Note: Descriptions are shown in the official language in which they were submitted.
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
USE OF SPIROSTENOLS TO TREAT
MITOCHONDRIAL DISORDERS
Related Application
This application claims priority from U.S. Provisional Application Serial
No. 60/667,229 filed April 1, 2005 and to U.S. Provisional Application Serial
No. 60/697,518 filed July 8, 2005, which applications are herein incorporated
by reference.
Background of the Invention
Mitochondria are specialized compartments present in every cell of the
body except red blood cells, and are responsible for generating, by oxidative
phosphorylation, more than 90% of the energy needed by the body to sustain
life
and support growth. When the mitochondrial respiratory chain fails, the energy
level of the cell will rapidly decrease leading to the cell death and organ
function
impairment. Depending on the organ considered, the impairment of the
mitochondrial physiological function is involved in neurodegenerative diseases
and disorders. These include those of the central nervous system, such as
Alzheimer's Disease and Parlcinson's Disease, and those affecting the
peripheral
nerves, such as muscular disorders (myopathies) and various kidney, liver and
respiratory disorders.
Depending on how severe the mitochondrial disorder is, the illness can
range in severity from mild to lethal. Whether the mitochondrial failure is a
cause or a consequence of the condition experienced, protecting respiratory
function is critical to restore the impaired physiological functions. In
addition,
the preservation of the mitochondrial respiratory function is essential for
the
conservation of tissue required for a successful graft.
There are no cures for mitochondrial diseases which have a genetic basis,
but treatment can help reduce symptoms or delay or prevent the progression of
the disease (M. Zeviani et al., Brain; 127: 2153 (2004)). Treatment is
iindividualized for each patient, depending on the nature and the severity of
the
disorder, although these palliative treatments will not reverse the damage
already
sustained. However, a number of experimental strategies are currently being
assessed, including gene therapy and pharmacological inteivention.
One strategy consists of the introduction of modified genes or gene
products into mitochondria via the protein import machinery (G. Manfredi et
al.,
1
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Natur. Genet., 30:394 (2002)) and inhibition of replication of mutant mtDNA by
sequence-specific antigenomic peptide-nucleic acids (R.W. Taylor et al.,
Natur.
Genet., 15:212 (1997)). These approaches are not yet employed clinically.
In selected, isolated myopathy cases, reduction of heteroplasmic mutant
load was obtained by controlled inuscle fiber damage and regeneration by
mutation-free satellite cells, using myotoxic drugs (W. Irwin et al., J. Biol.
Ch.ern., 277:2221 (2002)). However, this treatment was not effective in
improving ptosis in five patients with progressive external ophtllalmoplegia
(PEO).
Creatine is the substrate for the synthesis of phosphocreatine and is the
most abundant energy storage compound in muscle, heart and brain. An open
trial of 81 patients with various neuromuscular disorders (including 17 with
mitochondrial diseases) showed significant improvement of ischaemic isometric
handgrip strength and non-ischemic isometric dorsiflexion torque. Another
placebo-controlled, double-blind, randomized crossover trial in 16 patients
with
chronic PEO or mitochondrial myopathy, however, did not find significant
effects on exercise performance, eye movements, or activities of daily life
(P.F.
Chinnery et al., Anz. J Med. Genet., 106:94 (2001)). Taken together, these
data
suggest that creatine may be effective in some, but not all mitochondrial
diseases.
While Coenzyme Q10 is not effective in mtDNA-associated
mitochondrial disease (N. Bresolin et al., J. Neurol. Sci., 100:70 (1990)) it
leads
to marked improvement in 'primary' CoQ10 deficiency. Idebenone, a shorter
chain analogue of Coenzyme Q10, appears to be effective in improving the
hypertrophic cardiomyopathy in Friedreich's ataxia (P. Rustin et al., Lancet,
354:
477 (1999); C. Mariotti et al., Neurology, 60:1676 (2003)).
Thus, there is a continuing need for methods to treat mitrochondrial
disorders, including mitochondrial diseases.
Summary of the Invention
The present invention provides a therapeutic method comprising
administering to a mammal, such as a huinan, afflicted with or threatened by,
a
mitochondrial disorder that is not a neuropathology of the central nervous
system
2
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
comprising administering to said mammal an effective amount of a compound of
formula (I):
R16 X R17
R12 R13
R11 0
R2
R1 R1o Rs R14 R15
~ 9
R3 R7
R4 R6 (I)
wherein each of Ri, R2, R4, R7, Rl l, R12, and R15, independently, is
hydrogen,
(C1-C8)alkyl, that is optionally inserted with -NR'-, -0-, -S-, -SO-, -SO2-, -
O-
SoZ-, -S02-0-, -C(O)-, -C(o)-0-, -0-C(O)-, -C(O)-NR'-, or -NR'-C(O)-;
hydroxy, N(R')2, carboxyl, oxo, or sulfonic acid; R3 is hydroxy, (C1-
C8)alkoxy,
(C1-C22)alkylCO2 or R'02C(CH2)2_8C02-, wherein (C1-CZ2)alkyl or (CH2)2_$ can
optionally comprise 1-2 CH=CH units, 1-2 OH, and/or 1-2 epoxy substituents,
toluene-4-sulfonyloxy or benzoyloxy; each of R6, R8, R9, Rlo, R13, R14, R16
and
R17, independently, is hydrogen, (Cl-C$)alkyl, hydroxyl(C1-C8)alkyl, (C1-
C8)allcoxy, or hydroxy; and X is 0, S, S(O), N(R'), N(Ac) or N(toluene-4-
sulfonyloxy),
wherein each R' is individually H, (C1-Cg)alkyl, phenyl or benzyl; or a
phannaceutically acceptable salt thereof.
R16 and/or R17 are preferably CH3.
Alkyl is preferably (C2-C8)alkyl.
Therefore, the present invention includes therapeutic methods of treating
a mitochondrial disorder in a subject afflicted with or threatened by said
disorder, wherein said disorder is not a neuropathology of the central nervous
system, comprising administering to the subject an effective amount of a
compound of formula (II):
H3C X CF-l3
R12 R13
R11 O
R1 R1o Rs
R2
R15
~9 R14
R3 R7
R4 R6 (R)
3
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
wherein each of Rl, R2, R4, R7, Rl l, R12, and R15, independently, is
hydrogen, (C1-C8)allcyl, hydroxy, amino, carboxyl, oxo, sulfonic acid, or (C1-
C$)alkyl that is optionally inserted with -NH-, -N((C1-C$)alkyl)-, -0-, -S-, -
SO-,
-SOz-, -O-SO2-, -SOz-O-, -C(O)-, -C(0)-0-, -O-C(O)-, -C(O)-NR'-, or -NR'-
C(O)-, wherein R' is H or (C1-C8)alkyl; R3 is hydroxy, (C1-C6)a1ky1CO2-,
HO2C(CH2)2C02-, toluene-4-sulfonyloxy or benzoyloxy; each of R6, R8, R9, Rlo,
R13, and R14, independently, is hydrogen, (C1-C8)alkyl, hydroxyl(Ct-C8)alkyl,
(C1-C$)alkoxy or hydroxy; and X is 0, N(H), N(Ac) or N(toluene-4-
sulfonyloxy); or a pharmaceutically acceptable salt thereof.
As discussed in detail below, spirostenols, particularly 5,6 unsaturated
spirostenols such as (22S,25S)-(20S)-spirost-5-en-3(3-y1 hexanoate, a 22R-
hydroxycholesterol derivative naturally occurring in Gynura japonica
(asteraceae), can provide a novel therapeutic strategy that works by its
direct
targeting of the mitochondrial respiratory chain, as well as its targeting of
A(3
which is toxic to mitochondria. (22S,25S)-(20S)-Spirost-5-en-3(3-yl hexanoate
also abolished the induced uncoupling of oxidative phosphorylation and
amplified the effect of cyclosporin A, a potent blocker of mitochondrial
membrane permeability transition pore, wllich suggest an effect on membrane
permeability transition pore.
Mitochondrial disorders, including mitochondrial diseases that are not
considered to be CNS neuropathologies, and that can be treated with the
present
method include the following: eye: retinitis pigmentosa, optical nerve
atrophy;
muscles: disorders of the extraoccular muscles (ptosis, acquired strabismus,
ophthalmoplegia), crushed muscle s}nldrome (compartment syndrome);
mitochondrial dysfunction as a result of drug side-effect (i.e., highly active
anti-
retroviral therapy, HAART); myopathy; heart: heart stroke, heart attack, heart
conditions, heart disease, cardiomyopathy, and any cardiac mitochondrial
dysfunction as a result of drug side-effect (i.e., HAART, mega-HAART, anti-
cancer drugs like anthracyclins); liver: hepatocellular dysfunctions; kidney
and
the endocrine system: tubulopathy, diabetes; and respiratory disorders.
The present invention also provides novel compounds of formulas I or II
and compositions, such as pharmaceutical compositions comprising an effective
amount of a compound of formula I and/or II in combination with a
pharmaceutically acceptable carrier.
4
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Brief Description of the Figures
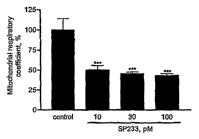
Fig.1 Effect of increasing concentrations of SP-233 (10 to 100 pM) on
the mitochondrial respiratory chain, assessed by monitoring the evolution of
the
respiratory coefficient in rat brain mitochondria. This mitochondrial
respiratory
coefficient (MRC) is V3/V4. V4 is the basal 02 consumption, V3 is the 02
consumption after adding ADP (ATP production). The mitochondrial
concentration was adjusted to 0.4 mg protein/ml. It is noteworthy that
concentrations of SP-233 as low as 10 pM induced a 50% decrease in the MRC,
compared to control (p<0.001). Results are expressed as mean SD;
comparisons between groups were made by an ANOVA followed by a Dunnett's
test.
Fig. 2 (a): Effect of SP-233 on CCCP-induced uncoupling of oxidative
phosphorylation in rat brain mitochondria. To assess the effect of SP-233 on
CCCP-induced uncoupling of oxidative phosphorylation, the mitochondrial
fraction was incubated for 3 inin at 37 C in presence of SP-233 before adding
malate/glutainate, followed 1 min later by the addition of CCCP. Exposure to
the uncoupling agent CCCP (1 M) increased the 02 consumption to 150% of
the basal value (p<0.01), a change that reflected an increase in oxygen
consumption. At all concentrations tested (even at 1 pM), SP-233 abolished the
metabolic effect of CCCP (p<0.001). This inhibitory effect of SP-233 on
"uncoupling" was associated with decreases in the 02 consumption to 60-65% of
the basal level and was concentration-independent. (b): Effect of SP-233 on
CsA-induced hypoxia. Hypoxia was induced by adding CsA (1 M) to the
medium bathing the mitochondria. This test was controlled by adding ADP in
presence of CsA and absence of SP-233. SP-233, or its vehicle, was added
first,
and incubation was continued for 1.5 min before CsA was gently added. Then,
the substrate malate/glutamate was added 1.5 min later and ADP 2.5 min later.
Exposure to SP-233 did not inhibit the decrease in the MRC induced by CsA,
but on the contrary, the effect of CsA was amplified in a concentration-
dependent manner. Results are expressed as mean SD; comparisons between
groups were made by an ANOVA followed by a Dunnett's test.
Fig. 3 Effects of fresh and aged A(3i-42 on the mitochondrial respiratory
coefficient of rat brain mitochondria. Mitochondria were incubated in the
5
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
presence of A(3i--42 for 1.5 min at 37 C before adding the substrates
malate/glutamate. ADP was added 1 min after malate/glutamate. This
experiment was controlled by omitting ADP. A(3i-42 significantly decreased the
MRC even at the low concentration of 0.1 pM. The effect of the fresh amyloid
peptide on the MCR was more pronounced than that of the aged form, the
respective decreases in MRC being 55-63% and 43-53%. Results are expressed
as mean + SD; comparisons between groups were made by an ANOVA followed
by a Dunnett's test.
Fig. 4 Effects of SP-233 on modifications of the mitochondrial
respiratory coefficient induced by fresh or aged A(3i-42 in rat brain
mitochondria.
Mitochondria were incubated in the presence of SP-233 for 3 min and in the
presence of freshly prepared (a) or aged (b) A(3i-42 for 1.5 min at 37 C
before
addition of malate/glutamate. ADP was added 1 min after the substrates
malate/glutamate. The control for the effect of SP-233 versus AOi-42 was the
same as for the experiment without ADP. (a): Fresh A(3i-42 reduced the MRC by
71% compared to the control, and this effect was partially inhibited by SP-
233.
The most potent effect was observed with the lowest concentration of SP-233
which restored the MRC to 39% of the control value (38.56 0.34 versus 28.93
::L1.75, p<0.001). (b): Aged A(3i-42 reduced the MRC by 51% compared to the
control value, but SP-233 did not prevent this effect. Results are expressed
as
mean SD; comparisons between groups were made by an ANOVA followed
by a Dunnett's test.
Fig. 5 Assessment of the protective effect of SP-233 against A(3i-42-
induced toxicity in human neuroblastoma cells. The cellular toxicity of AO was
assessed after 72 h incubation of SK-N-AS neuroblastoma cells with Aoi-42
(0.1,
1 and 10 M) or vehicle and in the presence or absence of SP-233 (1 M) using
the MTT assay. SP-233 (1 M) prevented the neurotoxicity induced by the three
concentrations ofAoi-42. Results are expressed as mean ~:L SD; comparisons
between groups were made by an ANOVA followed by Dunnett's test.
Neuroblastoma cells treated with A,6i-42 displayed strong
immunoreactivity (which co-localized with complex II of the mitochondrial
respiratory chain, indicating that A01-42 was present inside the
mitochondria).
Treatment with SP-233 abolished A(3i-42 immunoreactivity, indicating its
ability
to block the entry of A(.ii-42 into the mitochondria.
6
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Fig. 6 Effect of SP-233 on mitochondrial respiration. The Chemical
formula of the spirostenol derivative (22R,25R)-20a-spirost-5-en-3B-yl
hexanoate (SP-233) is shown in (a). The effect of increasing concentrations of
SP-233 (1 aM to 100 pM) on the mitochondrial respiratory chain, assessed by
monitoring the evolution of the respiratory coefficient in isolated rat brain
mitochondria, is shown in (b). This mitochondrial respiratory coefficient
(MRC)
is defined as V3/V4 where V4 is the basal 02 consumption and V3 is the 02
consumption after adding ADP (ATP production). The mitochondrial
concentration was adjusted to 0.4 mg protein/ml. Note that concentrations of
SP-233 as low as 1 fM resulted in a 40% decrease in the MRC, compared to the
control (p<0.001). Results are expressed as means SD from tliree independent
experiments performed in triplicate.
Fig. 7 Effect of SP-233 on CCCP-induced uncoupling of oxidative
phosphorylation and on CsA-induced hypoxia. (a) To assess the effect of SP-233
on CCCP-induced uncoupling of oxidative phosphorylation, the mitochondrial
fraction was incubated for 3 min at 37 C in presence of SP-233 before adding
malate/glutamate. CCCP was added 1 min later. Exposure to the uncoupling
agent 1 M CCCP increased the 02 consumption to 150% of the basal value
(p<0.01). At all concentrations tested, SP-233 abolished the metabolic effect
of
CCCP (p<0.001). SP-233's inhibitory effect on "uncoupling" was associated
with a decrease in 02 consumption to 60-65% of the basal level and was
concentration-independent. (b) Hypoxia was induced by adding 1 M CsA to the
medium batliing the mitochondria. As a control, ADP was added in presence of
CsA and in the absence of SP-233. SP-233, or its vehicle, was added first, and
incubation was continued for 1.5 min before CsA was gently added. The
substrate malate/glutamate was added 1.5 min later, and ADP was added 2.5 min
later. Exposure to SP-233 did not inhibit the decrease in the MRC induced by
CsA. On the contrary, CsA's effect was amplified in a concentration-dependent
manner. Results are expressed as means SD from three independent
experiments performed in triplicate.
Fig. 8 Effects of fresh and aged A131-42 on the mitochondrial respiratory
coefficient of rat brain mitochondria. Mitochondria were incubated in the
presence of AB1-42 for 1.5 min at 37 C before adding the substrates
inalate/glutainate. ADP was added 1 min after malate/glutamate. In these
7
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
experiments ADP was omitted from the incubation mixture. AB1_42 significantly
decreased the MRC, even at a concentration of 0.1 pM. The effect of the fresh
amyloid peptide on the MCR was more pronounced than that of the aged form,
the respective decreases in MRC being 55-63% and 43-53%. Results are
expressed as means :L SD from three independent experiments performed in
triplicates.
Fig. 9 Effects of SP-233 on changes in the MRC induced by fresh or
aged AB1_42. Mitochondria were incubated in the presence of SP-233 for 3 min
and in the presence of freshly prepared (a) or aged (b) Af31-42 for 1.5 min at
37 C before addition of malate/glutainate. ADP was added 1 inin after the
addition of malate/glutamate. Controls reactions were performed in the absence
of ADP. Fresh Af31_42 reduced the MRC by 71% compared to the control, and
this effect was partially inhibited by SP-233.
The most potent effect was observed with the lowest concentration of SP-
233 examined, which restored the MRC to 39% of the control value (38.56 :h
0.34 vs. 28.93 ::L 1.75, p<0.001). Aged A131_42 reduced the MRC by 51%
compared to the control value, but SP-233 did not prevent this effect. Results
are expressed as means SD from three independent experiments performed in
triplicate.
Fig. 10 Assessment of the protective effect of SP-233 against Af31_42-
induced toxicity in human neuroblastoma cells. The cellular toxicity of AB was
assessed 72 h after incubation of SK-N-AS cultures with Af31-42 (0.1, 1 and 10
RM) or vehicle and in presence or absence of SP-233 (1 M) using the MTT
assay. Results are expressed as means SD from three independent experiments
performed in triplicate.
Fig. 11 Confocal inicroscopy analysis of the effect of SP-233 on
mitochondrial uptake of A131-42 in SK-N-AS human neuroblastoma cells.
Representative images taken from healthy, uninjured (control) cells are shown
in
(al-4). These cultures did not receive any A131_42 or SP-233. Representative
images taken from SK-N-AS cells treated with AB1_42 10 M for 3 hours are
shown in (b 1-4). Representative images talcen from SK-N-AS cells treated with
Af31_42 10 M and SP-233 1 M for 3 hours are shown in (cl-4). Cells were
stained with DAPI nuclear counterstain (al, b 1, and c1) and labeled for
AB1_42
(FITC; a2, b2, and c2) and complex II 70-kDa subunit (Texas red; a3, b3, and
8
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
c3). Merged images are shown in a4, b4, and c4. Neuroblastoma cells treated
with A131_42 displayed strong immunoreactivity (b2) that co-localized with
complex II of the mitochondrial respiratory chain (b4), and treatment with SP-
233 abolished A131_42 iinmunoreactivity (c2).
Fig. 12 Neuroprotective effects of SP-233 against mitochondrial complex
inhibitors in SK-N-AS cells. Five different mitochondrial toxins were added to
SK-N-AS cultures 1 hour after the addition of SP-233 or its solvent. Rotenone,
malonate, and inyxothiazol were incubated for 24 hours in absence or presence
of SP-233 before the cell viability measures were carried out. The duration of
incubation for KCN and oligomycin was 6 hours. Results are expressed as
means ~: SD from three independent experiments performed in triplicate. *
p<0.05, ** p<0.01, *** p<0.001 compared to cultures not treated with SP-233.
Fig. 13 Neuroprotective effects of SP-233 on SK-N-AS neuronal cells
against PAO. PAO was added to cultures 1 hour after the addition of SP-233 or
its solvent. Cultures were incubated in PAO for 24 hours in the absence or
presence of SP-233, prior to cell viability measurements. Results are
expressed
as means SD from three independent experiments performed in triplicate. **
p<0.01 compared to cells not treated with SP-233.
Fig. 14 Effect of SP-233 on FCCP-induced uncoupling of the
mitochondrial respiratory chain in SK-N-AS cells. FCCP was added to SK-N-
AS cultures 1 hour after the addition of SP-233 or its solvent. Cultures were
incubated in FCCP for 24 hours in the absence or presence of SP-233, before
the
cell viability measurements were performed. Results are expressed as means ~
SD from three independent experiments performed in triplicate. ** p<0.01
compared to cells not treated with SP-233.
Fig. 15 Schematic representation of the mitochondrial sites targeted by
SP-233. The elements of the respiratory chain targeted by SP-233 are indicated
with a green arrow. Dotted arrows denote a poor neuroprotective effect, and
full
green arrows denote a strong protective effect. The most pronounced effect was
observed against KCN, an inhibitor of complex IV, oligomycin, an inhibitor of
the complex V, and PAO, a promoter of the MPT.
9
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Detailed Description of the Invention
As discussed hereinbelow, impairment of the mitochondrial respiratory
chain has been extensively described in relation to Alzheimer's disease (AD),
and it has been reported that Af3i-42 uncouples the mitochondrial respiratory
chain and promotes the opening of the membrane permeability transition (MPT)
pores of mitochondria, leading to cell death. The spirostenol (22S,25S)-(20S)-
spirost-5-en-30-yl hexanoate (SP-233) has been reported to protect neuronal
cells against A,61-42 toxicity by binding and inactivating the peptide. The
present
invention is based on the discovery that the protective effect of SP-233 also
results in protection of mitochondrial function. It was found that picomolar
concentrations of A(31-42 decreased the mitochondrial respiratory coefficient
in
mitochondria isolated from rat forebrain by a protective effect that was
partially
reversed by SP-233. This protective effect of SP-233 probably results from its
direct targeting of the respiratory chain, as well as its targeting of Afll-
42.
SP-233 also abolished the uncoupling of oxidative phosphorylation
induced by carbonyl cyanide 3-chlorophenylhydrazone (CCCP) and amplified
the effect of cyclosporin A (a potent blocker of mitochondrial MPT pores),
suggesting an effect on MPT. These properties of SP-233 might contribute to
its
neuroprotective effect, since the uncoupling of the respiratory chain and the
opening of mitochondrial MPT pores are two deleterious events that could be
triggered by A(31-42.
Using human SK-N-AS neuroblastoma cells, it was further observed,
using confocal microscopy, that Ao1-42 accumulated in the mitochondrial matrix
and that SP-233 completely scavenged A,61-42 from the matrix. These results
indicate that A,61-42 and SP-233 exert direct effects on the mitochondrial
function
of human neuronal cells, and that SP-233 (when present in the incubation
mixture together with the amyloid peptide) can protect these cells against A(3-
induced toxicity. Collectively, these findings indicate that A,f31-42 may
exert a
direct toxic effect on mitochondrial function, and that SP-233 exerts its
neuroprotective effects by targeting Ao directly, thereby protecting the
respiratory chain, and providing a family of compounds useful to treat
initochondrial diseases and disorders other than AD and other CNS
neuropathologies.
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
As used herein, the term mitochondrial disorder encompasses art
recognized mitochondrial diseases as well as conditions in which mitochondrial
disorders or dysfunction plays a role in the onset symptomology, progression
and/or outcome. The latter conditions include neurodegenerative diseases such
as Alzheimer's disease, Parkinson's disease, multiple sclerosis, or myotrophic
lateral sclerosis as well as neuronal death induced by stroke, cerebral
ischemia
and other brain and spinal cord injuries. The use of spirostenols including
(22S,25S)-(20S)-spirost-5-en-3-yl hexanoate to protect against A-induced
neurotoxicity and their potential use in AD therapy has been reported (L.
Lecanu
et al., Steroids, 69:1 (2004)) and is the subject of published PCT application
WO/03/077869. The use of (22S,25S)-(20S)-spirost-5-en-3-yl hexanoate to
protect mitochondria against a variety of stressors and its use in the therapy
of
various mitochondrial diseases has not been previously known.
Mitochondrial diseases or mitochondrial myopathies are a group of
diseases affecting the mitochondria, that also interfere with the function of
muscles. The group includes Kearns-Sayre syndrome, Leigh's syndrome,
mitochondrial DNA depletion syndrome (MDS), mitochondrial
encephaloinyopathy, lactic acidosis and strokelike episodes (MELAS),
inyoclonus epilepsy with ragged red fibers (MERFF), mitochondrial
neurogastrointestinal encephalomyopathy (MNGIE), neuropathy, ataxia and
retinitis piginentosa (NARP), and progressive external ophthalmoplegia (PEO).
The symptoms of mitochondrial myopathies include muscle weakness or
exercise intolerance, heart failure (cardiomyopathy) or rhythm disturbances,
dementia, movement disorders, stroke-like episodes, deafiiess, blindness,
droopy
eyelids, limited mobility of the eyes, vomiting, and seizures. The prognosis
for
these disorders ranges in severity from progressive weakness to death. Thus,
mitochondrial disorders can include disorders of the eye, such as optic nerve
atrophy, ptosis, acquired stabimus, and ophthalomplegia. Mitochondrial
dysfunction can be caused by crushed muscle syndrome (or compartment
syndrome), the side effects of drug therapies such as HAART and anti-cancer
therapies involving drugs that damage the heart (e.g., anthracycline),
hepatocellular dysfunction, endocrine tubulopathy and respiratory disorders.
Most mitochondrial myopathies occur before the age of 20, and often
begin with exercise intolerance or muscle weakness. During physical activity,
11
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
muscles may become easily fatigued or weak. Muscle cramping is rare, but may
occur. Nausea, headache, and breathlessness are also associated with these
disorders.
The term "treat" or "treatment" as used herein refers to any treatment of a
disorder or disease associated with a disease or disorder related to
mitochondria,
in a subject, and includes, but is not limited to, preventing the disorder or
disease
from occurring in a subject who may be predisposed to the disorder or disease,
but has not yet been diagnosed as having the disorder or disease; inhibiting
the
disorder or disease, for example, arresting the development of the disorder or
disease; relieving the disorder or disease, for exainple, causing regression
of the
disorder or disease; or relieving the condition caused by the disease or
disorder,
for example, stopping the symptoms of the disease or disorder. As used herein,
"mitochondrial disorder" or "mitochondrial disease" is intended to encompass
all
disorders disclosed herein.
The present metllod can also be used to prevent or reduce mitochondrial
disorders in vitro, as in organs or tissues awaiting or undergoing
transplantation.
The term "prevent" or "prevention," in relation to a disease or disorder
related to nlitochondria, in a subject, means no disease or disorder
development
if none had occurred, or no further disorder or disease development if there
had
already been development of the disorder or disease, or no symptoms to
logically
observable signs of the disease.
Preferred stereoisomers are 3S, as well as 10R and 13S, and are also 20S,
22S and 25S wherein the carbon skeleton is numbered in accord with spirosten-
3-ol numbering. Thus, a preferred compound of formula (I or II) is (22S,25S)-
(20S)-spirost-5-en-3p-yl hexanoate (SP233). Note that the carbon atoms shown
in formula (I) or (II) are saturated with hydrogen unless otherwise indicated.
Unless defined otherwise, each of the term "alkyl," the prefix "alk" (as in
alkoxy), and the suffix "-alkyl" (as in hydroxyalkyl) refers to a C1-8
hydrocarbon
chain, linear (e.g., butyl) or branched (e.g., isobutyl). Alkylene,
alkenylene, and
alkynylene refer to divalent C1_$ allcyl (e.g., ethylene), alkene, and alkyne
radicals, respectively. Preferably, alkyl is (C1-C6)alkyl, such as butyl,
hexyl,
methyl, ethyl, propyl or isopropyl. The term "alkyl" includes cycloakyl,
(cycloalkyl)alkyl and alkyl(cycloalkyl)alkyl. The term "alkenyl" likewise
12
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
includes alkyl comprising 1-2 CH=CH units, as well as the corresponding
cycloalkenyl moieties.
Specifically, (C1-C$)alkyl can be methyl, ethyl, propyl, isopropyl, butyl,
iso-butyl, sec-butyl, pentyl, 3-pentyl, heptyl or octyl; (C3-C8)cycloalkyl can
be
monocyclic, bicyclic or tricyclic and includes cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, bicyclo[2.2.2] octanyl or norbomyl, as well as
various
terpene and terpenoid structures. (C3-C6)cycloalkyl(C1-CZ)alkyl includes the
foregoing cycloalkyl and can be cyclopropylmethyl, cyclobutylmethyl,
cyclopentylmethyl, cyclohexylmethyl, 2-cyclopropylethyl, 2-cyclobutylethyl, 2-
cyclopentylethyl, or 2-cyclohexylethyl. Heterocycloalkyl includes cycloalkyl
wherein the cycloalkyl ring system is monocyclic, bicyclic or tricyclic and
optionally comprises 1-2 S, non-peroxide 0 or N(R') as well as 4-8 ring carbon
atoms; such as morpholinyl, piperidinyl, piperazinyl, indanyl, 1,3-dithian-2-
yl,
and the like. Any cycloalkyl or heterocycloalkyl ring system optionally
includes
1-3 double bonds or epoxy moieties and optionally is substituted with 1-3 OH,
(Ci-C6)alkanoyloxy, (CO), (C1-C6)alkyl or (C2-C6)alkynyl. (C1-C$)Alkoxy can
be methoxy, ethoxy, propoxy, isopropoxy, butoxy, iso-butoxy, sec-butoxy,
pentoxy, 3-pentoxy, or hexyloxy; (C2-C6)alkenyl can be vinyl, allyl, 1-
propenyl,
2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 1,-pentenyl, 2-pentenyl, 3-
pentenyl,
4-pentenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, or 5-hexenyl;
hydroxy(C1-C8)alkyl or hydroxy (C1-C8)alkoxyl can be alkyl substituted with 1
or 2 OH groups, such as alkyl substituted with 1 or 2 OH groups such as
hydroxymethyl, 1-hydroxyethyl, 2-liydroxyethyl, 1-hydroxypropyl, 2-
hydroxypropyl, 3-hydroxypropyl, 1-hydroxybutyl, 4-hydroxybutyl, 3,4-
dihydroxybutyl, 1-hydroxypentyl, 5-hydroxypentyl, 1-hydroxyhexyl, or 6-
hydroxyhexyl; (C1-C22)a1ky1CO2- can be acetoxy, propanoyloxy, butanoyloxy,
isobutanoyloxy, pentanoyloxy or hexanoyloxy, or (C8-C22)CO2- can represent
the residue of a naturally occurring fatty acid.
In one embodiment of the invention, Rlo and/or R12 are CH3.
In another embodiment of the invention, R16 and R17 are CH3.
In a further embodiment of the invention, Rl, R2 and/or R12 can be H or
OH.
In another embodiment, Rl, R2, R4, R6, R7, R8, R9, Rll, R12, R14 and R15
are H.
13
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
R2 can be (C1-C2a)alkylCOa-, including (C$-Ca2)alkylCO2- or (Cl-
C6)alkylCO2-, or can be OH or O(C1-C$)alkyl, or HO2C(CH2)2C02-.
X can be 0 or NR' including NH or NAc.
R3 may also be OR23 wherein R23 is a removable hydroxy-protecting
group such as tosyl, mesyl, tri(C1-C4)alkylsilyl, THP, Eto(Et), benzyl,
benzoyloxycarbony and the like. C3-(C8-C22) fatty acid esters of compounds in
which C3 is hydroxysubstituted are also within the invention, wherein the
fatty
acid is preferably naturally occurring.
Shown below in Table 1 are several compounds of formula (I) and (II)
described above that can be used to practice this invention:
Table 1.
Denomination Chemical Name Origin
SP224 (20(x)-25~-methyl-(22R,26)-azacyclofurost-5-en-3~- Solanuna asperum
ol (solanaceae)
SP226 (204)-254-methyl-N-acetyl-(22R,26)-azacyclofiuost- Solanum asperurn
5-en-34-o1 (solanaceae)
SP227 (22R,254)-(20a)-spirost-5-en-(2(x,3~)-diol Gynura japonica
(asteraceae)
SP229 (20a)-254-methyl-N-paratoluenesulfonyl-(22R,26)- Solanuna aviculare
azacyclofurost-5-en-3~-y1 paratoluenesulfonate (solanaceae)
SP230 (22R,254)-(20a)-(14a,20a)-spirost-5-en-(3 p,12(3)- Gynurajaponica
diol (asteraceae)
SP231 (22R,25S)-(204)-spirost-5-en-3~-o1 Gynurajaponica
(asteraceae)
SP232 (22R,254)-(20a)-spirost-5-en-3p-y1 benzoate Gynura sp. (asteraceae)
SP233 (22S,25S)-(20S)-spirost-5-en-3(3-yl hexanoate Gynura sp. (asteraceae)
SP234 (22R,254)-(20(x)-spirost-5-en-(1~,3~)-diol Gyizura japonica
(asteraceae)
SP235 (22R,25S)-(20a)-spirost-5-en-3R-ol Gynura japonica
(asteraceae)
SP236 (22R,25S)-(20a)-spirost-5-en-3(3-y1 succinate Gynura sp. (asteraceae)
SP238 (20a)-25S-methyl-N-acetyl-(22S,26)-azacyclofurost- Solanuin asperum
5-en-3(3-ylpropanoate (solanaceae)
An effective amount of an efficacious compound of the invention can be
formulated with a pharmaceutically acceptable carrier to form a pharmaceutical
composition before being administered for treatment of a disease related to
14
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
impaired mitochondrial function. "An effective amount" or "pharmacologically
effective amount" refers to the amount of the compound which is required to
confer therapeutic effect on the treated subject. The interrelationship of
dosages
for animals and humans (based on milligrams per square meter of body surface)
is described by Freireich et al., Cancer Chemother. Rep., 50, 219 (1966). Body
surface area may be approximately determined from height and weight of the
patient. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardley, New
York,
1970, 537. Effective doses can be based on in vitro concentrations of the
present
compounds found to be effective to inhibit the toxicity of A(3 or to otherwise
protect mitochondria. Doses of the present compounds useful to treat a model
neuronal cell line are disclosed below. Effective doses will also vary, as
recognized by those skilled in the art, depending on the route of
administration,
the excipient usage, and the optional co-administration with other therapeutic
agents.
Toxicity and therapeutic efficacy of the active ingredients can be
determined by standard pharmaceutical procedures, e.g., for deterinining LD50
(the dose lethal to 50% of the population) and the ED50 (the dose
therapeutically
effective in 50% of the population). The dose ratio between toxic and
therapeutic effects is the therapeutic index and it can be expressed as the
ratio
LD50/ED50. Compounds which exhibit large therapeutic indices are preferred.
While compounds that exhibit toxic side effects may be used, care should be
taken to design a delivery system that targets such compounds to the site of
affected tissue in order to minimize potential damage to uninfected cells and,
thereby, reduce side effects.
Included in the methods, kits, combinations and pharmaceutical
compositions of the present invention are the crystalline forms (e.g.,
polymorphs), enantiomeric forms, isomeric forms and tautomers of the described
compounds and the pharinaceutically-acceptable salts thereof. Illustrative
pharmaceutically acceptable salts are prepared from formic, acetic, propionic,
succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic,
glucuronic,
maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, mesylic,
stearic, salicylic, p-hydroxybenzoic, phenylacetic, mandelic, embonic
(pamoic),
methanesulfonic, ethanesulfonic, benzenesulfonic, pantothenic,
toluenesulfonic,
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
2-hydroxyethanesulfonic, sulfanilic, cyclohexylaminosulfonic, algenic, b-
hydroxybutyric, galactaric and galacturonic acids.
The term "prodrug" refers to a drug or compound (active moiety) that
elicits the pharmacological action results from conversion by metabolic
processes within the body. Prodrugs are generally considered drug precursors
that, following administration to a subject and subsequent absorption, are
converted to an active or a more active species via some process, such as a
metabolic process. Other products from the conversion process are easily
disposed of by the body. Prodrugs generally have a chemical group present on
the prodrug which renders it less active and/or confers solubility or some
other
property to the drug, such as an ester or acyl group. Once the chemical group
has been cleaved from the prodrug the more active drug is generated. Prodrugs
may be designed as reversible drug derivatives and utilized as modifiers to
enhance drug transport to site-specific tissues. The design of prodrugs to
date
has been to increase the effective water solubility of the therapeutic
compound
for targeting to regions where water is the principal solvent. For example,
Fedorak, et al., Am. J. Physiol., 269, G210-218 (1995), describe
dexainethasone-
beta-D-glucuronide. McLoed, et al., Gastroenterol., 106, 405-413 (1994),
describe dexamethasone-succinate-dextrans. Hochhaus, et al., Biomed. Chrom.,
6, 283-286 (1992), describe dexamethasone-21-sulphobenzoate sodium and
dexamethasone-21-isonicotinate. Additionally, J. Larsen and H. Bundgaard, Int.
J. Pharmaceutics, 37, 87 (1987) describe the evaluation of N-acylsulfonamides
as potential prodrug derivatives. J. Larsen et al., Int. J. Pharmaceutics, 47,
103
(1988) describe the evaluation of N-methylsulfonamides as potential prodrug
derivatives. Prodrugs are also described in, for example, Sinkula et al., J.
Pharm.
Sci., 64, 181-210 (1975). Prodrugs are also useful as synthetic intermediates
in
the preparation of other compounds of formulas (I) or (II), by synthetic
interconversions known to the art. For example, see, I.T. Harrison,
Compendium of Organic Synthetic Metllods, Wiley-Interscience (1971), for
methods useful to interconvert spirostenol substituents.
The term "derivative" refers to a compound that is produced from another
compound of similar structure by the replacement or substitution of one atom,
molecule or group by anotlier. For example, a hydrogen atom of a compound
16
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
may be substituted by alkyl, acyl, amino, etc., to produce a derivative of
that
compound.
"Plasma concentration" refers to the concentration of a substance in
blood plasma or blood serum.
"Drug absorption" or "absorption" refers to the process of moveinent
from the site of administration of a drug toward the systemic circulation, for
example, into the bloodstream of a subject.
"Bioavailability" refers to the extent to which an active moiety (drug or
metabolite) is absorbed into the general circulation and becomes available at
the
site of dr-ug action in the body. "Metabolism" refers to the process of
chemical
transforinations of drugs in the body.
"Pharmacodynamics" refers to the factors which determine the biologic
response observed relative to the concentration of drug at a site of action.
"Pharmacokinetics" refers to the factors which determine the attainment
and maintenance of the appropriate concentration of drug at a site of action.
"Plasma half-life" refers to the time required for the plasma drug
concentration to decrease by 50% from its maximum concentration.
The use of the term "about" in the present disclosure means
"approximately," and encoinpasses variations in parameters that would arise
during practice of the relevant art. Illustratively, the use of the term
"about"
indicates that dosages outside the cited ranges may also be effective and
safe,
and such dosages are also encompassed by the scope of the present claims.
The term "measurable serum concentration" means the serum
concentration (typically measured in mg, g, or ng of therapeutic agent per
ml,
dl, or 1 of blood serum) of a therapeutic agent absorbed into the bloodstream
after administration.
The term "pharmaceutically acceptable" is used adjectivally herein to
mean that the modified noun is appropriate for use in a pharmaceutical
product.
Pharmaceutically acceptable salts include metallic ions and organic ions. More
preferred metallic ions include, but are not limited to appropriate alkali
metal
(Group Ia) salts, allcaline earth metal (Group IIa) salts and other
physiological
acceptable metal ions. Exemplary ions include aluminum, calcium, lithium,
magnesiuin, potassiuin, sodium and zinc in their usual valences. Preferred
organic ions include protonated tertiary amines and quatemary ammonium
17
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
cations, including in part, trimethylamine, diethylamine, N,N'-
dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamine, meglumine (N-methylglucamine) and procaine. Exemplary
pharmaceutically acceptable acids include without limitation hydrochloric
acid,
hydrobromic acid, phosphoric acid, sulfuric acid, methanesulfonic acid, acetic
acid, formic acid, tartaric acid, maleic acid, malic acid, citric acid,
isocitric acid,
succinic acid, lactic acid, gluconic acid, glucuronic acid, pyruvic acid
oxalacetic
acid, fumaric acid, propionic acid, aspartic acid, glutamic acid, benzoic
acid, and
the like.
The compositions of the present invention are usually adininistered in the
form of pharmaceutical compositions. These compositions can be adininistered
by any appropriate route including, but not limited to, oral, nasogastric,
rectal,
transdermal, parenteral (for example, subcutaneous, intramuscular,
intravenous,
intramedullary and intradermal injections, or infusion techniques
administration), intranasal, transmucosal, implantation, vaginal, topical,
buccal,
and sublingual. Such preparations may routinely contain buffering agents,
preservatives, penetration enhancers, compatible carriers and other
therapeutic or
non-therapeutic ingredients.
The present invention also includes methods employing a phannaceutical
composition that contains one or more compound of formula I or II associated
with phannaceutically acceptable carriers or excipients. As used herein, the
terms "pharmaceutically acceptable carrier" or "pharmaceutically acceptable
excipients" includes any and all solvents, dispersion media, coatings,
antibacterial and antifungal agents, isotonic and absorption delaying agents,
and
the like. The use of such media and agents for ingestible substances is well
known in the art. Except insofar as any conventional media or agent is
incompatible witli the compositions, its use is conteinplated. Supplementary
active ingredients can also be incorporated into the compositions. In making
the
compositions of the present invention, the compositions(s) can be mixed with a
pharmaceutically acceptable excipient, diluted by the excipient or enclosed
within such a carrier, which can be in the form of a capsule, sachet, or other
container. The carrier materials that can be employed in making the
composition
of the present invention are any of those commonly used excipients in
18
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
phaimaceutics and should be selected on the basis of compatibility with the
active drug and the release profile properties of the desired dosage form.
Illustratively, pharmaceutical excipients are chosen below as exainples:
(a) Binders such as acacia, alginic acid and salts thereof, cellulose
derivatives, methylcellulose, hydroxyethyl cellulose, hydroxypropyl cellulose,
magnesium aluminum silicate, polyethylene glycol, gums, polysaccharide acids,
bentonites, llydroxypropyl methylcellulose, gelatin, polyvinylpyrrolidone,
polyvinylpyrrolidone/vinyl acetate copolymer, crospovidone, povidone,
polymethacrylates, hydroxypropylmethylcellulose, hydroxypropylcellulose,
starch, pregelatinized starch, ethylcellulose, tragacanth, dextrin,
cyclodextrins,
microcrystalline cellulose, sucrose, or glucose, and the like.
(b) Disintegration agents such as starches, pregelatinized corn starch,
pregelatinized starch, celluloses, cross-linked carboxymethylcellulose, sodium
starch glycolate, crospovidone, cross-linked polyvinylpyrrolidone,
croscarmellose sodium, microcrystalline cellulose, a calcium, a sodium
alginate
complex, clays, alginates, gums, or sodium starch glycolate, and any
disintegration agents used in tablet preparations.
(c) Filling agents such as lactose, calcium carbonate, calcium phosphate,
dibasic calcium phosphate, calcium sulfate, microcrystalline cellulose,
cellulose
powder, dextrose, dextrates, dextran, starches, pregelatinized starch,
sucrose,
xylitol, lactitol, mannitol, sorbitol, sodium chloride, polyethylene glycol,
and the
like.
(d) Surfactants such as sodiuin lauryl sulfate, sorbitan monooleate,
polyoxyetllylene sorbitan monooleate, polysorbates, polaxomers, bile salts,
glyceryl monostearate, PluronicTM line (BASF), and the like.
(e) Solubilizer such as citric acid, succinic acid, fumaric acid, malic acid,
tartaric acid, maleic acid, glutaric acid sodium bicarbonate and sodium
carbonate
and the like.
(f) Stabilizers such as any antioxidation agents, buffers, or acids, and the
like, can also be utilized.
(g) Lubricants such as magnesium stearate, calcium hydroxide, talc,
sodium stearyl fumarate, hydrogenated vegetable oil, stearic acid, glyceryl
behapate, magnesium, calcium and sodium stearates, stearic acid, talc, waxes,
19
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
boric acid, sodium benzoate, sodium acetate, sodium chloride, DL-leucine,
polyethylene glycols, sodiuin oleate, or sodium lauryl sulfate, and the like.
(h) Wetting agents such as oleic acid, glyceryl monostearate, sorbitan
monooleate, sorbitan monolaurate, triethanolamine oleate, polyoxyethylene
sorbitan monooleate, polyoxyethylene sorbitan monolaurate, sodium oleate, or
sodium lauryl sulfate, and the like.
(i) Diluents such lactose, starch, mannitol, sorbitol, dextrose,
microcrystalline cellulose, dibasic calcium phosphate, sucrose-based diluents,
confectioner's sugar, monobasic calcium sulfate monohydrate, calcium sulfate
dihydrate, calcium lactate trihydrate, dextrates, inositol, hydrolyzed cereal
solids,
amylose, powdered cellulose, calcium carbonate, glycine, or bentonite, and the
like.
(j) Anti-adherents or glidants such as talc, corn starch, DL-leucine,
sodium lauryl sulfate, and magnesium, calcium, or sodium stearates, and the
like.
(k) Pharmaceutically compatible carrier comprises acacia, gelatin,
colloidal silicon dioxide, calcium glycerophosphate, calcium lactate,
maltodextrin, glycerine, magnesiuin silicate, sodium caseinate, soy lecithin,
sodiuin chloride, tricalcium phosphate, dipotassium phosphate, sodium stearoyl
lactylate, carrageenan, monoglyceride, diglyceride, or pregelatinized starch,
and
the like.
Additionally, drug formulations are discussed in, for example,
Remington's The Science and Practice of Pharmacy (2000). Anotller discussion
of drug forinulations can be found in Liberman, H.A. and Lachinan, L., Eds.,
Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980. The
tablets or granules comprising the inventive compositions may be film coated
or
enteric-coated.
Besides being useful for human treatment, the present invention is also
useful for otlier subjects including veterinary animals, reptiles, birds,
exotic
animals and farm animals, including mammals, rodents, and the like. Mammal
includes a primate, for example, a monkey, or a lemur, a horse, a dog, a pig,
or a
cat. A rodent includes a rat, a mouse, a squirrel, or a guinea pig.
The pharmaceutical compositions of the present invention are useful
where administration of an inhibitor of mitochondrial toxicity is indicated.
It is
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
believed that these compositions will be particularly effective in the
treatment of
mitochondrial diseases.
For treatment of a neurodegenerative disorder/mitochondrial disorder,
compositions of the invention can be used to provide a dose of a coinpound of
the present invention in an amount sufficient to elicit a tllerapeutic
response, e.g.,
reduction of drug-induced mitochondrial toxicity, for example a dose of about
5
ng to about 1000 mg, or about 100 ng to about 600 mg, or about 1 mg to about
500 mg, or about 20 mg to about 400 mg. Typically a dosage effective amount
will range from about 0.000 1 mg/kg to 1500 mg/kg, more preferably 1 to 1000
mg/kg, more preferably from about 1 to 150 mg/kg of body weight, and most
preferably about 50 to 100 mg/kg of body weight. A dose can be administered
in one to about four doses per day, or in as many doses per day to elicit a
therapeutic effect. Illustratively, a dosage unit of a composition of the
present
invention can typically contain, for example, about 5 ng, 50 ng 100 ng, 500
ng, 1
mg, 10 mg, 20 mg, 40 mg, 80 mg, 100 mg, 125 mg, 150 mg, 200 mg, 250 mg,
300 mg, 350 mg, 400 mg, 450 mg, 500 mg, 550 mg, 600 mg, 700 mg, 800 mg,
900 mg, or 1000 mg of a compound of the present invention. The dosage form
can be selected to accominodate the desired frequency of administration used
to
achieve the specified dosage. The amount of the unit dosage form of the
composition that is administered and the dosage regimen for treating the
condition or disorder depends on a variety of factors, including, the age,
weight,
sex and medical condition, of the subject, the severity of the condition or
disorder, the route and frequency of administration, and this can vary widely,
as
is well known.
In one embodiment of the present invention, the composition is
administered to a subject in an effective ainount, that is, the composition is
administered in an amount that achieves a therapeutically effective dose of a
compound of the present invention in the blood serum of a subject for a period
of
time to elicit a desired therapeutic effect. Illustratively, in a fasting
adult human
(fasting for generally at least 10 hours) the composition is administered to
achieve a therapeutically effective dose of a compound of the present
invention
in the blood serum of a subject from about 5 minutes after administration of
the
composition. In another embodiment of the present invention, a therapeutically
effective dose of the compound of the present invention is achieved in the
blood
21
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
serum of a subject at about 10 minutes from the time of administration of the
composition to the subject. In another embodiment of the present invention, a
therapeutically effective dose of the compound of the present invention is
achieved in the blood serum of a subject at about 20 minutes from the time of
administration of the composition to the subject.
In yet another embodiment of the present invention, a tllerapeutically
effective dose of the compound of the present invention is achieved in the
blood
serum of a subject at about 30 minutes from the time of administration of the
composition to the subject. In still anotller embodiment of the present
invention,
a therapeutically effective dose of the compound of the present invention is
achieved in the blood serum of a subject at about 40 minutes from the time of
adininistration of the composition to the subject. In one embodiment of the
present invention, a therapeutically effective dose of the compound of the
present invention is achieved in the blood serum of a subject at about 20
minutes
to about 12 hours from the time of administration of the composition to the
subject. In anotller embodiment of the present invention, a therapeutically
effective dose of the compound of the present invention is achieved in the
blood
serum of a subject at about 20 minutes to about 6 hours from the time of
administration of the composition to the subject. In yet another embodiment of
the present invention, a therapeutically effective dose of the compound of the
present invention is achieved in the blood serum of a subject at about 20
minutes
to about 2 hours from the time of administration of the composition to the
subj ect.
In still another embodiment of the present invention, a therapeutically
effective dose of the compound of the present invention is achieved in the
blood
serum of a subject at about 40 minutes to about 2 hours from the time of
administration of the composition to the subject. And in yet another
embodiment of the present invention, a therapeutically effective dose of the
compound of the present invention is achieved in the blood seruin of a subject
at
about 40 minutes to about 1 hour from the time of administration of the
composition to the subject.
In one embodiment of the present invention, a composition of the present
invention is administered at a dose suitable to provide a blood serum
concentration with a half maxiinum dose of a compound of the present
22
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
invention. Illustratively, a blood serum concentration of about 0.01 to about
1000 nM, or about 0.1 to about 750 nM, or about 1 to about 500 nM, or about 20
to about 1000 nM, or about 100 to about 500 nM, or about 200 to about 400 nM
is achieved in a subject after administration of a composition of the present
invention.
Contemplated compositions of the present invention provide a
therapeutic effect over an interval of about 5 minutes to about 24 hours after
administration, enabling once-a-day or twice-a-day administration if desired.
In
one embodiment of the present invention, the composition is administered at a
dose suitable to provide an average blood serum concentration with a half
maximuin dose of a compound of the present invention of at least about 1
g/ml,
or at least about 5 g/ml, or at least about 10 g/ml, or at least about 50
g/ml, or
at least about 100 g/ml, or at least about 500 ,ug/ml, or at least about 1000
g/ml in a subject about 10, 20, 30, or 40 minutes after administration of the
composition to the subject.
The amount of therapeutic agent necessary to elicit a therapeutic effect
can be experimentally detennined based on, for example, the absorption rate of
the agent into the blood serum, the bioavailability of the agent, and the
potency
for treating the disorder. It is understood, however, that specific dose
levels of
the therapeutic agents of the present invention for any particular subject
depends
upon a variety of factors including the activity of the specific compound
employed, the age, body weight, general health, sex, and diet of the subject
(including, for example, whether the subject is in a fasting or fed state),
the time
of administration, the rate of excretion, the drug combination, and the
severity of
the particular disorder being treated and form of administration. Treatment
dosages generally may be titrated to optimize safety and efficacy. Typically,
dosage-effect relationships from in vitro and/or in vivo tests initially can
provide
useful guidance on the proper doses for subject administration. Studies in
animal models generally may be used for guidance regarding effective dosages
for treatment of gastrointestinal disorders or diseases in accordance witli
the
present invention. In terms of treatment protocols, it should be appreciated
that
the dosage to be administered will depend on several factors, including the
particular agent that is administered, the route administered, the condition
of the
particular subject, etc. Generally speaking, one will desire to administer an
23
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
amount of the compound that is effective to achieve a serum level commensurate
witli the concentrations found to be effective ira vitro for a period of time
effective to elicit a therapeutic effect. Thus, where a compound is found to
demonstrate irt vitro activity at, for example, a half-maximum effective dose
of
200 nM, one will desire to administer an amount of the drug that is effective
to
provide about a half-maximum effective dose of 200 nM concentration iyz vivo
for a period of time that elicits a desired therapeutic effect, for example,
treating
a disorder related to high beta-amyloid-induced neurotoxicity and other
indicators as are selected as appropriate measures by those skilled in the
art.
Determination of these parameters is well within the skill of the art. These
considerations are well known in the art and are described in standard
textbooks.
In order to measure and determine the effective ainount of a compound
of the present invention to be delivered to a subject, serum compound of the
present invention concentrations can be measured using standard assay
techniques.
Conteinplated compositions of the present invention provide a
tllerapeutic effect over an interval of about 30 minutes to about 24 hours
after
administration to a subject. In one embodiment compositions provide such
therapeutic effect in about 30 minutes. In another einbodiment compositions
provide therapeutic effect over about 24 hours, enabling once-a-day
adininistration to improve patient compliance.
The present methods, kits, and compositions can also be used in
coinbination ("combination therapy") with another pharmaceutical agent that is
indicated for treating or preventing a mitochondrial disease, such as, for
example, creatinine. When used in conjunction with the present invention, that
is, in combination therapy, an additive or synergistic effect may be achieved
such that many if not all of unwanted side effects can be reduced or
eliminated.
The reduced side effect profile of these drugs is generally attributed to, for
example, the reduced dosage necessary to achieve a therapeutic effect with the
administered combination.
The phrase "combination therapy" embraces the administration of a
composition of the present invention in conjunction with another
pharmaceutical
agent that is indicated for treating or preventing a mitochondrial disorder in
a
subject, as part of a specific treatment regimen intended to provide a
beneficial
24
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
effect from the co-action of these therapeutic agents for the treatment of a
neurodegenerative and/or a mitochondrial disorder. The beneficial effect of
the
combination includes, but is not limited to, pharmacokinetic or
pharmacodynamic co-action resulting from the combination of therapeutic
agents. Administration of these therapeutic agents in combination typically is
carried out over a defined time period (usually substantially simultaneously,
minutes, hours, days, weeks, months or years depending upon the combination
selected). "Combination therapy" generally is not intended to encompass the
administration of two or more of these therapeutic agents as part of separate
monotherapy regimens that incidentally and arbitrarily result in the
combinations
of the present invention. "Combination therapy" is intended to embrace
administration of these therapeutic agents in a sequential manner, that is,
where
each therapeutic agent is administered at a different time, as well as
administration of these therapeutic agents, or at least two of the therapeutic
agents, in a substantially simultaneous manner. Substantially simultaneous
administration can be accomplished, for example, by administering to the
subject
a single tablet or capsule having a fixed ratio of each therapeutic agent or
in
multiple, single capsules, or tablets for each of the therapeutic agents.
Sequential or substantially simultaneous administration of each therapeutic
agent
can be effected by any appropriate route. The composition of the present
invention can be administered orally or nasogastric, while the other
therapeutic
agent of the combination can be administered by any appropriate route for that
particular agent, including, but not limited to, an oral route, a percutaneous
route,
an intravenous route, an intramuscular route, or by direct absorption through
mucous membrane tissues. For example, the coinposition of the present
invention is administered orally or nasogastric and the therapeutic agent of
the
combination may be administered orally, or percutaneously. The sequence in
which the therapeutic agents are administered is not narrowly critical.
"Combination therapy" also can embrace the administration of the therapeutic
agents as described above in further combination with other biologically
active
ingredients, such as, but not limited to, an analgesic, for example, and with
non-
drug therapies, such as, but not limited to, surgery.
The therapeutic compounds which make up the combination therapy may
be a combined dosage form or in separate dosage forms intended for
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
substantially simultaneous administration. The therapeutic compounds that
make up the combination therapy may also be administered sequentially, with
either therapeutic compound being administered by a regimen calling for two
step administration. Thus, a regimen may call for sequential administration of
the therapeutic compounds with spaced-apart administration of the separate,
active agents. The time period between the multiple adininistration steps may
range from, for example, a few minutes to several hours to days, depending
upon
the properties of each therapeutic compound such as potency, solubility,
bioavailability, plasma half-life and kinetic profile of the therapeutic
compound,
as well as depending upon the effect of food ingestion and the age and
condition
of the subject. Circadian variation of the target molecule concentration may
also
determine the optimal dose interval.
The tlierapeutic compounds of the combined therapy whether
administered simultaneously, substantially simultaneously, or sequentially,
may
involve a regimen calling for administration of one therapeutic compound by
oral route and another therapeutic compound by an oral route, a percutaneous
route, an intravenous route, an intramuscular route, or by direct absorption
through mucous membrane tissues, for exainple. Whether the therapeutic
compounds of the combined therapy are administered orally, by inhalation
spray,
rectally, topically, buccally, sublingually, or parenterally (for example,
subcutaneous, intramuscular, intravenous and intradermal injections),
separately
or together, each such therapeutic compound will be contained in a suitable
pharmaceutical formulation of pharmaceutically-acceptable excipients, diluents
or other formulations components.
For oral administration, the pharmaceutical composition can contain a
desired amount of a compound of fonnula (I) or (II) and be in the form of, for
example, a tablet, a hard or soft capsule, a lozenge, a cachet, a troche, a
dispensable powder, granules, a suspension, an elixir, a liquid, or any other
form
reasonably adapted for oral administration. Illustratively, such a
pharmaceutical
coinposition can be made in the form of a discrete dosage unit containing a
predetermined amount of the active compound such as a tablet or a capsule.
Such oral dosage forms can further coinprise, for example, buffering agents.
Tablets, pills and the like additionally can be prepared with enteric
coatings.
26
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Pharmaceutical compositions suitable for buccal or sublingual
administration include, for example, lozenges comprising the active compound
in a flavored base, such as sucrose, and acacia or tragacanth, and pastilles
comprising the active compound in an inert base such as gelatin and glycerin
or
sucrose and acacia.
Liquid dosage forms for oral administration can include pharmaceutically
acceptable emulsions, solutions, suspensions, syrups, and elixirs containing
inert
diluents commonly used in the art, such as water. Such compositions can also
comprise, for example, wetting agents, emulsifying and suspending agents, and
sweetening, flavoring, and perfuming agents.
Examples of suitable liquid dosage forms include, but are not limited to,
aqueous solutions comprising the active compound and beta-cyclodextrin or a
water soluble derivative of beta-cyclodextrin such as sulfobutyl ether beta-
cyclodextrin; heptakis-2,6-di-O-methyl-beta-cyclodextrin; hydroxypropyl-beta-
cyclodextrin; and dimethyl-beta-cyclodextrin.
The pharmaceutical compositions of the present invention can also be
administered by injection (intravenous, intramuscular, subcutaneous). Such
injectable compositions can employ, for example, saline, dextrose, or water as
a
suitable carrier material. The pH value of the composition can be adjusted, if
necessary, with suitable acid, base, or buffer. Suitable bulking, dispersing,
wetting or suspending agents, including mannitol and polyethylene glycol (such
as PEG 400), can also be included in the composition. A suitable parenteral
composition can also include an active compound lyophilized in injection
vials.
Aqueous solutions can be added to dissolve the composition prior to injection.
The phannaceutical compositions can be administered in the form of a
suppository or the like. Such rectal formulations preferably contain the
active
compound in a total amount of, for example, about 0.075 to about 75% w/w, or
about 0.2 to about 40% w/w, or about 0.4 to about 15% w/w. Carrier materials
such as cocoa butter, theobroma oil, and other oil and polyethylene glycol
suppository bases can be used in such compositions. Other carrier materials
such as coatings (for example, hydroxypropyl methylcellulose film coating) and
disintegrants (for example, croscarmellose sodiuin and cross-linked povidone)
can also be employed if desired.
27
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
The subject compounds may be free or entrapped in microcapsules, in
colloidal drug delivery systems such as liposomes, microemulsions, and
macroemulsions.
These pharmaceutical compositions can be prepared by any suitable
method of pharmaceutics, which includes the step of bringing into association
active compound of the present invention and a carrier material or carriers
materials. In general, the compositions are uniformly and intimately adinixing
the active compound with a liquid or finely divided solid carrier, or both,
and
then, if necessary, shaping the product. For example, a tablet can be prepared
by
compressing or molding a powder or granules of the compound, optionally with
one or more accessory ingredients. Compressed tablets can be prepared by
compressing, in a suitable machine, the compound in a free-flowing form, such
as a powder or granules optionally mixed with a binding agent, lubricant,
inert
diluent and/or surface active/dispersing agent(s). Molded tablets can be made
by
molding, in a suitable machine, the powdered compound moistened with an inert
liquid diluent.
Tablets of the present invention can also be coated with a conventional
coating material such as OpadryTM White YS-1-18027A (or another color) and
the weight fraction of the coating can be about 3% of the total weight of the
coated tablet. The compositions of the present invention can be formulated so
as
to provide quick, sustained or delayed release of the compositions after
administration to the patient by employing procedures known in the art.
When the excipient serves as a diluent, it can be a solid, semi-solid or
liquid material, which acts as a vehicle, carrier or medium for the active
ingredient. Thus, the compositions can be in the forin of tablets, chewable
tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions,
emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium),
soft and
hard gelatin capsules and sterile packaged powders.
In one einbodiment of the present invention, the manufacturing processes
may employ one or a combination of methods including: (1) dry mixing, (2)
direct compression, (3) milling, (4) dry or non-aqueous granulation, (5) wet
granulation, or (6) fusion. Lachman et al., The Theory and Practice of
Industrial
Pharmacy (1986).
28
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
In another embodiment of the present invention, solid compositions, such
as tablets, are prepared by mixing a therapeutic agent of the present
invention
with a pharmaceutical excipient to form a solid preformulation composition
containing a homogeneous mixture of the therapeutic agent and the excipient.
When referring to these preformulation compositions(s) as homogeneous, it is
meant that the therapeutic agent is dispersed evenly throughout the
composition
so that the composition may be readily subdivided into equally effective unit
dosage forms, such as tablets, pills and capsules. This solid preformulation
is
then subdivided into unit dosage forms of the type described herein.
Compressed tablets are solid dosage forms prepared by compacting a
formulation containing an active ingredient and excipients selected to aid the
processing and improve the properties of the product. The term "compressed
tablet" generally refers to a plain, uncoated tablet for oral ingestion,
prepared by
a single compression or by pre-compaction tapping followed by a final
compression.
The tablets or pills of the present invention may be coated or otherwise
coinpounded to provide a dosage form affording the advantage of prolonged
action. For example, the tablet or pill can comprise an inner dosage and an
outer
dosage component, the latter being in the form of an envelope over the former.
A variety of materials can be used for such enteric layers or coatings,
including a
nuinber of polymeric acids and mixtures of polymeric acids with such materials
as shellac, cetyl alcohol and cellulose acetate.
Use of a long-term sustained release implant may be suitable for
treatment of mitochondrial disorders in patients who need continuous
administration of the compositions of the present invention. "Long-term"
release, as used herein, means that the implant is constructed and arranged to
deliver therapeutic levels of the active ingredients for at least 30 days, and
preferably 60 days. Long-term sustained release implants are well known to
those of ordinary skill in the art and include some of the release systems
described above.
In another embodiment of the present invention, the compound for
treating a mitochondrial disorder comes in the form of a kit or package
containing one or more of the therapeutic compounds of the present invention.
These therapeutic compounds of the present invention can be packaged in the
29
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
form of a kit or package in which hourly, daily, weekly, or monthly (or other
periodic) dosages are arranged for proper sequential or simultaneous
administration. The present invention further provides a kit or package
containing a plurality of separately-packaged dosage units, adapted for
successive daily administration, each dosage unit comprising at least one of
the
therapeutic compounds of the present invention. This drug delivery system can
be used to facilitate administering any of the various embodiments of the
therapeutic compounds of the present invention. In one embodiment, the system
contains a plurality of dosages to be administered daily or weekly. The kit or
package can also contain the agents utilized in combination therapy to
facilitate
proper administration of the dosage forms. The kits or packages also contain a
set of instructions for the subject.
The invention will be further described by reference to the following
detailed examples wherein the following abbreviations are used:
AChEI, acetylcholinesterase inhibitor; A,6, 0-amyloid peptide; ADDLs,
amyloid-derived diffusible ligands; ANT, adenine nucleotide translocase; APP,
amyloid precursor protein; CCCP, carbonyl cyanide 3-chlorophenylhydrazone;
FCCP, carbonyl cyanide p-trifluoromethoxy-phenylhydrazone; CsA, cyclosporin
A; MPT, membrane permeability transition; MRC, mitochondrial respiratory
coefficient; PAO, phenylarsine oxide; SP-233, (22S,25S)-(20S)-spirost-5-en-3(3-
yl hexanoate; SP-233, (22R,25R)-(20a)-spirost-5-en-30-yl hexanoate.
Mitochondrial permeability transition (MPT) may be further defined as a
nonspecific increase in the permeability of the inner mitochondrial membrane
that occurs under adverse conditions, such as an increase in mitochondrial Ca
a
content of the mitochondrial matrix or oxidative stress, and which causes the
assembly (opening) of non-specific pores ("megachannels") in the mitochondrial
inner membrane, leading to a loss of mitochondrial membrane potential, an
uncoupling of mitochondrial respiration, and cellular energy failure, all of
which
can contribute to cell death.
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Example 1. Use of SP-233 to Protect Mitochondrial Function
A. Materials and methods
1. Materials
A(31-42 peptide was purchased from American Peptide Company
(Sunnyvale, CA, USA) and stored at -80 C. The antibody anti-coinplex II was
purchased from Molecular Probes (Eugene, OR, USA) and the antibody anti-
Ab1_42 was from Signet Laboratories (Dedham, MA, USA). The spirostenol,
(22S,25S)-(20S)-spirost-5-en-30-y1 hexanoate (SP-233) was purchased from
Interbioscreen (Moscow, Russia). Cyclosporin A (CsA) and carbonyl cyanide 3-
chlorophenylhydrazone (CCCP) were obtained from Sigma (St. Louis, MO,
USA). Dulbecco's Minimum Essential Medium (DMEM), fetal bovine serum
(FBS) and SK-N-AS human neuroblastoma cells were purchased form ATCC
(Manassas, VA, USA).
2. Mitochondrial isolation procedure
Male Long-Evans rats weighing 230-280 g were used. After decapitation,
the brain was rapidly removed and homogenized on ice in a Potter-Elvejhem
homogenizer (Eurostar, IKA-Verke, Staufen, Germany) containing 6 ml of
isolation buffer (TRIS 20 mM, sucrose 250 mM, KC140 mM, EGTA 2 mM,
bovine serum albumin 1 mg/ml; pH 7.2 at 4 C). The homogenate was
centrifuged at 2000xg for 8 min to remove cell debris and nuclei. The
mitochondria-containing supernatant was centrifuged at 12000xg for 10 min.
The pellet was resuspended in 300 l of respiratory buffer (D-mannito1300 mM,
KC1 10 mM, KH2PO4 10 mM, MgC12 5 mM; pH 7.2 at 37 C). This pellet is
highly enriched in mitochondria (Zini et al. 1996). The protein concentration
of
the mitochondrial suspension was determined by the method of Lowry et al.
1951.
3. Preparation of the different solutions
A(31-42 solution was reconstituted with fresh distilled water to provide a
concentration of 500 M. A(31.42 was used as freshly reconstituted or in an
aggregated form after having been "aged" by incubating the solution for 48 h
at
4 C. Both solutions were diluted with the respiratory buffer (see above)
before
use and then added directly to the mitochondria at the desired concentration.
In
31
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
experiments conducted with neuroblastoma cells (see below), only the freshly
reconstituted A(31.42 solution was used. SP-233, CCCP and CsA were first
dissolved in N,N-dimethylformamide (DMF) and then diluted in fresh distilled
water.
4. Monitoring of mitochondrial respiration
Mitochondrial funetions were studied using an Oxytherm (Hansatech,
Norfolk, UK) linked to a PC (Coinpaq, PIII 400 MHz). The 37 C thermostated
incubation chamber of the Oxytherm was connected to a platinum-silver Clarck
electrode for 02 detection. 02 consumption was monitored as a marker of
mitochondrial function and viability in the presence of ADP (precursor for
ATP)
in the presence or absence of CCCP (an uncoupling agent for oxidative
phosphorylation), or CsA (an inhibitor of the MPT pore). Measureinent of 02
consumption permitted calculation of the mitochondrial respiratory coefficient
(MRC) which is V3/V4. V4 is the basal 02 consumption, V3 is the 02
consumption after adding ADP (ATP production). The effect of increasing
concentrations of SP-233 (1 pM to 1 nM) on the mitochondrial respiratory chain
was assessed by monitoring the evolution of MRC. The concentration of
mitochondria was adjusted to 0.4 mg protein/ml using respiratory buffer.
Mitochondria were activated by addition of malate/glutamate as substrates of
complex I of the respiratory chain. State 2 is the baseline rate of
mitochondrial
oxygen consumption. When ATP synthesis was initiated by addition of ADP, the
rate of oxygen consumption increased dramatically to reach state 3. When the
supply of ADP had been depleted, the rate of 02 consumption retunled to
baseline (state 2) wliich was called state 4 (Chance and Williams 1956).
Controls
for each experiment corresponded to basal respiration in the presence of ADP.
5. Effect of SP-233 on CCCP-induced uncoupling of oxidative
phosphorylation
Addition of CCCP (1 M) uncouples the respiratory chain reaction and
increases 02 consumption. To assess the effect of SP-233 on CCCP-induced
uncoupling, the mitochondrial fraction was incubated for 3 min at 37 C in
presence of SP-233 before adding malate/glutamate, followed 1 min later by the
addition of CCCP.
32
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
6. Assessment of the effect of SP-233 on CsA-induced hypoxia
Hypoxia was induced by adding CsA (1 M) to the incubation medium
bathing the mitochondria (Zini et al. 1996). This test was controlled by
adding
ADP in the presence of CsA and absence of SP-233. As described above, SP-
233 or its vehicle was added first and incubation was carried out for 1.5 min
before addition of CsA. Then, the substrate malate/glutamate was added 1.5 min
later, and ADP was added 2.5 min later.
7. Assessment of the protective effect of SP-233 against A(31_42-induced
mitochondrial dysfunction
SP-233 was present in the incubation mixture for 3 min and A(31-42 for
1.5 min at 37 C before addition of the substrates malate/glutamate. ADP was
added 1 min after addition of malate/glutamate. Control for the effect of SP-
233
versus A(31-42 was the same as for the experiment conducted in the absence of
ADP.
8. Assessment of the protective effect of SP-233 against A,61_42-induced
toxicity in human neuroblastoma cells
Human neuroblastoma SK-N-AS cells were seeded in 96-well plates
(7x104 cells/well) in DMEM containing 10% FBS and cultured in 5% COZ and
95% humidity. A(31-4Z (0.1, 1 and 10 gM) or its vehicle was then added and
incubation was continued for 72 h in the presence or absence of SP-233 (1 M).
The cellular toxicity of AO was assessed using the 3-(4,5-dimethylthiazol-2-
yl)-
2,5-diphenyl tetrazolium bromide (MTT) assay (Trevigen, Gaithersburg, MD).
9. Immunocytochemical analysis of A(31-42 uptake by mitochondria of
human
neuroblastoma cells
Human neuroblastoma SK-N-AS cells were seeded on 13-mm diameter
coverslips (20,000 cells/coverslip) and incubated overnight at 37 C, 5% C02,
in
DMEM containing 10% FBS. A(31-42 (10 M) or its vehicle was then added
and incubation was continued for 3 h in the presence or absence of SP-233 (1
M). To assess mitochondrial uptake of A(31-42, a co-localization study was
33
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
performed using a mouse monoclonal antibody Anti-OxPhos Complex II 70-kDa
subunit raised against complex II of the mitochondrial respiratory chain
(Molecular Probes, Eugene, OR, USA) and a rabbit polyclonal antibody raised
against amino acid residues 33-42 of A(31-42 (Signet Laboratories, Dedham, MA,
USA). Neuroblastoma cells were washed with phosphate-buffered saline (PBS)
1X, fixed with methanol at -20 C and rinsed with PBS 1X. Cells were then
blocked with PBS 1X containing 10 % donkey serum for 30 min and then
incubated for 24 h at 4 C with the first primary antibody raised against
complex
11(1/200) in PBS 1X solution containing 1.5% donkey serum and 0.01% Triton
X100. After washing the preparation 3 times with PBS 1X, a donkey anti-mouse
rhodamine-labeled secondary antibody (Jackson ImmunoResearch, West Grove,
PA, USA) was added at a 1/200 dilution and incubation was continued for 2 h at
room temperature. The primary antibody raised against amino acids 33-42 of
A(31-4Z was then used following the same protocol at a 1/500 dilution.
Positive
staining was revealed using a donkey-anti-rabbit secondary antibody labeled
witll the fluorescent marker Alexa Fluor 488 (Molecular Probes, Eugene, OR,
USA). Coverslips were then mounted with an aqueous mounting medium
containing 4',6-diamidino-2-phenylindole-2-hydrochloride (DAPI) (Vector
Laboratories. Burlingame, CA, USA). Confocal images were taken using an
Olympus Fluoview BX61 Laser Scanning microscope.
10. Statistical analysis
For each experiment, the results are expressed as means SD.
Comparisons between groups were made by an ANOVA followed by a
Dunnett's test.
B. Results
1. Effect of SP-233 on mitochondrial respiratory control
SP-233 was studied at concentrations of 10, 30 and 100 pM. At a
concentration as low as 10 pM, SP-233 induced a significant 50% decrease in
the MRC of rat brain mitochondria, as compared to the control group (p<0.001)
(Fig. 1). Although the differences in the effects induced by various
concentrations of SP-233 are not significant, the results do tend to display a
34
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
concentration-effect relationship since the MRC was progressively decreased
over the concentration range of 10-100 pM.
2. Effect of SP-233 on CCCP- or CsA-induced modification of the
mitochondrial respiratory coefficient
The uncoupling agent CCCP at 1 M increased the 02 consumption of
rat brain mitochondria to 150% of the basal value (p<0.01) (Fig. 2a). All
concentrations of SP-23 3 that were tested (1-1000 pM) completely inhibited
this
metabolic effect of CCCP (p<0.001). The inhibition of CCCP-induced
uncoupling by SP-233 was associated with decreases in 02 consumption values
to 60-65% of the basal level, and was concentration-independent. SP-233 did
not counteract the decrease in the MRC induced by CsA, but on the contrary, it
amplified this effect of CsA in a concentration-dependent manner (Fig. 2b).
3. Effects of fresh and aged Ao1_42 on the mitochondrial respiratory
coefficient
A,(31_42 significantly decreased the MRC of rat brain initochondria even
when it was present at a concentration as low as 0.1 pM (Fig. 3). This effect
was
more pronounced with the fresh amyloid peptide than with the aged form, the
respective inhibitory effects being 55-63% and 43-53%. This difference may be
revealing that the non-aggregated form of AO1_42 penetrates into the
mitochondria
to a greater extent than the aged, aggregated form of the peptide.
4. Effects of SP-233 on modifications of the mitochondrial respiratory
coefficient induced by fresh and aged A,61_42
Fresh A(.i1_42 reduced the MRC by 71 % compared to the control, and this
effect was partially inhibited by SP-233 (Fig. 4a). At the lowest
concentration of
SP-233 that was tested (1 pM), the MRC was restored to about 40% of the
control value, and the MRC was significantly increased in comparison with the
value obtained with A,C31_42 alone (38.56 ~: 0.34 versus 28.93 1.75,
p<0.001).
Aged A(31_42 reduced the MRC by 51% compared to the control but SP-233 did
not significantly prevent this effect (Fig. 4b).
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
5. Effect of SP-233 on A,61.42-induced toxicity on SK-N-AS human
neuroblastoma cells
Fig. 5 shows the effect of SP-233 on the A(31_42 induced decrease in SK-
N-AS cell viability. SP-233 1 M prevented the neurotoxicity induced by the
three concentrations of A(31_42 used by 19%, 26% and 28%, respectively
(p<0.01).
6. Effect of SP-233 on A(31_42 entry into the mitochondria of SK-N-AS
human neuroblastoma cells
Neuroblastoma cells treated with Af31_42 displayed strong
immunoreactivity that co-localized with complex II of the mitochondrial
respiratory chain, providing evidence that A(31-42 entered, and was present
inside,
the mitochondria. Treatment with SP-233 abolished A,61_42 immunoreactivity,
indicating that it blocked the entry of Ao1_42 into the cell and the
mitochondria.
C. Discussion
It is well known that AD is clinically characterized by a progressive
impairment of cognitive processes and memory loss. Since the early days, this
"mnesic" aspect of the disease has been related to the degeneration of central
cholinergic pathways, reflected as a decrease in synaptic concentrations of
acetylcholine (ACh). Thus, drug development research has been focused mainly
on restoring the ACh levels in the brain, and has led to the development of
the
acetylcholinesterase inhibitors (AChEIs), tacrine being the prototype of this
class
of compounds. However, despite promising clinical data, the beneficial effects
of
tacrine have been modest, and despite the development of a new generation of
AChEIs, represented by rivastigmine, galantamine and donepezil, the delay of
symptom onset in AD patients has not been shortened, as compared to tacrine.
This short (1 to 2 years) delay in the further progression of AD symptoms
(Tariot and Winblad, 2001; Waldemar et al. 2001; Grossberg et al. 2004),
although precious for the patients and their relatives, is probably due to the
progressive degeneration of cholinergic neurons and is a limitation to the use
of
the AChEIs. Moreover, a recent study performed on 565 community-resident
patients with mild-to-moderate AD indicated that donepezil is not cost
effective,
with benefits below minimally relevant thresholds (Courtney et al. 2004).
Since,
36
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
no major advance has been made in AD drug development, even though
memantine, an N-methyl-D-aspartate (NMDA) receptor antagonist has recently
been approved for treating moderately to severe AD (Livingston and Katona
2004), there remains an urgent need to develop agents that are more efficient
than AChEIs.
The etiology of AD is well documented only for the familial early-onset
form of the disease, which involves a mutation of the APP, presenilin-1 or
presenilin-2 gene. In contrast, the cause of the late-onset sporadic form of
AD,
which represents about 95% of all AD cases, remains to be established. Many
theories have emerged, and among these it has been proposed that an energetic
failure due to mitochondrial impairment and oxidative damage may be at the
origin of AD (Schulz et al. 1997; Beal, 2000).
Evidence that mitochondrial dysfunction is involved in AD etiology,
together with the recent finding that SP-233 (a naturally occurring
spirostenol)
can protect neuronal PC12 cells against AO1_42-induced neurotoxicity (Lecanu
et
al. 2004), that led us to examine the capacity of SP-233 to restore the
function of
isolated mitochondria that were impaired by exposure to A(31_42. Taken
together,
the present findings indicate that SP-233 has the potential to influence basic
mechanisms which are associated with A,6 and which have been implicated in
the pathogenesis of AD.
The effect of Af3 on mitochondrial function has been extensively
investigated during the past ten years and the data generated by these studies
has
indicated various effects of the peptide that could lead to mitochondria
dysfunction. A,61_42 and the fragment A(.325_35 have been shown to inhibit
respiration and the activities of key enzymes, such as succinate
dehydrogenase,
a-ketoglutarate dehydrogenase, pyruvate dehydrogenase and cytochrome
oxidase, in neuronal mitochondria (Kaneko et al. 1995; Casley et al. 2002a).
Inhibition of the different complexes of the respiratory chain has also been
reported to occur in neuronal mitochondria exposed to A(325_35 (Pereira et al.
1998; Canevari et al. 1999;Casley et al. 2002b) and this same peptide fragment
promoted pore-opening that is associated with the MPT (Moreira et al. 2001;
Moreira et al. 2002; Bachurin et al. 2003).
Results presented herein above show further that A(31_42 inhibits
mitochondrial respiration of rat brain mitochondria, as revealed by a decrease
in
37
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
the MRC, and that this inhibition occurs at Aol_42 concentrations as low as
0.1
pM with both freshly prepared and aged forms of the peptide (see Fig. 3). The
inhibitory effect is more pronounced with the fresh form probably because
polymeric forms of A(31_42 would traverse mitochondrial membranes to a lesser
extend than the monomeric form or oligoineric forms such as ADDLs, these
latter forms therefore being more likely to exert their detrimental effects on
the
respiratory chain and being defacto more toxic. Also, the lack of protection
by
SP-233 of mitochondria submitted to the aged/aggregated form of A,61_42 (see
Fig. 4) might be due to an inability of SP-233 to bind the aggregated form of
the
ainyloid peptide in the same way that it binds the monomeric form.
SP-233 was able to partially prevent the decrease in MRC induced by
freshly prepared Ao1_42 and protect mitochondrial function, confirming
previous
data which showed a restoring effect of SP-233 on the ATP synthesis of PC12
cells exposed to A(31_42 (Lecanu et al. 2004). As SP-233 was active in very
low
concentrations (picomolar range), its mechanism of action might be relatively
specific. The present data also confirms recent results which showed that SP-
233
protected neuronal cells against A,61_42 neurotoxicity by binding the
monomeric
form of the peptide and inllibiting the fonnation of the neurotoxic oligomeric
ADDLs (Lecanu et al. 2004).
The confocal microscopy analysis performed on human neuroblastoma
cells, in showing that Ao1_42 co-localized witll complex II of the respiratory
chain, indicates that A,61_42 can cross both cell and the mitochondrial
membranes.
When SP-233 and A,61_42 were both present in the incubation medium
iinmunocytochemical staining corresponding to the amyloid peptide was
completely suppressed, and this "scavenging" effect of SP-233 was accompanied
by a partial restoration of the MRC. In addition, SP-233 prevented, although
partially, the neurotoxic effect of A,61_42 on the same huinan neuroblastoma
cells.
This finding is of particular interest in view of recent results which have
indicated that amyloid-deposits are present in the mitochondria of cortical
pyramidal neurons of post-mortem AD brain (Fernandez-Vizarra et al. 2004).
These histological features have been described as being a very early event in
the
progression of the disease, appearing before the formation of paired-helical
filaments and leading to neuronal death.
38
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
After the binding and "scavenging" effects of SP-233 on A,61_42 had been
established, the possibility that a direct effect of SP-233 on the
mitochondria
might result in an additive protective effect against A(.i1_42 was
investigated. In
this regard, it had been previously demonstrated that A,6 promotes the opening
of
MPT pores in brain mitochondria, leading to functional impairment, and that
this
effect is abolished by CsA (Moreira et al. 2001; Bachurin et al. 2003; Abramov
et al. 2004). The present experiments show that SP-233, like CsA, decreases
the
MRC. SP-233 also amplifies the effect of the CsA, suggesting that it has CsA-
like properties and thereby inhibits MPT pore-opening, which could contribute
to its protective effect against Ao1_42 -induced mitochondrial dysfunction.
SP-233 was also able to abolish the uncoupling of oxidative
phosphorylation induced by CCCP (see Fig. 2a). It has been shown that A(31_42
disrupts mitochondrial membraiie structure (Kremer et al. 2001; Rodrigues et
al.
2001) and that mitochondrial membrane fluidity may be altered in AD brain
(Mecocci et al, 1996), botl7 phenomena leading to mitochondrial uncoupling.
Therefore, even though the "recoupling" effect of SP-233 cannot yet be fully
clarified, it might contribute to the restoration of mitochondrial function
described herein. Moreover, a combination of the scavenging effect of SP-233
(presumably involving its modulation of MPT pores) and its re-coupling effect
might explain why SP-233 counteracted the effect of A(31_42 at concentrations
as
low as 1 pM.
Example 2. The Spirostenol (22R,25R)-20a-spirost-5-en-3B-yl Hexanoate
Blocks Mitochondrial Uptake of A13 in Neuronal Cells and Prevents AB-
Induced Impairment of Mitochondrial Function
A. Materials and methods
1. Materials
AB1_42 peptide was purchased from American Peptide Company
(Sunnyvale, CA). The anti-complex II antibody was purchased from Molecular
Probes (Eugene, OR), and the antibody anti-Af31-42 was from Signet
Laboratories (Dedhain, MA). Spirostenol (SP-233) was purchased from
Interbioscreen (Moscow, Russia). Cyclosporin A (CsA), carbonyl cyanide 3-
chlorophenylhydrazone (CCCP), carbonyl cyanide p-trifluoromethoxy-
phenylhydrazone (FCCP), rotenone, malonate, myxothiazol, potassium cyanide
39
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
(KCN), oligomycin, and phenylarsine oxide (PAO) were obtained from Sigma
(St Louis, MO). Dulbecco's Minimum Essential Medium (DMEM), fetal bovine
serum (FBS), and SK-N-AS human neuroblastoma cells were purchased form
ATCC (Manassas, VA).
2. Isolation of mitochondrial fractions
To isolate mitochondrial fractions, rats weighing 230-350 g were
decapitated, and the brain was rapidly removed and homogenized on ice in a
Potter-Elvejhem homogenizer (Eurostar, IKA-Verke, Staufen, Gennany)
containing 6 ml of ice-cold isolation buffer (TRIS 20 mM, sucrose 250 mM, KCl
40 mM, EGTA 2 mM, bovine serum albumin 1 ing/ml; pH 7.2). The
homogenate was centrifuged at 2000xg for 8 min at 4 C to remove cell debris
and nuclei. The mitochondria-containing supematant was centrifuged at
12000xg for 10 min at 4 C. The pellet was resuspended in 300 1 of respiratory
buffer (D-mannitol 300 mM, KCl 10 mM, KH2PO4 10 mM, MgC12 5 mM; pH
7.2 at 37 C). This pellet is highly enriched in mitochondria (Zini et al.
1996).
The protein concentration of the mitochondrial suspension was determined by
the method of Lowry.
3. Measurement of mitochondrial respiration
Mitochondrial respiration was measured using an Oxytherm (Hansatech,
Norfolk, UK) linked to a PC (Compaq, PIII 400 MHz). The 37 C incubation
chamber of the Oxytherm was connected to a platinum-silver Clarck electrode
for 02 detection. 02 consumption was employed as a marker of mitochondrial
respiration in the presence of ADP (precursor for ATP) and in the presence or
absence of the following: CCCP (an uncoupling agent for oxidative
phosphorylation), CsA (an inhibitor of the meinbrane permeability transition
pore), or AJ31_42. Measurement of 02 consumption allowed for calculation of
the
mitochondrial respiratory coefficient (MRC)- defined as V3/V4 where
V4 is the basal 02 consumption and V3 is the 02 consumption after adding ADP
(ATP production). The final mitochondrial concentration was adjusted to 0.4 mg
protein/mL using respiratory buffer. Mitochondria were activated by the
addition of malate/glutamate, substrates of complex I of the respiratory
chain.
State 2 is defined as the baseline rate of mitochondrial oxygen consumption.
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
When ATP synthesis was initiated by addition of ADP, the rate of oxygen
consumption increased dramatically to reach state 3. When the supply of ADP
had been depleted, the rate of 02 consumption returned to baseline (state 2),
which was called state 4 (Chance and Williams 1956). The affects of all
experimental treatments on respiration were compared to baseline respiration,
or
mitochondrial respiration in the presence of only ADP.
To test the effects of SP-233 on mitochondrial dysfunction induced by
CCCP, isolated mitochondria were incubated in SP-233 for 3 min at 37 C before
adding malate/glutamate. One minute later, CCCP was added. To test the effects
of SP-233 on mitochondrial dysfunction induced by CsA, isolated mitochondria
were incubated in SP-233 for 1.5 min before the addition of CsA.
Malate/glutamate was added 1.5 min later, and ADP was added 2.5 min later.
Finally, to test the affect of SP-233 on A131-42-induced mitochondrial
dysfunction, mitochondrial fractions were incubated in SP-233 for 3 min and
A131-42 for 1.5 min at 37 C before addition of malate/glutamate. ADP was added
1 min after addition of malate/glutamate. In all experiments, SP-233, CCCP,
and CsA were first dissolved in N,N-dimethylformamide (DMF) and then
diluted in fresh distilled water. A131-42 employed in these experiments was
reconstituted with fresh distilled water to a concentration of 500 M. A131-42
was
used as freshly reconstituted or in an aggregated form after having been
"aged"
by incubating the solution for 48 h at 4 C. Both solutions were diluted with
the
respiratory buffer before use and then added directly to the mitochondria to
achieve the desired concentration.
4. Cell Culture
Human neuroblastoma SK-N-AS cells were seeded in 96-well plates
(7x104 cells/well) in DMEM containing 10% FBS and were cultured in 5% COa
and 95% humidity. Cultures were treated with the following in the presence or
absence of varying concentrations of SP-233: A131-42 (0.1, 1, and 10 M),
rotenone (50 nM), malonate (100 mM), myxothiazol (0.3 M), KCN (12 mM),
oligomycin (0.5 g/ml), FCCP (3 M), and PAO (0.25 M). Cell viability was
measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium
bromide (MTT) assay according to manufacturer's instructions (Trevigen,
Gaithersburg, MD). For experiments involving A131-42, cell viability was
41
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
measured 72 hours after the insult in the presence or absence of 1 M At31-az.
Only the freshly reconstituted A131-4Z solutions were used. For experiments
involving mitochondrial toxins (rotenone, malonate, myxothiazol, KCN, and
oligomycin), cell viability was measured 6 or 24 hours after the insult in the
presence or absence of 1, 3, 10, 30, or 100 M A131-42. SP-233 was added to
cultures 1 hour prior to the addition of the mitochondrial toxins. For
experiments involving FCCP and PAO, parallel cell cultures were incubated
with or without increasing concentrations of SP-233, ranging from 1 to
100 M, one hour prior to the addition of FCCP or PAO. Cell viability was
assessed 24 hours later. FCCP, rotenone, malonate, myxothiazol, KCN,
oligoinycin, and PAO were all first dissolved as a stock solution in ethanol
before being diluted in culture medium to reach the indicated final
concentrations. The percentage of ethanol in the culture medium was 0.09%.
5. Immunocytochemical analysis of A131. a2
Hunlan neuroblastoma SK-N-AS cells were seeded on 13-mm diameter
coverslips (20,000 cells/coverslip) and incubated overnight at 37 C, at 5%
C02,
in DMEM containing 10% FBS. Cultures were then incubated with AB1-42 (10
M) or its vehicle for 3 h in the presence or absence of SP-233 (1 M). To
assess mitochondrial uptake of AB1-42, a co-localization study was performed
using an Anti-OxPhos Complex II 70-kDa subunit mouse monoclonal antibody
raised against complex II of the mitochondrial respiratory chain (Molecular
Probes, Eugene, OR) and a rabbit polyclonal antibody raised against ainino
acid
residues 33-42 of AB1-42 (Signet Laboratories, Dedham, MA). Neuroblastoma
cells were washed with phosphate-buffered saline (PBS), fixed with methanol at
-20 C, and rinsed with PBS. Cells were then blocked with PBS containing 10%
donkey serum for 30 min and then incubated for 24 h at 4 C with the antibody
raised against complex II (1:200 in PBS containing 1.5% donkey serum and
0.01% Triton X100). Cells were then washed with PBS and incubated with a
donkey anti-mouse rhodamine-labeled secondary antibody (1:200; Jackson
IinmunoResearch, West Grove, PA) for 2 h at room temperature. The same
sequential steps were then followed using the antibody raised against amino
acids 33-42 of A131-42 (1:500) and a donkey-anti-rabbit secondary antibody
labeled with the fluorescent marker Alexa FluorO 488 (Molecular Probes,
42
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Eugene, OR). Coverslips were mounted with an aqueous mounting medium
containing 4',6-diamidino-2-phenylindole-2-hydrochloride (DAPI; Vector
Laboratories, Burlingame, CA). Confocal images were acquired using an
Olyinpus Fluoview BX61 Laser Scanning microscope.
6. Statistical analysis
All results are expressed as mean SD. Comparisons between groups
were carried out using an Analysis of Variance (ANOVA) followed by
Duimett's test. P values < 0.05 were accepted as significant.
B. Results
1. Effect of SP-233 on CCCP- or CsA-induced changes in mitochondrial
respiration
First, the effects of SP-233 at concentrations ranging from 1 fM to 100
pM on mitochondrial respiration were assessed by monitoring the evolution of
MRC (Fig. 6). Increasing concentrations of SP-233 resulted in a progressive
decrease in the MRC. At concentrations as low as 1 nM, SP-233 induced a
significant decrease in the MRC of rat brain mitochondria, as compared to the
control group (p<0.001; Fig. 6b). Concentrations of SP-233 at or above 10 pM,
induced a 50% decrease in the MRC.
CCCP uncouples oxidative phosphorylation and increases 02
consumption. As shown in Figure 7a, 1 M CCCP increases 02 consumption in
rat brain mitochondria to 150% of the basal value (p<0.01). Concentrations of
SP-233 ranging from 1-1000 pM completely abolished this effect (p<0.001 for
all), and the inhibition of CCCP-induced uncoupling in the presence of all of
these SP-233 concentrations was associated with a decrease in 02 consumption
values to 60-65% of the basal level.
Hypoxia was induced by adding 1 M CsA to the incubation mediuin
bathing the mitochondria (Zini et al. 1996). SP-233 did not counteract the
decrease in the MRC induced by CsA. On the contrary, it amplified this effect
in
a concentration-dependent manner (Fig. 7b).
43
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
2. Effects of SP-233 on changes in mitochondrial respiration induced by
fresh and aged A01_42
Addition of A131_42 to isolated mitochondria significantly decreased the
MRC, even when present at concentrations of 0.1 pM (Fig. 8). This effect was
more pronounced with the fresh amyloid peptide than with the aged fonn; the
inhibitory effect of the former was approximately 10% greater. It was
hypothesize that the non-aggregated form of AB1_42 penetrates into the
mitochondria to a greater extent than the aged, aggregated form of the
peptide.
As shown in Figure 9a, addition of fresh A131_42 to isolated mitochondria
resulted
in a decrease in the MRC to 71% of the control values, and this decrease was
partially reversed by SP-233. At the lowest concentration of SP-233 tested (1
pM), the MRC was restored to approximately 39% of the control value. The
MRC was significantly increased in comparison to the value obtained with AB1_
42 alone (38.56 ::L 0.34 vs. 28.93 1.75, p<0.001). Aged A131_42 reduced the
MRC
by 51 % compared to the control, but SP-233 did not significantly affect this
aged
AB1_42-induced change in the MRC (Fig. 9b).
3. Effect of SP-233 on A131-42-induced toxicity on SK-N-AS human
neuroblastoma cells
Figure 10 shows the effect of SP-23 3 on AB1_42-induced neurotoxicity in
SK-N-AS cells. In the presence of all three AB1_42 concentrations tested,
addition of 1 M SP-233 resulted in a significant increase in cell viability.
SP-
233 decreased neurotoxicity induced by 0.1, 1, and lO M AB1_42, by 19%
(p<0.01, compared to AB1_42 alone), 26% (p<0.01) and 28% (p<0.01),
respectively.
4. Effect of SP-233 on AB1_42 entry into the mitochondria of SK-N-AS cells
Figure 11 shows representative images of the DAPI staining (al, b 1 and
cl), the immunofluorescent labeling of A131_~2 (a2, b2 and c2), and the
immunofluorescent labeling of the complex II 70-kDa subunit (a3, b3 and c3) in
SK-N-AS human neuroblastoma cells. Merged images are shown in Figure a4,
b4, and c4. Neuroblastoma cells treated with AB1_42 displayed strong At31_42
immunoreactivity (Fig. 11b2) that co-localized with labeling of coinplex II of
the
mitochondrial respiratory chain (Fig. 11b4), providing evidence that A(31_42
44
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
entered, and was present inside, the mitochondria. Asl-4z immunoreactivity was
abrogated in the presence of SP-233 (Fig. 11c2), indicating that SP-233
blocked
the entry of AB1_42 into the cell and the mitochondria.
5. Neuroprotective effect of SP-233 against inhibitors of the mitochondrial
complexes in SK-N-AS cells
SP-233 did not display any neuroprotective effect against the inhibitors
of complexes I, II, and III of the mitochondria respiratory chain (Fig. 12 a,
b, and
c). At concentrations of 1 M, SP-233 significantly protected against rotenone
and 100 M SP-233 significantly protected against inyxoyhiazol (p<0.05 for
both). Although this protective effect was significant, in both cases the
magnitude of the effect was small. In contrast, SP-233 exerted a pronounced
beneficial effect against toxicity induced by KCN, a complex IV inhibitor, and
oligomycin, a complex V iiihibitor (Fig. 12d and 12e). Low concentrations of
SP-233 (1, 3, and 10 M) were active against KCN; whereas, high doses (30 and
100 M) offered no beneficial effect. The protective effect of SP-233 against
oligomycin was dose-dependent, although 10 M SP-233 did not demonstrate
statistically significant neuroprotection. These data demonstrate that SP-233
protects SK-N-AS cells against inhibition of complexes IV and V.
6. Neuroprotective effect of SP-233 on PAO and FCCP in SK-N-AS cells
Adenine nucleotide translocase (ANT) is a channel protein located in the
inner membrane of the mitochondria that, under physiological conditions,
exports ATP and imports ADP with a 1:1 ratio. The inhibition of ANT by PAO
promotes the opening of the permeability transition pore, which leads to
apoptosis. One micro molar SP-233 reduced the toxic effect of PAO on SK-N-
AS neuronal cells by 25% (Fig. 13, p<0.01). Concentrations of SP-233 above
1 M were not associated with any neuroprotective effects. In addition, SP-233
exerted a small, but significant (p<0.01), effect on FCCP-induced uncoupling
of
the respiratory chain when administered at the concentration of 100 M (Fig.
14).
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
C. Discussion
AD is clinically characterized by a progressive impairment of cognitive
processes and memory loss. Early on, this "mnesic" aspect of the disease was
related to the degeneration of central cholinergic pathways, as reflected by
decreases in synaptic concentrations of acetylcholine (ACh). Thus, drug
research has focused on restoring the ACh levels in the brain, and this has
led to
the development of the acetylcholinesterase inhibitors (AChEIs). Although
these drugs are invaluable for AD patients and their caregivers, AchEIs are
symptomatic drugs that only slow down the progression of the disease for a
limited period of time without stopping it (Tariot and Winblad, 2001; Waldemar
et al. 2001; Grossberg et al. 2004). Even though memantine, an N-methyl-D-
aspartate (NMDA) receptor antagonist, has recently been approved for treating
moderately to severe AD (Livingston and Katona 2004), there is an urgent need
for new drugs and new tlierapeutic strategies that target the etiological
causes of
the AD.
The etiology of AD is well documented for the familial, early-onset form
of the disease, which involves a mutation of the APP (presenilin-1 or
presenilin-
2) gene. In contrast, the cause of the late-onset sporadic form of AD, which
represents about 95% of all AD cases, remains to be established. Many
tlieories
have emerged, and among these is the proposal that an energetic failure due to
mitochondrial impairment and oxidative damage may be at the origin of AD
(Schulz et al. 1997; Beal, 2000). This mitochondrial dysfunction theory,
together with the recent finding that the spirostenol derivative, SP-233, can
protect neuronal PC12 cells against AB1_42-induced neurotoxicity (Lecanu et
al.
2004), led to the examination of whether SP-233 is capable of restoring the
function of isolated mitochondria exposed to A131_42. The results provided
herein
demonstrate that SP-233 can protect mitochondrial functions against the toxic
effects of Af31_42, both in isolated rat brain mitochondria and in human
neuroblastoma cells. These observations suggest that
SP-233 has the potential to influence basic mechanisms that are associated
with
AB-induced neurotoxicity and have been implicated in the pathogenesis of AD.
The effect of AB on mitochondrial function has been extensively
investigated during the past ten years, and the data generated by these
studies
indicates that various effects of the peptide could lead to mitochondrial
46
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
dysfunction. A131-42 and the fragment A132s-3s have been shown to inhibit
respiration and the activities of key enzymes such as succinate dehydrogenase,
a-ketoglutarate dehydrogenase, pyruvate dehydrogenase, and cytochrome
oxidase, in neuronal mitochondria (Kaneko et al. 1995; Casley et al. 2002a).
Inhibition of the respiratory chain complexes has also been reported in
neuronal
mitochondria exposed to AB25-35 (Pereira et al. 1999; Canevari et al. 1999;
Casley et al. 2002b), and this same peptide fragment promoted pore-opening
that
is associated with the MPT (Moreira et al. 2001; Moreira et al. 2002; Bachurin
et
al. 2003). In addition, the results presented herein revealed that AB1-42
inhibited
mitochondrial respiration of rat brain mitochondria, as revealed by a decrease
in
the MRC. Moreover, this inhibition occurred in response to AB1-42
concentrations as low as 0.1 pM, witli both freshly prepared and aged forms of
the peptide. It is likely that the inhibitory effect was more pronounced with
the
fresh form since the polymeric fonns of AB1-42 would traverse mitochondrial
membranes to a lesser extent than the monomeric form or oligomeric forms such
as ADDLs. These latter forms are more likely to exert their detrimental
effects
on the respiratory chain and are defacto more toxic. Also, the lack of
protection
imparted by SP-233 on mitochondria exposed to the aged/aggregated form of
AB1-42 might be due to the inability of SP-233 to bind the aggregated form of
the
amyloid peptide in the same way that it binds the monomeric form.
Interestingly, SP-233 was able to partially prevent the decrease in MRC
induced by freshly prepared A131-42, confirming previous results showing a
restorative effect of SP-233 on ATP synthesis in PC12 cells exposed to A131_42
(Lecanu et al. 2004). The finding that SP-233 is active at very low
concentrations suggests that its mechanism of action might be relatively
specific.
The data described herein also confirmed recent results showing that SP-233
protects neuronal cells against Af31-42 neurotoxicity by binding to the
monomeric
form of the peptide and inhibiting the formation of the neurotoxic oligomeric
ADDLs (Lecanu et al. 2004).
The confocal microscopy analysis performed on human neuroblastoma
cells, in showing that A131-42 co-localized with complex II of the respiratory
chain, indicated that A131-42 is capable of traversing both the outer cell
membrane
and the mitochondrial membrane. When SP-233 and A131-42 were both present in
the incubation medium, amyloid peptide labeling was abolished, and this
47
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
"scavenging" effect of SP-233 was accompanied by a partial restoration of the
MRC. In addition, SP-233 partially prevented the neurotoxic effects of AB1_42
on
the same human neuroblastoma cells. This finding is of particular interest in
view of recent results showing that amyloid-deposits are present in the
mitochondria of cortical pyramidal neurons of post-mortem AD brain
(Femandez-Vizarra et al. 2004). These histological features have been
described
as being a very early event in the progression of the disease, appearing
before the
formation of paired-helical filaments and leading to neuronal death. On this
basis, the inactivation of Af31_42 by SP-233, as described herein, may justify
use
of SP-233 in the treatment of AD (Lecanu et al. 2004).
After the binding and "scavenging" effects of SP-233 on AI31_42 had been
established, the possibility that a direct effect of SP-233 on mitochondria
might
result in an additive protective effect against Af31-42 was examined. In this
regard, it has been shown that AB promotes the opening of MPT pores in brain
mitochondria, leading to functional impairment, and this affect is abolished
by
CsA (Moreira et al. 2001; Bachurin et al. 2003; Abramov et al. 2004). The
experiments revealed that SP-233, like CsA, decreased the MRC. SP-233 also
amplified the CsA's effects, suggesting that it might have CsA-like
properties.
That is, it might inhibit MPT pore opening, which could contribute to its
protective effect against A131_42 -induced mitochondrial dysfunction. This
hypothesis is supported by SP-233's protective effect against PAO, a protein
tyrosine phosphatase inhibitor 1uZown to promote the MPT (Korge et al., 2001).
Thus, SP-233, at concentrations of 1 M, restored neuronal cell viability by
25%; whereas, higller concentrations of SP-233 were not associated with a
protective effect. SP-233's effect on MPT-linked pore opening may occur via
direct and/or indirect action (e.g., by allosteric modification after its
insertion
into mitochondrial membranes).
SP-233 was also able to abolish CCCP-induced uncoupling of oxidative
phosphorylation in isolated mitochondria and in neuroblastoma cells. It has
been
shown that Af31_42 disrupts mitochondrial membrane structure (Kremer et al.
2001; Rodrigues et al. 2001) and that mitochondrial membrane fluidity may be
altered in AD brain (Mecocci et al, 1996); both phenomena lead to
mitochondrial
uncoupling. Therefore, even though the "re-coupling" effect of SP-233 has not
been fully clarified, it might contribute to the restoration of mitochondrial
48
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
function described herein. Moreover, a combination of the scavenging effect of
SP-233 (presumably involving its modulation of MPT pores) and its re-coupling
effect might account for SP-233's ability to counteract the effects of Al31-
42, at
concentrations as low as 1 pM.
In an attempt to identify other targets of SP-233 in the mitochondrial
respiratory chain, it was demonstrated that SP-233 is able to protect
neuroblastoma cells against inhibitors of complexes I, III, IV, and V, but not
against inhibitors of complex II (Fig. 15). The most pronounced effect was
observed in coinplexes III and IV, although SP-233, at concentrations of 1 M
and 100 M, also exerted a small but significant neuroprotective effect
against
rotenone and myxothiazol-induced toxicity, respectively. The magnitude of the
cell viability rescue against KCN and oligomycin compared to that against AB1-
42
suggests that complexes IV and V are the main targets for the amyloid peptide-
induced impairment of mitochondrial functions. These findings confirm
previous results showing that the fragment A1325-35 inhibits complex IV in
isolated brain mitochondria (Canevari et al., 1999). However, in contrast to
the
data, the same authors reported that A1325-35 has no effect on the other
complexes
of the respiratory chain. This apparent discrepancy could be explained by
differences in the amyloid species used, and it raises questions about the
role of
Af325-35 and A131-42 in neuropathology.
As AD is an incurable neurodegenerative disorder and as the existing
treatinents, most of which are based solely on the inhibition of the AChE,
have
serious limitations, there is an urgent need to develop more effective drugs.
Here it is reported that the small molecule, SP-233, which has previously been
shown inliibits .Af31-42-induced neurotoxicity by blocking the formation of
the
ADDLs, protects brain mitochondria against the deleterious effects of Af31-42
exposure. The molecular mechanisms underlying SP-233's neuroprotective
effects might be due, at least in part, to its "scavenging" of ABt-42, its
"anti-
uncoupling" effect, its protective effect toward mitochondrial complexes IV
and
V, and its ability to inhibit the opening of MPT pores. In conclusion, the
data
provided herein have confirmed and extended the previous findings showing that
SP-233 possesses anti-amyloid properties, and they identify the mitochondria
and the respiratory chain as direct targets for A131-42 and SP-233.
49
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Bibliography
The publications listed below are incorporated by reference herein as
though set forth in full.
Abramov A.Y., Canevari L. and Duchen M. R. (2004) Beta-amyloid peptides
induce mitochondrial dysfunction and oxidative stress in astrocytes and death
of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565-575.
Bachurin S. 0., Shevtsova E. P., Kireeva E. G., Oxenkrug G. F. and Sablin S.
O.
(2003) Mitochondria as a target for neurotoxins and neuroprotective agents.
Ann. N.Y. Acad. Sci. 993, 334-344.
Beal M. F. (1992) Does impairment of energy metabolism result in excitotoxic
neuronal death in neurodegenerative disease? Ann. Neurol. 31, 119-130.
Beal M. F. (2000) Energetics in the pathogenesis of neurodegenerative
diseases.
Trends Neurosci. 23, 298-304.
Busciglio J., Pelsman A., Wong C., Pigino G., Yuan M., Mori H. and Yankner
B. A. (2002) Altered metabolism of the ainyloid beta precursor protein is
associated with mitochondrial dysfunction in Down's syndrome. Neuron 33,
677-688.
Canevari L., Clark J. B. and Bates T. E. (1999) 0-amyloid fragment 25-35
selectively decreases complex IV activity in isolated mitochondria. FEBS
Lett. 457, 131-134.
Casley C. S., Canevari L., Land J. M., Clark J. B. and Sharpe M.A. (2002a) (3-
Amyloid inhibits integrated mitochondrial respiration and key enzyme
activities. J. Neuf-ochefn. 80, 91-100.
Casley C. S., Land J. M., Sharpe M. A., Clark J. B., Duchen M. R. and Canevari
L. (2002b) Beta-ainyloid fragment 25-35 causes mitochondrial dysfunction
in primary cortical neurons. Neurobiol. Dis. 10, 258-267.
Courtney C., Farrell D., Gray R. et al.; AD2000 Collaborative Group. (2004)
Long-term donepezil treatment in 565 patients witll Alzheiiner's disease
(AD2000): randomised double-blind trial. Lancet 363, 2105-2115.
Eckert G. P., Wood W. G. and Muller W. E. (2001) Effects of aging and (3-
amyloid on the properties of brain synaptic and mitochondrial membranes. J
Neural. Transin. 108, 1051-1064.
Fernandez-Vizarra P., Fernandez A. P., Castro-Blanco S., Serrano J., Bentura
M.
L., Martinez-Murillo R., Martinez A. and Rodrigo J. (2004) Intra- and
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
extracellular A,(3 and PHF in clinically evaluated cases of Alzheimer's
disease. Histol. Histopatliol. 19, 823-844.
Grossberg G., Irwin P., Satlin A., Mesenbrink P. and Spiegel R. (2004)
Rivastigmine in Alzheimer disease: efficacy over two years. Am. J Geriatr.
Psychiatfy 12, 420-43 1.
Kaneko I., Yainada N., Sakuraba Y., Kamenosono M. and Tutumi S. (1995)
Suppression of mitochondrial succinate dehydrogenase, a primary target of
beta-amyloid, and its derivative receinized at Ser residue. J. Neurochem. 65,
2585-2593.
Kim S. H., Vlkolinsky R., Cairns N. and Lubec G. (2000) Decreased levels of
complex III core protein 1 and complex V beta chain in brains from patients
with Alzheimer's disease and Down syndrome. Cell. Mol. Life Sci. 57, 1810-
1816.
Kremer J. J., Sklansky D. J., Murphy R. M. (2001) Profile of changes in lipid
bilayer structure caused by beta-amyloid peptide. Biochemistry 40, 8563-
8571.
Kroemer G. and Reed J. C. (2000) Mitochondrial control of cell death. Nat.
Med.
6, 513-519.
Lecanu L., Yao W., Teper G. L., Yao Z. X., Greeson J. and Papadopoulos V.
(2004) dentification of naturally occurring spirostenols preventing beta-
amyloid-induced neurotoxicity. Steroids 69, 1-16.
Livingston G. and Katona C. (2004) The place of memantine in the treatment of
Alzheimer's disease: a number needed to treat analysis. Int. J Geriatr.
PsyehiatJy 19, 919.
Lowry O. H., Rosebrough N. J., Farr A. L. and Randall R. J. (1951) Protein
measurement with the folin phenol reagent. J. Biol. Chem. 93, 265-275.
Lustbader J. W., Cirilli M., Lin C. et al. (2004) ABAD directly links A(3 to
mitochondrial toxicity in Alzheimer's disease. Science 304, 448-452.
Manfredi G. and Beal M. F. (2000) The role of mitochondria in the pathogenesis
of neurodegenerative diseases. Brain Patlzol. 10, 462-472.
Mecocci P., Cherubini A., Beal M. F., Cecchetti R., Chionne F., Polidori M.
C.,
Romano G. and Senin U. (1996) Altered mitochondrial membrane fluidity in
AD brain. Neurosci. Lett. 207, 129-132.
51
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Moreira P. I., Santos M. S., Moreno A. and Oliveira C. (2001) Amyloid beta-
peptide promotes penneability transition pore in brain mitochondria. Biosci.
Rep. 21, 789-800.
Moreira P. I., Santos M. S., Moreno A., Rego A. C. and Oliveira C. (2002)
Effect of amyloid beta-peptide on permeability transition pore: a comparative
study. J. Neurosci. Res. 69, 257-267.
Pereira C., Santos M. S. and Oliveira C. (1999) Involvement of oxidative
stress
on the iinpairment of energy metabolism induced by Abeta peptides on PC12
cells: protection by antioxidants. Neurobiol. Dis. 6, 209-219.
Rodrigues C. M. P., Sola S., Brito M. A., Brondino C. D., Brites D. and Moura
J.
J. G. (2001) Amyloid 0-peptide disrupts mitochondrial membrane lipid and
protein structure: protective role of tauroursodeoxycholate. Biocheni.
Biophys. Res. Comntun. 281, 468-474.
Sergeant N., Wattez A., Galvan-Valencia M. et al (2003) Association of ATP
synthase alpha-chain with neurofibrillary degeneration in Alzheimer's
disease. Neuroscience 117, 293-303.
Schulz J. B., Matthews R. T., Klockgether T., Dichgans J. and Beal M. F.
(1997)
The role of mitochondrial dysfunction and neuronal nitric oxide in animal
models of neurodegenerative diseases. Mol. Cell. Biochem. 174, 193-197.
Tariot P. and Winblad B. (2001) Galantamine, a novel treatment for Alzheimer's
disease: a review of long-tenn benefits to patients and caregivers in
Alzheimer's Disease: Advances in Etiology, Pathogenesis and Therapeutics
(Iqbal K., Sisodia S. S. and Winblad B., eds.), pp. 705-723. John Wiley &
Sons, Chichester.
Volicer L. and Crino P. B. (1990) Involvement of free radicals in dementia of
the
Alzheimer type: a hypothesis. Neurobiol. Agiizg 11, 567-571.
Waldemar G., Winblad B., Engedal K., Soininen H., Verhey F., Wimo A.,
Wetterholm A. L., Zhang R., Haglund A. and Subbiah P. (2001) Benefits of
donepezil on cognition, function and neuropsychiatric symptoms in patients
with mild and moderate Alzheimer's disease over one year, in Alzheimer's
Disease: Advances in Etiology, Pathogenesis and Therapeutics (Iqbal K.,
Sisodia S. S. and Winblad B., eds.), pp. 725-738. John Wiley & Sons,
Chichester.
52
CA 02603127 2007-09-28
WO 2006/107902 PCT/US2006/012380
Zini R., Simon N., Morin C., d'Athis P. and Tillement J. P. (1996) Inhibition
of
rat cerebral mitochondrial respiration by cyclosporins A, D, and G and
restoration with trimetazidine. C. R. Acad Sci. III. 319, 1087-1092.
All publications, patents and patent applications are incorporated herein
by reference. While in the foregoing specification this invention has been
described in relation to certain preferred embodiments thereof, and many
details
have been set forth for purposes of illustration, it will be apparent to those
skilled
in the art that the invention is susceptible to additional embodiments and
that
certain of the details described herein may be varied considerably without
departing from the basic principles of the invention.
53