Note: Descriptions are shown in the official language in which they were submitted.
CA 02600189 2007-09-06
WO 2006/100026 PCT/EP2006/002557
-1-
90721pct
2-Oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl-piperidines used as
CGRP antagonists
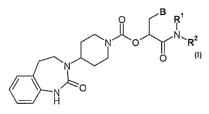
The present invention relates to new CGRP antagonists of general formula I
O B R~
I
~ O N, R2
N O
7 N
NO
H
wherein B, R' and R2 are defined as in claim 1, the tautomers, the isomers,
the
diastereomers, the enantiomers, the hydrates, the mixtures thereof and the
salts
thereof as well as the hydrates of the salts, particularly the physiologically
acceptable salts thereof with inorganic or organic acids or bases,
pharmaceutical
compositions containing these compounds, the use thereof and processes for the
preparation thereof.
BACKGROUND TO THE INVENTION
In International Patent Applications PCT/EP03/11762 and PCT/EP2005/003094
and in US 2005/0215576, CGRP antagonists have already been described for the
treatment of migraine.
DETAILED DESCRIPTION OF THE INVENTION
In the above general formula I in a first embodiment
B denotes a group selected from
CA 02600189 2007-09-06
WO 2006/100026 PCT/EP2006/002557
-2-
CI Br CI Br
OH ~ OH OH OH
CI * I / Br
CH3 CH3 CF3
OH NH2
CH3 CH3 CF3
JaCF3 ~
* * I / CH
and 3,
R' and R2 together with the enclosed nitrogen atom denote a group of general
formula II
R
Y"3
R
N
wherein
Y' denotes the carbon atom or, if R4 denotes a free pair of electrons, it
also denotes the nitrogen atom,
R3 denotes a cyclopentyl, cyclohexyl or cycloheptyl group or
R3 denotes a heterocycle selected from a morpholin-4-yl, 1,1-dioxo-
thiomorholin-4-yi, piperidin-1-yl, piperidin-4-yl, piperazin-1-yl or
pyrrolidin-1-yl group,
wherein the above-mentioned monocyclic heterocycles in the ring may be
mono- or disubstituted by hydroxy, methyl, ethyl, trifluoromethyl,
hydroxymethyl or hydroxyethyl groups, or may optionally additionally be
monosubstituted by a hydroxycyclopropyl, trifluoromethylcarbonylmethyl,
amino, carboxy-carbonyl, methoxycarbonyl, ethoxycarbonyl-carbonyl,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-3-
carboxymethyl, carboxyethyl, ethoxyca rbo nyl m ethyl, ethoxyca rbo nyl ethyl,
carboxy-ethylcarbonyl, ethoxycarbonyl-ethylcarbonyl, aminosulphonyl,
methylaminosulphonyl, dimethylaminosulphonyl, methylsulphonyl,
ethylsulphonyl, isopropylsulphonyl, cyclopropylsulphonyl, (hydroxyamino)-
carbonylmethyl, hydroxy-(methyl)-aminocarbonyl-methyl or
methoxyaminocarbonyl-methyl group, wherein the substituents may be
identical or different and may be bound to a ring carbon or ring nitrogen
atom, or
the above-Ynentioned monocyclic heterocycles in the ring may be
monosubstituted by a carboxy group, if this carboxy group is not bound via
a nitrogen atom, and
R4 denotes the hydrogen atom, if Y' denotes the carbon atom, or
R4 denotes a free pair of electrons, if Y' denotes the nitrogen atom,
the tautomers, the diastereomers, the enantiomers, the hydrates, the mixtures
thereof and the salts thereof as well as the hydrates of the salts,
particularly the
physiologically acceptable salts thereof with inorganic or organic acids or
bases.
The following compounds are mentioned for example as most particularly
preferred compounds of the above general formula I:
No. Structure
/ r OH
1 O ~~&
( ) C ~ ~NAp~N~N_ O
N p ~
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-4-
No. Structure
r
OH
/
/n ~
(2) CQ ~ NAO~0
N4
H O
40H
(3) I 0
N
H O
H
/r O Br
(4) \ (4) I \ ~ ~N~OyN N n
~/
/ O
N
H O
Br
(5)
I J~NIO~
N O
H O
OH
LBr
( np \6) I Vv
N
H O
/ ~
/ \ I Br
(7) ~NJ101~NN~~
/ 101 ~/
N
H O
Br
OH
8 0 -f('~) Br
N_CNxO'yN''//OH
H O O
T'
~~OH
&
(9) NO~N~N~/,OH
~J '~ T
H 0 0 CF,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-5-
No. Structure
Br
OH
Br
(10)
N Oy N\N'\,OH
N'\1\ 0
H O
Br
OH
JI"\~~~&
11 CC N~N O~N % N ~~N,\~OH
0
N
H O
OH
(12) Br
O
H O
~91 OH
(13) \ N~NXO~N~,N
~NHZ
~ O
N
H O
bOH
er
0 ~ OH
N'~ ~NIO~N~N'~
H O
k
15 \ ( ) O
H o
Br
~OH
\~
(16) I N~N x O'N\~N
Q 1( 'OH
0
N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-6-
No. Structure
Br
nOH
0 (17) N-CNx01"YN,~~N OH
~ 0
N
H O
Br
OH
~
o Br
(18) OCN_(JNAOYNDOH
0
.H 0
L ~
(19) Q er\ OH
N
H O
Br
~OH
20 &
~ ) OCNjNONN~OH
N O
H O
&__ Br
~
(21) ~ O
N~NI O = N S
ll ~ O ~'~ N \
N
H O
/ OH
22 R Br
~ ) \ - ~NJIO~N ry ~ NOS
/ ~ 0 Vv
N4 ~
H O
OH
n Br
(23) O N %N'~ N'S\
N O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-7-
No. Structure
OH
~
n '~ Br Nzt ~L4) N OS NH
2
N4 O ~N
H O
~OH
0
(25) CC4 ~\ ' N Br
N-( N O~ j N.SNHz
N~ J
~ J
H O
Br
i OH
(26) ' I Br
~
NIp N % N ~/~N \S NH
~L" CC
N 2
H O
~
/n ' ~ er
lG~) OIiCOr
j OH
q~ -0
(28) C"C N Br
~N O~_~N jN~ S,NH
O
H H O
i ~
n ' I Br
/~ O
-{ N O~N~N'~N-S NH
~L~) /H O
~_/ O
OH
L
I Br
(30) O / N~N O~N''% / N'SNi
H_ \\O
L ~
31) - Br
(
N \
Q / NN O~ / y ~N'S, Ni
''[\ ~~~/// 0 ~
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-8-
No. Structure
Br
b ~+
Br
(32) ~ ~O O N~~N~~NOS Ni
H O ~
OH
Br
(33) N Q o
H O
(
Br
\34) ~
S
C ~~NIO O N''~ V'N~ O
H O
L ~
/~ 0
(35) ~-{ N O O N; Br 0
H O tJ
~
' I Br
~ = H O
(36) N
CN_JNOYN,kN..SN
H O
OH
Br
(37) \ N-(:~lO,1~yN\
/~ N~ fls0
~
0 H O 0
OH
Br
(38) F
OCN_JNOYNNN,YkF
O 0
H
Br
/ I OH
' Br
(39) N~ ~N'O
0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-9-
No. Structure
Br
(40) ~OH
&
~ ~NXO~N~O,H
O O
H O
/ ~
~Ier
(41) a N
4 ~NIO O N~ ~N'yO'-"
c-) 0
H O
/ X
OH
42 /--~ ~
( ) N~ NIO~
O N~J, H
H O O
/ ~
(43) ~ I Br N4 N % N' /~~N~O~/
H O
OH
Br
(44)
N~N O O ~ ~~N~N~ o. H
H O O
Br
OH
~
(45) Q N~
H o
Br
nOH
Br
O Q
/~ lI
(46) ~ N ilN\'~ /'Nl/'o.H
N Il
H O
~OH
(47) N-CNIO~y " ~:~N
N4 O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-10-
No. Structure
OH
Br 0
L(48) H
0("
N
4
H O
k
(49) & o
~
CCN ~ N O ~N~ ~N
O
H O
~OH (50) B' 0
~ ~N O.~/N~ ~N f~' O'H
N 101 v
H O
L ~
Br o
~~\ II
(51) N~ ~N O~N~'~ O",
O II
H O
r
OH
(52) N~o~ er
//~~~ 0II
-{ N~~aT/\ /~/~O=H
LJ O N _ II
~
(53) o
~I &
YN~ ~N~01
N~N O O I
A
H O
L ~
54 / B'
C ~~ o
( ) N-( N O~N/~ ~N" O.H
~/ O
H O
Br
nOH
(55) Br
o
flN_(JNAONN)L(0
\ 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-11-
No. Structure
Ar H
(56
\
) I / N~ ~N O~ \ N''/.~N JL ~ 'O,H
O ~/ v ~
H 0
Br
oti
& H (57)
~
0JNiOYN
N~,NH
0
N4
0
H O
i
~ ~ Q
) & H
58
CC N~N O~~ NN,
H O 0
Q
(59) OH
cpj N~NxO~'N'~N~N H
H ~ ( 0
/r
OH
(60) Q ~ ~ Br H
\ ~N N~NXO~N/~ H
'1\ 0
H O 0
Br
OH
(61) Q H
\ N~NJ~O~ 'N~ ~~N\
~O( 0
H O
/ H
~
n Br ~
(62) / N~NI ~N~ ~N--fN,H
lkN4 O 0
H O
r
i OH
(63) B r
/~ ~
\ ~ NI O~ \NN H
N IOI
O
H 0
CA 02600189 2007-09-06
WO 2006/100026 PCT/EP2006/002557
-12-
No. Structure
~ ~ H
&
(64) -CIN I O~ N,
'O o 0
H O
~
&
(65) I ~ ~ ,
H O 0
r
i OH
(66) OCN_JNAOYNN,0,-
0
H O Br
(67) OH
O \ Br
\ N \ N H
~N~N O~ /~1. ,~/~
~/ / _ ~
H O 0
Br
OH
(68) Br
X I H N~N O~N % N~'\1\ 0
H
AOr
( 69 ~N
\ ) ~N~N O II 0 /~~~H
/ / _ ~
H O 0
L ~
O Br
(70) (1) ~~Nxo'1(" N
O
H 0 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP20061002557
-13-
No. Structure
L ~
O Br
(71)
C N-( NO~N N
,,\( ~J o
H O O H
0
r
OH
/
(72 ' I Br
CC ~\
) N-( N O O ~N\~~N~~/
H. 0 0
OH
Br
(73) =
CC /~N O N
N-( ~ ~N OH
~1 O
H O 0
Br
OH
~
(74) 0 Br
CQ
N
~N~oN/ ~N
H~O o o O
Br
OH
~
(75) o = ~ Br
N-CNJ~ o~N~=N
O
H 0 0 O
Br
OH
0 ~
Br
(76) = ~
C_CNAOYN'O
N O o
0
CC
H 0 Br
~/OH
(77) 0 = ~f'~~L
Br
C /~~ \
O
N-( _N
~N~/\l~N
NN \~ 0 .9'O
4
H 0 0
H
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-14-
No. Structure
Br
-OH
/78) o I Br
l (XI N~NxN'~ N
\V
O O O,
4
H O
Br
OH
/79) o Br
N
1 Nz~ N,-
/
O O OH
H 0
Br
-~OH
~
/$O) O \ Br
' \ N-CNxO~N~N
H~O O O O
Br
OH
O
Br
(81)
O OO.H
H O
Br
OH
(82)
~\
N-( N O~N~ ~O
0
~/
N
H O
Br
~OH
(83) p O f'"\~~i
-(~I
\\ N /~NxO~ /'N~
N O
H O
OH
(p O \ ' /~
4) ~ ~NxON~N I )
\v
N O '~/
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-15-
No. Structure
Br
OH
(85) ~ '
(VCC~NxO~N% Nn
N4 0 /
H O
r
OH
\
(86)
~
N
~~ N~N O~N,./~
0 / IY
H O
k
(87) cri N~N O~N~N~OH
0
N
H O
Br
/ I ~
~
~
(88)
CC~-C N ON,/~i~OH
0
H O
Br
~ H
(89) \ I \
OCO(H O
Br
OH
b
(90) \
-( 0
H 0 CF3
r
, OH
\ I
91
( ) I ~ ~ Oy N,~~N\,OH
N 0
H O
Br
OH
(92)
\
~ / N~N 0~ '~N'~~N~_OH
0
N'\!\ 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-16-
No. Structure
OH
(93) CC
I O~0H
~N
~ o
N
H O
Br
OH
(94) / N~ ~H O
i OH
~
(95)
N~N O~ ~/v Nv I
OH
0 OH
H O
Br
OH
(96)
OflN_JNO(NXoH
O
N
H O
Br
/ I oti
(97) ~ ~ N--( N" O~N/ N OH
N ~\' ~/ O
H O
Br
&OH
(98) o
GflN_JNAO)OH
O
H O
Br
oH
(99) o
QflN_JNAO(AOOH
O
N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-17-
No. Structure
Br
~1OH
(100) N~N O~~N\~\
/N'1\\ O OH
H O
Br
OH
(101) ~\N O _~
CC~-(
~/ O OH
N4
H O
Br
OH
(102) /~~ ~ q~ ,0
N--( N O~N~'~~N'8
N_~\' ~/ O
H O
Br
OH
(103) 1 ~ ~N~OYN ~NOS\
N 0
H O
~OH
~ I
(104) ~~N~O~N~ N~N~S\
N O
H O
OH
(105) L ~ ;~N~~ o o
N N O S
Q
\ ,
/ ~ O N- NHZ
N
H O
k OH
(106) I (N_(JN-O-YN~N~ N ~S O
/N4 O NHz
H O
OH
R
(107) N~NJlO O \% N ~/~NOS NH
N4 z
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-18-
No. Structure
Br
oH
p
(108) ~ ~ ~NON- NOS NH
H O II
Br
OH
(109) CC /~N~pY N~ ~\NO O
-(v' p i H
H p
~
~ I
(110) OH ~~NXp O N j N\/~NO NH
H O
L OH
(111) CCN]\ N N~O~ N''%~ 'S ~ ~ O N 'N/
H O ~
Br
, I OH
~
(112) ~ ~ /~ ~NJIOi~N_/~ ~S ~
- (v 01 N
H O ~
nON
(113) ~ ~NXO O ~/'N'~NOSpi
H O ~
Br
, I OH
~
(114) ~ o p
~
H O
Ofi
(115) ~N~O~N''% NOS p
H4 o
0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-19-
No. Structure
~~
Q ~
(116) ~ ~NnO~N''~~OSO
H~ o N
O
OH
(117) N4_CjON''NOS\
0
H O
OH
(118) ~\ 1
N-{ N O O
'1\\
H O ~_l IOi
Br
OH
(119) = ~ F
I
11x '
I/ N~ ~N~ O O ~N/N j N/~ X F
H p 0
r
\ I OH
~
(120) GflN_JNO(N
~O~
O 0
H O
Br
OH
(121) ~ = ~
I j ~N p~N~~: O,H
"
A "D' O
H O O
Br
OH
~
(122) O
~\NI ~
~ A N/y ~N~O~
H~-( O
~/ O O
OH
(123) ~ = '~
N N ~ ~N~O,H
~ O - V.
O
H 0 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-20-
No. Structure
\I oH
(124)
-{ /~N I O=~
~~// O 0
4
H O
OH
\ I =
(125) ~
j ~ NIp'Y /~N.~/~N~O.H
O O ~/'
C(---
H O o
o
(126) OH
\ a~
N~N o~N'%% ~/ N~0~
O
N
H O
Br
~ I OH
\ o
(127)
~ ~Nõp~N~'-~ N ~ xO H
N o
H O
k
(128) o
\
0
N
H O
OH
(129) C ~ /~NI pAX\N 1~N~p'H
N -{v p /
H O
Br
OH
(130) \ \ o
\ -CNI O~N~ ~N II p~\
'1\ O
N
H O
r
~ OH
\ I
(131) = 0
I / N~N O-~-yN\
, ~N,)~ 0
'H
N'\!\ p
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-21 -
No. Structure
&
/ I OH
(132) N~N/~O O N~'~ N"
0
H4O O
/r OH
~
(133) ~
N O,H
~
N-CN Oy N\''
-- O
H O
Br
/ I OH
(134) ~_~ 0II
N4 ~~N O~N~~ O1
O II
H O
Br
~
(135) CQ-1 o
~N O _~ Y N , ~N~OH
O
H O
OH
(136) ~ 0
/ N~ ~N O~ ~N ~~N"O I
O
H O
Br
OH
(137) 0
O o.
.N~N
O
H O
Br
OH
(138) ~ 4H
H N~N O~ ~
N~N N, H
''!\ 0
r
, I OH
(139) CC 4H
N~NION ~N0
H 0 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-22-
No. Structure
Br
~ ~
~~ .
(140) I =Y"~'~ " ~/' ~~/~ 4
N ",H
~"
~ 1(
o
H O 0
(141) OH ~\ ; N Q'H
~"" p~ ', ~N~ N, H
H O 0
Br
~
Q H
(142) N~NXpi'~N\/~
./~ N~
ol
H O 0
Br
&OH
(143) cc-]\ - N o
~" p~ \~ ~"~N.H
O
H O
Br
, I OH
(144) % N~NIO N/~ ; ~N 4 H
'~/~ N
~/N H
H~ 0
Br
OH
//f"~~~
/-~ '
(145)
/ , i~I
N~ /N ~~N",
p
_.\(' v 0
H O 0
OH ~
(146) N-CN' O"'YN " \/'N_-,(N,H
H O 0 0
L OH
(147) N-cly ",~ N\~
H4O 0 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-23-
No. Structure
OH
(148) CC N~NO O OH
H O / ~ ~ 0
Br
, ( OH
(149)
'I\\ ~VJ O IOI
H O
OH
(150) ~ =
O
N~N O~ /'N " u H
H 0 O
Br
OH
~ ~
0 -
xo~(N! N
(151) ~ 0
H 0
0
Br
OH
(152) C ~~N~p~
0
H 0 O H
0
Br
OH
(153) _ 0
/~
N--( .N O~N\'
/ ~1\ ~/ O NAYO"'
H 0 0
Br
- \ OH
(154) /~\ ~
0 ~ O,H
N\'~
-{ N
I O N
~/ ~1 ~ O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-24-
No. Structure
Br
~OH
(155) N0
N4 H O 0 ~
Br
~OH
I
(156) /~\
N-{ NxOy N~jJ.N
~/ 0
N4
H 0
Br
~OH
~
(157) - QflN_JNAO(NN
H ;,-
Br
yOH
(158)
/~
~
N-( N
O~N
N ~/ 0 0
C(I
H 0
Br
~OH
(159) -
H O O 0 O,
Br
OH
~ I
(160) I 0 H
0,
H \O O
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-25-
No. Structure
Br
OH
I
(161) - x ~/\~ N
~ N~NO ~ N~ "'
O 041-1
H ~
Br
OH
I
~\
(162) x N
CC-i N--( N
~N N4 ~/ / O OO.H
H O
~OH
(163) ~ ~N~Oi~NO
/ '~1\ IOI ~/
N
H O
~OH
(164) N--CN'I O~~%N%~ N'~~
N~ /~ O
H O
OH
/~
(165) ~ % ~ ~Nl~O~N % N'~J
N O
H O
~OH
r"~~
(166) I / ~ 0
-(~l ~\Nx0 N~
N
H O
(167) N'~~N~O
0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-26-
No. Structure
R
(168) /~NXj~ .õ N~OH
-(~/. ~0( /
N4
H O
Q
(169) C(----), ~ NJ~ON~OH
o
N
H O
OH
(170)
Xl N~N O~N~NOH
\\ O
H
(171) 4
oH
H 0 O CF3
H
(172)
OflN_(JN1OrNOH
O
N
H O
OH
\I
(173) ~ ~NJIOi,N ~OH
N 'OI
H O
H
(174) N~~NIO~ oN~ /~N~_OH
H 0
OH
///I"~~~
(175) ~ ~ ~NI O o ~N~N'/~i/NH2
N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-27-
No. Structure
OH
I\
(176) N-(~/ Jl /-\N Q O'~'YN\~N~
o
H O H pH
(177) NN4
H O
OH
(178)
OH
02~-) ~ NxO~N~N
O
H O
OH
~
(179) OCNJNAO(NDOH
-( ~/~
H O
OH
(180) \
0N_(JNAO(NDOH
O
N
H O
(181) N-CNIO-IyN\
N O OH
H \'O
I
~
(182) N /~N~O~N~N~
Nõ( -{~/ O OH
H \'O
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-28-
No. Structure
OH
.~ 0
(183) C(--j-l ~
N O~N~i~~N'SNH O
Ofi
a
(184) CQ ~N~O~N~~N % NOS\
ol
N
H O
(185) O
~~N O~N% N'~N'S\
N O
H O
I
~~
(186) 111 /~ I = N o\ o
~ N- ( N O O~N'SNHz
N'1\\ ~J
H O
~ I
~
~/~ O
(187) NN O~NV~N j N.S NH
N~ \~ ~~~/// O x
H O
pp ~~~ I\\
(1 (JC7) , ~NJlO p N~N'~NOS NHz
N4 ~
H O
OH
(189) N-- \ ,'N~O~N OS O
~ v O ~N' H
H O
~ I
(190) ~~N O~N~N j N O S N
0 ~
H H
0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-29-
No. Structure
~~
~~
(191) 0
I N N OYN % N~S O
N\\~(~ 0 iH
H O
(192) O o
N OII N N
CC]\ ~ l=\ N~''S~
O
H H O
\ '
(193)
Q~
1 N~N OYN~ ~N,S.O
O N
H'\1\
O
\ I
~ _ 0
(194) o
N N~~N ~/~N'3 N/
H O
(195) O O
~ ~\N~O O N~'~~NB
H o-{~/
~ ~o~
(196) O O
~N O~N~'~ NI3
H O
(197) lll~ -CNlO ON''N~SO
H O
(198) A ~NIO~N''%~ N~S
N4
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-30-
No. Structure
R
(199) ]~INXO~N~ O
H O IOf
~
R ~
(200) -(~l ~ /-\NJIOi 1N\
0 N~ N ~( F
N fl
H O 0
~ I
(201)
N~N O~-N~'N'yO"'
O
H O
(202) ~N~O O N''% N~O.H
N4 H O
~ I
(203) _ N~ N,~N"~O""-
H O 0
(204) =
YN~ ~N~O.H
\ N
'~ ~N O
/N O
H O
~
(205) ~
-CN OyN ';Z-,~N'yO"/
N4
H O O
OH
f~~/I
0.
(206) N~ ~Nl O~ /\N V'N~ H
O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-31-
No. Structure
i
~
~
(207)
N4
H O
0
(208) C ~N0 O N\'~~N~O H
N4
H O
~ I
(209) ~\ ~J
N-( N OYN % N'~N~/~0~
N,,( ~1 0
H O
(210)
~N
I
0~ ~N ~j N~p'H
N4 0
H O
(211) O o
I ~ ~l("/~''" ~ N'J~O'~
N~N
O
N
H O
H
(212) 0
N-CN O0 ~N~N/'N~0 H
H O
0 (213) N~ ~N O~N~'~
o
H O
~OH
//I
(214) ~O~N\/~~O.H
0
N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-32-
No. Structure
OH
(215)
O
N~
H O
(216) _
N~N O~N~ :'N~O,H
0
H O
~I
(217) O
N~ ~N O~ ~N ~N"
O
H O
~OH
//I"\\~~~i\\
(218) J=
\ /~ II ~
I N-{ N O~N~N ~~N/\/~/O=H
N''(\ ~~// O 'I
/
H O
(219) H
0 N\~ NH
~ N~N N-\/
/N'1\\ O I' 0
H O
~
~
(220) /~ = N o H
N-( N O~ ;'% N,
~/ O 0
H O
, I OH
(221) N N N H
N~N 0~~~
O 0
H O
H
H
(222) ' 0
~N~N O~ /, H
O
0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-33-
No. Structure
/ a+
~ I H
(223) / N-~ ~NIOo N,
O
H O
(224) /~ Q - o'
\ N
N N( N O~ ~N~N,H
/ '\(\ \V~ O
O 0
H O
~OH
H
(225) N4 ~\N=~/~N~N,H
0
0
H O
(226) o H
O Ii
N4
0
H O
/ I OH
~ i
(227) ~ -y \ N 4
~{N, H
N~N O '~N I'
O 0
H O
~OH
(228)
Ol
O
/ N~ ~NION~N 0
H O
H
(229) ~ ~N~O~N~N, O,H
N O /
H O 0
(230)
I O-;,y
NQ-CN
O _ II
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-34-
No. Structure
nH
(231)
Cr N ~N O ONjN OH
H O
0 ~ I
(232) 4 ~ 0
H 0 0,
0
0 k
(233) CC N-( Nxp~N j N
~/ p
H~ ~H
0
~OH
(234)
O Ilppll
N4
H O
~OH
~ I
(235) II
012 N~N O'N/-O,H
O
II
H 0
&OH
(236) -
I
p p
H O
~ OH
~ I
(237) NIt -
I O p
H
0
H
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-35-
No. Structure
OH
(238) QflN_JNAOTh(NO HO O
\\ \
XOH 0
(239)
OCN_JNAOfO
0
H 0
OH
(240) CC ~NxON
O i
H ~'O O O
, OH
(241) N~N~ON_ N
\% ~( O . H
H"~O O O
OH
(242) /~ -
CQ N-( NxO~NN
N ~/ O O OO
H 0
, OH
I
(243) /~ _ ~ "
{ N
xO~N~/v/N
N-
C10::~
N ~/ 0 OO.H
H~O
O
i
/ oH
(244) 0 - ~ ci
'~~
~NxO~NV ~' ~N'
~ O
N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-36-
No. Structure
OH
L (
245) 0 a
N % N N~~~/// O
CQ-~ NNxO~
/// O
H O
b OH
6) 0 ci
(24
~ ~NxO~N\'~ /'Ni
/ O
N
N
H O
I
, OH
N
(247) ~\ ~ ci
~N-{ N O~ ~N,H
''!\ ~/ O
N
H O
I
, OH
(248) ~ o - ci
N-( NxO~ ~N ~N,H
C2 \
\\ ~/ 0
N
H O
, OH
(249) 0 - ci
NNxO O N~'~ /'N,H
N'\!\ ~~JJ
H O
I
, OH
~
(250) 0I1 - ~ ci
~ ~N~//
NNxO~N ~
N ~
-~ / 0
H O
I
, OH
~
(251) ~ ~ = N c'
% ~N O~ V ~ ~~O
N'D 0
H O
I
, I OH
(252) N--CNxo~
N4 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-37-
No. Structure
OH
(253) O - ' ci
I j ~NxO~N' - /'N~O~
O
H O O
OH
(254) / O
I N- ( NxO~N\'~ /'N~O.H
~!\ ~/ O
H O 0
OH
~
(255) ~ ~ = N C'
N~N O~ ~N~
N OH
''!\ O
H O
OH
(256) N
N N~N O~ ~N~
O OH
'\!\
H O
OH
ci
(257) O
OflN_JNAOYNN( H O
OH
~ x N c '
(258) NHZ
N~ O~ N~
O
H O
1
/ OH
(259) ~ //~~ ~ = N
~ ~N O~ / ~N
/ O ~OH
H O OH
\~
(260) ~ ~ 0 0
\~ N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-38-
No. Structure
\~
//----~~
(261) o
N O~N a ~NOS\
/
N
H O
~ ~
Q ~
(262) /~
/~NJlO- / .,NOS O
-(~/ 101 / 'N~ \
N
H O
I
OH
(263)
/~
N
-( NxO~
\\ ~/ 0
CC
N
N
H O
OH
(264) , ~NJ~O~N~
N4 ~
H O
I
\ I OH
(265)
~NxO~N''~~N'
O
N
H O
OH
(266) OCN_CNY.O.YN,
NH
0
H O
OH
(267) O
~NO~N N H
N
H O
, OH
\~
~
(268) N
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-39-
No. Structure
b OH (269) -
OCN_JNAO(N,NA
O
N
H O
OH
(270) \ ~ ~Np~N~ ~
/ p
N
H O
OH
(271) ~Nxp~N~
/
N4 0
H O
b OH
(272) J~ = N
N~N O~ '%O'/
II
~/ 4 p N~
H O 0
b OH
(273) ~ H O O
OH
~ 'N~N~OH
(274) I / N ~ ~ p
(
N
H O
~OH
(275) /~~ 0
N--( N O~ N~OH
/ ~/ p
H O
OH
(276)
k ~ ~NxO~N \/~V
/N O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-40-
No. Structure
(277) 5OH
CQ N\ \ ~N NHZ
0
0
H O
b OH
(278) ~
\ ( ~ ~ ~N~O~N~N~ ~ OH
O ~/~ IY
H O OH
(279) OH \ ~ a~ N O O
\
=S0
N
H O
p k
O
(2U0) Cri ~N O~N~ ~N_S\
0
N
H O
OH
(281) QN_JNO'(N~N'~N; S'0
N4 0
H O
~NHz
(282) /~ x = N
N-( N O~ ~ / 0
O
~~//
N
H O
~NH2
(283) I ~NxON~N
/
N4 O
H O
~NHz
(284) N
\ ~~ _\
N~NO 0 ~\'~ /'N~
'1\\
H 0 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-41 -
No. Structure
NHZ
p O -
(285)
~H O O
NH,
(286) =
\ N
O ,OH
N
N
H O
NH,
O
(287)
Xl ,~N~ OH
~N x ~ N
( NO
H O
~NHZ
~
(288) O
N~NxON O
CC ~
O
H O
NH,
N'\\
(289) -CNxO~yN ~ N I NH
~/~1.
O
H O
I
(290) NHZ
I / ~ ~N O~N~N,/~/~
O '~ T OH
H O OH
NHZ
(291) /~ N Q~ 0
N-( NI O~ ~~%~~N'S~
0 ~/
N
H O
NH,
(292) j ~ ~N O'y"~ Os~
N 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-42-
No. Structure
)6( HZ
(293) I % N ~ IO;~ ~N/\N QS o
'~ 0
N
H O
~
CF3
(294) OflN_JNO(NO
O
N
H O
F3
GF~
(295) __~-CNOi~
o
H O
F3
/ I
(296) _/--~ o CF,
N vNJtO~
O
H O O
F'
\ '
4
(297) ~ = O CF3
N~N ON~'N~o.H
'\!\
H O O
LFI,
(298) ~ = N CF3
NN O~ ~N~OH
0
~~~///
N
H O
F,
CF3
(299) CN_CNOy,,OH
N~ o '
H O
/I
(300) O -F~
~ CF,
~
N
~ ~NxO~N V
O
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-43-
No. Structure
~
(301) ~ = N CF3
NN OY~N~NH2
~~~~~/// O
H O
(302) LCF,
/~CC N ~NxO ~ N-N~OH
H(\0 OH
F3
(303) CF' o N_CN 0
O N \
N
H O
F,
(304) I A ~ ~NO~N\ CF3 ~NQS\
N
H O
F,
\I
(305) CF3 o O
~ ~
~N 0 II N~
~ O
N
H O
/I
(306) N~NxO'~N/,N %
0
N
H O
/I
/~ \ CF
(307) 0
--( N O~N % N /~N~
~J
N
H O
/I
p O - \ CF~
(308)
--( Ny'0
N II O ~N'
~/
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-44-
No. Structure
/~
0 - \ CF
(309) ~NxO~\/~ ~N'H
0 N
H O
a
~NxOH
(310) X-)\
o
N
H O
/ I
O ~ CF,
(311) cc_-~ -( Nx0 O N\'~ /'N'H
N ~/
H O
/I
(312) N~NxO-yN,~~N/~,OH
0
N
H O
/ I
(313)
I N~NxO~N~N~OH
AN4 0
H O
/I
3
(314)
H O
/ I
0 (315) N~NxON\/~N NHz
AN4 o ~
H O
aCF,
(316) I ~ ~ ~A 0 ~ ~
H 0 OH
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
' -45-
No. Structure
p
(317) 4 -CNxpN
N 0
H O
(318) p
N-CNxp~
N'1\\ 0
H 0
/I
(319) p \
flN_JNAON~ O
N4
H O
/I
(320) N--( NxO~N\/vN~N,H
~( ~/ 0
N_\'
H O
(321) ~'N--CNpy ~N,H
IAN4 0
H O
O - ~
(322) I O
H 0
/I
(323) aThN_cNAO(NNOH
0
H O
O
(324) CN_(JNAO(OH
N_\\ 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-46-
No. Structure
(325) I -( NxO~N /vN
N~ ~1 O
H 0
i I
(326) CC N-{ NxONHZ
N~l 0 I
H 0
i I
(327) 0~' N~N~OiN/~N
~ 10' \_OH
0 OH
the enantiomers, the diastereomers, the hydrates, the mixtures thereof and the
salts thereof as well as the hydrates of the salts, particularly the
physiologically
acceptable salts thereof with inorganic or organic acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(1), (2), (3), (4), (5), (6), (7), (8), (9), (10), (11), (12), (13), (14),
(15), (16), (17),
(18), (19), (20), (21), (22), (23), (24), (25), (26), (27), (28), (29), (30),
(31), (32),
(33), (34), (35), (36), (37), (38), (39), (40), (41), (42), (43), (44), (45),
(46), (47),
(48), (49), (50), (51), (52), (53), (54), (55), (56), (57), (58), (59), (60),
(61), (62),
(63), (64), (65), (66), (67), (68), (69), (70), (71), (72), (73), (74), (75),
(76), (77),
(78), (79), (80) and (81),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-47-
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(82), (83), (84), (85), (86), (87), (88), (89), (90), (91), (92), (93), (94),
(95), (96),
(97), (98), (99), (100), (101), (102), (103), (104), (105), (106), (107),
(108), (109),
(110), (111), (112), (113), (114), (115), (116), (117), (118), (119), (120),
(121),
(122), (123), (124), (125), (126), (127), (128), (129), (130), (131), (132),
(133),
(134), (135), (136), (137), (138), (139), (140), (141), (142), (143), (144),
(145),
(146), (147), (148), (149), (150), (151), (152), (153), (154), (155), (156),
(157),
(158), (159), (160), (161), and (162),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(163), (164), (165), (166), (167), (168), (169), (170), (171), (172), (173),
(174),
(175), (176), (177), (178), (179), (180), (181), (182), (183), (184), (185),
(186),
(187), (188), (189), (190), (191), (192), (193), (194), (195), (196), (197),
(198),
(199), (200), (201), (202), (203), (204), (205), (206), (207), (208), (209),
(210),
(211), (212), (213), (214), (215), (216), (217), (218), (219), (220), (221),
(222),
(223), (224), (225), (226), (227), (228), (229), (230), (231), (232), (233),
(234),
(235), (236), (237), (238), (239), (240), (241), (242) and (243),
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-48-
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(244), (245), (246), (247), (248), (249, (250), (251), (252), (253), (254),
(255),
(256), (257), (258), (259), (260), (261) and (262),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(263), (264), (265), (266), (267), (268), (269), (270), (271), (272), (273),
(274),
(275), (276), (277), (278), (279), (280) and (281),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(282), (283), (284), (285), (286), (287), (288), (289), (290), (291), (292)
and (293),
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-49-
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(294), (295), (296), (297), (298), (299), (300), (301), (302), (303), (304)
and (305),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(306), (307), (308), (309), (310), (311), (312), (313), (314), (315) and
(316),
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
Examples of other preferred compounds of the above general formula I are the
following compounds, which are selected from the group consisting of:
(317), (318), (319), (320), (321), (322), (323), (324), (325), (326) and
(327),
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-50-
the enantiomers thereof, the diastereomers thereof, the hydrates thereof, the
mixtures thereof and the salts thereof, as well as the hydrates of the salts,
particularly the physiologically acceptable salts thereof with inorganic or
organic
acids or bases.
TERMS AND DEFINITIONS USED
Within the scope of this application, in the definition of possible
substituents, these
may also be represented in the form of a structural formula. An asterisk (*)
in the
structural formula'of the substituent is to be understood as being the linking
point
to the rest of the molecule.
Also included in the subject matter of this invention are the compounds
according
to the invention, including the salts thereof, in which one or more hydrogen
atoms,
for example one, two, three, four or five hydrogen atoms, are replaced by
deuterium.
Compounds of general formula I may have acid groups, mainly carboxyl groups,
and/or basic groups such as e.g. amino functions. Compounds of general formula
I
may therefore be present as internal salts, as salts with pharmaceutically
useable
inorganic acids such as for example hydrobromic acid, phosphoric acid, nitric
acid,
hydrochloric acid, sulphuric acid, methanesulphonic acid, ethanesulphonic
acid,
benzenesulphonic acid, p-toluenesulphonic acid or organic acids such as for
example malic acid, succinic acid, acetic acid, fumaric acid, maleic acid,
mandelic
acid, lactic acid, tartaric acid, citric acid or as salts with
pharmaceutically useable
bases such as alkali or alkaline earth metal hydroxides, for example sodium
hydroxide or potassium hydroxide, or carbonates, ammonia, zinc or ammonium
hydroxides or organic amines such as e.g. diethylamine, triethylamine,
ethanolamine, diethanolamine, triethanolamine, cyclohexylamine,
dicyclohexylamine, inter alia.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-51-
As mentioned hereinbefore, the compounds of formula I may be converted into
the
salts thereof, particularly for pharmaceutical use, into the physiologically
and
pharmacologically acceptable salts thereof. These salts may on the one hand be
in the form of the physiologically and pharmacologically acceptable acid
addition
salts of the compounds of formula I with inorganic or organic acids. On the
other
hand, if B contains a phenolic OH group, the compound of formula I may also be
converted by reaction with inorganic bases into physiologically and
pharmacologically acceptable salts with alkali or alkaline earth metal cations
as
counter ion. The acid addition salts may be prepared for example using
hydrochloric acid; hydrobromic acid, sulphuric acid, phosphoric acid,
methanesulphonic acid, acetic acid, fumaric acid, succinic acid, lactic acid,
citric
acid, tartaric acid or maleic acid. It is also possible to use mixtures of the
above-
mentioned acids. The alkali and alkaline earth metal salts of the compound of
formula I are preferably the alkali and alkaline earth metal hydroxides and
hydrides
thereof, of which the hydroxides and hydrides of the alkaline earth metals,
particularly of sodium and potassium, are preferred and sodium and potassium
hydroxide are particularly preferred.
The compounds according to the invention may occur as racemates if they have
only one chiral element, but they may also be obtained as pure enantiomers,
i.e. in
the (R) or (S) form. Preferred compounds are those which occur as racemates or
as the (R) form.
However, the application also includes the individual diastereomeric pairs of
antipodes or the mixtures thereof which are present when there is more than
one
chiral element in the compounds of general formula I, as well as the
individual
optically active enantiomers of which the above-mentioned racemates are made
up.
The invention relates to the compounds in question, optionally in the form of
the
individual optical isomers, mixtures of the individual enantiomers or
racemates, in
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-52-
the form of the tautomers as well as in the form of the free bases or the
corresponding acid addition salts with pharmacologically acceptable acids -
such
as for example acid addition salts with hydrohalic acids - for example
hydrochloric
or hydrobromic acid or organic acids - such as for example oxalic, fumaric,
diglycolic or methanesulphonic acid.
PREPARATION METHODS
The compounds of general formula I are prepared by methods known in principle.
The following methods have proved particularly suitable for preparing the
compounds of general formula I according to the invention:
(a) In order to prepare compounds of general formula I wherein all the groups
are as hereinbefore defined:
reacting a piperidine of formula III
N
NH
O
H
with a carbonic acid derivative of general formula IV
O
y2)L'~
wherein Y2 and Y3 represent nucleofugic groups which may be identical or
different, preferably the chlorine atom, the p-nitrophenoxy or
trichloromethoxy
group,
and with a compound of general formula V
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-53-
B
H.O O.Z
O
wherein B is as hereinbefore defined and Z' denotes a protective group for a
carboxy group, for example a C1_6-alkyl or benzyl group, wherein the alkyl
groups
may be straight-chain or branched and the benzyl group may be substituted by
one or two methoxy groups. Preferably Z' denotes the methyl, ethyl, tert-butyl
or
benzyl group.
In a first step the compounds of general formula III are reacted in a solvent,
for
example in dichloromethane, THF, pyridine or mixtures thereof, at a
temperature
from -20 to 50 C in the presence of a base, for example triethylamine,
pyridine or
ethyidiisopropylamine, with the carbonic acid derivatives of general formula
IV.
The resulting intermediate may be purified or further reacted without
purification.
The reaction of these intermediates with compounds of general formula V is
also
carried out in one of the above-mentioned solvents, and at the temperatures
specified hereinbefore, in the presence of a base, such as triethylamine or
pyridine, with or without the addition of an activating reagent, such as e.g.
4-
dimethylaminopyridine. To activate them the compounds of general formula V may
also be deprotonated by means of a metal hydride such as e.g. NaH or KH, in
which case there is no need for the presence of the base or the activating
reagent.
The starting compounds of formula III and IV are either commercially
available,
known from the literature or may be prepared using methods known from the
literature.
Processes for preparing compounds of general formula III are described for
example in EP 1 619 187 A1.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-54-
(b) In order to prepare compounds of general formula I wherein all the groups
are as hereinbefore defined:
coupling a carboxylic acid of general formula VI
B
O
~O O, H
N O
N
C N~O
H
wherein B is as hereinbefore defined, with an amine of general formula VII
H-NR'RZ,
wherein R' and R2 are as hereinbefore defined. Before the reaction is carried
out
any carboxylic acid functions, primary or secondary amino functions or hydroxy
functions present in the groups R' and R2 of the amine of general formula VII
may
be protected by conventional protecting groups and any protecting groups used
may be cleaved again after the reaction using methods familiar to the skilled
man.
The coupling is preferably carried out using methods known from peptide
chemistry (cf. e.g. Houben-Weyl, Methoden der Organischen Chemie, Vol. 15/2),
for example using carbodiimides such as e.g. dicyclohexylcarbodiimide (DCC),
diisopropyl carbodiimide (DIC) or ethyl-(3-dimethylaminopropyl)-carbodiimide,
O-(1 H-benzotriazol-1 -yl)- N, N-N', N' tetramethyluronium hexafluorophosphate
(HBTU) or tetrafluoroborate (TBTU) or 1 H-benzotriazol-1 -yl-oxy-
tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP). By adding
1-hydroxybenzotriazole (HOBt) or 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-
benzotriazine
(HOObt) the reaction speed can be increased. The couplings are normally
carried
out with equimolar amounts of the coupling components as well as the coupling
W0 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-55-
reagent in solvents such as dichloromethane, tetrahydrofuran, acetonitrile,
dimethyl formamide (DMF), dimethyl acetamide (DMA), N-methylpyrrolidone
(NMP) or mixtures thereof and at temperatures between -30 C and +30 C,
preferably -20 C and +25 C. If necessary, N-ethyl-diisopropylamine (Hunig
base)
is preferably used as an additional auxiliary base.
The so-called "anhydride process" is used as a further coupling method for
synthesising compounds of general formula I (cf. also: M. Bodanszky, "Peptide
Chemistry", Springer-Verlag 1988, p. 58-59; M. Bodanszky, "Principles of
Peptide
Synthesis", Springer-Verlag 1984, p. 21-27). The Vaughan variant of the "mixed
anhydride process" is preferred (J.R. Vaughan Jr., J. Amer. Chem. Soc. 73,
3547
(1951)), in which the mixed anhydride of the carboxylic acid of general
formula VI
which is to be coupled and monoisobutyl carbonate is obtained, using isobutyl
chlorocarbonate in the presence of bases such as 4-methylmorpholine or
4-ethylmorpholine. The preparation of this mixed anhydride and the coupling
with
amines of general formula VII are carried out in a one-pot process, using the
above-mentioned solvents and at temperatures between -20 and +25 C,
preferably between 0 C and +25 C.
(c) In order to prepare compounds of general formula I wherein all the groups
are as hereinbefore defined:
coupling a compound of general formula VIII
B
0
Nu
N N
O
cc
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-56-
wherein B is as hereinbefore defined and Nu denotes a leaving group, for
example
a halogen atom, such as the chlorine, bromine or iodine atom, an
alkylsulphonyloxy group with 1 to 10 carbon atoms in the alkyl moiety, a
phenylsulphonyloxy or naphthylsulphonyloxy group optionally mono-, di- or
trisubstituted by chlorine or bromine atoms or by methyl or nitro groups,
wherein
the substituents may be identical or different, a 1 H-imidazol-1 -yl,
optionally
substituted by one or two methyl groups in the carbon skeleton, a 1 H-pyrazol-
1 -yl,
a 1 H-1,2,4-triazol-1-yl, 1 H-1,2,3-triazol-1-yl, 1 H-1,2,3,4-tetrazol-1-yl, a
vinyl,
propargyl, p-nitrophenyl, 2,4-dinitrophenyl, trichlorophenyl,
pentachlorophenyl,
pentafluorophenyl, pyranyl or pyridinyl, a dimethylaminyloxy, 2(1H)-oxopyridin-
1-
yl-oxy, 2,5-dioxopyrrolidin-1-yloxy, phthalimidyloxy, 1 H-benzo-triazol-1 -
yloxy or
azide group, with an amine of general formula VII
H-NR1R2,
wherein R' and R2 are as hereinbefore defined.
Before the reaction any carboxylic acid functions, primary or secondary amino
functions or hydroxy functions present in the groups R' and R2 of the amine of
general formula VII may be protected by conventional protecting groups and
after
the reaction any protecting groups used may be cleaved again using methods
familiar to those skilled in the art.
The reaction is carried out under Schotten-Baumann or Einhorn conditions, i.e.
the
components are reacted in the presence of at least one equivalent of an
auxiliary
base at temperatures between -50 C and +120 C, preferably -10 C and +30 C,
and optionally in the presence of solvents. The auxiliary bases used are
preferably
alkali metal and alkaline earth metal hydroxides, e.g. sodium hydroxide,
potassium
hydroxide or barium hydroxide, alkali metal carbonates, e.g. sodium carbonate,
potassium carbonate or caesium carbonate, alkali metal acetates, e.g. sodium
or
potassium acetate, as well as tertiary amines, e.g. pyridine, 2,4,6-
trimethylpyridine,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-57-
quinoline, triethylamine, N-ethyl-diisopropylamine, N-ethyl-dicyclohexylamine,
1,4-diazabicycfo[2,2,2]octane or 1,8-diazabicyclo[5,4,0]undec-7-ene, the
solvents
used may be, for example, dichloromethane, tetrahydrofuran, 1,4-dioxane,
acetonitrile, dimethyl formamide, dimethyl acetamide, N-methyl-pyrrolidone or
mixtures thereof; if alkali metal or alkaline earth metal hydroxides, alkali
metal
carbonates or acetates are used as the auxiliary bases, water may also be
added
to the reaction mixture as cosolvent.
The new compounds of general formula I according to the invention contain one
or
more chiral centres. If for example there are two chiral centres the compounds
may occur in the form of two pairs of diastereomeric antipodes. The invention
covers the individual isomers as well as the mixtures thereof.
The diastereomers may be separated on the basis of their different physico-
chemical properties, e.g. by fractional crystallisation from suitable
solvents, by high
pressure liquid or column chromatography, using chiral or preferably non-
chiral
stationary phases.
Racemates covered by general formula I may be separated for example by HPLC
on suitable chiral stationary phases (e.g. Chiral AGP, Chiralpak AD).
Racemates
which contain a basic or acidic function can also be separated via the
diastereomeric, optically active salts which are produced on reacting with an
optically active acid, for example (+) or (-)-tartaric acid, (+) or (-)-
diacetyl tartaric
acid, (+) or (-)-monomethyl tartrate or (+) or (-)-camphorsulphonic acid, or
an
optically active base, for example with (R)-(+)-1-phenylethylamine, (S)-(-)-1-
phenylethylamine or (S)-brucine.
According to a conventional method of separating isomers, the racemate of a
compound of general formula I is reacted with one of the above-mentioned
optically active acids or bases in equimolar amounts in a solvent and the
resulting
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-58-
crystalline, diastereomeric, optically active salts thereof are separated
using their
different solubilities. This reaction may be carried out in any type of
solvent
provided that it is sufficiently different in terms of the solubility of the
salts.
Preferably, methanol, ethanol or mixtures thereof, for example in a ratio by
volume
of 50:50, are used. Then each of the optically active salts is dissolved in
water,
carefully neutralised with a base such as sodium carbonate or potassium
carbonate, or with a suitable acid, e.g. dilute hydrochloric acid or aqueous
methanesulphonic acid, and in this way the corresponding free compound is
obtained in the (+) or (-) form.
The (R) or (S) enantiomer alone or a mixture of two optically active
diastereomeric
compounds covered by general formula I may also be obtained by performing the
syntheses described above with a suitable reaction component in the (R) or (S)
configuration.
The hydroxycarboxylic acids of general formula V needed for the synthesis
B
H.O O, Z~
O
wherein B is as hereinbefore defined and Z' denotes a protective group for a
carboxy group, for example a C1_6-alkyl or benzyl group, wherein the alkyl
groups
may be straight-chain or branched and the benzyl group may be substituted by
one or two methoxy groups, wherein Z1 preferably denotes the methyl, ethyl,
tert-
butyl or benzyl group, may be obtained from compounds of general formula IX
B
H, N O, H
I
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-59-
wherein B is as hereinbefore defined.
By diazotising compounds of general formula IX with a suitable diazotising
reagent, preferably sodium nitrite in an acid medium, it is possible to obtain
the
compounds of general formula V. When enantiomerically pure compounds are
used the corresponding enantiomerically pure hydroxycarboxylic acid compounds
are obtained, the reaction taking place with retention of configuration.
1o An alternative method of obtaining compounds of general formula V comprises
reacting aidehydes of general formula X
Hy B
O
wherein B is as hereinbefore defined, with N-acetylglycine in acetic anhydride
as
solvent in the presence of alkali metal acetate, preferably sodium or
potassium
acetate, at a suitable temperature, preferably at 80 to 130 C.
The azlactones obtained as primary product are hydrolysed without being
isolated
to form the compounds of general formula XI
O
AN, O, H
I
H O
wherein B is as hereinbefore defined.
By further reaction in the presence of aqueous inorganic acids, such as for
example sulphuric, phosphoric or hydrochloric acid, but preferably
hydrochloric
acid, compounds of general formula Xii
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-60-
B
O O, H
O
are obtained wherein B is as hereinbefore defined. These are then converted
with
suitable reducing agents into the compounds of general formula V.
Suitable reducing agents are alkali metal borohydrides, such as sodium or
potassium borohydride. Other suitable reducing agents are
chlorodialkylboranes,
such as chlorodicyclohexylborane. If chiral chlorodialkylboranes, such as e.g.
B-
chlorodiisopinocampheylborane, are used, the compounds of general formula V
may be isolated in enantiomerically pure form.
The further reaction of compounds of general formula V, wherein Z' denotes the
hydrogen atom, to compounds of general formula V, wherein Z' is as
hereinbefore
defined, takes place in an alcoholic medium, preferably in methanol or
ethanol, in
the presence of a suitable acid, such as hydrochloric acid. The reaction may
alternatively be carried out by reaction in alcoholic solvents, preferably
methanol,
with thionyl chloride.
2o Another method of obtaining compounds of general formula V comprises
alkylating
a compound of formula XIII
0
c,ojo
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-61 -
with an aryl- or heteroaryl-methylhalide of general formula XIV
r B
Hal
wherein Hal denotes a chlorine, bromine or iodine atom, and B is as
hereinbefore
defined, analogously to methods known from the literature (Michael T.
Crimmins,
Kyle A. Emmitte and Jason D. Katz, Org. Left. 2, 2165-2167 [2000]).
The diastereomeric products obtained may then be separated using
physicochemical methods, preferably by chromatographic methods or
recrystallisation. The hydrolytic cleaving of the chiral auxiliary and
cleaving the
benzyl protecting group also provides a method of obtaining enantiomerically
pure
hydroxycarboxylic acid compounds of general formula V.
All the compounds of general formula I which contain primary or secondary
amino,
hydroxy or hydroxycarbonyl functions are preferably obtained from precursors
provided with protective groups. Examples of protective groups for amino
functions
include for example a benzyloxycarbonyl, 2-nitrobenzyloxycarbonyl, 4-nitro-
benzyloxycarbonyl, 4-methoxy-benzyloxycarbonyl, 2-chloro-benzyloxycarbonyl,
3-chloro-benzyloxycarbonyl, 4-chloro-benzyloxycarbonyl,
4-biphenylyl-a,a-dimethyl-benzyloxycarbonyl or 3,5-
dimethoxy-a,a-dimethyl-benzyloxycarbonyl group, an alkoxycarbonyl group with a
total of 1 to 5 carbon atoms in the alkyl moiety, for example the
methoxycarbonyl,
ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl, n-butoxycarbonyl,
1-methylpropoxycarbonyl, 2-methylpropoxy-carbonyl or tert-butyloxycarbonyl
group, the allyloxycarbonyl, 2,2,2-trichloro-(1,1-dimethylethoxy)carbonyl or
9-fluorenylmethoxycarbonyl group or a formyl, acetyl or trifluoroacetyl group.
Examples of protective groups for hydroxy functions include a trimethylsilyl,
triethylsilyl, triisopropyl, tert-butyldimethylsilyl or tert-
butyldiphenylsilyl group, a tert-
butyl, benzyl, 4-methoxybenzyl or 3,4-dimethoxybenzyl group.
A protective group for hydroxycarbonyl functions might be, for example, an
alkyl
group with a total of 1 to 5 carbon atoms, for example the methyl, ethyl, n-
propyl,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-62-
isopropyl, n-butyl, tert-butyl, allyl, 2,2,2-trichloroethyl, benzyl or 4-
methoxybenzyl
group.
The new compounds of general formula I and the physiologically acceptable
salts
thereof have valuable pharmacological properties, based on their selective
CGRP-
antagonistic properties. The invention further relates to pharmaceutical
compositions containing these compounds, their use and the preparation
thereof.
The new compounds mentioned above and the physiologically acceptable salts
thereof have CGRP-antagonistic properties and exhibit good affinities in CGRP
receptor binding studies. The compounds display CGRP-antagonistic properties
in
the pharmacological test systems described hereinafter.
The following experiments were carried out to demonstrate the affinity of the
above-mentioned compounds for human CGRP-receptors and their antagonistic
properties:
A. Binding studies with SK-N-MC cells (expressing the human CGRP receptor)
SK-N-MC cells are cultivated in "Dulbecco's modified Eagle medium". The medium
is removed from confluent cultures. The cells are washed twice with PBS buffer
(Gibco 041-04190 M), detached by the addition of PBS buffer mixed with 0.02%
EDTA, and isolated by centrifuging. After resuspension in 20 ml of "Balanced
Salts
Solution" [BSS (in mM): NaCI 120, KCI 5.4, NaHCO3 16.2, MgSO4 0.8, NaHPO4
1.0, CaC12 1.8, D-glucose 5.5, HEPES 30, pH 7.40] the cells are centrifuged
twice
at 100 x g and resuspended in BSS. After the number of cells has been
determined, the cells are homogenised using an Ultra-Turrax and centrifuged
for
10 minutes at 3000 x g. The supernatant is discarded and the pellet is
recentrifuged in Tris buffer (10 mM Tris, 50 mM NaCI, 5 mM MgC12, 1 mM EDTA,
pH 7.40) enriched with 1% bovine serum albumin and 0.1 % bacitracin, and
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-63-
resuspended (1 ml / 1000000 cells). The homogenised product is frozen at -80
C.
The membrane preparations are stable for more than 6 weeks under these
conditions.
After thawing, the homogenised product is diluted 1:10 with assay buffer (50
mM
Tris, 150 mM NaCl, 5 mM MgCI2, 1 mM EDTA, pH 7.40) and homogenised for 30
seconds with an Ultra-Turrax. 230 pl of the homogenised product are incubated
for
180 minutes at ambient temperature with 50 pM 1251-iodotyrosyl-Calcitonin-Gene-
Related Peptide (Amersham) and increasing concentrations of the test
substances
in a total volume of 250 NI. The incubation is ended by rapid filtration
through GF/B
glass fibre filters treated with polyethyleneimine (0.1 /a) using a cell
harvester. The
protein-bound radioactivity is measured using a gamma counter. Non-specific
binding is defined as the bound radioactivity in the presence of 1 pM human
CGRP-alpha during incubation.
The concentration binding curves are analysed using computer-aided non-linear
curve matching.
The compounds mentioned hereinbefore show IC50 values <_ 10000 nM in the test
described.
B. CGRP Antagonism in SK-N-MC cells
SK-N-MC cells (1 million cells) are washed twice with 250 pI incubation buffer
(Hanks' HEPES, 1 mM 3-isobutyl-l-methylxanthine, 1% BSA, pH 7.4) and pre-
incubated at 37 C for 15 minutes. After the addition of CGRP (10 pI) as
agonist in
increasing concentrations (10"11 to 10"6 M), or additionally the substance in
3 to 4
different concentrations, the mixture is incubated for another 15 minutes.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-64-
Intracellular cAMP is then extracted by the addition of 20 NI of 1 M HCI and
centrifugation (2000 x g, 4 C, for 15 minutes). The supernatants are frozen in
liquid nitrogen and stored at -20 C.
The cAMP contents of the samples are determined by radioimmunoassay
(Messrs. Amersham) and the pA2 values of antagonistically acting substances
are
determined graphically.
The compounds according to the invention exhibit CGRP-antagonistic properties
in the in vitro test model described, in a dosage range between 10-12 and 10-5
M.
TYPES OF INDICATIONS
In view of their pharmacological properties the compounds according to the
invention and the salts thereof with physiologically acceptable acids are thus
suitable for the acute and prophylactic treatment of headaches, particularly
migraine, cluster headaches and tension headaches. Moreover, the compounds
according to the invention also have a positive effect on the following
diseases:
non-insulin-dependent diabetes mellitus ("NIDDM"), cardiovascular diseases,
morphine tolerance, diarrhoea caused by clostridium toxin, skin diseases,
particularly thermal and radiation-induced skin damage including sunburn,
lichen,
prurigo, pruriginous toxidermies and severe itching, inflammatory diseases,
e.g.
inflammatory diseases of the joints (osteoarthritis, rheumatoid arthritis,
neurogenic
arthritis), generalised soft tissue rheumatism (fibromyalgia), neurogenic
inflammation of the oral mucosa, inflammatory lung diseases, allergic
rhinitis,
asthma, COPD, diseases accompanied by excessive vasodilatation and resultant
reduced blood supply to the tissues, e.g. shock and sepsis, chronic pain, such
as
e.g. diabetic neuropathies, neuropathies induced by chemotherapy, HIV-induced
neuropathies, post-herpetic neuropathies, neuropathies induced by tissue
trauma,
trigeminal neuralgias, temporomandibular dysfunctions, CRPS (complex regional
pain syndrome), back pain and visceral diseases such as for example irritable
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-65-
bowel syndrome (IBS), inflammatory bowel syndrome. In addition, the compounds
according to the invention have a general pain-relieving effect. The symptoms
of
menopausal hot flushes caused by vasodilatation and increased blood flow in
oestrogen-deficient women and hormone-treated patients with prostate carcinoma
and castrated men are favourably affected by the CGRP antagonists of the
present application in a preventive and acute-therapeutic capacity, this
therapeutic
approach being distinguished from hormone replacement by the absence of side
effects.
Preferably, the compounds according to the invention are suitable for the
acute
and preventive treatment of migraine and cluster headaches, for treating
irritable
bowel syndrome (IBS) and for the preventive and acute therapeutic treatment of
hot flushes in oestrogen-deficient women.
The dosage required to achieve a corresponding effect is conveniently 0.0001
to 3
mg/kg of body weight, preferably 0.01 to 1 mg/kg of body weight, when
administered intravenously or subcutaneously, and 0.01 to 10 mg/kg of body
weight, preferably 0.1 to 10 mg/kg of body weight when administered orally,
nasally or by inhalation, 1 to 3 times a day in each case.
If the treatment with CGRP antagonists and/or CGRP release inhibitors is given
as
a supplement to conventional hormone replacement, it is advisable to reduce
the
doses specified above, in which case the dosage may be from 1/5 of the lower
limits mentioned above up to 1/1 of the upper limits specified.
The invention further relates to the use of the compounds according to the
invention as valuable adjuvants for the production and purification (by
affinity
chromatography) of antibodies as well as in RIA and ELISA assays, after
suitable
radioactive labelling, for example by tritiation of suitable precursors, for
example
by catalytic hydrogenation with tritium or replacing halogen atoms with
tritium, and
as a diagnostic or analytical adjuvant in neurotransmitter research.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-66-
COMBINATIONS
Categories of active substance which may be used in the combination include
e.g.
antiemetics, prokinetics, neuroleptics, antidepressants, neurokinin
antagonists,
anticonvulsants, histamine-H1-receptor antagonists, R-blockers, a-agonists and
a-antagonists, ergot alkaloids, mild analgesics, non-steroidal
antiinflammatories,
corticosteroids, calcium antagonists, 5-HTIBilp-agonists or other anti-
migraine
agents which may be formulated together with one or more inert conventional
carriers and/or diluents, e.g. with corn starch, lactose, glucose,
microcrystalline
cellulose, magnesium stearate, polyvinyl pyrrolidone, citric acid, tartaric
acid,
water, water/ethanol, water/glycerol, water/sorbitol, water/polyethylene
glycol,
propylene glycol, cetylstearyl alcohol, carboxymethylcellulose or fatty
substances
such as hard fat or suitable mixtures thereof, into conventional galenic
preparations such as plain or coated tablets, capsules, powders, suspensions,
solutions, metered dose aerosols or suppositories.
Thus, other active substances which may be used for the combinations mentioned
above include for example the non-steroidal antiinflammatories aceclofenac,
acemetacin, acetylsalicylic acid, acetaminophen (paracetamol), azathioprine,
diclofenac, diflunisal, fenbufen, fenoprofen, flurbiprofen, ibuprofen,
indometacin,
ketoprofen, leflunomide, lornoxicam, mefenamic acid, naproxen, phenylbutazone,
piroxicam, sulphasalazine, zomepirac or the pharmaceutically acceptable salts
thereof as well as meloxicam and other selective COX2-inhibitors, such as for
example rofecoxib, valdecoxib, parecoxib, etoricoxib and celecoxib, as well as
substances which inhibit earlier or later stages in prostaglandin synthesis or
prostaglandin receptor antagonists such as e.g. EP2-receptor antagonists and
IP-
receptor antagonists.
It is also possible to use ergotamine, dihydroergotamine, metoclopramide,
domperidone, diphenhydramine, cyclizine, promethazine, chlorpromazine,
vigabatrin, timolol, isometheptene, pizotifen, botox, gabapentin, pregabaline,
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-67-
duloxetine, topiramate, riboflavin, montelukast, lisinopril, micardis,
prochloroperazine, dexamethasone, flunarizine, dextropropoxyphene, meperidine,
metoprolol, propranolol, nadolol, atenolol, clonidine, indoramin,
carbamazepine,
phenytoin, valproate, amitryptiline, imipramine, venlafaxine, lidocaine or
diltiazem
and other 5-HT1B/1D-agonists such as, for example, almotriptan, avitriptan,
eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan and
zolmitriptan.
In addition, CGRP-antagonists may be combined with vanilloid receptor
antagonists, such as e.g. VR-1 antagonists, glutamate receptor antagonists,
such
as e.g. mGIu5-receptor antagonists, mGlu1-receptor antagonists, iGiu5-receptor
antagonists, AMPA-receptor antagonists, purine receptor blockers, such as e.g.
P2X3 antagonists, NO-synthase inhibitors, such as e.g. iNOS inhibitors,
calcium
channel blockers, such as e.g. PQ-type blockers, N-type blockers, potassium
channel openers, such as e.g. KCNQ channel openers, sodium channel blockers,
such as e.g. PN3 channel blockers, NMDA-receptor antagonists, acid-sensing ion
channel antagonists, such as e.g. ASIC3 antagonists, bradykinin receptor
antagonists such as e.g. B1-receptor antagonists, cannabinoid receptor
agonists,
such as e.g. CB2 agonists, CB1 agonists, somatostatin receptor agonists, such
as
e.g. sst2 receptor agonists.
The dosage for these active substances is conveniently 1/5 of the normally
recommended dose to 1/1 of the normally recommended dose, i.e. for example 20
to 100 mg sumatriptan.
FORMULATIONS
The compounds prepared according to the invention may be administered either
on their own or optionally in combination with other active substances for the
treatment of migraine by intravenous, subcutaneous, intramuscular, intra-
articular,
intrarectal or intranasal route, by inhalation, topically, transdermally or
orally, while
aerosol formulations are particularly suitable for inhalation. The
combinations may
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-68-
be administered either simultaneously or sequentially.
Suitable forms for administration include for example tablets, capsules,
solutions,
syrups, emulsions or inhalable powders or aerosols. The proportion of
pharmaceutically active compound or compounds should be in the range from 0.1
to 90% by weight, preferably 0.5 to 50% by weight of the total composition,
i.e. in
amounts which are sufficient to achieve the dosage range mentioned
hereinbefore.
The preparations may be given orally in the form of tablets, powders, powders
in
capsules (e.g. hard gelatine capsules), or as solutions or suspensions. When
taken by inhalation the active substance combination may be administered as a
powder, an aqueous or aqueous-ethanolic solution or by means of a propellant
gas formulation.
Preferably, therefore, pharmaceutical formulations are characterised in that
they
contain one or more compounds of formula I according to the preferred
embodiments described hereinbefore.
It is particularly preferable if the compounds of formula I are administered
orally,
and it is most preferable if they are administered once or twice a day.
Suitable
tablets may be obtained, for example, by mixing the active substances with
known
excipients, for example inert diluents such as calcium carbonate, calcium
phosphate or lactose, disintegrants such as corn starch or alginic acid,
binders
such as starch or gelatine, lubricants such as magnesium stearate or talc
and/or
agents for delaying release, such as carboxymethyl cellulose, cellulose
acetate
phthalate, or polyvinyl acetate. The tablets may also comprise several layers.
Coated tablets may be prepared accordingly by coating cores produced
analogously to the tablets with substances normally used for tablet coatings,
for
example collidone or shellac, gum arabic, talc, titanium dioxide or sugar. To
achieve delayed release or prevent incompatibilities the core may also consist
of a
number of layers. Similarly the tablet coating may consist of a number or
layers to
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-69-
achieve delayed release, possibly using the excipients mentioned above for the
tablets.
Syrups containing the active substances or combinations thereof according to
the
invention may additionally contain a sweetener such as saccharine, cyclamate,
glycerol or sugar and a flavour enhancer, e.g. a flavouring such as vanillin
or
orange extract. They may also contain suspension adjuvants or thickeners such
as
sodium carboxymethyl cellulose, wetting agents such as, for example,
condensation products of fatty alcohols with ethylene oxide, or preservatives
such
as p-hydroxybenzoates.
Capsules containing one or more active substances or combinations of active
substances may for example be prepared by mixing the active substances with
inert carriers such as lactose or sorbitol and packing them into gelatine
capsules.
Suitable suppositories may be made for example by mixing with carriers
provided
for this purpose, such as neutral fats or polyethyleneglycol or the
derivatives
thereof.
Excipients which may be used include, for example, water, pharmaceutically
acceptable organic solvents such as paraffins (e.g. petroleum fractions),
vegetable
oils (e.g. groundnut or sesame oil), mono- or polyfunctional alcohols (e.g.
ethanol
or glycerol), carriers such as e.g. natural mineral powders (e.g. kaolins,
clays, talc,
chalk), synthetic mineral powders (e.g. highly dispersed silicic acid and
silicates),
sugars (e.g. cane sugar, lactose and glucose), emulsifiers (e.g. lignin, spent
sulphite liquors, methylcellulose, starch and polyvinylpyrr:olidone) and
lubricants
(e.g. magnesium stearate, talc, stearic acid and sodium lauryl sulphate).
For oral use the tablets may obviously contain, in addition to the carriers
specified,
additives such as sodium citrate, calcium carbonate and dicalcium phosphate
together with various additional substances such as starch, preferably potato
starch, gelatin and the like. Lubricants such as magnesium stearate, sodium
lauryl
sulphate and talc may also be used to produce the tablets. In the case of
aqueous
suspensions the active substances may be combined with various flavour
enhancers or colourings in addition to the abovementioned excipients.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-70-
It is also preferred if the compounds of formula I are administered by
inhalation,
particularly preferably if they are administered once or twice a day. For this
purpose, the compounds of formula I have to be made available in forms
suitable
for inhalation. Inhalable preparations include inhalable powders, propellant-
containing metered-dose aerosols or propellant-free inhalable solutions which
are
optionally in admixture with conventional physiologically acceptable
excipients.
Within the scope of the present invention, the term propellant-free inhalable
solutions also includes concentrates or sterile inhalable solutions ready for
use.
The formulations which may be used within the scope of the present invention
are
described in more detail in the next part of the specification.
EXPERIMENTAL SECTION
As a rule, 1H-NMR and/or mass spectra have been obtained for the compounds
prepared. Unless otherwise stated, Rf values are obtained using ready-made
silica
gel TLC plates 60 F254 (E. Merck, Darmstadt, Item no. 1.05714) without chamber
saturation.
The ratios given for the eluants relate to units by volume of the solvents in
question. The units by volume specified for NH3 refer to a concentrated
solution of
NH3 in water. '
Unless otherwise stated, the acid, base and salt solutions used for working up
the
reaction solutions are aqueous systems of the specified concentrations.
For chromatographic purification, silica gel made by Millipore (MATREXTM, 35-
70
m) is used.
The HPLC data given are measured using the parameters shown below:
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-71-
Method A:
time (min) percent by volume of water percent by volume of
(with 0.1 % formic acid) acetonitrile
(with 0.1 % formic acid)
0 95 5
4.5 10 90
10 90
5.5 90 10
Analytical column: Zorbax column (Agilent Technologies), SB (Stable Bond) C18;
3.5 pm; 4.6 x 75 mm; column temperature: 30 C; flow: 1.6 mL / min; injection
5 volume: 5 pL; detection at 254 nm
Method B:
time (min) percent by volume of water percent by volume of
(with 0.1 % formic acid) acetonitrile
(with 0.1 % formic acid)
0 95 5
9 10 90
10 90
11 95 5
Analytical column: Zorbax column (Agilent Technologies), SB (Stable Bond) C18;
10 3.5 pm; 4.6 x 75 mm; column temperature: 30 C; flow: 0.8 mL / min;
injection
volume: 5 pL; detection at 254 nm
Method C:
time (min) percent by volume of water percent by volume of
(with 0.1 % formic acid) acetonitrile
(with 0.1 % formic acid)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-72-
0 95 5
4 50 50
4.5 10 90
10 90
5.5 90 10
Analytical column: Zorbax column (Agilent Technologies), SB (Stable Bond) C18;
3.5 pm; 4.6 x 75 mm; column temperature: 30 C; flow: 1.6 mL / min; injection
volume: 5 NL; detection at 254 nm
5
Method D:
time (min) percent by volume of water percent by volume of
(with 0.04% TFA) acetonitrile
(with 0.04% TFA)
0 80 20
30 20 80
Analytical column: Waters Symmetry C8, 5 m, 4.6 X 150 mm; column
temperature: 25 C, flow: 1.3 mi/min, injection volume: 5 l, detection at 254
nm.
Method E:
time (min) percent by volume of water percent by volume of
(with 0.04% TFA) acetonitrile
(with 0.04% TFA)
0 80 20
20 80
17 20 80
Analytical column: Symmetry C8 Waters - 4.6 X 150 mm; 5 micron, flow: 1.3
ml/min, column temperature: 25 C, detection at 254 nm.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-73-
In preparative HPLC purifications as a rule the same gradients are used as
were
used to collect the analytical HPLC data.
The products are collected under mass control and the fractions containing the
product are combined and freeze-dried.
If no detailed information is given as to the configuration, it is not clear
whether it is
a pure enantiomer or whether partial or even complete racemisation has
occurred.
The following abbreviations are used in the description of the experiments:
Cyc cyclohexane
DCM dichloromethane
DIPE diisopropylether
DMF N,N-dimethylformamide
EtOAc ethyl acetate
EtOH ethanol
HATU O-(7-azabenzotriazol-1-yl)-N,N,N,M-tetramethyluronium-hexafluoro-
phosphate
AcOH acetic acid
i.vac. in vacuo (under vacuum)
MeOH methanol
MTBE tert-butylmethylether
NaOAc sodium acetate
NMP N-methylpyrroldinone
PE petroleum ether
RT ambient temperature
TBTU O-(benzotriazol-1-yl)-N,N,N,M-tetramethyluronium-tetrafluoroborate
TFA trifluoroacetic acid
THF tetrahydrofuran
Preparation of the starting compounds:
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-74-
Amine Al
Ethyl [4,4'lbipiperidinyl-1-yl-oxo-acetate
0
H'N N ~0
O
A1 a) tert-butyl 1'-ethoxyoxalyl-[4,4'lbipiperidinyl-1-carboxylate
1.68 mL (15.0 mmol) ethyl chloro-oxo-acetate were slowly added dropwise to a
solution of 4.0 g (14.9 mmol) tert-butyl [4,4']bipiperidinyl-l-carboxylate and
2.15
mL (15.4 mmol) triethylamine in 80 mL DCM while cooling with ice and the
reaction mixture was stirred for 1 h at RT. A little water was added to the
reaction
solution, the organic phase was separated off and dried over Na2SO4. After the
desiccant and solvent had been eliminated the residue was taken up in EtOAc
and
filtered through silica gel, wherein the product was eluted with EtOAc. After
evaporation i.vac. the product was obtained, which was further reacted without
purification.
Yield: 3.1 g (57% of theory)
ESI-MS: (M+NH4)+ = 386
A1 b) Ethyl [4,4'1 bipiperidinyl-1-yl-oxo-acetate
5 mL TFA were added dropwise to a solution of 3.08 g(8,.36 mmol) tert-butyl 1'-
ethoxyoxalyl-[4,4']bipiperidinyl-1-carboxylate in 40 mL DCM and the reaction
mixture was stirred for 4 h at RT. The mixture was evaporated down i.vac., the
residue was taken up in 50 mL DCM and this solution was slowly stirred into an
ice-cooled solution of 4 g Na2CO3 in 20 mL water. After the addition had ended
the
mixture was stirred for a further 15 min, the organic phase was separated off,
the
aqueous phase was extracted twice more with DCM and the combined organic
phases were dried over Na2SO4. After the desiccant and solvent had been
eliminated the product was obtained as the hydrogen carbonate salt.
Yield: 2.33 g (84% of theory)
ESI-MS: (M+H) + = 269
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-75-
Amine A2
ethyl 4-[4,4'lbipiperidinyl-1-yl-4-oxo-butanoate
0
H'N N 1[,_",y 0
O
A2a) tert-butyl 1'-(3-carboxy-propionyl)-f4,4'1bipiperidinyl-1-carboxylate
A solution of 4.07 g (41.0 mmol) succinic anhydride in 50 mL THF was slowly
added dropwise to a solution of 10.0 g (37.0 mmol) tert-butyl
[4,4']bipiperidinyl-l-
carboxylate in 100 mL THF and the reaction mixture was stirred overnight at
RT.
To complete the reaction a further 2.0 g (20.0 mmol) succinic anhydride were
added, the reaction mixture was stirred for 4 h at 50 C and overnight at RT.
The
mixture was carefully combined with 200 mL 7.5% K2CO3 solution and 200 mL
EtOAc, the organic phase was separated off, extracted again with 100 mL 7.5%
K2CO3 solution and the combined aqueous phases were acidified with 10% citric
acid solution. The aqueous phase was extracted with 300 mL EtOAc, the organic
phase was separated off and the solvent was eliminated i.vac..
Yield: 11.7 g (85% of theory)
ESI-MS: (M-H)" = 367
2o A2b) ethyl 4-[4,4'lbipiperidinyl-1-yl-4-oxo-butanoate
A solution of 11.7 g (31.7 mmol) tert-butyl 1'-(3-carboxy-propionyl)-
[4,4']bipiperidinyl-1-carboxylate in 250 mL ethanolic HCI (1.25 M) was stirred
overnight at RT. The mixture was evaporated to dryness i.vac., the residue was
taken up in 100 mL EtOAc and 100 mL 15% K2C03 solution, the organic phase
was separated off and dried over Na2SO4. After the desiccant and solvent had
been eliminated the product was further reacted without purification.
Yield: 4.3 g (46% of theory)
ESI-MS: (M+H)+ = 297
Preparation of the final compounds:
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-76-
Example 1
(R)-2-[4-(4-methyl-piperazin-1 -yl)-piperidin-1 -yl]-2-oxo-1 -(3-
trifluoromethyl-benzyl)-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yi)-piperidine-l-
carboxylate
\ CF3
N-CN O II N~/ CHa
O
H O
1 a) (Z,E)-2-acetylamino-3-(3-trifluoromethyl-phenyl)-acrylic acid
A solution of 15.8 g (115 mmol) 3-trifluoromethyl-benzaldehyde and 21.3 g (182
mmol) N-acetylglycine in 60 mL acetic anhydride was heated for 4 h to 120 C
(bath temperature). After cooling 40 mL water were slowly added and the
mixture
was stirred for 1 h at 80 C. After cooling it was combined with 400 mL water
and
150 mL toluene, stirred for 1 h, the precipitate formed was filtered off,
washed with
toluene and dried at 50 C.
Yield: 16.7 g (53% of theory)
ESI-MS: (M+H)+ = 274
Rf = 0.4 (silica gel, DCM/MeOH/AcOH 90:10:1)
1 b) 2-oxo-3-(3-trifluoromethyl-phenyl)-propionic acid
2o 200 mL 4 M HCL were added dropwise to a suspension of 16.6 g (60.8 mmol)
(Z,E)-2-acetylamino-3-(3-trifluoromethyl-phenyl)-acrylic acid in 200 mL 1,4-
dioxane
heated to 80 C (bath temperature), the reaction mixture was refluxed for 1.5 h
and
then stirred overnight at RT. To complete the reaction the mixture was
refluxed for
a further 4 h. The organic solvent was eliminated i.vac., the aqueous residue
was
combined with NaOH until an alkaline reaction occurred, washed with MTBE and
acidified with HCI. The mixture was extracted exhaustively with EtOAc and the
combined organic phases were dried over MgSO4. After the desiccant and solvent
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-77-
had been eliminated the desired product was obtained, which was further
reacted
without purification.
Yield: 5.7 g (40% of theory)
ESI-MS: (M-H)- = 231
Rf = 0.19 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
1 c) (R)-2-hydroxy-3-(3-trifluoromethyl-phenyl)-propionic acid
Under a nitrogen atmosphere a solution of 11.8 g (36.8 mmol) (1 R)-B-
chlorodiisopinocampheylborane in 40 mL THF was added dropwise to a solution,
cooled to -35 C, of 5.70 g (24.6 mmol) 2-oxo-3-(3-trifluoromethyl-phenyl)-
propionic
acid and 4.4 mL (31.4 mmol) triethylamine in 60 mL THF, within 15 min, and the
reaction solution was stirred overnight at RT. Then the reaction solution was
carefully made alkaline with 20 mL 1 M NaOH while cooling with ice and stirred
for
30 min. 40 mL EtOAc was added, the aqueous phase was separated off, the
organic phase was washed three times with 15 mL 1 M NaOH solution and once
with 15 mL water. The combined aqueous phases were acidified with
semiconcentrated HCI, extracted twice with 40 mL EtOAc and the combined
organic phases were dried over MgSOa. After the desiccant and solvent had been
eliminated the product was obtained, which was further reacted without
purification.
Yield: 4.25 g (74% of theory)
ESI-MS: (M-H)- = 233
Rf = 0.35 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
1 d) methyl (R)-2-hyd roxy-3-(3-trifl uo romethyl-p he nyl)-pro p ion ate
A solution of 4.20 g (17.9 mmol) (R)-2-hydroxy-3-(3-trifluoromethyl-phenyl)-
propionic acid in 80 mL methanolic HCI was stirred for 70 h at RT. The
reaction
solution was evaporated down i.vac., the residue was taken up in water and
exhaustively extracted with EtOAc. The combined organic phases were washed
three times with 15% K2C03 solution and once with water and dried over MgSO4.
CA 02600189 2007-09-06
WO 2006/100026 PCT/EP2006/002557
-78-
After the desiccant and solvent had been eliminated the residue was further
reacted without purification.
Yield: 2.47 g (55% of theory)
ESI-MS: (M+H)+ = 249
Rf = 0.73 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
1 e) (R)-1-methoxycarbonyl-2-(3-trifluoromethyl-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
Under a nitrogen atmosphere 2.10 g (10.4 mmol) 4-nitrophenyl chloroformate
were
added to a solution of 1.64 g (13.4 mmol) 4-dimethylaminopyridine in 40 mL
pyridine and stirred for 3 h at RT. Then 2.47 g (9.95 mmol) methyl (R)-2-
hydroxy-
3-(3-trifluoromethyl-phenyl)-propionate in 20 mL pyridine were added and the
reaction mixture was stirred for 2.5 h at RT. The reaction solution was
combined
with 4.1 g (10.9 mmol) 3-piperidin-4-y1-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-
one
(purity 65%) and stirred for 20 h at RT. The mixture was evaporated down
i.vac.,
the residue was taken up in 60 mL EtOAc, the organic phase was washed twice
with 15% K2C03 solution, four times with saturated NaHCO3 solution and dried
over Na2SO4. After the desiccant and solvent had been eliminated the residue
was
purified by chromatography (silica gel, Cyc/EtOAc 1:2).
Yield: 3.16 g(61 % of theory)
ESI-MS: (M+H)+ = 520
Rf = 0.93 (silica gel, EtOAc/MeOH/NH3 80:20:2)
1 f) (R)-1-carboxy-2-(3-trifluoromethyl-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
A solution of 0.22 g (9.00 mmol) LiOH in 40 mL water was added to a solution
of
3.10 g (5.97 mmol) (R)-1-methoxycarbonyl-2-(3-trifluoro-methyl-phenyl)-ethyl 4-
(2-
oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate in 80
mL
THF and the reaction mixture was stirred for 1 h at RT. The organic solvent
was
eliminated i.vac., the aqueous residue was combined with 80 mL water and
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-79-
acidified with 1 M HCI. The precipitate formed was suction filtered and dried
overnight at 35 C.
Yield: 2.80 g (93% of theory)
ESI-MS: (M+H)+ = 506
Rf = 0.58 (silica gel, EtOAc/MeOH/NH3 70:30:3)
1 g) (R)-244-(4-methyl-piperazin-1-yl)-piperidin-1-yll-2-oxo-1-(3-
trifluoromethyl-
benzyl)-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-carboxylate
30 mg (0.16 mmol) 1-methyl-4-piperidin-4-yl-piperazine were added at RT to a
solution of 80.0 mg (0.16 mmol) (R)-1-carboxy-2-(3-trifluoromethyl-phenyl)-
ethyl 4-
(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate, 61
mg
(0.19 mmol) TBTU and 27 pL (0.20 mmol) triethylamine in 1 mL DMF and the
reaction solution was stirred for 70 h at RT. This was purified by HPLC
without any
further working up; the fractions containing the product were combined and
lyophilised.
Yield: 67 mg (63% of theory)
ESI-MS: (M+H) + = 671
Rf = 0.4 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
The following compounds were prepared analogously from in each case 80 mg
(Examples 1.1 to 1.4) or 140 mg (Examples 1.5 and 1.6) (R)-1-carboxy-2-(3-
trifluoro-methyl-phenyl)-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-l-carboxylate and the corresponding amount of amine:
cILCF3
~\N O~R
-{~/.
N~ 0
H
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-80-
Example R Yield (%) Mass Rf )
spectrum (method)
1.1 93 656 0.68
[M+H]+
1.2 64 671 0.35
[M+H]+
1.3 74 670 0.53
[M+H]+
1.4 " 48 685 5.1 min
NHZ [M+H]+ (B)
1.5 * 62 757 0.51
[M+H]+
1.6 * 71 757 0.50
[M+H]+
)(silica gel, DCM/MeOH/ Cyc/NH3 70:15:15:2) or retention time HPLC
Example 1.7
(R)-1-(3-methyl-benzyl)-2-oxo-2-(4-piperazin-1-yl-piperidin-1-yl)-ethyl 4-(2-
oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
a
CF3
~NON~~H
N~ 0 H O
A solution of 131 mg (0.17 mmol) tert-butyl 4-{1-[(R)-2-[4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1 -carbonyloxy]-3-(3-
trifluoromethyl-
phenyl)-propionyl]-piperidin-4-yl}-piperazine-l-carboxylate (Example 1.5) in
1.5 mL
4 M HCI was stirred overnight at RT. The reaction solution was purified by
HPLC
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-81-
without any further working up. The fractions containing the product were
combined and lyophilised.
Yield: 75 mg (67% of theory)
ESI-MS: (M+H) + = 657
Rf = 0.38 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 1.8
(R)-2-oxo-2-(4-piperidin-4-yl-piperazin-1-yl)-1-(3-trifluoromethyl-benzyl)-
ethyl 4-(2-
oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
I~
/ CF3
~I \ N1OyN~~N~ \N'H
0
H 0
Analogously to Example 1.7 the product was obtained from 149 mg (0.20 mmol)
(R)-1 -(3-trifluoromethyl-benzyl)-2-[4-(1 -tert-butoxy-carbonyl-piperidin-4-
yl)-
piperazin-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-l-carboxylate (Example 1.6).
Yield: 66 mg (51 % of theory)
ESI-MS: (M+H)+ = 657
Rf = 0.18 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 2
(R)-2-[4-(4-methyl-piperazin-1-yl)-piperidin-1-yl]-2-oxo-1-(3-methyl-benzyl)-
ethyl 4-
(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
~I \ N NO~N\~/
-C
H 0
~
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-82-
2a) (Z,E)-2-acetylamino-3-m-tolyl-acrylic acid
Analogously to Example 1a the product could be obtained from 25.0 g (212 mmol)
3-methyl-benzaldehyde and 24.9 g (212 mmol) N-acetylglycine.
Yield: 26.0 g (56% of theory)
ESI-MS: (M+H) + = 220
retention time (HPLC): 5.4 min (method B)
2b) 2-oxo-3-m-tolyl-propionic acid
400 mL 4 M HCI were added to a solution of 13.0 g (59.3 mmol) (Z,E)-2-
acetylamino-3-m-tolyl-acrylic acid in 200 mL 1,4-dioxane and the reaction
mixture
was refluxed for 2.5 h. The organic solvent was evaporated down i.vac., the
precipitate formed was suction filtered and dried. This was taken up in 300 mL
EtOAc, the organic phase was washed twice with 200 mL water and dried over
Na2SO4. After the desiccant and solvent had been eliminated the residue was
reacted further without purification.
Yield: 9.2 g (88% of theory)
ESI-MS: (M-H)- = 177
retention time (HPLC): 7.3 min (method B)
2c) (R)-2-hydroxy-3-m-tolyl-propionic acid
Analogously to Example 1 c the product could be obtained from 9.24 g (51.9
mmol)
2-oxo-3-m-tolyl-propionic acid and 24.0 g (74.8 mmol) (1 R)-B-
chlorodiisopinocampheylborane.
Yield: 8.4 g (90% of theory)
ESI-MS: (M-H)- = 179
retention time (HPLC): 7.2 min (method B)
2d) methyl (R)-2-hydroxy-3-m-tolyl-propionate
3.74 mL (51.28 mmol) SOCI2 were slowly added dropwise to a solution, cooled to
0 C, of 8.40 g (46.6 mmol) (R)-2-hydroxy-3-m-tolyl-propionic acid in 200 mL
MeOH and after the addition had ended the reaction mixture was stirred for 1 h
at
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-83-
RT. The mixture was evaporated down i.vac. and the residue was purified by
chromatography (silica gel, Cyc/EtOAc 3:1).
Yield: 6.28 g (69% of theory)
ESI-MS: (M+H)+ = 195
retention time (HPLC): 6.9 min (method B)
2e) (R)-1-methoxycarbonyl-2-m-tolyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-1-carboxylate
Analogously to Example le the product could be obtained from 1.12 g (5.76
mmol)
methyl (R)-2-hydroxy-3-m-tolyl-propionate and 1.41 g (5.76 mmol) 3-piperidin-4-
yl-
1,3,4,5-tetrahydro-1,3-benzodiazepin-2-one.
Yield: 2.07 g (77% of theory)
ESI-MS: (M+H) + = 466
retention time (HPLC): 9.0 min (method B)
2f) (R)-1-carboxy-2-m-tolyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate
Analogously to Example 1f the product could be obtained from 2.07 g (4.45
mmol)
(R)-1-methoxycarbonyl-2-m-tolyl-ethyi 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate and 0.16 g (6.72 mmol) LiOH.
Yield: 1.86 g (93% of theory)
ESI-MS: (M+H) + = 452
retention time (HPLC): 8.0 min (method B)
2g) (R)-244-(4-methyl-piperazin-1-yl)-piperidin-1-yll-2-oxo-1-(3-methyl-
benzyl)-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
A solution of 80.0 mg (0.18 mmol) (R)-1-carboxy-2-m-tolyl-ethyl 4-(2-oxo-
1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1 -carboxylate, 57 mg (0.18
mmol)
TBTU and 31 pL (0.22 mmol) triethylamine in 1 mL DMF were stirred for 1 h at
RT.
Then 33 mg (0.18 mmol) 1-methyl-4-piperidin-4-yl-piperazine were added to the
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-84-
reaction solution, which was then stirred overnight at RT. It was purified by
HPLC
without any further working up; the fractions containing the product were
combined
and lyophilised.
Yield: 46.7 mg (43% of theory)
ESI-MS: (M+H)+ = 617
retention time (HPLC): 5.5 min (method B)
The following compounds were prepared analogously from in each case 80 mg
(R)-1-carboxy-2-m-tolyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-l-carboxylate and the corresponding amount of amine:
R
N-CN O",-r
O
H O
Example R Yield (%) Mass retention
spectrum time
HPLC
(method)
2.1 80 617 5.0 min
IM+HI+ (B)
2.2 75 616 6.2 min
N~ [M+H]+ (B)
Example 3
(R)-1-(3,5-bis-trifluoromethyl-benzyl)-2-(1'-ethoxycarbonylmethyl-4,4'-
bipiperidinyl-
1-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-
carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-85-
0 LCF,
N-CINON N~O
N" o lol 0
H
3a) methyl 2-acetylamino-3-(3,5-bis-trifluoromethyl-phenyl)acrylate
Under a nitrogen atmosphere 50.0 g (171 mmol) 3,5-bis-(trifluoromethyl)-
bromobenzene and 25.0 g (171 mmol) methyl 2-acetylamino-acrylate in 475 mL
triethylamine and 250 mL acetonitrile were combined with 3.9 g (12.4 mmol) tri-
o-
tolyl-phosphane and 2.8 g (12.5 mmol) Pd(OAc)2 and stirred for 18 h at 80 C.
After
the reaction had ended the reaction mixture was evaporated down i.vac. to
approx. 200 mL, combined with 400 mL EtOAc and 400 mL water, the precipitate
was suction filtered and the phases were separated. The organic phase was
dried
over Na2SO4, combined with activated charcoal, filtered and evaporated to
dryness. The residue was stirred with DIPE, suction filtered and dried i.vac..
Yield: 19.5 g(32 % of theory)
ESI-MS: (M+H) + = 356
Rf = 0.76 (silica gel, PE/EtOAc 1:1)
3b) 3-(3,5-bis-trifluoromethyl-phenyl)-2-oxo-propionic acid
19.5 g (54.9 mmol) methyl 2-acetylamino-3-(3,5-bis-triflu6romethyl-phenyl)-
acrylate in 100 mL 1,4-dioxane were heated to 100 C bath temperature, combined
with 100 mL 4 M HCI and stirred for 8 h at 100 C bath temperature. The
reaction
mixture was evaporated down i.vac., the crystals were suction filtered, washed
with water and dried in the drying cupboard at 50 C.
Yield: 16.1 g(98 % of theory)
ESI-MS: (M-H)- = 299
Rf = 0.18 (silica gel, EtOAc)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-86-
3c) (R)-3-(3,5-bis-trifluoromethyl-phenyl)-2-hydroxy-propionic acid
16.1 g (53.6 mmol) 3-(3,5-bis-trifluoromethyl-phenyl)-2-oxo-propionic acid in
9.5
(70.0 mmol) triethylamine and 100 mL THF were combined at -35 C with a
solution of 26.0 (81.1 mmol) (1 R)-B-chlorodiisopinocampheylborane in 40 mL
THF
within 30 min, stirred for 1 h at this temperature and stirred overnight at
RT. After
the reaction had ended the reaction mixture was made alkaline at 0 C with 160
mL
1 M NaOH, stirred for 15 min, combined with 100 mL MTBE and the phases were
separated. The organic phase was washed with 50 mL water and 50 mL 1 M
NaOH. The combined aqueous phases were acidified with 4 M HCI, exhaustively
extracted with MTBE, the combined organic phases were dried over Na2SO4,
suction filtered through activated charcoal and evaporated down i.vac.. The
product was reacted further without purification.
Yield: 12.5 g (77% of theory)
ESI-MS: (M-H)- = 301
Rf = 0.45 (silica gel, EtOAc)
3d) methyl (R)-3-(3 5-bis-trifluoromethyl-phenyl)-2-hydroxy-propionate
12.5 g (41.4 mmol) of (R)-3-(3,5-bis-trifluoromethyl-phenyl)-2-hydroxy-
propionic
acid in 150 mL methanolic HCI (1.25 M) were stirred for 4 h at RT and then
evaporated down i.vac.. The residue was taken up in EtOAc and washed with
saturated NaHCO3 solution, the organic phase was dried over Na2SO4, suction
filtered through activated charcoal and evaporated down i.vac.. The residue
was
stirred with PE, suction filtered and evaporated down i.vac.. The product was
reacted further without purification.
Yield: 11.4 g (87% of theory)
ESI-MS: (M+H)+ = 316
Rf = 0.80 (silica gel, PE/EtOAc 1:1)
3e) (R)-2-(3 5-bis-trifluoromethyl-phenyl)-1-methoxycarbonyi-ethyl 4-(2-oxo-
1 2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
Analogously to Example 1 e the product was obtained from 6.0 g (8.2 mmol)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
" = -87-
methyl (R)-3-(3,5-bis-trifluoromethyl-phenyl)-2-hydroxy-propionate and 5.13 g
(20.9 mmol) 3-piperidin-4-yl-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-one.
Purification was carried out by chromatography (silica gel, gradient PE/EtOAc
1:1
to 1:9).
Yield: 5.1 g (46 % of theory)
ESI-MS: (M+H)+ = 588
Rf = 0.63 (silica gel, DCM/MeOH/cyc/NH3 70:15:15:2)
3f) (R)-2-(3 5-bis-trifluoromethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
A solution of 307 mg (12.8 mmol) LiOH in 5 mL water was added to a solution of
5.0 g (8.5 mmol) of (R)-2-(3,5-bis-trifluoromethyl-phenyl)-1-methoxy-carbonyl-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
in 50
mL THF and the reaction mixture was stirred overnight at RT. The mixture was
evaporated down i.vac., the residue was taken up in water, acidified with 1 M
HCI,
the precipitate was filtered off and dried in the vacuum drying cupboard at 40
C.
Yield: 4.5 g (92% of theory)
ESI-MS: (M+H) + = 574
Rf = 0.32 (silica gel,DCM/MeOH/cyc/NH3 70:15:15:2)
3g) (R)-1-(3 5-bis-trifluoromethyl-benzyl)-2-(1'-ethoxycarbonylmethyl-4,4'-
bipiperidinyl-l-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-1-carboxylate
50 mg (0.20 mmol) ethyl [4,4']bipiperidinyl-1-yl-acetate were added at RT to a
solution of 100 mg (0.17 mmol) (R)-2-(3,5-bis-trifluoromethyl-phenyl)-1-
carboxy-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-benzodiazepin-3-yl)-piperidine-1-
carboxylate, 61
mg (0.20 mmol) TBTU and 33 pL (0.16 mmol) ethyldiisopropylamine in 1 mL DMF
and the reaction mixture was stirred overnight at RT. The reaction solution
was
filtered through a syringe filter and purified directly by HPLC without any
further
working up. The fractions containing the product were combined and
lyophilised.
Yield: 55 mg (39% of theory)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= -88-
ESI-MS: (M+H) + = 810
retention time (HPLC-MS): 7.3 min (method B)
Example 3.1
(R)-1-(3,5-bis-trifluoromethyl-benzyl)-2-(1'-carboxymethyl-4,4'-bipiperidinyl-
1-yl)-2-
oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
0II LCF,
N~NJ-O~N OH
N O O 0
H
A solution of 1.5 mg (0.06 mmol) LiOH in 1 mL water was added to a solution of
35
mg (0.04 mmol) (R)-1-(3,5-bis-trifluoromethyl-benzyl)-2-(1'-
ethoxycarbonylmethyl-
4,4'-bipiperidinyl-1-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-l-carboxylate (Example 3g) in 5 mL THF and the reaction
solution
was stirred overnight at RT. The mixture was evaporated down i.vac., the
residue
was taken up in water, acidified with 1 N HCI, the precipitate was filtered
off and
dried in the vacuum drying cupboard.
Yield: 15 mg (44% of theory)
ESI-MS: (M+H)+ = 782
Rf = 0.41 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 3.2
(R)-2-(4-amino-4-methyl-1,4'-bipiperidinyl-1'-yl)-1-(3,5-bis-trifluoromethyl-
benzyl)-
2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
' = -89-
F3
~ \
/ CF3
~I \ N ~N
N-~ ~0/ I ~N\/ _ l ~ /N NHZ
IO ~ CH3
H O
52 mg (0.14 mmol) tert-butyl (4-methyl-[1,4']bipiperidinyl-4-yl)-carbamate
(used as
the bis-hydrochloride salt) were added at RT to a solution of 80.0 mg (0.14
mmol)
(R)-2-(3, 5-bis-trifluoromethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4, 5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate, 58 mg (0.18 mmol) TBTU and
140 pL (1.00 mmol) triethylamine in 1.8 mL DMF and the reaction solution was
shaken overnight at RT. It was purified by HPLC without any further working
up;
the fractions containing the product were combined and lyophilised. The
coupling
product was dissolved in 4 mL DCM, the solution was combined with 0.5 mL TFA,
stirred for 5 h at RT and then left overnight at RT during which time the
solvent
evaporated. The residue was taken up in 2 mL 15% K2C03 solution, extracted
twice with 2 mL DCM and the combined organic phases were shaken overnight
during which time the solvent evaporated. The residue was purified by HPLC;
the
fractions containing the product were combined and lyophilised, and the
product
was obtained as the formate salt.
Yield: 47 mg (42% of theory)
ESI-MS: (M+H)+ = 753
retention time (HPLC): 5.4 min (method B)
Example 4
(R)-1 -(4-amino-3,5-dimethyl-benzyl)-2-(4-morpholin-4-yl-piperidin-1 -yl)-2-
oxo-ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
H3
NH2
CH3
Gfl0 .~/ "'/ %/~
O~ 0
N 0
H 0
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-90-
4a) methyl (Z E)-2-acetylamino-3-(4-amino-3,5-dimethyl-phenyl)-acrylate
Under a nitrogen atmosphere first of all a solution of 90.0 g (441 mmol) 4-
bromo-
2,6-dimethyl-phenylamine in 200 mL acetonitrile was added to a mixture of 7.2
g
(32.1 mmol) Pd(OAc)2 and 10.1 g (32.1 mmol) tri-o-tolyl-phosphane in 1.2 L
triethylamine and 600 mL acetonitrile and then a solution of 65.0 g (445 mmol)
methyl 2-acetylamino-acrylate in 200 mL acetonitrile was added dropwise. After
the addition had ended the mixture was stirred for 18 h at 80 C. To complete
the
reaction the reaction mixture was again combined with 4.0 g (17.8 mmol)
Pd(OAc)2 and 5.0 g(16.4 mmol) tri-o-tolyl-phosphane and kept for another 5 h
at
80 C. The mixture was evaporated down i.vac to approx. 200 mL, the residue was
combined with 400 mL EtOAc, the residue (A) was filtered and the organic phase
was dried over Na2SO4. After elimination of the desiccant by filtration over
activated charcoal the filtrate was evaporated down to about 100 mL, the
precipitated substance was suction filtered, washed with 30 mL EtOAc and
dried.
The above residue A was combined with 1 L DCM, Na2SO4 and activated charcoal
and filtered through Celite. The filtrate was evaporated down, the residue was
combined with 350 mL diethyl ether, the precipitate formed was suction
filtered,
then washed with 100 mL diethyl ether and dried. The two product fractions
were
combined.
Yield: 74.8 g (65% of theory)
ESI-MS: (M+H)+ = 263
Rf = 0.51 (silica gel, EtOAc)
4b) 3-(4-amino-3,5-dimethyl-phenyl)-2-oxo-propionic acid
A suspension of 74.0 g (282 mmol) methyl (Z,E)-2-acetylamino-3-(4-amino-3,5-
dimethyl-phenyl)-acrylate in 500 mL 1,4-dioxane was heated to 100 C and
combined with 460 mL 4 M HCI, whereupon a solution was formed. The mixture
was heated for another 8 h at 100 C and the cooled solution was evaporated
down i.vac. to approx. 200 mL, during which time the product crystallised out.
It
was filtered, the residue was washed with 50 mL water and the product was
dried
at 50 C.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= = -91 -
Yield: 43.6 g (63% of theory)
ESI-MS: (M+H) + = 208
Rf = 0.68 (silica gel, PE/EtOAc 1:1)
4c) methyl (R)-3-(4-amino-3,5-dimethyl-phenyl)-2-hydroxy-propionate
Under a nitrogen atmosphere a mixture of 20.0 g (82.1 mmol) 3-(4-amino-3,5-
dimethyl-phenyl)-2-oxo-propionic acid and 25.7 mL (189 mmol) triethylamine in
400 mL THF was cooled to -35 C. Then a solution of 40.0 g (125 mmol) (1 R)-B-
chlorodiisopinocampheyfborane in 100 mL THF was added dropwise so that the
reaction temperature remained between -35 C and -25 C. The reaction mixture
was kept for 1 h at this temperature, the cooling bath was removed and the
reaction mixture was stirred overnight at RT. THF was evaporated off i.vac.,
the
residue was combined with methanolic HCI (1.25 M) and stirred for 2 h at RT.
It
was evaporated down i.vac., the residue was taken up in 2 M HCI and extracted
exhaustively with EtOAc. The aqueous phase was made alkaline with
semiconcentrated NaOH and exhaustively extracted with EtOAc. The combined
organic phases were dried over Na2SO4, suction filtered through activated
charcoal and evaporated down. The product was obtained as a brown oil.
Yield: 8.3 g (45% of theory)
ESI-MS: (M+H)+ = 224
Rf = 0.46 (silica gel, PE/EtOAc 1:1)
4d) (R)-2-(4-amino-3 5-dimethyl-phenyl)-1-methoxycarbonyl-ethyl 4-(2-oxo-
1,2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
Analogously to Example 1 e the desired product was obtained from 4.0 g (17.9
mmol) methyl (R)-3-(4-amino-3,5-dimethyl-phenyl)-2-hydroxy-propionate and 4.8
g
(19.6 mmol) 3-piperidin-4-y1-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-one.
Yield: 3.2 g (36% of theory)
ESI-MS: (M+H)+ = 495
Rf = 0.35 (silica gel, DCM/MeOH/NH3 90:10:1)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-92-
4e) (R)-2-(4-amino-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
A solution of 500 mg (20.9 mmol) LiOH in 10 mL water was added to a solution
of
6.7 g (13.6 mmol) of (R)-2-(4-amino-3,5-dimethyl-phenyl)-1-methoxycarbonyl-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
in 50
mL THF and the reaction mixture was stirred overnight at RT. To complete the
reaction another 300 mg (12.5 mmol) LiOH were added and the reaction solution
was stirred for 3 h at 40 C. It was evaporated down i.vac., the residue was
taken
up in 15% K2C03 solution and extracted exhaustively with DCM. The aqueous
phase was acidified with 4 M HCI, exhaustively extracted with DCM and the
combined organic phases were dried over Na2SO4. After the desiccant and
solvent
had been eliminated the residue was reacted further without purification.
Yield: 4.2 g (65% of theory)
ESI-MS: (M+H) + = 481
Rf = 0.21 (silica gel, DCM/MeOH/cyc/NH3 70:15:15:2)
4f) (R)-1-(4-amino-3 5-dimethyl-benzyl)-2-(4-morpholin-4-yl-piperidin-1-yl)-2-
oxo-ethyl 4-(2-oxo-1 2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
Analogously to Example 3g the product was obtained from 80 mg (0.17 mmol) (R)-
2-(4-amino-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-
1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate and 32 mg (0.19 mmol) 4-piperidin-
4-
yl-morpholine.
Yield: 52 mg (49% of theory)
ESI-MS: (M+H)+ = 633
retention time (HPLC): 4.9 min (method B)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-93-
Example 4.1
(R)-1-(4-amino-3,5-dimethyl-benzyl)-2-(1'-ethoxycarbonylmethyl-4,4'-
bipiperidinyl-
1-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-l-
carboxylate
O NHZ
N~NON N~ /O
N' ~O O O
H
Analogously to Example 3g the product was obtained from 100 mg (0.21 mmol)
(R)-2-(4-amino-3, 5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate and 59 mg (0.23 mmol) ethyl
[4,4']bipiperidinyl-1 -yl-acetate.
Yield: 58 mg (39% of theory)
ESI-MS: (M+H) + = 717
retention time (HPLC): 4.7 min (method B)
Example 4.2
(R)-1 -(4-amino-3,5-dimethyl-benzyl)-2-(1'-carboxymethyl-4,4'-bipiperidinyl-1-
yl)-2-
oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yI)-piperidine-1-
carboxylate
NHZ
/
0
I ~ N~NJ~O/~N N~ OH
/ ( TI'f
O O
H O
A solution of 3.1 mg (0.13 mmol) LiOH in 1 mL water was added to a solution of
55
mg (0.08 mmol) (R)-1-(4-amino-3,5-dimethyl-benzyl)-2-(1'-ethoxycarbonylmethyl-
4,4'-bipiperidinyl-1-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-l-carboxylate (Example 4.1) in 5 mL THF and the reaction
mixture
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-94-
was stirred overnight at RT. The mixture was evaporated down i.vac., the
residue
was taken up in 1 mL DMF and the crude product was purified by HPLC. The
fractions containing the product were combined and lyophilised.
Yield: 22 mg (42% of theory)
ESI-MS: (M+H) + = 689
retention time (HPLC): 4.8 min (method B)
Example 5
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-(4-morpholin-4-yl-piperidin-1-yl)-2-
oxo-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
O ~ OH
~ /
N~N~O--,iy N\ '
O
1 O
H
5a) 2-benzyloxy-5-bromo-1,3-dimethylbenzene
39.9 g (286 mmol) K2C03 were added to a solution of 50.0 g (249 mmol) 2,6-
dimethyl-4-bromophenol in 500 mL DMF and stirred for 20 min. Then 34.0 mL
(286 mmol) benzylchloride were slowly added dropwise and the reaction mixture
was stirred for 3 h at 100 C bath temperature. After the reaction had ended
the
mixture was poured onto 500 mL water and exhaustively extracted with EtOAc.
The organic phases were combined, dried over Na2SO4 and evaporated down
i.vac..
Yield: quantitative
GC-MS: (M+) = 290/292 (Br)
Rf = 0.87 (silica gel, cyc/EtOAc 3:1)
5b) methyl 2-acetylamino-3-(4-benzyloxy-3,5-dimethyl-phenyl)-acrylate
Under a nitrogen atmosphere a mixture of 40.0 g (137 mmol) 2-benzyloxy-5-
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-95-
bromo-1,3-dimethylbenzene and 24.1 g (165 mmol) methyl 2-acetylamino-acrylate
in 420 mL triethylamine and 200 mL acetonitrile was combined with 3.5 g(11.2
mmol) tri-o-tolyl-phosphane and 2.5 g (11.1 mmol) Pd(OAc)2 and the mixture was
stirred for 18 h at 80 C. The precipitate was suction filtered, the filtrate
was
evaporated down i.vac. and combined with 800 mL DCM and 800 mL water. The
organic phase was separated off, suction filtered over Na2SO4, the solvent was
removed i.vac., the residue was stirred with EtOAc, suction filtered and dried
i.vac..
Yield: 31.1 g (64% of theory)
ESI-MS: (M+H) + = 354
retention time (HPLC-MS): 8.6 min (method B)
5c) 3-(4-benzyloxy-3,5-dimethyl-phenyl)-2-oxo-gropionic acid
31.1 g (88.1 mmol) methyl 2-acetylamino-3-(4-benzyloxy-3,5-dimethyl-phenyl)-
acrylate in 150 mL 1,4-dioxane were combined with 125 mL 4 M HCI, stirred for
7
h at reflux temperature and stirred overnight at RT. The precipitate was
suction
filtered, washed with water and dried at 45 C in the vacuum drying cupboard.
Yield: 14.3 g(54 % of theory)
El-MS: (M) + = 298
retention time (HPLC-MS): 9.0 min (method B)
5d) (R)-3-(4-benzoyl-3 5-dimethyl-phenyl)-2-hydroxy-propionic acid
Under a nitrogen atmosphere a solution of 14.3 g (47.8 mmol) 3-(4-benzyloxy-
3,5-
dimethyl-phenyl)-2-oxo-propionic acid and 8.3 mL (59.8 mmol) triethylamine in
170
mL THF at -35 C was combined with a solution of 22.1 (69.0 mmol) (1R)-B-
chlorodiisopinocampheylborane in 70 mL THF within 30 min. After the addition
had
ended the cooling bath was removed and the reaction solution was stirred
overnight at RT. The reaction mixture was made alkaline at 0 C with 70 mL 1 M
NaOH, combined with 100 mL MTBE, stirred for 15 min and the phases were
separated. The organic phase was washed with 50 mL water and three times with
50 mL 1 M NaOH. The combined aqueous phases were acidified with semiconc.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-96-
HCI, exhaustively extracted with EtOAc and the combined organic phases were
dried over Na2SO4. After the desiccant and solvent had been eliminated the
residue was reacted further without purification.
Yield: 14.0 g (98% of theory)
ESI-MS: (M-H)- = 299
retention time (HPLC-MS): 7.9 min (method B)
5e) methyl (R)-3-(4-benzyloxy-3,5-dimethyl-phenyl)-2-hydroxy-propionate
To a solution cooled to 0 C of 14.0 g (23.3 mmol) of (R)-3-(4-benzoyl-3,5-
dimethyl-
phenyl)-2-hydroxy-propionic acid in 150 mL MeOH, 2.0 mL (27.4 mmol) SOC12
were added dopwise and the reaction mixture was stirred for 1 h at RT. The
reaction solution was evaporated down i.vac. and the residue was purified by
chromatography (silica gel, cyc/EtOAc 3:1).
Yield: 5.7 g (78% of theory)
ESI-MS: (M+NH4)+ = 332
retention time (HPLC-MS): 9.1 min (method B)
5f) (R)-2-(4-benzyloxy-3 5-dimethyl-phenyl)-1-methoxycarbonyl-ethyl 4-(2-oxo-
1 2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
Under a nitrogen atmosphere 1.93 g (9.58 mmol) 4-nitrophenyl chloroformate was
added to a solution of 1.17 g (9.58 mmol) 4-dimethylaminopyridine in 50 mL
pyridine, stirred for 1.5 h at RT, combined with 3.0 g (9.58 mmol) methyl (R)-
3-(4-
benzyloxy-3,5-dimethyl-phenyl)-2-hydroxy-propionate and stirred for 20 min at
RT.
Then 2.35 g (9.58 mmol) 3-piperidin-4-yl-1,3,4,5-tetrahydro-1,3-benzodiazepin-
2-
one were added and the mixture was stirred for 20 h at RT. The reaction
mixture
was evaporated down i.vac., the residue was taken up in EtOAc, the organic
phase was washed with 10% KHSO4 and saturated NaHCO3 solution and dried
over Na2SO4. After the desiccant and solvent had been eliminated the residue
was
purified by chromatography (silica gel, gradient cyc/EtOAc 1:1 to 1:2 ).
Yield: 3.21 g (57% of theory)
ESI-MS: (M+H) + = 586
. . WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
' -97-
retention time (HPLC-MS): 10.4 min (method A)
5g) (R)-2-(4-benzyloxy-3 5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
A solution of 3.21 g (5.48 mmol) of (R)-2-(4-benzyloxy-3,5-dimethyl-phenyl)-1-
methoxy-carbonyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yi)-
piperidine-l-carboxylate in 80 mL THF was combined with a solution of 200 mg
(8.35 mmol) LiOH in 40 mL water and stirred for 1 h at RT. The reaction
mixture
was evaporated down i.vac., the residue was taken up in 100 mL water,
acidified
with 2 M HCI, the precipitate was suction filtered and dried in the vacuum
drying
cupboard at 40 C.
Yield: quantitative
ESI-MS: (M+H)+ = 572
retention time (HPLC-MS): 9.2 min (method B)
5h) (R)-2-(4-hydroxy-3 5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetra hyd ro-1 3-be nzod iazepi n-3-yi )-pi pe rid i ne-l-ca rboxylate
3.72 g (6.51 mmol) of (R)-2-(4-benzyloxy-3,5-dimethyl-phenyl)-1-carboxy-ethyi
4-
(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate in
50
mL DCM were combined with 300 mg 10% Pd/C and shaken at RT and 3000 hPa
hydrogen pressure until the reaction came to a stop. The.catalyst was suction
filtered and the solvent was evaporated down i.vac.. The residue was
triturated
with DIPE and suction filtered.
Yield: 2.41 g (77% of theory)
ESI-MS: (M+H)+ = 482
retention time (HPLC-MS): 7.0 min (method B)
5i) (R)-1 -(4-hydroxy-35-dimethyl-benzyl)-2(4-morpholin-4-yl-piperidin-1-yl)-2-
oxo-ethyl 4-(2-oxo-1 2 4,5-tetrahydro-1,3-benzodiazepin-3- rLl)-piperidine-1-
carboxylate
. WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
' -98-
A solution of 70 mg (0.15 mmol) of (R)-2-(4-hydroxy-3,5-dimethyl-phenyl)-1-
carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate, 51 mg (0.16 mmol) TBTU and 26 pL (0.18 mmol) triethylamine in 1
mL DMF was stirred for 1 h at RT. Then 25 mg (0.15 mmol) 4-piperidin-4-yl-
morpholine was added to the reaction solution, which was then stirred for 16 h
at
RT. The reaction solution was purified by HPLC without any further working up;
the fractions containing the product were combined and lyophilised.
Yield: 35 mg (38% of theory)
ESI-MS: (M+H) + = 634
retention time (HPLC-MS): 5.6 min (method B)
Example 5.1
(R)-2-(4-hydroxy-1,4'-bipiperidinyl-1'-yi)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-
oxo-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
OH
I /
N~/ ~ N OH
N-CNO-
~! O
N'~\
H O
Analogously to Example 5i the product was obtained from 70 mg (0.15 mmol) (R)-
2-(4-hydroxy-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-
1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate and 27 mg (0.15 mmol)
[1,4']bipiperidinyl-4-ol.
Yield: 35 mg (37% of theory)
ESI-MS: (M+H) + = 648
retention time (HPLC): 5.5 min (method B)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-99-
Example 5.2
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-(4-hydroxy-4-methyl-
[1,4']bipiperidinyl-1'-
yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-
1-
carboxylate
O &OH
NNON\~ OH
~~~///
'~
N4 0
H O
5.2a) (R)-1-(4-hydroxy-3 5-dimethyl-benzyl)-2-oxo-2-(4-oxo-1) iperidin-1-yl)-
ethyl 4-
(2-oxo-1 2 4 5-tetrahydro-1 3-benzodiazepin-3-yl)-piperidine-1-carboxylate
130 mg (0.83 mmol) piperidin-4-one (used as the hydrate of the hydrochloride
salt)
were added at RT to a solution of 400 mg (0.83 mmol) (R)-2-(4-hydroxy-3,5-
dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-
3-
yl)-piperidine-l-carboxylate, 320 mg (1.00 mmol) TBTU and 260 pL (1.87 mmol)
triethylamine in 10 mL DMF and the reaction mixture was stirred for 16 h at
RT.
The mixture was evaporated down i.vac., the residue was taken up in EtOAc, the
organic phase was washed with semisaturated NaHCO3 solution and dried over
Na2SO4. After the desiccant and solvent had been eliminated the residue was
purified by chromatography (silica gel, EtOAc). The fractions containing the
product were evaporated down, the residue stirred with DIPE, suction filtered
and
dried.
Yield: 333 mg (71 % of theory)
ESI-MS: (M+H) + = 563
retention time (HPLC-MS): 6.9 min (method B)
5.2b) (R)-1-(4-hydroxy-3 5-dimethyl-benzyl)-2-(4-hydroxy-4-methyl-
L1 4'lbipiperidinyl-1'-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-100-
A solution of 50 mg (0.09 mmol) (R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-2-
(4-
oxo-piperidin-1-yl)-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-l-carboxylate in 1.5 mL DCM was combined with 21 mg (0.18 mmol) 4-
methyl-piperidin-4-ol and 10.3 pL (0.19 mmol) AcOH, cooled to 0 C cooled and
stirred for 2 h. Then 28 mg (0.19 mmol) sodium-triacetoxyborohydride were
added
and the mixture was stirred overnight at 0 C. After the solvent had been
eliminated
the residue was combined with 2 mL DMF and purified by HPLC. The fractions
containing the product were combined and lyophilised.
Yield: 25 mg (42% of theory)
ESI-MS: (M+H) + = 662
retention time (HPLC-MS): 2.90 min (method A)
Example 5.3
(R)-2-(4,4-dimethyl-[1,4']bipiperidinyl-1'-yl)-1-(4-hydroxy-3,5-dimethyl-
benzyl)-2-
oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
~ OH
I /
I
~/\ N l ~\
O 0
H
Analogously to Example 5.2b the product could be obtained from 50.0 mg (0.09
mmol) (R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-2-(4-oxo-piperidin-1-yl)-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
(Example 5.2a) and 31.0 mg (0.18 mmol) 4,4-dimethylpiperidin.
Yield: 18.3 mg (31 % of theory)
ESI-MS: (M+H)+ = 660
retention time (HPLC-MS): 3.2 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
- 101 -
Example 5.4
(R)-2-(4-amino-4-methyl-[1,4']bipiperidinyl-1'-yl)-1-(4-hydroxy-3,5-dimethyl-
benzyl)-
2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
OH
~
N- ( N O~ N NH
~ ~ O s
N
H O
A solution of 150 mg (0.27 mmol) (R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-2-
(4-oxo-piperidin-1-yl)-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-l-carboxylate (Example 5.2a) in 4 mL DCM was combined with 120 mg
(0.53 mmol) tert-butyl (4-methyl-piperidin-4-yl)-carbamate and 31 pL (0.56
mmol)
AcOH, cooled to 0 C and stirred for 2 h. Then 85 mg (0.56 mmol) sodium
triacetoxyborohydride were added and the mixture was stirred overnight at 0 C.
Then the reaction solution was combined with 0.5 mL TFA and again stirred
overnight at RT. After elimination of the solvents the residue was dissolved
in 2 mL
DMF and purified by HPLC. The fractions containing the product were combined
and lyophilised, wherein the product was obtained as the TFA salt.
Yield: 94 mg (46% of theory)
ESI-MS: (M+H)+ = 661
retention time (HPLC-MS): 2.5 min (method A)
Example 5.5
(R)-2-(1'-ethoxycarbonylmethyl-4,4'-bipiperidinyl-1-yl)-1-(4-hydroxy-3, 5-d
imethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahyd ro-1, 3-benzod iazepin-3-yl)-
piperid ine-
1-carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= = -102-
c H
O J
N~NO
CQ
N N O O O
H
117 mg (0.46 mmol) ethyl [4,4']bipiperidinyl-l-yl-acetate were added at RT to
a
solution of 200 mg (0.42 mmol) (R)-2-(4-hydroxy-3,5-dimethyl-phenyl)-1 -
carboxy-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
(Example 5h), 148 mg (0.46 mmol) TBTU and 64 pL (0.46 mmol) triethylamine in
mL THF and 1 mL DMF and the reaction mixture was shaken overnight at RT.
The reaction solution was evaporated down i.vac., the residue was taken up in
MeOH and purified by HPLC. The fractions containing the product were combined,
evaporated down i.vac., the residue was triturated with DIPE, suction filtered
and
10 dried.
Yield: 222 mg (74% of theory)
ESI-MS: (M+H)+ = 718
retention time (HPLC): 3.1 min (method A)
Example 5.6
(R)-2-(1'-carboxymethyl-4,4'-bipiperidinyl-1-yl)-1-(4-hydroxy-3,5-dimethyl-
benzyl)-
2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
~ OH
~ /
nN-CN N 0, H
O~
O O
H
A solution of 3.8 mg (0.16 mmol) LiOH in 1 mL water was added to a solution of
100 mg (0.14 mmol) (R)-2-(1'-ethoxycarbonylmethyl-4,4'-bipiperidinyl-1-yl)-1-
(4-
hydroxy-3,5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate in 3 mL THF and the reaction
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-103-
solution was stirred overnight at RT. The organic solvent was removed in the
nitrogen stream, the residue was combined with 1 mL water and acetonitrile and
acidified with formic acid. The product was purified by HPLC; the fractions
containing the product were combined and lyophilised.
Yield: 56 mg (58% of theory)
ESI-MS: (M-H)- = 688
retention time (HPLC): 3.0 min (method A)
Example 5.7
(R)-2-(4-cyclohexyl-piperazin-1-yl)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-
ethyi
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
OH
~ N
(N~N O~ 0
0
Analogously to Example 3g the product could be obtained from 69 mg (0.14 mmol)
(R)-2-(4-hydroxy-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate (Example 5h) and 24 mg (0.14
mmol) 1-cyclohexyl-piperazine.
Yield: 51 mg (91 % of theory)
ESI-MS: (M+H)+ = 632
retention time (HPLC): 3.1 min (method A)
Example 5.8
(R)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)-piperidin-1 -yl]-1-(4-hydroxy-
3,5-
dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-1-carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-104-
O OH
(XIN-CINOON O O 0
H
Analogously to Example 5i the product could be obtained from 150 mg (0.31
mmol) (R)-2-(4-hydroxy-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate (Example 5h) and
87
mg (0.34 mmol) ethyl (4-piperidin-4-yl-piperazin-1-yl)-acetate.
Yield: 64 mg (28% of theory)
ESI-MS: (M+H)+ = 719
retention time (HPLC): 3.6 min (method A)
Example 5.9
(R)-2-[4-(4-ca rboxymethyl-pi pe razi n- 1 -yl)-p i pe rid i n- 1 -yl]- 1 -(4-
hyd roxy-3,5-d imethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate
OH
I\ NO~N\/~
0~N-Co 0
, ~
H
A solution of 2.3 mg (0.09 mmol) LiOH in 5 mL water was added to a solution of
40.0 mg (0.06 mmol) (R)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)-piperidin-
1-yl]-1-(4-hydroxy-3, 5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4, 5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate in 5 mL THF and the reaction
mixture was stirred for 2 h at RT. The mixture was evaporated down i.vac., the
residue was taken up in 1 mL DMF and purified by HPLC; the fractions
containing
the product were combined and lyophilised.
Yield: 20 mg (38% of theory)
ESI-MS: (M+H) + = 691
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-105-
retention time (HPLC): 2.6 min (method A)
Example 5.10
(R)-2-[4-(1-ethoxycarbonylmethyl-piperidin-4-yl)-piperazin-1-yl]-1-(4-hydroxy-
3,5-
dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperid i ne-1-carboxylate
J OH
N~ ~N 0~
O 0
i 0
H
5.10a) (R)-1 -(4-benzyloxy-35-dimethyl-benzyl)-2-[4-(1-ethoxycarbonyimethyl-
piperidin-4-yl)-piperazin-l-yll-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-1 -carboxylate
A solution of 8.00 g (14.0 mmol) (R)-2-(4-benzyloxy-3,5-dimethyl-phenyl)-1-
carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate (Example 5g), 5.17 g (16.1 mmol) TBTU and 8.84 mL (63.0 mmol)
triethylamine in 100 mL DMF was stirred for 1 h at RT. Then 5.28 g (16.1 mmol)
ethyl (4-piperazin-1 -yl-piperidin-1 -yl)-acetate (used as the bis-
hydrochloride salt)
was added to the reaction mixture, which was then stirred for 1 h at RT. 150
mL of
15% K2C03 solution were added, the mixture was extracted with 200 mL EtOAc,
the organic phase was separated off and extracted with 150 mL 10% citric acid
solution. The aqueous phase was made alkaline with K2CO3, extracted with 200
mL EtOAc and the organic phase was dried over Na2SO4. After the desiccant and
solvent had been eliminated the residue was purified by chromatography (silica
gel, EtOH). The fractions containing the product were combined, evaporated
down
i.vac., the residue was stirred with DIPE, suction filtered and dried.
Yield: 8.58 g (76% of theory)
ESI-MS: (M+H)+ = 809
retention time (HPLC): 3.7 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-106-
5.10b) (R)-2-[4-(1-ethoxycarbonylmethyl-piperidin-4-yl)-piperazin-1-yl1-1-(4-
hydroxy-3,5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-1-carboxylate
A suspension of 4.00 g (4.94 mmol) (R)-1-(4-benzyloxy-3,5-dimethyl-benzyl)-2-
[4-
(1-ethoxycarbonylmethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate and 400 mg
10% Pd/C in 50 mL EtOH was hydrogenated at RT and 3000 hPa hydrogen
pressure until the theoretical hydrogen uptake had occurred. The catalyst was
filtered off, the filtrate was evaporated down i.vac., the residue was stirred
with
DIPE, suction filtered and dried.
Yield: 3.40 g (96% of theory)
ESI-MS: (M+H) + = 719
retention time (HPLC): 2.5 min (method A)
Example 5.11
(R)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-1-(4-hydroxy-3,5-
dimethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate
jOH
(::CN N~O~N\%
~o 0
, ~
H
A solution of 126 mg (5.25 mmol) LiOH in 10 mL water was added at RT to a
solution of 2.50 g (3.48 mmol) (R)-2-[4-(1-ethoxycarbonylmethyl-piperidin-4-
yl)-
piperazin-1-yl]-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate in 30 mL THF and
the
reaction mixture was stirred overnight at RT. The mixture was evaporated to
dryness i.vac. and the residue was purified by chromatography (silica gel,
gradient
DCM to DCM/MeOH/NH3 70:30:3). The fractions containing the product were
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-107-
combined, evaporated down i.vac., the residue was triturated with DIPE,
suction
filtered and dried.
Yield: 1.80 g (75% of theory)
ESI-MS: (M+H)+ = 691
Rf = 0.10 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 5.12
(R)-2-{4-[1-(2-ethoxycarbonyl-ethyl)-piperidin-4-yl]-piperazin-1-yl}-1-(4-
hydroxy-
3,5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-
3-
yl)-piperidine-1-carboxylate
~ OH
O O
N N O~ ~~N~~~~Nv 0~~
~ -~ O
O
H
352 mg (0.93 mmol) ethyl 3-(4-piperazin-1-yl-piperidin-1-yl)-propionate (used
as
the bis-Hydrochloride salt) were added to a solution of 400 mg (0.83 mmol) (R)-
2-
(4-hydroxy-3,5-dimethyl-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-
l,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate, 299 mg (0.93 mmol) TBTU and 477
pL (3.40 mmol) triethylamine in 5 mL DMF and the reaction mixture was stirred
for
2 h at RT. The reaction solution was combined with 15%'K2C03 solution,
exhaustively extracted with DCM and the combined organic phases were dried
over Na2SO4. After the desiccant and solvent had been eliminated the residue
was
purified by HPLC; the fractions containing the product were combined and
lyophilised.
Yield: 305 mg (50% of theory)
ESI-MS: (M+H) + = 733
retention time (HPLC): 2.6 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-108-
Example 5.13
(R)-2-{4-[1-(2-carboxy-ethyl)-piperidin-4-yl]-piperazin-1-yl}-1-(4-hydroxy-3,5-
d imethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1, 3-benzodiazepin-3-
yl)-
piperidine-1 -carboxylate
OH
O O
cr- ~NON~~NO~H
cj O
N O
H
Analogously to Example 5.11 the product could be obtained from 2.60 g (3.55
mmol) (R)-2-{4-[1-(2-ethoxycarbonyl-ethyl)-piperidin-4-yl]-piperazin-1-yl}-1-
(4-
hydroxy-3,5-dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate and 128 mg (5.33 mmol) LiOH.
Yield: 1.60 g (64% of theory)
ESI-MS: (M+H) + = 705
Rf = 0.07 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 5.14
(R)-2-[1'-(2-ethoxycarbonyl-ethyl)-4,4'-bipiperidinyl-1-yl]-1-(4-hydroxy-3,5-
dimethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benz6diazepin-3-yl)-
piperidine-
1-carboxylate
OH
O
N N
CQ- ~
N 0 0
H O
A solution of 80 mg (0.17 mmol) (R)-2-(4-hydroxy-3,5-dimethyl-phenyl)-1-
carboxy-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
(Example 5h), 59 mg (0.18 mmol) TBTU and 76 pL (0.54 mmol) triethylamine in 1
mL DMF was stirred for 1 h at RT. Then 49 mg (0.18 mmol) ethyl 3-
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-109-
[4,4']bipiperidinyl-l-yl-propionate were added to the reaction mixture, which
was
then stirred for 2 h at RT. This was purified by HPLC without any further
working
up; the fractions containing the product were combined and lyophilised.
Yield: 11 mg (9% of theory)
ESI-MS: (M+H) + = 732
retention time (HPLC): 3.4 min (method C)
Example 5.15
(R)-2-[1'-(2-carboxy-ethyl)-4,4'-bipiperidinyl-1-yl]-1-(4-hydroxy-3,5-dimethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate
OH
O O
~~ ~
I N
~ N O~ N O
'(
O O
H
Analogously to Example 5.11 the product could be obtained from 2.50 g (3.42
mmol) (R)-2-[1'-(2-ethoxycarbonyl-ethyl)-4,4'-bipiperidinyl-1-yl]-1-(4-hydroxy-
3,5-
dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-l-carboxylate and 128 mg (5.33 mmol) LiOH.
Yield: 1.70 g(71 % of theory)
ESI-MS: (M+H)+ = 704
Rf = 0.20 (silica gel, DCM/MeOH/Cyc/NH3 70:15:15:2)
Example 5.16
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-{1'-[(hydroxy-methyl-carbamoyl)-
methyl]-
4,4'-bipiperidinyl-l-yl}-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-1-carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= -110-
~ OH
/~
-( N
CC O~N N~N I
O ~/ 0 H
H
A solution of 80.0 mg (0.12 mmol) (R)-2-(1'-carboxymethyl-4,4'-bipiperidinyl-1-
yl)-
1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-ethyi 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-l-carboxylate (Example 5.6), 44.6 mg (0.14
mmol)
TBTU and 64.6 pL (0.46 mmol) triethylamine in 1.2 mL DMF was stirred for 1 h
at
RT. Then 29.1 mg (0.35 mmol) N-methylhydroxylamine (used as the hydrochloride
salt) was added to the reaction mixture, which was then stirred for 20 h at
RT. To
the reaction mixture were added 5 drops of AcOH, the mixture was filtered
through
a syringe filter and purified by HPLC. The fractions containing the product
were
combined, 30 mL EtOAc and 5% NaHCO3 solution were added, the organic phase
was separated off and dried over Na2SO4. After the desiccant and solvent had
been eliminated the residue was purified by chromatography (silica gel,
DCM/MeOH/NH3 90:10:1). The fractions containing the product were combined,
evaporated down i.vac., the residue was triturated with DIPE, suction filtered
and
dried in the air until a constant weight was obtained.
Yield: 12.4 mg (15% of theory)
ESI-MS: (M+H) + = 719
Rf = 0.16 (silica gel, DCM/MeOH/NH3 90:10:1)
retention time (HPLC): 3.0 min (method A)
Example 5.17
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-[1'-(methoxycarbamoyl-methyl)-4,4'-
bipiperidinyl-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-
3-yl)-
piperidine-1 -carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
- 111 -
OH
H
C N N~N
I Q ~II(
H 0 O
0
Analogously to Example 5.16 the product could be obtained from 80.0 mg (0.12
mmol) (R)-2-(1'-carboxymethyl-4,4'-bipiperidinyl-1-yl)-1-(4-hydroxy-3,5-
dimethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate (Example 5.6) and 29.1 mg (0.35 mmol) O-methylhydroxyamine
(used as the hydrochloride salt).
Yield: 13.0 mg (16% of theory)
ESI-MS: (M+H)+ = 719
Rf = 0.27 (silica gel, DCM/MeOH/NH3 90:10:1)
retention time (HPLC): 3.0 min (method A)
Example 5.18
(R)-2-(4-cyclopentyl-piperazin-1-yl)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-
ethyi
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
OH
NO N~N~
N~
~ ~
O
H O
5.18a)(R)-1-(4-benzyloxy-3 5-dimethyl-benzyl)-2-(4-benzyl-piperazin-1-yl)-2-
oxo-
ethyl 4-(2-oxo-1 2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
A solution of 1.54 g (2.69 mmol) (R)-2-(4-benzyloxy-3,5-dimethyl-phenyl)-1-
carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate (Example 5g), 0.95 g (2.96 mmol) TBTU and 0.47 mL (3.37 mmol)
triethylamine in 20 mL THF and 2 ml DMF was stirred for 1 h at RT. Then 0.52
mL
(2.96 mmol) N-benzylpiperazine were added to the reaction mixture, which was
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-112-
then stirred for 14 h at RT. 30 mL EtOAc were added to the reaction mixture,
it
was washed with semisaturated NaHCO3 solution and the organic phase was
dried over Na2SO4. After the desiccant and solvent had been eliminated the
residue was purified by chromatography (silica gel, EtOAc/Cyc 95:5). The
fractions
containing the product were evaporated down, the residue was triturated with
DIPE, suction filtered and dried.
Yield: 1.62 g (82% of theory)
ESI-MS: (M+H) + = 730
retention time (HPLC): 4.3 min (method A)
5.18b)(R)-1-(4-hydroxy-3 5-dimethyl-benzyl)-2-oxo-2-piperazin-l-yl-ethyi 4-(2-
oxo-
1 2 4 5-tetrahydro-1 3-benzodiazepin-3-yl)-piperidine-l-carboxylate
A suspension of 1.62 g (2.22 mmol) (R)-1-(4-benzyloxy-3,5-dimethyl-benzyl)-2-
(4-
benzyl-piperazin-1-yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-1-carboxylate and 150 mg 10% Pd/C in 40 mL MeOH was
hydrogenated at RT and 3000 hPa hydrogen pressure until the theoretical
hydrogen uptake had occurred. The catalyst was suction filtered, the solvent
was
evaporated down i.vac. and the residue was purified by chromatography (silica
gel, EtOAc + 15% MeOH/NH3 9:1).
Yield: 1.03 g (85% of theory)
ESI-MS: (M+H)+ = 550
retention time (HPLC): 2.7 min (method A)
5.18c) (R)-2-(4-cyclopentyl-piperazin-1-yl)-1-(4-hydroxy-3,5-dimethyl-benzyl)-
2-
oxo-ethyl 4-(2-oxo-1 2 4 5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
A solution of 100 mg (0.18 mmol) (R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-2-
piperazin-1-yl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate, 48 pL (0.55 mmol) cyclopentanone and 20 pL (0.37 mmol) AcOH in
2 mL THF/MeOH (2:1) was stirred overnight at RT. Then 24 mg (0.36 mmol)
sodium cyanoborohydride was added to the reaction solution which had been
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= -113-
cooled to 0 C, and this was then stirred for 4 h at 0 C and overnight at RT.
The
solvents were eliminated at 40 C, the residue was dissolved in 1 mL DMF and
purified by HPLC; the fractions containing the product were combined and
lyophilised.
Yield: 48 mg (43% of theory)
ESI-MS: (M+H)+ = 618
retention time (HPLC): 3.2 min (method A)
Example 5.19
(R)-2-(4-cycloheptyl-piperazin-1-yl)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
OH
\ ~\ ~ N/\ ~
0
O
H
Analogously to Example 5.18c the product could be obtained from 100 mg (0.18
mmol) (R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-oxo-2-piperazin-1-yl-ethyl -(2-
oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate, 40.8 mg
(0.36
mmol) cycloheptanone and 12 mg (0.18 mmol) sodium cyanoborohydride.
Yield: 21 mg (18% of theory)
ESI-MS: (M+H) + = 646
retention time (HPLC): 3.5 min (method A)
Example 5.20
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-(1'-methanesulphonyl-4,4'-
bipiperidinyl-1-
yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-
1-
carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-114-
O OH
0~ 0
~ -~
N 0
H O
A solution of 100 mg (0.21 mmol) (R)-1-carboxy-2-(4-hydroxy-3,5-dimethyl-
phenyl)-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate, 73 mg (0.23 mmol) TBTU and 36 pL (0.26 mmol) triethylamine in 1
mL DMF was stirred for 1 h at RT. Then 56 mg (0.23 mmol) 1-methanesulphonyl-
[4,4']bipiperidinyl was added to the reaction mixture, which was then stirred
for 5 h
at RT. The reaction mixture was purified by HPLC without any further working
up;
the fractions containing the product were combined and lyophilised.
Yield: 63 mg (43% of theory)
ESI-MS: (M-H)" = 708
retention time (HPLC): 4.0 min (method A)
Example 5.21
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-[4-(4-methanesulphonyl-piperazin-1-yl)-
piperidin-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-
piperidine-1-carboxylate
OH
O
flN_JNAON S\
N-~ O
H O
Analogously to Example 5.20 the product could be obtained from 100 mg (0.21
mmol) (R)-1-carboxy-2-(4-hydroxy-3,5-dimethyl-phenyl)-ethyl and 57 mg (0.23
mmol) 1-methanesulphonyl-4-piperidin-4-yl-piperazine 4-(2-oxo-1,2,4,5-
tetrahydro-
1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate.
Yield: 43 mg (29% of theory)
ESI-MS: (M+H) + = 711
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-115-
retention time (HPLC): 3.1 min (method A)
Example 5.22
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-{4-[4-(2-hydroxy-ethyl)-piperazin-1-
yl]-
piperidin-1-yl}-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-l,3-benzodiazepin-3-
yl)-
piperidine-1-carboxylate
OH
II I -CN 0~~~%:5~N/~O-H
N-~ 0
H O
Analogously to Example 5.20 the product could be obtained from 80 mg (0.17
mmol) (R)-1-carboxy-2-(4-hydroxy-3,5-dimethyl-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate and 52 mg (0.18
mmol) 2-(4-piperidin-4-yl-piperazin-1-yl)-ethanol.
Yield: 63 mg (56% of theory)
ESI-MS: (M+H)+ = 677
retention time (HPLC): 2.4 min (method A)
Example 5.23
(R)-2-[1'-(3-ethoxycarbonyl-propionyl)-4,4'-bipiperidinyl-1-yl]-1-(4-hydroxy-
3,5-
dimethyl-benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-l,3-benzodiazepin-3-
yl)-
piperidine-1-carboxylate
~ OH
~ /
O
N-CIN O-'yN O,,,,-
O O 0
H
207 mg (0.70 mmol) ethyl 4-[4,4']bipiperidinyl-1-yl-4-oxo-butyrate (amine A2)
were
added at RT to a solution of 300 mg (0.62 mmol) (R)-1-carboxy-2-(4-hydroxy-3,5-
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-116-
d imethyl-phenyl )-ethyl 4-(2-oxo-1,2,4, 5-tetra hyd ro-1, 3-benzod iazepi n-3-
yl)-
piperidine-l-carboxylate, 225 mg (0.70 mmol) TBTU and 97 pL (0.70 mmol)
triethylamine in 5 mL DMF and the reaction mixture was shaken overnight at RT.
It
was directly purified by HPLC without any further working up; the fractions
containing the product were combined and lyophilised.
Yield: 170 mg (36% of theory)
ESI-MS: (M+H)+ = 760
retention time (HPLC): 4.1 min (method A)
Example 5.24
(R)-2-[1'-(3-carboxy-propionyl)-4,4'-bipiperidinyl-1-yl]-1-(4-hydroxy-3,5-
dimethyl-
benzyl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-
1-carboxylate
~OH
O O
/~
~O~ N' v ~( , H
(~N-( N O
~/
N O O O
H
A solution 0.96 mg (0.04 mmol) LiOH in 1 mL water was added to a solution of
20.0 mg (0.03 mmol) (R)-2-[1'-(3-ethoxycarbonyl-propionyl)-4,4'-bipiperidinyl-
1-yl]-
1-(4-hyd roxy-3, 5-d i methyl-be nzyl )-2-oxo-ethyl 4-( 2-oxo-1', 2, 4, 5-
tetra hyd ro-1, 3-
benzodiazepin-3-yl)-piperidine-1-carboxylate in 1 mL THF and the reaction
mixture
was stirred for 2 h at RT. The solvents were eliminated in a nitrogen stream,
the
residue was taken up in water/acetonitrile and lyophilised. The product was
obtained as the Li salt.
Yield: 17 mg (88% of theory)
ESI-MS: (M+H) + = 732
Rf = 0.13 (silica gel, DCM/MeOH/NH3 90:10:1)
. . WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
- 117 -
Example 5.25
(R)-2-(1'-ethoxyoxalyl-4,4'-bipiperidinyl-1-yl)-1-(4-hydroxy-3,5-d imethyl-
benzyl)-2-
oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-l,3-benzodiazepin-3-yl)-piperidine-l-
carboxylate
OH
O I /
N-CINO~N N O~~
O O 0
H
231 mg (0.70 mmol) ethyl [4,4']bipiperidinyl-l-yl-oxo-acetate (amine Al, used
as
the hydrogen carbonate salt) were added at RT to a solution of 300 mg (0.62
mmol) (R)-1-carboxy-2-(4-hydroxy-3,5-dimethyl-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate, 225 mg (0.70
mmol)
TBTU and 197 pL (1.40 mmol) triethylamine in 5 mL DMF and the reaction mixture
was stirred for 3 h at RT. The mixture was evaporated down i.vac., the residue
was taken up in DCM, the organic phase was washed with 15% K2C03 solution
and dried over Na2SO4. After the desiccant and solvent had been eliminated the
residue was purified by HPLC; the fractions containing the product were
combined, evaporated down i.vac., the residue was triturated with DIPE,
suction
filtered and dried.
Yield: 360 mg (79% of theory)
ESI-MS: (M+H)+ = 732
retention time (HPLC): 4.0 min (method A)
Example 5.26
(R)-1-(4-hydroxy-3,5-dimethyl-benzyl)-2-(1'-oxalyl-4,4'-bipiperidinyl-1-yl)-2-
oxo-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-l,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
- WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-118-
O OH
N
0, N~N O N H
H O O O
A solution 0.96 mg (0.04 mmol) LiOH in 1 mL water was added to a solution of
20.0 mg (0.03 mmol) (R)-2-(1'-ethoxyoxalyl-4,4'-bipiperidinyl-1-yi)-1-(4-
hydroxy-
3, 5-d imethyl-benzyl )-2-oxo-ethyl 4-(2-oxo-1,2,4, 5-tetrahyd ro-1, 3-benzod
iazepin-3-
yl)-piperidine-l-carboxylate in 1 mL THF and the reaction mixture was stirred
for 2
h at RT. The solvents were eliminated in a nitrogen stream, the residue was
taken
up in water/acetonitrile and lyophilised. The product was obtained as the Li
salt.
Yield: 19 mg (99% of theory)
ESI-MS: (M+H)+ = 704
Rf = 0.10 (silica gel, DCM/MeOH/NH3 90:10:1)
Example 6
(R)-2-(4-cyclohexyl-piperazin-1-yl)-1-(3,5-dibromo-4-hydroxy-benzyl)-2-oxo-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
r
~ OH
~ /
Br
O- y N
.,\(\ 0
H O
6a) (Z,E)-3-(4-acetoxy-3,5-dibromo-phenyl)-2-acetylamino-acrylic acid
Under a nitrogen atmosphere a mixture of 30.0 g (107 mmol) 3,5-dibromo-4-hy-
droxy-benzaldehyde, 18.8 g (268 mmol) N-acetylglycine and 13.2 g (161 mmol)
NaOAc in 120 mL acetic anhydride was heated to 130 C for 1.5 h. The mixture
was cooled to 90 C and 15 mL water was slowly added dropwise thereto in such a
way that the temperature did not exceed 100 C. After the addition had ended
the
mixture was kept for a further 2 h at 90 C, cooled to 70 C, combined with 300
mL
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-119-
water and stirred for 30 min. The precipitate was filtered off, washed with
water
and dried at 50 C. The crude product was further reacted without purification.
Yield: 35.7 g (79% of theory)
ESI-MS: (M+H)+= 420/422/424 (2 Br)
Rf = 0.20 (silica gel, DCM/MeOH/AcOH 90:10:1)
6b) 3-(3,5-dibromo-4-hydroxy-phenyl)-2-oxo-propionic acid
While cooling with ice 325 mL of 4 M HCI was added to a solution of 35.7 g
(84.8
mmol) (Z,E)-3-(4-acetoxy-3,5-dibromo-phenyl)-2-acetylamino-acrylic acid in 290
mL NMP and the 'reaction mixture was heated to 120 C (bath temperature) for
1.5
h. The mixture was cooled to 0 C, combined with 1.4 L water and stirred for a
further 30 min. The precipitate formed was filtered off and dried.
Yield: 20.5 g (72% of theory)
ESI-MS: (M-H)- = 335/337/339 (2 Br)
Rf = 0.35 (silica gel, DCM/MeOH/AcOH 80:20:2)
6c) (R)-3-(3,5-dibromo-4-hydroxy-Dheny)-2-hydroxy-gropionic acid
Under an argon atmosphere 6 mL (43.1 mmol) triethylamine was added to a
solution of 14.5 g (42.9 mmol) 3-(3,5-dibromo-4-hydroxy-phenyl)-2-oxo-
propionic
acid in 250 mL THF and the mixture was cooled to -32 C. Then a solution of
22.6
g (70.5 mmol) (1 R)-B-chlorodiisopinocampheylborane in 90 mL THF was added
dropwise such that the temperature did not exceed -20 C. The reaction mixture
was stirred for 30 min at -30 C and allowed to heat up to 0 C within 2.5 h. To
complete the reaction the mixture was again cooled to -32 C, a solution of 5.8
g
(18.1 mmol) (1R)-8-chlorodiisopinocampheylborane in 40 mL THF was added
dropwise such that the temperature did not exceed -20 C and then the reaction
mixture was stirred overnight in the ice bath. After cooling again to -32 C a
further
2.5 g (7.8 mmol) (1 R)-8-chlorodiisopinocampheylborane in 20 mL THF were
added dropwise, the reaction mixture was stirred for 30 min at this
temperature,
heated to 0 C within 2.5 h and then stirred for 66 h at RT. 100 mL 10% NaOH
were added to the reaction solution such that the temperature did not exceed
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-120-
25 C, stirred for a further 30 min, combined with MTBE, the organic phase was
separated off and again extracted with 20 mL 10% NaOH solution. The combined
aqueous phases were washed several times with MTBE, acidified with 20% HCI
and exhaustively extracted with diethyl ether/EtOAc (1:1). The combined
organic
phases were combined with activated charcoal and filtered. The product was
further reacted without purification.
Yield: 12.7 g (87% of theory)
ESI-MS: (M-H)- = 337/339/341 (2 Br)
Rf = 0.4 (silica gel, DCM/MeOH/AcOH 80:20:2)
retention time (HF5LC-MS): 6.4 min (method D)
6d) methyl (R)-3-(3,5-dibromo-4-hydroxy-phenyl)-2-hydroxy-propionate
A solution of 14.0 g (34.8 mmol) (R)-3-(3,5-dibromo-4-hydroxy-phenyl)-2-
hydroxy-
propionic acid in 100 mL methanolic HCI (6 M) was stirred for 3 h at RT. The
mixture was evaporated down i.vac. and the residue was purified by
chromatography (silica gel, n-hexane/EtOAc 7:3).
Yield: 7.0 g (57% of theory)
ESI-MS: (M-H)-= 351/353/355 (2 Br)
retention time (HPLC-MS): 9.8 min (method D)
6e) methyl (R)-3-f3,5-dibromo-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-2-
hydroxy-propionate
Under a nitrogen atmosphere 11.1 g (76.6 mmol) 40% KF/AI203 were added to a
solution of 6.78 g (19.2 mmol) methyl (R)-3-(3,5-dibromo-4-hydroxy-phenyl)-2-
hydroxy-propionate in 100 mL acetonitrile and the resulting suspension was
stirred
for a few min at RT. Then a solution of 4.07 mL (23.0 mmol) (2-chloromethoxy-
ethyl)-trimethylsilane in 20 mL acetonitrile was added and the reaction
mixture was
stirred for 20 h at RT. The mixture was filtered through Celite, the solvent
was
evaporated down i.vac. and the residue was purified by chromatography (silica
gel, n-hexane/EtOAc 7:3).
Yield: 5.49 g (59% of theory)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
- 121 -
Rf = 0.45 (silica gel, n-hexane/EtOAc 1:1)
6f) (R)-2-[3,5-dibromo-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-1-methoxy-
carbonyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3 yI)-
piperidine-1-carboxylate
Under a nitrogen atmosphere 1.23 g (10.0 mmol) 4-dimethylaminopyridine was
added to a solution of 1.99 g (9.56 mmol) 4-nitrophenyl chloroformate in 80 mL
acetonitrile cooled to 15 C. The resulting suspension was cooled to -7 C and
slowly combined with a solution of 4.63 g (9.56 mmol) methyl (R)-3-[3,5-
dibromo-
4-(2-trimethylsilariyl-ethoxymethoxy)-phenyl]-2-hydroxy-propionate in 20 mL
acetonitrile. The mixture was stirred for a further 15 min at this
temperature, 2.35 g
(9.56 mmol) 3-piperidin-4-yl-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-one was
added and the reaction mixture was stirred for 2.5 h at RT. The mixture was
evaporated down i.vac., the residue was taken up in EtOAc, the organic phase
was washed with 10% citric acid and 10% Na2CO3 solution and dried over
Na2SO4. After the desiccant and solvent had been eliminated the residue was
purified by chromatography (silica gel, gradient n-hexane/EtOAc 1:1 to 2:8).
Yield: 4.35 g (69% of theory)
ESI-MS: (M+H)+ = 754/756/758 (2 Br)
retention time (HPLC): 29.2 min (method D)
6g) (R)-2-(3,5-dibromo-4-hydroxy-phenyl)-1-methoxycarbonyl-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate
Under a nitrogen atmosphere 5.46 mL methanolic H2SO4 (0.5 M) were added to a
solution of 4.30 g (5.69 mmol) (R)-2-[3,5-dibromo-4-(2-trimethylsilanyl-
ethoxymethoxy)-phenyl]-1-methoxy-carbonyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-
1,3-
benzodiazepin-3-yi)-piperidine-l-carboxylate in 40 mL THF and 40 mL MeOH and
the reaction solution was stirred for 6 h at RT. The reaction mixture was
evaporated down i.vac. and the residue was further reacted without
purification.
Yield: quantitative
ESI-MS: (M+H)+ = 624/626/628 (2 Br)
CA 02600189 2007-09-06
WO 2006/100026 PCT/EP2006/002557
-122-
retention time (HPLC): 17.3 min (method D)
6h) (R)-1-carboxy-2-(3,5-dibromo-4-hydroxy-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahyd ro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
A solution of 0.51 g (21.3 mmol) LiOH was added to a solution of (R)-2-(3,5-
d i bromo-4-hyd roxy-phenyl)-1-methoxycarbonyl-ethyl 4-(2-oxo-1, 2,4, 5-tetra
hyd ro-
1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate (crude product from Example
6g)
in 80 mL THF and the reaction mixture was stirred for 3 h at RT. The THF was
eliminated i.vac., the aqueous phase was washed with EtOAc, acidified with 10%
HCI and the aqueous phase was exhaustively extracted with EtOAc. The
combined organic phases were evaporated down i.vac., suspended in diethyl
ether, filtered, the residue was dried and then purified by chromatography
(silica
gel, DCM/MeOH/AcOH 90:10:1).
Yield: 3.5 g (100% of theory)
ESI-MS: (M+H) + = 610/612/614 (2 Br)
retention time (HPLC): 14.1 min (method D)
6i) (R)-2-(4-cyclohexyl-piperazin-1-yl)-1-(3,5-dibromo-4-hydroxy-benzyl -2-oxo-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
mg (0.18 mmol) 1-cyclohexyl-piperazine were added at RT to a solution of 100
mg (0.16 mmol) (R)-1-carboxy-2-(3,5-dibromo-4-hydroxy-phenyl)-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate, 58 mg
(0.18
mmol) TBTU and 25 pL (0.18 mmol) triethylamine in 1 mL DMF and the reaction
25 mixture was stirred overnight at RT. The reaction solution was filtered
through a
syringe filter and directly purified by HPLC without any further working up.
The
fractions containing the product were combined and lyophilised.
Yield: 64 mg (51 % of theory)
ESI-MS: (M+H)+= 760/762/764 (2 Br)
30 retention time (HPLC-MS): 3.3 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= -123-
Example 6.1
(R)-1-(3,5-dibromo-4-hydroxy-benzyl)-2-(4-morpholin-4-yl-piperidin-1-yl)-2-oxo-
ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
Br
~ OH
~ /
Br
NN O 0
~~~///
CQ
N \\
\
0
Analogously to Example 6i the product could be obtained from 100 mg (0.16
mmol) (R)-1-carboxy-2-(3,5-dibromo-4-hydroxy-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate and 31 mg (0.18
mmol) 4-piperidin-4-yl-morpholine.
Yield: 72 mg (58% of theory)
ESI-MS: (M+H)+ = 762/764/766 (2 Br)
retention time (HPLC-MS): 3.1 min (method A)
Example 6.2
(R)-1-(3,5-dibromo-4-hydroxy-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-
yl)-piperidin-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-
3-yl)-
piperidine-1 -carboxylate
r
~ OH
~ /
Br
N-CN O~N\~
O 0 0
H
Analogously to Example 6i the product could be obtained from 100 mg (0.16
mmol) (R)-1-carboxy-2-(3,5-dibromo-4-hydroxy-phenyl)-ethyi 4-(2-oxo-1,2,4,5-
WO 2006/100026 CA 02600169 2007-09-06 PCT/EP2006/002557
-124-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-l-carboxylate and 46 mg (0.18
mmol) ethyl (4-piperidin-4-yl-piperazin-1-yl)-acetate.
Yield: 5 mg (4% of theory)
ESI-MS: (M+H) + = 847/849/851 (2 Br)
retention time (HPLC-MS): 2.9 min (method A)
Example 7
(R)-1-(3-bromo-4-hydroxy-benzyl)-2-(4-cyclohexyl-piperazin-l-yl)-2-oxo-ethyl 4-
(2-
oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
OH
~ /
Br
c~-o H O
7a) (Z,E)-3-(4-acetoxy-3-bromo-phenyi)-2-acetylamino-acrylic acid
Prepared analogously to Example 6a from 75.0 g (366 mmol) 3-bromo-4-hydroxy-
benzaldehyde and 64.2 g (548 mmol) N-acetylglycine. After cooling of the
reaction
mixture the product precipitated out and was filtered, washed with water and
dried.
Yield: 69.8 g (56% of theory)
retention time (HPLC): 7.6 min (method D)
7b) 3-(3-bromo-4-hydroxy-phenyl)-2-oxo-propionic acid
While cooling with ice, 750 mL 4 M HCI was added to a solution of 69.7 g (204
mmol) (Z,E)-3-(4-acetoxy-3-bromo-phenyl)-2-acetylamino-acrylic acid in 300 mL
NMP and the reaction mixture was heated to 95 C heated (bath temperature) for
2.5 h. It was cooled overnight to RT, combined with 2 L water, extracted three
times with 300 mL EtOAc, the combined organic phases were washed twice with 1
L water and dried over Na2SO4. After the desiccant and solvent had been
eliminated the residue was reacted further without purification.
Yield: 45.8 g (87% of theory)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-125-
retention time (HPLC): 7.8 min (method D)
7c) (R)-3-(3-bromo-4-hydroxy-phenyl)-2-hydroxy-propionic acid
Under an argon atmosphere 29 mL (356 mmol) triethylamine was added to a
solution of 45.0 g (174 mmol) 3-(3-bromo-4-hydroxy-phenyl)-2-oxo-propionic
acid
in 350 mL THF and the mixture was cooled to -27 C. Then a solution and 114 g
(356 mmol) (1 R)-B-chlorodiisopinocampheylborane in 200 mL THF was added
dropwise such that the temperature did not exceed -20 C. The reaction mixture
was stirred for 15 min at -30 C and allowed to warm up to RT within 1 h. 200
mL
10% NaOH were added to the reaction solution such that the temperature did not
exceed 25 C, stirred for a further 15 min, diluted with 400 mL water, combined
with 400 mL MTBE and the aqueous phase was separated off. It was washed with
400 mL MTBE, acidified with 150 mL 4 M HCI, extracted twice with 400 mL EtOAc,
the combined organic phases were washed with saturated NaCI solution and dried
over Na2SO4. After the desiccant and solvent had been eliminated the residue
was
reacted further without purification.
Yield: 53.7 g (89% of theory)
retention time (HPLC): 4.0 min (method D)
7d) methyl (R)-3-(3-bromo-4-hydroxy-phenyl)-2-hydroxy-propionate
2.5 mL concentrated sulphuric acid were added to a solution of 53.6 g (154
mmol)
(R)-3-(3-b ro mo-4-hyd roxy-p henyl)-2-hyd roxy- prop ion i c acid in 250 mL
MeOH and
the reaction mixture was stirred for 4 h at RT. The mixture was evaporated
down
i.vac., the residue was taken up in 250 mL EtOAc, the organic phase was washed
twice with 100 mL saturated NaHCO3 and saturated NaCI solution and dried over
Na2SO4. After the desiccant and solvent had been eliminated the residue was
reacted further without purification.
Yield: quantitative
retention time (HPLC): 6.8 min (method D)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-126-
7e) methyl (R)-3-[3-bromo-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-2-
hydroxy-propionate
6.7 mL (39.1 mmol) ethyidiisopropylamine were added to a solution of 10.2 g
(34.6
mmol) methyl (R)-3-(3-bromo-4-hydroxy-phenyl)-2-hydroxy-propionate in 100 mL
DCM and the reaction mixture was cooled in the ice bath. Then a solution of
7.9
mL (44.6 mmol) (2-chloromethoxy-ethyl)-trimethylsilane in 20 mL DCM was added.
The reaction mixture was stirred for 3 h at RT and then to complete the
reaction
combined with another 0.67 mL ethyldiisopropylamine and 0.8 mL (4.5 mmol) (2-
chloromethoxy-ethyl)-trimethylsilane and stirred for 1.5 h at RT. The reaction
mixture was washed with 5% Na2CO3 and saturated NaCi solution and dried over
Na2SO4. After the desiccant and solvent had been eliminated the residue was
purified by chromatography (silica gel, Cyc/EtOAc 75:25).
Yield: 9.6 g (68% of theory)
retention time (HPLC): 15.1 min (method E)
7f) (R)-2-[3-bromo-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-1-
methoxycarbonyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-carboxylate
Prepared analogously to Example 6f from 4.55 g (11.2 mmol) methyl (R)-3-[3-
bromo-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyl]-2-hydroxy-propionate and
2.75 g (11.2 mmol) 3-piperidin-4-y1-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-
one.
Yield: 5.46 g (72% of theory)
retention time (HPLC): 16.5 min (method E)
7g) (R)-2-(3-bromo-4-hydroxy-phenYl)-1-methoxycarbonyl-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
Prepared analogously to Example 6g from 5.40 g (7.98 mmol) (R)-2-[3-bromo-4-
(2-trimethylsilanyl-ethoxy-methoxy)-phenyl]-1-methoxycarbonyl-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate and 7.7 mL
(4.2 mmol) methanolic sulphuric acid (0.5 M). The crude product (5.44 g) was
further reacted without purification.
= t WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-127-
retention time (HPLC): 9.9 min (method E)
7h) (R)-2-(3-bromo-4-hydroxy-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
A solution of 0.84 g (34.1 mmol) LiOH in 20 mL water was added to a solution
of
5.44 g of the crude product from Example 7g in 80 mL THF and the reaction
mixture was stirred for 1 h at RT. The organic solvent was eliminated i.vac.,
the
aqueous phase was washed with EtOAc, acidified with 10% HCI and exhaustively
extracted with EtOAc. The combined organic phases were washed with saturated
NaCI solution and dried over Na2SO4. After the desiccant and solvent had been
eliminated the residue was triturated with 90 mL diethyl ether, filtered, the
solid
was washed with diethyl ether and dried at 45 C.
Yield: 4.10 g (89% of theory over 2 steps)
retention time (HPLC): 8.2 min (method E)
7i) (R)-1-(3-bromo-4-hydroxy-benzyl)-2-oxo-2-f4-(tetrahydro-pyran-4-yl)-
piperazin-1-yil-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-carboxylate
Prepared analogously to Example 5.20 from 100 mg (0.19 mmol) (R)-2-(3-bromo-
4-hydroxy-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-l-carboxylate and 35 mg (0.21 mmol) 1-cyclohexyl-piperazine.
Yield: 62 mg (49% of theory)
ESI-MS: (M+H)+ = 682/684 (Br)
retention time (HPLC-MS): 3.2 min (method A)
Example 7.1
(R)-1-(3-bromo-4-hydroxy-benzyl)-2-(4-morpholin-4-yl-piperid in-1-yl)-2-oxo-
ethyl 4-
(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-128-
~OH
Br
~N~ ~
cc---), N--( .N O
~/ / O
H O
Prepared analogously to Example 5.20 from 100 mg (0.19 mmol) (R)-2-(3-bromo-
4-hydroxy-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-1-carboxylate and 36 mg (0.21 mmol) 4-piperidin-4-yl-
morpholine.
Yield: 66 mg (51 % of theory)
ESI-MS: (M+H)+ = 684/686 (Br)
retention time (HPLC-MS): 2.9 min (method A)
Example 7.2
(R)-1-(3-bromo-4-hydroxy-benzyl)-2-(1'-ethoxycarbonylmethyl-4,4'-bipiperidinyl-
1-
yl)-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-
1-
carboxylate
OH
0 Br
~
/
~N~N~O~ N~
O O 0
H
Prepared analogously to Example 5.20 from 150 mg (0.28 mmol) (R)-2-(3-bromo-
4-hydroxy-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-l-carboxylate and 79 mg (0.31 mmol) ethyl [4,4']bipiperidinyl-
1-yl-
acetate.
Yield: 63 mg (29% of theory)
ESI-MS: (M+H) + = 768/770 (Br)
retention time (HPLC-MS): 3.2 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-129-
Example 7.3
(R)-1-(3-bromo-4-hydroxy-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1 -yl)-
piperidin-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-l,3-benzodiazepin-3-
yl)-
piperidine-1-carboxylate
OH
0 Br
N~NON\ ~~N
I r-j
N O O O
H
Prepared analogously to Example 5.20 from 150 mg (0.28 mmol) (R)-2-(3-bromo-
4-hydroxy-phenyl)-1-carboxy-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-
3-yl)-piperidine-1 -carboxylate and 79 mg (0.31 mmol) ethyl (4-piperidin-4-yl-
piperazin-1 -yl)-acetate.
Yield: 82 mg (38% of theory)
ESI-MS: (M+H)+= 769/771 (Br)
retention time (HPLC-MS): 3.1 min (method A)
Example 7.4
(R)-1-(3-bromo-4-hydroxy-benzyl)-2-[4-(4-carboxymethyl-piperazin-1 -yi)-
piperidin-
1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-
carboxylate
OH
~ /
Br
I / N-CN O--_rN\~ H
N4\O O O
H
A solution of 3.0 mg (0.12 mmol) LiOH in 5 mL water was added at RT to a
solution of 50 mg (0.07 mol) (R)-1-(3-bromo-4-hydroxy-benzyl)-2-[4-(4-ethoxy-
carbonylmethyl-piperazin-1-yl)-piperidin-1-yl]-2-oxo-ethyl 4-(2-oxo-1,2,4,5-
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-130-
tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate in 5 mL THF and
the
reaction mixture was stirred for 1 h. The organic solvent was eliminated
i.vac., the
aqueous residue was acidified with 1 M HCI and again evaporated down i.vac..
The residue was purified by HPLC; the fractions containing the product were
combined and lyophilised.
Yield: 28 mg (59% of theory)
ESI-MS: (M+H)+= 741/743 (Br)
retention time (HPLC-MS): 2.5 min (method A)
Example 8
(R)-1 -(3,5-dichloro-4-hydroxy-benzyl)-2-[4-(4-methyl-piperazin-1 -yl)-
piperidin-1 -yl]-
2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-
carboxylate
I
~ OH
~ /
CI
0~
0
H O
8a) 2,6-dichloro-4-iodo-phenol
Solutions of 40.7 g (245 mmol) sodium iodide in 160 mL water and 9.6 mL (143
mmol) ethane-1,2-diamine in 16 mL water were added to a solution of 40.0 g
(245
mmol) 2,6-dichloro-phenol in 180 mL EtOH and the mixture was stirred for 15
min
at RT. Then 62.3 g (245 mmol) iodine was added in small batches to the
reaction
mixture. To complete the reaction, after 3 h at RT, a further 31.1 g (122
mmol)
iodine and 4.8 mL (72 mmol) ethane-1,2-diamine were added and the reaction
mixture was stirred overnight at RT. Saturated NaHSO3 solution was added until
an acidic reaction occurred, the mixture was extracted three times with 400 mL
EtOAc, the combined organic phases were washed with saturated NaCI solution
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
- 131 -
and dried over Na2SO4. After the desiccant and solvent had been eliminated the
residue was reacted further without purification.
Yield: 69.0 g (97% of theory)
Rf = 0.5 (silica gel, n-hexane/EtOAc 4:1)
8b) [2-(2,6-dichloro-4-iodo-phenoxymethoxy)-ethyll-trimethylsilane
Under a nitrogen atmosphere 33.5 g (242 mmol) K2C03 and 20.7 mL (117 mmol)
(2-chloromethoxy-ethyl)-trimethylsilan were added to a solution of 28.0 g
(96.9
mmol) 2,6-dichloro-4-iodo-phenol in 800 mL acetonitrile and the reaction
mixture
was refluxed for I h. The reaction mixture was evaporated down i.vac., the
residue
was taken up in 300 mL water, extracted three times with 300 mL EtOAc and
dried
over Na2SO4. After the desiccant and solvent had been eliminated the residue
was
reacted further without purification.
Yield: 38.4 g (95% of theory)
Rf = 0.83 (silica gel, n-hexane/EtOAc 4:1)
8c) 3,5-dichloro-4-hydroxy-benzaldehyde
Under an argon atmosphere 39.4 mL (78.8 mmol) isopropylmagnesium chloride (2
M in THF), diluted with 50 mL THF, was slowly added dropwise to a solution of
30.0 g (71.6 mmol) [2-(2,6-dichloro-4-iodo-phenoxymethoxy)-ethyl]-
trimethylsilane
in 200 mL THF, cooled to -20 C. After the addition had ended 11.0 mL (143
mmol)
DMF were added at -10 C and the reaction mixture was allowed to come up slowly
to RT. After the reaction had ended (monitored by TLC) 100 mL of 2 M HCL were
added and the reaction solution was stirred overnight at RT. It was extracted
twice
with 300 mL EtOAc, the combined organic phases were washed with saturated
NaCI solution and dried over Na2SO4. After the desiccant and solvent had been
eliminated the residue was purified by recrystallising twice from Cyc/EtOAc
(1:1).
Yield: 12.0 g (88% of theory)
= WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-132-
8d) (Z,E)-3-(4-acetoxy-3,5-dichloro-phenyl)-2-acetylamino-acrylic acid
A mixture of 12.2 g (64 mmol) 3,5-dichloro-4-hydroxy-benzaldehyde, 11.2 g (96
mmol) N-acetylglycine and 7.86 g (96 mmol) sodium acetate in 50 mL acetic
anhydride was heated to 130 C (bath temperature) for 3 h. The reaction mixture
was cooled to 90 C and 5 mL water were added such that the internal
temperature
did not exceed 100 C. The mixture was stirred for 2 h at 90 C and 200 mL water
were added, whereupon a precipitate formed. This was filtered off, thoroughly
washed with water and dried.
Yield: 12.0 g (57% of theory)
Rf = 0:84 (silica gel, Cyc/EtOAc 1:1)
8e) 3-(3,5-dichloro-4-hydroxy-phenyl)-2-oxo-propionic acid
131 mL 4 M HCI were added at RT to a solution of 12.0 g (36.1 mmol) (Z,E)-3-(4-
acetoxy-3,5-dichloro-phenyl)-2-acetylamino-acrylic acid in 70 mL NMP and the
reaction mixture was heated to 130 C (bath temperature) for 4 h. After cooling
to
0 C 200 mL water were added and the mixture was stirred overnight, whereupon a
precipitate formed. This was filtered off and dried (2.9 g; purity 95%). The
filtrate
was extracted three times with 300 mL Cyc/EtOAc (1:3), the combined organic
phases were dried and evaporated down i.vac.. After the desiccant and solvent
had been eliminated 4 g of the product (purity: 80%) were obtained.
Yield: 6.0 g (66% of theory)
ESI-MS: (M+H)+= 250/252/254 (2CI)
Rf = 0.30 (silica gel, DCM/MeOH/AcOH 90:10:1)
8f) (R)-3-(3,5-dichloro-4-hydroxy-phenyl)-2-hydroxy-propionic acid
Prepared analogously to Example 7c from 3.00 g (10.0 mmol) 3-(3,5-dichloro-4-
hydroxy-phenyl)-2-oxo-propionic acid (purity: 80%) and 6.34 g (20.0 mmol) (1
R)-B-
chlorodiisopinocampheylborane. The crude product (1.9 g) was further reacted
without purification.
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
= -133-
8g) methyl (R)-3-(3,5-d ich lo ro-4-hyd roxy-phe nyl)-2-hyd roxy-propio n ate
2 mL concentrated sulphuric acid were added to a solution, cooled to 0 C, of
the
crude product from Example 8f in 30 mL MeOH and the reaction mixture was
stirred for 2 h at this temperature. The mixture was neutralised with solid
KHCO3,
diluted with water and extracted exhaustively with EtOAc. The combined organic
phases were washed with saturated NaCl solution and dried over Na2SO4. After
the desiccant and solvent had been eliminated the product was further reacted
without purification.
Yield: 1.8 g (75% of theory over 2 steps)
8h) methyl (R)-3-f 3,5-dichloro-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-2-
hydroxy-propionate
Under a nitrogen atmosphere 5.79 g (76.6 mmol) 40% KF/AI2O3 were added to a
solution of 2.64 g (9.96 mmol) methyl (R)-3-(3,5-dichloro-4-hydroxy-phenyl)-2-
hydroxy-propionate in 20 mL acetonitrile and the resulting suspension was
stirred
for a few min at RT. Then a solution of 2.12 mL (12.0 mmol) (2-chloromethoxy-
ethyl)-trimethylsilane in 20 mL acetonitrile was added and the reaction
mixture was
stirred for 20 h at RT. The mixture was filtered through Celite and the
solvent was
evaporated down i.vac.. The residue was further reacted without purification.
Yield: 3.80 g (97% of theory)
8i) (R)-2-[3,5-dichloro-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyll-1-
methoxycarbonyl-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-
piperidine-1-carboxylate
Prepared analogously to Example 6f from 3.80 g (8.65 mmol) methyl (R)-3-[3,5-
dichloro-4-(2-trimethylsilanyl-ethoxymethoxy)-phenyl]-2-hydroxy-propionate and
2.12 g (8.65 mmol) 3-piperidin-4-yl-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-
one.
The product was purified by chromatography (silica gel, DCM/MeOH 98:2).
Yield: 3.10 g (54% of theory)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-134-
8k) (R)-2-(3,5-dichloro-4-hydroxy-phenyl)-1-methoxycarbonyl-ethyl 4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
Prepared analogously to Example 6g from 3.10 g (4.65 mmol) (R)-2-[3,5-dichloro-
4-(2-tri methyl-silanyl-ethoxy-methoxy)-phenyl]-1-methoxycarbonyl-ethyl 4-(2-
oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate and 4.48
mL
methanolic sulphuric acid (0.5 M).
Yield: 2.49 g (100% of theory)
81) (R)-1-carboxy-2-(3,5-dichloro-4-hydroxy-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahydro=1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
A solution of 0.42 g (17.3 mmol) LiOH in 20 mL water was added to a solution
of
2.49 g (4.64 mmol) (R)-2-(3,5-dichloro-4-hydroxy-phenyl)-1-methoxycarbonyl-
ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
in 50
mL THF and the reaction mixture was stirred for 1 h at RT. The organic solvent
was eliminated i.vac., the aqueous phase was washed with EtOAc, acidified with
10% HCI, exhaustively extracted with EtOAc, the combined organic phases were
washed with saturated NaCI solution and dried over Na2SO4. After the desiccant
and solvent had been eliminated the residue was triturated with diethyl ether,
suction filtered and dried.
Yield: 1.60 g (66% of theory)
Rf = 0.05 (silica gel, DCM/MeOH 9:1)
8m) (R)-1-(3,5-dichloro-4-hydroxy-benzyl)-2-[4-(4-methyl-piperazin-l-yi)-
piperidin-l-yll-2-oxo-ethyl 4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-
yl)-piperidine-1-carboxylate
Under a nitrogen atmosphere a mixture of 150 mg (0.29 mmol) (R)-1-carboxy-2-
(3,5-dichloro-4-hydroxy-phenyl)-ethyi 4-(2-oxo-1,2,4,5-tetrahydro-1,3-
benzodiazepin-3-yl)-piperidine-1-carboxylate, 63.8 mg (0.34 mmol) 1-methyl-4-
piperidin-4-yl-piperazine and 0.17 mL (0.98 mmol) ethyldiisopropylamine in 10
mL
DMF was stirred for 5 min at RT. Then 124 mg (0.32 mmol) HATU was added to
the reaction mixture, which was then stirred for 4 h. This was evaporated down
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-135-
i.vac. and the residue was purified by chromatography (silica gel, gradient
DCM to
DCM/MeOH 85:15). The fractions containing the product were combined,
evaporated down i.vac. and filtered through aluminium oxide, wherein the
product
was eluted with DCM/MeOH 9:1. The mixture was evaporated down i.vac., the
residue was triturated with diethyl ether, and the product was filtered and
dried.
Yield: 43 mg (22% of theory)
ESI-MS: (M+H)+= 687/689 (2CI)
retention time (HPLC-MS): 2.8 min (method A)
The following compounds were prepared analogously from in each case 150 mg
(R)-1-carboxy-2-(3, 5-dichloro-4-hydroxy-phenyl)-ethyl 4-(2-oxo-1,2,4,5-
tetrahyd ro-
1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate and the corresponding amount
of
amine:
~
\ OH
0 I ~ CI
~NN~O R
~
~~~///
H O
Example R Yield (%) Mass retention
spectrum time HPLC
(method)
8.1 21 687/689 2.6
/ N~N~ [M+H]+ (A)
8.2 39 672/74 3.1
[M+H]+ (A)
8.3 = 38 686/688 3.1
[M+H]+ (A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-136-
Example 8.4
(R)-1-(3,5-dichloro-4-hydroxy-benzyl)-2-oxo-2-(4-piperidin-4-yl-piperazin-1-
yl)-ethyl
4-(2-oxo-1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
I
~ OH
~ /
CI
/~ N~/\
C(N-~ -{ N O H
~/ O
1 0
H
Under a nitrogen atmosphere a mixture of 150 mg (0.29 mmol) (R)-1-carboxy-2-
(3, 5-d ichloro-4-hyd roxy-phenyl )-ethyl 4-(2-oxo-1,2,4, 5-tetrahyd ro-1, 3-
benzodiazepin-3-yl)-piperidine-l-carboxylate, 105 mg (0.34 mmol) tert-butyl 4-
piperazin-1-yl-piperidine-1-carboxylate (used as the hydrochloride salt) and
0.17
mL (0.98 mmol) ethyldiisopropylamine in 10 mL DMF was stirred at RT for 5 min.
Then 124 mg (0.32 mmol) HATU was added to the reaction mixture, which was
then stirred for 4 h. This was evaporated down i.vac. and the residue was
purified
by chromatography (silica gel, gradient DCM to DCM/MeOH 85:15). The fractions
containing the product were combined and evaporated down i.vac.. The residue
was taken up in 10 mL formic acid and stirred for 3 h at RT. The mixture was
evaporated down i.vac., the residue was taken up in saturated NH3 solution,
extracted exhaustively with EtOAc, the combined organic phases were washed
with saturated NaCi solution and dried over Na2SO4. After the desiccant and
solvent had been eliminated the residue was triturated with diethyl ether,
suction
filtered and dried.
Yield: 47 mg (24% of theory)
ESI-MS: (M+H)+ = 673/675 (2C1)
retention time (HPLC-MS): 2.6 min (method A)
WO 2006/100026 CA 02600189 2007-09-06 PCT/EP2006/002557
-137-
Example 8.5
(R)-2-4,4'-bipiperid inyl-l-yl-1-(3, 5-dichloro-4-hydroxy-benzyl)-2-oxo-ethyl
4-(2-oxo-
1,2,4,5-tetrahydro-1,3-benzodiazepin-3-yl)-piperidine-1-carboxylate
I
\ OH
~ /
CI
O~N N_H
0
. H O
Prepared analogously to Example 8.4 from 150 mg (0.29 mmol) (R)-1 -carboxy-2-
(3,5-d ich lo ro-4-hyd roxy-phe nyl )-ethyl 4-(2-oxo-1, 2, 4, 5-tetra hyd ro-
1, 3-
benzodiazepin-3-yl)-piperidine-1-carboxylate and 92 mg (0.34 mmol) tert-butyl
[4,4']bipiperidinyl-1-carboxylate.
Yield: 84 mg (44% of theory)
ESI-MS: (M+H) + = 672/674 (2CI)
retention time (HPLC-MS): 3.1 min (method A)