Note: Descriptions are shown in the official language in which they were submitted.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
TRIAZOLE DERIVATIVES AS VASOPRESSIN ANTAGONISTS
This invention relates to triazole derivatives and to processes for the
preparation of,
intermediates used in the preparation of, compositions containing and the uses
of, such
derivatives.
The triazole derivatives of the present invention are vasopressin antagonists.
In
particular they are antagonists of the V1 a receptor and have a number of
therapeutic
applications, particularly in the treatment of dysmenorrhoea (primary and
secondary).
There is a high unmet need in the area of menstrual disorders and it is
estimated that
up to 90% of all menstruating women are affected to some degree. Up to 42% of
women miss work or other activities due to menstrual pain and it has been
estimated
that around 600 million work hours a year are lost in the US as a result
{Coco, A.S.
(1999). Primary dysmenorrhoea. [Review] [30 refs]. American Family Physician,
60,
489-96.}.
Menstrual pain in the lower abdomen is caused by myometrial hyperactivity and
reduced uterine blood flow. These pathophysiological changes result in
abdominal pain
that radiates out to the back and legs. This may result in women feeling
nauseous,
having headaches and suffering from insomnia. This condition is called
dysmenorrhoea
and can be classified as either primary or secondary dysmenorrhoea.
Primary dysmenorrhoea is diagnosed when no abnormality causing the condition
is
identified. This affects up to 50% of the female population {Coco, A.S.
(1999). Primary
dysmenorrhoea. [Review] [30 refs]. American Family Physician, 60, 489-96.;
Schroeder,
B. & Sanfilippo, J.S. (1999). Dysmenorrhoea and pelvic pain in adolescents.
[Review]
[78 refs]. Pediatric Clinics of North America, 46, 555-71}. Where an
underlying
gynaecological disorder is present, such as endometriosis, pelvic inflammatory
disease
(PID), fibroids or cancers, secondary dysmenorrhoea will be diagnosed.
Secondary
dysmenorrhoea is diagnosed in only approximately 25% of women suffering from
dysmenorrhoea. Dysmenorrhoea can occur in conjunction with menorrhagia, which
accounts for around 12% of referrals to gynaecology outpatients departments.
Currently, women suffering from primary dysmenorrhoea are treated with non-
steroidal
anti-inflammatory drugs (NSAID's) or the oral contraceptive pill. In cases of
secondary
dysmenorrhoea surgery may be undertaken to correct the underlying
gynaecological
disorder.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-2-
Women suffering from dysmenorrhoea have circulating vasopressin levels which
are
greater than those observed in healthy women at the same time of the menstrual
cycle.
Inhibition of the pharmacological actions of vasopressin, at the uterine
vasopressin
receptor, may prevent dysmenorrhoea.
The compounds of the present invention are therefore potentially useful in the
treatment
of a wide range of disorders, particularly aggression, Alzheimer's disease,
anorexia
nervosa, anxiety, anxiety disorder, asthma, atherosclerosis, autism,
cardiovascular
disease (including angina, atherosclerosis, hypertension, heart failure,
edema,
hypernatremia), cataract, central nervous system disease, cerebrovascular
ischemia,
cirrhosis, cognitive disorder, Cushing's disease, depression, diabetes
mellitus,
dysmenorrhoea (primary and secondary), emesis (including motion sickness),
endometriosis, gastrointestinal disease, glaucoma, gynaecological disease,
heart
disease, intrauterine growth retardation, inflammation (including rheumatoid
arthritis),
ischemia, ischemic heart disease, lung tumor, micturition disorder,
mittlesmerchz,
neoplasm, nephrotoxicity, non-insulin dependent diabetes, obesity,
obsessive/compulsive disorder, ocular hypertension, preclampsia, premature
ejaculation,
premature (preterm) labour, pulmonary disease, Raynaud's disease, renal
disease, renal
failure, male or female sexual dysfunction, septic shock, sleep disorder,
spinal cord
injury, thrombosis, urogenital tract infection or urolithiasis.
Particularly of interest are the following diseases or disorders:
anxiety, cardiovascular disease (including angina, atherosclerosis,
hypertension, heart
failure, edema, hypernatremia), dysmenorrhoea (primary and secondary),
endometriosis, emesis (including motion sickness), intrauterine growth
retardation,
inflammation (including rheumatoid arthritis), mittlesmerchz, preclampsia,
premature
ejaculation, premature (preterm) labour and Raynaud's disease.
The compounds of the invention, and their pharmaceutically acceptable salts
and
solvates, have the advantage that they are selective inhibitors of the V1 a
receptor (and
so are likely to have reduced side effects), they may have a more rapid onset
of action,
they may be more potent, they may be longer acting, they may have greater
bioavailability or they my have other more desirable properties than the
compounds of
the prior art.
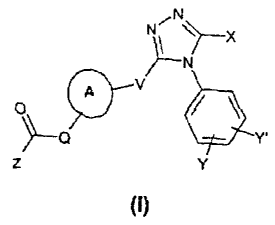
According to the present invention there is provided a compound of formula
(I),
CA 02551038 2006-06-21
64680-1603
-3-
,N
Nõ X
o / V
--'
z a Y.
Y
or a pharmaceutically acceptable derivative thereof, wherein:
X represents -[CHz]e-R or -[CHz]e O-[CH2]b-R;
a represents a number selected from 0 to 6;
b represents a number selected from 0 to 6;
R represents H, CF3 or Het;
Het represents a 5- or 6-membered saturated, partially saturated or aromatic
heterocyclic ring comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen
atom or 1
sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen
atoms,
optionally substituted with one or more groups independently selected from W;
Y and Y independently represent one or more substituents independently
selected from -[O]c[CHz]rR',
which may be the same or different at each occurrence;
c at each occurrence independently represents a number selected from 0 or 1;
d at each occurrence independently represents a number selected from 0 to 6;
R' at each occurrence independently represents H, halo, CF3, CN or Het';
Het' at each occurrence independently represents a 5- or 6-membered
unsaturated heterocyclic ring, comprising either (a) I to 4 nitrogen atoms,
(b) 1 oxygen
atom or 1 sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2
nitrogen
atoms;
V represents a direct link or -0-;
Ring A represents a 5- to 7-membered saturated heterocyclic ring comprising
either (a)
1 to 4 nitrogen atoms, (b) 1 oxygen atom or 1 sulphur atom, or (c) 1 oxygen
atom or 1
sulphur atom and 1 or 2 nitrogen atoms, or it represents a phenylene group;
ring A
being optionally substituted with one or more groups selected from C,-8 alkyl,
phenyl or
hydroxy;
Q represents a direct link or -N(R2)-;
R 2 represents hydrogen or C,$ alkyl;
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-4-
Z represents -[O]e [CHJrR3, a phenyl ring (optionally fused to a benzene ring
or Het2,
and the group as a whole being optionally substituted with one or more groups
independently selected from W), or Het3 (optionally fused to an benzene ring
or Het4,
and the group as a whole being optionally substituted with one or more groups
independently selected from W);
R3 represents C1_6 alkyl (optionally substituted with one or more groups
independently selected from W), C3_6 cycloalkyl, C3_6 cycloalkenyl, phenyl
(optionally
substituted with one or more groups independently selected from W), Het5 or
NR4R5;
e represents a number selected from 0 or 1;
f represents a number selected from 0 to 6;
Het2 and Het5 independently represent a 5- or 6-membered saturated, partially
saturated or aromatic heterocyclic ring, comprising either (a) 1 to 4 nitrogen
atoms, (b) 1
oxygen atom or 1 sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or
2
nitrogen atoms, optionally substituted with one or more groups selected from
W;
Het3 represents a 4 to 6-membered saturated, partially saturated or aromatic
heterocyclic ring, comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen
atom or 1
sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen
atoms,
optionally substituted with one or more groups selected from W;
Het4 represents a 6-membered aromatic heterocyclic ring, comprising either (a)
1
to 4 nitrogen atoms, (b) 1 oxygen atom or 1 sulphur atom, or (c) 1 oxygen atom
or 1
sulphur atom and 1 or 2 nitrogen atoms, optionally substituted with one or
more groups
selected from W;
R4 and R5 independently represent hydrogen, C1_6 alkyl, Cl-6 alkyloxy, C3-8
cycloalkyl (optionally fused to C3$ cycloalkyl) or Het6;
R4 and R5 being optionally independently substituted with one of more groups
selected from C,-6 alkyl, C1_6 alkyloxy, C3$ cycloalkyl (optionally fused to
C3$ cycloalkyl),
or phenyl;
Hets represents a 5- or 6-membered saturated, partially saturated or aromatic
heterocyclic ring, comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen
atom or 1
sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen
atoms,
optionally substituted with one or more groups selected from W;
W independently at each occurrence represents halo, [O]gRs, SO2 R6, SR6,
SO2NR6R',
[O]n[CHz];CF3, [O];CHF2i phenyl (optionally substituted with halo, C,-6 alkyl
or C,-6
alkyloxy), CN, phenoxy (optionally substituted with halo), OH, benzyl, NR6R7 ,
NCOR 6,
benzyloxy, oxo, CONHR6, NSO2R6 R7, COR 6, C1_ealkylene-NCOR7 , Het7;
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-5-
R6 represents hydrogen, C,_salkyl, C3_6cycloalkyl, C3-6 cycloalkenyl or C,-
6alkylene-
O-C1_6alkyl;
R' represents hydrogen or C,_galkyl;
i represents a number selected from 0 to 6
h represents a number selected from 0 or 1;
g represents a number selected from 0 or 1;
j represents a number selected from 0 or 1;
Het7 represents a 5- or 6-membered saturated, partially saturated or aromatic
heterocyclic ring, comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen
atom or 1
sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen
atoms,
optionally substituted by R6 and/or R' and/or an oxo group.
In an alternative embodiment there is provided a compound of formula (I'):
N
NI ~-x
N
A
N
Z_ Y.
0 Y
(I')
or a pharmaceutically acceptable derivative thereof, wherein:
X represents -[CHJa-R or -[CH2]a-O-[CH2]b-R;
a represents a number selected from 0 to 6;
b represents a number selected from 0 to 6;
R represents H, CF3 or Het;
Het represents a 5- or 6-membered heterocyclic ring comprising either (a) 1
to 4 nitrogen atoms, (b) 1 oxygen atom or 1 sulphur atom, or (c) 1 oxygen atom
or 1
sulphur atom and 1 or 2 nitrogen atoms;
Y represents -[O]c-[CHz]d-R';
Y' represents -[O]c'-[CH2]d'-R1';
c and c' independently represent a number selected from 0 or 1;
d and d' independently represent a number selected from 0 to 6;
R' and R" independently represent H, halo, CF3 or Het';
Het' represents a 5- or 6-membered unsaturated heterocyclic ring
comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen atom or 1 sulphur
atom, or (c) I
oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms;
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-6-
Ring A represents a 5- or 6-membered saturated heterocyclic ring comprising at
least
one nitrogen atom;
Z represents -[O]e [CHz]f-R2, a phenyl ring (optionally fused to a phenyl ring
or a 5- or 6-
membered saturated, partially unsaturated or aromatic heterocyclic ring,
and/or
optionally substituted with one or more groups independently selected from W),
or a 6-
membered aromatic heterocyclic ring (optionally fused to an phenyl ring or a 6-
membered aromatic heterocyclic ring, and/or optionally substituted with one or
more
groups independently selected from W);
R2 represents C1_6 alkyl or C3_6 cycloalkyl;
e represents a number selected from 0 or 1;
f represents a number selected from 0 to 6;
W represents halo, [O]9R3, S02R3, SR3, SO2NR3R4, [O]h[CH2];CF3, OCHF2, phenyl,
CN,
phenoxy (optionally substituted with halo), OH, benzyl, NCOR3, benzyloxy, oxy,
CONHR3, NSO2R3R4, COR3, C1_6alkylene-NCOR3, Het2;
R3 represents hydrogen, C1_6alkyl, C3_6cycloalkyl or C,-6alkylene-O-C,-6alkyl;
R4 represents hydrogen or C1_6alkyl;
i represents a number selected from 0 to 6
h represents a number selected from 0 or 1;
g represents a number selected from 0 or 1;
Het2 represents a 5- or 6-membered saturated, partially unsaturated or
aromatic
heterocyclic group comprising either (a) 1 to 4 nitrogen atoms, (b) 1 oxygen
atom or 1
sulphur atom, or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen
atoms, the
heterocyclic group being optionally substituted by R3 and/or R4 and/or an oxy
group.
In the above definitions, halo means fluoro, chloro, bromo or iodo. Alkyl,
alkylene and
alkyloxy groups, containing the requisite number of carbon atoms, can be
unbranched or
branched. Examples of alkyl include methyl, ethyl, n-propyl, i-propyl, n-
butyl, i-butyl,
sec-butyl and t-butyl. Examples of alkyloxy include methoxy, ethoxy, n-
propoxy, i-
propoxy, n-butoxy, 1-butoxy, sec-butoxy and t-butoxy. Examples of alkylene
include
methylene, 1,1-ethylene, 1,2-ethylene, 1,1-propylene, 1,2-propylene, 1,3-
propylene and
2,2-propylene. Het represents a heterocyclic group, examples of which include
tetrahydrofuranyl, tetrahydrothiophenyl, pyrrolidinyl, tetrahydropyranyl,
tetrahydrothiopyranyl, piperidinyl, 1,4-dioxanyl, 1,4-oxathianyl, morpholinyl,
1,4-dithianyl,
piperazinyl, 1,4-azathianyl, 3,4-dihydro-2H-pyranyl, 5,6-dihydro-2H-pyranyl,
2H-pyranyl,
1,2,3,4-tetrahydropyridinyl, 1,2,5,6-tetrahydropyridinyl, pyrrolyl, furanyl,
thiophenyl,
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-7-
pyrazolyl, imidazolyl, isoxazolyl, oxazolyl, isothiazolyl, thiazolyl, 1,2,3-
triazolyl, 1,3,4-
triazolyl, 1-oxa-2,3-diazolyl, 1-oxa-2,4-diazolyl, 1-oxa-2,5-diazolyl, 1-oxa-
3,4-diazolyl, 1-
thia-2,3-diazolyl, 1-thia-2,4-diazolyl, 1-thia-2,5-diazolyl, 1-thia-3,4-
diazolyl, tetrazolyl,
pyridinyl, pyridazinyl, pyrimidinyl and pyrazinyl.
A preferred compound is one in which X represents CH2OCH3. More preferred is a
compound in which X represents -[CHz]aR.
A preferred compound is one in which a represents a number selected from 0 to
5. More
preferred is a compound in which a represents a number selected from 0 to 4.
Still more
preferred is a compound in which a represents a.number selected from 0 to 3.
Still more
preferred is a compound in which a represents a number selected from 0 to 2.
Most
preferred is a compound in which a represents the number 1.
A preferred compound is one in which R represents H. A more preferred compound
is
one in which R represents Het. Stoll more preferred is a compound in which R
represents triazolyl
A preferred compound is one in which Y represents one or two substituents. A
more
preferred compound is one in which Y represents a single substituent.
A preferred compound is one in which Y represents halo. A more preferred
compound is
one in which Y represents chloro and/or fluoro.
A preferred compound is one in which V represents a direct link. A preferred
compound
is one in which Q is a direct link. A more preferred compound is one in which
both V and
Q represent a direct link.
A preferred compound is one in which Ring A contains 2 nitrogen atoms. A more
preferred compound is one in which Ring A contains I nitrogen atom.
A preferred compound is one in which Ring A represents a 5-membered ring. A
more
preferred compound is one in which Ring A represents a 6-membered ring. A
still more
preferred compound is one in which Ring A represents piperidinylene.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-8-
A preferred compound is one in which Ring A is attached to V via a nitrogen
atom. A
more preferred compound is one in which Ring A is attached to Q via a nitrogen
atom.
A preferred compound is one in which Ring A is attached to both Q and V via a
nitrogen
atom.
A preferred compound is one in which Z represents Het3. Het3 may represent an
optionally substituted group selected from indazolyl, indolyl, indenyl,
pyrazolyl,
piperidinyl, pyridinyl, pyrimidinyl, pyrrolyl, thiazolyl, benzothienyl,
benzothiazolyl,
quinolinyl, benzoxazinyl, isoxazolyl, imidazolyl, furyl, benzofuryl,
cinnolinyl, morpholinyl,
chromenyl, or derivatives thereof. A more preferred compound is one in which Z
represents phenyl.
A preferred compound is one in which Z is mono or di substituted. A more
preferred
compound is one in which Z is mono substituted.
A preferred compound is one in which Z is substituted by tri-fluoromethyl. A
more
preferred compound is one in which Z is substituted by halo. A more preferred
compound is one in which Z is substituted by chloro and/or fluoro.
Specific preferred compounds according to the invention are those listed in
the
Examples section below, and the pharmaceutically acceptable salts thereof. In
particular:
(3-Chloro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
(4-Chloro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
(5-Chloro-2-fluoro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-
4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(3,5-difluoro-phenyl)-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yI}-(3-fluoro-phenyl)-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(2,3-difluoro-phenyl)-methanone;
(3-Chloro-2-fluoro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazoi-2-ylmethyl-
4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-9-
(3-Chloro-4-fluoro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-
4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-l-
yl}-(4-trifluoromethyl-phenyl)-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(3-trifluoromethyl-phenyl)-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(2-trifluoromethyl-phenyl)-methanone;
(3-Chloro-5-fluoro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-
4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(4-difluoromethyl-phenyl)-methanone;
{4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-
piperidin-1-
yl}-(1 H-indazol-3-yl)-methanone;
and pharmaceutically acceptable derivatives thereof.
Pharmaceutically acceptable derivatives of the compounds of formula (I)
according to
the invention include salts, solvates, complexes, polymorphs, prodrugs,
stereoisomers,
geometric isomers, tautomeric forms, and isotopic variations of compounds of
formula
(I). Preferably, pharmaceutically acceptable derivatives of compounds of
formula (I)
comprise salts, solvates, esters and amides of the compounds of formula (I).
More
preferably, pharmaceutically acceptable derivatives of compounds of formula
(I) are salts
and solvates.
The pharmaceutically acceptable salts of the compounds of formula (I) include
the acid
addition and base salts thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts.
Examples
include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate,
bisulphate,
borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate,
gluconate,
glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride,
hydrobromide/bromide, hydroiodide/iodide, isethionate, D- and L-lactate,
malate,
maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate,
nicotinate,
nitrate, orotate, oxalate, palmitate, palmoate, phosphate, hydrogen phosphate,
dihydrogen phosphate, saccharate, stearate, succinate, sulphate, D- and L-
tartrate,
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-10-
tosylate and trifluoroacetate salts. A particularly suitable salt is the
besylate derivative of
the compounds of the present invention.
Suitable base salts are formed from bases, which form non-toxic salts.
Examples
include the aluminium, arginine, benzathine, calcium, choline, diethylamine,
diolamine,
glycine, lysine, magnesium, meglumine, olamine, potassium, sodium,
tromethamine and
zinc salts.
For a review on suitable salts see Stahl and Wermuth, Handbook of
Pharmaceutical
Salts: Properties, Selection and Use, Wiley-VCH, Weinheim, Germany (2002).
A pharmaceutically acceptable salt of a compound of formula (I) may be readily
prepared by mixing together solutions of the compound of formula (I) and the
desired
acid or base, as appropriate. The salt may precipitate from solution and be
collected by
filtration or may be recovered by evaporation of the solvent. The degree of
ionisation in
the salt may vary from completely ionised to almost non-ionised.
The compounds of the invention may exist in both unsolvated and solvated
forms. The
term "solvate" is used herein to describe a molecular complex comprising the
compound
of the invention and one or more pharmaceutically acceptable solvent
molecules, for
example, ethanol. The term "hydrate" is employed when said solvent is water.
Included within the scope of the invention are complexes such as clathrates,
drug-host
inclusion complexes wherein, in contrast to the aforementioned solvates, the
drug and
host are present in stoichiometric or non-stoichiometric amounts. Also
included are
complexes of the drug containing two or more organic and/or inorganic
components
what may be in stoichiometric or non-stoichiometric amounts. The resulting
complexes
may be ionised, partially ionised, or non-ionised. For a review of such
complexes, see J
Pharm Sci, 64 (8), 1269-1288 by Haleblian (August 1975).
Hereinafter all references to compounds of formula (I) and pharmaceutically
acceptable
derivatives include references to salts, solvates and complexes thereof and to
solvates
and complexes of salts thereof.
The compounds of the invention include compounds of formula (I) as
hereinbefore
defined, polymorphs, prodrugs, and isomers thereof (including optical,
geometric and
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-11-
tautomeric isomers) as hereinafter defined and isotopically-labelled compounds
of
formula (I).
As stated, the invention includes all polymorphs of the compounds of formula
(I) as
hereinbefore defined.
Also within the scope of the invention are so-called "prodrugs" of the
compounds of
formula (I). Thus certain derivatives of compounds of formula (I) which may
have little or
no pharmacological activity themselves can, when administered into or onto the
body, be
converted into compounds of formula (I) having the desired activity, for
example,
hydrolytic cleavage. Such derivatives are referred to as "prodrugs". Further
information
on the use of prodrugs may be found in "Pro-drugs as Novel Delivery Systems,
Vol. 14,
ACS Symposium Series (T Higuchi and W Stella) and "Bioreversible Carriers in
Drug
Design", Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical
Association).
Prodrugs in accordance with the invention can, for example, be produced by
replacing
appropriate functionalities present in the compounds of formula (I) with
certain moieties
know to those skilled in the art as "pro-moieties" as described, for example,
in "Design of
Prodrugs" by H Bundgaard (Elsevier, 1985).
Some examples of prodrugs in accordance with the invention include:
(i) where the compound of formula (I) contains a carboxylic acid functionality
(-
COOH), an ester thereof, for example, replacement of the hydrogen with C,-8
alkyl;
(ii) where the compound of formula (I) contains an alcohol functionality (-
OH), an
ether thereof, for example, replacement of the hydrogen with C,-6
alkanoyloxymethyl; and
(iii) where the compound of formula (I) contains a primary or secondary amino
functionality (-NH2 or -NHR where R# H), an amide thereof, for example,
replacement of one or both hydrogens with C,-,o alkanoyl.
Further examples of replacement groups in accordance with the foregoing
examples and
examples of other prodrug types may be found in the aforementioned references.
Finally, certain compounds of formula (I) may themselves act as prodrugs of
other
compounds of formula (I).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-12-
Also within the scope of the invention are the metabolites of the compounds of
formula
(I) when formed in vivo.
Compounds of formula (I) containing one or more asymmetric carbon atoms can
exist as
two or more stereoisomers. Where a compound of formula (I) contains an alkenyl
or
alkenylene group, geometric cis/trans (or Z/E) isomers are possible, and where
the
compound contains, for example, a keto or oxime group or an aromatic moiety,
tautomeric isomerism ('tautomerism') may occur. It follows that a single
compound may
exhibit more than one type of isomerism.
Included within the scope of the present invention are all stereoisomers,
geometric
isomers and tautomeric forms of the compounds of formula (I), including
compounds
exhibiting more than one type of isomerism, and mixtures of one or more
thereof. Also
included are acid addition or base salts wherein the counter ion is optically
active, for
example, D-lactate or L-lysine, or racemic, for example, DL-tartrate or DL-
arginine.
Cis/trans isomers may be separated by conventional techniques well known to
those
skilled in the art, for example, fractional crystallisation and
chromatography.
Conventional techniques for the preparation/isolation of individual
enantiomers include
chiral synthesis from a suitable optically pure precursor or resolution of the
racemate (or
the racemate of a salt or derivative) using, for example, chiral HPLC.
Alternatively, the racemate (or racemic precursor) may be reacted with a
suitable
optically active compound, for example, an alcohol, or, in the case where the
compounds
of formula (i) contains an acidic or basic moiety, an acid or base such as
tartaric acid or
1 -phenylethylamine. The resulting diastereomeric mixture may be separated by
chromatography and/or fractional crystallisation and one or both of the
diastereomers
converted to the corresponding pure enantiomer(s) by means well known to a
skilled
person.
Chiral compounds of the invention (and chiral precursors thereof) may be
obtained in
enantiomerically-enriched form using chromatography, typically HPLC, on an
asymmetric resin with a mobile phase consisting of a hydrocarbon, typically
heptane or
hexane, containing from 0 to 50% isopropanol, typically from 2 to 20%, and
from 0 to 5%
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-13-
of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate
affords the
enriched mixture.
Stereoisomeric conglomerates may be separated by conventional techniques known
to
those skilled in the art - see, for example, "Stereochemistry of Organic
Compounds" by
E L Eliel (Wiley, New York, 1994).
The present invention also includes all pharmaceutically acceptable isotopic
variations of
a compound of the formula (I) one or more atoms is replaced by atoms having
the same
atomic number, but an atomic mass or mass number different from the atomic
mass or
mass number usually found in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention
include
isotopes of hydrogen such as 2H and 3H, carbon such as "C, 13C and 14C,
nitrogen such
as13N and15N, oxygen such as150, "O and180, phosphorus such as 32P, sulphur
such
as 35S, fluorine such as'$F, iodine such as1231 and'251, and chlorine such as
36CI.
Certain isotopically-labelled compounds of formula (I), for example those
incorporating a
radioactive isotope, are useful in drug and/or substrate tissue distribution
studies. The
radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are
particularly useful for this
purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford
certain
therapeutic advantages resulting from greater metabolic stability, for
example, increased
in vivo half-life or reduced dosage requirements, and hence may be preferred
in some
circumstances.
Substitution with positron emitting isotopes, such as "C, 18F, 150 and 13N,
can be useful
in Positron Emission Topography (PET) studies for examining substrate receptor
occupancy.
Isotopically-labelled compounds of formula (I) can generally be prepared by
conventional
techniques known to those skilled in the art or by processes analogous to
those
described in the accompanying Examples and Preparations using appropriate
isotopically-labelled reagents in place of the non-labelled reagent previously
employed.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-14-
Pharmaceutically acceptable solvates in accordance with the invention include
those
wherein the solvent of crystallisation may be isotopically substituted, e.g.
D20, d6-
acetone and ds-DMSO.
The compounds of the invention are useful in therapy. Therefore, a further
aspect of the
invention is the use of a compound of formula (I), or a pharmaceutically salt
or solvate
thereof, as a medicament.
The compounds of the invention show activity as V1a antagonists. In particular
they are
useful in the treatment of a number of conditions including aggression,
Alzheimer's
disease, anorexia nervosa, anxiety, anxiety disorder, asthma, atherosclerosis,
autism,
cardiovascular disease (including angina, atherosclerosis, hypertension, heart
failure,
edema, hypernatremia), cataract, central nervous system disease,
cerebrovascular
ischemia, cirrhosis, cognitive disorder, Cushing's disease, depression,
diabetes mellitus,
dysmenorrhoea (primary and secondary), emesis (including motion sickness),
endometriosis, gastrointestinal disease, glaucoma, gynaecological disease,
heart
disease, intrauterine growth retardation, inflammation (including rheumatoid
arthritis),
ischemia, ischemic heart disease, lung tumor, micturition disorder,
mittlesmerchz,
neoplasm, nephrotoxicity, non-insulin dependent diabetes, obesity,
obsessive/compulsive disorder, ocular hypertension, preciampsia, premature
ejaculation,
premature (preterm) labor, pulmonary disease, Raynaud's disease, renal
disease, renal
failure, male or female sexual dysfunction, septic shock, sleep disorder,
spinal cord
injury, thrombosis, urogenital tract infection or urolithiasis.sleep disorder,
spinal cord
injury, thrombosis, urogenital tract infection, urolithiasis. Particularly of
interest is
dysmenorrhoea (primary or secondary), more particularly, primary
dysmenorrhoea.
Therefore, a further aspect of the invention is the method of treatment of a
mammal,
including a human being, to treat a disorder for which a V1a antagonist is
indicated,
comprising administering a therapeutically effective amount of a compound of
formula
(I), or a pharmaceutically acceptable salt or solvate thereof, to the mammal.
In
particular, the compounds of formula (I) are useful in treating anxiety,
cardiovascular
disease (including angina, atherosclerosis, hypertension, heart failure,
edema,
hypernatremia), dysmenorrhoea (primary and secondary), endometriosis, emesis
(including motion sickness), intrauterine growth retardation, inflammation
(including
rheumatoid arthritis), mittlesmerchz, preclampsia, premature ejaculation,
premature
(preterm) labour or Raynaud's disease. Even more particularly, they are useful
in
treating dysmenorrhoea (primary or secondary).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-15-
A further aspect of the present invention is the use of a compound of formula
(I), or a
pharmaceutically acceptable salt or solvate thereof, in the manufacture of a
medicament
for the treatment of a disorder for which a V1a receptor antagonist is
indicated.
All of the compounds of the formula (I) can be prepared by the procedures
described in
the general methods presented below or by the specific methods described in
the
Examples section and the Preparations section, or by routine modifications
thereof. The
present invention also encompasses any or one or more of these processes for
preparing the compounds of formula (I), in addition to any novel intermediates
used
therein.
Unless otherwise provided herein:
WSCDI means 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, DCC
means N,N'-dicyclohexylcarbodiimide, HOAT means 1-hydroxy-7-aza benzotriazole,
and
HOBT means 1-hydroxybenzotriazole hydrate;
PyBOP means Benzotriazol-1-yloxytris(pyrrolidino)phosphoniumhexa fluoro
phosphate, PyBrOP means bromo-tris-pyrrolidino-phosphoniumhexafluoro
phosphate,
and HBTU means O-benzotriazol-l-yl-N,N,N',N'-tetramethyluronium hexafluoro
phosphate;
mCPBA means meta-chloroperbenzoic acid, AcOH means acetic acid, HCI
means hydrochloric acid, TFA means trifluoroacetic acid, and p-TSA means p-
toluenesulphonic acid;
Et3N means triethylamine and NMM means N-methylmorpholine;
K2CO3 means potassium carbonate and KO -'Bu mean potassium tert-butoxide;
NaOH, KOH and LiOH mean sodium, potassium and lithium hydroxide
respectively;
Boc means tert-butoxycarbonyl and CBz means benzyloxycarbonyl;
PTFE means polytetrafluoroethane;
Mel means methyl iodide;
MeTosylate means methyl p-toluenesulphonate;
MeOH means methanol, EtOH means ethanol and n-BuOH means n-butyl
alcohol;
EtOAc means ethyl acetate, MeCN means acetonitrile, THF means
tetrahydrofuran, DMSO means dimethyl sulphoxide, DCM means dichloromethane,
DMF
means N,N-dimethylformamide, NMP means N-methyl-2-pyrrolidinone, and DMA means
dimethylacetamide;
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-16-
Me means methyl, Et means ethyl, Cl means chloro, and OH means hydroxy;
cat means catalyst or catalytic.
In the following general methods, R, R', R2, R3, ring A, V, X, Q, Z, Y, Y',
Het, Het', and
HetZ are as previously defined for a compound of the formula (I) unless
otherwise stated.
When Q represents NR2, or Q represents a direct link attached to a nitrogen
atom within
ring A, then compounds of formula (I) may be prepared according to Scheme 1.
ON N N~X N X
`1 N /--'
NHz ~ Q
N
O () (b)~
A A Y
PG-Q Y
(11) PG-Q (III) PG-Q (IV) N
Ni
~X
N
N\ N ~X ID (d)
o q Q (V) Y
z (~) Y
PG represents a suitable N protecting group, typically a benzyl, BOC or CBz
group, and
preferably BOC.
Scheme 1
Compounds of formula (II) may be obtained as described in WO 9703986 Al
19970206,
or by reaction of the corresponding lower alkyl ester (e.g. methyl or ethyl)
with hydrazine
under standard conditions, as exemplified in the preparations below.
Step (a): Compounds of formula (III) may be prepared by reaction of hydrazine
(I1) with
a suitable acetal (e.g. N,N-dimethylacetamide dimethyl acetal) in a suitable
solvent such
as THF, or DMF, at between room temperature and about 60 C, for up to 18
hours. The
resulting intermediate may then be treated under acid catalysis (e.g. p-TSA,
or TFA) in a
high boiling solvent (e.g. toluene, or xylene) for about 18 hours, to provide
the compound
of formula (III). Preferred conditions: 1.5-2.0 eq. of acetal (e.g. N,N-
dimethylacetamide
dimethyl acetal, triethyl orthopropionate), in THF or DMF at room temperature
to 60 C
for about 18 hours, followed by p-TSA, or TFA (cat), in toluene at reflux for
18 hours.
Step (b): Formation of triazole (IV) may be achieved by reaction of compound
(III) with
a suitable aniline, in the presence of a suitable acid catalyst, such as TFA
or p-TSA, in a
suitable high boiling solvent (e.g. toluene, or xylene), at an elevated
temperature.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-17-
Preferred conditions: 0.5-1.0 eq. TFA, 1.0-2.0 eq. aniline in toluene at about
reflux
temperature for up to 18 hours.
Step (c): Deprotection of compound (IV) is undertaken using standard
methodology, as
described in "Protecting Groups in Organic Synthesis" by T.W. Greene and P.
Wutz.
When PG represents BOC, the preferred conditions are: 4M HCI in dioxan in
MeOH,
dioxan or DCM at between room temperature and about 50 C, for up to 18 hours;
Or, 2.2M HCI in MeOH for up to 18 hours at room temperature;
Or, TFA in DCM at room temperature for about 1 hour.
Alternatively, when PG represents BOC, compound (V) may be prepared directly
from
compound (III) by treatment with an excess of TFA, typically 1.1-1.5 eq.) and
the
appropriate aniline in toluene, at the reflux temperature of the reaction, for
up to 4 days.
Step (d): Compounds of formula (I) may be prepared by reaction of amine (V)
with a
~
suitable acid or acid chloride (Z r, where T represents OH or CI). The
coupling may
be undertaken by using either:
0
(i) an acyl chloride, z1kci + amine (V), with an excess of acid base in a
suitable
solvent; or
(ii) an acid ZCO2H with a conventional coupling agent + amine (V) optionally
in
the presence of a catalyst, with an excess of base in a suitable solvent.
Typically the conditions are as follows:
(i) acid chloride z 1, ci, amine (V) (optionally with an excess of 3 amine
such as
Et3N, Hunig's base or NMM) in DCM or THF, without heating, for 1 to 24 hours;
or
(ii) acid ZCO2H, WSCDI /DCC and HOBT /HOAT, amine, an excess of NMM,
Et3N, or Hunig's base, in THF, DCM, DMA or EtOAc, at room temperature for 4 to
48
hours; or
acid ZCO2H, PYBOP /PyBrOP /O-benzotriazol-1-yl-N,N,N',N'-tetra methyl
uronium hexafluorophosphate, excess amine, excess of NMM, Et3N, or Hunig's
base, in
THF, DCM, DMA or EtOAc, at room temperature for 4 to 24 hours.
Preferred conditions are:
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-18-
0
~
1 eq. amine (V), 1.0-1.5 eq. z 1.5-5 eq. NMM, Et3N or Hunig's base in DCM
at room temperature for up to 18 hours;
Or, 1 eq. amine (V), 1.2 eq. ZCOzH, 1.2-1.5 eq. HOBT, 1.2-1.5 eq. WSCDI, 2-4
eq.
Et3N, in DCM at room temperature for 24 hours;
Or, 1 eq. amine (V), 1.2-1.5 eq. ZCO2H, 1.2-2.0 eq. HBTU, 5eq. Et3N or NMM in
DMA or
DCM, at between room temperature and 60 C for up to 24 hours.
Compounds of formula (IV), wherein Q represents NR2, or Q represents a direct
link
attached to a nitrogen atom within ring A which is in turn attached to the
triazole "ring
through a nitrogen atom, may alternatively be prepared as described in Scheme
2
below, and are represented as (IVA).
PG
PG
S=\ (l ~
PG~Q~ (VII) A N S G A N~S~CH3
N
Y' (e) Y /
r r
(VI) (VIIIA) (IXA)
N
N~ ~YX (8)
~~f\\_ Nl
~ ^ /
PG~Q~~~
(IVA) Y
Scheme 2.
Step (e): Compounds of formula (VIIIA) may be prepared by reaction of
approximately
equimolar amounts of isothiocyanate (VI) and amine (VII), in a suitable
solvent (e.g.
EtOH, or DCM) at room temperature for between 2 and 72 hours. Preferred
conditions:
1-1.1 eq. (VI), 1 eq. (VII), in EtOH or DCM, at room temperature for 0.5-2
hours.
Compounds of formulae (VI) and (VII) are commercially available, or may be
prepared
from known compounds using standard chemical transformations.
Step (f): Compounds of formula (IXA) may be prepared by methylation of
thiourea
(VIIIA), using a suitable methylating agent (e.g. Mel, or MeTosylate), in the
presence of
a suitable base (e.g. KOt-Bu) in a suitable solvent (e.g. THF, or ether) at
between 0 C
and the reflux temperature of the reaction, for about 18 hours. Preferred
conditions:
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-19-
1eq. (VIIIA), 1-1.2 eq. KOt-Bu, 1-1.2 eq. Mel or MeTosylate, in THF, at
between 10 C
and room temperature for up to 18 hours.
Step (g): Compounds of formula (IVA) may be prepared by reaction of compounds
(IXA) with a suitable hydrazide (XCONHNH2), optionally under acidic catalysis
(e.g. TFA,
or p-TSA) in a suitable solvent (e.g. THF, or n-BuOH), at between room
temperature and
the reflux temperature of the reaction. Preferred conditions: 0.5 eq. TFA,
excess
hydrazide (XCONHNH2), in THF at reflux for up to 18 hours.
Compounds of formula (VIIIA), wherein Q represents NR2, or Q represents a
direct link
attached to a nitrogen atom within ring A, which in turn is attached to the
triazole ring
through a nitrogen atom, may alternatively be prepared as shown in Scheme 3.
N PG~
A
S%\N Q Q/ N S Q A
(X) S
lb ~ N _ N
Y Y (e) (h) ~
Y ~ Y ~
Y'
r
(VI) (XI) (VIIIA)
Scheme 3.
Compounds of formula (X) are commercially available, or may be prepared from
known
compounds using standard chemical transformations.
Compounds of formula (XI) may be prepared from isothiocyanate (VI) and amine
(X), by
analogy with the methods previously described for Step (e) above.
Step (h): Compounds of formula (VIIIA) may be obtained by protection of the
reactive
nitrogen atom, using standard methodology, as described in "Protecting Groups
in
Organic Synthesis" by T.W. Greene and P. Wutz. When PG is BOC, the preferred
conditions are: 1 eq. amine (XI), 1 eq. di-t-butyl dicarbonate, in DCM and
dioxan, at
room temperature, for about 3 hours.
Compounds of formula (VIII), wherein Q represents NR2, or Q represents a
direct link
attached to a nitrogen atom within ring A, which in turn is attached to the
triazole ring
through a carbon atom, may be prepared as described in Scheme 4., and are
represented as (VIIIB).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-20-
OH
A O s
HZN PG,~
Q 1 _(XI I) N N
A
\\a \ -' \
PG Q
Y PG Y. (XIII) (XIV) Y (VIIIB) Y
Scheme 4.
Step (i): Compounds of formula (XIV) may be prepared by coupling of aniline
(XIII) with
acid (XII), by analogy with the methods previously described in Step (d).
Preferred
conditions: leq. acid (XII), 1.1eq. amine (XIII), 1.2 eq. WSCDI, 3 eq. Et3N in
MeCN at
room temperature, for about 3 days.
Step (j): The compound of formula (VIIIB) is prepared by thionation of
compound (XIV)
by treatment with a suitable thionating agent, such as Lawesson's reagent, in
a high
boiling solvent (e.g. toluene) at between room temperature and the reflux
temperature of
the reaction. Preferred conditions: 1 eq. (XIV), 0.5 eq. Lawesson's reagent in
toluene,
for between room temperature and reflux, for up to 18 hours.
Compounds of formula (III), wherein Q represents NR2, or Q represents a direct
link
which is attached to a nitrogen atom within ring A, may alternatively be
prepared as
shown in Scheme 5.
0 x
H H ~-x N-
0 N~NH2 O N H N \O
A ~ --
(k) A (q A
~'Q
PG PG,Q PG-Q
(II) (XV) (III)
Scheme 5.
Step (k): Di-acylhydrazides (XV) may be prepared by coupling hydrazides (II)
with acid
0
or acid chloride ( z'J~ T , where T represents Cl or OH), by analogy with the
methods
previously described in Step (d). Preferred conditions: 1 eq. hydrazide (II),
1.1 eq.
XCO2H, 1.1 eq. WSCDI, 1.1 eq. HOBT, 1.2 eq. Et3N, in DMF at room temperature
for 18
hours.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-21-
Step (I): Oxadiazole (III) may be prepared by cyclisation of compound (XV),
typically
under acid catalysis (e.g. polyphosphoric acid, POCI3, triflic
anhydride/pyridine, or 1-
methylimidazole), optionally in a suitable solvent (e.g. DCM), at between 0 C
and the
reflux temperature of the reaction. Preferred conditions: 1eq. (XV), excess
pyridine or
1-methylimidazole, 1.5-2eq triflic anhydride, in DCM, at between 0 C and room
temperature for up to 3 hours.
Compounds of formula (XV) may alternatively be prepared by coupling acid (XII)
with a
suitable hydrazide (XCONHNH2), by analogy with the methods previously
described in
Step (d). Preferred conditions: 1 eq. acid (XII), 1 eq. hydrazide, 1.02 eq.
WSDCI, in
DCM at between 0 C and room temperature.
Compounds of formula (III), wherein X represents CH2N-linked-Het, may
alternatively be
prepared as shown in Scheme 6.
o ci x
O N_ H N--~ N
NHZ N-N ~ O N
O
0 H cl N
A
I (m) A (~) A (n) A
PG' Q
Q I I
PG PG"O PGiQ
00 (XVI) (XVII) (111)
Scheme 6
Step (m): The compounds of formula (XVI) may be prepared by reaction of
hydrazide
(II) with chloroacetyl chloride, in the presence of a suitable 3 amine base
(e.g. Et3N, or
NMM) in a suitable solvent (e.g. EtOAc, or DCM) at between 0 C and room
temperature,
for about 18 hours. Preferred conditions: 1 eq. (II), 1 eq. acetyl chloride,
1.1 eq. NMM,
in DCM, at 10 C to room temperature, for up to 18 hours.
Compounds of formula (XVII) may be prepared by cyclisation compound (XVI), by
analogy with the methods previously described in Step (I) above.
Step (n): Compounds of formula (III) may be prepared by reaction of compound
(XVII)
with a suitable Het (containing a reactive N atom), in the presence of a
suitable base
(e.g. Et3N, or K2CO3), in a suitable solvent (e.g. DMF, or MeCN), at between
room
temperature and the reflux temperature of the reaction, for about 18 hours.
Preferred
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-22-
conditions: 1 eq. (XVII), 1.4 eq. K2CO3, 2 eq. Het, in DMF at room
temperature, for 18
hours.
Compounds of formula (I), wherein Q represents NR2, or Q represents a direct
link which
is attached to a nitrogen atom within ring A, may alternatively be prepared as
shown in
Scheme 7.
0
H H
O ORa O~ORa O N~ O N
NHZ T ~H
A -a A -' A -jw
(o) (p) (k)
O 0-1:::::::~ O O O O Q (I
z
z z
i N x
(XVIII) (XIX) (XX) (XXI) 0
N-
I ~ A (XXII)
(b) Q
N X E O~
z O A / z
I
(~) Y Y.
Ra represents CI-4 alkyl or benzyl, and is preferably Me or Et.
Scheme 7
Step (o): The compound of formula (XIX) may be prepared by reaction of amine
(XVIII)
0
~
with a suitable acid or acid chloride (z T, where T represents OH or CI), by
analogy
with the methods previously described in Step (d). Preferred conditions: 1 eq.
(XVIII),
0.9 eq. ZCOCI, 1.1 eq. Et3N, in DCM, at between 10 C and room temperature, for
about
3 hours.
Step (p): The hydrazide of formula (XX) may be prepared by treating ester
(XIX) with
excess hydrazine in a suitable solvent (e.g. EtOH, or MeOH), at the reflux
temperature
of the reaction, for up to 18 hours. Preferred conditions: 1 eq. (XIX), 2-4
eq. hydrazine,
in MeOH at reflux, for betweenlO and 48 hours.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-23-
Compounds of formula (XXI) may be prepared by reaction of hydrazide (XX) with
0
~
z T, using the methods previously described in Step (k).
Compounds of formula (XXII) may be prepared by cyclisation of compound (XXI),
using
the methods previously described in Step (I).
Compounds of formula (I) may be prepared from oxadiazole (XXII) with a
suitable
aniline, as previously described in Step (b).
Alternatively, compounds of formula (XXII) may be prepared directly from
compound
(XX) by reaction with an appropriate acetal (e.g. triethyl orthopropionate,
N,N-
dimethylacetamide dimethyl acetal) by analogy with the methods previously
described in
Step (a).
Compounds of formula (XXII), wherein X represents CH2-N-linked-Het, may
alternatively
be prepared as shown in Scheme 8.
a ci x
0 N~ H
NH2 ~-o ~ O N O N O
H
A --------- 4-
(m) A A (n) A
OYQ
OYO OYQ OYQ
z
z z z
(XX) (XXIII) (XXIV) (XXII)
Scheme 8
Compounds of formula (XXIII) may be prepared by reaction of hydrazide (XX)
with
chloroacetyl chloride, by analogy with the methods previously described in
Step (m).
Oxadiazole (XXIV) may be prepared by cyclisation of compound (XXIII), by
analogy with
the methods previously described in Step (I).
Compound (XXII) may be prepared by reaction of compound (XXIV) with a suitable
Het
(containing a reactive N atom), as previously described in Step (n).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-24-
Compounds of formula (I), wherein ring A is attached to the triazole ring
through a
nitrogen atom, may alternatively be prepared as shown in Scheme 9.
z~-- O z
O N ~ --O
zQ 'CA (~/)
A
N N
"rg A
(f) ' ~S
Y' (e) Y \ N
Y YN
/
r
(VI) (XXVI) (XXVII)
N'~N ~YX (9)
\\rNl
N
O ~
~O Y
z (q
Scheme 9
Compounds of formula (XXV) are either commercially available or may be
prepared from
commercially available compounds, using standard chemical transformations.
Compounds of formula (XXVI) may be prepared by reaction of compound (XXV) with
the
appropriate isothiocyanate (VI), by analogy with the methods previously
described in
Step (e).
The compound of formula (XXVII) may be prepared by alkylation of compound
(XXVI),
by analogy with the methods previously described in Step (f).
The compound of formula (I) may be prepared by reaction of compound (XXVII)
with a
suitable hydrazide, as previously described in Step (g).
Compounds of formula (I), wherein Q represents a direct link which is attached
to a
carbon atom in ring A, which in turn is attached to the triazole ring via a
nitrogen atom in
4
ring A, and Z represents NRR5, may be prepared as shown in Scheme 10.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-25-
RaO
RaO
s 0 N O O
\ A
~ O
N Ra0 O(XXVIII) ZA A
~
S(e) (f) ui" (
9)
Y/
Y ~ N ~
Y
(VI) ~ (~IX) / (~
Y. yRaO
Z HO
~-- O )==O O
O
nA `- ~ a
(d) A (q)
N N
~ N
N
N Y \ N /
N
N /N Y ~ ~--
, 1'
Y
x Y X Y. x
Y.
(I) (XXXII) (XXXI)
Ra represents C,-Q alkyl or benzyl, and is preferably Me or Et.
Scheme 10
Compounds of formula (XXVIII) are either commercially available or may be
prepared
from commercially available compounds, using standard chemical
transformations.
Compounds of formula (XXIX) may be prepared by reaction of compound (XXVIII)
with
the appropriate isothiocyanate (VI), by analogy with the methods previously
described in
Step (e).
The compound of formula (XXX) may be prepared by alkylation of compound
(XXIX), by
analogy with the methods previously described in Step (f).
The compound of formula (XXXI) may be prepared by reaction of compound (XXX)
with
a suitable hydrazide, as previously described in Step (g).
Step (q): Hydrolysis of ester (XXXI) using a suitable acid or base catalyst,
preferably an
alkali metal base (e.g. NaOH, KOH, or LiOH) in a suitable aqueous solvent
(e.g. dioxan,
or MeOH) at between room temperature and the reflux temperature of the
reaction, for
between 2 and 48 hours. Preferred conditions: 1 eq. (XXXI), 5-10 eq. NaOH
solution, in
dioxan at between room temperature and reflux, for between 2 and16 hours.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-26-
Compounds of formula (I) may be prepared by reaction of Z-H (containing a
reactive N
atom) with acid (XXXII), by analogy with the methods previously described for
Step (d).
Preferred conditions: 1 eq. acid (XXXII), 1.5 eq. amine (ZH), 4 eq. Et3N, 1.5
eq. WSCDI,
1.5 eq. HOBT, in DCM at room temperature, for 24 hours.
Compounds of formula (XXXII) may alternatively be prepared by hydrolysis of
the
corresponding nitrile compound (XXXIII), under standard conditions (e.g. 5 eq.
KOH, 1
eq. nitrile (XXXIII), in ethanol/ethylene glycol dimethyl ether at reflux).
Compounds of formula (XXXIII) may be prepared as shown in Scheme 11.
N-N\\_x A ~
O \ N
(b N/-`x
A /
N
N
(XXXIV) I
y Y'
(XXXIII)
Scheme 11
Compounds of formula (XXXIII), wherein Q is a direct link and ring A is
attached to the
triazole ring through a nitrogen atom, may be prepared from the appropriate
N~
isothiocyanate (VI) and an A ring containing nitrile, , by analogy with the
methods described in Scheme 9.
Compounds of formula (I), wherein V represents an oxygen atom, may be prepared
as
shown in Scheme 12.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-27-
x
s\ ~
N N s S~Nx H3C\
s~N~X
- -- y -- ->
(r) \N (s)
/ (t) /
Y / I I
Y 11V Y r Y y
(VI) (X)OCV) (XXXVI) (XXXVII) (u)
N X N-N
N y (v) ~g'\ \\'X
O O\\~- H3C \\ N/
O
r Z Q A OH
Z ~O y
(XXXIX) Y Y
(I) (XXXVIII)
Scheme 12
Step (r): The compound of formula (XXXV) may be prepared by reaction of
hydrazide
(XCONHNH2) with isothiocyanate (VI) by analogy with the methods previously
described
in Step (e). Preferred conditions: 1 eq. isothiocyanate, 1 eq. hydrazide, in
EtOH at room
temperature for 72 hours.
Step (s): Compounds of formula (XXXVI) may be prepared by cyclisation of
compound
(XXXV) under acid or base conditions, preferably base catalysis (e.g. alkali
metal
hydroxide) in aqueous solvent (e.g. water/EtOH), at an elevated temperature,
for about
24 hours. Preferred conditions: 1 eq. (XXXV), 10 eq. NaOH(aq) in EtOH at 80 C
for 18
hours.
Step (t): Alkylation of compound (XXXVI) to provide compound (XXXVII) may be
achieved by treatment with a suitable alkylating agent (e.g. Mel, or Me-
Tosylate), by
analogy with the methods previously described in Step (f). Preferred
conditions: 1 eq.
(XXXVI), 1 eq. KOt-Bu, 1 eq. Me-Tosylate, in THF at between room temperature
and
reflux for 3 hours.
Step (u): Compounds of formula (XXXVIII) may be obtained by oxidation of
compound
(XXXVII) by treatment with a suitable oxidising agent (e.g. mCPBA, or hydrogen
peroxide) in a suitable solvent (e.g. DCM) at room temperature for about
18hours.
Preferred conditions: 1 eq. (XXXVII), 4 eq. mCPBA, in DCM at room temperature
for 18
hours.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-28-
Step (v): Compounds of formula (I) may be prepared by reaction of sulphoxide
(XXXVIII) with an excess of alcohol (XXXIX) in the presence of a suitable base
(e.g.
NaH, or KOt-Bu), in a suitable solvent (e.g. THF, or ether) at between 00 and
room
temperature for up to 18 hours. Preferred conditions: 1 eq. (XXXVIII), 2 eq.
NaH, 2 eq.
alcohol (XXXIX), in THF for 18 hours at room temperature.
Compounds of formula (XXXIX) are either commercially available or may be
prepared
from commercially available compounds, using standard chemical
transformations.
Certain compounds of formulae (I) (III), (IV), (V), (XXII), and (XXXI) may
undergo
functional group interconversions (e.g. alkylation, or hydrolysis) to provide
alternative
compounds of formulae (I) (III), (IV), (V), (XXII), or (XXXI), respectively.
Compounds of the invention intended for pharmaceutical use may be administered
as
crystalline or amorphous products. They may be obtained, for example, as solid
plugs,
powders, or films by methods such as precipitation, crystallisation, freeze
drying, spray
drying, or evaporative drying. Microwave or radio frequency drying may be used
for this
purpose.
They may be administered alone or in combination with one or more other
compounds of
the invention or in combination with one or more other drugs (or as any
combination
thereof). Generally, they will be administered as a formulation in association
with one or
more pharmaceutically acceptable excipients. The term 'excipient' is used
herein to
describe any ingredient other than the compound(s) of the invention. The
choice of
excipient will to a large extent depend on factors such as the particular mode
of
administration, the effect of the excipient on solubility and stability, and
the nature of the
dosage form.
A further aspect of the invention is a pharmaceutical formulation including a
compound
of formula (I), or a pharmaceutically acceptable salt or solvate thereof,
together with a
pharmaceutically acceptable excipient, diluent or carrier. In a further
embodiment there
is provided the pharmaceutical formulation for administration either
prophylactically or
when pain commences.
Pharmaceutical compositions suitable for the delivery of compounds of the
present
invention and methods for their preparation will be readily apparent to those
skilled in the
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-29-
art. Such compositions and methods for their preparation may be found, for
example, in
Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing Company,
1995).
The compounds of the invention may be administered orally. Oral administration
may
involve swallowing, so that the compound enters the gastrointestinal tract, or
buccal or
sublingual administration may be employed by which the compound enters the
blood
stream directly from the mouth.
Formulations suitable for oral administration include solid formulations such
as tablets,
capsules containing particulates, liquids, or powders, lozenges (including
liquid-filled),
chews, multi- and nano-particulates, gels, solid solution, liposome, films,
ovules, sprays
and liquid formulations.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such
formulations
may be employed as fillers in soft or hard capsules and typically comprise a
carrier, for
example, water, ethanol, polyethylene glycol, propylene glycol,
methylcellulose, or a
suitable oil, and one or more emulsifying agents and/or suspending agents.
Liquid
formulations may also be prepared by the reconstitution of a solid, for
example, from a
sachet.
The compounds of the invention may also be used in fast-dissolving, fast-
disintegrating
dosage forms such as those described in Expert Opinion in Therapeutic Patents,
11 (6),
981-986, by Liang and Chen (2001).
For tablet dosage forms, depending on dose, the drug may make up from 1 weight
% to
80 weight % of the dosage form, more typically from 5 weight % to 60 weight %
of the
dosage form. In addition to the drug, tablets generally contain a
disintegrant. Examples
of disintegrants include sodium starch glycolate, sodium carboxymethyl
cellulose,
calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone,
polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower
alkyl-substituted
hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
Generally,
the disintegrant will comprise from 1 weight % to 25 weight %, preferably from
5 weight
% to 20 weight % of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet
formulation. Suitable
binders include microcrystalline cellulose, gelatin, sugars, polyethylene
glycol, natural
and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl
cellulose
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-30-
and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as
lactose
(monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol,
xylitol,
dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic
calcium
phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium
lauryl
sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
When present,
surface active agents may comprise from 0.2 weight % to 5 weight % of the
tablet, and
glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium
stearate,
zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate
with sodium
lauryl sulphate. Lubricants generally comprise from 0.25 weight % to 10 weight
%,
preferably from 0.5 weight % to 3 weight % of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring
agents,
preservatives and taste-masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 weight % to
about 90
weight % binder, from about 0 weight % to about 85 weight % diluent, from
about 2
weight % to about 10 weight % disintegrant, and from about 0.25 weight % to
about 10
weight % lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet
blends or
portions of blends may alternatively be wet-, dry-, or melt-granulated, melt
congealed, or
extruded before tabletting. The final formulation may comprise one or more
layers and
may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in Pharmaceutical Dosage Forms:
Tablets, Vol.
1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Consumable oral films for human or veterinary use are typically pliable water-
soluble or
water-swellable thin film dosage forms which may be rapidly dissolving or
mucoadhesive
and typically comprise a compound of formula (I), a film-forming polymer, a
binder, a
solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-
modifying agent
and a solvent. Some components of the formulation may perform more than one
function.
CA 02551038 2006-06-21
WO 2005/063754 -3 ~- PCT/IB2004/004059
The compound of formula (I) may be water-soluble or insoluble. A water-soluble
compound typically comprises from 1 weight % to 80 weight %, more typically
from 20
weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a
greater proportion of the composition, typically up to 88 weight % of the
solutes.
Alternatively, the compound of formula (I) may be in the form of
multiparticulate beads.
The film-forming polymer may be selected from natural polysaccharides,
proteins, or
synthetic hydrocolloids and is typically present in the range 0.01 to 99
weight %, more
typically in the range 30 to 80 weight %.
Other possible ingredients include anti-oxidants, colorants, flavourings and
flavour
enhancers, preservatives, salivary stimulating agents, cooling agents, co-
solvents
(including oils), emollients, bulking agents, anti-foaming agents, surfactants
and taste-
masking agents.
Films in accordance with the invention are typically prepared by evaporative
drying of
thin aqueous films coated onto a peelable backing support or paper. This may
be done
in a drying oven or tunnel, typically a combined coater dryer, or by freeze-
drying or
vacuuming.
Solid formulations for oral administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are
described in
US Patent No. 6,106,864. Details of other suitable release technologies such
as high
energy dispersions and osmotic and coated particles are to be found in
Pharmaceutical
Technology On-line, 25(2), 1-14, by Verma et al (2001). The use of chewing gum
to
achieve controlled release is described in WO 00/35298.
The compounds of the invention may also be administered directly into the
blood stream,
into muscle, or into an internal organ. Suitable means for parenteral
administration
include intravenous, intraarterial, intraperitoneal, intrathecal,
intraventricular,
intraurethral, intrasternal, intracranial, intramuscular and subcutaneous.
Suitable
devices for parenteral administration include needle (including microneedle)
injectors,
needle-free injectors and infusion techniques.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-32-
Parenteral formulations are typically aqueous solutions which may contain
excipients
such as salts, carbohydrates and buffering agents (preferably to a pH of from
3 to 9),
but, for some applications, they may be more suitably formulated as a sterile
non-
aqueous solution or as a dried form to be used in conjunction with a suitable
vehicle
such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for
example, by
lyophilisation, may readily be accomplished using standard pharmaceutical
techniques
well known to those skilled in the art.
The solubility of compounds of formula (I) used in the preparation of
parenteral solutions
may be increased by the use of appropriate formulation techniques, such as the
incorporation of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release. Thus compounds of the invention
may
be formulated as a solid, semi-solid, or thixotropic liquid for administration
as ari
implanted depot providing modified release of the active compound. Examples of
such
formulations include drug-coated stents and poly(d/-lactic-coglycolic)acid
(PGLA)
microspheres.
The compounds of the invention may also be administered topically to the skin
or
mucosa, that is, dermally or transdermally. Typical formulations for this
purpose include
gels, hydrogels, lotions, solutions, creams, ointments, dusting powders,
dressings,
foams, filrris, skin patches, wafers, implants, sponges, fibres, bandages and
microemulsions. Liposomes may also be used. Typical carriers include alcohol,
water,
mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene
glycol and
propylene glycol. Penetration enhancers may be incorporated - see, for
example, J
Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation,
iontophoresis,
phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderjectr"'
BiojectT"", etc.) injection.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-33-
Formulations for topical administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
The compounds of the invention can also be administered intranasally or by
inhalation,
typically in the form of a dry powder (either alone, as a mixture, for
example, in a dry
blend with lactose, or as a mixed component particle, for example, mixed with
phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an
aerosol
spray from a pressurised container, pump, spray, atomiser (preferably an
atomiser using
electrohydrodynamics to produce a fine mist), or nebuliser, with or without
the use of a
suitable propellant, such as 1,1,1,2-tetrafluoroethane or 1,1,1,2,3,3,3-
heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive
agent,
for example, chitosan or cyclodextrin.
The pressurised container, pump, spray, atomizer, or nebuliser contains a
solution or
suspension of the compound(s) of the invention comprising, for example,
ethanol,
aqueous ethanol, or a suitable alternative agent for dispersing, solubilising,
or extending
release of the active, a propellant(s) as solvent and an optional surfactant,
such as
sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is
micronised to
a size suitable for delivery by inhalation (typically less than 5 microns).
This may be
achieved by any appropriate comminuting method, such as spiral jet milling,
fluid bed jet
milling, supercritical fluid processing to form nanoparticles, high pressure
homogenisation, or spray drying.
Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose),
blisters
and cartridges for use in an inhaler or insufflator may be formulated to
contain a powder
mix of the compound of the invention, a suitable powder base such as lactose
or starch
and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
The
lactose may be anhydrous or in the form of the monohydrate, preferably the
latter.
Other suitable excipients include dextran, glucose, maltose, sorbitol,
xylitol, fructose,
sucrose and trehalose.
A suitable solution formulation for use in an atomiser using
electrohydrodynamics to
produce a fine mist may contain from 1 pg to 20mg of the compound of the
invention per
actuation and the actuation volume may vary from 1 NI to 100pl. A typical
formulation
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-34-
may comprise a compound of formula (I), propylene glycol, sterile water,
ethanol and
sodium chloride. Alternative solvents which may be used instead of propylene
glycol
include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as
saccharin
or saccharin sodium, may be added to those formulations of the invention
intended for
inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be
immediate
and/or modified release using, for example, PGLA. Modified release
formulations include
delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined
by
means of a valve, which delivers a metered amount. The overall daily dose will
typically
be in the range 0.01 pg to 15 mg which may be administered in a single dose
or, more
usually, as divided doses throughout the day.
The compounds of the invention may be administered rectally or vaginally, for
example,
in the form of a suppository, pessary, or enema. Cocoa butter is a traditional
suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be
immediate and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
The compounds of the invention may also be administered directly to the eye or
ear,
typically in the form of drops of a micronised suspension or solution in
isotonic, pH-
adjusted, sterile saline. Other formulations suitable for ocular and aural
administration
include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and
non-
biodegradable (e.g. silicone) implants, wafers, lenses and particulate or
vesicular
systems, such as niosomes or liposomes. A polymer such as crossed-linked
polyacrylic
acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example,
hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a
heteropolysaccharide polymer, for
example, gelan gum, may be incorporated together with a preservative, such as
benzalkonium chloride. Such formulations may also be delivered by
iontophoresis.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-35-
Formulations for ocular/aural administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted, or programmed release.
The compounds of the invention may be combined with soluble macromolecular
entities,
such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-
containing
polymers, in order to improve their solubility, dissolution rate, taste-
masking,
bioavailability and/or stability for use in any of the aforementioned modes of
administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for
most
dosage forms and administration routes. Both inclusion and non-inclusion
complexes
may be used. As an alternative to direct complexation with the drug, the
cyclodextrin
may be used as an auxiliary additive, i.e. as a carrier, diluent, or
solubiliser. Most
commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins,
examples of which may be found in International Patent Applications Nos. WO
91/11172, WO 94/02518 and WO 98/55148.
Inasmuch as it may desirable to administer a combination of active compounds,
for
example, for the purpose of treating a particular disease or condition, it is
within the
scope of the present invention that two or more pharmaceutical compositions,
at least
one of which contains a compound in accordance with the invention, may
conveniently
be combined in the form of a kit suitable for coadministration of the
compositions.
Thus the kit of the invention comprises two or more separate pharmaceutical
compositions, at least one of which contains a compound of formula (I) in
accordance
with the invention, and means for separately retaining said compositions, such
as a
container, divided bottle, or divided foil packet. An example of such a kit is
the familiar
blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different
dosage forms,
for example, oral and parenteral, for administering the separate compositions
at different
dosage intervals, or for titrating the separate compositions against one
another. To
assist compliance, the kit typically comprises directions for administration
and may be
provided with a so-called memory aid.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-36-
For administration to human patients, the total daily dose of the compounds of
the
invention is typically in the range 0.01 mg to 15 mg depending, of course, on
the mode
of administration. The total daily dose may be administered in single or
divided doses
and may, at the physician's discretion, fall outside of the typical range
given herein.
These dosages are based on an average human subject having a weight of about
60kg
to 70kg. The physician will readily be able to determine doses for subjects
whose weight
falls outside this range, such as infants and the elderly.
For the avoidance of doubt, references herein to "treatment" include
references to
curative, palliative and prophylactic treatment.
The compounds of the present invention may be tested in the screens set out
below:
1.0 VlA Filter Binding Assay
1.1 Membrane Pregaration
Receptor binding assays were performed on cellular membranes prepared from CHO
cells stably expressing the human V,A receptor, (CHO-hV,A). The CHO-hV1A cell
line was
kindly provided under a licensing agreement by Marc Thibonnier, Dept. of
Medicine,
Case Western Reserve University School of Medicine, Cleveland, Ohio. CHO-hV1A
cells
were routinely maintained at 37 C in humidified atmosphere with 5% CO2 in
DMEM/Hams F12 nutrient mix supplemented with 10 % fetal bovine serum, 2 mM L-
glutamine, 15 mM HEPES and 400 Ng/mI G418. For bulk production of cell
pellets,
adherent CHO-hV1A cells were grown to confluency of 90-100% in 850 cm2 roller
bottles
containing a medium of DMEM/Hams F12 Nutrient Mix supplemented with 10 % fetal
bovine serum, 2 mM L-glutamine and 15 mM HEPES. Confluent CHO-hV1A cells were
washed. with phosphate-buffered saline (PBS), harvested into ice cold PBS and
centrifuged at 1,000 rpm. Cell pellets were stored at -80 C until use. Cell
pellets were
thawed on ice and homogenised in membrane preparation buffer consisting of 50
mM
Tris-HCI, pH 7.4, 5 mM MgCI2 and supplemented with a protease inhibitor
cocktail,
(Roche). The cell homogenate was centrifuged at 1000 rpm, 10 min, 4 C and the
supernatant was removed and stored on ice. The remaining pellet was
homogenised
and centrifuged as before. The supernatants were pooled and centrifuged at
25,000 x g
for 30 min at 4 C. The pellet was resuspended in freezing buffer consisting of
50 mM
Tris-HCI, pH 7.4, 5 mM MgCI2 and 20 % glycerol and stored in small aliquots at
-80 C
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-37-
until use. Protein concentration was determined using Bradford reagent and BSA
as a
standard.
1.2 V,A Filter binding
Protein linearity followed by saturation binding studies were performed on
each new
batch of membrane. Membrane concentration was chosen that gave specific
binding on
the linear portion of the curve. Saturation binding studies were then
performed using
various concentrations of [3H]-arginine vasopressin, [3H]-AVP (0.05 nM - 100
nM) and
the Kd and Bmax determined.
Compounds were tested for their effects on [3H]-AVP binding to CHO-hV1A
membranes,
(3H-AVP; specific activity 65.5 Ci / mmol; NEN Life Sciences). Compounds were
solubilised in dimethylsulfoxide (DMSO) and diluted to working concentration
of 10%
DMSO with assay buffer containing 50 mM Tris-HCL pH 7.4, 5 mM MgCIz and 0.05%
BSA. 25 pl compound and 25 pl [3H]-AVP, (final concentration at or below Kd
determined
for membrane batch, typically 0.5 nM - 0.6 nM) were added to a 96-well round
bottom
polypropylene plate. The binding reaction was initiated by the addition of 200
NI
membrane and the plates were gently shaken for 60 min at room temperature. The
reaction was terminated by rapid filtration using a Filtermate Cell Harvester
(Packard
Instruments) through a 96-well GF/B UniFilter Plate which had been presoaked
in 0.5%
polyethyleneimine to prevent peptide sticking. The filters were washed three
times with 1
ml ice cold wash buffer containing 50 mM Tris-HCL pH 7.4 and 5 mM MgCI2. The
plates
were dried and 50 pl Microscint-0 (Packard instruments) was added to each
well. The
plates were sealed and counted on a TopCount Microplate Scintillation Counter
(Packard Instruments). Non-specific binding (NSB) was determined using 1 pM
unlabelled d(CH2)5Tyr(Me)AVP ([R-mercapto-R,R-cyclopentamethylenepropionyl,O-
Me-
Tyr2,Arg8]-vasopressin )(RMCPVP), (Sigma). The radioligand binding data was
analysed
using a four parameter logistic equation with the min forced to 0%. The slope
was free
fitted and fell between -0.75 and -1.25 for valid curves. Specific binding was
calculated
by subtracting the mean NSB cpm from the mean Total cpm. For test compounds
the
amount of ligand bound to the receptor was expressed as % bound = (sample cpm -
mean NSB cpm)/specific binding cpm xlOO. The % bound was plotted against the
concentration of test compound and a sigmoidal curve was fitted. The
inhibitory
dissociation constant (K;) was calculated using the Cheng-Prusoff equation:
K;=IC50/(1+[L]/Kd) where [L] is the concentration of ligand present in the
well and Kd is the
dissociation constant of the radioligand obtained from Scatchard plot
analysis.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-38-
2.0 V,A Functional Assay; Inhibition of AVP / Võ -R mediated Ca2+ mobilization
by
FLIPR (Fluorescent Imaging Plate Reader) (Molecular Devices)
Intracellular calcium release was measured in CHO-hVIA cells using FLIPR,
which allows
the rapid detection of calcium following receptor activation. The CHO-hV1A
cell line was
kindly provided under a licensing agreement by Marc Thibonnier, Dept. of
Medicine,
Case Western Reserve University School of Medicine, Cleveland, Ohio. CHO-V,A
cells
were routinely maintained at 37 C in humidified atmosphere with 5% CO2 in
DMEM/Hams F12 nutrient mix supplemented with 10 % fetal bovine serum, 2 mM L-
glutamine, 15 mM HEPES and 400 Ng/mI G418. On the afternoon before the assay
cells
were plated at a density of 20,000 cells per well into black sterile 96-well
plates with
clear bottoms to allow cell inspection and fluorescence measurements from the
bottom
of each well. Wash buffer containing Dulbecco's phosphate buffered saline
(DPBS) and
2.5 mM probenecid and loading dye consisting of cell culture medium containing
4 pM
Fluo-3-AM (dissolved in DMSO and pluronic acid),(Molecular Probes) and 2.5 mM
probenecid was prepared fresh on the day of assay. Compounds were solubilised
in
DMSO and diluted in assay buffer consisting of DPBS containing 1% DMSO, 0.1%
BSA
and 2.5 mM probenecid. The cells were incubated with 100 pl loading dye per
well for 1
hour at 37 C in humidified atmosphere with 5% CO2. After dye loading the cells
were
washed three times in 100 pl wash buffer using a Denley plate washer. 100 NI
wash
buffer was left in each well. Intracellular fluorescence was measured using
FLIPR.
Fluorescence readings were obtained at 2s intervals with 50 pl of the test
compound
added after 30s. An additional 155 measurements at 2s intervals were then
taken to
detect any compound agonistic activity. 50 pl of arginine vasopressin (AVP)
was then
added so that the final assay volume was 200 NI. Further fluorescence readings
were
collected at 1s intervals for 120s. Responses were measured as peak
fluorescence
intensity (FI). For pharmacological characterization a basal Fl was subtracted
from each
fluorescence response. For AVP dose response curves, each response was
expressed
as a % of the response to the highest concentration of AVP in that row. For
IC5o
determinations , each response was expressed as a % of the response to AVP.
IC50
values were converted to a modified Kb value using the Cheng-Prusoff equation
which
takes into account the agonist concentration, [A], the agonist EC50 and the
slope:
Kb=IC50/(2+[A]/A50]")'/"-1 where [A] is the concentration of AVP, A50 is the
EC50 of AVP
from the dose response curve and n=slope of the AVP dose response curve.
The compounds of the invention may be administered alone or in combination
with one
or more other compounds of the invention or in combination with one or more
other
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-39-
drugs (or as any combination thereof). The compounds of the present invention
may be
administered in combination with an oral contraceptive. Thus in a further
aspect of the
invention, there is provided a pharmaceutical product containing an V1a
antagonist and
an oral contraceptive as a combined preparation for simultaneous, separate or
sequential use in the treatment of dysmenorrhoea.
The compounds of the present invention may be administered in combination with
a
PDE5 inhibitor. Thus in a further aspect of the invention, there is provided a
pharmaceutical product containing a V1 a antagonist and a PDEV inhibitor as a
combined preparation for simultaneous, separate or sequential use in the
treatment of
dysmenorrhoea.
PDEV inhibitors useful for combining with V1a antagonists include, but are not
limited to:
(i) The PDE5 inhibitors mentioned in International Patent Application
publication
nos. W003/000691; W002/64590; W002/28865; W002/28859;
W002/38563; W002/36593; W002/28858; W002/00657; W002/00656;
W002/10166; W002/00658; W001/94347; W001/94345; W000/15639 and
W 000/15228;
(ii) The PDE5 inhibitors mentioned in US Patents 6,143,746; 6,143,747 and
6,043,252;
(iii) the pyrazolo [4,3-d]pyrimidin-7-ones disclosed in EP-A-0463756; the
pyrazolo
[4,3-d]pyrimidin-7-ones disclosed in EP-A-0526004; the pyrazolo [4,3-
d]pyrimidin-7-ones disclosed in published international patent application WO
93/06104; the isomeric pyrazolo [3,4-d]pyrimidin-4-ones disclosed in
published international patent application WO 93/07149; the quinazolin-4-
ones disclosed in published international patent application WO 93/12095; the
pyrido [3,2-d]pyrimidin-4-ones disclosed in published international patent
application WO 94/05661; the purin-6-ones disclosed in published
international patent application WO 94/00453; the pyrazolo [4,3-d]pyrimidin-7-
ones disclosed in published international patent application WO 98/49166; the
pyrazolo [4,3-d]pyrimidin-7-ones disclosed in published international patent
application WO 99/54333; the pyrazolo [4,3-d]pyrimidin-4-ones disclosed in
EP-A-0995751; the pyrazolo [4,3-d]pyrimidin-7-ones disclosed in published
international patent application WO 00/24745; the pyrazolo [4,3-d]pyrimidin-4-
ones disclosed in EP-A-0995750; the hexahydropyrazino [2',1':6,1]pyrido [3,4-
b]indole-1,4-diones disclosed in published international application
W095/19978; the pyrazolo [4,3-d]pyrimidin-4-ones disclosed in W000/27848;
the imidazo[5,1-t][1,2,4]triazin-ones disclosed in EP-A-1092719 and in
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-40-
published international application WO 99/24433 and the bicyclic compounds
disclosed in published international application WO 93/07124; the pyrazolo
[4,3-d]pyrimidin-7-ones disclosed in published international application WO
01/27112; the pyrazolo [4,3-d]pyrimidin-7-ones disclosed in published
international application WO 01/27113; the compounds disclosed in EP-A-
1092718 and the compounds disclosed in EP-A-1092719; the tricyclic
compounds disclosed in EP-A-1241170; the alkyl sulphone compounds
disclosed in published international application WO 02/074774; the
compounds disclosed in published international application WO 02/072586;
the compounds disclosed in published international application WO
02/079203 and the compounds disclosed in WO 02/074312.
( iv ) Preferably 5-[2-ethoxy-5-(4-methyl-1-piperazinylsulphonyl)phenyl]-1-
methyl-3-
n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil, e.g. as
sold
as Viagra ) also known as 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-
pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulphonyl]-4-methylpiperazine
(see EP-A-0463756);5-(2-ethoxy-5-morpholinoacetylphenyl)-1-methyl-3-n-
propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see EP-A-0526004);3-
ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxyphenyl]-2-(pyridin-2-
yl)methyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see
W098/49166);3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-
methoxyethoxy)pyridin-3-yl]-2-(pyridin-2-yl)methyl-2,6-dihydro-7H-
pyrazolo[4,3-d]pyrimidin-7-one(see W099/54333); (+)-3-ethyl-5-[5-(4-
ethylpiperazin-1 -ylsulphonyl)-2-(2-methoxy-1(R)-methylethoxy)pyridin-3-yl]-2-
methyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, also known as 3-ethyl-
5-{5-[4-ethylpiperazin-1 -ylsulphonyl]-2-([(1 R)-2-methoxy-1-
methylethyl]oxy)pyrid i n-3-yl}-2-methyl-2, 6-d i hyd ro-7 H-pyrazolo[4, 3-d]
pyrimidin-7-one (see W099/54333);5-[2-ethoxy-5-(4-ethylpiperazin-l-
ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-
pyrazolo[4,3-d]pyrimidin-7-one, also known as 1-{6-ethoxy-5-[3-ethyl-6,7-
dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-
pyridylsulphonyl}-4-ethylpiperazine (see WO 01/27113, Example 8);5-[2-iso-
Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-
methylpiperidin-4-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one(see WO
01/27113, Example 15);5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-
3-yl]-3-ethyl-2-phenyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see WO
01/27113, Example 66);5-(5-Acetyl-2-propoxy-3-pyridinyl)-3-ethyl-2-(1-
isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-41-
WO 01/27112, Example 124); 5-(5-Acetyl-2-butoxy-3-pyridinyl)-3-ethyl-2-(1-
ethyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (see WO
01/27112, Example 132); (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-
(3,4-methylenedioxyphenyl)pyrazino[2',1':6,1 ]pyrido[3,4-b]indole-1,4-dione
(tadalafil, IC-351, Cialis ), i.e. the compound of examples 78 and 95 of
published international application W095/19978, as well as the compound of
examples 1, 3, 7 and 8; 2-[2-ethoxy-5-(4-ethyl-piperazin-1-yl-l-sulphonyl)-
phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one (vardenafil,
LEVITRA ) also known as 1-[[3-(3,4-dihydro-5-methyl-4-oxo-7-
propylimidazo[5,1-f]-as-triazin-2-yl)-4-ethoxyphenyl]sulphonyl]-4-
ethylpiperazine, i.e. the compound of examples 20, 19, 337 and 336 of
published international application W099/24433;the compound of example 11
of published international application W093/07124 (EISAI); compounds 3 and
14 from Rotella D P, J. Med. Chem., 2000, 43, 1257; 4-(4-
chlorobenzyl)amino-6,7,8-trimethoxyquinazoline; N-[[3-(4,7-dihydro-1-methyl-
7-oxo-3-propyl-1 H-pyrazolo[4,3-d]-pyrimidin-5-yl)-4-propxyphenyl]sulfonyl]-1-
methyl2-pyrrolidinepropanamide ["DA-8159" (Example 68 of W000/27848)];
and 7,8-dihydro-8-oxo-6-[2-propoxyphenyl]-1 H-imidazo[4,5-g]quinazoline and
1-[3-[1-[(4-fluorophenyl)methyl]-7,8-dihydro-8-oxo-1 H-imidazo[4,5-
g]quinazolin-6-yl]-4-propoxyphenyl]carboxamide.
(v) 4-bromo-5-(pyridylmethylamino)-6-[3-(4-chlorophenyl)-propoxy]-
3(2H)pyridazinone; 1-[4-[(1,3-benzodioxol-5-ylmethyl)amiono]-6-chloro-2-
quinozolinyl]-4-piperidine-carboxylic acid, monosodium salt; (+)-cis-
5,6a,7,9,9,9a-hexahydro-2-[4-(trifluoromethyl)-phenylmethyl-5-methyl-
cyclopent-4,5]imidazo[2,1-b]purin-4(3H)one; furazlocillin; cis-2-hexyl-5-
methyl-
3,4,5,6a,7,8,9,9a- octahydrocyclopent[4,5]-imidazo[2,1-b]purin-4-one; 3-
acetyl-1-(2-chlorobenzyl)-2-propylindole-6- carboxylate; 3-acetyl-1 -(2-
chlorobenzyl)-2-propylindole-6-carboxylate; 4-bromo-5-(3-
pyridylmethylamino)-6-(3-(4-chlorophenyl) propoxy)-3- (2H)pyridazinone; I-
methyl-5(5-morpholinoacetyl-2-n-propoxyphenyl)-3-n-propyl-1,6-dihydro- 7H-
pyrazolo(4,3-d)pyrimidin-7-one; 1-[4-[(1,3-benzodioxol-5-ylmethyl)arnino]-6-
chloro-2- quinazolinyl]-4-piperidinecarboxylic acid, monosodium salt;
Pharmaprojects No. 4516 (Glaxo Wellcome); Pharmaprojects No. 5051
(Bayer); Pharmaprojects No. 5064 (Kyowa Hakko; see WO 96/26940);
Pharmaprojects No. 5069 (Schering Plough); GF-196960 (Glaxo Wellcome);
E-8010 and E-4010 (Eisai); Bay-38-3045 & 38-9456 (Bayer); FR229934 and
FR226807 (Fujisawa); and Sch-51866.
CA 02551038 2009-05-06
69387-703
-42-
Preferably the PDEV inhibitor is selected from sildenafil, tadalafit,
vardenafil, DA-8159
and 5-[2-ethoxy-5-(4-ethyipiperazin-1-yisuiphonyl)pyridin-3-ytJ-3-ethyl-2-[2-
methoxyethyi]-
2,6-dihydro-7H-pyrazolo[4,3-dJpyrimidin-7-one.
Most preferably the PDE5 inhibitor is sifdenafil and pharmaceutically
acceptable salts
thereof. Sildenafil citrate is a preferred salt.
The compounds of the present invention may be administered in combination with
an
NO donor. Thus in a further aspect of the invention, there is provided a
pharmaceutical
product containing a V1a antagonist and a NO donor as a combined preparation
for
simultaneous, separate or sequential use in the treatment of dysmenorrhoea.
The compounds of the present invention may be administered in combination with
L-
arginine, or as an arginate salt. Thus in a further aspect of the invention,
there is
provided a pharmaceuticai product containing a V 1 a antagonist and L-arginine
as a
combined preparation for simultaneous, separate or sequential use in the
treatment of
dysmenorrhoea.
The compounds of the present invention may be administered in combination with
a
COX inhibitor. Thus in a further aspect of the invention, there is provided a
pharmaceutical product containing a V1a antagonist and a COX inhibitor as a
combined
preparation for simultaneous, separate or sequential use in the treatment of
dysmenorrhoea.
COX inhibitors useful for combining with the compounds of the present
invention include,
but are not limited to:
(i) ibuprofen, naproxen, benoxaprofen, flurbiprofen, fenoprofen, fenbufen,
ketoprofen, indoprofen, pirprofen, carprofen, oxaprozin, prapoprofen,
miroprofen, tioxaprofen, suprofen, alminoprofen, tiaprofenic acid, fluprofen,
bucloxic acid, indomethacin, sulindac, tolmetin, zomepirac, diciofenac,
fenclofenec, alclofenac, ibufenac, isoxepac, furofenac, tiopinac,
zidometacin, acetyl salicylic acid, indometacin, piroxicam, tenoxicam,
nabumetone, ketorolac, azapropazone, mefenamic acid, tolfenamic acid,
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-43-
diflunisal, podophyllotoxin derivatives, acemetacin, droxicam, floctafenine,
oxyphenbutazone, phenylbutazone, proglumetacin, acemetacin, fentiazac,
clidanac, oxipinac, mefenamic acid, meclofenamic acid, flufenamic acid,
niflumic acid, flufenisal, sudoxicam, etodolac, piprofen, salicylic acid,
choline magnesium trisalicylate, salicylate, benorylate, fentiazac, clopinac,
feprazone, isoxicam and 2-fluoro-a-methyl[1,1'-biphenyl]-4-acetic acid, 4-
(nitrooxy)butyl ester (See Wenk, et al., Europ. J. Pharmacol. 453:319-324
(2002));
(ii) meloxicam, (CAS registry number 71125-38-7; described in U.S. Patent
No. 4,233,299), or a pharmaceutically acceptable salt or prodrug thereof;
(iii) Substituted benzopyran derivatives that are described in U.S. Patent No.
6,271,253. Also benzopyran derivatives described in U.S. Patent Nos.
6,034,256 and 6,077,850 along with International Publication No's WO
98/47890 and WO 00/23433;
(iv) Chromene COX2 selective inhibitors described in U.S. Patent No.
6,077,850 and U.S. Patent No. 6,034,256;
(v) The compounds described in International Patent Application Publication
No's WO 95/30656, WO 95/30652, WO 96/38418 and WO 96/38442, and
the compounds described in European Patent Application Publication No.
799823, along with the pharmaceutically acceptable derivatives thereof;
(vi) celecoxib (US Patent No. 5,466,823), valdecoxib (US Patent No.
5,633,272), deracoxib (US Patent No. 5,521,207), rofecoxib (US Patent No.
5,474,995), etoricoxib (International Patent Application Publication No. WO
98/03484), JTE-522 (Japanese Patent Application Publication No.
9052882), or a pharmaceutically acceptable salt or prodrug thereof;
(vii) Parecoxib (described in U.S. Patent No. 5,932,598), which is a
therapeutically effective prodrug of the tricyclic Cox-2 selective inhibitor
valdecoxib (described in U.S. Patent No. 5,633,272), in particular sodium
parecoxib;
(viii) ABT-963 (described in International Patent Application Publication No.
WO
00/24719)
(ix) Nimesulide (described in U.S. Patent No. 3,840,597), flosulide (discussed
in J. Carter, Exp.Opin.Ther.Patents, 8(1), 21-29 (1997)), NS-398 (disclosed
in U.S. Patent No. 4,885,367), SD 8381 (described in U.S. Patent No.
6,034,256), BMS-347070 (described in U.S. Patent No. 6,180,651), S-2474
(described in European Patent Publication No. 595546) and MK-966
(described in U.S. Patent No. 5,968,974);
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-44-
(x) The compounds and pharmaceutically acceptable derivatives described in
U.S. Patent No. 6,395,724, U.S. Patent No. 6,077,868, U.S. Patent No.
5,994,381, U.S. Patent No. 6,362,209, U.S. Patent No. 6,080,876, U.S.
Patent No 6,133,292, U.S. Patent No. 6, 369,275, U.S. Patent No.
6,127,545, U.S. Patent No. 6,130,334, U.S. Patent No. 6,204,387, U.S.
Patent No. 6,071,936, U.S. Patent No. 6,001,843, U.S. Patent No.
6,040,450, International Patent Application Publication No WO 96/03392,
International Patent Application Publication No WO 96/24585, U.S. Patent
No. 6,340,694, U.S. Patent No. 6,376,519, U.S. Patent No. 6,153,787, U.S.
Patent No. 6,046,217, U.S. Patent No. 6,329,421, U.S. Patent No.
6,239,137, U.S. Patent No. 6,136,831, U.S. Patent No. 6,297,282, U.S.
Patent No. 6,239,173, U.S. Patent No. 6,303,628, U.S. Patent No.
6,310,079, U.S. Patent No. 6,300,363, U.S. Patent No. 6,077,869, U.S.
Patent No. 6,140,515, U.S. Patent No. 5,994,379, U.S. Patent No.
6,028,202, U.S. Patent No. 6,040,320, U.S. Patent No. 6,083,969, U.S.
Patent No 6,306,890, U.S. Patent No. 6,307,047, U.S. Patent No.
6,004,948, U.S. Patent No. 6,169,188, U.S. Patent No. 6,020,343, U.S.
Patent No. 5,981,576, U.S. Patent No. 6,222,048, U.S. Patent No.
6,057,319, U.S. Patent No. 6,046,236, U.S. Patent No. 6,002,014, U.S.
Patent No. 5,945,539, U.S. Patent No. 6,359,182, International Patent
Application Publication No. WO 97/13755, International Patent Application
Publication No. WO 96/25928, International Patent Application Publication
No. WO 96/374679, International Patent Application Publication No. WO
95/15316, International Patent Application Publication No. WO 95/15315,
International Patent Application Publication No. WO 96/03385, International
Patent Application No. WO 95/00501, International Patent Application No.
WO 94/15932, International Patent Application Publication No. WO
95/00501, International Patent Application Publication No. WO 94/27980,
International Patent Application Publication No. WO 96/25405, International
Patent Application Publication No. WO 96/03388, International Patent
Application Publication No. WO 96/03387, U.S. Patent No. 5,344,991,
International Patent Application Publication No. WO 95/00501, International
Patent Application Publication No. WO 96/16934, International Patent
Application Publication No. WO 96/03392, International Patent Application
Publication No. WO 96/09304, International Patent Application Publication
No. WO 98/47890, and International Patent Application Publication No. WO
00/24719.
CA 02551038 2009-05-06
69387-703
-45-
The following Preparations and Examples illustrate the preparation of
compounds of
formula (I).
'H Nuclear magnetic resonance (NMR) spectra were in all cases consistent with
the
proposed structures. Characteristic chemical shifts (6) are given in parts-per-
million
downfield from tetramethytsilane using conventional abbreviations for
designation of
major peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; m,
multiplet; br, broad. The
mass spectra (m/z) were recorded using either electrospray ionisation (ESI) or
atmospheric pressure chemical ionisation (APCI). The following abbreviations
have
been used for common solvents: CDCI3, deuterochloroform; DB-DMSO,
deuterodimethylsulphoxide; CD3OD, deuteromethanol; THF, tetrahydrofuran.
"Ammonia" refers to a concentrated solution of ammonia in water possessing a
specific
gravity of 0.88. Where thin layer chromatography (TLC) has been used it refers
to silica
gel TLC using silica gel 60 F254 plates, Rf is the distance traveled by a
compound
divided by the distance traveled by the solvent front on a TLC plate. When
microwave
radiation is employed, the two microwaves used are the Emrys Creator and the
Emrys
Liberator, both supplied by Personal Chemistry Ltd. The power range is 15-300W
at
2.45GHz. The actual power supplied varies during the course of the reaction in
order to
maintain a constant temperature.
Preparation 1: 4-(5-Methyl-[1,3,4]oxadiazol-2-yl)-pipe6dine-1-carboxylic acid
tert-butyl
ester.
N_N
~N1\~O ~
O
To a solution of 9.0 g of 4-Hydrazinocarbonyl-piperidine-1 -carboxylic acid
tert-butyl ester
(see reference WO 9703986 Al 19970206) (37 mmoles, 1 eq.) in 40 ml of THF,
were
added 8.1 ml of dimethylformamide dimethyl acetal (55.4 mmoles, 1.5 eq.). The
reaction mixture was then stirred at 50 C for 4 hours under nitrogen. The
solvent was
removed under reduced pressure, the residue dissolved in 40 ml of toluene, and
400 mg
of para toluene sulfonic acid were added. The mixture was then heated at 100 C
under
nitrogen for 18 hours, the volatiles were removed under reduced pressure and
the
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-46-
residue was partitioned between methylene chloride and an aqueous solution of
sodium
bicarbonate. The organic phase was dried over magnesium sulfate and filtered.
The
volatiles were removed under reduced pressure and the residue was purified by
column
chromatography on silica gel using methylene chloride/methanol as eluant (98:2
v/v to
95:5 v/v) to afford 8.07 g of the title compound as a white solid (81 %).
'H NMR (400MHz, CD3OD): S 1.42 (s, 9H), 1.70 (m, 2H), 2.05 (m, 2H,), 2.50 (s,
3H),
3.00 (m, 2H), 3.15 (m, 1 H), 4.05 (m, 2H); LCMS: m/z APCI+ , 268 [MH]+; Found;
C,
58.26%; H, 7.96%; N, 15.78%; C13H21N303 requires C, 58.41%, H, 7.92%, N,
15.72%
Preparation 2a: 4-[4-(4-Chloro-phenyl)-5-methyl-4H-[1,2,4]triazol-3-yl]-
piperidine:
/N
N ~
N N
H cl
4.0 g of the compound of preparation 1 (15 mmoles, 1 eq.) were dissolved in
100 ml of
toluene. 2.1 g of para chloro aniline (16.5 mmoles, 1.1 eq.) were added,
followed by 2
ml of TFA. The solution was heated at 110 C for 16 hours, 2 ml of TFA were
added, and
the solution was heated at 110 C for a further 48 hours. The reaction mixture
was then
cooled, an aqueous solution of sodium bicarbonate added and the organic phase
was
decanted. The aqueous phase was basified with potassium carbonate and
extracted
four times with methylene chloride (50 mi). The methylene chloride solution
was dried
over magnesium sulfate and the solvent was removed in vacuo, to afford 2.90 g
of the
title compound as a white solid.
'H NMR (400MHz, CDCI3): S 1.60-2.00 (m), 2.20 (s, 3H), 2.40-2.80 (m, 5H,),
3.10 (m,
2H), 7.10 (d, 2H), 7.55 (d, 2H); LCMS: m/z APCI+ , 277 [MH]+
Preparation 2b: 4-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidine
hydrochloride
N/N\YCH3
N
N CI
HCI
Hydrochloric acid in dioxan (4M, 10 mL) was added to a cooled (5 C) solution
of the
compound of preparation 1 (1.32 g, 4.76 mmol) in methanol (30 mL), and the
solution
was allowed to warm to room temperature with stirring for a further 90
minutes. Tic
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-47-
analysis showed that starting material remained, so additional hydrochloric
acid in dioxan
(4M, 10 mL) was added and the reaction was stirred for a further 4 hours. The
mixture
was concentrated under reduced pressure and the residue was azeotroped with
toluene
(3x) to afford the title compound, 1.4 g.
'HNMR (400MHz, DMSO-d6): 8 1.86 (m, 4H), 2.25 (m, 3H), 2.80-2.97 (m, 3H), 3.22
(m,
2H), 7.64 (m, 2H), 7.77 (d, 2H).
Preparation 3: 1-(3-Chloro-benzoyl)-piperidine-4-carboxylic acid ethyl ester
N
I \ `
ci cH3
0
A solution of ethyl isonipecotate (27.6 g, 175 mmol) in dichloromethane (50
ml) was
added dropwise over 10 minutes to a solution of 3-chlorobenzoyl chloride (20
ml, 160
mmol) and triethylamine (28 ml, 200 mmol) in dichloromethane (500 mi) cooled
to
between 10 and 15 C. The reaction was then stirred at room temperature for 3
hours
and concentrated under reduced pressure. The residue was diluted with ether,
the
solution washed with 1 N hydrochloric acid, sodium carbonate solution (x3) and
brine. It
was then dried over MgSO4 and evaporated under reduced pressure, to give the
title
compound as a solid, 44.4 g.
'H NMR (400MHz, CDCI3): 8 1.24 (t, 3H), 1.62-2.10 (m, 4H), 2.58 (m, 1 H), 2.98-
3.16 (m,
2H), 3.70 (m, 1 H), 4.15 (q, 2H), 4.49 (m, 1 H), 7.24 (m, 1 H), 7.31-7.40 (m,
3H). LRMS :
m/z (APCI+) 296 [MH]+
Preparation 4: 1-(3-Chloro-benzoyl)-piperidine-4-carboxylic acid hydrazide
o
ci ~
~ ~ Cr'
N NH
HZN
O
A mixture of the ester of preparation 3 (44.4 g, 0.15 mol) and hydrazine
hydrate (30 ml,
0.58 mol) in methanol (120 ml) was heated under reflux for 10 hours, and then
allowed
to cool to room temperature. The mixture was concentrated under reduced
pressure
and the product crystallised from ethyl acetate/ether to afford the title
compound as a
solid, 32.5 g.
'H NMR (400MHz, CDCI3): S 1.62-1.98 (m, 4H), 2.36 (m, 1H), 2.78-3.09 (m, 2H),
3.64-
4.00 (m, 2H), 4.65 (m, 1 H), 7.04 (m, 1 H), 7.26 (m, 1 H), 7.36 (m, 3H). LRMS
: m/z
(APCI+) 282 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-48-
Preparation 5: 1-(3-Chloro-benzoyl)-piperidine-4-carboxylic acid N'-(2-chloro-
acetyl)-
hydrazide
0
I ~ ~H
CI ~ N HN` ^CI
O IxOI
Acetyl chloride (4.3 ml, 53 mmol) was added dropwise, over 30 minutes, to an
ice-cooled
solution of the hydrazide of preparation 4 (10 g, 35.5 mmol) and N-methyl
morpholine
(5.4 g, 53 mmol) in dichloromethane (200 ml), in order to maintain the
internal
temperature below 10 C. The reaction mixture was then allowed to warm to room
temperature and was stirred for a further 18 hours. The reaction was diluted
with ethyl
acetate, washed with saturated sodium bicarbonate solution, and then brine.
The
solution was dried over MgSO4, concentrated under reduced pressure and the
residue
was triturated with ether to afford the title compound as a white solid, 10.2
g.
'H NMR (400MHz, CD3OD): 8 1.66-1.84 (m, 3H), 1.98 (m, 1H), 2.61 (m, 1 H), 2.99
(m,
1 H), 3.19 (m, 1 H), 3.74 (m, 1 H), 4.14 (s, 2H), 4.60 (m, 1 H), 7.35 (dd, 1
H), 7.46 (m, 3H).
LRMS : m/z (ES+) 358 [MH]+
Preparation 6: [4-(5-Chloromethyl-[1,3,4]oxadiazol-2-yl)-piperidin-1-yl]-(3-
chloro-
phenyl)-methanone
CI / _ N
N 0__/0~
O CI
Trifluoroacetic anhydride (9.45 ml, 57 mmol) was added dropwise over 30
minutes to a
cooled solution (0 to 5 C) of the compound of preparation 5 (10.2 g, 28.5
mmol) and
pyridine (11.5 ml, 142.5 mmol) in dichloromethane (300 ml). Once the addition
was
complete, the resulting pink suspension was stirred for a further 90 minutes
at 10 C.
The reaction mixture was poured carefully into saturated sodium bicarbonate
solution
(600 ml), and the layers were separated. The organic phase was washed with
further
saturated sodium bicarbonate solution (x2), dried over MgSO4, and treated with
decolourising charcoal. The mixture was then filtered and the filtrate
evaporated under
reduced pressure to afford the title compound, 13 g.
'H NMR (400MHz, CD3OD): 8 1.80-1.97 (m, 2H), 2.08 (m, 1H), 2.22 (m, 1 H), 3.15-
3.40
(m, 3H), 3.76 (m, 1 H), 4.56 (m, 1 H), 4.84 (s, 2H), 7.37 (m, 1 H), 7.48 (m,
3H). LRMS :
m/z (APCI+) 340 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-49-
Preparation 7 : 4-[N'-(2-Chloro-acetyl)-hydrazinocarbonyl]-piperidine-l-
carboxylic acid
tert-butyl ester
0
H3C O ~N
~N N
H3C CH3 O// H CI
4-Hydrazinocarbonyl-piperidine-l-carboxylic acid tert-butyl ester (see
reference WO
2000039125, prep 27)(25 g, 103 mmol) was dissolved in dichloromethane (300 ml)
and
4-methylmorpholine (12.5 ml, 113 mmol) was then added. The mixture was cooled
using an ice bath and chloroacetyl chloride (8.2 ml, 103 mmol) was added
dropwise.
The reaction was then allowed to warm to room temperature and was stirred for
4 hours.
The reaction mixture was partitioned with aqueous sodium hydrogen carbonate
solution,
dried over magnesium sulphate, filtered and the filtrate was evaporated to
give the title
compound as an off white solid (29.6 g).
LRMS m/z APCI" 318 [M-H]-
Preparation 8: 4-(5-Chloromethyl-[1,3,4]oxadiazol-2-yl)-piperidine-1-
carboxylic acid tert-
butyl ester
p N_N
xo
CI
The hydrazide of preparation 7 (5.0 g, 15.6 mmol) was suspended in
dichloromethane
(200 ml) and then pyridine (6.4 ml, 78 mmol) was added before cooling the
mixture to
10 C. Trifluoroacetic anhydride (6.6 ml, 39 mmol) was added dropwise over 15
minutes
and then the mixture was stirred at room temperature for 3 hours. The reaction
was
then partitioned with water (50m1), the organic layer was dried over magnesium
sulphate,
filtered and the filtrate was evaporated under reduced pressure. The residue
was
purified by chromatography on silica gel using methanol in dichloromethane as
eluant
(2:98) to afford the title compound as a white solid (2.95 g).
'H NMR (400MHz, CD3OD): S 1.45 (s, 9H), 1.74 (m, 2H), 2.19 (m, 2H), 3.04 (m,
2H),
3.24 (m, 1 H), 4.09 (m, 2H), 4.85 (s, 2H)
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-50-
Preparation 9a: 4-(5-[1,2,3]Triazol-2-ylmethyl-[1,3,4]oxadiazol-2-yl)-
piperidine-1-
carboxylic acid tert-butyl ester and
4-(5-[1,2,3]Triazol-1-ylmethyl-[1,3,4]oxadiazol-2-yl)-piperidine-1-carboxylic
acid tert-butyl
ester
N-N
I ~ N \
O~ ' O~
H3CON ~N~IN H3C` _ N N~11
CH3 O N \`/ H3C/X\CH N
30
A mixture of the compound of preparation 8 (8 g, 26.5 mmol), triazole (3.7 g,
53 mmol)
and potassium carbonate (5.2 g, 38 mmol) in N,N-dimethylformamide (60 ml) was
stirred
at room temperature for 18 hours. The mixture was then filtered, and the
filtrate was
concentrated under reduced pressure. The residue was partitioned between ethyl
acetate and brine, the layers were separated and the organic solution was
dried over
MgSO4 and concentrated under reduced pressure to afford the title compounds as
a
mixture of isomers.
'H NMR (400MHz, CD3OD): 8 1.43 (s, 9H), 1.62-1.78 (m, 2H), 2.02 (m, 2H), 3.00
(m,
2H), 3.19 (m, 1 H), 4.03 (m, 2H), 5.95, 5.99 (2xs, 2H), [7.77 (s), 7.80 (d),
8.18 (s) total
2H].
Preparation 9b: 4-(5-[1,2,3]Triazol-2-ylmethyl-[1,3,4]oxadiazol-2-yl)-
piperidine-l-
carboxylic acid tert-butyl ester
Dichloromethane (500 mL) was added to a suspension of the hydrazide of
preparation
170 (132.3 g, 375 mmol) in 1-methylimidazole (120 mL), and the resulting
solution was
cooled in an ice/acetone-bath. Triflic anhydride (92 mL, 561 mmol) was added
dropwise
over 2.5 hours, in order to maintain the reaction temperature below 0 C. Once
the
addition was complete, the reaction was stirred for a further 20 minutes. It
was then
quenched by the addition of 2M hydrochloric acid (350mL). The phases were
separated
and the aqueous layer was extracted with dichloromethane (200 mL). The
combined
organic solutions were washed with brine, dried over MgSO4 and evaporated
under
reduced pressure. The residue was purified by column chromatography on silica
gel
using dichloromethane as eluant to afford the title compound as a viscous oil.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-51-
Preparation 10: (3-Chloro-phenyl)-[4-(5-[1,2,3]triazol-2-ylmethyl-
[1,3,4]oxadiazol-2-yl)-
piperidin-1-yl]-methanone
N-N
1 ~
~ \ o
CI ~ N N~- N
O 0
A mixture of the compound of preparation 6 (2 g, 5.9 mmol), triazole (810 mg,
11.75
mmol) and potassium carbonate (1.2 g, 8.85 mmol) in acetonitrile (20 ml) was
stirred at
room temperature for 30 minutes, followed by a further hour at 50 C. The
reaction
mixture was filtered, washed through with ethyl acetate and the filtrate was
concentrated
under reduced pressure. The residual brown oil was partitioned between ethyl
acetate
and water, the layers were separated, and the organic phase was washed with
additional
water, then brine. The solution was dried over MgSO4 and evaporated under
reduced
pressure. The crude product was purified by column chromatography on silica
gel using
dichloromethane:methanol (100:0 to 95:5) to afford the title compound, (202
mg).
'H NMR (400MHz, CD3OD): 8 1.76-1.90 (m, 2H), 2.02 (m, 1 H), 2.18 (m, 1 H),
3.12-3.38
(m, 3H), 3.72 (m, 1 H), 4.50 (m, 1 H), 5.97 (s, 2H), 7.37 (dd, 1 H), 7.41-7.51
(m, 3H), 7.78
(s, 2H). LRMS : m/z (ES+) 373, 375 [MH]+
Preparation 11 a: 4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-
yl]-piperidine-l-carboxylic acid tert-butyl ester
N,N
~-~
N
H,c -x O 7_
HaC CHs 101 CI
Trifluoroacetic acid (1 ml, 13.2 mmol) was added to a solution of the
compounds of
preparation 9a (8.8 g, 26.5 mmol) and 4-chloroaniline (5 g, 39.75 mmol) in
toluene (200
ml) and the reaction mixture was stirred at reflux for 5 hours, The cooled
mixture was
diluted with dichloromethane, then washed with 1 N sodium hydroxide solution
and brine,
and evaporated under reduced pressure. The residual brown oil was purified by
column
chromatography on silica gel using an elution gradient of
dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) and then re-
columned
using ethyl acetate:methanol:0.88 ammonia (100:0:0 to 90:10:1) to afford the
title
compound, (2.3 g).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-52-
'H NMR (400MHz, CD3OD): 8 1.42 (s, 9H), 1.68-1.82 (m, 4H), 2.62-2.78 (m, 3H),
4.08
(m, 2H), 5.70 (s, 2H), 7.24 (d, 2H), 7.56 (d, 2H), 7.59 (s, 2H); LRMS : m/z
(APCI+) 444
[MH]+
Preparation 11 b: 4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-yl]-
piperidine-l-carboxylic acid tert-butyl ester
N-Methylimidazole (4.66 g, 56.75 mmol) and dichloromethane (20 ml) were added
to the
bis-acyl hydrazide from preparation 170 (5.00 g, 14.19 mmol), and the
resulting solution
was cooled to -20 C. Trifluoromethanesulfonic acid anhydride (6.00 g, 21.28
mmol) was
added, keeping the temperature below 0 C. Upon completion of the addition, the
reaction was warmed to ambient temperature and stirred for 15 hours. The
reaction was
quenched with H20 (10 ml), the phases were separated and the aqueous layer was
re-
extracted with dichloromethane (10 ml). The combined organic phases were dried
over
magnesium sulphate, filtered, and the dichloromethane was distilled and
replaced with
toluene under vacuum to give a toluene solution of the intermediate oxadiazole
(-20 ml
volume). 4-Chloroaniline (1.90 g, 14.90 mmol) was added to the toluene
solution
followed by trifluoroacetic acid (0.81 g, 7.09 mmol) and the reaction was
stirred at 85 C
for 5.5 hours. The mixture was cooled to ambient temperature and stirred with
1.8N
aqueous ammonia (14 ml) for 5 minutes. The phases were separated, the organic
phase was diluted with tert-butyl methyl ether (20m1) and then stirred for 15
hours. The
resulting solid precipitate was collected by filtration, washing with tert-
butyl methyl ether
(2 x 5 ml), to give the title compound as a beige coloured solid (2.72 g). 'H
NMR
(400MHz, CDCI3) : 8 1.43 (s, 9H), 1.72 (d, 2H), 1.85 (bm, 2H), 2.56 (m, 1 H),
2.66 (bm,
2H), 4.09 (bd, 2H), 5.64 (s, 2H), 7.01 (d, 2H), 7.43 (d, 2H), 7.50 (s, 2H).
Preparation 12a: 4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-
yl]-piperidine
"\I-
N N
H cl
N 0
4M Hydrochloric acid in dioxan (10 ml) was added to a solution of the compound
of
preparation 11a (2.3 g, 5.2 mmol) in methanol (30 ml), and the reaction was
stirred at
room temperature for 2 hours. The solution was concentrated under reduced
pressure,
the residue was diluted with dichloromethane and basified with 1 N sodium
hydroxide
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-53-
solution to pH 10, and the layers were separated. The aqueous phase was re-
extracted
with dichloromethane and the combined organic solutions were dried over MgSO4
and
concentrated under reduced pressure to afford the title compound as a foam,
(1.65 g).
'H NMR (400MHz, CD3OD): S 1.79 (m, 4H), 2.48 (m, 2H), 2.65 (m, 1H), 3.02 (m,
2H),
5.70 (s, 2H), 7.22 (d, 2H), 7.55 (d, 2H), 7.59 (s, 2H); LRMS : m/z (APCI+) 344
[MH]+
Preparation 12b: 4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-
yl]-piperidine bis p-toluenesulfonate salt
N\N ~N-\
N NJ
TsOH
o = 2
N H cl
Ethyl acetate (30 ml) was added to a mixture of the compound from preparation
11 b
(6.5g, 14.6mmol) and p-toluenesulfonic acid monohydrate (8.4g, 44.2mmol) and
the
reaction was stirred at room temperature for 17 hours. The solid precipitate
was
collected by filtration, washing with ethyl acetate (20 ml) to give the title
compound as a
white solid, (9.32g).
'H NMR (400MHz, DMSO-d6): 6 1.85 (m, 4H), 2.26 (s, 6H), 2.83 (m, 3H), 3.24 (m,
2H),
5.68 (s, 2H), 7.10 (d, 4H), 7.32 (d, 2H), 7.47 (d, 4H), 7.54 (d, 2H), 7.65 (s,
2H), 8.29 (m,
1 H), 8.49 (m, 1 H).
LRMS : m/z (APCI+) 344 [MH]+
Preparation 13: Methyl 1H-tetrazol-1-ylacetate
o N=N
F13C'~ O~N,"//N
A mixture of tetrazol-1-yl acetic acid (5 g, 39 mmol) and 4M hydrochloric acid
in dioxan
(100 L) in methanol (50 mL) was heated under reflux for 18 hours. The cooled
mixture
was evaporated under reduced pressure to provide the title compound.
'H NMR (400MHz, DMSO-d6): 8 3.74 (s, 3H), 5.58 (s, 2H), 9.39 (s, 1H); LCMS:
m/z
APCI+ 143 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-54-
Preparation 14: (3-Methyl-isoxazol-5-yl)-acetoyl chloride
o O- ~
CI CH3
N,N-Dimethylformamide (few drops), followed by oxalyl chloride (9.5 mL, 106
mmol)
were added dropwise to a cooled (10 C) solution of (3-methyl-isoxazol-5-yl)-
acetic acid
(5 g, 35.4 mmol) in dichloromethane (50 mL), and the solution was allowed to
warm to
room temperature. The reaction was stirred for a further 3 hours, then
concentrated
under reduced pressure. The residue was azeotroped with toluene to afford the
title
compound.
'H NMR (400MHz, CDCI3): 8 2.30 (s, 3H), 4.32 (s, 2H), 6.18 (s, 1 H).
Preparation 15: Ethyl (2-methyl-1 H-imidazol-1 -yl)acetate
H3C
CH3 O N
ON
Potassium carbonate (8.42 g, 61 mmol) was added to a solution of 2-
methylimidazole (5
g, 61 mmol) in tetrahydrofuran (100 mL) and the suspension was stirred for 30
minutes.
Ethyl bromoacetate (6.75 mL, 61 mmol) was added and the reaction was stirred
for a
further 30 minutes at room temperature. The mixture was filtered, washing
through with
dichloromethane:methanol (90:10). The filtrate was evaporated under reduced
pressure
and the crude product was purified by column chromatography on silica gel
using
dichloromethane:methanol:0.88 ammonia (93:7:0.5) as eluant to afford the title
compound as an oil, 5.28 g.
'H NMR (400MHz, CDCI3): 8 1.26 (t, 3H), 2.35 (s, 3H), 4.22 (q, 2H), 4.58 (s,
2H), 6.81 (s,
1 H), 6.94 (s, 1 H). LCMS: m/z APCI+ 169 [MH]+
Preparation 16: 2-(1 H-Tetrazol-l-yl)acetohydrazide
N=N
H2N~N'J~N
H
Hydrazine hydrate (3.2 g, 63 mmol) was added to a solution of the ester of
preparation
13 (3 g, 21.1 mmol) in methanol (18 mL) and the mixture was heated under
reflux for 18
hours. The cooled reaction was concentrated under reduced pressure and the
residue
was azeotroped with toluene to afford the title compound.
'H NMR (400MHz, DMSO-d6): S 5.18 (s, 2H), 9.38 (s, 1 H). LCMS: m/z APCI+ 143
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-55-
Preparation 17: [1,2,3]Triazol-1-yl-acetic acid ethyl ester and [1,2,3]triazol-
2-yl-acetic
acid ethyl ester
N, i N~
' N
~
Et0~ N Et0~ N
1,2,3-Triazole (19.00 kg, 275 mol) was charged over 30 minutes to a suspension
of
potassium carbonate (42.15 kg, 305 mol) in ethanol (80 L), and was rinsed in
with
ethanol (2 L). A solution of ethyl bromoacetate (45.8 kg, 274 mol) in ethanol
(30 L) was
added slowly and was rinsed in with ethanol (2L). During this time the
reaction
temperature was maintained at <20 C. The reaction mixture was then warmed to
room
temperature and stirred overnight. The suspension was filtered; washing the
residue
with ethanol (25 L and 17 L) and then the filtrate was concentrated under
reduced
pressure. The concentrate was dissolved in ethyl acetate (120 L) and the
solution was
washed with 1 N hydrochloric acid (1 x 40 L, 7 x 20 L, 4 x 15 L). The aqueous
washings
were combined and extracted with ethyl acetate (3 x 21 L). The organic phases
were
combined, dried over magnesium sulphate, filtered and concentrated to dryness
giving a
mixture of the title compounds (25 kg). 'H NMR spectroscopic analysis
indicated that
this was a 6:5 mixture of N-2/N-1 isomers.
'H NMR (400MHz, CDCI3): 6 1.25 (m, 3H), 4.13 (q, 2H, N-1 isomer), 4.25 (q, 2H,
N-2
isomer), 5.20 (s, 2H, N-1 isomer), 5.22 (s, 2H, N-2 isomer), 7.70 (s, 2H, N-2
isomer),
7.77 (s, 2H, N-1 isomer).
Preparation 18: [1,2,3]Triazol-2-yl-acetic acid hydrazide
HZN~H~N\N/
%~
Hydrazine hydrate (8.65 kg, 270 mol) was added to a cooled (<10 C) solution of
the
mixture of esters of preparation 17 (19 kg) in ethanol (69 L) keeping the
temperature
below 20 C throughout the addition. The reaction mixture was stirred at
between 14 and
19 C for 3 hours, then more ethanol (25 L) was added and the product was
collected by
filtration, washing with ethanol (10 L). The crude solid was purified by
recrystallisation
from ethanol (120 L), followed by three recrystallisations from methanol (105
L, 120 L
and 90 L) to give the title compound, (4.53 kg) after drying in vacuo.
'H NMR (400MHz, DMSO-d6): 6 4.33 (s, 2H), 5.02 (s, 2H), 7.77 (s, 2H), 9.40 (s,
1 H).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-56-
Preparation 19: 2-(2-Methyl-1 H-imidazol-1-yl)acetohydrazide
H3C~
O N
HZN~HN~
The title compound was obtained as a white solid from the compound of
preparation 15
and hydrazine following a similar procedure to that described for preparation
16, except
that 5 equivalents of hydrazine were used, and isopropanol was used as the
reaction
solvent.
'H NMR (400MHz, CD3OD): S 2.35 (s, 3H), 4.60 (s, 2H), 6.81 (s, 1 H), 6.99 (s,
1 H).
LCMS: m/z APCI+ 155 [MH]+
Preparation 20: 2-(3-Methyl-1,2,4-oxadiazol-5-yl)acetohydrazide
\
HZN'~ N ~ \
H N CHs
The title compound was obtained from 3-methyl 1,2,4-oxadiazol-5-yl-acetic acid
methyl
ester (NL 7807076) and hydrazine following a similar procedure to that
described for
preparation 16, except that 8 equivalents of hydrazine were used, and
isopropanol was
used as the reaction solvent.
'H NMR (400MHz, CDCI3): 8 2.42 (s, 3H), 3.86 (s, 2H), 6.89 (br s, 1 H), 8.18
(br s, 1 H).
Preparation 21: 2-(Pyrimidin-2-yloxy)acetohydrazide
O N
HZN, N~O 11 ~
H N
A mixture of ethyl 2-pyrimidinyloxyacetate (GB2373186, step i ex 368) (4.4 g,
24.15
mmol) and hydrazine hydrate (5 mL, 160 mmol) in isopropanol (30 mL) was heated
under reflux for 1 hour. The mixture was then cooled and concentrated under
reduced
pressure and the residue was purified by column chromatography on silica gel
using
dichloromethane:methanol:0.88 ammonia (100:0:0 to 100:10:1) to provide the
title
compound, 600 mg.
'H NMR (400MHz, CDCI3): 8 4.98 (s, 3H), 7.04 (m, 1 H), 8.58 (d, 2H); LCMS: m/z
APCI+
169 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-57-
Preparation 22: tert-Butyl 2-[(3-methylisoxazol-5-
yl)acetyl]hydrazinecarboxylate
H3C ~N
CH3 3"CH3
H,C H NI-N
H
0
Triethylamine (24 mL, 17 mmol) was added slowly to a cooled (10 C) solution of
the acid
chloride of preparation 14 (5.64 g, 35.4 mmol) in dichloromethane (200 mL),
followed
by tert-butyl carbazate (5.6 g, 42.5 mmol) and the reaction was stirred at
room
temperature for 18 hours. The reaction was diluted with ethyl acetate and the
precipitate
was filtered off. The filtrate was concentrated under reduced pressure and the
residue
was purified by column chromatography on silica gel using an elution gradient
of ethyl
acetate:pentane (50:50 to 100:0) to provide the title compound.
'H NMR (400MHz, CDCI3): S 1.47 (s, 9H), 2.30 (s, 3H), 3.77 (s, 2H), 6.15 (s, 1
H), 6.45
(br s, 1 H), 7.59 (br s, 1 H).
Preparation 23: tert-Butyl 2-[3-(3,5-dimethylisoxazol-4-yl)propanoyl]
hyd razi n eca rboxyl ate
H H3C
H,C,_~ 3 CH3 0
H j
O_'~N-N
H 0
0 H,c
Oxalyl chloride (5.16 mL, 59.2 mmol) was added to a solution of R-(3,5-
dimethyl-4-
isoxazolyl)propionic acid (J. Org. Chem. 59(10); 1994; 2882) (2.5 g, 14.8
mmol) in
dichloromethane (50 mL) and N,N-dimethylformamide (1 drop), and the solution
was
stirred at room temperature for 30 minutes. The mixture was concentrated under
reduced pressure and the residue was azeotroped with dichloromethane (5x) to
provide
a brown liquid. This was dissolved in dichloromethane (25 mL), and tert-butyl
carbazate
(2.93 g, 22.2 mmol) was added portionwise. The mixture was diluted with
further
dichloromethane (23 mL) and the reaction was stirred for 18 hours at room
temperature.
The mixture was concentrated under reduced pressure, the residue was suspended
in
dichloromethane, the resulting precipitate was filtered off and the filtrate
was evaporated
under reduced pressure. The residue was purified by column chromatography on
silica
gel using an elution gradient of dichloromethane:methanol:0.88 ammonia
(95:5:0.5 to
90:10:1) to provide the title compound as an oil, 3.08 g.
'H NMR (400MHz, CDCI3): S 1.45 (s, 9H), 2.21 (s, 3H), 2.36 (m, 4H), 2.45 (m,
1H), 2.60-
2.73 (m, 2H), 6.48 (br s, 1 H), 7.42 (br s, 1 H). LCMS: m/z ES+ 306 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-58-
Preparation 24: 2-(3-Methylisoxazol-5-yl)acetohydrazide hydrochloride
0 0N
HCIN\H CH3
A mixture of the compound of preparation 22 (1.6 g, 6.3 mmol) in 4M
hydrochloric acid
in dioxan (20 mL) and methanol (60 mL) was stirred at room temperature for 3
hours.
The solution was concentrated under reduced pressure to low volume, the
resulting
precipitate was filtered off, washed with dichloromethane and dried to afford
the title
compound, 810 mg.
'H NMR (400MHz, DMSO-d6): 5 2.20 (s, 3H), 3.86 (s, 2H), 6.24 (s, 1H).
LCMS: m/z APCI+ 156 [MH]+
Preparation 25: 3-(3,5-Dimethylisoxazol-4-yl)propanohydrazide hydrochloride
O H3
HZN\H /
.HCI 0
H3C
A solution of the compound of preparation 23 (3.08 g, 10.87 mmol) in 2.2M
methanolic
hydrochloric acid (50 mL) was stirred at room temperature for 18 hours. The
solution
was concentrated under reduced pressure and the residue was azeotroped with
toluene.
The crude product was triturated with pentane/ether and then ether, and the
resulting
solid was filtered off to provide the title compound as an off-white solid,
1.39 g.
'H NMR (400MHz, DMSO-d6): S 2.22 (s, 3H), 2.27 (t, 2H), 3.35 (s, 3H), 2.66 (t,
2H), 6.75
(br s, 1 H). LCMS: m/z APCI+ 184 [MH]+
Preparation 26: tert-Butyl 2-(hydrazinocarbonyl)morpholine-4-carboxylate
o ~--~
H3C ~- N O
H3C+-0
H3C 0
H2N-N
A mixture of 4-(tert-butyl) 2-methyl 2,4-morpholinecarboxylate (WO 03/018576,
ex 1 part
i)(2.03 g, 8.3 mmol), hydrazine hydrate (1.2 mL, 24 mmol) and methanol (50 mL)
was
heated under reflux for 4 days. The cooled mixture was evaporated under
reduced
pressure, the residue was partitioned between water and ethyl acetate, and the
layers
were separated. The organic phase was dried over MgSO4 and evaporated under
reduced pressure to provide the title compound, 1.92 g.
LCMS: m/z ES+ 268 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-59-
Preparation 27: tert-Butyl 3-(hydrazinocarbonyl)piperidine-l-carboxylate
0
H3C N
H3C~)-0
H3C 0
HZN-H
Hydrazine hydrate (75 mL, 1.5 mol) was added to a solution of piperidine-1-3-
dicarboxylic acid 1 tert-butyl 3-ethyl ester (US 2002 0099035, ex 12) (72 g,
280 mmol) in
ethanol (250 mL) and the reaction was heated under reflux for 18 hours. The
cooled
mixture was concentrated under reduced pressure and the residue was
partitioned
between dichloromethane and water, and the layers were then separated. The
aqueous
phase was extracted with dichloromethane, and the combined organic solutions
were
dried over MgSO4 and evaporated under reduced pressure. The product was
azeotroped with ether to afford the title compound as a colourless gum, 59.8
g.
'H NMR (400MHz, CDCI3): 8 1.40-1.50 (m, 11 H), 1.63 (m, 1 H), 1.83 (m, 2H),
2.25 (m,
1 H), 2.97 (m, 1 H), 3.16 (m, 1 H), 3.78-3.98 (m, 3H), 7.40-7.60 (br s, 1 H).
Preparation 28: Ethyl 1 -(4-ch lorobe nzoyl)pi pe rid i ne-4-ca rboxylate
ci 0
C N 0/\CH3
0
The title compound was obtained as a yellow oil from ethyl isonipecotate and 4-
chlorobenzoyl chloride, following a similar procedure to that described for
preparation 3.
'H NMR (400MHz, CDCI3): 8 1.24 (t, 3H), 1.62-2.06 (m, 4H), 2.59 (m, 1H), 3.03
(m, 2H),
3.72 (m, 1 H), 4.17 (q, 2H), 4.54 (m, 1 H), 7.36 (d, 2H), 7.39 (d, 2H). LCMS:
m/z APCI+
296 [MH]+
Preparation 29: 1-(4-Chlorobenzoyl)piperidine-4-carbohydrazide
ci o
N--NH2
N H
0
A solution of the compound of preparation 28 (148 g, 0.5 mol) in methanol (400
mL)
was heated at 70 C for 30 minutes. Hydrazine hydrate (50 g, 1.0 mol) was then
added
and the reaction was stirred at 60 C for a further 3 hours. Tic analysis
showed that
starting material remained, so additional hydrazine hydrate (50 mL, 1.0 mol)
was added
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-60-
and the reaction was stirred for a further 48 hours at 75 C. The cooled
mixture was
concentrated under reduced pressure, the residue was suspended in
dichloromethane (1
L) and washed with water (2x). The organic solution was dried over MgSO4 and
evaporated under reduced pressure to afford the title compound as a white
solid, 119 g.
'HNMR (400MHz, CDCI3): S 1.65-1.94 (m, 4H), 2.35 (m, 1H), 2.80-3.06 (m, 2H),
3.79
(m, 1 H), 4.65 (m, 1 H), 7.10 (s, 1 H), 7.38 (m, 4H). LCMS: m/z APCI+ 282
[MH]+
Preparation 30: 1-(4-Chlorobenzoyl)-M-(trifluoroacetyl)piperidine-4-
carbohydrazide
CI F
N~_N
H F
H F
__~
O
0
Trifluoroacetic anhydride (1.56 mL, 11.18 mmol) was added dropwise to an ice-
cooled
solution of the compound of preparation 29 (3.0 g, 10.65 mmol) and N-
methylmorpholine (1.29 mL, 11.7 mmol) in dichloromethane (50 mL), and the
reaction
was stirred at room temperature for 18 hours. The resulting precipitate was
filtered off,
washed with dichloromethane and dried to afford the title compound, 1.78 g.
'H NMR (400MHz, DMSO-d6): 6 1.56 (m, 2H), 1.64-1.84 (m, 2H), 2.56 (m, 1H),
2.85 (m,
1 H), 3.08 (m, 1 H), 3.58 (m, 1 H), 4.40 (m, 1 H), 7.40 (d, 2H), 7.50 (d, 2H),
10.20 (s, 1 H),
11.15 (br s, 1 H).
Preparation 31: 1-(4-Chlorobenzoyl)-M-(ethoxyacetyl)piperidine-4-
carbohydrazide
ci H
)a,NCr'H N~O CH3
O
0
N-Methylmorpholine (2.60 g, 26.6 mmol), and then ethoxyacetyl chloride (WO
01/46150
ex 33A) (1.09 g, 8.87 mmol) were added to a solution of the compound of
preparation
29 (2.5 g, 8.87 mmol) in dichloromethane (70 mL), and the reaction was stirred
at room
temperature for 18 hours. The mixture was washed with water, then ammonium
chloride
solution and finally sodium carbonate solution. It was dried over MgSO4 and
evaporated
under reduced pressure to afford the title -compound.
'H NMR (400MHz, CDCI3): 8 1.22 (t, 3H), 1.72-1.99 (m, 4H), 2.56 (m, 1H), 2.86-
3.06 (m,
2H), 3.60 (q, 2H), 3.80 (m, 1 H), 4.04 (s, 2H), 4.62 (m, 1 H), 7.38 (m, 4H),
8.90 (d, 1 H),
8.99 (d, 1 H). LCMS: m/z ES+ 368, 370 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-61-
Preparation 32: 1-(3-Chlorobenzoyl)-M-(ethoxyacetyl)piperidine-4-
carbohydrazide
o
CI
N
0_N
H- CH3
O O
The title compound was obtained in 91 % yield from the compound of preparation
4 and
ethoxyacetyl chloride (WO 01/46150 ex 33A) following the procedure described
for
preparation 31.
'H NMR (400MHz, CDCI3): 8 1.22 (t, 3H), 1.72-2.00 (m, 4H), 2.56 (m, 1H), 2.84-
3.10 (m,
2H), 3.60 (q, 2H), 3.79 (m, 1 H), 4.04 (s, 2H), 4.62 (m, 1 H), 7.26 (m, 1 H),
7.38 (m, 3H),
8.61 (d, 1 H), 8.75 (d, 1 H). LCMS: m/z APCI+ 368, 370 [MH]+
Preparation 33: tert-Butyl 4-{[(4-chlorophenyl)amino]carbonyl}piperidine-l-
carboxylate
H,c o
H,c-~-o
-
H3C N~\__~N \ / CI
O H __
1-BOC-piperidine-4-carboxylic acid (100 g, 437 mmol), 4-chloroaniline (61.2 g,
480
mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (100 g, 524
mmol)
and triethylamine (182.6 mL, 1.31 mol) were dissolved in cold (10 C)
acetonitrile (1.75
L). The reaction mixture was stirred for 54 hours at room temperature and then
concentrated under reduced pressure. The residue was dissolved in ethyl
acetate and
then partitioned with 2N hydrochloric acid. The resulting precipitate was
filtered off, re-
dissolved in dichloromethane, the solution was dried over MgSO4 and evaporated
under
reduced pressure. The residue was triturated with ether to afford the desired
compound
as a white solid.
The filtrate was separated and the organic layer was washed with 2N
hydrochloric acid
(2x), dried over MgSO4 and evaporated under reduced pressure. The solid was
triturated with ether to afford further compound as a white solid, combined
yield 99.4 g.
'H NMR (400MHz, CDCI3): 8 1.46 (s, 9H), 1.68-1.80 (m, 2H), 1.90 (m, 2H), 2.39
(m, 1H),
2.79 (m, 2H), 4.19 (m, 2H), 7.10 (s, 1 H), 7.26 (d, 2H), 7.46 (d, 2H). LCMS:
m/z APCI+
339 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-62-
Preparation 34: tert-Butyl 4-{[(4-chlorophenyl)amino]carbonothioyl}piperidine-
1-
carboxylate
H3c s
H3C--~-0 -
H3C I7~N\\--~N / CI
O H
A solution of the compound of preparation 33 (99.4 g, 294 mmoL) and Lawesson's
reagent (30 g, 74.3 mmol) in toluene (1 L) was heated under reflux for 1 hour,
then
stirred at room temperature for a further 18 hours. Tlc analysis showed that
starting
material remained, so additional Lawesson's reagent (11.1 mmol) was added and
the
reaction was heated under reflux for a further hour. The cooled mixture was
concentrated under reduced pressure and the residue was azeotroped with ethyl
acetate. The crude product was triturated with hot ethyl acetate, the
resulting solid was
filtered off and dried to afford the title compound as a white solid, 53 g.
LCMS: m/z APCI" 353 [M-H]"
Preparation 35: Ethyl 1-{[(4-chlorophenyl)amino]carbonothioyl}piperidine-4-
carboxylate
~s
~o _
N CI
H3C N ~ ~
o H
A mixture of 4-chlorophenyl isothiocyanate (3.5 g, 20.7 mmol), and ethyl
isonipecotate
(3.19 mL, 20.7 mmol) in dichloromethane (30 mL) was stirred at room
temperature for 1
hour. The mixture was concentrated under reduced pressure, the residue was
triturated
with ether and the resulting solid was filtered off and dried to afford the
title compound
as a white solid, 6.27 g.
'H NMR (400MHz, CDCI3): S 1.24 (t, 3H), 1.82 (m, 2H), 1.99 (m, 2H), 2.60 (m,
1H), 3.34
(m, 2H), 4.19 (q, 2H), 4.38 (m, 2H), 7.09 (d, 2H), 7.17 (br s, 1 H), 7.30 (d,
2H).
LCMS: m/z APCI+ 327 [MH]+
Preparation 36: N-(4-Chlorophenyl)-3-methylpiperazine-1-carbothioamide
HN N--Y _
H3C~ H CI
~ ~
A solution of 4-chlorophenyl isothiocyanate (8.0 g, 47.17 mmol) in
dichloromethane (250
mL) was added dropwise over 30 minutes to an ice cooled solution of 2-
methylpiperazine (9.45 g, 94.33 mmol) in dichloromethane (250 mL). Once the
addition
was complete, the reaction was stirred at room temperature for an hour. The
reaction
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-63-
was then washed with water (3x), dried over MgSO4 and concentrated under
reduced
pressure, to give the title compound as a white solid, 11.8 g.
'H NMR (400MHz, CDCI3): S 1.08 (d, 3H), 2.70 (m, 1H), 2.88 (m, 2H), 3.02 (m,
2H), 4.43
(m, 2H), 7.10 (m, 2H), 7.29 (m, 2H). LCMS: m/z ES+ 270.1 [MH]+
Preparation 37: N-(4-Chlorophenyl)-4-(2,2-dimethylpropanoyl)-3-
methylpiperazine-l-
carbothioamide
H3C S
H C ~N-~ CI
~N\ N ~ ~
H'C H
O
H3C
Di-tert-butyl dicarbonate (9.30 g, 42.6 mmol) was added to a solution of the
compound
of preparation 36 (11.5 g, 42.6 mmol) in dichloromethane (300 mL) and dioxan
(100
mL) and the reaction was stirred at room temperature for 3 hours. The mixture
was
concentrated under reduced pressure and the product was recrystallised from
methanol.
The resulting solid was filtered off, and the filtrate was evaporated under
reduced
pressure. The residue was recrystallised again from methanol to afford the
title
compound, 9.64 g.
'H NMR (400MHz, DMSO-d6): 8 1.05 (d, 3H), 1.39 (s, 9H), 3.17-3.36 (m, 2H),
3.58 (m,
1 H), 3.77 (m, 1 H), 4.14 (m, 1 H), 4.40 (m, 2H), 7.32 (m, 4H), 9.36 (s, 1 H).
LCMS: m/z
APCI+ 370 [MH]+
Preparation 38: N-(4-Chlorophenyl)-4-(2,2-dimethylpropanoyl)-2-
methylpiperazine-l-
carbothioamide
H3C S
H3C-C NN~ / CI
H3C ~ H__
C CH3
A solution of 4-chlorophenylisothiocyanate (5.1 g, 30 mmol) and 4-N-BOC-2-
methylpiperazine (6.0 g, 30 mmol) in dichloromethane (250 mL) was stirred at
room
temperature for 2 hours. The reaction mixture was evaporated under reduced
pressure
to afford the title compound as a white foam.
LCMS: m/z APCI+ 370 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-64-
Preparation 39: Ethyl 1-{[(4-chlorophenyl)amino]carbonothioyl}-4-
methylpiperidine-4-
carboxylate
/-p H S
-
H3C N4
p H ~ ~ CI
A mixture of 4-chlorophenyl isothiocyanate (2.36 g, 13.98 mmol), and ethyl 4-
methylpiperidine-4-carboxylate (US 2002/0086887, example 532C) (2.17 g, 12.71
mmol)
in dichloromethane (50 mL) was stirred at room temperature for 30 minutes. The
mixture was partitioned between dichloromethane and brine, and the layers were
separated. The organic phase was dried over MgS04 and evaporated under reduced
pressure. The crude product was purified by column chromatography on silica
gel using
dichloromethane:ethyl acetate (100:0 to 90:10) to provide the title compound
as a white
solid, 3.46 g.
'H NMR (400MHz, CDCI3): 8 1.22 (m, 6H), 1.58 (m, 2H), 2.18 (m, 4H), 3.30 (m,
2H),
4.19 (q, 2H), 4.24 (m, 1 H), 7.09 (d, 2H), 7.28 (d, 2H); LCMS: m/z APCI+ 341
[MH]+
Preparation 40: N-(4-Chlorophenyl)-4-cyano-4-phenylpiperidine-l-carbothioamide
NC S
N
IC
Triethylamine (1.4 mL, 10 mmol) was added to a suspension of 4-chlorophenyl
isothiocyanate (1.69 g, 10 mmol) and 4-cyano-4-phenylpiperidine hydrochloride
(2.22 g,
10 mmol) in dichloromethane (100 mL), and the reaction was then stirred at
room
temperature for 20 minutes. The mixture was washed with 2N hydrochloric acid,
then
brine, it was dried over MgS04 and evaporated under reduced pressure to afford
the title
compound as a white solid, 3.62 g.
'H NMR (400MHz, CDCI3): 8 2.18 (m, 4H), 3.50 (m, 2H), 4.78 (m, 2H), 7.08 (d,
2H),
7.27-7.48 (m, 7H).
Preparations 41 to 44:
s
- / cl
R~Q A N
H
A mixture of 4-chlorophenyl isothiocyanate (1 eq.) and the appropriate amine
(1 eq.) in
ethanol (0.8-1.28 mLmmol"') was stirred at room temperature for 30 minutes.
The
reaction mixture was then evaporated under reduced pressure to afford the
title
compounds as a white solid.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-65-
Prep. x ~ Data
R~
No Q
41 cH3 r 'H NMR (400MHz, CDC13): 8 1.45 (s, 9H), 3.50
N-
H3C-~ C ~N~ (m, 4H), 3.80 (m, 4H), 7.10 (d, 2H), 7.25 (d,
3 0 2H), 7.80 (s, 1 H). LCMS: m/z APCI+ 356 [MH]+
42 HC 'H NMR (400MHz, CDCI3): 8 2.10 (s, 3H), 3.60
r N (m, 2H), 3.78 (m, 4H), 4.02 (m, 2H), 7.14 (d,
0 2H), 7.30 (d, 2H). LCMS: m/z APCI+ 298 [MH]+
43 0 'H NMR (400MHz, CDCI3): S 2.04-2.24 (m, 4H),
H3cl~N 2.85 (m, 1 H), 2.94 (s, 3H), 3.62 (m, 2H), 3.95
CH3 (m, 2H), 5.25 (m, 1 H), 6.60 (s, 1 H), 7.30 (s,
4H). LCMS: m/z APCI+ 312 [MH]+
44 0 'H NMR (400MHz, CDCI3): S 1.29-1.54 (m,
C p H
H ~~ 11 H), 2.01 (m, 2H), 3.20 (m, 2H), 3.74 (br s,
3c~
H3C 1 H), 4.35-4.55 (m, 3H), 7.09 (d, 2H), 7.17 (br s,
1 H), 7.32 (d, 2H).
Preparation 45: tert-Butyl 4-{[(4-chlorophenyl)amino]carbonothioyl}-1,4-
diazepane-l-
carboxylate
H3C
S
H3C C z~ ~ ~ CI
~N
H3C O// H ~ ~
A mixture of 4-chlorophenyl isothiocyanate (5.0 g, 29.95 mmol), and BOC-
homopiperazine (6.0 g, 29.95 mmol) in ethanol (50 mL) was stirred at room
temperature
for 2 hours. The mixture was evaporated under reduced pressure, the residue
was
partitioned between ethyl acetate and water and the layers were separated. The
organic
phase was dried over MgSO4 and evaporated under reduced pressure to give the
title
compound as a white foam.
'H NMR (400MHz, CD3OD): 8 1.45 (s, 9H), 1.98 (m, 2H), 3.43 (m, 2H), 3.64 (m,
2H),
3.94-4.10 (m, 4H), 7.28 (s, 4H). LCMS: m/z APCI+ 370 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-66-
Preparation 46: tert-Butyl 4-[(Z)-[(4-
chlorophenyl)imino](methylthio)methyl]piperidine-1-
carboxylate
0 ~ /S-CH3
H3C N\ j--(\
H3C ~-~ // \\N CI
3
Potassium tert-butoxide (20.1 g; 0.18 mol) was added portionwise to a cooled
(10 C)
solution of the compound of preparation 34 (53 g, 0.15 mol) in tetrahydrofuran
(1 L), in
order to maintain the temperature at 10 C. Methyl iodide (11.2 mL, 0.18 mol)
was added
dropwise, in order to maintain the temperature at 10 C, and the reaction was
then
allowed to warm slowly to room temperature. The reaction was stirred for a
further 90
minutes, then it was quenched by the addition of water. The reaction was
diluted with
ethyl acetate and washed with water. The phases were separated, the aqueous
layer
was extracted with further ethyl acetate, and the combined organic solutions
were dried
over MgSO4 and evaporated under reduced pressure. The residual oil was
adsorbed
onto silica gel and purified by column chromatography on silica gel using
pentane:ethyl
acetate (75:25) as eluant to afford the title compound as an oil that
crystallised upon
standing.
'H NMR (400MHz, CDCI3): 8 1.43 (s, 9H), 1.57-1.82 (m, 5H), 2.35 (s, 3H), 2.42-
2.62 (m,
1 H), 2.78 (m, 1 H), 4.16 (m, 2H), 6.65 (d, 2H), 7.26 (d, 2H). LCMS: m/z ES+
391 [MNa]+
Preparation 47: Ethyl 1-[(Z)-[(4-
chlorophenyl)imino](methylthio)methyl]piperidine-4-
carboxylate
O ``-~'"H9
H3C ~-CN
`' -
~O N \ /
CI
Potassium tert-butoxide (2.58 g, 23.1 mmol) was added portionwise to a
solution of the
compound of preparation 35 (6.27 g, 19.2 mmol) in tetrahydrofuran (100 mL) and
the
reaction was stirred for 10 minutes. Methyl iodide (1.44 mL, 23.1 mmol) was
added and
the reaction was stirred at room temperature for a further 30 minutes. The
reaction was
diluted with ether, and washed with brine. The organic solution was evaporated
under
reduced pressure and the resulting orange solid was purified by column
chromatography
on silica gel using dichloromethane as eluant to afford the title compound as
an oil, 3.6
9=
'H NMR (400MHz, CDCI3): S 1.25 (t, 3H), 1.78 (m, 2H), 1.98 (m, 2H), 2.04 (s,
3H), 2.56
(m, 1 H), 3.01 (m, 2H), 4.12-4.23 (m, 4H), 6.80 (d, 2H), 7.20 (d, 2H); LCMS:
m/z ES+
341 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-67-
Preparation 48: Ethyl 1-[(Z)-[(4-chlorophenyl)imino](methylthio)methyl]-4-
methylpiperidine-4-carboxylate
0 /S-CH3
H3C ~~-~N--(\ D \-O H3C \N < CI
The title compound was obtained as an oil in 75% yield from the compound of
preparation 39 and methyl iodide, following a similar procedure to that
described for
preparation 47, except that the product was not purified by column
chromatography on
silica gel.
'H NMR (400MHz, CDCI3): S 1.25 (m, 6H), 1.50 (m, 2H), 2.04 (s, 3H), 2.18 (m,
2H), 3.19
(m, 2H), 3.98 (m, 2H), 4.19 (q, 2H), 6.80 (m, 2H), 7.20 (d, 2H); LCMS: m/z ES+
355
[MH]+
Preparations 49 to 55:
Rx \ S-CH3
Q A ~
N \ / CI
Potassium tert-butoxide (1.1 eq.), followed by methyl p-toluenesulphonate (1.1
eq.) was
added to a solution of a compound selected from preparations 37, 38, 40-42 and
45 (1
eq.) in tetrahydrofuran and the reaction was stirred at room temperature for 2
hours.
The mixture was concentrated under reduced pressure and the residue was
partitioned
between ethyl acetate and water, the layers were separated and the organic
solution
was washed with water (3x), dried over MgSO4 and evaporated under reduced
pressure
to give the title compounds.
Prep. x ~ Data
No R~Q
49 ~H3 ~ 'H NMR (400MHz, CDCI3): 8 1.20 (d, 3H), 1.44 (s,
H3C~0ff-N W*I
9H), 2.00 (s, 3H), 2.99 (m, 1 H), 3.19 (m, 2H), 3.88
H3C p ~
H 3 c (m, 1 H), 4.16 (m, 1 H), 4.20-4.36 (m, 2H), 6.83 (m,
2H), 7.21 (m, 2H). LCMS: m/z APCI+ 384 [MH]+
50 H3c CHO ~~ 'H NMR (400MHz, CDCI3): S 1.21 (d, 3H), 1.42 (s,
H3c~ 0 9H), 2.00 (s, 3H), 2.80-3.30 (m, 3H), 3.80-4.18 (m,
CH3
3H), 4.54 (m, 1 H), 6.80 (m, 2H), 7.20 (m, 2H).
LCMS: m/z APCI+ 384 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-68-
51 Ph - 'H NMR (400MHz, CDCI3): 8 2.06 (s, 3H), 2.17 (m,
2H), 3.38-3.52 (m, 4H), 4.50 (m, 2H), 6.83 (d, 2H),
N 7.22 (d, 2H), 7.38 (m, 1H), 7.42 (m, 2H), 7.52 (d,
2H).
52 cH3 ~~ H NMR (400MHz, CDCI3): S 1.46 (s, 9H), 2.02 (s,
H3c H3C ~N\,J 3H), 3.48 (m, 4H), 3.58 (m, 4H), 6.80 (d, 2H), 7.20
(d, 2H).
53 H C r~\N` 'H NMR (400MHz, CDC13): S 2.01 (s, 3H), 2.10 (s,
. 3\lr N 3H), 3.48-3.76 (m, 8H), 6.80 (d, 2H), 7.19 (d, 2H).
o LCMS: m/z APCI+ 312 [MH]+
54 H3C 'H NMR (400MHz, CDCI3): S 1.98 (s, 3H), 2.09 (s,
H3c--~3H), 2.84 (m, 1 H), 2.90 (s, 3H), 3.38 (m, 1 H), 3.46
o (m, 1 H), 3.77 (m, 2H), 5.22 (m, 1 H), 6.82 (d, 2H),
7.19 (d, 2H). LCMS: m/z APCI+ 326 [MH]+
55 cH30 'H NMR (400MHz, CD3OD) : 5 1.45 (2xs, 9H), 1.90
H3o \ff-N(m, 5H), 3.44 (m, 2H), 3.60 (m, 2H), 3.76 (m, 2H),
H3C o 3.80 (m, 2H), 6.85 (d, 2H), 7.20 (d, 2H).
A = the reaction was stirred at room temperature for 18 hours, and the product
was
additionally purified by column chromatography on silica gel using
dichloromethane:methanol as eluant.
Preparation 56: Methyl 4-[(tert-butoxycarbonyl)amino]-N-(4-
chlorophenyl)piperidine-l-
carbimidothioate
H3L` H3 oII S-(':H3
N~N CI
H3C /\ O jj H-0
Potassium tert-butoxide (12.8 g, 114 mmol) was added to a suspension of the
compound of preparation 44 (42.3 g, 114 mmol) in tetrahydrofuran (400 mL) and
the
suspension was stirred for 10 minutes at room temperature. Methyl p-
toluenesulphonate
(21.29 g, 114 mmol) was added and the reaction was stirred for 10 minutes.
Additional
potassium tert-butoxide (641 mg, 5.7 mmol) and methyl p-toluenesulphonate
(1.08 g, 5.7
mmol) were added and the reaction was stirred for a further 10 minutes. The
mixture
was diluted with ether, washed with water (200 mL) and brine, then dried over
MgSO4
and evaporated under reduced pressure to afford the title compound.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-69-
'H NMR (400MHz, CDCI3): 8 1.34-1.52 (m, 11H), 2.00 (m, 2H), 2.05 (s, 3H), 3.04
(m,
2H), 3.68 (br s, 1 H), 4.19 (m, 2H), 4.50 (br s, 1 H), 6.80 (d, 2H), 7.20 (d,
2H). LCMS: m/z
ES+ 384 [MH]+
Preparation 57: tert-Butyl 4-[5-(methoxymethyl)-1,3,4-oxadiazol-2-
yl]piperidine-1-
carboxylate
H 3 NO~N
~
H C ~N O
H3C
O CH3
Potassium tert-butoxide (3.40 g, 30.3 mmol) was added to a solution of the
compound of
preparation 8 (7.62 g, 25.25 mmol) in methanol (120 mL) and the reaction was
stirred
at room temperature for 18 hours. Tlc analysis showed that starting material
remained,
so additional potassium tert-butoxide (1 g, 8.9 mmol) was added and the
reaction was
stirred at 50 C for 2 hours. The mixture was concentrated under reduced
pressure and
the residue was partitioned between ethyl acetate and ammonium chloride
solution. The
layers were separated, the organic phase was dried over MgSO4 and evaporated
under
reduced pressure to afford the title compound as a yellow oil, 7.30 g.
'H NMR (400MHz, CDC13): S 1.45 (s, 9H), 1.82 (m, 2H), 2.08 (m, 2H), 2.96 (m,
2H), 3.08
(m, 1 H), 3.44 (s, 3H), 4.10 (m, 2H), 4.61 (s, 2H). LCMS: m/z APCI+ 298 [MH]+
Preparation 58: tert-Butyl 4-{5-[(2,2,2-trifluoroethoxy)methyl]-1,3,4-
oxadiazol-2-
yl}piperidine-1-carboxylate
N~N
H3C~O~_N O I F
H3C /
O~ 1iF
H3C
O F
Potassium tert-butoxide (1.8 g, 41.8 mmol) was added to a solution of 2,2,2-
trifluoroethanol (4.64 g, 46.4 mmol) in tetrahydrofuran (100 mL) and the
solution was
stirred at room temperature for 10 minutes. The compound of preparation 8 (7.0
g,
23.2 mmol) was added and the mixture was then heated at 50 C for 2 hours. The
reaction was quenched by the addition of ammonium chloride solution, then the
organic
layer was decanted off and evaporated under reduced pressure. The residue was
re-
dissolved in ethyl acetate, the solution was washed with brine, dried over
MgSO4 and
then evaporated under reduced pressure to afford the title compound as a
yellow oil,
8.15 g.
'H NMR (400MHz, CDCI3): 8 1.43 (s, 9H), 1.80 (m, 2H), 2.06 (m, 2H), 2.97 (m,
2H), 3.10
(m, 1 H), 3.96 (q, 2H), 4.12 (m, 2H), 4.82 (s, 2H).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-70-
Preparation 59: tert-Butyl 4-[5-(hydroxymethyl)-1,3,4-oxadiazol-2-
yl]piperidine-l-
carboxylate
H 3 C N-1
-O N
H3C I
H3C ~N O
0 OH
Potassium acetate (5.2 g, 53.0 mmol) was added to a solution of the compound
of
preparation 8 (8 g, 26.5 mmol) in acetonitrile (150 mL), and the reaction was
heated at
80 C for 18 hours. The cooled mixture was concentrated under reduced pressure
and
the residue was partitioned between water and ethyl acetate, and the layers
were
separated. The organic phase was washed with brine, dried over MgSO4 and
evaporated under reduced pressure. The residual oil was dissolved in methanol
(120
mL), and sodium carbonate (5.6 g, 53.0 mmol) and water (1 mL) were added. The
mixture was stirred at room temperature for 2 hours and then it was evaporated
under
reduced pressure. The residue was partitioned between ethyl acetate and water.
The
organic layer was washed with brine, dried over MgSO4 and evaporated under
reduced
pressure to afford the title compound as an off-white solid, 7.16 g.
'H NMR (400MHz, CDCI3): S 1.45 (s, 9H), 1.81 (m, 2H), 2.04 (m, 2H), 2.17 (m, 1
H), 2.97
(m, 2H), 3.08 (m, 1 H), 4.11 (m, 2H), 4.82 (s, 2H). LCMS: m/z ES+ 306 [MNa]+
Preparation 60: tert-Butyl 4-[5-(morpholin-4-ylmethyl)-1,3,4-oxadiazol-2-
yl]piperidine-1-
carboxylate
H3C j ~N
O
H3C~ N O~ ~O
H3C
O
A mixture of the compound of preparation 8 (10 g, 33.1 mmol), morpholine (4.3
mL,
49.7 mmol) and potassium carbonate (9.2 g, 66.2 mmol) in acetonitrile (300 mL)
was
stirred at 80 C for 4 hours. The cooled mixture was concentrated under reduced
pressure and the residue was partitioned between water and ethyl acetate. The
organic
phase was dried over MgSO4 and evaporated under reduced pressure to provide
the title
compound as an orange oil, 12.06 g.
'H NMR (400MHz, CD3OD): 8 1.45 (s, 9H), 1.65-1.78 (m, 2H), 2.04 (m, 2H), 2.58
(m,
4H), 3.00 (m, 2H), 3.10 (m, 1 H), 3.68 (t, 2H), 3.81 (s, 2H), 4.06 (m, 2H).
LCMS: m/z ES+
353 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-71-
Preparation 61: tert-Butyl 4-{[2-(ethoxyacetyl)hydrazino]carbonyl}piperidine-1-
carboxylate
0
H3C H
H3C_~_O~N N~N~
H3C crk H CH3
O O
A mixture of 4-hydrazinocarbonyl-piperidine-l-carboxylic acid tert-butyl ester
(WO
2000039125, prep 27) (3 g, 12.33 mmol), ethoxyacetic acid (1.28 mL, 13.56
mmol), 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (2.6 g, 13.56 mmol), 1-
hydroxybenzotriazole hydrate (1.83 g, 13.56 mmol) and triethylamine (2.1 mL,
14.8
mmol) in N,N-dimethylformamide (15 mL) was stirred at room temperature for 18
hours.
The mixture was concentrated under reduced pressure and the residue was
partitioned
between ethyl acetate and aqueous sodium carbonate solution. The layers were
separated, the organic phase was dried over MgSO4 and evaporated under reduced
pressure to provide the title compound as an oil, that crystallised on
standing.
'H NMR (400MHz, CDCI3): S 1.24 (t, 3H), 1.45 (s, 9H), 1.58-1.78 (m, 2H), 1.81
(m, 2H),
2.38 (m, 1 H), 2.74-2.82 (m, 2H), 3.60 (q, 2H), 4.04-4.21 (m, 2H), 8.26 (br s,
1 H), 8.82 (br
s, 1 H). LCMS: m/z APCI+ 330 [MH]+
Preparation 62: tert-Butyl 4-{[2-(3,3,3-trifluoropropanoyl)hydrazino]
carbonyl}piperidine-
1 -carboxylate
O
H3C H
H C O N~N\~F
3 N H
H3C
O O F
The title compound was obtained from 4-hydrazinocarbonyl-piperidine-l-
carboxylic acid
tert-butyl ester (WO 2000039125, prep 27) and 3,3,3-trifluoropropionic acid,
following a
similar procedure to that described for preparation 61.
'H NMR (400MHz, CD3OD): S 1.44 (s, 9H), 1.60 (m, 2H), 1.80 (m, 2H), 2.43 (m,
1H),
2.81 (m, 2H), 3.22 (q, 2H), 4.10 (m, 2H). LCMS: m/z APCI- 352 [M-H]"
Preparation 63: tert-Butyl 4-[5-(ethoxymethyl)-1,3,4-oxadiazol-2-yl]piperidine-
1-
carboxylate
H3C
H3C~0 / ~I
H3C ~N OO,,~CH3
O
Pyridine (4 mL, 49.3mmol) was added dropwise to an ice-cooled solution of the
compound of preparation 61 (4.06 g, 12.33 mmol) in dichloromethane (60 mL).
Triflic
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-72-
anhydride (4.2 mL, 24.6 mmol) was then added dropwise over 20 minutes and the
solution was stirred for an hour at 0 C. It was then stirred for a further
hour at room
temperature. The mixture was basified to pH 4 using aqueous sodium bicarbonate
solution, and extracted with dichloromethane. The combined organic extracts
were dried
over MgSO4, and concentrated under reduced pressure. The crude product was
purified
by column chromatography on silica gel using dichloromethane:methanol:0.88
ammonia
(97:3:0.3) to provide the title compound as a yellow oil, 1.25 g.
'H NMR (400MHz, CDC13): 5 1.23 (t, 3H), 1.44 (s, 9H), 1.81 (m, 2H), 2.04 (m,
2H), 2.96
(m, 2H), 3.10 (m, 1 H), 3.60 (q, 2H), 4.14 (m, 2H), 4.66 (s, 2H). LCMS: m/z
APCI+ 312
[MH]+
Preparation 64: tert-Butyl 4-[5-(2,2,2-trifluoroethyl)-1,3,4-oxadiazol-2-
yl]piperidine-l-
carboxylate
H3c N_N
H3C F
3C-~-0~j-
O F
The title compound was obtained as a pale yellow solid, from the compound of
preparation 62, following a similar procedure to that described for
preparation 63.
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.81 (m, 2H), 2.04 (m, 2H), 2.98 (m,
2H), 3.08
(m, 1 H), 3.76 (q, 2H), 4.10 (m, 2H). LCMS: m/z APCI+ 358 [MNa]+
Preparation 65: 1-(4-Chlorobenzoyl)-4-(1,3,4-oxadiazol-2-yl)piperidine
0
CI
N,N-Dimethylformamide dimethyl acetal (5.71 g, 47.9 mmol) was added to a
solution of
the compound of preparation 29 (9.0 g, 31.94 mmol) in N,N-dimethylformamide
(100mL), and the reaction was stirred for 3 hours at 50 C. The mixture was
concentrated under reduced pressure and the residue was suspended in toluene
(150mL). p-Toluene sulphonic acid (1 g, 5.26 mmol) was added and the reaction
was
heated at 110 C for 18 hours. The reaction was diluted with ethyl acetate (100
mL),
washed with brine, water and then brine again. The solution was dried over
MgSO4 and
then evaporated under reduced pressure. The crude product was purified by
column
chromatography using a silica gel cartridge and dichloromethane:methanol
(100:0 to
90:10) as eluant to afford the title compound as a white solid, 4.25 g.
LCMS: m/z APCI+ 292 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-73-
Preparation 66: 1-(3-Chlorobenzoyl)-4-(1,3,4-oxadiazol-2-yl)piperidine
0
N
i
N
CI C-'//
N,N-Dimethylformamide dimethyl acetal (5.71 g, 47.9 mmol) was added to a
solution of
the compound of preparation 4 (9.0 g, 31.94 mmol) in tetrahydrofuran (6 mL),
and the
reaction was stirred for 18 hours at 50 C. TIc analysis showed that starting
material
remained, so additional N,N-dimethylformamide dimethyl acetal (15 mmol) was
added
and stirring was continued for a further 2 hours. The mixture was concentrated
under
reduced pressure and the residue was suspended in toluene (32 mL). p-Toluene
sulphonic acid (1 g, 5.26 mmol) was added and the reaction was heated at 110 C
for 18
hours. The reaction was diluted with ethyl acetate (100 mL), washed with
saturated
sodium bicarbonate solution (2x) and brine, then dried over Na2SO4 and
evaporated
under reduced pressure. The crude product was purified by column
chromatography on
silica gel using an elution gradient of dichloromethane:methanol:0.88 ammonia
(100:0:0
to 90:10:1) to afford the title compound.
'H NMR (400MHz, CDCI3): S 1.95 (m, 2H), 2.04-2.28 (m, 2H), 3.12-3.30 (m, 3H),
3.80
(m, 1 H), 4.58 (m, 1 H), 7.28 (m, 2H), 7.39 (m, 2H), 8.40 (s, 1 H).
LCMS: m/z APCI+ 292 [MH]+
Preparation 67: 1-(4-Chlorobenzoyl)-4-(5-methyl-1,3,4-oxadiazol-2-
yl)piperidine
0
_ N C~~'\
\ ~ o~ N
CI CH3
N,N-Dimethylacetamide dimethyl acetal (6.38 g, 47.9 mmol) was added to a
solution of
the compound of preparation 29 (9.0 g, 31.94 mmol) in N,N-dimethylformamide
(20
mL), and the reaction was stirred at room temperature for 1 hour. It was then
stirred for
a further 2 hours at 40 C. The mixture was diluted with toluene (150 mL),
heated to
110 C and then p-toluene sulphonic acid (400 mg, 2.22 mmol) was added. The
reaction
was heated at 110 C for 18 hours, then cooled and concentrated under reduced
pressure. The residue was dissolved in ethyl acetate, washed with ammonium
chloride
solution and brine, then it was dried over MgSO4 and evaporated under reduced
pressure to afford the title compound as an oil, 9.75 g.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-74-
'H NMR (400MHz, CDCI3): 8 1.81-1.97 (m, 2H), 2.00-2.22 (m, 2H), 2.56 (s, 3H),
3.18 (m,
3H), 3.90 (m, 1 H), 4.58 (m, 1 H), 7.38 (m, 4H). LCMS: m/z APCI+ 306 [MH]+
Preparation 68: 1-(4-Chlorobenzoyl)-4-(5-ethyl-1,3,4-oxadiazol-2-yl)piperidine
0
_ N /
N
\
Ci H,C
Triethyl orthopropionate (1.63 g, 9.23 mmol) was added to a solution of the
compound of
preparation 29 (2.0 g, 7.1 mmol) in N,N-dimethylformamide (10 mL), and stirred
at 60 C
for 3 hours. Tic analysis showed that starting material remained, so
additional triethyl
orthopropionate (0.5 g, 2.83 mmol) was added and the reaction was stirred at
60 C for a
further 18 hours. The mixture was concentrated under reduced pressure, the
residue
was suspended in toluene (15 mL) and trifluoroacetic acid (5 drops) was added.
The
reaction was heated under reflux for 18 hours, then cooled and concentrated
under
reduced pressure. The crude product was purified by column chromatography
using a
silica gel cartridge and dichloromethane:methanol (100:0 to 95:5) as eluant to
afford the
title compound as an oil, 1.6 g.
'H NMR (400MHz, CDC13): 8 1.38 (t, 3H), 1.90 (m, 2H), 2.00-2.21 (m, 2H), 2.85
(q, 2H),
3.19 (m, 3H), 3.80 (m, 1 H), 4.58 (m, 1 H), 7.38 (m, 4H). LCMS: m/z ES+ 320,
322 [MH]+
Preparation 69: 1-(3-Chlorobenzoyl)-4-[5-(ethoxymethyl)-1,3,4-oxadiazol-2-
yl]piperidine
0
N N
/ N
CI
O
~ \_-CH3
Pyridine (1.8 g, 22.84 mmol) and then triflic anhydride (3.22 g, 11.42 mmol)
were added
to an ice-cooled solution of the compound of preparation 32 (2.80 g, 7.61
mmol) in
dichloromethane (50 mL). The reaction was then stirred at room temperature for
2
hours. The mixture was washed with ammonium chloride solution (3x), then with
saturated aqueous sodium carbonate solution, dried over MgSO4 and treated with
activated carbon. This mixture was then filtered and the filtrate was
evaporated under
reduced pressure. The crude product was purified by column chromatography
using a
silica gel cartridge and an elution gradient of dichloromethane:methanol
(100:0 to 95:5)
to provide the title compound as a yellow oil, 566 mg.
CA 02551038 2006-06-21
WO 2005/063754 -75- PCT/IB2004/004059
'H NMR (400MHz, CDCI3): S 1.23 (t, 3H), 1.83-2.24 (m, 3H), 3.14-3.26 (m, 4H),
3.62 (q,
2H), 3.80 (m, 1 H), 4.59 (m, 1 H), 4.67 (s, 2H), 7.25 (m, 1 H), 7.40 (m, 3H).
LCMS: m/z
APCI+ 350 [MH]+
Preparation 70: 1-(4-Chlorobenzoyl)-4-[5-(ethoxymethyl)-1,3,4-oxadiazol-2-
yl]piperidine
O
N~N\N
O__~_O
c,.i \-cH3
The title compound was obtained as a crystalline solid from the compound of
preparation 31 following a similar procedure to that described for preparation
69.
'H NMR (400MHz, CDCI3): 5 1.23 (t, 3H), 1.83-1.99 (m, 2H), 2.04-2.22 (m, 2H),
3.14-
3.26 (m, 3H), 3.62 (q, 2H), 3.79-3.90 (m, 1 H), 4.59 (m, 1 H), 4.67 (s, 2H),
7.40 (m, 4H).
LCMS: m/z APCI+ 350 [MH]+
Preparation 71: 1-(4-Chlorobenzoyl)-4-[5-(trifluoromethyl)-1,3,4-oxadiazol-2-
yl]piperidine
N~N\N
0 I F F
~ /
_ F
ci
Triflic anhydride (1.98 mL, 11.7 mmol) was added to an ice-cooled solution of
the
compound of preparation 30 (1.77 g, 4.69 mmol) and pyridine (1.53 mL,- 18.74
mmol) in
dichloromethane (40 mL). The mixture was then allowed to warm to room
temperature
and stirred for 18 hours. The reaction was diluted with dichloromethane,
washed with
2N hydrochloric acid, then saturated aqueous sodium bicarbonate solution. It
was dried
over MgSO4 and evaporated under reduced pressure. The crude product was
purified
by column chromatography on silica gel using an elution gradient of
dichloromethane:methanol (99:1 to 96:4) to provide the title compound as a
brown oil,
620 mg.
'H NMR (400MHz, CDCI3): S 1.60 (m, 1 H), 1.97 (m, 3H), 2.20 (m, 2H), 3.20 (m,
2H),
3.34 (m, 1 H), 7.39 (m, 4H). LCMS: m/z APCI+ 360 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-76-
Preparations 72 to 74:
The compounds of the following general structure shown below:
0
H3C3 `~
O A
Hc 0cH,
were prepared from N,N-dimethylacetamide dimethyl acetal and the appropriate
hydrazide, following a similar procedure to that described for preparation 67.
Prep. Data
A
No
72 'H NMR (400MHz, CDCI3): S 1.46 (s, 9H), 2.58 (s, 3H), 3.15
N (m, 1 H), 3.37 (m, 1 H), 3.68 (m, 1 H), 3.92 (m, 1 H), 4.00 (m,
1 H), 4.21 (m, 1 H), 4.70 (m, 1 H). LCMS: m/z APCI+ 270
[MH]+
73 'H NMR (400MHz, DMSO-d6): 8 1.38 (m, 11H), 1.73 (m,
"N 2H), 2.06 (d, 1 H), 2.46 (s, 3H), 3.04 (m, 2H), 3.63 (s, 1 H),
3.95 (s, 1 H).
74 ~ 'H NMR (400MHz, CDCI3): 8 1.44 (s, 9H), 2.19-2.38 (m,
'rN 2H), 2.54 (s, 3H), 3.44 (m, 1 H), 3.60 (m, 3H), 3.80 (m, 1 H).
A = 3-hydrazinocarbonyl-pyrrolidine-1-carboxylic acid tert-butyl ester was
used (obtained
from CB Research and Development Inc.).
Preparation 75: 1-(3-Chlorobenzoyl)-4-[5-(1 H-pyrazol-1-ylmethyl)-1,3,4-
oxadiazol-2-
yl]piperidine
0
N N N
N
cl
A mixture of the compound of preparation 6 (1 g, 2.94 mmol), pyrazole (400 mg,
5.9
mmol) and potassium carbonate (610 mg, 4.4 mmol) in acetonitrile (10 mL) and
N,N-
dimethylformamide (10 mL) was stirred at 100 C for 18 hours. The cooled
mixture was
filtered, and the filtrate was concentrated under reduced pressure. The
residue was
diluted with ethyl acetate, washed with water (2x), and brine. It was then
dried over
MgSO4. The crude product was purified by column chromatography on silica gel
using
dichloromethane:methanol:0.88 ammonia (100:0:0 to 95:5:0.5) to afford the
title
compound, 550 mg.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-77-
'H NMR (400MHz, CD3OD): 8 1.76-1.92 (m, 3H), 1.99-2.22 (m, 3H), 3.18 (m, 1H),
3.70
(m, 1 H), 4.50 (m, 1 H), 5.63 (s, 2H), 6.35 (d, 1 H), 6.38 (d, 1 H), 7.37 (d,
1 H), 7.44 (m, 2H),
7.52 (s, 1 H), 7.80 (s, 1 H); LCMS: m/z ES+ 372 [MH]+
Preparation 76: tert-Butyl 4-[4-(4-chlorophenyl)-5-(2-morpholin-4-ylethyl)-4H-
1,2,4-
triazol-3-yl]piperidine-1-carboxylate
N-N
N
N O
O
--\
H3C O
H3C CHs ci
Trifluoroacetic acid (0.35 mL, 4.3 mmol) was added to 4-morpholinpropanoic
acid
hydrazide (Comptes Rendus des Seances de I'acadamie des Sciences, Serie
C;Sciences Chimiques 1976; 282 (17); 857-60) (1.5 g, 8.7 mmol) and the
compound of
preparation 46 (2.7 g, 7.25 mmol) in tetrahydrofuran (18 mL), and the reaction
was
heated under reflux for 8 hours. The cooled mixture was diluted with
dichloromethane,
washed with 1 N sodium hydroxide solution, and evaporated under reduced
pressure.
The crude product was purified by column chromatography on silica gel using an
elution
gradient of dichloromethane:methanol:0.88 ammonia (100:0:0 to 10:10:1) to give
the title
compound, 2.2 g.
'H NMR (400MHz, CDCI3): S 1.45 (s, 9H), 1.59-1.95 (m, 4H), 2.40 (m, 4H), 2.58
(m, 1H),
2.61-2.80 (m, 6H), 3.64 (m, 4H), 4.10 (m, 2H), 7.19 (d, 2H), 7.57 (d, 2H);
LCMS: m/z
APCI+ 476, 478 [MH]+
Preparations 77 to 87:
The compounds of the general formula shown below:
N-N
I
N x
0
--<\
H3C O
H3C CH3 cl
were prepared from the compound of preparation 46 and the appropriate
hydrazide,
following a similar procedure to that described for preparation 76.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-78-
Prep. Data
No.
77 X = 2-oxo-1-pyrrolidinylmethyl
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.71 (m, 2H), 1.77-2.00 (m, 4H),
2.20 (t, 2H), 2.56-2.74 (m, 3H), 3.45 (t, 2H), 4.06 (m, 2H), 4.42 (s, 2H),
7.19
(d, 2H), 7.54 (d, 2H). LCMS: m/z APCI+ 460 [MH]+
78 X = imidazol-1-ylmethyl
'H NMR (400MHz, CDCI3): S 1.41 (s, 9H), 1.70 (m, 2H), 1.83 (m, 2H), 2.55
(m, 1 H), 2.65 (m, 2H), 4.10 (m, 2H), 5.17 (s, 2H), 6.78 (s, 1 H), 6.90 (d,
2H),
7.01 (s, 1 H), 7.19 (s, 1 H), 7.52 (d, 2H). LCMS: m/z APCI+ 443 [MH]+
79 X = 2-methyl-1H-imidazol-1-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.43 (s, 9H), 1.70 (m, 2H), 1.84 (m, 2H), 2.01
(s, 3H), 2.52 (m, 1 H), 2.66 (m, 2H), 4.10 (m, 2H), 5.02 (s, 2H), 6.55 (s, 1
H),
6.85 (m, 3H), 7.53 (d, 2H). LCMS: m/z APCI+ 457 [MH]+
80 X = 1,2,4-triazol-1-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.76 (m, 2H), 1.86 (m, 2H), 2.55-
2.75 (m, 3H), 4.12 (m, 2H), 5.39 (s, 2H), 7.12 (d, 2H), 7.57 (d, 2H), 7.82 (s,
1 H), 8.05 (s, 1 H).
LCMS: m/z APCI+ 444 [MH]+
81 X = tetrazol-1-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.43 (s, 9H), 1.63-1.95 (m, 4H), 2.58-2.75 (m,
3H), 4.11 (m, 2H), 5.60 (s, 2H), 7.15 (d, 2H), 7.59 (d, 2H), 8.81 (s, 1 H).
LCMS: m/z ES+ 445 [MH]+
82 X = 3-methylisoxazol-5-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.37 (s, 9H), 1.60-1.82 (m, 4H), 2.14 (s, 3H),
2.45-2.65 (m, 3H), 3.96-4.05 (m, 4H), 5.82 (s, 1 H), 7.06 (d, 2H), 7.44 (d,
2H). LCMS: m/z APCI+ 458 [MH]+
83 X = 3-methyl-1,2,4-oxadiazol-5-ylmethyi
'H NMR (400MHz, CDCI3): S 1.44 (s, 9H), 1.62-1.95 (m, 4H), 2.32 (s, 3H),
2.70 (m, 3H), 4.10 (m, 2H), 4.24 (s, 2H), 7.18 (d, 2H), 7.54 (d, 2H). LCMS:
m/z APCI+ 459 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-79-
84 X = pyrimidin-2-yloxymethyl
'H NMR (400MHz, CDCI3): S 1.41 (s, 9H), 1.59-76 (m, 2H), 1.86 (m, 2H),
2.58-2.76 (m, 3H), 4.10 (m, 2H), 5.39 (s, 2H), 6.95 (m, 1 H), 7.26 (d, 2H),
7.43 (d, 2H), 8.42 (s, 2H). LCMS: m/z APCI+ 471, 473 [MH]+
85 X = 2-piperidin-1-yiethyl
'H NMR (400MHz, CDCI3): S 1.39-1.78 (m, 19H), 1.78-1.86 (m, 2H), 2.28-
3.04 (m, 3H), 2.56-2.82 (m, 6H), 4.08 (m, 2H), 7.19 (d, 2H), 7.58 (d, 2H).
LCMS: m/z APCI+ 474 [MH]+
86 X = 2-pyridin-2-yiethyl
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.66 (m, 2H), 1.78-1.90 (m, 2H),
2.56 (m, 1 H), 2.60-2.74 (m, 2H), 2.98 (t, 2H), 3.26 (t, 2H), 4.06 (m, 2H),
7.03
(d, 2H), 7.12 (m, 1 H), 7.18 (d, 1 H), 7.50 (d, 2H), 7.58 (m, 1 H), 8.42 (d, 1
H).
LCMS: m/z APCI+ 468 [MH]+
87 X = 2-(3,5-dimethylisoxazol-4-yl)ethyl
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.70 (m, 2H), 1.79-1.93 (m, 5H),
2.04 (s, 3H), 2.54 (m, 1 H), 2.60-2.80 (m, 4H), 4.10 (m, 2H), 5.32 (s, 2H),
6.90 (d, 2H), 7.56 (d, 2H). LCMS: m/z APCI+ 486 [MH]+
A = 1-imidazol-1-yl acetic acid hydrazide was used; prepared as described in
Boll. Chim.
Farm. 114(2); 70-72; 1975.
B = the reaction was stirred for 18 hours under reflux.
C = I eq. hydrazide was used.
Preparation 88: tert-Butyl {1-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidin-
4-yl}carbamate
'N
~ \CH3
N
N
H,c
N
HsC 0 H
HsC ci
Acetic hydrazide (16.9 g, 228 mmol) followed by trifluoroacetic acid (4.4 mL,
57.1 mmol)
were added to a solution of the compound of preparation 56 (43.6 g, 114 mmol)
in
tetrahydrofuran (250 mL) and the mixture was heated under reflux for 7 hours.
The
cooled mixture was then washed with dilute ammonia solution, the layers were
separated and the aqueous phase was extracted further with ethyl acetate. The
combined organic solutions were dried over MgSO4 and evaporated under reduced
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-80-
pressure. The residue was triturated with ether (100 mL) and the resulting
crystals were
filtered off and dried in vacuo to afford the title compound, 32.4 g.
'H NMR (400MHz, CDCI3): 6 1.32 (m, 2H), 1.40 (s, 9H), 1.85 (m, 2H), 2.22 (s,
3H), 2.84
(m, 2H), 3.24 (m, 2H), 3.52 (m, 1 H), 4.44 (m, 1 H), 7.24 (d, 2H), 7.51 (d,
2H); LCMS: m/z
APCI+ 392 [MH]+
Preparation 89: tert-butyl 4-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]-1,4-
diazepane-1-carboxylate
N
N
-CH3
/
Oh
H3C O ~
H3C CH O CI
3
The title compound was obtained in 75% yield from the compound of preparation
55
and acetic hydrazide, following a similar procedure to that described for
preparation 88,
except 2 equivalents of acetic hydrazide were used.
'H NMR (400MHz, CDCI3): S 1.41 (s, 9H), 1.58 (m, 1 H), 1.72 (m, 1 H), 3.02 (m,
1 H), 3.20
(m, 1 H), 3.24-3.44 (m, 5H), 3.52 (m, 2H), 4.24 (s, 2H), 7.38 (m, 2H), 7.50
(d, 2H);
LCMS: m/z APCI+ 422 [MH]+
Preparation 90: Ethyl 1 -[4-(4-ch lorop henyl)-5-methyl-4H- 1, 2,4-triazol-3-
yl] pipe rid i ne-4-
carboxylate
N
N, CH3
11r N
N
H3C--/
o ci
Trifluoroacetic acid (0.84 mL, 10.85 mmol) followed by acetic hydrazide (2.41
g, 32.6
mmol) were added to a solution of the compound of preparation 47 (7.38 g, 21.7
mmol)
in tetrahydrofuran (100 mL), and the reaction was heated under reflux for 3
hours. The
cooled mixture was partitioned between ethyl acetate and aqueous ammonia and
the
layers were separated. The organic phase was washed with brine, dried over
MgSO4
and evaporated under reduced pressure. The crude product was triturated with
ether to
provide the title compound as a white solid, 5.04 g.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-81-
'H NMR (400MHz, CDCI3): S 1.22 (t, 3H), 1.60 (m, 2H), 1.83 (m, 2H), 2.22 (s,
3H), 2.38
(m, 1 H), 2.82 (m, 2H), 3.28 (m, 2H), 4.14 (q, 2H), 7.25 (d, 2H), 7.55 (d,
2H); LCMS: m/z
APCI+ 349 [MH]+
Preparation 91: Ethyl 1-[4-(4-chlorophenyl)-5-(2H-1,2,3-triazol-2-ylmethyl)-4H-
1,2,4-
triazol-3-yl]piperidine-4-carboxylate
N-N
H3C\-0 N
N
O
ci
Trifluoroacetic acid (0.41 mL, 5.3 mmol) was added to a solution of the
hydrazide of
preparation 18 (3.6 g, 10.6 mmol) and the compound of preparation 47 (2.24 g,
15.9
mmol) in tetrahydrofuran (50 mL), and the reaction was heated under reflux for
15 hours.
The cooled mixture was partitioned between ethyl acetate and brine and the
layers were
separated. The organic phase was filtered, dried over MgSO4 and evaporated
under
reduced pressure to provide the title compound as a gum.
'H NMR (400MHz, CDCI3): 5 1.21 (t, 3H), 1.58 (m, 2H), 1.82 (m, 2H), 2.38 (m,
1H), 2.86
(m, 2H), 3.38 (m, 2H), 4.12 (q, 2H), 5.59 (s, 2H), 7.15 (d, 2H), 7.40 (d, 2H),
7.50 (s, 2H).
Preparation 92: Ethyl 1-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]-4-
methylpiperidine-4-carboxylate
>)CH3
-N
N
H,C~/0 CH,
0 Ci
The title compound was obtained as a clear oil in 86% yield, from the compound
of
preparation 48 and acetic hydrazide, following the procedure described for
preparation
91.
'H NMR (400MHz, CDCI3): 5 1.18 (s, 3H), 1.23 (t, 3H), 1.40 (m, 2H), 2.00 (m,
2H), 2.23
(s, 3H), 2.90 (m, 2H), 3.18 (m, 2H), 4.14 (q, 2H), 7.26 (d, 2H), 7.57 (d, 2H);
LCMS: m/z
APCI+ 363 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-82-
Preparation 93: tert-Butyl 4-[4-(4-chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-
triazol-3-
yl]-2-methylpiperazine-1-carboxylate
NI N\ ,O-CH3
N/?--/
N
N / \
H,C O
CH3
HsC CH3 ~ CI
Methoxyacetic acid hydrazide (1.95 g, 18.75 mmol) was added to a solution of
the
compound of preparation 49 (4.80 g, 12.50 mmol) in tetrahydrofuran (200 mL)
and the
solution was stirred for 10 minutes. Trifluoroacetic acid (710 mg, 6.25 mmol)
was added
and the reaction was stirred at room temperature for 18 hours. The mixture was
concentrated under reduced pressure and the crude product was purified by
column
chromatography using a silica gel cartridge and dichloromethane:methanol
(100:0 to
90:10) as eluant, and repeated using ethyl acetate as eluant to afford the
title compound
as a foam, 1.84 g.
'H NMR (400MHz, CDCI3): 5 0.98 (d, 3H), 1.42 (s, 9H), 2.82-3.05 (m, 4H), 3.24
(m, 1H),
3.34 (s, 3H), 3.80 (m, 1 H), 4.18 (m, 1 H), 4.34 (s, 2H), 7.40 (d, 2H), 7.64
(d, 2H); LCMS:
m/z APCI+ 363 [MH]+
Preparation 94: tert-Butyl 4-[4-(4-chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-
triazol-3-
yI]-3-methylpiperazine-1-carboxylate
N-,N` 'O-CH3
H3C
N
O` NJ
H3C~ l1 ~
H3C CH3 O CI
The title compound was obtained in 67% yield, from the compound of preparation
50,
following a similar procedure to that described for preparation 93, except
that the
reaction was heated under reflux.
LCMS: m/z APCI+ 363 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-83-
Preparation 95: tert-Butyl 4-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperazine-1-carboxylate
-,N
CH3
N
H3C`/ O~
H3C/~CH3 0 Ci
Acetic hydrazide (6.51 g, 88 mmol) was added to a solution of the compound of
preparation 52 (32.46 g, 88 mmol) in n-butanol (250 mL) and the reaction was
heated
under reflux for 18 hours. The reaction was heated for a further 5 days under
reflux with
additional acetic hydrazide (36.5 g in total) added periodically. The cooled
mixture was
concentrated under reduced pressure and the residue was purified by column
chromatography on silica gel using dichloromethane:methanol:0.88 ammonia
(95:5:0.5)
to provide the title compound as a white foam.
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 2.24 (s, 3H), 3.01 (m, 4H), 3.38 (m,
4H), 7.25
(d, 2H), 7.54 (d, 2H). LCMS: m/z APCI+ 378 [MH]+
Preparation 96: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]-4-
phenylpiperidine-4-carbonitrile
~N
))CH3
_N
N
CN
cl
Acetic hydrazide (1.65 g, 22.3 mmol) was added to a solution of the compound
of
preparation 51 (3.3 g, 8.93 mmol) in n-butanol (5 mL) and the reaction was
heated
under reflux for 2 days. The cooled mixture was concentrated under reduced
pressure
and the residue was pre-adsorbed onto slica gel. It was then purified by
column
chromatography on silica gel using dichloromethane:methanol:0.88 ammonia
(95:5:0.5),
and the product was triturated with ethyl acetate to provide the title
compound as a solid.
'H NMR (400MHz, CDC13): 8 1.97-2.16 (m, 4H), 2.24 (s, 3H), 3.35 (m, 2H), 3.42
(m, 2H),
7.22-7.45 (m, 7H), 7.56 (d, 2H); LCMS: m/z ES+ 400 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-84-
Preparation 97: tert-Butyl 4-[4-(4-chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-
triazol-3-
yl]piperidine-1-carboxylate
~N O-CH
NI 3
N / \
H3C`/ O~
H3c/~cH3 o c;i
Trifluoroacetic acid (2.14 g, 18.83 mmol) was added to a solution of the
compound of
preparation 57 (7.0 g, 23.54 mmol) and 4-chloroaniline (3.60 g, 28.24 mmol) in
toluene
(50 mL), and the reaction mixture was heated under reflux for 18 hours. The
cooled
solution was concentrated under reduced pressure and the residue was purified
by
column chromatography using a silica gel cartridge and an elution gradient of
dichloromethane:methanol (100:0 to 90:10) to afford the title compound as an
oil, 4.25 g.
'H NMR (400MHz, CD3OD): S 1.45 (s, 9H), 1.67-1.83 (m, 4H), 2.68-2.83 (m, 3H),
3.32
(s, 3H), 4.08 (m, 2H), 4.39 (s, 2H), 7.46 (d, 2H), 7.63 (d, 2H); LCMS: m/z
APCI+ 407
[MH]+
Preparation 98: tert-Butyl 4-[4-(4-chlorophenyl)-5-(hydroxymethyl)-4H-1,2,4-
triazol-3-yl]
piperidine-1 -carboxylate
~-N OH
~ ~
N
N / \
H3C`/ O~
H3C/~CH3 O Ci
A mixture of the compound of preparation 59 (6.8 g, 24 mmol), 4-chloroaniline
(4.3 g,
33.7 mmol) and trifluoroacetic acid (0.9 mL, 12 mmol) in toluene (40 mL) was
stirred at
100 C for 18 hours. The cooled mixture was evaporated under reduced pressure
and
the residue was dissolved in dichloromethane and washed with 2M sodium
hydroxide
solution. The organic solution was dried over MgSO4 and concentrated under
reduced
pressure. The crude product was purified by column chromatography on silica
gel using
ethyl acetate:methanol:0.88 ammonia (95:5:0.5 to 90:10:1) to afford the title
compound
as an off-white solid, 7.1 g.
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.75 (m, 2H), 1.82 (m, 2H), 2.59-2.76
(m, 3H),
3.00 (br s, 1 H), 4.10 (m, 2H), 4.58 (s, 2H), 7.30 (d, 2H), 7.58 (d, 2H);
LCMS: m/z APCI+
393 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 -85- PCT/IB2004/004059
Preparations 99 to 101:
The compounds of the general formula shown below:
N ~-N
N I
H3 O
C ~
HsCCH3 CI
were prepared from the appropriate compound selected from preparations 60, 63
and
64 and 4-chloroaniline, following a similar procedure to that described for
preparation
98.
Prep.
No. Data
99 X = ethoxymethyl
'H NMR (400MHz, CDCI3): 8 1.09 (t, 3H), 1.43 (s, 9H), 1.68 (m, 2H), 2.59-
2.78 (m, 3H), 3.42 (q, 2H), 4.14 (m, 2H), 4.42 (s, 2H), 7.24 (d, 2H), 7.57 (d,
2H). LCMS: m/z APCI` 421 [MH]+
100 X = 2,2,2-trifluoroethyl
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.72 (m, 2H), 1.87 (m, 2H), 2.58
(m, 1 H), 2.66 (m, 2H), 3.45 (q, 2H), 4.10 (m, 2H), 7.19 (d, 2H), 7.60 (d,
2H).
LCMS: m/z APCI+ 445 [MH]+
101 X = morpholin-4-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.41 (s, 9H), 1.65-1.96 (m, 4H), 2.38 (m, 4H),
2.58-2.77 (m, 3H), 3.57 (m, 4H), 4.08 (m, 2H), 7.23 (d, 2H), 7.56 (d, 2H).
LCMS: m/z APCI+ 462 [MH]+
A = 0.8 eq. of trifluoroacetic acid were used.
8= 1 eq. of trifluoroacetic acid was used.
Preparation 102: tert-Butyl 2-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]
morpholine-4-carboxylate
(_JN)CH3
-~
H,c-x
H3C CH3
ci
The title compound was obtained as an oil in 75% yield from the compound of
preparation 72 and 4-chloroaniline, following a similar procedure to that
described for
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-86-
preparation 98, except that 1 eq. of trifluoroacetic acid and 2 eq. of 4-
chloroaniline were
used.
LCMS: m/z APCI+ 393 [MH]+
Preparation 103: tert-Butyl 3-[4-(4-chloro-2-methylphenyl)-5-methyl-4H-1,2,4-
triazol-3-
yl]piperidine-1-carboxylate
N-\
D CH3
N
/N
O-(~ CH3
H3C~ \`O
H3C CH3
ci
4-Chloro-2-methylaniline (3.78 g, 26.3 mmol) and p-toluene sulphonic acid (50
mg) were
added to a solution of the oxadiazole of preparation 73 (2.33 g, 8.9 mmol) in
xylene (10
mL) and the reaction was heated under reflux for 24 hours. The cooled reaction
was
purified directly by column chromatography on silica gel using ethyl
acetate:methanol:dichloromethane (100:0:0 to 0:5:95). The product was
azeotroped
with dichloromethane to afford the title compound as a purple crystalline
solid.
'H NMR (400MHz, CDCI3): S 1.39 (m, 10H), 1.69 (m, 1H), 1.80-1.97 (m, 2H),
1.98, 2.01
(s, 2xs, 3H), 2.17 (s, 3H), 2.32 (m, 1 H), 2.59-3.17 (m, 2H), 4.10 (m, 2H),
7.05, 7.12 (m,
1 H), 7.38 (t, 1 H), 7.44 (d, 1 H); LCMS: m/z APCI+ 391 [MH]+
Preparation 104: tert-Butyl 3-[4-(4-chloro-2-methylphenyl)-5-methyl-4H-1,2,4-
triazol-3-
yl]pyrrolidine-1-carboxylate
N-N
~-CH3
N CH3
O ~
O-~\
H3C
H3C CH3 Ci
A mixture of the compound of preparation 74 (1.50 g, 5.92 mmol),
trifluoroacetic acid
(528 L, 7.1 mmol) and 4-chloroaniline (1.68 g, 11.8 mmol) in toluene (20 mL)
was
heated at 110 C for 18 hours. The cooled mixture was concentrated under
reduced
pressure and the residue was purified by column chromatography on silica gel
using an
elution gradient of dichloromethane:methanol:triethylamine (98:1.5:0.5 to
90:10:1 to
80:20:1) to provide the title compound, 810 mg.
'H NMR (400MHz, CDCI3): 8 1.44 (s, 9H), 2.01 (s, 3H), 2.22 (m, 5H), 2.94-3.70
(m, 5H),
7.08 (m, 1 H), 7.37-7.46 (m, 2H); LCMS: m/z APCI+ 377 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-87-
Preparation 105: tert-Butyl 4-[4-(4-chloro-2-methoxyphenyl)-5-methyl-4H-1,2,4-
triazol-3-
yl]piperidine-1-carboxylate
N-N
>-CH3
N
/ O CH3
N 1
H3C~~
H3C CH3 O ci
A mixture of the compound of preparation 1 (2 g, 7.5 mmol), 4-chloro-2-
methoxyaniline
(Bioorganic and Medicinal Chemistry Letters, 1999; 9(19); 2805-2810) (1.77 g,
11.2
mmol) and trifluoroacetic acid (0.29 mL, 3.7 mmol) in toluene (20 mL) was
stirred at
85 C for 5 hours. The cooled mixture was diluted with ethyl acetate and washed
with 2M
sodium hydroxide solution. The aqueous solution was extracted with
dichloromethane
(2x) and the combined organic solutions were dried over Na2SO4 and then
concentrated
under reduced pressure. The crude product was purified by column
chromatography on
silica gel using dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) to
afford
the title compound, 2 g.
'H NMR (400MHz, CD30D): S 1.46 (s, 9H), 1.63-1.84 (m, 4H), 2.18 (s, 3H), 2.59-
2.81
(m, 3H), 3.86 (s, 3H), 4.05 (m, 2H), 7.20 (dd, 1 H), 7.39 (m, 2H); LCMS: m/z
APCI+ 407
[MH]+
Preparation 106: tert-Butyl 4-[4-(2,4-dichlorophenyl)-5-methyl-4H-1,2,4-
triazol-3-yl]
piperidine-1 -carboxylate
N1N
(1N\>CH3
CI
H3C- O__~N
/
H3C CH O
' ci
The title compound was prepared from the oxadiazole of preparation 1 and 2,4-
dichloroaniline, following a similar procedure to that described for
preparation 105,
except that 2 equivalents of aniline were used.
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.65-1.94 (m, 4H), 2.20 (s, 3H), 2.42
(m, 1H),
2.61-2.78 (m, 2H), 4.10 (m, 2H), 7.22 (d, 1 H), 7.46 (d, 1 H), 7.66 (s, 1 H).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-88-
Preparations 107 to 116:
N
H C-- NN (N
/
H3C O ~N
Y
The appropriate aniline (Y'Y-PhNH2) (1-1.1 eq.) followed by trifluoroacetic
acid (0.5 eq.)
were added to a solution of the compound of preparation 9 (1 eq.) in toluene
(1.6
mlmmol"'), and the reaction mixture was heated under reflux. The reaction was
monitored by tlc and upon completion (45 minutes to 9 hours) the mixture was
allowed to
cool. The reaction was washed with dilute ammonia solution and brine, then
dried over
MgSO4 and evaporated under reduced pressure. The crude product was purified by
column chromatography on silica gel using dichloromethane:methanol (95:5) as
eluant
to afford the title compounds.
Prep Data
No
107 Y=H;Y'=H;
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.74 (m, 2H), 1.87 (m, 2H),
2.51-2.73 (m, 3H), 4.08, (m, 2H), 5.63 (s, 2H), 7.05 (d, 2H), 7.40-7.55 (5H,
m). LCMS: m/z ES+ 432 [MNa]+
108 Y = 4-OCH3; Y' = H;
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.74 (m, 2H), 1.86 (m, 2H),
2.53-2.71 (m, 3H), 3.84 (s, 3H), 4.08 (m, 2H), 5.61 (s, 2H), 6.91 (d, 2H),
6.96 (d, 2H), 7.50 (s, 2H) LCMS: m/z ES+ 462 [MNa]+
109 Y= 4-F; Y'= H;
'H NMR (400MHz, CDCI3): S 1.42 (s, 9H), 1.72 (m, 2H), 1.89 (m, 2H), 2.56
(m, 1 H), 2.65 (m, 2H), 4.09 (m, 2H), 5.62 (s, 2H), 7.06 (d, 1 H), 7.13 (m,
2H), 7.48 (s, 2H). LCMS: m/z ES+ 428 [MNa]+
110 Y= 4-Br; Y'= H;
'H NMR (400MHz, CDCI3): 8 1.46 (s, 9H), 1.74 (m, 2H), 1.92 (m, 2H), 2.59
(m, 1 H), 2.68 (m, 2H), 4.12 (m, 2H), 5.66 (s, 2H), 6.98 (d, 2H), 7.52 (s,
2H), 7.61 (d, 2H). LCMS: m/z ES+ 510 [MNa]+
111 Y= 4-CF3i Y'= H;
1 H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.74 (m, 2H), 1.90 (m, 2H),
2.54 (m, 1 H), 2.65 (m, 2H), 4.10 (m, 2H), 5.67 (s, 2H), 7.21 (d, 2H), 7.47
(s, 2H), 7.73 (d, 2H). LCMS: m/z ES+ 500 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 -89- PCT/1B2004/004059
112 Y= 4-CH3; Y' = H;
'H NMR (400MHz, CDC13): 8 1.42 (s, 9H), 1.74 (m, 2H), 1.87 (m, 2H), 2.41
(s, 3H), 2.54-2.73 (m, 3H), 4.07 (m, 2H), 6.94 (d, 2H), 7.26 (d, 2H), 7.52
(s, 2H). LCMS: m/z ES+ 446 [MNa]+
113 Y=4-CN;Y'=H;
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.72 (m, 2H), 1.92 (m, 2H), 2.55
(m, 1 H), 2.66 (m, 2H), 4.09 (m, 2H), 5.68 (s, 2H), 7.28 (d, 2H), 7.52 (s,
2H), 7.79 (d, 2H). LCMS: mlz ES+ 435 [MH]+
114 A Y = 4-CI; Y' = 2-CH3;
'H NMR (400MHz, CDCI3): 8 1.45 (s, 9H), 1.62-1.78 (m, 3H), 1.86 (s, 3H),
2.00 (m, 1 H), 2.41 (m, 1 H), 2.67 (m, 2H), 4.01-4.18 (m, 2H), 5.52 (d, 1 H),
5.67 (d, 1 H), 6.87 (d, 1 H), 7.25 (d, 1 H), 7.34 (s, 1 H), 7.48 (s, 2H)
LCMS: m/z ES+ 458 [MH]+
115 Y= 4-CI; Y' = 3-F;
'H NMR (400MHz, CDC13): 8 1.44 (s, 9H), 1.73 (m, 2H), 1.89 (m, 2H), 2.57
(m, 1 H), 2.68 (m, 2H), 4.11 (m, 2H), 5.67 (s, 2H), 6.84-6.92 (m, 2H), 7.46-
7.54 (m, 3H). LCMS: m/z ES+ 462 [MH]+
116 Y= 4-CI; Y' = 3-CI;
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 1.73 (m, 2H), 1.88 (m, 2H), 2.55
(m, 1 H), 2.70 (m, 2H), 4.08 (m, 2H), 5.67 (s, 2H), 6.97 (d, 1 H), 7.07 (d,
1 H), 7.52 (s, 2H), 7.55 (d, 1 H). LCMS: m/z ES+ 478 [MH]+
A = 1.5 eq of aniline were used in the reaction.
Preparation 117: tert-Butyl 4-[4-(4-chlorophenyl)-5-
({[(methylthio)carbonothioyl]oxy}methyl)-4H-1,2,4-triazol-3-yl]piperidine-1-
carboxylate
H3C N~N
H3C-~-0 N~
H3C ~ 0 S~
0 CH3
S
Ci
Sodium hydride (60% dispersion in mineral oil, 112 mg, 2.8 mmol) was added to
an ice-
cooled solution of the compound of preparation 98 (1 g, 2.55 mmol) in
tetrahydrofuran
(20 mL), and the solution was stirred for an hour at room temperature. Carbon
disulfide
(230 L, 3.83 mmol) and then methyl iodide (238 L, 3.83 mmol) were added, and
the
reaction was stirred at room temperature for a further 2 hours. Water (1 mL)
was added,
the mixture was concentrated under reduced pressure and the residue was
partitioned
CA 02551038 2006-06-21
WO 2005/063754 -90- PCT/1B2004/004059
between dichloromethane and aqueous sodium bicarbonate solution. The organic
phase was dried over MgSO4 and evaporated under reduced pressure. The crude
product was purified by column chromatography on silica gel using an elution
gradient of
dichloromethane:methanol:0.88 ammonia (97:3:0.3 to 95:5:0.5) to afford the
title
compound as a pale yellow solid, 460 mg.
'H NMR (400MHz, CDC13): 8 1.42 (s, 9H), 1.77 (m, 2H), 1.90 (m, 2H), 2.54 (s,
3H), 2.58-
2.77 (m, 3H), 4.13 (m, 2H), 5.56 (s, 2H), 7.20 (d, 2H), 7.57 (d, 2H); LCMS:
m/z APCI+
483 [MH]+
Preparations 118 to 137:
N\ N I~rx
N Y
H Y.
4M Hydrochloric acid in dioxan (8 to 30 eq.) was added to a solution of the
appropriate
protected piperidine selected from preparations 76, 77, 80 to 85, and 105 to
116 (1
eq.) in methanol (9 to 22.5 mLmmol"') and the reaction was stirred at room
temperature
for between 1.5 and 3 hours. The mixture was concentrated under reduced
pressure,
the was residue partitioned between dichloromethane and 1 M sodium hydroxide
solution
and the layers were separated. The organic solution was dried over MgSO4 and
evaporated under reduced pressure to afford the title compounds.
Prep Data
No
1 1 8 Y X 'H NMR (400MHz, CD3OD): S 2.05-2.22 (m, 4H), 2.58 (s, 3H), 3.10 (m,
3H),
3.45 (m, 2H), 7.78 (d, 1 H), 7.84 (m, 1 H), 8.00 (s, 1 H).
LCMS: m/z APCI+ 311 [MH]+
1 1 9 Y= 4-CI; Y' = 2-OCH3; X= CH3
'H NMR (400MHz, CD3OD): 8 2.00-2.22 (m, 4H), 2.62 (s, 3H), 3.08 (m, 3H),
3.44 (m, 2H), 3.95 (s, 3H), 7.30 (d, 1 H), 7.50 (s, 1 H), 7.61 (d, 1 H).
LCMS: m/z APCI+ 307 [MH]+
120 Y = 4-C1; Y' = H; X = 2-oxo-1-pyrrolidinylmethyl
'H NMR (400MHz, CDCI3): S 1.70-1.90 (m, 4H), 1.98 (m, 2H), 2.21 (m, 2H),
2.58 (m, 3H), 3.14 (m, 2H), 3.45 (t, 2H), 4.42 (s, 2H), 7.18 (d, 2H), 7.58 (d,
2H).LCMS: m/z APCI+ 360 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-91-
121 Y= 4-Cl; Y' = H; X = 1,2,4-triazol-1-ylmethyl
'H NMR (400MHz, CDCI3): S 1.80-1.98 (m, 4H), 2.17-2.30 (m, 1 H), 2.62 (m,
2H), 3.22 (m, 2H), 5.38 (s, 2H), 7.04 (d, 2H), 7.52 (d, 2H), 7.82 (s, 1 H),
8.03
(s, 1 H). LCMS: m/z APCI+ 344 [MH]+
122 Y = H; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CD3OD): S 1.83-1.96 (m, 4H), 2.65-2.85 (m, 3H), 3.25 (d,
2H), 5.66 (s, 2H), 7.23 (d, 2H), 7.43-7.60 (5H, m)
LCMS: m/z ES+ 310 [MH]+
123 Y = 4-F; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDC13): S 1.75 (m, 2H), 1.85 (m, 2H), 2.53 (m, 3H), 3.16
(d, 2H), 5.62 (s, 2H), 7.02 (d, 2H), 7.12 (t, 2H), 7.49 (s, 2H). LCMS: m/z ES+
328 [MH]+
124 Y = 4-Br; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.79 (m, 2H), 1.87 (m, 2H), 2.54-2.65 (m, 3H),
5.64 (s, 2H), 6.93 (d, 2H), 7.52 (s, 2H), 7.59 (d, 2H). LCMS: m/z ES+ 388
[MH]+
125 Y = 4-Cl; Y' = 3-F; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.80 (m, 2H), 1.93 (m, 2H), 2.54-2.66 (m, 3H),
3.22 (d, 2H), 5.66 (s, 2H), 6.80-6.88 (m, 2H), 7.47-7.53 (m, 3H). LCMS: m/z
ES+ 362 [MH]+
126 Y = 4-Cl; Y' = 3-Cl; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): 5 1.80 (m, 2H), 1.91 (m, 2H), 2.52-2.66 (m, 3H),
3.20 (d, 2H), 5.66 (s, 2H), 6.94 (d, 1 H), 7.07 (d, 1 H), 7.52 (s, 2H), 7.54
(d,
1 H). LCMS: m/z ES+ 378 [MH]+
127 Y= 4-CF3; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.76 (m, 2H), 1.87 (m, 2H), 2.56 (m, 3H), 3.14
(d, 2H), 5.66 (s, 2H), 7.20 (d, 2H), 7.46 (s, 2H), 7.64 (d, 2H). LCMS: m/z ES+
378 [MH]+
128 Y= 4-Cl; Y' = 2-CH3; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.68-1.87 (m, 5H), 1.91-2.15 (m, 2H), 2.45 (m,
1 H), 1.55-1.72 (m, 2H), 3.17 (d, 1 H), 3.28 (d, 1 H), 5.47 (d, 1 H), 5.64 (d,
1 H),
6.84 (d, 1 H), 7.20 (d, 1 H), 7.31 (d, 1 H), 7.46 (s, 2H). LCMS: m/z ES+ 358
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-92-
129 Y = 4-CH3; Y' = H; X = 1,2,3-triazol-2-yimethyl
'H NMR (400MHz, CDCI3): 6 1.74-1.98 (m, 4H), 2.43 (s, 3H), 2.55-2.68 (m,
3H), 3.20 (d, 2H), 5.60 (s, 2H), 6.92 (d, 2H), 7.24 (d, 2H), 7.51 (s, 2H).
LCMS: m/z ES+ 324 [MH]+
130 Y = 4-OCH3; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CD3OD): S 1.72-1.79 (m, 4H), 2.45 (m, 2H), 2.65 (m, 1H),
3.00 (d, 2H), 3.82 (s, 3H), 5.62 (s, 2H), 7.00 (d, 2H), 7.11 (d, 2H), 7.57 (s,
2H). LCMS: m/z ES+ 340 [MH]+
131 Y = 4-CN; Y' = H; X = 1,2,3-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.61-1.95 (m, 4H), 2.44-2.60 (m, 3H), 3.16 (m,
2H), 5.66 (s, 2H), 7.21 (d, 2H), 7.48 (s, 2H), 7.76 (d, 2H). LCMS: mlz ES+
335 [MH]+
132 A Y = 4-Cl; Y' = H; X = tetrazol-1-ylmethyl
'H NMR (400MHz, DMSO-d6): S 1.80-1.98 (m, 4H), 2.80 (m, 3H), 3.20 (m,
2H), 3.56 (s, 4H), 5.77 (s, 2H), 7.43 (d, 2H), 7.60 (d, 2H), 8.75 (br s, 1 H),
8.94 (br s, 1 H), 9.18 (s, 1 H). LCMS: m/z ES+ 345, 347 [MH]+
133 Y = 4-Cl; Y' = H; X = 3-methylisoxazol-5-ylmethyl
'H NMR (400MHz, CD3OD): 6 2.05 (m, 2H), 2.20 (m, 2H), 3.00 (m, 2H), 3.18
(m, 1 H), 3.38 (m, 2H), 3.54 (s, 3H), 4.25 (s, 2H), 7.48 (m, 2H), 7.58 (d,
2H).
LCMS: m/z APCI+ 358 [MH]+
134 A Y = 4-Cl; Y' = H; X = 3-methyl-1,2,4-oxadiazol-5-ylmethyl
'H NMR (400MHz, CD3OD): S 2.12 (m, 4H), 2.34 (s, 3H), 3.02 (m, 2H), 3.15
(m, 1 H), 3.43 (m, 2H), 3.64 (s, 2H), 7.57 (m, 2H), 7.68 (d, 2H). LCMS: m/z
APCI+ 359 [MH]+
135 Y = 4-Cl; Y' = H; X = pyrimidin-2-yloxymethyl
'H NMR (400MHz, CDCI3): 6 2.05 (m, 4H), 2.80-2.95 (m, 3H), 3.46 (m, 2H),
5.40 (m, 2H), 6.98 (m, 1 H), 7.20-7.34 (m, 1 H), 7.41-7.58 (m, 3H), 8.44 (m,
2H). LCMS: m/z APCI+ 371 [MH]+
136 Y= 4-Cl; Y' = H; X = 2-piperidin-1-yiethyl
'H NMR (400MHz, CDCI3): S 1.41 (m, 2H), 1.56 (m, 4H), 1.78-1.97 (m, 4H),
2.20-2.40 (m, 6H), 2.60-2.78 (m, 5H), 3.22 (m, 2H), 7.18 (d, 2H), 7.57 (d,
2H).LCMS: m/z APCI+ 374 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-93-
137 Y= 4-CI; Y' = H; X= 2-morpholin-4-ylethyl
'H NMR (400MHz, CDCI3): 8 1.70-1.90 (m, 4H), 2.38 (m, 4H), 2.58 (m, 3H),
2.70 (s, 4H), 3.14 (m, 2H), 3.61 (m, 4H), 7.18 (d, 2H), 7.57 (d, 2H). LCMS:
m/z APCI+ 376 [MH]+
A= The reaction mixture was evaporated under reduced pressure, prior to work-
up to
isolate the hydrochloride salt of the title compound.
Preparations 138 to 141:
;\/x
N\ `T
N
N CI
H
A solution of the appropriate protected piperidine selected from preparations
78 to 79,
and 86 to 87 (1 eq.) in 2.2M methanolic hydrochloric acid (13 mlmmol"') was
stirred at
room temperature for 18 hours. The mixture was concentrated under reduced
pressure
and the residue was azeotroped with toluene. The crude product was partitioned
between dichloromethane and 1M sodium hydroxide solution and the layers were
separated. The organic solution was dried over MgSO4 and evaporated under
reduced
pressure to afford the title compounds.
Prep X Data
No
138 /--N~ 'H NMR (400MHz, CDCI3): 8 1.78 (m, 2H), 1.80-1.98 (m,
N 2H), 2.56 (m, 3H), 3.16 (m, 2H), 5.12 (s, 2H), 6.75 (s, 1 H),
6.84 (d, 2H), 7.00 (d, 2H), 7.50 (d, 2H). LCMS: m/z APCI+
343 [MH]+
139 'H NMR (400MHz, CDCI3): 8 1.74-1.97 (m, 4H), 2.04 (m,
N 3H), 2.48-2.64 (m, 3H), 3.19 (m, 2H), 5.00 (s, 2H), 6.46 (s,
H3C 1 H), 6.82 (s, 2H), 6.85 (d, 2H), 7.48 (d, 2H). LCMS: m/z
APCI+ 357 [MH]+
140 N 'H NMR (400MHz, CDCI3): 8 2.06-2.19 (m, 4H), 3.03 (m,
I i 2H), 3.18-3.22 (m, 1H), 3.30 (m, 2H), 3.44 (m, 2H), 3.60
(m, 2H), 7.77 (s, 4H), 7.98 (m, 1 H), 8.05 (d, 1 H), 8.58 (m,
1 H), 8.78 (d, 1 H).
LCMS: m/z APCI+ 368 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-94-
141 CH3 'H NMR (400MHz, CD3OD): 6 1.99 (s, 3H), 2.12 (m, 7H),
N 2.78 (t, 2H), 2.97 (t, 2H), 3.00-3.18 (m, 3H), 3.43 (m, 2H),
co 7.54 (d, 2H), 7.78 (d, 2H).
H3C
LCMS: m/z APCI+ 386 [MH]+
A = The reaction mixture was evaporated under reduced pressure, and azeotroped
with
toluene. The product was triturated with ethyl acetate, filtered and dried to
afford the
hydrochloride salt of the title compound.
Preparation 142: 4-[4-(4-Chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-triazol-3-
yl]
piperidine
HN
N
N
N
O
CH3
CI C
4M Hydrochloric acid in dioxan (60 mL) was added to a solution of the compound
of
preparation 97 (3.75 g, 9.22 mmol) in dioxan (50 mL) and the reaction was
stirred at
room temperature for 3 hours. The mixture was evaporated under reduced
pressure
and the residue was re-dissolved in dichloromethane and washed with aqueous
ammonia, then brine. The organic solution was dried over MgSO4 and evaporated
under
reduced pressure. The crude product was purified by column chromatography
using a
silica gel cartridge and dichloromethane:methanol:0.88 ammonia (90:10:1) as
eluant to
afford the title compound, 1.99 g.
'H NMR (400MHz, CDCI3): S 1.80-1.98 (m, 4H), 2.57-2:70 (m, 3H), 3.20 (m, 2H),
3.25 (s,
3H), 4.38 (s, 2H), 7.22 (d, 2H), 7.57 (d, 2H). LCMS: m/z APCI+ 307 [MH]+
Preparation 143: 4-[4-(4-Chlorophenyl)-5-(ethoxymethyl)-4H-1,2,4-triazol-3-yl]
piperidine hydrochloride
HN
N
HCI / \ N
Nto
~CH3
CI
4M Hydrochloric acid in dioxan (20 mL) was added to a solution of the compound
of
preparation 99 (990 mg, 2.35 mmol) in dioxan (20 mL) and the reaction was
stirred at
room temperature for 18 hours. The mixture was evaporated under reduced
pressure,
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-95-
and the residue was azeotroped with dichloromethane to afford the title
compound, 910
mg.
'H NMR (400MHz, CD3OD): 8 1.02 (t, 3H), 1.80 (m, 4H), 2.55 (m, 2H), 2.76 (m,
1H),
3.05 (m, 2H), 3.38 (q, 2H), 4.42 (s, 2H), 7.45 (d, 2H), 7.63 (d, 2H); LCMS:
m/z APCI+
321 [MH]+
Preparation 144: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperazine
dihydrochloride
NiN\iCH3
~-N
cD .2HCI
4M Hydrochloric acid in dioxan (13.75 mL) was added to a solution of the
compound of
preparation 95 (4.12 g, 110 mmol) in dichloromethane (50 mL) and the reaction
was
stirred at room temperature for 30 minutes. The mixture was evaporated under
reduced
pressure and dried in vacuo to afford the title compound as a white solid, 3.8
g
'H NMR (400MHz, CDC13): 8 2.23 (s, 3H), 2.80 (m, 4H), 3.05 (m, 4H), 7.25 (d,
2H), 7.50
(d, 2H).
Preparation 145: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidin-4-amine
dihydrochloride
~)CH3
N p
H2N .2HCI CI
A suspension of the compound of preparation 88 (32.3 g, 82.5 mmol) in methanol
(250
mL) and 4N hydrochloric acid in dioxan (40 mL) was warmed to 50 C for 3 hours.
The
mixture was concentrated under reduced pressure and the residue was slurried
in
tetrahydrofuran (50 mL). The resulting solid was filtered off and dried in
vacuo to
provide the title compound.
'H NMR (400MHz, CD3OD): S 1.65 (m, 2H), 1.96 (m, 2H), 2.36 (s, 3H), 3.07 (m,
2H),
3.36 (m, 1 H), 3.47 (m, 2H), 7.66 (d, 2H), 7.75 (d, 2H). LCMS: m/z APCI+ 292
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 -96- PCT/1B2004/004059
Preparation 146: 3-[4-(4-Chloro-2-methyiphenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]
piperidine
/ ~YCH3
I CH3
N
NH CI
4M Hydrochloric acid (5 mL) was added to a solution of the compound of
preparation
103 (846 mg, 2.16 mmol) in dioxan (10 mL) and the reaction was stirred at room
temperature for 18 hours. Tlc analysis showed that starting material remained,
so
additional 4M hydrochloric acid in dioxan (5 mL) was added and the reaction
was stirred
for a further hour at room temperature. The reaction mixture was then
concentrated
under reduced pressure and the crude product was purified by column
chromatography
on silica gel using dichloromethane:methanol:0.88 ammonia (90:10:1). The
product was
azeotroped with dichloromethane and ether to afford the title compound as an
off-white
foam.
'H NMR (400MHz, CDC13): S 1.49 (m, 1 H), 1.68-1.93 (m, 3H), 1.97 (s, 3H), 2.16
(s, 3H),
2.59 (m, 1 H), 2.80 (m, 1 H), 3.03 (m, 1 H), 3.16 (m, 1 H), 7.06 (2xd, 1 H),
7.35 (2xm, 1 H),
7.42 (2xd, 1 H); LCMS: m/z APCI+ 291 [MH]+
Preparation 147: 4-(4-Chioro-2-methylphenyl)-3-methyl-5-pyrrolidin-3-yi-4H-
1,2,4-
triazole
/ \~CH3
CH3
N
N CI
H
A solution of the compound of preparation 104 in 4M hydrochloric acid in
dioxan (20
mL) was stirred at room temperature for 4 hours. The mixture was partitioned
between
ethyl acetate and 2N sodium hydroxide solution, and the layers were separated.
The
organic solution was dried over MgSO4 and evaporated under reduced pressure to
provide the title compound.
'H NMR (400MHz, CDC13): 8 2.00 (m, 5H), 2.20 (m, 5H), 2.80-3.02 (m, 2H), 3.10-
3.27
(m, 1 H), 7.05 (d, 1 H), 7.38 (d, 1 H), 7.41 (s, 1 H); LCMS: m/z APCI+ 277
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-97-
Preparation 148: 1-[4-(4-Chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-triazol-3-
yl]-1,4-
diazepane
O-CH3
/N
N ~
N
~N Z
HN CI
4M Hydrochloric acid in dioxan (25 mL) was added to a solution of the compound
of
preparation 89 (5.45 g, 12.93 mmol) in dioxan (30 mL) and the reaction was
stirred at
room temperature for 18 hours. The mixture was concentrated under reduced
pressure
and the residue was partitioned between water and ether. The layers were then
separated, the aqueous phase was basified using sodium hydroxide and the
solution
was extracted with dichloromethane (3x). The combined organic extracts were
dried
over MgSO4 to afford the title compound as a foam, 3.84 g.
'H NMR (400MHz, CD3OD): S 1.78 (m, 2H), 2.84 (m, 4H), 3.21 (s, 3H), 3.30 (m,
4H),
4.24 (s, 2H), 7.50 (d, 2H), 7.60 (d, 2H); LCMS: m/z APCI+ 322 [MH]+
Preparation 149: 1-[4-(4-Chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-triazol-3-
yl]-2-
methylpiperazine
O-CH3
N
N ~
H3C N
d N 0
H CI
The title compound was obtained as a yellow oil in 95% yield from the compound
of
preparation 94, following the procedure described for preparation 148.
'H NMR (400MHz, CDCI3): 5 1.14 (d, 3H), 2.10 (br s, 1 H), 2.60 (m, 1 H), 2.82
(m, 2H),
2.97 (m, 1 H), 3.07 (m, 2H), 3.35 (s, 3H), 4.28 (d, 1 H), 4.40 (d, 1 H), 7.38
(d, 2H), 7.50 (d,
2H); LCMS: m/z APCI+ 322 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-98-
Preparation 150: 1-[4-(4-Chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-triazol-3-
yl]-3-
methylpiperazine
HN_"~ N
N
H3C ~
~
\ O
~ CH3
CI
Trifluoroacetic acid (25 mL) was added to an ice-cooled solution of the
compound of
preparation 93 (2.80 g, 6.63 mmol) in dichloromethane (25 mL) and the solution
was
stirred at room temperature for an hour. The mixture was concentrated under
reduced
pressure and the residue was re-dissolved in ethyl acetate, then washed with 1
N sodium
hydroxide solution. The organic solution was dried over MgSO4 and evaporated
under
reduced pressure. The crude product was purified by column chromatography
using a
silica gel cartridge and an elution gradient of dichloromethane:methanol:0.88
ammonia
(100:0:0 to 90:10:1) to afford the title compound as an oil, 780 mg.
'H NMR (400MHz, CDCI3): 8 1.19 (d, 3H), 2.98 (m, 2H), 3.02-3.23 (m, 5H), 3.36
(s, 3H),
4.34 (s, 2H), 7.40 (d, 2H), 7.52 (d, 2H); LCMS: m/z APCI+ 322 [MH]+
Preparation 151: 4-[4-(4-Chloro-2-methylphenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidine
N/N~CH3
~ N CH3
N
H CI
A mixture of the compound of preparation 1 (3 g, 11.2 mmol), 4-chloro-2-
methylaniline
(2.4 g, 16.8 mmol) and trifluoroacetic acid (0.8 mL, 11.2 mmol) in toluene (30
mL) was
stirred at room temperature for 110 C for 18 hours. The cooled mixture was
concentrated under reduced pressure, 2M sodium hydroxide solution (10 mL)
added and
the mixture was azeotroped with toluene. The crude product was purified by
column
chromatography on silica gel using dichloromethane:methanol:0.88 ammonia
(95:5:0.5
to 80:20:3) to afford the title compound as an off-white foam, 1.45 g.
'H NMR (400MHz, CD3OD): S 1.81 (m, 2H), 1.92 (m, 2H), 2.00 (s, 3H), 2.19 (s,
3H),
2.60-2.78 (m, 3H), 3.20 (m, 2H), 7.38 (d, 1 H), 7.46 (d, 1 H), 7.59 (s, 1 H);
LCMS: m/z
APCI+ 291 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-99-
Preparation 152: 4-[4-(4-Chloro-2-fluorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidine
ditrifluoroacetate
i ~~CH3
N \ N F
z
H 2CF3COZH CI
A mixture of the compound of preparation 1 (900 mg, 3.4 mmol), 4-chloro-2-
fluoroaniline (109.1 mg, 0.75 mmol) and trifluoroacetic acid (288 L, 3.74
mmol) in
toluene (8 mL) was stirred at 110 C for 72 hours. The cooled mixture was
concentrated
under reduced pressure, 0.88 ammonia added and the mixture was concentrated
under
reduced pressure. The residue was azeotroped with toluene and the crude
product was
purified by column chromatography on silica gel using an elution gradeint of
dichloromethane:methanol:0.88 ammonia (90:10:1 to 80:20:3) to afford the title
compound as an off-white foam, 985 mg.
'H NMR (400MHz, CDCI3): 8 2.02-2.30 (m, 7H), 2.80 (m, 1 H), 3.10 (m, 2H), 3.59
(m,
2H), 3.86 (m, 1 H), 7.26 (m, 1 H), 7.40 (m, 2H), 9.34 (br s, 1 H), 10.10 (br
s, 1 H); LCMS:
m/z APCI+ 295 [MH]+
Preparation 153: 4-{4-(4-Chlorophenyl)-5-[(2,2,2-trifluoroethoxy)methyl]-4H-
1,2,4-
triazol-3-yl}piperidine
HN
N' ~ N
_ N--~-O F
F
CI F
Trifluoroacetic acid (2.49 g, 21.8 mmol) was added to a solution of 4-
chloroaniline (3.63
g, 28.4 mmol) and the compound of preparation 58 (8.0 g, 21.9 mmol) in toluene
(50
mL), and the reaction heated was under reflux for 48 hours. TIc analysis
showed that
starting material remained, so additional trifluoroacetic acid (8 mL) was
added and the
reaction was heated for a further 4 hours under reflux. The cooled mixture was
extracted with water, the aqueous solution was basified using potassium
hydroxide and
then extracted with dichloromethane (4x100 mL). The combined organic extracts
were
dried over MgSO4 and evaporated under reduced pressure. The crude product was
purified by column chromatography using a silica gel cartridge and an elution
gradient of
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-100-
dichloromethane:methanol:0.88 ammonia (100:0:0 to 85:15:1.5) to give the title
compound, 4.34 g.
'H NMR (400MHz, CDCI3): 8 1.86-2.04 (m, 4H), 2.78 (m, 3H), 3.37 (m, 2H), 3.82
(q, 2H),
4.22-4.41 (br s, 1 H), 4.58 (s, 2H), 7.22 (d, 2H), 7.58 (d, 2H); LCMS: m/z ES+
375.1
[MH]+
Preparation 154: 4-{4-(4-Chlorophenyl)-5-[(trifluoromethoxy)methyl]-4H-1,2,4-
triazol-3-
yl}piperidine
HN ~N11 N~ N
O
/~-F
F' \
ci F
Hydrogen fluoride-pyridine (0.35 mL, 13.2 mmol), followed by a solution of the
compound of preparation 117 (160 mg, 0.33 mmol) in dichloromethane (1 mL) were
added to a solution of 1,3-dibromo-2,4-dimethylhydantoin (283 mg, 1.0 mmol) in
dichloromethane (5 mL) at -78 C. The reaction was allowed to warm to room
temperature over 30 minutes then stirred for a further hour. The mixture was
washed
with 1 N sodium hydroxide solution, dried over MgSO4 and evaporated under
reduced
pressure. The crude product was purified by column chromatography on silica
gel using
dichloromethane:methanol:0.88 ammonia (90:10:1) as eluant to provide the title
compound as a yellow oil, 45 mg.
LCMS: m/z APCI+ 361 [MH]+
Preparation 155: 4-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]benzonitrile
NZ CHs
N
NC CI
Trifluoroacetic acid (600 L, 8.1 mmol) was added to a suspension of 4-
chloroaniline
(2.1 g, 16.2 mmol) and 4-(5-methyl-1,3,4-oxadiazol-2-yl)benzonitrile (Journal
fur
Praktische Chemie, 1994; 336(8); 678-85) (3.0 g, 16.2 mmol) in tetrahydrofuran
(50 mL)
and the reaction was heated at reflux for 22 hours. The cooled mixture was
partitioned
between ethyl acetate (300 mL) and 20% aqueous 0.88 ammonia (120 mL), and then
the layers were separated. The organic phase was dried over MgSO4 and
evaporated
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-101-
under reduced pressure. The residue was triturated with ether and a minimum
volume
of ethyl acetate to afford the title compound as a white crystalline solid,
2.82 g.
'H NMR (400MHz, CDCI3): S 2.38 (s, 3H), 7.19 (d, 2H), 7.55 (m, 4H), 7.60 (d,
2H);
LCMS: m/z ES+ 295 [MH]+
Preparation 156: 4-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]benzoic
acid
~N
N1 ~-CH3
N
o
OH CI
A solution of potassium hydroxide (2.6 g, 46.0 mmol) in water (10 mL) was
added to a
solution of the compound of preparation 155 (2.7 g, 9.2 mmol) in ethylene
glycol
dimethyl ether (40 mL) and the reaction was heated under reflux for 18 hours.
Ethanol
(50 mL) was added and the reaction was heated under reflux for a further 72
hours. The
cooled mixture was acidified to pH 6 and concentrated under reduced pressure.
The
residue was extracted using dichloromethane:methanol:0.88 ammonia (84:14:2),
the
suspension was then filtered and the filtrate was evaporated under reduced
pressure.
The product was triturated with ether, filtered off and dried to afford the
title compound
as a white solid.
'H NMR (400MHz, CD3OD): S 2.38 (s, 3H), 7.19 (m, 4H), 7.58 (d, 2H), 7.90 (d,
2H);
LCMS: m/z ES" 312 [M-H]"
Preparation 157: 4-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]benzoyl
chloride
~N
N1 ~-CH3
N
o
ci ci
Oxalyl chloride (3 mL, 32 mmol) followed by N,N-dimethylformamide (5 drops)
were
added to a solution of the acid of preparation 156 (2 g, 6.4 mmol) in
dichloromethane
(200 mL) and the mixture was stirred at room temperature for 2 hours. The
mixture was
filtered, the filtrate was evaporated under reduced pressure and the residue
was
azeotroped with dichloromethane (3x200 mL) to afford the title compound as an
orange
oil, 2.01 g.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-102-
'H NMR (400MHz, CDC13): 8 2.78 (s, 3H), 7.58 (d, 2H), 7.63 (d, 2H), 7.74 (m,
2H), 8.06
(d, 2H).
Preparation 158: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]piperidine-4-
carboxylic acid
~N
N11 CH3
/~~~N
N
O
OH cl
A solution of the ester of preparation 90 (4 g, 10.4 mmol) and 4N sodium
hydroxide (13
mL, 52 mmol) in dioxan (20 mL) was stirred at room temperature for 2 hours.
The
mixture was partitioned between water and ethyl acetate and the phases were
separated. The aqueous layer was acidified to pH 4 using 2N hydrochloric acid,
the
resulting precipitate was filtered off and washed with water. The solid was
triturated with
ether, filtered and dried in vacuo to afford the title compound as a white
solid.
'H NMR (400MHz, CD3OD): 5 1.52-1.61 (m, 2H), 1.81 (m, 2H), 2.21 (s, 3H), 2.40
(m,
1H), 2.81 (m, 2H), 3.21-3.36 (m, 2H), 7.47 (d, 2H), 7.62 (d, 2H).
Preparation 159: 1-[4-(4-Chlorophenyl)-5-(2H-1,2,3-triazol-2-ylmethyl)-4H-
1,2,4-triazol-
3-yl]piperidine-4-carboxylic acid
O N-N
~-CN--~/ ~ /
N
HO N~~
N
p
The title compound was obtained as a white solid, from the ester of
preparation 91,
following a similar procedure to that described for preparation 158, except
that 10 eq. of
sodium hydroxide were used in the reaction.
'H NMR (400MHz, CD3OD): S 1.48-1.60 (m, 2H), 1.81 (m, 2H), 2.40 (m, 1H), 2.82
(m,
2H), 3.32 (m, 2H), 5.64 (s, 2H), 7.30 (d, 2H), 7.50 (d, 2H), 7.58 (s, 2H);
LCMS: m/z
APCI+ 388 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-103-
Preparation 160: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]-4-
methylpiperidine-4-carboxylic acid
,N
))CH3
' N
PCH3 ~ ~
Ozz~ O
H CI
A mixture of the ester of preparation 92 (1.45 g, 4.0 mmol) and 4N sodium
hydroxide
solution (5 mL, 20 mmol) in dioxan (50 mL) was stirred under reflux for 16
hours. Tic
analysis showed that starting material remained, so additional sodium
hydroxide (4N, 5
mL, 20 mmol) was added, and the reaction was heated under reflux for a further
18
hours. The cooled mixture was partitioned between ethyl acetate and water, and
the
layers were separated. The aqueous phase was acidified to pH 3.5 using 2N
hydrochloric acid and extracted with ethyl acetate (2x). These combined
organic
extracts were dried over MgSO4 and evaporated under reduced pressure. The
product
was triturated with ethyl acetate. The resulting solid was filtered off and
dried to provide
the title compound as a white solid, 800 mg.
'H NMR (400MHz, CD3OD): 8 1.18 (s, 3H), 1.38 (m, 2H), 1.99 (m, 2H), 2.21 (s,
3H), 2.92
(m, 2H), 3.16 (m, 2H), 7.49 (d, 2H), 7.63 (d, 2H). LCMS: m/z APCI+ 335 [MH]+
Preparation 161: N-(4-Chlorophenyl)-2-(2H-1,2,3-triazol-2-
ylacetyl)hydrazinecarbothioamide
\ ~N O
N N-
I NN.
CI ~ S H N
4-Chlorophenylisothiocyanate (8.58 g, 50.6 mmol) was added portionwise to a
suspension of the hydrazide of preparation 18 (7.0 g, 50.6 mmol) in ethanol
(200 mL)
and the mixture was stirred at room temperature for 72 hours. The resulting
precipitate
was filtered off, washed with ether and dried to afford the title compound as
a white
solid, 14.5 g.
'H NMR (400MHz, CD3OD): 5 5.30 (s, 2H), 7.36 (d, 2H), 7.44 (d, 2H), 7.78 (s,
2H).
CA 02551038 2006-06-21
WO 2005/063754 PCT/1B2004/004059_
-104-
Preparation 162: 4-(4-Chlorophenyl)-5-(2H-1,2,3-triazol-2-ylmethyl)-2,4-
dihydro-3H-
1,2,4-triazole-3-thione
H
SN\
/N
N~
/N
N _
cl N
A solution of the compound of preparation 161 (14.5 g, 46.5 mmol) and 2M
sodium
hydroxide solution (232 mL, 465 mmol) in ethanol (36 mL) was stirred at 80 C
for 18
hours. The cooled mixture was acidified to pH 9 using concentrated
hydrochloric acid,
then it was extracted with dichloromethane (6x250 mL). The combined organic
solutions
were evaporated under reduced pressure to afford the title compound as a white
solid,
7.5 g.
'H NMR (400MHz, CDCI3): S 5.54 (s, 2H), 7.05 (d, 2H), 7.42 (d, 2H), 7.58 (s,
2H).
Preparation 163: 2-{[4-(4-Chlorophenyl)-5-(methylthio)-4H-1,2,4-triazol-3-
yl]methyl}-2H-
1,2,3-triazole
Ha
ISN\N
Nt
~
1 /N
~ N
~
cl N
Potassium tert-butoxide (2.9 g, 25.6 mmol) was added to a solution of the
compound of
preparation 162 (7.5 g, 25.6 mmol) in tetrahydrofuran (250 mL) and the
suspension was
stirred at room temperature for 30 minutes. Methyl p-toluenesulphonate (4.8 g,
25.7
mmol) was added and the mixture was heated under reflux for 45 minutes, then
at room
temperature for a further 2 hours. The mixture was diluted with
dichloromethane (1000
mL), washed with saturated ammonium chloride solution (300 mL) and brine (300
mL)
then it was dried over MgSO4 and evaporated under reduced pressure. The crude
material was purified by column chromatography on silica gel using
dichloromethane:methanol (85:15) as eluant to provide the title compound, 4.9
g.
'H NMR (400MHz, CDCI3): 8 2.70 (s, 3H), 5.64 (s, 2H), 7.02 (d, 2H), 7.41 (d,
2H), 7.54
(s, 2H). LCMS: m/z ES+ 329 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-105-
Preparation 164: 2-{[4-(4-Chlorophenyl)-5-(methylsulfonyl)-4H-1,2,4-triazol-3-
yl]methyl}-
2H-1,2,3-triazole
0
H3C-IS N
O /
/ N ~ \N
\
,N
N CI N~
Meta-chloroperbenzoic acid (3.4 g, 19.56 mmol) was added to a solution of the
compound of preparation 163 (1.5 g, 4.90 mmol) in dichloromethane (60 mL) and
the
reaction was stirred at room temperature for 18 hours. The mixture was diluted
with
dichloromethane, then washed with saturated sodium bicarbonate solution (300
mL) and
brine (200 mL). The organic solution was dried over Na2SO4 and concentrated
under
reduced pressure. The residual solid was washed with ethanol, then dried in
vacuo to
afford the title compound as a white solid, 1.40 g.
'H NMR (400MHz, CDCI3): S 3.46 (s, 3H), 5.69 (s, 2H), 7.18 (d, 2H), 7.41 (d,
2H), 7.57
(s, 2H); LCMS: m/z ES+ 361 [MNa]+
Preparation 165: 4-Chloro-2-ethoxynitrobenzene
0 N+p
O
cI \-CH3
Sodium ethoxide (21 % in ethanol, 8.6 mL, 26 mmol) was added dropwise to a
solution of
4-chloro-2-fluoronitrobenzene (3 g, 17.1 mmol) in ethanol (20 mL), and once
addition
was complete the reaction was stirred for a further hour. The mixture was
concentrated
under reduced pressure, the residue was diluted with ethyl acetate, and the
solution was
washed with water (x2), then brine. The solution was dried over MgS04 and
evaporated
under reduced pressure to afford the title compound as a solid, 3.45 g.
'H NMR (400MHz, CDCI3): S 1.49 (t, 3H), 4.19 (q, 2H), 7.00 (d, 1 H), 7.05 (s,
1 H), 7.81
(d, 1 H).
Preparation 166: 4-Chloro-2-ethoxyaniline
NHZ
~ ~
_ O
CI \_ CHa
A mixture of the nitro compound of preparation 165 (3.30 g, 16.4 mmol), iron
powder
(2.7 g, 49 mmol) and calcium chloride (810 mg, 7.4 mmol) in water (5 mL) and
ethanol
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-106-
(30 mL) was heated under reflux for 3.5 hours. The cooled mixture was filtered
through
Celite and the filtrate was concentrated under reduced pressure. The residue
was
partitioned between water and ethyl acetate, the layers were separated and the
organic
phase was washed further with brine. The solution was dried over MgSO4 and
evaporated under reduced pressure. The crude product was purified by column
chromatography on silica gel using an elution gradient of pentane;ethyl
acetate (100:0 to
0:100) to afford the title compound as an oil, 2.4 g.
'H NMR (400MHz, CDC13): S 1.42 (t, 3H), 402 (q, 2H), 6.61 (d, 1H), 6.76 (m,
2H).
Preparation 167: 1-(5-Chloro-2-nitrobenzyl)pyrrolidine
ci
p= O
Pyrrolidine (4 mL, 48.5 mmol) was added to a solution of 5-chloro-2-
nitrobenzaldehyde
(6 g, 32.2 mmol) in dichloromethane (150 mL) and the solution was stirred at
room
temperature for 30 minutes. The solution was then cooled in ice, and sodium
triacetoxyborohydride (10.3 g, 48.5 mmol) was added portionwise. Once addition
was
complete, the reaction was stirred at room temperature for 4 hours. The
reaction was
washed with sodium carbonate solution, dried over MgSO4 and evaporated under
reduced pressure. The residual oil was purified by column chromatography on
silica gel
using ethyl acetate:pentane (86:14) to afford the title compound as a pale
yellow solid,
6.3 g.
'H NMR (400MHz, CDC13): S 1.82 (m, 4H), 2.58 (m, 2H), 3.98 (s, 2H), 7.37 (d, 1
H), 7.80
(s, 1 H), 7.87 (d, 1 H). LCMS: m/z APCI+ 241 [MH]+
Preparation 168: 4-Chloro-2-(pyrrolidin-1-ylmethyl)aniline
NHZ
ci
A mixture of the compound of preparation 167 (6.2 g, 25.8 mmol) and Raney
Nickel
(400 mg) in ethanol (200 mL) was hydrogenated at 40 psi and room temperature
for 2
hours. The mixture was filtered through Arbocel and the filtrate was
evaporated under
reduced pressure. The residue was purified by column chromatography on silica
gel
CA 02551038 2006-06-21
WO 2005/063754 -107 PCT/1B2004/004059
-
using an elution gradient of dichloromethane:methanol:0.88 ammonia (95:5:0.5
to
90:10:1) to afford the title compound as a solid, 3.95 g.
'H NMR (400MHz, CDCI3): S 1.78 (m, 4H), 2.48 (m, 2H), 3.59 (s, 2H), 4.65-4.90
(br s,
2H), 6.56 (d, 1 H), 6.98 (s, 1 H), 7.00 (d, 1 H). LCMS: m/z APCI+ 211 [MH]+
Preparation 169: 4-{[4-(4-Chlorophenyl)-5-piperidin-4-yl-4H-1,2,4-triazol-3-
yl] methyl}morphol i ne
/~
HN\ J~~ N'N
~-~ N ^
N\ O
CI
A solution of the compound of preparation 101 (8.6 g, 18.6 mmol) in dioxan (50
mL)
and 4M hydrochloric acid in dioxan (30 mL) was stirred at room temperature for
18
hours. The solution was evaporated under reduced pressure and the residue was
partitioned between 2N sodium hydroxide solution and ethyl acetate. The
resulting solid
was filtered off and dried to afford the title compound as a white solid, 1.2
g. The filtrate
was separated, the aqueous layer was extracted with dichloromethane and the
combined organic solutions were dried over MgSO4 and evaporated under reduced
pressure to afford additional product, 1.11 g.
LCMS: m/z APCI+ 362 [MH]+
Preparation 170: tert-Butyl 4-{[2-(2H-1,2,3-triazol-2-ylacetyl)hydrazino]
carbonyl}piperidine-1-carboxylate
H3C N-~ N
H C O NN/
H
O
A suspension of N-Boc-isonipecotic acid (30.0 g, 130.8 mmol) and the hydrazide
of
preparation 18 (18.5 g, 130.8 mmol) in dichloromethane (150 mL) was cooled in
an ice
bath under N2. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
(25.6 g,
133.5 mmol) was added via an addition funnel, and the funnel was washed with
additional dichloromethane (10 mL). The resulting solution was warmed to
ambient
temperature. Once the reaction was complete, iso-propanol (150 mL) was added
and
the homogeneous solution was concentrated in vacuo to approx 165 mL, heated to
approx. 70 C, and allowed to cool to ambient temperature with stirring. The
resulting
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-108-
thick white slurry was filtered in vacuo, washing with iso-propanol (15 mL and
30 mL)
and dried in vacuo at 50 C to give the title compound as a white solid, 25.3
g(55 /o).
The liquors from the filtration were concentrated under reduced pressure to
low volume,
the resulting syrup was treated with water (50 mL) and vigorously stirred to
give a thick
slurry within two minutes. After overnight granulation, the slurry was
filtered (rapid
filtration), the residues washed with water (2 x 10 mL) and dried in vacuo at
50 C to give
additional product, 8.5 g (18%).
'H NMR (CDCI3) 8: 1.42 (s, 9H), 1.50-1.85 (m, 4H), 2.41 (m, 1H), 2.73 (t, 2H),
4.12 (d,
2H), 5.21 (s, 2H), 7.70 (s, 2H), 9.15 (d, 1 H), 9.75 (d, 1 H).
Examples 1 to 162:
,,N
NI \Me
N
Z~ -
~ ci
Examples 1 to 162 illustrated below were synthesised as a library. The
following
solutions were used:
carboxylic acid, ZCO2H, was dissolved in dimethylacetamide (anhydrous) plus 3
.75% triethylamine at 0.2M concentration.
the amine of preparation 2a was dissolved in DMA (anhydrous) plus 3.75%
triethylamine at 0.2M concentration.
HBTU was dissolved in DMA (anhydrous) at 0.2M concentration.
(N.B. Gentle sonication, in a warm water bath (temp <40 C), was used to
dissolve the
monomers where necessary.)
Experimental Procedure:
The reaction Scale was between 20 and 30 micromoles per well (experimental
details
shown for 20 Nmole reaction, scale can be adjusted accordingly within this
range).
Reactions were performed in a polypropylene 96 well plate.
a) Amine solutions (0.1 ml, 20,umoles, 1 eq.) were added to the wells
b) Carboxylic acid solutions (0.15mi, 30,umoles, 1.5 eq.) were added to the
wells
c) HBTU Solution (0.15 ml, 30 Nmoles, 1.5 eq.) was added to each well
d) The polypropylene 96 well plate was sealed with a PTFE and Rubber gasket
and
clamped between a pair of metal plates.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-109-
e) The plate was heated in an oven for 6 h at 60 C and then left to cool down
in the
oven overnight.
f) When cool, the plate was unclamped and placed in a Genevac to remove the
solvent.
g) The samples were re-dissolved in DMSO/water (9:1) (5001I) and any
particulate
matter was removed by filtration.
h) Purification was carried out by RP-HPLC.
HPLC purification conditions:
Column: Phenomenex Luna C18, 10um, 150 x 10 mm id
Temperature: ambient
Eluent A: 0.05% Diethylamine in water
Eluent B: Acetonitrile
Samples dissolved in: 90% Dimethylsulphoxide in water.
Sample loaded using Gilson Autosampler with Injection Volume of 550 I
Gilson LC Pump Initial Conditions:
Solvents
A% 80.0
B% 20.0
Flow (mI/min) 8.000
Gilson LC Pump Gradient Timetable:
Time A% B% Flow (mI/min)
0.00 80.0 20.0 8.000
0.20 80.0 20.0 8.000
7.00 5.0 95.0 8.000
9.00 5.0 95.0 8.000
9.10 80.0 20.0 8.000
10.5080.0 20.0 8.000
Gilson 119 uv detector monitoring at 254nm:
Collector set at 225nm
Dual sensitivity 200
Peak sensitivity 80
Peak width 0.3 min.
CA 02551038 2006-06-21
WO 2005/063754 -110- PCT/IB2004/004059
HPLC analysis conditions and Mass Spectrometer details:
Column: Phenomenex Luna C18, 5um, 30 x 4.6 mm id.
Eluent A: 0.05% Diethylamine in water
Eluent B: Acetonitrile
Samples dissolved in: 90% Dimethylsulphoxide in water
Sample loaded using Gilson Quad Z with Injection Volume of 5 l
Waters 1525 binary LC Pump Initial Conditions:
Solvents
A% 95.0
B% 5.0
Flow (mI/min) 2.5 (per channel)
Temperature ( C) ambient
LC Pump Gradient Timetable:
The gradient Timetable contains 4 entries, which are:
Time A% B% Flow
0.00 95.0 5.0 2.500
3.00 5.0 95.0 2.500
3.50 95.0 5.0 2.500
Total run time 4.50 mins
Detection:
Waters 2488 dual wavelength detector
UV1 (nm) 225
UV2 (nm) 255
and
ELSD: PolymerLabs, Temperature: 75 C, Gas flow: 1.2 bar
Mass Spectrometer:
Waters ZQ 2000 4 way MUX,
ES+ Cone voltage: 26 v Capillary: 3.85 kv
ES- Cone voltage:-30 v Capillary:-3.00 kv
Desolvation gas: 800 I/min
Source Temp: 300 C.
Scan range 160-1000 Da
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-111-
Ex. Z Mass ion of
HPLC Ret"
No. product
time (Mins)
found
I 2-methoxy-pyridin-3-yl 412.15 1.55
2 3-trifluoromethyl-phenyl 449.13 2
3 2-methoxy-phenyl 411.15 1.35
4 2-methanesulfonyl-phenyl 459.12 1.6
3-methanesulfonyl-phenyl 459.12 1.55
6 phenyl 381.14 1.72
7 3-methoxy-phenyl 411.15 1.67
8 4-fluoro-phenyl 399.13 1.74
9 2-Chloro-phenyl 415.1 1.8
4-Chloro-phenyl 415.1 1.92
11 4-methanesulfonyl-phenyl 459.12 1.49
12 2,4-dichloro-phenyl 449.06 1.99
13 3,4-dichloro-phenyl 449.06 2.04
14 2,5-dichloro-phenyl 449.06 1.99
4-ethoxy-phenyl 425.17 1.77
16 4-methylsulfanyl-phenyl 427.13 1.85
17 4-Chloro-2-methoxy-phenyl 445.11 1.89
18 2-ethoxy-phenyl 425.17 1.84
19 isoquinolin-1-yi 432.15 1.6
2,6-dimethyl-phenyl 409.17 1.82
21 quinolin-2-yl 432.15 1.74
22 quinolin-4-yl 432.15 1.6
23 quinolin-3-yl 432.15 1.57
24 2-Chloro-6-fluoro-phenyl 433.09 1.54
2,3-dichloro-phenyl 449.06 1.95
26 2,5-difluoro-phenyl 417.12 1.82
27 2,5-dimethoxy-phenyl 441.16 1.55
28 2,3-difluoro-phenyl 417.12 1.8
29 2,4-difluoro-phenyl 417.12 1.8
3,4-difluoro-phenyl 417.12 1.85
31 4-isopropyl-phenyl 423.19 2
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-112-
32 6-methyl-pyridin-3-yl 396.15 1.45
33 4-fluoro-naphthalen-1-yl 449.15 2
34 3,5-difluoro-phenyl 417.12 1.87
35 3-aminosulfonyl-4-chloro-phenyl 494.07 1.5
36 1 H-Benzoimidazol-5-yl 421.15 1.37
37 2-Chloro-4-fluoro-phenyl 433.09 1.85
38 4-trifluoromethoxy-phenyl 465.12 2.07
39 1 H-Benzotriazol-5-yl 422.14 1.34
40 4-methoxy-quinolin-2-yl 462.16 1.82
41 2-fluoro-4-trifluoromethyl-phenyl 467.12 2.04
42 2,3,6-trifluoro-phenyl 435.11 1.87
43 2-methyl-pyridin-3-yl 396.15 1.32
44 2,4,5-trifluoro-phenyl 435.11 1.85
45 4-propyl-phenyl 423.19 2.15
46 2-fluoro-3-trifluoromethyl-phenyl 467.12 1.92
47 3-fluoro-2-methyl-phenyl 413.15 1.82
48 2,4-dichloro-5-fluoro-phenyl 467.05 2.05
49 3-fluoro-4-methoxy-phenyl 429.14 1.8
50 4-isopropoxy-phenyl 439.18 1.89
51 4-propoxy-phenyl 439.18 2.02
52 3-Chloro-4-fluoro-phenyl 433.09 1.95
53 2,6-dimethoxy-pyridin-3-yl 442.16 1.7
54 2-fluoro-5-methyl-phenyl 413.15 1.85
55 3-fluoro-5-trifluoromethyl-phenyl 467.12 2.09
56 4-difluoromethoxy-phenyl 447.13 1.79
57 Biphenyl-2-yl 457.17 2.02
58 4-aminosulfonyl-phenyl 460.11 1.45
59 3-Chloro-phenyl 415.1 1.9
60 4-cyano-phenyl 406.14 1.6
61 2,3-dimethoxy-phenyl 441.16 1.68
62 2,6-dimethoxy-phenyl 441.16 1.68
63 3,5-dimethoxy-phenyl 441.16 1.82
64 3-fluoro-4-methyl-phenyl 413.15 1.8
65 3-fluoro-phenyl 399.13 1.75
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-113-
66 3-methoxy-4-methyl-phenyl 425.17 1.9
67 naphthalen-1-yl 431.16 1.95
68 pyridin-3-yl 382.14 1.3
69 pyridin-2-yl 382.14 1.42
70 6-methyl-pyridin-2-yl 396.15 1.52
71 m-tolyl 395.16 1.85
72 p-tolyl 395.16 1.74
73 4-fluoro-3-methoxy-phenyl 429.14 1.79
74 3-Chloro-4-methyl-phenyl 429.12 2
75 5-Chloro-2-methyl-phenyl 429.12 1.97
76 3-Chloro-2,6-dimethoxy-phenyl 475.12 1.82
77 3-Chloro-2-fluoro-phenyl 433.09 1.89
78 2-phenoxy-pyridin-3-yl 474.16 1.82
79 2-trifluoromethoxy-phenyl 465.12 1.99
80 3-ethoxy-phenyl 425.17 1.79
81 3-Chloro-4-methoxy-phenyl 445.11 1.85
82 3,5-dimethoxy-4-methyl-phenyl 455.18 1.99
83 4-Chloro-3-methyl-phenyl 429.12 2.05
84 2-Chloro-3,4-dimethoxy-phenyl 475.12 1.72
85 3-cyclopentyloxy-4-methoxy-phenyl 495.21 2
86 4-methoxy-3-propoxy-phenyl 469.19 1.89
87 3-isopropoxy-4-methoxy-phenyl 469.19 1.85
88 3-Butoxy-4-methoxy-phenyl 483.21 1.95
89 4-trifluoromethyl-pyridin-3-yl 450.12 1.65
90 6-(2,2,2-trifluoro-ethoxy)-pyridin-3-yI 480.13 1.95
91 2-(4-fluoro-phenoxy)-pyridin-3-yI 492.15 1.9
92 2-chloro-3-trifiuoromethyl-phenyl 483.09 1.97
93 2-difluoromethoxy-phenyl 447.13 1.82
94 3-difluoromethoxy-phenyl 447.13 1.89
95 6-trifluoromethyl-pyridin-3-yl 450.12 1.84
96 2-methyl-[1,8]naphthyridin-3-yl 447.16 1.32
97 2-methyl-[1,6]naphthyridin-3-yl 447.16 1.42
98 2,3-dihydro-benzofuran-7-yl 423.15 1.57
99 2-Chloro-3-methyl-phenyl 429.12 1.92
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-114-
100 4-methoxy-3-methyl-phenyl 425.17 1.92
101 2-ethoxy-pyridin-3-yl 426.16 1.6
102 2-ethoxy-naphthalen-1-yl 475.18 2.02
103 3-(dimethylamino)sulfonyl-phenyl 488.14 1.72
104 2-propoxy-pyridin-3-yl 440.18 1.84
105 2-(4-Chloro-phenoxy)-pyridin-3-yl 508.12 1.93
106 2-methyl-1 H-benzoimidazol-5-yi 435.16 1.4
107 6-hydroxy-pyridin-2-yl 398.13 1.32
108 2-(5-methyl-[1,2,4]oxadiazol-3-yl)-pyridin-4-yl 464.15 1.45
109 4-(5-methyl-[1,2,4]oxadiazol-3-yl)-phenyl 463.16 1.67
110 4-(5-ethyl-[1,2,4]oxadiazol-3-yl)-phenyl 477.17 1.92
111 3-(5-methyl-[1,2,4]oxadiazol-3-yl)-phenyl 463.16 1.68
112 3-(5-ethyl-[1,2,4]oxadiazol-3-yl)-phenyl 477.17 1.9
113 2-hydroxy-pyridin-4-yl 398.13 1.25
114 2-Benzyl-phenyl 471.19 2.17
115 3,5-dichloro-phenyl 449.06 2
116 3-Chloro-2-methyl-phenyl 429.12 1.93
117 2,3-dihydro-benzofuran-5-yl 423.15 1.74
118 2-(3-methyl-[1,2,4]oxadiazol-5-yl)-pyridin-4-yI 464.15 1.49
119 3-hydroxy-2-methyl-phenyl 411.15 1.54
120 2-fluoro-5-trifluoromethyl-phenyl 467.12 2.02
121 4-methoxy-2-methyl-phenyl 425.17 1.82
122 3-methoxy-2-methyl-phenyl 425.17 1.82
123 2-hydroxy-5-methyl-phenyl 411.15 1.68
124 3,5-dichloro-4-hydroxy-phenyl 465.06 1.57
125 2-hydroxy-3-isopropyl-phenyl 439.18 2.93
126 1 H-indol-6-yl 420.15 1.75
127 3-hydroxy-phenyl 397.14 1.6
128 3-methoxy-naphthalen-2-yl 461.17 1.9
129 3-hydroxy-4-methoxy-phenyl 427.15 1.54
130 4-Chloro-2-hydroxy-phenyl 431.1 1.77
131 3,4-dimethoxy-2-methyl-phenyl 455.18 1.79
132 6-(acetylamino)-pyridin-3-yl 439.16 1.35
133 2,6-dimethoxy-4-methyl-phenyl 455.18 1.8
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-115-
134 2-Benzyloxy-phenyl 487.18 2.07
135 6-methoxy-quinolin-2-yi 462.16 1.82
136 quinoxalin-6-yl 433.15 1.42
137 1,2-dimethyl-1 H-benzoimidazol-5-yl 449.18 1.49
138 1 H-indol-5-yl 420.15 1.7
139 2-(3,5-dimethyl-1 H-pyrazol-4-yl)-5-methoxy-
505.2 1.6
phenyl
140 8-methyl-2-oxo-1,2-dihydroquinolin-6-yI 462.16 1.45
141 3-aminosulfonyl-phenyl 460.11 1.5
142 4-(3-methyl-6-oxo-3-piperidinyl)-phenyl 492.21 1.54
143 2-(2-methoxy-ethoxy)-phenyl 455.18 1.62
144 2-hydroxy-4-methyl-phenyl 411.15 1.68
145 3-Chloro-4-hydroxy-phenyl 431.1 1.64
146 3-hydroxy-4-methyl-phenyl 411.15 1.75
147 3-methoxy-5-(methylsulfonyl)amino-phenyl 504.14 1.57
148 2-{[(2,2-dimethylpropyl)amino]carbonyl}-phenyl 494.22 1.85
149 5-acetyl-2-ethoxypyridin-3-yl 468.17 1.7
150 2-(2-methoxy-ethoxy)-pyridin-3-yI 456.17 1.62
151 isoquinolin-4-yl 432.15 1.52
152 2-ethoxy-3-methoxy-phenyl 455.18 1.8
153 4-[(1R)-1-(acetylamino)ethyl]-phenyl 466.19 1.47
154 4-pyrimidin-4-yl-phenyl 459.16 1.6
155 3-methyl-2-propoxy-phenyl 453.2 2
156 4-ethoxy-pyridin-3-yl 426.16 1.5
157 2-chloro-4-methylsulfonylamino-phenyl 508.09 1.64
158 2-ethoxy-5-methanesulfonyl-phenyi 503.14 1.68
159 4-hydroxy-2-(2,2,2-trifluoro-ethoxy)-phenyl 495.13 1.65
160 4-hydroxy-2-methoxy-phenyl 427.15 1.52
161 4-cyano-pyridin-2-yl 407.13 1.55
162 2-pyridin-4-yi-3H-benzoimidazol-5-yl 498.17 1.5
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-116-
Example 163: (3-Chloro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-
ylmethyl-4H-
[1,2,4]triazol-3-yl]-piperidin-1-yl}-methanone
N ~,N
\
N-\
N N
1 / N
CI O CI
A mixture of the compound from preparation 10 (202mg, 0.54mmol), 4-
chloroaniline
5(140mg, 1.1 mmol) and trifluoroacetic acid (42 1, 0.54mmol) in toluene (2ml)
was heated
at 170 C for 20 minutes under microwave radiation. The cooled mixture was
diluted with
ethyl acetate, washed with 1 N sodium hydroxide solution and brine, then dried
over
Na2SO4 and concentrated under reduced pressure. The product was purified by
column
chromatography on silica gel using dichloromethane:methanol:0.88 ammonia
(100:0:0 to
90:10:1) to afford the title compound, (234mg).
'H NMR (400MHz, CD3OD): 8 1.80-1.97 (m, 4H), 2.86 (m, 2H), 3.08 (m, 1H), 3.70
(m,
1 H), 4.58 (m, 1 H), 5.72 (s, 2H), 7.26 (m, 2H), 7.32 (m, 1 H), 7.41-7.54 (m,
5H), 7.59 (s,
2H).
LRMS : m/z (APCI+) 482 [MH]+
Example 164: (4-Chloro-phenyl)-{4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-
ylmethyl-4H-
[1,2,4]triazol-3-yl]-piperidin-l-yl}-methanone
N
i 'J
N \
O CI
4-Chlorobenzoyl chloride (49 1, 0.38mmol) was added to a mixture of the
compound
from preparation 12a (120mg, 0.35mmol) and N-methyl morpholine (77 1,
0.70mmol) in
dichloromethane (2ml), then the mixture was stirred at room temperature for 2
hours.
The reaction was diluted with dichloromethane, washed with 1 N sodium
hydroxide
solution and the aqueous wash re-extracted with dichloromethane. The combined
organic solutions were evaporated under reduced pressure to give an oil. This
was
purified by column chromatography on silica gel using
dichloromethane:methanol:0.88
ammonia (90:10:1) to afford the title compound as a white solid, (160mg).
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-117-
'H NMR (400MHz, DMSO-d6): S 1.60-1.82 (m, 4H), 2.70-3.04 (m, 3H), 3.54 (m,
1H), 4.33
(m, 1 H), 5.66 (s, 2H), 7.34 (d, 2H), 7.39 (d, 2H), 7.48 (d, 2H), 7.57 (d,
2H), 7.64 (s, 2H);
LRMS : m/z (APCI+) 482 [MH]+
Example 165a: {4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-yl]-
piperidin-1-yl}-(3,5-difluoro-phenyl)-methanone
N ,N
'
\ N
F
'J
\ N
F O cl
The title compound was obtained in 92% yield from the compound from
preparation 12a
and 2,4-difluorobenzoyl chloride, following the procedure described in example
164.
'H NMR (400MHz, CD3OD): S 1.80-1.98 (m, 4H), 2.84 (m, 2H), 3.09 (m, 1H), 3.65
(m,
1 H), 4.58 (m, 1 H), 5.72 (s, 2H), 7.05 (m, 3H), 7.24 (m, 2H), 7.57 (d, 2H),
7.59 (s, 2H).
LRMS : m/z (APCI+) 484 [MH]+
Example 165b: {4-[4-(4-Chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-
[1,2,4]triazol-3-yl]-
piperidin-1-yl}-(3,5-difluoro-phenyl)-methanone
Triethylamine (3.2ml, 23.Ommol) was added to a slurry of the bis salt from
preparation
12b (4.89g, 7.12mmol) in dichloromethane (25ml) giving a pale yellow solution.
The
solution was cooled in an ice-bath and then 3,5-difluorobenzoyl chloride
(0.95m1,
8.09mmol) was added. The reaction was stirred for 30 minutes, and then water
(20m1)
was added. After a further 20 minutes of stirring, the phases were separated
and the
organic phase was washed successively with aqueous citric acid, water, aqueous
sodium hydrogen carbonate and half-saturated brine. The clear dichloromethane
solution was then dried over magnesium sulphate and concentrated to give a
white
foam. Recrystallisation from ethyl acetate gave the title compound as a white
solid,
(2.69g), identical to material prepared as described in Example 165a.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-118-
Examples 166 to 167:
N ,N
CH3
N
N / \
O CI
The appropriate acid chloride (1.2 eq.) was added to a solution of the amine
of
preparation 2 (1 eq.) and N-methylmorpholine (1.5 eq.) in dichloromethane (5.5
mLmmof') and the reaction was stirred at room temperature for 4 hours. Tris-(2-
aminoethyl)amine polystyrene (3.85 mmol/g) was added and the reaction was
stirred for
a further hour. Saturated ammonium chloride solution was added, the mixture
was then
stirred for 20 minutes and the layers were separated using a hydrophobic
membrane.
The organic phase was washed with saturated sodium bicarbonate solution, the
layers
were separated and the organic solution was evaporated under reduced pressure
to
provide the title compounds.
Ex Z Yield Data
No (%)
166 Neopentvl 39 'H NMR (400MHz, CDCI3): 8 1.00 (s, 9H), 1.71 (m, 2H),
1.82 (m, 1 H), 1.98 (m, 1 H), 2.20 (s, 2H), 2.24 (s, 3H),
2.57 (m, 1 H), 2.65 (m, 1 H), 3.00 (m, 1 H), 3.98 (m, 1 H),
4.57 (m, 1 H), 7.20 (d, 2H), 7.57 (d, 2H). LCMS: m/z
APCI+ 375 [MH]+
167 Cyclopropyl 39 'H NMR (400MHz, CDCI3): S 0.73 (m, 2H), 0.94 (m, 2H),
1.70 (m, 3H), 1.88 (m, 1 H), 2.02 (m, 1 H), 2.22 (s, 3H),
2.58-2.75 (m, 2H), 3.08 (m, 1 H), 4.23 (m, 1 H), 4.44 (m,
1 H), 7.20 (d, 2H), 7.58 (d, 2H). LCMS: m/z APCI+ 347
[MH]+
Examples 168 to 173:
N
\ X
N
N
W O y Y'
A mixture of the appropriate amine, or amine salt, selected from preparations
12a, 100,
118, 119, 143, and 152 (1 eq.), the appropriate acid chloride (W-PhCOCI) (1.2
to 1.4
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-119-
eq.) and N-ethyidiisopropylamine (4 eq.) in dichloromethane (16 mLmmol-') was
stirred
at room temperature for 2 hours. Tris-(2-aminoethyl)amine polystyrene was then
added
and the mixture was stirred for another hour. The mixture was then washed with
1 N
sodium hydroxide solution, the aqueous solution was extracted with
dichloromethane
(2x) and the combined organic solutions were concentrated under reduced
pressure.
The crude products were purified by column chromatography on silica gel using
ethyl
acetate:methanol:0.88 ammonia (90:10:1) as eluant, to afford the title
compounds.
Ex Data
No
168 W = 3-F, 4-CH3; Y = 4-Cl; Y' = 2-Cl; X = CH3
'H NMR (400MHz, CDCI3): 8 1.77-1.99 (m, 4H), 2.19 (s, 3H), 2.26 (s, 3H),
2.59 (m, 1 H), 2.84-3.02 (m, 2H), 3.83 (m, 1 H), 4.55 (m, 1 H), 7.02 (m, 2H),
7.18-7.28 (m, 2H), 7.48 (m, 1 H), 7.66 (s, 1 H). LCMS: m/z APCI+ 447 [MNa]+
169 W = 3-F, 4-CH3; Y = 4-Cl; Y' = 2-F; X= CH3
'H NMR (400MHz, CDCI3): 8 1.75-2.00 (m, 4H), 2.24 (m,.6H), 2.63 (m, 1 H),
2.92 (m, 2H), 3.81 (m, 1 H), 4.56 (m, 1 H), 7.02 (m, 2H), 7.19 (m, 1 H), 7.40
(m,
3H). LCMS:. m/z APCI+431 [MH]+
170 W = 3,5-di-Cl; Y = 4-Cl; Y' = 2-OCH3; X = CH3
'H NMR (400MHz, CD3OD): S 1.70-2.00 (m, 4H), 2.17 (s, 3H), 2.72-2.92 (m,
2H), 3.1 (m, 1 H), 3.62 (m, 1 H), 3.84 (m, 3H), 4.56 (m, 1 H), 7.20 (m, 1 H),
7.39
(m, 4H), 7.58 (s, 1 H). LCMS: m/z APCI+ 480 [MH]+
171 W = 2-F, 3-CI; Y = 4-CI; Y' = H; X = CH2OCH2CH3;
'H NMR (400MHz, CDCI3): 8 1.08 (t, 3H), 1.64-1.81 (m, 2H), 1.84-2.01 (m,
2H), 2.79 (m, 1 H), 2.85-3.17 (m, 2H), 3.41 (q, 2H), 3.62 (m, 1 H), 4.41 (s,
2H),
4.62 (m, 1 H), 7.17 (m, 1 H), 7.23 (m, 3H), 7.43 (m, 1 H), 7.58 (d, 2H). LCMS:
m/z APCI+ 477 [M]+
172 W = 4-Cl; Y = 4-Cl; Y' = H; X = CH2CF3;
'H NMR (400MHz, CDCI3): 8 1.81 (m, 2H), 1.98 (m, 2H), 2.70 (m, 1H), 2.80-
3.02 (m, 2H), 3.44 (q, 2H), 3.81 (m, 1 H), 4.59 (m, 1 H), 7.19 (d, 2H), 7.38
(m,
4H), 7.60 (d, 2H). LCMS: m/z APCI' 483 [M]+
173 W= 2-OCF3; Y = 4-Cl; Y' = H; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.76 (m, 1 H), 1.79-2.02 (m, 3H), 2.68 (m, 1 H),
2.82-3.02 (m, 2H), 3.58 (m, 1 H), 4.59 (m, 1 H), 5.62 (s, 2H), 6.99 (m, 2H),
7.22-7.42 (m, 6H), 7.50 (s, 2H). LCMS: m/z APCI+ 532 [MH]+
A = 4 eq. of triethylamine were used, and the crude product was not treated
with
polymer supported amine.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-120-
8= 10 eq. of polymer supported N-ethyldiisopropylamine was used in place of N-
ethyldiisopropylamine.
Examples 174 to 187:
N~-N
i `~-x
PlY 5 The appropriate acid chloride (W-PhCOCI) (1.0 to 1.5 eq.) was added to a
solution of
the appropriate amine hydrochloride, or amine, selected from preparations 120
to 121,
132, 134 to 135, 137, 139 to 142, 151, 153 to 154, and 169 (1 eq.), and
triethylamine
(1.2 to 5 eq.) in dichloromethane (10 to 25 mLmmol"'). The reaction was
stirred at room
temperature for 18 hours. The mixture was then diluted with dichloromethane,
it was
washed with saturated sodium carbonate solution, followed by ammonium chloride
solution, and then it was concentrated under reduced pressure. The crude
products
were purified by column chromatography on silica gel using
dichloromethane:methanol
(100:0 to 90:10) as eluant to afford the title compounds.
Ex No Data
174 W = 3-Cl; Y = 4-Cl; Y' = 2-CH3; X = CH3
'H NMR (400MHz, CDCI3): S 1.65-2.02 (m, 7H), 2.18 (s, 3H), 2.55 (m, 1H),
2.78-3.01 (m, 2H), 3.79 (m, 1 H), 4.58 (m, 1 H), 7.03 (m, 1 H), 7.20-7.39 (m,
5H), 7.41 (s, 1 H). LCMS: m/z APCI+ 451 [MNa]+
175 W= 3,5-di-Cl; Y = 4-Cl; Y' = H; X = CHZOCH3
'H NMR (400MHz, CD3OD): S 1.81-1.98 (m, 4H), 2.86 (m, 2H), 3.12 (m,
1 H), 3.20 (s, 3H), 3.64 (m, 1 H), 4.38 (s, 2H), 4.58 (m, 1 H), 7.40 (s, 2H),
7.46 (d, 2H), 7.58 (s, 1 H), 7.62 (d, 2H). LCMS: m/z APCI+ 479, 481 [MH]+
176 W = 4-Cl; Y = 4-Cl; Y' = H; X = CH2OCF3
'H NMR (400MHz, CDCI3): 8 1.81 (m, 2H), 1.98 (m, 2H), 2.78 (m, 1 H),
2.96 (m, 2H), 3.75-3.90 (m, 1 H), 4.40-4.60 (m, 1 H), 4.97 (s, 2H), 7.21 (m,
2H), 7.37 (m, 4H), 7.59 (d, 2H). LCMS: m/z APCI+ 499 [M]+
177 W = 3,5-di-F; Y = 4-Cl; Y' = H; X = CH2OCH2CF3
'H NMR (400MHz, CDCI3): 8 1.80-2.02 (m, 4H), 2.79 (m, 1 H), 2.83-3.05
(m, 2H), 3.80 (m, 3H), 4.58 (m, 3H), 6.85 (m, 3H), 7.22 (d, 2H), 7.57 (d,
2H). LCMS: m/z APCI+ 515 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-121-
178 W = 3-CI; Y = 4-Cl; Y' = H; X = (2-oxopyrrolidin-1 -yl)methyl;
'H NMR (400MHz, CDCI3): 8 1.78-2.00 (m, 6H), 2.22 (t, 2H), 2.75 (m, 1H),
2.97 (m, 2H), 3.48 (t, 2H), 3.80 (m, 1 H), 4.42 (s, 2H), 4.58 (m, 1 H), 7.00
(d, 2H), 7.23 (d, 1 H), 7.38 (m, 3H), 7.58 (d, 2H). LCMS: m/z APCI+ 499,
501 [MH]+
179A W= 3,5-di-Cl; Y = 4-Cl; Y' = H; X = morpholin-4-ylmethyl;
'H NMR (400MHz, CDCI3): 8 1.78-2.02 (m, 4H), 2.40 (m, 4H), 2.78 (m,
1 H), 2.78-3.04 (m, 2H), 3.41 (s, 2H), 3.58 (m, 4H), 3.78 (m, 1 H), 4.58 (m,
1 H), 7.23 (m, 5H), 7.56 (d, 2H). LCMS: m/z APCI+ 534 [M]+
180 W = 3-F; Y = 4-CI; Y' = H; X = 1 H-tetrazol-1-ylmethyl;
'H NMR (400MHz, CDC13): S 1.60-1.83 (m, 4H), 2.60-2.78 (m, 2H), 3.00
(m, 1 H), 3.90 (m, 1 H), 4.50 (m, 1 H), 5.58 (s, 2H), 6.98 (m, 2H), 7.50 (m,
2H), 7.29 (m, 1 H), 7.60 (d, 2H), 8.79 (s, 1 H). LCMS: m/z ES+ 481, 483
[MH]+
181 W = 3-Cl; Y= 4-Cl; Y' = H; X = 2-methyl-imidazo-3-ylmethyl;
'H NMR (400MHz, CDCI3): S 1.77-1.83 (m, 2H), 1.89-2.00 (m, 2H), 2.14 (s,
3H), 2.64 (m, 2H), 2.94 (m, 1 H), 3.82 (m, 1 H), 4.58 (m, 1 H), 5.08 (s, 2H),
6.60 (s, 1 H), 6.96 (m, 3H), 7.24 (m, 1 H), 7.32-7.40 (m, 3H), 7.58 (d, 2H).
LCMS: m/z APCI+ 495, 497 [MH]+
182 W = 3-Cl; Y = 4-Cl; Y' = H; X = 3-methyl-[1,2,4]oxadiazol-5-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.80 (m, 2H), 1.98 (m, 2H), 2.30 (s, 3H), 2.76
(m, 1 H), 2.95 (m, 2H), 3.80 (m, 1 H), 4.22 (s, 2H), 4.58 (m, 1 H), 7.19 (d,
2H), 7.24 (d, 1 H), 7.37 (m, 3H), 7.52 (d, 2H). LCMS: m/z APCI+ 497, 499
[MH]+
183 W = 3,5-di-F; Y = 4-Cl; Y' = H; X = [1,2,4]-triazol-1-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.82 (m, 2H), 2.00 (m, 2H), 2.75 (m, 1H),
2.80-3.08 (m, 2H), 3.80 (m, 1 H), 4.58 (m, 1 H), 5.40 (s, 2H), 6.89 (m, 3H),
7.15 (m, 2H), 7.58 (d, 2H), 7.82 (s, 1 H), 8.04 (s, 1 H). LCMS: m/z APCI+
484, 486 [MH]+
184 W = 2-F, 3-Cl; Y = 4-CI; Y' = H; X = pyrimidin-2-yloxymethyl
'H NMR (400MHz, CDCI3): 8 1.75 (m, 2H), 1.97 (m, 2H), 2.81 (m, 1H),
2.88-3.15 (m, 2H), 3.62 (m, 1 H), 4.62 (m, 1 H), 5.43 (s, 2H), 6.98 (m, 1 H),
7.16 (m, 1 H), 7.25 (m, 1 H), 7.36 (d, 2H), 7.44 (m, 3H), 8.44 (d, 2H).
LCMS: m/z APCI+ 527 [M]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-122-
185 W = 2-F, 3-Cl; Y = 4-Cl; Y' = H; X = 2-pyridin-2-yl-ethyl
'H NMR (400MHz, CDCI3): 8 1.58-1.98 (m, 4H), 2.75 (m, 1H), 2.90-3.00
(m, 2H), 3.04-3.18 (m, 2H), 3.42 (m, 2H), 3.61 (m, 1 H), 4.60 (m, 1 H), 7.10-
7.27 (m, 6H), 7.42 (m, 1 H), 7.56 (d, 2H), 7.82 (m, 1 H), 8.48 (s, 1 H).
LCMS: m/z APCI+ 524 [MH]+
186 W = 3-Cl; Y = 4-Cl; Y' = H; X = 2-morpholin-4-yiethyl
'H NMR (400MHz, CDC13): 8 1.80 (m, 2H), 1.97 (m, 2H), 2.40 (m, 4H),
2.64-3.02 (m, 7H), 3.59-3.90 (m, 5H), 4.58 (m, 1 H), 7.19 (d, 2H), 7.24 (m,
2H), 7.36 (m, 2H), 7.58 (d, 2H). LCMS: m/z APCI+ 514 [M]+
187 W = 5-Cl, 2-F; Y= 4-Cl; Y' = H; X = 2-(3,5-dimethyl-isoxazol-4-yl)-ethyl;
'H NMR (400MHz, CDCI3): S 1.75-1.98 (m, 7H), 2.04 (s, 3H), 2.63-2.80 (m,
5H), 2.86-3.08 (m, 2H), 3.62 (m, 1 H), 4.59 (m, 1 H), 6.75 (m, 2H), 7.01 (m,
1 H), 7.37 (m, 2H), 7.57 (d, 2H). LCMS: m/z APCI+ 542, 544 [MH]+
A= ethyl acetate:methanol:0.88 ammonia was used as the column eluant.
8= 2 eq N-methylmorpholine was used instead of triethylamine.
C = 3 equivalents of N-methylmorpholine were used in place of triethylamine.
Example 188: 4-[4-(4-Chlorophenyl)-5-(1H-imidazol-1-ylmethyl)-4H-1,2,4-triazol-
3-yl]-1-
(3,3-dimethylbutanoyl)piperidine
O ~N
CH N
3
H3C CH3
CI
The title compound was obtained as a white solid, from the compound of
preparation
138 and isovaleryl chloride, following the procedure described for examples
174 to 187.
'H NMR (400MHz, CDCI3): 8 1.01 (s, 9H), 1.70-1.85 (m, 2H), 1.98 (m, 3H), 2.22
(m, 2H),
2.55-2.66 (m, 2H), 3.00 (m, 1 H), 4.00 (m, 1 H), 4.59 (m, 1 H), 5.18 (s, 2H),
6.60 (s, 1 H),
6.90 (m, 2H), 7.00 (s, 1 H), 7.20 (s, 1 H), 7.52 (m, 2H); LCMS: m/z APCI+ 441
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 -123- PCT/IB2004/004059
Examples 189 to 198:
~N\~/
0N
z N N
N
Y'
Y
The appropriate acid chloride, ZCOCI, (1.0 eq.) was added to a solution of the
appropriate amine selected from those of preparations 122 to 131 (1 eq.), and
triethylamine (1.1 eq.) in dichloromethane (4 mLmmol-'). The reaction was
stirred at
room temperature for 1 hour. It was then diluted with water, stirred for 5
minutes, and
then filtered through a phase separation cartridge. The organic solution was
concentrated under reduced pressure and the crude product was purified by
column
chromatography on silica gel using ethyl acetate:dichloromethane:methanol
(100:0:0 to
0:95:5) as eluant. The products were azeotroped with ether to afford the title
compounds as white foams.
Ex No Data
189 Z = 3-Chloro-2-fluorophenyl; Y = H; Y' = H;
'H NMR (400MHz, CDCI3): S 1.68-2.07 (m, 4H), 2.75 (m, 1H), 2.92 (m,
1 H), 3.01 (m, 1 H), 3.61 (m, 1 H), 4.60 (m, 1 H), 5.64 (s, 2H), 7.06 (d, 2H),
7.13 (t, 1 H), 7.38-7.57 (m, 7H). LCMS: m/z ES+ 488 [MNa]+
190 Z= 3-Chlorophenyl Y = 4-OCH3; Y' = H;
'H NMR (400MHz, CDCI3): 8 1.75-2.20 (m, 4H), 2.79 (m, 1H), 2.93 (m,
2H), 3.80 (m, 1 H), 3.84 (s, 3H), 4.56 (m, 1 H), 5.66 (s, 2H), 6.94 (m, 2H),
7.01 (m, 2H), 7.26 (m, 1 H), 7.28-7.40 (m, 3H), 7.51 (s, 2H). LCMS: m/z
ES+ 500 [MNa]+
191 Z = 3-Chloro-2-fluorophenyl; Y = 4-F; Y'= H;
'H NMR (400MHz, CDCI3): S 1.72-2.07 (m, 4H), 2.77 (m, 1H), 2.90 (m,
1 H), 3.02 (m, 1 H), 3.62 (m, 1 H), 4.61 (m, 1 H), 5.64 (s, 2H), 7.05-7.19 (m,
6H), 7.41 (t, 1 H), 7.49 (s, 2H). LCMS: m/z ES+ 503 [MNa]+
192 Z = 3-Chloro-2-fluorophenyl; Y = 4-Br; Y'= H;
'H NMR (400MHz, CDCI3): 6 1.71-2.06 (m, 4H), 2.72 (m, 1H), 2.94 (m,
1 H), 3.05 (m, 1 H), 3.63 (m, 1 H), 4.62 (m, 1 H), 5.65 (s, 2H), 6.95 (d, 2H),
7.14 (t, 1 H), 7.27 (m, 1 H), 7.43 (t, 1 H), 7.49 (s, 2H), 7.60 (d, 2H). LCMS:
m/z ES+ 546 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-124-
193 Z = 3-Chloro-2-fluoro-phenyl; Y = 4-CF3; Y'= H;
1 H NMR (400MHz, CDCI3): 8 1.75-2.15 (m, 4H), 2.76-2.94 (m, 2H), 3.05
(m, 1 H), 3.62 (m, 1 H), 4.67 (m, 1 H), 5.76 (s, 2H), 7.14 (dd, 1 H), 7.20-
7.30
(m, 2H), 7.36-7.50 (m, 4H), 7.75 (d, 2H). LCMS: m/z ES+ 556 [MNa]+
194 Z = 3-Chlorophenyl; Y = 4-CH3; Y' = H;
'H NMR (400MHz, CDCI3): S 1.88 (m, 2H), 2.06 (m, 2H), 2.42 (s, 3H),
2.79-3.00 (m, 3H), 3.80 (m, 1 H), 4.59 (m, 1 H), 5.70 (s, 2H), 7.07 (d, 2H),
7.23-7.40 (m, 6H), 7.53 (s, 2H). LCMS: m/z ES+ 484 [MNa]+
195 Z= 3-Chloro-2-fluoro-phenyl; Y = 4-CN; Y' = H;
'H NMR (400MHz, CDCI3): S 1.67-2.00 (m, 4H), 2.67 (m, 1H), 2.80-3.08
(m, 2H), 3.60 (m, 1 H), 4.58 (m, 1 H), 5.64 (s, 2H), 7.11 (t, 1 H), 7.16-7.25
(m, 3H), 7.41 (t, 1 H), 7.46 (s, 2H), 7.75 (d, 2H). LCMS: m/z ES+ 513
[MNa]+
196 Z= 3-Chloro-2-fluorophenyl; Y = 4-CI; Y' = 2-CH3;
'H NMR (400MHz, CDCI3): 8 1.68-1.98 (m, 6H), 2.07 (m, 1 H), 2.60 (m,
1 H), 1.80-3.13 (m, 2H), 3.60 (d, 1 H), 4.60 (d, 1 H), 5.55 (d, 1 H), 5.67 (d,
1 H), 6.86-7.50 (m, 8H). LCMS: m/z ES+ 536 [MNa]+
197 Z = 3-chlorophenyl; Y = 4-CI; Y' = 3-F;
'H NMR (400MHz, CDCI3): 8 1.84 (m, 2H), 2.00 (m, 2H), 2.72 (m, 1H),
2.94 (m, 2H), 3.83 (m, 1 H), 4.55 (m, 1 H), 5.67 (s, 2H), 6.90 (d, 2H), 7.26-
7.41 (m, 4H), 7.50-7.56 (m, 3H). LCMS: m/z ES+ 500 [MH]+
198 Z = 3-Chloro-2-fluoro-phenyl; Y = 4-CI; Y' = 3-CI;
'H NMR (400MHz, CDCI3): 8 1.80 (m, 1H), 1.86-2.05 (m, 3H), 2.74 (m,
1 H), 2.92 (m, 1 H), 3.04 (m, 1 H), 3.61 (m, 1 H), 4.61 (m, 1 H), 5.70 (s,
2H),
7.05-7.16 (m, 3H), 7.27 (br s, 1 H), 7.40 (t, 1 H), 7.50 (s, 2H), 7.55 (d, 1
H)
LCMS: m/z ES+ 534 [MNa]+
Examples 199 to 201:
N~N
`}-x
N
N / \
oz=~
z cl
A mixture of the appropriate acid, ZCOZH, (1.2 eq.), 1-hydroxybenzotriazole
hydrate (1.2
eq.), triethylamine (2 to 4 eq.) and 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-125-
hydrochloride (1.2 eq.) and the appropriate amine, or amine hydrochloride,
selected from
preparations 2 and 143 (1 eq.) in dichloromethane (26 mimmol-') was stirred at
room
temperature for 24 hours. The reaction was then washed with 2N sodium
hydroxide
solution and the organic solution was evaporated under reduced pressure. The
crude
product was purified by column chromatography on silica gel using an elution
gradient of
ethyl acetate:methanol:0.88 ammonia (100:0:0 to 95:5:0.5) to afford the title
compounds.
Ex Data
No
199 Z =Cyclopropylmethyl; X = CH2OCH2CH3
'H NMR (400MHz, CDCI3): 8 0.18 (m, 2H), 0.58 (m, 2H), 1.06 (t, 3H),
1.70-1.90 (m, 3H), 2.00 (m, 2H), 2.22 (m, 2H), 2.59-2.77 (m, 2H), 3.02
(m, 1 H), 3.40 (q, 2H), 3.92 (m, 1 H), 4.40 (s, 2H), 4.55 (m, 1 H), 7.22 (d,
2H), 7.54 (d, 2H). LCMS: m/z APCI+ 403 [MH]+
200 Z= 5-Trifluoromethylpyridin-2-yl; X = CH2OCH2CH3
'H NMR (400MHz, CDCI3): S 1.08 (t, 3H), 1.79-2.04 (m, 4H), 2.60 (m,
1 H), 2.95-3.17 (m, 2H), 3.20 (q, 2H), 3.78 (m, 1 H), 4.41 (s, 2H), 4.59
(m, 1 H), 7.22 (d, 2H), 7.57 (d, 2H), 7.77 (d, 1 H), 7.95 (d, 1 H), 8.55 (s,
1 H). LCMS: m/z APCI+ 494 [MH]+
201 Z = 1 H-Indazol-3-yl; Y = 4-Cl; Y' = H; X = CH3
'H NMR (400MHz, CDCI3): 8 1.65-1.88 (m, 4H), 2.15 (s, 3H), 2.80-2.97
(m, 2H), 3.08 (m, 1 H), 4.42-4.62 (m, 2H), 7.20 (dd, 1 H), 7.40 (dd, 1 H),
7.57 (m, 3H), 7.65 (d, 2H), 7.90 (d, 1 H), 13.42 (s, 1 H). LCMS: m/z
APCI+ 421 [MH]+
A = 5-(trifluoromethyl)-2-pyridinecarboxylic acid was used and may be prepared
as
described in J. Org. Chem. (European) 2003; (8); 1559-1568.
8= reaction was performed in the absence of 1-hydroxybenzotriazole hydrate and
triethylamine.
Examples 202 to 204:
x
N/~-
N
N / \
0::=<
Z cl
A solution of the appropriate acid, ZCOzH, (1.5 eq.), O-benzotriazol-1-yl-
N,N,N;N'-
tetramethyluronium hexafluorophosphate (2 eq.), N-methylmorpholine (5 eq.) and
the
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-126-
appropriate amine hydrochloride selected from preparations 2 and 133 (1 eq.)
in
dichloromethane (8 mLmmol"') was stirred at room temperature for 24 hours. The
reaction was then washed with sodium hydroxide solution and the organic
solution was
evaporated under reduced pressure. The crude product was purified by column
chromatography on silica gel using an elution gradient of
dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) to afford the title
compounds.
Ex Data
No
202 Z = 4-Chloro-3-fluorophenyl; X = CH3;
'H NMR (400MHz, CD3OD): 8 1.79-1.99 (m, 4H), 2.23 (s, 3H), 2.85 (m,
2H), 3.11 (m, 1 H), 3.70 (m, 1 H), 4.58 (m, 1 H), 7.22 (d, 1 H), 7.37 (d,
1 H), 7.45 (d, 2H), 7.58 (m, 1 H), 7.64 (d, 2H). LCMS: m/z APCI+ 433
[M]+
203 Z = 2,3,4-Trifluorophenyl; X = CH3;
'H NMR (400MHz, CD3OD): 8 1.82 (m, 3H), 1.98 (m, 1H), 2.22 (s, 3H),
2.89 (m, 2H), 3.15 (m, 1 H), 3.60 (m, 1 H), 4.60 (m, 1 H), 7.12 (m, 2H),
7.44 (m, 2H), 7.66 (m, 2H). LCMS: m/z APCI+ 435 [M]+
204 Z = 1 H-Indazol-3-yl; X = 3-methyl-isoxazol-5-ylmethyl;
'H NMR (400MHz, CDCI3): S. 1.80-2.15 (m, 5H), 2.18 (s, 3H), 2.75 (m,
1 H), 2.80-2.92 (m, 2H), 3.17 (m, 1 H), 4.04 (s, 2H), 4.62-4.81 (m, 2H),
4.86 (s, 1 H), 7.15 (m, 3H), 7.30 (dd, 1 H), 7.50 (m, 3H), 8.00 (d, 1 H),
11.90 (br s, 1 H). LCMS: m/z ES+ 500, 502 [M]+
Examples 205 to 207:
N-N
~-x
N
N / \
Ozz:<
z CI
A solution of the appropriate acid, ZCO2H (1.2 eq.), O-benzotriazol-1-yl-
N,N,N;N'-
tetramethyluronium hexafluorophosphate (1.2 eq.), N-methylmorpholine (1.4 eq.)
and
the appropriate amine selected from preparations 12 and 136 (1 eq.) in
dichloromethane (7 to 10 mLmmol"') was stirred at room temperature for 24
hours. The
reaction was then partitioned between sodium hydroxide solution and
dichloromethane,
and the layers were separated. The organic solution was washed with ammonium
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-127-
chloride solution, dried over MgSO4 and then it was evaporated under reduced
pressure.
The crude product was purified by column chromatography on silica gel using an
elution
gradient of dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) to
afford the
title compounds.
Ex Data
No
205 Z = 3-Difluoromethyl-phenyl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): 8 1.78-2.02 (m, 4H), 2.70 (m, 1H), 2.95 (m,
2H), 3.80 (m, 1 H), 4.58 (m, 1 H), 5.63 (s, 2H), 6.50-6.80 (t, 1 H), 7.01 (d,
2H), 7.42-7.58 (m, 8H). LCMS: m/z APCI+ 498, 500 [MH]+
206 ' Z = 4-Difluoromethyl-phenyl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.78 (m, 2H), 1.97 (m, 2H), 2.66 (m, 1 H),
2.90 (m, 2H), 3.78 (m, 1 H), 4.58 (m, 1 H), 5.61 (s, 2H), 6.46-6.78 (t, 1 H),
6.98 (d, 2H), 7.22 (s, 2H), 7.39-7.57 (m, 8H). LCMS: m/z APCI+ 498, 500
[MH]+
207 Z= 1H-Indazol-3-yl; X = 2-piperidin-1-yl-ethyl;
'H NMR (400MHz, CDCI3): 8 1.38 (m, 2H), 1.50 (m, 4H), 1.74-2.18 (m,
4H), 2.30 (m, 4H), 2.72 (m, 6H), 2.88 (m, 1 H), 3.19 (m, 1 H), 4.61-4.88
(m, 2H), 7.18 (m, 3H), 7.30 (m, 1 H), 7.54 (m, 3H), 8.02 (d, 1 H). LCMS:
m/z APCI+ 518 [M]+
A = 3-difluoromethyl benzoic acid was used. It can be prepared according to
Tetrahedron 31; 1977; 391-401.
8= 4-difluoromethyl benzoic acid was used. It can be prepared according to
Tetrahedron 31; 1977; 391-401.
C = product additionally recrystallised from isopropyl alcohol, and 2.8 eq of
N-
methylmorpholine was used.
Examples 208 to 210:
N-N
~-x
N
N
z Y Y
A mixture of the appropriate oxadiazole selected from preparations 66 and 67
(1 eq.),
the appropriate aniline from preparations 166 and 168 or commercially
available 4-
chloro-2-(trifluoromethoxy)phenylamine (1.5 to 2.0 eq.) and trifluoroacetic
acid (0.5 to
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-128-
1.0 eq.) in toluene (2.5 to 9.5 mLmmol-') was heated at 110 C for 18 hours.
The cooled
mixture was partitioned between dichloromethane and sodium carbonate solution,
and
the layers were then separated. The organic phase was dried over MgSO4 and
evaporated under reduced pressure. The crude product was purified by column
chromatography on silica gel using dichloromethane:methanol:0.88 ammonia
(100:0:0 to
90:10:1) as eluant to afford the title compounds.
Ex No Data
208 Z = 3-Chlorophenyl; Y = 4-CI; Y' = 2-OCH2CH3; X = H;
'H NMR (400MHz, CD3OD): S 1.23 (t, 3H), 1.78-1.98 (m, 4H), 2.92 (m,
2H), 3.16 (m, 1 H), 3.70 (m, 1 H), 4.14 (q, 2H), 4.58 (m, 1 H), 7.18 (d, 1 H),
7.34 (m, 2H), 7.42 (m, 3H), 8.43 (s, 1 H). LCMS: m/z APCI+ 445 [M]+
209 Z = 3-Chlorophenyl; Y= 4-Cl; Y' = 2-OCF3; X = H;
'H NMR (400MHz, CD3OD): 6 1.80-1.98 (m, 4H), 2.84-2.99 (m, 2H),
3.10-3.20 (m, 1 H), 3.72 (m, 1 H), 4.60 (m, 1 H), 7.37 (d, 1 H), 7.44 (m,
3H), 7.70 (m, 2H), 7.78 (s, 1 H), 8.62 (s, 1 H). LCMS: m/z APCI+ 485 [M]+
210 Z = 4-Chlorophenyl; Y = 4-Cl; Y' = 2-pyrrolidin-1-ylmethyl ; X = CH3;
'H NMR (400MHz, CDCI3): 8 1.64-1.84 (m, 7H), 2.18 (s, 3H), 2.37 (m,
4H), 2.58 (m, 1 H), 2.78-2.99 (m, 2H), 3.16-3.20 (m, 2H), 3.64-3.95 (m,
1 H), 4.45-4.70 (m, 1 H), 7.09 (m, 1 H), 7.30-7.44 (m, 5H), 7.62 (s, 1 H).
LCMS: m/z APCI+ 498 [M]+
Examples 211 to 216:
I-~'x
N
o~N
z Y Y
A mixture of the appropriate oxadiazole selected from preparations 66 to 69,
71 and 75
(1 eq.), aniline (1.5 to 2.0 eq.) and trifluoroacetic acid (0.5 to 1.0 eq.) in
toluene (1.0 to
2.5 mLmmol"') was heated at 170 to 185 C for 20 minutes under microwave
radiation.
The crude solution was purified by column chromatography on a silica gel
cartridge
using dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) as eluant to
afford
the title compounds.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-129-
Ex No Data
211 Z = 3-Chlorophenyl; Y = 4-CI; Y' = 2-OCH2CF3; X = H;
'H NMR (400MHz, CDCI3) : 8 1.78-2.02 (m, 4H), 2.80 (m, 1H), 2.95-3.06
(m, 2H), 3.81 (m, 1 H), 4.40 (q, 2H), 4.58 (m, 1 H), 7.17 (s, 1 H), 7.25 (m,
3H), 7.38 (m, 3H), 8.18 (s, 1 H). LCMS: m/z APCI+ 499 [M]+
212 A Z= 4-Chlorophenyl; Y = 4-CI; Y' = H; X = CF3;
'H NMR (400MHz, CDCI3) : 8 1.82 (m, 2H), 2.00 (m, 2H), 2.76 (m, 1H),
2.83-3.02 (m, 2H), 3.83 (m, 1 H), 4.60 (m, 1 H), 7.22 (d, 2H), 7.37 (d, 2H),
7.40 (d, 2H), 7.60 (d, 2H). LCMS: m/z APCI+ 469 [M]+
213 Z = 3-Chlorophenyl; Y = 4-CI; Y' = H; X = CH2OCH2CH3;
'H NMR (400MHz, CDCI3): 8 1.08 (t, 3H), 1.78-1.90 (m, 2H), 1.92-2.02
(m, 2H), 2.78 (m, 1 H), 2.80-3.02 (m, 2H), 3.41 (q, 2H), 3.80 (m, 1 H),
4.41 (s, 2H), 4.60 (m, 1 H), 7.24 (m, 3H), 7.36 (m, 2H), 7.57 (d, 2H).
LCMS: m/z APCI+ 459 [M]+
214 Z = 4-Chlorophenyl; Y = 4-CH3; Y' = 2-CH3; X = CH3;
'H NMR (400MHz, CDCI3): 8 1.68-2.00 (m, 7H), 2.14 (s, 3H), 2.40 (s,
3H), 2.58 (m, 1 H), 2.78-2.98 (m, 2H), 3.78 (m, 1 H), 4.54 (m, 1 H), 6.97
(m, 1 H), 7.15 (m, 1 H), 7.20 (s, 1 H), 7.30 (d, 2H), 7.36 (d, 2H). LCMS:
m/z APCI+ 409 [MH]+
215 Z = 4-Chlorophenyl; Y = 4-CI; Y' = 2-CH3; X = CH2CH3;
'H NMR (400MHz, CDCI3): 8 1.20 (t, 3H), 1.68-2.02 (m, 6H), 2.14 (m,
1 H), 2.38-2.58 (m, 3H), 2.79-2.98 (m, 2H), 3.80 (m, 1 H), 4.58 (m, 1 H),
7.04 (m, 1 H), 7.26-7.39 (m, 5H), 7.41 (s, 1 H). LCMS: m/z ES+ 443, 446
[MH]+
216 Z = 3-chlorophenyl; Y = 4-CI; Y' = H; X = pyrazol-1-ylmethyl;
'H NMR (400MHz, CD3OD): 8 1.80-1.97 (m, 4H), 2.82 (m, 2H), 3.06 (m,
1 H), 3.69 (m, 1 H), 4.58 (m, 1 H), 5.41 (s, 2H), 6.18 (s, 1 H), 7.20 (m, 2H),
7.30 (m, 2H), 7.38-7.57 (m, 5H). LCMS: m/z APCI+ 481 [M]+
A = Crude reaction mixture was partitioned between ethyl acetate and 2N
hydrochloric
acid, then the organic solution washed with saturated sodium bicarbonate
solution and
evaporated under reduced pressure.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-130-
Examples 217 to 222:
N-N
N 'IIN x
o ~ I
e/N~
R R ci
A solution of the appropriate acid selected from preparation 158 and 159 (1
eq.), 1-
hydroxybenzotriazole hydrate (1.5 eq.), triethylamine (4 eq.) and 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.5 eq.) in
dichloromethane
(3.5 mLmmol"') was added to a solution of the appropriate amine (HNR4R5) (1.5
eq.) in
dichloromethane (2.5 mLmmol-') and the reaction was stirred at room
temperature for 24
hours. The reaction was then washed with ammonium chloride solution, and the
organic
solution was evaporated under reduced pressure. The crude product was purified
by
column chromatography on silica gel using an elution gradient of
dichloromethane:methanol:0.88 ammonia (100:0:0 to 95:5:0.5) to afford the
title
compounds.
Ex Data
No
217 R4 = H; R= bicyclo[1.1.1]pent-1-yl; X = CH3;
'H NMR (400MHz, CDCI3): S 1.58-1.78 (m, 4H), 1.86 (m, 3H), 2.02 (m, 5H),
2.22 (s, 3H), 2.79 (m, 2H), 3.32 (m, 2H), 5.90 (s, 1 H), 7.24 (d, 2H), 7.54
(d,
2H). LCMS: m/z APCI+ 386 [MH]+
218 R= H; R= t-butyl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDC13): 81.29 (s, 9H), 1.55-1.74 (m, 4H), 1.99-2.09 (m,
1 H), 2.80 (m, 2H), 3.38 (m, 2H), 5.23 (m, 1 H), 5.59 (s, 2H), 7.10 (d, 2H),
7.40 (d, 2H), 7.52 (s, 2H). LCMS: m/z APCI+ 443 [MH]+
219 NR R= azetidin-l-yl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.56-1.68 (m, 4H), 2.18-2.30 (m, 3H), 2.80 (m,
2H), 3.37 (m, 2H), 3.98 (t, 2H), 4.16 (t, 2H), 5.59 (s, 2H), 7.10 (d, 2H),
7.40
(d, 2H), 7.55 (s, 2H). LCMS: m/z ES+ 427 [MH]+
220 NR R= pyrrolidin-1-yl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.59-1.76 (m, 4H), 1.82 (m, 2H), 1.95 (m, 2H),
2.41 (m, 1 H), 2.82 (m, 2H), 3.36-3.46 (m, 6H), 5.59 (s, 2H), 7.15 (d, 2H),
7.40 (d, 2H), 7.55 (s, 2H). LCMS: m/z ES+ 441 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-131-
221 NR 4 R= morpholin-4-yl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDCI3): S 1.58-1.78 (m, 4H), 2.56 (m, 1 H), 2.82 (m, 2H),
3.38 (m, 2H), 3.44 (m, 2H), 3.60.(m, 2H), 3.62 (m, 4H), 5.60 (s, 2H), 7.12 (d,
2H), 7.40 (d, 2H), 7.52 (s, 2H). LCMS: m/z ES+ 457 [MH]+
222 R= H; R= 2-phenylethyl; X = [1,2,3]-triazol-2-ylmethyl
'H NMR (400MHz, CDC13): S 1.52-1.68 (m, 4H), 2.04 (m, 1 H), 2.79 (m, 4H),
3.36 (m, 2H), 3.47 (m, 2H), 5.40 (m, 1 H), 5.59 (s, 2H), 7.10 (d, 2H), 7.17
(d,
2H), 7.20-7.36 (m, 3H), 7.40 (d, 2H), 7.54 (s, 2H). LCMS: m/z ES+ 491 [MH]+
A 1-bicyclo[1.1.1]pentylamine hydrochloride (see ref. J.O.C. 2001; 66(19);
6282-6285).
Example 223: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]-N-isopropyl-
4-
methylpiperidine-4-carboxamide
N~ ~
N CH3
0
CH3
H3C N
I
CH3 Ci
Oxalyl chloride (0.04 mL, 0.55 mmol) was added to a solution of the acid of
preparation
160 (50 mg, 0.15 mmol) in dichloromethane (50 mL), and the solution was
stirred at
room temperature for 20 minutes. Additional oxalyl chloride (0.02 mL, 0.27
mmol) was
added and the solution was stirred for a further 10 minutes. The solution was
then
evaporated under reduced pressure and the residue was azeotroped with
dichloromethane (3x). The oily residue was dissolved in dichloromethane (10
mL).
Isopropylamine (0.19 mL, 2.25 mmol) was added to the solution and the mixture
was
then stirred at room temperature for 18 hours. The reaction was then washed
with
ammonium chloride solution, dried over MgSO4 and then evaporated under reduced
pressure. The residue was purified by column chromatography on silica gel
using
dichloromethane:methanol:0.88 ammonia (95:5:1) as eluant. The product was
triturated
with ether to afford the title compound as a solid, 30 mg.
'H NMR (400MHz, CDCI3): S 1.15 (m, 9H), 1.42 (m, 2H), 1.95 (m, 2H), 2.22 (s,
3H), 3.02
(m, 2H), 3.18 (m, 2H), 4.03 (m, 1 H), 5.38 (m, 1 H), 7.26 (d, 2H), 7.57 (d,
2H); LCMS: m/z
APCI+ 376 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 -132- PCT/IB2004/004059
Example 224: N-{1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]piperidin-
4-yl}
benzamide
N-N
N~N~CH3
a ci
Triethylamine (105 L, 0.75 mmol) and then benzoyl chloride (79.6 L, 0.69
mmol) were
added to a solution of the amine of preparation 145 (200 mg, 0.69 mmol) in
dichloromethane (5 mL), and the reaction was stirred at room temperature for 5
minutes.
Water (5 mL) was added and the mixture was stirred vigorously for 5 minutes.
The
mixture was then filtered using a phase separation cartridge and the organic
layer was
concentrated under reduced pressure. The residue was then azeotroped with
ether to
provide the title compound as an off-white solid, 278 mg.
'H NMR (400MHz, CD3OD): 8 1.55 (m, 2H), 1.86 (m, 2H), 2.22 (s, 3H), 2.93 (m,
2H),
3.52 (m, 2H), 3.97 (m, 1 H), 7.42 (t, 2H), 7.45-7.53 (m, 3H), 7.63 (d, 2H),
7.75 (d, 2H);
LCMS: m/z APCI+ 418 [MNa]+
Example 225: 1-Benzoyl-4-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]
piperazine
O N
%N
CH3
- ~/
ci
1-Hydroxybenzotriazole hydrate (150 mg, 1.1 mmol), 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride (225 mg, 1.17 mmol), triethylamine (0.4 mL,
2.7 mmol)
and the amine of preparation 144 (250 mg, 0.9 mmol) were added sequentially to
a
solution of benzoic acid (110 mg, 0.9 mmol) in dichloromethane (10 mL). The
reaction
was then stirred at room temperature for 18 hours. The mixture was partitioned
between
2M sodium hydroxide solution and dichloromethane, and then the phases were
separated. The aqueous layer was further extracted with dichloromethane, and
the
combined organic solutions were evaporated under reduced pressure. The crude
product was purified by column chromatography on silica gel using an elution
gradient of
dichloromethane:methanol:0.88 ammonia (100:0:0 to 95:5:0.5) to afford the
title
compound as a white foam, 182 mg.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-133-
'H NMR (400MHz, CDCI3): S 2.22 (s, 3H), 3.08 (m, 4H), 3.38-3.78 (m, 4H), 7.25
(d, 2H),
7.39 (m, 4H), 7.54 (d, 3H). LCMS: m/z APCI+ 382 [MH]+; Microanalysis found: C,
61.66;
H, 5.32; N, 17.42. C20H20CIN50;0.14CH2C12 requires C, 61.43; H, 5.19; N,
17.79%.
Example 226: 4-Benzoyl-l-[4-(4-chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-
triazol-3-yl]-
2-methylpiperazine
~ CH
_ N /
O
C~"~3
C~
Triethylamine (100 L, 0.71 mmol), and then benzoyl chloride (82 L, 0.71
mmol) were
added to a solution of the compound of preparation 149 (150 mg, 0.47 mmol) in
dichloromethane (10 mL), and the reaction was then stirred at room temperature
for 18
hours. The mixture was washed with sodium bicarbonate solution, the layers
separated
and the organic solution evaporated under reduced pressure. The crude product
was
purified by column chromatography on silica gel using ethyl
acetate:methanol:0.88
ammonia (95:5:0.5) as eluant to afford the title compound as a white solid,
120 mg.
'H NMR (400MHz, CDCI3): S 1.01-1.16 (m, 3H), 3.18-3.23 (m, 3H), 3.30 (s, 3H),
3.39-
3.48 (m, 3H), 3.83 (m, 1 H), 4.04 (m, 1 H), 4.28 (d, 1 H), 4.38 (d, 1 H), 7.39
(m, 7H), 7.52
(d, 2H); LCMS: m/z APCI+ 426 [MH]+
Example 227: 1-(4-Chlorobenzoyl)-4-[4-(4-chlorophenyl)-5-(methoxymethyl)-4H-
1,2,4-
triazol-3-yl]-2-methylpiperazine
0 --\
N N---~ N ~N
Y N
H3C _ CH3
cii
ci
The title compound was prepared from the compound of preparation 150 and 4-
chlorobenzoyl chloride in 37% yield, following the procedure described for
example 226.
'H NMR (400MHz, CDCI3): 6 1.08 (d, 3H), 2.94-3.39 (m, 8H), 4.34 (s, 2H), 7.25
(d, 2H),
7.39 (m, 4H), 7.54 (d, 2H); LCMS: m/z APCI+ 460 [M]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-134-
Example 228: 1-[4-(4-Chlorophenyl)-5-(methoxymethyl)-4H-1,2,4-triazol-3-yl]-4-
(3-
fluorobenzoyl)-1,4-diazepane
N-N
flNNoCH
3
O N~/
F cl
The title compound was prepared from the compound of preparation 148 and 3-
fluorobenzoyl chloride in 84% yield, following the procedure described for
example 224.
'H NMR (400MHz, CDCI3): 8 1.68-1.80 (m, 2H) 3.17 (m, 1 H), 3.25 (s, 3H), 3.39
(m, 4H),
3.58 (m, 1 H), 3.65 (m, 1 H), 3.78 (m, 1 H), 4.22 (d, 2H), 7.02 (d, 1 H), 7.10
(m, 2H), 7.22-
7.42 (m, 3H), 7.49 (m, 2H); LCMS: m/z APCI+ 444 [M]+
Example 229: 4-(2-Chlorobenzoyl)-2-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-
triazol-3-
yl]morpholine
N \
a c
0 \
JJNff
('
..H3
CI
Trifluoroacetic acid (5 mL) was added to a cooled (5 C) solution of the
compound of
preparation 102 (1.3 g, 3.43 mmol) in dichloromethane (5 mL), and the solution
was
then stirred at room temperature for 2 hours. The mixture was concentrated
under
reduced pressure, and then triethylamine (450 mg, 4.47 mmol) and
dichloromethane (30
mL) were added.
A portion of this solution (10 mL) was treated with 2-benzoyl chloride (1.26
mmol) and
then stirred for 2 hours at room temperature. Tris-(2-aminoethyl)amine
polystyrene (500
mg) was added and the mixture stirred for a further 24 hours. The mixture was
then
diluted with aqueous ammonium chloride solution, the layers were separated
using a
hydrophobic membrane and the organic solution was purified directly using a
silica gel
cartridge and dichloromethane:methanol (100:0 to 95:5) as eluant to provide
the title
compound.
'H NMR (400MHz, CDC13)(rotamers): 8 2.23, 2.34 (2xs, 3H), 3.00-3.62 (m, 3H),
3.64-
4.00 (m, 2H), 4.11-4.40 (m, 1 H), 4.58, 4.90 (2xm, 1 H), 7.17-7.40 (m, 6H),
7.56 (m, 2H);
LCMS: m/z ES+ 439 [MNa]+
CA 02551038 2006-06-21
WO 2005/063754 -135- PCT/IB2004/004059
Example 230: 1-(2-Chlorobenzoyl)-3-[4-(4-chloro-2-methylphenyl)-5-methyl-4H-
1,2,4-
triazol-3-yl]piperidine
N
py N
\ N
CI O a'zz~ \//
IC(H3
CI CH3
Triethylamine (79 L, 0.57 mmol) and then 2-benzoyl chloride (66 L, 0.52
mmol) were
added to a solution of the compound of preparation 146 (150 mg, 0.52 mmol) in
dichloromethane (5 mL). The mixture was then stirred at room temperature for
18
hours. After this time the reaction was diluted with water (5mL) and the
mixture was
stirred rapidly for 30 minutes. The layers were then separated, the organic
solution
evaporated under reduced pressure and the product was azeotroped from ether to
afford the title compound as a white foam, 193 mg.
'H NMR (400MHz, CDCI3): 5 1.38-1.82 (m, 3H), 1.82-2.22 (m, 8H), 2.54-2.87 (m,
1 H),
3.03-3.50 (m, 2H), 4.80 (m, 1 H), 6.94-7.50 (m, 7H); LCMS: m/z ES+ 451 [MNa]+
Example 231: 3-[1-(2-Chlorobenzoyl)pyrrolidin-3-yl]-4-(4-chloro-2-
methylphenyl)-5-
methyl-4H-1,2,4-triazole
N N
~
N
9--~
CI 0 N
CH3
CI CH3
A mixture of the compound of preparation 147 (97 mg, 0.35 mmol), 2-
chlorobenzoyl
chloride (40.4 L, 0.32 mmol) and N-methylmorpholine (58 L, 0.53 mmol) in
dichloromethane (5 mL) was stirred at room temperature for 18 hours. The
reaction was
then diluted with dichloromethane (20 mL), washed with 2N hydrochloric acid
(20 mL)
and saturated sodium bicarbonate solution (20 mL). The organic solution was
then dried
over MgSO4 and concentrated under reduced pressure. The residual oil was
triturated
with ether, the resulting solid filtered off and then dried to afford the
title compound, 55
mg.
'H NMR (400MHz, CDCI3)(rotamers): 8 1.84-2.62 (m, 8H), 3.00-3.20 (m, 1H), 3.22-
3.42
(m, 1 H), 3.44-3.79 (m, 2H), 3.81-3.98 (m, 1 H), 7.20-7.50 (m, 7H); LCMS: mlz
APCI+ 415
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-136-
Example 232: N-{1-[4-(4-Chlorophenyl)-5-(2H-1,2,3-triazol-2-ylmethyl)-4H-1,2,4-
triazol-
3-yl]pyrrolidin-3-yl}-N-methylacetamide
N N
3
H ~NN
3C N /
C
~
' N
CIH3 N
~ N
The title compound was obtained as a solid in 50% yield from the compounds of
preparations 54 and 18, following the procedure described for preparation
93.'H NMR
(400MHz, CDCI3)(rotamers): 8 1.78-1.90 (m, 2H), 2.04 (s, 3H), 2.74 (s, 1 H),
2.80 (s, 3H),
2.98 (m, 1 H), 3.04-3.18 (m, 2H), 3.22-3.38 (m, 2H), 4.40, 5.21 (2xm, 1 H),
5.55 (m, 2H),
7.03 (d, 2H), 7.38 (d, 2H), 7.52 (s, 2H); LCMS: m/z ES+ 423 [MNa]+
Example 233: 1-[4-(4-Chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]-4-
phenylpiperidine-
4-carboxamide
N~N
CH3
N
NH2
0 CI
Sulphuric acid (930 mg, 95%, 9.5 mmol) was added to a solution of the compound
of
preparation 96 (700 mg, 1.9 mmol) in acetic acid (1.5 mL), and the reaction
was then
heated at 100 C for 3 days. The cooled mixture was carefully quenched by the
addition
of 0.88 ammonia and then extracted with dichloromethane (4x). The combined
organic
layers were washed with brine, then dried over MgSO4 and evaporated under
reduced
pressure. The product was crystallised from ethyl acetate to afford the title
compound,
282 mg.
'H NMR (400MHz, CDCI3): 8 2.06 (m, 2H), 2.20 (s, 3H), 2.32 (m, 2H), 3.05-3.20
(m, 4H),
5.20 (m, 2H), 7.24 (m, 3H), 7.38 (m, 4H), 7.52 (d, 2H); LCMS: m/z ES+ 396
[MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-137-
Example 234: tert-Butyl 4-{[4-(4-chlorophenyl)-5-(2H-1,2,3-triazol-2-ylmethyl)-
4H-1,2,4-
triazol-3-yl]oxy}piperidine-1-carboxylate
H3C CH3 C N
A N~N N_
1 N
H3C C N
N~ I
O-
CI
Tetrahydrofuran (2 mL) was added to sodium hydride (24 mg, 60% in mineral
oil), which
had been pre-washed with pentane (2 mL), and the suspension was stirred at
room
temperature. tert-Butyl 4-hydroxy-1-piperidinecarboxylate (119 mg, 0.6 mmol)
was then
added and the mixture was stirred at room temperature for a further 30
minutes. The
compound of preparation 164 (100 mg, 0.3 mmol) was added and the reaction was
stirred at room temperature for a further 18 hours. The reaction was then
partitioned
between dichloromethane (20 mL) and brine (20 mL), the layers separated and
the
organic phase evaporated under reduced pressure. The residue was dissolved in
dichloromethane (6 mL), PS-DIEA (Argonaut Technologies) (638 mg) and
triethylamine
(0.5 mL, 3.6 mmol) was added. The mixture was then stirred for 18 hours. The
mixture
was filtered, the filtrate was washed with saturated aqueous potassium
carbonate
solution and evaporated under reduced pressure. The crude product was purified
by
column chromatography on silica gel using dichloromethane:methanol (99:1) as
eluant.
The product was further purified by HPLC using a Phenomenex Luna C18 column
and
0.1 % aqueous formic acid:acetonitrile/0.1 % formic acid (80:20 to 5:95) to
afford the title
compound, 34 mg.
'H NMR (400MHz, CD3OD): S 1.40 (m, 9H), 1.62 (m, 2H), 1.98 (m, 2H), 3.30-3.59
(m,
4H), 4.90-5.02 (m, 1 H), 5.61 (s, 2H), 7.18 (d, 2H), 7.40 (d, 2H), 7.50 (m,
2H).
Example 235: N-(tert-Butyl)-4-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-
yl]
benzamide
N~N
~_CH3
N
~
N \ ~ / `
H3C~ ~
H3c CH3 ~ ci
tert-Butylamine hydrochloride (223 mg, 2.0 mmol), followed by a solution of
the acid
chloride of preparation 157 (150 mg, 0.4 mmol) in dichloromethane (3 mL), was
added
CA 02551038 2006-06-21
WO 2005/063754 -138- PCT/IB2004/004059
to a solution of triethylamine (300 L, 2.0 mmol) in dichloromethane (2 mL)
and the
reaction was stirred at room temperature for an hour. The mixture was then
partitioned
between dichloromethane and aqueous citric acid solution, and the phases were
separated. The aqueous layer was further extracted with dichloromethane (2x25
mL)
and the combined organic solutions were dried over MgSO4 and evaporated under
reduced pressure. The crude product was purified by column chromatography on
silica
gel using dichloromethane:methanol:0.88 ammonia (93:7:1) as eluant to afford
the title
compound, 122 mg.
'H NMR (400MHz, CDCI3): 8 1.42 (s, 9H), 2.40 (s, 3H), 5.98 (br s, 1 H), 7.19
(d, 2H),
7.41 (d, 2H), 7.50 (d, 2H), 7.61 (d, 2H); LCMS: m/z ES+ 391 [MNa]+
Examples 236 to 395:
C~- N\)--~ /N
'
\
Z NN/ N
N_
51 N_
CI
The appropriate acid, ZCO2H, (0.25 mL, 0.2M solution in N,N-dimethylformamide,
50
mol) was, if necessary, neutralised with triethylamine (7 l, 50 mol per salt
equivalent)
and then treated with O-(7-azabenzotriazol-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate solution (0.1 mL, 0.525M, 52.5 mol). The solution was then
treated with triethylamine (28 l, 0.20 mmol) and the amine of preparation 12
(0.25 mL,
0.2M solution in N,N-dimethylformamide, 50 mol) in a 96 deep-well
polypropylene
microtitre plate. The plate was sealed and agitated for 16 hours at 40 C. The
reaction
mixtures were then evaporated under reduced pressure and the residues were
purified
by HPLC using a Waters XTerra MS C18 column, and acetonitrile:lOmM ammonium
hydrogen carbonate (adjusted to pH 10 with ammonium hydroxide) (5:95 to 98:2),
to
provide the desired compounds.
Time %A %B %D
0 min 94 5 1
3.5 min 4.5 95 0.5
4.0 min 4.5 95 0.4
CA 02551038 2006-06-21
WO 2005/063754 -139- PCT/IB2004/004059
Ex No Z MS Retention
[MH]+ time (min)
236 5-methyl-1 -phenyl-1 H-pyrazol-4-yl 529 2.26
237 5-methyl-1 H-indol-2-yl 502 2.56
238 3-(1H-indol-3-yl)propyl 530 2.17
239 1-benzyl-1 H-pyrazol-4-yl 529 2
240 3-(2-fluorophenyl)-1 H-pyrazol-5-yl 533 2.08
241 3-(4-methylphenyl)-1 H-pyrazol-5-yl 529 2.17
242 2-(phenylmethyl)thiazol-4-yl 546 2.26
243 5-methyl-1 H-indazol-3-yl 503 2.04
244 (2,5,7-trimethyl-1 H-indol-3-yl)methyl 544 2.3
245 4,5,6,7-tetrahydro-2-methyl-2H-indazol-3-yl 507 1.95
246 1 H-indazol-1-ylmethyl 503 1.95
247 3-(2,5-dimethylphenyl)propyl 519 2.47
248 (1S)-1-(dimethylamino)-2-phenylethyl 520 2.04
249 (2,5-dimethyl-1,3-thiazol-4-yl)methyl 498 1.87
250 (4-methoxyphenyl)-5-methyl-1 H-pyrazol-4-yi] 559 2.04
251 1-(1-cyclopenten-1-yl)butyl 495 2.47
252 cyclohex-3-en-1-yl 453 2.04
253 (1,3-dimethyl-1 H-thieno[2,3-c]pyrazol-5-yl) 523 1.91
254 (2S)-1-(tert-butoxycarbonyl)piperidin-2-yl 556 2.3
255 benzo[b]thien-3-yl 505 2.21
256 4-(5-oxazolyl)phenyl 516 1.87
257 3-(2-methyl-4-thiazolyl)phenyl 546 2.13
258 2-phenoxypyridin-5-yl 542 2.21
259 2-methyl-4-phenylpyrimidin-5-yl 541 1.95
260 5-methoxy-indol-2-yl 518 2.13
261 4-chlorobenzyl 497 2.43
262 1-phenylcyclopentyl 517 2.69
263 3-(4-fluorophenyl)-3-oxopropyl 523 2.03
264 1,2,3,4-tetrahydronaphthalen-2-yl 503 2.23
265 cyclopentyl(phenyl)methyl 531 2.52
266 biphenyl-4-ylmethyl 539 2.37
CA 02551038 2006-06-21
WO 2005/063754 -140 PCT/1B2004/004059
-
267 1-(4-chlorophenoxy)-1-methylethyl 541 2.5
268 1-(3-chlorophenoxy)ethyl 527 2.23
269 2-methyl-1-phenylbutyl 519 2.49
270 (1-naphthyloxy)methyl 529 2.13
271 (2,3-dimethylphenoxy)methyl 507 2.2
272 3-(4-methylphenyl)propyl 505 2.18
273 1 H-indol-1-ylethyl 516 2.12
274 (phenylthio)methyl 495 2.08
275 1-(4-chlorophenyl)ethyl 511 2.27
276 2,3-dihydro-1 H-inden-2-ylmethyl 503 2.23
277 2-[(4-chlorophenyl)thio]propyl 558 2.47
278 2-chloro-4-fluorobenzyl 515 2.13
279 (1R)-1-phenyl-propyl 491 2.22
280 3-methoxycyclohexyl 485 1.81
281 1 -benzyl-2,2-dimethylpropyl 533 2.52
282 1-methyl-2-phenylethyl 491 2.15
283 (benzyloxy)methyl 493 1.96
284 3-(4-chlorophenyl)-3-oxopropyl 539 2.17
285 [(4-chlorophenyl)thio]methyl 529 2.27
286 (benzylthio)methyl 509 2.12
287 3-chlorobenzyl 497 2.1
288 1,1-diphenylethyl 553 2.5
289 2,2-diphenylethyl 553 2.39
290 (2,3-dichlorophenoxy)methyl
291 4-fluorobenzyl 548 2.29
292 2-methoxybenzyl 481 1.98
293 2-(2-methoxyphenyl)ethyl 493 1.91
294 2,8-dimethylquinolin-4-yl 507 2.16
295 (2-naphthyloxy)methyl 528 2.21
296 2-naphthylmethyl 529 2.29
297 2-phenoxyethyl 513 2.26
298 1-(2-fluorophenyl)cyclopentyl 493 2.11
299 5-methoxy-1-oxoindan-2-ylmethyl 535 2.36
300 (3-methylbenzoyl)aminomethyl 520 1.89
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-141-
301 Anti-4-methylcyclohexyl 469 2.26
302 3-phenyl-3-oxo-1 methyl-propyl 519 2.09
303 2-(2-methylphenyl)ethyl 491 2.21
304 1-methyl-indol-3-ylmethyl 516 2.14
305 diphenylmethyl 539 2.38
306 1-(4-chlorophenyl)-1-methylethyl 525 2.41
307 1-methyl-1-phenylethyl 491 2.26
308 1 -acetyloxy-1 -phenylmethyl 521 2.01
309 cyclohex-l -en-l -ylmethyl 467 2.12
310 (2R)-2-phenylpropyl 491 2.19
311 [(4-fluorophenyl)thio]methyl 513 2.14
312 (2R)-1-(tert-butoxycarbonyl)piperidin-2-yI 556 2.26
313 3-(2,3-dihydro-1,4-benzodioxin-6-yl)propyl 549 2.14
314 6-chloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yI 568 2.11
315 2-(4-fluorophenoxy)ethyl 511 2.16
316 2-hydroxyquinoxalin-3-yl 517 1.57
317 5-methyl-3-phenylisoxazol-4-yl 530 2.07
318 isoquinolin-1-yl 500 1.87
319 2-phenoxyphenyl 541 2.29
320 quinolin-2-yl 500 1.97
321 quinolin-4-yl 500 1.8
322 quinolin-3-yl 500 1.87
323 2-naphthyl 499 2.21
324 5-butylpyridin-2-yl 506 2.22
325 3-benzoylphenyl 553 2.21
326 1 H-benzimidazol-6-yl 489 1.55
327 9-oxo-9H-fluoren-1 -yl 551 2.16
328 4-methoxyquinolin-2-yl 530 2.07
329 1-benzofuran-2-yl 489 2.16
330 2-(4-methylbenzoyl)phenyl 567 2.31
331 7-methoxy-1-benzofuran-2-yl 519 2.17
332 2,6-dimethoxypyridin-3-yl 510 2.3
333 2,5-dimethyl-3-furyl 467 2.26
334 biphenyl-2-yl 525 2.52
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-142-
335 5-methyl-2-thienyl 469 2.3
336 2-phenoxypyridin-3-yl 542 2.17
337 1,3-benzothiazol-6-yl 506 2.08
338 3-phenylcinnolin-4-yl 577 1.76
339 3-tert-butyi-1-methyl-1 H-pyrazol-5-yl 509 2.26
340 2,1,3-benzoxadiazol-5-yl 491 2.26
341 5-bromo-2,3-dihydro-1-benzofuran-7-yl 570 2.43
342 1-(4-chlorophenyl)cyclopropyl 523 2.27
343 5-isobutylisoxazol-3-yl 496 2.23
344 3-(1 H-pyrazol-1-yl)phenyl 515 1.91
345 3,5-dimethylindol-2-yl 516 2.32
346 4-(tert-butoxycarbonyl)morpholin-3-ylmethyl 572 1.98
347 3-isobutyl-1 H-pyrazol-5-yl 495 1.96
348 5-propylisoxazol-3-yi 482 2.1
349 2,4-dimethyl-1,3-thiazol-5-yl 498 1.61
350 2-methylquinolin-4-yl 514 1.81
351 6-chloro-imidazo[1,2-a]pyridin-2-yI 523 1.71
352 3-phenyl-1 H-pyrazol-5-yl 515 1.96
353 3-isopropyl-1 H-pyrazol-5-yl 481 1.66
354 4,5,6,7-tetrahydro-2-indazol-3-yI 493 1.81
355 2,3-dimethyl-1 H-indol-5-yl 516 2.05
356 3,5-dimethyl-1 H-pyrrol-2-yi 466 1.96
357 3-(3,5-dimethyl-1 H-pyrazol-1 -yl)phenyl 543 2
358 3-[(3,5-dimethyl-1 H-pyrazol-1-yl)methyl]phenyl 557 1.98
359 3'-fluorobiphenyl-4-yl 543 2.37
360 4-phenyl-1,3-thiazol-2-yl 546 2.17
361 4-(1 H-pyrazol- 1 -yl)phenyl 515 1.9
362 5,7-dimethyl-pyrazolo[1,5-a]pyrimidin-2-yI 518 1.76
363 3-phenylisoxazol-5-yl 516 2.2
364 2,3-dihydro-1-benzofuran-2-yl 491 2
365 1 H-benzimidazol-1 -ylmethyl 503 1.73
366 5-(4-methoxyphenyl)-2-furyl 545 2.25
367 1,3-benzothiazol-2-ylethyl 534 2.03
368 2-methyl-5-propylpyrazol-3-yl 495 1.96
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-143-
369 1 -benzyl-2-oxo-1,2-dihydropyridin-3-yI 556 1.85
370 1-benzyl-6-oxo-1,6-dihydropyridin-3-yl 556 1.75
371 2-phenyl-1,3-thiazol-4-yl 532 2.22
372 4-methyl-2-phenyl-1,3-thiazol-5-yI 546 2.18
373 8-methoxy-2H-chromen-3-yl 533 2.02
374 2H-chromen-3-yl 503 2.12
375 6-methoxy-2H-chromen-3-yl 533 2.1
376 4-(tert-butoxycarbonyl)morpholin-3-yI 558 1.95
377 4-methoxy-3-thienyl 485 1.85
378 4-(1H-imidazol-1-yl)phenyl 515 1.68
379 2-methyl-1 H-benzimidazol-5-yl 503 1.51
380 2,3-dihydro-1H-inden-2-yl 489 2.15
381 Trans-2-phenylcyclopropan-1-yl 489 2.15
382 1-methyl-1 H-indol-2-yl 502 2.18
383 1-methyl-1 H-indol-3-yl 502 2.02
384 5-fluoro-1 H-indol-2-yl 506 2.13
385 6-methyl-4-oxo-4H-chromene-2-yl 531 2.02
386 3-isopropyl-1 -methylpyrazol-5-yl 495 1.95
387 5-bromo-2-methoxypyridin-3-yl 559 2.03
388 5-methyl-2-phenyl-1 H-imidazol-4-yl 529 1.95
389 3-(1-oxo-1,3-dihydro-2H-isoindol-2-yl)propyl 532 1.82
390 1-ethylpiperidin-2-yl 484 1.91
391 3-[(pyrimidin-2-ylthio)methyl]phenyl 573 2.08
392 4-[(pyridin-2-ylthio)methyl]phenyl 572 2.17
393H 6-cyclohexyl-2-oxo-1,2,3,6-tetrahydropyrimidin-4-yl 551 2.04
394 5-oxo-1-propylpyrrolidin-3-yl 498 1.74
395 5-propylisoxazol-4-yl 482 2
A = 3-isopropyl-l-methyl-1H-pyrazole-5-carboxylic acid was used; see DE
3029281.
8= 5-bromo-2-methoxynicotinic acid was used, see EP 306251, preparation I.
C = 5-methyl-2-phenyl-IH-imidazole-4-carboxylic acid was used, see J. Chem
Soc.
1948; 1969.
D= 3-(1-oxo-1,3-dihydro-2H-isoindol-2-yl)propanoic acid was used, see J. Med.
Chem.
83; 26(2); 243.
E = 1-ethylpiperidine-2-carboxylic acid was used, see Journal of Inorganic and
Nuclear
medicine; 1978; 40(6); 1103-6.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-144-
F = 3-((pyrimidin-2-ylthio)methyl]benzoic acid was used, see J. Indian Chem.
Soc. (97);
74(7); 575.
G = 4-((pyridin-2-ylthio)methyl]benzoic acid was used, see US 4325959, example
2.
H = 6-cyclohexyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid was
used, see
J. 0. C. 2000; 65(20); 6777.
1= 5-oxo-l-propylpyrrolidine-3-carboxylic acid was used, see WO 200202614.
J= 5-propylisoxazole-4-carboxylic acid was used, see J. Het. Chem. 1991; 28(2)
453.
Examples 396 to 403:
0 N-N
N\~ ~
N
N, \ ~ ~
N
w
ci
A mixture of the appropriate amine from preparation 12a (1 eq.), the
appropriate acid
chloride (1.2 to 1.4 eq.) and polymer supported N-ethyldiisopropylamine (10
eq.) in
dichloromethane (16 mLmmol-') was stirred at room temperature for 2 hours.
Tris-(2-
aminoethyl)amine polystyrene was added and the mixture was stirred for an
hour. It was
then washed with 1 N sodium hydroxide solution. The aqueous solution was
extracted
with dichloromethane (2x) and the combined organic solutions were concentrated
under
reduced pressure. The crude products were purified by column chromatography on
silica gel using ethyl acetate:methanol:0.88 ammonia (90:10:1) as eluant, to
afford the
title compounds.
Ex Data
No
396 W = 2-CF3;
'H NMR (400MHz, CDCI3): 8 1.61-1.76 (m, 1H), 1.79-1.98 (m, 3H), 2.62-2.80
(m, 1 H), 2.84-2.97 (m, 2H), 3.42 (m, 1 H), 4.60 (m, 1 H), 5.62 (d, 2H), 6.99
(m,
2H), 7.20-7.55 (m, 6H), 7.59 (m, 1 H), 7.66 (m, 1 H). LCMS: m/z APCI+ 516
[MH]+
397 W = 3-C F3;
'H NMR (400MHz, CDCI3): S 1.80 (m, 2H), 1.98 (m, 2H), 2.70 (m, 1 H), 2.88-
3.02 (m, 2H), 3.72 (m, 1 H), 4.57 (m, 1 H), 5.62 (s, 2H), 7.00 (m, 2H), 7.22
(d,
2H), 7.25-7.59 (m, 4H), 7.65 (m, 2H). LCMS: m/z APCI+ 516 [MH]+
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-145-
398 W = 4-CF3;
'H NMR (400MHz, CD3OD): S 1.78-1.96 (m, 4H), 2.82 (m, 2H), 3.00-3.14 (m,
1 H), 3.62 (m, 1 H), 4.58 (m, 1 H), 5.67 (s, 2H), 7.22 (m, 2H), 7.50 (d, 2H),
7.58
(m, 4H), 7.77 (d, 2H). LCMS: m/z APCI+ 516 [MH]+
399 W = 2-F, 5-CI;
'H NMR (400MHz, CDCI3): S 1.78-2.00 (m, 4H), 2.70 (m, 1 H), 2.91 (m, 1 H),
3.01 (m, 1 H), 3.62 (m, 1 H), 4.58 (m, 1 H), 5.63 (s, 2H), 7.01 (m, 3H), 7.30
(m,
2H), 7.42 (d, 2H), 7.50 (s, 2H). LCMS: m/z APCI+ 500 [MH]+
400 W = 3-F
'H NMR (400MHz, CDCI3): S 1.66-1.81 (m, 2H), 1.98 (m, 2H), 2.66 (m, 1 H),
2.80-2.96 (m, 2H), 3.74-3.82 (m, 1 H), 4.45-4.60 (m, 1 H), 5.62 (s, 2H), 6.99-
7.12 (m, 4H), 7.15 (s, 1 H), 7.37 (m, 1 H), 7.42 (d, 2H), 7.49 (s, 2H). LCMS:
m/z APCI+ 466 [MH]+
401 W = 2,3-di-F
'H NMR (400MHz, CDCI3): S 1.74-2.00 (m, 4H), 2.72 (m, 1 H), 2.79 (m, 1 H),
3.01 (m, 1 H), 3.62 (m, 1 H), 4.60 (m, 1 H), 5.62 (s, 2H), 7.01 (m, 2H), 7.07-
7.21 (m, 3H), 7.42 (d, 2H), 7.52 (s, 2H). LCMS: m/z APCI+ 484 [MH]+
402 W = 2-F, 3-Cl
'H NMR (400MHz, CDCI3): S 1.62-2.00 (m, 4H), 2.70 (m, 1 H), 2.82-3.06 (m,
2H), 3.60 (m, 1 H), 4.60 (m, 1 H), 5.62 (s, 2H), 7.00 (m, 2H), 7.15 (m, 1 H),
7.24
(m, 1 H), 7.41 (m, 3H), 7.45 (s, 2H). LCMS: m/z APCI+ 500 [MH]+
403 W = 4-F, 3-CI;
'H NMR (400MHz, CDCI3): 5 1.74-1.85 (m, 2H), 1.98 (m, 2H), 2.70 (m, 1H),
2.82-2.98 (m, 2H), 3.70-3.88 (m, 1 H), 4.40-4.58 (m, 1 H), 5.63 (s, 2H), 7.02
(m, 2H), 7.17 (m, 1 H), 7.23 (m, 1 H), 7.42 (m, 3H), 7.49 (s, 2H). LCMS: m/z
APCI+ 500 [MH]+
A = 2.5 eq. of triethylamine were used instead of polymer supported N-ethyl
diisopropylamine.
8= 2 eq. of N-methyl morpholine used instead of polymer supported N-ethyl
diisopropylamine.
CA 02551038 2006-06-21
WO 2005/063754 PCT/IB2004/004059
-146-
Examples 404 to 405:
O j N
N~~N
z
~N,
N
~-~0 ci
A solution of the appropriate acid, ZCOZH (1.2 eq.), O-benzotriazol-1-yl-
N,N,N',N'-
tetramethyluronium hexafluorophosphate (1.2 eq.), N-methylmorpholine (1.4 eq.)
and
the amine from preparation 12a (1 eq.) in dichloromethane (7-10 mlmmol"') was
stirred
at room temperature for 24 hours. The reaction was partitioned between sodium
hydroxide solution and dichloromethane, and the layers were then separated.
The
organic solution was washed with ammonium chloride solution, dried over MgSO4
and
evaporated under reduced pressure. The crude product was purified by column
chromatography on silica gel using an elution gradient of
dichloromethane:methanol:0.88 ammonia (100:0:0 to 90:10:1) to afford the title
compounds.
Ex Data
No
404 Z = 3-fluoro-5-chlorophenyl;
'H NMR (400MHz, CDCI3): S 1.80 (m, 2H), 1.98 (m, 2H), 2.70 (m, 1H),
2.95 (m, 2H), 3.78 (m, 1 H), 4.48 (m, 1 H), 5.63 (s, 2H), 6.99 (m, 3H), 7.15
(m, 2H), 7.42 (d, 2H), 7.50 (s, 2H). LCMS: m/z APCI+ 500 [M]+
405 Z = indazol-3-yl;
'H NMR (400MHz, DMSO-d6): 8 1.63-1.82 (m, 4H), 2.78-2.90 (m, 2H),
3.18 (m, 1 H), 4.45 (m, 1 H), 4.62 (m, 1 H), 5.77 (s, 2H), 7.18 (m, 1 H),
7.32-7.40 (m, 3H), 7.58 (m, 3H), 7.63 (s, 2H), 7.90 (d, 1 H). LCMS: m/z
ES- 486 [M-H]-
AII of the compounds exemplified above showed a Ki value of less than 500 nM
when
tested in screen 1 .0 (V,A filter binding assay) as described above.
CA 02551038 2006-06-21
WO 2005/063754 -147- PCT/IB2004/004059
Examples of specific compounds are illustrated below:
Example No. Ki (nM)
165 2.98
206 2.43
399 1.99
405 1.11