Note: Descriptions are shown in the official language in which they were submitted.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
THIAZOLOPYRIDINONE DERIVATES AS MCH RECEPTOR ANTAGONISTS
Fxeld of InFentiuA
The present invenLicsn is in the field of viedic}n% parlicu,Wly ilti the
twatrmnt of
s obesity and discascs causcd by or exacerbated by obesity. More spec'-
f'ically, the pre,wut
iavention relates to antagonists +af ,nelftnin concentrating honnone useful in
fihc
prevention nd troatrrxnt of obesity and related diseases.
Background of the Invetttiors
The affluence of the 19Ws Wcmg with the exponantxe1 inctem in fwd pTuductfon
parficulatly x,n Westem and Asian tooaoniies has rmlW in feeding patwns that
lead to
obesity. Obesity is defined as being excessively overwui&t. Excessive weight
is
generaly eharacterize'd by cxcrssive body fat, because unused energy is stored
io the
adipose iissueg as faL
1s Obesity has associated with it, twiaomzc and saciW costs. Obese people, an
inmasing proportion qf deve,loped and developing smieties. am regarded as
having out
of contml feeding habits often associated with low self-esmern. Mareaver,
obese persons
are mom likely to have medical pmbiams associated w9th or exacerbated by tle
extess
body wexgiat Examptes of medical conditions caused, exace"ed or triggered by
excesiye weight include bone Oaccun~.et pains in the kne+e joints, arfttas,
ancreased risk
of hypcrt,cns,im $ttxerasclexosis, stmke, diabetes, ccc,
Melanin cvncentraating hanmae (NiCH) is oL 19 amin4 acid nouroMkide produced
in the lawral hypothalaruie a= and zona xnca-ta. Although. MCH-expressing
neurons
ptoject to nutxtemus mgi.ons of the b,raixz. MCH is processed from a]$rger pm-
pro]xarmcane
that also includes a s=nd peptl~~, NEI, and possibly a tliird, N'GS (Nahon,
Crit. Rev in
Ncumbiolagy, 8:221-262, 1994). MCH mediates its effects flue+mgh at least two
t,~
protein-coupled receptors, ?+vlCHRl, and M.CM (Saito ot al.l+latitte 400: 265-
269, 1999;
3 o IT'i11 et al., J Diuf Chem. 276: 20125-20129, 2001). Both reteptrars are
expressd in
regions of the brain consistent with MCH neuronal projection and known MCH
pbysiolagxc fitoctaon [Hervieu ot al.,.Euf I T+1'enr+aswience .1~:. 7194-1216,
2000; Will et al.,
RECTIFIED SHEET (RULE 91)
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
2
J Biol Chem. 276: 20125-20129, 2001; Sailer et al., Proc Nat Acad Sci. 98:
7564-7569,
2001).
Extensive evidence exists to support the orexigenic activity of MCH. MCH
mRNA is elevated in rodent models of obesity and in the fasted state (Qu et
al., Nature
380: 243-247, 1996). Intra-cerebroventricularly administered MCH increases
feeding and
blocks the anorexic effect of a-melanocyte stimulating hormone (Ludwig et al.,
Am J
Physiol 274: E627-E633, 1998). MCH knockout mice (MCH4- mice) are lean,
hypophagic
and hypometabolic (Shimada et al., Nature 396: 670-674, 1998), while MCH over-
expressing transgenic mice are obese and insulin resistant (Ludwig et al., J
Clin Invest
107: 379-386, 2001). MCHR1 4- mice have recently been reported to be lean and
hypermetabolic, indicating that the R1 isoform mediates at least some of the
metabolic
effects of MCH (Marsh et al., Proc Nat Acad Sci 99: 3240-3245, 2002).
In addition to its effects on feeding, MCH has been implicated in regulation
of the
hypothalamic-pituitary-adrenal axis through modulation of CRF and ACTH release
(Bluet-Pajot et al., J Neuroendocrinol 7: 297-303, 1995). MCH may also play a
role in the
modulation of reproductive function (Murray et al., J Neuroendocrinol 12: 217-
223, 2000)
and memory (Monzon et al., Peptides 20: 1517-1519, 1999).
The current preferred treatment for obesity as well as Type II non-insulin
dependent diabetes is diet and exercise with a view toward weight reduction
and
improved insulin sensitivity for diabetics. Patient compliance, however, is
usually poor.
The problem is compounded by the fact that there are currently only two
medications
approved for the treatment of obesity (sibutramine, or Meridiam and orlistat,
or
Xenicaff.
PCT application number WO 01/21577 (JPOO/06375) filed September 19, 2000,
discloses compounds reportedly useful as antagonists of the MCH receptor. In
particular
the WO 01/21577 application claims a compound of formula A
Ar' X- r'Y\N-"R 1
2
(A)
wherein:
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
3
Arl is a cyclic group that may have substituents;
X is a spacer having a main chain of 1 to 6 atoms;
Y is a bond or a spacer having a main chain of 1 to 6 atoms;
Ar is a monocyclic aromatic ring which may be condensed with a 4 to 8 membered
non-
aromatic ring, and may have further substituents;
Rl and R2 are independently hydrogen atom or a hydrocarbon group which may
have
substituents;
Rl and R2 together with the adjacent nitrogen atom may form a nitrogen-
containing hetero
ring which may have substituent; R2 may form a spiro ring together with Ar; or
R2,
together with the adjacent nitrogen atom and Y, may form a nitrogen-containing
hetero
ring which may have substituents; or salts thereof.
PCT application number WO 01/82925, filed April 26, 2001, also discloses
compounds reportedly useful as antagonists of the MCH receptor. In particular
the WO
01/82925 application claims a compound of formula B
Ari X- r' \N~R 1
\2
(B)
wherein:
Arl is an optionally substituted cyclic group;
X and Y are independently a spacer having a C1_6 main chain;
Ar is an optionally substituted fused polycyclic aromatic ring;
Rl and R2 are independently hydrogen atom or an optionally substituted
hydrocarbon
group; or alternatively Rl and R2 together with the nitrogen atom adjacent
thereto may
form a nitrogenous heterocycle, or R2 together with the nitrogen atom adjacent
thereto and
Y may-form an optionally substituted nitrogenous heterocycle, or R2 together
with the
nitrogen atom adjacent thereto, Y, and Ar may form a fused ring.
PCT application number WO 01/87834, filed May 15, 2001, also discloses
compounds reportedly useful as antagonists of the MCH receptor. In particular
the WO
01/87834 application claims a compound of formula C.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
4
, / R2
R-X-N B \ Y N
R1
(C)
wherein;
R represents hydrogen, halogen, or an optionally substituted cyclic group; X
represents a
bond or a spacer in which the main chain has one to ten atoms; Y represents a
spacer in
which the main chain has one to six atoms; ring A represents a benzene ring
which may
have other substituents; ring B represents a five- to nine-membered nitrogen
containing
nonaromatic heterocycle which may have other substituents; and Rl and Ra are
the same
or different and each represents hydrogen, an optionally substituted
hydrocarbon group, or
an optionally substituted heterocyclic group, or Rl and R2 may form an
optionally
substituted nitrogenous heterocycle in cooperation with the adjacent nitrogen
atom and R2
may form an optionally substituted nitrogenous heterocycle in cooperation with
the
adjacent nitrogen atom and Y.
DE2502588 describes a compound of the formula:
CH3
R4 A, N. R2
\
R 3
R\IV N
R' R5 R1
Wherein the variables are as defined therein.
PCT International publication WO 03/033476 Al discloses a compound of the
formula (Ia) :
R7)t O
~R8) 3)a M\I/NRiR2
S\Q~ N
/ (R6)"
~
A N R 5
1
(R7)t
comprising a pharmaceutically acceptable salt, solvate, or physiologically
functional
derivative thereof, wherein the variables are as described therein.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Current treatments targeted at obesity have side effects. Examples of such
treatments include various over-the-counter appetite suppressants. These
agents have not
been proven effective for all patients and for sustainable periods of time.
Similarly, the
approved treatments, sibutramine (MeridiaTM) and orlistat (XenicalTM) have
been
5 associated with side effects which may compromise compliance and may
preclude long
term use for sustained weight loss for certain patient populations.
Therefore, there is a need for new and/or improved therapeutically effective
agents
useful as antagonists of melanin concentrating hormone to better control the
dietary
habits, minimize the preponderance of obesity and treat, prevent and/or
ameliorate the
effects of obesity, including for example diabetes.
Summary of Invention
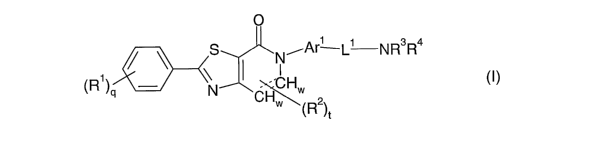
The present invention relates to a compound of formula I:
O
S N-Ar L' NR3R4
,
(R )q N W
CH,,,~ (R2)t
M
wherein:
"----" is optionally a bond to form a double bond
q is 0, 1, 2, or 3; wherein other positions on the phenyl ring have hydrogen
atoms;
t is 1 or 2;
w is 1 or 2 depending on substitution pattern and/or the presence of a double
bond;
Rl is independently selected from the group consisting of hydrogen, Cl-C8
alkyl, C2-C8
alkenyl, C2-C8 alkynyl, halo, hydroxy, Cl-C8 haloalkyl, Cl-C8 alkoxy, -CI-Cg
alkyl
alcohol, Cl-C$ haloalkoxy, aryl, -O-aryl, -O-heteroaryl, -OC1-C8 alkylaryl, -
Cl-C8
alkylaryl, -C1-C8 alkylheteroaryl, heterocyclic, -Cl-C8 alkylheterocyclic,
cycloalkyl, -Cl-
2 5 C8 alkylcycloalkyl, amino, and C1-C8 a1ky1NR6R6', Co-Cg alkylCOOR6, Co C8
alkylCONR6R6';
R2 is independently selected from the group consisting of hydrogen, halo, C1-
C6 alkyl, Cl-
C4 haloalkyl, C2-C4 alkenyl, phenyl, and Cl-C4 alkylaryl;
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
6
Arl is a cyclic group optionally substituted with one to three groups
independently
selected from the group consisting of C1-C8 alkyl, C2-C8 alkenyl, C2-C8
alkynyl, hydroxy,
-OC1-C8 alkyl, Cl-C8 alkylaryl, C1-C8 alkylheteroaryl, phenyl, -0-aryl, -0-
heteroaryl,
heterocyclic, Cl-C4 alkylheterocyclic, cycloalkyl, Cl-C8 alkylcycloalkyl,
cyano, -Cl-C8
alkylNR6R6', C1-C8 haloalkyl, C1-C8 alkyl alcohol, C1-C8 haloalkoxy, halo,
(CH2)nCOR6,
-O(CH2)nCHR 6R6' , NR6SO2R6', (CH2)n NR6SO2R6', and -(CH2)nC(O)NR6R6';
Ll is a bond or a divalent linker selected from the group consisting of C1-C5
alkyl, C2-C5
alkynyl, C2-C5 alkenyl, Co-C5 alkyl-S-Co-C5 alkyl, Co-C5 alkyl-S- C1-C5
alkylhalide, Co-
C5 alkyl-NR6-CO-C5 alkyl, CO-C5 alkyl-NR6- C1-CS alkyl-S- Co-C5 alkyl wherein
each Ll
group has a maximum of 6 carbon atoms in the main chain and wherein each alkyl
is
optionally substituted with 1 to 3 groups independently selected from halo,
cyano, and
hydroxy;
R3 and R4 are independently selected from the group consisting of hydrogen, Cl-
C8 alkyl,
C2-C8 alkenyl, C3-C8 cycloalkyl, aryl, heteroaryl, heterocyclic, Cl-C8
alkylaryl, Cl-C8
alkylcycloalkyl, Cl-C8 alkylheteroaryl, Cl-C4 alkylheterocyclic; wherein each
of the alkyl,
alkenyl, cycloalkyl, aryl, heteroaryl, or heterocyclic group or subgroup is
optionally
substituted with one to three groups independently selected from C1-C8 alkyl,
C2-C8
alkenyl, phenyl, alkylaryl, (CH2)nNSO2CI-Cg alkyl, (CH2)õNSOaphenyl,
(CHZ)nNSO2ary1,
=C(O)Cl-Cg alkyl, COOH, -C(O)OC1-C8 alkyl and Co-C4 a1ky1NR6R6'; and wherein
R3
and R4 optionally combine together with the nitrogen atom to which they are
attached to
form an optionally substituted nitrogen containing 5 to7-member heterocyclic,
or one or
both of R3 and R4 combine with Ll at a position a, (3, y, or, S(e.g. 1, 2, 3,
or 4 positions
adjacent) to the nitrogen of NR3R4 to form a nitrogen containing 5 to7-member
heterocyclic group with Ll said heterocyclic groups optionally having one to
three
substituents independently selected from oxo, hydroxy, cyano, Cl-C8 alkyl, C2-
C8 alkenyl,
C3-C8 cycloalkyl, Cl-C8 alkylaryl, Cl-C8 alkylcycloalkyl, C1-C4
alkylheterocyclic, Cl-C4
alkylheteroaryl, halo, (CH2)õNSO2Cl-C8 alkyl, (CH2)õNSO2phenyl,
(CH2)nNSO2ary1, -
C(O)C1-C$ alkyl, -C(O)OCl-C8 alkyl and Co-C4 alkylNR6R6';
R6 and R6' are independently selected from the group consisting of hydrogen,
C1-C8 alkyl,
phenyl, aryl, C1-C8 alkylaryl, C3-C8 cycloalkyl, or Cl-C6 alkylcycloalkyl; and
wherein R6
and R6' may combine to form a 5-7 member nitrogen-containing heterocycle
optionally
having one to three substituents independently selected from oxo, hydroxy,
cyano, C1-C8
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
7
alkyl, C2-C8 alkenyl, C3-C8 cycloalkyl, Cl-C8 alkylaryl, Cl-C8
alkylcycloalkyl, Ci-C4
alkylheterocyclic, halo, (CH2)nNSO2C1-C8 alkyl, (CH2)õNSO2phenyl,
(CH2)õNSO2ary1, -
C(O)Cl-C$ alkyl, COOH, or -C(O)OCl-C$ alkyl and Co-C4 alkylNR7R8;
R7 and R8 are each independently selected from hydrogen, and Cl-C4 alkyl; n is
an integer
from 0 to 4 wherever it ocurrs; or a pharmaceutically acceptable salt,
solvate, enantiomer,
diastereomer or mixture of or diastereomer thereof.
The present invention also relates to pharmaceutical compositions comprising a
compound of formula I.
In another embodiment, the pharmaceutical composition of the present invention
may be adapted for use in treating obesity and related diseases.
The present invention also relates to a method for treating and/or preventing
obesity in a patient in need thereof, wherein such treatment comprises
administering to
said patient a therapeutically effective amount of a compound of formula I in
association
with a pharmaceutically acceptable carrier, diluent or excipient.
The present invention also relates to a method for antagonizing the binding of
MCH to MCH receptors for the treatment of diseases caused, or exacerbated by
melanin
concentrating hormone.
The present invention provides the use of a compound of formula I as an
appetite
suppressant and/or as a weight loss agent.
The present invention is related to the use of a compound of formula I for the
manufacture of a medicament for treating obesity and related diseases.
Detailed Description
For the purposes of the present invention, as disclosed and/or claimed herein,
the
following terms are defined below.
The term "main chain" as used herein describes the number of atoms in the
shortest distance between two ends of a variable or radical or linker and
includes the
distance in number of atoms when traversing a straight chain, branched chain
or atoms in
a mono or bicyclic ring from one end of the variable or radical to the other.
As used
herein the radical or group -CH2CHaOCH2CH(CH2CHZCH3)CH2- has a chain length of
6.
General chemical terms used in the description of compounds herein described
bear their usual meanings. For example, the term "Cl-8 alkyl," or "(Cl-
C8)alkyl" or "Cl-
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
8
C8 alkyl" or as indicated refers to a straight or branched aliphatic chain of
1 to 8 carbon
atoms including but not limited to methyl, ethyl, propyl, iso-propyl, n-butyl,
pentyl, and
and the like as indicated. Unless otherwise stated, the term "alkyl" means Cl-
C8 alkyl.
Similarly, the term "Co-C8 alkyl" implies an alkyl group as indicated wherein
when the
term Co applies, the alkyl group is not present, and the remaining groups
attach directly to
the substrate. For example, the group -Co-CB alkylCONR10Rll implies that when
Co
applies, the group -Co-C8 alkylCONRlORII becomes to -CONR1oR11
The invention also contemplates that the term C1-C6 alkyl or C2-C6 alkenyl or
similar terms encompass the specified alkyl or alkenyl or similar group, which
may be
chiral, regio or steroisomeric. Such chiral or regio or stereoisomeric groups
are also
objects of the present invention.
The term "C3-C8 cycloalkyl" ? as used herein refers to a cyclic hydrocarbon
radical
or group having from 3 to 8 carbon atoms and having no double bonds. Examples
of C3-
C8 cycloalkyl groups include but are not limited to cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, cycloheptyl, and cyclooctyl.
The term "C3-Cg cycloalkenyl" as used herein refers to a cyclic hydrocarbon
radical or group having from 3 to 8 carbon atoms and having from 1 to 3 double
bonds.
Specific examples of C3-C8 cycloalkenyl include cyclopropenyl, cyclobutenyl,
cyclopentenyl, cyclohexenyl, cycloheptenyl, and cyclooctenyl.
The term "halo" means halogens including iodo, chloro, bromo and fluoro.
The term "Cl-C4 haloalkyl" refers to a Ci-C4 alkyl (or as indicated) group
substituted with one, two three or more halogen atoms as possible and
chemically
appropriate. Examples of C1-C4 haloalkyl include but are not limited to
trifluoromethyl,
chloroethyl, and 2-chloropropyl. Similarly, a"C17Cg haloalkyl" group is a C1-
C8 alkyl
moiety substituted with up to six halo atoms, preferably one to three halo
atoms.
A"Cl-C$ alkoxy" group is a C1-C8 alkyl moiety connected through an oxy
linkage. Examples of alkoxy groups include but are not limited to methoxy,
ethoxy,
propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, pentyloxy, and hexyloxy.
The term "haloalkoxy", "C1-C$ haloalkyloxy", -OC2-C8 haloalkyl" or
"halogenated C1-C8 alkoxy" means an alkoxy group having halogen substituents
at one or
more carbon atoms of the group. The term encompasses groups including for
example,
difluoromethoxy, trifluoromethoxy, 2-haloethoxy, 2,2,2-trifluoroethoxy, 4,4,4-
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
9
trifluorobutoxy, up to and including the like groups having the indicated
number of
carbon atoms.
The term "cyclic" as used herein refers to substituted or unsubstituted
aromatic
(including heteroaromatic) and non-aromatic, carbocyclic or heterocyclic ring
structures.
Cyclic groups may also be monocyclic or bicyclic unless otherwise specified.
Aromatic
groups include, for example, benzene, thiophene, furan, pyrrole, imidazole,
pyrazole,
thiazole, isothiazole, oxazole, isoxazole, pyridine, pyrimidine, pyrazine,
pyrimidine,
pyridazine, napthyl, 1,2,4-oxadiazole, 1,3,4-oxadiazole, 1,2,4,-thiadiazole,
1,3,4-
thiadiazole, pyrrolidine, imidazoline, imidazolidine, pyrazoline,
pyrazolidine,
tetrahydrothiazole, tetrahydroisothiazole, tetrahydrooxazole,
tetrahydroisoxazole,
piperidine, tetrahydropyridine, dihydropyridine, piperazine, morpholine,
thiomorpholine,
tetrahydropyrimidine, tetrahydropyridazine, and hexamethyleneimine. Examples
of
bicyclic groups within the ambit of cyclic groups as used herein include
benzofuran,
benzimidazole, benzoxazole, benzothiophene, benzothiazole, benzisothiazole,
naphtho[2,3-b]thiophene, naphthyl, isoquinoline, quinoline, indole, indazole,
quinoxaline,
phenanthridine, phenothiazine, phenoxathlin, phenoxazine, naphthylidene,
quinazoline,
carbazole, (3-carboline, acridine, phenazine, phthalimide, and thioxanthene
each of which
may be optionally substituted. Cyclic groups as defined by Arl are optionally
substituted
with one to five groups independently selected from Cl-C8 alkyl, C2-C8
alkenyl, C2-C8
alkynyl, hydroxy, Cl-C$ alkoxy, Cl-C8 alkylaryl, phenyl, -0-aryl, heteroaryl,
cycloalkyl,
C1-C8 alkylcycloalkyl, cyano, -(CH2)nNR6R6', Cr-C8 haloalkyl, -OCl-C8
haloalkyl, halo,
(CH2)õCOR6, (CH2)õ NR6SO2R6, -(CH2)õC(O)NR6R6, heterocyclic, and Cl-C8
alkylheterocyclic; wherein the cycloalkyl, phenyl, aryl, and heterocyclic
substituents are
each optionally substituted with one to three groups independently selected
from hydroxy,
Cl-C$ alkoxyalkyl, Cl-C8 haloalkoxy, C1-C8 alkyl, halo, Cj-C8 haloalkyl,
nitro, cyano,
amino, carboxamido, phenyl , aryl, alkylheterocyclic, heterocyclic, and oxo.
The term "alkylcycloalkyl" as used herein refers to an alkyl group on which a
cycloalkyl group is substituted. Exemplary of alkylcycloalkyl groups are
methylcyclopropyl, methylcyclohexyl, methylcycloheptyl, ethylcyclopropyl, etc.
The
alkylcycloalkyl group may optionally be substituted with one to five groups
independently
selected from Cl-C8 alkyl, phenyl, aryl, halo, amino, alkylsulfonyl, alkyl
sulfonamide,
haloalkyl, carboxyalkyl, carboxamide, alkoxy, and perfluoroalkoxy.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
The term "optionally substituted" as used herein and unless otherwise
specified,
means an optional substitution of one to five (or as specified), preferably 1
or 2 groups
independently selected from halo, hydroxy, oxo, cyano, amino, alkylamino,
nitro, phenyl,
benzyl, aryl, -0-aryl, triazolyl, tetrazolyl, 4,5-dihydrothiazolyl, Cl-C6
alkyl, Ci-C4
5 haloalkyl, -(CH2)nNR6R6', C1-C8 haloalkyl, Cl-C8 haloalkoxy, (CH2)õCOR6,
(CH2)n
NR6SO2R6', -(CH2)õC(O)NR6R6', heterocyclic, and C1-C8 alkylheterocyclic on the
subject
group, subgroup, or substituent and wherein R6 , R6' and n are as defined
herein.
The term "heterocycle" or "heterocyclic" represents a stable, saturated,
partially
unsaturated, fully unsaturated, or aromatic 4, 5, or 6 or 7 membered ring or
as otherwise
10 specified. Such heterocyclic ring has from one to three heteroatoms that
are
independently selected from the group consisting of sulfur, oxygen, and
nitrogen. The
heterocycle may be attached at any point which affords a stable structure.
Representative
heterocycles include 1,3-dioxolane, 4,5-dihydro-lH-imidazole, 4,5-
dihydrooxazole, furan,
imidazole, imidazolidine, isothiazole, isoxazole, morpholine, oxadiazole,
oxazole,
oxazolidinedione, oxazolidone, piperazine, piperidine, pyrazine, pyrazole,
pyrazoline,
pyridazine, pyridine, pyrimidine, pyrrole, pyrrolidine, tetrazole,
thiadiazole, thiazole,
thiophene and triazole.
The heterocyclic group or heterocyle according to the present invention unless
otherwise indicated is optionally substituted with one to three, preferably
one or two
groups independently selected from oxo, hydroxy, cyano, Cl-C8 alkyl, C2-C8
alkenyl, C3-
C8 cycloalkyl, C1-C$ alkylaryl, C1-C8 alkylcycloalkyl, Cl-C4
alkylheterocyclic, Cl-C4
alkylheteroaryl, halo, (CH2)nNHSO2C1-C8 alkyl, (CH2)nNHSO2phenyl,
(CH2)õNHSO2aryl,
-C(O)Cl-C8 alkyl, -C(O)OC1-C8 alkyl and Co-C4 alkylNR6R6' wherein R6, R6'and n
are
as defined herein.
The term "alkylheterocyclic" as used herein refers to an alkyl group further
substituted with a heterocyclic group. Examples of alkylheterocyclic include
but are not
limited to 2-methylimidazoline, N-methylmorpholinyl, N-methylpyrrolyl and 2-
methylindolyl.
The term "nitrogen containing heterocyclic" means a heterocyclic ring having
at
least one nitrogen and include heterocyclic groups optionally having in
addition to a
nitrogen atom one or more of oxygen and sulfur atoms.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
11
The term "oxo" as used herein implies an oxygen atom attached to a carbon atom
which is part of a ring or a chain to form a carbonyl group.
The term "basic group" refers to an organic radical which is a proton
acceptor.
The term "basic group" also refers to an organic group containing one or more
basic
radicals. Illustrative basic radicals are amidino, guanidino, amino,
piperidyl, pyridyl, etc,
and exclude amides.
The term "suitable solvent" refers to any solvent, or mixture of solvents,
inert to
the ongoing reaction, that sufficiently solubilizes the reactants to afford a
medium within
which to effect the desired reaction.
As used herein, the term "patient" includes human and non-human animals such
as companion animals (dogs and cats and the like) and livestock animals.
Livestock
animals are animals raised for food production. Ruminants or "cud-chewing"
animals
such as cows, bulls, heifers, steers, sheep, buffalo, bison, goats and
antelopes are
examples of livestock. Other examples of livestock include pigs 'and avians
(poultry)
such as chickens, ducks, turkeys and geese. Also included are exotic animals
used in food
production such as alligators, water buffalo and ratites (e.g., emu, rheas or
ostriches). The
preferred patient of treatment is a human.
The terms "treating" and "treat", as used herein, include their generally
accepted
meanings, e.g., preventing, prohibiting, restraining, alleviating,
ameliorating, slowing,
stopping, or reversing the progression or severity of a pathological
condition, or sequela
thereof.
The terms "preventing", "prevention of ', "prophylaxis", "prophylactic" and
"prevent" are used herein interchangeably and refer to reducing the likelihood
that the
recipient of a compound of formula I will incur or develop any of the
pathological
conditions, or sequela thereof, described herein.
As used herein, the term "effective amount" means an amount of a compound of
formula I that is sufficient for treating or preventing a condition, or
detrimental effects
thereof herein described; or an amount of a compound of formula I that is
sufficient for
antagonizing the MCHR1 receptor to achieve the objectives of the invention.
The term "pharmaceutically acceptable" is used herein as an adjective and
means
substantially non-deleterious to the recipient patient.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
12
The term "formulation", as in pharmaceutical formulation, is intended to
encompass a product comprising the active ingredient(s) (compound(s) of
formula I), and
the inert ingredient(s) that make up the carrier, as well as any product which
results,
directly or indirectly, from combination, complexation or aggregation of any
two or more
of the ingredients, or from dissociation of one or more of the ingredients, or
from other
types of reactions or interactions of one or more of the ingredients.
Accordingly, the
pharmaceutical formulations of the present invention encompass any composition
made
by admixing a compound of the present invention and a pharmaceutical carrier,
or a
compound of formula I and a pharmaceutically acceptable co-antagonist of MCHR1
useful for the treatment and/or prevention of obesity or a related disease
where
antagonism of a MCH receptor may be beneficial.
The terms "diseases related to obesity" or "related diseases" as used herein
refer to
such symptoms, diseases or conditions caused by, exacerbated by, induced by,
or adjunct
to the condition of being obese. Such diseases, conditions and/or symptoms
include but
are not limited to eating disorders (bulimia, anorexia nervosa, etc.),
diabetes, diabetic
complications, diabetic retinopathy, sexual/reproductive disorders,
depression, anxiety,
epileptic seizure, hypertension, cerebral hemorrhage, congestive heart
failure, sleeping
disorders, atherosclerosis, rheumatoid arthritis, stroke, hyperlipidemia,
hypertriglycemia,
hyperglycemia, and hyperlipoproteinenamia, stress related disorders including
post
traumatic stress disorder, substance abuse, including alcohol and drug abuse,
and
nonpharamcologic disorders such as gambling, sex and internet related
addictions.
The term "unit dosage form" refers to physically discrete units suitable as
unitary
(i.e. individual, separate or separate able) dosages for human subjects and
other non-
human animals (as described above), each unit containing a predetermined
quantity of
active material/ingredient (compound of formula I) calculated to produce the
desired
therapeutic effect, in association with a suitable pharmaceutical carrier.
Certain compounds of the invention may contain an acidic moiety (e.g.,
carboxylic
acid). Therefore, certain compounds of formula I may exist as a pharmaceutical
base
addition salts or ionic salts.. Such salts include those derived from
inorganic bases such
as ammonium and alkali and alkaline earth metal hydroxides, carbonates,
bicarbonates,
and the like, as well as acid addition salts derived from basic organic amines
such as
aliphatic and aromatic amines, aliphatic diamines, hydroxy alkylamines, and
the like.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
13
Methods of preparing and isolating salts are known to one of skill in the art.
Pharmaceutically acceptable salts and common methodology for preparing them
are well
known to one of skill in the art. See, e.g. P. Stahl, et al. Handbook of
Pharmaceutical
Salts: Properties, Selections and Use (VCHA/Wiley-VCH, 200); S. M. Berge, et
al.,
"Pharmaceutical Salts" Journal of Pharmaceutical Sciences, Vol. 66, No. 1,
January 1977.
Preferred Compounds of the Invention
Certain compounds of the invention are particularly interesting and preferred.
The
following listing sets out several groups of preferred compounds. It will be
understood
that each of the listings may be combined with other listings or groupings
described
herein to create additional groups of preferred compounds.
Preferred Rl Groups
Preferred RI groups are independently selected from the group consisting of
hydrogen, halo, hydroxy, C1-C6 alkyl, C2-C6 alkenyl, Cl-C6 haloalkyl, Cl-C6
alkoxy, C1-
C6 haloalkoxy, C3-C8 cycloalkyl, C3-C8 alkylcycloalkyl, heterocyclic, Cl-C6
alkylheterocyclic, phenyl, benzyl, cyano, and Cl-C4 a1ky1NR6R6', and wherein
each
phenyl, aryl, cycloalkyl or heterocyclic group or subgroup is optionally
substituted with I
to 2 groups independently selected from halo, Cl-C4 alkyl, amino, cyano,
nitro, C1-C6
haloalkyl, or Cl-C6 alkoxy haloalkyl.
Preferred R2 Groups
Preferred R2 groups are independently selected from the group consisting of
hydrogen, or Cl-C6 alkyl.
Preferred Arl
Preferred Arl groups are selected from optionally substituted C3-C8
cycloalkyl,
pyridinyl, indolyl, benzthiazolyl, pyrrolidinyl, imidazolinyl, imidazolidinyl,
pyrazolinyl,
pyrazolidinyl, phenyl, piperidinyl, benzothiophenyl, benzofuranyl, naphthyl,
benzimidazolyl, indolinyl, indazolyl, benztriazolyl, quinolinyl,
isoquinolinyl,
tetrahydroquinolinyl, tetrahydroisoquinolinyl, benzo[1,3]dioxolyl, dihydro-
benzo[1,4]dioxinyl, 3,4-dihydro-2H-benzo[1,4]-oxazinyl, each optionally
substituted with
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
14
1-3 groups independently selected from C1-C6 alkyl, C1-C6 alkylcycloalkyl, C1-
C6
haloalkyl, hydroxy, alkoxyalkyl, cyano, halo, aryl, COOR6, and CONR6R6'.
Particularly
preferred Arl groups include phenyl, indolyl, benzthiazolyl, benzimidazolyl,
benzotriazolyl, imidazolyl, indazolyl, quinolinyl, isoquinolinyl,
tetrahydroquinolinyl,
tetrahydroisoquinolinyl, benzo[1,3]dioxolyl, dihydro-benzo[1,4]dioxinyl, and
3,4-
dihydro-2H-benzo[1,4]-oxazinyl optionally substituted with 1-3 groups
independently
selected from halogen, -OC1-C4 alkyl, Cl-C4 haloalkyl, and -CO-C4 alkylamine.
Preferred Ll Groups
A preferred Ll group is selected from the group consisting of -CH2-, -C(O)-
,CH2CH2-, -CH2CH2CH2-, -CH2CH2Oalkyl, -SCH2CH2-, -OCH2CH2-, -OCH2CH2CH2-,
-O(CH2)3CH2-, -OCH(Et)CH2CH2CH2 , -OCH(iPr)CH2CH2CH2-, -acetylene-CH2-,
-OCH(CH3)CH2CH2SCH2-, -O(CH2)3SCH(CH3)-, -O(CH2)2SCH(CF3)-,
-OCH(CN)CH2CH2-, -NR6CH2CH2-, -NR6CH2CH2CH2-, -NR6(CH2)3CH2-,
-NR6CH(Et)CH2CH2CH2 , -NR6CH(iPr)CH2CH2CHa, -NR6CH(CH3)CH2CH2SCH2-, -
NR6(CH2)2SCH(CF3)-, -OCH(CH3)CH(CH3)-, -OC(CH3)2CH2-, -OCH2C(CH3)2-, -
C(CH3)2CH-2CH2-, and - CH2CH2C(CH3)2-, and -NR6CH(CN)CH2CH2-.
Preferred R3 and R4 Groups
Preferred R3 and R4 groups are independently selected from the group
consisting
of hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C3-C8 cycloalkyl, Cl-C8
alkylcycloalkyl, phenyl,
aryl, C1-C6 alkylaryl, heterocyclic, Cl-C6 alkylheterocyclic, COR6, S02R6 and
(CH2)nSO2R6.
Also preferred are R3 and R4 groups which combine with each other and the
nitrogen atom to which they are attached to form an optionally substituted 5-7
member
heterocyclic ring; or where one or both of R3 and R4 combine with Ll at a
position a, (3, or
,y to the nitrogen of NR3R4 to form an optionally substituted heterocyclic
group selected
from the group consisting of optionally substituted morpholino,
thiomorpholino, pyrrole,
2H-pyrrole, 2-pyrroline, pyrrolidine, oxazole, oxadiazolyl, thiazole,
imidazoline,
imidazolidine, pyrazole, pyrazoline, piperazinyl, piperidinyl, pyrazinyl,
pyrimidine,
azepine, diazepine, pyridinyl, indolyl, N-methylpyrrolidinyl, benzthiazolyl,
benzimidazolyl, and benzthiopheneyl.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Most preferred are R3 and R4 groups which singly or in combination with each
other and the nitrogen atom to which they are attached form or are respesented
by groups
independently selected from methyl, ethyl, propyl, isopropyl, isobutyl,
cyclopentyl,
cyclohexyl, N-morpholinyl, benzyl, pyridinyl, pyrrolidinyl, piperidinyl, N-
5 methylpiperidinyl, and N-methylpiperazinyl, 2-methylthiazolyl, N-
methylimidazolyl, and
4-piperidinylpiperidine.
Preferred R6 groups
A preferred R6 or R6' is independently selected from hydrogen, C1-C8 alkyl,
10 phenyl, aryl, alkylaryl, and C3-C8 cycloalkyl.
A more preferred compound of the invention is a compound of formula I wherein
Rl is methyl, chloro, methoxy, fluoro, trifluoromethyl, dichloro, N,N-
dimethyl, or
methylsulfonate; .
15 W is 1 and p is 0 or 1;
R2 is hydrogen; t is 0;
Arl is selected from a group consisting of phenyl, benzimidazolyl, 1H-
insazolyl, 2-
methylindolyl, 3-methoxyphenyl, 2,3-dimethylindolyl, 1-methylindoluyl, benzo-
1,4-
oxazin, 4-methylquinolinyl-6yl, 2,3-dihydroindolyl, oxazolyl, 3-chlorophenyl,
Li is selected from the group consisting of a bond, -C(O)-, -CH2-, -CH2CH2-, -
CH2CH2CH2, -NHCH2CH2, -N(CH3)CH2CH2, -OCH2, -OCH2CH2, -OCH2CH2CH2, and -
acetyleneCH2;
Preferably, R3 and R4 are independently selected from the group consisting of
methyl,
ethyl, isopropyl, cyclohexyl; or R3 and R4 combine with each other or with a
carbon atom
one to four atoms removed ((x, P, or y position) from the nitrogen of NR3R4 to
form a
cyclic ring selected from pyrrole, morpholino, piperidinyl, 4-bipiperidinyl,
piperazinyl,
pyridinyl, -morpholinyl-2y1, N-methylmorpholinyl-2y1, 3-hydroxypyrrloidin-l-
yl, 3-
methyl,-3H-imidazole, 1H-1-methylimidazolyl, pyridine-4-one, 4-hydroxy-
piperidin-1-yl,
pyridinyl, optionally containing 1 or 2 heteroatoms selected from 0, N, or S.
An example of a preferred compound of the present invention is a compound
selected from the group consisting of:
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
16
2-(4-Chloro-phenyl)-5- { 4-[2-(isopropyl-methyl-amino)-ethoxy]-3-methoxy-
phenyl } -6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[ 1-((S)-pyrrolidine-3-carbonyl)-2,3-dihydro-lH-indol-5-
yl]-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one, triflate salt,
2-(4-Chloro-phenyl)-5-[4-(2-diethylamino-ethoxy)-3-methoxy-phenyl]-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-l-yl-ethoxy)-phenyl]-6, 7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one,
5-[3-Methoxy-4-(3-methyl-3H-imidazol-4-ylmethoxy)-phenyl]-2-(4-
trifluoromethoxy-
phenyl)-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5- { 2-[methyl-(1-methyl-piperidin-4-yl)-amino]-
benzooxazol-5-yl }-
6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt
5-[3-Methoxy-4-(3-methyl-3H-imidazol-4-ylmethoxy)-phenyl]-2-(4-methoxy-phenyl)-
5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Methoxy-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-
5H-thiazolo [5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-piperidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-{ 3-methoxy-4-[2-(3-oxo-morpholin-4-yl)-ethoxy]-phenyl }
-5H-
2 0 thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5- [4-(2-pyrrolidin-1-yl-ethyl)-3,4-dihydro-2H-benzo [
1,4] oxazin-7-
yl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one,
2-(2,4-Dichloro-phenyl)-5- [3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -
6,7-dihydro-
5H-thiazolo [5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-{2-[(2-dimethylamino-ethyl)-methyl-amino]-benzooxazol-5-
yl}-
6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5- { 4-[2-(cyclohexyl-methyl-amino)-ethoxy]-3-methoxy-
phenyl }-6,7-
dihydro-5H-thiazolo [5,4-c] pyridin-4-one,
2-(4-Chloro-phenyl)-5-[4-(3-dimethylamino-propoxy)-3-methoxy-phenyl]-6,7-
dihydro-
3 0 5H-thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5- [4-methyl-2-(2-morpholin-4-yl-ethylamino)-quinolin-6-
yl] -6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
17
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -5H-
thiazolo [5,4-
c]pyridin-4-one,
2-(4-Chloro-phenyl)-5- [4-(2-dimethylamino-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[1-(2-pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one, 2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-
l-yl-
ethoxy)-phenyl]-5H-thiazolo[5,4-c]pyridin-4-one citrate salt,
2-(4-Chloro-phenyl)-5-[3-chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-5H-
thiazolo[5,4-
c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-5H-
thiazolo [5,4-
c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-5H-
thiazolo[5,4-
c]pyridin-4-one,
2-(4-Chloro-phenyl)-5- [3-methoxy-4-(3-pyrrolidin-l-yl-propyl)-phenyl] -SH-
thiazolo [5,4-
c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-morpholin-4-yl-ethoxy)-phenyl]-5H-
thiazolo [5,4-
c]pyridin-4-one, hydrochloride salt,
2-(4-Methoxy-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -5H-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt
2-(4-Chloro-phenyl)-5-[1-methyl-3-(2-pyrrolidin-1-yl-ethyl)-1H-indol-6-yl]-6,7-
dihydro-
5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
5- [3 -Methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -2-(4-trifluoromethoxy-
phenyl)-5H-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl]-5H-
thiazolo[5,4-
2 5 c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[4-(2-dimethylamino-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-methyl-3H-imidazol-4-ylmethoxy)-phenyl]-
5H-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[1-(2-pyrrolidin-l-yl-ethyl)-1H-indol-5-yl]-6,7-dihydro-
5H-
thiazolo [5,4-c] pyridin-4-one,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
18
2-(4-Chloro-phenyl)-5-14-[2-(2,2-dimethyl-morpholin-4-yl)-ethoxy]-3-methoxy-
phenyl } -
5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
5- [4-(2-Dimethylamino-ethoxy)-3-methoxy-phenyl] -2-(4-methoxy-phenyl)-6,7-
dihydro-
5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[1-methyl-3-(2-pyrrolidin-1-yl-ethyl)-1H-indol-6-yl]-6,7-
dihydro-
5H-thiazolo [5,4-c] pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3 -methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl] -6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5- [2-methyl-l-(2-pyrrolidin-1-yl-ethyl)-1 H-indol-5-yl] -
6,7-dihydro-
5H-thiazolo [5,4-c] pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-prop-1-ynyl)-phenyl]-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
5- [3-Methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -2-(4-trifluoromethyl-
phenyl)-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-13-methoxy-4-[2-(2,2,6,6-tetramethyl-morpholin-4-yl)-
ethoxy]-
phenyl}-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[ 1-(2-pyrrolidin-1-yl-ethyl)-1 H-benzoimidazol-5-yl]-
6,7-dihydro-
5H-thiazolo [5,4-c] pyridin-4-one,
2-(4-Chloro-phenyl)-5-[3-methoxy-4-((R)-1-morpholin-2-ylmethoxy)-phenyl]-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt,
2-(4-Chloro-phenyl)-5-[2,3-dimethyl-l-(2-pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-
6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
5-[4-(2-[ 1,4']Bipiperidinyl-1'-yl-ethoxy)-3-methoxy-phenyl]-2-(4-chloro-
phenyl)-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one,
2-(4-Chloro-phenyl)-5-[ 1-(2-morpholin-4-yl-ethyl)-1 H-indol-5-yl] -6,7-
dihydro-5H-
3 0 thiazolo[5,4-c]pyridin-4-one, hydrochloride salt, or a pharmaceutically
acceptable salt,
solvate, enantiomer, or mixture of enantiomers thereof.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
19
Preparim Compounds of the Invention
Scheme 1 shows a synthetic route for preparing a common intermediate VI
generally utilized in the preparation of compounds of the invention.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Scheme 1
S O O Step 1 N O,
Ari< __j + R1.0'~X Ar</ 1'Or R1
S
1 II I11
Step 2
N Step 4 N O ~Step 3 N
Ar1-~S O Ari---~S Ari</ ,J
S
O O
VI V IV
Preparation of the intermediate VI starts with the condensation of thioamide I
and
5 (3-keto ester II as shown in step 1. This can be achieved in polar solvent
(such as MeOH,
EtOH or DMF) from about 2 to 24 hours (h) at a temperature range from about
room
temperature to 80 C to give a thiazole of formula III.
In step 2, reduction of the ester III to the alcohol IV can be achieved using
one of
several methods well known in the literature. For example, ester III can be
reduced with
10 DIBAL (or other suitable reducing agent like LiAlH4, NaBH4, and LiBH4) in
THF (or
other aprotic solvent such as ether or toluene) from about 1 to 8h at a
temperature range
from about -78 C to 60 C. Compound IV is isolated by aqueous work-up and
purified
by means known in the art.
As shown in step 3, carboxylic acid V can be prepared from alcohol IV by
15 dissolving in THF (or ether) at about -78 C, slowly treating with a
solution of n-BuLi (or
other suitable base such as LDA or HMDA) over about 2 to 4 hours, then
treating with a
solution of CO2 (g) in THF (or ether). Compound V is isolated by precipitation
from a
dilute aqueous solution and purified by means known in the art.
The use of a Dean-Stark trap accelerates the reaction by removing H20 as it is
produced.
20 Step 4 involves lactone formation to give VI using anhydrous conditions.
For
example, a solution of alcohol V in anhydrous toluene (or THF, benzene, etc.)
is treated
with an acid catalyst (ex. para-toluenesulfonic acid) and heated to reflux for
4 to 24 hours
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
21
to cyclize to VI. The use of a Dean-Stark trap accelerates the reaction by
removing H20
as it is produced.
02N,Ar' St~ 02N,Ar' N
P Ar S
VII ~ N,Ar i,L: AB
VIII
,
O D
XIII
Step 6
Step 10
HZN=Ar'
i
P
Ar~N ~ IX Ar~N I O Step 8 Ar~N I Step 9 Ar~N I
S O Ste 7 S N-Ar' S Nr S N Ar'
O p O p O p O
VI X XI XII
Scheme 2. Synthesis of lactam compounds of formula XIII (Route 1).
Lactone VI can be elaborated to provide compounds of formula XIII as shown in
Scheme 2. In step 5, a nitro compound of formula VII that contains a free OH
or NH
group is protected with an appropriate group, to give compound of formula VIII
that can
be removed later in the synthetic sequence. For example, 2-methoxy-4-nitro-
phenol is
protected as a silyl ether by dissolving the phenol in a polar solvent such as
DMF or THF,
treating with a base such as sodium hydride, and then adding triisopropylsilyl
triflate (or
similar silyl reagent like TBSC1, TIPSCI, or TBSOTf). The reactions is stirred
within a
temperature range of about RT to 50 C for 1 to 24 hours then isolated via
aqueous work-
up and purified by means known in the art. Other protecting groups for an OH
or NH
group can be employed and are familiar to those skilled in the art (see Philip
J. Kocienski,
"Protecting Groups," Thieme: New York 1994 or Theodora W. Green, "Protective
Groups in Organic Synthesis," John Wiley and Sons: New York, 1981 for
additional
examples).
In step 6, a compound of formula VIII is prepared by reduction of the nitro
group
to give an amine of formula IX by treatment with 5-10% Pd/C under H2
atmosphere
(latm) in a suitable solvent (like THF, EtOAc, EtOH or MeOH) from about 2 to
24 hours
at room temperature. Several other nitro reduction techniques known in the art
can be
employed.
Amide formation, as shown in step 7, is accomplished using a typical Weinreb
protocol (see Basha, Anwer; Lipton, M.; Weinreb, Steven M. Tetrahedron
Letters, 1977,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
22
48, 4171-4174). For example, amine IX is dissolved in an aprotic solvent (such
as
CH2Cl2 or toluene) and treated with a 2-2.5M solution of Me3A1 in hexanes. The
resulting solution is stirred at a temperature from about 0 C to room
temperature for
about 5 to 60 minutes, and then treated with lactone VI. The resulting
solution is stirred
at a range of between about room temperature and 110 C for about 3 to 24
hours to give
amide X which is isolated by aqueous work-up and purified by trituration with
ether or by
flash chromatography.
In step 8, lactam XI is prepared under Mitsunobu conditions (Maligres, P. E.;
Waters, M. S.; Weissman, S. A.; McWilliams, J. C.; Lewis, S.; Cowen, J.;
Reamer, R. A.;
Volante, R. P.; Reider, P. J.; Askin, D. J. Het. Cliem. 2003, 40(2), 229-241).
For
example, amide X is dissolved in a suitable anhydrous solvent (ex. THF,
CH2C12, toluene,
etc.) and treated with a trialkyl- or triarylphosphine (ex. Me3P, Bu3P, or
Ph3P) and
dialkylazo-dicarboxylate (ex. DEAD or DIAD) at a suitable temperature (about 0
C to
RT) for about 4 to 24 hours. Compound XI is isolated by aqueous workup and
chromatographic purification.
In step 9, the protecting group that was installed in Step 5 is removed using
conditions that are appropriate for the type of protecting group used to give
compound of
formula XII. For example, removal of a silyl ether, such as a
triisopropylsilyl group, is
achieved by dissolving the silyl ether in a polar solvent like THF or CHaC12
and treating
with a fluoride source such as nBu4NF or HF=pyridine. The reaction is stirred
from about
15 minutes to 4 hours at a temperature within a range of about 0 to 50 C and
is isolated
by aqueous work-up and purified by means known in the art.
Compounds of formula XIII can be prepared by the alkylation of an NH or OH
group (see step 10) by dissolving in a polar solvent (like THF, DMF, DMSO, and
NMP)
and treating with a base such as NaH or K2C03 and an electrophile (e.g. alkyl
halide, alkyl
mesylate, or alkyl tosylate). The reaction is stirred within a range of about
room
temperature to about 100 C from 4-24 hours and then isolated by aqueous work-
up and
purified by means known in the art.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
23
N O
O2N'~'Ar' Step 11 O2N, l"B N, Ar~~L~A~6 Step 13 Ar--(i ' N' L: .B
Ar I -- ID S Ar1~ A
D Step 12 p
VII XIV XV XVI O
Step 14/
N
~
Ar S ~ N. i~L:AB
Ar
O D
XIII
Scheme 3. Synthesis of lactam compounds of formula XIII (Route 2).
Scheme 3 shows an alternative route to compounds of formula XIII. In this
approach, the alkylation of an NH or OH group occurs early in the synthetic
sequence.
For example, alkylation of VII as shown in Step 11 occurs under conditions
similar to
step 8 above to give compounds of formula XIV.
In step 13, the nitro group is reduced to an amine as described in step 5.
Also, step
13 and 14 proceed under similar conditions as described in steps 7 and 8,
respectively, to
ultimately provide compounds of formula XIII.
Ar-{~N ~ O Step 13 Ar~N Step 14 Ar~N I\
S N, Ar1 S N, Ar' S N'Ar1
O p O P O
X XVI I XVI I I
Step 15
N
Ar--i I N '
S Ar'
0 L~.A.B
XIX p
Scheme 4. Synthesis of pyridone compounds of formula XIX (Route 1).
Schemes 4 and 5 show synthetic routes for preparing thiazole-pyridone
compounds of the invention and/or precursors thereof.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
24
In step 13 of scheme 4, pyridone XVII is prepared in one step by the oxidation
of
intermediate alcohol X. For example compound X is dissolved in a suitable
polar solvent
(e.g. CH2CI2, THF) and treated with an oxidizing reagent (e.g. Dess-Martin
periodinane,
pyridine=S03, PDC, or under Swern-oxidation conditions). Oxidation conditions
are
abundantly known to those skilled in the art and can be found in Comprehensive
Organic
Transforfyiations, by R.C. Larock, VCH Publishers, 1989, p. 604-614. Dess-
Martin
periodinane is the reagent of choice for this transformation and the oxidation
is performed
at about 0 C to room temperature from about 1 hour to 3 days. Pyridone XVII is
isolated
by aqueous workup and chromatographic purification.
In step 14, analogous to step 9 above, removal of the protecting group to
reveal an
NH or OH group is achieved under similar conditions and the compound of
formula
XVIII is isolated by aqueous work-up and purified by means known in the art.
Alkylation of the OH or NH group of XVIII (step 15) can occur under basic
conditions with an alkylating reagent, as described in step 10 above, or under
Mitsunobu
conditions to provide compounds of formula XIX.
Alternatively, and as shown in step 16 of Scheme 5, intermediate XVI can be
oxidized with the sidechain already installed using similar conditions as
described in step
13 above to afford thiazole-pyridone compounds of formula XIX.
N O Step 16 N
Ar-_<I S ~ N' L: B ~ Ar-~ ~ N, ,L: .B
Ar1 A S Ar1 A
O D p D
XVI XIX
Scheme 5. Synthesis of pyridone compounds of formula XIX (Route 2).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Scheme 6 shows a synthetic route for preparing compounds of the invention from
an intermediate acetal wherein Ll is an alkylene of varying carbon chain
lengths.
N
Ar i N
~
S N, Step 17 Ar--~
O Ar n --~ S N'Ari n CH
O
XX, n= 0, 1, 2 O
XXI, n = 0, 1, 2
3
Step 18 Ar--~~
31. S N'Ari-~/N1-1 Ra
\ ,n
O
XXII, n = 0, 1, 2
Scheme 6. Synthesis of amines of formula XXII.
5
If groups A, B and D (Compound XIII) together define an acetal group (such as
A= CH and B=D=OMe or OEt), then hydrolysis to an aldehyde group is performed
according to conditions recognized by persons skilled in the art (Scheme 6).
For example,
in step 17 acetal XX is dissolved in a suitable solvent (e.g. THF, acetone,
MeOH) and
10 treated with water and an acid catalyst (e.g. p-toluenesulfonic acid) at
reflux for about 4 to
24 hours to give aldehyde XXI. Reductive amination (step 18) is performed by
dissolving
the aldehyde XXI in dichloroethane or another suitable solvent such as for
example,
CH2Cl2 or THF and treated with an 1 or 2 amine and a reducing reagent such
as for
example NaCNBH3, or NaBH(OAc)3. The mixture is stirred at about RT to 80 C
from
15 about 30 min to 8 hours. Amines of foimula XXII are isolated by aqueous
workup and
purified by means known in the art.
Scheme 7 shows an alternative synthetic route for preparing compounds of the
invention and/or precursors thereof. In step 19, lactone VI is treated with a
protected
20 amine using conditions previously described in step 7 to give amide XXIII.
In step 19,
Lactam XXIV is prepared using conditions previously described in step 8. The
lactam
nitrogen is deprotected, as shown in step 21, using conditions consistent with
the type of
protecting group that is used. For example, a 3,4-dimethoxy benzyl group is
removed
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
26
under acidic conditions (e.g. p-toluene sulfonic acid or TFA) in a solvent
such as toluene
at a temperature range of RT to reflux for 0.5 to 4h. Lactam XXIV is isolated
by
precipitation from water and purified by means known in the art.
In step 22, the lactam is coupled to an aryl bromide using catalytic cross-
coupling
conditions such as Buchwald arylation of an amide (see Yin, J.; Buchwald, S.J.
T. Am.
Chem. Soc. 2002, 124 (21), 6043-6048). For example, lactam of formula XXIV is
coupled to bromide XXV (where P2 is a protecting group for an OH or NH group)
using a
base such as for example, Cs2CO3), a palladium reagent such as. Pd2dba3, and a
phosphine ligand such as XantphosTM in a non-protic solvent (ex. dioxane,
toluene,
benzene etc.). The reaction is performed at a temperature range of about RT to
reflux
from about 3 to 24h and is then isolated by aqueous work-up and purified by
means
known in the art.
As shown in step 23, the protecting group of XXVII is removed using conditions
consistent with the type of protecting group that is used. For example, a
silyl ether is
removed using a Bu4NF. In addition, a p-toluene sulfonate ester is removed
under basic
conditions using for example, LiOH in 2:1 dioxane water, to afford a compound
of
formula XII.
In step 24, alkylation of the free NH or OH group of XII is achieved using
conditions previously described in step 8 to afford a compound of formula
XIII.
N NPi N 0 N
Step 21 Ar~ I
Ar~S p ~ ~ Ar~ ~ Step 20 Ar- rr I N
S N= \S =
N -~ SN
0 Step 19 p Pi 0 P1 O
VI xxlll xxlV xxV
Br, Ar'
p xxVl
Step 22
N Step 24 N Step 23 N
r
r r
Ar-~S ~ N,Ar'L:A.6 Ar- ~S ~ N' .- Ar-{S N Ar,
p Ar
~ 0 o Pz
xiu xu xxvu
Scheme 7. Synthesis of lactam compounds of formula XIII (Route 3).
Scheme 8 shows the preferred synthesis of substituted morpholines that are
used
as reagents in the synthesis of compounds of the invention.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
27
Ste 25 OH
P Step 26
~ -' HN N
H 2 N 0-1
XXVIII XXIX xxx
Step 27
Step 29 Step 28
N N
H N
HCI I \ \
/
XXXIII XXXII XXXI
Scheme 8. Synthesis of substituted morpholine analogs.
In step 25, the amino group of methallyl amine (XXVIII) is protected with a
benzyl group via a reductive amination. Amine XXVIII is dissolved in a polar
aprotic
solvent like and treated with benzaldehyde. The imine intermediate is then
reduced with a
reducing reagent like NaBH4 for 10-24 hours at a temperature range of room
temperature
to 50 C to give an amine of formula XXIX that is isolated by aqueous work-up
and
purified by means known in the art.
In step 26, an amine of formula XXIX is alkylated by treating with an epoxide
(for
example, isobutylene oxide) and Lewis acid such as LiBr at a temperature range
from
room temperature to 60 C for 1 to 8 hours to give alcohol of formula XXX. The
product
is isolated by aqueous work-up and purified by means known in the art.
In step 27, the preferred method for forming the substituted morpholine is via
halo-etherification methodology. In this approach, an alcohol of formula XXX
is treated
with iodine. The reaction is performed in a biphasic mixture of a nonpolar
aprotic
solvent, such as MTBE, and an aqueous basic solution (for example, 1M NaHCO3)
for 12
to 24h. The iodide of formula XXXI is then isolated by aquoues work-up and
purified by
means known in the art.
In step 28, the iodide is removed under reducing conditions to give the benzyl
morpholine of formula XXXII. Typical conditions to remove an alkyl iodide
group are to
dissolve the iodide XXXI in a polar solvent such as DMSO and treat with a
reducing
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
28
reagent like NaBH4 for 2 to 6 hours. Morpholine of formula XXXII is isolated
by
aqueous work-up and purified by means known in the art.
In step 29, the benzyl protecting group is removed under typical reductive
conditions that are recognized by persons skilled in the art. For example,
compound of
formula XXXII is dissolved in suitable solvent (example THF, ETOH), treated
with 3%
palladium on activated carbon under hydrogen atmosphere that is pressurized up
to 60psi
at 40 C for up to 24h. Morpholine of formula XXXIII is purified by means known
in the
art and can be isolated as the hydrochloride salt by treating with an HCl
source (ex. 1.OM
HCI in ether).
Demonstration of Function
In order to demonstrate that compounds of the present invention have the
capacity
to bind to and inhibit the function of MCHR1, binding and functional assays
were
established. All ligands, radioligands, solvents and reagents employed in
these assays are
readily available from commercial sources or can be readily prepared by those
skilled in
the art.
The full-length cDNA for human MCHR1 was cloned from a human adult brain
cDNA library (Edge Biosystems, Cat. 38356) by standard polymerase chain
reaction
(PCR) methodology employing the following primers: sense, 5'-GCCACCATGGACCT
GGAAGCCTCGCTGC-3'; anti-sense, 5'-TGGTGCCCTGACTTGGAGGTGTGC-3'.
The PCR reaction was performed in a final volume of 50 l containing 5 l of a
lOx stock
solution of PCR buffer, 1 l of 10 mM dNTP mixture (200 M final), 2 1 of 50
mM
Mg(S04) (2 mM final), 0.5 l of 20 pM solutions of each primer (0.2 M final),
5 l of
template cDNA containing 0.5 ng DNA, 0.5 l of Platinum Taq High Fidelity DNA
polymerase (Gibco Life Technologies) and 36 l of H20. PCR amplification was
performed on a Perkin Elmer 9600 thermocycler. After denaturation for 90 sec
at 94 C,
the amplification sequence consisting of 94 C for 25 sec, 55 C for 25 sec
and 72 C for 2
min was repeated 30 times, followed by a final elongation step at 72 C for 10
min. The
desired PCR product (1.1 Kb) was confirmed by agarose gel electrophoresis and
the band
was extracted from the gel by Geneclean (Biol01) following the manufacturer's
instructions. Following extraction, the cDNA fragment was cloned into pCR2.1-
TOPO
plasmid (Invitrogen Corp) to confirm the identity and sequence.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
29
In order to generate cell lines stably expressing MCHR1, the insert was then
subcloned into the Xba I and Not I sites of pcDNA(+)-3.1-neomycin
(Invitrogen). After
purification by Qiagen Maxi-prep kit (QIAGEN, Inc.), the plasmid was
transfected by
Fugene 6 (Roche Applied Science) into AV 12 cells that had been previously
transfected
with the promiscuous G protein Ga15. The transfected cells were selected by
G418 (800
g/ml) for 10-14 days and single colonies were isolated from culture plates.
The G418-
resistant colonies were further selected for MCHRI expression by measuring MCH-
stimulated Ca2+ transients with a fluorometric imaging plate reader (FLIPR,
Molecular
Devices).
Typically, individual clones are plated out in 96-well plates at 60,000 cells
per
well in 100 l of growth medium (Dulbecco's modified Eagle's medium (DMEM), 5%
fetal bovine serum, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 0.5
mg/ml Zeocin, and 0.5 mg/ml Geneticin). After 24 hrs at 37 C, medium is
removed and
replaced with 50 l of dye loading buffer (Hank's balanced salt solution
(HBSS)
containing 25 mM HEPES, 0.04% Pluronate 127 and 8 M Fluo3 Both from Molecular
Probes)). After a 60 min loading period at room temperature, dye loading
buffer is
aspirated and replaced with 100 l of HEPES/HBBS. Plate is placed in FLIPR and
basal
readings are taken for 10 sec, at which point 100 1 of buffer containing 2 M
MCH (1
pM final) is added and measurements are taken over 105 sec. To correct for
variations
between clones in numbers of cells per well, the MCH response is normalized to
the
response induced by epinephrine.
Both the 125I-MCH binding and functional GTPJ5S binding assays employed
membranes isolated from a clone designated as clone 43. Typically, cells from
20
confluent T225 flasks were processed by washing the monolayers in cold
phosphate-
buffered saline (PBS), scraping the cells into same and re-suspending the cell
pellet in 35
ml of 250 mM Sucrose, 50 mM HEPES, pH 7.5, 1 mM MgC12, 24 g/m1 DNase I, and
protease inhibitors (1 Complete tablet, per 50 ml of buffer prepared, Roche
Diagnostics). Alternatively, greater levels of cells could be generated by
adapting cell
growth to suspension culture in 20 L stirred vessel bioreactors. After
incubation on ice
for 5 min, cells were disrupted with 20-25 strokes of a Teflon/Glass
homogenizer attached
to an overhead motorized stirrer, and the homogenate was centrifuged at 40,000
rpm in
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Beckman Type 70.1 Ti rotor. The pellets were re-suspended in 250 mM Sucrose,
50 mM
HEPES, pH 7.5, 1.5 mM CaC12, 1 mM MgSO4 and protease inhibitors by
Teflon/Glass
homogenization to achieve a protein concentration of -3-5 mg/ml (Pierce BCA
assay with
Bovine serum albumin as standard). Aliquots were stored at -70 C.
5 Binding of compounds to MCHR1 was assessed in a competitive binding assay
employing 125I-MCH, compound and clone 43 membranes. Briefly, assays are
carried out
in 96-well Costar 3632 white opaque plates in a total volume of 200 l
containing 25 mM
HEPES, pH 7.0, 10 mM CaC12, 2 mg/nil bovine serum albumin, 0.5% dimethyl
sulfoxide
(DMSO), 5 g of clone 43 membranes, 200 pM 125I-MCH (NEN), 0.625 mg/ml of
wheat
10 germ agglutinin scintillation proximity assay beads (WGA-SPA beads,
Amersham Inc.,
now GE Healthcare) and a graded dose of test compound. Non-specific binding is
assessed in the presence of 0.1 pM unlabeled MCH. Bound 125I-MCH is determined
by
placing sealed plates in a Microbeta Trilux (Perkin Elmer Life and Analytical
Sciences
Inc.) and counting after a 12 hr delay.
15 IC50 values (defined as the concentration of test compound required to
reduce
specific binding of 125I-MCH by 50%) are determined by fitting the
concentration-
response data to a 4-parameter model (max response, min response, Hill
coefficient, IC50)
using Excel (Microsoft Corp.). K; values are calculated from IC50 values
using the
Cheng-Prusoff approximation as described by Cheng et al. ( Relationship
between the
20 inhibition constant (Ki) and the concentration of inhibitor which causes
50% inhibition
(IC50) of an enzymatic reaction, Biochem. Pharmacol., 22: 3099-3108 (1973)).
The Kd
for 125I-MCH is determined independently from a saturation binding isotherm.
Exemplified compounds showed a Ki of < 1 M under the binding assay
conditions.
Specifically, a sample of observed Ki values is provided in Table 1 (below)
for
25 demonstration purposes only.
Table 1
Example # Average MCHR 1 Ki (nM)
2 39.7
5 10.2
15 19.0
33 5.13
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
31
47 3.16
65 35.8
Functional antagonism of MCH activity is assessed by measuring the ability of
test
compound to inhibit MCH-stimulated binding of GTPy35S to clone 43 membranes.
Briefly, assays are carried out in Costar 3632 white opaque plates in a total
volume of 200
l containing 50 mM Hepes, pH 7.4, 5 mM MgC12, 10 g/mi saponin, 1.0 mg/ml
bovine
serum albumin, 100 mM NaCl, 3 M GDP, 0.3 nM GTPJSS, 10 nM MCH
(approximately equal to EC90), 20 g of clone 43 membranes, 5.0 mg/ml of wheat
germ
agglutinin scintillation proximity assay beads (WGA-SPA beads, Amersham Inc.,
now
GE Healthcare) and a graded dose of test compound. The plates are sealed and
left for
16-18 hrs at 4 C. After a 1 hr delay to allow plates to equilibrate to ambient
temperature,
bound GTPy35S is determined by counting in a Microbeta Trilux (Perkin Elmer
Life and
Analytical Sciences Inc).
IC50 values (defined as the concentration of test compound required to reduce
MCH-stimulated GTPJ5S binding by 50%) are determined by fitting the
concentration-
response data to a 4-parameter model (max response, min response, Hill
coefficient, IC50)
using Excel (Microsoft). After verifying competitive antagonism by Schild
analysis, Kb
values are calculated from the IC50 values for each antagonist and the EC50
for MCH
(determined independently) using a modification of the Cheng-Prusoff
approximation as
described by Leff and Dougal (Trends Plzarnaac l. Sci. (1993) 14: 110-112).
Exemplified compounds showed IC50 values of < 1 M under the functional assay
conditions disclosed herein.
In order to demonstrate in vivo efficacy, compounds of the invention were
administered by oral gavage to diet-induced obese male Long-Evans rats
(Harlan, IN)
weighing 500-550g. Vehicle consisted of 1% CMC and 0.25% PS-80 in water.
Animals were individually housed in a temperature regulated room (240C) with a
reverse 12 hour light/dark cycle (dark 10:00/22:00). Water and food (Teklad
95217,
Harlan, WI) were available ad libitum. Compounds were dosed orally once a day
before
onset of dark for 3 days. Daily food intake and body weight change were
measured for
the 3 day period. Exemplified compounds tested at 10 mg/kg showed reduction of
3 day
cumulative body weight gain when compared with vehicle-treated controls.
Specifically,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
32
a sample of observed 3 day cumulative body weight reduction, relative to
control, is
provided in Table 2 (below) for demonstration purposes only.
Table 2
Example # Body weight reduction @ 10 mg/Kg versus vehicle
control. Data expressed in grams.
42 1.7
47 7.2
52 9.6
Utility
As antagonists of the MCHRlbinding, a compound of the present invention is
useful in treating conditions in human and non-human (especially companion)
animals in
which the MCHR1 receptor has been demonstrated to play a role. The diseases,
disorders
or conditions for which compounds of the present invention are useful in
treating or
preventing include, but are not limited to, diabetes mellitus, hyperglycemia,
obesity,
hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, atherosclerosis of
coronary,
cerebrovascular and peripheral arteries, gastrointestinal disorders including
peptic ulcer,
esophagitis, gastritis and duodenitis, (including that induced by H. pylori),
intestinal
ulcerations (including inflammatory bowel disease, ulcerative colitis, Crohn's
disease and
proctitis) and gastrointestinal ulcerations, neurogenic inflammation of
airways, including
cough, asthma, depression, prostate diseases such as benign prostate
hyperplasia, irritable
bowel syndrome and other disorders needing decreased gut motility, diabetic
retinopathy,
neuropathic bladder dysfunction, elevated intraocular pressure and glaucoma
and non-
2 0 specific diarrhea dumping syndrome. The diseases, disorders or conditions
for which
compounds of the present invention are useful in treating or preventing also
include,stress
related disorders including post traumatic stress disorder, substance abuse,
including
alcohol and drug abuse, and nonpharamcologic disorders such as gambling, sex
and
internet related addictions. By inhibiting MCH activity the compounds of the
present
invention provide anorexic effects. That is, the compounds of the invention
are useful as
appetite suppressants and/or weight loss agents. The compounds of the
invention may
also be used in combination with other approved therapeutic agents for the
treatment,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
33
prevention and/or amelioration of obesity and related diseases. In this
format, the
compounds of the present invention enhance the positive effects of such
approved
combination treatments while minimizing the side effects due to the potential
requirement
of lower doses of such combination compounds. Such combination therapies may
be
delivered individually or in a combined formulation. Examples of compounds
useful in
combination with a compound of formula I include weight loss agents
(MeridiaTM,
XenicalTM), cholesterol lowering agents (such as for example lovastatin,
simvastatin
pravastatin, fluvastatin, and atorvastatin), glucose level control or
modulating agents,
nerve growth factor agonists (such as for example, axokine), cannabinoid CB-1
antagonist
compounds (such as for example rimonanbant) and the like.
In treating non-human, non-companion animals, the compounds of the present
invention are useful for reducing weight gain and/or improving the feed
utilization
efficiency and/or increasing lean body mass.
Formulation
The compound of fozmula I is preferably formulated in a unit dosage form prior
to
administration. Therefore, yet another embodiment of the present invention is
a
pharmaceutical formulation comprising a compound of formula I and a
pharmaceutical
carrier.
The present pharmaceutical formulations are prepared by known procedures using
well-known and readily available ingredients. In making the formulations of
the present
invention, the active ingredient (formula I compound) will usually be mixed
with a
carrier, or diluted by a carrier, or enclosed within a carrier which may be in
the form of a
liquid, tablet, capsule, sachet, paper or other container. When the carrier
serves as a
diluent, it may be a solid, semisolid or liquid material which acts as a
vehicle, excipient or
medium for the active ingredient. Thus, the compositions can be in the form of
tablets,
pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions,
solutions,
syrups, aerosol (as a solid or in a liquid medium), soft and hard gelatin
capsules,
suppositories, sterile injectable solutions and sterile packaged powders.
One of skill in the art is aware of methods, reagents and conditions for
preparing various
standard formulations or can assess such information without undue
experimentation.
The compositions of the invention may be formulated so as to provide quick,
sustained or
delayed release of the active ingredient after administration to the patient.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
34
Dose
The specific dose administered is determined by the particular circumstances
surrounding each situation. These circumstances include, the route of
administration, the
prior medical history of the patient, the pathological condition or symptom
being treated,
the severity of the condition/symptom being treated, and the age and sex of
the recipient.
However, it will be understood that the therapeutic dosage administered will
be
determined by the physician in the light of the relevant circumstances, or by
the
veterinarian for non-human recipients.
Generally, an effective minimum daily dose of a compound of formula I is about
to 200 mg. Typically, an effective maximum dose is about 200 to 1000 mg. The
exact
dose may be determined, in accordance with the standard practice in the
medical arts of
"dose titrating" the recipient; that is, initially administering a low dose of
the compound,
and gradually increasing the dose until the desired therapeutic effect is
observed.
Route of Administration
The compounds may be administered by a variety of routes including the oral,
rectal, transdermal, subcutaneous, topical, intravenous, intramuscular or
intranasal routes.
Ap referred route of administration is oral.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Combination Therapy
A compound of formula I may be used in combination with other drugs or
therapies that have been approved for the treatment/prevention/suppression or
amelioration of the diseases or conditions for which compounds of formula I
are useful.
5 Such other drug(s) may be administered, by a route and in an amount commonly
used
therefor, contemporaneously or sequentially with a compound of formula I. When
a
compound of formula I is used contemporaneously with one or more other drugs,
a
pharmaceutical unit dosage form containing such other drugs in addition to the
compound
of formula I is preferred. Accordingly, the pharmaceutical compositions of the
present
10 invention include those that also contain one or more other active
ingredients, in addition
to a compound of formula I. Examples of other active ingredients that may be
combined
(upon approval) with a compound of formula I, and either administered
separately or in
the same pharmaceutical composition, include, but are not limited to:
15 (a) insulin sensitizers including (i) PPARy agonists such as the glitazones
(e.g.
troglitazone, pioglitazone, englitazone, MCC-555, BRIA9653 and the like), and
compounds disclosed in W097/27857, 97/28115, 97/28137 and 97/27847; (ii)
biguanides such as metformin;
(b) insulin or insulin mimetics;
20 (c) sulfonylureas such as tolbutamide and glipizide;
(d) alpha-glucosidase inhibitors (such as acarbose);
(e) cholesterol lowering agents such as
i. HMG-CoA reductase inhibitors (lovastatin, simvastatin pravastatin,
fluvastatin, atorvastatin, and other statins),
25 ii. sequestrants (cholestyramine, colestipol and a dialkylaminoalkyl
derivatives of a cross-linked dextran),
iii. nicotinyl alcohol nicotinic acid or a salt thereof,
iv. proliferator-activator receptor a agonists such as fenofibric acid
derivatives
(gemfibrozil, clofibrat, fenofibrate and benzafibrate),
30 v. inhibitors of cholesterol absorption for example (3-sitosterol and (acyl
CoA:cholesterol acyltransferase) inhibitors for example melinamide,
vi. probucol,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
36
vii. vitamin E, and
viii. thyromimetics;
(f) PPARS agonists such as those disclosed in W097/28149;
(g) Anti obesity compounds such as fenfluramine, dexfenfluramine, phentermine,
sibutramine, orlistat, axokine, rimonanbant, etc;
(h) feeding behavior modifying agents such as neuropeptide Y antagonists (e.g.
neuropeptide Y5) such as those disclosed in WO 97/19682, WO 97/20820, WO
97/20821, WO 97/20822 and WO 97/20823;
(i) PPARa agonists such as described in WO 97/36579 by Glaxo;
(j) PPARy antagonists as described in W097/10813; and
(k) serotonin reuptake inhibitors such as fluoxetine and sertraline
(1) antipsychotic agents such as for example olanzapine.
Examples
The following examples are only illustrative of the preparation protocols and
Applicants' ability to prepare compounds of the present invention based on the
schemes
presented or modifications thereof. The examples are not intended to be
exclusive or
exhaustive of compounds made or obtainable.
Materials and Method
Solvents and reagents were used as purchased from chemical suppliers and
reactions were conducted at ambient atmosphere unless otherwise stated. Mass
spectrum
data was obtained on a Microrimass Platform LCZ spectrometer using
electrospray (ES)
ionization. NMR data was obtained on a Varian 400 MHz spectrometer and is
reported in
ppm. A CEM Discover microwave reactor was used where indicated. Common
abbreviations used throughout the experimentals are: methanol (MeOH), ethanol
(EtOH),
ethyl acetate (EtOAc), dichloromethane (CH2C12), dimethylformamide (DMF),
tetrahydrofuran (THF), and room temperature (RT).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
37
Preparation 1
Triisopropyl-(2-methoxy-4-nitro-phenoxy)-silane
N02
O
.O
Si
Dissolve 4-nitroguiacol (50.0 g, 295.6 mmol) in DMF (anhydrous, 1000 mL) and
cool the solution to 0-5 C then slowly treat with NaH (60% in mineral oil,
13.4 g, 335.0
mmol) keeping the temp. < 10 C. Stir the yellow-orange solution mechanically
at room
temp. for ca. 30 min. then cool to 0-5 C. Treat the mixture with TIPS triflate
(90.0 mL,
334.8 mmol), keeping the temp. < 10 C, then stir at room temp. overnight.
Quench the
mixture with 14% aqueous NH4C1(1000 mL) then extract with EtOAc (3 x 1000 mL).
Combine the organic solutions, wash with brine (1000 mL), and concentrate in
vacuo to
give a light yellow oil that was purified by flash chromatography, using 100%
hexanes
then 10% EtOAc/hexanes, to give the title compound as a yellow oil (95.8 g,
99.6%
yield). MS (ES+)326.2 (M+1)+.
Preparation 2
3-Methoxy-4-triisopropylsilanyloxy-Phenylamine
N H2
~ O
~.O
~Sr
Dissolve triisopropyl-(2-methoxy-4-nitro-phenoxy)-silane (95.7 g, 294.0 mmol)
in
EtOH (1800 mL) and add 5% Pd/C (10.0 g). Hydrogenate the slurry at room
temperature
under 50 psi hydrogen for 8 h. Filter the slurry through a pad of Celite and
rinse with
EtOH. Concentrate the filtrate in vacuo to give a brown oil. Purify by flash
chromatography, using a gradient from 100% hexanes to 20% EtOAc/hexanes, to
give the
title compound as a brown solid (67.4 g, 77.6% yield). MS (ES+) 296.2 (M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
38
Preparation 3
[2-(4-chloro-phenyl)-thiazol-4-yl] -acetic acid ethyl ester
ci
O
OEt
Dissolve 4-chlorothiobenzamide (74.0 g, 431.1 mmol) in absolute EtOH (470 ml,
absolute). Add ethyl-4-chloroacetoacetate (58.0 ml, 70.1 g, 426.0 mmol) to the
solution.
stir mechanically at reflux for 2 h. Allow the reaction to cool to room
temperature and
dilute with water (1000 ml). Extract the mixture with Et20 (2000 ml, then 2 x
500 ml).
Combine the organic layers and wash with brine (950 ml). Concentrate the
organic layer
in vacuo to give an oil weighing 121.8 g. The oil solidifies on standing.
Suspend the solid in isopropyl alcohol (610 ml) and heat the slurry to 35 C
at
which temperature all the solids dissolve. Charge the solution with water
(1830 mL) and
allow to cool to room temperature. At approximately 32 C, precipitation
occurs. Stir the
resulting slurry mechanically at room temperature for 4.5 h and filter. Dry
the solid in a
vacuum oven at 35 C for 2 days to give a solid weighing 107.3 g (89.4%
yield). MS
(ES+) 282.1 (M)+.
Preparation 4
[2-(4-Methoxy-phenyl)-thiazol-4-yl]-acetic acid ethyl ester
MeO O
OEt
Prepare the title compound by essentially following the procedure as described
in
Preparation 7, using 4-methoxythiobenzamide. MS (ES+) 278.2(M+1)+. 1H NMR (400
MHz, CDC13): Fi 7.82 (d, J= 8.8 Hz, 2H), 7.11 (s, 1H), 6.93 (d, J= 8.8 Hz,
2H), 4.21 (q,
J= 7.0 Hz, 2H), 3.87 (s, 2H), 3.85 (s, 3H), 1.29 (t, J= 7.0 Hz, 3H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
39
Preparation 5
2-[2-(4-chloro-phenyl)-thiazol-4-yl] -ethanol
CI ~
OH
S
Dissolve [2-(4-chloro-phenyl)-thiazol-4-yl] -acetic acid ethyl ester (107.4 g,
381.2
mmol) in THF (800 mL) and cool to 0-5 C. Add DIBAL (1.0 M in THF, 800 mL, 800
mmol) slowly over approximately 3.5 h (somewhat exothermic) keeping the temp.
< 5 C.
Allow the reaction to warm to room temperature with mechanical stirring
ovemight. Cool
the reaction to 0-5 C and slowly add more DIBAL (150 mL) over approximately
15 min
keeping the temperature < 5 C. Stir the reaction solution at room temperature
for 2.5 h.
Cool to 0-5 C and slowly add over 5 h aqueous saturated Rochelle's salt (2900
mL, very
exothermic at first, minor gas evolution) keeping the temperature < 10 C. The
mixture
solidifies after approximately 150 mL has been added. It becomes more fluid
and then
solidifies again as the addition continues. Extract the mixture with EtOAc (2
x 3300 mL).
Combine the organic layers and concentrate in vacuo to give an oil weighing
112.9 g.
Take the oil up in toluene (600 mL), concentrate in vacuo and repeat. Dry the
residue on
a vacuum pump for 6 h to give a residue weighing 107.4 g (110% yield). MS
(ES+) 240.1
(M)+. 1H NMR (400 MHz, CDC13): 8 7.84 (dt, J = 8.4, 2.2 Hz, 2H), 7.39 (dt, J =
8.4, 2.2
Hz, 2H), 6.98 (s, 1H), 3.98 (m, 2H), 3.44 (bs, 1H), 3.02 (t, J= 5.5 Hz, 2H).
Preparation 6
2- [2-(4-Methoxy-phenyl)-thiazol-4-yl] -ethanol
MeO C>rOH
Prepare the title compound by essentially following the procedure as described
in
Preparation 5, using [2-(4-methoxy-phenyl)-thiazol-4-yl]-acetic acid ethyl
ester. MS
(ES+) 236.2(M+1)+. IH NMR (400 MHz, CDC13): S 7.88 (d, J= 8.8 Hz, 2H), 6.95
(d, J=
8.8 Hz, 2H), 6.91 (s, 1H), 3.98 (t, J= 5.3 Hz, 2H), 3.85 (s, 3H), 3.03 (t, J=
5.3 Hz, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Preparation 7
2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid
CI ~
~ OH
S rO
O
Suspend 2- [2-(4-chloro-phenyl)-thiazol-4-yl] -ethanol (107 g gross, 91 g net,
380
5 mmol) in THF (1210 mL). Decant the solution from the undissolved solids.
Cool the
THF solution to -75 C. Evacuate under vacuum and purge with nitrogen three
times.
Add n-butyl lithium (1.6 M in hexanes, 530 mL, 848 mmol) slowly over 4 h
keeping the
temp. <-70 C. Then add the cold solution (at -75 C) slowly via cannulae over
3.5 h to a
flask containing THF at -75 C that has been saturated with COa gas
(approximately 390
10 g) keeping the temp. <-60 C (addition is very exothermic). Charge the
resulting brown
slurry with additional CO2 gas (approximately 355 g). Allowed the reaction to
come to
room temperature while stirring mechanically at room temperature overnight.
Add 1N HCl (2100 mL + 900 mL), cool the slurry to 16 C and filter. Rinse the
resulting solid with hexane (1400 mL) and dry on the filter funnel witli
vacuum and a
15 stream of nitrogen to give a solid weighing 81.3 g (75.4% yield). MS (ES+)
284.0
(M+1)+. 1H NMR (400 MHz, DMSO-d6): S 7.96 (dt, J = 8.8, 2.2 Hz, 2H), 7.55 (dt,
J
8.4, 2.2 Hz, 2H), 3.74 (t, J = 7.0 Hz, 2H), 3.35 (s, 1H), 3.26 (t, J= 7.0 Hz,
2H).
Preparation 8
20 4-(2-Hydroxy-ethyl)-2-(4-methoxy-phenyl)-thiazole-5-carboxylic acid
MeO
N OH
S
OH
0
Prepare the titled compound by essential following the procedure as described
in
Preparation 7, using 2- [2-(4-methoxy-phenyl)-thiazol-4-yl] -ethanol. MS (ES+)
280.2(M+1)+. 1H NMR (400 MHz, CD30D): 8 7.92 (d, J = 8.8 Hz, 2H), 7.02 (d, J =
8.8
25 Hz, 2H), 3.92 (t, J= 7.0 Hz, 2H), 3.86 (s, 3H), 3.38 (t, J= 7.0 Hz, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
41
Preparation 9
2-(4-Chloro-phenyl)-4-(2-methoxy-ethyl)-thiazole-5-carboxylic acid methyl
ester
O
g
0
Add a 1.0 M solution of sufuryl chloride in dichloromethane (20.0 mL, 20.0
mmol) dropwise to a solution of 5-methoxy-3-oxo-pentanoic acid methyl ester
(3.0 g,
18.8 mmol) in dichloromethane (20.0 mL) at 0 C and stir under nitrogen at 0 C
for 2 h.
Concentrate the reaction mixture on a rotavap (rotary evaporator), keeping the
bath
temperature at RT. Add 4-chlorothiobenz-amide (3.67 g, 21.5 mmol) to the
residue,
followed by methanol (30.0 mL) and heat to 60 C for 18 h. Quench the reaction
with
water and extract with EtOAc (2x). Combine the organic portions, wash with
brine, dry
over MgSO4, filter, and concentrate under vacuum. Purify by flash
chromatography on
silica gel, using a gradient of EtOAc/Hexane (0-60%) to give the title
compound (3.3 g,
57%). Exact mass = 311.0, MS (ES+) 312.0 (M+1). 'H NMR (CDC13): 8 7.89 (d, 2H,
J
= 8.8 Hz), 7.40 (d, 211, J= 8.8 Hz), 3.88 (s, 3H), 3.82 (t, 2H, J= 6.8 Hz),
3.47 (t, 2H, J=
6.8 Hz), 3.38 (s, 3H).
Prepare the compounds below, Preparations 9b to 9f, by essentially following
the
procedure as described in Preparation 13, using the appropriate thiobenzamide
as starting
material.
Prep Product Structure MS (ES+)
(Chemical Name) or NMR
9b 4-(2-Methoxy-ethyl)-2-phenyl- CHS N I oMe 278.2(M+1)+.
thiazole-5-carboxylic acid oMe
0
methyl ester
9c 2-(3-Chloro-phenyl)-4-(2- - N oMe 312.3(M+1)+
r I
methoxy-ethyl)-thiazole-5- s oMe
ci o
carboxylic acid methyl ester
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
42
Prep Product Structure MS (ES+)
(Chemical Name) or NMR
9d 4-(2-Methoxy-ethyl)-2-(4- F N OMe 346.3(M+1)+
FF ~ ~ S I OMe
trifluoromethyl-phenyl)-
0
thiazole-5-carboxylic acid
methyl ester
9e 2-(2,4-Dichloro-phenyl)-4-(2- N oMe
CI H NMR (400
OMe
methoxy-ethyl)-thiazole-5- cl o MHz, CDC13)
carboxylic acid methyl ester S: 8.34 (d, 1H, J
= 7.3 Hz), 7.52
(d, 1H,J=2.0
Hz), 7.36 (dd,
1H,J=2.0,9.0
Hz), 3.90 (s,
3H), 3.83 (t,
2H, J = 6.9 Hz),
3.49 (t, 2H, J =
6.9 Hz).
9f 2-(4-Fluoro-phenyl)-4-(2- N OMe 296.3(M+1)+
S OMe
methoxy-ethyl)-thiazole-5-
0
carboxylic acid methyl ester
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
43
Preparation 10
5-Acetoxy-3-oxo-pentanoic acid ethyl ester
O O O
In a 2 L round bottom flask with stir bar, dissolve acetic acid 3-buten-1-yl
ester
(50 g, 438.1 mmol) in 1.5 L of dichloromethane and cool to -78 C. Vigorously
bubble
ozone through the reaction solution for about 2 h at which time the solution
becomes very
deeply colored (blue/purple). Bubble ozone through for an additional 5 min.
Discontinue
the ozone and bubble in oxygen until the color fades completely (about 15
min). To the
reaction, which is maintained at a temperature of -78 C, add dimethyl sulfide
(83.8 g,
99.0 mL, 1.35 mole). Allow to warm to ambient temperature overnight.
Concentrate the
reaction in vacuo to provide ccetic acid 3-oxo-propyl ester. Use the material
as is, with
no further purification or characterization.
Charge a round-bottom flask with tin(II) chloride (16.6 g, 0.088 mol), purge
with
nitrogen, and add dichloroethane (300 mL) by cannula. Add ethyl diazoacetate
(92 mL,
0.88 mol) by cannula and stir 10 min. Add a solution of acetic acid 3-oxo-
propyl ester
(0.44 mol) in CH2C12 (600 mL) slowly by cannula over 1 h, then stir the
reaction in a 50
C oil bath for 3 h. Concentrate under vacuum, add saturated aqueous NaHCO3 and
remove the organic phase. Extract the aqueous portion with EtOAc (2x). Wash
the
combined organic portions with brine, dry over MgSO4, filter through Celite ,
and
concentrate under vacuum. Purify by flash chromatography on silica gel,
eluting with a
gradient of EtOAc/hexane 8%-25% to give the title compound (27.8 g, 31%),
exact mass
202.08, mass spectrum (ES) 225.1 (M + Na). 1H NMR (CDC13): 8 4.34 (t, J= 6.1
Hz,
2H), 4.20 (q, J= 7.1 Hz, 2H), 3.46 (s, 2H), 2.89 (t, J= 6.1 Hz, 2H), 2.03 (s,
3H), 1.28 (t, J
= 7.1 Hz, 3H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
44
Preparation 11
5-Acetoxy-2-bromo-3-oxo-pentanoic acid ethyl ester
O O O
AO O'~
Br
Purge a round-bottom flask containing 5-acetoxy-3-oxo-pentanoic acid ethyl
ester
(11.3 g, 55.9 mmol) with nitrogen, add acetonitrile (250 rnL) by cannula and
chill in an
ice water bath. Add copper(II) bromide (13.1 g, 58.7 mmol) neat and stir 5 min
under
nitrogen. Add [hydroxy(tosyloxy)iodo]benzene (23.0 g, 58.7 mmol) neat, stir 5
min and
quench with water. Extract with ether (3x), wash combined organics with brine,
dry over
MgSO4, filter and concentrate under vacuum. Purify by flash chromatography on
silica
gel, using a gradient of EtOAc/hexane (8%-30%) to give the title compound
(6.56 g,
42%). 1H NMR (CDC13): 8 4.78 (s, 1H), 4.35 (t, J = 6.2 Hz, 2H), 4.29 (q, J =
7.1 Hz,
2H), 3.12 (q, J= 5.7 Hz, 2H), 2.04 (s, 3H), 1.32 (t, J= 7.1Hz, 3H).
Preparation 12
2-(4-Chloro-phenyl)-6,7-dihydro-pyrano[4,3-d]thiazol-4-one
O
S O
CI
Method 1: Purge a round-bottom flask containing 4-chloro-thiobenzamide (5.23
g, 18.6
mmol), with nitrogen, and add acetonitrile (50 mL) by syringe. Add a solution
of 5-
acetoxy-2-bromo-3-oxo-pentanoic acid ethyl ester (3.83 g, 22.3 mmol) in
acetonitrile (15
mL) by syringe and stir at RT under nitrogen for 1 h. Concentrate under vacuum
to a
solid, dilute with toluene (100 mL), water (5 drops) and add p-toluenesulfonic
acid
monohydrate (7.08 g, 37.2 mmol) neat. Attach a fractional distillation
apparatus with
collection flask and set in 120 C oil bath. After first distillate is
collected at
approximately 80 C (monitored at head of distillation column) increase oil
bath
temperature in 5 degree increments to 140 C until reaction has been
concentrated to one-
half volume. Remove from heat, neutralize with saturated aqueous NaHCO3,
extract with
EtOAc (3x), dry over MgSO4, filter and concentrate under vacuum. Purify by
flash
chromatography on silica gel, using a gradient of EtOAc in CH2C12 (0%-10%) to
give the
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
title compound (2.48 g, 48%). Exact mass = 265.0, MS (ES+) 266.0 (M+1)+. 1H
NMR
(CDC13): S 7.93 (dt, J= 8.4, 2.1 Hz, 2H), 7.46 (dt, J= 8.4, 2.2 Hz, 2H), 4.67
(t, J= 6.4
Hz, 2H), 3.23 (t, J= 6.4 Hz, 2H).
Method 2: Add 1.0 M solution of boron tribromide in dichloromethane (21.0 mL,
21.0
5 mmol) dropwise to a solution of 2-(4-chloro-phenyl)-4-(2-methoxy-ethyl)-
thiazole-5-
carboxylic acid methyl ester (6.0 g, 19.3 mmol) in dichloromethane (60.0 mL)
at -78 C
and stir under nitrogen at 0 C for 3 h. Quench reaction mixture with ether
(50.0 mL) and
water (50.0 mL), stir for additiona130 min and concentrate. Dilute residue
with water and
extract EtOAc (2x). Combine EtOAc, wash with brine, dry over MgSO4, filter,
and
10 concentrate under vacuum. Add p-TsOH (7.0 g, 36.8 mmol) and toluene (100.0
mL) to
the residue, reflux at 110 C for 18 h, and concentrate the reaction mixture.
Add saturated
NaHCO3 solution and extract with EtOAc (2x). Combine EtOAc, wash with brine,
dry
over MgSO4, filter, and concentrate under vacuum. Purify by flash
chromatography on
silica gel, using a gradient of MeOH/dichloromethane (0-5%) to give the title
compound
15 (1.5 g, 29%).
Method 3: Combine 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-
carboxylic acid
(81.2 g, 286.2 mmol) with p-TsOH monohydrate (32.0 g, 168.2 mmol) in toluene
(1200
mL). Heat the resulting slurry to reflux, eventually reaching a temperature of
approximately 112 C. Stir the resulting tan solution mechanically at reflux
for 2 h while
20 using a Dean-Stark trap to collect water. Allow the reaction to cool to
room temperature
and add saturated aqueous NaHCO3 (1700 mL) and EtOAc (1700 mL). Separate the
layers and extract the aqueous layer with EtOAc (2 x 1700 mL). Combine the
organic
layers, wash with brine (1700 mL) and concentrate in vacuo. Take up the
resulting solid
in CH2C12 (500 mL) and concentrate in vacuo, repeating with CH2C12 twice more
to
25 obtain 60.1 g (79.2% yield).
Prepare the compounds below, Preparations 12b to 12f, by essentially following
the
procedure as described in Preparation 12, Method 2, using the appropriate (2-
methoxy-
ethyl)-thiazole-5-carboxylic acid methyl ester as starting material.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
46
Prep Product Structure MS (ES+)
(Chemical Name)
12b 2-Phenyl-6,7-dihydro- C)~'Njl 232.2 (M+1)+
pyrano[4,3-d]thiazol-4-one s o
0
12c 2-(3-Chloro-phenyl)-6,7- p N I 266.2 (M+1)+
dihydro-pyrano[4,3- s 0
d]thiazol-4-one a o
12d 2-(4-Trifluoromethyl- F N 300.3 (M+1)+
F S
phenyl)-6,7-dihydro-
0
pyrano [4, 3 -d] thi azol-4-one
12e 2-(2,4-Dichloro-phenyl)- N 300.0 (M+1)"'
6,7-dihydro-pyrano[4,3-
d]thiazol-4-one Ci 0
12f 2-(4-Fluoro-phenyl)-6,7- FN 250.2 (M+1)+
dihydro-pyrano[4,3- s 0
d]thiazol-4-one 0
12g: 2-(4-Methoxy-phenyl)-6,7-dihydro-pyrano [4, 3-d] thiazol-4-one
~ O
MeO ~ N~
S
O
Prepare the title conipound by essentially following the procedure of Example
12,
Method 3, us'ing 4-(2-hydroxy-ethyl)-2-(4-methoxy-phenyl)-thiazole-5-
carboxylic acid.
MS (ES+) 262.2 (M+1)+. 1H NMR (400 MHz, CDC13): 8 7.94 (d, J= 9.2 Hz, 2H),
6.98
(d, J= 9.2 Hz, 2H), 4.66 (t, J= 6.2 Hz, 2H), 3.88 (s, 3H), 3.21 (t, J= 6.2 Hz,
2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
47
Preparation 13
tert-Butyl-(2-methoxy-4-nitro-phenoxy)-dimethyl-silane
02N I ,,Tt:::~
O,S i
1-1O
Add tert-butyl-dimethylsilylchloride ( 14 g, 90 mmol) to a solution of 4-
nitroguaiacol (5 g, 30 mmol) in DMF (250 mL) and then add imidazole (6.13 g,
90
mmol). Stir the mixture at room temperature for 16 h. Quench the reaction
mixture with
water (150 mL). Extract with diethyl ether (3 x 200 mL). Wash the combined
organic
portions with water, brine, and dry over MgSO4. Filter and concentrate to a
residue.
Purify the residue by silica gel flash chromatography, eluting with 15%
ethylacetate:hexanes to give the title compound (8.053 g, 95%) as a pale
yellow oil. MS
(ES+) 284.1 (M+1)+. 1H NMR(CDC13): S 7.79 (dd, J= 7.8 Hz, 2.7 Hz, 1H), 7.26
(d, J=
2.7 Hz, 1H), 6.69 (d, J= 7.8 Hz 1H), 3.85 (s, 3H), 0.96 (s, 9H), 0.18 (s, 6H).
Preparation 14
1-(2,2-Dimethoxy-ethoxy)-4-nitro-benzene
02N )::~O,,-yoMe
OMe
Dissolve glycolaldehyde dimethylacetal (5 g, 47.12 mmol) in dry DMF (100 mL)
and cool to 0 C. Add portion-wise NaH (60% dispersion, 1.88 g, 47.12 mmol).
Heat the
reaction mixture to 100 C overnight. Add water (200 mL) and extract with
EtOAc (3 x
50 mL). Dry the organic layer with Na2SO4, filter, and concentrate. Purify by
silica gel
chromatography, eluting with 0-50% EtOAc in hexanes to give the title compound
as a
wet yellow solid (7.96 g, 74%). 1H NMR(CDC13): 8 8.23 (d, J = 8.8 Hz, 2H),
7.02 (d, J
8.8 Hz, 2H), 4.77 (t, J= 4.8 Hz, 1H), 4.12 (d, J= 5.3 Hz, 2H), 3.50 (s, 6H).
Preparation 15
2,2-Dimethyl-propionic acid 2-methoxy-4-nitro-phenyl ester
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
48
O2N OMe
O
O
Dissolve trimethylacetyl chloride (3.64 mL, 29.56 mmol) in dry pyridine (100
mL). Add 4-nitroguaiacol (5.0 g, 29.56 mmol) followed by addition of DMAP (100
mg)
and stir overnight. Remove the pyridine via reduced pressure and then add 1N
HCl
solution to give a white solid precipitate which is collected by vacuum
filtration and
washed with water to give the title compound as a white solid (7.4 g, 99%).
'H NMR(CDC13): b 7.87 (dd, J = 8.8, 2.6 Hz, 1H), 7.82 (d, J = 2.6 Hz, 1H),
7.15 (d, J
8.8 Hz, 1H), 3.91 (s, 3H), 1.37 (s, 9H).
Preparation 16
1-(2,2-Dimethoxy-ethoxy)-2-methoxy-4-nitro-benzene
02N '_Z~ OMe
O"
01"
To an oven-dried round bottom flask, add 2-methoxy-4-nitro-phenol (2.45 g,
14.5
mmol) and purge with nitrogen. Add DMF (25 mL) by syringe, followed by K2C03
(3.0
g, 21.7 mmol) and KI (catalytic) neat. Stir 30 min at room temperature and add
2-bromo-
1,1-dimethoxy-ethane (1.9 mL, 15.9 mmol) by syringe. Attach a reflux condenser
and stir
overnight in a 120 C oil bath. Quench with water, extract with ether (3x),
dry over
MgSO4, filter and concentrate under vacuum. Add xylenes and concentrate again
under
vacuum. Purify by flash chromatography on silica gel using a gradient of
EtOAc/hexane
(20% to 60%) to give the title compound as a white residue (2.55 g, 68%).
Exact mass =
257.1, MS (ES+) 258.2 (M+1)+. 1H NMR (CDC13): S 7.88 (dd, J= 9.1, 2.8 Hz, 1H),
7.74
(d, J= 2.8 Hz, 1H), 6.94 (d, J= 9.1 Hz, 1H), 4.77 (t, J= 5.2 Hz, 1H), 4.13 (d,
J= 4.9 Hz,
2H), 3.93 (s, 3H), 3.48 (s, 6H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
49
Preparation 17
4-(2-Methoxy-4-nitro-phenyl)-morpholine
02N ~ O
~ N~
~O
Mix morpholine (1.50 mL, 17.20 mmol) and 1-chloro-2-methoxy-4-nitro-benzene
(1.06 g, 5.65 mmol) and heat to 100 C for 4 h while stirring. Cool the
solution to room
temperature, then partition between EtOAc (40mL) and 1N HCl (20mL). Wash the
organic solution with water (20 mL) and brine (20 mL), dry, filter, and
concentrate.
Purify the crude material by flash chromatography, using a linear gradient of
100%
hexanes to 50% EtOAc/hexanes, to give the title compound as a yellow solid
(250 mg,
18%). MS (ES+) 239.0 (M+l)+. 1H NMR (400 MHz, CDC13) 8: 7.86 (dd, 1H, J=8.8,
2.6
Hz), 7.71 (d, 1H, J=2.2 Hz), 6.87 (d, 1H, J=9.2 Hz), 3.94 (s, 3H), 3.87 (m,
4H), 3.21 (m,
4H).
Preparation 18
1-(2-Methoxy-4-nitro-phenyl)-piperidin-4-ol
02N I O~
aOH
Prepare the title compound by essentially following the procedure as described
for
Preparation 17, using 4-hydroxypiperidine. MS (ES+) 253.0 (M+1)+. 1H NMR (400
MHz, CDC13) S 7.83 (dd, 1H, J=8.8, 2.6 Hz), 7.69 (d, 1H, J=2.6 Hz), 6.88 (d,
1H, J=9.2
Hz), 3.99 (s, 1H), 3.94 (s, 3H), 3.90 (m, 1H), 3.56-3.50 (m, 2H), 2.99-2.91
(m, 2H), 2.07-
2.00 (m, 2H), 1.79-1.69 (m, 2H).
Preparation 19
1-Prop-2-ynyl-pyrrolidine
Add propargyl bromide (18.0 g, 120.0 mmol) dropwise at 0 C to a solution of
pyrrolidine (23.0 g, 323.0 mmol) in ether (50 mL). Stir for 18 h at room
temperature and
filter the reaction to remove the solids. Dilute the filtrate with water and
extract with
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
ether. Dry the ether with brine, then Na2SO4, and concentrate on a rotary
evaporator at
low temperature to give the title compound (10.0 g, 77%). MS (ES+) 110 (M+l)+.
1H
NMR (400 MHz, DMSO-d6): S 3.33 (d, 2H, J=2.2 Hz), 2.53 (m, 4H), 2.12 (t, 1H,
J=2.4
Hz), 1.72 (m, 4H).
5
Preparation 20
1- [3 -(2-Methoxy-4-nitro-phenyl)-prop-2-ynyl] -pyrrolidine
02N I O'll
N
Dissolve 1-iodo-2-methoxy-4-nitro-benzene (618 mg, 2.21 mmol) in acetonitrile
10 (10 mL) and treat sequentially with 1-prop-2-ynyl-pyrrolidine (352 mg, 3.22
mmol), Et3N
(2 mL), CuI (77 mg, 0.404 mmol) and Pd(PPh3)4 (360 mg, 0.311 mmol). Stir the
mixture
at room temperature for 3 h, then dilute with EtOAc (50 mL) and wash with
saturated
NaHCO3 (30 mL). Dry, filter and concentrate the organic solution. Purify the
crude
material by flash chromatography, using a linear gradient of 50% EtOAc/hexanes
to 100%
15 EtOAc, to give the title compound as an orange oil (292 mg, 51%). MS (ES+)
261.1
(M+1)+. 1H NMR (400 MHz, CDC13) S: 7.77 (dd, 1H, J=8.3, 2.2 Hz), 7.70 (d, 1H,
J=2.2
Hz), 7.50 (d, 1H, J=8.3 Hz), 3.95 (s, 3H), 3.72 (s, 2H), 2.75-2.70 (m, 4H),
1.87-1.83 (m,
4H).
20 Preparation 21
1 -(3, 3 -Diethoxy-prop-1-ynyl)-2-methoxy-4-nitro-benzene
O2N
O\
Prepare the title compound by essentially following the procedure as described
for
Preparation 20, using propargylaldehyde diethylacetal. IH NMR (400 MHz, CDC13)
8:
25 7.78 (dd, 1H, J=8.6, 2.0 Hz), 7.70 (d, 1H, J = 2.0 Hz), 7.55 (d, 1H, J=8.8
Hz), 5.52 (s,
1H), 3.95 (s, 3H), 3.87-3.78 (m, 2H), 3.71-3.63 (m, 2H), 1.27 (t, 6H, J=7.0
Hz).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
51
Preparation 22
1-(2-Methoxy-4-nitro-phenyl)-4-triisopropylsilanyloxy-piperidine
O2N + ~ O
/ N
O'S~
Dissolve 1-(2-methoxy-4-nitro-phenyl)-piperidin-4-ol (1.19 g, 4.72 mmol) in
DMF (25 mL), followed by addition of triisopropylsilyl-
trifluoromethanesulfonate (1.50
mL, 5.56 mmol) and Et3N (0.80 mL, 5.87 mmol). Stir the solution at room
temperature
for 2 h, then add water (50 mL) and extract with EtOAc (2 x 50 mL). Combine
the
organic solutions and wash with water (2 x 30 mL) and brine (30 mL), then dry,
filter, and
concentrate. Purify the crude material by flash chromatography, using a linear
gradient of
100% hexanes to 20% EtOAc/hexanes, to give the title compound as a yellow
solid (1.55
g, 80%). MS (ES+) 409.3 (M+1)+. 1H NMR (400 MHz, CDC13) S: 7.84 (dd, 1H,
J=8.8,
2.6 Hz), 7.68 (d, 1H, J=2.6 Hz), 6.89 (d, 1H, J=8.8 Hz), 4.07-4.01 (m, 1H),
3.93 (s, 3H),
3.45-3.38 (m, 2H), 3.13-3.06 (m, 2H), 1.99-1.91 (m, 2H), 1.81-1.73 (m, 2H),
1.07-1.06
(m, 21H).
Preparation 23
3-Methoxy-4-(3-pyrrolidin-1-yl-propyl)phenylamine
H2N O~
(C~ N
Dissolve 1-[3-(2-methoxy-4-nitro-phenyl)-prop-2-ynyl]-pyrrolidine (292 mg,
1.12
mmol) in EtOH (5 mL) and treat with 5% Pd/C. Purge the black mixture with
hydrogen,
then stir overnight at room temperature under a hydrogen atmosphere (1 atm).
Filter the
black mixture through a pad of Celite and wash the solids with additional
EtOH (20
rnL). Concentrate the filtrate to give the title compound as an oil (240 mg,
91%). MS
(ES+) 235.2 (M+1)+. 1H NMR (400 MHz, CDC13) 8: 6.88 (d, 1H, J=8.3 Hz), 6.22-
6.18
(m, 2H), 3.73 (s, 3H), 3.55 (s, 2H), 2.53-2.41 (m, 8H), 1.79-1.72 (m, 6H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
52
Preparation 24
H2N I
0
4-(3,3-Diethoxy-propyl)-3-methoxy-phenylamine
Prepare the title compound using procedures as essentially described for
Preparation 23. 1H NMR (400 MHz, CDC13) S: 6.89 (d, 1H, J=8.3 Hz), 6.23-6.20
(m,
2H), 4.48 (t, 1H, J=5.9 Hz), 3.75 (s, 3H), 3.68-3.60 (m, 2H), 3.64 (br s, 2H),
3.52-3.44
(m, 2H), 2.57-2.52 (m, 2H), 1.80-1.89 (m, 2H), 1.20 (t, 6H, J=7.0 Hz).
Preparation 25
7-Nitro-4H-benzo [ 1,41 oxazin-3-one
02N ~ O
I/
NI O
H
Mix 2-amino-5-nitro-phenol (10.0 g, 64.9 mmol) and NaHCO3 (13.1 g, 155.7
mmol) in 4-methyl-pentan-2-one (40 mL) and water (40 mL). Cool the m.ixture to
0 C
and slowly add chloroacetyl chloride (6.0 mL, 75.3mmo1) with stirring. After
the addition
is complete, reflux the mixture for 5h. Cool the mixture to room temperature
and let
stand for 2.5 days. Collect the light yellow solid, wash with water and dry in
a vacuum
oven at 80 C for 3 h. MS (ES-) 193.1 (M-1)-. 1H NMR (400 MHz, DMSO-d6): b
11.31
(s, 1H), 7.90 (dd, 1H, J=8.8, 2.2 Hz), 7.76 (d, 1H, J=2.6 Hz), 7.06 (d, 1H,
J=8.8 Hz), 4.72
(s, 2H),
Preparation 26
7-Nitro-3,4-dihydro-2H-benzo [ 1,4] oxazine
02N,:: O
N
H
Mix 7-nitro-4H-benzo[1,4]oxazin-3-one (2.00 g, 10.3 mmol) in THF (10 mL) and
treat with BH3=THF (l.OM in THF, 35 mL). Heat the solution to reflux for 30
min, then
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
53
cool to 0 C and quench with 1N HCl (20 mL). Stir the solution for 30 min, then
concentrate to 1/2 volume. Collect the orange solid, wash with water, and dry
under
vacuum to give the title compound (1.66 g, 89%). MS (ES+) 181.1 (M+1)+, MS (ES-
)
179.2 (M-1)". 1H NMR (400 MHz, DMSO-d6): S 7.68 (dd, 1H, J=8.8, 2.6 Hz), 7.53
(s,
1H), 7.47 (d, 1H, J=2.6 Hz), 6.63 (d, 1H, J=9.2 Hz), 4.15 (t, 2H, J=4.4 Hz),
3.44-3.40 (m,
2H),
Preparation 27
5-nitro-l-(2-pyrrolidin-1-yl-ethyl)-1H-indole
02N "Z~
N
a
Dissolve 1-(2-chloro-ethyl)-pyrrolidine hydrochloride (2.36 g, 13.9 mmol) and
5-
nitro-lH-indole (1.50 g, 9.23 mmol) in DMF (25 rnL) and carefully treat with
sodium
hydride (60% dispersion, 1.50 g, 37.5 mmol). Stir the mixture at room
temperature
overnight, then dilute with cold water (100 mL) and extract with EtOAc (3 x 50
mL).
Was the combined organic portions with water (2 x 50 mL) and brine (50 mL).
Dry, filter
and concentrate under vacuum. Purify the crude material by flash
chromatography, using
100% acetone as eluant, to give the title compound as a yellow oil (2.08 g,
87%). MS
(ES+) 260.1 (M+1)+. 1H NMR (400 MHz, CDC13): S 8.57 (d, 1H, J= 2.2 Hz), 8.10
(dd,
1H, J = 9.2, 2.2 Hz), 7.37 (d, 1H, J = 9.2 Hz), 7.30 (d, 1H, J= 3.1 Hz), 6.67
(d, 1H, J
3.1 Hz), 4.29 (t, 2H, J= 7.3 Hz), 2.89 (t, 2H, J= 7.0 Hz), 2.54 (m, 4H), 1.78
(m, 4H).
Prepare the compounds below, Preparations 28 to 36, by essentially following
the
procedure as described in Preparation 27, using the appropriate nitroaryl or
nitroheterocycle.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
54
Prep Product Structure MS (ES+)
(Chemical Name)
28 5-Nitro-1-(2-pyrrolidin-l-yl- 02N llz~ N> 261.1 (M+1)+
~
ethyl)-1H-benzoimidazole
~
G
29 5-Nitro-1-(2-pyrrolidin-l-yl- 02N N 261.1 (M+1)+
ethyl)-1H-indazole
CN'
~
v
30 5-Nitro-2-(2-pyrrolidin-l-yl- 02N 261.1 (M+1)+
ethyl)-2H-indazole NN
N~
31 2-Methyl-5-nitro-l-(2- 02N I~\ 274.2 (M+1)+
pyrrolidin-l-yl-ethyl)-1H- N
indole
v
32 2,3-Dimethyl-5-nitro-l-(2- 288.1 (M+l)+.
02N
pyrrolidin-l-yl-ethyl)-1H-
indole
C
33 1-[2-(2-Chloro-4-nitro- O2N CI 271.0 (M+1)+
phenoxy)-ethyl]-pyrrolidine
O
34 5-Nitro-1-(2-pyrrolidin-l-yl- 02N N~F 329.1 (M+1)+
~
ethyl)-2-trifluoromethyl-1 H- N F F
benzoimidazole
C
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
rep Product Structure MS (ES+)
(Chemical Name)
35 7-Nitro-4-(2-pyrrolidin-l-yl- 02N ~ O 278.2 (M+1)+
ethyl)-3,4-dihydro-2H- ~ i ~
N
benzo[1,4]oxazine
a_____
36 1-[2-(4-Nitro-phenoxy)-ethyl]- 02N alz:~ 237.2 (M+1)+
pyrrolidine _'N
Preparation 37
(R)-1-(2-Methoxy-4-nitro-phenyl)-3-triisopropylsilanyloxy-pyrrolidine
O2N OMe
N~õ0
5 Combine 1-chloro-2-methoxy-4-nitro-benzene (10 g, 53.3 nunol) and (3R)-3-
pyrrolidinol (9.3 g, 106.6 mmol). Heat the mixture to 100 C overnight. Cool
the
mixture and dissolve in CH2C12 (200 rnL) and wash with 1N NaOH (100 mL). Wash
the
extract with brine (3 x 50 mL). Dry the organic layer with Na2SO4, filter, and
concentrate
to give the intermediate pyrrolidinol as a crude dark reddish wet solid (12.17
g, 95%).
1 MS (ES+) 239.1 (M+1)+.
Dissolve the crude (R)-1-(2-methoxy-4-nitro-phenyl)-pyrrolidin-3-ol (10.9 g,
45.5
mmol) in dry pyridine (50 mL) and chill to 0 C. Add chloro-triisopropyl-silane
(19.8
mL, 91 mmol) dropwise and then heat to 80 C overnight. Remove the pyridine
via
reduced pressure and then wash the crude material with NaHSO3 solution and
extract
15 with EtOAc (3 x 100 mL). Combine the organic solutions, then dry and
concentrate to
give the crude product. Purify over a silica plug with hexanes (300 mL) and
flush with
10% EtOAc in hexanes (800 mL) to give the title compound as a reddish oil
(17.85 g,
99%). MS (ES+) 395.2 (M+1)+. 1H NMR (400 MHz, CDC13): 8 7.81 (dd, J= 8.8, 2.2
Hz, 1H), 7.63 (d, J = 2.2 Hz, 1H), 6.45 (d, J= 88 Hz, 1H), 4.57-4.52 (m, 1H),
3.84 (s,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
56
3H), 3.84-3.78 (m, 1H), 3.72-3.64 (m, 1H), 3.61-6.53 (m, 1H), 3.45 (dd, J
11.0, 2.2 Hz,
111), 2.06-1.92 (m, 2H), 1.04-1.01 (m, 21H).
Preparation 38
1-(6-Nitro-1 H-indol-3 -yl)-2-pyrrolidin-1-yl-ethane-1,2-dione
'
J
O N
O
02N N
H
Add oxalyl chloride (11.6 g, 90.6 mmol) dropwise to a solution of 6-
nitroindole
(10.6 g, 65.4 mmol) in ether (100 mL). Stir at room temperature for 18 h,
filter the
precipitate formed, and dry. Dissolve the precipitate in CH2C12 (100 mL), cool
to -20 C,
and add pyrrolidine (16.0 mL, 191.5 nunol) dropwise. Warm to room temperature
and
stir for 2 h. Filter the solid from the reaction, wash several times with
ether, and dry to
give the title compound (8.5 g, 45%). MS (ES+) 288 (M+1)+.
Preparation 39
1-(1-Methyl-6-nitro-1 H-indol-3-yl)-2-pyrrolidin-1-yl-ethane-1,2-dione
'
N
J
O
O
02N N
Add NaH (0.83 g, 20.8 mmol) to a solution of 1-(6-nitro-lH-indol-3-yl)-2-
pyrrolidin-1-yl-ethane-1,2-dione (5.0 g, 17.42 nunol) in THF (60 mL). Stir at
room
temperature for 10 min, add iodomethane (1.18 mL, 19.2 mmol), and continue
stirring for
18 h. Dilute with water and extract with EtOAc (2x). Filter the solid that
formed
between the layers during the extraction. Dry organic portion, concentrate,
and combine
the solids. Triturate the solid with ether, filter, and dry to obtain the
title compound (5.20
g, 99%). 'H NMR (400 MHz, DMSO-d6): 5 8.59 (m, 2H), 8.30 (d, 1H, J=8.8 Hz),
8.17
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
57
(dd, 1H, J=8.8, 2.2 Hz), 4.01 (s, 3H), 3.48 (t, 2H, J=6.8 Hz), 3.41 (t, 2H,
J=6.4 Hz), 1.85
(m, 4H).
Preparation 40
1-Methyl-6-nitro-3-(2-pyrrolidin-1-yl-ethyl)-1 H-indole
c
02N
~
Treat a solution of 1-(1-methyl-6-nitro-lH-indol-3-yl)-2-pyrrolidin-1-yl-
ethane-
1,2-dione (5.0 g, 17.4 mmol) in THF (20 mL) with BH3.THF (70 mL of 1N in THF,
70
mmol) and stir at room temperature for 18 h. Concentrate the reaction mixture
and add
EtOH (100 mL) followed by 5N HCl (20 mL) and reflux for 6 h. Concentrate and
dilute
with 1N NaOH (100 mL). Extract with CH2C12 (2x), then extract with EtOAc (2x).
Combine the organics, dry, and concentrate. Purify by flash chromatography
using 0 -
10% 2N NH3/MeOH in CH2C12, to give the title compound (2.5 g, 53%). 1H NMR
(400
MHz, DMSO-d6): S 8.26 (d, 1H, J=2.2 Hz), 7.98 (dd, 1H, J=8.8, 1.8 Hz), 7.61.
(d, 1H,
J=8.8 Hz), 7.19 (s, 1H), 3.84 (s, 3H), 2.97 (t, 2H, J=8.1 Hz), 2.76 (t, 2H,
J=8.1 Hz), 2.61
(m, 4H), 1.83 (m, 4H).
Preparation 41
1-(2-Methoxy-4-nitro-benzyl)-4-methyl-piperazine
NO2
O
N
To a round bottom flask or vial containing 2-methoxy-4-nitro-benzaldehyde (1.0
g, 5.5 mmol), add dichloroethane (40 mL), 1-methylpiperazine (1.0 ml, 8.3
mmol), and
sodium triacetoxyborohydride (3.5 g, 16.5 mmol). Stir at room temperature
overnight.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
58
Quench with saturated aqueous NaHCO3 and extract with CH2C12 (lx) and EtOAc
(2x).
Combine the organic portions, dry over MgSO4, filter, and concentrate under
vacuum.
Purify the residue by flash chromatography on silica gel using a gradient of
MeOH(0.005% NH4OH)/CH2C12 (5% to 10%) to give the title compound. MS (ES+)
266.0 (M+1)+. 1H NMR(CDC13): S 7.80 (dd, J = 8 Hz, 2 Hz, 1H), 7.66 (d, J = 2
Hz, 1H),
7.56 (J= 8 Hz, 1H), 3.89 (s, 3H), 3.58(s, 2H), 2.52 (br. 4H), 2.45 (br, 2H),
2.28 (s, 3H).
Preparation 42
1-(2-Pyrrolidin-l-yl-ethyl)-1H-indol-5-ylamine
H2N I ~ \
N
C
Dissolve 5-nitro-l-(2-pyrrolidin-1-yl-ethyl)-1H-indole (375 mg, 1.45 mmol) in
ethanol (15 mL) and add 5% Pd/C (149 mg). Purge the black mixture with
hydrogen (1
atm) and stir overnight under a hydrogen atmosphere. Filter the black mixture
through
Celite and wash the solids with additional ethanol (-lOmL). Concentrate the
filtrate to
give the title compound as a yellow solid. MS (ES+) 230.2 (M+1)+. 1H NMR
(400MHz,
CDC13): S: 7.17 (d, 1H, J=8.8 Hz), 7.05 (d, 1H, J=3.1 Hz), 6.92 (d, 1H, J=2.2
Hz), 6.67
(dd, 1H, J=8.3, 2.2 Hz), 6.29 (d, 1H, J=3.1 Hz), 4.21 (t, 2H, J=7.5 Hz), 3.37
(s, 2H), 2.86
(t, 2H, J=7.5 Hz), 2.55 (m, 4H), 1.79 (m, 4H).
Prepare the compounds below, Preparations 43 to 59, essentially following the
procedure as described in Preparation 42 using the appropriate nitro compound
which is
previously prepared or commercially available.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
59
rep Product Structure MS (ES+)
(Chemical Name) and/or NMR
43 1-(2-Pyrrolidin-1-yl-ethyl)- H2N IIZZ~ N> 231.2 (M+1)+
1H-benzoimidazol-5- ~ N
ylamine
44 1-(2-Pyrrolidin-1-yl-ethyl)- H2N ';Zzz 231.2 (M+1)+
1H-indazol-5-ylamine N
v
45 2-(2-Pyrrolidin-1-yl-ethyl)- 231.2 (M+1)+
2H-indazol-5-ylamine H2N ~~ 1H NMR (400
MHz, CDC13): S
7.70 (s, 1 H), 7.52
(d, 1H, J = 9.2
Hz), 6.78 (dd, 1H,
J = 9.2, 2.2 Hz),
6.73 (d, 1H, J =
2.2 Hz), 4.46 (t,
2H, J = 7.0 Hz),
3.53 (s, 2H), 3.05
(t, 4H, J = 6.8
Hz), 2.53 (m,
4H).
46 2-Methyl-1-(2-pyrrolidin-l- H2N 244.2 (M+1)+.
yl-ethyl)-1H-indol-5- I ~ N
ylamine
G
47 4-(2,2-Dimethoxy-ethoxy)- H2N ~ OMe 227.1 (M)+,
3-methoxy- hen lamine o~
P Y p'y'
O~1
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
rep Product Structure MS (ES+)
(Chemical Name) and/or NMR
48 3-Methoxy-4-(4-methyl- NH2 236.1 (M+1)+
piperazin-l-ylmethyl)- I ~
phenylamine 0
~\
49 3-Methoxy-4-(2-pyiTolidin- H2N ~ o~ 237.3 (M+1)+
1-yl-ethoxy)-phenylamine I i N
50 2,3-Dimethyl-l-(2- 258.3 (M+1)+
pyrrolidin-1-yl-ethyl)-1H- H2N I \
~
indol-5-ylamine N
51 6-(4-Methyl-piperazin-l- H2N N 193.3 (M+1)+
yl)-pyridin-3-ylamine f
52 1-(2-Pyrrolidin-1-yl-ethyl)- H2N N~F 299.1 (M+1)+.
2-trifluoromethyl-lH- N F F
benzoimidazol-5-ylamine
v
53 1-Methyl-3-(2-pyrrolidin-l- 244.2 (M+1)+.
yl-ethyl)-1H-indol-6- H2N c N
ylaniine
N
0
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
61
rep Product Structure MS (ES+)
(Chemical Name) and/or NMR
54 3-Methoxy-4- H2N ~ O 296.1 (M+l)+
triisopropylsilanyloxy- I i
O /
phenylamine Si--(
\
55 4-(2-Pyrrolidin-1-yl-ethyl)- H2N a,, 0 248.2 (M+1)+
3,4-dih dro-2H- J
y N
benzo [ 1,4]oxazin-7- J
ylamine 0N
56 -(2-Pyrrolidin-1-yl-ethoxy)- H2N 207.5 (M+1)+
phenylamine O~~N
57 3-Methoxy-4-morpholin-4- H2N I--' o 209.3 (M+1)+.
yl-phenylamine N
Oo
58 4-(2,2-Dimethoxy-ethoxy)- H2N ~ H NMR (400
hen lamine I/ OMe MHz, CDC13) S:
p Y o"."y 6.76 (m, 2H),
OMe 6.21 (m, 2H),
4.68 (t, 1H, J=
5.4 Hz), 3.93 (d,
2H,J=5.OHz),
3.44 (s, 3H), 3.38
(br s, 2H).
59 4-(tert-Butyl-dimethyl- H2N O~ H NMR (400
silanyloxy)-3-methoxy- MHz, CDC13) S:
O-S\ 6.65 (d, 1H, J=7.9
phenylamine Hz), 6.30 (d, 1H,
J=2.6 Hz), 6.21
(dd, 1H, J=8.4,
2.6 Hz), 3.79 (s,
2H), 3.74 (s, 3H),
0.97 (s, 9H), 0.11
(s, 6H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
62
Preparation 60
5-Nitro-l-triisopropylsilanyl-lH-indole
2N I
N
Dissolve 5-nitro-lH-indole (5.00 g, 30.8 mmol) in DMF (100 mL) and treat with
NaH (1.62 g, 40.5 mmol). Stir the mixture at room temperature for 1 h and then
add
triisopropyl-silyl-trifluoromethanesulfonate (9.15 mL, 33.9 mmol). Stir the
mixture for
an additional 2 h then dilute with water (100 mL) and 1N HCl (40 mL), then
extract with
EtOAc (3X100mL). Combine the organic solutions and wash with water (2X5OmL)
and
brine (50mL). Dry, filter and concentrate the organic solution and purify the
crude
material by flash chromatography, using a linear gradient of 100% hexanes to
20%
EtOAc/hexanes as eluant, to give the title compound as a clear yellow oil
(6.30g, 64%).
1H NMR (400 MHz, CDC13) 8:8.56 (d, 1H, J=2.6 Hz), 8.05 (dd, 1H, J=9.0, 2.4
Hz), 7.51
(d, 1H, J=9.2 Hz), 7.38 (d, 1H, J=3.1 Hz), 6.78 (d, 1H, J=3.5 Hz), 1.74-1.66
(m, 3H),
1.14 (d, 18H, J=7.9 Hz).
Preparation 61
5-Nitro-l-triisopropylsilanyl-2,3-dihydro-1 H-indole
02N ~
N
~Si
Prepare the title compound by essentially following procedures as described
for
Preparation 60, using 5-nitroindoline. MS (ES+) 320.1 (M)+. 1H NMR (400 MHz,
CDC13): S 7.95 (dd, 1H, J=8.8, 2.6 Hz), 7.92-7.90 (m, 1H), 6.56 (d, 1H, J=9.2
Hz), 3.86
(t, 2H, J=8.8 Hz), 3.08 (t, 2H, J=8.8 Hz), 1.46 (m, 3H), 1.14 (d, 18H, J=7.5
Hz).
Prepare the compounds below, Preparations 62-65, by essentially following the
procedure
as described in Preparation 42.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
63
Preparation 62
1-Triisopropylsilanyl-2,3-dihydro-lH-indol-5-ylamine
H2N\~~ N
MS (ES+) 290.2 (M)+. 'H NMR (400 MHz, CDC13): S 6.62-6.27 (m, 3H), 3.66 (s,
2H),
2.89 (s, 2H), 1.45-1.33 (m, 3H), 1.10 (d, 18H, J=7.5 Hz).
Preparation 63
1-Triisopropylsilanyl-lH-indol-5-yl amine
H2N \
~~\%~ N
Si
1H NMR (400 MHz, CDC13): S 7.29 (d, 1H, J=8.8 Hz), 7.16 (d, 1H, J=3.1 Hz),
6.92 (d,
1H, J=2.6 Hz), 6.59 (dd, 1H, J=8.8, 2.2 Hz), 6.43 (d, 1H, J=3.1 Hz), 1.69-1.61
(m, 3H),
1.12 (d, 18H, J=7.5 Hz).
Preparation 64
2,2-Dimethyl-propionic acid 4-amino-2-methoxy-phenyl ester
H2N OMe
/
O
O
1H NMR (400 MHz, CDC13): 6.76 (d, J=8.8 Hz, 1H), 6.31 (d, J=2.6 Hz, 1H), 6.25
(dd,
J= 8.8, 2.2 Hz, 1H), 3.74 (s, 3H), 1.34 (s, 9H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
64
Preparation 65
(R)-3-Methoxy-4-(3-triisopropylsilanyloxy-pyrrolidin-1-yl)-phenylamine
H2N ~ OMe
~ /
Nõ111p
Si
--("r
Dissolve (R)-1-(2-Methoxy-4-nitro-phenyl)-3-triisopropylsilanyloxy-pyrrolidine
(12 g, 30.4 mmol) in EtOH (200 mL) and add 5% Pd/C (1.26 g). Purge the black
mixture
with hydrogen (1 atm) and stir overnight under a hydrogen atmosphere at
ambient
temperature at 60 psi. Filter the black mixture through Celite and wash the
solids with
additional EtOH (1 OmL). Concentrate the filtrate to give the title compound
as a dark
brown oil. 1H NMR (400 MHz, CDC13): S 6.76 (d, 1H, J=8.8 Hz), 6.31 (d, 1H,
J=2.6
Hz), 6.25 (dd, 1H, J= 8.8, 2.6 Hz), 3.74 (s, 3H), 1.34 (s, 9H). Note: title
compound
decomposed rapidly. Store compound in freezer immediately after use. MS (ES+)
365.2
(M+1)+.
Preparation 66
3-Chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine
H2N q C-11-iN
CI
Add sodium borohydride (0.58 g, 15.26 mmol) to a solution of 1-[2-(2-chloro-4-
nitro-phenoxy)-ethyl]-pyrrolidine (0.83 g, 3.07 mmol) and NiC12.6H20 (1.45 g,
6.12
mmol) in MeOH (20 mL). Stir at room temperature for 2 h and add 10% NH4OH
solution. Extract with CH202 and then EtOAc, combine the organics, dry, and
concentrate. Purify by flash chromatography using 0 - 10% 2N NH3/MeOH in
CH2C22, to
give the title compound (0.5 g, 69%). MS (ES+) 241.2 (M+1)+. 1H NMR (400 MHz,
CDC13): S 6.78 (d, 1H, J=8.4 Hz), 6.72 (d, 1H, J=3.1 Hz), 6.51 (dd, 1H, J=8.4,
3.1 Hz),
4.07 (t, 2H, J=6.2 Hz), 3.47 (s, 2H), 2.90 (t, 2H, J=6.2 Hz), 2.64 (m, 4H),
1.79 (m, 4H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Preparation 67
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-methoxy-
4-(4-
triisopropylsilanyloxy-piperidin-1-yl)-phenyl]-amide
N OH
CI ~~ S ~ O N o,~
I ~ N
O,Si
r
5 Dissolve 1-(2-Methoxy-4-nitro-phenyl)-4-triisopropylsilanyloxy-piperidine
(1.53g,
3.74 mmol) in THF (30 mL) and add 5% Pd/C then stir the slurry at room
temperature
under a hydrogen atmosphere for 3 h. Filter the black mixture through a pad of
Celite
and concentrate the filtrate in vacuo to give 3-methoxy-4-(4-
triisopropylsilanyloxy-
piperidin-l-yl)-phenylamine (1.42 g, 100%) that was used immediately.
10 Dissolve the above 3-methoxy-4-(4-triisopropylsilanyloxy-piperidin-l-yl)-
phenylamine (1.41 g, 3.72 mmol) in CH2C12 and add a trimethylaluminum solution
(2.OM
in hexanes, 2.25 mL, 4.50 mmol). Stir the solution at room temperature for 1
h, then add
solid 2-(4-chloro-phenyl)-6,7-dihydro-pyrano[4,3-d]thiazol-4-one (1.01 g, 3.80
mmol)
and continue stirring at room temperature overnight. Carefully quench reaction
with
15 saturated Rochelle's salt solution (15 mL) and stir at room temperature for
1 h. Extract
the mixture with CH2C12 (3 x 20mL). Combine all organic solutions, dry,
filter, and
concentrate in vacuo. Purify the crude material by flash chromatography using
2N
NH3/MeOH in CH2C12 as eluent to give the title compound as a sold (1.00 g,
42%). MS
(ES+) 644.0 (M+1)+, (ES-) 642.3 (M-1)-. 1H NMR (400 MHz, CDC13): S 10.76 (s,
1H),
20 8.00 (d, 2H, J=8.4 Hz), 7.60 (d, 2H, J=8.8 Hz), 7.35 (d, 1H, J=2.2 Hz),
7.13 (dd, 1H,
J=8.8, 2.2 Hz), 6.88 (d, 1H, J=8.8 Hz), 5.78 (t, 1H, J=4.4 Hz), 3.94-3.86 (m,
3H), 3.78 (s,
3H), 3.21-3.13 (m, 4H), 2.78-2.70 (m, 2H), 1.93-1.85 (m, 2H), 1.67-1.57 (m,
2H), 1.06-
1.04 (m, 21H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
66
Preparation 68
(R)-2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-
methoxy-4-(3-
triisopropylsilanyloxy-pyrrolidin-1-yl)-phenyl] -amide
OH
GI H
S N ~ OMe
0
N.,0
Charge an oven-dried round bottom flask with (R)-3-methoxy-4-(3-
triisopropylsilanyloxy-pyrrolidin-1-yl)-phenylamine (750 mg, 2.05 nunol),
purge with
nitrogen, and dilute with CH2C12 (11 mL). Add trimethylaluminum (2M in
hexanes, 1.03
mL, 2.05 mmol) dropwise by syringe and stir 20 min at room temperature. Add
solid 2-
(4-chloro-phenyl)-6,7-dihydro-pyrano[4,3-d]thiazol-4-one (364 mg, 1.37 mmol)
to the
reaction mixture and stir overnight at ambient temperature. Absorb the
reaction mixture
on silica gel and purify by silica gel flash chromatography, using a gradient
of
EtOAc/hexane (0-100%) to give the title compound (1.02 g, 73%). MS (ES+) 630.1
(M+1)+. 'H NMR (CDC13): S 9.88 (bs, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.48 (bs,
1H), 7.37
(d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.4 Hz, 1H), 6.69 (bs, 1H), 4.60-4.52 (m,
1H), 4.19-4.11
(m, 2H), 3.81 (s, 3H), 3.69-3.58 (m, 1H), 3.39-3.29 (m, 1H), 3.26 (t, J = 5.3
Hz, 2H),
3.20-3.02 (m, 2H), 2.21-2.08 (m, 1H), 1.93-1.83 (m, 1H), 1.59 (bs, 1H), 1.12-
0.95 (m,
21H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
67
Preparation 69
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [1-(2-
pyrrolidin-l-yl-
ethyl)-1 H-indol-5-yl] -amide
N OH
CI H
O N
N
Dissolve 1-(2-pyrrolidin-l-yl-ethyl)-1H-indol-5-ylamine (247 mg, 1.08 mmol) in
CH2Cl2 (5 mL), cool to 0 C, and treat with a solution of trimethylaluminum
(2.0 M in
hexanes, 0.7 mL, 1.40 mmol). Stir the solution at 0 C for 15 min and then at
room
temperature for 30 min. Add 2-(4-chloro-phenyl)-6,7-dihydro-pyrano[4,3-
d]thiazol-4-one
(272 mg, 1.02 mmol) neat and stir the reaction at room temperature overnight.
Carefully
quench the mixture with saturated Rochelles salt solution (5 mL) and stir at
room
temperature for 1 h. Dilute with additional saturated Rochelles salt solution
(10 mL) and
extract with CH2C12 (3 x 20 mL). Combine the organic portions, dry, filter,
and
concentrate under vacuum. Triturate the crude solid with diethyl ether to give
the title
compound as a white powder (400 mg, 75%). MS (ES+) 495.1 (M+1)+MS (ES-) 493.2
(M-1)-. 1H NMR (400 MHz, DMSO-d6): S 10.78 (s, 1H), 8.01 (d, 2H, J= 8.3 Hz),
7.92
(s, 1H), 7.60 (d, 2H, J= 8.8 Hz), 7.46 (d, 1H, J= 8.8 Hz), 7.40 (d, 1H, J= 3.1
Hz), 7.34
(dd, 1H, J= 8.8, 1.8 Hz), 6.41 (d, 1H, J= 3.1 Hz), 5.84 (m, 1H), 4.26 (t, 2H,
J= 6.6 Hz),
3.91 (q, 2H, J= 5.3 Hz), 3.21 (t, 2H, J= 5.9 Hz), 2.78 (t, 2H, J= 6.8 Hz),
2.46 (s, 4H),
1.65 (m, 4H).
Prepare the compounds below, Preparations 70 to 88, by essentially following
the
procedure as described in Preparation 69. Preparation 82 was made using
4-methyl-N2-(2-morpholin-4-yl-ethyl)quinoline-2,6-diamine (Krahler, S. E.;
Burger, A. J.
Am. Chern. Soc., 1941, 63 2367-71).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
68
Preparation 70
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [1-(2-
pyrrolidin-l-yl-
ethyl)-1 H-benzoimidazol-5-yl]-amide
OI \ N ( H OH
~ N
O , N\>
MS (ES+) 496.0 (M+1)+, (ES-) 494.2 (M-1)".
Preparation 71
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [1- (2-
pyrrolidin-l-
yl-ethyl)-1 H-indazol-5-yl] -amide
CI N I H OH
S
NN
O
MS (ES+) 496.0 (M+1)+, (ES-) 494.2 (M-1)".
Preparation 72
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [2-(2-
pyrrolidin-l-yl-
ethyl)-2H-indazol-5-yl]-amide
OH
CI ~ ~ H
N
S N /
O N~N~
MS (ES+) 496.0 (M+1)+, (ES-) 494.2 (M-1)-.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
69
Preparation 73
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [2-methyl-l-
(2-
pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-amide
CI ~~ N I H OH
N
O N
v
MS (ES+) 509.0 (M+1)+, (ES-) 507.0 (M-1)-.
Preparation 74
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [2,3-
dimethyl-l-(2-
pyrrolidin-l-yl-ethyl)-1 H-indol-5-yl]-amide
N OH
CI H
S I N
O / N
MS (ES+) 523.1 (M+1)+.
Preparation 75
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [1-methyl-3-
(2-
pyrrolidin-1-yl-ethyl)-1 H-indol-6-yl]-amide
H OH
CI N I
::;
N
O
~.J
MS (ES+) 509.1 (M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Preparation 76
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [ 1-(2-
pyrrolidin-l-yl-
ethyl)-2-trifluoromethyl-1 H-benzoimidazol-5-yl]-amide
N OH
CI I H
g
N NF F
-~~
0
N F
v
5 MS (ES+) 564.1 (M+1)+.
Preparation 77
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (3-methoxy-
4-
triisopropylsilanyloxy-phenyl)-amide
HN OH
CI S N ~
O / O.Si
MS (ES+) 561.1 (M+l)+.
Preparation 78
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [6-(4-
methyl-
piperazin-1-yl)-pyridin-3-yl]-amide
CI NOH
S N N
O ONII
MS (ES+) 458.0 (M+1)+.
Preparation 79
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-chloro-4-
(2-
pyrrolidin-1-yl-ethoxy)-phenyl] -amide
CI N H OH
CI
S I N ao,~No
0 MS (ES+) 506.0 (M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
71
Preparation 80
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(2-
pyrrolidin-1-yl-
ethyl)-3,4-dihydro-2H-benzo [ 1,4]oxazin-7-yl]-amide
OH
N
O
O l
MS (ES+) 513.0 (M+l)+, (ES-) 511.2 (M-1)-.
Preparation 81
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-methoxy-
4-(3-
pyrrolidin-1-yl-propyl)-phenyl]-amide
N OH
CI 0 S qN O
O N\
MS (ES+) 500.4 (M+1)+.
Preparation 82
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-methyl-2-
(2-
morpholin-4-yl-ethylamino)-quinolin-6-yl]-amide
N I OH
01 ~ ~ S N
0 N NN
H
MS (ES+) 552.1 (M+1)+.
Preparation 83
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(3,3-
diethoxy-
propyl)-3-methoxy-phenyl]-amide
N O
CI ~N, O"
O
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
72
1H NMR (400 MHz, CDC13): b 9.99 (s, 1H), 7.85 (d, 2H, J=8.8 Hz), 7.54 (d, 1H,
J=1.8
Hz), 7.41 (d, 2H, J=8.3 Hz), 7.04 (d, 1H, J=7.9 Hz), 6.87 (dd, 1H, J=8.1, 2.0
Hz), 4.50 (t,
1H, J= 5.9 Hz), 4.19 (t, 2H, J=5.3 Hz), 3.82 (s, 3H), 3.69-3.60 (m, 2H), 3.53-
3.45 (m,
2H), 3.29 (t, 2H, J=5.3 Hz), 2.62 (t, 2H, J=7.9 Hz), 1.91-1.81 (m, 2H), 1.23-
1.17 (m, 6H).
Preparation 84
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(2-
pyrrolidin-l-yl-
ethoxy)-phenyl]-amide
N OH
CI ~ ~ S ~ N 010 O,,,N
MS (ES+) 472.0 (M+1)+, 470.0 (M-1)".
Preparation 85
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (3-methoxy-
4-
morpholin-4-yl-phenyl)-amide 15
N OH
CI 3 I N
O N~
~O
MS (ES+) 474.0 (M+1)+, (ES-) 472.3 (M-1)-.
Preparation 86
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (1-
triisopropylsilanyl-
1 H-indol-5-yl)-amide
N OH
CI ~ ~ ~ N
C I ~ N
MS (ES+) 554.1 (M+1)+, 552.3 (M-1)".
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
73
Preparation 87
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (1-
triisopropylsilanyl-2,3-dihydro-1 H-indol-5-yl)-amide
N OH
cl N
o ~ i N
MS (ES+) 556.0 (M+1)+.
Preparation 88
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid 3,4-
dimethoxy-
benzylamide
N OH O-
CI ~~ S ' N ~
O
O 1
MS (ES+) 433.0 (M+1)+, MS (ES-) 431.0 (M-1)-. 1H NMR(CDC13): S 9.13 (t, 1H,
J=5.7
Hz), 7.96 (d, 2H, J--8.8 Hz), 7.58 (d, 2H, J=8.8 Hz), 6.94 (d, 1H, J=1.8 Hz),
6.91 (d, 1H,
J=7.9 Hz), 6.85 (dd, 1H, J=8.4, 1.8 Hz), 5.29 (t, 1H, J=4.6 Hz), 4.40 (d, 2H,
J=5.7 Hz),
3.82-3.76 (m, 2H), 3.71 (s, 3H), 3.70 (s, 3H), 3.15 (t, 2H, J=6.2 Hz).
Preparation 89
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(2,2-
dimethoxy-
ethoxy)-3-methoxy-phenyl]-amide
o~_
a ' S 0 ~ c 0,
~ / ~ N ~ ~ ~O~
N
OH
Method 1: Charge an oven-dried round bottom flask with 4-(2,2-dimethoxy-
ethoxy)-3-
methoxy-phenylamine (0.65 g, 2.88 mmol), purge with nitrogen, and dilute with
toluene
(5 mL). Add trimethylaluminum (2 M in hexanes, 1.44 mL, 2.88 mmol) dropwise by
syringe and stir 5 min at room temperature. Add 2-(4-chloro-phenyl)-6,7-
dihydro-
pyrano[4,3-d]thiazol-4-one (0.51g, 1.91 mmol) in toluene (20 mL), attach a
reflux
condenser and stir overnight in an 80 C oil bath. Allow to cool to ambient
temperature
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
74
and add 1N HCI, extracting with EtOAc (3x). Dry the combined organic portions
over
MgSO4, filter, and concentrate under vacuum. Purify by flash chromatography on
silica
gel, using a gradient of EtOAc/hexane (20%-70%) to give the title compound
(0.78 g,
83%). Exact mass = 492.1, MS (ES+) 493.4 (M+1)+. 1H NMR (CDC13): S 9.91 (s,
1H),
7.88 (dt, J= 8.5, 2.2 Hz, 2H), 7.57 (ap d, 1H), 7.42 (dt, J = 8.5, 2.2 Hz,
2H), 6.94 (dd, J
8.8, 2.4 Hz, 111), 6.87 (d, J= 8.8 Hz, 1 H), 4.75 (t, J = 5.2 Hz, 1H), 4.22
(t, J = 5.2 Hz,
2H), 4.03 (d, J= 5.2 Hz, 2H), 3.87 (s, 3H), 3.46 (s, 6H), 3.31 (t, J= 5.2 Hz,
2H).
Prepare the compounds Preparations 90 and 91, by essentially following the
procedures
as described in Preparation 89, Method 1.
Preparation 90
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(tert-
butyl-
dimethyl-silanyloxy)-3 -methoxy-phenyl] -amide
o~
cl \ ~ s O ~ .
N ~ / si
H
OH
Exact mass 518, mass spectrum (ES) 519.3 (M+1)+.
Preparation 91
2-(4-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-methoxy-
4-(4-
methyl-piperazin-l-ylmethyl)-phenylj-amide
g ON :r O ON",
OH
Exact mass: 500.0, MS(ES+): 501.3 (M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Preparation 92
2,2-Dimethyl-propionic acid 4-{ [4-(2-hydroxy-ethyl)-2-(4-methoxy-phenyl)-
thiazole-5-
carbonyl]-amino } -2-methoxy-phenyl ester
OH
Me0 / I
N N~ OMe
O
O
5 Method 2: Prepare the title compound by essentially following procedures as
described in
Preparation 89, Method 1, except the reaction n-iixture is run overnight at
ambient
temperature. Quench the reaction mixture with 1N HCl (20 mL) and extract with
CH2C12
(3 x10 mL). Dry the combined organic portions with Na2SO4, filter, and
concentrate.
Purify by flash chromatography on silica gel, using a gradient of EtOAc/hexane
(20%-
10 70%) to give the title compound. MS (ES+) 485.2 (M+1)+. 1H NMR (CDC13): S
10.01
(s, 1H), 7.90 (d, J= 9.2 Hz, 2H), 7.70 (d, J= 1.8 Hz, 1H), 6.95 (dd, J= 8.4;
2.2 Hz, 2H),
6.92 (d, J= 1.8 Hz, 1H), 6.91 (d, J= 8.4 Hz, 1H), 4.18 (t, J= 5.3 Hz, 2H),
3.85 (s, 3H),
3.79 (s, 3H), 3.29 (t, J= 5.3 Hz, 2H), 1.36 (s, 9H).
15 Preparation 93
4-(2-Hydroxy-ethyl)-2-(4-methoxy-phenyl)-thiazole-5-carboxylic acid [4-(2,2-
dimethoxy-
ethoxy)-3-methoxy-phenyl]-amide
/ ~ N OH
Me0 S N OMe
O I
ly OMe
OMe
Method 3: Prepare the titled compound by essentially following procedures as
described
20 in Preparation 89, Method 1, except run the reaction overnight at ambient
temperature.
Cool the reaction mixture and add 1N NaOH (25 mL), extract with EtOAc (3 x 10
mL).
Filter the solid precipitate from the partitioned aqueous/organic layer to
give the title
compound as a fine yellow powder. MS (ES+) 489.2 (M+1)+. 1H NMR (d4_MeOH): 8
7.92 (d, J= 8.8 Hz, 2H), 7.44 (d, J= 2.2 Hz, 1H), 7.11 (dd, J= 8.8, 2.2 Hz,
1H), 7.03 (d,
25 J = 8.8 Hz, 2H), 6.95 (d, J = 8.8 Hz, 1 H), 4.70 (t, J = 5.3 Hz, 1 H), 4.05
(t, J = 5.3 Hz,
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
76
2H), 3.99 (d, J = 5.3 Hz, 2H), 3.86 (d, J = 4.4 Hz, 6H), 3.44 (s, 6H), 3.25
(t, J = 5.7 Hz,
2H).
Preparation 94
4-(2-Hydroxy-ethyl)-2-phenyl-thiazole-5-carboxylic acid [4-(2,2-dimethoxy-
ethoxy)-
phenyl]-amide
OH
S 1 N OMe
0 OMe
Method 4: Prepare the title compound by essentially following the procedures
as
described in Preparation 89, Method 1, using 4-(2,2-dimethoxy-ethoxy)-
phenylamine (385
mg, 1.95 mmol) and 2-phenyl-6,7-dihydro-pyrano[4,3-d]thiazol-4-one (300 mg,
1.30
mmol). Heat the reaction mixture for 1 h at 70 C (no reflux condenser is
needed). Cool
the reaction mixture and add water (20 mL), then extract with EtOAc (3 x 10
mI.). Dry
the organic layer with Na2SO4, filter, and concentrate. Purify on silica gel
chromatography with 0-100% EtOAc in hexanes to give the title compound as a
light
brown solid. MS (ES+) 429.2 (M+1)+, (ES-) 427.2 (M-1)-.
Preparation 95
2-(3-Chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [4-(2,2-
dimethoxy-
ethoxy)-3-methoxy-phenyl]-amide
OH
2,NJMeoMe
O
OMe
cl
Method 5: Prepare the title compound by essentially following procedures as
described
in Preparation 89, Method 1, using the following alternate work-up. Dilute
with 1N
NaOH, and extract with EtOAc (3 x 10 mL). Dry with Na2SO4, filter, and
concentrate.
Add minimal amounts of CH2C12 to extract color and then add hexanes to give a
solid
precipitate. Collect the solid via vacuum filtration. Wash the solid with
hexanes to give
the title compound. MS (ES+) 493.2 (M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
77
Prepare the following compounds, Preparations 96 to 98, by essentially
following the
procedures as described in Preparation 95, Method 5.
Prep Product Structure MS (ES+)
(Chemical Name)
96 4-(2-Hydroxy-ethyl)-2-(4- OH 527.2 (M+1)+
trifluoromethyl-phenyl)- N OMe
F oMe
thiazole-5-carbox lic acid F'' 'J S o
y F OMe
[4-(2,2-dimethoxy-
ethoxy)-3-methoxy-
phenyl]-amide
97 2-(2,4-Dichloro-phenyl)- CI ~ " H 527 (M+1)+
~
4-(2-hydroxy-ethyl)= S N OMe
ci o
thiazole-5-carboxylic acid I
SOMe
[4-(2,2-dimethoxy- ~O"Me
ethoxy)-3-methoxy-
phenyl]-amide
98 2-(4-Fluoro-phenyl)-4-(2- F N " H 477.2 (M+1)+
hydroxy-ethyl)-thiazole-5- s N oMe
o
carboxylic acid [4-(2,2-
OMe
dimethoxy-ethoxy)-3- OMe
methoxy-phenyl]-amide
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
78
Example 1
2-(4-Chloro-phenyl)-5-[ 1-(2-pyrrolidin-1-yl-ethyl)-1 H-indol-5-yl]-6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one
GI N X
S N ~ ~
O I /
N
a
Method 1: Dissolve 2-(4-chloro-phenyl)-4-(4-hydroxy-ethyl)-thiazole-5-
carboxylic acid
[1-(2-pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-amide (390 mg, 0.79 mmol) in THF
(8.0 mL)
and cool to 0 C. Treat the solution with tributylphosphine (0.255 mL, 1.03
mmol) and
diisopropylazodicarboxylate (0.205 mL, 1.04 mmol). Warm the solution to room
temperature and stir overnight. Dilute the solution with EtOAc (50 mL) and
wash with
water (25 mL) and brine (25 mL). Dry the organic portion, filter and
concentrate under
vacuum. Purify the crude material by flash chromatography, using 8% 2N
NH3/MeOH in
CHC13, to give a foam. Triturate the foam with ether to give the title
compound as a
yellow solid (289 mg, 77%). MS (ES+) 477.4 (M+1)+. 1H NMR (400MHz, DMSO-d6):
8 8.06 (d, 2H, J = 8.8 Hz), 7.62 (d, 2H, J = 8.8 Hz), 7.49-7.53 (m, 2H), 7.44
(d, 1H, J =
3.1 Hz), 7.13 (dd, 1H, J= 8.8, 2.2 Hz), 6.43 (d, 1H, J= 3.1 Hz), 4.29 (t, 2H,
J= 6.6 Hz),
4.11 (t, 2H, J= 6.8 Hz), 3.29 (t, 2H, J= 6.8 Hz), 2.80 (t, 2H, J= 6.8 Hz),
2.49 (m, 4H),
1.66 (m, 4H).
Prepare Example 2 to 13 and Preparations 99 to 106 by essentially following
the
procedures as described in Example 1, Method 1, using the appropriate
intermediate 4-
hydroxy-ethyl-thiazole.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
79
Example 2
2-(4-Chloro-phenyl)-5-[ 1-(2-pyrrolidin-1-yl-ethyl)-1 H-benzoimidazol-5-yl]-
6,7-dihydro-
5H-thiazolo [5,4-c]pyridin-4-one
CI N
N N
S
C I i
(D
MS (ES+) 478.4 (M+1)+. 1H NMR (400 MHz, CDC13): S 8.03 (s, 1H), 7.93 (d, 2H,
J=
8.8 Hz), 7.72 (d, 1H, J= 1.8 Hz), 7.43-7.46 (m, 3H), 7.34-7.38 (m, 1H), 4.37
(s, 2H), 4.18
(t, 2H, J= 7.0 Hz), 3.32 (t, 2H, J= 7.0 Hz), 2.99 (s, 2H), 2.61 (s, 4H), 1.82
(s, 4H).
Example 3
2-(4-Chloro-phenyl)-5-[1-(2-pyrrolidin-1-yl-ethyl)-1H-indazol-5-yl]-6, 7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
CI / \ N
S N C 0 NN
V
MS (ES+) 478.4 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.98 (s, 1H), 7.93 (d, 2H, J
8.8 Hz), 7.65 (d, 1H, J= 1.3 Hz), 7.48 (d, 1H, J= 8.8 Hz), 7.44 (d, 2H, J= 8.8
Hz), 7.39
(dd, 1H, J= 9.0, 2.0 Hz), 4.55 (t, 2H, J= 7.5 Hz), 4.16 (t, 2H, J= 7.0 Hz),
3.32 (t, 2H, J=
7.0 Hz), 3.01 (t, 2H, J= 7.3 Hz), 2.58 (s, 4H), 1.78 (m, 4H).
Example 4
2-(4-Chloro-phenyl)-5-[2-(2-pyrrolidin-1-yl-ethyl)-2H-indazol-5-yl]-6, 7-
dihydro-5H-'
thiazolo[5,4-c]pyridin-4-one
-XI
N
C' / \
N 31
0
MS (ES+) 478.4 (M+1)+. 1H NMR (400 MHz, DMSO-d6): b 8.42 (d, 1H, J = 0.9 Hz),
8.06 (d, 2H, J= 8.8 Hz), 7.67-7.69 (m, 1H), 7.60-7.64 (m, 3H), 7.25 (dd, 1H,
J= 9.0, 2.0
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Hz), 4.54 (t, 2H, J= 6.4 Hz), 4.14 (t, 2H, J= 6.8 Hz), 3.28 (t, 2H, J= 7.0
Hz), 2.97 (t, 2H,
J = 6.4 Hz), 2.47 (s, 4H), 1.65 (m, 4H).
Example 5
5 2-(4-Chloro-phenyl)-5-[2-methyl-l-(2-pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-
6,7-dihydro-
5H-thiazolo [5,4-c]pyridin-4-one
ci / \ N
S N
0 I ~ N
a
MS (ES+) 491.1 (M+1)+. 1H NMR (400 MHz, DMSO-d6): S 8.05 (d, 2H, J= 7.9 Hz),
7.62 (d, 2H, J = 8.3 Hz), 7.37-7.42 (m, 2H), 7.06 (d, 1H, J = 8.3 Hz), 6.22
(s, 1H), 4.23 (s,
10 2H), 4.09 (t, 2H, J= 6.6 Hz), 3.27 (s, 2H), 2.69 (s, 2H), 2.43 (s, 3H),
1.69 (s, 4H), 1.69 (s,
4H).
Example 6
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(4-methyl-piperazin-1-ylmethyl)-phe
15 nyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one
oll
N~
S N ~N"
cl
MS (ES+) 483.3 (M+1)+. 1HNMR(CDC13): b 7.92 (d, J= 7.4 Hz, 2H), 7.44 (d, J=
7.4
Hz, 2H), 7.39 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 1.8Hz, 1H), 6.86 (dd, J= 8.0Hz,
1.8Hz,
1H), 4.11 (t, J= 5.2.Hz, 2H), 3.82 (s, 3H), 3.55 (s, 2H), 3.28 (t, J= 5.2 Hz,
2H), 2.62-2.44
20 (m, 8H), 2.29 (s, 3H).
Example 7
2-(4-Chloro-phenyl)-5-[2,3-dimethyl-l-(2-pyrrolidin-1-yl-ethyl)-1H-indol-5-yl]-
6,7-
dihydro-5H-thiazolo [5,4-c] pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
81
CI N
S N
O N
a
MS (ES+) 505 (M+1)+. 1H NMR (400 MHz, CDC13: S 7.94 (d, 2H, J=8.3 Hz), 7.44
(m,
3H), 7.27 (d, 1 H, J=10.1 Hz), 7.10 (dd, 1 H, J= 8.6, 2.0 Hz), 4.24 (brs, 2H),
4.15 (t, 2H,
J=6.8 Hz), 3.31 (t, 2H, J=7.0 Hz), 2.77 (brs, 2H), 2.63 (brs, 4H), 2.37 (s,
3H), 2.22 (s,
3H), 1.85 (brs, 4H).
Example 8
2-(4-Chloro-phenyl)-5-[ 1-methyl-3-(2-pyrrolidin-1-yl-ethyl)-1H-indol-6-yl]-
6,7-dihydro-
5H-thiazolo [5,4-c]pyridin-4-one
N
CI ~
N ~
0 ~ / /
V
MS (ES+) 491.1 (M+1)+. 1H NMR (400 MHz, CDC13): 8 7.93 (d, 2H, J=8.3 Hz), 7.63
(d,
1H, J=8.3 Hz), 7.45 (d, 2H, J=8.3 Hz), 7.29 (s, 1H), 7.04 (d, 1H, J=8.3 Hz),
6.92 (s, 1H),
4.17 (t, 2H, J=7.0 Hz), 3.72 (s, 3H), 3.32 (t, 2H, J=6.8 Hz), 2.99 (t, 2H,
J=8.1 Hz), 2.78
(t, 2H, J=8.1 Hz), 2.63 (m, 4H), 1.84 (m 4H).
Example 9
2-(4-Chloro-phenyl)-5- [4-(2-pyrrolidin-1-yl-ethyl)-3,4-dihydro-2H-benzo [
1,4] oxazin-7-
N~/~
CI SN ~ C
C I / "
yl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one ~"
MS (ES+) 495.0 (M+1)+. 1H NMR (400 MHz, DMSO-d6): 8 8.03 (d, 2H, J=8.4 Hz),
7.61 (d, 2H, J=8.4 Hz), 6.78 (dd, 1H, J=8.6, 2.4 Hz), 6.73-6.67 (m, 2H), 4.16
(t, 2H,
J=4.0 Hz), 3.99 (t, 2H, J=7.0 Hz), 3.42-3.37 (m, 4H), 3.22 (t, 2H, J=7.0 Hz),
2.61 (t, 2H,
J=6.8 Hz), 2.52-2.47 (m, 4H), 1.68 (s, 4H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
82
Example 10
2-(4-Chloro-phenyl)-5- [3-methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
N
CI ~ S I
N cL MS (ES+) 482.0 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.93 (d, 2H, J=8.3 Hz),
7.45 (d,
2H, J=7.9 Hz), 7.16 (d, 1H, J=7.9 Hz), 6.89 (d, 1H, J=1.8 Hz), 6.82 (dd, 1H,
J=7.9, 2.2
Hz), 4.12 (t, 2H, J=6.8 Hz), 3.82 (s, 3H), 3.29 (t, 2H, J=7.0 Hz), 2.65 (t,
2H, J=7.7 Hz),
2.54 (s, 6H), 1.87-1.77 (m, 6H).
Example 11
2-(4-Chloro-phenyl)-5-[4-methyl-2-(2-moipholin-4-yl-ethylamino)-quinolin-6-yl]-
6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
N
CI ~
N ~ O
0 N N--,,N,/
H
MS (ES+) 534.0 (M+1)+. 1H NMR (400 MHz, DMSO-d6): S 8.07 (d, 2H, J=8.3 Hz),
7.71 (s, 1H), 7.63 (d, 2H, J=8.3 Hz), 7.49 (s, 2H), 6.84 (m, 1H), 6.67 (s,
1H), 4.18 (t, 2H,
J=6.8 Hz), 3.59 (t, 4H, J=4.4 Hz), 3.54-3.48 (m, 2H), 3.35-3.28 (m, 2H), 2.51
(m, 2H),
2.44 (m, 7H).
Example 12
2-(4-Chloro-phenyl)-5-[4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
CI S ~ N \
0 C~,N
MS (ES+) 454.0 (M+1)+. 'H NMR (400 MHz, CDC13): 8 7.93 (d, 2H, J=8.8 Hz), 7.45
(d,
2H, J=8.8 Hz), 7.25 (d, 2H, J=8.8 Hz), 6.96 (d, 2H, J=8.8 Hz), 4.15 (t, 2H,
J=5.7 Hz),
4.08 (t, 2H, J=7.0 Hz), 3.28 (t, 2H, J=6.8 Hz), 2.94 (s, 2H), 2.67 (s, 4H),
1.83 (s, 4H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
83
Example 13
2-(4-Chloro-phenyl)-5-(3-methoxy-4-morpholin-4-yl-phenyl)-6,7-dihydro-5H-
thiazolo [5,4-c]pyridin-4-one, hydrochloride salt
N
cl ~
N ~ 0
O I N~
HCI ~p
Prepare the hydrochloride salt of the free base by mixing 2-(4-chloro-phenyl)-
5-(3-
methoxy-4-moipholin-4-yl-phenyl)-6,7-dihydro-5H-thiazolo [5,4-c]pyridin-4-one
(273
mg, 0.599 mmol) in MeOH (4 mL) and adding a l.OM HCl/ether (0.7 mL, 0.70 mmol)
solution. After all solids dissolve, cool the solution to -20 C for 4 days.
Collect the
white precipitate by filtration, wash with ether, and dry under vacuum to give
the title
compound as a white solid (275 mg, 57%). MS (ES+) 456.0 (M+1)+. 1H NMR (400
MHz, DMSO-d6): S 8.05 (d, 2H, J=8.4 Hz), 7.62 (d, 2H, J=8.4 Hz), 7.18 (s, 1H),
7.12 (s,
1H), 7.00-6.96 (m, 1H), 5.69 (s, 1H), 4.10 (t, 2H, J=7.0 Hz), 3.86-3.80 (m,
7H), 3.27 (t,
2H, J=7.0 Hz), 3.20-3.12 (m, 4H).
Preparation 99
2-(4-Chloro-phenyl)-5- [4-(2,2-dimethoxy-ethoxy)-3 -methoxy-phenyl] -6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one
o-
o o o-
N~N p-
Exact mass = 474.1, MS (ES+) 475.2 (M+1)+. 1H NMR (CDC13): S 7.93 (d, J= 8.5
Hz,
2H), 7.45 (d, J= 8.5 Hz, 2H), 6.97-6.93 (m, 2H), 6.83 (dd, J= 8.5, 2.5 Hz,
1H), 4.76 (t, J
= 5.2 Hz, 1H), 4.09 (t, J= 7.0 Hz, 2H), 4.06 (d, J= 5.2 Hz, 2H), 3.86 (s, 3H),
3.46 (s,
6H), 3.29 (t, J = 7.0 Hz, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
84
Preparation 100
5- [4-(2,2-Dimethoxy-ethoxy)-3 -methoxy-phenyl] -2 -(4-methoxy-phenyl)-6,7-
dihydro-5 H-
thiazolo[5,4-c]pyridin-4-one
Me0 ~ ~
N OMe
0
/OMe
~O"Me
MS (ES+) 471.3 (M+1)+. 1H NMR (CDC13): S 8.01 (d, J= 8.8 Hz, 2H), 7.00 (d, J=
9.2
Hz, 2H), 6.96-6.99 (m, 2H), 6.81-6.87 (m, 1H), 4.76 (t, J= 5.3 Hz, 1H), 4.09
(t, J= 7.0
Hz, 2H), 4.06 (d, J= 5.3 Hz, 2H), 3.89 (s, 3H), 3.86 (s, 3H), 3.46 (s, 6H),
3.34 (t, J= 7.0
Hz, 2H).
Preparation 101
5-[4-(2,2-Dimethoxy-ethoxy)-phenyl]-2-phenyl-6,7-dihydro-5H-thiazolo[5,4-
c]pyridin-4-
one
/ \ N I
0 \ O~ ~,OMe
N
~O"Me
MS (ES+) 411.2 (M+1)+. 1H NMR (CDC13): 8 8.02 (d, J= 7.9 Hz, 2H), 7.52-7.49
(m,
3H), 7.31 (d, J= 9.2 Hz, 2H), 7.00 (d, J= 8.8 Hz, 2H), 4.77 (t, J= 5.3 Hz, 1
H), 4.12 (t, J
= 6.6 Hz, 2H), 4.06 (d, J= 5.3 Hz, 2H), 3.50 (s, 6H), 3.33 (t, J= 7.0 Hz, 2H).
Preparation 102
5- [4-(tert-Butyl-dimethyl-silanyloxy)-3-methoxy-phenyl]-2-(4-chloro-phenyl)-
6,7-
dihydro-5H-thiazolo [5,4-c] pyridin-4-one
o-
ci o.
j i
N~
Exact mass = 500.1, MS (ES+) 501.3 (M+1)+. 1H NMR (CDC13): cS 7.93 (d, J= 8.6
Hz,
2H), 7.44 (d, J= 8.6 Hz, 2H), 6.90 (d, J= 8.4 Hz, 1 H), 6.86 (d, J= 8.4 Hz, 1
H), 6.75 (dd,
J= 8.4, 2.5 Hz, 1H), 4.09 (t, J= 7.0 Hz, 2H), 3.81 (s,3H), 3.28 (t, J= 7.0 Hz,
2H), 1.00 (s,
9H), 0.17 (s, 6H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Preparation 103
2-(4-Chloro-phenyl)-5-(3-methoxy-4-triisopropylsilanyloxy-phenyl)-6,7-dihydro-
5H-
thiazolo [5,4-c]pyridin-4-one
ci ~\N S
N
o (
"O 0
5 MS (ES+) 543.4 (M+1)+. 1H NMR (400 MHz, CDC13): 5 7.92 (d, 2H, J=8.4 Hz),
7.43 (d,
2H, J=8.8 Hz), 6.86 (m, 2H), 6.72 (dd, 1H, J=8.4, 2.6 Hz), 4.07 (t, 2H, J=6.8
Hz), 3.78
(s, 3H), 3.26 (t, 2H, J=6.8 Hz), 1.23 (m, 3H), 1.08 (d, 18H, J=7.5 Hz).
Preparation 104
10 2-(4-Chloro-phenyl)-5-[3-methoxy-4-(4-triisopropylsilanyloxy-piperidin-1-
yl)-phenyl]-
6,7-dihydro-5H-thiazolo [5,4-c]pyridin-4-one
N
CI
o N I:10,
O-cL.
MS (ES+) 626.0 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.92 (d, 2H, J=8.8 Hz), 7.44
(d,
2H, J=8.4 Hz), 6.96 (d, 1H, J=8.4 Hz), 6.88 (d, 1H, J=2.2 Hz), 6.84 (dd, 1H,
J=8.4, 2.6
15 Hz), 4.09 (t, 2H, J=7.0 Hz), 4.00-3.94 (m, 1H), 3.86 (s, 3H), 3.27 (t, 4H,
J=7.0 Hz), 2.90-
2.83 (m, 2H), 2.01-1.93 (m, 2H), 1.84-1.75 (m, 2H), 1.08-1.06 (m, 21H).
Preparation 105
2-(4-Chloro-phenyl)-5-(1-triisopropylsilanyl-2,3-dihydro-lH-indol-5-yl)-6,7-
dihydro-5H-
2 0 thiazolo[5,4-c]pyridin-4-one
N
cl
o N
Si
MS (ES+) 538.0 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.91 (d, 2H, J=8.3 Hz), 7.43
(d, 2H, J=8.3 Hz), 7.05-7.03 (m, 1H), 6.86 (dd, 1H, J=8.3, 2.2 Hz), 6.61 (d,
1H, J=8.8
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
86
Hz), 4.04 (t, 2H, J=6.8 Hz), 3.74 (t, 2H, J=8.6 Hz), 3.23 (t, 2H, J=6.8 Hz),
3.00 (t, 2H,
J=8.8 Hz), 1.47-1.38 (m, 3H), 1.13 (d, 18H, J=7.5 Hz).
Preparation 106
2-(4-Chloro-phenyl)-5-(3,4-dimethoxy-benzyl)-6,7-dihydro-5H-thiazolo [5,4-
c]pyridin-4-
one
N
CI
I N ~ I
0
MS (ES+) 415.0 (M+1)+. 1H NMR(CDC13): 8 7.88 (d, 2H, J=8.4 Hz), 7.41 (d, 2H,
J=8.8
Hz), 6.88-6.85 (m, 2H), 6.81 (d, 1H, J=8.8 Hz), 4.66 (s, 2H), 3.86 (s, 3H),
3.85 (s, 3H),
3.58 (t, 2H, J=7.0 Hz), 3.07 (t, 2H, J=7.0 Hz).
Preparation 107
2-(3-Chloro-phenyl)-5- [4-(2,2-dimethoxy-ethoxy)-3 -methoxy-phenyl] -6,7-
dihydro-5H-
thiazolo [5,4-c] pyridin-4-one
/ ~ / q oMe
~ g
OMe
O ~ O
01 oMe
Method 2: Prepare the title compound by essentially following procedures as
described
for Example 1, Method 1, with the following exceptions. When the reaction is
complete
remove the solvent via reduced pressure. Dissolve the residue in minimal
amounts of
CH2Cla, then add hexanes until a solid precipitates. Collect the solid via
vacuum
filtration. Wash the solid with hexanes several times to give the title
compound. MS
(ES+) 475.2 (M+1)}. 1HNMR(CDC13): S 8.02 (s, 1H), 7.85 (d, J= 7.5 Hz, 1H),
7.49-7.39
(m, 2H), 6.97-6.93 (m, 2H), 6.87-6.82 (m,1 H), 4.77 (t, J= 5.4 Hz, 1 H), 4.10
(t, J= 7.0
Hz, 2H), 4.07 ( d, J = 4.8 Hz, 2H), 3.86 (s, 3H), 3.46 (s, 6H), 3.30 (t, J =
7.0 Hz, 2H).
Prepare the compounds below, Preparation 108 to 110, by essentially following
the
procedure as described in Preparation 107, Method 2, using the appropriate 4-
hydroxy-
ethyl-thiazole intermediate.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
87
Preparation 108
5- [4-(2,2-Dimethoxy-ethoxy)-3 -methoxy-phenyl]-2-(4-trifluoromethyl-phenyl)-
6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
FF ~-~ ~S N I OMe OMe
F O \ O~
OMe
MS (ES+) 509.2 (M+1)+.
Preparation 109
2-(2,4-Dichloro-phenyl)-5- [4-(2,2-dimethoxy-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-
5H-thiazolo [5,4-c]pyridin-4-one
OMe
cl S Q N ~ I OMe
OI OMe
MS (ES+) 509.0 (M+1)+.
Preparation 110
5-[4-(2,2-Dimethoxy-ethoxy)-3-methoxy-phenyl]-2-(4-fluoro-phenyl)-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one
F ~ \ 1SI N OMe OMe
O
OMe
MS (ES+) 459.2 (M+1)+.
Preparation 111
{4-[2-(4-Chloro-phenyl)-4-oxo-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-2-
methoxy-
phenoxy } -acetaldehyde
o-
ci ;~~ t o ~
\ N N
~
Method 1: Combine 2-(4-chloro-phenyl)-5-[4-(2,2-dimethoxy-ethoxy)-3-methoxy-
phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (0.695 g, 1.46 mmol), p-
toluenesulfonic acid (0.224 g, 1.16 mmol), acetone (10 mL) and water (2 mL).
Attach a
reflux condenser and stir at 70 C overnight. Concentrate under vacuum,
neutralize with
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
88
saturated aqueous NaHCO3, and extract with EtOAc (3x). Wash the combined
organic
portions with brine, dry over MgSO4, and concentrate under vacuum to give the
title
compound. IH NMR (CDC13): S 9.90 (s, 1H), 7.93 (d, J= 8.6 Hz, 2H), 7.45 (d, J=
8.6
Hz, 2H), 7.01-6.84 (m, 3H), 4.62 (d, J= 1.2 Hz, 2H), 4.13-4.05 (m, 2H), 3.86
(s, 3H),
3.30 (t, J= 6.1 Hz, 2H).
Preparation 112
{ 2-Methoxy-4-[2-(4-methoxy-phenyl)-4-oxo-6,7-dihydro-4H-thiazolo[5,4-
c]pyridin-5-
yl ] -phenoxy } -acetaldehyde
OMe
Me0 S 0 p //0
~ /
~
Method 2: Dissolve 5-[4-(2,2-Dimethoxy-ethoxy)-3-methoxy-phenyl]-2-(4-methoxy-
phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (145 mg, 0.309 mmol) in
THF (2
ml) and 1N HC1 solution (360 l). Heat to 50-60 C overnight (no reflux
condenser
used). Cool reaction mixture, filter solid via vacuum filtration and wash
solid with H20
to give the title compound. MS (ES+) 425.4 (M+1)+.
Prepare the compounds in the table below, Preparations 113 to 117, by
essentially
following the procedure as described in Preparation 112, Method 2 using the
appropriate
starting acetal.
Prep Name Structure MS
{4-[2-(4-Methoxy- MS (ES+) 397.2
phenyl)-4-oxo-6,7- So NO H(M+MeOH)+.
113 dihydro-4H- ~~
thiazolo[5,4-c]pyridin-5-
yl]-phenoxy } -
acetaldehyde
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
89
Prep Name Structure MS
{4 [2 (3 Chloro- ~ ~ ~ N oMe MS (ES+) 429.0
phenyl)-4-oxo-6,7- S o o_)f" (M+1)+.
ci o
114 dihydro-4H-
thiazolo[5,4-c]pyridin-5-
yl]-2-methoxy-
phenoxy } -acetaldehyde
{2-Methoxy-4-[4-oxo-2- FF MS (ES+) 463.3
(4-trifluoromethyl- F 0'1(" (M+1)+.
phenyl)-6,7-dihydro-4H-
115 thiazolo[5,4-c]pyridin-5-
yl]-phenoxy}-
acetaldehyde; compound
with methane
{ 4-[2-(2,4-Dichloro- oMe MS (ES+) 463.0
phenyl)-4-oxo-6,7- c, c, s ~
o ~ o1(H (M+1)+.
0
116 dihydro-4H-
thiazolo[5,4-c]pyridin-5-
yl]-2-methoxy-
phenoxy } -acetaldehyde
{4-[2-(4-Fluoro-phenyl)- F / ~ ~SN ~ oMe MS (ES+) 413.3
4-oxo-6,7-dihydro-4H- o 0'1(" (M+1)}.
0
117 thiazolo [5,4-c]pyridin-5-
yl]-2-methoxy-
phenoxy}-acetaldehyde
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
General Procedure 1
R5
R3 Q S O
A ~ / O~"N.R1
U N ~ R2
N
R4
To a round bottom flask or vial containing {4-[2-(4-chloro-phenyl)-4-oxo-6,7-
dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-2-methoxy-phenoxy}-acetaldehyde (0.064
g,
5 0.15 mmol) add dichloroethane (1.5 mL), a secondary amine (1.2 molar
equivalent), and
sodium triacetoxyborohydride (1.1 molar equivalent). Stir at room temperature
overnight.
Quench with saturated aqueous NaHCO3, extract with CH2C12 (lx), EtOAc (2x),
dry over
MgSO4, filter and concentrate under vacuum. Purify by flash chromatography on
silica
gel, using a gradient of MeOH (2 N NH3)/EtOAc (5%-15%) to give the title
compound.
Prepare Examples 14 to 29 by essentially following the general procedure as
described
above, using the appropriate amine reagent. For Examples 28 and 29 prepare the
citrate
salt by dissolving the free base in acetone and treating with a stoichiometric
amount of
citric acid.
Example 14
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
CI ~ \~ N
$ N ~ O~
31 0 I / C~,N
Exact mass = 483.1, MS (ES+) 484.2 (M+1)+. 'H NMR (CDC13): S 7.93 (dt, J= 8.5,
2.1
Hz, 2H), 7.45 (dt, J= 8.5, 2.1 Hz, 2H), 6.93 (m, 2H), 6.84 (dd, J= 8.6, 2.4
Hz, 1H), 4.18
(t, J = 6.6 Hz, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.86 (s, 3H), 3.28 (t, J = 7.0
Hz, 2H), 2.96 (t,
J= 6.6 Hz, 2H), 2.64 (br s, 4H), 1.81 (m, 4H).
Example 15
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-piperidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c] pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
91
CI / \ N-Q
N
S
0 Exact mass = 497.1, MS (ES+) 498.3 (M+1)+. 1H NMR (CDC13): 7.93 (d, J = 8.7
Hz,
2H), 7.44 (d, J= 8.7 Hz, 2H), 6.92 (m, 2H), 6.84 (m, 1H), 4.20 (t, J= 5.5 Hz,
2H), 4.09 (t,
J = 6.9 Hz, 2H), 3.86 (s, 3H), 3.28 (t, J = 7.0 Hz, 2H), 2.87 (br s, 2H), 2.58
(br s, 4H),
1.65 (br s, 4H), 1.47 (br s, 2H).
Example 16
2-(4-Chloro-phenyl)-5- [3 -methoxy-4-(2-morpholin-4-yl-ethoxy)-phenyl] -6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
CI / \ N 31 /~
s N I~ p
0 O~,N J
Exact mass = 499.1, MS (ES+) 500.3 (M+1)+. 1H NMR (CDC13): S 7.92 (d, J= 8.4
Hz,
2H), 7.44 (d, J = 8.4 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J = 8.6, 2.6 Hz,
1H), 4.18 (t, J
= 5.8 Hz, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.86 (s, 3H), 3.75 (m, 4H), 3.28 (t,
J = 7.0 Hz,
2H), 2.87 (ap t, 2H), 2.62 (br s, 411).
Example 17
2-(4-Chloro-phenyl)-5- [4-(2-diethylamino-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
CI ~ \ N ~
N OfN,,,-
0 / O
Exact mass = 485.1, MS (ES+) 486.3 (M+1)+. 1H NMR (CDC13): S 7.93 (d, J= 8.6
Hz,
2H), 7.45 (d, J= 8.6 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.6, 2.4 Hz,
1H), 4.15-4.07
(m, 411), 3.86 (s, 3H), 3.27 (t, J= 6.9 Hz, 2H), 2.95 (ap d, 2H), 2.67 (ap t,
4H), 1.09 (t, J=
7.1 Hz, 6H).
Example 18
2-(4-Chloro-phenyl)-5- {4-[2-(cyclohexyl-methyl-amino)-ethoxy]-3-methoxy-
phenyl }-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
92
CI ~ ~ N '
S N ::,~ ~~No I O
Exact mass = 525.2, MS (ES+) 526.3 (M+1)+. 1H NMR (CDC13): 8 7.93 (d, J= 8.6
Hz,
2H), 7.45 (d, J= 8.6 Hz, 2H), 6.93-6.90 (m, 2H), 6.84 (dd, J= 8.6, 2.4 Hz,
1H), 4.14-4.07
(m, 4H), 3.86 (s, 3H), 3.28 (t, J= 7.0 Hz, 2H), 2.95 (ap t, 2H), 2.45 (br s,
1H), 2.40 (s,
3H), 1.90-1.77 (m, 4H), 1.64 (br s, 2H), 1.29-1.20 (m, 4H).
Example 19
2-(4-Chloro-phenyl)-5-{ 3-methoxy-4-[2-(4-methyl-piperazin-1-yl)-ethoxy]-
phenyl } -6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one
CI N ~
S N ~ 0~ N
0 I ~ ~~iN~
Exact mass = 512.2, MS (ES+) 513.3 (M+1)+. 1H NMR (CDC13): S 7.92 (d, J= 8.7
Hz,
2H), 7.44 (d, J= 8.7 Hz, 2H), 6.93-6.89 (m, 2H), 6.83 (dd, J= 8.6, 2.3 Hz,
1H), 4.16 (t, J
= 6.2 Hz, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.85 (s, 3H), 3.28 (t, J= 7.0 Hz,
2H), 2.86 (t, J
6.2 Hz, 2H), 2.64 (br s, 4H), 2.50 (br s, 4H), 2.30 (s, 3H).
Example 20
2-(4-Chloro-phenyl)-5- { 4-[2-(isopropyl-methyl-amino)-ethoxy]-3-methoxy-
phenyl } -6,7-
dihydro-5H-thiazolo [5,4-c] pyridin-4-one
CI
S N ~ O N"
O OJr
Exact mass = 485.2, MS (ES+) 486.3 (M+1)+. 1H NMR (CDC13): S 7.93 (d, J = 8.5
Hz,
2H), 7.45 (d, J= 8.5 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.4, 2.5 Hz,
1H), 4.15-4.07
(m, 4H), 3.86 (s, 3H), 3.28 (t, J = 7.0 2H), 2.94-2.85 (m, 3H), 2.35 (br s,
3H), 1.05 (d, J
6.5 Hz, 6H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
93
Example 21
5-[4-(2-[ 1,4']Bipiperidinyl-1'-yl-ethoxy)-3-methoxy-phenyl]-2-(4-chloro-
phenyl)-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
CI ~ ~ N
S N ~ O" c
0 I ~ oi~N
Exact mass = 580.2, MS (ES+) 581.4 (M+1)+. 1H NMR (CDC13): S 7.93 (d, J= 8.6
Hz,
2H), 7.45 (d, J= 8.6 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.5, 2.5 Hz,
1H), 4.15 (t, J
= 6.4 Hz, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.86 (s, 3H), 3.28 (t, J = 7.0 Hz,
2H), 3.06 (d, J =
5.7 Hz, 2H), 2.83 (t, J= 6.4 Hz, 2H), 2.52 (br s, 4H), 2.12 (t, J= 11.7 Hz,
2H), 1.80 (ap d,
2H), 1.70-1.52 (m, 7H), 1.43 (br s, 2H).
Ex Product Structure Physical Data
(Chemical Name)
22 2-Phenyl-5-[4-(2- N MS (ES+) 420.2
pyrrolidin-1-yl-ethoxy)- S' o"~ ~ f" (M+1)+.
0
phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-
one
23 2-(4-Methoxy-phenyl)- Meo i~ "~ MS (ES+) 480.2
5-[3-methoxy-4-(2- '~ (" ~ ~ aMti"~ (M+1)+.
pyrrolidin-1-yl-ethoxy)-
phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-
one
24 2-(3-Chloro-phenyl)-5- ~~ s" oMe MS (ES+) 484.2
[3-methoxy-4-(2- ci 0 o" (M+1)+.
pyrrolidin-1-yl-ethoxy)-
phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-
one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
94
Ex Product Structure Physical Data
(Chemical Name)
25 5-[3-Methoxy-4-(2- MS (ES+) 518.2
F ~
F S N / OMe
pyrrolidin-1-yl-ethoxy)- (M+l)+.
phenyl]-2-(4-
trifluoromethyl-phenyl)-
6,7-dihydro-SH-
thiazolo[5,4-c]pyridin-4-
one
26 2-(2,4-Dichloro-phenyl)- , N MS (ES+) 518.2
5-[3-methoxy-4-(2- ' S 0 ~ ~OMe (M+1)+.
pyrrolidin-1-yl-ethoxy)-
phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-
one
27 2-(4-Fluoro-phenyl)-5- ~~ . MS (ES+) 468.2
F I N OMe
[3-methoxy-4-(2- s
~ ~ ,,N (M+l)+.
pyrrolidin-1-yl-ethoxy)-
phenyl]-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-
one
28 5-[3-Methoxy-4-(2- Meo ~_~ ; jn MS (ES+) 496.0
S'~N ~ OMe
morpholin-4-yl-ethoxy)- (M+1)+.
phenyl]-2-(4-methoxy-
0
phenyl)-6,7-dihydro-5H- o
thiazolo[5,4-c]pyridin-4- 00 0
one
MS (ES+) 454.0
29 5-[4-(2-Dimethylamino- Me ~~ sN ~ OMe
ethoxy)-3-methoxy- I ~ (M+1)+.
phenyl]-2-(4-methoxy-
phenyl)-6,7-dihydro-5H- a
0
thiazolo[5,4-c]pyridin-4- 00
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
Ex Product Structure Physical Data
(Chemical Name)
one
Preparation 118
2,2-Dimethyl-propionic acid 2-methoxy-4-[2-(4-methoxy-phenyl)-4-oxo-6,7-
dihydro-4H-
thiazolo[5,4-c]pyridin-5-yl]-phenyl ester
N
Me0 ~/ \' S~N OMe
O O
5 O~*
Dissolve 2,2-dimethyl-propionic acid 4-{ [4-(2-hydroxy-ethyl)-2-(4-methoxy-
phenyl)-thiazole-5-carbonyl]-amino }-2-methoxy-phenyl ester (480 mg, 0.98
mmol) and
NEt3 (177 mL, 1.27'mmol) in dry CH2C12 and cool to 0 C. Add dropwise
methanesulfonyl chloride (98.1 mL, 1.27 mmol) and stir for 30 min. Quench the
reaction
10 mixture with saturated NH4C1 solution and extract with CH2C12 (3 x 10 mL).
Dry, filter,
and concentrate. Redissolve the crude material in dry DMF (6.5 mL) and chill
to 0 C.
Add portionwise NaH (60% dispersion, 51 mg, 1.27 mmol) then warm to ambient
temperature overnight. Add 1N HCl (20 mL) and extract with EtOAc (3 x 10 mL).
Collect insoluble solid from portioned layers via filtration. Wash the
filtrate with water
15 (40 mL), dry, filter, and concentrate. Combine the resulting material with
the collected
solid to give the title compound as a yellow solid (749 mg, 99%). MS (ES+)
467.3
(M+1)+. 1H NMR (400 MHz, CD3OD): 8 7.96 (d, 2H, J = 8.4 Hz), 7.14 (d, 1H, J
2.2
Hz), 7.05 (d, 2H, J = 8.4 Hz), 7.04 (dd, 1H, J = 8.8, 2.2 Hz), 6.96 (dd, 1H,
J.= 8.4, 2.2
Hz), 4.15 (t, 2H, J= 7.0 Hz), 3.87 (s, 3H), 3.81 (s, 3H), 3.27 (t, 2H, J= 7.5
Hz), 1.35 (s,
20 9H).
Preparation 119
5-(4-Hydroxy-3-methoxy-phenyl)-2-(4-methoxy-phenyl)-6,7-dihydro-5H-thiazolo
[5,4-
c]pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
96
N
Me0 S~N oMe
ll0lf OH
Dissolve 2,2-dimethyl-propionic acid 2-methoxy-4-[2-(4-methoxy-phenyl)-4-oxo-
6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-phenyl ester (749 mg, 1.61 mmol)
in absolute
ethanol (18 mL) and add NaOMe (183.1 mg, 6.44 mmol). Allow the reaction
mixture to
stir for 4 h at ambient temperature. Quench the reaction mixture with 1N HCl
solution. to
pH = 7. Add a small amount of EtOAc (15 mL) and filter the solid precipitate
via
vacuum filtration to give the title compound as a yellow solid (430 mg, 70%).
MS (ES+)
383.3 (M+1)+. 'HNMR(CDC13): b 7.95 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz,
2H), 6.94
(d, J = 6.2 Hz, 1H), 6.93 (s,1H), 6.80 (dd, J = 8.8, 2.2 Hz, 1H), 4.07 (t, J =
7ØHz, 2H),
3.90 (s, 3H), 3.87 (s, 3H), 3.28 (t, J= 7.0 Hz, 2H).
Preparation 120
2-(4-Chloro-phenyl)-6,7-dihydro-5H-thiazolo [5,4-c]pyridin-4-one
N
CI a S qNH
Dissolve 2-(4-chloro-phenyl)-5-(3,4-dimethoxy-benzyl)-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-one (376 mg, 0.91mmo1) in toluene (5.0 mL) and treat
with para-
toluene sulfonic acid (176 mg, 0.92 mmol). Stir the solution at reflux for 2
d, then
concentrate and purify the crude material by flash chromatography, using 5%
MeOH (2N
NH3)/CH2C12 as eluent, to give the title compound as a white solid (170 mg,
70%). MS
(ES+) 265.0 (M+1)+. 1H NMR(CDC13): S 8.02 (d, 2H, J=8.8 Hz), 7.94 (s, 1H),
7.60 (d,
2H, J=8.4 Hz), 3.52 (dt, 2H, J=7.1, 2.5 Hz), 3.04 (t, 2H, J=7.3 Hz).
Preparation 121
Toluene-4-sulfonic acid 4- [2-(4-chloro-phenyl)-4-oxo-6,7-dihydro-4H-thiazolo
[5,4-
2 5 c]pyridin-5-yl]-2-methoxy-phenyl ester
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
97
CI N O
O
O
0=S=0
I
Mix 2-(4-chloro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (128 mg,
0.48 mrnol), toluene-4-sulfonic acid 4-bromo-2-methoxy-phenyl ester (218 mg,
0.61
mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (17.2 mg, 0.030 mmol),
Cs2CO3 (0.123 mg, 0.377 mmol) in dioxane (13 mL). Purge the solution with
nitrogen
for 30 min and then add tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3)
(6.7 mg,
0.0073 mmol). Stir the mixture at reflux overnight, then cool to room
temperature.
Dilute the mixture with EtOAc (50 mL) and wash with water (2 x 30mL) and brine
(30
mL). Dry, filter and concentrate the organic solution and purify the residue
by flash
chromatography, using a linear gradient of 100% hexanes to 80% EtoAc/hexanes
as
eluent, to give the title compound as a light brown solid (155 mg, 60%). MS
(ES+) 541.0
(M+1)+. 1H NMR(CDC13): 5 7.92 (d, 2H, J=8.4 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.44
(d, 2H,
J=8.8 Hz), 7.31 (d, 2H, J=8.4 Hz), 7.17 (d, 1H, J=8.8 Hz), 6.96 (d, 1H, J=2.6
Hz), 6.82
(dd, 1H, J=8.6, 2.4 Hz), 4.11 (t, 2H, J=6.8 Hz), 3.57 (s, 3H), 3.28 (t, 2H,
J=6.8 Hz), 2.44
(s, 3H).
Preparation 122
2-(4-Chloro-phenyl)-5-(4-hydroxy-3 -methoxy-phenyl)-6,7-dihydro-5H-thiazolo
[5,4-
c]pyridin-4-one
N
CI S N
O I /
OH
Method 1. Mix toluene-4-sulfonic acid 4-[2-(4-chloro-phenyl)-4-oxo-6,7-dihydro-
4H-thiazolo[5,4-c]pyridin-5-yl]-2-methoxy-phenyl ester (110 mg, 0.20mmo1) in
dioxane
(mL) and water (mL) and treat with LiOH=H20 (44 mg, 1.0 mmol). Stir the
mixture at
reflux for 3 h, cool to room temperature, neutralize with 1N HCl (1.0 mL), and
dilute with
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
98
additional water. Collect the solid by filtration and purify by flash
chromatography, using
a 5% MeOH (2N NH3)/CHZC12 as eluent, to give the title compound as an off-
white solid
(29 mg, 37%). MS (ES+) 387.0 (M+l)+, MS (ES-) 385.0 (M-1)-. 1H NMR(CDC13): S
7.92 (d, 2H, J=8.4 Hz), 7.44 (d, 2H, J=8.4 Hz), 6.95-6.92 (m, 2H), 6.80 (dd,
1H, J=8.6,
2.4 Hz), 5.61 (s, 1H), 4.08 (t, 2H, J=7.0 Hz), 3.90 (s, 3H), 3.28 (t, 2H,
J=7.0 Hz).
Method 2. Combine 5-[4-(tert-butyl-dimethyl-silanyloxy)-3-methoxy-phenyl]-2-
(4-chloro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (0.92 g, 1.84
mmol), THF
(10 mL), and tetrabutylammonium flouride (1M in THF, 2.0 mL, 2.0 mmol and stir
at
room temperature overnight. Neutralize with saturated aqueous NH4C1, extract
with
diethyl ether (lx), EtOAc (2x), dry over MgSO4, filter, and concentrate under
vacuum.
Purify by flash chromatography on silica gel, eluting with a gradient of
EtOAc/hexane
20%-45% to give the title compound as a yellow residue (0.28 g, 40%). Exact
mass =
386.0, MS (ES+) 387.1 (M+1)+.
Preparation 123
(R)-2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-triisopropylsilanyloxy-pyrrolidin-l-
yl)-
phenyl]-6,7-dihydro-5H-thiazolo [5,4-c]pyridin-4-one
cl 0 ) /
S ~)N N~ O
O N
,.,, s
~
Prepare the title compound by essentially following the procedures as
described
for Preparation 118, with the following alternate workup. Quench the reaction
mixture
with saturated NH4C1 solution (10 mL) and extract with EtOAc (3 x 20 mL). Wash
the
organic layer with water (2 x 20 mL). Dry the organic layer with Na2SO4,
filter, and
concentrate. Purify on silica gel chromatography using 0-25% EtOAc in hexanes
to give
the title compound. MS (ES+) 612.1(M+1)+. 1HNMR(CDC13): 8 7.90 (d, J= 8.4 Hz,
2H),
7.42 (d, J= 8.8 Hz, 2H), 6.85 (bs,1 H), 6.80 (dd, J= 8.4, 2.2 Hz, 1 H), 6.75-
6.68 (m, 1 H),
4.57 (bs, 1H), 4.06 (t, J= 7.0 Hz, 2H), 3.81 (s, 3H), 3.72-3.63 (m, 1H), 3.43-
3.29 (m, 2H),
3.25 (t, J= 7.0 Hz, 2H), 3.17-3.09 (m, 1H), 2.20-2.06 (m, 1H), 1.96-1.86 (m,
1H), 1.12-
0.98 (m, 21H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
99
Preparation 124
5-Chloromethyl-l-methyl-1 H-imidazole
Cl N HCI
"
N
Add thionyl chloride (4.00 ml, 53.8 mmol) to a solution of (3-methyl-3H-
imidazol-4-yl)-methanol (4.0 g, 35.7 mmol) in dichloroethane (30 mL) and stir
at room
temperature for 18 h. Concentrate the reaction mixture and add ether to the
residue.
Sonicate for 5 min, filter, and dry to give the title compound (5.8 g, 98%).
MS (ES+) 131
(M+1)+. 'H NMR (400 MHz, DMSO-d6): 8 14.99 (s, 1H), 9.18 (s, 1H), 7.75 (s,
1H),
5.00 (s, 2H), 3.85 (s, 3H).
Preparation 125
4-chloromethyl-lH-imidazole
Cl N HCI
~NH
Prepare the title compound by essentially following the procedure as described
for
Preparation 124, using (3H-imidazol-4-yl)-methanol. MS (ES+) 117.1 (M+1)+. 1H
NMR
(400 MHz, DMSO-d6): b 15.03 (s, 1 H), 9.12 (d, 1 H, J=1.3 Hz), 7.71 (d, 1 H,
J=1.3 Hz),
4.85 (s, 2H)
General Procedure 2
O--
CI \~ S O 0'r lR
N N ~ ~ / M
n = 1-3
To a vial containing 2-(4-chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one ( 0.050 g, 0.13 mmol) add DMF (1 mL),
K2C03
(3 molar equivalents), potassium iodide (catalytic), and an alkyl halide (1.2
molar
equivalents). Stir overnight at room temperature. If reaction is not complete,
heat in a
microwave reactor at 100 C for 10 min, or heat in a 100 C oil bath until the
phenol
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
100
starting material is consumed. Add water, extract with EtOAc (3x), dry by
elution
through a Na2SO4 drying tube and concentrate under vacuum. Purify by flash
chromatography on silica gel to give the title compound.
Prepare Examples 30 to 36 as essentially described according to the general
procedure,
above, using the appropriate alkyl halide reagent.
Example 30
2-(4-Chloro-phenyl)-5- j4-(3-dimethylamino-propoxy)-3-methoxy-phenyl] -6,7-
dihydro-
5H-thiazolo[5,4-c]pyridine-4-one
CI SN N~ o
r I
O I
Exact mass = 471.1, MS (ES+) 472.3 (M+1)+. 1H NMR (CDC13): 8 7.93 (d, J= 8.6
Hz,
2H), 7.45 (d, J= 8.6 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.5, 2.4 Hz,
1H), 4.12-4.07
(m, 4H), 3.87 (s, 3H), 3.29 (t, J = 6.9 Hz, 2H), 2.57 (br s, 2H), 2.34 (br s,
6H), 2.07 (m,
2H).
Example 31
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-piperidin-1-yl-propoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one
cl / \ N~
N O'~
O I / O,-U~N
Exact mass = 511.2, MS (ES+) 512.3 (M+1)+. 1H
2 0 NMR (CDC13): S 7.93 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 6.94-
6.91 (m, 2H),
6.84 (dd, J= 8.7, 2.3 Hz, 1H), 4.11-4.06 (m, 4H), 3.86 (s, 3H), 3.28 (t, J=
7.0 Hz, 2H),
2.53 (br s, 2H), 2.45 (br s, 4H), 2.06 (m, 2H), 1.62 (br s, 4H), 1.45 (br s,
2H).
Example 32
2-(4-Chloro-phenyl)-5-[4-(3-diethylamino-propoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
101
cl / ~ ~ I J
S O N I OfN
Exact mass = 499.2, MS (ES+) 500.3 (M+1)+. 1H NMR (CDC13): 8 7.93 (d, J = 8.4
Hz,
2H), 7.44 (d, J= 8.4 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.5, 2.5 Hz,
IH), 4.11-4.06
(m, 4H), 3.86 (s, 3H), 3.28 (t, J = 7.0 Hz, 2H), 2.61 (t, J= 7.2 Hz, 2H), 2.54
(t, J= 7.2 Hz,
4H), 2.02-1.94 (m, 2H), 1.02 (t, J= 7.2 Hz, 6H).
Example 33
2-(4-Chloro-phenyl)-5- [4-(2-dimethylamino-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c] pyridin-4-one
cl N
N ~ O N~
O ~ O,J(
Exact mass = 457.1, MS (ES+) 458.2 (M+1)+. 1H NMR (CDC13): 8 7.93 (d, J= 8.5
Hz,
2H), 7.45 (d, J= 8.5 Hz, 2H), 6.94-6.91 (m, 2H), 6.84 (dd, J= 8.5, 2.5 Hz, 1
H), 4.13 (t, J
= 6.1 Hz, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.86 (s, 3H), 3.28 (t, J = 7.0 Hz,
2H), 2.79 (t, J
6.1 Hz, 2H), 2.35 (s, 6H).
Example 34
2-(4-Chloro-phenyl)-5- [ 3 -methoxy-4-(pyridin-4-ylmethoxy)-phenyl] -6,7-
dihydro-5H-
thiazolo [5,4-c] pyridin-4-one
S N
0 (
OI / \ N
~ N
Exact mass = 477.1, MS (ES+) 478.2 (M+1)+. 1H NMR (CDC13): S 8.64 (br s, 2H),
7.93
(d, J = 8.5 Hz, 2H), 7.49 (ap d, 2H), 7.45 (d, J = 8.5 Hz, 2H), 7.00 (d, J =
2.5 Hz, 1H),
6.86 (d, J= 8.5 Hz, 1H), 6.81 (dd, J= 8.5, 2.2 Hz, 1H), 5.21 (s, 211), 4.09
(t, J= 7.0 Hz,
2H), 3.91 (s, 311), 3.29 (t, J= 7.0 Hz, 211).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
102
Example 35
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(1-methyl-1 H-imidazol-2-ylmethoxy)-phenyl]-
6,7-
dihydro-5 H-thiazol o [5,4-c] pyridin-4-one
N 0
CI N
0 I O/-N
;N~
Exact mass = 480.1, MS (ES+) 481.2 (M+1)+. 1H NMR (CDC13): S 7.92 (dt, J= 8.4,
2.2
Hz, 2H), 7.44 (dt, J= 8.7, 2.2 Hz, 2H), 7.18 (d, J= 8.7 Hz, 1H), 7.00 (s, 1H),
6.95 (d, J=
2.5 Hz, 1H), 6.89 (s, 1H), 6.81 (dd, J= 8.4, 2.5 Hz, 1H), 5.26 (s, 2H), 4.08
(t, J= 7.0 Hz,
2H), 3.86 (s, 3H), 3.79 (s, 3H), 3.28 (t, J= 7.0 Hz, 2H).
Example 36
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-methyl-thiazol-5-ylmethoxy)-phenyl]-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
CI / l N
g N O
O I O I' N
S
Exact mass = 497.1, MS (ES+) 498.2 (M+1)+. 1H NMR (CDC13): S 7.93 (dt, J= 8.7,
2.2
Hz, 2H), 7.45 (dt, J= 8.7, 2.2 Hz, 2H), 7.19 (ap s, 1 H), 6.99-6.95 (m, 2H),
6.81 (dd, J=
8.7, 2.2 Hz, 1H), 5.25 (ap d, 2H), 4.09 (t, J = 7.0 Hz, 2H), 3.89 (s, 3H),
3.28 (t, J = 7.0
Hz, 2H), 2.73 (s, 3H).
Example 37
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(morpholin-2-ylmethoxy)-phenyl]-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one
cl i j
N ~ OMe
O /
O\
NJl
H
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
103
Dissolve 2-(4-chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-one (103 mg, 0.267 mmol), 2-hydroxymethyl-morpholine-
4-
carboxylic acid tert-butyl ester (Pharmacore, CAS: 135065-69-9)(70 mg, 0.324
mmol),
and PBu3 (84 l, 0.324 mmol) in dry toluene (1.2 mL). Cool to 0 C then add
1,1'-
(azodicarbonyl)piperidine (84 l, 0.324 nunol). Let stir for 10 min at 0 C,
then warm to
ambient temperature overnight. Reaction mixture thickens and turns gel like.
Add
hexanes and collect solid via vacuum filtration. Wash the solid with hexanes
several
times. Dissolve the crude material in dry CH2C12 (500 1) and TFA (200 l) and
stir
overnight. Add 1N NaOH until the reaction is pH = 10 and extract with EtOAc (3
x 10
mL). Dry the combined organic portions with Na2SO4, filter, and concentrate to
give the
title compound. MS (ES+) 486.0 (M+1)+.
Example 38
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(4-methyl-morpholin-2-ylmethoxy)-phenyl]-
6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt
ci / \ i I
S N OMe
Q
)
i HCI
Dissolve 2-(4-chloro-phenyl)-5-[3-methoxy-4-(morpholin-2-ylmethoxy)-phenyl]-
2 0 6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (460 mg, 0.928 mmol) in dry
acetone (3 mL)
under nitrogen. Add K2C03 (321 mg, 1.11 mmol) and NaI (14 mg, 0.092 mmol).
Evacuate under vacuum and charge the reaction mixture with nitrogen. Mix well
and
then add Mel (70 mL, 1.11 mmol). Stir the reaction mixture overnight. Add
saturated
NH4Cl solution (5 mL) and extract with EtOAc (3 x 10 mL). Wash the combined
organic
layers with water (10 mL), dry with NaaS04, filter, and concentrate. Purify
the resulting
residue with silica gel chromatography, using 0-10% MeOIi/CHC13 to give the
title
compound. Dissolve the compound in minimal CH2C12 and add HCl/Et2O to make the
HCl salt as a yellow-orange solid (74 mg, 15%). MS (ES+) 500.0 (M+l)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
104
Example 39
(R)-2-(4-Chloro-phenyl)-5-[4-(3-hydroxy-pyrrolidin-1-yl)-3-methoxy-phenyl]-6,7-
dihydro-
5H-thiazolo[5,4-c]pyridin-4-one, hydrochloride salt
ci
i
N ~ OMe
O I /
N
--,,OH
HCI
Dissolve (R)-2-(4-chloro-phenyl)-5-[3-methoxy-4-(3-triisopropylsilanyloxy-
pyrrolidin-1-yl)-phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (370 mg,
0.610
mmol) in dry THF (2 mL). Add TBAF (1.OM in THF, 610 l, 0.610 mmol) and stir 2
h.
Absorb the reaction mixture on silica gel and remove organic solvent via
reduce pressure.
Purify by silica gel chromatography using 0-100% EtOAc in hexanes to give the
title
compound. Dissolve the compound in a minimal amount of CH2C12 and add HCl/Et2O
solution to give precipitated product. Remove the organic solvent via reduced
pressure
and triturate with MeOH to give the desired product as white solid HCl salt
(144 mg,
48%). MS (ES+) 456.0 (M+1)+. 1H NMR (CD30D): S 8.01 (d, J= 8.8 Hz, 2H), 7.71
(br
d, J= 8.8 Hz, 1H), 7.51 (d, J= 8.4 Hz, 2H), 7.35 (br s, 1H), 7.16 (br d, J=
8.8 Hz, 1H),
4.73-4.67 (m, 1H), 4.19 (br t, J= 6.6 Hz, 2H), 4.02 (br s, 3H), 3.97-3.85 (m,
3H), 3.66 (br
d, J= 11.0 Hz, 1H), 3.31 (t, J= 7.0 Hz, 2H), 2.49-2.36 (m, 1H), 2.26-2.17 (m,
11-1).
Preparation 126
2-(4-Chloro-phenyl)-5-(3-methoxy-4-triisopropylsilanyloxy-phenyl)-5H-thiazolo
[5,4-
c]pyridin-4-one
Ci N
N ~ O
O I/ O
-(\
>7,
Treat a solution 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic
acid (3-methoxy-4-triisopropylsilanyloxy-phenyl)-amide (1.0 g, 1.79 mmol) in
CH2C12
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
105
(30 mL) with Dess- Martin periodinane (1.13 g, 2.67 mmol). Stir at room
temperature for
18 h, dilute with 1N NaOH, and extract with CH2C12 (2x). Dry, filter, and
concentrate the
organic solution and purify the crude material by flash chromatography, using
a gradient
of 0 - 10% MeOH in CH2C12 to give the title compound (0.47 g, 48%). MS (ES+)
541.0
(M+1)+. 1H NMR (400 MHz, CDC13) 5: 8.01 (d, 2H, J=8.8 Hz), 7.46 (d, 2H, J=8.4
Hz),
7.41 (d, 1H, J=7.5 Hz), 6.93 (m, 3H), 6.81 (dd, 1H, J=8.4, 2.6 Hz), 3.80 (s,
3H), 1.25 (m,
3H), 1.09 (d, 18H, J=7.5 Hz).
Preparation 127
2-(4-Chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-5H-thiazolo[5,4-c]pyridin-4-
one
N I ~
CI
- S N O"
O
OH
Treat a solution of 2-(4-chloro-phenyl)-5-(3-methoxy-4-triisopropylsilanyloxy-
phenyl)-5H-thiazolo[5,4-c]pyridin-4-one (0.47 g, 0.87 mmol) in THF ( 5.0 mL)
with
TBAF (1.3 mL of 1N in THF) and stir for 4 h. Acidify reaction mixture to pH 4
with 1N
HCI. Filter precipitate, wash several times with water, and dry to give title
compound
(0.23 g, 69%). MS (ES ) 385 (M+1)+. 1H NMR (400MHz, DMSO-d6) S: 9.33 (s, 1H),
8.11 (d, 2H, J=8.4 Hz), 7.69 (d, 1H, J=7.0 Hz), 7.62 (d, 2H, J=8.8 Hz), 7.02-
6.97 (m,
2H), 6.86-6.81 (m, 2H), 3.74 (s, 3H).
Example 40
2-(4-Chloro-phenyl)-5- [3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl] -5H-
thiazolo [5,4-
c]pyridin-4-one
N I \
cl~
S N ~ 0
0 I / O~iNfD
Mix 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [3-
methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amide (80 mg, 0.16 mmol) and Dess-
Martin periodinane (70 mg, 0.1 6mmol) in CH2C12 and stir at RT for 48 h.
Dilute the
mixture with aqueous 1 N NaOH and extract with CH2C12. Dry, filter, and
concentrate
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
106
the organic solution. Purify the crude material by flash chromatography, using
a gradient
of 100% EtOAc to 12% 2 N NH3/MeOH in EtOAc, to give the title compound (12 mg,
16%). MS (ES+) 482.0 (M+1)+. IH NMR (400 MHz, DMSO-d6): S 8.01 (d, 2H, J = 8.8
Hz), 7.46 (d, 2H, J = 8.3 Hz), 7.40 (d, 1H, J = 7.0 Hz), 7.34 (s, 1H), 6.89-
6.99 (m, 3H),
4.20 (t, 2H, J= 6.4 Hz), 3.86 (s, 3H), 2.96 (t, 2H, J= 6.4 Hz), 2.64 (s, 4H),
1.80 (m, 4H).
Example 41
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-5H-
thiazolo[5,4-c]pyridin-4-one hydrochloride
CI HCI
N , 0"
0 I iN
Add NaH (0.7 g, 17.5 mmol) to a solution of 2-(4-chloro-phenyl)-5-(4-hydroxy-3-
methoxy-phenyl)-5H-thiazolo[5,4-c]pyridin-4-one (1.6 g, 4.2 mmol) in DMF (15
mL) at
room temperature. Stir for 10-30 min, add 1-(2-chloro-ethyl)-pyrrolidine
hydrochloride
(2.1 g, 12.4 mmol), and warm to 90 C for 1-2 days. Cool the reaction mixture,
dilute
with water, and extract with CHaC12 (2x). Combine the organic portions, dry,
and
concentrate. Purify by flash chromatography, using 0 - 10% 2N NH3/MeOH in
CH2C12,
to give the free amine. Dissolve the free amine in MeOH (10.0 mL) and add 1N
HCl in
ether (10.0 mL), sonicate for 5 min, and concentrate. Triturate the solid with
ether, filter
the solid, and dry to give the title compound (0.97 g, 45%). MS (ES+) 481.8
(M+1)+; free
amine)+. 1H NMR (400 MHz, DMSO-86): b 10.76 (s, IH, HCI), 8.11 (d, 2H, J=8.8
Hz),
7.72 (d, 1H, J=7.0 Hz), 7.63 (d, 2H, J=8.4 Hz), 7.17-7.14 (m, 2H), 7.03-
6.99.(m, 2H),
4.37 (t, 2H, J=5.1 Hz), 3.77 (s, 3H), 3.61 (m, 4H), 3.10 (m, 2H), 1.99 (m,
2H), 1.86 (m,
2H).
Prepare Example 42 to 44 by essentially following the procedures as described
for
Example 41, using the appropriate alkylating agent.
Example 42
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-methyl-3H-imidazol-4-ylmethoxy)-phenyl]-
6,7-
3 o dihydro-5H-thiazolo[5,4-c]pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
107
/ ~ N I ~
CI
S N
O I / O N
~ /
i0 HCI N
MS (ES+) 479.0 (M+1)+. 1H NMR (400MHz, DMSO-d6): S 14.56 (s, 1H), 9.18 (s,
1H),
8.16 (d, 2H, J=8.4 Hz), 7.86 (d, 1H, J=1.3 Hz), 7.76 (d, 1H, J=7.5 Hz), 7.67
(d, 2H,
J=8.4 Hz), 7.32 (d, 1H, J=8.8 Hz), 7.20 (d, 1H, J=2.6 Hz), 7.06 (m, 2H), 5.32
(s, 2H),
3.93 (s, 3H), 3.79 (s, 3H).
Example 43
2-(4-Chloro-phenyl)-5- [ 3 -methoxy-4-(2-morpholin-4-yl-ethoxy)-phenyl] -5H-
thiazolo [5,4-
c]pyridin-4-one hydrochloride
~/ \ N I ~
CI
N /~ O
O ( / O,,,,NJ
iO HCI
MS (ES+) 498.0 (M+1)+. 1H NMR (400MHz, DMSO-d6): 5 11.03 (s, 1H), 8.11 (d, 2H,
J=8.8 Hz), 7.71 (d, 1H, J=7.3 Hz), 7.63 (d, 2H, J=8.8 Hz), 7.16 (m, 2H), 7.02
(m, 2H),
4.44 (t, 2H, J=4.9 Hz), 3.96 (d, 2H, J=10.5 Hz), 3.77 (m, 5H), 3.56-3.52 (m,
4H), 3.21
(m, 2H).
Example 44
2-(4-Chloro-phenyl)-5-[4-(1 H-imidazol-4-ylmethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one hydrochloride
Cl ~ ~ N~
S N
0 I O N
O ~ \>
.~ HCI N
1H NMR (400 MHz, CD30D): S 8.92 (d, 1H, J=1.3 Hz), 8.00 (d, 2H, J=8.8 Hz),
7.62 (s,
1H), 7.51 (d, 2H, J=8.8 Hz), 7.08 (m, 2H), 6.91 (dd, 1H, J=8.4, 2.4 Hz), 5.19
(s, 2H),
4.11 (t, 2H, J=7.0 Hz), 3.83 (s, 3H), 3.28 (t, 2H, J=7.0 Hz).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
108
Preparation 128
4-(2-B enzyloxy-ethyl)-morpholin-3-one
O")
NO
O
Add NaH (0.47 g, 11.8 mmol) to a solution of morpholin-3-one (Vieles, P.;
Seguin, J., Bulletiia de la Societe Chimique de France, 1953, 287-9) (1.0 g,
9.9 mmol) in
DMF (10 ml) at room temperature. Stir for 30 min, add (2-bromo-ethoxymethyl)-
benzene
(2.2 g, 10.2 mmol), and stir at room temperature for 18 h. Dilute with water
and extract
with EtOAc (2x). Combine the organics, dry, and concentrate. Purify by flash
chromatography using 0 - 5% MeOH in CH~C12, to give the product as an oil.(1.7
g,
74%). 'H NMR (400 MHz, CDC13): 5 7.28 (m, 5H), 4.48 (s, 2H), 4.13 (s, 2H),
3.80 (t,
2H, J=5.1 Hz), 3.65 (m, 2H), 3.59 (dd, 2H, J=7.5, 2.6 Hz), 3.48 (t, 2H, J=5.1
Hz).
Preparation 129
4-(2-Hydroxy-ethyl)-morpholin-3-one
f O
HO_-_~N
0
Dissolve 4-(2-benzyloxy-ethyl)-morpholin-3-one (1.7 g, 7.23 mmol) in ethanol
(25 mL) and add 5% Pd/C (0.30 g). Hydrogenate at 60 psi overnight, filter the
black
mixture through Celite0, and wash the Celite with additional ethanol
(approximately
lOmL). Concentrate the filtrate to give the title compound as an oil (0.7 g,
70%). MS
(ES+) 146.3 (M+1)+. 1H NMR (400 MHz, CDC13): S 4.11 (s, 2H), 3.83 (t, 2H,
J=5.1 Hz),
3.73 (t, 2H, J=5.3 Hz), 3.49 (t, 2H, J=5.3 Hz), 3.43 (t, 2H, J=5.1 Hz), 3.12
(s, 1H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
109
Example 45
2-(4-Chloro-phenyl)-5-{ 3-methoxy-4-[2-(3-oxo-morpholin-4-yl)-ethoxy]-phenyl }-
5H-
~ ~ N I
CI
N O O
0 ):::~O___~N
thiazolo[5,4-c]pyridin-4-one 0
Combine 2-(4-chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-5H-thiazolo[5,4-
c]pyridin-4-one (0.70 g, 1.82 mmol), 4-(2-hydroxy-ethyl)-morpholin-3-one (0.50
g, 3.45
mmol) and triphenylphosphine (0.50 g, 1.90 mmol) in THF (10.0 mL), stir for 10
min and
add DIAD (0.77 g, 3.81 mmol). Heat to 80 C for 2 days, cool the reaction
mixture, and
dilute with water. Extract with CHZC12 (2x), combine the organics, dry, and
concentrate
under vacuum. Purify the product by flash chromatography using 0 - 10% MeOH in
CH2C12 to give the title compound (0.40 g, 43%). MS (ES+) 512.0 (M+1)+. 1H NMR
(400 MHz, CDC13): S 8.01 (d, 2H, J=8.6 Hz), 7.47 (d, 2H, J=8.6 Hz), 7.40 (d,
1H, J=7.5
Hz), 6.98-6.95 (m, 3H), 6.91 (dd, 1H, J=8.6, 2.4 Hz), 4.27 (t, 2H, J=5.2 Hz),
4.17 (s, 2H),
3.89-3.81 (m, 7H), 3.68 (t, 2H, J=5.1 Hz).
Example 46
2-(4-Chloro-phenyl)-5- [3 -chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-5H-
thiazolo [5,4-c]pyridin-4-one,
C~ N f
S N CI
0 N
Dissolve 3-chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.20 g, 0.83 mmol)
in CH2C12 (10.0 mL) and treat with trimethylaluminum (2.OM in hexanes, 0.6 mL,
1.20
mmol). Stir at room temperature for 15 min and add 2-(4-chloro-phenyl)-6,7-
dihydro-
pyrano[4,3-d]thiazol-4-one (0.22 g, 0.83 mmol) neat and stir the reaction at
room
temperature for 2 h. Carefully quench the mixture with saturated Rochelles
salt solution
and stir at room temperature for 1 h. Dilute with water and extract with
CH2C12 (2x).
Combine the organic portions and dry, filter and concentrate. Dissolve the
residue in
CH2C12, and treat with triethylamine (0.50 mL, 3.56 nunol) followed by
methanesulfonyl
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
110
chloride (0.05 mI.,, 0.65 mmol). Stir for 1 h at room temperature, dilute with
water and
extract with CH2C12 (2x). Combine the organic portions and dry, filter, and
concentrate.
Dissolve the residue in THF and treat with NaH (0.03 g, 0.75 mmol) and stir at
room
temperature for 18 h. Dilute the reaction with water and extract with CHZC12
(2x).
Combine the organic portions, dry, filter, and concentrate. Purify the crude
material by
flash chromatography, using a gradient of 0% to 10% 2N NH3/MeOH in CH2C12, to
give
the title compound (80 mg, 37%). MS (ES+) 488.0 (M+1)+. 'H NMR (400 MHz,
CDC13): b 7.93 (d, 2H, J=8.3 Hz), 7.45 (d, 2H, J=8.8 Hz), 7.39 (d, 1H, J=2.2
Hz), 7.22
(dd, 1H, J=8.8, 2.6 Hz), 6.97 (d, 1H, J=9.2 Hz), 4.22 (t, 2H, J=5.9 Hz), 4.07
(t, 2H,
J=7.0 Hz), 3.28 (t, 2H, J=7.0 Hz), 3.01 (t, 2H, J=5.9 Hz), 2.73 (m 4H), 1.84
(m 4H).
Example 47
2-(4-Chloro-phenyl)-5-[ 1-methyl-3-(2-pyrrolidin-1-yl-ethyl)-1 H-indol-6-yl]-
6,7-dihydro-
5H-thiazolo[5,4-c]pyridin-4-one hydrochloride
CI ~ ~ N
g N
O N
HCI N
(v~
Dissolve 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid [1-
methyl-3-(2-pyrrolidin-1-yl-ethyl)-1H-indol-6-yl]-amide (0.92 g, 1.81 mmol) in
THF (20
mL) and treat the solution with tributylphosphine (1.0 mL, 3.47 mmol) and
diisopropylazodicarboxylate (0.73 mL, 3.61 mmol). Stir the reaction at room
temperature
for 18 h. Concentrate and purify the crude material by flash chromatography,
using 0-
10% 2N NH3/MeOH in CH2C12, to give the free amine. Dissolve the free amine in
MeOH
(10.0 mL) and add iN HCl in ether (5.0 mL), sonicate for 5 min, and
concentrate.
Triturate the solid with ether, filter the solid, and dry to give the title
compound (0.64 g,
69%). MS (ES+) 491.1 (M+1)+. 1H NMR (400 MHz, DMSO-d6): S 10.39 (s, 1H), 8.03
(d, 2H, J=8.6 Hz), 7.63 (d, 1H, J=8.6 Hz), 7.59 (d, 2H,J=8.6 Hz), 7.44 (d, 1H,
J=1.8
Hz), 7.26 (s, 1H), 7.06 (dd, 1H, J=8.5, 1.6 Hz), 4.11(t, 2H, J=7.0 Hz), 3.72
(s, 3H), 3.54
(m, 2H), 3.33 (m, 2H), 3.27 (t, 2H, J=6.9 Hz), 2.97 (m, 4H), 1.98 (m, 2H),
1.85 (m, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
111
Example 48
2-(4-Chloro-phenyl)-5-[6-(4-methyl-piperazin-1-yl)-pyridin-3 -yl] -6,7-dihydro-
5H-
thiazolo [5,4-clpyridin-4-one, dihydrochloride salt
CI ~ \ N 1
S N N
O /
N
HCI NNI
Prepare the title compound by essentially following procedures as described
for
Example 46 and isolating as the dihydrochloride salt. MS (ES+) 439.8 (M+1)+.
1H NMR
(400 MHz, DMSO-d6): S 11.38 (s, 1H), 8.19 (d, 1H, J=2.6 Hz), 8.01 (d, 2H,
J=8.6 Hz),
7.80 (dd, 1H, J=9.1, 2.5 Hz), 7.58 (d, 2H, J=8.8 Hz), 7.14 (d, 1H, J=9.4 Hz),
4.42 (d, 2H,
J=13.8 Hz), 4.04 (t, 2H, J=6.9 Hz), 3.42 (m, 4H), 3.24 (t, 2H, J=6.9 Hz), 3.08
(m, 2H),
2.75 (d, 3H, J=4.2 Hz).
Example 49
2-(4-Chloro-phenyl)-5- [3-methoxy-4-(3-methyl-3H-imidazol-4-ylmethoxy)-
phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one
CI ~ \ N 1
- S N p
O N
O, - ~' tN
Treat a solution of (3-methyl-3H-imidazol-4-yl)-methanol (60.0 mg, 0.54 mmol)
in CH2C12 with oxalyl chloride (0.15 g, 1.2 mmol) and 2 drops of DMF. Stir at
room
temperature for 4 h, concentrate, and dissolve in DMF (5.0 mL). Add this
solution to a
suspension of NaH (62.5 mg, 1.6 mmol) and 2-(4-chloro-phenyl)-5-(4-hydroxy-3-
methoxy-phenyl)-5H-thiazolo[5,4-c]pyridin-4-one (200.0 mg, 0.5 mmol) in DMF (5
mL).
Stir at room temperature for 2 h, dilute with water, and extract with CH2C12
(2x).
Combine the organics, dry, and concentrate. Purify by flash chromatography,
using 0 -
10% 2N NH3/MeOH in CH2C12, to give the title compound (100.0 mg, 40%). MS
(ES+)
481.0 (M+1)+. 1H NMR (400 MHz, CDC13): 8 7.90 (d, 2H, J=8.4 Hz), 7.44 (d, 2H,
J=4.0 Hz), 7.41 (s, 1H), 7.07 (s, 1H), 6.97 (d, 1H, J=8.8 Hz), 6.94 (d, 1H,
J=2.2 Hz),
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
112
6.81 (dd, 1H, J=8.6, 2.4 Hz), 5.04 (s, 2H), 4.07 (t, 2H, J=7.0 Hz), 3.83 (s,
3H), 3.72 (s,
3H), 3.27 (t, 2H, J=6.8 Hz).
Preparation 130
2-(4-Chloro-phenyl)-5-[4-(3,3-diethoxy-propyl)-3-methoxy-phenyl]-5H-
thiazolo[5,4-
c]pyridin-4-one
N ~
CI ~ ~ S I N x ~
O I O'/
0 1
Prepare the title compound by essentially following the procedures as
described for
Preparation 132. 1H NMR (400 MHz, CDC13): 5 8.02 (d, 2H, J=8.3 Hz), 7.48 (d,
2H,
J=8.3 Hz), 7.42 (d, 1H, J=7.0 Hz), 7.25 (d, 1H, J=7.9 Hz), 6.96 (d, 1H, J=7.5
Hz), 6.93-
6.87 (m, 2H), 4.54 (t, 1H, J=5.7 Hz), 3.83 (s, 3H), 3.72-3.63 (m, 2H), 3.56-
3.47 (m, 2H),
2.74-2.68 (m, 2H), 1.97-1.90 (m, 2H), 1.22 (t, 6H, J=7.0 Hz).
Preparation 131
3-{ 4-[2-(4-Chloro-phenyl)-4-oxo-4H-thiazolo[5,4-c]pyridin-5-yl]-2-methoxy-
phenyl ]-
propionaldehyde
N Y ~
cl
_ S N
O H
O
Dissolve 2-(4-chloro-phenyl)-5-[4-(3,3-diethoxy-propyl)-3-methoxy-phenyl]-5H-
thiazolo[5,4-c]pyridin-4-one (180 mg, 0.36 mmol) in THF (2.0 mL) and water
(1.0 mL)
then add glacial acetic acid (0.6 mL). Stir the solution at 45 C overnight.
Dilute the
solution with EtOAc (50 mL), wash with saturated NaHCO3 (20 mL), then dry,
filter and
concentrate the solution. Purify the crude material by flash chromatography,
using a
linear gradient of 100% hexanes to 80% EtOAc/hexanes, to give the title
compound (94
mg, 61%). MS (ES+) 425.0 (M+1)+. 1H NMR (400 MHz, CDC13): b 9.83 (s, 1H), 8.03
(d, 2H, J=8.3 Hz), 7.49 (d, 2H, J=8.8 Hz), 7.41 (d, 1H, J=7.5 Hz), 7.26 (m,
1H), 6.99-
6.89 (m, 3H), 3.84 (s, 3H), 2.99 (t, 2H, J=7.3 Hz), 2.77 (t, 2H, J=7.3 Hz).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
113
Example 50
2-(4-Chloro-phenyl)-5-[3 -methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl] -5H-
thiazolo [5,4-
c]pyridin-4-one
/ \ N I ~
CI
- S N ~ O~
I / N
Dissolve 3-{4-[2-(4-chloro-phenyl)-4-oxo-4H-thiazolo[5,4-c]pyridin-5-yl]-2-
methoxy-phenyl}-propionaldehyde (94mg, 0.22mmol) in 1,2-dichloroethane (2.2
mL) and
add pyrrolidine (20 L, 0.24 mmol), AcOH (19 L, 0.33 mmol), and NaHB(OAc)3
(70
mg, 0.33 mmol). Stir the yellow solution at room temperature for 1 h, then add
1N NaOH
(5 mL), and extract the mixture with CH2C12 (2 x 10 mL). Combine the organic
portions,
then dry, filter, and concentrate. Purify the crude material by flash
chromatography, using
8% 2N NH3 in MeOH/CHC13 as eluent, to give the title compound (75 mg, 71%). MS
(ES+) 480.1 (M+1)+. 1H NMR (400 MHz, CDC13): S 8.03 (d, 2H, J=8.3 Hz), 7.48
(d, 2H,
J=8.8 Hz), 7.43 (d, 1H, J=7.5 Hz), 7.25 (t, 1H, J=3.7 Hz), 6.97 (d, 1H, J=7.5
Hz), 6.93-
6.88 (m, 2H), 3.83 (s, 3H), 2.70 (t, 2H, .1=7.7 Hz), 2.60 (br s, 6H), 1.96-
1.79 (m, 6H).
Example 51
2-(4-Chloro-phenyl)-5-[4-(4-hydroxy-piperidin-1-yl)-3-methoxy-phenyl]-6,7-
dihydro-SH-
thiazolo[5,4-c]pyridin-4-one, hydrochloride salt
N
CI ~~ S N O\
O N
HCI
OH
Dissolve 2-(4-chloro-phenyl)-5-[3-methoxy-4-(4-triisopropylsilanyloxy-
piperidin-
1-yl)-phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (681 mg, 1.09 mmol)
in THF
(10 mL) then add tert-butylammonium fluoride (1.0 M solution in THF, 1.30 mL,
1.30
mmol). Stir the solution at room temperature for 2 h, then dilute with EtOAc
(50 mL) and
wash with 2N NH4C1 (20 mL). Concentrate the organic solution and purify the
crude
material by flash chromatography, using 8% MeOH (2N NH3)/CHC13 as eluent, to
give
semi-pure material. Triturate the solids with ether to give the title compound
as the free
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
114
base (265 mg, 52%). Mix 2-(4-chloro-phenyl)-5-[4-(4-hydroxy-piperidin-l-yl)-3-
methoxy-phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (53 mg, 0.11 mmol)
in
MeOH (1 mL) and add 1N HCl (2.0 mL, 2.0 mmol). Stir the mixture at room
temperature
until all the solids dissolve and then cool to -20 C overnight. Collect the
precipitate by
filtration, wash with ether, and dry under vacuum to give the title compound
(40 mg,
70%). MS (ES+) 470.0 (M+1)+. 1H NMR (400 MHz, DMSO-d6): S 11.69 (s, 1H), 8.06
(d, 2H, J=8.4 Hz), 7.70 (s, 1H), 7.63 (d, 2H, J=8.4 Hz), 7.34 (s, 1H), 7.14
(s, 1H), 4.65 (s,
4H), 4.16 (t, 2H, J=6.8 Hz), 3.95 (s, 3H), 3.64-3.32 (m, 2H), 3.29 (t, 2H,
J=7.0 Hz), 2.11-
1.76 (m, 4H),
Example 52
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl]-5H-
thiazolo[5,4-
c]pyridin-4-one hydrochloride
CI ~ \ N ~ \
N 0
~
O ()~ N
HCI
Mix 2-(4-chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-propyl)-phenyl]-5H-
thiazolo[5,4-c]pyridin-4-one (833 mg, 1.74 mmol) in MeOH (10 mL) and add 1N
HCI
(2.0 mL, 2.0 mmol). Stir the mixture at room temperature until all the solids
dissolve
then cool to -20 C overnight. Collect the precipitate by filtration, wash
with ether, and
20, dry under vacuum to give the title compound (725 mg, 81%). MS (ES+) 480.0
(M+1)+.
1H NMR (400 MHz, CDC13): 810.14 (s, 1H), 8.16 (d, 2H, J=8.8 Hz), 7.77 (d, 1H,
J=7.5
Hz), 7.67 (d, 2H, J=8.8 Hz), 7.34 (d, 1H, J=8.4 Hz), 7.15 (d, 1H, J--1.8 Hz),
7.09-7.02 (m,
2H), 3.83 (s, 3H), 3.58-3.50 (m, 2H), 3.20-3.13 (m, 2H), 3.02-2.94 (m, 2H),
2.68 (t, 2H,
J=7.7 Hz), 2.03-1.93 (m, 4H), 1.90-1.83 (m, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
115
Example 53
2-(4-Chl oro-phenyl)-5 -[ 3-metho xy-4-( 3-pyrrol idin-l-yl-propyl )-phenyl] -
6,7 -dihydro-5H-
CI S I N ~ O
0
thiazolo[5,4-c]pyridin-4-one HCI
Prepare the titled compound by essentially following procedures as described
for
Example 52. MS (ES+) 482.0 (M+1)+. 1H NMR (400 MHz, CDC13) S: 10.45 (s, 1H),
8.05 (d, 2H, J=8.4 Hz), 7.62 (d, 2H, J=8.8 Hz), 7.22 (d, 1H, J=7.9 Hz), 7.05
(d, 1H, J=1.8
Hz), 6.93 (dd, 1H, J=7.9, 1.8 Hz), 4.11 (t, 2H, J=7.0 Hz), 3.80 (s, 3H), 3.55-
3.47 (m, 2H),
3.27 (t, 2H, J=6.8 Hz), 3.15-3.08 (m, 2H), 3.00-2.91 (m, 2H), 2.62 (t, 2H,
J=7.5 Hz),
2.02-1.81 (m, 6H).
Preparation 132
Methanesulfonic acid 2-[2-(4-chloro-phenyl)-5-(1-triisopropylsilanyl-lH-indol-
5-
ylcarbamoyl)-thiazol-4-yl]-ethyl ester
o=s=o
N O
CI ~ ~ S I N
C ~ N
Dissolve 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (1-
triisopropylsilanyl-lH-indol-5-yl)-amide (1.54g, 2.77mmol) in CH2C12 (25 mL)
and add
Et3N (0.33 mL, 2.36 mmol) and methanesulfonyl chloride (0.16 mmol, 2.13 mmol).
Stir
the mixture at room temperature for 2 h, then add additional Et3N (0.33 mL,
2.36 mmol)
and methanesulfonyl chloride (0.16 mmol, 2.13 mmol). Stir the mixture for an
additional
2 h, dilute with EtOAc (50 mL), then wash with water (20 mL) and brine (20
mL). Dry,
filter and concentrate the organic solution. Purify the crude material by
flash
chromatography, using a linear gradient of 100% hexanes to 50% EtOAc/hexanes
as
eluent, to give the title compound (1.15 g, 100%). MS (ES+) 632.1 (M+1)+. 1H
NMR
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
116
(400 MHz, CDC13): S 7.96-7.84 (m, 4H), 7.50-7.43 (m, 3H), 7.30-7.26 (m, 2H),
6.62 (d,
1H, J=3.1 Hz), 4.77 (t, 2H, J=6.2 Hz), 3.60 (t, 2H, J=6.4 Hz), 2.99 (s, 3H),
1.73-1.65 (m,
3H), 1.14 (d, 18H, J=7.5 Hz).
Preparation 133
2-(4-Chloro-phenyl)-5-(1-triisopropylsilanyl-1 H-indol-5-yl)-6,7-dihydro-5H-
thiazolo [5,4-
c]pyridin-4-one
N
CI
S N ~ ~
O
\rS
~
Dissolve 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-carboxylic acid (1-
triisopropylsilanyl-lH-indol-5-yl)-amide in CH2C12 (25 mL) and add Et3N (0.33
mL, 2.36
mmol) and methanesulfonyl chloride (0.16 mmol, 2.13 nunol). Stir the mixture
at room
temperature for 2h, then add additional Et3N (0.33 mL, 2.36 mmol) and
methanesulfonyl
chloride (0.16 mmol, 2.13 mmol). Stir the mixture for an additional 2 h,
dilute with
EtOAc (50 mL), then wash with water (20 mL) and brine (20 mL). Dry, filter and
concentrate the organic solution then and purify the crude material by flash
chromatography, using a linear gradient of 100% hexanes to 50% EtOAc/hexanes
as
eluent, to give the title compound (1.15 g, 100%). MS (ES+) 536.1 (M+1)+. 1H
NMR
(400 MHz, CDC13) 8: 7.93 (d, 2H, J=8.3 Hz), 7.56 (d, 1H, J=1.8 Hz), 7.50 (d,
1H, J=8.8
Hz), 7.44 (d, 2H, J=8.3 Hz), 7.27 (d, 1H, J=3.1 Hz), 7.14-7.10 (m, 1H), 6.61
(d, 1H,
J=2.6 Hz), 4.16 (t, 2H, J=6.8 Hz), 3.29 (t, 2H, J=7.0 Hz), 1.73-1.65 (m, 3H),
1.14 (d,
18H, J=7.5 Hz).
Preparation 134
2-(4-Chloro-phenyl)-5-(1 H-indol-5-yl)-6,7-dihydro-5H-thiazolo [5,4-c] pyridin-
4-one
N
S ~ N
O N
H
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
117
Mix 2-(4-chloro-phenyl)-5-(1-triisopropylsilanyl-1 H-indol-5-yl)-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one (4.23 g, 7.89 mmol) in THF (50 mL) and add
tetrabutyl-
ammonium fluoride (1.OM in THF, lOvmL, 10 mmol). Stir the red solution at room
temperature for 2 h, then quench with aqueous 2M NH4C1(50 mL) and extract with
CH2Cla (3 x 50mL). Dry, filter, and concentrate the organic solution. Purify
the crude
material by flash chromatography, using 8%MeOH (2N NH3)/CHC13 as eluent, then
triturate the resulting yellow solid with ether to give the title compound
(2.68 g, 89%).
MS (ES+) 380.0 (M+1)+. 1H NMR (400 MHz, CDC13): S 11.20 (s, 1H), 8.05 (d, 2H,
J=8.4 Hz), 7.62 (d, 2H, J=8.4 Hz), 7.53 (d, 1H, J=1.8 Hz), 7.43-7.38 (m, 2H),
7.09 (dd,
1H, J=8.6, 2.0 Hz), 6.45-6.43 (m, 1H), 4.11 (t, 2H, J=7.0 Hz), 3.28 (t, 2H,
J=7.0 Hz).
Preparation 135
2-(4-Chloro-phenyl)-5-(2,3-dihydro-1 H-indol-5-yl)-6,7-dihydro-5H-thiazolo
[5,4-
c]pyridin-4-one
cl
S
O N
H
Prepare the title compound by essentially following procedure as described in
Preparation
134. MS (ES+) 381.9 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.91 (d, 2H, J=8.3 Hz),
7.43 (d, 2H, J=8.8 Hz), 7.13 (s, 1H), 6.99 (dd, 1H, J=8.1, 2.0 Hz), 6.76 (d,
1H, J=8.3 Hz),
4.04 (t, 2H, J=6.8 Hz), 3.64 (t, 2H, J=8.3 Hz), 3.26 (t, 2H, J=6.8 Hz), 3.08
(t, 2H, J=8.3
2 0 Hz).
Preparation 136
( )-3- { 5-[2-(4-Chloro-phenyl)-4-oxo-6,7-dihydro-4H-thiazolo [5,4-c]pyridin-5-
y1]-2, 3-
dihydro-indole-l-carbonyl}-pyrrolidine-l-carboxylic acid tert-butyl ester
N
CI ~ ~ <
S N \
O I ~ ON 0
~NAO" \
O
Dissolve 2-(4-chloro-phenyl)-5-(2,3-dihydro-lH-indol-5-yl)-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-one (163 mg, 0.43 mmol) in CH2C12 (4.0 mL) and add (
)-
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
118
pyrrolidine-1,3-dicarboxylic acid 1-tert-butyl ester (140 mg, 0.65 mmol), Et3N
(0.09 mL,
0.64 mmol), and [dimethylamino-([1,2,3]triazolo[4,5-b]pyridin-3-yloxy)-
methylene]-
dimethyl-amrnonium hexafluoro phosphate (246 mg, 0.65 mmol). Stir the solution
at
room temperature for 2 h. Concentrate and purify by flash chromatography,
using a 8%
MeOH (2N NH3)/CHC13 as eluent, to give the title compound (231 mg, 93%). IH
NMR
(400 MHz, DMSO-d6): 8 8.10 (d, 1H, J=8.8 Hz), 8.05 (d, 2H, J=8.8 Hz), 7.62 (d,
2H,
J=8.3 Hz), 7.29 (s, 1H), 7.19-7.15 (m, 1H), 4.22 (t, 2H, J=8.8 Hz), 4.07 (t,
2H, J=7.0 Hz),
3.54 (t, 1H, J=8.8 Hz), 3.47-3.36 (m, 3H), 3.29 (m, 1H), 3.26 (t, 2H, J= 6.2
Hz), 3.18 (t,
2H, J=8.3 Hz), 2.16 (m, 1 H), 2.01 (m, 1 H), 1.41 (s, 9H).
Example 54
( )-2-(4-Chloro-phenyl)-5-[ 1-(pyrrolidine-3-carbonyl)-2,3-dihydro-1 H-indol-5-
yl]-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-onium trifluoroacetate
N
cl
S N'\
O I /N F O
Om~NH F~OH
F
Dissolve ( )-3-{5-[2-(4-chloro-phenyl)-4-oxo-6,7-dihydro-4H-thiazolo[5,4-
c]pyridin-5-yl]-2,3-dihydro-indole-l-carbonyl}-pyrrolidine-l-carboxylic acid
tert-butyl
ester (225 mg, 0.39 mmol) in TFA (2 mL) and stir at room temperature for 1 h.
Concentrate the solution and re-dissolve the crude material in MeOH. Remove
the light
yellow solid by filtration and wash with ether. Dry under vacuum to give the
title
compound (188 mg, 82%). MS (ES+) 479.0 (M+l)+. 1H NMR (400 MHz, DMSO-d6): S
8.89 (s, 2H), 8.10 (d, 1H, J=8.8 Hz), 8.05 (d, 2H, J=8.8 Hz), 7.62 (d, 2H,
J=8.8 Hz), 7.31
(s, 1H), 7.22-7.18 (m, 1H), 4.22 (t, 2H, J=9.4 Hz), 4.07 (t, 2H, J=7.0 Hz),
3.56-3.47 (m,
2H), 3.42-3.35 (m, 1H), 3.29-3.20 (m, 6H), 2.35-2.27 (m, 1H), 2.13-2.03 (m,
1H).
Example 55
( )-2-(4-Chloro-phenyl)-5-[ l -(1-methyl-pyrrolidine-3-carbonyl)-2,3-dihydro-1
H-indol-5-
yl] -6,7-dihydro-5H-thiazolo [5,4-c] pyridin-4-one
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
119
CI S N \
0 I ~ N
I-CN -
O
Dissolve ( )-2-(4-chloro-phenyl)-5-[1-(pyrrolidine-3-carbonyl)-2,3-dihydro-1H-
indol-5-yl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-onium trifluoroacetate
(167 mg, 0.28
mmol) in 1,2-dichloroethane (3.0 mL) and add paraformaldehyde (203 mg), acetic
acid
(0.02 mL, 0.35 mmol), and sodium triacetoxyborohydride (78 mg, 0.37 mmol).
Stir the
mixture at room temperature for 4 h, then dilute with CH2Cl2 (20 mL) and wash
with 1N
NaOH (10 mL). Dry, filter and concentrate the organic solution then and purify
the crude
material by flash chromatography, using a 8% MeOH (2N NH3)/CHC13 as eluent, to
give
the title compound as a yellow solid (86 mg, 62%). MS (ES+) 493.0 (M+1)+. 1H
NMR
(400 MHz, DMSO-d6): S 8.30 (d, 1H, J=8.8 Hz), 7.95 (d, 2H, J=8.3 Hz), 7.47 (d,
2H,
J=8.3 Hz), 7.28 (s, 1H), 7.15 (d, 1H, J=10.1 Hz), 4.17 (t, 2H, J=8.8 Hz), 4.12
(t, 2H,
J=6.8 Hz), 3.33-3.22 (m, 5H), 3.17-3.10 (m, 1H), 3.01-2.90 (m, 1H), 2.85-2.76
(m, 1H),
2.64-2.56 (m, 1H), 2.50 (s, 3H), 2.29-2.21 (m, 2H).
Example 56
2-(4-Chloro-phenyl)-5-(1-pyridin-2-ylmethyl-1 H-indol-5-yl)-6,7-dihydro-5H-
thiazolo[5,4-c]pyridin-4-one
N
CI
N
~ /
~
Dissolve 2-(4-chloro-phenyl)-5-(1H-indol-5-yl)-6,7-dihydro-5H-thiazolo[5,4-
2 0 c]pyridin-4-one (49 mg, 0.13 mmol) in DMF (1 mL) and add sodium hydride
(17 mg,
0.42 mmol). Stir the mixture at room temperature for 30 min then add 2-
bromomethyl-
pyridine hydrobromide (35 mg, 0.14 mmol). Stir the mixture at room temperature
for 5 h,
then dilute with EtOAc (30 mL) and wash with saturated NaHCO3 (10 mL). Dry,
filter
and concentrate the organic solution. Purify the crude material by flash
chromatography,
using a linear gradient of 20% to80% EtOAc/hexanes as eluent, to give the
title
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
120
compound (32 mg, 52%). MS (ES+) 471.0 (M+l)+. 'H NMR (400MHz, CDC13) 8: 8.55-
8.53 (m, 1H), 8.05 (d, 2H, J=8.4 Hz), 7.73 (dt, 1H, J=7.7, 1.8 Hz), 7.62 (d,
2H, J=8.8 Hz),
7.57-7.55 (m, 2H), 7.45 (d, iH, J=8.8 Hz), 7.30-7.26 (m, 1H), 7.10 (dd, 1H,
J=8.6, 2.0
Hz), 7.03 (d, 1H, J=7.9 Hz), 6.52 (d, 1H, J=3.5 Hz), 5.53 (s, 2H), 4.10 (t,
2H, J=7.0 Hz),
3.27 (t, 2H, J=7.0 Hz).
Prepare Examples 57 and 58 by essentially following the procedure as described
for
Example 56, using the appropriate alkyl halide.
Example 57
2-(4-Chloro-phenyl)-5 -(1-pyridin-4-ylmethyl-1 H-indol-5-yl)-6,7-dihydro-5H-
thiazolo [5,4-c]pyridin-4-one
N
CI C S I N
O I N
N
MS (ES+) 471.0 (M+1)+. 1H NMR (400 MHz, CDC13): $ 8.50 (d, 2H, J=5.7 Hz), 8.05
(d,
2H, J=8.8 Hz), 7.62 (d, 2H, J=8.8 Hz), 7.58 (d, 2H, J=2.6 Hz), 7.42 (d, 1H,
J=8.8 Hz),
7.14-7.09 (m, 3H), 6.56 (d, 1H, .1=3.1 Hz), 5.53 (s, 2H), 4.11 (t, 2H, J=6.8
Hz), 3.28 (t,
2H, J=7.0 Hz).
Example 58
2-(4-Chloro-phenyl)-5-[1-(2-morpholin-4-yl-ethyl)-1H-indol-5-yl]-6,7-dihydro-
5H-
thiazolo[5,4-c]pyridin-4-one
N
CI ~ f S ]
N \
O
O I / \N NJ
MS (ES+) 493.0 (M+l)+. 'H NMR (400MHz, DMSO-d6): S 8.06 (d, 2H, J=8.8 Hz),
7.68
(d, 1H, J=8.8 Hz), 7.62 (d, 2H, J=8.8 Hz), 7.58 (d, 1H, J=2.2 Hz), 7.50 (d,
1H, J=3.1 Hz),
7.22 (dd, 1H, J=8.6, 2.0 Hz), 6.53 (d, 1H, J=2.6 Hz), 4.71 (s, 2H), 4.12 (t,
2H, J=7.0 Hz),
4.02-3.94 (m, 2H), 3.84-3.75 (m, 2H), 3.56-3.42 (m, 4H), 3.29 (t, 2H, J=7.0
Hz), 3.20-
3.08 (m, 2H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
121
Preparation 137
5-nitro-3H-benzoox azole-2 -thione
I ~ 0; ~ s
02N N
H
Combine 2-amino-4-nitrophenol (15.8 g, 102 mmol) and potassium ethyl xanthate
(18.3 g, 114 mmol) in pyridine (200 mL). Heat the reaction at reflux for 1 h.
Allow the
reaction to cool to room temperature and pour into concentrated HCl (100 mL)
and ice.
Filter and wash the solids with 1N HCl to remove excess pyridine. Dry the
solids under
house vacuum at 50 C for 2 days to obtain the title compound (15.85 g, 79%).
IH NMR
(400 MHz, DMSO-d6): S 8.18 (dd, 1H, J = 8.8, 2.2 Hz), 7.93 (d, 1H, J = 2.2
Hz), 7.73 (d,
1H, J= 8.8 Hz).
Preparation 138
11 !C 's \-
2-Ethylsulfanyl-5-nitrobenzooxazole 02N N
Dissolve 5-Nitro-3H-benzooxazole-2-thione (10.58 g, 53.9 mmol) in anhydrous
THF
(300mL). Cool the mixture to 0 C in an ice bath. Add NaH (4.90 g, 60%
dispersion in
mineral oil) slowly. Stir the resulting mixture at 0 C for 10 min. Add
iodoethane (20.0
mL, 0.250 mmol) to the stirring mixture. Allow the mixture to warm to room
temperature
and stir overnight. Adsorb the reaction mixture onto silica gel and subject to
flash column
chromatography in 2 batches (330 g, 120 g columns, eluting with 10-50% ethyl
acetate/n-
hexane both times) to yield the desired product (4.93g, 41%). 1H NMR (400 MHz,
DMSO-d6): S 8.47 (d, J=2.4 Hz, 1H), 8.23 (dd, J= 9.2, 2.6Hz, 1H), 7.88 (d,
J=8.8 Hz,
1H), 3.37 (q, J= 6.8 Hz, 2H), 1.45 (t, J=7.6 Hz, 3H).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
122
Preparation 139
Methyl-(1-methyl-piperidin-4-yl)-(5-nitro-benzooxazol-2-yl)-amine
/
N
O
/N~
O2N N
Dissolve 2-ethylsulfanyl-5-nitro-benzooxazole (1.17 g, 5.23 mmol) in anhydrous
THF (10
mL) in a reaction tube and blow nitrogen into the vessel for 10 s. Add methyl-
(1-methyl-
piperidin-4-yl)-amine (1.37 mL, 9.42 mmol) to the solution. Quickly seal the
vessel and
immerse into a pre-heated oil bath (100 C) and stir for 24 h. Concentrate the
reaction
mixture in vacuo, wash with 1.OM NaOH(aq) (2 x 50mL), dry over Na2SO4, filter,
and
concentrate in vacuo. Subject the residue by silica gel flash column
chromatography,
eluting with 2N NH3 in MeOH/CH2C12, to yield the desired product (0.608 g,
40%).
MS(ES+) 291.0 (M+1)+.
Preparation 140
N2-Methyl-N2-(1-methyl-piperidin-4-yl)-benzooxazole-2,5-diamine
/
N
~ O
~ , N\
H2N N
Dissolve methyl-(1-methyl-piperidin-4-y1)-(5-nitro-benzooxazol-2-yl)-amine
(0.583 g, 2.01 mmol), in acetic acid (8 mL), and add iron (1.12 g, 20.1 mmol)
to the
solution. Stir the mixture at 40 C for 3 h. Filter the reaction mixture
through Celite
and wash with water/MeOH. Concentrate the reaction mixture in vacuo. Subject
the
residue to silica gel flash column chromatography, eluting with 10% 2N NH3 in
MeOH/CH2C12, to yield the desired product (0.474 g, 91%). MS(ES+) 261.2
(M+1)+.
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
123
Preparation 141
N,N,N'-Trimethyl-N-(5-nitro-benzooxazol-2-yl)-ethane-1,2-diamine
N-
)Zz~ O
//\- N
02N N
Prepare the title compound by essentially following the procedure as described
in
Preparation 139, using 2-methylsulfanyl-5-nitro-benzooxazole (5.0 g, 23.8
mmol) and
N,N,N'-Trimethyl-ethane-1,2-diamine (15.4 mL, 118.9mmo1) at 140 C. The
product is
purified by silica gel flash column chromatography (330 g column, eluting with
5% 2N
NH3 in MeOH/CH2C2) to yield the desired product (2.8 g, 44%). MS(ES+) 265.3
(M+ W.
Preparation 142
NZ-(2-Dimethylamino-ethyl)-NZ-methyl-benzooxazole-2,5-diamine
\
N-
O
/N
H2N N
The title compound was prepared according to the procedure described in
General
Method B using N,N,N'-Trimethyl-N'-(5-nitro-benzooxazol-2-yl)-ethane-1,2-
diamine
(4.131g, 15.63mmol), acetic acid (50 mL), and Fe (8.72 g, 78.15 mmol),
stirring for 3h:
(3.57g, 98%): mass spectrum (m/e): 265.3 (M+1).
Example 59
2-(4-Chloro-phenyl)-5-{2-[(2-dimethylamino-ethyl)-methyl-amino]-benzooxazol-5-
yl}-
6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one hydrochloride
CI ~\ N ] HCI N-
_ s N I~ N
\>-
O N
Dissolve Na-(2-dimethylamino-ethyl)-Na-methyl-benzooxazole-2,5-diamine (0.50
g, 2.14 mmol) in CH2C12 (10.0 mL) and treat with 2N aluminum trimethyl in
hexanes (2.0
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
124
mL, 4.0 mmol). Stir at room temperature for 15 min and add 2-(4-chloro-phenyl)-
6,7-
dihydro-pyrano[4,3-d]thiazol-4-one (0.60 g, 2.26 mmol) neat and stir the
reaction at room
temperature for 2 h. Carefully quench the mixture with saturated Rochelles
salt solution
and stir at room temperature for 1 h. Dilute with water, filter the
precipitate and dry.
Dissolve the solid (0.30 g, 0.60 mmol) and treat with tributylphophine (0.26
mL, 0.90
mmol) and diisopropylazodicarboxylate (0.18 rnL, 0.09 mmol). Stir the reaction
at room
temperature for 18 h and concentrate. Purify the crude material by flash
chromatography,
using 0 - 10% 2N NH3/MeOH in CH2C12 to give the free amine. Dissolve the free
amine
in MeOH (2.0 mL) and add 1N HCl in ether (1.0 mL), sonicate for 5 min, and
concentrate. Triturate the solid with ether, filter, and dry to give the title
compound (0.13
g). MS (ES+) 482 (M+1)+. IH NMR (400 MHz, DMSO- d6): S 10.59 (s, 1H), 8.01 (d,
2H, J=8.4 Hz), 7.58 (d, 2H, J=8.4 Hz), 7.44 (d, 1H,J=8.4 Hz), 7.30 (d, 1H,
J=2.2 Hz),
7.03 (dd, 1H, J=8.4, 2.2 Hz), 4.07 (t, 2H, J=6.8 Hz), 3.92 (t, 2H, J=5.3 Hz),
3.39 (t, 2H,
J=5.3 Hz), 3.24 (t, 2H, J=6.8 Hz), 3.17 (s, 3H), 2.81 (d, 6H, J=4.8 Hz).
Example 60
2-(4-Chloro-phenyl)-5- { 2-[methyl-(1-methyl-piperidin-4-yl)-amino]-
benzooxazol-5-yl } -
N
cl
C N N~N
HCI (N~
6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one
Prepare the title compound by essentially following the procedure as described
in
Example 59, using N2-methyl-N2-(1-methyl-piperdin-4-yl)-benzooxazole-2,5-
diamine.
MS (ES+) 508 (M+1, free amine)+. 1H NMR (400 MHz, DMSO-d6): S 10.79 (brs, 1H),
8.04 (d, 2H, J=8.4 Hz), 7.62 (d, 2H, J=8.8 Hz), 7.45 (d, 1H, J=8.8 Hz), 7.31
(d, 111,
J=2.2 Hz), 7.04 (dd, 1H, J=8.6, 2.2 Hz), 4.39 (m, 1H), 4.10 (t, 2H, J=7.0 Hz),
3.48 (d,
2H, J=11.4 Hz), 3.28 (t, 2H, J= 2.0 Hz), 3.17 (m, 2H), 3.04 (s, 3H), 2.73 (d,
311, J=4.8
Hz), 2.27 (m, 2H), 1.94 (d, 2H, J=13.2 Hz).
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
125
Example 61
2-(4-Chloro-phenyl)-5-[3-methoxy-4-(3-pyrrolidin-1-yl-prop-1-ynyl)-phenyl]-6,7-
dihydro-5H-thiazolo [5,4-c]pyridin-4-one
CI ~ \ N
S ) N 0
NI
I /
N
Treat a suspension of 5-(4-bromo-3-methoxy-phenyl)-2-(4-chloro-phenyl)-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one (0.25 mg, 0.56 mmol), 1-prop-2-ynyl-
pyrrolidine (0.12 g, 1.10 mmol), dichlorobis(triphenylphosphine)palladium (Il)
(12.0 mg,
0.02 mmol), triethylamine (0.5 mL) in DMF with CuI (4.0 mg, 0.02 mmol). Stir
at 80 C
under nitrogen for 2 days. Dilute the reaction with water and extract with
CH2C12 (2x).
Dry, filter, and concentrate the organic solution and purify the crude
material by flash
chromatography, using a gradient of 0 - 10% MeOH in CHZC12 to give the title
compound
(30.0 mg, 12%). MS (ES+) 478.0 (M+1)+. 1H NMR (400 MHz, CDC13): S 7.89 (d, 2H,
J=8.8 Hz), 7.40 (m, 3H), 6.95 (d, 1H, J=2.2 Hz), 6.82 (dd, 111, J=8.1, 2.0
Hz), 4.10 (t,
2H, J=6.8 Hz), 3.84 (m, 5H), 3.26 (t, 2H, J=6.8 Hz), 2.88 (m, 4H), 1.89 (m,
4H).
Example 62
2-(4-Chloro-phenyl)-5-[4-(2-imidazol-1-yl-ethoxy)-3-methoxy-phenyl]-6,7-
dihydro-5H-
thiazolo[5,4-c]pyridin-4-one hydrochloride
CI ~ \'N :1( HCI
S
0
Treat a solution of 2-(4-chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-6,7-
dihydro-5H-thiazolo[5,4-c]pyridin-4-one (0.20 g, 0.52 mmol), 2-imidazol-1-yl-
ethanol
(0.09 g, 0.80 mmol), and triphenylphosphine (0.27 g, 1.03 rnmol) with
diisopropylazodicarboxylate (0.27 g, 1.34 mmol). Warm the solution to 80 C
and stir for
18 h. Concentrate the reaction and purify the residue by flash chromatography,
using 0 -
10% 2N NH3/MeOH in CH2Cl2, to give the free amine. Dissolve the free amine in
MeOH
(2.0 mL) and add 1N HC1 in ether (2.0 mL), sonicate for 5 min, and
concentrate. Triturate
the solid with ether, filter, and dry to give the title compound (0.15 g,
58%). MS (ES+)
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
126
481 (M+1, free amine)+. 1H NMR (400 MHz, DMSO-d6).: b 14.84 (s, 1H), 9.17 (s,
1H),
8.00 (d, 2H, J=8.4 Hz), 7.79 (t, 1H, J=1.8 Hz), 7.67 (t, 1H, J=1.8 Hz), 7.57
(d, 2H, J=8.8
Hz), 7.00 (m, 2H), 6.86 (dd, 1H, J=8.6, 2.4 Hz), 4.58 (t, 2H, J=4.8 Hz), 4.35
(t, 2H,
J=4.8 Hz), 4.01 (t, 2H, J=7.0 Hz), 3.70 (s, 3H), 3.21 (t, 2H, J=7.0 Hz).
Example 63
2-(4-Chloro-phenyl)-5-[3-chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-6,7-
dihydro-SH-
thiazolo[5,4-c]pyridin-4-one hydrochloride
CI ~ ~ N HCI
S N ~OCI
0 I / ~,~N
Treat a solution of 2-(4-chloro-phenyl)-5-[3-chloro-4-(2-pyrrolidin-1-yl-
ethoxy)-
phenyl]-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (60.0 mg, 0.12 mmol) in
MeOH
(2.0 mL) with 1N HCl in ether (1.0 mL). Sonicate at room temperature for 15
min,
concentrate, and dry to give the title compound (50 mg, 78%). MS (ES+) 488
(M+l; free
amine)+. 1H NMR (400 MHz, DMSO-d6): cS 10.44 (s, 1H), 8.02 (d, 2H, J=8.6 Hz),
7.59
(m, 2H), 7.55 (d, 1H, J=2.4 Hz), 7.36 (dd, 1H, J=8.8, 2.4 Hz), 7.25 (d; 1H,
J=8.8 Hz),
4.43 (t, 2H, J=4.8 Hz), 4.05 (t, 2H, J=7.0 Hz), 3.62 (m, 4H), 3.24 (t, 2H,
J=7.0 Hz), 3.14
(m, 2H), 2.01 (m, 2H), 1.86 (m, 2H).
Example 64
2-(4-Chloro-phenyl)-5-[3-chloro-4-(2-pyrrolidin-l-yl-ethoxy)-phenyl]-5H-
thiazolo[5,4-c]pyridin-4-one
Cf 0 N
S N Cl HCI
0 0 i~N
Treat a solution of 2-(4-chloro-phenyl)-4-(2-hydroxy-ethyl)-thiazole-5-
carboxylic
acid [3-chloro-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amide (0.15 g, 0.30 mmol)
in CH2Cla
(20 ml) with pyridinium dichromate (0.33 g, 0.88 mmol) and stir suspension at
room
temperature for 3 days. Apply reaction mixture onto silica gel chromatography
column
and purify using 0 - 10% 2N NH3/MeOH in CH202, to give the free amine.
Dissolve the
free amine in MeOH (1.0 mL) and add 1N HCl in ether (0.5 mL), sonicate for 5
min, and
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
127
concentrate. Triturate the solid with ether, filter, and dry to give the title
compound (16
mg, 10%). MS (ES+) 486 (M+1, free amine)+. 1H NMR (400 MHz, DMSO-d6): S 10.59
(s, 1H), 8.13 (d, 2H, J=8.8 Hz), 7.75 (d, 1H, J=7.3 Hz), 7.70 (d, 1H,J=2.6
Hz), 7.64 (d,
2H, J=8.8 Hz), 7.48 (dd, IH, J=8.8, 2.4 Hz), 7.34 (d, 1H, J=8.8 Hz), 7.04 (d,
1H, J=7.5
Hz), 4.49 (t, 2H, J=4.9 Hz), 3.62 (m, 4H), 3.15 (m, 2H), 2.02-1.87 (m, 4H).
Preparation 143
Benzyl-(2-methyl-allyl)-amine
HN
Add benzaldehyde (14.5 mL, 143 mmol) to a mixture of inethallylamine (9.73 g,
137 mmol) and MgSO4 (15.0 g, 125 mmol) in THF (180 mL). Stir for 22 h, filter
the
mixture, and concentrate the filtrate. Dissolve the residue in EtOH (200 mL)
and treat
with NaBH4 (5.00 g, 132 mmol) in 3 portions. After 19 h, remove the solvent by
rotary
evaporation. Treat the residue with 1 M HCl (200 mL) then 5 M HCl (20 mL).
Wash the
solution with tert-butyl methyl ether (250 mL) and then treat with 5 M NaOH
(50 mL) to
make basic. Extract the mixture with CH2Cl2 (200 mL followed by 100 mL). Dry,
filter
and concentrate the organic solution to give the title compound (20.3 g, 92%)
as a
colorless liquid. 1H NMR (400 MHz, DMSO-d6): S 7.2-7.4 (5H, m), 4.84 (1H, s),
4.79
(1H, s), 3.63 (2H, s), 3.03 (2H, s), 1.69 (3H, s).
Preparation 144
1-[Benzyl-(2-methyl-allyl)-amino]-2-methyl-propan-2-ol
HO
N
Add lithium bromide (955 mg, 11.0 mmol) to a mixture of isobutylene oxide
(6.20 mL, 68.8 mmol) and benzyl-(2-methyl-allyl)-amine (9.51 g, 59.0 mmol).
Stir the
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
128
mixture for 3.5 h at room temperature then treat with additional epoxide (1.5
mL, 16.6
mmol) and heat at 60 C for 1.7 h. Dilute the mixture with CH2C12 (200 mL) and
wash
with water (200 mL). Dry, filter and concentrate the organic solution. Dry the
residue at
80 C under vacuum to give the title compound (13.5 g, 98%) as a colorless
oil. 1H NMR
(400 MHz, DMSO-d6): S 7.2-7.4 (5H, m), 4.90 (1H, s), 4.83 (1H, s), 4.18 (1H,
s), 3.59
(2H, s), 2.98 (2H, s), 2.27 (2H, s), 1.71 (3H, s), 1.05 (6H, s).
Preparation 145
4-B enzyl-2-iodomethyl-2,6,6-trimethyl-morpholine
O)<
N
&
Add solid I2 (21.1 g, 83.1 mmol) to a biphasic mixture of 1-[benzyl-(2-methyl-
allyl)-amino]-2-methyl-propan-2-ol (17.6 g, 75.4 mmol) in tert-butyl methyl
ether(250
mL) and 1 M NaHCO3 (100 mL). Stir for 18 h and then add 1 M Na2S2O3 (100mL).
Dilute the mixture with additional tert-butyl methyl ether (200 mL) and
separate the
organic solution. Wash the organic solution with a mixture of 1 M Na2S2O3 (100
mL)
and 1M NaHCO3 (100 mL). Dry, filter, and concentrate the organic solution. Dry
the
residue at 60 C under vacuum to give the title compound (25.2 g, 93%) as a
golden oil.
1H NMR (400 MHz, DMSO-d6): 6 7.2-7.4 (5H, m), 3.49 (1H, d), 3.47 (2H, s), 3.41
(1H,
s), 2.49 (1H, d), 2.23 (1H, s), 2.20 (1H, s), 2.10 (1H, d), 1.24 (3H, s), 1.22
(3H, s), 1.15
(3H, s).
Preparation 146
4-Benzyl-2,2,6,6-tetramethyl-morpholine
>(O)<
N
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
129
Add solid NaBH4 (776 mg, 20.5 mmol) to a solution of 4-benzyl-2-iodomethyl-
2,6,6-trimethyl-morpholine (6.22 g, 17.3 mmol) in DMSO (20 mL) and then heat
the
mixture at 100 C. After 2 h, add additional DMSO (10 rnT.). After an
additional 1.25 h,
add extra NaBH4 (120 mg, 3.17mmo1). Remove the heat after an additional 1.25 h
(total
reaction time = 4.5 h). Quench the excess NaBH4 with 5 M HCl (20 ml). After 15
min,
add 5 M NaOH (20 mL) and 1 M Na2S2O3 (20 mL) and then stir the mixture
overnight.
Dilute the mixture with tert-butyl methyl ether (250 mL) and water (100 mL).
Separate
the organic solution and wash with additional water (4 x 100 mL). Dry, filter
and
concentrate the organic solution. Purify the residue by flash chromatography,
using a
gradient from 50% to 100% CH2Cl2 in pentane as eluent. Dry the product so
obtained
briefly at 60 C under vacuum to give the title compound (2.43 g, 60%) as a
colorless
liquid. 1H NMR (400 MHz, DMSO-d6): 8 7.2-7.4 (5H, m), 3.45 (2H, s), 2.12 (4H,
s),
1.15 (12H, s).
Preparation 147
2,2,6,6-Tetramethymorpholine
>(O)<
N
H
Dissolve 4-benzyl-2,2,6,6-tetramethylmorpholine (Bennett, G.B.; Houlihan,
W.J.;
Mason, R.B.; Engstrom, R.G. J. Med. Chem. 1976, 19, 709-714) (11.0 g, 47.1
mmol) in
EtOH (650 mL) and add 3% Pd/C (8.61 g). Shake the mixture under hydrogen (60
psi) at
40 C for 24 h. Filter the mixture to remove Pd catalyst, and treat the
filtrate with 2M
HC1 in ether, then concentrate. Dry the residue at 80 C under vacuum to give
the title
compound (6.79 g) as a white solid. iH NMR (DMSO-d6, 400 MHz) 8 9.8 (2H, br
s),
2.88 (4H, s), 1.25 (12H, s).
Preparation 148
4-(2-Benzyloxy-ethyl)-2,2,6,6-tetramethyl-morpholine
4-\ j-o -
~ ~
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
130
Dissolve 500 mg (2.79 mmol) of 2,2,6,6-tetramethyl-morpholine (500 mg, 2.79
mmol) in dichloroethane (10 mL). Add benzyloxy-acetaldehyde (470 l, 3.35
mmol) and
stir at room temperature for 20 min. Add sodium triacetoxyborohydride (770 mg,
3.63
mmol) and continue stirring at room temperature for 20 h. Pour the reaction
mixture into
100 mL of 1N NaOH (100 mL) and extract with CH2C12 (2 x 100 mL). Wash the
combined organic layers with brine (100 mL). Purify using silica gel
chromatography,
using a gradient of 0% to 10% (2N NH3 in MeOH)/CHC13 as eluent, to give 490 mg
(63%) of the desired product. MS (ES+) 278.3 (M+1)+.
Preparation 149
2-(2,2,6,6-Tetramethyl-morpholin-4-yl)-ethanol
HO--\- ~-~
N O
Dissolve 4-(2-benzyloxy-ethyl)-2,2,6,6-tetramethyl-morpholine (490 mg, 1.77
mmol)
MeOH (40 mL). Add to a pressure vessel containing a slurry of 10% Pd/C (100
mg) in
MeOH (20 mL). Pressurize with 45 psi hydrogen gas. Monitor the reaction by MS.
After 48 h, add another portion of 10% Pd/C (100 mg )and re-pressurize to 45
psi
hydrogen. Stir an additional 3 days. Filter the reaction mixture through
Celite eluting
with MeOH. Concentrate to give the desired product in quantitative yield. MS
(ES+)
188.3 (M+1)+.
Example 65
2-(4-Chloro-phenyl)-5-{ 3-methoxy-4-[2-(2,2,6,6-tetramethyl-morpholin-4-yl)-
ethoxy]-
phenyl}-5H-thiazolo[5,4-c]pyridine-4-one Hydrochloride
O-
-
N
~ S O
Cj I / HCI
Dissolve 2-(2,2,6,6-tetramethyl-morpholin-4-yl)-ethanol (100 mg, 0.53 mmol)
CH2Cla (5
mL). Add triethylamine (96 L, 0.69 mmol) and then cool the reaction to 0 C.
Add
methanesulfonyl chloride (53 L, 0.69 mmol) and stir for 2 h. Add more
methanesulfonyl
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
131
chloride (53 L, 0.69 mmol) and stir I h. Add more methanesulfonyl chloride
(53 L,
0.69 mmol) and triethyl amine (96 L, 0.69 mmol). Store in freezer (-4 C)
overnight.
Pour the reaction mixture into 1N NaOH (100 mL) and extract with CH2Cla (2 x
100
mL). Wash the combined organics with brine (100 mL). Concentrate the organic
portion
to give crude mesylate which is dissolved in 1-methyl-2-pyrrolidinone (2 mL).
Add this
solution to a room temperature slurry of 2-(4-chloro-phenyl)-5-(4-hydroxy-3-
methoxy-
phenyl)-SH-thiazolo[5,4-c]pyridin-4-one (204 mg, 0.53 mmol) and NaH (21 mg,
0.53
mmol) in 1-methyl-2-pyrrolidinone (6 mL). Stir at room temperature for 2 h and
then
warm to 80 C for 48 h. Cool to room temperature and pour into 1N NaOH (200
mI.) and
extract with EtOAc (2 x 200 mL). Purify via silica gel chromatography, using a
gradient
of 0% to 10% (2N NH3 in MeOH)/CHC13 as eluent to give a mixture of product and
recovered phenol. Dissolve the mixture in CH2Cla (100 mL) and extract with 1N
NaOH
(5 x 100 mL). Concentrate to give the pure product as the free amine. Dissolve
in
CH2C12 (20 mL) and add 4M HCl in dioxane (200 L). Concentrate to give the
product
as the hydrochloride salt. MS (ES+) 554.3 (M+1)}, 1H NMR (400 MHz, DMSO-d6) fi
10.11 (bs, 1H), 8.12 (d, J= 9.0 Hz, 2H), 7.70 (d, J= 7.2 Hz, 1H), 7.63 (d, J =
9.0 Hz, 2H),
7.19-7.16 (m, 2H), 7.02 (d, J= 7.2 Hz, 2 H), 4.52 (bs, 2H), 3.77 (s, 3H), 3.59-
3.54 (m,
4H), 2.95 (t, J= 10.0 Hz, 2H), 1.41 (s, 6H), 1.16 (s, 6H).
Preparation 150
2- [B enzyl-(2-methyl-allyl)-amino]-ethanol
~ OH
N
Add methallyl chloride (68.8 g, 0.760 mol, Aldrich) to a mixture of N-
benzylethanolamine (100 g, 0.663 mol) and potassium carbonate (139 g, 1.00
mol) in
water (600 mL). Heat the mixture to 62 C for 23 h and then transfer to a
separatory
funnel. Extract the product with tert-butyl methyl ether (500 mL). Dry,
filter, and
concentrate the organic solution to give the title compound (131g, 96%) as a
colorless
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
132
liquid. 1H NMR (DMSO-d6, 400 MHz) S 7.20-7.33 (5H, m), 4.92 (1H, br s), 4.83
(1H, br
s), 4.35 (1H, t), 3.53 (2H, s), 3.44-3.50 (2H, m), 2.94 (1H, s), 2.42 (2H, t),
1.69 (1H, s).
Preparation 151
4-Benzyl-2,2-dimethyl-morpholine
>(C
)
N
Add 2- [benzyl-(2-methyl-allyl)-amino] -ethanol (13.0 g, 63.2 mmol) to a
slurry of
mercury (Il) acetate (20.7 g, 65.0 mmol) in water (45 mL) and THF (45 mL).
After 3 h,
treat the mixture with NaOH (25 mL, 2.5 M aqueous, 125 mmol) followed by NaBH4
(2.72 g, 71.9 mmol). After 19 h, decant the mixture away from the metallic
mercury and
add to a separatory funnel with tert-butyl methyl ether (250 mL). Separate the
organic
solution, wash with water (250 mL), filter through a silica plug, and
concentrate. Purify
the residue by flash chromatography using a gradient from 5% to 10% tert-butyl
methyl
ether in CH2C12. Collect and concentrate the fractions containing product then
dissolve
the residue in hexanes (100 mL). Filter the solution through Celite to remove
metallic
mercury and then concentrate the filtrate to give the title compound (7.01 g,
54%) as a
colorless liquid. 1H NMR (DMSO-d6, 400 MHz) 8 7.20-7.40 (5H, m), 3.60 (2H, m),
3.42
(2H, s), 2.29 (2H. m), 2.10 (2H, s), 1.14 (6H, s).
Preparation 152
2,2-Dimethylmorpholine hydrochloride
H HCI
Dissolve 4-benzyl-2,2-dimethyl-morpholine (5.67 g, 27.6 mmol) in CH2C12 (50
mL) and add 1-chloroethyl chloroformate (4.60 mL, 42.2 mmol) while stirring at
room
temperature. After 4 h, concentrate the solution and treat the residue with
MeOH (60
mL). Heat the mixture at 60 C for 2 h, then concentrate again. Dissolve the
residue in
water (125 mL) and wash with tert-butyl methyl ether (125 mL). Concentrate the
aqueous
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
133
layer and dry the resulting residue at 80 C under vacuum to give the title
compound (3.91
g, 93%) as a white solid. 'H NMR (DMSO-d6, 400 MHz) S 9.52 (2H, br s), 3.75
(2H, m),
2.89-2.96 (4H, m), 1.25 (6H, s).
Preparation 153
2-(2,2-Dimethyl-morpholin-4-yl)-ethanol
o
Ho~~N--+
Dissolve 2,2-dimethylmorpholine (151 mg, 1.0 mmol) in 1,2-dichloroethane (3
mL) and
add glycolaldehyde (60 mg, 1.0 mmol). Stir at room temperature for 30 min
followed by
addition of NaBH(OAc)3 (233 mg, 1.1 mmol). Stir 3 h, then quench by adding 30
mL of
1N NaOH. Pour into a separatory funnel and extract with EtOAc (2 x 50 mL).
Wash the
combined organic layers with brine (50 mL). The crude alcohol was used as is
without
further purification. MS (ES+) 160.2 (M+1)+.
Example 66
2-(4-Chloro-phenyl)-5- { 4- [2-(2,2-dimethyl-morpholin-4-yl)-ethoxy] -3-
methoxy-phenyl } -
5H-thiazolo [5,4-c]pyridin-4-one
N o-- C-
CI~S1 C HCI
0
Dissolve 2-(2,2-dimethyl-morpholin-4-yl)-ethanol (88 mg, 0.55 mmol) in 4.5 mL
THF
2 0 (4.5 mL). Add 2-(4-chloro-phenyl)-5-(4-hydroxy-3-methoxy-phenyl)-5H-
thiazolo[5,4-
c]pyridin-4-one (211 mg, 0.55 mmol). This forms a slurry to which is added 217
mg
(0.83 mmol) of triphenylphosphine (217 mg, 0.83 mmol) followed by 161 pL (0.83
mmol) of diisopropyl azodicarboxylate (DIAD). The reaction then becomes a
solution.
Heat the reaction to 80 C for 16 h. Pour into 1N NaOH (200 mL) and extract
with
CH2C12 (2 x 150 mL). Purify via silica gel chromatography, using a gradient of
0% to
10% (2N NH3 in MeOH)/CHC13 as eluent, to obtain a mixture of product and
starting
phenol. Dissolve the mixture in CH2C12 (300 mL) and extract with 5N NaOH (5 x
100
mL) until all the phenol is removed from the organic layer. Wash the organic
layer with
CA 02589695 2007-06-01
WO 2006/066174 PCT/US2005/045866
134
brine (100 mL) and concentrate. Dissolve the residue in CH2C12 (30 mL) and
treat with
4M HCl in dioxane (100 L). Diethyl ether is added until the solution becomes
cloudy.
Let sit at room temperature for 1.5 h then filter the resulting precipitate to
give 18 mg
(6%) of the desired product. MS (ES+) 526.0 (M+1)+, 1H NMR (400 MHz, (CD3OD):
S
8.11 (d, J= 8.5 Hz, 2H), 7.66 (d, J= 7.3 Hz, 1 H), 7.56 (d, J= 8.5 Hz, 2H),
7.19 (d, J= 8.5
Hz, 1H), 7.17 (d, J= 2.0 Hz, 1 H), 7.09 (d, J= 7.3 Hz, 1H), 7.01 (dd, J= 8.5,
2.0 Hz, 1H),
4.51-4.43 (m, 2H), 4.07-4.00 (m, 2H), 3.93-3.90 (m, 1H), 3.89 (s, 3H), 3.70-
3.56 (m, 4H),
3.26-3.19 (m, 1H), 3.07 (d, J= 12.2 Hz, 1H), 1.44 (s, 3H), 1.30 (s, 3H).