Note: Descriptions are shown in the official language in which they were submitted.
CA 02560002 2006-10-05
52574-4D
1
DESCRIPTION
COMPOUNDS ACTIVE AT A NOVEL SITE ON RECEPTOR-OPERATED
CALCIUM CHANNELS USEFUL FOR TREATMENT OF NEUROLOGICAh
DISORDERS AND DISEASES
This is a divisional application of Canadian
Patent Application No. 2,257,234 filed December 11, 1996.
The subject matter of the parent application was
restricted generally to those compounds of the formula (I)
described hereinunder, and their use.
The subject matter of this divisional application
are restricted generally to those compounds of the
formulae (VIII) and (IX) described hereinunder and their use.
This specification discloses other subject matters
claimed in none of the parent and this divisional
application.
It should be understood that the expression "this
invention" or the like encompasses the subject matters of the
parent and this divisional applications as well as those
disclosed but not claimed in the parent and divisional
applications.
Field of the Invention
This invention relates to compounds useful as
neuroprotectants, anticonvulsants, anxiolytics, analgesics,
muscle relaxants or adjuvants to general anesthetics. The
invention relates as well to methods useful for the treatment
of neurological disorders and diseases, including, but not
limited to, global and focal ischemic and hemorrhagic stroke,
head trauma, spinal cord injury, hypoxia-induced nerve cell
damage such as in cardiac arrest or neonatal distress,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
2
epilepsy, anxiety, and neurodegenerative diseases such
as Alzheimer's Disease, Huntington's Disease,
Parkinson's Disease, and amyotrophic lateral sclerosis
(ALS). The invention relates as well to methods of
screening for compounds active at a novel site on
receptor-operated calcium channels, and thereby
possessing therapeutic utility as neuroprotectants,
anticonvulsants, anxiolytics, analgesics, muscle
relaxants or adjuvants to general anesthetics, and/or
possessing potential therapeutic utility for the
treatment of neurological disorders and diseases as
described above.
Background of the Invention
The following is a description of relevant
art, none of which is admitted to be prior art to the
claims.
Glutamate is the major excitatory
neurotransmitter in the mammalian brain. Glutamate
binds or interacts with one or more glutamate receptors
which can be differentiated pharmacologically into
several subtypes. In the mammalian central nervous
system (CNS) there are three main subtypes of ionotropic
glutamate receptors, defined pharmacologically by the
selective agonists N-methyl-D-aspartate (NMDA), kainate
(KA), and
a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
(AMPA). The NMDA receptor has been implicated in a
variety of neurological pathologies including stroke,
head trauma, spinal cord injury, epilepsy, anxiety, and
neurodegenerative diseases such as Alzheimer's Disease
(Watkins and Collingridge, The 1V1~A Receptor, Oxford:
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
3
IRL Press, 1989). A role for NMDA receptors in
nociception and analgesia has been postulated as well
(Dickenson, A cure for wind-up: NMDA receptor
antagonists as potential analgesics. Trends Pharmacol.
Sci. 11: 307, 1990). More recently, AMPA receptors have
been widely studied for their possible contributions to
such neurological pathologies (Fisher and Bogousslavsky,
Evolving toward effective therapy for acute ischemic
stroke. J. Amer. Med. Assoc. 270: 360, 1993; Yamaguchi
et al., Anticonvulsant activity of AMPA/kainate
antagonists: Comparison of GYKI 52466 and NBQX in
maximal electroshock and chemoconvulsant seizure models.
Epilepsy Res. 15: 179, 1993).
When activated by glutamate, the endogenous
neurotransmitter, the NMDA receptor permits the influx
of extracellular calcium (Caz') and sodium (Na') through
an associated ion channel. The NMDA receptor allows
considerably more influx of Ca2' than do kainate or AMPA
receptors (but see below), and is an example of a
receptor-operated Caz' channel. Normally, the channel is
opened only briefly, allowing a localized and transient
increase in the concentration of intracellular Caz'
([Cap'];), which, in turn, alters the functional activity
of the cell. However, prolonged increases in (Cap']i,
resulting from chronic stimulation of the NMDA receptor,
are toxic to the cell and lead to cell death. The
chronic elevation in [Ca2'];, resulting from stimulation
of NMDA receptors, is said to be a primary cause of
neuronal degeneration following a stroke (Choi,
Glutamate neurotoxicity and diseases of the nervous
system. Neuron 1: 623, 1988). Overstimulation of NMDA
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
4
receptors is also said to be involved in the
pathogenesis of some forms of epilepsy (Dingledine et
al., Excitatory amino acid receptors in epilepsy.
Trends Pharmacol. Sci. 11: 334, 1990), anxiety (Wiley
and Balster, Preclinical evaluation of
N-methyl-D-aspartate antagonists for antianxiety
effects: A review. In: Multiple Sigma and PCP Receptor
Ligands: Mechanisms for Neuromodulatzon and
Neuroprotection? NPP Books, Ann Arbor, Michigan, pp.
80I-815, 1992), neurodegenerative diseases (Meldrum and
Garthwaite, Excitatory amino acid neurotoxicity and
neurodegenerative disease. Trends Pharmacol. Sci. 11:
379, 1.990), and hyperalgesic states (Dickenson, A cure
for wind-up: NMDA receptor antagonists as potential
analgesics. Trends Pharmacol. Sci. 11: 307, 1990).
The activity of the NMDA receptor-ionophore
complex is regulated by a variety of modulatory sites
that can be targeted by selective antagonists.
Competitive antagonists, such as the phosphonate APS,
act at the glutamate binding site, whereas
noncompetitive antagonists, such as phencyclidine (PCP),
MK-801 or magnesium (Mgr';, act within the associated ion
channel (ionophore). There is also a glycine binding
site that can be blocked selectively with compounds such
as 7-chlorokynurenic acid. There is evidence suggesting
that glycine acts as a co-agonist, so that both
glutamate and glycine are necessary to fully elicit NMDA
receptor-mediated responses. Other potential sites for
modulation of NMDA receptor function include a zinc
(Znz') binding site and a sigma ligand binding site.
Additionally, endogenous polyamines such as spermine are
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
believed to bind to a specific site and so potentiate
NMDA receptor function (Ransom and Stec, Cooperative
modulation of ('H]MK-801 binding to the NMDA receptor-ion
channel complex by glutamate, glycine and polyamines.
5 J. Neurochem. 51: 830, 1988). The potentiating effect
of polyamines on NMDA receptor function may be mediated
via a specific receptor site for polyamines; polyamines
demonstrating agonist, antagonist, and inverse agonist
activity have been described (Reynolds, Arcaine is a
competitive antagonist of the polyamine site on the NMDA
receptor. Europ. J. Pharmacol. 177: 215, 1990; Williams
et al., Characterization of polyamines having agonist,
antagonist, and inverse agonist effects at the polyamine
recognition site of the NMDA receptor. Neuron 5: 199,
1990). Radioligand binding studies have demonstrated
additionally that higher concentrations of polyamines
inhibit NMDA receptor function (Reynolds and Miller,
Ifenprodil is a novel type of NMDA receptor antagonist:
Interaction with polyamines. Molec. Pharmacol. 36: 758,
1989; Williams et al., Effects of polyamines on the
binding of ('H]MK-801 to the NMDA receptor:
Pharmacological evidence for the existence of a
polyamine recognition site. Molec. Pharmacol. 36: 575,
1989; Sacaan and Johnson, Characterization of the
stimulatory and inhibitory effects of polyamines on
('H]TCP binding to the NMDA receptor-ionophore complex.
Molec. Pharmacol. 37: 572, 1990). This inhibitory
effect of polyamines on NMDA .receptors is probably a
nonspecific effect (i.e., not mediated via the polyamine
- 30 receptor) because patch clamp electro-physiological
studies have demonstrated that this inhibition is
CA 02560002 2006-10-05
WO 97/46511 PCTIIJS96/20697
6
produced by compounds previously shown to act at the
polyamine receptor as either agonists or antagonists
(Donevan et al., Arcaine Blocks N-Methyl-D-Aspartate
Receptor Responses by an Open Channel Mechanism:
Whole-Cell and Single-Channel Recording Studies in
Cultured Hippocampal Neurons. Molec. Pharmacol. 41:
727, 1992; Rock and Macdonald, Spermine and Related
Polyamines Produce a Voltage-Dependent Reduction of NMDA
Receptor Single-Channel Conductance. Molec. Pharmacol.
42: 157, 1992).
Recent studies have demonstrated the molecular
diversity of glutamate receptors (reviewed by Nakanishi,
Molecular Diversity of Glutamate Receptors and
Implications for Brain Function. Science 258: 597,
1992). At least five distinct NMDA receptor subunits
(NMDAR1 and NMDAR2A through NMDAR2D), each encoded by a
distinct gene, have been identified to date. Also, in
NMDAR1, alternative splicing gives rise to at least six
additional isoforms. It appears that NMDAR1 is a
necessary subunit, and that combination of NMDAR1 with
different members of NMDAR2 forms the fully functional
NMDA receptor-ionophore complex. The NMDA
receptor-ionophore complex, thus, can be defined as a
hetero-oligomeric structure composed of at least NMDAR1
and NMDAR2 subunits; the existence of additional, as yet
undiscovered, subunits is not excluded by this
definition. NMDAR1 has been shown to possess binding
sites for glutamate, glycine, Mgr', MK-801, and Zn2~. The
binding sites for sigma ligands and polyamines have not
yet been localized on NMDA receptor subunits, although
ifenprodil recently has been reported to be more potent
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
7
at the NMDAR2B subunit than at the NMDAR2A subunit
(Williams, Ifenprodil discriminates subtypes of the
N-methyl-D-aspartate receptor: selectivity and
mechanisms at recombinant heteromeric receptors. Mol.
Pharmacol. 44: 851, 1993).
Several distinct subtypes of AMPA and kainate
receptors have been cloned as well (reviewed by
Nakanishi, Molecular diversity of glutamate receptors
and implications for brain function. Science 258: 597,
1992). Of particular relevance are the AMPA receptors
designated GluRl, GluR2, GluR3, and GluR4 (also termed
GluRA through GluRD), each of which exists in one of two
forms, termed flip and flop, which arise by RNA
alternative splicing. GluRl, GluR3 and GluR4, when
expressed as homomeric or heteromeric receptors, are
permeable to Ca2', and are therefore examples of
receptor-operated Caz' channels. Expression of GluR2
alone or in combination with the other subunits gives
rise to a receptor which is largely impermeable to Ca2'.
As most native AMPA receptors studied in situ are not
Cap"-permeable (discussed above), it is believed that
such receptors in situ possess at least one GluR2
subunit.
Furthermore, it is hypothesized that the GluR2
subunit is functionally distinct by virtue of the fact
that it contains an arginine residue within the putative
pore-forming transmembrane region II; GluRl, GluR3 and
GluR4 all contain a glutamine residue in this critical
region (termed the Q/R site, where Q and R are the
. 30 single letter designations for glutamine and arginine,
respectively). The activity of the AMPA receptor is
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
8
regulated by a number of modulatory sites that can be
targeted by selective antagonists (Honore et al.,
Quinoxalinediones: potent competitive non-NMDA glutamate
receptor antagonists. Science 241: 701, 1988; Donevan
and Rogawski, GYKI 52466, a 2,3-benzodiazepine, is a
highly selective, noncompetitive antagonist of
AMPA/kainate receptor responses. Neuron 10: 51, 1993).
Competitive antagonists such as NBQX act at the
glutamate binding site, whereas compounds such as GYKI
52466 appear to act noncompetitively at an associated
allosteric site.
Compounds that act as competitive or
noncompetitive antagonists at the NMDA receptor are said
to be effective in preventing neuronal cell death in
various in vitro neurotoxicity assays (Meldrum and
Garthwaite, Excitatory amino acid neurotoxicity and
neurodegenerative disease. Trends Pharmacol. Sca. 11:
379, 1990) and in in vivo models of stroke (Scatton,
Therapeutic potential of NMDA receptor antagonists in
ischemic cerebrovascular disease in Drug Strategies in
the Prevention and Treatment of Stroke, IBC Technical
Services Ltd., 1990). Such compounds are also effective
anticonvulsants (Meldrum, Excitatory amino acid
neurotransmission in epilepsy and anticonvulsant therapy
in Excitatory Amino Acids. Meldrum, Moroni, Simon, and
Woods (Eds.), New York: Raven Press, p. 655, 1991),
anxiolytics (Wiley and Balster, Preclinical evaluation
of N-methyl-D-aspartate antagonists for antianxiety
effects: A review. In: Multiple Sigma and PCP Receptor
Ligands: Mechanisms for Neurornodulation and
Neuroprotection? NPP Books, Ann Arbor, Michigan, pp.
CA 02560002 2006-10-05
WO 97146511 PCTIUS96/20697
9
BO1-815, 1992), and analgesics (Dickenson, A cure for
wind-up: NMDA receptor antagonists as potential
analgesics. Trends Pharmacol. Sci. 11: 307, 1990), and
certain NMDA receptor antagonists may lessen dementia
associated with Alzheimer's Disease (Hughes, Merz' novel
approach to the treatment of dementia. Script No. 1666:
24, 1991).
Similarly, AMPA receptor antagonists have come
under intense scrutiny as potential therapeutic agents
for the treatment of such neurological disorders and
diseases. AMPA receptor antagonists have been shown to
possess neuroprotectant (Fisher and Bogousslavsky,
Evolving toward effective therapy for acute ischemic
stroke. J. Amer. Med. Assoc. 270: 360, 1993) and
anticonvulsant (Yamaguchi et al., Anticonvulsant
activity of AMPA/kainate antagonists: comparison of GYKI
52466 and NBQX in maximal electroshock and
chemoconvulsant seizure models. Epilepsy Res. 15: 179,
1993) activity in animal models of ischemic stroke and
epilepsy, respectively.
The nicotinic cholinergic receptor present in
the mammalian CNS is another example of a
receptor-operated Caz' channel (Deneris et al.,
Pharmacological and functional diversity of neuronal
nicotinic acetylcholine receptors. Trends Pharmacol.
Sci. 12: 34, 1991). Several distinct receptor subunits
have been cloned, and these subunits can be expressed,
- in Xenopus oocytes for example, to form functional
receptors with their associated cation channels. It is
hypothesized that such receptor-ionophore complexes are
heteropentameric structures. The possible role of
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
nicotinic receptor-operated Caz~ channels in the
pathology of neurological disorders and diseases such as
ischemic stroke, epilepsy and neurodegenerative diseases
has been largely unexplored.
5 It has been demonstrated previously that
certain spider and wasp venoms contain arylalkylamine
toxins (also called polyamine toxins, arylamine toxins,
acylpolyamine toxins or polyamine amide toxins) with
activity against glutamate receptors in the mammalian
10 CNS (for reviews see Jackson and Usherwood, Spider
toxins as tools for dissecting elements of excitatory
amino acid transmission. Trends Neurosci. 11: 278,
1988; Jackson and Parks, Spider Toxins: Recent
Applications In Neurobiology. Annu. Rev. Neurosci. 12:
405, 1989; Saccomano et al., Polyamine spider toxins:
Unique pharmacological tools. Annu. Rep. Med. Chem. 24:
287, 1989; Usherwood and Blagbrough, Spider Toxins
Affecting Glutamate Receptors: Polyamines in Therapeutic
Neurochemistry. Pharmacol. Therap. 52: 245, 1991;
Kawai, Neuroactive Toxins of Spider Venoms. J. Toxicol.
Toxin Rev. 10: 131, 1991). Arylalkylamine toxins were
initially reported to be selective antagonists of the
AMPA/kainate subtypes of glutamate receptors in the
mammalian CNS (Kawai et al., Effect of a spider toxin on
glutaminergic synapses in the mammalian brain. Biomed.
Res. 3: 353, 1982; Saito et al., Spider Toxin (JSTX)
blocks glutamate synapse in hippocampal pyramidal
neurons. Brain Res. 346: 397, 1985; Saito et al.,
Effects of a spider toxin (JSTX) on hippocampal CA1
neurons in vitro. Brain Res. 481: 16, 1989; Akaike et
al., Spider toxin blocks excitatory amino acid responses
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
11
in isolated hippocampal pyramidal neurons. Neurosci.
Lett. 79: 326, 1987; Ashe et al., Argiotoxin-636 blocks
excitatory synaptic transmission in rat hippocampal CA1
pyramidal neurons. Brain Res. 480: 234, 1989; Jones et
al., Philanthotoxin blocks quisqualate-induced,
AMPA-induced and kainate-induced, but not NMDA-induced
excitation of rat brainstem neurones in vivo. Br. J.
Pharmacol. 101: 968. 1990). Subsequent studies have
demonstrated that while certain arylalkylamine toxins
are both nvnpotent and nonselective at various glutamate
receptors, other arylalkylamines are both very potent
and selective at antagonizing responses mediated by NMDA
receptor activation in the mammalian CNS (Mueller et
al., Effects of polyamine spider toxins on NMDA
receptor-mediated transmission in rat hippocampus in
vitro. Soc. Neurosci. A.bst. 15: 945, 1989; Mueller et
al., Arylamine spider toxins antagonize NMDA
receptor-mediated synaptic transmission in rat
hippocampal slices. Synapse 9: 244, 1991; Parks et al.,
Polyamine spider toxins block NMDA receptor-mediated
increases in cytosolic calcium in cerebellar granule
neurons. Soc. Neurosci. Abst. 15: 1169, 1989; Parks et
al., Arylamine toxins from funnel-web spider
(Agelenopsis aperta) venom antagonize N-methyl-D-
aspartate receptor function in mammalian brain. J.
Biol. Chem. 266: 21523, 1991; Priestley et al.,
Antagonism of responses to excitatory amino acids on rat
cortical neurones by the spider toxin, argiotoxin-636.
Br. J. Pharmacol. 97: 1315, 1989; Draguhn et al.,
Argiotoxin-636 inhibits NMDA-activated ion channels
expressed in Xenopus oocytes. Neurosci. Lett. 132: 187,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
12
1991; Kiskin et al., A highly potent and selective
N-methyl-D-aspartate receptor antagonist from the venom
of the AgeIenopsis aperta spider. Neuroscience 51: 11,
1992; Brackley et al., Selective antagonism of native
and cloned kainate and NMDA receptors by
polyamine-containing toxins. J. Pharmacol. Exptl.
Therap. 266: 1573, 1993; Williams, Effects of
Age.Ienopsis aperta toxins on the N-methyl-D-aspartate
receptor: Polyamine-like and high-affinity antagonist
actions. J. Pharmacol. Exptl. Therap. 266: 231, 1993).
Inhibition of nicotinic cholinergic receptors by the
arylalkylamine toxin philanthotoxin has also been
reported (Rozental et al., Allosteric inhibition of
nicotinic acetylcholine receptors of vertebrates and
insects by philanthotoxin. J. Pharmacol. Bxptl. Therap.
249: 123, 1989).
Parks et aI. (Arylamine toxins from funnel-web
spider (Agelenopsis aperta) venom antagonize
N-methyl-D-aspartate receptor function in mammalian
brain. J. viol. Chem. 266: 21523, 1991), describe
arylalkylamine spider toxins ( a-agatoxins) which
antagonize NMDA receptor function in mammalian brain.
The authors discuss the mechanism of action of
arylalkylamine toxins, and indicate that an NMDA
receptor-operated ion channel is the probable site of
action of the a-agatoxins, and most probably other
spider venom arylalkylamines. They state:
The discovery that endogenous
polyamines in the vertebrate brain
modulate the function of NMDA
receptors suggests that the
arylamine toxins may produce their
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
13
antagonism via a polyamine-binding
site on glutamate receptors.
Brackley et a1. studied the effects
of spermine and philanthotoxin 433
S on the responses evoked by
application of excitatory amino
acids in Xenopus oocytes injected
with mRNA from rat or chick brain.
These authors reported that, at
concentrations below those that
antagonize glutamate receptor
function, both spermine and
philanthotoxin potentiate the
effects of excitatory amino acids
and some other neurotransmitters.
On the basis of these and other
data, Brackley et a1. conc';uded that
the arylamine toxins may, by binding
nonspecifically to the membranes of
excitable cells, reduce membrane
fluidity and alter receptor
function. The validity of this
intriguing idea for NMDA receptor
function is not well supported by
two recent binding studies.
Reynolds reported that argiotoxin
636 inhibits the binding of
['H]MK-801 to rat brain membranes in
a manner that is insensitive to
glutamate, glycine, or spermidine.
This author concluded that
argiotoxin 636 exerts a novel
inhibitory effect on the NMDA
receptor complex by binding to one
of the Mg 2' sites located within the
NMDA-gated ion channel. Binding
data reported by Williams et a1.
also support the conclusion that
argiotoxin 636 does not act
primarily at the polyamine
modulatory site on the NMDA
receptor, but rather acts directly
to produce an activity-dependent
block of the ion channel. It is
already known that compounds such as
phencyclidine and ketamine can block
CA 02560002 2006-10-05
WO 97146511 PCTIUS96120697
14
the ion channels associated with
both arthropod muscle glutamate
receptors and mammalian NMDA
receptors. Thus, it seems possible
that vertebrate and invertebrate
glutamate receptors share additional
binding sites for allosteric
modulators of receptor function,
perhaps related to divalent
cation-bending sites. Clearly,
considerable additional work will be
needed to determine if the
arylamines define such a novel
regulatory site.
Usherwood and Blagbrough (Spider Toxins
Affecting Glutamate Receptors: Polyamines in Therapeutic
Neurochemistry. Pharmacol. Therap. 52: 245, 1991)
describe a proposed intracellular binding site for
arylalkylamine toxins (polyamine amide toxins) located
within the membrane potential field referred to as the
QUIS-R channel selectivity filter. The authors
postulate that the binding site for polyamine amide
toxins may occur close to the internal entrance of the
channel gated by the QUIS-R of locust muscle. The
authors also note that one such toxin, argiotoxin-636,
selectively antagonizes the NMDA receptor in cultured
rat cortical neurons.
Gullak et a1. (CNS binding sites of the novel
NMDA antagonist Arg-636. Soc. Neurosci. Abst. 15: 1168,
1989), describe argiotoxin-636 (Arg-636) as a polyamine
(arylalkylamine) toxin component of a spider venom.
This toxin is said to block NMDA-induced elevation of
cGMP in a noncompetitive fashion. The authors state
that:
[lzSI] Arg-636 bound to rat forebrain
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
membranes with Kd and BmaX values of
11.25 ACM and 28.95 pmol/mg protein
(80's specific). The ability of
other known polyamines and recently
5 discovered polyamines from
' Agelenopsis aperta to inhibit
binding paralleled neuroactivity as
functional NMDA antagonists. No
other compounds tested were able to
10 block specific binding.
The authors then stated that polyamines
(arylalkylamines) may antagonize responses to NMDA by
interacting with membrane ion channels.
Seymour and Mena (In vivo NMDA antagonist
15 activity of the polyamine spider venom component,
argiotoxin-636. Soc. Neurosci. Abst. 15: 1168, 1989)
describe studies that are said to show that
argiotoxin-636 does not significantly affect locomotor
activity at doses that are effective against audiogenic
seizures in DBA/2 mice, and that it significantly
antagonizes NMDA-induced seizures with a minimal
effective dose of 32 mg/kg given subcutaneously (s.c.).
Herold and Yaksh (Anesthesia and muscle
relaxation with intrathecal injections of AR636 and
AG489, two acylpolyamine spider toxins, in rats.
Anesthesiology 77: 507, 1992) describe studies that are
said to show that the arylalkylamine argiotoxin-636
(AR636), but not agatoxin-489 (AG489), produces muscle
relaxation and anesthesia following intrathecal
administration in rats.
williams (Effects of Agelenopsis aperta toxins
on the N-methyl-D-aspartate receptor: Polyamine-like and
high-affinity antagonist actions, J. Pharmacol. Exptl.
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96I2069?
16
Therap. 266: 231, 1993) reports that the a-agatoxins
(aryialkylamines) Agel-489 and Agel-505 enhance the
binding of ['H)MK-801 to NMDA receptors on membranes
prepared from rat brain by an action at the stimulatory
polyamine receptor; polyamine receptor agonists occluded
the stimulatory effects of Agel-489 and Agel-505 and
polyamine receptor antagonists inhibited the stimulatory
effect of Agel-505. Higher concentrations of Agel-489
and Agel-505, and argiotoxin-636 at al/ concentrations
tested, had inhibitory effects on the binding of
['H]MK-801. In Xenopus oocytes voltage-clamped at -70
mV, Agel-505 inhibited responses to NMDA with an ICso of
13 nM; this effect of Agel-505 occurred at
concentrations approximately 10,000-fold lower than
those that affected ('H)MK-801 binding. Responses to
kainate were inhibited only 11% by 30 nM Agel-505. The
antagonism of L~1MDA-induced currents by Agel-505 was
strongly voltage-dependent, consistent with an
open-channel blocking effect of the toxin. williams
states:
Although a-agatoxins can interact
at the positive allosteric polyamine
site on the NMDA receptor,
stimulatory effects produced by this
interaction may be masked in
functional assays due to a separate
action of the toxins as
high-affinity, noncompetitive
antagonists of the receptor.
Brackley et al. (Selective antagonism of
native and cloned kainate and NMDA receptors by
polyamine-containing toxins, ,T. Pharmacol. Exp. Therap.
266: 1573, 1993) report that the polyamine-containing
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
17
toxins (arylalkylamines) philanthotoxin-343 (PhTX-343)
and argiotoxin-636 (Arg-636) produce reversible,
noncompetitive, partly voltage-dependent antagonism of
kainate- and NMDA-induced currents in Xenopus oocytes
injected with rat brain RNA. Arg-636 was demonstrated
to be selective for NMDA-induced responses (ICSO =
0.04 ~M) compared to kainate-induced responses (ICso =
0.07 ACM), while PhTX-343 was selective for
kainate-induced responses (ICSO = 0.12 ~cM) compared to
NMDA-induced responses (ICSa = 2.5 uM). Arg-636 more
potently antagonized responses to NMDA in Xenopus
oocytes expressing cloned NMDAR1 subunits (ICso =
0.09 ~cM) than responses to kainate in oocytes expressing
either cloned GluR1 (ICso = 3.4 ~cM) or GluRi+GluR2
subunits (ICso = 300 ~M). PhTX-343, on the other hand,
was equipotent at antagonizing NMDAR1 (ICSO = 2.19 ~cM)
and GluR1 (ICso = 2.8 ~M), but much less potent against
GluRl+GluR2 subunits ( ICSO = 270 ~cM) .
Raditsch et a1. (Subunit-specific block of
cloned NMDA receptors by argiotoxin-636. FEBS Lett.
324: 63, 1993) report that Arg-636 more potently
antagonizes responses in Xenopus oocytes expressing
NMDAR1+NMDAR2A subunits (ICSO = 9 nM) or NMDARl+NMDAR2B
subunits (ICso = 2.5 nM) than NMDAR1+NMDAR2C subunits
(ICso = 460 nM), even though all of the receptor subunits
contain an asparagine residue in the putative
pore-forming transmembrane region II (the Q/R site, as
discussed above). The authors state that the large
difference in Arg-636 sensitivity between NMDARl+NMDAR2A
_ 30 and NMDAR1+NMDAR2C channels "must be conferred by other
structural determinants."
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
18
Herlitz et a1. (Argiotoxin detects molecular
differences in AMPA receptor channels. Neuron 10: 1131,
1993) report that Arg-636 antagonizes subtypes of AMPA
receptors in a voltage- and use-dependent manner
consistent with open-channel blockade. Arg-636 potently
antagonizes Ca2'-permeable AMPA receptors comprised of
GluRAi (Ki= 0.35 ~M), GluRCi (K; = 0.23 ~M), or GluRDi
subunits (Ki= 0.43 ~cM), while being essentially
ineffective against Caz'-impermeable GluRBi subunits at
concentrations up to 10 ACM.
Other data reported by these investigators
strongly suggest that the Q/R site in the putative
pore-forming transmembrane region II is of primary
importance in determining Arg-636 potency and Caz'
permeability.
Blaschke et a1. (A single amino acid
determines the subunit-specific spider toxin block of
a-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate
receptor channels. Proc. Natl. Acad. Sci, USA 90: 6528,
1993) report that the arylalkylamine JSTX-3 potently
antagonizes responses to kainate in Xenopus oocytes
expressing GluR1 ( ICSO = 0 . 04 ~cM) or GluR3 ( ICSO =
0.03 P.M) subunits, but that expressed receptors in which
a GluR2 subunit is present are essentially unaffected by
the toxin. Site-directed mutagenesis studies strongly
implicate the Q/R site as the primary site influencing
toxin potency.
Nakanishi et al. (Bioorganic studies of
transmitter receptors with philanthotoxin analogs. Pure
Appl. Chem., in press) have synthesized a number of
highly potent photoaffinity labeled philanthotoxin
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
19
(PhTX) analogs. Such analogs have been studied on
expressed nicotinic cholinergic receptors as a model
system for receptor-operated calcium channels. These
investigators suggest that these PhTX analogs block the
ion channel with the hydrophobic headpiece of the toxin
binding to a site near the cytoplasmic surface while the
polyamine tail extends into the ion channel from the
cytoplasmic side.
Summary of the Invention
Applicant has examined the structural
diversity and biological activity of arylalkylamines
(sometimes referred to as arylamine toxins, polyamine
toxins, acylpolyamine toxins or polyamine amide toxins)
in spider and wasp venoms, and determined that some of
the arylalkylamines present in these venoms act as
potent noncompetitive antagonists of glutamate
receptor-operated Caz' channels in the mammalian CNS.
Although these arylalkylamine compounds contain within
their structure a polyamine moiety, they are unlike
other known simple polyamines in possessing extremely
potent and specific effects on certain types of
receptor-operated Caz' channels.
Using native arylalkylamines as lead
structures, a number of analogs were synthesized and
tested. Initial findings on arylalkylamines isolated
and purified from venom were confirmed utilizing
synthetic arylalkylamines. These compounds are small
molecules (mol. wt. <800) with demonstrated efficacy in
in vivo models of stroke and epilepsy. The NMDA
receptor-ionophore complex was used as a model of
CA 02560002 2006-10-05
WO 97/46511 PCT/I1S96/20697
receptor-operated Cap' channels. Selected
arylalkylamines were shown to block NMDA
receptor-mediated responses by a novel mechanism.
Moreover, the unique behavioral pharmacological profile
5 of these compounds suggests that they are unlikely to
cause the PCP-like psychotomimetic activity and
cognitive deficits that characterize other inhibitors of
the NMDA receptor. Finally, the arylalkylamines are
unique amongst NMDA receptor antagonists in that they
10 are able to antagonize certain subtypes of cloned and
expressed AMPA receptors, namely, those permeable to
Cap'. The arylalkylamines, therefore, are the only known
class of compounds able to antagonize glutamate
receptor-mediated increases in cytosolic Ca2' regardless
15 of the pharmacological definition of receptor subtype.
Additionally, the arylalkylamines inhibit another
receptor-operated CaZ~ channel, the nicotinic cholinergic
receptor. Given that excessive and prolonged increases
in cytosolic CaZ' have been implicated in the etiology of
20 several neurological disorders and diseases, such
arylalkylamines are valuable small molecule leads for
the development of novel therapeutics for various
neurological disorders and diseases.
Applicant has determined that the selected
arylalkylamines bind with high affinity at a novel site
on the NMDA receptor-ionophore complex which has
heretofore been unidentified, and that said
arylalkylamines do not bind with high affinity at any of
the known sites (glutamate binding site, glycine binding
site, MK-801 binding site, Mg2' binding site, Zn2' binding
site, polyamine binding site, sigma binding site) on
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
21
said NMDA receptor-ionophore complex. This
determination has allowed applicant to develop methods
and protocols by which useful compounds can be
identified which provide both therapeutically useful
compounds and lead compounds for the development of
other therapeutically useful compounds. These compounds
can be identified by screening for compounds that bind
at this novel arylalkylamine binding site, and by
determining whether such a compound has the required
biological, pharmacological and physiological
properties.
The method includes the step of identifying a
compound which binds to the receptor-operated Ca2'
channel at that site bound by the arylalkylamine
compounds referred to herein as Compound 1, Compound 2
or Compound 3, and having the structures shown below.
H N H H N~ H
N~, N~ N~ N~ N' - NHS
~ I~IN
OH H 0 0 Wf
Compound 1
H
O ~~ ~ ~~ N~ ~~~ NHS
N
H
Compound 2
CA 02560002 2006-10-05
WO 97/46511 PC1YUS96/20697
22
H
N~ N~ N~ N~ N~ NHS
~I~ H H H H
Fs
Compound 3
By "therapeutically useful compound" is meant
a compound that is potentially useful in the treatment
of a disorder or disease state. A compound uncovered by
the screening method is characterized as having
potential therapeutic utility in treatment because
clinical tests have not yet been conducted to determine
actual therapeutic utility.
By "neurological disorder or disease" is meant
a disorder or disease of the nervous system including,
but not limited to, global and focal ischemic and
hemorrhagic stroke, head trauma, spinal cord injury,
spinal cord ischemia, ischemia- or hypoxia-induced nerve
cell damage, hypoxia-induced nerve cell damage as in
cardiac arrest or neonatal distress, epilepsy, anxiety,
neuropsychiatric or cognitive deficits due to ischemia
or hypoxia such as those that frequently occur as a
consequence of cardiac surgery under cardiopulmonary
bypass, and neurodegenerative disease. Also meant by
"neurological disorder or disease" are those disease
states and conditions in which a neuroprotectant,
anticonvulsant, anxiolytic, analgesic, muscle relaxant
and/or adjunct in general anesthesia may be indicated,
useful, recommended or prescribed.
By "neurodegenerative disease" is meant
diseases including, but not limited to, Alzheimer's
Disease, Huntington's Disease, Parkinson's Disease, and
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
23
amyotrophic lateral sclerosis (ALS).
By "neuroprotectant" is meant a compound
capable of preventing the neuronal damage or death
associated with a neurological disorder or disease.
By "anticonvulsant" is meant a compound
capable of reducing convulsions produced by conditions
such as simple partial seizures, complex partial
seizures, status epilepticus, and trauma-induced
seizures such as occur following head injury, including
l0 head surgery.
By "anxiolytic" is meant a compound capable of
relieving the feelings of apprehension, uncertainty and
fear that are characteristic of anxiety.
By "analgesic" is meant a compound capable of
relieving pain by altering perception of nociceptive
stimuli without producing anesthesia or loss of
consciousness.
By "muscle relaxant" is meant a compound that
reduces muscular tension.
By "adjunct in general anesthesia" is meant a
compound useful in conjunction with anesthetic agents in
producing the loss of ability to perceive pain
associated with the loss of consciousness.
By "potent" or "active" is meant that the
compound has activity at receptor-operated calcium
channels, including NMDA receptors, Ca2'-permeable AMPA
receptors, and nicotinic cholinergic receptors, with an
ICso value less than 10 ~M, more preferably less than
100 nM, and even more preferably less than 1 nM.
By "selective" is meant that the compound is
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
24
potent at receptor-operated calcium channels as defined
above, but is less potent by greater than 10-fold, more
preferably 50-fold, and even more preferably 100-fold,
at other neurotransmitter receptors, neurotransmitter
receptor-operated ion channels, or voltage-dependent ion
channels.
By "biochemical and electrophysiological
assays of receptor-operated calcium channel function" is
meant assays designed to detect by biochemical or
electrophysiological means the functional activity of
receptor-operated calcium channels. Examples of such
assays include, but are not limited to, the fura-2
fluorimetric assay for cytosolic calcium in cultured rat
cerebellar granule cells (see Example 1 and Example 2),
patch clamp electrophysiolocial assays (see Example 3
and Example 27), rat hippocampal slice synaptic
transmission assays (see Example 5), radioligand binding
assays (see Example 4, Example 24, Example 25, and
Example 26), and in vitro neuroprotectant assays (see
Example 6).
By "efficacy" is meant that a statistically
significant level of the desired activity is detectable
with a chosen compound; by "significant" is meant a
statistical significance at the p < 0.05 level.
By "neuroprotectant activity" is meant
efficacy in treatment of neurological disorders or
diseases including, but not limited to, global and focal
ischemic and hemorrhagic stroke, head trauma, spinal
cord injury, spinal cord ischemia, ischemia- or hypoxia-
induced nerve cell damage, hypoxia-induced nerve cell
damage as in cardiac arrest or neonatal distress,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
neuropsychiatric or cognitive deficits due to ischemia
or hypoxia such as those that frequently occur as a
consequence of cardiac surgery under cardiopulmonary
bypass, and neurodegenerative diseases such as
5 Alzheimer's Disease, Huntington's Disease, Parkinson's
Disease, and amyotrophic lateral sclerosis (ALS) (see
Examples 7 and 8, below).
By "anticonvulsant activity" is meant efficacy
in reducing convulsions produced by conditions such as
10 simple partial seizures, complex partial seizures,
status epilepticus, and trauma-induced seizures such as
occur following head injury, including head surgery (see
Examples 9 and 10, below).
By "anxiolytic activity" is meant that a
15 compound reduces the feelings of apprehension,
uncertainty and fear that are characteristic of anxiety.
By "analgesic activity" is meant that a
compound produces the absence of pain in response to a
stimulus that would normally be painful. Such activity
20 would be useful in clinical conditions of acute and
chronic pain including, but not limited to the
following: preemptive preoperative analgesia; peripheral
neuropathies such as occur with diabetes mellitus and
multiple sclerosis; phantom limb pain; causalgia;
25 neuralgias such as occur with herpes zoster; central
pain such as that seen with spinal cord lesions;
hyperalgesia; and allodynia.
By "causalgia" is meant a painful disorder
associated with injury of peripheral nerves.
By "neuralgia" is meant pain in the
distribution of a nerve or nerves.
CA 02560002 2006-10-05
WO 97/A6511 PCTNS96IZ0697
26
By "central pain" is meant pain associated
with a lesion of the central nervous system.
By "hyperalgesia" is meant an increased
response to a stimulus that is normally painful.
By "allodynia" is meant pain due to a stimulus
that does not normally provoke pain (see Examples 11
through 14, below).
By "induction of long-term potentiation in rat
hippocampal slices" is meant the ability of tetanic
electrical stimulation of afferent Schaffer collateral
fibers to elicit long-term increases in the strength of
synaptic transmission at the Schaffer collateral-CA1
pyramidal cell pathway in rat hippocampal slices
maintained in vitro (see Example 19).
By "therapeutic dose" is meant an amount of a
compound that relieves to some extent one or more
symptoms of the disease or condition of the patient.
Additionally, by "therapeutic dose" is meant an amount
that returns to normal, either partially or completely,
physiological or biochemical parameters associated with
or causative of the disease or condition. Generally, it
is an amount between about 1 nmole and 1 .mole of the
compound, dependent on its ECSO (ICso in the case of an
antagonist) and on the age, size, and disease associated
with the patient.
By "impair cognition" is meant the ability to
impair the acquisition of memory or the performance of a
learned task (see Example 20). Also by "impair
congnition" is meant the ability to interfere with
normal rational thought processes and reasoning.
By "disrupt motor function" is meant the
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
27
ability to significantly alter locomotor activity (see
Example 15) or elicit significant ataxia, loss of the
righting reflex, sedation or muscle relaxation (see
Example 16).
By "locomotor activity" is meant the ability
to perform normal ambulatory movements.
By "loss of the righting reflex" is meant the
ability of an animal, typically a rodent, to right
itself after being placed in a supine position.
By "neuronal vacuolization" is meant the
production of vacuoles in neurons of the cingulate
cortex or retrosplenial cortex (see Example 18).
By "cardiovascular activity " is meant the
ability to elicit significant changes in parameters
including, but not limited to, mean arterial blood
pressure and heart rate (see Examples 21 and 22).
By "hyperexcitability" is meant an enhanced
susceptibility to an excitatory stimulus.
Hyperexcitability is often manifested as a significant
increase in locomotor activity in rodents administered a
drug (see Example 15).
Hy "sedation" is meant a calmative effect, or
the allaying of activity and excitement. Sedation is
often manifested as a significant decrease in locomotor
activity in rodents administered a drug (see Example
15) .
By "PCP-like abuse potential" is meant the
potential of a drug to be wrongfully used, as in the
recreational use of PCP (i.e., "angel dust") by man. It
_ 30 is believed that PCP-like abuse potential can be
predicted by the ability of a drug to generalize to PCP
CA 02560002 2006-10-05
WO 97/46511 PCT1US96/20697
28
in rodents trained to discriminate PCP from saline (see
Example 17.)
By "generalization to PCP" is meant that a
compound is perceived as being PCP in rodents trained to
discriminate PCP from saline (see Example 17).
By "PCP-like psychotomimetic activity" is
meant the ability of a drug to elicit in man a
behavioral syndrome resembling acute psychosis,
including visual hallucinations, paranoia, agitation,
and confusion. It is believed that PCP-like
psychotomimetic activity can be predicted in rodents by
the ability of a drug to produce PCP-like stereotypic
behaviors including ataxia, head weaving,
hyperexcitability, and generalization to PCP in rodents
trained to discriminate PCP from saline (see Example 15,
Example 16, and Example 17).
By "ataxia" is meant a deficit in muscular
coordination.
By "head weaving" is meant the stereotypic
behavior elicited in rodents by PCP in which the head is
repeatedly moved slowly and broadly from side to side.
By "pharmaceutical composition" is meant a
therapeutically effective amount of a compound of the
present invention in a pharmaceutically acceptable
2S carrier, i.e., a formulation to which the compound can
be added to dissolve or otherwise facilitate
administration of the compound. Examples of
pharmaceutically acceptable carriers include water,
saline, and physiologically buffered saline. Such a
pharmaceutical composition is provided in a suitable
dose. Such compositions are generally those which are
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
29
approved for use in treatment of a specified disorder by
the FDA or its equivalent in non-U. S. countries.
In a related aspect, the invention features a
method for treating a neurological disease or disorder,
comprising the step of administering a pharmaceutical
composition comprising a compound which binds to a
receptor-operated calcium channel at the site bound by
one of the arylalkylamines Compound 1, Compound 2 and
Compound 3, said compound being a potent and selective
noncompetitive antagonist at such a receptor-operated
calcium channel, and having one or more of the following
pharmacological and physiological properties: efficacy
in in vitro biochemical and electrophysiological assays
of receptor-operated calcium channel function, in vivo
anticonvulsant activity, in vivo neuroprotectant
activity, in vivo anxiolytic activity, and in vivo
analgesic activity; said compound also possessing one or
more of the following pharmacological effects: the
compound does not interfere with the induction of
long-term potentiation in rat hippocampal slices, and,
at a therapeutic dose, does not impair cognition, does
not disrupt motor performance, does not produce neuronal
vacuolization, has minimal cardiovascular activity, does
not produce sedation or hyperexcitability, has minimal
PCP-like abuse potential, and has minimal PCP-like
psychotomimetic activity. By "minimal" is meant that
any side effect of the drug is tolerated by an average
individual, and thus that the drug can be used for
therapy of the target disease. Such side effects are
well known in the art and are routinely regarded by the
FDA as minimal when it approves a drug for a target
CA 02560002 2006-10-05
WO 97/46511 PCT/ITS96120697
disease.
Treatment involves the steps of first
identifying a patient that suffers from a neurological
disease or disorder by standard clinical methodology and
5 then treating such a patient with a composition of the
present invention.
In a further aspect, the invention features
compounds useful for treating a patient having a
neurological disease or disorder wherein said compound
10 is a polyamine-type compound or an analog thereof (i.e.,
a polyheteroatomic molecule) having the formula
R1 R~ R' R1
I I ~
15 Ar- ( i ) m-CO-NR'- (C) ~- [ZR1- (C) ~] k-N
Rz Rz Rz ~R2
wherein Ar is an appropriately substituted aromatic
ring, ring system or other hydrophobic entity; Ar can be
an aromatic (e.g., carbocyclic aryl groups such as
20 phenyl and bicyclic carbocyclic aryl ring systems such
as naphthyl, 1,2,3,4-tetrahydronaphthyl, indanyl, and
indenyl), heteroaromatic (e. g., indolyl, dihydroindolyl,
quinolinyl and isoquinolinyl, and their respective
1,2,3,4-tetrahydro- and 2-oxo- derivatives), alicyclic
25 (cycloaliphatic), or heteroalicyclic ring or ring system
(mono-, bi-, or tricyclic), having 5- to 7-membered
rings) optionally substituted with 1 to 5 substituents
independently selected from lower alkyl of 1 to 5 carbon
atoms, lower haloalkyl of 1 to 5 carbon atoms
30 substituted with 1 to 7 halogen atoms, lower alkoxy of 1
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
31
tv 5 carbon atoms, halogen, nitro, amino, lower
alkylamino of 1 to 5 carbon atoms, amido, lower
alkylamido of 1 to 5 carbon atoms, cyano, hydroxyl,
sulfhydryl, lower acyl of 2 to 4 carbon atoms,
sulfonamido, lower alkylsulfonamido of 1 to 5 carbon
atoms, lower alkylsulfoxide of 1 to 5 carbon atoms,
lower hydroxyalkyl of 1 to 5 carbon atoms, lower
alkylketo of 1 to 5 carbon atoms, or lower thioalkyl of
1 to 5 carbon atoms,
each m is an integer from 0 to 3, inclusive,
eack k is an integer from 1 to 10, inclusive,
each j is the same or different and is an integer
from 1 to 12, inclusive,
each Rl and RZ independently is selected from the
group consisting of hydrogen, lower alkyl of 1 to 5
carbon atoms, lower alkylamino of 1 to 5 carbon atoms,
lower alkylamido of 1 to 5 carbon atoms, lower mono-,
di-, or trifluoroalkyl of 1 to 5 carbon atoms, hydroxy,
amidino, guanidino, or typical common amino acid side
chain or with an associated carbon atom R1 and Rz taken
together form a carbonyl, and
each Z is selected from the group consisting of
nitrogen, oxygen, sulfur, amido, sulfonamido, and
carbon.
CA 02560002 2006-10-05
WO 97/46511 PGT/US96/20697
32
Preferred aromatic headgroups include, but are
not limited to, the following:
OCH3 R Y
Y \ ~ /
Y
OH
Headgroup B ( Headgroup C
R \ Y R Y H2~ .. O
~ R
/ p ~ / ~ O \~ Y
Headgraup D /
Headgroup E
eadaroun F
R Y
OR
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
33
where Y = R1 R' R1 R'
Ar- (C) m-CO-NRl- (C) j- (ZR~- (C) j~ k-N
i
Rz Rz R2 R2
Preferably the claims claiming a .compound
exclude known compounds whose chemical structures are
enabled.
In further preferred embodiments, the compound
is selected from the group of Compounds 4 through 18,
1. 5
where such compounds have the formulae:
F ~ ~~~~~~~~~~NH2
I HI
0
Compound 4
pCH3 O O NH
~~~~H ~~NHZ
1l
/ .; O O NHZ
NH2
Compound 5
OCH3 O O NH
NHZ
I HI
/ O NH2
/
Compound 6
OCH3 O O NH
~/~~~H~~ ~~NH2
/ O CH3 NH2
CH3
Compound 7
CA 02560002 2006-10-05
WO 97!46511 PCTIUS96/20697
34
OCH3 O O
\ ~ ~~.~~~~~.~~ NH2
/ O O NHz
NHZ
Compound B
F \ \ a ~ a w/~,., a \-~ NHz
O
r
Compound 9
OH O O NH
,~~ N N
/ 0 ~~~~H H~NH2
O NHz
NHz
Compound ZO
F \ ~ a.~-w. a ~ a ~,~.. a., CHa
..
0
Compound 11
F \ ~~O~O~NH
..
r o
Compound I2
OH
a
I \ \ O ~..O~O~NHz
r N~
Compound 13
\ \ ,
15 ~ ~'~.'' O ~ O ~~ NH2
O
Compound 14
CA 02560002 2006-10-05
WO 97146511 PCT/US96120697
OH
\ ~ ~/\~ 0 w/\/~ 0 ~ NH2
I I
/ O
Br
Compound 15
Br
\ ~ ~~O~O~NHZ
/ ~ O
Compound 16
5
/ \ ~ ~/\~ 0~ 0 ~ NH2
\ I / O
Compound 17
OCH3
/ ~~/\iO~O~NH2
II
0
Compound 18
Applicant has also determined (see Example 23
10 below) that simplified arylalkylamines (see below) are
potent, noncompetitive antagonists of the NMDA
receptor-ionophore complex. The simplified
arylalkylamines are distinct from the arylalkylamines
exemplified by Compounds 4-18 as described above. For
15 example, such compounds bind to the site labeled by
['HJMK-801 at concentrations ranging approximately 1 to
400-fold higher than those which antagonize NMDA
receptor-mediated function. Such simplified
arylalkylamines possess one or more of the following
20 additional biological properties: significant
neuroprotectant activity, significant anticonvulsant
CA 02560002 2006-10-05
WO 97146511 PCT1US96/10697
36
activity, significant analgesic activity, no PCP-like
stereotypic behavior in rodents (hyperexcitability and
head weaving) at effective neuroprotectant,
anticonvulsant and analgesic doses, no generalization to
PCP in a PCP discrimination assay at effective
neuroprotectant, anticonvulsant and analgesic doses, no
neuronal vacuolization at effective neuroprotectant,
anticonvulsant and analgesic doses, significantly less
potent activity against voltage-sensitive calcium
ZO channels, and minimal hypotensive activity at effective
neuroprotectant, anticonvulsant and analgesic doses.
Such compounds may, however, inhibit the induction of
LTP in rat hippocampal slices and may produce motor
impairment at neuroprotectant, anticonvulsant and
analgesic doses.
One aspect of the invention features a method
for treating a patient having a neurological disease or
disorder, comprising administering a compound of Formula
I:
Y
FORMULA I
wherein:
R1 and RS are independently selected from the group
consisting of phenyl, benzyl, and phenoxy (each of which
is optionally substituted with alkyl, hydroxyalkyl, -OH,
-O-alkyl, -0-acyl, -F, -C1, -Br, -I, -CF3, or -OCF3), -H,
alkyl, hydroxyalkyl, -OH, -O-alkyl, and 0-acyl;
RZ and R6 are independently selected from the group
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
37
consisting of -H, alkyl, and hydroxyalkyl; or Rz and R6
together are imino; or R1 and RZ together are - (CH?) ~- or
-(CH2)n-N(R3)-(CHz)n-;
R' is independently selected from the group
consisting of -H, alkyl, 2-hydroxyethyl and alkylphenyl;
n is an integer from 0 to 6, but at least one n
must be greater than 0;
R° is selected from the group consisting of
thiofuran, pyridyl, phenyl, benzyl, phenoxy, and
phenylthio (each of which is optionally substituted
with alkyl, -F, -C1, -Br, -I, -CFA, -OH, -OCF~, -O-alkyl,
or -O-aryl), -H, alkyl and cycloalkyl;
X is independently selected from the group
consisting of phenyl, benzyl, and phenoxy (each of which
is optionally substituted with -F, -C1, -Br, -I, -CFA,
alkyl, -OH, -OCF3, -0-alkyl, or -O-acyl) -F, -C1, -Br,
-I, CF3, alkyl, -OH, -OCF3, -0-alkyl, and O-acyl;
m is independently an integer from 0 to S;
Y is -NR~R', except when R1 and RZ together are
- (CHz) ~-N (R') - (CH2) n-, then Y is -H;
and pharmaceutically acceptable salts and complexes
thereof, wherein the compound is active at an NMDA
receptor.
By "patient" is meant any animal that has a
cell with an NMDA receptor. Preferably, the animal is a
mammal. Most preferably, the animal is a human.
By "alkyl" is meant a branched or unbranched
hydrocarbon chain containing between 1 and 6, preferably
between 1 and 4, carbon atoms, such as, e.g., methyl,
ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl,
iso-butyl, tert-butyl, 2-methylpentyl,
CA 02560002 2006-10-05
WO 97146511 PCT/US96120697
38
cyclopropylmethyl, allyl, and cyclobutylmethyl.
By "lower alkyl" is meant a branched or
unbranched hydrocarbon chain containing between 1 and 4
carbon atoms, of which examples are listed herein.
By "hydroxyalkyl" is meant an alkyl group as
defined above, substituted with a hydroxyl group,
By "alkylphenyl" is meant an alkyl group as
defined above, substituted with a phenyl group.
By "acyl" is meant -C(O)R, where R is H or
alkyl as defined above, such as, e.g., formyl, acetyl,
propionyl, or butyryl; or,
R is -0-alkyl such as in alkyl carbonates or R is
N-alkyl such as in alkyl carbamates.
By "cycloalkyl" is meant a branched or
unbranched cyclic hydrocarbon chain containing between 3
and 12 carbon atoms.
In preferred aspects of the invention,
Y is selected from the group consisting of -NHz and
-NH-methyl;
R4 is thiofuran, pyridyl, phenyl, benzyl, phenoxy,
or phenylthio, each of which is optionally substituted
with -F, -C1, -Br, -I, -CF3, alkyl, -OH, -OCF3, -O-alkyl,
or -O-acyl;
X is independently selected from the group
consisting of meta-fluoro, mesa-chloro, ortho-O-lower
alkyl, ortho-methyl, ortho-fluoro, ortho-chloro, meta-O-
lower alkyl, meta-methyl, ortho-OH, and meta-OH; and
either
R', R~~ RS, and R6 are -H;
or R2 is methyl, and Rl, R5, and R6 are -H;
or Rl is methyl, and RZ, R5, and R6 are -H.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
39
In other preferred aspects of the present
invention,
R1 and RS are independently selected from the group
consisting of -H, lower alkyl, hydroxyalkyl, -OH,
-O-alkyl, and -O-acyl;
Rz and R6 are independently selected from the group
consisting of -H, lower alkyl, and hydroxyalkyl;
or R1 and RZ together are - (CHZ) "- or
-(CH2)n-N(R')-, and Y is H;
R' is independently selected from the group
consisting of -H and lower alkyl;
R" is selected from the group consisting of
thiofuran, pyridyl, phenyl, benzyl, phenoxy, and
phenylthio (each of which is optionally substituted with
lower alkyl, -F, -C1, -Br, -I, -CF3, -OH, -OCF~,
-0-alkyl, or -O-acyl), -H, lower alkyl, and cycloalkyl;
X is independently selected from the group
consisting of -F, -C1, -Br, -I, -CF3, lower alkyl, -OH,
and -OCF3 ;
m is independently an integer from 0 to 5;
Y is -NHR', or hydrogen when R1 and RZ together are
-(CHZ)~-N(R')-, and pharmaceutically acceptable salts and
complexes thereof, with the provisos that
(a) when R1 and RZ together are - (CH2)~-N(R') -,then
R5, R6, and Y are hydrogens; and
(b) when Rl and R~ together are not (CHZ) n-N (R') -,
then Y is -NHR' .
In one preferred aspect, the invention
features a method for treating a patient having a
neurological disease or disorder comprising
administering a compound of Formula II:
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
' ~ R~ 2
~X)m ;
R NR3R3
R~ i2
R
~~m
Formula II
wherein:
X is independently selected from the group
5 consisting of -H, -Br, -C1, -F, -I, -CF" alkyl, -OH,
-OCF" -0-alkyl, and -0-acyl;
R1 is independently selected from the group
consisting of -H, alkyl, hydroxyalkyl, -OH, -O-alkyl,
and -O-acyl;
10 Rz is independently selected from the group
consisting of -H, alkyl, and hydroxyalkyl, or both RZs
together are imino; R' is independently selected from the
group consisting of -H, alkyl, 2-hydroxyethyl, and
alkylphenyl; and m is independently an integer from 0 to
15 5.
Thus, in this preferred aspect, the compounds
include the compound of Formula I, wherein:
X is independently selected from the group
consisting of -F, -C1, -Br, -I, -CF3, alkyl, -OH, -OCF3,
20 -O-alkyl, and -O-acyl;
R1 is selected from the group consisting of -H,
alkyl, hydroxyalkyl, -OH, -O-alkyl, and -O-acyl;
Rz and R6 are independently selected from the group
consisting of -H, alkyl, and hydroxyalkyl, or RZ and R6
25 together are imino;
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
41
RS is selected from the group consisting of -H,
alkyl, hydroxyalkyl, -OH, -0-alkyl, and -O-acyl;
Y is NR3R'; and
R4 is phenyl, optionally substituted with alkyl, -F,
-C1, -Br, -I, -CF3, -OH, -OCF" -O-alkyl, or -0-acyl.
In another preferred aspect, the administered
compound has the structure of Formula III:
R' 2
Ra R NRa R3
R
~X)m
Formula III
wherein:
X is independently selected from the group
consisting of -H, -Br, -C1, -F, -I, -CF" alkyl, -OH,
-OCF" -O-alkyl, and -O-acyl.
R1 is independently selected from the group
consisting of -H, alkyl, hydroxyalkyl, -OH, -O-alkyl,
and -O-acyl;
Rz is independently selected from the group
consisting of -H, alkyl, and hydroxyalkyl, or both RZs
together are imino;
R' is independently selected from the group
consisting of -H, alkyl, 2-hydroxyethyl, and
alkylphenyl;
R4 is selected from the group consisting of
thiofuran, pyridyl, phenyl, benzyl, phenoxy, and
phenylthio, (each of which is optionally substituted
CA 02560002 2006-10-05
WO 97146511 PCT/US96120697
42
with (X)m), alkyl, and cycloalkyl; and, m is
independently an integer from 0 to 5.
Thus, in the preferred aspect, the compounds
include the compound of Formula I, wherein:
X is independently selected from the group
consisting of -F, -Cl, -Br, -I, -CF3, alkyl, -OH, -OCF3,
-O-alkyl, and -O-acyl;
R1 is selected from the group consisting of -H,
alkyl, hydroxyalkyl, -OH, -O-alkyl, and -O-acyl;
R2 and R6 are selected from the group consisting of
-H, alkyl, and hydroxyalkyl, or Rz and R6 together are
ammo;
RS is independently selected from the group
consisting of -H, alkyl, hydroxyalkyl, -OH, -O-alkyl,
and -O-acyl; and
Y is NR3R' .
In another preferred aspect, the administered
compound has the structure of Formulas IV and V.
(Xjm ° (CH ~n G ~ (CH ~ n
~ ~m ~ /~ ~ \ NRa
NR3 R3
(X)m y ~ (X)m ;'
Formula V
Formula IV
wherein:
n is an integer from 1 to 6;
X is independently selected from the group
consisting of -H, -Br, -C1, -F, -I -CF" alkyl, -OH,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
43
-OCF3, -O-alkyl, and -O-acyl;
R' is independently selected from the group
consisting of -H, alkyl, 2-hydroxyethyl, and
alkylphenyl; and m is independently an integer from 0 to
5.
Thus, in this preferred aspect, the
administered compounds include the compound of Formula
I, wherein:
R' is independently selected from the group
consisting of -H, and alkyl;
R4 is phenyl, optionally substituted with alkyl, -F,
-C1, -Br, -I, -CF,, -OH, -OCF3, -O-alkyl, or -O-acyl: and
R1 and R2 together are - (CHz)"- or - (CHZ)~-N(R') -.
In another preferred aspect, the administered
compound has the structure of Formulas VI and VII:
(C;H 2)n
NR3
~3 R3
Formula VI
Formula VII
wherein:
n is an integer from 1 to 6;
X is independently selected from the group
consisting of -H, -Br, -C1, -F, -I, -CF3, alkyl, -OH,
-OCF3, -O-alkyl, and -O-acyl;
R3 is independently selected from the group
CA 02560002 2006-10-05
WO 97146511 PGT/US96120697
44
consisting of -H, alkyl, 2-hydroxyethyl, and
alkylphenyl;
R' is selected from the group consisting of
thiofuran, pyridyl, phenyl, benzyl, phenoxy, and
phenylthio (each of which is optionally substituted with
(X)m), alkyl, and cycloalkyl; and m is independently an
integer from 0 to 5.
Thus, in this preferred aspect, the
administered compounds include the compound of
Formula I, wherein:
X is independently selected from the group
consisting of -F, -C1, -Br, -I, CF" alkyl, -OH, -OCF3,
-0-alkyl, and -O-acyl-; and
Rl and R~ together are - (CH2) "- or - (CH2) n-N (R') -
More preferred aspects are those embodiments
in which:
X is independently selected from the group
consisting of meta-fluoro, meta-chloro, ortho-O-lower
alkyl, ortho-methyl, ortho-fluoro, ortho-chloro,
meta-O-lower alkyl, meta-methyl, ortho-OH, and meta-OH;
NR' is selected from the group consisting of NH,
N-methyl, and N-ethyl;
NR'R' is selected from the group consisting of NHz,
NH-methyl, and NH-ethyl;
R' is selected from the group consisting of -H and
methyl;
R2 is selected from the group consisting of -H and
methyl; and
R° is selected from the group consisting of phenyl,
benzyl, and phenoxy, each of which is optionally
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
substituted with alkyl, -F, -C1, -Br, -F, -CF" -OH,
-OCF" -O-alkyl, or -O-acyl.
Especially preferred aspects are those
embodiments in which:
5 X is meta-fluoro;
each R= and Rz is -H;
NR' is selected from the group consisting of NH and
N-methyl;
NR'R3 is selected from the group consisting of NHZ
10 and NH-methyl; and
RQ is selected from the group consisting of phenyl,
benzyl, and phenoxy, each of which is optionally
substituted with alkyl, -F, -Cl, -Br, -I, -CFA, -OH,
-OCF3, -O-alkyl, or -O-acyl.
15 In a further aspect, the invention features a
method for treating a patient having a neurological
disease or disorder comprising administering the
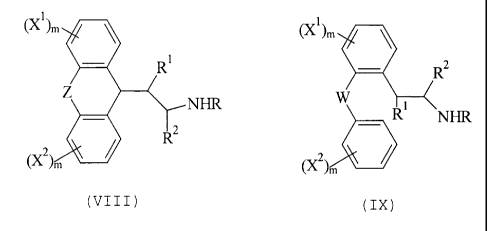
compounds of Formula VIII:
2 3
(Xl>m
1
NHR
FORMULA VIII
20 wherein:
CA 02560002 2006-10-05
WO 97146511 PCT/US96120697
46
Z is selected from the group consisting of
-CHZCHz-, -CHZCH (CHI) -, -CH=CH-, -O-CH2-, -S-CH2-, -0-,
and -S-;
X1 and X~ are independently selected from the group
consisting of -F, -C1, -CH3, -OH, and lower O-alkyl in
the 1-, 3-, 7-, or 9-substituent positions;
m is independently an integer from 0 to 2;
-NHR is selected from the group consisting of
-NH2, -NHCH" and -NHCzHs;
R1 is selected from the group consisting of -H,
alkyl, hydroxyalkyl, -OH, -O-alkyl, and -O-acyl, and
R~ is selected from the group consisting of -H,
alkyl, hydroxyalkyl, and pharmaceutically acceptable
salts and complexes thereof, wherein the compound is
active at an NMDA receptor.
Especially preferred aspects are those
embodiments in which:
Z is -CH2CH2-;
X1 or XZ is -F, or both X1 and XZ are -F;
either R1 or R~ is methyl or both R' and RZ are -H;
and
-NHR is selected from the group consisting of
-NHZ or -NHCH3.
In other preferred embodiments, the invention
features a method for treating a patient having a
neurological disease or disorder comprising
administering the compounds of Formula IX:
CA 02560002 2006-10-05
WO 97/46511 PCT/IJS96120697
47
tXl)m_
NHR
R1
W
~ Xz ) m I FORMULA IX
wherein:
W is selected from the group consisting of -CHz-,
-O-, and -S-;
X1 and X~ are independently selected from the group
consisting of -F, -C1, -CH" -GH, and lower O-alkyl;
m is independently an integer from 0 to 2;
-NHR is selected from the group consisting of -NH~,
-NHCH3 , and -NHCZHS ;
R1 is selected from the group consisting of -H,
alkyl, hydroxyalkyl, -OH, -O-alkyl, and -O-acyl; and
Rz is selected from the group consisting of -H,
alkyl, hydroxyalkyl, and pharmaceutically acceptable
salts and complexes thereof, wherein the compound is
active at an NMDA receptor.
In preferred aspects, the administered
compound is selected from the group consisting of
Compound 128, 129, I30, 131, 132, 200, 201, 202, 203,
204, 205, 206, 207, 208, 209, 210, 211, 212, 213, 214,
and 215.
In preferred embodiments, the methods of
treatment include administration of a compound selected
from Compounds 19 through 215, or pharmaceutically
acceptable salts and complexes thereof. Preferably, the
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
48
compound s 10 ~M at an NMDA receptor,
has more
an
ICSo
pref erably s 2.5 and most preferably s 0.5 ~.M
~M, at an
NMDA receptor.
In further preferred embodiments, the methods
of reatment includeadministration of a compound
t
sele cted from the oup consisting of Compound 19,
gr 20,
21, 22, 23, 24, 27, 28, 29, 30, 31, 32, 33, 34,
25, 37,
38, 39, 40, 41, 43, 44, 45, 46, 47, 48, 49, 50,
42, 51,
52, 53, 54, 55, 57, 58, 59, 60, 61, 62, 63, 64,
56, 65,
66, 67, 68, 69, 71, 72, 73. 75, 76, 77, 78, 79,
70, 81,
82, 83, 84, 85, 87, 88, 89, 90, 91, 92, 93, 94,
86, 95,
96, 97, 98, 100, , 102, 103, 105, 106, 107, 108,
101 109,
111, 114, 1I5, 116, 117, 118, 119, 120, 121, 122,
123,
124, 125, 126, 127, 128, 129, 130, 131, 132, 133,
134,
135, 136, 137, 138 potential prodrug), 139, 141,
( 142,
143, 144, 145, 146, 147, 148, 149, and 150, and
pharmaceutically eptable salts and complexes thereof.
acc
Thes e compounds an ICSO s l0um at an NMDA receptor.
have
In more pr eferred embodiments, the methods
of
trea tment include administration of a compound
sele cted from the oup consisting of Compound 19,
gr 20,
21, 22, 23, 24, 27, 28, 29, 30, 31, 32, 33, 34,
25, 37,
38, 39, 40, 41, 43, 44, 45, 46, 47, 48, 49, 50,
42, 51,
52, 53, 54, 55, 57, 58, 59, 60, 61, 62, 63, 64,
56, 65,
66, 69, 70, 75, 81, 82, 83, 85, B6, 87, 88, 89,
76, 90,
91, 92, 93, 94, 96, 97, 100, 101, 102, 103, 105,
95,
106, 108, 109, 111, 115, 118, 119, 120, 121, 122,
125,
126, 127, 128, 129, 130, 131, 132, 133. 135, 136,
137,
138 (potential prodrug), 139, 142, 144, 145, 146,
147,
148, 149, and 150, nd pharmaceutically acceptable
a salts
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
49
and complexes thereof. These compounds have an ICSO
s
2.5 uM at an NMDA receptor
In other embodiments, the compound is selected
from the group consisting of Compound 54, 55, 56,
57,
58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, B3,
88,
89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 109,
111, 115, 118, 119, 120, 121, 122, 125, 126, 127,
129,
130, 131, 135, 136, 137, 138, 139, 142, 144, 145,
148,
I49, and 150, and pharmaceutically acceptable salts
and
comp lexes thereof.
In particularly preferred embodiments, the
meth ods of treatment include administration of a
comp ound selected from the group consisting of Compound
I9, 20, 21, 22, 23, 24, 25, 27, 28, 30, 31, 32, 33,
38,
39, 43, 44, 46, 47, 49, 50, 52, 53, 54, 55, 56, 57,
58,
59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90,
91,
93, 94, 95, 96, 97, 103, 111, 118, 119, 120, 122,
126,
135, 136, 137, 138 (potential prodrug), 142, 144,
145,
147, 148, 149, and 150, and pharmaceutically acceptable
salt s and complexes thereof. These compounds have
an
ICSO s 0.5 ACM at an NMDA receptor.
In more preferred embodiments, the methods of
trea tment include administration of a compound selected
from the group consisting of Compound 20, 24, 25,
33,
50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136,
137,
138 (potential prodrug), 142, 144, 145, 148, 149,
and
150, and pharmaceutically acceptable salts and complexes
ther eof.
In particularly preferred embodiments, the
methods of treatment include administration of a
compound selected from the group consisting of Compound
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96IZ0697
20, 33, 50, 60, 119, and 144, and pharmaceutically
acceptable salts and complexes thereof.
In other particularly preferred embodiments,
the methods of treatment include administration of a
5 compound selected from the group consisting of Compound
33, 50, 60, 119, and 144, and pharmaceutically
acceptable salts and complexes thereof.
In preferred aspects, the invention provides a
method for treating a patient having a neurological
10 disease or disorder, comprising administering a compound
which is selected from the group consisting of Compound
I56, 157, 158, 159, 160, 16I, 162, 163, 164, 165, 166,
167, 168, 169, 170, 171, 181, 182, 183, 184, 185, 186,
187, and pharmaceutically acceptable salts and complexes
15 thereof. These compounds have an ICso s 10~M at an NMDA
receptor.
In further preferred aspects, the invention
provides a method for treating a patient having a
neurological disease or disorder, comprising
20 administering a compound which is selected from the
group consisting of Compound 157, 158, 159, 163, 164,
166, 167, 168, 169, 170, 171, 181, 185, 186, and
pharmaceutically acceptable salts and complexes thereof.
These compounds have an ICsfl s 10~.M at an NMDA receptor.
25 In other more preferred aspects, the invention
provides a method for treating a patient having a
neurological disease or disorder, comprising
administering a compound which is selected from the
group consisting of Compound 156, 157, 158, 159, 161,
30 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 181,
183, 184, 185, 186, 187, and pharmaceutically acceptable
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
51
salts and complexes thereof. These compounds have an
ICSO s 2.5 ~M at an NMDA receptor.
In further preferred aspects, the invention
provides a method for treating a patient having a
neurological disease or disorder, comprising
administering a compound which is selected from the
group consisting of Compound 157, 158, 159, 163, 164,
166, 167, 168, 169, 170, 171, 181, 185, 186, and
pharmaceutically acceptable salts and complexes thereof.
These compounds have an ICSo s 2.5 ~eM at an NMDA
receptor.
In other particularly preferred aspects, the
invention provides a method for treating a patient
having a neurological disease or disorder, comprising
administering a compound which is selected from the
group consisting of Compound 156, 157, 158, 159, 161,
163, 164, 165, 167, 168, 169, 170, 171, 181, 186 and
pharmaceutically acceptable salts and complexes thereof.
These compounds have an ICSO s 0.5 ~.M at an NMDA
receptor.
In further preferred aspects, the invention
provides a method for treating a patient having a
neurological disease or disorder, comprising
administering a compound which is selected from the
group consisting of Compound 157, 158, 159, 163, 164,
167, 168, 169, 170, 171, 181, 186 and pharmaceutically
acceptable salts and complexes thereof. These compounds
have an ICSo s 0.5 ACM at an NMDA receptor.
In other preferred aspects. the invention
provides a method for treating a patient having a
neurological disease or disorder comprising
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
52
administering a compound selected from the group
consisting of Compounds 151 - 215, and pharmaceutically
acceptable salts and complexes thereof.
In more preferred aspects, the invention
provides a method for treating a patient having a
neurological disease or disorder comprising
administering a compound selected from the group
consisting of Compound 151, 152, 153, 154, 155, 157,
158, 159, 163, 164, 166, 167, 168, 169, 170, 171, 172,
173, 174, 175, 176, 181, 185, 186, 188, 189, 190, 191,
192, 193, 194, 195, 196, 197, 198, 199, 200, 201, 202,
203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213,
214, 215 and pharmaceutically acceptable salts and
complexes thereof.
The present invention provides simplified
arylalkylamines comprising the compounds of Formulas
I-IX and all preferred aspects of Formulas I-IX as set
out above.
Examples of such simplified arylalkylamines
include, but are not limited to, Compounds 19 - 215,
whose structures are provided below. Preferably, the
compound has an ICso s 10 ~M at an NMDA receptor. More
preferably, the compound has an ICSO s 5 ~cM, more
preferably s 2.5 ~M, and most preferably s 0.5 ~cM at an
NMDA receptor. ,
In preferred embodiments, the compound is
selected from the group consisting of Compound 21, 22,
23, 24, 25, 26, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41,
42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55,
56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 78,
79, 82, 83, 84, 88, 89, 90, 92, 93, 94, 95, 96, 98, 101,
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96I20697
53
102, 103, 105, 107, 108, 109, 111, 115, 116, 118, 119,
120, 121, 122, 124, 125, 126, 127, 129, 130, 131, 134,
135, 136, 137, 138 (potential prodrug), 139, 141, 142,
143, 144, 145, 148, 149, and 150. These compounds have
an ICso s IO ~.M at an NMDA receptor.
In other embodiments, the compound is selected
from the group consisting of Compound 54, 55, 56, 57,
58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88,
89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 109,
111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129,
130, 131, 135, 136, 137, 138, 139, 142, 144, 145, 148,
149, and 150.
In more preferred embodiments, the compound is
selected from the group consisting of Compound 21, 22,
23, 24, 25, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41, 42,
43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56,
57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83,
88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 108,
109, 111, 115, 118, 119, 120, 121, I22, 125, 126, 127,
129, 130, 131, 135, 136, 137, 138 (potential prodrug),
139, 142, 144, 145, 148, 149, and 150. These compounds
have an ICSO s 2.5 ~M at an NMDA receptor.
In particularly preferred embodiments, the
compound is selected from the group consisting of
Compound 21, 22, 23, 24, 25, 27, 28, 33, 38, 39, 43, 44,
46, 47, 49, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61,
62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 93, 94, 95, 96,
103, 111, 118, 119, 120, 122, 126, 135, 136, 137, 138
(potential prodrug), 142, 144, 145, 148, 149, and 150.
These compounds have an ICSO s 0.5 ~cM at an NMDA
receptor.
CA 02560002 2006-10-05
WO 97/46511 PG"f/US96/20697
54
In preferred embodiments, the compound is
selected from the group consisting of Compound 24, 25,
33, 50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136,
137, 138, 142, 144, 145, 148, 149, and 150.
In particularly preferred embodiments, the
compound is selected from the group consisting of
Compound 20, 33, 50, 60, 119, and 144.
In more particularly preferred embodiments,
the compound is selected from the group consisting of
Compound 33, 50, 60, 119, and 144.
In other preferred aspects, the compound is
selected from the group consisting of Compound 151, 152,
153, 154, 155, 157, 158, 159, 163, 164, 166, 167, 168,
~69, 170, 171, 172, 173, 174, 175, 176, 181, 185, 186,
188, 189, 190, 191, 192, 193, 194, 195, 196, 197, 198,
199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209,
210, 211, 212, 213, 214, 215 and pharmaceutically
acceptable salts and complexes thereof.
In other preferred aspects, the compound is
selected from the group consisting of Compound 157, 158,
159, 163, I64, I66, 167, 168, 169, 170, 171, 181, 185,
186, and pharmaceutically acceptable salts and complexes
thereof. These compounds have an ICSO s 10 uM at an
NMDA receptor.
In more preferred aspects, the compound is
selected from the group consisting of Compound 157, 158,
159, 163, 164, 167, 168, 169, 170, 171, 181, 185, 186,
and pharmaceutically acceptable salts and complexes
thereof. These compounds have an ICSO s 2.5 ~M at an
NMDA receptor.
CA 02560002 2006-10-05
WO 97!46511 PCTlUS96/20697
In most preferred aspects, the compound is
selected from the group consisting of Compound 157, 158,
159, 163, 164, 167, 168, 169, 170, 171, 181, 186, and
pharmaceutically acceptable salts and complexes thereof.
5 These compounds have an ICso s 0.5 ACM at an NMDA
receptor.
Excluded from the composition of matter
aspect of the present invention are known compounds
whose chemical structures are covered by the generic
10 formulae presented above.
Also provided in an aspect of the invention
are pharmaceutical compositions useful for treating a
patient having a neurological disease or disorder. The
pharmaceutical compositions are provided in a
15 pharmaceutically acceptable carrier and appropriate
dose. The pharmaceutical compositions may be in the
form of pharmaceutically acceptable salts and complexes,
as is known to those skilled in the art.
The pharmaceutical compositions comprise the
20 compounds of Formulas I-IX and all preferred aspects of
Formulas I-IX as set out above.
Preferred pharmaceutical compositions comprise
Compounds 19 - 215. Preferably, the compound has an ICSo
s 10 ~M at an NMDA receptor. More preferably the
25 compound has an ICso s 5 ~M, more preferably s 2.5 ~M,
and most preferably s 0.5 ACM at an NMDA receptor.
In further preferred embodiments, the
pharmaceutical composition comprises a compound selected
from the group consisting of Compound 20, 21, 22, 23,
30 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40,
41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
56
55, 56, 57, 58. 59, 60, 61, 62, 63, 64, 65, 66, 67, 68,
69, 70, 71, 72, 73, 75, 76, 77, 78, 79, 81, 82, 83, 84,
85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98,
100, 101, 102, 103, 105, 106, 107, 108, 109, 111, 114,
115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125,
126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136,
137, 138 (potential prodrug), 139, 141, 142, 143, 144,
145, 146, 147, 148, 149, and 150. These compounds have
an ICSo s lO~cM at an NMDA receptor.
Preferably, the compound is selected from the
group consisting of 21, 22, 23, 24, 25, 26, 27, 28, 29,
33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48,
49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62,
63, 64, 65, 66, 69, 76, 78, 79, 82, 83, 84, 88, 89, 90,
92, 93, 94, 95, 96, 98, 101, 102, 103, 105, 107, 108,
109, 111, 115, 116, 118, 119, 120, 121, 122, 124, 125,
126, 127, 129, 130, 131, 134, 135, 136, 137, 13B
(potential prodrugl, 139, 141, 142, 143, 144, 145, 148,
149, and 150.
In other embodiments, the compound is selected
from the group consisting of 54, 55, 56, 57, 58, 59, 60,
61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92,
93, 94, 95, 96, 101, 102, 103, 105, 109, 111, 115, 118,
119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135,
136, 137, 138, 139, 142, 144, 145, 148, 149, and 150.
In more preferred embodiments, the
pharmaceutical composition comprises a compound selected
from the group consisting of Compound 20, 21, 22, 23,
24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40,
41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54,
55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 70,
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
57
75, 76, 81, 82, 83, 85, 86, 87, 88, 89, 90, 91, 92, 93,
94, 95, 96, 97, 100, 101, 102, 103, 105, 106, 108, 109,
111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 128,
129, 130, 131, 132, 133, 135, 136, 137, 138 (potential
prodrug), 139, 142, 144, 145, 146, 148, 149, and 150.
These compounds have an TCSa s 2.5 ACM at an NMDA
receptor.
Preferably, the compound is selected from the
group consisting of 21, 22, 23, 24, 25, 27, 28, 29, 33,
34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49,
50, 51, 52, 53, 54, 55, 56, 57, S8, 59, 60, 61, 62, 63,
64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95,
96, 101, 102, 103, 105, 108, 109, 111, 115, 118, 119,
120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136,
137, 138 (potential prodrug), 139, 142, 144, 145, 148,
149, and 150.
In particularly preferred embodiments, the
pharmaceutical composition comprises a compound is
selected from the group consisting of Compound 20, 21,
22, 23, 24, 25, 27, 28, 30, 31, 32, 33, 38, 39, 43, 44,
46, 47, 49, 50, 52, 53, S4, 55, 56, 57, 58, 59, 60, 61,
62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 91, 93, 94, 95,
96, 97, 103, 111, 118, 119, 120, 122, 126, 135, 136,
137, 138 (potential prodrug), 142, 144, 145, 148, 149,
and 150. These compounds have an ICso s 0.5 ~.M at an
NMDA receptor.
Preferably, the compound is selected from the
group consisting of 21, 22, 23, 24, 25, 27, 28, 33, 38,
39, 43, 44, 46, 47, 49, 50, 5'~, 53, 54, 55, 56, 57, 58,
59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 93,
94, 95, 96, 103, 111, 118, 119, 120, 122, 126, 135, 136,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
58
137, 138 (potential prodrug), 142, 144, 145, 148, 149,
and 150.
In more preferred embodiments, the
pharmaceutical composition comprises a compound selected
from the group consisting of Compound 20, 24, 25, 33,
50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136, 137,
138, 142, 144, 145, 148, 149, and 150.
Preferably, the compound is selected from the
group consisting of Compound 24, 25, 33, 50, 60, 66, 69,
103, 111, 118, 119, 120, 122, 136, 137, 138, 142, 144,
145, 148, 149, and 150.
In most particularly preferred embodiments,
the pharmaceutical composition comprises a compound
selected from the group consisting of Compound 20, 33,
50, 60, 119, and 144.
Preferably, the compound is selected from the
group consisting of 33, 50, 60, 119, and 144.
In other preferred aspects, the pharmaceutical
composition comprises a compound selected from the group
consisting of compound 151, 152, 153, 154, 155, 157,
158, 159, 163, 164, 166, 167, 168, 169, 170, 171, 172,
173, I74, 175, 176, 181, 185, 186, 188, 189, 190, 191,
192, 193, 194, 195, 196, 197, 198, 199, 200, 201, 202,
203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213,
214, 215 and pharmaceutically acceptable salts and
complexes thereof, and a pharmaceutically acceptable
carrier.
In other preferred aspects the pharmaceutical
composition comprises a compound which is selected from
the group consisting of Compound 157, 158, 159, 163,
164, 166, 167, 168, 169, 170, 171, 181, 185, 186, and
CA 02560002 2006-10-05
WO 97146511 PCTIUS96120697
59
pharmaceutically acceptable salts and complexes thereof,
and a pharmaceutically acceptable carrier. These
compounds have an ICso s 10 ACM at an NMDA receptor.
In more preferred aspects, the pharmaceutical
composition comprises a compound which is selected from
the group consisting of Compound 157, 158, 159, 163,
164, 166, 167, 168, 169, 170, 171, 181, 185, 186, and
pharmaceutically acceptable salts and complexes thereof,
and a pharmaceutically acceptable carrier. These
compounds have an ICSO s 2.5 uM at an NMDA receptor.
In most preferred aspects, the pharmaceutical
composition comprises a compound which is selected from
the group consisting of Compound 157, 158, 159, 163,
164, 167, 168, 169, 170, 171, 181, 186, and
pharmaceutically acceptable salts and complexes thereof,
and a pharmaceutically acceptable carrier. These
compounds have an ICSO s 0.5 ~M at an NMDA receptor.
Structural modifications can be made to
compounds such as 20 or 60 which do not add materially
to the structure-activity relationships (SAR)
illustrated here. For example, successful bioisosteric
replacement or substitution of optionally substituted
phenyl groups, such as those occurring in Compounds 20
or 60, can be accomplished with other lipophilic or
semi-polar aromatic (e. g., naphthyl, naphthoxy, benzyl,
phenoxy, phenylthio), aliphatic (alkyl, e.g.,
isopropyl), cycloaliphatic (cycloalkyl, e.g.,
cyclohexyl), heterocyclic [e. g., pyridyl, furanyl,
thiofuranyl (thiophenyl)], or other functional groups or
systems, as is well known in the art, will afford
clinically useful compounds (structural homologs,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
analogs, and/or congeners) with similar
biopharmaceutical properties and activity at the NMDA
receptor (e. g., cf. Compounds 37, 75, 79, 83, 89, 119-
122, 125, 126, 128, 130, 132, 137, 144, and 145). For
5 example, such replacements or substitutions have been
used to great effect in the development of SAR among
other groups of highly clinically and commercially
successful synthetic pharmaceutical agents such as the
classical H,-antihistamines, anticholinergics
10 (antimuscarinics; e.g., anti-Parkinsonians),
antidepressants (including tricyclic compounds), and
opioid analgesics [See, Foye et a1. (Eds.), Principles
of Medicinal Chemistry, 4th ed., Lea and
Febiger/Williams and Wilkins, Philadelphia, PA, 1995,
15 pp. 233, 265, 281-282, 340-341, 418-427, and 430; Prous,
J.R., The Year's Drug News, Therapeutic Targets - 1995
Edition, Prous Science Publishers, Barcelona, Spain,
1995, pp. 13, 55-56, 58-59, 74, 89, 144-145, 152, 296-
297, and 317]. Similarly, bioisosteric replacement or
20 substitution of the methylene or methine groups in the
propyl backbone of compounds such as 20 or 60 with,
e.g., oxygen, sulfur, or nitrogen, will afford
clinically useful NMDA receptor-active compounds with
similarly useful biopharmaceutical properties, such as
25 Compound 88 (a modified "classical H1-antihistamine-type"
structure), which can be further optimized for activity
at the NMDA receptor by preparing, e.g., the
corresponding compounds) containing, e.g., (bis)(3-
fluorophenyl) group(s), as taught by the present
30 invention. The propyl backbone of compounds such as 20
and 60 may also be modified successfully by the
CA 02560002 2006-10-05
WO 97146511 PCTlUS96/20697
61
incorporation of ring systems (as in Compounds 102 and
111) and/or unsaturation (e.g., a double bond, as in
Compounds 81, 106, 109, and 139) to afford further
clinically useful NMDA receptor-active compounds of the
present invention (cf. compounds cited above).
In a related aspect, the invention features a
method for making a therapeutic agent comprising the
steps of screening for said agent by determining whether
said agent is active on a receptor-operated calcium
channel, and synthesizing said therapeutic agent in an
amount sufficient to provide said agent in a
therapeutically effective amount to a patient. Said
screening may be performed by methods known to those of
ordinary skill in the art, and may, for example be
performed by the methods set out herein. Those skilled
in the art are also familiar with methods used to
synthesize therapeutic agents in amounts sufficient to
be provided in a therapeutically effective amount.
In a preferred aspect, said receptor-operated
calcium channel is an I~1MDA receptor. In a more
preferred aspect, said method further comprises the step
of adding a pharmaceutically acceptable carrier to said
agent. In a further preferred aspect said therapeutic
agent comprises a compound of Formula I, as set out
herein. In a further preferred aspect said therapeutic
agent comprises a compound of Formula II, III, IV, V,
VI, VII, VIII, or IX, as set out herein. In
particularly preferred aspects, said therapeutic agent
comprises a compound having a structure selected from
the group consisting of Formulas I-IX, and all preferred
aspects of said formulas as set out herein. In further
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
62
preferred aspects, said therapeutic agent is selected
from the group consisting of Compounds 19-215. In a
particularly preferred aspect, said therapeutic agent is
provided to a patient having a neurological disease or
S disorder. In a related aspect, said screening comprises
the step of identifying a compound which binds to said
receptor-operated calcium channel at a site bound by one
of the arylalkylamines Compound 1, Compound 2, and
Compound 3.
CHs
NHz F ~ ~ NH2 F ~ i NH2
F F
Compound I9 Compound 20 ComDOUnd 21
OCH3
F ( i NH2 F ~ i NH2 ~ r NHZ
~ CH3 \ I lCH3
F F F w
(Compound 22 Compound 23
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
63
H3C0 ' \ CH3
H3C0 I ~ NH2 I ~ NHZ I / NHZ
~I
F F
Compound 25 Compound 26 Compound 27
H3C ~ I w
H3C I / NH2 I / NH2 CI ~ NH2
F w I F w I /
Compound 28 Compound 29 Compound 30
CH3
GI I / NHZ I / NH2 F ~ NHZ
/
/ I
CI ~ I F ~ I F
Compound 31 Compound 32 Compound 33
CH3 H3C NH2 H3C NH2
I / NH2
F . w ( w
/ I F F
F \ Compound 35 Compound 36
Compound 34
H3C ~ CH3 ~ CH3 H
H3C NH2 I , NHZ F I / NCH;,
,- F w.
CH3 /
I I
F F ~ F
Compound 37 Com pond 38 Compound 39
w CH3 H ~ CHg ( ~ CN3 H
F I / NvCH3 F I / NHp F / NvCH3
- CH3 CH3
I F ~ I F
F
Compound 40 Compound 41 ComDOUnd 42
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
64
w w
C1 I / NH2 C7 i / NH2 CI I i NH2
/ CH3 / CH3 , G~H3
I I
CI ~ CI ~ CI ~ .
ComDOUnd 43 ComDOUnd 44 ComDOUnd 45
CHg I ~ CHg I ~ CH3
C1 / NH2 C1 / NH2 CI / NH2
/ ( / I / I
CI ~ CI ~ CI
Compound 46 Comflound 47 Compound 48
w CH3 w w
CI i / NH2 F i / NHZ F i / NH2
a
/ I CH3 / I CHg / I CHg
CI ~ F ~ F
ComDOUnd 49 Compound 50 Cottroound 51
F ~ F ~ OH
/ NH2 i / NH2 F I / NH2
F
F w I w I F w i
Compound 52 Compound 53 ComDOUnd 54
CH3
F I / NH2 F I / NHZ F I / NH2
H3C / / CH3
/ I F ~ I F ~ I
F
Com ound 55 Compound 56 Compound 57
CH3 ~ H I
H
F I / NH2 F I / N vCH3 / N' CH
3
CH3 / /
F ' I
F
Compound 58 Compound 59 Compound 60
CA 02560002 2006-10-05
WO 97/46511
PCTIUS96I20697
\ OCtig ~ OCH3
F I , NH2 I , NH2 F /
~.. V - a.
HgCO , , I NHZ
F ~ I ~, F ~
Compound 61 Compound 62 Compound 63
I ~ I \ CHI I ' CH3 H
F / r N~ F / N.CH3
NH2 H /
F ~ I ~ ~ F
Compound 66
Compound 64 Compound 65_
H
I / N. I / NvCH3 F I / N'CH
CH3 '~' I
CH3
I .F
Compound 67 Compound 68 Compound 69
W . ~ H
I / r NHZ I r NY CH3
,,,, _ N~ ,-
/ / I CHI
I
~orn~ound 70 Compound 71 ComDOUnd 72
~H3 I / . ty CHy I r NHZ
/ N. CH3
/ / I
Compound 73 Comvound 74 Compound 75
F F
w I
/
I / N~ I r NHy CFA ~ I
I
CF3
p F Compound 78
C~~:~ound 76 ~______~ Compound ?Z
CA 02560002 2006-10-05
WO 97146511
66
PCTlUS96/20697
N \ ~ CHa \
/ NH2 F I / NH2 I / / NHZ
~CH3
\I ~1 \I
F
Compound ?9 Compound 80 Compound 81
\ cH3 / 1 \
F I / NH2 g NHZ F ' ~ N H
/ OH
F \ I F \ I F ,.. I
Compound 82 Compound 83 Comr~ound 84
\ CH3 ' \ CH3 ~ '~ CH3
/ NH2 ~ NH2 / NH2
.
/ CH3 / CH3 / CH3
\ I \ I \. I
I \ CH3 I \ CH3 Compound 87
/ NH2 / NH2
/ I CH3 / I CH3
\ W
Compound 85 Compound 86
(mixture of 2 (mixture of 2
compounds) compounds)
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
67
F - F
H
I~ ~I
.. I I / N~
CH3 F ~/ v
i
/ , /
Compound 94 F ~ I F l
Compound 95 Compound 96
OCH3 F
I I
I / NHZ ~ ~pi2 / NHZ
~I ~I ~I
Compound 97 Compound 98 F
Compound 99
l / ( / ~ NH
F v ~ N~"~ F v v F
l l
(
F H~
Compound 100 Compound 101 ComDOUnd I02
F \ _ I ~ CHI I ~
i N, i
F I / N~ I /
I l F
Compound 105
Compound 103 F
Compound 104
H I \ I \ H
I / / ~ CH3 / ~ / ~ CHs
~l ~l ~l
Compound 206 I ~ Compound 108
/ /
I
F
Compound I07
(mixture of 2
CA 02560002 2006-10-05
WO 97/46511 PCT/US96IZ0697
68
W w W
/ NH I / / / ~NH
F ~ ~ ~ ~. _
.) F
COmI~OUnd 109 COmDOUnd 110 COmDOUnd, 111
w w ~ w
I/ I~ I/
F
I ~ I
F
112 Compound 113 Compound 114
w w
~ NH2 ' / , NHz 1 / ~ ~2
H3C0
~I ~i
H3C0
Com~ourd 115 Comnou__~_d 116 Compound 117
w w w
F I / NHz F I / NHz F I / NHz
~~O F ~~O C ,\~O
I/ f/
iromnound 118 IC'om~ound 119 lComDOUnd 120
CHI
F I / NHZ I / ~2 I / t'~~ CH3
w O w i0 i
I / I ~ F ~ I
OCH3
ComDOUnd 121 Compound 122 ComDOUnd 123
F
( / NH2 ' ~ NH2 / I
I ~ , F I / NHz
F
ComDOUnd 124 Compound 125 Compound 1.26
~1
/ NH2 I / NH NH2
- - 2
I
/ I/
Compound i27 Compound'1Z8 Compound 129
CA 02560002 2006-10-05
WO 97/46511
69
PCT/iJS96/20697
/ I
I I
w NE"Ix \ NH2 NHp
I/ I/ /
Compound 131 Compound 132
130
W
/ I / ~ I NHZ H3CH2C0 I / NHZ
w I NHZ
Compound 133 ~ I ~ I
Compound 134 IC
w
H
I / NHy I / NH2 I / N" CH3
H~CO " " - --
F O O
w1 I/ y
C '~
Compound 137 IComnou.-~d 138
-\ w
I / NH= 1 ~ / ''".~ F I / , /
,. -
~ I ~ I ~
H9C1"12C0 Ii~CO
Compound 139 Compound 140 Comflou.-:d 141
H I , ,,,.~ I ~ NH:
/ N.~, C ~ v v
/
w1 w1 I/
H~CO
. ~w f.~.,..,nl~T!~ ia3 Compound laa
Comnouna
w
I ~ H J , H
N. ~' ~ I / ~ CHI
i
F \ ~O ' O I ~ 0
I/ I/
Co:noound 145 Contoound 146 Coa~ouad i47
\ w
w
I / ~ I / NHZ C I / NHZ
C
C \~p F ~~0
I ~~ I/
~o~mavnd 14g Connacund 1s9 Comcouad 150
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
H
r NHZ ~ r t~t~ I r N\/
v \ F ~ v \
F/ I ~ F
Compound 151 Compound 152 Compound 153
cH, ~ . H
~ CHs F r ~ CHs ~ /~ NHz
\ o '1~r 1
y
F/
Compound 154 Compound 155 Compound 156
\ H / ~ / ~
r ~ \ I ~ r ~ \ ( ~ r ~ \ I
CI
\ \ \
I i ~ / I i
Compound 157 Compound 158 Compound 159
H / ( CI ' \ H / I I ' H / I Br
i ~ \ i
Br
\ \ \
I, J I,
Compound 160 Compound 161 Compound 162
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
71
H / \ H ~ H ~F
I / ~ \ ~ I / ~ \ ~ I /
f h F !~
\ F \ \
Compound 163 Compound 164 Compound 165
OCH
I ~ H / I~ ~ I \ H / I I ~ H I
~ \ / \ ~~ N \ CHI
\ \ ~ OCH~
I / ~ ~ \J
Compound 166 Compound 167 Compound 168
CHI ~ /
I ~ /\. N \ I I ~ ~ N \ I NO~ I / H \
v
I / I / I /
Compound 169 Compound 170 Compound 171
H
F I / / N~ I N~ F I / ~ ~ CH
/ / / i
F
OCH~ OCH~ OCH~
Compound 172 Compound 173 Compound 174
\ \ H \
I / N hit F ( / /~ N, C H I / /\~ N H2
3
~ \ O
/ / /
F/~ CHy
OH OH
Compound 175 Compound 176 Compound 177
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
72
H H
I .. NH, I ~ N- I ,~ ~ N,
I I CHs I CHs
\ .O \ .O \ O
/ OCH I / CH
s s CFs
Compound 178 Compound 179 Compound 180
I ~ I N"~ I ~ i\.NHz I I N~
o.° ~ ~ \
/, ~~ I,
CHs
Compound 181 Compound 182 Compound 183
Co
NHz I ~ ~NHZ mp I ~ N~
1
/ ' I \ ° ou ~ \
I nd I
F F
18
Compound 184 Compound 185 6
NKt CI I y N~ C~ I H~,/\i NI'~t
\ \
HsCO ~ i HsCO I /
Compound 187 Compound 188 Compound 189
H
/\~ WHt I ~ /\~ NHz I r - NH2
1
\ \ 0 \,O
I I
F F
Compound 190 Compound 191 Compound 192
CA 02560002 2006-10-05
WO 97/46511 PGT/US96/Z0697
73
\ F F
t
F i ~N~ ' \
NHy j NH2
w
F '~ ~~ ~F
Compound 193 Compound 194 Compound 195
\ ~ \ ~ ~ w
/ /~ N~'~2 ~ ~/~ N~'~t ~ CHI
~~ 0
~~ ~ /
F
F
Compound 196 Compound 197 Compound 198
F F
i N, I ~ NH2 I ~ /~/ Nf'~2
CH3
p / r
I
~F /
~I ~I
Compound 199 Compound 200 Compound 201
F ~ NHZ ~~~ NHp
~ /u N~h I /
I /
F
I I ~ I F
F
Compound 202 Compound 203 Compound 204
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
74
F\/~/~ NHZ I ~ NHZ ~ ~ /~ Nit
/ / / /
F
F
\I \I \I
I
F
Compound 205 Compound 206 Compound 207
~ ~ NH2 ~ Nhh ~ NHZ
/ / ( / I /
F
I F ~ I /F F I
I F
F
Compound 208 Compound 209 Compound 210
N~ ~ F F
I / I '~ NH z I
F / ~ ~ NH 2
~I I/ I\
r
F
Compound 211 Compound 212 Compound 213
F
\ \
I / NH2 F I / NH2
I\ I\
/ /
Compound 214 Compound 215
Other features and advantages of the invention
will be apparent from the following description of the
preferred embodiments thereof, and from the claims.
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
Description of the Preferred Embodiments
The following is a detailed description of the
methods and tests by which therapeutically useful
compounds can be identified and utilized for the
5 treatment of neurological disorders and diseases. The
tests are exemplified by use of Compound 1, Compound 2
or Compound 3, but other compounds which have similar
biological activity in these assays can also be used (as
discovered) to improve on the tests. Lead compounds
10 such as Compound 1, Compound 2 or Compound 3 can be used
for molecular modeling using standard procedures, or
existing or novel compounds in natural product libraries
can be screened by the methods described below.
One key method is the means by which compounds
15 can be quickly screened with standard radioligand
binding techniques (a radiolabeled arylalkylamine
binding assay) to identify those which bind at the same
site on receptor-operated Cap' channels as Compound 1,
Compound 2 or Compound 3. Data from such radioligand
20 binding studies will also confirm that said compounds do
not inhibit ('H]arylalkylamine binding via an action at
the known sites on receptor-operated Caz' channels (such
as the glutamate binding site, glycine binding site,
MK-801 binding site, Zn2' binding site, Mgz' binding site,
25 sigma binding site, or polyamine binding site on the
NMDA receptor-ionophore complex). This screening test
allows vast numbers of potentially useful compounds to
be identified and screened for activity in the other
assays. Those skilled in the art will recognize that
30 other rapid assays for detection of binding to the
CA 02560002 2006-10-05
WO 97!46511 PCTIUS96/20697
76
arylalkylamine site on receptor-operated Caz' channels
can be devised and used in this invention.
Additional testing utilizes
electrophysiological (patch clamp) methodology to extend
the results obtained with the above-mentioned
radioligand binding assay. Such results will confirm
that compounds binding to the arylalkylamine site are
functional, noncompetitive antagonists of
receptor-operated Ca2' channels with the following
properties in common with the arylalkylamines
themselves: open-channel block manifested as
use-dependent block, and voltage-dependent onset and
reversal from block. Such results will also confirm
that said compounds do not have their primary activity
at the previously described sites on receptor-operated
Caz' channels (such as the glutamate binding site,
glycine binding site, MK-801 binding site, Zn2~ binding
site, Mgz' binding site, sigma binding site, or polyamine
binding site on the NMDA receptor-ionophore complex).
In addition, recombinant DNA technology can be
used to make such testing even more rapid. For example,
using standard procedures, the genes) encoding the
novel arylalkylamine binding site (i.e., receptor) can
be identified and cloned. This can be accomplished in
one of several ways. For example, an arylalkylamine
affinity column can be prepared, and solubilized
membranes from cells or tissues containing the
arylalkylamine receptor passed over the column. The
receptor molecules bind to the column and are thus
isolated. Partial amino acid sequence information is
then obtained which allows for the isolation of the gene
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
77
encoding the receptor. Alternatively, cDNA expression
libraries are prepared and subfractions of the library
are tested for their ability to impart arylalkylamine
receptors on cells which do not normally express such
receptors (e.g., CHO cells, mouse L cells, HEK 293
cells, or Xenopus oocytes). In this way, the library
fraction containing the clone encoding the receptor is
identified. Sequential subfractionation of active
library fractions and assay eventually results in a
single clone encoding the arylalkylamine receptor.
Similarly, hybrid-arrest or hybrid-depletion cloning can
be used. Xenopus oocytes are injected with mRNA from an
appropriate tissue or cell source (e. g., human brain
tissue). Expression of the arylalkylamine receptor is
detected as, for example, an NMDA- or
glutamate-stimulated influx of calcium which can be
blocked by Compound 1, Compound 2 or Compound 3. cDNA
clones are tested for their ability to block expression
of this receptor when cDNA or cRNA are hybridized to the
mRNA of choice, prior to injection into Xenopus oocytes.
The clone responsible for this effect is then isolated
by the process described above. Once the receptor gene
is isolated, standard techniques are used to identify
the polypeptide or portions) thereof which is (are)
sufficient for binding arylalkylamines (the
arylalkylamine binding domains]). Further, using
standard procedures, the entire receptor or
arylalkylamine binding domains) can be expressed by
recombinant technology. Said receptor or binding
domains) can be isolated and used as a biochemical
reagent such that, rather than using a competitive assay
CA 02560002 2006-10-05
WO 97/46511 PCT/U596/20697
78
exemplified below, a simple direct binding assay can be
used. That is, a screen is set up for compounds which
bind at the novel arylalkylamine receptor. In this way
large numbers of compounds can be simultaneously
screened, e.g., by passage through a column containing
the novel arylalkylamine receptor or arylalkylamine
binding domain, and analysis performed on compounds
which bind to the column.
Additional testing utilizes the combination of
l0 molecular biological techniques (expression of cloned
NMDA, AMPA or nicotinic cholinergic receptors) and patch
clamp electrophysiological techniques. Specifically,
arylalkyl-amine analogs can be rapidly screened for
potency at cloned and expressed subunits of the
above-mentioned receptor-ionophore complexes.
Site-directed mutagenesis can be utilized in an effort
to identify which amino acid residues may be important
in determining arylalkylamine potency.
Assays for Potent and Selective Antagonists
of Receptor-Operated Calcium Channels
in the Mammalian CNS
Desired properties of a drug include: high
affinity and selectivity for receptor-operated Ca2,
channels, such as those present in NMDA, AMPA and
nicotinic cholinergic receptor-ionophore complexes
(compared to responses mediated via other
neurotransmitter receptors, neurotransmitter
receptor-operated ion channels, or voltage-dependent ion
channels) and a noncompetitive antagonism of said
receptor-operated CaZ' channels.
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
79
The NMDA receptor-ionophore complex is
utilized as an example of a receptor-operated Ca2'
channel. Activation of the NMDA receptor opens a
cation-selective channel that allows the influx of
extracellular Ca2' and Na', resulting in increases in
[Caz');and depolarization of the cell membrane.
Measurements of [Ca2'];were used as primary assays for
detecting the activity of arylalkylamine compounds on
NMDA receptors. Purified arylalkylamines, synthetic
aryl-alkylamines, and synthetic analogs of
arylalkylamines were examined for activity in in vitro
assays capable of measuring glutamate receptor activity.
Selected for detailed study were the arylalkylamines
present in the venom of various spider species. The
arylalkylamines present in these venoms are structurally
distinct but have the basic structure of the class
represented by Compounds 1 through 3. Other more
simplified synthetic analogs generally consist of
suitably substituted aromatic chromophoric groups
attached to an alkyl(poly)amine moiety (see Compounds 19
through 215 below).
A primary assay that provides a functional
index of glutamate receptor activity and that allows
high-throughput screening was developed. Primary
cultures of rat cerebellar granule cells loaded with the
fluorimetric indicator fura-2 were used to measure
changes in [Caz'];elicited by NMDA and its coagonist
glycine. This assay provides an extremely sensitive and
precise index of NMDA receptor activity. Increases in
[Cap') i evoked by 1~1MDA are dependent on the presence of
glycine, and are blocked by extracellular Mgr' or
CA 02560002 2006-10-05
WO 97/46511 PCTNS96l20697
ao
antagonists acting at the glutamate, glycine, or MK-SOl
binding sites. Increases in [Ca2'];elicited by
NMDA/glycine are readily distinguished from those
resulting from depolarization by their refractoriness to
inhibition by blockers of voltage-sensitive Caz'
channels. The fidelity with which measurements of
[Ca2~]; corroborate results obtained by
electrophysiological and ligand-binding studies suggests
that such measurements mirror closely activation of the
NMDA receptor-ionophore complex.
Exaanple l: Potent noncompetitive inhibition of NINA
receptor function
Preferential inhibitory effects of
arylalkylamines on NMDA receptor-mediated increases in
[Ca2'];in cultured rat cerebellar granule cells were
measured. Increases in [Caa'];were elicited by the
addition of NMDA/glycine (50 ~cM/1 E.cM) in the presence or
absence of different concentrations of each test
compound. The ICso values were derived for each test
compound using from 2 to 8 separate experiments per test
compound, and the standard error level was less than l00
of the mean value for each compound.
All of the arylalkylamines tested blocked
increases in LCa'~];in cerebellar granule cells elicited
by NMDA/glycine. Certain arylalkylamines similar in
structure to Compound 1 or Compound 2 were nearly as
potent as MK-801 (ICSO = 34 nM) which is the most potent
compound in the literature known to preferentially block
NMDA receptors. Compound 3 had an ICSO = 2 nM, that is,
17-fold more potent than MK-801. Many of the
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
81
arylalkylamines tested were more potent than competitive
antagonists such as APS (ICso = 15 ~.M). The inhibitory
effects of the arylalkylamines were not overcome by
increasing the concentrations of NMDA or glycine. That
is, no change was observed in the ECso for either NMDA or
glycine. The arylalkylamines are thus noncompetitive
antagonists at the NMDA receptor-ionophore complex, and
act neither at the glutamate nor the glycine binding
sites.
Example 2: Activity against Kainate and AMPA receptor
function
Measurements of [Caz'];in cerebellar granule
cells can also be used to monitor activation of the
native kainate or AMPA receptors present in this tissue.
Although the increases in [Cap'];evoked by these agonists
are of a lesser magnitude than those evoked by
NMDA/glycine, such responses are robust and can be used
to precisely assess the specifv:.city of action of
arylalkylamines on pharmacologically defined glutamate
receptor subtypes. Comparative measurements of [Caz']i
revealed a clear distinction in the receptor selectivity
of the arylalkylamines. Some, like JSTX-3 (,7oro Spider
toxin from the spider Nephila clavata), were more potent
antagonists of responses elicited by kainate (100 ~.M) or
AMPA (30 ~,cM). On the other hand, arylalkylamines within
the two structural classes defined by Compound 1 and by
Compound 2 were found to inhibit preferentially
responses evoked by NMDA (showing about a 100-fold
difference in potency). Thus, arylalkylamines such as
Compound 1 and Compound 2 are potent and selective
CA 02560002 2006-10-05
WO 97146511 PCT/US961Z0697
82
inhibitors of NMDA receptor-mediated responses in
cerebellar granule cells.
Example 3: Patch clamp electrophysiology studies
Patch clamp electrophysiological studies on
isolated cortical or hippocampal neurons from adult rat
brain have provided additional insight into the
mechanism of action of Compound 1, Compound 2 and
Compound 3. These studies revealed potent and selective
inhibitory effects of arylalkylamines on responses
mediated by NMDA receptors. Thus, compounds such as
Compound 1 blocked responses to NMDA at nanomolar
concentrations without affecting the responses to
kainate. These results, which show selective inhibitory
effects of the arylalkylamines in cortical and
hippocampal neurons, indicate that the arylalkylamines
target NMDA receptors in different regions within the
mammalian CNS. Moreover, it was found that the
inhibitory effects of these compounds were use- and
voltage-dependent. This strongly suggests that these
compounds are blocking the open channel and, by this
action, behave as noncompetitive NMDA receptor
antagonists. Importantly, however, the arylalkylamines
could be distinguished from both Mgr' and MK-801,
especially with respect to the voltage-dependence of
their onset of action and reversibility of effect.
Example 4: Radioligand binding assays
Radioligand binding studies have demonstrated
that arylalkylamines such as Compound 1 and Compound 2
have a unique site of action. Although they act like
CA 02560002 2006-10-05
WO 97/46511 PCT/U596IZ0697
83
MK-801 in some respects (noncompetitive open-channel
blockade, discussed above), they fail to displace
['H]MK-801 binding at concentrations that completely
block NMDA receptor-mediated responses. Assays such as
these also demonstrate that the arylalkylamines do not
bind with high affinity to the known MK-801, Mgr', or
polyamine binding sites on the NMDA receptor-ionaphore
complex. Neither do the arylalkylamines bind directly
to either the glutamate, glycine or sigma binding sites
at concentrations that block NMDA receptor-mediated
responses. ['H]Compound 2 was synthesized as a
radioligand for use in binding studies to further
explore the mechanism of action of Compound 2 and
particularly for use in a high-throughput screen to
assess the activity of other analogs and to detect new
lead structures. A similar approach was taken for
['H]Compound 5. It is clear that compounds like Compound
1 and Compound 2 target a site on the NMDA
receptor-ionophore complex for which no other known
compounds presently exist. The novel site of action of
the arylalkylamines at the molecular level translates
into pronounced therapeutic advantages at the behavioral
level. As described below, the arylalkylamines possess a
quite different behavioral profile from other
noncompetitive antagonists of the NMDA receptor.
Example 5: Synaptic transmission studies
The above findings demonstrate that certain
arylalkylamines, specifically those related in structure
to Compound 1 and Compound 2, act through a novel
mechanism and site of action to potently and selectively
CA 02560002 2006-10-05
WO 97/46511 PCT/US96l20697
84
inhibit NMDA receptor-mediated responses on neurons from
several different brain areas. To further assess the
selective inhibitory actions of the arylalkylamines,
their effects on synaptic transmission mediated by NMDA
S or AMPA receptors were assessed.
Glutamate-mediated transmission at synapses of
Schaffer collateral fibers and CA1 pyramidal cells was
measured in slices of rat brain containing the
hippocampus. This assay measures electrophysiologically
the postsynaptic depolarization caused by the
presynaptic release of glutamate, and can readily
distinguish synaptic transmission mediated by NMDA or
AMPA receptors. Arylalkylamines like Compound 1,
Compound 2 and Compound 3 were again found to exert
preferential inhibitory effects on NMDA
receptor-mediated responses, and depressed responses
mediated by AMPA receptors only at much higher
concentrations. For example, Compound 1 had an ICso for
the NMDA receptor-mediated response of 20 E.cM, but an ICso
for the AMPA receptor-mediated response of 647 ~M. These
results show that arylalkylamines can selectively
inhibit synaptic transmission mediated by NMDA
receptors. Other naturally occurring arylalkylamines
present in the venom of Agelenopsis aperta likewise
exert potent and selective inhibitory effects on NMDA
receptor-mediated responses in the rat hippocampus.
In the aggregate, then, the results of these
various studies are complementary and together identify
a structurally novel class of compounds with potent and
selective inhibitory activity on NMDA receptors in the
mammalian CNS. Additionally, these compounds target a
CA 02560002 2006-10-05
WO 97/46511 PCT/US96I20697
unique site on the NMDA receptor-ionophore complex.
Compound 1, Compound 2 and Compound 3 were selected for
additional study in a variety of in vitro and in vivo
assays that model therapeutically important endpoints.
5 Neuroprotectant activity
Desired properties of a neuroprotectant drug
include the following. (1) The drug can be administered
by oral or injectable routes (i.e., it is not
significantly broken down in the stomach, intestine or
10 vascular system and thus reaches the tissues to be
treated in a therapeutically effective amount). Such
drugs are easily tested in rodents to determine their
bioavailability. (2) The drug exhibits neuroprotectant
activity (i.e., efficacy) when given after an ischemic
15 insult (stroke, asphyxia) or traumatic injury (head
trauma, spinal cord injury). (3) The drug is devoid of
or has minimal side effects such as impairment of
cognition, disruption of motor performance, sedation or
hyperexcitability, neuronal vacuolization,
20 cardiovascular activity, PCP-like abuse potential, or
PCP-like psychotomimetic activity.
Although glutamate is the physiological
synaptic transmitter, chronic exposure to glutamate
leads to neuronal cell death. Much of the
25 neurodegeneration caused by glutamate appears to be
mediated by NMDA receptors and results directly from
chronically elevated levels of cytosolic Caz'. There is
now extensive experimental support for the view that
NMDA and AMPA receptors play a major role in mediating
30 the neuronal degeneration following a stroke and other
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
86
ischemic/hypoxic events (Choi, Glutamate neurotoxicity
and diseases of the nervous system. Neuron 1: 623,
1988). Most of this evidence is based on the ability of
competitive or noncompetitive antagonists of the NMDA or
AMPA receptor to effectively block neuronal cell death
in both in vitro and in vivo models of stroke. Compound
1, Compound 2 and Compound 4 were therefore examined for
neuroprotectant effects in standard assays designed to
detect such activity.
Example 6: Cortical neuron protection
To assess the in vitro neuroprotectant effect
of arylalkylamines, mouse cortical neurons grown in
culture were exposed for S minutes to NMDA, and cell
death after 24 hours was monitored by measuring the
release of lactate dehydrogenase (LDH), a cytoplasmic
enzyme that is released from dying cells (Choi et al.,
Glutamate neurotoxicity in cortical cell culture. J.
Neurosci. 7: 357, 1987). Exposure to NMDA killed about
80% of the cortical neurons. Compound 1 or Compound 2,
included along with NMDA, prevented cell death with ICso
values of 70 ~cM and 30 ~.M, respectively. The effective
concentrations of the arylalkylamines are higher than
those of other noncompetitive NMDA receptor antagonists,
but similar to those of competitive antagonists. The
effective concentrations of NMDA receptor antagonists
vary depending on the particular experimental conditions
and the type of cell studied (cortical, hippocampal,
striatal). This neuroprotectant effect likely results
from the ability of these compounds to block the influx
of extracellular Caz' triggered by the NMDA receptor.
CA 02560002 2006-10-05
WO 97!46511 PCTlUS96120697
87
More rigorous testing to determine potential
therapeutic efficacy involved in vivo stroke models. In
these models, the blood supply is temporarily blocked by
clamping the main arteries to the brain. Two in vivo
models of this sort were used to determine the ability
of Compound 1, Compound 2 and Compound 4 to prevent
neuronal cell loss.
Example 7: Bilateral carotid artery occlusion
The first assay was the bilateral common
carotid artery occlusion model of forebrain ischemia
performed in the gerbil (Karpiak et al., Animal models
for the study of drugs in ischemic stroke. Ann. Rev.
Pharrr~acol. Toxicol. 29: 403, 1989; Ginsberg and Busto,
Rodent models of cerebral ischemia. Stroke 20: 1627,
1989). Hlood flow to the brain was interrupted for 7
minutes by clamping the carotid arteries. The test
compounds were administered as a single dose given
intraperitoneally (i.p.) 30 minutes after reinstating
blood flow. During the course of these experiments, the
core body temperature of the animals was maintained at
37°C to prevent any hypothermic reaction. It has been
shown that many NMDA receptor antagonists cause
hypothermia and this effect can account for much of the
protective effect of these compounds. The brains were
examined for neuronal cell death 4 days later by silver
staining sections of the brain and quantifying death by
morphometric analysis. Compound 2 (20 mg/kg)
significantly (p < 0.05) protected against neuronal cell
death in all areas of the brain examined (region CA1 of
hippocampus, striatum and neocortex). Doses as low as 1
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
88
mg/kg afforded complete (>98%) protection of the
striatum. The degree of protection is comparable to
that achieved with similar doses of the noncompetitive
NMDA antagonist, MK-801.
In subsequent experiments, Compound 1
(10 mg/kg) produced a 23% reduction in the amount of
neuronal death in region CA1 of the gerbil hippocampus
measured at 7 days post-ischemia, while Compound 4
(10 mg/kg) provided 90% protection.
Example 8: Middle cerebral artery occlusion
The middle cerebral artery model of stroke
performed in the rat (Karpiak et al., Animal models for
the study of drugs in ischemic stroke. Ann. Rev.
Pharmacol. Toxicol. 29: 403, 1989; Ginsberg and Busto,
Rodent models of cerebral ischemia. Stroke 20: 1627,
1989) is different from the gerbil model because it
results in a more restricted brain infarct, and thereby
approximates a different kind of stroke (focal
thrombotic stroke). In the first study using this
stroke model, one cerebral artery was permanently
occluded by surgical ligation. The test compounds were
administered 30 minutes after the occlusion by a single
intraperitoneal (i.p.) injection. During the course of
these experiments, the core body temperature of the
animals was maintained at 37°C to prevent any
hypothermic reaction. Brains were assessed
histologically for neuronal cell loss 24 hours later.
Infarct volumes were calculated using the area of
histological pallor from 10 slides and integrating the
distance between each successive section. A single dose
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
89
(30 mg/kg) of Compound 1 was found to significantly (p <
0.05) protect against neuronal cell loss equally as well
as a maximally effective dose (10 mg/kg) of MK-801
(approximately 15% protection). Preliminary studies
with Compound 2 (20 mg/kg) indicated a similar trend.
In the second study of focal cerebral ischemia
in the rat, the middle cerebral artery was permanently
occluded by passing a small piece of suture thread
through the carotid artery to the region of the middle
cerebral artery. Core body temperature was maintained
at 37°C. Compound 4, 10 mg/kg i.p. administered
immediately after the onset of the ischemic event,
produced a statistically significant reduction in the
volume of the brain infarct (20%) recorded 24 hr later.
In a third model of focal cerebral ischemia in
the rat, an ischemic infarct was produced by a
photothrombotic method using the dye Rose Bengal.
Compound 4, 10 mg/kg i.p. administered 30 min after the
ischemic event, produced a 20% reduction in the volume
of the infarct, similar to that seen with the
noncompetitive NMDA receptor antagonist, MK-BO1.
In a fourth model of focal cerebral ischemia
in the rat, the middle cerebral artery was temporarily
occluded by passing a small piece of suture thread
through the carotid artery to the region of the middle
cerebral artery. The suture thread was withdrawn after
an ischemic period of 2 hr. Core body temperature was
maintained at 37°C. Compound 4 administered at l0 mg/kg
i.p. immediately after the onset of the ischemic event,
produced a statistically significant reduction in the
volume of the brain infarct (37%) recorded 72 hr later.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
Several important features of the lead
compounds emerge from these in vivo results. First, and
most importantly, Compound 1, Compound 2 and Compound 4
demonstrate neuroprotectant effects in several different
S in vivo models of stroke. The gerbil assay is a model
for transient global cerebral ischemia and hypoxia such
as cardiac arrest or perinatal hypoxia. The rat assays
are models of permanent and temporary focal cerebral
ischemia. The finding that Compound 1 and Compound 4
10 are neuroprotective in the permanent focal stroke models
is surprising because the accessibility of the drug to
the site of infarction. is limited to the penumbral
region which generally is not large. Nonetheless,
Compound 1 and Compound 4 significantly (p < 0.05)
15 limited the extent of damage. Second, the compounds are
effective when administered after the ischemic event.
This is important because there is believed to be a
"window of opportunity" following an infarct during
which drugs may effectively halt necrotic damage. How
20 long this time is in humans has not been defined
precisely, and will likely vary depending upon the type
of infarct. The essential observation, however, is that
these compounds can prevent the spread of neuronal cell
death once the degenerative process has commenced.
25 Finally, Compounds 1, 2, and 4 are effective when
administered parenterally, demonstrating that they
penetrate the blood-brain barrier.
Anticonvulsant activity
Desired properties of an anticonvulsant drug
30 include: the drug can be administered by oral or
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
91
injectable routes, the drug exhibits effective
anticonvulsant activity against several seizure types,
including, but not limited to, simple partial seizures,
complex partial seizures, status epilepticus, and
trauma-induced seizures such as occur following head
injury, including head surgery; and the drug is devoid
of or has minimal side effects such as impairment of
cognition, disruption of motor performance, sedation or
hyperexcitability, neuronal vacuolization,
cardiovascular activity, PCP-like abuse potential, or
PCP-like psychotomimetic activity.
Glutamate is the major excitatory transmitter
in the brain, and thus may play a major role in seizure
activity, and contribute to the pathogenesis of
epilepsy. Much of the evidence favoring a major role for
glutamate receptors in epilepsy derives from
pharmacological studies demonstrating that glutamate
receptor agonists elicit seizures, and that NMDA and
AMPA receptor antagonists are effective anticonvulsants
when administered in vivo. There are numerous in vivo
models involving different kinds of seizures and
behavioral effects that are relevant for clinically
distinct forms of epilepsy. It is thus prudent to test
for effects in several models, because it may be an
oversimplification to suppose that the same mechanism
underlies all forms of seizure activity.
Example 9: Convulsant blocking activity
In initial studies, the ability of
arylalkylamines to block seizures induced by kainate,
picrotoxin or bicuculline were examined. Each of these
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
92
convulsants acts through a different mechanism and
seizures elicited by kainate are qualitatively different
from those elicited by picrotoxin or bicuculline. In
these experiments, a fraction of Agelenopsis aperta
venom containing several arylalkylamine toxins was
administered intravenously (iv) 5 min before picrotoxin
or bicuculline, and 5 min after kainate administration.
The arylalkylamines diminished the seizures induced by
all three of these agents. The effects of picrotoxin or
bicuculline were so severe that all 19 control animals
died within 25 minutes. In contrast, there were no
deaths in the 9 animals pretreated with the
arylalkylamines. In fact, only about half the animals
treated with the arylalkylamines showed any convulsions
at all and those symptoms abated within an hour. These
results demonstrate clear anticonvulsant effects of
arylalkylamines and prompted further studies using
purified arylalkylamines and their analogs.
Example 10: Seizure stimuli
Three different seizure-inducing test
paradigms were used initially in this second group of
studies and arylalkylamines such as Compound 1 proved to
be effective anticonvulsants in two such paradigms. The
first two models used DBA/2 mice which are prone to
audiogenic seizures. Seizures were elicited by sound
(bell tone at 109 dBs) or the intraperitoneal (ip)
administration of NMDA (56 mg/kg). The test substances
were administered 15-30 min before the convulsant
stimulus. The number of clonic seizures was recorded
for 1 min following the audiogenic stimulus or for 15
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
93
min following the administration of NMDA. Compound 1,
Compound 2, and several other arylalkylamines such as
Compound 3 and Compound 4 depressed seizures evoked by
either stimulus. For example, Compound 2 had an EDso of
O.I3 mg/kg s.c. for audiogenic stimulus and 0.083 mg/kg
s.c. for NMDA stimulus. Similarly, the ECso for Compound
4 in the audiogenic seizure model (0.08 mg/kg)
approached that for MK-801 (0.02 mg/kg). In contrast,
neither Compound 1 nor Compound 2 was effective at doses
up to 50 mg/kg s.c. in reducing seizures in CF-1 mice
elicited by i.p. NMDA.
In a second independent series of experiments,
Compound 1 and Compound 4 were found to prevent seizures
induced by sound in another genetically susceptible
mouse model of reflex epilepsy (Frings mice) following
intraperitoneal injection with ICso values of 14.3 mg/kg
and -15 mg/kg, respectively. These compounds were
considerably more potent against audiogenic seizures in
Frings mice following intracerebroventricular (i.c.v.)
injection, with ICso values of 0.63 ~Cg (Compound 1) and
4.77 /.cg (Compound 4). Compound 1 was also found to be
effective against seizures elicited by maximal
electroshock in CF1 mice at a dose of 4 ~cg i.c.v.
In further studies using the genetically
susceptible mouse model of reflex epilepsy (Frings
mice), Compound 9, Compound 12 and Compound 14,
administered by i.c.v. injection, prevented
sound-induced seizures with ICso values of 4.77 fig, 12.2
~cg and 13.9 fig, respectively.
These collective findings demonstrate that
arylalkylamines such as Compound 1, Compound 2 and
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
94
Compound 4 are effective in preventing epileptic
(audiogenic) and nonepileptic (chemoconvulsant)
seizures. This generalized pattern of activity suggests
that arylalkylamines are clinically useful in
S controlling seizure activity. In addition, the potency
of Compound 1, Compound 2 and especially Compound 4 in
in vivo models of seizure activity shows that these
compounds can have the therapeutically relevant effects
when administrated parenterally in low doses, and are
especially potent when administered directly into the
cerebral ventricles.
Analgesic activity
Desired properties of an analgesic drug
include: the drug can be administered by oral or
injectable routes, the drug exhibits analgesic activity,
the drug is devoid of or has minimal side effects such
as impairment of cognition, disruption of motor
performance, sedation or hyperexcitability, neuronal
vacuolization, cardiovascular activity, PCP-like abuse
potential, or PCP-like psychotomimetic activity.
Glutamate and NMDA receptor-mediated responses
may play a role in certain kinds of pain perception
(Dickenson, A cure for wind up: NMDA receptor
antagonists as potential analgesics. Trends Pharmaeol.
Sci. 11: 302, 1990). The possible analgesic effects of
Compound 1, Compound 2, Compound 3 and Compound 4 were
therefore examined.
CA 02560002 2006-10-05
WO 97/46511 PCT/LTS96/20697
Example I1: Writhing response test
In the first series of experiments, the
animals were administered an unpleasant stimulus
(2-phenyl-1,4-benzoquinone, PBQ) which elicits a
5 writhing response (abdominal stretching). Typically,
the number of writhes occurring in a 5 min observation
period is recorded. Classic analgesic drugs, such as
morphine, are effective at decreasing the number of
PBQ-elicited writhes (1000 block of the writhing
10 response at 4 mg/kg i.p.). Nonsteroidal
antiinflammatory agents axe likewise effective in this
model. Compound 1 (2 mg/kg), Compound 2 (2 mg/kg) and
Compound 3 (1 mg/kg) depressed the writhing response by
greater than 95% when administered s.c. or i.p. 30
15 minutes before PBQ. These results demonstrate that
Compound 1, Compound 2 and Compound 3 alleviate visceral
pain.
In a similar series of studies, Compound 1 and
Compound 4 were found to inhibit acetic acid-induced
20 writhing in mice following i.p. injection with ICso
values of 10 mg/kg and 1 mg/kg, respectively.
Example 12: Hot plate test
Compound 1 was tested for analgesic activity
in an additional assay. In this model of analgesic
25 activity, mice were administered test substances s.c.
30 min before being placed on a hot plate (50°C). The
time taken to lick the feet or jump off the plate is an
index of analgesic activity, and effective analgesics
increase the latency to licking or jumping. Morphine
30 (5.6 mg/kg) increased the latency to jump by 765%.
CA 02560002 2006-10-05
WO 97!46511 PCT/US96120697
96
Compound 1 was likewise effective in this assay and, at
doses of 4 and 32 mg/kg, increased the latency to foot
licking by 136% and the latency to jumping by 360%,
respectively.
It is noteworthy that the analgesic effects of
Compound 1 in the hot plate assay were not accompanied
by a decreased performance in the inverted grid assay
(see below). This shows that the increase in the
latency to jump off the hot plate does not simply
reflect impaired motor capabilities. Together, these
data suggest that Compound 1 possesses significant
analgesic activity.
In a later series of experiments, Compound 1
and Compound 4 were demonstrated to possess significant
analgesic activity in rats when administered by the
intrathecal (i.th.) route. In these experiments, a 52°C
hot plate was used as the nociceptive stimulus.
Compound 1 (0.3 - 3 nmol) and Compound 4 (0.3 - 3 nmol)
produced dose- and time-dependent antinociceptive
effects; these arylalkylamines were similar to morphine
(0.3 - 3 nmol) in terms of potency and efficacy. The
NMDA receptor antagonist, MK-801, on the other hand, was
ineffective in this assay (3-30 nmol).
Example 13. Tail flick test
In this standard assay, the thermal
nociceptive stimulus was 52°C warm water with the
latency to tail flick or withdrawal taken as the
endpoint. Compound 1 (0.3 - 3 nmol) and Compound 4 (0.3
- 3 nmol) produced a dose- and time-dependent analgesic
effect following i.th. administration. These
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
97
arylalkylamines were similar to morphine (0.3 - 3 nmol)
in terms of potency and efficacy. The NMDA receptor
antagonist, MK-801, on the other hand, was ineffective
in this assay (3-30 nmol).
Example 14. Formalin test
Male Sprague-Dawley rats were habituated to an
observation chamber for at least 1 hr before receiving
an injection of dilute formalin (5~) in a volume of
50 ~cl into the left rear paw. Behavioral responses were
monitored immediately after s.c. injection of formalin
into the dorsal surface of the paw by counting the
number of flinches exhibited by the animal. Behaviors
were monitored for at least 50 min after formalin
injection and were recorded as early phase responses
(0 - 10 min post-formalin) and late phase responses
(20 - 50 min post-formalin). Compounds were injected
intrathecally (i.th.) l0 min prior to formalin
(pre-treatment) or 10 min after formalin
(post-treatment) in a volume of 5 ~cl.
Intraplantal administration of formalin
produced a typical biphasic response of flinching
behavior, commonly described as the early and late phase
responses. Intrathecal administration of Compound 1
(0.3 - 10 nmol) or Compound 4 (0.3 - 10 nmol) given as a
pretreatment to formalin effectively inhibited both
early- and late-phase flinching behaviors. This effect
of pretreatment with the arylalkylamines was similar to
that seen with pretreatment with morphine (1 - 10 nmol)
or MK-801 (1 - 30 nmol).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
98
Compound 1 (0.3 - 10 nmol i.th.) administered
after the formalin produced some inhibition of
late-phase flinching, though significance was achieved
only at the 10 nmol dose. Compound 4 (0.3 - 10 nmol
i.th.) administered after the formalin produced
significant inhibition of late-phase flinching, with
significance achieved at the 3 and l0 nmol doses. This
analgesic profile of activity of the arylalkylamines is
similar to that seen with post-formalin administration
of morphine (1 - 10 nmol); post-formalin administration
of MK-BO1 (1 - 30 nmol), however, failed to affect
late-phase flinching.
Taken together, the results obtained with the
hot plate, tail flick and formalin assays demonstrate
that arylalkylamines such as Compound 1 and Compound 4
have significant analgesic activity in several rodent
models of acute pain. The formalin assay additionally
demonstrates that arylalkylamines are effective in an
animal model of chronic pain. Importantly, the
arylalkylamines possess significant analgesic activity
when administered after the formalin stimulus. This
profile of activity clearly distinguishes the
arylalkylamines from standard NMDA receptor antagonists
such as MK-801.
Side effects of arylalkylamiaes
Given the important role NMDA receptors play
in diverse brain functions, it is not surprising to find
that antagonists of this receptor are typically
associated with certain unwelcome side effects. In
fact, it is this property that provides the major
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
99
obstacle to developing therapies that target NMDA
receptors. The principal side effects, which
characterize both competitive and noncompetitive
antagonists, are a PCP-like psychotomimetic activity,
impairment of motor performance, sedation or
hyperexcitability, impairment of cognitive abilities,
neuronal vacuolization, or cardiovascular effects
(Willetts et al., The behavioral pharmacology of NMDA
receptor antagonists. Trends Pharmacol. Sci. 11: 423,
l0 1990; Olney et al., Pathological changes induced in
cerebrocortical neurons by phencyclidine and related
drugs. Science 244: 1360, 1989). The psychotomimetic
effect associated with inhibition of NMDA
receptor-mediated responses is epitomized in the
response to phencyclidine (PCP) or "angel dust" which
acts at the MK-801 binding site. Impairment of
cognitive ability is associated with the important role
that NMDA receptors normally play in learning and
memory.
Relatively less is known concerning the side
effect profile of AMPA receptor antagonists. However,
it is becoming clear that such compounds also elicit
motor impairment, ataxia and profound sedation.
The activity of arylalkylamines was examined
in animal models that index motor impairment, sedation
and psychotomimetic activity as well as both in vitro
and .in vivo models of learning and memory.
(a) PCP-like Peychotomimetic Activity
In rodents, both competitive and
noncompetitive antagonists of the NMDA receptor produce
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
100
a PCP-like stereotypic behavior characterized by
hyperactivity, head-weaving, and ataxia (Willetts et
al., The behavioral pharmacology of NMDA receptor
antagonists. Trends Pharmacol. Sci. 11: 423, 1990;
Snell and Johnson, In: Excitatory Amino Acids in Health
and Disease, John Wiley & Sons, p. 261, 1988). We
investigated whether the arylalkylamines would elicit
such behaviors. In addition, we investigated whether
the arylalkylamines would substitute for PCP in rats
trained to discriminate PCP from saline (Willetts et
al., The behavioral pharmacology of NMDA receptor
antagonists. Trends Pharmacol. Sci. 11: 423, 1990), and
whether the arylalkylamines would elicit a PCP-like
neuronal vacuolization (Olney et al., Pathological
changes induced in cerebrocortical neurons by
phencyclidine and related drugs. Science 244: 1360,
1989) .
Example 15: Locomotor activity
The first assay simply monitors locomotor
activity during the first hour following peripheral
(s. c. or i.p.) administration of test substance. Mice
received a dose of Compound 1 15 min before being placed
into activity chambers. Activity was quantified by
counting the number of breaks in a phototube grid in a
60 min period. In this assay, MK-801 (0.25 mg/kg p.o.)
causes a 2- to 3-fold increase in locomotor activity.
However, Compound 1, even when tested at 32 mg/kg s.c.,
did not elicit hyperactivity and, in fact, tended to
depress it. This result, using a purified
arylalkylamine in mice, complements earlier results
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
101
obtained in rats where the entire
arylalkylamine-containing fraction from Agelenopsis
aperta, when injected intravenously, did not elicit a
PCP-like behavioral syndrome but seemed to produce a
S mild sedative effect.
Example 16: Motor impairment
In the first assay for generalized motor
impairment, Compound 1 was examined in the inverted grid
assay. In this assay, animals are placed on a
wire-holed grid suspended from a rotating metal bar
which can be inverted. The animals are then scored for
their ability to climb to the top or hang on to the
grid. Animals with severe motor impairment fall off the
grid. This assay provides an index of "behavioral
disruption" that may result from ataxia, loss of the
righting reflex, sedation, or muscle relaxation. In
these tests, Compound 1, administered at 32 mg/kg s.c.,
did not lessen the ability of DBA/2 mice to right
themselves when the grid was inverted (p > 0.05).
Compound 2 was likewise without effect (p > 0.05) on
motor performance in DBA/2 mice when administered at a
dose of 20 mg/kg s.c. These doses are considerably
higher than those required to prevent sound-induced
seizures in DBA/2 mice (see Example 10 above).
The second assay of acute motor impairment was
the rotorod assay. In this assay, Frings and CF1 mice
were injected with test compound and placed on a knurled
rod which rotated at a speed of 6 rpm. The ability of
the mice to maintain equilibrium for long periods of
time was determined; those mice that were unable to
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
102
maintain equilibrium on the rotorod for 1 min in each of
3 trials were considered impaired. Compound 1 produced
acute motor impairment in Frings mice with a TDSo (that
dose which produced motor toxicity in 50s of the test
animals) of 16.8 mg/kg i.p. This dose is similar to
that which prevents sound-induced seizures in Frings
mice (see Example 10 above). There is a much clearer
separation between effective and toxic doses of
Compound 1 in Frings mice, however, when the Compound is
administered i.c.v. In this case, no apparent motor
toxicity was evident until the dose of Compound 1
exceeded 1.56 ~g i.c.v. (>2 times the EDso of 0.63 fig).
Finally, motor impairment in CF1 mice was noted with
Compound 1 following i.c.v. administration of 4 fig.
Compound 4, Compound 9, Compound 12 and
Compound 14 were administered to Frings mice by i.c.v.
injection, and acute motor impairment was measured. The
TDSa values for Compounds 4,~ 9, 12 and 14 were 8-16 ~cg,
14.8 ug, 30.2 E.tg and 30.8 E.eg, respectively. These TDso
values were 2-3 times higher than the effective ICso
values for anticonvulsant potency (see Example 10
above); a clear separation between effective and toxic
doses was noted.
Example 17. PCP discrimination
In this assay, rats who have been trained to
lever press for food reinforcement must select which of
two levers in their cages is correct. The only stimulus
they have for selecting the correct lever is their
ability to detect whether they received a PCP or vehicle
injection. After about two months of training, rats
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
103
become very good at discriminating PCP from vehicle
injections and can then be tested with other drugs to
determine if they are discriminated as PCP. When tested
in this procedure, other drugs which are known to
produce a PCP-like intoxication substitute for PCP.
These drugs include various PCP analogs such as ketamine
and the noncompetitive NMDA receptor antagonist, MK-801.
Compound 1 (1 - 30 mg/kg i.p.) did not
substitute for PCP, and thus was completely devoid of
PCP-like discriminative stimulus effects. At 30 mg/kg
i.p., only 1 of the 7 animals tested responded at all on
either lever. It is thus clear that a behaviorally
effective dosage range of Compound 1 was evaluated. As
the ability of test compounds to produce PCP-like
effects in rats is believed to be predictive of their
ability to produce PCP-like psychotomimetic activity and
abuse liability in humans, these results strongly
suggest that the arylalkylamines such as Compound 1 will
lack such deleterious side effects in man.
Example 18
The administration of compounds such as PCP
and MK-801 to rats produces a neurotoxic effect termed
neuronal vacuolization. Following a single dose of such
compounds, vacuoles are found in particular central
neurons, especially those in the cingulate cortex and
retrosplenial cortex. No such vacuolization was present
in rats treated with Compound 1 at the single high dose
of 100 mg/kg i.p.
Taken together, the results on locomotor
activity, motor impairment, PCP discrimination and
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
104
neuronal vacuolization strongly suggest that
arylalkylamines will be devoid of PCP-like side effects
in man.
(b) Cognitive impairment
One of the major reasons for postulating a
role of NMDA receptors in memory and learning derives
from cellular studies on long-term potentiation (LTP) in
the rat hippocampus. LTP is a long-lasting increase in
the magnitude of synaptic responses produced by brief
yet intense synaptic stimulation. Since the discovery
of this phenomenon, it has become the preeminent
cellular model of learning in the vertebrate brain
(Teyler and Discenna, Long-term potentiation. Anna.
Rev. Neurosci. 10: 131, 1987). Transmission at synapses
formed by Schaffer collaterals onto CA1 pyramidal cells
is mediated by NMDA and AMPA receptors. Following a
brief tetanizing stimulus, the magnitude of the
population spike (a measure of synaptic transmission) is
greatly increased and remains so for hours. It has been
shown that all known competitive and noncompetitive
antagonists of NMDA receptors block LTP in the rat
hippocampus, whereas antagonists of non-NMDA receptors
are without effect (Collingridge and Davis, In: The NNmA
Receptor, IRL Press. p. 123, 1989). This supports a
role of NMDA receptors in memory and learning.
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
105
Example 19: LTP assay
The effects of selected arylalkylamines and
literature standards were examined for effects on LTP in
slices of rat hippocampus. As anticipated, all the
conventional competitive (AP5 and AP7) and
noncompetitive (MK-801 and ifenprodil) NMDA receptor
antagonists inhibited the induction of LTP in the
hippocampus. Slices of rat hippocampus were superfused
for 30-60 min with a test compound before delivering a
tetanizing stimulus consisting of 3 trains, separated by
500 msec, of 100 Hz for 1 sec each. The response
amplitude was monitored for an additional 15 minutes
post-tetanus. The tetanizing stimulus caused a mean 95%
increase in the amplitude of the synaptic response. The
induction of LTP was significantly blocked (p < 0.05) by
competitive (AP5, AP7) or noncompetitive (MK-801,
ifenprodil) NMDA receptor antagonists. Quite
surprisingly, none of the arylalkylamines tested
(Compound 1, Compound 2, Compound 3 and others) blocked
the induction of LTP (p > 0.05), even when used at high
concentrations (100-300 E.cM) that caused some inhibition
of the control response.
These results highlight yet another unique and
important feature of arylalkylamines. Arylalkylamines
are the first, and at present the only, class of
compounds shown to be selective and potent antagonists
of the NMDA receptor that do not block the induction of
LTP. This likely reflects the novel mechanism and site
of action of arylalkylamines and suggests that drugs
which target the novel site on the NMDA receptor will
similarly lack effects on LTP. As LTP is the primary
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
106
cellular model for learning and memory in the mammalian
CNS, it additionally suggests that such drugs will lack
deleterious effects on cognitive performance.
Exempla 20: Learning tests
Preliminary experiments using one of the more
potent synthetic arylalkylamine analogs, Compound 3, in
an in vivo learning paradigm demonstrate that these
drugs lack effects on cognitive performance. In this
test, rats were trained to alternate turning in a T maze
l0 for a food reward. MK-801 was included for comparison.
Test compounds were administered i.p. 15 min before
testing. Control animals made the correct choice about
BO% of the time. Increasing doses of MK-801
progressively decreased the number of correct choices
and this decrement in behavior was accompanied by
hyperactivity. In contrast, Compound 3 did not impair
the ability of the animals to make the correct choices
(p > 0.05). At the highest doses tested, Compound 3
caused some decrease in locomotor activity, exactly the
opposite effect observed with MK-801.
Although MK-801 decreased learning performance
in parallel with increases in locomotor activity, other
studies using different paradigms in rodents and
primates have shown a clear dissociation between the
effects on learning and locomotion. Thus, both
competitive and noncompetitive NMDA receptor antagonists
impair learning at doses which. do not cause any overt
change in motor behavior. This demonstrates that
conventional NMDA receptor antagonists impair learning
independently of other side effects. The results of the
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
107
T-maze assay demonstrate that Compound 3, and other
arylalkylamines, do not impair learning even at doses
that cause some decrease in locomotor activity.
One additional observation emerged from these
S learning tests. The animals' first response on the
second day of testing was random and was therefore not
dependent on the last response of the previous day's
testing. Control animals thus correctly made the first
choice about 50% of the time. MK-801 has no effect on
this first choice. However, animals administered
Compound 3 on the previous day made the first choice
correctly considerably more often. Unlike control
animals then, the animals treated with Compound 3
behaved as if they remembered the last choice of the
previous day.
In a second series of experiments, the effect
of Compound 4 on learning in the Morris water maze task
was evaluated. In this test, a hidden platform was
placed in a fixed location in a circular steel tank, and
submerged 2 cm below the surface of the water. Each rat
was given 3 trials per day with a 10 min intertrial
interval for 5 days. A trial was initiated by placing
the rat in the water, nose facing the wall of the tank,
at one of three predetermined starting locations. The
order of the start location was varied daily. Learning
was measured as a decrease in time required to swim to
the platform. If an animal failed to locate the
platform within 60 sec after the start of the trial, the
rat was hand-guided to it. The animals remained on the
platform for 10 sec before being removed from the tank.
Ten min after the last training trial on day 5, the
CA 02560002 2006-10-05
WO 97/46511 PCTI~JS96/20697
108
animals received a probe test. The platform was removed
for this 1 trial task and the animals were allowed to
swim for 60 sec to assess the spatial bias for the
platform location. Two measures were recorded from this
task: latency to first crossing the area where the
platform had been, and total number of crossings. A
total of 5 injections of Compound 4 were given to each
rat. In the first series of experiments, Compound 4 was
administered at IO mg/kg i.p. daily for 5 days. This
treatment regimen impaired learning; however, these
animals experienced significant weight loss and unusual
behavioral signs ("shivering," motor impairment,
difficulty in swimming) with repeated dosing of Compound
4. In a subsequent study, six animals received 1 mg/kg
i.p. for the first 4 days of training, while two animals
received 5 mg/kg i.p. during this period. On the last
day of training, both groups received 10 mg/kg. Neither
the 1 mg/kg nor the 5 mg/kg animals showed any
impairment in learning the location of the hidden
platform, nor did the final 10 mg/kg dose produce any
impairment in the ability of the animal to perform the
already learned task.
The results of these learning tasks are
encouraging. They suggest that arylalkylamines lack the
learning and memory deficits that typify other NMDA
receptor antagonists. In fact, there is a suggestion
that the arylalkylamines may even be nootropic (memory
enhancers).
CA 02560002 2006-10-05
WO 9'1146511 PCT/US96/20697
109
(c) Cardiovascular effects
In vivo studies with certain arylalkylamines
revealed a hypotensive effect of these compounds,
- especially at high doses. On the basis of these
results, a systematic study of the effects of
arylalkylamines on cardiovascular function was
performed.
Example 21: Ca'' channel inhibition
We have discovered that some of the
l0 arylalkylamines are quite potent inhibitors of
voltage-sensitive Ca2~ channels, specifically those
sensitive to inhibition by dihydropyridines (L-type
channels). Such effects on vascular smooth muscle would
be expected to dilate blood vessels and cause a drop in
blood pressure, thus producing hypotension.
The ability of arylalkylamines to inhibit
dihydropyridine-sensitive Cap' channels was examined in
cerebellar granule cells and a rat aortic smooth muscle
cell line, A,r5 cells. In cerebellar granule cells,
Compound 2 inhibited depolarization-induced increases in
[Ca2~]iat concentrations 100-fold higher than those
required to block responses to NMDA (ICso values of 24 p.M
and 161 nM, respectively). Overall, we have observed a
wide range of potencies against voltage-sensitive Caz'
channels that does not correlate with potency against
NMDA receptors. This strongly suggests that further
structure-activity work based on chemical modification
of the arylalkylamine molecule will lead to the
development of compounds that are very potent NMDA
antagonists with low potency against voltage-sensitive
Cap' channels . Indeed, Compound 1 (with an ICSO of 102 nM
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
110
against NMDA receptor-mediated responses in cerebellar
granule cells) is a relatively poor inhibitor of
voltage-sensitive Ca2' channels in cerebellar granule
cells (ICSO = 257 ,uM) and is virtually without effect on
voltage-sensitive Cap' influx in A,rS cells (ICso = 808
~.M ) .
Arylalkylamines are not, however,
indiscriminate blockers of voltage-sensitive Cap
channels. They do not, for example, affect
voltage-sensitive Ca2' channels in cerebellar Purkinje
cells (P-type channels) or those channels thought to be
involved in neurotransmitter release (N-channels). The
arylalkylamines that do block voltage-sensitive Ca2'
channels appear to target specifically L-type Ca2'
channels. Moreover, as mentioned above, there is a high
degree of structural specificity in this effect. For
example, one arylalkylamine is 57 times more potent than
another arylalkylamine in blocking Ca2' influx through
L-type channels, where the only structural difference
between the compounds is the presence or absence of a
hydroxyl group.
Example 22: Ia vivo cardiovascular studies
The arylalkylamines Compound 1 and Compound 2
produce moderate drops (20-40 mm Hg) in mean arterial
blood pressure (MABP) in anesthetized rats at doses
which are effective in the in vivo stroke models (10-30
mg/kg s.c.). The hypotensive effect of Compound 4 has
been evaluated in greater detail. Compound 4 elicited a
marked drop (40 mm Hg) in mean arterial pressure which
persisted for approximately 90-120 min when administered
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
111
at the dose of ZO mg/kg i.p.; it was in this same group
of rats that Compound 4 afforded significant
neuroprotection in the suture model of middle cerebral
artery occlusion (see Example 8 above). Similar results
were obtained in the rat study in which Compound 4
demonstrated neuroprotectant activity in the Rose Bengal
photothrombotic model of focal ischemic stroke (see
Example 8 above). Further studies using the pithed rat
preparation strongly suggest that the hypotensive
activity of Compound 4 is a peripherally mediated
effect. The hypotension and bradycardia produced by
Compound 4 was maintained in rats pretreated with
atropine, suggesting that these effects are not mediated
by a cholinergic mechanism. Similarly, Compound 4
elicited hypotension and bradycardia in chemically
sympathectomized rats (pretreated with a ganglionic
blocker), suggesting that these effects are not mediated
via the sympathetic nervous system.
On the basis of these findings, it is
anticipated that chemical efforts will minimize the
cardiovascular side effects by (1) enhancing the uptake
of arylalkylamine into the brain such that lower doses
are required for neuroprotection, and (2) increasing the
selectivity (potency ratio) of arylalkylamines for
receptor-operated CaZ' channels over voltage-sensitive
Ca~~ channels .
Example 23: Biological activity of Compound 19 and
analogs
Compounds 19 - 215 had high potencies against
NMDA-induced increases in [Ca2']i in rat cerebellar
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
112
granule cells grown in culture (Table 1). The
inhibitory effect of Compound 19 on responses to NMDA
was noncompetitive. Compounds 19 - 215 inhibited
['H]MK-801 binding in membranes prepared from rat
hippocampal and cortical tissue (Table 1).
Compound 19 possessed the following additional
biological activities. significant (p < 0.05 compared to
control) anticonvulsant activity against maximal
electroshock-induced seizures in mice following i.p.
administration (EDSo = 26.4 mg/kg and TDso (rotorod) -
43.8 mg/kg); significant anticonvulsant activity against
maximal electroshock-induced seizures in mice following
oral (p. o.) administration (EDSO = 35 mg/kg), but with
motor impairment at 30 mg/kg; significant analgesic
activity in the hot-plate and PBQ-induced writhing
assays at 16 mg/kg i.p.; no PCP-like stereotypic
behavior (hyperexcitability and head weaving) at
30 mg/kg i.p. in rats; no generalization to PCP in the
PCP discrimination assay in rats at doses up to the
behaviorally active dose of 30 mg/kg i.p. Compound 19
was significantly less potent in antagonizing increases
in (Ca2'];elicited by depolarizing concentrations of KC1
in rat cerebellar granule cells (ICso = 10.2 uM), and was
without effect on blood pressure when administered s.c.
in rats at doses up to 100 mg/kg. Compound 19, however,
did block the induction of LTP in rat hippocampal slices
when tested at 100 ~,cM.
Compound 20 possessed the following additional
biological activities: significant anticonvulsant
activity against maximal electroshock-induced seizures
in mice following i.p. administration (EDso = 20.1 mg/kg
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
113
and TDso (rotorod) - 20.6 mg/kg); no significant
anticonvulsant activity against maximal
electroshock-induced seizures in mice following oral
(p. o.) administration at doses up to 30 mg/kg, but with
motor impairment at 30 mg/kg; significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p. (EDso = 2.1 mg/kg and TDSp = 19.9 mg/kg)
and oral (EDso = 9.7 mg/kg and TDsfl = 21.8 mg/kg)
administration; significant anticonvulsant activity
against maximal electroshock-induced seizures in rats
following oral administration with an EDSo value of
33.64 mg/kg and an TDSO value of 55.87 mg/kg; an increase
in seizure threshold as indexed by the i.v. Metrazol
test in mice at the dose of 10 mg/kg i.p.; significant
neuroprotectant activity in a rat model of temporary
focal ischemia (a 51o reduction in the infarct volume
following the administration of two doses of 1 mg/kg
i.p., the first given immediately after middle cerebral
artery occlusion and the second given 6 hr later; a 430
reduction in the infarct volume following the
administration of two doses of 1 mg/kg i.p., the first
given 2 hr after middle cerebral artery occlusion (i.e.,
at the time of reperfusion) and the second given 6 hr
later); significant neuroprotectant activity (a 24%
reduction in the infarct volume) in a rat model of
permanent focal ischemia following the administration of
1 mg/kg i.p. at 30 min and again 4 hr post-occlusion;
significant neuroprotectant activity (a 50% reduction in
the infarct volume) in a rat photothrombotic model of
focal ischemia following the administration of 10 mg/kg
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
114
i.p. at 15 min, 3 hr, and again 6 hr post-occlusion; no
significant analgesic activity at the dose of 25 mg/kg
i.p. in the rat 52°C hot plate test or the rat 48°C tail
flick test; significant analgesic activity, not blocked
by the opiate receptor antagonist naloxone, in the rat
formalin test at the dose of 10 mg/kg i.p.; significant
analgesic activity, not blocked by naloxone, against
acetic acid-induced abdominal writhing in mice at the
dose of 10 mg/kg i.p.; no generalization to PCP in the
PCP discrimination assay in rats at doses up to the
behaviorally active dose of 10 mg/kg i.p.; no neuronal
vacuolization in rats when administered at doses of 10
and 30 mg/kg i.p.; no significant cardiovascular
activity in anesthetized rats at doses up to
15 ~.cmoles/kg i.v. or 10 mg/kg i.p.; no significant
cardiovascular activity in conscious beagle dogs at
doses of 0.3 or 1 mg/kg i.v. (60 sec bolus injection);
transient increases in mean arterial pressure and heart
rate in conscious beagle dogs at the dose of 3 mg/kg
i.v., with larger magnitude and longer duration effects
seen at the dose of 10 mg/kg i.v. (60 sec bolus
injection); increased motor activity, agitation and
anxiousness, slight tremors, licking of the mouth,
whining,and urination in conscious beagle dogs at the
dose of 3 mg/kg i.v. (60 sec bolus injection); dilated
pupils, whole body tremors, incoordination, licking of
the mouth, salivation, panting, rapid blinking of the
eyes, whining, anxiousness, seizures, and death in
conscious beagle dogs at the dose of 10 mg/kg i.v. (60
sec bolus injection); no behavioral effects in conscious
male NMRI mice at the doses of 2 and 4 mg/kg i.p.;
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
115
excitation and increased reactivity to touch in
conscious male NMRI mice at the dose of 8 mg/kg i.p.;
excitation, Straub tail, tremor, stereotypies,
hypothermia, and mydriasis in conscious male NMRI mice
at the doses of 16 and 32 mg/kg i.p.; convulsions and
death in conscious male NMRI mice at the dose of
64 mg/kg i.p.; convulsions and death in conscious male
NMRI mice at the doses of 128 and 256 mg/kg i.p; no
behavioral effects in conscious male Wistar rats at the
dose of 2 mg/kg i.v.; excitation, stereotypies,
increased reactivity to touch, increased muscle tone,
and tremor in conscious male Wistar rats at doses
ranging from 4 to 16 mg/kg i.v.; Straub tail,
convulsions, and death in conscious male Wistar rats at
the dose of 32 mg/kg i.v.
Compound 21 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p. administration (EDso = 3.41 mg/kg and TDso
(motor impairment= 15.3 mg/kg).
Compound 33 (an enantiomer of Compound 21)
possessed the following additional biological
activities: significant anticonvulsant activity against
sound-induced seizures in a genetically susceptible
mouse model of reflex epilepsy (Frings mice) following
i.p. administration (EDso = 4.6 mg/kg and TDSO (motor
impairment) - 27.8 mg/kg); significant anticonvulsant
activity against maximal electroshock-induced seizures
in rats following oral administration at the dose of
25 mg/kg, with no motor toxicity apparent at this dose;
CA 02560002 2006-10-05
WO 97/46511 PCT/fJS96/20697
116
significant neuroprotectant activity in a rat model of
focal ischemic stroke following i.p. administration of
2 mg/kg 30 min prior to vessel occlusion and 2 mg/kg
3 hr post-occlusion; no significant analgesic activity
at the dose of 25 mg/kg i.p. in the rat 52°C hot plate
test or the rat 48°C tail flick test; significant
analgesic activity in a rat model of chronic neuropathic
pain following i.th. administration of doses ranging
from 15 to 80 ~.cg; significant analgesic activity in a
rat model of chronic neuropathic pain following i.p.
administration of doses of 3-10 mg/kg; no neuronal
vacuolization when administered to rats at the dose of
30 mg/kg i.p.; nv significant cardiovascular activity in
anesthetized rats at doses up to 3 mg/kg i.v.; no
significant cardiovascular activity in conscious beagle
dogs at the dose of 0.3 mg/kg i.v. (60 sec bolus
injection); transient increases in mean arterial
pressure in conscious beagle dogs at the dose of 1 mg/kg
i.v., with larger magnitude and longer duration effects
seen at the doses of 3 and 10 mg/kg i.v. (60 sec bolus
injection); a transient increase in heart rate in
conscious beagle dogs at the dose of 10 mg/kg i.v.
(60 sec bolus injection); licking of the mouth in
conscious beagle dogs at the dose of 3 mg/kg i.v.
(60 sec bolus injection); dilated pupils, whole body
tremors, incoordination, licking of the mouth,
salivation, and panting in conscious beagle dogs at the
dose of 10 mg/kg i.v. (60 sec bolus injection); no
significant drug-induced changes in the ECG in conscious
beagle dogs at doses up to 10 mg/kg i.v. (60 sec bolus
injection); no behavioral effects in conscious male NMRI
CA 02560002 2006-10-05
WO 97/46511 PCTNS96/20697
117
mice at the doses of 2 and 4 mg/kg i.p.; excitation,
increased reactivity to touch, and hypothermia in
conscious male NMRI mice at the dose of 8 mg/kg i.p.;
excitation, Straub tail, tremor, jumping, stereotypies,
hypothermia, and mydriasis in conscious male NMRI mice
at the doses of 16 and 32 mg/kg i.p.; convulsions in
conscious male NMRI mice at the dose of 64 mg/kg i.p.;
convulsions and death in conscious male NMRI mice at the
doses of 128 and 256 mg/kg i.p.
Compound 34 (an enantiomer of Compound 21)
possessed the following additional biological
activities: significant anticonvulsant activity against
sound-induced seizures in a genetically susceptible
mouse model of reflex epilepsy (Frings mice) following
i.p. administration (EDSO = 22 mg/kg and TDSO (motor
impairment) between 10 and 15 mg/kg); hyperthermia in
conscious male NMRI mice at the dose of 2 mg/kg i.p.; no
behavioral effects in conscious male L~1MRI mice at the
dose of 4 mg/kg i.p.; excitation, increased reactivity
to touch, and hypothermia in conscious male NMRI mice at
the dose of 8 mg/kg i.p.; excitation, Straub tail,
tremor, jumping, stereotypies, hypothermia, and
mydriasis in conscious male NMRI mice at the doses of 16
and 32 mg/kg i.p.; convulsions in conscious male NMRI
mice at the dose of 64 mg/kg i.p.; convulsions and death
in conscious male NMRI mice at the doses of 128 and
256 mg/kg i.p.
Compound 22 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
CA 02560002 2006-10-05
WO 97146511 PCT/I1S96/20697
118
following i.p. (EDso = 4.9 mg/kg and TDSO (inverted grid)
- 26.8 mg/kg) and oral (EDSO = 5.1 mg/kg and LDso =
18.3 mg/kg) administration; and no significant
cardiovascular activity in anesthetized rats at doses up
to 15 ~moles/kg (4.47 mg/kg) i.v.
Compound 50 (an enantiomer of Compound 22)
possessed the following additional biological
activities: significant anticonvulsant activity against
sound-induced seizures in a genetically susceptible
l0 mouse model of reflex epilepsy (Frings mice) following
i.p. administration (EDSO = 2.7 mg/kg and TDSO (motor
impairment) = 17.4 mg/kg); significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following p.o. administration (EDSp = 9.0 mg/kg and TDso
(motor impairment) - 18.9 mg/kg); significant
anticonvulsant activity against maximal
electroshock-induced seizures in rats following oral
administration with EDSO = 28 mg/kg and TDso = 20 mg/kg;
significant neuroprotectant activity in a rat model of
focal ischemic stroke following i.p. administration of
2 mg/kg 30 min prior to vessel occlusion and 2 mg/kg
3 hr post-occlusion; no significant analgesic activity
at the dose of 25 mg/kg i.p. in the rat 52°C hot plate
test or the rat 48°C tail flick test; and no significant
cardiovascular activity in anesthetized rats at doses up
to 5 mg/kg i.v.
Compound 51 (an enantiomer of Compound 22)
possessed the following additional biological
activities: significant anticonvulsant activity against
sound-induced seizures in a genetically susceptible
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
119
mouse model of reflex epilepsy (Fringe mice) following
i.p. administration (EDSo = 9.1 mg/kg and TDSO (motor
impairment) - 13.6 mg/kg).
Compound 24 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Fringe mice)
following i.p. administration (EDSa = S mg/kg and TDSo
(motor impairment) - 16 mg/kg); significant
anticonvulsant activity against maximal
electroshock-induced seizures in rats following oral
administration with EDSO = 46 mg/kg and TDso = S1 mg/kg;
no significant neuroprotectant activity in a rat model
of focal ischemic stroke following i.p. administration
of 2 mg/kg 30 min prior to vessel occlusion and 2 mg/kg
3 hr post-occlusion; and no significant cardiovascular
activity in anesthetized rats at doses up to l0 mg/kg
i.v.
Compound 25 possessed the following additional
biological activities: significant anticonvulsant
activity against maximal electroshock-induced seizures
in mice following i.p, administration with an EDso =
12.47 mg/kg and a TDSO = 32.18 mg/kg; significant
anticonvulsant activity against maximal
electroshock-induced seizures in rats following oral
administration with an EDSO = 46.43 mg/kg and a TDSo
between 163 and 326 mg/kg.
Compound 31 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Fringe mice)
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
120
following i.p. administration (EDSO = 6 mg/kg and TDSo
(motor impairment) between to and 20 mg/kg).
Compound 46 possessed the follawing additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p, administration (EDso = 25 mg/kg and TDso
(motor impairment) between 18 and 21 mg/kg); and no
significant neuroprotectant activity in a rat model of
focal ischemic stroke following i.p. administration of
2 mg/kg 30 min prior to vessel occlusion and 2 mg/kg 3
hr post-occlusion.
Compound 57 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p. administration (EDso = 1 mg/kg and TDso
(motor impairment) between~6 and s mg/kg).
Compound 58 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p. administration (EDSo = 0.9 mg/kg and TDso
(motor impairment) - 14.5 mg/kg); no significant
neuroprotectant activity in a rat model of focal
ischemic stroke following i.p. administration of 2 mg/kg
min prior to vessel occlusion and 2 mg/kg 3 hr
post-occlusion; and no significant cardiovascular
activity in anesthetized rats at doses up to 2 mg/kg
30 i.v.
CA 02560002 2006-10-05
WO 97146511 PCTIUS96120697
121
Compound 59 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of reflex epilepsy (Frings mice)
following i.p. administration (EDso = 2.7 mg/kg and TDso
(motor impairment) - 7.8 mg/kg); a reduction in seizure
threshold as indexed by the i.v. Metrazol test in mice
at the dose of 11.7 mg/kg i.p.; no significant
neuroprotectant activity in a rat model of focal
ischemic stroke following i.p. administration of 2 mg/kg
30 min prior to vessel occlusion and 2 mg/kg 3 hr
post-occlusion; and no significant cardiovascular
activity in anesthetized rats at doses up to 10 mg/kg
i.v.
Compound 60 possessed the following additional
biological activities: significant anticonvulsant
activity against sound-induced seizures in a genetically
susceptible mouse model of 'reflex epilepsy (Frings mice)
following i.p. administration (EDso = 4.4 mg/kg and TDS~
(motor impairment) - 9.2 mg/kg); significant
anticonvulsant activity against sound-induced seizures
in a genetically susceptible mouse model of reflex
epilepsy (Frings mice) following oral administration
(EDSO = 10 mg/kg and TDSO (motor impairment) -
25.6 mg/kg); significant anticonvulsant activity against
maximal electroshock-induced seizures in mice following
i.p. administration (EDSO = 8.17 mg/kg and TDso (rotorod)
- 17.30 mg/kg); no effect on seizure threshold as
indexed by the i.v. Metrazol test in mice at the doses
of 1 and 4 mg/kg i.p.; a reduction in seizure threshold
as indexed by the i.v. Metrazol test in mice at the
CA 02560002 2006-10-05
WO 97146511 PCT/LTS96/20697
122
doses of 8 and 17 mg/kg i.p.; significant
neuroprotectant activity in a rat model of temporary
focal ischemic stroke following i.p. administration of
2 mg/kg 30 min prior to vessel occlusion and 2 mg/kg
3 hr post-occlusion; significant neuroprotectant
activity in a rat madel of temporary focal ischemic
stroke following i.p. or i.v. administration of 1 mg/kg
2 hr and again 8 hr post-occlusion; significant
neuroprotectant activity in a rat model of temporary
focal ischemic stroke following i.v. administration of
1 mg/kg 2 hr post-occlusion; no significant
neuroprotectant activity in a rat photothrombotic model
of focal ischemia following the administration of
10 mg/kg i.p. at 15 min, 3 hr, and again 6 hr
post-occlusion; no neuronal vacuolization when
administered at doses of 20 mg/kg i.p. or l0 mg/kg i.v.;
no significant cardiovascular activity in conscious
beagle dogs at the dose of 0.3 mg/kg i.v. (60 sec bolus
injection); transient increases in mean arterial
pressure in conscious beagle dogs at the doses of 1 and
3 mg/kg i.v., with larger magnitude and longer duration
effects seen at the dose of 10 mg/kg i.v. (60 sec bolus
injection); transient increases in heart rate in
conscious beagle dogs at the doses of 3 and 10 mg/kg
i.v. (60 sec bolus injection); no significant changes in
the ECG in conscious beagle dogs at doses ranging from
0.3 to 10 mg/kg i.v. (60 sec bolus injection); no
significant behavioral effects in conscious beagle dogs
at the doses of 0.3 and 1 mg/kg i.v. (60 sec bolus
injection); a slight increase in respiratory rate in
conscious beagle dogs at the dose of 3 mg/kg i.v.
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
123
(60 sec bolus injection); dilated pupils, whole body
tremors, salivation, and urination in conscious beagle
dogs at the dose of 10 mg/kg i.v. (60 sec bolus
injection); no significant behavioral effects in
conscious male Wistar rats at doses up to 4 mg/kg i.v.;
excitation, stereotypies, increased reactivity to touch,
increased muscle tone, and tremor in conscious male
Wistar rats at the dose of 8 mg/kg i.v.; Straub tail,
convulsions, and death in conscious male wistar rats at
the dose of 16 mg/kg i.v.
Compound 119 possessed the following
additional biological activities: significant
anticonvulsant activity against sound-induced seizures
in a genetically susceptible mouse model of ref lex
epilepsy (Frings mice) following i.p. administration
with an EDSp = 7.0 mg/kg and TD5o (motor impairment) -
26.3 mg/kg.
Compound 120 possessed the following
additional biological activities: significant
anticonvulsant activity against sound-induced seizures
in a genetically susceptible~mouse model of reflex
epilepsy (Frings mice) following i.p. administration
with an EDSp = 4.77 mg/kg and TDSp (motor impairment)
between 20 and 30 mg/kg.
Compound 122 possessed the following
additional biological activities: significant
anticonvulsant activity against sound-induced seizures
in a genetically susceptible mouse model of reflect
epilepsy (Frings mice) following i.p. administration
with an EDSp = 4.7 mg/kg and TDgp (motor impairment) -
15.3 mg/kg.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
124
Compound 138 possessed the following
additional biological activities: significant
anticonvulsant activity against maximal electroshock-
induced seizures in mice following i.p. administration
with an EDSp = 51.9 mg/kg and TDSp (motor impairment) -
100.7 mg/kg.
Compound 151 possessed the following
additional biological activities: significant
anticonvulsant activity against maximal electroshock-
induced seizures in mice following i.p. administration
with an EDSp = 36.5 mg/kg and TDSp (motor impairment) -
108.4 mg/kg; a significant increase in seizure threshold
as indexed by the i.v. Metrazol test in mice at the
doses of 36.5 and 108 mg/kg i.p.
Compound 156 possessed the following
additional biological activites: significant
anticonvulsant activity against sound-induced seizures
in a genetically susceptible mouse model of reflect
epilepsy (Frings mice) following i.p. administration
with an EDSp = 5.0 mg/kg and TDSQ (motor impairment) -
17.4 mg/kg.
Taken together, the results obtained with
these simplified synthetic arylalkylamines suggest that
such simplified molecules do not interact specifically
with the arylalkylamine binding site on
receptor-operated Ca2' channels as do Compounds 1, 2 and
3. Specifically, Compounds 19 - 215 bind to the site
labeled by ['H]MK-801 at concentrations ranging
approximately 1 to 400-fold higher than those which
antagonize the function of the NMDA receptor-ionophore
complex. The fact that Compounds 19 - 215 at
CA 02560002 2006-10-05
WO 97/46511 PCT/US96l20697
125
therapeutic doses do not generally produce PCP-like
stereotypic behavior, substitute for PCP in drug
discrimination assays, or elicit neuronal vacuolization
suggests, however, that such compounds might be useful
S either as lead compounds or drug candidates for
neurological disorders and diseases. It has been
reported that compounds which bind with low affinity
(relative to MK-801) to the site labeled by ('H]MK-801
might possess therapeutic utility and possess a more
favorable side effect profile than that possessed by a
high affinity antagonist such as MK-801 itself
(Rogawski, Therapeutic potential of excitatory amino
acid antagonists: channel blockers and
2,3-benzodiazepines. Trends Pharmacol. Sci. 14: 325,
1993). The low affinity of certain compounds within the
group of Compounds 19 - 215 (relative to MK-801) for the
site labeled by ['H]MK-801 may place these compounds into
this general class of low affinity noncompetitive
antagonists.
Identification of a novel modulatory site on
receptor-operated calcium channels
Having identified arylalkylamines which have
therapeutically useful properties as defined above,
compounds can now be identified which act at the
critical arylalkylamine binding site on
receptor-operated Ca" channels, such as those present
within NMDA, AMPA and nicotinic cholinergic
receptor-ionophore complexes.
Examples of suitable tests now follow:
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
126
Example 24: Radioligand binding in rat cortex or
cerebellum.
The following assay can be utilized as a high
throughput assay to screen product libraries (e. g.,
natural product libraries and compound files at major
pharmaceutical companies) to identify new classes of
compounds with activity at this unique arylalkylamine
site. These new classes of compounds are then utilized
as chemical lead structures for a drug development
program targeting the arylalkylamine binding site on
receptor-operated Caz' channels. The compounds
identified by this assay offer a novel therapeutic
approach to treatment of neurological disorders or
diseases. Examples of such compounds include those
provided in the generic chemical formulae above.
Routine experiments can be performed to identify those
compounds having the desired activities.
Rat brain membranes are prepared according to
the method of Williams et al. (Effects of polyamines on
the binding of ['H]MK-801 to the NMDA receptor:
Pharmacological evidence for the existence of a
polyamine recognition site. Mo.Iec. Pharmacol. 36: 575,
1989) with the following alterations: Male
Sprague-Dawley rats (Harlan Laboratories) weighing
100-200 g are sacrificed by decapitation. The cortex or
cerebellum from 20 rats are cleaned and dissected. The
resulting brain tissue is homogenized at 4°C with a
polytron homogenizer at the lowest setting in 300 ml
0.32 M sucrose containing 5 mM K-EDTA (pH 7.0). The
homogenate is centrifuged for 10 min at 1,000 x g and
the supernatant removed and centrifuged at 30,000 x g
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
127
for 30 minutes. The resulting pellet is resuspended in
250 ml 5 mM K-EDTA (pH 7.0) stirred on ice for 15 min,
and then centrifuged at 30,000 x g for 30 minutes. The
pellet is resuspended in 300 ml 5 mM K-EDTA (pH 7.0) and
incubated at 32°C for 30 min. The suspension is then
centrifuged at 100,000 x g for 30 min. Membranes are
washed by resuspensi.on in 500 ml 5 mM K-EDTA (pH 7.0),
incubated at 32°C for 30 min, and centrifuged at
100,000 x g for 30 minutes. The wash procedure,
including the 30 min incubation, is repeated. The final
pellet is resuspended in 60 ml 5 mM K-EDTA (pH 7.0) and
stored in aliquots at -80°C. The extensive washing
procedure utilized in this assay was designed in an
effort to minimize the concentrations of glutamate and
glycine (co-agonists at the NMDA receptor-ionophore
complex) present in the membrane preparation.
To perform a binding assay with
['H]arylalkylamine, aliquots of SPMs (Synaptic Plasma
Membranes) are thawed, resuspended in 30 mls of 30 mM
EPPS/1mM K-EDTA, pH 7.0, and centrifuged at 100,000 x g
for 30 minutes. SPMs are resuspended in buffer A (30 mM
EPPS/1 mM K-EDTA, pH 7.0). The ['H]arylalkylamine is
added to this reaction mixture. Binding assays are
carried out in polypropylene test tubes. The final
incubation volume is 500 ~cl. Nonspecific binding is
determined in the presence of 100 ~M nonradioactive
arylalkylamine. Duplicate samples are incubated at 0°C
for 1 hour. Assays are terminated by the addition of
3 ml of ice-cold buffer A, followed by filtration over
glass-fiber filters (Schleicher & Schuell No. 30) that
are presoaked in 0.33% polyethyleneimine (PEI). The
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
128
filters are washed with another 3 x 3 ml of buffer A,
and radioactivity is determined by scintillation
counting at an efficiency of 35-40% for 'H.
In order to validate the above assay, the
following experiments are also performed:
(a) The amount of nonspecific binding of the
[3H)arylalkylamine to the filters is determined by
passing 500 ~.1 of buffer A containing various
concentrations of ['H)arylalkylamine through the
. presoaked glass-fiber filters. The filters are washed
with another 4 x 3 ml of buffer A, and radioactivity
bound to the filters is determined by scintillation
counting at an efficiency of 35-40% for 'H. In filters
that are not pretreated with 0.33% PEI, it was found
that 87% of the 'H-ligand was bound to the filter.
Presoaking with 0.33% PEI reduces the nonspecific
binding to 0.5 - 1.0% of the total ligand added.
(b) A saturation curve is constructed by
resuspending SPMs in buffer A. The assay buffer
(500 ~cl) contains 60 ~cg of protein. Concentrations of
['H)arylalkylamine are used, ranging from 1.0 nM to
400 ~M in half-log units. A saturation curve is
constructed from the data, and an apparent KD value and
B"~x value determined by Scatchard analysis (Scatchard,
The attractions of proteins for small molecules and
ions. Ann. N.Y. Acad. Sci. 51: 660, 1949). The
cooperativity of binding of the ['H)arylalkylamine is
determined by the construction of a Hill plot (Hill, A
new mathematical treatment of changes of ionic
concentrations in muscle and nerve under the action of
CA 02560002 2006-10-05
WO 97/46511 PCTJUS96/20697
129
electric currents, with a theory to their mode of
excitation. J. Physiol. 40: 190, 19101.
(c) The dependence of binding on protein
(receptor) concentration is determined by resuspending
SPMs in buffer A. The assay buffer (500 /,c1) contains a
concentration of ['H]arylalkylamine equal to its Ko value
and increasing concentrations of protein. The specific
binding of ['H]arylalkylamine should be linearly related
to the amount of protein (receptor) present.
(d) The time course of ligand-receptor
binding is determined by resuspending SPMs in buffer A.
The assay buffer (500 ~cl) contains a concentration of
['H]arylalkylamine equal to its Kp value and 100 ~g of
protein. Duplicate samples are incubated at 0°C for
varying lengths of time; the time at which equilibrium
is reached is determined, and this time point is
routinely used in all subsequent assays.
(e) The pharmacology of the binding site can
be analyzed by competition experiments. In such
experiments, the concentration of ['H]arylalkylamine and
the amount of protein are kept constant, while the
concentration of test (competing) drug is varied. This
assay allows for the determination of an ICso and an
apparent Kp for the competing drug (Cheng and Prusoff,
Relationship between the inhibition constant (K;) and the
concentration of inhibitor which causes 50 percent
inhibition (ICSo) of an enzymatic reaction. J. Biochem.
Pharmacol. 22: 3099, 1973). The cooperativity of
binding of the competing drug is determined by Hill plot
analysis.
CA 02560002 2006-10-05
WO 97/46511 PCTILJS96/20697
130
Specific binding of the [3H]arylalkylamine
represents binding to a novel site on receptor-operated
Ca2' channels such as those present within NMDA-, AMPA-
and nicotinic cholinergic receptor-ionophore complexes.
As such, other arylalkylamines should compete with the
binding of ['H]arylalkylamine in a competitive fashion,
and their potencies in this assay should correlate with
their inhibitory potencies in a functional assay of
receptor-operated Ca2' channel antagonism (e. g.,
l0 inhibition of NMDA receptor-induced increases in [Ca2");
in cultures of rat cerebellar granule ce-.~ls).
Conversely, compounds which have activity at the other
known sites on receptor-operated Ca2' channels (e. g.,
MK-801, Mg2', polyamines) should not displace
[3H]arylalkylamine binding in a competitive manner.
Rather, complex allosteric modulation of
[3H]arylalkylamine binding, indicative of noncompetitive
interactions, might be expected to occur. In
preliminary experiments, MK-801 did not displace
['H]arylalykylamine binding at concentrations up to
10 0 E.cM .
(f) Studies to estimate the dissociation
kinetics are performed by measuring the binding of
['H]arylalkylamine after it is allowed to come to
equilibrium (see (d) above), and a large excess of
nonradioactive competing drug is added to the reaction
mixture. Binding of the ['H]arylalkylamine is then
assayed at various time intervals. With this assay, the
association and dissociation rates of binding of the
['H) arylalkylamine are determined (Titeler, Mu1 tiple
Dopamine Receptors: Receptor Binding Studies in
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
131
Dopamine Pharmacology. Marvel Dekker, Inc., New York,
1983). Additional experiments involve varying the
reaction temperature (0°C to 37°C) in order to
understand the temperature dependence of this parameter.
Example 25: Radioligasd binding in cerebellar granule
cells
Primary cultures of cerebellar granule neurons
are obtained from 8-day-old rats and plated onto squares
of Aclar plastic coated with poly-L-lysine. The plastic
l0 squares are placed in 24-well culture plates, and
approximately 7.5 X 105 granule cells are added to each
well. Cultures are maintained in Eagles' medium
(HyClone Laboratories) containing 25 mM KC1, 10°s fetal
calf serum (HyClone Laboratories), 2 mM glutamine,
100 ~eg/ml gentamicin, 50 U/ml penicillin, and 50 ,ug/ml
streptomycin at 37°C in a humid atmosphere of 5o COz in
air for 24 hr before the addition of cytosine
arabinoside (10 ~cM, final). No changes of culture medium
are made until the cells are used for receptor binding
studies 6-8 days after plating.
To perform a binding assay with
('H]arylalkylamine, the reaction mixture consists of
200 ,u1 of buffer A (20 mM K-HEPES, 1 mM K-EDTA, pH 7.0)
in each well of the 24-well plate. The
['H]arylalkylamine is added to this reaction mixture.
Nonspecific binding is determined in the presence of
100 ~cM nonradioactive arylalkylamine. Triplicate
samples are incubated at 0°C for 1 hour. Assays are
terminated by manually scraping the cells off the Aclar
squares and placing them into polypropylene test tubes.
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
132
The membranes prepared from whole cells in this manner
are suspended in 10 ml of ice-cold buffer A, and
filtered over glass-fiber filters (Schleicher & Schuell
No. 30) that are presoaked in 0.33% PEI. The filters
are washed with another 3 x 3 ml of buffer A, and
radioactivity on the filters is determined by
scintillation counting at an efficiency of 35-40% for 'H.
The assay may be terminated by centrifugation rather
than filtration in order to minimize nonspecific.
binding.
Specific experiments to characterize and
validate the assay are performed essentially as above,
except that cells are used in place of membranes for the
initial binding. The binding assay allows for the
determination of an ICSo value and an apparent Kp for the
competing drug as described by Scatchard analysis (The
attractions of proteins for small molecules and ions.
Ann. N.Y. Acad. Sci. 51: 660, 1949). Cooperativity of
binding of the competing drug is determined by Hill plot
analysis (A new mathematical treatment of changes of
ionic concentrations in muscle and nerve under the
action of electric currents, with a theory to their mode
of excitation. J. Physiol. 40: 190, 1910). The
specific binding of the ['H]arylalkylamine represents
binding to a novel site on receptor-operated calcium
channels.
Example 26: Recombinant receptor binding assay
The following is one example of a rapid
screening assay for useful compounds of this invention.
In this assay, a cDNA or gene clone encoding the
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
133
arylalkylamine binding site (receptor) from a suitable
organism such as a human is obtained using standard
procedures. Distinct fragments of the clone are
expressed in an appropriate expression vector to produce
the smallest polypeptide(s) obtainable from the receptor
which retain the ability to bind Compound 1, Compound 2
or Compound 3. In this way, the polypeptide(s) which
includes the novel arylalkylamine receptor for these
compounds can be identified. Such experiments can be
facilitated by utilizing a stably transfected mammalian
cell line (e.g., HEK 293 cells) expressing the
arylalkylamine receptor.
Alternatively, the arylalkylamine receptor can
be chemically reacted with chemically modified
Compound 1, Compound 2 or Compound 3 in such a way that
amino acid residues of the arylalkylamine receptor which
contact (or are adjacent to) the selected compound are
modified and thereby identifiable. The fragments) of
the arylalkylamine receptor containing those amino acids
which are determined to interact with Compound 1,
Compound 2 or Compound 3 and are sufficient for binding
to said molecules, can then be recombinantly expressed,
as described above, using a standard expression
vector(s).
The recombinant polypeptide(s) having the
desired binding properties can be bound to a solid phase
support using standard chemical procedures. This solid
phase, or affinity matrix, may then be contacted with
Compound 1, Compound 2 or Compound 3 to demonstrate that
3o those compounds can bind to the column, and to identify
conditions by which the compounds may be removed from
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
134
the solid phase. This procedure may then be repeated
using a large library of compounds to determine those
compounds which are able to bind to the affinity matrix,
and then can be released in a manner similar to
Compound 1, Compound 2 or Compound 3. However,
alternative binding and release conditions may be
utilized in order to obtain compounds capable of binding
under conditions distinct from those used for
arylalkylamine binding (e. g., conditions which better
mimic physiological conditions encountered especially in
pathological states). Those compounds which do bind can
thus be selected from a very large collection of
compounds present in a liquid medium or extract.
Once compounds able to bind to the
arylalkylamine binding polypeptide(s) described above
are identified, those compounds can then be readily
tested in the various assays described above to
determine whether they, or~simple derivatives thereof,
are useful compounds for therapeutic treatment of
neurological disorders and diseases described above.
In an alternate method, native arylalkylamine
receptor can be bound to a column or other solid phase
support. Those compounds which are not competed off by
reagents which bind other sites on the receptor can then
be identified. Such compounds define novel binding
sites on the receptor. Compounds which are competed off
by other known compounds thus bind to known sites, or
bind to novel sites which overlap known binding sites.
Regardless, such compounds may be structurally distinct
from known compounds and thus may define novel chemical
classes of agonists or antagonist which may be useful as
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
135
therapeutics. In summary, a competition assay can be
used to identify useful compounds of this invention.
Example 27: Patch-clamp electrophysiology assay
The following assay is performed for selected
compounds identified in the above-mentioned radioligand
binding assays as interacting in a highly potent and
competitive fashion at the novel arylalkylamine binding
site on receptor-operated Caz~ channels, such as those
present in NMDA-, AMPA- or nicotinic cholinergic
receptor-ionophore complexes. This patch-clamp assay
provides additional relevant data about the site and
mechanism of action of said previously selected
compounds. Specifically, the following pharmacological
and physiological properties of the compounds
interacting at the arylalkylamine binding site are
determined, utilizing the NMDA receptor-ionophore
complex as an example of receptor-operated Caz' channels:
potency and efficacy at blocking NMDA receptor-mediated
ionic currents, the noncompetitive nature of block with
respect to glutamate and glycine, use-dependence of
action, voltage-dependence of action, both with respect
to onset and reversal of blocking, the kinetics of
blocking and unblocking (reversal), and open-channel
mechanism of blocking. Such data confirm that the
compounds interacting at the arylalkylamine binding site
retain the unique biological profile of the
arylalkylamines, and do not have their primary activity
at the known sites on the NMDA receptor-ionophore
complex (glutamate binding site, glycine binding site,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
136
MK-801 binding site, Mgr' binding site, Znz' binding site,
sigma binding site, polyamine binding site).
Patch-clamp recordings of mammalian neurons
(hippocampal, cortical, cerebellar granule cells) are
carried out utilizing standard procedures (Donevan et
al., Arcaine blocks N-methyl-D-aspartate receptor
responses by an open channel mechanism: whole-cell and
single-channel recording studies in cultured hippocampal
neurons. Molec. Pharmacol. 41: 727, 1992; Rock and
Macdonald, Spermine and related polyamines produce a
voltage-dependent reduction of NMDA receptor
single-channel conductance. Molec. Pharmacol. 42: 157,
1992) .
Alternatively, patch-clamp experiments can be
performed on Xenopus oocytes or on a stably transfected
mammalian cell line (e. g., HEK 293 cells) expressing
specific subunits of receptor-operated Cap' channels. In
this manner, for example, potency and efficacy at
various glutamate receptor subtypes (e. g., NMDAR1,
NMDAR2A through NMDAR2D, GluR1 through GluR4) can be
determined. Further information regarding the site of
action of the arylalkylamines on these glutamate
receptor subtypes can be obtained by using site-directed
mutagenesis.
Example 28: Synthesis of arylalkylamines
Arylalkylamines such as Compound 1, Compound 2
and Compound 3 are synthesized by standard procedures
(Jasys et al., The total synthesis of argiotoxins 636,
659 and 673. Tetrahedron Lett. 29: 6223, 1988; Nason et
al., Synthesis of neurotoxic Nephila spider venoms:
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
137
NSTX-3 and JSTX-3. Tetrahedron Lett. 30: 2337, 1989).
Specific examples of syntheses of arylalkylamine analogs
4-18 are provided in co-pending application U.S. Serial
No. 08/485,038, filed June 7, 1995, and co-pending
International Patent Application No. PCT/US94/12293,
published as W095/21612,filed October 26, 1994, hereby
incorporated by reference herein in their entirety.
Example 29: Synthesis of simplified arylalkylamines
Synthesis of Compound 20 was accomplished as
follows.
A solution of sodium hydride (1.21 g, 50 mmol)
in dimethoxyethane was treated with diethyl cyanomethyl-
phosphonate (8.86 g, 50 mmol) and the reaction stirred
4 hr at room temperature. To this was added
3,3~-difluorobenzophenone (10 g, 46 mmol) in DME. The
reaction was stirred 24 hr at room temperature, quenched
with HzO, and partitioned between diethyl ether and
water. The ether fraction was dried over NaZS04 and
concentrated. GC/MS of this material showed 90% of the
2o product A and 10% starting benzophenone.
A solution of this material in ethanol with a
catalytic amount of Pd(OH)2 was hydrogenated at 55 psi
hydrogen for 4 hr at room temperature. The reaction was
filtered and the catalyst washed with ethanol (3x). The
filtrate and ethanol washes were combined and
concentrated. GC/MS of this material showed 90% of the
product B and 10% of the starting benzophenone.
A solution of this material in THF was treated
with 70 ml 1 M BZH6 (70 mmol) in THF and refluxed 1 hr.
After cooling the reaction was treated with 6 N HC1
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
138
(50 ml) and refluxed an additional hour. After cooling
the reaction was basified to pH 14 with 10 N NaOH and
equilibrated with ether. The ether layer was removed
and washed with 10% HC1 (3x). The acidic washes were
combined, basified to pH 14 with 10 N NaOH and extracted
with dichloromethane (3x). The organic washes were
combined, dried over Na2S04, and concentrated to yield an
oil. GC/MS of this material showed 100°s Compound 20.
GC/EI-MS (R~ =7.11 min) m/z (relative intensity) 247 (M',
31), 230 (100), 215 (30), 201 (52), 183 (63), 134 (23),
121 (16), 101 (21), 95 (15), 77 (15). This material in
diethyl ether was filtered and treated with 35 ml 1 M
HC1 in ether. The precipitate was collected, dried, and
recrystallized from water-ethanol to afford 1.045 g of
Compound 20, as the hydrochloride salt. 'H-NMR (CDC1,) d
8.28 (3H, br s), 7.28-7.17 (2 H, m), 7.02-6.86 (6 H, m),
4.11 (1H, t, J=8 Hz), 2.89 (2H, br t, J=8 Hz), 2.48 (2H,
br t, J=7 Hz) ; i'C-NMR (CDCi,) d 164.6, 161.3, 144.8,
144.7, 130.4, 130.3, 123.3, 123.2, 114.7, 114.5, 114.1,
113.8, 47.4, 38.4, 32.7.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
139
I ~ ~ ~ off I ~ on
p ~ o Lj' (H~CCN)~ F ~ CN -H,/Nj~AI F i NII;
i i
W ~ I ~ I
A p
HCI/G j ~ ~ NHyCt HZ/catalyst F I ~ NFI,CI
i
C Compound 20
(hydrochloride saltf
Synthesis of Compound 21, Compound 33 and
Compound 34 was accomplished as follows.
A 100 ml round-bottomed flask equipped with
stir bar, septa, and argon source was charged with
Compound 1 (2.43 g, 10 mmol) in 30 ml THF. The
solution was cooled to -78°C and treated dropwise with
11 ml lithium bis(trimethylsilyl)amide (1M in THF)
(11 mmol). The reaction was stirred at -78°C for 30 min
and treated dropwise with excess iodomethane (3.1 ml,
50 mmol). The reaction was stirred 30 min at -SB°C.
GC/EI-MS analysis of an aliquot from the reaction showed
consumption of the starting nitrile 1. The reaction was
quenched with water, diluted with diethyl ether and
transferred to a separatory funnel. The ether layer was
washed with 10's HCl (3X), brine (1X), dried with
anhydrous MgS04, and concentrated to a brown oil. This
material was distilled (Kugelrohr, 100°C) at reduced
pressure to afford 1.5 g of a clear oil. GC/EI-MS of
this material showed it to contain the desired
product 2, (R~=7.35 min) m/z (rel. int. ) 257 (M', 3) ,
203 (100), 183 (59), 170 (5), 133 (4), 109 (3); 1H-NMR
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
140
(CDC13) d 7.4-6.9 (8H, m), 4.01 (1H, d, J=10 Hz), 3.38
(1H, dq, J=7, 10 Hz), 1.32 (3H, d, J=7 Hz); =3C-NMR
(CDC13) d 19.4, 30.5, 54.2, 114.5, 114.6, 114.7, 114.9,
115.0, 115.3, 123.3, 123.4, 123.6, 123.7, 130.5, 130.6,
131.7.
Product 3 was synthesized by the catalytic
reduction of 2 using Raney nickel in 95:5 EtOH:aqueous
sodium hydroxide (2 Eq.) under 60 psi hydrogen.
GC/EI-MS (R'=7.25 min) m/z (rel. irt. ) 261 (M', 20) , 244
(35), 229 (16), 215 (17), 201 (80), 183 (100), 133 (42),
115 (27), 109 (47), 95 (20); 1H-NMR (CDC13) d 7.3-6.8
(8H, m), 3.62 (1H, d, J=10 Hz), 2.70 (1H, M), 2.40
(2H, m) , 1.73 (2H, m) , 0.91 (3H, d, J=7 Hz) . Note that
product 3 in this reaction sequence corresponds to
Compound 21.
Product 2 in 10% IPA-hexane (100 mg/ml) was
chromatographed, in 500 ~cl aliquots, through Chiral Cel
OD (2.0 x 25 cm) using 10% IPA-hexane at 10 ml/min
measuring optical density at 254 nm. This afforded the
two optically pure enantiomers 4 and 5 (as determined by
analytical chiral HPLC; Note, the stereochemistry of
these two compounds has not been assigned at this time).
These two compounds were identical in their GC/EI-M5 and
1H-NMR spectra as product 2 (data above).
Each of the enantiomers 4 and 5 was reduced
separately using dimethyl sulfideborane complex in the
following manner. A solution of compound (4 or S ) in
THF was heated to reflux and treated with excess (2 Eq.)
1M (in THF) dimethyl sulfideborane complex and the
CA 02560002 2006-10-05
WO 97!46511 PCT/US96l20697
141
reaction refluxed 30 min. After this time the reaction
was cooled to 0°C and treated with 6 N HC1. The
reaction was set to reflux for 30 min. After this time
the reaction was transferred to a separatory funnel,
basified to pH > 12 with !ON NaOH, and the product (6 or
7) extracted into ether. The ether layer was washed
with brine, dried over anhydrous MgS04 and concentrated
to an oil. The product was purified by prep-TLC using
5o methanol-chlorform. Each of the individual
enantiomers (6 and 7) were found to be identical in
their GC/EI-MS and 1H-NMR spectra as product 3 (data
above). Note that products 6 and 7 in this scheme
correspond to Compounds 33 and 34. Compound 33~HC1: mp
- 260-270°C (dec), [«]3ss26 = +6.6 (c 1.0 in EtOH),
[«)p26 = +0.4 (c 1.0 in EtOH). Compound 34~HC1: C«]35523
- -6.1 (c 1.0 in EtOH), [«]023 = +0.1 (c 1.0 in EtOH).
Compound 33~HI: The free base of Compound 33 was
dissolved in EtOH and 47% hydriodic acid (1.1 equivt.)
was added. The solvent was evaporated under vacuum and
the resulting solid hydroiodide was recrystallized twice
from heptane/EtOAc by slow evaporation: mp = 195-197°C.
The absolute configuration of Compound 33~HI was
determined to be R by single-crystal (monoclinic
colorless needle, 0.50 x 0.05 x 0.03 mm) X-ray
diffraction analysis using a Siemens R3m/V
diffractometer (3887 observed reflections).
CA 02560002 2006-10-05
WO 97/4b511 PCT/US96/2Ub97
142
I! IMeIS~):NL~ I ~ CHI Rorer nictel 1 ~ CH!
F ~ CN I! Mel CN ;h F NH=
F I \ I P \ 1
1 Z
1
Compound I1
Chitd Cel OD I ~ H! ~ ~ ~H!
IOR IPA / He:ene F ~ CN . F ~ CN
I 1
F ~ F
i ~ 3
I1 Me,S~BH~
Il HCI
HI w ~H!
I i NH1 ~~NII!
F ' I
F
6 I
iCompaund 331 ~Campound 36l
Synthesis of Compound 22 was accomplished as
described below. Compound 23 was synthesized in a
similar manner.
Triethyl phosphonoacetate (17.2 g, 76.8 mmol)
was slowly added to a suspension of sodium hydride (3.07
g, 76.8 mmol) in N,N-dimethylformamide (350 ml). After
minutes 3,3'-difluorobenzophenone (15.2 g, 69.8 mmol)
was added to the solution and stirred an additional 28
hr. The reaction mixture was quenched with water and
10 partitioned between water and ether. The combined
organic layers were washed with brine and dried over
anhydrous magnesium sulfate. The solvent was evaporated
in vacuo to give 19.7 g of ethyl
3,3-bis(3-fluorophenyl>acrylate as a yellow oil.
15 To a solution of ethyl 3,3-bis(3-fluorophenyl)
-acrylate (19.7 g, 68.4 mmol) in 200 ml of ethanol was
added palladium hydroxide on carbon (3.5 g). The
mixture was shaken under 60 psi of hydrogen for 3 hours,
CA 02560002 2006-10-05
WO 97/46511 PCTILTS96120697
143
then filtered and evaporated in vacuo to give 19.5 g of
product A as a colorless oil.
The ethyl ester A (19.2 g) was hydrolyzed by
stirring for 6 days with 50 ml of 10 N sodium hydroxide.
The reaction mixture was then diluted with 50 ml of
water and acidified to pH 0 with concentrated HC1. The
aqueous mixture was extracted 3 times with ether and the
ether extracts dried over magnesium sulfate and
evaporated to give 3,3-bis(3-fluorophenyl)propionic acid
l0 as a white powder.
3,3-bis(3-fluorophenyl)propionic acid (13 g,
49.6 mmol) was dissolved in 50 ml (685 mmol) of thionyl
chloride and stirred overnight at room temperature. The
excess thionyl chloride was removed in vacuo on a rotary
evaporator to give 13.7 g of product B as a yellow oil.
To acid chloride B (13.7 g, 49 mmol) dissolved
in 100 ml of dry THF was added iron(III) acetylacetonate
(0.52 g, 1.47 mmol). Methylmagnesium chloride (16.3 ml,
49 mmol) was then added over a period of 1 hr by syringe
pump. The reaction was stirred for an additional hour,
then quenched by pouring into ether/5% HC1. The ether
layer was separated, washed with 5o HC1 and saturated
NaCl, and dried over sodium sulfate. The solvent was
evaporated in vacuo to give
4,4-bis(3-fluorophenyl)-2-butanone as a yellow oil. The
crude oil was purified on silica gel using heptane/ethyl
acetate as the elutant.
To 4,4-bis(3-fluorophenyl)-2-butanone (5.7 g,
21.9 mmol) in 25 m1 of ethanol was added pyridine
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
144
(1.91 g, 24.1 mmol) and methoxylamine hydrochloride
(2.01 g, 24.1 mmol). The reaction was stirred overnight
at room temperature, then poured into ether/5% HC1. The
ether layer was separated, washed with 5% HC1 and
saturated NaCI, and dried over sodium sulfate. The
solvent was evaporated in vacuo to give 6.26 g of the
O-methyl oxime of 4,4-bis(3-fluorophenyl)-2-butanone.
To sodium borohydride (4.1 g, 108.3 mmol) in 15 ml of
THF was slowly added zirconium tetrachloride (6.31 g,
27.1 mmol). This mixture was stirred for 15 min, then
the oxime (6.26 g, 21.7 mmol) in 6 ml of THF was added
over 5 min. After 3 hours of stirring at room
temperature, the reaction was worked up by slowly adding
50 mM sodium hydroxide followed by ether. The aqueous
layer was extracted 4 times with ether, and the combined
ether extracts were dried over sodium sulfate. The
solvent was evaporated in vacuo to give 5.3 g of
Compound 22.
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
145
O
II
I 0 Ec0' P"COOEt i I
F 1.
NaH , DMF _ F \ COOEt
2. H2 / Pd(OH)~ - Ethanol
F
F
A
CI
1. NaOH/Mo0HIH20 F
2. SOCl2 i
F
B
1. Fe(Acac)3 , MeMgBr F ~ I NHS
2. HEN-OMe , pyridine CH3
i
3. NaHH4 I ZrCl4 , THF F
Compound 22
Synthesis of Compound 24 was accomplished as
described below. Compounds 25-29, 52-53, 65, 76-78, 83,
90, 96-97, 115, and 135-136 were prepared in a similar
manner.
A suspension of magnesium turnings (0.95 g,
39.2 mmol) in 150 ml anhydrous diethyl ether was treated
with 1-bromo-3-fluorobenzene (6.83 g, 39.2 mmol)
dropwise via syringe. After 1.5 hr the solution was
transfered via cannula to a flask containing
o-anisaldehyde (5.0 g, 36.7 mmol) in 100 ml anhydrous
diethyl ether at 0°C and stirred 2hr. The reaction
mixture was quenched with water and partitioned between
water and ether. The combined organic layers were
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
146
washed with brine and dried over anhydrous magnesium
sulfate to afford 7.90g (93% yield) of product A.
Pyridinium dichromate (16.0 g, 42.5 mmol) was
added to a solution of the alcohol A (7.90 g, 34.0 mmol)
in dichloromethane (100 ml), and the reaction was
stirred 12 hr. Diethyl ether (300 ml) was added to the
reaction mixture and the black solution was filtered
through a silica gel plug (30 cm) and washed with an
additional 500 ml ether. After evaporation of the
solvent in vacuo, the solid was recrystallized from
acetor_e to give 7.45 g (95% yield) of product B.
Diethyl cyanomethylphosphonate (7.0 g,
39.5 mmol) was slowly added to a suspension of sodium
hydride (1.58 g, 39.5 mmol) in 100 ml
N,N-dimethylformamide. After 30 minutes the ketone B
was added to the solution and stirred an additional
2 hr. The reaction mixture.was quenched with water, and
partitioned between water and ether. The combined
organic layers were washed with brine and dried over
anhydrous magnesium sulfate. The solvent was evaporated
in vacuo to give a pale yellow oil.
In a glass bomb, the oil was dissolved in
100 ml ethanol and 20 ml 10N NaOH. A catalytic amount
of Raney Nickel suspened in water (ca. 15 mol percent)
was added to the solution. The reaction mixture was
shaken under 60 psi HZ for 12 hr on a Parr Hydrogenator.
After filtering off excess Raney Nickel, the solution
was extracted with chloroform. The combined organic
layers were washed with brine and dried over anhydrous
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
147
magnesium sulfate. After filtration, the oil was run
through a silica gel column in chloroform and methanol.
The solvent was evaporated in vacuo to give a pale
yellow oil. GC/EI-MS (R~=8.10 min) m/z (rel. intensity)
259 (100) , 242 (44) , 213 (48) , 183 (42) , 136 (50) , 109
(94), 91 (60), 77 (25). The oil was then acidified with
hydrogen chloride in diethyl ether. Evaporation of the
ether afforded a pale yellow solid that was
recrystallized in hot acetonitrile to afford 3.45 g
(42.1°s yield) white needles of: Compound 24, as the
hydrochloride salt.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
148
1) Mg, ether
2), / H / I OCH3 / OCH3
PCC or PDC \ I O
OCH~ \ OH CHZCI,
F \ Br
F F
A B
EtO~ P~ CN, NaH-DMF
2) Rancy Ni. EtOH, NaOH / OCH3
HZ 60 p.s.i. \ ~ NH3Cl
3) HCl-ether -
F
Compound 24 (HC1 salt)
Compounds 101 and 103 were synthesized from
Compounds 25 and 24, respectively, by cleavage of their
0-methyl ethers with borane tribromide in the normal
manner.
Synthesis of Compound 30 was accomplished as
described below. Compound 31 was prepared in a similar
manner.
A suspension containing magnesium turnings
(0.95 g, 39.1 mmol) in 150 ml anhydrous diethyl ether
was treated with 1-bromo-3-fluorobenzene (6.85 g,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
149
39.1 mmol) dropwise via syringe. After 1.5 hr the
solution was transfered via cannula to a flask
containing 3-chlorobenzaldehyde (S.0 g, 35.6 mmol) in
100 ml anhydrous diethyl ether at 0°C and stirred 2 hr.
The reaction mixture was quenched with water and
partitioned between water and ether. The combined
organic layers were washed with brine and dried over
anyhydrous magnesium sulfate to afford 8.40 g (>99s
yield) of product A.
Pyridinium chlorochromate (15.0 g, 39.8 mmol)
was added to a solution of the alcohol A (8.40 g,
35.5 mmol) in 100 ml dichloromethane and stirred 18 hr.
Diethyl ether (300 ml) was added to the reaction mixture
and the black solution was filtered through a silica gel
plug (30 cm), and washed with an additional 500 ml
ether. After evaporation of the solvent the solid was
recrystallized from acetone to give 6.31 g (76% yield)
of product B.
Diethyl cyanomethylphosphonate (5.2 g,
29.6 mmol) was slowly added to a suspension of sodium
hydride (1.2 g, 29.6 mmol) in N,N-dimethylformamide
(100 ml). After 3o minutes the ketone B was added to
the solution and stirred an additional 6 hr. The
reaction mixture was quenched with water and partitioned
between water and ether. The combined organic layers
were washed with brine and dried over anhydrous
magnesium sulfate. The solvent was evaporated in vacuo
to give a yellow oil.
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
150
In a glass bomb, the oil was dissolved in
ethanol (100 ml) and !ON NaOH (20 ml). A catalytic
amount of rhodium suspended on alumina (ca. 35 mol
percent) was added to the solution. The reaction
mixture was shaken under 60 psi HZ for 24 hr on a Parr
Hydrogenator. After filtering off excess catalyst, the
solution was extracted with chloroform. The combined
organic layers were washed with brine and dried over
anhydrous magnesium sulfate. After filtration and
evaporation of the solvent in vacuo, the oil was taken
up in tetrahydrofuran (100 ml). Diborane (23.4 ml,
1.0 M) was added and the solution was refluxed for
1.5 hr. The solvent was evaporated in vacuo and 6N HC1
(50 ml) was added carefully. The solution was refluxed
for 1 hr. After cooling, the mixture was basified with
!ON NaOH to pH 14 and partitioned between
dichloromethane and water. The combined organic layers
were dried over anhydrous magnesium sulfate and
filtered. After evaporation of the solvent, the yellow
oil was run through a silica gel column in chloroform
and methanol. The solvent was evaporated in vacuo to
give a yellow oil. GC/EI-MS (Rr=8.15 min) m/z (re!.
intensity) 263 (17), 246 (21), 211 (84), 196 (33), 183
(100>, 165 (19), 133 (19). The oil was then acidified
with hydrogen chloride in diethyl ether. Evaporation of
the ether afforded 0.96 g of a white solid, Compound 30,
as the hydrochloride salt.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
151
1) Mg, ether
r r
z)/ / H C1 \ I OH PCC-CHZCiz CI \ I 0
C1 0
W
F I3r
F F
A B
I ~~O~P~CN, NaH-DMF
2) Rh/Alumina, EtOH
HZ 60 p.s.i.
3) BzH6-THF
4) 6N HCI NH3Cl
5) HCi-ether Ci
c
Compound 30 (HC1 salt)
Synthesis of Compound 35 was accomplished as
described below. Compounds 36-37 were prepared in a
similar manner.
A solution of 3-fluorobenzaldehyde (3.0 g,
24.2 mmol) at 0°C in 150 ml diethyl ether was treated
with 3.0 M ethyl magnesium chloride (12.7 ml, 25.4 mmol)
in tetrahydofuran (THF) via syringe. After 4 hr, the
reaction mixture was quenched with water and partitioned
between water and ether. The combined organic layers
CA 02560002 2006-10-05
WO 97/465!1 PCTlUS96/20697
152
were washed with brine and dried over anyhydrous
magnesium sulfate to afford 4.25 g of product A.
Pyridinium chlorochromate (6.53 g, 30.3 mmol)
was added to a solution of A in dichloromethane (100 ml)
and stirred 18 hr. Diethyl ether (300 ml) was added to
the reaction mixture and the black solution was filtered
through a silica gel plug (30 cm) and washed with an
additional 500 ml ether. After evaporation of the
solvent the solid was recrystallized from acetone to
give 3.05 g of product B. The solvent was evaporated in
vacuo to give a pale yellow oil.
Diethyl cyanomethylphosphonate (4.7 g,
26.4 mmol) was slowly added to a suspension of sodium
hydride (1.1 g, 26.4 mmol) in 100 ml
N,N-dimethylformamide. After 30 minutes the ketone B
was added to the solution and stirred an additional 6
hr. The reaction mixture was quenched with water and
partitioned between water and ether. The combined
organic layers were washed with brine and dried over
anhydrous magnesium sulfate. The solvent was evaporated
in vacuo to give a yellow oil.
In a glass bomb, the oil was dissolved in
100 ml ethanol and 20 ml !ON NaOH. A catalytic amount
of Raney Nickel suspended in water (ca. 15 mol percent)
was added to the solution. The reaction mixture was
shaken under 60 psi HZ for 24 hr on a Parr Hydrogenator.
After filtering off excess catalyst, the solution was
extracted with chloroform. The combined organic layers
were washed with brine and dried over anhydrous
CA 02560002 2006-10-05
4
WO 97/46511 PCT/US96/20697
153
magnesium sulfate. After filtration, the oil was run
through a silica gel column in chloroform and methanol.
The solvent was evaporated in vacuo to give a pale
yellow oil. GC/EI-MS (R~=3.45 min} m/z (rel. intensity)
167 (4) , 150 (63) , 135 (58) , 7.09 (100) , 96 (53) ,
75 (48). The oil was then acidified with hydrogen
chloride in diethyl ether. Evaporation of the ether
left a pale yellow solid that was recrystallized in hot
acetonitrile to afford 2.2 g of Compound 35, as the
hydrochloride salt.
OH 0
HOC HOC
I CH3CH~MgBr, ether PCC-CHZC12
F \ H /
\ \
0 F F
A B
1) 0
EtOi~P~CN, NcH-DMF
Et0
2) Raney Ni, EtOH, NaOH
HZ 60 p.s.i.
NH3CI
3) HCl-ether H3C
F
Compound 35 (HC1 salt)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96I20697
154
Synthesis of Compound 38 was accomplished as
described below.
To a solution of 3,3-bis(3-fluorophenyl)
-propionitrile (1.5 g, 6.17 mmol) in 250 ml of THF at
-70°C was added butyl lithium (4,25 ml in hexanes, 6.8
mmol) by syringe over 5 minutes. The solution was
stirred for 5 min then methyl iodide (1.75 g, 12.3 mmol)
was added over 1 min. The reaction mixture was then
allowed to warm up to room temperature and worked up by
diluting with ether and washing with So HC1 and water.
The ether layer was dried over sodium sulfate and
evaporated to give 1.5 g of the methylated nitrile as a
yellow oil.
To the 3,3-bis(3-fluorophenyl)
2-methyl-propionitrile (1.46 g, S.7 mmol) in 50 ml of
dichloromethane at 0°C was added diisobutylaluminum
hydride (1.02 ml, 5.7 mmol) by syringe over a 10 min
period. The reaction was stirred for 30 min at 0°C
followed by 2 additional hours at room temperature. The
reaction was worked up by adding 200 ml of 10a HC1 and
stirring at 40°C for 30 min followed by extraction of
the product with dichloromethane. The organic layer was
dried over sodium sulfate and evaporated to give 1.36 g
of the product A.
To a solution of the aldehyde A (1.36 g,
5.23 mmol) in 40 ml of ether at 0°C was added
methylmagnesium bromide (5.23 ml in ether, 5.23 mmol).
The reaction was stirred for 3 hr at room temperature,
and then quenched with dilute HC1. The ether layer was
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
155
separated, dried over sodium sulfate and evaporated to
give 1.48 g of 4,4-bis(3-fluorophenyl)
-3-methylbutan-2-ol.
To a solution of the alcohol (1.4 g,
5.07 mmol) in 300 ml of dichloromethane was added
pyridinium chlorochromate (1.2 g, 5.58 mmol), and the
mixture was stirred overnight. The reaction was then
diluted with 100 ml of ether and filtered through a
silica plug. The solvent was evaporated to give 1.39 g
of product B.
The ketone B (1.3 g, 4.9 mmol) was added to a
solution of methoxylamine hydrochloride (0.45 g, 5.38
mmol) and pyridine (0.44 ml, 5.38 mmol) in 30 ml of
ethanol, and stirred overnight. The ethanol was then
Z5 evaporated, and the residue taken up in ether and 10%
HCl. The ether layer was separated, washed once with
10% HC1, dried over sodium'sulfate and evaporated to
give 1.4 g of the O-methyl oxime.
To a suspension of sodium borohydride (0.87 g,
23.1 mmol) in 5 ml of THF was added zirconium
tetrachloride (1.35 g, 5.8 mmol), and the solution was
stirred for 15 min followed by the addition of another
5 ml of THF. The O-methyl oxime (1.4 g, 4.6 mmol) in
5 ml of THF was then added, and the mixture stirred
overnight. The THF was removed by evaporation in vacuo,
and the residue treated with 10% sodium hydroxide.
After the bubbling ceased ether was added and the layers
separated. The aqueous layer was extracted four times
with ether, and the combined ether extracts were dried
CA 02560002 2006-10-05
WO 97/46511 PCT/ITS96/20697
156
over sodium sulfate. The ether was evaporated to give
1.25 g of Compound 38.
CHI
CN 1~ BuLi , MeI \
CHO
2. DIBAH , CHZCIZ
r-
A
CH3
1. MeMg$r
2. PCC / CHzCIz
B
CH3
1. H2N.DIv(~ , pyridine ~ \ i HHz
~BH4 /~ CH3
i
\O
Compound 38
CA 02560002 2006-10-05
WO 9'7/46511 PCTlUS96/20697
157
Compound 32 and Compounds 39 - 53 were
synthesized according to standard procedures as
described above.
Compounds 107, 116, 139, and 143 were prepared
as synthetic intermediates used in the preparation of
Compounds 32, 115, 20, and 25, respectively.
Compound 50 was also prepared using the chiral
synthesis described below.
To an ice-cold solution of
N-benzyl-(S)-a-methylbenzylamine (18.0 g, 85.2 mmol) in
THF (75 ml) was added butyl lithium (2.5 M in hexane;
37.5 ml, 93.8 mmol) via a syringe over a period of
10 min at such a rate as to keep the reaction
temperature below 10°C during the addition. The
reaction was then stirred at 0°C for 15 min. The
reaction was cooled to -78°C in a dry ice/isopropanol
bath and then a solution of benzyl crotonate (15.0 g,
85.2 mmol) in THF (100 ml) was added dropwise over a
period of 45 min. The reaction was stirred at -78°C for
15 min, and then saturated NH4C1 (50 ml) was added. The
reaction mixture was then quickly transferred to a
separatory funnel containing saturated NaCl (500 ml) and
ether (200 ml). The layers were separated and the
aqueous layer extracted with ether (200 ml). The
combined organic layers were dried, evaporated, and
chromatographed on silica gel (50 mm x 30 cm) (hexane-
ethyl acetate [20:1]) to yield 21.0 g, 63.70 of product
A. 1H-NMR showed that the diastereoselectivity of the
reaction is > 90%.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96I20697
158
A mixture of magnesium (2.58 g, 106 mmol), THF
(200 ml), and 1-bromo-3-fluorobenzene (18.60 g,
106.3 mmol) was refluxed for 45 min. While still under
reflux, product A (16.45 g, 42.45 mmol) was added via
syringe with THF (25 ml) over a 2 min period. The
reaction was refluxed for 1 hr, and then allowed to cool
to room temperature. Saturated NH4C1(aq., 200 ml) was
added. The reaction mixture was then transferred to a
separatory funnel containing saturated NaCl(aq) (500 ml)
and diethyl ether (200 ml). The layers were separated
and the aqueous layer extracted with ether (200 ml).
The combined organic layers were dried over sodium
sulfate and evaporated to give 21.41 g of product B as a
yellow liquid.
Product B (20.02 g, 42.45 mmol, theoretical)
was dissolved in acetic acid (120 ml) and sulfuric acid
(30 ml). The reaction was stirred at 90°C for 1 hr.
The acetic acid was rotary evaporated giving a brown
sludge. This material was placed in an ice bath and
cold water (400 ml) was added. The product immediately
precipitated. To the reaction was slowly added 10 N
NaOH (150 ml) to neutral pH. Diethyl ether (200 ml) was
added to this mixture. The mixture was shaken until
there was no undissolved material. The ether layer was
separated, washed with water (2 x 100 ml), dried over
sodium sulfate, and rotary evaporated yielding 13.14 g
(68.2% based on ester) of a thick brown oil. This oil
was taken up in ether and converted to the hydrochloride
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
159
salt with hydrogen chloride in diethyl ether to give
product C as a yellow-white solid.
Product C (7.17 g, 14.6 mmol) was taken up in
absolute ethanol (200 ml). Pearlman's catalyst
(Pd(OH)z/C; 2.00 g) was added. The reaction was shaken
under 70 psi hydrogen gas at 70°C for 20 hr, and the
reaction mixture was filtered through Celite. The
filtrate was rotary evaporated to give 3.54 g of a light
yellow glass. This material was taken up in diethyl
ether (100 ml) and was basified with 1 N NaOH (25 ml).
The ether layer was washed with water (1 x 25 ml), dried
over sodium sulfate, and rotary evaporated to give
2.45 g of a light yellow oil. This material was
Kugelrohr distilled (90-100°C, 1 mm Hg) to give 1.17 g
of a colorless liquid. This material was taken up in
diethyl ether and converted to the hydrochloride salt
with ethereal hydrogen chloride. After rotary
evaporation, the salt was recrystallized from 0.12 N HC1
(200 mg/ml). The crystals were filtered off and were
washed with cold 0.12 N HC1 yielding 0.77 g (180) of
Compound 50 as silvery white ~~rystals (as the
hydrochloride salt).
Compound 51 was synthesized in a similar
manner to Compound 50 utilizing N-benzyl-(R)-a-
methylbenzylamine as a chiral starting material.
CA 02560002 2006-10-05
WO 97/46511
PCT/US96I20697
160
I ~ KFlCeiiic / w
Br HyN / t)M1'U w I N I
/
CH3 CH3
i
I Mgl3r
1) BuLi, THF, -78'C /
I
~0 N I ,.~ THF, reilux
O"~
0 ~ 3 CH3
A
/
I I
r
i
I OH N . I ~ AcOH, HZS04 I
. HZO F w r N w I
CH3 CH3 ~ CH3 CHI
F /
F
B
C
/
W
t) t'd(OH)zIC, EtOH F NHz ~ HC!
2) HCt~EtZO ~ CH3
I
r
F
Compound 50 (HC1 salC)
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
161
Synthesis of Compound 54 was accomplished as
described below.
To a solution of 3,3'-difluorobenzophenone
(5 g, 22.9 mmol) and methyl cyanoacetate (3.4 g,
34.4 mmol) in 15 ml of ether was added titanium
isopropoxide (16.9 ml, 57.25 mmol). This solution was
stirred for 6 days at room temperature then quenched
with 0.5 mol of HCl in 300 ml of water. The mixture was
diluted with 100 ml of ether, and the layers separated.
The ether layer was washed with 5o HC1 and saturated
brine, then dried over sodium sulfate. The solvents
were evaporated in vacuo to give 8 g of product A.
Compound A was dissolved in 50 ml of
isopropanol, followed by the addition of a small amount
of bromocresol green. Sodium cyanoborohydride (1.52 g,
24.2 mmol) was added all at once followed immediately
with the dropwise addition of concentrated HC1, added at
such a rate as to keep the solution yellow. After
2 hours the reaction was worked up by partitioning
between ether and water. The ether layer was washed
with water and saturated brine, dried over sodium
sulfate, and concentrated to give the product B.
To a solution of lithium aluminum hydride
(30.4 ml, 30.4 mmol) in THF was added product B (1 g,
3.04 mmol) in 2 ml of THF over a period of 30 seconds.
This solution was stirred overnight at room temperature,
then quenched with the addition of 20 ml of ethyl
acetate. The solvents were then removed in vacuo, and
the resulting oil was dissolved in aqueous HC1 and
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
162
acetonitrile. The product was then purified on a C-18
column with a gradient of 0.1°s HC1 to acetonitrile to
give 82 mg of Compound 54, as the hydrochloride salt.
EI-MS m/z (relative intensity) 277 (M', 100), 260 (2.4),
242 (8.6), 229 (28), 215 (11.7), 204 (16), 183 (12), 133
(9.5), 124 (14), 109 (6.8), 30 (22).
r CN
w I O NC~COONie F ~ I , DYCH3
F
r Ti(i-OPr)4 , Evhcr r O CH3
p ~ F
A
OH
r N
pYCH~ ~ ) NH~CI
NaCNBH~ F !. LAN F
Isopropanol. HC/ ~ 0 CH3 z, Chromatography r
in HCUAcCN
F ~ F
D
Compound 54 (HC1 salt)
Compound 55 was synthesized analogously to
Compound 21 except that ethyl iodide was used in the
alkylation step. GC/EI-MS (RC = 7.43 min) m/z (relative
intensity) 275 (M', 100), 258 (66), 229 (63), 204 (57),
201 (72), 183 (84), 134 (57), 124 (68), 109 (98), 72
(72) .
The synthesis of Compound 56 was accomplished
as follows.
The alcohol A was synthesized from
3-fluorobromobenzene and 3-fluoro-2-methylbenzaldehyde
CA 02560002 2006-10-05
WO 97/46511 PCT/IJS96/20697
163
as described for product A in the synthesis of
Compound 24.
The alcohol A (8.4 g, 36.2 mmol) was stirred
with manganese dioxide (12.6 g, 144.8 mmol) in 100 ml of
dichloromethane for 4 days. The reaction mixture was
then diluted with ether and filtered through a
0.2 micron teflon membrane filter. The filtrate was
concentrated to give 7.6 g of the ketone B.
The substituted acrylonitriTe C was
synthesized as described for product A in the
Compound 20 synthesis.
To the nitrile C (4 g, 15.7 mmol) in 240 ml of
ethanol was added 2 g of l0% palladium dihydroxide on
carbon. This mixture was hydrogenated at 60-40 psi for
3 days. The reaction mixture was then filtered and
concentrated. The resulting oil was dissolved in
chloroform and chromatographed on silica gel (300
methanol/5% isopropylamine in chloroform) to give the
amine. This amine was dissolved in aqueous
HC1/acetonitrile and purified via HPLC on C-18 (10%
acetonitrile/0.1% HC1 to 50% acetonitrile/0.1% HC1 over
60 min) then lyophilized to give 800 mg of Compound 56,
as the hydrochloride salt. GC/EI-MS (Rt = 7.39 min) m/z
(relative intensity) 261 (M', 64), 244 (56), 229 (57),
215 (100), 203 (53), 183 (21), 133 (39), 122 (31), 109
(32) .
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
164
I) M g, ether
H w I OH ~ I O
F F
F O H3 , Mn---~ H~ i
F Br F ~ ~ CH2ClZ F ~, I
A B
i
Et0 R ~ I ~ I NH3C!
Et0'p~CN F V ~~CN I) pd(OH)Z , Hy, Ethanol F
H3 i H3
I 2) Chromatography in I
NaH-DMF w HCUAcCN p
F
C Compound 56 (HC1 salt)
The synthesis of Compound 57 was accomplished
as follows.
To a solution of 5-fluoro-2-methylbenzonitrile
(5 g, 37 mmol) in 50 ml of THF was added
3-fluorophenylmagnesium bromide (46 ml, 40 mmol) and
copper (I) cyanide (0.072 g, 0.8 mmol). This solution
was refluxed for 4 hours, then poured into ether/20% HC1
and stirred for a further 2 hours. The layers were
separated, and the ether layer washed with water and
saturated brine. The solution was dried over sodium
sulfate and concentrated. The crude oil was purified on
silica (hexane to 50% dichloromethane in hexane over
60 min) to give 6.7 g of the ketone A.
The ketone A was converted to Compound 57 as
described for Compound 56. GC/EI-MS (RC = 7.35 min) m/z
(relative intensity) 261 (M', 52), 244 (41), 229 (67),
215 (100), 203 (42), 201 (42), 183 (21), 133 (45), 122
(28), 109 (26).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
165
O
l) Mg,ethcr F
CH3
F ~ Br 2)
l CN . CuCN F
F A
) NH3Cl
a
Et0 RvCN
Et0 F ) ~ CN 1) Pd(OH)q , Hz, Ethanol F
CH3
NaH~DMF ~ CH3 2) Chromatography in I
HCI/AcCN F
F
Compound 57
(HC1 salt)
The synthesis of Compound 58 was accomplished as
follows.
To a solution of 5-fluoro-2-methylbenzoyl
chloride (2.24 g, 13 mmol) in 10 ml of dry THF was added
iron III acetyiacetonate (0.16 g, 0.44 mmol). The
solution was cooled to 0°C,' and a THF solution of
5-fluoro-2-methylphenylmagnesium bromide (20 ml,
15.5 mmol) was added by syringe over a period of 30 min.
The reaction was stirred for another 30 min, then poured
slowly into ether/5% HC1. The ether layer was
separated, washed with saturated brine, dried over
sodium sulfate, and concentrated to give 3.2 g of
ketone A.
Dry THF (30 ml)~ was cooled to -78°C followed
by the addition of butyl lithium (5.85 ml, 14.6 mmol,
2.5 M solution in hexanes). Acetonitrile (0.76 ml,
14.62 mmol) was then added over a period of 2 min, then
allowed to stir at -78°C for 15 min. To this solution
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
166
was added ketone A (3 g, 12.2 mmol) in S ml of THF. The
solution was stirred for 30 min at -78°C then allowed to
warm to room temperature and stirred overnight. The
reaction mixture was partitioned between ether and 50
S HC1. The ether layer was separated, washed with
saturated brine, dried over sodium sulfate, and
concentrated to give 2.2 g of the nitriie B.
The nitrile B (1 g, 3.48 mmol) was dissolved
in 30 ml cf ethanol and 3 ml of 10 N sodium hydroxide.
To this solution was added 1 g of a SO°s aqueous slurry
of Raney nickel, and the mixture was hydrogenated at
60 psi for 20 hours. The reaction was filtered and
concentrated to a white solid. This residue was taken
up in ether/water and the ether layer separated. The
ether solution was dried over sodium sulfate and
concentrated to give 0.96 g of the hydroxyamine C.
The hydroxyamine~C ;0.96 g, 3.3 mmol) was
taken up in concentrated HC1 and heated to 70°C which
caused brief solution, and then precipitation of the
alkene D. The alkene was collected by filtration and
dissolved in 30 ml of ethanol and 1 ml of conc. HC1.
Palladium dihydroxide on carbon (0.4 g) was added to the
solution;=and the mixture hydrogenated at 60 psi for
24 hours. The product was isolated by filtering off the
catalyst and evaporating the solvent. The residue was
dissolved in 0.1% HC1 and acetonitrile, and purified on
C-18 (15% acetonitrile/0.1% HC1 to acetonitrile) to give
0.6 g of Compound 58, as the hydrochloride salt.
GC/EI-MS (RC = 7.82 min) m/z (relative intensity) 275
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
167
(M', 100), 258 (20), 243 (74). 229 (38), 214 (65), 201
(31), 196 (32), 183 (20), 148 (35), 138 (42), 133 (48),
122 (69), 109 (41).
CH3
3 w ~ I
CH CH l;c(Acac)3 \ O
( + I
F ~ MgBr ~ CI THF , CH3
o ~I
F
A
CH; ~ CHI
~ I H ~ I H
BuLi . AcCN ~ ~CN Raney Ni , H2, NaOH F
CHj ~ CHI
F ~ I Ethanol F
Ii C
CH3 , CH3
F w I ~ NH3CI F '~ I NH3C1
conc: HCI , heat ~ CH3 Pd(OH),1C , HZ , HCI ~ CH3
Ethanol ~ I
F
Compound 58
(HC1 salt)
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
168
Synthesis of Compound 59 was accomplished as
follows.
Compound 20 (2.0 g, 7.05 mmol) was dissolved
in abs. EtOH (200 ml) and cooled to 5-10°C in an ice
bath. Acetaldehyde (0.395 ml, 7.05 mmol, cooled to
-4°C) was added followed by nickel-aluminum alloy
(200 mg, Fluka Chemika), and the reaction was
hydrogenated on a Parr apparatus at SO psi for 2 hr.
GC/MS showed 75% yield of the product and 20 of the
N,N-diethyl side-reaction product. The reaction mixture
was filtered.through diatomaceous earth and the filtrate
was evaporated under reduced pressure. The crude
product was dissolved in isopropanol (5 ml)/ether (60
ml)/ethereal HCl (1 M), and then hexane (5 ml) was added
to the cloud point. The cloudy mixture was filtered
through paper, then hexane (10 ml) was added to the
cloud point, and the solution was filtered again. The
filtrate was stoppered and the product was allowed to
crystallize at room temperature. The crystals were
collected and dried to provide 0.325 g (14.8% yield) of
Compound 59, as the hydrochloride salt (colorless
needles).
The synthesis of Compound 60 was accomplished
as follows. Compounds 66, 69, 108, 123, 142, and 145
can be synthesized in a similar manner starting from
Compounds 33, 50, 32, 60, 25 and 119, respectively.
Compound 20 (as the free base) (1.0 g,
4.0 mmol) was refluxed in ethyl formate (150 ml) for
2 hr. The solvent was then removed under reduced
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
169
pressure to provide 1.1 g, 99% yield of formamide A as a
colorless oil. GC/MS showed the product to be 100.0%
pure and was used in the following step without further
purification.
The formamide A (1.1 g, 4.0 mmol) was
dissolved in dry THF (100 ml) and heated to reflux (no
condenser). Borane-methyl sulfide complex (1.2 ml, 12
mmol, 10.5 M) was added dropwise over a period of 3 min
to the refluxing solution. Reflux was maintained for
approximately 15 min, open to the air, until the
reaction volume was reduced to approximately 30 ml. The
reaction was then cooled in an ice bath, and ice (5 g,
small pieces) was carefully added followed by H~O (25 ml)
and cons. HC1 (25 m1). The acidic solution was refluxed
for 30 min. The reaction mixture was then cooled in an
ice bath, basified with NaOH (10N), extracted with ether
(3 X 100 ml), dried (Na2S0" anhydrous), and evaporated
under reduced pressure. The crude product was dissolved
in ether t10 m1)/hexane (50 ml) and ethereal HCL (1 M)
was added dropwise to precipitate the hydrochloride
salt. The salt was collected and recrystallized from
isopropanol (3 ml)/ether (40 ml) to provide 0.5 g of
Compound-.~0, as the hydrochloride salt.
CA 02560002 2006-10-05
WO 97/46511 PGT/US96/20697
170
Ethyl formate
f: ~ ~z ~ I ~ N H
retlux
i I i O
F ~
F \
Compound 20 (as free base) p
1) Borane-methyl sulfide , THF ~ I CH3
F w NHZCI
2) HC1
i
F ~ I
Compound 60 (HCl salt)
Alternatively, Compound 60 was synthesized
from commercially available starting materials in the
following four step reaction sequence. The first
intermediate in this synthetic route,
ethyl-N-benzyl-N-methyl-3-aminopropionate, was prepared
by conjugate addition of N-benzylmethylamine to ethyl
acrylate. The ester functionality of the first
intermediate was then reacted with two equivalents of
Grignard reagent (prepared from 1-bromo-3-fluorobenzene)
to provide N-benzyl-N-methyl-3-hydroxy-3-
(bis-3-fluorophenyl) propylamine. The Grignard reaction
product was then dehydrated in a mixture of 6N
HC1/acetic acid to yield
N-benzyl-N-methyl-3-(bis-3-fluorophenyl)-2-propenamine.
Catalytic hydrogenation of this material as its
hydrochloride salt in ethanol over Pearlman~s catalyst
[Pd(OHZ)/C] provided, after recrystallization from ethyl
CA 02560002 2006-10-05
WO 97/46511 PCT'/US96/Z069'7
171
acetate, colorless, needles of Compound 60 as the
hydrochloride salt.
In a 500-mL, 3-necked flask equipped with
thermometer, reflux condenser, and a 125-mL addition
funnel [charged with ethyl acrylate (88.3 mL, 81.5 g,
0.815 mol)] was placed N-benzylmethylamine (100 mL,
94.0 g, 0.776 mol). The ethyl acrylate was added
dropwise to the stirring reaction mixture over a period
of e0 min. After stirring for 18 h at room temperature,
the product was vacuum distilled and the fraction
containing product was collected at 78-95°C
(0.12-0.25 mm Hg), (138 g, 80% yield): Bp 78-95°C
(0.12-0.25 mm Hg); TLC, Rf = 0.23 [hexane-EtOAc (5:1)],
Rf = 0.57 [MeOH-CHC1, (100:5)]; GC, tR - 6.06 min; MS,
221 (M') , 206 (M-CH,) , 192 (M-C2H5) , 176 (M-OCzHS) ,
144 (M-C5H5) , 134 [CHzN(CH3) CHzPh] , 120 (N(CH3) CH~Ph] ,
91 (C,H,) , 77 (CbHs) , 42 (CHzCH2N) ; -H NMR (free base,
CDC13) d 1.25 ppm (t, J = 7.1, 3H, CHzCh,) , 2.20 (S, 3H,
NC~j3) , 2.5I (t, J = 7.3, 2H, COC~z) , 2.74 (t, J = 7.2,
2H, C~iN), 3.51 (s, 2H, NC$zPh), 4.13 (q, J = 7.1, 2H,
OCgZCH,) , 7.18-7.35 (m, 5H, ArH) ; 1'C NMR (free base,
CDC1~) d 15.2 (CH~~Hi) , 34 .0 (CO~HZ) , 42.9 (N~H3) ,
53.8 (N~F~Z) , 61.4 (O~HzCH3) , 63.1 (~HZPh) , 128.0 (CH) ,
129.2 (CH), 130.0 (CH), 139.9 (q), 173.7 (q).
In a 5-L, four-necked, round-bottom flask,
under nitrogen, were placed Mg (51.5 g, 2.12 mol,
turnings, washed with THF (2 x 300 mL)] and THF (2 L).
_ An addition funnel was charged with
1-bromo-3-fluorobenzene (neat, 392.8 g, 2.24 mol).
CA 02560002 2006-10-05
WO 97!46511 PCT/US96120697
172
One-twentieth of the bromide was added to the magnesium
suspension followed by one crystal of iodine. After
initiation of the Grignard reaction the remaining
1-bromo-3-fluorobenzene was then added to the refluxing
mixture over a period of 50 min. The reaction was
refluxed for an additional 45 min. To the refluxing
solution of Grignard reagent was added a solution of
ethyl N-benzyl-N-methyl-3-aminopropionate (187.5 g,
0.847 mol) in THF (100 mL) over a period of 20 min.
After the ester addition was complete, the reaction was
refluxed for !h. The reaction was then cooled in an ice
bath. Saturated NH4C1 (aq., 400 mL) and Hz0 (400 mL)
were added and the mixture was transferred to a
separatory funnel. The organic layer was separated and
the aqueous layer was extracted once with THF (400 mL).
The combined organic layers were washed with satd. NaCl
(2 x 200 mL, aq.), dried (anh~ Na2S04), filtered through
paper, and rotary evaporated vacuum to yield 281.5 g
(90a) of crude product as an orange, viscous oil. This
material (281.6 g, 0.766 mol) was dissolved in
acetonitrile (1.4 L). Concentrated hydrochloric acid
(65.0 mL, 0.786 mol, 12N) was added to the stirring
filtrate.-The crystallizing mixture was then cooled to
-20 °C for 17 h. The product was collected, washed with
cold acetonitrile (800 mL~, and dried to provide a white
solid, 235.6 g (69% yield from the ester). For
analytical purposes, the hydrochloride salt was further
purified by recrystallization from acetonitrile:
mp 194-197°C (uncorr.); TLC, Rf = 0.23 [hexane-EtOAc
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
173
(5:1) ] , Rf = 0.85 (MeOH-CHC1, (100:5) J , R~ = 0.72
[MeOH-CHC13 (100:3)]; GC, tR = 10.93 min; MS, 367 (M'),
272 (M-CSH4F) , 258 (M-CHZPh-Hz0) . 219 ( (C6H4F) ZCH] ,
148 [CHzCHZN(CH3) CHZPh] , 134 (CHzN (CH3) CHZPh] , 91 (C,H,) ,
42 (CHzCHzN) ; 1H NMR (free base, CDClj) d 2.18 (s, 3H,
NC$3) . 2.41 (m, 2H, CHC$z) , 2. SS (m, 2H, C$ZN) , 3 .42 (s,
2H, CHzPh) , 6.86 (dt, Jl = 8.5, Jz = 1.8, 2H, Ar-H) ,
7.18-7.30 (m, 10H, Ar-j~), 8.33 (bs, 1H, O$); wC NMR
(free base, CDC1,) d 35.6 (CH~H2) . 41.5 (CH3, NCH,) ,
54.3 (CHz, ~HZN) , 62.6 (CH2, ~HZPh) , 113.1 (d, J = 23, CH,
Ar-C;,;.) , 113.5 (d, J = 23, CH) , 121 .2 (d, J = 3, CH) ,
127.5 (CH), 128.5 (CH), 129.2 (CH), 129.5 (CH),
129.6 (CH), 137.0 (q), 150.2 (q), 162.8 (d, J = 243, q,
Ar-C,.3~ )
In a 5-L, 3-necked reaction vessel, equipped
with an overhead mechanical stirrer, reflux condenser,
and thermometer, was placed
N-benzyl-N-methyl-3-hydroxy-3-bis
(3-fluorophenyl)propylamine hydrochloride (225.4 g,
0.559 mol), 6N HC1 (1392 mL) and glacial HOAc (454 mL).
The suspension was heated in a water bath (80-85 °C) and
stirred for 18 h. After 18 h of heating, the reaction
mixture was cooled in an ice/MeOH bath. Ethyl acetate
(500 mL) was added to the cooled reaction mixture. NaOH
(10N, 1.7 L) was then added to the cooled mixture over a
period of 25 min at such a rate as to keep the
temperature below 40 °C. The mixture was transferred to
a 6-L separatory funnel. The organic layer was
separated and the aqueous layer was extracted with ethyl
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
174
acetate (2 x 500 mL). The combined organic layers were
washed with satd. NaCl (2 x 100 mL, aq.), dried NazS04
(250 g), rotary evaporated, and then dried under vacuum
to provide 185.6 g (9~o yield) of the free base as a
fluid, brownish-colored oil.
The material above was stirred with hexane
(1.5 L). The resulting solution was filtered through
paper. 4M HC1 ~in dioxane (146 mL) was added dropwise
with stirring to the filtrate over a period of 5 min.
The semi-translucent solvent was then decanted away from
the light-yellow colored, semisolid precipitate. The
crude hydrochloride salt was dissolved in refluxing
ethyl acetate (600 mL) and was filtered. The filtrate
was then thoroughly cooled in an ice bath, and hexane
(110 mL) was slowly added, with vigorous stirring.
After cooling in an ice bath for 2 h, the entire flask
filled with a white crystalline solid. '='his material
was collected on a filter funnel, washed with ice-cold
hexane/ethyl acetate [(1:4), 400 mL], and dried to yield
128.7 g, 59.7°s of a white solid. On standing the mother
liquor precipitated another 14.8 g of an off-white
solid. Total yield 128.7 g + 14.8 g = 143.5 (67%). Mp
141-142 °.C (uncorr.); TLC, Rf = 0.20 [hexane-EtOAc
(5:1) ] , RF = 0.75 [MeOH-CHC13 (100:5) ] , Rf = 0.49
[MeOH-CHC13 (100:3)]; GC; tR = 10.40 min; MS, 349 (M'),
330, 301, 281, 258 (M-CH~Ph) , 240, 229 [M-N (CHI) CHzPh] ,
201, 183, 146, 133, 109, 91 (CHZC5H5) , 65, 42 (CHzNHCH3) ;
=H NMR (free base, CDCl,) d 2.20 ppm (s, 3H, NCI-~,) ,
3.08 (d, J = 6.8, 2H, C~zN) , 3.47 (d, J < 1, 2H, C~ZPh) ,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
175
6.29 (t, J = 6.8, 1H, Cg), 6.85-7.04 (m, 6H, ArH),
7.19-7.35 (m, 7H, ArH).
N-Henzyl-N-methyl-3-bis(3-fluorophenyl)allylamine
hydrochloride (120.0 g, 0.311 m01) was dissolved in abs.
EtOH (1250 mL). Pd(OH)z/charcoal (10.0 g, -.20% Pd, Fluka
Chemical) was added. The reaction mixture was stirred
under a steady flow of hydrogen gas for 18 h at 25 °C
(atmospheric pressure). The mixture was then filtered
through Celite'/fritted glass, the catalyst was washed
with EtOH (2 x 50 mL), and the solvent was removed under
reduced pressure to yield 95.4 g, 103% of crude product.
This material was dissolved in refluxing ethyl acetate
(300 mL) with vigorous stirring and filtered. The flask
was allowed to stand for 2 h at 25 °C, during which time
the hydrochloride salt began to crystallize as needles.
The flask was then cooled, the produce was collected,
washed with ice-cold ethyl acetate (20 mL), and dried to
yield 73.7 g, 80%, of Compound 60 as a white,
crystalline solid. Mp 129-130°C; L'V/Vis, a = 2.1 x 10'
L~mol'1-cm'~ (264 nm, EtOH, 25 °C, linear range: O.OS-0.20
mg/mL); TLC, Rf = 0.00 (hexane-EtOAc (5:1)), Rf = 0.07
[MeOH-CHC13 (100:5) J , Rf = 0.19 [MeOH-CHC1,-NH,OH
(100:5:1)]; GC, tR = 7.45 min; MS, 261 (M'), 229, 215,
201, 183, 164, 150, 138,122, 101, 83, 75, 57, 42
(CHZNHCH3) ; =H NMR (HC1 salt, CDC1, + 1 gtt MeOD) b 2.56
(m, 2H, NC~jz) , 2.60 (s, 3H, NCjj,) , 2.85 (t, J = 8.0,
2H, CHCHZ), 4.11 (t, J = 8.0, 1H, C~), 6.87-6.98 (m, 4H,
ArH) , 7 . 06 (d, J = 7 . 7, 2H, Arz,~~H) , 7. 25 (dd, J1 = 6 , Jz
CA 02560002 2006-10-05
WO 97146511 PCT/US96l20697
176
- 8, ArH); ~'C NMR (HC1 salt, CDC13 + 1 gt MeOD) 3 30.9
( CHZ , CH~HZ ) , 3 2 . 7 ( CH3 , N~H3 ) , 4 7 . 6 ( CH, ~HCH~ ) , : 7 . 8
(CHz, ~H~N) , 113 . 9 (J = 21, ArC~,Z, or ArC4,4, ) , 114 . S (d, J
- 22, ArCz,~, or ArC,,4~} , 123.2 (d, J = 3, Ar-CS,b.; , 130.3
(d, J = 9, Ar-C5,5.) , 144.7 (d, J = 7, Ar-C1.:.) , ? 52.9 (d,
J = 245, Ar-C~,3,) ; IR: KBr pellet (cm-1} , 3436.9,
2963.4, 2778.5. 2453.7, 1610.6, 1589.3, 1487.0, ,445.3,
1246.0, 764.5; solubility: 2 g/mL (Hz0), 1 g/mL (EtOH);
anal. calcd. for C16H1~NF_.HC1 (Karl Fischer: 0.25's Hz0)
C, 64.37; H, 6.11; N, 4.69; found: C, 64.14; H,
6.13; N, 4.69.
/
G2H5 ~~HJ~1B9~
--~ CtHI~~~~
0 HiNy H 6 ~~CH~ (2equvt.)
w W W W
pH / NHCH~-HCI
/ NHCH~.HCI H~ F / / NNCN~.NCI
., ~ ~ Hi
'-'~ /
/ ~ Pd(OH)=/C
F F
Canpound 60
(NCI tilt)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
177
Compound 105 was prepared by selective
reduction of its corresponding alkene by catalytic
hydrogenation over Pd/C.
Compound 61 was prepared from
2-bromo-4-fluoroanisole and 3-fluorobenzaldehyde as
described for Compound 24. 3C/EI-MS (R~ = 9.22 min) m/z
(relative intensity) 277 (M', 74), 260 (46), 245 (35),
231 (44), 229 (34), 217 (24), 203 (28), 201 (31), 183
(28) , 154 (24) , 133 (1 9) , 109 (100) .
Compound 62 was prepared from 2-bromoanisole
and 2-methoxybenzaldehyde as described for Compound 24.
GC/EI-MS (R~ = 9.30 min) rn/z (relative intensity) 271
(M', 100), 254 (17), 240 (23), 225 (40), 223 (45), 207
(22), 181 (32), 165 (31), 136 (48), 121 (98), 91 (83).
The synthesis of Compound 63 was accomplished
as follows.
Alcohol A was obtained from
3-fluorobenzaldehyde as described for product A of the
Compound 24 synthesis.
To alcohol A (10.275 g, 47 mmol) in 200 ml of
ethanol was added 1.6 g of loo Pd/C and 1 ml of
concentrated HC1. This mixture was hydrogenated for
3 hr ~~-60 psi, then filtered and concentrated to give
the diphenylmethane B.
Product B (2.01 g, 9.86 mmol) was dissolved in
20 ml of THF and cooled to -78°C. Butyl lithium
(4.4 ml. 10.8 mmol, 2.5 M in hexanes) was added slowly
by syringe, and then the reaction stirred for another 30 min
at -7s°C. To this orange solution was added cyclopentene
oxide (0.9 ml, 10.3 mmol). The reaction was allowed to
stir 3 hours while warming slowly to room temperature.
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
178
The reaction was quenched with 150 ml of 10% HC1 and
extracted 3 times with ether. The ether layer was dried
over sodium sulfate and concentrated to give 2.5 g of
the alcohol C.
To the alcohol C (1 g, 3.5 mmol) in 10 ml of
dry THF was added triphenylphosphine (1.37 g, 5.2 mmol)
in 5 ml of THF and p-nitrobenzoic acid (0.87 g, 5.2
mmol) in 5 ml of THf. This solution was cooled to 0°C
followed by the addition of DEAD (0.82 ml, 5.2 mmol),
and allowed to stir overnight. The reaction was
partitioned between water and ether. The ether was
removed in vacuo and the resulting oil was
chromatographed on silica gel in hexane/ethyl acetate to
yield 365 mg of tre cis-ester. This ester was
hydrolyzed in methanol with potassium carbonate by
stirring overnight. After removal of the methanol, the
residue was taken up in ether, washed with water, dried
over sodium sulfate and concentrated to give 250 mg of
the cis alcohol D.
To the alcohol D (.25 g, 0.9 mmol) in 5 ml of
dry THF was added triphenylphosphine (342 mg, 1.3 mmol)
in 5 ml of THF and phthalimide (191.3 mg, 1.3 mmol) in
5 ml of THF. This solution was cooled to 0°C followed
by the addition of DEAD (0.205 ml, 1.3 mmol), and
allowed to stir overnight. The reaction was partitioned
between water and ether. The ether was removed in vacuo
and the resulting oil was chromatographed on silica gel
in hexane/ethyl acetate to yield 100 mg of the
phthalimide E.
To a solution of the phthalimide E (100 mg) in
20 ml of ethanol was added 8.8 mg of hydrazine hydrate.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96I20697
179
The solution was refluxed for 5 hours then stirred at
room temperature overnight. The reaction was worked up
by adding 1 ml of cone. FiCl and filtering off the white
solid. The resulting solution was concentrated to
S dryness and the solid taken up in ether and aqueous
sodium hydroxide. The ether layer was dried over sodium
sulfate and concentrated to a white solid. This was
taken up in a small amount of ether and treated with
drops of 1M HC1 in ether. After stirring overnight,
10 the white solid was collected by filtration and dried to
give 50 mg of Compound 63, as the hydrochloride salt.
GC/EI-MS (R~ = 9.22 min) rrr/z (relative intensity)
2B7 (M', 45), 270 (12), 201 (63), 183 (81), 133 (38),
109 (43), 83 (441, 56 (100), 43 (37).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
180
1) M, eti;er ~ i
F \ I OH ~ I
F
F 0 Pd/C , Hz
w ~ I
F Br ~ I E.tOH , HCl
F F
A B
i I
1) DuLi , THF F = 1 ) p~Nitrobenzoic Acid, F v ~(
OH Ph3P , DEAD , 'I'HF ~ OH
i C
2) Cyclopentene Oxide ~ ~ 2) Na~C03 , MeOH
F F
C D
i I
Phthalimide , Ph3P, F \ 0 t) Hydrazine , EtOH F
NH~CI
i / 2) HCI , Ether
DEAD, THF ~ I 0~ F
F
E Compound 63
(HC1 salt)
The synthesis of Compound 64 was done as described for
Compound 53 except that the inversion step (product C to
D) was omitted in order to obtain the cis amine as the
final product. GC/EI-MS (R~ = 8.28 min) m/z (relative
intensity) 287 (M', 15), 270 (4), 201 (13), 183 (15),
133 (11), 109 (16), 84 (43), 56 (100), 43 (32).
The synthesis of Compound 65 was accomplished
as follows.
The ketone A was synthesized similarly to
ketone B in the Compound 24 synthesis using
2-methylphenylmagnesium bromide and 2-methylbenzaldehyde
as starting materials. This ketone was converted to the
final product using the procedure outlined for Compound
58. GC/EI-MS (R~ = 7.84 min) m/z (relative intensity)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
181
239 (M', 88), 222 (14), 207 (100), 193 (46), 178 (71),
165 (60), 130 (39), 120 (40), 115 (51), 104 (40),
91 (38) , 77 (21) .
1) Mg, ether , CH3 CH3
I
2) Q1 i~( H ~ I OH w 0
~0 PCC
H3 -~ H3
Br ' I CH2Clz ~ I
CH3
A
CH3 , CH3
OH ~ I OH
BuLi , AcCN CN NH2
Raney Ni , H2, NaOH
H3 i H3 i
THF
Ethanol
CH3 ~ CH3
v I ~ NH~C! w I NH~CI
conc. HCl , teat H3 Pd(OH)2/C , HZ , HCI H3
Ethanol ~ I
Compound 65
;HC1 salt)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
182
Ccmpound 119 was synthesized in a seven-step reaction
sequence starting from commercially-available trans-3-
fluorocinnamic acid. This synthetic route is
conceptually similar to that reported in the literature
[U. S. Patent 4,313,896 (1982)] for related analogs.
However, the three final steps were performed using a
significantly different reaction sequence than that
reported. The cinnamic acid was reduced and chlorinated
in three steps to the corresponding
3-(3-fluorophenyl)propylchloride. This compound was
brominated with NBS (N-bromosuccinimide) and the
resulting trihalide was then reacted with
3-fluorophenol. The resulting ether was converted to
the final product using a Gabriel synthesis.
Trans-3-f~uorocinnamic acid (25.0 g, 150.4 mmol)
was dissolved in abs. EtOH (250 mL) and hydrogenated over
loo Pd/C (2.5 g) in a Parr apparatus at 60 psig, 50°C, for
1 h (hydrogen uptake: calcd. 245 psig; found 260 psig). The
reaction mixture was filtered and evaporated to yield a
crystalline product (23.0 g, 89%). GC, tk = 4.43 min; MS,
168 (M') .
Under' a stream of dry nitrogen, at 0-10°C, a
solution of 3-fluorohydrocinnamic acid (22.0 g, 131 mmol) in
THF (100 mL) was added dropwise, over a period of 15 min, to
a suspension of LiAlH~ (4.23 g, 111 mmol) in THF (200 mL).
The reaction was heated to reflux for a period of 1 h and
then worked-up according to Fieser & Fieser's Reagents for
Organic Synthesis (Vol. 1, 1967) to provide a white solid
(20.1 g, 99%). GC, tR = 3.74 min; MS, 154 (M').
A solution of 3-(3-fluorophenyl)-1-propanol
(15.0 g, 97.4 mmol) and triphenylphosphine (36.0 g,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
183
137.3 mmol) in CC14 (150 mL) was refluxed for 19 h.
Additional P(C5H5)3 (3 x 3.0 g, 3 x 11.4 mmol) was added
periodically over a period of 24 h. The resulting
precipitate was removed by filtration and the solids were
washed with hexane. The filtrate was evaporated under
vacuum and the residue was suspended it hexane (200 mL) and
then filtered. Evaporation of the filtrate provided 16.0 g
(95.10) of crude product which was purification by silica
gel flash chromatography, elution with hexane, to provide
14.7 g (870) of a colorless liquid. GC, t,R = 3.63 min;
MS, 172/174 (M') .
A solution of the above chloride (12.0 g,
69.5 mmol), N-bromosuccinimide (17.3 g, 97.2 mmol), and
dibenzoyl peroxide (0.06 g) in CC1~ (75 mL) was refluxed for
1 h. The reaction mixture was then cooled in an ice bath,
filtered, and the solids were washed with hexane. The
filtrate was evaporated to provide 17.9 g (100x) of product.
GC, t.q = 5.21 min; MS, 251/253 (M') .
A mixture of 3-bromo-3-(3-fluorophenyl)
-1-propylchloride (4.0 g, 15.9 mmol), 3-fluorophenol
(1.98 g. 17.7 mmol), and KZC03 (2.65 g, 19.2 mmol) suspended
in acetone (80 mL) was refluxed for 15 h. The volatiles were
them-removed under vacuum and the resulting residue was
suspended_in a mixture of hexane (200 mL) and NaOH (0.1N,
100 mL). The layers were separated and the organic layer
washed, O.1N NaOH (100 mL) and Hz0 (100 mL), dried
(anh. NaZS04), and evaporated in vacuuo. The resulting
residue was chromatographed on silica gel, elution with
hexane followed by hexane/EtOAc [100:1) then [40:1] to
provide 1.64 g (370) of product as a colorless oil. GC,
tR = 7.28 min; MS, 282/283 (M'); TLC rF = 0.3, hexane/EtOAc
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
184
[40:1] .
A solution of 3-(3-fluorophenyl)-3-
(3-fluorophenoxy)-1-propylchloride (1.52 g, 5.38 mmol) and
potassium phthalate (1.20 g, 6.48 mmol) was heated to 90°C
in DMF (30 mL) for a period of 2 h in a nitrogen. atmosphere.
The reaction mixture was then cooled and poured _:~to H~O
(100 mL). The resulting solution was extracted with Et~O
(2 x 100 mL). The organic extract was washed, sat.
NaCl (100 mL) and H_,O (2 x 100 mL) , dried (anh. ':a,S04) , and
evaporated under vacuum to provide 2.17 g of cr~.:e product.
The material was chromatographed on silica gel, elution with
hexane/EtOAc [40:1] and then (20:1] to provide G~ter
evaporation 1.81 g (86%) of product as a glass.
A solution of N-phthaloyl-3 - ( 3 -f luorop:-:enyl ) - 3
-(fluorophenoxy)-1-propylamine (1.74 g, 4.42 mmo-) and
anh. hydrazine (1.43 g, 44.6 mmol) in abs. EtOH '30 mL) was
refluxed for 1 h. The reaction was cooled and evaporated
under vacuum. The resulting material was suspe.~.ded in Et~O
(75 mL) and washed with 0.2N NaOH (2 x 25 mL). .'he organic
layer was dried (anh. NazS04), and evaporated under vacuum to
provide 1.04 g (89.3%) which was purified by reverse-phase
chromatography (Vydac Prep. C18; 264 nm; 50 mL/min;
gradi.e~ elution ACN/O.lo HC1 aq., 10%-50% over 20 min;
rt = 17.4 min], to yield 0.89 g (67%) of Compound 119 as a
hygroscopic hydrochloride salt.
CA 02560002 2006-10-05
WO 97!46511 PCT/US96120697
185
ON I / ON ~ / ON F ~ / CI
0 0
\ \
/ CI ~ / 0 H ~ ~ / NN7CI
F F
/ CI 0 ~ O \ 0
F~ \ \
Br ~ / ~ / ~ /
F G F
Cartpa~lW 111
pG y
Compounds 118, 120-122 and 137 were prepared in a manner
similar to the procedures used for the preparation of
Compound 119.
Compound 113 was synthesized from commercially
S available .4,4-diphenylcyclohexenone in three steps. First,
the alkene in the starting material was reduced by means of
catalytic hydrogenation. Methoxylamine formation followed by
reduction using standard procedures.
The synthesis of Compounds 188 and 189 was
l0 accomplished as follows.
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
186
\ \ \
/ JNH7 / NH= / NH=
CI " C6.r.!-HPLC ~~ " H,"~ ~'' ~~ H
\) \
' NCO H~CO NCO
nd 1~t Compound 11t Compound 1~!
Compounds 188 and 189
The enantiomers of Ccmpound 136 were separated by
analytical chirai HPLC. Aliquots (20 fig) were injected onto
a Chiralcel-OD-R (Chiral Technologies, Inc., Exton, PA)
reversed-phase HPLC column (0.46 x 250 mm) using the
following conditions: gradient elution, 40%-70% ACN (60-
30% 0.5N KTFA) over 30 min; flow rate, 1 mL/min; detector,
264 nm, Two identically-sized peaks were collected at
22.0 and 24.4 min. GC/MS analysis of the two samples
indicate that both materials have identical GC retention
times as well as identical mass spectra.
The synthesis of Compound 151 was accomplished as
follows.
\ \
/ ~~ NH / NH2
j[ ' II
o ~ o
\ F \
F
Compound 151
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
187
3,3-Bis(3-fluorophenyl)propanamide (Compound I51)
A solution of liquid anh. ammonia (10 mL) in
CHZClz (50 mL) at -78°C was treated with a solution of
3,3-bis(3-fluorophenyl)propionyl chloride (2.19 g,
7.81 mmol) in CHzClz (25 mL). The reaction was then stirred
at ambient temperature for 15 min and was then diluted with
diethyl ether (500 mL), washed three times with 10% HC1,
three times with 1N NaOH, and finally once with HBO. The
organic layer was dried (anh. Na,S04) and evaporated to give
the primary amide as a white solid (2.01 g, 98°s).
The synthesis of Compound 156 was accomplished as
follows.
/ ~ VCCIi_POfOEtI~ / / CN H. / NI tRsnevt / NHp
Compound 158
5-Cyanomethylidino-10,11-dihydrodibenzo(a,d)cycloheptene
To a solution of diethyl cyanomethylphosphonate
(9.66 g, 54.5 mmol) in dry N,N-dimethylformamide (DMF,
40 mL) was added NaH (60% dispersion, 2.20 g, 55.0 mmol)
over a period of 2 min. The reaction was stirred for 10 min
and then a solution of dibenzosuberone (10.3 g, 49.6 mmol)
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
188
in dry DMF (10 mL) was added over a period of 2 min. The
reaction was stirred at 80°C for 4 h under N,. Water
(200 mL) was added and the reaction mixture was extracted
with Et20 (2 x 100 mL). The combined organic layers were
rotary evaporated to less than 50 mL. The resulting
crystals were collected and washed with cold Et20 (2 x 50 mL)
to yield 7.48 g (65.30).
5-(2-Aminoethyl)-5H-10,11-dihydrodibenzo(a,dJcycloheptene
hydrochloride (Compound 156)
5-Cyanomethylidino-10,11-dihydrodibenzo[a,d]cycloheptene
was dissolved in EtOH (100 mL). 1N NaOH (10 mL) and Raney°
nickel (aq. suspension, 0.50 g) were added. The reaction
mixture was shaken under 60 psig H2 at 50°C for 22 h, and was
then filtered through Celite~. The filtrate was rotary
evaporated and the residue was dissolved in Et20 (100 mL),
washed with satd. aq. NaC1 (50 mL) and H20 (50 mL). The Et20
layer was dried (anh. Na2S04) and rotary evaporated to give
the crude product (850 mg) as a colorless oil. This oil was
dissolved in EtOAc (5 mL) and filtered. 1.0M HC1 (5 mL) in
Et~O was added to the filtrate and a white, crystalline solid
precipitated. This material was recrystallized from EtOH
(S mL)-Et20 (12 mL) to yield 600 mg (50.7%) of product as a
white, powder.
The synthesis of Compound 167 was accomplished as
follows.
N !4i
o ~ j cH,c. ~cco, o I ~ ~ ~ I b
11 Tili-Pr0) OCH~
OH OCH~ 21 NaBIjCN
Compound 167
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
189
2-Methoxypropiophenone
A mixture of 2-hydroxypropiophenone (3.00 g,
20.0 mmol), iodomethane (3.40 g, 24.0 mmol), and KzC03
(granular, anh.; 13.8 g, 99.9 mmol) was refluxed in acetone
(75 mL) for 18 h. The reaction mixture was cooled to room
temperature and the inorganic salts were removed by
filtration. The filtrate was evaporated under vacuum to
give an oil which was subsequently dissolved in diethyl
ether (200 mL) and then washed with O.1N NaOH (3 x 50 mL)
followed by Hz0 (50 mL). The organic layer was dried (anh.
NaZS04), filtered, and evaporated to an orange oil (3.17 g,
96.9%). This material was used in the following step
without further purification. TLC, Rf 0.55 (1o MeOH:io
IPA:CHC1,); GC, tr 4.58 min; MS, m/z 164 (M+1).
(R,S)-N-1-(2-rnethoxyphenylpropyl)-3,3-diphenylpropylamine
(Compound 167)
A solution of 2-methoxypropiophenone (0.848 g,
5.17 mmol), 3,3-diphenylpropylamine (1.00 g, 4.70 mmol), and
titanium(IV) isopropoxide [Ti (OCH (CH3) Z) 4; (1 . 76 mL,
5.88 mmol, 1.25 equiv)] was stirred at room temperature for
6 h. EtOH- (2 mL) was then added, followed by sodium
eyanoborohydride (0.295 g, 4.70 mmol) in portions over a
period pf 10 min, and the reaction was then stirred for
18 h. The reaction mixture was then poured into diethyl
ether (200 mL) and the resulting suspension was centrifuged
to remove the titanium precipitate. The supernatant was
collected and the pellet was rinsed with diethyl ether
(200 mL). The combined organic washings were evaporated
under vacuum to give a crude oil which was chromatographed
on silica gel (elution with 4% MeOH-CHZClZ) to provide 647 mg
(38%) of product. The material was then dissolved in
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
190
diethyl ether (50 mL), filtered, and excess ethereal HC1 was
added to precipitate the hydrochloride salt (125 mg, 7.40)
as a white solid; TLC, Rf 0.25 (4% MeOH-CH,C12) ; GC,
tr = 11.2 min; MS, m/z 359 (M+).
The synthesis of Compounds 172 - 176 was accomplished as
follows.
/ /
OH
CHO F \ I OH \ O F \
\ m-FC,,H~MgBr PCC ~ CH,CN
FI/ ~ / FI/ FI/
OCH~
OCH~ OCFi~ OCH~
OH
\ NFiZ~NCI
\ I CN F \ I / NHZ.HCI / I
4 BH~SfCH,): H:, Pd/C
I \ 2) HC1 r \ I \
F / F / /
OCH~ OCH~ OCHz
R HCO,Et Compound 173
.) Btu S(Ctlj)=
BHy
/I /I H /
b
\ ~CH~ BB \ ~~CH~ \ NH2
5 - -
\ \
F I / ~ / I /
OCH~ OH OH
Compound 171 Compound 176 Compound 175
R,S)-3,3'-Difluoro-4-methoxybenzhydrol
A mixture of Mg° turnings (2.45 g, 101 mmol),
1-bromo-3-fluorobenzene (17.6 g, 100 mmol), and dry THF
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
191
(200 mL) was carefully heated to reflux for 30 min. While
still refluxing, 3-fluoro-p-anisaldehyde (15.3 g, 99.3 mmol)
in THF (100 mL) was added over a period of 5 min. The
reaction temperature was maintained for 30 min, cooled to
room temperature, and then the reaction was quenched with
satd. aq. NHqCl (200 mL). The organic layer was separated,
washed with satd. aq. NaCl (2 x 200 mL), dried (anh. Na~S04),
and rotary evaporated to yield 23.5 g (94.4%) of product as
an orange-brown oil.
3,3'-Difluoro-4-methoxybenzophenone
Pyridinium chlorochromate (22.3 g, 103 mmol) was
added to a solution of 3,3'-difluoro-4-methoxybenzhydrol.
(23.5 g, 93.8 mmol) in CH2C12 (300 mL) and the reaction
mixture was stirred for 16 h. Diethyl ether (500 mL) was
added and the reaction mixture was filtered through Celite°.
The filtrate was rotary evaporated and t::e resulting oil was
flash chromatographed (gradient elution of hexanes to
1:1 hex-EtOAci. The TLC-pure fractions were rotary
evaporated to yield 1.58 g of a white sciid_ The rest of
the impure fractions containing product were combined and
rotary evaporated to the point where crystals began to form.
Additional hexane (300 mL) was added and the crystallizing
solution was allowed to stand. The resulting crystals were
collected and washed with hexanes (2 x SO mL) to yield
6.81 g of product. The two batches were combined to afford
a total yield of 8.39 g (36.1%).
(R,S)-a-Cyanomethyl-3,3'-difluoro-4-methoxybenzhydrol
To dry THF (100 mL) was added butyllithium (2.6M
in heptane; 16.0 mL, 41.6 mmol) at -78°C. Acetonitrile
(2.20 mL, 42.1 mmol) was added over a period of 1 min and
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/10697
192
the reaction was stirred at -78°C under N~ for 30 min. A
solution of 3,3'-difluoro-4-methoxybenzophenone (8.38 g,
33.8 mmol) in anh. THF (50 mL) was added to the reaction
over a period of 5 min and the solution was stirred at -78°C
for 30 min. The cold bath was removed and the reaction was
allowed to warm for 30 min. Satd. aq. NHqCl (100 mL) was
added to quench the reaction. The THF layer was separated,
washed with satd. aq. NaCl (2 x 25 mL), dried (anh. NazS04),
rotary evaporated, and dried under vacuum to yield 10.1 g
(1030) of product as a yellow oil.
(E) - and (Z) -3- (.3-Fluoro-4-methoxy) -3-
(3-fluorophenyl)a11y1amine hydrochloride (Compound
I72)
(R,S)-a-Cyanomethyl-3,3'-difluoro-4-methoxybenzhydrol
(9.77 g, 33.8 mmol) was dissolved in dry THF (200 mL) and
heated to boiling (ao condenser). Under a stream of
nitrogen, borane-dimethyl sulfide complex (BH,~S (CH;) 2, 10 . 1M;
16.8 mL, 170 mmol) was added carefully over a period of
2 min to the boiling solution. Boiling was then maintained
for 15 min until most of the THF was gone. The reaction
mixture was then cooled in an ice bath. Ice (10 g) was
carefully added, followed by H20 (50 mL). The reaction was
then.heated to near boiling and 12.1N HC1 (100 mL) was
added. The reaction was boiled (no condenser) for 30 min and
was then cooled in an ice bath, basified with lON NaOH
(100 mL), and extracted twice with EtzO (200 mL, 100 mL).
The combined ether layers were washed with 1N NaOH (50 mL)
and H~O (50 mL), dried (anh. NaaS04), and rotary evaporated.
The resulting oil was flash chromatographed (CHC1,; 1:100
MeOH-CHC1,; 1:10 MeOH-CHC1,) through flash silica gel to
afford 6.83 g of a yellow oil. This oil was dissolved in
CA 02560002 2006-10-05
WO 97/46511 PCTI~JS96/10697
193
EtOH (2 mL) and EtzO (10 mL). 1.0M HC1 in Et~O (27 mL) was
added and the solution was rotary evaporated to yield 7.10 g
(67.5%) of product as a solid, yellow foam.
(R, S) -3- (3-Fluoro-4-methoxyJ -3- (3-fluorophenylJpropylamine
maleate lCompound 173)
The mixture of (E)- and (Z)-3-(3-fluoro-4-
methoxy)-3-(3-fluorophenyl)-allylamine hydrochlorides
(7.10 g, 22.8 mmol) was dissolved in EtOH (200 mL) and a
suspension of palladium on charcoal (10% Pd; 0.71 g) in HZO
(3.5 mL) was added. The reaction mixture was then shaken
under 60 psig Hz for 18 h and subsequently filtered through
Celite9. The filtrate was rotary evaporated, the residue was
dissolved in EtOAc (25 mL) and Et20 (100 mL), and was
basified with sat. aq. NaHC03 (25 mL). The organic layer was
separated, dried (anh. Na~S04), and rotary evaporated to
yield 6.28 g of an oil. This ail and malefic acid (2.59 g)
were dissolved into hot EtOAc (100 mL). Diethyl ether
(70 mL) was added and crystals soon began to form. The
crystals were collected and dried to yield 2.45 g (27.3%) of
a white powder. The combined filtrate and washings afforded
more crystalline product out upon standing. The second crop
was filtered, washed with 1:1 EtOAc-EtzO (2 x 25 mL) and
EtzO-11:x 25 mL), and dried to provide 3.69 g (41.2%) of a
white powder. The total yield was thus 6.14 g (68.5%).
(R, S) -3- (3-Fluoro-4-methoxy) -3- (3-
fluorophenyl)propylformamide
(R, S) -3- (3-Fluoro-4-methoxy) -3- (3-
fluorophenyl)propyl amine maleate (3.12 g, 7.93 mmol) was
free-based in a mixture of EtOAc (25 mL), EtZO (100 mL), and
satd. aq. NaHC03 (25 mL). The organic layer was separated,
dried (anh. NazSOa), and rotary evaporated. A solution of
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
194
the amine in ethyl formate (75 mL, 930 mmol) was refluxed
for 17 h. The reaction solution was rotary evaporated to
yield 2.38 g (98.3%) of formamide as a light-orange, viscous
oil.
(R,S)-N-Methyl-.3-(3-fluoro-4-methoxy)-3-(3-
fluorophenyl)propyl amine maleate (Compound 174)
(R,S)-3-(3-Fluoro-4-methoxy)-3-(3-
fluorophenyl)propyl formamide (2.27, 7.43 mmol) in THF
(100 mL) was heated to boiling (no condenser). Borane-
dimethyl sulfide complex (10.1M; 2.30 mL, 23.2 mmol) was
added carefully over a period of 2 min to the boiling
solution. Boiling was then maintained for 15 min. The
reaction was then cooled in an ice bath. Ice (10 g) was
carefully added, followed by H20 (30 mL), followed by 12.1N
HC1 (50 mL). The reaction was then boiled (no condenser)
for 30 min. The reaction was subsequently cooled in an ice
bath, basified with lON NaOH (50 mL), and extracted with EtzO
(200 mL). The ether layer was washed with satd. aq. NaCl
(100 mL), dried (anh. Na~SO,), and rotary evaporated to yield
2.03 g of a yellow oil. This material was purified by RP-
HPLC !20-60o acetonitrile-0.1% aq. HCl over 20 min). The
collected fractions were frozen and lyophilized to yield
1.28 g of a white solid. The free-base of the purified
amine was dissolved in EtOAc. Malefic acid (305 mg) was
added and the mixture was heated until everything had
dissolved. The product was crystallized by adding Et20
(5 mL). The crystals were filtered and washed with
CA 02560002 2006-10-05
WO 97/46511 PCT/L1S96/20697
195
1:1 EtOAc-Et20 (10 mL) followed by EtzO (10 mL) to yield
967 mg of product as a white, finely crystalline solid.
(R,S)-3-(3-Fluoro-4-hydroxy)-3-(3-fluorophenyl)propylamine
maleate (Compound 175)
(R, S)-3- (3-~luoro-4-methoxy) -3- (3-
fluorophenyl)propyl-amine maleate (2.45 g, 6.23 mmol) was
free-based in the normal manner and dissolved in CHzClz
(25 mL). The resulting solution was cooled to -78°C. Under
N2 flow, boron tribromide (1. OM in CH,C1,; 15 mL, 15 mmol)
was added over a period of 5 min. The cold bath was removed
and the reaction mixture was allowed to warm to room
temperature. After 30 min at 25°C, the reaction was
hydrolyzed with 12.1N HC1 (l0 mL). The aqueous layer was
neutralized (pH 7) by the careful addition of 10N NaOH
(14 mL). Satd. aq. NaHC03 (50 mL) was added along with
Et20 (100 mL) and EtOAc (20 mL). This mixture was shaken
vigorously and the organic layer was separated. The aqueous
layer was extracted with EtOAc (20 mL). The combined
organic layers were dried (anh. Na=SO~) and rotary
evaporated. The resulting oil was dissolved in EtOH,
1.0M HC1 in EtzO (7 mL) was added, and the solution was
rotary evaporated. This material was then purified by
RP-F~RL~ (20-60% acetonitrile-0.1% HCl (aq.) over 20 min].
The collected fractions were frozen and lyophilized,
affording 716 mg of a white solid. The free-base of the
purified amine (315 mg) was dissolved in EtOAc. Malefic acid
(138 mg) was added and the mixture was heated until
everything dissolved. The EtOAc was rotary evaporated to
give a hard glass which was dissolved in MeOH (5 mL). Water
(100 mL) was then added and the solution was subsequently
frozen and lyophilized. The above procedure yielded 445 mg
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
196
of product as a white solid.
(R, S) -N-Methyl-3- (3-fluoro-4-hydroxy) -3- (3-
fluorophenyl)propyl amine hydrochloride (Compound 376)
A solution of (R,S)-N-methyl-3-(3-fluoro-4-
methoxy)-3-(3-fluorophenyl)propylamin a (703 mg, 2.41 mmol)
in CH~Clz (10 mL) was cooled to -78°C. Under nitrogen, boron
tribromide (1. OM in CHzCl~>; 6.0 mL, 6.0 mmol) was added over
a period of 5 min. The cooling bath was then removed and
the reaction was allowed to warm to room temperature. After
z0 1 h, the reaction was quenched with 12.1N HC1 (5 mL). The
aqueous layer was then neutralized (pH 7) by the careful
addition of lON NaOH (.-7 mL) . Satd. aq. NaHCO, (25 mL) vras
added along with EtzO (50 mL), EtOAc (15 mL), and CHC1,
(5 mL). This mixture was shaken vigorously, and the organic
layer was separated, dried (anh. Na2S04), and filtered
through paper. The crude product was then purified by
RP-HPLC (20-60% acetonitrile-O. to aq. HCI gradient over
min). The fractions were frozen and lyophilized to
afford 602 mg of product as a white solid.
20 The synthesis of Compound 185 was accomplished as
follows.
\ ~ NHZ
CI NHjO ~ Y1 vH
m-FG"~ F O
~C6ti~~P. DEAD F \
OH
Comb°und 185
(R)-3-(3-Fluorophenoxy)-3-phenylpropylchloride
Following a similar procedure for the chiral
synthesis of fluoxetine [Srebnik, M. et al., J. Org. Chem.
53(13), 2916-20 (1988), hereby incorporated by reference
herein], a solution of (S)-(-)3-chloro-1-phenyl-1-propanol
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
197
(4.00 g, 23.4 mmol), 3-fluorophenol (2.63 g, 23.4 mmoi), and
diethyl azodicarboxylate (4.00 g, 23.4 mmol) were dissolved
in THF (200 mL). The mixture was cooled to 0°C and
triphenylphosphine (6.77 g, 25.8 mmol, 1.1 equiv) was added
slowly over 10 min. The reaction mixture was then stirred
at room temperature for 18 h. The THF was subsequently
evaporated under vacuum to afford a gel which was washed
with pentane (3 x 50 mL). The pentane washings were
filtered and the filtrate was evaporated under vacuum to
give a clear oil. This oil was dissolved in diethyl ether
(150 mL) and washed with 1% HC1-satd. NaCl (25 mL), O.1N
NaOH-satd. NaCl (2 x 2S mL), and finally H20 (2 x 25 mL).
The organic layer was then dried (anh. NazSOq), filtered, and
evaporated to dryness under vacuum to give an orange oil.
The crude product was chromatographed on silica gel
(25 x 180 mm, gravity column), elution with 40:1
hexane-EtOAc, to provide 971 mg (15.7%) of product as a
colorless oil.
(R)-.3-(3-Fluorophenoxy)-3-phenylpropylamine
(Compound 185)
A solution of (R)-3-(3-fluorophenoxy)-3-
phenylpropyl chloride (0.971 g, 3.96 mmol), cone. NH90H
(30-mL;., and EtOH (20 mL) were shaken at 90°C on a Parr9
apparatu~_(SO-90 psig) for 18 h. The mixture was then
evaporated under vacuum and the residue was dissolved in Et20
(100 mL) and washed with HZO (2 x 25 mL). The organic layer
was dried (anh. Na~S04), filtered, and evaporated under
vacuum to provide a yellow oil. This material was then
dissolved in EtOAc (50 mL) and filtered. A solution of
malefic acid (0.272 g, 2.6 mmol, 0.93 equiv) dissolved in hot
EtOAc (5 mL) was added to precipitate the maleate salt
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
198
(519 mg, 53.5%) as a white solid: TLC RF 0.25 (1%
MeOH-CHC13); GC, t. 7.37 min; MS, m/z 245 (M+).
The synthesis of Compound 187 was accomplished as follows.
NHS
VCCH;POfOEy ( ~ ~ ~ ..~I(Hgl ~ / CN ~~H
--~ ---s
O ~ ~ ~ ~ O ~ ~ O
Compound 167
11-Cyanoirre thyl ene-1 ~ , 11-dihydrodibenzo (b, c] oxepine
To a solution of diethyl cyanomethylphosphonate
(5.06 g, 28.6 mmol) in dry DMF (15 mL) was added NaH (60%
mineral oil dispersion; 1.14 g, 28.5 mmol) over a period-of
2 min. The reacticn was stirred for 10 min and then a
solution of 6,11-dihydrodibenzo[b,c]oxepin-11-one [Kurokawa
M. et al., Chem. Pharm. Bull. 39(10), 2564-2573 (1991),
hereby incorporated by reference herein] (4.00 g, 19.0 mmol)
in dry DMF (5 mL) was added. The reaction mixture was
stirred under argon for 21 h. water (l00 mL) was then added
and the product was extracted with EtOAc (2 x 50 mL). The
combined organic payers were washed with satd. aq. NaCl
(2 x 50 mL~, dried (ann. NazS04), and rotary evaporated. The
resulting solid was recrystallized from hot EtOAc (10 mL)-
hexanes (40 mL) to provide 2.43 g (54.7x) of product.
11-Cyanomethyl-11H-10,11-dihydrodibenzo(b,c)oxepine
Following a procedure described in Great Britain
Patent 1,129,029 (1968) CChem. Abstr. x:37664), hereby
incorporated by reference herein], for the preparation of
aluminum amalgam, A1° granules (2.00 g, 74.1 mmol) were
first etched with 0.5N NaOH (100 mL) and then washed with Hz0
(100 mL) followed by EtOH (100 mL). A solution of HgCl2
(2.00 g, 7.37 mmol) in EtzO (100 mL) was added. The reaction
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
199
mixture was stirred for 5 min and the supernatant was
decanted. The solid A1(Hg) amalgam was washed with H20
(100 mL), EtOH (100 mL), and then EtzO (100 mL). The amalgam
was covered with Et20 (I00 mL) and a solution of
11-cyanomethylene-10,11-dihydrodibenzo[b,c)oxepine (2.00 g,
8.57 mmol) in EtOAc (30 mL) and EtOH (20 mL) was added.
Water (2 mL) was added and the reaction mixture was stirred
for 18 h and then filtered. The filtrate was rotary
evaporated to yield 1.65 g (81.8%) of product as a white,
crystalline solid.
11-(2-Aminoethyl)-IIH-10,II-dihydrodibenzo(b,c]oxepine
hydrochloride (Compound 187J
To a stirring suspension of lithium aluminum
hydride (0.67 g, 18 mmol) in anh. Et,O (30 mL) was added a
solution of 11-cyanomethyl-11H-10,11-dihydrodibenzo[b, c]
oxepine (1.65 g, 7.01 mmol) in dry THF i5 mL)/anh. Et20
(10 mL) over a period of 2 min. The reaction was stirred
for 30 min. In the following order, HBO (0.7 mL), 5N NaOH
(0.7 mL), and HBO (2.1 mL) were added to the reaction
mixture. Diethyl ether (30 mL) was added and the mixture was
filtered. The filtrate was rotary evaporated and the
resulting oil was dissolved in EtOH (10 mL)-Et20 (65 mL).
1.01 liC.l in EtzO (10 mL) was added and the solution was
allowed to crystallize, giving 1.42 g (73.4 %) of the title
compound.
Compounds 67-68, 70-75, 79-82, 84-89, 91-95, 98-
100, 102, 104-106, 109-114, 117, 124-134, 138, and 140-150
were synthesized by standard procedures known to those
skilled in the art, as described above.
Gae Chromatography of Simplified Arylalkylamines
Gas chromatographic and mass spectral data were
CA 02560002 2006-10-05
WO 97/46511 PCTNS96/20697
200
obtained or. Hewlett-Packard 5890
a Series
II Gas
Chromatograph equipped with 5971 es Mass Selective
a Seri
Detector [Ultra-2 UltraPerformance illary Column (cross-
Cap
linked nyl methylsilicone); mn length, 25 m,
5o phe colu
column d., 0.20 mm; The 0 mL/min; injector
i. flow
rate,
6
temp., gradient temperature ram, 20oC/min from
250C; prog 125
to 325C for 10 min, constantat 325C for 6 min].
then
held
Compound 19.~(Rt = min), m/z (rel.int.) 211 (M+,13),
7.40
195 (16),194 (100), 3 (73),180 (8),179 (331, 178 (19),
19
168 (24),167 (50), (23), 165 (72),164 (B), 153 (10),
166
152 (3I),117 (13), (38), 115 (26),106 (14), 104 (14),,
116
103 (24) 102 (8) , 11) (14) (29) , 63 (9) , 51-
, 91 ( , 78 , 77
(17)
Compound 20. (Rt = min), m/z (rel.int.) 247 (M+,27),
7.34
231 (16),230 (100), 9 (45),215 (29), 214 (14), 204 (43),
22
203 (37) 202 (13) , (47) 184 (14)183 (58) , 181 (8)
, 201 , , ,
151 (9), 135 (13), (31), 33 (25),124 (18), 122 (16),
134 1
121 (19),109 (15), (29), 96 (18),95 (11), 83 (11),
101 75
(20), (10), 42 (9)
57
Compound 21. (Rt = min), m/z (rel.int.) 261 (M+,69),
7.53
262 (13),245 (17), (100), 230 (11), 229 (42), 216 (11),
244
215 (15),214 (14), (45), 203 (35),202 (16), 201 (63),
204
184 -(-12.~183 (61) , (11) 136 (9) 135 (27) , 133 (36)
, 148 , , ,
124 (21) ,115(16) , (43) 83 (12) 74 (8) , 58 (14)
,_ 109 , , , 57
(11)
Compound 22. (Rt = min), m/z (rel.int.) 261 (M+,4),
7.37
244 (14),229 (7), 204 (10), 03 (16),201 (12), 183 (16),
2
138 14), 133 (5), 109 4), (7), (4), 58 (8), 57 (4),
( 101 75
44 (100) 42 (7)
,
Compound 24. (Rt = min), m/z (rel.int.) 259 (M+,122),
8.21
260 (23),242 (44), (15), 22B (15).227 (49), 216 (1S),
241
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
201
213 (56), 212 (16), 211 (55), 199 (32), 196 (22), 185 (34),
184 (19), 183 (67}, 171 (161, 170 (38), 165 (44), 151 (20),
150 (16) , 146 (13) , 136 (46) , 134 (17) , 133 (37) , 123 (15) ,
121 (22), 120 (13), 109 (100), 91 (34), 77 (29), 51 (15)
Compound 25. (Rt = 8.49 min), m/z (rel. int.) 259 (M+,39),
243 (16), 242 (95), 241 (25), 227 (27), 217 (15), 216 (100),
215 (27), 212 (13), 211 (50), 201 (14), 200 (11), 199 (15),
196 (15), 185 (20), 184 (19), 183 (50), 171 (24), 170 (28),
165 (15) , 146 (10) , 136 (11) , 134 (I2) , 133 (23) , 121 (21) ,
77 (9)
Compound 26. (Rt = 8.69 min), m/z (rel. int.) 259 (M+,11),
243 (17), 242 (100), 241 (69), 227 (10), 215 (31), 212 (11),
211 (52} , 184 (14) , 183 (31) , 172 (13) , 171 (35) , 170 (23) ,
165 (13) , 147 (21) , 146 (12) , 134 (19) , 133 (23) , 121 (13) ,
91 (11), 77 (10)
Compound 27. (Rt = 8.80 min), m/z (rel. int.) 243 (M+,54),
226 (36), 212 (12), 211 (69), 200 (14), 199 (16), 198 (20),
197 (100), 196 (39), 185 (35), 184 (30), 183 (50), 179 (13),
178 (14) , 165 (13) , 134 (15) , 133 (19) , 120 (29) , 117 (16) ,
115 (27) , 104 (13) , 101 (11) , 91 (23) , 77 (13)
Compound 2.8. (Rt = 8.77 min), m/z (Yel. int.) 243 (M+,25),
227 (15), 226 (85), 225 (26), 212 (19), 211 (100), 200 (22),
199 (_17~ , 197 (18) , 196 (29) , 185 (46) , 184 (35) , 183 (64) ,
179 (9), 165 (11), 134 (19), 133 (23), 12l (12}, 120 (18),
117 (14), 115 (24), 101 (12), 91 (25), 77 (12), 65 (11), 51
(9)
Compound 29. (Rt = 7.89 min), m/z (rel. int.) 243 (M+,12),
227 (9), 226 (52), 225 (17), 212 (19), 211 (100), 199 (13),
197 (12) , 196 (21) , 185 (19) , 184 (24) , 183 (43) , 179 (7) ,
134 (11), 133 (15), 120 (9), 117 (10), 115 (17), 91 (14)
Compound 30. (Rt = 8.36 min), m/z (rel. int.} 263 (M+,21),
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
202
246 (26),220 (13),212 (17), 211 (100), 196 (25),
197
(10),
185 (43),184 I30),183 (69), 181 (9),165 (12),
133 (I8),
115 (14),101 (15),75 (15)
Compound 31. (Rt 9.31min), m/z (rel.int.) 279 (M+,18),
=
281 (11),262 (10),236 (10), 229 (33),228 (17), 227 (100),
203 (9), 201 (33),199 (15), 15),178 (19),
192 166 (18),
(
165 (53),164 (13),163 (16), 140 (12),115 (13), 103 (9)
Compound 32. (Rt 7.30min), m/z (rel.int.) 229 (M+,21),
=
213 (16),212 (100), 197 (33), 196 (19),194 (14),
211
(61),
186 (26),185 (30),184 (19), 183 (69),I70 (17), 166 (16),
165 (77),134 (25),133 (23), 116 (17),115 (17), 103 (18),
101 (11),78 (13),77 ), (18), 43 13), 42
(23), 51 (
75
(13
(13)
Compound 33. (Rt 7.56min), m/z (rel.int.) 261 (M+,68),
=
245 (18),244 (100), 215 (16), 214 (15),204 (57),
229
(43),
203 (43),202 (15),201 (64), 184 (14),183 (73), 148 (16),
136 (13),135 (46),133 (601, 124 (51),115 (27), 111 (14),
109 (96),107 (16),96 (14), 7), 5 (20), (96), 57
83 7 58
(2
(33), (23), 35)
56 41
(
Compound 34. (Rt 7.39min), m/z (rel.int.) 261 (M+,72),
=
262 (14) 245 (18) 244 (100) 229 (42), 216 (9) 215 (15)
, , , , ,
214 (14),204 (521,203 (38), 202 (14),201 (54), 184 (12),
183 (,6~ 181 (10) 148 (13) 136 (9) 135 (31) 33 (40)
, , , , , 1 ,
124 (30),115 (18),109 (57), 107 (9),83 (13), (21), 57
58
(11)
Compound 35. (Rt 4.45min), m/z (rel.int.) 181 (M+,8),
=
165 (10),164 (76),138 (48), 136 (11),135 (63), 133 (12),
123 (22),122 (22),121 (11), 110 (21),109 (100),101 (13),
96 27), 83 5 (15), (21), 44 0), 42
( (14), (11), 45 (4
7 56
(9),41 15)
(
Compound 37. (Rt min), m/z (rel.int.) 196 (M+,4),
=
4.87
CA 02560002 2006-10-05
WO 97146511 PCTNS96/20697
203
195 (17), 178 (76), 163 (20), 152 (41), 150 (22), 137 (12),
136 (29), 135 (60), 133 (19), I24 (13), 123 (20), 122 (49),
121 (17), 110 (78), 109 (100), 101 (17), 96 (29), 83 (i7),
75 (12), 56 (29), 55 (12), 45 (53), 44 (45), 43 (39), 41
(30)
Compound 38. (Rt = min),m/z (rel. int.) (M+,.1),
7.68 275
203 (5), 201 (6), 183 8), 5 ), (4), 109 (8), 71
( 13 (4 I33
(3) , 3) 44 (100) 42
45 ( , , (4)
Compound 39. (Rt = min),m/z (rel. nt.) 289 (M+,6),
7.67 i 203
(3), 201 (5),183 (6), 135 ), 33 109 (7), 85 (3),
(2 1 (3), 70
(3) . 4) 58 (100)
59 ( ,
Compound 40. (Rt = m/z (rel. nt.) 289 (M+,19);
7.63min), i
203 (6), 201 (13), (17),152 (5), 5 (6), 3 (8),
183 13 13 109
(15), (5),70 (4), 8 )
85 5 (100
Compound 41. (Rt = min),m/z (rei. int.) (M+,23),
7.93 275
258 (20) 203 (27) , (14) 201 (52) 184 (9) 183 (59)
, 202 , , , ,
181 (10) 150 (11) , (18) 147 (11) 135 (24) 134 (14)
, 149 , , , ,
133 (40) 124 (12) , (I9) I09 (76) 107 (9) 103 (I0)
, 123 , , , ,
83 (15) 75 10) , 00) 1 2) (18) , (21) ,
, ( 72 (1 , (1 , 56 55
7 57
(41)
Compound 43. (Rt = min),m/z (rel. int.) (M+,11),
9.18 293
276 (I0),243 (11), (31),236 (11), 235 (16),201 (18),
241
199-(-2?.-)179 (11) , (25) 176 (10) 156 (16) 165 (70)
, 178 , , , ,
164 (19),I63 (24), (9), 102 (9), (12), (100),
103 75 44 43
(11), (15)
42
Compound 46. (Rt = min),m/z (rel. int.) (M+,46),
9.34 293
295 (28),276 (16), (24),242 (15), 241 (75),237 (12),
243
236 (18),201 (33), (31),178 (26), 176 (I3),166 (31),
199
165 (100), , I51 (13) , 149 (12),
164 152
(32), (11),
163
(43)
140 (30) 139 (11) , (12) 127 (20) 125 (31) 117 (26)
, 129 , , , ,
116 (26),115 (64), (12),89 (13), (22), 63
91 (17), 75
77
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
204
(14), 58 (51), 57 (15), 56 (191, 41 (19)
Compound 50. (Rt = 7.37 min), m/z (rel. int.) 261 (M+,2),
244 (9), 229 (4), 204 (7), 203 (11), 201 (8), 183 (11), 101
(5), 58 (7), 44 (100), 42 (7)
Compound 51. (Rt = 7.30 min), m/z (rel. int.) 261 (M+,5),
244 (20), 229 (9), 204 (14), 203 (23), 202 (6), 201 (20),
183 (27) , 133 (7) , 121 (6) , 101 (9) , 75 (6) , 58 (7) , 44
(100), 43 (6), 42 (11)
Compound 52. (Rt = 7.24 min), m/z (rel. int.) 247 (M+,56),
231 (13), 230 (81), 229 (47), 216 (12), 215 (32), 214 (16),
204 (29) , 203 (31) , 202 (16) , 201 (63) , 196 (21) , 184 (20)~
183 (100), 182 (11), 181 (15), 170 (13), 151 (13), 150 (11),
135 (13), 134 (29), 133 (25), 124 (14), 122 (20), 121 (21),
109 (13) , 101 (27) , 96 (21) , 75 (23) , 43 (14) , 42 (15)
Compound 53. (Rt = 7.21 min), m/z (rel. int.) 247 (M+,98),
248 (17), 231 (13), 230 (84), 229 (56), 215 (38), 214 (16),
203 (33), 202 (16), 201 (68), 196 (26), 184 (16), 183 (100),
181 (15), 151 (21), 150 (15), 135 (14), 134 (35), 133 (24),
124 (19), 122 (23), 121 (25), 111 (13), 101 (31), 96 (19),
75 (19)
Compound 5~. (Rt = 7.86 min), m/z (rel. int.) 275 (M+,98),
276 (20), 258 (59), 229 (58), 216 (31), 215 (22), 214 (19),
204 (,4.2~ , 203 (41) , 202 (21) , 201 (82) , 184 (18) , 183 (100) ,
181 (14) , 150 (21) , 135 (33) , 133 (55) , 124 (41) , 115 (13) ,
109 (90), 101 (15), 83 (20), 75 (16), 72 (23), 57 (13), 56
(24)
Compound 56. (Rt = 7.79 min), m/z (rel. int.) 261 (M+,67),
262 (12), 244 (54), 229 (56), 218 (27), 217 (16), 216 (19),
215 (100), 214 (45), 203 (50), 202 (32), 201 (51), 197 (16),
196 (26), 183 (24), 138 (17), 135 (20), 134 (17), 133 (39),
122 (26), 121 (13), 109 (30), 101 (17), 96 (14), 83 (16), 75
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
205
(13)
Compound 57. (Rt 7.65min),m/z (rel.int.) 261 (M+,62),
=
244 (50),229 (50),218 (24),217 (13),216 (18), 215 (100),
214 (36) 203 (42) 202 (19) 201 (33) 197 (14) 196 (19)
, , , , , ,
183 (17),138 (19),135 (16),134 (12),133 (29), 122 (29),
109 (25) 101 (13)
,
Compound 58. (Rt 8.15min),m/z (rel.int.) 275 (M+,134),
=
276 (26),258 (23),244 (19),243 (100), 232 (25), 229
(53),
217 (51),216 (23),215 (67),214 (97),201 (44), 197 (21),
196 (43),183 (23),148 (38),147 (21),138 (46), 135 (46),
134 (18),133 (64),125 (25),123 (28),122 (81), 115 (27),
109 (54),107 (17),83 (27),44 19), 3 (19)
( 4
Compound 59. (Rt 7.61min),m/z (rel.int.) 275 (M+,27),
=
204 (8), 203 (10),201 (19),183 (25),109 (8), 01 (7),
1 58
(100) (8) , 8)
, 57 56 ,
( 44
(9)
Compound 60. (Rt 7.34min),m/z (rel.int.) 261 (M+,55),
=
262 (10),204 (16),203 (15),201 (31),183 (35), 133 (11),
122 (11),121 (10),109 (9), 101 (16),96 (11), 5 (10),
7 57
(9), 44 100), 11)
( 42
(
Compound 61. (Rt 8.07min), m/z (rel.int.) 277 (M+,68),
=
278 (13),260 (31),246 (11),245 (25),234 (12), 231 (32),
229 (26),217 (20),203 (23),201 (24),188 (12), 183 (22),
154 (24) 151 (15) 150 (10) 133 (18) 124 (10) 109 (100)
, , , , , ,
95 (11) 44
, (14)
Compound 62. (Rt 8.93min),m/z (rel.int.) 271 (M+,115),
=
272 (22),254 (16),239 (22),225 (36),223 (40), 181 (33),
165 (34) 153 (13) 152 (24) 136 (39) 132 (13) 131 (16)
, , , , , ,
123 (20),122 (13),121 (89),119 (13),115 (23), 105 (17),
91 (100),77 (22)
Compound 63. (Rt 8.47min), (rel.int.) 287 (M+,31),
= m/z
241 (9). 204 (27),203 (20),202 (9), 01 (30),
2 183 (38),
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
206
150 (13),133 (20), 109 (27), (45),83 (43), 82 (11),
84 57
(18), (100), (25)
56 43
Compound 64. (Rt 8.57 min), (rel.int.) 287 (M+,63),
= m/z
288 (13),270 (14), 242 (16), (17),215 (17), 214 (18),
241
204 (35),203 (27), 202 (18), (70),183 (86), 150 (18),
201
147 (16) 146 (17) 135 (16) , (45) 109 (45) , 84 (31)
, , 133 , ,
83 (38), 82 13), 5 (15), 57 ), (100), 43 (44)
( 7 (21 56
Compound 65. (Rt 8.18 min), (rel.int.) 239 (M+,88),
= m/z
240 (17) 222 (12) 208 (18) , (100), 195 (24) , 193
, , 207 (48) ,
10192 (11),181 (33), 180 (32), (57),178 (72), 166 (16),
179
165 (60),152 (13), 130 (36), (17),120 (40), 117 (34),
129
116 (14) 115 (53) 107 (20) , (19) 104 (42) , 103 (11)
, , 105 , ,
91 (37), 77 20), 5 (17)
( 6
Compound 66. (Rt 7.46 min), (rel.int.) 275 (M+,7),
= m/z
15201 (5), 183 (6), 33 (3), 109 ), (3), 45 (3), 44
1 (6 71
(100) (3)
, 42
Compound 67. (Rt 7.56 min), (rel.int.) 225 (M+,24),
= m/z
194 (8), 193 (12), 179 (6), 168 67 (12), 166 (6),
(10), 1 165
(20) , (9) , 120 (8) , 116 115 ) , 103 (7) , 77
152 (6) , (7 (8) ,
2051 (5), 4
4 (100)
Compound 6B. (Rt 7.85 min), (rel.int.) 239 (M+,22),
= m/z
194 (5), 193 (10), 168 (10}, (12),166 (6), 165 (19),
167
152 (9) 134 (6) 16 (5) , 115 (7) , 77 (6) , 59
, , (7) , 91 (5) ,
1
58 (100),44 (8)
25Compound 69. (Rt 7.35min), (rel.int.) 275 (M+,11),
= m/z
203 (24),202 (7), 201 (23), (35),122 (6), 121 (6),
183 101
(9). 58 100), 57 8), 56 (10)
( (
Compound 72. (Rt 7.90 min), (rel.int.) 253 (M+,25),
= m/z
238 (9) 193 (7} 68 (8) , 167 5 (17) , 152 (9)
, , (14) , 16 , 115
1
30(7), 91 11),73 ), 72 (100), , 56 (7), 44 (6),
( (8 58 (45) 43
(9) , 8)
42 (
CA 02560002 2006-10-05
WO 97/46511 PC'T/US96/20697
207
Compound 73.(Rt 7.29 min),m/z (rel. int.) 239 (M+,9),
=
240 (2), 167(2),
16S
(5),
152
(2),
115
(2),
77
(2),
59
{5),
58 (100),44 (3),
42
(S)
Compound 74.(Rt 8.01 min),m/z (rel. int.) 267 (M+,7),
=
S 167 (3) 165(6)
, ,
152
(3)
,
91
(4)
,
87
(7)
,
86
(100)
,
72
(13) , (10), 56 (4) ,
58 42 (4)
Compound 79.(Rt 7.89 min),m/z (rel. int.) 230 (M+,37),
=
214 (15),213(100) , 212 , 201 (26), 200 (72), 198
(62) (21),
195 (12),188(17), 187 (85),186 (46), 185 (42), 184 (9},
157 (12),135(9), 133 (24),109 (10), 107 (20), 106 (62),
80 {14) 79 32) (20)
, ( ,
78
(9)
,
51
Compound 81.(Rt 7.40 min),m/z (rel. int.) 209 (M+,891,
=
210 (14),208(100) , 193 , 192 (56), 191 (42), 189
(17) (12),
178 (20),166(11), 165 (45),152 (12), 132 (86), 131 (10),
130 (53),117(22), 115 (48),106 (22), 105 (10), 104 (12),
103 (16),91 (16), 77 (22),
51 (15)
Compound 82.(Rt 7.93min),m/z (rel. int.) 275 (M+,124),
=
276 (25),232(33), 215 (12),214 {16), 204 (14), 203 (100),
201 (24),196(8), 183 (20),150 (14), 138 (9), 136 (14),
135 (44),133(26), 125 (9), 124 (71), 123 (29), 121 (14),
115 (14),111(72), 110 (9), 109 (84), 101 (14), 83 (9),
75
(8)
Compound 83.(Rt 7.22 min),m/z (rel. int.) 235 (M+,10),
=
219 (17),218(100) , 217 , 203 (20), 192 (10), 191
(62) (38),
190 (7), 189(14), 185 (17),183 (7), 171 (9), 165 (B),
147
(10) , (11) , (12) , (17) , 121 (8) , 109 (8)
146 134 133 , 97
(8) , 7)
45 (
Compound 85.(Rt 7.73 min),m/z (rel. int.) 239 (M+,7),
=
222 (15),179(8), 178 (9), 68 (16), 167 (33), 166 (12),
1
165 (43) 161(9) 152 (20) 146 (17) , 129 (7) , 120
, , , (15) ,
118 (7), 117(19), 115 (25),91 (25), 77 (7), 72 (9),
44
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
208
(100), 42 (6)
Compound 86. (Rt = 7.66 min), m/z (rel. int.) 239 (M+,3),
222 (4) 168 (4) , I67 (11) , 166 (4) , 165 (14) , 252
, (7) , 120
(6) , (6) 115 (8) , 91 (9) , 72 (5) , 44 (100) , 42
117 , (3)
Compound 87. (Rt = 7.33 min), m/z (rel. int.) 239 (M+,4),
222 (9), 179 (9), 178 (1I), I68 (11), 167 (27), 166 (13),
165 (48),161 (7), 152 (22), 146 (14), 128 (7), 120 (I1),
118 (8), 117 (21), 7.15 (31), 91 (29), 77 (9), 72 (8),
S1
(7) , 100), 42 (9)
44 (
10Compound 88. (Rt = 7.4 min), m/z (rel. int.) 227 (M+,.0),
183 (10),168 (18), 167 (100), 166 (32), 165 (83), 164
(10),
163 (6) I53 (6) , 152 (35) , 139 (6) , 115 (8) , 105
, (9) , 77
(12) , (7) 45 (23)
51 ,
Compound 89. (Rt = 8.74 min), m/z (rel. int.) 260 (M+,220),
1526I (39),259 (89), 242 (18), 203 (17}, 202 (16), 201
(61),
183 (58),165 (100), 1S0 (20), 148 (25), 138 (24), 137
(61),
122 (73),121 (31), lIl (47), 101 (23), 96 (26), 75 (16),
44
(17) , (29)
43
Compound 90. (Rt = 7.32min), m/z (rel. int.) 235 (M+,9),
219
20(16), (10 0), 217 (42), 206 (17), 205 (9), 204 (7),
218 203
(21), (8} , 193 (12), 192 (71), 191 (62), 190 (9),
202 189
(19), (13 ), 171 (14), 159 (9), 147 (14), 146 (16),
185 134
(10), (17 ), 121 (14), I09 (11), I01 (8), 97 (17),
133 45
(15)
25Compound 91. (Rt = 10.67 min), m/z (rel. int.) 329 (M+,6),
301 (20),300 (81), 167 (18), 166 (6), 165 (18), 152 (10),
132 (5), 120 (45), 119 (21), 118 (11), 117 (9), 115 (11),
106 (6), 105 (5), 104 (12), 103 (5), 92 (8), 91 (100),
77
(10), (6)
41
30Compound 92. (Rt = 10.37min), m/z (rel. int.) 337 (M+,30),
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
209
338 (7) , 204 (7) , 203 (7) , 201 (7) , 183 (10) , 133 (6) , 121
(8) , 120 (70) , 106 (6) , 92 (9) , 91 (100)
Compound 93. (Rt = 10.25 min), m/z (rel. int.) 351 (M+,28),
352 (7), 337 (9), 336 (39), 203 (10), 201 (11), 183 (17),
S 135 (6) , 134 (20) , 133 (6) , 132 (6) , 120 (111 , 118 (5) , 109
(18), 106 (12), 105 (100), 104 (13), 103 (8), 91 (14), 79
(11) , 77 (12)
Compound 94. (Rt = lU.4B min), m/z (rel. int.) 365 (M+,2),
337 (25), 336 (100), 203 (8), 201 (8), 183 (14), 133 (5),
132 (6). 120 (14}, 119 (13), 118 (9l, 115 (5), 109 (20}, 106
(5), 104 (10), 91 (52)
Compound 95. (Rt = 6.68min), m/z (rel. int.) 283 (M+,59)~
284 (11), 267 (11), 266 (71), 265 (19), 251 (24), 250 (9),
241 (14), 240 (100), 239 (48), 237 (30), 232 (10), 220 (17),
219 (65) , 199 (9) , 152 (12) , 151 (18) , 142 (20) , 140 (13) ,
139 (20), 127 (22), 119 (24), 114 (12), 101 (10), 63 (10),
44 (9)
Compound 96. (Rt = 6.93 min), m/z (rel. int.) 265 (M+,46),
249 (16), 248 (100), 247 (34), 233 (27), 232 (11), 223 (9),
222 (65), 221 (39), 220 (10), 219 (36), 202 (14), 201 (54),
152 (15), 151 (14),I 133 (9), 124 (12), 119 (9), 109 (9), 101
(14), 75 (9)
Compound 97. (Rt = 8.10 min), m/z (rel. int.) 241 (M+,101),
242 (18), 224 (50), 223 (19), 210 (11), 209 (37), 197 (12),
196 (10), 195 (55), 194 (16), 193 (60), 181 (29), 7.78 (20),
167 (38) , 166 (16) , 165 (52) , 153 (i2) , 152 (36) , 136 (27) ,
133 (12) , 132 (14) , 116 (12) , 115 (25) , 103 (13) . 91 (100) ,
77 (18)
Compound 98. (Rt = 6.69 min), m/z (rel. int.) 232 (M+,3),
204 (11), 203 (37), 202 (30), 201 (100), 188 (9), 184 (14},
183 (84), 182 (10), 181 (15), 170 (9), 109 (17), 107 (10),
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
210
83 (10), 75 (7)
(8),
57
Compound 99. (Rt 6.75min), m/z (rel.int.) (M+,2),
= 233
204 (12) 203 (68) 202 (26) 201 (100)200 (6) I88 (9)
, , , , , ,
184 (13) 183 (86) 182 (8) 14)
, , , ,
181 I70
( (9)
,
I33
(6)
,
109
(15) , ) , (12)8I 5 57 (9)
107 (11 83 , (7) (7)
, ,
7
Compound 100.(Rt 7.66min), m/z (rel.int.)
= 261 (M+,150),
262 (29),217 (11), 216 (70), 215 (28),214 (11),203 (30),
202 (31),201 (100) , (10), 184 (15),183 (90),181 (11),
196
133 (20) 124 (12) 122 (20) 109 (39) 101 (14) 83 (10)
, , , , , ,
75 (10) 45 43)
, (
Compound 101.(Rt 7.?2min), m/z (rel.int.)
= 245 (M+,20),
229 (16),228 (100) , (36), 213 (21),211 (22),202 (S7),
227
201 (30),199 (21), 183 (50), 181 (14),171 (15),170 (26),
165 (12) 152 (21) 134 (19) 133 (35) 122 (28) 120 (19)
, , , , , ,'
120 (13),119 (I2), 109 (20), 107 (20),106 (18),101 (15),
94 (15), 91 20), ), (15), 20), 63 4), 55
( 77 74 65 (1
(18 (
(14), (15), 44 43 ), 2
51 (27), (17 4 (14)
Compound 102.(Rt 8.33min), m/z (rel.int.) (M+,19),
= 273
204 (16) 203 (16) 201 (15) 183 (18) '177 (9) 33 (8)
, , , , , 1 ,
109 (13) 70 (41) 00) 8 0) (25) , (5) ,
, , , (2 , 42 41
69 6 43
(1
(5)
Compound 103.(Rt 8.59min), m/z (rel.int.) (M+,118),
= 245
246 -(-2Ø)229 (15) 228 (100) 227 (85) 213 (27) 21I (23)
, , , , , ,
209 (15),_207(12), 202 (19), 201 (32),200 (17),199 (84),
196 (10),183 (38), 181 (15), 171 (13),170 (23),152 (19).
151 (15) 150 (~~.0)134 (18) 133 (32) 131 (12) 122 (36)
, , , , , ,
119 (15),109 (24), 107 (10), 106 (12),91 (19), 7 (12)
7
Compound 104.(Rt 7.72min), m/z (rel.int.) (M+,94),
= 261
262 (17),217 (15), 216 (92), 215 (18),204 (12),203 (86),
202 (25),201 (100),184 (10), 183 (69),148 (12),133 (13),
122 (8), 109 (26), (8),
101 45
(9), (33)
83
CA 02560002 2006-10-05
WO 97146511 PCT/US96I20697
211
Compound 105.(Rt 10.24
= min),
m/z (rel.
int.)
351 (M+,7),
201 (5) 183 (7) ) (4) , (5) , 92
, , , 109
135 133
(9)
,
134
(79
(8) , , 65
91 (100) (8)
,
42
(7)
Compound 106.(At 7.52 min),m/z (rel.int.)
= 259 (M+,77),
260 (14),258 (31), 244 (30),228 (13),227 (28),214 (14),
201 (24) x.65(12) 164 (100)162 (29) 133 (56) 109 (44)
, , , , , ,
75 (13) 44 80)
, ( ,
42
(56)
Compound 107.(Rt 7.45 mir.),m/z (rel.int.) (M+,101),
= 227
228 (16),226 f100),211 (22),210 (68),209 (49),207 (13),
196 (22),184 (15), 183 (62),150 (50),148 (31),133 (44),
132 (53) 130 (45) 117 (15) 115 (29) i06 (14) 77 (18)
, , , , , ,
75 (13) 51 14)
, (
Compound 108.(Rt 7.46 min),m/z (rel.int.) (M+,34),
= 243
244 (6) 2;2 (6) (6) , (12) , (10) ,
, , 186 185 184
211
(9)
,
197
(5} , (19), 165 6) 120 ) , 103 , 77 (6)
183 (15) , (6 (5) ,
,
133
(
44 (100),.2 (6)
Compound 1~~9.(At 8.68 min),m/z (rel.int.) (M+,110),
= 285
286 (22),284 (27), 256 (16),228 (37),227 (27),225 (10),
220 (11),207 (15), 201 (27),191 (14),190 (100),163 (11),
162 (85),,.~61(10), 147 (11),146 (11),133 (32),109 (20),
83 (12) 82
, (36)
Compound 1~0.(Rt 8.66 min),m/z (rel.int.) (M+,91),
= 285
286 (16),284 (100),243 (16),227 (26),225 (11),221 (10),
220 (17),214 (12), 207 (15),201 (23),147 (25),146 (16),
133 (17),109 (20), 42 (15)
Compound 1~.1.(Rt 8.81 min),m/z (rel.int.) (M+,29),
= 287
214 (9), 204 (15), 03 (18), 02 3 (42),
2 2 (9),
201
(34),
18
135 (9), ,33 (28), 09 (28), 4 (100}, (19), 75
1 8 (47), 82
83
(8) , 16) 68 ) , 57 , (28) 44 (16) 43 (25)
70 ( , (13 (18) 56 , , ,
42 (14)
Compound 1.2.(Rt 8.85 min),m/z (rel.int.) (M+,141),
= 287
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
212
288 (29),286 (22),202 (21),201 (62), 183 (64),133 (23),
109 (27),84 100),83 18), (14), (58),
( ( 82 56 55
(31),
57
{53), (14),42 35)
43 (
Compound 1I3. (Rt 9.08 min),m/z (re!. int.) (M+,27),
= 251
180 (38) 179 (36)178 (39) 174 (15) 173 (100)166 (11)
, , , , , ,
165 (53),158 (12),152 (10),132 (9), 15 (28), 1 (31),
1 9 82
(18), (16),56 45), 51 , 43 (23)
77 ( (9)
Compound 114. (Rt 8.71 min),m/z (re!. int.) (M+,197),
= 237
238 (37),236 (67),193 (15),179 (30), 178 (40),165 (41),
159 (43) 158 (26)132 (24) 130 (16) 116 (17) 115 (37)
, , , , , ,
106 (21),103 (34),91 50), (68), (100),
( 77 56 51
(48),
57
(32), (50),42 34)
43 (
Compound 115. (Rt 9.45 min),m/z (re!. int.) (M+,34),
= 271
255 (12),254 (67),253 (14),239 (23), 229 (16),228 (100),
227 {18),224 (16),223 (68),213 (9), 12 (10), 11 (10),
2 2
197 (34),196 (17),195 (11),181 (18), 169 (10),165 (22),
153 (19) 152 (27)146 (16) 145 (13) 141 (12) 139 (10)
, , , , , ,
a 136 (22),I34 (11),133 (41),122 (16), 121 (31),115 (30),
91 (18), 77 5), 5 ), (10), 44 10)
(1 6 (11 63 (
Compound 116. (Rt 9.50 min),m/z (re!. int.) (M+,41),
= 269
268 (32),254 (8),253 21), 52 (100), 251 (14),238 (23),
( 2
237 (18) 221 (10)209 (9) 78 (8) 5 (19) 2 (22)
, , , , 16 , 16 ,
1
160 (19),152 (18},147 (11),146 (8), 45 (18), 39 (9),
1 1
130 (11),115 (10)
Compound 117. (Rt 7.64 min),m/z (re!. int.) (M+,13),
= 212
183 (I6),182 (100), (7), 167 (7), 4 (27),
180 152 (3), 91
10
(7) , ) (13)
78 (4) ,
, 77 51
(41
Compound 118. (Rt 7.46 min),m/z (re!. int.) {M+,4),
= 245
153 (8) 152 150 5 (6) , (10) , (5) ,
, (43) (9) 133 124 123
, ,
13
(36), (38) , (17) , (16), 101 (14),
122 121 109 96 (24),
95
(16) , (100), (7) , 77 (21) 75 (11) 66 (15)
94 93 , , , ,
83
(7)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
213
65 (30) 63
, (10)
,
51
(14)
,
50
(6)
Compound 119. (Rt = 7.39 min), m/z (rel. int.) (M+,7),
263
171 (14),170 (14), 152 (74), 151 (13), 150 141 (55),
(20),
135 (10) 133 (23) , 123 (20) , 122 (100) , 120 (11)
, 121 (49) , ,
113 (9), 112 92), 111 (9), 109 (41), 107 (12),3 (13),
( 10
1.02 (11)101 (40) , 97 (9) , 96 (66) , 95 (51)9) , 84
, , 94 (
(28) , (88) 82 (8) , 81 (16) , 77 (14) , 75 74 (10)
83 , (54} , ,
70 (10), 69 0), 64 (10), 63 (23), 57 (62}, 3), 51
(1 56 (1
(15), (12),42 (8)
50
10Compound 120. (Rt = 8.48 min), m/z (rel. int.) (M+,4),
279
159 (16),157 (49), 153 (11), 152 (100), 150 133 (11~,
(12),
130 (27),128 (73), 123 (12), 122 (57), 121 111 (10),
(23},
109 (25),101 (23), 99 (16), 96 (26), 95 (I0), (9), 75
83
(28), (10),65 (12), 64 (11), 63 (22), 51 50 (8)
73 (9),
15Compound 121. (Rt = 8.30 min), m/z (rel. int.) (M+,2),
275
152 (15),125 (8), 124 (100), 122 (14), 121 09 (35),
(7), 1
96 (7) ) , 81 (14) , 77 (9) , 65 (7)
, 95 , 52 (11)
(10
Compound 122. (Rt = 7.39 min), m/z (rel. int.) (M+,.1},
263
170 (12),152 (66), 151 (10), 150 (18), 141 135 (10),
(68),
20133 (19),123 (16), 122 (76), 121 (39), 112 111 (18),
(100),
109 (36) '107 (11) , 103 (11) , 102 (9) , 101 6 (56)
, (33) , 9 , 95
(32), (11),83 (96), 81 (13), 77 (13), 75 64 (25),
92 (43),
63 ~r26a 57 1) , 56 (14) , 51 (14) , 50 (11)
, (6
Compound_123. (Rt = 5.88 min), m/z (rel. int.) (M+,46),
275
25276 (9), 202 (9), 101
(8),
201
(30),
183
(2B),
133
(8),
109
(9), 71 9), 9 (12), 58 .(100), 44 (8), 42
( 5 (26)
Compound 124. (Rt = 7.05 min), m/z (rel. int.) (M+,15),
229
213 (15),212 (89), 211 (13), 198 (20), 197 196 (24),
(100),
186 (12) 185 (21) , 184 (29) , 183 (87) , 179
, (7) , 17B (8) ,
30177 (13) 176 (5) , 171 (7) , 170 (18} , 169 (5) ,
, (4) , 166 165
(20), (5), 133 (7), 75 (4), 63 (4), 57 (9), (4)
152 56
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
214
Compound 125. (Rt = 7.54 min), m/z (rel. int.) 225 (M+,57),
226 (13), 209 (13), 208 (75), 193 (13), 180 (14), 179 (21),
178 (20), 165 (22), 130 (34), 117 (59), 115 (28), 105 (18),
104 (94), 103 (45), 91 (100), 78 (30), 77 (38), 6S (36), 63
(13), 51 (20), 45 (17}
Compound 126. (Rt = 7.81 min), m/z (rel. int.) 261 (M+,12),
244 (31), 152 (27), 151 (17), 150 (9), 136 (11), 135 (100),
133 (21), 122 (24), 115 (9), 110 (13), 109 (90), 107 (6), 96
(7) , 83 (27) , 56 (7)
Compound 127. (Rt = 7.93 min), m/z (rel. int.) 225 (M+,23),
208 (20), 207 (6), 193 (13), 181 (7), 180 (37), 179 (100),
178 (36), 167 (9), 166 (12), 165 (36), 152 (9), 134 (30);
130 (26), 129 (9), 117 (18), 1I5 (22), 104 (6), 9I (38), 77
(7) , 65 (7)
Compound 128. (Rt = 7.42 min), m/z (rel. int.) 211 (M+,83),
212 (15), 194 (36), 193 (18), 182 (62), 181 (20), 180 (17),
179 (53), 178 (60), 176 (11), 167 (57), 166 (44), 165 (100),
152 (24), 120 (39), 116 (12), 115 (28), 104 (22), 103 (15),
91 (46) , 89 (16) , 78 (10) , 77 (20) , 65 (15) , 63 (12) , 51
(12)
Compound 129. (Rt = 7.39 min), m/z (rel. int.) 229 (M+,104),
230 (19), 212 (28), 211 (14), 201 (13), 200 (85), 199 (22),
198 (.14..1 , 197 (501 , 196 (58) , 185 (73) , 184 (45) , 183 (100) ,
179 (43), 178 (55), 177 (17), 176 (17), 170 (18), 165 (33),
152 (12) , 133 (22) , 120 (57) , 115 (17) . 109 (44) , 104 (23) ,
103 (17), 91 (32), 89 (16), 83 (20), 78 (12), 77 (22), 63
(16), 51 (13)
Compound 130. (Rt = 7.38 min), m/z (rel. int.? 229 (M+,133),
230 (24), 212 (27), 211 (14), 200 (54), 199 (17), 198 (16),
197 (53), 196 (64), 185 (49), 184 (43), 183 (100), 179 (28),
178 (29), 177 (14), 170 (19), 165 (26), 133 (22), 120 (35),
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/Z0697
215
115 (19), 109 (32), 104 (17), 103 (18), 91 (38), 89 (17), 83
(18) , 77 (24) , 63 (16)
Compound 131. (Rt = 7.40 min), m/z (rel. int.) 229 (M+,146),
230 (26), 212 (48), 211 (23), 200 (51), 199 (17), 198 (16),
197 (61), 196 (70), 185 (50), 184 (43), 183 (100), 179 (28),
I78 (28), 170 (20), 165 (23), 133 (21), 120 (35), 115 (20},
109 (59), 104 (25), 103 (17), 91 (27), 89 (17), 83 (22), 77
(22)
Compound 132. (Rt 7.03min),m/z (rel.int.)
= 0 (M+,.0),
185 (14),184 (100),183 (23),181 (17),165 (18),155 (12),
153 (14),152 (12),120 (85),119 (67),115 (10),106 (16)
91 (19) 89 (12 ) (25), 16)
, (14) , 51
, 77 (
78
Compound 133. (Rt 7.09min),m/z (rel.int.)
= 211 (M+,13),
195 (16),194 (100),181 (27),180 (70),179 (31),178 (28),
166 (25),165 (40),152 (9), 14), 19 (14),
120 1 118 (12),
(
I15 (10),104 (26),103 (53},102 (12),91 (62),
89 (10),
78
(13) , (42) 65 7) 51
77 , (1 , (13)
Compound 134. (Rt 7.45min},m/z (rel.int.) (M+,14),
= 211
183 (15) 182 (100)181 (14) 179 (13) 178 (18) 167 (27)
, , , ,' , ,
166 (18) 165 (46) 152 (10) 115 (8) 04 (8) 3 (6) ,
, , , , , 10 91
1
(29) , f'7) 78 , (7) 65 7)
89 , (5) 77 , (
Compound 135. (Rt 8.60min),m/z (rel.int.) (M+,34),
= 273
257 (-14-)256 (76) 231 (16) 230 (100)228 (18) 227 (57)
, , , , , ,
213 (14),211 (37),202 (30),201 (40),199 (26},184 (13),
183 (50),181 (12),171 (17),170 (20),152 (15),150 (19),
134 (15) 133 (31) 122 (14) 121 (29) 109 (16) 107 (13)
. , , , , ,
106 (17),91 12), 5
( 6 (12)
Compound 136. (Rt 9.26min),m/z (rel.int.) (M+,44),
= 275
277 (15),260 (28),259 (19},258 (81),257 (13),243 (15),
234 (33),233 (19),232 (100),231 (13),229 (15),227 (42),
224 (15),223 (86),208 (13),197 (45),196 (26),195 (13),
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
216
182 (14},181 (33),179 (11), 178 (18),166 (22), 165 (60),
164 (12),163 (10),153 (32), 152 (55),151 (18), 149 (10),
139 (11),137 (17),136 (19), 121 (13),115 (25), 102 (11),
91 (16),77
(17)
Compound 137. (Rt 7.42 min), (rel.int.) 245 (M+,1),
= m/z
153 (8) I52 (7) 0)
, , ,
141 134
(64) (100)
, ,
135 132
(1 (11)
,
117 (6) 115 (12) 112 (56) , 15)
, , 105 ( ,
104
(55)
,
103
(32)
,
95 (8) (8) , 83 (15) 78
, , (24)
91 ,
(16) 77
, (24)
84 ,
75
(9)
,
65 (6) (10) , 51 (9)
,
63
(8)
,
57
10Compound 138. (Rt 9.24 min), (rel.int.) 289 (M+,77),
= m/z
290 (16),230 (20),229 (21), 215 (15),203 (22), 201 (32)
183 (36),134 (10),133 (13), 124 (10),121 (9), 109 (10r,
101 (10),73 (100),43 (23}
Compound 139. (Rt 7.25 min), (rel.int.) 245 (M+,92),
= m/z
15246 (15),244 (67),229 (16), 228 (63),227 (46), 225 (10),
224 (15),214 (13),201 (39), 183 (13),151 (13), 150 (100),
149 (14)148 (58) 135 (22) , (54) 124 (14) , 122
, , 133 , (12) ,
109 (18),101 (15),75 (13)
Compound 140a.(Rt = 8.64 min),
m/z (rel.
int.) 271
(M+,72),
20272 (14),270 (37),255 (21), 254 (100),242 (19), 227 (14),
226 (63),225 (50),199 (19), 197 (30),196 (25), 183 (32),
176 (27)170 (20) 150 (44) , (34) 146 (14) , 133
, , 148 , (32} ,
131 (-14}121 (11)
,
Compound_ 140b.(Rt 8.68 min), . int.) 271 (M+,57),
= m/z (rel
25272 (10),270 (33),255 (20), 254 (100),242 (15), 227 (12),
226 (54),225 (40).209 (8), 199 97 (22), 196 (19),
(14), 1
183 (25), (21) I70 (16) , (33) 148 (22) , 146
176 , 150 , (9) ,
133 (20),131 (10)
Compound 141. (RC 8.44 min), (rel.int.) 257 (M+,48),
= m/z
30258 (8),256 36), 239 (19), 226 (22),
( 241
(21),
240
(100),
225 (20),209 (11),197 (14), 196 (18),183 (25), 170 (16),
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
217
162 (19) 160 (10) 150 (28) , 148 (26) , 147
, , (9) , 146 (8) ,
145 (13),133 (20),130 (8), 121 (10)
Compound 142.(R~ 8.47 min), m/z (rel. int.) (M+,14),
= 273
217 (5), 216 (31),215 (5), 183 (8), 170 (4), (5), 121
150
(4) , 5) 45 , 44 (100)
58 ( , (5)
Compound 143.(R. 9.39 min), m/z (rel. int.) (M+,47),
= 273
275 (16),274 (19),272 (36), 258 (39), 257 (26),256 (100),
255 (17},242 (25),241 (15), 221 (23), 178 (25),177 (11),
176 (14),168 (14),167 (11), 166 (54), 165 (34),164 (34),
10163 (16},162 (4S),150 (19), 152 (28), 151 (22),149 (19),
147 (18) 145 (24) 139 (11) , 136 (15) , 13I 130 (35)~
, , (15) ,
121 (15),115 (14),111 (11), 103 (13), 102 (19),89 (11~,
77 (16) 75 14) 3 (16) , 51 (12)
, ( .
6
Compound 145.(Rt 7.35 min), m/z (rel. int.) (M+,7),
= 277
15122 (10) 109 (8) 96 (6) , 95 (6) , 83 (10) , 63 (2)
, , , 75 (6) ,
57 (7) 4 00) 2 (9) .
, 4 (1 ,
4
Compound 148.(Rt = 8.43 min), m/z (rel. int.) 1 (M+,
26 3),
170 (I4) 169 (5) 168 (44) , 153 (4) , 151 (4) (6) ,
, , , 140 139
(4), 138 (15), (6), 125 (7), 123 (40), 115 , 103
132 (6)
20(24), (8) , (5), 95 (7), 94 (100), 89 77 (22),
102 101 (5),
75 (6) 6- ) (16) , 63 ( 7 ) , 51 110)
, 6 (8 , , 50 (4) .
65
Compound 149.(Rt 9.28 min), m/z (rel. int.) (M+,4),
= 295
170 (.321169 (12) 168 (100) , 166 (8) , 159 157 (66)
, , (22) , ,
152 (11) 140 (16) 139 (11) , 138 (41) , 132 130 (32)
, , (11) , ,
25129 (8), 128 (82),127 (10), 125 (16), 115 (12),11 (15),
1
103 (55),102 (18),101 (15}, 99 (19), 89 (10), (26),
77 76
(8) , 27) 73 1) , 65 (11) , 64 (10) , 63 51 (11)
75 ( , (1 (22) , .
Compound 150.(Rt 8.32 min), m/z (rel. int.) (M+,4),
= 279
171 (9), 170 (37),169 (13), 168 (100), 166 (8),.42 (8),
1
30141 (88),140 (19),139 (12), 138 (42), 132 (12),130 (7),
125 (16),1I5 (12),113 (10), 112 (89), 111 (11),104 (8),
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
218
203 (60), 102 (19), 101 (12), 95 (14), 89 (11), 84 (11),
83
(24), 77 (29), 76 (6), 75 (24), 63 (13), 57 (17), 51
(11).
Compound 151. (Rt = 7.68 min), m/z (rel. int.) 261 (M+,62),
244 (8), 216 (79), 203 (65), 201 (82), 183 (100), 121
(50),
101 (40) 75 (35) , 44 (52) .
,
Compound 152. (Rt = 8.097 min), m/z (rel. int.) 42 (34),
43
(10), 44 (42), 56 (13), 57 (10), 58 (72), 71 (6), 72
(74),
73 (14) 74 (14) , 75 (14) , 86 (15) , 95 (9) , 96 (10)
, , 100
(42), 101 (31), 114 (90), 115 (8), 120 (10), 121 (20),
122
10(9) , 123 (7) , 138 (10) , 149 (9) , 164 (7) , 170 (8)
, 181 (7) ,
183 (100) , 184 (15) , 188 (8) , 194 (6) , 195 (7) , 196
(10) ,
201 (64), 202 (21), 203 (63), 204 (10), 214 (12), 215
(12),
216 (12), 317 (92), 318 (20).
Compound 153. (Rt = 7.88 min), m/z (rel. int.) 42 (7),
43
15(8), 44 32), 46 (16), 72 (24), 75 (11), 86 (21), 95
( (8), 96
(9), 101 (20), 109 (14), 121 (30), 122 (14), 123 (6),
139
(6), 149 (10), 170 (6), 181 (6), 183 (59), 184 (9), 188
(6),
194 (1I), 195 (7), 196 (10), 201 (51), 202 (17), 203 (56),
204 (8), 214 (10), 215 (18), 216 (52), 217 (13), 289
(100),
20290 {20).
Compound 154. (Rt = 7.74 min), m/z (rel. int.) 42 (16),
44
(23), 45 (35), 46 (I5), 58 (20), 72 (59), 73 {17), 75
(12),
86 L231-, 96 (9) , 101 (18) , 121 (22) , 183 (52) , 194
(9) , 201
(44), 202 (15), 203 (41), 214 (9), 215 (11), 216 (20),
217
25(10) , (100) , 290 (20) .
289
Compound 155. (Rt = 7.67 min), m/z (rel. int.) 58 (44),
75
{15), 95 (9), 96 (11), 101 (22), 109 (16), 121 {33),
122
(15), 125 (12), 149 (9), 183 (62), 184 (10), 196 (12),
201
(57), 202 (19), 203 (53), 214 (11), 215 (19), 217 (13),
275
30(100) , 6 (18) .
27
Compound 156. (Rt = 8.93 min), m/z !rel. int.) 237 (M+,11),
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
219
220 (41),219 (30), 219 (30), 206 (7), 205 (39), 204 (5),
194 (28),193 (100),192 (21), 191 (31), 190 (9), 189 (I7),
179 (15),178 (50), 177 (5), 176 (5), 165 (20), 152 (7),
128
(5),116 (7),115
(39),
91
(11}.
Compound 157.(Rt 10.88 min), m/z (rel. int.) 343 (M+,6),
=
300 (100), 7 (16),166 (6), 166 (6), 165 (16), 152 (9),
16
133 (8), 120 (28),
118
(8),
117
(6),
115
(e),
104
(11),
92
(5),91 62),77 .
( (7)
Compound 158.(Rt 10.74 min), m/z (rel. int.) 342 (M+,.0),
=
300 (100), 7 (13),166 (5), 165 (13), 152 (7), 120 (22),
16
118 (6) 115 (6) 6 (3) , 104 (7) , 91 (31) .
, ,
10
Compound 159.(Rt 11.41 min), m/z (rel. int.) 363 (M+,I3),
=
336 (33),335 (24), 334 (95), 182 (10), 181 (9), 168 (14),
167 (40),166 (18), 165 (36), 156 (27), 155 (15), 154
(85);
153 (27),152 (29), 140 (9), 139 (7), 138 (14), 127 (32),
126 (9), 125 (100),117 (19), 116 (7), 115 (24), 103 (11),
91 43) 77 (7) , 41 (12) .
( , (15)
,
72
Compound 160.(Rt 11.48 min), m/z (rel. int.) 363 (M+,8),
=
336 (35),335 (25), 334 (100), 182 (5), 181 (11), 168
(9),
167 (29),166 (13), 165 (29), 156 (11), 155 (11), 154
(37),
153 (26) '152(23) 140 (8) , 139 (6) , 138 (12) , 127
, , (26) ,
126 (7), 125 (81), 25 (81), 117 (13), 115 (17), 103 (7),
1 91
(26),-- (5) 77 .
&9 . (9)
Compound_161.(Rt 11.83 min), m/z (rel. int.) 408 (M+,4),
=
- 25 407 (12),381 (24), 380 (98), 379 (25), 378 (100), 200
(77),
299 (31),198 (87), 197 (24), 184 (17), 182 (15), 181
(16),
171 (75),169 (77), 168 (18), 167 (60), I66 (22), I65
(58),
152 (32),118 (27), 117 (47), 116 (13), 115 (37), 104
(13),
103 (19) 91 (64) 0 (17) , 77 (25) .
, ,
9
Compound 162.(Rt 12.02 min), m/z (rel. int.) 408 (M+,3),
=
380 (100), 378 (99), 200 (40), 199 (32), 198
379 (48),
(25),
CA 02560002 2006-10-05
WO 97/46511 PCT/U596/20697
220
197 (30),184 (18),182 (16), 181 (23),
171 (83), 169 (85),
168 (16) 167 (49) 166 (18) , 165 (50) 52 (28) 9 (11)
, , , 1 , 11 ,
118 (32),117 (46),116 (12), 115 (34), 04 (11), 3 (17),
1 10
91 (63),90 6), 9 (10), 77 (23).
(1 8
Compound 163. (Rt 10.58 min), m/z (rel.int.) 347 (M+,14),
=
318 (100), (5) 168 (6) , 167 (24) 6 (17) (26)
181 , , 16 , 165 ,
152 (16) 150 (6) 139 (8) , 138 (66) (23) , (23)
, , , 137 137 ,
136 (10),I24 (6), 122 (13), 117 (8), (14), 110 (8),
115 109
(100), 3 , (22), 77 (7).
(6) 91
10 Compound 164. (Rt 10.59 min), m/z (rel.int.) 347 (M+,14?.
=
318 (100), (5), 178 (5), 168 (7), (27), 166 (17),
181 167
165 (28),152 (16),150 (6), 139 (8), (70), 137 (18);
138
136 (11),122 (14),117 (6), 115 (13), 0 (7), (79),
11 109
103 (6) 91 0) 1 (20) , 77 (6) .
, (2 ,
9
Compound 165. (Rt 10.61 min), m/z (rel.int.) 347 (M+,8),
=
318 (95),181 (8), 167 (20), 166 (10), 5 (21), (13),
16 152
138 (34),137 (27),136 (11), 136 (11), 22 (14), 7 (6),
1 11
115 (11),110 (8), 109 (100), 91 (22), (6).
77
Compound 166. (Rt 11.62 min), m/z (rel.,int.) 359 (M+,2),
=
330 (100), (13) , 165 (14), 152 (8),50 (6), (38),
167 1 149
148 (7) 135 8) 34 (13) , 122 (7) (7) , 121 3) ,
, ( , , 122 (7 117
1
(6) 115 (8) 91 3) , 77 (7) .
, , (2
Compou~i 167 (Rt 11 . 18 min) , m/z int . ) (M+,
. = (rel . 359 4)
,
330 (100)., (0) 121 (6) .
136 ,
Compound 168. (Rt 10.86 min), m/z (rel.int.) 343 (M+,6),
=
314 (100), (16) , 166 .(6) , 165 52 (9) (17)
167 (16) , 1 , 134 ,
133 (13) 132 (6) 118 (14) , i17 (9) (10) , (7)
, , , 115 106 , 106
(7) 105 (59) 91 20) , 77 (6) .
, , (
Compound 169. (Rt 10.94 min), m/z (rel.int.) 343 (M+,4),
=
314 (100), (14) , 166 (5) , 165 (15) (9)
167 , 152 (8) , 134 ,
133 (16) 132 (7) 132 (7) , 118 (14) (8) , 115 9) ,
, , , 117 ( 106
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
221
(6) , (62)91
105 , (18)
,
77
(5)
.
Compound 170.(Rt 12.52 min), m/z (rel. int.) 374 (M+,13),
=
345 (50),315 (5), 207 (12), 194 (8), 193 (16), 179 (5),
168
(9) , (22)166 (16) , 165 (100) , 164 (15) , 164 (15)
167 , , 152
(121 , (45), (13) , 115 (11) , 104 (6) , 103 (6)
136 117 , 91
(26) , (8) 77 ) .
90 , (7
Compound 171.(Rt 11.16 min), m/z (rel. int.) 341 (M+,14),
=
182 (8), 181 53), 168 (6), 167 (18), 167 (18), 166 (8),
( 165
(21) , (11), (8) , 132 (18) , 131 (100) , 129 (10)
152 144 , 128
(5), 120 (12),118 (5), 117 (9), 116 (10), 115 (15), 106
(5) , (9) 103 9) , 91 (48) , 77 (10) .
104 , (
Compound 172.(Rt 8.53 min), m/z (rel. int.) 275 (M+,165),
=
274 (95),260 (18),259 (24), 258 (87), 257 (2$), 254 (17),
244 (41) 243 (46) 242 (18) , 233 (30) , 214 (26) , 201
, , (46) ,
189 (18) 188 (36) 183 (27) , 'w81 (22) , 180 (100) ,
, , 178 (24) ,
165 (38) 163 (28) 154 (17) , 150 (39) , 149 (25) , 148
, , (82) ,
139 (58),135 (20),133 (35), 109 (20).
Compound 173.(Rt 8.44 min), m/z (rel. int.) 277 (M+,11),
=
260 (80),259 (34),245 (8), 241 (7), 234 (8), 233 (23),
230
(19), (100), 4 (9), 203 (12), 202 (17), 201 (25),
229 21 190
(8), 189 (25),188 (18), 183 (18), 171 (7), 170 (15),
165
(14) , (7) 154 (7) , 152 (7) . 151 (8) , 134 (12)
164 , , 133
(23) , (7) 109 (13) , 101 (8) .
121 ,
Compound 174.(Rt 8.49 min), m/z (rel. int.) 291 (M+,59),
=
260 (27),259 (17),234 (18), 233 (14), 230 (10), 229 (55),
203 (12),202 (10),201 (18), 189 (201, 188 (14), 183 (15),
170 (12) 169 (6) 168 (13) , 165 (14) , 164 (8) , 152
, , (8) , 151
(6), 138 (7),137
(8),
134
(?),
133
(13),
109
{12),
101
(7),
57 (6), 0),
44 (10 42
(6).
Compound (Rt 9.64 min), m/z (rel. int.) 303 (M+,8),
175. =
123 (47),109 (6), 96 (7), 95 (40), 85 (26), 84 (100),
82
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
222
(7), 75 (9), 68 (4), S6 (25), 55 (29), 43 (9), 42 (16).
Compound 176. (Rt = 8.35 min), m/z (rel. int.) 277 (M+,24),
245 (8), 220 (6), 219 (5), 183 (4), 171 (6), 170 (10), 151
(5) , 138 (7) , 109 (5) , 57 (6) , 44 (100) , 42 (6) .
Compound 177. (Rt = 7.83 min), m/z (rel. int.) 241 (M+,.1),
134 (30), 109 (8), 108 (100), 107 (16), 104 (17), 103 (9),
91 (12), 90 (4), 79 (10), 78 (14), 77 (24), 65 (8), 51 (9).
Compound 178. (Rt = 8.29 min), m/z (rel. int.) 257 (M+,.1),
134 (17) , 125 (8) , 124 (100) , 109 (23) , 104 (10) , 103 (6) ,
95 (4), 91 (5), 81 {9), 78 (7), 77 (11), 65 (7), 52 (9), 51
(6)
Compound 179. (Rt = 7.88 min), m/z (rel. int.? 255 (M+,61,
148 (12) , 115 (5) , 108 (8) , 107 (7) , 104 (12) , 103 (7) , 91
(11), 79 (5), 78 (11), 77 (19), 65 (6), S1 (S), 44 (100), 42
1 S (8) .
Compound 180. (Rt = 7.28 min), m/z (rel. int.) 29S (M+,1),
183 (10), 162 (22), 145 (11), 143 (28), 135 (10), 134 (100),
133 (11) , 132 (13) , 117 (6) , 115 (12) , 114 (6) , 113 (9) , 112
(7) , 105 (17) , 104 (57) , 103 (32) , 102 (6) , 95 (7) , 91 (18) ,
89 (5), 83 (8), 79 (6), 78 (28), 77 (28), 75 (6), 65 (8), 63
(11) , 51 (11) .
Compound 181. (Rt = 7.7 min), m/z (rel. int.) 259 (M+,3),
137 (16), 135 (5), 122 (1S), 121 (6), 109 (15), 108 (100),
107 (18), 96 (7), 91 (S), 79 (7), 78 (5), 77 (i7), 65 (5),
S1 (5) .
Compound 182. (Rt = 8.00 min), m/z (rel. int.) 225 (M+,2),
208 (51), 207 (40), 182 (14), 181 (100), 165 (7), 152 (24),
151 (8) , 74 (2) .
Compound 183. (Rt = 8.98 min), m/z (rel. int.) 241 (M+,7),
224 (33), 223 (46), 199 (6), 198 (15), 197 (100), 178 (6),
165 (28) , 152 (13) , 150 (2) .
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
223
Compound 184. (Rt = 8.90 min), m/z (rel. int.) 235 (M+,3),
218 (30), 217 (13), 203 (8), 202 (9), 193 (10), 192 (67),
191 (100) , 190 (11) , 189 (29) , 178 (7) , 165 (18) , 152 (3) .
Compound 185. (Rt = 7.37 min), m/z (rel. int.) 245 (M+,.1),
152 (8) , 141 (61) , 135 (10) , 134 (100) , 132 (11) , 115 (14) ,
112 (64), 105 (19), 104 (71), 103 (45), 102 (8), 95 (13), 91
(24), 89 (8), 84 (12), 83 (28), 79 (10), 78 (42), 77 (44),
75 (16) , 6S (12) , 64 (7) , 63 (1.6) , 57 (24) , 56 (7) , 52 (7) ,
51 (22), 50 (9).
Compound 186. (Rt = 7.31 min), m/z (rel. int.) 245 (M+,.1),
152 (8), 141 (64), 135 (10), 134 (10C), 132 (11), 117 (6),.
115 (13) , 112 (38) , 77 (40) , 75 (14) , 65 (11) , 64 (6) , 63
(14) , 57 (21) , 52 (6) , 51 (18) , 50 (7) .
Compound 187. (Rt = 8.64 min), m/z (rel. int.) 239 (M+,.0),
221 (17), 220 (6), 207 (8), 196 (11), 195 (39), 194 (14),
193 (11), 192 (38), 191 (7), 181 (6), 179 (21), 178 (100),
168 (7), 167 (41), 166 (17), 165 (53), 164 (6), 153 (6), 152
(26), 139 (6), 128 (7), 115 (18), 91 (6), 89 (6), 77 (6), 63
(6) , 51 (5) , 44 (6) .
Example 30: Biological properties of synthesized
arylalkylamines
Compounds synthesized as described in Example 28 and
Example 29 were tested for various biological properties
detailed in the examples.
CA 02560002 2006-10-05
WO 97!46511 PCT/US96l20697
224
Table 1
Compound ICSp (l.~M) IC$p (l,cM)
vs. NNmAa vs. [3H)MK-801
Compound 1 0.102 (7) 126 (4)
Compound 2 0.192 (4) not tested
Compound 3 0.003 (7) not tested
Compound 4 0.184 (5) 89 (1)
Compound 5 0.102 (1) 15.2 (2)
0.070 (3)b
Compound 6 0.129 (1) > 100 (1)
(0o at 100 ~M)d
Compound 7 I 0.163 (2) 129 (1)
Compound 8 0.099 (2) 219 (1)
Compound 9 1.2 (5) > 100 (2)
(!0a at 100 ~M)d
Compound 10 0.082 (2) -. 80 (1)
(57o at 80 uM)d
Compound 11 4.0 (2) not tested
Compound 12 6.0 (11) 98 (1)
Compound 13 not tested not tested
Compound 14 8.8 (2) - 100 uM
Compound 15 4.9 (3) - 100 ,uM
..Compound 16 5.1 (1) 28.8 (1)
Compound 17 9.6 (1) 36.3 (1)
Compound 18 5.1 (3) 34 (1)
Compound 19 0:435 (11) 2.1 (5)
Compound 20 0.070 (15) 0.252 (9)
Compound 21 0.038 (3) 0.457 (2)
Compound 22 0.145 (6) 3.45 (2)
Compound 23 0.267 (3) 5.4 (1)
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
225
Compound 24 0.206 (6) 0.591 (6)
Compound 25 0.279 (2) 0.871 (2)
Compound 26 27 (2) 34 (2)
Compound 27 0.071 (1) 0.180 (2)
Compound 28 0.380 (1) 2.3 (3)
Compound 29 1.9 (2) 5.8 (3)
Compound 30 0.035 (2) 0.407 (2)
Compound 31 0.052 (7) 1.3 (2)
Compound 32 0.284 (5) 0.799 (3)
Compound 33 0.060 (9) 0.181 (6)
Compound 34 0.426 (6) 2.7 (3)
Compound 35 6.2 (1) 25.1 (1)
Compound 36 rot tested nat tested
Compound 37 0.944 (2) 11.1 (2)
Compound 38 0.407 (2) 2.3 (2)
Compound 39 0.251 (1) 2.9 (3)
Compound 40 0.933 (1) 18.1 (3)
Compound 41 0.724 (1) 14.0 (3)
Compound 42 not tested not tested
Compound 43 0.232 (4) 7.5 (2)
Compound 44 not tested not tested
Compound 45 not tested not tested
Compound 46 O.OI3 (3) 5.2 (2)
Compound 47 not tested not tested
Compound 48 not tested not tested
not tested not tested
Compound 49
Compound 50 0.089 (6) 0.762 (4)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
226
Compound 51 1.1 (4) 4.5 (2)
Compound 52 0.102 (3) 0.380 (2)
Compound 53 0.217 (3) 4.2 (2)
Compound 54 0.036 (4) 0.046 (3)
Compound 55 0.035 (3) 0.153 (2)
Compound 56 0.218 (4) 0.955 (2)
Compound 57 0.028 (4) 0.063 (2)
Compound 58 0.028 (2) 0.203 (3)
Compound 59 0.272 (2) 0.453 (3)
Compound 60 0.416 (11) 0.641 (9)
Compound 61 0.134 (4) 0.324 (2)
Compound 62 0.177 (5) 0.617 (1)
Compound 63 0.093 (6) 0.245 (3)
Compound 64 0.309 (3) 0.851 (2)
Compound 65 0.167 (3) 2.0 (1)
Compound 66 0.236 (4) 1.2 (2)
Compound 67 10.95 (2) 2.9 (1)
Compound 68 2.9 (1) not tested
Compound 69 0.224 (2) 0.366 (1)
Compound 70 1.7 (1) not tested
Compound 71 6.35 (2) not tested
Compound 72 7.4 (1) not tested
Compound 73 12.6 (1) not tested
Compound 74 27.5 (1) not tested
Compound 75 0.94 (2) not tested
Compound 76 0.73 (2) not tested
Compound 77 5.5 (2) not tested
Compound 78 10.2 (1) not tested
CA 02560002 2006-10-05
WO 97/46511 PCT/US96l20697
227
Compound 79 12.6 (4) 10.2 (2)
Compound 80 28 (1) 182 (1)
Compound 81 1.4 (1) 6.1 (2)
Compound 82 0.106 (5) 0.794 (1)
Compound 83 0.342 (4) 0.794 (1)
Compound 84 7.9 (2) 23.4 (1)
Compound 85 1.2 (3) 3.5 (1)
Compound 86 1.2 (3) 6.0 (1)
Compound 87 0.657 (4) 3.0 (1)
Compound 88 2.5 (3) 10.6 (2)
Compound 89 0.240 (3) 1.2 (2)
Compound 90 0.270 (4) 1.4 (2)
Compound 91 0.162 (3) 14.1 (2)
Compound 92 1. 3 ( 3 ) 20 . 2 ( 2 )
Compound 93 0.486 (3) 26.9 (2)
Compound 94 0.248 (4) 22.6 (2)
Compound 95 0.311 (3) 3.0 (2)
Compound 96 0.187 (5) 1.1 (2)
Compound 97 0.410 (3) 2.6 (1)
Compound 98 7.9 (1) 52.5 (2)
Compound 99 > 100 (1) 105 (2)
Compound 100 0.602 (2) 3.2 (1)
Compound 101 0.912 (2) 2.0 (1)
Compound 102 1.01 (2) 3.3 (1)
Compound 103 0.380 (4) 0.661 (2)
Compound 104 7.983 (3) > 10 (1)
Compound 105 1.03 (1) > 3 (1)
Compound 106 0.767 (1) 1.31 (1)
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96120697
228
Compound 107 2.67 (1) 3.83 (?)
Compound 108 1.06 (1) 0.942 (1)
Compound 109 1.95 (1) 1.08 (3)
Compound IIO 42.7 (1) 13.3 (')
Compound 111 0.645 (3) 0.167 (2)
Compound 112 28.0 (2) 21.0 (~)
Compound 113 13.5 (1) not tested
Compound 114 3.4 (1) not tested
Compound 115 1.4 (1) '_.0 (1)
Compound 116 3.6 (1) not tested
Compound 117 19.6 (2) 6.0 (2)
Compound 118 0_409 (2) 0.240 (3)
Compound 119 C.115 (4) 0.087 (3)
Compound 120 0.101 (3) 0.074 (3)
Compound 121 0.656 (3) 0.670 (3)
Compound 122 0.209 (2) 0.342 (2)
Compound I23 9.6 (7) > 3 (2)
Compound 124 3.5 (1) 14.3 (3)
Compound 125 1.7 (1) 6.7 (2)
Compound 126 0.398 (3) 6.0 (1)
Compound 127 1.2 (3) 17.5 (2)
Compound 128 0.646 (4) 5.5 (1)
Compound 129 1.26 (2) not tested
Compound 130 0-.851 (2) not tested
Compound 131 1.23 (2) not tested
Compound 132 1.3 (1) 6.4 (1)
Compound 133 0.760 (1) 3.0 (1)
Compound 134 2.5 (1) > 10 (1)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
229
Compound 135 0.244 (2) 1.185 (2)
Compound 136 0.139 (2) 0.706 (1)
Compound 137 0.232 (3) 0.074 (2)
Compound 138 107 (1) > 100 (1)
Compound 139 1.97 (2) 5.6 (2)
Compound 140 20.8 (1) not tested
Compound 141 4.26 (1) 8.97 (1)
Compound 142 1.013 (3) 1.54 (2)
Compound 143 2.82 (1) not tested
Compound 144 not tested not tested
Compound 145 0.098(1) 0.626(1)
Compound 146 0.829 (3) 0,372 (1)
Compound 147 0.894 (2) not tested
Compound 148 0.549 (2) 0.373 (2)
Compound 149 0.085 (3) 0.150 (3)
Compound 150 0.195 (2) 0.351 (2)
Compound 151 54.9 (1) > 100 (1)
Compound 152 not tested not tested
Compound 153 not tested not tested
Compound 154 not tested not tested
Compound 155 not tested not tested
Compound 156 0.069 (3) 0.090 (2)
Compound 157 0.142 (2) 23.16 (2)
Compound I58 0.351 (2) 39.64 (1)
Compound 159 0.185 (2) 10.41 (1)
Compound 160 7.35 (3) 48.94 (1)
Compound 161 0.247 (2) 5.62 (1)
Compound 162 1.138 (2) 76.41 (1)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
230
Compound I63 0.326 (2) 10.34 (1)
Compound 164 0.475 (2) 18.30 (1)
Compound 165 0.337 (2) 171 (1)
Compound 166 0.619 (2) 36.7 (1)
Compound 167 0.080 (2) 14.5 (1)
Compound 168 0.092 (2) 17.4 (1)
Compound 169 0.298 (2) 26.7 (1)
Compound 170 0.238 (2) 57.0 (1)
Compound 171 0.310 (3) 39.6 (1)
Compound 172 38.0 (1) 37.3 (1)
Compound 173 22.9 (1) 24.1 (1)
Compound 174 not tested 57.0 (1)
Compound 175 not tested 5.1 (1)
Compound 176 not tested 10.0 (1)
Compound 177 not tested 0.754 (1)
Compound 178 not tested 1.25 (1)
Compound 179 not tested 1.67 (1)
Compound 180 <100 (1) <10(1)
Compound 181 0.081 (1) 0.632(1)
Compound 182 2.6 (1) 7.05(1)
compound 183 0.676 (1) 5.01(1)
Compound 184 1.5 (1) 1.51(1)
Compound 185 0.646 (1) 0.639(1)
Compound 186 0..155 (1) 0.123 (1)
Compound 187 1.78 (1) 2.01(1)
Compound 188 not tested not tested
Compound 189 ~ not tested not tested
Compound 190 not tested not tested
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
231
Compound 191 not tested not tested
Compound 192 not tested not tested
Compound 193 not tested not tested
Compound 194 not tested not tested
Compound 195 not tested not tested
Compound 196 not tested not tested
Compound 197 not tested not tested
Compound 198 not tested not tested
Compound 199 not tested not tested
Compound 200 not tested not tested
Compound 201 not tested not tested
Compound 202 not tested not tested
Compound 203 not tested not tested
Compound 204 not tested not tested
Compound 205 not tested not tested
Compound 206 not tested not tested
Compound 207 not tested not tested
Compound 208 not tested not tested
Compound 209 not tested not tested
Compound 210 not tested not tested
Compound 211 not tested not tested
Compound 212 not tested not tested
Compound 213 not tested not tested
Compound 214 not tested not tested
Compound 215 not tested not tested
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1). (# in parentheses
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
232
indicates the number of experiments).
b:TFA salt.
:Inhibition of ['H]MK-801 binding in rat cortical/
hippocampal washed membrane preparations (see
Example 4).
d:IC4~ study incomulete. % inhibition at the stated
concentration.
A comparison of the ICSO values in the RCGC
assay with the ICS, values in the ['H]MK-801 binding
assay (Table 1) illustrates that the arylalkylamines of
the invention inhibit NMDA receptor activity by a
mechanism different than that of binding to the MK-801
binding site; the concentration of the compound that
inhibits NMDA receptor function is several orders of
magnitude less than the concentration that competes at
the site labeled by ['H]MK-801. This is not the case,
however, with the simplified arylalkylamines exemplified
by Compounds 19 - 215. Such compounds bind to the site
labeled by ['H]MK-801 at concentrations ranging
app~oximateiy 1 to 400-fold higher than those which
antagonize-NMDA receptor-mediated function in the rat
cerebellar granule cell assay.
Some of the simplified arylalkylamines
disclosed have structural features similar to portions
of other compounds which are utilized as, for example,
anticholinergics, antiparkinsonians, antihistamines,
antidepressants, calcium channel blockers, coronary
vasodilators, opiate analgesics, and antiarrhythmics.
However, when certain of these compounds were evaluated
for :~MDA receptor antagonist potency (Example 1), as can
be seen in Table 2, none of the compounds tested, with
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
233
the exception of (g)- and (~)-fendiline, nisoxetine, and
the Eli Lilly compound, had ICso values less than 1 ~M.
These data are summarized in Table 2.
TaDie
Compound Structure ICSO (w
and vs. NMDA$
Therapeutic
Vtilit
($)-fendiline H ~ 0.719
~
I
(calcium channel ;
w N ~
blocker; coronary
vasodilator)
(~)-fendiline \ I H \ I 0.686
(calcium channel " -
blocker; coronary / I
vasodilator)
prenylamine ~ ~ H -10
(calcium channel
/ ~3 I /
blocker; coronary \ I
vasodilator)
pheniramine i 4 a..v3 22
(antihistamine) \
3
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
234
Table 2
chlorpheniramine ~I i I ~3 >100
i
(antihistamine) \
3
brompheniramine B~ i ~3 138
iantihistamine)
3
1~
diphenhydramine i 26
(antihistamine) I
\
~3
doxylamine i . I G-~3 62
(antihistamine; \ I ~/~~~s
hypnotic) ~ G-t3
chlorcyclizine I \ ~N~CH3 -10
~ N
(antihistamine)
i
CI
cyclizine I \ ~N~CH3 28
i ~ N
i
(ant i
emet \
c)
nor-cyclizine
f NH 23
(pharmaceutical N
intermediate)
\
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
.35
lidoflazine F ~
N~.-~ p ~ 3
(calcium c annel
blocker; coronary ~ I H ~
3
vasodilator) F
pimozide F , p~-NH >10
(antipsychotic) ~~ ~'
~I
F
disopyramide p N 3C~~3 )100
~3
(antiarrhy~hmic)
~s
isopropamide p N 3C~~3 87
z
(anticholinergic) ~ ~ ~ Crl3
/ fl3C CH 3
pridinol I % ~H N~ 10.7
(anticholiergic;
antiparki~sonian) /
chloropyr~~tine ~3 76
N.
(antihista.~.~ine) / I ~ ~s
CI \
trihexyplienidyl , ~ off 5.13
(antichol_aergic;
antiparki:sonian)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/2U697
2'i5
f luoxetine ~ 3 . :~
H
I
(antidepressant) ~
~ N~~
3
0
F3C
zimeldine 8r i ~3 ? 26
I '
antidepressant) \
3
methadone 0 ~3 not tested
S
(opiate analgesic)
\ ~
CH
3
~3
I
~stra compoundb \ > 30
(antidepressant) I
~3
~I
OCH3
Novo-Nordisk ~ ~~3 not tested
compound
(calcium channel 0
blocker; ~ /
neuroprotectant) F3C
Novfl~N~'rdisk I \ ~3 28.8
c ompoundd ~ N' Cf~-~
s
(calcium channel 0
blocker; I i
neuroDrotectant)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
23~
nisoxetine ~ 0.894
~
H
(monaamine uptake
3
inhibitor;
antidepressant) ~ i
~H
3
terodiline ~ ~ H a..13 not tested
(calcium channel
~3
blocker;
anticholinergic; w
vasodilator)
tomoxetine ~ not tested
-
~
H
(monoamine uptake
3
inhibitor;
antidepressant) ~ i
3
amitriptyline ~ ~ not tested
~
(serotonin uptake
/ ~3
inhibitor; \
antidepressant)
imipramine ~ not tested
(serotonin uptake ~
~ t~~N'~3
i
inhibitor;
/ \
antidepressant)
c~.omipramine ~ not tested
( serotanin uptakei
~ r~N'~3
inhibitor;
/ \
antidepressant)
CI
CA 02560002 2006-10-05
WO 97/46511 PCT/(1S96/20697
238
~'oxepine ~ not tested
~~3
serotonin uptake /
N
~
,
~3
_nhibitor;
antidepressant)
~'.~_~rorpromazineI not tested
~
aopamine ~
N~
r~1'~
3
Ip :_~agonist;
neuroleptic) w
CI
desipramine 2.3
(antidepressant) ~
~
I
15
3
H
o_ rotriptyline <_ 10
!antidepressant) ~
~
~
,
~~3
H
_..~_. 0.609
Lilly
Compound
NMDA
receptor
antagonist ~
'
NH2
20
a: Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
239
b: Disclosed as compound 2 in Table 4 in Marcusson et
al., Inhibition of [3H]paroxetine binding by various
serotonin uptake inhibitors: structure-activity
relationships. Europ. J. Pharmacol. 215: 191-198, 1992.
c: Disclosed as compound 17 in Jakobsen et al.,
Aryloxy-phenylpropylamines and their calcium overload
blocking compositions and methods of use. U.S. Patent
No. 5,310,756, May 10, 1994.
d: Disclosed as compound 25 in Jakobsen et al.,
Aryloxy-phenylpropylamines and their calcium overload
blocking compositions and methods of use. U.S. Patent
No. 5,310,756, May 10, 1994.
e: Disclosed as Compound 1 in McQuaid et al., Inhibition
of ['H]-MK801 binding and protection against NMDA-induced
lethality in mice by a series of imipramine analogs Res.
Comm. in Pathol. and Pharm. 77:171-178, 1992.
Structure-activity relationship studies were
initiated using Compound 19 as the lead structure. An
examination of the side chain demonstrated that the
propyl side chain was optimal for NMDA receptor
antagonist. potency (Table 3). This finding was verified
using Compound 20 as the lead structure (Table 3).
CA 02560002 2006-10-05
WO 97/46511
~4C
Table 3
PCT/US96l20697
Compound Structure '-.IC3o
( N,bt ) vs .
NMDAa
2,2-diphenylethylamine I w 24.5
i
- Nhiz
(
3,3-diphenylpropylamine I ~ 0.435
(Compound 19)
i I
w
4,4-diphenylbutylamine I ~ 1.7
(Compound 70) ~ N~
5,5-diphenylpentylamine I ~ 6.4
(Compound 71) ~ N~
i
~I
2,2-bis(3-fluorophenyl)-1-
i
ethylamine " ~ N~'~z
(.Compound 981 i I
w
3,3-bis(3~-fluorophenyl)-1- ~ 0.070
propylamine
(Compound 20)
I
4, 4-bis (3-fluorophenyl)-1- ~ ~ :9: 602
_ N~2
butylamine
(Compound 100) i
F
CA 02560002 2006-10-05
WO 97146511 PCT/US961Z0697
241
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
Further SAR studies examined the optimal
pattern of phenyl ring substitution. Initial studies
demonstrated that substitution of a halogen group
(fluoro or chloro> at the meta position was optimal for
NMDA receptor antagonist potency (Table 4). Increasing
the number of fluoro substituents led to an apparent
decrease in potency (Table 4).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
2~~
Table 4
Compound Structure =C5o
Ed.M )
vs .
N1~A
3,3-diphenyl-1-propylamineI ~ 0.435.
(Compound 19) ~ NHZ
W
3-(2-fluorophenyl)-3-(4- w ~ 0.730
fluorophenyl)-1- ~ i t~z
propylamine
i
(Compound 76)
F
3,3-bis(4-fluorophenyl)-~-~ 5.5
propylamine
(Compound 77)
~i~
F
3,3-bis(3-fluorophenyl)-1-' ~ 0.070
propylamine
(Compound 20)
3-(2-fluorophenyl)-3-(3- ~ F 0.102
f luorophenyl ) -1- ~ i Nt~
propylamine
i
(Compound 52)
CA 02560002 2006-10-05
WO 97146511 PCT/US96lZ0697
243
3,3-bis(2-fluorophenyl)-1-w F 0.217
propylamine I i
.(Compound 53) / F
3,3-bis(3-chlorophenyl)-1-~ ~ 0.052
propylamine C / Nt~
( Co.:ound 31 )
I
C
3-(3-fluorophenyl)-3-(3- I ~ 0.035
chlorophenyl ) -1- C / Nt~
propylamine
/
I
( Compound 3 0
3-(3-fluorophenyl)-3- I ~ 0.284
phenyl-1-propylamine / NH2
(Compound 32) / I
F
3-(3,5-difluorophenyl)-3-F 0.187
(3-fluoropheny_)-1-
w
I
/ NHZ
propylamine
(Compound 96) /
I
3,3- F t~410
bis ( 3 , 5 , diluo.~ophenylI
) -1-
propylamine F /
(Compound 97) / I
F \ F
3,3-bis[3- I ~ 10.2
(trifluoromethyl)phenyll-CFA /
1-propylamine
/
I
(Compound 78)
CFA
CA 02560002 2006-10-05
WO 97!46511 PCT/US96/20697
244
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
Replacement of one of the fluoro groups on one
phenyl ring with a methyl, methoxy or hydroxy group led
to no change or a decrease in the in vitro NMDA receptor
antagonist potency. The ortho position was optimal for
this methyl, methoxy or hydroxy group, and the rank
order of potency for this substitution was methyl >
methoxy > hydroxy (Table 5). Also illustrated i: Table
5 are those compounds possessing the 3,3-bis(3-
fluorophenyl) moiety with additional methyl or methoxy
substitutions on the phenyl rings, often leading to an
increase in NMDA receptor antagonist potency. Table 5
also illustrates those compounds possessing the 3,3-
bis(2-methylphenyl) or 3,3-bis(2-methoxyphenyl) moiety
in place of the 3,3-bis(3-fluorophenyl) moiety; these
substitutions are acceptable, although a decrease in
potency is noted.
CA 02560002 2006-10-05
WO 97146511
245
Table 5
PCTIUS96/20697
Compouad Structure ICSp
(ELM)
PS.
NMDAa
3,3-bis(3-fluorophenyl)-1-' ~ O:a'70
propylamine
(Coaroound ZO)
i I
3-(3-fluorophenyl)-3-(2- w CH3 0.071
methylphenyl)-I- I i
propylamine
i
(Compound 27)
3-(3-fluorophenyl)-3-(3- I ~ 0.380
methylphenyl)-1- H C
a
propylamine
(Compound 28)
I
3-(3-fluorophenyl)-3-(4- H3 ~ 1.9
(
methylphenylj-I- i ~x
propylamine
i
(Compound 29j
3-(3-fluorophenyl)-3-(2- ~ oCH3 0.206
methoxyphenyl)-1- I i NHz
,propylamine
(Compound 24j
CA 02560002 2006-10-05
WO 97/46511 PCT/US96l20697
246
?-(3-fluorophenyl)-3-(3-I ~ 0.279
me thoxyphenyl ) -1- H~~ ~ N~
propylamine
i
(Compound 25)
3-(3-fluorophenyl)-3-(4-H~~ ~ 27
methoxyphenyl)-1- ~ i NH2
propylamine
i
(Compound 26)
3 - ( 2-methoxyphenyl ~ ~ ~H' '~T~:.41Q
) -3 -
phenyl-1-propylamine
(Compound 97)
3-(2-hydroxyphenyl)-3-(3-I ~ 0.380
fluorophenyl)-1- F ~ N~
propylamine
(Compound 103) w
3-(3-hydroxyphenyl)-3-3-~ ~ 0.912
fluorophenyl)-1- F ~ NHz
propylamine
(Compound 101) ~ w
3-(3-fluorophenyl)-3-(2-I w 0.218
methyl-3-fluozophenyl)-1-~ NH2
propylamine H3
(Compound 56) ~
3-(3-fluoraphenyl)-3-(3-I ~ 0.028
fluoro-6-methylphenyl)-1-~ NH2
propylamine ~ CHI
(Compound 57)
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/Z0697
247
3,3-bis(3-fluoro-6- ~ ~ 0.028
methylphenyl)-1-
propylamine
(Compound 58)
3-(3-fluorophenyl)-3-(3- ~ ~~ 0.134
fluoro-6-methoxyphenyl)-1-~ ~ t~z
propylamine
i
(Compound 61)
3,3-bis(2-methylphenyl)-1-~ CH3 "~:2b7
propylamine
(Compound 65) H
3,3-bis(2-methoxyphenyl)-~ ~3 0.177
1-propylamine I i t~z
(Compound 62) ~C
3,3-bis(3-methoxyphenyl)-~ ~ 1.9
1-propylamine H3Co
(Compound L15)
w
H~CO
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
248
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
The next series of SAR experiments
S investigated the effect of alkyl chain substitutions
(branching patterns) on NMDA receptor antagonist potency
in vitro. The addition of a methyl group on either the a
or 13 carbon on the propyl side chain led to a decrease
or no change in potency, respectively (Table 6).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
249
Table 6
Cosapound Structure IC50
( ~1M )
v s .
N1~A a
3,3-bis(3-fluorophenyl)-1-~ 0.070
I
propylamine ~ N~
(Compound 20)
i
3, 3-bis (3-fluorophenyl)I ~ CHI '$~0~8
-2-
methyl-1-propylamine
(Compound 21)
3,3-bis(3-fluorophenyl)-2-I ~ CH3 0.060
methyl-1-propylamine ~ NHz
(Compound 33)
3,3-bis(3-fluorophenyl)-2-I ~ CHz 0.426
methyl-1-propylamine
(Compound 34)
3,3-bis(3-fluorophenyl)-1-I ~ 0.145
methyl-1-propylamine ~ NHz
(Compound 22) ~ CH3
3,3-bis(3-fluorophenyl)-1-I ~ 0.089
methyl-1-propylamine ~ NHz
(Compound 50)
CA 02560002 2006-10-05
WO 9'7/46511 PCT/US96/20697
250
3,3-bis(3-fluorophenyl)-1-I ~ 1.I
methyl-1-propylamine r
(Compound 51) ~ CH3
3,3-bis(3-fluorophenyl)-2-~ ~H3 0.035
ethyl-1-propylamine F I r NH2
(Compound 55)
I
F ~
3 , 3-bis (3-fluorophenyl)I ~ ~-,2.67
-1-
ethyl-1-propylamine r
(Compound 23) r
3,3-bis(3-fluorophenyl)-2-~ ~ H 0.036
hyd~oxyethyl-1-propylamineF I r NH2
(Compound 54)
I
F ~
3,3-bis(3-fluorophenyl)-3-~ ~ CH3 0.106
ethyl-1-propylamine F r NH2
(Compound 82) r I
F
3,3-bis(3-fluorophenyl)- I ~ CH3 0.40?
1,2-dimethyl-1-propylamine
(Compound 38) r
3,3-bis(3-fluorophenyl)- I ~ CHI 0.724
2,2-dimethyl-1-propylaminer
(Compound 41)
w CH3
3,3-bis(3-fluorophenyl)- I 28
2,2-diethyl-1-propylamineF ~ NHZ
(Compound 80) ~ I
F
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
251
a-Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
The next series of SAR experiments
investigated the effect of incorporation of a double
bond within the propyl chain on NMDA receptor antagonist
potency in vitro (Table 7). As can be seen in Table 7,
the incorporation of a double bond decreased potency in
a consistent manner.
Table 7
Compound Structure ICSo
( ~1M ) v s .
_.
~A a
3,3-bis(3-fluorophenyl)-1- I ~ 0.070
propylamine ~ NHZ
(Compound 20)
3,3-bis(3-fluorophenyl)- ~ ~ 1.4
prop-2-ene-1-amine ~ , NHZ
(Compound 139)
W
3,3-diphenyl-1-propylamine I ~ 0.435
(Compound I9)
CA 02560002 2006-10-05
WO 97146511 PCT/US96120697
252
3,3-diphenyl-prop-2-ene-1- I ~ 1.4
am? ne ~ ~ NH2
(Compound 81)
3-(3-fluorophenyl)-3- I ~ 0.284
phenyl-1-propylamine ~ NH2
(Compound 32) i I
F
3-(3-fluorophenyl)-3- I ~ 2.67
phenyl-prop-2-ene-1-amine ~ i
(Compound 107)
I
NH-z
i I
w
F
(mixture of 2 compounds)
3,3-bis(3-methoxyphenyl)- ~ ~ 1.9
1-propylamine H3C0
( Compound 115 ) , I
H3C0
3,3-bis(3-methoxyphenyl)- ( ~ 4.47
prop-2-ene-1-amine H3Cp
(Compound 116)
H~CO
CA 02560002 2006-10-05
WO 97/46511 PCT/~JS96/20697
253
3:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
The next series of SAR experiments
investigated the effect of incorporation of the
propylamine chain into a ring structure on NMDA receptor
antagonist potency in vitro (Table 8).
CA 02560002 2006-10-05
WO 97!46511
254
Table 8
PCT/US961Z0697
Compound Structure ICso
( ~1M ) vs .
a
3,3-bis(3-fluorophenyl)-1- I ~ 0.070
propylamine
(Compound 20)
0.093
Compound 63
NHZ
F
0.309
Compound 64 F
NHZ
F
1.01
Compound 102
F
NH 7
Compound 84 F
OH
i
F
0.790
i ~N~
Compound 111 F
F
28.9
Compound 112 F
F
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
255
d:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
The next series of SAR experiments
investigated the effect of simple alkyl substitution on
the nitrogen on NMDA receptor antagonist potency in
vitro (Table 9).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
256
Table 9
Compound Structure ICSo
( ~tM )
vs .
~' NINA
3,3-bis(3-zluoroohe.~_y1)-1-, ~ 0.070
propylam.ir~e
(Compound 20)
N-methyl-3,3-bis(3- ~ 0.416
~
H
fluorophenyl)-I-
propylamine
(Compound 60) w
N-ethyl-3,3-bis(3- , ~ H 0.272
f Iuorophenyl ) -1 - F ~' N vCH3
propylamine i I
(Compound S9) F
N, N-diethyl-3 , 3-bis ( 3- ~ ~ ~~~ 9 . 6
=luorophenyl)-1- ~ ~ CNy
propylamine
(Compound Z23) w
3-(3-fluorophenyl)-3- I ~ 0.284
phenyl-1-propylamine ~ NH2
(Compound 32) i
F
N-methyl-3-(3- ( ~ H 1.06
fluorophenyl)-3-phenyl-1-
propylamine
(Compound 108)
3,3-diphenylpropylamine ~ ~ 0.435
(Compound 19)
CA 02560002 2006-10-05
PCT/US96/20697
WO 97/46511
257
N-methyl-3,3- I ~ H 10.95
di.phenyipropylzmir_e ~ N'CH
3
(Compound 67) i '
N-ethyl-3,3- ~ ~ H ~' 2.9
diphenylpropylamine i N~CH3
(Coamound s8 )
N,N-dimethyl-3,3- ~ ~ CHa 12.6
dipheaylpropylamine ~ '~'CNz
(Compound 73)
N-isopropyl-3,3- ~ ~ H 7.4
diphenylpropylamine
(Compound 72) ~ C
CH
N,N-diethyl-3,3- ( ~ ~ 27.5
diphenylpropylamine ~ t
(Compound 74)
w ~
CA 02560002 2006-10-05
WO 97!46511 PCT/11S96/20697
258
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
Certain simplified arylalkylamine compounds
were selected for evaluation of activity in a battery of
neurotransmitter receptor binding assays, and for
activity against the L-type calcium channel and delayed
rectifier potassium channel. The compounds were
inactive (less than 50% inhibition at concentrations up
l0 to l0 uM) in the following assays: nonselective a2
adrenergic receptor (('H]RX 821002 binding in rat
cortex), H1 histamine receptor (['H]pyrilamine binding in
bovine cerebellum), nonselective sigma receptor (['H]DTG
binding in guinea pig brain), nonselective opiate
receptor (['H]naloxone binding in rat forebrain),
monoamine oxidase (MAO) activity, both MAO-A
((=;C]serotonin metabolism in rat liver mitochondria) and
MAO-B (['aC)phenylethylamine metabolism in rat liver
mitochondria)
As can be seen in Table 10, activity was noted
for several compounds at concentrations below 10 E.cM in
the following assays: L-type calcium channel, delayed
rectifier potassium channel, central muscarinic
cholinergic receptor binding, and monoamine (dopamine,
norepinephrine, and serotonin) uptake binding assays.
This profile of activity in the central muscarinic
cholinergic receptor and monoamine uptake binding assays
is not unexpected,. given the chemical structures of our
simplified arylalkylamines (refer to Table 2 above).
With the exceptions, however, of the activity of
Compound 19 in the serotonin uptake binding assay, the
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
259
activity of Compound 34 in the dopamine uptake binding
assay, the activity of Compound 50 in the serotonin
uptake binding assay, the activity of Compounds 63 and
64 in the dopamine uptake binding assay, and the
activity of Compound 60 in the dopamine and serotonin
uptake binding assays, the simplified arylalkylamine
compounds were most potent at the NMDA receptor.
Table 10
Compound IC,a t~M)L-type Delayed Central Mono~ine
vs. NL~A calcium rectifiermuscarinic uptake
channel potassiumcholinergicbinding
channel receptor assays
Compound 0.435 10.2 1-10 4% at 7% at
19 0.174% at 0.175% at
10 103 % at
0.1953% at
10918% at
0.1"89% at
10"
Compound 0.070 2.2 1-10 8% at 6% at
20 0.190% at 0.181% at
10 10f
5% at
0.1958% at
10928 % at
0 . 1"94
% at
10"
Compound 0.060 1.6 > 10 42% at 23% at
33 0.199% at 0.186% at
10 102% at
0 . 1954
% at
10914s at
0 . 1"8 9
% a t
10"
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
260
Compound 0.426 nct ~ 10 250 at 60s at
34 tested 0.199% at 0.199s at
10 10f10 o at
0 . 1964
o at
10g12% at
0 . 1"79
o at
10"
Compound 0.089 not ~ 10 110 at 17% at
50 tested 0.1840 at 0.1=930 at
10 10f
100 at
O.1g78% at
1075_% at
0.1"97% at
1 ~"
Ccmpound 0.013 0.675 ~ 3 33% at 400 at
46 0.1890 at 0.1197% at
10 107% at
0 . 1964
o at
10310 o at
0 . 1"75
o at
10"
Compound 0.093 1.9 not 11s at 64% at
63 tested 0.181% at 0.1~98o at
10 107% at
0 . 1876
o at
10913 o at
0.1"85% at
10"
Compound 0.309 not not llo at 500 at
64 tested tested 0.183% at O.1f99% at
10 10L
8% at
0 . 1965
o at
10929 a at
C . 1"6 8
o at
10"
CA 02560002 2006-10-05
WO 97/46511 PCTlUS96/20697
261
Compound 0.028 1.6 not 1% at 0% at
5g tested 0.148% at 0.1=45%
at
10 10'1 % at
0.1867%
at
10327% at
0 . 1"95
% at
10"
Compound 0.272 not not 9% at 2% at
59 tested tested 0.187% at 0.1'-78%
at
10 10f7% at
0 . 151
% at
1014% at
0.1"86%
at
10~
Compound 0.416 2.3 not 13% at 0.9116%
60 tested 0.193% at at 0.1964%
l0 at 109
0.068"
a:Inhibition of NMDA/glycine-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGC's) (see Example 1).
°:Inhibition of KC1 depolarization-induced increases in
intracellular calcium in cultured rat cerebellar granule
cells (RCGCs) ; estimated ICSO value in ~cM.
:Inhibition of delayed rectifier potassium channel in
cultured N1E-115 neuroblastoma cells; estimated ICso
value in ~M.
':Inhibition of the binding of ['H]quinuclidinylbenzilate
(QNB) to rat cortical membranes; percent block at
indicated concentration in ~M.
e:Inhibition of the binding of ['H]WIN-35,428 to guinea
pig striatal membranes (dopamine uptake binding assay),
['H]desipramine to rat cortical membranes (norepinephrine
uptake binding assay), or ['H]citalopram to rat forebrain
membranes (serotonin uptake binding assay); percent
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
262
block at indicated concentration in uM, or ICSO when
available.
:dopamine uptake binding assay
9:norepinephrine uptake binding assay
":serotonin uptake binding assay
Advantageous properties of the arylalkylamine
ccmpounds of the present invention are illustrated by
the fact that concentrations which suppress NMDA
receptor-mediated synaptic transmission fail to inhibit
LTP. Furthermore, while compounds such as Compound 9,
and 11 do produce a hypotensive respcnse following
systemic administration in rats, the hypotensive effect
produced by these compounds is of a relatively short
duration (approximately 30 min). Additionally,
Compounds 12 and 14 have no cardiovascular activity at
doses up to 37.3 ~cmoles/kg i.v. and 15 ~cmoles/kg i.v.,
respectively.
CA 02560002 2006-10-05
WO 97/46511 PCT/LJS96I20697
263
Table 11
Compound Suppression of LTP Assayb Drop in Mean
Nt~A
Receptor-Mediated Arterial Blood
Synaptic Pressure'
Transmission'
Compound 1 10 - 30 ,uM no block at 65 mm Hg at
300 ACM 1.5 ~Cmoles/kg
i.v., 60 min
duration
l0 - 30 ACM no block at 40 mm Hg at
S Compound 2 100 ~cM 1.5 ~cmoles/kg
i.v., 120 min
duration
Compound 3 10 - 30 ACM not tested 20 mm Hg at3
mg/kg s.c.,>
60 min
duration
Compound 4 10 - 100 E.cM no block at 40 mm Hg at
100 uM 1.5 ~cmoles/kg
i.v., 120 min
duration
Compound 9 10 - 100 ACM no block at 75 mm Hg at
300 ACM 4.5 ,umoles/kg
i.v., 90 min
duration
Compound 11 not tested not tested 20 mm Hg atl
mg/kg i.v.,
30
min duration
not tested not tested no effect at
Compound 12 doses up to
37.3
~cmoles/kgi.v.
Compound 14 not tested not tested no effect at
doses up to
15
~.cmoles/kg
i.v.
Compound 19 100 - 300 ACM block at 100 not tested
~M
CA 02560002 2006-10-05
WO 97146511 PCT/US96/20697
264
Compound 20 30 - 300 ~.M block at 100 no effect at
~cM doses up to
15
~molesjkg i.v.
Compound 22 not tested not tested no effect at
doses up to
15
~.cmoles/kg
i.v.
a:Concentration which suppresses NMDA receptor-mediated
synaptic transmission (see Example 5).
b:Concentration that does not block the induction of LTP
(see Example 19) .
:Drop in systemic blood pressure produced by
administration of compound in rats (see Example 22).
CA 02560002 2006-10-05
WO 97/46511 PCT/US96120697
265
Formulation and Administration
As demcnstrated herein, useful compounds of
this invention and their pharmaceutically acceptable
salts may be used to treat neurological disorders or
diseases. While these compounds will typically be used
in therapy for human patients, they may also be used to
treat similar or identical diseases in other vertebrates
such as other primates, farm animals such as swine,
cattle and poultry, and sports animals and pets such as
horses, dogs and cats.
In therapeutic and/or diagnostic applications,
the compounds of the invention can be formulated for a
variety of modes of administration, including systemic
and topical or localized administration. Techniques and
formulations generally may be found in Remington's
Pharmaceutical Sciences, Mack Publishing Co., Easton PA
(18th ed. 1990).
Pharmaceutically acceptable salts are
generally well known to those of ordinary skill in the
art, and may include, by way of example but not
limitation, acetate, benzenesulfonate, besylate,
benzoate, bicarbonate, bitartrate, calcium edetate,
camsylate, carbonate, citrate, edetate, edisylate,
estolate, esylate, fumarate, gluceptate, gluconate,
glutamate, glycollylarsanilate, hexylresorcinate,
hydrabamine, hydrobromide, hydrochloride,
hydroxynaphthoate, iodide, isethionate, lactate,
lactobionate, malate, maleate, mandelate, mesylate,
mutate, napsylate, nitrate, pamoate (embonate),
3o pantothenate, phosphate/disphosphate, polygalacturonate,
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
266
salicylate, stearate, subacetate, succinate, sulfate,
tannate, tartrate, or teoclate. Other pharmaceutically
acceptable salts may be found in, for example,
Remington's Pharmaceutical Sciences, Mack Publishing
Co., Easton, PA (18th ed. 1990).
Preferred pharmaceutically acceptable salts
include, fox example, acetate, benzoate, bromide,
carbonate, citrate, gluconate, hydrobromide,
hydrochloride, maleate, mesylate, napsylate pamoate
(embonate), phosphate, salicylate, succinate, sulfate,
or tartrate.
The useful compounds of this invention may
also be in the form of pharmaceutically acceptable
complexes. Pharmaceutically acceptable complexes are
known to those of ordinary skill in the art and include,
by way of example but not limitation,
8-chlorotheophyllinate (teoclate).
The exact formulation, route of administration
and dosage can be chosen by the individual physician in
view of the patient's condition. (See e.a. Fingl et
al., in The Pharmacological Basis of Therapeutic,, 1975,
Ch. 1 p. 1).
It should be noted that the attending
physician would know how and when to terminate,
interrupt, or adjust administration due to toxicity or
organ dysfunction. Conversely, the attending physician
would also know to adjust treatment to higher levels if
the clinical responses were not adequate (precluding
toxicity). The magnitude of an administered dose in the
management of the disorder of interest will vary with
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96/20697
267
the severity of the condition to be treated and to the
route of administration. The severity of the condition
may, for example, be evaluated in part, by standard
prognostic evaluation methods. Further, the dcse and
perhaps dose frequency, will also vary according to the
age, body weight, and response of the individual
patient. A program comparable to that discussed above
may be used in veterinary medicine.
Depending on the specific conditions being
treated, such agents may be formulated into liquid or
solid dosage forms and administered systemically or
locally. The agents may be delivered, for example, in a
timed or sustained-release form as is known to those
skilled in the art. Techniques for formulation and
administration may be found in Remington's
Pharmaceutical Sciences, Mack Publishing Co., Easton, PA
(18th ed. 1990). Suitable routes may include oral,
buccal, sublingual, rectal, transdermal, vaginal,
transmucosal, nasal or intestinal administration;
parenteral.delivery, including intramuscular,
subcutaneous, intramedullary injections, as well as
intrathecal, direct intraventricular, intravenous,
intraperitoneal, intranasal, or intraocular injections,
just to name a few.
For injection, the agents of the invention may
be formulated in aqueous solutions, preferably in
physiologically compatible buffers such as Hank's
solution, Ringer's solution, or physiological saline
buffer. For such transmucosal administration,
penetrants appropriate to the barrier to be permeated
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
268
are used in the formulation. Such penetrants are
generally known in the art.
Use of pharmaceutically acceptable carriers to
formulate the compounds herein disclosed for the
practice of the invention into dosages suitable for
systemic administration is within the scope of the
invention. With proper choice of carrier and suitable
manufacturing practice, the compositions of the present
invention, in particular, those formulated as solutions,
may be administered parenterally, such as by intravenous
injection. The compounds can be formulated readily
using pharmaceutically acceptable carriers well known in
the art into dosages suitable for oral administration.
Such carriers enable the compounds of the invention to
be formulated as tablets, pills, capsules, liquids,
gels, syrups, slurries, suspensions and the like, for
oral ingestion by a patient to be treated.
Agents intended to be administered
intracellularly may be administered using techniques
well known.to those of ordinary skill in the art. For
example, such agents may be encapsulated into liposomes,
then administered as described above. Liposomes are
spherical lipid bilayers with aqueous interiors. All
molecules present in an aqueous solution at the time of
liposome formation are incorporated into the aqueous
interior. The liposomal contents are both protected
from the external-microenvironment and, because
r
liposomes fuse with cell membranes, are efficiently
delivered into the cell cytoplasm. Additionally, due to
their hydrophobicity, small organic molecules may be
CA 02560002 2006-10-05
WO 97/46511 PCTIUS96I20697
- 269
directly administered intracellularly.
Pharmaceutical compositions suitable for use
in the present invention include compositions wherein
the active ingredients are contained in an effective
amount to achieve its intended purpose. Determination
of the effective amounts is well within the capability
of those skilled in the art, especially in light of the
detailed disclosure provided herein.
In addition to the active ingredients, these
pharmaceutical compositions may contain suitable
pharmaceutically acceptable carriers comprising
excipients and auxiliaries which facilitate processing
of the active compounds into preparations which can be
used pharmaceutically. The preparations formulated for
oral administration may be in the form of tablets,
dragees, capsules, or solutions.
The pharmaceutical compositions of the present
invention may be manufactured in a manner that is itself
known, e.g., by means of conventional mixing,
dissolving., granulating, dragee-making, levigating,
emulsifying, encapsulating, entrapping or lyophilizing
professes .
Pharmaceutical formulations for parenteral
administration include aqueous solutions of the active
compounds in water-soluble form. Additionally,
suspensions of the active compounds may be prepared as
appropriate oily injection suspension. Suitable
lipophilic solvents or vehicles include fatty oils such
as sesame oil, or synthetic fatty acid ester, such as
ethyl oleate or triglycerides, or liposomes. Aqueous
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
270
injection suspensions may contain substances which
increase the viscosity of the suspension, such as sodium
carboxymethyl cellulose, sorbitol, or dextran.
Optionally, the suspension may also contain suitable
stabilizers or agents which increase the solubility of
the compounds to allow for the preparation of highly
concentrated solutions.
Pharmaceutical preparations for oral use can
be obtained by combining the active compounds with solid
to excipients, optionally grinding a resulting mixture, and
processing the mixture of granules, after adding
suitable auxiliaries, if desired, to obtain tablets or
dragee cores. Suitable excipients are, in particular,
fillers such as sugars, including lactose, sucrose,
mannitol, or sorbitol; cellulose preparations, for
example, maize starch, wheat starch, rice starch, potato
starch, gelatin, gum tragacanth, methyl cellulose,
hydroxypropylmethyl-cellulose, sodium
carboxymethylcellulose (CMC), and/or
polyvinylpyrrolidone (PVP: povidone). If desired,
disintegrating agents may be added, such as the
cross-linked polyvinylpyrrolidone, agar, or alginic acid
or a salt thereof such as sodium alginate.
Dragee cores are provided with suitable
coatings. For this purpose, concentrated sugar
solutions may be used, which may optionally contain gum
arabic, talc, polyvinylpyrrolidone, carbopol gel,
polyethylene glycol (PEG), and/or titanium dioxide,
lacquer solutions, and suitable organic solvents or
solvent mixtures. Dye-stuffs or pigments may be added
CA 02560002 2006-10-05
WO 97/46511 PCT/US96/20697
_ 271
to the tablets or dragee coatings for identification or
to characterize different combinations of active
compound doses.
Pharmaceutical preparations which can be used
orally include push-fit capsul~ss made of gelatin, as
well as soft, sealed capsules made of gelatin, and a
plasticizer, such as glycerol or sorbitol. The push-fit
capsules can contain the active ingredients in admixture
with filler such as lactose, binders such as starches,
and/or lubricants such as talc or magnesium stearate
and, optionally, stabilizers. In soft capsules, the
active compounds may be dissolved or suspended in
suitable liquids, such as fatty oils, liquid paraffin,
or liquid polyethylene glycols (PEGs). In addition,
stabilizers may be added.
Other embodiments are within the following
claims.