Note: Descriptions are shown in the official language in which they were submitted.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
1
2,4-DIAMINO-PYRIDOPYRIMIDINE DERIVATIVES AND THEIR USE AS MTOR INHIBITORS
The present invention relates to compounds which act as mTOR inhibitors, their
use and
their synthesis.
Background
Growth factor/mitogenic activation of the phosphatidylinositol 3-kinase
(PI3K)/AKT
signalling pathway ultimately leads to the key cell cycle and growth control
regulator
mTOR, the mammalian target of rapamycin (alternatively referred to as FRAP
(FKBP12
and rapamycin associated protein), RAFT1 (rapamycin and FKBP12 target 1),
RAPT1
(rapamycin target 1) - all derived from the interaction with the FK-506-
binding protein
FKBP12, and SEP (sirolimus effector protein)). mTOR is a mammalian
serine/threonine
kinase of approximately 289 kDa in size and a member of the evolutionary
conserved
eukaryotic TOR kinases (refs. 1-4). The mTOR protein is a member of the P13-
kinase like
kinase (PIKK) family of proteins due to its C-terminal homology (catalytic
domain) with
P13-kinase and the other family members, e.g. DNA-PKcs (DNA dependent protein
kinase), ATM (Ataxia-telangiectasia mutated). In addition to a catalytic
domain in the C-
terminus, mTOR contains a FKBP12/rapamycin complex binding domain (FRB). At
the N-
terminus up to 20 HEAT (Huntingtin, EF3, alpha regulatory subunit of PP2A and
TOR)
motifs are found whilst more C-terminal is a FAT (FRAP-ATM-TRRAP) domain, and
at the
extreme C-terminus of the protein an additional FAT domain is found (FAT-C)
(refs. 5,6).
TOR has been identified as a central regulator of both cell growth (size) and
proliferation,
which is in part governed by translation initiation. TOR dependant
phosphorylation of S6-
kinase (S6K1) allows translation of ribosomal proteins involved in cell cycle
progression
(refs. 7-9).Cap-dependant translation is regulated by the phosphorylation of
the eukaryotic
translation initiation factor 4E (eIF4E)-binding protein 1(4E-BP1 (PHAS-1)).
This
modification prevents PHAS-1 binding eIF4E, thereby permitting formation of an
active
eIF4F translation complex (reviewed in refs. 10,11,12). Activation of these
signalling
elements is dependant on insulin, other growth factors and nutrients
suggesting a
gatekeeper role for mTOR in the control of cell cycle progression only under
favourable
environmental conditions. The P13K/AKT signalling cascade lies upstream of
mTOR and
this has been shown to be deregulated in certain cancers and results in growth
factor
independent activation in, for example, PTEN deficient cells. mTOR lies at the
axis of
control for this pathway and inhibitors of this kinase (e.g. sirolimus
(rapamycin or
RapamuneT"') and everolimus (RAD001 or CerticanT"')) are already approved for
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
2
immunosuppression and drug eluting stents (reviewed in refs. 13, 14), and are
now
receiving particular interest as novel agents for cancer treatment.
Tumour cell growth arises from the deregulation of normal growth control
mechanisms
such as the loss of tumour suppressor function(s). One such tumour suppressor
is the
phosphatase and tensin homologue. deleted from chromosome ten (PTEN). This
gene,
also known as mutated in multiple advanced cancers (MMAC), has been shown to
play a
significant role in cell cycle arrest and is the most highly mutated tumour
suppressor after
p53. Up to 30% of glioblastoma, endometrial and prostate cancers have somatic
mutations or deletions of this locus (refs. 15,16).
P13K converts phosphatidylinositol 4,5, bisphosphate (PIP2) to
phosphatidylinositol 3,4,5,
triphosphate (PIP3) whilst PTEN is responsible for removing the 3' phosphate
from PIP3
producing PIP2. P13-K and PTEN act to maintain an appropriate level of PIP3
which
recruits and thus activates AKT (also known as PKB) and the downstream
signalling
cascade that is then initiated. In the absence of PTEN, there is inappropriate
regulation of
this cascade, AKT becomes effectively constitutively activated and cell growth
is
deregulated. An alternative mechanism for the deregulation of this cell
signalling process
is the recent identification of a mutant form of the P13K isoform, p110alpha
(ref. 17). The
apparent increased activity of this mutant is thought to result in increased
PIP3
production, presumably in excess of that which the function of PTEN can
counteract.
Increased signalling from P13K, thus results in increased signalling to mTOR
and
consequently, its downstream activators.
In addition to the evidence linking mTOR with cell cycle regulation (from G1
to S-phase)
and that inhibition of mTOR results in inhibition of these regulatory events
it has been
shown that down regulation of mTOR activity results in cell growth inhibition
(Reviewed in
refs. 7,18,19). The known inhibitor of mTOR, rapamycin, potently inhibits
proliferation or
growth of cells derived from a range of tissue types such as smooth muscle, T-
ceils as
well as cells derived from a diverse range of tumour types including
rhabdomyosarcoma,
neuroblastoma, glioblastoma and medulloblastoma, small cell lung cancer,
osteosarcoma,
pancreatic carcinoma and breast and prostate carcinoma (reviewed in ref. 20).
Rapamycin has been approved and is in clinical use as an immunosuppressant,
its
prevention of organ rejection being successful and with fewer side effects
than previous
therapies (refs. 20, 21). Inhibition of mTOR by rapamycin and its analogues
(RADOOI,
CCI-779) is brought about by the prior interaction of the drug with the FK506
binding
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
3
protein, FKBP12. Subsequently, the complex of FKBP12/rapamycin then binds to
the
FRB domain of mTOR and inhibits the downstream signalling from mTOR.
The potent but non-specific inhibitors of P13K, LY294002 and wortmannin, also
have been
shown to inhibit the kinase function of mTOR but act through targeting the
catalytic
domain of the protein (ref. 21). Further to the inhibition of mTOR function by
small
molecules targeted to the kinase domain, it has been demonstrated that kinase
dead
mTOR cannot transmit the upstream activating signals to the downstream
effectors of
mTOR, PHAS-1 or p70S6 kinase (ref. 22). It is also shown that not all
functions of mTOR
are rapamycin sensitive and this may be related to the observation that
rapamycin alters
the substrate profile of mTOR rather than inhibiting its activity per se (ref.
23). Therefore,
it is proposed that a kinase domain directed inhibitor of mTOR may be a more
effective
inhibitor of mTOR.
In addition to rapamycin's ability to induce growth inhibition (cytostasis) in
its own right,
rapamycin and its derivatives have been shown to potentiate the cytotoxicity
of a number
of chemotherapies including cisplatin, camptothecin and doxorubicin (reviewed
in ref. 20).
Potentiation of ionising radiation induced cell killing has also been observed
following
inhibition of mTOR (ref. 24) Experimental and clinical evidence has shown that
rapamycin
analogues are showing evidence of efficacy in treating cancer, either alone or
in
combination with other therapies (see refs. 10,18,20).
The vast majority of mTOR pharmacology to date has focused on inhibition of
mTOR via
rapamycin or its analogues. However, as noted above, the only non-rapamycin
agents
that have been reported to inhibit mTOR's activity via a kinase domain
targetted
mechanism are the small molecule LY294002 and the natural product wortmannin
(ref.
21).
Summary of the Invention
The present inventors have identified compounds which are ATP-competitive
inhibitors of
mTOR, and hence are non-rapamycin like in their mechanism of action.
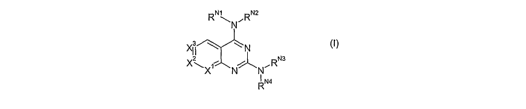
Accordingly, the first aspect of the present invention provides a compound of
formula I:
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
4
RN~NRNa
XII ~ L
N
X~X NN"RNa
RN4
and isomers, salts, solvates, chemically protected forms, and prodrugs
thereof, wherein:
one of X', X2, and X3 is N, and the others are CH;
RN' and RNZ , together with the nitrogen atom to which they are attached form
a nitrogen-
containing heterocyclic ring having from 4 to 8 ring atoms;
RN3 and RN4 together with the nitrogen atom to which they are attached form a
nitrogen-
containing heterocyclic ring having from 4 to 8 ring atoms.
A second aspect of the present invention provides a pharmaceutical composition
comprising a compound of the first aspect and a pharmaceutically acceptable
carrier or
diluent.
A third aspect of the present invention provides a compound of the first
aspect for use in a
method of treatment of the human or animal body.
A fourth aspect of the present invention provides the use of a compound of
formula II:
N1 N2
R 1-1 NR
a
si ~ N
2 ~ ~ N3
X~X1 N N'R
RN4
and isomers, salts, solvates, chemically protected forms, and prodrugs thereof
in the preparation of a medicament for treating a disease ameliorated by the
inhibition of
mTOR, wherein:
one of X', X2, X3 and X4 is N, and the others are CH
RN' and RN2 , together with the nitrogen atom to which they are attached form
a nitrogen-
containing heterocyclic ring having from 4 to 8 ring atoms;
RN3 and RN4 together with the nitrogen atom to which they are attached form a
nitrogen-
containing heterocyclic ring having from 4 to 8 ring atoms.
Further aspects of the invention provide the use of a compound as defined in
the fourth
aspect of the invention in the preparation of a medicament for the treatment
of: cancer,
immuno-suppression, immune tolerance, autoimmune disease, inflammation, bone
loss,
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
bowel disorders, hepatic fibrosis, hepatic necrosis, rheumatoid arthritis,
restinosis, cardiac
allograft vasculopathy, psoriasis, beta-thalassaemia, and ocular conditions
such as dry
eye. mTOR inhibitors may also be effective as antifungal agents
5 Another further aspect of the invention provides for the use of a compound
as defined in
the fourth aspect of the invention in the preparation of a medicament for use
as an adjunct
in cancer therapy or for potentiating tumour cells for treatment with ionizing
radiation or
chemotherapeutic agents.
Other further aspects of the invention provide for the treatment of disease
ameliorated by
the inhibition of mTOR, comprising administering to a subject in need of
treatment a
therapeutically-effective amount of a compound as defined in the fourth
aspect, preferably
in the form of a pharmaceutical composition and the treatment of cancer,
comprising
administering to a subject in need of treatment a therapeutically-effective
amount of a
compound as defined in the fourth aspect in combination, preferably in the
form of a
pharmaceutical composition, simultaneously or sequentially with ionizing
radiation or
chemotherapeutic agents.
Definitions
Nitrogen-containing heterocyclic ring having from 4 to 8 ring atoms: The term
"Nitrogen-
containing heterocyclic ring having from 4 to 8 ring atoms" as used herein
refers to a 4 to
8 membered heterocylic ring containing at least one nitrogen ring atom.
Examples of
these groups include, but are not limited to:
Nl: azetidine (C4), pyrrolidine (tetrahydropyrrole) (C5), pyrroline (e.g., 3-
pyrroline,
2,5-dihydropyrrole) (C5), 2H-pyrrole or 3H-pyrrole (isopyrrole, isoazole)
(C5), piperidine
(C6), dihydropyridine (C6), tetrahydropyridine (C6), azepine (C7);
N2: imidazolidine (CS), pyrazolidine (diazolidine) (C5), imidazoline (C5),
pyrazoline
(dihydropyrazole) (C5), piperazine (C6);
NIOI: tetrahydrooxazole (CS), dihydrooxazole (C5), tetrahydroisoxazole (C5),
dihydroisoxazole (C5), morpholine (C6), tetrahydrooxazine (C6), dihydrooxazine
(C6),
oxazine (C6);
NIS1: thiazoline (C5), thiazolidine (C5), thiomorpholine (C6);
N2O1: oxadiazine (Cs);
NIOIS1: oxathiazine (C6).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
6
Alkyl: The term "alkyl" as used herein, pertains to a monovalent moiety
obtained by
removing a hydrogen atom from a carbon atom of a hydrocarbon compound having
from 1
to 20 carbon atoms (unless otherwise specified), which may be aliphatic or
alicyclic, and
which may be saturated or unsaturated (e.g. partially unsaturated, fully
unsaturated).
Thus, the term "alkyl" includes the sub-classes alkenyl, alkynyl, cycloalkyl,
cycloalkyenyl,
cylcoalkynyl, etc., discussed below.
In the context of alkyl groups, the prefixes (e.g. Cl_4r Cl_7, C1_20, C2_7,
C3.7, etc.) denote the
number of carbon atoms, or range of number of carbon atoms. For example, the
term
"C1_4 alkyl", as used herein, pertains to an alkyl group having from 1 to 4
carbon atoms.
Examples of groups of alkyl groups include C1_4 alkyl ("lower alkyl"), Cl_7
alkyl, and Ci_20
alkyl. Note that the first prefix may vary according to other limitations; for
example, for
unsaturated alkyl groups, the first prefix must be at least 2; for cyclic
alkyl groups, the first
prefix must be at least 3; etc.
Examples of (unsubstituted) saturated alkyl groups include, but are not
limited to, methyl
(CI), ethyl (CA propyl (C3), butyl (C4), pentyl (C5), hexyl (C6), heptyl (CA
octyl (C8), nonyl
(C9), decyl (Clo), undecyl (Cll), dodecyl (C12), tridecyl (C13), tetradecyl
(C14), pentadecyl
(C15), and eicodecyl (C20).
Examples of (unsubstituted) saturated linear alkyl groups include, but are not
limited to,
methyl (Ci), ethyl (CA n-propyl (C3), n-butyl (C4), n-pentyl (amyl) (C5), n-
hexyl (C6), and n-
heptyl (C7).
Examples of (unsubstituted) saturated branched alkyl groups include iso-propyl
(C3),
iso-butyl (C4), sec-butyl (C4), tert-butyl (Ca), iso-pentyl (C5), and neo-
pentyl (C5).
Alkenyl: The term "alkenyl", as used herein, pertains to an alkyl group having
one or more
carbon-carbon double bonds. Examples of groups of alkenyl groups include Ca4
alkenyl,
C2_7 alkenyl, C2_20 alkenyl.
Examples of (unsubstituted) unsaturated alkenyl groups include, but are not
limited to,
ethenyl (vinyl, -CH=CH2), 1-propenyl (-CH=CH-CH3), 2-propenyl (allyl, -CH-
CH=CH2),
isopropenyl (1-methylvinyl, -C(CH3)=CH2), butenyl (C4), pentenyl (C5), and
hexenyl (C6).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
7
Alkynyl: The term "alkynyl", as used herein, pertains to an alkyl group having
one or more
carbon-carbon triple bonds. Examples of groups of alkynyl groups include CZ4
alkynyl,
C2.7 alkynyl, C2.2o alkynyl.
Examples of (unsubstituted) unsaturated alkynyl groups include, but are not
limited to,
ethynyl (ethinyl, -C=CH) and 2-propynyl (propargyl, -CH2-C CH).
Cycloalkyl: The term "cycloalkyl", as used herein, pertains to an alkyl group
which is also
a cyclyl group; that is, a monovalent moiety obtained by removing a hydrogen
atom from
an alicyclic ring atom of a carbocyclic ring of a carbocyclic compound, which
carbocyclic
ring may be saturated or unsaturated (e.g. partially unsaturated, fully
unsaturated), which
moiety has from 3 to 20 carbon atoms (unless otherwise specified), including
from 3 to 20
ring atoms. Thus, the term "cycloalkyl" includes the sub-classes cycloalkenyl
and
cycloalkynyl. Preferably, each ring has from 3 to 7 ring atoms. Examples of
groups of
cycloalkyl groups include C3.2o cycloalkyl, C3.15 cycloalkyl, C3.10
cycloalkyl, C3.7 cycloalkyl.
Examples of cycloalkyl groups include, but are not limited to, those derived
from:
saturated monocyclic hydrocarbon compounds:
cyclopropane (C3), cyclobutane (C4), cyclopentane (C5), cyclohexane (C6),
cycloheptane
(C7), methylcyclopropane (C4), dimethylcyclopropane (C5), methylcyclobutane
(C5),
dimethylcyclobutane (C6), methylcyclopentane (C6), dimethylcyclopentane (CA
methylcyclohexane (CA dimethylcyclohexane (C8), menthane (Clo);
unsaturated monocyclic hydrocarbon compounds:
cyclopropene (C3), cyclobutene (C4), cyclopentene (C5), cyclohexene (C6),
methylcyclopropene (C4), dimethylcyclopropene (C5), methylcyclobutene (C5),
dimethylcyclobutene (C6), methylcyclopentene (C6), dimethylcyclopentene (CA
methylcyclohexene (CA dimethylcyclohexene (C8);
saturated polycyclic hydrocarbon compounds:
thujane (CIo), carane (Clo), pinane (Clo), bornane (CIo), norcarane (C,),
norpinane (C,),
norbornane (C,), adamantane (Clo), decalin (decahydronaphthalene) (Cio);
unsaturated polycyclic hydrocarbon compounds:
camphene (C1o), limonene (Clo), pinene (Clo);
polycyclic hydrocarbon compounds having an aromatic ring:
indene (C9), indane (e.g., 2,3-dihydro-1 H-indene) (C9), tetraline
(1,2,3,4-tetrahydronaphthalene) (Clo), acenaphthene (C12), fluorene (C13),
phenalene
(C13), acephenanthrene (CI5), aceanthrene (C16), cholanthrene (C20).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
8
Heterocyclyl: The term "heterocyclyP", as used herein, pertains to a
monovalent moiety
obtained by removing a hydrogen atom from a ring atom of a heterocyclic
compound,
which moiety has from 3 to 20 ring atoms (unless otherwise specified), of
which from 1 to
are ring heteroatoms. Preferably, each ring has from 3 to 7 ring atoms, of
which from 1
5 to 4 are ring heteroatoms.
In this context, the prefixes (e.g. C3.20i C3-7i C5.(3, etc.) denote the
number of ring atoms, or
range of number of ring atoms, whether carbon atoms or heteroatoms. For
example, the
term "C5_6heterocyclyP", as used herein, pertains to a heterocyclyl group
having 5 or 6 ring
10 atoms. Examples of groups of heterocyclyl groups include C3.20
heterocyclyl, C5_20
heterocyclyl, C3.15 heterocyclyl, C5_15 heterocyclyl, C3.12 heterocyclyl,
C5.12 heterocyclyl, C3-10
heterocycly4, C5.1o heterocyclyl, C3_7 heterocycly{, C5.7 heterocyclyl, and
C5_6 heterocyclyl.
Examples of monocyclic heterocyclyl groups include, but are not limited to,
those derived
from:
N1: aziridine (C3), azetidine (C4), pyrrolidine (tetrahydropyrrole) (C5),
pyrroline (e.g.,
3-pyrroline, 2,5-dihydropyrrole) (C5), 2H-pyrrole or 3H-pyrrole (isopyrrole,
isoazole) (C5),
piperidine (Cs), dihydropyridine (Cs), tetrahydropyridine (C6), azepine (C7);
O1: oxirane (C3), oxetane (C4), oxolane (tetrahydrofuran) (C5), oxole
(dihydrofuran) (C5),
oxane (tetrahydropyran) (C6), dihydropyran (C6), pyran (C6), oxepin (C7);
S1: thiirane (C3), thietane (C4), thiolane (tetrahydrothiophene) (C5), thiane
(tetrahydrothiopyran) (C6), thiepane (C7);
02: dioxolane (C5), dioxane (C6), and dioxepane (C7);
03: trioxane (C6);
N2: imidazolidine (C5), pyrazolidine (diazolidine) (C5), imidazoline (C5),
pyrazoline
(dihydropyrazole) (C5), piperazine (C6);
N1O1: tetrahydrooxazole (C5), dihydrooxazole (C5), tetrahydroisoxazole (CS),
dihydroisoxazole (C5), morpholine (Cs), tetrahydrooxazine (C6), dihydrooxazine
(C6),
oxazine (C6);
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
9
NIS1: thiazoline (C5), thiazolidine (C5), thiomorpholine (C6);
N201: oxadiazine (C6);
OIS1: oxathiole (C5) and oxathiane (thioxane) (C6); and,
N1O1SI: oxathiazine (C6).
Examples of substituted (non-aromatic) monocyclic heterocyclyl groups include
those
derived from saccharides, in cyclic form, for example, furanoses (C5), such as
arabinofuranose, lyxofuranose, ribofuranose, and xylofuranse, and pyranoses
(Cs), such
as allopyranose, altropyranose, glucopyranose, mannopyranose, gulopyranose,
idopyranose, galactopyranose, and talopyranose.
Spiro-C3_7 cycloalkyl or heterocyclyl: The term "spiro C3-7 cycloalkyl or
heterocyclyl" as
used herein, refers to a C3_7 cycloalkyl or C3_7 heterocyclyl ring joined to
another ring by a
single atom common to both rings.
C5_20 aryl: The term "C5_20 aryl" as used herein, pertains to a monovalent
moiety obtained
by removing a hydrogen atom from an aromatic ring atom of a C5_2o aromatic
compound,
said compound having one ring, or two or more rings (e.g., fused), and having
from 5 to
20 ring atoms, and wherein at least one of said ring(s) is an aromatic ring.
Preferably,
each ring has from 5 to 7 ring atoms.
The ring atoms may be all carbon atoms, as in "carboaryl groups" in which case
the group
may conveniently be referred to as a"C5_2o carboaryl" group.
Examples of C5_20 aryl groups which do not have ring heteroatoms (i.e. Cs-2o
carboaryl
groups) include, but are not limited to, those derived from benzene (i.e.
phenyl) (C6),
naphthalene (Clo), anthracene (Ctia), phenanthrene (CI4), and pyrene (C16).
Alternatively, the ring atoms may include one or more heteroatoms, including
but not
limited to oxygen, nitrogen, and sulfur, as in "heteroaryl groups". In this
case, the group
may conveniently be referred to as a"C5_2o heteroaryl" group, wherein "C5_20"
denotes ring
atoms, whether carbon atoms or heteroatoms. Preferably, each ring has from 5
to 7 ring
atoms, of which from 0 to 4 are ring heteroatoms.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
Examples of C5_20 heteroaryl groups include, but are not limited to, C5
heteroaryl groups
derived from furan (oxole), thiophene (thiole), pyrrole (azole), imidazole
(1,3-diazole),
pyrazole (1,2-diazole), triazole, oxazole, isoxazole, thiazole, isothiazole,
oxadiazole,
tetrazole and oxatriazole; and C6 heteroaryl groups derived from isoxazine,
pyridine
5 (azine), pyridazine (1,2-diazine), pyrimidine (1,3-diazine; e.g., cytosine,
thymine, uracil),
pyrazine (1,4-diazine) and triazine.
The heteroaryl group may be bonded via a carbon or hetero ring atom.
10 Examples of C5_2o heteroaryl groups which comprise fused rings, include,
but are not
limited to, C9 heteroaryl groups derived from benzofuran, isobenzofuran,
benzothiophene,
indole, isoindole; CIo heteroaryl groups derived from quinoline, isoquinoline,
benzodiazine,
pyridopyridine; C14 heteroaryl groups derived from acridine and xanthene.
The above alkyl, heterocyclyl, and aryl groups, whether alone or part of
another
substituent, may themselves optionally be substituted with one or more groups
selected
from themselves and the additional substituents listed below.
Halo: -F, -CI, -Br, and -I.
Hydroxy: -OH.
Ether: -OR, wherein R is an ether substituent, for example, a Cl_7 alkyl group
(also
referred to as a Cl_7 alkoxy group), a C3_2o heterocyclyl group (also referred
to as a C3.20
heterocyclyloxy group), or a C5_20 aryl group (also referred to as a C5_2o
aryloxy group),
preferably a Cl_7 alkyl group.
Nitro: -NO2.
Cyano (nitrile, carbonitrile): -CN.
Acyl (keto): -C(=O)R, wherein R is an acyl substituent, for example, H, a C1_7
alkyl group
(also referred to as Cl_7 alkylacyl or CI_7 alkanoyl), a C3_20 heterocyclyl
group (also referred
to as C3_20 heterocyclylacyl), or a C5_2o aryl group (also referred to as
C5_2o arylacyl),
preferably a Cl_7 alkyl group. Examples of acyl groups include, but are not
limited to,
-C(=O)CH3 (acetyl), -C(=O)CH2CH3 (propionyi), -C(=O)C(CH3)3 (butyryl), and -
C(=O)Ph
(benzoyl, phenone).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
11
Carboxy (carboxylic acid): -COOH.
Ester (carboxylate, carboxylic acid ester, oxycarbonyl): -C(=0)OR, wherein R
is an ester
substituent, for example, a CI-7 alkyl group, a C3_20 heterocyclyl group, or a
C5.2o aryl group,
preferably a CI-7 alkyl group. Examples of ester groups include, but are not
limited to,
-C(=O)OCH3, -C(=O)OCH2CH3, -C(=O)OC(CH3)3, and -C(=O)OPh.
Amido (carbamoyl, carbamyl, aminocarbonyl, carboxamide): -C(=O)NR'R2, wherein
R'
and R2 are independently amino substituents, as defined for amino groups.
Examples of
amido groups include, but are not limited to, -C(=0)NH2, -C(=0)NHCH3, -
C(=0)N(CH3)2,
-C(=O)NHCH2CH3i and -C(=O)N(CH2CH3)2, as well as amido groups in which R' and
R2,
together with the nitrogen atom to which they are attached, form a
heterocyclic structure
as in, for example, piperidinocarbonyl, morpholinocarbonyl,
thiomorpholinocarbonyl, and
piperazinylcarbonyl.
Amino: -NR1R2, wherein R' and R2 are independently amino substituents, for
example,
hydrogen, a CI-7 alkyl group (also referred to as Cl_7 alkylamino or di-CI_7
alkylamino), a
C3_2o heterocyclyl group, or a C5_20 aryl group, preferably H or a CI-7 alkyl
group, or, in the
case of a "cyclic" amino group, R' and R2, taken together with the nitrogen
atom to which
they are attached, form a heterocyclic ring having from 4 to 8 ring atoms.
Examples of
amino groups include, but are not limited to, -NH2, -NHCH3, -NHCH(CH3)2, -
N(CH3)2,
-N(CH2CH3)2, and -NHPh. Examples of cyclic amino groups include, but are not
limited to,
aziridinyl, azetidinyl, pyrrolidinyl, piperidino, piperazinyl,
perhydrodiazepinyl, morpholino,
and thiomorpholino. The cylic amino groups may be substituted on their ring by
any of the
substituents defined here, for example carboxy, carboxylate and amido.
Acylamido (acylamino): -NR'C(=O)Ra, wherein R' is an amide substituent, for
example,
hydrogen, a CI-7 alkyl group, a C3_20 heterocyclyl group, or a C5_20 aryl
group, preferably H
or a CI-7 alkyl group, most preferably H, and R2 is an acyl substituent, for
example, a CI-7
alkyl group, a C3_2o heterocyclyl group, or a C5_2Q aryl group, preferably a
CI-7 alkyl group.
Examples of acylamido groups include, but are not limited to, -NHC(=0)CH3 ,
-NHC(=O)CH2CH3, and -NHC(=O)Ph. R' and R 2 may together form a cyclic
structure, as
in, for example, succinimidyl, maleimidyl, and phthalimidyl:
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
12
O N O
OO 0;~,/N\GO
succinimidyl maleimidyl phthalimidyl
Ureido: -N(R')CONR2 R3 wherein R2 and R3 are independently amino substituents,
as
defined for amino groups, and RI is a ureido substituent, for example,
hydrogen, a
Ci-7alkyl group, a C3-20heterocyclyl group, or a Cs-2oaryl group, preferably
hydrogen or a
C1-7alkyl group. Examples of ureido groups include, but are not limited to, -
NHCONH2, -
NHCONHMe, -NHCONHEt, -NHCONMe2, -NHCONEt2, -NMeCONH2, -NMeCONHMe,
-NMeCONHEt, -NMeCONMe2, -NMeCONEt2 and -NHC(=0)NHPh.
Acyloxy (reverse ester): -OC(=O)R, wherein R is an acyloxy substituent, for
example, a
Cry-7 alkyl group, a C3-2o heterocyclyl group, or a C5-20 aryl group,
preferably a CI_7 alkyl
group. Examples of acyloxy groups include, but are not limited to, -OC(=O)CH3
(acetoxy),
-OC(=O)CH2CH3, -OC(=O)C(CH3)3, -OC(=O)Ph, -OC(=O)C6H4F, and -OC(=0)CH2Ph.
Thiol : -SH.
Thioether (sulfide): -SR, wherein R is a thioether substituent, for example, a
Ci-7 alkyl
group (also referred to as a Cl_7 alkylthio group), a C3-2o heterocyclyl
group, or a C5-20 aryl
group, preferably a Cl-7 alkyl group. Examples of CI-7 alkylthio groups
include, but are not
limited to, -SCH3 and -SCH2CH3.
Sulfoxide (sulfinyl): -S(=O)R, wherein R is a sulfoxide substituent, for
example, a Cti-7 alkyl
group, a C3-20 heterocyclyl group, or a C5-20 aryl group, preferably a C1-7
alkyl group.
Examples of sulfoxide groups include, but are not limited to, -S(=0)CH3 and
-S(=O)CH2CH3.
Sulfonyl (sulfone): -S(=O)2R, wherein R is a sulfone substituent, for example,
a CI-7 alkyl
group, a C3-20 heterocyclyl group, or a C5-2o aryl group, preferably a C1-7
alkyl group.
Examples of sulfone groups include, but are not limited to, -S(=0)ZCH3
(methanesulfonyl,
mesyl), -S(=O)2CF3, -S(=O)2CH2CH3, and 4-methylphenylsulfonyl (tosyl).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
13
Thioamido (thiocarbamyl): -C(=S)NR'R2, wherein R' and R2 are independently
amino
substituents, as defined for amino groups. Examples of amido groups include,
but are not
limited to, -C(=S)NH2, -C(=S)NHCH3, -C(=S)N(CH3)2, and -C(=S)NHCH2CH3.
Sulfonamino: -NR'S(=O)2R, wherein R' is an amino substituent, as defined for
amino
groups, and R is a sulfonamino substituent, for example, a CI_7alkyl group, a
C3_
20heterocyclyl group, or a C5_20aryl group, preferably a CI_7alkyl group.
Examples of
sulfonamino groups include, but are not limited to, -NHS(=O)2CH3, -NHS(=O)2Ph
and
-N(CH3)S(=O)2C6H5.
As mentioned above, the groups that form the above listed substituent groups,
e.g. CI_7
alkyl, C3_20 heterocyclyl and C5_20 aryl, may themselves be substituted. Thus,
the above
definitions cover substituent groups which are substituted.
Further Preferences
The following preferences can apply to each aspect of the present invention,
where
applicable. The preferences for each group may be combined with those for any
or all of
the other groups, as appropriate.
X', X2, X3 and X''
Preferably one of X', X2 and X4 (where present) is N, and more preferably one
of Xl and
X2 is N. It is most preferred that X' is N.
R"' and RN2
R"' and RN2, together with the nitrogen atom to which they are attached,
preferably form a
nitrogen-containing heterocyclic ring having from 5 to 7 ring atoms. Preferred
optionally
substituted groups include, but are not limited, to morpholino,
thiomorpholino, piperadinyl,
piperazinyl (preferably N-substituted), homopiperazinyl (preferably N-
substituted) and
pyrrolidinyl. An additional preferred optionally substituted group is
oxazepanyl.
Preferred N-substituents for the piperazinyl and homopiperazinyl groups
include esters, in
particular, esters bearing a Cl_7 alkyl group as an ester substituent, e.g. -
C(=0)OCH3,
-C(=O)OCH2CH3 and -C(=O)OC(CH3)3.
More preferred groups are morpholino and pyrrolidinyl, with morpholino being
the most
preferred. These groups are preferably unsubstituted. In some embodiments,
they may
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
14
be substituted by one or more Ci 4 alkyl groups (e.g. methyl). A preferred
group may be
(3-methyl-morpholin-4-yl).
R'3 and RN4
RN3 and RN4 preferably, together with the nitrogen atom to which they are
attached, form a
nitrogen-containing heterocyclic ring having from 5 to 7 ring atoms. Preferred
optionally
substituted groups include, but are not limited, to morpholino,
thiomorpholino, piperadinyl,
piperazinyl (preferably N-substituted), homopiperazinyl (preferably N-
substituted) and
pyrrolidinyl.
Preferred substituents for the groups include Cl_7 alkyl (e.g. methyl), amido
(e.g.
-C(=O)NH2), hydroxy, ether, amino and esters, of which methyl, -C(=0)NH2 and
hydroxy
are more preferred. The groups may bear 1, 2 or more substituents and these
substituents may be in any position.
Preferred N-substituents for the piperazinyl and homopiperazinyl groups
include esters, in
particular, esters bearing a CI_7 alkyl group as an ester substituent, e.g. -
C(=O)OCH3,
-C(=O)OCH2CH3 and -C(=O)OC(CH3)3.
More preferred groups are morpholino (e.g. 3, 5-dimethyl-morpholino) and
piperadinyl
(e.g. 4-amido-piperadinyl, 2-methyl-piperadinyl, 4-hydroxy-piperadinyl).
A particularly preferred set of groups are those defined by formula III:
N'~Rl Ill
~O
wherein R' is either:
(i) NRN5RN6, where R"5 and R"6 are independently selected from H, optionally
substituted
Cl_, alkyl, optionafiy substituted C3_20 heterocyclyl and optionaily
substituted C5_20 aryl, or
together with the nitrogen atom to which they are attached form a nitrogen-
containing
heterocyclic ring having from 4 to 8 ring atoms; or(ii) OR ', where RO1 is
selected from the
group consisting of optionally substituted CI_7 alkyl, optionally substituted
C3_20 heterocyclyl
and optionally substituted C5_20 aryl.
RN5 and RN6 may have the same preferences as RN3 and RN4, except for being
another
group of formula II.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
RO1 is preferably selected from optionally substituted C5_20 aryl.
Particularly preferred compounds are shown in the examples. Other compounds of
interest may include:
C:)
(N) 5 NR
where R is selected from:
*~~NH *~N ~ / *~ ~, ~
NH ~NH
N~ ' * N~ H*-N ' O
/ H *~
N
N \ I N O N~
~NN ~NH II I
N
Includes Other Forms
Included in the above are the well known ionic, salt, solvate, and protected
forms of these
10 substituents. For example, a reference to carboxylic acid (-COOH) also
includes the
anionic (carboxylate) form (-COO"), a salt or solvate thereof, as well as
conventional
protected forms. Similarly, a reference to an amino group includes the
protonated form
(-N+HR'R2), a salt or solvate of the amino group, for example, a hydrochloride
salt, as well
as conventional protected forms of an amino group. Similarly, a reference to a
hydroxyl
15 group also includes the anionic form (-O"), a salt or solvate thereof, as
well as
conventional protected forms of a hydroxyl group.
Isomers, Salts, Solvates, Protected Forms, and Prodrugs
Certain compounds may exist in one or more particular geometric, optical,
enantiomeric,
diasteriomeric, epimeric, stereoisomeric, tautomeric, conformational, or
anomeric forms,
including but not limited to, cis- and trans-forms; E- and Z-forms; c-, t-,
and r-forms; endo-
and exo-forms; R-, S-, and meso-forms; D- and L-forms; d- and /-forms; (+) and
(-) forms;
keto-, enol-, and enolate-forms; syn- and anti-forms; synclinal- and
anticlinal-forms; a- and
(3-forms; axial and equatorial forms; boat-, chair-, twist-, envelope-, and
halfchair-forms;
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
16
and combinations thereof, hereinafter collectively referred to as "isomers"
(or "isomeric
forms").
If the compound is in crystalline form, it may exist in a number of different
polymorphic
forms.
Note that, except as discussed below for tautomeric forms, specifically
excluded from the
term "isomers", as used herein, are structural (or constitutional) isomers
(i.e. isomers
which differ in the connections between atoms rather than merely by the
position of atoms
in space). For example, a reference to a methoxy group, -OCH3i is not to be
construed as
a reference to its structural isomer, a hydroxymethyl group, -CH2OH.
Similarly, a
reference to ortho-chlorophenyl is not to be construed as a reference to its
structural
isomer, meta-chlorophenyl. However, a reference to a class of structures may
well
include structurally isomeric forms falling within that class (e.g., Ci_7
alkyl includes n-propyl
and iso-propyl; butyl includes n-, iso-, sec-, and tert-butyl; methoxyphenyl
includes ortho-,
meta-, and para-methoxyphenyl).
The above exclusion does not pertain to tautomeric forms, for example, keto-,
enol-, and
enolate-forms, as in, for example, the following tautomeric pairs: keto/enol,
imine/enamine, amide/imino alcohol, amidine/amidine, nitroso/oxime,
thioketone/enethiol,
N-nitroso/hyroxyazo, and nitro/aci-nitro.
Note that specifically included in the term "isomer" are compounds with one or
more
isotopic substitutions. For example, H may be in any isotopic form, including
'H, 2H (D),
and 3H (T); C may be in any isotopic form, including 12C, 13C, and 14C; 0 may
be in any
isotopic form, including160 and'80; and the like.
Unless otherwise specified, a reference to a particular compound includes all
such
isomeric forms, including (wholly or partially) racemic and other mixtures
thereof.
Methods for the preparation (e.g. asymmetric synthesis) and separation (e.g.
fractional
crystallisation and chromatographic means) of such isomeric forms are either
known in
the art or are readily obtained by adapting the methods taught herein, or
known methods,
in a known manner.
Unless otherwise specified, a reference to a particular compound also includes
ionic, salt,
solvate, and protected forms of thereof, for example, as discussed below, as
well as its
different polymorphic forms.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
17
It may be convenient or desirable to prepare, purify, and/or handle a
corresponding salt of
the active compound, for example, a pharmaceutically-acceptable salt. Examples
of
pharmaceutically acceptable salts are discussed in ref. 25.
For example, if the compound is anionic, or has a functional group which may
be anionic
(e.g., -COOH may be -COO"), then a salt may be formed with a suitable cation.
Examples
of suitable inorganic cations include, but are not limited to, alkali metal
ions such as Na+
and K+, alkaline earth cations such as Ca2+ and Mg2+ , and other cations such
as AI3+.
Examples of suitable organic cations include, but are not limited to, ammonium
ion (i.e.,
NH4+) and substituted ammonium ions (e.g., NH3R+, NHZRZ+, NHR3+, NR4+).
Examples of
some suitable substituted ammonium ions are those derived from: ethylamine,
diethylamine, dicyclohexylamine, triethylamine, butyiamine, ethylenediamine,
ethanolamine, diethanolamine, piperazine, benzylamine, phenylbenzylamine,
choline,
meglumine, and tromethamine, as well as amino acids, such as lysine and
arginine. An
example of a common quaternary ammonium ion is N(CH3)4+.
If the compound is cationic, or has a functional group which may be cationic
(e.g., -NH2
may be -NH3}), then a salt may be formed with a suitable anion. Examples of
suitable
inorganic anions include, but are not limited to, those derived from the
following inorganic
acids: hydrochloric, hydrobromic, hydroiodic, sulfuric, sulfurous, nitric,
nitrous, phosphoric,
and phosphorous. Examples of suitable organic anions include, but are not
limited to,
those derived from the following organic acids: acetic, propionic, succinic,
gycolic, stearic,
palmitic, lactic, malic, pamoic, tartaric, citric, gluconic, ascorbic, maleic,
hydroxymaleic,
phenylacetic, glutamic, aspartic, benzoic, cinnamic, pyruvic, salicyclic,
sulfanilic,
2-acetyoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethanesulfonic,
ethane
disulfonic, oxalic, isethionic, valeric, and gluconic. Examples of suitable
polymeric anions
include, but are not limited to, those derived from the following polymeric
acids: tannic
acid, carboxymethyl cellulose.
It may be convenient or desirable to prepare, purify, and/or handle a
corresponding
solvate of the active compound. The term "solvate" is used herein in the
conventional
sense to refer to a complex of solute (e.g. active compound, salt of active
compound) and
solvent. If the solvent is water, the solvate may be conveniently referred to
as a hydrate,
for example, a mono-hydrate, a di-hydrate, a tri-hydrate, etc.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
18
It may be convenient or desirable to prepare, purify, and/or handle the active
compound in
a chemically protected form. The term "chemically protected form," as used
herein,
pertains to a compound in which one or more reactive functional groups are
protected
from undesirable chemical reactions, that is, are in the form of a protected
or protecting
group (also known as a masked or masking group or a blocked or blocking
group). By
protecting a reactive functional group, reactions involving other unprotected
reactive
functional groups can be performed, without affecting the protected group; the
protecting
group may be removed, usually in a subsequent step, without substantially
affecting the
remainder of the molecule. See, for example, ref. 26.
For example, a hydroxy group may be protected as an ether (-OR) or an ester
(-OC(=0)R), for example, as: a t-butyl ether; a benzyl, benzhydryl
(diphenylmethyl), or
trityl (triphenylmethyl) ether; a trimethylsilyl or t-butyldimethylsilyl
ether; or an acetyl ester
(-OC(=O)CH3, -OAc).
For example, an aldehyde or ketone group may be protected as an acetal or
ketal,
respectively, in which the carbonyl group (>C=0) is converted to a diether
(>C(OR)2), by
reaction with, for example, a primary alcohol. The aidehyde or ketone group is
readily
regenerated by hydrolysis using a large excess of water in the presence of
acid.
For example, an amine group may be protected, for example, as an amide or a
urethane,
for example, as: a methyl amide (-NHCO-CH3); a benzyloxy amide (-NHCO-
OCH2C6H5, -
NH-Cbz); as a t-butoxy amide (-NHCO-OC(CH3)3, -NH-Boc); a 2-biphenyl-2-propoxy
amide (-NHCO-OC(CH3)2C6H4C6H5, -NH-Bpoc), as a 9-fluorenylmethoxy amide (-NH-
Fmoc), as a 6-nitroveratryloxy amide (-NH-Nvoc), as a 2-trimethylsilylethyloxy
amide (-
NH-Teoc), as a 2,2,2-trichloroethyloxy amide (-NH-Troc), as an allyloxy amide
(-NH-Alloc), as a 2(-phenylsulphonyl)ethyloxy amide (-NH-Psec); or, in
suitable cases, as
an N-oxide (>NO = ).
For example, a carboxylic acid group may be protected as an ester for example,
as: an
CI_7 alkyl ester (e.g. a methyl ester; a t-butyl ester); a Cl_7 haloalkyl
ester (e.g. a Cl_7
trihaloalkyl ester); a triCj_7 alkylsilyl-Cl_7 alkyl ester; or a C5_2o aryl-
CI_7 alkyl ester (e.g. a
benzyl ester; a nitrobenzyl ester); or as an amide, for example, as a methyl
amide.
For example, a thiol group may be protected as a thioether (-SR), for example,
as: a
benzyl thioether; an acetamidomethyl ether (-S-CH2NHC(=0)CH3).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
19
It may be convenient or desirable to prepare, purify, and/or handle the active
compound in
the form of a prodrug. The term "prodrug", as used herein, pertains to a
compound which,
when metabolised (e.g. in vivo), yields the desired active compound.
Typically, the
prodrug is inactive, or less active than the active compound, but may provide
advantageous handling, administration, or metabolic properties.
For example, some prodrugs are esters of the active compound (e.g. a
physiologically
acceptable metabolically labile ester). During metabolism, the ester group (-
C(=0)OR) is
cleaved to yield the active drug. Such esters may be formed by esterification,
for
example, of any of the carboxylic acid groups (-C(=O)OH) in the parent
compound, with,
where appropriate, prior protection of any other reactive groups present in
the parent
compound, followed by deprotection if required. Examples of such metabolically
labile
esters include those wherein R is C1_2o alkyl (e.g. -Me, -Et); Cl_7 aminoalkyl
(e.g.
aminoethyl; 2-(N,N-diethylamino)ethyl; 2-(4-morpholino)ethyl); and acyloxy-
C1_7 alkyl (e.g.
acyloxymethyl; acyloxyethyl; e.g. pivaloyloxymethyl; acetoxymethyl; 1-
acetoxyethyl; 1-(1-
methoxy-l-methyl)ethyl-carbonxyloxyethyl; 1-(benzoyloxy)ethyl; isopropoxy-
carbonyloxymethyl; 1-isopropoxy-carbonyloxyethyl; cyclohexyl-
carbonyloxymethyl;
1-cyclohexyl-carbonyloxyethyl; cyclohexyloxy-carbonyloxymethyl; 1-
cyclohexyloxy-
carbony4oxyethyl; (4-tetrahydropyranyloxy) carbonyloxymethyl; 1-(4-
tetrahydropyranyloxy)carbonyloxyethyl;
(4-tetrahydropyranyl)carbonyloxymethyl; and 1-(4-
tetrahydropyranyl)carbonyloxyethyl).
Further suitable prodrug forms include phosphonate and glycolate salts. In
particular,
hydroxy groups (-OH), can be made into phosphonate prodrugs by reaction with
chlorodibenzylphosphite, followed by hydrogenation, to form a phosphonate
group -O-
P(=O)(OH)a. Such a group can be cleared by phosphotase enzymes during
metabolism
to yield the active drug with the hydroxy group.
Also, some prodrugs are activated enzymatically to yield the active compound,
or a
compound which, upon further chemical reaction, yields the active compound.
For
example, the prodrug may be a sugar derivative or other glycoside conjugate,
or may be
an amino acid ester derivative.
Acronyms
For convenience, many chemical moieties are represented using well known
abbreviations, including but not limited to, methyl (Me), ethyl (Et), n-propyl
(nPr), iso-
propyl (iPr), n-butyl (nBu), tert-butyl (tBu), n-hexyl (nHex), cyclohexyl
(cHex), phenyl (Ph),
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
biphenyl (biPh), benzyl (Bn), naphthyl (naph), methoxy (MeO), ethoxy (EtO),
benzoyl (Bz),
and acetyl (Ac).
For convenience, many chemical compounds are represented using well known
5 abbreviations, including but not limited to, methanol (MeOH), ethanol
(EtOH), iso-propanol
(i-PrOH), methyl ethyl ketone (MEK), ether or diethyl ether (Et20), acetic
acid (AcOH),
dichloromethane (methylene chloride, DCM), trifluoroacetic acid (TFA),
dimethylformamide (DMF), tetrahydrofuran (THF), and dimethylsulfoxide (DMSO).
10 General Synthesis
O O CI R2
41
X11 a
ZO + X N -~ XII ~ N
2 ~ ~ 2 . ~
X~X' NH2 X'X'p H X~X' N CI X' Xi N~RI
2 3 4
Compounds of formulae I and II can be represented by Formula 1:
R2
a
XII X \ N Formula 1
2 ~
X \X~ N RI
15 wherein in compounds of formula I, X4 = CH, R' represents NRN3R"a and R2
represents
NR"'R"Z. Compounds of Formula 1 can be synthesized from compounds of Formula
2:
CI
4
3 ~
I ~ Formula 2
.
X' N CI
by reaction with HNRN'R"2 (HR2) followed by reaction with HNRN3RNa (HR').
20 Compounds of Formula 2 can be synthesised from compounds of Formula 3:
0
a
3,1 X~I K+
Formula 3
X~X' . N N-o
H
by treatment with POCI3 and N,N-diiospropylamine, for example.
Compounds of Formula 3 can be synthesized from compounds of Formula 4:
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
21
0
4
X~) ~ OH
z Formula 4
~
X' NH2
by treatment with potassium cyanate and ammonium chloride, for example.
Use
The present invention provides active compounds, specifically, active in
inhibiting the
activity of mTOR.
The term "active" as used herein, pertains to compounds which are capable of
inhibiting
mTOR activity, and specifically includes both compounds with intrinsic
activity (drugs) as
well as prodrugs of such compounds, which prodrugs may themselves exhibit
little or no
intrinsic activity.
One assay which may conveniently be used in order to assess the mTOR
inhibition
offered by a particular compound is described in the examples below.
The present invention further provides a method of inhibiting the activity of
mTOR in a cell,
comprising contacting said cell with an effective amount of an active
compound,
preferably in the form of a pharmaceutically acceptable composition. Such a
method may
be practised in vitro or in vivo.
For example, a sample of cells may be grown in vitro and an active compound
brought
into contact with said cells, and the effect of the compound on those cells
observed. As
examples of "effect", the inhibition of cellular growth in a certain time or
the accumulation
of cells in the G1 phase of the cell cycle over a certain time may be
determined. Where
the active compound is found to exert an influence on the cells, this may be
used as a
prognostic or diagnostic marker of the efficacy of the compound in methods of
treating a
patient carrying cells of the same cellular type.
The term "treatment", as used herein in the context of treating a condition,
pertains
generally to treatment and therapy, whether of a human or an animal (e.g. in
veterinary
applications), in which some desired therapeutic effect is achieved, for
example, the
inhibition of the progress of the condition, and includes a reduction in the
rate of progress,
a halt in the rate of progress, amelioration of the condition, and cure of the
condition.
Treatment as a prophylactic measure (i.e. prophylaxis) is also included.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
22
The term "adjunct" as used herein relates to the use of active compounds in
conjunction
with known therapeutic means. Such means include cytotoxic regimes of drugs
and/or
ionising radiation as used in the treatment of different cancer types.
Examples of adjunct
anti-cancer agents that could be combined with compounds from the invention
include,
but are not limited to, the following: alkylating agents: nitrogen mustards,
mechlorethamine, cyclophosphamide, ifosfamide, melphaian, chlorambucil:
Nitrosoureas:
carmustine (BCNU), lomustine (CCNU), semustine (methyl-CCNU),
ethylenimine/methylmelamine, thriethylenemelamine (TEM), triethylene
thiophosphoramide (thiotepa), hexamethylmelamine (HMM, altretamine): Alkyl
sufonates;
busulfan; Triazines, dacarbazine (DTIC): Antimetabolites; folic acid analogs,
methotrexate, trimetrexate, pyrimidine analogs, 5-fluorouracil,
fluorodeoxyuridine,
gemcitabine, cytosine arabinoside (AraC, cytarabine), 5-azacytidine, 2,2'-
difluorodeoxycytidine: Purine analogs; 6-mercaptopurine, 6-thioguanine,
azathioprine, 2'-
deoxycoformycin (pentostatin, erythrohydroxynonyladenine (EHNA), fludarabine
phosphate, 2-Chlorodeoxyadenosine (cladribine, 2-CdA): Topoisomerase I
inhibitors;
camptothecin, topotecan, irinotecan, rubitecan: Natural products; antimitotic
drugs,
paclitaxel, vinca alkaloids, vinblastine (VLB), vincristine, vinorelbine,
TaxotereTM
(docetaxel), estramustine, estramustine phosphate; epipodophylotoxins,
etoposide,
teniposide: Antibiotics; actimomycin D, daunomycin (rubidomycin), doxorubicin
(adriamycin), mitoxantrone, idarubicin, bleomycins, plicamycin (mithramycin),
mitomycin
C, dactinomycin: Enzymes; L-asparaginase, RNAse A: Biological response
modifiers;
interferon-alpha, IL-2, G-CSF, GM-CSF: Differentiation Agents; retinoic acid
derivatives:
Radiosensitizers;, metronidazole, misonidazole, desmethylmisonidazole,
pimonidazole,
etanidazole, nimorazole, RSU 1069, E09, RB 6145, SR4233, nicotinamide, 5-
bromodeozyuridine, 5-iododeoxyuridine, bromodeoxycytidine: Platinium
coordination
complexes; cisplatin, carboplatin: Anthracenedione; mitoxantrone, AQ4N
Substituted
urea, hydroxyurea; Methylhydrazine derivatives, N-methylhydrazine (MIH),
procarbazine;
Adrenocortical suppressant, mitotane (o.p' DDD), aminoglutethimide: Cytokines;
interferon (a, (3, y), interleukin; Hormones and antagonists;
adrenocorticosteroids/antagonists, prednisone and equivalents, dexamethasone,
aminoglutethimide; Progestins, hydroxyprogesterone caproate,
medroxyprogesterone
acetate, megestrol acetate; Estrogens, diethylstilbestrol, ethynyl
estradiol/equivalents;
Antiestrogen, tamoxifen; Androgens, testosterone propionate,
fluoxymesterone/equivalents; Antiandrogens, flutamide, gonadotropin-releasing
hormone
analogs, leuprolide; Nonsteroidal antiandrogens, flutamide; EGFR inhibitors,
VEGF
inhibitors; Proteasome inhibitors.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
23
Active compounds may also be used as cell culture additives to inhibit mTOR,
for
example, in order to sensitize cells to known chemotherapeutic agents or
ionising
radiation treatments in vitro.
Active compounds may also be used as part of an in vitro assay, for example,
in order to
determine whether a candidate host is likely to benefit from treatment with
the compound
in question.
Cancer
The present invention provides active compounds which are anticancer agents or
adjuncts
for treating cancer. One of ordinary skill in the art is readily able to
determine whether or
not a candidate compound treats a cancerous condition for any particular cell
type, either
alone or in combination.
Examples of cancers include, but are not limited to, lung cancer, small cell
lung cancer,
gastrointestinal cancer, bowel cancer, colon cancer, breast carinoma, ovarian
carcinoma,
prostate cancer, testicular cancer, liver cancer, kidney cancer, bladder
cancer, pancreas
cancer, brain cancer, sarcoma, osteosarcoma, Kaposi's sarcoma, melanoma and
leukemias.
Any type of cell may be treated, including but not limited to, lung,
gastrointestinal
(including, e.g., bowel, colon), breast (mammary), ovarian, prostate, liver
(hepatic), kidney
(renal), bladder, pancreas, brain, and skin.
Administration
The active compound or pharmaceutical composition comprising the active
compound
may be administered to a subject by any convenient route of administration,
whether
systemically/ peripherally or at the site of desired action, including but not
limited to, oral
(e.g. by ingestion); topical (including e.g. transdermal, intranasal, ocular,
buccal, and
sublingual); pulmonary (e.g. by inhalation or insufflation therapy using, e.g.
an aerosol,
e.g. through mouth or nose); rectal; vaginal; parenteral, for example, by
injection,
including subcutaneous, intradermal, intramuscular, intravenous,
intraarterial, intracardiac,
intrathecal, intraspinal, intracapsular, subcapsular, intraorbital,
intraperitoneal,
intratracheal, subcuticular, intraarticular, subarachnoid, and intrasternal;
by implant of a
depot, for example, subcutaneously or intramuscularly.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
24
The subject may be a eukaryote, an animal, a vertebrate animal, a mammal, a
rodent
(e.g. a guinea pig, a hamster, a rat, a mouse), murine (e.g. a mouse), canine
(e.g. a dog),
feline (e.g. a cat), equine (e.g. a horse), a primate, simian (e.g. a monkey
or ape), a
monkey (e.g. marmoset, baboon), an ape (e.g. gorilla, chimpanzee, orangutang,
gibbon),
or a human.
Formulations
While it is possible for the active compound to be administered alone, it is
preferable to
present it as a pharmaceutical composition (e.g., formulation) comprising at
least one
active compound, as defined above, together with one or more pharmaceutically
acceptable carriers, adjuvants, excipients, diluents, fillers, buffers,
stabilisers,
preservatives, lubricants, or other materials well known to those skilled in
the art and
optionally other therapeutic or prophylactic agents.
Thus, the present invention further provides pharmaceutical compositions, as
defined
above, and methods of making a pharmaceutical composition comprising admixing
at
least one active compound, as defined above, together with one or more
pharmaceutically
acceptable carriers, excipients, buffers, adjuvants, stabilisers, or other
materials, as
described herein.
The term "pharmaceutically acceptable" as used herein pertains to compounds,
materials,
compositions, and/or dosage forms which are, within the scope of sound medical
judgement, suitable for use in contact with the tissues of a subject (e.g.
human) without
excessive toxicity, irritation, allergic response, or other problem or
complication,
commensurate with a reasonable benefit/risk ratio. Each carrier, excipient,
etc. must also
be "acceptable" in the sense of being compatible with the other ingredients of
the
formulation.
Suitable carriers, diluents, excipients, etc. can be found in standard
pharmaceutical texts.
See, for example, refs. 27 to 29.
The formulations may conveniently be presented in unit dosage form and may be
prepared by any methods well known in the art of pharmacy. Such methods
include the
step of bringing into association the active compound with the carrier which
constitutes
one or more accessory ingredients. In general, the formulations are prepared
by uniformly
and intimately bringing into association the active compound with liquid
carriers or finely
divided solid carriers or both, and then if necessary shaping the product.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
Formulations may be in the form of liquids, solutions, suspensions, emulsions,
elixirs,
syrups, tablets, losenges, granules, powders, capsules, cachets, pills,
ampoules,
suppositories, pessaries, ointments, gels, pastes, creams, sprays, mists,
foams, lotions,
5 oils, boluses, electuaries, or aerosols.
Formulations suitable for oral administration (e.g., by ingestion) may be
presented as
discrete units such as capsules, cachets or tablets, each containing a
predetermined
amount of the active compound; as a powder or granules; as a solution or
suspension in
10 an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or
a water-in-oil
liquid emulsion; as a bolus; as an electuary; or as a paste.
A tablet may be made by conventional means, e.g. compression or molding,
optionally
with one or more accessory ingredients. Compressed tablets may be prepared by
15 compressing in a suitable machine the active compound in a free-flowing
form such as a
powder or granules, optionally mixed with one or more binders (e.g. povidone,
gelatin,
acacia, sorbitol, tragacanth, hydroxypropylmethyl cellulose); fillers or
diluents (e.g.
lactose, microcrystalline cellulose, calcium hydrogen phosphate); lubricants
(e.g.
magnesium stearate, talc, silica); disintegrants (e.g. sodium starch
glycolate, cross-linked
20 povidone, cross-linked sodium carboxymethyl cellulose); surface-active or
dispersing or
wetting agents (e.g., sodium lauryl sulfate); and preservatives (e.g., methyl
p-hydroxybenzoate, propyl p-hydroxybenzoate, sorbic acid). Molded tablets may
be made
by molding in a suitable machine a mixture of the powdered compound moistened
with an
inert liquid diluent. The tablets may optionally be coated or scored and may
be formulated
25 so as to provide slow or controlled release of the active compound therein
using, for
example, hydroxypropylmethyl cellulose in varying proportions to provide the
desired
release profile. Tablets may optionally be provided with an enteric coating,
to provide
release in parts of the gut other than the stomach.
Formulations suitable for topical administration (e.g. transdermal,
intranasal, ocular,
buccal, and sublingual) may be formulated as an ointment, cream, suspension,
lotion,
powder, solution, past, gel, spray, aerosol, or oil. Alternatively, a
formulation may
comprise a patch or a dressing such as a bandage or adhesive plaster
impregnated with
active compounds and optionally one or more excipients or diluents.
Formulations suitable for topical administration in the mouth include losenges
comprising
the active compound in a flavored basis, usually sucrose and acacia or
tragacanth;
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
26
pastilles comprising the active compound in an inert basis such as gelatin and
glycerin, or
sucrose and acacia; and mouthwashes comprising the active compound in a
suitable
liquid carrier.
Formulations suitable for topical administration to the eye also include eye
drops wherein
the active compound is dissolved or suspended in a suitable carrier,
especially an
aqueous solvent for the active compound.
Formulations suitable for nasal administration, wherein the carrier is a
solid, include a
coarse powder having a particle size, for example, in the range of about 20 to
about 500
microns which is administered in the manner in which snuff is taken, i.e. by
rapid
inhalation through the nasal passage from a container of the powder held close
up to the
nose. Suitable formulations wherein the carrier is a liquid for administration
as, for
example, nasal spray, nasal drops, or by aerosol administration by nebuliser,
include
aqueous or oily solutions of the active compound.
Formulations suitable for administration by inhalation include those presented
as an
aerosol spray from a pressurised pack, with the use of a suitable propellant,
such as
dichlorodifluoromethane, trichlorofluoromethane, dichoro-tetrafluoroethane,
carbon
dioxide, or other suitable gases.
Formulations suitable for topical administration via the skin include
ointments, creams,
and emulsions. When formulated in an ointment, the active compound may
optionally be
employed with either a paraffinic or a water-miscible ointment base.
Alternatively, the
active compounds may be formulated in a cream with an oil-in-water cream base.
If
desired, the aqueous phase of the cream base may include, for example, at
least about
30% w/w of a polyhydric alcohol, i.e., an alcohol having two or more hydroxyl
groups such
as propylene glycol, butane-1,3-diol, mannitol, sorbitol, glycerol and
polyethylene glycol
and mixtures thereof. The topical formulations may desirably include a
compound which
enhances absorption or penetration of the active compound through the skin or
other
affected areas. Examples of such dermal penetration enhancers include
dimethylsulfoxide and related analogues.
When formulated as a topical emulsion, the oily phase may optionally comprise
merely an
emulsifier (otherwise known as an emulgent), or it may comprises a mixture of
at least
one emulsifier with a fat or an oil or with both a fat and an oil. Preferably,
a hydrophilic
emulsifier is included together with a lipophilic emulsifier which acts as a
stabiliser. It is
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
27
also preferred to include both an oil and a fat. Together, the emulsifier(s)
with or without
stabiliser(s) make up the so-called emulsifying wax, and the wax together with
the oil
and/or fat make up the so-called emulsifying ointment base which forms the
oily dispersed
phase of the cream formulations.
Suitable emulgents and emulsion stabilisers include Tween 60, Span 80,
cetostearyl
alcohol, myristyl alcohol, glyceryl monostearate and sodium lauryl sulphate.
The choice of
suitable oils or fats for the formulation is based on achieving the desired
cosmetic
properties, since the solubility of the active compound in most oils likely to
be used in
pharmaceutical emulsion formulations may be very low. Thus the cream should
preferably be a non-greasy, non-staining and washable product with suitable
consistency
to avoid leakage from tubes or other containers. Straight or branched chain,
mono- or
dibasic alkyl esters such as di-isoadipate, isocetyl stearate, propylene
glycol diester of
coconut fatty acids, isopropyl myristate, decyl oleate, isopropyl paimitate,
butyl stearate,
2-ethylhexyl paimitate or a blend of branched chain esters known as Crodamol
CAP may
be used, the last three being preferred esters. These may be used alone or in
combination depending on the properties required. Alternatively, high melting
point lipids
such as white soft paraffin and/or liquid paraffin or other mineral oils can
be used.
Formulations suitable for rectal administration may be presented as a
suppository with a
suitable base comprising, for example, cocoa butter or a salicylate.
Formulations suitable for vaginal administration may be presented as
pessaries, tampons,
creams, gels, pastes, foams or spray formulations containing in addition to
the active
compound, such carriers as are known in the art to be appropriate.
Formulations suitable for parenteral administration (e.g., by injection,
including cutaneous,
subcutaneous, intramuscular, intravenous and intradermal), include aqueous and
non-
aqueous isotonic, pyrogen-free, sterile injection solutions which may contain
anti-oxidants,
buffers, preservatives, stabilisers, bacteriostats, and solutes which render
the formulation
isotonic with the blood of the intended recipient; and aqueous and non-aqueous
sterile
suspensions which may include suspending agents and thickening agents, and
liposomes
or other microparticulate systems which are designed to target the compound to
blood
components or one or more organs. Examples of suitable isotonic vehicles for
use in
such formulations include Sodium Chloride Injection, Ringer's Solution, or
Lactated
Ringer's Injection. Typically, the concentration of the active compound in the
solution is
from about 1 ng/mi to about 10 g/ml, for example from about 10 ng/ml to about
1 g/ml.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
28
The formulations may be presented in unit-dose or multi-dose sealed
containers, for
example, ampoules and vials, and may be stored in a freeze-dried (lyophilised)
condition
requiring only the addition of the sterile liquid carrier, for example water
for injections,
immediately prior to use. Extemporaneous injection solutions and suspensions
may be
prepared from sterile powders, granules, and tablets. Formulations may be in
the form of
liposomes or other microparticulate systems which are designed to target the
active
compound to blood components or one or more organs.
Dosage
It will be appreciated that appropriate dosages of the active compounds, and
compositions
comprising the active compounds, can vary from patient to patient. Determining
the
optimal dosage will generally involve the balancing of the level of
therapeutic benefit
against any risk or deleterious side effects of the treatments of the present
invention. The
selected dosage level will depend on a variety of factors including, but not
limited to, the
activity of the particular compound, the route of administration, the time of
administration,
the rate of excretion of the compound, the duration of the treatment, other
drugs,
compounds, and/or materials used in combination, and the age, sex, weight,
condition,
general health, and prior medical history of the patient. The amount of
compound and
route of administration will ultimately be at the discretion of the physician,
although
generally the dosage will be to achieve local concentrations at the site of
action which
achieve the desired effect without causing substantial harmful or deleterious
side-effects.
Administration in vivo can be effected in one dose, continuously or
intermittently (e.g., in
divided doses at appropriate intervals) throughout the course of treatment.
Methods of
determining the most effective means and dosage of administration are well
known to
those of skill in the art and will vary with the formulation used for therapy,
the purpose of
the therapy, the target cell being treated, and the subject being treated.
Single or multiple
administrations can be carried out with the dose level and pattern being
selected by the
treating physician.
In general, a suitable dose of the active compound is in the range of about
100 g to
about 250 mg per kilogram body weight of the subject per day. Where the active
compound is a salt, an ester, prodrug, or the like, the amount administered is
calculated
on the basis of the parent compound and so the actual weight to be used is
increased
proportionately.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
29
Examples
General Experimental Methods
Thin Layer chromatography was carried out using Merck Kieselgel 60 F254 glass
backed
plates. The plates were visualized by the use of a UV lamp (254 nm). Silica
gel 60
(particle sizes 40-63 m) supplied by E.M.Merck was employed for flash
chromatography.
'H NMR spectra were recorded at 300 MHz on a Bruker DPX-300 instrument.
Chemical
shifts were referenced relative to tetramethylsilane.
Purification and identification of library samples
The samples were purified on Gilson LC units. Mobile phase A - 0.1% aqueous
TFA,
mobile phase B - Acetonitrile; flow rate 6 ml/min; Gradient - typically
starting at 90%
A/10% B for 1 minute, rising to 97% after 15 minutes, holding for 2 minutes,
then back to
the starting conditions. Column: Jones Chromatography Genesis 4 m, C18 column,
10
mm x 250 mm. Peak acquisition based on UV detection at 254 nm.
Mass spectra were recorded on a Finnegan LCQ instrument in positive ion mode.
Mobile
phase A - 0.1 % aqueous formic acid. Mobile phase B - Acetonitrile; Flowrate 2
ml/min;
Gradient - starting at 95% A/5% B for 1 minute, rising to 98% B after 5
minutes and
holding for 3 minutes before returning to the starting conditions. Column:
Varies, but
always C18 50 mm x 4.6 mm (currently Genesis C18 4 m. Jones Chromatography).
PDA detection Waters 996, scan range 210-400 nm.
Microwave synthesis
Reactions were carried out using a Personal ChemistryTM Emrys Optimiser
microwave
synthesis unit with robotic arm. Power range between. 0-300 W at 2.45 GHz.
Pressure
range between 0-20 bar; temperature increase between 2-5 C/sec; temp range 60-
250 C.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
Example I
0 0 CI R2
a
XII OH XII N-K+ XII X
X' x N ws~X ~ N
2 ~ -~ Z ~ -~ p ~ ~--
XI NHZ ~Xl H O X~X' N CI X'
Xi N~R~
2 3 4
5 Starting materials:
1 a: X'=N, X2=CH, X3=CH, X4=CH : 2-amino-nicotinic acid
1b: X'=CH, X2=CH, X3=N, X4=CH : 3-amino-isonicotinic acid
1c: X'=CH, X2=CH, X3=CH, X4=N : 3-Amino-pyridine-2-carboxylic acid
10 (i) IH-Pyridopyrimidine-2,4-diones (2)
The appropriate amino acid (1)(1 equivalent), potassium cyanate (5
equivalents) and
ammonium chloride (10 equivalents) were suspended in water. The slurry was
heated
(160 C) and mixed manually for 2 hours as water vapour was expelled from the
reaction
vessel. The reaction temperature was then raised to 200 C for 40 minutes
before being
15 cooled to 90 C whereupon hot water was added and then the mixture was
allowed to cool
to room temperature. The precipitate that formed during cooling was removed by
filtration, washed with water (twice), and diethyl ether (once) before being
dried in a
desiccator to give the desired product in suitably clean form to be used
without further
purification.
20 2a: 1 H-Pyrido[2,3-d]pyrimidine-2,4-dione: mlz (LC-MS, ESP): does not
ionise R/T = 0.76
mins
2b: 1 H-Pyrido[3,4-d]pyrimidine-2,4-dione: m/z (LC-MS, ESP): 164 [M-K+H]+, R/T
= 0.38
mins
2c: 1H-Pyrido[3,2-d]pyrimidine-2,4-dione: m/z (LC-MS, ESP): 164 [M-K+H]+,R/T =
0.45
25 mins
(ii) 2,4-Dichloro-pyridopyrimidines (3)
The appropriate 1 H-pyridopyrimidine-2,4-dione (2)(1 equivalent) was dissolved
in POCI3
(44 equivalents). To this mixture was added N,N-diisopropylamine (2.8
equivalents) in a
30 dropwise fashion. The reaction was stirred at room temperature under an
inert
atmosphere for 5 hours. After this time the mixture was concentrated in vacuo
while taking
care to keep the temperature below 30 C. The resulting black residue was
poured onto
crushed ice. The mixture was extracted with CH2CI2 (x2) and the organic
extracts then
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
31
washed with water, dried (MgSO4), filtered and concentrated in vacuo to
provide a tar like
material that corresponded to the desired product in suitably clean form to be
used without
any further purification.
3a: 2,4-Dichloro-pyrido[2,3-d]pyrimidine: m/z (LC-MS, ESP): 200 [M+H]+ R/T =
3.60 mins
3b: 2,4-Dichloro-pyrido[3,4-d]pyrimidine: m/z (LC-MS, ESP): 200 [M+H]+, R/T =
3.82
mins
3c: 2,4-Dichloro-pyrido[3,2-d]pyrimidine: m/z (LC-MS, ESP): 200 [M+H]+,R/T =
3.80 mins
(iii) Pyridopyrimidine-2,4-diamines (4)
The appropriate 2,4-dichloro-pyridopyrimidine (3)(1 equivalent) was suspended
in CH2CI2
(4 ml of solvent per mmol of material) and to this mixture was added
triethylamine (1
equivalent). The resultant orange solution was then cooled to 0 C and the
appropriate
amine (R2H) (1 equivalent) added dropwise as a 0.1 M solution in CH2CI2 over 5
minutes.
The mixture was stirred for a further 45 minutes before it was diluted with
water and
extracted with CH2CI2 (x2). The organic extracts were dried using MgSO4,
filtered and
concentrated in vacuo to give a crude solid that was purified by flash
chromatography
(Si02) using Hexanes:EtOAc (2:3) as eluent to give the desired product (1
equivalent)
which was diluted in dimethylacetamide (0.7 M) and the appropriate amine (R'H)
(2.5
equivalents) added. The reaction mixture was heated to 60 C for 16 hours. Upon
completion the reaction mixture was submitted for preparative HPLC
purification to give
the desired pyridopyrimidine-2,4-diamines, as detailed below:
X XZ x3 X4 R R 2 Purity m/z RT
% [M+H]+ (mins)
4a N CH CH CH "-N
La (0)
oH N 85 317 2.51
4b N CH CH CH (0)
N 99 314 3.11
4c N CH CH CH
~ (0)
90 330 2.97
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
32
X XZ X3 X R R Purity m/z RT
% [nn+Hl+ (mins)
4d CH N CH CH N o ( ) - - -
NH2 *
4e CH CH CH N
~N' 100 416 3.4
4f N CH CH CH EN) 96.0 314.2 3.92
9 N CH CH CH *-~N (0) 100.0 314.3 3.95
4h N CH CH CH *~N o
CN 100.0 368.2 4.18
CF3
4i N CH CH CH N (0)
N
100.0 390.3 4.60
i I
~
4j N CH CH CH C )
N
100.0 344.3 3.60
OH
4k N CH CH CH *~N JI '
C 100.0 316.2 3.43
N
OH
41 N CH CH CH '-N o C o\
Jl 98.0 358.2 3.70
O'~ N
4m N CH CH CH *~N (0) 98.0 330.2 3.67
N
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
33
X XZ X X R R2 Purity mlz RT
% [M+H]+ (mins)
4n N CH CH CH (0) 91.0 328.3 4.22
N
4o N CH CH CH ~N Ho
99.0 344.3 3.70
(N0)
4p N CH CH CH C'N 91.0 362.2 4.30
4q N CH CH CH N EN) oH 100.0 392.3 4.05
i =
4r N CH CH CH (0)H02cJ91.0 N 344.2 3.70
4s N CH CH CH N (0)
N
100.0 438.2 4.56
~ I .
ci
4t N CH CH CH o
~N
96.0 376.2 4.22
=~ .
N
4u N CH CH CH EN) o
99.0 376.2 4.24
4v N CH CH CH =~N (0) 100.0 314.5 3.16
N
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
34
xl X2 X X4 R R Purity mlz RT
% [M+H]+ (mins)
4w N CH CH CH NHZ N (0)
N 85.0 328.5 3.30
4x N CH CH CH (0)
N 99.0 354.5 3.58
4y N CH CH CH N (oJl \
99.0 300.4 2.98
N
4z N CH CH CH =~N COZEt (0) 85.0 372.4 3.18
N
4aa N CH CH CH 0
N 100.0 314.3 3.90
4ab N CH CH CH o
C)
100.0 328.3 3.94
N
4ac N CH CH CH N--')
(0)."", 100.0 316.0 3.34
4ad N CH CH CH N o
C J..., 100.0 330.1 3.38
~O N
4ae N CH CH CH "-,N~ o
0
~N~'=.
93.3 474.9 7.30
N
O
4af N CH CH CH N co)..,." 100.0 344.1 3.62
=
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
X X X3 X R Rz Purity mlz RT
% [M+Hl + (mins)
4ag N CH CH CH N-')
o
100.0 392.1 4.09
I
4ah N CH CH CH N-')
o N95.0 392.0 7.17
4ai N CH CH CH
(0) 99.0 350.3 3.57
N
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
36
Example 2
(a)
C )....,,
N N
I ~ ~N
~ ~ ~~
R
NN ~ NCN N ~R
(N)-
~O
4aj 5
To a solution of 2-(2-Chloromethyl-morpholin-4-yl)-4-((S)-3-methyl-morpholin-4-
yl)-
pyrido[2,3-d]pyrimidine (4aj)(36 mg, 0.1 mmol) and the appropriate amine (0.5
mmol) in
dimethylacetamide (2.5 ml) was added Nal (3 mg, 0.02 mmol) and K2CO3 (14 mg,
0.1
mmol). The reaction vessel was sealed and heated under the influence of
microwave
radiation (low absorption setting, 200 C, 20 minutes). After this the crude
reaction mixture
was concentrated in vacuo and purified by preparative HPLC to give the desired
compounds (5).
NR'R" Purity m/z RT
% [M+H]+ (mins)
5a
*\N / \ 100.0 461.3 3.91
0
5b H'~~ I~ 99.0 451.4 3.43
5c N
y 87.1 520.6 3.54
OMe
5d '\NLa 96.0 485.5 3.31
COaEt
5e Q
NA S
~ / - 97.0 513.5 -4.81
Br
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
37
5f 0
*\N
95.0 559.3 4.65
Br
5g o
N
0 96.3 490.5 4.05
HZN
5h Q
=\NA 0 96.9 463.5 4.17
5i o
H 99.3 449.4 3.82
5j o
N~ 99.2 413.5 3.54
H
5k o
H 98.3 455.5 3.76
51 0
98.8 485.5 3.97
H aF
F 5m o
H-Y oMe 83.1 431.4 3.48
O
5n ko
N
98.0 463.4 3.84
H
50 0
N
H 100.0 471.5 4.20
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
38
5p o
H \
, 479.4 99.4 3.89
OMe
(b)
O
C J .,..
N N
N
~
(N) N N " -N CI N N" N~\OR
~
4aj 5
To a solution of 2-(2-Chloromethyl-morpholin-4-yl)-4-((S)-3-methyl-morpholin-4-
yl)-
pyrido[2,3-d]pyrimidine (4aj)(36 mg, 0.1 mmol) (11 mg, 0.03 mmol) in anhydrous
dimethylacetamide (0.5 ml) was added tert-BuOK (6.8 mg, 0.6 mmol), and 18-
crown-6
ether (0.006 mmol, 1.6 mg). The appropriate alcohol was then added to the
reaction
mixture and each reaction heated to 110 C for 15 hours. After this the crude
reaction
mixture was concentrated in vacuo and purified by preparative HPLC to give the
desired
compounds (5).
OR"' Purity mlz RT
% [M+Hl+ (mins)
5r o
NHPh 97.0 513.5 4.81
5s
92.0 461.2 4.26
5t OH
' 98.0 492.1 4.18
O
5u / F
~ ~ 78.0 440.1 4.46
0
5v
100.0 422.2 4.40
0
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
39
5w MeO /
*\ 96.0 452.1 4.32
5x
*"o 91.0 437.1 3.69
NH2
5y H2N
/
N -- 93.0 488.1
3.58
O
5z
98.0 473.1 4.17
O N
5aa N
o 90.0 473.1 6.30
Br
5ab YN~N
o 100.0 568.0 4.68
H
5ac ni
*"o yN 78.0 448.1 4.04
CN
5ad C02Me
82.9 480.1 4.39
5ae ~
69.0 480.1 4.43
O COaMe
5af F , ar
*~o '~ I 95.4 436.0 4.85
F
Example 3: Biological Assay
For mTOR enzyme activity assays, mTOR protein was isolated from HeLa cell
cytoplasmic extract by immunoprecipitation, and activity determined
essentially as
described previously using recombinant PHAS-1 as a substrate (ref. 21).
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
All the compounds tested exhibited IC50 values less than 15 pM.
The following compounds exhibited IC50 values less than 1.5 pM: 4c, 4d, 4h,
4n, 4o, 4y,
5 4ab, 4af, 4ag, 4ah, 5e, 5g, 5h, 5k, 5m, 5z.
CA 02599212 2007-08-24
WO 2006/090169 PCT/GB2006/000671
41
Reference List
The following documents are all herein incorporated by reference.
1) Brown, et al., Nature, 369, 756-758 (1994)
2) Chiu, et al., Proc Natl Acad Sci, 91, 12574-12578 (1994)
3) Sabatini, et al., Cell, 78, 35-43, (1994)
4) Sabers, et al., J Biol Chem, 270, 825-822 (1995)
5) Abraham, Curr Opin Immunol, 8, 412-418 (1996)
6) Schmelze and Hall, Cell, 103, 253-262 (2000)
7) Burnett, et al., Proc Natl Acad Sci, 95, 1432-1437 (1998)
8) Terada, et al., Proc Natl Acad Sci, 91,11477-11481 (1994)
9) Jeffries, et al., EMBO J, 16,3693-3704 (1997)
10) Bjornsti and Houghton, Nat Rev Cancer, 4, 335-348 (2004)
11) Gingras, et al., Genes Dev, 13, 1422-1437 (1999)
12) Gingras, et al., Genes Dev, 15, 807-826 (2001)
13) Neuhaus, et al., Liver Transplantation, 7, 473-484 (2001)
14) Woods and Marks, Ann Rev Med, 55, 169-178 (2004)
15) Dahia, Endocrine-Related Cancer, 7, 115-129 (2000)
16) Cristofano and Pandolfi, Cell, 100, 387-390 (2000)
17) Samuels, et al., Science, 304, 554 (2004)
18) Huang and Houghton, CurrOpin Pharmacol, 3, 371-377 (2003)
19) Sawyers, Cancer Cell, 4, 343-348 (2003)
20) Huang and Houghton, Curr Opin in Invest Drugs, 3, 295-304 (2002)
21) Brunn, et al., EMBO J, 15, 5256-5267 (1996)
22) Edinger, et al., Cancer Res, 63, 8451-8460, (2003)
23) Lawrence, et al., Curr Top Microbiol Immunol, 279, 199-213 (2004)
24) Eshleman, et al., Cancer Res, 62, 7291-7297 (2002)
25) Berge, et al., J. Pharm. Sci., 66, 1-19 (1977).
26) Green, T. and Wuts, P., "Protective Groups in Organic Synthesis", 3rd
Edition, John
Wiley and Sons (1999)
27) "Handbook of Pharmaceutical Additives", 2nd Edition (eds. M. Ash and I.
Ash), 2001
(Synapse Information Resources, Inc., Endicott, New York, USA),
28) "Remington's Pharmaceutical Sciences", 20th edition, pub. Lippincott,
Williams &
Wilkins, 2000
29) "Handbook of Pharmaceutical Excipients", 2nd edition, 1994.