Note: Descriptions are shown in the official language in which they were submitted.
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Pharmaceutical Dosage Forms Having Immediate Release and/or Controlled Release
Properties That Contain A GABAB Receptor Agonist
Background of the Invention
The present invention relates generally to pharmaceutical dosage forms having
immediate release and controlled release properties that contain a y-
aminobutyric acid
(GABAB) receptor agonist, e.g., baclofen, for the treatment of medical
conditions, which
include spasms, cramping, tightness of muscles, or spasticity associated with
ailments such as
multiple sclerosis, spinal cord diseases or certain spinal injuries.
Multiple sclerosis is considered to be an autoimmune disease. In this regard,
an
individual's immune system can attack the rnyelin sheath that surrounds nerve
cells. This
damage leads to muscle weakness, paralysis, poor coordination, balance
problems, fatigue,
and possible blindness. The GABAB agonist baclofen can be used to treat these
symptoms.
Baclofen can also facilitate adjunct medical treatment, such as physical
therapy, to improve
the condition of a patient with multiple sclerosis or certain spinal injuries.
Baclofen is also
used to reduce the number and severity of attacks of trigeminal neuralgia in
patients who are
not able to tolerate, or who have become refractory to, the effects of
carbamazepine.
Baclofen, or 4-amino-3-(4-chlorophenyl)-butanoic acid, is a muscle relaxant
and
antispastic. Its mechanism of action appears unclear. Baclofen seems capable
of inhibiting
both monosynaptic and polysynaptic reflexes at the spinal level, possibly by
hyperpolarization of afferent terminals, although actions at supraspinal sites
may also occur
and contribute to its clinical effect. In studies with animals, baclofen has
been shown to have
general central nervous system (CNS) depressant properties as indicated by the
production of
sedation with tolerance, somnolence, ataxia, and respiratory and
cardiovascular depression.
The absorption of baclofen is site specific. Baclofen is primarily absorbed in
the upper
gastrointestinal (GI) tract, with the extent of absorption of baclofen
substantially reduced in
the lower GI tract. Baclofen is rapidly and extensively absorbed. Absorption
may be dose-
dependent, being reduced with increasing doses. An improved method of
administering
baclofen to a patient would include the delivery of effective amounts of the
drug to the upper
GI tract for an extended period.
Several side effects are possibly associated with the administration of
baclofen to
mammals. These problems include nausea, vomiting, diarrhea, dizziness, daytime
sedation,
and less frequently, psychotic states such as depressive mood disorder. In
addition, patient
1
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
compliance with a dosing regimen can be suboptimal where frequent doses are
required, such
as the need for administering a dosage form three or four times a day. A
pharmaceutical
dosage form that requires less frequent dosing, such as once or twice a day,
thus would be
preferable. Furthermore, a pharmaceutical dosage form capable of establishing
and
maintaining stable plasma levels of baclofen for a prolonged period of time
may benefit
patients by requiring less frequent dosing and by minimizing side effects.
Certain baclofen pharmaceutical formulations, including Baclofen Tablet, 10/20
mg
(Watson Pharmaceuticals, Inc., Corona, CA) and the orally disintegrating
tablet marketed as
KEMSTROTM (Schwarz Pharma, Monheim, Germany), are marketed commercially, but
do
not provide controlled release of baclofen. For example, following a single 20
mg oral dose
of KEMSTROTM, the peak plasma concentration is reached about 1'/2 hours after
administration.
Baclofen has a serum half-life of 2.5 to 4 hours. (Drug Monograph: Baclofen
American Hospital Formulary Service (AHFS) American Society of Hospital
Pharmacists,
Inc., Bethesda, MD 2003). Following oral administration of existing immediate
release
baclofen formulations, minimum therapeutic plasma levels are typically reached
at about four
to eight hours following administration. Therefore, existing immediate-release
formulations
typically require dosing three to four times daily.
Various controlled release baclofen compositions have been reported. For
example,
U.S. Patent No. 5,091,184, issued Feb. 25, 1992, to Khanna describes adhesive
tablets that
stick to the oral mucosa and deliver drug through the mucous membrane. These
compositions
have one or more of the problems associated with adhesive tablets and deliver
the drug to a
less than optimal site for GABA related drugs. Additionally, U.S. Patent No.
5,651,985,
issued Jul. 29, 1997, to Penners et al, refers to matrix dosage forms having
extended gastric
residence time. Dosage forms made according to the Penners reference are
described as
having marked swelling and high dimensional stability in the swollen state. In
addition, an
osmotic pump type dosage form for delivering baclofen is referred to in U.S.
Patent
No. 4,764,380, issued Aug. 16, 1988, to Urquhart et al., which describes the
continuous
administration of drug over a prolonged period of time.
Nevertheless, there remains a significant and continuing need for
pharmaceutical
dosage forms having controlled release properties that contain a GABAB
receptor agonist,
such as baclofen, to treat medical conditions like multiple sclerosis or
certain spinal injuries
by establishing and maintaining stable plasma levels of the drug for a
prolonged period of
2
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
time to achieve less frequent dosing and to minimize side effects. These and
other objectives
are accomplished by the present invention.
Summary of the Invention
The present invention relates generally to pharmaceutical dosage forms having
controlled release properties that contain a GABAB receptor agonist, such as
baclofen. These
dosage forms can be used in the treatment of medical conditions, like spasms,
cramping, and
tightness of muscles, which are associated with ailments such as multiple
sclerosis or certain
spinal injuries.
For example, the pharmaceutical dosage forms of the present invention may
involve a
controlled release dosage form, where the controlled release dosage f rm
includes a GABAB
agonist and a pharmaceutically acceptable excipient, and the dosage Eorm
exhibits an in vitro
dissolution profile in simulated intestinal fluid medium comprising le:ss than
about 70%
GABAB agonist release after 1 hour, at least about 20% GABAB agoraist release
after 4 hours,
and at least about 30% GABAB agonist release after 6 hours. In this
e;mbodiment, the
controlled release dosage form exhibits an in vitro dissolution profile in
simulated gastric
fluid/simulated intestinal fluid (1 hour switchover) medium comprisimg less
than about 80%
GABAB agonist release after 1 hour, at least about 30% GABAB agoraist release
after 4 hours,
and at least about 40% GABAB agonist release after 6 hours.
In a preferred embodiment, the controlled release dosage fonrn including the
GABAB
agonist and pharmaceutically acceptable excipient exhibits an in vitro
dissolution profile in
simulated intestinal fluid medium comprising less than about 50% GABAB agonist
release
after 1 hour, at least about 40% GABAB agonist release after 4 hours, and at
least about 50%
GABAB agonist release after 6 hours. In this preferred embodiment, t11e
controlled release
dosage form exhibits an in vitro dissolution profile in simulated gastrsc
fluid/simulated
intestinal fluid (1 hour switchover) medium comprising less than about 70%
GABAB agonist
release after 1 hour, at least about 40% GABAB agonist release after 4 hours,
and at least
about 50% GABAB agonist release after 6 hours
In another embodiment, the controlled-release GABAB agonist dosage form is
combined with an immediate release GABAB agonist component. In this
embodiment, the
immediate release component exhibits an in vitro dissolution profile i-n
simulated gastric fluid
comprising at least about 80% GABAB agonist release after 1 hour. T'he ratio
of the
immediate-release component to the controlled-release component will be from
about 1:10 to
3
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
about 10:1, preferably from about 1:4 to about 4:1, more preferably from about
1:3 to about
3:1, and most preferably from about 1:2 to about 2:1.
In another embodiment, the pharmaceutical dosage forms of the pre: sent
invention
contain an enteric-coated controlled release component, where the enteric-
coated controlled
release component includes a GABAB agonist and a pharmaceutically acceptable
excipient,
and the enteric-coated controlled release component exhibits an in vitro
dissolution profile in
simulated gastric fluid/simulated intestinal fluid (2 hour switchover) mediuin
comprising less
than about 10% GABAB agonist release after 2 hours, at least about 40% GABAB
agonist
release after 3 hours, and at least about 70% GABAB agonist release after &
hours.
Preferably, the enteric-coated controlled release component exhibits an in
vitro dissolution
profile in simulated gastric fluid/simulated intestinal fluid (2 hour switchov-
er) medium
comprising less than about 10% GABAB agonist release after 2 hours, at least
about 50%
GABAB agonist release after 3 hours, and at least about 80% GABAB agon3st
release after 6.
Most preferably, the enteric-coated controlled release component exhibits an
in vitro
dissolution profile in simulated gastric fluid/simulated intestinal fluid (2
ho-ur switchover)
medium comprising less than about 10% GABAB agonist release after 2 hours, at
least about
60% GABAB agonist release after 3 hours, and at least about 90% GABAB agonist
release
after 6 hours.
In a further preferred embodiment, the dosage form also contains ari immediate
release component, in combination with the enteric-coated controlled release
component. For
example, the GABAB agonist may be formulated as a combination of immediate-
release
beads and controlled-release beads, compressed into a tablet or contained ir3
a capsule dosage
form. The ratio of the immediate-release component to the controlled-release
component will
be from about 1:10 to about 10:1, preferably from about 1:4 to about 4:1,
nzore preferably
from about 1:3 to about 3:1, and most preferably from about 1:2 to about 2: 1.
The present invention includes pharmaceutical dosage forms having both
immediate
release and extended release properties. In this embodiment, the
pharmaceiutical dosage form
comprising a GABAB agonist and a pharmaceutically acceptable excipient
exhibits an in vitro
dissolution profile in simulated gastric fluid/simulated intestinal fluid (2
hour switchover)
medium comprising less than about 75% GABAB agonist release after 2 hours, and
at least
about 80% GABAB agonist release after 3 hours. Preferably, the pharmaceutical
dosage form
exhibits an in vitro dissolution profile in simulated gastric fluid/simulated
intestinal fluid (2
4
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
hour switchover) medium comprising less than about 65% GABAB agonist release
after 2
hours, and at least about 90% GABAB agonist release after 3 hours
The pharmaceutical dosage forms of the present invention are adapted to be
administered twice daily in patients requiring chronic baclofen therapy. As
such, the iri vivo
absorption is prolonged as compared to immediate release baclofen
formulations, such that
the median time period at which at least 80% of said baclofen is absorbed, in
vivo, under
fasting conditions, is greater than 2.5 hours. The dosage forms of the present
invention will
typically exhibit an in vivo plasma profile comprising mean maximum baclofen
levels (C,,,aX)
from about 30 minutes to about 7 hours after administration to a fasting
patient, often
between 2.5 and 5.5 hours after administration.
Brief Description of the Drawings
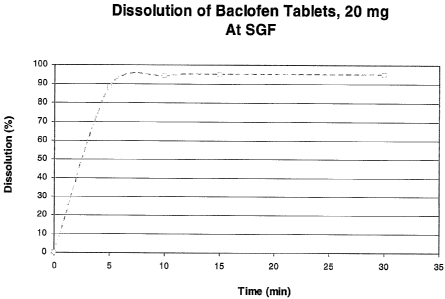
Figure lA is a graph of the in vitro dissolution profile of a baclofen tablet
formulation, 20 mg, prepared according to Example 8, according to measurements
under the
USP paddle method of 50 rpm in 900 ml simulated gastric fluid (pH 1.2) at 37
C.
Figure 1B is a graph of the in vitro dissolution profile of a baclofen tablet
formuelation,
mg, prepared according to Example 11, according to measurements under the USP
paddle
method of 50 rpm in 900 ml simulated intestinal fluid (pH 6.8) at 37 C.
Figure 2 is a graph of the in vitro dissolution profiles of baclofen tablet
fonnulations,
20 20 mg, prepared according to Example 11, according to measurements under
the USP paddle
method of 50 rpm in 900 ml simulated gastric fluid (pH 1.2) at 37 C for 1 hour
with a
switchover to simulated intestinal fluid (pH 6.8).
Figure 3 is a graph of the in vitro dissolution profile of a baclofen capsule
formulation, 20 mg, prepared according to Example 2, according to measurements
unde:r the
USP paddle method of 75 rpm in 900 ml simulated gastric fluid (pH 1.2) at 37
C.
Figure 4 is a graph of the in vitro dissolution profiles of baclofen capsule
formulations, 20 mg, prepared according to Example 12, according to
measurements under
the USP paddle method of 75 rpm in 900 ml simulated gastric fluid (pH 1.2) at
37 C foir- 2
hours with a switchover to simulated intestinal fluid (pH 6.8).
Figure 5 is a graph of the in vitro dissolution profiles of baclofen capsule
formulations, 30 mg, prepared according to Examples 13 and 14, according to
measurernents
5
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
under the USP paddle method of 75 rpm in 900 ml simulated gastric fluid (pH
1.2) at 37 C
for 2 hours with a switchover to simulated intestinal fluid (pH 6.8).
Figure 6 is a graph of the in vivo plasma profiles of baclofen tablet
formulations,
determined as described in Example 16.
Figure 7 is a graph simulating steady-state baclofen plasma levels determined
as
described in Example 17, where (C) represents the 40 mg dosage form of the
present
invention and (D) represents the reference 20 mg immediate-release dosage
form.
Detailed Description of the Invention
The present invention relates to pharmaceutical dosage forms comprising a
controlled-release GABAB agonist (preferably baclofen, a baclofen prodrug, a
baclofen
analog, or a mixture thereof, as well as a racemic baclofen mixture or a
substantially pure L-
baclofen enantiomeric product) formulation. The controlled-release GABAB
agonist
formulation may be in the form of an enteric-coated controlled-release
formulation. In
addition, the controlled-release GABAB agonist formulations, including enteric-
coated
controlled-release formulations, may be combined with immediate release GABAB
agonist
formulations in the final pharmaceutical dosage forms.
It has been found that the formulations of the present invention allow for
less frequent
dosing as compare to existing immediate release formulations. For example, for
patients
requiring chronic GABAB agonist therapy, twice daily administration of the
formulations of
the present invention is bioequivalent to three times daily administration of
an existing
immediate release formulation. This reduced dosing frequency is more
convenient for
patients, and typically leads to better patient compliance. In addition, it
reduces the number
of plasma peaks and troughs, which is typically associated with improved
efficacy and
reduced side effects.
As used herein and in the claims, the singular forms "a," "an," and "the"
include the
plural reference unless the context clearly indicates otherwise. Thus, for
example, the
reference to a profile is a reference to one or more such profiles, including
equivalents thereof
known to those skilled in the art. Other than in the operating examples, or
where otherwise
indicated, all numbers expressing quantities of ingredients or reaction
conditions used herein
should be understood as modified in all instances by the term "about." The
term "about"
when used in connection with percentages can mean 1%.
6
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
All patents and other publications identified are incorporated herein by
reference for
the purpose of describing and disclosing, for example, the methodologies
described in such
publications that might be used in connection with the present invention.
Unless defined otherwise, all technical and scientific terms used herein have
the same
meaning as those commonly understood to one of ordinary skill in the art to
which this
invention pertains. Although any known methods, devices, and materials may be
used in the
practice or testing of the invention, the preferred methods, devices, and
materials in this
regard are described here.
The baclofen, also known as butanoic acid or 4-amino-3-(4-chlorophenyl)
butanoic
acid, of the present invention includes racemic baclofen, enantiomerically
pure L-baclofen,
and analogs, derivatives, prodrugs, metabolites thereof, and any
pharmaceutically acceptable
salts thereof.
Baclofen is a GABAB receptor agonist, and thus other GABAB receptor agonists
are
envisioned within the scope of the invention. These may include 4-
aminobutanoic acid
(GABA); 3-aminopropyl)methylphosphinic acid; 4-amino-3-phenylbutanoic acid; 4-
amino-3-
hydroxybutanoic acid; 4-amino-3-(4-chlorophenyl)-3-hydroxyphenylbutanoic acid;
4-amino-
3-(thien-2-yl)butanoic acid; 4-amino-3-(5-chlorothien-2-yl)butanoic acid; 4-
amino-3-(5-
bromothien-2-yl)butanoic acid; 4-amino-3-(5-methylthien-2-yl)butanoic acid; 4-
amino-3-(2-
imidazolyl)butanoic acid; 4-guanidino-3-(4-chlorophenyl)butanoic acid; 3-amino-
2-(4-
chlorophenyl)-1-nitropropane; (3-aminopropyl)phosphonous acid; (4-aminobut-2-
yl)phosphonous acid; (3-amino-2-methylpropyl)phosphonous acid; (3-
aminobutyl)phosphonous acid; (3-amino-2-(4-chlorophenyl)propyl)phosphonous
acid; (3-
amino-2-(4-chlorophenyl)-2-hydroxypropyl)phosphonous acid; (3-amino-2-(4-
fluorophenyl)propyl)phosphonous acid; (3-amino-2-phenylpropyl)phosphonous
acid; (3-
amino-2-hydroxypropyl)phosphonous acid; (E)-(3-aminopropen-1-yl)phosphonous
acid; (3-
amino-2-cyclohexylpropyl)phosphonous acid; (3-amino-2-benzylpropyl)phosphonous
acid;
[3-amino-2-(4-methylphenyl)propyl]phosphonous acid; [3-amino-2-(4-
trifluoromethylphenyl)propyl]phosphonous acid; [3-amino-2-(4-
methoxyphenyl)propyl]phosphonous acid; [3-amino-2-(4-chlorophenyl)-2-
hydroxypropyl]phosphonous acid; (3-amino propyl)methylphosphinic acid; (3-
amino-2-
hydroxypropyl)methylphosphinic acid; (3-aminopropyl)(difluoromethyl)phosphinic
acid; (4-
aminobut-2-yl)methylphosphinic acid; (3-amino-l-hydroxypropyl)methylphosphinic
acid; (3-
amino-2-hydroxypropyl)(difluoromethyl)phosphinic acid; (E)-(3-aminopropen-l-
7
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
yl)methylphosphinic acid; (3-amino-2-oxo-propyl)methyl phosphinic acid; (3-
aminopropyl)hydroxymethylphosphinic acid; (5-aminopent-3-yl)methylphosphinic
acid; (4-
amino- 1, 1, 1-trifluorobut-2-yl)methylphosphinic acid; (3-amino-2-(4-
chlorophenyl)propyl)sulfinic acid; 3-aminopropylsulfinic acid, 1-
(aminomethyl)cyclohexaneacetic acid, and the like. See, e.g., U.S. Patent No.
6,664,069.
The term "analog" means a compound which comprises a chemically modified form
of a specific compound or class thereof, and which maintains the
pharmaceutical and/or
pharmacological activities characteristic of said compound or class. For
example, baclofen
analogs include 3-thienyl- and 3-furylaminobutyric acids.
The term "derivative" means a chemically modified compound wherein the
modification is considered routine by the ordinary skilled chemist, such as an
ester or an
amide of an acid, protecting groups, such as a benzyl group for an alcohol or
thiol, and tert-
butoxycarbonyl group for an amine.
The term "prodrug", as used herein, includes any covalently bonded carriers
which
release an active parent drug of the present invention in vivo when such
prodrug is
administered to a patient. Because prodrugs are known to enhance numerous
desirable
qualities of pharmaceuticals (i.e., solubility, bioavailability,
manufacturing, etc.) the
compounds of the present invention may be delivered in prodrug form. Prodrugs
of the
present invention may be prepared by modifying functional groups present in
the compound
in such a way that the modifications are cleaved, either in routine
manipulation or in vivo, to
the parent compound. The transformation in vivo may be, for example, as the
result of some
metabolic process, such as chemical or enzymatic hydrolysis of a carboxylic,
phosphoric or
sulphate ester, or reduction or oxidation of a susceptible functionality.
Prodrugs within the
scope of the present invention include compounds wherein a hydroxy, amino, or
sulfhydryl
group is bonded to any group that, when the prodrug of the present invention
is administered
to a mammalian subject, it cleaves to form a free hydroxyl, free amino, or
free sulfhydryl
group, respectively. Functional groups that may be rapidly transformed, by
metabolic
cleavage, in vivo form a class of groups reactive with the carboxyl group of
the compounds of
this invention. They include, but are not limited to, such groups as alkanoyl
(such as acetyl,
propionyl, butyryl, and the like), unsubstituted and substituted aroyl (such
as benzoyl and
substituted benzoyl), alkoxycarbonyl (such as ethoxycarbonyl), trialkysilyl
(such as
trimethyl- and triethysilyl), monoesters formed with dicarboxylic acids (such
as succinyl),
and the like. Because of the ease with which metabolically cleavable groups of
the
8
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
compounds useful according to this invention are cleaved in vivo, the
compounds bearing
such groups act as prodrugs. The compounds bearing the metabolically cleavable
groups have
the advantage that they may exhibit improved bioavailability as a result of
enhanced
solubility and/or rate of absorption conferred upon the parent compound by
virtue of the
presence of the metabolically cleavable group.
A discussion of prodrugs is provided in the following: DEsiGN OF PRODRUGS, H.
Bundgaard, ed. (Elsevier, 1985); METHODS IN ENZYMOLOGY, K. Widder et al.,
eds., vol. 42,
309-96 (Academic Press 1985); A TExTBOOK OF DRUG DESIGN AND DEVELOPMENT,
Krogsgaard-Larsen & H. Bundgaard, ed., Chapter 5; Design and Applications of
Prodrugs,
113-91 (1991); H. Bundgard, Advanced Drug Delivery Reviews, 1-38 (1992); 8 J.
PHARM.
SCIENCES 285 (1988); N. Nakeya et al., 32 CHEM. PHARM. BuLL. 692 (1984); T.
Higuchi and
V. Stella, Prodrugs as Novel Delivery Systems, 14 A.C.S. SYMPOSIUM SERIES:
BIOREVERSIBLE CARRIERS IN DRUG DESIGN, Edward B. Roche, ed. (Am. Pharm. Assoc.
&
Pergamon Press 1987), each of which is incorporated herein by reference.
Thus, the present invention contemplates the use of prodrugs of GABAB receptor
agonists (including baclofen), methods of delivering the same, and
compositions containing
the same. For example, baclofen prodrugs have been described in Leisen et al.,
Lipophilicities of Baclofen Ester Prodrugs Correlate witla Affirzities to tlae
ATP-dependent
Efflux Pump P-glycoprotein, 20 PHARM. REs. 772-78 (2003).
The term "metabolite" refers to a form of a compound obtained in a human or
animal
body by action of the body on the administered form of the compound, for
example a de-
methylated analog of a compound bearing a methyl group which is obtained in
the body after
administration of the methylated compound as a result of action by the body on
the
methylated compound. Metabolites may themselves have biological activity.
The phrase "pharmaceutically acceptable" is employed herein to refer to those
compounds, materials, compositions, and/or dosage forms which are, within the
scope of
sound medical judgment, suitable for use in contact with the tissues of human
beings and
animals without excessive toxicity, irritation, allergic response, or other
problem or
complication commensurate with a reasonable benefit/risk ratio.
For example, "pharmaceutically acceptable salts" refer to derivatives of the
disclosed
compounds wherein the specified compound is converted to an acid or base salt
thereof. Such
pharmaceutically acceptable salts include, but are not limited to, mineral or
organic acid salts
of basic residues such as amines; alkali or organic salts of acidic residues
such as carboxylic
9
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
acids; and the like. The pharmaceutically acceptable salts include the
conventional non-toxic
salts or the quaternary ammonium salts of the parent compound formed, for
example, from
non-toxic inorganic or organic acids. For example, such conventional non-toxic
salts include
those derived from inorganic acids such as hydrochloric, hydrobromic,
sulfuric, sulfamic,
phosphoric, nitric and the like; and the salts prepared from organic acids
such as acetic,
propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric,
ascorbic, pamoic, maleic,
hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-
acetoxybenzoic,
fumaric, toluensulfonic, methanesulfonic, ethane dislfonic, oxalic,
isethionic, and the like.
For purposes of the present invention, the term "controlled release" refers to
part or
all of a dosage form that can release one or more active pharmaceutical agents
over a
prolonged period of time (i.e., over a period of more than 1 hour), or delays
the release of
active agent for a prolonged period of time. The characteristic of controlled
release (CR) may
also be referred to as sustained release (SR), prolonged release (PR),
modified release (MR),
delayed release (DR) or extended release (ER). When used in association with
the dissolution
profiles discussed herein, the term "controlled release" refers to that
portion of a dosage form
according to the present invention that delivers active agent over a period of
time greater than
1 hour.
"Immediate release" refers to part or all of a dosage form that releases
active agent
substantially immediately upon contact with gastric juices and that results in
substantially
complete dissolution within about 1 hour. The characteristic of immediate
release (IR) may
also be referred to as instant release (IR). When used in association with the
dissolution
profiles discussed herein, the term "immediate release" refers to that portion
of a dosage form
according to the present invention that delivers active agent over a period of
time less than 1
hour.
The term "CMAX" is the peak blood plasma concentration exhibited by the
compositions of the present invention. "TMAX" refers to the time that CMAX
occurs in the
plasma concentration-time profile. "CMIN" is the minimum plasma concentration.
"C' is
shorthand for concentration, "T" for time, "max" for maximum, and "min" for
minimum.
Initial peak plasma level refers to the first rise in blood plasma level of
the active agent and
may be followed by one or more additional peaks, one of which may be CMAX. As
used
herein, "mean maximum GABAB agonist level" refers to the mean GABAB agonist
CMAX.
The blood plasma concentrations described herein are typically determined
across a
population of at least 12 subjects.
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
The blood plasma concentrations described above may refer to plasma levels
after a
single oral administration of the dosage form, or may refer to levels obtained
at steady state.
As used herein, "steady state" blood plasma concentrations refers to the
plasma levels
obtained upon the repeated dosing of a drug until it reaches a stable level of
absorption and
elimination such that the amount of drug in the body is substantially
constant.
As used herein, the term "patient" means any mammal including humans.
The term "effective amount" means an amount of a compound/composition
according
to the present invention effective in producing the desired therapeutic
effect.
The term "excipients" refer to phannacologically inert ingredients that are
not active
in the body. See HANDBOOK OF PHARMACEUTICAL EXCIPIENTS (Am. Pharm. Ass'n
1986). The
artisan of ordinary skill in the art will recognize that many different
excipients can be used in
formulations according to the present invention and the list provided herein
is not exhaustive.
The active ingredients of the present invention may be mixed with
pharmaceutically
acceptable carriers, diluents, adjuvants, excipients, or vehicles, such as
preserving agents,
fillers, polymers, disintegrating agents, glidants, wetting agents,
emulsifying agents,
suspending agents, sweetening agents, flavoring agents, perfuming agents,
lubricating agents,
acidifying agents, and dispensing agents, depending on the nature of the mode
of
administration and dosage forms. Such ingredients, including pharmaceutically
acceptable
carriers and excipients, may be used to formulate oral dosage forms.
Pharmaceutically
acceptable carriers include water, ethanol, polyols, vegetable oils, fats,
waxes polymers,
including gel forming and non-gel forming polymers, and suitable mixtures
thereof.
Examples of excipients include starch, pregelatinized starch, Avicel, lactose,
milk sugar,
sodium citrate, calcium carbonate, dicalcium phosphate, and lake blend.
Examples of
disintegrating agents include starch, alginic acids, and certain complex
silicates. Examples of
lubricants include magnesium stearate, sodium lauryl sulphate, talc, as well
as high molecular
weight polyethylene glycols.
"Dosing under fasting conditions" is defined as when the dosage is
administered
orally with 240 ml of room temperature water after subjects are fasted
overnight for at least
10 hours. No fluid, except that given with drug administration, will be
allowed from 1 hour
prior to dose administration until 1 hour after dosing. At 2 hours post-dose,
subjects may
consume 240 ml of room temperature water.
The pharmaceutical dosage forms of the present invention may involve a
controlled
release dosage form, where the controlled release dosage form includes a GABAB
agonist and
11
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
a pharmaceutically acceptable excipient, and the dosage form exhibits an in
vitro dissolution
profile in simulated intestinal fluid medium comprising less than about 70%
GABAB agonist
release after 1 hour, at least about 20% GABAB agonist release after 4 hours,
and at least
about 30% GABAB agonist release after 6 hours. In this embodiment, the
controlled release
dosage form exhibits an in vitro dissolution profile in simulated gastric
fluid/simulated
intestinal fluid (1 hour switchover) medium comprising less than about 80%
GABAB agonist
release after 1 hour, at least about 30% GABAB agonist release after 4 hours,
and at least
about 40% GABAB agonist release after 6 hours.
Preferably, the controlled release dosage form exhibits an in vitro
dissolution profile
in simulated intestinal fluid medium comprising less than about 50% GABAB
agonist release
after 1 hour, at least about 40% GABAB agonist release after 4 hours, and at
least about 50%
GABAB agonist release after 6 hours. In this preferred embodiment, the
controlled release
dosage form exhibits an in vitro dissolution profile in simulated gastric
fluid/simulated
intestinal fluid (1 hour switchover) medium comprising less than about 70%
GABAB agonist
release after 1 hour, at least about 40% GABAB agonist release after 4 hours,
and at least
about 50% GABAB agonist release after 6 hours
In another embodiment, the controlled-release GABAB agonist dosage form is
combined with an immediate release GABAB agonist component. In this
embodiment, the
immediate release component exhibits an in vitro dissolution profile in
simulated gastric fluid
comprising at least about 80% GABAB agonist release after 1 hour.
In another embodiment, the pharmaceutical dosage forms of the present
invention
contain an enteric-coated controlled release component, where the enteric-
coated controlled
release component includes a GABAB agonist and a pharmaceutically acceptable
excipient,
and the enteric-coated controlled release component exhibits an in vitro
dissolution profile in
simulated gastric fluid/simulated intestinal fluid (2 hour switchover) medium
comprising less
than about 10% GABAB agonist release after 2 hours, at least about 40% GABAB
agonist
release after 3 hours, and at least about 70% GABAB agonist release after 6
hours.
Preferably, the enteric-coated controlled release component exhibits an in
vitro dissolution
profile in simulated gastric fluid/simulated intestinal fluid (2 hour
switchover) medium
comprising less than about 10% GABAB agonist release after 2 hours, at least
about 50%
GABAB agonist release after 3 hours, and at least about 80% GABAB agonist
release after 6.
Most preferably, the enteric-coated controlled release component exhibits an
in vitro
dissolution profile in simulated gastric fluid/simulated intestinal fluid (2
hour switchover)
12
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
medium comprising less than about 10% GABAB agonist release after 2 hours, at
least about
60% GABAB agonist release after 3 hours, and at least about 90% GABAB agonist
release
after 6 hours.
In a further preferred embodiment, the dosage form also contains an immediate
release component, in combination with the enteric-coated controlled release
component.
The present invention includes pharmaceutical dosage forms having both
immediate
release and extended release properties. In this embodiment, the
pharmaceutical dosage form
comprising a GABAB agonist and a pharmaceutically acceptable excipient
exhibits an in vitro
dissolution profile in simulated gastric fluid/simulated intestinal fluid (2
hour switchover)
medium comprising less than about 75% GABAB agonist release after 2 hours, and
at least
about 80% GABAB agonist release after 3 hours. Preferably, the pharmaceutical
dosage form
exhibits an in vitro dissolution profile in simulated gastric fluid/simulated
intestinal fluid (2
hour switchover) medium comprising less than about 65% GABAB agonist release
after 2
hours, and at least about 90% GABAB agonist release after 3 hours
Appropriate in vitro dissolution testing methods for the dosage forms of the
present
invention are known to those of skill in the art and include those described
in the Examples
herein. The USP paddle method refers to the Paddle and Basket Method as
described in
United States Pharmacopoeia, Edition XXII (1990). In particular, the USP
paddle method of
50 rpm or 75 rpm in 900 mi simulated gastric fluid (SGF) (pH 1.2) or simulated
intestinal
fluid (SIF) (pH 6.8) at 37 C may be used to detennine the in vitro dissolution
profiles
according to the present invention.
When the dosage forms of the present invention include a controlled release
component, including enteric-coated controlled release, as well as an
immediate release
component, the ratio of the immediate release component to the controlled
release component
is from about 1:10 to about 10:1, preferably about 1:4 to about 4:1, more
preferably from
about 1:3 to about 3:1, and most preferably from about 1:2 to about 2:1.
The phannaceutical dosage forms of the present invention are adapted to allow
prolonged absorption of the active agent, which allows less frequent
administration as
compared to existing immediate-release formulations. As used herein,
"prolonged
absorption" means that the active agent is absorbed in vivo, under fasting
conditions, over an
extended period of time. In particular, the time period over which the
majority (i.e., 80-90%)
of the absorption occurs extends to about 7 or 8 hours after administration of
the dosage
form. Specifically, the median time period at which at least 80% of the active
agent=is
13
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
absorbed from the dosage forms of the present invention is greater than 2.5
hours after
administration, typically three to 4.5 hours after administration. By
comparison, the median
time period at which at least 80% of the active agent is absorbed from
existing immediate-
release formulations is 1.5 to two hours after administration. The period over
which an active
agent is absorbed from a dosage form can be calculated by deconvolution, using
mathematical methods known to those of skill in the art.
The dosage forms of the present invention will exhibit an in vivo plasma
profile
comprising mean maxinnum GABAB agonist levels from about 30 minutes to about 7
hours,
often from about 2.5 hours to about 5.5 hours, after administration of a
single dose to a
fasting patient. At steady-state, the pharmaceutical dosage forms of the
present invention
will reach a Cmw comparable to that obtained at steady-state from an immediate-
release
dosage form at a later tirne point, which will allow less frequent dosing. In
particular, a 40
mg dosage form of the present invention, when administered twice daily, will
deliver mean
steady-state area under the plasma concentration-time curve (AUC), maximum
plasma
concentration (CMAx), and minimum plasma concentration (CMIN) similar to that
of an
immediate-release tablet formulation administered three times daily.
These dosage forms (preferably a tablet or capsule, which may contain beads,
granules, particles, or a mixture thereof) may contain baclofen in the amount
of from about 2
mg to about 150 mg (preferably from about 2.5 mg to about 100 mg) and can be
used in the
treatment of medical conditions, which includes spasms, cramping, and
tightness of muscles,
that are associated with ailments such as multiple sclerosis or certain spinal
injuries.
Total daily dosages of the compounds useful according to this invention
administered
to a host in single or divided doses are generally in amounts of from about
0.01 mg/kg to
about 100 mg/kg body weight daily, and preferably from about 0.05 mg/kg to
about 50 mg/kg
body weight daily. It should be understood, however, that the specific dose
level for any
particular patient will depend upon a variety of factors including body
weight, general health,
gender, diet, time and route of administration, rates of absorption and
excretion, combination
with other drugs, and the severity of the particular disease being treated.
Actual dosage levels
of active ingredient in the compositions of the present invention may be
varied so as to obtain
an amount of active ingredient that is effective to obtain a desired
therapeutic response for a
particular composition and method of administration.
Total daily dose of the compounds useful according to this invention
administered to
a host in single or divided doses may be in amounts, for example, of from
about 0.01 mg/kg
14
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
to about 20 mg/kg body weight daily and preferably 0.02 to 10 mg/kg/day. The
preferred
dosage range of baclofen is between 2.5 mg and 100 mg per dosage form. Dosage
forms
according to the present invention may contain such amounts or fractions
thereof as may be
used to make up the daily dose.
Preferred dosage strengths for the formulations of the present invention
include those
having 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg and 40 mg baclofen. Typically,
the
optimal dosage for a patient will be determined by titration, whereby the
patient is initially
given small doses, which are then gradually increased until the patient
reaches the dosage
level that achieves maximum therapeutic efficacy with minimum side effects.
One embodiment of the present invention provides a controlled release solid
oral
dosage form in which there is immediate release of baclofen and delayed or
delayed-
sustained release of baclofen. Dosages according to the present invention may
include an
immediate release component and a delayed or delayed-sustained release
component. The
combination of these two components can release the drug in a pulsed release
fashion or a
continuous fashion upon oral administration of the dosage form.
In one aspect, the invention relates to a controlled release baclofen solid
oral dosage
form comprising an immediate release baclofen component and a delayed or
delayed-
sustained, or sustained release baclofen component. The immediate release
baclofen
component comprises baclofen formulated with one or more pharmaceutically
acceptable
excipients that allow for immediate release of the baclofen, and the delayed,
or delayed-
sustained, or sustained release baclofen component comprises baclofen
formulated with one
or more excipients that allow for delayed, or delayed-sustained, or sustained
release of the
baclofen. For example, see U.S. Patent No. 6,372,254 that refers to
formulations, such as
tablets, having both an immediate release component and an extended release
component.
Among other dosage forms apparent to the skilled artisan, the solid oral
dosage form
according to the present invention may be a tablet formulation, or a discrete
unit-filled
capsule formulation, or a sachet forrnulation. The discrete units of the
present invention
include beads, granules, pellets, spheroids, particles, tablets, pills, etc.
Specifically, the immediate release, delayed release, delayed-sustained
release, and
sustained release components of the dosage form can take any form known to a
skilled
pharmaceutical formulator, including one component of a multi-component tablet
such as
described in U.S. Patent 6,372,254, issued Apr. 16, 2002, pending U.S. Patent
Application
Serial No. 10/241,837, filed Sept. 12, 2002, and published International
Patent Application
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
No. WO 03/101432, filed Dec. 11, 2003, each assigned to Impax Laboratories,
Inc. The
controlled release baclofen dosages according to the present invention may be
in the form of
cores comprising baclofen.
Dosage forms can be made according to known methods in the art. Some preferred
methods are described below.
Matrix Dosage Forms. The term matrix, as used herein, refers to a solid
material
having an active agent incorporated therein. Upon exposure to a dissolution
media, channels
are formed in the solid material so that the active agent can escape. Dosage
forms according
to the present invention may be in the form of coated or uncoated matrices. A
coating, for
example may contain immediate release baclofen, or in the alternative, and the
matrix itself
can contain controlled release baclofen. Drug release from the delayed or
delayed-sustained,
or sustained release component can be immediate or sustained, for example
within 7 hours
after oral administration of the oral dosage form to ensure effective
absorption of the drug.
The controlled release baclofen component may be comprised of baclofen coated
with
at least one delayed release layer. The delayed-sustained release baclofen
component may be
comprised of sustained-release-coated baclofen coated with at least one
delayed release layer.
The sustained release baclofen component may be comprised of baclofen coated
with at least
one sustained-release polymer, or a matrix-controlled release polymer.
The skilled artisan should appreciate that the matrix material can be chosen
from a
wide variety of materials that can provide the desired dissolution profiles.
Materials can
include, for example, one or more gel forming polymers such as polyvinyl
alcohol, cellulose
ethers including, for example, hydroxyl propyl alkyl, celluloses such as
hydroxypropyl
methyl cellulose, hydroxy alkyl celluloses such as hydroxy propyl cellulose,
natural or
synthetic gums such as guar gum, xanthum gun-i, and alginates, as well as,
ethyl cellulose,
polyethylene oxide, polyvinyl pyrrolidone, fats, waxes, polycarboxylic acids
or esters such as
the Carbopol0 series of polymers, methacrylic acid copolymers, and
methacrylate polymers.
Methods of making matrix dosages are known in the art and any such method that
can
yield the desired dissolution profiles and/or plasma profiles may be relied
upon according to
the present invention. One such method involves baclofen with a solid
polymeric material
and one or more pharmaceutically acceptable excipients that are then blended
and
compressed in controlled release tablet cores. Such tablet cores can be used
for further
processing as bilayer tablets, press coated tablets, or film coated tablets.
16
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
A coating containing the immediate release baclofen can be added to the
outside of
the controlled release tablet cores to produce a final dosage form. Such a
coating can be
prepared by mixing baclofen with polyvinylpyrrolidone (PVP) 29/32 or
hydroxypropyl
methylcellulose (HPMC) and water/isopropyl alcohol and triethyl acetate. Such
an immediate
release coating can be spray coated onto the tablet cores. The immediate
release coating may
also be applied using a press-coating process with a blend consisting of 80%
by weight
baclofen and 20% by weight of lactose and hydroxypropyl methylcellulose type
2910. Press
coating techniques are known in the art and are described in U.S. Patent No.
6,372,254 (Ting
et al.), incorporated herein by reference in its entirety.
In addition, the formulation of respective release components can occur by
appropriate granulation methods as is well known in the art. In wet
granulation, solutions of
the binding agent (polymer) are added with stirring to the mixed powders. The
powder mass
is wetted with the binding solution until the mass has the consistency of damp
snow or brown
sugar. The wet granulated material is forced through a sieving device. Moist
material from
the milling step is dried by placing it in a temperature controlled container.
After drying, the
granulated material is reduced in particle size by passing it through a
sieving device.
Lubricant is added, and the final blend is then compressed into a matrix
dosage form.
In fluid-bed granulation, particles of inert material and/or active agent are
suspended
in a vertical column with a rising air stream. While the particles are
suspended, a common
granulating material in solution is sprayed into the column. There is a
gradual particle
buildup under a controlled set of conditions resulting in tablet granulation.
Following drying
and the addition of lubricant, the granulated material is ready for
compression.
In dry-granulation, the active agent, binder, diluent, and lubricant are
blended and
compressed into tablets. The compressed large tablets are comminuted through
the desirable
mesh screen by sieving equipment. Additional lubricant is added to the
granulated material
and blended gently. The material is then compressed into tablets.
Particle Based Dosage Forms, Immediate Release Particles. The immediate
release/controlled release dosage forms of the present invention can also take
the form of
pharmaceutical particles. The dosage forms can include immediate release
particles in
combination with controlled release particles in a ratio sufficient to deliver
the desired release
of active agents. The controlled release particles can be produced by coating
the immediate
release particles.
17
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
The term "particle" as used herein means a granule having a diameter of
between
about 0.01 mm and about 5.0 mm, preferably between about 0.1 mm and about 2.5
mm, and
more preferably between about 0.5 mm and about 2 mm. The skilled artisan
should
appreciate that particles according to the present invention can be any
geometrical shape
within this size range and so long as the mean for a statistical distribution
of particles falls
within the particle sizes enumerated above, they will be considered to fall
within the
contemplated scope of the present invention. Particles can assume any standard
structure
known in the pharmaceutical arts. Such structures include, for example, matrix
particles, non-
pareil cores having a drug layer and active or inactive cores having multiple
layers thereon. A
controlled release coating can be added to any of these structures to create a
controlled
release particle.
The particles can be produced according to any of a number of known methods
for
making particles. The immediate release particles comprise the active agent
combination and
a disintegrant. Suitable disintegrants include, for example, starch, low-
substitution
hydroxypropyl cellulose, croscarmellose sodium, calcium carboxymethyl
cellulose,
hydroxypropyl starch, sodium starch glycolate, and microcrystalline cellulose.
In addition to the above-mentioned ingredients, the matrix may also contain
suitable
quantities of other materials, for example, diluents, lubricants, binders,
granulating aids,
colorants, flavorants, and glidants that are conventional in the
pharmaceutical arts. The
quantities of these additional materials are sufficient to provide the desired
effect to the
desired formulation. A matrix incorporating particles may also contain
suitable quantities of
these other materials such as diluents, lubricants, binders, granulating aids,
colorants,
flavorants, and glidants that are conventional in the pharmaceutical arts in
amounts up to
about 75% by weight of the particulate, if desired.
In one preferred embodiment, oral dosage forms are prepared to include an
effective
amount of particles as described above within a capsule. For example, melt-
extruded particles
may be placed in a gelatin capsule in an amount sufficient to provide an
effective controlled
release dose when ingested and contacted by gastric fluid. In another
preferred embodiment,
a suitable amount of the particles are compressed into an oral tablet using
conventional
tableting equipment using standard techniques. Techniques and compositions for
making
tablets (compressed and molded), capsules (hard and soft gelatin), and pills
are also described
in REMINGTON'S PHARMACEUTICAL SCIENCES, Arthur Osol, ed., 1553-93 (1980),
incorporated herein by reference. The particles can be made by mixing the
relevant
18
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
ingredients and granulating the mixture. The resulting particles are dried and
screened, and
the particles having the desired size are used for drug formulation.
Controlled Release Particles. The controlled release particles of the present
invention
slowly release baclofen when ingested and exposed to gastric fluids, and then
to intestinal
fluids. The controlled release profile of the formulations of the invention
can be altered, for
example, by increasing or decreasing the thickness of the retardant coating,
i.e., by varying
the amount of overcoating. The resultant solid controlled release particles
may thereafter be
placed in a gelatin capsule in an amount sufficient to provide an effective
controlled release
dose when ingested and contacted by an environmental fluid, e.g., gastric
fluid, intestinal
fluid or dissolution media. The particles may be overcoated with an aqueous
dispersion of a
hydrophobic or hydrophilic material to modify the release profile. The aqueous
dispersion of
hydrophobic material preferably further includes an effective amount of
plasticizer, e.g.
triethyl citrate. Preformulated aqueous dispersions of ethylcellulose, such as
Aquacoat or
Surelease , may be used. If Surelease is used, it is not necessary to
separately add a
plasticizer.
The release of the therapeutically active agent from the controlled release
formulation
of the present invention can be further influenced, i.e., adjusted to a
desired rate, by the
addition of one or more release-modifying agents. The release-modifying agent
may be
organic or inorganic and include materials that can be dissolved, extracted,
or leached from
the coating in the environment of use. The pore-formers may comprise one or
more
hydrophilic materials such as hydroxypropyl methylcellulose. The release-
modifying agent
may also comprise a semi-permeable polymer. In certain preferred embodiments,
the release-
modifying agent is selected from hydroxypropyl methylcellulose, lactose, metal
stearates, and
mixtures thereof.
The controlled-release component may also include a combination of hydrophilic
and
hydrophobic polymers. In this embodiment, once administered, the hydrophilic
polymer
dissolves away to weaken the structure of the controlled-release component,
and the
hydrophobic polymer retards the water penetration and helps to maintain the
shape of the
drug delivery system.
The hydrophobic material may be selected from the group consisting of
alkylcellulose, acrylic and methacrylic acid polymers and copolymers, shellac,
zein,
hydrogenated castor oil, hydrogenated vegetable oil, or mixtures thereof. In
certain preferred
embodiments, the hydrophobic material is a pharmaceutically acceptable acrylic
polymer,
19
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
including but not limited to acrylic acid and methacrylic acid copolymers,
methyl
methacrylate, methyl methacrylate copolymers, ethoxyethyl methacrylates,
cyanoethyl
methacrylate, aminoalkyl methacrylate copolymer, poly(acrylic acid),
poly(methacrylic acid),
methacrylic acid alkylamine copolymer, poly(methyl methacrylate),
poly(methacrylic acid
anhydride), polymethacrylate, polyacrylamide, poly(methacrylic acid
anhydride), and
glycidyl methacrylate copolymers. In alternate embodiments, the hydrophobic
material is
selected from materials such as one or more hydroxyalkyl celluloses such as
hydroxypropyl
methylcellulose. The hydroxyalkyl cellulose is preferably a hydroxy (Cl to C6)
alkyl
cellulose, such as hydroxypropylcellulose, hydroxypropylmethylcellulose, or
preferably
hydroxyethylcellulose. The amount of the hydroxyalkyl cellulose in the present
oral dosage
form is determined, inter alia, by the precise rate of active agents desired
and may vary from
about 1% to about 80%.
In embodiments of the present invention where the coating comprises an aqueous
dispersion of a hydrophobic polymer, the inclusion of an effective amount of a
plasticizer in
the aqueous dispersion of hydrophobic polymer can further improve the physical
properties
of the film. For example, because ethylcellulose has a relatively high glass
transition
temperature and does not form flexible films under normal coating conditions,
it is necessary
to plasticize the ethylcellulose before using it as a coating material.
Generally, the amount of
plasticizer included in a coating solution is based on the concentration of
the film-former,
e.g., most often from about 1 percent to about 50 percent by weight of the
film-former.
Concentration of the plasticizer, however, is preferably determined after
careful
experimentation with the particular coating solution and method of
application.
Examples of suitable plasticizers for ethylcellulose include water-insoluble
plasticizers such as dibutyl sebacate, diethyl phthalate, triethyl citrate,
tributyl citrate, and
triacetin, although other water-insoluble plasticizers (such as acetylated
monoglycerides,
phthalate esters, castor oil, etc.) rnay be used. Triethyl citrate is an
especially preferred
plasticizer for the aqueous dispersions of ethyl cellulose of the present
invention.
Examples of suitable plasticizers for the acrylic polymers of the present
invention
include, but are not limited to, citric acid esters such as triethyl citrate
NF XVI, tributyl
citrate, dibutyl phthalate, and possibly 1,2-propylene glycol. Other
plasticizers which have
proved to be suitable for enhancing the elasticity of the films formed from
acrylic fihns such
as Eudragit0 RL/RS lacquer solutions include polyethylene glycols, propylene
glycol,
diethyl phthalate, castor oil, and triacetin. Triethyl citrate is an
especially preferred plasticizer
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
for aqueous dispersions of ethyl cellulose. It has further been found that
addition of a small
amount of talc reduces the tendency of the aqueous dispersion to stick during
processing and
acts a polishing agent.
One commercially available aqueous dispersion of ethylcellulose is Aquacoat
which
is prepared by dissolving the ethylcellulose in a water-immiscible organic
solvent and then
emulsifying the ethylcellulose in water in the presence of a surfactant and a
stabilizer. After
homogenization to generate submicron droplets, the organic solvent is
evaporated under
vacuum to form a pseudolatex. The plasticizer is not incorporated into the
pseudolatex during
the manufacturing phase. Thus, prior to using the pseudolatex as a coating,
the Aquacoat is
mixed with a suitable plasticizer.
Another aqueous dispersion of ethylcellulose is commercially available as
Surelease
(Colorcon, Inc., West Point, PA, USA). This product is prepared by
incorporating plasticizer
into the dispersion during the manufacturing process. A hot melt of a polymer,
plasticizer
(dibutyl sebacate), and stabilizer (oleic acid) is prepared as a homogeneous
mixture which is
then diluted with an alkaline solution to obtain an aqueous dispersion which
can be applied
directly onto substrates.
In one preferred embodiment, the acrylic coating is an acrylic resin lacquer
used in
the form of an aqueous dispersion, such as that which is commercially
available from Rohni
Pharma under the trade name Eudragit . In additional preferred embodiments,
the acrylic
coating comprises a mixture of two acrylic resin lacquers commercially
available from Rohrn
Pharma under the trade names Eudragit@ RL 30 D and Eudragit RS 30 D. Eudragit
RL
D and Eudragit@ RS 30 D are copolymers of acrylic and methacrylic esters with
a low
content of quaternary ammonium groups, the molar ratio of ammonium groups to
the
remaining neutral (meth)acrylic esters being 1:20 in Eudragit RL 30 D and
1:40 in
25 Eudragit@ RS 30 D. The mean molecular weight is about 150,000 Daltons. The
code
designations RL (high permeability) and RS (low permeability) refer to the
permeability
properties of these agents. Eudragit RL/RS mixtures are insoluble in water
and in digestive
fluids, however, coatings formed fr rn them are swellable and permeable in
aqueous
solutions and digestive fluids.
30 The Eudragit@ RL/RS dispersions may be mixed together in any desired ratio
in order
to ultimately obtain a controlled release formulation having a desirable
dissolution profile.
Desirable controlled release formulations may be obtained, for instance, from
a retardant
coating derived from one of a variety of coating combinations, such as 100%
Eudragit@ RL;
21
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
50% Eudragit RL and 50% Eudragit RS; or 10% Eudragit RL and 90% Eudragit
RS.
One skilled in the art should recognize that other acrylic polymers may also
be used, for
example, Eudragit L. In addition to modifying the dissolution profile by
altering the relative
amounts of different acrylic resin lacquers, the dissolution profile of the
ultimate product may
also be modified, for example, by increasing or decreasing the thickness of
the retardant
coating.
In preferred embodiments of the present inventiori, the stabilized product is
obtained
by subjecting the coated substrate to oven curing at a temperature above the
Tg of the
plasticized acrylic polymer for the required time period, t3ze optimum values
for temperature
and time for the particular formulation being determined experimentally. In
certain
embodiments of the present invention, the stabilized product is obtained via
an oven curing
conducted at a temperature of about 45 C for a time period from about 1 to
about 48 hours. It
is also contemplated that certain products coated with the controlled release
coating of the
present invention may require a curing time longer than 24 to 48 hours, e.g.,
from about 48 to
about 60 hours or more.
The coating solutions preferably contain, in addition to the film-fomier,
plasticizer,
and solvent system (i.e., water), a colorant to provide elegance and product
distinction. Color
may be added to the solution of the therapeutically active agent instead of,
or in addition to
the aqueous dispersion of hydrophobic material. For example, color may be
added to
Aquacoat via the use of alcohol or propylene glycol based color dispersions,
milled
aluminum lakes and opacifiers such as titanium dioxide by adding color with
shear to the
water soluble polymer solution and then using low shear to the plasticized
Aquacoat0.
Alternatively, any suitable method of providing color to t3ie formulations of
the present
invention may be used. Suitable ingredients for providing color to the
formulation when an
aqueous dispersion of an acrylic polymer is used include -titanium dioxide and
color
pigments, such as iron oxide pigments. The incorporation of pigments, may,
however,
increase the retardant effect of the coating.
Spheroids or beads coated with the therapeutically active agents can be
prepared, for
example, by dissolving the therapeutically active agents in water and then
spraying the
solution onto a substrate, for example, non pareil 18/20 beads, using a Wuster
insert.
Optionally, additional ingredients are also added prior to coating the beads
in order to assist
the binding of the active agents to the beads, and/or to color the solution,
etc. For example, a
product that includes hydroxypropyl methylcellulose witth or without colorant
(e.g.,
22
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Opadry , commercially available from Colorcon, Inc.) may be added to the
solution and the
solution mixed (e.g., for about 1 hour) prior to application onto the beads.
The resultant
coated substrate, beads in this example, may then be optionally overcoated
with a barrier
agent to separate the therapeutically active agent from the hydrophobic
controlled release
coating. An example of a suitable barrier agent is one that comprises
hydroxypropylmethylcellulose. However, any film-foriraer known in the art may
be used. It is
preferred that the barrier agent does not affect the dissolution rate of the
final product.
Press Coated, Pulsatile Dosa eg Form. In another embodiment of the present
invention,
baclofen is administered via a press coated, pulsatile drug delivery system
suitable for oral
administration with a controlled release component, which contains a
compressed blend of an
active agent and one or more polymers, substantially enveloped by an immediate
release
component, which contains a compressed blend of the a_ctive agent and
hydrophilic and
hydrophobic polymers. The immediate release component preferably comprises a
compressed
blend of active agent and one or more polymers with disintegration
characteristics such that
the polymers disintegrate rapidly upon exposure to the a.queous medium.
The controlled release component preferably cornprises a combination of
hydrophilic
and hydrophobic polymers. In this embodiment, once administered, the
hydrophilic polymer
dissolves away to weaken the structure of the controlledt release component,
and the
hydrophobic polymer retards the water penetration and helps to maintain the
shape of the
drug delivery system.
In accordance with the present invention, the terrn "polymer" includes single
or
multiple polymeric substances, which can swell, gel, degrade or erode on
contact with an
aqueous environment (e.g., water). Examples include alginic acid,
carboxymethylcellulose
calcium, carboxymethylcellulose sodium, colloidal silicon dioxide,
croscarmellose sodium,
crospovidone, guar gum, magnesium aluminum silicate, methylcellulose,
microcrystalline
cellulose, polacrilin potassium, powdered cellulose, pregelatinized starch,
sodium alginate,
sodium starch glycolate, starch, ethylcellulose, gelatin, hydroxyethyl
cellulose,
hydroxypropyl cellulose, hydroxypropyl methylcellulose, polymethacrylates,
povidone,
pregelatinized starch, shellac, zein, and combinations thcreof.
The term "hydrophilic polymers" as used herein includes one or more of
carboxymethylcellulose, natural gums such as guar gurn- or gum acacia, gum
tragacanth, or
gum xanthan, hydroxyethyl cellulose, hydroxypropyl cellulose, hydroxypropyl
methylcellulose, methylcellulose, and povidone, of which hydroxypropyl
methylcellulose is
23
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
further preferred. The term "hydrophilic polymers" can also include sodium
carboxymethycellulose, hydroxymethyl cellulose, polyethelene oxide,
fiydroxyethyl methyl
cellulose, carboxypolymethylene, polyethelene glycol, alginic acid, gelatin,
polyvinyl
alcohol, polyvinylpyrrolidones, polyacrylamides, polymethacrylamides,
polyphosphazines,
polyoxazolidines, poly(hydroxyalkylcarboxylic acids), an alkali metal or
alkaline earth metal,
carageenate alginates, ammonium alginate, sodium alganate, or mixtures
thereof.
The "hydrophobic polymer" of the drug delivery system can be any hydrophobic
polymer which will achieve the goals of the present invention including, but
not limited to,
one or more polymers selected from carbomer, carnauba wax, ethylcellulose,
glyceryl
palmitostearate, hydrogenated castor oil, hydrogenated vegetable oil type 1,
microcrystalline
wax, polacrilin potassium, polyethylene oxide, polymethacrylates, or stearic
acid, of which
hydrogenated vegetable oil type 1 is preferred. Hydrophobic polymers can
include, for
example, a pharmaceutically acceptable acrylic polymer, including, but not
limited to, acrylic
acid and methacrylic acid copolymers, methyl methacrylate copolymers,
ethoxyethyl
methacrylates, cyanoethyl methacrylate, aminoalkyl methacrylate copolymer,
poly(acrylic
acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer,
Toly(methyl
methacrylate), poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl
methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl
methacrylate
copolymers. Additionally, the acrylic polymers may be cationic, anionic, or
non-ionic
polymers and may be acrylates, methacrylates, formed of methacrylic acid or
methacrylic
acid esters. The polymers may also be pH dependent.
Enteric Coated Controlled Release. In one embodiment, the delayed or delayed-
sustained release coating is an enteric coating. All commercially available pH-
sensitive
polymers may be used to form the enteric coating. The drug coated witlh the
enteric coating is
minimally or not released in the acidic stomach environment of approximately
below pH 4.5.
The drug should become available when the enteric layer dissolves at the
higher pH present
in the intestine; after a suitable delayed time; or after the unit passes
through the stomach.
The preferred duration of drug release time is in the range of up to 7 h urs
after dosing under
fasting conditions.
Enteric polymers include cellulose acetate phthalate, cellulose a.cetate
trimellitate,
hydroxypropyl methylcellulose phthalate, polyvinyl acetate phthalate,
carboxymethylethylcellulose, co-polymerized methacrylic acid/methacaylic acid
methyl
esters such as, for instance, materials known under the trade name Eudragit
L12.5,
24
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Eudragit0 L 100, or Eudragit0 S 12.5, S 100 (Rohm GmbH, Darmstadt, Germany) or
similar
compounds used to obtain enteric coatings. Aqueous colloidal polymer
dispersions or re-
dispersions can be also applied, e.g., Eudragit0 L 30D-55, Eudragit0 L100-55,
Euciragit0
S100, Eudragit0 preparation 4110D c; Aquateric0, Aquacoat0 CPD 30 (FMC Corp.);
Kollicoat MAEO 30D and Kollicoat MAEO 30DP (BASF); Eastacryl0 30D (Eastrrnan
Chemical, Kingsport, TN).
The enteric polymers used in this invention can be modified by mixing with
other
known coating products that are not pH sensitive. Examples of such coating
products include
the neutral methacrylic acid esters with a small portion of
trimethylammonioethyl
methacrylate chloride, sold currently under the trade names E Eudragit0,
Eudragit RL,
Eudragit0 RS; a neutral ester dispersion without any functional groups, sold
under the trade
names Eudragit NE30D and Eudragit0 NE30; and other pH independent coating
products.
The enteric coating will substantially envelop the controlled-release
component. The
term "substantially envelop" is intended to define the total or near-total
enclosure of a
component. Such an enclosure includes, preferably, at least about 80%
enclosure, n-kore
preferably at least about 90% enclosure, and even more preferably at least
about 99 10
enclosure.
An embodiment of the present invention provides for a free flowing
formula_tion
comprising baclofen. The term "free flowing" as used herein, means dosage
forms tliat pass
through a patient's digestive system without impediment or mechanism to slow
passage.
Thus, for example, the term "free flowing" would exclude gastric raft type
dosage forms,
which are designed to reside in the stomach for extended periods as in, e.g.,
U.S. Patent No.
5,651,985.
Dosage forms according to the present invention can also include a combination
of
baclofen and at least one additional active agent, such as tizanidine,
dantrolene, nonsteroidal
anti-inflammatory agents (NSAIDs), opioids, and COX-2 inhibitors. The other
active agents
can be co-formulated in the immediate-release or delayed-release, delayed-
sustained release,
or sustained-release components to provide desirable therapeutic effects.
Dosage forms according to the present invention can also apply to pure
racernic, L-
baclofen, and other GABA related active agents as referred to in U.S. Patent
6,350,769,
issued February 26, 2002 to Kaufman et al.
Dosage levels of baclofen (racemic or L-baclofen), as well as any active agent
that is
to be used in combination with baclofen, in the compositions may be varied so
as to obtain an
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
amount of baclofen, and, when used as a combination product, an amount of
active ingredient
that is effective to obtain a desired therapeutic response for a particular
composition and
method of administration.
An object of the present invention provides for controlled bioavailability of
baclofen
as desired by health providers. Bioavailability refers to the degree to which
the
therapeutically active medicainent becomes available in the body after
administration.
Typically, bioavailability is measured in patients who fasted overnight before
being dosed
with the test preparation. Plasma samples are then taken and analyzed for the
plasma
concentration of the parent compound and/or its active metabolite. These data
may be
expressed as CmAx, the maximum amount of active ingredient found in the
plasma, or as
AUC, the area under the plasma concentration time curve. Shargel & Yu, APPLIED
BIOPHARMACEUTICS AND PHARMACOKINETICS ch. 10 (3d ed. 1996); see also APPLIED
PHARMACOKINETICS: PRINCIPLES OF THERAPEUTIC DRUG MONITORING, Evans et al.,
eds. (3 d
ed. 1992).
For example, baclofen formulations may be used in a comparative
bioavailability
study in subjects. Subjects fast over night prior to drug administration.
Plasma samples are
then taken at dosing, and every hour for twelve hours after dosing, and then
at sixteen and
twenty-four hours after dosing, and analyzed for the ng/ml concentration of
baclofen or a
baclofen metabolite.
Without further elaboration, one skilled in the art having the benefit of the
preceding
description can utilize the present invention to the fullest extent. The
following examples are
illustrative only and do not limit the remainder of the disclosure in any way.
EXAMPLES
Example 1. Active baclofen-coated seeds.
FORMULATION
INGREDIENT % mg
Sugar Spheres, NF (mesh 20-25) 81.4 250.0
Micronized Baclofen, USP 13.0 40.0
Povidone, USP (Plasdone K-29/32) 5.6 17.14
Purified Water, USP N/A N/A
TOTAL: 100.0 307.14
26
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Povidone (Plasdone K-29/32 ) is added to purified water and mixed until the
povidone is fully dissolved. Baclofen is mixed in the above solution until
uniformly
dispersed. A fluidized bed coating apparatus is then used to coat the sugar
spheres with the
baclofen suspension to produce active coated seeds.
Example 2. Active baclofen-coated seeds.
FORMULATION
INGREDIENT % mg
Sugar Spheres, NF (mesh 20-25) 81.4 250.0
Micronized Baclofen, USP 13.0 40.0
Hypromellose, Type 2910, USP 5.6 17.14
(Pharmacoat 606, 6cps)
Purified Water, USP N/A N/A
TOTAL: 100.0 307.14
Hypromellose, Type 2910 , USP (Pharmacoat 606, 6cps) is added to a suitable
amount of purified water and mixed until the Hypromellose is fully dissolved.
Baclofen is
mixed in the above solution until uniformly dispersed. A fluidized bed coating
apparatus is
then used to coat the sugar spheres with the baclofen suspension to produce
active coated
seeds.
The dissolution profile of this formulation is shown in Figure 3.
Example 3. Active baclofen-containing granules.
FORMULATION
INGREDIENT % mg
Baclofen, USP 7.4 20.0
Pregelatinized Starch, NF 21.3 57.5
(Starch 1500)
Microcrystalline Cellulose, NF 70.8 191.3
(Avicel PH-102)
Magnesium Stearate, NF 0.5 1.3
Purified Water, USP N/A N/A
TOTAL: 100.0 270.1
27
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Mix Baclofen, Starch 1500 (pregelatinized starch) and Avicel PH-102
(microcrystalline cellulose). Charge the baclofen mixture into a Hobart niixer
and blend to
form a uniform mixture. Granulate the mixture with purified water to form a
granulate. Dry
the granulate in an oven at a temperature of 60 C to form granules. Screen the
granules using
a #30 mesh screen. Mix magnesium stearate to form active granules.
Example 4. Enteric-coated seeds containing baclofen.
FORMULATION
INGREDIENT % mg
Active coated seeds 76.5 153.61
(containing 13.02% Baclofen)
Hypromellose, Type 2910, USP 8.5 17.07
(Pharmacoat 606, 6cps)
Hypromellose Phthalate, NF 13.5 27.11
(HPMCP; HP-50)
Acetyltributyl Citrate, NF 1.5 3.01
Acetone, NF N/A N/A
Purified Water, USP N/A N/A
TOTAL: 100.0 200.8
Charge purified water into a stainless steel container and mix in hypromellose
until
completely dissolved. Then charge purified water and acetone into another
stainless steel
container and then mix in acetyltributyl citrate to form an acetyltributyl
citrate solution. To
this, add hypromellose phthalate to form an enteric coat solution.
Film coat the baclofen active coated seeds as produced in any of examples 1-3
with
the seal coat solution to form sealed baclofen beads. Then film coat the
sealed baclofen beads
with the enteric coat solution to produce enteric-coated seeds.
Example 5. Enteric-coated seeds containing baclofen.
FORMULATION
INGREDIENT A B
% mg % mg
Active coated seeds 90.0 149.4 90.0 149.4
(containing 13.42% Baclofen)
Methacrylic Acid Co ol mer Type 8.0 13.28 - -
28
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
A, NF (Eudragit L 100)
Methacrylic Acid Copolymer Type
C, NF (Eudragit L 100-55) - - 8A 13.28
Talc, USP 1.0 1.66 1.0 1.66
Triethyl Citrate, NF 1.0 1.66 1.0 1.66
Iso ro yl Alcohol, USP N/A N/A N/A N/A
Purified Water, USP N/A N/A N/A N/A
TOTAL: 100.0 166.00 100.0 166.0
Charge isopropyl alcohol and purified water into a stainless steel container
and then
mix in triethyl citrate. Add in methacrylic acid copolymer Type A, NF
(Eudragit L 100) or
methacrylic acid copolymer Type C, NF (Eudragit L 100-55) to form a Eudragit
suspension. Disperse talc into the Eudragit suspension. Film coat the
baclofen active coated
seeds from example 4 with the Eudragit suspension to form enteric-coated
seeds.
Example 6. Composition containing baclofen active coated and enteric-coated
seeds.
FORMULATION
Ingredient Immediate release Delayed release TOTAL
component component
Baclofen 10 mg 20 mg 30 mg
Pharmacoat 606 2 m 4 mg 6 mg
Talc 0.4 m 12.1 mg 12.5 mg
Sugar Spheres 62.5 mg 125 mg 187.5 mg
Eudragit L100-55 0 22.32 mg 22.32 mg
Triethyl Citrate 0 3.72 mg 3.72 mg
Water N/A N/A N/A
Iso ro 1 Alcohol N/A N/A N/A
Acetone N/A N/A N/A
TOTAL: 74.9 187.14 262.04
Designated portions of active coated seeds and eiiteric-coated seeds are mixed
together to form dosage forms. In the case of capsules, the seeds are mixed
and added to
gelatin capsules. In the case of tablets, the seeds are compressed to form a
tablet. In the case
of sachets, the seed are mixed and filled into the pouch.
29
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Example 7. Enteric-coated seeds containing baclofen.
FORMULATION
INGREDIENT Weight %
Baclofen 10.56
Sugar Spheres 65.97
Pharmacoat 606 4.52
Eudra it RL 100 0.60
Eudra it RS 100 1.39
Dibutyl Sebacate 0.20
Talc 1.39
Magnesium Stearate 0.40
HPMCP HP-50 13.50
Triethyl Citrate 1.50
TOTAL: 100.00
Pharmacoat 606 is dissolved in purified water and baclofen is then dispersed
into this
aqueous solution to make an aqueous suspension. A fluidized bed coating
equipment is used
to coat the sugar sphere with the baclofen suspension to produce active coated
seeds.
Eudragit RL 100, RS 100, and dibutyl sebacate are dissolved in a mixture of
acetone
and isopropyl alcohol. Talc and magnesium stearate are then dispersed into the
solution. A
fluidized bed coating equipment is used to coat the active coated seeds with
the above
suspension to produce sustained-release coated seeds.
HPMCP and triethyl citrate are dissolved in a mixture of acetone and purified
water.
A fluidized bed coating equipment is used to coat the sustained-release coated
seeds with the
above solution to produce enteric-coated seeds.
Example 8. Baclofen tablets.
FORMULATION
INGREDIENT Weight (mg)
Baclofen 20
Sodium Starch Glycolate 20
Dicalcium Phosphate Anhydrous 26.5
Lactose Anhydrous 132.5
Mg stearate 1
TOTAL: 200
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Mix baclofen, sodium starch glycolate, dicalcium phosphate anhydrous, and
lactose
anhydrous in a high-shear granulator. Wet granulate the mixture with purified
water and dry
the granulate in an oven at a temperature of 60 C for at least 16 hours.
Screen the granules
using a #25 mesh screen. Mill the oversized granules by a Fitzpatric
comminuting machine
equipped with a #18 mesh screen. Blend the screened and milled granules with
magnesium
stearate and compress the blend into tablets using a rotary tablet press.
Example 9. Baclofen tablets.
FORMULATION
INGREDIENT Weight (mg)
Baclofen 20
Hydroxypropyl Methylcellulose, type
2910, USP (Methocel K100LV) 60
Lactose Monohydrate or Mannitol 39.60
Microcrystalline Cellulose, NF (Avicel
PH 101 79.40
Ma nesium Stearate 1.00
TOTAL: 200
Mix baclofen, hydroxypropyl methylcellulose, lactose monohydrate or mannitol,
and
microcrystalline cellulose in a high-shear granulator. Wet granulate the
mixture with purified
water and dry the granulate in an oven at a temperature of 60 C for at least
16 hours. Screen
the granules using a #25 mesh screen. Mill the oversized granules by a
Fitzpatric
comminuting machine equipped with a #18 mesh screen. Blend the screened and
niilled
granules with magnesium stearate and compress the blend into tablets using a
rotary tablet
press.
Example 10. Composition containing baclofen active coated and enteric-coated
seeds.
Formulation
Ingredient IR Per Capsule EC Per Capsule Total Per Capsule
% Amount % Amount % Amount
(w/w) (mg) (w/w) (mg) (w/w) (m )
Micronized Baclofen 13.36 19.00 21.87 21.00 16.79 40.00
Sugar Spheres, NF (Mesh 83.48 118.73 34.11 32.75 63.58 151.48
20-25)
Hypromellose, Type 2910, 2.67 3.80 4.37 4.20 3.36 8.00
USP (Pharmacoat 606, 6
31
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
c s )
Talc, USP (ALTALC 0.49 0.70 9.60 9.22 4.16 9.92
500V USP BC (*1814))
Methacrylic Acid -- -- 15.53 14.91 6.26 14.91
Copolymer, Type C, NF
(Eudragit L100-55)
Methacrylic Acid -- -- 10.61 10.19 4.28 10.19
Copolymer, Type A, NF
(Eudragit L100-55)
Triethyl Citrate NF -- -- 3.91 3.75 1.57 3.75
Total 100.0 142.23 100.00 96.02 100.00 238.25
Hypromellose, Type 2910, USP is added to a suitable amount of purified water
and
mixed until the hypromellose is fully dissolved. Baclofen is then mixed in the
above solution
until uniformly dispersed. The suspension is passed through a #40 mesh sieve
into a stainless
steel container. Sugar spheres are charged into a fluid-bed coater equipped
with a Wurster
insert and heated until exhaust air temperature reaches 50 5 C. The active
suspension from
above is sprayed to coat the sugar spheres, which are then dried at a
temperature of 60 10 C
for 5 minutes. The IR seeds are passed through a #16 mesh stainless steel
screen. Acceptable
IR seeds are collected and niixed with talc, USP in a slant cone blender for
one minute.
An enteric solution is prepared by mixing purified water and acetone. Triethyl
citrate
and methacrylic acid copolymer, type C, are stirred into the mixture until
completely
dissolved. Talc is mixed in the above solution until completely dispersed. A
fluidized bed
coating apparatus is then used to coat IR seeds prepared as above with the
enteric solution to
produce enteric-coated seeds. The enteric-coated seeds are passed through a
#14 mesh
stainless steel screen. Acceptable enteric-coated seeds are collected and
mixed with talc, USP
in a slant cone blender for one minute.
An appropriate amount of IR seeds plus the appropriate amount of enteric-
coated seeds
are encapsulated to yield Baclofen ER capsules.
Example 11. Baclofen Tablets
Tablets having the following compositions were prepared according to the
process
described in Example 9.
Ingredients 579-7C 579-10A PX01802 PX02002
Baclofen, USP 20 20 20 20
Hydroxypropyl
Meth lcellulose,ty e 0 60 0 0
32
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
2910,USP(Pharmacoat
606)
Hydroxypropyl
Methylcellulose,type
2280,USP(Methocel
K100LV) 20 0 0 0
Hydroxypropyl
Methylcellulose,type
2280,USP(Methocel
K100M) 0 0 60 120
Microcrystalline
Cellulose, NF (Avicel
PH101) 59 59 59 59
Mg stearate, NF 1 1 1 1
Purified Water, USP
tablet wt. 100 140 140 200
Dissolution profiles of the above formulations are shown in Figure 1B (in SIF)
and in
Figure 2 (SGF/SIF switchover method).
Example 12. Baclofen ER Capsules.
Baclofen extended release capsules (20 mg) were prepared having the following
formulations, using the process described in Example 7.
Ingredients PX01903 PX02103 PX02503 PB01403 PB00903(A)
Micronized Baclofen,
USP 20.0 20.0 20.0 20 20
Sugar Spheres 125.0 125.0 125.0 125.0 125.0
Hydroxypropyl
Methylcellulose,type
2910,USP(Pharmacoat
606) 8.6 8.6 8.6 4.0 4.0
Hydromellose
Phthalate, NF
(HPMCP; HP-5O) 24.4 25.6 25.6 0.0 0.0
Ammonio
Methacrylate
Copolymer Type A, NF
(Eudragit RL100) 0.0 1.5 1.1 0.0 0.0
Ammonio
Methacrylate
Copolymer Type B, NF
(Eudra it RS 100) 0.0 2.3 2.6 0.0 0.0
33
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Methacrylic Acid
Copolymer, Type A,
NF (Eudragit L100) 0.0 0.0 0.0 13.2 13.2
Methacrylic Acid
Copolymer, Type C,
NF (Eudragit L100-55) 0.0 0.0 0.0 8.1 0.0
Triethyl Citrate, NF 2.7 2.8 2.8 3.0 3.0
Dibut l Sebacate, NF 0.0 0.4 0.4 0.0 0.0
Talc, USP 0.0 2.6 2.6 5.7 5.7
Magnesium Stearate,
NF 0.0 0.8 0.8 0.0 0.0
Isopropyl Alcohol,
USP 0.0
Acetone, NF
Purified Water, USP
Dissolution profiles of the above formulations are shown in Figure 4.
Example 13. Baclofen ER Capsules.
Baclofen ER capsules having the following formulations were prepared according
to
the process described in Example 10.
Composition of Baclofen ER (ER1A) Capsules 30 mg (Lot PB01903)
IR/ER (EC1) = 2:1
Ingredient IR Per Ca sule ECI Per Capsule Total Per Capsule
% (w/w) Amount % (w/w) Amount % (w/w) Amount
(mg) (mg) (mg)
Micronized Baclofen 8.22 20.0 4.11 10.0 12.33 30.0
Sugar Spheres, NF (Mesh 20-25) 51.36 125.0 25.68 62.5 77.04 187.5
Hypromellose, Type 2910, USP 1.64 4.0 0.82 2.0 2.47 6.0
(Pharmacoat 606, 6 cps)
Talc, USP (ALTALC 500V USP BC 0.33 0.8 2.49 6.05 2.81 6.85
(*1814))
Methacrylic Acid Copolymer, Type C, -- -- 4.59 11.16 4.59 11.16
NF (Eudragit L100-55)
Trieth I Citrate NF -- -- 0.76 1.86 0.76 1.86
Total 61.55 149.8 38.45 93.57 100.00 243.37
Composition of Baclofen ER (ER1B) Capsules 30 mg (Lot PB01803)
IR/ER (EC1) = 1:2
Ingredient IR Per Ca sule EC1 Per Capsule Total Per Capsule
% (w/w) Amount % (w/w) Amount % (w/w) Amount
(m ) (mg) (mg)
Micronized Baclofen 3.82 10.0 7.63 20.0 11.45 30.0
Sugar Spheres, NF (Mesh 20-25) 23.85 62.5 47.70 125.0 71.55 187.5
Hypromellose, Type 2910, USP 0.76 2.0 1.53 4.0 2.29 6.0
(Pharmacoat 606, 6 cps)
Talc, USP (ALTALC 500V USP BC 0.15 0.4 4.62 12.1 4.77 12.5
34
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
(*1814))
Methacrylic Acid Copolymer, Type -- -- 8.52 22.32 8.52 22.32
C, NF (Eudragit L100-55)
Triethyl Citrate NF -- -- 1.42 3.72 1.42 3.72
Total 28.58 74.9 71.42 187.14 100.00 262.04
Composition of Baclofen ER (ER2A) Capsules 30 mg (Lot PB02003)
Il2/ER (EC2) = 2:1
Ingredient IR Per Ca sule EC2 Per Capsule Total Per Capsule
% (w/w) Amount % (w/w) Amount % (w/w) Amount
(mg) (mg) (mg)
Micronized Baclofen 8.34 20.0 4.17 10.0 12.51 30.0
Sugar Spheres, NF (Mesh 20-25) 52.14 125.0 26.07 62.5 78.21 187.5
Hypromellose, Type 2910, USP 1.67 4.0 0.83 2.0 2.50 6.0
(Pharmacoat 606, 6 cps)
Talc, USP (ALTALC 500V USP BC 0.33 0.8 1.38 3.29 1.71 4.09
(*1814)) .
Methacrylic Acid Copolymer, Type -- -- 1.68 4.03 1.68 4.03
C, NF (Eudragit L100-55)
Methacrylic Acid Copolymer, Type -- -- 2.76 6.62 2.76 6.62
A, NF Eudra it L100
Triethyl Citrate NF -- -- 0.63 1.5 0.63 1.5
Total 62.48 149.8 37.52 89.94 100.00 239.74
Composition of Baclofen ER (ER2B) Capsules 30 mg (Lot PB02103)
Il2/ER (EC2) = 1:2
Ingredient IR Per Ca sule EC2 Per Capsule Total Per Capsule
% (wlw) Amount % (w/w) Amount % (w/w) Amount
(mg) (mg) (mg)
Micronized Baclofen 3.92 10 7.85 20.0 11.78 30.0
Sugar Spheres, NF (Mesh 20-25) 24.53 62.5 49.06 125.0 73.59 187.5
Hypromellose, Type 2910, USP 0.78 2.0 1.57 4.0 2.35 6.0
(Pharmacoat 606, 6 cps)
Talc, USP (ALTALC 500V USP BC 0.16 0.4 2.58 6.58 2.74 6.98
(* 1814))
Methacrylic Acid Copolymer, Type -- -- 3.16 8.06 3.16 8.06
C, NF (Eudragit L100-55)
Methacrylic Acid Copolymer, Type -- -- 5.20 13.24 5.20 13.24
A, NF (Eudragit L100
Triethyl Citrate NF -- -- 1.18 3.0 1.18 3.0
Total 29.39 74.9 70.60 179.88 100.00 254.78
The dissolution profile of the above formulations is shown in Figure 5.
Example 14. Baclofen ER Capsules.
Baclofen ER capsules of the following compositions were prepared according to
the
process described in Example 10, with the exception that the enteric materials
are
Acetyltributyl Citrate and Hydromellose Phthalate instead of Triethyl Citrate
and Methacrylic
Acid Copolymer, type C.
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
PX03503-30 Formulation
IR Per Capsule EC Per Capsule Total Per Capsule
Ingredient % Amount % Amount % Amount
(w/w) (mg) (w/w) (mg) (w/w) (mg)
Micronized 12.37 20.00 9.17 10.00 11.06 30.0
Balcofen
Sugar Spheres, NF 77.33 125.03 57.31 62.50 69.12 187.53
(Mesh 20-25)
Hypromellose, 10.30 16.65 7.63 8.32 9.20 24.97
Type 2910, USP
(Pharmacoat 606,
6cps)
Hypromellose - - 11.77 12.83 4.73 12.83
Phthalate, NF
(HPMCP; HP-55)
Acetyltributyl - - 0.47 0.51 0.19 0.51
Citrate, NF
Talc, USP - - 13.65 14.89 5.70 15.46
Total 100.0 161.68 100.0 109.05 100.0 271.30
PX03403-30 Formulation
IR Per Capsule EC Per Capsule Total Per Capsule
Ingredient % Amount % Amount % Amount
(w/w) (mg) (w/w) (mg) (w/w) (mg)
Micronized 12.37 6.00 9.17 24.00 9.65 30.0
Balcofen
Sugar Spheres, NF 77.32 37.50 57.31 149.98 60.30 187.48
(Mesh 20-25)
Hypromellose, 10.31 5.00 7.63 19.98 8.03 24.98
Type 2910, USP
(Pharmacoat 606,
6cps)
Hypromellose - - 11.77 30.80 9.91 30.80
Phthalate, NF
(HPMCP; HP-55)
Acetyltributyl - - 0.47 1.23 0.40 1.23
Citrate, NF
Talc, USP - - 13.65 35.73 11.71 36.41
Total 100.0 48.50 100.0 261.72 100.0 310.9
Dissolution profiles of the above formulations are shown in Figure 5.
36
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Example 15. Baclofen ER Capsules.
Baclofen ER capsules having the following composition were prepared according
to
the method described in Example 10. Capsules were prepared having 10 mg, 15
mg, 20 mg,
25 mg, 30 mg, 35 mg and 40 mg baclofen, with the different dosage strengths
being directly
proportional.
Composition of Baclofen ER Capsules 40 mg (Lot RB04042-60A)
IR/EC = 19:21
Ingredient IR Per Ca sule EC Per Ca sule Total Per Capsule
% (w/w) Amount % (w/w) Amount % (w/w) Amount
(mg) (mg) (mg)
Micronized Baclofen 13.36 19.00 21.87 21.00 16.79 40.00
Sugar Spheres, NF (Mesh 20-25) 83.48 118.73 34.11 32.75 63.58 151.48
Hypromellose, Type 2910, USP 2.67 3.80 4.37 4.20 3.36 8.00
(Pharmacoat 606, 6 cps)
Talc, USP (ALTALC 500V USP BC 0.49 0.70 9.60 9.22 4.16 9.92
(*1814))
Methacrylic Acid Copolymer, Type -- -- 15.53 14.91 6.26 14.91
C, NF (Eudragit L100-55)
Methacrylic Acid Copolymer, Type -- -- 10.61 10.19 4.28 10.19
A, NF (Eudragit L100-55)
Triethyl Citrate NF -- -- 3.91 3.75 1.57 3.75
Total 100.0 142.23 100.00 96.02 100.00 238.25
Example 16. Determining plasma profiles for baclofen-containing formulations.
A bioavailability study was done in 20 healthy volunteers comparing a 36mg
baclofen
formulation prepared according to Example 15, with the exception that the
immediate-release
component contained 12mg baclofen and the enteric-coated controlled release
component
contained 24mg baclofen, and the remaining excipients were adjusted dose
proportionally.
The formulation was compared with a 20mg immediate release reference tablet
(Watson
Laboratories, Inc.) under fasting conditions. Test samples were administered
orally with 240
ml of room temperature water after subjects are fasted overnight for at least
10 hours. No
fluid, except that given with drug administration, is allowed from 1 hour
prior to dose
administration until 1 hour after dosing. At 2, 6, 8 and 12 hours post-dose,
subjects consumed
240 ml of room temperature water. In addition, subjects consumed 480 ml of
fluid with
lunch and dinner. Blood samples were drawn at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4,
4.5, 5, 6, 7, 8, 10,
12, 16, and 24 hours after administration. The results are shown in Figure 6.
In addition,
Figure 6 shows simulated blood plasma levels for 30 mg immediate-release
baclofen, based
on the data obtained from administration of the 20 mg dosage strength.
37
CA 02560995 2006-09-20
WO 2005/097079 PCT/US2005/011032
Example 17. Determining steady state plasma profiles for baclofen-containing
formulations.
Based on single-dose bioavailability data, steady-state mean baclofen plasma
levels
were calculated for a 40 mg baclofen formulation prepared according to Example
15
administered every 12 hours and an immediate-release 20 mg baclofen
formulation (Watson
Laboratories, Inc.) administered every 8 hours. The results are shown in
Figure 7 (where (C)
represents the 40 mg dosage form of the present invention and (D) represents
the reference 20
mg immediate-release dosage form). The results show that, at steady-state, the
40 mg dosage
form of the present invention will reach a CmiN at 12 hours after
administration comparable to
the CmIN obtained by the immediate-release formulation eight hours after
administration.
Having now fully described this invention, it will be understood to those of
ordinary
skill in the art that the methods of the present invention can be carried out
with a wide and
equivalent range of conditions, formulations, and other parameters without
departing from
the scope of the invention or any embodiments thereof.
38