Note: Descriptions are shown in the official language in which they were submitted.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
1
MODULATEURS DES RECEPTEURS LXR
La présente invention a pour objet de nouveaux composés capables de
moduler l'activité des récepteurs nucléaires LXR, leur procédé de fabrication
ainsi
que des compositions pharmaceutiques les contenant.
Les récepteurs « liver X receptor » (LXR) sont des facteurs de
transcription qui appartiennent à la super famille des récepteurs nucléaires
dont
font également partie les récepteurs e< retinoic acid receptor >a (RXR), «
farnesoid
X receptor » (FXR) et « peroxisome proliferator-activated receptors >a
(PPARs).
Les récepteurs LXR forment en se liant au récepteur RXR un hétérodimère qui se
fixe de façon spécifique aux éléments de réponse de l'ADN (LXRE) pour conduire
à la transactivation de gènes cibles (Genes dev 1995 9 : 1033 - 4S).
Ces récepteurs sont impliqués dans de multiples voies métaboliques et
participent notamment à l'homéostase du cholestérol, des acides biliaires, des
triglycérides et du glucose.
La modulation de l'activité de ces récepteurs nucléaires influence la
progression des désordres métaboliques tels que le diabète de type II, les
dyslipidémies et le développement de l'athérosclérose.
L'hétérodimère LXR/RXR peut être activé par des ligands LXR et/ou RXR. La
transactivation des gènes cibles nécessite le recrutement de co-activateurs
tel
que Grip-1. (Nature 1996 383:728:31).
Les deux types de récepteurs LXR aujourd'hui identifiés, à savoir LXRa et
LXR~3, présentent un haut degré de similarité au niveau de leur séquence en
acides aminés mais difFèrent quant à leur distribution tissulaire. Le LXRa est
fortement exprimé dans le foie et dans une moindre mesure dans les reins,
l'intestin, le tissu adipeux et la rate. Le LXR(3 est distribué de façon
ubiquitaire
(Gene 2000; 243 : 93 -103, N. Y. Acad. Sci. 1995; 761 : 38 - 49).
Si le cholestérol n'active pas directement les récepteurs LXR, des dérivés
mono-oxydés du cholestérol (oxystérols) le font et plus particulièrement les
22
(R)-hydroxycholestérol, 24(S)-hydroxycholestérol et 24(S),25-époxycholestérol.
Ces oxystérols sont considérés comme les ligands physiologiques des récepteurs
LXR (Nature 1996; 383 : 728 - 31, J.Biolchem 1997; 272 : 3137 - 40). En
outre, il a été montré que l'oxystérol 5,6,24(S),25-diépoxycholestérol est un
ligand spécifique du LXRa, ce qui suggëre qu'il est possible de développer des
ligands spécifiques du LXRa et/ou LXR~i (Proc. Natl. Acad Sci USA 1999; 96:
26 - 71, Endocrino%gy 2000; 141: 4180 - 4).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
2
Par ailleurs, on a pu démontrer que le plasma humain contient des
antagonistes naturels des LXRa et [3 (Steroicls 2001; 66 ; 473 - 479).
A partir d'hépatocytes de rat, il a pu être montré que des acides gras non
saturés augmentent de façon importante l'expression du LXRa sans affecter
LXR~i
(Mol Endoerinol 2000 ; 14 :161-171). De plus, les activateurs des PPARs a et y
induisent également l'expression du LXRa dans des macrophages primaires
humains.
Les fortes concentrations de LXRa dans le foie et l'identification des
ligands endogènes du LXR ont suggéré que ces récepteurs jouent un rôle
essentiel dans le métabolisme du cholestérol. Dans des conditions
physiologiques, l'homéostase du cholestérol est maintenue, via la régulation
des
voies de synthèse de novo et de catabolisme. L'accumulation de stérols dans le
foie conduit par un mécanisme de feed-back impliquant des facteurs de
transcription tels que SREBP-1 et SREBP-2 à une inhibition de la biosynthèse
de
cholestérol. (Cell 1997; 39 ; 331-40). L' excès de cholestérol active
également
une autre voie métabolique qui conduit à la conversion du cholestérol en
acides
biliaires. La conversion du cholestérol en 7a-hydroxy-cholestérol est réalisée
par
une enzyme localisée dans le foie (CYP7A : 7a-hydroxylase) (J.Biol, Chem.
1997;
272 : 3137 - 40).
L'implication du LXR dans la synthèse d'acides biliaires et donc dans la
régulation de l'homéostase du cholestérol a été démontrée au moyen de souris
déficientes en LXRa qui, lorsqu'elles sont soumises à un régime gras,
accumulent
de grandes quantités d'esters de cholestérol au niveau hépatique (Cell 1993;
93
693-704). Les souris déficientes en LXR~i ont la même résistance physiologique
que les souris normales vis-à-vis d'un régime enrichi en graisses.
L'expression du
LXR(3 inchangé chez les souris déficientes en LXR tend à démontrer que le
LXR~i
est incapable à lui seul d'augmenter de façon importante le métabolisme du
cholestérol (J, Clin. Invest. 2001; 107; 565-573).
Les récepteurs LXR exprimés au niveau du macrophage jouent également
un rôle important dans la régulation de certaines fonctions de celui-ci. Ils
sont
plus particulièrement impliqués dans le contrôle du transport inverse du
cholestérol qui permet d'exporter l'excès de cholestérol des tissus
périphériques
vers le foie . Le cholestérol est pris en charge par des pré-bHDL via l'apoA1
et
ABCAi pour être transporté vers le foie où il est catabolisé en acides
biliaires puis
éliminé.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
3
L'ABCA1 est un membre de la super famille des protéines de transport
(ATP - binding cassette) dont l'importance est illustrée par le fait qu'une
mutation au niveau du gène de l'ABCA1 est responsable de la maladie de Tangier
(Nat. Genet. 1999; 22: 336-45),
L'expression de l'ABCA1 et l'efflux de cholestérol sont induits par le
chargement des macrophages humains en cholestérol et l'activation des
récepteurs LXR (Biochem. Biophys Res Comm. 1999 257;29-33). II a été
également démontré ultérieurement que l'expression au niveau intestinal de
l'ABCG1, l'ABCGS et ABCGB, autres membres de la famille des transporteurs de
type ABC, est régulée également par l'hétérodimère RXR/LXR (J. Biol, Chem.
2000; 275 : 14700-14707, Proc, Natl. Acad. Sci. USA 2000; 97 : 817 22, J.
Bi~l.
Chem. 2002; 277.' 18793-18800, Proe. Natl, Acad Sci. USA 2002; 99: 16237
16242).
II a aussi été montré que des ligands agonistes LXR réduisaient les lésions
athéromateuses dans deux différents modèles murins (souris ApoE-/- et souris
LDLR-/-) (Proc, Natl. Acad. Sci. USA 2002 ; 99 :7604-7609, FEBS Letters 2003 ;
536 :6-11). Ces résultats suggèrent que les ligands LXR peuvent constituer des
agents thérapeutiques pour traiter l'athérosclérose.
Enfin, on sait que les macrophages jouent un rôle important dans
l'inflammation en particulier dans la pathogénèse de l'athérosclérose. II a
été
montré que l'activation des LXRs inhibe l'expression des gènes impliqués dans
l'inflammation au niveau du macrophage. (Nature Medecine, 2003; 9 : 213-219).
In vitro, l'expression des médiateurs, tels que la nitric oxide synthase, la
cyclo
oxygénase -2(COX-2) et l'interleukine-6 (IL-6) est inhibée. In vivo, les
agonistes
LXR réduisent l'inflammation dans un modèle de dermatite et inhibent
l'expression des gènes impliqués dans l'inflammation des aortes de souris
athéromateuses.
Parce que l'homéostase du cholestérol semble jouer également un rôle
essentiel dans le fonctionnement du système nerveux central et les mécanismes
de la neurodégénérescence, l'expression de l'ABCA1 a également été étudiée
dans des cultures de neurones primaires, d'astrocytes et de microglia isolés à
partir de cerveaux de rats embryonnaires. Les résultats de ces études montrent
que l'activation des LXRs conduit à une diminution de la sécrétion d'amyldide
~i et
par voie de conséquence à une réduction des dépôts d'amylôide au niveau
cérébral. Ces travaux suggèrent que l'activation de LXRs peut constituer une
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
4
nouvelle approche pour traiter la maladie d'Alzheimer (J. Biol. Chem. 2003,
275
(15) :13244-13256, J. Biol. Chem.2003, 278 (30): 27688 2769.
Les récepteurs LXR sont également impliqués dans la régulation de
l'expression de l'apolipoprotéine E (ApoE). Cette protéine est largement
impliquée dans la clairance hépatique des lipoprotéines et favorise l'efFlux
de
cholestérol à partir de macrophages riches en lipides. II a été démontré que
l'activation des récepteurs LXR conduit à une augmentation de l'expression de
l'ApoE via un élément de réponse du LXR (LXRE) situé dans la séquence du
promoteur de l'ApoE (Pros. Natl. Acad Sci, USA 2001 ; 98 : 507-512).
L'activation de récepteurs LXR favoriserait également le transport inverse
du cholestérol via la modulation de l'expression de la protéine CETP
(cholestérol
ester transfer protein) qui est impliquée dans le transfert du cholestérol
estérifié
des lipoprotéines HDL aux lipoprotéines riches en triglycérides éliminées par
le
foie (J. Clin. Invest. 2000 ; 105 : 513-520).
En résumé, l'activation des récepteurs LXR conduit à une augmentation
de l'expression de nombreux gènes favorisant l'élimination du cholestérol en
excès des tissus périphériques. Dans le macrophage chargé en cholestérol,
l'activation des récepteurs LXRs augmente l'expression de l'ABCAi, l'ABCG1,
l'ABCGS, l'ABCG8 et l'ApoE entraînant une augmentation de l'efflux de
cholestérol
des macrophages au foie où il est excrété sous forme d'acides biliaires.
L induction de l'expression de la CETP et de la CYP7A dans le foie conduit
respectivement à une augmentation de la clairance hépatique des esters de
cholestérol des lipoprotéines HDL et au catabolisme du cholestérol.
Par ailleurs, il a été également démontré que les récepteurs LXRs jouent
un rôle important dans le métabolisme du glucose. Le traitement de rongeurs
diabétiques avec un agoniste LXR conduit à une diminution drastique des taux
de
glucose plasmatique. En particulier chez le rat Zucker (fa/fa) insulino-
résistant,
l'activation du LXR inhibe l'expression des gènes impliqués dans la
gluconéogénèse dont plus particulièrement la phosphoénolpyruvate
carboxykinase (PEPCK) (J. Biol. Chem. 2003, 278 (2) : 1131-1136). II est aussi
décrit que le traitement de souris par un agoniste LXR conduit à une
diminution
des taux de glucose plasmatique et de la production de glucose hépatique par
inhibition des enzymes jouant un rôle clef dans la gluconéogénèse (Diabetes,
53,suppl 1, S36-S42 feb 2004)
En outre , il a été montré qu'un agoniste LXR augmente la tolérance au
glucose dans un modèle murin d'insulino-résistance et d'obésité (Proc. Natl.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Acad. Sci, USA 2003 100: 5419-5424), L'analyse de l'expression des gènes
démontre une régulation des gènes impliqués dans le métabolisme du glucose
dans le foie
o diminution de l'expression du « peroxisome proliferator-activated
5 receptor coactivator-1a » (PGC-1), de la « phosphoenol pyruvate
carboxykinase » (PEPCIC) et de la ee glucose-6-phosphatase »
o induction de l'expression de la glucokinase qui favorise l'utilisation du
glucose hépatique,
Une induction transcriptionnelle du transporteur de glucose sensible à
l'insuline (GLUT4) dans le tissu adipeux a également été démontrée.
Ces résultats soulignent l'importance des LXRs dans la coordination du
métabolisme du glucose.
On sait aussi que les récepteurs LXR interviennent dans les processus de
régulation de l'inflammation (Nature Meeiecine 2003 9, 213-219 ).
Des composés modulateurs de l'activité des récepteurs LXR sont connus
dans l'état de la technique notamment des documents WO 03/090869, WO
03/90746, WO 03/082192 ou WO 03/082802 ; ou encore des documents WO
03/043985 et WO 04/005253 qui décrivent des composés de type
benzènesulfonamide agonistes des récepteurs PPAR.
Dans ce contexte, il existe un intérêt significatif à trouver de nouveaux
composés modulateurs de l'activité des récepteurs LXR qui seraient utiles dans
le
traitement de certaines pathologies telles que les maladies cardiovasculaires,
les
hypercholestérolémies, les dyslipidémies, l'infarctus du myocarde,
l'athérosclérose, le diabète, l'obésité, l'inflammation et les maladies
neurodégénératives.
La présente invention est précisément fondée sur la découverte de
nouveaux composés modulateurs de l'activité des récepteurs LXR.
Ainsi, selon un premier aspect, la présente invention vise à protéger, en
tant que produit industriel nouveau, un composé de benzènesulfonamide,
caractérisé en ce qu'il est choisi parmi
i) les composés de formule
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
6
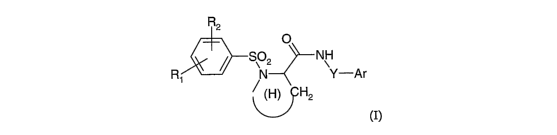
R2
O
S02 NH
Ri / N Y-är
(H) H2
(I)
dans laquelle
- (H) représente un hétérocycle saturé azoté à 5 ou 6 chaînons condensé avec
un
noyau phényle ou cyclohexyle, éventuellement substitué par un halogène , un
groupe alcoxy en Ci-C4 ou un groupe N(R)~ dans lequel R représente l'atome
d'hydrogène ou un groupe alkyle en Ci-C4,
- Ri représente
o un atome de chlore,
o un groupe alkyle en C3-C6, ramifié ou cyclisé,
o un groupe alkoxy en Cz-C6 linéaire, ramifié ou en C3-C6 cyclisé,
o un groupe phénoxy éventuellement substitué par un halogène,
o un groupe phényle, ou
o un groupe aminométhyle éventuellement substitué par un
groupe acétyle ou trifluoroacétyle,
- R2 représente un atome d'hydrogène ou un halogène, ou,
- Rl et Ra forment ensemble un hétérocycle oxygéné ou azoté, éventuellement
substitué par un ou plusieurs groupes alkyle en Cl-C3, par un groupe acyle ou
par un groupe perFluoroacyle en C~-C3,
- Y représente
o une liaison simple,
o un groupe alkylène en Cl-C4, linéaire ou ramifié ou en C3-C4
cyclisé, éventuellement substitué par un groupe alkoxy en Cl-
C3, un groupe phényle, un groupe N(R)2 ou un groupe COOH,
o un groupe -(CH~)~-O-,
o un groupe -(CHz)n S-, ou
o un groupe -(CHZ)m CO-,
n est égal à 2 ou 3,
mestégalàl,2ou3,
R représente l'atome d'hydrogène ou un groupe alkyle en Cl-C4,
- Ar représente un noyau aromatique ou hétéroaromatique choisi parmi les
groupes phényle, naphtalényle, tétrahydronaphtalényle, pyridinyle ou indolyle,
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
7
éventuellement substitué par un ou deux substituants R3, R4 identiques ou
différents choisi parmi un halogène, un groupe alkyle en Cl-C4, alkoxy en Ci-
C4,
nitro, phényle, phénoxy, trifluorométhyle, amino, hydroxy, ou un groupe de
formule -X-[C(R)2]P CORS dans laquelle
o X représente une liaison simple, un atome d'oxygène, un atome
de soufre ou un groupe NH,
o R5 représente OR ou N(R)Z,
o R représente l'atome d'hydrogène ou un groupe alkyle en Cl-C4,
o pestégalà0,iou2;
lesdits substituants R3 et R4 pouvant en outre former ensemble un groupe
méthylènedioxy ;
ü) les sels pharmaceutiquement acceptables desdits composés de formule (I).
Selon un deuxième aspect, l'invention concerne les composés précités
pour leur utilisation en tant que substance pharmacologiquement active.
En particulier, l'invention concerne l'utilisation d'au moins un composé de
la formule (I) ou l'un de ses sels pharmaceutiquement acceptable en tant que
principe actif pour la préparation d'un médicament destiné à une utilisation
en
thérapeutique, notamment pour lutter contre les hypercholestérolémies, les
dyslipidémies, le diabète, l'obésité ainsi que les maladies cardiovasculaires
qui
sont la conséquence d'un déséquilibre des lipoprotéines sériques. D'une façon
plus générale, les composés de formule I selon l'invention trouvent leur
utilité
pour corriger la dérive des paramètres annonciateurs d'un syndrôme
métabolique. Les composés selon l'invention sont également utiles comme
principes actifs de médicaments destinés à prévenir ou traiter
l'athérosclérose,
l'infarctus du myocarde, certaines maladies inflammatoires comme par exemple
les dermatites, et les neurodégénérescences comme par exemple la maladie
d'Alzheimer.
Descriation détaillée
Dans la présente description, on entend par hétérocycle saturé azoté à 5
ou 6 chaînons condensé avec un noyau phényle ou un cyclohexyle, un
hétérocycle tel que 2,3-dihydroindole, octahydroindole, 1,2,3,4-
tétrahydroquinolëine, décahydroquinolëine, 1,2,3,4-tétrahydroisoquinolëine et
décahydroisoquinolëine. On entend par groupe alkyle en C3-C6 ramifié ou
cyclisé,
une chaîne hydrocarbonée ayant de 3 à 6 atomes de carbone, ramifiée ou
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
ô
cyclique, par exemple et sans limitation, les groupes 1-méthyléthyle, 1-
méthylpropyle, 2-méthylpropyle, 1,1-diméthyléthyle, 1-méthylbutyle, 1,1-
diméthylpropyle, 1-méthylpentyle, 1,1-diméthylbutyle, cyclopropyle,
cyclobutyle,
cyclopentyle, cyclohexyle ou cyclopentylméthyle. On entend par groupe alkyle
en Cl-C4 une chaîne hydrocarbonée ayant de 1 à 4 atomes de carbone, linéaire
ou ramifiée, ou bien encore cyclique ayant 3 ou 4 atomes de carbone. Des
exemples de groupes alkyle en Cl-C4 comprennent les groupes méthyle, éthyle,
propyle, butyle, 1-méthyléthyle, 1-méthylpropyle, 2-méthylpropyle, 1,1-
diméthyléthyle, cyclopropyle, méthylcyclopropyle ou cyclopropylméthyle. Par
groupe alkoxy en CZ-C6 linéaire, ramifié ou cyclique, on entend notamment les
groupes éthoxy, propoxy, butoxy, pentyloxy, hexyloxy, 1-méthyléthoxy, 1-
éthyléthoxy ou cylohexyloxy. Par groupe alkoxy en Ci-C4 linéaire ou ramifié,
on
entend notamment les groupes méthoxy, éthoxy, propoxy, butoxy, 1-
méthyléthoxy, 1-éthyléthoxy, 1- ou 2-méthylpropoxy.
Par groupe alkylène en Cl-C4, linéaire ou ramifié, on entend une chaîne
saturée disubstituée comprenant 1 à 4 atomes de carbone, par exemple les
groupes -CHZ-, -(CH2)~-, -(CHZ)3-, -CH(CH3)-CHa-, ou -CHZ-CH(CH3)-CHI-.
Par halogène, on comprend un atome de fluor, chlore, brome ou iode, les atomes
de fluor et de chlore étant préférés.
Les composés selon I invention comprennent un carbone asymétrique
(porteur de la fonction carboxamide) qui peut être soit racémique, soit de
configuration R ou, préférentiellement, de configuration S (fig. Ia)
~NH
\Y-Ar
H2
( Ia)
Les composés selon l'invention peuvent être préparés selon un procédé
faisant appel aux étapes consistant à
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
9
a) faire réagir un acide de formule
C(CH3)3
O O
OH
N
(H) H2
II
dans laquelle (H) représente un hétérocycle saturé azoté à 5 ou 6 chaînons
condensé avec un noyau phényle ou cyclohexyle, éventuellement substitué par
un halogène ou un groupe alcoxy en Ci-C4, ou un groupe N (R)~ dans lequel R
représente l'atome d'hydrogène ou un groupe alkyle en Ci-C4,
avec une amine de formule
NH2 Y-Ar III
dans laquelle
Y représente
o une liaison simple,
o un groupe alkylène en C1-C4, linéaire ou ramifié ou en C3-C4
cyclisé, éventuellement substitué par un groupe alkoxy en Cl-C3,
un groupe phényle, un groupe amino protégé par un groupe
aminoprotecteur (différent de Boc) ou un groupe N(R)Z, dans
lequel R représente l'atome d'hydrogène ou un groupe alkyle en
C1-C4,
o un groupe -(CHa)~-O-,
o un groupe -(CHZ)~-S-, ou
o un groupe -(CH~)m CO-,
n est égal à 2 ou 3,
m est égal à 1, 2 ou 3,
Ar représente un noyau aromatique ou hétéroaromatique choisi parmi les
groupes phényle, naphtalényle, tétrahydronaphtalényle, pyridinyle ou indolyle,
éventuellement substitué par un ou deux substituants R3, R4 identiques ou
différents choisi parmi un halogène, un groupe alkyle en Cl-C4, alkoxy en Cl-
C4,
nitro, phényle, phénoxy, trifluorométhyle, hydroxy, un groupe amino protégé
par
un groupe amino protecteur (différent de Boc), ou un groupe de formule -X-
[C(R)Z]P CORS dans laquelle
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
o X représente une liaison simple, un atome d'oxygène ou un atome
de soufre
o R5 représente OH, OR ou N(R)Z
o R représente un groupe alkyle en Cl-C4,
5 o p est égal à 0, 1 ou 2 ;
lesdits substituants R3 et R4 pouvant en outre former ensemble un groupe
méthylènedioxy,
dans un solvant anhydre tel que par exemple le dichlorométhane et en présence
10 d'un catalyseur tel que par exemple le DCC (dicyclohexylcarbodümide) libre
ou
greffé sur une résine ou le HOAT (1-hydroxy-7-azabenzotriazole), à une
température proche de la température ambiante et pendant 2 à 20 heures, pour
obtenir l'amide de formule IV
,C(CH3)3
C) C1
0
N ~ N H-Y-Ar
~ (H) H2
IV
dans laquelle (H), Y et Ar conservent la même signification que dans les
composés de départ,
b) faire réagir le composé de formule IV obtenu précédemment avec l'acide
trifluoroacétique, dans un solvant tel que le dichlorométhane, à température
ambiante pendant 2 à 20 heures pour obtenir le composé de formule V
O
H
~ N H-Y-Ar
/ ~H) H2
V
dans laquelle (H), Y et Ar conservent la même signification que dans le
composé
(IV),
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
11
c) faire réagir le composé de formule V avec un chlorure de benzènesulfonyle
de
formule VI
R2
S02C1
R1 VI
dans laquelle
- Rl représente un atome de chlore, un groupe alkyle en C3-C6, ramifié ou
cyclisé,
un groupe alkoxy en CZ-C6 linéaire, ramifié ou cyclisé, un groupe phénoxy
éventuellement substitué par un halogène, un groupe phényle, un groupe
aminométhyle éventuellement substitué par un groupe acétyle ou
trifluoroacétyle,
- R2 représente un atome d'hydrogène ou un halogène, ou,
- Rl et R~ forment ensemble un hétérocycle oxygéné ou azoté, éventuellement
substitué par un ou plusieurs groupes alkyle en Cl-C3, par un groupe acyle ou
par un groupe perfluoroacyle en Cz-C3.
dans un solvant tel que par exemple le dichlorométhane, à température
ambiante pendant 2 à 20 heures pour obtenir le composé de formule I
R2
O
S02 NH
R1 ~ N Y-Ar
(H) H2
I
dans laquelle Ri, RZ, (H), Y et Ar conservent la même signification que dans
les
composés de départ,
d) si nécessaire, dans le cas où l'un des substituants R3 ou R4 du groupe Ar
représente un groupe amino protégé, éliminer le groupement aminoprotecteur
pour obtenir R3 ou R4 sous forme d'une amine libre.
En variante du procédé décrit ci-dessus, les composés de formule I selon
l'invention peuvent être obtenus selon un procédé consistant à
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
12
a) faire réagir un chlorure de benzènesulfonyle de formule VI
R2
S02C1
R~ VI
dans laquelle
- Ri représente un atome de chlore, un groupe alkyle en C3-C6, ramifié ou
cyclisé,
un groupe alkoxy en CZ-C6 linéaire ramifié ou cyclisé, un groupe phénoxy
éventuellement substitué par un halogène, un groupe phényle, ou un groupe
aminométhyle substitué par un groupe acétyle ou trifluoracétyle,
- RZ représente un atome d'hydrogène ou un halogène, ou
- Rl et RZ forment ensemble un hétérocycle oxygéné ou azoté, éventuellement
substitué par un ou plusieurs groupes alkyle en Ci-C3, par un groupe acyle ou
par un groupe perfluoroacyle en C2-C3,
avec un ester de formule VII
H C02CH3
N
CH) H2
VII
dans laquelle (H) représente un hétérocycle saturé azoté à 5 ou 6 chaînons
condensé avec un noyau phényle ou cyclohexyle, éventuellement substitué par
un halogène ou un groupe alcoxy en Cl-C4,
dans un solvant anhydre tel que par exemple le dichlorométhane, à température
ambiante et pendant 2 à 10 heures pour obtenir le composé de formule VIII
R2
R1 S~2 C02CH3
N
(H) H2
VIII
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
13
dans laquelle (H), Ri et RZ conservent la même signification que dans les
composés de départ,
b) transformer l'ester VIII en acide, par action d'une base en milieu
hydroalcoolique selon des techniques bien connues de l'homme de métier
pour obtenir l'acide de formule IX
R2
R1 /
C02H
N
(H) H2
IX
dans laquelle (H), Ri et Rz restent inchangés,
c) faire réagir le composé acide (IX) avec une amine primaire de formule III
NH2 Y-Ar III
dans laquelle
Y représente
o une liaison simple,
o un groupe alkylène en Ci-C4, linéaire ou ramifié ou en C3-C4
cyclisé, éventuellement substitué par un groupe alkoxy en Ci-C3,
un groupe phényle , un groupe N(R)z ou un groupe COOH,
o un groupe -(CHZ)~ O-,
o un groupe -(CHZ)~-S-, ou
o un groupe -(CHZ)m-CO-,
n est égal à 2 ou 3 ;
m est égal à 1, 2 ou 3 ;
R représente l'atome d'hydrogène ou un groupe alkyle en Ci-C4,
Ar représente un noyau aromatique ou hétéroaromatique choisi parmi les
groupes phényle, naphtalényle, tétrahydronaphtalényle, pyridinyle ou indolyle,
éventuellement substitué par un ou deux substituants R3, R4 identiques ou
différents choisi parmi un halogène, un groupe alkyle en Cl-C4, alkoxy en Ci-
C4,
vitro, phényle, phénoxy, trifluorométhyle, hydroxy, N(R)2, un groupe amino
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
14
protégé par un groupe aminoprotecteur, ou un groupe de formule
-X-[C(R)Z]p CORS dans laquelle
o X représente une liaison simple, un atome d'oxygène ou un atome
de soufre ,
o RS représente OH, OR ou N(R)2,
o R représente un groupe alkyle en Ci-C4,
o p est égal à 0, 1 ou 2 ;
lesdits substituants R3 et R~ pouvant en outre former ensemble un groupe
méthylènedioxy,
selon un mode opératoire analogue à celui décrit à l'étape (a) du procédé
précédent, pour obtenir le composé de formule (I)
R2
O
S02 NH
R1 ~ N Y-är
(H) H2
I
dans laquelle (H), Rl, Rz, Y et Ar conservent la même signification que dans
les
composés de départ.
Les exemples suivants de préparation de composés selon la formule (I)
permettront de mieux comprendre l'invention.Dans ces exemples, on désigne par
«préparation» les exemples décrivant la synthèse de composés intermédiaires et
par «exemples» ceux décrivant la synthèse de composés de formule (I) selon
l'invention. Les points de fusion sont mesurés au banc Kofler et les valeurs
spectrales de Résonance Magnétique Nucléaire sont caractérisées par le
déplacement chimique calculé par rapport au TMS, par le nombre de protons
associés au signal et par la forme du signal (s pour singulet, d pour doublet,
dd
pour doublet dédoublé, t pour triplet, q pour quadruplet, m pour multiplet).
La
fréquence de travail et le solvant utilisé sont indiqués pour chaque composé.
La
température ambiante est de 20°C ~ 2°C.
Dans ces exemples, les abréviations suivantes ont été utilisées
- mmol» signifie millimole
- Boc signifie tbutoxycarbonyl (ou : 1,1-diméthyléthoxycarbonyl)
- « HOAT » signifie 1-hydroxy-7-azabenzotriazole
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Préparation I:
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-1,2,3,4-tétrahydro-2-
quinolinylcarboxylique, méthyl ester
On prépare une solution de 300 mg (1,32 mmol) d'ester méthylique de
5 l'acide 1,2,3,4-tétrahydro-2-quinolinylcarboxylique dans 20 ml
d'acétonitrile et on
ajoute, à température ambiante, 309 mg (1,32 mmol) de chlorure de 4-(1,1-
diméthyléthyl)benzènesulfonyle, puis 0,29 ml (2,64 mmol) de N-
méthylmorpholine. Le mélange réactionnel est agité pendant 18 heures à reflux
du solvant puis concentré sous pression réduite. Le résidu d'évaporation est
10 repris dans 50 ml d'acétate d'éthyle et lavé par une solution d'acide
chlorhydrique 1 N puis par une solution de bicarbonate de sodium saturée. La
phase organique est séchée sur sulfate de magnésium et concentrée sous
pression réduite. Le produit brut est purifié par chromatographie sur gel de
silice
en éluant à l'aide d'un mélange méthylcyclohexane/acétate d'éthyle (99/1 puis
15 95/5 ; v/v). On obtient ainsi le produit attendu sous forme d'un solide
blanc
(rendement = 22 %).
RMN 1H (DMSO, 300 MHz) ~ : 7.52-7.61 (m,SH); 7.19-7.24 (m,iH); 7.05-
7.10 (m,2H); 5.03 (t,iH); 3.65 (s,3H); 2.41-2.49 (m,lH); 2.12-2.17 (m,lH);
1.89-1.95 (m,iH); 1.73-1.80 (m,iH); 1.26 (s,9H).
Préparation II
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-1,2,3,4-tétrahydro-2-
quinolinylcarboxylique
On mélange 112 mg (0,289 mmol) du composé obtenu selon la
préparation I avec 6 ml de tétrahydrofurane et 4 ml d'eau et on ajoute, sous
agitation et à température ambiante, 18 mg (0,429 mmol) de lithine. Ce mélange
réactionnel est agité à température ambiante pendant 2 heures, puis acidifié
jusqu'à pH 1 par une solution d'acide chlorhydrique, dilué à l'eau puis
extrait par
3 fois 25 ml d'acétate d'éthyle. Les phases organiques rassemblées sont
séchées
sur sulfate de magnésium et concentrées sous pression réduite. On obtient
ainsi
le produit attendu sous forme d'un solide orange (rendement = 86%).
RMN iH (DMSO, 300 MHz), 8 : 7.49-7.74 (m,6H) ; 7.16-7.23 (m,iH); 7.05
(dd,iH); 4.9 (t,lH); 2.42-2.51 (m,lH); 1.80-2.07 (m,3H); 1.27 (s,9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
16
Exemple 1
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-N-(2-phényléthyl)-1,2,3,4-tétrahydro-
2-
quinolinylcarboxamide
On conditionne 150 mg (0,20 mmol) de résine PS-carbodümide (résine
polystyrène fonctionnalisée avec un groupe carbodümide fournie par la société
Argonaut Technologies) dans 5 ml de dichlorométhane pendant 30 mn puis on
élimine le solvant par filtration. La résine est rincée par 4 ml de
dichlorométhane,
puis on ajoute 50 mg (0,134 mmol) de l'acide obtenu selon la préparation II en
solution dans 5 ml de dichlorométhane, 25 pl (0,2 mmol) de 2
phényléthanamine et 5 mg de HOAT. Ce mélange réactionnel est agité à
température ambiante pendant 18 heures, puis on ajoute 200 mg de résine
IR120 et on agite à nouveau le mélange réactionnel pendant 3 heures à
température ambiante. La phase liquide du mélange est séparée par filtration
et
concentrée sous pression réduite. Le produit brut est purifié par
chromatographie
sur gel de silice en éluant à l'aide d'un mélange méthylcyclohexane/acétate
d'éthyle (70/30 ; v/v). on obtient ainsi le produit attendu sous forme d'un
solide
blanc (rendement = 55 %).
F = 142-143°C
RMN 1H (DMSO, 300 MHz) 8 : 7.94 (t,iH); 7.65 (dd,lH); 7.54 (d,2H);
7.44 (d,2H); 7.04-7.24 (m,BH); 4.66 (t,lH); 3.29 (m,2H); 2.66 (t,2H); 2.27-
2.34
(m,iH); 1.87-1.92 (m,lH); 1.48-1.66 (m,2H); 1.26 (s,9H).
Préparation III
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-(2S)-lffindole-2-
carboxylique, méthyl ester
On prépare une solution de 4,25 g (19,9 mmol) du chlorhydrate de l'ester
méthylique de l'acide 2,3-dihydro-(2S)-lffindole-2-carboxylique dans 100 ml de
THF (tétrahydrofurane) et on ajoute 5,55 ml (39,8 mmol) de triéthylamine. Le
mélange est agité et refroidi à 0° C et on ajoute lentement une
solution de 6,02
g (25,86 mmol) de chlorure de 4-(1,1-diméthyléthyl)benzènesulfonyle dans 40 ml
de THF. Le mélange réactionnel est maintenu sous agitation pendant 20 heures à
température ambiante puis hydrolysé avec 140 ml d'eau, et extrait 3 fois par
200
ml d'acétate d'éthyle. Les phases organiques rassemblées sont lavées 3 fois
par
50 ml d'eau, puis séchées sur sulfate de magnésium et concentrées sous
pression réduite. Le résidu d'évaporation est purifié par chromatographie sur
gel
de silice en éluant à l'aide d'un mélange méthylcyclohexane/acétate d'éthyle
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
17
(90/10 puis 80/20 ; v/v). on obtient ainsi le produit attendu sous forme de
cristaux blancs (rendement = 53 %).
F = 122° C
RMN iH (DMSO, 300 MHZ) 8 : 7.78 (d, 2H, Harom.)~ 7.58 (d, 2H, Harom.)
7.38 (d, 1H, Harom.)i 7.23-7.14 (m, 2H, Harom.). 6.99 (t, 1H, Harom.)i 5.02
(dd, 1H,
NChCO), 3.71 (s, 3H, OCH3), 3.40 (dd, 1H, CHzCHCO), 3.06 (dd, 1H, CHzCHCO),
1.25 (s, 9H, tBu).
Préparation IV
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-(2S)-1/f-indole-2-
carboxylique
On dissout 2 g (5,36 mmol) de l'ester obtenu selon la préparation III dans
ml de dioxane et on ajoute 5 ml d'eau, puis 0,43 g (10,7 mmol) d'hydroxyde
de sodium. Le mélange réactionnel est concentré sous pression réduite. Le
résidu
15 blanc obtenu est dissout dans 30 ml d'eau et la solution obtenue est
acidifiée par
une solution d'acide chlorhydrique 1M, jusqu'à pHi. Le précipité blanc est
séparé
par filtration puis séché dans un dessicateur. On obtient ainsi l'acide
attendu sous
forme d'une poudre blanche (rendement = 85 %).
RMN iH (DMSO, 300 MHz) 8 : 13.10 (s, 1H, C00/~, 7.77 (d, 2H, Harom.).
20 7.57 (d, 2H, Harom.)~ 7.36 (d, 1H, Harom.). 7.20-7.13 (m, 2H, Harom.)i 6.98
(t, 1H,
Harom.)i 4.88 (dd, 1H, NChCO), 3.35 (dd, 1H, CHzCHCO), 3.02 (dd, 1H,
CH~CHCO), 1.25 (s, 9H, tBu).
Exemale 2
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(2-phénoxy-éthyl)-(2S)-
lffindole-2-carboxamide
On conditionne 400 mg (équivalent à 0,556 mmol) de résine PS-EDC (N-
(3-diméthylaminopropyl)-N'-éthylcarbodümide fixé sur résine polystyrène) dans
4 ml de dichlorométhane sous agitation pendant 15 mn, puis on élimine le
solvant par filtration. La résine est rincée plusieurs fois avec 4 ml de
dichlorométhane, puis on ajoute une solution de 100 mg (0, 278 mmol) d'acide
obtenu selon la préparation IV et 25,4 mg (0,185 mmol) de 2-
phénoxyéthanamine en solution dans 6 ml de dichlorométhane. Le mélange est
agité pendant 20 heures à température ambiante et on ajoute 0,278 mmol de
résine isocyanate. Le mélange est agité pendant 2 heures à température
ambiante et on ajoute 0,278 mmol de résine Amberlite IRA 400. On agite le
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
18
mélange à nouveau pendant 2 heures puis on élimine les résines par filtration.
Le
filtrat est ensuite concentré sous pression réduite. On obtient ainsi le
composé
attendu sous forme d'une poudre blanche (rendement = 58 %).
RMN iH (DMSO, 250 MHZ) 8 : 8.37 (t, 1H, NhCO), 7.70 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.32-7.20 (m, 3H, Harom.)i 7.10
(d, 1H,
Harom.)~ 7.03-6.93 (m, 4H, Ha,-om.)i 4.80 (dd, 1H, NChCO), 4.02 (t, 2H,
CHzOPh),
3.50 (m, 2H, CHzNCO), 3.11 (dd, iH, CH~CHCO), 2.91 (dd, iH, CHzCHCO), 1.25
(s, 9H, tBu).
En opérant de façon analogue à l'exemple 2, on obtient les composés
suivants
Exemple 3
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-nitro-
phényl)éthyl]-
2S-Iffindole-2-carboxamide
Rdt = 17% ; poudre blanche ;
RMN 1H (DMSO, 300 MHZ) 8 : 8.22 (t, 1H, NhCO), 8.07 (d, 2H, Harom.)r
7.66 (d, 2H, Harom.)i 7.54 (d, 2H, Harom.). 7.45 (m, 3H, Harom.). 7.22 (t, 1H,
Harom.)i
7.08 (d, 1H, Harom.)i 7.02 (t, 1H, Harom.)i 4.68 (dd, 1H, NCf~CO), 3.41 (m,
2H,
CHzNCO), 2.99 (dd, 1H, CH~CHCO), 2.89 (t, 2H, CHzPh), 2.79 (dd, 1H,
CH~CHCO), 1.24 (s, 9H, tBu).
MS (ESI+) m/z508 (MH+).
Exemple 4
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3-fluoro-
phényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 26% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) 8 : 8.22 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.32-7.20 (m, 2H, Harom.)i 7.10-
6.99 (m,
5H, Harom.)i 4.69 (dd, 1H, NChCO), 3.37 (m, 2H, CHzNCO), 3.04 (dd, 1H,
CHzCHCO), 2.81 (dd, iH, CHzCHCO), 2.75 (t, 2H, CH~,Ph), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z481 (MH+).
Exemple 5
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2,6-
dichlorophényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 23% ; poudre blanche ;
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
19
RMN iH (DMSO, 300 MHZ) 8 : 8.38 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)
7.55 (d, 2H, Harom.)i 7.47-7.40 (m, 3H, Harom.)i 7.29-7.22 (m, 2H, Harom.)i
7.10 (d,
1H, Harom.)i 7.03 (t, 1H, Harom.)i 4.69 (dd, 1H, NChCO), 3.33 (m, 2H, CHzNCO),
3.09-3.00 (m, 3H, CH~CHCO, CHzPh), 2.87 (dd, iH, Cf~CHCO), 1.25 (s, 9H,
tBu) ;
MS (ESI+) m/z 531 (MH+).
Exemple 6
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(2-phényl-propyl)-2S-
.off
indole-2-carboxamide
Rdt = 15% ; huile incolore ;
RMN iH (DMSO, 300 MHz), ~ : 8.08 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i 7.55 (d,
2H, Harom.). 7.44 (t, 1H, Harom.)~ 7.30-7.18 (m, 6H, Harom.)~ 7.09 (t, 1H,
Harom.)i
7.01 (td, 1H, Harom.)i 4.72 (dd, 1H, NChCO), 3.27 (m, 2H, CHzNCO), 3.05-2.90
(m, 2H, CHzCHCO, CfyPh), 2.75 (dd, 1H, CHzCHCO), 1.25 (s, 9H, tBu) ; 1.18 (d,
3H, CH3).
MS (ESI+) m/z477 (MH+).
Exemale 7
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-méthyl-
phényl)éthyl]-2S-1/findole-2-carboxamide
Rdt = 19% ; huile incolore ;
RMN 1H (DMSO, 300 MHZ), 8 : 8.17 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i 7.55 (d,
2H, Harom.). 7.45 (d, 1H, Harom.)~ 7.22-7.10 (m, 3H, Harom.)~ 7.04-6.96 (m,
4H,
Harom.)i 4.71 (dd, 1H, NChCO), 3.32 (m, 2H, CHzNCO), 3.05 (dd, 1H, CHzCHCO),
2.84 (dd, iH, CHzCHCO), 2.69 (t, 2H, CHzPh), 2.27 (s, 3H, CH3), 1.25 (s, 9H,
tBu) ;
MS (ESI+) m/z477 (MH+).
Exemale 8
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3,4-
dichlorophényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 18% ; huile incolore ;
RMN iH (DMSO, 300 MHZ), 8 : 8.21 (t, 1H, NhCO), 7.69 (d, 2H, Harom.). 7.55 (d,
2H, Harom.)i 7.47 (m, 3H, Harom.)~ 7.22-7.09 (m, 3H, Harom.)i 7.01 (t, 1H,
Harom.)i
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
4.68 (dd, 1H, NChCO), 3.35 (m, 2H, CHzNCO), 3.03 (dd, 1H, CHzCHCO), 2.83-
2.78 (m, 3H, CHzCHCO, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z531 (MH+).
5 Exem~ole 9
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-fluoro-
phényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 92% ; huile incolore ;
RMN iH (DMSO, 300 MHZ), ~ : 8.25 (t, 1H, NhCO), 7.69 (d, 2H, Harom.). 7.55 (d,
10 2H, Harom.). 7.45 (d, 1H, Harom.)i 7.27-7.20 (m, 3H, Harom.)i 7.15-7.01 (m,
4H,
Harom.), 4.68 (dd, 1H, NChCO), 3.35 (m, 2H, Cf~NCO), 3.03 (dd, 1H, CHzCHCO),
2.85-2.74 (m, 3H, Cff~CHCO, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z481 (MH+).
15 Exemple 10
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(méthoxy)-1-
(phénylméthyl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 11% ; huile incolore ;
RMN iH (DMSO, 300 MHZ), ~ : 8.01 (d, 1H, N/'~CO), 7.68 (d, 2H, Harom.), 7.55
(d,
20 2H, Harom.)i 7.47 (d, 1H, Harom.)~ 7.18-7.03 (m, 8H, Harom.). 4.74 (dd, 1H,
NChCO),
4.05 (m, 1H, ChNCO), 3.31 (d, 2H, OC/~,CHN), 3.28 (s, 3H, OCH3), 3.02 (dd, 1H,
CHzCHCO), 2.78-2.71 (m, 3H, CHzCHCO, CH~Ph), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z 507 (MH+).
Exemple 11
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[E-phényl-cyclopropyl]-
2S-1/findole-2-carboxamide
Rdt = 24% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ), 8 : 8.48 (t, 1H, NhCO), 7.73 (d, 2H, Harom.)i 7.57
(dd,
2H, Harom.)i 7.42 (dd, 1H, Harom.)i 7.26-7.11 (m, 7H, Harom.)i 7.00 (t, 1H,
Harom.),
4.67 (dd, iH, NChCO), 3.16 (m, iH, ChNCO), 2.96-2.83 (m, 2H, CHzCHCO), 2.00
(m, 1H, ChPh), 1.25 (s, 9H, tBu), 1.18 (m, 2H, PhCHCHz) ;
MS (ESI+) m/z475 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
21
Exemale 1~
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(3,3-diphénylpropyl)-
2S-
rffindole-2-carboxamide
Rdt = 34% ; huile incolore ;
RMN iH (DMSO, 250 MHZ), 8 : 8.22 (t, 1H, NfiCO), 7.72 (d, 2H, Harom.). 7.55
(d,
2H, Harom.). 7.45 (d, 1H, Harom.). 7.29-7.11 (m, 12H, Harom.). 7.01 (t, 1H,
Harom.)i
4.71 (dd, 1H, NChCO), 4.01 (t, iH ; C/~Ph)Z), 3.45 (m, 2H, CHzNCO), 3.17-2.86
(m, 2H, CHzCHCO), 2.22 (m, 2H, CHz-CH), 1.25 (s, 9H, tBu).
MS (ESI+) m/z553 (MH+).
Exemple 13
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-chloro-
phényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 31% ; poudre blanche ;
RMN 1H (DMSO, 300 MHZ) ~ : 8.26 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.40 (d, 1H, Harom.)i 7.25-7.20
(m, 4H,
Harom.)~ 7.10 (d, 1H, Harom.)r 7.03 (t, 1H, Harom.)i 4.69 (dd, 1H, NChCO),
3.35 (m,
2H, CH~NCO), 3.03 (dd, 1H, CffzCHCO), 2.89-2.80 (m, 3H, CffzCHCO, CH~Ph),
1.25 (s, 9H, tBu).
MS (ESI+) m/z497 (MH+).
Exemple 14
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3-chloro-
phényl)éthyl]-2S-Iff-indole-2-carboxamide
Rdt = 26% ; poudre blanche ;
RMN 1H (DMSO, 300 MHZ) 8 : 8.20 (t, 1H, NhCO), 7.70 (d, 2H, H~rom.)i
7.56 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.29-7.10 (m, 6H, Harom.)i 7.03
(t, 1H,
Harom.)i 4.69 (dd, 1H, NChCO), 3.35 (m, 2H, CHzNCO), 3.03 (dd, 1H, CHzCHCO),
2.84-2.72 (m, 3H, C/~CHCO, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z497 (MH+).
Exemale 15
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-chloro-
phényl)éthyl]-2S-1/findole-2-carboxamide
Rdt = 24% ; poudre blanche ;
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
22
RMN iH (DMSO, 300 MHz), 8 : 8.17 (t, 1H, NhCO), 7.68 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)~ 7.45 (d, 1H, Harom.)i 7.30-7.18 (m, 5H, Harom.). 7.10
(d, 1H,
Harom.). 7.05 (t, 1H, Harom.)i 4.69 (dd, 1H, NChCO), 3.35 (m, 2H, Cf~,NCO),
3.02
(dd, 1H, Cf~CHCO), 2.80 (dd, 1H, CHzCHCO), 2.72 (t, 2H, CHzPh), 1.25 (s, 9H,
tBu).
MS (ESI+) m/z497 (MH+)
Exemple 16
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2,4-
dichlorophényl)éthyl]-2S-lffindole-2-carboxamide
Rdt = 24% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) b : 8.24 (t, 1H, NhCO), 7.68 (d, 2H, Harom.).
7.57-7.54 (m, 3H, Harom.)i 7.46 (d, 1H, Harom.)i 7.24 (m, 3H, Harom.). 7.11
(d, 1H,
Harom.)i 7.02 (t, 1H, Harom.)i 4.69 (dd, 1H, NCf7C0), 3.34 (m, 2H, CH~NCO),
3.01
(dd, iH, CHzCHCO), 2.87-2.78 (m, 3H, CHzCHCO, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z 531 (MH+).
Exemple 17
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(3-phényl-propyl)-2S-
Ifi~
indole-2-carboxamide
Rdt = 19% ; poudre blanche ;
RMN iH (DMSO, 300 MHz) 8 : 8.20 (t, 1H, N/~CO), 7.73 (d, 2H, Harom.)i
7.57 (d, 2H, Harom.). 7.45 (d, 1H, Harom.)i 7.30-7.11 (m, 7H, Harom.)i 7.01
(t, 1H,
Harom.). 4.72 (dd, 1H, NChCO), 3.16-3.08 (m, 3H, Cf/zNCO, CHzCHCO), 2.91 (dd,
1H, CHzCHCO), 2.58 (t, 2H, CHzPh), 1,73 (m, 2H, CHzCH2Ph)1.25 (s, 9H, tBu).
MS (ESI+) m/z477 (MH+).
Exemple 18
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(4-phényl-butyl)-2S-
Iff
indole-2-carboxamide
Rdt = 25% ; huile incolore ;
RMN 1H (DMSO, 300 MHz) 8 : 8.12 (t, 1H, NhCO), 7.71 (d, 2H, Harom.)
7.55 (d, 2H, Harom.)i 7.44 (d, 1H, Harom.), 7.29-7.10 (m, 7H, Harom.). 7.00
(t, 1H,
Harom.)i 4.71 (dd, 1H, NChCO), 3.15-3.04 (m, 3H, Cff~NCO, Cf/zCHCO), 2.88 (dd,
1H, CHzCHCO), 2.58 (t, 2H, CHzPh), 1.60-1.42 (m, 4H, Cf~C/~,CHZPh), 1.25 (s,
9H, tBu).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
23
MS (ESI+) m/z491 (MH+).
Exemple 19
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(phényl-méthyl)-2S-1ff
indole-2-carboxamide
Rdt = 42% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) 8 : 8.72 (t, 1H, NhCO), 7.72 (d, 2H, Ha,.om.)i
7.55 (d, 2H, Harom.)i 7.'46 (d, 1H, Harom.). 7.32-7.22 (m, 6H, Harom.). 7.12
(d, 1H,
Harom.). 7.01 (t, 1H, Harom.)i 4.82 (dd, 1H, NChCO), 3.34 (t, 2H, CHaNCO),
3.16
(dd, 1H, CHzCHCO), 2.94 (dd, 1H, Cf~CHCO), 1.25 (s, 9H, tBu).
MS (ESI+) m/z449 (MH+).
Exemple 20
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(Iffindol-3-
yl)éthyl]-
2S-Iffindole-2-carboxamide
Rdt = 17% ; poudre blanche ;
RMN 1H (DMSO, 300 MHZ) 8 : 8.23 (t, 1H, NhCO), 7.71 (d, 2H, Harom.)i
7.55 (m, 3H, Harom.r nl~, 7.46 (d, 1H, Harom.)i 7.33 (d, 1H, Harom.)i 7.20 (t,
1H,
Harom.)i 7.14-6.95 (m, 6H, Harom.)i 4.74 (dd, 1H, NCfiCO), 3.38 (m, 2H,
Cf~NCO),
3.04 (dd, 1H, CH~CHCO), 2.92-2.82 (m, 3H, CHzCHCO, CHzPh), 1.25 (s, 9H,
tBu) ;
MS (ESI+) m/z 502 (MH+).
Exemale 21
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3-pyridinyl)éthyl]-
2S-
Iffindole-2-carboxamide
Rdt = 25% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) ô : 8.42 (m, 2H, Harom.)~ 8.24 (t, 1H, NhCO),
7.69 (d, 2H, Harom.). 7.59-7.54 (m, 3H, Harom.). 7.45 (d, 1H, Harom.)~ 7.28-
7.22 (m,
2H, Harom.)i 7.10 (d, 1H, Harom.). 7.02 (t, 1H, Harom.)i 4.69 (dd, 1H, NChCO),
3.38
(m, 2H, CHzNCO), 3.03 (dd, iH, CHzCHCO), 2.83-2.73 (m, 3H, CHzCHCO,
CHz,Ph), 1.25 (s, 9H, tBu).
MS (ESI+) m/z464 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
24
Exemple 22
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(2-oxo-2-phényléthyl)-
2S-Ih~indole-2-carboxamide
Rdt = 33% ; huile incolore ;
RMN 1H (DMSO, 300 MHZ) 8 : 8.46 (t, 1H, NhCO), 7.99 (d, 2H, Harom.)i
7.72 (d, 2H, Harom.)i 7.66 (d, 1H, Harom.). 7.58-7.46 (m, 5H, Harom.). 7.23
(t, 1H,
Harom.), 7.14 (d, 1H, Harom.)~ 7.03 (t, 1H, Harom.)~ '1'.95 (dd, 1H, NChCO),
4.74 (dd,
1H, CHzNCO), 4.62 (dd, 1H, CHzNCO), 3.15 (dd, 1H, CHz,CHCO), 3.01 (dd, 1H,
CH~CHCO), 1.25 (s, 9H, tBu).
MS (ESI+) m/z477 (MH+).
Exemple 23
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-fluoro-
phényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 11% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) 8 : 8.17 (t, 1H, Nf~CO), 7.68 (d, 2H, Harom.).
7.55 (d, 2H, Harom.)i 7.'45 (d, 1H, Harom.)i 7.22-7.02 (m, 7H, Harom.). '4.69
(dd, 1H,
NChCO), 3.35 (m, 2H, CHzNCO), 3.02 (dd, 1H, Cff~CHCO), 2.81 (dd, iH,
CHzCHCO), 2.71 (t, 2H, CHzPh), 1.25 (s, 9H, tBu).
MS (ESI+) m/z481 (MH+).
Exemple 24
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-pyridinyl)éthyl]-
2S-
Iff-indole-2-carboxamide
Rdt = 20% ; poudre blanche ;
RMN 1H (DMSO, 300 MHZ) & : 8.49 (dd, 1H, Harom.). 8.30 (t, 1H, NhCO),
7.70-7.66 (m, 3H, Harom.)i 7.55 (d, 2H, Harom.). 7.45 (d, 1H, Harom.). 7.23-
7.19 (m,
3H, Harom.)i 7.10 (d, 1H, Harom.), 7.02 (t, 1H, Harom.). 4.71 (dd, 1H, NChCO),
3.47
(m, 2H, Cf/~NCO), 3.03 (dd, iH, CHzCHCO), 2.91-2.82 (m, 3H, CHzCHCO,
CH2Ph), 1.25 (s, 9H, tBu).
MS (ESI+) m/z464 (MH+).
Exemple 25
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3,5-
diméthoxyphényléthyl]-2S-Iffindole-2-carboxamide
Rdt = 21% ; huile incolore ;
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
2S
RMN iH (DMSO, 300 MHZ) 8 : 8.17 (t, 1H, NhCO), 7.70 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)i 7.44 (d, 1H, Harom.)i 7.21 (t, 1H, Harom.). 7.10 (d, 1H,
Harom.).
7.01 (t, 1H, Harom.)i 6.39 (d, 2H, Harom.)i 6.33 (t, 1H, Harom.). 4~72 (dd,
1H,
NChCO), 3.70 (s, 6H, OCH3), 3.32 (m, 2H, CHzNCO), 3.02 (dd, 1H, Cf~CHCO),
2.85 (dd, iH, CHzCHCO), 2.69 (t, 2H, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z523 (MH+).
Exemple 26
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-
éthylphényl)éthyl]-
2S-Iffindole-2-carboxamide
Rdt = 19% ; poudre blanche ;
RMN iH (DMSO, 300 MHZ) 8 : 8.16 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.), 7.22 (t, 1H, Harom.)r 7.10 (d, 1H,
Harom.)i
7.09 (s, 4H, Harom.)i 7.01 (t, 1H, Harom.)i 4.71 (dd, 1H, NChCO), 3.31 (m, 2H,
CHzNCO), 3.03 (dd, 1H, CHzCHCO), 2.82 (dd, 1H, CH~CHCO), 2.68 (t, 2H,
CHzPh), 2.56 (q, 2H, CHzCH3), 1.25 (s, 9H, tBu), 1.15 (t, 3H, CH3).
MS (ESI+) m/z491 (MH+).
Exemele 27
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-
phénoxyphényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 11% ; huile incolore ;
RMN iH (DMSO, 250 MHZ) 8 : 8.21 (t, 1H, NhCO), 7.68 (d, 2H, Harom.)
7.55 (d, 2H, Harom.). 7.45 (d, 1H, Harom.)i 7.40-6.84 (m, 11H, Harom.)i 6.84
(t, 1H,
Harom.)i 4.67 (dd, 1H, NChCO), 3.36 (m, 2H, CHzNCO), 3.02 (dd, 1H, CHzCHCO),
2.86-2.73 (m, 3H, CHzCHCO, CHzPh), 1.25 (s, 9H, tBu).
MS (ESI+) m/z 555 (MH+).
Exemple 28
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[(1-
naphtalènyl)méthyl]-
2S-Iffindole-2-carboxamide
Rdt = 13% ; poudre blanche ;
RMN iH (DMSO, 250 MHZ) 8 : 8.73 (t, 1H, NhCO), 8.05 (m, 1H, Harom.)i
7.94 (m, 1H, Harom.)~ 7.85 (m, 1H, Harom.). 7.73 (dd, 2H, Harom.)i 7.57-7.44
(m, 7H,
Harom.)i 7.22 (t, 1H, Harom.)i 7.12 (d, 1H, Harom.)i 7.01 (t, 1H, Harom.)i
4.92-4.83 (m,
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
26
2H, NChCO, CHzNCO), 4.72 (dd, 1H, CHzNCO), 3.15 (dd, 1H, CHzCHCO), 2.94
(dd, iH, CHzCHCO), 1.25 (s, 9H, tBu).
MS (ESI+) m/z499 (MH+).
Exemple 29
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-méthyl-
phényl)éthyl]-2S-llfindole-2-carboxamide
Rdt = 12% ; huile incolore ;
RMN iH (DMSO, 250 MHZ) b : 8.14 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i
7.55 (d, 2H, Ha,.om.)i 7.45 (d, 1H, Harom.)i 7.22 (t, 1H, Harom.). 7.12-7.01
(m, 6H,
Harom.)i 4.70 (dd, 1H, NChCO), 3.29 (m, 2H, CH~NCO), 3.03 (dd, 1H, CHzCHCO),
2.83 (dd, 1H, CHzCHCO), 2.67 (t, 2H, CHzPh), 2.26 (s, 3H, CH3), 1.25 (s, 9H,
tBu).
MS (ESI+) m/z477 (MH+)
Exemple 30
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-Z,3-dihydro-N-(1,2,3,4-tétrahydro-1-
naphtalényl)-2S-Iffindole-2-carboxamide
Rdt = 7% ; huile incolore ;
RMN iH (DMSO, 250 MHz) ~ : 8.52 (t, 1H, NhCO), 7.76 (dd, 2H, Harom.).
7.56 (dd, 2H, Harom.). 7.4'1 (dd, 1H, Ha,.om.)i 7.24-7.12 (m, 6H, Harom.).
6.99 (t, 1H,
Harom.)~ 4.96 (m, 1H, ChNCO), 4.79 (m, 1H, NChCO), 3.17 (m, 1H, Cf~CHCO),
2.97 (m, 1H, CHzCHCO), 2.76 (m, 2H, CHZCH~CHZCHNCO), 1.92-1.69 (m, 4H,
CHzCHzCHNCO), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z489 (MH+).
Exemple 3i
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2-
méthoxyphényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 7% ; huile incolore ;
RMN iH (DMSO, 300 MHz) 8 : 8.14 (t, 1H, NhCO), 7.69 (d, 2H, Harom.),
7.55 (d, 2H, Harom.). 7.45 (d, 1H, Harom.)i 7.25-6.93 (m, 6H, Harom.)i 6.84
(t, 1H,
Harom.)i '4.69 (dd, 1H, NChCO), 3.78 (s, 3H, OCH3), 3.29 (m, 2H, C//zNCO),
3.03
(dd, 1H, CHzCHCO), 2.84 (dd, iH, CHzCHCO), 2.72 (m, 2H, Cf~Ph), 1.25 (s, 9H,
tBu).
MS (ESI+) m/z493 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
27
Exemele 32
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-pyridinyl)éthyl]-
2S-
1/findole-2-carboxamide
Rdt = 14% ; huile incolore ;
RMN iH (DMSO, 300 MHZ) 8 : 8.64 (m, 2H, Harom.)i 8.29 (t, 1H, NhCO),
7.68-7.63 (m, 4H, Harom.)i 7.55 (d, 2H, Harom.)o 7.45 (d, 1H, Harom.). 7.23
(t, 1H,
Harom.)i 7.10 (d, 1H, Harom.)i 7.02 (t, 1H, Harom.)i 4.66 (dd, 1H, NChCO),
3.35 (m,
2H, CHzNCO), 3.06-2.94 (m, 3H, CHzCHCO, CHaPh), 2.80 (dd, iH, CHzCHCO),
1.25 (s, 9H, tBu).
MS (ESI+) m/z464 (MH+).
Exemple 33
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(1,3-benzodioxol-5-
yl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 10% ; poudre blanche ;
RMN iH (DMSO, 300 MHz) 8 : 8.14 (t, 1H, NhCO), 7.70 (d, 2H, Harom.).
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.22 (t, 1H, Harom.)~ 7.10 (d, 1H,
Harom.).
7.01 (t, 1H, Harom.)i 6.78 (m, 2H, Harom.)r 6.62 (d, 1H, Harom.)i 5.95 (S, 2H,
OCHzO), 4.71 (dd, 1H, NChCO), 3.26 (m, 2H, CHzNCO), 3.04 (dd, 1H,
CHzCHCO), 2.84 (dd, iH, CHzCHCO), 2.64 (t, 2H, CH~Ph), 1.25 (s, 9H, tBu).
MS (ESI+) m/z 507 (MH+).
Exemple 34
1-[[4-( 1,1-diméthyléthyl)phényl]sulfonyl]-2,3-di hydro-N-[2-(4-phényl-
phényl)éthyl]-2S-Ilfindole-2-carboxamide
Rdt = 11% ; poudre blanche ;
RMN 1H (DMSO, 300 MHz) 8 : 8.22 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)i
7.63 (d, 2H, Harom.)i 7. 55 (d, 4H, Harom.)i 7.45 (t, 3H, Harom.). 7.37-7.22
(m, 4H,
Harom.)i 7.09 (d, 1H, Harom.)i 7.01 (t, 1H, Harom.). 4.74 (dd, 1H, NChCO),
3.36 (m,
2H, CHzNCO), 3.03 (dd, 1H, CHzCHCO), 2.88-2.75 (m, 3H, C/~CHCO, Cff~Ph),
1.25 (s, 9H, tBu).
MS (ESI+) m/z539 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
28
Exemple 35
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-[4-(1,1-
diméthyléthyl)phényl]éthyl]-2S-1H-indole-2-carboxamide
Rdt = 10% ; poudre blanche ;
RMN iH (DMSO, 300 MHz) 8 : 8.17 (t, 1H, Nf~CO), 7.69 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.)~ 7.45 (d, 1H, Harom.)i 7.28-7.22 (m, 3H, Harom.), 7.10
(d, 3H,
Harom.). 7.01 (t, 1H, Harom.). 4.71 (dd, 1H, NChCO), 3.31 (m, 2H, CH~NCO),
3.03
(dd, iH, CHzCHCO), 2.82 (dd, 1H, CHaCHCO), 2.68 (t, 2H, CHzPh), 1.25 (s, 18H,
tBu).
MS (ESI+) m/z519 (MH+).
Exemple 36
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3,4-
diméthylphényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 10% ; huile incolore ;
RMN 1H (DMSO, 300 MHz), ~ : 8.14 (t, 1H, NhCO), 7.70 (d, 2H, Harom.)
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.22 (t, 1H, Harom.)i 7.10 (d, 1H,
Harom.)i
7.03-6.97 (m, 3H, Harom.)r 6.89 (d, 1H, Harom.)i 4.71 (dd, 1H, NChCO), 3.31
(m,
2H, CHzNCO), 3.05 (dd, 1H, CHzCHCO), 2.84 (dd, 1H, CHZCHCO), 2.65 (t, 2H,
CHZPh), 2.18 (s, 6H, CH3), 1.25 (s, 9H, tBu).
MS (ESI+) m/z491 (MH+).
Exemple 37
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2,3-
diméthoxyphényl)éthyl]-2S-lffindole-2-carboxamide
Rdt = 6% ; huile incolore ;
RMN iH (DMSO, 300 MHZ), 8 : 8.22 (t, 1H, NhCO), 7.69 (d, 2H, Harom.)
7.55 (d, 2H, Harom.)i 7.46 (d, 1H, Harom.)i 7.22 (t, 1H, Harom.), 7.10 (d, 1H,
Harom.).
7.04-6.90 (m, 3H, Harom.)i 6.70 (d, 1H, Harom.)i 4.70 (dd, 1H, NChCO), 3.78
(s,
3H, OCH3), 3.73 (s, 3H, OCH3), 3.29 (m, 2H, Cf/zNCO), 3.04 (dd, iH, CHzCHCO),
2.86 (dd, 1H, CHzCHCO), 2.72 (t, 2H, CHzPh), 1.25 (s, 9H, tBu) ;
MS (ESI+) m/z 523 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
29
Exemple 38
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(3-méthyl-
phényl)éthyl]-2S-Iffindole-2-carboxamide
Rdt = 14% ; huile incolore ;
RMN iH (DMSO, 300 MHz), 8 : 8.17 (t, 1H, NhCO), 7.70 (d, 2H, Harom.)i
7.55 (d, 2H, Harom.). 7.45 (d, 1H, Harom.)i 7.22-7.10 (m, 3H, Harom.)i 7.03-
6.98 (m,
4H, Harom.)~ 4.71 (dd, 1H, NChCO), 3.31 (m, 2H, CH~NCO), 3.05 (dd, 1H,
CH~CHCO), 2.84 (dd, 1H, CH~CHCO), 2.69 (t, 2H, CH~Ph), 2.27 (s, 3H, CH3), 1.25
(s, 9H, tBu).
MS (ESI+) m/~477 (MH+).
Exemple 39
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-[3-
(trifluorométhyl)phényl]éthyl]-2S-1H-indole-2-carboxamide
Rdt = 9% ; huile incolore ;
RMN iH (DMSO, 300 MHz), 8 : 8.22 (t, 1H, NhCO), 7.69 (d, 2H, Harom.).
7.57-7.46 (m, 7H, Harom.)r 7.22 (t, 1H, Harom.)i 7.08 (d, 1H, Harom.)i 7.01
(t, 1H,
Harom.)i 4.69 (dd, 1H, NChCO), 3.39 (m, 2H, CHzNCO), 3.03 (dd, 1H, CHzCHCO),
2.87-2.75 (m, 3H, Cf~fzCHCO, CHZPh), 1.25 (s, 9H, tBu).
MS (ESI+) m/z 531 (MH+).
Exemple 40
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(2,5-
diméthoxyphényl)éthyl]-2S-.~ffindole-2-carboxamide
420 mg (0,556 mmol) de résine PS-carbodümide sont conditionnés dans
4 ml de dichlorométhane. Après 10 minutes d'agitation, la résine est filtrée
puis
rincée 3 fois avec 4 ml de dichlorométhane. On ajoute 4 ml de dichlorométhane
et 4 ml de tétrahydrofurane (THF), puis on introduit successivement 100 mg
(0,278 mmol) d'acide obtenu selon la préparation IV, 33,5 mg (0,185 mmol) de
2,5-diméthoxybenzèneéthanamine et 3,8 mg (0,019 mmol) de HOAT. Le milieu
réactionnel est alors agité à température ambiante. Après 16h d'agitation, on
introduit dans le milieu réactionnel 0,278 mmol de résine IRA400 puis, après 2
heures d'agitation, 0,278 mmol de résine isocyanate. Après 1 heure
d'agitation,
le milieu réactionnel est filtré puis les résines sont lavées 3 fois avec 3 ml
de
dichlorométhane. Les filtrats rassemblés sont concentrés puis séchés sous
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
pression réduite. On obtient ainsi le composé attendu sous forme d'une huile
incolore (rendement = 60 %).
RMN iH (DMSO, 300 MHZ) 8 : 8.14 (t, 1H, NhCO), 7.69 (d, 2H, Harom.),
7.55 (d, 2H, Harom.)i 7~'44 (d, 1H, Harom.)i 7.21 (t, 1H, Harom.)i 7.10 (d,
1H, Harom.)i
5 7.00 (t, 1H, Harom.)i 6.87 (d, 1H, Harom.)i 6.74 (m, 2H, Harom.)~ 4.69 (dd,
1H,
NChCO), 3.73 (s, 3H, OCH3), 3.68 (s, 3H, OCH3), 3.29 (m, 2H, CHzNCO), 3.05
(dd, 1H, CHaCHCO), 2.84 (dd, 1H, CHaCHCO), 2.69 (m, 2H, Cff~Ph), 1.25 (s, 9H,
tBu).
MS (ESI+) m/z523 (MH+).
Exemele 41
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-amino-
phényl)éthyl]-2S-Iffindole-2-carboxamide
On prépare une solution de 100 mg (0,278 mmol) de l'acide obtenu selon
la préparation IV dans 4 ml de dichlorométhane et 4 ml de THF puis on ajoute
64 mg (0,334 mmol) de EDCI (chlorhydrate de 1-(3-diméthylaminopropyl)-3
éthylcarbodümide) et 7,8 mg (0,028 mmol) de HOAT. Après 10 minutes sous
agitation à température ambiante, on ajoute 45,5 mg (0,334 mmol) de 4
aminobenzèneéthanamine, ainsi que 4 ml de mélange dichlorométhane/THF
(50/50) et 58 p.l (0,682 mmol) de triéthylamine. Le milieu réactionnel est
ensuite
agité à température ambiante pendant 2 heures. Le milieu est alors traité par
addition de 20 ml d'eau puis extraction 3 fois avec 50 ml d'acétate d'éthyle.
Les
phases organiques sont ensuite regroupées et lavées 3 fois avec 50 ml d'eau.
La
phase organique est ensuite séchée sur sulfate de magnésium, puis concentrée
sous pression réduite. Le produit brut obtenu est ensuite purifié par
chromatographie sur gel de silice en éluant à l'aide d'un mélange
dichlorométhane/acétate d'éthyle ( 95/5 puis 90/10 ; v/v). On obtient ainsi le
produit attendu sous forme d'une mousse blanche (rendement = 30 %).
RMN iH (DMSO, 300 MHZ) F : 8.09 (t, 1H, NhCO), 7.68 (d, 2H, Harom.)r
7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.). 7.22 (t, 1H, Harom.)i 7.11 (d, 1H,
Harom.)i
7.01 (t, 1H, Harom.)r 6.82 (d, 2H, Harom.)i 6.47 (d, 2H, Harom.)i 4.84 (S, 2H,
PhNHz),
4.71 (dd, 1H, NChCO), 3.23 (m, 2H, CHzNCO), 3.04 (dd, 1H, Cf~CHCO), 2.85
(dd, 1H, Cf~CHCO), 2.53 (t, 2H, CH~Ph), 1.25 (s, 9H, tBu).
MS (ESI+) m/z478 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
31
Exemple 42
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-[2-(4-
hydroxyphényl)éthyl]-2S-lffindole-2-carboxamide
On prépare une solution de 100 mg (0,278 mmol) de l'acide obtenu selon
la préparation IV dans 4 ml de dichlorométhane et 4 ml de THF puis on ajoute
64 mg (0,334 mmol) de EDCI (chlorhydrate de 1-(3-diméthylaminopropyl)-3
éthylcarbodümide) et 7,8 mg (0,028 mmol) de HOAT. Après 10 minutes sous
agitation à température ambiante, on ajoute 45,8 mg (0,334 mmol) de 4-(2
aminoéthyl)phénol, ainsi que 4 ml de mélange dichlorométhane/THF (50/50) et
58 p.l (0,682 mmol) de triéthylamine. Le milieu réactionnel est ensuite agité
à
température ambiante pendant 2 heures. Le milieu est alors traité par addition
de 20 ml d'eau puis extraction 3 fois avec 50 ml d'acétate d'éthyle. Les
phases
organiques sont ensuite regroupées et lavées 3 fois avec 50 ml d'eau. La phase
organique est ensuite séchée sur sulfate de magnésium, puis concentrée sous
pression réduite. Le produit brut obtenu est ensuite purifié par
chromatographie
sur gel de silice en éluant à l'aide d'un mélange dichlorométhane/acétate
d'éthyle ( 95/5 puis 90/10 ; v/v). On obtient ainsi le produit attendu sous
forme
d'un solide blanc (rendement = 40 %).
RMN iH (DMSO, 300 MHz) 8 : 9.15 (s, 1H, PhOf~, 8.12 (t, 1H, NhCO),
7.69 (d, 2H, Harom.). 7.55 (d, 2H, Harom.)i 7.45 (d, 1H, Harom.)i 7.22 (t, 1H,
Harom.)i
7.10 (d, 1H, Harom.)i 7.04-6.95 (m, 3H, Harom.)i 6.65 (d, 2H, Harom.). 4.71
(dd, 1H,
NChCO), 3.25 (m, 2H, Cf~NCO), 3.04 (dd, 1H, CH~CHCO), 2.84 (dd, 1H,
Cf~CHCO), 2.60 (t, 2H, CHZPh), 1.25 (s, 9H, tBu).
MS (ESI+) m/z479 (MH+).
Pré~uaration V
Acide 2,3-dihydro-2-[[(2-phényléthyl)amino]carbonyl]-(2S)-lffindole-1-
carboxylique, 1,1-diméthyléthyl ester
On conditionne pendant 30 mn à température ambiante 564 mg
(0,75 mmol) de résine DCC (cyclohexylcarbodümide) dans 8 ml de
dichlorométhane. On élimine le solvant par filtration, puis ajoute 8 ml de
dichlorométhane, 132 mg (0,5 mmol) d'ester 1-(1,1-diméthyléthylique) de
l'acide
2,3-dihydro-(2S)iH-indole-1,2-dicarboxylique, 10 mg de HOAT, puis 94 pl
(0,75 mmol) de 2-phényléthanamine . Le mélange réactionnel est agité pendant
16 heures à température ambiante, puis on ajoute 1 g de résine IR 120 ; Le
mélange est à nouveau agité pendant 3 heures, puis les résines sont éliminées
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
32
par filtration. Le filtrat est ensuite agité pendant 2 heures en présence de 1
g de
résine IRA 400. La résine est éliminée par filtration et le filtrat est
concentré sous
pression réduite. On obtient ainsi l'amide attendue sous forme d'une huile
incolore (rendement = 88%).
RMN iH (DMS~, 300 MHZ) 8 : 8.14 (s, 1H, NhCO), 7.72 (s, 1H, Harom.).
7.31-7.12 (m, 7H, Harom.)~ 7.89 (t, 1H, Harom.)i '4.68 (dd, 1H, NChCO), 3.44-
3.33
(m, 2H, CHzCHCO, CHzNCO), 3.23 (m, 1H, Cf~NCO), 2.82-2.71 (m, 3H, CHzPh,
CHaCHCO), 1.42 (s, 9H, tBu).
MS (ESI+) m/z367 (MH+).
Préparation VI
2,3-dihydro-N-(2-phényléthyl)-(2S)-Iffindole-2-carboxamide
On prépare une solution de 4,3 g (11,7 mmol) d'ester obtenu selon la
préparation V dans 5 ml de dichlorométhane. On ajoute ensuite 4 ml d'acide
trifluoroacétique concentré, puis le milieu réactionnel est agité à
température
ambiante pendant 24h. Le mélange réactionnel est ensuite concentré sous
pression réduite puis est neutralisé jusqu'à pH 8 par addition d'une solution
aqueuse de bicarbonate de sodium à 10%. La solution obtenue est ensuite
extraite 3 fois avec 50 ml d'acétate d'éthyle, puis la phase organique est
lavée 3
fois avec 50 ml d'eau et séchée sur sulfate de magnésium, puis concentrée sous
pression réduite. Le résidu obtenu est ensuite purifié par chromatographie sur
gel de silice en éluant avec un mélange toluène/isopropanol ( 100/5 ; v/v). On
obtient ainsi le composé attendu sous forme d'un solide beige (rendement =
82%).
F = 112-116°C
Exemple 43
1-[[4-(1-méthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(2-phényléthyl)-2S-Iff
indole-2-carboxamide
On conditionne 96 mg (0,38 mmol) de résine morpholine (Résine
morpholinométhyl PS-HL 2% DVB Novabiochem) dans 3 ml de dichlorométhane.
Après élimination du solvant, on ajoute successivement 1 ml d'acétonitrile,
52,4
mg (0,24 mmol) de chlorure de 4(1-méthyléthyl)benzènesulfonyle, 50 mg
(0,187 mmol) du composé obtenu selon la préparation VI, puis 3 mg (0,19
mmol) de fluorure de césium . Le milieu réactionnel est alors agité à
température
ambiante. Après 20h d'agitation, la résine est filtrée et le filtrat est
repris dans 3
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
33
ml de dichlorométhane et traité avec 92 mg (0,24 mmol) de résine polyamine
(Résine polyamine HL Novabiochem). Après 20 heures d'agitation, la résine est
filtrée et le filtrat est repris dans 3 ml de dichlorométhane et traité avec
0,37
mmol de résine IR120 Amberlite. Après 20 heures d'agitation, la résine est
filtrée
et le filtrat est concentré puis séché sous pression réduite. On obtient ainsi
le
produit attendu sous forme d'un solide blanc (rendement = 45%).
RMN iH (DMSO, 300 MHz) 8 : 8.17 (t, 1H, NhCO), 7.68. (d, 2H, Harom.).
7.47-7.40 (m, 3H, Harom.). 7.27-7.17 (m, 6H, Harom.)i 7.10 (d, 1H, Harom.)i
7.01 (t,
1H, Harom.)i 4.71 (dd, 1H, NChCO), 3.34 (m, 2H, CH~NCO), 3.02-2.91 (m, 2H,
CH~CHCO, Cf~CH3)2), 2.82 (dd, 1H, CHzCHCO), 2.73 (t, 2H, CHzPh), 1.16 (d, 6H,
CH(CH3)2).
MS (ESI+) m/z449 (MH+).
En opérant de façon analogue à l'exemple 43, on obtient les composés
suivants
Exemple 44
1-[(3,4-dichlorophényl)sulfonyl]-2,3-dihydro-N-(2-phényléthyl)-2S-Iffindole-2-
carboxamide
Rdt = 46% ; solide blanc ;
RMN iH (DMSO, 300 MHz) cS: 8.25 (t, 1H, NhCO), 8.02 (d, 1H, Harom.). 7.83 (d,
1H, Harom.)i 7.72 (dd, 1H, Harom.)i 7.43 (d, 1H, Harom.)i 7.27-7.14 (m, 7H,
H~rom.)i
7.04 (t, 1H, Harom.)i 4.84 (dd, 1H, NChCO), 3.36 (m, 2H, Cff~NCO), 3.13 (dd,
1H,
CHzCHCO), 2.85 (dd, 1H, CHzCHCO), 2.73 (t, 2H, CHaPh).
Rf = 0,8 (CH~CIZ/AcOEt : 9/1) ;
MS (ESI+) m/z475 (MH+).
Exemple 45
1-[(4-phénylphényl)sulfonyl]-2,3-dihydro-N-(2-phényléthyl)-2S-lEfindole-2-
carboxamide
Rdt = 28% ; solide blanc ;
RMN iH (DMSO, 250 MHz) 8 : 8.22 (t, 1H, NhCO), 7.84 (s, 4H, Harom.)i
7.70 (d, 2H, Harom.)~ 7.51-7.45 (m, 4H, Harom.). 7.27-7.03 (m, 8H, Harom.)i
4.79 (dd,
1H, NC/~CO), 3.36 (m, 2H, CHzNCO), 3.08 (dd, 1H, CHzCHCO), 2.85 (dd, 1H,
CHzCHCO), 2.74 (t, 2H, Cf~Ph).
Rf = 0,6 (CHIC h/AcOEt : 9/1) ;
MS (ESI+) m/z483 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
34
Exemple 46
2,3-dihydro-1-[(4-phénoxyphényl)sulfonyl]-N-(2-phényléthyl)-(2S)-Iffindole-2-
carboxamide
RMN iH (DMSO, 250 MHz) 8 : 8.16 (t,lH) ; 7.75 (d,2H); 7.42-7.46
(m,3H); 7.01-7.30 (im,3H); 4.71 (dd,lH); 3.33 (m,2H); 3.06 (dd,lH); 2.84
(dd,lH); 2.72 (t,2H).
Exemple 47
1-[(3,4-dihydro-2,2-diméthyl-~ffbenzopyran-6-yl)sulfonyl]-2,3-dihydro-N-(2-
phényléthyl)-(2S)-Iffindole-2-carboxamide
RMN 1H (DMSO, 250 MHz) 8 : 8.13 (t,lH) ; 7.54 (d,iH); 7.44(d,lH); 7.25
(dd,iH); 7.15-7.25 (m,6H); 7.12 (d,lH); 7.02 (td,lH); 6,76 (d,lH); 4.68
(dd,lH);
3.33 (m,2H); 3.00(dd,lH); 2.62-2.83 (dd,lH); 2.78 (m,4H); 1.75 (t,2H); 1.26
(d,6H).
Exemple 48
2,3-dihydro-N-(2-phényléthyl)-1-[[1,2,3,4-tétrahydro-2-(trifluoro-acétyl)-7-
isoquinolinyl]sulfonyl]-(2S)-Iffindole-2-carboxamide
RMN iH (DMSO, 250 MHz) b : 8.15(m,lH) ; 7.80 (dd,lH); 7.55 (m,lH);
7.46 (d,lH); 7.34 (d,lH); 7.09-7.32 (m,7H); 7.01(td,lH); 4.65-4.91 (m,3H);
3.78
(t,2H); 3.35 (q,2H); 2.81-3.06 (m,4H); 2.73 (t,2H).
Exemple 49
2,3-dihydro-1-[[4-(1-méthyléthoxy)phényl]sulfonyl]-N-(2-phényl-éthyl)-(2S)-Iff
indole-2-carboxamide
RMN iH (DMSO, 250 MHz) b : 8.15 (t,lH) ; 7.64 (d,lH); 7.44(d,2H); 7.10-
7.26 (m,7H); 6.98-7.05 (m,3H); 4.68 (m,2H); 3.33(m,2H); 3.00(dd,lH); 2.83
(dd,lH); 2.72 (t,2H); 1.24 (d,6H).
En opérant de façon analogue à la préparation V, on obtient les composés
suivants
Préparation VII
Acide octahydro-2-[[(2-phényléthyl)amino]carbonyl]-(2S)-1H-indole-1-
carboxylique, 1,1-diméthyléthyl ester.
Rdt = 48% ; solide blanc ;
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
RMN iH (DMSO, 250 MHz) S : 7.82 (s, 1H, NhCO), 7.32-7.19 (m, 5H,
Harom.)~ 3.99 (dd, 1H, NChCO), 3.64 (m, 1H, ChNCOOtBu), 3.37-3.20 (m, 2H,
CH~NCO), 2.72 (t, 2H, CHzPh), 2.21 (m, 1H, ChCHZCHCO), 1.90-1.11 (m, 19H,
CHzCHzCHzCHzCHCHzCHCO, tBu).
5 MS (ESI+) m/z373 (MH+).
Préparation VIII
2,3-dihydro-N-(2-phényléthyl)-(2S)-Iffindole-2-carboxamide, trifluoroacétate .
On prépare une solution de 182 mg (0,36 mmol) du composé obtenu
10 selon la préparation V dans 1,5 ml de dichlorométhane et on ajoute 1,5 ml
d'acide trifluoroacétique. Le mélange réactionnel est agité à température
ambiante pendant 16 heures puis le dichlorométhane est chassé sous pression
réduite. Le résidu d'évaporation est ensuite repris dans 5 ml d'un mélange eau
acétonitrile (90/10 ; v/v) et la solution obtenue est lyophilisée. On obtient
ainsi le
15 composé attendu sous forme d'un solide blanc (rendement = 93 %).
RMN iH (DMSO, 250 MHz) S : 7.99 (t, iH, NhCO), 7.30-7.15 (m, 5H,
Harom.)i 7.06-6.97 (m, 2H, Harom.), 6.68 (t, 2H, Harom.). 4.40 (S, 1H,
NhCHCO), 4.24
(dd, 1H, NChCO), 3.37-3.25 (m, 3H, Cf~CHCO, CHzNCO), 2.88 (dd, iH,
CH2,CHC0), 2.73 (t, 2H, CHzPh).
20 MS (ESI+) m/z267 (MH+).
En opérant de façon analogue à la préparation VIII, on obtient le
composé suivant
Préparation IX
25 Octahydro-N-(2-phényléthyl)-lff(2S)-indole-2-carboxamide, trifluoro-acétate
Rdt = 91% ; solide blanc ;
RMN 1H (DMSO, 250 MHz), S : 9.57 (s, iH, Nf~+), 8.55 (t, 1H, NhCO),
7.91 (m, 1H, Nff~,+), 7.33-7.18 (m, 5H, Harom.). 4.13 (m, 1H, NChCO), 3.61-
3.48
(m, 2H, ChNH2+, CHzNCO), 3.34 (m, 1H, CHzNCO), 2.79 (td, 2H, CHzPh), 2.23
30 (m, 2H, CHCHzCHCO), 1.76-1.23 (m, 9H, CHZCf~CH2CH2ChCHZCHCO).
MS (ESI+) m/z 273 (MH+).
Exemale 50
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-N-(2-phényl-éthyl)-(2S)-
35 Iffindole-2-carboxamide
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
36
On prépare un mélange de 144 mg (0,39 mmol) du composé obtenu
selon la préparation VIII et de 0,5 g de résine IRA400 dans 2 ml de
dichlorométhane et on maintient ce mélange sous agitation à température
ambiante pendant 3 heures. La résine est ensuite éliminée par filtration et le
filtrat est ajouté à une suspension de 202 mg (0,787 mmol) de résine
morpholine
préalablement conditionnée dans 3 ml de dichlorométhane. On ajoute 101 mg
(0,432 mmol) de chlorure de 4-(1,1-diméthyléthyl)benzènesulfonyle et on agite
le mélange réactionnel à température ambiante pendant 16 heures. La résine est
éliminée et le filtrat est agité pendant 2 heures, à température ambiante,
avec
0,5 g (1,3 mmol) de résine polyamine. Cette résine est ensuite éliminée par
filtration et le filtrat est agité en présence de 0,5 g de résine IR120,
pendant 2
heures à température ambiante. Après séparation de la résine, le filtrat est
concentré sous pression réduite et on obtient ainsi le produit attendu sous
forme
d'un solide brun (rendement = 49%).
RMN iH (DMSO, 300 MHz) ~ : 7.93 (m, 1H, NhCO), 7.66 (d, 2H, Harom.)i
7.54 (d, 2H, Harom.). 7.43 (d, 1H, Harom.)r 7.25-6.99 (m, 8H, Harom.). 4.70
(dd, 1H,
NChCO), 3.35 (m, 2H, CH~NCO), 3.01 (dd, iH, CHz,CHCO), 2.89 (dd, 1H,
CHzCHCO), 2.73 (t, 2H, CHzPh), 1.24 (s, 9H, tBu).
MS (ESI+) m/~463 (MH+).
En opérant de façon analogue à l'exemple 50, au départ du composé
obtenu selon la préparation IX, on obtient le composé suivant
Exemule 51
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-octahydro-N-(2-phényl-éthyl)-2S-Ih~
indole-2-carboxamide
Rdt = 76% ; huile incolore ;
RMN 1H (DMSO, 300 MHz) S : 7.82-7.78 (m, 3H, NhCO, Harom.)i 7.62 (d,
2H, Harom.)i 7.32-7.20 (m, 5H, Harom.). 3.93 (t, 1H, NChCO), 3.60 (m, 1H,
ChNS02), 3.37 (m, 2H, CHzNCO), 2.75 (t, 2H, Cf~Ph), 1.76 -1.10 (m, 20H,
CHzCHzCHzCHzChCHzCHCO, tBu).
MS (ESI+) m/z469 (MH+).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
37
Exemple 52
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-1H-
indol-2-yl]carbonyl]amino]éthoxy]benzdique, méthyl ester
En opérant de façon analogue à l'exemple 2, au départ du composé
obtenu selon la préparation IV et d'ester méthylique de l'acide 4-(2
aminoéthoxy)benzdique, on obtient le composé attendu sous forme d'une huile
incolore. (rendement = 85%).
RMNiH (DMSO, 300 MHz) : 8,40 (t,lH) ; 7,90 (d,2H) ; 7,70 (d,2H) ; 7,55
(d,2H) ; 7,44 (d,iH) ; 7,21 (t,lH) ; 7,15-6,95 (m,4H); 4,79 (dd,lH); 4,12
(t,2H);
3,81 (s,3H); 3,65-3,35 (m,2H) ; 3,11 (dd,iH) ; 2,90 (dd,lH) ; 1,25 (s,9H).
Exemple 53
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iH-
indol-2-yl]carbonyl]amino]éthoxy]benzôique
En opérant de façon analogue à la préparation II, au départ du composé
obtenu selon l'exemple 52 et après purification par chromatographie sur gel de
silice en éluant avec un mélange dichlorométhane/éthanol (98/2 ; v/v), on
obtient le composé attendu sous forme d'un solide blanc (rendement = 33,5%)
RMN1H (DMSO, 300 MHz) : 12,57 (s Iarge,iH) ; 8,40 (t,lH) ; 7,88
(d,2H) ; 7,71 (d,2H) ; 7,55 (d,2H) ; 7,44 (d,lH) ; 7,21 (t,lH); 7,10 (d,lH);
7,05
6,9 (m,3H); 4,79 (dd,iH); 4,11 (t,2H) ; 3,65-3,35 (m,2H) ; 3,11 (dd,lH) ; 2,9
(dd,iH) ; 1,25 (s,9H).
Exemple 54
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iH-
indol-2-yl]carbonyl]amino]éthoxy]benzèneacétique, méthyl ester
En opérant de façon analogue à l'exemple 2, au départ du composé
obtenu selon la préparation IV et d'ester méthylique de l'acide 4-(2
aminoéthoxy)benzèneacétique, on obtient le composé attendu sous forme d'une
huile (rendement = 35%)
RMNiH (DMSO, 300 MHz) : 8,37 (t,iH) ; 7,71 (d,2H) ; 7,55 (d,2H) ; 7,44
(d,iH) ; 7,25-7,05 (m,4H) ; 7,01 (t,iH) ; 6,88 (d,2H); 4,79 (dd,lH); 4,00
(t,2H) ;
3,60 (s,3H); 3,59 (s,2H) ; 3,6-3,3 (m,2H) ; 3,10 (dd,iH) ; 2,90 (dd,lH) ; 1,25
(s,9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
38
Exemple 55
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iH-
indol-2-yl]carbonyl]amino]éthoxy]benzèneacétique
En opérant de façon analogue à la préparation II, au départ du composé
obtenu selon l'exemple 54, on obtient le composé attendu sous forme d'une
huile
(rendement = 81%)
RMN1H (DMSO, 300 MHz) 8: 12,20 (s Iarge,iH); 8,37 (t,lH) ; 7,71 (d,2H)
7,55 (d,2H) ; 7,44 (d,lH) ; 7,25-7,05 (m,4H) ; 7,01 (t,lH); 6,88 (d,2H); 4,80
(dd,lH); 4,00 (t,2H); 3,6-3,3 (m,4H) ; 3,10 (dd,lH) ; 2,90 (dd,lH) ; 1,25
(s,9H).
Préaaration X
1-[(3-cyanophényl)sulfonyl]-2,3-dihydro-N-(2-phényléthyl)-(2S)-1/findole-2-
carboxamide
En opérant de façon analogue à l'exemple 43, au départ de chlorure de 3-
cyanobenzènesulfonyle, on obtient le composé attendu sous forme d'un solide
beige. (rendement = 88%).
F = 50°C
Préparation XI
1-[[3-(aminométhyl)phényl]sulfonyl]-2,3-dihydro-N-(2-phényléthyl)-(2S)-1/f
indole-2-carboxamide
On prépare une solution de 1 g (2,31 mmol) du composé obtenu selon la
préparation X dans 55 ml d'éthanol anhydre et on ajoute 100 mg de Nickel de
Raney. Le mélange est agité sous atmosphère d'hydrogène, à température
ambiante et à pression atmosphérique pendant 8 heures. Le catalyseur est
ensuite éliminé par filtration et le filtrat est concentré sous pression
réduite ; Le
résidu obtenu est purifié par chromatographie sur gel de silice en éluant avec
un
mélange dichlorométhane/méthanol/ammoniaque ( 95/5/0,5 ; v/v/v). On obtient
ainsi le composé attendu sous forme d'un solide beige (rendement = 48%).
RMN1H (DMSO, 300 MHz) &: 8.16 (t,lH) ; 7.76 (s,lH); 7.42-7.60
(m,4H); 7.15-7.30 (m,6H); 7.09 (dd,iH); 7.02 (td,iH); 4.76 (dd,lH); 3.71
(s,2H); 3.34 (m,2H); 2.97 (dd,lH); 2.83 (dd,iH); 2.73 (t,2H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
39
Exemple 56
2,3-dihydro-N-(2-phényléthyl)-1-[[3-[[(trifluoroacétyl)amino]méthyl]
phényl]sulfonyl]-(2S)-Iffindole-2-carboxamide
On dissout 474 mg (1,1 mmol) du composé obtenu selon la préparation
XI dans 7,5 ml de dichlorométhane et on ajoute 300 pl de triéthylamine et 185
pl
d'anhydride trifluoroacétique. Le mélange réactionnel est agité pendant
heures à température ambiante, puis hydrolysé sur 5ml d'eau. La phase
organique est séparée, séchée sur sulfate de magnésium et concentrée sous
pression réduite. Le produit brut est purifié par chromatographie sur gel de
silice
10 en éluant avec un mélange dichlorométhane/acétate d'éthyle ( 85/15; v/v).
On
obtient ainsi le composé attendu sous forme d'un solide blanc(rendement =
80%).
F= 70°C
RMN1H (DMSO, 300 MHz) : 10.00 (s,iH) ; 8.18 (t,iH); 7.75 (s,iH); 7.63
15 (dt,lH); 7.47-7.57 (m,2H); 7.43 (d,lH); 7.15-7.28 (m,6H); 7.09 (dd,iH);
7.01
(td,iH), 4.76 (dd,lH); 4.41 (s,2H); 3.33 (m,2H); 2.98 (dd,iH); 2.84 (dd,lH);
2.72 (t,2H).
Exemale 57
/IE[2-(diméthylamino)-2-phényléthyl]-1-[[4-(1,1-diméthyléthyl)
phényl]sulfonyl]-2,3-dihydro-(2S)- 1/findole-2-carboxamide
On prépare une solution de 100 mg (0,278 mmol) de l'acide obtenu selon la
préparation IV dans 4 ml de dichlorométhane et on ajoute 59 mg (0,306 mmol)
de EDCI (chlorhydrate de 1-(3-diméthylaminopropyl)-3-éthylcarbodümide) et 2,8
mg (0,028 mmol) de HOAT. Après 10 minutes sous agitation à température
ambiante, on ajoute 51 mg (0,31 mmol) de 2-(diméthylamino)-2-
phényléthanamine dissoute dans 1 ml de dichlorométhane. Le milieu réactionnel
est ensuite agité à température ambiante pendant 20 heures. Le milieu est
alors
traité par addition de 20 ml d'eau puis extrait 3 fois avec 50 ml de
dichlorométhane. Les phases organiques sont ensuite regroupées et lavées 3
fois
avec 50 ml d'eau, puis séchées sur sulfate de magnésium, et concentrées sous
pression réduite. Le produit brut obtenu est ensuite purifié par
chromatographie
sur gel de silice en éluant à l'aide d'un mélange dichlorométhane/éthanol
(95/5 ;
v/v). On obtient ainsi le produit attendu sous forme d'une mousse
blanche (rendement = 55 %).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
RMN iH (DMSO, 250 MHz) 8 : 7,9-7,75 (m, NH); 7,68 (d, 2H); 7,54 (d, 2H); 7,42
(dd, 1H); 7,40-7,15 (m, 6H); 7,15-6,95 (m, 2H); 4,76 (dd, iH); 3,75-3,25 (m,
3H); 3,05-2,85 (m, iH); 2,85-2,55 (m, iH); 2,10 (s, 3H); 2,09 (s, 3H); 1,25
(s,
9H).
5
Préaaration XII
N,/IEdiméthyl-4-[2-[[(1,1-diméthyléthoxy)carbonyl]amino]éthoxy]
benzèneacétamide
On prépare une solution de 1,2 g (4,06 mmol) d'acide 4-[2-(Boc-
10 amino)éthoxy]benzèneacétique dans 4 ml de dichlorométhane et puis on ajoute
932 mg (4,87 mmol) de EDCI et 55 mg (0,406 mmol) de HOAT. Après 10
minutes sous agitation à température ambiante, on ajoute 400 mg (4,87 mmol)
de chlorhydrate de diméthylamine. Le milieu réactionnel est ensuite agité à
température ambiante pendant 24 heures. Le milieu est alors traité par
addition
15 de 20 ml d'eau puis extrait 3 fois avec 50 ml de dichlorométhane. Les
phases
organiques sont ensuite regroupées et lavées 3 fois avec 50 ml d'eau, puis
séchées sur sulfate de magnésium, et concentrées sous pression réduite. Le
produit brut obtenu est ensuite purifié par chromatographie sur gel de silice
en
éluant à l'aide d'un mélange dichlorométhane/éthanol (95/5 ; v/v). On obtient
20 ainsi le produit attendu sous forme d'une huile incolore (rendement = 46
%).
RMN 1H (DMSO, 300 MHz) 8 : 7,11 (d, 2H); 7,05-6,9 (m, NH); 6,85 (d, 2H); 3,92
(t, 2H); 3,59 (s, 2H); 3,35-3,20 (m, 2H); 2,97 (s, 3H); 2,81 (s, 3H); 1,38 (s,
9H).
Préaaration XIII
25 4-(2-aminoéthoxy)- N,IIEdiméthyl-benzèneacétamide
On dissout 200 mg (0,62 mmol) du produit obtenu selon la préparation XII dans
5 ml de dichlorométhane, on ajoute 940 pl d'acide trifluoroacétique. On agite
1
heure à température ambiante. On évapore le dichlorométhane et l'acide
trifluoroacétique, on reprend le produit à l'eau, on lave la phase aqueuse
obtenue
30 à l'aide de dichlorométhane. On amène la phase aqueuse à pH alcalin par
ajout
d'une solution saturée de bicarbonate de sodium. On extrait au
dichlorométhane,
lave la phase organique avec un minimum d'eau, sèche sur sulfate de sodium, et
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
41
concentre sous pression réduite. On obtient le produit attendu sous forme d'un
solide jaune pâle avec un rendement de 20%.
RMN iH (DMSO, 250 MHz) 8: 7,11 (d, 2H); 6,85 (d, 2H); 3,88 (t, 2H); 3,59 (s,
2H); 2,97 (s, 3H); 2,85 (t, 2H); 2,81 (s, 3H).
Exemple 58
/IE[2-[4-[2-(diméthylamino)-2-oxoéthyl]phénoxy]éthyl]-1-[[4-(1,1-
diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-(25)-Iffindole-2-carboxamide
En opérant de faon analogue à l'exemple 57, au départ de l'acide obtenu selon
la préparation IV et du produit obtenu selon la préparation XIII, on obtient
le
composé attendu sous forme d'une huile incolore (rendement = 48%).
RMN 1H (DMSO, 250 MHz) 8 : 8,37 (t, NH); 7,72 (d, 2H); 7,54 (d, 2H); 7,45 (d,
1H); 7,21 (t, 1H); 7,2-7,1 (m, 3H); 7,00 (t, 1H); 6,88 (d, 2H); 4,80 (dd, 1H);
4,00 (t, 2H); 3,60 (s, 2H); 3,6-3,35 (m, 2H); 3,11 (dd, 1H); 2,98 (s, 3H);
2,91
(dd, iH); 2,81 (s, 3H); 1,25 (s, 9H).
Préaaration XIV
Acide 4-méthoxy-2,3-dihydro-ih~indole-2-carboxylique, méthyl ester
A une solution de 500 mg (2,44 mmol) d'acide 4-méthoxy-llfindole-2-
carboxylique dans 10 ml de méthanol, on ajoute 119 mg (4,90 mmol) de
tournures de magnésium. Le mélange est agité 3 heures dans un bain à
10°C
puis 1 heure à température ambiante. On additionne 20 ml d'acide chlorhydrique
à 0°C et on agite une heure. On additionne ensuite une solution
d'ammoniaque
3N jusqu'à pH 10 et on extrait 3 fois par 50 ml d'acétate d'éthyle. Les phases
organiques sont séchées sur sulfate de magnésium, filtrées et concentrées sous
pression réduite. Le résidu d'évaporation est purifié par chromatographie sur
colonne de silice en éluant à l'aide d'un mélange dichlorométhane/acétate
d'éthyle (98/2 puis 95-5 ; v/v). On obtient le produit attendu sous forme
d'une
huile marron avec un rendement de 64%.
RMN 1H (DMSO, 250 MHz) 8: 6,91 (t, iH); 6,24-6,20 (m, 2H); 5,97 (d, 1H); 4,41-
4,34 (m; iH); 3,70 (s, 3H); 3,65 (s, 3H); 3,17 (dd, 1H); 3,01 (dd, 1H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
42
Préearation XV '
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-4-méthoxy-2,3-dihydro-1/~-
indole-2-carboxylique, méthyl ester
En opérant de façon analogue à l'exemple 43, au départ du produit obtenu selon
la préparation XIV et du chlorure de 4-(1,1-diméthyléthyl)benzènesulfonyle, on
obtient le composé attendu sous forme d'un solide marron (rendement = 79%).
RMN iH (DMSO, 300 MHz) 8: 7,77 (d, 2H); 7,58 (d, 2H); 7,20 (t, iH); 7,01 (d,
1H); 6,67 (d, 1H); 5,02 (dd, iH); 3,72 (s; 6H); 3,26 (dd, iH); 2,92 (dd, iH);
1,27 (s, 9H).
Préparation XVI
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-4-méthoxy-2,3-dihydro-1ff
indole-2-carboxylique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon la préparation XV, on obtient le composé attendu sous forme d'un solide
noir (rendement = 89 %).
RMN 1H (DMSO, 250 MHz) â: 13,10 (s large, 1H); 7,76 (d, 2H); 7,57 (d, 2H);
7,19 (t, 1H); 6,99 (d, 1H); 6,66 (d, iH); 4,99 (dd, iH); 3,72 (s; 3H); 3,24
(dd,
iH); 2,87 (dd, 1H); 1,25 (s, 9H).
Exemple 59
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-4-méthoxy-/I~(2-phényl éthyl)-2,3-
dihydro-iH )indole-2-carboxamide
En opérant de façon analogue à la préparation V, au départ du produit obtenu
selon la préparation XVI et de la benzèneéthanamine, on obtient le composé
attendu sous forme d'un solide blanc (rendement = 72 %).
F = 188-192 °C.
RMN iH (DMSO, 250 MHz) 8: 8,15 (t, iH); 7,70 (d, 2H); 7,56 (d, 2H); 7,28-7,16
(m, 6H); 7,08 (d, iH); 6,68 (d, iH); 4,72 (dd, iH); 3,72 (s; 3H); 3,35-3,29
(m,
2H); 2,92 (dd, 1H); 2,76-2,65 (m, 3H); 1,25 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
43
Préparation XVII
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-5-fluoro-2,3-dihydro-ih~indole-
2-
carboxylique, méthyl ester
En opérant de façon analogue à la préparation XV, au départ de l'ester
méthylique de l'acide 2,3-dihydro-5-fluoro-iffindole-2-carboxylique, on
obtient le
composé attendu sous forme d'un solide rose (rendement = 72 %).
RMN iH (DMSO, 300 MHz) 8: 7,74 (d, 2H); 7,58 (d, 2H); 7,40-7,35 (m, iH);
7,07-7,02 (m, 2H); 5,05 (dd, iH); 3,72 (s; 3H); 3,26 (dd, 1H); 3,06 (dd, iH);
1,26 (s, 9H).
Préparation XVIII
Acide 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-5-fluoro-2,3-dihydro-iffindole-
2-
carboxylique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon la préparation XVII, on obtient le composé attendu sous forme d'un
solide
rouge (rendement = 84 %).
RMN 1H (DMSO, 250 MHz) 6: 13,10 (s large, iH); 7,74 (d, 2H); 7,56 (d, 2H);
7,38-7,33 (m, 1H); 7,07-7,00 (m, 2H); 4,91 (dd, iH); 3,28 (dd, 1H); 3,02 (dd,
iH); 1,25 (s, 9H).
Exemule 60
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-5-fluoro-ll~(2-phényléthyl)-2,3-
dihydro-
llfindole-2-carboxamide
En opérant de façon analogue à la préparation V, au départ du produit obtenu
selon la préparation XVIII et de la benzèneethanamine, on obtient le composé
attendu sous forme d'un solide jaune (rendement = 75 %).
F = 168-172 °C.
RMN 1H (DMSO, 300 MHz) b: 8,19 (t, 1H); 7,66 (d, 2H); 7,56 (d, 2H); 7,47-7,43
(m, iH); 7,26-6,98 (m, 7H); 4,72 (dd, 1H); 3,37-3,27 (m, 2H); 2,93 (dd, 1H);
2,82 (dd, 1H); 2,72 (t, 2H); 1,25 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
44
Préparation XIX
Chlorure de 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-(2,~-1ff
indole-2-carbonyle
A une solution de 20 g (55,6 mmol) de l'acide obtenu selon la préparation IV
dans 150 ml de toluène, on ajoute goutte à goutte 14,11 g (111 mmol) de
chlorure d'oxalyle, puis on agite une heure à température ambiante. On évapore
le solvant, puis on reprend le produit dans 100m1 de toluène et on chasse à
nouveau le solvant sous pression réduite. On obtient le produit attendu sous
forme d'un solide beige avec un rendement de 95%.
F = 148 °C.
Exemple 61
4-[2-[[[(25~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-ir~indol-2-
yl]carbonyl]amino]éthoxy]-N,/IEdiméthyl-benzeneéthane-
aminium, trifluoroacétate
On prépare une solution de 132mg (1,27 mmol) de N,/I~diméthyl-4-(2-
aminoéthoxy) benzèneéthanamine, bis trifluoroacétate dans 10 ml de
dichlorométhane, on ajoute 200 mg (0,529 mmol) du produit obtenu selon la
préparation XIX et 402 mg (3,96 mmol) de triéthylamine. On agite 16 heures à
température ambiante. On ajoute de l'eau au milieu réactionnel, puis on
extrait
au dichlorométhane. On sèche la phase organique sur sulfate de magnésium et
on évapore les solvants. Le produit brut est purifié une première fois par
chromatographie sur gel de silice en éluant à l'aide d'un mélange
toluène/éthanol/ammoniaque (8/2/0,1 ;v/v/v), puis une seconde fois par
chromatographie en phase inverse en éluant à l'aide d'un mélange
acétonitrile/eau/acide trifluoroacétique (gradient volumétrique de 20/80/1 à
80/20/1). On obtient le produit attendu sous forme d'une huile incolore avec
un
rendement de 26%.
RMN iH (DMSO, 250 MHz) ~ : 9,32 (sl, NH); 8,35 (t, NH); 7,71 (dd, 2H); 7,55
(dd, 2H); 7,45 (d, iH); 7,30-7,15 (m, 3H); 7,11 (d, iH); 7,01 (t, 1H); 6,92
(d,
2H); 4,79 (dd, 1H); 3,99 (dd, 2H); 3,6-3,4 (m, 2H); 3,3-3,2 (m, 2H); 3,2-3,0
(m,
1H); 3,0-2,75 (m, 3H); 2,83 (s, 3H); 2,81 (s, 3H); 1,25 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Préaaration XX
Acide 4-[2-[[(1,1-diméthyléthoxy)carbonyl]amino]éthoxy]-3-fluoro
benzôique, méthyl ester
5 On mélange 227 mg (1,32 mmol) d'ester méthylique de l'acide 3-fluoro-4-
hydroxybenzôique, 278 mg (1,72 mmol) d'ester 1,1-diméthyléthylique de l'acide
(2-hydroxyéthyl)carbamique, 452 mg (2,12 mmol) de triphénylphosphine dans 10
ml de toluène anhydre. On refroidit à - 10 °C puis on ajoute lentement
349 mg
(1,72 mmol) de düsopropyl-azodicarboxylate (DIAD). On agite à -10 °C
pendant
10 15 minutes puis 16 heures à température ambiante. On évapore les solvants
puis
on purifie le résidu d'évaporation par chromatographie sur colonne de silice
en
éluant à l'aide d'un mélange toluène/acétate d'éthyle (100/0 pendant 30
minutes
puis 80/20 ; v/v). On obtient le produit attendu sous forme d'un solide blanc
avec
un rendement de 93%.
15 RMN iH (DMSO, 250 MHz) 8 : 7,78-7,66 (m, 2H); 7,30 (t, 1H); 7,02 (t, 1H);
4,14
(t, 2H); 3,82 (s, 3H); 3,32 (q, 2H); 1,37 (s, 9H).
Préparation XXI
Acide 4-(2-aminoéthoxy)-3-fluorobenzdique, méthyl ester
20 On dissout 387 mg (1,235 mmol) du produit obtenu selon la préparation XX
dans
2 ml de dichlorométhane puis on ajoute 1 ml d'acide trifluoroacétique et on
agite
16 heures à température ambiante. On évapore les solvants, on ajoute de
l'acétate d'éthyle et on lave la phase organique par une solution de carbonate
de
sodium à 10 %. La phase organique est ensuite séchée sur sulfate de
25 magnésium, filtrée et concentrée sous pression réduite. On obtient le
produit
attendu sous forme d'un solide blanc avec un rendement de 89 %.
RMN 1H (DMSO, 300 MHz) ~ : 7,78 (d, 1H); 7,70 (d, iH); 7,29 (t, 1H); 4,09 (t,
2H); 3,82 (s, 3H); 2,91 (t, 2H); NH2 non visibles
30 Exemple 62
Acide 4-[2-[[[(2.~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff-
indol-2-yl]carbonyl]amino]éthoxy]-3-fluorobenzdique, méthyl ester
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
46
On dissout 230 mg (1,079 mmol) du produit obtenu selon la préparation XXI
dans 10 ml de dichlorométhane anhydre puis on ajoute sous argon 408 mg
(1,079 mmol) de chlorure de 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-
dihydro-(2S)-lffindole-2-carbonyle et 180,5 pl (1,29 mmol) de triéthylamine.
On
agite 16 heures à température ambiante. On évapore le dichlorométhane sous
pression réduite, on reprend le résidu d'évaporation en solution dans
l'acétate
d'éthyle et on lave la phase organique à l'eau. On sèche la phase organique
sur
sulfate de magnésium, on filtre, on évapore. Le résidu d'évaporation est
purifié
par chromatographie sur colonne de silice en éluant à l'aide d'un mélange
toluène/isopropanol (95/5 ; v/v). On obtient le produit attendu sous forme
d'un
solide blanc, avec un rendement de 46%.
RMN iH (DMSO, 250 MHz) 8 : 8,42 (t, 1H); 7,80-7,67 (m, 4H); 7,54 (d, 2H); 7,44
(d, 1H); 7,32 (t, 1H); 7,20 (t, 1H); 7,10 (d, 1H); 6,99 (t, 1H); 4,78 (dd,
1H); 4,21
(t, 2H); 3,82 (s, 3H); 3,60-3,48 (m, 2H); 3,11 (dd, iH); 2,90 (dd, 1H); 1,24
(s,
9H).
Exemule 63
Acide 4-[2-[[[(25~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff
indol-2-yl]carbonyl]amino]éthoxy]-3-fluorobenzdique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 62, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 79 %).
RMN 1H (DMSO, 300 MHz) ~: 12,95 (s large, iH); 8,43 (t, iH); 7,76-7,64 (m,
4H); 7,56 (d, 2H); 7,44 (d, 1H); 7,29 (t, 1H); 7,21 (t, 1H); 7,10 (d, 1H);
6,99 (t,
iH); 4,78 (dd, iH); 4,20 (t, 2H); 3,60-3,47 (m, 2H); 3,11 (dd, 1H); 2,90 (dd,
1H); 1,26 (s, 9H).
Préparation XXII
Acide 4-[2-[[(1,1-diméthyléthoxy)carbonyl]amino]éthoxy]-3-fluoro
benzèneacétique, méthyl ester
En opérant de façon analogue à la préparation XX, au départ de l'ester
méthylique de l'acide 3-fluoro-4-hydroxybenzèneacétique, on obtient le composé
attendu sous forme d'un solide blanc (rendement = 92 %).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
47
RMN iH (DMSO, 300 MHz) 8: 7,18-6,98 (m, 4H); 4,01 (t, 2H); 3,62 (s, 2H); 3,60
(s, 3H); 3,29 (q, 2H); 1,37 (s, 9H).
Préparation XXIII
Acide 4-(2-aminoéthoxy)-3-fluorobenzèneacétique, méthyl ester
En opérant de façon analogue à la préparation XXI, au départ du produit obtenu
selon la préparation XXII, on obtient le composé attendu sous forme d'une
huile
incolore(rendement = 91 %).
RMN iH (DMSO, 300 MHz) 8: 7,16-6,99 (m, 3H); 4,35 (s large, 2H); 4,03 (t, 2H);
3,63 (s, 2H); 3,61 (s, 3H); 2,99 (t, 2H).
Exemale 64
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff-
indol-2-yl]carbonyl]amino]éthoxy]-3-fluoro-benzèneacétique, méthyl ester
En opérant de façon analogue à l'exemple 62, au départ du produit obtenu selon
la préparation XXIII, on obtient le composé attendu sous forme d'une pâte
beige
(rendement = 61 %).
RMN iH (DMSO, 300 MHz) ~: 8,40 (t, 1H); 7,71 (d, 2H); 7,55 (d, 2H); 7,44
(d,lH); 7,21 (t, 1H); 7,16-6,98 (m, 5H); 4,81 (dd; 1H); 4,08 (t, 2H); 3,63 (s,
2H); 3,61 (s, 3H); 3,58-3,44 (m, 2H); 3,11 (dd, 1H); 2,90 (dd, iH); 1,25 (s,
9H).
Exemple 65
Acide 4-[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff-
indol-2-yl]carbonyl]amino]éthoxy]-3-fluoro-benzèneacétique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 64, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 34 %).
RMN iH (DMSO, 300 MHz) â: 12,40 (s large, 1H); 8,40 (t, 1H); 7,71 (d, 2H);
7,55
(d, 2H); 7,44 (d,lH); 7,21 (t, 1H); 7,16-6,98 (m, 5H); 4,79 (dd; iH); 4,08 (t,
2H); 3,63-3,40 (m, 4H); 3,11 (dd, 1H); 2,90 (dd, iH); 1,25 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
48
Exemple 66
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-/I~[(3-hydroxyphényl) méthyl]-2,3-
dihydro-(25)-lffindole-2-carboxamide
A une suspension de 42,26 mg (0,265 mmol) de chlorhydrate de 3-
(aminométhyl)phénol dans 8 ml de dichlorométhane à température ambiante, on
additionne 54 mg (0,53 mmol) de triéthylamine, puis on ajoute une solution de
100 mg (0,265 mmol) de chlorure de 1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-
2,3-dihydro-(2,~-1/findole-2-carbonyle dans 2 ml de dichlorométhane en 10
minutes. On laisse agiter à température ambiante pendant 1 heure. On lave le
milieu réactionnel à l'eau, on sèche sur sulfate de magnésium, on évapore les
solvants et on purifie le résidu d'évaporation par chromatographie sur colonne
de
silice en éluant à l'aide d'un mélange toluène/acétate d'éthyle (8/2 ; v/v).
On
obtient le produit attendu sous forme d'un solide blanc avec un rendement de
51%.
F=94°C.
Exemale 67
1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-/If-[(4-chlorophényl)méthyl] -2,3-
dihydro-(25)-iffindole-2-carboxamide
En opérant de façon analogue à l'exemple 66, au départ de chlorhydrate de 4-
chloro- benzèneméthanamine, on obtient le composé attendu sous forme d'un
solide blanc (rendement = 98%).
F = 69 °C.
Exemple 68
Acide 4-[[[[(25)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-
lffindol-
2-yl]carbonyl]amino]méthyl]benzdique, méthyl ester
En opérant de façon analogue à l'exemple 66, au départ du chlorhydrate de
l'ester méthylique de l'acide 4-(aminométhyl)benzôique, on obtient le composé
attendu sous forme d'un solide blanc (rendement = 80%).
F = 190 °C.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
49
Exemple 69
Acide 4-[[[[(2~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iffindol-
2-yl]carbonyl]amino]méthyl]benzôique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 68, on obtient le composé attendu sous forme d'un solide blanc
(rendement quantitatif).
F = 105-120 °C.
Exemple 70
Acide 4-[[[[(25~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-
lffindol-
2-yl]carbonyl]amino]méthyl]benzènepropandique, méthyl ester
En opérant de façon analogue à l'exemple 66, au départ du chlorhydrate de
l'ester méthylique de l'acide (4-aminométhyl)benzènepropandique, on obtient le
composé attendu sous forme d'un solide blanc (rendement = 89%).
F = 56 °C.
Exemple 71
Acide 4-[[[[(2.5)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-
iffindol-
2-yl]carbonyl]amino]méthyl]benzènepropanôique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 70, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 97%).
F = 80-85 °C.
Exemale 72
Acide 4-[[[[(25)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-
lffindol-
2-yl]carbonyl]amino]méthyl]benzèneacétique, méthyl ester
En opérant de façon analogue à l'exemple 67, au départ du chlorhydrate de
l'ester méthylique de l'acide (4-aminométhyl)benzèneacétique, on obtient le
composé attendu sous forme d'une huile incolore (rendement = 89 %).
RMN iH (DMSO, 300 MHz) â: 8,69 (t, NH); 7,72 (d, 2H); 7,56 (d, 2H); 7,46 (d,
iH); 7,3-7,15 (m, 5H); 7,13 (d, 1H); 7,01 (t, iH); 4,81 (dd, 1H); 4,4-4,2 (m,
2H); 3,65 (s, 2H); 3,60 (s, 3H); 3,13 (dd, iH); 2,94 (dd, iH); 1,25 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
S0
Exemple 73
Acide 4-[[[[(25)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-
iffindol-
2-yl]carbonyl]amino]méthyl]benzèneacétique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 72, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 94 %).
F = 80 - 85 °C.
Préparation X70CIV
Acide (25)-1-[(3,4-dihydro-2,2-diméthyl-2/f1-benzopyran-6-yl)
sulfonyl]-2,3-dihydro-lffindole-2-carboxylique, méthyl ester
En opérant de façon analogue à la préparation I, au départ du chlorhydrate de
l'ester méthylique de l'acide (2S)-2,3-dihydro-1/findole-2-carboxylique et du
chlorure de 3,4-dihydro-2,2-diméthyl-2h~1-benzopyran-6-yl-sulfonyle, on
obtient
le composé attendu sous forme d'une huile jaune (rendement = 93 %).
RMN 1H (DMSO, 300 MHz) 8: 7,62 (d, 1H); 7,46 (dd, iH); 7,36 (d, 1H); 7,25-
7,13 (m, 2H); 6,99 (t, 1H); 6,78 (d, 1H); 4,99 (dd, 1H); 3,71.(s, 3H); 3,33
(dd,
1H); 3,04 (dd, 1H); 2,83-2,66 (m, 2H); 1,75 (t, 2H); 1,25 (d, 6H).
Préparation )O(XV
Acide (2,5~-1-[(3,4-dihydro-2,2-diméthyl-2ff1-benzopyran-6-yl)
sulfonyl]-2,3-dihydro-lffindole-2-carboxylique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon la préparation ?OOCIV, on obtient le composé attendu sous forme d'un
solide blanc (rendement = 91 %).
RMN iH (DMSO, 300 MHz) â: 13,05 (s Iarge,lH); 7,62 (s, 1H); 7,46 (dd, iH);
7,34 (d, iH); 7,23-7,13 (m, 2H); 7,00 (t, iH); 6,78 (d, 1H); 4,85 (dd, 1H);
3,31
(dd, 1H); 3,00 (dd, iH); 2,83-2,66 (m, 2H); 1,75 (t, 2H); 1,25 (d, 6H).
Exemale 74
Acide 4-[2-[[[(25~-1-[(3,4-dihydro-2,2-diméthyl-2ff1-benzopyran-6-yl)sulfonyl]-
2,3-dihydro-lffindol-2-yl]carbonyl]amino]éthoxy]
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
51
benzoïque, méthyl ester
En opérant de façon analogue à la préparation V, au départ de l'acide obtenu
selon la préparation XXXV et de l'ester méthylique de l'acide 4-(2-
aminoéthoxy)benzoïque, on obtient le composé attendu sous forme d'un solide
blanc (rendement = 82 %).
RMN iH (DMSO, 300 MHz) 8: 8,34 (t,lH); 7,89 (d, 2H); 7,55 (d, 1H); 7,45-7,36
(m, 2H); 7,20 (t, 1H); 7,11-6,95 (m, 4H); 6,76 (d, 1H); 4,75 (dd, iH); 4,11
(t,
2H); 3,81 (s, 3H); 3,60-3,34 (m, 2H); 3,06 (dd, iH); 2,88 (dd, 1H); 2,90-2,63
(m, 2H); 1,74 (t, 2H); 1,25 (d, 6H).
Exemale 75
Acide 4-[2-[[[(2.~-1-[(3,4-dihydro-2,2-diméthyl-2ff1-benzopyran-6-yl)sulfonyl]-
2,3-dihydro-lffindol-2-yl]carbonyl]amino]éthoxy] benzdique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 74, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 32 %).
RMN iH (DMSO, 300 MHz) 8: 12,57 (s large, 1H); 8,34 (t,lH); 7,87 (d, 2H); 7,55
(d, 1H); 7,45-7,36 (m, 2H); 7,21 (t, iH); 7,22 (d, iH); 7,03-6,98 (m, 3H);
6,75
(d, 1H); 4,75 (dd, 1H); 4,10 (t, 2H); 3,60-3,34 (m, 2H); 3,03 (dd, 1H); 2,89
(dd,
1H); 2,84-2,58 (m, 2H); 1,74 (t, 2H); 1,25 (d, 6H).
Préaaration )OOCVI
Chlorhydrate de l'acide 3-[3-(aminométhyl)phénoxy]propandique, méthyl ester
On dissout 0,8 g (3,9 mmol) du chlorhydrate de l'ester méthylique de l'acide 3-
(3-cyanophénoxy)propandique dans 20 ml de méthanol. On additionne 0,4 ml~
d'acide chlorhydrique 10 N puis, sous atmosphère inerte, 100 mg de palladium
sur charbon. Le mélange est agité sous atmosphère d'hydrogène à température
ambiante et pression atmosphérique. On élimine le catalyseur par filtration,
puis
on concentre le filtrat sous pression réduite. On reprend le résidu au toluène
et
on évapore à nouveau. On recristallise le produit dans un mélange
méthanol/éther éthylique (10/50 ; v/v), et on sèche sous vide. On obtient le
produit attendu sous forme d'un solide blanc avec un rendement de 84%.
F = 134 °C.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
S2
Exemple 76
Acide 3-[3-[[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-1fE
indol-2-yl]carbonyl]amino]méthyl]phénoxy]propandique, méthyl ester
En opérant de façon analogue à l'exemple 66, au départ du produit obtenu selon
la préparation )OCXVI, on obtient le composé attendu sous forme d'un solide
blanc (rendement = 97%).
F = 103 °C.
Exemple 77
Acide 3-[3-[[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff
indol-2-yl]carbonyl]amino]méthyl]phénoxy]propandique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 76, on obtient le composé attendu sous forme d'une mousse
blanche (rendement = 33%).
F = 122 °C.
RMN iH (DMSO, 250 MHz) ~: 5,69 (t, NH); 7,72 (d, 2H); 7,55 (d, 2H); 7,46 (d,
1H); 7,3-7,1 (m, 3H); 7,02 (t, iH); 6,9-6,7 (m, 3H); 4,52 (dd, 1H); 4,4-4,2
(m,
2H); 4,12 (t, 2H); 3,13 (dd, iH); 2,93 (dd, 1H); 2,56 (t, 2H); 1,25 (s, 9H).
Préparation X)OCVII
Acide (2,5)-2,3-dihydro-1-[[4-(1-méthyléthoxy)phényl]sulfonyl]-iffindol-2-
carboxylique, méthyl ester
En opérant de façon analogue à la préparation I, au départ de l'ester
méthylique
de l'acide (2~-2,3-dihydro-lffindole-2-carboxylique et du chlorure de [4-(1
méthyléthoxy)phényl]sulfonyle, on obtient le composé attendu sous forme d'un
solide blanc (rendement = 76 %).
RMN iH (DMSO, 300 MHz) 8: 7,73 (d, 2H); 7,36 (d, 1H); 7,22-7,13 (m, 2H);
7,05-6,97 (m, 3H); 4,99 (dd, iH); 4,69 (hep, 1H); 3,72 (s, 3H); 3,32 (dd, iH);
3,04 (dd, 1H); 1,24 (d, 6H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
53
Préparation X)O(VIII
Acide (2.5)-2,3-dihydro-1-[[4-(1-méthyléthoxy)phényl]sulfonyl]-iffindol-2-
carboxylique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon la préparation )OOCVII, on obtient le composé attendu sous forme d'un
solide blanc (rendement = 98 %).
RMN 1H (DMSO, 300 MHz) â: 13,11 (s large, 1H); 7,72 (d, 2H); 7,35 (d, 1H);
7,22-7,12 (m, 2H); 7,02-6,95 (m, 3H); 4,84 (dd, 1H); 4,68 (hep, iH); 3,31 (dd,
1H); 3,01 (dd, 1H); 1,24 (d, 6H).
Exemple 78
Acide 4-[2-[[[(2.~-2,3-dihydro-1-[[4-[(1-méthyléthoxy)phényl] sulfonyl]-
iffindol-
2-yl]carbonyl]amino]éthoxy]benzèneacétique
On mélange 50 mg (0,139 mmol) d'acide obtenu selon la préparation )OOCVIII et
1 ml de dichlorométhane. On agite à 0°C et on additionne 42 pl de
triéthylamine
et 22 pl (0,3 mmol) de chlorure de thionyle. On agite 30 minutes à 0
°C. On
laisse la température du milieu réactionnel remonter à température ambiante et
on évapore les solvants. On additionne sur le résidu d'évaporation une
suspension de 30 mg d'acide 4-(2-aminoéthoxy)benzèneacétique dans 43 pl de
triéthylamine et 2 ml de dichlorométhane, puis on agite une nuit à température
ambiante. On reprend le milieu réactionnel dans 10 ml de dichlorométhane, et
on
lave avec de l'eau. La phase organique est séchée sur sulfate de sodium,
filtrée
et concentrée sous pression réduite. Le résidu d'évaporation est purifié par
chromatographie sur colonne de silice en éluant à l'aide d'un mélange
dichlorométhane/méthanol (95/5 ; v/v). On obtient le produit attendu sous
forme
d'un solide blanc avec un rendement de 21 %.
RMN iH (DMSO, 250 MHz) &: 12,25 (s large, 1H); 8,33 (t, iH); 7,66 (d, 2H);
7,44
(d, iH); 7,23-6,86 (m, 9H); 4,79-4,63 (m, 2H); 3,99 (t, 2H); 3,55-3,35 (m,
4H);
3,07 (dd, 1H); 2,90 (dd, 1H); 1,24 (d, 6H)
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
54
Préparation XXXIX
Acide 7-[[(phénylméthoxy)carbonyl]amino]-iffindole-2-carboxylique, éthyl ester
On mélange 0,8 g (3,925 mmol) d'ester éthylique de l'acide 7-amino-1/findole-2-
carboxylique avec 16 ml de tétrahydrofurane et 1,08 g (7,83 mmol) de carbonate
de potassium. Le milieu réactionnel est refroidi à 0 °C et on ajoute
0,615 ml
(4,30 mmol) de chloroformate de benzyle. On agite 1 heure 30 à 0 °C,
puis on
laisse la température du mélange remonter à température ambiante pendant une
heure. On ajoute ensuite 30 ml d'eau et on extrait le produit par de l'acétate
d'éthyle. Les phases organiques réunies sont séchées sur sulfate de magnésium,
filtrées et concentrées sous pression réduite. Le résidu d'évaporation est
purifié
par chromatographie sur colonne de silice en éluant à l'aide d'un mélange
méthylcyclohexane/acétate d'éthyle (90/1 ; v/v). On obtient le produit attendu
sous forme de cristaux blancs avec un rendement de 61 %.
RMN 1H (DMSO, 250 MHz) 8: 11,67 (s large, 1H); 9,68 (s large, 1H); 7,82
(d,iH); 7,49-7,34 (m, 6H); 7,16 (s, 1H); 7,05 (t, 1H); 5,21 (s, 2H); 4,35 (q,
2H);
1,34 (t, 3H).
Préparation XL
Acide 7-[[(phénylméthoxy)carbonyl]amino]-2,3-dihydro-1/findole-2-
carboxylique, méthyl ester
En opérant de façon analogue à la préparation XIV, au départ du produit obtenu
selon la préparation XXXIX, on obtient le composé attendu sous forme d'un
solide marron (rendement = 59 %).
RMN iH (DMSO, 250 MHz) S: 9,07 (s large, iH); 7,47-7,31 (m, 5H); 7,28 (d, iH);
6,81 (d, iH); 6,59 (t, iH); 5,78 (d, 1H); 5,14 (s, 2H); 4,48-4,41 (m, iH);
3,67 (s,
3H); 3,32 (dd, 1H); 3,14 (dd, 1H).
Préparation XLI
Acide 1-[[4-(diméthyléthyl)phényl]sulfonyl]-7-[[(phénylméthoxy)
carbonyl]amino]-2,3-dihydro-iffindole-2-carboxylique, méthyl ester
En opérant de façon analogue à la préparation I, au départ du produit obtenu
selon la préparation XL, on obtient le composé attendu sous forme d'une pâte
jaune (rendement = 71 %).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
SS
RMN 1H (DMSO, 250 MHz) b: 8,86 (s, iH); 7,76 (d, iH); 7,79-7,32 (m, 9H); 7,16
(t, iH); 6,86 (d, iH); 5,20-5,15 (m, 3H); 3,64 (s, 3H); 2,86 (d, iH); 2,36
(dd,
1H); 1,24 (s, 9H).
Préparation XLII
Acide 1-[[4-(diméthyléthyl)phényl]sulfonyl]-7-[[(phénylméthoxy)
carbonyl]amino]-2,3-dihydro-iffindole-2-carboxylique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon la préparation XLI, on obtient le composé attendu sous forme d'un solide
blanc (rendement = 84 %).
RMN 1H (DMSO, 250 MHz) 8: 13,13 (s large, iH); 8,92 (s, 1H); 7,77 (d, 1H);
7,72-7,35 (m, 9H); 7,15 (t, 1H); 6,85 (d, iH); 5,20 (s, 2H); 5,03 (dd, 1H);
2,83
(d, iH); 2,28 (dd, iH); 1,24 (s, 9H).
Préparation XLIII
1-[[4-(diméthyléthyl)phényl]sulfonyl]-N-(2-phényléthyl)-7-[[(phényl
méthoxy)carbonyl]amino]-2,3-dihydro-iffindole-2-carboxamide
En opérant de façon analogue à l'exemple 57, au départ du produit obtenu selon
la préparation XLII, on obtient le composé attendu sous forme d'un solide
blanc
(rendement = 67 %).
RMN 1H (DMSO, 250 MHz) 8: 9,57 (s, iH); 8,87 (t, iH); 7,63 (d, iH); 7,50-7,31
(m, 9H); 7,16 (t, iH); 7,09-7,06 (m, 3H); 6,97-6,94 (m, 2H); 6,79 (d, iH);
5,22
(s, 2H); 4,68 (d, iH); 3,32-3,20 (m, 2H); 2,77 (d, iH); 2,62 (t, 2H); 2,06
(dd,
iH); 1,25 (s, 9H)
Exemgle 79
7-amino-1-[[4-(diméthyléthyl)phényl]sulfonyl]-N-(2-phényléthyl)-2,3-dihydro-
1/f
indole-2-carboxamide
On mélange 30 mg (0,049 mmol) du produit obtenu selon la préparation XLIII et
3 ml de méthanol. Sous atmosphère protégée, on additionne 3 mg de palladium
sur charbon et le mélange réactionnel est agité sous atmosphère d'hydrogène à
température ambiante et pression atmosphérique pendant 5 heures. Le milieu
réactionnel est filtré, le catalyseur lavé au méthanol et les filtrats sont
concentrés
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
56
sous pression réduite. Le résidu d'évaporation est purifié par chromatographie
sur colonne de silice en éluant à l'aide d'un mélange
méthylcyclohexane/acétate
d'éthyle (75/25 ; v/v). On obtient le produit attendu sous forme d'un solide
blanc
avec un rendement de 83 %.
RMN iH (DMSO, 300 MHz) 8: 8,12 (t, 1H); 7,55-7,49 (m, 4H); 7,22-7,03 (m, 5H);
6,84 (t, iH); 6,59 (d, 1H); 6,22 (d, 1H); 5,49 (s, 2H); 4,67 (d, iH); 3,31-
3,24
(m, 2H); 2,73-2,63 (m, 3H); 2,07 (dd, iH); 1,25 (s, 9H).
Exemple 80
Acide 4-[2-[[[(25)-1-[(3,4-dihydro-2,2-diméthyl-2/fi-benzopyran-6-yl)sulfonyl]-
2,3-dihydro-lffindol-2-yl]carbonyl]amino]éthoxy] benzèneacétique
En opérant de faon analogue à l'exemple 78, au départ de l'acide obtenu selon
la préparation X?OCV et de l'acide 4-(2-aminoéthoxy)benzèneacétique, on
obtient
le composé attendu sous forme d'un solide jaune (rendement = 74 %).
RMN iH (DMSO, 250 MHz) 8: 12,23 (s large, 1H); 8,30 (t,iH); 7,55 (s, iH); 7,45-
7,36 (m, 2H); 7,22-7,09 (m, 4H); 7,00 (t, 1H); 6,87 (m, 2H); 6,76 (d, 1H);
4,76
(dd, 1H); 3,99 (t, 2H); 3,52-3,41 (m, 4H); 3,07-2,62 (m, 4H); 1,75 (t, 2H);
1,25
(d, 6H).
Préparation XLIV
Chlorhydrate de l'acide 4-[(2-aminoéthyl)thio]-3-chloro-benzène
acétique, méthyl ester
On dissout 50 mg (0,144 mmol) d'ester méthylique de l'acide 3-chloro-4-[2
[[(1,1-diméthyléthoxy)carbonyl]amino]éthylthio] benzèneacétique dans 322 pl
d'acide trifluoroacétique. On agite 2 heures à température ambiante. On chasse
l'acide trifluoroacétique par coévaporation avec du méthylcyclohexane. On
dissout le résidu d'évaporation dans de l'acétate d'éthyle, et on neutralise
par
ajout d'une solution de carbonate de sodium à 10 %. On sèche la phase
organique sur sulfate de magnésium, on filtre et on concentre sous pression
réduite. On obtient le produit attendu sous forme d'une huile orange avec un
rendement quantitatif.
RMN 1H (DMSO, 250 MHz) 8: 8,24 (s large, 3H); 7,49-7,43 (m, 2H); 7,27 (d, 1H);
3,71 (s, 2H); 3,62 (s, 3H); 3,32-3,21 (m, 2H); 3,00 (t, 2H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
S7
Exemple 81
Acide 3-chloro-4-[[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl] sulfonyl]-2,3-
dihydro-lffindol-2-yl]carbonyl]amino]éthyl]thio] benzèneacétique, méthyl ester
On dissout 53,6 mg (0,142 mmol) du produit obtenu selon la préparation XLIV
dans 4 ml de dichlorométhane anhydre, puis on ajoute 109 mg (3,42 mmol) de
morpholine supportée sur résine et 40 mg (0,142 mmol) de chlorure de [4-(1,1-
diméthyléthyl)phényl]sulfonyle, on agite 4 jours à température ambiante. On
filtre la résine, on concentre le filtrat sous pression réduite et on obtient
le
produit attendu sous forme d'un solide gris avec un rendement de 95 %.
RMN 1H (DMSO, 250 MHz) 8: 8,46 (t, iH); 7,71 (d, 2H); 7,55 (d, 2H); 7,49-7,46
(m, 2H); 7,38 (s, 1H); 7,26-7,20 (m, 2H); 7,12 (d, iH); 7,01 (t, 1H); 4,73
(dd,
1H); 3,68 (s, 2H); 3,61 (s, 3H); 3,42-3,02 (m, 5H); 2,94 (dd, iH); 1,25 (s,
9H).
Exemale 82
Acide 3-chloro-4-[[2-[[[(2S)-1-[[4-(1,1-diméthyléthyl)phényl) sulfonyl]-2,3-
dihydro-iffindol-2-yl]carbonyl]amino]éthyl]thio] benzèneacétique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 81, 'on obtient le composé attendu sous forme d'un solide
blanc
(rendement = 60 %).
RMN iH (DMSO, 300 MHz) 8: 12,40 (s large, 1H); 8,46 (t, 1H); 7,71 (d, 2H);
7,55
(d, 2H); 7,48-7,45 (m, 2H); 7,37 (s, 1H); 7,26-7,20 (m, 2H); 7,12 (d, iH);
7,01
(t, iH); 4,73 (dd, 1H); 3,56 (s, 2H); 3,42-3,02 (m, 5H); 2,94 (dd, 1H); 1,25
(s,
9H).
Préparation XLV
Acide 3-(4-cyanophénoxy)propandique, méthyl ester
On refroidit par un bain de glace une solution de 3,80 g (19,9 mmol) d'acide 3-
(4-cyanophénoxy)propandique dans 10 ml de méthanol. On additionne goutte à
goutte 3 ml (40 mmol) de chlorure de thionyle, on agite ensuite le milieu
réactionnel à reflux du solvant pendant 2 heures. Le mélange réactionnel est
ensuite concentré sous pression réduite et on purifie le résidu d'évaporation
par
chromatographie sur colonne de silice en éluant à l'aide d'un mélange
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Ss
dichlorométhane/méthanol (99/1 ; v/v). On obtient le produit attendu sous
forme
d'un solide blanc avec un rendement de 79%.
F=51°C.
Préuaration XLVI
Acide 3-[4-(aminométhyl)phénoxy]prôpanoïque, méthyl ester (chlorhydrate)
En opérant de façon analogue à la préparation XXXVI, au départ du produit
obtenu selon la préparation XLV, on obtient le composé attendu sous forme d'un
solide blanc (rendement = 60%).
F=217°C.
Exemple 83
Acide 3-[4-[[[[(25>-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-iff
indol-2-yl]carbonyl]amino]méthyl]phénoxy]propandique, méthyl ester
En opérant de façon analogue à l'exemple 66, au départ du produit obtenu selon
la préparation XLVI, on obtient le composé attendu sous forme d'un solide
blanc
(rendement = 83%).
F = 50-52 °C.
Exemule 84
Acide 3-[4-[[[[(2,5)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-1h~
indol-2-yl]carbonyl]amino]méthyl]phénoxy]propanoïque
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 83, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 41%).
F = 75 °C.
Exemple 85
(2.5)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-/IE[(4-
nitrophényl)méthyl]-lffindole-2-carboxamide
En opérant de façon analogue à l'exemple 66, au départ de 4-(aminométhyl)-
nitrobenzène, on obtient le composé attendu sous forme d'un solide blanc
(rendement = 68%).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
59
F = 75-80 °C.
Exemple 86
(2~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-/I~[(4-
aminophényl)méthyl]-iffindole-2-carboxamide
En opérant de façon analogue à la préparation XXXVI, au départ du produit
obtenu selon l'exemple 85, on obtient le composé attendu sous forme d'un
solide
blanc (rendement = 82%).
F = 83-87 °C
Exemple 87
/I~[4-[[[[(25)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-ih~-indol-
2-
yl]carbonyl]amino]méthyl]phényl]glycine, méthyl ester
On mélange, dans un tube de réaction sous micro-ondes, 400 mg (0,864 mmol)
du produit obtenu selon l'exemple 86, 264 mg (1,728 mmol) de bromoacétate de
méthyle et 120 mg (0,864 mmol) de carbonate de potassium dans 8 ml
d'acétone. Le mélange est chauffé à 130 °C pendant une heure à l'aide
de micro-
ondes. On filtre le milieu réactionnel, puis on concentre le filtrat sous
pression
réduite. Le résidu d'évaporation est purifié par chromatographie sur colonne
de
silice en éluant à l'aide d'un mélange toluène/acétate d'éthyle (95/5 puis
90/10 ;
v/v). On obtient le produit attendu sous forme d'un solide blanc avec une
rendement de 31%.
F = 70 °C.
Exemple 88
/IE[4-[[[[(2,5~-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-ih~indol-
2-
yl]carbonyl]amino]méthyl]phényl]glycine
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 87, on obtient le composé attendu sous forme d'un solide
jaunâtre (rendement = 24%).
F = 110 °C.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Préparation XLVII
Acide 2,3-dihydro-2-[[[2-[4-(2-méthoxy-2-oxoéthyl)phénoxy]éthyl]
amino]carbonyl]-(2,~-iffindole-1-carboxylique, 1,1-diméthyléthyl ester
En opérant de façon analogue à la préparation XII, au départ d'acide 1-[(1,1
5 diméthyléthoxy)carbonyl]-2,3-dihydro-(2S)-lffindole-2-carboxylique, on
obtient
le composé attendu sous forme d'une mousse blanche (rendement = 48 %).
Préparation XLVIII
Acide 4-[2-[[[(2S~-2,3-dihydro-1/findol-2-yl]carbonyl]amino] éthoxy]
10 benzèneacétique, méthyl ester
En opérant de façon analogue à la préparation XXI, au départ du produit obtenu
selon la préparationXLVII, on obtient le composé attendu sous forme d'un
solide
beige (rendement = 89 %).
RMN 1H (DMSO, 250 MHz) &: 8,03 (t, NH) ; 7,16 (dd, 2H) ; 7,05-6,80 (m, 4H) ;
15 6,65-6,5 (m, 2H) ; 5,93 (d, NH) ; 4,22 (ddd, 1H) ; 3,99 (t, 2H) ; 3,59 et
3,58 (2s, 5H) ; 3,55-3,4 (m, 2H) ; 3,31 (dd, iH) ; 2,90 (dd, 1H).
Exemple 89
Acide 4-[2-[[[(2S)-1-[[4-(4-fluorophénoxy)phényl]sulfonyl]-2,3-dihydro-
iffindol-
20 2-yl]carbonyl]amino]éthoxy]benzèneacétique, méthyl ester
On dissout 0,2 g (0,564 mmol) du produit obtenu selon la préparation XLVIII
dans 4 ml de dichlorométhane. On ajoute 0,086 g (0,846 mmol) de triéthylamine
et 0,178 g (0,621 mmol) de chlorure de (4-fluorophénoxy)benzènesulfonyle, on
agite 20 heures à température ambiante. On évapore le dichlorométhane, on
25 purifie le résidu d'évaporation par chromatographie sur colonne de silice
en
éluant à l'aide d'un mélange dichlorométhane/acétate d'éthyle (9/1 ;v/v). On
obtient le produit attendu sous forme d'une huile incolore avec un rendement
de
76%.
RMN iH (DMSO, 300 MHz) 8 : 8,36 (t, NH); 7,77 (d, 2H); 7,43 (d, iH); 7,35-7,1
30 (m, 8H); 7,1-6,95 (m, 3H); 6,88 (d, 2H); 4,79 (dd, iH); 4,00 (t, 2H); 3,60
(s,
5H); 3,65-3,35 (m, 2H); 3,12 (dd, iH); 2,92 (dd, iH).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
61
Exemule 90
Acide 4-[2-[[[(25~-1-[[4-(4-fluorophénoxy)phényl]sulfonyl]-2,3-dihydro-
ilfindol-
2-yl]carbonyl]amino]éthoxy]benzèneacétique
En opérant de façon analogue à la préparation II, au départ du produit obtenu
selon l'exemple 89, on obtient le produit attendu sous forme d'un solide blanc
avec un rendement de 65%.
F = 65 °C.
Préuaration IL:
Acide (25)-2,3-dihydro-1-[(3,4-dihydro-2,2-diméthyl-2ff1-benzopyran-6-
yl)sulfonyl]indole-2-carboxylique
On solubilise 1 g (6,13 mmol) d'acide 2,3-dihydro-(2S)-lffindole-2-
carboxylique,
0,97 g (7,05 mmol) de carbonate de potassium dans 2 ml d'eau. On ajoute 1,6 g
(6,13 mmol) de chlorure de 3,4-dihydro-2,2-diméthyl-2/fi-benzopyran-6-
sulfonyle solubilisé dans 12 ml d'acétonitrile et 2 ml d'eau. On laisse sous
agitation pendant 5 heures à température ambiante. On ajoute de l'eau au
milieu
réactionnel, on lave à l'acétate d'éthyle, on acidifie la phase aqueuse
jusqu'à pH
3 par une solution d'acide chlorhydrique 1 N, puis on extrait à l'acétate
d'éthyle.
Les phases organiques sont rassemblées, séchées sur sulfate de magnésium,
filtrées et concentrées sous pression réduite. Le résidu d'évaporation est
purifié
par chromatographie sur colonne de silice en éluant à l'aide d'un mélange
dichlorométhane/méthanol (9/1 ; v/v). On obtient le produit attendu sous forme
d'une mousse beige avec un rendement de 73%.
RMN 1H (DMSO, 300 MHz) 8: 7,59 (s, iH); 7,44 (m, 1H); 7,31 (m, 1H); 7,20
7,05 (m, 2H); 6,96 (t, iH); 6,75 (d, iH); 7,74 (s large , 1H); 3,1-2,85 (m,
2H);
2,85-2,60 (m, 2H); 1,73 (t, 2H); 1,25 (m, 6H).
Exemple 91
Acide (~3S)-[i-[[[(2,~-1-[(3,4-dihydro-2,2-diméthyl-2ff1-benzopyran-6-
yl)sulfonyl]-2,3-dihydro-l/findol-2-yl]carbonyl]amino] benzène
butanôique, 1,1-diméthyléthyl ester
En opérant de façon analogue à l'exemple 87, au départ du produit obtenu selon
la préparation IL et de l'ester tbutylique de l'acide (~S)-[i-amino-
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
62
benzènebutanôique, on obtient le produit attendu sous forme d'une mousse
blanche avec un rendement de 15 %.
RMN 1H (DMSO, 300 MHz) 8: 8,02 (d, iH); 7,51-7,46 (m, 2H); 7,34 (dd, 1H);
7,25 (t, iH); 7,16-7,01 (m, 7H); 6,76,(d,lH); 4,63 (dd, 1H); 4,25 (hex, iH);
2,94
(dd, 1H); 2,75-2,65 (m, 5H); 2,46-2,30 (m, 2H); 1,75 (t, 2H); 1,39 (s, 9H);
1,26
(m, 6H).
Exemple 92
Acide ([iS)-[i-[[[(25~-1-[(3,4-dihydro-2,2-di méthyl-2ff 1-benzopyran-6-
yl)sulfonyl]-2,3-dihydro-lh~indol-2-yl]carbonyl]amino] benzène
butanôique
En opérant de façon analogue à la préparation XIII, au départ du produit
obtenu
selon l'exemple 91, on obtient le produit attendu sous forme d'une mousse
blanche avec un rendement de 34 %.
RMN iH (DMSO, 300 MHz) 8: 12,30 (s large, 1H); 8,04 (d, 1H); 7,52 (d, 1H);
7,46 (d, 1H); 7,34 (dd, iH); 7,24 (t, 1H); 7,18-7,01 (m, 7H); 6,76 (d, 1H);
4,65
(dd, 1H); 4,25 (hex, 1H); 2,96 (dd, 1H); 2,81-2,63 (m, 5H); 2,54-2,30 (m, 2H
);
1,75 (t, 2H); 1,26 (m, 6H).
Préparation L
Acide (25)-2,3-dihydro-1-[[4-(phénylméthoxy)phényl]sulfonyl]indole-2-
carboxylique, méthyl ester
En opérant de façon analogue à la préparation IL, au départ du chlorure de 4-
(phénylméthoxy)benzènesulfonyle et d'ester méthylique de l'acide 2,3-dihydro-
(2S)-lffindole-2-carboxylique, on obtient le produit attendu sous forme d'une
poudre blanche avec un rendement de 81 %.
RMN 1H (DMSO, 300 MHz) 8: 7,73 (d, 2H); 7,55 (d, 1H); 7,37-7,33 (m, 5H); 7,20
(t, iH); 7,07-6,95 (m, 4H); 5,06 (s, 2H); 4,78 (dd, 1H); 3,79 (s, 3H); 3,25-
3,05
(m, 2H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
63
Préaaration LI
Acide (25)-2,3-dihydro-1-[(4-hydroxyphényl)sulfonyl]indole-2-carboxylique,
méthyl ester
En opérant de façon analogue à la préparation XLIII, au départ du composé
obtenu selon la préparation L, on obtient le produit attendu sous forme d'un
solide beige avec un rendement de 98 %.
RMN iH (DMSO, 300 MHz) 8: 10,67 (s large, iH); 7,66-7,61 (m, 2H); 7,36-7,33
(m, 1H); 7,22-7,12 (m, 2H); 7,02-6,96 (m, iH); 6,85-6,81 (m, 2H); 4,97-4,90
(m, iH); 3,72 (s, 3H); 3,5-3,20 (m, 1H); 3,05-2,95 (m, 1H).
Préparation LII
Acide (2~-2,3-dihydro-1-[[4-(1,1-diméthyléthoxy)phényl]sulfonyl] indole-2-
carboxylique, méthyl ester
On prépare une solution de 2,4 g (7,2 mmol) d'ester obtenu selon la
préparation
LI dans 140 ml de toluène à chaud et on ajoute lentement 9,6 ml (40 mmol) de
di-t butylacétal de N,N-diméthylformamide. Le mélange réactionnel est maintenu
sous agitation à 100 °C pendant 2 heures, puis concentré sous pression
réduite.
Le résidu huileux est purifié par chromatographie sur gel de silice en éluant
à
l'aide d'un mélange cyclohexane/acétate d'éthyle (85/15 ; v/v). On obtient
ainsi
le produit attendu sous forme d'une poudre grise avec un rendement de 38 %.
RMN iH (CDCL3, 300 MHz) 8: 7,72 (d, 2H); 7,55 (d, 1H); 7,22 (t, 1H); 7,12-6,95
(m, 4H); 4,81 (dd, 1H); 3,80 (s, 3H) 3,25-3,07 (m, 2H); 1,27 (s, 9H).
Préparation LIII
Acide (2~-2,3-dihydro-1-[[4-(1,1-diméthyléthoxy)phényl]sulfonyl] indole-2-
carboxylique
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon la préparation LII, on obtient le produit attendu sous forme d'un solide
blanc avec un rendement de 91 %.
RMN iH (CDCL3, 300 MHz) â: 7,65-7,58 (m, 3H); 7,26-7,20 (m, 1H); 7,06-6,95
(m, 4H); 4,77 (dd, 1H); 3,27-3,06 (m, 2H);1,39 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
64
Préparation LIV
Chlorure de (2.~-2,3-dihydro-1-[[4-(1,1-diméthyléthoxy)phényl] sulfonyl]indole-
2- carbonyle
En opérant de façon analogue à la préparation XIX, au départ du composé
obtenu selon la préparation LIII, on obtient le produit attendu avec un
rendement de 98 % sous forme d'un solide jaune que l'on remet en réaction
rapidement pour la synthèse de l'exemple 93.
Exemple 93
Acide 4-[2-[[[(2,5)-1-[[4-(1,1-diméthyléthoxy)phényl]sulfonyl]-2,3-dihydro-1ff
indol-2-yl]carbonyl]amino]éthoxy]benzèneacétique, méthyl ester
En opérant de façon analogue à l'exemple 61, au départ du composé obtenu
selon la préparation LIV et d'ester méthylique de l'acide 4-(2-
aminoéthoxy)benzèneacétique, on obtient le produit attendu sous forme d'un
solide jaune clair avec un rendement de 50 %.
RMN 1H (DMSO, 300 MHz) 8: 7,71 (d, iH); 7,46 (d, 2H); 7,40 (t, iH); 7,26-7,01
(m, 5H); 7,87 (d, 2H); 7,80 (d, 2H); 4,61 (dd, 1H); 4,01(m, 2H); 3,80-3,60 (m,
5H); 3,56 (s, 2H); 3,24 (dd, 1H); 2,73 (dd, 1H); 1,36 (s, 9H).
F = 45-48°C.
Exemple 94
Acide 4-[2-[[[(2,~-1-[[4-(1,1-diméthyléthoxy)phényl]sulfonyl]-2,3-dihydro-1ff-
indol-2-yl]carbonyl]amino]éthoxy]benzèneacétique
En opérant de façon analogue à la préparation II, au départ du composé obtenu
selon l'exemple 93, on obtient le produit attendu sous forme d'un solide blanc
avec un rendement de 68 %.
F = 68-71°C.
RMN iH (DMSO, 300 MHz) 8: 12,5 (s large, iH); 8,37 (t, 1H); 7,66 (d, 2H); 7,44
(d, iH); 7,24-6,99 (m, 7H); 6,88 (d, 2H); 4,78 (dd, 1H); 3,99 (dd, 2H); 3,60-
3,40
(m, 4H); 3,09-2,85 (m, 2H); 1,34 (s, 9H).
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
Exemule 95
Sodium 4-[2-[[[(2,5)-1-[[4-(1,1-diméthyléthyl)phényl]sulfonyl]-2,3-dihydro-1ff
indol-2-yl]carbonyl]amino]éthoxy]benzèneacétate
On dissout 960,65 mg (1,791 mmol) du produit obtenu à l'exemple 55 dans 1 ml
5 d'acétonitrile, puis on ajoute 3,58 ml (1,791 mmol) d'une solution de soude
à 0,5
N et on agite 30 minutes à température ambiante. On lyophilise le milieu
réactionnel, on recristallise le lyophilisat dans l'isopropanol et on obtient
le
produit désiré sous forme d'un solide blanc avec un rendement quantitatif.
RMN iH (DMSO, 250 MHz) 8: 8,42 (t, 1H); 7,72 (d, 2H); 7,55 (d, 2H); 7,44(d,
10 2H); 7,21 (t, iH); 7,14-7,10 (m, 3H); 6,99 (t, 1H); 6,80 (d, 1H); 4,81 (dd,
1H);
3,97 (t, 2H); 3,63-3,35 (m, 2H); 3,20 (s, 2H); 3,11 (dd, iH); 2,90 (dd, iH);
1,25
(s, 9H).
Activité biologique
15 Test de transactivation des chimeres GAL4-LXR après transfection
transitoire en
cellules COS7
Les tests de transactivation reposent sur la capacité des récepteurs
nucléaires
(1) à se lier à une séquence ADN spécifique (RE = Response Element)
20 située devant un promoteur, via leur domaine de liaison à l'ADN (ou DBD =
DNA
Binding Domain).
(2) et à augmenter la transcription d'un gène sous contrôle de ce
promoteur en présence d'un ligand agoniste, via leur domaine de liaison au
ligand (ou LBD = Ligand Binding Domain).
25 Le test de transactivation en cellules COS7 développé ici vise à évaluer
l'effet de composés sur l'activité des LXRs humains : il permet de valider
l'interaction des composés avec les LXRs et de déterminer l'EC50 de
l'interaction.
Ce test est basé sur l'utilisation de protéines chimères « Gal4-LXR »
contenant le
LBD du LXR (LXRa humain ou LXR~3 humain) fusionné avec le DBD de Gal4. Les
30 cellules COS7 sont ainsi co-transfectées de façon transitoire avec
- un vecteur d'expression codant pour la protéine chimère « Gal4(DBD)-
LXRa(LBD) a> ou un vecteur d'expression codant pour la chimère « Gal4(DBD)-
LXR[3(LBD) »
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
66
- un vecteur rapporteur comportant l'élément de réponse Gal4-RE (Gal4-
Response Element) reconnaissant le DBD du Gal4, et situé devant le promoteur
minimal PTK qui contrôle le gène de la luciférase.
L'activité de la luciférase ainsi produite génère de la luminescence en
présence d'un excès de substrat, donnée quantifiable qui reflète l'interaction
du
composé avec le LBD du LXR.
Les composés selon l'invention sont évalués par rapport à un composé de
référence (T-0901317, CAS RN : 293754-55-9).
Selon ce test les composés selon l'invention présentent une EC 50
inférieure à 1 ~,M .
Les propriétés biologiques des composés selon l'invention démontrent leur
intérêt potentiel et leur utilité pour leur application en tant que substances
actives de médicaments destinés au traitement ou à la prévention des maladies
dépendantes d'une dérégulation des fonctions des récepteurs LXR oc et LXR Vii,
notamment les hypercholestérolémies, les dyslipidémies, ainsi que l'obésité,
le
diabète, les maladies cardiovasculaires, certaines neurodégénérescences et les
maladies inflammatoires. Les composés selon l'invention présentent également
un intérêt en thérapeutique quand il est nécessaire de corriger la dérive des
paramètres annonciateurs d'un syndrôme métabolique.
En fonction des pathologies à traiter, les composés selon l'invention
peuvent être utilisés seuls ou en association avec les traitements connus du
diabète comme par exemple la metformine, les sulfonylurées, l'acarbose, les
activateurs PPARy, l'insuline ou encore en association avec les composés
analogues de GLP-1 (Glucagon-like Peptide), les inhibiteurs de DPP-IV,
PPARa/y,
PPARb/y, PPARB, panPPAR, les inhibiteurs de 11~i-HSD1(11 beta hydroxysteroid
dehydrogenase), les inhibiteurs de PTP-iB (Protein Tyrosin Phosphatase), les
antagonistes du récepteur CB1 (Cannabinoide), les antagonistes du récepteur au
glucagon, les inhibiteurs de PDK (Pyruvate Dehydrogenase Kinase), les
activateurs de Glucokinase Hépatique et les inhibiteurs de GSK-3 (Glycogen
Synthase Kinase).
L'invention concerne également les compositions pharmaceutiques
destinées à la prévention ou au traitement des maladies précédemment citées
lorsqu'elles contiennent en tant que principe actif au moins l'un des composés
de
formule I selon l'invention.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
67
Ces compositions pharmaceutiques font appel à des formulations
classiques comprenant des excipients pharmaceutiquement acceptables afin
d'obtenir des formes administrables de préférence par voie orale, par exemple
des comprimés ou des gélules.
De façon pratique, en cas d'administration du composé par voie orale, la
posologie quotidienne chez l'homme sera de préférence comprise entre 5 et
500 mg.
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
68
R2
R1
S02 0
~N ,,.~~N~A~B~Ar
(H) CH H
z
Ex (H) R1 R2 A B Ar
1 Q C(CHs)s H (CH~)2 -
2 I C(CHs)s H (CH~)z
3 I C(CH3)3 H (CHZ)z -
N0~
4 I C(CH3)3 H (CH~)2 - F
I C(CH3)3 H (CHZ)~ - Cl
Cl
6 I C(CH3)3 H -CHz-CH(CH3)-
7 I C(CH3)3 H (CHZ)z - H3C
8 I C(CH3)3 H (CH2)2
Cl
Cl
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
69
9 I C(CH3)3 H (CHz)z - F
I C(CH3)s H OMe -
11 I C(CH3)3 H '''~.. -
12 I C(CH3)3 H -
-(CHZ)2
13 I C(CH3)3 H (CHz)z - Cl
14 I C(CH3)3 H (CHz)z - Cl
I C(CH3)3 H (CHz)z -
Cl
16 I C(CH3)3 H (CHz)z - Cl
Cl
17 I C(CH3)3 H (CHz)3 -
18 I C(CH3)s H (CHz)4 -
19 I C(CH3)3 H _CHz_ -
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
20 I C(CH3)3 H (CHz)z -
N
21 I C(CH3)3 H (CHz)z -
22 I C(CH3)3 H -CHz- -CO-
23 I C(CH3)3 Fi (CHz)z -
F
24 I C(CH3)3 H (CHz)z - /
25 I C(CH3)3 H (CHz)z - OMe
OMe
26 I C(CH3)3 H (CHz)z
C~H5
27 I C(CH3)3 H (CHz)z -
O
28 I C(CH3)3 H _CHz_ -
29 I C(CH3)3 H (CHz)z -
CH3
30 I C(CH3)3 H - -
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
71
31 I C(CH3)s H (CHz)z - Me0
32 I C(CH3)3 H (CHz)z
N
33 I C(CH3)3 H (CHz)z - O
0
34 I C(CH3)s H (CHz)z -
35 I C(CH3)3 H (CHz)z - / \ CH3
CH3
CH3
36 I C(CH3)3 H (CHz)z -
CH3
CH3
37 I C(CH3)3 H (CHz)z
MeO~ OMe
38 I C(CH3)3 H (CHz)z
CH3
39 I C(CH3)3 H (CHz)z
CF3
40 I C(CH3)3 H (CHz)z - Me0
OMe
41 I C(CH3)3 H (CHz)z -
NHz
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
72
42 I C(CH3)3 H (CHz)z _ e \
OH
43 I CH(CH3)z H (CHz)z - e
44 I 4-CI 3-CI (CHz)z - e
45 I e \ H (CHz)z - e
46 I ~ e \ H (CHz)z - e
O
47# I ~,, O CH3 (CHz)z _
~I CH3 o v
.~,..
48# I ~ (CHz)z - e \
y
N
O~CF
3
49 I -O-CH(CH3)z H (CHz)z - e
50 I -C(CH3)3 H (CHz)z - e \
51 HI -C(CH3)3 H (CHz)z - e
52 I -C(CH3)3 H (CHz)z -O- e \
COOCH3
53 I -C(CH3)3 H (CHz)z -O- e
COOH
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
73
54 I -C(CH3)3 H (CHz)z -O-
~OOCH3
55 I -C(CH3)3 H (CHz)z -O-
1:00H
56 I H FAM (CHz)z -
57 I -C(CH3)3 H H3C~ ~CH3 -
N
I _C(CH ) H (CHz)z -0- ~ 0
N
~ H3Ci wCH3
59* 4- -C(CH3)3 H (CHz)z -
* OMe-
I
60* 5-F-I -C(CH3)3 H (CHz)z -
61 I -C(CH3)3 H (CHz)z -0-
N
~ H3Ci wCH3
62 I -C(CH3)3 H (CHz)z -0- F
OMe
63 I -C(CH3)3 H (CHz)z -0- F
0
~OH
64 I -C(CH3)3 H (CHz)z -O- F ~ OMe
/ O
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
74
65 I -C(CH3)3 H (CHz)z -0- F OH
/ O
66 I -C(CH3)s H CHz -
OH
67 I -C(CH3)s H CHz -
Cl
68 I -C(CH3)3 H CHz - / \ O
v
OMe
69 I -C(CH3)s H CHz - ~ \ O
OH
70 I -C(CH3)s H CHz -
O OMe
71 I -C(CH3)s H CHz -
O OH
72 I -C(CH3)3 H CHz - ~ 0
OMe
73 I -C(CH3)s H CHz - ~ O
OH
74# I ~~. O C CH (CHz)z -~- ~ ~ O
' 3
i
. '~.- OMe
75# I y. O C CH (CHz)z -~- ~ ~ O
' 3
. '~.~ OH
76 I -C(CH3)s H CHz -
0
0 OMe
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
7S
77 I -C(CH3)3 H CHz -
O
0 OH
78 I -O-CH(CH3)z H (CHz)z -O-
L:OOH
79* 7- -C(CHs)s H (CHz)z -
* NHz-I
80# I ~,, 0 CH3 (CHz)z O
CH3
-- L:OOH
81 I -C(CH3)3 (CHz)z -S- Cl ~ OMe
/ 0
82 I -C(CH3)3 H (CHz)z -S- Cl ~ OH
/ 0
83 I -C(CH3)3 H CHz - / O
OMe
84 I -C(CH3)3 H CHz - / O
OH
85 I -C(CH3)3 H CHz -
NOz
86 I -C(CH3)3 H CHz -
NH2
87 I -C(CH3)3 H CHz - H
/ N
O OMe
88 I -C(CH3)3 H CHz - H
/ N
\
O OH
CA 02564642 2006-10-25
WO 2005/121093 PCT/FR2005/001139
76
89 I / F H (CHZ)~ -O- \ O
\ \ / OMe
0
90 I / F ~ H (CH2)2 -O- \ 0
\ \ ~ ~ / Ö
0
91# I ~,, O CH3 COOtBu -
CHs
.\
92# I ~,, 0 CH3 COOH -
CH3
- \
-. ~.-- ,,,,,
93 I -O-C(CH3)3 H (CH~)Z -O- \
r
/ OMe
94 I -O-C(CH3)3 H (CH2)~ -0- \
r
/ OH
95 I -C(CH3)3 H (CH~)2 -0- \
r
/ ONa
Me = CH3
Q = 1,2,3,4-tétrahydroquinoléine
I = indoline (4-OMe-I = 4-méthoxy-indoline)
HI = octahydroindole
*3-FAM = 3-(trifluoroacétylaminométhyl)
** : composé racémique
Ri
~I
# : dans cet exemple la formule R1 et RZ représente l'ensemble R2