Note: Descriptions are shown in the official language in which they were submitted.
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
PROCESS FOR THE DE-ENRICHMENT OF ENANTIOMERICALLY ENRICHED
SUBSTRATES
The invention concerns a process for the de-enrichment of enantiomerically
enriched substrates, especially alcohols and sulphides.
According to a first aspect of the present invention, there is provided a
process for
the de-enrichment of enantiomerically enriched compositions which comprises
reacting an
enantiomerically enriched composition comprising at least a first enantiomer
or
diastereomer of a substrate comprising a carbon-heteroatom bond, wherein the
carbon is
a chiral centre and the heteroatom is a group VI heteroatom, in the presence
of a catalyst
system and optionally a reaction promoter to give a product composition
comprising first
and second enantiomers or diastereomers of the substrate having a carbon-
heteroatom
bond, the ratio of second to first enantiomer or diastereomer in the product
composition
being greater than the ratio of second to first enantiomer or diastereomer in
the
enantiomerically enriched composition.
Preferably the product composition is a racemic mixture of the first and
second
enantiomers of the substrate comprising a carbon-heteroatom bond, wherein the
carbon
is a chiral centre.
Substrates which may be enantiomerically de-enriched by the process of the
present invention include alcohols at a chiral secondary carbon atom and
sulphides chiral
at a secondary carbon atom.
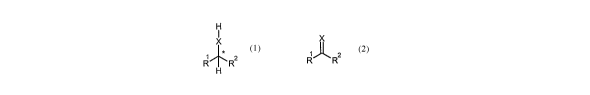
Preferably, in the process of the present invention, the substrate comprising
a
carbon-heteroatom bond, the carbon atom being a chiral centre, is a compound
of
formula (1):
H
I
x
kR2
H
(~)
wherein:
X represents 0, S;
R1, R2 each independently represents an optionally substituted hydrocarbyl, a
perhalogenated hydrocarbyl, an optionally substituted heterocyclyl group; or
R' & R2 are optionally linked in such a way as to form an optionally
substituted
ring(s);
provided that R' and R2 are selected such that * is a chiral centre.
Hydrocarbyl groups which may be represented by R1-2 independently include
alkyl,
alkenyl and aryl groups, and any combination thereof, such as aralkyl and
alkaryl, for
example benzyl groups.
Alkyl groups which may be represented by R'-2 include linear and branched
alkyl
groups comprising up to 20 carbon atoms, particularly from 1 to 7 carbon atoms
and
CONFIRMATION COPY
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
2
preferably from I to 5 carbon atoms. When the alkyl groups are branched, the
groups
often comprising up to 10 branch chain carbon atoms, preferably up to 4 branch
chain
atoms. In certain embodiments, the alkyl group may be cyclic, commonly
comprising
from 3 to 10 carbon atoms in the largest ring and optionally featuring one or
more bridging
rings. Examples of alkyl groups which may be represented by R1-4 include
methyl, ethyl,
propyl, 2-propyl, butyl, 2-butyl, t-butyl and cyclohexyl groups.
Alkenyl groups which may be represented by R'-2 include C2_20, and preferably
C2_6
alkenyl groups. One or more carbon - carbon double bonds may be present. The
alkenyl
group may carry one or more substituents, particularly phenyl substituents.
Examples of
alkenyl groups include vinyl, styryl and indenyl groups. When either of R' or
R2
represents an alkenyl group, a carbon - carbon double bond is preferably
located at the
position R to the C-heteroatom moiety.
Aryl groups which may be represented by R'-2 may contain 1 ring or 2 or more
fused rings which may include cycloalkyl, aryl or heterocyclic rings. Examples
of aryl
i5 groups which may be represented by R'-Z include phenyl, tolyl,
fluorophenyl, chlorophenyl,
bromophenyl, trifluoromethylphenyl, anisyl, naphthyl and ferrocenyl groups.
Perhalogenated hydrocarbyl groups which may be represented by R'-2
independently include perhalogenated alkyl and aryl groups, and any
combination thereof,
such as aralkyl and alkaryl groups. Examples of perhalogenated alkyl groups
which may
be represented by R'-2 include -CF3 and -C2FS.
Heterocyclic groups which may be represented by R1-2 independently include
aromatic, saturated and partially unsaturated ring systems and may constitute
1 ring or 2
or more fused rings which may include cycloalkyl, aryl or heterocyclic rings.
The
heterocyclic group will contain at least one heterocyclic ring, the largest of
which will
commonly comprise from 3 to 7 ring atoms in which at least one atom is carbon
and at
least one atom is any of N, 0, S or P. When either of R' or R2 represents or
comprises a
heterocyclic group, the atom in R' or R2 bonded to the C-heteroatom group is
preferably a
carbon atom. Examples of heterocyclic groups which may be represented by R1-2
include
pyridyl, pyrimidyl, pyrrolyl, thiophenyl, furanyl, indolyl, quinolyl,
isoquinolyl, imidazoyl and
triazoyl groups.
When any of R''2 is a substituted hydrocarbyl or heterocyclic group, the
substituent(s) should be such so as not to adversely affect the rate or
stereoselectivety of
the reaction. Optional substituents include halogen, cyano, nitro, hydroxy,
amino, thiol,
acyl, hydrocarbyl, perhalogentated hydrocarbyl, heterocyclyl, hydrocarbyloxy,
mono or di-
hydrocarbylamino, hydrocarbylthio, esters, carbonates, amides, sulphonyl and
sulphonamido groups wherein the hydrocarbyl groups are as defined for R'
above. One
or more substituents may be present.
When R' & R 2 are linked in such a way that when taken together with either
the
carbon atom and/or atom X of the compound of formula (1) that a ring is
formed, it is
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
3
preferred that these be 5, 6 or 7 membered rings and optionally containing one
or more
ring heteroatoms, preferably 0, S or N ring atoms. Examples of such compounds
of
formula (1) include 2-methylcyclohexanol.
In certain preferred embodiments, R' and R2 are both different and selected to
both be different C,_s alkyl groups, both be different aryl groups,
particularly where one is
a phenyl group, or are selected such that one is aryl, particularly phenyl and
one is C,_s
alkyl. Substituents may be present, particularly substituents para to the C-X
group when
one or both of R' and R2 is a substituted phenyl group.
Examples of compounds of formula (1) include 1-phenylethan-l-ol, 1-(2-
naphthyl)ethan-l-ol, 1-(1-naphthyl)ethan-1-oI 1-phenylethan-l-thiol, 1-(2-
naphthyl)ethan-
1-thiol, and 1 -(1 -naphthyl)ethan-1 -thiol.
The catalyst system preferably comprises a transition metal catalyst and
optionally
a ligand.
Ligands which optionally may be present include amines, alcohols and
sulphides.
When a ligand is used, optionally the ligand and the transition metal catalyst
may
be pre-mixed or pre-coordinated prior to the reaction with the substrate.
Examples of
such pre-coordinated ligand and the transition metal catalysts include those
catalysts
disclosed in the International patent applications with publication numbers
W097/20789,
W098/42643, and W002/441 11, each of which is incorporated herein by
reference.
Transition metal catalysts include transition metal halides, transition metal
halide
complexes and transition metal complexes wherein the transition metal is
optionally
complexed by a displaceable ligand.
Displaceable ligands include phosphines, such as tri-hydrocarbyl phosphines
for
example Ph3P, carbenes such as imidazole carbene, nitriles such as
acetonitrile, carbon
monoxide, triflate, alkenes and dienes. Examples of transition metal complexes
wherein
the transition metal is optionally complexed by a displaceable ligand include
complexes of
the formula MnLoXpYr
Wherein
M is a transition metal;
L is a displaceable ligand;
X is a halide;
Y is a neutral optionally substituted hydrocarbyl complexing group, a
neutral optionally substituted perhalogenated hydrocarbyl complexing
group, or an optionally substituted cyclopentadienyl complexing group; and
n is an integer; and
each of o, p, and r is 0 or an integer provided that o+p+r is an integer.
Preferably, the transition metal catalyst is a transition metal halide or
transition
metal halide complex based on the transition metals in Group VIII of the
Periodic Table,
especially ruthenium, rhodium or iridium.
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
4
More preferably, the transition metal catalyst is a transition metal halide
complex
of the formula MnXPYr
Wherein
M is a transition metal;
X is a halide;
Y is a neutral optionally substituted hydrocarbyl complexing group, a
neutral optionally substituted perhalogenated hydrocarbyl complexing
group, or an optionally substituted cyclopentadienyl complexing group; and
n, p and r are integers.
Although transition metal catalyst is believed to be substantially as
represented in
the above formula, in some circumstances the transition metal catalyst may
also exist as
a dimer, trimer or some other polymeric species.
Metals which may be represented by M include metals which are capable of
catalysing transfer hydrogenation. Preferred metals include transition metals,
more
preferably the metals in Group VIII of the Periodic Table, (iron, cobalt,
nickel, ruthenium,
rhodium, palladium, osmium, iridium and platinum), more preferably ruthenium,
rhodium
or iridium, most preferably iridium.
Typically, the integers n, p, r are selected such that the transition metal
halide
complex is overall a neutral species. Therefore, the selection of n, p, r are
directly related
to the valance state of the metal and the number of halides present and the
nature of the
complexing group Y. For example, where Y is a negatively charged
cyclopentadienyl
complexing group, the number of negatively charged halides required to balance
the
valence state of the metal will be less than when Y is a neutral hydrocarbyl
complexing
group.
When the metal is ruthenium it is preferably present in valence state II. When
the
metal is rhodium or iridium it is preferably present in valence state I when Y
is a neutral
optionally substituted hydrocarbyl or a neutral optionally substituted
perhalogenated
hydrocarbyl ligand, and preferably present in valence state III when Y is an
optionally
substituted cyclopentadienyl ligand. An especially preferred metal is iridium.
The neutral optionally substituted hydrocarbyl or perhalogenated hydrocarbyl
complexing group which may be represented by Y includes optionally substituted
aryl and
alkenyl complexing group.
Optionally substituted aryl complexing groups which may be represented by Y
may contain 1 ring or 2 or more fused rings which include cycloalkyl, aryl or
heterocyclic
rings. Preferably, the complexing group comprises a 6 membered aromatic ring.
The
ring or rings of the aryl complexing group are often substituted with
hydrocarbyl groups.
The substitution pattern and the number of substituents will vary and may be
influenced
by the number of rings present, but often from 1 to 6 substituents are
present.
Substituents may include halogen, cyano, nitro, hydroxy, amino, thiol, acyl,
hydrocarbyl,
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
perhalogentated hydrocarbyl, heterocyclyl, hydrocarbyloxy, mono or di-
hydrocarbylamino,
hydrocarbylthio, esters, carbonates, amides, sulphonyl and sulphonamido groups
wherein
the hydrocarbyl groups are as defined for R' above. Typically, the 1 to 6
substituents are
each independently hydrocarbyl groups, preferably 2, 3 or 6 hydrocarbyl groups
and more
5 preferably 6 hydrocarbyl groups. Preferred hydrocarbyl substituents include
methyl, ethyl,
iso-propyl, menthyl, neomenthyl and phenyl. Particularly when the aryl
complexing group
is a single ring, the complexing group is preferably benzene or a substituted
benzene.
When the complexing group is a perhalogenated hydrocarbyl, preferably it is a
polyhalogenated benzene such as hexachlorobenzene or hexafluorobenzne. When
the
hydrocarbyl substitutents contain enantiomeric and/or diastereomeric centres,
it is
preferred that the enantiomerically and/or diastereomerically purified forms
of these are
used. Benzene, p-cymyl, mesitylene and hexamethylbenzene are especially
preferred
complexing group.
Optionally substituted alkenyl complexing groups which may be represented by Y
include Ca_30, and preferably C6_,a, alkenes or cycloalkenes with preferably
two or more
carbon-carbon double bonds, preferably only two carbon-carbon double bonds.
The
carbon-carbon double bonds may optionally be conjugated to other unsaturated
systems
which may be present, but are preferably conjugated to each other. The alkenes
or
cycloalkenes may be substituted preferably with hydrocarbyl substituents. When
the
alkene has only one double bond, the optionally substituted alkenyl complexing
group
may comprise two separate alkenes. Preferred hydrocarbyl substituents include
methyl,
ethyl, iso-propyl and phenyl. Examples of optionally substituted alkenyl
complexing
groups include cyclo-octa-1,5-diene and 2,5-norbornadiene. Cyclo-octa-1,5-
diene is
especially preferred.
Optionally substituted cyclopentadienyl complexing groups which' may be
represented by Y includes cyclopentadienyl groups capable of eta-5 bonding.
The
cyclopentadienyl group is often substituted with from 1 to 5 substituents.
Substituents
may include halogen, cyano, nitro, hydroxy, amino, thiol, acyl, hydrocarbyl,
perhalogentated hydrocarbyl, heterocyclyl, hydrocarbyloxy, mono or di-
hydrocarbylamino,
hydrocarbylthio, esters, carbonates, amides, sulphonyl and sulphonamido groups
wherein
the hydrocarbyl groups are as defined for R' above. Preferably, the
cyclopentadienyl
group is substituted with 1 to 5 hydrocarbyl groups, more preferably with 3 to
5
hydrocarbyl groups and most preferably with 5 hydrocarbyl groups. Preferred
hydrocarbyl
substituents include methyl, ethyl and phenyl. When the hydrocarbyl
substitutents
contain enantiomeric and/or diastereomeric centres, it may be advantageous
that the
enantiomerically and/or diastereomerically purified forms of these are used.
Examples of
optionally substituted cyclopentadienyl complexing groups include
cyclopentadienyl,
pentamethyl-cyclopentadienyl, pentaphenylcyclopentadienyl,
tetraphenylcyclopentadienyl,
ethyltetramethylpentadienyl, menthyltetraphenylcyclopentadienyl, neomenthyl-
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
6
tetraphenylcyclopentadienyl, menthylcyclopentadienyl,
neomenthylcyclopentadienyl,
tetrahydroindenyl, menthyltetrahydroindenyl and neomenthyltetrahydroindenyl
groups.
Pentamethylcyclopentadienyl is especially preferred.
Transition metal halide complexes of the formula M,XpY, wherein M is Rh or Ir,
and Y is an optionally substituted cyclopentadienyl group are preferred.
Transition metal
halide complexes of the formula MnXPYr wherein M is Ir and Y is an optionally
substituted
cyclopentadienyl group are most preferred.
Examples of transition metal halide complexes include RuZCI4(cymyl)2,
Rh2CI4(Cp')z, Rh2Br4(Cp*)2, Rh2l4(Cp*)2, Ir2C14(Cp')2, Ru214(cymyl)2,
RhCI2Cp', RhBr2Cp*,
RhI2Cp*, and Ir2l4(Cp')2wherein Cp* is a pentamethylcyclopentadienyl group.
In certain preferred embodiments, the catalyst system is preferably a
composition
obtainable by contacting a transition metal halide complex of the formula
M,,XPYrwherein
M is a transition metal; X is a halide; Y is a neutral optionally substituted
hydrocarbyl
complexing group, a neutral optionally substituted perhalogenated hydrocarbyl
complexing group, or an optionally substituted cyclopentadienyl complexing
group; and n,
p and r are integers with an alcohol or sulphide ligand of formula (1).
The catalytic system may advantageously be introduced, at least in part, on a
solid
support or as'an encapsulated system. Where the catalytic system is present on
a solid
support or as an encapsulated system, such supported catalyst systems may be
of
assistance in performing separation operations which may be required, and may
facilitate
the ease of cycling of materials between steps, especially when repetitions
are envisaged.
Examples of solid support or encapsulation technology that may be employed to
support
or encapsulate the catalytic system are described in W003/006151 and
W005/016510.
Reaction promoters, which optionally may be present, include halide salts, for
example metal halides. Preferred reaction promoters include bromide and
especially
iodide salts. Highly preferred are potassium iodide and caesium iodide.
Preferably, the process of the present invention is carried out in the
presence of a
base. Examples of bases include potassium carbonate and sodium carbonate.
In a further aspect of the present invention, the corresponding ketones or
thioketones of formula (2)
x
R~ R~
(2)
wherein:
X represents 0, S;
R1, R 2 each independently represents an optionally substituted hydrocarbyl, a
perhalogenated hydrocarbyl, an optionally substituted heterocyclyl group; or
R' & R2 are optionally linked in such a way as to form an optionally
substituted
ring(s);
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
7
provided that R' and R2 are different,
derived by deprotonation of the starting alcohols or sulphides of formula (1)
may be
produced.
Where it is desired to suppress or promote the production of the corresponding
ketones or thioketones of formula (2) derived by deprotonation of the starting
alcohols or
sulphides of formula (1), the use of hydrogen acceptors and/or hydrogen donors
may
advantageously be employed.
Hydrogen acceptors which may be present in the process of the present
invention
include the proton from an acid, oxygen, aldehydes and ketones, imines and
imminium
salts, readily hydrogenatable hydrocarbons, dyes, clean oxidising agents,
carbonates,
bicarbonates and any combination thereof.
The proton may emanate from any convenient and compatible acid such as formic
acid, acetic acid, hydrogen carbonate, hydrogen sulfate, ammonium salt or
alkyl
ammonium salt. Conveniently the proton may emanate from the substrate itself.
Aldehydes and ketones which may be employed as hydrogen acceptors comprise
commonly from 1 to 20 carbon atoms, preferably from 2 to 15 carbon atoms, and
more
preferably 3 to 5 carbon atoms. Aldehydes and ketones include alkyl, aryl,
hetroaryl
aidehydes and ketones, and ketones with mixed alkyl, aryl or hetroaryl groups.
Examples of aldehydes and ketones which may be represented as hydrogen
acceptors
include formaldehyde, acetone, methylethylketone and benzophenone. When the
hydrogen donor is an aldehyde or ketone, acetone is especially preferred.
Readily hydrogenatable hydrocarbons which may be employed as hydrogen
acceptors comprise hydrocarbons which have a propensity to accept hydrogen or
hydrocarbons which have a propensity to form reduced systems. Examples of
readily
hydrogenatable hydrocarbons which may be employed by as hydrogen donors
include
quinones, dihydroarenes and tetrahydroarenes.
Clean oxidising agents which may be represented as hydrogen acceptors
comprise reducing agents with a high reduction potential, particularly those
having an
oxidation potential relative to the standard hydrogen electrode of greater
than about
0.1 eV, often greater than about 0.5eV, and preferably greater than about 1eV.
Examples
of clean oxidising agents which may be represented as hydrogen acceptors
include
oxidising metals and oxygen.
Dyes include Rose Bengal, Proflavin, Ethidium Bromide, Eosin and
Phenolphthalein.
Carbonates and bicarbonates include alkali metal, alkaline earth metal,
ammonium and quaternary amine salts of carbonate and bicarbonate.
The most preferred hydrogen acceptors are protons from acids, acetone, oxygen,
the substrate amine and carbonate and bicarbonate salts.
Hydrogen donors include hydrogen, primary and secondary alcohols, primary
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
8
secondary and tertiary amines, carboxylic acids and their esters and amine
salts, readily
dehydrogenatable hydrocarbons, clean reducing agents, and any combination
thereof.
Primary and secondary alcohols which may be employed as hydrogen donors
comprise commonly from 1 to 10 carbon atoms, preferably from 2 to 7 carbon
atoms, and
more preferably 3 or 4 carbon atoms. Examples of primary and secondary
alcohols which
may be represented as hydrogen donors include methanol, ethanol, propan-l-ol,
propan-
2-ol, butan-l-ol, butan-2-ol, cyclopentanol, cyclohexanol, benzylalcohol, and
menthol.
When the hydrogen donor is an alcohol, secondary alcohols are preferred,
especially
propan-2-ol and butan-2-ol.
Primary secondary and tertiary amines which may be employed as hydrogen
donors comprise commonly from 1 to 20 carbon atoms, preferably from 2 to 14
carbon
atoms, and more preferably 3 or 8 carbon atoms. Examples of primary, secondary
and
tertiary amines which may be represented as hydrogen donors include
ethylamine,
propylamine, isopropylamine, butylamine, isobutylamine, hexylamine,
diethylamine,
1.5 dipropylamine, di-isopropylamine, dibutylamine, di-isobutylamine,
dihexylamine,
benzylamine, dibenzylamine, piperidine, (R) or (S) 6,7-dimethoxy-l-
methyidihydroisoquinoline, triethylamine. When the hydrogen donor is an amine,
primary
amines are preferred, especially primary amines comprising a secondary alkyl
group,
particularly isopropylamine and isobutylamine.
Carboxylic acids or their esters or salts which may be employed as hydrogen
donors comprise commonly from 1 to 10 carbon atoms, preferably from 1 to 3
carbon
atoms. In certain embodiments, the carboxylic acid is advantageously a beta-
hydroxy-
carboxylic acid. Esters may be derived from the carboxylic acid and a C,_,o
alcohol.
Examples of carboxylic acids which may be employed as hydrogen donors include
formic
acid, lactic acid, ascorbic acid and mandelic acid. When a carboxylic acid is
employed as
hydrogen donor, at least some of the carboxylic acid is preferably present as
a salt.
Amine salts may be formed. Amines which may be used to form such salts include
both
aromatic and non-aromatic amines, also primary, secondary and tertiary amines
and
comprise typically from 1 to 20 carbon atoms. Tertiary amines, especially
trialkylamines,
are preferred. Examples of amines which may be used to form salts include
trimethylamine, triethylamine, di-isopropylethylamine and pyridine. The most
preferred
amine is triethylamine. When at least some of the carboxylic acid is present
as an amine
salt, particularly when a mixture of formic acid and triethylamine is
employed, the mole
ratio of acid to amine is commonly about 5: 2. This ratio may be maintained
during the
course of the reaction by the addition of either component, but usually by the
addition of
the carboxylic acid. Other preferred salts include sodium, potassium,
magnesium
Readily dehydrogenatable hydrocarbons which may be employed as hydrogen
donors comprise hydrocarbons which have a propensity to aromatise or
hydrocarbons
which have a propensity to form highly conjugated systems. Examples of readily
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
9
dehydrogenatable hydrocarbons which may be employed by as hydrogen donors
include
cyclohexadiene, cyclohexene, tetralin, dihydrofuran and terpenes.
Clean reducing agents which may be represented as hydrogen donors comprise
reducing agents with a high reduction potential, particularly those having a
reduction
potential relative to the standard hydrogen electrode of greater than about -
0.1eV, often
greater than about -0.5eV, and preferably greater than about -1eV. Examples of
clean
reducing agents which may be represented as hydrogen donors include hydrazine
and
hydroxylamine.
The most preferred hydrogen donors are (R) or (S) 6,7-dimethoxy-l-
methyldihydroisoquinoline propan-2-ol, butan-2-ol, triethylammonium formate,
sodium
formate, potassium formate and a mixture of triethylammonium formate and
formic acid.
Although gaseous hydrogen may be present, the process is normally operated in
the absence of gaseous hydrogen since it appears to be unnecessary.
Typically, inert gas sparging may be employed.
is Suitably the process is carried out at temperatures in the range of from
minus 78
to plus 150 C, preferably from minus 20 to plus 110 C and more preferably from
minus
10 to plus 40 C.
The initial concentration of the substrate, a compound of formula (1), is
suitably in
the range 0.05 to 1.0 and, for convenient larger scale operation, can be for
example up to
6.0 more especially 0.75 to 2.0, on a molar basis. The 'molar ratio of the
substrate to the
catalyst system is suitably no less than 50:1 and can be up to 50000:1,
preferably
between 250:1 and 5000:1 and more preferably between 500:1 and 2500:1.
If a reaction promoter is present, the reaction promoter is preferably
employed in a
molar excess over the substrate, especially from 1 to 5 fold or, if
convenience permits,
greater, for example up to 20 fold.
If a hydrogen donor and/or acceptor is present, the hydrogen donor and/or
acceptor is preferably employed in a molar excess over the substrate,
especially from 5 to
20 fold or, if convenience permits, greater, for example up to 500 fold.
Reaction times are typically in the range of from 1.0 min to 24h, especially
up to
8h and conveniently about 3h. After reaction, the mixture is worked up by
standard
procedures.
A reaction solvent may be present, for example dimethylformamide,
acetonitrile,
tetrahydrofuran, toluene, chloroform, dichloromethane or, conveniently, the
substrate
alcohol or sulphide when the substrate alcohol or sulphide is liquid at the
reaction
temperature. Usually it is preferred to operate in substantial absence of
water, but water
does not appear to unduly inhibit the reaction. If the substrate amine or the
reaction
solvent is not miscible with water and the desired product is water soluble,
it may be
desirable to have water present as a second phase. The concentration of
substrate may
be chosen to optimise reaction time, yield and de-enrichment of enantiomeric
excess.
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
In a yet further aspect of the present invention, a composition comprising the
ketones or thioketones of formula (2) resulting from reacting an
enantiomerically enriched
composition comprising at least a first enantiomer or diastereomer of a
substrate
comprising a carbon-heteroatom bond, wherein the carbon is a chiral centre and
the
5 heteroatom is a group VI heteroatom, in the presence of a catalyst system
and optionally
a reaction promoter is then contacted with a transfer hydrogenation catalyst
and a
hydrogen donor to give a product composition comprising first and second
enantiomers or
diastereomers of the substrate having a carbon-heteroatom bond, the ratio of
second to
first enantiomer or diastereomer in the product composition being greater than
the ratio of
so second to first enantiomer or diastereomer in the enantiomerically enriched
composition.
Hydrogen donors are as defined hereinbefore above.
The reduction of compounds of Formula 2 is preferably accomplished employing a
stereoselective reduction system. It is most preferred that the
stereoselective reduction
employs a chiral coordinated transition metal catalysed transfer hydrogenation
process.
i.s Examples of such processes, and the catalysts, reagents and conditions
employed
therein include those disclosed in International patent application
publication numbers
W097/20789, W098/42643, and W002/441 11 each of which is incorporated herein
by
reference. Preferred transfer hydrogenation catalysts for, use in the process
of the
present invention have the general formula (a):
E
A B
\ M
Y~ 'R3
(a)
wherein:
R' represents a neutral optionally substituted hydrocarbyl, a neutral
optionally
substituted perhalogenated hydrocarbyl, or an optionally substituted
cyclopentadienyl
ligand;
A represents an optionally substituted nitrogen;
B represents an optionally substituted nitrogen, oxygen, sulphur or
phosphorous;
E represents a linking group;
M represents a metal capable of catalysing transfer hydrogenation; and
Y represents an anionic group, a basic ligand or a vacant site;
provided that at least one of A or B comprises a substituted nitrogen and the
substituent has at least one chiral centre; and
provided that when Y is not a vacant site that at least one of A or B carries
a
hydrogen atom.
Particularly preferred transfer hydrogenation catalysts are those Ru, Rh or Ir
catalysts of the type described in W097/20789, W098/42643, and W002/441 11
which
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
11
comprise an optionally substituted diamine ligand, for example an optionally
substituted
ethylene diamine ligand, wherein at least one nitrogen atom of the optionally
substituted
diamine ligand is substituted, preferably with a group containing a chiral
centre, and a
neutral aromatic ligand, for example p-cymene, or an optionally substituted
cyclopentadiene ligand, for example pentamethylcyclopentadiene.
Highly preferred transfer hydrogenation catalysts for use in the process of
the
present invention are of general Formula (A):
E
i ~
A\ /B
Y/ %3
Formula (A)
wherein:
R3 represents a neutral optionally substituted hydrocarbyl, a neutral
optionally
substituted perhalogenated hydrocarbyl, or an optionally substituted
cyclopentadienyl
ligand;
A represents -NR4-, -NRS-, -NHR4, -NR4R5 or -NR4R5 where R4 is H, C(O)Rs,
S02R6, C(O)NR6R'0, C(S)NR6R10, C(=NR'0)SR" or C(=NR'0)OR", R5 and R6 each
independently represents an optionally substituted hydrocarbyl, perhalogenated
hydrocarbyl or an optionally substituted heterocyclyl group, and R'0 and R"
are each
independently hydrogen or a group as defined for R6;
B represents -0-, -OH, OR', -S-, -SH, SR', -NR'-, -NR8-, -NHR8, -NR7R 8, -
NR'R9,
-PR'- or -PR'R9 where R$ is H, C(O)R9, SOZR9, C(O)NR9R12, C(S)NR9R12,
C(=NR12)SR'3
or C(=NR'2)OR13, R7 and R9 each independently represents an optionally
substituted
hydrocarbyl, perhalogenated hydrocarbyl or an optionally substituted
heterocyclyl group,
and R12 and R13 are each independently hydrogen or a group as defined for R9;
E represents a linking group;
M represents a metal capable of catalysing transfer hydrogenation; and
Y represents an anionic group, a basic ligand or a vacant site; and
provided that when Y is not a vacant site that at least one of A or B carries
a
hydrogen atom.
Highly preferred are transfer hydrogenation catalysts of Formula (A) wherein
at
least one of A or B comprises a substituted nitrogen and the substituent has
at least one
chiral centre.
The catalytic species is believed to be substantially as represented in the
above
formula. It may be introduced on a solid support.
Optionally substituted hydrocarbyl groups represented by R5-' or R9'" include
alkyl,
alkenyl, alkynyl and aryl groups, and any combination thereof, such as aralkyl
and alkaryl,
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
12
for example benzyl groups.
Alkyl groups which may be represented by R5-' or R9'" include linear and
branched
alkyl groups comprising I to 20 carbon atoms, particularly from 1 to 7 carbon
atoms and
preferably from I to 5 carbon atoms. In certain embodiments, the alkyl group
may be
cyclic, commonly comprising from 3 to 10 carbon atoms in the largest ring and
optionally
featuring one or more bridging rings. Examples of alkyl groups which may be
represented by R5"7 or R9'" include methyl, ethyl, propyl, 2-propyl, butyl, 2-
butyl, t-butyl
and cyclohexyl groups.
Alkenyl groups which may be represented by one or more of R5-' or R9-" include
C2_2o, and preferably C2_6 alkenyl groups. One or more carbon - carbon double
bonds may
be present. The alkenyl group may carry one or more substituents, particularly
phenyl
substituents.
Alkynyl groups which may be represented by one or more of R5-' or Rs-" include
C210, and preferably C2_,o alkynyl groups. One or more carbon - carbon triple
bonds may
be present. The alkynyl group may carry one or more substituents, particularly
phenyl
substituents. Examples of alkynyl groups include ethynyl, propyl and
phenylethynyl
groups.
Aryl groups which may be represented by one or more of RS-' or R9-" may
contain
1 ring or 2 or more fused or bridged rings which may include cycloalkyl, aryl
or
heterocyclic rings. Examples of aryl groups which may be represented by R5-'
or R9-"
include phenyl, tolyl, fluorophenyl, chlorophenyl, bromophenyl,
trifluoromethylphenyl,
anisyl, naphthyl and ferrocenyl groups.
Perhalogenated hydrocarbyl groups which may be represented by one or more of
R5-' or R9-" independently include perhalogenated alkyl and aryl groups, and
any
combination thereof, such as aralkyl and alkaryl groups. Examples of
perhalogenated
alkyl groups which may be represented by R5'' or R9-1' include -CF3 and -C2F5.
Heterocyclic groups which may be represented by one or more of R5-' or RQ-"
independently include aromatic, saturated and partially unsaturated ring
systems and may
comprise 1 ring or 2 or more fused rings which may include cycloalkyl, aryl or
heterocyclic
rings. The heterocyclic group will contain at least one heterocyclic ring, the
largest of
which will commonly comprise from 3 to 7 ring atoms in which at least one atom
is carbon
and at least one atom is any of N, 0, S or P. Examples of heterocyclic groups
which may
be represented by R5-7 or Rs-" include pyridyl, pyrimidyl, pyrrolyl,
thiophenyl, furanyl,
indolyl, quinolyl, isoquinolyl, imidazolyl and triazolyl groups.
When any of R5-' or R9-" is a substituted hydrocarbyl or heterocyclic group,
the
substituent(s) should be such so as not to adversely affect the rate or
stereoselectivity of
the reaction. Optional substituents include halogen, cyano, nitro, hydroxy,
amino, imino,
thiol, acyl, hydrocarbyl, perhalogenated hydrocarbyl, heterocyclyl,
hydrocarbyloxy, mono
or di-hydrocarbylamino, hydrocarbylthio, esters, carboxy, carbonates, amides,
sulphonyl
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
13
and sulphonamido groups wherein the hydrocarbyl groups are as defined for R5-'
or R9-"
above. One or more substituents may be present. R5-' or R9-" may each contain
one or
more chiral centres.
The neutral optionally substituted hydrocarbyl or perhalogenated hydrocarbyl
ligand which may be represented by R3 includes optionally substituted aryl and
alkenyl
ligands.
Optionally substituted aryl ligands which may be represented by R3 may contain
1
ring or 2 or more fused rings which include cycloalkyl, aryl or heterocyclic
rings.
Preferably, the ligand comprises a 6 membered aromatic ring. The ring or rings
of the
aryl ligand are often substituted with hydrocarbyl groups. The substitution
pattern and the
number of substituents will vary and may be influenced by the number of rings
present,
but often from 1 to 6 hydrocarbyl substituent groups are present, preferably
2, 3 or 6
hydrocarbyl groups and more preferably 6 hydrocarbyl groups. Preferred
hydrocarbyl
substituents include methyl, ethyl, iso-propyl, menthyl, neomenthyl and
phenyl.
Particularly when the aryl ligand is a single ring, the ligand is preferably
benzene or a
substituted benzene. When the ligand is a perhalogenated hydrocarbyl,
preferably it is a
polyhalogenated benzene such as hexachlorobenzene or hexafluorobenzne. When
the
hydrocarbyl substitutents contain enantiomeric and/or diastereomeric centres,
it is
preferred that the enantiomerically and/or diastereomerically purified forms
of these are
used. Benzene, p-cymyl, mesitylene and hexamethylbenzene are especially
preferred
ligands.
Optionally substituted alkenyl ligands which may be represented by R3 include
C2_30, and preferably C6_12, alkenes or cycloalkenes with preferably two or
more carbon-
carbon double bonds, preferably only two carbon-carbon double bonds. The
carbon-
carbon double bonds may optionally be conjugated to other unsaturated systems
which
may be present, but are preferably conjugated to each other. The alkenes or
cycloalkenes may be substituted preferably with hydrocarbyl substituents. When
the
alkene has only one double bond, the optionally substituted alkenyl ligand may
comprise
two separate alkenes. Preferred hydrocarbyl substituents include methyl,
ethyl, iso-propyl
and phenyl. Examples of optionally substituted alkenyl ligands include cyclo-
octa-1,5-
diene and 2,5-norbornadiene. Cyclo-octa-1,5-diene is especially preferred.
Optionally substituted cyclopentadienyl groups which may be represented by R3
include cyclopentadienyl groups capable of eta-5 bonding. The cyclopentadienyl
group is
often substituted with from 1 to 5 hydrocarbyl groups, preferably with 3 to 5
hydrocarbyl
groups and more preferably with 5 hydrocarbyl groups. Preferred hydrocarbyl
substituents include methyl, ethyl and phenyl. When the hydrocarbyl
substitutents
contain enantiomeric and/or diastereomeric centres, it is preferred that the
enantiomerically and/or diastereomerically purified forms of these are used.
Examples of
optionally substituted cyclopentadienyl groups include cyclopentadienyl,
pentamethyl-
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
14
cyclopentadienyl, pentaphenylcyclopentadienyl, tetraphenylcyclopentadienyl,
ethyltetramethylpentadienyl, menthyltetraphenylcyclopentadienyl, neomenthyl-
tetraphenylcyclopentadienyl, menthylcyclopentadienyl,
neomenthylcyclopentadienyl,
tetrahydroindenyl, menthyltetrahydroindenyl and neomenthyltetrahydroindenyl
groups.
Pentamethylcyclopentadienyl is especially' preferred.
When either A or B is an amide group represented by -NR4-, -NHR4, NR4R5, -NRB-
-NHRa or NR'R8 wherein R5 and R' are as hereinbefore defined, and where R4 or
R$ is
an acyl group represented by -C(O)R6 or -C(O)R9, R6 and R9 independently are
often
linear or branched C,_,alkyl, C,_$-cycloalkyl or aryl, for example phenyl.
Examples of acyl
groups which may be represented by R6 or R10 include benzoyl, acetyl and
halogenoacetyl, especially trifluoroacetyl groups.
When either A or B is present as a sulphonamide group represented by -NR4-,
-NHR4, NR4R4, -NR8-, -NHR$ or NR'R8 wherein R5 and R' are as hereinbefore
defined,
and where R4 or R$ is a sulphonyl group represented by -S(O)2R6 or -S(O)2R9,
R6 and R9
independently are often linear or branched C,_12alkyl, C,_12cycloalkyl or
aryl, for example
phenyl. Preferred sulphonyl groups include methanesulphonyl,
trifluoromethanesulphonyl, more preferably p-toluenesulphonyl groups and
naphthylsulphonyl groups and especially camphorsulphonyl.
When either of A or B is present as a group represented by -NR4-, -NHR4,
NR4R5,
-NR$-, -NHRB or NR'R8 wherein R5 and R' are as hereinbefore defined, and where
R4 or
R 8 is a group represented by C(O)NR6R10, C(S)NR6R'0, C(=NR'0)SR11,
C(=NR'0)OR1'
,
C(O)NR9R'2, C(S)NR9R12, C(=NR'2)SR13 or C(=NR'Z)OR13, R6 and R9 independently
are
often linear or branched C,_salkyl, such as methyl, ethyl, isopropyl,
C,_$cycloalkyl or aryl,
for example phenyl, groups and R10"13 are often each independently hydrogen or
linear or
branched C,_8alkyl, such as methyl, ethyl, isopropyl, C,_acycloalkyl or aryl,
for example
phenyl, groups.
When B is present as a group represented by -OR', -SR7, -PR'- or -PR 7R9,
R'
and R9 independently are often linear or branched C,_8alkyl, such as methyl,
ethyl,
isopropyl, C,_8cycloalkyl or aryl, for example phenyl.
It will be recognised that the precise nature of A and B will be determined by
whether A and/or B are formally bonded to the metal or are coordinated to the
metal via a
lone pair of electrons.
The groups A and B are connected by a linking group E. The linking group E
achieves a suitable conformation of A and B so as to allow both A and B to
bond or
coordinate to the metal, M. A and B are commonly linked through 2, 3 or 4
atoms. The
atoms in E linking A and B may carry one or more substituents. The atoms in E,
especially the atoms alpha to A or B, may be linked to A and B, in such a way
as to form
a heterocyclic ring, preferably a saturated ring, and particularly a 5, 6 or 7-
membered ring.
Such a ring may be fused to one or more other rings. Often the atoms linking A
and B will
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
be carbon atoms. Preferably, one or more of the carbon atoms linking A and B
will carry
substituents in' addition to A or B. Substituent groups include those which
may substitute
R5-' or R9-" as defined above. Advantageously, any such substituent groups are
selected
to be groups which do not coordinate with the metal, M. Preferred substituents
include
5 halogen, cyano, nitro, sulphonyl, hydrocarbyl, perhalogenated hydrocarbyl
and
heterocyclyl groups as defined above. Most preferred substituents are C1_6
alkyl groups,
and phenyl groups. Most preferably, A and B are linked by two carbon atoms,
and
especially an optionally substituted ethyl moiety. When A and B are linked by
two carbon
atoms, the two carbon atoms linking A and B may comprise part of an aromatic
or
10 aliphatic cyclic group, particularly a 5, 6 or 7-membered ring. Such a ring
may be fused to
one or more other such rings. Particularly preferred are embodiments in which
E
represents a 2 carbon atom separation and one or both of the carbon atoms
carries an
optionally substituted aryl group as defined above or E represents_ a 2 carbon
atom
separation which comprises a cyclopentane or cyclohexane ring, optionally
fused to a
15 phenyl ring.
E preferably comprises part of a compound having at least one stereospecific
centre. Where any or all of the 2, 3 or 4 atoms linking A and B are
substituted so as to
define at least one stereospecific centre on one or more of these atoms, it is
preferred
that at least 'one of the stereospecific centres be located at the atom
adjacent to either
group A or B. When at least one such stereospecific centre is present, it is
advantageously present in an enantiomerically purified state.
When B represents -0- or -OH, and the adjacent atom in E is carbon, it is
preferred that B does not form part of a carboxylic group.
Compounds which may be represented by A-E-B, or from which A-E-B may be
derived by deprotonation, are often aminoalcohols, including 4-aminoalkan-l-
ols,
1-aminoalkan-4-ols, 3-aminoalkan-l-ols, 1-aminoalkan-3-ols, and especially
2-aminoalkan-l-ols, 1-aminoalkan-2-ols, 3-aminoalkan-2-ols and 2-aminoalkan-3-
ols, and
particularly 2-aminoethanols or 3-aminopropanols, or are diamines, including
1,4-diaminoalkanes, 1,3-diaminoalkanes, especially 1,2- or 2,3- diaminoalkanes
and
particularly ethylenediamines. Further aminoalcohols that may be represented
by A-E-B
are 2-aminocyclopentanols and 2-aminocyclohexanols, preferably fused to a
phenyl ring.
Further diamines that may be represented by A-E-B are 1,2-diaminocyclopentanes
and
1,2-diaminocyclohexanes, preferably fused to a phenyl ring. The amino groups
may
advantageously be N-tosylated. When a diamine is represented by A-E-B,
preferably at
3 5 least one amino group is N-tosylated. The aminoalcohols or diamines are
advantageously substituted, especially on the linking group, E, by at least
one alkyl group,
such as a C,-4-alkyl, and particularly a methyl, group or at least one aryl
group, particularly
a phenyl group.
Specific examples of compounds which can be represented by A-E-B and the
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
16
protonated equivalents from which they may be derived are:
H3 H Ph PhH Ph Ph- Ph Ph- Ph
H2N OH H2N NH-tosyl HZN/~-/\NH2 HzN/v~NH-SOZ naphthyl NH-tos I
y
Ph CH3 Ph Ph PhCHZ C6H4OMe ~
u,~f ~ CH40Me NHZ
H2N OH HO NH2 HO H2N NH2
oN 1 - - - -
H OH
H N tosyl-HN
HzN HO z H2N 3
OH NH2 HO H2 NHZ
Preferably, the enantiomerically and/or diastereomerically purified forms of
these
are used. Examples include (1 S,2R)-(+)-norephedrine, (1 R,2S)-(+)-cis-1-amino-
2-
indanol, (1 S,2R)-2-amino-1,2-diphenylethanol, (1 S,2R)-(-)-cis-1 -amino-2-
indanol,
(1 R,2S)-(-)-norephedrine, (S)-(+)-2-amino-1 -phenylethanol, (1 R,2S)-2-amino-
1,2-
diphenylethanol, N-tosyl-(1 R,2R)-1,2-diphenylethylenediamine, N-tosyl-(1
S,2S)-1,2-
diphenylethylenediamine, (1 R,2S)-cis-1,2-indandiamine, (1 S,2R)-cis-1,2-
indandiamine,
(R)-(-)-2-pyrrolidinemethanol and (S)-(+)-2-pyrrolidinemethanol.
Metals which may be represented by M include metals which are capable of
catalysing transfer hydrogenation. Preferred metals include transition metals,
more
preferably the metals in Group VIII of the Periodic Table, especially
ruthenium, rhodium or
iridium. When the metal is ruthenium it is preferably present in valence state
II. When
is the metal is rhodium or iridium it is preferably present in valence state I
when R3 is a
neutral optionally substituted hydrocarbyl or a neutral optionally substituted
perhalogenated hydrocarbyl ligand, and preferably present in valence state III
when R3 is
an optionally substituted cyclopentadienyl ligand.
It is preferred that M, the metal, is rhodium present in valence state I I I
and R3 is an
optionally substituted cyclopentadienyl ligand.
Anionic groups which may be represented by Y include hydride, hydroxy,
hydrocarbyloxy, hydrocarbylamino and halogen groups. Preferably when a halogen
is
represented by Y, the halogen is chloride. When a hydrocarbyloxy or
hydrocarbylamino
group is represented by Y, the group may be derived from the deprotonation of
the
hydrogen donor utilised in the reaction.
Basic ligands which may be represented by Y include water, C,-4 alcohols, C,_8
primary or secondary amines, or the hydrogen donor which is present in the
reaction
system. A preferred basic ligand represented by Y is water.
Most preferably, A-E-B, R3 and Y are chosen so that the catalyst is chiral.
When
such is the case, an enantiomerically and/or diastereomerically purified form
is preferably
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
17
employed. Such catalysts are most advantageously employed in asymmetric
transfer
hydrogenation processes. In many embodiments, the chirality of the catalyst is
derived
from the nature of A-E-B.
Especially preferred are catalysts of Formula B(i-iv):
CH3 3
C O CH3
PhXN>H3
N ci Ph~,,= ~ N ci
I So
s02 CH3 2 CH3
CH3 CH3
O O
B(i) B(ii)
CH3 CH3
H3C CH3 H3C CH3
Ph Ph ,
R HsC CH3 \Rh HsC CH3
Ph' N CI PhN ci
I I
CH3 S02 CH3 SO2
H3C H3C
B(iii) B(iv)
The transfer hydrogenation catalysts may be prepared in advance or in-situ by
combining a ligand, preferably a chiral bidentate nitrogen ligand, with a
metal complex, for
example a Ru, Rh or Ir metal complex containing a neutral optionally
substituted
hydrocarbyl complexing group, a neutral optionally substituted perhalogenated
hydrocarbyl complexing group, or an optionally substituted cyclopentadienyl
complexing
group. Preferably a solvent is present in this operation. The solvent used may
be anyone
which does not adversely effect the formation of the catalyst. These solvents
include
acetonitrile, ethylacetate, toluene, methanol, tetrahydrofuran, ethylmethyl
ketone.
Preferably the solvent is methanol.
Advantageously, the process of the present invention may find use in recycling
unwanted isomers obtained from chiral processes, such as chiral separations,
chemical
and enzymic chiral resolutions and the likes. Typically, in chiral separations
or
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
18
resolutions, racemic mixtures are subjected to physical, chemical or
biochemical
treatments which result in the separation of a desired enantiomer or
enatiomeric product
while often leaving behind an unreacted or unwanted enatiomers or enatiomeric
bi-
products. The process of the present invention provides a method for
converting the
unreacted enatiomers to usable feedstocks containing wanted enatiomers.
The invention is illustrated by the following Examples.
Example 1
Procedure
Rxn 1: Std Conditions + 0.5eq 2.4-Dimethyl-3-pentanol
To a 10 ml round bottom flask was added (S)-1-phenylethanol (246.8 mg 99%,
is 244.3 mg, 2.0 mmol), pentamethylcyclopentadienyliridium(III) chloride dimer
(16.6 mg
96%, 15.9 mg, 0.02 mmol), tridecane (372.4 mg 99%, 368.7mg, 2.0 mmol),
potassium
iodide (335.4 mg 99%, 332.0 mg, 2.0 mmol), 2,4-dimethyl-3-pentanol (117.4 mg
99%,
116.2 mg, 1.0 mmol), potassium carbonate (279.2 mg 99%, 276.4 mg, 2.0 mmol)
and
toluene (4 ml) resulting in a pale orange solution. A water condenser was
attached and
the reaction vessel was placed in an oil bath at 80 C and a timer started,
within one
minute of being in the oil bath the reaction solution became a dark orange and
darkened
gradually to a dark brown after 2hrs and remained this colour throughout.
Samples were
taken (-100 1) at regular intervals and quenched into dichloromethane (2 ml)
and 0.5M
sodium hydroxide solution (2 ml), the organic layer was separated, dried using
sodium
sulphate, filtered and analysed by achiral and chiral g.c..
Rxn 2 : Std Conditions + 1.Oeq 2,4-Dimethyl-3-pentanol
To a 10 ml round bottom flask was added (S)-1-phenylethanol (246.8 mg 99%,
244.3 mg, 2.0 mmol), pentamethylcyclopentadienyliridium(III) chloride dimer
(16.6 mg
96%, 15.9 mg, 0.02 mmol), tridecane (372.4 mg 99%, 368.7mg, 2.0 mmol),
potassium
iodide (335.4 mg 99%, 332.0 mg, 2.0 mmol), 2,4-dimethyl-3-pentanol (234.7 mg
99%,
232.4 mg, 2.0 mmol), potassium carbonate (279.2 mg 99%, 276.4 mg, 2.0 mmol)
and
toluene (4 ml) resulting in a pale orange solution. A water condenser was
attached and
the reaction vessel was placed in an oil bath at 80 C and a timer started,
within one
minute of being in the oil bath the reaction solution became a dark orange and
darkened
gradually to a dark brown after 2hrs and remained this colour throughout.
Samples were
taken (-100 i) at regular intervals and quenched into dichloromethane (2 ml)
and 0.5M
sodium hydroxide solution (2 ml), the organic layer was separated, dried using
sodium
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
19
sulphate, filtered and analysed by achiral and chiral g.c..
Analysis
Achiral g.c.
Chrompac 7680 CP SIL 5CB column. Length = 25.0 m, Diameter 320 m, Film
thickness
= 5.0 m.
Pressure = 8.0 psi.
Flow= 1.1 mI/min.
Temperature = 250 C for 22.5 mins then ramp at 20 C/min to 300 C.
1-Phenylethanol = 13.1 mins.
Acetophenone = 13.7 mins.
Tridecane = 27.5 mins
2,4-Dimethyl-3-pentanol = 5.7 mins
Chiral g.c.
CP-Chirasil-Dex-CB column. Length = 25.0 m, Diameter = 250 m, Film thickness
= 0.25
m.
Pressure = 10.0 psi.
Flow = 0.7 mI/min.
Temperature = 110 C for 40 mins then ramp at 20 C/min to 190 C and hold for 5
mins.
(R)-1-Phenylethanol= 23.7 mins.
(S)-1-Phenylethanol= 26.0 mins.
Acetophenone = 10.0 mins.
Tridecane = 21.0 mins.
2,4-Dimethyl-3-pentanol = 4.6 mins.
1-Phenylethanol racemisation using [IrCp*CI2]2/ KI + 1eq K2C03 in toluene at
80degC.
Rxnl + 0.5eg 2,4-Dimethylpentan-3-ol
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
Area
Area Area Tridecane % % (R) (S)
Time Alcohol Ketone Std Alcohol Ketone %std area area %ee
0 100 0 0 100.0 0.0 0.0 0 100 100.0
10 1163.67 23.761 2237.92 98.0 2.0 65.3 1.298 391.097 99.3
1019.62 24.127 2014.6 97.7 2.3 65.9 1.124 343.047 99.3
60 944.357 31.175 1832.72 96.8 3.2 65.3 2.436 317.466 98.5
120 1028.81 93.275 2110.7 91.7 8.3 65.3 24.911 296.958 84.5
185 957.324 239.739 2282.01 80.0 20.0 65.6 47.163 233.936 66.4
240 733.704 298.108 1955.44 71.1 28.9 65.5 49.046 171.742 55.6
305 518.397 353.609 1641.57 59.4 40.6 65.3 49.494 117.522 40.7
365 586.707 663.707 2378.319 46.9 53.1 65.5 64.354 111.557 26.8
425 465.977 771.361 2361.16 37.7 62.3 65.6 70.318 98.47 16.7
1440 371.226 928.745 2467.85 28.6 71.4 65.5 49.098 60.26 10.2
Rxn2 + 1.Oeq 2,4-Dimethylpentan-3-ol
Area %
Area Area Tridecane Alcoh % (R) (S)
Time Alcohol Ketone Std ol Ketone %std area area %ee
0 100 0 0 100.0 0.0 0.0 0 100 100.0
10 1149.75 24.886 2200.81 97.9 2.1 65.2 1.641 421.902 99.2
30 926.296 23.157 1812.66 97.6 2.4 65.6 1.351 312.695 99.1
60 892.692 28.24 1727.72 96.9 3.1 65.2 1.986 347.031 98.9
120 1030.14 93.317 2110.7 91.7 8.3 65.3 24.279 296.685 84.9
185 952.602 182.593 2156.04 83.9 16.1 65.5 60.163 279.616 64.6
240 739.385 243.95 1888.64 75.2 24.8 65.8 56.662 180.176 52.2
305 397.693 223.874 1197.56 64.0 36.0 65.8 35.522 85.438 41.3
365 371.698 321.486 1326.93 53.6 46.4 65.7 44.073 79.6 28.7
425 404.798 551.429 1834.782 42.3 57.7 65.7 60.164 82.946 15.9
1440 317.004 898.786 2365.34 26.1 73.9 66.1 47.624 53.189 5.5
Example 2
Procedure
5
Rxn 2 : + potassium carbonate
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
21
To a 10 ml round bottom flask was added (S)-1-phenylethanol (246.8 mg 99%,
244.3 mg, 2.0 mmol), pentamethylcyclopentadienyliridium(III) chloride dimer
(16.6 mg
96%, 15.9 mg, 0.02 mmol), tridecane (372.4 mg 99%, 368.7mg, 2.0 mmol),
potassium
iodide (335.4 mg 99%, 332.0 mg, 2.0 mmol), potassium carbonate (279.2 mg 99%,
276.4
mg, 2.0 mmol) and toluene (4 ml) resulting in a pale orange solution. A water
condenser
was attached and the reaction vessel was placed in an oil bath at 80 C and a
timer
started, within one minute of being in the oil bath the reaction solution
became a dark
orange which became increasingly darker and was a dark brown after 2 hrs and
remained
this colour throughout. Samples were taken (-100 1) at regular intervals and
quenched
into dichloromethane (2 ml) and 2.5M sodium hydroxide solution (2 ml), the
organic layer
was separated, dried using sodium sulphate, filtered and analysed by achiral
and chiral
g.c..
Analysis
Achiral g.c. '
Chrompac 7680 CP SIL 5CB column. Length = 25.0 m, Diameter 320 m, Film
thickness
= 5.0 m.
Pressure = 8.0 psi.
Flow = 1.1 mI/min.
Temperature = 250 C for 22.5 mins then ramp at 20 C/min to 300 C:
1-Phenylethanol = 13.1 mins.
Acetophenone = 13.7 mins.
Tridecane = 27.5 mins
Chiral g.c.
CP-Chirasil-Dex-CB column. Length = 25.0 m, Diameter = 250 m, Film thickness
= 0.25
m.
Pressure = 10.0 psi.
Flow = 0.7 ml/min.
Temperature = 110 C for 40 mins then ramp at 20 C/min to 190 C and hold for 5
mins.
(R)-1-Phenylethanol= 23.7 mins.
(S)-1-Phenylethanol= 26.0 mins.
Acetophenone = 10.0 mins.
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
22
Tridecane = 21.0 mins.
Rxn2 Std Conditions + leg K2C03
Area
Area Area Tridecane % % (R) (S)
Time Alcohol Ketone Std Alcohol Ketone %std area area %ee
0 100 0 0 100.0 0.0 0.0 0.000 324.281 100.0
12 1385.06 48.979 2681.29 96.6 3.4 65.2 2.096 396.069 98.9
30 1076.38 42.302 2104.06 96.2 3.8 65.3 1.662 334.271 99.0
60 1505.995 76.598 2992.3 95.2 4.8 65.4 3.767 393.765 98.1
122 1052.644 102.146 2206.188 91.2 8.8 65.6 113.431 199.7487.4
180 1002.368 142.23 2156 87.6 12.4 65.3 68.342 366.146 68.5
240 1032.22 273.729 2434.783 79.0 21.0 65.1 77.619 243.057 51.6
300 918.881 409.19 2521.09 69.2 30.8 65.5 82.965189.190 39.0
375 479.918 471.136 1804.79 50.5 49.5 65.5 55.711 95.038 26.1
1380 0 1178.13 2215.49 0.0 100.0 65.3 * 0
Example 3
Preparation of catalyst and reduction of acetophenone.
Reactant Wt used Mo1.Wt Mol ratio
[Rh(Cp*)CI2]z** 0.0254g 618.08 1.0 41.2 mol
(1 S,2R)-(+)-
Norephedrine 0.0209g 151.21 3.36 138.2 mol
2-propanol (anhydrous) 100mi 60.10 31677 1.305mo1
KOH 0.1 M in
2-propanol 3.3ml 56.11 4.01 0.33mmol
Acetophenone 2.06g 120.15 209 17mmol
Notes: ** purchased from STREM Chemicals
Prior to the reaction, the solvent was degassed:
100ml of anhydrous 2-propanol was added by syringe to a sealed clean dry round
bottomed flask and degassed in vacuo at under 20 C for 30 min.
(a) Catalyst Preparation
The (+)-norephedrine and rhodium compound were weighed out into a clean dry
Schlenk flask. The flask was stoppered with a 'Suba-seal' (RTM). Its contents
were
evacuated, then purged at room temperature by 15 changes of nitrogen. Then 2-
propanol (20m1) was added by cannula. The flask tap was closed and the flask
swirled
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
23
until the starting solids dissolved. The result was an orange-coloured
supernatant and a
dark solid. The flask tap was re-opened, a current of nitrogen fed in, and the
flask
contents heated at 60 C for 2h 5min. The catalyst was checked at 30 min
intervals. At
each interval it was a dark brown solution, with a black solid at the bottom.
(b) Hydrogenation
The acetophenone was dissolved in 2-propanol (80m1) then degassed for 40min.
This solution was added to the catalyst-containing flask by cannula, followed
via syringe
by the degassed 0.1 M solution of KOH in 2-propanol. The mixture was left at
room
temperature, samples being taken at intervals and examined by gas
chromatography. At
the small scale of operation the reaction mixture was not sparged with the
nitrogen, but
sparging would be used in larger scale production. After lh, (R)-1-
phenylethanol was
obtained conversion 92%, 84% ee.
Example 4:
Reactant Wt used Mol wt mol ratio
[Rh(Cp*)CI2]Z 6.3mg 618.08 1.0 10.2 mol
(1 S,2R)-(-)-cis-
1-amino-2-indanol 3.1 mg 149.19 2.0 20.8 mol
acetophenone 1.29g 120.15 1039 10.6mmol
2-propanol 63857
(a) Catalyst Preparation
The rhodium compound was suspended in 50ml of 2-propanol and degassed by 3
cycles
of vacuum and nitrogen flush. The mixture was heated to gentle reflux until
the solid
dissolved, then cooled to ambient temperature. (1S,2R)-(-)-cis-l-amino-2-
indanoi was
added to the solution with stirring. The mixture was degassed by cycles of
vacuum and
nitrogen flush and warmed at 30 C for 30min. The resulting orange-red solution
of the
catalyst was passed to the next stage but could be stored under argon or
nitrogen.
(b) Hydrogenation
The acetophenone was added to the catalyst solution. The mixture was stirred
at
ambient temperature for lh. Sodium 2-propoxide (0.25ml of freshly prepared 0.1
M
solution in 2-propanol) was added. The mixture was stirred for 2h and sampled;
57% of
the acetophenone had reacted to give (R)-1-phenylethanol of 79% ee.
CA 02583821 2007-04-11
WO 2006/046062 PCT/GB2005/004179
24
Example 5:
Reactant Wt used Mol wt mol ratio
[Ir(Cp*)CIz]Z 32.8mg 796.67 1.0 41.2 mol
(1 S,2R)-(+)-
norephedrine 20mg 151.21 3.2 132 mol
acetophenone 2m! 120.15 413 17mmo!
2-propanol 100m1
(a) Catalyst Preparation
The iridium compound and (+)-norephedrine were suspended in degassed 2-
propanol (20ml) under nitrogen, and the reaction purged with nitrogen for 30
minutes.
The mixture was heated to 60 C for 90min, then cooled to ambient temperature.
The
resulting solution of the catalyst: was passed to the next stage but could be
stored under
argon or nitrogen.
1s
(b) Hydrogenation
Acetophenone (2m1, 17mmol) was dissolved in 2-propanol (80m1) and purged- with
nitrogen. Then the catalyst solution was added followed by potassium hydroxide
solution
(3.3ml of 0.1 M solution in 2-propanol). The mixture was stirred at ambient
temperature
under nitrogen for 1 0h. This gave 1-phenylethanol. Yield 68%, ee 49%.