
6.55 (3H), 5.10
(s, 2H, CH2), 5.07 (s, 2H, CHZ), 5.34 (s, 3H, CH3)
CA 02550627 2006-06-20
-27-
Comuarative Example 2.4: (2-~3-~2-(4-Fluoronhenyl)oxazol-4-ylmethoxylphenoxy-
methyl?-6-methylbenzoic acid
1 g (6.$ mmol) of 7-methylphthalide and 2 g (7.0 mmol) of 3-[2-(4-
fluorophenyl)oxazol-4-
ylmethoxy]phenol are dissolved in NMP. 3.9 ml of methanolic sodium methoxide
(30%
strength) are added, the methanol is distilled off, and the mixture is heated
to 130°C. No
formation of 2-{3-[2-(4-fluorophenyl)oxazol-4-ylmethoxy]phenoxymethyl}-6-
methyl-
benzoic acid is observed. The 3-[2-(4-fluorophenyl)oxazol-4-ylmethoxy]phenol
decomposes.
to