Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME DE _2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
IMMUNOGLOBULINS COMPRISING PREDOMINANTLY A MAN3GLCNAC2
GLYCOFORM
RELATED APPLICATIONS
This application claims the benefit of U.S. Provisional Application No.
60/589,913, filed July 21, 2004 and U.S. Provisional Application No.
60/589,937,
filed July 21, 2004; and is a continuation-in-part of U.S. Application Serial
No.
10/500,240, filed June 25, 2004, which is a national stage filing of
International
Application No. PCT/US02/41510, filed December 24, 2002, which claims the
benefit
of U.S. Provisional Application No. 60/344,169, filed December 27, 2001. Each
of
the above cited applications is incorporated herein by reference in its
entirety.
FIELD OF THE INVENTION
The present invention relates to compositions and methods for producing
glycoproteins having specific N-linked glycosylation patterns. Particularly,
the
present invention relates to compositions of immunoglobulin glycoproteins
comprising a plurality of N-glycans having specific N-glycan structures, and
more
particularly, to compositions comprising immunoglobulin glycoproteins wherein
within the plurality there are one or more predominant glycoform structures on
the
immunoglobulins that regulate, e.g., promote a specific effector function.
1
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
BACKGROUND OF THE INVENTION
Glycoproteins mediate many essential functions in humans and other
mannnals, including catalysis, signaling, cell-cell communication, and
molecular
recognition and association. Glycoproteins make up the majority of non-
cytosolic
proteins in eukaryotic organisms (Lis and Sharon, 1993, Eur. J. Biochem. 218:1-
27).
Many glycoproteins have been exploited for therapeutic purposes, and during
the last
two decades, recombinant versions of naturally-occurring glycoproteins have
been a
major part of the biotechnology industry. Examples of recombinant glycosylated
proteins used as therapeutics include erythropoietin (EPO), therapeutic
monoclonal
antibodies (mAbs), tissue plasminogen activator (tPA), interferon-(3 (IFN-
(3),
granulocyte-macrophage colony stimulating factor (GM-CSF), and human chorionic
gonadotrophin (hCH) (Cumming et al., 1991, Glycobiology 1:115-130). Variations
in
glycosylation patterns of recombinantly produced glycoproteins have recently
been
the topic of much attention in the scientific community as recombinant
proteins
produced as potential prophylactics and therapeutics approach the clinic.
Antibodies or immunoglobulins (Ig) are glycoproteins that play a central role
in the humoral immune response. Antibodies may be viewed as adaptor molecules
that provide a link between humoral and cellular defense mechanisms. Antigen-
specific recognition by antibodies results in the formation of immune
complexes that
may activate multiple effector mechanisms, resulting in the removal and
destruction
of the complex. Within the general class of immunoglobulins, five classes of
antibodies-IgM, IgD, IgG, IgA, and IgE--can be distinguished biochemically as
well as functionally, while more subtle differences confined to the variable
region
account for the specificity of antigen binding. Amongst these five classes of
Igs, there
are only two types of light chain, which are termed lambda (X) and kappa (K).
No
2
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
functional difference has been found between antibodies having 2~ or x chains,
and the
ratio of the two types of light chains varies from species to species. There
are five
heavy chain classes or isotypes,and these determine the functional activity of
an
antibody molecule. The five functional classes of inununoglobulin are:
immunoglobulin M(IgM), immunoglobulin D (IgD), immunoglobulin G (IgG),
immunoglobulin A (IgA) and inimunoglobulin E(IgE). Each isotype has a
particular
function in immune responses and their distinctive functional properties are
conferred
by the carboxy-terminal part of the heavy chain, where it is not associated
with the
light chain. IgG is the most abundant immunoglobulin isotype in blood plasma,
(See
for example, I~nmuraobiology, Janeway et al, 6th Edition, 2004, Garland
Publishing,
New York).
The immunoglobulin G (IgG) molecule comprises a Fab (fragment antigen
binding) domain with constant and variable regions and an Fc (fragment
crystallized)
domain. The CH2 domain of each heavy chain contains a single site for N-linked
glycosylation at an asparagine residue linking an N-glycan to the Ig molecule,
usually
at residue Asn-297 (Kabat et al., Sequences of proteins of immunological
interest,
Fifth Ed., U.S. Department of Health and Human Services, NIH Publication No.
91-
3242).
Analyses of the structural and functional aspects of the N-linked
oligosaccharides are of biological interest for three main reasons: (1) the
glycosylation of the CH2 domain has been conserved throughout evolution,
suggesting an important role for the oligosaccharides; (2) the immunoglobulin
molecule serves as a model system for the analysis of oligosaccharide
heterogeneity
(Rademacher and Dwek, 1984; Rademacher et al., 1982); and (3) antibodies
comprise
dimeric associations of two heavy chains which place two oligosaccharide units
in
3
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
direct contact with each other, so that the inimunoglobulin molecule involves
both
specific protein-carbohydrate and carbohydrate-carbohydrate interactions.
It has been shown that different glycosylation patterns of Igs are associated
with different biological properties (Jefferis and Lund, 1997, Antibody Eng.
Claem.
Immunol., 65: 111-128; Wright and Morrison, 1997, Trends Biotechnol., 15: 26-
32).
However, only a few specific glycoforms are known to confer desired biological
functions. For example, an immunoglobulin composition having decreased
fucosylation on N-linked glycans is reported to have enhanced binding to human
FcyRIII and therefore enhanced antibody-dependent cellular cytotoxicity (ADCC)
(Shields et al., 2002, J. Biol Claern, 277: 26733-26740; Shinkawa et al.,
2003, J. Biol.
Chem. 278: 3466-3473). And, compositions of fucosylated G2 (Gal2GIcNAc2_
Man3GlcNAc2) IgG made in CHO cells reportedly increase complement-dependent
cytotoxicity (CDC) activity to a greater extent than compositions of
heterogenous
antibodies (Raju, 2004, US Pat. Appl. No. 2004/0136986). It has also been
suggested
that an optimal antibody against tumors would be one that bound preferentially
to
activate Fc receptors (FcyRI, FcyRIIa, FcyRIII) and minimally to the
inhibitory
FcyRIIb receptor (Clynes et al., 2000, Nature, 6:443-446). Therefore, the
ability to
enrich for specific glycoforms on Ig glycoproteins is highly desirable.
In general, the glycosylation structures (oligosaccharides) on glycoprotein
will
vary depending upon the expression host and culturing conditions. Therapeutic
proteins produced in non-human host cells are likely to contain non-human
glycosylation which may elicit an immunogenic response in humans-e.g.
hypermannosylation in yeast (Ballou, 1990, Metlaods Enz,ymol. 185:440-470);
a(1,3)-
fucose and (3(1,2)-xylose in plants, (Cabanes-Macheteau et al., 1999,
Glycobiology, 9:
365-372); N-glycolylneuraminic acid in Chinese hamster ovary cells (Noguchi et
al.,
4
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
1995. J. Biochem. 117: 5-62) and Gala-1,3Gal glycosylation in mice (Borrebaeck
et
al., 1993, Immun. Today, 14: 477-479). Furthermore, galactosylation can vary
with
cell culture conditions, which may render some immunoglobulin compositions
immunogenic depending on their specific galactose pattern (Patel et al., 1992.
Biochem J. 285: 839-845). The oligosaccharide structures of glycoproteins
produced
by non-human mammalian cells tend to be more closely related to those of human
glycoproteins. Thus, most commercial immunoglobulins are produced in mammalian
cells. However, mammalian cells have several important disadvantages as host
cells
for protein production. Besides being costly, processes for expressing
proteins in
mammalian cells produce heterogeneous populations of glycoforms, have low
volumetric titers, and require both ongoing viral containment and significant
time to
generate stable cell lines.
It is understood that different glycoforms can profoundly affect the
properties
of a therapeutic, including pharmacokinetics, pharmacodynamics, receptor-
interaction
and tissue-specific targeting (Graddis et al., 2002, Curr Pharm Biotechnol. 3:
285-
297). In particular, for antibodies, the oligosaccharide structure can affect
properties
relevant to protease resistance, the serum half-life of the antibody mediated
by the
FcRn receptor, binding to the complement complex Cl, which induces complement-
dependent cytoxicity (CDC), and binding to FcyR receptors, which are
responsible for
modulating the antibody-dependent cell-mediated cytoxicity (ADCC) pathway,
phagocytosis and antibody feedback. (Nose and Wigzell, 1983; Leatherbarrow and
Dwek, 1983; Leatherbarrow et al.,1985; Walker et al., 1989; Carter et al.,
1992, Proc.
Natl. Acad. Sci. USA, 89: 4285-4289).
Because different glycoforms are associated with different biological
properties, the ability to enrich for one or more specific glycoforms can be
used to
5
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
elucidate the relationship between a specific glycoform and a specific
biological
function. After a desired biological function is associated with a specific
glycoform
pattern, a glycoprotein composition enriched for the advantageous glycoform
structures can be produced. Thus, the ability to produce glycoprotein
compositions
that are enriched for particular glycoforms is highly desirable.
SUMMARY OF THE INVENTION
The present invention provides a composition comprising a plurality of
immunoglobulins, each immunoglobulin comprising at least one N-glycan attached
thereto wherein the composition thereby comprises a plurality of N-glycans in
which
the predonlinant N-glycan consists essentially of Man3GlcNAc2. In preferred
embodiments, greater than 50 mole percent of said plurality of N-glycans
consists
essentially of Man3G1cNAc2. More preferably, greater than 75 mole percent of
said
plurality of N-glycans consists essentially of Man3GlcNAc2. Most preferably,
greater
than 90 percent of said plurality of N-glycans consists essentially of
Man3GlcNAc2.
In other preferred embodiments, said Man3GlcNAc2 N-glycan structure is present
at a
level that is from about 5 mole percent to about 50 mole percent more than the
next
most predominant N-glycan structure of said plurality of N-glycans.
The present invention also provides methods for increasing binding to
FcyRIIIa and FcyRIIIb receptor and decreasing binding to FcyRIlb receptor by
enriching for a specific glycoform (e.g. Man3GlcNAc2) on an immunoglobulin. A
preferred embodiment provides a method for producing a composition comprising
a
plurality of immunoglobulins, each immmunoglobulin comprising at least one N-
glycan attached thereto wherein the composition thereby comprises a plurality
of N-
glycans in which the predominant N-glycan consists essentially of Man3GlcNAc2,
said
6
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
method comprising the step of culturing a host cell that has been engineered
or
selected to express said immunoglobulin or fragment thereof. Another preferred
embodiment provides a method for producing a composition comprising a
plurality of
immunoglobulins, each immunoglobulin comprising at least one N-glycan attached
thereto wherein the composition thereby comprises a plurality of N-glycans in
which
the predominant N-glycan consists essentially of Man3GlcNAc2, said method
comprising the step of culturing a lower eukaryotic host cell that has been
engineered
or selected to express said immunoglobulin or fragment thereof. In other
embodiments of the present invention, a host cell comprises an exogenous gene
encoding an immunoglobulin or fragment thereof, said host cell is engineered
or
selected to express said immunoglobulin or fragment thereof, thereby producing
a
composition comprising a plurality of immunoglobulins, each immunoglobulin
comprising at least one N-glycan attached thereto wherein the composition
thereby
comprises a plurality of N-glycans in which the predominant N-glycan consists
essentially of Man3GlcNAc2. In still other embodiments of the present
invention, a
lower eukaryotic host cell comprises an exogenous gene encoding an
immunoglobulin
or fragment thereof, said host cell is engineered or selected to express said
immunoglobulin or fragment thereof, thereby producing a composition comprising
a
plurality of immunoglobulins, each immunoglobulin comprising at least one N-
glycan
attached thereto wherein the composition thereby comprises a plurality of N-
glycans
in which the predominant N-glycan consists essentially of Man3GlcNAc2.
In preferred embodiments of the present invention, a composition comprising
a plurality of immunoglobulins, each immunoglobulin comprising at least one N-
glycan attached thereto wherein the composition thereby comprises a plurality
of N-
glycans in which the predominant N-glycan consists essentially of Man3GlcNAc2
7
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
wherein said immunoglobulins exhibit decreased binding affinity to FcyRIlb
receptor.
In other preferred embodiments of the present invention, a composition
comprising a
plurality of immunoglobulins, each immunoglobulin comprising at least one N-
glycan
attached thereto wherein the composition thereby comprises a plurality of N-
glycans
in which the predominant N-glycan consists essentially of Man3GlcNAc2 wherein
said
immunoglobulins exhibit increased binding affinity to FcyRIIIa and FcyRIIIb
receptor. In still another preferred embodiment of the present invention, a
composition comprising a plurality of immunoglobulins each immunoglobulin
comprising at least one N-glycan attached thereto wherein the composition
thereby
comprises a plurality of N-glycans in which the predominant N-glycan consists
essentially of Man3GlcNAc2 wherein said immunoglobulins exhibit increased
antibody-dependent cellular cytoxicity (ADCC).
In one embodiment the composition of the present invention comprises
immunoglobulins which are essentially free of fucose. In another embodiment,
the
composition of the present invention comprises immunoglobulins which lack
fucose.
The composition of the present invention also comprises a pharmaceutical
composition and a pharmaceutically acceptable carrier. The composition of the
present invention also comprises a pharmaceutical composition of
immunoglobulins
which have been purified and incorporated into a diagnostic kit.
Accordingly, the present invention provides materials and methods for
production of compositions of glycoproteins having predetermined glycosylation
structures, in particular, immunoglobulin or antibody molecules having N-
glycans
consisting essentially of Man3GlcNAc2.
8
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
BRIEF DESCRIPTION OF THE DRAWINGS
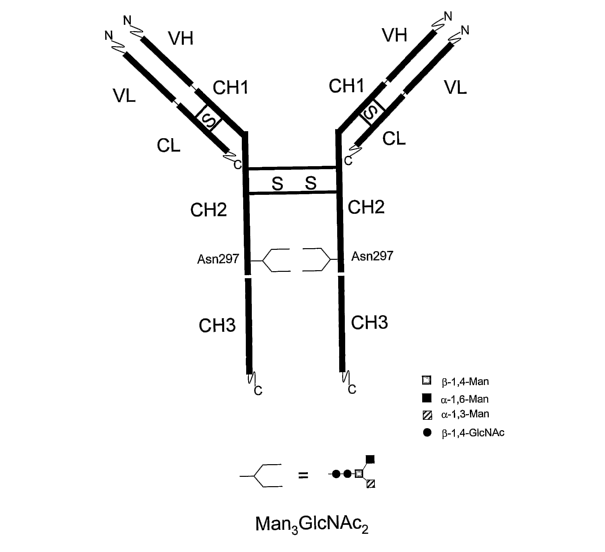
Figure 1. Schematic representation of an IgG molecule having a Man3GlcNAc2 N-
glycan structure.
Figure 2. Coomassie blue stained SDS-PAGE gel of JC-IgG expressed in YAS309
(as described in Example 2 and purified from the culture medium (as described
in
Example 3) over a Protein A column (lane 1) and a phenyl sepharose column
(lane
2). (3.0 g protein/lane.)
Figure 3. Coomassie blue stained SDS-PAGE gel of DX-IgG expressed in YAS309
(as described in Example 2) and purified from the culture medium (as described
in
Example 3) over a Protein A column (lane 1) and a phenyl sepharose column
(lane
2). (3.5 g protein/lane).
Figure 4A. MALDI-TOF spectra of JC-IgG expressed in YAS309, treated with
galactosidase and hexosaminidase showing predominantly Man3GlcNAc2 N-glycans.
Figure 4B. MALDI-TOF spectrum of DX-IgG expressed in YAS309, treated with
galactosidase and hexosaminidase showing predominantly Man3GlcNAc2 N-glycans.
Figure 5A. ELISA binding assay of FcyRIIIb with JC-IgG and Rituximab . Figure
5B. ELISA binding assay of FcyRIIIb with DX-IgG and Rituximab . (M3=
Man3G1cNAc2 N-glycan).
Figure 6. ELISA binding assay of FcyRIIIa-158F with JC-IgG and Rituximab .
(M3= Man3GlcNAc2 N-glycan).
Figure 7A. ELISA binding assay of FcyRIIb with JC-IgG and Rituximab . Figure
7B. ELISA binding assay of FcgRIIb with DX-IgG and Rituximab . (M3=
Man3GlcNAc2 N-glycan).
BRIEF DESCRIPTION OF THE SEQUENCES
SEQ ID NO: 1 encodes the nucleotide sequence of the murine variable and human
constant regions of DX-IgGl light chain.
SEQ ID NO: 2 encodes the nucleotide sequence of the murine variable and human
constant regions of DX-IgGl heavy chain.
SEQ ID NO: 3 encodes the nucleotide sequence of the human constant region of
an
IgGl light chain.
SEQ ID NO: 4 encodes the nucleotide sequence of the human constant region of
an
IgG 1 heavy chain.
9
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
SEQ ID NO: 5 to 19 encode 15 overlapping oligonucleotides used to synthesize
by
polymerase chain reaction (PCR) the murine light chain variable region of DX-
IgGl.
SEQ ID NO: 20 to 23 encode four oligonucleotide primers used to ligate the DX-
IgGl murine light chain variable region to a human light chain constant
region.
SEQ ID NO: 24 to 40 encode 17 overlapping oligonucleotides used to synthesize
by
PCR the murine heavy chain variable region of DX-IgGl.
SEQ ID NO: 41 to 44 encode four oligonucleotide primers used to ligate the DX-
IgGl murine heavy chain variable region to a human heavy chain constant
region.
SEQ ID NO: 45 encodes the nucleotide sequence encoding the Kar2 (Bip) signal
sequence with an N-terminal EcoRl site.
SEQ ID NO: 46 to 49 encode four oligonucleotide primers used to ligate the
Kar2
signal sequence to the light and heavy chains of DX-IgGI.
SEQ ID NO: 50 encodes the nucleotide sequence corresponding to the murine IgGl
variable region of the JC-IgGl light chain (GenBank #AF013576).
SEQ ID NO: 51 encodes the nucleotide sequence corresponding to the murine IgGI
variable region of the JC-IgGl heavy chain (GenBank #AF013577).
SEQ ID NO: 52 to 63 encode 12 overlapping oligonucleotide sequences used to
PCR-synthesize the murine light chain variable region of JC-IgGI.
SEQ ID NO: 64 to 75 encode 12 overlapping oligonucleotides used to PCR-
synthesize the murine heavy chain Fab fragment of JC-IgG1.
SEQ ID NO: 76 to 87 encode 12 overlapping oligonucleotides used to synthesize
by
PCR the murine heavy chain Fc fragment of JC-IgGl.
SEQ ID NO: 88 encodes a 3' Kpnl primer corresponding to the 3' end of the Fc
fragment.
SEQ ID NO: 89 encodes the nucleotide sequence for human serum albumin (HSA).
SEQ ID NO: 90 encodes the nucleotide sequence for thrombin cleavage used in
the
present invention.
DETAILED DESCRIPTION OF THE INVENTION
Unless otherwise defined herein, scientific and technical terms and phrases
used in connection with the present invention shall have the meanings that are
commonly understood by those of ordinary skill in the art. Further, unless
otherwise
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
required by context, singular terms shall include the plural and plural terms
shall
include the singular. Generally, nomenclatures used in connection with, and
techniques of biochemistry, enzymology, molecular and cellular biology,
microbiology, genetics and protein and nucleic acid chemistry and
hybridization
described herein are those well known and commonly used in the art. The
methods
and techniques of the present invention are generally performed according to
conventional methods well known in the art and as described in various general
and
more specific references that are cited and discussed throughout the present
specification unless otherwise indicated. See, e.g., Sambrook et al. Molecular
Cloning: A Labonatofy Manual, 2d ed., Cold Spring Harbor Laboratory Press,
Cold
Spring Harbor, N.Y. (1989); Ausubel et al., Current Protocols in Molecular
Biology,
Greene Publishing Associates (1992, and Supplements to 2002); Harlow and Lane,
Antibodies: A Laboratofy Manual, Cold Spring Harbor Laboratory Press, Cold
Spring
Harbor, N.Y. (1990); Taylor and Drickamer, Introduction to Glycobiology,
Oxford
Univ. Press (2003); Wof thington Enzyme Manual, Worthington Biochemical Corp.,
Freehold, NJ; Handbook of Biochemistry: Section A Proteins, Vol I, CRC Press
(1976); Handbook ofBiochernistry: Section A Proteins, Vol II, CRC Press
(1976);
Essentials of Glycobiology, Cold Spring Harbor Laboratory Press (1999);
Immunobiology, Janeway et al, 6th Edition, 2004, Garland Publishing, New York)
All publications, patents and other references mentioned herein are hereby
incorporated by reference in their entireties.
The following terms, unless otherwise indicated, shall be understood to have
the following meanings:
As used herein, the terms "N-glycan","glycan" and "glycoform" are used
interchangeably and refer to an N-linked oligosaccharide, e.g., one that is or
was
11
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
attached by an N-acetylglucosamine residue linked to the amide nitrogen of an
asparagine residue in a protein. The predominant sugars found on glycoproteins
are
glucose, galactose, mannose, fucose, N-acetylgalactosamine (GaINAc), N-
acetylglucosamine (G1cNAc) and sialic acid (e.g.,1V-acetyl-neuraminic acid
(NANA)). The processing of the sugar groups occurs cotranslationally in the
lumen
of the ER and continues in the Golgi apparatus for N-linked glycoproteins.
N-glycans have a common pentasaccharide core of Man3GlcNAc2 ("Man"
refers to mannose; "Glc" refers to glucose; and "NAc" refers to N-acetyl;
GIcNAc
refers to N-acetylglucosamine). N-glycans differ with respect to the number of
branches (antennae) comprising peripheral sugars (e.g., G1cNAc, galactose,
fucose
and sialic acid) that are added to the Man3GlcNAc2 ("Man3") core structure
which is
also referred to as the "trimannose core", the "pentasaccharide core" or the
"paucimannose core". N-glycans are classified according to their branched
constituents (e.g., high mannose, complex or hybrid). A "high mannose" type N-
glycan has five or more mannose residues. A "complex" type N-glycan typically
has
at least one G1cNAc attached to the 1,3 mannose arm and at least one G1cNAc
attached to the 1,6 mannose arm of a "trimannose" core. Complex N-glycans may
also have galactose ("Gal") or N-acetylgalactosamine ("Ga1NAc") residues that
are
optionally modified with sialic acid or derivatives (e.g., "NANA" or "NeuAc",
where
"Neu" refers to neuraminic acid and "Ac" refers to acetyl). Complex N-glycans
may
also have intrachain substitutions comprising "bisecting" G1cNAc and core
fucose
("Fuc"). Complex N-glycans may also have multiple antennae on the "trimannose
core," often referred to as "multiple antennary glycans." A "hybrid" N-glycan
has at
least one GIcNAc on the terminal of the 1,3 mannose arm of the trimannose core
and
12
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
zero or more mannoses on the 1,6 mannose arm of the trimannose core. The
various
N-glycans are also referred to as "glycoforms."
Abbreviations used herein are of common usage in the art, see, e.g.,
abbreviations of sugars, above. Other common abbreviations include "PNGase",
or
"glycanase" or "glucosidase" which all refer to peptide N-glycosidase F (EC
3.2.2.18).
An "isolated" or "substantially pure" nucleic acid or polynucleotide (e.g., an
RNA, DNA or a mixed polymer) is one which is substantially separated from
other
cellular components that naturally accompany the native polynucleotide in its
natural
host cell, e.g., ribosomes, polymerases and genomic sequences with which it is
naturally associated. The term embraces a nucleic acid or polynucleotide that
(1) has
been removed from its naturally occurring environment, (2) is not associated
with all
or a portion of a polynucleotide in which the "isolated polynucleotide" is
found in
nature, (3) is operatively linked to a polynucleotide which it is not linked
to in nature,
or (4) does not occur in nature. The term "isolated" or "substantially pure"
also can
be used in reference to recombinant or cloned DNA isolates, chemically
synthesized
polynucleotide analogs, or polynucleotide analogs that are biologically
synthesized by
heterologous systems.
However, "isolated" does not necessarily require that the nucleic acid or
polynucleotide so described has itself been physically removed from its native
environment. For instance, an endogenous nucleic acid sequence in the genome
of an
organism is deemed "isolated" herein if a heterologous sequence is placed
adjacent to
the endogenous nucleic acid sequence, such that the expression of this
endogenous
nucleic acid sequence is altered. In this context, a heterologous sequence is
a
sequence that is not naturally adjacent to the endogenous nucleic acid
sequence,
13
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
whether or not the heterologous sequence is itself endogenous (originating
from the
same host cell or progeny thereof) or exogenous (originating from a different
host cell
or progeny thereof). By way of example, a promoter sequence can be substituted
(e.g., by homologous recombination) for the native promoter of a gene in the
genome
of a host cell, such that this gene has an altered expression pattern. This
gene would
now become "isolated" because it is separated from at least some of the
sequences
that naturally flank it.
A nucleic acid is also considered "isolated" if it contains any modifications
that do not naturally occur to the corresponding nucleic acid in a genome. For
instance, an endogenous coding sequence is considered "isolated" if it
contains an
insertion, deletion or a point mutation introduced artificially, e.g., by
human
intervention. An "isolated nucleic acid" also includes a nucleic acid
integrated into a
host cell chromosome at a heterologous site and a nucleic acid construct
present as an
episome. Moreover, an "isolated nucleic acid" can be substantially free of
other
cellular material, or substantially free of culture medium when produced by
recombinant techniques, or substantially free of chemical precursors or other
chemicals when chemically synthesized.
As used herein, the phrase "degenerate variant" of a reference nucleic acid
sequence encompasses nucleic acid sequences that can be translated, according
to the
standard genetic code, to provide an amino acid sequence identical to that
translated
from the reference nucleic acid sequence. The tenn "degenerate
oligonucleotide" or
"degenerate primer" is used to signify an oligonucleotide capable of
hybridizing with
target nucleic acid sequences that are not necessarily identical in sequence
but that are
homologous to one another within one or more particular segments.
14
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The term "percent sequence identity" or "identical" in the context of nucleic
acid sequences refers to the residues in the two sequences which are the same
when
aligned for maximum correspondence. The length of sequence identity comparison
may be over a stretch of at least about nine nucleotides, usually at least
about 20
nucleotides, more usually at least about 24 nucleotides, typically at least
about 28
nucleotides, more typically at least about 32 nucleotides, and preferably at
least about
36 or more nucleotides. There are a number of different algorithms known in
the art
which can be used to measure nucleotide sequence identity. For instance,
polynucleotide sequences can be compared using FASTA, Gap or Bestfit, which
are
programs in Wisconsin Package Version 10.0, Genetics Computer Group (GCG),
Madison, Wisconsin. FASTA provides alignments and percent sequence identity of
the regions of the best overlap between the query and search sequences.
Pearson,
Methods Enzymal. 183:63-98 (1990) (hereby incorporated by reference in its
entirety).
For instance, percent sequence identity between nucleic acid sequences can be
determined using FASTA with its default parameters (a word size of 6 and the
NOPAM factor for the scoring matrix) or using Gap with its default parameters
as
provided in GCG Version 6.1, herein incorporated by reference. Alternatively,
sequences can be compared using the computer program, BLAST (Altschul et al.,
J.
Mol. Biol. 215:403-410 (1990); Gish and States, Nature Genet. 3:266-272
(1993);
Madden et al., Meth. Enz,ymol. 266:131-141 (1996); Altschul et al., Nucleic
Acids
Res. 25:3389-3402 (1997); Zhang and Madden, Genonae Res. 7:649-656 (1997)),
especially blastp or tblastn (Altschul et al., Nucleic Acids Res. 25:3389-3402
(1997)).
The term "substantial homology" or "substantial similarity," when referring to
a nucleic acid or fragment thereof, indicates that, when optimally aligned
with
appropriate nucleotide insertions or deletions with another nucleic acid (or
its
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
complementary strand), there is nucleotide sequence identity in at least about
50%,
more preferably 60% of the nucleotide bases, usually at least about 70%, more
usually
at least about 80%, preferably at least about 90%, and more preferably at
least about
95%, 96%, 97%, 98% or 99% of the nucleotide bases, as measured by any well-
known algorithm of sequence identity, such as FASTA, BLAST or Gap, as
discussed
above.
Alternatively, substantial homology or similarity exists when a nucleic acid
or
fragment thereof hybridizes to another nucleic acid, to a strand of another
nucleic
acid, or to the complementary strand thereof, under stringent hybridization
conditions.
"Stringent hybridization conditions" and "stringent wash conditions" in the
context of
nucleic acid hybridization experiments depend upon a number of different
physical
parameters. Nucleic acid hybridization will be affected by such conditions as
salt
concentration, temperature, solvents, the base composition of the hybridizing
species,
length of the complementary regions, and the number of nucleotide base
mismatches
between the hybridizing nucleic acids, as will be readily appreciated by those
skilled
in the art. One having ordinary skill in the art knows how to vary these
parameters to
achieve a particular stringency of hybridization.
In general, "stringent hybridization" is performed at about 25 C below the
thermal melting point (Tm) for the specific DNA hybrid under a particular set
of
conditions. "Stringent washing" is performed at temperatures about 5 C lower
than
the Trõ for the specific DNA hybrid under a particular set of conditions. The
T,,, is the
temperature at which 50% of the target sequence hybridizes to a perfectly
matched
probe. See Sambrook et al., Molecular Clorting: A Laboratory Manual, 2d ed.,
Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1989), page 9.51,
hereby
incorporated by reference. For purposes herein, "stringent conditions" are
defined for
16
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
solution phase hybridization as aqueous hybridization (i.e., free of
formamide) in 6X
SSC (where 20X SSC contains 3.0 M NaCl and 0.3 M sodium citrate), 1% SDS at 65
C for 8-12 hours, followed by two washes in 0.2X SSC, 0.1% SDS at 65 C for 20
minutes. It will be appreciated by the skilled worker that hybridization at 65
C will
occur at different rates depending on a number of factors including the length
and
percent identity of the sequences which are hybridizing.
The term "mutated" when applied to nucleic acid sequences means that
nucleotides in a nucleic acid sequence may be inserted, deleted or changed
compared
to a reference nucleic acid sequence. A single alteration may be made at a
locus (a
point mutation) or multiple nucleotides may be inserted, deleted or changed at
a single
locus. In addition, one or more alterations may be made at any number of loci
within
a nucleic acid sequence. A nucleic acid sequence may be mutated by any method
known in the art including but not limited to mutagenesis techniques such as
"error-
prone PCR" (a process for performing PCR under conditions where the copying
fidelity of the DNA polymerase is low, such that a high rate of point
mutations is
obtained along the entire length of the PCR product; see, e.g., Leung et al.,
Technique, 1:11-15 (1989) and Caldwell and Joyce, PCR Methods Applic. 2:28-33
(1992)); and "oligonucleotide-directed mutagenesis" (a process which enables
the
generation of site-specific mutations in any cloned DNA segment of interest;
see, e.g.,
Reidhaar-Olson and Sauer, Science 241:53-57 (1988)).
The term "vector" as used herein is intended to refer to a nucleic acid
molecule capable of transporting another nucleic acid to which it has been
linked. One
type of vector is a "plasmid", which refers to a circular double stranded DNA
loop
into which additional DNA segments may be ligated. Other vectors include
cosmids,
bacterial artificial chromosomes (BAC) and yeast artificial chromosomes (YAC).
17
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
Another type of vector is a viral vector, wherein additional DNA segments may
be
ligated into the viral genome (discussed in more detail below). Certain
vectors are
capable of autonomous replication in a host cell into which they are
introduced (e.g.,
vectors having an origin of replication which functions in the host cell).
Other vectors
can be integrated into the genome of a host cell upon introduction into the
host cell,
and are thereby replicated along with the host genome. Moreover, certain
preferred
vectors are capable of directing the expression of genes to which they are
operatively
linked. Such vectors are referred to herein as "recombinant expression
vectors" (or
simply, "expression vectors").
As used herein, the term "sequence of interest" or "gene of interest" refers
to a
nucleic acid sequence, typically encoding a protein, that is not normally
produced in
the host cell. The methods disclosed herein allow one or more sequences of
interest
or genes of interest to be stably integrated into a host cell genome. Non-
limiting
examples of sequences of interest include sequences encoding one or more
polypeptides having an enzymatic activity, e.g., an enzyme which affects N-
glycan
synthesis in a host such as mannosyltransferases, N-
acetylglucosaminyltransferases,
UDP-N-acetylglucosamine transporters, galactosyltransferases, UDP-N-
acetylgalactosyltransferase, sialyltransferases and fucosyltransferases.
The term "marker sequence" or "marker gene" refers to a nucleic acid
sequence capable of expressing an activity that allows either positive or
negative
selection for the presence or absence of the sequence within a host cell. For
example,
the P. pastoris ZIRA5 gene is a marker gene because its presence can be
selected for
by the ability of cells containing the gene to grow in the absence of uracil.
Its
presence can also be selected against by the inability of cells containing the
gene to
grow in the presence of 5-FOA. Marker sequences or genes do not necessarily
need
18
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
to display both positive and negative selectability. Non-limiting examples of
marker
sequences or genes from P. pastoris include ADEl, ARG4, HIS4 and URA3. For
antibiotic resistance marker genes, kanamycin, neomycin, geneticin (or G41 8),
paromomycin and hygromycin resistance genes are commonly used to allow for
growth in the presence of these antibiotics.
"Operatively linked" expression control sequences refers to a linkage in which
the expression control sequence is contiguous with the gene of interest to
control the
gene of interest, as well as expression control sequences that act in trans or
at a
distance to control the gene of interest.
The term "expression control sequence" as used herein refers to
polynucleotide sequences which are necessary to affect the expression of
coding
sequences to which they are operatively linked. Expression control sequences
are
sequences which control the transcription, post-transcriptional events and
translation
of nucleic acid sequences. Expression control sequences include appropriate
transcription initiation, termination, promoter and enhancer sequences;
efficient RNA
processing signals such as splicing and polyadenylation signals; sequences
that
stabilize cytoplasmic mRNA; sequences that enhance translation efficiency
(e.g.,
ribosome binding sites); sequences that enhance protein stability; and when
desired,
sequences that enhance protein secretion. The nature of such control sequences
differs
depending upon the host organism; in prokaryotes, such control sequences
generally
include promoter, ribosomal binding site, and transcription termination
sequence.
The term "control sequences" is intended to include, at a minimum, all
components
whose presence is essential for expression, and can also include additional
components whose presence is advantageous, for example, leader sequences and
fusion partner sequences.
19
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The term "recombiriant host cell" ("expression host cell", "expression host
system", "expression system" or simply "host cell"), as used herein, is
intended to
refer to a cell into which a recombinant vector has been introduced. It should
be
understood that such terms are intended to refer not only to the particular
subject cell
but to the progeny of such a cell. Because certain modifications may occur in
succeeding generations due to either mutation or environmental influences,
such
progeny may not, in fact, be identical to the parent cell, but are still
included within
the scope of the term "host cell" as used herein. A recombinant host cell may
be an
isolated cell or cell line grown in culture or may be a cell which resides in
a living
tissue or organism.
The term "eukaryotic" refers to a nucleated cell or organism, and includes
insect cells, plant cells, mammalian cells, animal cells and lower eukaryotic
cells.
The term "lower eukaryotic cells" includes yeast, fungi, collar-flagellates,
microsporidia, alveolates (e.g., dinoflagellates), stramenopiles (e.g, brown
algae,
protozoa), rhodophyta (e.g., red algae), plants (e.g., green algae, plant
cells, moss) and
other protists. Yeast and fungi include, but are not limited to: Pichia sp.,
such as
Pichia pastoYis, Pichia ftnlandica, Pichia trehalophila, Pichia koclanaae,
Pichia
menzbranaefaciens, Pichia minuta (Ogataea minuta, Pichia lindneri), Pichia
opuntiae, Pichia tlterntotolerans, Pichia salictaria, Pichia guercuum, Pichia
puperi,
Pichia stiptis andPichia ntethanolica;. Saccharomyces sp., such as
Saccharom.yces
cerevisiae; Hansenula polymorpha, Kluyverornyces sp., such as Kluyverontyces
lactis;
Candida albicans, Aspergillus nidulans, Aspergillus ttiger, Aspergillus
otyzae,
Trichodernaa reesei, ClatysospoYiunz luckttowense, Fusariuna sp., such as
Fusariutn
gramineunt, Fusarium venen.aturn; Pltyscomitrella patens and Neurospora
crassa.
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The term "peptide" as used herein refers to a short polypeptide, e.g., one
that
is typically less than about 50 amino acids long and more typically less than
about 30
amino acids long. The term as used herein encompasses analogs and mimetics
that
mimic structural and thus biological function.
The term "polypeptide" encompasses both naturally-occurring and non-
naturally-occurring proteins, and fragments, mutants, derivatives and analogs
thereof.
A polypeptide may be monomeric or polymeric. Further, a polypeptide may
comprise
a number of different domains each of which has one or more distinct
activities.
The temi "isolated protein" or "isolated polypeptide" is a protein or
polypeptide that by virtue of its origin or source of derivation (1) is not
associated
with naturally associated components that accompany it in its native state,
(2) exists in
a purity not found in nature, where purity can be adjudged with respect to the
presence of other cellular material (e.g., is free of other proteins from the
same
species) (3) is expressed by a cell from a different species, or (4) does not
occur in
nature (e.g., it is a fragment of a polypeptide found in nature or it includes
amino acid
analogs or derivatives not found in nature or linkages other than standard
peptide
bonds). Thus, a polypeptide that is chemically synthesized or synthesized in a
cellular
system different from the cell from which it naturally originates will be
"isolated"
from its naturally associated components. A polypeptide or protein may also be
rendered substantially free of naturally associated components by isolation,
using
protein purification techniques well known in the art. As thus defined,
"isolated"
does not necessarily require that the protein, polypeptide, peptide or
oligopeptide so
described has been physically removed from its native environment.
The term "polypeptide fragment" as used herein refers to a polypeptide that
has a deletion, e.g., an amino-terminal and/or carboxy-terminal deletion
compared to
21
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
a full-length polypeptide. In a preferred embodiment, the polypeptide fragment
is a
contiguous sequence in which the amino acid sequence of the fragment is
identical to
the corresponding positions in the naturally-occurring sequence. Fragments
typically
are at least 5, 6, 7, 8, 9 or 10 amino acids long, preferably at least 12, 14,
16 or 18
amino acids long, more preferably at least 20 amino acids long, more
preferably at
least 25, 30, 35, 40 or 45, amino acids, even more preferably at least 50 or
60 amino
acids long, and even more preferably at least 70 amino acids long.
A "modified derivative" refers to polypeptides or fragments thereof that are
substantially homologous in primary structural sequence but which include,
e.g., in
vivo or in vitro chemical and biochemical modifications or which incorporate
amino
acids that are not found in the native polypeptide. Such modifications
include, for
example, acetylation, carboxylation, phosphorylation, glycosylation,
ubiquitination,
labeling, e.g., with radionuclides, and various enzymatic modifications, as
will be
readily appreciated by those skilled in the art. A variety of methods for
labeling
polypeptides and of substituents or labels useful for such purposes are well
known in
the art, and include radioactive isotopes such as 1u1, 32P, 35S, and 3H,
ligands which
bind to labeled antiligands (e.g., antibodies), fluorophores, chemiluminescent
agents,
enzymes, and antiligands which can serve as specific binding pair members for
a
labeled ligand. The choice of label depends on the sensitivity required, ease
of
conjugation with the primer, stability requirements, and available
instrumentation.
Methods for labeling polypeptides are well known in the art. See, e.g.,
Ausubel et al.,
Currefat Protocols in Molecular Biology, Greene Publishing Associates (1992,
and
Supplements to 2002) (hereby incorporated by reference).
The term "fusion protein" refers to a polypeptide comprising a polypeptide or
fragment coupled to heterologous amino acid sequences. Fusion proteins are
useful
22
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
because they can be constructed to contain two or more desired functional
elements
from two or more different proteins. A fusion protein comprises at least 10
contiguous amino acids from a polypeptide of interest, more preferably at
least 20 or
30 amino acids, even more preferably at least 40, 50 or 60 amino acids, yet
more
preferably at least 75, 100 or 125 amino acids. Fusions that include the
entirety of the
proteins of the present invention have particular utility. The heterologous
polypeptide
included within the fusion protein of the present invention is at least 6
amino acids in
length, often at least 8 amino acids in length, and usefully at least 15, 20,
and 25
amino acids in length. Fusions that include larger polypeptides, such as an
immunoglobulin Fc fragment, or an immunoglobulin Fab fragment or even entire
proteins, such as the green fluorescent protein ("GFP") chromophore-containing
proteins or a full length immunoglobulin having particular utility. Fusion
proteins can
be produced recombinantly by constructing a nucleic acid sequence which
encodes
the polypeptide or a fragment thereof in frame with a nucleic acid sequence
encoding
a different protein or peptide and then expressing the fusion protein.
Alternatively, a
fusion protein can be produced chemically by crosslinking the polypeptide or a
fragment thereof to another protein.
As used herein, the terms "antibody", "immunoglobulin","Ig" and "Ig
molecule" are used interchangeably. Each antibody molecule has a unique
structure
that allows it to bind its specific antigen, but all
antibodies/immunoglobulins have the
same overall structure as described herein. The basic antibody structural unit
is
known to comprise a tetramer of subunits. Each tetramer has two identical
pairs of
polypeptide chains, each pair having one "light" chain (about 25 kDa) and one
"heavy" chain (about 50-70 kDa). The amino-terminal portion of each chain
includes
a variable region of about 100 to 110 or more amino acids primarily
responsible for
23
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
antigen recognition. The carboxy-terminal portion of each chain defines a
constant
region primarily responsible for effector function. Light chains are
classified as either
kappa or lambda. Heavy chains are classified as gamma, mu, alpha, delta, or
epsilon,
and define the antibody's isotype as IgG, IgM, IgA, IgD and IgE, respectively.
The
light and heavy chains are subdivided into variable regions and constant
regions (See
generally, Fundamental Immunology (Paul, W., ed., 2nd ed. Raven Press, N.Y.,
1989), Ch. 7 (incorporated by reference in its entirety for all purposes). The
variable
regions of each light/heavy chain pair form the antibody binding site. Thus,
an intact
antibody has two binding sites. Except in bifunctional or bispecific
antibodies, the two
binding sites are the same. The chains all exhibit the same general structure
of
relatively conserved framework regions (FR) joined by three hypervariable
regions,
also called complementarity determining regions or CDRs. The CDRs from the two
chains of each pair are aligned by the framework regions, enabling binding to
a
specific epitope. The terms include naturally occurring forms, as well as
fragments
and derivatives. Included within the scope of the term are classes of Igs,
namely, IgG,
IgA, IgE, IgM, and IgD. Also included within the scope of the terms are the
subtypes
of IgGs, namely, IgGI, IgG2, IgG3 and IgG4. The term is used in the broadest
sense
and includes single monoclonal antibodies (including agonist and antagonist
antibodies) as well as antibody compositions which will bind to multiple
epitopes or
antigens. The terms specifically cover monoclonal antibodies (including full
length
monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g.,
bispecific antibodies), and antibody fragments so long as they contain or are
modified
to contain at least the portion of the CH2 domain of the heavy chain
immunoglobulin
constant region which comprises an N-linked glycosylation site of the CH2
domain, or
a variant thereof. Included within the terms are molecules comprising the Fc
region,
24
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
such as immunoadhesins (US Pat. Appl. No. 2004/0136986), Fc fusions and
antibody-
like molecules. Alternatively, these terms can refer to an antibody fragment
of at
least the Fab region that at least contains an N-linked glycosylation site.
The term "Fc" fragment refers to the 'fragment crystallized' C-terminal region
of the antibody containing the CH2 and CH3 domains (Figure 1). The term "Fab"
fragment refers to the 'fragment antigen binding' region of the antibody
containing
the VH, CH1, VL and CL domains (Figure 1).
The term "monoclonal antibody" (mAb) as used herein refers to an antibody
obtained from a population of substantially homogeneous antibodies, i.e., the
individual antibodies comprising the population are identical except for
possible
naturally occurring mutations that may be present in minor amounts. Monoclonal
antibodies are highly specific, being directed against a single antigenic
site.
Furthermore, in contrast to conventional (polyclonal) antibody preparations
which
typically include different antibodies directed against different determinants
(epitopes), each mAb is directed against a single determinarit on the antigen.
In
addition to their specificity, monoclonal antibodies are advantageous in that
they can
be synthesized by hybridoma culture, uncontaminated by other immunoglobulins.
The term "monoclonal" indicates the character of the antibody as being
obtained from
a substantially homogeneous population of antibodies, and is not to be
construed as
requiring production of the antibody by any particular method. For example,
the
monoclonal antibodies to be used in accordance with the present invention may
be
made by the hybridoma method first described by Kohler et al., (1975) Nature,
256:495, or may be made by recombinant DNA methods (see, e.g., U.S. Pat. No.
4,816,567 to Cabilly et al.).
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The monoclonal antibodies herein include hybrid and recombinant antibodies
produced by splicing a variable (including hypervariable) domain of an
antibody with
a constant domain (e.g. "humanized" antibodies), or a light chain with a heavy
chain,
or a chain from one species with a chain from another species, or fusions with
heterologous proteins, regardless of species of origin or immunoglobulin class
or
subclass designation, (See, e.g., U.S. Pat. No. 4,816,567 to Cabilly et al.;
Mage and
Lamoyi, in Monoclonal Antibody Production Techniques and Applications, pp. 79-
97
(Marcel Dekker, Inc., New York, 1987).) The monoclonal antibodies herein
specifically include "chimeric" antibodies (immunoglobulins) in which a
portion of
the heavy and/or light chain is identical with or homologous to corresponding
sequences in antibodies derived from a first species or belonging to a
particular
antibody class or subclass, while the remainder of the chain(s) is identical
with or
homologous to corresponding sequences in antibodies derived from a different
species
or belonging to a different antibody class or subclass, as well as fragments
of such
antibodies, so long as they contain or are modified to contain at least one
CH2 .
"Humanized" forms of non-human (e.g., murine) antibodies are specific chimeric
immunoglobulins, immunoglobulin chains or fragments thereof (such as Fv, Fab,
Fab', F(ab')2, or other antigen-binding subsequences of antibodies) which
contain
sequences derived from human immunoglobulins. An Fv fragment of an antibody is
the smallest unit of the antibody that retains the binding characteristics and
specificity
of the whole molecule. The Fv fragment is a noncovalently associated
heterodimer of
the variable domains of the antibody heavy chain and light chain. The F(ab)'2
fragment is a fragment containing both arms of Fab fragments linked by the
disulfide
bridges.
26
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The most common forms of humanized antibodies are human
immunoglobulins (recipient antibody) in which residues from a complementary-
determining region (CDR) of the recipient are replaced by residues from a CDR
of a
non-human species (donor antibody) such as mouse, rat, or rabbit having the
desired
specificity, affinity, and capacity. In some instances, Fv framework residues
of the
human immunoglobulin are replaced by corresponding non-human residues.
Furthermore, humanized antibodies can comprise residues which are found
neither in
the recipient antibody nor in the imported CDR or framework sequences. These
modifications are made to further refine and maximize antibody performance. In
general, the humanized antibody will comprise substantially all of at least
one, and
typically two, variable domains, in which all or substantially all of the CDR
regions
correspond to those of a non-human immunoglobulin and all or substantially all
of the
CDR regions are those of a human immunoglobulin consensus sequence. The
humanized antibody optimally also will comprise at least a portion of an
immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
For
further details see Jones et al., 1986, Nature 321:522-524; Reichmann et al.,
1988,
Nature 332:323-327, and Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
"Fragments" within the scope of the terms antibody or immunoglobulin
include those produced by digestion with various proteases, those produced by
chemical cleavage and/or chemical dissociation and those produced
recombinantly, so
long as the fragment remains capable of specific binding to a target molecule.
Among
such fragments are Fc, Fab, Fab', Fv, F(ab')2, and single chain Fv (scFv)
fragments.
Targets of interest for antibodies of the invention include growth factor
receptors (e.g., FGFR, PDGFR, EGFR, NGFR, and VEGF) and their ligands. Other
targets are G protein receptors and include substance K receptor, the
angiotensin
27
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
receptor, the a- and (3-adrenergic receptors, the serotonin receptors, and PAF
receptor.
See, e.g., Gilman, Ann. Rev.Biochem. 56:625-649 (1987). Other targets include
ion
channels (e.g., calcium, sodium, potassium channels), muscarinic receptors,
acetylcholine receptors, GABA receptors,glutamate receptors, and dopamine
receptors (see Harpold, U.S. 5,401,629 and U.S. 5,436,128). Other targets are
adhesion proteins such as integrins, selectins, and immunoglobulin superfamily
members (see Springer, Nature 346:425-433 (1990). Osbom, Cell 62:3 (1990);
Hynes, Cell 69:11 (1992)). Other targets are cytokines, such as interleukins
IL-1
through IL- 13, tumor necrosis factors a&(3, interferons a, 0 and y, tumor
growth
factor Beta (TGF-0), colony stimulating factor (CSF) and granulocyte monocyte
colony stimulating factor (GMCSF). See Human Cytokines: Handbook for Basic &
Clinical Research (Aggrawal et al. eds., Blackwell Scientific, Boston, MA
1991).
Other targets are hormones, enzymes, and intracellular and intercellular
messengers,
such as, adenyl cyclase, guanyl cyclase, and phospholipase C. Other targets of
interest are leukocyte antigens, such as CD20, and CD33. Drugs may also be
targets
of interest. Target molecules can be human, mammalian or bacterial. Other
targets are
antigens, such as proteins, glycoproteins and carbohydrates from microbial
pathogens,
both viral and bacterial, and tumors. Still other targets are described in
U.S.
4,366,241.
Immune Fc receptors discussed herein, may include: FcyRI, FcyRIIa, FcyRIIb,
FcyRIIIa, FcyRIIIb and FcRn (neonatal receptor). The term FcyRI can refer to
any
FcyRI subtype unless specified otherwise. The term FcyRII can refer to any
FcyRII
receptor unless specified otherwise. The term FcyRIII refers to any FcyRIII
subtype
unless specified otherwise.
28
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
"Derivatives" within the scope of the term include antibodies (or fragments
thereof) that have been modified in sequence, but remain capable of specific
binding
to a target molecule, including: interspecies chimeric and humanized
antibodies;
antibody fusions; heteromeric antibody complexes and antibody fusions, such as
diabodies (bispecific antibodies), single-chain diabodies, and intrabodies
(see, e.g.,
Intracellular Antibodies: Research and Disease Applications, (Marasco, ed.,
Springer-Verlag New York, Inc., 1998).
The term "non-peptide analog" refers to a compound with properties that are
analogous to those of a reference polypeptide. A non-peptide compound may also
be
termed a"peptide mimetic" or a "peptidomimetic". See, e.g., Jones, Amino Acid
and
Peptide Synthesis, Oxford University Press (1992); Jung, Combinatorial Peptide
and
Nonpeptide Libraries: A Handbook, John Wiley (1997); Bodanszky et al., Peptide
Chemistry--A Practical Textbook, Springer Verlag (1993); Synthetic Peptides: A
Users Guide, (Grant, ed., W. H. Freeman and Co., 1992); Evans et al., J. Med.
Chena.
30:1229 (1987); Fauchere, J. Adv. Drug Res. 15:29 (1986); Veber and
Freidinger,
Trends Neurosci., 8:392-396 (1985); and references sited in each of the above,
which
are incorporated herein by reference. Such compounds are often developed with
the
aid of computerized molecular modeling. Peptide mimetics that are structurally
similar to useful peptides of the invention may be used to produce an
equivalent effect
and are therefore envisioned to be part of the invention.
Amino acid substitutions can include those which: (1) reduce susceptibility to
proteolysis, (2) reduce susceptibility to oxidation, (3) alter binding
affinity for
forming protein complexes, (4) alter binding affinity or enzymatic activity,
and (5)
confer or modify other physicochemical or functional properties of such
analogs.
29
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
As used herein, the twenty conventional amino acids and their abbreviations
follow conventional usage. See Immunology - A Synthesis (Golub and Gren eds.,
Sinauer Associates, Sunderland, Mass., 2d ed. 1991), which is incorporated
herein by
reference. Stereoisomers (e.g., D-amino acids) of the twenty conventional
amino
acids, unnatural amino acids such as a-, a-disubstituted amino acids,lV alkyl
amino
acids, and other unconventional amino acids may also be suitable components
for
polypeptides of the present invention. Examples of unconventional amino acids
include: 4-hydroxyproline, y-carboxyglutamate, $-N,N,N-trimethyllysine,
s-N-acetyllysine, O-phosphoserine, N-acetylserine, N-formylmethionine,
3-methylhistidine, 5-hydroxylysine, N-methylarginine, and other similar amino
acids
and imino acids (e.g., 4-hydroxyproline). In the polypeptide notation used
herein, the
left-hand end corresponds to the amino terminal end and the right-hand end
corresponds to the carboxy-terminal end, in accordance with standard usage and
convention.
A protein has "homology" or is "homologous" to a second protein if the
nucleic acid sequence that encodes the protein has a similar sequence to the
nucleic
acid sequence that encodes the second protein. Alternatively, a protein has
homology
to a second protein if the two proteins have "similar" amino acid sequences.
(Thus,
the term "homologous proteins" is defined to mean that the two proteins have
similar
amino acid sequences.) In a preferred embodiment, a homologous protein is one
that
exhibits at least 65% sequence homology to the wild type protein, more
preferred is at
least 70% sequence homology. Even more preferred are homologous proteins that
exhibit at least 75%, 80%, 85% or 90% sequence homology to the wild type
protein.
In a yet more preferred embodiment, a homologous protein exhibits at least
95%,
98%, 99% or 99.9% sequence identity. As used herein, homology between two
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
regions of amino acid sequence (especially with respect to predicted
structural
similarities) is interpreted as implying similarity in function.
When "homologous" is used in reference to proteins or peptides, it is
recognized that residue positions that are not identical often differ by
conservative
amino acid substitutions. A "conservative amino acid substitution" is one in
which an
amino acid residue is substituted by another amino acid residue having a side
chain (R
group) with similar chemical properties (e.g., charge or hydrophobicity). In
general, a
conservative amino acid substitution will not substantially change the
functional
properties of a protein. In cases where two or more amino acid sequences
differ from
each other by conservative substitutions, the percent sequence identity or
degree of
homology may be adjusted upwards to correct for the conservative nature of the
substitution. Means for making this adjustment are well known to those of
skill in the
art. See, e.g., Pearson, 1994,11Iethods Mol. Biol. 24:307-31 and 25:365-89
(herein
incorporated by reference).
The following six groups each contain amino acids that are conservative
substitutions for one another: 1) Serine (S), Threonine (T); 2) Aspartic Acid
(D),
Glutamic Acid (E); 3) Asparagine (N), Glutamine (Q); 4) Arginine (R), Lysine
(K);
5) Isoleucine (I), Leucine (L), Methionine (M), Alanine (A), Valine (V), and
6)
Phenylalanine (F), Tyrosine (Y), Tryptophan (W).
Sequence homology for polypeptides, which is also referred to as percent
sequence identity, is typically measured using sequence analysis software.
See, e.g.,
the Sequence Analysis Software Package of the Genetics Computer Group (GCG),
University of Wisconsin Biotechnology Center, 910 University Avenue, Madison,
Wisconsin 53705. Protein analysis software matches similar sequences using a
measure of homology assigned to various substitutions, deletions and other
31
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
modifications, including conservative amino acid substitutions. For instance,
GCG
contains programs such as "Gap" and "Bestfit" which can be used with default
parameters to determine sequence homology or sequence identity between closely
related polypeptides, such as homologous polypeptides from different species
of
organisms or between a wild-type protein and a mutein thereof. See, e.g., GCG
Version 6.1.
A preferred algorithm when comparing a particular polypepitde sequence to a
database containing a large number of sequences from different organisms is
the
computer program BLAST (Altschul et al., J. Mol. Biol. 215:403-410 (1990);
Gish
and States, Nature Genet. 3:266-272 (1993); Madden et al., Meth. Enz.ymol.
266:131-
141 (1996); Altschul et al., Nucleic Acids Res. 25:3389-3402 (1997); Zhang and
Madden, Genorne Res. 7:649-656 (1997)), especially blastp or tblastn (Altschul
et al.,
Nucleic Acids Res. 25:3389-3402 (1997)).
Preferred parameters for BLASTp are: Expectation value: 10 (default); Filter:
seg (default); Cost to open a gap: 11 (default); Cost to extend a gap:
1(default);
Max. alignments: 100 (default); Word size: 11 (default); No. of descriptions:
100
(default); Penalty Matrix: BLOWSUM62.
The length of polypeptide sequences compared for homology will generally be
at least about 16 amino acid residues, usually at least about 20 residues,
more usually
at least about 24 residues, typically at least about 28 residues, and
preferably more
than about 35 residues. When searching a database containing sequences from a
large
number of different organisms, it is preferable to compare amino acid
sequences.
Database searching using amino acid sequences can be measured by algorithms
other
than blastp known in the art. For instance, polypeptide sequences can be
compared
using FASTA, a program in GCG Version 6.1. FASTA provides alignments and
32
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
percent sequence identity of the regions of the best overlap between the query
and
search sequences. Pearson, Methods Enz,ymol. 183:63-98 (1990) (herein
incorporated
by reference). For example, percent sequence identity between amino acid
sequences
can be determined using FASTA with its default parameters (a word size of 2
and the
PAM250 scoring matrix), as provided in GCG Version 6.1, herein incorporated by
reference.
"Specific binding" refers to the ability of two molecules to bind to each
other
in preference to binding to other molecules in the environment. Typically,
"specific
binding" discriminates over adventitious binding in a reaction by at least two-
fold,
more typically by at least 10-fold, often at least 100-fold. Typically, the
affinity or
avidity of a specific binding reaction, as quantified by a dissociation
constant, is about
10-7 M or stronger (e.g., about 10"$ M, 10"9 M or even stronger).
The term "region" as used herein refers to a physically contiguous portion of
the primary structure of a biomolecule. In the case of proteins, a region is
defined by
a contiguous portion of the amino acid sequence of that protein.
The term "domain" as used herein refers to a structure of a biomolecule that
contributes to a known or suspected function of the biomolecule. Domains may
be
co-extensive with regions or portions thereof; domains may also include
distinct, non-
contiguous regions of a biomolecule.
As used herein, the term "molecule" means any compound, including, but not
limited to, a small molecule, peptide, protein, glycoprotein, sugar,
nucleotide, nucleic
acid, lipid, etc., and such a compound can be natural or synthetic.
As used herein, the term "comprise" or variations such as "comprises" or
"comprising", will be understood to imply the inclusion of a stated integer or
group of
integers but not the exclusion of any other integer or group of integers.
33
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
As used herein, the term 'consisting essentially of' will be understood to
imply the inclusion of a stated integer or group of integers; while excluding
modifications or other integers which would materially affect or alter the
stated
integer. With respect to species of N-glycans, the term "consisting
essentially of' a
stated N-glycan will be understood to include the N-glycan whether or not that
N-
glycan is fucosylated at the N-acetylglucosamine (GlcNAc) which is directly
linked to
the asparagine residue of the glycoprotein.
As used herein, the term "predominantly" or variations such as "the
predominant" or "which is predominant" will be understood to mean the glycan
species that has the highest mole percent (%) of total N-glycans after the
glycoprotein
has been treated with PNGase and released glycans analyzed by mass
spectroscopy,
for example, MALDI-TOF MS. In other words, the phrase "predominantly" is
defined as an individual entity, such as a specific glycoform, is present in
greater mole
percent than any other individual entity. For example, if a composition
consists of
species A in 40 mole percent, species B in 35 mole percent and species C in 25
mole
percent, the composition comprises predominantly species A, and species B
would be
the next most predominant species.
As used herein, the term "essentially free of' a particular sugar residue,
such
as fucose, or galactose and the like, is used to indicate that the
glycoprotein
composition is substantially devoid of N-glycans which contain such residues.
Expressed in terms of purity, essentially free means that the amount of N-
glycan
structures containing such sugar residues does not exceed 10%, and preferably
is
below 5%, more preferably below 1%, most preferably below 0.5%, wherein the
percentages are by weight or by mole percent. Thus, substantially all of the N-
glycan
34
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
structures in a glycoprotein composition according to the present invention
are free of
fucose, or galactose, or both.
As used herein, a glycoprotein composition "lacks" or "is lacking" a
particular
sugar residue, such as fucose or galactose, when no detectable amount of such
sugar
residue is present on the N-glycan structures at any time. For example, in
preferred
embodiments of the present invention, the glycoprotein compositions are
produced by
lower eukaryotic organisms, as defined above, including yeast [e.g., Pichia
sp.;
Saccharomyces sp.; Klu,yveromyces sp.; Aspergillus sp.], and will "lack
fucose,"
because the cells of these organisms do not have the enzymes needed to produce
fucosylated N-glycan structures. Thus, the term "essentially free of fucose"
encompasses the term "lacking fucose." However, a composition may be
"essentially
free of fucose" even if the composition at one time contained fucosylated N-
glycan
structures or contains limited, but detectable amounts of fucosylated N-glycan
structures as described above.
As used herein, the phrase "increased binding activity" is used
interchangeably with "increased binding affinity" referring to an increase in
the
binding of the IgG molecule with a receptor--or otherwise noted molecule.
As used herein, the phrase "decreased binding activity" is used
interchangeably with "decreased binding affinity" referring to a decrease in
the
binding of the IgG molecule with a receptor--or otherwise noted molecule.
As used herein, the phrase, "phagocytosis" is defined to be clearance of
immunocomplexes. Phagocytosis is an immunological activity of immune cells -
including but not limited to, macrophages and neutrophils.
The interaction of antibodies and antibody-antigen complexes with cells of the
immune system and the variety of responses, including antibody-dependent cell-
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC),
clearance of immunocomplexes (phagocytosis), antibody production by B cells
and
IgG serum half-life are defined respectively in the following: Daeron et al.,
1997,
Annu. Rev. Immunol. 15: 203-234; Ward and Ghetie, 1995, Therapeutic Inamunol.
2:77-94; Cox and Greenberg, 2001, Semin. Imnaunol. 13: 339-345; Heyman, 2003,
Immunol. Lett. 88:157-161; and Ravetch, 1997, Curr. Opin. Imrnunol. 9: 121-
125.
Unless otherwise defined, all technical and scientific terms used herein have
the same meaning as commonly understood by one of ordinary skill in the art to
which this invention pertains. Exemplary methods and materials are described
below,
although methods and materials similar or equivalent to those described herein
can
also be used in the practice of the present invention and will be apparent to
those of
skill in the art. All publications and other references mentioned herein are
incorporated by reference in their entirety. In case of conflict, the present
specification, including definitions, will control. The materials, methods,
and
examples are illustrative only and not intended to be limiting.
Recombinant I -g Man3GlcNAc2 molecules
The present invention provides compositions comprising a population of
glycosylated Igs having a predominant Man3GlcNAc2 N-linked glycoform. The
present invention also provides Igs and Ig compositions having a predominant
Man3GlcNAc2 N-linked glycoform that mediates antibody effector functions, such
as
receptor binding. Preferably the interaction between an Ig of the present
invention
and an FcyRIII receptor provides an increase in direct binding activity. And,
preferably the interaction between an Ig of the present invention and the
FcyRIIb
receptor provides a decrease (or lack of) direct binding activity. In another
36
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
embodiment, an Ig or Ig composition of the present invention exhibits
increased
binding activity conferred by the enrichment/predominance of a glycoform
structure.
A salient feature of the present invention is that it provides Igs and Ig
compositions
having a predominant, specific glycoform that mediates antibody effector
functions,
such as an increase in ADCC activity or an increase in antibody production by
B cells.
In another embodiment, an Ig or Ig composition of the present invention
exhibits
increased ADCC activity or antibody production by B cells conferred by the
enrichment/predominance of one glycoform. Furthermore, it will be readily
apparent
to a skilled artisan that one advantage of producing Ig compositions having a
predominant glycoform is that it avoids production of Igs having undesired
glycoforms and/or production of heterogeneous mixtures of Igs which may induce
undesired effects and/or dilute the concentration of the more effective Ig
glycoform(s). It is, therefore, contemplated that a pharmaceutical composition
comprising Igs having predominantly Man3GlcNAc2 glycoforms will have
beneficial
features, including but not limited to, decreased binding to FcyRIIb and
increased
binding to FcyRIIIa and FcyRIIIb, and therefore may well be effective at lower
doses,
thus having higher efficacy/potency.
In one embodiment, an Ig molecule of the present invention comprises at least
one Man3GlcNAc2 glycan structure at Asn-297 of a CH2 domain of a heavy chain
on
the Fc region mediating antibody effector function in an Ig molecule.
Preferably, the
Man3GlcNAc2 glycan structure is on each Asn-297 of each CH2 region in a
dimerized
Ig (Figure 1). In another embodiment, the present invention provides
compositions
comprising Igs which are predominantly glycosylated with an N-glycan
consisting
essentially of Man3GlcNAc2 glycan structure at Asn-297 (Figure 1).
Alternatively,
one or more carbohydrate moieties found on an Ig molecule may be deleted
and/or
37
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
added to the molecule, thus adding or deleting the number of glycosylation
sites on an
Ig. Further, the position of the N-linked glycosylation site within the CH2
region of a
Ig molecule can be varied by introducing asparagines (Asn) or N-glycosylation
sites
at varying locations within the molecule. While Asn-297 is the N-glycosylation
site
typically found in murine and human IgG molecules (Kabat et al., Sequences of
Proteins oflmmunologicallnterest, 1991), this site is not the only site that
can be
envisioned, nor does this site necessarily have to be maintained for function.
Using
known methods for mutagenesis, the skilled artisan can alter a DNA molecule
encoding an Ig of the present invention so that the N-glycosylation site at
Asn-297 is
deleted, and can further alter the DNA molecule so that one or more N-
glycosylation
sites are created at other positions within the Ig molecule. It is preferred
that N-
glycosylation sites are created within the CH2 region of the Ig molecule.
However,
glycosylation of the Fab region of an Ig has been described in 30% of serum
antibodies-commonly found at Asn-75 (Rademacher et al., 1986, Biochem.. Soc.
Syrnp., 51: 131-148). Glycosylation in the Fab region of an Ig molecule is an
additional site that can be combined in conjunction with N-glycosylation in
the Fc
region, or alone.
In one embodiment, the present invention provides a recombinant Ig
composition having a predominant Man3GlcNAc2 N-glycan structure, wherein said
Man3GlcNAc2 glycan structure is present at a level that is at least about 5
mole
percent more than the next predominant glycan structure of the recombinant Ig
composition. In a preferred embodiment, the present invention provides a
recombinant Ig composition having a predominant Man3GlcNAc2 glycan structure,
wherein said Man3GlcNAc2 glycan structure is present at a level of at least
about 10
mole percent to about 25 mole percent more than the next predominant glycan
38
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
structure of the recombinant Ig composition. In a more preferred embodiment,
the
present invention provides a recombinant Ig composition having a predominant
Man3GlcNAc2 glycan structure, wherein said Man3GlcNAc2 glycan structure is
present at a level that is at least about 25 mole percent to about 50 mole
percent more
than the next predominant glycan structure of the recombinant Ig composition.
In a
preferred embodiment, the present invention provides a recombinant Ig
composition
having a predominant Man3GlcNAc2 glycan structure, wherein said Man3GlcNAc2
glycan structure is present at a level that is greater than about 50 mole
percent more
than the next predominant glycan structure of the recombinant Ig composition.
In
another preferred embodiment, the present invention provides a recombinant Ig
composition having a predominant Man3GlcNAc2 glycan structure, wherein said
Man3GlcNAc2 glycan structure is present at a level that is greater than about
75 mole
percent more than the next predominant glycan structure of the recombinant Ig
composition. In still another embodiment, the present invention provides a
recombinant Ig composition having a predominant Man3GlcNAc2 glycan structure,
wherein said Man3GlcNAc2 glycan structure is present at a level that is
greater than
about 90 mole percent more than the next predominant glycan structure of the
recombinant Ig composition. MALDI-TOF analysis of N-glycans of JC-IgG having a
predominant Man3GlcNAc2N-glycan (75%) is shown in Figure 4A. MALDI-TOF
analysis of N-glycans of DX-IgG having a predominant Man3GlcNAc2 (64%) is
shown in Figure 4B.
Increased binding of Ig-Man~GlcNAc2 to Fc7RIII receptor
The effector functions of Ig binding to FcyRIIIa and FcyRIllb, such as
activation of ADCC, are mediated by the Fc region of the Ig molecule.
Different
39
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
functions are mediated by the different domains in this region. Accordingly,
the
present invention provides Ig molecules and compositions in which an Fc region
on
an Ig molecule has a predominant Man3GlcNAc2N-glycan capable of carrying out
an
effector function. In one embodiment, the Fc region having a predominant
Man3GlcNAc2 N-glycan confers an increase in binding to FcyRIIIa (Figure 6) and
FcyRIIIb (Figure 5) receptors. In another embodiment, an Fc has a predominant
Man3GlcNAc2 N-glycan. It will be readily apparent to the skilled artisan that
molecules comprising the Fc region, such as immunoadhesions (Chamow and
Ashkenazi, 1996, Trends Biotechnol. 14: 52-60; Ashkenazi and Chamow, 1997,
Curr.
Opin. Irnmunol. 9: 195-200), Fc fusions and antibody-like molecules are also
encompassed in the present invention.
Binding activity (affinity) of an Ig molecule to an Fc receptor may be
determined by an assay. An example of an FcyRIII binding assay with IgG is
described in Example 6. One skilled in the art recognizes that this assay can
be easily
adapted for use in conjunction with assays for any immunoglobulin molecule.
JC-IgG (an Ig made according to the present invention) having predominantly
Man3GlcNAc2 N-glycans has 50-100 fold increased binding activity to FcyRIllb
and
FcyRIIIa compared with Ritaximab as shown in Figure 5A and Figure 6. DX-IgG
(another Ig made according to the present invention) having predominantly
Man3GlcNAc2 N-glycans also has 50-100 fold increased binding activity to
FcyRIIIb
compared with Rituximab as shown in Figure 5B.
Most interestingly, FcyRIIIa gene dimorphism generates two allotypes:
FcyRIIIa-158V and FcyRIIIa-158F (Dall'Ozzo et al., 2004, Cancer Res. 64: 4664-
4669). The genotype homozygous for FcyRIIIa-158V is associated with a higher
clinical response to Rituximab (Cartron et al., 2002, Blood, 99: 754-758).
However,
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
most of the population carries one FcyRIIIa-158F allele, rendering Rituximab
less
effective for most of the population for induction of ADCC through FcyRIIIa
binding.
However, when a Rituximab -like anti-CD20 antibody is expressed in a host cell
which lacks fucosyltransferase activity, this antibody is equally effective
for
enhancing ADCC through both FcyRIIIa -1 58F and FcyRIIIa-158V (Niwa et al.,
2004, Clin. Canc Res. 10: 6248-6255). The antibodies of certain preferred
embodiments of the present invention are expressed in host cells that do not
add
fucose to N-glycans (e.g., P. pastoris, a yeast host lacking fucose; see
Examples 1
and 2). Therefore, it is contemplated that the antibodies of the present
invention that
lack fucose and have enhanced binding to FcyRIIIa- 15 8F may be especially
useful for
treating many patients exhibiting a reduced clinical response to Rituximab .
Decreased binding of I-g Man3GlcNAc, to FcyRIIb receptor
The effector functions of Ig binding to FcyRIIb, such as increased antibody
production by B cells and increased ADCC activity, are mediated by the Fc
region of
the Ig molecule. Different functions are mediated by the different domains in
this
region. Accordingly, the present invention provides Ig molecules and
compositions in
which an Fc region on an Ig molecule has a predominant Man3GlcNAc2 N-glycan
capable of carrying out an effector function. In one embodiment, an Fc region
of an
Ig having a predominant Man3GlcNAc2 N-glycan confers a decrease in binding to
an
FcyRIIb receptor. It will be readily apparent to the skilled artisan that
molecules
comprising an Fc region, such as immunoadhesions (Chamow and Ashkenazi, 1996,
Trends Biotechnol. 14: 52-60; Ashkenazi and Chamow, 1997, Curr Opin.
Irnrnunol. 9:
195-200), Fc fusions and antibody-like molecules are also encompassed in the
present
invention.
41
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
Binding activity (affinity) of an Ig molecule to an Fc receptor may be
determined by an assay. An example of an FcyRIIb binding assay with IgGl is
disclosed in Example 6. One skilled in the art recognizes that this disclosed
assay
can be easily adapted for use in connection to any immunoglobulin molecule.
JC-IgG (an Ig of the present invention) having predominant Man3GlcNAc21V-
glycans, has 2-3 fold decreased binding activity to FcyRIIb compared with
Rituximab as shown in Figure 7A. DX-IgG (another Ig of the present invention)
having predominant Man3GlcNAc2 N-glycans, also has 2-3 fold decreased binding
activity to FcyRIIb compared with Rituximab as shown in Figure 7B.
Increased antibody-dependent cell-mediated cytoxicity
In yet another embodiment, the increase in FcyRIIIa or FcyRIIIb binding of an
Ig molecule or composition having Man3GlcNAc2 as the predominant N-glycan may
confer an increase in FcyRIII-mediated ADCC. It is well established that the
FcyRIII
(CD 16) receptor is responsible for ADCC activity (Daeron et al., 1997, Annu.
Rev.
Ibnmunol. 15: 203-234). In another embodiment, the decrease in FcyRIIb binding
of
an Ig molecule or composition having Man3GlcNAc2 as the predominant N-glycan
confers an increase in ADCC (Clynes et al., 2000, supra). In another
embodiment, an
Ig molecule or composition of the present invention exhibits increased ADCC
activity
conferred by the presence of a predominant Man3GlcNAc2 glycan.
An example of in vitro assays measuring B-cell depletion and fluorescence
release ADCC assays are disclosed in Example 7. One skilled in the art
recognizes
that these disclosed assays can be easily adapted for use in conjunction with
assays for
any Ig molecule. Furthermore, an in vivo ADCC assay in an animal model can be
42
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
adapted for any specific IgG from Borchmann et al., 2003, Blood, 102: 3737-
3742,
Niwa et al., 2004, Cancer Research, 64: 2127-2133 and Example 7.
Increased antibody production by B cells
Antibody engagement against tumors through the regulatory FcyR pathways
has been shown (Clynes et al., 2000, Nature, 6: 443-446). Specifically, it is
known
when FcyRIIb is co-cross-linked with immunoreceptor tyrosine based activation
motifs (ITAM)-containing receptors such as the B cell receptor (BCR), FcyRI,
FcyRIII, and FcERI, it inhibits ITAM-mediated signals (Vivier and Daeron,
1997,
Iinmunol. Today, 18: 286-291). For example, the addition of FcgRII-specific
antibodies blocks Fc binding to the FcgRIIB, resulting in augmented B cell
proliferation (Wagle et al., 1999, J of bninunol. 162: 2732-2740).
Accordingly, in
one embodiment, an Ig molecule of the present invention can mediate a decrease
in
FcyRIIb receptor binding resulting in the activation of B cells which in turn,
catalyzes
antibody production by plasma cells (Parker, D.C. 1993, Annu. Rev. Immunol.
11:
331-360). An example of an assay measuring antibody production by B cells with
IgGl is described in Example 6. One skilled in the art recognizes that this
assay can
be easily adapted for use in conjunction with assays for any immunoglobulin
molecule.
Other immunoloizical activities
Altered surface expression of effector cell molecules on neutrophils has been
shown to increase susceptibility to bacterial infections (Ohsaka et al., 1997,
Br. J.
Haenaatol. 98: 108-113). It has been further demonstrated that IgG binding to
the
FcyRIIIa effector cell receptors regulates expression of tumor necrosis factor
alpha
43
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
(TNF-a) (Blom et al., 2004, Arthritis Rheuna., 48: 1002-1014). Furthermore,
FcyR-
induced TNF-a also increases the ability of neutrophils to bind and
phagocytize IgG-
coated erythrocytes (Capsoni et al., 1991, J. Clin. Lab Ihnnaunol. 34: 115-
124). It is
therefore contemplated that the Ig molecules and compositions of the present
invention that show an increase in binding to FcyRIII, may confer an increase
in
expression of TNF-a.
An increase in FcyRIII receptor activity has been shown to increase the
secretion of lysosomal beta-glucuronidase as well as other lysosomal enzymes
(Kavai
et al., 1982, Adv. Exp Med. Biol. 141: 575-582; Ward and Ghetie, 1995,
Therapeutic
linmunol., 2: 77-94). Furthermore, an important step after the engagement of
immunoreceptors by their ligands is their internalization and delivery to
lysosomes
(Bonnerot et al., 1998, EMBO J., 17: 4906-4916). It is therefore contemplated
that an
Ig molecule or composition of the present invention that shows an increase in
binding
to FcyRIIIa and FcyRIIIb may confer an increase in the secretion of lysosomal
enzymes.
Present exclusively on neutrophils, FcyRIIIb plays a predominant role in the
assembly of immune complexes, and its aggregation activates phagocytosis,
degranulation, and the respiratory burst leading to destruction of opsonized
pathogens.
Activation of neutrophils leads to secretion of a proteolytically cleaved
soluble form
of the receptor corresponding to its two extracellular domains. Soluble
FcyRIIIb
exerts regulatory functions by competitive inhibition of FcyR-dependent
effector
functions and via binding to the complement receptor CR3, leading to
production of
inflammatory mediators (Sautes-Fridman et al., 2003, ASHI Quarterly, 148-151).
The present invention thus provides an immunoglobulin molecule comprising
an N-glycan consisting essentially of Man3GlcNAc2; and provides a composition
44
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
comprising immunoglobulins and a plurality of N-glycans attached thereto
wherein
the predominant N-glycan within said plurality of N-glycans consists
essentially of
Man3GlcNAc2. In either embodiment, the predominance of said Man3GlcNAc2 N-
glycan on an immunoglobulin preferably confers desired therapeutic effector
activity
in addition to the improved binding to FcyRIIIa and FcyRIIIb and decreased
binding
to FcyRIIb, as shown herein.
Immunoglobulin subclasses
The IgG subclasses have been shown to have different binding affinities for Fc
receptors (Huizinga et al., 1989, J. oflbnmunol., 142: 2359-2364). Each of the
IgG
subclasses may offer particular advantages in different aspects of the present
invention. Thus, in one aspect, the present invention provides an IgGl
composition
that comprises Man3GlcNAc2 as the predominant N-glycan attached to IgGl
molecules. In another aspect, the present invention comprises an IgG2
composition
that comprises Man3GlcNAc2 as the predominant N-glycan attached to IgG2
molecules. In yet another aspect, the present invention comprises an IgG3
composition that comprises Man3GlcNAc2 as the predominant N-glycan attached to
IgG3 molecules. In another aspect, the present invention comprises an IgG4
composition that comprises Man3GlcNAc2 as the predominant N-glycan attached to
IgG4 molecules.
Alternatively, the present invention can be applied to all of the five major
classes of immunoglobulins: IgA, IgD, IgE, IgM and IgG. A preferred
immunoglobulin of the present invention is a human IgG and preferably from one
of
the subtypes IgGl, IgG2, IgG3 or IgG4. More preferably, an immunoglobulin of
the
present invention is an IgGl molecule.
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
Production of recombinant immunoglobulin (Ig) molecules mediating antibody
effector function and activity
In one aspect, the invention provides a method for producing a recombinant Ig
molecule having an N-glycan consisting essentially of a Man3GlcNAc2 glycan
structure at Asn-297 of the CH2 domain, wherein the Ig molecule mediates
antibody
effector function and activity, and similarly, an immunoglobulin composition
wherein
the predominant N-glycan attached to the immunoglobulins is Man3GlcNAc2. In
one
embodiment, the heavy and light chains of the Ig are synthesized using
overlapping
oligonucleotides and are separately cloned into an expression vector (Example
1) for
expression in a host cell. In a preferred embodiment, recombinant Ig heavy and
light
chains are expressed in a host strain which catalyzes predominantly the
addition of
Man3GlcNAc2. In one embodiment, this glycoform structure is more specifically
denoted as [(Mana1,3)(Mana1,6) Man(31,4-G1cNAc (31,4-G1cNAc] forming a linkage
between the nitrogen of the amino acid Asn-297 of the Fc region on an Ig and
the
hydroxyl group of N-acetyl-(3-D-glucosamine on the Man3GlcNAc2 glycan. In yet
another embodiment, this predominant glycan can be added to an asparagine at a
different site within the Ig molecule (other than Asn-297), or in combination
with the
N-glycosylation site in the Fab region.
Production of Ig having predominantly Man3GlcNAc, in Lower Eukaryotes
One aspect of the present invention provides recombinant lower eukaryotic
host cells which may be used to produce immunoglobulin or antibody molecules
with
predominantly the Man3GlcNAc2 glycoform, which is an advantage compared with
46
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
compositions of glycoproteins expressed in mammalian cells which naturally
produce
said glycoform in low yield.
It is another advantage of the present invention that compositions of
glycoproteins are provided with predetermined glycosylation patterns that are
readily
reproducible. The properties of such compositions are assessed and optimized
for
desirable properties, while adverse effects may be minimized or avoided
altogether.
The present invention also provides methods for producing recombinant host
cells that are engineered or selected to express one or more nucleic acids for
the
production of Ig molecules comprising an N-glycan consisting essentially of
Man3GlcNAc2 and Ig compositions having predominantly a Man3GlcNAc2 glycan
structure. In certain preferred embodiments of the present invention,
recombinant
host cells, preferably recombinant lower eukaryotic host cells, are used to
produce
said Ig molecules and compositions having predominantly Man3GlcNAc2 glycan.
In other preferred embodiments, the invention comprises the glycoproteins
obtainable from recombinant host cells or by the methods of the present
invention.
The host cells of the invention may be transformed with vectors encoding the
desired Ig regions, and with vectors encoding one or more of the glycosylation-
related
enzymes described herein, and then selected for expression of a recombinant Ig
molecule or composition having a predominant Man3GlcNAc2 N-glycan. The
recombinant host cell of the present invention may be a eukaryotic or
prokaryotic host
cell, such as an animal, plant, insect, bacterial cell, or the like which has
been
engineered or selected to produce an Ig composition having predominantly
Man3GlcNAc21V-glycan structures.
Preferably, the recombinant host cell of the present invention is a lower
eukaryotic host cell which has been genetically engineered as described in the
art
47
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
(WO 02/00879, WO 03/056914, WO 04/074498, WO 04/074499, Choi et al., 2003,
PNAS, 100: 5022-5027; Hamilton et al., 2003, Nature, 301: 1244-1246 and
Bobrowicz et al., 2004, Glycobiology, 14: 757-766). Specifically, WO 03/056914
discloses methods to obtain 75% Man3GIcNAc2 in Fig. 22, as well as disclosure
of
immunoglobulins in Fig. 30, 31 and paragraphs 207-211.
In one embodiment of the present invention, a vector encoding an IgGl, for
example an AOX1/pPICZA vector containing JC-IgG (Example 1) is introduced into
the yeast P. pastoris YAS309 strain. This YAS309 strain is similar to the
YSH44
strain with the K3 reporter protein removed (Hamilton et al., 2003, Science,
301:
1244-1246), and has had the PNO1 and MNN4b genes disrupted as described (US
Pat.
Appl. No. 11/020808), as well as a(3-1,4 galactosyltransferase I gene
introduced as
described (US Pat. Appl. No. 11/108088). The dpnoldmnn4b double disruption
results in the elimination of mannosphosphorylation. The mannosidase II gene
which
was introduced as described for YSH44 (Hamilton et al., 2003) flanked by the
URA5
gene, was knocked out by growing the strain on 5-Fluoroorotic acid (5-FOA)
(Guthrie
and Fink, 1991, Guide to Yeast Geiaetics and Molecular Biology, Methods in
Enzynaology, Vol. 169, Academic Press, San Diego). The mannosidase II activity
was
then reintroduced at the AMR2 locus, resulting in the reintroduction of the
mannosidase II activity and the loss of the AMR2 gene, thus eliminating (3-
mannosylation as described (US Pat. Appl. No. 11/118008). Glycoproteins from
this
YAS309 strain upon treatment with (3-galactosidase and (3-N-
acetylhexosaminidase
have predominantly Man3GlcNAc2 N-glycans. Thus, JC-IgG expressed in YAS309
and treated with (3-galactosidase and (3-N-acetylhexosaminidase (Example 3)
has
predominantly Man3GlcNAc2 N-glycans (Figure 4A).
48
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
In another embodiment, the vector encoding an IgGl, for example an
AOX1/pPICZA containing DX-IgG (Example 1) was also introduced into the yeast
P. pastoris T'AS309 strain (supra), purified and then treated with (3-N-
acetylhexosaminidase (Example 3), resulting in DX-IgG having predominantly
Man3GlcNAc2 N-glycans (Figure 4B).
Alternatively, an antibody of the present invention can be expressed using
several methods known in the art (Monoclonal Antibody Production Techniques
and
Applications, pp. 79-97 (Marcel Dekker, Inc., New York, 1987).
Production of Ig avingpredominantly Man3GlcNAc2 in an dalg3 yeast host
Alternatively, an Ig of the present invention can be expressed in a lower
eukaryotic host which synthesizes the Man3GlcNAc2 N-glycans in vivo. Such host
would be engineered in an Aalg3 mutant as described in WO 03/056914 with an a-
1,2
mannosidase gene introduced as also described. An immunoglobulin introduced
into
such a host would express predominantly Man3GlcNAc2 N-glycans by in vivo
methods.
Expression of glycosyltransferases and stable genetic inteQ;ration in lower
eukaryotes
Methods for introducing and contirming integration of heterologous genes in a
lower eukaryotic host strain (e.g. P. pastoris) using selectable markers such
as URA3,
URA5, HIS4, SUC2, G418, BLA or SHBLA have been described. Such methods may
be adapted to produce an Ig of the present invention when the expression
system is
produced in a lower eukaryote. Additionally, methods have been described that
allow
for repeated use of the URA3 marker to eliminate undesirable
mannosyltransferase
activities. Alani et al., 1987, Genetics, 116: 541-545 and US Pat. No.
6,051,419
49
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
describe a selection system based on disrupting the URA3 gene in P. pastoris.
Preferably, the PpUR,43- or PpURA5-blaster cassettes are used to disrupt the
URA3,
URA5 or any gene in the uracil biosynthesis pathway, allowing for both
positive and
negative selection, based on auxotrophy for uracil and resistance to 5-
fluoroorotic
acid (5FOA) (Boeke, et al., 1984, Mol. Gen. Genet., 197: 345-346). A skilled
artisan,
therefore, recognizes that such a system allows for insertion of multiple
heterologous
genes by selecting and counterselecting.
Further enzymatic modiflcations
Further enzymatic deletions may be beneficial or necessary to isolate an Ig
free of mannosylphosphorylation or (3-mannosylation which may confer aberrant
immunogenic activities in humans. As mentioned, US Pat. Appl. No. 11/020808
discloses a method for the elimination of mannosylphosphorylation, and US Pat.
Appl. No. 11/118008 discloses a method for the elimination of P-mannosylation.
Production of Ig having predominantly Man3GlcNAc,glycan structure in other
protein expression systems
It is understood by the skilled artisan that an expression host system
(organism) is selected for heterologous protein expression that may or may not
need
to be engineered to express Igs having a predominant glycan structure. The
Examples
provided herein are examples of one method for carrying out the expression of
Ig with
a particular glycan at Asn-297 or another 1V-glycosylation site, or both. One
skilled in
the art can easily adapt these details of the invention and examples for any
protein
expression host system (organism).
Other protein expression host systems including animal, plant, insect,
bacterial
cells and the like may be used to produce Ig molecules and compositions
according to
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
the present invention. Such protein expression host systems may be engineered
or
selected to express a predominant glycoform or alternatively may naturally
produce
glycoproteins having predominant glycan structures. Examples of engineered
protein
expression host systems producing a glycoprotein having a predominant
glycoform
include gene knockouts/mutations (Shields et al., 2002, JBC, 277: 26733-
26740);
genetic engineering in (Umana et al., 1999, Nature Biotech., 17: 176-180) or a
combination of both. Alternatively, certain cells naturally express a
predominant
glycoform-for example, chickens, humans and cows (Raju et al., 2000,
Glycobiology, 10: 477-486). Thus, the expression of an Ig glycoprotein or
composition having predominantly one specific glycan structure according to
the
present invention can be obtained by one skilled in the art by selecting at
least one of
many expression host systems. Further expression host systems found in the art
for
production of glycoproteins include: CHO cells: Raju W09922764A1 and Presta
W003/035835A1; hybridroma cells: Trebak et al., 1999, J. Immunol. Methods,
230:
59-70; insect cells: Hsu et al., 1997, JBC, 272:9062-970, and plant cells:
Gerngross et
al., W004/074499A2.
Purification of IgG
Methods for the purification and isolation of antibodies are known and are
disclosed in the art. See, for example, Kohler & Milstein, (1975) Nature
256:495;
Brodeur et al., Monoclonal Antibody Production Techniques and Applications,
pp.5 1-
63, Marcel Dekker, Inc., New York, 1987);. Goding, Monoclonal Antibodies:
Principles and Practice, pp.59-104 (Academic Press, 1986); and Jakobovits et
al.
(1993) Proc. Natl. Acad. Sci. USA 90:2551-255 and Jakobovits et al, (1993)
Nature
362:255-258. In a further embodiment, antibodies or antibody fragments can be
51
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
isolated from antibody phage libraries generated using the techniques
described in
McCafferty et al. (1990) Nature, 348:552-554 (1990), using the antigen of
interest to
select for a suitable antibody or antibody fragment.
Recombinant Ig molecules produced according to the methods of the present
invention can be purified according to methods outlined in Example 3. Figure 2
shows an SDS-PAGE Coomassie stained gel of JC-IgG purified from YAS309.
Figure 3 shows an SDS-PAGE Coomassie stained gel of DX-IgG purified from
YAS309. In one embodiment, a purified Ig antibody has Man3GlcNAc2 as the
predominant N-glycan. The glycan analysis and distribution on any Ig molecule
can
be determined by several mass spectroscopy methods known to one skilled in the
art,
including but not limited to: HPLC, NMR, LCMS and MALDI-TOF MS. In a
preferred embodiment, the glycan distribution is determined by MALDI-TOF MS
analysis as disclosed in Example 5. Figure 4A shows a MALDI-TOF spectra of JC-
IgG purified from YAS309 and treated with galactosidase and hexosaminidase
(Example 3). This MALDI-TOF shows approximately 75 mole % of the total N-
glycans are Man3GlcNAc2. Figure 4B shows a MALDI-TOF spectra of DX-IgG
purified from YSH44 and treated with galactosidase and hexosaminidase. This
MALDI-TOF shows approximately 64 mole % of the total N-glycans are
Man3GlcNAc2.
Pharmaceutical Compositions
Antibodies of the invention can be incorporated into pharmaceutical
compositions comprising the antibody as an active therapeutic agent and a
variety of
other pharmaceutically acceptable components. See Remington's Pharmaceutical
Science (15th ed., Mack Publishing Company, Easton, Pennsylvania, 1980). The
52
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
preferred form depends on the intended mode of administration and therapeutic
application. The compositions can also include, depending on the formulation
desired,
pharmaceutically-acceptable, non-toxic carriers or diluents, which are defined
as
vehicles commonly used to formulate pharmaceutical compositions for animal or
human administration. The diluent is selected so as not to affect the
biological activity
of the combination. Examples of such diluents are distilled water,
physiological
phosphate-buffered saline, Ringer's solutions, dextrose solution, and Hank's
solution.
In addition, the pharmaceutical composition or formulation can also include
other
carriers, adjuvants, or nontoxic, nontherapeutic, nonimmunogenic stabilizers
and the
like.
Pharmaceutical compositions for parenteral administration are sterile,
substantially isotonic, pyrogen-free and prepared in accordance with GMP of
the FDA
or similar body. Antibodies can be administered as injectable dosages of a
solution or
suspension of the substance in a physiologically acceptable diluent with a
pharmaceutical carrier that can be a sterile liquid such as water, oils,
saline, glycerol,
or ethanol. Additionally, auxiliary substances, such as wetting or emulsifying
agents,
surfactants, pH buffering substances and the like can be present in
compositions.
Other components of pharmaceutical compositions are those of petroleum,
animal,
vegetable, or synthetic origin, for example, peanut oil, soybean oil, and
mineral oil. In
general, glycols such as propylene glycol or polyethylene glycol are preferred
liquid
carriers, particularly for injectable solutions. Antibodies can be
administered in the
form of a depot injection or implant preparation which can be formulated in
such a
manner as to permit a sustained release of the active ingredient. Typically,
compositions are prepared as injectables, either as liquid solutions or
suspensions;
solid forms suitable for solution in, or suspension in, liquid vehicles prior
to injection
53
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
can also be prepared. The preparation also can be emulsified or encapsulated
in
liposomes or micro particles such as polylactide, polyglycolide, or copolymer
for
enhanced adjuvant effect, as discussed above (see Langer, Science 249, 1527
(1990)
and Hanes, Advanced Drug Delivezy Reviews 28, 97-119 (1997).
Diagnostic Products
Antibodies of the invention can also be incorporated into a variety of
diagnostic kits
and other diagnostic products such as an array. Antibodies are often provided
prebound to a solid phase, such as to the wells of a microtiter dish. Kits
also often
contain reagents for detecting antibody binding, and labeling providing
directions for
use of the kit. Immunometric or sandwich assays are a preferred format for
diagnostic
kits (see US 4,376,110, 4,486,530, 5,914,241, and 5,965,375). Antibody arrays
are
described by e.g., US 5,922,615, US 5,458,852, US 6,019,944, and US 6,143,576.
Therapeutic applications
The present invention provides glycoprotein compositions which comprise
predominantly a particular glycoform on the glycoprotein. It is a feature of
the
present invention that when administered to mammals including humans,
pharmaceutical compositions comprising the novel glycoprotein compositions, in
preferred embodiments, advantageously exhibit superior in vivo properties when
compared to other glycoprotein compositions having similar primary structure.
Thus,
the novel compositions of the invention may be used wherever the glycoprotein
pharmaceutical agent is presently used and may advantageously provide improved
properties as well as increased uniformity between and throughout production
lots.
The preparations of the invention can be incorporated into solutions, unit
dosage
54
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
forms such as tablets and capsules for oral delivery, as well as into
suspensions,
ointments and the like, depending on the particular drug or medicament and its
target
area.
In a particular aspect, the present invention provides novel compositions for
glycoprotein pharmaceutical agents, drugs or medicaments wherein the
glycoprotein
comprises an immunoglobulin molecule and the composition comprises
predominantly particular glycoforms of the glycoprotein agent. According to a
particular aspect of the invention, compositions are provided comprising an
immunoglobulin glycoprotein having predominantly an N-linked oligosaccharide
of
the Man3GlcNAc2 glycan structure as described herein. In preferred aspects,
the
glycoprotein is an antibody and especially may be a monoclonal antibody. The
invention further provides methods and tools for producing the compositions of
the
invention.
The invention further encompasses pharmaceutical compositions comprising
the glycoform preparations of the invention. The compositions are preferably
sterile.
Where the composition is an aqueous solution, preferably the glycoprotein is
soluble.
Where the composition is a lyophilized powder, preferably the powder can be
reconstituted in an appropriate solvent.
In other aspects, the invention involves a method for the treatment of a
disease
state comprising administering to a mammal in need thereof a therapeutically
effective dose of a pharmaceutical composition of the invention. It is a
further object
of the invention to provide the glycoform preparations in an article of
manufacture or
kit that can be employed for purposes of treating a disease or disorder.
The Ig molecules of the present invention having predominantly
Man3GlcNAc2 N-glycans have many therapeutic applications for indications such
as
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
cancers, inflammatory diseases, infections, immune diseases, autoimmune
diseases
including idiopathic thrombocytopenic purpura, arthritis, systemic lupus
erythrematosus, and autoimmune hemolytic anemia.
The following are examples which illustrate the compositions and methods of
this invention with reference to production of an Ig glycoprotein composition.
These
examples should not be construed as limiting-the examples are included for the
purposes of illustration only. The skilled artisan recognizes that numerous
modifications and extensions of this disclosure including optimization are
possible.
Such modifications and extensions are considered part of the invention.
Example 1
Cloning o{DX-IgGl for expression in P. pastoris
The light (L) and heavy (H) chains of DX-IgG1 (an anti-CD20 IgGl) consists
of mouse variable regions and human constant regions. The light chain is
disclosed as
SEQ ID NO: 1 and heavy chain as SEQ ID NO: 2. The heavy and light chain
sequences were synthesized using overlapping oligonucleotides purchased from
Integrated DNA Technologies (IDT). For the light chain variable region, 15
overlapping oligonucleotides (SEQ ID NOs: 5-19) were purchased and annealed
using
Extaq (Takada) in a PCR reaction to produce the light chain variable region
fragment
having a 5' M1yI site. This light chain variable fragment was then joined with
the
light chain constant region (SEQ ID NO: 3) (Gene Art, Toronto, Canada) by
overlapping PCR using the 5' Mlyl primer CD20L/up (SEQ ID NO: 20), the 3'
variable/5' constant primer LfusionRTVAAPS/up (SEQ ID NO: 21), the 3' constant
region primer Lfusion RTVAAPS/lp (SEQ ID NO: 22) and 3' CD20L/lp (SEQ ID
56
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
NO: 23). The final MlyI-light chain fragment (which included 5'AG base pairs)
was
then inserted into pCR2.1 topo vector (Invitrogen) resulting in pDX343. For
the
heavy chain, 17 overlapping oligonucleotides (SEQ ID NOs: 24-40) corresponding
to
the mouse heavy chain variable region were purchased from IDT and annealed
using
Extaq. This heavy chain variable fragment was then joined with the heavy chain
constant region (SEQ ID NO: 4) (Gene Art) by overlapping PCR using the 5' MlyI
primer CD20H/up (SEQ ID NO: 41), the 5' variable/constant primer
HchainASTKGPS/up (SEQ ID NO: 42), the 3' variable/constant primer
HchainASTKGPS/lp (SEQ ID NO: 43) and the 3' constant region primer HFckpnl/lp
(SEQ ID NO: 44). The final Mlyl-heavy chain fragment (which included 5'AG base
pairs) was inserted into pCR2.1 topo vector (Invitrogen) resulting in pDX360.
The
full length light chain and full length heavy chain were isolated from the
respective
topo vectors as Mlyl and Notl fragments. These light chain and heavy chain
fragments were then ligated to a Kar2(Bip) signal sequence (SEQ ID NO: 45)
using 4
overlapping oligonucleotides-P.BiPss/UPl-EcoRI, P.BiPss/LP1, P.BiPss/UP2 and
P.BiP/LP2 (SEQ ID NOS: 46-49, respectively), and then ligated into the EcoRI-
Notl
sites of pPICZA resulting in pDX344 carrying the Kar2-light chain and pDX468
carrying the Kar2-heavy chain. A Bg1II-BamHI fragment from pDX344 was then
subcloned into pBK85 containing the AOX2 promoter gene for chromosomal
integration, resulting in pDX458. A Bg1II-BamHI fragment from pDX468 carrying
the heavy chain was then subcloned into pDX458, resulting in pDX478 containing
both heavy and light chains of CD20 under the AOX1 promoter. This plasmid was
then linearized with SpeI prior to transformation into YAS309 for integration
into the
AOX2 locus with transformants selected using Zeocin resistance. (See Example
2)
57
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
Cloning ofJC-IjzG for expression in P. pastoris
The light (L) and heavy (H) chains of the JC-IgGl consists of mouse variable
regions and human constant regions. The mouse variable light chain is
disclosed as
SEQ ID NO: 50 (GenBank #AF013576) and mouse variable heavy chain as SEQ ID
NO: 51 (GenBank #AF013577). The heavy and light chain sequences were
synthesized using overlapping oligonucleotides purchased from Integrated DNA
Technologies (IDT). For the light chain, 12 overlapping oligonucleotides (SEQ
ID
NOs: 52-63) were purchased and annealed using Extaq (Takada) in a PCR reaction
to
produce the 660 base pair light chain having a 5' EcoRl site and a 3' Kpnl
site. This
light chain was then subcloned into a pPICZa vector (Invitrogen) as an EcoRI-
KpnI
fragment. For the heavy chain, 12 overlapping oligonucleotides (SEQ ID NOs: 64-
75) corresponding to the Fab fragment were purchased and annealed using Extaq
to
produce the 660 base pair Fab fragment. The Fc fragment was synthesized using
12
overlapping oligonucleotides (SEQ ID NOs: 76-87) which were annealed in a
similar
overlapping PCR reaction. Both Fab and Fc fragments of the heavy chain were
then
annealed using a 5' EcoRI primer (SEQ ID NO: 64) corresponding to the 5' end
of the
heavy Fab fragment and a 3' Kpnl primer (SEQ ID NO: 88) corresponding to the
3'
end of the Fc fragment using pFU Turbo polymerase (Stratagene) producing the
1,330
base pair heavy chain. Using 5' EcoRI and 3' Kpnl sites encoded in the
primers, the
heavy chain was cloned into a pPICZa vector. The AOX2 promoter sequence, which
functions as an integration locus, was subcloned into a final pPICZa vector.
Next, a
Bg1II-BstB1 fragment containing the AOX1 promoter and a BstB 1 -BamHI fragment
containing an HSA sequence from a human liver cDNA library (SEQ ID NO:89),
thrombin site (SEQ ID NO:90) and JC light chain were both subcloned into the
BamHI site of this AOX2/pPICZa vector. Then another a B1gII-BstBI fragment
58
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
containing the AOX1 promoter and a BstIBl-BamHI fragment containing an HSA
sequence, thrombin site and JC heavy chain were subcloned into the BamHI site
of
this same pPICZa vector. This final vector contains the AOX2 integration
locus,
HSA-tagged JC light chain and HSA-tagged JC heavy chain, resulting in pJC140.
This expression cassette was integrated into the AOX2 locus of a P. pastoris
strain
with transformants selected for zeocin resistance. (See Example 2).
RituximabQ/Rituxan is an anti-CD20 mouse/ human chimeric IgGl purchased from
Biogen-IDEC/Genentech, San Francisco, CA.
PCR am,plification. An Eppendorf Mastercycler was used for all PCR reactions.
PCR
reactions contained template DNA, 125 M dNTPs, 0.2 M each of forward and
reverse primer, Ex Taq polymerase buffer (Takara Bio Inc.), and Ex Taq
polymerase
or pFU Turbo polymerase buffer (Stratagene) and pFU Turbo polymerase. The DNA
fragments were amplified with 30 cycles of 15 sec at 97 C, 15 sec at 55 C and
90 sec
at 72 C with an initial denaturation step of 2 min at 97 C and a final
extension step of
7 min at 72 C.
PCR samples were separated by agarose gel electrophoresis and the DNA
bands were extracted and purified using a Gel Extraction Kit from Qiagen. All
DNA
purifications were eluted in 10 mM Tris, pH 8.0 except for the final PCR
(overlap of
all three fragments) which was eluted in deionized H20.
Example 2
Transformation oflQG (pD~'478 and pJC140) vectors into P. pastoris strain
YAS309.
The vector DNA of pDX478 and pJC140 was prepared by adding sodium
acetate to a final concentration of 0.3 M. One hundred percent ice cold
ethanol was
59
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
then added to a final concentration of 70% to the DNA sample. The DNA was
pelleted by centrifugation (12000g x 10min) and washed twice with 70% ice cold
ethanol. The DNA was dried and resuspended in 50 l of 10mM Tris, pH 8Ø The
YAS309 yeast culture (supra) to be transformed was prepared by expanding a
smaller
culture in BMGY (buffered minimal glycerol: 100 mM potassium phosphate, pH
6.0;
1.34% yeast nitrogen base; 4x10-5% biotin; 1% glycerol) to an O.D. of -2-6.
The
yeast cells were then made electrocompetent by washing 3 times in 1M sorbitol
and
resuspending in -1-2 mis 1M sorbitol. Vector DNA (1-2 g) was mixed with 100
1
of competent yeast and incubated on ice for 10 min. Yeast cells were then
electroporated with a BTX Electrocell Manipulator 600 using the following
parameters; 1.5 kV, 129 ohms, and 25 F. One milliliter of YPDS (1% yeast
extract,
2% peptone, 2% dextrose, 1M sorbitol) was added to the electroporated cells.
Transformed yeast was subsequently plated on selective agar plates containing
zeocin.
Culture conditions for I-zGI production in P. pastoris
A single colony of YAS309 transformed with pDX478 or pJC140 was
inoculated into 10ml of BMGY media (consisting of 1% yeast extract, 2%
peptone,
100mM potassium phosphate buffer (pH 6.0), 1.34% yeast nitrogen base, 4x10-5%
biotin, and 1% glycerol) in a 50m1 Falcon Centrifuge tube. The culture was
incubated
while shaking at 24 C/170-190 rpm for 48 hours until the culture was
saturated.
100m1 of BMGY was then added to a 500m1 baffled flask. The seed culture was
then
transferred into a baffled flask containing the 100m1 of BMGY media. This
culture
was incubated with shaking at 24 C/170-190rpm for 24 hours. The contents of
the
flask was decanted into two 50m1 Falcon Centrifuge tubes and centrifuged at
3000rpm
for 10 minutes. The cell pellet was washed once with 20m1 of BMGY without
glycerol, followed by gentle resuspension with 20m1 of BMMY (BMGY with 1%
MeOH instead of 1% glycerol). The suspended cells were transferred into a
250m1
baffled flask. The culture was incubated with shaking at 24 C/170-190rpm for
24
hours. The contents of the flask was then decanted into two 50m1 Falcon
Centrifuge
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
tubes and centrifuged at 3000rpm for 10 minutes. The culture supernatant was
analyzed by ELISA to determine approximate antibody titer prior to protein
isolation
(see Example 3).
Quanti acatiora of antibody in culture supematants was performed by enzyme
linked
immunosorbent assays (ELISAs): High binding microtiter plates (Costar) were
coated with 24 g of goat anti-human Fab (Biocarta, Inc, San Diego, CA) in 10
ml
PBS, pH 7.4 and incubate over night at 4 C. Buffer was removed and blocking
buffer (3% BSA in PBS), was added and then incubated for 1 hour at room
temperature. Blocking buffer was removed and the plates were washed 3 times
with
PBS. After the last wash, increasing volume amounts of antibody culture
supematant
(0.4, 0.8, 1.5, 3.2, 6.25, 12.5, 25 and 50 gl) was added and incubated for 1
hour at
room temperature. Plates were then washed with PBS + 0.05% Tween20. After the
last wash, anti-human Fc-HRP was added in a 1:2000 PBS solution, and then
incubated for 1 hour at room temperature. Plates were then washed 4 times with
PBS-
Tween20. Plates were analyzed using TMB substrate kit following manufacturer's
instructions (Pierce Biotechnology).
Example 3
Purification oflgGl
Monoclonal antibodies were captured from the culture supernatant using a
Streamline Protein A column. Antibodies were eluted in Tris-Glycine pH 3.5 and
neutralized using 1M Tris pH 8Ø Further purification was carried out using
hydrophobic interaction chromatography (HIC). The specific type of HIC column
depends on the antibody. For the JC-IgG and the DX-IgG a phenyl sepharose
column (can also use octyl sepharose) was used with 20mM Tris (7.0), 1M
(NH4)2SO4
buffer and eluted with a linear gradient buffer of 1M to OM (NH4)2SO4. The
antibody
61
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
fractions from the phenyl sepharose column were pooled and exchanged into 50mM
NaOAc/Tris pH 5.2 buffer for final purification through a cation exchange (SP
Sepharose Fast Flow) (GE Healthcare) column. Antibodies were eluted with a
linear
gradient using 50mM Tris, 1M NaCI (pH 7.0)
Treatment of JC-IgG or DX I G with ,(3 galactosidase and fl--N-acetyl-
hexosaminidase.
5mg of purified IgG (JC-IgG or DX-IgG) was buffer exchanged into 50 mM
NH4Ac pH 5Ø In a siliconized tube, 0.3 U(3-N-acetylhexosaminidase and 0.03U
(3-
1,4 galactosidase (EMD Biosciences, La Jolla, CA) was added to the purified
IgG in
50mM NH4Ac pH 5.0 and incubated for 16-24 hours at 37 C. A sample of this was
evaporated to dryness, resuspended in water and analyzed by MALDI-TOF. The
antibody was then purified from the (3-N-acetylhexosaminidase and (3-1,4
galactosidase using a phenyl sepharose purification as described above.
Example 4
Detection ofpurifzed I~
Purified JC-IgG or DX-IgG were mixed with an appropriate volume of
sample loading buffer and subjected to sodium dodecyl sulfate-polyacrylamide
gel
electrophoresis (SDS-PAGE) with precast gels according to the manufacturer's
instructions (NuPAGE bis-Tris electrophoresis system; Invitrogen Corporation,
Carlsbad, Calif.). The gel proteins were stained with Coomassie brilliant blue
stain
(Bio-Rad, Hercules, CA). See Figures 2 and 3.
Antibody concentrations
62
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
The concentration of protein chromatography fractions were determined using
a Bradford assay (Bradford, M. 1976, Anal. Biochem. (1976) 72, 248-254) using
albumin as a standard (Pierce, Rockford, IL)
Example 5
IgGl carbohydrate analysis
Matrix Assisted Laser Desorption Ionization Time of Flight Mass
Spectrometry (MALDI-TOF MS). MALDI-TOF analysis of aspargine-linked
oligosaccharides: N-linked glycans were released from JC-IgG and DX-IgG using
a
modified procedure of Papac et al., Glycobiology 8, 445-454 (1998). A sample
of the
antibodies was reduced and carboxymethylated and the membranes were blocked,
the
wells were washed three times with water. The IgG proteins were deglycosylated
by
the addition of 30 ul of 10 mM Nh4HCO3 (pH 8.3) containing 1 mU of N-glycanase
(EMD Biosciences, La Jolla, CA). After 16 hours at 37 C, the solution
containing the
glycans was removed by centrifugation and evaporated to dryness. The dried
glycans
from each well were dissolved in 15 l of water, and 0.5 l was spotted on
stainless-
steel sample plates and mixed with 0.5 l of S-DHB matrix (9 mg/ml of
dihydroxybenzoic acid/1 mg/ml of 5-methoxy-salicylic acid in 1:1
water/acetonitrile/0. 1 % trifluoroacetic acid) and allowed to dry. Ions were
generated
by irradiation with a pulsed nitrogen laser (337 nm) with a 4-ns pulse time.
The
instrument was operated in the delayed extraction mode with a 125-ns delay and
an
accelerating voltage of 20 W. The grid voltage was 93.00%, guide wire voltage
was
0.1%, the internal pressure was <5x 10-7 torr (1 torr=133 Pa), and the low
mass gate
was 875 Da. Spectra were generated from the sum of 100-200 laser pulses and
acquired with a 500-MHz digitizer. (Man)5(G1cNAc)2 oligosaccharide was used as
an
63
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
external molecular weight standard. All spectra were generated with the
instrument in
the positive-ion mode.
Example 6
Antijzen binding ELISA assay
High binding microtiter plates (Costar) were coated with 10ug of antigen in
PBS, pH 7.4 and incubate over night at 4 C. Buffer was removed and blocking
buffer (3% BSA in PBS), was added and then incubated for 1 hour at room
temperature. Blocking buffer was removed and the plates were washed 3 times
with
PBS. After the last wash, increasing amounts of purified antibody were added
from
0.2ng to 100ng and incubated for 1 hour at room temperature. Plates were then
washed with PBS + 0.05% Tween2O. After the last wash, anti-human Fc-HRP was
added in a 1:2000 PBS solution, and then incubated for 1 hour at room
temperature.
Plates were then washed 4 times with PBS-Tween2O. Plates were analyzed using
TMB substrate kit following manufacturer's instructions (Pierce
Biotechnology).
Fc Receptor binding assays
Fc receptor binding assays for FcyRIIb, FcyRIIIa and FcyRI1Ib were carried
out according to the protocols previously described (Shields et al., 2001,
J.Biol. Chein,
276: 6591-6604). For FcyRIII binding: FcyRIIIb (Figure 5) and F yRIIb (Figure
7)
fusion proteins at 1 g/ml or FcyRIIIa-LF (Figure 6) fusion proteins at 0.8
g/m in
PBS, pH 7.4, were coated onto ELISA plates (Nalge-Nunc, Naperville, IL) for 48
h at
4 C. Plates were blocked with 3% bovine serum albumin (BSA) in PBS at 25 C for
1 h. JC-IgG or DX-IgG dimeric complexes were prepared in 1% BSA in PBS by
mixing 2:1 molar amounts of JC-IgG or DX IgG and HRP-conjugated F(Ab')2anti-
F(Ab')2 at 25 C for 1 h. Dimeric complexes were then diluted serially at 1:2
in 1%
BSA/PBS and coated onto the plate for 1 hour at 25 C. The substrate used is
3,3',5,5'-tetramethylbenzidine (TMB) (Vector Laboratories). Absorbance at 450
nm
was read following instructions of the manufacturer (Vector Laboratories).
64
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
ELISPOT assay for antibody feedback in B cells.
This assay is conducted as described ira Westman, et al., 1997, Scand. J.
Immunol. 46: 10-15. BSA (bovine serum albumin) is first conjugated to an IgG
antibody resulting in a BSA-IgG complex. The number of B cells secreting BSA-
specific IgG is determined using an ELISPOT assay. Spleens are removed from
injected mice and cell suspensions are prepared in DMEM (Gibco, New York) with
0.5% normal mouse serum. One hundered microliter cell suspensions are applied
to
BSA-coated microtiter plates (see ELISA protocol above) and incubated at 37 C,
5%
CO2 for 3.5 h. Plates are washed and incubated at 4 C o.n. with 50 l of
alkaline
phosphatase-conjugated sheep anit-mouse IgG dilute 1/100 in PBS-Tween. Spots
are
developed for 1 hour at room temperature in 50 gl of 5 bromo-4-chloro-3-indoyl
phosphate (Sigma-Aldrich) and counted under a stereomicroscope.
Example 7
For ADCC assayed using a blood matrix study (e.g. B-cell depletion) as
described in
Vugmeyster and Howell, 2004, Int. Immunopharm. 4: 1117-1124. Whole blood
depleted of plasma and red blood cells (RBCs) is reconstituted in stain buffer
(Hank's
balanced salt solution (HBSS) with 1% BSA and 0.1% sodium azide) leading to
leukocyte suspension in stain buffer. Whole blood sample is then spun for 5
minutes
at 1000 g, the supernatant (plasma) is discarded and the pellet is treated
with
ammonium chloride lysing (ACL) reagent, washed, and resuspended in an
equivalent
volume of stain buffer. For B-cell depletion assay: 10 l of 100 g/mi
solution of
antibody or stain buffer is added to 90 l of SB matrix and incubated for 1
hour at
37 C. Samples are stained immediately with anti-CD19-FITC and anti-CD45-PE for
30 minutes at 25 C. Samples are then fixed in 1% formaldehyde and run in
triplicate.
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
Quantification of B-cell depletion is obtained by flow cytometry. Flow
cytometric
analysis ofB-cell depletion: A FACS Calibur (BD Biosciences) instrument
equipped
with an automated FACS Loader and Cell Quest Software is used for acquisition
and
analysis of all samples. Cytometer QC and setup include running CaliBrite
beads and
SpheroTech rainbow beads (BD Biosciences) to confirm instrument functionality
and
detector linearity. Isotype and compensation controls are run with each assay
to
confirm instrument settings. Percent of B cells of total lymphocytes is
obtained by
the following gating strategy. The lymphocyte population is marked on the
forward
scatter/side scatter scattegram to define Region 1(Rl). Using events in Rl,
fluorescence intensity dot plots are displayed for CD 19 and CD45 markers.
Fluorescently labeled isotype controls are used to determine respective cutoff
points
for CD19 and CD45 positivity. %B is determined using CellQuest as a fraction
of
cells in Rl region that have CD19-positive, CD45-positive phenotype.
Triplicate
samples are run for each treatment group. The percent B cell depletion is
calculated
using the formula average [100*(1 - %B treated with control antibody/average
[%B
treated with SB]). Fluorescent dye release ADCC assay: PBMC isolation:
Peripheral
venous blood from healthy individuals or blood donors (10-20) is collected
into
heparinised vacutainer tubes (Becton Dickinson Vacutainer Systems, Rutherford,
NJ,
USA). Approximately 5m1 of blood is required for implanting 2 mice. Peripheral
blood mononuclear cells (PBMCs) are separated by centrifugation using OptiPrep
following manufacturer's instructions. PBMCs are washed once with complete
culture media (CM) consisting of RPMI 1640, 2mM L-glutamine, 100 IU/ml
penicillin, 100g/mi streptomycin (Gibco/BRL) and supplemented with 20% fetal
calf
serum, and then resuspended at a concentration of 1x106/ml CM and transfered
to a
250 ml culture flask (Falcon, NJ, USA) for monocyte depletion. After 1 hour of
66
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
incubation at 37 C and 5% C02, non-adherent cells are recovered, washed once
with
culture media and the peripheral blood lymphocytes (PBLs) are adjusted to a
concentration of 2.5x107 /ml CM. Fluorescent dye-release ADCC. The premise
behind the ADCC assay is that antibody binding to CD20 or CD40 antigen
presenting
target cells (Raji cell line or BCL1-3B3 cells, respectively) stimulates
target cell
binding to Fcy receptors on the effector cells. This in turn promotes lysis of
the target
cells presenting the CD20 or CD40 antigen, releasing an internal fluorescent
dye that
can be quantified. Alamar-blue fluorescence is used in place of 51Cr labeling
of the
target cells. 50u1 of CD20-presenting Raji cell suspension (1x104 cells) is
combined
with 50u1 amount of anti-DX-IgG or anti-JC-IgG mAb (various concentrations)
and
50 ul amount of PBMC effector cells isolated as described above (effector to
target
cell ratio can be 100:1, 50:1. 25:1 and 12.5:1) in 96 well tissue culture
plates and
incubated for 4h hours at 37 temperature and 5% C02 to facilitate lysis of the
Raji or
BCL1-3B3 cells. 50 g1 of Alamar blue is added and the incubation is continued
for
another 5 hours to allow for uptake and metabolism of the dye into its
fluorescent
state. The plates cool to room temperature on a shaker and the fluorescence is
read in
a fluorometer with excitation at 530 nm and emission at 590 nm. Relative
fluorescence units (RFU) are plotted against mAb concentrations and sample
concentrations are computed from the standard curve using a control antibody-
e.g.
Rituximab . In viyo ADCC usinz Severe Combined ImnZunode acient (SCID) mice
(Niwa et al., 2004, Cancer Research, 64: 2127-2133). In vivo ADCC activity can
be
assayed using a mouse model engrafted with human peripheral blood mononuclear
cells (PMBCs) from healthy donors which include heterozygous (FcyRIIIa-
LF/FcyRIIIa-LV) and homozygous (FcyRIIIa-LV/FcyRIIIa-LV and FcyRIIIa-
LF/FcyRIIIa-LF) genotypes. Using this model system, Igs having a predominant N-
67
CA 02573541 2007-01-11
WO 2006/014685 PCT/US2005/025662
glycan are assayed for enhanced ADCC activity compared with Rituximab or any
other control antibody. A detailed and sufficient protocol for this in vivo
ADCC
assay is found in Niwa et al., 2004, supra.
68
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.