Note: Descriptions are shown in the official language in which they were submitted.
CA 02592124 2007-06-19
1
Metal Catalysts with Perm-selective Coatings,
Methods of Making Same and Uses Thereof
Background
The world-wide reserves of conventional crude oil are steadily decreasing and
heavy
oil, such as those obtained from oil sands, are becoming an increase source
for producing oil
and oil-derived products. Heavy oil has different properties than those of
conventional crude
oil. In particular, heavy gas oil (HGO) produced from the oil sands contains
about 50% more
cyclic aromatic compounds and significantly higher concentrations of sulfur
(40 000 ppm) and
nitrogen than conventional crude-derived HGO. The current technology to reduce
the aromatic
content of oil uses conventional non-noble metal sulfide catalysts, often
referred to as
hydrodearomatization (HDA) catalysts. Noble metal catalysts are also known,
but are sensitive
to sulfur poisoning.
Summary
This invention is based, in part, on protecting a metal in sulfur-resistant
metal
containing catalyst that are active and selective under mild operating
conditions, for the
removal of aromatic compounds in heavy oil derived from oil sands. Such
catalysts may
contain protected noble metals. The metal resides in the core of the catalyst.
The core is
sometimes refered to as a catalyst and this core is covered with a coating
that is permeable only
to hydrogen. Larger molecules are excluded from the core do not deactive the
metals. The
hydrogen that can penetrate to the core, adsorbs and dissociates on the metals
to form hydrogen
atoms or ions that are highly reactive. The hydrogen atoms or ions can then
spillover from the
core through the coating to the external surface of the catalyst where other
reactions, such as
hydrogenation, occur.
In various illustrative embodiments of the present invention, there is
provided a catalyst
comprising: a core comprising a metal catalyst dispersed on a support, an
inner shell adsorbed
on the core, and an outer shell adjacent and in contact with the inner shell.
In various other illustrative embodiments of the present invention, there is
provided a
catalyst as described herein further comprising a hydrogenation site adsorbed
on, embedded in
or embedded in part in the outer shell.
CA 02592124 2007-06-19
2
In various other illustrative embodiments of the present invention, there is
provided a
catalyst described herein wherein the metal is a noble metal.
In various other illustrative embodiments of the present invention, there is
provided a
method of hydrogenating unsaturated hydrocarbons comprising exposing an
unsaturated
hydrocarbon to a catalyst described herein.
Brief Description of the Drawings
Figure 1 is a schematic of an illustrative embodiment of a catalyst according
to
the present invention.
Figure 2 is a chart illustrating pore size distribution and adsorption
isotherms of
samples A1-1 (see example 1) and commercial gamma-A1203 (Alfa
Aesar). Sample A1-1 is representative of all in-house prepared samples
with or without platinum as described in Example 1.
Figure 3 is a chart illustrating XRD patterns of alumina samples and Pt/A12O3
catalysts. All samples were calcinated. Catalysts were reduced at 300
C for 2h with flowing H2.
Figure 4 is a chart illustrating XRD patters of reduced Pt/A12O3 catalysts
obtained
with slow scans (0.2 /min).
Figure 5 is a chart illustrating influence of platinum precursors on
dispersion in
Pt/Al203 catalysts. The dry gels were calcinated in a muffle furnace at
550 C for 2h with a heating rate of 2 C/min, and reduced to 300 C for
2h in flowing H2.
Figure 6 (a and b) are TEM images of catalyst BA-1.
Figure 7 is a chart illustrating influence of heating rate during calcination
on Pt
dispersion of Pt/A1203 catalysts. The dry gel was calcinated in flowing
gas at a heating rate of 2 or 10 C/min.
Figure 8 is a chart illustrating influence of gas flow during calcination on
Pt
dispersion of Pt/A1203 catalysts. All the catalysts were calcinated in
flowing air or in static air (muffle furnace at a heating rate of 2 Clmin.
Figure 9 is a chart illustrating CO2 and NO2 evolution monitored by mass
spectrometry during calcination of dry gel alumina (Al-200) in flowing
CA 02592124 2007-06-19
3
02 or He (20m1/min, 10 C/min).
Figure 10 is a chart illustrating CO2 evolution monitored by mass spectrometry
during calcination of Al-200, N-200 and Py-200 in flowing 02 or He
(20m1/min, 10 C/min).
Figure 11 is a chart illustrating thermogravimetric analysis of Al-200, N-200
and
Py-200 heated at 2 C/min in air.
Figure 12 is a chart illustrating differential thermal analysis of Al-200, N-
200 and
Py-200 heated at 2 C/min in air and also for N-200 heated at 2 C/min
in He and at 10 C/min in air.
Figure 13 is a chart illustrating toluene hydrogenation activity of N-4, Py-4,
MA-1,
and BA-1 catalysts at 240 C for three different runs: (=) run 1(o) run 2,
and (V) run 3. The numbers given in brackets on the x-axis are the Pt
dispersions of each catalyst.
Figure 14 is a schematic illustration of chemical vapour deposition apparatus.
Figure 15 is a chart illustrating effect of deposition temperature on measured
surface area of A1203 for a deposition time of lh.
Figure 16 is a chart illustrating change in measured surface area of Ni/A12O3
(~)
and A1203 (o) with deposition time, at a deposition temperature of
350 C.
Figure 17 is a chart illustrating change in pore size distribution of NilA12O3
with
deposition time, at a deposition temperature of 350 C.
Figure 18 is a chart illustrating amount of Si02 on Ni/A12O3 as a function of
deposition time at a deposition temperature of 350 C.
Figure 19 is a chart illustrating change in H2 (s) and CO (o) uptakes on
NI/A1203
with deposition time at a deposition temperature of 350 C.
Figure 20 is a chart illustrating XRD spectra for Ni/A12O3: (a) reduced, with
no
deposition, (b) 3-h deposition at 350 C, before reduction, and (c) 3-h
deposition at 350 C, after calcination and reduction.
Figure 21 is a chart illustrating Temperature-programmed desorption (10
C/min)
of NH3 on Ni/A12O3: (a) no deposition, (b) 1-h deposition, (c) 1.5-h
deposition, (d) 2-h deposition, (e) 2.5-h deposition, and (f) 3-h
CA 02592124 2007-06-19
4
deposition, all at a deposition temperature of 350 C.
Figure 22 is a chart illustrating change in n-octane conversion as a function
of Si02
deposition time on Ni/A12O3 during hydrocracking of n-octane at 400 C
and atmospheric pressure.
Figure 23 is a schematic illustration of the structure of Ni/A12O3 catalyst
coated
with Si02.
Figure 24 is an Arrhenius plot of ln k versus 1/T for the hydrogenation of
toluene
with spillover hydrogen. Plot shows two distinct regimes of reaction and
diffusion control.
Figure 25 is a chart illustrating a change in H2 (e) and CO (o) uptakes on
Ni/A1203
with deposition time at a deposition temperature of 350 C
Figure 26 is a chart illustrating a change of n-octane conversion as a
function of
Si02 deposition time on Ni/A1203 during hydrocracking of n-octane at
400 C and atmospheric pressure
Figure 27 is a schematic illustration of Fluidized Chemical Vapour Deposition
(CVFD) apparatus.
Figure 28 is a chart illustrating N2 uptake and Si02 content of Mo,,Oy/A12O3
after
FCVD for 0.5h to 2h at 350 C.
Figure 29 is a chart illustrating change in H2 (=) and CO (o) uptake on
Ni/A12O3
with deposition time at a deposition temperature of 350 C.
Figure 30 is a chart illustrating Si02 deposition on A1203 being more rapid
than on
Ni/A1203 and N2 uptake of coated A1203 decreasing faster than coated
Ni/A1203.
Figure 31 is an illustration of an MoOy/Al2O3 particle before SiO2 deposition.
Figure 32 is an illustration of an MoxOy/A12O3 particle after Si02 deposition.
Figure 33 is a chart illustrating an Energy Dispersive Spectroscopy (EDS)
spectra
of the uncoated Mo,{Oy/A12O3 shown in Figure 31.
Figure 34 is a chart illustrating an EDS spectra of the coated Mo,tOy/Al2O3
particle
shown in Figure 32.
CA 02592124 2007-06-19
Figure 35 is a chart illustrating 29Si MAS NMR of MoXOy/Al2O3 after FCVD for
(a) 0.5h, and (b) 2.5h. Both samples were calcinated after the FCVD
process.
Figure 36 is a chart illustrating 29Si MAS NMR of MoROy/Al2O3 after FCVD for
0.5h.
Figure 37 is a chart illustrating 29Si MAS NMR of Mo,,Oy/Al2O3 after FCVD for
2.5h.
Figure 38 is a chart illustrating activity of Ni/A12O3 towards n-octane
cracking.
Figure 39 is a chart illustrating the percent conversion of benzene to
cyclohexane
at varying temperatures using catalysts, diluents and mixtures thereof.
Figure 40 is a chart illustrating the percent conversion of toluene to
methylcyclohexane at varying temperatures using catalysts, diluents and
mixtures thereof.
Figure 41 is a chart illustrating the percent conversion of O-xylene to
di-methylcyclohexane and trimethylcyclopentane at varying
temperatures using mixtures of catalysts and diluents.
Figure 42 is a chart illustrating the percent conversion to hydrogenated
species
using varying amounts of diluent with Pt/gamma-A1203 at varying
temperatures.
a
Figure 43 is a chart illustrating the Dv(d) [cc/A/g] vs Diameter (A) for the
distribution of pore size of Ni containing catalysts deposited with Si02
under varying conditions.
Figure 44 is a chart illustrating the H2 and CO uptake ( Ug) of Ni containing
catalysts deposited with Si02 under varying deposition times.
Figure 45 is a chart illustrating the Mass of Si02 (g)/g of sample of Ni
containing
catalysts deposited with Si02 under varying deposition times.
Figure 46 is a chart illustrating the NH3 uptake ( mol/g) of Ni containing
catalysts
deposited with Si02 under varying deposition times.
Figure 47 is a chart illustrating the n-octane conversion (%) of Ni containing
catalysts deposited with Si02 under varying deposition times.
CA 02592124 2007-06-19
6
Figure 48 is a chart illustrating the 29 Si MAS NMR data for two Ni containing
catalysts coated with Si02, the lower line corresponds to the 0.5 hour
coating and the higher line corresponds to the 2.5h coating.
Figure 49 is a chart illustrating the BET surface area (m2/g) of Ni containing
catalysts deposited with Si02 under varying deposition times.
Detailed Description
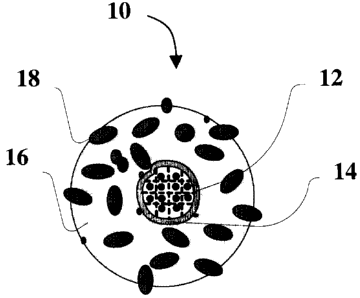
Referring to Figure 1, there is an illustrative embodiment of the invention
shown
generally at 10. A core 12 is provided that is adsorbed on an inner shell 14.
In the examples
and Figures, the core 12 is sometimes referred to as a catalyst. An outer
shell 16 is adsorbed
on the inner shell 14. In the examples and Figures, the outer shell 16 is
sometimes referred to
as a diluent. Hydrogenation sites 18 may be adsorbed, embedded or embedded in
part in or on
the outer shell 16 or may be absent. Catalysts of the present invention may be
used to catalyze
hydrogenation reactions, for example, hydrogenation of aliphatic or aromatic,
unsaturated
organic molecules, including, but not limited to those found in crude oil,
heavy oil, bitumen
and other organic molecules and mixtures used in the production of oil and oil
products.
The core 12 may comprise at least one metal or metal complex of two or more
metals
on a support. The metal may be a noble metal. The support may be an alumina,
often a
gamma-alumina support. The core 12 provides highly reactive spillover hydrogen
which is
able to exit from the core 12 through the inner shell 14 to the outer shell 16
and the
hydrogenation sites 18.
The inner shell 14 may be a silica coating that overlays, at least in part,
the core such
that a shape selective sulfur barrier is formed around the core 12. The inner
shell 14 comprises
pores that are sized so that hydrogen molecules may pass from the outer shell
16, through the
inner shell 14 to the core 12 and so that hydrogen atoms and/or ions may pass
from the core 12,
through the inner shell 14 to the outer shell 16 and the hydrogenation sites
18.
The outer shell 16 may be acidic. The outer shell may be an acidic alumina or
an acidic
silica-alumina support. The support may provide a site for the opening of
cyclic aromatic
compounds to aid hydrogenation of the cyclic aromatic compounds.
The hydrogenation sites 18 may be conventional non-noble metal catalyst. In
some
embodiments, the hydrogenation sites 18 may be Ni-Mo-S particles.
The core 12 may be made by using the sol-gel method. The sol gel method may be
used prepare of a core 12 that comprises pores. The sol-gel method is known in
the art and
CA 02592124 2007-06-19
7
may be found, for example, in Romero-Pascual et al., Journal of Solid State
Chemistry, vol.
168, pp. 343-353 (2002) and Shubert et al, New J. Chem., pp. 721-724 (1998),
which are herein
incorporated by reference.
The sol-gel method allows the preparation of mono disperse nanosized metal
oxide or
metal particles in oxide matrices for many different metals and oxides. The
sol-gel method can
be used to produce a core 12 with uniform metal distribution, tunable particle
size, high surface
area, and stable dispersion. The sol gel method comprises four main steps: 1)
Hydrolysis, 2)
"SoP', 3) "Gel" and 4) Calcination. The resulting properties of the core 12
are sensitive to the
particular processing conditions used in the sol gel method. The dispersion of
the noble metal
within the core 12 may vary between 1% and 100%, between 5% and 70%, between
11% and
100% or may be 80%, depending on the calcination procedure used and the noble
metal
precursor used. The Examples section describes sol-gel synthesis conditions of
the present
invention which are suitable for making a core 12 of the present invention and
for use in the
present invention.
Once a core 12 is prepared an inner shell 14 may be applied to the core 12 to
produce a
perm-selective core. Chemical vapour deposition (CVD) may be used to apply the
inner shell
14 to the core 12. CVD is known in the art and examples may be found in: Niwa,
M., et al., "A
Shape-Selective Platinum-Loaded Mordenite Catalyst For The Hydrocracking Of
Paraffins By
The Chemical Vapor-Deposition Of Silicon Alkoxide." Journal Of The Chemical
Society-
Faraday Transactions I, vol. 81, pp. 2757-2761, (1985); Hibino, Takashi, et
al., "Shape-
selectivity over HZSM-5 modified by chemical vapor deposition of silicon
alkoxide." Journal
of Catalysis, vol. 128, pp. 551-558, (1991); Katada, N., et al., "A continuous-
flow method for
chemical vapor deposition of tetramethoxysilane on gamma-alumina to prepare
silica
monolayer solid acid catalyst." Journal of Chemical Engineering of Japan, vol.
34, pp.
306-311, (2001). Sato, S., et al. "Catalytic and Acidic Properties of Silica-
Alumina Prepared
by Chemical Vapor-Deposition." Applied Catalysis, vol. 62, pp. 73-84, (1990).
CVD may be used to precisely control the pore-opening size of the pores in the
core 12.
In other words the surface of the core 12 is coated such that some of the
inner shell 16 is
positioned, relative to the core 12, to restrict access to the pores of the
core 12 in a manner that
permits hydrogen atoms and/or ions and hydrogen molecules to enter and exit
the pores of core
12 and does not permit some other larger compounds to enter or exit the pores.
For example,
fine control of the pore-opening of NilAl2O3 using CVD for the selective
chemisorption of Hz
(2.9 angstroms) and exclusion of larger molecules (N2 - 3.6 angstroms and CO -
3.8
CA 02592124 2007-06-19
8
angstroms) may be provided by CVD. The pore-openings of a commercial gamma-
A1203
impregnated with Ni may be modified by depositing Si02 on the external surface
in a fluidized
bed using tetramethoxysilane (TMOS) as the Si02 precursor. The pore volume of
Ni/A1203
decreases with increasing deposition time. The modal pore diameter may not
change
significantly and internal pore structure may not be modified. For example, a
sample coated
for 2.5 hours (about 14 wt% Si02), may reduce the uptake of CO and N2 by about
95% and
about 87%, respectively, while the uptake of H2 remains constant. The NH3
uptake may
decrease by about 80%. The Examples section describes CVD conditions of the
present
invention which are suitable for making an inner shell 14 of the present
invention and for use
in the present invention. CVD conditions may be adjusted to provide the
desired deposition of
the inner shell 14 on the core 12 to provide the desired characteristics of
the perm-selective
core.
Once an inner shell 14 is adsorbed on a core 12, an outer shell 16 may be
added to the
inner shell 14. This can be achieved by mechanically mixing the outer shell 16
with the perm-
selected core to produce a shelled core. The mass ratio of the outer shell 16
to the perm-
selective core for mixing can be 1:1, 2:1 or 4:1 or any mass ratio in between
1:1 and 4:1. The
mass ratio may be selected to suit the particular desired characteristics of
the shelled core. In
some embodiments, the hydrogenation sites 18 may be mixed with the outer shell
16 to provide
a primed outer shell. The primed outer shell may then be mixed with the perm-
selective core
in a desired mass ratio to provide a catalyst of the present invention. In
other embodiments, the
shelled core is mixed in a desired mass ratio with hydrogenation sites 18 to
provide a catalyst
of the present invention.
Examples
Example 1
Catalyst preparation
The platinum precursors were prepared by dissolving PtCl2 (Sigma-Aldrich, +99%
purity) in an aqueous solution of NH3, CH3NH2, n-butylamine or pyridine. The
solvent and
excess ligands were removed by open dish drying in a fume hood. The resulting
Pt precursors
were Pt(NH3)4C12, Pt(C5HSN)4C12, Pt(CH3NH2)4C12, and Pt(C4H9NH2)4C12. The
platinum
content in the Pt precursors was determined using ICP-MS (Galbraith Labs
Inc.). Elemental
analysis for C, H, and N content in the Pt precursors were performed using a
Perkin-Elmer
CA 02592124 2007-06-19
9
2400 CHN Analyzer. Proton and carbon NMR(Bruker AMX300) were also performed to
confirm the identities of the groups present in the precursor using the BBI5
probe with the
sample dissolved in dimethyl sulfoxide (DMSO) or deuterated chloroform
(CDC13). Pt-
containing dry alumina gels were prepared using a sol-gel method similar to
the procedure by
Cho et al. Deionized water was mixed with aluminium tri-sec-butoxide (ATB) in
an H20/ATB
molar ratio of 100, and then stirred for 30 min at room temperature. Next, a
0.1 g/ml HNO3
solution was added drop-wise to the mixture, and stirred for 10 min. During
the stirring the
ATB decomposed resulting in a phase containing sec-butanol forming on top of a
phase
containing the sol. After separating sec-butanol from the mixture, additional
HNO3 solution
was added to the sol until the HNO3/Al ratio reached 0.5. Finally, the Pt
precursor was added
to the alumina sol, which was stirred at room temperature for 1 h, and then
sonicated for 30
min. The sol was then placed in the fume hood for 48 h to allow the gel to
form and the
solvent water to evaporate. The dry gel was further dried at 110 C for 12 h,
and then at 200 C
for 2 h. The final material consisted of yellow cubic particles.
Catalyst calcination
Several methods of calcination can be used. A portion of the dry gel was
calcined by a
one-step process. This dry gel was calcined at 550 C in one of three ways: (1)
in flowing
oxygen for 2 h in a U-tube flow reactor heated on the outside by an electric
furnace; (2) in
flowing air for 2 h in the same U-tube flow reactor or (3) in static air in a
muffle furnace for 2
h. In order to investigate the influence of heating rate, two ramping rates
were used-2 or
C/min. The other portion of the dry gel was calcined by a two-step process.
The gel was
first calcined at 550 C (2 or 10 C/min heating rate) in flowing helium for 0.5
h. After cooling
to 50 C, the flow was switched to pure oxygen. The temperature was then ramped
to 550 C at
2 or 10 C/min and held for 2 h.
For some of the calcinations, the exhaust gas composition was monitored during
the
temperature ramp using a Cirrus 200 Quadrupole Mass Spectrometer system (MKS)
to
determine which products were being produced during the calcination. The
catalysts have been
named according to the Pt precursor and the calcination treatment. For
instance, Al-1 refers to
a catalyst containing only alumina and calcined in oxygen at 550 C, while Py-2
refers to a
catalyst prepared with a Pt-pyridine precursor and calcined in two steps with
helium first and
then oxygen (see Tables 1 and 2).
CA 02592124 2007-06-19
Catalyst characterization
The N2 adsorption-desorption isotherms for the catalysts were measured on an
AUTOSORB-1C (Quantachrome) instrument. All samples were evacuated at 120 C
until the
outgas rate was below 15 mHg/min (or 2 Pa/min) prior to analysis. The
specific surface area
was calculated using the BET method. The total pore volume was determined at a
relative
pressure P1P = 0.99. Pore size distributions were calculated from the
desorption isotherms
using the Barrett, Joyner, and Halenda (BJH) method. The desorption leg of the
isotherm is
preferred for pore analysis because it is thermodynamically more stable than
the adsorption leg
due to the lower Gibb's free energy change.
H2 chemisorption measurements were carried out on the same AUTOSORB-1C
instrument. For these measurements, approximately 1.0 g of catalyst was placed
in a quartz U-
tube (i.d. = 10 mm), and reduced in a H2 flow of 15 ml/min at 300 C for 2 h.
After reduction,
the sample cell was evacuated at 300 C for 2 h, and then cooled to 40 C for
the H2
chemisorption measurement. The H2 monolayer uptake of the catalysts was
calculated by
extrapolating the H2 adsorption isotherm to zero pressure. The Pt particle
diameter (dpt) was
calculated using the formula, dp, =6V/S, where V is the volume of total
metallic Pt, and S is the
active Pt surface area, assuming the Pt2' ions were reduced completely and the
Pt particles
were spherical in shape. An adsorption stoichiometry of one hydrogen atom
adsorbed per
surface Pt atom (H/Pts = 1) was assumed. The percent Pt dispersion was
calculated by dividing
the number of exposed surface Pt atoms (as determined by H2 chemisorption) by
the total
amount of Pt in the catalyst.
Powder X-ray diffraction (XRD) spectra were recorded on a Multiflex X-ray
diffractometer (Rigaku) using CuKal radiation (A = 1.54056 A) at 40 kV tube
voltage and
40mA tube current with a scanning speed of either 0.2 or 2 /min. The XRD
patterns were
referenced to the powder diffraction files (ICDD-FDP database) for
identification. If possible,
the average crystallite diameter of metallic Pt was calculated using Scherer's
method, Dpt
=KV,8cos9, where the constant K was taken as 0.9 and,8 was the full width at
half maximum
(FWHM) of the Pt(3 1 1) peak at 20 = 81.3 .
Transmission electron microscopy (TEM) images were recorded on an H-7000
transmission electron microscope (Hitachi) at 75 kV. The samples were ground
to a fine
powder, and mixed with acetone to make a suspension. A drop of the suspension
was placed
on a lacey carbon nickel grid, which was subsequently dried at room
temperature before the
measurement.
CA 02592124 2007-06-19
11
Differential thermal and thermogravimetric analyses
Differential thermal and thermogravimetric analyses (DTAPTGA) were performed
on
three samples (A1-200, N-200, and Py-200) to examine the thermal and
gravimetric changes
that occur in those samples during calcination. A DSC/TGA Q600 instrument (TA
Instruments) was used for this analysis. The analysis conditions were selected
so as to mimic
the calcination procedure. Three different types of tests were performed as
follows: (1) all
samples (A1-200, N-200, and Py-200) were heated under air flow from room
temperature to
550 C at 2 C/min and held at 550 C for 30 min; (2) sample N-200 was heated
under air flow
from room temperature to 550 C at 10 C/min and held at 550 C for 30 min and
(3) sample N-
200 was heated under He flow from room temperature to 550 C at 10 C/min and
held at 550 C
for 30 min. Heat flow, mass loss, and differential temperatures were recorded
during the
analyses.
Reactivity testing
Four catalyst samples, N-4, Py-4, MA-1, and BA-1 (see Table 2) were tested for
reactivity in a fixed bed reactor using hydrogenation of toluene at
atmospheric pressure as a
model reaction. The reactor was a quartz tube with inner diameter of 7 mm.
Approximately
400 mg of catalyst was used for each run. All the catalysts particles were
sieved to the same
size range, 90-250 p,m. Reactions were conducted at temperatures between 60
and 270 C at
30 C intervals. A liquid hourly space velocity (LHSV) of 1 h-1 was used, with
a H2 to toluene
volumetric ratio of 1250. The catalysts were reduced in flowing H2 for 2 h at
300 C and then
the temperature was reduced to the reaction temperature. The reactor effluent
was analyzed
using a gas chromatograph (Agilent 6890) equipped with a GS-GasPro PLOT column
and a
flame ionization detector (FID). The reactor came to steady state after
approximately 30 min
on stream. The steady-state compositions were used to calculate activities.
After 120 min on
stream at one temperature, the temperature was increased by 30 C. Once at 270
C, the reactor
was cooled to 60 C and the testing and temperature cycle repeated. One
complete temperature
cycle between 60 and 270 C constituted one run. Three runs were done for each
of the four
catalysts.
Results and discussion
CA 02592124 2007-06-19
12
Physical properties of catalysts
rahle I
Properttes of eol-Wl atumina and Pt-containing sampks before and after
caktnatiaa
Sample Pt prrcurstx Trc,ument Snrface wYa (m2lg) Pote volume (mllg)
AI 200 Al2(h only Ikying in au at 200-C t2h) 94 0.012
Al-1 A12Oa only Oi 550 - C(2 h) 281 035
Al-2 AI20j only F(u 550'C (0,5h), 02 550 C 12h) 294 R i0
N-200 Pt(NH3)4Cl2 Urying in air at 200 C(2 h) 0 -
N-t Pt(NH0;)aCh O, 550 C (2 h) 269 0.34
N-2 PttNH4)4Cl2 He 550 C(0,5h), 02 550C (2h) 262 033
Py-200 Pt{tCsHsM+Ci2 Dry'ing in air at 2ri) C {3li) 0 -
Py-1 !rt(CsHst=1)4('12 04 550'C(2h) 272 0a2
Py ? Pt(CSH:N);C12 He 550-C (0.5h). O2 550'C t2h) 254 0 72
C.onturxciai y-Ai.O, - - 208 0 11
koru-: &rrcv is 15% in the sYtrface area arxl the }wm valturLL nttarummc.~nis.
Table 1 contains the surface areas and pore volumes for catalyst samples
prepared using
different Pt precursors and treatment procedures. The samples Al-200, N-200,
and Py-200
exhibit very low surface areas (<10m2/g), which indicates that aluminium
hydroxide and nitrate
were not decomposed to refractory oxide A1203 after being dried in air at 200
C for 2 h. The
remaining catalysts had surface areas between 254 and 281m2/g, and pore
volumes between
0.30 and 0.35 ml/g. The pore size distributions were similar for all of the
calcined catalysts,
with mean pore diameters of 3.8 nm. These results indicate that the precursor
and calcination
procedure had little effect on the bulk physical structure of the catalysts.
For comparison, a commercial yA12O3 (Alfa Aesar) was characterized. The
surface
area of the commercial alumina is slightly lower (208m2/g) than the surface
areas of the
prepared catalysts, while the pore volume is similar (Table 1). Figure 2
compares the pore size
distributions and adsorption isotherms of the commercial alumina and sample Al-
1, which is
representative of all the calcined catalysts. The in-house prepared alumina is
similar to the
commercial alumina in terms of its pore structure.
Based on the elemental analysis performed by ICP-MS (Galbraith Labs), the
platinum
content of the catalysts are as follows: 1.58% for N-batch catalysts, 1.43%
for Py-batch
samples, 1.54% for MA-1, and 1.19% for BA-1. These Pt metal contents are shown
in Table 2,
with the Pt dispersions as determined by H2 chemisorption. The dispersions
range between 11
and 106%. The metal surface areas used to calculate the dispersions are also
shown in Table 2.
Dispersions above 100% may be attributed to errors in the Pt metal content,
errors in the
adsorbed H2 determination, or hydrogen spillover from the Pt. The Autosorb-1C
was
calibrated with the Quantachrome standard reference (1%PVA1203), and had an
error of 0.5%.
CA 02592124 2007-06-19
13
TaMe 2
pt dispersians of R/A1301, sampFes after rginus calcinadoo procedures
Sample R precurscu Calcinaticm pracedure Heatitv, raw Pt metal Actite meal
sarface Dispersion Partide
(C7mik~ wnteN (4) area (m2/g) ("k) dianrctcr (nm)
N-1 Pt(NHz);C'Iõ 0. 550 =C (2h) 10 1.58 1.4 35 3.2
N-2 Pt(NH3kC12. 1-Ic 5.50'C (0.5 h). Ch 550 C(2 h) 10 1.58 23 59 1.9
N-3 Pt(NH34C1+. 0Z5=.S0 C(2h) 2 1.58 4.i 105 1.1
N4 Pt(NHzkCI+_ He550^C(0.5h).C?1550 C(2h) 2 i.58 4.l 104 i.i
N-5 PHNH;}yCt, Static air in muffle furoace, 550'C (2h) 2 1 58 3,3 87 1.3
N-fi Pt(NH;e l{CI3 Flowing uir, 550 C(2 h) 2 1.58 4.1 106 1.1
Py-1 Pt(C.HsN)4Cl2 (), 550 C (2 h) 10 1.43 2.2 6i3 1.8
p} -2 Pt(CsHcN)yCi2 He .5.i0'C (Ø3 h), (.)a 550 C (2 h) 10 1.43 2.0 56 2.0
Py-3 4't(C,HcN)aClz Oi 5.50'C (2h) 2 1_43 3.1 88 1.3
Py-4 Pt(C.1IiN)aCl7 lie 5.50 C (0.5 h). (?z 550 =C (2 h) 2 1.43 2.7 77 1.3
Py-5 R(C.HSN){C1Z Static air in rrwtfte fumace, 550 C (.2 h) 2 1.43 2.0 58
2.0
Py-6 pt(CsH!;N){Cii Flowing air, 550 C(2 h) 2 1.43 3.1 89 1.3
MA-1 PE(Cll; ~iF12)aC}1 Slraic air in nwtlie furnxce, 550 C(2 h) 2 1.54 ~ 2 59
1.9
BA-1 Pt(C4}{9NHj)rC.l2 Static air in nmf0e fumace, 550 C (2 h) 2 1.19 0.3d 11
9.9
Notr. (1) H2 cheme: tvption was performed after reducing ihe cntaiyxt on lire
at 30(I C fcx 2 h in fiowing Hc. (2,) F:rrar is f5 % in the disperston
measurements.
In general, a lower heating rate (2 C/min versus 10 C/min) results in an
increased
dispersion. In addition, the precursor may also significantly influence the
dispersion. In this
example, all comparisons are made between the same batches of catalyst.
Despite batch to
batch variability, the overall trends are consistent.
The diameter of the Pt particles can be estimated from the hydrogen uptake and
is given
in Table 2. For all catalysts, except BA-1, the average particle sizes of Pt
are less than 3.2 nm.
X-ray diffraction can also be used to estimate the particle size provided that
the particles are
larger than approximately 4-5 nm. Figure 3 shows the XRD spectra of the
prepared catalysts
and a commercial yAl2O3. The XRD pattern of aluminium oxide can be quite
different
depending on the preparation method and crystalline phase according to the
ICDD-FDP
database. The spectra in Figure 3 indicate that the commercial yA12O3 sample
and the in-house
prepared samples have similar structures although the latter alumina has
smaller crystalline size
as indicated by the broader peaks at 29 = 67.3 . The XRD patterns of the
samples are all quite
similar despite different platinum particle sizes, calcination procedures, and
platinum
precursors. This result indicates that the alumina crystalline structure is
not sensitive to these
parameters, which is consistent with the BET measurements with respect to the
constant pore
structure and surface area.
The peaks for Pt overlap, at least partially, with alumina at most diffraction
angles,
except for the Pt(3 1 1) peak at 20 = 81.3 . In order to obtain accurate Pt
particle size
information, slow scans (0.2 /min) were performed in the range of 78-90 20.
The results are
shown in Figure 4 and are consistent with the average particle size of Pt
calculated from the
chemisorption results. Pt is undetectable for catalysts N-5 and N-6, while Py-
5 and MA-1 both
have a peak at 20 = 81.3 that is too small for estimation of particle size.
The calculated
CA 02592124 2007-06-19
14
average particle size of Pt in catalyst BA-1 is 23 nm, which is larger than
that estimated by
hydrogen chemisorption (10 nm).
Effect of platinum precursor
The platinum precursors - Pt(NH3)4C12, Pt(CH3NH2)4C12, Pt(C5H5N)4C12, and
Pt(C4H9NH2)4C12 - were analyzed with CHN analysis to obtain the C, H, and N
ratios
contained in the catalysts. Based on the CHN analysis results, it was
confirmed that the desired
precursors were obtained. Carbon NMR of the Pt(C4H9NH2)4C12 precursor showed a
CH3
chemical shift at 13.7 ppm and CH2 at 19.4, 32.7, and 46.1 ppm. Proton NMR of
the same
Pt(C4H9NH2)4C12 confirmed the presence of the CH3 and CH2 peaks in addition to
the NH2
peak at ca. 5.6 ppm chemical shift. These results in addition to the CHN
analysis confirmed
the presence of a (C4H9NH2) group in the precursor. Proton NMR of the
Pt(C5H5N)4C12
precursor revealed an aromatic structure with the first CH at 8.9 ppm, the
second at 7.5 ppm,
and the third at 7.9 ppm. Combined with results of CHN analysis, these results
confirmed the
presence of a (C5H5N) group in the precursor.
The selection of platinum precursor was based on the hypothesis that the
larger the
precursor the better the resulting Pt dispersion. During calcination, platinum
(oxide) atoms
may agglomerate into clusters. Assuming that the agglomeration occurs as a
series of binary
interactions, the rate of agglomeration is related to the distance between any
two platinum
atoms. Thus, the ligand of the Pt(II) precursor would act as space holder for
the platinum
atoms. As shown in Table 2, the precursors are all nitrogen-containing
molecules and soluble
in water. The molecular diameters of the precursors were estimated by bond
lengths and are
approximately 5, 7, 12, and 13 A for Pt(NH3)4C12, Pt(CH3NH2)4C12,
Pt(C5H5N)4C12i and
Pt(C4H9NH2)4C12, respectively.
Figure 5 illustrates that the results were exactly the opposite of what was
expected.
That is, the smaller the precursor, the better the dispersion. The results
shown in Figure 5 are
those taken from Table 2 for samples N-5, Py-5, MA-1, and BA-1. The Pt
dispersion is
strongly dependent on the precursor and increases from 11% for BA-1 (largest
precursor) to
87% for N-5 (smallest precursor). These samples were calcined in a muffle
furnace in static
air. Although calcining in static air results in lower dispersions than
flowing air, the trend of
increasing dispersion with decreasing precursor size did not change with a
change in the
calcination procedure.
CA 02592124 2007-06-19
The extremely low dispersion of BA-1, obtained using Pt(C4H9NH2)4C12 as
precursor,
may be caused partly by the relatively poor solubility of the precursor in
water. During the
precursor preparation, the water solution (15 ml water, containing 0.1 g of
Pt) had to be heated
to 60 C in order to dissolve this precursor. The precursor did not precipitate
in the sol or wet
gel because of the presence of sufficient water (ca. 50 ml). The precursor,
Pt(C4H9NH2)4C12,
probably precipitated, however, during water removal. If the precipitate
separated from the
alumina gel during subsequent heat treatments, large Pt particles may be
formed. TEM analysis
of the reduced BA-1 catalyst (Figure 6) indicated that large Pt particles have
formed. The
darker particles in Figure 6(a) are on the order of 200 nm in size and are
likely Pt particles or
agglomerates. The lighter particles are alumina. In Figure 6(b), Pt particles
are visible at the
edges of the alumina particles with sizes of 10-50 nm. A homogenous Pt
distribution was not
obtained from this Pt precursor as the majority of the alumina particles did
not contain any
visible Pt particles. On all other catalysts, no Pt particles were visible,
consistent with the
hydrogen chemisorption and XRD results.
The chemical nature of the Pt precursors and their interaction with the
support
precursor (i.e., silane as precursor of silica) may also be a factor in the
resulting metal
dispersion. The effect of Pt precursor on the Pt dispersion and particle size
has been studied
over sol-gel processed Pt/Si02 by Lembacher. The studied precursors included
Pt(AcAc)Z,
PtC12, H2PtC16, Na2PtC16, Pt(CN)2, and Pt(NH3)4(NO3)2. PtC12 led to the
highest Pt dispersion
when a nitrogen-containing silane, H2NCH2CH2NH(CH2)3Si(OEt)3 (AEAPTS), was
used as
silica precursor. This result may stem from the strong interaction of Pt(II)
and the silica
precursor, while Pt(IV) cannot form a complex with nitrogen-containing
molecules with a
silica precursor. In this example, the Pt precursors will not react with the
aluminium precursors
and so anchoring of some of the Pt precursors cannot explain the different
dispersions that
were obtained.
The effect of calcination procedure on Pt dispersion
A heating rate of 2 C/min under all calcination conditions resulted in higher
Pt
dispersions than using a heating rate of 10 C/min. Figure 7 presents the
results for the catalysts
derived from Pt(NH3)4C12. Samples N-1 (35% dispersion) and N-3 (105%
dispersion) were
both calcined in 02 at 550 C for 2 h, while samples N-2 (59% dispersion) and N-
4 (104%
dispersion) were calcined in a two-step process involving helium and then
oxygen. Clearly a
slower heating rate results in higher dispersions. The same trend is observed
for the catalysts
CA 02592124 2007-06-19
16
prepared with Pt(C5H5N)4C12 as the precursor (Py-catalysts). For example, the
Pt dispersion for
Py-3, heated at 2 C/min, was 88%, compared to a Pt dispersion of 63% for Py-1,
which was
heated at 10 C/min.
The atmosphere during calcination also affected the final Pt dispersion, but
the
magnitude of the effect depended on the heating rate and Pt precursor. For
example, samples
N-1 and N-2, were both prepared from the same precursor and heated at the same
rate
(10 C/min) during calcination, but had significantly different Pt dispersions
(35% versus 59%).
In this case, a higher dispersion was obtained by heating in He before heating
in 02.
Conversely, for samples N-3 and N-4 (Table 2), heated at 2 C/min, the Pt
dispersion was the
same regardless of the calcination procedure. When Pt(C5H5N)4ClZ was used as
the Pt
precursor, a comparison of Py-1 with Py-2, and Py-3 with Py-4 (Table 2),
indicated that He
pretreatment before calcination in 02 resulted in a lower Pt dispersion
compared to no He
pretreatment. Lembacher has shown that pretreatment in an inert atmosphere is
beneficial for
Pt dispersion on silica supports. For Pt/SiO2 the average particle size of Pt
decreased from 22
nm to less than 3 nm by pretreating the catalyst with argon before exposure to
air at 400 C.
During the one-step calcination, the Pt particles may have sintered due to
local overheating
caused by rapid oxidation of organic groups in the silane precursor.
Heating in a muffle furnace is a much simpler method than heating a catalyst
in a flow
apparatus. In these experiments, however, treatment in static air (i.e., the
muffle furnace)
resulted in much lower Pt dispersions than treatment in a U-tube on a flow
apparatus. For
example, as shown in Figure 8 and Table 2, sample N-5 was calcined in the
muffle furnace and
had a Pt dispersion of 87% compared to a dispersion of 106% for sample N-6,
calcined in
flowing air. Similar results were obtained for samples Py-5 and Py-6. Likely
the water
produced during the calcination was not removed quickly enough without flowing
gases. The
presence of water may have promoted the sintering of the Pt particles.
Calcination studies performed by mass spectrometry
Mass spectrometry (MS) was used to better understand what species are being
formed
during the calcination. This analysis was used in conjunction with XRD
analysis. The alumina
support was analyzed first, and then several samples containing Pt were
analyzed. The alumina
gel is soluble in water after drying at 200 C for 2 h, indicating the dry gel
has the same or
similar composition with the sol or wet gel except for solvent (H20) content.
In order to
investigate the structure of the dry gel (A1-200 in Table 2), the XRD patterns
were recorded
CA 02592124 2007-06-19
17
before and after calcinations (spectra not shown). The XRD pattern of the dry
gel was
different from that of the sol-gel A1203 (Al-1 and Al-2). Referencing the ICDD
database, the
dry gel had a similar structure to Al(OH)3, indicating the dry gel did not
decompose after
drying for 2 h at 200 C in air. The composition of the dry gel can be
represented by
A1(N03)b(OH)3-6, or more precisely Al(NO3)sOx(OH)3.6-2x because of
condensation. Here, d
0.5 since the molar ratio of HNO3/Al is 0.5 during peptization. After
calcination, the mass loss
of the dry gel is 45 4%. From the mass loss, the value of x can be
calculated. The calcined
alumina samples (Al-1 and Al-2) had structures similar to a commercial yA12O3
as described
previously.
Figure 9 shows the evolution of CO2 (mass 44) and NO2 (mass 46) during the
calcination of the dry gel in oxygen or in helium. Masses 18 (H20) and 30 were
also
monitored during the experiment. During calcination water was produced from
the
decomposition of Al(OH)3. The water, however, condensed before the inlet to
the mass
spectrometer and, thus, the water signal is not reliable and not shown. The
signals from masses
46 and 30 were identical in shape and, thus, mass 30 is attributed to the
cracking pattern of
NO2. The plots in Figure 9 are nearly identical whether the atmosphere is
helium or oxygen.
In both cases, COZ is detected at temperatures between 240 and 470 C, while
NO2 is detected
at temperatures above 250 C. The CO2 originated from the oxidation of an
organic compound
in the gel produced during the hydrolysis of ATB. The organic compound is
likely 2-butanol;
however, 2-butanol was not detected in the emissions. The organics were
oxidized by oxygen
or the produced NOx in the absence of oxygen (i.e., helium atmosphere).
Following the
treatment in flowing He, the same sample was monitored during treatment in
flowing 02 and
no NO2 or CO2 was detected. These results suggest that complete decomposition
occurred
during the pre-treatment in helium and that treatment with oxygen was
unnecessary.
The NO2, NO, and H2O emissions during the calcination of Pt-containing dry
gels (N-
200 and Py-200) were similar to that for blank alumina gel (Al-200) in either
oxygen or
helium. The CO2 evolution during the calcination of Al-200, N-200, and Py-200
is shown in
Figure 10. The plots are similar for a helium or oxygen atmosphere. The
quantity of CO2
evolved, however, depended on the presence of the precursor. Emissions of CO2
for the gel
derived from Pt(NH3)4C12 (N-200) and for a blank alumina gel (Al-200) were
similar. In
comparison, the gel derived from Pt(C5H5N)4C12 (Py-200) produced approximately
20 times
more CO2 than either N-200 or Al-200, consistent with the carbon content of
the precursor.
CA 02592124 2007-06-19
18
The additional emitted CO2 came from the oxidation of the pyridine contained
in the dry gel
for Py-200.
Chemical reactions during calcination
The composition of the alumina dry gel before calcination can be represented
by
Al(NO3)aOx(OH)3_a-a, 8z 0.5. The value of x can then be calculated based on
mass loss during
calcination. The final solid product after calcination is A1203, and the mass
losses are 4 5 4%
during calcination. Thus, the reaction during calcination can be described as
follows:
2A1(NOo)o.5Ox(OH)2.s-2z
= A1Z03 +N02 +(2.5-2x)H20 + 0.2502 (1)
The calculated value of x is between 0 and -0.8. On average, if the mass loss
is 45%, x
0.4. That is, lOOg of the dry gel would produce 25g of NO2, 4.3g of 02, and
16g of H20.
Water condensation in the lines after the calcination vessel was evident. The
condensed water
had a yellow color and was strongly acidic (pH < 1), indicating that
HNO3likely has been
formed from the following reactions in the exit stream:
4NO2 + 02 +2H20 = 4HN03 (2)
3NO2 + H2O = NO + 2HN03 (3)
Water vapor has been reported to enhance Pt sintering in Pt1AlZO3 during
calcination
and reduction. The presence of excess water vapor may have been a factor in
the lower
dispersions obtained when the calcination was performed in static air (muffle
furnace) rather
than flowing air, in which the water would have been removed and not
accumulated on the
surface of the catalyst. Similarly, the lower heating rate (2 C/min) may be
beneficial because
of the slower decomposition, and hence water evolution, of the alumina gel.
The gas flow rate
and amount of catalyst was kept constant throughout the calcination tests.
The effect of the Pt precursor on dispersion may be related to the composition
of the
precursor. In this example, the Pt precursors with organic ligands yielded
lower Pt dispersions
than the precursor containing ammonia. The dry gel containing a pyridine
Pt(II) ligand (Py-
CA 02592124 2007-06-19
19
200) produces approximately 20 times the amount of CO2 during calcination than
the dry gel
containing an ammonia Pt(II) ligand (N-200). The localized heating from the
oxidation of the
organic ligands may have been sufficient to result in sintering of the Pt
particles. Lembacher
suggested that localized heating caused by rapid oxidation of the support
precursor, silane,
resulted in sintering in Pt/Si02 catalysts. In contrast to the work on
Pt/SiO2, with Pt/A1203 He
treatments before calcination in oxygen only improved the dispersion for the
ammonia
precursor with a heating rate of 10 C/min. The mass spectrometer results
indicated that the
pyridine and ammonia precursors were completely oxidized in He and a second
treatment in 02
was not required. The decomposition of NO3-groups produced significant amounts
of 02 and
NOx which can act as oxidants. Likely these species were sufficient to
completely decompose
the precursors.
Results of differential thermal and thermogravimetric analyses
The thermogravimetric and differential thermal analysis of samples Al-200, N-
200, and
Py-200 are shown in Figures 11 and 12. In agreement with the mass spectrometry
results, the
mass loss in an air atmosphere corresponded to decomposition within the
temperature range of
200-450 C (Figure 11). The differential thermal analysis is shown for two
heating rates (2 and
C/min) as well as two atmospheres (helium and air) for sample N-200. More heat
was
evolved during the calcination of Py-200 (comparable to Py-3 with a dispersion
of 88%) than
for the calcination of N-200 (comparable to N-3 with a dispersion of 105%).
The largest
change in heat flow occurred on sample N-200 heated in air at 10 C/min, which
is consistent
with the lower dispersions obtained for samples heated at 10 C/min than 2
C/min (Table 2).
These results support the theory that localized heating during calcination may
contribute to
sintering of the Pt resulting in lower dispersions.
Reactivity tests-toluene hydrogenation
Figure 13 shows a comparison of the production of methylcyclohexane (MCH) over
catalysts produced from the different precursors (N-4, Py-4, MA-1, and BA-1)
at 240 C and
atmospheric pressure. Three runs were performed for each catalyst and in each
case, the only
product was methylcyclohexane. Catalyst BA-1 had the lowest Pt dispersion
(11%) and the
lowest production of MCH. In contrast, catalyst N-4 had the highest Pt
dispersion (104%) and
the highest production rate. Catalysts Py-4 and MA-1 had intermediate Pt
dispersions (77 and
CA 02592124 2007-06-19
59%) and intermediate activities. The activity of N-4 stayed within 2.5% of
the mean activity
of this catalyst. The other catalysts, however, had larger decreases in
activity over time.
Conclusions
Pt dispersions ranging between 11 and 106% were obtained for 1.5 wt% Pt/A12O3
catalysts prepared by sol-gel synthesis. The Pt dispersion of the catalyst was
found to be
strongly dependent on the platinum precursor, and a larger precursor molecule
did not result in
better dispersion. Specifically, in terms of highest Pt dispersion,
Pt(NH3)4C12 was the best
precursor followed by Pt(CH3NH2)4C12, Pt(CSH5N)4C12, and finally
Pt(C4H9NH2)aC12. The
latter precursor resulted in a very poor dispersion, likely because of its
poor solubility. The
dispersion also depended on the calcination procedure. The use of flowing gas
instead of static
air, and a lower heating rate (2 C/min rather than 10 C/min) resulted in
higher Pt dispersions.
The presence of accumulated water (from treatment in static air or from too
high a heating rate)
and/or localized heating effects (dependent on the decomposition of the
precursor) resulted in
sintering of the Pt and lower dispersions. Pretreatment in helium before
oxygen did not
improve the dispersion. Toluene hydrogenation experiments indicated that
higher activities and
selectivities can be achieved with catalysts containing more highly dispersed
Pt. Thus, it is
desirable to optimize catalyst preparation to achieve high dispersion.
Example 2
This example demonstrates the use of CVD to control the pore openings of
Ni/A12O3
catalysts for the selective chemisorption of H2 (2.9 A) and exclusion of
larger molecules (N2,
3.6 A; CO, 3.8 A). A fluidized bed reactor was used with steam injection
through an annulus
of the reactor, and silicon alkoxide introduction through the bottom of the
reactor was carried
by an inert gas. The effect of deposition time on the subsequent adsorption of
H2, CO, and N2
was investigated. The catalysts were characterized before and after
modification with N2
physisorption, H2 and CO chemisorption, temperature-programmed desorption of
NH3, X-ray
diffraction, and inductively coupled plasma. In addition, the catalysts have
been tested with a
model reaction of n-octane hydrocracking to demonstrate the influence of the
coating on the
access to the active sites as well as on the acidity of the catalyst. The
results indicate that CVD
is an appropriate method for reducing the size of the pore openings in
Ni/A1203 catalysts. The
modified catalysts will be useful in a catalytic reaction in which H2 is
competing with larger
molecules for adsorption sites.
CA 02592124 2007-06-19
21
Experimental Method
Apparatus. The CVD apparatus used in this work is shown in Figure 14. The
fluidized bed reactor consisted of a quartz tube mounted vertically inside an
electric furnace.
The reactor tube had an inside diameter of 1 cm and overall length of about 41
cm. One
feature of the reactor is the attachment of an annular tube made of '/g-in.
stainless steel tubing
for steam injection into the reaction zone inside the bed (Figure 14). A 1/32-
in. thermocouple is
also extended through the annulus to measure the temperature of the bed.
Quartz frits with
openings of 15-40 micrometers were used as the distributor plate. The reactor
operated at
atmospheric pressure. A piston pump (Ailtech 426 HPLC pump) pumped water
through an
evaporator into the fluidized bed with flowing N2 (20 sccm) as the carrier.
The TMOS was
pumped by a syringe infusion pump (Cole Parmer), was evaporated, and was
carried to the
reactor by N2 flowing at 60 sccm. The flow of N2 was controlled by a mass flow
controller
(Type 1179A by MKS Instruments). FieldPoint and LabView (National Instruments)
were used
for data acquisition and readout.
Preparation of Ni/A1203. A 25-g batch of Ni/A12O3 was prepared by the wetness
impregnation method. The y-A1203 (60 mesh, Alfa Aesar) was impregnated with an
aqueous
solution of Ni(N03)2-6H20. The mixture was dried at room temperature for about
16 h in a
fume hood and then was transferred to a muffle furnace and was treated at 80
C for 2 h
followed by drying at 110 C for 10 h. The impregnated A1203 was then calcined
in the muffle
furnace at 550 C for 8 h. On the basis of temperature-programmed reduction, a
reduction
temperature of 550 C was chosen. Thus, the catalysts were reduced in flowing
H2 by heating
to 550 C at 10 C/min and were held at 550 C for 4 h. The resulting Ni/A12O3
catalysts had
Ni loadings of 17% (Galbraith Laboratories Inc.) and a surface area of 129
m2/g ((2 m2/g). The
surface area of the purchased A1203 was 208 m2/g as measured by N2
physisorption using an
Autosorb-1C adsorption instrument (Quantachrome Instruments).
Silica Deposition. The Si02 deposition was performed using the CVD apparatus
described above (Figure 14). One gram of Ni/A1203 was placed in the fluidized
bed reactor
and was fluidized with N2 (60 sccm). TMOS (1.75 mol %) was vaporized and
injected into the
bottom of the reactor while 14 mol % H2O was evaporated with N2 (20 sccm) as
carrier gas and
was injected into the annulus of the reactor. The purpose of steam injection
was to suppress
carbon formation and at the same time hydrolyze the TMOS.
CA 02592124 2007-06-19
22
Preliminary experiments were done using the blank A1203 support to develop the
CVD
procedure. Si02 deposition experiments were done with temperatures between 150
C and
500 C. After Si02 deposition, the samples were calcined in flowing air at 500
C for 2 h to
remove any traces of carbon and organic matter remaining in the catalyst
caused by side
reactions. The Ni catalysts were coated for times of 0.5, 1, 1.5, 2, 2.5, or
3h at 350 C and were
identified according to this deposition time. That is, Ni-0 refers to an
uncoated catalyst while
Ni-2.5 refers to a catalyst treated in the CVD apparatus for 2.5h.
Catalyst Characterization. The characterization techniques used were N2
physisorption, H2 and CO chemisorption, X-ray diffraction (XRD), and NH3
temperature-
programmed desorption (TPD). The coated samples were analyzed for H2
chemisorption
before and after coating. As such, the coated samples have been reduced twice
while the
uncoated samples have only been reduced once. N2 physisorption was performed
using an
Autosorb-1C adsorption apparatus (Quantachrome Instruments) to determine
surface area, pore
volume, and pore size distribution. The surface area was calculated using the
Brunauer,
Emmett, and Teller (BET) method, while the pore volume and pore size
distribution were
calculated by the Barret-Joyner-Halenda (BJH) method using the desorption leg
of the
isotherm. The desorption leg of the isotherm is preferred for pore analysis
because it is
thermodynamically more stable than the adsorption leg because of the lower
Gibb's free-
energy change. The error in the surface area measurements is 2% on the basis
of repeat
analysis of the samples.
H2 and CO chemisorption were performed on a ChemBET 3000 (Quantachrome
Instruments) to determine the H2 and CO uptakes before and after Si02 coating
of the
Ni/A12O3. All catalysts (with and without Si02 deposition) were pretreated by
reduction in
flowing H2 at 550 C for 4 h before the chemisorption measurements. To test
whether CO can
penetrate the SiO2 coating, each of the samples that were coated for 2 and 2.5
h was exposed to
60 mUmin of pure CO for 30 min at 40 C. Following the CO exposure, each of the
samples
was purged with flowing N2 for 1 h to remove any physically adsorbed CO. Each
of the
samples was tested for H2 uptake following the exposure to CO. An uncoated
Ni/Al203 catalyst
was also tested for H2 uptake after CO exposure to obtain a baseline for
comparison.
Powder X-ray diffraction (XRD) spectra were recorded on a Rigaku Multiflex X-
ray
diffractometer using Cu1Ka1 radiation (y) 1.54056 A at 40 kV tube voltage and
40 mA tube
current with a scanning speed of 2 lmin. The XRD patterns were referenced to
the powder
CA 02592124 2007-06-19
23
diffraction files (ICDD-FDP database) for identification. XRD was performed to
monitor
changes in the oxidation state of the Ni phase during the coating procedure.
NH3 TPD was performed using the ChemBET 3000 instrument with 10% NH3 diluted
in He, before and after Si02 deposition, to determine the effect of the
deposition on the acidity
of the A1203 support. The NH3 was adsorbed at 40 C and desorption was
performed in the
temperature range of 40-550 C with a heating rate of 10 C/min.
Silicon elemental analysis of the coated Ni/A12O3 catalysts was performed
using
inductively coupled plasma-mass spectroscopy (ICP-MS, Galbraith Laboratories).
The ICP-
MS technique used to perform the Si elemental analysis has a relative error
margin of 10%.
Carbon elemental analysis was performed using a Perkin-Elmer 2400 CHN Analyzer
to
determine the carbon content before and after calcination of coated catalysts.
n-Octane Hydrocracking Procedure. The hydrocracking of n-octane was carried
out
in a quartz fixed bed reactor with 100 mg of catalyst at 400 C, a weight-
hourly space velocity
(WHSV) of 2.0 h-1, and H2/n-octane molar ratio of 20 under atmospheric
pressure. Before
reaction, the catalyst was reduced at 550 C under flowing H2 for 4 h. The
reaction products
were analyzed online using a gas chromatograph (Agilent 6890 GC) with a 60-m
long, 0.32-
mm i.d. GS-GasPro PLOT column and a flame ionization detector (FID). A mass
spectrometry
(Cirrus by MKS Instruments) was also used to analyze the product stream.
Results
Development of CVD Procedure. The Si02 deposition procedure was developed by
coating plain commercial y-A1203 with Si02 for different deposition times and
temperatures.
Figure 15 illustrates the change in measured surface area with deposition
temperatures 150 C,
200 C, 250 C, 300 C, 350 C, 400 C, and 500 C at a constant deposition time of
1 h. The
surface area is calculated directly from the nitrogen uptake and, thus,
representative of the
accessibility of the pores to nitrogen. The surface area decreases as the
deposition temperature
increases.
The measured surface areas in Figure 15 steadily decreased until a temperature
of
400 C at which point the surface area increased from 80 m2/g to 100 m2/g. With
a further
increase in temperature to 500 C, the surface area decreased to 83 m2/g. This
increase in
nitrogen adsorption at 400 C may be due to surface coverage by methoxy species
and other
decomposition products, which are removed from the surfaces and pores upon
calcination. At
deposition temperatures of 400 C and above, significant carbon formation was
visible on the
CA 02592124 2007-06-19
24
A1203 after the deposition. That is, the white A1203 powder turned black in
color. The carbon
was removed by calcination in flowing air at 500 C (the powder turned white or
slightly beige
in color). At deposition temperatures of 350 C and below, there was no visible
carbon
formation. Because of increased carbon formation at higher temperatures, a
deposition
temperature of 350 C was used for the silica deposition on the Ni/A1203
catalysts.
Nitrogen Physisorption on Ni/A1203. The Ni1A1203 catalysts were reduced
immediately before N2 physisorption measurements. Figure 16 shows the change
in measured
surface area as a function of Si02 deposition time for an A1203 sample and a
Ni/A1203 catalyst.
The nitrogen uptake decreased with increasing deposition time. The rate of
decrease in surface
area is different for the Ni/A12O3 than for the blank A1203 support. After 1.5
h of deposition,
the A1203 surface area is -3 m2/g while that of Ni/A12O3 is -35 mz/g,
indicating that the
deposition of Si02 on the A1203 is more rapid than the deposition on Ni/A1203.
The difference
in behavior is ascribed to the stronger affinity of Si02 for the alumina
surface. In the case of
Ni/A1203, some of the surface has likely been covered by Ni deposited on the
exterior surface
of the alumina particles.
The pore volumes of Ni/A1203 as a function of deposition time at 350 C are
given in
Table 3. Consistent with the surface area measurements, the pore volume
decreased as the
deposition time increased. After 2.5 h of deposition, the pore volume had
decreased to
essentially zero compared to a value of 0.193 cm3/g before deposition. Figure
17 shows that the
pore size distribution changes as the amount of Si02 deposition increases;
that is, the pores
decrease in diameter as the amount of Si02 deposition increases. The reduction
in pore size
0
may be a result of Si02 deposition within the pores since the original
Ni/A1203 pores (38 A
modal pore diameter) are large enough for the silicon alkoxide molecule to
penetrate the
0
catalyst (TMOS has a kinetic diameter of 8.9 A).
CA 02592124 2007-06-19
Table 3. Pore Volumes of Ni/A1203 Coated with Si02 for Various
Times at 350 Ca
Sample ID Deposition Time (h) Average pore volume cm /g
Ni-0 0 0.193
Ni-1 1.0 0.107
Ni-1.5 1.5 0.022
Ni-2 2.0 0.005
Ni-2.5 2.5 0.0007
Ni-3 3.0 0.0005
aMeasurements were performed by N2 physisorption.
Amount of Deposition and Carbon Formation. Figure 18 shows the amount of Si02,
as determined by ICP-MS, deposited on Ni/A12O3 as a function of deposition
time. After 1 h of
deposition, the Si02 fraction was 16%. The amount of silica deposited
increased to 30% after
1.5 h and then the amount deposited remained constant up to 2.5 h of
deposition. The surface
may have been saturated after 1.5 h with physisorbed species that hindered the
growth of Si-O-
Si bonds. To verify if other species were deposited on the surface, the carbon
content of a
Ni/A12O3 catalyst was determined by carbon, hydrogen, and nitrogen (CHN)
analysis after
coating the catalyst with Si02 for 2 h at 400 C. The Ni/A1203 catalyst was
gray in color after
the deposition, and the CHN analysis revealed a carbon content of 0.7%. This
catalyst was
then calcined and the carbon content was reduced to 0.2%. N2 physisorption was
also
performed on the same sample before and after calcination. The surface area of
the sample
before calcination was 50 m2/g compared to 4 m2/g after calcination.
H2 and CO Chemisorption. Figure 19 shows H2 and CO uptakes on Ni/A1203 after
Si02 deposition for various times. The H2 uptake actually increased after 1 h
of deposition
from 398 pUg to 493 ,uUg. This increase is probably due to a further reduction
of NiA12O4
within the structure during the second reduction after the coating. After 2.5
h of deposition, the
average H2 uptake was 430,uUg. In contrast, the CO uptake decreased from 405
,uUg before
coating to 5.8 pLlg, after 2.5 h of deposition, indicating that the deposited
silica had reduced
the pore openings and that the technique was successful.
To further test the size-exclusion properties of the coated Ni/A12O3
catalysts, three
catalysts were exposed to pure flowing CO for 30 min. The H2 uptakes, before
and after this
exposure, are listed in Table 4. The uncoated catalyst (Ni-0) was severely
poisoned by
CA 02592124 2007-06-19
26
exposure to CO with the H2 uptake decreasing from 398 pUg before exposure to
96 L/g after
exposure (76% change). The second and third samples, coated for 2 and 2.5 h,
respectively,
were less affected by the exposure to CO with decreases in H2 uptakes of 68%
and 28%,
respectively. These results indicate that the pores reduced the accessibility
for CO.
Table 4. H2 Uptake on Ni/A12O3 after Exposure of Catalysts to 100% CO flow for
30 Min.
Ni/A1203 Was Coated with Si02 at 350 C
Sample ID Deposition time H2 uptake ( L1g) Percent change
(h) Before exposure After exposure
Ni-0 0 398 96 -76
Ni-2 2 441 139 -68
Ni-2.5 2.5 430 308 -28
X-ray Diffraction. The XRD spectra for Ni/A1203 at various stages in the
deposition
process are shown in Figure 20. After reduction (Figure 20a), the spectrum had
peaks at 44.5,
51.8, and 76.4 20 corresponding to Ni and peaks at 37.3 and 67.3 20
corresponding to A1203
(matched to ICDD-FDP database). After deposition (Figure 20b), most of the Ni
has been
oxidized, as evidenced by peaks at 37.2, 43.3, and 62.9 28 corresponding to
NiO. The peaks
around 37 20 overlap; 37.2 20 is associated with NiO, 37.0 28 is associated
with NiA12O4,
and 37.3 20 is associated with A12O3. After deposition and reduction (Figure
20c), the XRD
spectra is very similar to the spectra of the originally reduced Ni/A12O3
catalyst (Figure 20a),
except that the Ni peak intensities have decreased. This decrease is due to
the silica coating.
NH3 Temperature-Programmed Desorption. Temperature programmed desorption
(TPD) of NH3 on the NilA1203 catalyst was done to monitor changes in the
accessibility of the
acid sites on the alumina support. The TPD spectra are shown in Figure 21 for
six different
samples with deposition times ranging from 0 to 3 h. The total ammonia uptake
was
determined by integrating the area under the TPD curves, and these areas are
listed in Table 5.
Two main peaks are observed in the TPD spectra around 160-180 C and 400-440 C.
These
peaks correspond to weak and strong acid sites, respectively, and are
consistent with the
literature. The ammonia uptake decreased with increasing deposition time,
indicating that the
acid sites were progressively blocked. Si02 is significantly less acidic than
A1203. After 0-3 h
of deposition, the uptake decreased from 461 ymol/g to 4,umollg (Table 5). The
NH3 uptake
decreased because of coverage of the external sites on the particles as well
as narrowing of the
pores preventing NH3 from accessing internal sites.
CA 02592124 2007-06-19
27
Table 5. Ammonia Uptake on Ni/A1203 as a Function
of Si02 Deposition Time at 350 C
Sample ID Deposition time (h) Total NH3 uptake
( mol/g)
Ni-0 0 461
Ni-0.5 0.5 448
Ni-1 1.0 246
Ni-1.5 1.5 224
Ni-2 2.0 143
Ni-2.5 2.5 113
Ni-3 3.0 4.1
Catalytic Performance for Hydrocracking of n-Octane. Figure 22 shows the
conversion of n-octane as a function of the Si02 deposition time. The
conversions shown in
Figure 22 are taken after 20 min on stream. With no Si02 deposition, n-octane
conversion was
29%. The maximum conversion (67%) was obtained on the Ni/A1203 catalyst coated
for 0.5 h.
The conversion decreased to zero for catalysts coated for 1.5 h or longer. The
product stream
consisted of only one C4 species that is likely 1-butene. The stabilities of
the catalysts varied.
That is, the loss of activity over 3 h on stream was 47%, 23%, and 0% for the
uncoated
catalyst, the catalyst coated for 0.5 h, and the catalyst coated for 1 h,
respectively.
Deposition Process. The surface areas of the A1203 and Ni/A1ZO3 samples
decreased
with increasing deposition temperature and time (Figures 15 and 16),
consistent with the results
reported by Sato et al. for a Si02/Al203 system and by Fodor et al. for a
Si02/MCM-41 system.
During the deposition, some additional carbonaceous material was deposited on
the
surface. The CHN analysis and N2 physisorption results before and after
calcination are
consistent with a porous layer of contamination being formed during the
deposition. This layer
was removed with a mild calcination, leaving behind a compact Si02 structure.
In an inert
carrier gas, the thermal decomposition of silicon alkoxide tends to produce
carbon and other
undesired organic products. The addition of steam helps to reduce the
formation of undesirable
products and also helps to propagate the growth of Si-O-Si bonds so that the
growth of Si02
can be faster. The carbonaceous layer likely prevented additional silica from
being deposited
beyond a deposition time of 1.5 h (Figure 18). Additional silica can be
deposited by calcining
the catalysts between deposition runs.
CA 02592124 2007-06-19
28
The silica layer narrowed the pore openings of the catalyst and prevented the
penetration of the larger molecules (Figures 16, 17, and 19). That is, CO (3.8
A) and N2 (3.6
A) were excluded from the pores, while H2 (2.9 A) could still penetrate to the
Ni sites. In
catalyst reaction systems of noble metals using H2/CO mixture as feedstock (or
H2 with CO as
contaminant), the CO tends to decrease the reactivity of the noble metal by
strongly adsorbing
on the surface and by inhibiting further adsorption of H2. This technique may
be a new way
for separating H2 from CO in the reaction systems involving noble metals,
thereby preserving
the reactivity of noble metal catalyst against the poisoning effect of CO.
The silica precursor, TMOS, has a molecular dimension of approximately 8.9 A,
which
0
is significantly smaller than the modal pore diameter of the A1203 support (-
38 A). Therefore,
the TMOS likely penetrated some of the pores of the alumina creating a
framework through
which H2 could still penetrate. The presence of Ni in the A1203 structure
influenced the
deposition process (Figure 16). The silica will interact more strongly with
the A1203 than the
Ni. Thus, a complete shell of silica is not formed on the NiJA1203 catalyst
and the measured
surface area remains at 17 m2/g after 1.5 h of deposition rather than being
reduced to zero as
for the A1203 sample. Addition of Ni to the A1203 resulted in a decrease in
surface area from
208 m2/g to 129 m2/g, indicating that the Ni is situated at the pore mouths
and is blocking
access to some of the internal pores. The low Ni dispersion of 1% is likely a
result of some Ni
being inaccessible because of this pore-mouth blocking, as well as some Ni
being situated on
the external surface of the particles, or some Ni being associated with the
alumina in a spinel
phase. Interestingly, the N2 uptake reached a minimum after 1.5 h of
deposition (Figure 15),
corresponding to a maximum silica uptake at 1.5 h (Figure 18), while the CO
uptake (Figure
19) and ammonia uptake (Table 5) continued to decrease up to 3 h of
deposition. The activity
of the catalyst (Figure 22) reached a minimum (at zero conversion) after 1.5 h
of deposition.
These results may indicate that the silica shell was not uniformly formed.
That is, after 1.5 h of
deposition, sufficient silica had been deposited to remove the activity of the
catalyst but not to
completely exclude all molecules larger than H2. Further deposition may have
filled in some
gaps in the coating so that ammonia and CO adsorption continued to decrease.
The N2
physisorption and ICP measurements will be less sensitive to small changes in
the coating than
the selective chemisorption measurements. Initial analysis of the thickness of
the coating
indicated that the coating could not be detected by scanning electron
microsopy (SEM). The
fact that the Ni peaks are still visible in the XRD spectra (Figure 20)
suggests that the coating
CA 02592124 2007-06-19
29
is relatively thin (<1,um). A schematic representation of the coated catalyst
is given in Figure
23.
Acidity and Reactivity of Coated Ni/A1203 Catalysts. NH3 is an excellent probe
molecule for the measurement of acidic properties of catalysts. The
temperature-programmed
desorption (TPD) results (Figure 21 and Table 5) indicated that the silica
deposition
significantly reduced the acid nature of the NiJA1203 catalysts. The acid
sites were covered on
the exterior of the particles and the pore openings were narrowed (Figure 17)
so that the
ammonia could not penetrate into the interior of the particles. The peaks in
the TPD spectra
(Figure 21) around 150 C correspond to weaker acid sites while the peaks above
400 C
correspond to stronger acid sites. According to Sato and co-workers and Katada
et al., the
nature of acidity formed on A1203 by the deposition of Si02 is of the Bronsted
type created at
the interface between the Si02 and A1203. In this work, the higher temperature
NH3 TPD peak
shifts to higher temperatures as additional silica is deposited indicating
that stronger acid sites
are created by the deposition of silica on alumina, in agreement with previous
work. As the
layer of Si02 becomes thicker, however, the acidity declines because access to
the interface is
blocked.
The balance between an increase in strong acid sites and a decrease in
accessibility of
the Si0z/AlZ03 interface is consistent with a maximum in the n-octane
conversion (Figure 22).
That is, as Si02 is deposited on the Ni/A12O3 catalyst, the number of Bronsted
sites, which
favor the cracking of molecules, increases because of the contribution of
acidity from the
Si02/Al203 interface. Further deposition of Si02 prevents access to the
interface because of
the large kinetic diameter (6.2 A) of n-octane and, thus, the conversion
decreases. The role of
Ni is to provide activated H species which reduce carbon formation. The effect
of the presence
of Ni on carbon formation was not directly investigated in this work, although
the stability of
the catalysts increased with increasing deposition time. The role of the
coating may have been
to limit the access of n-octane to the Ni particles, on which the hydrocarbon
may crack and
form carbon.
Conclusions
The pore size distribution of a Ni/A12O3 catalyst was modified by depositing
Si02 on
the outer surface in a fluidized bed using TMOS as the Si02 precursor. The
amount of
deposition increased with increasing time and temperature. A temperature of
350 C was
chosen for the deposition because at this temperature the rate of deposition
was sufficiently fast
CA 02592124 2007-06-19
without significant carbon formation. After 1.5 h of deposition at 350 C, the
pores had been
narrowed such that the measured surface area (by N2 uptake) of the Ni/Al2O3
catalyst
decreased to a minimum value of 17 m2/g compared to an initial surface area of
129 m2/g. At
these deposition conditions, CO uptake and NH3 uptake were also significantly
reduced and
continued to decrease up to a deposition time of 3 h. In contrast, the H2
uptake remained
relatively constant for Si02 deposition times up to 3 h. Even after exposure
to pure CO for 30
min, the H2 uptake on a Ni/A12O3 catalyst coated with Si02 for 2.5 h decreased
by only 28%
compared to a decrease of 76% for an uncoated Ni/Al2O3 catalyst. NH3 TPD and n-
octane
hydrocracking reactions demonstrated that a deposition time of 30 min at 350 C
was optimal in
terms of the number of Bronsted acids sites and accessibility to these sites.
Overall, these
results indicate that the pore openings and acidity of Ni/A12O3 catalysts can
be modified by the
deposition of Si02 with a fluidized CVD technique.
Example 3
Sol-gel Synthesis of PtlA12O3 catalysts: effect of precursor and calcination
procedure on
Pt dispersion
Currently there is much interest in tailored catalyst design. Sol-gel
synthesis can be
used to produce catalysts with uniform metal distribution, tunable particle
size, high surface
area, and stable dispersion. The resulting catalyst properties are sensitive
to changes in the
processing conditions. In this example, 2 wt% PtIA12O3 catalysts were prepared
using sol-gel
synthesis with various precursors including [Pt(C5H5N)4]C12 and
[Pt(CõH2õ+2NH2)4]C12. After
drying, calcination and reduction, the Pt particle size and dispersion were
determined by TEM,
XRD and H2 chemisorption.
The dispersions obtained varied between 5% and 70%. The effect of calcination
procedure on the Pt dispersion depended on the precursor that was used. That
is, when organic
ligands were present in the precursor, the dispersion varied considerably with
the calcination
conditions. For example, using [Pt(C5H5N)4]C1z as a precursor, the Pt
dispersion decreased
from 41% to 28% when treatment in flowing He before calcination in air was
removed. In
contrast, the dispersions were relatively constant regardless of calcination
procedure if a non-
organic precursor such as [Pt(NH3)4]CI2 was used. It is possible that
localized heating occurred
when the organic ligands of the precursor were oxidized in air, and this
temperature increase
resulted in sintering of the Pt particles.
CA 02592124 2007-06-19
31
Example 4
Chemical vapor deposition of Si02 on Ni/A1203 catalysts in a fluidized-bed
reactor
The modification of catalyst structures can be very important for achieving
high
activities, selectivities, and stabilities. Chemical vapour deposition (CVD)
is one method that
can be used for this purpose. Fine control of the pore-opening of Ni/A12O3 for
the selective
chemisorption of H2 (2.9 angstroms) and exclusion of larger molecules (N2 -
3.6 angstroms,
NH3 - 3.6 angstroms, and CO - 3.8 angstroms) was achieved by depositing Si02
on the
external surface of Ni/A1203 in a fluidized bed reactor using
tetramethoxysilane (TMOS) as
precursor. TMOS (1.75 mol%) was hydrolyzed with steam (14 mol%), using N2 as
the carrier
gas. The catalyst was characterized using N2 physisorption, H2 and CO
chemisorption, NH3
temperature-programmed desorption, and 29Si NMR. The effects of Si02
deposition time and
reaction temperature were investigated. The surface areas and the pore volumes
of Ni/A12O3
decreased as the deposition time increased. For a sample coated for 2.5 hours
(0.31 g Si02 per
g of sample), CO and N2 uptakes reduced significantly while H2 uptake remained
constant.
This result was an indication that the Ni sites were still accessible to H2
while CO and N2 were
excluded. Similarly the NH3 uptake diminished to near zero for a sample coated
for 3 hours
(0.37 g Si02 per g of sample). The reduction in acidity was ascribed to the
covering of acid
sites on the external surface by the Si02 coating and reduced penetration into
the pores by NH3
because of the reduced size of the pore-openings. The activity of the coated
catalysts was
tested for n-octane hydrocracking. The catalyst coated for 30 minutes showed
the maximum
conversion towards n-octane cracking, while the catalysts coated for 1.5 hours
or longer
showed no reactivity due to decreased acidity and narrowing of pores for n-
octane penetration.
An investigation of the microstructure of the Si02 coating using 29Si NMR
suggests that the
coating contains at least two layers of Si after 30 minutes of coating.
Example 5
Controlled pore-opening of Ni/A1203 using chemical vapor deposition in a micro
fluidized-bed reactor
Chemical vapor deposition (CVD) provides an efficient technique to precisely
control
the pore-openings of porous catalysts. In this example, the fine control of
the pore-opening of
NilA12O3 using CVD for the selective chemisorption of H2 (2.9 angstroms) and
exclusion of
larger molecules (N2 - 3.6 angstroms and CO - 3.8 angstroms) is shown.
The pore-openings of a commercial gamma-A1203 impregnated with Ni were
modified
CA 02592124 2007-06-19
32
by depositing Si02 on the external surface in a fluidized bed using
tetramethoxysilane (TMOS)
as the Si02 precursor. TMOS (1 mol%) was hydrolyzed with steam(10 mol%), using
N2 as the
carrier gas. The effect of deposition time was investigated. The catalysts
were characterized
using N2 physisorption, H2 and CO chemisorption, and NH3 temperature-
programmed
desorption (TPD).
The surface areas measured by N2 physisorption and the pore volumes of the
NilA12O3 samples decreased as the deposition time increased. The modal pore
diameter,
however, did not change significantly, indicating that the internal pore
structure was not
modified. For a sample coated for 2.5 hours (about 14 wt% SiO2), the uptakes
of CO and N2
were reduced by 95% and 87%, respectively, while the uptake of H2 remained
constant. This
result was an indication that the Ni sites were still accessible to H2 while
CO and N2 were
excluded. Similarly, the NH3 uptake decreased by 80%. The reduction in acidity
was ascribed
to the covering of acid sites on the external surface by the silica coating,
and reduced
penetration into the pores by NH3 because of the reduced size of the pore-
openings.
Example 6
Diffusion-controlled hydrogen spillover from modified Pt/gamma-A1Z03:
application to
toluene hydrogenation
Spillover is defined as the activation of species on a surface followed by the
transport
of those active species on an adjoining surface, which under the same
conditions will not be
able to activate that species. Hydrogen spillover is very important in the
sense that dihydrogen
(H2) can be activated on a catalyst surface to produce very reactive H species
for reaction to
increase the activity, selectivity, or stability of the catalyst. Surface
diffusion can significantly
control the rate of a reaction involving hydrogen spillover, especially when
the reaction
proceeds at a very rapid rate. The migration of the activated H species can
play a very
important role in the overall reaction kinetics.
In this example, hydrogen spillover has been demonstrated in the temperature
range of
90-240 C at atmospheric pressure using toluene hydrogenation as a model
reaction and
modified Pt/gamma-A1203. The Pt/gamma-A1203 catalysts were modified by coating
with
Si02. Only H2 (kinetic diameter - 2.9 angstroms) could access the Pt sites,
while toluene
molecules (kinetic diameter of 6.7 angstroms) were excluded. The kinetics of
the reaction
were studied in the temperature range of 120-240 C to determine whether the
reactions were
influenced by diffusion. The theoretical model of Freeman and Doll was applied
to the
CA 02592124 2007-06-19
33
experimental data to obtain an estimate of the diffusion coefficients for H2
spillover from the
modified Pt/gamma-A1203. A reaction mechanism was proposed.
A differential reactor was used for the reactions. The modified Pt/gamma-A1203
(40
mg) catalysts were mechanically mixed with various diluents (zeolite 13X and
gamma-A1203)
in a ratio of 1:1 in the reactor operating at atmospheric pressure. The
reaction temperature was
varied from 90 - 240 C to demonstrate H2 spillover from the modified Pt/gamma-
A1203
catalyst. To study the reaction kinetics the reaction temperature was varied
between
120-240 C. Toluene mole fraction was varied between 0.08 and 0.19, while H2
mole fraction
was varied from 0.26 to 0.60.
The degree of conversion depended on the type of diluent, with the more acidic
diluent,
zeolite 13X, showing a higher conversion of toluene. Neither the modified
Pt/gamma-A1203
nor the diluents alone were active towards aromatic hydrogenation. Therefore
it was
concluded that H2 spillover was responsible for the reactivity. The reactions
were determined
to be diffusion-controlled with activation energy EA of 12 kJlmol in the range
of 120-180 C.
For 210-240 C the activation energy increased to 86 kJlmol suggesting a change
in
mechanism, possibly reaction control. Figure 24 depicts the change in reaction
rate constant
with temperature. The model of Freeman and Doll, which provides a relationship
between
diffusion-controlled rate constant kd, and diffusion coefficient D, was
applied to the
experimental data to estimate the D as well as the activation energy for
diffusion, Ed,: In the
range of 120-180 C, D values were between 7.1x10-3 and 1.3x10-2 m2/s, the
average surface
residence time was 2.2x10-15 s. Edt~ was 15 kJ/mol, which is of the same
magnitude as EA and
confirms diffusion-controlled reaction. The reactions were tested to see
whether Eley-Rideal
(ER) mechanism played any role in the mechanisms. The proposed mechanism
involved the
dissociation of H2 on Pt, followed by the surface migration of the activated H
to attack
adsorbed toluene on the diluent surface, and the surface reaction of spillover
H with the
adsorbed toluene. No reaction between adsorbed species and gas phase species
(ER
mechanism) occurred.
Example 7
Evidence of H2 Spillover from Pt1A1203 with Protected Active Sites
Spillover involves the activation of species and transport of those active
species from
one surface to another surface on which the active species will not form under
the same
conditions. In H2 spillover, dihydrogen (H2) is dissociated into very reactive
H atoms (or ions)
CA 02592124 2007-06-19
34
for reaction. Hydrogen spillover from modified Pt/A12O3 catalysts has been
demonstrated
using toluene hydrogenation at atmospheric pressure in the temperature range
of 90 - 240 C, as
a model reaction. The Pt/A12O3 catalysts were modified so that only H2 could
access the Pt
sites. The modified Pt/A1203 catalysts were then mechanically mixed with
various diluents
(zeolite 13X, gamma-A1203, W03/A1203 and Si02). A 1:1 weight ratio of Pt/A12O3
and
diluents showed toluene conversion with methylcyclohexane as the only product
in all cases.
The degree of conversion depended on the type of diluent, with the most acidic
diluent, zeolite
13X, showing the highest conversion of approximately 20% at 180 C. Neither the
modified
Pt/A12O3 nor the diluents alone were active towards toluene hydrogenation when
tested under
the same conditions. In addition, in the absence of Pt, toluene conversion was
not observed.
Therefore, hydrogen spillover from the modified Pt/A12O3 catalysts was
responsible for the
hydrogenation of toluene in this reaction system.
Example 8
Evidence of H2 Spillover from Pt1A1203 With Protected Active Sites
Spillover applies to the activation of species and transport of those active
species from
one surface to another in which the second surface does not adsorb or form the
active species
under the same conditions. Hydrogen spillover from PtlA12O3 catalyst with
protected active
sites, in which the pores have been narrowed to restrict access to reactant
molecules except H2,
was demonstrated using the hydrogenation of toluene at atmospheric pressure in
the
temperature range of 90 - 240 C. The Pt/A12O3 with narrowed pores (hereby
called NP-
Pt/A1203) was mechanically mixed with various diluents (zeolite 13X, gamma-
A1203, Ti02,
W03/A1203 and Si02). Neither NP-Pt/A12O3 nor the diluents alone were active
towards toluene
hydrogenation when tested under the same conditions. A 1:1 weight ratio of NP-
Pt/A1203 and
diluents showed toluene conversion with methylcyclohexane as the only product
in all cases.
The degree of conversion depended on the type of diluent, with the most acidic
diluent, zeolite
13X, showing the highest conversion of about 20% at 150 C. The confirmation of
methylcyclohexane as the only product was evidence of toluene hydrogenation
caused by H2
activation on Pt sites located inside the NP-Pt/A1203 catalyst, and subsequent
spillover of
highly reactive atomic hydrogen (H), interparticle migration to toluene
molecules adsorbed on
the diluent. In the absence of Pt, toluene conversion was not observed. The
role of the diluent
was to provide the sites for toluene adsorption. Therefore spillover hydrogen
from Pt was
responsible for the conversion of toluene on NP-Pt/A12O3 and diluent mixtures.
CA 02592124 2007-06-19
Example 9
Characterization of Si02 deposition on Ni/A1203 and Mo03/Al203 catalysts
The modification of catalyst structures can be very important for achieving
high
activities, selectivities, and stabilities. Chemical vapor deposition (CVD) is
one method that
can be used for this purpose. Fine control of the pore-opening of Ni/Al2O3 for
the selective
chemisorption of H2 (2.9 angstroms) and the exclusion of larger molecules (N2-
3.6 angstroms,
NH3-3.6 angstroms, and CO-3.8 angstroms) was demonstrated by depositing Si02
on the
external surface of Ni/A12O3 in a fluidized bed reactor. N2 physisorption, H2
and CO
chemisorption, and NH3 TPD were used to characterize the catalysts to
investigate the effect of
deposition time and temperature. The CVD technique was also applied to
Mo03/A12O3
catalysts to study the mechanism, microstructure, and thickness of the Si02
deposition using
solid state 29Si NMR, ToF-SIMS, and ICP-MS. The activity of the coated
Ni/Al2O3 catalysts
for n-octane cracking was tested to see the effect of amount of Si02
deposition on acidity and
reactivity.
The Ni1A1203 (17% Ni loading) and Mo03/A12O3 (10% Mo loading) catalysts were
prepared by wet impregnation using Ni(N03)2=6H20 and (NH4)6Mo7O24=4H20
respectively as
precursors. Si02 was deposited on the catalysts using the hydrolysis of
tetramothoxysilane
(TMOS, 1-1.75%) with steam (10-14%) in a fluidized bed reactor at atmospheric
pressure
using N2 as a carrier gas. The cracking of n-octane was performed in a fixed
bed reactor at
400 C and atmospheric pressure.
The BET surface areas and the pore volumes of the Ni/A12O3 and MoO3/A1203
catalysts
decreased as the deposition time increased. For the Ni/Al2O3 catalyst coated
for 2.5 hours
(0.31 g Si02 per g of sample), CO uptake reduced significantly while H2 uptake
remained
constant, as shown in Figure 25. This result was an indication that the Ni
sites were still
accessible to H2 while CO was excluded. Similarly the NH3 uptake diminished to
near zero for
a sample coated for 3 hours (0.37 g Si02 per g of sample). The reduction in
acidity was
ascribed to the covering of acid sites on the external surface by the Si02
coating and reduced
penetration into the pores by NH3 because of the reduced size of the pore-
openings.
Figure 26 shows the conversion of n-octane as it changes with SiO2 deposition
time
during n-octane hydrocracking on Ni/A12O3. The catalyst coated for 30 minutes
showed the
maximum conversion towards n-octane cracking, while the catalysts coated for
1.5 hours or
longer showed no reactivity due to decreased acidity and narrowing of pores
for n-octane
CA 02592124 2007-06-19
36
penetration.
The 29Si NMR of the coated Mo03/Al2O3 catalysts suggested that the coating
contained
at least two layers of Si after 30 minutes of coating. ToF-SIMS results also
suggested a thin
layer of Si02 (2-3 monolayers), or an uneven distribution of Si02 on the
surface which is due
to the rough surface topology of the particles. Both positive and negative ToF-
SIMS spectra
revealed the presence of SiOXH (x=1,2,3) and CmHn (m,n=1,2,3, etc) fragments
which indicated
that silanols and methoxy species are produced during the hydrolysis of TMOS.
The
mechanism of Si02 deposition was proposed to be first the adsorption of Si-
OCH3 species on
the substrate, followed by hydrolysis to form silanol and methanol, and the
oxidation of the
silanol to form the Si-O-Si network.
Example 10
Kinetics of Toluene Hydrogenation on Modified gamma-A1203 Supported Catalysts
with
H2 Spillover
Hydrogen spillover from modified gamma-A1203 supported metal catalysts was
demonstrated in the range of 90 - 240 C at atmospheric pressure using toluene
hydrogenation
as the model reaction. The catalysts were modified by Si02 deposition so that
only H2 (kinetic
diameter, k.d., 2.7 angstroms) could access the metal sites, while toluene
molecules (k.d. 6.7
angstroms) were excluded. A mechanical mixture of the modified catalysts and
zeolite 13X or
c-A1203 was active for toluene hydrogenation, with approximately 20%
conversion for 13X
and 8% for c-A1203 at 180 C. Neither the modified catalysts nor the diluents
alone were
active. On the metal surface, H2 is activated into highly reactive H, which
then spills over onto
the diluent for reaction with adsorbed toluene molecules. The kinetics of the
reaction was
studied in the range of 120-240 C using the 13X diluent. The reactions were
modeled using a
power law model of the type r=kPTPH' and a Langmuir-Hinshelwood type
mechanistic model.
The order of the reaction with respect to toluene, n, ranged from -3 to 0.8.
For H2, the order m
ranged from 0.8 to 1.7. The activation energy, EA, of approximately 45 kJ/mol
suggests a
kinetics controlled reaction. The proposed mechanism involves activation of H2
on the metal
surface followed by the migration of the activated H to the diluent,
competitive adsorption of
toluene and H on the diluent, and sequential addition of adsorbed H to the
toluene to form the
product.
CA 02592124 2007-06-19
37
Example 11
Characterization of Si02 deposition on Ni/A1203 and MNOy/A12O3 catalysts
The modification of catalyst structures can be very important for achieving
high
activities, selectivities, and stabilities. Chemical vapor deposition (CVD) is
one method that
can be used for this purpose. Silica (SiO2) was deposited onto the external
surface of Ni/A1203
and Mo,,Oy/Al2O3 samples using chemical vapor deposition in a fluidized bed
(FCVD).
Tetramethyl oxysilane (TMOS) was used as silica precursor and was hydrolyzed
with steam
and nitrogen carrier gas at atmospheric pressure. The resulting deposit was
characterized using
various techniques including N2 physisorption, inductively coupled plasma
(ICP), and solid
state 29Si nuclear magnetic resonance (NMR). Due to the ferromagnetic behavior
of Ni, the
Ni/A12O3 was not used for NMR experiments. MoXOy/Al2O3 was used for NMR
experiments.
The Ni/A1203 catalysts with Si02 deposition were used for n-octane cracking to
determine the
effect of deposition on the cracking activity and the results compared to the
activity of an
uncoated Ni/A12O3 catalyst.
Si02 Formation:
Si(OCH3)4 (g) + 2H20 (g) 4 Si02 (s) + 4CH3OH (g) (overall reaction)
Mechanism of Si02 formation by TMOS hydrolysis:
=A1-OH + Si(OCH3)4 4 =Al-O-Si(OCH3)3 + CH3OH [1] (adsorption)
=Si-OCH3 + H20 4 =Si-OH + CH3OH [2] (hydrolysis)
=Si-OH + OH-Si= 4 =Si-O-Si- + H20 [3] (condensation)
=Si-OCH3 + OH-Si- 4 -Si-O-Si=_ + CH3OH [4] (condensation)
EXPERIMENTAL
Catalyst preparation:
- Ni/Al2O3 (17% Ni loading) - wet impregnation of gamma-A1203 with
Ni(N03)=6H20
- Mo,,Oy/Al2O3 (32% Mo loading) - wet impregnation of gamma-A1203 with
(NH4)6Mo7O24=4H20
- Calcination at 550 C for 2 hours, 2 C/min ramping rate
- Si02 deposition - TMOS (1-1.75%) hydrolyzed with steam (10%) at 350 C and
atmospheric pressure, followed by calcination at 500 C in air.
CA 02592124 2007-06-19
38
Characterization (before and after Si02 deposition)
Figure 27 illustrates a Schematic of the apparatus used for FCVD.
- SEM/EDX - morphology/composition; gold-coated
- N2, H2 and CO chemisorption to determine the number of active sites
- 29Si NMR - Si02 microstructure and bonding characteristics
Amount of deposition and shape-selectivity:
Figures 28, 29 and 30 illustrate the results.
- NZ uptake of Mo,,Oy/Al2O3 decreases with increasing deposition time.
- Amount of deposition for Mo,,Oy/Al2O3 increases at the rate of 0.3 g/h of
Si02
per g of sample for first 0.5 h, and then slows down to 0.025 g/h of Si02 per
g
of sample for more than 0.5 h of coating.
- H2 uptake of Ni/A12O3 remains almost constant as the deposition time
increases.
- CO uptake however, decreases with increasing deposition time, indicating a
better selective chemisorption of H2 (kinetic diameter, k.d. of 2.7 A) than CO
0
(k.d. of 3.8 A).
- Surface area of Ni/A12O3 and R-A1203 decrease with increasing deposition
time.
Surface area of R-A1203 decreases more rapidly than Ni/Al2O3. Bonding
between R-A1203 and Si02 is stronger than that of Ni and Si02.
Energy Dispersive Spectroscopy:
Figures 31 and 32 illustrate the particles and spots selected for energy
dispersive
spectroscopy (EDS). Figures 33 and 34 illustrate the results of the EDS for
each particle
shown in Figures 31 and 32.
CA 02592124 2007-06-19
39
Several spots were selected for EDS
Spots measured by EDS on MoXOy/Al2O3 particles in Figure 32
Element 1 2 3 4 5 6 7
0 48.3 43.0 38.4 28.7 46.4 26.5 36.3
Al 37.1 41.4 43.8 18.6 19.5 37.7 36.9
Si 14.6 15.6 17.7 9.0 6.3 11.2 14.1
Mo 0.0 0.0 0.0 43.7 27.8 24.6 12.7
AI:Si 2.5 2.7 2.5 2.1 3.1 3.4 2.6
Mo:Si 0.0 0.0 0.0 4.9 4.4 2.2 0.9
- Concentration of each species varies at different points on the surface.
- Mo is not uniformly distributed on the particle.
- Concentration of Si on the surface correlates more with Al than with Mo
concentration. That is, AI:Si ratio varies between 2.1 and 3.4, while Mo:Si
ratio
varies between 0 and 4.9.
29Si NMR:
Figures 35, 36 and 37 illustrate the results.
Si(4Si) -110 ppm
Si(3Si, Al) -105 ppm
Si(2Si, 2Al) -100 ppm
Si(3Si, H) -100 ppm
Si(2Si, Al, H) -95 ppm
Si(Si, 3A1) -95 ppm
- Chemical shifts increase to a higher field as Si concentration increases.
- Bonding characteristics of Si on A1203 coated for 2.5 hours consistently
shows
deposition thickness of at least a bilayer, without the presence of a
monolayer.
- At least 2 monolayers are present after 0.5 h deposition (10% Si02
deposition)
due to the presence of -109 ppm chemical shift. Low shift signal is also
present
(-94 ppm) indicative of a monolayer.
- Deposition of Si02 on Mo,,Oy/A12O3 greater extent of non-uniformity than
A1203.
CA 02592124 2007-06-19
n-Octane Cracking:
Figure 38 illustrates the results.
- Maximum activity for n-octane cracking observed for 0.5 h coating.
- Activity for n-octane cracking decreases with increasing deposition time.
- Cracking activity enhanced by the acid sites created at the Al-O-Si-OH
interface.
- Interface of AI-O-Si-OH is covered as deposition amount increases, leading
to
decrease in reactivity
- In addition, as the deposition time increases, the pore-opening decreases in
size
relative to size of n-octane molecule resulting in decrease in n-octane
penetration and loss of reactivity
CONCLUSIONS
- Pore characteristics can be changed using FCVD.
- At least two Si monolayers can be achieved after 0.5 hr coating.
- SiO2 deposition may not be uniform for both A12O3 and MoROy/Al2O3, but
Mo,{Oy/A12O3 shows greater extent of non-uniformity.
- Shape-selectivity is achievable by depositing Si on Ni/A1203.
- ICP and physisorption results showed that the deposition increased rapidly
initially at a rate of 0.3 g/h Si02 per g of sample, and reached a plateau as
deposition time increased. The decrease in deposition with time may be
attributed to coverage of surface by methoxy (-OCH3) and other organic species
that can be removed by calcination.
- N2 uptake decreased with increasing deposition time as a result of narrowing
of
pores to N2 penetration.
Example 12
Evidence of H2 Spillover from Pt/gamma-A1203 with protected Metal Active Sites
EXPER04ENTAL METHOD
Reactions:
CA 02592124 2007-06-19
41
Benzene hydrogenation:
/ I
+ 3H2 --~-
\
Toluene hydrogenation:
+ 3H2 -
O-xylene hydrogenation:
(trace)
2 + 6H2 -~- +
Reactivity Tests:
- Catalyst with protected metal sites only
- Pt loading -0.8% (measured by ICP)
- Diluent only (acidity: Zeolitel3X > gamma-A1203> Si02)
- Catalysts with protected metal sites + diluent, 1:1 mass ratio
- 50 mg catalyst (undiluted)
- 0.6 mlJhhydrocarbon, 30 mL/min H2 flow
- 1 atm, 90 -240 C
Effect of Amount of Diluent:
- Diluent-catalyst ratio of 1:1, 2:1, 4:1
RESULTS
Benzene Hydrogenation:
Figure 39 graphically illustrates the results.
- Synergism observed with bifunctional modified Pt/gamma-A1203 + diluents.
- Reactivity increases with increasing diluent acidity (Zeolite 13X >
gamma-A1203)
- Diluent shows no reactivity in the absence of noble metal. Spillover works!
CA 02592124 2007-06-19
42
Toluene Hydrogenation:
Figure 40 graphically illustrates the results.
- Synergism observed with bifunctional modified Pt/gamma-A1203 + diluents.
- Reactivity increases with increasing diluent acidity (Zeolitel3X >
gamma-A1203 > Si02).
O-xylene Hydrogenation:
Figure 41 graphically illustrates the results.
> Synergism observed with bifunctional modified Pt/gamma-A1203 + diluents.
> Reactivity decreases in order: Benzene > Toluene > O-xylene.
Effect of Diluent Amount:
Figure 42 graphically illustrates the results.
- Reactivity increases with increasing diluent amount.
> Optimal diluent-catalyst ratio is 2:1
CONCLUSIONS
- Spillover hydrogen can migrate from modified Pt/gamrna-A1203 catalyst and
react with species adsorbed on diluent
> Mixture of modified Pt/gamma-A1203 and the most acidic diluent(Zeolitel3X)
gives highest conversion
- Reactivity decreases with increasing aromatic substituents (benzene >
toluene >
o-xylene)
- Optimal diluent-catalyst ratio is 2:1
Example 13
Chemical Vapour Deposition of Si02 on Ni/A1203 catalyst in a fluidized-bed
reactor
EXPERIMENTAL METHODS
FCVD:
- Reduction of Ni1A1203 at 550 C for 4 hours in ChemBET 3000
> Hydrolysis of silicon alkoxide at latm, 350 C
> 1-2 mol% tetramethyl-oxysilane (TMOS), 10 mol% steam
- 1-3 hours deposition time
> Calcination in dry air at 500 C for 2 hours
Si(OCH3)4 + 2H20 4Si02 + 4CH3OH
CA 02592124 2007-06-19
43
- Characterization: H2 + CO chemisorption, N2 physisorption, NH3, TPD, ICP,
SEM, NMR
- Fluidized bed reactor - quartz tube, 1 cm ID, 41 cm length
Mechanism of Deposition:
=AL-OH + Si(OCH3)4 a =Al-O-Si(OCH3)3 + CH3OH
-Si-OCH3 + H20 <=> =Si-OH + CH3OH (Hydrolysis)
=Si-OH + OH-Si=_ a -Si-O-Si= + H20 (Condensation)
-Si-OCH3 + OH-Si= ~#* =Si-O-Si= + CH3OH (Condensation)
RESULTS:
Pore Size Distribution:
Figure 43 graphically illustrates the results.
- Pore sizes decrease as Si02 deposition increases
Deposition Time:
Figure 44 graphically illustrates the results.
- CO (3.8 Angstroms) uptake decreases with increasing duration of deposition.
HZ (2.9 Angstroms) uptake remains almost constant.
Amount of Deposition:
Figure 45 graphically illustrates the results.
- SiO2 deposition increases with increasing deposition time - 37% deposition
after 3h.
- Si Content measured by ICP-MS (Galbraith Labs).
Total Acidity:
Figure 46 graphically illustrates the results.
- SiO2 deposition covers acid sites and narrows pore openings. NH3 uptake
decreases with increasing Si02 deposition.
- Acidity is created by induced effect of -Al-O-Si-OH species.
n-Octane Cracking:
Figure 47 graphically illustrates the results.
- Maximum reactivity for n-octane cracking observed after 0.5h coating.
- Si02 monolayer on gamma-A1203 increases Bronsted acidity.
29Si MAS NMR:
CA 02592124 2007-06-19
44
Figure 48 graphically illustrates the results.
- 29Si chemical shift of -122 3 ppm indicates Si(OSi)4 network - at least 2
layers of Si02.
N2 Uptake:
Figure 49 graphically illustrates the results.
- Deposition is more rapid on plain A1203 compared to Ni (17%) loaded A1203.
CONCLUSIONS
- Selective chemisorption of H2 and rejection of larger molecules (CO, NH3) is
possible on Ni/A12O3 coated with Si02.
- A deposition time of 2 hours at 350 C is sufficient to block majority of
pores to
CO adsorption.
- At least 2 layers of Si02 are present after 0.5h of deposition.