Note: Descriptions are shown in the official language in which they were submitted.
CA 02572505 2007-O1-22
PROCESS AND INTERMEDIATE COMPOUNDS FOR THE
PREPARATION OF Y-LACTONES
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a division of Canadian
Patent Application No. 2,415,097, filed May 14, 2001.
BACKGROUND OF THE INVENTION
(1) Field of the Invention
The present invention relates to the
preparation of Y-lactones from 2,3,5-tri-0-acetyl L-4
pentulsonic acid or ester.
(2) Description of Related Art
Aza-sugar analogs of D-ribofuranosides are
important targets for the synthesis of drugs that
regulate nucleic acid synthesis. (3R,4R,5R)-3,4-
dihydroxy-5-hydroxymethyl-2-pyrrolidone is an important
aza-sugar intermediate.
The current routes (Fleet, G.W.J., et al.,
Tetrahedron 44 (9) 2637-2647 (1988); and Fleet, G.W.J.,
et al., Tetrahedron 44 (9) 2649-2655 (1988) to 1,4-
CA 02572505 2007-O1-22
-2-
dideoxy-1,4-imino-D-ribitol (a pyrrolidine) and its
derivatives employ hexose sugars and require the
removal of 1 carbon atom (usually by an oxidative
process) that is difficult on large scale. One of the
methods uses the L-gulono lactone which is a rare sugar
and not a regular article of commerce available in
significant quantities. There is no relatively simple
and economic synthesis available.
OBJECTS
It is therefore an object of the present
invention to provide novel processes for the
preparation of y-lactones. It is further an object of
the present invention to provide a process which is
relatively easy to perform and economical. These and
other objects of the present invention will become
increasingly apparent by reference to the following
description and the drawings.
SUMMARY OF THE INVENTION
The present invention relates to the
preparation of a first intermediate to the pyrrolidines
by a process fox the preparation of a 2,3,5-tri-0
acetyl-4-ketopentulosonic acid-1-methyl ester which
comprises:
(a) reacting a pentose sugar with methanol in
the presence of an acid to form a 1-methyl pentose
sugar;
(b) reacting the 1-methyl pentose sugar with
acetic anhydride in the presence of an amine to form
a 1-methyl-2,3,4-tri-0-acetyl pentose sugar; and
CA 02572505 2007-O1-22
-3-
(c) reacting the 1-methyl-2,3,5-tri-O-acetyl
1-methyl pentose sugar with an oxidizing agent to form
the 2,3,5-tri-O-acetyl-4-ketopentulosonic acid-1-methyl
ester.
In particular the present invention relates to
a process for the preparation of 2,3,5-tri-O-acetyl-D-
erythro-4-pentulosonic acid methyl ester which
comprises:
(a) reacting D-ribose with an acidic solution
of methanol to form 1-methyl D-ribofuranoside;
(b) reacting the 1-methyl D-ribose with acetic
anhydride in the presence of pyridine to form 1-methyl-
2,3,5 tri-O-acetyl-D- riboside in the reaction mixture;
and
(d) reacting 1-methyl-2,3,5-tri-O-acetyl-D-
riboside with an oxidizing agent to form the 2,3,5-tri-
O-acetyl-D-erythro-4-pentulosonic acid methyl ester.
The oxidizing agent is preferably chromium trioxide in
acetic anhydride_ The process is specifically shown in
. Scheme I I I .
The present invention also relates to a
process for the preparation of a second intermediate to
the pyrrolidines which is a process which comprises:
(a) reacting tri-O-acetyl-4-pentulosonic acid
methyl ester with hydroxylamine, an amine or an ammonium
ion in the presence of pyridine with the hydroxylamine
to form an oxime or imine of the formula:
CA 02572505 2007-O1-22
R
a
2
3
wherein R is selected from the group consisting of
acyloxy, alkyloxy, hydroxyl, alkyl, aryl and hydrogen
and R1 to R3 are hydrogen .or a protecting group;
(b) separating the oxime or imine from the
reaction mixture. The reaction is conducted in a non-
reactive solvent with an amine base at low temperatures
-lOoC to 10~C and then poured over ice containing an
acid to trap the excess amine base or hydroxylamine. In
this and the following reactions, R preferably contains
0 to 10 carbon atoms and R1 contains 0 to 10 carbon
atoms. R and R1 are generally groups which are non-
labile under the reaction conditions.
The present invention also relates to a
process for the preparation of a third intermediate to
the pyrrolidines which is a process for the preparation
of a pyrrolidone lactam of the formula:
R
R3 N 0
R2 R1
which comprises reducing an oxime or imine of the
CA 02572505 2007-O1-22
formula:
R1
-5-
R
with a source of singlet hydrogen (H) or a hydride to
form the pyrrolidone lactam, wherein R is selected from
the group consisting of acyloxy, alkyloxy, hydroxyl,
alkyl, aryl, and hydrogen, and wherein R1 to R3 are
hydrogen or a protecting group and Me is methyl. The
reaction is conducted in a non-reactive solvent,
preferably methanol, at -lOoC to 30~C.
The present invention also relates to a
process for the preparation of a 2,3,5-tri-0-acetyl-1,4-
dideoxy-1,4-iminopentitol which comprises:
reacting a pyrrolidone lactam of the formula:
R
R30 N O
R20 OR1
with a source of singlet hydrogen (H) or a hydride to
form the pentitol, wherein R is selected from the group
consisting of alkyl, aryl and hydrogen and R1 to R3 are
hydrogen or a protecting group. The reaction is
preferably conducted at -20 to 40~C.
The present invention also relates to a
process for the preparation of a lactone which
CA 02572505 2007-O1-22
-6-
comprises:
(a) reacting in a reaction mixture 2, 3, 5-tri-
0-acetyl-4-pentulosonic acid or ester with a hydride or
hydrogen and a catalyst to produce 2,3,5-tri-0-acetyl-
pentonic acid or ester in a reaction mixture; and
(b) reacting the 2,3,5-tri-O-acetyl-pentonic
acid or ester with an acid in water to form a lactone.
A preferred lactone is L-lyxono-y-lactone.
The present invention also relates to a
process for the preparation of a 1,4-dideoxy-1,4-imino
pentitol which comprises:
(a) reacting tri-0-acetyl -4-pentulosonic acid
methyl ester in methanol ammonium acetate and acetic
acid in the presence of a hydride reducing agent to form
an ammonium compound which spontaneously cyclizes to a
lactam;
(b) reacting the lactam with a hydride to form
2,3,5-tri-O-acetyl 1,4-dideoxy-1,4-imino pentitol; and
(c) deacylating the tri-O-acetyl-1,4-dideoxy
1,4-iminopentitol to form the 1,4-dideoxy-1,4
iminopentitol.
The present invention also relates to a
process for the preparation of 1,4-dideoxy-1,4-
aminopentitol which comprises:
(a) reductive cyclization of tri-O-acetyl-4-
amino pentonic acid methyl ester with a reducing agent
to form 2,3,5-tri-0-acetyl 1,4-dideoxy-1,4-iminopentitol
via an intermediate lactam; and
(b) deacylating the 2,3,5-tri-O-acetyl-1,4-
dideoxy-1,4-iminopentitol to form 1,4-dideoxy-1,4-imino
CA 02572505 2007-O1-22
_')_
pentitol.
The present invention also relates to a
pentulosonic acid methyl ester which comprises:
0
OMe
R10
OR2
O
R3
where R1 to R3 is a protecting group or hydrogen and Me
is methyl.
The present invention also relates to a
pentulosonic acid methyl ester oxime or imine of the
formula
20
wherein R is selected from the group consisting of
acyloxy, alkoxy, hydroxyl, alkyl, aryl and hydrogen, R1
to R3 are protecting groups or hydrogen and Me is
methyl.
CA 02572505 2007-O1-22
_g_
The present invention also relates to a
pyrrolidone of the formula:
R
R30 N O
R20 OR1
wherein R1 to R3 is a prot~eeting group or hydrogen, and
R is selected from the group consisting of acyloxy,
alkyloxy, hydroxy,alkyl, aryl and hydrogen.
The present invention also relates to a
pyrrolidine of the formula:
R
Rg0 N O
R20 OR1
where R is selected from the group consisting of
acyloxy, alkyloxy, hydroxy, alkyl, aryl and hydrogen and
R1 to R3 is a protecting group.
The specific novel compounds are:
2,3,5-Tri-O-acetyl-D-erythro-4-oximyl
pentulosonic acid methyl ester.
2,3,5-Tri-0-acetyl-D-erythro-4-pentulosonic
acid methyl ester.
3,4-Dihydroxy-5-hydroxymethyl-2-pyrrolidone.
(3R,4R,5R)-3,4-Dihydroxy-S-hydroxymethyl-2
pyrrolidone.
2,3,5-Tri-O-acetyl-1,4-Dideoxy-1,4-imino-D-
CA 02572505 2007-O1-22
-9-
ribitol.
2,3,5-Tri-O-acetyl-4-amino-4-deoxy-D-erythro-
pentonic acid methyl ester.
N-benzyl (3R,4R,5R) 3,4-dihydroxy-5-
S hydroxymethyl 2-pyrrolidone.
3,4-dihydroxy-5-hydroxymethyl-N-benzyl-2-
pyrrolidone.
The present invention further relates to
2,3,5-tri-0-acetyl-L-lyxonic acid methyl ester.
The present invention also relates to lyxono-
y-lactone.
The present invention also relates to L-
lyxono-Y-lactone.
BRIEF DESCRIPTION OF DRAWINGS
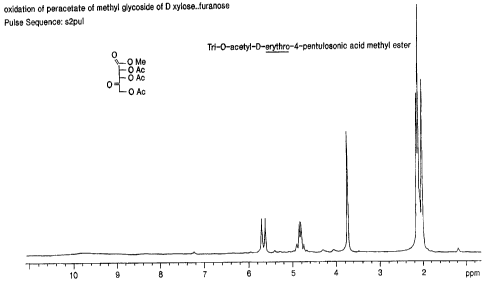
Figure 1 is a proton NMR spectra for tri-0-
acetyl-D-erythro-4-pentulosonic acid methyl ester 6.
Figure 2 is a 13C NMR spectra for the compound
6 of Figure 1.
DESCRIPTION OF PREFERRED EMBODIMENTS
1,4-dideoxy-1,4-imino pentitols from
triacetoxy keto pentonic acids (tr~i-O-acetyl
pentulosonic acid esters).
The process preferably starts from the pentose
D-ribose which is available in ton quantities and has
the correct number of carbons and the correct
stereochemistries. It is much shorter and more
efficient than the other routes. Other pentoses could
be used such as L-ribose, D or L arabinose, xylose or
lyxose.
1,4-Dideoxy-1,4-imino-D-ribitol is made from
CA 02572505 2007-O1-22
-10-
tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
or a related molecule by one of several possible
methods, the first two of which are:
( 1 ) Reductive amination with an amine or ammonia to form
S a 4-amino-4-deoxy pentonic acid compound that can then
be cyclized to a lactam. Reduction of the lactam with
borane or lithium aluminum hydride yields the desired
1,4-dideoxy-1,4-imino-D-ribitol.
(2) Formation of an oxime which can be reduced by one of
several possible methods to yield a 4-amino-4-deoxy
pentonic acid compound that can then be cyclized to the
lactam. Reduction of the lactam with borane or lithium
aluminum hydride will yield the desired l, 4-dideoxy-l, 4
imido-D-ribitol.
The tri-O-acetyl D-erythro-4-pentulosonic acid
methyl ester, the oxime and the lactam (in these
examples (3R, 4R, 5R)-3,4-dihydroxy-5-hydroxymethyl-2-
pyrrolidone and its N-alkyl derivatives) have not been
previously described. Once these compounds can be
prepared, the subsequent process step for transformation
to the desired 1, 4-Dideoxy-1, 4-imino-D-ribitol is in the
known art.
Tri-0-acetyl D-erythro-4-pentulosonic acid
methyl ester, its oxime and (3R, 4R, 5R)-3,4-dihydroxy-
5-hydroxymethyl-2-pyrrolidone and its N-benzyl
derivative (formed if benzylamine is used instead of
ammonia in the reductive amination) are new compounds.
The pyrrolidines are derived from an
appropriately protected (R1 to R3) or unprotected R1 to
R3 is hydrogen 2,3,5-trihydroxy 4-ketopentulosonic acid
CA 02572505 2007-O1-22
esters I by any of several routes as shown in Scheme I.
O O O
S OR OR / OR
R20 ~H R20 2H Rz0
R1 --~ OR1 =~ ORS
0 RN NHz
OR3 OR3 OR3
1 2 3
- -
R R
R30 N HO
0 2H N
IS
RIO OR2 HO OH
4 5
ZO
Scheme I
wherein R is OH. Steps 2 and 3 combine together, where
R is hydrogen or alkyl, aryl, acyloxy, alkoxy then the
process follows each of the steps. Generally R1 to R3
is acetyl. Other groups are benzoyl, propanoyl and
ZS trifluoroacetyl.
s0
It should be noted that in the present
application the compounds can be numbered using the
carbohydrate system wherein the carboxyl group is 1 and
the compounds are "pyrrolidines. Scheme I uses this
carbohydrate system to show the position of the carbons.
CA 02572505 2007-O1-22
-12-
In the pyrrolidone system the N in the ring is 1 in
naming the various compounds. The pyrrolidone system is
preferred for purposes of claiming the compounds.
In this scheme the, protected trihydroxy 4
ketopentulosonic acid ester 1 is reacted with ammonia or
a primary amine or ammonium ion or with hydroxylamine to
form an imine (in the former case) or an oxime 2 where
R is OH which is then hydrogenated or reduced with a
metal or a metal hydride reagent to form an amine 3.
The amine spontaneously cyclizes to a lactam 4 which can
be reduced with ~borane or a hydride reagent to the
desired pyrrolidine 5.
Starting with the previously unknown compound
tri-O-acetyl-D-erythro-4-pentulosonic acid methyl ester
(R=methyl, R1 to R3=acetyl in Scheme I) (6), direct
syntheses of the tri and di hydroxypyrrolidines (9 and
10 respectively) is obtained with the D-ribo
configuration (scheme II). The deoxygenation of the 5-
position to form 10 was produced by reduction of the
triacetate of the oxime (2) with hydrogen on palladium
in acetic acid and thus this combination is not used as
a reducing agent. Under these conditions the amino
group was also introduced by reduction of the oxime 2.
The amine cyclized to form the intermediate amide 8
(lactam) which was reduced to the pyrrolidine 10 with
borane or lithium aluminum hydride. Deoxygenation of
the 5-position did not occur if the molecule was
deactylated first or if an imine was used instead of an
oxime for introducing the nitrogen.
CA 02572505 2007-O1-22
-13-
6
r
H
N 0
Ac0 OAc
7 8
Hydride
H H
HO
0 N O
HO OH HO OH
30 Scheme II
CA 02572505 2007-O1-22
-14-
Tri-0-acetyl-D-erythro-4-pentulosonic acid
methyl ester (6) was prepared by two routes as outlined
in Schemes III and IV.
HO HO Ac0
O
OH H+/jqeOH 0 OMe 1~c20/pyridine 0 OMe
HO pH HO OH Ac0 OAc
crog/Ac2o Ac0 0 OMe
0
Ac ~ (~Ac
6
Scheme III
CA 02572505 2007-O1-22
-15-
HO HO
O ~ Ac0
OH H+~MeOH OMe A~2o/pyridine O OMe
HO OH HO OH Ac0 OAC
Ac0 OH OMe ~o~ Ac0 0 OMe
p ~O
Ac0 OAc Ac0 OAc
Scheme IV
In the first route (Example l, Scheme III), D-
CA 02572505 2007-O1-22
-16-
ribose is converted to a mixture of its a and (3
furanosides by treatment with methanol in the presence
of a catalytic amount of sulfuric acid. The methyl
glycosides are peracetylated and then oxidized with
chromium trioxide in acetic anhydride (Example 2) . This
yields the Tri-O-acetyl-D-erythro-4-pentulosonic acid
methyl ester (6) in very pure state as evidenced by the
proton (Figure 1) and 13C NMR spectra (Figure 2).
In the second route (Example 6, Scheme IV) the
peracetylated glycosides are oxidized with ozone to give
the 2,3,5-triacetyl aldonic acid methyl ester which is
then oxidized to the tri-O-acetyl-D-erythro-4
pentulosonic acid methyl ester 6 by treatment with DMSO
and acetic anhydride or DMSO and trifluoroacetic
anhydride.
The pentulosonic acid methyl ester 6 can be
converted_to the pyrrolidine nucleus by several routes:
(1) Conversion to the oxime 2 and reduction to the 4-
amino-4-deoxy ester 3 with hydrogen Pd/C with
concomitant deoxygenation at the 5 position followed by
cyclization to form 10 (Scheme II) where R = H and R1 =
R2 = Ac .
(2) Deacetylation by acid methanolysis, oxime 2
formation, and reduction with Pd/C to form 7 where R =
R1 = R2 = R3 = H .
(3) Reductive amination with ammonia and a reductant to
form the 4-amino-4-deoxy ester 3 followed by cyclization
to form 7 where R = H Rl = R2 = R3 = Ac.
(4) Conversion to the oxime 2, deacetylation with
CA 02572505 2007-O1-22
-17-
hydrazine, reduction to the 4-amino-4-deoxy ester 3 with
hydrogen Pd/C with concomitant deoxygenation at the 5
position followed by cyclization to from 7 where R = R1
- R2 = R3 = H .
(5) Reductive amination with benzylamine and a reductant
to form the 4-amino-4-deoxy ester. 3 followed by
cyclization to form 7 where R = Benzyl and Rl = R2 = R3
- Ac.
(6) Reductive amination with 2,4-dimethoxybenzylamine
and a reductant to form the 4-amino-4-deoxy ester 3
followed by cyclization to form 11 where R = Benzyl and
R1 = R2 = R3 = Ac .
Tri-0-acetyl D-erythro-4-pentulosonic acid
methyl ester 6 is thus a key intermediate in the
synthesis of (3R,4R,5R)-3,4-dihydroxy-5-hydroxymethyl-2
pyrrolidone as a 1,4-dideoxy-1,4-imino-D-ribitol (9).
These compounds are valuable intermediates in the
synthesis of "aza-sugar" analogs of D-ribofuranose.
R R
R10 N 0 R10 N
0
R 0 .''~OR3 R2p .'''0R3
2
11
CA 02572505 2007-O1-22
-18-
The transformation of tri-O-acetyl D-erythro-
4-pentulosonic acid methyl ester 6 and its oxime 2 to 9
via 7 and its per-O-acetate was achieved via various
chemical transformations. Typical strategies are:
(1) Reduction of the oxime to an amine and cyclization
to the pyrrolidone with expulsion of methanol with
reagents such as hydrogen and palladium, hydrogen and
platinum, hydrogen and Raney nickel, zinc and acetic
acid and sodium cyanoborohydride.
(2) Reductive amination of the ketone function of tri-O-
acetyl D-erythro-4-pentulosonic acid methyl ester 6
with ammonia or an amine using reagents such as sodium
cyanoborohydride, sodium borohydride or hydrogen and a
catalyst followed by cyclization to the pyrrolidone.
The pyrrolidone is reduced to the 1,4-dideoxy-1,4-imino-
D-ribitol with reagents such as lithium aluminum hydride
or borane.
EXAMPLE 1
Preparation of tri-O-acetyl D-erythro-4-
pentulosonic acid methyl ester 6
6
3
There are two efficient routes to the preparation of
CA 02572505 2007-O1-22
-19-
tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
6. The first route is by the oxidation of tri-0-acetyl
methyl a,(3-ribofuranoside with chromium trioxide in
acetic acid/acetic anhydride. The second method is by
the oxidation of tri-0-acetyl methyl a,(3-ribofuranoside
with ozone to produce 2,3,5-tri-O-acetyl D-ribo-pentonic
acid methyl ester which is then oxidized with a reagent
such as DMSO/TFAA or DMSO/Ac20.
Tri-O-acetyl methyl cx, ~3-ribofuranoside
Procedure 1.
D-ribose (100 g) was dissolved in methanol (1000 ml) and
conc sulfuric acid (2 ml) added. The mixture was left
at room temperature for 24 hours and then the solvent
was removed at a bath temperature of less than 30-35~C.
Pyridine (400 ml) was added and the mixture cooled in
ice to ~S~C. Acetic anhydride (300) was then added over
a 20 minute period. The mixture was allowed to come to
room temperature and left there for 10 hours after which
the solvents were removed by rotary evaporation at a
bath temperature of 45-50oC. The syrup was dissolved in
ethyl acetate (1000 ml) and washed twice with cold
saturated sodium chloride (200 ml) containing ~ 30 ml
of conc HCl. After 1 wash with cold saturated sodium
chloride (100 ml), the solution was dried (sodium
sulfate) and concentrated to an oil. The crude tri-O-
acetyl methyl a,(3-D-ribofuranoside that was so produced
was used without further purification.
Tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
The tri-O-acetyl methyl a,~3-ribofuranoside prepared
CA 02572505 2007-O1-22
from 100g of D-ribose by -procedure 1 above was dissolved
in acetic acid (1500 ml) and acetic anhydride (330 ml)
added. The mixture was cooled in ice to 0-5oC and a
stream of nitrogen passed over the surface. Chromium
trioxide (130 g) was added over a period of 40 minutes
and the temperature~never allowed to exceed 10~C. The
mixture was stirred at this temperature for 1 hour then
allowed to reach room temperature over a 30 minute
period. It was stirred at room temperature for 5 hours.
The solvents were then rapidly removed under vacuum at
a temperature not to exceed 50~C. It was then diluted
with 2000 ml of ethyl acetate, stirred vigorously for 30
minutes and filtered. The filter cake was washed with
a further 500 m1 of ethyl acetate. The combined ethyl
acetate extracts was washed with 2 X 300 ml of cold
water, dried and the solvent removed to yield the
desired product in over 92o yield (>92o pure by NMR
spectroscopy). 1H NMR in chloroform, 2.0 - 2.3 (3 X 3H
singlets), 4.8 (dd, 2H, J = 12 Hz), 5.61 (s, 1H), 5.71
(s, 1H). 13C NMR 30-31 ppm (3 signals), 53.2, 66.8,
71.3, 76.0, 160.7, 169.5, 170.5, 19?.8.
Preparation of tri-O-acetyl D-erythro-4-pentuloson~c
acid methyl ester oxime (2), where R = H and R1 to R3 =
acetyl
30
2
CA 02572505 2007-O1-22
-21-
Tri-0-acetyl D-erythro-4-pentulosonic acid methyl ester
(5.5 g) was dissolved in pyridine (16 ml) and the
solution cooled to O~C. Hydroxyamine hydrochloride (2g,
29 mmol) was added and the mixture was kept at O~C for
S a further 15 minutes.and then at room temperature for 2
hours. It was poured into ice containing 18 ml of
concentrated HC1 (sufficient to neutralize the pyridine)
and extracted with 3 times with 60 mol of chloroform.
The combined chloroform extracts were washed once with
15 ml of cold saturated sodium chloride, dried
(anhydrous sodium sulfate) and concentrated to yield a
colorless syrup which slowly formed white crystals.
Yield - 5 . 7 g ( 97 0 ) . 13 C NMR- (d-chloroform) 21. 0, 53 . 5,
57.8, 62.0, 68.3, 70.8, 72.0, 151.6, 168.0, 170.1,
171.1, 172Ø
EXAMPLE 3
N-benzyl (3R,4R,5R)-3,4-dihydroxy-5-hydroxymethyl-2-
pyrrolidone
Tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
6 (15.2g) was dissolved in methanol (85 ml) and acetic
acid (3.1 g) and benzylamine (5.4 g) added. Sodium
cyanoborohydride (3.1g) was then added and the mixture
kept at room temperature for 24 hours to reduce the
imine to an amine 3. Sodium bicarbonate (6 g) and water
20 ml was added and the mixture heated for 4 hours at
70~C to effect cyclization to the lactam 7. The mixture
was concentrated to a syrup and partitioned between
ethyl acetate (300 ml) and cold saturated sodium
chloride (100 ml). The ethyl acetate layer was
recovered, dried (sodium sulfate) and concentrated to a
CA 02572505 2007-O1-22
-22-
syrup. The syrup was dissolved in methanol (200 ml) to
which was added potassium carbonate 20g and water 2 ml.
The resulting mixture was stirred at room temperature
for 14 hours, filtered, the filtrate concentrated and
the resulting syrup dissolved in methanol (400 ml).
Concentrated HC1 (4.1 ml) was added. A white solid was
formed. This was removed by filtration and the filtrate
concentrated to dryness. Methanol was added again and
the solution again concentrated. This was repeated one
more time to give the crude N-benzyl pyrrolidone which
can be converted to the pyrrolidine to reduction.
EXAMPLE 4
(3R,4R,5R)-3,4-dihydroxy-5-hydroxymethyl-2-pyr~olidone
Procedure 1
Tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
6 (15.2g) was dissolved in methanol (100 ml) and
ammonium acetate (3.0 g) and acetic acid (0.2 ml) added.
Sodium cyanoborohydride (3.1 g) was then added and the
mixture kept at room temperature for 24 hours to reduce
the ammoniated compound to an amino group which are
rearranged to the tri-acetylated product 4. The
triacetylated product was deacetylated with potassium
carbonate-methanol to form the pyrrolidone.
Procedure 2
Tri-O-acetyl D-erythro-4-pentulosonic acid methyl ester
oxime wherein R = H and R1 to R3 = acetyl (3.1 g) was
dissolved in methanol (40 ml) and Raney nickel (0.5 g)
added. The mixture was hydrogenated at 2 atmospheres
for 6 hours, filtered and concentrated to give the crude
triacetylated product. The product was deactylated with
CA 02572505 2007-O1-22
-23-
potassium carbonate-methanol to form the pyrrolidone.
Procedure 3
The oxime derivative formed above was treated with 4
equivalents of hydrazine in methanol for 4 hours and
then hydrogenated with loo Pd/C in ethanol containing
loo acetic acid at 50 psi and room temperature for 5
hours. The product was deacetylated with potassium
carbonate - methanol to form the pyrrolidone.
In these procedures, the intermediate steps of
3 and 4 Scheme I are by-passed to produce the tri-0
acetylated intermediate pyrrolidone and the intermediate
tri-O-acetylate pyrrolidone is then deacylated and
reduced to the pyrrolidine (pentitol 5 in Scheme I).
EXAMPLE 5
The following is an additional procedure
(Scheme V) for using the tri-0-acetyl-D-erythro-4-
pentulosonic acid methyl ester 6 to form the
pyrrolidine.
CA 02572505 2007-O1-22
-24-
S
0 0
Me OMe
14 OAc -
BH4 or BHg or H2/catalyst OAC H+
OAc - Ac
O HO
Ac Ac
11
15 - -
2,3,5-Tri-O-acetyl-4-pentulosonic acid
2,3,5-Tri-O-Acetyl-L-Lyaconic acid methyl ester
20 0 0
Fleet et a1. procedure
--
HO
HD OH
1~
25 L-Lyx ono-y-lactose 1,4=dideoxy-1,4-imino-D-ribitol
30 Scheme V
CA 02572505 2007-O1-22
-25-
In a typical step, the 4-pentulosonic acid (30 g) is
dissolved in 150 ml of methanol and 0.5 molar
equivalents of sodium borohydride is added after the
solution is cooled to OoC. The mixture is maintained at
S 0-5~ for 2 hours and then 4 equivalents of acetic acid
are added to decompose the borohydride. The methanol is
removed by rotary evaporation. 200 ml of methanol is
added and removed and this process of adding method and
removing repeated four times to remove all borate
esters. The product 11 is refluxed in 300 ml of
methanol containing to HCl for 3 hours, to .effect
deacylation and concentrated to effect lactonization.
The crude L-lyxono-Y-lactone 12 so obtained is converted
to the iminopentitol 9 using procedures such as that
1$ described by Fleet et al, cited previously.
EXAMPT~E 6
Methyl tri-0-acetyl-a, (3, D-ribofuranoside (2g)
was dissolved in ethyl acetate (30 ml) and the solution
was cooled to 0-lOoC. Ozone was passed through for 2
hours at the rate of 20 mM per hour. The ethyl acetate
was then removed and the product dissolved in dimethyl
pentoxide (30 ml) and acetic anhydride (2 ml) added.
The mixture was left at room temperature for 24 hours.
The keto ester was isolated by concentration, and
partitioning between water/ethyl acetate. The product
was recovered from the ethyl acetate layer.
It is intended that the foregoing description
be only illustrative of the present invention and that
the present invention be limited only by the hereinafter
appended claims.