Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 397
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 397
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
ANTIINFECTIVE LIPOPEPTIDES
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] The present application claims the benefit of United States Provisional
Application
Numbers. 60/710,705, filed August 23, 2005 and 60/627, 056, filed November 12,
2004, which
are hereby incorporated by reference in their entirety.
GOVERNMENT SUPPORT
[0002] Portions of the work described herein were made with government support
under
Small Business Innovation Research (SBIR) Grant No. 5R44GM068173-03 and Grant
No.1R43A156858-1. The government may have certain rights to such work.
FIELD OF THE INVENTION
[0003] The present invention relates to novel depsipeptides compounds. The
invention also
relates to pharmaceutical coinpositions of these compounds and methods of
using these
compounds as antibacterial agents.
BACKGROUND OF THE INVENTION
[0004] The rapid increase in the incidence of gram-positive infections -
including those
caused by resistant bacteria - has sparked renewed interest in the development
of novel classes
of antibiotics. A class of compounds that has shown potential as useful
antibiotic agents is the
cyclic depsipeptides. A notable member of the cyclic depsipeptides is the
A21978C lipopeptides
described in, for example, United States Patents RE 32,333; RE 32,455; RE
32,311; RE 32,310;
4,482,487; 4,537,717; 5,912,226; 6,911,525; and 6,794,490 and International
Patent Applications
WO01/44272; WO01/44274; and WO01/44271. Additionally, the A54145 class of
compounds
described in United States Patents 4,994,270; 5,039,789; and 5,028,590 have
also been shown to
possess antibiotic activity.
[0005] Daptomycin, also known as LY146032, is comprised of an n-decanoyl side
chain
linked to the N-terminal tryptophan of a three-amino acid chain linked to a
cyclic 10-amino acid
peptide. Daptomycin has potent bactericidal activity in vitro and in vivo
against clinically
relevant gram-positive bacteria that cause serious and life-threatening
diseases. These bacteria
1
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
include resistant pathogens, such as vancomycin-resistant enterococci (VRE),
methicillin-
resistant Staphylococcus aureus (MRSA), glycopeptide intermediate susceptible
Staphylococcus
aureus (GISA), vancomycin-resistant Staphylococcus aureus (VRSA), coagulase-
negative
staphylococci (CNS), and penicillin-resistant Stf eptococcus pneunaoniae
(PRSP), for which there
are few therapeutic alternatives. See, e.g., Tally et al., 1999, Exp. Opin.
Invest. Drugs 8:1223-
1238.
[0006] Despite the promise that existing antibacterial agents have shown, the
need for novel
antibiotics continues. Many pathogens have been repeatedly exposed to commonly
used
antibiotics. This exposure has led to the selection of variant antibacterial
strains resistant to a
broad spectrum of antibiotics. The loss of potency and effectiveness of an
antibiotic caused by
resistant mechanisms renders the antibiotic ineffective and consequently can
lead to some life-
threatening infections that are virtually untreatable. As new antibiotics come
to market,
pathogens may develop resistance or intermediate resistance to these new
drugs, effectively
creating a need for a stream of new antibacterial agents to combat these
emerging strains. In
addition compounds that exhibit bactericidal activity offer advantages over
present bacteriostatic
compounds. Thus, novel antibacterial agents would be expected to be useful to
treat not only
"natural" pathogens, but also intermediate drug resistant and drug resistant
pathogens because
the pathogen has never been exposed to the novel antibacterial agent. New
antibacterial agents
may exhibit differential effectiveness against different types of pathogens.
SUMMARY OF THE INVENTION
[0007] The present invention provides novel compounds that have antibacterial
activity
against a broad spectrum of bacteria, including drug-resistant bacteria, and
processes for making
these compounds.
2
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0008] The present invention provides, in one aspect, compounds of Formula I:
R12 R13
HO2C
NH
HN 0 0 R2 0
00 O N R1
R11 N N
NR11' O H R3 0 R2=
O NRS
R5* N
HN O H
O HN
Rs R8 0 H Rs
HN
N
YI__ 1
0 Rs* O I
HO2C
and salts thereof; wherein:
a) R2 is an amino acid side chain,
OH O
0 or NH2.
b) R2* is H or alternatively R2 together with R2* forms a five or six-member
heterocyclic
ring;
NH2
O _\__~/OH NH2
~If( .~s
c) R3 is OH 0 or a non-proteinogenic amino acid side
chain;
d) R5 is H or methyl;
e) R5* is H or an amino acid side chain derived from an N-methylamino acid.
Alternatively R5 together with R5* forms a five or six-member heterocyclic
ring;
f) R6 is methyl or
R6'
NH2
8*
~ O, OH or
g) R a is an amino acid side chain, methyl, ~
3
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
h) R8* is H or, alternatively, R8 together with R8* forms a five or six-member
heterocyclic ring;
OMe OH
i) R9 is CO2H CO2H CO2H , or an amino acid side chain
substituted with at least one carboxylic acid;
j) R" is an amino acid side chain, methyl,
O
.
~OH or NHz -
k) R1 is H or, alternatively, R11 together with R" forms a five or six-member
heterocyclic ring;
1) R12 is H or CH3
in) R13 is CH(CH3)2, CH(CH2CH3)CH3,
O NH2
H
or and
n) each of R1, R6* and R8** is independently amino, monosubstituted amino,
disubstituted amino, NH-amino protecting group, acylamino, ureido, guanidino,
carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
4
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0009] In another aspect, the invention provides a compound of the Formula F
1:
HO2C R12 R13
HN NH O CONH2
O O
00 O N R1
R11 N N
NH 0 H H
O NH CO2H
N
HN O H
O HN
R8 0
HO2C HN
Y1--- N R6*
H
0
HO2C (F 1)
and salts thereof; wherein:
NH2
~
a) R8 is hydrogen, O, or '~ OH
0
.
b) Rl l is methyl, '~õ~OH , or NH2
c) R12 is H or CH3;
d) R13 is CH(CH3)2, CH(CH2CH3)CH30 0 NH2
H
or and
e) each of RI and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino;
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0010] The present invention provides, in another aspect, compounds of Formula
F2:
H02C R12 R13
HN NH O CONH2
O O
00 O N R1
H3C H H
NH O O
O NH C02H
N
HN O H
O HN
R$ O
HO2C HN N R6*
H O
O
HO2C (F2)
and salts thereof; wherein:
NH2
a) R 8 is hydrogen, methyl, \111,1~O ,'~"~OH or \s' R8-=
b) R12 is H or CH3i
c) R13 is CH(CH3)2, CH(CH2CH3)CH3,
O NH2
H
or ; and
d) each of R', R6*and R$** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino;
6
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00111 In another aspect, the invention provides compounds of Formula F3:
R12 NH
HO2C
HN NH 0 CONH2
O 0
00 0 N RI
R" N N
H
NH O H
\
0
O NH CO2H
N ~
HN O H
O HN
R8 0
HO2C HN N R Ir, N
H O
O
HO2C (F3)
and salts thereof; wherein:
NH2
$ ,~ ~ R8.*
a) R is hydrogen, O T OH or ;
0
.
b) R11 is methyl, '~r"~OH or NH2 ;
c) R1a is H or CH3i and
d) each of R1, R6*and R 8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
7
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0012] The present invention provides, in another aspect, compounds of Formula
F4:
~
~/
R12 NH
HO2C
HN NH O CONH2
O O
00
HO2C HN N R6*
H
O
HOzC (F4)
and salts thereof; wherein:
NH2 ~.~ ~.-~ ~~ R8.,
a) R8 is hydrogen, methyl, O~~ OH or
O
.~
b) Rl l is methyl, or ~ NH2 ;
c) R12 is H or CH3; and
d) each of RI, R6*and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
8
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0013] In another aspect, the invention provides compounds of Formula F5:
9~'NH
H02C
HN NH O CONH2
O O
00
HO2C HN
Ir, N Rs*
H
O
HO2C (F5)
and salts thereof; wherein:
NH2
a) R8 is hydrogen, methyl, \'" _ O,'~" H , or R$**
O
.
b) Rll is methyl, '~'~OH , or NH2 ; and
c) each of R1, R6*and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
9
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0014] The present invention provides, in another aspect, compounds of Formula
F6:
HO2C CO2H
NH
HN O
0 O
0
00 N R~
R~~ N N
H
NH 0 H
0
O NCH3 HO CONH2
N
HN O H
O HN
R9 R8 0 H
HN N
H 0
0
HO2C (F6)
and salts thereof; wherein:
NH2
.
"~
a) R8 is O or OH
OMe OH
b) R9 is C02H C02H or ~"t \C02H
>
c) Rll is, methyl,
0
.
or ~ NH2
d) R12 is H or CH3; and
e) R' is amino, monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0015] In another aspect, the invention provides compounds of Formula F7:
HO2C CO2H
NH
HN O O
00 O N R'
H3C H H
NH 0 O
O NCH3 HO CONH2
N
HN O H
O HN
R9 R8 O
H
HN
N N
H O
O
HO2C (F7)
and salts thereof; wherein:
NH2
a) R8 is methyl, \'" O,~'~OH or R8.* =
OMe OH
b) R9 is ''" CO2H '~~ CO2H ~~ \CO~H =
, or
c) R12 is H or CH3; and
d) each of R' and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
I1
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
(00161 The present invention provides, in another aspect, compounds of Formula
F8:
HO2C CO2H
NH
HN O
O
00 O N AR1
R~~ N N
NH O H
O H
O NCH3 Rs** CONH2
N
HN O H
HO O HN
HO R$ O
H
O HN
H N
O
HO2C (F8)
and salts thereof; wherein:
a) R3** is hydroxyl or hydrogen
NH2
b) R8 is methyl, 0OH or R8-* =
c) Rll is an amino acid side chain, methyl,
0
OH , or NH2 ;
d) R12 is H or CH3; and
e) each of R' and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
12
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00171 In another aspect, the invention provides compounds of Formula F9:
HO2C R12 CO2H
NH
HN O 0
H2NOC O 0 0
N R~
N
H
NH 0 H 0
111 N
O NCH3 CONH2
N
HN (CH2)4Ra*. O H
O HN
HO O
H
HN N
0 H
0
HOzC (F9)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of RI, and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-aanino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
13
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00181 The present invention provides, in another aspect, compounds of Formula
F10:
R13*
HO2C H CO2H
NH
HN O O O
HO 00 O kc1 RI
N N
O H
NH 0 H
O NH CONH2
N
HN O H
O HN
HO O
H
HN N
O
ir, H O R6.
0
H02C (F10)
and salts thereof; wherein:
a) R13* is H or CH3i and
'b) each of Rl, and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iininoamino, or phosphonamino.
14
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00191 In another aspect, the invention provides compounds of Formula F11:
CH3 R13*
HO2C
HN NH O CONH2
O O
00 O N R1
N N
HO NH O H
O H O NH C02H ~
/
N
HN O H
O HN
CH3 O
HO2C HN N R6= Y-1- H
O
HO2C (F 1 l )
and salts thereof; wherein:
a) R13*isHorCH3;and
b) each of R1, and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0020] The present invention provides, in another aspect, compounds of Formula
F 12:
HO2C Me R13
HN NH O CONH2
O O
00 O N R1
N N
H2NOC NH O H
O H
O NH C02H
N
HN O H
O HN
Z-t CH3 O
HO2C HN
NJtf N Rs*
H O
O
HO2C (F12)
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
and salts thereof; wherein:
0 NH2
a) R13 is CH(CH2CH3)CH3 or and
b) each of Ri and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0021] In another aspect, the invention provides compounds of Formula F13:
O NH2
H02C Me (
NH
HN O CONH2
O O
00 O N
N N
H2NOC NH O H
O H
O NH C02H
N
HN (CH2)4R** O H
O HN
O
-t 1 HO2C HN N Rs*
H O
O
HO2C (F13)
and salts thereof; wherein each of Rl, R6* and R8** is independently amino,
monosubstituted
amino, disubstituted amino, NH-amino protecting group, acylamino, ureido,
guanidino,
carbamoyl, sulfonamino, thioacylamino, thioureido, iminoamino, or
phosphonamino.
16
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00221 The present invention provides, in another aspect, compounds of Formula
F14:
R12
HO2C
HN NH O CONH2
O O
O O O N R
1
H2NOC NH O O H
)~11 N 11 1 J~~
O NH CO2H
N
HN O H
MeO 0 HN
0
HO2C HN N RV
H O
0
HO2C (F14)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of R' and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
17
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00231 In another aspect, the invention provides compounds of Fonnula F15:
HO2C R12 CO2H
NH
;HN O O
O O O N R'
N N
HO
NH O H p H
O NCH3 HO CONH2 0 I~
N
HN O H
MeO O (CH2)4R8** HN
O
HO HN N
H
O
H02C (F15)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of Rl and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
18
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0024] The present invention provides, in another aspect, compounds of Formula
F16:
HO2C R12 CO2H
NH
HN O O O
O O 0 N R~
N N
H2NOC NH O H O H
O NCH3 HO CONH2
N
HN O H
O (CH2)4R$" HN
O
HO OHN N
H
H02C (F16)
and salts thereof; wherein:
a) R12 is H or CH3, and
b) each of Rl and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
19
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00251 In another aspect, the invention provides compounds of Formula F17:
HO2C R12 CO2H
T~N)IT 0 O
00 O N RI
N N
H2NOC NH O H
O H
O NCH3 HO CONH2
N
HN O H
MeO 0 HN
O
HO O HN N
H
O
HO2C (1717)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) Rl is amino, monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonainino.
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[00261 The present invention provides, in another aspect, compounds of Formula
F18:
HO2C Me CO2H
NH
HN O O O
00 0 N Ri
N N
H
H2NOC NH O H
0
O NH HO CONH2
HN O H
MeO O (CH2)4R8- HN
O
HO HN N
O
H
HO2C (F18)
and salts thereof; wherein each of Rl and Rg** is independently amino,
monosubstituted amino,
disubstituted amino, NH-amino protecting group, acylamino, ureido, guanidino,
carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
[00271 In another aspect, the invention provides compounds of Formula F19:
HO2C
NH.
HN O 0 R2 0
00 0 N RI Y-'- N N
HO NH O H H
0 O NCH3 CONH2
N
HN O H
O HN
R$ O Rs
HO OHN H
H O
O
HO2C (F19)
and salts thereof; wherein:
21
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
O O
a) R2 is ~ NH2 or
~./~~ R6*
b) R6 is methyl or
c) R8 is methyl or\~ R8~ ; and
d) each of R', R6*, and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
[0028] The present invention provides, in another aspect, compounds of Formula
F20:
HO2C R12
HN NH 0 CONH2
O O
O 0 0 N R~
N N
H2NOC NH O H
O H
O NCH3 HO CONH2
N
HN O H
MeO 0 (CH2)4R 8** HN
O
HO HN N
O
Y H
O
HO2C (F20)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of Rl and R8** is ainino, monosubstituted amino, disubstituted amino,
NH-amino
protecting group, acylamino, ureido, guanidino, carbamoyl, sulfonamino,
thioacylamino,
thioureido, iminoamino, or phosphonamino.
22
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0029] In another aspect, the invention provides compounds of Formula F21
HOZC R12
HN NH O CONH2
O O
H2NOC O O O N R'
N N
NH O O H
O NC3 C02H I
N
HN (CH2)4Rs*# O H
MeO O HN =
HO HN N
H
11-f
HO2C
(F21)
and salts thereof; wherein:
a) Rl is
C N (CH2)6CH(CH3)2 N (CH2)6CH(CH3)CHZCH3
\
N~(CHZ8CH3 ~ y
H 0 0
> > ,
H H
Ny(CH2)BCH(CH3)CH2CH3 ~Ny(CH2)8CH(CH3)2
0 or 0
b) R 12 is H or CH3, and
c) R8** is amino, monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
23
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0030] In another aspect, the invention provides compounds of Formula F22
H02C CH3 NH
HN NH O CONH2 O
O O
N J.~
00 O '' CH2)8CH(CH3)CH2CH
NH ~ s
N N
HO NH O H H
O
O NH CO2H
N
HN O H
O HN
CH3 O
HO2C HN N R
O
HO2C
(F22)
and salts thereof; wherein:
R6*is amino, monosubstituted amino, disubstituted amino, NH-amino protecting
group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido, iminoamino,
or phosphonamino.
[0031] In another aspect, the present invention also provides pharmaceutical
compositions
including compounds of Formula I and compounds of Formula Fl-F22, and methods
of use
thereof.
[00321 In yet another aspect, the present invention also provides
antibacterial compositions
including compounds of Formula I and compounds of Formula F 1-F22, and methods
of use
thereof.
[0033] In a further aspect the present invention provides a process for
preparing the
compounds of Formula I and compounds of Formula F1-F22.
BRIEF DESCRIPTION OF THE DRAWINGS
[00341 Figure 1 shows a depiction of the biosynthetic genes cluster for
daptomycin, A54145,
and CDA. The numbers in parenthesis denote the amino acid number. The
following
abbreviations are used: Trp: tryptophan; Asn: asparagine; Asp: aspartic acid;
Thr: threonine; Gly:
24
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
glycine; Orn: ornithine; Ala: alanine; Ser: serine; MeGlu: 3-methylglutamic
acid; Kyn:
kynurenine; Glu: glutamic acid; hAsn: 3-hydroxyasparagine; Sar: sarcosine;
Lys: lysine;
OMeAsp: 3-methoxyaspartic acid; Ile: isoleucine; Val: valine; D-HPG:D-
hydroxyphenyl
gly.cine.
[0035] Figure 2 depicts the deletion of dptA-H in S. roseosporus whereby a
dptA-H deletion
was constructed in S. roseosporus, by exchanging the tsr (thiostrepton
resistance) and cat
(chloramphenicol) for the dptA H locus to construct the deletion in the
chromosome of S.
roseosporus.
[0036] Figure 3 depicts the general method for "Red-mediated" gene replacement
in the
daptomycin NRPS pathway. The bacteriophage X-induced "hyper-recombination"
state (the
"Red" system or Red-mediated recombination) was used to construct both
deletions within
dptBC and to clone the replacement modules via a technique called "gap-
repair". Abbreviations:
"C", condensation domain; "ASe1.", adenylation domain for serine; "T",
thiolation domain; "E",
epimerase domain.
[0037] Figure 4 depicts constructs from S. roseosporus combinatorial library.
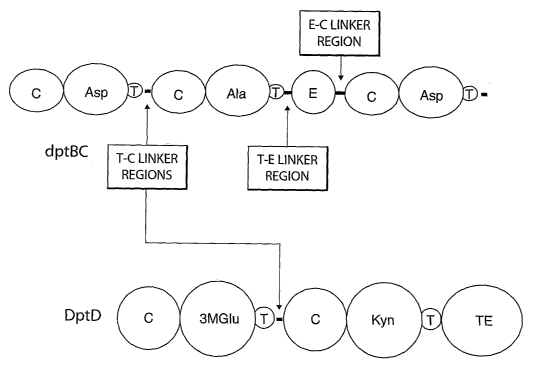
[0038] Figure 5 depicts the module organization in dptBC (internal module for
a D- amino
acid in dptBC)'and the terminal amino acid module (kynurenine) in dptD
associated with the
thioesterase. C:is a condensation domain. Circles containing amino acid 3
letter codes are
adenylation domains specific to the amino acid: Asn: asparagines; Ala:
alanine; Asp: aspartic
acid; 3MGlu: 3-methylglutamic acid; and Kyn: kynurenine. T is a thiolation
domain. E is an
epimerization domain. TE is a thioesterase domain.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
[0039] The term "acyl" denotes a carbonyl radical attached to an alkyl,
alkenyl, alkynyl,
cycloalkyl, heterocycyl, aryl or heteroaryl group, examples including, without
limitation, such
radicals as 8-methyldecanoyl, 10-methylundecanoyl, 10-methyldodecanoyl, n-
decanoyl, 8-
methylnonanoyl, dodecanoyl, undecanoyl, acetyl and benzoyl. In one embodiment
of the
invention, the acyl group is an "alkanoyl" group which is defined as a
carbonyl radical attached
to an alkyl group. In another embodiment of the invention, the alkanoyl group
is a"Cl-CZO-
alkanoyl" group which is defined as an alkanoyl group containing a total of 1
to 20 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
branched chain. In another embodiment of the invention, the alkanoyl group is
a"C1-C15-
alkanoyl" group which is defined as an alkanoyl group containing a total of 1
to 15 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
branched chain. In another embodiment of the invention, the alkanoyl group is
a"CI-C13-
alkanoyl" group which is defined as an alkanoyl group containing a total of 1
to 13 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
branched chain. In another embodiment of the invention, the alkanoyl group is
a"Cs-C20-
alkanoyl" group which is defined as an alkanoyl group containing a total of 5
to 20 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
branched chain. In another embodiment of the invention, the alkanoyl group is
a"CIO-C20-
alkanoyl" group which is defined as an alkanoyl group containing a total of 10
to 20 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
branched chain. In another embodiment of the invention, the alkanoyl group is
a"Cio-C13-
alkanoyl" group which is defined as an alkanoyl group containing a total of 1
to 13 carbon
atoms, including the carbonyl carbon. The carbon atoms can be arranged in a
straight chain or
branched chain. In another embodiment of the invention, the alkanoyl group is
C (CH2)6CH(CH3)2 (CH2)6CH(CH3)CH2CH3
'L, \
~ '~ (CH2)aCHs ~ > >
II II
(CH2)8CH(CH3)CH2CH3 (CH2)8CH(CH3)Z
Y
or 0
In another embodiment of the invention, the subsets of the term acyl are (1)
"Unsubstituted
alkanoyl" which is defined as carbonyl radical attached to an unsubstituted
alkyl group and (2)
"unsubstituted alkenoyl" which is defined as carbonyl radical attached to an
unsubsituted alkenyl
group.
[0040] The term "acylamino" is defined as a nitrogen radical adjacent to an
acyl group. In
one embodiment of the invention, the acylamino group is an "alkanoylamino"
group which is
defined as a nitrogen radical attached to an alkanoyl group. In another
embodiment of the
invention, the alkanoylamino group is a"CI-C20-alkanoylamino" group which is
defined as a
alkanoylamino group containing a total of 1 to 20 carbon atoms, including the
carbonyl carbon.
26
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
The carbon atoms can be arranged in a straight chain or branched chain. In
another embodiment
of the invention, the alkanoylamino group is a"C1-C15- alkanoylamino" group
which is defined
as an alkanoylamino group containing a total of 1 to 15 carbon atoms,
including the carbonyl
carbon. The carbon atoms can be arranged in a straight chain or branched
chain. In another
embodiment of the invention, the alkanoylamino group is a"Cl-C13-
alkanoylamino" group
which is defined as an alkanoylamino group containing a total of 1 to 13
carbon atoms, including
the carbonyl carbon. The carbon atoms can be arranged in a straight chain or
branched chain. In
another embodiment of the invention, the alkanoylamino group is a"C5-CZO-
alkanoylamino"
group which is defined as a alkanoylamino group containing a total of 5 to 20
carbon atoms,
including the carbonyl carbon. The carbon atoms can be arranged in a straight
chain or branched
chain. In another embodiment of the invention, the alkanoylamino group is
a"Clo-C20-
alkanoylamino" group which is defined as an alkanoylamino group containing a
total of 10 to 20
carbon atoms, including the carbonyl carbon. The carbon atoms can be arranged
in a straight
chain or branched chain. In another embodiment of the invention, the
alkanoylamino group is a
"Clo-CI3- alkanoylamino" group which is defined as an alkanoylamino group
containing a total
of 1 to 13 carbon atoms, including the carbonyl carbon. The carbon atoms can
be arranged in a
straight chain or branched chain. In another embodiment of the invention, the
alkanoylamino
group is
C N (CH2)6CH(CH3)2 N (CHZ)6CH(CH3)CHzCH3
\ ~L L
' N~ (CH28CH3 ~
H O O
H
1ACH2)8CH(CH3)CH2CH3 N (CHZ)8CH(CH3)2
o or o
[0041] The term "acyloxy" denotes an oxygen radical adjacent to an acyl group.
[0042] The term "alkenyl" is defined as linear or branched radicals having two
to about
twenty carbon atoms, preferably three to about ten carbon atoms, and
containing at least one
carbon-carbon double bond. One or more hydrogen atoms can also be replaced by
a substituent
group selected from acyl, acylamino, acyloxy, alkenyl, alkoxy, alkyl, alkynyl,
amino, aryl,
aryloxy, carbamoyl, carboalkoxy, carboxy, carboxyamido, carboxyamino, cyano,
disubstituted
27
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
amino, formyl, guanidino, halo, heteroaryl, heterocyclyl, hydroxy, iminoamino,
monosubstituted
amino, nitro, oxo, phosphonamino, sulfinyl, sulfonamino, sulfonyl, thio,
thioacylamino,
thioureido, or ureido. The double bond port ion(s) of the unsaturated
hydrocarbon chain may be
either in the cis or trans configuration. Examples of alkenyl groups include,
without limitation,
ethylenyl or phenyl ethylenyl. A subset of tenn alkenyl is "unsubstituted
alkenyl" which is
defined as an alkenyl group that bears no substituent groups.
[0043] The term "alkoxy" denotes oxygen radical substituted with an alkyl,
cycloalkyl or
heterocyclyl group. Examples include, without limitation, methoxy, tert-
butoxy, benzyloxy and
cyclohexyloxy.
[0044] The term "alkyl" is defined as a linear or branched, saturated radical
having one to
about twenty carbon atoms unless otherwise specified. The term "lower alkyl"
is defined as an
alkyl group containing 1-4 carbon atoms. One or more hydrogen atoms can also
be replaced by a
substitutent group selected from acyl, acylamino, acyloxy, alkenyl, alkoxy,
alkyl, alkynyl,
amino, aryl, aryloxy, carbamoyl, carboalkoxy, carboxy, carboxyamido,
carboxyarnino, cyano,
disubstituted amino, formyl, guanidino, halo, heteroaryl, heterocyclyl,
hydroxy, iminoamino,
monosubstituted amino, nitro, oxo, phosphonamino, sulfinyl, sulfonamino,
sulfonyl, thio,
thioacylamino, thioureido, or ureido. Examples of alkyl groups include,
without limitation,
methyl, butyl, tert-butyl, isopropyl, trifluoromethyl, nonyl, undecyl, octyl,
dodecyl,
methoxymethyl, 2-(2'-aminophenacyl), 3-indolylmethyl, benzyl, and
carboxymethyl. Subsets of
the term alkyl are (1) "unsubstituted alkyl" which is defined as an alkyl
group that bears no
substituent groups and (2) "substituted alkyl" which denotes an alkyl radical
in which one or
more hydrogen atoms is replaced by a substitutent group selected from acyl,
acylamino, acyloxy,
alkenyl, alkoxy, alkyl, alkynyl, amino, aryl, aryloxy, carbamoyl, carboalkoxy,
carboxy,
carboxyamido, carboxyamino, cyano, disubstituted amino, formyl, guanidino,
halo, heteroaryl,
heterocyclyl, hydroxy, iminoamino, monosubstituted amino, nitro, oxo,
phosphonamino,
sulfinyl, sulfonamino, sulfonyl, thio, thioacylamino, thioureido, or ureido.
In another
embodiment of the invention, the alkyl group is a"CI-CZO-alkyl" group which is
defined as a
alkyl group containing a total of 1 to 20 carbon atoms. The carbon atoms can
be arranged in a
straight chain or branched chain. In another embodiment of the invention, the
alkyl group is a
"C1-CI5- alkyl" group which is defined as a alkyl group containing a total of
1 to 15 carbon
atoms. The carbon atoms can be arranged in a straight chain or branched chain.
In another
embodiment of the invention, the alkyl group is a"Cl-C13- alkyl" group which
is defined as an
28
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
alkyl group containing a total of 1 to 13 carbon atoms. The carbon atoms can
be arranged in a
straight chain or branched chain. In another embodiment of the invention, the
alkyl group is a
"C5-C20-alkanoyl" group which is defined as a alkyl group containing a total
of 5 to 20 carbon
atoms. The carbon atoms can be arranged in a straight chain or branched chain.
In another
embodiment of the invention, the alkyl group is a"Clo-C20- alkyl" group which
is defined as a
alkyl group containing a total of 10 to 20 carbon atoms. The carbon atoms can
be arranged in a
straight chain or branched chain. In another embodiment of the invention, the
alkyl group is a
"Clo-C13- alkyl" group which is defined as a alkyl group containing a total of
10 to 13 carbon
atoms. In another embodiment of the invention, the alkyl group is a "C9-C12-
alkyl" group which
is defined as a alkyl group containing a total of 9 to 12 carbon atoms. The
carbon atoms can be
arranged in a straight chain or branched chain. In another embodiment of the
invention, the alkyl
group is nonyl, 7-methyloctyl, 7-methylnonyl, n-decyl, 9-methylundecyl, 9-
methyldecyl, n-
undecyl.
[0045] The term "alkylidenyl" is defined as a carbon radical of the formula
RX
Rx1
wherein R" and R"1 are independently selected from hydrido or C7-C17
unsubstituted alkyl,
wherein the total number of carbons from Rx and R"1 does not exceed 17.
[0046] The term "alkynyl" denotes linear or branched radicals having from two
to about ten
carbon atoms, and containing at least one carbon-carbon triple bond. One or
more hydrogen
atoms can also be replaced by a substituent group selected from acyl,
acylamino, acyloxy,
alkenyl, alkoxy, alkyl, alkynyl, amino, aryl, aryloxy, carbamoyl, carboalkoxy,
carboxy,
carboxyamido, carboxyamino, cyano, disubstituted amino, formyl, guanidino,
halo, heteroaryl,
heterocyclyl, hydroxy, iminoamino, monosubstituted amino, nitro, oxo,
phosphonamino,
sulfinyl, sulfonamino, sulfonyl, thio, thioacylamino, thioureido, or ureido.
An example of
alkynyl group includes, without limitation, propynyl.
[0047] The term "amino" is defined as an NH2 radical.
[0048] The term "amino acid" denotes a compound of the formula
29
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
O
HO NH2
Raa
wherein Raa is an amino acid side chain. A "naturally occurring amino acid" is
an amino acid
that is found in nature. An "essential amino acid" is one of the twenty common
amino acids:
alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic
acid, glycine, histidine,
isoleucine, leucine, lysine, methionine, phenyalanine, proline, serine,
threonine, tryptophan,
tyrosine and valine. A "non-proteinogenic amino acid" is any amino acid other
than an essential
amino acid. In this specification, the following abbreviations are used to
describe specific amino
acids:
Abbreviation(s) Amino acid
(MeO)Asp or (m)Asp or mAsp 3-methoxy-aspartic acid
or moAsp or mo(Asp)
(OH)Asn or h(Asn) or hAsn or 3-hydroxy-asparagine
h-Asn
(OH)Asp or h(Asp) or hAspor 3-hydroxy-aspartic acid
h-Asp
3-MG 3-methylglutamic acid
D-HPG D-hydroxyphenyl glycine
Ala Alanine
Asn Asparagines
Asp Aspartic acid
Glu Glutamic acid
Gly Glycine
Ile Isoleucine
Kyn Kynurinine
Lys Lysine
Om Ornithine
Sar Sarcosine
Ser Serine
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Abbreviation(s) Amino acid
Thr Threonine
Trp Tryptophan
Val Valine
In one aspect of the invention amino acids are 3-methoxy-aspartic acid, 3-
hydroxy-asparagine,3-
hydroxy-aspartic acid, 3-methylglutamic acid, Alanine, Asparagine, Aspartic
acid, Glutamic
acid, Glycine, Isoleucine, Kynurinine, Lysine, Omithine, Sarcosine, Serine,
Threonine,
Tryptophan, and Valine.
[0049] It will be understood by those of skill in the art, that peptides are
described by the
joining of the three letter codes above. For example, Asp-Asn-Trp refers to
the compound
COzH
O NHZ
HO N
N
H
O O
HZNOC
N
H
Alternatively, the compound above could also be described as Asp-Asn-Trp-NH2.
It will also be
understood by one of skill in the art that the peptides of the invention may
contain protecting
groups (vide infra). When an amino acid contains a protecting group, the three
letter code will
be adapted to indicate the protecting group. For example,
Thr-Asp(OtBu)-Asn(NHTrt)-Trp-NH2, refers to the following compound:
0
OtBu
0 O NH2
N N
HO N
H
HO O TrtHN O / ( \
O H
[0050] Common protecting groups for the amino acids of this invention include
tert-butoxy
(tBu), trityl (Trt) and tert-butoxy carbonyl (BOC) protecting groups.
[0051] It will also be understood by one of skill in the art that cyclic
peptides may also be
described by three letter codes. For example, the three letter structure
31
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
R'(Trp)-Asn-h-Asn- -Sar-Ala-Asp-Lys-omAsp-Gly-Asn-Glu-Ile is identical with
the
structure: -
HO2C
HN NH O CONH2
JY O O
00 O N C
N
N
HZNOC NH O H
O H
O NCH3 HO CONH2
I ly """
N
HN O H
Me0 O (CH2)4R8*f HN
O
HO O HN N
H
HO2C
[0052] It will also be understood by one of skill in the art that amino acids
can exist in either
the L or D configuration. When it is desirable to indicate the configuration
of the amino acid, the
D or L designation is placed before the three letter code.
[0053] The term "amino acid residue" denotes a compound of the formula
O
NH2
Raa
wherein Raa is an amino acid side chain. In one aspect of the invention, the
amino acid residue is
derived from a natural amino acid. In another aspect of the invention, the
amino acid residue is
derived from the amino acids 3-methoxy-aspartic acid, 3-hydroxy-asparagine,3-
hydroxy-aspartic
acid, 3-methylglutamic acid, Alanine, Asparagine, Aspartic acid, Glutamic
acid, Glycine,
Isoleucine, Kynurinine, Lysine, Ornithine, Sarcosine, Serine, Threonine,
Tryptophan, and
Valine.
[0054] The term "amino acid side chain" denotes any side chain (R group) from
a naturally-
occurring or synthetic amino acid. For example, 3-indolylmethyl could also be
called a
tryptophan side chain. Examples of amino acid side chains include, without
limitation,
32
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
N I \ ~ I v N \ N
N
F OH F OH
F
HN F
OH
I ~ Me OH OMe
-,~CO2H ' -I~CO2H ' CO2H
N
GH3
HO
H HO OH HO
~ HO OH
O / I / HO
OH
OH OH H2N
O OH
HO
OH
NH2 0 NH2
OMe
HO2C
O , , > >
'
C02H
OH
33
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
0
Raa~ ~ 1----~OH , ./OH
NH O O
~1
OH' NNH2 NH , ~~
H 2 NH2 ,
0
~~\
'~/\~~Raa2 ~ ~i~/S\ a I NH ~
, ~" OH ,
N NH OH OH
O CI H
OH 1~~ N
N >=NH ~
H NH2 N
CI H
fl N~ \k hydrido and methyl,
wherein each of Raal and Ra2 is independently amino, monosubstituted amino,
disubstituted
amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino. A"non-proteinogenic amino acid side chain" is an
amino acid
side chain derived from a non-proteinogenic amino acid (vide supra). Examples
of a non-
proteinogenic amino acid side chains include, without limitation,
HO
HO OH HO
OH I HO OH
) I >
HO
OH
/ OH \ OH HzN
\ I , I / OH
HO
OH
NH2 0 NHz
OMe
O I \ , '~ , and HOzC
?;\COzH
OH
34
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
In one aspect of the invention, the amino acid side chain is derived from a
natural amino acid. In
another aspect of the invention, the amino acid side chain is derived from the
amino acids 3-
methoxy-aspartic acid, 3-hydroxy-asparagine,3-hydroxy-aspartic acid, 3-
methylglutamic acid,
Alanine, Asparagine, Aspartic acid, Glutamic acid, Glycine, Isoleucine,
Kynurinine, Lysine,
Ornithine, Sarcosine, Serine, Threonine, Tryptophan, and Valine.
[0055] The term "2-(2'-aminophenacyl)" refers to a radical of the formula
NH2
O
.Mr
[0056] The term "aryl" or "aryl ring" is defined as an aromatic radical in a
single or fused
carbocyclic ring system, having from five to fourteen ring members. In a
preferred embodiment,
the ring system has from six to ten ring meinbers. One or more hydrogen atoms
may also be
replaced by a substituent group selected from acyl, acylamino, acyloxy,
alkenyl, alkoxy, alkyl,
alkynyl, amino, aryl, aryloxy, azido, carbamoyl, carboalkoxy, carboxy,
carboxyamido,
carboxyamino, cyano, disubstituted amino, formyl, guanidino, halo, heteroaryl,
heterocyclyl,
hydroxy, iminoamino, monosubstituted amino, nitro, oxo, phosphonamino,
sulfinyl,
sulfonamino, sulfonyl, thio, thioacylamino, thioureido, or ureido. Examples of
aryl groups
include, without limitation, phenyl, naphthyl, biphenyl, terphenyl.
[0057] The term "aryloxy" denotes oxy-containing radicals substituted with an
aryl or
heteroaryl group. Examples include, without limitation, phenoxy.
[0058] The term "carbamoyl" denotes a nitrogen radical of the formula
0
~
N ORx3
ix2
wherein R"2 is selected from hydrido, alkyl, aryl, cycloalkyl, heteroaryl or
heterocyclyl and R"3 is
selected from alkyl, aryl, cycloalkyl, heteroaryl or heterocyclyl.
[0059] The term "carboalkoxy" is defined as a carbonyl radical adjacent to an
alkoxy or
aryloxy group.
[0060] The term "carboxy" denotes a COOH radical.
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0061] The term "carboxyamino" denotes a CONH2 radical.
[0062] The term "carboxyarnido" is defined as a carbonyl radical adjacent to a
monosubstitnted amino or disubstituted amino group.
[0063] The term "a-carboxy amino acid side chain" is defined as a carbon
radical of the
formula
Rx4
OH
Z O
wherein Ri4 is defined as an amino acid side chain.
[0064] The term "carboxymethyl" denotes a CH2CO2H radical.
[0065] The term "cycloalkyl" or "cycloalkyl ring" denotes a saturated or
partially unsaturated
carbocyclic ring in a single or fused carbocyclic ring system having from
three to twelve ring
members. In a preferred embodiment, a cycloalkyl is a ring system having three
to seven ring
members. One or more hydrogen atoms may also be replaced by a substituent
group selected
from acyl, acylamino, acyloxy, alkenyl, alkoxy, alkyl, alkynyl, amino, aryl,
aryloxy, carbamoyl,
carboalkoxy, carboxy, carboxyamido, carboxyamino, cyano, disubstituted ainino,
formyl,
guanidino, halo, heteroaryl, heterocyclyl, hydroxy, iminoamino,
monosubstituted amino, nitro,
oxo, phosphonamino, sulfinyl, sulfonamino, sulfonyl, thio, thioacylamino,
thioureido, or ureido.
Examples of a cycloalkyl group include, without limitation, cyclopropyl,
cyclobutyl, cyclohexyl,
and cycloheptyl.
[0066] The term "disubstituted amino" is defined as a nitrogen radical
containing two
substituent groups independently selected from, alkyl, cycloalkyl,
heterocyclyl, aryl, or
heteroaryl. Preferred disubstituted amino radicals are "lower disubstituted
amino" radicals,
whereby the substituent groups are lower alkyl. Also preferred disubstituted
amino radicals are
amino radicals wherein one substituent is a lower alkyl group and the other
substituent is an a-
carboxy amino acid side chain.
[0067] The group "Fmoc" is a 9-fluorenylmethoxycarbonyl group.
[0068] The term "guanidino" is defined as a nitrogen radical of the formula
36
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Rx5 R X7
I /
Rx6
I I
N
\Rx6
wherein each of R"5, R"7 and R"8 is independently selected from hydrido,
alkyl, aryl, cycloalkyl,
heteroaryl or heterocyclyl group; and R"6 is selected from alkyl, aryl,
cycloalkyl, heteroaryl or
heterocyclyl group.
[0069] The term "halo" denotes a bromo, chloro, fluoro or iodo radical.
[0070] "Heteroaryl" or "heteroaryl ring" is defined as an aromatic radical
which contain one
to four hetero atoms or hetero groups selected from 0, N, S, or SO in a single
or fused
heterocyclic ring system, having from five to fifteen ring members. In a
preferred embodiment,
the heteroaryl ring system has from six to ten ring members. One or more
hydrogen atoms may
also be replaced by a substituent group selected from acyl, acylamino,
acyloxy, alkenyl, alkoxy,
alkyl, alkynyl, amino, aryl, aryloxy, carbainoyl, carboalkoxy, carboxy,
carboxyamido,
carboxyamino, cyano, disubstituted amino, formyl, guanidino, halo, heteroaryl,
heterocyclyl,
hydroxy, iminoamino, monosubstituted amino, nitro, oxo, phosphonamino,
sulfinyl,
sulfonamino, sulfonyl, thio, thioacylamino, thioureido, or ureido. Examples of
heteroaryl groups
include, without limitation, pyridinyl, thiazolyl, thiadiazoyl, isoquinolinyl,
pyrazolyl, oxazolyl,
oxadiazoyl, triazolyl, and pyrrolyl groups.
[0071] The term "heterocyclyl," "heterocyclic" or "heterocyclyl ring" denotes
a saturated or
partially unsaturated ring containing one to four hetero atoms or hetero
groups selected from 0,
N, NH, N(lower alkyl), S, SO or SO2, in a single or fused heterocyclic ring
system having from
three to twelve ring members. In a preferred embodiment, a heterocyclyl is a
ring system having
three to seven ring members. One or more hydrogen atoms may also be replaced
by a substituent
group selected from acyl, acylamino, acyloxy, alkenyl, alkoxy, alkyl, alkynyl,
amino, aryl,
aryloxy, carbamoyl, carboalkoxy, carboxy, carboxyamido, carboxyamino, cyano,
disubstituted
amino, formyl, guanidino, halo, heteroaryl, heterocyclyl, hydroxy, iminoamino,
monosubstituted
amino, nitro, oxo, phosphonamino, sulfinyl, sulfonamino, sulfonyl, thio,
thioacylamino,
thioureido, or ureido. Examples of a heterocyclyl group include, without
limitation,
morpholinyl, piperidinyl, and pyrrolidinyl.
[0072] The term "hydrido" is defined as a single hydrogen atom (H).
[0073] The term "iminoamino" denotes a nitrogen radical of the formula:
37
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Rx9
Rx10
Rx"
wherein each of R"9 and R"11 is independently selected from a hydrido, alkyl,
cycloalkyl, aryl,
heteroaryl or heterocyclyl group; and R"io is selected from an alkyl,
cycloalkyl, aryl, heteroaryl
or heterocyclyl group.
[00741 The term "N-methyl amino acid" denotes a compound of the formula
O
HO NHMe
Raa
wherein Raa is an amino acid side chain. Examples of amino acid side chains of
an N-methyl
amino acid include
38
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
4CH(CH3)2 -~-CH2CH(CH3)2 , -~-(CHZ)2CO2H , 4CH2OH
4CH2SCH3 > 4CH(CH3)Et > -~-CH(CH3)OH 4(CH2)4CHs
OH OH
NA N I
HN ~NH
F OH F
/ HN
OH
OH
IF
( and
N
CH3
[0075] The term "monosubstituted amino" denotes a nitrogen radical containing
a hydrido
group and a substituent group selected from alkyl, cycloalkyl, heterocyclyl,
aryl, or heteroaryl.
Preferred monosubstituted amino radicals are "lower monosubstituted amino"
radicals, whereby
the substituent group is a lower alkyl group. More preferred monosubstituted
amino radicals are
amino radicals containing an a-carboxy amino acid side chain.
[0076] The term "phosphonamino" is defined as a nitrogen radical of the
formula:
0
II/Rx13
N i~ ~Rx14
Rx12
wherein Ri12 is selected from hydrido, alkyl, aryl, cycloalkyl, heteroaryl or
heterocyclyl; wherein
each of R"13 and R"Ia is independently selected from alkyl, alkoxy, aryl,
aryloxy, cycloalkyl,
heteroaryl and heterocyclyl.
[0077] The term "protecting group" refers to any chemical compound that may be
used to
prevent a group on a molecule from undergoing a chemical reaction while
chemical change
39
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
occurs elsewhere in the molecule. Groups that may need protecting include
hydroxyl, amino,
carboxylic acids and carboxyamino groups. Numerous protecting groups are known
to those
skilled in the art and examples can be found in "Protective Groups in Organic
Synthesis" by
Theodora W. Greene and Peter G. M. Wuts, John Wiley and Sons, New York, 3d
Edition 1999,
hereafter Greene.
[0078] The term "amino protecting group" refers to any chemical compound that
may be
used to prevent an amino group on a molecule from undergoing a chemical
reaction while
chemical change occurs elsewhere in the molecule. Numerous amino protecting
groups are
known to those skilled in the art and examples can be found in Greene.
Examples of "amino
protecting groups" include phthalimido, trichloroacetyl, STA-base,
benzyloxycarbonyl, t-
butoxycarbonyl, t-amyloxycarbonyl, isobomyloxycarbonyl, adamantyloxycarbonyl,
chlorobenzyloxycarbonyl, nitrobenzyloxycarbonyl or the like. Preferred amino
protecting
groups are "carbamate amino protecting groups" which are defined as an amino
protecting group
that when bound to an amino group forms a carbamate, or- the azido group.
Preferred amino
carbamate protecting groups are allyloxycarbonyl (alloc), carbobenzyloxy
(CBZ), 9-
fluorenylmethoxycarbonyl (Fmoc) and tert-butoxycarbonyl protecting groups.
[0079] The term "hydroxyl protecting group" refers to any chemical compound
that may be
used to prevent a hydroxyl group on a molecule from undergoing a chemical
reaction while
chemical change occurs elsewhere in the molecule. Numerous hydroxyl protecting
groups are
known to those skilled in the art and examples can be found in Greene (vide
supra) Examples of
hydroxyl protecting groups include esters such as, but not limited to formate,
acetate, substituted
acetate, crotonate, benzoate, substituted benzoates, methyl carbonate, ethyl
carbonate, alkyl and
aryl carbonates, borates, and sulphonates. Examples of hydroxyl protecting
groups also include
ethers such as, but not limited to methyl, benzyloxylmethyl, siloxymethyl,
tetrahydropyranyl,
substituted tetrahydropyranyl, ethyl, substituted ethyl, allyl, tert-butyl,
propargyl, phenyl,
substituted phenyl , benzyl, substituted benzyl, alkyl silyl and silyl ethers
or the like. Preferred
hydroxyl protecting groups are "acid labile ethers" which are defined as an
ether protecting
group that may be removed by treatment with acid. Preferred hydroxyl ether
protecting groups
are trityl (Trt), tert-butyl (tBu), benzyl (Bzl) and tert-butyldimethylsilyl
(TBDMS) protecting
groups.
[0080] The term "carboxylic acid protecting group" refers to any chemical
compound that
may be used to prevent a carboxylic acid on a molecule from undergoing a
chemical reaction
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
while chemical change occurs elsewhere in the molecule. Numerous carboxylic
acid protecting
groups are known to those skilled in the art and examples can be found in
Greene (vide supra).
Examples of carboxylic acid protecting groups include, but are not limited to
amides,
hydrazides, and esters such as, methyl esters, substituted methyl, phenacyl,
tetrahydropyranyl,
tetrahydrofuranyl, cyanomethyl, triisopropylsilylmethyl, desyl, ethyl2-
substituted ethyl, phenyl,
2,6 dialkyl phenyl, benzyl, substituted benzyl, silyl, and stannyl, or the
like. Preferred carboxylic
acid ester protecting groups are allyl (All), tert-butyl (tBu), benzyl (Bzl),
4- {N-[ 1-(4,4-dimethyl-
2,6-dioxocyclohexylidinene)-3-methylbutyl]-amino}benzyl (ODmab), 1-adamantyl
(lAda) and
2-phenylisopropyl (2-PhiPr) protecting groups.
[0081] The term "sulfinyl" denotes a tetravalent sulfur radical substituted
with an oxo
substituent and a second substituent selected from the group consisting of
alkyl, cycloalkyl,
heterocyclyl, aryl, or heteroaryl group.
[0082] The tenn sulfonamino is defined as an amino radical of the formula:
Rx15
N Rx16
00
wherein R"ls is selected from a hydrido, alkyl, cycloalkyl, aryl, heteroaryl
or heterocyclyl group;
and R"16 is selected from alkyl, cycloalkyl, aryl, heteroaryl or heterocyclyl
group.
[0083] The term "sulfonyl" denotes a hexavalent sulfur radical substituted
with two oxo
substituents and a third substituent selected from alkyl, cycloalkyl,
heterocyclyl aryl, or
heteroaryl.
[0084] The term "thio" is defined as a radical containing a substituent group
independently
selected from hydrido, alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,
attached to a divalent
sulfur atom, such as, methylthio and phenylthio.
[0085] The term "thioacylamino" denotes an amino radical of the formula
Rx17
Rx18
S
wherein R"17 is selected from a hydrido, alkyl, aryl, cycloalkyl, heteroaryl
or heterocyclyl group;
and wherein R"18 is selected from an alkyl, aryl, cycloalkyl, heteroaryl or
heterocyclyl group.
[0086] The term "thioureido" is defined as a sulfur radical of the formula
41
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Rx19 Rx20
I /
~N N
/ Rx21
s
wherein each of R"I9and R"20 is independently selected from hydrido, alkyl,
aryl, cycloalkyl,
heteroaryl or heterocyclyl group; and R"21 is selected from an alkyl, aryl,
cycloalkyl, heteroaryl
or heterocyclyl group.
[0087] The group trityl is a triphenylmethyl group.
[0088] The term "ureido" is defined as a nitrogen radical of the formula
Rx21 Rx22
~N N
Rxzs
O
wherein each of R"21and R"22 is independently selected from hydrido, alkyl,
aryl, cycloalkyl,
heteroaryl or heterocyclyl group; and R"23 is selected from an alkyl, aryl,
cycloalkyl, heteroaryl
or heterocyclyl group.
[0089] The terms "ZptA", "lptB" "lptC" and "lptD" refer to nucleic acid
molecules that
encode subunits of the A54145 NRPS. In a preferred embodiment, the nucleic
acid molecule is
derived from Streptomyces, more preferably the nucleic acid molecule its
derived from S. fradiae.
The lptA nucleic acid encodes for amino acids 1-5. The lptB nucleic acid
encodes for amino
acids 6 and 7. The lptC nucleic acid encodes for amino acids 8-11. The lptD
nucleic acid
encodes for amino acids 12 and 13 (Figurel). The terms "ZptA", "lptB, "lptC '
and "lptD" also
refer to allelic variants of these genes, which may be obtained from other
species of
Streptonzyces or from other S. fradiae strains.
[0090] The terms "dptA", "dptBC' and "dptD" refer to nucleic acid molecules
that encode
subunits of the daptomycin NRPS. In a preferred embodiment, the nucleic acid
molecule is
derived from Streptoinyces, more preferably the nucleic acid molecule is
derived from S.
roseosporus. The dptA nucleic acid encodes amino acids 1-5. The dptBC nucleic
acid encodes
amino acids 6-11. The dptD nucleic acid encodes amino acids 12-13 (Figure 1).
The terms
42
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
"dptA", "dptBC' and "dptD" also refer to allelic variants of these genes,
which may be obtained
from other species of Sts eptonzyces or from other S. roseosporus strains.
[0091] The salts of the compounds of the invention include acid addition salts
and base
addition salts. In a preferred embodiment, the salt is a pharmaceutically
acceptable salt of the
compound of Formula I or the compound of any of Formula Fl-F22. The term
"pharmaceutically acceptable salts" embraces salts commonly used to form
alkali metal salts and
to form addition salts of free acids or free bases. The nature of the salt is
not critical, provided
that it is pharmaceutically-acceptable. Suitable pharmaceutically acceptable
acid addition salts
of the compounds of the invention may be prepared from an inorganic acid or an
organic acid.
Examples of such inorganic acids include, without limitation, hydrochloric,
hydrobromic,
hydroiodic, nitric, carbonic, sulfuric and phosphoric acid. Appropriate
organic acids may be
selected from aliphatic, cycloaliphatic, aromatic, arylaliphatic,
heterocyclic, carboxylic and
sulfonic classes of organic acids, examples of which include, without
limitation, formic, acetic,
propionic, succinic, glycolic, gluconic, maleic, embonic (pamoic),
methanesulfonic,
ethanesulfonic, 2-hydroxyethanesulfonic, pantothenic, benzenesulfonic,
toluenesulfonic,
sulfanilic, mesylic, cyclohexylaminosulfonic, stearic, algenic,l3-
hydroxybutyric, malonic,
galactic, and galacturonic acid. Suitable pharmaceutically-acceptable base
addition salts of
compounds of the invention include, but are not limited to, metallic salts
made from aluminum,
calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made
from N,N'-
dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamine, N-
methylglucamine, lysine and procaine. All of these salts may be prepared by
conventional
means from the corresponding compound of the invention by treating, for
example, the
compound of the invention with the appropriate acid or base.
[0092] The compounds of the invention can possess one or more asymmetric
carbon atoms
and are thus capable of existing in the form of optical isomers as well as in
the form of racemic
or non-racemic mixtures thereof. The compounds of the invention can be
utilized in the present
invention as a single isomer or as a mixture of stereochemical isomeric forms.
Diastereoisomers,
i.e., nonsuperimposable stereochemical isomers, can be separated by
conventional means such as
chromatography, distillation, crystallization or sublimation. The optical
isomers can be obtained
by resolution of the racemic mixtures according to conventional processes, for
example by
formation of diastereoisomeric salts by treatment with an optically active
acid or base. Examples
of appropriate acids include, without limitation, tartaric, diacetyltartaric,
dibenzoyltartaric,
43
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
ditoluoyltartaric and camphorsulfonic acid. The mixture of diastereomers can
be separated by
crystallization followed by liberation of the optically active bases from the
optically active salts.
An alternative process for separation of optical isomers includes the use of a
chiral
chromatography column optimally chosen to maximize the separation of the
enantiomers. Still
another method involves synthesis of covalent diastereoisomeric molecules by
reacting
compounds of the invention with an optically pure acid in an activated form or
an optically pure
isocyanate. The synthesized diastereoisomers can be separated by conventional
means such as
chromatography, distillation, crystallization or sublimation, and then
hydrolyzed to obtain the
enantiomerically pure compound. The optically active coinpounds of the
invention can likewise
be obtained by utilizing optically active starting materials. These isomers
may be in the form of
a free acid, a free base, an ester or a salt.
[0093] The invention also embraces isolated compounds, preferably compounds of
Formula I
or compounds of any of Formulas F1-F22. An isolated compound refers to a
compound,
preferably a compound of Formula I or a compound of any of Formulas F1-F22,
which
represents at least about 1%, preferably at least about10 1o, more preferably
at least about 20%,
even more preferably at least about 50%, yet more preferably at least about
80%, yet even more
preferably at least about 90% and most preferably at least about 99% of the
compound present in
the mixture. In one embodiment of the invention the compound, preferably a
compound of
Formula I or a compound of any of Formulas F l-F22õ is present in at least
about 80% to about
90% of the composition. In another embodiment the compound, preferably a
compound of
Formula I or a compound of any of Formulas F1-F22, is present in at least 90%
of the
composition. In another embodiment the compound, preferably a compound of
Formula I or
compound of any of Formulas F1-F22, is is present in greater than 90% of the
composition.
[0094] The percentation of the compound, preferably a compound of Formula I or
a
compound of any of Formulas F1-F22, may be measured by any means including
nuclear
magnetic resonance (NMR), gas chromatography/mass spectroscopy (GC/MS), liquid
chromatography/mass spectroscopy (LC/MS) or microbiological assays. A
preferred means for
measuring the purity of the compound is by analytical high pressure liquid
chromatography
(HPLC) or LC/MS.
[0095] In one embodiment of the invention, the compound, a pharmaceutically
acceptable
salt thereof or a pharmaceutical composition comprising the compound exhibits
a detectable (i.e.
44
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
statistically significant) antimicrobial activity when tested in conventional
biological assays such
as those described herein.
Depsipeptide Compounds
[00961 In one aspect, the invention provides compounds of Formula I
R12 R13
HO2C
NH
HN O O R2 O
00 O N R1
R11 N N
NR11" H O R3 O R2.
O NR5 /
R5" N
HN O H
O HN
Rs R$ 0 Rs
HN N
N
O
O RW
HO2C
and salts thereof.
[0097] The group R2 of Formula I is an amino acid side chain,
OH O
0 or NH2. In embodiment one of the invention the amino acid side
Of..l O
chain is 0 or NH2 . In another embodiment of the invention, the amino
acid side chain is derived from a D- amino acid. In another embodiment of the
invention, the
amino acid side chain is
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
O
OH /OH
NH O O
OH N'k NH2 4')~
H NH2 NH2
O
~ /~~ aa2 ~
OH OH
~ N=:-\
N H CI H
OH
~
NH2 NH
H N
CI H
X___SH ND or k
wherein each of Raal and Raa2 is independently amino, monosubstituted amino,
disubstituted
amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
[0098] Substituent R2* is H. Alternatively, Ra and R2* together with the atoms
to which they
are attached, form a five or six-member heterocyclic ring. In one embodiment
of Formula I, R2
and R2* together with the atoms to which they are attached, form a pyrrolidine
ring.
NH2
fro ''\-y oH NH2
~s
[0099] The group R3 of Formula I is OH , 0 ,\c~ O or a non-
proteinogenic amino acid side chain. In one embodiment of the invention the
group R3 of
NH2
-1-rl~o '\-y OH NH2
Formula I is OH , 0 , or I O. In another embodiment of the invention, the
non-proteinogenic amino acid is
46
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
HO
HO \ OH ::. HO OH OH
/ OH OH H2N
\ ( , I O OH , or
HO
OH
[0100] Substituent R5 of Formula I is H or methyl and substiuent R5* of
Formula I is H or an
amino acid side chain derived from an N-methylamino acid. In one embodiment of
the
invention, R5* is methyl,
4CH(CH3)2 -~-CHZCH(CH3)2 > -~-(CHZ)2CO2H , 4CH20H
4CHZSCH3 , 4CH(CH3)Et -~-CH(CH3)OH -~-(CHZ)4CH3
OH OH
N N I
HN ~ ~NH
N
F OH F
/ HN
OH
OH
F
or
N~
CH3
Alternatively, R5 and R5* together with the atoms to which they are attached,
form a five or six-
member heterocyclic ring. In one embodiment of Formula I, R5 and R5* together
with the atoms
to which they are attached, form a piperidine or a pyrrolidine ring.
47
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0101] Group R6 of Formula I is methyl or
R6*
[0102] Substituent R8 of Formula I is an amino acid side chain, hydrogen,
methyl,
NH2
%I~OH or In one embodiment of the invention, substituent R8
NH2
of Formula I is hydrogen, methyl, ~~OH or In another
embodiment of the invention, the amino acid side chain is derived from a D-
amino acid. In
another embodiment of the invention substituent R8 is the amino acid side
chain derived from
glycine, D-alanine, D-asparagine, D-serine or D-lysine. In another embodiment
of the invention,
the amino acid side chain is
O
'-,-AOH ,,</OH
NH O O
~~ OH NNH2
H -ANH2 4NH
2 ~
~ \ O
~ I
I ~~
NH 3 OH
OH OH
~.~NH~
O CI H
OH ~N
'.~-\/~ ~NH
H NH2 N
'~ CI H
XSH ND or kk
wherein each of Raal and Ra2 is independently amino, monosubstituted amino,
disubstituted
amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
48
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0103] Substituent R8* of Formula I is H. Alternatively, R8 and R8* together
with the atoms
to which they are attached, form a five or six-member heterocyclic ring. In
one embodiment of
Formula l, R8 and R$* together with the atoms to which they are attached, form
a pyrrolidine
ring.
OMe OH
~ ~
[0104] Group R9 of Formula I is /~~co~H ~/~ co2H ~~ co2H or an amino acid
side chain substituted with at least one carboxylic acid. In one embodiment of
the invention
OMe OH
~
group R9 of Formula I is /~ co2H ~/~~COaH "~
or CO2H hi another embodiment of
the invention, the amino acid side chain is
OMe Me OH
\CO2H
/ 2 COZH / L CO2H COaH
Me OMe OH
COZH
COZH COZH ' or CO2H
[0105] Substituent Rl l of Formula I is an amino acid side chain, methyl,
O
.
%~~OH , or NH2 . In one embodiment of the invention substituent Rl l of
Formula I is
O
.
methyl, '~"~OH , or NH2. In one embodiment of the invention, the amino acid
side
chain is derived from a D- amino acid. In another embodiment of the invention
Rll of Formula I
is an amino acid side chain derived from D-alanine, D-serine, or D-asparagine.
In another
embodiment of the invention, the amino acid side chain is
49
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
O
aal ,<~,OH
NH O O
~~ OH N""-""NNH2 NH _~~
H 2 NH
~ 2
O
~
~~/\/-Raa2, i- NH _1 OH ~
N-\ OH OH
NH~
CI H O
OH N
NNH2 >==NH , ~NH2
OH
H CI N
'~~SH r ND or k
wherein each of Raal and Raa2 is independently amino, monosubstituted amino,
disubstituted
amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylainino,
thioureido,
iminoamino, or phosphonamino.
[0106] Substituent Rll* is H. Alternatively, Rll and Rli* together with the
atoms to which
they are attached, form a five or six-member heterocyclic ring. In one
embodiment of Formula I,
R11 and Rl l* together with the atoms to which they are attached, form a
pyrrolidine ring.
[0107] Group R12 of Formula I is H or CH3.
[0108] Substituent R13 of Formula I is CH(CH3)2, CH(CH2CH3)CH3,
O NH2
H
or
O NH2
In one embodiment of the invention, R13 is CH(CH2CH3)CH3 or ~.
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0109] Each of R', R6* and R8** is independently amino, monosubstituted amino,
disubstituted amino, NH-amino protecting group, acylamino, ureido, guanidino,
carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino. In one
embodiment of
the invention RI is amino, NH-amino protecting group, or acylamino. In another
embodiment of
the invention R' is amino. In another embodiment of the invention, R' is NH-
amino protecting
group. In another embodiment of the invention Rl is acylamino. In another
embodiment of the
invention Rl is alkanoylamino. In yet another embodiment of the invention R'
is Cio-Cl3
alkanoylamino. In still another embodiment of the invention, R' is
O N (CHa)sCH(CH3)2 N (CH2)6CH(CH3)CH2CH3
\I I ~2 ' L
' N/x\ II H H2aCHs O
> > >
~ N (CH2)gCH(CH3)CH2CH3 Ny(CH2)8CH(CH3)2
y
O or O
[0110] In another embodiment of the invention each of R6* and R$** is
independently amino,
or NH-amino protecting group. In another embodiment of the invention each of
R6* and R$** is
independently amino. In yet another embodiment of the invention each of R6*
and R 8** is
independently NH-amino protecting group.
[01111 Table I provides exemplary compounds of Formula I.
Table I
Conapoufads of Fornzula I
# Compound
NH
HO2C
HN NH O CONH2
O 0
HO~O 0 0 )~11 N N N-CO(CHZ6CH(CH3)CH2CH3
H
NH O O
cl 0 NH CO2H
N
HN ~=O H
HO2 C' ~O HN
~/\ O
HN N NH2
H O
O
HO2C
51
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HOzC
NH CONH2
HN 0 O 0
HOO 0 0 N N N NH
-CO(CH2)8CH(CH3)2
)~H H
C2 NH 0 0
O=~ NH CO2H
N 14
HN O H
HOZCO HN
NH2
HN N 4
H 0
0 HO2C
NH
HOzC
NH CONH2
HN O 0 0
HO' O 0 0 N N N N-CO(CHZ)$CH(CH3)CH2CH3
NH 0 H H
C3 0
O=~ NH CO2H
N
HN - 0 H
HOaC~O HN
O
HN N NHa
H 0
O
HOZC
HO2C
NH CONH2
HN O 0 0
HO' O 0 O N N N N-CO(CH2)6CH(CH3)CH2CH3
~f\NH 0 H H
I I
0 C4 O=~ NH CO2H
N
HN O H
HOZCO HN
0
HN N NH2
H O
O
OZC
H
52
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC
HN NH p CONH2
O O
HO' O 0 0 N N N N-CO(CHa)8CH(CH3)2
~(\ H H
NH 0 0
C5 O=~ NH COZH
N
HN 0 H
HO2C' O HN
v\ O
HN N NHZ
N O
YIH
0
HO2C
HO2C
HN NH O CONH2
O 0
HOO 0 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
0 C6 0=~ NH COzH
N
HN 0 H
HO2C' ~O HN
0
NH2
HN N 4
H 0
0 HOZC
HO2C
NH CONHZ
HN O O 0
HO O 0 0 N N N N-CO(CHZ)6CH(CH3)CHZCH3
NH 0 H H
0 C7 O~ NH CO2H
N
HN 0 H
HOzCO HN
O
HN N NHZ
H 0
0 HOZC
53
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
HO 1 O 0 0 N N N N-CO(CH2)aCH(CH3)2
NH 0 H H
0 C8 O=~ NH COaH
N
HN ~=o H
HOaC' O HN
0
HN N NH2
- --~
-~~ H O
0
HO2C
HOaC
HN NH O CONH2
O O
HOO 0 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
0 C9 O=~ NH CO2H
N
HN ~=o H
HOzC' O HN
HN N NH~
H O
O
HOaC
0 NH2
HO2C '
NH
HN O CONH2
0 O
HO O 0 0 N N 'r~ N N-CO(CH2)6CH(CH3)CH2CH3
NH O H H
O
ClO O=~ NH C02H N
HN ~=o H
HO2 C' ~O HN
~J\ 0
HN N NH2
H O
O
HO2C
54
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
0 NH2
HO2C HN NH O CONH2
0 0
HO O 0 O N N N N-CO(CHa)8CH(CH3)2
NH 0 H H
0 C11 O=~ NH CO2H
N
HN O H
HO2 C' O HN
~/\ O
HN N NHZ
ir, H O
0 HOzC
O NH2
HO2C NH
HN O CONH2
O O
HO' O 0 0 N N N N-CO(CHz)8CH(CH3)CH2CH3
NH 0 H H
O
C12 O=~ NH COzH
N
HN O H
H02C' ~O HN
~/\ O
HN N NHZ
H O
0
HO2C
NH
HO2C
HN NH 0 CONH2
0 O
HO' O 0 0 N N N N-CO(CHZ)6CH(CH3)CH2CH3
~f\ H 0 H
C13 NH 0
O NH CO2H
~
HN ~0 H
HOzC' ~O HN
~/\ O
HN N NHZ
H 0
O
HO2C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HOZC
NH CONH2
HN O 0 0 Hy~ HO1O 0 0 N N N N-CO(CHZ)6CH(CH3)CHzCH3
NH O H H
C14 0 O) NH CO2H
N
HN O H
HOZC~O HN
0
HN N NH2
H O
O
HOZC
NH
HO2C
HN NH 0 CONH2
1 O 0
H
HO O 0 0 N H N-CO(CH2)6CH(CH3)CH2CH3
C15 NH O 0
O=~ NH CO2H
N
HN O H
H02C' ~O HN
~/\ 0
HN N NHZ
H 0
O
HO2C
HO2C
HN NH 0 CONH2
0 0 Hy~ H
HO' O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH O H H
O
C16 O=~ NH CO2H
N
HN 0 H
HOZC' ~O HN
~/\ O
HN N NH2
H O
O
HO2C
56
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC
HN NH 0 CONH2
O O
HOO O 0 H N H NH
-CO(CH2)8CH(CH3)2
NH O O
C17 O=~ NH CO2H
N
HN 0 H
HOZC' ~O HN
0
HN N NH2
YIH O
0
HOaC
HO2C
HN NH O CONH2
0 O
H0 00 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
0 C18 O~ NH CO2H
N
HN O H
HO2 C' O HN
0
HN N NH2
- 4
H O
0
HO2C
HO2C
NH CONH2
HN 0 0 O
H0 O O 0 N N N N-CO(CH2)sCH(CH3)CH2CH3
H H
NH 0 0 C19 O~ NH CO2H
N 14,
HN 0 H
HOZC' O HN
~/\ O
HN N NH2
H O
O
HOzC
57
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC -"'r NH CONHZ
HN O
O 0
HO ~O 0 0 N N N N-CO(CHZ)8CH(CH3)2
NH 0 H H
O
C20 O:::::~ NH CO2H
N
HN 0 H
HOaC~O HN
O
HN H
NH2
4
H O
O
HO2C
HO2C
HN NH 0 CONH2
1 O O
HO O 0 0 N
I N-CO(CH2)8CH(CH3)CHZCH3
NH 0 H H
0 C21 O=~ NH CO2H
N
HN O H
H02C~0 HN
O
HN N NH2
H 0
0
T
H02C
0 NHZ
HO2G X(NHIO
0 CONH2
0 O N A~ HO O 0 0 H N H N-CO(CH2)sCH(CH3)CHZCH3
NH O 0
C22 O~ NH CO2H
N
HN 0 H
HOZC' O OH HN
0
HN N NH2
-4
H O
O
HO2C
58
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
O NH2
HOaC HN T~NHI~
O CONH2
O Q
HO' 00 0 N N N N-CO(CH2)8CH(CH3)2
NH O )~H H
O
C23 Q=~ NH CO2H
N
HN O H
H02C0 OH HN
O
HN N NH2
H O
O
HOzC
O NH2
HQ2C I ~
i
HN NH 0 CONH2
O O
HO~O O 0 N N N N-CO(CHz8CH(CH3)CHZCH3
NH O H H
O
C24 O) NH CO2H
N
HN O H
HOZC' O OH HN
~J\ O
HN N NH2
H O
O
HOZC
/ \
NH
HO2C
HN NH 0 CONH2
O O
HO' 00 O N N N N-CO(CH2)6CH(CH3)CH2CH3
NH O H O H
C25 O NH COZH
~ N
HN O H
HOzC' O OH HN
O
HN N NHZ
H O
O
HO2C
59
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C \ NH
HN NH O CONH2
O O
HO~O O O N N N N-CO(CHZ8CH(CH32
NH O H H
C26 0
O~ NH COZH
N
HN O H
HO2C0 OH HN
H 4-~ HN N NH2
H O
O
HO2C
/ \
HO2C NH
HN NH O CONH2
O O
H H
HO 00 0 N N-CO(CHaeCH(CH3)CH2CH3
C27 NH O O
O=~ NH CO2H /
N
HN O H
HOZC' O OH HN
0
HN N NH2
H O
O
HO2C
HO2C
HN NH p CONHZ
O 0
HO O 0 0 N N-CO(CH2)6CH(CH3)CHZCH3
NH 0 H H
O
C28 0=~ NH CO2H
N
HN 0 H
HOZC' O OH HN
~.-(\ O
HN N NHZ
N
H O
0
HO2C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOzC
HN NH O CONH2
O 0
HO' O 0 0 N N N N-CO(CH2)BCH(CH3)2
~f\NH 0 H H
0 C29 O~ NH CO2H
N
HN O H
HOZC' O OH HN
~(\ O
HN N NH2
H 0
0
HOzC
HOZC
HN NH 0 CONH2
0 0
HO' O 0 0 N N N N-CO(CH2)$CH(CH3)CHZCH3
NH 0 0
)~, H
C30 O) NH COZH
N
HN O H
H02C' ~O OH HN
v\ O
HN N NHZ
H O
O
HO2C
HOZC
HN NH 0 CONH2
O O Hy~ H
HO1O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
0 C31 O~ NH CO2H
N
HN O H
HO2C0 OH HN
HN N NH2
H 0
0
HOZC
61
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
HO' ~O 0 0 N N N NH
-CO(CH2)8CH(CH3)2
~/\ H H
NH 0 p
C32 O) NH C02H N
HN O H
HOZC' O OH HN
~/\ O
HN N NHZ
O
0
HO2C
HO2C
HN NH O CONH2
O O
HO 1 O O O N N N N-CO(CH2)8CH(CH3)CHzCH3
NH 0 H H
O
C33 O=~ NH CO2H
N 14
HN 0 H
H02C' O OH HN
0
HN
I N NHa
H O
0
HOaC
0 NH2
HO2C V,," O
N NH CONH2
H
O
HOI 00 0 N N H N N-CO(CHZ)6CH(CH3)CH2CH3
NH 0 H H
O
C34 O) NH CO2H
N
HN 0 H
HOZC' ~O OH { HN
O
HN
I N NH2
H O
0
HO2C
62
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
O NHZ
HOaG HN NH O CONHz
O O
HO ~O O 0 N N N N-CO(CH2)$CH(CH3)Z
~f\NH 0 H H
O
C35 O=~ NH CO2H
N
HN O H
HO2C0 OH HN
HN N NH2
H O
O
HO2C
O NH2
HO2C NH
HN 0 CONH2
O O
HO ~O 0 0 H N H N-CO(CHa)$CH(CH3)CH2CH3
NH 0 O
C36 O~ NH CO2H
N
HN 0 H
HOaCO OH HN
HN N NH2
H O
0
HOzC
~ NH
HOzC
NH CONH2
HN O O O
H0~0 0 0 N N N N-CO(CHa)6CH(CH3)CH2CH3
NH 0 H O H
C37 O NH COzH
~ a N
HN ~=O H
HOZC' ~O OH HN
HN N NHZ
H O
O
HO2C
63
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HO2C
HN NH 0 CONH2
O O
HO' O O O N N N N-CO(CHZ)BCH(CH3)2
NH O H H
C38
O=~ NH CO2H
N
HN O H
HOaC' ~O OH HN
O
HN N NHa
H O
O
HO2C
NH
HOZC
HN NH 0 CONH2
O O
HO O O O N N N NH
-CO(CHZ)8CH(CH3)CH2CH3
NH O H H
C39
O=~ NH COzH
N
HN O H
HO2 C' O OH HN
O
HN N NH2
H O
O
HOzC
HO2C
NH CONHZ
HN O O O
HO' O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C40 0=~ NH COaH
HN 0 H
HOZC' O OH HN
\_J\ O
HN N NH2
H O
0
HO2C
64
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC
HN NH 0 CONH2
0 O
HO' O 0 0 N N N N-CO(CH2)8CH(CH3)a
NH 0 H H
O
C41 O~ NH COaH
N
HN O H
HOZC~O OH HN
HN N NH2
N
H O
O
HOaC
HO2C
HN NH O CONH2
~ O O
HO O 0 0 H N H NH
-CO(CHZ)8CH(CH3)CHZCH3
NH 0 O
C42 O) NH CO2H
N
HN O H
HOaC' ~O OH HN
O
HN N
NHz
4
H O
O
HO2C
HOZC
NH CONH2
HN O O 0
HO 10 0 0 N N HY~ H
N N-CO(CH2)6CH(CH3)CH2CH3
~f\NH 0 H H
O
C43 O~ NH COzH
N
HN O H
HO2C' O OH HN
HN N NH2 --~~~
H 0
O
OZC
H
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HOZC "-)y HN NH O CONH2
O O
HOO O 0 N N N N-CO(CHZ)8CH(CH3)2
NH 0 H H
0 C44 O~ NH COaH
N
HN 0 H
HOaCO OH HN
O
I NHa
HN N-
H O
0
HOZC
HO2C
HN NH O CONH2
O O
HO O O O N N N N-CO(CHZ)8CH(CH3)CH2CH3
NH 0 H H
0 C45 O~ NH CO2H
N
HN ~=O H
HOZC' O OH HN
O
HN N NH2
I H 0
O
HO2C
0 NH2
HOzC XT(NHO
O CONH2
0 O
O 0 O N N N N-CO(CH2)6CH(CH3)CHZCH3
NH O H H
O
C46 O~ NH CO2H
N
HN 0 H
HOZC' O HN
~1\ O
HN N NH2
H O
0
HO2C
66
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
O NH2
HO2C I
HN NH O CONH2
O O
00 O N N N N-CO(CH2)8CH(CH3)2
NH O H 0 H
C47 O=~ NH CO2H
N
HN ~=O H
HOZC' O HN
O
HN N NH2
O
O
HO2C
O NH2
HO2C I ~
/
HN NH O CONH2
O O
00 O N N N N-CO(CH2)8CH(CH3)CH2CH3
NH O H H
O
C48 O=~ NH CO2H
N
HN ~=O H
HOZCO HN
O
HN
I N NH2
H O
O
HO2C
NH
HO2C
HN NH O CONH2
H -CO(CH2)6CH(CH3)CH2CH3
O 0 O 0 O N -,k H N
NH O 0
C49 O NH COZH
~ N
HN ~=O H
HO2 C' O HN
~J\ O
HN N NHZ
H 0
O
HO2C
67
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOzC NH
HN NH 0 CONH2
0 0
O 0 )~N N N N-CO(CH2)8CH(CH3)Z
NH p H H
C50 0
O=~ NH COZH
N
HN O H
HOZC~O HN
O
HN N NHa
H O
O
HO2C
NH
HO2C
HN NH O CONH2
0 0
O O O N N N N-CO(CH2)8CH(CH3)CH2CH3
NH
C51 O H H
0
O~ NH CO2H
N
HN O H
HOZCO HN
O
HN N NH~
H O
O
HO2C
H02C
HN NH O CONH2
0 O
O O O N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C52 O=~ NH COZH
N 14,
HN 0 H
HO2 C' O HN
~/\ O
HN N NH~
H 0
O
HO2C
68
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH 0 CONH2
O O
O O 0 N N N N-CO(CH2)8CH(CH3)2
NH O H H
O
C53 O~ NH CO2H
N
HN O H
HOZC~O HN
HN N NH2
H 0
0
HO2C
HO2C
HN NH O CONH2
O O
~O 0 0 N N N NH
-CO(CH2)8CH(CH3)CHZCH3
_)~
NH 0 H H
O
C54 0~ NH CO2H
HN O H
H02C' ~O HN
HN N NHZ
H
O
HOzC
HO2C --'- T
NH CONH2
HN ~-O O O
~O O O N N N N-CO(CHZ)6CH(CH3)CHZCH3
NH 0 H H
O
C55 0) NH COpH
N
HN O H
HOyC' O HN
~/\ O
HN N NH2
H 0
O
HOzC
69
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC
HN NH O CONH2
O O
O O O N N N N-CO(CH2)gCH(CH3)2
NH 0 H H
0 C56 O=~ NH COzH
N
HN O H
HOZC' ~O HN
0
HN N NH2
H O
0
HO2C
HO2C
NH CONH2
HN O O O
~O O O N N N N-CO(CHz)8CH(CH3)CH2CH3
H
NH 0 H
0
C57 O=~ NH CO2H
N
HN O H
HO2C' O HN
~/\ O
HN N NH2
H 0
0
HO2C
0 NH2
H02C I
HN NH O CONHZ
O 0
~O O 0 N N N N-CO(CH2)sCH(CH3)CH2CH3
NH 0 H
0 C58 O=~ NH CO2H
N
HN ~=O H
HOZC' ~O HN
0
HN
I N NHZ
H O
0
HO2C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
O NH2
H02C HN NH 0 CONHa
O O
H -CO(CH2)8CH(CH3)2
O O O N N N
NH O O
C59 O=~ NH COaH
N
HN O H
HOzC' O HN
O
HN N NHa
H O
O
HOzC
O NHa
HO2C V,,, HN
NH CONH2
O O
O O O N N N N-CO(CH2)8CH(CH3)CH2CH3
NH O H H
O
C60 O) NH CO2H
N
HN ~=O H
HO2C' ~O HN
~/\ O
HN N NHZ
H O
O
HOzC
NH
HOzC
HN NH O CONH2
O O
-CO(CH2)6CH(CH3)CH2CH3
4=0 O O N N N NH
C61 NH O H 0 H
O=~ NH CO2H
N
HN O H
HOzC' ~O HN
O
HN N NH2
H O
O
HO2C
71
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HOzC
NH CONH2
HN O
O O
00 O N N N
H -CO(CH2)8CH(CH3)2
C62 NH O O
O=~ NH CO2H
N
HN O H
HO2C' O HN
\-/\ O
HN N NHa
- 4
H O
O
HO2C
~ NH
HO2C
HN NH O CONH2
O O
O O O N N N N-CO(CH2)8CH(CH3)CH2CH3
NH O H O H
C63 O NH COzH
~ N
HN O H
HOZC' O HN
~(\ O
HN N NH2
- 4
H O
O
HOzC
HO2C
HN NH 0 CONH2
O O
O O O N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C64 O=~ NH COZH
N
HN 0 H
HOzC' O HN
~/\ O
HN N NH2
H O
O
HO2C
72
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC
HN NH 0 CONH2
O O
O O O N H N-CO(CH2H )8CH(CH3)2
NH O 0
C65 O=~ NH COaH
N
HN O H
HOzC' O HN
0 H HN N NHZ
H O
0
HO2C
H02C
NH
HN O CONHZ
0 O
~O 0 0 N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
O
C66 O=~ NH COaH
HN a H
HO2 C' O HN
0
HN
I N NH2
H O
O
HO2C
HO2C
HN NH 0 CONH2
o a
00 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C67 O=~ NH CO2H
N
HN O H
HOzC' ~O HN
~(\ 0
HN
I N NH2
H a
O
HO2C
73
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C
HN NH O CONH2
O O
00 O N N N N-CO(CHz)8CH(CH3)2
NH O H H
0 C68 O=~ NH COZH
N
HN O H
H02C0 HN
O
HN N NHZ
H O
O
HO2C
HO2C
HN NH O CONH2
O O
4=00 0 N N N N-CO(CHa)8CH(CH3)CHZCH3
NH 0 )~H H
0 C69 O=~ NH CO2H
N
HN O H
HO2 C' O HN
0
HN N NH2
H O 4
O
HOZC
HO2C
NH CONH2
HN O 0 O
H2NOC~O O 0 N N N N-CO(CH2)sCH(CH3)CH2CH3
NH 0 H H
0 C72 O=~ NH COZH
N
HN 0 H
HO,C' 0 HN
O
HN N NH2
H 0
0
HO2C
74
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
HO~H
O 0 O H N H N-CO(CHZ)6CH(CH3)CH2CH3
NH O 0
C73 0=~ NH COzH
N .4:
HN ~=O H
HOzC' O (CH2)4NH2 HN
0
HN N NHZ
H O
0 HO2C
HOZC --),
NH O CONH2
HN O O
HO~O O O
I N N-CO(CHZ)6CH(CH3)CH2CH3
NH 0 0
~11 H
C74 O=~ NH CO2H
N
HN O H
HOZC' O HN
\-/\ O
HN N
H O
O
HO2C
HO2C
NH CONH2
HN O O O
HO~O O 0 N H N-CO(CHz
H )6CH(CH3)CH2CH3
NH 0 0
C75 O) NCH3 CO2H
N
HN O H
HOzC' O HN
~J\ O
HN N NHZ
H O
O
02C
H
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC COaH
NH
HN O 0 O
HO' O 0 0 N N-CO(CHa)6CH(CH3)CH2CH3
~-(\ H H
NH 0 0 C76 O=~ NH CO2H
N
HN O H
HO2C O HN
0
HN N NH2
O
O
HO2C
H02C
NH CONH2
HN 0 0 O
~O 0 0 N N-CO(CH2)6CH(CH3)CH2CH3
H2NOC, H H
~(\
NH 0 0
C77 O NH CO2H
~ N
HN O H
HOZCO (CH2)4NH2 HN
NHa
HN N
H O
O
HO2C
H02C COZH
NH
HN 0 O O
H2NOC, 0 0 0 N N
-CO(CH2)6CH(CH3)CHZCH3
H H
~--(\
NH 0 0
C78 O=~ NH CO2H
N
HN ~=O H
HOZC' O HN
v\ O
HN N NHz
H O
O
HO2C
76
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
H02C CO2H
NH
HN O O 0
HO~O 0 0 N N N N-CO(CH2)6CH(CH3)CHZCH3
NH 0 H H
0 C79 O=~ NCH3 C02H N
HN 0 H
HOZC' O HN
0
HN N NHz
H O
0
HO2C
HOaC CO2H
NH
HN O 0 0
HO~O 0 0 N N N N-CO(CH2)66CH(CH3)CH2CH3
NH 0 H H
0
C80 O=~ NCH3 CO2H
N
HN O H
HO2 C' O HN
0
HN N
H O
O
H02C
HOzC CO2H
NH
HN 0 O O
HO~O 0 0 N N N N-CO(CH2)6CH(CH3)CHZCH3
NH 0 H H
0 C81 O=~ NH CO2H
N
HN 0 H
HOzCO HN
O
HN N
H O
O
HO2C
77
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
H~NOC' O O O N N N N-CO(CH2)6CH(CH3)CHzCHg
NH 0 H H
O
C82 0~ NCH3 COZH
N
HN O H
HO2C' O HN
\-/\ O
HN N
H O
O
HO2C
HOzC CO2H
NH
HN O O O
H2NOC~O O O N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C83 O) NCH3 CO2H
N
HN ~=O H
HO2C' ~O HN
0
HN N
Yt N H O NHa
0
HOZC
H02C CO2H
NH
HN O 0 O
HZNOC~O O O N N N N-CO(CH2)6CH(CH3)CHZCH3
NH 0 H H
O
C84 0=~ NH CO2H
N
HN ~=O H
HOZC' ~O HN
0
HN N
H 0
0 HO2C
78
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
## Compound
HO2C
NH CONH2
HN O O O
C' O 0 0 N N-CO(CH2)6CH(CH3)CH2CH3
HaNO H
1 H
NH 0 O
C85 O=~ NCH3 CO2H
N
HN O H
HOaC' O HN
O
HN
H 0 NH2
0 HOZC
HO2C
HN NH O CONH2
O
0
HO~O 0 0 N Hy~ H N
H -CO(CH2)6CH(CH3)CH2CH3
"--~~
NH 0 O
C86 O=~ NCH3 COzH
N
HN 0 H
HOaC' O HN
O
HN N
H 0
0 HO2C
HO2C COzH
NH
HN 0 O O
H2NOC~0 O O N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
~ I\
C87 O NH NH2 O NCH3HO
CONH2
~
HN O H
HOZCVH HN
HO HN N
0
HO2C 79
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O 0
HZNOC~O O 0 N H N-CO(CH2)6CHZCHZCH3
H
NH O 0
C88 0~ NH2 NCH3HOCONH2
N
HN 0 H
HOaC~O HN
O
HO HN N
H O
0
HO2C
HO2C
HO2C
NH
HN O O HZNOCO O O :OO
-CO(CH2)6CH(CH3)CH3
N
NH O I C49 O~ NH2 NCH3H0 NH2
N
HN O H
HOZCO HN
O
HO HN N
H O
0
HO2C
HO2C HO2C
NH
HN 0 O O
H2NOC' O 0 O N H N
H -CO(CHz)6CH(CH3)CH2CH3
~(\
NH O 0
C90 O~ NH2 NCH30 CONH2 2
N
HN 0 H
HOZCO HN
O O
HN N
N
H
O
H02C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO~C
NH
HN O O O
H2NOC~O 0 0 N N N N-CO(CH2)6CH2CH2CH3
NH O H H
C91 O~ NH2 NCH3HO CONH2 N
HN 0 H
HOZC' O HN
O
HN N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
HzNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C92 O) NH2 NCH3HO CONH2 ~ I\
N
HN O H
HO2 C' O HN
O
HN N N
O
H
O
HO2C
HO2C HO2C
NH
HN 0 0 O
HZNOC~O 0 O N N N N-CO(CH~)6CH(CH3)CH2CH3
NH 0 H H
O
C93 O~ NH2 NCH3 CONH2
N
HN O H
HOZCO HN
O
0 HN N
CH3 H 0
O
HOzC
81
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O O
H2NOC~0 O O N N-CO(CH~}6CH2CH2CH3
NH 0 H H
0 C94 O~ NH2 NCH3 CONH2 N
HN O H
HOZCX-= O HN
0
0 HN N
CH3 N
0
O
HOZC
HO2C HO2C
NH
HN O O O
H2NOC, 00 0 N N-CO(CH~)6CH(CH3)CH3
~-- NH 0 H H
0 C95 O~ NH2 NCH3 CONH2 N
HN O H
H02C~0 HN
0
O HN
CH3 H O
O
HO2C
CH3
HO2C HOZC
NH
HN O 0 0
H2NOC~0 0 0 N H N
H -CO(CHa)6CH(CH3)CH2CH3
NH 0 0
C96 O~ NH2 NCH3 CONH2 N Is,
HN 0 H
HOaC~O HN
0
O HN N N
CH3 H 0
O
HO2C
82
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HOZC
NH
HN O O O
HZNOC~O O O N H H N
H -CO(CHZ)6CHaCH2CH3
NH 0 0
C97 O~ NH2 NCH3 CONH2
N
HN O H
H02CX-= 0 HN
O
O HN N
CH3 H O
0
HOaC
HOZC CH3 HO2C
NH
HN O 0 0
HzNOC~O 0 0 N N-CO(CH2)6CH(CH3)CH3
-it, NH 0 H H
O
C98 O~ NH2 NCH3 CONH2
N
HN O H
HOzCO HN
O
O HN N N
CH3 H O
O
HOaC
CH3
HO2C HO2C
NH
HN O 0 0
H2NOC~0 0 0 N H N-CO(CHZ)6CH(CH3)CH2CH3
NH 0 p
H
C99 O~ NH2 NCH3 CONH2 N
HN 0 H
HOZCVH HN
HO HN N
O
HO2
C
83
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HO2C
NH
HN O O O
HZNOC~O O 0 H N H N-CO(CHZ)6CHZCHaCH3
NH 0 O
C100 O~ NH2 NCH3 CONH2
N
HN O H
HOZC~O HN
O
HO HN - N--~
N
H O
O
HO2C
HOZC CH3 COzH
NH
HN O O O
HZNOC~O 0 0 N H N
H -CO(CH~)sCH(CH3)CH3
NH 0 O
C101 O=~ NH2 NCH3 CONH2
N
HN ~=O H
HO2 C~O HN
O
HO HN N
,,H O
HOaC
HO2C COaH
NH
HN 0 O O
H2NOC~0 O O N H N
H -CO(CH2)6CH(CH3)CH2CH3
NH 0 O
C102 O~ NH2 NCH3 CONH2
N
HN 0 H
HOZC>-~ VN HN
HO HN N
O
O
2C
H
84
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C HOZC
NH
HN O O O
H2NOC~0 O O N N-CO(CH~)6CH2CH2CH3
NH 0 H H
O
C103 O~ NH2 NCH3 CONH2
N
HN 0 H
HOpCO HN
O
HO HN N
H 0
0
HOaC
HO2C HOaC
NH
HN O O O
HZNOC' O O O N N N
N -CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C104 O~ NH2 NCH3 CONH2
HN 0 H
HOZC~O HN
O
HO HN N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
HZNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH,CH3
NH 0 H H
C105 O ~
~ NH2 NCH3 CONH2
N
HN O H
HOZC' O HN
O
HN
N N
H 0
O
HOzC
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOzC HO2C
NH
HN O O O
HZNOC, O 0 0 H N H N-CO(CH2)6CH2CHzCH3
NH 0 0
C106 O~ NH2 NCH3 CONH2 N
HN O H
HO2 CO HN
0
HN N
H O
0
H02C
HOZC HO2C
NH
HN O 0 0
HZNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C107 O=~ NH2 NCH3 CONH
2 N
HN O H
HOZCO HN
0
HN N N-~
H O
HO2C
CH3
HOZC HOaC
NH
HN O 0 0
0
HpNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H O H
C 108 0~ NHa NCH3 CONH2
N
HN O H
HOZC' O HN
O
HN N
H 0
O
OzC
H
86
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C COZH
NH
HN O O O
H2NOC~0 O 0 N H H N
H -CO(CHz)6
C109 O CH2CHZCH3
NH O 0
~ NHZ
NCH3 CONH2 HN O H
HOzC= O HN
O
HN N
N
H 0
O
HO2C
CH3
HO2C HO2C
NH
HN O O O
H2NOC~O 0 0 N H N-CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C110 O) NH2 NCH3 CONH2
N
HN 0 H
HOZC' O HN
\-/\ O
HN N
H
O 0
HOZC
CH3
HO2C HO2C
NH
HN O O O
H2NOC~00 0 N N N N-CO(CH~)6
C111 O CH(CH3)CH2CH3
NH 0 H H
O
~ NH2
NCH30 CONH2 N
HN O H
HOaCO HN
O
HO HN N
H 0
O
HOaC
87
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HO2C
NH
HN O 0 O
H2NOC' O 0 0 N N N-CO(CH2)6CHzCH,CH3
~-(\ H
NH O 0
CONH2 I\
C112 O~ NH2 NCH3HO
N
HN O H
HOzC>-= O HN
O
HO HN N
H
O O
HOzC
CH3
HO2C HO2C
NH
HN O 0 H2NOCO O O :OO
N-CO(CH~)6CH(CH3)CH3
NH C113 ONH2 NCH3H0 NHa I\
N
HN O H
HO2 C~O HN .
O
HO HN N
H 0
O
HO2C
CH3
HO2C HOZC
NH
HN O 0 O
HZNOC' O 0 0 N N-CO(CH2)sCH(CH3)CH2CH3
~(\NH O H H
O
CONH2 I\
C114 0~ NH2 NCH3HO
N
HN O H
H02C' O HN
O
HN N N
H 0
O
HO2C
88
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HOzC
NH
HN 0 O O
HaNOC~O 0 0 N N H N N-CO(CH2)6CH2CH2CH3
NH 0 H H
O
C115 O~ NH2 NCH3H0 CONH2
N
HN 0 H
HO2 CO HN
0
HN N--~
H O
0
HO2C
CH3
HO2C HOZC
NH
HN O 0
O
HZNOC~O 0 0 N N H N N-CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C116 O~ NH2 NCH3H0 CONH2 N
HN 0 H
HO2C' O . HN
O
HN N
H 0
0
HO2C
CH3
HO2C HO2C
NH
HN 0 0
H2NOCO 0 0 N N H N N-CO(CH~)6CH(CH3)CH2CH3
NH 0 H H
O
C117 O~ NH2 NCH3H0 CONH2
HN 0 H
HOZCO HN
0
HN N
H 0
0
HOZC
89
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HOzC
NH
HN O O 0
HZNOC, O O O N H N
H -CO(CH2)6CH2CH2CH3
~-(\
NH 0 p
CONH2 I\
Ci 18 O~ NH2 NCH3HO
N
HN O H
HOZCO HN
O
HN N
H O
O
HO2C
CH3
HO2C HO2C
NH
HN O O 0
H2NOC' O 0 0 N N-CO(CHZ)6CH(CH3)CH3
~(\NH 0 H H
O
C119 O) NH2 NCH3HO CONHZ (\
N
HN O H
HOaC ~O HN
\-/\ O
HN
H O
O
HO2C
HO2C HOzC
NH
HN O O O
H2NOC, O 0 0
N -CO(CHZ)6CH(CH3)CHaCH3
N N N
H H
~(\NH 0 O
C120 O~ NH2 NCH3HO CONH2
N
HN O H
HOaC' O HN
\-/\ O
HN N
H O
O
HO2C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOzC
NH
HN O O 0
HZNOC' O 0 0 N H N
H -CO(CH2)6CHZCHZCH3
~(\
NH O O
C121 O~ NH2 NCH3H0 CONH2 \
N
HN O H
HOzC' VH HN
0
HN N
O
4
HO2C HOZC HO2C
NH
HN 0 0 0
HZNOCO 0 0 N N-CO(CH2)6CH(CH3)CH3
NH 0 H H
0
CONH2
C122 O~ NH2 NCH3HO
N I
HN O H
HOZC' O HN
\-J\ 0
HN N
H O
0
HOzC
HO2C HOzC
NH
HN O O 0
HZNOC~O 0 0 N N-CO(CHZ)6CH(CH3)CHZCH3
NH 0 H H
O
C123 O~ NH2 NCH3HO CONH2 ~ I\
N
HN O H
HOZCO HN
O
HO HN N
H O
O
HO2C
91
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC HOaC
NH
HN O O O
HzNOC~O 0 0 N N N N-CO(CH2)6CH2CH2CH3
H H
NH 0 O
CONH2 I\
C124 O~ NH2 NCH3HO
HN O H
HOZC~O HN
O
HO HN N
H O
O
HO2C
HO2C HO2C
NH
HN O O
H2NOC~0 0 0 N N N N-CO(CH~)6CH(CH3)CH3
NH 0 H H
O
CONH2 I\
C125 0~ NH2 NCH3HO
N
HN O H
HOzCX-= O HN
O
HO HN N
H O
O
HO2C
CH3
HO2C HO2C
NH
HN O O
HZNOC~O 0 0 N H H N-CO(CH2)6CH(CH3)CHZCH3
NH O 0
H
CONH2 I\
C126 O~ NH2 NCH3HO
N
HN O H
HO2C>-~ VN HN
HO HN N
O
C
HO2
92
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC CH3 HO2C
NH
HN O O 0
H,NOC~O 0 0 H N H N-CO(CH2)6CHzCH2CH3
NH O O
/ I\
CONH2
C127 O~ NH2 NCH3HO
N
HN O H
HO2C>-~ VH HN
HO HN N
O
HO2C
CH3
HO2C HO2C
NH
HN O O 0
HaNOC' O 0 0 N N N-CO(CH2)6CH(CH3)CH3
~(\ H H
NH O O
CONH2 / I\
C128 O~ NH2 NCH3HO
N
HN O H
HO2 C~O HN
O
HO HN N
N
H O
O
HO2C
HO2C CH3 HOZC
NH
HN O O 0
HZNOC~O O 0 N H N-CO(CH2)6
C129 CH(CH3)CH2CH3
NH O O
H
NCH CONH2 N
O~ NH2 3
HN O H
HOaC>-= O HN
O
HO HN N
H O
O
HO2C
93
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HO2C
NH
HN
O O O
~
H2NOC' O 0 0 H N H H N-CO(CH216CHaCHzCH3
~(\
NH 0 O
C130 O NH2 NCH3 CONH2 X
I N
HN O H
HO2 CO HN
O
HO HN N
H 0
O
HOZC
CH3
HO2C HO2C
NH
HN O O O H
H2NOC~0 0 0 N N-CO(CH2)6CH(CH3)CH3
NH 0 H H
O
C131 O~ NH2 NCH3 CONH2
N
HN O H
HOZC~O HN
O
HO HN N
H 0
O
HO2C
HOZC HO2C
NH
HN O O O
HZNOC' /~O O 0 N N H N N-CO(CH2)6CH(CH3)CH2CH3
~1\NH 0 H O H
C132 O~ NH2 NCH3 CONH2
N
HN O H
HOZCO HN
O
HO HN N
H O
O
HO2C
94
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOZC
NH
HN O 0 O
H2NOC~O 0 0 N H N-CO(CH2)6CH2CH2CH3
H NH O O
C133 O~ NH2 NCH3 CONHZ
N
HN O H
HOzCO HN
O
HO HN N
N
H O
0
HO2C
HO2C HOaC
NH
HN O O O
H2NOC, O 0 0 N N-CO(CH2)6CH(CH3)CH3
N NH 0 H H
~(\
O
C134 O) NH2 NCH3 CONH2
N
HN 0 H
HO2 CO HN
O
HO HN N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
HZNOC~O O 0 N N H N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
Z
C135 O~ NH2 NCH3 CONH
HN O H
HOzCO HN
O
HN N N
H O
O
HO2C
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C H0ZC
NH
HN O O O
HZNOC' O O O N H N H
N -CO(CH2)6CH2CH2CH3
NH 0 H H
O
C136 O~ NH2 NCH3 CONHZ
N
HN O H
HO2 CO HN
O
HN N
H O
0
HOaC
HOZC HOZC
NH
HN O 0 O
H2NOC, /~O 0 0 N N N N-CO(CH2)6CH(CH3)CH3
~(\NH 0 H O H
C137 O~ NH2 NCH3 CONH2
N
HN O H
HOZC' O HN
\-/\ 0
HN
H O
0
HO2C
CH3
HO2C HO2C
NH
HN O O O H
H
HaNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 )~H O H
C138 O~ NH2 NCH3 CONH2 N
HN O H
HO2 C' O HN
0
HN N
H O
O
HO2C
96
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C CH3 HO2C
NH
HN O O O
H2NOC' {~O 0 0 N N H N-CO(CH2)6CHZCH2CH3
~(\
NH 0 p
C139 O~ NH2 NCH3 CONH2
N
HN 0 H
HO2 C= O HN
0
HN N
N
H O
0
HO2C
HO2C CH3 HO2C
NH
HN O 0 O
H2NOC~0 0 0 N N N N-CO(CH2)sCH(CH3)CH3
NH 0 O
C140 O~ NH2 NCH3 CONH2
N
HN O H
HOZC' O HN
0
HN N
H 0
O
H02C
HO2C CH3 HOZC
NH
HN O 0 O
H2NOCI0 0 0 N N
H p N -CO(CH2)6CH(CH3)CH2CH3
NH 0
C141 O~ NHZ NCH3 CONHZ
N
HN O H
H02C>-~ 0 HN
0
HN N N-~
CH3 H O
O
HO2C
97
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
HO2C HO2C
NH
HN O O O
H2NOC~O 0 0 N N N N-CO(CH2)6CH2CHzCH3
NH 0 p
C142 O~ NH2 NCH3 CONH2
N
HN ~=O H
HOZC~O HN
0
O HN N
CH3 H 0
0
HOaC
CH3
HO2C HO2C
NH
HN 0 0 O
HaNOC~O 0 0 N N N NH
-CO(CH2)6CH(CH3)CH3
H H
NH 0 p
C143 O~ NH2 NCH3 CONH2 N
HN O H
HOZCHl=O HN
0 H
0 HN N
CH3 N
0
0
HO2C
HO2C HO2C
NH
HN O 0 0
H2NOC0 0 0 N N N N-CO(CH2)6CH(CH3)CHaCH3
H H
NH 0 p
C144 O~ NH2 NCH3 CONH2 N
HN ~=O H
HOZC>-~ O HN
0
0 HN N
CH3 OH 0
HO2C
98
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
H02C HO2C
NH
HN O O 0
HaNOC~O 0 0 N H H N
H -CO(CH2)6CHzCH2CH3
NH 0 O
C145 O~ NH2 NCH3 CONH2
N
HN 0 H
HO2 CO HN
O
O HN N
CH3 N
0
O
H02C
HO2C H02C
NH
HN O 0 0
H2NOC' O 0 0 N N-CO(CH2)6CH(CH3)CH3
~(\ N
NH 0 O
H
C146 O) NH2 NCH3 CONH2
N
HN ~=O H
HOZC~O HN
O
0 HN N
CH3 N
0
O
HO2C
HO2C HO2C
NH
HN O O O
HO~O O 0 N N H N N-CO(CH~)6CH(CH3)CH2CH3
NH 0 H O H
CONH2
C147 O~ NH2 NCH3HO
HN O H
HO2C0 HN
O
0 HN N N
CH3 H O
O
HO2C
99
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC HO2C
NH
HN O O O
HO~O 0 0 H H
NH O N-CO(CHz)6CH2CH2CH3
N H
H
CONH2
C148 O NH2 NCH3HO
~
HN ~=O H
HOZCO HN
O
O HN N
N
CHs H
O
O
HO2C
HOZC HO2C
NH
HN O 0 O
HO~O 0 0 N N-CO(CH~)sCH(CH3)CH3
NH 0 H H
O
CONH2 I\
C149 O NH2 NCH3HO
~ N
HN ~=O H
HOaC~O HN
O
O HN N
CH3 N
O
O
HO2C
CH3
HO2C HO2C
NH
HN 0 O 0
HO' O 0 0 N N-CO(CH2)6CH(CH3)CH2CH3
~(\NH 0 H H
O
CONH2 I\
C150 O NH2 NCH3HO
I N
HN O H
HOaCO HN
O
O HN N
CH3 H O
O
HO2C
100
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
CH3
H02C H02C
NH
HN O O O
HO, O 0 0 N N-CO(CH~)sCH2CH2CH3
~(\NH 0 H H
O
C151 O~ NH2 NCH3HO CONH2 I\
N
HN O H
H02CX-~=0 HN
O
O HN N
CH3 OH O
H02C
CH3
HO2C HO2C
NH
HN O O O
HO~O O 0 N N N N-CO(CH2)6CH(CH3)CH3
H H
NH 0 O
CONH2 f\
C152 OI ~ NH2 NCH3HO
N
HN O H
H02G>_~ 0 HN
O
O HN N N
CH3 H O
O
H02C
CH3
HO2C HO2C
NH
HN O O O
HO~O 0 0 N H N-CO(CH2)6CH(CH3)CH2CH3
H "----~~
NH 0 O
C153 O~ NH2 NCH3HO
CONH2 (\
N
HN O H
HO2 CP=O HN
O
O HN N
CH3 H O
O
HO2C
101
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C CH3 HO2C
NH
HN O 0 O
HO~O 0 0 H N H N-CO(CH2)6CHZCHaCH3
NH O 0
C154 O~ NH2 NCH3HO CONH2 ~ I\
N
HN 0 H
HOZC~O HN
O
O HN N
CH3 H 0
O
HO2C
HO2C CH3 HO2C
NH
HN O O O
HO' O 0 0 N N N-CO(CH~)sCH(CH3)CH3
H H
NH O 0
C155 O~ NH2 NCH3H0 CONH2
N
HN O H
H02CX-= 0 HN
O
O HN N N
CH3 H 0
O
HO2C
HO2C HO2C
NH
HN O 0 0
HZNOC' O 0 0 N N N-CO(CH~)g
CH(CH3)CH2CH3
~(\ H H
NH 0 -) , 0
~ I\
C180 O= NH HO CONH2
NH2
N
HN O H
HOZCX-VH HN
O HN N
CH3 0
HOZC 102
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C HOaC
NH
HN 0 O HZNOCO O O :OO
-CO(CH2)6CH2CH2CH3
N
NH O C181 NHNH HO NH2 ~ I\
N
HN O H
HOZCX-VN HN
O HN N
CH3 O
HO
2C
HOaC HOaC
NH
HN O 0 0
H,NOC, O 0 O N N N
N -CO(CH2)6CH(CH3)CH3
NH O H H
O
C182 O~ NH2 NH HO CONH2
N
HN O H
HN
HOzCVN
HN N
CH3 0
HO2C
HO2C HO2C
NH
HN O O O
H2NOC, 00 0 N N-CO(CHz)6CH(CH3)CH2CH3
~(\NH O H H
C183 O~ NH2 NH HO CONHz / (\
N
HN O H
HOzCX-= O HN
O HN N
CH3 H O
O
HOzC
103
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O O
HZNOC' ~O O O N H N
CHZCHzCH3
H -CO(CH2)6
~(\
NH 0 0
~ NH NH HO CONH2 / I\
C184 p
N ~
HN O H
HOaC~O HN
O
O HN N
CH3 H O
O
H02C
HO2C HO2C
NH
D O O 0
HzNOC~O O O N N-CO(CHZ)6CH(CH3)CH3
NH O H H
O
C185 O~ NH2 NH HO CONH2
N ~
HN ~=O H
HOZC~VN HN
O HN N
CH3 O
H
O2C
/ NHZ
HO2C 0
HN NH O CONHZ
O O
HO' ~O O O N N N N-CO(CH2)6CH(CH3)CHZCH3
C189 v\NH O H O H
0=~ NH C02H ~ I \
N ~
HN O H
HO2 C' ~O CONH2 HN
~/\ O
HN N-
I NHZ
H 0
O
02C
H
104
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
~ \
/
NH2
HOZC O
HN N0 CONH2
0 0
HO I 00 0 N N N N-CO(CHZ)8CH(CHA
C190 NH O H 0 H
O~ NH CO~H
N
HN O H
HOZC' O CONH2 HN
HN N NH2
H O
O
HOZC
~ '
/ NHZ
HO2C O
HN NH 0 CONH2
I O O
HO O 0 0 N N-CO(CH2)$CH(CH3)CH,CH3
C191 NH O H H
o
O=~ NH CO2H
N
HN O H
HOZC' O CONH2 HN
\-/\ O
HN N NHZ
H O
O
HOaC
NH2
HO2C O
NH CONH2
HN O O
HO' O 0 O. N N N N-CO(CH2)6CH(CH3)CH2CH3
C192 NH 0 H H
O
O=~ NH C02H
N
HN O H
H02C0 CONH2 HN
HN N NH~
H O
O
HO2C
105
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
f~ =
NH2
HOaC O
HN NH O CONH2
O O
H0- O 0 0 N N-CO(CH2)8CH(CH3)2
C193 NH 0 H o H
O=~ NH COzH
N
HN ~=O H
HO2C' ~O CONH2 HN
NHZ
HN N--
H O
O
HO2C
NH2
HOZC O
HN NH O CONH2
0 0 H HO0 O 0 O N N-CO(CHz)8CH(CH3)CHaCH3
C194 NH 0 H 0 H
O=~ NH CO2H
N
HN ~=O H
HOaC' O CONH2 HN
~/\ O
NH2
HN N
H O
O
HOaC
NH
HOzG
NH CONH2
HN 0 0 O
HO1O O O N N-CO(CHZ)6CH(CH3)CH2CH3
C195 NH 0 )~H 0 "
O~ NH COzH
N
HN O H
HOZC' O CONH2 HN
~(\ O
HN N NH2
H O
0
HO2C
106
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
\ .
NH
HOZC
HN NH 0 CONH2
O O
HO O 0 0 N N-CO(CH2)8CH(CH3)2
C196 NH 0 H p H
O~ NH COZH
N
HN 0 H
HO2C' O CONOHZ HN
HN N--~ NH2
1 N O
O
HOZC
/
NH
HO2C
HN NH O CONH2
O O H HOO 0 O )~N N N N-CO(CH2)8CH(CH3)CH,CH3
C197 NH 0 H H
o
O~ NH CO2H ~ .
N
HN O H
HOZC' O CONH2 HN
t_J\ O
HN N NHa
1 H O
O
HO2C
NH
HO2C
HN NH 0 CONH2
O O
HO' O 0 0 N N N N-CO(CH2)sCH(CH3)CHZCH3
C198 NH O H O H
O~ NH CO2H
N
HN O H
HOZCO CONH2 HN
O
HN N NHZ
H O
O
HO2C
107
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
. ~ \
NH
HOZC
HN N" O CONH2
O O
HOO O O N N-CO(CH2)BCH(CH3)2
C199 NH O " 0 "
0=~ NH COZH
N
HN 0 H
HO2 C' O CONH2 HN \-/\ O
HN N NH~
H O
O
HO
~C
I \
NH
H
HO2C
N" CONH2
O
O O H
HO' 00 O N N N N-CO(CH2)$CH(CH3)CH2CH3
C200 NH O H H
O
O=~ NH COzH
N
HN ~=O H
HOZC' O CONH2 HN
O
HN
I N NH2
I H O
O
HO2C
HO2C
NH CONH2
HN O O O
HO' O O O N N N N-CO(CH2)6CH(CH3)CHZCH3
H H
NH O 0
C201 O=~ NH CO2H ~ ( \
N
HN O H
HOZC' O CONH2 HN
~(\ O
HN N NHZ
H 0
O
HO2C
108
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC
HN NH p CONH2
O O
HO' ~O O O N N )r( N N-CO(CHa)8CH(CH3)2
NH 0 H H
O
C202 O=~ NH CO2H
N
HN ~=O H
HOzCO CONH2 HN
O
HN
I N NHa
I H O
0
HO2C
HO2C
NH CONH2
HN O O 0
HO' ~O 0 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
O
C203 O=~ NH CO2H
N
HN O H
HOZC' O CONH2 HN
~J\ 0 =
HN N NH2
H O
0
HO2C
H02C
HN NH 0 CONHZ
O 0
HO' ~O 0 0 N N H
lr( N N-CO(CH2)6CH(CH3)CH2CH3
~f\NH 0 H H
O
C204 O=~ NH CO2H
N
HN ~=O H
HOZC' O CONH2 HN
O
HN N NH2
0
0 HO2C
109
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
NH CONH2
HN O O 0
HO' O 0 O N H N-CO(CHz)aCH(CH3)2
\-/\ H
NH 0 0
C205 O) NH COZH
N
HN O H
FiOzC' O CONH2 HN
HN N NHa
H O
O
HO2C
HO2C
NH CONH2
HN O 0 0
HO' O 0 0 N N N N-CO(CH2)aCH(CH3)CH2CH3
~f\ H H
NH 0 0
C206 O=~ NH CO2H I\
N 14
HN O H
HOaC' O CONHa HN
0
HN N NH2
H O
0
HO2C
HO2C
NH CONH2
HN O 0 O
HO' O 0 0 N N N N-CO(CH2)6CH(CH3)CHaCH3
~/\ H H
NH 0 0
C207 O NH CO2H I~
/ N
HN O H
H02C' O CONH2 HN
~J\ 0
HN N NH2
H O
0 HO2C
110
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
HO1O 0 0 N N-CO(CHz)BCH(CH3)Z
~ )~H H
NH 0 0
C208 O=~ NH CO2H
N
HN ~=O H
HO2 C' ~O CONH2 HN
O
HN N NH2
I H O
O
HO2C
HO2C
HN NH 0 CONH2
0 O
HOIO 0 0 )~H N H N-CO(CHZ)8CH(CH3)CH2CH3
NH 0 0 C209 0 NH CO2H
N
HN 0 H
HOZC' ~O CONH2 HN
O
HN N NH2
I N O
H .
O
HO2C
HOaC
HN NH O CONH2
0 O
HO 1 O 0 0
1 N N-CO(CHz)6CH(CH3)CH2CH3 "r( ~J\ H H
NH 0 0
C210 O=~ NH CO2H
N
HN O H
H02C0 CONH2 HN
O
NH2
HN N 4
0 H O
HO2C
111
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC
HN NH O CONH2
O 0
HO' O 0 0 N N N N-CO(CH2)BCH(CH3)2
NH 0 H H
O
C211 0=~ NH CO2H
N
HN ~=O H
HOZC' O CONH2 HN
~(\ O
HN N NHZ
H O
0
H02C
HOaC
HN T~NH1 O CONHZ
0 0 'J'~ HO' O 0 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH O H H
O
C212 O=~ NH CO2H
N
HN ~=O H
HO2 C' O CONH2 HN
~(\ 0
HN N NH2
H O
O
HO2C
HO2C
HN NH O CONH2
O 0
HzNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH O H H
O
C213 O=~ NH CO2H
N
HN ~=O H
HOZC' O HN
0
HN N NHZ
H O
0
HOaC
112
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C
HN NH O CONH2
O O
HZNOC' O 0 0 N N N N-CO(CH2)8CH(CHg)2
NH 0 H H
O
C214 O=~ NH C02H N
HN ~=O H
HOpC' 0 HN
~/\ O
HN N NHz
H O
0
HO2C
HOZC
NH CONHa
HN O
0 0
H2NOC~O 0 0 N N N N-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
O
C215 O=~ NH CO2H
N
HN ~=O H
HOZC' 0 HN
~J\ O
HN N NH2 - 4
H O
0
HO2C
H02C
NH CONH2
HN O 0 0
H2NOC'~ O 0 0 N N lr( N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 )~H H
O
C216 O=~ NH COaH / I
N
HN ~=o H
HO2C' 0 HN
~/\ O
HN N NH2
H O
O
HO2C
113
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C
HN NH CON2
O O
HaNOC~O 0 0 N N N N-CO(CH2)8CH(CH3)2
NH 0 H H
0 C217 O=~ NH CO2H
N
HN o H
H02C' ~O HN
~/\ O
HN N NHZ
H O
O
HOzC
HOaC
HN NH 0 CONH2
O O
H2NOC ~O 0 0 N N N NH
-CO(CH2)8CH(CH3)CH2CH3
NH 0 H H
O
C218 0=~ NH CO2H
N
HN O H
HOZC' ~O HN
HN N NHZ
H O
O
HO2C
H02C
HN NH 0 CONHZ
O O
HZNOC' O 0 0 N N N N-CO(CH2)BCH(CH3)2
NH 0 H H
0 C219 O~ - NH COzH
N
HN ~=O H
H02C' O HN
0
HN N NHz
H O
0 HO2C
114
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O H H2NOC' ~O O O N N N N-CO(CHz)8CH(CH3)CH2CH3
H H
NH 0 0
C220 0=~ NH CO2H
N
HN O H
H02C 0 HN
O
HN N NH2
N 0
H
0
HOaC
HOzC
HN NH 0 CONH2
O O
H2NOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
NH 0 H H
O
C221 O~ /NH CO2H
N
HN /--0 H
HOZC' O HN
~/\ O
HN N NH~
H 0
0
HO2C
HOaC
NH CONH2
HN O O O
H2NOC' ~O 0 0 N N N N-CO(CHp)8CH(CH3)2
~f\NH 0 H H
O
C222 O=~ NH CO2H
N
HN O H
H02C' O HN
~/\ O
HN N NH2
H O
0
HO2C
115
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOaC
NH CONH2
HN O O 0
HZNOC' O O O N -r( N N-CO(CHz)BCH(CH3)CH2CH3
NH O ~,, H
O
C223 O=~ NH Co2H
N
HN O H
H02C0 HN
HN N NHZ
H O
0
HO2C
NH
H02C
HN NH 0 CONH2
1 0 0
H2NOC O 0 0 N N-CO(CHZ)6CH(CH3)CH2CH3
C224 NH O H H
o
O=~ NH CO2H
N
HN 0 H
HOZC' O HN
\-/\ O
HN' ~ N NH~
TI( H O
0 HO2C
NH
HO2C
NH CONH2
HN O 0 0
HZNOC O 0 0 N N N N-CO(CHz)8CH(CH3)2
C225 NH O H H
o
O~ NH CO2H
N
HN O H
HO2C' O HN
~/\ O
HN' ~ N NHa
H
~''I(
O
0 HOZC
116
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HOZC
HN NH O CONHa
O O
HZNOC' O 0 0 N N-GO(CH2)BCH(CH3)CHZCH3
C226 NH O H 0 H
O~ NH COZH
N
HN O H
H02C' ~O HN
~(\ 0
HN N NH2
O
YIH -1- 4
0
HO2C
NH
HO2C
NH CONH2
HN O 0 0
HZNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
C227 NH O H H
0 O=~ NH COaH
N 14-
HN O H
HO2C' ~O HN
~/\ O
NHa
HN N 4
N O
0 HO2C
NH
HO2C
HN NH O CONH2
1~ 0 O
H H
HZNOC 0 0 0 N N-CO(CH2)eCH(CH3)2
C228 NH 0 0 ~
O~ NH COZH / I
N
HN O H
HOZC' ~O HN
\-/\ O
HN' , N NHZ
TI'(
H O
0
HO2C
117
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH
HO2C
HN NH O CONH2
O O
HZNOC~O 0 0 N N N N-CO(CHZ)8CH(CH3)CH2CH3
C229 NH 0 H H
O
O=~ NH CO2H
N
HN 0 H
HOZC' O HN
~(\ O
HN N NH2
H O
O
HO2C
NH2
HO2C O
NH CONH2
HN 0
0 O H Y~ H
H2NOC 10 O 0 N N N N-CO(CHZ)6CH(CH3)CH2CH3
C230 NH 0 H H
O
O~ NH COzH
N
HN 0 H
H02C' O HN
NHZ
HN N 4
H O
0
HO2C
NH2
H02C 0
HN NH p CONH2
0 O
H2NOC O 0 0 ) ) H N N-CO(CHZ)8CH(CH3)2
C231 NH 0 H H
O
O) NH CO2H
N
HN O H
HOZC' O HN
~/\ O
NHZ
HN' ~ N 4
TI'( H O
0
HO2C
118
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NH2
H02C O
HN NH 0 CONH2
0 0
H~NOC' O 0 0 N N N-CO(CH2)8CH(CH3)CH2CH3
C232 NH O )~,, p H
O=~ NH CO2H
N ~
HN 0 H
HOZC' O HN
~/\ O
HN N NH2
N O
H
0
H02C
\
NH2
HO2C 0
NH CONH2
HN O 0 0
H2NOC10 0 0
: A~ N N N N-CO(CHZ)sCH(CH3)CH2CH3
C233 NH O H O H
O=~ NH CO2H
N ~
HN 0 H
HOzC' O HN
~/\ O
HN N NH2
N
H O
0
HO2C
NH2
HO2C O
HN NH O CONH2
O O
HZNOC' O 0 0 N N-CO(CH2)8CH(CH3)z
C234 NH O H p H
O) NH CO2H
N
HN 0 H
HO2C~0 HN
HN N NHZ
N O
H
0 O2C
H
119
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
~
~
NH2
HOzC 0
HN NH O CONH2
O O
HZNOC' O 0 0 N
N N N-CO(CH2)8CH(CH3)CH2CH3
C235 NH 0 " 0 "
i -,--( -1
O=~ NH CO2H
N
HN O H
HOzCO HN
O
HN N NHz
H 0
0
HOZC
NH2
HO2C 0
NH CONHz
HN O O 0
H2NOC0 0 0 N N N N-CO(CHz)6CH(CH3)CHzCH3
C236 NH 0 " "
NH CO2H
0 O~ NH
2
N
HN O H
HOzC' O HN
O
HN N NHz
H
O 0
HOpC
NH2
HO2C O
HN N" 0 CONH2
O O
HzNOC' O 0 0 N N N N-CO(CH2)8CH(CH3)z
C237 NH 0 H H
0 O=~ NH
z NH COzH
N
HN 0 "
HOzC' O HN
~.(\ O
HN N
NHz
H O
0
HO2C
120
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
NHz
HOzC 0
HN TI~(NHz' 0 CONH2
0 0
H2NOC0 0 0 N N-CO(CHz)8CH(CH3)CHzCH3
C238 NH 0 " 0 H
O) NH2 NH COzH
N
HN O H
HOzC' O HN
O O
HN N NHz
N
H
O
HOZC
HO2C HOzC
NH
HN O O O
HO~O 0 0 N N N-CO(CHz)6CH(CH3)CHzCH3
)~H H
NH 0 0
O~ NH CONHz I ( ~
C259
N
HN O H
HOpC' O HN
v\ O
H HN N NHz
H O
O
HO2C
HOzC HOzC
NH
HN O O O H
HOO 0 0 N N N-CO(CH2)8CH(CH3)2
H H
NH O 0
C260 O~ NH CONH2 N
HN 0 H
HOzC' O HN
~/\ O
H HN N NHz
H O
O
O2C
H
121
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O O
HO' O 0 0 N H N-CO(CH2)BCH(CH3)CH2CH3
H H
NH O O
C261 O=~ NH CONH2
N
HN O H
HOZC~O HN
O
HN N NHz
H 0
O
HO2C
HO2C HOaC
NH
HN O 0 O
HO' O 0 0 N H N-CO(CH2)6CH(CH3)CHZCH3
N H H
~f\
NH 0
O
C262 O=~ NH CONH2 N
HN 0 H
H02C' HN
~/\ O
NH~
HN N 4
YIH O
O
HO2C
H02C HOzC
NH
'~'y
HN O 0 O H HO' O 0 0 N NH
-CO(CH2)BCH(CH3)2
~f\ H H
NH 0 p
C263 O~ NH CONH2
HN 0 H
H02C' O HN
~J\ O
HN N NH~
H O
0 HOzC
122
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O
H
HO' O 0 0 N H N-CO(CH2)8CH(CH3)CH2CH3
~J\NH 0 0
C264 O NH CONH2
/ N
HN O H
HOZC' O HN
~/\ O
HN N NHz
H O
0
HO2C
HOZC
x1NH~
C
ONH2
O O
C~0 0 0 N H N-CO(CH2)sCH(CH3)CH2CH3
H2NO H
NH 0 0 C265 O~ NH2 NCH3 CO2H
N
HN O H
HO2 C>-~O HN
0
0 HN N
CH3 OH O 4
HOaC
HO2C
T~NHTIT CONH2
HN O O 0
H2NOC\ 0 0 0 N H N-CO(CH2)6CH2CH2CH3
NH 0 0
H
C266 O~ NH2 NCH3 COzH
N
HN 0 H
HOzCX-VH HN
O HN N CH3 0
OzC
H
123
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HOZC
HN NH O CONH2
O O
C' ~O O O N N-CO(CHZ)6CH(CH3)CH3
H2NO H H
_--(\
NH O 0
C267 O~ NH2 NCH3 CO2H
N
HN ~=O H
HOaC~O HN
O
O HN N
CH3 H O
O
HO2C
HOZC
HN NH O CONH2
O O
H~r~ H
H2NOC~00 O )~N N
N N-CO(CH~)6CH(CH3)CHzCH3
NH O 0
C268 O~ NH2 NCH3 CO2H
N
HN O H
HO2 CO HN
O
O HN N N
CH3 H O
O
HOZC
HO2C
HN NH O CONH2
O O
HzNOC~O O O N N-CO(CHz)6CHZCH2CH3
NH O H H
O
NCH CO2H
C269 O~ NH2 3
N
HN O H
HOzC~O HN
O
0 HN N
CH3 OH O 4
HO2C
124
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HOaC
HN NH O CONH2
O O
H2NOC' 00 0 N N-CO(CHZ6CH(CH3)CH3
NCH3 COZH
O 11H C270 O~ NH2
N
HN O H
HOZC~O HN
O
HN N
CH3 H O
HO2C
HO2C
HN NH v 0 CONH2
O 0
H2NOC, ~O 0 0 N N-CO(CH2)6CH(CH3)CHZCH3
~--(\ H H
NH O O
C271 O~ O NHZ NCH3 COzH ~ I
N
HN O H
HOZC~O HN
O H
O HN CH3 OH O
HO2C
HO2C
NH CONH2
HN O 0 O
H2NOC' ~0 0 0 N N-CO(CH~)6CH2CH2CH3
-(\ H H
NH O NCH3 CO H N
C272 0~ NH2
HN O
H
DHOZCO HN
O
H
O HN N
CH3 H O
O
HO2C
125
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
NH CONH2
HN V 0 O O
HaNOC~0 0 0 N NH
-CO(CH2)6CH(CH3)CH3
H A~ Y~
H
NH 0 O
C273 O=~ NH2 NCH3 CO2H
N
HN 0 H
HO,CX_~=O O HN
O HN N
CH3 H 0
O
HO2C
HOzC
HN NH O CONH2
O O
HZNOC~O O O N N-CO(CH2)6CH(CH3)CH2CH3
H N
NH O 0
O~ NH2 NCH3 CO2H I\
C274
N
HN ~=O H
HO2 C~O HN
O
0 HN N
N
O
CH3 H
O
HO2C
HO2C
N CONH2
O O
HzNOC~O 0 O H N N N-CO(CH~)6CH2CH2CH3
NH O 0
3 3 CO2H
C275 O~ NH2 NCH
N
HN O H
HO2 C~O HN
O
O HN N
CH3 H O
0
HO2C
126
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
C~O 0 0 N H N-CO(CHZ)6CH(CH3)CH3
HzNO H
NH 0 O
C276 O~ NH2 NCH3 COzH
N
HN O H
H02CX-= O HN
0
0 HN N
CH3 OH O
HO2C
HOzC
HN NH V CONH2
O 0
H2NOC~0 0 0 N N-CO(CH2)6CH(CH3)CH2CH3
H H
NH 0 0
C-,277 O=~ NH CO2H ~ I\
N
HN ~=O H
HOzC~-tO HN
0
\ HN N
H O NH2
O
HOaC
HO2C
HN NH O CONH2
O O
HZNOC~O O 0 H N H N-CO(CH2)sCHzCHZCH3
NH 0 O
C278 O~ NH CO2H
N
HN O H
HOZCk~=O HN
0
\ HN N
H O NH2
O
HO2C
127
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
NH CONH2
HN V 0 0 O
0 0 N H N-CO(CHZ)6CH(CH3)CH3
HzNOC H
NH 0 O
C279 O=~ NH CO2H
N
HN O H
HO2C~0 HN
0
\ HN N
H O NHZ
O
HO2C
HO2C
NH CONH2
HN O O O
HaNOC~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
NH 0 0
L-,280 O=~ NH CO2H ~ I\
N
HN O H
HO2C~0 HN
0
0 HN N4
H 0 NH2
O
HO2C
HO2C
HN NH O CONH2
O O
HzNOC~O 0 0 N N
-CO(CH2)6CH2CH2CH3
H H
NH O 0
C281 0=~ NH COpH
N
HN O H
H02C~0 HN
0
0 HN N
'flN
O NH2
O
HOzC
128
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH 0 CONH2
O O
HZNOC' 1 O 0 0 H
I N H N-CO(CH2)6CH(CH3)CH3
~(\
NH O 0
C282 0=~ NH CO2H \
N
HN O H
HO2 C~O HN
0
0 HN N
H O NH2
O
HO2C
HO2C
NH CONH2
HN V 0 0 0
HZNOC' ~O 0 0 N N N-CO(CH2)6CH(CH3)CHaCH3
~ (\ H H
NH 0 0
C283 O=~ NH CO2H
N
HN O H
HOaC~O HN
0
0 HN N
H O NH2
O
HO2C
HO2C -~-)y NH CONH2
HN V 0 0 O
H
HZNOC~O 0 0 N H N
-CO(CH2)6CH2CH2CH3
NH 0 0
C284 O=~ NH CO2H \
H
HN ~=O
HOaC~O HN
0
\ HN N
H NHZ
0
HO2C
129
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HOaC
NH CONH2
HN v 0 O O
H2NOC~O 0 0 N H N-CO(CH2)6CH(CH3)CH3
H
NH O O
C285 O=~ NH CO2H Z'
N
HN ~=O H
HO2C~0 NN
O
\ HN N
H O NH2
O
HOaC
HO2C
HN NH O CONH2
O O
)~H H
H2NOC~0 O O N N-CO(CH2)sCH(CH3)CH2CH3
NH O 0
C286 O=~ NH CO2H
N
HN O H
HOZCO HN
O
H
\ HN N
H O NHz
O
HO2C
HOZC
NH CONH2
HN O O O
HZNOC' ~O O O N N-CO(CHZ)6CHZCHZCH3
~(\ H H
NH O 0
C287 O=~ NH CO2H
N
HN O H
HOZCO HN
O
\ HN N
H O NH2
O
02C
H
130
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC
HN NH O CONH2
O O
H2NOC' 1 O 0 0 N Hy~ H
H H
N-CO(CH2)6CH(CH3)CH3
NH O 0 C288 O=~ NH CO2H
N
HN O H
HO2C~O HN
0
0 HN
H O NH2
O
HOaC
HOzC CO2H
NH
HN V 0 O O
H2NOC' IO 0 0 H N N N-CO(CHz)6CH(CH3)CH2CH3
~(\
NH O 0
CONH2 I\
C289 O~ NCH3HO
N
HN ~=O H
HOZC~O OH HN
O
\ HN N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
HzNOC~O 0 0 N N-CO(CH2)6CH2CH2CH3
H N
NH 0 0
CONH2 I\
C290 O=~ NCH3HO
N
HN O H
H02C~0 OH HN
O
\ HN N
N 4
H O
O
I
HOzC
131
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC HO2C
NH
HN V 0 O 0
HZNOC~O O O H N N N-CO(CH2)6CH(CH3)CH3
NH O 0
C291 O~ NCH3H0 CONH2 N
HN O H
HO2 C~O 1OH HN
O
\ HN N
N
H 0
HOaC
HO2C COzH
NH
HN V 0 0 0
HzNOC~O 0 0 N H N
H -CO(CH2)6CH(CH3)CH2CH3
NH O 0
~ I\
C292 O~ NCH3HOCONH2
N ~
HN O H
HOZG~O OH HN
O
0 HN N
H O
O
HOZC
HO2C HO2C
NH
HN V 0 O 0
HaNOC~O 0 0 N N
-CO(CH2)6CH2CHZCH3
H H
NH O 0
C293 O NCH3HOCONH2
N ~
HN O H
HOZCO OH HN
O
\ HN N
I N
H O
O
HO2C
132
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
-- ~ .
HN V 0 O O
HZNOC~O 0 0
I N N-CO(CH2)6CH(CH3)CH3
H H
NH O HO 0
C294 0=~ NCH3 CONH2
N
HN ~=O H
HOzC~O OH HN
O
O HN N
N
H O
0
HO2C
HO2C CO2H
NH
HN O O O
H2NOC' O 0 0 N N-CO(CHz)6CH(CH3)CHaCH3
~(\ H H
NH 0 0
C295 O=~ NCH3 O CONH2 \
HN ~=O H
HOZC~O OH HN
O
\ HN N N
H O
0
HOzC
HOzC HO2C
NH
HN O O O
HZNOC~O 0 0 N H H N-CO(CH2)sCH2CHzCH3
H
NH O 0
CONHa
C296 O) NCH3HO
N
HN ~=O H
HOZC~O OH HN
O
\ HN N N
H O
O
HOzC
133
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOaC
NH
H N-CO(CHz)6CH(CH3)CH3
HN O O H2NOC~O O O :OO
NH O HO C297 NCH3 NH2
N
HN O H
HOZC~O OH HN
0
\ HN N
N
H O
O
HOzC
HO2C CO2H
NH
'~y
N N-CO(CHZ)sCH(CH3)CH2CH3
HN O O H2NOC~O 0 0 :OO
H
NH O HO C298 0~ NCH3 NH2
N
HN 0 H
HOzC>-tO OH HN
0 H
\ HN N
H O
O
HO2C
HO2C HO2C
NH
H N-CO(CHa)6CH~CH2CH3
HN O O H2NOC~0 0 0 :OO
NH O C299 ~ NCHaHO NH2 I\
N
HN O H
H02C~0 OH HN
O
\ HN N
N
H O
O
HO2C
134
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
H NCO(CHa)6CH(CH)CH3
HN O O HzNOC~O O O :Oo
NH O HO G.,300 0~ NCH3 NH2
N
HN 0 H
HO2 C~O O--I HN
O
\ HN N
N
H O
O
HO2C
HOaC COZH
NH
-CO(CH2)gCH(CH3)CH2CH3
NH
HN O O H2NOC~0 0 0 :OO
NH O C301 ~ NCH3H0 NH2 ( \
N
HN ~=O H
HOzC~O CONH2 HN
O
\ HN N
H O
O
HOzC
HO2C HOpC
NH
-CO(CHZ)6CHZCHZCH3
NH
HN O O HZNOC~O 0 0 :OO
NH O C302 =~ NCH3H0 NHZ
N
HN ~=O H
HO2 C~O CONH2 HN
O
H
\ HN N
H O
O
02C
H
135
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2 G H02C
NH
HN O O
H
H N-CO(CHZ)sCH(CH3)CH3
HZNOC~O 0 0 H :OO
NH O C303 NCH3H0 NH2
N
HN O H
HOpG~O CONH2 HN
O
\ HN N
H O
O
HO2C
HO2C COzH
NH
HN V 0 O 0
H2NOC' ~O 0 0 N H N
H -CO(CHI)sCH(CH3)CHZCH3
~(\
NH O 0
C304 O=~ NCH3 O CONH2 I\
N
HN O H
HO2 C~-tO CONH2 HN
O
\ HN N
H O
O
HOzC
HO2C HO2C
NH
HN v O 0
HZNOC~O 0 0 N H N
H -CO(CH2)sCHZCH2CH3
NH O 0
C305 O~ NCH3HO
CONH2 N 141
HN O H
HOZC~O CONH2 HN
O
\ HN N
N
H O
O
HO2C
136
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN V 0 O 0
H2NOC~0 0 0 H N H N-CO(CH2)6CH(CH3)CH3
NH O O
C306 O=~ NCH3HO CONH2
N
HN O H
HOaC~O CONH2 HN
O
\ HN N
N O
H
O
HO2C
HO2C CO2H
NH
HN V 0 O O
H2NOlO 0 0 N H N-CO(CH2)6CH(CH3)CH2CH3
H NH O 0
C307 O NCH3H0 CONH2
~ N
HN ~0 H
HO2 C~O CONH2 HN
O
\ HN N
N O
H
O
HO2C
HO2C HO2C
NH
HN ' O 0 HzNOC0 rOO
-CO(CH2)6CH2CH2CH3
NH C30$ NCH3H0 NH2 \
N
HN O H
HOpC~O CONH2 HN
O
\ HN N
H 0
O
O2C
H
137
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
--'r .
HN V 0 O O
HZNOC' O 0 0 N H N-CO(CH2)6CH(CH3)CH3
H
NH O HO O
C309 O=~ NCH3 CONH2
N
HN ~=O H
HOZC~O CONH2 HN
O
\ HN
N
H O
0
HO2C
HO2C CO2H
NH
HN 0 O HZNOC~O 0 0 :OO
H -CO(CH2)6CH(CH3)CH2CH3
NH O C310 NCH3H0 NH2 I\
N
HN ~=O H
H02C~0 CONH2 HN
O
\ HN
N
H O
O
HO2C
HOzC HOZC
NH
HN O O H2NOC' O 0 0 :OO
N-CO(CHZ)6CH2CH2CHH
NH O C311 NCH3H0 NH2 I\
N
HN ~=O H
HO2 C~O CONH2 HN
O
\ HN N
N O
H
O
HOZC
138
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOaC
NH
N-CO(CH2)6CH(CH3)CH3
HN O O H2NOC~O O O :Oo
H
NH HO C312 NCH3 NH2
N
HN O H
HO2 C~O CONH2 HN
O
\ HN N
N
H O
O
HO2C
HO2C CO2H
NH
N-CO(CH,)6CH(CH3)CHZCH3
HN O O HZNOC~O O O :OO
NH 0 HO C313 NCH3 NH2
N
HN O H
HO2C~0 HN
O
\ HN N
H O
O
HO2C
HOaC HO2C
NH
N-CO(CH2)6CH2CH2CH3
HN O O HZNOCO :OO
NH O C314 NCH3H0 NH2 I\
N
HN O H
HOZC~O HN
O
\ HN N N
H 0
Q
02C
H
139
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOzC
NH
HN O O O
H NOC
2 O 0 0 N N-CO(CH2)6CH(CH3)CH3
H H
NH O HO 0 C315 O~ NCH3 CONH2
N
HN O H
HOZC~O HN
O
\ HN N
H O
O
HO2C
HO2C CO2H
--~y NH
HN V 0 O O
H2NOC~O 0 0 N N N-CO(CH2)6CH(CH3)CH2CH3
H
NH O HO 0 ~
C316 O=~ NCH3 CONH2 ~ I
N ~
HN ~=O H
HOZC~O HN
O
\ HN
N O
I N
H
O
HO2C
HOzC HOZC
NH
HN V 0 O O
HZNOC~O 0 0 N NH
-CO(CH2)6CH2CH2CH3
H H
NH O 0
CONHz ~ I\
C317 O=~ NCH3HO
HN O H
HO2C~O HN
O
\ HN N
N
H O
O
HO2C
140
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN V 0 O 0
H2NOC~0 0 0 H N H N-CO(CH2)6CH(CH3)CH3
NH O 0 C318 0=~ NCH3 H O CONH2
N
HN O H
HOZC~O HN
O
\ HN N
N O
H
O
HO2C
HO2C COaH
NH
HN V 0 O O
HZNOC' ~O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
~(\ H H
NH O 0
C319 O NCH3HO CONH2
~
HN ~=O H
HO2C~O HN
O
\ HN N
H O
0
HO2C
HOZC HO2C
NH
HN V 0 0 H2NOCO O O rOO
-CO(CHZ)6CHzCH2CH3
NH O HO C320 NCH3 NH2
N
HN ~=O H
HOZC~O HN
\ HN N N
H O
O
HOZC
141
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Compound
HO2C HO2C
NH
HN V 0 O H2NOCO O O :Oo
H N-CO(CH2)6CH(CH3)CH3
NH O C321 O=~ NCHsHO NH2
N
HN O H
HOZC~O HN
O
; HN N
H O
O
HO2C
HOZC CO2H
NH
HN 0 O O
HZNOC~O 0 0 N N N
H -CO(CHa)6CH(CH3)CHaCH3
NH O 0
CONH2
C322 O=~ NCH3HO
N
HN O H
HO2C~0 HN
O
\ HN N
N O
H
O
HO2C
H02C HO2C
NH
HN O O O
HpNOC~O 0 0 H N N-CO(CHz)6CH2CH2CH3
H
NH O 0
C323 O=~ NCH3HO CONH2
N
HN 0 H
HOzC~O HN
O
\ HN N
H 0
0
HO2C
142
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HO2C
NH
HN O O O
HZNOC' O 0 0 N H N
H -CO(CH2)6CH(CH3)CH3
NH O HO O
C324 O=~ NCH3 CONHa
N
HN O H
HO2C~O HN
O
\ HN N
N
H O
O
HOzC
HO2C CO2H
NH
HN O O 0
HO' O 0 0
-CO(CHZ)6CH(CH3)CH2CH3
I N NH
~(\ H H
NH O 0
C325 O~ NH2 NCH3HOCONH2
N
HN ~=O H
HOZCX-~ O HN
O
; HN N
N
H O
O
HOaC
HO2C HO2C
NH
HN O O O
HO~O 0 0 N N-CO(CHz)6CH2CH2CH3
H H
NH O 0
C326 O~ NH2 NCH3HOCONHa
N '41
HN O H
HOZCO HN
O
0 HN N
N
H O
O
OaC
H
143
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC HO2C
NH
HN O O O
HO' =O 0 0 N N-CO(CHZ)6
CH(CH3)CH3
'-(\ H H
NH 0
HO
O
C327 O NH2 NCH3 CONH2
I N
HN O H
HOZCVN HN
H
\ HN N
O HOaC
HOzC CO2H
NH
HN O O O
O 0 0 N N-CO(CH2)6CH(CH3)CH2CH3
H H
NH O 0
C328 O= NH2 NCH3HO CONHZ
N
HN O H
HOaC>-tO HN
O
H
\ HN N N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
IO 0 0 N N-CO(CH~)6CHZCH2CH3
H H
NH O 0
C329 O NH2 NCH3HO CONHZ
I
HN O H
HO2 C~-tO HN
O O
\ HN N
N
H
O
HO2C
144
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C H02C
NH
HN O 0 O
O 0 O N H N-CO(CH2)6CH(CH3)CH3
H NH O O
C330 O~ NH2 NCH3HO CONH2
N
HN O H
HOzCO HN
O
\ HN N
H 0
O
HO2C
HO2C CO2H
NH
HN O O 0
O 0 0 N N N N-CO(CH~)6CH(CH3)CHaCH3
H H
NH O 0
C331 O~ NH2 NCH3HO CONH2
N
HN 0 H
HOzCO HN
O
\ HN N
N
H O
O
HOZC
HO2C HOzC
NH
HN O O O
H -CO(CH2)6CH2CH2CH3
O 0 0 N H N
NH O 0
C332 O~ NH2 NCH3HO
CONH2 N
HN O H
HO2 CV HN
\ HN N
O
H
OZC
145
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C H02C .
NH
HN O 0 O
O 0 O N N N N-CO(CH2)6CH(CH3)CH3
H H
NH O O
CONH2
C333 O~ NH2 NCH3HO
N
HN ~=O H
H02CX-= 0 HN
0
0 HN N
N
0
H
O
H02C
HO2C CO2H
NH
HN O O O
O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
NH 0 0
C334 O~ NH2 NCH3 O CONH2
N
HN ~=O H
H02C0 HN
O
\ HN N
N
H O
0
HO2C
HO2C HO2C
NH
HN O 0 O
O O 0 N N N-CO(CH2)6CH2CH2CH3
H NH O 0
C335 O~ NH2 NCH3HO CONH2
N
HN ~=O H
H02C~0 HN
O
0 HN N
H 0
0
HO2C
146
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
## Compound
HO2C HOaC
NH
HN 0 O O
O 0 0 N N-CO(CH2)6CH(CH3)CH3
H H
NH 0 O
C336 O NH2 NCH3HO CONH2
I N
HN O H
HO2C0 HN
O O
\ HN N
N
H
O
HO2C
HO2C CO2H
NH
HN 0 O O
O 0 O N N N N-CO(CH2)sCH(CH3)CH2CH3
H H
NH O 0
C337 O~ NH2 NCH3HOCONH2 I\
N
HN O H
HO2 C~O HN
O
\ HN N N
H O
O
HO2C
HO2C HO2C
NH
HN O O O
O 0 0 N N-CO(CH2)6CH2CH2CH3
N H
NH O 0
H
CONH2
C338 O~ NH2 NCH3O
N
HN O H
HO2 C>-~ O HN
O O
0 HN N
N
H
O
HOaC
147
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C HOaC
NH
HN O 0 O
IO 0 0 N H N-CO(CH2)6CH(CH3)CH3
NH O HO O
C339 O~ NH2 NCH3 CONH2
H HN 0
H
HO2C0 HN
0
O H
0 HN N
N
H
O
HO2C
HO2C
HN NH O H2NOC
O 0
O 0 0 rOIt N -CO(CH2)6CH(CH3)CHZCH3
H
H2NOC NH 0 C340 O~ NHZ NCH3 HO NH2
N
HN O H
HOZC~O HN
O
\ HN N N .
H O
O
HOzC
HO2C
NH CONH2
HN O O O
O 0 0 N N N-CO(CHz)6CH2CHaCH3
H
H2NOC NH O O
C341 O~ NH2 NCHsHO CONHz
N
HN O H
HO2C0 HN
O
\ HN N
H
0
HO2C
148
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
NH CONH2
HN O O O
O 0 0 N N-CO(CH2)61CH(CH3)CH3
H H
H2NOC NH O 0
C342 O=~ NH2 NCH3HO CONH2 N
HN ~=O H
HO2 C~O HN
O
\ HN N
N
H O
HO2C
HO2C
HN NH 0 HzNOC
O
0
O 0 0
N N N N-CO(CHZ)6CH(CH3)CHzCH3
0 N
H H
H2NOC NH O 0
C343 0 NH2 NCHsHO CONH2
HN ~=O H
H02C1\-t0 HN
O
\ HN
N N H O
0
HO2C
HOZC
NH CONH2
HN V 0 O O
~O 0 0 N N N NH
-CO(CHACHZCH2CH3
)~IH H
H2NOC NH O 0
C344 O~ NH2 NCH3H0 CONH2
HN O H
HOZC>-tO HN
O
\ HN Nl-~-
H -11 O
O
H
OzC
149
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH O CONH2
O O
O 0 0 N H N
-CO(CHa)6CH(CH3)CH3
H N A~ I J~
H2NOC NH O O
I~
C345 O=~ NH2 NCH3HO CONHa /
N
HN O H
HO2 C~O HN
O
\ HN N
N
H O
O
HN HOaC
HO2C
NH O H2NOC
O O
O 0 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
H2NOC NH 0 p
C346 O=~ NH2 NCH3HO CONHz
N
HN O H
HOaC~O HN
O
\ HN N
N
H O
HO2C
HO2C
NH CONH2
HN O O O
O 0 0 N N N-CO(CH2)6CH2CH
H 2CH3
HZNOC NH O 0
C347 O=~ NH2 NCH3HO CONH2
N
HN O H
HO2C~0 HN
O
0 HN N
N O
H
O
HO2C
150
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOzC
NH CONHZ
HN O O O
O 0 O N N N N-CO(CHZ)6CH(CH3)CH3
H H
H2NOC NH O 0
C348 O) NH2 NCH3HO CONH2
N
HN O H
HOaCO HN
O
\ HN N
H O
O
HO2C
HO2C
HN NH O O HzNOC O
O 0 0 N N N N-CO(CH2)6CH(CH3)CHZCH3
H2NOC NH O H H
O
C349 O~ NH2 NCH3HO CONHp
N
HN O H
HOZC~O HN
O
\ HN
H O
O
HO2C
HOzC
NH CONH2
HN O O O
~O O O N N N N-CO(CH2)6CH2CH2CH3
H H
H2NOC NH O 0
C350 O~ NH2 NCH3HO CONHZ
N
HN 0 H
HOZC~O HN
O
0 HN N
H O
O
HO2C
151
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOzC
HN NH O CONH2
Q O
O O 0 N N-CO(CH2)6CH(CH3)CH3
H H
H2NOC NH O HO 0 C351 Q=~ NH2 NCH3 CONH2
HN O H
HOaCO HN
O
\ HN N
N
H Q
HO2C
NH
2
NH2
O O
0
HO zo
O N 0
H O O
HN O L N N N Ny(CH2)8CHs
C352 HO
Q H H
O O O
0
NH O OH HN~
OH
HOO NH H 0 HN O N
N H
H
0 NH2
HO2C --), HN NH O CONH2
O O
O O 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
H2NOC NH 0 p
C353 O=~ NH COZH
N /
HN O H
HOzCO HN
O
HN N
H O
O
02C
H
152
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C CO2H
HN NH O O O
-CO(CHZ)sCH(CH3)CHaCH3
O 0 0 N 'J~, N N X NH
H H
H2NOC NH O 0
C354 0=~ NCH3 COaH
N
HN O H
HOZC' ~O HN
0
HN N
H 0
0 HO2C
HO2C C02H
HN NH O O
H -CO(CH2)6CH(CH3)CHZCH3
O O N N N
Y HO NH 0 O
C355 O~ NH2 NH COzH
N
HN O H
HOZC' O HN
~-J\ O 4\-~NHZ
HN N
H O
O
HOaC
HOzC
HN NH O CONH2
O O
O O O N N N N-CO(CH2)6CH(CH3)CH2CH3
H H
HO NH 0 O
C356 O~ NH2 NCH3 COZH
N
HN 0 H
HOZC' O HN
O --~,- NHz
HN N N
H O
O
HOaC
153
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH p CONH2
O 0
N -CO(CHz)yCH3
O O 0 N N N
HO NH O H 0 H
C357 O~ NH2 NH COzH
HN 0 H
HOZC' O HN
O O
HN N N
H
O
HO2C
HO2C
HN NH p CONH2
O 0
H -CO(CHZ)9CH3
O O O N H N
HO NH O 0
C358 0) NH2 NCH3 COzH
N
HN 0 H
HO2 C' O HN
O
HN N N
H O
O
HO2C
HO2C CO2H
HN NH O 0 O
O O O N N N N-CO(CH~)sCH(CH3)CH2CH3
H H
HO NH O 0
C359 O~ NH2 NCH3 COZH
N
HN 0 H
HO2 C' O HN
NH2
O O
HN N
N
H
O
HOZC
154
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C COZH
HN NH O O O
O O 0 EN N N N-CO(CHZ)9CH3
HO NH 0 H H
0 C360 O~ NHZ NH COZH
N
HN O H
H02C' O HN
~-J\ O
HN N N
H O
O
HO2C
HOaC CO2H
HN NH 0 O
O O O H H
N N N N-CO(CH2)sCH(CH3)CH2CH3
HO NH 0 H H
O
C361 O~ NH2 NCH3 CO2H
N
HN O H
HO2 C' O HN
O
HN N N
H O
O
HO2C
HO2C COZH
HN NH 0
O O
O O 0 N N N N-CO(CH2)6CH(CH3)CH2CH3
H2NOC NH 0 H O H
C362 O=~ NH2 NH CO2H
N
HN O H
HOZC' O HN
O NHZ
HN N
H O
O
HOzC
155
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO2C
HN NH 0 CONH2
O 0
H )6CH(CH3)CH2CH3
zl~O O O N H N-CO(CHz
H2NOC NH O 0
C363 0~ NH2 NCH3 COaH
N
HN O H
HOZC' O HN
0 NHZ
HN N N
H O
O
HO2C
H02C
HN NH O CONH2
O 0
O O O N H N-CO(CH2
H )1,CH3
H2NOC NH 0 0
C364 O~ NH2 NH COZH
HN ~=O H
HO2C' O HN
O
HN
N
O H O
HO2C
HOzC
HN NH 0 CONH2
O 0
O 0 O N N N N-CO(CH2)
6CH(CH3)CH2CH3
H H
H2NOC NH O 0
C365 O~ NH2 NCH3 CO2H
N
HN 0 H
HOZC' O HN
0
HN N
N
H O
O
HO2C
156
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HOZC CO2H
HN NH O O O
H -CO(CH~)6CH(CH3)CHZCH3
O 0 0 N H N
H2NOC NH O 0
C366 O~ NH2 NCH3 CO2H
N
HN ~=O H
H02C' O HN
0 ~NHZ
HN N --~
H O
O
HO2C
HO2C CO2H
HN NH O 0 0
O 0 O N N N N-CO(CH~)6CH(CH3)CHzCH3
H H
H2NOC NH O 0
C367 O~ NH2 NH CO2H
N
HN ~=O H
HO2C' O HN
O
HN N
H O
O
HOaC
H02C CO2H
HN NH O 0 0
O 0 0 N N N N-CO(CH~)6CH(CH3)CH2CH3
H H
H2NOC NH 0 0
C368 0~ NH2 NCH3 CO2H
HN 0 H
HOpC' VN HN
HN N
O HO2C
157
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Compound
HO
NH2
O O
O N N O O
H O O
HO HN O N H TI I N\ /(C
C369 H2)8CHa
0~ o " f
0 0
O 0 'll'~NH OH HN~ ~
101 OH
HO NH 0 HN O N
H
N N H H )L-~ O NH2
[0112] In one embodiment of the invention, each of R2*, RS*, R8*, Rlland R12
is H R9 is
OMe
~~ C02H ~ ''2,/~CO2H ,
or
and R13 is CH(CH2CH3)CH3. This embodiment provides a compound of Formula II.
H02C
NH
HN O O R2 O
O O N RI
R~~ N jj"~'N
H H
NH O R3 O
O NR5
N
HN O H
HO2C O HN
R8 0 Rs
R9* HN N
H O
O
HO2C
wherein R9* is H or OMe and Rl, R2, R3, R5, R6, R8, and Ri 1 are as previously
defined.
[0113] Table II provides exemplary compounds of Formula II.
158
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Table II
Compounds of Formula II
HO2C
NH
HN O 0 R2 0
00 0 jly N R'
Ril N jf~' N
NH 0 H R3 0 H
O NR5
N
HN O H
HO2C O HN
R8 0 Rs
R9* HN N
Y-IH 0
0
HO2C
# R 2 R3 RS R R8 R9* R
TII1 0 OH H +(CH2)3NH2 CH3 H
V,)J-NH2 0
T112 ~~/~CO2H =\'Y OH H +(CH2)3NH2 CH3 H ~~OH
0
T113 0 NH2 H -J-(CH2)3NH2 CH3 H ~~OH
V-IKNH2
OH
T114 0 .'~OH CH3 -~-(CH2)3NH2 CH3 H .~,.'~OH
~~NH2 O
T115 0 ~'~oH H CH3 CH3 H I QH
V~IkNH2 O
T116 ~~/~CO H NH2 H --(CH2)sNH2 CH3 H ~-~OH
z
~~o
OH
159
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R RS R 6 R8 R9 R11
TII7 0 NH2 CH3 +(CH2)3NH2 CH3 H ~Lr,~OH
\l-ANHz \s~~O
OH
T118 0 oH CH3 CH3 CH3 H ~,'~oH
\,,,kNHz 0
T119 ~~/~COzH =~~OH CH3 -~-(CH2)3NH2 CH3 H ~.~"~'OH
0
oH
TII10 - oH H CH3 CH3 H ~Lr"
~~C02H
0
TIIll O NH2 H CH3 CH3 H ~L~oH
--~i-NH2 ~c~~0
s~ OH
T1112 - ~/~CO NH2 H CH3 CH3 H oH
zH
OH
T1113 - ~~CO ~oH CH3 CH3 CH3 H oH
zH
0
T1114 -/"-\COzH NH2 CH3 __(CH2)3NH2 CH3 H OH
-Y-I~o
OH
T1115 O NH2 CH3 CH3 CH3 H ~~oH
~~NHz ~~0
OH
TII16 -rS '~~oo2H NH2 CH3 CH3 CH3 H ~~oH
OH
TII17 0 .'.'( /OH H --(CHz)aNHz CH3 H o
~4NHz lo --,,IKNHz
TII18 '~/~COzH =~, ~( OH H --(CHz)sNH2 CH3 H O
'Of Ij-~NHz
160
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# RZ R3 ]-~R5 R R R Rll
T1119 O NH2 H -1-(CH2)3NH2 CH3 H 0
--4NH2 o NHz
OH
T1120 \~ H CH3 --(CHz)aNHz CH3 H \~
NHz 0 NHz
T1121 0 -\'~y oH H CH3 CH3 H
--,-I-kNHz o --41~-NH2
T1122 0
.~,s~'/~COzH NH2 H - -(CHz)sNH2 CH3 H
\ Y--I~ \41KNH2
OH
T1123 0 NH2 CH3 +(CHz)sNHz CH3 H ~
~/JL'NHz ~~o ~s~ NHz
OH
T1124 H CH3 CH3 CH3 H 0
--F,IKNHz o \IIKNHz
T1125 -~+~'.~ o2H ='~~oH CH3 +(CHz)aNH2 CH3 H O
0 Y"KNHz
T1126 r '-"C02H H H CH3 CH3 H
lot V'ANHz
TI127 0 NH2 H CH3 CH3 H 0
---NHz o NHZ
OH
T1128 -,~o'~~'~CO H NH2 H CH3 CH3 H 0
z
. Y-I~o V- NHz
OH
T1129 /"' \C02H .~ H CH3 CH3 CH3 H
lof Y-IKNHz
TII30 -.'COzH NH2 CH3 +(CHz)sNH2 CH3 H 0
-/o \4NHz
IOH
161
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R3 R5 R 6 R 8 R9* R 1
TII31 0 NH2 CH3 CH3 CH3 H 0
--,-,J~NH2 NH2
OH
T1132 -"r"-"CO H NH2 CH3 CH3 CH3 H 0
2 \C
P,S O V-IKNH2
OH
T1133 0 .V~Ilr OH H -1-(CH2)3NH2 +(CH2)4NH2 H
\,-IKNHZ O
T1134 -'Sr/-"C02H =~y OH H +(CH2)aNH2 +(CH2)4NH2 H OH
0
T1135 0 NH2 H - -(CH2)sNH2 - -(CH2)aNH2 H
--,-IKNH2 -I-rl~o
OH
T1136 0 .\'ifOH CH3 -~-(CH2)3NH2 -1-(CH2)4NH2 H ~'L""OH
NH2 0
TII37 0 ~'.~ pH H CH3 --(CH2)4NH2 H
,J~NH2 10(
T1138
-//"CO2H -- NH2 H (CH2)sNH2 --(CH2)4NH2 H %IC--OH
O
OH
TII39 0 NH2 CH3 -1-(CH2)3NH2 -J-(CH2)4NH2 H
--,,IKNH2 -Y-I~o
OH
TII40 0 -\'~y OH CH3 CH3 +(CH2)4NH2 H
--./KNH2 0
T1141 - S-~'C02H ""Y OH CH3 +(CH2)3NH2 +(CH2)4NH2 H 'V--OH
0
T1142 -/"'-"CO2H OH H CH3 +(CH2)4NH2 H 'It~OH
0
162
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
RZ R3 R R R 8 Ry* R'
T1143 0 NH2 H CH3 --(CH2)4NH2 H ILLt-
~OH
~,NHz =c~~0
c~ OH
TII44 -/"'~COzH NH2 H CH3 --(CHz)aNHz H :z+-"'OH
O
OH
T1145 -//""COzH y OH CH3 CH3 +(CH2)4NH2 H ~"'~'OH
0
~
T1146 -"'CO H NH2 CH3 _ CH H '''L,.=
2 ~-( z)sNHz --(CHz)4NHz OH
O
OH
TII47 0 NH2 CH3 CH3 -1-(CH2)4NH2 H '%~-~OH
V"J~NHz -I-rk0
OH
T1148 -"CoZH NH2 CH3 CH3 --(CHz)4NHz H %~~OH
~o
OH
TII49 0 .~~oH H -J-(CH2)3NH2 CH3 OMe OH
--,-IKNHz O
T1150
'//-~'CO2H oH H -J-(CH2)3NH2 CH3 OMe
0
T1151 0 NH2 H -1-(CH2)3NH2 CH3 OMe ~~OH
--,,,J~NHz ~_~~O
c~ OH
T1152 0 .'~~oH CH3 -~-(CHz)sNHz CH3 OMe :~~OH
\I~ NHz O
T1153 0 -\'-r oH H CH3 CH3 OMe ~,,,--"OH
NH2 O
T1154 -'/"'-"COzH NH2 H +(CHz)sNHz CH3 OMe ~'~OH
O
OH
163
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R 3 R R R R y R
T1155 0 NH2 CH3 _-(CH2)3NH2 CH3 OMe
~~OH
NHZ -~0
F OH
T1156 0 -\-~y oH CH3 CH3 CH3 OMe
lIKNH2 0
T1157 -//-'CO2H -~~ ,OH CH3 -1-(CH2)3NH2 CH3 OMe 'ZI~OH
0
T1158 '~/~CO H '~4~oH H CH3 CH3 OMe ~,~OH
a
0
T1159 O NH2 H CH3 CH3 OMe ~Ll-~"oH
--,-IKNH2 -0
OH
T1160 - ~'~/~co2H NH2 H CH3 CH3 OMe ~õ~oH
-I-rko
OH
T1161 - ~''~/~co H oH CH3 CH3 CH3- OMe :L~OH
2
0
T1162 -/"'-"COzH NH2 CH3 __(CH2)3NH2 CH3 OMe ~,-"OH
-1-rl~O
OH
T1163 0 NH2 CH3 CH3 CH3 OMe
~41KNH2 ~_c~0
c~ OH
T1164 /"co2H NH2 CH3 CH3 CH3 OMe ~õ~oH
OH
T1165 0 -\' OH H _-(CH2)3NHz --(CH2)4NHZ H 0
~4NH2 101 V-IKNH2
T1166 -//"CO2H ~\'Y OH H +(CH2)3NH2 +(CH2)4NHZ H 0
0 \IIKNH2
164
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
6
R8 Rv R'
# R2 R3 RS R
T1167 0 NHz H --(CH2)3NH2 --(CH2)4NH2 H 0
NHz V"KNHz
OH
T1168 O .\""Y OH CH3 +(CH2)3NH2 +(CH2)4NH2 H O
V-IKNH2 0 \l-ANH2
T1169 \~ -'z~~oH H CH3 --(CH2)aNH2 H \~s (~
NHz O /~NH2
-~s~/~ NH2 H - -(CH2)sNH2 - -(CH2)aNH2 H 0
T1170 Co2H
\s' 7 O \~NHz.
OH
T1171 ~ 0 NH2 CH3 _-(CH2)aNH2 -1-(CH2)aNHz H ~ 0
~ NHz ~~o ~ NHz
OH
T1172 0 -\'~y oH CH3 CH3 -J-(CH2)4NH2 H o
VIKNH2 O V"KNHz
T1173 ~~/~C02H ~~OH CH3 - -(CHz)aNHz - -(CHz)aNH2 H O
0 V,)I,-NHz
T1174 -/ /~C02H ='~~oH H CH3 --(CH2)aNH2 H o
0 \~~NHz
T1175 0 NH2 H CH3 _ H 0
-(CH2)aNHz
~Il,kNHz -I-rl~0 \,,IKNHz
OH
TII76 -/"-~"CO H NH2 H CH3 -
_(CH
2 2)aNHz H 0
\ ~O V-IKNHz
OH
T1177 -/"'-"oo2H .'.~( /OH CH3 CH3 --(CH2)aNH2 H o
lo V-IKNHz
T1178 -/"--'CO2H NH2 CH3 +(CH2)3NH2 +(CH2)4NH2 H 0
.S-~o V~NH2
OH
165
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R3 R R 6 R 8 R9* R 11
T1179 O NH2 CH3 CH3 _ H 0
-(CHa)4NH2
--l-ANH2 -I0 \'NH2
OH
T1180 -//-"COaH NH2 CH3 CH3 --(CH2)4NHa H 0
C
.~s'
p --,-NH2
OH
T1181 p -\''/pH H -J-(CHZ)3NHa CH3 OMe 0
~-NH2 10( V-ANH2
T1182 CO2H =\"Y OH H +(CH2)3NH2 CH3 OMe 0
p V"KNHZ
T1183 0 NH2 H +(CHZ)3NHa CH3 OMe 0
--,-IKNH~ -Y-I~p V-IKNH2
OH
\~ .'zz~~OH CH3 --(CH2)aNH2 CH3 OMe \~ 0
T1184
V-IKNH2 O V-IKNH2
T1185 p pH H CH3 CH3 -OMe 0
--FIKNH2 p V-IKNH2
TII86 -/"-\C02H NH2 H +(CH2)3NH2 CH3 OMe 0
.~S~p ~-NH2
s~ OH
T1187 0 NH2 CH3 +(CH2)3NH2 CH3 OMe 0
V-IKNH2 -1O \-NH2
fOH
TII88 p -\"'Y pH CH3 CH3 CH3 OMe 0
\l'ANH2 O V-IKNH2
T1189 -pH CH 0
//~COZH ~ 3 - -(CH2)sNH2 CH3 OMe
0 V-IKNH2
T1190 ~/"-"COZH pH H CH3 CH3 OMe 0
~
0 V-IKNH2
166
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R3 R5 R R8 R R
T1191 0 NH2 H CH3 CH3 OMe 0
--,-.IKNH~ -Y'~o \41kNH2
OH
T1192 S-"CO H NH2 H CH3 CH3 OMe 0
z
-Y-I~p --4 NHZ
OH
T1193 ~ "'-"co2H 'Y pH CH3 CH3 CH3 OMe 0
0 \,-IKNH2
T1194 '~"-~'CO2H NH2 CH3 +(CH2)3NH2 CH3 OMe 0
~_~So ~~NH2
s~ OH
T1195 0 NH2 CH3 CH3 CH3 OMe 0
NHZ -o --,NHZ
OH
T1196 - NH2 CH3 CH3 CH3 OMe 0
~~C02H
-I O \~NH2
IOH
T1197 0 .~~OH H +(CH20H2* +(CH2)4NH2 OMe 'LC'-OH
--4lKNH2 O
T1198 ~/~COZH =~OH H -1-(CH2)3NH2 --(CH2)4NH2 OMe --'oH
0
T1199 0 NH2 H +(CH2)3NH2 +(CH2)4NH2 OMe "I~OH
V-IKNH2 -I-rk0
OH
TII100 0 .\'~'Y OH CH3 +(CH2)3NH2 +(CH2)4NHa OMe "I~OH
--,-IKNH2 O
TII101 p pH H CH3 _-(CH2)4NH2 OMe "I~OH
--,-IKNH2 O
_(CH r,~~
TII102 '~+~'~/-"co H NH2 H -
Z 2)sNH2 - -(CH2)aNH2 OMe 'OH
o
OH
167
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R3 RS R R8 R R 1
T11103 0 NH2 CH3 _-(CH2)3NHz --(CHz)aNHz OMe OH
\~~NHz ~~~0
OIH
T11104 0 ~'~oH CH3 CH3 (CHz)aNHz OMe ~,~OH
\~NHz O --
TII105 -ss's~/-"COZH OH CH3 -~-(CHz)aNHz -J-(CHz)aNHz OMe 'LL'--OH
0
TII106 '/"'-"COzH \"'YoH H CH3 --(CHz)aNHz OMe
0
T11107 0 NH2 H CH3 -1-(CH2)4NHz OMe oH
--41kNH2 ~_c~0
s~ OH
T11108 - s'~~ NH2 H CH3 -~-(cH2)aNHz OMe L,'~OH
COZH ~
' O
OH
T11109 /~"CO H ~ _( oH CH3 CH3 --(CHz)aNH2 OMe OH
2 tr
0
N OMe '~~
TII110 /~CO H NH2 CH3 _ CH '-
z ~~O -( z)s Hz (CHz)aNHz OH
~~s'
OH
TII111 0 NH2 CH3 CH3 (CH2)4NH2 OMe %~-'~OH
\~NH2 \cs~0 --
OH
TII112 -/"-"CO2H NH2 CH3 CH3 --(CHz)aNHz OMe
\-r-k0
OH
TIIl13 0 -\-,~y OH H _J-(CH2)3NH2 +(CHz)aNHz OMe O
\-NHZ 0 \,-NHz
TII114 -s's~/-"CO H OH H --(CHz)aNHz --(CHz)aNHz OMe 0
2 'I
0 --/ NHz
168
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
6 8
# RZ R RS R
R R9 R'
T11115 0 NH2 H - CH NH
-( 2)a z '-(CH2)4NH2 OMe 0
--,-IKNHz 0 Y-ANHz
OH
TII116 0 -\,,( /oH CH3 =~-(CHz)aNHz -1-(CH2)aNH2 OMe 0
NHz 10 Y"kNHz
TII117 0 ~'~oH H CH3 _-(CHZ)4NH2 OMe 0
1-1
NHz 0
~~~NHz
TII118
-/"'-"'COzH NH2 H - -(CHz)sNHz -J-(CHz)aNHz OMe 0
O Y-IKNHz
s~ OH
TII119 O NH2 CH3 -
_ CH NH ( z)s z '-(CHz)aNHz OMe o \-NHz -I0 ~c~~NHz
IOH
TII120 \~ -'~~oH CH3 CH3 --(CHz)aNHz OMe \~ 0
NHz 0 NHz
T11121 ~~/~COzH =~~OH CH3 _-(CH2)sNHz -J-(CH2)4NH2 OMe 0
0 Y-)~NH2
TII122 CO H ~( OH H CH3 --(CHz)aNHz OMe 0
z 'I
0 \,- NHz
TII123 0 NH2 H CH3 _ OMe 0
-(CHz)aNHz
--41~-NH2 0 Y-ANHz
OH
T11124 ~~/-"COzH NH2 H CH3 _(CH2)aNHz OMe 0
-
-I-rl~O Y-IKNHz
OH
T11125 cozH oH CH3 CH3 --(CH2)4NH2 OMe 0
0 \,- NH2
T11126 S -\COzH NH2 CH3 +(CHz)sNHz --(CHz)aNH2 OMe 0
~~S~O ~'NHz
s~ OH
169
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R2 R R5 R R R9 R11
T11127 0 NH2 CH3 CH3 +(CH2)4NH2 OMe 0
-1-1KNH2 NH2
OH
T11128 -'SS'-~C02H NH2 CH3 CH3 -J-(CH2)aNH2 OMe 0
--,' NH2
OH
[0114] In another embodiment of the invention,
O
.
R2 is N H 2
-\--,y OH
R3is 0
R9 is ~ \CO2H
R2*, R5, RS*, R$*, and Rll* are each H; and
=~.~~
R6is Rs'
This embodiment gives a compound of Formula III.
R12 R13
HO2C
NH CONH2
HN O 0 0
00 0 N R1
R11 )~N N
H H
NH 0 0
O NH CO2H
N
HN O H
HO2C O HN
R8 0
s*
HN N R
Y-1 H 40
0 III
HO2C
wherein Rl, R6*, R8, R11, Rla and R13 are as previously defined.
170
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01151 Table III provides exemplary compounds of Formula III.
Table III
Compounds of Fornzula III
R12 R13
HO2C
NH CONH2
HN O 0 O
00 0 N RI
R11 H H
NH 0 0
O NH CO2H 0 I~
N
HN O H
HO2C 0 HN
Rg 0
H R6
HN N
N
Y-1H
0
III
HO2C
# R 8 Rll R 12 R 3
TIIIl CH3 %L~OH CH3 0 NH2
I \
T1112 CH3 LI~ H CH3 -
N
H
T1113 CH3 %?.'\OH CH3
TIII4 CH3 %~OH CH3 ~
T1115 CH3 '."'oH H 0 NH2
T1116 CH3 %?+-'\OH H
N
H
171
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R R 12 R s
T1117 CH3 H
T1118 CH3 %L+-'~OH H
TIII9 +(CH2)3NH2 CH3 O NH2
OH
TIIIlO +(CHz)sNH2 %1~OH CH3
N
H
TIII11 -1-(CH2)3NH2 'tt~OH CH3
T11112 +(CHO3NH2 ~L'~OH CH3
T11113 _J-(CH2)3NH2 LI~OH H 0 NH2
llvvv
T11114 +(CHz)sNH2 %%+-~OH H
N
H
T11115 -J-(CH2)3NH2 ~'~OH H
TIII16 _-(cH2)3NH2 ~zr~OH H
T11117 +(CH2)4NH2 'LL~OH CH3 0 NH2
lr%rLlv
TIII18 +(GH2)4NH2 %'~.'~OH CH3
H
TIII19 +(cH2)4NH2 =~'~OH CH3
172
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R Rl RZ R3
T11120 +(cH2)4NH2 %zr~OH CH3
T11121 +(CH2)4NH2 OH H 0 NH2
T11122 +(cH2)4NH2 H
OH
H
T11123 +(CHZ)4NH2 ~OH H
T11124 --(cH2)4NH2 :%~H
OH
T11125 0 ~LL ~OH CH3 0 NH2
.~NH2 \
11%/VV
T11126 ~ 0 '~~OH CH3
'
NH2
N
H
T11127 0 ~'~OH CH3
NHz
T11128 \~ 0 '~kOH CH3 11
NH2
TIII29 0 OH H 0 NH2
~ NH2
T11130 '~ OH H
NH2
N
H
TIII31 0
-4A "-~OH H
NH2
173
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R R1 R2 R13
\~ H
T11132
NH2
T11133 OH %~OH CH3 0 NH2
T11134 oH OH CH3
N
H
T11135 OH ~L~OH CH3
TIII36 OH %~OH CH3
~
TIII37 OH '~"OH H 0 NH2
\ ( \
/
T11138 OH
'I~OH H
H
T11139 OH s '"~oH H
T11140 OH '~"OH H
~
TIII41 NH ~OH CH3 0 NH2
HNHz
~
T11142
\N NH ~.~r,.~OH CH3
2
H N
H
TIII43 NH %~OH CH3
.~~/,--H~2
174
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R Rii R 12 13
T11144
I ~~OH CH3
Hl~~z
TIII45 NH %~-~OH H 0 NH2
-,-~H)\NH2
T11146
~ :~~OH H
1N H ~2 H
T11147
~~~ I~ :~.~OH H
Hl~~z
TIII48 NH '?%~oH H
N H ~2
T11149 '%L~OH %~OH CH3 0 NH2
T11150 '~'+z.'~OH %'~OH CH3 -
N
H
TIII51 ~OH CH3
T11152 'I~OH '~OH CH3
T11153 'L~OH 'I~OH H 0 NH2
T11154 ~OH H
N
H
T11155 "~OH -'~OH H
175
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R Ru R 12 Ri3
T11156 '~"OH %~-'"~OH H
T11157 \ 'x---OH CH3 O NH2
NH
T11158 OuN %~'~OH CH3 N
NH H
T11159 "~OH CH3 NH
T11160 \~OH CH3
NH
TIII61 'L ~OH H o NH2
NH
TIII62 \ \ %'~,-~OH H
N
NH H
T11163 OC ~~OH H NH
T11164 ~L~OH H
\
~
T11165 N "'~'OH CH3 0 NH2
NH
T11166 N \~OH CH3
)-:NH
NH H
176
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R R Rl R 3
T11167 N CH3
= -~ ~~ '~ v
NH
T11168 N H %~'~~\OH CH3
.-~ )--NH
NH
T11169 N ~OH H 0 NH2
)-:NH
NH
TIII70 N H
NH N
H
TIII71 N %~OH H
NH
T11172 N %~OH H
. -~ )---NH
NH
T11173 ~OH CH3 0 NH2
HNH2
T11174 :I,.'~OH CH3
xNH2 N
H
T11175 :L,.\OH CH3
HNH2
T11176 ~L~OH CH3
HNH2
T11177 "'~'OH H 0 NH2
HNH2
177
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R 8 R R12 R13
T11178 ~~\ !o ~L,.~OH H
H NH2 N
H
T11179 0 'I~OH H
HNH2
TIII80
'L""OH H
H NH2
T11181 Cl ~~OH CH3 0 NH2
OH
C1
TIII82 Cl ~~OH CH3
\ OH H
cl
c
TIII83 I l ~~oH CH3
\ OH
C1
T11184 - Cl ~~OH CH3
- \ / OH
C1
TIII85 Ci 'LL ~OH H 0 NH2
~ \ /C1OH I I
TIII86 Cl ''~,,~~OH H
-
\ / OH H
C1
T11187 - ci
~OH H
\ / OH
C1
178
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R Ri R12 R13
T11188 - Cl ~OH H
- ~ / OH
C1
T11189 CH3 CH3 CH3 0 NH2
TIII90 CH3 CH3 CH3
N
H
T11191 CH3 CH3 CH3
T11192 CH3 CH3 CH3
T11193 CH3 CH3 H 0 NH2
T11194 CH3 CH3 H ,~ -
N
H
T11195 CH3 CH3 H
T11196 CH3 CH3 H
T11197 CH3 -J-(CH2)3NH2 CH3 0 NH2
T11198 CH3 +(CH2)3NH2 CH3
N
H
T11199 CH3 +(CH2)3NH2 CH3
179
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R 8 R" R 12 R 13
TIII100 CH3 +(CHZ)3NHZ CH3
TIII101 CH3 +(CH2)3NHa H 0 NH2
TIII102 CH3 +(CH2)3NH2 H
N
H
TIII103 CH3 +(CH2)3NH2 H
T111104 CH3 -J-(cH2)3NH2 H
TIII105 CH3 +(CH2)4NH2 CH3 O NH2
TIII106 CH3 +(CH2)4NH2 CH3 -
N
H
TIII107 CH3 +(CH2)4NHZ CH3
TIII108 CH3 +(CH2)4NH2 CH3
T111109 CH3 +(CH2)4NH2 H 0 NH2
TIII110 CH3 +(CHZ)4NHZ H
rNp
H
TIII111 CH3 +(cH2)4NH2 H
TIII112 CH3 +(CH2)4NH2 H
180
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R Ri R 12 R 13
TIIIl13 CH3 0 CH3 0 NH2
:kANH2
TIII114 CH3 0 CH3
,41K NH2
N
H
T111115 CH3 0 CH3
NH2
T111116 CH3 0 CH3
Nl-12
TIII117 CH3 0 H 0 NH2
Nf32
TIII118 CH3 0 H
.~~NH '~-
a
N
H
TIII119 CH3 0 H
Nx2
T111120 CH3 'k 0 H
Nl-12
T111121 CH3 ox CH3 0 NH2
~ \
T111122 CH3 / ox CH3
N
H
T111123 CH3 / ox CH3
TIII124 CH3 / ox CH3
. :~.
181
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R R1 RZ 13
T111125 CH3 11OH H 0 NH2
\ \
TIII126 CH3 / OH H
1N
H
TIII127 CH3 oH H
TIII128 CH3 / oH H
T111129 CH3 NH CH3 0 NH2
H' \NH2
T111130 CH3 CH3
N~
=''~r,.
H 2 N
H
T111131 CH3 NH CH3
H'\~2
T111132 CH3 NH CH3
H~2
TIII133 CH3 NH H 0 NH2
H~NH2 \
T111134 CH3 NH H
NNHz
H N
H
T111135 CH3 NH H
H~~2
182
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R R1Y R Z R'3
T111136 CH3 NH H
N)~NH
x 2
TIII137 CH3 CH3 0 NH2
H
T111138 CH3 CH3
H H
T111139 CH3 CH3
N
H
T111140 CH3 CH3
N
H
TIII141 CH3 H O NH2
H
T111142 CH3 H
H H
TIII143 CH3 H
N
H
TIII144 CH3 H
N
H
TIII145 CH3 N CH3 0 NH2
~=NH
NH
TIII146 CH3 N CH3
~=NH -
NH N
H
183
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# R8 R" R 12 R 13
T111147 CH3 N CH3
~=NH
NH
T111148 CH3 N CH3 31k,
~=NH
NH
T111149 CH3 N H 0 NH2
~=NH
NH
T111150 CH3 H H
N~=NH
NH N
H
T111151 CH3 N H
NH
T111152 CH3 H H
,~~)-NH
NH
T111153 CH3 0 (~H3 0 NH2
HNH2 \
T111154 CH3 CH3
H NH2
H
T111155 CH3 o CH3
H NHZ
TIII156 CH3 z, 0 CH3
H NH2
T111157 CH3 o H 0 NH2
H NH2
184
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
R R R12 R13
TIII158 CH3
H
HNH2
N
H
TIII159 CH3 H
g NH2
T111160 CH3 H
HNH2
TIII161 CH3 Cl CH3 0 NH2
OH
cl
TII1162 CH3 cl CH3
OH N
H
C1
TIII163 CH3 Cl CH3
OH
C1
T111164 CH3 cl CH3
OH
cl
TIII165 CH3 Cl H 0 NH2
4 \ / OH
C1
TIII166 CH3 Cl H
~
4 ~ OH N
H
C1
TIII167 CH3 - cl H
- \ / OH
C1
185
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
# Ril Ri2 R 13
T111168 CH3 - Cl H
CI
186
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0116] In another embodiment of the invention each of R2*, R8* and R11* is H.
This
embodiment gives a compound of Formula IV.
R12 R13
HO2C
NH
HN O 0 R2 0
)ty
N R'
O 0 0
R~ ~ N N
NH O H R3 0 H
O NR5
R5' N
HN O H
O HN
Rs R$ 0 Rs
HN N
H O IV
O
HO2C
[0117] Table IV provides exemplary compounds of Formula IV.
187
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w
U U
S 2
U U
a .nnr nnr
a x x
i 0 = 0
z~
N N
z= 0 0
O
Z2 ~ z z
N
~ N N
U U
O i i
=Z a ~~ ,vlv
~
O
z2 O ~
a ~~ U
~Az)yo
U p O -W 2z
~2 O
p
z Z2 = ~ x x
N CC)
O
0 O O
= = a U U
_ 0
o 0 =
_
. Ma O
N N
O
0
U U
~ H H
188
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w w w w w w
x x = = x x x
U U U U U U U
e=> U U U U U U U
a nnr nnr .nrv .nrv .nr nni nnr
c~ x x x x x x x
0 0 o 0 0 0 0
Z Z Z Z Z~
N N N N N N N
0 0 0 0 0 0 0
0-- O O 0~ O O
_
z z z z z z z
N N N N N N N
U U U U U U U
VVv ,n i r ~w
U U U U U U U
x x x x x x x x
M M M M M M M
U U U U U U U
O
0 0
2 ;_\
0 O 0 0 Z O
O O O O S x
x x x x
O
ILYf ~ ~ ~
M d' N 00 01
~k E H E~ FEH E~
189
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w w w w w w w
x x x x x x x x
U U U U U U U U
M U U U U U U U U
.rvv ivv nnr ~nr ~nnr nn! ~v nnr
a x x x x x x x x
0 U 0 N 0 U 0 0 0
Z Z Z Z Z Z Z Z
N N N N N N N
0 0 0 0 0 0 O O
U O O O--( O O 0~,~
Z z z z z z z z
N N N N N N N N
U U U U U U U U
Jti ~ r ,n 1~ .n i n .n i r ~w~ ,n ~ r .n i r ,n vv
M M M M M M M M
ai U U U U U U U U
x x
M O x
U N N 0 N
x x x x x
U U
U M U U x / \
O
~a x '~,r~
~ M M M M M M
U U U U U U U
O O O O O O O
U Z~U U Z40 Z~U Z~U Z~U ZU
x x x x x x x x
oN 0N 0N 0N 0N 0N 0N 0N
N N N N N N 0 N
N N N N N N N N
U U U U U U U U
~ v .i'~nr e'~r ,n v ~ v ,rv r .nnr nnr
F~1 O ID l~
.--~ .--~ ~ --~
~k F~ E~ E~ f+ F~ H F~ F+
190
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w w w w w w u~
x x s x x x x "
U U U U U U U
en U U U U U U U U
~/ nnr .nrv nrv nn~ nn~ fuv nnr ,Myr
~W I1'I Fil W Fi'd W W W Fy
O U = U = U U = U
U = U =
z~ Z Z z~ Z
N N N N N N
O 0 O O O O O O
o4 04 04 04 4 4 4 o
sN z z = _ _ = z
z z z z z z z z
i s x x i i x x
U U U U U U U U
~ 1 1 1 1 1
a1 t1p vl lJl vl Itfl ~J1 IV~ JWI_ JWt~
M M M M M M M M
ai U U G U U U U U
w 0 s
s x p
U
2 x O \ U
U U
~~ ~r ,rw+ ~ z ~ z A z z
~ M M M r M
U U U U U U x
U O U U U U O
0
x x x //
z z z ~z s z z z zJf
0 ~,, 0 0 _ 0 y,, 0 ~,, 0 ~,, 0
/ ~.ti, / W /.,~, / "1~,
' x x x x x x x x
0 0 0 0 O 0 0 0
N N N N N N N N
s x x x x x x =
~ vy-,-r IrIff 'Aff lryv ~
oO O\ O N M It v) N N N N N
191
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w w w w w
U U U U U U
U U U U U U
õ/ .tiv ruu tiv / ~rv J ~nr .~u1.r
NF~i
a x x x x x x
0 0 0 0 0 0
Z~
N N N N N N
O 0 0 0 0 0
o o o o
o o4
z z z z z z
N N N N N N
U U U 2 U U
a n i r .n'nr+ ~v~v.n i r w-ll
m M M m M M
0.~i U U U U U U
I \ ~-~ \
~\ LL\
~ z'\
M M M M
U U U U U U
If,
0 Q 0 0 N ~ N 0
M Z~~ Z~o Z4~ 0 z~o Z~0
;~,.. ;~.
N N N N N N
0 0V 0U 0 0 0
U U U U U U
~ nnr r ti~ ~v r nnr n~~ev nnr
oo rn o ~.
N >
192
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
w w w w w w w
m o S 2 S S
U U U U U U U
M U U U U U U U
a r~nr vv nn~ r~nr nnr
.r ~rv n~ tr
a x x x x x x x
0 O O O O 0 0
Z-~( Z-"C Z~ ZZ Zi
w N N
O O 0
U U U
2 2 2 2
O O O O
U U U U
O--{
Z Z Z Z z Z z
U U U U U U U
I I 1 1 1
vv vv
M M M M M M M
f~i U U U U U U U
A O LL__~ 0 / \ Z
~~ ~ x x x x
U U U U U U U
N ~ N O O 0 0 0 O
M Z~ ~ ~o ZO Z~0 Z~y,,0 Z~0
~ "t..
~i=~ S 2 S S = = I
0 0 0 0 O C 0
U U U U U U
C)
~ ~ ~ ~ ~
2 S 2 S S S 2
N
Vl/\f JlN IJYV /l/lf ~/lJ\! all~ lf OU'l!
f~'1 N M c} 00
en > > cn > m >
~ F~ F F+ H H H E~
193
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
z Z w w = "'
U U
U U
M M M
fYi U x U x U x
0 0 0 0 0 0
N N N N N N
O o 0 0 o O
o o o o o o
M M M M M M
U U U U U U
z z z z z z
N N N N N N
U_ U U_ U_ U_ U_
.nr tfxr .n i vI .rwu+ .nr ~ v~ .nr f v~ rw~r
a x x x x x x
x x x x x x
N N N N N N
0 0 0 0 0
U U U U U U
0 0 0 o 0 0
~
N
Nj~=
M
M d' 'cP d' 'V' 'ch
194
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Z z W W
I U U
c~ U U
aI - - n; u .n; v nnl r n,w,
M M M
x U x U x
0 0 0 0 0 0
_ _ _
0 0 o a o
U U U U U U
O O O O O O
W - '
M M M M M M
f~i U U U U U U
x = = x x x
z z z z z z
U_ U U U_ U U_
~ lrtfxfxp lfxfvlul -V'U'XP
"a x x x x x x
x x x x x x
_ _ _
0 0N 0N o o" o
U U U U U U
S 2 2 2 2 2
U U U U U U
l/1 IlP !7 fV' JVl/' uUitf'
O O N O N O N O N 0
Z- Z- Z- Z- Z-
o
~k F~ F~ H E~ F
195
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
z z W W = = Z
~ ~ U U
/ \ o / \ o = = U V / \ o
c, ~ I I
M M M M
~z ~ x ~ x U x
0 0 0 0 N 0 0 N 0
Zl~ Z~ Z~ Z~ Z~ Z- z-
j~. j~t..
~
2 S 2 S 2 = V
O O O O O O
U U U U U U
0 O~ O~ O~ O--( O
Z ~ Z Z Z Z Z
N N N N N N N
U U U U U U U
M M M M M M M
0.~i U U U U U U U
a x x x x x x x
~'r~ x x x x x x x
0 0N o 0 0N 0N o
N N N N N N 0
U U U U U U U
Ma .rvir Irvir .niv "nr .ntr
N 0 N ~ N 0 N 0 N 0 N /0 N 0
Z~ Z~ Z~ Z~ z_ Z Z~
~j'h, s w
cn kn
196
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Z
w W x x
= S V V
U U
V U rv I ti fv l~+ -
44
O N 0 N 0 N 0 N 0 N 0
Z- Z- Z- Z Z~ Z~O
0 0 0 0 U U 0 0 0
0
S
U
~ ~ ~ ~ ~~
Z z z z z
U U U U U
I t t t t 'n
1 1 U
x
z
U
M M M M M I
U U U U ~I
"a x x x x x x
r~ x x x x x x
N = N N N
O 0 0 0 U 0 C) 0U
U U U U U U
N 0 0 N 0 0 N 0 N 0
N Z~ Z~ Z~ Z~ Z~ Z~
W ~j~= ~j~=. j~ j~ j~
00 ~ ~ N cM
197
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
z Z z Z z Z
4 0 0
a - - - - - -
4
M M M
~ U x U x U x
= 0 = 0 = 0 = 0 = 0 = 0
Z~ Z~ Z~ ~
z~
;~.. ;ti,.
o ~ 0
-~
z z z z z
N N N N N
U U U U U
z z z z z z
N N N N N N
_ _ _ _
U_ U U U U U
rv i n nr i r rwv~ nr i n nr i r .nr i n
a x x x x x x
~~ x x x x x x
oN o 0N o o" o
N N N N N N
U U U U U U
M~ JVV' af1.l1!' Jll'l!' JVV' JVV' J11'L!'
I I i
0 0 0 0 0 0
Z Z_~ Z_~ Z~ Z~ Z_
j~'4 ,"4 oo
198
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
2 S = S 2 T
6~6~6c6~6~
M M M
u u ~ x x
N 0 0 0 0 0 0
Z~ Z~ Z~
O O ~
U U U ~ 0 0
0
O O O--( [ [ C
z z z z
N N N N
U U U U
I I i
U U ~
_
z z z z z
N N N N N
U U U U U
~l11ULf~ JlNIJ~ JLNI./~ J1Ml~ ~M11l~ M
a ~ ~ ~ ~ - ~
"a x x x x x x
~ x x x x x v
N N N N
0V 0 0V = 0 = 0 0
Z z
_ _ _ _
U U U c?
N N
= 0 = 0 _ 0 0 = 0
z Z~ Z~ ~ fv ! Z
;~-
~
199
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
2 2 =
z Z z
~6~6~
o 0 0
N N
0 0 O
\ \ \
Z z Z
U U U
1 1
M M M
U U U
O
U N Q N 0
= Z~ Z~
U
M~ JVt! ~,, ~,,
N N
0 0
N 0
N N
Z
U U
~k E'-H H
200
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01181 In another embodiment, the invention provides a compound of the Formula
Fl:
R12 R13
HO2C
HN NH 0 CONH2
O O
00 O N R1
R11 N N
NH O 0
O NH C02H
N
HN O H
O HN
R$ 0
1-t H02C HN N Rs=
Ir, H 40
O
HO2C (F1)
and salts thereof; wherein:
NH2
.
a) R8 is hydrogen, or
O
.
A
b) R" is methyl, ~OH or NH2;
c) R12 is H or CH3;
d) R13 is CH(CH3)2, CH(CH2CH3)CH3,
O NH2
~
H or I and
e) each of Ri and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0119] In one embodiment of the invention, substituent R13 of Formula F1 is
O NH2
N
CH(CH2CH3)CH3, H or
201
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0120] In another embodiment of the invention, a compound of Formula Fl is
selected from
Rl(L-Trp)-D-Asn-L-Asp-L- hr-Gly-L-Orn-L-Asp-D-Ser-L-Asp-Gly-D-Ser-1.-3mGlu-L-
&yn
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-Gly-L-Asp-GIy-D-Ser-L-3mGlu-L-Kyn
R'(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Asn-L-Asp-Gly-D-Ser-L-3mGlu-L-
Kyn
RI (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Asn-L-Asp-Gly-D-Ser-L-3mGlu-L-
Ile
Rl (L-Trp)-D -Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Asn-L-Asp-Gly-D-Ser-L-3mGlu-L-
Val
Rl(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ser-L-Asp-Gly-D-Ser-L-Glu-L-Trp
, and
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ser-L-Asp-Gly-D-Ser-L-Glu-L-Trp
[0121] In one embodiment of the invention, substituent R' of Formula Fl is not
Clo-aikanoyl when substitutent R$** is hydrogen or "'~'OH .
[0122] Exemplary compounds Formula F1 include, without limitation, compounds
C22,
C189, C201, C210, C37 and C39 (vide supra).
202
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01231 In another embodiment, the invention provides a compound of the Formula
F2:
R12 R13
HO2C
HN NH 0 CONH2
O O
00 O N R'
H3C H H
NH O 0
O NH C02H
N
HN O H
O HN
R$ O
HO2C HN Y-I N N R6*
H 40
O
HO2C (F2)
and salts thereof; wherein:
NH2
,~ '~~ R8,.
a) R8 is hydrogen, methyl, OOH or
b) R1aisHorCH3;
c) R13 is CH(CH3)2, CH(CH2CH3)CH3,
O NH2
f ~ \
H
or and
d) each of Rl, R6*and Rg** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
[0124] In another embodiment of the invention, a compound of Formula F2 is
selected from
R1(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-AIa-L-Asp-Gly-D-Ala-L-3mGlu-L-
Kyn
R1(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Ala-L-3mGlu-L-
Trp , and
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Ala-L-Glu-L-Trp
203
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0125] Exemplary compounds Formula F2 include, without limitation, compounds
C46,
C49, and C61 (vide supra).
[0126] In another embodiment, the invention provides a compound of the Formula
F3:
R12 N NH
HO2C
HN NH 0 CONH2
O 0
00 O N RI
Rll N N
O H
NH 0 H
O NH CO2H
N
HN O H
O HN
R8 0
1-t HO2C HN N Rs* N H
O
HO
2C (F3)
and salts thereof; wherein:
NH2
~.~ .-~ Ra,~
a) R8 is hydrogen, O~OH ' or ;
0
b) RI 1 is methyl, '~õ~OH or NH2 ;
c) R12 is H or CH3; and
d) each of Rl, R6*and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
204
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01271 The present invention provides, in another aspect, compounds of Formula
F4:
= / ~
R12 NH
HO2C
HN NH 0 CONH2
O O
00
1-t HO2C HN N Rs*
Ir, N
H
O
HO2C (F4)
and salts thereof; wherein:
NH2
.~ \=-~ ~~ a) R8 is hydrogen, T/0 ethyl, OH or R
O
~s
b)Rli is methyl, or '~ NH2;
c) R12 is H or CH3i and
d) each of Rl, R6*and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
205
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01281 In another embodiment, the invention provides a compound of the Formula
F5:
y \
\ NH
HO2C
HN NH 0 CONH2
O 0
00 O N R'
R" H H
NH O 0
O NH CO2H
N
HN O H
O HN
R8 0
HO2C HN
Y I N Rs=
N H 40
0
HO2C (F5)
and salts thereof; wherein:
NH2
~~~ '~ R8.,
,' OH , ar ;
a) R8 is hydrogen, methyl, O ~'~
0
.
b) Rl l is methyl, , ' ~ " ~ O H , or ~ N H2 ; and
c) each of R', R6*and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
206
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0129] In another embodiment, the invention provides a compound of the Formula
F6:
H02C C02H
NH
HN O
O O
00
O
N RI
R" N N
O H
NH O H
O NCH3 HO CONH2
N
HN O H
O HN
R9 R8 O
HN N
H
O
HO2C (F6)
and salts thereof; wherein:
NH2
a) R8 is . O or
OMe OH
b) R9 is CO2H CO2H or CO2H ,
> > >
c) Ri 1 is, methyl,
0
.
'~r..~OH , or ~ NH2 ~
d) R 12 is H or CH3; and
e) RI is amino, monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
[0130] In another embodiment of the invention, a compound of Formula F6 is
selected from
Rl (L-Trp)-D-GIu-L-h-Asn-LThr-Sar-L-AIa-L-Asp-D-Ser-L-omAsp-Gly-D-Asn-L-GIu-L-
Ile
207
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Rl(L-Trp)-D-Glu-L-h-Asn-L- hr-Sar-L-Ala-L-Asp-D-Ser-L-omAsp-Gly-D-Asn-L-3mGlu-
L-Ile
Rl(L-Trp)-D-Glu-L-h-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Asn-L-omAsp-Gly-D-Asn-L-Glu-L-
Ile' and
Rl (L-Trp)-D-GIu-L-h-Asn-L-Thr-Sar-L-AIa-L-Asp-D-Asn-L-omAsp-Gly-D-Asn-L-3mGlu-
-Ile
[0131] Exemplary compounds Fonnula F6 include, without limitation, compounds
C292,
C289, C307 and C304 (vide supra).
[01321 In another embodiment, the invention provides a compound of the Formula
F7:
H02C CO2H
NH
HN O
O O
O
O N R'
H3C H H
NH O O
O NCH3 HO CONH2
N
HN O H
HN
R9 R8 0
H
HN N
H
O
HO2C (F7)
and salts thereof; wherein:
NH2
a) R8 is methyl,'~O ,~~OH or R$'~ =
OMe OH
b) R9 is CO2H CO2H or CO2H =
> > >
c) R12 is H or CH3i and
d) each of Rl and R$** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
208
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0133] In another embodiment of the invention, a compound of Formula F7 is
selected from
RI(L-Trp)-D-GIu-L-h-Asn-LThr-Sar-L-AIa-L-Asp-D-Lys-L-omAsp-Gly-D-AIa-L-GIu-L-
Ile and
R'(L-Trp)-D-GIu-L-h-Asn-LThr-Sar-L-AIa-L-Asp-D-Lys-L-omAsp-Gly-D-AIa-L-3mGlu-L-
Ile
[0134] Exemplary compounds Formula F7 include, without limitation, compounds
C337,
and C328 (vide supra).
[01351 In another embodiment, the invention provides a compound of the Formula
F8:
HO2C C02H
NH
HN O
O O
O
00 N
Ril N N
NH O H H
O
O NCH3 R3*= CONH2
N
HN O H
HO O HN
HO R8 0
g HN N
H O
O
HO2C (F8)
and salts thereof; wherein:
a) R3** is hydroxyl or hydrogen
NH2
b) R8 is methyl, or
c) Rll is an amino acid side chain, methyl,
O
.~s
or c~ NH2 ;
d) R12 is H or CH3i and
e) each of R' and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
209
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0136] In one embodiment of the invention group R3** of Formula F8 is
hydroxyl. This
gives a compound of Formula F8A:
H02C C02H
NH
HN O O O
00 0 N Ri
R11 H H
NH O 0
O NCH3 HO CONH2
N
HN O H
HO 0 HN
HO R8 0 H
O HN N
H
O
HO2C (F8A)
wherein R1,R8, R8**, Rll, and R12, are as described for Formula F8.
[0137] In another embodiment of the invention, a compound of Formula F8A is
selected
from,
Rl(L-Trp)-D-GIu-L-h-Asn-L-Thr-Sar-L-AIa-L-Asp-D-Lys-L-hAsp-Gly-D-Asn-L-GIu-
LIle , and
Rl (L-Trp)-D-G1u-L-h-Asn-L-Thr-S ar-L-AIa-L-Asp-D-Lys-L-hAsp-Gly-D-Asn-L-3mGlu-
L-Ile
[0138] Exemplary compounds Formula F8A include, without limitation, compounds
C87
and Cl 11 (vide supra).
210
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0139] In another embodiment of the invention group R3** of Formula F8 is
hydrogen. This
gives a compound of Formula F8B:
HO2C CO2H
NH
HN O O O
00 O N Ri
R11 H H
NH O 0
O NCH3 CONH2
N
HN O H
HO 0 HN
HO R8 O
H
O HN N
N O
H
O
HO2C (F8B)
wherein RI,R8, R$**, Rll, and R12, are as described for Formula F8.
[0140] In another embodiment of the invention, a compound of Formula F8B is
selected
from
Rl(L-Trp)-D-Glu-L-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-hAsp-Gly-D-Asn-L-Glu-L-Ile
and
Rl (L-Trp)-D-Glu-L-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-hAsp-Gly-D-Asn-L-3mGlu-L-
Ile
[0141] Exemplary compounds Formula F8B include, without limitation, compounds
C102,
and C99 (vide supra).
211
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0142] In another embodiment, the invention provides a compound of the Formula
F9:
H02C R12 C02H
NH
HN O O O
H2NOC 00 O N Ri
N N
H H
NH O 0
O NCH3 CONH2
N
HN (CH2)4Raõ O H
O HN
HO O
H
HN N
O H
HO2C (F9)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of Rl, and R$** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
[0143] In one embodiment of the invention, substituent group R12 of Formula F9
is methyl.
[0144] In another embodiment of the invention, a compound of Formula F9 is
selected from
Ri(L-Trp)-D-GIu-L-Asn-LThr-Sar-L-AIa-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-GIu-L-Ile
and
R1(L-Trp)-D-GIu-L-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-3mGlu-L-
Ile
[0145] Exemplary compounds Formula F2 include, without limitation, compounds
C105,
and C108 (vide supra).
212
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01461 In another embodiment, the invention provides a compound of the Formula
F10:
R13*
HO2C H C02H
NH
HN O O O
HO 00 O N R1
N N
111
NH O H O H
O NH CONH2
N
HN O H
O HN
HO O
H
HN N
H O R6'
O
HO2C (F10)
and salts thereof; wherein:
a) R13* is H or CH3; and
b) each of R1, and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0147] In another embodiment of the invention, a compound of Formula F10 is
selected from
R1(L-Trp)-D-GIu-L-Asn-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Ser-L-3mGlu-L-
Ile and
Rl (L-Trp)-D-Glu-L-Asn-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Ser-L-3mGlu-L-
VaI
[0148] Exemplary compounds Formula F10 include, without limitation, compounds
C259,
and C262 (vide supra).
213
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0149] In another embodiment, the invention provides a compound of the Formula
F11:
CH
ONH2
g R 7;HNHNf-jr
NH O C
O O
O O
00 O 11 N f2i
)~11 N
HO NH O O H
O NH C02H
N
HN O H
O HN
CH3 O
HO2C HN
Y-, I N Rs.
H O
O
HO2C (F11)
and salts thereof; wherein:
a) R13*isHorCH3iand
b) each of R1, and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0150] In another embodiment of the invention, a compound of Formula F11 is
selected from
R1(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Om-L-Asp-D-AIa-L-Asp-Gly-D-Ser-L-3mGlu-LIle
and
R1(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-AIa-L-Asp-Gly-D-S er-L-3mGlu-L-
Val
[0151) Exemplary compounds Formula F11 include, without limitation, compounds
C4, and
C8 (vide supra).
214
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0152] In another embodiment, the invention provides a compound of the Formula
F12:
HO2C Me R13
HN NH O CONH2
O O
00 O N R1
N N
H2NOC NH O H O H
O NH C02H ~ I \
N
HN O H
O HN
CH3 O
1-t HO2C HN N Rs*
Yl- H O
O
HO2C (F12)
and salts thereof; wherein:
0 NH2
a) R13 is CH(CH2CH3)CH3 or and
b) each of Rl and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0153] In another einbodiment of the invention, a compound of Formula F12 is
selected from
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Asn-L-3mGlu-L-
Kyn and
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-Asp-Gly-D-Asn-L-3mGlu-L-
Ile
[0154] Exemplary compounds Formula F12 include, without limitation, compounds
C233,
and C221 (vide supra).
215
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01551 In another embodiment, the invention provides a compound of the Formula
F13:
O NH2
H02C Me I
HN NH O CONH2
O O
00 O N R'
N N
H2NOC NH O H
O H
O NH C02H
N
H
HN (CH2)4R8- O
O HN
H02C HN N Rs=
H O
O
HO2C (F13)
and salts thereof; wherein each of R', R6* and R$** is independently amino,
monosubstituted
amino, disubstituted amino, NH-amino protecting group, acylamino, ureido,
guanidino,
carbamoyl, sulfonamino, thioacylamino, thioureido, iminoamino, or
phosphonamino.
[0156] In another embodiment of the invention, a compound of Formula F 13 is
selected from
Rl (L-Trp)-D-Asn-L-Asp-LThr-Gly-L-Orn-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-3mGlu-L-
Kyn
Rl(L-Trp)-D-Asn-L-Asp-LThr-Gly-L-Orn-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-3mGlu-L-Kyn
, and
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-3mGlu-L-
Kyn
[0157] Exemplary compounds Formula F13 include, without limitation, compounds
C236,
C237, and C238 (vide supra).
216
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0158] In another embodiment, the invention provides a compound of the Formula
F14:
R12
HO2C
HN NH O CONH2
O O
00 O N Ri
N N
H2NOC NH O H
O H
O NH C02H
N
HN O H
Me0 O HN
O
HO2C HN N 4 Rs-
jr~ H O
HO2C (F14)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of Rl and R6* is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0159] In another embodiment of the invention, a compound of Formula F14 is
selected from
Rl(L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-omAsp-Gly-D-Asn-L-Glu-L-
Ile and
Rl (L-Trp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-Ala-L-omAsp-Gly-D-Asn-L-3 mGlu-
L-Ile
[0160] Exemplary compounds Formula F14 include, without limitation, compounds
C283,
and C277 (vide supra).
217
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0161] In another embodiment, the invention provides a compound of the Formula
F15:
H02C R12 C02H
NH
HN O O O
00 O N
N N
HO NH O H H
O
O NCH3 HO CONH2 / I
N
HN O H
Me0 0 (CH2)4R$'* HN
O
HO HN N
O H
O
HO2C (F15)
and salts thereof; wherein:
a) R12 is H or CH3i and
b) each of Rl and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0162] In one embodiment of the invention, substituent group R12 of Formula
F15 is methyl.
[0163] In another embodiment of the invention, a compound of Formula F15 is
selected from
Rl(L-Trp)-D-GIu-L-h-Asn-LThr-Sar-L-AIa-L-Asp-D-Lys-L-omAsp-Gly-D-Ser-L-Glu-L-
Ile and
Rl(L-Trp)-D-Glu-L-h-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-omAsp-Gly-D-Ser-L-3mGlu-
L-Ile .
[0164] Exemplary compounds Formula F15 include, without limitation, compounds
C325,
and C153 (vide supra).
218
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0165] In another embodiment, the invention provides a compound of the Formula
F16:
HO2C R12 C02H
NH
HN O O O
00 0 N R~
N N
H2NOC NH O H 0 H
O NCH3 HO CONH2
X I~
N
HN O H
0 (CH2)4R8** HN
O
HO HN N
O N
H
HO2C (F16)
and salts thereof; wherein:
a) R12 is H or CH3, and
b) each of Rl and R8** is independently amino, monosubstituted amino,
disubstituted amino,
NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[01661 In one embodiment of the invention, substituent group R12 of Formula
F16 is methyl.
[0167] In another embodiment of the invention, a compound of Formula F16 is
selected from
Rl(L-Trp)-D-GIu-L-h-Asn-L-Thr-Sar-L-AIa-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-GIu-L-
Ile and
R1(L-Trp)-D-GIu-L-h-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-Asp-Gly-D-Asn-L-3mGlu-L-
Ile
[0168] Exemplary compounds Formula F16 include, without limitation, compounds
C90,
and C114 (vide supra).
219
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
--- -- ..... ......
[0169] In another embodiment, the invention provides a compound of the Formula
F17:
H02C R12 C02H
NH
HN O O O
00 0 N Ri
N N
H2NOC NH O H H
O NCH3 HO CONH2 / I
N
HN O H
Me0 O HN
O
HO O HN N
H
O
HOaC (F17)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) R' is amino; monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
[0170] In another embodiment of the invention, a compound of Formula F 17 is
selected from
Rl(L-Trp)-D-GIu-L-h-Asn-LThr-Sar-L-AIa-L-Asp-D-AIa-L-omAsp-Gly-D-Asn-L-GIu-
LIle , and
R l(L-Trp)-D-GIu-L-h-Asn-LThr- S ar-L-AIa-L-Asp-D-AIa-L-omAsp-Gly-D-Asn-L-3
mGlu-L-Ile
[0171] Exemplary compounds Formula F17 include, without limitation, compounds
C316,
and C319 (vide supra).
220
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
..... .....
[01721 In another embodiment, the invention provides a compound of the Fonnula
F18:
H02C Me C02H
NH
HN O 0 O
00 0 N R
N N
H2NOC NH O H 0 H
O NH HO CONH2
N
HN O H
Me0O 0 (CH2)4R8** HN
O
HO HN N
H
HO2C (F18)
and salts thereof;
wherein each of Rl and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylamino, ureido, guanidino, carbamoyl,
sulfonamino,
thioacylamino, thioureido, iminoamino, or phosphonamino.
[0173] In another embodiment of the invention, a compound of Formula F 18 is
RI (L-Trp)-D-GIu-L-h-Asn-L-Thr-Gly-L-Ala -L-Asp-D-Lys-L-omAsp-Gly-D-Asn-L-
3mGlu-L-Ile
[0174] An exemplary compound of Formula F18 is, without limitation, compound
C180
(vide supra).
221
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0175] In another embodiment, the invention provides a compound of the Formula
F19:
H02C
NH
HN O O R2 O
00
O N RI
N )rN
HO NH O H O H
O NCH3 CONH2 ~ I \
N
HN O H
O HN
R8 0 R6
HO HN N
O N
H
O
HO2C (F19)
and salts thereof; wherein:
O O
'j
~ 0 H
a) R2 is NH2 or.
=~.~~ R6*
b) R6 is methyl or ;
c) R8 is methyl or\~ R8- ; and
d) each of Rl, R6*, and R8** is independently amino, monosubstituted amino,
disubstituted
amino, NH-amino protecting group, acylainino, ureido, guanidino, carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino.
[0176] In another embodiment of the invention, a compound of Formula F19 is
selected from
Rl(L-Trp)-D-Asn-L-Asp-L-Thr-Sar-L-AIa-L-Asp-D-AIa-L-Asp-Gly-D-Ser-L-GIu-L- le
Rl(L-Trp)-D-GIu-L-Asp-L-Thr-Sar-L-Orn-L-Asp-D-Lys-L-Asp-Gly-D-Ser-L-Glu-L-Ile
, and
RI (L-Trp)-D-Asn-L-Asp-L-Thr-S ar-L-Orn-L-Asp-D-Lys-L-Asp-Gly-D-S er-L-GIu-L-
Ile
[0177] Exemplary compounds Formula F19 include, without limitation, compounds
C86,
C359, and C356 (vide supra).
222
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[01781 In another embodiment, the invention provides a compound of the Formula
F20:
HO2C R12
HN NH 0 CONH2
O O
00 O N RI
N
)~11
H2NOC NH O O H
)y 1---(
O NCH3 HO CONH2 y
N
HN O H
MeO 0 (CH2)4R8** HN
O
HO HN N
O H
HO2C (F20)
and salts thereof; wherein:
a) R12 is H or CH3; and
b) each of Rl and R8** is amino, monosubstituted amino, disubstituted amino,
NH-amino
protecting group, acylamino, ureido, guanidino, carbamoyl, sulfonainino,
thioacylamino,
thioureido, iminoamino, or phosphonamino.
[0179] In another embodiment of the invention, a compound of Formula F20 is
selected from
Rl(L-Trp)-D-Asn-L-h-Asn-L-Thr-Sar-L-Ala-L-Asp-D-Lys-L-omAsp-Gly-D-Asn-L-Glu-L-
Ile and
Rl (L-Trp)-D-Asn-L-h-Asn-L-Thr-Sar-L-AIa-L-Asp-D-Lys-L-omAsp-Gly-D-Asn-L-3mGlu-
LIle
[0180] Exeinplary compounds Formula F20 include, without limitation, compounds
C343,
and C340 (vide supra).
223
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0181] In another embodiment, the invention provides a compound of the Formula
F21
H02C R12
HN NH O CONH2
O O
H2NOC O O O N R1
N
N
H H
NH O O
11 ~r( "'
O NCH3 C02H /
N
HN (CH2)4R8** O H
MeO 0 HN
HO H
HN N
O H
O
HO2C
(F21)
and salts thereof; wherein:
a) RI is
C N (CH2)6CH(CH3)2 N (CH2)sCH(CH3)CH2CH3
N (CH2)sCHa y = ~
H 0 0
> > >
Ny(CH2)8CH(CH3)CHZCH3 Ny(CHZ)8CH(CH3)a
o , or o
b) R1a is H or CH3, and
c) R8** is amino, monosubstituted amino, disubstituted amino, NH-amino
protecting group,
acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino.
[0182] In another embodiment of the invention, a compound of Formula F21 is
selected from
Rl-(L-Trp)-D-Asn-L-Asp-L-Thr-Sar-L-AIa-L-Asp-D-Lys-L-omAsp-Gly-D-Asn-L-3mGlu-L-
Ile and
Rl-(L-Trp)-D-Asn-L-Asp-L-Thr-Sar-L-A1a-L-Asp-D-Lys-L-omAsp-Gly-D-Asn-L-Glu-
LIle.
[0183] Exemplary compounds Formula F21 include, without limitation, compounds
C265,
and C271 (vide supra).
224
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0184] In another embodiment, the invention provides a compound of the Formula
F22
HOZC ~ ~
CH3 \ NH
HN NH p CONH2 O
O O
00 O N NH~(CH2)$CH(CH3)CH2CH3
N
N
HO NH O H 0 H
I I
O NH C02H ~ I \
N
HN O H
O HN
CH3 O
HO2C HN N Rs= 1-1 H
-,If O
HO2C
(F22)
and salts thereof; wherein:
R6*is amino, monosubstituted amino, disubstituted amino, NH-amino protecting
group,
acylamino, ureido, guanidino, carbamoyl, sulfoinamino, thioacylamino,
thioureido, iminoamino,
or phosphonamino.
[0185] In another embodiment of the invention, a compound of Formula F22 is
(Li rp)-D-Asn-L-Asp-L-Thr-Gly-L-Orn-L-Asp-D-AIa-L-Asp-Gly-D-Ser-L-3mGlu-L- rp
NH(CO)(CH2)$CH(CH3)CH2CH3
[0186] An exemplary compound Formula F22 includes, without limitation,
compound C3
(vide supra).
[0187] In one embodiment of the invention, substituent R' of any of the
compounds of
Formula F1-F20 is amino, acylamino, NH-amino protecting group or carbamoyl. In
another
embodiment of the invention, substituent Rl of any of the compounds of Formula
F1-F20 is a
Clo-C13 alkanoylamino. In yet another embodiment of the invention, substituent
Rl of any of the
compounds of Formula F1-F20 is
225
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
C N (CH2)6CH(CH3)2 N (CHz)aCH(CH3)CHpCH3
..SS' N CH CH ~ = y
H ( z)a s 0 0
> > >
~õ-N (CHz)aCH(CH3)CH2CH3 õ~N ~L.\/ c.~y(cH2)acH(cH3)2
I0 I , or In yet another embodiment of the invention, substituent Rl of any of
the compounds of Formula
Fl-F20is
H
(CH2)6CH(CH3)CH2CH3
y
0
[0188] In one embodiment of the invention, substituent R6* of any of the
compounds of
Formula Fl-F5, F10-F14, F19 and F22 is amino, NH-amino protecting group or
carbamoyl. In
another embodiment of the invention, substituent R6* of any of the compounds
of Formula of F1-
F5, F10-F14, F19 and F22 is amino.
[0189] In one embodiment of the invention, substituent R8** of any of the
compounds of
Formula F2-F5, F7-F9, F13, F15, F16, F18 and F20-F21 is amino, NH-amino
protecting group
or carbamoyl. In another embodiment of the invention, substituent R8** of any
of the compounds
of Formula F2-F5, F7-F9, F13, F15, F16, F18 and F20-F21 is amino.
[01901 It will be understood by one of skill in the art that the compounds of
the invention,
particularly compounds of Formula I and Formula Fl-F22, are useful as
intermediates for the
preparation of other compounds of Formula I and Formula F1-F22. Particularly
useful
compounds that are also intermediates are compounds of Formula I, F2-F5, F13
and F19 wherein
at least one of R1, R6* or R 8** is amino, NH-amino protecting group or
carbamoyl; compounds of
Formula F1 or F10-F14 wherein at least one of R' or R6* is amino, NH-amino
protecting group
or carbamoyl; compounds of Formula F7-9, F15-16, F18 and F20 wherein at least
one of Ri or
R8** is amino, NH-amino protecting group or carbamoyl; compounds of Formula
F22 wherein
R6* is amino, NH-amino protecting group or carbamoyl; compounds of Formula F21
wherein
R$** is amino, NH-amino protecting group or carbamoyl;and compounds of Formula
F6 and F17
wherein R' is amino, NH-amino protecting group or carbamoyl.
Pharmaceutical Compositions and Methods of Use Thereof
226
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0191] The instant invention provides pharmaceutical compositions or
formulations
comprising, in one embodiment, compounds of Formula I or compounds of any of
Formula Fl-
F22, or salts thereof.
[0192] Compounds of the present invention, preferably compounds Formula I or
compounds
of any of Formula F1-F22, or pharmaceutically acceptable salts thereof, can be
formulated for
oral, intravenous, intramuscular, subcutaneous or parenteral administration
for the therapeutic or
prophylactic treatment of diseases, particularly bacterial infections. For
oral or parenteral
administration, compounds of the present invention can be mixed with
conventional
pharmaceutical carriers and excipients and used in the form of tablets,
capsules, elixirs,
suspensions, syrups, wafers and the like. The compositions comprising a
compound of this
invention will contain from about 0.1 to about 99% by weight of the active
compound, and more
generally from about 10 to about 30%.
[0193] The pharmaceutical preparations disclosed herein are prepared in
accordance with
standard procedures and are administered at dosages that are selected to
reduce, prevent or
eliminate the infection (See, e. g., "Remington's Pharmaceutical Sciences",
Mack Publishing
Company, Easton, PA and "Goodman and Gilman's The Pharmaceutical Basis of
Therapeutics",
Pergamon Press, New York, NY, the contents of which are incorporated herein by
reference, for
a general description of the methods for adniinistering various antimicrobial
agents for human
therapy). The compositions of the present invention, preferably compositions
of Formulas I or
compositions of any of Formulas Fl-F22, can be delivered using controlled
(e.g., capsules) or
sustained release delivery systems (e.g., bioerodable matrices). Exemplary
delayed release
delivery systems for drug delivery that are suitable for administration of the
compositions of the
invention, preferably compositions of Formula I or any of Formulas Fl-F22, are
described in
U.S. Patent Nos. 4,452,775 (issued to Kent), 5,239,660 (issued to Leonard),
and 3,854,480
(issued to Zaffaroni).
[0194] The pharmaceutically-acceptable compositions of the present invention
comprise one
or more compounds of the invention, preferably compounds of Formula I or
compounds of any
of Formulas F1-F22, in association with one or more nontoxic, pharmaceutically-
acceptable
carriers and/or diluents and/or adjuvants and/or excipients, collectively
referred to herein as
"carrier" materials, and if desired other active ingredients. The compositions
may contain
common carriers and excipients, such as corn starch or gelatin, lactose,
sucrose, microcrystalline
cellulose, kaolin, mannitol, dicalcium phosphate, sodium chloride and alginic
acid. The
227
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
compositions may contain croscarmellose sodium, microcrystalline cellulose,
corn starch,
sodium starch glycolate and alginic acid.
[0195] Tablet binders that can be included are acacia, methylcellulose, sodium
carboxymethylcellulose, polyvinylpyrrolidone (Povidone), hydroxypropyl
methylcellulose,
sucrose, starch and ethylcellulose.
[0196] Lubricants that can be used include magnesium stearate or other
metallic stearates,
stearic acid, silicone fluid, talc, waxes, oils and colloidal silica.
[0197] Flavoring agents such as peppermint, oil of wintergreen, cherry
flavoring or the like
can also be used. It may also be desirable to add a coloring agent to make the
dosage form more
aesthetic in appearance or to help identify the product.
[0198] For oral use, solid formulations such as tablets and capsules are
particularly useful.
Sustained release or enterically coated preparations may also be devised. For
pediatric and
geriatric applications, suspensions, syrups and chewable tablets are
especially suitable. For oral
administration, the pharmaceutical compositions are in the form of, for
example, a tablet,
capsule, suspension or liquid. The pharmaceutical composition is preferably
made in the form of
a dosage unit containing a therapeutically-effective amount of the active
ingredient. Examples of
such dosage units are tablets and capsules. For therapeutic purposes, the
tablets and capsules
which can contain, in addition to the active ingredient, conventional carriers
such as binding
agents, for example, acacia gum, gelatin, polyvinylpyrrolidone, sorbitol, or
tragacanth; fillers, for
example, calcium phosphate, glycine, lactose, maize-starch, sorbitol, or
sucrose; lubricants, for
example, magnesium stearate, polyethylene glycol, silica, or talc;
disintegrants, for example,
potato starch, flavoring or coloring agents, or acceptable wetting agents.
Oral liquid preparations
generally are in the form of aqueous or oily solutions, suspensions,
emulsions, syrups or elixirs
may contain conventional additives such as suspending agents, emulsifying
agents, non-aqueous
agents, preservatives, coloring agents and flavoring agents. Examples of
additives for liquid
preparations include acacia, almond oil, ethyl alcohol, fractionated coconut
oil, gelatin, glucose
syrup, glycerin, hydrogenated edible fats, lecithin, methyl cellulose, methyl
or propyl para-
hydroxybenzoate, propylene glycol, sorbitol, or sorbic acid.
[0199] For intravenous (IV) use, a compound of the present invention can be
dissolved or
suspended in any of the commonly used intravenous fluids and administered by
infusion.
Intravenous fluids include, without limitation, physiological saline or
Ringer's solution.
228
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Intravenous administration may be accomplished by using, without limitation,
syringe,
minipump or intravenous line.
[0200] Formulations for parenteral administration can be in the form of
aqueous or non-
aqueous isotonic sterile injection solutions or suspensions. These solutions
or suspensions can
be prepared from sterile powders or granules having one or more of the
carriers mentioned for
use in the formulations for oral administration. The compounds can be
dissolved in polyethylene
glycol, propylene glycol, ethanol, corn oil, benzyl alcohol, sodium chloride,
and/or various
buffers.
[0201] For intramuscular preparations, a sterile formulation of a compound of
the present
invention, or a suitable soluble salt form of the compound, for example the
hydrochloride salt,
can be dissolved and administered in a pharmaceutical diluent such as Water-
for-Injection
(WFI), physiological saline or 5% glucose. A suitable insoluble form of the
compound may be
prepared and administered as a suspension in an aqueous base or a
pharmaceutically acceptable
oil base, e.g., an ester of a long chain fatty acid such as ethyl oleate.
[0202] A dose of an intravenous, intramuscular or parental formulation of a
compound of the
present invention may be adminstered as a bolus or by slow infusion. A bolus
is a dose that is
administered in less than 30 minutes. In a preferred embodiment, a bolus is
administered in less
tharn 15 or less than 10 minutes. In a more preferred embodiment, a bolus is
administered in less
than 5 minutes. In an even more preferred embodiment, a bolus is administered
in one minute or
less. An infusion is a dose that is administered at a rate of 30 minutes or
greater. In a preferred
embodiment, the infusion is one hour or greater. In another embodiment, the
infusion is
substantially constant.
[0203] For topical use the compounds of the present invention, preferably
compounds of
Formula I or compounds of any of Formula Fl-F22, can also be prepared in
suitable forms to be
applied to the skin, or mucus membranes of the nose and throat, and can take
the form of creams,
ointments, liquid sprays or inhalants, lozenges, or throat paints. Such
topical formulations
further can include chemical compounds such as dimethylsulfoxide (DMSO) to
facilitate surface
penetration of the active ingredient.
[0204] For application to the eyes or ears, the compounds of the present
invention, preferably
compounds Formula I or compounds of any of Formula F1-F22, can be presented in
liquid or
semi-liquid form formulated in hydrophobic or hydrophilic bases as ointments,
creams, lotions,
paints or powders.
229
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0205] For rectal administration the compounds of the present invention,
preferably
compounds Formula I or compounds of any of Formula FI-F22, can be administered
in the form
of suppositories admixed with conventional carriers such as cocoa butter, wax
or other glyceride.
[0206] Alternatively, the compounds of the present invention, in one
embodiment,
compounds of Formula I or compounds of any of Formulas F1-F22, can be in
powder form for
reconstitution in the appropriate pharmaceutically acceptable carrier at the
time of delivery. In
another embodiment, the unit dosage form of the compound can be a solution of
the compound
or preferably a salt thereof in a suitable diluent in sterile, hermetically
sealed ampoules or sterile
syringes. The concentration of the compound in the unit dosage may vary, e.g.
from about 1
percent to about 50 percent, depending on the compound used and its solubility
and the dose
desired by the physician. If the compositions contain dosage units, each
dosage unit preferably
contains from 1-500 mg of the active material. For adult human treatment, the
dosage employed
preferably ranges from 5 mg to 10 g, per day, depending on the route and
frequency of
administration.
[0207] In another aspect, the invention provides a method for inhibiting the
growth of
microorganisms, preferably bacteria, comprising contacting said organisms with
a compound of
the present invention under conditions which permit contact of the compound
with said organism
and with said microorganism. Such conditions are known to one skilled in the
art and are
exemplified in the Examples. This method involves contacting a microbial cell
with a
therapeutically-effective amount of compound(s) of the invention, preferably
compound(s) of s
Formula I or compound(s) of any of Formula F1-F22 in vivo or in vitro.
[0208] According to this aspect of the invention, the novel compositions
disclosed herein are
placed in a pharmaceutically acceptable carrier and are delivered to a
recipient subject
(preferably a human) in accordance with known methods of drug delivery. In
general, the
methods of the invention for delivering the compositions of the invention in
vivo utilize art-
recognized protocols for delivering the agent with the only substantial
procedural modification
being the substitution of the compounds of the present invention, preferably
compounds of
Formula I or compounds of any of Formula Fl-F22, for the drugs in the art-
recognized protocols.
Likewise, the methods for using the claimed composition for treating cells in
culture, for
example, to eliminate or reduce the level of bacterial contamination of a cell
culture, utilize art-
recognized protocols for treating cell cultures with antibacterial agent(s)
with the only substantial
procedural modification being the substitution of the compounds of the
invention, preferably
230
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
compounds of Formula I or compounds of any of Formula F 1-F22, for the agents
used in the art-
recognized protocols.
[0209] In one embodiment, the invention provides a method for treating an
infection,
especially those caused by gram-positive bacteria, in a subject with a
therapeutically-effective
amount of a compound of the invention. Exemplary procedures for delivering an
antibacterial
agent are described in U.S. Patent Number 5,041,567, and PCT patent
application number
EP94/02552 (publication number WO 95/05384), the entire contents of which
documents are
incorporated in their entirety herein by reference. As used herein, the phrase
"therapeutically-
effective amount" means an amount of a compound of the present invention that
prevents the
onset, alleviates the symptoms, or stops the progression of a bacterial
infection. The term
"treating" is defined as administering, to a subject, a therapeutically-
effective amount of a
compound of the invention both to prevent the occurrence of an infection and
to control or
eliminate an infection. The term "subject," as described herein, is defined as
a mammal, a plant
or a cell culture. In a preferred embodiment, a subject is a human or other
animal patient in need
of antibacterial treatment.
[0210] The method comprises administering to the subject an effective dose of
a compound
of the present invention. An effective dose is generally between about 0.1 and
about 100 mg/kg
of a compouncl of the invention or a pharmaceutically acceptable salt thereof.
A preferred dose
is from about 0.1 to about 50 mg/kg of a compound of the invention or a
pharmaceutically
acceptable salt thereof. A more preferred dose is from about 1 to 25 mg/kg of
a compound of the
invention or a pharmaceutically acceptable salt thereof. An effective dose for
cell culture is
usually between 0.1 and 1000 g/mL, more preferably between 0.1 and 200 g/mL.
[0211] Compositions containing the compounds of the invention can be
administered as a
single daily dose or in multiple doses per day. The treatment regime may
require administration
over extended periods of time, e.g., for several days or for from two to four
weeks. The amount
per administered dose or the total amount administered will depend on such
factors as the nature
and severity of the infection, the age and general health of the patient, the
tolerance of the patient
to the compound and the microorganism or microorganisms involved in the
infection. A method
of administration to a patient of daptomycin, another member of the
depsipeptide compound
class, is disclosed in United States Patent Numbers 6,468,967 and 6,852,689,
the contents of
which are herein incorporated by reference.
231
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0212] A compound of the present invention may also be administered in the
diet or feed of a
patient or animal. If administered as part of a total dietary intake, the
amount of compound
employed can be less than 1% by weight of the diet and preferably no more than
0.5% by weight.
The diet for animals can be normal foodstuffs to which the compound can be
added or it can be
added to a premix.
[0213] The present invention also provides methods of administering a compound
of the
invention, preferably a compound of Formula I or a compound of any of Formulas
F1-F22, or a
pharmaceutical composition thereof to a subject in need thereof in an amount
that is efficacious
in reducing, ameliorating or eliminating the bacterial infection. The compound
may be
administered orally, parenterally, by inhalation, topically, rectally,
nasally, buccally, vaginally,
or by an implanted reservoir, external pump or catheter. The compound may be
prepared for
opthalmic or aerosolized uses. The compounds of the present invention can be
administered as
an aerosol. A preferred aerosol delivery vehicle is an anhydrous or dry powder
inhaler.
Compounds of Formula I or compounds of any of Formula F1-F22, or a
pharmaceutical
composition thereof may also be directly injected or administered into an
abscess, ventricle or
joint. Parenteral administration includes subcutaneous, intravenous,
intramuscular, intra-
articular, intra-synovial, cistemal, intrathecal, intrahepatic, intralesional
and intracranial injection
or infusion. In a preferred embodiment, the compounds of the present invention
are administered
intravenously, subcutaneously or orally. In a preferred embodiment for
administering a
compound according to Formula I or a compound of any of Formula F1-F22 to a
cell culture, the
compound may be administered in a nutrient medium.
[0214] The method of the instant invention may be used to treat a subject
having a bacterial
infection in which the infection is caused or exacerbated by any type of
bacteria, particularly
gram-positive bacteria. In one embodiment, a compound of the present invention
or a
pharmaceutical composition thereof is administered to a patient according to
the methods of this
invention. In a preferred embodiment, the bacterial infection may be caused or
exacerbated by
gram-positive bacteria. These gram-positive bacteria include, but are not
limited to, methicillin-
susceptible and methicillin-resistant staphylococci (including Staphylococcus
aureus, S.
epiderinidis, S. haemolyticus, S. hominis, S. saprophyticus, and coagulase-
negative
staphylococci), glycopeptide intermediary- susceptible S. aureus (GISA),
vancomycin-resistant
Staphylococcus aureus (VRSA), penicillin-susceptible and penicillin-resistant
streptococci
(including Streptococcus pneumoniae, S. pyogenes, S. agalactiae, S. avium, S.
bovis, S. lactis, S.
232
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
sangius and Streptococci Group C, Streptococci Group G and viridans
streptococci), enterococci
(including vancomycin-susceptible and vancomycin-resistant strains such as
Enterococcus
faecalis and E. faecium), Clostridium difficile, C. clostridiiforme, C.
innocuum, C. pe~fringens,
C. ramosuna, Haemophilus influenzae, Listeria monocytogenes, Corynebacter=ium
jeikeium,
BifidobacteYium spp., Eubacterium aerofaciens, E. lentum, Lactobacillus
acidophilus, L. casei,
L. plantarum, Lactococcus spp., Leuconostoc spp., Pediococcus,
Peptostreptococcus anaerobius,
P. asaccarolyticus, P. magnus, P. micros, P. prevotii, P. productus,
Propionibacterium acnes,
Actinonayces spp., Moraxella spp. (including M. catarrhalis) and Escherichia
spp. (including E.
coli).
[0215] In a preferred embodiment, the antibacterial activity of compounds of
Formula I or
compounds of any of Formula F1-F22 against classically "resistant" strains is
comparable to that
against classically "susceptible" strains in in vitro experiments. In another
preferred
embodiment, the minimum inhibitory concentration (MIC) value for compounds
according to
this invention, against susceptible strains, is typically the same or lower
than that of vancomycin
or daptomycin. Thus, in a preferred embodiment, a compound of this invention
or a
pharmaceutical composition thereof is administered according to the methods of
this invention to
a patient who exhibits a bacterial infection that is resistant to other
compounds, including
vancomycin or daptomycin. In addition, unlike glycopeptide antibiotics,
depsipeptide
compounds such as those disclosed in the present invention, exhibit rapid,
concentration-
dependent bactericidal activity against gram-positive organisms. Thus, in a
preferred
embodiment, a compound according to this invention or a pharmaceutical
composition thereof is
administered according to the methods of this invention to a patient in need
of rapidly acting
antibiotic therapy.
[0216] The method of the instant invention may be used for any bacterial
infection of any
organ or tissue in the body. In a preferred embodiment, the bacterial
infection is caused by
gram-positive bacteria. These organs or tissue include, without limitation,
skeletal muscle, skin,
bloodstream, kidneys, heart, lung and bone. The method of the invention may be
used to treat,
without limitation, skin and soft tissue infections, bacteremia and urinary
tract infections. The
method of the invention also may be used to treat mixed infections that
comprise different types
of gram-positive bacteria, or which comprise both gram-positive and gram-
negative bacteria.
These types of infections include intra-abdominal infections and
obstetrical/gynecological
infections. The method of the invention also may be used to treat an infection
including, without
233
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
limitation, endocarditis, nephritis, septic arthritis, intra-abdominal sepsis,
bone and joint
infections. and osteomyelitis. In a preferred embodiment, any of the above-
described diseases
may be treated using compounds according to this invention or pharmaceutical
compositions
thereof.
[0217] The method of the present invention may also be practiced while
concurrently
administering one or more other antimicrobial agents, such as antibacterial
agents (antibiotics) or
antifungal agents. In one aspect, the method may be practiced by administering
more than one
compound according to this invention. In another embodiment, the method may be
practiced by
administering a compound according to this invention with a lipopeptide
compound, such as
daptomycin or the lipopeptide compounds described, for example in United
States Patents
6,911,525; and 6,794,490 and in International Patent Applications WO01/44272;
WO01/44274;
WO01/44271 and W003/014147.
[0218] Antibacterial agents and classes thereof that may be co-administered
with a
compound according to the invention include, without limitation, penicillins
and related drugs,
carbapenems, cephalosporins and related drugs, aminoglycosides, bacitracin,
gramicidin,
mupirocin, chloramphenicol, thiamphenicol, fusidate sodium, lincomycin,
clindamycin,
macrolides, novobiocin, polymyxins, rifamycins, spectinomycin, tetracyclines,
vancomycin,
teicoplanin, streptogramins, anti-folate agents including sulfonamides,
trimethoprim and its
combinations and pyrimethamine, synthetic antibacterials including
nitrofurans, methenamine
mandelate and methenamine hippurate, nitroimidazoles, quinolones,
fluoroquinolones, isoniazid,
ethambutol, pyrazinamide, para-atninosalicylic acid (PAS), cycloserine,
capreomycin,
ethionamide, prothionamide, thiacetazone, viomycin, everninomycin,
glycopeptide,
glycylcylcline, ketolides, oxazolidinone; imipenen, amikacin, netilmicin,
fosfomycin,
gentamicin, ceftriaxone, ZIRACIN , LY 333328, CL 331002, HMR 3647, ZYVOX ,
SYNERCID , aztreonam metronidazole, epiroprim, OCA-983, GV-143253, sanfetrinem
sodium,
CS-834, biapenem, A-99058.1, A-165600, A-179796, KA 159, dynemicin A, DX8739,
DU
6681; cefluprenam, ER 35786, cefoselis, sanfetrinem celexetil, HGP-31,
cefpirome, HMR-3647,
RU-59863, mersacidin, KP 736, rifalazil; AM 1732, MEN 10700, lenapenem, BO
2502A, NE-
1530, PR 39, K130, OPC 20000, OPC 2045, veneprim, PD 138312, PD 140248, CP
111905,
sulopenem, ritipenam acoxyl, RO-65-5788, cyclothialidine, Sch-40832, SEP-
132613,
micacocidin A, SB-275833, SR-15402, SUN A0026, TOC 39, carumonam, cefozopran,
cefetamet pivoxil, and T 3811.
234
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0219] Antifungal agents that may be co-administered with a compound according
to the
invention include, without limitation, caspofungen, voriconazole,
sertaconazole, IB-367, FK-
463, LY-303366, Sch-56592, sitafloxacin, DB-289 polyenes, such as
amphotericin, nystatin,
primaricin; azoles, such as fluconazole, itraconazole, and ketoconazole;
allylamines, such as
naftifine and terbinafine; and anti-metabolites such as flucytosine. Other
antifungal agents
include without limitation, those disclosed in Fostel, et al., 2000, Drug
Discovery Today 5: 25-
32, herein incorporated by reference. Fostel et al. discloses antifungal
compounds including
corynecandin, Mer-WF3010, fusacandins, artrichitin/LL 15G256, sordarins,
cispentacin,
azoxybacillin, aureobasidin and khafrefungin.
[0220] A compound according to this invention may be administered according to
this
method until the bacterial infection is eradicated or reduced. In one
embodiment, a compound of
Formula I or a compound of any of Formulas Fl-F22 is administered for a period
of time from 2
days to 6 months. In a preferred embodiment, a compound of Formula I or a
compound of any
of Formulas Fl-F22 is administered for 7 to 56 days. In a more preferred
embodiment a
compound of Formula I or a compound of any of Formulas Fl-F22 is administered
for 7 to 28
days. In an even more preferred embodiment, a compound of Formula I or a
compound of any
of Formulas Fl-F22 is administered for 7 to 14 days. A compound of Formula I
or or a
compound of any of Formulas F1-F22 may be administered for a longer or shorter
time period if
it is so desired.
[0221] The instant invention provides antibacterial compositions or
formulations comprising,
in one embodiment, compounds of Formula I or compounds of any of Formula F1-
F22, or salts
thereof. In one embodiment the antibacterial compositions may be contained in
an aqueous
solution. In another embodiment the aqueous solution may be buffered. In
another embodiment
the buffer may hav an acidic, neutral, or basic pH.
Preparation of Novel Depsipeptides
1. Synthetic Processes
[0222] In one embodiment of the invention, the compounds of Formula I or
Formula Fl-F22
may be prepared using solid support chemistry. Three preferred methods,
Methods A-C,
produce resin bound linear precursor nn3, nn3 a or nn3b.
235
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0223] As outlined in Scheme I, Method A utilizes a resin-bound 7 amino acid -
derived
polypeptide fragment, nnl, and a six amino acid-derived polypeptide fragment,
nn2. This
method is referred to as a "7 + 6 fragment synthesis".
236
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme I
Method A "7 + 6" fragment synthesis
R12 R13A
ResinO2C
P1SHN
HN OP1 p 0 WA 0
H
R11A p 0 0 H)y NYJ,~N R1A
\NR11. O R3A p Ra.
O + RyA NR5A ,
/ N
O H
HN
~O P7HN HO
RsA R8A o RHN' s N
1''1( N p
p Ps02C (nn1) (nn2)
R12
ResinO2C
R13A
HN OP1
I
R11A O 0 P18HN O 0 RZA O
O ~N R1A
NR11R N N
H
p) O R3A p Rz.
NRSA
HN R5'A
N
O R6A ~=O H
RaA p NH
WA-~=
HN~ N
N p
O RB'
PBOZC (nn3)
ResinO2C R12 R13A
HN
HN O 0 R2A 0
R11A ~O O 0 ~N~ N R1A
N
NR11. p H R3A O RZ
p=~ R5'A NR5A
N
HN ~=O H
~p HN
R9A RSA o RsA
HN,l N N
p
O Rs
P80ZC (nn4)
237
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0224] Alternatively, as described in Scheme II, Method B utilizes a resin-
bound 6 amino
acid -derived polypeptide fragment, nnl a, and a seven amino acid-derived
polypeptide fraglnent,
nn2a. This method is referred to as a "6 + 7 fragment synthesis".
238
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme II
Method B. "6 + 7" fragment synthesis
R12 R13A
Resin02C
OP1 P18HN
HN O 0 R2A O
IO 0 0
~N R1A
H I NH
R11A NR11' O R3A 0 R2'
~ + NR5
O
R5'
HN -~=O H
HN 6A
R9A 0 RBA O R
HN' ~N NHP20 HO 0
~if( RB.
O 802C (nn1a) (nn2a)
R12
Resin02C
R13A
HN OP1
~
R11A O 0 P16HN O 0 R2A O
0 N R1q
NR11' H N)y N
O=~ O R3A 0 R2.
NR5
HN e-~= N
~O R6A O H
R9A RBA 0 NH
HN~ N
O
0 Re
P802C (nn3a)
R12 R13A
Resin02C
HN
1
HN O 0 R2A 0
O O 0 N~N
R11A --~N R1A
NR11. O H R3A O R2.
O=~ NR5
e
HN ~O N
H
~O HN
Rsq RA 0 RsA
HN N
N O
O RB'
P8O2C (nn4a)
239
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0225] Another method, Method C, utilizes a 6 amino acid derived polypeptide,
a resin
bound-amino acid, and a second 6 amino acid derived polypepetide. This method
is referred to
as a"1 + 6 + 6 fragxnent synthesis".
240
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme III
Method C. "1 + 6 + 6" fragment synthesis.
R12A
ResinO2C
OP R12A
HN 25 ResinO2C
OH
O O OK OP25
H2N 0 ~Q
R11A R11A
NR11'
p=~ (n23) + p NR11
HN
Ry~O R8A p P28HN RsA HN
A 0 P28HN
R9A RBA p RsA -~= O
HNN HN HN N~
O Rs 0 R8 O
Pg02C (nnlb) P802C (n26)
R13A
NHP1s +
O 0 R2A 0
~ N R1A R12A
N N Resin02C
H R13A
Q R3A O R2= OP1
NRSA / HN
Rs=A ~ O 0 P18HN O O R2A O
~O H N R71A O N H R
1A
HO (nn2) NR11 N-ly T~-~NN
Q~ Q H R3A NRSA HN RsA
R9A~O R8A o RsA O H
NH
HN_ N
lN O
R12A R13A R$=
ResinO2C Pa02C (nn3b)
HN
HN O 0 R2P' 0
O O 0 ~N R1A
R11A N ~N
NR11 O H R3A Q R2.
O R5 A NR5A
N I / I
~ -~
HN ~=O H
~p HN
sPsR RBA o RsA
HN~ N
O Rs= O
Pa02C (nn4b)
Solid Support Synthesis of Depsipeptide Compounds Method A: 7 + 6 Frament
Synthesis
241
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0226] The depsipeptide compounds of Formula I may be synthesized on a solid
support as
outlined in Scheme IV, Scheme V and Scheme VI as follows.
242
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme IV
R12
ResinO2C
HN OP1
R12 O O
R12 ResinO2C IIq
Resin02C
HN pP~ R NR11;
P2HN ~
p OP, R1~a Op ~ O
NR11*P3 HN
~O P7HN
Rgq R8A p RsA
(nn5) (nn6) HN~N * N
i O
O Ra
P$O2C (nn1)
[0227] In the first step, a protected glutamic acid-derivative such as
commercially available
N-a-Fmoc-L-glutamic acid a-allyl ester or N-Fmoc-L-3-methyl glutamic acid a-
allyl ester (See
Examples 1-68 and 1-69, vide infra) is coupled to a resin to give Compound
nn5, wherein R12 is
as defined previously. A resin or solid support, such as, but not limited to,
Wang, HMPA, Safety
Catch, Rink Acid, 2-chlorotrityl-chloride resin, trityl-chloride resin, 4-
methyltrityl-chloride resin,
4-methoxytrityl-chloride resin or PAM resin may be used in this reaction.
Protecting groups P1
and P2 are chosen so that they may be removed independently of one another and
without
effecting cleavage of the peptide from the resin. Examples of protecting
groups can be found in
"Protecting Groups in Organic Synthesis" by Theodora W. Greene, (vide supra),
hereafter
"Greene", incorporated herein by reference. A protecting group combination,
such as, but not
limited to P1 is allyl ester and P2 is Fmoc is suitable for this reaction.
[0228] Deprotection of the amine of Compound nn5, followed by coupling of the
free amino
with an amino acid or a protected amino acid affords Compound nn6, wherein P3
is a protecting
group that can be removed independently of P1 and without effecting cleavage
of the peptide
from the resin; R' lA is an amino acid side chain, a protected amino acid side
chain, methyl, CH2-
OP4, or CH2-CONHP5; each of P4 and P5 .is independently a suitable protecting
group and each
of PI and R' 1* is as defined previously. This peptide coupling process, i.e.,
deprotection of the
alpha-amino group, followed by coupling to a protected amino acid, is repeated
until the desired
number of amino acids has been coupled to the resin. In Scheme IV, a total of
seven amino acids
have been coupled to give compound nnl wherein,
243
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
6A
R6A is methyl orR ; R6*A is a protected amino, monosubstituted amino,
disubstituted amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino,
thioacylamino,
thioureido, iminoamino, or phosphonamino, provided that R6*A is compatible
with the conditions
required to remove the resin from the peptide;
R8A is an amino acid side chain, a protected amino acid side chain, methyl,
CHa-OP6 , CH2-
CONHP5* or R$**A ; wherein each of P5* and P6 is independently a suitable
protecting group; wherein R8**A is a protected amino, monosubstituted amino,
disubstituted
amino, acylamino, ureido, guanidino, carbamoyl, sulfonamino, thioacylamino,
thioureido,
iminoamino, or phosphonamino, provided that R8**A is compatible with the
conditions required
to remove the resin from the peptide;
OMe
OP9 OP9
wherein R9A is 0 , 0 , or an amino acid side chain substituted with at least
OP9
one carboxylic acid group of the formula, 0 ; P7 is a protecting group that
can be
removed independently of PI without effecting cleavage of the peptide from the
resin; each of P8
and P9 is independently a suitable protecting group such that Pi and P7 may be
removed
independently of each of P8 and P9 and that each of P8 and P9 is cleaved upon
cleavage from the
resin; and P1, R8*, R9A, Rll* R11A and R12 are as defined previously.
[0229] A second peptide is coupled to a resin in a similar fashion, as
outlined in Scheme V.
244
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme V
HO HO O
NR5AP10 NHP11 NHP12
step I Rs a~=O step 2 p step 3 p H 3A
Resin-OH ~
NR 5A NR5a R
Resin-O R5a R5=A
O O
Resin-O Resin-O
(nn7) (nn8) (nn9)
step 4
HO O R2A HO O R2a 0
,~yN~ H~N y R1A
N NR2*P13 N " N
i
R3A O O R3A O R2.
/NRSA step NRsa ~ I \
RS=A RS=a N
p p H
Resin-O (n10) Resin-O (n11)
step 6
R13A R13A
P18HN p o R2A o P18HN-1V p O Rza O
H I
N~NN R1A Ste N I R
O O N~ N 1a
p R 3A O I R Z= H R3a R~.
NR5A / I \ NRSA
RS a R5 a N
O
H p H
Resin-O (n12) HO (nn2)
[0230] In step 1, an N-protected-glycine, such as commercially available Fmoc-
N-glycine, is
coupled to a resin to give Compound nn7 wherein R5A and R5*A are independently
hydrido and
Plo is a protecting group chosen so that it may be removed without effecting
cleavage of the
peptide from the resin. The choice of resin used in step 1 is dependent upon
the nature of the
amino acid that is coupled in steps 2-6. If the amino acid side chains contain
protecting groups, a
resin must be chosen such that the protecting groups remain intact when the
resin is removed
from the peptide in step 7. Resins that can be cleaved while preserving the
protecting groups of
peptides include, but are not limited to, Safety Catch, Rink Acid, 2-
chlorotrityl-chloride resin,
trityl-chloride resin, 4-methyltrityl-chloride resin, 4-methoxytrityl-chloride
resin or PAM resin.
[0231] Deprotection of the protected amino of Compound nn7, followed by
coupling of the
free amino with n14
245
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
HO CH3
HO
NHP11
0
(n14)
affords Compound nn8, wherein P11 is a protecting group chosen so that it may
be removed
without effecting cleavage of the peptide from the resin. This peptide
coupling process, i.e.,
deprotection of the alpha-amino group, followed by coupling to a protected
amino acid, is
repeated until the desired number of amino acids has been coupled to the
resin. In Scheme V,
five amino acids have been coupled to give Compound nl l wherein R1A is a
protected amino,
monosubstituted amino, disubstituted amino, acylamino, ureido, guanidino,
carbamoyl,
sulfonamino, thioacylamino, thioureido, iminoamino, or phosphonamino, provided
that R1A is
compatible with the conditions required to remove the resin from the peptide;
R2A is an amino
acid side chain, a protected amino acid side chain, CHZ-CH2-C02P14, or CH2-
CONHP15; R3A 1S
CH2-COZP16, CH(OP17)CONH2, CH2CONH2, a non-protienogenic amino acid side
chain, or a
protected non-proteinogenic amino acid side chain; each of P 12 and P13 is a
protecting group
chosen so that it may be removed without effecting cleavage of the peptide
from the resin; each
P14, P15, P 16 and P17 is independently a suitable protecting group; and R2*,
RSA and R5*A is as
previously defined.
[0232] Compound nl l is coupled with
NHP18
HO
R13A
O
(n15)
to give Compound n12, wherein P18 is a suitable protecting group and R13A is
CH(CH3)2,
CH(CH2CH3)CH3,
O NHP19 O N3
N
H , or
[0233] The peptide n12 is then removed from the resin to give compound nn2
wherein P19 is
a suitable protecting group.
246
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0234] Coupling of the peptide fragments nnl and nn2 is outlined in Scheme VI.
Scheme VI
R12 R13A
ResinOzC
P18HN
R2A O
HN OPt O 0
1 O O 0 N~N
R11A --N R1A
H
NRtti O R3A 0 Rz=
~ I \
O + R5*A NR5A
~
N
HN ~=o H
~O P7HN HO
Ryq R8A o RBq
N
HN' ~ 8 O
~ijf' N
O 802C (nnl) (nn2)
R12
ResinOZC
R73A
HN OPt
O O P18HN O O R2A O
R11A O H
N R1A
NRtt' H N
O=~ Q R3A 0 R2.
NR5A ,
HN R5A N
O RsA ~O H
RsA R8A o NH
HN N~
1 O
0 Rg.
~ Peo2C (nn3)
R12 R13A
ResinO2C
HN
HN O 0 R2A O
1 O O 0 ~N N
R11A R1A
N
NR11 O H R3A O R2.
O=~ NR5A
RVA I
N
HN O H
O HN
Rsq R8A O
-~= HN O N N~RM
O
RgP802C (nn4)
[0235] The peptide fragments nnl and nn2 are coupled to yield the resin bound
peptide nn3
wherein, R2*, R3A R5A R5*A R6A R8* R8A R9A R11A Rll* R1z R13A p p and P18
> > > > > > > > > > > > > > > 1~ 8~ 247
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
are as previously defined. Deprotection of the P 1 and P18 protecting groups,
followed by
cyclization affords a resin-bound depsipeptide nn4 wherein, R1A, R2*, R2A >
R3A > RsA R5*A,
> > >
R8*, R8A, R9A, R11A, Rll*, R1a, R13A, and P8 are as previously defined.
Cleavage of the
depsipeptide from the resin and deprotection of any remaining protecting
groups yields
compounds of Formula I.
Solid Support Synthesis of Depsipeptide ComDounds Method B:
6+ 7 Fragment Synthesis
[0236] The depsipeptide compounds of Formula I may be synthesized on a solid
support as
described in Schemes VII, VIII and IX.
Scheme VII
R12
ResinO2C
HN OPJ
R12 00
ResinO2C R~2 ResinO2C 11q
HN Xopi R NR1
P2HN OP1 00 O
0 R11q ~
t,I*P3 HN
O
Rgq R8A o __~= (nn5) (nn6) HN,Irj., N NHP20
O R$*
Pgo2C (nn1a)
[0237] Compound nn6 is prepared as described in Method A. The peptide coupling
process
(vide supra), i.e., deprotection of the alpha-amino group, followed by
coupling to a protected
amino acid, is repeated until the desired number of amino acids has been
coupled to the resin. In
Scheme VII, a total of six amino acids have been coupled to give compound nnla
wherein, R8*,
RsA, R9A, Rll*, R11A, R12, P1, and P8 are as defined previously and P20 is a
protecting group that
can be removed independently of P1 and without effecting cleavage of the
peptide from the resin,
such as Pi is allyl and P20 is Fmoc.
[0238] A second peptide is coupled to a resin in a similar fashion, as
outlined in Scheme
VIII.
248
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme VIII
NR5P22 HO HO Q
Re' ~ NP71 NP23
N 7
Resin-OH Step lu. P21HN R6A Step HN Q' f-D~ O NR5 Sjep4~ O NR5
ri H R3A
O ._..[ / 0 ~R 6A 5 R~Q R5~Q
Resi O
Resin 0 HN HN
/ ~R6A / ~R6A
0-
Resin 0 Resin 0
(n16) (n17) (n18) (n19)
Step 5
HO 0 R2A 0 HO 0 R2A
H
)yN N
R H I1A NR2"Pz4
H ~N
Q R3A 0 Rz= Q R3A o
NR5 NR5
Rs' N Step 6 R5-=0 H O
HN R6A (n21) HN R6A (n20)
% --- / 0___'~
Resin O Resin O
Step 7
R13A R13A
P18HN Q 0 R2A O P18HN O zA
I' 0 R 0
O N ~ R1A O N ~ R1A
H ~N Step H~ 11 N"
Q R3A O Rz' O R3A O R2
NR5 ~ t \ NR5 ~ I \
R5"~ N R5" N /
H
)=o H
HN (n22) HN R
RsA sA (nn2a)
0- HO
Resiri 0 O
[0239] In step 1, a N-protected-amino acid is coupled to a resin to give
Compound nl6
wherein P21 is a protecting group that can be removed without effecting
cleavage of the peptide
from the resin and R6A is as defined previously. The choice of resin used in
step 1 is dependent
upon the nature of the amino acid that is coupled in steps 2-6. If the amino
acid side chains
contain protecting groups, a resin must be chosen such that the protecting
groups remain intact
when the resin is removed from the peptide in step 8. Resins that can be
cleaved while
249
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
preserving the protecting groups of peptides include, but are not limited to,
Safety Catch, Rink
Acid, 2-chlorotrityl-chloride resin, trityI-chloride resin, 4-methyltrityl-
chloride resin, 4-
methoxytrityl-chloride resin or PAM resin.
[0240] Deprotection of the protected amino of Compound n16, followed by
coupling of the
free amino with a second protected amino acid affords Compound nl7 wherein P22
is a
protecting group that can be removed without effecting cleavage of the peptide
from the resin;
and R5, RS*, and R6 are as defined previously.
[0241] Deprotection of the protected amino of Compound n17, followed by
coupling of the
free amino with n14 (vide supra) affords Compound nl 8, wherein, R5, R5*, R6A
and P9 are as
described previously. The peptide coupling process, i.e., deprotection of the
alpha-amino group,
followed by coupling to a protected amino acid, is repeated until the desired
number of amino
acids has been coupled to the resin. In Scheme VIII, six amino acids have been
coupled to give
Compound n2 1, wherein each of P23 and P24 is a protecting group that can be
removed without
effecting cleavage of the peptide from the resin; R1A, RZA, R2*, R3A, R5, R5*,
and R6A are as
described previously.
[0242] Compound n21 is coupled with n15 (vide supra) to give Compound n22,
wherein
R1A, RzA~ R2*' R3A, R5, Rs*, R6A, Ri3A and P18 are as described previously.
[0243] The peptide n22 is then removed from the resin to give compound nn2a,
wherein RiA,
RzA, Ra*' R3A' Rs, Rs*' R6A' R 13A and P18 are as described previously.
[0244] Coupling of the peptide fragments nnl a and nn2a is outlined in Scheme
IX.
250
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme IX
R12 R13A
Resin02C
PtaHN
R2A 0
HN OP1 O 0
R11A ~O O 0 N"-r NY-'- N R1A
H
NR11O R3A 0 Rz'
+ NR5 ~ I \
R5*
H
HN -~=o
HN
R9A RaA O R6A
HNy N NHP20 HO O
i
a
802C (nnla) (nn2a)
R12
ResinO2C
R13A
HN OPt
I
R11A O 0 P18HN O 0 H R2A 0
O 1A
N R
NR11* N
0=~ O R3A O Rz.
NR5
HN R5~ N /
O R6A H
R9A~ R8A 0 NH
HN
N N~
i O
0 Ra"
/ PaOpC (nn3a)
',// =
ResinO2C R12 R13A
HN
HN O 0 R2A 0
R1tA ~O O 0 N -"Y N N RtA
NR11" 0 H R3A 0 R2.
O=~ NR5
R5'
N
HN O H
0 HN
R9A RaA 0 RsA
HN~ N N
O
O RaPaOZC (nn4a)
[0245] The peptide fragments nnl a and nn2a are coupled to yield the resin
bound peptide
nn3a wherein R1Aa R2A, R2*, R3A a Rsa RS*a R6A a R8*a R8A a R 9A, Ril*a R11A a
R1a a R 13A, pla p8a p9 and
P18 are as described previously.
251
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0246] Deprotection of the PI and P18 protecting groups, followed by
cyclization affords a
resin-bound de si eptide nn4a, wherein R1A> R2A, R2*> R3A> Rs> Rs*, R6A , a
R8* RsA R9A Rii*
p p > > >
R1]A, R12, R13A, and Pg are as described previously.
[0247] Cleavage of the depsipeptide from the resin and deprotection of any
remaining
protecting groups yields compounds of Formula I.
Solid Support Synthesis of Depsipeptide Compounds Method C
1+ 6 + 6 Fra ili~ynthesis.
[0248] In an alternative embodiment of the invention, the depsipeptide
compounds of
Formula I may be synthesized as described in Scheines X-XII.
Scheme X
0
Step 1
Resin Resin-O O
R12A
OP25
P26HN
(n23)
[0249] In step 1, a protected-(3-methyl glutamic acid derivative such as
commercially
available N-a-Fmoc-L-glutamic acid a-allyl ester or N-Fmoc-L-3-methyl glutamic
acid a-allyl
ester (See Examples 1-68 and 1-69, vide infta) is coupled to a resin to give
Compound n23
wherein RI2A is methyl. A resin or solid support, such as, but not limited to,
Wang, HMPA,
Safety Catch, Rink Acid, 2-chlorotrityl-chloride resin, trityl-chloride resin,
4-methyltrityl-
chloride resin, 4-methoxytrityl-chloride resin or PAM resin may be used in
this reaction.
Protecting groups P25 and P26 are chosen so that they can be removed
independently of one
another and without effecting cleavage of the peptides from the resin. A
protecting group
combination, such as, but not limited to P25 is allyl ester and P26 is Fmoc is
suitable for this
reaction.
[0250] A second peptide is coupled to a resin in a similar fashion, as
outlined in Scheme XI.
Scheme XI
252
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Resin- OH
Resin-p HO
W/ p ~
R11A R11A
Resin~p \N-R11= N-R11*
R11A~p )W
\NR11 Pz7 HN HN
O PasHN
Rya ~p Rsa p P28HN -
Rsa Rsa RaA p Rsa
HN N~ HN N
(n24) ~N p Ns. 0
O Rs O R
PaO2C Psp2C
(n25) (n26)
[0251] In step 1, a protected amino acid is coupled to a resin to give
Compound n24, wherein
P27 is a protecting group that can be removed without effecting cleavage of
the peptide from the
resin; Rl l* and Rl l' are as previously defined. The choice of resin used in
the first step is
dependent upon the nature of the amino acid that is coupled in the proceeding
steps. If the amino
acid side chains contain protecting groups, a resin must be chosen such that
these protecting
groups remain intact when the peptide is removed from the resin. Resins that
can be cleaved
while preserving the protecting groups of peptides include, but are not
limited to, Safety Catch,
Rink Acid, 2-chlorotrityl-chloride resin, trityl-chloride resin, 4-
methyltrityl-chloride resin, 4-
methoxytrityl-chloride resin or PAM resin.
[0252] This peptide coupling process, i.e., deprotection of the alpha-amino
group, followed
by coupling to a protected amino acid, is repeated until the desired number of
amino acids has
been coupled to the resin. In Scheme XI, a total of six amino acids have been
coupled to give
compound n25 wherein, P28 is a protecting group that can be removed without
effecting cleavage
of the peptide from the resin; R6A, Ra*, RsA, R9A, Rli*, Ri1A, and P8 are as
previously defined.
[0253] Cleavage of the peptide from the resin affords compound n26. Coupling
of the 3
peptide fragments is outlined in Scheme XII.
253
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Scheme XII
R1zA
ResinOZC
OP25 R12A
HN ResinO2C
~O O HzN OP25 OH
R1tA ~O
NR11 E 0 R11A
o=~ (n23) + NR91
o=~
HN
~O P28HN HN
R9A R8A 0 Rsq R9A O R8A p P28HN
RsA
HN HN
N HN
N
0 Rs. O ),--, N O
PsozC (nnlb) p Rs
R13A PSOzC (n26)
NHPts +
0 p R2A 0
~N R1A R12A
H N ResinO2C
p R3A p Rz. pP1 R13A
NRSq / HN
R5'A I ~O 0 P18HN O 0 RzA O
p H
~ R11A O R1A
HO (nn2) NR11 N~N T~--,Nx
O= p H R3A NR5A '~=
HN Rsq
O RsA O H
R9A RBA p ~~NH
HN~ N
R12A 13A p R8= p
ResinO2C R ~ P802C (nn3b)
HN
HN O 0 R2A 0
O O O ~N' ~ RtA
R11A N ~ N
NR11. O H R3A 0 Rz.
p=~ NR5A
RS'A
N
HN 0 H
~p HN
Rgq RBA 0 RsA H HN'Ix' ~Ra N N
O
O
PaoZC (nn4b)
[0254] The resin bound 3-methylglutamate n23, where R12A is as described
previously is
deprotected to give the free amine then coupled to fragment n26 to give resin
bound fragment
nnlb, wherein R11A, RI1*, R9A, R8A, R8*, R6A, P8, P25, and P28, are as
previously described. This
is then coupled to the previously described fragment nn2, to give nn3b wherein
R1A, RaA, R2*,
254
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
R3AI R5*A, RS*A, R6A, R8*, R8A, R9A' Rll*' RIIA' R12A' R 13A, pl, P8, and P18
are as described
previously. Deprotection and cyclization as described in Methods A affords a
resin-bound
depsipeptide nn4b wherein RIA, R2A, R2*, R3A5 R5*A, R5*A, R6A, Rs*, R8A' R9AI
Rl l*' RIIAa R12A,
R13A' and P8 are as described previously. Cleavage of the depsipeptide from
the resin followed
by deprotection of any remaining protecting groups yields compounds of Formula
I.
[0255] Following the synthetic schemes above (Schemes IV-XII), it is
understood that both
the amino acid amino group and the amino acid side chain functional groups
must be
orthogonally protected prior to attaching them to the growing peptide chain.
Suitable protecting
groups can be any protecting group useful in peptide synthesis. Such pairings
of protecting
groups are well known. See, e.g., "Synthesis Notes" in the Novabiochem Catalog
and Peptide
Synthesis Handbook, 1999, pages S1-S93 and references cited therein.
[0256] It will also be understood by those skilled in the art that the choice
of protecting
group on the amino acid side chain functional groups will either result or not
result in the
protecting group being cleaved concomitantly with the peptide's final cleavage
from the resin,
which will give the natural amino acid functionality or a protected derivative
thereof,
respectively. When the protecting groups are not concomitantly cleaved when
the depsipeptide
is cleaved from the resin, additional deprotection may be necessary.
[0257] It would be clear to one of skill in the art that the linear precursor
nn3 nn3a or nn3b
and hence intermediate nn4 nn4a and nn4b and final product I can be obtained
not only by
Methods A-C as described above, but also, by combining any two fragment pairs.
These
fragment pairs can be envisioned by fragmenting the compound of Formula I
between any two
amino acids in the sequence, i.e. 1+12, 2+11, 3+10, etc.
R12 R13
HO2C
NH
~-
N''" \ O RZ . O
O \ O H;
R'
R" N N"'=
H
NR'O , R3 p RZ-
O "" , NR5
R5'
HN,/.= O H
O HN -'1.
R9 R$ 0 R6 -~=t ~ N~
HN%% '.
O
O R8-
HOZC
255
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0258] Alternatively, the compounds can be formed by linear assembly prior to
ester
formation by the methods described in United States Patent Numbers 6,911,525
and 6,794,490,
and International Patent Application Numbers WO01/44272, WO01/44274,
WO01/44271 and
W003/014147. Alternatively, the compounds can be formed by assembly of
multiple fragments.
[0259] Although the methods described above employ resin chemistry, the
methods would
also be suitable for solution-phase peptide chemistry.
[0260] Alternatively, the compounds of the present invention can be formed by
the methods
described in International Patent Application Number W02005/012541.
2. Biosynthetic Process
Non-Ribosomal Pe tip 'de Synthetases Pathways
[0261] Bacteria, including actinomycetes, and fungi synthesize a diverse array
of low
molecular weight peptide and polyketide compounds (approx. 2-48 residues in
length). The
biosynthesis of these compounds is catalyzed by non-ribosomal peptide
synthetases (NRPSs) and
by polyketide synthetases (PKSs). The NRPS process, which does not involve
ribosome-
mediated RNA translation according to the genetic code, is capable of
producing peptides that
exhibit enormous structural diversity, compared to peptides translated from
RNA templates by
ribosomes. These include the incorporation of D- and L-amino acids and hydroxy
acids;
variations within the peptide backbone which form linear, cyclic or branched
cyclic structures;
and additional structural modifications, including oxidation, acylation,
glycosylation, N-
methylation and heterocyclic ring formation. Many non-ribosomally synthesized
peptides have
been found which have useful pharmacological (e.g., antibiotic, antiviral,
antifungal,
antiparasitic, siderophore, cytostatic, immunosuppressive, anti-
cholesterolemic and anticancer),
agrochemical or physicochemical (e.g., biosurfactant) properties.
[0262] Non-ribosomally synthesized peptides are assembled by large (e.g.,
about 200-2000
kDa), multifunctional NRPS enzyme complexes comprising one or more subunits.
Examples
include daptomycin, A54145, vancomycin, echinocandin and cyclosporin.
Likewise, polyketides
are assembled by large multifunctional PKS enzyrne complexes comprising one or
more
subunits. Examples include erythromycin, tylosin, monensin and avernlectin. In
some cases,
complex molecules can be synthesized by mixed PKS/NRPS systems. Examples
include
rapamycin, bleomycin and epothilone.
256
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0263] An NRPS usually consists of one or more open reading frames that make
up an NRPS
complex. The NRPS complex acts as a protein template, comprising a series of
protein
biosynthetic units configured to bind and activate specific building block
substrates and to
catalyze peptide chain formation and elongation. (See, e.g., Konz and
Marahiel, 1999, Chem.
Biol. 6: 39-48 and references cited therein; von D6hren et al., 1999, Chem.
Biol. 6: 273-279, and
references cited therein; and Cane and Walsh, 1999, Chem. Biol. 6: 319-325,
and references
cited therein - each hereby incorporated by reference in its entirety). Each
NRPS or NRPS
subunit comprises one or more modules. A "module" is defined as the catalytic
unit that
incorporates a single building block (e.g., an amino acid) into the growing
peptide chain. The
order and specificity of the biosynthetic modules that form the NRPS protein
template dictates
the sequence and structure of the ultimate peptide products.
[0264] Each module of an NRPS acts as a semi-autonomous active site containing
discrete,
folded protein domains responsible for catalyzing specific reactions required
for peptide chain
elongation. A minimal module (in a single module complex) consists of at least
two core
domains: 1) an adenylation domain responsible for activating an amino acid
(or, occasionally, a
hydroxy acid); and 2) a thiolation or acyl carrier domain responsible for
transferring activated
intermediates to an enzyme-bound pantetheine cofactor. Most modules also
contain 3) a
condensation domain responsible for catalyzing peptide bond formation between
activated
intermediates. Supplementing these three core domains are a variable number of
additional
domains which can mediate, e.g., N-methylation (M or methylation domain) and L-
to D-
conversion (E or epimerization domain) of a bound amino acid intermediate, and
heterocyclic
ring formation (Cy or cyclization domain). The domains are usually
characterized by specific
amino acid motifs or features. It is the combination of such auxiliary domains
acting locally on
tethered intermediates within nearby modules that contributes to the enormous
structural and
functional diversity of the mature peptide products assembled by NRPS and
mixed NRPS/PKS
enzyme complexes.
[0265] The adenylation domain of each minimal module catalyzes the specific
recognition
and activation of a cognate amino acid. In this early step of non-ribosomal
peptide biosynthesis,
the cognate amino acid of each NRPS module is bound to the adenylation domain
and activated
as an unstable acyl adenylate (with concomitant ATP-hydrolysis). See, e.g.,
Stachelhaus et al.,
1999, Chem. Biol. 6: 493-505 and Challis et al., 2000, Chem. Biol. 7: 211-224,
each
incorporated herein by reference in its entirety. In most NRPS modules, the
acyl adenylate
257
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
intermediate is next transferred to the T (thiolation) domain (also referred
to as a peptidyl carrier
protein or PCP domain) of the module where it is converted to a thioester
intermediate and
tethered via a transthiolation reaction to a covalently bound enzyme cofactor
(4'-
phosphopantetheinyl (4'-PP) intermediate). Modules responsible for
incorporating D-configured
or N-methylated amino acids may have extra modifying domains which, in several
NRPSs
studied, are located between the A and T domains.
[0266] The enzyme-bound intermediates in each module are then assembled into
the peptide
product by stepwise condensation reactions involving transfer of the thioester-
activated carboxyl
group of one residue in one module to, e.g., the adjacent amino group of the
next amino acid in
the next module while the intermediates remain linked covalently to the NRPS.
Each
condensation reaction is catalyzed by a condensation domain which is usually
positioned
between two minimal modules. The number of condensation domains in a NRPS
generally
corresponds to the number of peptide bonds present in the final (linear)
peptide. An extra C
domain has been found in several NRPSs (e.g., at the amino terminus of
cyclosporin synthetase
and the carboxyl terminus of rapamycin; see, e.g., Konz and Marahiel, supra)
that has been
proposed to be involved in peptide chain termination and cyclization
reactions. Many other
NRPS complexes, however, release the full length chain in a reaction catalyzed
by a C-terminal
thioesterase (Te) domain (of approximately 28K-35K relative molecular weight).
[0267] Thioesterase domains of most NRPS complexes use a catalytic triad
(similar to that of
the well-known chymotrypsin mechanism) which includes a conserved serine (less
often a
cysteine or aspartate) residue in a conserved three-dimensional configuration
relative to a
histidine and an acidic residue. See, e.g. V. De Crecy-Lagard in
"Comprehensive Natural
Products Chemistry", Volume 4, ed.by J.W. Kelly, Elsevier, New York, 1999, pp.
221-23 8, each
incorporated herein by reference in its entirety. Thioester cleavage is a two
step process. In the
first (acylation) step, the full length peptide chain is transferred from the
thiol tethered enzyme
intermediate in the thiolation domain (see above) to the conserved serine
residue in the Te
domain, forming an acyl-O-Te ester intermediate. In the second (deacylation)
step, the Te
domain serine ester intermediate is either hydrolyzed (thereby releasing a
linear, full length
product) or undergoes cyclization, depending on whether the ester intermediate
is attacked by
water (hydrolysis) or by an activated intramolecular nucleophile
(cyclization).
[0268] Sequence comparisbns of C-terminal thioesterase doinains from diverse
members of
the NRPS superfamily have revealed a conserved motif comprising the serine
catalytic residue
258
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
(GXSXG motif), often followed by an aspartic acid residue about 25 amino acids
downstream
from the conserved serine residue. A second type of thioesterase, a free
thioesterase enzyme, is
known to participate in the biosynthesis of some peptide and polyketide
secondary metabolites.
See e.g., Schneider and Marahiel, 1998, Arch. Microbiol. 169: 404-410, and
Butler et al., 1999,
Chem. Biol. 6: 87-292, each incorporated herein by reference in its entirety.
These thioesterases
are often required for efficient natural product synthesis (See United States
Patent Application
Number 20020192773). Butler et al. have postulated that the free thioesterase
found in the
polyketide tylosin gene cluster - which is required for efficient tylosin
production - may be
involved in editing and proofreading functions.
[0269] The modular organization of the NRPS multienzyme complex is mirrored at
the level
of the genomic DNA encoding the modules. The organization and DNA sequences of
the genes
encoding several different NRPSs have been studied. (See, e.g., Marahiel,
1997, Chem. Biol. 4:
561-567, incorporated herein by reference in its entirety). Conserved
sequences characterizing
particular NRPS functional domains have been identified by comparing NRPS
sequences derived
from many diverse organisms and those conserved sequence motifs have been used
to design
probes useful for identifying and isolating new NRPS genes and modules.
[0270] The modular structures of PKS and NRPS enzyme complexes can be
exploited to
engineer novel enzymes having new specificities by changing the numbers and
positions of the
modules at the DNA level by genetic engineering and recombination in vivo.
Functional hybrid
NRPSs have been constructed, for example, based on whole-module fusions. See,
e.g., Gokhale
et al., 1999, Science 284: 482-485; Mootz et al., 2000, Proc. Natl. Acad. Sci.
U.S.A. 97: 5848-
5853, incorporated herein by reference in their entirety. Recombinant
techniques maybe used to
successfully swap domains originating from a heterologous PKS or NRPS complex.
See, e.g.,
Schneider et al., 1998, Mol. Gen. Genet. 257: 308-318; McDaniel et al., 1999,
Proc. Natl. Acad.
Sci. U.S.A. 96: 1846-1851; United States Patent Nos. 5,652,116 and 5,795,738;
and International
Patent Number WO 00/56896; incorporated herein by reference in their entirety.
[0271] Engineering a new substrate specificity within a module, by altering
residues which
form the substrate binding pocket of the adenylation domain, has also been
described. See, e.g.,
Cane and Walsh, 1999, Chem. Biol. 6: 319-325; Stachelhaus et al., 1999, Chem.
Biol. 6: 493-
505; and International Patent Application NumberWO 00/52152; each incorporated
herein by
reference in its entirety. By comparing the sequence of the B. subtilis
peptide synthetase GrsA
adenylation domain (PheA, whose structure is known) with sequences of 160
other adenylation
259
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
domains from pro- and eukaryotic NRPSs, for example, Stachelhaus et al.
(supra) and Challis et
al., 2000, Chem. Biol. 7: 211-224 defined adenylation (A) domain signature
sequences
(analogous to codons of the genetic code) for a variety of amino acid
substrates. From the
collection of those signature sequences, a putative NRPS selectivity-
conferring code (with
degeneracies like the genetic code) was formulated.
[02721 The ability to engineer NRPSs having new modular template structures
and new
substrate specificities by adding, deleting or exchanging modules (or by
adding, deleting or
exchanging domains within one or more modules) will enable the production of
novel peptides
having altered and potentially advantageous properties. A combinatorial
library comprising over
50 novel polyketides, for example, was prepared by systematically modifying
the PKS that
synthesizes an erythromycin precursor (DEBS) by substituting counterpart
sequences from the
rapamycin PKS (which encodes alternative substrate specificities). See, e.g.,
International Patent
Application NumberWO 00/63361 and McDaniel et al., 1999, supra, each
incorporated herein by
reference in its entirety.
[0273] Daptomycin is an example of a non-ribosomally synthesized peptide made
by a
NRPS (Figure 1). Modification of the genes encoding the proteins involved in
the daptomycin
biosynthetic pathway, including the daptomycin NRPS, provide a first step in
producing
modified Streptomyces roseosporus (NRRL 11379) as well as other host strains
which can
produce an improved antibiotic (for example, having greater potency); which
can produce
natural or new antibiotics in increased quantities; or which can produce other
peptide products
having useful biological properties. Compositions and methods relating to the
Streptornyces
roseosporus daptomycin biosynthetic gene cluster, including isolated nucleic
acids and isolated
proteins, are described in International Patent Application Number
W003/014297; hereby
incorporated by reference.
[02741 A54145 is another example of a non-ribosomally synthesized peptide made
by a
NRPS. A54145 is a cyclic lipopeptide antibiotic that is produced by the
fermentation of
Streptomycesfradiae (NRRL 18158). A54145 comprises a fatty acid chain linked
via a three-
amino acid chain to the N-terminal tryptophan of a cyclic 10-amino acid
peptide (Figure 2). The
compound has similar in vitro anti-bactericidal activity to A21978C/daptomycin
factors against
various strains of S. aureus, S. epiderrnidis, Streptococcus pyogesaes, and
enterococci.
Compositions and methods relating to the Streptorrayces fradiae A54145
biosynthetic gene
260
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
cluster, including isolated nucleic acids and isolated proteins, are described
in International
Patent Application Number W003/060127; hereby incorporated by reference.
[0275] The genes encoding the proteins involved in the A54145 biosynthetic
pathway,
including the A54145 NRPS, provide a first step in producing modified
Streptofnyces f adiae as
well as other host strains which can produce an improved antibiotic (for
example, having greater
potency); which can produce natural or new antibiotics in increased
quantities; or which can
produce other peptide products having useful biological properties.
Methods of Altering Gene Clusters for Production of Novel Compounds by NRPS
Alteration ofNRPS Polypeptide Modules and Domains
[0276] In one aspect, the invention provides a method of altering the number
or position of
the modules in an NRPS to obtain the compounds of Formula I or compounds of
any of Formula
Fl-F22. In one embodiment, one or rriore domains may be deleted from the NRPS.
In this case,
the product produced by the NRPS will have a chemical change relative to the
peptide produced
in the absence of the deletion, e.g., if an epimerization and/or methylation
domain is deleted.
[0277] In another embodiment, one or more domains may be added to the NRPS. In
this
case, the peptide synthesized by the NRPS will have an additional chemical
change. For
instance, if an epimerization domain or a methylation domain is added, the
resultant peptide will
contain an extra D-amino acid or will contain a methylated amino acid,
respectively. In a yet
further embodiment, one or more modules may be mutated, e.g., an adenylation
domain may be
mutated such that it has a different amino acid specificity than the naturally-
occurring
adenylation domain. With the amino acid code in hand, one of skill in the art
can perform
mutagenesis, by a variety of well known techniques, to exchange the code in
one module for
another code, thus altering the ultimate amino acid composition and/or
sequence of the resulting
peptide synthesized by the altered NRPS. In another embodiment, one or more
subunits may be
added or deleted to the NRPS.
[0278] In a still further embodiment, one or more domains, modules or subunits
may be
substituted with another domain, module or subunit in order to produce novel
peptides by
complementation (See International Patent Application Number WO 01/30985,
providing, intet
alia, methods for substituting modules). In this case, the peptide produced by
the altered NRPS
will have, e.g., one or more different amino acids compared to the naturally-
occurring peptide.
261
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
In addition, different combinations of insertions, deletions, substitutions
and mutations of
domains, modules or subunits may be used to produce a peptide of interest. For
instance, one
may substitute a modified module, domain or subunit for a naturally-occurring
one, or may
substitute a naturally-occurring module, domain or subunit from the NRPS from
one organism
for a module, domain or subunit of an NRPS from another organism.
Modifications of the
modules, domains and subunits may be performed by site-directed mutagenesis,
domain
exchange (for module or subunit modification), deletion, insertion or
substitution of a domain in
a module or subunit, or deletion, insertion or substitution of a module in a
subunit. Further, a
domain, module or subunit may be disrupted such that it does not function
using any method
known in the art. These disruptions include, e.g., such techniques as a single
crossover
disruptant or replacement through homologous recombination by another gene
(e.g., a gene that
permits selection or screening).
[0279] The products produced by the modified NRPS complexes will have
different
incorporated amino acids, different chemical alterations of the amino acids
(e.g., methylation and
epimerization). The domains, modules or subunits may be derived from any
number of NRPS
desired, including two, three or four NRPS. Further, the invention
contemplates these altered
NRPS complexes with and without an integral thioesterase domain.
[0280] The source of the modules, domains and/or subunits may be derived from
the
daptomycin biosynthetic gene cluster NRPS, the A54145 biosynthetic gene
cluster NRPS, or
may be derived from any NRPS that encodes another lipopeptide or other peptide
source. These
peptide sources include glycopeptide gene clusters, mixed pathway gene
clusters and siderophore
gene clusters. Artificial NRPSs' and methods for making them, have been
desribed in
International Patent Application Number WO01/30985, herein incorporated by
reference.
Further, the source of the modules, domains and/or subunits may be obtained
from any
appropriate source, including both streptomycete and non-streptomycete
sources. Non-
streptomycete sources include actinomycetes, e.g., Amycolatopsis; prokaryotic
non-
actinomycetes, e.g., Bacillus and cyanobacteria; and non-bacterial sources,
e.g., fungi.
[0281] An NRPS or portion thereof may be heterologous to a host cell of
interest or may be
endogenous to the host cell. In one embodiment, the NRPS or a portion thereof
(e.g., a domain,
module or subunit thereof) is introduced into the host cell on any vector
known to one having
ordinary skill in the art, e.g., a plasmid, a cosmid, bacteriophage or BAC.
The host cell into
which the NRPS or portion thereof is introduced may contain an endogenous NRPS
or portion
262
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
thereof (e.g., a domain, module or subunit thereof). Alternatively, a
heterologous NRPS or
portion thereof may be introduced into the host cell containing the
heterologous NRPS or portion
thereof. The first NRPS, or another NRPS, or domain, module or subunit of an
NRPS may have
either a naturally-occurring sequence or a modified sequence. In another
embodiment, the NRPS
or portion thereof is endogenous to the host cell, e.g., the host cell is S.
fradiae in the case of
A54145 or is S. roseosporus in the case of daptomycin. A naturally-occuring or
modified NRPS,
or a domain, module or subunit thereof may be introduced into the host cell
comprising the
endogenous NRPS or portion thereof. The heterologous domains, modules,
subunits or NRPS
may comprise a constitutive or regulatable promoter, which are known to those
having ordinary
skill in the art. The promoter can be either homologous or heterologous to the
nucleic acid
molecule being introduced into the cell. In certain embodiments, the promoter
may be from the
A54145 biosynthetic gene cluster or the daptomycin biosynthetic gene cluster,
as described
above.
[0282] The nucleic acid molecule comprising the NRPS or portion thereof (e.g.,
a domain,
module or subunit) may be maintained episomally or integrated into the genome.
The nucleic
acid molecule may be introduced into the genome at, e.g., phage integration
sites. Further, the
nucleic acid molecule may be introduced into the genome at the site of an
endogenous or
heterologous NRPS or portion thereof or elsewhere in the genome. The nucleic
acid molecule
may be introduced in such a way to disrupt all or part of the function of a
domain, module or
subunit of an NRPS already present in the genome, or may be introduced in a
manner that does
not disturb the function of the NRPS or portion thereof.
[0283] The peptides produced by these NRPSs may be useful as new compounds or
may be
useful in producing new compounds. In a preferred embodiment, the new
compounds are useful
as or may be used to produce antibiotic compounds. In another preferred
embodiment, the new
compounds are useful as or may be used to produce other peptides having useful
activities,
including but not limited to antibiotic, antifungal, antiviral, antiparasitic,
antimitotic, cytostatic,
antitumor, immuno-modulatory, anti-cholesterolemic, siderophore, agrochemical
(e.g.,
insecticidal) or physicochemical (e.g., surfactant) properties.
[0284] Further diversity of non-ribosomally synthesized peptides and
polyketides may also
be achieved by expressing one or more NRPS and PKS genes (encoding natural,
hybrid or
otherwise altered modules or domains) in heterologous host cells, i.e., in
host cells other than
those from which the NRPS and PKS genes or modules originated.
263
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
3. Post Peptide Modification
[0285] The compounds of the present invention may be obtained by first
assembling the core
of the molecule by any of the methods described above followed by synthetic
manipulation of all
or some of the remaining primary amino groups as described in United States
Patent Numbers
6,911,525; and 6,794,490 and in International Patent Application
NumbersWO01/44272;
W001/44274; and W001/44271.
[0286] Treatment of the primary amino group(s) with reagents such as
isocyanates,
isothiocyanates, activated esters, acid chlorides, sulfonylchlorides or
activated sulfonamides,
heterocycles bearing readily displaceable groups, imidates, lactones or
reductively with
aldehydes affords compounds in which one or more of substituents Rl, Raal,
Raa2, R6*,and R8** is
independently monosubstituted amino, disubstituted amino, acylamino, ureido,
guanidino,
carbamoyl, sulfonamino, thioacylamino, thioureido, iminoamino, or
phosphonamino.
[0287] In order to achieve these modifications, it may be necessary to protect
certain
functionalities in the molecule. Protecting these functionalities should be
within the expertise of
one skilled in the art following the disclosure of this invention. See, e.g.,
Greene, supra.
Cells and Methods for Making Cells that Can Express Recombinant NRPS
[0288] The present invention includes cells and methods for making cells that
can express
recombinant NRPS gene clusters that are capable of expressing the recombinant
NRPS and
capable of producing the various compounds of the invention. In certain
specific embodiments,
the cells are gram positive cells, including Streptonayces lividans,
Streptomyces coelicolor, or
Streptomyces roseosporus.. In other specific embodiments of the invention, a
recombinant
NRPS is assembled from modules from a daptomycin or A54145 NRPS gene cluster.
These
genes may be "swapped" using recombination techniques known in the art or
exemplified herein.
In other embodiments, certain genes in the recombinant NRPS are deactivated or
"knocked out"
to avoid the expression product and its activity in the cell. [JILL, SHOULD WE
MENTION
3MG HERE AND lptl?]
[0289] In a preferred embodiment, bacterial host cells are used to express the
nucleic acid
molecules of the instant invention. Useful expression vectors for bacterial
hosts include bacterial
plasmids, such as those from E. coli or Streptomyces, including pBluescript,
pGEX-2T, pUC
vectors, col El, pCRl, pBR322, pMB9 and their derivatives, wider host range
plasmids, such as
264
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
RP4, phage DNAs, e.g., the numerous derivatives of phage lambda, e.g., NM989,
?GT10 and
?GT11, and other phages, e.g., M13 and filamentous single stranded phage DNA.
A preferred
vector is a bacterial artificial chromosome (BAC). A more preferred vector is
pStreptoBAC, as
described in Example 2 of International Patent Application Number 03/014297.
[0290] In other embodiments, eukaryotic host cells, such as yeast, insect or
mammalian cells,
may be used. Yeast vectors include Yeast Integrating plasmids (e.g., YIp5) and
Yeast
Replicating plasmids (the YRp and YEp series plasmids), Yeast centromere
plasmids (the YCp
series plasmids), pGPD-2, 2 plasmids and derivatives thereof, and improved
shuttle vectors
such as those described in Gietz and Sugino, Geine, 74, pp. 527-34 (1988)
(YIplac, YEplac and
YCplac). Expression in mammalian cells can be achieved using a variety of
plasmids, including
pSV2, pBC12BI, and p91023, as well as lytic virus vectors (e.g., vaccinia
virus, adeno virus, and
baculovirus), episomal virus vectors (e.g., bovine papillomavirus), and
retroviral vectors (e.g.,
murine retroviruses). Useful vectors for insect cells include baculoviral
vectors and pVL 941.
[0291] Other aspects of the invention provide compounds and methods for making
the
compounds from recombinant cells described herein. The compounds can be
produced by
culturing the cells using techniques and conditions that are known in the art
or described herein.
The conditions for culturing the cells may include fermenting the cells with a
lipopeptide tail
precursor that promotes the production of a particular compound of the
invention. This
precursor may be taken up by the cell during fermentation and increase the
production of a
particular compound in the cell. A precursor provided to the cell during
fermentation is
sometimes called a fermentation feed and the resulting compound a feed
product. The
compounds of the invention produced by culturing or fermenting the cells of
the invention may
be farther isolated from the fermentation product and/or purified.
Preparation of Novel Depsippptides
1. Synthetic Processes
[0292] In order that this invention may be more fully understood, the
following examples are
set forth. These examples are for illustrative purposes only and are not to be
construed as
limiting the scope of the invention in any way.
[0293] Examplel -1: Synthesis of Peptide Resin Conzpound 1:
Resin-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-NH2 (1)
265
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0294] Reaction 1: Preparation of Resin-Gly-Thr-NHFmoc (2)
[0295] A solution of conunercially available Na-(9-Fluorenylmethoxycarbonyl)-L-
threonine
(2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2 mL of a
0.5 molar solution in N-inethylpyrolidine), and 1-hydroxy-benzotriazole (2 mL
of a 0.5 molar
solution in N-methylpyrolidine) was added to commercially available glycine 2-
chlorotrityl resin
(334 mg). The mixture was shaken for one hour, filtered through a glass sinter
funnel and a few
beads were tested for the presence of a free amine using the standard Kaiser
test (see E. Kaiser,
et al., 1970, Anal. Biochem. 34: 595; and "Advanced Chemtech Handbook of
Combinatorial,
Organic and Peptide Chemistry" 2003-2004, page 208). The Kaiser test gave a
blue color
indicating that the reaction was incomplete therefore the coupling conditions
above was
repeated. After filtration through a glass sinter funnel the product bearing
resin was washed with
N-methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with N-
methylpyrolidine (3 x 6
mL ) to give compound 2.
[0296] Reaction 2: Preparation of Resin-Gly-Thr-NH,(3)
[0297] Compound 2 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 3.
[0298] Reaction 3: Preparation of Resin-Gly-Thr-Asp(OtBu)-NHFmoc (4)
[0299] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
0-tert-
butyl ester (2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2
mL of a 0.5 molar solution in N-methylpyrolidine), and 1-hydroxy-benzotriazole
(2 mL of a 0.5
molar solution in N-methylpyrolidine) were added to compound 3. The mixture
was shaken for
one hour, filtered through a glass sinter funnel and the coupling was
repeated. The reaction
mixture was filtered through a glass sinter fannel then the solid was washed
with N-
methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL ), and again with N-
methylpyrolidine (3 x 6
mL) to give compound 4.
[0300] Reaction 4: Preparation of Resin-Gly-Thr-Asp(OtBu)-NH, 5
[0301] Compound 4 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
266
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then washed with N-methylpyrolidine (3 x 6 mL),
methanol (3 x 6
mL), and again with N-methylpyrolidine (3 x 6 mL) to give compound 5.
[0302] Reaction 5: Preparation of Resin-Gly-Thr-Asp(OtBu -DAsn HTrt)-NHFmoc(6)
[0303] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-
asparagine 8-N-trityl (2 mL of a 0.5 molar solution in N-methylpyrolidine),
1,3-
diisopropylcarbodiimide (2 mL of a 0.5 molar solution in N-methylpyrolidine),
and 1-hydroxy-
benzotriazole (2 mL of a 0.5 molar solution in N-methylpyrolidine) was added
to resin 5. The
reaction mixture was shaken for one hour, filtered through a glass sinter
funnel and the coupling
was repeated. The reaction mixture was filtered through a glass sinter funnel
then the solid was
washed with N-methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with
N-
methylpyrolidine (3 x 6 mL) to give compound 6.
[0304] Reaction 6: Preparation of Resin-Gly-Thr-Asp(OtBu)-DAsn(NHTrt-NH, 7
[0305] Compound 6 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 7.
[0306] Reaction 7: Preparation of Resin-Gly-Thr-Asp(OtBu -DAsn(NHTrtLrp-NHFmoc
(8)
[0307] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan (2
mL of a
0.5 molar solution in N-methylpyrolidine), 1,3-diisopropylcarbodiimide (2 mL
of a 0.5 molar
solution in N-methylpyrolidine), and 1-hydroxy-benzotriazole (2 mL of a 0.5
molar solution in
N-methylpyrolidine) were added to resin 7. The reaction mixture was shaken for
one hour, then
filtered through a glass sinter funnel and the coupling was repeated. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6
mL), methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 8.
[0308] Reaction 8: Preparation of Resin-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-TM-NH, 1
[0309] Compound 8 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter fitnnel and re-
suspended in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
267
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
resin peptide
compound 1.
[0310] Example 1-2: Synthesis of Peptide Resin Compound 9:
Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-DAIa-Asp-Orn(NHBoc)-NHa (9)
[0311] Reaction 1: Preparation of Resin-Glu(aOAllyl)-NHFmoc (10)
[0312] To a suspension of commercially available 4-hydroxymethylphenoxy resin
(Wang
resin, 5 g, 0.4 mmol/g) in dichloromethane (60 mL) was added 1,3-
diisopropylcarbodiimide
(0.940 mL), 4-dimethylaminopyridine (24 mg in N-methylpyrolidine (1 mL)), and
commercially
available Na-(9-Fluorenylmethoxycarbonyl)-L-glutamic acid a-allyl ester (2.46
g in N-
methylpyrolidine (9 mL)). The reaction mixture was stirred for 16 hours,
filtered through a glass
sinter funnel, and the solid was washed with N-methylpyrolidine and
dichloromethane and dried
under reduced pressure to give compound 10.
[0313] Reaction 2: Preparation of Resin-Glu(aOAllyl)-NH, (11)
[0314]] Compound 10 (526 mg) was agitated in 20% piperidine in N-
methylpyrolidine (6
mL) for 30 minutes. The resin was filtered through a glass sinter funnel and
re-suspended in
20% piperidine in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 6 mL), methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to
give compound
11.
[0315] Reaction 3: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-NHFmoc(12)
[0316] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-serine-tert-
butyl ether
(2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2 mL of a
0.5 molar solution in N-methylpyrolidine), and 1 -hydroxy-benzotriazole (2 mL
of a 0.5 molar
solution in N-methylpyrolidine) were added to resin 11. The reaction mixture
was shaken for
one hour, then filtered through a glass sinter funnel and the coupling was
repeated. The reaction
mixture was filtered through a glass sinter funnel then the solid was washed
with N-
methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with N-
methylpyrolidine (3 x 6
mL) to give compound 12.
268
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0317] Reaction 4: Preparation of Resin-Glu(aOAllvl -DSer(OtBu)-NH, 13
[0318] Compound 12 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 13.
[0319] Reaction 5: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-NHFmoc(14)
[0320] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-glycine (2 mL
of a 0.5
molar solution in N-methylpyrolidine), 1,3-diisopropylcarbodiimide (2 mL of a
0.5 molar
solution in N-methylpyrolidine), and 1-hydroxy-benzotriazole (2 mL of a 0.5
molar solution in
N-methylpyrolidine) were added to resin 13. The reaction mixture was shaken
for one hour, then
filtered through a glass sinter furmel and the coupling was repeated. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6
mL), methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 14.
[0321] Reaction 6: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gl -, (15)
[0322] Compound 14 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 15.
269
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0323] Reaction 7: Preparation of Resin-Glu(aOAllyI)-DSer(OtBu)-Gly-AsR(OtBu)_
NHFmoc (16)
[0324] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
(3-tert-
butyl ester (2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2
mL of a 0.5 molar solution in N-methylpyrolidine), and 1-hydroxy-benzotriazole
(2 mL of a 0.5
molar solution in N-methylpyrolidine) were added to resin 15. The reaction
mixture was shaken
for one hour, through a glass sinter fiuuiel and the coupling was repeated.
The reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 6 mL), methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to
give compound
16.
[0325] Reaction 8: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gl y -
Asp(OtBu)-NH2
17
[0326] Compound 16 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 17.
[0327] Reaction 9: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-
DAla-
NHFmoc (18)
[0328] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-
alanine
((2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2 mL of a
0.5 molar solution in N-methylpyrolidine), and 1 -hydroxy-benzotriazole (2 mL
of a 0.5 molar
solution in N-methylpyrolidine) was added to resin 17. The reaction mixture
was shaken for one
hour, filtered through a glass sinter fumiel and the coupling was repeated.
The reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 6 mL), methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to
give compound
18.
[0329] Reaction 10: Preparation of Resin-Glu((xOAllYI)-DSer(OtBu)-Gly-
Asp(OtBu)-DAla-
NH, 19
[0330] Compound 18 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
270
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 19.
[0331] Reaction 11: Preparation of Glu(aOAllyl)-DSer ,OtBuLGly-As (p OtBu -
DAla-
Asp(OtBu)-NHFmoc (20)
[0332] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
[i-
tertbutyl ester ((2 mL of a 0.5 molar solution in N-methylpyrolidine), 1,3-
diisopropylcarbodiimide (2 mL of a 0.5 molar solution in N-methylpyrolidine),
and 1-hydroxy-
benzotriazole (2 mL of a 0.5 molar solution in N-methylpyrolidine) was added
to resin 19. The
reaction mixture was shaken for one hour, filtered through a glass sinter
funnel and the coupling
was repeated. The reaction mixture was filtered through a glass sinter funnel
then the solid was
washed with N-methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with
N-
methylpyrolidine (3 x 6 mL) to give compound 20.
[0333] Reaction 12: Preparation of Resin-Glu(aOAllyI)-DSer(OtBu)-Gly-Asp(OtBu)-
DAla-
AsR(OtBu)-NH, 21
[0334] Compound 20 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound
21. [0335] Reaction 13: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-
Asp(OtBu)-
DAIa-Asp(OtBu)-Orn-NHFmoc (22)
[0336] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-NS-
(tertbutoxycarbonyl)-L-ornithine (2 mL of a 0.5 molar solution in N-
methylpyrolidine), 1,3-
diisopropylcarbodiimide (2 mL of a 0.5 molar solution in N-methylpyrolidine),
and 1-hydroxy-
benzotriazole (2 mL of a 0.5 molar solution in N-methylpyrolidine) was added
to resin 21. The
reaction mixture was shaken for one hour, then filtered through a glass sinter
funnel and the
coupling was repeated. The reaction mixture was filtered through a glass
sinter funnel then the
solid was washed with N-methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and
again with N-
methylpyrolidine (3 x 6 mL) to give compound 22.
271
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0337] Reaction 14: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-Gly-Asp(OtBu)-
DAIa-
Asp(OtBu)-Orn(NHBoc)-NH, 9
[0338] Compound 22 was agitated in 20% piperidine in N-methylpyrolidine (6 mL)
for 30
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (6 mL) and agitated for 30 minutes. The reaction mixture
was filtered
through a glass sinter furmel then the solid was washed with N-
methylpyrolidine (3 x 6 mL),
methanol (3 x 6 mL), and again with N-methylpyrolidine (3 x 6 mL) to give
compound 9.
[0339] Example 1-3: Synthesis of Peptide Resin Compound 23:
~ Ph
HN Ph
HO CH3 0
O H O H
N N N-,.,(CH2)8CH3
O H N
O H O
HN~ O
Resin-O O O~ tBu N
H
(23)
[0340] Reaction 1: Preparation of Compound 24
F F F F
F * OH F Oy (CH2)8CHs
F F F F O
24
[0341] Pentafluorophenol (3.68 g) was dissolved in dichloromethane (40 mL) and
cooled to
0 C in an ice/NaCl bath. Decanoylchloride (4.15 mL) was added dropwise such
that the
temperature remained below 2 C. Once addition was complete, the reaction was
stirred for an
additiona12.5 hours at 0 C. The cooling bath was then removed and the reaction
warmed to
ambient temperature and stirred for 17 hours. The volatiles were removed under
reduced
pressure to give the crude product pentafluorophenyl ester 24, which could be
used subsequently
without further purification.
272
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0342] Reaction 2 Preparation of Compound 23
Ph
,~-Ph
HN Ph
F F
HO CH3 O
O O
N N N NH2 F Oy (CHZ)sCH3
O
"Ti, H O H F F O
HN~ O
O-tBu 24
Resin-O 0
N
H
1
Ph Ph
HN Ph
HO CH3 0
O H O
N N N N~(CH2)aCHs
O H O H O
O
HN
Resin-O10 O~'tBu
N
H
23
[0343] Resin peptide compound 1 (2 g) was added to a solution of the
pentafluorophenyl
ester of decanoic acid, 24, (440 mg) in dichloromethane. The mixture was
shaken for 17 hours,
filtered through a glass sinter funnel, and the reaction was judged to be
incomplete using the
Kaiser Test (vide supra). Decanoic acid (517 mg), 1-hydroxy-benzotriazole (446
mg), and 1,3-
diisopropylcarbodiimide (438 L) were dissolved in N-methylpyrolidine (8 mL)
and stirred for
one hour. The resin was then added to the decanoic acid mixture then stirred
for 8 hours, filtered
through a glass sinter funnel and washed with N-methylpyrolidine (3 x 6 mL),
methanol (3 x 6
mL), and again with N-methylpyrolidine (3 x 6 mL). The reaction was found to
be complete
using the Kaiser Test, yielding the resin bound lipopeptide 23.
[0344] Example 1-4: Synthesis of Compound C352:
HO NH2
O O NH2
O
~N N O O O
HO HN H O N N N N~(CH2)aCH3
O O H O H O
O NH HN
O
~ O OH ~ OH I\ I
HO NH H O HN 0
N
--'-TN
O O H~NH2 H
(C352)
273
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0345] Reaction 1: Preparation of Compound (25)
Q-N3
NH2 O O O
H N OH N OH
2 O H O
[0346] Commercially available Kynurenine (3 g) was suspended in acetonitrile
(100 mL) and
water (30 mL). Diisopropylethylamine (DIPEA, 5.01mL) was added dropwise to the
solution
and stirring was continued until the solution was homogeneous. The solution
was then cooled to
0 C in an ice/sodium chloride bath and a solution of allyloxycarbonyl
oxysuccinimide
(AllocOSu, 4.3 g) in acetonitrile (30 mL) was added. The reaction mixture was
stirred for 3
hours then concentrated to remove acetonitrile, basified with 5% K2C03
solution (220 mL) and
washed with ethyl acetate (5 x 90 mL) and dichloromethane (1 x 90 mL). The
aqueous portion
was then acidified to pH 1 and extracted with ethyl acetate (4 x 90 mL).
Combined acidic
organic washes were dried with anhydrous MgSO4 and evaporated to give crude
product
(4.85 g). Purification by column chromatography on silica gel, eluting with
dichloromethane
methanol 19:1, gave the desired intermediate, L-2-N-(allyloxycarbonyl)-4-(2-
aminophenyl)-4-
oxobutanoic acid, after evaporation of the solvent as a yellow solid 2.92 g.
This solid (2.9 g) was
dissolved in 4N HCl (100 mL) and cooled to 0 C in an ice/sodium chloride bath.
A solution of
NaNO2 (0.76 g) in water (10 mL) was added dropwise such that the temperature
remained below
3 C, and the resultant solution was stirred for 2.5 hours at 0 C. A solution
of NaN3 (1.95 g) in
water (10 mL) was added dropwise such that the temperature remained below 3 C
and the
resultant solution was warmed to ambient temperature and stirred over 19
hours. The reaction
mixture was poured into water (250 mL) and extracted with dichloromethane (4 x
I OOml). The
combined organic washes were dried with anhydrous MgSO4 and evaporated to the
desired
product compound 25 (2.86 g).
274
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0347] Reaction 2: Preparation of Compound (26)
~Ph
HN Ph
HO CH3 0
O H O H N3
N N N N-,,,(CH2)aCH3 O
O Q O O o N OH
HN O~tBu ~\ ~ II H O
Resi rt-O~O N
H 25
23
Ph
N3 J<Ph
O 0 HN Ph
~O~IN O CH3 O
O 0
H O N N N N~(CH2)aCHs
O H O H O
HN O
O-tBu
HO O N
H
26
[0348] L-2-N-(allyloxycarbonyl)-4-(2-azidophenyl)-4-oxobutanoic acid 25 (636
n1g), 4-
dimethylaminopyridine (25 mg), and N-methyl-2-chloropyridinium iodide (511 mg)
were
flushed well with argon, then suspended in dichloromethane (10 mL).
Triethylamine (560 L)
was added and the reaction mixture was stirred to give a homogeneous solution.
Resin
lipopeptide 23 (667 mg) was added to the solution and the flask was flushed
again with argon
and shaken for 17 hours. A 20 mg sample of the resin was removed to test the
reaction for
completion (20 mg of resin in dichloromethane (0.6 mL) was treated with 2,2,2-
trifluoroethanol,
(0.2 mL) and acetic acid (0.2 mL) and stirred for 3 hours. The reaction
mixture was filtered
through a glass sinter funnel, and the solvent was evaporated to give a
residue. Liquid
Chromatography/Mass Spectral analysis of the residue indicated the reaction
was incomplete).
Coupling was judged to be incomplete so the resin was dried under reduced
pressure for 5 days,
and the above coupling was repeated over another 17 hours. The reaction
mixture was filtered
through a glass sinter funnel and the solid was washed well with
dichloromethane. The solid
was then suspended in dichloromethane (6 mL), 2,2,2-trifluoroethanol (2 mL),
acetic acid (2
mL), and shaken for 5 hours. The reaction mixture was filtered through a glass
sinter funnel and
evaporation of the filtrate gave the crude desired peptide 26 (44 mg). The
crude product was
purified by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting
with a
275
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
gradient from 20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid
to 80%
acetonitrile 0.5% formic acid : 20 % water 0.5% formic acid over 25 minutes.
The product
bearing fractions were freeze-dried to give the pure product 26 (10.6 mg).
[0349] Reaction 3: Prenaration of Compound(27)
Resin O
I Ph
O O N O N3 ~Ph
tBu-O O~ O O HN Ph
~ i1
H tBu 9xN O CH3 O O
O
O ( -- I H O N N (CH2)8CH3
U 0 NH O N N ~
O 0 H O H O
O NH N H 0 NH2 HN O
tBu 0 NNBoc HOIO O-tBu
O H H
9 26
0
Resin-O Z~Z~OA NH 0 N3 Ph
O O , I HN~Ph
O N O ~ 0
But-O~ O O H O H
HN N N N N(CH2$CH3
O tBu 0 0 O H O
O O NH 0 HN -
O~ \ /
But-O NH ~ O HN'O tBu ~ N
N H" NBoc H
O H
27
[0350] Hydroxy-benzotriazole (5 mg), 1,3-diisopropylcarbodiimide (6 L), and
peptide resin
compound 9 (12.3 mg) were added to a solution of compound 26 (10.6 mg) in N-
methylpyrolidine (0.7 mL) then shaken for 22 hours. The resin was filtered
through a glass
sinter funnel and the coupling was judged to be complete using the Kaiser Test
(vide supra),
yielding resin bound lipopeptide 27.
276
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[03511 Reaction 4: Preparation of Compound f 28)
I
Resin-O N3 Ph
O ~
0 p O1 N O HN" Ph Ph
O N oJ H O O
O
tBu ___ _ N N N (CHz)aCHs
O~ tBu O ~ H O
O NH i HN -~
O O
O--
0 NH H O HN'O tBu N
I
tBu N Nfl -, Boc H
O H H
27
~
Ph
Resin-O NH2 O
O O HN~Ph
N N N O O
H O N
HN N (CH2)gCH3
tBu N ~(
O tBu O ~ O H O
O NH i HN
O O
O~
O NH o O HN~O tBu N
tBu O~N N~ Boc H
O H N
28
[0352] The dried resin 27 was placed under an argon atmosphere, and treated
with a solution
of tetrakis-(triphenylphosphine)palladium(0) (19 mg) in dichloromethane (1.47
mL), acetic acid
(74 L), and N-methylmorpholine (37 L). The mixture was shaken for 4 hours at
ambient
temperature, filtered-through a glass sinter funnel, and the solid was washed
with two times with
N-methyhuorpholine, two times with methanol, and again two times with N-
methylmorpholine .
1-Hydroxy-benzotriazole (0.5 mL of a 0.5 molar solution in N-methylmorpholine)
and 1,3-
diisopropylcarbodiimide (0.5 mL of a 0.5 molar solution in N-methylmorpholine)
were added to
the resin. The reaction was shaken for 17 hours, filtered through a glass
sinter funnel, and
washed well with N-methylmorpholine to give the resin bound cyclized
depsipeptide 28.
277
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0353] Reaction 5: Preparation of Compound (C352)
Ph
Resin-O NH2 ~Ph
O O O HN Ph
H
ri~'N N 0 O O O
HNy H O N N N N,(CH2)8CH3
tBu
O~ tBu O 0 O H O
O O NH 0 HN
O'
1
tBu
O NH H O~HtN~ O N
tBu O N~N" ~~ ~Boc H
~ O H H
28
NH2
HO
O O NH2
O
~N N O O O O
HO HNy H O N N N-Ity Ny(CH2)eCH3
O O H O H O
O O NH OH HN O
~ OH
HO NH H O HN O N
~N H
O O H NH2
C352
[0354] The dried resin 28 was suspended in dichloromethane, (4 mL)
trifluoroacetic acid, (6
mL) ethanedithiol (250 l), and triisopropylsilane (250 l), and the reaction
mixture was stirred
for 3 hours at ambient temperature. The resin was filtered through a glass
sinter funnel and the
combined filtrates were evaporated under reduced pressure. Crude product was
then partitioned
between diethyl ether (6 mL), and water (3 mL). The aqueous layer was freeze-
dried to give
crude product. The crude product was purified by reverse phase HPLC (C18 10 M
Jupiter
column 250 x 21.2mm) eluting with a gradient from 20% acetonitrile 0.5% formic
acid: 80 %
water 0.5% formic acid to 80% acetonitrile 0.5% formic acid : 20 % water 0.5%
formic acid over
25 minutes. The product bearing fractions were combined and freeze-dried to
give the pure
product C352(1.0 mg).
278
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0355] Exanaple 1-5: Synthesis of Compound C369 :
HO
N HZ
O 0
O N N O O
~ O O
HO HNy H O N N N Ny (CH2)sCHa
O O H H 0
O
O NH O OH HN OH
~
HO~
~-H O HN O N
N L~L H
O O H NH2
(C369)
[0356] Reaction 1: Preparation of Compound (30)
~Ph
HN Ph
HO CH3 0
O H O H
N N N N~(CH2)$CH3 Fmoc
O O O H O ,H OH
HN~ -
O~tBu 1~ /
Resin O O
0 N
H
23
Ph
J<Ph
HN Ph
Fmoc,, N O CH3 O
O O
H O N N N N-,(CH2)8CHs
O H O O
HN O -
:L O-tBu
HO O N
H
Compound 30 is obtained from compound 23 using either Method D or Method
E(vide infra).
Method D
[0357] To the resin bound lipopeptide 23 (1 g) was added a solution of
commercially
available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine (618 mg), bromo-tris-
pyrrolidinophosphonium hexafluorophosphate (PyBrOP, 815 mg), and Di-
isopropylethylamine
279
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
(914 gL), in dichloromethane (5 mL). Dimethylaminopyridine (5 mg) was added
and the
mixture was shaken for 2 hours. After 2 h, the mixture was filtered through a
glass sinter farmel
and washed with dichloromethane (3 x 10 mL) and the coupling procedure was
repeated. The
resulting resin was filtered through a glass sinter funnel, washed with
dichloromethane (3 x 10
mL) and methanol (3 x 10 mL), and dried under diminished pressure over
potassium hydroxide
pellets. This dried resin was suspended in dichloromethane (3 mL), 2,2,2-
trifluoroethanol (1
mL), and acetic acid (1 mL), and shaken for 3 hours. The resin was filtered
through a glass
sinter fumiel and evaporation of the filtrate gave the desired peptide 30 (400
mg) as a white solid.
Method E
[0358] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine (95
mg), 4-
dimethylaminopyridine (6 mg), and N-methyl-2-chloropyridinium iodide (69 mg)
were flushed
well with argon then suspended in dichloromethane (2.7 mL). Triethylamine (76
gL) was added
and the reaction mixture was stirred to give a homogeneous solution. Resin
lipopeptide 23 (200
mg) was added to the solution, the flask was flushed again with argon and then
the reaction
mixture was shaken for 14 hours. The resulting resin was then filtered through
a glass sinter
funnel and washed well with dichloromethane. The solid was suspended in
dichloromethane (6
mL), 2,2,2-trifluoroethanol (2 mL), and acetic acid (2 mL), and shaken for 3
hours. The resin
was filtered through a glass sinter funnel and evaporation of the filtrate
gave the desired peptide
30 (54 mg) as a white solid.
280
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[03591 Reaction 2: Preparation of Com op und (31)
Resin O Ph
~Ph
tBu-O O O~ N O~ 1 HN Ph
HN FmocHN O CH3 O O O
O~ ~Bu + O N H N N (CHzaCHs
00 NH O O H H a
O~NH o 0 NH2 HN O
~~i~ O- /
tBu O~N N" ~~ Boc HO 1 O tBu
O H H H
9
Ph
Resin-O
O O FmocHN O HN Ph Ph
O N oJ O O O
O
But-O HN N N N N~(CHZ8CH3
O~ tBu O H O H O
O NH i HN O -
O' /
But-OO NH ~ 0 HN O tBu N
O~N H" NBoc H
O H
31
[0360] 1 -Hydroxy-benzotriazole (26 mg), 1,3-diisopropylcarbodiimide (30 L),
and peptide
resin compound 9 (64 mg) were added to a solution of the depsipeptide 30 (54
mg) in N-
methylmorpholine (3.8 mL), and the resulting mixture was shaken for 22 hours.
The resin was
filtered through a glass sinter funnel, and the coupling was judged to be
complete using the
Kaiser Test (vide supra), yielding the resin bound depsipeptide 31.
281
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0361] Reaction 3: Preparation of Compound (32)
Ph
Resin-O ~ FmocHN 0 HN" Ph Ph
O O~
O
N O O
tBu HN N O N N O N~(CH2aCH3
O~ tBu O ~ O H O
O NH Oi HN -
O-
OO NH ~ O HNIO tBu N
tBu O N~N~ Boc H
~ O H H
31
Ph
Resin-O HN~Ph
O
O
O N N O O O O
Bu~ H O N N N NY(CH2)aCH3
O-:I-) tBu O H O H O
O NH i HN O
x ~ o-
0 " v'NHH OHN~O tBu N
tBu O N~N~ Boc H
~ O H N
32
[0362] The dried resin 31 was'placed under an argon atmosphere, and treated
with a solution
of tetrakis-(triphenylphosphine)palladium(0) (48 mg in dichloromethane (7.63
mL)), acetic acid
(0.38 mL), and N-methylmorpholine (0.19 mL). The mixture was shaken for 4
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
two times with N-
methylmorpholine, two times with methanol, and again two times with N-
methylmorpholine.
The solid resin was suspended in 20% piperidine in N-methylmorpholine (7 mL)
for 105
minutes, filtered through a glass sinter funnel and the solid was washed well
with N-
methylmorpholine. 1-Hydroxy-benzotriazole (0.3 mL of a 0.5 molar solution in N-
methylmorpholine) and 1,3-diisopropylcarbodiimide (0.3 mL of a 0.5 molar
solution in N-
methylmorpholine) were added to the resin. The reaction mixture was shaken for
17 hours,
filtered through a glass sinter funnel, and the precipitate was washed well
with N-
methylmorpholine to give the resin bound cyclized depsipeptide 32.
282
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0363] Reaction 4: Preparation of Campound C369
Ph
Resin-O HNIA'Ph
O
O
~N N O O O O
H O H H
HN N N N Ny(CHzaCH3
tBu
O~ tBu O p H O
O 1NH I HN
O
O~ tBu
O NH H O~ HN 0 N
tBu N H" N Boc H
O H
32
HO
O NHZ
O
NIfl, N O O
O O
HO HN H O N N N Ny(CH2$CH3
Oll O H O H O
5NHOH HN ~ OH =
HO O~N N O HN O H
O HJL NH2
C369
[0364] The dried resin 32 was suspended in dichloromethane (4 mL),
trifluoroacetic acid (6
mL), ethanedithiol (250 L), and triisopropylsilane (250 L), and stirred for
3 hours at ambient
temperature. The reaction mixture was filtered through a glass sinter funnel
and washed with
dichloromethane (2 x 2 mL) and the combined filtrates were evaporated under
reduced pressure.
Crude product was then partitioned between diethyl ether (6 mL) and water (3
mL). The
aqueous layer was separated and freeze dried to give the crude product 33
(21.5 mgs). The crude
product was then purified by reverse phase HPLC (C18 10 M Jupiter column 250
x 21.2mm)
eluting with a gradient from 20% acetonitrile 0.5% formic acid: 80 % water
0.5% formic acid to
80% acetonitrile 0.5% formic acid : 20 % water 0.5% formic acid over 25
minutes. The product
bearing fractions were combined and freeze-dried to give the pure product C369
(1.8 mg).
283
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0365] Exafnple 1-6: Synthesis of Peptide Resin Compound 34:
Resin-Glu(aOAllyl)-DSer(OtBu -Gly-Asp(OtBu)-DLys(NHBoc)-A~-(p OtBu)NH, (34)
[0366] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu -Gly-AM(OtBu)-
DLys(NHBoc)-NHFmoc (35)
[0367] Commercially available Na-(9-Fluorenylmethoxycarbonyl)- Ns-(t-
butyloxycarbonyl
D-lysine (1.48 g), 1,3-diisopropylcarbodiimide (0.49 mL), 1-hydroxy-
benzotriazole (425 mg)
and 4-dimethylaminopyridine (37 mg) as a solution in N-methylpyrolidine (20
mL) was added to
resin 17(vide supra). The reaction mixture was shaken for three hours,
filtered through a glass
sinter funnel and the coupling was repeated for 15 hours. The reaction mixture
was filtered,
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15 mL),
methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give
compound 35.
[0368] Reaction 2: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-Gl -Asp(OtBu)-
DLs(NHBoc)-NH, 36
[0369] Compound 35 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for one
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid was washed
with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine
(3 x 15 mL) to give compound 36.
[0370] Reaction 3: Prenaration of Resin-Glu(aOAl1yl)-DSer(OtBu -Gly-Asp(OtBu)-
DLys(NHBoc -Asp(OtBu)-NHFmoc (37)
[0371] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
0-tertbutyl
ester (2.16 g), 1,3-diisopropylcarbodiimide (822 L), and 1-hydroxy-
benzotriazole (710 mg) as a
solution in N-methylpyrolidine (20 mL) was added to resin 36. The reaction
mixture was shaken
for four hours. The reaction mixture was filtered through a glass sinter
funnel then the solid was
washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again
with N-
methylpyrolidine (3 x 15 mL) to give compound 37.
[0372] Reaction 4: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-G1y-Ap(OtBu)-
DLys(NHBoc)-Asp(OtBu)-NH, (34)
[0373] Compound 37 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for one
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid was washed
with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine
(3 x 15 mL) to give compound 34.
284
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0374] Example 1-7: Synthesis of Peptide Resin Compound 38:
Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Ala-NH2 (38)
[0375] Reaction 1: Preparation of Resin-Glu(aOAllyl -DSer OtBu)-Gly-AsR(OtBu)-
DAIa-
Asp(OtBu)-Ala-NHFmoc (39)
[0376] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-alanine (1.62
g), 1,3-
diisopropylcarbodiimide (825 L), and 1-hydroxy-benzotriazole (715 mg) as a
solution in N-
methylpyrolidine ( 20 mL) was added to resin 21 (vide supra). The reaction
mixture was shaken
for 17 hours. The reaction mixture was filtered through a glass sinter funnel
then the solid was
washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again
with N-
methylpyrolidine (3 x 15 mL) to give compound 39.
[0377] Reaction 2: Preparation of Resin-Glu(aOAllyl)-DSer OtBu-Gly-AsR(OtBu)-
DAla-
Asp(OtBu -Ala-NH2 38
[0378] Compound 39 (227mg) was agitated in 20% piperidine in N-
methylpyrolidine (1 mL)
for 0.5 hour. The reaction mixture was filtered through a glass sinter funnel
then the solid was
washed with N-methylpyrolidine (3 x 5 mL), methanol (3 x 5 mL), and again with
N-
methylpyrolidine (3 x 5 mL) to give 3 8
[0379] Example 1-8: Synthesis of Peptide Resin Conapound 40:
Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-NH2 (40)
[0380] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt )-NHFmoc (41)
[0381] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-
asparagine (NHTrt)OH (3.1 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-
tetramethyluronium
tetrafluroborate (TBTU, 1.67 g), Hydroxy-benzotriazole (0.56g) and
diisopropylethylamine
(DIPEA, 2.7 mL) as a solution in N-methylpyrrolidone (NMP, 40 mL) was added to
Resin-Glu-
NH2 (l l,vide supra, 4 g). The mixture was shaken for 30 minutes, filtered
through a glass sinter
funnel and a few beads were tested for the presence of a free amine using the
standard Kaiser test
(vide supra). The Kaiser test gave a yellow color so the coupling was deemed
complete. After
filtration through a glass sinter fiuuiel the product bearing resin was washed
with N-
methylpyrolidine (3 x 40 mL), methanol (3 x 40 mL), and again with N-
methylpyrolidine (3 x 40
mL ) to give compound 41.
285
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0382] Reaction 2: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt-NH, (42)
[0383] Compound 41 was agitated in 20% piperidine in N-methylpyrolidine (30
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter fmuiel and
was re-suspended in
20% piperidine in N-methylpyrolidine (30 mL) and was agitated for 30 minutes.
The reaction
mixture was filtered through a glass sinter funnel then the solid was washed
with N-
methylpyrolidine (3 x 30 mL), methanol (3 x 30 mL), and again with N-
methylpyrolidine (3 x 30
mL) to give compound 42.
[0384] Reaction 3: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-NHFmoc
(43)
[0385] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-glycine (1.55
g), 2-(IH-
Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 1.67 g),
Hydroxy-
benzotriazole (HOBt, 0.56g) and diisopropylethylamine (DIPEA, 2.7 mL) as a
solution in N-
methylpyrrolidone (NMP, 40 mL) was added to compound 42 (4 g). The mixture was
shaken for
30 minutes, filtered through a glass sinter funnel and a few beads were tested
for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 40 mL), methanol (3 x 40
mL), and
again with N-methylpyrolidine (3 x 40 mL ) to give compound 43.
[0386] Reaction 4: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-NH, 44
[0387] Compound 43 was agitated in 20% piperidine in N-methylpyrolidine (30
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel and
was re-suspended in
20% piperidine in N-methylpyrolidine (30 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 30 mL), methanol (3 x 30 mL), and again with N-methylpyrolidine (3 x 30 mL)
to give
compound 44.
[0388] Reaction 5: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-
Asp(OtBu)=
NHFmoc (45)
[0389] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
~i-
tertbutyl ester (2.14 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium
tetrafluroborate
(TBTU, 1.67 g), HOBt (0.56g) and diisopropylethylamine (DIPEA, 2.7 mL) as a
solution in N-
methylpyrrolidone (NMP, 40 mL) was added to compound 44 (4 g). The mixture was
shaken for
30 minutes, filtered through a glass sinter funnel and a few beads were tested
for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
286
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 40 mL), methanol (3 x 40
mL), and
again with N-methylpyrolidine (3 x 40 mL ) to give compound 45
[03901 Reaction 6: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu -
NH,
46
[0391] Compound 45 was agitated in 20% piperidine in N-methylpyrolidine (30
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter fiuinel and
was re-suspended in
20% piperidine in N-methylpyrolidine (30 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 30 mL), methanol (3 x 30 mL), and again with N-methylpyrolidine (3 x 30 mL)
to give
compound 46.
[0392] Reaction 7: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-
DAIa-
NHFmoc (47)
[0393] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-alanine (0.81
g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.84
g), HOBt (0.28g)
and diisopropylethylamine (DIPEA, 1.4 mL) as a solution in N-methylpyrrolidone
(NMP, 20
mL) was added to compound 46 (2 g). The mixture was shaken for 30 minutes,
filtered through
a glass sinter funnel and a few beads were tested for the presence of a free
amine using the
standard Kaiser test (vide supra). The Kaiser test gave a yellow color so the
coupling was
deemed complete. After filtration through a glass sinter funnel the product
bearing resin was
washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20 mL), and again
with N-
methylpyrolidine (3 x 20 mL) to give compound 47.
[0394] Reaction 8: Preparation of Glu(aOAlly1)-DAsn HTrt )-Gly-Asp(OtBu)-DAla-
NHa
48
[0395] Compound 47 was agitated in 20% piperidine in N-methylpyrolidine (15
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter fiuinel and
was re-suspended in
20% piperidine in N-methylpyrolidine (15 mL) and agitated for 30 minutes. The
reaction
mixture was filtered through a glass sinter funnel then the solid was washed
with N-
methylpyrolidine (3 x 10 mL), methanol (3 x 10 mL), and again with N-
methylpyrolidine (3 x 10
mL) to give compound 48.
287
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0396] Reaction 9: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt-Gl -Asp(OtBu)-
DAla-
As (p OtBu)-NHFmoc (49)
[0397] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
(3-
tertbutyl ester (1.07 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium
tetrafluroborate
(TBTU, 0.84 g), HOBt (0.28g) and diisopropylethylamine (DIPEA, 1.4 mL) as a
solution in N-
methylpyrrolidone (NMP, 20 mL) was added to compound 48 (2 g). The mixture was
shaken for
30 minutes, filtered through a glass sinter funnel and a few beads were tested
for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20
mL), and
again with N-methylpyrolidine (3 x 20 mL) to give compound 49
[0398] Reaction 10: Pre aration of Glu aOAll 1-DAsn HTrt-Gl -As OtBu -DAla-
Asp(OtBu)-NH2 40
[0399] Compound 49 was agitated in 20% piperidine in N-methylpyrolidine (15
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel and
was re-suspended in
20% piperidine in N-methylpyrolidine (15 mL) and agitated for 30 minutes. The
reaction
mixture was filtered through a glass sinter funnel then the solid was washed
with N-
methylpyrolidine (3 x 10 mL), methanol (3 x 10 mL), and again with N-
methylpyrolidine (3 x 10
mL) to give compound 40
[0400] Example 1-9: Synthesis ofPeptide Resin Compound 50:
Resin-Glu(aOA)Iyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Orn(NHBoc)-NH2 50
[0401] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn HTrt)-Glv-Asp(OtBu)-
DAla-
Asp(OtBu)-Orn(NHBoc)-NHFmoc (51)
[0402] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-ornithine
(Boc)-OH
(1.17 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate
(TBTU, 0.83 g),
HOBt (0.31 g) and diisopropylethylamine (DIPEA, 1.4 mL) as a solution in N-
methylpyrrolidone
(NMP, 20 mL) was added to compound 40 (2.8 g). The mixture was shaken for 30
minutes,
filtered through a glass sinter funnel and a few beads were tested for the
presence of a free amine
using the standard Kaiser test (vide supra). The Kaiser test gave a yellow
color so the coupling
was deemed complete. After filtration.through a glass sinter funnel the
product bearing resin
288
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
was washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20 mL), and
again with N-
methylpyrolidine (3 x 20 mL ) to give compound 51.
[0403] Reaction 2 Preparation of Resin-Glu(aOAlly1)-DAsn(NHTrt
)-G13L-ASp(OtBu)-DAIa-
Asp(OtBu)-Orn(NHBoc -NH, 50
[0404] Compound 51 was agitated in 20% piperidine in N-methylpyrolidine (15
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (15 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 10
mL), methanol (3 x 10 mL), and again with N-methylpyrolidine (3 x 10 mL) to
give compound
50.
[0405] Exarnple 1-10: Synthesis of Peptide Resin Compound 52:
Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-AIa-NH2 (52)
[0406] Reaction 1:Preparation of Resin-Glu(aOAllyI)-DAsn(NHTrt )-Gly A~(OtBu -
DAIa-
Asp(OtBu)-Ala-NHFmoc (53)
[0407] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-alanine (63
mg), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 64
mg), HOBt (27
mg) and diisopropylethylamine (DIPEA, 70 L) as a solution in N-
methylpyrrolidone (NMP, 1
mL) was added to compound 40 (340 mg). The mixture was shaken for 30 minutes,
filtered
through a glass sinter funnel and a few beads were tested for the presence of
a free amine using
the standard Kaiser test (vide supra). The Kaiser test gave a yellow color so
the coupling was
deemed complete. After filtration through a glass sinter funnel the product
bearing resin was
washed with N-methylpyrolidine (3 x 2 mL), methanol (3 x 2 mL), and again with
N-
methylpyrolidine (3 x 2 mL) to give compound 53.
[0408] Reaction 2: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt )-
Gly:Asp(OtBu)-DAIa-
AM(OtBu)-Ala-NH, 52
[0409] Compound 53 was agitated in 20% piperidine in N-methylpyrolidine (1.5
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (1.5 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 1 mL), methanol (3 x 1 mL), and again with N-methylpyrolidine (3 x 1 mL) to
give compound
52.
289
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0410] Example 1-11: Synthesis of Peptide Resin Compound 54:
Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-
NHa (54)
[0411] Reaction 1: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-Gl -Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-NHFmoc(55)
[0412] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-ornithine
(Boc)-OH
(0.44 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate
(TBTU, 0.31 g),
HOBt (0.13 g) and diisopropylethylamine (DIPEA, 0.3 mL) as a solution in N-
methylpyrrolidone (NMP, 20 mL) was added to compound 34 (vide supra, 0.8 g).
The mixture
was shaken for 30 minutes, filtered through a glass sinter funnel and a few
beads were tested for
the presence of a free amine using the standard Kaiser test (vide supra). The
Kaiser test gave a
yellow color so the coupling was deemed complete. After filtration through a
glass sinter funnel
the product bearing resin was washed with N-methylpyrolidine (3 x 20 mL),
methanol (3 x 20
mL), and again with N-methylpyrolidine (3 x 20 mL) to give compound 55.
[0413] Reaction 2: Preparation of Resin-Glu(aOAllyl -DSer OtBu)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-NH2 54
[0414] Compound 55 was agitated in 20% piperidine in N-methylpyrolidine (8 mL)
for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (8 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 8
mL), methanol (3 x 8 mL), and again with N-methylpyrolidine (3 x 8 mL) to give
compound 54.
[0415] Exarnple 1-12: Synthesis of Peptide Resin Compound 56
Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-NH2 (56)
[0416] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-NHFmoc (57)
[0417] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D- Na-(9-
Fluorenylmethoxycarbonyl)- Ns-(t-butyloxycarbonyl L-lysine (1.28 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.84 g), HOBt (0.28 g) and
diisopropylethylamine (DIPEA, 1.4 mL) as a solution in N-methylpyrrolidone
(NMP, 20 mL)
was added to compound 46 (2 g). The mixture was shaken for 30 minutes,
filtered through a
290
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
glass sinter funnel and a few beads were tested for the presence of a free
amine using the
standard Kaiser test (vide supra). The Kaiser test gave a yellow color so the
coupling was
deemed complete. After filtration through a glass sinter funnel the product
bearing resin was
washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20 mL), and again
with N-
methylpyrolidine (3 x 20 mL) to give compound 57.
[0418] Reaction 2: Preparation of Resin-Glu(aOAll~)-DAsn TrtLGly-As(OtBu)-
DLys(NHBoc)-NHa (58)
[0419] Compound 57 was agitated in 20% piperidine in N-methylpyrolidine (15
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (15 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 10
mL), methanol (3 x 10 mL), and again with N-methylpyrolidine (3 x 10 mL) to
give compound
58.
[0420] Reaction 3: Preparation of Resin-Glu(aOAllyl)-DAsn HTrt )-Gly-Asp(OtBu)-
DLys(NHBoc -Asp(OtBu)-NHFmoc (59)
[0421] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
(3-
tertbutyl ester (1.07 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium
tetrafluroborate
(TBTU, 0.84 g), HOBt (0.28 g) and diisopropylethylamine (DIPEA, 1.4 mL) as a
solution in N-
methylpyrrolidone (NMP, 20 mL) was added to compound 58 (2 g). The mixture was
shaken for
30 minutes, filtered through a glass sinter funnel and a few beads were tested
for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20
mL), and
again with N-methylpyrolidine (3 x 20 mL) to give compound 59 ,
[0422] Reaction 4: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-NH2 (56)
[0423] Compound 59 was agitated in 20% piperidine in N-methylpyrolidine (15
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (15 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 10
mL), methanol (3 x 10 mL), and again with N-methylpyrolidine (3 x 10 mL) to
give compound
56.
291
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0424] Example 1-13: Synthesis of Peptide Resin Compound 60:
Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-
Orn(NHBoc)-NH (60)
[0425] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn HTrt)-Glv-Asp(OtBu)=
DLysNHBoc)-Asp(OtBu)-Orn(NHBoc)-NHFmoc(61)
[0426] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-ornithine
(Boc)-OH
(0.54 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate
(TBTU, 0.38 g),
HOBt (0.12 g) and diisopropylethylamine (DIPEA, 0.63 mL) as a solution in N-
methylpyrrolidone (NMP, 12 mL) was added to compound 56 (1.2 g). The mixture
was shaken
for 30 minutes, filtered through a glass sinter funnel and a few beads were
tested for the presence
of a free amine using the standard Kaiser test (vide supra). The Kaiser test
gave a yellow color
so the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20
mL), and
again with N-methylpyrolidine (3 x 20 mL) to give compound 61.
[0427] Reaction 2: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gl -Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-NH2 (60)
[0428] Compound 61was agitated in 20% piperidine in N-methylpyrolidine (12 mL)
for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (12 mL) and agitated for 30minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 10
mL), methanol (3 x 10 mL), and again with N-methylpyrolidine (3 x 10 mL) to
give compound
60.
[0429] Example 1-14: Synthesis of Peptide Resin Compound 62:
Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-AIa-NH2
(62)
[0430] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gl -p(OtBu)-
DLys(NHBoc -As (OtBu)-Ala-NHFmoc(63)
[0431] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-alanine (0.78
g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.80
g), HOBt (0.27
g) and diisopropylethylamine (DIPEA, 0.81 mL) as a solution in N-
methylpyrrolidone (NMP, 20
mL) was added to compound 56 (2 g). The mixture was shaken for 30 minutes,
filtered through
a glass sinter funnel and a few beads were tested for the presence of a free
amine using the
292
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
standard Kaiser test (vide supra). The Kaiser test gave a yellow color so the
coupling was
deemed complete. After filtration through a glass sinter funnel the product
bearing resin was
washed with N-methylpyrolidine (3 x 20 mL), methanol (3 x 20 mL), and again
with N-
inethylpyrolidine (3 x 20 mL ) to give compound.63.
[0432] Reaction 2: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu -Ala-NH, 62
[0433] Compound 63 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (20 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 20
mL), methanol (3 x 20 mL), and again with N-methylpyrolidine (3 x 20 mL) to
give compound
62.
[0434] Example 1-15: Synthesis of Peptide Resin Compound 64:
Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-NH2 (64)
[0435] Reaction 1: Preparation of Resin-Ala-Sar-NMeFmoc (65)
[0436] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-
sarcosirie
(1.56 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate
(TBTU, 1.61 g),
and diisopropylethylamine (DIPEA, 871 l) as a solution in N-methylpyrrolidone
(NMP, 25 mL)
was added to commercially available alanine 2-chlorotrityl resin (66, 2.5 g).
The mixture was
shaken for 30 minutes, filtered through a glass sinter funnel and a few beads
were tested for the
presence of a free amine using the standard Kaiser test (vide supra). The
Kaiser test gave a
yellow color so the coupling was deemed complete. After filtration through a
glass sinter funnel
the product bearing resin was washed with N-methylpyrolidine (3 x 15 mL),
methanol (3 x 15
mL), and again with N-methylpyrolidine (3 x 15 mL ) to give compound 65.
[0437] Reaction 2: Preparation of Resin-Ala-Sar-NMeH (67)
[0438] Compound 65 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 67.
293
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0439] Reaction 3: Preparation of Resin-Ala-Sar-Thr-NHFmoc (68)
[0440] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-threonine (853
mg),
bromo-tris-pyrrolidinophosphonium hexafluorophosphate (PyBrOP, 1.165 g), and
DIPEA (1.31
mL) as a solution in dichloromethane (25 mL) was added to compound 67 (334
mg). The
mixture was shaken for one hour. The reaction mixture was filtered through a
glass sinter funnel
then the solid was washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x
15 mL ), and
again with N-methylpyrolidine (3 x 15 mL) to give compound 68.
[0441] Reaction 4: Preparation of Resin-Ala-Sar-Thr-NH, 69
[0442] Compound 38 (vide supra) was agitated in 20% piperidine in N-
methylpyrolidine (25
mL) for 1 hour. The reaction mixture was filtered through a glass sinter
funnel then the solid
was washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and
again with N-
methylpyrolidine (3 x 15 mL) to give compound 69.
[0443] Reaction 5: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-NHFmoc (70)
[0444] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
(3-tert-
butyl ester (2.06 g), TBTU (1.61 g), and DIPEA (871 gL) as a solution in NMP
(25 mL) were
added to compound 69. The mixture was shaken for three hours. The reaction
mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to
give compound
70.
[0445] Reaction 6: Preparation of Resin-Ala-Sar-Thr-AsR(OtBu)-NH2 71
[0446] Compound 70 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for one
hour. The reaction mixture was filtered through a glass sinter funnel then
washed with N-
methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x 15
mL) to give compound 71.
[0447] Reaction 7: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-
NHFmoc
72
[0448] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-asparagine S-N-
trityl
(1.49 g), TBTU (1.61 g), and DIPEA (871 L) as a solution in NMP (25 mL) was
added to
compound 71. The reaction mixture was shaken for seventeen hours. The reaction
mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to
give compound
72.
294
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0449] Reaction 8: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt-NH,
(73)
[0450] Compound 72 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for 2
hours. The reaction mixture was filtered through a glass sinter funnel then
the solid was washed
with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine
(3 x 15 mL) to give compound 73.
[0451] Reaction 9: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-
NHFmoc (74)
[0452] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(1.07 g),
TBTU (802 mg), and DIPEA (435 L) as a solution in NMP (10 mL) was added to
resin 73.
The reaction mixture was shaken for forty three hours. The reaction mixture
was filtered through
a glass sinter funnel then the solid was washed with N-methylpyrolidine (3 x
15 mL), methanol
(3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give compound
74.
[0453] Reaction 10: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu -DAsn(NHTrt)-Trp
NH2
64
[0454] Compound 74 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for one
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid was washed
with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine
(3 x 15 mL) to give resin peptide compound 64.
[0455] Example 1-16: Synthesis of Peptide Resin Compound (75):
Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-Undecanoic amide. (75)
[0456] Commercially available undecanoic acid (930 mg), 1,3-
diisopropylcarbodiimide (0.78
mL), and 1-hydroxy-benzotriazole (676 mg) as a solution in N-methylpyrolidine
(20 mL) was
added to compound 64. The mixture was shaken for 23 hours, filtered through a
glass sinter
fiuulel, and the reaction was judged to be incomplete using the Kaiser Test
(vide supra). The
resin was then filtered through a glass sinter funnel and washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL). The
reaction was
found to be complete using the Kaiser Test, yielding the resin bound compound
75.
295
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0457] Example 1-17. Synthesis ofPeptide Resin Compound (76)
Resin-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-8-Methyldecanoic amide (76)
0458 Commercially available 8-methyldecanoic acid (1.55 g), 2-(1H-Benzotriazol-
yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 2.67 g),
diisopropylethylamine (DIPEA, 2.9
mL), and 1-hydroxy-benzotriazole (1.12 g) as a solution in N-methylpyrolidine
(80 mL) was
added to compound 1 (7.6 g). The mixture was shaken for 18 hours, filtered
through a glass
sinter funnel, and the reaction was judged to be complete using the Kaiser
Test (vide supra),
yielding the resin bound compound 76.
[0459] Example 1-18: Synthesis of Peptide Resin Compound (77)
Resin-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-tridecanoic amide (77)
[0460] Commercially available tridecanoic acid (2.39 g), 2-(1H-Benzotriazol-
yl)-1,1,3,3-
tetramethyluronium tetrafluroborate (TBTU, 3.47 g), diisopropylethylamine
(DIPEA, 3.75 mL),
and 1-hydroxy-benzotriazole (1.46 g) as a solution in N-methylpyrolidine (80
mL) was added to
compound 1 (10 g). The mixture was shaken for 17 hours, filtered through a
glass sinter funnel,
and the reaction was judged to be complete using the Kaiser Test (vide supra),
yielding the resin
bound compound 77.
[0461] Example 1-19: Synthesis of Peptide Resin Compound (78)
Resin-Gly-Thr-Asp(OtBu)-DGIu(OtBu)-Trp-8-Methyldecanoic amide (78)
[0462] Reaction 1: Preparation of Resin-Gly-Thr-Asp(OtBu)-DGIu OtBu) NHFmoc
(79)
[0463] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-glutamic acid
7-t-butyl
ester (1.14 g), TBTU (0.87 g), HOBt (0.37 g) and DIPEA (940 L) as a solution
in NMP (20
mL) was added to compound 5. The reaction mixture was shaken for one hour. The
reaction
mixture was filtered through a glass sinter fu.nnel then the solid was washed
with N-
methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x 15
mL). The reaction was judged to be complete using the Kaiser Test (vide
supra), yielding the
resin bound compound 79.
[0464] Reaction 2: Preparation of Resin-Gly-Thr-Asp(OtBu)-DGlu(OtBu -NH, (80)
[0465] Compound 79 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 15
minutes. The resin was filtered through a glass sinter funnel and re-suspended
in 20% piperidine
in N-methylpyrolidine (20 mL) and agitated for 15 minutes. The reaction
mixture was filtered
296
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15 mL),
methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give
resin bound
compound 80.
[0466] Reaction 3: Preparation of Resin-Gly-Thr-Asp(OtBu -DGlu(OtBu)-Trp-
NHFmoc
81
[0467] Commercially available Na-(9-Fluorenylmethoxycarbonyl)- L-tryptophan
(1.15 g),
TBTU (0.87 g), HOBt (0.37 g) and DIPEA (940 L) as a solution in NMP (20 mL)
was added to
the compound 80. The reaction mixture was shaken for one hour. The reaction
mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL). The
reaction was
judged to be complete using the Kaiser Test (vide supra), yielding the resin
bound 81.
[0468] Reaction 4: Preparation of Resin-Gly-Thr-Asp(OtBu -DGlu(OtBu)-Trp-NH,
82
[0469] Resin bound compound 81 was agitated in 20% piperidine in N-
methylpyrolidine (20
mL) for 15 minutes. The resin was filtered through a glass sinter funnel and
re-suspended in
20% piperidine in N-methylpyrolidine (20 mL) and agitated for 15 minutes. The
reaction
mixture was filtered through a glass sinter farmel then the solid was washed
with N-
methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and agaiin with N-
methylpyrolidine (3 x 15
mL) to give resin bound compound 82.
[0470] Reaction 5: Preparation of Resin-Gly-Thr-Asp(OtBu)-DGlu(OtBuLr -8-
Methyldecanoic amide (78)
[0471] Commercially available 8-methyldecanoic acid (0.71 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 1.21 g),
diisopropylethylamine (DIPEA, 2.0
mL), and 1-hydroxy-benzotriazole (0.508 g) as a solution in N-methylpyrolidine
(80 mL) was
added to compound 82 (4.0 g). The mixture was shaken for 18 hours, filtered
through a glass
sinter funnel), and the reaction was judged to be complete using the Kaiser
Test (vide supra).
The reaction mixture was filtered through a glass sinter funnel then the solid
was washed with N-
methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with N-
methylpyrolidine (3 x 6
mL) to give resin bound compound 78.
297
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0472] Example 1-20: Synthesis of Peptide Resin Con2pound (83)
Resin-Ala-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-8-Methyldecanoic amide (83)
[0473] Commercially available 8-methyldecanoic acid (0.71 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.60 g),
diisopropylethylamine (DIPEA,
0.64 mL), and 1-hydroxy-benzotriazole (0.25 g) as a solution in N-
methylpyrolidine (20 mL)
was added to compound 34 (1.8 g). The mixture was shaken for 18 hours,
filtered through a
glass sinter funnel, and the reaction was judged to be complete using the
Kaiser Test (vide
supra). The reaction mixture was filtered through a glass sinter funnel then
the solid was washed
with N-methylpyrolidine (3 x 6 mL), methanol (3 x 6 mL), and again with N-
methylpyrolidine (3
x 6 mL) to give resin bound compound 83.
[0474] Example 1-21: Synthesis of Peptide Resin Compound (84)
Resin-Ala-Sar-Thr-Asp(OtBu)-DGIu(OtBu)-Trp-8-Methyldecanoic amide (84)
[0475] Reaction 1: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DGlu OtBu)-
NHFmoc (85)
[0476] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-glutamic acid
y-t-butyl
ester (0.98 g), TBTU (0.74 g), HOBt (0.31 g) and DIPEA (810 L) as a solution
in NMP (20
mL) was added to compound 71 (1.8 g). The reaction mixture was shaken for
seventeen hours.
The reaction mixture was filtered through a glass sinter fu.nnel then the
solid was washed with N-
methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x 15
mL) to give compound 85.
[0477] Reaction 2: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DGlu(OtBu)-NH,
86
[0478] Compound 85 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel , re-
suspended in 20%
piperidine in N-methylpyrolidine (20 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 20
mL), methanol (3 x 20 mL), and again with N-methylpyrolidine (3 x 20 mL) to
give compound
86.
[0479] Reaction 3: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DGlu(OtBu)-Trp-
NHFmoc
87
[0480] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(0.98 g),
TBTU (0.74 g), HOBt (0.31 g) and DIPEA (810 L) as a solution in NMP (25 mL)
was added to
compound 86 (2.2 g). The reaction mixture was shaken for seventeen hours. The
reaction
298
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
mixture was filtered through a glass sinter funnel then the solid was washed
with N-
methylpyrolidine (3 x 25 mL), methanol (3 x 25 mL), and again with N-
methylpyrolidine (3 x 25
mL) to give compound 87.
[0481] Reaction 4: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DGlu(OtBu)-Trp
NH, 88
[0482] Compound 87 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel , re-
suspended in 20%
piperidine in N-methylpyrolidine (25 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 30
mL), methanol (3 x 30 mL), and again with N-methylpyrolidine (3 x 30 mL) to
give compound
88.
[0483] Reaction 5: Preparation of Resin-Ala-Sar-Thr-Asp(OtBu)-DGIu(OtBu)-Trp-8-
Methyldecanoic amide (84)
[0484] Commercially available 8-methyldecanoic acid (0.34 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.60 g),
diisopropylethylamine (DIPEA,
0.64 mL), and 1-hydroxy-benzotriazole (0.25 g) as a solution in N-
methylpyrolidine (20 mL)
was added to compound 88 (2.0 g). The mixture was shaken for 18 hours,
filtered through a
glass sinter funnel, and the reaction was judged to be complete using the
Kaiser Test (vide
supra). The solid was washed with N-methylpyrolidine (3 x 15 mL), methanol (3
x 16 mL), and
again with N-methylpyrolidine (3 x 16 mL) to give resin bound compound 84.
[0485] Example 1-22: Synthesis of Peptide Resin Cornpound 89
Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-Undecanoic amide (89)
[0486] Reaction 1: Preparation of Resin-Ala-Gly-NHFmoc (90)
[0487] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-
glycine
(1.49 g), TBTU (1.61 g), and DIPEA (871 L) as a solution in NMP (25 mL) were
added to the
commercially available Alanine-2-cholrotrityl-resin (66, 2.5 g). The mixture
was shaken for
three hours, filtered through a glass sinter funnel and a few beads were
tested for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x 15
mL), and
again with N-methylpyrolidine (3 x 15 mL) to give compound 90.
299
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0488] Reaction 2: Preparation of Resin-Ala-Gl -NH, (91)
[0489] Compound 90 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 91.
[0490] Reaction 3: Preparation of Resin-Ala-Gly-Thr-NHFmoc (92)
[0491] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-threonine (853
mg),
bromo-tris-pyrrolidinophosphonium hexafluorophosphate (PyBrOP, 1.165 g), and
DIPEA (1.31
mL) as a solution in dichloromethane (25 mL) was added to compound 91 (334
mg). The
mixture was shaken for one hour. The reaction mixture was filtered through a
glass sinter funnel
then the solid was washed with N-methylpyrolidine (3 x 15 mL), methanol (3 x
15 mL), and
again with N-methylpyrolidine (3 x 15 mL) to give compound 92.
[0492] Reaction 4: Preparation of Resin-Ala-Gly-Thr-NH2 (93)
[0493] Compound 92 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1.5
hours. The reaction mixture was filtered through a glass sinter funnel then
the solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give resin bound compound 93.
[0494] Reaction 5: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-NHFmoc (94)
[0495] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
(3-tert-
butyl ester (2.06g), TBTU (1.61 g), and DIPEA (871 L) as a solution in NMP
(25 mL) was
added to compound 93. The mixture was shaken for three hours. The reaction
mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to
give compound
94.
[0496] Reaction 6: Preparation of Resin-Ala-Gly-Thr-AM(OtBu)-NH, 95
[0497] Compound 94 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 94.
300
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0498] Reaction 7: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-
NHFmoc
96
[0499] Commercially available Na-(9-Fluorenylmethoxycarbonyl)=D-asparagine 6-N-
trityl
(1.49 g), TBTU (0.80 g), and DIPEA (435 L) as a solution in DMF (10 mL) were
added to
compound 95. The mixture was shaken seventeen hours. The reaction mixture was
filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15 mL),
methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give
compound 96.
[0500] Reaction 8: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-NH,
97
[0501] Compound 96 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 2
hours. The reaction mixture was filtered through a glass sinter funnel then
the solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 97.
[0502] Reaction 9: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Tip-
NHFmoc (98)
[0503] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(1.07 g),
TBTU (0.80 g), and DIPEA (435 L) as a solution in NMP (25 mL) was added to
compound 97.
The mixture was shaken for forty three hours. The reaction mixture was
filtered through a glass
sinter funnel then the solid was washed with N-methylpyrolidine (3 x 15 mL),
methanol (3 x 15
mL), and again with N-methylpyrolidine (3 x 15 mL) to give compound 98.
[0504] Reaction 10: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-
NH,
99
[0505] Compound 98 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 99.
[0506] Reaction 11: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-
Undecanoic amide (89)
[0507] Commercially available undecanoic acid (930 mg), 1,3-
diisopropylcarbodiimide (0.78
mL), and 1-hydroxy-benzotriazole (676 mg) as a solution in N-methylpyrolidine
(20 mL) was
added to compound 99. The mixture was shaken for 23 hours, filtered through a
glass sinter
funnel, and the reaction was judged to be incomplete using the Kaiser Test
(vide supra). The
resin was then filtered through a glass sinter fumnel and washed with N-
methylpyrolidine (3 x 15
301
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL). The
reaction was
found to be complete using the Kaiser Test, yielding compound 89.
[0508] Example 1-23: Synthesis ofPeptide Resin Compound 100
Resin-Ala-Gly-Thr-Asp(OtBu)-DGIu(OtBu)-Trp-Undecanoic amide (100)
[0509] Reaction 1: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DGlu(OtBu)-
NHFmoc
(101)
[0510] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-glutamic acid
y-t-butyl
ester (1.06 g), TBTU (0.80 g), and DIPEA (435 L) as a solution in DMF (10 mL)
were added to
compound 95. The mixture was shaken seventeen hours. The reaction mixture was
filtered
through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 15 mL),
methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give
compound
compound 101.
[0511] Reaction 2: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DGlu(OtBu)-NH,
(102)
[0512] Compound 101 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 102.
[0513] Reaction 3: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu -DGlu(OtBu)-Trp-
NHFmoc
(103)
[0514] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(1.07 g),
TBTU (0.80 g), and DIPEA (435 L) as a solution in NMP (25 mL) was added to
compound
102. The mixture was shaken for forty three hours. The reaction mixture was
filtered through a
glass sinter funnel then the solid was washed with N-methylpyrolidine (3 x 15
mL), methanol (3
x 15 mL), and again with N-methylpyrolidine (3 x 15 mL) to give 103.
[0515] Reaction 4: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu)-DGlu(OtBuLrp-NH,
(104)
[0516] Compound 103 was agitated in 20% piperidine in N-methylpyrolidine (20
mL) for 1
hour. The reaction mixture was filtered through a glass sinter funnel then the
solid washed with
N-methylpyrolidine (3 x 15 mL), methanol (3 x 15 mL), and again with N-
methylpyrolidine (3 x
15 mL) to give compound 104.
302
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[05171 Reaction 5: Preparation of Resin-Ala-Gly-Thr-Asp(OtBu -DGlu(OtBu)-Trp-
Undecanoic amide. (100)
[0518] Commercially available undecanoic acid (930 mg), 1,3-
diisopropylcarbodiimide (0.78
mL), and 1-hydroxy-benzotriazole (676 mg) as a solution in N-methylpyrolidine
(20 mL) was
added to compound 104. The mixture was shaken for 23 hours, filtered through a
glass sinter
funnel, and the reaction was judged to be incomplete using the Kaiser Test
(vide supra). The
resin was then filtered through a glass sinter funnel and washed with N-
methylpyrolidine (3 x 15
mL), methanol (3 x 15 mL), and again with N-methylpyrolidine (3 x 15 mL). The
reaction was
found to be complete using the Kaiser Test, yielding the compound 100.
[0519] Example 1-24: Synthesis of f Peptide Resin 105
Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-DAsn(NHTrt)-Trp-8-Methyldecanoic amide
(105)
[05201 Reaction 1: Preparation of Resin-Orn(NHBoc)-NHFmoc (106)
[0521] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-N-B-
tertbutoxycarbonyl-
L-ornithine (8.73 g) as a solution in dichloromethane (100 mL) and
diisopropylethylamine
(DIPEA, 13.4 mL), were added to a pre-swollen commercially available 2-
chlorotrityl resin 107
(10.0 g). The mixture was shaken for 1 hour, filtered through a glass sinter
funnel, and the
reaction was judged to be complete using the Kaiser Test (vide supra). The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 100
mL), methanol (3 x 100 mL), and again with N-methylpyrolidine (3 x 100 mL) to
give
compound 106.
[0522] Reaction 2: Preparation of Resin-Orn(NHBoc -NH, (108)
[0523J Compound 106 was agitated in 20% piperidine in N-methylpyrolidine (100
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter funnel,
re-suspended in 20%
piperidine in N-methylpyrolidine (100 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 130 mL), methanol (3 x 130 mL), and again with N-methylpyrolidine (3 x 130
mL) to give
compound 108.
[05241 Reaction 3: Preparation of Resin-Orn(NHBoc)-Sar-NMeFmoc (109)
[0525] A solution of commercially available Na-(9-Fluorenylmethoxycarbonyl)-
sarcosine
(2.6 g), 2-(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate
(TBTU, 2.7 g),
HOBt (1.13 g) and diisopropylethylamine (DIPEA, 2.9 mL) as a solution in N-
303
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
inethylpyrrolidone (100 mL) was added to compound 108 (10 g). The mixture was
shaken for
60 minutes, filtered through a glass sinter furmel and a few beads were tested
for the presence of
a free amine using the standard Kaiser test (vide supra). The Kaiser test gave
a yellow color so
the coupling was deemed complete. After filtration through a glass sinter
funnel the product
bearing resin was washed with N-methylpyrolidine (3 x 115 mL), methanol (3 x
115 mL), and
again with N-methylpyrolidine (3 x 115 mL) to give compound 109.
[0526] Reaction 4: Preparation of Resin-Orn(NHBoc)-Sar-NMeH (110)
[0527] Compound 109 was agitated in 20% piperidine in N-methylpyrolidine (100
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter funnel,
re-suspended in 20%
piperidine in N-methylpyrolidine (100 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 130 mL), methanol (3 x 130 mL), and again with N-methylpyrolidine (3 x 130
mL) to give
compound 110.
[0528] Reaction 5: Preparation of Resin-Orn(NHBoc)-Sar-Thr NHFmoc (111)
[0529] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-threonine (2.9
g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 2.7
g), HOBt (1.13 g)
and diisopropylethylamine (DIPEA, 2.9 mL) as a solution in N-methylpyrrolidone
(100 mL) was
added to compound 110 (11 g), Tlie mixture was shaken for 60 minutes, filtered
through a glass
sinter funnel and a few beads were tested for the presence of a free amine
using the standard
Kaiser test (vide supra). The Kaiser test gave a yellow color so the coupling
was deemed
complete. After filtration through a glass sinter fumiel the product bearing
resin was washed
with N-methylpyrolidine (3 x 115 mL), methanol (3 x 115 mL), and again with N-
methylpyrolidine (3 x 115 mL) to give compound 111.
[0530] Reaction 6: Preparation of Resin-Orn(NHBoc)-Sar-Thr-NH2 112
[0531] Compound 111 was agitated in 20% piperidine in N-methylpyrolidine (110
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter funnel,
re-suspended in 20%
piperidine in N-methylpyrolidine (110 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 110 mL), methanol (3 x 110 mL), and again with N-methylpyrolidine (3 x 110
mL) to give
compound 112.
304
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0532] Reaction 7 Preparation of Resin-Orn(NHBoc)-Sar-Thr-As (p OtBu)-NHFmoc
(113)
[0533] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-aspartic acid
P-tert-
butyl ester, TBTU (2.7 g), HOBt (1.13 g) as a solution in N-methylpyrrolidone
(100 mL) was
added to compound 112 (11 g) followed by addition of diisopropylethylamine
(DIPEA, 2.9 mL).
The mixture was shaken for 60 minutes, filtered (through a glass sinter funnel
and a few beads
were tested for the presence of a free amine using the standard Kaiser test
(vide supra). The
Kaiser test gave a yellow color so the coupling was deemed complete. After
filtration v the
product bearing resin was washed with N-methylpyrolidine (3 x 115 mL),
methanol (3 x 115
mL), and again with N-methylpyrolidine (3 x 115 mL) to give 113.
[0534] Reaction 8: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-NH, (114)
[0535] Compound 113.was agitated in 20% piperidine in N-methylpyrolidine (115
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter fiuuiel,
re-suspended in 20%
piperidine in N-methylpyrolidine (115 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 120 mL), methanol (3 x 120 mL), and again with N-methylpyrolidine (3 x 120
mL) to give
compound 114.
[0536] Reaction 9: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-DAsn
HTrt)-
NHFmoc (115) '
[0537] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-asparagine
(5.0 g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 2.7
g), HOBt (1.13 g)
and diisopropylethylamine (DIPEA, 2.9 mL) as a solution in NMP (120 mL) was
added to
compound 114 (12 g). The mixture was shaken for 60 minutes, filtered through a
glass sinter
funnel and a few beads were tested for the presence of a free amine using the
standard Kaiser test
(vide supra). The Kaiser test gave a yellow color so the coupling was deemed
complete. After
filtration through a glass sinter funnel the product bearing resin was washed
with N-
methylpyrolidine (3 x 125 mL), methanol (3 x 125 mL), and again with N-
methylpyrolidine (3 x
125 mL) to give compound 115.
[0538] Reaction 10: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp OtBu -
DAsn(NHTrt)=
NH, 116
[0539] Compound 115 was agitated in 20% piperidine in N-methylpyrolidine (130
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter funnel,
re-suspended in 20%
piperidine in N-methylpyrolidine (130 mL) and agitated for 30 minutes. The
reaction mixture
305
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
was filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3
x 130 mL), methanol (3 x 130 mL), and again with N-methylpyrolidine (3 x 130
mL) to give
compound 116.
[0540] Reaction 11: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-
DAsn(NHTrt)-
Trp-NHFmoc (117)
[0541] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(3.57 g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 2.7
g), HOBt (1.13 g)
and diisopropylethylamine (DIPEA, 2.9 mL) as a solution in N-methylpyrrolidone
(130 mL) was
added to compound 116 (13 g). The mixture was shaken for 60 minutes, filtered
through a glass
sinter funnel and a few beads were tested for the presence of a free amine
using the standard
Kaiser test (vide supra). The Kaiser test gave a yellow color so the coupling
was deemed
complete. After filtration through a glass sinter funnel the product bearing
resin was washed
with N-methylpyrolidine (3 x 135 mL), methanol (3 x 135 mL), and again with N-
methylpyrolidine (3 x 135 mL ) to give compound 117.
[0542] Reaction 12: Preparation of Resin-Orn(NHBoc)-Sar-Thr-AsR(OtBu)-
DAsn(NHTrt)-
Trp-NHa (118)
[0543] Compound 117 was agitated in 20% piperidine in N-methylpyrolidine (130
mL) for
30 minutes. The reaction mixture was filtered through a glass sinter funnel,
re-suspended in 20%
piperidine in N-methylpyrolidine (130 mL) and agitated for 30 minutes. The
reaction mixture
was filtered through a glass sinter fiuinel then the solid was washed with N-
methylpyrolidine (3
x 130 mL), methanol (3 x 130 mL), and again with N-methylpyrolidine (3 x 130
mL) to give
compound 118.
[0544] Reaction 13: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-
DAsn(NHTrtZ
Trp-8-Methyldecanoic amide (105)
[0545] Commercially available 8-methyldecanoic acid (1.56 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 2.7 g),
diisopropylethylamine (DIPEA, 2.9
mL), and 1-hydroxy-benzotriazole (1.25 g) as a solution in N-methylpyrolidine
(120 mL) was
added to compound 118 (13.8 g). The mixture was shaken for 18 hours, filtered
through a glass
sinter funnel, and the reaction was judged to be complete using the Kaiser
Test (vide supra). The
reaction mixture was filtered through a glass sinter funnel then the solid was
washed with N-
methylpyrolidine (3 x 120 mL), methanol (3 x 120 mL), and again with N-
methylpyrolidine (3 x
120 mL) to give compound 105.
306
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0546] Example 1-25: Synthesis of Peptide Resin C'ompound 119
Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-DG1u(OtBu)-Trp-8-Methyldecanoic amide (119)
[0547] Reaction 1: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-DGlu
OtBu)-
NHFmoc. (120)
[0548] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-D-glutamic acid
y-t-butyl
ester (2.29 g), TBTU (1.73 g), HOBt (0.73 g) and DIPEA (1.9 mL) as a solution
in NMP (25
mL) were added to compound 114 (3.3 g). The mixture was shaken for three
hours. The
reaction mixture was filtered through a glass sinter funnel then the solid was
washed with N-
methylpyrolidine (3 x 25 mL), methanol (3 x 25 mL), and again with N-
methylpyrolidine (3 x 25
mL) to give compound 120.
[0549] Reaction 2: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-
DG1u(OtBu)-NH,
(121)
[0550] Compound 120 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (25 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 30
mL), methanol (3 x 30 mL), and again with N-methylpyrolidine (3 x 30 mL) to
give compound
121.
[0551] Reaction 3: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-DGlu
(OtBu)-Trp-
NHFmoc (122)
[0552] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-tryptophan
(2.30 g), 2-
(1H-Benzotriazol-yl)-1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 1.7
g), HOBt (0.73 g)
and diisopropylethylamine (DIPEA, 1.9 mL) as a solution in N-methylpyrrolidone
(25 mL) was
added to compound 121 (3.5 g). The mixture was shaken for 60 minutes, filtered
through a glass
sinter funnel and a few beads were tested for the presence of a free amine
using the standard
Kaiser test (vide supra). The Kaiser test gave a yellow color so the coupling
was deemed
complete. After filtration through a glass sinter funnel the product bearing
resin was washed
with N-methylpyrolidine (3 x 25 mL), methanol (3 x 25 mL), and again with N-
methylpyrolidine
(3 x 25 mL ) to give compound 122.
307
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0553] Reaction 4: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-
DG1u(OtBu)-Trp-
NH 123
[0554] Compound 122 was agitated in 20% piperidine in N-methylpyrolidine (25
mL) for 30
minutes. The reaction mixture was filtered through a glass sinter funnel, re-
suspended in 20%
piperidine in N-methylpyrolidine (25 mL) and agitated for 30 minutes. The
reaction mixture was
filtered through a glass sinter funnel then the solid was washed with N-
methylpyrolidine (3 x 30
mL), methanol (3 x 30 mL), and again with N-methylpyrolidine (3 x 30 mL) to
give compound
123.
[0555] Reaction 5: Preparation of Resin-Orn(NHBoc)-Sar-Thr-Asp(OtBu)-
DGIu(OtBu)-Tip-
8-Methyldecanoic amide (119)
[0556] Commercially available 8-methyldecanoic acid (0.50 g), 2-(1H-
Benzotriazol-yl)-
1,1,3,3-tetramethyluronium tetrafluroborate (TBTU, 0.86 g),
diisopropylethylamine (DIPEA,
0.94 mL), and 1-hydroxy-benzotriazole (0.35 g) as a solution in N-
methylpyrolidine (30 mL)
was added to compound 123 (3.8 g). The mixture was shaken for 18 hours,
filtered through a
glass sinter funnel, and the reaction was judged to be complete using the
Kaiser Test (vide
supra). The reaction mixture was filtered through a glass sinter funnel then
the solid was washed
with N-methylpyrolidine (3 x 36 mL), methanol (3 x 36 mL), and again with N-
methylpyrolidine
(3 x 36 mL) to give compound 119.
[0557] Exanaple 1-26: Esterification and Cleavage of Peptide Resin Compound 76
Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-8-Methyldecanoic amide (126)
[0555] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine
(1.7 g), 4-
dimethylaminopyridine (117 mg), and N-methyl-2-chloropyridinium iodide (1.23
g) were
flushed well with argon then suspended in dichloromethane (20 mL).
Triethylamine (76 L) was
added and the reaction mixture was stirred to give a homogeneous solution.
Compound 76 (2.0
g) was added to the solution. The flask was flushed again with argon and then
shaken for 14
hours. The resulting resin was then filtered through a glass sinter funnel and
washed well with
dichloromethane. The solid was suspended in dichloromethane (21 mL), 2,2,2-
trifluoroethanol
(7 mL), and acetic acid (7 mL), and shaken for 3 hours. The resin was filtered
through a glass
sinter funnel and evaporation of the filtrate gave the desired peptide 126
(285 mg) as a white
solid.
308
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0559] Example 1-27: Esterification and Cleavage ofPeptide Resin Compound 77
Preparation of Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-tridecanoic amide
(127)
[0560] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine
(1.48 g), 4-
dimethylaminopyridine (102 mg), and N-methyl-2-chloropyridinium iodide (1.07
g) were
flushed well with argon then suspended in dichloromethane (20 mL).
Triethylamine (1.17 mL)
was added and the reaction mixture was stirred to give a homogeneous solution.
Compound 77
(1.75 g) was added to the solution. The flask was flushed again with argon and
then shaken for
14 hours. The resulting resin was then filtered through a glass sinter funnel
and washed well
with dichloromethane. The solid was suspended in dichloromethane (15 mL),
2,2,2-
trifluoroethanol (5 mL), and acetic acid (5 mL), and shaken for 4 hours. The
resin was filtered
through a glass sinter funnel and evaporation of the filtrate gave the desired
peptide 127 (490
mg) as a white solid.
[0561] Example 1-28: Esterification and Cleavage ofPeptide Resin Conzpound 78
Preparation of Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DG1u(OtBu)-Trp-8-Methyldecanoic
amide (128)
[0562] To compound 78 (5.9 g) was added a solution of commercially available
Na-(9-
Fluorenylmethoxycarbonyl)-L-isoleucine (4.9 g), bromo-tris-
pyrrolidinophosphonium
hexafluorophosphate (PyBrOP, 6.5 g), and di-isopropylethylamine (7.3 mL), in
dichloromethane
(60 mL). Dimethylaminopyridine (25 mg) was added and the mixture was shaken
for 2 hours.
After 2 h, the mixture was filtered through a glass sinter funnel and washed
with
dichloromethane (3 x 60 mL) and the coupling procedure was repeated. The
resulting resin was
filtered through a glass sinter funnel, washed with dichloromethane (3 x 60
mL) and methanol (3
x 60 mL), and dried under diminished pressure over potassium hydroxide
pellets. This dried
resin was suspended in dichloromethane (27 mL), 2,2,2-trifluoroethanol (9 mL),
and acetic acid
(9 mL), and shaken for 3 hours. The resin was filtered through a glass sinter
funnel and
evaporation of the filtrate gave the desired peptide 128 (2.1 g) as a white
solid.
309
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0563] Example 1-29: Esterification and Cleavage ofPeptide Resin Compound 83
Preparation of Ala-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(ONHTrt)-Trp-S-
Methyldecanoic amide (129)
[0564] To compound 83 (3.3 g) was added a solution of commercially available
Na-(9-
Fluorenylmethoxycarbonyl)-L-isoleucine (3.2 g), bromo-tris-
pyrrolidinophosphonium
hexafluorophosphate (PyBrOP, 4.2 g), and Di-isopropylethylamine (4.7 mL), in
dichloromethane
(60 mL). Dimethylaminopyridine (23 mg), was added and the mixture was shaken
for 2 hours.
After 2 h, the mixture was filtered through a glass sinter funnel and washed
with
dichloromethane (3 x 30 mL) and the coupling procedure was repeated. The
resulting resin was
filtered through a glass sinter funnel, washed with dichloromethane (3 x 30
mL) and methanol (3
x 30 mL), and dried under diminished pressure over potassium hydroxide
pellets. This dried
resin was suspended in dichloromethane (24 mL), 2,2,2-trifluoroethanol (6 mL),
and acetic acid
(6 mL), and shaken for 3 hours. The resin was filtered through a glass sinter
funnel and
evaporation of the filtrate gave the desired peptide 129 (2.9 g) as a white
solid.
[0565] Example 1-30: Esterification and Cleavage ofPeptide Resin Compound 75
Preparation of Ala-Sar-Thr(OIleNI3Alloc)-Asp(OtBu)-DAsn(NHTrt)-Trp-Undecanoic
amide (130)
[0566] Na-(Allyloxycarbonyl)-L-isoleucine 124 (1.34 g, vide infra), 4-
dimethylaminopyridine (15 mg), and N-methyl-2-chloropyridinium iodide (1.59 g)
were flushed
well with argon then suspended in dichloromethane (30 mL). Triethylamine (1.74
mL) was
added and the reaction mixture was stirred to give a homogeneous solution.
Compound 75 (1.25
g) was added to the solution. The flask was flushed again with argon and then
shaken for 14
hours. The resulting resin was then filtered through a glass sinter funnel and
washed well with
dichloromethane. The solid was suspended in dichloromethane (12 mL), 2,2,2-
trifluoroethanol
(4 mL), and acetic acid (4 mL), and shaken for 3 hours. The resin was filtered
through a glass
sinter funnel and evaporation of the filtrate gave the desired peptide 130
(154 mg) as a white
solid.
310
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0567] Example 1-31: Estef ifacation and Cleavage of Peptide Resin Compound 84
Preparation of Ala-S ar-Thr(OIIeNHFmoc)-Asp(OtBu)-DGIu(OtBu)-Trp-8-
Methyldecanoic
amide (131)
[0568] To compound 84 (4.8 g) was added a solution of commercially available
Na-(9-
fluorenylmethoxycarbonyl)-L-isoleucine (3.2 g), bromo-tris-
pyrrolidinophosphonium
hexafluorophosphate (PyBrOP, 4.2 g), and Di-isopropylethylamine (4.7 mL), in
dichloromethane
(60 mL). Dimethylaminopyridine (23 mg) was added and the mixture was shaken
for 2 hours.
After 2 h, the mixture was filtered through a glass sinter funnel and washed
with
dichloromethane (3 x 30 mL) and the coupling procedure was repeated. The
resulting resin was
filtered through a glass sinter funnel, washed with dichloromethane (3 x 30
mL) and methanol (3
x 30 mL), and dried under diminished pressure over potassium hydroxide
pellets. This dried
resin was suspended in dichloromethane (24 mL), 2,2,2-trifluoroethanol (6 mL),
and acetic acid
(6 mL), and shaken for 3 hours. The resin was filtered through a glass sinter
funnel and
evaporation of the filtrate gave the desired peptide 131 (2.92 g) as a white
solid.
[0569] Example 1-32: Esterification and Cleavage ofPeptide Resin Compound 89
Preparation of A1a-Gly-Thr(OIIeNHAlloc)-Asp(OtBu)-DAsn(NHTrt)-Trp-Undecanoic
amide (132)
[0570] Na-(Allyloxycarbonyl)-L-isoleucine 124 (1.34 g, vide infra), 4-
dimethylaminopyridine (15 mg), and N-methyl-2-chloropyridinium iodide (1.59 g)
were flushed
well with argon then suspended in dichloromethane (30 mL). Triethylamine (1.74
mL) was
added and the reaction mixture was stirred to give a homogeneous solution.
Compound 89 (1.25
g) was added to the solution. The flask was flushed again with argon and then
shaken for 14
hours. The resulting resin was then filtered through a glass sinter funnel and
washed well with
dichloromethane. The solid was suspended in dichloromethane (12 mL), 2,2,2-
trifluoroethanol
(4 mL), and acetic acid (4 mL), and shaken for 3 hours. The resin was filtered
through a glass
sinter funnel and evaporation of the filtrate gave the desired peptide 132
(147 mg) as a white
solid.
311
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0571] Example 1-33: Esterification and Cleavage of Peptide Resin Compound 100
Preparation of Ala-Gly-Thr(OIIeNHAlloc)-Asp(OtBu)-DGIu(OtBu)-Trp-Undecanoic
amide (133)
[0572] Na-(Allyloxycarbonyl)-L-isoleucine 124 (1.34 g, vide infi a), 4-
dimethylaminopyridine (15 mg), and N-methyl-2-chloropyridinium iodide (1.59 g)
were flushed
well with argon then suspended in dichloromethane (30 mL). Triethylamine (1.74
mL) was
added and the reaction mixture was stirred to give a homogeneous solution.
Compound 100
(1.25 g) was added to the solution. The flask was flushed again with argon and
then shaken for
14 hours. The resulting resin was then filtered through a glass sinter funnel
and washed well
with dichloromethane. The solid was suspended in dichloromethane (12 mL),
2,2,2-
trifluoroethanol (4 mL), and acetic acid (4 mL), and shaken for 3 hours. The
resin was filtered
through a glass sinter funnel and evaporation of the filtrate gave the desired
peptide 133 (95 mg)
as a white solid.
[0573] Example 1-34: EsteYification and Cleavage of Peptide Resin Conapound
105
Preparation of Orn(NHBoc)-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-8-
Methyldecanoic amide (134)
[0574] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine
(2.0 g), 4-
dimethylaminopyridine (140 mg), and N-methyl-2-chloropyridinium iodide (1.46
g) were
flushed well with argon then suspended in dichloromethane (20 mL).
Triethylamine (1.6 mL)
was added and the reaction mixture was stirred to give a homogeneous solution.
Compound 105
(2.0 g) was added to the solution. The flask was flushed again with argon and
then shaken for 14
hours. The resulting resin was then filtered through a glass sinter funnel and
washed well with
dichloromethane. The solid was suspended in dichloromethane (21 mL), 2,2,2-
trifluoroethanol
(7 mL), and acetic acid (7 mL), and shaken for 3 hours. The resin was filtered
through a glass
sinter fu.nnel and evaporation of the filtrate gave the desired peptide 134
(890 mg) as a white
solid.
312
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0575] Example 1-35: Esterification and Cleavage of Peptide Resin Compound 119
Preparation of Orn(NHBoc)-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DG1u(OtBu)-Trp-8-
Methyldecanoic amide (135)
[0576] Commercially available Na-(9-Fluorenylmethoxycarbonyl)-L-isoleucine
(2.0 g), 4-
dimethylaminopyridine (140 mg), and N-methyl-2-chloropyridinium iodide (1.46
g) were
flushed well with argon then suspended in dichloromethane (20 mL).
Triethylamine (1.6 mL)
was added and the reaction mixture was stirred to give a homogeneous solution.
Compound 119
(1.75 g) was added to the solution. The flask was flushed again with argon and
then shaken for
14 hours. The resulting resin was then filtered through a glass sinter fu.nnel
and washed well
with dichloromethane. The solid was suspended in dichloromethane (21 mL),
2,2,2-
trifluoroethanol (7 mL), and acetic acid (7 mL), and shaken for 3 hours. The
resin was filtered
through a glass sinter funnel and evaporation of the filtrate gave the desired
peptide 135 (761
mg) as a white solid.
[0577] Example 1-36. Preparation of Compound C16
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Orn-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C16)
[0578] Reaction 1: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-Gly-Asp(OtBu)-
DA1a-
Asp(OtBu)-Orn(NHBoc)-Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn HTrt)-Trp-B-
Methyldecanoic amide (137)
[0579] Hydroxy-benzotriazole (17 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 55 mg), and diisopropylethylamine (22
L), were
added to a solution of compound 126 (174 mg) in dimethylformamide (3 mL), then
compound 9
(300 mg) was added and the mixture then shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding resin bound compound 137.
313
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0580] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Orn(NHBoc)-Gly-Thr- i
sp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(138)
[0581] Compound 137 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) and dried under reduced
pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL), and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
furmel, and washed
well with N-methylmorpholine to give compound 138.
[0582] Reaction 3: Preparation of compound (C16)
[0583] Dried compound 138 was suspended in dichloromethane, (2.5 mL)
trifluoroacetic
acid, (2.5 mL) ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give the pure product C16 (3.7 mg).
314
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0584] Example 1-3 7: Preparation of Conapound C76
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Orn-Gly-Thr-Asp-Dglu-Trp-8-Methyldecanoic amide
(C76)
[0585] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer OtBu)-Gly-Asp(OtBu -
DA1a-
Asp(OtBu)-Orn(NHBoc)-Gly-Thr(OIleNHFmoc)-A~p(OtBu -DGlu(OtBu)-_T rp-8-
Methddecanoic amide (140)
[0586] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 128 (174 mg) in dimethylformamide (3 mL).
Compound 9
(300 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding resin bound compound 140.
[0587] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Orn(NHBoc)-Gly-Thr-Asp(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu)
(141)
[0588] Compound 140 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hours then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL), and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL) then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157
gL) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 141.
315
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0589] Reaction 3: Preparation of compound (C76)
[0590] Dried compound 141 was suspended in dichloromethane, (2.5 mL)
trifluoroacetic
acid, (2.5 mL) ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter colunm 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give the pure product C76 (9.0 mg).
[0591] Example 1-38: Preparation of Con2pound C75
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Orn-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C75)
[0592] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer OtBu)-Gly-Asp(OtBu -
DAla-
Asp(OtBu)-Orn(NHBoc)-Sar-Thr(OIIeNHFmoc -Asp(OtBu -DAsn(NHTrt)-Trp-8-
Methyldecanoic amide (143)
[0593] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 134 (243 mg) in dimethylformamide (3 mL).
Compound 21
(322 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 143.
316
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0594] Reaction 2: Preparation of
Ile
Resin-Glu-D Ser(OtBu)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Orn(NHBoc)-S ar-Thr-
Asp(OtBu)-
I
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(144)
[0595] Compound 143 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel). The solid was washed with dimethylformamide (10 mL) and
dichioromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 144.
[0596] Reaction 3: Preparation of com op und (C75)
[0597] Dried compound 144 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
fu.nnel, washed with dichloromethane, and the combined filtrates were
evaporated under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
forinic acid : 20 % water 0.5% formic acid over 25 minutes. The product
bearing fractions were
combined and freeze-dried to give compound C75 (8.1 mg).
317
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0598] Example 1-39 Preparation of Compound C74
Ile
Glu-DSer-Giy-Asp-DAla-Asp-Ala-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C74)
[0599] Reaction 1: Preparation of Resin-Glu(aOA11y1)-DSer(OtBu)-Gly-Asp(OtBu)-
DAla-
Asp(OtBu -Ala-Gly_Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn HTrt)-Trp-8-Methyldecanoic
amide
(146)
[0600] Hydroxy-benzotriazole (27 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 88.5 mg), and diisopropylethylamine
(100 L), were
added to a solution of compound 126 (278 mg) in dimethylformamide (3 mL).
Compound 38
(227 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60 mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced presure,
yielding compound 146.
[0601] Reaction 2: Preparation of compound (147)
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAIa-Asp(OtBu) Ala-Gly-Thr-Asp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(147)
[0602] Compound 146 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichioromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
318
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 147.
[0603] Reaction 3 Preparation of compound (C74)
[0604] Dried compound 147 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 gM Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C74 (3.9 mg).
[0605] Example 1-40: Preparation of
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Ala-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic ainide
(C86)
[0606] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer OtBu)-Gl3L-Asp(OtBu)-
DAIa-
Asp(OtBu)-Ala-Sar-Thr(OIleNHFmoc)-Asp(OtBu)-DAsn(NHTrt )-Trp-8-Methyldecanoic
amide
(149)
[0607] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 129 (228 mg) in dimethylformamide (3 mL).
Compound 21
(280 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test(vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 149.
319
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0608] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Ala-Sar-Thr-A~ (OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(150)
[0609] Compound 149 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL),
dichloromethane (10
mL) and dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3 mL),
then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157 L)
were added.
The reaction was shaken for 7 hours, filtered through a glass sinter funnel,
and washed well with
N-methylmorpholine to give compound 150.
[0610] Reaction 3: Preparation of (C86) -
[0611] Dried compound 150 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C86 (2.8 mg).
320
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0612] Example 1-41: Preparation of Compound C79
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Orn-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C79)
[0613] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-
DA1a-
Asp(OtBu)-Orn(NHBoc)-Sar-Thr OIIeNHFmoc)-Asp(OtBu)-DG1u(OtBu)-Trp-8-
Methyldecanoic amide (152)
[0614] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (52
L), were
added to a solution of compound 135 (217 mg) in dimethylformamide (3 mL).
Coinpound 21
(278 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding resin bound compound 152.
[0615] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-AI
p(OtBu)
8-Methyldecanoic axnide-Trp-DG1u(OtBu)
(153)
[0616] Compound 151 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF:piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL) then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157
gL) were
added. The reaction was shaken for 7 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 153.
[0617] Reaction 3: Preparation of compound (C79)
[0618] Dried compound 153 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
321
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (Cl 8 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C79 (1.5 mg).
[0619] Example 1-42: Preparation of Compound C81
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Ala-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C81)
[0620] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-
DAIa-
Asp(OtBu)-Ala-Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DG1u(OtBu)-Trp-8-Methyldecanoic
amide
155
[0621] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 128 (183 mg) in dimethylformamide (3 mL).
Compound 38
(227 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried reduced pressure, yielding compound 155.
[0622] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DA1a-Asp(OtBu)-Ala-Gly-Thr-ASp(OtBu)
8-Methyldecanoic amide-Trp-D Glu(OtBu)
(156)
[0623] Compound 155 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
322
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
1;
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropvlethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter fumlel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 156.
[0624] Reaction 3: Preparation of (C81)
[0625] Dried compound 156 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 'riminutes. The product
bearing fractions were
combined and freeze-dried to give compound C81 (2.3 mg).
[0626] Example 1-43: Preparation of Compound C80
Ile
Glu-DSer-Gly-Asp-DAla-Asp-Ala-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C80)
[0627] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-As (p
OtBu)-DAIa-
Asp(OtBu)-Ala-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DGlu(OtBu)-Trp-8-Methyldecanoic
amide
(158)
[0628] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 131 (196 mg) in dimethylformamide (3 mL).
Compound 21
(278 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra) The
323
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
resin was filtered through a glass sinter funnel, washed with dicbloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 158.
[0629] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Ala-Sar-Thr-AI p(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu)
159
[0630] The resin 158 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 gL) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 159.
[0631] Reaction 3: Preparation of compound (C80)
[0632] Dried compound 159 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 gL), and triisopropylsilane(125 gL), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C80 (6.2 mg).
324
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0633] Example 1-44: Preparation of Compound C72
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Orn-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C72)
[0634] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn-Gly-AsR(OtBu)-DA1a-
Asp(OtBu -Orn(NHBoc)-Gly-Thr(OIleNHFmoc)-Asp(OtBu)-DAsn HTrt)-Trp-8-
Methyldecanoic amide (161)
[0635] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 126 (274 mg) in dimethylformamide (3 mL).
Compound 50
(303 mg) was added and the mixture then shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 161.
[0636] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Orn(NHBoc)-Gly-Thr-
Alsp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(162)
[0637] Compound 161 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL),
dichloromethane (10
mL) and dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3 mL)
and 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157 L)
were added.
The reaction was shaken for 17 hours, filtered through a glass sinter furmel,
and washed well
with N-methylmorpholine to give compound 162.
325
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[06381 Reaction 3: Preparation of (C72)
[0639] Dried compound 162 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid"to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C72 (2.9 mg).
[0640] Example 1-45: Preparation of C352
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Orn-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C352)
[0641] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-A~p(OtBu)-
DA1a-
-Thr(OIleNHFmoc)-A~p(OtBu -DGIu(OtBu)-Trp-8-
Asp(OtBu -Orn(NHBoc~y
Methyldecanoic amide (164)
[0642] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 127 (183 mg) in dimethylformamide (2 mL).
Compound 50
(265 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 164.
326
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0643] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Orn(NHBoc)-Gly-Thr-
A,Sp(OtBu)
8-Methyldecanoic amide-Trp-D IGlu(OtBu)
(165)
[0644] Compound 164 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 165.
[0645] Reaction 3: Preparation of compound (C352)
[0646] Dried compound 165 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichioromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C352 (4.7 mg).
327
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0647] Example 1-46: Preparation of Compound C85
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Orn-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C85)
[0648] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn Trt)-Gly-AsR(OtBu -
DAIa-
Asp(OtBu)-Orn(Boc -Sar-Thr OIIeNHFmoc)-Asp(OtBu -DAsn HTrt )-Trp-8-
Methyldecanoic
amide (167)
[0649] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (30
L), were
added to a solution of compound 134 (248 mg) in dimethylformamide (3 mL).
Compound 40
(238 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test(vide
supra). The
resin was filtered through a glass sinter fitnnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 167.
[0650] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DA1a-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-AI
p(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(168)
[0651] Compound 167 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0).(340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), 'and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL) then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157
L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 168.
328
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0652] Reaction 3: Preparation of compound (C85)
[0653] Dried compound 168 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C85 (3.7 mg).
[0654] Example I-47: Preparation of Compound C353
Ile
G1u-DAsn-Gly-Asp-DAla-Asp-Ala-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C353)
[06551 Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn HTrt)-Gly-Asp(OtBu -
DAla-
Asp(OtBu)-Ala-Gly-Thr(OIleNHFmoc)-AsR(OtBu)-DAsn(NHTrt)-_Trp-8-Methyldecanoic
amide
(170)
[0656] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (52
L), were
added to a solution of compound 126 (209 mg) in dimethylformamide (2 mL).
Compound 52
(340 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 gL), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 170.
329
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0657] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Ala-Gly-Thr-Ai p(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(171)
[0658] The resin 170 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel. The solid was washed with
0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 then filtered
through a glass sinter
funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL)
then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3 mL)
then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157 L)
were added.
The reaction was shaken for 17 hours, filtered through a glass sinter funnel,
and washed well
with N-methylmorpholine to give compound 171.
[0659] Reaction 3: Preparation of compound (C353)
[0660] Dried compound 171 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 tCL), and triisopropylsilane(125 L), and
the reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C353 (6.8 mg).
330
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0661] Example 1-48: Preparation of Compound C82
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Ala-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C82)
[0662] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn HTrt)-Gly-Asp(OtBu -
DAIa-
Asp(OtBu)-Ala-Sar-Thr(OIIeNHFmoc)-Asp(OtBu -DAsn(NHTrt)-Trp-8-Methyldecanoic
amide
(173)
[0663] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 129 (221 mg) in dimethylformamide (3 mL).
Compound 40
(238 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) then
dried under reduced pressure, yielding compound 173.
[0664] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAIa-Asp(OtBu)-Ala-S ar-Thr-Aip(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(174)
[0665] Compound 173 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered, through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 174.
331
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0666] Reaction 3: Preparation (C82)
[0667] Dried compound 174 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 gL), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
fu.nnel washed with dichloromethane, and the combined filtrates were
evaporated under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
coinbined and freeze-dried to give compound C82 (3.8 mg).
[0668] Example 1-49: Preparation of Compound C83
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Orn-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C83)
[0669] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu -
DAla-
Asp(OtBu)-Orn(NHBoc -Sar-Thr OIleNHFmoc)-&p(OtBu)-DGlu(OtBu)-Trp-8-
Methyldecanoic amide (176)
[0670] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 135 (221 mg) in dimethylformamide (3 mL).
Compound 40
(238 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test(vide
supra. The resin
was filtered, through a glass sinter funnel, washed with dichloromethane (3 x
3 mL) and dried
under reduced pressure, yielding compound 176.
332
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0671] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DA1a-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-A i
p(OtBu)
8-Methyldecanoic amide-Trp-DGIu(OtBu)
(177)
[0672] Compound 176 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 ml) for 4 hrs then filtered
through a glass
sinter fitmiel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 177.
[0673] Reaction 3: Preparation of Compound (C83)
[0674] Dried compound 177 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 gL), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 gM Jupiter colunm 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C83 (4.3 mg).
333
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0675] Example 1-50: Preparation of Compound C84
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Ala-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C84)
[0676] Reaction 1: Pre,paration of Resin-Glu(aOAlly1 -DAsn Trt ) -Gly-
Asp(OtBu)-
DAla-Asp(OtBu)-Ala-Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DGlu(OtBu)-Trp-8-
Methyldecanoic
amide (179)
[0677] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 128 (183 mg) in dimethylformamide (3 mL).
Compound 52
(212 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter fiuulel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 179.
[0678] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Ala-Gly-Thr-AI p(OtBu)
8-Methyldecanoic amide-Trp-D Glu(OtBu)
(180)
[0679] Compound 179 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methyhnorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylainine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 180.
[0680] Reaction 3: Preparation of compound (C84)
[0681] Dried compound 180 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
334
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C84 (6.6 mg).
[0682] Example 1-51: Preparation of Compound C354
Ile
Glu-DAsn-Gly-Asp-DAla-Asp-Ala-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C354)
[0683] Reaction 1: Preparation of Resin-Glu(aOAlly1)-DAsn(NHTrt )-Gly-Asp(OtBu
-DAIa-
Asp(OtBu)-Ala-Sar-Thr(OIleNHFmoc)-Asp(OtBu -DGlu(OtBu)-Trp-8-Methyldecanoic
amide
(182)
[0684] Hydroxy-benzotriazole (20 mg); benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 131 (196 mg) in dimethylformamide (2 mL).
Compound 40
(238mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 gL), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 182.
335
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0685] Reaction 2: Preparation of
Ile
Resin-Giu-DAsn(NHTrt)-Gly-Asp(OtBu)-DAla-Asp(OtBu)-Ala-Sar-Thr-A i p(OtBu)
8-Methyldecanoic amide-Trp-DG1u(OtBu)
(183)
[0686] Compound 182 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(O) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hrs then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL) then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide (157
L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
fu.nnel, and washed
well with N-methylmorpholine to give compound 183.
[0687] Reaction 3: Preparation of compound (C354)
[0688] Dried compound 183 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C354 (4.7 mg).
336
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0689] Example 1-52: Preparation of Compound C73
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Orn-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide :
(C73)
[0690] Reaction 1: Preparation of Resin-GIu(aOAllyl)-DSer OtBu)-Gly-Asp(OtBu)-
DLys(NHBoc)-AsR(OtBu -Orn(NHBoc)-Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-
8-Methyldecanoic amide (185)
[0691] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 126 (298 mg) in dimethylformamide (3 mL).
Compound 54
(312 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter fiumel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 185.
[0692] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Gly-Thr-AI
p(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(186)
[0693] Compound 185 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(O) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
fiuulel, and washed
well with N-methylmorpholine to give compound 186.
337
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0694] Reaction 3: Preparation of compound (C73)
[0695] Dried compound 186 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 gL), and triisopropylsilane(125 pL), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C73 (24.6 mg).
[0696] Example 1-53: Preparation of Compound C355
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Orn-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C355)
[0697] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer OtBu)-Gly-Asp(OtBu-
DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-GlY-Tbr(OIIeNHFmoc)-As-D(OtBu)-DGlu(OtBu)u-
Tr1a-
8-Methyldecanoic amide (188)
[0698] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
~tL), were
added to a solution of compound 128 (183 mg) in dimethylformamide (2 mL).
Compound 54
(322.6mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel), washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 188.
338
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0699] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Gly-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-JGlu(OtBu)
(189)
[0700] Compound 188 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformainide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 ing) and 1,3-
diisopropylcarbodiimide (157 RL) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 189.
[0701] Reaction 3:Preparation of (C355)
[0702] Dried compound 189 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 l), and triisopropylsilane(125 l), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C355 (8.2 mg).
339
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0703] Example 1-54: Preparation of Compound C356
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Orn-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C356)
[0704] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-AsR(OtBu)-
DLys(NHBoc)-Asp(OtBu -Orn(NHBoc)-Sar-Thr(OIleNHFmoc-&p(OtBu)-DAsn(NHTrt)-Trp-
8-Methyldecanoic amide (191)
[0705] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
gL), were
added to a solution of compound 134 (243 mg) in dimethylformamide (2 mL).
Compound 34
(263 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 191.
[0706] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Sar-Thr- i
sp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(192)
[0707] Compound 191 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The inixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
tliiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL)and
dichloromethane (10 mL) then dried under reduced pressure. T he resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
340
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 192.
[0708] Reaction 3: Preparation of (C356)
[0709] Dried compound 192 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 1), and triisopropylsilane(125 g1), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C356 (8.7 mg).
[0710] Example 1-55: Preparation of Compound C 357
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Ala-Gly-Thr-Asp-DAsn-Trp-undecanoic amide (C357)
[0711] Reaction 1: Preparation of Resin-Glu(aOA11y1)-DSer(OtBu)-Gly-&P(OtBu~
DLYs(NHBoc)-Asp(OtBu)-Ala-Gly-Thr(OIIeNHAlloc)-Asp(OtBu)-DAsn HTrt)-Trp-
undecanoic amide (194)
[0712] Hydroxy-benzotriazole (14 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 44 mg), and diisopropylethylamine (20
L), were
added to a solution of compound 132 (147 mg) in dimethylformamide (3 mL).
Compound 34
(200 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 194.
341
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0713] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr-A i
p(OtBu)
Undecanoic amide-Trp-DAsn(NHTrt)
195
[0714] Compound 194 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 inL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was suspended in N-methylmorpholine (3 mL), then 1-hydroxy-
benzotriazole (135
mg) and 1,3-diisopropylcarbodiimide (157 L) were added. The reaction was
shaken for 17
hours, filtered through a glass sinter funnel, and washed well with N-
methylmorpholine to give
compound 195.
[0715] Reaction 3: Preparation of (C357)
[0716] Dried compound 195 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 gL), and triisopropylsilane(125 - L); and
the reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product (43 mg). The
crude product was
purified by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting
with a
gradient from 20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid
to 80%
acetonitrile 0.5% formic acid : 20 % water 0.5% formic acid over 25 minutes.
The product
bearing fractions were combined and freeze-dried to give compound C357 (4.5
mg).
342
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0717] Example 1-56: Preparation of Compound C358
Ile
G1u-DSer-Gly-Asp=DLys-Asp-Ala-Sar-Thr-Asp-DAsn-Trp-undecanoic amide (C358)
[0718] Reaction 1: Preparation of Resin-G1u(aOA1ly1 -DSer(OtBu)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp-Ala-Sar-Thr(OIIeNHAlloc)-Asp(OtBu)-DAsn HTrt)-Trp-undecanoic
amide (197)
[0719] Hydroxy-benzotriazole (14 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 44 mg), and diisopropylethylamine (20
L), were
added to a solution of compound 130 (154 mg) in dimethylformamide (3 mL).
Compound 34
(200 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloroinethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 197.
[0720] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr-A i
p(OtBu)
Undecanoic amide-Trp-DAsn(NHTrt)
(198)
[0721] Compound 197 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was suspended in N-methylmorpholine (3 mL), then 1-hydroxy-
benzotriazole (135
mg) and 1,3-diisopropylcarbodiimide (157 L) were added. The reaction was
shaken for 17
hours, filtered through a glass sinter funnel, and washed well with N-
methylmorpholine to give
compound 198.
343
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0722] Reaction 3: Preparation of com-pound (C358)
[0723] Dried compound 198 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
fiuinel, washed with dichloromethane, and the combined filtrates were
evaporated under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C 358 (2.7 mg).
[0724] Example 1-57: Preparation of Compound C359
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Orn-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C359)
[0725] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer OtBu)-Gly-Asp(OtBu-
DLys(NHBoc)-Asp(OtBu)=Orn(NHBoc)-Sar-Thr(OIIeNHFmoc~-Asp(OtBu)-DGlu(OtBu)-Trp-
8-Methyldecanoic amide (200).
[0726] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 135 (217 mg) in dimethylformamide (2 mL).
Compound 34
(263 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 200.
344
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0727] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-A
~p(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu)
(201)
[0728] Compound 200 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 201.
[0729] - Reaction 3: Preparation of (C359)
[0730] Dried compound 201 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C 18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C359 (4.7 mg).
345
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0731] Exan-aple 1-58: Preparation of Compound C360
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Ala-Gly-Thr-Asp-DGlu-Trp-undecanoic amide (C360)
[07321 Reaction 1: Preparation of Resin-Glu(aOAllyl -DSer(OtBu)-GIy-Asp(OtBu)_
DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr(OIIeNHAlloc)-Asp(OtBu)-DG1u(OtBu)-Trp-
undecanoic amide (203)
[0733] Hydroxy-benzotriazole (14 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 44 mg), and diisopropylethylamine (20
L), were
added to a solution of compound 133 (95 mg) in dimethylformamide (3 mL).
Compound 34
(200 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 203.
[0734] Reaction 2: Preparation of
Ile
Resin-Glu-D Ser(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr-
Asp(OtBu)
Undecanoic amide-Trp-DGlu(OtBu)
204
[0735] Compound 203 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was suspended in N-methylmorpholine (3 mL), then 1-hydroxy-
benzotriazole (135
mg) and 1,3-diisopropylcarbodiimide (157 L) were added. The reaction was
shaken for 17
hours, filtered through a glass sinter funnel, and washed well with N-
methylmorpholine to give
compound 204.
346
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0736] Reaction 3: Preparation of compound (C360)
[0737] Dried compound 204 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 gL), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (Cl 8 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C360 (3.4 mg).
[0738] Example 1-59: Preparation of Compound C361
Ile
Glu-DSer-Gly-Asp-DLys-Asp-Ala-Sar-Thr-Asp-DGlu-Trp-B-Methyldecanoic amide
(C361)
[0739] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DSer(OtBu)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DGIu(OtBu)-Trp-8-
Methyldecanoic amide (206)
[0740] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 131 (196 mg) in dimethylformamide (2 mL).
Compound 34
(270 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser
Test(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 206.
347
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0741] Reaction 2: Preparation of
Ile
Resin-Glu-DSer(OtBu)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr-
Alsp(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu)
1207
[0742] Compound 206 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then driedunder reduced
pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter fiinnel, and washed well with N-methylmorpholine to
give compound 207.
[0743] Reaction 3: Pre-paration of compound (C361)
[0744] Dried compound 207 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel wa.shed with dichloromethane, and the combined filtrates were
evaporated under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C361 (6.3 mg).
348
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0745] Example 1-60: Preparation of Compound C77
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Orn-Gly-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(C77)
[0746] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn Trt)-Gly-ASp(OtBu)-
DLys(NHBoc)-ASp(OtBu -Orn(NHBoc)-Gly-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-
8-Methyldecanoic amide (209)
[0747] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
gL), were
added to a solution of compound 126 (243 mg) in dimethylformamide (3 mL).
Compound 60
(217 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter fimnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 209.
[0748] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NIiBoc)-Gly-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(210)
[0749] Compound 209 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 210.
349
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0750] Reaction 3: Preparation of com op und (C77)
[0751] Dried compound 210 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C77 (3.8 mg).
[0752] Example 1-61: Preparation of Compound C362
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Orn-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C362)
[0753] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt ~-Gly-
Asp(OtBu)-
DLtis(NHBoc)-Asp(OtBu)-Orn HBoc)-Gly-Thr(OIleNHFmoc)-Asp(OtBu)-DGlu(OtBu)-Trp-
8-Methyldecanoic amide (212)
[0754] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 128 (183 mg) in dimethylformamide (2 mL).
Compound 60
(217 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced presure,
yielding compound 212.
350
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0755] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NBBoc)-Gly-Thr-
A'sp(OtBu)
8-Methyldecanoic amide-Trp-DGIu(OtBu)
(213)
[0756] Compound 212 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
furuiel, and washed
well with N-methylmorpholine to give compound 213.
[0757] Reaction 3: Preparation of compound (C362)
[0758] Dried compound 213 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 pL), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
fiulnel washed with dichloromethane, and the combined filtrates were
evaporated under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C1 8 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C362 (3.1 mg).
351
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0759] Example 1-62: Preparation of Compound C363
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Orn-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide
(363)
[0760] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn HTrt)-Gly-AsR(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Sar-Thr(OIleNHFmoc)-Asp(OtBu)-DAsn(NHTrt )-
Trp-
8-Methyldecanoic amide (215)
[0761] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 134 (342 mg) in dimethylformamide (2 mL).
Compound 56
(416 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 215.
[0762] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(216)
[0763] Compound 215 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) 4 hours then filtered
through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
352
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 216.
[0764] Reaction 3: Preparation of compound (C363)
[0765] Dried compound 216 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
finnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C363 (6.1 mg).
[0766] Example 1-63: Preparation of Compound C364
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Ala-Gly-Thr-Asp-DAsn-Trp-tridecanoic amide (C364)
[0767] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr(OIIeNHFmoc)-Asp(OtBu -DAsn(NHTrt)-Trp-
Tridecanoic amide (218)
[0768] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 127 (146 mg) in dimethylformamide (3 mL).
Compound 62
(171 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter fumiel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 218.
353
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0769] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr-Af
p(OtBu)
Tridecanoic amide-Trp-DAsn(NIHTrt)
219
[0770] Compound 218 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4: 1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 219.
[0771] Reaction 3: Preparation of compound (C364)
[0772] Dried compound 219 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C364 (4.8 mg).
354
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0773] Example 1-64: Preparation of Compound C365
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Ala-Sar-Thr-Asp-DAsn-Trp-8-Methyldecanoic amide (C36
5)
[0774] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DAsn(NHTrt)-Trp-8-
Methyldecanoic amide (221)
[0775] Hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 129 (195 mg) in dimethylformamide (2 mL).
Compound 56
(400 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide-supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 221.
[0776] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-DAsn(NHTrt)
(222)
[0777] Compound 221 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4: 1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
355
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 222.
[0778] Reaction 3: Preparation of compound (C365)
[0779] Dried compound 222 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (C18 10 gM Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C365 (10.2 mg).
[0780] Example 1-65: Preparation of Compound C366
Ile
Giu-DAsn-Gly-Asp-DLys-Asp-Orn-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C366)
[07811 Reaction 1: Preparation of Resin-Glu(aOA11yI -DAsn Trt)-Gly-Asp(OtBu)-
DLys(NHBoc -Asp(OtBu -Orn(NHBoc)-Sar-Thr(OIIeNHFmoc)-Asp(OtBu)-DGIu(OtBu)-Trp-
8-Methyldecanoic amide (224)
[0782] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 135 (217 mg) in dimethylformamide (2 mL).
Compound 56
(217 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 L), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 224.
356
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0783] Reaction 2: Preparation of
I1e
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Orn(NHBoc)-Sar-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-D8lu(OtBu)
gL5)
[0784] Compound 224 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4 :1 (10mL) for 4 hours then
filtered through a glass
sinter funnel. The solid was washed with dimethylformamide (10 mL)and
dichloromethane (10
mL) then dried under reduced pressure. The resin was suspended in N-
methylmorpholine (3
mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-diisopropylcarbodiimide
(157 L) were
added. The reaction was shaken for 17 hours, filtered through a glass sinter
funnel, and washed
well with N-methylmorpholine to give compound 225.
[0785] Reaction 3: Preparation of Compound (C366)
[0786] Compound 225 was suspended in dichloromethane (2.5 mL), trifluoroacetic
acid (2.5
mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the reaction
mixture was stirred
for 4.5 hours at ambient temperature. The resin was filtered through a glass
sinter funnel washed
with dichloromethane, and the combined filtrates were evaporated under reduced
pressure.
Crude product was then partitioned between diethyl ether (10 mL), and water
(2.5 mL). The
aqueous layer was freeze-dried to give crude product. The crude product was
purified by reverse
phase HPLC (Cl 8 10 ttM Jupiter column 250 x 21.2mm) eluting with a gradient
from 20%
acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80% acetonitrile
0.5% formic acid
: 20 % water 0.5% formic acid over 25 minutes. The product bearing fractions
were combined
and freeze-dried to give compound C366 (1.1 mg).
357
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0787] Example 1-66: Preparation of Compound C367
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Ala-Gly-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C367)
[0788] Reaction 1: Preparation of Resin-Glu(aOAllyl)-DAsn(NHTrt)-Gly-Asp(OtBu)-
DLyss(NHBoc)-Asp(OtBu)-Ala-Gly-Thr(OIleNHFmoc)-Asp(OtBu)-DGlu OtBu)-Trp-8-
Methyldecanoic amide (227)
[0789] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 128 (183 mg) in dimethylformamide (3 mL).
Compound 62
(294 mg) was added and the mixture was shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be complete using the Kaiser Test (vide
supra). The
resin was filtered through a glass sinter funnel, washed with dichloromethane
(3 x 3 mL) and
dried under reduced pressure, yielding compound 227.
[0790] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)-Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Gly-Thr-
Asp(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu) (228)
[0791] Compound 227 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4: 1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel. The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure. The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered
through a glass sinter funnel, and washed well with N-methylmorpholine to give
compound 228.
358
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0792] Reaction 3: Preparation of (C367)
[0793] Dried compound 228 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 gL), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (Cl 8 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give product C366 (6.9 mg).
[0794] Example 1-67: Preparation of Compound C368
Ile
Glu-DAsn-Gly-Asp-DLys-Asp-Ala-Sar-Thr-Asp-DGlu-Trp-8-Methyldecanoic amide
(C368)
[0795] Reaction 1: Preparation of Resin-Glu(aOAllyl -DAsn(NHTrt)-Gly-Asp(OtBu)-
DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr(OIIeNHFmoc)-Ap(OtBu -DGlu OtBu)-Trp-8-
Methyldecanoic amide (230)
[0796] Hydroxy-benzotriazole (20 mg), benzotriazole-l-yl-oxy-tris-
(dimethylamino)-
phosphoniumhexafluorophosphonate (BOP, 66 mg), and diisopropylethylamine (26
L), were
added to a solution of compound 131(296 mg) in dimethylformamide (2 mL).
Compound 56
(416 mg) was added and the mixture then shaken for 17 hours. A portion of the
resin was
removed and the coupling was judged to be incomplete using the Kaiser Test
(vide supra). An
additional portion of hydroxy-benzotriazole (20 mg), benzotriazole-1-yl-oxy-
tris-
(dimethylamino)-phosphoniumhexafluorophosphonate (BOP, 60mg), and di-
isopropylethylamine (30 gL), were added and the mixture was then shaken for 26
hours.
Coupling was judged to be complete using the Kaiser Test so the resin was
filtered through a
glass sinter funnel, washed with dichloromethane (3 x 5 mL) and dried under
reduced pressure,
yielding compound 230.
359
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0797] Reaction 2: Preparation of
Ile
Resin-Glu-DAsn(NHTrt)=Gly-Asp(OtBu)-DLys(NHBoc)-Asp(OtBu)-Ala-Sar-Thr-
Isp(OtBu)
8-Methyldecanoic amide-Trp-DGlu(OtBu)
(231)
[0798] Compound 230 was placed under an argon atmosphere, and treated with a
solution of
tetrakis-(triphenylphosphine)palladium(0) (340 mg) in dichloromethane (9.25
mL), acetic acid
(0.5 mL), and N-methylmorpholine (0.25 mL). The mixture was shaken for 4.5
hours at ambient
temperature, filtered through a glass sinter funnel, and the solid was washed
with 0.5% sodium
thiocarbozoate in dimethylformamide (10 mL), 0.5% di-isopropylethylamine in
dimethylformamide (10 mL), and dichloromethane (10 mL) then dried under
reduced pressure.
The resin was washed with DMF: piperidine 4:1 (10 mL) for 4 hours then
filtered through a
glass sinter funnel The solid was washed with dimethylformamide (10 mL) and
dichloromethane (10 mL) then dried under reduced pressure The resin was
suspended in N-
methylmorpholine (3 mL), then 1-hydroxy-benzotriazole (135 mg) and 1,3-
diisopropylcarbodiimide (157 L) were added. The reaction was shaken for 17
hours, filtered,
through a glass sinter funnel and washed well with N-methylmorpholine to give
compound 231
[0799] Reaction 3: Preparation of compound (C368)
[0800] Dried compound 231 was suspended in dichloromethane (2.5 mL),
trifluoroacetic
acid (2.5 mL), ethanedithiol (125 L), and triisopropylsilane(125 L), and the
reaction mixture
was stirred for 4.5 hours at ambient temperature. The resin was filtered,
through a glass sinter
funnel washed with dichloromethane, and the combined filtrates were evaporated
under reduced
pressure. Crude product was then partitioned between diethyl ether (10 mL),
and water (2.5
mL). The aqueous layer was freeze-dried to give crude product. The crude
product was purified
by reverse phase HPLC (Cl 8 10 M Jupiter column 250 x 21.2mm) eluting with a
gradient from
20% acetonitrile 0.5% formic acid: 80 % water 0.5% formic acid to 80%
acetonitrile 0.5%
formic acid : 20 % water 0.5% formic acid over 25 minutes. The product bearing
fractions were
combined and freeze-dried to give compound C368 (11.8 mg).
360
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0801] Example 1-68: Stereoselective synthesis of 2S,3R-N-Fmoc-L-3-methyl
glutamic acid
alpha allyl ester 232
O HN~O ~
ZZ
_ OH
O
O
232
[0802] Reaction 1
O O
O N-~1O O N~O
H
O'~-
0 O
233 234
[0803] Tetra butyl ammonium iodide (39.4 g) was added to a solution of
commercially
available Garner's aldehyde 233 (98 g) in 3M potassium carbonate (K2CO3, 100
mL) under a
nitrogen atmosphere to give a heterogeneous solution. After 15 minutes tert-
butyl-diethyl
phosphonoacetate (130 g) was added and the reaction mixture was stirred
vigorously for 18
hours. Water (500 ml) was added and the resultant mixture was extracted with
methyl tert-butyl
ether (MTBE, 3 x 250 mL). The combined organic fractions were combined, washed
with
saturated sodium chloride (1 x 250 mL), dried over magnesium sulfate (MgSO4),
filtered, and
concentrated to give the crude product as a yellow oil. Purification by colunm
chromatography
on silica gel, eluting with ethyl acetate: hexane 1: 9, gave the desired
product 234 (95.3 g).
[0804] Reaction 2
O o
O N-"O O N-11O
O
O O
234 235
361
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0805] A solution of cuprous iodide (CuI, 137 g) in dry tetrahydrofuran (THF,
2250 mL)
under a nitrogen atmosphere was cooled to -10 C and stirred for 30 minutes.
To this solution
was added a 1.6 M solution of methyl lithium (MeLi) in diethyl ether (900 mL)
such that the
temperature remained below -10 C. The resultant mixture was stirred at -10 C
for 30 minutes
then cooled to -78 C and stirred for 45 minutes. Trimethyl silyl chloride
TMSCI, (91 mL) was
added such that the temperature remained below -78 C then the reaction
mixture was stirred for
15 minutes. A solution of the substrate ester 234 (85.45 g) in THF (250 ml)
was added dropwise
over one hour. The reaction mixture was stirred at -78 C for one hour and
allowed to warm to
-40 C before a quench solution of 90% saturated ammonium chloride (NH4C1):
10%
ammonium hydroxide (NH4OH, 1500 mL) was added slowly. The reaction mixture was
stirred
for 30 minutes and warmed to -30 C before being worked up in 3 separate 1500
mL portions.
Each portion was partitioned and the aqueous layer was extracted with MTBE
(500 mL). The
combined organic phases were filtered through celite and washed with the 90%
saturated
NH4C1:10% NH4OH solution (4 x 400 mL), dried over sodium sulfate (Na2SO4),
filtered, and
concentrated to give the product. The volatiles were removed from the product
under high
vacuum to give the product 235 (85.45 g). Compound 235 was used without
further purification.
[08061 Reaction 3
o O
~LliO HO HN-IJ-O
O N
,
SS
O O
O O
235 236
[0807] A solution of the oxazolidine 235 (70 g) in methanol (1800 mL) was
cooled to 0 C
and stirred for one hour. Boron trifluoride acetic acid complex (BF3.2HOAc,
450 mL) was
added dropwise over two hours such that the internal temperature remained
below 3 C. The
reaction mixture was then quenched by the addition of 20% sodium bicarbonate
(Na2CO3, 3000
mL) over two hours and the resultant solution was worked up in 5 separate 1000
mL portions.
Each 1000 mL portion was extracted with dichloromethane (3 x 300 mL), the
organic extracts
were combined, washed with NaHCO3, (1 x 300 mL) saturated sodium chloride (1 x
30 OmL),
dried over MgSO4, filtered, and concentrated to give the crude product. The
combined products
were purified by column chromatography on silica gel, eluting with a gradient
elution from 20%
362
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
ethyl acetate:80% hexane to 50% ethyl acetate:50% hexane. Combining and
evaporating the
product bearing fractions gave the desired product 236 (36.3 g).
[0808] Reaction 4
O o
HO HN---O HO HN~O
O
O O
236 237
[0809] A solution of the alcohol 236 (24 g) in acetonitrile (238 mL) and water
(29.7 mL) was
cooled to 0 C, and periodic acid (52.2 g) was added in portions to maintain a
temperature of
0 C. The reaction mixture was stirred at 0 C for 45 minutes and chromium
trioxide (Cr03, 460
mg) was added. The reaction mixture was stirred for 15minutes before being
quenched by the
slow addition of a 0.4 M dibasic sodium phosphate solution (Na2HPO4, 560 mL,
pH 9.0). The
resultant mixture was extracted with MBTE (4 x 300 mL), and the combined
organic extracts
were washed with saturated sodium chloride (1 x 250 mL), NaHCO3 (1 x 250 mL),
and saturated
sodium chloride (1 x 250 mL). The organic portion was then dried over MgSO4,
filtered, and
concentrated to give the crude product. Purification by preparative thin layer
chromatography on
silica gel, eluting with 20 %ethyl acetate:80% hexane and extraction from
silica gel with
dichloromethane, gave the desired product 237(15.32 g).
[0810] Reaction 5
o
O
HO HN---O O HN-~10
PT O o o
O
237 238
[0811] To a solution of the acid 237 (15.32 g) in N,N-dimethylformamide (DMF,
200 mL)
was added potassium bicarbonate (KHCO3, 9.66 g) and the resultant suspension
was stirred for
15 minutes. A solution of allyl bromide (21 mL) in DMF (200 mL) was then added
dropwise
over 30 minutes and the reaction mixture was stirred for 19 hours. Water (500
mL) was added
and the resultant mixture was extracted with ethyl acetate (5 x 200 mL), and
the combined
363
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
organic extracts were washed with water (2 x 200 mL), and saturated sodium
chloride (1 x 200
mL). The organic portion was then dried over Na2SO4, filtered, and
concentrated to give the
crude product as a yellow oil. Purification by column chromatography on silica
gel, eluting with
ethyl acetate:hexane 1: 4 gave the desired product 238(9.2 g).
[0812] Reaction 6
\ \ ~ /
O O
O HN---O 0 HN-l'O
~ =
O O OH
O -zz
O
238 232
[0813] Trifluoroacetic acid (TFA, 25 mL) and triisopropyl silane (TIPS, 1 mL)
was added to
a solution of the ester 238 (9.2 g) in dichlromethane and the reaction mixture
was stirred for 1
hour. The mixture was then concentrated under vacuum and the resultant residue
was dissolved
in hexane (100 mL) and re-evaporated three times. The residue was then
dissolved in saturated
NaHCO3 (53 mL) and 1,4-dioxane (50 mL) and a solution of 9-
Fluorenylmethoxycarbonyl-N-
hydroxysuccinimide (FmocOSu, 9.52 g) in 1,4-dioxane (50 mL) was added dropwise
over 30
minutes. During this time the reaction mixture became cloudy so a further
portion of 1,4-
dioxane (20 mL) added to give a heterogeneous solution that was stirred for a
further 17 hours.
The reaction mixture was filtered, and the residue was washed with 1,4-dioxane
(50 mL). The
combined organic fractions were evaporated and re-dissolved in ethyl acetate
(250 mL) and
acidified prior to washing with potassium sulfate (KHSO4, 3 x 50 mL), and
saturated sodium
chloride (1 x 50 mL). The organic portion was then dried over Na2SO4,
filtered, and
concentrated to give the crude product. The product was purified by column
chromatography on
silica gel, using a gradient elution from 20% ethyl acetate:80% hexane to 40%
ethyl acetate:60%
hexane. Combining and evaporating the product bearing fractions gave the
desired product 232
(6.32 g).
364
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[08141 Example 1-69: Stereoselective synthesis of 2S,3S'-N-Fmoc-L-3-methyl-
glutamic acid
alpha allyl ester 239
~
~ /
O / \
O HN~O '
O OH
O
239
[0815] Reaction 1
Ph ~ Ph Ph
) O=S=O
)_jHs N/ Ph
O
240 241
[0816] Glycine benzyl ester tosylate salt (6.75 g) was partitioned between
dichloromethane
(100 mL) and aqueous 10% w/v K2C03 (100 mL). The aqueous portion was extracted
with
dichloromethane (2 x 50 mL), and the combined organic fractions were dried
over MgSO4,
filtered and evaporated to a glassy solid (3.29 g). This solid was dissolved
in dry
dichloromethane (80 mL) and a solution of benzophenone imine (3.62 g) in
dichloromethane (20
mL) was added. The resultant mixture was stirred at ambient temperature for 17
hours. The
mixture was concentrated to an oil under vacuum, re-dissolved in ether (80
mL), and washed
with water (2 x 40 mL). The organic layer was dried over MgSO4, filtered and
evaporated to
give the crude product as a clear oil. Purification by recrystallization from
warm ether/hexane
gave pure 241 (3.82 g).
[0817] Reaction 2
Ph Ph Ph Ph
0 ) Ni \ OJ N" Ph
Ph
'_
O
O
241 242
365
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0818] To a suspension of benzyl-N-(diphenylmethylene) glycinate 241 (5.7 g)
and 0-allyl-
N-(9-anthracenylmethyl)cinchonidinium bromide (1.05 g) in dichloromethane (80
mL) cooled to
-78 C under a nitrogen atmosphere, was added cesium hydroxide (14.53 g). The
mixture was
stirred for 20 minutes and tert butyl crotonate (9.13 mL) was added dropwise
so that the
temperature remained at -78 C. After stirring at -78 C for 2 hours the
mixture was wanned to
-50 C for 30 minutes then the mixture was allowed to warm to ambient
temperature over 2
hours. The mixture was then poured into diethyl ether (600 mL) and water (200
mL),
partitioned, and the organic layer was washed with water (2 x 170 mL) and
saturated sodium
chloride (1 x 150 mL). The ether fraction was then dried over MgSO4, filtered
and evaporated to
give the product 242 (4.46 g), which was used subsequently without further
purification.
[0819] Reaction 3
Ph Ph
/ ~
O N/ \ HO HN~O ~
Ph
O O
O O O O~
242 243
[0820] To a solution of the protected 3-methyl glutamate 64 (4.46 g) in
tetrahydrofuran (250
mL) was added a solution of 10% w/v citric acid (120 mL) and the mixture was
stirred for 17
hours. The solution was then concentrated under vacuum to remove the
tetrahydrofuran and
diethyl ether (100 mL) and 1N HCl (250 mL) were added. After partitioning, the
aqueous layer
was washed with diethyl ether (2 x 100 mL), basified to pH 14 by the addition
of solid K2C03,
and extracted with ethyl acetate (4 x 100 mL). Acetic acid (3 mL), and 10%
palladium on
carbon (500 mg) were added to the combined ethyl acetate fractions and the
resultant suspension
was stirred under a hydrogen atmosphere for 16 hours. Methanol (300 mL) was
added, and the
reaction mixture was filtered through celite. The filtrate was evaporated to
an oil, which was
dissolved and evaporated first in ethyl acetate (300 mL) and then diethyl
ether (300 mL) to give
a white gel. This residue was dissolved in tetrahydrofuran (200 mL) and 10%
w/v K2C03 (100
mL), and 9-fluorenylmethoxycarbonyl-N-hydroxysuccinimide (5.83g) was added.
The reaction
mixture was stirred for 22 hours, and concentrated under vacuum to remove the
tetrahydrofuran.
To the concentrated solution, diethyl ether (170 mL) and water (300 mL) were
added. After
partitioning, the aqueous layer was washed with diethyl ether (3 x 130 mL),
acidified to pH 2
366
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
with concentrated HCI, and extracted with ethyl acetate (3 x 200 mL). The
ethyl acetate
fractions were then dried over MgSO4, filtered and evaporated to give the
product 243 (3.31 g),
which was used subsequently without further purification.
[08211 Reaction 4
~ ~
I /
O / \ O / \
~O ~ O) I /
HN~O ~
HO HN
. -'
O O O O
O O
243 244
[0822] To a solution of 2S,3S-N-Fmoc-L-3-methyl-glutamic acid y tert-butyl
ester 243 (3.3
g) in dichloromethane (150 mL) was added N,N'diisopropylcarbodiimide
polystyrene resin (10.8
g) and 4-dimethylaminopyridine (92 mg), and the reaction mixture was stirred
for 5 minutes.
Allyl alcohol (0.612 mL) was added, and the reaction mixture was stirred for a
further 90
minutes. -Filtration and evaporation of the solvent gave the desired diester
244 (2.02 g).
[0823] Reaction 5
a 0 I / \
~ \ ~
~O ~ O HN~O ~
O HN
O O O,f_ O
O OH
244 239
[0824] To a solution of the diester 244 in dichloromethane (42 mL), cooled to
0 C, was
added triisopropylsilane (0.82 mL) and trifluoroacetic acid (4 mL). The
reaction mixture was
stirred at 0 C for 10 minutes, warmed to ambient temperature, and stirred for
90 minutes.
Hexane (600 mL) was added and the mixture was evaporated, the residue was
dissolved in
diethyl ether (150 mL) and 5% w/v K2C03 (200 mL). The aqueous layer was washed
with
diethyl ether (2 x 80 mL), acidified to pH 2 with concentrated HCI, and
extracted with ethyl
367
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
acetate (3 x 100 mL). The ethyl acetate fractions were then dried over MgSO4,
filtered and
evaporated to give the product 239 (1.48 g).
[0825] Example 1-70: Preparation of 2S' N-Fmoc-L-3-O-(tert-butyldimethysilyl)-
asparagine
FmocHN H OH
TBSO O
NH2
245
[0826] Reaction 1
H H
CIH3N OH CIH3N Ot-Bu
O O
OCH3 OCH3
246 247
[0827] To a suspension of the commercially available aspartic acid ester 246
(2.75 g) in 70%
perchloric acid (HC1O4a 3 mL) was added t-butyl acetic acid ester (100 mL).
After 24 h, the
solution was poured into saturated K2C03 (200 mL). The resulting biphasic
mixture was
extracted with diethyl ether (3 x 100 mL) and the combined organic extract was
washed with
saturated K2C03 (3 x 50 mL), dried over Na2SO4, filtered and concentrated
under diminished
pressure to give a clear colorless oil. The oil was then dissolved in cold (0
C) diethyl ether (50
mL) and 1 N HCI in diethyl ether (15 mL) was added. After 20 min the solution
was
concentrated under diminished pressure to give 247 (2.0 g).
[0828] Reaction 2
H H
CIH3N Ot-Bu TrHN Ot-Bu
O O
OCH3 OCH3
247 248
[0829] To a suspension of the diester 247 (4.78 g) in methyl tert-butyl ether
(100 mL) was
added saturated aqueous K2C03 (100 mL). The resulting aqueous layer was washed
with methyl
tert-butyl ether (3 x 100 mL). The organic extract was combined and washed
with saturated
K2C03 (2 x 100 mL), dried over Na2SO4, filtered and concentrated under
diminished pressure.
368
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
The resulting colorless oil was mixed with trityl chloride (5.57 g), CH3CN
(100 mL), and
triethylamine (5.60 mL). After 16 h, the mixture was filtered and diluted with
methyl tert-butyl
ether (200 mL). The resulting organic extract was washed with 1N citric acid
(3 x 100 mL),
saturated NaHCO3 (3 x 100 mL), saturated NaCl (3 x 100 mL), dried over Na2SO4,
filtered, and
concentrated under diminished pressure. Chromatography on base washed flash
silica gel (20 x
4 cm) using 1:11 ethyl acetate-hexanes with 1% triethylamine present gave
product 248 (7.12 g).
[0830] Reaction 3
HO HO
TrHN Ot-Bu TrHN Ot-Bu
O HO O
OCH3 OCH3
248 249
[0831] To a cooled (-78 C) solution of 248 (2.22 g) in tetrahydrofuran (20
mL) was added
16.5 mL of 0.91 M potassium hexamethyldisilazane solution in tetrahydrofuran.
After 1 h at -78
C, oxodiperoxy(pyridine)(1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-
pyrimidinone)molybdenum
(IV) (MoOPD, 3.68 g, obtained from STREM Chemicals, Inc; see Anderson, JC. and
Smith,
SC., 1990, S ett 107-109 for the synthesis of this reagent) was added in
portions. The mixture
was stirred for 1 h at -78 C, allowed to warm up to -55 C, and stirred for
an additional hour.
The mixture was quenched with saturated aqueous Na2SO3 (20 mL) and warmed to
room
temperature. The resulting biphasic mixture was washed with methyl tert-butyl
ether (3 x 100
mL) and the organic layers were combined and washed with 1 N citric acid (3 x
50 mL),
saturated aqueous NaHCO3 (3 x 50), and saturated aqueous NaCl (3 x 50), dried
over Na2SO4,
filtered, and concentrated under diminished pressure. Chromatography on flash
silica gel (20 x 3
cm) using 1:5 ethyl acetate-hexanes gave product 249 (1.64g).
[0832] Reaction 4
H H
TrHN Ot-Bu TrHN Ot-Bu
HO O HO O
OCH3 OH
249 250
[0833] To a solution of 249 (650 mg) in 1:1 dioxane-water (50 mL) was added
lithium
hydroxide (507 mg). After 1 h the solution was washed with methyl tert-butyl
ether (3 x 50 mL)
and the resulting aqueous layer was acidified to pH -4 with 1N citric acid.
The resulting
369
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
solution was extracted with methyl tert-butyl ether (3 x 100 mL). The organic
phase was washed
with 1 N citric acid (3 x 100 mL), and saturated NaCI (3 x 100 mL), dried over
Na2SO4, filtered,
and concentrated under diminished pressure to give 250 (630mg).
[0834] Reaction 5
O H O
TrHN H Ot-Bu TrHN Ot-Bu
O -~ HO O
HO
OH NH2
250 251
[0835) To a solution of 250 (650 mg), benzotriazol-l-yloxy-tris(dimethylamino)-
phosphonium hexafluorophosphate (997 mg), and NH4C1(153 mg) in
dimethylformamide (30
mL) was added diisopropylethylamine (0.78 mL). After lh, ethyl acetate (150
mL) was added
and the resulting solution was washed with 10% K2C03 (3 x 100 mL), water (3 x
100 mL), 1 N
citric acid (3 x 100 mL), and saturated NaCl, dried over Na2SO4, filtered, and
concentrated under
diminished pressure to yield 251 (640 mg).
[0836] Reaction 6
HO HO
TrHN Ot-Bu H2N OH CF3COOH
HO HO
NH2 NH2
251 252
[0837] To a solution of 251 (1.91 g) in dichloromethane (5 mL) was added water
(1 mL)
followed by trifluoroacetic acid (10 mL). After 4 h, the solution was
concentrated under
diminished pressure and the remaining slurry was concentrated twice from
toluene. The
resulting solid was triturated with diethyl ether and the resulting solid was
filtered and washed
with diethyl ether. The resulting solid was dried under diminished pressure to
give 252 (952
mg).
370
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0838] Reaction 7
O O
H2N H OH FmocHN OH
CF3COOH
HO O HO O
NH2 NH2
252 253
[0839] To a solution of a solution of 9-Fluorenylmethoxycarbonyl-N-
hydroxysuccinimide
(3.37 g) in 1-4-dioxane (50 mL). After 16 h, the resulting solution was
diluted with aqueous 5 %
K2C03 solution (25 mL) and extracted with diethyl ether (3 x 50 mL). The
resulting aqueous
extract was acidified to pH -2 with a 1 N HCl solution and diethyl ether (50
mL) was added.
The resulting solid was partitioned between the acidic solution and diethyl
ether. The solid was
collected and washed with 1N HCl and diethyl ether to yield 253 (1.50 g).
[0840] Reaction 8
H H
FmocHN OH FmocHN OH
HO O TBSO O
NH2 NH2
253 245
[0841] To a solution of 253 (370 mg) in dimethylformamide (10 mL) was added
tert-
butyldimethylsilyl chloride (300 mg), followed by imidazole (200 mg). After 8
h, the solution
was diluted with ethyl acetate and washed with 1 N HCl (3 x 100 mL) and
saturated sodium
chloride, dried over Na2SO4, filtered, and concentrated under diminished
pressure.
Chromatography on flash silica gel (25 x 2 cm) using 19:1:0.1 CH2C12:MeOH:AcOH
as eluent
gave 245 (300 mg).
371
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0842] Example 1-71 = PYeparation of 2S-N-Fmoc-L-(3-metho.xy)-fl-tert-butyl
aspartic acid
ester 254
FmocHN H OH
H3CO O
Ot-Bu
254
[0843] Reaction 1
HO HO
CIH3N OCH TrHN OCH
3 3
O ~ O Ot-Bu Ot-Bu
255 256
[0844] To a suspension of commercially available diester 255 (4.78 g) in
methyl tert-butyl
ether (100 mL) was added saturated aqueous K2CO3 (100 mL). The resulting
aqueous layer was
washed with methyl tert-butyl ether (3 x 100 mL). The organic extract was
combined and
washed with saturated K2C03 (2 x 100 mL), dried over Na2SO4, filtered and
concentrated under
diminished pressure. The resulting colorless oil was dissolved in a solution
of trityl chloride
(5.57 g), and triethylamine (5.60 mL) in acetonitrile (100 mL). After 16 h,
the mixture was
filtered and diluted with methyl tert-butyl ether (200 mL). The resulting
organic extract was
washed with 1N citric acid (3 x 100 mL), saturated NaHCO3 (3 x 100 mL), and
saturated NaCl
(3 x 100 mL), dried over Na2SO4, filtered, and concentrated under diminished
pressure.
Chromatography on base washed flash silica gel (20 x 4 cm) using 1:11 ethyl
acetate-hexanes
with 1% triethylamine present gave 256 (7.12 g).
[0845] Reaction 2
HO HO
TrHN OCH TrHN OCH
3 ~ 3
O HO O
Ot-Bu Ot-Bu
256 257
[0846] To a cooled (-78 C) solution of 256 (2.22 g) in tetrahydrofuran (20
mL) was added
16.5 mL of 0.91 M potassium hexamethyldisilazane solution in tetrahydrofuran.
After 1 h at -78
372
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
C, oxodiperoxy(pyridine)(1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone)
molybdenum
(IV) (MoOPD, obtained from STREM Chemicals, Inc; 3.68 g, see Anderson, JC. and
Smith,
SC., 1990, S ett 107-109 for the synthesis of this reagent) was added in
portions. The mixture
was stirred for 1 h at -78 C, allowed to warm up to -55 C and stirred for an
additional hour.
The mixture was quenched with saturated aqueous Na2SO3 (20 mL) and warmed to
room
temperature. The resulting biphasic mixture was washed methyl tert-butyl ether
(3 x 100 mL).
The organic layers were combined and washed with 1 N citric acid (3 x 50 mL),
saturated
aqueous NaHCO3 (3 x 50), and saturated aqueous NaC1(3 x 50), dried over
Na2S04, filtered, and
concentrated under diminished pressure. Chromatography on flash silica gel (20
x 3 cm) using
1:5 ethyl acetate-hexanes gave 257 (1.64 g).
[0847] Reaction 3
HO HO
TrHN OCH CbzHN OCH
3 3
HO O HO O
Ot-Bu Ot-Bu
257 258
[0848] To a solution of 257 (4.61 g) in dichloromethane (100 mL) was added
trifluoroacetic
acid (2 mL). After 1 h, the resulting solution was concentrated under
diminished pressure, and
aqueous 5 % K2C03 (50 mL) was added followed by a solution of
benzyloxycarbonyl-N-
hydroxysuccinimide (2.49 g) in dioxane (50 mL). After 16 h, the resulting
solution was
extracted with ethyl acetate (3 x 100 mL). The organic extract was washed with
1 N citric acid
(3 x 50 mL), saturated NaHCO3 (3 x 50 mL) and NaC1(3 x 50 mL), dried over
Na2SO4, filtered
and concentrated under diminished pressure. Chromatography on flash silica gel
(25 x 3 cm)
using 1:3 ethyl acetate-hexanes gave 258 (2.01 g).
[0849] Reaction 4
H H
CbzHN OCH CbzHN OCH
3 3
HO O H3CO O
Ot-Bu Ot-Bu
258 259
[0850] To a cold (0 C) suspension of 258 (353 mg) and silver oxide (462 mg)
in
tetrahydrofuran (25 mL) was added iodomethane (0.62 mL). The mixture was
allowed to warm
373
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
up to room temperature over 4 h. After 48 h, the suspension was filtered
through Celite and
concentrated under diminished pressure. Chromatography on flash silica gel (25
x 2 cm) using
1:3 ethyl acetate-hexanes gave 259 (300 mg).
[0851] Reaction 5
O O
CbzHN H OCH CbzHN H OH
3
H3C0 O H3CO O
Ot-Bu Ot-Bu
259 260
[0852] To a cold (0 C) solution containing 259 (300 mg, 0.81 mmol) in 25 mL
of dioxane
was added 240 mg (10 mmol) of LiOH in 25 mL of water. After 1 h, the mixture
was acidified
to pH - 4 with 1 N citric acid, and extracted with ether (3 x 50 mL). The
resulting organic
extract was washed with 1 N citric acid (3 x 50 mL), and saturated NaCI (3 x
30 mL) dried over
Na2SO4, filtered and concentrated under diminished pressure to give 260 as a
clear, colorless oil
(280 mg).
[0853] Reaction 6
HO HO
CbzHN OH FmocHN OH
~
H3C0 O H3CO O
Ot-Bu Ot-Bu
260 254
[0854] Compound 260 is converted to 254 by treatment of an ethyl acetate
solution of 260
with 10% palladium on carbon, under a hydrogen atmosphere as previously
described for the
conversion of 242 to 243 followed by an amine protection as previously
described for conversion
of 252 to 253.
374
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0855] Example 1-72: Preparation ofNa-(Allyloxycarbonvl)-L-isoleucine 124
[0856] Reaction 1
O
H2N OH O~ H N OH
0 O
125 124
[0857] Commercially available Isoleucine (22 g) was added to a solution of
allyloxycarbonyl
oxysuccinimide (AllocOSu, 51 g) in tetrahyrofuran (150 mL). Ten percent K2C03
aqueous
solution (100 mL) was added to this suspension and the mixture was stirred for
17 hours before
concentrating to approximately 120 ml under reduced pressure. The solution was
added to 10%
K2C03 aqueous solution (100 mL) and water (200ml) and washed with diethyl
ether (4 x 150
mL). The aqueous portion was then acidified to pH 1 and extracted with
dichloromethane (4 x
200 mL). Combined acidic dichloromethane washes were dried with anhydrous
MgSO4 and
evaporated to crude product (38.1 g). Purification by column chromatography on
silica gel,
(eluting with dichloromethane/methanol gradient of 100% dichloromethane to
dichloromethane:
methanol 9:1) followed by evaporation of the solvent, gave the compound 124 as
a yellow oil
(36 g)
[0858] Example 2-1: Construction of an S. roseosporus-based in-trans
expression system for
the production of the novel biosynthetic pathways.
[0859] For the expression of the hybrid non-ribosomal polypeptide synthetase
(NRPS)
pathways, a version of the S. roseosporus high daptomycin-producing strain
(NRRL 11379) that
lacked all of the NRPS genes was constructed. The hybrid pathways were
conjugated into this
strain on BAC-based vectors which integrated site-specifically in a neutral
site of the S.
roseosporus genome at a~C31 attB site.
[0860] To delete all the proposed NRPS genes from S. roseosporus a deletion
cassette was
constructed that contained flanking DNA from upstream of dptEF and downstream
of dptH
(Figure 2).
[0861] Flanking regions from upstream of dptEF (5') and downstream of dptH
(3') were
cloned around a selection cassette containing tsr (thiostrepton resistance)
and cat
(chloramphenicol resistance). The 5' fragment was 1478 bp long and the 3'
fragment was 1862
375
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
bp long. These two fragments were cloned into a copy of pUC19 (New England
Biolabs) that
already contained the tsr and cat resistance cassettes to create the deletion
cassette. This cassette
was then transferred to a delivery plasmid called pRHB538 (Hosted, T.J. and
Baltz, R.H., 1997, J
Bacteriol. 179(1): 180-6), which contains a temperature sensitive origin of
replication and a
dominant allele of rpsL (streptomycin sensitive). This plasmid was introduced
into a S.
roseosporus strain carrying a recessive rpsL allele that confers streptomycin
resistance. This
recombinant strain was then incubated overnight in a broth culture before the
cells were spread
on plates containing streptomycin plus thiostrepton and incubated at 39 C.
Under these
conditions only those strains that have exchanged the deletion cassette
(containing tsr and cat)
for the dptA-H locus via homologous recombination survived the selection; all
other genotypes
were eliminated.
[0862] PCR and Southern blots confirmed the genotype of the dptA-H deletion
mutants. The
PCR fragments were designed to be amplified from primers that lay outside the
5' and 3'
flanking regions and inside the tsr and cat genes. In this way, the PCR
products can only be
formed when the cell has exchanged the deletion cassette for the dptA-H locus.
The Southern
blots provided further confirmation that dptA Hhad been deleted and that no
aberrant
integrations or recombination had occurred around this locus. Once the dptA-H
deletions were
confirmed genetically they were then tested to see if they were true null
mutants phenotypically.
This strain was then designated S. roseosporus UA43 1.
[0863] Example 2-2: Fermenting Streptomyces roseosporus
[0864] Spores of the Streptomyces roseosporus UA431 were harvested by
suspending a 10
day old slant culture of medium A (2% irradiated oats (Quaker), 0.7% tryptone
(Difco), 0.2%
soya peptone (Sigma), 0.5% sodium chloride (BDH), 0.1 % trace salts solution,
1.8% agar no. 2
(Lab M), 0.01 % apramycin (Sigma)) in 5 mL 10% aqueous glycerol (BDH)). One mL
of this
suspension, in a 1.5 mL cryovial, comprises the starting material, which was
retrieved from
storage at -135 C. A pre-culture was produced by aseptically placing 0.3 mL
of the starting
material onto a slant of medium A and incubating for 9 days at 28 C.
[0865] A seed culture was generated by aseptically treating the pre-culture
with 4 mL of a
0.1 % Tween 80 (Sigma) solution and gently macerating the slope surface to
generate a
suspension of vegetative mycelium and spores. A two mL aliquot of this
suspension was
transferred into a 250 mL baffled flask containing 40 mL of nutrient solution
S (1% D-glucose
376
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
(BDH), 1.5% glycerol (BDH), 1.5% soya peptone (Sigma), 0.3% sodium chloride
(BDH), 0.5%
malt extract (Oxoid), 0.5% yeast extract (Lab M), 0.1 % Junlon PW 100
(Honeywell and Stein
Ltd), 0.1 % Tween 80 (Sigma), 4.6% MOPS (Sigma) adjusted to pH 7.0 and
autoclaved)) and
shaken at 240 rpm for 44 hours at 30 C.
[0866] Production cultures were generated by aseptically transferring 5% of
the seed culture
to baffled 250 mL flasks containing 50 mL medium P(1% glucose (BDH), 2%
soluble starch
(Sigma), 0.5% yeast extract (Difco), 0.5% casein (Sigma), 4.6% MOPS (Sigma)
adjusted to pH 7
and autoclaved)) and shaken at 240 rpm for up to 7 days at 30 C.
[0867] Example 2-3: Analysis of the A219 78C Lipopeptides ft ~ om
fermentations of the
Streptomyces roseosporus
[0868] Production cultures described in Example 2-2 were sampled for analysis
by
aseptically removing 2 mL of the whole culture and centrifuging for 10 minutes
prior to analysis.
Volumes up to 50 microlitres of the supernatant were analyzed to monitor for
production of the
native lipopeptides (A21978C) as produced by Streptomyces roseosporus. This
analysis was
performed at ambient temperature using a Waters Alliance 2690 HPLC system and
a 996 PDA
detector with a 4.6 x 50 mm Symmetry C8 3.5 m column and a Phenomenex Security
Guard C8
cartridge. The gradient initially holds at 90% water and 10% acetonitrile for
2.5 minutes,
followed by a linear gradient over 6 minutes to 100% acetonitrile. The flow
rate is 1.5 mL per
minute and the gradient is buffered with 0.01 % trifluoroacetic acid. By day 2
of the fermentation,
production of three of the native lipopeptides, A21978C1, A21978C2 and
A21978C3, with
UV/visible spectra identical to that of daptomycin, was evident, as shown by
HPLC peaks with
retention times of 5.62, 5.77 and 5.90 minutes (?max 223.8, 261.5 and 364.5
nm) under the
analytical conditions stated. The lipopeptides then remained evident in the
fermentation at each
sample point during the 7-day period. Total yields of lipopeptides A21978C1a
A21978C2 and
A21978C3 ranged from 10-20 mg per liter of fermentation material.
[0869] Liquid chromatography-mass spectrometry (LC-MS) analysis was performed
on a
Finnigan SSQ710c LC-MS system using electrospray ionization in positive ion
mode, with a
scan range of 200-2000 daltons and 2 second scans. Chromatographic separation
was achieved
on a Waters Symmetry C8 column (2.1x 50mm, 3.5 m particle size) eluted with a
linear water-
acetonitrile gradient containing 0.01 % formic acid, increasing from 10% to
100% acetonitrile
over a period of six minutes after a initial delay of 0.5 minutes, then
remaining at 100%
377
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
acetonitrile for a further 3.5 minutes before re-equilibration. The flow rate
was 0.35 mL/minute
and the method was run at ambient temperature.
[0870] The identification of the three native lipopeptides was confirmed in
the controls (S.
roseosporus wild type), as indicated by molecular ions ([M+H]+) at m/z of
1634.7, 1648.7 and
1662.7, which is in agreement with the masses reported for the major A21978C
lipopeptide
factors A21978C1a A21978C2 and A21978C3, respectively, produced by
Streptomyces
roseosporus (Debono et al., 1987, J. Antibiotics 40: 761-777). The UA431
mutants failed to
produce any of the A21978C lipopeptides confirming that they were true null
mutants.
[0871] Example 2-4: Constructing pDA300 and complementing the S. roseosporus
dptA-H
deletions
[08721 Unlike yeast and some naturally competent bacteria, linear DNA
fragments do not
readily transform Escherichia coli. This is in part due to the degradation of
foreign DNA by
intracellular exonucleases such as RecBCD (Lorenz, M.G., and Wackernagel, W.,
1994,
Microbiol. Rev. 58: 563). Traditionally, homologous recombination was either
achieved by
using mutant strains lacking RecBCD (Jasin, M., and Schimmel, P., 1984, J.
Bacteriol 159: 783)
or by delivering DNA with the help plasmid vectors that cannot replicate in
the host under
restrictive conditions (Link, A.J. et al., 1997, J. Bacteriol. 179: 6228).
Recombination events
remain rare and require kilobases of homology.
[0873] Recently, several laboratories have developed strains that take
advantage of the
bacteriophage X-induced "hyper-recombination" state (Datsenko, K.A., and
Wanner, B.L., 2000,
Proc. Nat Acad Sci U.S.A. 97: 6640; United States Patent Numbers 6,355,412 and
6,509,156B;
Yu, D., et al., 2000, Proc. Nat Acad Sci U.S.A. 97: 5978). Recombination
between DNA
molecules with as little as 40-50 bp of identical sequence takes place even
when using linear
DNA. The ?. Red genes (exo, bet and gam) cause the enhancement of the
recombination rate.
The k-exonuclease and the (3-protein are responsible for recombination through
repair of double-
strand breaks, whereas the gam gene product binds to the host RecBCD complex
and inhibits its
functions (Murphy, K., 1998, J. Bacteriol. 180: 2063). We refer to this
technique as the "Red"
system or Red-mediated recombination system.
[0874] Using the "Red" system a pDA300 (a truncated version of B 12:03A05 that
contains
only the dptA-H genes) was constructed. This plasmid was constructed from B
12:03A05 (a
BAC plasmid that contains all of the dpt biosynthetic gene cluster, which was
isolated from a
378
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
chromosomal library of S. roseosporus (Miao et al, 2005, Microbiology 151:
1507-1523), all of
the genes upstream of dptA-H and all of genes downstream of dptA-H were
deleted using
homologous recombination via the Red-mediated recombination system. This was
achieved by
introducing B12:03A05 into an E. coli strain carrying the Red genes on a
plasmid (pKD78,
Datsenko, KA., and Wanner, BL., 2000, Proc. Nat Acad Sci U.S.A. 97: 6640).
This strain was
then transformed by PCR products for the tet resistance gene that were flanked
by
oligonucleotides with homology to either the upstream or downstream regions of
the dpt cluster.
Once constructed, pDA300 was introduced into UA431 by conjugation to create
strain UA493.
Plasmid pDA300 contains oriT from plasmid RK2 (Baltz, 1998, Trends in
Microbiol. 6: 76-83
(1998), incorporated herein by reference in its entirety) for conjugation from
E. coli to S.
roseosporus. Plasmid pDA300 is introduced into S. roseosporus by conjugation
from E. coli
S17.1, or a strain containing a self-replicating plasmid RK2 (Id.). S.
roseosporus UA493 was
fermented and analyzed using the techniques described in Examples 2-2 and 2-3
respectively.
[0875] The identification of the three native lipopeptides was confirmed, as
indicated by
molecular ions ([M+H]+) at m/z of 1634.7, 1648.7 and 1662.7, which is in
agreement with the
masses reported for the major A21978C lipopeptide factors A21978C1, A21978C2
and
A21978C3, respectively, produced by Streptomyces roseosporus (Debono et al.,
1987, J.
Antibiotics 40: 761-777). This demonstrated that the pDA300 was able to
successfully
complement the dptA-H deletion to restore lipopeptide production in UA493.
[08761 Example 2-5: Exchange of a non-ribosomal peptide synthetase (NRPS)
subunit fof
one that catalyzes the incorporation of different amino acid(s).
[0877] The gene that encodes the third subunit of the daptomycin NRPS (see
Figure 1)
contains two modules that encode the specificity for incorporation of amino
acids 12 (3-methyl-
glutamic acid (3-MeGlu)) and 13 (L-kynurenine (L-Kyn)). The gene that encodes
the third
subunit for the biosynthesis of the cyclic lipopeptide CDA (Kempter et al ,
1997, Angew. Chem.
Int. Ed. Engl. 36: 498-501; Chong et al., 1998, Microbiology 144: 193-199;
each of which is
incorporated by reference herein in its entirety) also encodes the last two
amino acids, in this
case amino acids 10 (3-MeGlu) and 11 (L-tryptophan (L-Trp); Figure 1). A
derivative of
daptomycin containing L-Trp instead of L-Kyn in position 13 was generated by
deleting gene
dptD, and by replacing it with the gene that encodes PS3 for CDA (Hojati et
al., 2002, Chem
Biol. 9(11):1175-87). The vector pMF23 expressed the PS3 gene from a strong
promoter (e.g.,
379
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
the ernaEp* promoter; Baltz, 1998, Trends Microbiol. 6: 76-83, incorporated
herein by reference
in its entirety), and when introduced in to S. roseosporus via interspecies
conjugation (Baltz,
1998, Trends Microbiol. 6: 76-83) before site-specifically inserting into a
neutral site in the S.
roseosporus genome, allowed cdaPS3 to complement the dptD mutation and
resulted in the
production of the altered daptomycin with L-Trp replacing L-Kyn to give
compound C1,
compound C2, and compound C3. The recombinant strain was fermented and the
product(s) of
the recombinant strain were analyzed by LC-MS as described in Examples 2-2 and
2-3. Similar
experiments were performed where the dptD deletion was complemented by the
gene that
encodes the third subunit for the biosynthesis of the cyclic lipopeptide
A54145 (pMF30 is a
derivative of pHMl 1 a that contains lptD expressed from ermEp* (Motamedi et
al., 1995, Gene
160: 25-31) which also encodes the last two amino acids, in this case amino
acids 12 (3-MeGlu)
and 13(L-isoleucine (L-Ile) or L-valine (L-Val)). Two derivatives of
daptomycin containing
either L-Ile or L-Val instead of L-Kyn in position 13 were generated by
disrupting gene dptD,
and by replacing it with the gene that encodes lptD for A54145 (compounds C4,
C5, C6, C7, C8,
C9).
[08781 Similar manipulations are performed for trans-complementation for other
subunits,
i.e. to generate a disruption or deletion in a subunit of the daptomycin
biosynthetic gene cluster
or the A54145 biosynthetic gene cluster, and then complement in trans by one
or more natural or
modified subunits from an NRPS (the latter can include trans-complementation
by modified
versions of daptomycin or A54145 biosynthetic gene cluster subunits). Trans-
complementation
between the NRPS subunits then leads to production of a novel nonribosomal
peptide which can
be analyzed for as described in previous examples.
[0879] To perfonn a trans-complementation experiment using portions of the
daptomycin or
A54145 biosynthetic gene cluster and the calcium dependent antibiotic (CDA)
biosynthetic gene
cluster, the set of daptomycin biosynthetic genes, or the set of daptomycin
biosynthetic genes
and accessory genes, such as those contained on the BAC clone B12:03A05, are
introduced by
transformation or conjugation into other natural or engineered strains or
species of
actinomycetes. The recipients may be known producers of secondary metabolites
or
uncharacterized strains, or may be generated by recombinant techniques to
carry biosynthetic
pathways other than that for biosynthesis of daptomycin. The transformants or
ex-conjugants are
fermented in a variety of media and whole broth or extracts thereof are
screened for either novel
daptomycin-like compounds or biological activity against daptomycin-resistant
tester organisms.
380
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0880] The complementation is often facilitated by inactivation of some of the
subunit genes
in the daptomycin or A54145 biosynthetic pathway (as is described above for
the deletion of
dptD and complementation by either cdaPS3 or lptD). Sequences encoding a
subunit of the
NRPS are deleted or replaced by a marker gene to form a modified NRPS
biosynthetic pathway;
this can be achieved either in the original producing strain (,S roseosporus
for daptomycin, S.
fradiae or S. refuineus for A54145, S. coelicolor for CDA) or plasmids
carrying these
biosynthetic pathways.
[0881] To produce the novel lipopeptide, homologous recombination across
flanking DNA
sequences was used to exchange the bulk of the coding region of dptD in pDA300
for a
heterologous marker gene. To perform the homologous recombination, two
oligonucleotides
were designed to amplify the regions directly upstream ("5' fragment") and
downstream ("3'
fragment") of dptD. The 5' and 3' fragments were amplified from chromosomal
DNA of S.
roseosporus using the following primer sets with 5'-terminal extensions in
which unique
restriction sites have been introduced (underlined):
5' fragment (1122 bp):
5' GCG AAG CTT CTG GTG GCG CAT CAC CTG G 3' (SEQ ID NO: 1)
5' GCT CTA GAT GGA AGT ATG TCC TCC ATC GC 3' (SEQ ID NO: 2)
3' fragment (1535 bp):
5' CGG ATC CCG CCG GCA CCT GAC CC 3' (SEQ ID NO: 3)
5' CCG AAT TCC GCC TCC GAG TAC ATC GAG G 3' (SEQ ID NO: 4)
[0882] The amplified fragments were cloned in succession into the
corresponding unique
sites in the multiple cloning site of pNEB 193 (New England Biolabs). The
resulting construct,
pSD002, was confirmed by restriction digest analysis for orientation, and by
sequencing for the
absence of errors in the portions generated by PCR. A Spel fragment containing
the marker
gene, ermE (erythromycin resistance gene; see Hopwood, supra) was inserted
into pSD002 at an
Xbal site and verified by restriction digest analysis. The resulting plasmid,
pSD005, thus
includes a cassette composed of eYmE flanked by DNA stretches homologous to
DNA sequences
upstream and downstream of dptD. Once inserted into the daptomycin
biosynthetic gene cluster
pathway by homologous recombination, this cassette would essentially replace
all of dptD,
except for the first 31 bp and the last 12 bp, with ernaE. The region
comprising the replacement
381
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
cassette was then subcloned into a vector (a cloning site-modified version of
pRHB538; (Hosted
and Baltz, 1997, J. Bacteriol. 179: 180-186) carrying a temperature-sensitive
replication origin
and rpsL (a gene conferring sensitivity to streptomycin) to create pSD030, the
final plasmid in
the series for introduction into S. roseosporus.
[0883] The plasmid, pSD030, was introduced into S. roseosporus by interspecies
conjugation
(Baltz, 1998, Trends Microbiol., 6: 76-83). Each plate was then flooded with 1
mL of water
containing 1.25 mg of erythromycin, resulting in a final concentration of 50
g/ml once the
liquid was absorbed into the media. Erythromycin-resistant colonies arising on
the
transformation plate after 7 days were inoculated into 25 mL of TSB (Hopwood,
supra) plus
erythromycin and incubated at 30 C for 48 hours. The mycelium was harvested,
and 1/10th of
the inycelial mass was macerated and transferred to a new aliquot of 25 mL TSB
plus
erythromycin. The resultant solution was then incubated at 40 C to select
against the
temperature-sensitive replicon of pSD030. After 48 hours, the mycelium was
harvested by
centrifugation, macerated and resuspended in a final volume of 2 mL TSB. This
suspension (100
L) was spread on SPMR plates (Babcock et al., 1988, J. Bacteriol. 170: 2802-
2808) containing
50 g/mL erythromycin and 30 g/mL of streptomycin. Colonies that survived
were screened
and shown to have the correct genotype by PCR to identify strains such as S.
roseosporus
UA378, in which ermE had successfully replaced dptD. This mutant was then
complemented in-
trans by initially dptD, where dptD was expressed from the expression plasmid
pHMI 1 a
(Motamedi H, et al., 1995, Gene 160(1): 25-31) under the control of the
constitutive promoter
erntEp*.
[0884] Starting material of UA378 was regenerated by suspending a 10 day old
slope culture
of medium A (see "Practical StYeptomyces Genetics" by Kieser T., et al., John
Innes Foundation,
Norwich, 2000, herein "Kieser"; 2% irradiate oats (Quaker), 0.7% tryptone
(Difco), 0.2% soya
peptone (Sigma), 0.5% sodium chloride (BDH), 0.1% trace salts solution, 1.8%
agar no. 2 (Lab
M), 0.01% apramycin (Sigma) in 5 mL 10% aqueous glycerol (BDH)). A 1.5 mL
cryovial
containing 1 mL of starting material was retrieved from storage at -135 C and
thawed rapidly.
A pre-culture was produced by aseptically placing 0.3 mL of the starting
material onto a slope of
medium A and incubating for 9 days at 28 C. Material for inoculation of the
seed culture was
generated by aseptically treating the preculture with 4 mL of a 0.1 % Tween 80
(Sigma) solution
and gently macerating the slope surface to generate a suspension of vegetative
mycelium and
spores.
382
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0885] A seed culture was produced by aseptically placing 1 mL of the
inoculation material
into a 2 L baffled Erlenmeyer flask containing 250 mL of nutrient solution S
(see Kieser, supra)
shaken at 240 rpm for 2 days at 30 C.
[0886] A production culture was generated by aseptically transferring the seed
culture to a 20
L fermenter containing 14 liters of nutrient solution P (see Kieser, supra).
The production
fermenter was stirred at 350 rpm, aerated at 0.5 vvm, and temperature
controlled at 30 C. After
20 hours incubation a 50% (w/v) glucose solution was fed to the culture at 5
g/hr throughout the
fermentation.
[0887] After 40 hours incubation, a 50:50 (w/w) blend of decanoic acid:methyl
oleate
(Sigma and Acros Organics, respectively) was fed to the fermenter at 0.5 g/hr
for the remainder
of fermentation. The culture was harvested after 112 hours, and the biomass
was removed from
the culture supematant by batch processing through a bowl centrifuge.
[0888] The biomass from the 20 L fermentation was discarded and the clarified
liquor was
applied to an open glass column, packed with Mitsubushi HP20 resin (60 x 300
mm) and
conditioned with methanol and water. Prior to elution, the column was washed
with 2 L of water
followed by 2 L of methanol/water (1:4). The column was then eluted with 2 L
of
methanol/water (4:1) followed by 1 L methanol, and collected as two separate
fractions.
[0889] Liquid chromatography-mass spectroscopy (LC-MS) electrospray ionization
(ESI)
analysis indicated that both fractions contained the A21978C/CDA hybrid
molecules, and the
less complex methanol/water (4:1) fraction was processed further. This was
evaporated under
vacuum to an aqueous residue and then made up to 500 mL with water. It was
then back
extracted with 3 x 500 mL of ethyl acetate in a 2 L separating funnel, to give
an aqueous and
organic fraction. LC-MS (ESI) indicated that the hybrid molecules were absent
from the organic
phase and it was discarded. The aqueous fraction was lyophilized overnight.
[0890] The hybrid molecules were purified by preparative high performance
liquid
chromatography (HPLC) using a Waters Prep LC system and a Waters 40 x 200mm
Nova-Pak
C18 60A 6 m radially-compressed double cartridge with 40 x 10mm guard. The
freeze-dried
material was dissolved in water and purified using a gradient method. This
method held at 90%
water and 10% acetonitrile for 2 minutes and was followed by a linear gradient
over 13 minutes
to 25% water and 75% acetonitrile. The flow was 55 mL/min and the whole
gradient was
buffered with 0.04% trifluoroacetic acid. Fractions were collected and
analyzed by LC-MS on a
Finnigan SSQ710c LC-MS system using electrospray ionisation (ESI) in positive
ion mode, with
383
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
a scan range of 200-2000 daltons and 2 second scans. Chromatographic
separation for this
LC-MS analysis was achieved on a Waters Symmetry C8 column (4.6x 50mm, 3.5 m
particle
size) eluted with a linear water-acetonitrile gradient containing 0.01% formic
acid, increasing
from 10% to 100% acetonitrile over a period of six minutes after an initial
delay of 0.5 minutes,
then remaining at 100% acetonitrile for a further 3.5 minutes before re-
equilibration. The flow
rate was 1.5 mL/minute and the method was run at ambient temperature.
[0891] The analysis identified compound Cl and compound C2. Both fractions
required
further purification prior to NMR studies. Compound Cl was further purified
using an isocratic
method with 60% water and 40% acetonitrile buffered with 0.04% trifluoroacetic
acid.
Approximately 1.8 mg of material was isolated. Final purification of compound
C2 used an
isocratic method with 58% water and 42% acetonitrile buffered with 0.04%
trifluoroacetic acid.
Approximately 1.5 mg of material was isolated. The UV maxima and ESI-MS
molecular ion
information (doubly-charged ions observed in negative ion mode) for compound
Cl and
compound C2 are presented below:
Compound Cl Compound C2
ESI -MS (m/z) 814 (M-2H)2- 821 (M-2H)2-
LN-vis ?,,,ax /nm 221, 280, 221, 280
j0892j Exafnple 2-6: Module exchanges constructed at positions 8 and 11 in
dptBC
[0893] A plasmid carrying dptBC pKN24 was constructed by truncation of B
12:03A05 that
carries daptomycin biosynthetic (dpt) gene cluster. The Red-mediated
recombination system
was employed to introduce linear PCR products of antibiotic resistance genes
flanked by 45 bp
sequences with homology to either upstream or downstream regions of the
interested dpt genes
(as described in Example 2-4). The upstream (5') region of dptBC (pKN24-26) or
dptD
(pKN27) was deleted by the spec-ernzEp * cassette that contains a
spectinomycin resistant gene
(spec) and strong, constitutively expressed errnEp*. This fragment was
amplified using the
primers Sp6De1-1-2 and dptBC-ermEp. The downstream (3') region of dptBC
(pKN24) was
deleted by a beta-lactamase gene (amp, from pBR322). this fragment was
amplified using
primers GTC del2 and DptD-3'::amp.
[0894] The selection cassette for the deletion of the CAT module was amplified
with PCR
primers that carry 50 bp of homology to the linker region of the module under
investigation (see
384
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Figure 5 for positions of linkers; International Patent Application Number WO
01/30985). When
these PCR fragments were introduced into electro-competent cells that
contained pKN24 (a
truncated version of pDA300 that contains only dptBC, which is expressed from
the constitutive
promoter ermEp*, Bibb, MJ. et al., 1985, Gene 38(1-3): 215-26) and induced Red-
system, the
resistance cassette was integrated site specifically at the target site in
pDA300 by homologous
recombination (Figure 3).
5' deletion, 3' deletion
Sp6Del-1-2
5'-GCCAGCATGGAGCCGAACTGCCGGAACACCGCGTCCCGGTCCACCTGTGTAGG
CTGGAGCTGCTTC-3', (SEQ ID NO: 5)
GTC de12
5'-GCCGACTGGGAGTGGGTCAAGTGGCTGCCGCACGTGCTGGATCCGCATATGAATA
TCCTCCTTA-3' (SEQ ID NO: 6)
dptBC-ermEp
5'-CCGAGACAGGCAGGATCTCCTCGACTACCTTCGACGGCGGTTCATATG TCC
GCCTCCTTTGGTCAC-3', (SEQ ID NO: 7)
DptD-3'::amp
5'-CATACTTCCTCTCACTCCGCTGCAGGAGGGACTGCTGTTCCACAGTGTGTAGGCTG
GAGCTGCTTC-3' (SEQ ID NO: 8)
[0895] These cells were then selected for the presence of the tet resistance
marker, and the
resulting colonies were analyzed genetically to validate the construction of
the appropriate
deletion or disruption. Part of the primer design involves placing a
restriction site within the
linker region of interest (Figure 3). Once the deletion BAC was verified, the
selection cassette
was excised using the unique restriction sites incorporated into the linker
regions (Figure 3 AvrII
and Pmel).
[0896] The replacement modules (Serine; Alanine),were subcloned into pBR322
(Yanisch-
Perron et al., 1985, Gene 33(1): 103-19; flanked by appropriate sites) again
using the Red-
mediated recombination. This technique is referred to as gap-filling, where
the primers include
the 50 bp overlap with the regions inside the linkers of the desired module
(Lee, EC. et al., 2001,
Genomics 73: 56). The primers were used to amplify a part of pBR322, including
the origin of
replication and ampr to generate a linear fragment flanked by the regions of
homology inside the
385
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
desired module (Serine; Alanine). These PCR fragments were introduced into
DH10B electro-
competent E. coli cells containing pKN24 (see above) and pKD78 (Datsenko, KA.,
and Wanner,
BL., 2000, Proc. Nat Acad Sci U.S.A. 97: 6640). Once recombination has
occurred through both
regions of homology the module will have been transferred from the original
vector to pBR322,
converting the linear PCR fragment into a circular version that can replicate
and be selected for
(Figure 3). It is preferred if the original vector that the module is cloned
from has an F-plasmid
origin of replication (as opposed to an origin of replication with a higher
copy number).
[08971 The cloned modules are excised from pBR322 and ligated into the deleted
versions of
pKN24 using the compatible restriction sites introduced around the deletion.
This produced 2
plasmids: 1) pDR2155 where the D-serine-11 of daptomycin had been replaced by
D-alanine by
module exchanges and 2) pDR2160 where D-alanine-8 of daptomycin had been
replaced by D-
serine. Both pDR2155 and pDR2160 were confirmed via PCR and sequencing.
[0898] A suitable expression host was then constructed in S. roseosporus for
these plasmids.
A dptB-D mutant KN100 which contains a chromosomal deletion that removes
dptBC, D was
constructed using the techniques described in Example 2-1. Both pKN24 and
pRB04 (a plasmid
constructed in the vector pHM11a which expresses the dptD subunit under the
control of enmE*
constitutive promoter) were added by interspecies conjugation to KN100 strains
to create
KN101. When fermented arid analyzed under the conditions described in Examples
2-2 and 2-3,
this strain was shown to produce the native lipopeptides A21978C1 A21978C2 and
A21978C3.
Once the S. roseosporus KN100 strain had been validated, then a second
derivative was created,
KN156 (KN100 carrying pRB04). This strain was then used as the host for all
module
exchanges perfornled in dptBC. PB103 was constructed by adding pDR2155 to
KN156. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C46, C47 and C48 Figure 4).
[0899] Strain PB118 was constructed by adding pDR2160 to KN156. When fermented
and
analyzed under the conditions described in Examples 2-2 and 2-3, this strain
was shown to
produce compounds C22, C23 and C24 (Figure 4).
[0900] The recombinant strain described above, were fermented and analyzed
under the
conditions described in Example 2-2 and then analyzed using the techniques
described in
Example 2-3 (The data is summarized in Table VI).
[0901] Derivatives having Asn at the position 8 or 11 were prepared by module
exchange
using fusion sites TC (B) and TE (CAT). The Red-mediated recombination system
was used to
386
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
replace module 8 or 11 on pKN24 by gentamycin resistance gene (ahp2) (Chow JW,
Kak V, You
I, Kao SJ, Petrin J, Clewell DB, Lemer SA, Miller GH, Shaw KJ. 2001,
Antimicrob. Agents
Chemother. 45, 2691-2694) flanked by engineered AvrII and Pmel restriction
sites. Since the
DNA sequences of the module 8 and 11 are highly homologous, the same primer
pair was used
for deletion of the two modules at the linkers B and CAT.
[0902] A DNA fragment coding for an Asn module (B-CAT), the 11th module from
A54145
NRPS was cloned by the gap-repair method. Gap-repair primers were used for PCR
amplification of a portion of pBR322 including amp resistance gene and origin
of replication to
generate a linear fragment flanked by incorporated Nhel and Hpal restriction
sites and 45 bp
with homology inside the desired module fragments. The linear PCR fragment was
transformed
into electro-competent E. coli carrying SF1:10D08 (a BAC clone of >100 kb DNA
encoding
parts of the A54145 biosynthetic gene cluster.(This clone was derived from a
genomic BAC-
based library of S. fradiae that was constructed using the protocols described
in Miao et al.,
2005, Microbiology 151: 1507-1523. Clone BAC-P13 was isolated from the library
using the
protocols in Miao et al., 2005, Microbiology 151: 1507-1523) and tetR- pKD119
(Datsenko,
KA., and Wanner, BL., 2000, Proc. Nat Acad Sci U.S.A. 97: 6640) coding for the
Red
recombination system.) Once the Red-induced recombination occurs at both
homologous
regions; the Asn module was transferred from BAC-P13 to the linear pBR322 to
generate a
circular and replicated plasmid. Module fragments with correct sequences (as
verified by
sequencing) were excised by Nhel and Hpal digestion and used for ligation with
appropriate
deleted pKN24 versions to generate hybrid plasmids.
[0903] Primers for deletion of module 8
pKN24-Mod8::Gen. B-CAT
8B
TTGTTCGAGGCGCCGACGGTGAGCCGTTTGGAGCGGTTGCTGCGGGAGCGCCTAGG
ACGTTGACACCATCGAATGG. (SEQ ID NO: 9)
8 CAT-Pnze
ACAATCTCAGCACCCCCCACCACACCAACCGCCCCAGCGTCCGAACCACGTTTAAAC
CCTCATTCATCGGGCGAAAG (SEQ ID NO: 10)
[0904] Primers for deletion of module 11
387
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
pKN24-Mod11::Gen. B-CAT
8 B
TTGTTCGAGGCGCCGACGGTGAGCCGTTTGGAGCGGTTGCTGCGGGAGCGCCTAGG
ACGTTGACACCATCGAATGG (SEQ ID NO: 11)
8 CAT-Pme
ACAATCTCAGCACCCCCCACCACACCAACCGCCCCAGCGTCCGAACCACGTTTAAAC
CCTCATTCATCGGGCGAAAG (SEQ ID NO: 12)
[0905] 'Primers for gap-repair of lpt4sn11.
LptN11 B P13
TCGGGGCGCGGGTCGGCGGGGCGCAGCCGGGGTCCGGCCTCGCCC
GCTAGCTTCTTAGACGTCAGGTGGCAC (SEQ ID NO: 13)
Lpt-N11-CAT-P14
CGCGACATCTTCGAACAGCGCACGCCCGCCGCCCTCGCCGGCCGC
GTTAACCGATACGCGAGCGAACGTGA (SEQ ID NO: 14)
[0906] Plasmids were screened by PCR and restriction digests, plasmids with
the correct
genotype were then designated as pKN45 (D-Asn module inserted at position 8)
and pKN47 (D-
Asn module inserted at position 11). These two plasmids were then conjugated
into the
expression host KN156 (AdptBCD + pRB04). Exconjugants were selected on ASI
plates
containing apramycin (50 g/mL) and recombinant strains selected from these
plates were then
fermented and analyzed using the protocols described in Example 2-2 and 2-3.
Novel
lipopeptides C189, C190 and C191 with molecular weights consistent with the
insertion of Asn
at position 8 in A21978C1,2,3 were detected by LC-MS from the fermentation
broth of KN392
(see table VI). Novel lipopeptides C233, C234 and C235 with molecular weights
consistent with
the insertion of Asn at position 11 in A21978C1,2,3 were detected by LC-MS
from the
fermentation broth of KN404 (see table VI).
388
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Table VI - Data from module exchanges at position 8, 11
Dpt
Replacement amino 5' 3'
amino Results
acid acid (source pathway) linker linker
#
Lipopeptide with molecular
mass of 1650 (compound
D-Ala-8 D-Ser (dpt) T-C# T-E# C22), 1664 (compound
C23) and 1678 (compound
C24) detected.
Lipopeptide with molecular
mass of 1677.72 (compound
D-Asn-8 D-Ser (dpt) T-C# T-E# C189), 1691.75 (compound
C190) and 1705.78
(compound C191) detected.
Lipopeptide with molecular
mass of 1618 (compound
D-Ser-1 1 Ala (dpt) T-C# T-E# C46), 1632 (compound
C47) and 1646 (compound
C48) detected.
Lipopeptide with molecular
mass of 1661.72 (compound
D-Asn-11 Ala (dpt) T-C# T-E# C243), 1675.75 (compound
C244) and 1689.78
(compound C245) detected.
(# See Figure 5 for positions of T-C and T-E)
[0907] Once the presence of the expected mass ions was confirmed PB 103, PB
118, KN392
and KN404 were fermented at large scale and compounds C22, C46, C189, C233
were purified
using the techniques described in Example 2.5.
389
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
Example 2-7: Module exchanges at position 13 in dptD
[0908] Module exchanges were constructed at position 13 in the dpt cluster to
replace
kynurenine. These constructs were made in the subunit expression plasmid pRB04
(a plasmid
constructed in the vector pHMl la which expresses the dptD subunit under the
control of ermE*
constitutive promoter described in Example 2-5). A unique AvrII site was
introduced inside the
T-C linker. A second unique PmeI was introduced just downstream of the coding
region of
dptD. This allowed the terminal module for Kyn to be removed from dptD along
with the
thioesterase. Two replacement modules containing the domain arrangement CATTe
were
prepared as fragments flanked by AvrII and Pmel sites. The isoleucine and
tryptophan modules
were responsible for the incorporation of the terminal amino acids in the
A54145 (Ile) and CDA
(Trp) pathways. After cloning the replacement modules into the deleted pRB04
the hybrid
constructs were introduced into a dptD deleted S. roseosporus, and
fermentation and analysis
were completed using the techniques described in Example 2-1. This data is
summarized in
Table VII.
Table VII - Data from module exchanges at position 13
Replacement
Dpt
amino acid 5' 3'
amino Results
(source acid # linker linker
pathway)
Lipopeptide with molecular mass
Trp (CDA) Kyn-13 T-C# 3' of of 1630 (compound Cl), 1644
dptD (compound C2) and 1658
(compound C3) detected.
Lipopeptide with molecular mass
Ile (A54145) Kyn-13 T-C4 3' of of 1557 (compound C4), 1571
dptD (compound C5) and 1585
(compound C6) detected.
#Linkers are defined in Figure 5
390
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0909] Example 2-8: Deleting the dptl gene fi~om Daptornycin NRPS Gene Cluster
results in
the production of lipopeptides with glutamate at position 12
[0910] Sequence comparisons between the dptl, lptI and glmT genes suggested
that dptl may
play a role in the methylation of the glutamate in position 12 (the glmT gene
product is believed
to methylate the glutamate in a similar position in the related lipopeptide
CDA; the lptl gene
product is believed to methylate glutamate in the synthesis of A54145). To
test this theory a
deletion was created in the dptl gene in S. roseosporus UA431 containing
pDA300. A deletion
plasmid was constructed that contained 2xlkb fragments that flanked dptl
upstream and
downstream. These fragments were ligated in such a way that they would create
an in-frame
deletion of dptl. This cassette was cloned into pRHB538 (see Example 2-1) and
introduced in S.
roseosporus UA431/pDA300. Under the appropriate selection conditions (see
Example 2-1) the
deletion cassette was exchanged for the dptl gene on the chromosome, thus
constructing an in-
frame deletion of dptl. The genotype of this mutant was confirmed by PCR and
Southern blots.
This mutant was fermented and analyzed using the techniques described in
Example 2-1. The
results of this analysis were that these strains were only able to produce
lipopeptides with masses
of 1620, 1634 and 1648, which corresponded to the predicted masses for
lipopeptides that
contain glutamate at position 12 instead of 3-methyl-glutamate: compound ClO,
compound C11
and compound C12 respectively. From this data it was concluded that dptl plays
a role in the
methylation of glutamate during the synthesis of daptomycin.
[0911] Example 2-9: Construction of a conabinatorial library of novel
lipopeptides from
recombinant Streptomyces roseosporus
[0912] Successful module exchanges produced from Example 2-5 were further
enhanced by
combining pDR2155 and pDR2160 with subunits exchanges for dptD that could
include lptD
(the terminal subunit from the S. jradiae A54145 biosynthetic pathway that
encodes for 3MeGlu
and Ile/Val cloned in an expression plasmid, supra) and cdaPS3 (the terminal
subunit from the S.
coelicolor calcium dependent antibiotic, CDA, biosynthetic pathway that
encodes for 3MeGlu
and Trp cloned in an expression plasmid, supra). These combinations were
further enhanced by
being expressed in hosts that contain a dptl (a putative methyl-transferase
involved in the
methylation of glutamate at position 12 of daptomycin) deletion which will
lead to the inclusion
of glutamate at position 12 instead of 3-methyl-glutamate.
One or more of the methods described above:
391
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
1. module exchanges to effect alterations at positions 8 and 11,
2. dptl deletion to effect alterations at position 12, and
3. subunit complementation to effect alterations at position 13
were combined to construct combinatorial libraries that contained 48 novel
lipopeptides.
[0913] In addition to the construction of KN100 described in Example 2-5 a
second S.
roseosporus mutant was constructed, designated KN125 (using the techniques
described in
Example 2-1) that contained a chromosomal deletion that removes dptBC, D, G,
H, I, J. After
KN125 was confirmed as a null mutant it was used to construct KN159 by adding
pKN24 and
pRB04 to KN125. When fermented and analyzed under the conditions described in
Examples 2-
2 and 2-3, this strain was shown to produce compounds C10, C11 and C12 which
all lack the
methyl group on glutamate 12 seen in A21978C.
[0914] Strain KN107 was constructed by adding pKN24 and pMF23 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds Cl, C2 and C3.
[0915] Strain KN110 was constructed by adding pKN24 and pMF30 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C4, C5, C6, C7, C8, and C9.
[0916] Strain KN160 was constructed by adding pKN24 and pMF23 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C13, C14 and C15.
[0917] Strain KN161 was constructed by adding pKN24 and pMF30 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C16, C17, C18, C19, C20, and C21.
[0918] The combinatorial approach described above, was then enhanced by the
addition of
the modified dptBC constructs in pDR2155 and pDR2160. Strain PB 105 was
constructed by
adding pDR2155 and pMF23 to KN100. When fermented and analyzed under the
conditions
described in Examples 2-2 and 2-3, this strain was shown to produce compounds
C49, C50 and
C51.
[0919] Strain PB108 was constructed by adding pDR2155 and pMF30 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C52, C53, C54, C55, C56, and C57.
392
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0920] Strain PB110 was constructed by adding pDR2155 and pRB04 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C58, C59 and C60.
[0921] Strain PB113 was constructed by adding pDR2155 and pMF23 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C61, C62 and C63.
[0922] Strain PB 116 was constructed by adding pDR2155 and pMF30 to KN125.
When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C64, C65, C66, C67, C68, and C69.
[0923] Strain PB120 was constructed by adding pDR2160 and pMF23 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C25, C26 and C27.
[0924] Strain PB123 was constructed by adding pDR2160 and pMF30 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C28, C29, C30, C31, C32, and C33.
[0925] Strain PB128 was constructed by adding pDR2160 and pRB04 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C34, C35 and C36.
[0926] Strain PB 13 0 was constructed by adding pDR2160 and pMF23 to KN 125.
When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C37, C38 and C39.
[0927] Strain PB131 was constructed by adding pDR2160 and pMF30 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C40, C41, C42, C43, C44, and C45
[0928] Strain KN393 was constructed by adding pKN45 and pMF23 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C198, C199 and C200.
[0929] Strain KN394 was constructed by adding pKN45 and pMF30 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C201, C202, C203, C210, C21 1, and C212.
393
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
[0930] Strain KN395 was constructed by adding pKN45 and pRB04 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C192 C193 and C 194.
[0931] Strain KN396 was constructed by adding pKN45 and pMF23 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C195, C196 and C197.
[0932] Strain KN397 was constructed by adding pKN45 and pMF30 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C204, C205, C206, C207, C208, and C209.
[0933] Strain KN405 was constructed by adding pKN47 and pMF23 to KN100. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C224, C225 and C226.
[0934] Strain KN406 was constructed by adding pKN47 and pMF30 to KN100. When
fennented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C221, C222, C223, C213, C214, and C215.
[0935] Strain KN407 was constructed by adding pKN47 and pRB04 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C230, C231 and C232.
[0936] Strain KN408 was constructed by adding pKN47 and pMF23 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C227, C228 and C229.
[0937] Strain KN409 was constructed by adding pKN47 and pMF30 to KN125. When
fermented and analyzed under the conditions described in Examples 2-2 and 2-3,
this strain was
shown to produce compounds C72, C219, C220, C216, C217, and C218
[0938] Example 2-10 Module exchanges constructed at positions 2 through 4 in
daptomycin
[0939] Multiple module exchanges were performed in either dptA, dptBC or a
combination
of dptA and BC in order to complete these exchanges a new expression plasmid
was needed as
pKN24 only contained dptBC. The new vector, pKN18, was a truncated product of
B12:03A05
(a BAC clone that contains entire daptomycin biosynthetic pathway, see Example
2-1) that was
able to express both dptA and dptBC. Plasmid pKN18 was constructed by
truncating
394
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
B12:03A05using the Red-mediated recombination (see Example 2-5) system through
two
sequential deletions of B 12: 03A05. The two deletions deleted all of genes
upstream of dptR and
all of the genes downstream of dptBC (pKN18 carries the locus dptR-drrAB-
dptEFABC).
Firstly, the upstream (5') region of the locus (insert coordinate 0.552 kb-
45,576 kb on
B12:03A05, see table VI for primers) was deleted by spectinoinycin resistance
gene. The region
downstream (3') of dptBC (insert coordinate 91,093 kb -127,392 kb, see table V
for primers)
was deleted by amp gene.
[0940] In order to combine module exchanges in dptA,BC with subunit swaps for
dptD (see
Example 2-6) and peptide tailoring methyl transferase dptl (see Example 2-8)
it was necessary to
construct a new expression plasmid that could express the dptlJ genes in the
dpt deletion host
UA431 (see Example 2-1) with the modified pKN18 plasmids. In order to express
a glutamate
methyltransferase in UA431 (AdptE-.I), pKN54, a plasmid that carries strong
promoter permEp*
and functions for integration on chromosome from phi-BT1 phage was constructed
based on
kanR pRT802 (Gregory, M.A.; Till, R.; Smith, M.C.;.2003. J Bacterio1.185: 5320-
5323.). The
1.8 kb BglII/Smal fragment from pHMl 1 a which carries ermEp* and a
transcriptional terminator
(.Integrative vectors for heterologous gene expression in Streptomyces spp.
Motamedi, H;
Shafiee, A; Cai, SJ; 1995, Geine.,160: 25-3 1) was cloned at BamHUEcoRV sites
ofpRT802
(Gregory, M.A.; Till, R.; Smith, M.C.;.2003. J Bacteriol. 185: 5320-5323),
which encodes for '
phi-BT1 integration system. The plasmid was multiplied in selective medium
with kanamycin
(50 Rg/mL).
[0941] A DNA fragment'coding for both dptl and dptJwas PCR amplified using
B12:03A05
as the template. Two primers (with engineered restriction sites underlined)
dptJ-C-HindIIl: 5'-
GGCGGAAGCTTACGGCACGGCAAGGCCGTTTC-3' (SEQ ID NO: 15) and dptI-N-Ndel:
5'-GGCGGCATATGACCGTGCACGACTACCAC-3' (SEQ ID NO: 16) were used for the PCR
amplification. The PCR fragment was cloned on pKN54 at NdeI and HindIII sites
to generate
pKN55.
[0942] Finally, a series of expression hosts were created that would be used
for the multi-
modular exchanges described in this Example. KN576 was constructed by
introducing pRB04
(expresses dptD from minicircle integration sites, see Example 2-6) into UA431
(AdptE-J).
KN580 was constructed by introducing pRB04 (see Example 2-6) and pKN55 (dptlJ
expressed
from phi-BT1 integration sites) into UA431 (AdptE-J). KN577 was constructed by
introducing
pMF30 (expresses lptD from minicircle integration sites, see Example 2-6) into
UA431 (AdptE-
395
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
J). KN587 was constructed by introducing pMF30 (see Example 2-6) and pKN55
into UA431
(AdptE J).
Sp6 del3
5'GCATCCGATGCAAGTGTGTCGCTGTCGACGGTGACCCTATAGTCGTGTAGGCTGGA
GCTGCTTC (SEQ ID NO: 17)
Sp6 del4
5'-CCGAGGAAAAGAGGGAACGGGACAGGTCAGTGACCGGCGACCGTGCATATGAAT
ATCCTCCTTA-3' (SEQ ID NO: 18)
DptD-3'::amp
5'-CATACTTCCTCTCACTCCGCTGCAGGAGGGACTGCTGTTCCACAGTGTGTAGGCTG
GAGCTGCTTC-3' (SEQ ID NO: 19)
GTC de12
5'-GCCGACTGGGAGTGGGTCAAGTGGCTGCCGCACGTGCTGGATCCGCATATGAATA
TCCTCCTTA-3' (SEQ ID NO: 20)
[0943] Multi module exchanges were completed on pKNl8 using the Red-mediated
recombination system to change several amino acid residues on the daptomycin
core
simultaneously. First, the genR (Wohlleben, W. et al., 1989, Mol. Gen. Genet.
217: 202-208)
gene was introduced into pKNl8 to replace the DNA fragment coding for modules
2-3-4 (2-4),
between the linker regions B and CAT (exchanges 2-4). The genR gene was then
removed by
AvrlUPmel digest.
[0944] DNA fragments coding for modules 2-4, from the A54145 pathway were
cloned onto
pBR322 by the gap-repair method as described for single module exchange in
Example 2-5.
This fragment was excised by Nhel and HpaI digests and ligated to the deleted
pKN18 to
generate pKN51 (carries D-Glu at position 2 and Asn at position 3 in
daptomycin). This
plasmid, pKN51, was introduced into expression hosts: KN576 to produce KN630,
KN580 to
produce KN631, KN577 to produce KN632 and KN587 to produce KN633 via
recombination.
These recombinant strains were fermented and analyzed using the techniques
described in
Example 2-2 and 2-3. The fermentation broth of KN633 was the only strain to
contain mass ions
396
CA 02587848 2007-05-10
WO 2006/110185 PCT/US2005/040919
consistent with the production of C259, C260, C261, C262, C263 and C264. LC/MS
analysis of
the fermentation broths from the other strains KN630, KN631 and KN632 did not
reveal the
presence of any novel lipopeptides.
[0945] Primers for deletion of
dpt2-4.
dpt-Asn2-Del-B:
GTTCGCCTTCCCCACCGTCGCCGGCCTTCTCCCGCTCCTGGACGACAA
CCTAGGTGTGTAGGCTGGAGCTGCTTCG (SEQ ID NO: 21)
dpt-Thr4-Del-CAT:
TCAGGGCGCCGGTCGATCCTGGTCACAGGTGGCAGGGCGGTGCCGG
GTTTAAACCATATGAATATCCTCCTTA (SEQ ID NO: 22)
[0946] Primers for gap repair cloning
lpt2-4
LptGlu2-Pickup-B:
5' TCC GGG CGG GGC CGG ACG GGA CGG ACG TGG TCG TCC GGC ACG GCC
GCTAGCTTCTTAGACGTCAGGTGGCAC 3' (SEQ ID NO: 23)
lpt-Thr4-pickup-CAT:
5' TTC GAG GCG CCC ACG CCC GCC GCG CTG TCC CGG CGC CTC GACACC
GTTAAC CGATACGCGAGCGAACGTGA 3' (SEQ ID NO: 24)
[0947] Example 2-11 Module exchanges constructed at positions 8 through 11 in
daptornycin
[0948] A daptomycin derivative containing 2 changes at positions 8 and 11 was
generated
using the Red-mediated recombination system as described in Example 2-5.
Briefly, a DNA
fragment coding for 4 modules (D-Ala8-Asp9-GlylO-D-Ser11) was deleted from
pKN24 by a
gentamycin resistance gene franked by Avrll and PmeI restriction sites. The
genR gene was then
removed by AvrIl/Pmel digest.
[0949] The corresponding DNA fragment coding for module 8-9-10-11 (D-Lys-Asp-
Gly-D-
Asn) from A54145 BAC-P13 that was subcloned on pBR322 by the gap-repair method
(Example
397
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 397
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 397
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: