Note: Descriptions are shown in the official language in which they were submitted.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-1-
NOUVEA.UX DERIVES Us'A.NRNOA.CIDEâ PHQSPHINIQUES,
LEUR PROCEDE DE PREPARATION
ET LES COMPOSITIONS PHARMACEUTIQUES QUI LES CONTIENNENT
La présente invention concerne de nouveaux dérivés d'aminoacides
phosphiniques, leur
procédé de préparation et 'les compositions pharmaceutiques qui les
contiennent.
Les nouveaux dérivés d'aminoacides phosphiniques selon l'invention présentent
la
remarquable propriété d'inhiber à la fois l'enzyme de conversion de
l'angiotensine I(ECA)
et l'enzyme de conversion de l'endothéline (ECE).
La balance existant entre les peptides vasoactifs, vasoconstricteurs
(angiotensine II,
endothéline 1) d'une part et vasodilatateurs (facteurs natriurétiques,
bradykinine) d'autre
part, constitue un élément majeur de régulation de la pression artérielle.
La synthèse et la dégradation de ces peptides sont sous le contrôle de
diverses enzymes
parmi lesquelles trois métalloprotéases à zinc semblent jouer un rôle
prépondérant :
- l'enzyme de conversion de l'angiotensine I (ECA) qui d'une part transforme
l'angiotensine
I, un décapeptide inactif, en angiotensine II, un octapeptide actif, et
d'autre part dégrade la
bradykinine en peptides inactifs,
- l'enzyme de conversion de l'endothéline (ECE) qui clive la big-endothéline 1
en
endothéline 1 et seinble participer dans de moindres proportions à la
dégradation de la
bradykinine,
- l'endopeptidase neutre (EPN) qui inactive le peptide atrial natriurétique
(ANP ou Atrial
Natriuretic Peptide) et la bradykinine en peptides inactifs.
L'angiotensine II est un octapeptide vasoconstricteur et antinatriurétique.
Les endothélines
sont des polypeptides vasoconstricteurs et antinatriurétiques d'une vingtaine
d'acides
aminés comportant deux ponts disulfures reliant des résidus de cystéine. La
bradykinine est
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-2-
un nonapeptide vasodilatateur et natriurétique.
L'angiotensine II, l'endothéline, et la bradykinine sont les polypeptides les
plus importants
à ce jour impliqués dans la régulation du tonus vasculaire, du remodelage
cardiovasculaire
et de l'homéostasie hydroélectrolytique. Leur métabolisme est essentiellement
contrôlé par
les trois enzymes ECA, ECE et EPN.
L'hypertension artérielle ainsi que d'autres pathologies cardiovasculaires
sont caractérisées
par un déséquilibre de la balance peptidergique en faveur des peptides
vasoconstricteurs
lesquels exercent une action globalement délétère sur la sphère réno-
cardiovasculaire
(rétention hydrosodée, hypertrophie cardiovasculaire, etc). Malgré les
avancées
thérapeutiques majeures réalisées dans les années 80 par les inhibiteurs
sélectifs de l'ECA,
le développement de nouvelles molécules est apparu nécessaire afin d'améliorer
encore le
contrôle tensionnel des patients hypertendus ainsi que leur pronostic vital
(action bénéfique
sur les facteurs majeurs de risque cardiovasculaire).
Les trois métalloprotéases ECA, ECE et EPN apparaissent donc comme des cibles
intéressantes pour le traitement des maladies cardiovasculaires. Ainsi, afin
de lutter contre
les effets vasoconstricteurs et délétères de l'angiotensine II et de
l'endothéline 1 et de
favoriser les effets vasodilatateurs et protecteurs de l'ANP et de la
bradykinine, des
inhibiteurs de l'ECA, de l'EPN et de l'ECE ont été développés.
De nombreux produits possédant l'une ou l'autre de ces activités sont connus.
De nombreuses demandes de brevets décrivent des dérivés d'acides aminés utiles
en tant
qu'inhibiteurs ECA, ECE et utiles en tant qu'inhibiteurs mixtes ECA/EPN et
ECE/EPN.
Des inhibiteurs sélectifs de l'ECA utilisés comme anti-hypertenseurs depuis
des années
sont décrits dans le brevet US 4 396 772. Les composés décrits admettent pour
formules :
II ~ o O
Ri-P-(CHz)n -CH- LRS LO-R4
1
ORZ
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-3-
Les brevets WO 97/32874 et US 5 476 847 décrivent des composés inhibiteurs de
l'ECE.
Les composés décrits admettent pour formules :
= pour le brevet WO 97/32874
~ R1
R1~
1HS-(CH2)a G-C -NH
A
= pour le brevet US 5 476 847
R O
i R2 p
R6\ .III/(11,)n /Rs
H P A'
OR Ra
D'après la demande de brevet WO 95/35302 on connaît certains dérivés de
l'acide
phosphinique possédant une activité inhibitrice ECE utile dans le traitement
des maladies
cardiovasculaires. Les composés décrits admettent pour formule :
11 1 1
RZ i - (CXZ)m - ;-CO-H i - COOH
OH
CF]~ R
Des inhibiteurs mixtes ECA/EPN (classe des "inhibiteurs de vasopeptidases")
ont aussi été
synthétisés et testés en clinique.
Des inhibiteurs mixtes ECA/EPN sont décrits dans les brevets WO 97/24341, WO
96/22998, WO 93/08142 et EP 0 723 974. Les composés décrits sont des dérivés
sulfurés
de peptides.
Dans Bioorganic and Medical Chenaistry Letters, 1996, 6(11), 1257-1260, sont
décrits des
dérivés de l'acide phosphinique possédant une activité inhibitrice mixte
ECA/EPN utiles
dans le traitement des maladies cardiovasculaires. Ces composés admettent pour
formule :
0
H il H 2Na+
RN P N COZ
YO-
Ri O
N
H
Ces molécules présentent une efficacité antihypertensive supérieure à celles
des inhibiteurs
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-4-
sélectifs de l'ECA. Néanmoins elles présentent des effets secondaires maj
eurs, notamment
à titre d'angioédème, vraisemblablement en relation avec un excès de
bradykinine (Trends
in Pharnaacological Sci., 2001, 22, 106-109 ; Tlae Lancet, 2001, 358, 1525-
1532). Ceci a
conduit à l'interruption du développement clinique des inhibiteurs mixtes
ECA/EPN les
plus avancés comme l'omapatrilate (Curr Opin Investig Drugs, 2001, 2, 1414-
1422).
Les propriétés pharmacologiques des inhibiteurs mixtes ECA/EPN décrits dans
l'art
antérieur négligent le rôle cardiovasculaire majeur du système endothéline
(Journal of
Hypertension, 1998, 16(8), 1081-1098) ainsi que l'implication de 1'EPN dans la
dégradation de l'endothéline-l (J. Biol. Chem., 1990, 265, 14150-14155).
Ainsi, le
traitement par des inhibiteurs mixtes ECA/EPN a pour conséquence
l'augmentation du
taux d'endothéline-1 qui à long terme, peut se révéler néfaste aux bénéfices
thérapeutiques
attendus.
Enfin des inhibiteurs mixte ECE/EPN sont décrits dans Life Sciences, 2000,
67(9), 1025-
1033 et Naunyn-Schmiedeberg s Arch. Pharmacol., 358 (1, suppl 1) : R 513-514
(Abstr).
Ces composés ont pour formules :
= pour Life Sciences
e,~4 O P
N HO OH rJ-H
= pour Naunyn-Schmiedeberg s Arch. Pharmacol.
OH
O') O
N
H
N
O
O
L'intérêt d'avoir des inhibiteurs mixtes ECE/EPN est de diminuer le taux
d'endothéline
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-5-
tout en augmentant le taux de peptides natriurétiques et ainsi obtenir un
effet additif ou
synergique bénéfique pour le traitement des maladies cardiovasculaires et
rénales.
Néanmoins l'EPN étant l'un des acteurs les plus importants de la dégradation
de la
bradykinine in vivo, l'arrêt clinique des inhibiteurs ECA/EPN a également
clairement
invalidé le développement des autres inhibiteurs multiples de vasopeptidases
combinant
une inhibition de l'EPN, à savoir les inhibiteurs mixtes ECE/EPN ou triples
ECE/ECA/EPN.
En revanche, l'alternative des inhibiteurs mixtes ECA/ECE reste prometteuse
laissant
présager d'une efficacité cardiovasculaire majorée et d'une bonne sécurité
d'emploi. Ces
molécules devraient permettre de diminuer la formation des deux peptides
vasoconstricteurs puissants que sont l'angiotensine II et l'endothéline-1 et
d'augmenter
raisonnablement les taux de bradykinine.
Par ailleurs, il est intéressant de noter que les systèmes angiotensinergique
et
endothélinergique fonctionnent certes de façon indépendante mais aussi de
façon
interactive. Ce "crosstalk" existant entre les deux systèmes a été étudié sur
le plan
expérimental mais également sur le plan clinique. Le rôle de l'endothéline 1,
comme
médiateur de certains effets cardiovasculaires de l'angiotensine II a été tout
particulièrement exploré (Hypertension, 1997, 30, 29-34 ; Cardiovasc. Res.,
1999, 43, 300-
307 ; Hypertension, 2002, 40, 840-846 ; Clin. Exp. Pharmacol. Physiol., 2003,
30, 278-283
; Hypertension, 2002, 39, 715-720 ; Hypertension, 2003, 42, 825-830 ; Bioorg.
Med.
Cliena. Letters, 2003, 13, 1093-1096).
L'ensemble des données obtenues suggère que l'inhibition d'un des systèmes
engendre une
hyperactivité de l'autre système, ce qui est en faveur de l'approche
"inhibition mixte" pour
renforcer le potentiel thérapeutique de chacune des deux propriétés, en
évitant ces contre-
régulations.
Enfin, la preuve du concept inhibition mixte ECA/ECE c'est-à-dire le bénéfice
thérapeutique attendu de cette approche à été a ce jour analysé sur le plan
expérimental,
dans trois axes thérapeutiques : l'hypertension artérielle, l'insuffisance
cardiaque et la
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-6-
protection rénale. La preuve du concept a généralement été réalisée en
combinant deux
molécules sélectives; d'une part un inhibiteur sélectif de l'ECA ou un
bloqueur des
récepteurs ATl de l'angiotensine II pour le blocage du système
angiotensinergique et
d'autre part un bloqueur des récepteurs ETA de l'endothéline 1 ou un bloqueur
mixte des
récepteurs ETA/ETB de l'endothéline 1 ou dans de rares cas un ECE inhibiteur,
pour le
blocage endothélinergique.
Cette littérature supporte largement le concept. Les bénéfices thérapeutiques
sont mis en
évidence chez l'animal que ce soit sur le plan structural ou sur le plan
fonctionnel dans
l'hypertension artérielle (J Cardiovasc Plaarmacol., 2000, 36, S337-S341 ;
Clin Sci., 2002,
103, 363S-366S ; Ain J Hypertens., 2003, 16, 324-328) et l'insuffisance
cardiaque
(Cardiovasc. Res., 2002, 54, 85-94 ; Circulation, 2002, 106, 1159-1164). La
protection de
certains organes cibles tels le rein et le cerveau est fortement anticipée (J.
Am. Soc.
Nephrol., 2001, 12, 2572-2584).
La transposition clinique de l'ensemble de ces résultats précliniques pourrait
être un plus
grand nombre de patients normalisés par le traitement mixte. Il est probable
qu'à ce
bénéfice "tensionnel" s'ajoutera sur le long terme une meilleure protection
des organes
cibles de l'hypertension artérielle (ie prévention du "target organ damage"
aux niveaux
cardiovasculaire, rénal et cérébral) ainsi que des effets favorables sur
certains facteurs de
risque, prévenant ainsi les complications de l'hypertension artérielle.
La présente invention a pour objet de fournir de nouveaux composés qui se
comportent
comme des inhibiteurs mixtes ECA/ECE sans exercer aucune inhibition sur l'EPN.
Ainsi, les composés de la présente invention sont très efficaces dans le
traitement des
hypertensions artérielles et leurs complications comprenant les hypertensions
artérielles
pulmonaires, des ischémies myocardiques, de l'angine de poitrine, des
insuffisances
cardiaques, des vasculopathies, néphropathies, rétinopathies diabétiques, de
l'athérosclérose et de la resténose post-angioplastie, des insuffisances
rénales aiguës ou
chroniques, des maladies cérébrovasculaires comprenant le stroke et les
hémorragies sous
arachnoïdiennes, des ischémies périphériques.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-7-
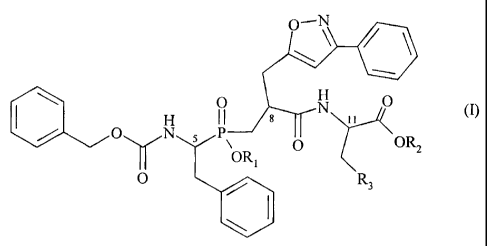
Plus particulièrement, la présente invention concerne les composés de formule
(I)
O-N
I \ O N O (~
p~ s ii
OR
z
N YoR,
O R3
dans laquelle :
Rl représente un atome d'hydrogène ou un groupement choisi parmi alkyl(Ci-
C6)carbonyloxyalkyl(C1-C6) linéaire ou ramifiée, la partie alkyle pouvant être
linéaire
ou ramifiée, ou alkyl(C1-C6)carbonylthioalkyl(C1-C6)linéaire ou ramifié, la
partie
alkyle pouvant être linéaire ou ramifiée,
R2 représente un atome d'hydrogène ou un groupement choisi parmi alkyl(C1-
C6)carbonyloxyalkyl(C1-C6)linéaire ou ramifié, la partie alkyle pouvant être
linéaire
ou ramifiée, arylcarbonylthioalkyl(C1-C6)linéaire ou ramifié, arylalkyl(C1-
C6)linéaire
ou ramifié éventuellement substitué sur sa partie aryle par un groupement
alkyl(Ci-
C6)carbonyloxy,
R3 représente un phényle éventuellement substitué par un groupement hydroxy ou
R3
représente un groupement 3-indolyle,
ou
Ri peut également représenter un groupement alkyl (Cl-C6) linéaire ou ramifié,
R2
représente un groupement alkyl (C1-C6) linéaire ou ramifié, lorsque R3
représente un
phényle substitué par un groupement hydroxy,
leurs énantiomères, diastéréoisomères, ainsi que leurs sels d'addition à une
base
pharmaceutiquement acceptable, ainsi que leurs hydrates et leurs solvats.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-8-
Parmi les bases pharmaceutiquement acceptables, on peut citer à titre non
limitatif
l'hydroxyde de sodium, l'hydroxyde de potassium, la triéthylamine, la
tertbutylamine, etc...
Selon une variante avantageuse de l'invention, les composés préférés sont les
composés
pour lesquels Rl et R2 représentent chacun un atome d'hydrogène.
Selon une deuxième variante avantageuse de l'invention, les composés préférés
sont les
composés pour lesquels Rl représente un atome d'hydrogène et R3 représente un
groupement phényle substitué par un groupement hydroxy.
Selon une troisième variante avantageuse de l'invention, les composés préférés
sont les
composés pour lesquels R2 représente un atome d'hydrogène et R3 représente un
groupement phényle substitué par un groupement hydroxy.
Parmi les composés préférés de l'invention, on peut citer plus
particulièrement :
- acide (5R,8R,11S)-5-benzyl-6-hydroxy-ll-(1H-indol-3-ylméthyl)-3,9-dioxo-1-
phényl-
8- [(3 -phényl-5 -i soxazolyl)méthyl] -2-oxa-4,10-di az a-6-pho sphadodéc an-
12-oique-6-
oxide,
- acide (5R,8R,11,S)-5,11-dibenzyl-6-hydroxy-3,9-dioxo-l-phényl-8-[(3-phényl-5-
isoxa-
zolyl)méthyl]-2-oxa-4,10-diaza-6-phosphadodécan-12-oique-6-oxide,
- acide (5R,8R,11S)-5-benzyl-6-hydroxy-11-(4-hydroxybenzyl)-3,9-dioxo-1-phényl-
8-
[(3-phényl-5-isoxazolyl)méthyl] -2-oxa-4,10-diaza-6-phosphadodécan-12-oique-6-
oxide.
Les énantiomères, diastéréoisomères, ainsi que leurs sels d'addition à une
base,
pharmaceutiquement acceptable, des composés préférés font partie intégrante de
l'invention.
La présente invention concerne également le procédé de préparation des
composés de
formule (I) caractérisé en ce qu'on utilise comme produit de départ le
chlorure de
diphénylméthanamine que l'on fait réagir avec du phénylacétaldéhyde en
présence d'acide
phosphonique, H3PO2, et d'acide chlorhydrique pour conduire au composé de
formule (II) :
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-9-
I
H O--H
N P (il)
OH
I \ / I
composé de formule (II) qui est soumis à l'action d'acide bromhydrique aqueux,
pour
conduire au composé de formule (III) :
O
H2N PI ~ H (III)
OH
composé de formule (III) que l'on fait réagir avec du chloroformiate de
benzyle en milieu
basique, pour conduire au composé de formule (IV) :
H 1 L-H
O N P\ (IV)
y OH
O
composé de formule (IV) qui est mis en présence de R-(+)-N,N-
(phényl)(éthyl)amine puis
soumis à l'action d'acide chlorhydrique pour conduire au composé de formule (R-
IV),
énantiomère R du composé de formule (IV) :
O
O N R \p'H (R-IV)
y OH
O
composé de formule (R-N) que l'on fait réagir, en présence de 1,1,1,3,3,3-
hexarnéthyldisilazane, avec un composé de formule (V) :
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-10-
(V)
O
pour conduire au composé de formule (R-VI) :
H 11
O N R i * O~~ (R-Vn
OH
O T O
composé de formule (R-VI) que l'on fait réagir avec une solution d'hydroxyde
de sodium,
pour conduire au composé de formule (R-VII) :
~
O
O N R PI ~ OH (R-VII)
y
OH
O T O
\ I .
composé de formule (R-VII) qui est soumis :
1) soit à l'action d'un composé de formule (VIII), en présence de
diisopropyléthylamine, de
1-hydroxybenzotriazole et de chlorure de 1-éthyl-3-[3-(diméthylamino)propyl]
carbodiimide :
O
H2N
OR'2 (VIII)
R'
3
dans laquelle R'2 représente un groupement alkyle (C1-C6) linéaire ou ramifié,
et R'3
représente un groupement choisi parmi 3-indolyle ou phényle éventuellement
substitué
par un groupement alkoxy (Cl-C6) linéaire ou ramifié,
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-11-
pour conduire au composé de formule (R,S-IX) :
~
I \ O O
H H
/ O N R * N S OR' (R,S-~
~ OH 2
O O R!3
dans laquelle R'2 et R'3 sont tels que définis précédemment,
composé de formule (R,S-IX) que l'on fait réagir avec du benzaldoxime en
présence de
N-chlorosuccinimide en milieu basique, pour conduire au composé de formule
(R,S-X) :
N
O~ ~N
0
O j
O N R
P N
I * TY OR~ (R,S-X)
1~ OH 2
O O R3
dans laquelle R'2 et R'3 sont tels que définis précédemment,
composé de formule (R,S-X) qui est placé en milieu acide, pour conduire au
composé
de formule (Ua), cas particulier des composés de formule (I) :
O~ \
O O
O N R I P N S (Ua)
y 1 * ORa
OH
O O
R3
dans laquelle R2 et R3 sont tels que définis dans la formule (I),
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-12-
2) soit aux mêmes conditions que le composé de formule (R,S-IX), pour conduire
au
composé de formule (R-XI) :
O_-NN
O
H 11
O N R i OH (R-XI)
OH
O O
composé de formule (R-XI) qui est soumis :
A/ soit à l'action d'un composé de formule (XII), en présence de
diisopropyléthylamine, de
1-hydroxybenzotriazole et de chlorure de 1-éthyl-3-[3-(diméthylamino)propyl]
carbodiimide :
O
H2N
ORZ (~
OPGA
dans laquelle R2 est tel que défini précédemment et PGA représente un
groupement
protecteur de la fonction phénol (T. W. Greene, "Protective Group in Organic
Synthesis", Wiley-Interscience, New-York, 1981) bien connu de l'homme du
métier,
pour conduire au composé de formule (R,S-XIII) :
O N
O O
O N R PI N S ~S-~
~ OR2
OH
O O
\ I \ I
OPGA
dans laquelle R2 et PGA sont tels que définis précédemment,
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-13-
composé de formule (R,S-XIII) dont la fonction phénol est déprotégée par des
techniques usuelles de la chimie organique et bien connu de l'homme du métier,
pour
conduire au composé de formule (I/b), cas particulier des composés de formule
(I) :
O.-N
O O
PI N
O ~ S O~ (~)
~N R OH
O O
\ \ I
OH
dans laquelle R2 est tel que défini précédemment,
B/ soit à l'action d'un composé de formule (XIV), dans les mêmes conditions
que
précédemment :
O
H2N
OR'2 (XIV)
OPGA
dans laquelle R'2 et PGA sont tels que définis précédemment, pour conduire au
composé
de formule (R,S XV) :
O,N
O O
PI N
O I S OR, (R,S-XV)
~N R OH 2
O O
\ ~ \ (
OPGA
dans laquelle R'2 et PGA sont tels que définis précédemment, composé de
formule
(R,S-XV) qui est soumis :
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-14-
a) soit à l'action d'un composé de formule (XVI), en présence d'iodure de
sodium, de
(nBu)4NHSO4 et de triéthylamine :
CI,,, R1-~O'-fr' Rb (XVI)
O
dans laquelle Ra et Rb, indépendamment les uns des autres, représentent un
groupement alkyle (Cl-C6) linéaire ou ramifié, pour conduire au composé de
formule
(R,S-XVII)
O--\
O O
I
O y N R P N S OR!2 (R,S-XVII)
O O
Ra O
Rb OPGA
O
dans laquelle Ra, Rb, R'2 et PGA sont tels que définis précédemment,
(3) soit à l'action d'un composé de formule (XVIII), en présence de PyBOP et
de
diisopropyléthylamine :
HO"-,Rl-.,S'~yRb (XVIR)
a
O
dans laquelle Ra et Rb sont tels que définis précédemment, pour conduire au
composé
de formule (R,S-XIX) :
O,\
O O
O N R PI N S (R,S-XTX)
~ OR2
O
O O
Ra S
~Rb
O OPGA
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-15-
dans laquelle Ra, Rb, R'2 et PGA sont tels que définis précédemment,
les composés de formules (R,S-XVII) et (R,S-XIX) formant le composé de formule
(R,S-XX) :
O-IN
O O
PI
O N R ~ N S OR! (R,S-XX)
y ORl z
O O
\ / \
OPGA
dans laquelle Rl est tel que défini dans la formule (I) et R'2 et PGA sont
tels que
définis précédemment,
composé de formule (R,S-XX) dont la fonction phénol et la fonction acide
carboxylique sont déprotégées par des techniques usuelles de la chimie
organique et
bien connu de l'homme du métier pour conduire au composé de formule (I/c), cas
particulier des composés de formule (I) :
O~N
O O
I/ O N R PI N S (Uc)
OH
ORl
O O
\ / \
OH
dans laquelle Rl est tel que défini précédemment,
composés de formule (Ua), (Ub) et (Uc) dont on sépare les stéréoisomères,
lorsqu'on le
souhaite par des techniques classiques de séparation, que l'on purifie le cas
échéant, par des
méthodes classiques de purification, et que l'on transforme, lorsqu'on le
souhaite, en leurs
sels d'addition à une base pharmaceutiquement acceptable.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-16-
Le composé de formule (XII) :
O
H2N
OR2 (XII)
/ I
\
OPGA
peut être obtenu à partir d'un composé de formule de (XXI) :
O
H
N
PGB OH (XXI)
a
OPGA
dans laquelle PGA est tel que défini précédemment et PGB représente un
groupement
protecteur de la fonction amine (T. W. Greene, "Protective Group in Organic
Synthesis",
Wiley-Interscience, New-York, 1981), bien connu de l'homme du métier,
composé de formule (XXI) qui est mis à réagir :
a) soit avec un composé de formule (XXII) en présence de NaI, (nBu)4NHS04,
NEt3 :
R2 - Cl (XXII)
dans laquelle R2 est tel que défini précédemment, pour conduire au composé de
formule
(XXIII) :
O
H
PGBN O_~ (XXII>)
OPGA
dans laquelle PGA, PGB et R2 sont tels que définis précédemment,
b) soit avec un composé de formule (XXIV) en présence de EDC.HC1 et de 4-
diméthyl-
aminopyridine :
R2 - OH (XXIV)
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-17-
dans laquelle R2 est tel que défini précédemment, pour conduire au composé de
formule
(XVIII) tel que défini précédemment,
composé de formule (XXIII) dont la fonction amine est déprotégée selon des
techniques
classiques de la synthèse organique bien connus de l'homme de l'art.
La présente invention a également pour objet les compositions pharmaceutiques
contenant
les composés de formule (I) en combinaison avec un ou plusieurs excipients
pharmaceutiquement acceptables.
Parmi les compositions pharmaceutiques selon l'invention, on pourra citer plus
particulièrement celles qui conviennent pour l'administration orale,
parentérale, nasale, per
ou transcutanée, rectale, perlinguale, oculaire ou respiratoire et notamment
les comprimés
simples ou dragéifiés, les comprimés sublinguaux, les sachets, les paquets,
les gélules, les
glossettes, les tablettes, les suppositoires, les crèmes, les pommades, les
gels dermiques et
les ampoules buvables ou injectables.
La posologie varie selon le sexe, l'âge et le poids du patient, la voie
d'administration, la
nature de l'indication thérapeutique, ou des traitements éventuellement
associés et
s'échelonne entre 0,1 mg et 1 g par 24 heures en 1 ou plusieurs prises.
Les exemples suivants illustrent l'invention et ne la limitent en aucune
façon. Les
préparations suivantes conduisent à des composés de l'invention ou à des
intermédiaires de
synthèse utiles dans la préparation de l'invention.
Les produits de départ utilisés sont des produits commerciaux ou préparés
selon des modes
préparatoires connus.
Les structures des composés décrits dans les exemples ont été déterminés selon
les
techniques spectrométriques usuelles (infrarouge, RMN, spectrométrie de
masse).
Par composé (5R,8*,11S), on entend mélange racémique de 2 diastéréoisomères de
!5 configurations absolues (5R,8S,11S) et (5R,8R,11S).
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-18-
Prêparation 1 : 2-Méthyléne-4-pentynoate d'éthyle
34,9 g de diéthyl malonate sont lentement ajoutés à une solution de EtONa
préparée par
dissolution de 5,0 g de sodium dans 280 ml d'éthanol absolue. Le mélange
réactionnel est
chauffé à 50 C pendant 1 heure puis 38,8 ml de 3-bromo-1-propyne dans 100 ml
d'éthanol
absolue sont ajoutés goutte à goutte au mélange. Le milieu réactionnel est
agité pendant
12 heures à cette température. Après évaporation sous pression réduite, le
résidu est repris
dans du diéthyléther et extrait avec de l'eau. La phase organique est séchée
sur sulfate de
sodium, filtrée puis évaporée sous pression réduite. L'huile obtenue est
reprise dans 100 ml
d'éthanol absolue puis une solution de KOH (12,2 g dans 340 ml d'éthanol) est
ajoutée
goutte à goutte. Après agitation à température ambiante pendant 1,5 heures,
l'éthanol est
évaporée sous pression réduite et le résidu est dissous dans de l'eau et
extrait avec du
diéthyléther. La phase aqueuse est refroidie à l'aide d'un bain de glace,
acidifiée avec HCI
2M jusqu'à pH-1 extrait avec du diéthyléther. La phase organique est séchée
sur sulfate de
sodium, filtrée puis évaporée sous pression réduite. 35,0 ml de pyridine, 13,4
g de
paraformaldéhyde et 1,85 ml de pipéridine sont ajoutés au résidu obtenu et le
mélange
réactionnel est agité à 100-105 C pendant 3 heures. Après refroidissement à
température
ambiante, le milieu est dilué avec 400 ml de diéthyléther et la phase
organique est
successivement lavée avec de l'eau, HC1 2M, une solution de NaHCO3 à 5% et une
solution saturée de NaC1. La phase organique est séchée sur sulfate de sodium,
filtrée puis
évaporée sous pression réduite. Une chromatographie sur gel de silice (éther
de pétrole
(40-60 C)/diéthyléther : 9,5/0,5 à 8/12) permet d'isoler le produit attendu.
Rf= 0,31 (éther de pétrole (40-60 C)/diéthyléther : 9,5/0,5)
Préparation 2 : Acide 2-fff1Rl-1-f f(benz-vk-xy)carbanyllamdnal-2-
tahényiélhyl)-
(hydraxy)uhasphoryllméthyll-4-pentyncaiïa u e
StadeA : Acide 1-(betzzlzydrylamifz )-2-plténylétlzylpliosplzifzigue
51,5 ml d'une solution aqueuse de H3PO2 50% sont ajoutée à une supension de
109,5 g de
chlorure de diphénylméthanamine dans 750 ml d'éthanol à 90% puis le milieu
reactionnel
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-19-
est chauffé à 85-90 C. A cette température, 58,4 ml de phénylacétaldéhyde dans
175 ml
d'éthanol sont additionnés sur une période de 3 heures et, le chauffage est
poursuivi
pendant 3 heures supplémentaires puis agité pendant 16 heures à température
ambiante. Le
précipité qui s'est formé est filtré, lavé avec de l'éthanol froid et du
diéthyléther puis séché
permettant ainsi d'isoler le produit attendu.
Stade B : Acide 1-anzino-2-plzénylétlzylplzosnhifzi9ue
181,1 g du composé du stade A précédent dans 500 ml d'acide bromhydrique sont
chauffés
à 110-120 C pendant 2 heures. Le mélange est alors concentré sous pression
réduite et le
résidu est dilué dans de l'eau et extrait avec du diéthyléther. La phase
aqueuse est
concentrée et 600 ml d'éthanol absolue sont ajoutés au résidu. 60 ml d'oxyde
de propylène
préalablement refroidi sont ajoutés lentement au mélange réactionnel maintenu
à 0 C. Le
produit attendu qui précipite lors du refroidissement est filtré, lavé au
diéthyléther puis
séché.
Stade C : Acide 1-{!(befzzyloxy)carbonyllaminol-2-pliénylétlzylphosnhifzigue
110 ml d'une solution d'hydroxyde de sodium 4M sont ajoutées a une suspension
de 49,4 g
du composé du stade B précédent dans 120 ml d'eau. Le milieu réactionnel est
amené à
0 C puis 45,5 ml de chloroformiate de benzyle sont additionnés sur une période
de 1 heure.
Le mélange est agité à 0 C pendant 1 heure et à température ambiante pendant 4
heures en
ramenant le pH de la solution à 9-10 par addition d'une solution d'hydroxyde
de sodium
2M. Le mélange est ensuite agité à température ambiante pendant une nuit, puis
extrait
avec du diéthyléther. La phase aqueuse est acidifiée par addition d'acide
chlorhydrique
6M. Le produit attendu précipite, est filtré, lavé avec de l'eau, du
diéthyléther puis séché
sur P205.
Stade D : Acide (1R)-1-{!(befzzyloxy)carbofzyllamitzo}-2-
plzéyzylétlzylplzosplzinigue
Une solution de 83,4 g du composé du stade C précédent dans 1000 ml d'éthanol
absolue
est chauffée sous reflux. Une solution de 34,4 ml de R-(+)-N,N-
(phényl)(éthyl)amine dans
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-20-
170 ml d'éthanol est lentement ajoutée au milieu réactionnel. Après 15 mn, le
milieu
réactionnel est ramené à température ambiante et refroidie à 4 C pendant la
nuit. Le
précipité formé est filtré, lavé avec de l'éthanol absolue et du diéthyléther.
Le solide est
recristallisé dans 445 ml d'éthanol absolue. Le sel obtenu est mis en
suspension dans
300 ml d'acide chlorhydrique 6M et agité pendant 2-3 heures. Le solide est
filtré, lavé avec
H20 et EtaO puis séchée sur P205.
Pouvoir rotatoire [a]20D= -46,7 (1% dans éthanol absolue)
Stade E : Acide (1R)-1-{!(benzyloxy)carbonylJamino)-2-phényléthyll2-(étlzoxy-
carbonyl)-4-pentyjzyllphosphinigue
1,3 mmol du composé de la préparation 1 sont additionnés goutte à goutte à un
mélange de
1 mmol du composé du stade D précédent et de 5 mmol de HMDS chauffé à 110 C
pendant 1 heure sous atmosphère d'argon. Le mélange réactionnel est chauffé à
100-105 C
pendant 3 heures supplémentaires. Il est alors refroidi à 70 C et de l'éthanol
absolue est
ajouté par petites portions toujours sous atmosphère d'argon. L'agitation est
poursuivie
pendant encore 15 min. à cette température. Le solvant est ensuite évaporé
sous pression
réduite. Une chromatographie sur gel de silice (chloroforme/méthanoUacide
acétique
7/0,3/0,3) permet d'isoler le produit attendu.
Stade F: Acide 2-!!(1R)-1-{!(benzyloxy)carbonylJamino~-2
nhénylétlayl)(hydroxy)
phosnhorylhnéthyll-4 nentynoïgue
Une solution de 1 mmol du composé du stade E précédent dans 9 ml d'éthanol est
refroidie
à 0 C. 5-6 mmol d'hydroxyde de sodium 1M sont additionnés par petites portions
puis le
mélange réactionnel est agité à température ambiante pendant 6-8 heures. Après
acidification avec HCl 2M, l'éthanol est évaporé et le résidu est repris dans
de l'eau et
extrait avec de l'acétate d'éthyle. La phase organique est lavée avec de
l'eau, une solution
saturée de chlorure de sodium puis séchée sur sulfate de sodium et évaporée
sous pression
réduite. Le produit attendu est obtenu par précipitation dans un mélange
diéthyléther/éther
de pétrole.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-21-
Préparation 3 : Acide 3- 1R .-1- 1(benzyloxy
carban I amîno -2- ,hën léth 1.
(hydraxv)nh asuhoryll-2,-f f 3-Rhényl-S-isoxaza-lyllméthvll Qronanaïa ue
Stade A : Acide 2-{%((1R)-1-{1(ben.zyloxy)carbonyliamino/-2-phénylétlayl)
(hydroxy)phosphoryl]méthyl/-4-pentynoïgue
Ce composé est obtenu selon un procédé décrit dans la littérature (A.
Makaritis et al.,
Chem. Eur. J., 2003, 9, 2079-2094).
Stade B : Acide 3-[((1R)-1-{f(benzyloxy)carbonylJamino~-2-
phényléthyl)(Izydroxy)
phosphorylJ-2-%(3-phényl-S-isoxazolyl)méthyl]propanoïgue
Une cycloaddition 1,3-dipolaire sur le composé du stade A précédent utilisant
du
benzaldéhyde oxime est effectuée selon un procédé décrit dans la littérature
(A. Makaritis
et al., Chem. Eur. J., 2003, 9, 2079-2094).
EXEMFLE 1 ;
Acide (5R,8R,11.S)-à-benzyI-6-hydraxy-11-(1H-indol-3-yIméthyl)-3,9-diaxn-1-
phényl-
8-[(3-phényl-5-isaxazc-lyl)méthyIj-2-ax.a.-4,1 Q-diaza-6-phosphadodëcan-12-
oique-h-
oxide
StadeA : Acide (1R)-1-ff(berzzyloxy)carbonylJamino}-2-phénylétlzyl-[2-([%(1S)-
1-(1H-
indol-3-ylméthyl)-2-méthoxy-2-oxoéthylJaminolcarbonyl)-4-pentynyll
hosphii:igue.
A une suspension de 1 mmol du composé du composé de la préparation 2 dans 20
ml de
dichlorométhane sont additionnés 3 mmol de diisopropyléthylamine, 1 mmol de
chlorure
de 1-tryptophane méthylester, 1 mmol de HOBt et 4 mmol de EDC.HCI. Le mélange
réactionnel est agité pendant 2 heures à température ambiante puis dilué avec
du
dichlorométhane. De l'acide chlorhydrique 1M est alors ajouté pour former deux
phases.
La phase organique est lavée avec HCl 1M , séchée sur sulfate de sodium puis
concentrée
sous pression réduite. Le produit attendu est obtenu par précipitation dans un
mélange
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-22-
diéthyléther/éther de pétrole.
Stade B: Acide (IR) -1-{f(benzyloxy)carbonyllamino)-2-phénylétlzyl -{3-{!(1S)-
1-(IH-
indol-3-ylméthyl)-2-méthoxy-2-oxoéthyllamino}-3-oxo-2-%(3-phényl-4-isoxa-
zolyl)métlzyl) jproyyl}phosphinigue.
6 mmol de benzaldoxime sont dissout dans 5 ml de chloroforme et 2 gouttes de
pyridine
sont additionnés à la solution. Puis, 6 mmol de N-chlorosuccinimide sont
ajoutés et après
mm d'agitation à température ambiante, le mélange réactionnel est agité à 45 C
pendant
3-4 heures. 1 mmol du composé du stade A précédent est alors ajouté ainsi que
7 mmoles
de triéthylamine. Le mélange réactionnel est agité à 45 C pendant 96 heures
puis concentré
10 sous pression réduite et le résidu est repris dans de l'acétate d'éthyle,
lavé avec HCl 1M et
une solution saturée de chlorure de sodium. La phase organique est séchée sur
sulfate de
sodium et concentrée sous pression réduite. Le produit attendu est obtenu par
précipitation
dans un mélange diéthyléther/éther de pétrole.
Stade C: Acide (SR,8*,11S)-S-benzyl-6-lzydroxy-ll-(IH-indol-3-ylméthyl)-3,9-
dioxo-1-
phényl-8-!(3-phenyl-4-isoxazolyl)métlzyl)J-2-oxa-4,10-diaza-6-phospha-
dodecan-12-oïgue-6-oxide.
Le produit est obtenu selon le procédé du stade F de la préparation 1 en
utilisant le
composé du stade B précédent à la place du composé du stade E.
Stade D : Acide (SR,8R,11S)-S-betzzyl-6-hydroxy-ll-(1H-indol-3-ylfnéthyl)-3,9-
dioxo-
1-phétzyl-8-[(3-plzenyl-4-isoxazolyl)nzéthyl)I-2-oxa-4,10-dia.za-6-phosplza-
dodecan-12-oïgue-6-oxide.
Le diastéréoisomère (R,R,S) a été obtenu par purification en mode isocratique
avec un
tampon conlposé de 40% d'acétonitrile et de 60% de formate d'ammonium 83,3 mM
à pH
6,4 en utilisant une colonne AIT de 250 x 30 mm (phase stationnaire Kromasil
C18 billes de
5 m, pores 100 A) du composé du stade C précédent.
Spectrométrie de masse (ES/MS) = 733,2 Da
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-23-
EXEMPLE 2 ;
Acide (SR,8R,l.1S)-5,11-diûenzyl-6-hydraxy-3,9-diaxo-1-phényl-&-[(3-phén.yl-5-
isaxa.-
zolyl)méthylj-2-axa-4a10-diaza-6-phasphadczdécan-1.2-oique-6-a,xide
Stade A: Acide 2-(f !(IS)-1-benzyl-2-tert-butoxy-2-oxoéthylJamino/carbonyl)-4-
pentynyl((IR)-I-{~(benzyloxy)caz=bonylJamino} 2 phényléthyl)-mhosphinipue.
Ce composé est obtenu selon le procédé décrit dans le stade A de l'exemple 1
en utilisant le
chlorure de l'ester L-phénylalanine-O-di-tertbutyl à la place du chlorure de 1-
tryptophane
méthylester.
Stade B: Acide 3-({((IS)-1-benzyl-2-tert-butoxy-2-oxoéthyllamino/-3-oxo-2-t(3-
plzényl-5-isoxazolyl)méthyl/nz=opyl((IR)-1-[I(benzyloxy)carborzyl/aminol-2-
phé,raylétliyl)-phosphinigue.
Ce composé est obtenu selon le procédé décrit dans le stade B de l'exemple 1
en utilisant le
composé du stade A précédent. Le produit est purifié par chromatographie sur
gel de silice
(chloroforme/métlianoUacide acétique : 7/0,3/0,3).
Stade C: Acide (5R,8*,11S)-5,11-dibenzyl-6-hydroxy-3,9-dioxo-l-phényl-8-!(3-
phényl-
5-isoxazolyl)méthyll2-oxa-4,10-diaza-6-phosplzadodécan-12-oipue-6-oxide.
Le produit est obtenu en faisant réagir le composé du stade B précédent avec
de l'acide
trifluoroacétique, triisopropylamine, dichlorométhane et de l'eau dans les
proportions
85/2,5/10/2,5.
0 Stade D : Acide (5R,8R,IIS)-5,11-dibenzyl-6-lzydroxy-3,9-dioxo-1-plzétzyl-8-
((3-nhényl-
5-isoxazolyl)rnétliylJ-2-oxa-4,10-diaza-6-phosphadodécan-12-oigue-6-oxide.
Le diastéréoisomère (R,R,S) a été obtenu par purification en mode isocratique
avec une
colonne semi-préparative AIT de 250 x 10 mm (phase stationnaire Kromasil C18
billes de
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-24-
m, pores 100 ~) du composé du stade C précédent. Elution isocratique en
condition
acide : 52% acétonitrile ; 0,1% acide trifluoroacétique.
Spectrométrie de masse (ES/MS) = 694,3 Da
EXEMPLE 3 =
5 AGïde (5R,xR,11S)-5-benzyl-6-hydroxy-ll-(4-hydroxybenzyl)-3x9-dïoxo-1-phényl-
û-
[(3-phényl-5-isoxazolyl)méthyI]-2-oxa-4,1 O-dïaza-6-phospbadodéean-12-oiclue-6-
oxide
StadeA : Acide (1R)-1-{!(benzyloxy)carbonyl]amino}-2-phényléthyll2-({!(1S)-2-
tert-
butoxy-l-(4-tert-butoxybenzyl)-2-oxoéthyl]aminolcarbonyl)-4 nentVnyl%
10 phosplzinigue.
Ce composé est obtenu selon le procédé décrit dans le stade A de l'exemple 1
en utilisant le
chlorure de l'ester L-tyrosine-O-di-tertbutyl à la place du chlorure de 1-
tryptophane
méthylester.
Stade B : Acide (IR)-1-{!(benzyloxy)carbonylJamino}-2-phényléthyl{3-{!(IS)-2-
tert-
butoxy-l-(4-tert-butoxybenzyl)-2-oxoéthyllaminol-3-oxo-2-!(3-phényl-5-
isoxazolyl)méthyllpropy0phosphinipue.
Ce composé est obtenu selon le procédé décrit dans le stade B de l'exemple 2
en utilisant le
composé du stade A précédent.
Stade C: Acide (5R, 8* 11 S)-5-benzyl-6-hydroxy-11-(4-hydroxybenzyl)-3, 9-
dioxo-1-
phényl-8-!(3-phényl-5-isoxazolyl)méthyll-2-oxa-4,10-diaza-6-
ph osph adodécan-12-oigue-6-oxide.
Ce composé est obtenu selon le procédé décrit dans le stade C de l'exemple 3
en utilisant le
composé du stade B précédent.
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-25-
Stade D = Acide (5R,8R.IIS)-5-benzyl-6-hydroxy7ll-(4-laydroxybenzyl-3,9-dioxo-
1-
plzényl 8-!(3-plzényl-5-isoxa,zolyl)métltyl J-2-oxa-4,10-diaza-6-
ph osph ado décan-12-oigue-6-oxide.
Le diastéréoisomère (R,R,S) a été obtenu par purification en mode isocratique
avec une
colonne semi-préparative AIT de 250 x 10 mm (phase stationnaire Kromasil C18
billes de
m, pores 100 A) du composé du stade C précédent. Elution isocratique en
condition
acide : 43% acétonitrile ; 0,1% acide trifluoroacétique.
Spectrométrie de masse (ES/MS) = 711,3 Da
EXElVIPLE 4 ;
10 Acide SR 8* 1.15 -5-hen 1-ll- 4-h drax benz i-â 91216-tétraaxa-l- hén VI-8-
3-
uhënyl S isa,xazalvl)méthyll-2,13,Ia-trïaxa-4,10-diaza-17,IZ-dïmëthyloctadëean-
6
phosuhinigue
Stade A = N-!(Benzyloxy)carbonyll-4-[(tert-butoxycarbonyl)oxylphénylalanine
A une solution de 10 mmol de N-[(benzyloxy)carbonyl]-4-hydroxyphénylalanine
sont
ajoutés de l'hydroxyde de potassium jusqu'à pH 12. Le milieu réactionnel est
refroidi dans
un bain de glace et 1,5 équivalents de Boc2O sont ajoutés, puis ramené à
température
ambiante tout en maintenant le pH de la solution à 12 par addition d'hydroxyde
de
potassium solide. Après 2 heures, 0,5 équivalent de Boc2O sont rajoutés. Après
18 heures,
le mélange est alors concentré sous pression réduite et, la phase aqueuse est
extraite avec
un mélange Et20/éther de pétrole (1/1). La phase aqueuse est acidifiée à froid
avec HCl
2M jusqu'à pH 1 puis extraite deux fois avec de l'acétate d'éthyle. Une
chromatographie
(chloroforme/méthanol : 9,5/0,5) permet d'isoler le produit attendu.
Stade B = Pivalate de !(2-f!(ben.zyloxy)carbonyllamino}-3-{4-[(tert-
butoxycarbotayl)
oxylphényl}propanoyl)oxylmétlzyle
A une solution de 2,5 minol du composé du stade A précédent dans 15 ml de
chloroforme
sont ajoutés, (nBu)4NHSO4 (0,5 équivalents), triéthylamine (2 équivalents),
iodure de
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
- 26 -
sodium (1 équivalent) et du pivalate de chlorométhyle (2 équivalents). Le
milieu
réactionnel est chauffé sous reflux pendant 18 heures puis concentré. Le
résidu est dissout
dans de l'éther diéthylique, extrait avec de l'eau, deux fois avec HC10,5 M,
deux fois avec
NaHCO3 5% et de l'eau. La phase organique est concentrée et une
chromatographie
(dichlorométhane/méthanol : 9,9/0,1 à 9,5/0,5) permet d'isoler le produit
attendu.
Stade C : Chlorhydrate de [(2-amino-3-{4-%(tert-butoxycarbonyl)oxyl
phétzyl}propanoyl)oxV]méthVl 2,2-diméthylnropanoate
2 mmol du composé du stade B précédent sont dissout dans un mélange
éthanol/eau (2/1)
et quelques gouttes de HCI 0,5 M sont ajoutées jusqu'à pH 1. Le mélange
réactionnel est
hydrogéné à l'aide d'un ballon à hydrogène en présence de Pd/C (300 mg) comme
catalyseur. Après 2,5 heures, le mélange est filtré sur célite puis évaporé à
sec pour obtenir
le produit attendu.
Stade D : Acide (SR,8* IIS)-S-benzyl-ll-{4-!(tert-butoxycarbonyl)oxylbenzyl}-
3,9,12,16-tétraoxo-1 nhényl-8-[(3 plzényl-5-isoxazolyl)méthyl12,13,15-
trioxa-4,10-diaza-17,17-dinzéthyloctadécan-6-phosphinigue
A une suspension de 1,3 mmol du composé de la préparation 3 dans 7 ml de
dichlorométhane sont ajoutés 3 équivalents de diisopropyléthylamine, 1
équivalent du
composé du stade C précédent, 1 équivalent de HOBt et 5 équivalents de
EDC.HCI. Le
mélange réactionnel est agité pendant 75 min puis concentré sous pression
réduite. Le
résidu est dissout dans 50 ml d'un mélange acétate d'éthyle/éther diéthylique
(9/1) puis
extrait quatre fois avec HCI 1M, H20, trois fois avec NH4HCO3, 5 %, H2O, deux
fois avec
NaHCO3 5 %, H20, HCI 1M puis de la saumure. La phase organique est concentrée
et une
chromatographie (chloroforme/méthanol//acide acétique : 9,5/0,4/0,1) permet
d'isoler le
produit attendu.
?5 Stade E : Acide (SR,8*,IIS)-5-benzyl-ll-(4-hydroxybenzyl)-3,9,12,16-
tétraoxo-1-
plzészyl-8-[(3-plzényl-S-isoxazolyl)rnétlzylJ-2,13,1 S-trioxa-4,10-diaza-17,17-
difnéthvloctadécan-6-plzosphinique
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-27-
1 mmol du composé du stade D précédent est dissout dans 10 ml d'acide formique
et
200 l de triisopropylsilane. Le mélange réactionnel est agité à température
ambiante
pendant 50 min puis concentré sous pression réduite. Le résidu est dissout
dans l'acétate
d'éthyle et extrait deux fois avec la saumure, deux fois avec NaHCO3 5 %,
saumure, HCl
1M et saumure. La phase organique est concentrée pour donner le produit
attendu.
Spectrométrie de masse (MS-ESl) = 826 3(M+H)+
EXEME 5 ;
Acide SR 8k 11 -5-ben 1-11- 4-h -drax beua1-3 912-trïaxo-1- .héu i-8- 3-
hêan 1-5-isoxazal 1 méth I-2 13-dioxa-41Q-diaza- entadécan-6- hos hinï ue
Stade A: Pro,nanoate de 2-{f(benzyloxy)carbonylJamino3-3-{4-!(tert-
butoxycarbonyl)oxyJphényl}éthyle
A une solution de 4 mmol du composé du stade A de l'exemple 4 dans 5 ml
d'éthanol
absolu sont ajoutés HOBt (1,1 équivalent), EDC.HCI (1,1 équivalent), DMAP
(quantité
catalytique) et DIPEA (1,1 équivalent). Le milieu réactionnel est agité à
température
ambiante pendant 18 heures puis concentré. Le résidu est dissout dans de
l'éther
diéthylique, extrait avec de l'eau, deux fois avec HCl 0,5 M, deux fois avec
NaHCO3 5 %
et de l'eau. La phase aqueuse est séchée sur NaZSO4 puis concentrée pour
donner le produit
attendu.
Stade B: Clzlorizydrate du propanoate de 2-anzino-3-{4-!(tert-
butoxycarbonyl)oxyJphényl}éthyle
Le produit est obtenu selon le procédé du stade C de l'exemple 4 en utilisant
le composé du
stade A précédent.
Stade C: Acide (5R,8*,115)-S-beizzyl-ll-{4-[(tert-butoxvcarbonyl)oxyJbenzyl3-
3,9,12-
trioxo-1-phényl-8-%(3-phényl-S-isoxazolyl)tnétlayll-2,13-dioxa-4,10-diaza-
pentadécan-6-nlzosphinipue
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-28-
Le produit est obtenu selon le procédé du stade D de l'exemple 4 en utilisant
le composé du
stade B précédent.
Stade D : Acide (5R,8*,11S)-5-beszzyl-ll-(4-hydroxybenzyl)-3,9,12-trioxo-1-
phényl-8-
[(3-phéiayl-5-isoxazolyl)méthyll-2,13-dioxa-4,10-diaza-pentadécan-6-
phosphinigue
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade C précédent.
Spectrométrie de masse (MS-ESI) = 740,2 (M+H)+
EXEIlIPIS 6 :
Acide (5R,8*,11S)-S-benzyI-11-(4-hydraxviaenz-vl)-3,9,12,17-tétraoxo-1,17-
diphénvl-8-
f(3-phényl-5-isaxazalyl)méth .=IT-2,1â-diaxa-4t1Q-diaza.-16-thiaheatadëcan-6-
phosphinigue
Stade A: Pronanoate de 2-(benzoylsulfanyl)-3-(4-tert-butoxyphényl)-2-{f(9H-
fluorèn-
9-ylméthoxy)carbonyl/aminoléthyle
Le composé est obtenu selon le procédé du stade A de l'exemple 5 en utilisant
FmocTyr
(OBu)(OH) à la place du composé du stade A de l'exemple 4 et PhCOSCH2CH2OH
(décrit
dans la littérature I. Lefebvre et al., J. Med. Chem., 1995, 38, 3941-3950) à
la place
d'éthanol absolu dans le dichlorométhane.
Stade B : Propanoate de 2-(benzoylsulfanyl)-2-amino-3-(4-tert-
butoxynhényl)éthyle
1,5 mmol du composé du stade A précédent sont dissout dans 12 ml de
diméthylformamide
et 3 équivalents de diéthylamine. Le milieu réactionnel est agité à
température ambiante
pendant 1 heure puis concentrée sous pression réduite. Le résidu est dissout
dans l'acétate
d'éthyle, extrait trois fois avec de l'eau et de la saumure. Une
chromatographie
(chloroforme/méthanol : 9,5/0,5) puis précipitation dans un mélange éther de
pétrole/éther
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-29-
diéthylique (2/1) permet d'isoler le produit attendu.
Stade C: Acide (5R,8*,115)-5-benzyl-ll-(4-tert-butoxybenzyl)-3,9,12,17-
tétraoxo-1,17-
diphényl-8-!(3-plzényl-5-isoxazolyl)métlzyll-2,13-dioxa-4,10-diaza-l6-
tlziaheptadécan-6-phosplzinigue
Le produit est obtenu selon le procédé du stade D de l'exemple 4 en utilisant
le composé du
stade B précédent.
Stade D : Acide (5R,8*,11S)-5-benzyl-ll-(4-hydroxybenzyl)-3,9,12,17-tétraoxo-
1,17-
diphényl-8-t(3 nlzényl-5-isoxazolyl)méthyl12,13-dioxa-4,10-diaza-16-
thiaheptadécan-6-phosphinigue
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade C précédent.
Spectrométrie de masse (MS-ESI) = 874,3 -H)=
EXEMPLE 7 ;
Acide 5R 8* 1IS -S-ben I-11-4- tert-butox ben I-6- 2 éthanethiaate éiha -3 9-
diaxo-1.- phëa I-8- 3- hën yl-5-isoxazol - i méth I-2-oxa-4 1Q-diaza-6-
nhosphadodécan-l2-aiaue- ("i.-oxide
Stade A : Acide (5R,8*,11S)-5-benzyl-11-(4-tert-butoxybenzyl)-3,9,12-trioxo-1-
phényl-
8-((3-plzényl-5-isoxazolyl)métlzyll-2,13-dioxa-4,10-diaza-14,14-
dimétJzylpentadécan-6-phosphinigue
Le produit est obtenu selon le procédé du stade D de l'exemple 4 en utilisant
le composé de
la préparation 3 et HCl.HTyr(OBut)OBut.
Stade B: tert-butyle de (5R,8*,IIS)-5-benzyl-11-4-(tert-butoxybe1iz0-6-
[2(étlaanetlzioate)éthoxyl-3,9-dioxo-1-phényl-8-[(3-phétzyl-5-
isoxazolyl)inéthyll-2-oxa-4,10-diaza-6-phosphadodécan-12-oate 6-oxide
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-30-
0,36 mmol de composé du stade A précédent sont dissouts dans 4 ml de
diméthylformamide puis sont ajoutés 3 équivalents de DIPEA, 3 équivalents
CH3COSCH2CH2OH (I. Lefebvre et al., J. Med. Chem., 1995, 38, 3941-3950) et 3
équivalents de PYPOB. Le mélange réactionnel est agité à température ambiante
pendant 48
heures puis dilué avec de l'acétate d'éthyle. La phase organique est extraite
quatre fois avec
HCl 1M, HZO, trois fois avec NH4HCO3 5 %, deux fois avec HCl 1M puis de la
saumure.
La phase organique est séchée puis concentrée. Une chromatographie
(chloroforme/méthanol : 9,8/0,2) permet d'isoler le produit attendu.
Stade C : Acide (5R,8*,11S)-5-benzyl-11-4-(tert-butoxybenzyl)-6-
l2(éthanethioate)
éthoxy]-3,9-dioxo-1-nhényl-8-[(3 phényl-5-isoxazolyl)néthylJ-2-oxa-4,10-
diaza-6 nhosyhadodécan-12-oipue-6-oxide
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade B précédent.
Spectrométrie de masse (MS-ESI) = 814,3 (M+H)+
EXEMPLEI :
Ethyle le de 5R. 8* 11 S-S-ben I-6-étbc-x -I l.- 4-h drox . ben I-3 9-dioxa-1-
hénI-L-
â- h.én yI-5-isoxazal. - I. .m6th I-2-axa-4 10-diaza-fi- has ,hadodêean.-1.2-
oate 6-oxide
StadeA : Ethyle de (5R,8*,11S)-5-benzyl-11-{4-[(tert-
butoxycarbonyl)oxy/benzyl/-6-
éthoxy-3,9-dioxo-1-phényl-8-!(3-phényl-5-isoxazolyl)méthylJ-2-oxa-4,10-
diaza-6nhosphadodécan-12-oate 6-oxide
Le produit est obtenu selon le procédé du stade B de l'exemple 7 en utilisant
le composé du
stade C de l'exemple 5 et de l'éthanol.
Stade B : Etlayle de (5R,8*,115)-5-benzyl-6-ététoxy-ll-(4-hydroxybenzyl)-3,9-
dioxo-l-
pliényi-8-!(3-phényr 5-isoxazolyl)niéthyl]-2-oxa-4,10-diaza-6-
phos,vhadodécan-12-oate 6-oxide
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-31-
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade A précédent.
Spectrométrie de masse (MS-ESI) = 768 3(M+H)+ = 790 3(M+Na) +
EXEMPLE 9 :
Ethyle de (5R,S*,11S)-5-benzvl-ll-(4hydrox-vbenzyl)-6-f2-méthyl-1-
(nronionyloxy)
pronoxyl-3,9-dioxo-l-phënyl-$-f(3-uhényl-5-isaxazol-vl)méthyll-2-oxa-4,10-
diaza-6-
phostahadodécan-l2-oate 6-oxide
StadeA : Etlzyle de (5R,8*,IIS)-5-benzyl-ll-{4-!(tert-
butoxycarbonyl)oxy]benzyl}-6-l2-
méthyl-l-(propionyloxy)pronoxyl-3,9-dioxo-1 nhényl-8-!(3 nhényl 5
isoxazolyl)méthylJ-2-oxa-4,10-diaza-6-nhosphadodécan-12-oate 6-oxide
Le produit est obtenu selon le procédé du stade B de l'exemple 7 en utilisant
le composé du
stade C de l'exemple 5 et BrCH(CH(CH3)2)OCOEt (M. Neuenschwander et al., Helv.
Chim. acta, 1978, 61, 2047-2058.
Stade B : Ethyle de (5R,8*,11S)-5-benzyl-ll-(4-hydroxybenzyl)-6-l2-nzéthyl-1-
(propionyloxy)propoxyl-3,9-dioxo-l-phényl-8-!(3-phényl-5-isoxazolyl)
méthyll-2-oxa-4,10-diaza-6-nhosphadodécan-12-oate 6-oxide
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade B précédent.
Spectrométrie de masse (MS-APCI) = 868 4(M+H)+ = 885 5(M+NH4+L
EXEMFLE 1 ~ ;
Acide (5R,8*,11S)-5-benzvl-ll-(4-hydraxybenzyl)-6-f2-méthv1-1-(uroAionyloxy)
propaxyl-3,9-diaxa-1-ohénYl-S-F(3-uhényl-5-isoxazolvl)mêthyll-2-oxa-4,10-diaza-
6-
phostahadndéécan-12-oipuc-6-oxide
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
-32-
StadeA : tert-butyle de (5R,8*,11S)-5-benzyl-ll-(4-tert-butoxybenzyl)-6-l2-
méthyl-1-
(propionyloxV)propoxy)-3,9-dioxo-1-phényl-8-[(3-phényl-5-
isoxazolyl)méthyl]-2-oxa-4,10-diaza-6-phosphadodécan-12-oate 6-oxide
Le produit est obtenu selon le procédé du stade A de l'exemple 9 en utilisant
le composé du
stade A de l'exemple 7 à la place du composé du stade C de l'exemple 5.
Spectrométrie de masse (MS-ESn = 974,5 (M+Na+)+
Stade B : Acide (SR,8*,IIS)-5-benzyl-ll-(4-hydroxybenzyl)-6-l2-métlzyl-l-
(propionyloxV)propoxyj-3,9-dioxo-1-phényl-8-[(3 phényl-5-isoxazolyl)
méthyll-2-oxa-4,10-diaza-6-phosphadodécan-12-oipue-6-oxide
Le produit est obtenu selon le procédé du stade E de l'exemple 4 en utilisant
le composé du
stade A précédent.
Spectrométrie de masse (MS-ESI) = 838,5 (M-H)-
ETUDE PHARMACOLOGIQUE DES COMPOSES DE L'INVENTION
EXEMPLE A =
Effet inhibiiteur in vitro sur l'enzyme de conversion de l'angiotensine (ECA)
et de
l'enzyme de conversion de l'endothéline (EGE)
Afin de comparer la spécificité, les composés des exemples ont été testés sur
2 enzymes
l'ECA (forme recombinante humaine) et l'ECE (forme recombinante humaine de
l'isoforme ECE- 1 c).
Les tests ont été réalisés en duplicate dans des plaques à 96 puits.
L'inhibiteur a été incubé
avec l'enzyme pendant 45 minutes avant l'ajout d'un substrat à fluorescence
éteinte. La
fluorescence émise a été détectée et mesurée dans un lecteur de plaques
Fluoroscan Ascent
(Thermo-Labsystems).
Les substrats fluorogéniques utiilsés sont : Mca-Arg-Pro-Pro-Gly-Phe-Ser-Pro-
Dpacoox
(5 M) avec l'ECA et Mca-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phc-Lys(Dnp) COOH (5 M ;
CA 02601025 2007-08-30
WO 2006/092495 PCT/FR2006/000446
- 33 -
RSzD Systems) avec l'ECE.
Les composés de l'invention montrent une bonne capacité d'inhibition de l'ECA
et de
l'ECE avec des IC50 de 25 nM et 8 nM respectivement.
Les résultats obtenus sont rassemblés dans le tableau suivant :
Composé IC50 (nM)
ECA ECE
..... Exemple 1... 36..~ .....___.._...~~.~.._.... .._....... _~__.....
...._ _......~
Exem-ple2 ....3;8_..._.... ___...___ .._ _ _._.._...._ ~...._..._. 7,7
. .....
Exemple 3 1,4 1,4
EXEMP'L.F R ~
Composition pharma.ceutique
1000 comprimé dosés à 5 mg du composé de l'exemple 1
................................... 5 g
Amidon de blé
...............................................................................
..................... 20 g
Amidon de maïs
...............................................................................
................... 20 g
Lactose
...............................................................................
................................. 30g
Stéarate de magnésium
...............................................................................
.......... 2 g
Silice
...............................................................................
......................................1 g
Hydroxypropylcellulose
...............................................................................
........ 2 g