Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 287
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 287
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
COMPOSITIONS AND METHODS FOR
TREATING DIVERTICULAR DISEASE
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates generally to pharmaceutical
compositions, methods and implants, and more specifically, to compositions,
implants, and methods for treating diverticular disease (e.g.,
diverticulitis).
Description of the Related Art
Diverticular disease is a condition whereby there is herniation of
the mucosa and submucosa of a hollow organ, such as the gastrointestinal (GI)
tract, urinary tract, or repiratory tract, which produces outpouchings through
the
muscular wall of the body passageway. Although diverticula can occur in any
tubular organ, diverticular disease is of greatest clinical relevance in the
lower
GI tract (large bowel or colon), where it can cause life threatening
inflammation
and infection (diverticulitis) or bleeding (lower GI hemorrhage). Typically
this
condition is treated medically or through open surgical removal of the
diverticula
and/or complete resection of the segment of the bowel that contains them.
Diverticular disease (which encompasses diseases such as
diverticulosis and diverticulitis) is an important medical condition and is
the
most common cause of large bleeds in the colon, accounting for 30% to 50% of
massive GI hemorrhage. Diverticular disease results when a small pouch
(referred to as a diverticulum) in the colon bulges outward through weak spot.
About 10% of Americans over the age 40 have diverticulosis (i.e., the
condition
of having diverticula), and the condition becomes more common as people age
(33% of the population over the age of 60 and 50% of people over 80 have
diverticular disease). In many patients, diverticulosis remains asymptomatic.
However, in about 10-25% of people with diverticulosis, the pouches become
1
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
infected or inflamed. This condition, referred to as diverticulitis, can cause
abdominal pain (in particular around the left side of the lower abdomen),
peritonitis, abscess formation, and lower GI bleeding. Perhaps the most
serious consequence of diverticular disease is lower intestinal hemorrhage
(blood passed via the rectum). As many as 15% to 40% of diverticulosis
patients experience an episode of bleeding, and 25% of those patients will
have
a recurrent bleeding episode. After a second hemorrhage, the chance of a third
bleed is approximately 50%. The combined mortality and significant morbidity
rate associated with diverticular hemorrhage is 10% to 20%, in part due to
patient age and comorbidity with other conditions such as cardiac, pulmonary,
or renal dysfunction.
Generally, diverticular bleeds are massive, painless, and self
limiting. In 5% of diverticular patients, however, the bleed is substantial
enough
to cause cardiovascular instablility and may require transfusion. Treatment of
diverticular disease generally involves resuscitation, which includes large
bore
intravenous access, placement of a foley catheter, placement of a nasogastric
tube to rule out upper gastrointestinal bleed, and administration of
intravenous
fluids. Patients are most frequently treated supportively with volume
resuscitation, correction of coagulation abnormatlities and blood transfusion,
if
required. Most active lower GI bleeds will stop spontaneously. However, 18%
to 25% of patients with diverticular bleed will become hemodynamically
unstable as a result of the hemorrhage and continue to be unstable despite
aggressive resuscitation. In cases of massive or severe bleeding, urgent
surgery (e.g., segmented colectomy, blind segment resection, abdominal
colectomy, total abdominal colectomy, and subtotal colectomy) may be required
to attempt to stop the bleeding.
Although several pharmacological approaches for treating
diverticulitis are described (see, e.g., U.S. Patent Nos. 4,837,229;
6,297,214;
6,114,304; and 4,455,305), none have proven to be particularly effective. For
example, hemodynamically stable, actively bleeding patients can be treated
2
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
with vasospastic substances such as vasopressin. Risks associated with
vasospastic substances include a re-bleed rate of 50% in patients after
withdrawl of the medication, decreased coronary perfusion, hypertension, and
cardiac arrythmias. Alternately, embolization (clogging the arteries that
supply
the bleeding bowel segment with small, locally injected particles) can
sometimes stop the bleeding but is associated with colon infarctions and is
preferably reserved only for patients who present a poor surgical risk.
Currently no reliable way exists to acutely treat diverticulitis, other
than supportive measures, or urgent surgery in severe cases. Even for patients
in whom symptoms spontaneously resolve (i.e., bleeding ceases), currently no
reliable nonsurgical interventions can be employed to prevent recurrent
bleeding. Also, many patients who would benefit from surgical resection of
their
diverticula are often not surgical candidates because of age, frailty as a
result of
blood loss, or other concurrent medical conditions. Therefore, a significant
unmet medical need remains to develop nonsurgical, minimally invasive
interventions that can eliminate diverticula and the morbidity and mortality
associated with them.
BRIEF SUMMARY OF THE INVENTION
Briefly stated, the present invention provides agents, implants,
compositions, and methods, for minimally invasive delivery of selected
therapeutic, fibrosis-inducing agents, implants, and compositions directly
into
diverticula for the treatment of patients with diverticular disease.
Compositions
and implants, including devices, are provided herein for delivery of selected
therapeutic agents into diverticula, as well as methods for making and using
these agents, compositions, and implants. In certain embodiments, therapeutic
agents or drug-impregnated implants (or biomaterials) are provided that induce
adhesion or fibrosis in the walls of the diverticula or facilitate "filling"
of the
diverticula in situ; thus obliterating the lumen of the diverticula and
relieving
symptoms or reducing the risk that subsequent complications will develop.
3
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Within various embodiments, fibrosis is induced within the diverticula by
local
(intraluminal) or regional release of specific pharmacological agents
delivered
to the site via endoscopy or catheter-based interventions.
In one aspect, the present invention provides a composition
comprising (a) a fibrosing agent and (b) a polymer or a compound that forms a
crosslinked polymer in situ.
In another aspect, the present invention provides a composition
comprising a composition comprising a fibrosing agent and a bulking agent.
In another aspect, the present invention provides a method for
treating a diverticular disease that comprises introducing into a diverticulum
in a
host, a therapeutically effective amount of a fibrosing agent or a composition
comprising a fibrosing agent, wherein the fibrosing agent induces a fibrotic
response within the diverticulum, thereby treating diverticular disease in the
host.
In another related aspect, the present invention provides a
method for inducing fibrosis in a diverticulum of a host in need thereof that
comprises introducing a composition into the diverticulum of the host, said
composition comprising a fibrosing agent, wherein the agent induces fibrosis
within the diverticulum.
In another aspect, the present invention provides a method for
treating a diverticular disease that comprises introducing into a diverticulum
in a
host a composition, said composition comprising (a) fibrosing agent and (b) a
polymer or a compound that forms a crosslinked polymer in situ, wherein the
compostion induces a fibrotic response within the diverticulum, thereby
treating
diverticular disease in the host.
In another related aspect, the present invention provides a
method for inducing fibrosis in a diverticulum of a host, comprising inserting
a
composition into the host, said composition comprising (a) a fibrosing agent
and
(b) a polymer or a compound that forms a crosslinked polymer in situ, wherein
the composition induces fibrosis in the host.
4
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
In another aspect, the present invention provides a method for
making an implant comprising combining (a) a fibrosing agent; (b) a polymer,
or
a composition comprising a polymer; and (c) an anti-infective agent, wherein
the fibrosing agent induces a fibrotic response within a diveriticulum.
In another aspect, the present invention provides a kit for use in
treating a diverticular disease, comprising: (a) a dry powder composition that
comprises (i) a first component having a core substituted with m nucleophilic
groups, where m>2; and (ii) a second component having a core substituted with
n electrophilic groups, where n>2 and m+n>4; wherein the nucleophilic and
electrophilic groups are non-reactive in a dry environment but are rendered
reactive upon exposure to an aqueous environment such that the componenets
inter-react in the aqueous environment to form a three-dimensional
composition; (b) a first buffer solution having a pH within the range of about
1.0
to 5.5; and (c) a second buffer solution having a pH within the range of about
6.0 to 11.0; and (d) a third component comprising a fibrosing agent, wherein
each component is packaged separately and admixed immediately prior to use.
In each of the above-mentioned aspects, the fibrosing agent may
be one or more of the following: a fibrosing agent that promotes cell
regeneration, a fibrosing agent that promotes angiogenesis, a fibrosing agent
that promotes fibroblast migration, a fibrosing agent that promotes fibroblast
proliferation, a fibrosing agent that promotes deposition of extracellular
matrix, a
fibrosing agent that promotes tissue remodeling, a fibrosing agent that is a
diverticular wall irritant, silk (such as silkworm silk, spider silk,
recombinant silk,
raw silk, hydrolyzed silk, acid-treated silk, and acylated silk), talc,
chitosan,
polylysine, fibronectin, bleomycin or an analogue or derivative thereof, a
fibrosing agent that connective tissue growth factor (CTGF), metallic
beryllium
or an oxide thereof, copper, saracin, silica, crystalline silicates, quartz
dust,
talcum powder, ethanol, a component of extracellular matrix, collagen, fibrin,
fibrinogen, poly(ethylene terephthalate), poly(ethylene-co-vinylacetate), N-
carboxybutylchitosan, an RGD protein, a polymer of vinyl chloride,
5
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
cyanoacrylate, crosslinked poly(ethylene glycol)-methylated collagen, an
inflammatory cytokine, TGFI3, PDGF, VEGF, TNFa, NGF, GM-CSF, IGF-a, IL-1,
IL-8, IL-6, a growth hormone, a bone nnorphogenic protein, a cell
proliferative
agent, dexamethasone, isotretinoin, 17-p-estradiol, estradiol,
diethylstibesterol,
cyclosporine a, a//-trans retinoic acid or an analogue or derivative thereof,
wool
(including animal wool, wood wool, and mineral wool), cotton, bFGF,
polyurethane, polytetrafluoroethylene, poly(alkylcyanoacrylate), activin,
angiopoietin, insulin-like growth factor (IGF), hepatocyte growth factor
(HGF), a
colony-stimulating factor (CSF), erythropoietin, an interferon, endothelin-1,
angiotensin II, bromocriptine, methylsergide, fibrosin, fibrin, an adhesive
glycoprotein, proteoglycan, hyaluronan, secreted protein acidic and rich in
cysteine (SPaRC), a thrombospondin, tenacin, a cell adhesion molecule, an
inhibitor of matrix metalloproteinase, a tissue inhibitor of matrix
metalloproteinase, methotrexate, carbon tetrachloride, and thioacetamide.
Additional fibrosing agents may also be used in the present invention are also
disclosed in the detailed description below.
In each of the above-mentioned aspect, one or more of the
following polymer may be used alone (as a fibrosing agent) or in combination
with each of the fibrosing agents listed above or otherwise described herein:
a
copolymer, a block copolymer, a random copolymer, a biodegradable polymer,
a non-biodegradable polymer, a hydrophilic polymer, a hydrophobic polymer, a
polymer having hydrophilic domains, a polymer having hydrophobic domains, a
non-conductive polymer, an elastomer, a hydroger, a silicone polymer, a
hydrocarbon polymer, a styrene-derived polymer, a butadiene-derived polymer,
a macromer, a poly(ethylene glycol), a collagen or a derivative thereof, a
methylated collagen, a combination of a collagen or a derivative thereof and a
fibrinogen, a combination of a collagen or a derivative thereof and a
thrombin, a
combination of (a) a collagen or a derivative thereof; (b) a fibrinogen; and
(c) a
thrombin, a combination of a methylated collagen and a poly(ethylene glycol)
or
a derivative thereof, CT3, COSTASIS, a poly(ethylene glycol), COSEAL,
6
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
TISSEAL, FLOSEAL, fibrin, an amorphous polymer, a cyanoacrylate, methyl
cyanoacrylate, ethyl cyanoacrylate, butyl cyanoacrylate, octyl cyanoacrylate,
methoxypropyl cyanoacrylate, a poly(alkylcyanoacrylate),
poly(methylcyanoacrylate), poly(ethylcyanoacrylate), poly(butylcyanoacrylate),
poly(isobutylcyanoacrylate), poly(hexylcyanoacrylate),
poly(octylcyanoacrylate),
a poly(carboxyalkylcyanoacrylate), poly(methoxypropylcyanoacrylate), a
crosslinked polymer, a polymer reacts with mammalian tissue, a naturally
occurring polymer, a protein, a carbohydrate, a both crosslinked and
biodegradable polymer, a nonbiodegradable polymer, a methylated collagen,
fibrinogen, thrombin, albumin, plasminogen, von Willebrands factor, Factor
VIII, hypoallergenic collagen, atelopeptidic collagen, telopeptide collagen,
crosslinked collagen, aprotinin, Gelatin, a protein conjugate, a gelatin
conjugate, hyaluronic acid, a hyaluronic acid derivative, a synthetic polymer,
a
polymer formed from reactants comprising a synthetic isocyanate-containing
compound, a synthetic isocyanate-containing compound, a polymer formed
from reactants comprising a synthetic thiol-containing compound, a synthetic
thiol-containing compound, a polymer formed from reactants comprising a
synthetic compound containing at least two thiol groups, a synthetic compound
containing at least two thiol groups, a polymer formed from reactants
comprising a synthetic compound containing at least three thiol groups, a
synthetic compound containing at least three thiol groups, a polymer formed
from reactants comprising a synthetic compound containing at least four thiol
groups, a synthetic compound containing at least four thiol groups, a polymer
formed from reactants comprising a synthetic amino-containing compound, a
synthetic amino-containing compound, a polymer formed from reactants
comprising a synthetic compound containing at least two amino groups, a
synthetic compound containing at least two amino groups, a polymer formed
from reactants comprising a synthetic compound containing at least three
amino groups, a synthetic compound containing at least three amino groups, a
polymer formed from reactants comprising a synthetic compound containing at
7
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
least four amino groups, a synthetic compound containing at least four amino
groups, a polymer formed from reactants comprising a synthetic compound
comprising a carbonyl-oxygen-succinimidyl group, a synthetic compound
comprising a carbonyl-oxygen-succinimidyl group, a polymer formed from
reactants comprising a synthetic compound comprising at least two carbonyl-
oxygen-succinimidyl groups, a synthetic compound comprising at least two
carbonyl-oxygen-succinimidyl groups, a polymer formed from reactants
comprising a synthetic compound comprising at least three carbonyl-oxygen-
succinimidyl groups, a synthetic compound comprising at least three carbonyl-
oxygen-succinimidyl groups, a polymer formed from reactants comprising a
synthetic compound comprising at least four carbonyl-oxygen-succinimidyl
groups, a synthetic compound comprising at least four carbonyl-oxygen-
succinimidyl groups, a polymer formed from from reactants comprising a
synthetic polyalkylene oxide-containing compound, a synthetic polyalkylene
oxide-containing compound, a polymer formed from reactants comprising a
synthetic compound comprising both polyalkylene oxide and biodegradable
polyester blocks, a synthetic compound comprising both polyalkylene oxide and
biodegradable polyester blocks, a polymer formed from reactants comprising a
synthetic polyalkylene oxide-containing compound having reactive amino
groups, a synthetic polyalkylene oxide-containing compound having reactive
amino groups, a polymer formed from reactants comprising a synthetic
polyalkylene oxide-containing compound having reactive thiol groups, a
synthetic polyalkylene oxide-containing compound having reactive thiol groups,
a polymer formed from reactants comprising a synthetic polyalkylene oxide-
containing compound having reactive carbonyl-oxygen-succinimidyl groups, a
synthetic polyalkylene oxide-containing compound having reactive carbonyl-
oxygen-succinimidyl groups, a polymer formed from reactants comprising a
synthetic compound comprising a biodegradable polyester block, a synthetic
compound comprising a biodegradable polyester block, a polymer formed from
reactants comprising a synthetic polymer formed in whole or part from lactic
8
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
acid or lactideA synthetic polymer formed in whole or part from lactic acid or
lactide, a polymer formed from reactants comprising a synthetic polymer formed
in whole or part from glycolic acid or glycolide, a synthetic polymer formed
in
whole or part from glycolic acid or glycolide, a polymer formed from reactants
comprising polylysine, Polylysine, a polymer formed from reactants comprising
(a) protein and (b) a compound comprising a polyalkylene oxide portion, a
polymer formed from reactants comprising (a) protein and (b) polylysine, a
polymer formed from reactants comprising (a) protein and (b) a compound
having at least four thiol groups, a polymer formed from reactants comprising
(a) protein and (b) a compound having at least four amino groups, a polymer
formed from reactants comprising (a) protein and (b) a compound having at
least four carbonyl-oxygen-succinimide groups, a polymer formed from
reactants comprising (a) protein and (b) a compound having a biodegradable
region formed from reactants selected from lactic acid, lactide, glycolic
acid,
glycolide, and epsilon-caprolactone, a polymer formed from reactants
comprising (a) collagen and (b) a compound comprising a polyalkylene oxide
portion, a polymer formed from reactants comprising (a) collagen and (b)
polylysine, a polymer formed from reactants comprising (a) collagen and (b) a
compound having at least four thiol groups, a polymer formed from reactants
comprising (a) collagen and (b) a compound having at least four amino groups,
a polymer formed from reactants comprising (a) collagen and (b) a compound
having at least four carbonyl-oxygen-succinimide groups, a polymer formed
from reactants comprising (a) collagen and (b) a compound having a
biodegradable region formed from reactants selected from lactic acid, lactide,
glycolic acid, glycolide, and epsilon-caprolactone, a polymer formed from
reactants comprising (a) methylated collagen and (b) a compound comprising a
polyalkylene oxide portion, a polymer formed from reactants comprising (a)
methylated collagen and (b) polylysine, a polymer formed from reactants
comprising (a) methylated collagen and (b) a compound having at least four
thiol groups, a polymer formed from reactants comprising (a) methylated
9
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
collagen and (b) a compound having at least four amino groups, a polymer
formed from reactants comprising (a) methylated collagen and (b) a compound
having at least four carbonyl-oxygen-succinimide groups, a polymer formed
from reactants comprising (a) methylated collagen and (b) a compound having
a biodegradable region formed from reactants selected from lactic acid,
lactide,
glycolic acid, glycolide, and epsilon-caprolactone, a polymer formed from
reactants comprising hyaluronic acid, a polymer formed from reactants
comprising a hyaluronic acid derivative, a polymer formed from reactants
comprising pentaerythritol poly(ethylene glycol)ether tetra-sulfhydryl of
number
average molecular weight between 3,000 and 30,000, Pentaerythritol
poly(ethylene glycol)ether tetra-sulfhydryl of number average molecular weight
between 3,000 and 30,000; a polymer formed from reactants comprising
pentaerythritol poly(ethylene glycol)ether tetra-amino of number average
molecular weight between 3,000 and 30,000; pentaerythritol poly(ethylene
glycol)ether tetra-amino of number average molecular weight between 3,000
and 30,000; a polymer formed from reactants comprising (a) a synthetic
compound having a number average molecular weight between 3,000 and
30,000 and comprising a polyalkylene oxide region and multiple nucleophilic
groups, and (b) a synthetic compound having a number average molecular
weight between 3,000 and 30,000 and comprising a polyalkylene oxide region
and multiple electrophilic groups; a mixture of (a) a synthetic compound
having
a number average molecular weight between 3,000 and 30,000 and comprising
a polyalkylene oxide region and multiple nucleophilic groups, and (b) a
synthetic compound having a number average molecular weight between 3,000
and 30,000 and comprising a polyalkylene oxide region and multiple
electrophilic groups. Additional polymers that may be used in the present
invention are also disclosed in the detailed description below.
In each of the above-mentioned aspect, one or more of the
following anti-infective agents may be used alone (if capable of functioning
as a
fibrosing agent), with each of the fibrosing agents listed above (or otherwise
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
described herein), with each of the polymers listed above (or otherwise
described herein), or with each of the combination of the fibrosing agents
listed
above (or otherwise described herein) and the polymers listed above (or
otherwise described herein): an anthracycline, doxorubicin, mitoxantrone, a
fluoropyrimidine, 5-fluorouracil (5-FU), a folic acid antagonist,
methotrexate, a
podophylotoxin, etoposide, camptothecin, a hydroxyurea, a platinum complex,
cisplatin, an antibiotic, doxycycline, metronidazole, trimethoprim-
sulfamethoxazole, a fourth generation penicillin (e.g., a ureidopenicillin a
carboxypenicillin, mezlocillin, piperacillin, carbenicillin, and ticarcillin,
and an
analogue or derivative thereof), a first generation cephalosporin (e.g.,
cephazolin sodium, cephalexin, cefazolin, cephapirin, and cephalothin), a
carboxypenicillin (e.g., ticarcillin), a second generation cephalosporin
(e.g.,
cefuroxime, cefotetan, and cefoxitin), a third generation cephalosporin (e.g.,
naxcel, cefdinir, cefoperazone, ceftazidime, ceftriaxone,and cefotaxime), a
fourth generation cephalosporin (e.g., cefepime), a monobactam (e.g.,
aztreonam), a carbapenem (e.g., imipenem, ertapenem and meropenem), an
aminoglycoside (e.g., streptomycin, gentamicin, tobramycin, and amikacin), an
MSL group member (e.g., a nnacrolide, a long acting macrolide, a lincosamide,
a streptogramin, Erythromycin, Azithromycin, Clindamycin, Syneroid,
clarithromycin, and kanamycin sulfate), a quinolone (e.g., ciprofloxacin,
ofloxacin, gatifloxacin, moxifloxacin, levofloxacin, and trovafloxacin), a DNA
synthesis inhibitor (e.g., metronidazole), and a sulfonamide (e.g.
trimethoprim,
including cefixime, spectinomycin, tetracycline, nitrofurantoin, polymyxin B,
and
neomycin sulfate).
In each of the above-mentioned aspect, one or more of the
following bulking agents may be used alone (if capable of functioning as a
fibrosing agent), with each of the fibrosing agents listed above (or otherwise
described herein), with each of the polymers listed above (or otherwise
described herein), with each of the combination of the fibrosing agents listed
above (or otherwise described herein) and the polymers listed above (or
11
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
otherwise described herein), with each of the combination of the polymers
listed
above (or otherwise described herein) and the anti-infective agents listed
above
(or otherwise described herein), with each of the combination of the fibrosing
agents listed above (or otherwise described herein) and the anti-infective
agents listed above (or otherwise described herein): an agent or a composition
that comprises microspheres, an agent or a composition that comprises a
hydroxyapatite loaded gel, an agent or a composition that comprises a
micronized alloderm acellular matrix, an agent or a composition that comprises
hyaluronic acid, an agent or a composition that comprises micro-beads in a
hydrogel, an agent or a composition that comprises a hylan polymer, and an
agent or a composition that comprises a silicon microballoon and biocompatible
polymer. Additional bulking agents that may be used in the present invention
are described in the detailed description below.
One aspect of the invention relates to a homogeneous dry powder
composition comprised of: a first component having a core substituted with m
nucleophilic groups, where m and a second component having a core
substituted with n electrophilic groups, where n and m+n>4; wherein the
nucleophilic and electrophilic groups are non-reactive in a dry environment
but
are rendered reactive upon exposure to an aqueous environment such that the
components inter-react in the aqueous environment to form a three-dimensional
matrix. A pharmaceutically acceptable carrier may also be included.
In one embodiment of the homogeneous dry powder composition,
the nucleophilic and electrophilic groups undergo a nucleophilic substitution
reaction, a nucleophilic addition reaction, or both. The nucleophilic groups
may
be selected from -NH2, -NHR1, -N(R1)2, -SH, -OH, -COOH, -C6H4-0H, -H, -PH2,
-PHR1, -P(R1)2, -NH-NH2, -CO-NH-NH2, and -05H4N, where R1 is a hydrocarbyl
group, and each R1 may be the same or different. The electrophilic groups may
be selected from -CO-CI, -(C0)-0-(C0)-R (where R is an alkyl group),
-CH=CH-CH=0 and -CH=CH-C(CH3)=0, halo, -N=C=O, -N=C=S,
-S02CH=CH2, -0(C0)-C=CH2, -0(C0)-C(CH3)=CH2, -S-S-(C5H4N),
12
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
-0(C0)-C(CH2CH3)=CH2, -CH=CH-C=NH, -COOH, -(CO)O-N(COCH2)2, -CHO,
-(CO)O-N(COCH2)2-S(0)20H, and -N(COCH)2.
In another embodiment of the homogeneous dry powder
composition, the nucleophilic groups are amino groups and the electrophilic
groups are amine-reactive groups. The amine-reactive groups may contain an
electrophilically reactive carbonyl group susceptible to nucleophilic attack
by a
primary or secondary amine. The amine-reactive groups may be selected from
carboxylic acid esters, acid chloride groups, anhydrides, ketones, aldehydes,
halo, isocyanato, thioisocyanato, epoxides, activated hydroxyl groups,
olefins,
carboxyl, succinimidyl ester, sulfosuccinimidyl ester, maleimido, epoxy, and
ethenesulfonyl.
In yet another embodiment of the homogeneous dry powder
composition, the nucleophilic groups are sulfhydryl groups and the
electrophilic
groups are sulfhydryl-reactive groups. The sulfhydryl-reactive groups may be
selected so as to form a thioester, imido-thioester, thioether, or disulfide
linkage
upon reaction with the sulfhydryl groups. Where the sulfhydryl-reactive groups
form a disulfide linkage, they may have the structure -S-S-Ar where Ar is a
substituted or unsubstituted nitrogen-containing heteroaromatic moiety or a
non-heterocyclic aromatic group substituted with an electron-withdrawing
moiety. Where the sulfhydryl-reactive groups form a thioether linkage, they
may be selected from maleimido, substituted maleimido, haloalkyl, epoxy,
imino, aziridino, olefins, and a,13-unsaturated aldehydes and ketones. The
sulfhydryl-reactive groups may be selected from mixed anhydrides; ester
derivatives of phosphorus; ester derivatives of p-nitrophenol, p-
nitrothiophenol
and pentafluorophenol; esters of substituted hydroxylamines, including N-
hydroxyphthalimide esters, N-hydroxysuccinimide esters, N-
hydroxysulfosuccinimide esters, and N-hydroxyglutarimide esters; esters of 1-
hydroxybenzotriazole; 3-hydroxy-3,4-dihydro-benzotriazin-4-one; 3-hydroxy-
3,4-dihydro-quinazoline-4-one; carbonylimidazole derivatives; acid chlorides;
ketenes; and isocyanates.
13
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In still another embodiment of the homogeneous dry powder
composition, the number of nucleophilic groups in the mixture is approximately
equal to the number of electrophilic groups in the mixture. For example, the
ratio of moles of nucleophilic groups to moles of electrophilic groups may be
about 2:1 to 1:2, with a ratio of 1:1 preferred.
In a further embodiment of the homogeneous dry powder
composition, the core is selected from hydrophilic polymers, hydrophobic
polymers, amphiphilic polymers, C2-14 hydrocarbyls, and heteroatom-containing
02-14 hydrocarbyls.
Where the core is a hydrophilic polymer, the core may be a
synthetic or naturally occurring hydrophilic polymer. The hydrophilic polymer
may be a linear, branched, dendrimeric, hyperbranched, or star polymer. The
hydrophilic polymer may be selected from polyalkylene oxides; polyols;
poly(oxyalkylene)-substituted diols and polyols; polyoxyethylated sorbitol;
polyoxyethylated glucose; poly(acrylic acids) and analogs and copolymers
thereof; polymaleic acids; polyacrylamides; poly(olefinic alcohols); poly(N-
vinyl
lactams); polyoxazolines; polyvinylamines; and copolymers thereof. The
hydrophilic polymer may also be selected from proteins, carboxylated
polysaccharides, aminated polysaccharides, and activated polysaccharides,
such as, for example, collagen and glycosaminoglycans.
Where the hydrophilic polymer is a polyalkylene oxide or polyols,
the hydrophilic polymer may be selected from polyethylene glycol and
poly(ethylene oxide)-poly(propylene oxide) copolymers. Where the hydrophilic
polymer is a polyols, the hydrophilic polymer may be selected from glycerol,
polyglycerol and propylene glycol. Where the hydrophilic polymer is a
poly(oxyalkylene)-substituted polyol, the hydrophilic polymer may be selected
from mono-, di- and tri-polyoxyethylated glycerol, mono- and
di-polyoxyethylated propylene glycol, and mono- and di- polyoxyethylated
trimethylene glycol. Where the hydrophilic polymer is a poly(acrylic acid),
analog or copolymer thereof, the hydrophilic polymer may be selected from
14
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
poly(acrylic acid), poly(methacrylic acid), poly(hydroxyethylmethacrylate),
poly(hydroxyethylacrylate), poly(methylalkylsulfoxide acrylates), and
poly(methylalkylsulfoxide methacrylates). Where the hydrophilic polymer is a
polyacrylamide, the hydrophilic polymer may be selected from polyacrylamide,
poly(methacrylamide), poly(dimethylacrylamide), poly(N-isopropylacrylamide),
and copolymers thereof. Where the hydrophilic polymer is a poly(olefinic
alcohol), the hydrophilic polymer may be selected from poly(vinyl alcohols)
and
copolymers thereof. Where the hydrophilic polymer is a poly(N-vinyl lactam),
the hydrophilic polymer may be selected from poly(vinyl pyrrolidones),
poly(vinyl caprolactanis), and copolymers thereof. Where the hydrophilic
polymer is a polyoxazoline, the hydrophilic polymer may be selected from
poly(methyloxazoline) and poly(ethyloxazoline).
Where the core is a hydrophobic polymer selected, the core may
be selected from polylactic acid and polyglycolic acid.
Where the core is a C2-14 hydrocarbyl, the core may be selected
from alkanes, diols, polyols, and polyacids.
Where the core is a heteroatom-containing C2_14 hydrocarbyl, the
core may be selected from di- and poly-electrophiles.
In another embodiment of the homogeneous dry powder
composition, the first component has the structure of formula (I)
(I) [X-(L1)0,-,,-R,
and the second component has the structure of formula (II)
(II) [Y-(L2)q]n-R',
wherein m and n are integers from 2-12 and m+n>4; R and R' are
independently selected from hydrophilic polymers, hydrophobic polymers,
amphiphilic polymers, C2-14 hydrocarbyls, and heteroatom-containing C2-14
hydrocarbyls; X is a nucleophilic group; Y is an electrophilic group; L1 and
L2
are linking groups; and p and q are integers from 0-1. The components may
inter-react to form covalent bonds, noncovalent bonds, or both. Noncovalent
bonds include ionic bonds, hydrogen bonds, or the association of hydrophobic
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
molecular segments. In one preferred embodiment, all of the molecular
segments are the same.
The homogeneous dry powder composition may further comprise a
biologically active agent with or without a pharmaceutically acceptable
carrier.
The pharmaceutically acceptable carrier may be a micelle, a microsphere, or a
nanosphere.
Where the pharmaceutically acceptable carrier is a microsphere
or a nanosphere, the pharmaceutically acceptable carrier may be a degradable
polymer, such as a polyester, and the polyester may be a glycolide/lactide
copolymer. The degradable polymer may also be comprised of residues of one
or more monomers selected from the group consisting of lactide, lactic acid,
glycolide, glycolic acid, e-caprolactone, gamma-caprolactone, hydroxyvaleric
acid, hydroxybutyric acid, beta-butyrolactone, gamma-butyrolactone, gamma-
valerolactone, y-decanolactone, ö-decanolactone, trimethylene carbonate, 1,4-
dioxane-2-one or 1,5-dioxepan-2one.).
The homogeneous dry powder composition may further comprise
a biologically active agent. In one embodiment, the biologically active agent
is
a fibrosing agent or a composition comprising a fibrosis agent. In certain
specific embodiments, the fibrosing agent is in another embodiment of the
invention, the homogeneous dry powder composition further comprises a
biologically active agent that is a fibrosing agent. As used in the
homogeneous
dry powder composition, the anti-fibrotic agent may be used to promote any of
the following; regeneration; angiogenesis; fibroblast migration; fibroblast
proliferation; deposition of extracellular matrix (ECM); and tissue
remodeling.
The fibrosing agent may also be used as a diverticular wall irritant.
Fibrosing agents that may be used in the homogeneous dry
powder composition may be or may be comprised of silk; silkworm silk; spider
silk; recombinant silk; raw silk; hydrolyzed silk; acid-treated silk; acylated
silk;
mineral particles; talc; chitosan; polylysine; fibronectin; bleomycin; or
CTGF.
The fibrosing agent may also be in the form of a particulate, which may be a
16
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
biodegradable particulate or a non-biodegradable particulate. Biodegradable
particulates may be comprised of a material selected from the group consisting
of polyester, polyanhydride, poly(anhydride ester), poly(ester-amide),
poly(ester-urea), polyorthoester, polyphosphoester, polyphosphazine,
polycyanoacrylate, collagen, chitosan, hyaluronic acid, chromic cat gut,
alginate, starch, cellulose and cellulose ester. Non-biodegradable
particulates
may be comprised of a material selected from the group consisting of
polyester,
polyurethane, silicone, polyethylene, polypropylene, polystyrene,
polyacrylate,
polymethacrylate, and silk. Examples of preferred particulates may be a
particulate form of a member selected from the group consisting of silk, talc,
starch, glass, silicate, silica, calcium phosphate, calcium sulfate, calcium
carbonate, hydroxyapatite, synthetic mineral, polymethylmethacrylate, silver
nitrate, ceramic and other inorganic particles.
In a further embodiment of the homogeneous dry powder
composition, the biologically active agent promotes bone growth. Within this
embodiment, the fibrosing agent may promote the bone growth. Fibrosing
agents that may promote bone growth may include a bone morphogenic protein
and an osteogenic growth factor, the latter which may be selected from
transforming growth factor, platelet-derived growth factor, and fibroblast
growth
factor.
In another embodiment of the invention, the homogeneous dry
powder composition with a fibrosing agent further comprises a pharmaceutical
agent that induces sclerosis (a sclerosant), wherein the sclerosant may be a
surfactant or it may be selected from the group consisting of ethanol,
dimethyl
sulfoxide, sucrose, sodium chloride, dextrose, glycerin, minocycline,
tetracycline, doxycycline, polidocanol, sodium tetradecyl sulfate, sodium
morrhuate, and sotradecol.
In a further embodiment of the invention, the homogeneous dry
powder composition with a fibrosing agent further comprises an inflammatory
cytokine, which may be selected from the group consisting of TGFI3, PDGF,
17
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
VEGF, bFGF, TNFa, NGF, GM-CSF, IGF-a, IL-1, IL-1-8, IL-8, IL-6, and growth
hormone.
In still another embodiment of the invention, the homogeneous dry
powder composition with a fibrosing agent further comprises an agent that
stimulates cell proliferation, which may be selected from the group consisting
of
dexamethasone, isotretinoin (13-cis retinoic acid), 17-13-estradiol,
estradiol, 1-a-
25 dihydroxyvitamin D3, diethylstibesterol, cyclosporine A, L-NAME, all-trans
retinoic acid (ATRA), and analogues and derivatives thereof.
In a further embodiment of the homogeneous dry powder
composition, the biologically active agent is mixed with the first and second
components to form a mixture.
In another embodiment of the homogeneous dry powder
composition, the biologically active agent is chemically coupled to the first
component or to the second component.
Another aspect of the invention relates to a crosslinkable
composition comprised of: (a) a first crosslinkable component having m
nucleophilic groups, wherein m and
(b) a second crosslinkable component
having n electrophilic groups capable of reaction with the m nucleophilic
groups
to form covalent bonds, wherein n and m+n the
first component comprises
two or more amino acid residues selected from the group consisting of amino
acids comprising primary amine groups and amino acids comprising thiol
groups, the second component comprises a polyethylene glycol moiety, and
each of the first and second crosslinkable components is biocompatible,
synthetic, and nonimmunogenic, and further wherein crosslinking of the
composition results in a biocompatible, nonimmunogenic, crosslinked matrix.
Any of the following are preferred embodiments of the
crosslinkable composition described immediately above: m > 3, m = 3, m = 4, n
= 4, the electrophilic groups are succinimidyl moieties, all n are identical,
and all
m are identical.
18
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
In one preferred embodiment, the selected amino acid residues
are lysine. Within this embodiment, any of the following is preferred: m > 3,
m
= 3, m = 4, n = 4, the electrophilic groups are succinimidyl moieties, all n
are
identical, and all m are identical.
In another preferred embodiment, the selected amino acid
residues are cysteine. Within this embodiment, any of the following is
preferred: m > 3, m = 3, m = 4, n = 4, the electrophilic groups are
succinimidyl
moieties, all n are identical, and all m are identical.
Yet another aspect of the invention relates to a crosslinkable
composition comprised of: (a) a first crosslinkable component having m
nucleophilic groups, wherein m and
(b) a second crosslinkable component
having n electrophilic groups capable of reaction with the m nucleophilic
groups
to form covalent bonds, wherein n and m+n, the first component comprises
two or more amino acid residues selected from the group consisting of amino
acids comprising primary amine groups and amino acids comprising thiol
groups, the second component comprises a polyethylene glycol moiety, the
electrophilic groups are succinimidyl moieties, and each of the first and
second
crosslinkable components is biocompatible, synthetic, and nonimmunogenic,
and further whererin crosslinking of the composition results in a
biocompatible,
nonimmunogenic, crosslinked matrix.
Any of the following are preferred embodiments of the
crosslinkable composition described immediately above: m > 3, m = 3, m = 4, n
= 4, the electrophilic groups are succinimidyl moieties, all n are identical,
and all
m are identical.
In one preferred embodiment, the selected amino acid residues
are lysine. Within this embodiment, any of the following is preferred: m > 3,
m
= 3, m = 4, n = 4, the electrophilic groups are succinimidyl moieties, all n
are
identical, and all m are identical.
In another preferred embodiment, the selected amino acid
residues are cysteine. Within this embodiment, any of the following is
19
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
preferred: m > 3, m = 3, m = 4, n = 4, the electrophilic groups are
succinimidyl
moieties, all n are identical, and all m are identical.
Still another aspect of the invention relates to a crosslinkable
composition comprised of: (a) a first crosslinkable component having m
nucleophilic groups, wherein m and (b) a second crosslinkable component
having n electrophilic groups capable of reaction with the m nucleophilic
groups
to form covalent bonds, wherein n and m+n, the first component comprises
two or more amino acid residues selected from the group consisting of amino
acids comprising primary amine groups and amino acids comprising thiol
groups, the second component comprises a multifunctionally activated
polyethylene glycol, and each of the first and second crosslinkable components
is biocompatible, synthetic, and nonimmunogenic, and further wherein
crosslinking of the composition results in a biocompatible, nonimmunogenic,
crosslinked matrix.
Any of the following are preferred embodiments of the
crosslinkable composition described immediately above: m > 3, m = 3, m = 4, n
= 4, the electrophilic groups are succinimidyl moieties, all n are identical,
all m
are identical, the multifunctionally activated polyethylene glycol is
tetrafunctionally activated polyethylene glycol, and the multifunctionally
activated polyethylene glycol is a star-branched polyethylene glycol.
In one preferred embodiment, the selected amino acid residues
are lysine. Within this embodiment, any of the following is preferred: m > 3,
m
= 3, m = 4, n = 4, the electrophilic groups are succinimidyl moieties, all n
are
identical, all m are identical, and the multifunctionally activated
polyethylene
glycol is tetrafunctionally activated polyethylene glycol or the
multifunctionally
activated polyethylene glycol is a star-branched polyethylene glycol.
In another preferred embodiment, the selected amino acid
residues are cysteine. Within this embodiment, any of the following is
preferred: m > 3, m = 3, m = 4, n = 4, the electrophilic groups are
succinimidyl
moieties, all n are identical, all m are identical, and the multifunctionally
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
activated polyethylene glycol is tetrafunctionally activated polyethylene
glycol or
the multifunctionally activated polyethylene glycol is a star-branched
polyethylene glycol.
Another aspect of the invention relates to a method of forming a
three-dimensional matrix comprising the steps of: (a) providing a composition
of
the invention; and (b) rendering the nucleophilic and electrophilic groups
reactive by exposing the composition to an aqueous environment to effect
inter-reaction; wherein said exposure comprises: (i) dissolving the
composition
in a first buffer solution having a pH within the range of about 1.0 to 5.5 to
form
a homogeneous solution, and (ii) adding a second buffer solution having a pH
within the range of about 6.0 to 11.0 to the homogeneous solution; and (c)
allowing a three-dimensional matrix to form. A preferred composition for use
in
this method is the homogeneous dry powder composition. The three-
dimensional matrix of the invention described immediately above may be
formed without input of any external energy or by polymerization.
In a preferred embodiment, the pH of the first buffer solution is
selected to retard the reactivity of the nucleophilic groups on the first
component by rendering the nucleophilic groups relatively non-nucleophilic. In
this preferred embodiment, the second buffer solution neutralizes the effect
of
the first buffer solution, so that the nucleophilic groups of the first
component
regain their nucleophilic character and inter-react with the electrophilic
groups
of the second component.
In another preferred embodiment, the composition, first buffer
solution and second buffer solution are housed separately in a multiple-
compartment syringe system having a multiple barrels, a mixing head, and an
exit orifice; step (b)(i) comprises adding the first buffer solution to the
barrel
housing the composition to dissolve the composition and form a homogeneous
solution, and extruding the homogeneous solution into the mixing head; step
(b)(ii) comprises simultaneously extruding the second buffer solution into the
21
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
mixing head; and step (c) further comprises extruding the resulting
composition
through the orifice onto a surface.
Yet another aspect of the invention relates to a method of sealing
tissue of a patient comprising the steps of: (a) providing a composition of
the
invention; (b) rendering the nucleophilic and electrophilic groups reactive by
exposing the composition to an aqueous environment to effect inter-reaction;
wherein said exposure comprises: (i) dissolving the composition in a first
buffer
solution having a pH within the range of about 1.0 to 5.5 to form a
homogeneous solution, and (ii) adding a second buffer solution having a pH
within the range of about 6.0 to 11.0 to the homogeneous solution to form a
mixture; and (c) placing the mixture into contact with tissue and allowing a
three-dimensional matrix to form and seal the tissue. A preferred composition
for use in this method is the homogeneous dry powder composition.
A further aspect of the invention relates to a method of forming a
three-dimensional matrix on a surface of a device comprising the steps of: (a)
providing a composition of the invention; and (b) rendering the nucleophilic
and
electrophilic groups reactive by exposing the composition to an aqueous
environment to effect inter-reaction; wherein said exposure comprises: (i)
dissolving the composition in a first buffer solution having a pH within the
range
of about 1.0 to 5.5 to form a homogeneous solution, and (ii) adding a second
buffer solution having a pH within the range of about 6.0 to 11.0 to the
homogeneous solution; and applying the homogeneous solution to a surface of
a device; and allowing the three-dimensional matrix to form. A preferred
composition for use in this method is the homogeneous dry powder
composition.
Yet another aspect of the invention relates to a method of
promoting scarring in the vicinity of a medical implant comprising the steps
of:
(a) providing a composition of the invention; (b) rendering the nucleophilic
and
electrophilic groups reactive by exposing the composition to an aqueous
environment to effect inter-reaction; wherein said exposure comprises: (i)
22
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
dissolving the composition in a first buffer solution having a pH within the
range
of about 1.0 to 5.5 to form a homogeneous solution, and (ii) adding a second
buffer solution having a pH within the range of about 6.0 to 11.0 to the
homogeneous solution; and (c) applying the mixture to a surface of a medical
implant and allowing a three-dimensional matrix to form on the surface of the
medical implant; and (d) placing the medical implant into an animal host,
wherein release of the fibrotic agent from the matrix inhibits scarring in the
animal host. In a preferred embodiment, the the fibrotic agent is released
into
tissue in the vicinity of the implant after deployment of the implant. A
preferred
composition for use in this method is the homogeneous dry powder composition
with a fibrosing agent.
A further aspect of the invention relates to a kit for use in medical
applications, comprising: (a) a homogeneous dry powder composition
comprised of: (i) a first component having a core substituted with m
nucleophilic
groups, where m ; and (ii) a second component having a core substituted with
n electrophilic groups, where n and m+n>4; wherein the nucleophilic and
electrophilic groups are non-reactive in a dry environment but are rendered
reactive upon exposure to an aqueous environment such that the components
inter-react in the aqueous environment to form a three-dimensional matrix; (b)
a
first buffer solution having a pH within the range of about 1.0 to 5.5; and
(c) a
second buffer solution having a pH within the range of about 6.0 to 11.0;
wherein each component is packaged separately and admixed immediately
prior to use. It is preferred of course that prior to use, each component is
in a
separate sterile package.
Another aspect of the invention relates to a kit for use in medical
applications, comprising: (a) a composition of the invention; (b) a first
buffer
solution having a pH within the range of about 1.0 to 5.5; and (c) a second
buffer solution having a pH within the range of about 6.0 to 11.0, wherein
each
component is packaged separately and admixed immediately prior to use. A
preferred composition of the invention for use in this kit is the homogeneous
dry
23
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
powder composition. It is preferred that each component of the kit is in a
separate sterile package.
The kit may further comprise a delivery device, which in one
embodiment, may be a multi-compartment device. A preferred multi-
compartment device of the invention is a multiple-compartment syringe system
having multiple barrels, a mixing head, and an exit orifice. Where the kit is
a
multiple-compartment syringe system, the homogeneous dry powder
composition, the first buffer solution, and the second buffer solution are
housed
separately in the multiple-compartment syringe system.
In another embodiment of the invention, the delivery device is a
pressurized delivery system. A preferred pressurized delivery system
comprises: a plurality of fluid component inlets each adapted to communicate
with a source of different fluid components; at least one carrier fluid inlet
adapted to communicate with a source of a pressurized carrier fluid; a
diffuser
surface located downstream from the plurality of fluid component inlets and
the
at least one carrier fluid inlet; and an outlet extending through the diffuser
surface, wherein the diffuser surface is adapted to receive fluid components
thereon and has a shape effective to direct and maintain each received fluid
component in a different flow path toward the outlet for mixing and dispensing
therethrough by the pressurized carrier fluid from the at least one carrier
fluid
inlet. Within this embodiment, a preferred pressurized carrier fluid is
pressurized air and the preferred fluid components are the first buffer
solution
and the second buffer solution of the invention.
Another embodiment of the kit for use in medical applications
further comprises a biologically active agent and the medical application
involves delivering the biologically active agent. The biologically active
agent
may be packaged with the homogeneous dry powder composition and may
further comprise a pharmaceutically acceptable carrier packaged with the
biologically active agent and the homogeneous dry powder composition. The
biologically active agent may also be packaged as a solution with the first
buffer
24
CA 02610948 2012-12-14
or as a solution with the second buffer. The kit may further comprise a
pharmaceutically acceptable carrier as a fourth component. The biologically
active agent is packaged with the pharmaceutically acceptable carrier.
These and other aspects of the present invention will become
evident upon reference to the following detailed description and the attached
drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is a graph showing the effect of cyclosporine A on
proliferation of human smooth muscle cells.
Figure 2 is a graph showing the effect of dexamethasone on
proliferation of human fibroblasts.
Figure 3 is a graph showing the effect of all-trans retinoic acid
(ATRA) on proliferation of human smooth muscle cells.
Figure 4 is a graph showing the effect of isotretinoin on
proliferation of human smooth muscle cells.
Figure 5 is a graph showing the effect of 17-(3-estradiol on
proliferation of human fibroblasts.
Figure 6 is a graph showing the effect of 1a,25-dihydroxy-vitamin
D3 on proliferation of human smooth muscle cells.
Figure 7 is a graph showing the effect of PDGF-BB on smooth
muscle cell migration.
Figure 8 is a bar graph showing the area of granulation tissue in
carotid arteries exposed to silk coated perivascular polyurethane (PU) films
relative to arteries exposed to uncoated PU films.
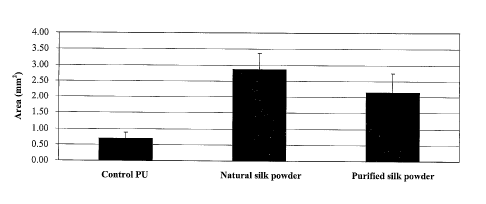
Figure 9 is a bar graph showing the area of granulation tissue in
carotid arteries exposed to silk suture coated perivascular PU films relative
to
arteries exposed to uncoated PU films.
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Figure 10 is a bar graph showing the area of granulation tissue in
carotid arteries exposed to natural and purified silk powder and wrapped with
perivascular PU film relative to a control group in which arteries are wrapped
with perivascular PU film only.
Figure ills a bar graph showing the area of granulation tissue (at
1 month and 3 months) in carotid arteries sprinkled with talcum powder and
wrapped with perivascular PU film relative to a control group in which
arteries
are wrapped with perivascular PU film only.
Figure 12 is a bar graph showing indicating the area of
perivascular granulation tissue quantified by computer-assisted morphometric
analysis in rat carotid arteries treated with control uncoated PU films and
with
PU films treated with degummed and virgin silk strands. As shown in the
figure,
both types of silk markedly increased granulation tissue growth around the
blood vessel to the same extent.
Figure 13 shows representative histology sections of rat carotid
arteries treated with PU films coated with degummed and virgin silk strands.
As
shown in the figure, both types of silk induced a marked tissue reaction
around
the treated blood vessel. Movat stain, 100X.
Figure 14 shows representative histology sections of rat carotid
arteries treated with PU films coated with degummed and virgin silk strands
showing the granulation tissue that had grown around the treated vessels. The
silk strands had broken down into small particles surrounded by giant cells
and
macrophages. The granulation tissue was highly vascularized and contained
numerous inflammatory cells and fibroblasts. Extracellular matrix deposition
was also extensive. H&E stain 200X.
Figure 15 shows the release profile for cyclosporine A from a
polyurethane film as analyzed by HPLC.
26
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides minimally invasive, endoluminal,
and endoscopic procedures that can be used to close, temporarily or
permanently, diverticula through the administration of pharmacological
compositions that induce scarring of the lumen of the diverticula and
elimination
of the diverticular sac.
As described herein, implants, procedures, and therapeutic
compositions are provided for treatment of diverticula using endoscopic and
imaging-guided interventions. For example, described herein are
pharmaceutical agents that promote one or more aspects of the production of
fibrous (scar) tissue or tissue regeneration. Furthermore, compositions and
methods are described for administering fibrosis-inducing agents and drug-
delivery compositions such that the pharmaceutical agent is delivered in
therapeutic levels over a time period sufficient for fibrosis and healing to
occur.
Numerous specific implants are described that produce superior clinical
outcomes by promotion of scarring and healing of diverticula.
Definitions
Prior to setting forth the invention, it may be helpful to an
understanding thereof to first set forth definitions of certain terms that are
used
hereinafter.
"Medical implant" refers to a device or object or composition that
is implanted (completely or partially) or inserted into a body. Accordingly,
an
implant refers to any object or composition placed in the body for the purpose
of
restoring physiological function, reducing/alleviating symptoms associated
with
disease, and/or repairing/replacing damaged or diseased organs and tissues.
"Diverticulitis" refers to diseases such as diverticulosis and
diverticulitis. Diverticular disease results when a small pouch (referred to
as a
diverticulum) in the colon bulges outward through weak spot. The condition of
having diverticula is called diverticulosis. When the pouches become infected
or
27
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
inflamed, the condition is called diverticulitis. Diverticulitis can cause
abdominal
pain, in particular, around the left side of the lower abdomen, and lower Cl
bleeding. Often the site of the herniations is the same site of penetration
for a
nutrient artery, leading to the approximation of the neck of the sack and
arterial
supply.
"Fibrosis," "scarring," or "fibrotic response" refers to the formation
of fibrous tissue in response to injury or medical intervention. Four general
components to the process of fibrosis (or scarring) include (1) formation of
new
blood vessels (angiogenesis); (2) migration and proliferation of fibroblasts;
(3)
deposition of extracellular matrix (ECM); and (4) remodeling (maturation and
organization of the fibrous tissue). Therapeutic agents which promote (also
referred to interchangeably herein as induce, stimulate, cause, increase,
accelerate, and the like) fibrosis or scarring are referred to interchangeably
herein as "fibrosis-inducing agents," "scarring agents," "fibrosing agents,"
"adhesion-inducing agents," and the like. These agents promote fibrosis
through one or more mechanisms including, for example, inducing or promoting
angiogenesis, stimulating migration or proliferation of connective tissue
cells
(such as fibroblasts, smooth muscle cells, vascular smooth muscle cells),
inducing extracellular matrix (ECM) production, and promoting tissue
remodeling. In addition, numerous therapeutic agents described herein can
have the additional benefit of promoting tissue regeneration (the replacement
of
injured cells by cells of the same type).
"Sclerosing" refers to a tissue reaction in which an irritant is
applied locally to a tissue that results in an inflammatory reaction and is
followed by scar tissue formation at the site of irritation. A pharmaceutical
agent that induces or promotes sclerosis is referred to as a "sclerosant," or
a
"sclerosing agent." Representative examples of sclerosants include ethanol,
dimethyl sulfoxide, surfactants (e.g., Triton X, sorbitan monolaurate,
sorbitan
sesquioleate, glycerol monostearate and polyoxyethylene, polyoxyethylene
cetyl ether, etc.), sucrose, sodium chloride, dextrose, glycerin, minocycline,
28
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
tetracycline, doxycycline, polidocanol, sodium tetradecyl sulfate, sodium
morrhuate, ethanolamine, phenol, sarapin and sotradecol.
"Radiographic guidance" refers to the placement of a drug
delivery cathether, medical device, implant, biomaterial, therapeutic agent,
access port or device and the like using medical imaging for guidance and to
confirm accurate placement. Imaging technology is used to allow manipulation
and intervention in a minimally invasive fashion (i.e., so as not to require
open
surgery). Any imaging technology can be used depending on the tissue being
treated, but includes, for example, x-ray, angiography, MRI, CT scanning,
ultrasound, PET scanning, and nuclear medicine scanning.
"Endoscopic guidance" refers to the placement of a drug delivery
cathether, medical device, implant, biomaterial, therapeutic agent, access
port
or device and the like using endoscopy for direct visualization of the target
tissue for guidance and to confirm accurate placement. Endoscopes are used
to allow direct visualization in a minimally invasive fashion (i.e., so as not
to
require open surgery) by inserting a small camera into the body via an orifice
(mouth, anus) or a small incision. Any endoscopic technology can be used
depending on the tissue being treated, but includes, for example, flexible
endoscopes, rigid endoscopes, gastroscopes, ERCP, bronchoscopes,
proctoscopes, angioscopes, and colonoscopes.
The terms "inter-react" and "inter-reaction" as used herein refer to
the formation of covalent bonds, noncovalent bonds, or both. The term thus
includes crosslinking, which involves both intermolecular crosslinks and
optionally intramolecular crosslinks as well, arising from the formation of
covalent bonds. Entanglement is another example of non-covalent bonding
that may result after inter-reaction between two or more reactive groups.
Covalent bonding between two reactive groups may bd direct in which case an
atom in reactive group is directly bound to an atom in the other reactive
group
or it may be indirect through a linking group. Noncovalent bonds include ionic
(electrostatic) bonds, hydrogen bonds, or the association of hydrophobic
29
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
molecular segments, which may be the same or different. A crosslinked matrix
may, in addition to covalent bonds, also include such intermolecular and/or
intramolecular noncovalent bonds.
When referring to polymers, the terms "hydrophilic" and
"hydrophobic" are generally defined in terms of an HLB value, i.e., a
hydrophilic
lipophilic balance. A high HLB value indicates a hydrophilic compound, while a
low HLB value characterizes a hydrophobic compound. HLB values are well
known in the art, and generally range from 1 to 18. Preferred multifunctional
compound cores are hydrophilic, although as long as the multifunctional
compound as a whole contains at least one hydrophilic component,
crosslinkable hydrophobic components may also be present.
The term "effective amount" refers to the amount of an agent or
composition that provides the effect desired. The actual amount that is
determined to be an effective amount will vary depending on factors such as
the size, general health and condition, sex and age of the patient and can be
more readily determined by the caregiver. The term "in situ" as used herein
means at the site of administration. Thus, agents and compositions described
can be delivered, injected, or otherwise applied to a specific site within a
patient's body, such as a diverticulum.
The term "aqueous medium" includes solutions, suspensions,
dispersions, colloids, and the like containing water. The term "aqueous
environment" means an environment containing an aqueous medium.
Similarly, the term "dry environment" means an environment that does not
contain an aqueous medium.
The terms "active agent," "biologically active agent," "therapeutic
agent," "pharmacologically active agent," and "drug" are used interchangeably
herein to refer to a chemical material or compound suitable for administration
to
a patient and that induces a desired effect. The terms include agents that are
therapeutically effective as well as prophylactically effective. Also included
are
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
derivatives and analogs of those compounds or classes of compounds
specifically mentioned that also induce the desired effect.
As discussed above, the present invention provides compositions,
implants, and methods for treating diverticular disease. Described in more
detail below are therapeutic agents; compositions and implants for delivering
the therapeutic agents; and methods for treating diverticulitis using the
agents
and compositions discussed herein.
Therapeutic Agents
Numerous therapeutic agents (also referred to herein as
'therapeutic agents' or 'drugs') have been identified that can be used to
induce
scarring and closure of a diverticula. The agent may be formulated with one or
more other materials, e.g., a polymeric carrier, which formulations are
discussed below. Many suitable therapeutic agents are specifically identified
herein, and others may be readily determined based upon in vitro and in vivo
(animal) models such as those provided in the Examples. Therapeutic agents
that promote fibrosis can be identified through in vivo models such as the rat
carotid artery model. One or more therapeutic agents may be introduced into a
host for treatment of diverticular disease. A host may be a mammal, which may
be a human (such as a patient or subject in need of treatment or a non-human
mammal. Exemplary non-human mammals include, but are not limited to, a
non-human primate, a rodent (e.g., rat, mouse, rabbit, hamster), a cat, dog,
horse, pig, bovine, sheep, or goat. A host in need of treatment is a host who
has developed or is at risk for developing a diverticular disease.
Fibrosis-Inducing Agents
Within one embodiment of the invention, a fibrosis-inducing
pharmacologic agent or an implant adapted to include or to release an agent
that induces fibrosis is administered onto or into diverticula. Thus, in one
embodiment, a medical implants is provided that comprises at least one of (i)
a
31
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
fibrosis-inducing agent (fibrosing agent) and (ii) a composition that
comprises a
fibrosis-inducing agent. When placed within diverticula, the fibrosing agent
is
capable of inducing fibrosis formation that would otherwise not occur. In
another embodiment, methods are provided for inducing a fibrosis in a
diverticulum and for treating a diverticular disease, wherein a fibrosis-
inducing
agent and/or an implant/composition that comprises a fibrosis-inducing agent,
are placed into a host (e.g., a mammal) having diverticula.
As discussed in further detail herein, a therapeutic agent includes
a fibrosis-inducing agent that is a diverticular wall irritant, for example,
talcum
powder, metallic beryllium and oxides thereof, copper, silk, coated silk
sutures,
uncoated silk sutures, virgin silk, degummed silk, saracin, silica,
crystalline
silicates, talc, quartz dust, and ethanol; a component of extracellular
matrix;
fibronectin, collagen, fibrin, or fibrinogen. A fibrosis-inducing agent may
also be
poly(ethylene terephthalate (Dacron), polylysine, poly(ethylene-co-
vinylacetate),
chitosan, N-carboxybutylchitosan, an RGD protein, or a polymer of vinyl
chloride. Therapeutic agents include adhesives, such as cyanoacrylates and
crosslinked poly(ethylene glycol) ¨ methylated collagen and may also include
an inflammatory cytokine (e.g., TGF8, PDGF, VEGF, bFGF, TNFa, NGF, GM-
CSF, IGF-a, IL-1, IL-8, 1L-6, and growth hormone); connective tissue growth
factor (CTGF); a bone morphogenic protein (BMP) (e.g., BMP-2, BMP-3, BMP-
4, BMP-5, BMP-6, or BMP-7); and bleomycin or an analogue or derivative
thereof. A fibrosis-inducing agent also includes a proliferative agent that
stimulates cellular proliferation, for example, dexamethasone, isotretinoin,
17-13-
estradiol, estradiol, diethylstibesterol, cyclosporine A, all-trans retinoic
acid
(ATRA), and analogues and derivatives thereof.
In one embodiment, the fibrosis or adhesion-inducing agent
suitable for use in the treatment of diverticula is silk. Silk refers to a
fibrous
protein, and may be obtained from a number of sources, typically spiders and
silkworms. Typical silks contain about 75% of actual fiber, referred to as
fibroin,
and about 35% sericin, which is a gummy protein that holds the filaments
32
CA 02610948 2012-12-14
together. Silk filaments are generally very fine and long - as much as 300-900
meters long. There are several species of domesticated silkworm that are used
in commercial silk production, however, Bombyx mori is the most common, and
most silk comes from this source. Other suitable silkworms include Philosamia
cynthia ricini, Antheraea yamamai, Antheraea pemyi, and Antheraea mylitta.
Spider silk is relatively more difficult to obtain, however, recombinant
techniques hold promise as a means to obtain spider silk at economical prices
(see, e.g., U.S. Patent Nos. 6,268,169; 5,994,099; 5,989,894; and 5,728,810,
which are exemplary only). Biotechnology has allowed researchers to develop
other sources for silk production, including animals (e.g., goats) and
vegetables
(e.g., potatoes). Silk from any of these sources may be used in the present
invention.
A commercially available silk protein is available from Croda, Inc.,
of Parsippany, N.J., and is sold under the trade marks CROSILK LIQUID (silk
amino acids), CROSILK 10,000 (hydrolyzed silk), CROSILK POWDER*
(powdered silk), and CROSILKQUAT (cocodiammonium hydroxypropyl silk
amino acid). Another example of a commercially available silk protein is
SERICIN, available from Pentapharm, LTD, a division of Kordia, By, of the
Netherlands. Further details of such silk protein mixtures can be found in
U.S.
Patent. No. 4,906,460, to Kim, et al., assigned to Sorenco. Silk useful in the
present invention includes natural (raw) silk, hydrolyzed silk, and modified
silk,
i.e., silk that has undergone a chemical, mechanical, or vapor treatment,
e.g.,
acid treatment or acylation (see, e.g., U.S. Patent No. 5,747,015).
Raw silk is typically twisted into a strand sufficiently strong for
weaving or knitting. Four different types of silk thread may be produced by
this
procedure: organzine, crepe, tram and thrown singles. Organzine is a thread
made by giving the raw silk a preliminary twist in one direction and then
twisting
two of these threads together in the opposite direction. Crepe is similar to
organzine but is twisted to a much greater extent. Twisting in only one
direction
two or more raw silk threads makes tram. Thrown singles are individual raw
*Trade mark
33
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
silk threads that are twisted in only one direction. Any of these types of
silk
threads may be used in the present invention.
The silk used in the present invention may be in any suitable form
that allows the silk to be joined with the medical implant applied to the
diverticula, for example, the silk may be in thread or powder-based forms. The
silk can be prepared in the powdered form by several different methods. For
example the silk can be milled (e.g., cryomill) into a powdered form.
Alternatively the silk can be dissolved in a suitable solvent (e.g., HFIP or
9M
LiBr) and then sprayed (electrospray, spray dry) or added to a non-solvent to
produce a powder. Furthermore, the silk may have any molecular weight,
where various molecular weights are typically obtained by the hydrolysis of
natural silk, where the extent and harshness of the hydrolysis conditions
determines the product molecular weight. For example, the silk may have an
average (number or weight) molecular weight of about 200 to 5,000. See, e.g.,
JP-B-59-29199 (examined Japanese patent publication) for a description of
conditions that may be used to hydrolyze silk.
A discussion of silk may be found in the following documents,
which are exemplary only, Hinman, M.B., et al. "Synthetic spider silk: a
modular
fibre" Trends in Biotechnology, 2000, 18(9) 374-379; Vollrath, F. and Knight,
D.P. "Liquid crystalline spinning of spider silk" Nature, 2001, 410(6828) 541-
548; and Hayashi, C.Y., et al. "Hypotheses that correlate the sequence,
structure, and mechanical properties of spider silk proteins" Int. J. Biol.
Macromolecules, 1999, 24(2-3), 265-270; and U.S. Patent No. 6,427,933.
The silk may be virgin silk, partially degummed, or degummed
silk. The silk can also further comprise a coating. The coating may be a
silicone-based coating, a wax based coating, or a degradable polymer based
coating.
In another embodiment, the fibrosis-inducing agent is a fibroin
protein, or a fragment or fragments thereof. In a certain embodiment, the
34
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
fibroin protein may be a synthetic analogue that is made and that has one or
more of the known repeat sequences of the fibroin protein.
In another embodiment, the fibrosing agent is sarecin. Sarecin is
a component of virgin silk that can be used to assist in the induction of a
fibrotic
response.
Other representative examples of fibrosis and adhesion-inducing
agents (fibrosing agents) suitable for administration to diverticula include
irritants (e.g., talc, talcum powder, copper, metallic beryllium (or its
oxides),
wool (e.g., animal wool, wood wolol, and synthetic wool), cotton, quartz dust,
silica, crystalline silicates), polymers (e.g., polylysine, polyurethanes,
poly(ethylene terephthalate), polytetrafluoroethylene (PTFE),
poly(alkylcyanoacrylates), and poly(ethylene-co-vinylacetate)); polymers of
vinyl
chloride; peptides with high lysine content; growth factors and inflammatory
cytokines involved in angiogenesis, fibroblast migration, fibroblast
proliferation,
ECM synthesis and tissue remodeling, such as epidermal growth factor (EGF)
family, transforming growth factor-a (TGF- a), transforming growth factor-8
(TGF-8-1, TGF-8-2, TGF-8-3, platelet-derived growth factor (PDGF), fibroblast
growth factor (acidic ¨ aFGF; and basic - bFGF), fibroblast stimulating factor-
1,
activins, vascular endothelial growth factor (including VEGF-2, VEGF-3, VEGF-
A, VEGF-B, VEGF-C, placental growth factor - P1GF), angiopoietins, insulin-
like
growth factors (IGF), hepatocyte growth factor (HGF), connective tissue growth
factor (CTGF), myeloid colony-stimulating factors (CSFs), monocyte
chemotactic protein, granulocyte-macrophage colony-stimulating factors (GM-
CSF), granulocyte colony-stimulating factor (G-CSF), macrophage colony-
stimulating factor (M-CSF), erythropoietin, interleukins (particularly IL-1,
1L-8,
and IL-6), tumor necrosis factor-a (TNFa), nerve growth factor (NGF),
interferon-a, interferon-13, histamine, endothelin-1, angiotensin II, growth
hormone (GH), and synthetic peptides, analogues or derivatives of these
factors are also suitable for release from specific implants described herein.
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Other examples of fibrosing agents include CTGF (connective
tissue growth factor); inflammatory microcrystals (e.g., crystalline minerals
such
as crystalline silicates); bromocriptine, methylsergide, methotrexate,
chitosan,
N-carboxybutyl chitosan, carbon tetrachloride, thioacetamide, fibrosin,
ethanol,
bleomycin, naturally occurring or synthetic peptides containing the Arg-Gly-
Asp
(RGD) sequence, generally at one or both termini (see, e.g., U.S. Patent No.
5,997,895), and tissue adhesives, such as cyanoacrylate and crosslinked
poly(ethylene glycol) ¨ methylated collagen compositions, such as described
below. Other examples of fibrosis-inducing agents include bone morphogenic
proteins (e.g., BMP-2, BMP-3, BMP-4, BMP-5, BMP-6 (Vgr-1), BMP-7 (0P-1),
BMP-8, BMP-9, BMP-10, BMP-11, BMP-12, BMP-13, BMP-14, BMP-15, and
BMP-16. Of these, BMP-2, BMP-3, BMP-4, BMP-5, BMP-6, and BMP-7 are of
particular utility. Bone morphogenic proteins are described, for example, in
U.S. Patent Nos. 4,877,864; 5,013,649; 5,661,007; 5,688,678; 6,177,406;
6,432,919; and 6,534,268 and Wozney, J.M., et al. (1988) Science: 242(4885);
1528-1534.
As described above, one fibrosis-inducing agent is a wool. The
term "wool" refers to an entangled mass of fibers without any ordered
arrangement, while the term "fiber" refers to a particle with a length to
diameter
ratio ("aspect ratio") of at least about 3:1 and roughly parallel edges.
Wool that may be used in the compositions and methods
described herein induces an enhanced fibrotic response between the medical
implant and the tissue adjacent to the in vivo medical implant. In other
words,
absent the wool, the medical implant would generate a "normal" adhesion
between the adjacent tissue and the medical implant, while in the presence of
the wool, the same medical implant is capable of generating an enhanced
adhesion (e.g., via an enhanced matrix deposition response to the presence of
the wool).
Wool useful as a fibrosis inducing agent may be obtained or
prepared from natural sources (e.g., animal wool and wood wool).
36
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Alternatively, it may be artificially synthesized (e.g., polymeric wool and
mineral
wool).
"Animal wool" refers to animal hair fibers, typically derived from
the fleece of sheep or lamb, goat (e.g., Angora and Cashmere), camel, alpaca,
llama, vicuna, or the like. Animal wool is a dead tissue that has a complex
morphological and chemical structure, which make it unique among textile
fibers. Morphologically, wool fibers are biological composites, with each
component having a different physical and chemical composition. Wool fibers
are generally composed of three different types of spindle-shaped cortical
cells
surrounded by a sheath of overlapping, rectangular cell known as the cuticle,
which forms the external layer of the fiber. Approximately 90% of the cortical
cell type is made up of longitudinally arrayed intermediate filaments with
accompanying matrix. The remainder includes membranes and remnants from
the nucleus and cytoplasm.
Animal wool fibers exhibit a range of diameters, lengths, and
crimp (i.e., a measure of fiber curvature), which allows the wool fibers to
entrap
air. Animal wool is also hygroscopic and is able to absorb and desorb large
amounts of water as the relative humidity surrounding the fiber changes.
Furthermore, animal wool liberates heat if it absorbs water. These properties
contribute to animal wool's extraordinary insulating quality.
Animal wool belongs to a family of proteins called a-keratins,
which also include materials such as hooves, horns, nails, claws, and beak. A
characteristic feature of a-keratins (also referred to as "hard" keratins) is
a
higher concentration of sulfur than "soft" keratins, such as those in the
skin.
Clean animal wool contains about 82% keratinous proteins that are high in
sulfur content, and about 17% of the fiber is protein with a relatively low
sulfur
content (<3%). The sulfur in wool occurs in the form of the amino acid
cysteine.
Due to the high cysteine content, animal wool is highly cross-linked by
disulfide
bonds that render it essentially insoluble. It is estimated that animal wool
contains about 170 different types of polypeptides varying in relative
molecular
37
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
mass from below 10,000 to greater than 50,000. The groups of proteins that
constitute animal wool are not uniformly distributed throughout the fiber but
are
aggregated within various regions. Animal wool also contains about 1% non-
proteinaceous material that consists mainly of free and structural lipids and
polysaccharide materials, trace elements, and pigments (e.g., melanin).
Animal wool is usually harvested from animals by annual
shearing. Thus, the fiber length is determined largely by the rate of growth,
which in turn depends on both genetic and environmental factors. For instance,
typical merino fibers are 50-125 mm long and have irregular crimp (curvature).
Animal wool fibers exhibit a range of diameters, which also depend on both
genetics and environment. For example, coarse wool fibers generally have a
diameter of 25-70 mm, while fine merino fibers typically have a diameter of 10-
25 mm.
Another example of a naturally derived wool is wood wool, which
is a specially prepared, non-compressed wood fiber frequently used in surgical
dressings and packaging materials. Wood wool fibers also can be obtained
from pine needles.
Although wool is usually associated with fibers derived from
natural sources, a variety of synthetic wool is also available. Synthetic wool
includes, for example, mineral wools, such as glass wool, stone wool, and slag
wool, and wool made from polymeric materials. Mineral wool may be formed,
for example, from a molten, inorganic material such as glass, stone, or slag
that
is spun into a fiber-like structure. Inorganic rock or slag is the main
component
(typically 98%) of stone wool. The remaining 2% organic content is generally a
thermosetting resin binder (an adhesive) and a small amount of oil. Glass wool
products usually contain about 95% inorganic material. Glass wool is made
from sand or recycled glass, limestone, and soda ash,which are the same
ingredients used for familiar glass objects such as window panes or glass
bottles. Glass fiber may, additionally, include a small amount of boron. Stone
'38
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
wool can be made from volcanic rock, typically basalt or dolomite. Slag wool
is
made from blast furnace slag (waste).
A discussion of wool may be found in the following documents,
which are exemplary only: Encyclopedia of Polymer Science and Technology,
John Wiley & Sons, Inc. (2003); Dowling and Sparrow, TIBS 16:115-118
(1991); Powman, J. Chromatogr. B 787:63-76 (2003); Hearle, Intl. J. Biol.
Macromol. 27:123-38 (2000); and Vuyst et al., Eur. Resp. J. 8:2149-73 (1995).
In certain embodiments, wool fibers have an average length of
about, or at least about, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35,
40, 45, 50
mm or longer. In certain embodiments, the lengths of wool fibers are in a
range
of about 1-5 mm, 5-10 mm, 10-50 mm, 50-100 mm, 1-10 mm, 1-50 mm, or 1-
100 mm. In certain embodiments, wool fibers have an average diameter of
about, or at least about, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35,
40, 45, or
50 mm. In certain embodiments, the diameters of wool fibers are in a range of
about 1-3 mm, 3-5 mm, 5-10 mm, 10-50 mm, 1-10 mm, or 1-50 mm. In certain
embodiments, the average length to diameter ratio of wool fibers is 3:1, 4:1,
5:1, 6:1, 7:1, 8:1, 9:1, 10:1 or larger.
In certain embodiments, wool may be further processed into other
forms or shapes, for example, sheet, powder, thread, braid, filament, fiber,
film,
foam, and the like. In certain embodiments, the wool is further processed into
threads or powder. In certain other embodiments, the wool is further processed
into the form of a spiral or coil.
Wool may be used alone or may be used in combination with a
medical implant, such as described herein. In certain embodiments, wool may
be used in combination with one or more of other fibrosis inducing agents
described herein. Wool may be secured to a medical implant by any of a
number of methods. Suitable methods include, without limitation, interweaving
the wool into the implant, interweaving the wool into the implant structure;
attaching the wool to the implant via knotting or suturing it around the
implant
structure; attaching the wool to the medical implant by means of an adhesive;
39
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
and using one or more sutures to "sew" the wool onto the medical implant. In
one aspect, a plurality of separated wool braids or threads is attached to the
medical implant.
In one embodiment, the wool is secured only to the outside of the
medical implant. In another embodiment, the wool is secured to distal regions
of the medical implant. The wool may be attached to the implant portion of the
medical implant, or it may be attached to the implant portion of the medical
implant, or it may be attached to both the implant and implant portions of the
medical implant.
The wool threads can be located on the implant in various
configurations that may result in either partial or complete coverage of the
exterior of the implant. The threads could be attached around the ends of the
implant. The wool threads can be attached in bands along the medical implant.
The attachment could be in a vertical, horizontal, or diagonal manner.
Depending on the specific design of the medical implant, the polymeric
thread(s) can be attached to either the implant component or the implant
component of the medical implant. Alternatively, or in addition, the wool
thread
may be allowed to extend some distance from the medical implant. For
,example, in certain embodiments, only one end of the wool threads may be
secured to the medical implant, thereby allowing the other end of the thread
to
extend away from the implant. Alternatively, both ends of the thread may be
secured to a medical implant, however, the mid-portion of the thread is not
secured to the medical implant, and the ends of the thread are secured at a
sufficiently short distance from one another that the mid-portion is free to
extend away from the medical implant.
In another embodiment, the ends of the wool threads can be
attached to the medical implant, and/or one or more points along the wool
thread can be attached to the medical implant. In yet another embodiment, the
ends of the wool thread are not attached to the medical implant. Rather, one
or
more points along the wool thread are attached to the medical implant. In yet
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
another embodiment, the wool thread(s) can be made into a preformed
structure (e.g., mesh, looped bundle, and the like) that is then attached to
the
medical implant.
Other representative examples of fibrosis-inducing agents suitable
for the induction of fibrosis within a divertiuculum include crosslinked
compositions that comprise amino-functional groups. For example, amino-
functionalized polyethylene glycol (e.g., 4-armed tetra-amino PEG [1011) can
be
reacted with a 4-armed NHS functionalized PEG (e.g., pentaerythritol
poly(ethylene glycol)ether tetra-succinimidyl glutarate) under basic buffer
conditions. In another example a 4-armed thiol functionalized PEG (e.g.,
pentaerythritol poly(ethylene glycol)ether tetra-thiol) can be substituted for
the
4-arm amino-functionalized PEG such that the amount of amino functional
groups in the final composition can be varied. These reagents can be mixed at
the time of application to provide an in situ forming crosslinked hydrogel.
These
reagents could be premixed to produce the crosslinked material. The material
can be made in various forms such as rods, tubes, films, meshes, screens,
threads, fibers, slabs, or spheres. The crosslinked material could also be
milled
to produce a particulate material. These materials can be dried (e.g., air,
vacuum, freeze-dried) and used as a dry powdered material. Alternatively the
materials can be hydrated just prior to application. These materials can
further
comprise one of the fibrosis-inducing agents described herein.
Other representative examples of fibrosis-inducing agents of
in the management of diverticular disease include components of extracellular
matrix (e.g., fibronectin, fibrin, fibrinogen, collagen (e.g., bovine
collagen),
fibrillar and non-fibrillar collagen, adhesive glycoproteins, proteoglycans
(e.g.,
heparin sulfate, chondroitin sulfate, dermatan sulfate), hyaluronan, secreted
protein acidic and rich in cysteine (SPARC), thrombospondins, tenacin, and
cell
adhesion molecules (including integrins, vitronectin, fibronectin, laminin,
hyaluronic acid, elastin, bitronectin), proteins found in basement membranes,
and fibrosin) and inhibitors of matrix metalloproteinases, such as T1MPs
(tissue
=
41
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
inhibitors of matrix metalloproteinases) and synthetic TIMPs, e.g.,
marimistat,
batimistat, doxycycline, tetracycline, minocycline, TROCADE, Ro-1130830,
CGS 27023A, and BMS-275291.
In separate embodiments, the agent is a diverticular vessel wall
irritant; the fibrosing agent is or comprises silk; the fibrosing agent is or
comprises silkworm silk; the fibrosing agent is or comprises spider silk; the
fibrosing agent is or comprises recombinant silk; the fibrosing agent is or
comprises raw silk; the fibrosing agent is or comprises hydrolyzed silk; the
fibrosing agent is or comprises acid-treated silk; the fibrosing agent is or
comprises acylated silk; the fibrosing agent is in the form of strands; the
fibrosing agent is in the form of tufts; the fibrosing agent is in the form of
microparticulates; the fibrosing agent is or comprises mineral particles; the
fibrosing agent is or comprises talc; the fibrosing agent is or comprises
chitosan; the fibrosing agent is or comprises polylysine; the fibrosing agent
is or
comprises fibronectin; the fibrosing agent is or comprises bleomycin; the
fibrosing agent is or comprises CTGF; the fibrosing agent is in the form of a
thread, or is in contact with a thread. Optionally, the thread is
biodegradable
(e.g., it comprises a material selected from the group consisting of
polyester,
polyanhydride, poly(anhydride ester), poly(ester-amide), poly(ester-urea),
polyorthoester, polyphosphoester, polyphosphazine, polycyanoacrylate,
collagen, chitosan, hyaluronic acid, chromic cat gut, alginate, starch,
cellulose
and cellulose ester); or the thread is non-biodegradable (e.g., it comprises a
material selected from the group consisting of polyester, polyurethane,
silicone,
polyethylene, polypropylene, polystyrene, polyacrylate, polymethacrylate,
polyacrylics, polymethacrylics, and silk); the thread is coated with a
polymer,
the thread is coated with a pharmaceutical agent that induces a fibrotic
response in the patient, the thread is coated with a pharmaceutical agent that
induces an osteogenic response in the patient; the composition further
comprises an agent that promotes bone growth. The agent that promotes bone
growth is a bone morphogenic protein or the agent that promotes bone growth
42
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
is an osteogenic growth factor (e.g., transforming growth factor, platelet-
derived
growth factor, and fibroblast growth factor); the composition further
comprises a
pharmaceutical agent that induces sclerosis (a sclerosant, e.g., a sclerosant
is
selected from the group consisting of ethanol, dimethyl sulfoxide, sucrose,
sodium chloride, dextrose, glycerin, minocycline, tetracycline, doxycycline,
polidocanol, sodium tetradecyl sulfate, sodium morrhuate, and sotradecol, or
the sclerosant may be a surfactant); the composition further comprises an
inflammatory cytokine (e.g., an inflammatory cytokine selected from the group
consisting of TGFI3, PDGF, VEGF, bFGF, TNFa, NGF, GM-CSF, IGF-a, IL-1,
IL-1-8, IL-8, IL-6, and growth hormone); the composition further comprises an
agent that stimulates cell proliferation (e.g., a cell proliferation agent
selected
from the group consisting of dexarnethasone, isotretinoin (13-cis retinoic
acid),
1713-estradiol, estradiol, 1-a-25 dihydroxyvitamin D3, diethylstibesterol,
cyclosporine A, L-NAME, all-trans retinoic acid (ATRA), and analogues and
derivatives thereof); the composition further comprises a bulking agent; the
composition further comprises a sealant; the composition further comprises a
polymeric carrier; the composition further comprises a resorbable ceramic; the
composition further comprises a contrast agent; the composition further
comprises a thread; the composition is in the form of a tuft; the composition
is in
the form of a gel; the composition is in the form of a paste; the composition
is in
the form of a spray; the composition is in the form of an aerosol; the
composition is in the form of a suspension; the composition is in the form of
an
emulsion or microemulsion; the composition is in the form of a microsphere;
the
composition is in the form of a microparticulate; the composition is in the
form of
a solid implant.
Within various embodiments of the invention, an implant or
composition may include an agent that promotes fibrosis and a second
composition or compound which acts to have an inhibitory effect on
pathological processes in or around the treatment site. Representative
examples of agents which can inhibit pathological processes (e.g.,
inflammation
43
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
associated with diverticultits) within the diverticula treatment site include,
but
not limited to, the following classes of compounds: anti-inflammatory agents
(e.g., dexamethasone, cortisone, fludrocortisone, prednisone, prednisolone, 6a-
methylprednisolone, triamcinolone, and betamethasone); matrix
metalloproteinase (MMP) inhibitors (e.g., marimistat, batimistat, TIMP's
representative examples of which are included in U.S. Patent Nos. 5,665,777;
5,985,911; 6,288,261; 5,952,320; 6,441,189; 6,235,786; 6,294,573; 6,294,539;
6,563,002; 6,071,903; 6,358,980; 5,852,213; 6,124,502; 6,160,132; 6,197,791;
6,172,057; 6,288,086; 6,342,508; 6,228,869; 5,977,408; 5,929,097; 6,498,167;
6,534,491; 6,548,524; 5,962,481; 6,197,795; 6,162,814; 6,441,023; 6,444,704;
6,462,073; 6,162,821; 6,444,639; 6,262,080; 6,486,193; 6,329,550; 6,544,980;
6,352,976; 5,968,795; 5,789,434; 5,932,763; 6,500,847; 5,925,637; 6,225,314;
5,804,581; 5,863,915; 5,859,047; 5,861,428; 5,886,043; 6,288,063; 5,939,583;
6,166,082; 5,874,473; 5,886,022; 5,932,577; 5,854,277; 5,886,024; 6,495,565;
6,642,255; 6,495,548; 6,479,502; 5,696,082; 5,700,838; 6,444,639; 6,262,080;
6,486,193; 6,329,550; 6,544,980; 6,352,976; 5,968,795; 5,789,434; 5,932,763;
6,500,847; 5,925,637; 6,225,314; 5,804,581; 5,863,915; 5,859,047; 5,861,428;
5,886,043; 6,288,063; 5,939,583; 6,166,082; 5,874,473; 5,886,022; 5,932,577;
5,854,277; 5,886,024; 6,495,565; 6,642,255; 6,495,548; 6,479,502; 5,696,082;
5,700,838; 5,861,436; 5,691,382; 5,763,621; 5,866,717; 5,902,791; 5,962,529;
6,017,889; 6,022,873; 6,022,898; 6,103,739; 6,127,427; 6,258,851; 6,310,084;
6,358,987; 5,872,152; 5,917,090; 6,124,329; 6,329,373; 6,344,457; 5,698,706;
5,872,146; 5,853,623; 6,624,144; 6,462,042; 5,981,491; 5,955,435; 6,090,840;
6,114,372; 6,566,384; 5,994,293; 6,063,786; 6,469,020; 6,118,001; 6,187,924;
6,310,088; 5,994,312; 6,180,611; 6,110,896; 6,380,253; 5,455,262; 5,470,834;
6,147,114; 6,333,324; 6,489,324; 6,362,183; 6,372,758; 6,448,250; 6,492,367;
6,380,258; 6,583,299; 5,239,078; 5,892,112; 5,773,438; 5,696,147; 6,066,662;
6,600,057; 5,990,158; 5,731,293; 6,277,876; 6,521,606; 6,168,807; 6,506,414;
6,620,813; 5,684,152; 6,451,791; 6,476,027; 6,013,649; 6,503,892; 6,420,427;
6,300,514; 6,403,644; 6,177,466; 6,569,899; 5,594,006; 6,417,229; 5,861,510;
44
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
6,156,798; 6,387,931; 6,350,907; 6,090,852; 6,458,822; 6,509,337; 6,147,061;
6,114,568; 6,118,016; 5,804,593; 5,847,153; 5,859,061; 6,194,451; 6,482,827;
6,638,952; 5,677,282; 6,365,630; 6,130,254; 6,455,569; 6,057,369; 6,576,628;
6,110,924; 6,472,396; 6,548,667; 5,618,844; 6,495,578; 6,627,411; 5,514,716;
5,256,657; 5,773,428; 6,037,472; 6,579,890; 5,932,595; 6,013,792; 6,420,415;
5,532,265; 5,639,746; 5,672,598; 5,830,915; 6,630,516; 5,324,634; 6,277,061;
6,140,099; 6,455,570; 5,595,885; 6,093,398; 6,379,667; 5,641,636; 5,698,404;
6,448,058; 6,008,220; 6,265,432; 6,169,103; 6,133,304; 6,541,521; 6,624,196;
6,307,089; 6,239,288; 5,756,545; 6,020,366; 6,117,869; 6,294,674; 6,037,361;
6,399,612; 6,495,568; 6,624,177; 5,948,780; 6,620,835; 6,284,513; 5,977,141;
6,153,612; 6,297,247; 6,559,142; 6,555,535; 6,350,885; 5,627,206; 5,665,764;
5,958,972; 6,420,408; 6,492,422; 6,340,709; 6,022,948; 6,274,703; 6,294,694;
6,531,499; 6,465,508; 6,437,177; 6,376,665; 5,268,384; 5,183,900; 5,189,178;
6,511,993; 6,617,354; 6,331,563; 5,962,466; 5,861,427; 5,830,869; and
6,087,359), cytokine inhibitors (chlorpromazine, mycophenolic acid, rapamycin,
la-hydroxy vitamin D3), IMPDH (inosine monophosplate dehydrogenase)
inhibitors (e.g., mycophenolic acid, ribaviran, aminothiadiazole,
thiophenfurin,
tiazofurin, viramidine) (Representative examples are included in U.S. Patent,
Nos. 5,536,747; 5,807,876; 5,932,600; 6,054,472; 6,128,582; 6,344,465;
6,395,763; 6,399,773; 6,420,403; 6,479,628; 6,498,178; 6,514,979; 6,518,291;
6,541,496; 6,596,747; 6,617,323; and 6,624,184, U.S. Patent Application Nos.
2002/0040022A1, 2002/0052513A1, 2002/0055483A1, 2002/0068346A1,
2002/0111378A1, 2002/0111495A1, 2002/0123520A1, 2002/0143176A1,
2002/0147160A1, 2002/0161038A1, 2002/0173491A1 , 2002/0183315A1,
2002/0193612A1, 2003/0027845A1, 2003/0068302A1, 2003/0105073A1,
2003/0130254A1, 2003/0143197A1, 2003/0144300A1, 2003/0166201A1,
2003/0181497A1, 2003/0186974A1, 2003/0186989A1, and 2003/0195202A1,
and PCT Publication Nos. WO 00/24725A1, WO 00/25780A1, WO 00/26197A1,
WO 00/51615A1, WO 00/56331A1, WO 00/73288A1, WO 01/00622A1, WO
01/66706A1, WO 01/79246A2, WO 01/81340A2, WO 01/85952A2, WO
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
02/16382A1, WO 02/18369A2, WO 02/051814A1, WO 02/057287A2, WO
02/057425A2, WO 02/060875A1, WO 02/060896A1, WO 02/060898A1, WO
02/068058A2, WO 03/020298A1, WO 03/037349A1, WO 03/039548A1, WO
03/045901A2, WO 03/047512A2, WO 03/053958A1, WO 03/055447A2, WO
03/059269A2, WO 03/063573A2, WO 03/087071A1, WO 99/001545A1, WO
97/40028A1, WO 97/41211A1, WO 98/40381A1, and WO 99/55663A1), p38
MAP kinase inhibitors (MAPK) (e.g., GW-2286, CGP-52411, BIRB-798,
SB220025, RO-320-1195, RWJ-67657, RWJ-68354, SC10-469)
(Representative examples are included in U.S. Patent Nos. 6,300,347;
6,316,464; 6,316,466; 6,376,527; 6,444,696; 6,479,507; 6,509,361; 6,579,874,
and 6,630,485, and U.S. Patent Application Publication Nos. 2001/0044538A1,
2002/0013354A1, 2002/0049220A1, 2002/0103245A1, 2002/0151491A1,
2002/0156114A1, 2003/0018051A1, 2003/0073832A1, 2003/0130257A1,
2003/0130273A1, 2003/0130319A1, 2003/0139388A1, 2003/0139462A1,
2003/0149031A1, 2003/0166647A1, and 2003/0181411A1, and PCT
Publication Nos. WO 00/63204A2, WO 01/21591A1, WO 01/35959A1, WO
01/74811A2, WO 02/18379A2, WO 02/064594A2, WO 02/083622A2, WO
02/094842A2,WO 02/096426A1, WO 02/101015A2, WO 02/103000A2, WO
03/008413A1, WO 03/016248A2, WO 03/020715A1, WO 03/024899A2, WO
03/031431A1, WO 03/040103A1, WO 03/053940A1, WO 03/053941A2, WO
03/063799A2, WO 03/079986A2, WO 03/080024A2, WO 03/082287A1, WO
97/44467A1, WO 99/01449A1, and WO 99/58523A1), and immunomodulatory
agents (rapamycin, everolimus, ABT-578, azathioprine azithromycin, analogues
of rapamycin, tacrolimus and derivatives thereof (e.g., EP 0184162B1 and
those described in U.S. Patent No. 6,258,823) and everolimus and derivatives
thereof (e.g., U.S. Patent No. 5,665,772). Further representative examples of
sirolimus analogues and derivatives include ABT-578 and those found in PCT
Publication Nos. WO 97/10502, WO 96/41807, WO 96/35423, WO 96/03430,
WO 96/00282, WO 95/16691, WO 95/15328, WO 95/07468, WO 95/04738,
WO 95/04060, WO 94/25022, WO 94/21644, WO 94/18207, WO 94/10843,
46
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
WO 94/09010, WO 94/04540, WO 94/02485, WO 94/02137, WO 94/02136, WO
93/25533, WO 93/18043, WO 93/13663, WO 93/11130, WO 93/10122, WO
93/04680, WO 92/14737, and WO 92/05179 and in U.S. Patent Nos. 6,342,507;
5,985,890; 5,604,234; 5,597,715; 5,583,139; 5,563,172; 5,561,228; 5,561,137;
5,541,193; 5,541,189; 5,534,632; 5,527,907; 5,484,799; 5,457,194; 5,457,182;
5,362,735; 5,324,644; 5,318,895; 5,310,903; 5,310,901; 5,258,389; 5,252,732;
5,247,076; 5,225,403; 5,221,625; 5,210,030; 5,208,241; 5,200,411; 5,198,421;
5,147,877; 5,140,018; 5,116,756; 5,109,112; 5,093,338; and 5,091,389.
Other examples of drugs that may be included in the compositions
and implants of the invention include tyrosine kinase inhibitors, such as
imantinib, ZK-222584, CGP-52411, CGP-53716, NVP-AAK980-NX, CP-
127374, CP-564959, PD-171026, PD-173956, PD-180970, SU-0879, and SKI-
606. Other examples of drugs that may be included in the compositions and
implants of the invention include MMP inhibitors such as nimesulide, PKF-241-
466, PKF-242-484, CGS-27023A, SAR-943, primomastat, SC-77964, PNU-
171829, AG-3433, PNU-142769, SU-5402, and dexlipotam. Other examples of
drugs that may be included in the compositions and implants of the invention
include p38 MAP kinase inhibitors such as CGH-2466 and PD-98-59. Other
examples of of drugs that may be included in the compositions and implants of
the invention include immunosuppressants such as argyrin B, macrocyclic
lactone, ADZ-62-826, CC1-779, tilomisole, amcinonide, FK-778, AVE-1726, and
MDL-28842. Other examples of cytokine inhibitors include TNF-484A, PD-
172084, CP-293121, CP-353164, and PD-168787. Other examples of drugs
that may be included in the compositions and implants of the invention include
include NFKB inhibitors, such as, AVE-0547, AVE-0545, and IPL-576092.
Other examples of drugs that may be included in the compositions and implants
of the invention include include HMGCoA reductase inhibitors, such as,
pravestatin, atorvastatin, fluvastatin, dalvastatin, glenvastatin,
pitavastatin, CP-
83101, U-20685, apoptosis antagonist (e.g., troloxamine, TCH-346 (N-methyl-
N-propargy1-10-aminomethyl-dibenzo(b,Doxepin), caspase inhibitors (e.g., PF-
47
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
5901 (benzenemethanol, alpha-penty1-3-(2-guinolinylmethoxy)-), and JNK
inhibitor (e.g., AS-602801).
Within various embodiments, a diverticular implant or composition
incorporates or is coated with a composition that promotes fibrosis, as well
as a
composition or compound that acts to stimulate cellular proliferation.
Representative examples of agents that stimulate cellular proliferation
include,
pyruvic acid, naltrexone, leptin, D-glucose, insulin, amlodipine, alginate
oligosaccharides, minoxidil, dexamethasone, isotretinoin (13-cis retinoic
acid),
17-3-estradiol, estradiol, 1-a-25 dihydroxyvitamin D3, diethylstibesterol,
cyclosporine A, L-NAME (L-NG-nitroarginine methyl ester (hydrochloride)), all-
trans retinoic acid (ATRA), and analogues and derivatives thereof. Other
examples of agents that stimulate cellular proliferation include sphingosine 1-
phosphate receptor agonist (e.g., FTY-720 (1,3-propanediol, 2-amino-2-(2-(4-
octylphenypethyl)-,hydrochloride; immunostimulants, such as Imupedone
(methanone, [5-amino-2-(4-methy1-1-piperidinyl)phenyl](4-chlorophenyl)-,
DIAPEP227 synthetic peptide (Peptor Ltd., Israel)); and nerve growth factor
agonist, e.g., NG-012 (5H,9H,13H,21H,25H,-dibenzo[k,u][1,5,9,15,19]
pentaoxacyclotetracosin-5,9,13,21,25-pentone, 7,8,11,12,15,16,23,24,27,28-
decahydro-2 ,4,18,20-tetrahydroxy-11-(hydroxymethyl)-7,15,23,27-tetrarnethyl-,
NG-121, SS-701 (2,2':6',2"-terpyridine, 4'-(4-methylpheny1)-,
trihydrochloride,
AMPAlex (piperidine, 1-(6-guinoxalinylcarbonyI)-, RGH-2716 (844,4-bis(4-
fluorophenyl)buty1]-3-(1,1-dimethylethyl)-4-methylene-1-oxa-3,8-diaza-
spiro[4.5]
decan-2-one, and TDN-345 (1-oxa-3,8-diazaspiro[4.5]decan-2-one, 844,4-
bis(4-fluorophenyl)buty11-3-(1,1-dimethylethyl)-4-methylene-).
Haemostatic Agents
For managing bleeding, including lower gastrointestinal
hemorrhage, which is the result of diverticular bleeding, the diverticular
implant
or composition may include a fibrosing agent and a haemostatic agent (a
thrombotic or clotting agent) that promotes clotting and hemostatis upon
48
CA 02610948 2012-12-14
implantation of a medical implant (or composition). A haemostat or haemostatic
agent is any agent that arrests chemically or mechanically the flow of blood
from an open vessel. A haemostatic agent can be applied directly to a bleeding
site, and the agent functions in the presence of actively flowing blood.
Accordingly, a haemostatic agent may chemically or biologically arrest the
flow
of blood by interfering with one or more steps in the clotting cascade, such
as
by accelerating the clotting mechanism (e.g., COSTASIS). The increased
clotting then serves as a physical barrier to the flow of blood. Alternatively
a
haemostatic agent may in a more direct manner mechanically or physically
block the flow of blood (e.g., COSEAL) and thus may be referred to as a
sealant (an agent used to prevent leakage of liquids gases or solids). A
haemostatic agent may be applied or claped to a tissue surface to provide a
barrier to the flow of blood.
Within various embodiments, a diverticular implant contains a
haemostatic agent or composition comprising a haemostatic agent and/or an
agent or composition that promotes fibrosis. In certain alternative
embodiments, an implant is coated on one aspect with a composition that
promotes fibrosis, as well as being coated with a composition or agent that is
hemostatic on another aspect of the implant. Representative examples of
hemostatic agents include fibrin; aminocaproic acid; tranexamic acid;
aprotinin;
desmopressin; vitamin K; Tisseel0 and FloSeal (which are fibrinogen-
containing formulations) (Baxter Healthcare Corp., Glendale, CA); CrossSeal
(American Red Cross); CoSeal() (PEG-containing formulation) and CoStasis
(collagen-containing formulation) (Angiotech BioMaterials Corp., Palo Alto,
CA);
and CryoSeal AHS (Thermogenesis, Sacramento, CA).
Anti-Infective Agents
In one embodiment, a composition and medical implant is
provided that includes a fibrosing agent and an anti-infective agent, which
reduces the likelihood of infections in medical implants. Infection is a
common
49
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
complication that results from the implantation of foreign bodies such as
medical devices and implants into a host or patient. Foreign materials provide
an ideal site for microorganisms to attach and colonize. In addition,
according
to non-limiting theory, the risk that a host will develop an infection may be
increased as a consequence of an impairment of host defenses against
infection in the microenvironment surrounding a foreign material. These
factors
make medical implants particularly susceptible to infection.
A composition for treating and/or managing diverticular infection
(diverticulitis) may include an anti-infective agent, antibiotic, or other
agent that
inhibits (impairs or decreases) the growth or division rate or kills a
microorganism (for example, bacteria and yeast). Diverticulitis can occur when
inspissated stool, a fecalith trapped within a diverticulum, results in local
infection (or abscess formation) within the diverticulum. When severe, this
can
lead to perforation and the formation of generalized peritonitis. Even in the
management of diverticula that are not acutely infected, diverticula lumen may
be sterilized such that bacteria are not contained within the developing scar
tissue. This will lessen the possibility that an infection or abscess will
develop
at a later point in time.
An anti-infective agent refers to an agent that reduces the
likelihood of an infection. An agent is demonstrated to be an active anti-
infective agent toward a microorganism by assays routinely practiced by
persons skilled in the art, for example, an in vitro assay determining
inhibition of
bacterial growth as indicated by the M.I.C. (minimimum inhibitory
concentration). In certain embodiments, anti-infective agents are
chemotherapeutic agents that have antimicrobial activity at low doses (e.g.,
anthracyclines, fluoropyrimidines, folic acid antagonists, podophylotoxins,
camptothecins, hydroxyureas, and platinum complexes.
An anti-septic agent refers to an agent or substance that is
capable of effective antisepsis, that is, prevention of infection by
inhibiting the
growth of an infectious organism without necessarily killing the organism.
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Representative examples of anti-septic agents include chlorhexadine,
triclosan,
and chloroxylenol.
Antibiotic refers to an agent that kills or inhibits the growth of
microorganisms. Antibiotics may have a narrow or wide range of activity
against either one or both of Gram-positive and Gram-negative organisms.
Antibiotic agents can be identified through in vitro inhibition of bacterial
growth
as shown in the M.I.C. assay described herein. Representative examples of
antibiotics include gentamicin sulfate, amikacin sulfate, kanamycin sulfate,
polymyxin B, neomycin sulfate, cephazolin sodium, metronidazole,
Ciprofloxacin, piperacillin, Cefoxitin, Cefepime,Azithromycin, andTrimethoprom-
sulfamethoxazole.
Within various embodiments of the invention, a composition
comprises, or an implant is loaded or coated with a composition that promotes
fibrosis, as well as being loaded or coated with an an anti-infective agent
(e.g.,
antibiotic, chemotherapeutic agent, and/or antiseptic agent) or a composition
that includes an antibiotic (or antiseptic agent). Representative examples are
provided herein of agents such as chemotherapeutic agents that can be
released from a composition, and which have potent antimicrobial activity at
extremely low doses. A wide variety of anti-infective agents can be used in
combination with the present compositions. Suitable anti-infective agents may
be readily determined by methods practiced in the art and as exemplified in
the
assays provided in Example 41. Described in greater detail below are several
representative examples of anati-infective agents: (A) anthracyclines (e.g.,
doxorubicin and mitoxantrone); (B) fluoropyrimidines (e.g., 5-FU); (C) folic
acid
antagonists (e.g., nnethotrexate); (D) podophylotoxins (e.g., etoposide); (E)
camptothecins; (F) hydroxyureas; and (G) platinum complexes (e.g., cisplatin).
Anti-infective agents have the capability to prevent infection,
reduce the incidence of abscess formation, treat diverticulitis, and/or
contribute
to sterilization of the lumen of the diverticula during the scarring process.
Such
anti-infective agents include, but are not limited to antibiotics and
anticancer
51
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
agents. In addition, implants may be coated with antimicrobial drugs.
Representative examples of implants and coating of implants with antimicrobial
drugs are provided in U.S. Patent No. 5,520,664, U.S. Patent No. 5,709,672,
U.S. Patent No. 6,361,526, U.S. Patent No. 6,261,271, U.S. Patent No.
5,902,283, U.S. Patent No. 5,624,704, and U.S. Patent No. 5,709,672.
Anti-Infective Agents ¨ Antibiotics
Antibiotics and combinations of antibiotics that are used by those
skilled in the medical art include the following exemplary antibiotics: fourth
generation penicillins such as mezlocillin and piperacillin
(ureidopenicillins),
carbenicillin and ticarcillin (carboxypenicillins), and analogues and
derivatives
thereof; first generation cephalosporins such as cephazolin, Cephazolin
Sodium, Cephalexin (Keflex), Cefazolin (Ancef), Cephapirin (Cefadyl), and
Cephalothin (Keflin), and analogues and derivatives thereof; Ticarcillin;
second
generation cephalosporins such as Cefuroxime (Ceftin (oral) andZinocef),
Cefotetan (Cefotan), and Cefoxitin (Mefoxin), and analogues and derivatives
thereof; third generation cephalosporin such as Naxcel (Ceftiofur Sodium),
Cefdinir (Omnicef), Cefoperazone (Cefobid), Ceftazidime (Fortaz), and
Ceftriaxone (Rocephin), and Cefotaxime (Claforan), and analogues and
derivatives thereof; and fourth generation cephalosporins such as Cefepime
(Maxipime) and analogues and derivatives thereof; monobactams such as
aztreonam and analogues and derivatives thereof; carbapenems such as
imipenem, ertapenem and meropenem, and analogues and derivatives thereof.
Also included are inhibitors of protein synthesis such as aminoglycosides
including streptomycin, gentamicin, gentamicin sulfate, tobramycin, and
amikacin, amikacin sulfate, and analogues and derivatives thereof; inhibitors
of
protein synthesis such as the MSL group including macrolides (Erythromycin),
long acting macrolides (Azithromycin) and lincosamides (Clindamycin) and
streptogramins (Syneroid), clarithromycin, kanamycin, kanamycin sulfate, and
analogues and derivatives thereof. Other exemplary antibiotics include
52
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
inhibitors of DNA snthesis such as the quinolones including ciprofloxacin,
ofloxacin, gatifloxacin, moxifloxacin, levofloxacin, trovafloxacin, and
analogues
and derivatives thereof, as well as other inhibitors of DNA synthesis such as
metronidazole and analogues and derivatives thereof. Other antibiotics include
inhibitors of folate metabolism such as sulfonamides and trimethoprim, and
analogues and derivatives thereof. Additional agents include but are not
limited
to cefixime, spectinomycin, tetracycline, nitrofurantoin, doxycycline,
polymyxin
B, neomycin, neomycin sulfate, and analogues and derivatives thereof. In
certain embodiments, the anti-infective agent is gentamicin sulfate, amikacin
sulfate, kanamycin sulfate, polymyxin B, neomycin sulfate, cephazolin sodium,
metronidazole, ciprofloxacin, piperacillin, cefoxitin, cefepime, azithromycin,
or
trimethoprim-sulfamethoxazole.
Furthermore, additional therapeutic agents may be delivered in
combinations. Such combinations include, by way of example, but are not
limited to amoxicillin and clavulanate, ampicillin and sulbactam, trimethoprom-
sulfamethoxazole, ampicillin and probenecid, amoxicillin and probenecid,
penicillin G and probenecid, and penicillin and a penicillinase inhibitor.
Antibiotics described herein and used by those skilled in the
medical art may be administered orally (1-2 grams per day depending upon
factors such as age and/or weight and/or mass of the patient). As described
herein, one or more antibiotics may also be administered parenterally or an
antibiotic may be administered in a composition that includes the fibrosis
agent,
or may be administered in combination with an implant.
Anti-Infective Agents ¨ Chemotherapeutic Agents
Also provided herein are agents (e.g., chemotherapeutic agents)
that can be incorporated onto or into, or released from, an implant or a
composition implanted within a diverticulum, and which have potent
antimicrobial activity at extremely low doses. A wide variety of anti-
infective
agents can be used in combination with a fibrosing agent. Described in more
53
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
detail below are several representative examples of chemotehrapeutidanti-
infective agents: (A) anthracyclines (e.g., doxorubicin and mitoxantrone), (B)
fluoropyrimidines (e.g., 5-FU), (C) folic acid antagonists (e.g.,
methotrexate),
(D) podophylotoxins (e.g., etoposide), (E) camptothecins, (F) hydroxyureas,
and (G) platinum complexes (e.g., cisplatin).
(A) Anthracyclines
Anthracyclines have the following general structure, where the R
groups may be a variety of organic groups:
0
R6 0 R4
R1
R7
404000,,, OH
R8
7
R5 0 R3 6-R2
According to U.S. Patent 5,594,158, suitable R groups are as
follows: R1 is CH3 or CH2OH; R2 is daunosamine or H; R3 and R4 are
independently one of OH, NO2, NH2, F, Cl, Br, I, CN, H or groups derived from
these; R5 is hydrogen, hydroxyl, or methoxy; and R6-8 are all hydrogen.
Alternatively, R5 and R6 are hydrogen and R7 and R8 are alkyl or halogen, or
vice versa.
According to U.S. Patent 5,843,903, R1 may be a conjugated
peptide. According to U.S. Patent 4,296,105, R5 may be an ether linked alkyl
group. According to U.S. Patent 4,215,062, R5 may be OH or an ether linked
alkyl group. R1 may also be linked to the anthracycline ring by a group other
than C(0), such as an alkyl or branched alkyl group having the C(0) linking
moiety at its end, such as ¨CH2CH(CH2-X)C(0)-R1, wherein X is H or an alkyl
group (see, e.g., U.S. Patent 4,215,062). R2 may alternately be a group linked
by the functional group =N-NHC(0)-Y, where Y is a group such as a phenyl or
substituted phenyl ring. Alternately R3 may have the following structure:
54
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
H3C 0
NH
R9
R10
in which R9 is OH either in or out of the plane of the ring, or is a second
sugar
moiety such as R3. R10 may be H or form a secondary amine with a group such
as an aromatic group, saturated or partially saturated 5 or 6 membered
Exemplary anthracyclines are doxorubicin, daunorubicin,
idarubicin, epirubicin, pirarubicin, zorubicin, and carubicin. Suitable
compounds
have the structures:
0 OH
R2
01110140101," OH
0 OH 6
1
H3C 0
NH2
R3
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
R1 R2 R3
OH out of ring
Doxorubicin: OCH3 C(0)CH2OH
plane
Epirubicin:
OH in ring
(4' epimer of OCH3 C(0)CH2OH
plane
doxorubicin)
OH out of ring
Daunorubicin: OCH3 C(0)CH3
plane
OH out of ring
ldarubicin: H C(0)CH3
plane
0
Pirarubicin: OCH3 C(0)CH2OH
Zorubicin: OCH3 C(CH3)(=N)NHC(0)C6H5 OH
OH out of ring
Carubicin: OH C(0)CH3
plane
Other suitable anthracyclines are anthramycin, mitoxantrone,
menogaril, nogalamycin, aclacinomycin A, olivomycin A, chromomycin A3, and
plicamycin having the structures:
R, H, Hz
Aknogaril H OCH, H
OH 0 HNõ,.....õOH
Nogalamycin 0-sugar 14 COOCH,
SOO sugar: 1.1.3c
OH 0 H co OCH3
CH,
Mtoxantrone
0 OCH,
0
CH,
R2 CH3
Ps CE4' 0 out OH
)71
CH, **OS
OH 0 OH 0
40001, 0 OH
R4 H3C\A>
OH OH 0 Aclacinomycin A
N(CF142
H3c 0
Ho
OH
HOR2 R,
Olivomycin A COCH(CH,), CH, COCH, H
Chromarnycin A, COCFt, CH, COCK, CH,
Plicamycin H H H CH,
Other representative anthracyclines include, FOE 23762, a
doxorubicin derivative (Quaglia etal., J. Lig. Chromatogr. 17(18):3911-3923,
56
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
1994), annamycin (Zou etal., J. Pharm. Sci. 82(11):1151-1154, 1993), ruboxyl
(Rapoport etal., J. Controlled Release 58(2):153-162, 1999), anthracycline
disaccharide doxorubicin analogue (Pratesi et al., Clin. Cancer Res.
4(11):2833-2839, 1998), N-(trifluoroacetyl)doxorubicin and 4'-0-acetyl-N-
(trifluoroacetyl)doxorubicin (Berube & Lepage, Synth. Commun. 28(6)1109-
1116, 1998), 2-pyrrolinodoxorubicin (Nagy etal., Proc. Nat'l Acad. ScL U.S.A.
95(4):1794-1799, 1998), disaccharide doxorubicin analogues (Arcamone et al.,
J. Nat'l Cancer Inst. 89(16):1217-1223, 1997), 4-demethoxy-7-012,6-dideoxy-
4-0-(2,3,6-trideoxy-3-amino-a-L-Iyxo-hexopyranosyl)-a-L-lyxo-hexopyranosyll-
adriamicinone doxorubicin disaccharide analogue (Monteagudo etal.,
Carbohydr. Res. 300(1)11-16, 1997), 2-pyrrolinodoxorubicin (Nagy et al., Proc.
Nat'/Acad. Sci. U. S. A. 94(2):652-656, 1997), morpholinyl doxorubicin
analogues (Duran etal., Cancer Chemother. Pharmacol. 38(3):210-216, 1996),
enaminomalonyl-p-alanine doxorubicin derivatives (Seitz et al., Tetrahedron
Lett. 36(9):1413-16, 1995), cephalosporin doxorubicin derivatives (Vrudhula et
al., J. Med. Chem. 38(8):1380-5, 1995), hydroxyrubicin (Solary etal., mt. J.
Cancer 58(1):85-94, 1994), methoxymorpholino doxorubicin derivative (Kuhl et
al., Cancer Chemother. Pharmacol. 33(1):10-16, 1993), (6-
maleimidocaproyl)hydrazone doxorubicin derivative (Willner et al., Bioconju
gate
Chem. 4(6):521-7, 1993), N-(5,5-diacetoxypent-1-y1) doxorubicin (Cherif &
Farquhar, J. Med. Chem. 35(17):3208-14, 1992), FCE 23762
methoxymorpholinyl doxorubicin derivative (Ripamonti et al., Br. J. Cancer
65(5):703-7, 1992), N-hydroxysuccinimide ester doxorubicin derivatives
(Demant etal., Biochim. Biophys. Acta 1118(1):83-90, 1991),
polydeoxynucleotide doxorubicin derivatives (Ruggiero et al., Biochim.
Biophys.
Acta 1/29(3):294-302, 1991), morpholinyl doxorubicin derivatives (EPA
434960), mitoxantrone doxorubicin analogue (Krapcho et al., J. Med. Chem.
34(8):2373-80. 1991), AD198 doxorubicin analogue (Traganos etal., Cancer
Res. 5/(14):3682-9, 1991), 4-demethoxy-3'-N-trifluoroacetyldoxorubicin (Horton
etal., Drug Des. Delivery 6(2):123-9, 1990), 4'-epidoxorubicin (Drzewoski
etal.,
57
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Pol. J. Pharmacol. Pharm. 40(2):159-65, 1988; Weenen etal., Eur. J. Cancer
an. Oncol. 20(7):919-26, 1984), alkylating cyanomorpholino doxorubicin
derivative (Scudder et al., J. Nat'l Cancer Inst. 80(16):1294-8, 1988),
deoxydihydroiodooxorubicin (EPA 275966), adriblastin (Kalishevskaya etal.,
Vestn. Mosk. Univ., /6(Biol. 1):21-7, 1988), 4'-deoxydoxorubicin (Schoelzel et
al., Leuk. Res. 10(12):1455-9, 1986), 4-demethyoxy-4'-o-methyldoxorubicin
(Giuliani etal., Proc. Int. Congr. Chemother. 16:285-70-285-77, 1983), 3'-
deamino-3'-hydroxydoxorubicin (Horton etal., J. Ant/blot. 37(8):853-8, 1984),
4-
demethyoxy doxorubicin analogues (Barbieri at al., Drugs Exp. Clin. Res.
10(2):85-90, 1984), N-L-leucyl doxorubicin derivatives (Trouet etal.,
Anthracyclines (Proc. Int. Symp, Tumor Pharmacother.), 179-81, 1983), 3'-
deamino-3'-(4-methoxy-1-piperidinyl) doxorubicin derivatives (U.S. 4,314,054),
3'-deamino-3'-(4-mortholinyl) doxorubicin derivatives (U.S. 4,301,277), 4'-
deoxydoxorubicin and 4'-o-methyldoxorubicin (Giuliani et al., Int. J. Cancer
27(1):5-13, 1981), aglycone doxorubicin derivatives (Chan & Watson, J. Pharm.
ScL 67(12):1748-52, 1978), SM 5887 (Pharma Japan 1468:20, 1995), MX-2
(Pharma Japan 1420:19, 1994), 4'-deoxy-13(S)-dihydro-4'-iododoxorubicin (EP
275966), morpholinyl doxorubicin derivatives (EPA 434960), 3'-deamino-3'-(4-
methoxy-1-piperidinyl) doxorubicin derivatives (U.S. 4,314,054), doxorubicin-
14-valerate, morpholinodoxorubicin (U.S. 5,004,606), 3'-deamino-3'-(3"-cyano-
4"-morpholinyl doxorubicin; 3'-deamino-3'-(3"-cyano-4"-morpholiny1)-13-
dihydoxorubicin); (3'-deamino-3'-(3"-cyano-4"-morpholinyl) daunorubicin; 3'-
deamino-3'-(3"-cyano-4"-morpholiny1)-3-dihydrodaunorubicin, and 3'-deamino-
3'-(4"-morpholiny1-5-iminodoxorubicin and derivatives (U.S. 4,585,859), 3'-
deamino-3'-(4-methoxy-1-piperidinyl) doxorubicin derivatives (U.S. 4,314,054)
and 3-deamino-3-(4-morpholinyl) doxorubicin derivatives (U.S. 4,301,277).
(B) Fluoropyrimidine analogues
In another aspect, the therapeutic agent is a fluoropyrimidine
analog, such as 5-fluorouracil, or an analogue or derivative thereof,
including
58
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
carmofur, doxifluridine, emitefur, tegafur, and floxuridine. Exemplary
compounds have the structures:
A
0
R2 F
N
1
0 N
I
R1
R1 R2
5-Fluorouracil H H
Carmofur C(0)NH(CH2)5CH3 H
Doxifluridine A1 H
Floxuridine A2 H
Emitefur CH2OCH2CH3 B
Tegafur C H
B
I 0
0 o'le"-o 410
Co
Other suitable fluoropyrimidine analogues include 5-FudR (5-
fluoro-deoxyuridine), or an analogue or derivative thereof, including 5-
iododeoxyuridine (5-ludR), 5-bromodeoxyuridine (5-Bud R), fluorouridine
triphosphate (5-FUTP), and fluorodeoxyuridine monophosphate (5-dFUMP).
Exemplary compounds have the structures:
59 '
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
q
R'i NH
I
HOm\"0
0
OH
5-Fluoro-2'-deoxyuridine: R = F
5-Bromo-2'-deoxyuridine: R = Br
5-lodo-2'-deoxyuridine: R =I
Other representative examples of fluoropyrimidine analogues
include N3-alkylated analogues of 5-fluorouracil (Kozai et al., J. Chem. Soc.,
Perkin Trans. /(19):3145-3146, 1998), 5-fluorouracil derivatives with 1,4-
oxaheteroepane moieties (Gomez etal., Tetrahedron 54(43):13295-13312,
1998), 5-fluorouracil and nucleoside analogues (Li, Anticancer Res. /7(1A):21-
27, 1997), cis- and trans-5-fluoro-5,6-dihydro-6-alkoxyuracil (Van der Wilt et
al.,
Br. J. Cancer 68(4):702-7, 1993), cyclopentane 5-fluorouracil analogues
(Hronowski & Szarek, Can. J. Chem. 70(4)1162-9, 1992), A-0T-fluorouracil
(Zhang etal., Zongguo Yiyao Gongye Zazhi 20(11):513-15, 1989), N4-
trimethoxybenzoy1-5'-deoxy-5-fluorocytidine and 5'-deoxy-5-fluorouridine (Miwa
etal., Chem. Pharm. Bull. 38(4):998-1003, 1990), 1-hexylcarbamoy1-5-
fluorouracil (Hoshi etal., J. Pharmacobio-Dun. 3(9):478-81, 1980; Maehara et
al., Chemotherapy (Basel) 34(6):484-9, 1988), B-3839 (Prajda et al., In Vivo
2(2):151-4, 1988), uracil-1-(2-tetrahydrofury1)-5-fluorouracil (Anal etal.,
Oncology 45(3):144-7, 1988), 1-(2'-deoxy-2'-fluoro-13-D-arabinofuranosyl)-5-
fluorouracil (Suzuko etal., Mol. Pharmacol. 31(3):301-6, 1987), doxifluridine
(Matuura et al., Oyo Yakuri 29(5):803-31, 1985), 5'-deoxy-5-fluorouridine
(Bollag & Hartmann, Eur. J. Cancer 16(4):427-32, 1980), 1-acety1-3-0-toluy1-5-
fluorouracil (Okada, Hiroshima J. Med. ScL 28(1):49-66, 1979), 5-fluorouracil-
m-formylbenzene-sulfonate (JP 55059173), N'-(2-furanidyI)-5-fluorouracil (JP
'
53149985) and 1-(2-tetrahydrofuryI)-5-fluorouracil (JP 52089680).
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
These compounds are believed to function as therapeutic agents
by serving as antimetabolites of pyrimidine.
(C) Folic acid antagonists
In another aspect, the therapeutic agent is a folic acid antagonist,
such as methotrexate or derivatives or analogues thereof, including
edatrexate,
trimetrexate, raltitrexed, piritrexim, denopterin, tomudex, and pteropterin.
Methotrexate analogues have the following general structure:
R1
R5
4
R6
R3 R3 R10
R7
R3
The identity of the R group may be selected from organic groups, particularly
those groups set forth in U.S. Patent Nos. 5,166,149 and 5,382,582. For
example, R1 may be N, R2 may be N or C(CH3), R3 and R3' may H or alkyl, e.g.,
CH3, R4 may be a single bond or NR, where R is H or alkyl group. R5, R6,
and/or
R8 may be H, OCH3, or alternately they can be halogens or hydro groups. R7 is
a side chain of the general structure:
-o
NH
HO
0 OH
_ n
wherein n = 1 for rnethotrexate, n = 3 for pteropterin. The carboxyl groups in
the side chain may be esterified or form a salt such as a Zn2+ salt. R9 and
Rio
can be NH2 or may be alkyl substituted.
61
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Exemplary folic acid antagonist compounds have the structures:
R H 2
R5
R6 01
R3 Ro
R7
R8
Ro R1 R2 R3 R4 R5 R6 R7 R8
Methotrexate NH2 N N H N(CH3) H H A (n=1) H
Edatrexate NH2 N N H CH(CH2CH3) H H A (n=1) H
Trimetrexate NH2 CH C(CH3) H NH H OCH3 OCH3 OCH3
Pteropterin OH N N H NH H H A (n=3) H
Denopterin OH N N CH3 N(CH3) H H A (n=1) H
Peritrexim NH2 N C(CH3) H single bond OCH3 H H OCH3
A: cl
NH
HO
0 OH ¨n
HOOV- 0 CH,
S NI NCH,
I NH
0
Tomudex
Other representative examples include 6-S-aminoacyloxymethyl
mercaptopurine derivatives (Harada etal., Chem. Pharm. Bull. 43(10):793-6,
1995), 6-mercaptopurine (6-MP) (Kashida etal., Biol. Pharm. Bull. 18(11):1492-
7, 1995), 7,8-polymethyleneimidazo-1,3,2-diazaphosphorines (Nilov et al.,
Mendeleev Commun. 2:67, 1995), azathioprine (Chifotides etal., J. lnorg.
Biochem. 56(4):249-64, 1994), methyl-D-glucopyranoside mercaptopurine
derivatives (Da Silva etal., Eur. J. Med. Chem. 29(2):149-52, 1994) and s-
alkynyl mercaptopurine derivatives (Ratsino etal., Khim.-Farm. Zh. 15(8):65-7,
1981); indoline ring and a modified ornithine or glutamic acid-bearing
62
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
methotrexate derivatives (Matsuoka etal., Chem. Pharm. Bull. 45(7):1146-
1150, 1997), alkyl-substituted benzene ring C bearing methotrexate derivatives
(Matsuoka et al., Chem. Pharm. Bull. 44(12)2287-2293, 1996), benzoxazine or
benzothiazine moiety-bearing methotrexate derivatives (Matsuoka et al., J.
Med. Chem. 40(1):105-111, 1997), 10-deazaaminopterin analogues (DeGraw
etal., J. Med. Chem. 40(3):370-376, 1997), 5-deazaaminopterin and 5,10-
dideazaaminopterin methotrexate analogues (Piper etal., J. Med. Chem.
40(3):377-384, 1997), indoline moiety-bearing methotrexate derivatives
(Matsuoka et al., Chem. Pharm. Bull. 44(7)1332-1337 , 1996), lipophilic amide
methotrexate derivatives (Pignatello et al., World Meet. Pharm. Biopharm.
Pharm. Technol., 563-4, 1995), L-threo-(2S,4S)-4-fluoroglutamic acid and DL-
3,3-difluoroglutamic acid-containing methotrexate analogues (Hart et al., J.
Med. Chem. 39(1):56-65, 1996), methotrexate tetrahydroquinazoline analogue
(Gangjee, et al., J. HeterocycL Chem. 32(1):243-8, 1995), N-(a-aminoacyl)
methotrexate derivatives (Cheung etal., Pteridines 3(1-2):101-2, 1992), biotin
methotrexate derivatives (Fan et al., Pteridines 3(1-2):131-2, 1992), D-
glutamic
acid or D-erythrou, threo-4-fluoroglutarnic acid methotrexate analogues
(McGuire et at., Biochem. PharmacoL 42(12):2400-3, 1991), (3,y-methano
methotrexate analogues (Rosowsky et al., Pteridines 2(3):133-9, 1991), 10-
deazaaminopterin (10-EDAM) analogue (Braakhuis etal., Chem. Biol.
Pteridines, Proc. mt. Symp. Pteridines Folic Acid Deny., 1027-30, 1989), 7-
tetrazole methotrexate analogue (Kalman et a/.1 Chem. Biol. Pteridines, Proc.
mt. Symp. Pteridines Folic Acid Deny., 1154-7, 1989), N-(L-a-aminoacyl)
methotrexate derivatives (Cheung et aL, Heterocycles 28(2):751-8, 1989), meta
and ortho isomers of aminopterin (Rosowsky etal., J. Med. Chem. 32(12):2582,
1989), hydroxymethylmethotrexate (DE 267495), y-fluoromethotrexate
(McGuire etal., Cancer Res. 49(16):4517-25, 1989), polyglutamyl methotrexate
derivatives (Kumar etal., Cancer Res. 46(10):5020-3, 1986), gem-
diphosphonate methotrexate analogues (WO 88/06158), a- and y-substituted
methotrexate analogues (Tsushima et al, Tetrahedron 44(17):5375-87, 1988),
63
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
5-methyl-5-deaza methotrexate analogues (4,725,687), 1\18-acyl-Na-(4-amino-4-
deoxypteroy1)-L-ornithine derivatives (Rosowsky et aL, J. Med. Chem.
3/(7)1332-7, 1988), 8-deaza methotrexate analogues (Kuehl et al., Cancer
Res. 48(6):1481-8, 1988), acivicin methotrexate analogue (Rosowsky etal., J.
Med. Chem. 30(8):1463-9, 1987), polymeric platinol methotrexate derivative
(Carraher etal., Polym. Sci. Technol. (Plenum), 35(Adv. Biomed. Poiym.):311-
24, 1987), methotrexate-y-dimyristoylphophatidylethanolamine (Kinsky et aL,
Biochim. Biophys. Acta 917(2):211-18, 1987), methotrexate polyglutamate
analogues (Rosowsky at al., Chem. Biol. Pteridines, Pteridines Folic Acid
Deny., Proc. Int. Symp. Pteridines Folic Acid Deny.: Chem., Biol. Clin.
Aspects:
985-8, 1986), poly-y-glutamyl methotrexate derivatives (Kisliuk at al., Chem.
Biol. Pteridines, Pteridines Folic Acid Deny., Proc. Int. Symp. Pteridines
Folic
Acid Deny.: Chem., Biol. Clin. Aspects: 989-92, 1986), deoxyuridylate
methotrexate derivatives (Webber et al., Chem. Biol. Pteridines, Pteridines
Folic
Acid Deny., Proc. Int. Symp. Pteridines Folic Acid Deny.: Chem., Biol. Clin.
Aspects: 659-62, 1986), iodoacetyl lysine methotrexate analogue (Delcamp et
al., Chem. Biol. Pteridines, Pteridines Folic Acid Deny., Proc. Int. Symp.
Pteridines Folic Acid Deny.: Chem., Biol. Clin. Aspects: 807-9, 1986),
2,.omega.-diaminoalkanoid acid-containing methotrexate analogues (McGuire
et al., Biochem. Pharmacol. 35(15):2607-13, 1986), polyglutamate
methotrexate derivatives (Kamen & Winick, Methods Enzymol. /22(Vitam.
Coenzymes, Pt. G):339-46, 1986), 5-methyl-5-deaza analogues (Piper etal., J.
Med. Chem. 29(6):1080-7, 1986), quinazoline methotrexate analogue
(Mastropaolo etal., J. Med. Chem. 29(1):155-8, 1986), pyrazine methotrexate
analogue (Lever & Vestal, J. HeterocycL Chem. 22(1):5-6, 1985), cysteic acid
and homocysteic acid methotrexate analogues (4,490,529), y-tert-butyl
methotrexate esters (Rosowsky etal., J. Med. Chem. 28(5):660-7, 1985),
fluorinated methotrexate analogues (Tsushima etal., Heterocycles 23(1):45-9,
1985), folate methotrexate analogue (Trombe, J. BacterioL 160(3):849-53,
1984), phosphonoglutamic acid analogues (Sturtz & Guillamot, Eur. J. Med.
64
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Chem.--Chim. Ther. 19(3):267-73, 1984), poly (L-lysine) methotrexate
conjugates (Rosowsky et aL, J. Med. Chem. 27(7):888-93, 1984), dilysine and
trilysine methotrexate derivates (Forsch & Rosowsky, J. Org. Chem.
49(7):1305-9, 1984), 7-hydroxymethotrexate (Fabre etal., Cancer Res.
43(10):4648-52, 1983), poly-y-glutamyl methotrexate analogues (Piper &
Montgomery, Adv. Exp. Med. Biol., 163(Foly1 Antifolyl Polyglutamates):95-100,
1983), 3',5'-dichloromethotrexate (Rosowsky & Yu, J. Med. Chem. 26(10)1448-
52, 1983), diazoketone and chloromethylketone methotrexate analogues
(Gangjee et al., J. Pharm. Sci, 71(6):717-19, 1982), 10-propargylaminopterin
and alkyl methotrexate homologs (Piper et al., J. Med. Chem. 25(7):877-80,
1982), lectin derivatives of methotrexate (Lin etal., JNCI 66(3):523-8, 1981),
polyglutamate methotrexate derivatives (Galivan, Mol. Pharmacol, 17(1):105-
10, 1980), halogentated methotrexate derivatives (Fox, JNCI 58(4):J955-8,
1977), 8-alkyl-7,8-dihydro analogues (Chaykovsky etal., J. Med. Chem.
20(10):J1323-7, 1977), 7-methyl methotrexate derivatives and
dichloromethotrexate (Rosowsky & Chen, J. Med. Chem. 17(12):J1308-11,
1974), lipophilic methotrexate derivatives and 3',5'-dichloronnethotrexate
(Rosowsky, J. Med. Chem. 16(10):J1190-3, 1973), deaza amethopterin
analogues (Montgomery etal., Ann. N.Y. Acad. Sci. 186:J227-34, 1971),
MX068 (Pharma Japan, 1658:18, 1999) and cysteic acid and homocysteic acid
methotrexate analogues (EPA 0142220);
These compounds are believed to act as antimetabolites of folic
acid.
CD) PodophvIlotoxins
In another aspect, the therapeutic agent is a Podophyllotoxin, or a
derivative or an analogue thereof. Exemplary compounds of this type are
etoposide or teniposide, which have the following structures:
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
R
0
HO 0
OH
0
(0 la*
0
Etoposide CH,
Teniposide vs
rH3c0 OCH3
OH
Other representative examples of podophyllotoxins include Cu(II)-
VP-16 (etoposide) complex (Tawa etal., Bioorg. Med. Chem. 6(7):1003-1008,
1998), pyrrolecarboxamidino-bearing etoposide analogues (Ji etal., Bioorg.
Med. Chem. Lett. 7(5):607-612, 1997), 413-amino etoposide analogues (Hu,
University of North Carolina Dissertation, 1992), y-lactone ring-modified
arylamino etoposide analogues (Zhou et aL, J. Med. Chem. 37(2):287-92,
1994), N-glucosyl etoposide analogue (Allevi et al., Tetrahedron Lett.
34(45):7313-16, 1993), etoposide A-ring analogues (Kadow etal., Bioorg. Med.
Chem. Lett. 2(1):17-22, 1992), 4'-deshydroxy-4'-methyl etoposide (Saulnier et
Bioorg. Med. Chem. Lett. 2(10)1213-18, 1992), pendulum ring etoposide
analogues (Sinha etal., Eur. J. Cancer 26(5):590-3, 1990) and E-ring desoxy
etoposide analogues (Saulnier etal., J. Med. Chem. 32(7)1418-20, 1989).
These compounds are believed to act as topoisomerase II
inhibitors and/or DNA cleaving agents.
(E) Camptothecins
In another aspect, the therapeutic agent is camptothecin, or an
analogue or derivative thereof. Camptothecins have the following general
structure.
R2 R3 0
Ri
X
R4
H3c---= OH
66
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In this structure, X is typically 0, but can be other groups, e.g., NH
in the case of 21-lactam derivatives. R1 is typically H or OH, but may be
other
groups, e.g., a terminally hydroxylated C1-3 alkane. R2 is typically H or an
amino containing group such as (CH3)2NHCH2, but may be other groups e.g.,
NO2, NH2, halogen (as disclosed in, e.g., U.S. Patent 5,552,156) or a short
alkane containing these groups. R3 is typically H or a short alkyl such as
C2H5.
R4 is typically Fl but may be other groups, e.g., a methylenedioxy group with
Rt
Exemplary camptothecin compounds include topotecan,
irinotecan (CPT-11), 9-aminocamptothecin, 21-lactam-20(S)-camptothecin,
10,11-methylenedioxycamptothecin, SN-38, 9-nitrocamptothecin, 10-
hydroxycamptothecin. Exemplary compounds have the structures:
R2R3
R1 N
X
I E
0
H30¨'* OH
R2 R,
Camptothecin:
Topotecan: OH (CH3)2NHCH2 H
SN-38: OH H C2H5
X: 0 for most analogs, NH for 21-lactam analogs
Camptothecins have the five rings shown here. The ring labeled
E must be intact (the lactone rather than carboxylate form) for maximum
activity
and minimum toxicity.
Camptothecins are believed to function as topoisomerase I
inhibitors and/or DNA cleavage agents.
(F) Hydroxyureas
The therapeutic agent of the present invention may be a
hydroxyurea. Hydroxyureas have the following general structure:
67
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
0
R3
-0 X
1\1
R2 R1
Suitable hydroxyureas are disclosed in, for example, U.S. Patent
No. 6,080,874, wherein R1 is:
S
2
R3 =
7
and R2 is an alkyl group having 1-4 carbons and R3 is one of H, acyl, methyl,
ethyl, and mixtures thereof, such as a methylether.
Other suitable hydroxyureas are disclosed in, e.g., U.S. Patent
No. 5,665,768, wherein R1 is a cycloalkenyl group, for example N4345-(4-
fluorophenylthio)-fury1]-2-cyclopenten-1-y1}N-hydroxyurea; R2 is H or an alkyl
group having 1 to 4 carbons and R3 is H, X is H or a cation.
Other suitable hydroxyureas are disclosed in, e.g., U.S. Patent
No. 4,299,778, wherein R1 is a phenyl group substituted with one or more
fluorine atoms; R2 is a cyclopropyl group; and R3 and X is H.
Other suitable hydroxyureas are disclosed in, e.g., U.S. Patent
No. 5,066,658, wherein R2 and R3 together with the adjacent nitrogen form:
(C12)n
N¨
/
(CI-12)m
wherein m is 1 or 2, n is 0-2 and Y is an alkyl group.
In one aspect, the hydroxyurea has the structure:
0
OH
H2N NH
Hydroxyurea
68
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
These compounds are thought to function by inhibiting DNA
synthesis.
(G) Platinum complexes
In another aspect, the therapeutic agent is a platinum compound.
In general, suitable platinum complexes may be of Pt(II) or Pt(IV) and have
this
basic structure:
1
R1
Pt
vY
R2
Z2
wherein X and Y are anionic leaving groups such as sulfate, phosphate,
carboxylate, and halogen; R1 and R2 are alkyl, amine, amino alkyl may be
further substituted, and are basically inert or bridging groups. For Pt(II)
complexes Z1 and Z2 are non-existent. For Pt(IV) Z1 and Z2 may be anionic
groups such as halogen, hydroxy, carboxylate, ester, sulfate or phosphate.
See, e.g., U.S. Patent Nos. 4,588,831 and 4,250,189.
Suitable platinum complexes may contain multiple Pt atoms. See,
e.g., U.S. Patent Nos. 5,409,915 and 5,380,897. For example bisplatinum and
triplatinum complexes of the type:
69
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
ZI Zi
x ,RI x I
Pt Pt
.)K AflY
Z2 Z2
Zi
X RI
X
Pt,X
Pt Pt
Y R2 Y
Z2 Z2 Z2
R2 R7 x
Pt Pt
1
Z2 Z2
Z,)
R3
Pt
y'7
Exemplary platinum compounds are cisplatin, carboplatin,
oxaliplatin, and miboplatin having the structures:
NH3
NH3
0 oj
CI ________________________ Pt NH3 (t NH3
CI
0
Cisplatin Carboplatin
0 H
0
0 NH2
Pt
:10 /
0 /11...7H
0 NH
0
0
Oxaliplatin Miboplatin
Other representative platinum compounds include
(CPA)2Pt[DOLYM] and (DACH)Pt[DOLYM] cisplatin (Choi of aL, Arch.
Pharmacal Res. 22(2):151-156, 1999), Cis-[PtC12(4,7-H-5-methyl-7-
oxo}1,2,4[triazolo[1,5-a]pyrimidine)2] (Navarro et aL, J. Med. Chem. 41(3):332-
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
338, 1998), [Pt(cis-1,4-DACH)(trans-C12)(CBDCA)] =1/2Me0H cisplatin
(Shamsuddin et al., lnorg. Chem. 36(25):5969-5971, 1997), 4-pyridoxate
diammine hydroxy platinum (Tokunaga etal., Pharm. ScL 3(7):353-356, 1997),
Pt(II) ...13411) (Pt2[NHCHN(C(CH2)(CH3))]4) (Navarro etal., In'org. Chem.
35(26):7829-7835, 1996), 254-S cisplatin analogue (Koga etal., NeuroL Res.
18(3):244-247, 1996), o-phenylenediamine ligand bearing cisplatin analogues
(Koeckerbauer & Bednarski, J. lnorg. Biochem. 62(4):281-298, 1996), trans,
cis-[Pt(OAc)212(en)] (Kratochwil etal., J. Med. Chem. 39(13):2499-2507, 1996),
estrogenic 1,2-diarylethylenediamine ligand (with sulfur-containing amino
acids
and glutathione) bearing cisplatin analogues (Bednarski, J. lnorg. Biochem.
62(1):75, 1996), cis-1,4-diaminocyclohexane cisplatin analogues (Shamsuddin
etal., J. Inorg. Biochem. 6/(4):291-301, 1996), 5' orientational isomer of cis-
[Pt(NH3)(4-aminoTEMP-0){d(GpG)}] (Dunham & Lippard, J. Am. Chem. Soc.
117(43):10702-12, 1995), chelating diamine-bearing cisplatin analogues
(Koeckerbauer & Bednarski, J. Pharm. ScL 84(7):819-23, 1995), 1,2-
diarylethyleneamine ligand-bearing cisplatin analogues (Otto et al., J. Cancer
Res. Clin. OncoL 121(1):31-8, 1995), (ethylenediannine)platinum(II) complexes
(Pasini etal., J. Chem. Soc., Dalton Trans. 4:579-85, 1995), CI-973 cisplatin
analogue (Yang etal., Int. J. Oncol. 5(3):597-602, 1994), cis-
diaminedichloroplatinum(II) and its analogues cis-1,1-
cyclobutanedicarbosylato(2R)-2-methyl-1,4-butanediamineplatinum(II) and cis-
diammine(glycolato)platinum (Claycamp & Zimbrick, J. lnorg. Biochem.
26(4):257-67, 1986; Fan etal., Cancer Res. 48(11):3135-9, 1988; Heiger-
Bernays etal., Biochemistry 29(36):8461-6, 1990; Kikkawa etal., J. Exp. Clin.
Cancer Res. 12(4):233-40, 1993; Murray etal., Biochemistry 3/(47):11812-17,
1992; Takahashi etal., Cancer Chemother. Pharmacol. 33(1):31-5, 1993), cis-
amine-cyclohexylamine-dichloroplatinum(II) (Yoshida et al., Biochem.
PharmacoL 48(4):793-9, 1994), gem-diphosphonate cisplatin analogues (FR
2683529), (meso-1,2-bis(2,6-dichloro-4-hydroxyplenyl)ethylenediamine)
dichloroplatinum(II) (Bednarski etal., J. Med. Chem. 35(23):4479-85, 1992),
71
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
cisplatin analogues containing a tethered dansyl group (Hartwig et aL, J. Am.
Chem. Soc. 114(21):8292-3, 1992), platinum(II) polyamines (Siegmann etal.,
lnorg. Met-Containing Polym. Mater., (Proc. Am. Chem. Soc. Int. Symp.), 335-
61, 1990), cis-(3H)dichloro(ethylenediamine)platinum(II) (Eastman, Anal.
Biochem. 197(2):311-15, 1991), trans-diamminedichloroplatinum(II) and cis-
(Pt(NH3)2(N3-cytosine)C1) (Bellon & Lippard, Biophys. Chem. 35(2-3):179-88,
1990), 3H-cis-1,2-diaminocyclohexanedichloroplatinum(II) and 31-1-cis-1,2-
diaminocyclohexanemalonatoplatinum (II) (Oswald etal., Res. Commun. Chem.
Pathol. Pharmacol. 64(1):41-58, 1989), diaminocarboxylatoplatinum (EPA
296321), trans-(D,1)-1,2-diaminocyclohexane carrier ligand-bearing platinum
analogues (Wyrick & Chaney, J. Labelled Compd. Radiopharm. 25(4):349-57,
1988), aminoalkylaminoanthraguinone-derived cisplatin analogues (Kitov et al.,
Eur. J. Med. Chem. 23(4):381-3, 1988), spiroplatin, carboplatin, iproplatin
and
JM40 platinum analogues (Schroyen etal., Eur. J. Cancer Clin. OncoL
24(8):1309-12, 1988), bidentate tertiary diamine-containing cisplatinum
derivatives (Orbell et al., lnorg. Chim. Acta 152(2):125-34, 1988),
platinum(II),
platinum(IV) (Liu & Wang, Shandong Yike Daxue Xuebao 24(1):35-41, 1986),
cis-diammine(1,1-cyclobutanedicarboxylato-)platinum(II) (carboplatin, JM8) and
ethylenediammine-malonatoplatinum(II) (JM40) (Begg etal., Radiother. OncoL
9(2):157-65, 1987), JM8 and JM9 cisplatin analogues (Harstrick etal., mt. J.
AndroL 10(1); 139-45, 1987), (NPr4)2((PtCL4).cis-(PtC12-(NH2Me)2))
(Brammer etal., J. Chem. Soc., Chem. Commun. 6:443-5, 1987), aliphatic
tricarboxylic acid platinum complexes (EPA 185225), and cis-dichloro(amino
acid)(tert-butylamine)platinum(II) complexes (Pasini & Bersanetti, lnorg.
Chim.
Acta 107(4):259-67, 1985). These compounds are thought to function by
binding to DNA, i.e., acting as alkylating agents of DNA.
Dosing of Therapeutic Agents
As described herein, various compositions and implants may be
used for treating a diverticular disease. Because medical implants and
72
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
compositions are made in a variety of configurations, forms, and sizes, the
exact dose of a therapeutic agent administered will vary with the implant
size,
surface area, design, and portions of the implant coated. In addition, the
number and size of diverticula present may be considered when determining
the total amount of drug and material administered to a host. However, certain
principles can be applied in the application of this art. Drug dose can be
calculated as a function of dose per unit area (for example, the portion of
the
implant being coated), total drug dose administered can be measured, and
appropriate surface concentrations of active drug can be determined.
Regardless of the method of application of the drug, whether in a composition
or applied to the medical implant, the anticancer/anti-infective agents
described
herein, used alone or in combination, may be administered under the following
dosing guidelines.
(a) Anthracyclines. Utilizing the anthracycline doxorubicin as an
example, whether applied as a polymer coating, incorporated into the polymers
which make up the implant components, or applied with or without a carrier
polymer, the total dose of doxorubicin applied to the implant preferably does
not
exceed 25 mg (range of 0.1 idg to 25 mg). In a particularly preferred
embodiment, the total amount of drug applied is in the range of 1 vg to 5 mg.
The dose per unit area (i.e., the amount of drug as a function of the surface
area of the portion of the implant to which drug is applied and/or
incorporated)
falls within the range of 0.01 lig - 100 [tg per mm2 of surface area. In
another
particularly preferred embodiment, doxorubicin is applied to the diverticular
surface at a dose of 0.1 lig/mm2¨ 10 g/mm2. Because different implants will
release doxorubicin at differing rates, the above dosing parameters are
preferably used in combination with the release rate of the drug from the
implant surface such that a minimum concentration of 10-7- 104 M of
doxorubicin is maintained on the surface. Preferably, surface drug
concentrations exceed concentrations of doxorubicin known to be lethal to
multiple species of bacteria and fungi (i.e., are in excess of 104 M) although
for
73
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
some embodiments lower concentrations are sufficient. In a preferred
embodiment, doxorubicin is released from the surface of the implant such that
anti-infective activity is maintained for a period ranging from several hours
to
several months. In a particularly preferred embodiment the drug is released in
effective concentrations for a period ranging from 1 week ¨ 6 months.
Analogues and derivatives of doxorubicin (as described previously) with
similar
functional activity can also be utilized for the purposes of this invention;
the
above dosing parameters are then adjusted according to the relative potency of
the analogue or derivative as compared to the parent compound (e.g., a
compound twice as potent as doxorubicin is administered at half the above
parameters, a compound half as potent as doxorubicin is administered at twice
the above parameters, etc.).
Utilizing mitoxantrone as another example of an anthracycline,
whether applied as a polymer coating, incorporated into the polymers which
make up the implant, or applied with or without a carrier polymer, the total
dose
of mitoxantrone applied preferably does not exceed 5 mg (range of 0.01 ,g to 5
mg). In a particularly preferred embodiment, the total amount of drug applied
is
in the range of 0.1 jig to 1 mg. The dose per unit area (i.e., the amount of
drug
as a function of the surface area of the portion of the implant to which drug
is
applied and/or incorporated) falls within the range of 0.01 g - 20 jig per mm2
of
surface area. In a particularly preferred embodiment, mitoxantrone is applied
to
the diverticular surface at a dose of 0.051,tg/mm2¨ 3 j.tg/mm2. Because
different implants will release mitoxantrone at differing rates, the above
dosing
parameters are preferably utilized in combination with the release rate of the
drug from the implant surface such that a minimum concentration of 10-5- 10-6M
of mitoxantrone is maintained. Preferably, drug concentrations on the implant
surface exceed concentrations of mitoxantrone known to be lethal to multiple
species of bacteria and fungi (i.e., are in excess of 10-5M) although for some
embodiments lower drug levels will be sufficient. In one embodiment,
mitoxantrone is released from the surface of the implant such that anti-
infective
74
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
activity is maintained for a period ranging from several hours to several
months.
In another embodiment the drug is released in effective concentrations for a
period ranging from 1 week ¨ 6 months. On the basis of the disclosure
provided herein analogues and derivatives of mitoxantrone (as described
previously) with similar functional activity can be used for the methods and
compositions described herein; the above dosing parameters are then adjusted
according to the relative potency of the analogue or derivative as compared to
the parent compound (e.g., a compound twice as potent as mitoxantrone is
administered at half the above parameters, a compound half as potent as
mitoxantrone is administered at twice the above parameters, etc.).
(b) Fluoropyrimidines Utilizing the fluoropyrimidine 5-
fluorouracil as an example, whether applied as a polymer coating, incorporated
into the polymers which make up the implant, or applied with or without a
carrier
polymer, the total dose of 5-fluorouracil applied preferably does not exceed
250
mg (range of 1.01..ig to 250 mg). In a particularly preferred embodiment, the
total amount of drug applied is in the range of 10 lag to 25 mg. The dose per
unit area (i.e., the amount of drug as a function of the surface area of the
portion of the implant to which drug is applied and/or incorporated) falls
within
the range of 0.11.ig ¨ 1 mg per mm2 of surface area. In one embodiment, 5-
fluorouracil is applied to the diverticular or impint surface at a dose of 1.0
lAg/mm2¨ 501..tg/mm2. Because different implants will release 5-fluorouracil
at
differing rates, the above dosing parameters can be used in combination with
the release rate of the drug from the implant surface such that a minimum
concentration of 104- 10-7 M of 5-fluorouracil is maintained. Preferably,
surface
drug concentrations exceed concentrations of 5-fluorouracil known to be lethal
to numerous species of bacteria and fungi (i.e., are in excess of 104 M)
although for some embodiments lower drug levels will be sufficient. In another
embodiment, 5-fluorouracil is released from the implant surface such that anti-
infective activity is maintained for a period ranging from several hours to
several
months. In still another embodiment the drug is released in effective
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
concentrations for a period ranging from 1 week ¨ 6 months. Analogues and
derivatives of 5-fluorouracil (as described previously) with similar
functional
activity can be used. The above dosing parameters are then adjusted
according to the relative potency of the analogue or derivative as compared to
the parent compound (i.e., a compound twice as potent as 5-fluorouracil is
administered at half the above parameters, a compound half as potent as 5-
fluorouracil is administered at twice the above parameters, etc.).
(c) Podophylotoxins Utilizing the podophylotoxin etoposide as
an example, whether applied as a polymer coating, incorporated into the
polymers which make up the implant, or applied with or without a carrier
polymer, the total dose of etoposide applied preferably does not exceed 25 mg
(range of 0.1 pg to 25 mg). In a particularly preferred embodiment, the total
amount of drug applied is in the range of 1 lig to 5 mg. The dose per unit
area
(i.e., the amount of drug as a function of the surface area of the portion of
the
implant to which drug is applied and/or incorporated) falls within the range
of
0.01 ,g - 100 ytg per mm2 of surface area. In one embodiment, etoposide is
applied to the diverticular surface at a dose of 0.1 pg/mm2¨ 10 [Lg/mm2. The
above dosing parameters should be utilized in combination with the release
rate
of the drug from the implant surface such that a concentration of 10-5- 10-6M
of
etoposide is maintained. It is necessary to insure that surface drug
concentrations exceed concentrations of etoposide known to be lethal to a
variety of bacteria and fungi (i.e., are in excess of 10-5M; although for some
embodiments lower drug levels will be sufficient). In one embodiment,
etoposide is released from the surface of the implant such that anti-infective
activity is maintained for a period ranging from several hours to several
months.
In another embodiment the drug is released in effective concentrations for a
period ranging from 1 week ¨ 6 months. On the basis of the description
provided herein analogues and derivatives of etoposide (as described
previously) with similar functional activity can be used in the compositions
and
methods described herein. The above dosing parameters are then adjusted
'
76
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
according to the relative potency of the analogue or derivative as compared to
the parent compound (Le., a compound twice as potent as etoposide is
administered at half the above parameters, a compound half as potent as
etoposide is administered at twice the above parameters, etc.).
(d) Haemostatic Agents. Utilizing CoStasis as an exemplary
hemostatic agent, whether it is applied as a polymer coating, incorporated
into
the polymers which make up an implant, applied as an implant, or applied to
tissue, the total volume of CoStasis delivered as an implant or from an
implant
or composition, or coated onto the surface of an implant or tissue, preferably
does not exceed 25 mL (range of 0.2 mL to 25 mL). In one embodiment, the
total amount of CoStasis() in the composition or implant is in the range of
0.5
mL to 15 mL. In another embodiment, the amount per unit area of the implant
or tissue (i.e., the amount of CoStasis as a function of the surface area of
the
portion of the implant or tissue to which it is applied and/or incorporated)
falls
within the range of 0.01 mL - 5.0 mL per cm2 of surface area coated. In
another
embodiment, CoStasis is applied on an implant or tissue surface at a dose of
0.1 mL/cm2 ¨0.5 mL/cm2 of surface area coated. Under circumstances where
one or more pharmacological agents is (are) included in the composition,
CoStasis() may release such pharmacologically active agent(s) at differing
rates, as such, the above dosing parameters are preferably used in
combination with the release rate of the drug from the composition or implant
such that a minimum concentration of 0.01 nM -1000 pM of pharmacologically
active agent is delivered to the tissue. In one embodiment, an agent is
released from the surface of an implant such that fibrosis in the diverticulum
is
promoted for a period ranging from several hours to several months. In another
embodiment, an agent is released from the surface of an implant such that
bacterial growth is inhibited for a period ranging from several hours to
several
months. For example, an agent may be released in effective concentrations for
a period ranging from 1 hour ¨ 30 days. The above dosing parameters of
CoStasis may be adjusted for an analogue or derivative of CoStasis or of a
77
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
component of the composition according to the relative potency of the analogue
or derivative of the pharmacologically active agent as compared to the parent
pharmacologically active agent (e.g., a compound twice as potent is
administered at half the above parameters, a compound half as potent may be
administered administered at twice the above parameters, etc.).
Utilizing Tisseel as an exemplary hemostatic agent, whether it is
applied as a polymer coating, incorporated into the polymers which make up an
implant, applied as an implant, or applied to tissue, the total volume of
Tisseel
delivered as an implant or from an implant or composition, or coated onto the
surface of an implant or tissue, preferably does not exceed 25 mL (range of
0.2
mL to 25 mL). In one embodiment, the total amount of Tisseel in the
composition or implant is in the range of 0.5 mL to 15 mL. In another
embodiment, the amount per unit area of the implant or tissue (i.e., the
amount
of Tisseel as a function of the surface area of the portion of the implant or
tissue to which it is applied and/or incorporated) falls within the range of
0.01
mL - 5.0 mL per cm2 of surface area coated. In another embodiment, Tisseel
is applied on an implant or tissue surface at a dose of 0.1 mL/cm2 ¨0.5 mL/cm2
of surface area coated. Under circumstances when one or more
pharmacological agents is (are) included in the composition, Tisseel may
release such pharmacologically active agent(s) at differing rates, as such,
the
above dosing parameters are preferably used in combination with the release
rate of the drug from the composition or implant such that a minimum
concentration of 0.01 nM -1000 pM of pharmacologically active agent is
delivered to the tissue. In one embodiment, an agent is released from the
,
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. In another embodiment,
an agent is released from the surface of an implant such that bacterial growth
is
inhibited for a period ranging from several hours to several months. For
example, an agent may be released in effective concentrations for a period
ranging from 1 hour ¨30 days. The above dosing parameters of Tisseel may
78
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
be adjusted for an analogue or derivative of Tisseel or of a component of
Tisseel according to the relative potency of the analogue or derivative of
the
pharmacologically active agent as compared to the parent pharmacologically
active agent (e.g., a compound twice as potent is administered at half the
above parameters, a compound half as potent may be administered
administered at twice the above parameters, etc.).
Utilizing FloSeal as an exemplary hemostatic agent, whether it is
applied as a polymer coating, incorporated into the polymers which make up an
implant, applied as an implant, or applied to tissue, the total volume of
FloSeal
delivered as an implant or from an implant or composition, or coated onto the
surface of an implant or tissue, preferably does not exceed 25 mL (range of
0.2
mL to 25 mL). In one embodiment, the total amount of FloSeal in the
composition or implant is in the range of 0.5 mL to 15 mL. In another
embodiment, the amount per unit area of the implant or tissue (i.e., the
amount
of FloSeal as a function of the surface area of the portion of the implant or
tissue to which it is applied and/or incorporated) falls within the range of
0.01
mL - 5.0 mL per cm2 of surface area coated. In another embodiment, FloSeal
is applied on an implant or tissue surface at a dose of 0.1 mUcm2-0.5 mL/cm2
of surface area coated. Under circumstances when one or more
pharmacological agents is (are) included in the composition, FloSeal may
release such pharmacologically active agent(s) at differing rates, as such,
the
above dosing parameters are preferably used in combination with the release
rate of the drug from the composition or implant such that a minimum
concentration of 0.01 nM -1000 pM of pharmacologically active agent is
delivered to the tissue. In one embodiment, an agent is released from the
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. In another embodiment,
an agent is released from the surface of an implant such that bacterial growth
is
inhibited for a period ranging from several hours to several months. For
example, an agent may be released in effective concentrations for a period
79
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
ranging from 1 hour ¨ 30 days. The above dosing parameters of FloSeal may
be adjusted for an analogue or derivative thereof according to the relative
potency of the analogue or derivative of the pharmacologically active agent
when compared to the parent pharmacologically active agent (e.g., a compound
twice as potent is administered at half the above parameters, a compound half
as potent may be administered administered at twice the above parameters,
etc.).Utilizing CoSeal as an exemplary hemostatic agent, whether it is
applied
as a polymer coating, incorporated into the polymers which make up an
implant, applied as an implant, or applied to tissue, the total volume of
CoSeal
delivered as an implant or from an implant or composition, or coated onto the
surface of an implant or tissue, preferably does not exceed 30 mL (range of
0.2
mL to 30 mL). In one embodiment, the total amount of CoSeal in the
composition or implant is in the range of 0.5 mL to 15 mL. In another
embodiment, the amount per unit area of the implant or tissue (i.e., the
amount
of CoSeal as a function of the surface area of the portion of the implant or
tissue to which it is applied and/or incorporated) falls within the range of
0.01
mL - 5.0 mL per cm2 of surface area coated. In another embodiment, CoSeal
is applied on an implant or tissue surface at a dose of 0A mL/cm2 ¨0.5 mL/cm2
of surface area coated. Under circumstances when one or more
pharmacological agents is (are) included in the composition, CoSeal may
release such pharmacologically active agent(s) at differing rates, as such,
the
above dosing parameters are preferably used in combination with the release
rate of the drug from the composition or implant such that a minimum
concentration of 0.01 nM -1000 pM of pharmacologically active agent is
delivered to the tissue. In one embodiment, an agent is released from the
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. In another embodiment,
an agent is released from the surface of an implant such that bacterial growth
is
,
inhibited for a period ranging from several hours to several months. For
example, an agent may be released in effective concentrations for a period
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
ranging from 1 hour ¨30 days. The above dosing parameters of CoSeal0 may
be adjusted for a derivative or analogue of a physiologically active agent of
CoSeal according to the relative potency of the analogue or derivative of the
pharmacologically active agent as compared to the parent pharmacologically
active agent (e.g., a compound twice as potent is administered at half the
above parameters, a compound half as potent may be administered
administered at twice the above parameters, etc.).
On the basis of the disclosure provided herein, combinations of
anthracyclines (e.g., doxorubicin or mitoxantrone), fluoropyrimidines (e.g., 5-
fluorouracil), folic acid antagonists (e.g., methotrexate and/or
podophylotoxins
(e.g., etoposide) can be used to enhance the antibacterial activity of the
diverticular implant. Similarly, anthracyclines (e.g., doxorubicin or
mitoxantrone), fluoropyrimidines (e.g., 5-fluorouracil), folic acid
antagonists
(e.g., methotrexate and/or podophylotoxins (e.g., etoposide) can be combined
with traditional antibiotic and/or antifungal agents to enhance efficacy. In
another embodiment, the anti-infective agent may be further combined with a
fibrosing agent and/or hemostatic agent for the comprehensive management of
acute diverticulitis.
Methods for Generating Compositions and Medical Implants That Include
a Fibrosis-Inducing Agent
Drug-coated, drug-impregnated, or drug containing implants are
provided herein that induce adhesion or fibrosis in the diverticular, or
facilitate
"filling" of the diverticular sac with scar tissue or fibrotic tissue in situ.
Within
various embodiments, fibrosis is induced by local or regional release of
specific
pharmacological agents that become localized within the diverticular. Nmerous
methods are available for optimizing delivery of the fibrosis-inducing agent
to
the diverticula, and several of these are described below.
81
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Implants That Contain or Release Fibrosis-Inducing Agents
Medical implants as described herein contain and/or are adapted
to release an agent which induces fibrosis or adhesion to the surrounding
tissue. Medical implants may be adapted to have incorporated into their
structure a fibrosis-inducing agent, adapted to have a surface coating of a
fibrosis-inducing agent and/or adapted to release a fibrosis-inducing agent by
(a) directly affixing to the implant a desired fibrosis-inducing agent or
composition containing the fibrosis-inducing agent (e.g., by either spraying
the
medical implant with a drug and/or carrier (polymeric or non-polymeric)-drug
composition to create a film or coating on all, or parts of the internal or
external
surface of the implant; by dipping the implant into a drug and/or carrier
(polymeric or non-polymeric)-drug solution to coat all or parts of the
implant; or
by other covalent or non-covalent (e.g., mechanically attached via knotting or
the use of an adhesive or thermal treatment, electrostatic, ionic, hydrogen
bonded or hydrophobic interactions) attachment of the therapeutic agent to the
implant surface); (b) by coating the medical implant with a substance such as
a
hydrogel which can in turn absorb the desired fibrosis-inducing agent or
composition; (c) by interweaving a "thread" composed of, or coated with, the
fibrosis-inducing agent into the medical implant (e.g., a polymeric strand
composed of materials that induce fibrosis (e.g., silk, collagen, EVA, PLA,
polyurethanes, polymerized drug compositions) or polymers which comprise
and/or release a fibrosis-inducing agent from the thread); (d) by covering
all, or
portions of the implant with a sleeve, cover or mesh containing a fibrosis-
inducing agent (i.e., a covering comprised of a fibrosis-inducing agent -
polymers such as silk, collagen, EVA, PLA, polyurethanes or polymerized
compositions containing fibrosis-inducing agents); (e) constructing all, or
parts
of the implant itself with the desired agent or composition (e.g.,
constructing the
implant from polymers such as silk, collagen, EVA, PLA, polyurethanes or
polymerized compositions of fibrosis-inducing agents); (f) otherwise
impregnating the implant with the desired fibrosis-inducing agent or
82
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
composition; (g) scoring (i.e., creating ridges or indentations) on all, or
parts, of
the implant surface to produce irritation of the tissue and ultimately
fibrosis; (h)
composing all, or parts, of the implant from metal alloys that induce fibrosis
(e.g., copper); (i) constructing all, or parts of the implant itself from a
degradable
or non-degradable polymer that releases one or more fibrosis-inducing agents;
(j) utilizing specialized multi-drug releasing medical implant systems
(described,
e.g., in U.S. Patent No. 6,562,065; U.S. Patent Application Publication Nos.
2003/0199970 and 2003/0167085; and in PCT Publication Nos. WO 03/015664
and WO 02/32347) to deliver fibrosis-inducing agents alone or in combination.
Bulking Agents. In one embodiment, an implant is or comprises
a bulking agent. A bulking agent refers to a liquid, solid or semi-solid
ingredient
used either alone or in combination with another material (e.g., a polymer) to
partially or fully seal or fill a void (e.g., a diverticulum) or lumen within
a host. A
bulking agent may be applied directly into the treatment site or may be
injected
into the tissue immediately surrounding the treatment area. Bulking agent also
refers to compound and mixtures that undergo a chemical reaction,
precipitation, or crystallization in situ, which can partially or fully seal
or fill a
void or lumen within a host. Bulking agents can be used to increase the
volume, extend, or dilute other solids. A bulking agent may have a fixed
volume or may increase in volume as it comes into contact with body fluids in
the host and begins to swell. Depending on the method of use, a bulking agent
may be in an injectable form (e.g., solution, gel, paste, and the like) or in
the
form of an implant. For example, the bulking agent may be in the form of a
three dimensional object, such as, a film, mesh, microsphere, bead, or another
shape). In certain embodiments, the bulking agent may be combined with a
polymeric composition (e.g., a gel or hydrogel) to facilitate delivery of the
agent
into the host.
Representative examples of bulking agents include inorganic
materials such as minerals, glasses, ceramics (e.g., ground and powdered
ceramics and glasses), clays, calcium carbonate, magnesium carbonate,
83
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
pumice, talc, zinc oxide, hydroxyapatite, cornstarch, cellulose, wood (e.g.,
saw
dust), naturally occurring materials such as bone, leather, horn, hair,
various
proteinaceous materials, such as collagen and collagen containing materials
(e.g., collagen based injectable products, including those derived from non-
bovine, human, or recombinant sources), polysaccharides (e.g., hyaluronic
acid), and synthetic polymers (e.g., ethylene vinyl alcohol polymer implant,
acrylates, methacrylates, acrylics, polydimethylsiloxane, silicone, and the
like).
Bulking agents for use in treating diverticulitis may be combined
with one or more fibrosis-inducing agents as described herein. Bulking agents
include but are not limited to commercially available products such as
collagen-
based injectable products, including those derived from non-bovine, human, or
recombinant sources; injectable microspheres from Artes Medical, Inc. (San
Diego, CA); ENTERYX (ethylene vinyl alcohol polymer implant from Boston
Scientific Corporation); hydroxyapatite loaded gel (COAPATITE from BioForm
Medical, Inc., San Mateo, CA); micronized alloderm acellular matrix (CYMETRA
from LifeCell Corporation, Branchburg, NJ); non-animal stabilized hyaluronic
acid (NASHA and DEFLUX from Q-Med); pyrolytic carbon-coated micro-beads
in hydrogel containing beta-glucan (DURASPHERE from Carbon Medical
Technologies, Inc. St. Paul, MN and Boston Scientific Corporation, Natick,
MA);
engineered collagen fibrils (Organogenesis, Inc., Canton, MA); hylan polymer
(HYLAGEL URO from Genzyme); MACROPLASTIQUE (polydimethylsiloxane
in hydrogel carrier) from Uroplasty, Inc. (Minneapolis, MN); microspheres
(e.g.,
acrylic beads, such as those available from Biosphere Medical, Inc.
Marlborough, MA); urethral bulking agents containing silk and elastin proteins
(Protein Polymer Technologies, San Diego, CA); cross-linked silicon
microballoon filled with biocompatible polymer (UROVIVE from American
Medical Systems, Minnetonka, MN); and URYX bulking agent and Embolyx
from Microtherapeutics, Inc., San Clemente, CA and Genyx Medical, Inc., Aliso
Viejo, CA. Other manufacturers of carriers suitable for use in bulking
compositions include C.R. Bard, Inc. (Murray Hill, NJ), Collagenesis, Inc.
84
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
(Acton, MA), American Medical Systems, Mentor, Uromed Corporation
(Norwood, MA), Boston Scientific Corporation, Johnson & Johnson (Ethicon,
Inc.), Cook Group, Inc. (Bloomington, IN), W.L. Gore & Associates, and SURx,
Inc. (Pleasonton, CA).
Regional and Local Delivery of Fibrosis-Inducing Agents to Diverticucla
A variety of drug-delivery technologies are available for regional
and local delivery of therapeutic agents. Several of these techniques may be
suitable to achieve preferentially elevated levels of fibrosis-inducing agents
within the diverticula, including: (a) using drug-delivery catheters for local
or
regional delivery of fibrosing agents into the diverticula (typically, drug
delivery
catheters are advanced into tissues under endoscopic or radiological guidance
until they reach the opening of the diverticula; the fibrosing agent can then
be
released from the catheter lumen in high local concentrations in order to
deliver
therapeutic doses of the drug to the diverticula); (b) drug localization
techniques
such as magnetic, ultrasonic or MRI-guided drug delivery; (c) chemical
modification of the fibrosis-inducing drug or formulation designed to increase
uptake of the agent into the diverticula (e.g., modification of the drug or
formulation to include antibodies directed against damaged or healing tissue
components such as macrophages, neutrophils, smooth muscle cells,
fibroblasts, extracellular matrix components, fibrin, components of the
clotting
cascade); (d) chemical modification of the fibrosis-inducing drug or
formulation
designed to localize the drug to areas of bleeding or disrupted vasculature
such
as encapsulation of the drug into site directed liposomes; and/or (e)
microparticulate silk and/or silk strands (e.g., linear, branched, and/or
coiled)
are also useful for directed delivery (via endoscope or guided catheter) into
the
diverticula; (f) injectable collagen-containing hemostatic formulations such
as
COSTASIS (Angiotech Pharmaceuticals, Inc., Vancouver, BC) or materials
made from 4-armed thiol PEG (10K), a 4-armed NHS PEG(10K) and
methylated collagen (described below), or materials made from 4-armed thiol
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
PEG (10K), a 4-armed NHS PEG(10K) and collagen or gelatin, either alone, or
loaded with a fibrosis-inducing agent and/or an anti-infective agent, applied
to
the diverticula; (g) sprayable in situ forming PEG-containing formulations
such
as COSEAL (Angiotech Pharmaceuticals, Inc., Canada), FOCALSEAL
(Genzyme Corporation, Cambridge, MA), SPRAYGEL or DURASEAL (both
from Confluent Surgical, Inc., Waltham, MA), either alone, or loaded with a
fibrosis-inducing agent and/or an anti-infective agent and/or a hemostatic
agent,
applied to the diverticula; (h) fibrinogen-containing formulations such as
FLOSEAL or TISSEAL (both from Baxter Healthcare Corporation; Fremont,
CA), either alone, or loaded with a fibrosis-inducing agent and/or an anti-
infective agent, applied to the diverticula; (i) hyaluronic acid-containing
formulations (either non-crosslinked, crosslinked or chemically modified) such
as PERLANE or RESTYLANE (both from Q-Med AB, Sweden), HYLAFORM
(Inamed Corporation; Santa Barbara, CA), SYNVISC (Biomatrix, Inc.;
Ridgefied, NJ), SEPRAFILM or SEPRACOAT (both from Genzyme
Corporation; Cambridge, MA) loaded with a fibrosis-inducing agent and/or an
anti-infective agent and/or a hemostatic agent applied to the diverticula; (j)
polymeric gels for surgical implantation such as REPEL (Life Medical Sciences,
Inc.; Princeton, NJ) or FLOWGEL (Baxter Healthcare Corporation, Deerfield, IL)
loaded with a fibrosis-inducing agent and/or an anti-infective agent and/or a
hemostatic agent applied to the diverticula; (k) surgical adhesives containing
one or more cyanoacrylate monomers (e.g., methyl cyanoacrylate, ethyl
cyanoacrylate, butyl cyanoacrylate, octyl cyanoacrylate, methoxypropyl
cyanoacrylate) such as DERMABOND (Johnson & Johnson, Inc.), INDERMIL
(United States Surgical; Norwalk, CT), GLUSTITCH (Blacklock Medical
Products, Inc., Canada) or TISSU MEND II (Veterinary Products Laboratories;
Phoenix, AZ), VETBOND (3M Company; St. Paul, MN), TISSUEMEND (TEI
Biosciences, Inc.; Boston, MA), HISTOACRYL or HISTOACRYL BLUE (Davis &
Geck; St. Louis, MO) and ORABASE SOOTHE-N-SEAL LIQUID
PROTECTANT (Colgate-Palmolive Company; New York; NY), either alone, or
86
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
loaded with a fibrosis-inducing agent and/or an anti-infective agent and/or a
hemostatic agent, applied to the diverticula; (I) other biocompatible tissue
fillers
loaded with a fibrosis-inducing agent and/or an anti-infective agent and/or a
hemostatic agent, such as those made by BioCure, Inc. (Norcross, GA), 3M
Company and Neomend, Inc. (Sunnyvale, CA), applied to the diverticula; (m)
polysaccharide gels such as the ADCON series of gels (Gliatech, Inc.;
Cleveland, OH) either alone, or loaded with a fibrosis-inducing agent and/or
an
anti-infective agent and/or a hemostatic agent, applied to the diverticula;
(n)
films, sponges or meshes such as INTERCEED or VICRYL mesh (Ethicon, Inc.,
a Johnson & Johnson Company, Somerville, NJ), and GELFOAM (Pharmacia &
Upjohn Company; Kalamazoo, MI) either alone, or loaded with a fibrosis-
inducing agent and/or an anti-infective agent and/or a hemostatic agent,
applied
to the diverticula; (o) polymeric (non-crosslinked and crosslinked) strands,
braids, fibers, particles, polymeric knitted, woven , non-woven or
electrosprayed material (e.g., linear, branched, and/or coiled; hydrogel and
non
hydrogel coated) are also useful for directed delivery (via endoscope or
guided
catheter) into the diverticula;and (p) a hydrogel that is formed from an amino-
functionalized polyethylene glycol (e.g., 4-armed tetra-amino PEG [10k]) and a
4-armed NHS functionalized PEG (e.g., pentaerythritol poly(ethylene
glycol)ether tetra-succinimidyl glutarate [10K]). This hydrogel may further
contain collagen, methylated collagen and/or gelatin. Other hydrogels can
include crosslinked polysaccharides (e.g,. carboxymethyl cellulose, dextran,
hyaluronic acid, chitosan, alignate etc), vinyl based crosslinked hydrogels
(e.g.,
polyacrylates, polyacrylic acids, polymethacrylic acids,
poly(hydroxyethylmethacrylate)).These hydrogels can further comprise a
fibrosis-inducing agent and/or an anti-infective agent and/or a hemostatic
agent,
and can be applied to the diverticula.
In one embodiment, a mesh or film or other similar material may
be inserted or applied to a diverticulum. This mesh, film, or similar material
is
capable, at least in part, to fill the diverticulum. An in-situ sealant, glue,
or
87
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
embolic agent may be used to maintain the position of the mesh or film in the
diverticulum. In other embodiments, a fibrosis-inducing agent, an anti-
infective
agent, and/or a hemostatic agent, may also be used in combination with the
mesh or film. The one or more agents may be applied directly to the
diverticular tissue or to the mesh or film according to any of the methods
described herein.
In one embodiment, the fibrosis-inducing agent may be delivered
to the diverticula as a solution via a catheter inserted into the diverticula
under
endoscopic or radiographic guidance. The fibrosis-inducing agent can be
incorporated directly into the solution to provide a homogeneous solution or
dispersion. In certain embodiments, the solution is an aqueous solution. The
aqueous solution may further include buffer salts, as well as viscosity
modifying
agents (e.g., hyaluronic acid, alginates, CMC, and the like). In another
aspect
of the invention, the solution can include a biocompatible solvent, such as
ethanol, DMSO, glycerol, PEG-200, PEG-300 or NMP.
Sustained-Release and Coating Preparations of Fibrosis-Inducing Agents
For many of the aforementioned therapeutic agents, the fibrosis-
inducing agent can be incorporated into a carrier so that therapeutic levels
can
be delivered locally into the diverticula for periods long enough for complete
healing and fibrosis to occur (weeks to months). For example, a desired
fibrosis-inducing agent may be admixed with, blended with, conjugated to, or,
otherwise modified to contain a polymeric composition (which may be either
biodegradable or non-biodegradable) or non-polymeric composition in order to
release the fibrosis-inducing agent over a period of time. For the above
embodiments, biodegradable and non-biodegradable polymers, polymer
conjugates as well as non-polymeric materials can be used to accomplish the
local delivery of the fibrosis-inducing agent, hemostatic agent and/or anti-
infective agent into the diverticula.
88
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Representative examples of biodegradable polymers suitable for
the delivery of fibrosis-inducing agents, hemostatic agents and/or anti-
infective
agents into diverticula include albumin, collagen, gelatin, hyaluronic acid,
aliphatic, heteroatomic and aromatic esters of hyaluronic acid, thiol
containing
hyaluronic acid derivatives, starch, cellulose and cellulose derivatives
(e.g.,
methylcellulose, hydroxypropylcellulose,
hydroxypropylmethylcellulose,
carboxymethylcellulose, cellulose acetate phthalate, cellulose acetate
succinate, hydroxypropylmethylcellulose phthalate), casein, dextrans,
carboxymethyl dextran, amino-dextran, polysaccharides, fibrinogen, poly(ether
ester) multiblock copolymers, based on poly(ethylene glycol) and poly(butylene
terephthalate), tyrosine-derived polycarbonates (e.g., U.S. Patent No.
6,120,491), poly(hydroxyl acids), poly(D,L-lactide), poly(D,L-lactide-co-
glycolide), poly(glycolide), poly(hydroxybutyrate),
polydioxanone,
poly(alkylcarbonate) and poly(orthoesters), polyesters, poly(hydroxyvaleric
acid), polydioxanone, poly(ethylene terephthalate), poly(malic acid),
poly(tartronic acid), poly(acrylamides), polyanhydrides, poly(ester-amides),
poly(ester-imid es), poly(ester-ureas),
poly(ester-urethane-ureas),
poly(anhydride-esters), poly(anhydride-imides), polyphosphazenes, poly(amino
acids), poly(alkylene oxide)-poly(ester) block copolymers (e.g., X-Y, X-Y-X or
Y-
X-Y, where X is a polyalkylene oxide and Y is a polyester (e.g., PLGA, PLA,
PCL, polydioxanone and copolymers thereof), and copolymers as well as
blends thereof. (see generally, Illum, L., Davids, S.S. (eds.) "Polymers in
Controlled Drug Delivery" Wright, Bristol, 1987; Arshady, J. Controlled
Release
17:1-22, 1991; Pitt, mt. J. Phar. 59:173-196, 1990; Holland et al., J.
Controlled
Release 4:155-0180, 1986).
Representative examples of non-degradable polymers suitable for
the delivery of fibrosis-inducing agents, hemostatic agents and/or anti-
infective
agents into diverticula include poly(ethylene-co-vinyl acetate) ("EVA")
copolymers, silicone rubber, acrylic polymers (e.g., polyacrylic acid,
polymethylacrylic acid, polymethylmethacrylate, poly(butyl methacrylate)),
89
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
poly(alkylcyanoacrylate) (e.g.,
poly(ethylcyanoacrylate),
poly(butylcyanoacrylate), poly(hexylcya noacrylate), and
poly(octylcyanoacrylate)), polyethylene, polypropylene, polyamides (nylon
6,6),
polyurethanes (including hydrophilic polyurethanes), poly(ester-urethanes),
poly(ether-urethanes), poly(ester-urea), poly(carbonate urethane)s, polyethers
(poly(ethylene oxide), poly(propylene oxide), polyoxyalkylene ether block
copolymers based on ethylene oxide and propylene oxide such as PLURONICs
and PLURONICs R and poly(tetramethylene glycol)), styrene-based polymers
(polystyrene, poly(styrene sulfonic acid), poly(styrene)-block-
poly(isobutylene)-
block-poly(styrene), poly(styrene)-poly(isoprene) block copolymers], and vinyl
polymers (polyvinylpyrrolidone, poly(vinyl alcohol), poly(vinyl acetate
phthalate)
as well as copolymers and blends thereof. Polymers may also be developed
which are either anionic (e.g., alginate, carrageenan, carboxymethyl
cellulose,
poly(acrylamido-2-methyl propane sulfonic acid) and copolymers thereof,
poly(methacrylic acid) and copolymers thereof, and poly(acrylic acid) and
copolymers thereof, as well as blends thereof) or cationic (e.g., chitosan,
poly-
L-lysine, polyethylenimine, and poly(ally1 amine) and blends thereof (see
generally, Dunn et at., J. Applied Polymer ScL 50:353-365, 1993; Cascone et
al., J. Materials Sc.: Materials in Medicine 5:770-774, 1994; Shiraishi et
al.,
Biol. Pharm. Bull. 16(11):1164-1168, 1993; Thacharodi and Rao, Intl J. Pharm.
120:115-118, 1995; Miyazaki et at., Intl J. Pharm. 118:257-263, 1995).
Particularly preferred polymeric carriers for sustained delivery of
the afformentioned therapeutic agents into diverticula include poly(ethylene-
co-
vinyl acetate), cellulose esters (nitrocellulose), poly(hydroxymethacrylate),
poly(methylmethacrylate), poly(ethylene-co-acrylic acid),
poly(vinylpyrrolidone)
polyurethanes (e.g., CHRONOFLEX AL and CHRONOFLEX AR (both from
CardioTech International, Inc., Woburn, MA) and BIONATE (Polymer
Technology Group, Inc., Emeryville, CA), poly (D,L-lactic acid) oligomers and
polymers, poly (L-lactic acid) oligomers and polymers, poly (glycolic acid),
copolymers of lactic acid and glycolic acid, poly (caprolactone), poly
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
(valerolactone), polyanhyd rides, poly(anhydride esters), poly(ester-amides),
poly(ester-ureas), copolymers of poly (caprolactone) or poly (lactic acid)
with a
polyethylene glycol (e.g., MePEG), polymers that comprise the residues of one
or more of the monomers selected from lactide, lactic acid, glycolide,
glycolic
acid, e-caprolactone, gamma-caprolactone, hydroxyvaleric acid, hydroxybutyric
acid, beta-butyrolactone, gamma-butyrolactone, gamma-valerolactone, y-
decanolactone, ö-decanolactone, trimethylene carbonate, 1,4-dioxane-2-one or
1,5-dioxepan-2one, poly(alkylene oxide)-poly(ester) block copolymers (e.g., X-
Y, X-Y-X, Y-X-Y, R-(Y-X), or R-(X-Y), where X is a polyalkylene oxide (e.g.,
poly(ethylene glycol, poly(propylene glycol) and block copolymers of
poly(ethylene oxide) and poly(propylene oxide) (e.g., PLURONIC and
PLURONIC R series of polymers from BASF Corporation, Mount Olive, NJ) and
Y is a polyester, wherein the polyester may comprise the residues of one or
more of the monomers selected from lactide, lactic acid, glycolide, glycolic
acid,
e-caprolactone, gamma-caprolactone, hydroxyvaleric acid, hydroxybutyric acid,
beta-butyrolactone, gamma-butyrolactone, gamma-valerolactone, y-
decanolactone, 5-decanolactone, trimethylene carbonate, 1,4-dioxane-2-one or
1,5-dioxepan-2one, silicone rubbers, poly(styrene)block-poly(isobutylene)-
block-poly(styrene), poly(acrylate) polymers, and blends, admixtures, or co-
polymers of any of the above. Other preferred polymers include collagen,
poly(alkylene oxide)-based polymers, polysaccharides such as hyaluronic acid,
chitosan and fucans, and copolymers of polysaccharides with degradable
polymers, as well as crosslinked compositions of the above.
Other representative polymers capable of sustained localized
delivery of fibrosis-inducing agents, hemostatic agents and/or anti-infective
agents into diverticula include carboxylic polymers, polyacetates,
polyacrylamides, polycarbonates, polyethers, substituted polyethylenes,
polyvinylbutyrals, polysilanes, polyureas, polyoxides, polystyrenes,
polysulfides,
polysulfones, polysulfonides, polyvinylhalides, pyrrolidones, isoprene
rubbers,
thermal-setting polymers, cross-linkable acrylic and methacrylic polymers,
91
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
ethylene acrylic acid copolymers, styrene acrylic copolymers, vinyl acetate
polymers and copolymers, vinyl acetal polymers and copolymers, epoxies,
melamines, other amino resins, phenolic polymers, and copolymers thereof,
water-insoluble cellulose ester polymers (including cellulose acetate
propionate,
cellulose acetate, nitrocellulose, cellulose acetate butyrate, cellulose
nitrate,
cellulose acetate phthalate, and mixtures thereof), polyvinylpyrrolidone
(pvp),
polyethylene glycols, polyethylene oxides, polyvinyl alcohol, polyethers,
polyhydroxyacrylate, dextran, xanthan, hydroxypropyl cellulose, methyl
cellulose, and homopolymers and copolymers of N-vinylpyrrolidone, N-
vinyllactam, N-vinyl butyrolactam, N-vinyl caprolactam, other vinyl compounds
having polar pendant groups, acrylate and methacrylate having hydrophilic
esterifying groups, hydroxyacrylate, and acrylic acid, and combinations
thereof;
cellulose esters and ethers, ethyl cellulose, nitro-cellulose, hydroxyethyl
cellulose, cellulose nitrate, cellulose acetate, cellulose acetate butyrate,
cellulose acetate propionate, polyacrylate, natural and synthetic elastomers,
acetal, styrene polybutadiene, acrylic resin, polyvinylidene chloride,
polycarbonate, homopolymers and copolymers of vinyl compounds,
polyvinylchloride, and polyvinylchloride acetate.
Representative examples of patents relating to drug-delivery
polymers and their preparation include PCT Publication Nos. WO 98/19713,
WO 01/17575, WO 01/41821, WO 01/41822, and WO 01/15526 (as well as
their corresponding U.S. applications), and U.S. Patent Nos. 4,500,676,
4,582,865, 4,629,623, 4,636,524, 4,713,448, 4,795,741, 4,913,743, 5,069,899,
5,099,013, 5,128,326, 5,143,724, 5,153,174, 5,246,698, 5,266,563, 5,399,351,
5,525,348, 5,800,412, 5,837,226, 5,942,555, 5,997,517, 6,007,833, 6,071,447,
6,090,995, 6,106,473, 6,110,483, 6,121,027, 6,156,345, 6,214,901, 6,368,611
6,630,155, 6,528,080, RE37,950, 6,46,1631, 6,143,314, 5,990,194, 5,792,469,
5,780,044, 5,759,563, 5,744,153, 5,739,176, 5,733,950, 5,681,873, 5,599,552,
5,340,849, 5,278,202, 5,278,201, 6,589,549, 6,287,588, 6,201,072, 6,117,949,
6,004,573, 5,702,717, 6,413,539, and 5,714,159, 5,612,052 and U.S. Patent
92
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Application Publication Nos. 2003/0068377, 2002/0192286, 2002/0076441, and
2002/0090398.
The polymers as described herein can also be blended or
copolymerized in various compositions appropriately to deliver therapeutic
doses of fibrosis-inducing agents, hemostatic agents, and/or anti-infective
agents to diverticula.
Polymeric carriers for fibrosis-inducing agents, hemostatic agents,
and/or anti-infective agents can be fashioned in a variety of forms, with
desired
release characteristics and/or with specific properties. For example,
polymeric
carriers may be fashioned to release a therapeutic agent upon exposure to a
specific triggering event such as pH (see, e.g., Heller et at., "Chemically
Self-
Regulated Drug Delivery Systems," in Polymers in Medicine III, Elsevier
Science Publishers B.V., Amsterdam, 1988, pp. 175-188; Kang et al., J. Applied
Polymer Sci. 48:343-354, 1993; Dong et al., J. Controlled Release 19:171-178,
1992; Dong and Hoffman, J. Controlled Release /5:141-152, 1991; Kim et al.,
J. Controlled Release 28:143-152, 1994; Cornejo-Bravo et al., J. Controlled
Release 33:223-229, 1995; Wu and Lee, Pharm. Res. 10(10)1544-1547, 1993;
Serres et at., Pharm. Res. /3(2)196-201, 1996; Peppas, "Fundamentals of pH-
and Temperature-Sensitive Delivery Systems," in Gurny et al. (eds.), Pulsatile
Drug Delivery, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, 1993, pp.
41-55; Doelker, "Cellulose Derivatives," 1993, in Peppas and Langer (eds.),
Biopolymers I, Springer-Verlag, Berlin). Representative examples of pH-
sensitive polymers include poly(acrylic acid) and its derivatives (including
for
example, homopolynners such as poly(aminocarboxylic acid); poly(acrylic acid);
poly(methyl acrylic acid), copolymers of such homopolymers, and copolymers
of poly(acrylic acid) and acrylmonomers such as those discussed above. Other
pH sensitive polymers include polysaccharides such as cellulose acetate
phthalate; hydroxypropylmethylcellulose
phthalate;
hydroxypropylmethylcellulose acetate succinate; cellulose acetate
trimellilate;
93
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
and chitosan. Yet other pH sensitive polymers include any mixture of a pH
sensitive polymer and a water-soluble polymer.
Likewise, fibrosis-inducing agents, hemostatic agents, and/or anti-
infective agents can be delivered to diverticula via polymeric carriers which
are
temperature sensitive (see, e.g., Chen et at., "Novel Hydrogels of a
Temperature-Sensitive Pluronic Grafted to a Bioadhesive Polyacrylic Acid
Backbone for Vaginal Drug Delivery," in Proceed. Intern. Symp. Control. Rel.
Bioact, Mater. 22:167-168, Controlled Release Society, Inc., 1995; Okano,
"Molecular Design of Stimuli-Responsive Hydrogels for Temporal Controlled
Drug Delivery," in Proceed. Intern, Symp. Control. Rel. Bioact. Mater. 22:111-
112, Controlled Release Society, Inc., 1995; Johnston et al., Pharm. Res.
9(3):425-433, 1992; Tung, Intl J. Pharm. 107:85-90, 1994; Harsh and Gehrke,
J. Controlled Release /7:175-186, 1991; Bae et al., Pharm. Res. 8(4):531-537,
1991; Dinarvand and D'Emanuele, J. Controlled Release 36:221-227, 1995; Yu
and Grainger, "Novel Thermo-sensitive Amphiphilic Gels: Poly N-
isopropylacrylamide-co-sodium acrylate-co-n-N-alkylacrylamide Network
Synthesis and Physicochemical Characterization," Dept. of Chemical &
Biological Sci., Oregon Graduate Institute of Science & Technology, Beaverton,
OR, pp. 820-821; Zhou and Smid, "Physical Hydrogels of Associative Star
Polymers," Polymer Research Institute, Dept. of Chemistry, College of
Environmental Science and Forestry, State Univ. of New York, Syracuse, NY,
pp. 822-823; Hoffman et al., "Characterizing Pore Sizes and Water 'Structure'
in
Stimuli-Responsive Hydrogels," Center for Bioengineering, Univ. of
Washington, Seattle, WA, p. 828; Yu and Grainger, "Thermo-sensitive Swelling
Behavior in Crosslinked N-isopropylacrylamide Networks: Cationic, Anionic and
Ampholytic Hydrogels," Dept. of Chemical & Biological Sc., Oregon Graduate
Institute of Science & Technology, Beaverton, OR, pp. 829-830; Kim et al.,
Pharm. Res. 9(3):283-290, 1992; Bae et at., Pharm. Res. 8(5):624-628, 1991;
Kono et al., J. Controlled Release 30:69-75, 1994; Yoshida et al., J.
Controlled
Release 32:97-102, 1994; Okano et al., J. Controlled Release 36:125-133,
94
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
1995; Chun and Kim, J. Controlled Release 38:39-47, 1996; D'Emanuele and
Dinarvand, Intl J. Pharm. /18:237-242, 1995; Katono et at., J. Controlled
Release /6:215-228, 1991; Hoffman, "Thermally Reversible Hydrogels
Containing Biologically Active Species," in Migliaresi et at. (eds.), Polymers
in
Medicine III, Elsevier Science Publishers B.V., Amsterdam, 1988, pp. 161-167;
Hoffman, "Applications of Thermally Reversible Polymers and Hydrogels in
Therapeutics and Diagnostics," in Third International Symposium on Recent
Advances in Drug Delivery Systems, Salt Lake City, UT, Feb. 24-27, 1987, pp.
297-305; Gutowska et al., J. Controlled Release 22:95-104, 1992; Palasis and
Gehrke, J. Controlled Release 18:1-12, 1992; Paavola et al., Pharm. Res.
12(12):1 997-2002, 1995).
Representative examples of thermogelling polymers, and their
gelatin temperature [LCST ( G)] include homopolymers such as
poly(N-methyl-N-n-propylacrylamide), 19.8; poly(N-n-propylacrylamide), 21.5;
poly(N-methyl-N-isopropylacrylamide), 22.3; poly(N-n-propylmethacrylamide),
28.0; poly(N-isopropylacrylamide), 30.9; poly(N, n-diethylacrylamide), 32.0;
poly(N-isopropylmethacrylamide), 44.0; poly(N-cyclopropylacrylamide), 45.5;
poly(N-ethylmethyacrylamide), 50.0; poly(N-methyl-N-ethylacrylamide), 56.0;
poly(N-cyclopropylmethacrylamide), 59.0; and poly(N-ethylacrylamide), 72Ø
Moreover, thermogelling polymers may be made by preparing copolymers
between (among) monomers of the above, or by combining such
homopolymers with other water-soluble polymers such as acrylmonomers (e.g.,
acrylic acid and derivatives thereof, such as methylacrylic acid, acrylate and
derivatives thereof, such as butyl methacrylate, acrylamide, and N-n-butyl
acrylamide).
Other representative examples of thermogelling polymers include
cellulose ether derivatives such as hydroxypropyl cellulose, 41 C; methyl
cellulose, 55 C; hydroxypropylmethyl cellulose, 66 C; and ethylhydroxyethyl
cellulose, polyalkylene oxide-polyester block copolymers of the structure X-Y,
Y-X-Y and X-Y-X where X is a polyalkylene oxide and Y is a biodegradable
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
polyester (e.g., PLG-PEG-PLG) and PLURONICS such as F-127, 10 - 15 C;
L-122, 19 C; L-92, 26 C; L-81, 20 C; and L-61, 24 C.
Representative examples of patents and patent applications
relating to thermally gelling polymers and their preparation include U.S.
Patent
Nos. 6,451,346; 6,201,072; 6,117,949; 6,004,573; 5,702,717; and 5,484,610;
and PCT Publication Nos. WO 99/07343; WO 99/18142; WO 03/17972; WO
01/82970; WO 00/18821; WO 97/15287; WO 01/41735; WO 00/00222; and WO
00/38651.
Another representative example of a gel composition is a gel
formed by the combination of a chitosan solution with glycerol phosphate.
Fibrosis-inducing agents, hemostatic agents, and/or anti-infective
agents may be linked by occlusion in the matrices of the polymer, bound by
covalent linkages, or encapsulated in microcapsules. Within certain preferred
embodiments of the invention, therapeutic compositions are provided in non-
capsular formulations such as microspheres (ranging from nanometers to
micrometers in size), pastes, and threads of various size, films and sprays.
Within certain embodiments, therapeutic compositions may be
fashioned in any size ranging from 50 nm to 500 pm, depending upon the
particular use (diverticula can occur in a variety of anatomical sites and
sizes to
be described below). These compositions can be in the form of microspheres
(porous or non-porous), microparticles, and/or nanoparticles.
These
compositions can be formed, for example, by spray-drying methods, milling
methods, coacervation methods, W/O (water-oil) emulsion methods, W/O/W
emulsion methods, and solvent evaporation methods. In some embodiments,
these compositions can include microemulsions, emulsions, liposomes and
micelles. Alternatively, such compositions may also be readily applied as a
"spray", which solidifies into a film or tissue surface coating at the
implantation
site. Such sprays may be prepared from microspheres of a wide array of sizes,
including for example, from 0.1 pm to 3 p.m, from 10 tAm to 30 [im, and from
30
[im to 100 pm, and are ideal for delivery via the delivery port of an
endoscope.
96
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Therapeutic compositions of the present invention may also be
prepared in a variety of paste or gel forms. For example, within one
embodiment of the invention, therapeutic compositions are provided which are
liquid at one temperature (e.g., temperature greater than 37 C, such as 40 C,
45 C, 50 C, 55 C or 60 C), and solid or semi-solid at another temperature
(e.g.,
ambient body temperature, or any temperature lower than 37 C). Such
"thermopastes" may be readily made utilizing a variety of techniques (see,
e.g.,
PCT Publication WO 98/24427). Other pastes may be applied as a liquid,
which solidify in vivo due to dissolution of a water-soluble component of the
paste and precipitation of encapsulated drug into the aqueous body
environment. These pastes and gels containing fibrosis-inducing agents,
hemostatic agents and/or anti-infective agents are particularly useful for
application to the lumen of a divertica under radiographic or endoscopic
guidance.
Within further aspects of the present invention, polymeric carriers
are provided which are adapted to contain and release a hydrophobic fibrosis-
inducing, hemostatic and/or anti-infective compound, and/or the carrier
containing the hydrophobic compound(s), in combination with a carbohydrate,
protein or polypeptide. In certain embodiments, the polymeric carrier provides
sustained release for a therapeutic agent (e.g., a fibrosis-inducing agent,
anti-
infective agent, an antibiotic, or another type of agent) from a composition
comprising the carrier and an agent. Within certain embodiments, the
polymeric carrier contains or comprises regions, pockets, or granules of one
or
more hydrophobic compounds. For example, within one embodiment of the
invention, hydrophobic compounds may be incorporated within a matrix which
contains the hydrophobic therapeutic compound, followed by incorporation of
the matrix within the polymeric carrier. A variety of matrices can be utilized
in
this regard, including for example, carbohydrates and polysaccharides such as
starch, cellulose, dextran, methylcellulose, sodium alginate, heparin,
chitosan
and hyaluronic acid and proteins or polypeptides such as albumin, collagen,
97
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
fibrin, and/or gelatin. Within alternative embodiments, hydrophobic compounds
may be contained within a hydrophobic core, and this core contained within a
hydrophilic shell.
Within another embodiment, the polymeric carriers used to deliver
therapeutic agents into the diverticula can be materials that are formed in
situ.
In one embodiment, the precursors can be monomers or macromers that
contain unsaturated groups which can be polymerized or crosslinked. The
monomers or macromers can then, for example, be injected into the diverticular
sac or onto the surface of the diverticula and polymerized or crosslinked in
situ
using a radiation source (e.g., visible or UV light) or a free radical system
(e.g.,
potassium persulfate and ascorbic acid or iron and hydrogen peroxide). The
polymerization or crosslinking step can be performed immediately prior to,
simultaneously to, or post injection of the reagents into the diverticula.
Representative examples of compositions that undergo free radical
polymerization or crosslinking reactions are described in WO 01/44307, WO
01/68720, WO 02/072166, WO 03/043552, WO 93/17669, and WO 00/64977,
U.S. Patent Nos. 5,900,245; 6,051,248; 6,083,524; 6,177,095; 6,201,065;
6,217,894; 6,639,014; 6,352,710; 6,410,645; 6,531,147; 5,567,435; 5,986,043;
and 6,602,975, and U.S. Patent Application Publication Nos. 2002/012796,
2002/0127266, 2002/0151650, 2003/0104032, 2002/0091229, and
2003/0059906.
In another embodiment, the reagents can undergo an
electrophilic-nucleophilic reaction to produce a crosslinked matrix. Polymers
terminated with nucleophilic groups such as amine, sulfhydryl, hydroxyl, -PH2
or
CO-NH-NH2 can be used as the nucleophilic reagents and polymers terminated
with electrophilic groups such as succinimidyl, carboxylic acid, aldehyde,
epoxide, isocyanate, vinyl, vinyl sulfone, maleimide, -S-S-(C5H4N) or
activated
esters, such as are used in peptide synthesis can be used as the electrophilic
reagents. For example, a 4-armed thiol derivatized poly(ethylene glycol)
(e.g.,
pentaerythritol poly(ethylene glycol)ether tetra-sulfhydryl) can be reacted
with a
98
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
4 armed NHS-derivatized polyethylene glycol (e.g., pentaerythritol
poly(ethylene glycol)ether tetra-succinimidyl glutarate) under basic
conditions
(pH > about 8). Representative examples of compositions that undergo such
electrophilic-nucleophilic crosslinking reactions are described, for example,
in
U.S. Patent. Nos. 5,752,974; 5,807,581; 5,874,500; 5,936,035; 6,051,648;
6,165,489; 6,312,725; 6,458,889; 6,495,127; 6,534,591; 6,624,245; 6,566,406;
6,610,033; 6,632,457; U.S. Patent Application Publication No.
2003/0077272A1; and co-pending patent applications entitled "Tissue Reactive
Compounds and Compositions and Uses Thereof' (U.S. Serial No. 60/437,384,
filed December 30, 2002, and U.S. Serial No. 60/44,924, filed January 17,
2003) and "Drug Delivery from Rapid Gelling Polymer Composition" (U.S. Serial
No. 60/437,471, filed December 30, 2002, and U.S. Serial No. 60/440,875, filed
January 17, 2003).
In another embodiment, the electrophilic- or nucleophilic-
terminated polymers can further comprise a polymer that can enhance the
mechanical and/or adhesive properties of the in situ forming compositions.
This
polymer can be a degradable or non-degradable polymer. For example, the
polymer may be collagen or a collagen derivative, for example methylated
collagen. An example of an in situ forming composition uses pentaerythritol
poly(ethylene glycol)ether tetra-sulfhydryl] (4-armed thiol PEG),
pentaerythritol
poly(ethylene glycol)ether tetra-succinimidyl glutarate] (4-armed NHS PEG) and
methylated collagen as the reactive reagents. This composition, when mixed
with the appropriate buffers can produce a crosslinked hydrogel. (See, e.g.,
U.S. Patent Nos. 5,874,500; 6,051,648; 6,166,130; 5,565,519 and 6,312,725).
In another embodiment, the in situ forming material polymer can
be a polyester. Polyesters that can be used in in situ forming compositions
include poly(hydroxyesters). In
another embodiment, the polyester can
comprise the residues of one or more of the monomers selected from lactide,
lactic acid, glycolide, glycolic acid, e-caprolactone, gamma-caprolactone,
hydroxyvaleric acid, hydroxybutyric acid, beta-butyrolactone, gamma-
99
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
butyrolactone, gamma-valerolactone, y-decanolactone, ö-decanolactone,
trimethylene carbonate, 1,4-dioxane-2-one or 1,5-dioxepan-2-one.
Representative examples of these types of compositions are described in U.S.
Patent. Nos. 5,874,500; 5,936,035; 6,312,725; 6,495,127 and PCT Publication
Nos. WO 2004/028547.
In another embodiment, the electrophilic-terminated polymer can
be partially or completely replaced by a small molecule or oligomer that
comprises an electrophilic group (e.g., disuccinimidyl glutarate).
In another embodiment, the nucleophilic¨terminated polymer can
be partially or completely replaced by a small molecule or oligomer that
comprises a nucleophilic group (e.g., dicysteine, dilysine, trilysine, etc.).
Other examples of in situ forming materials that can be used
include those based on the crosslinking of proteins (described in, for
example,
U.S. Patent Nos. RE38158; 4,839,345; 5,514,379, 5,583,114; 6,310,036;
6,458,147; 6,371,975; US Patent Application Publication Nos.
2004/0063613A1, 2002/0161399A1, and 2001/0018598A1, and PCT
Publication Nos. WO 03/090683, WO 01/45761, WO 99/66964, and WO
96/03159) and those based on isocyanate or isothiocyanate capped polymers
(see, e.g., PCT Publication No. WO 04/021983).
First and Second Synthetic Polymers
In one embodiment, crosslinked polymer compositions (in other
words, crosslinked matrices) are prepared by reacting a first synthetic
polymer
containing two or more nucleophilic groups with a second synthetic polymer
containing two or more electrophilic groups, where the electrophilic groups
are
capable of covalently binding with the nucleophilic groups. In one embodiment,
the first and second polymers are each non-immunogenic. In another
embodiment, the matrices are not susceptible to enzymatic cleavage by, e.g., a
matrix metalloproteinase (e.g., collagenase) and are therefore expected to
have
greater long-term persistence in vivo than collagen-based compositions.
100
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
As used herein, the term "polymer" refers inter alia to polyalkyls,
polyamino acids, polyalkyleneoxides and polysaccharides. Additionally, for
external or oral use, the polymer may be polyacrylic acid or carbopol. As used
herein, the term "synthetic polymer" refers to polymers that are not naturally
occurring and that are produced via chemical synthesis. As such, naturally
occurring proteins such as collagen and naturally occurring polysaccharides
such as hyaluronic acid are specifically excluded. Synthetic collagen, and
synthetic hyaluronic acid, and their derivatives, are included. Synthetic
polymers containing either nucleophilic or electrophilic groups are also
referred
to herein as "multifunctionally activated synthetic polymers." The term
"multifunctionally activated" (or, simply, "activated") refers to synthetic
polymers
which have, or have been chemically modified to have, two or more nucleophilic
or electrophilic groups which are capable of reacting with one another (i.e.,
the
nucleophilic groups react with the electrophilic groups) to form covalent
bonds.
Types of multifunctionally activated synthetic polymers include difunctionally
activated, tetrafunctionally activated, and star-branched polymers.
Multifunctionally activated synthetic polymers for use in the
present invention must contain at least two, more preferably, at least three,
functional groups in order to form a three-dimensional crosslinked network
with
synthetic polymers containing multiple nucleophilic groups (i.e., "multi-
nucleophilic polymers"). In other words, they must be at least difunctionally
activated, and are more preferably trifunctionally or tetrafunctionally
activated.
If the first synthetic polymer is a difunctionally activated synthetic
polymer, the
second synthetic polymer must contain three or more functional groups in order
to obtain a three-dimensional crosslinked network. Most preferably, both the
first and the second synthetic polymer contain at least three functional
groups.
Synthetic polymers containing multiple nucleophilic groups are
also referred to generically herein as "multi-nucleophilic polymers." For use
in
the present invention, multi-nucleophilic polymers must contain at least two,
more preferably, at least three, nucleophilic groups. If a synthetic polymer
101
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
containing only two nucleophilic groups is used, a synthetic polymer
containing
three or more electrophilic groups must be used in order to obtain a three-
dimensional crosslinked network.
Preferred multi-nucleophilic polymers for use in the compositions
and methods of the present invention include synthetic polymers that contain,
or have been modified to contain, multiple nucleophilic groups such as primary
amino groups and thiol groups. Preferred multi-nucleophilic polymers include:
(i) synthetic polypeptides that have been synthesized to contain two or more
primary amino groups or thiol groups; and (ii) polyethylene glycols that have
been modified to contain two or more primary amino groups or thiol groups. In
general, reaction of a thiol group with an electrophilic group tends to
proceed
more slowly than reaction of a primary amino group with an electrophilic
group.
In one embodiment, the multi-nucleophilic polypeptide is a
synthetic polypeptide that has been synthesized to incorporate amino acid
residues containing primary amino groups (such as lysine) and/or amino acids
containing thiol groups (such as cysteine). Poly(lysine), a synthetically
produced polymer of the amino acid lysine (145 MW), is particularly preferred.
Poly(lysine)s have been prepared having anywhere from 6 to about 4,000
primary amino groups, corresponding to molecular weights of about 870 to
about 580,000.
Poly(lysine)s for use in the present invention preferably have a
molecular weight within the range of about 1,000 to about 300,000; more
preferably, within the range of about 5,000 to about 100,000; most preferably,
within the range of about 8,000 to about 15,000. Poly(lysine)s of varying
molecular weights are commercially available from Peninsula Laboratories, Inc.
(Belmont, Calif.) and Aldrich Chemical (Milwaukee, WI).
Polyethylene glycol can be chemically modified to contain multiple
primary amino or thiol groups according to methods set forth, for example, in
Chapter 22 of Poly(ethylene Glycol) Chemistry: Biotechnical and Biomedical
Applications, J. Milton Harris, ed., Plenum Press, N.Y. (1992). Polyethylene
102
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
glycols which have been modified to contain two or more primary amino groups
are referred to herein as "multi-amino PEGs." Polyethylene glycols which have
been modified to contain two or more thiol groups are referred to herein as
"multi-thiol PEGs." As used herein, the term "polyethylene glycol(s)" includes
modified and or derivatized polyethylene glycol(s).
Various forms of multi-amino PEG are commercially available
from Shearwater Polymers (Huntsville, Ala.) and from Huntsman Chemical
Company (Utah) under the name "Jeffamine." Multi-amino PEGs useful in the
present invention include Huntsman's Jeffamine diamines ("D" series) and
triannines ("T" series), which contain two and three primary amino groups per
molecule, respectively.
Polyamines such as ethylenediamine (H2N-CH2-CF12-NF12),
tetramethylenediamine (H2N-(CH2)4-NH2), pentamethylenediamine (cadaverine)
(H2N-(CH2)5-NH2), hexamethylenediamine (H2N-(CH2)6-NH2), di(2-
aminoethyl)amine (HN-(CH2-CH2-NH2)2), and tris(2-aminoethyl)amine (N-(CH2-
CH2-NH2)3) may also be used as the synthetic polymer containing multiple
nucleophilic groups.
Synthetic polymers containing multiple electrophilic groups are
also referred to herein as "multi-electrophilic polymers." For use in the
present
invention, the multifunctionally activated synthetic polymers must contain at
least two, more preferably, at least three, electrophilic groups in order to
form a
three-dimensional crosslinked network with multi-nucleophilic polymers.
Preferred multi-electrophilic polymers for use in the compositions of the
invention are polymers which contain two or more succinimidyl groups capable
of forming covalent bonds with nucleophilic groups on other molecules.
Succinimidyl groups are highly reactive with materials containing primary
amino
(NH2) groups, such as multi-amino PEG, poly(lysine), or collagen. Succinimidyl
groups are slightly less reactive with materials containing thiol (SH) groups,
such as multi-thiol PEG or synthetic polypeptides containing multiple cysteine
residues.
103
= CA 02610948 2012-12-14
As used herein, the term "containing two or more succinimidyl
groups" is meant to encompass polymers which are preferably commercially
available containing two or more succinimidyl groups, as well as those that
must be chemically derivatized to contain two or more succinimidyl groups. As
used herein, the term "succinimidyl group" is intended to encompass
sulfosuccinimidyl groups and other such variations of the "generic"
succinimidyl
group. The presence of the sodium sulfite moiety on the sulfosuccinimidyl
group serves to increase the solubility of the polymer.
Hydrophilic polymers and, in particular, various derivatized
polyethylene glycols, are preferred for use in the compositions of the present
invention. As used herein, the term "PEG" refers to polymers having the
repeating structure (OCH2-CH2). Structures for some specific,
tetrafunctionally
activated forms of PEG are shown in FIGS. 4 to 13 of U.S. Patent 5,874,500,
Examples of suitable PEGS include PEG
succinimidyl propionate (SE-PEG), PEG succinimidyl succinamide (SSA-PEG),
and PEG succinimidyl carbonate (SC-PEG). In one aspect of the invention, the
crosslinked matrix is formed in situ by reacting pentaerythritol poly(ethylene
glycol)ether tetra-sulfhydryl] (4-armed thiol PEG) and pentaerythritol
poly(ethylene glycol)ether tetra-succinimidyl glutaratei (4-armed NHS PEG) as
reactive reagents. Structures for these reactants are shown in U.S. Patent
5,874,500. Each of these materials has a core with a structure that may be
seen by adding ethylene oxide-derived residues to each of the hydroxyl groups
in pentaerythritol, and then derivatizing the terminal hydroxyl groups
(derived
from the ethylene oxide) to contain either thiol groups (so as to form 4-armed
thiol PEG) or N-hydroxysuccinimydyl groups (so as to form 4-armed NHS
PEG), optionally with a linker group present between the ethylene oxide
derived
backbone and the reactive functional group, where this product is commercially
available as COSEAL from Angiotech Pharmaceuticals Inc. Optionally, a group
"D" may be present in one or both of these molecules, as discussed in more
detail below.
104
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
As discussed above, preferred activated polyethylene glycol
derivatives for use in the invention contain succinimidyl groups as the
reactive
group. However, different activating groups can be attached at sites along the
length of the PEG molecule. For example, PEG can be derivatized to form
functionally activated PEG propionaldehyde (A-PEG), or functionally activated
PEG glycidyl ether (E-PEG), or functionally activated PEG-isocyanate (I-PEG),
or functionally activated PEG-vinylsulfone (V-PEG).
Hydrophobic polymers can also be used to prepare the
compositions of the present invention. Hydrophobic polymers for use in the
present invention preferably contain, or can be derivatized to contain, two or
more electrophilic groups, such as succinimidyl groups, most preferably, two,
three, or four electrophilic groups. As used herein, the term "hydrophobic
polymer" refers to polymers which contain a relatively small proportion of
oxygen or nitrogen atoms.
Hydrophobic polymers which already contain two or more
succinimidyl groups include, without limitation, disuccinimidyl suberate
(DSS),
bis(sulfosuccinimidyl) suberate (BS3), dithiobis(succinimidylpropionate)
(DSP),
bis(2-succinimidooxycarbonyloxy) ethyl sulfone (BSOCOES), and 3,3'--
dithiobis(sulfosuccinimidylpropionate (DTSPP), and their analogs and
derivatives. The above-referenced polymers are commercially available from
Pierce (Rockford, Ill.), under catalog Nos. 21555, 21579, 22585, 21554, and
21577, respectively.
Preferred hydrophobic polymers for use in the invention generally
have a carbon chain that is no longer than about 14 carbons. Polymers having
carbon chains substantially longer than 14 carbons generally have very poor
solubility in aqueous solutions and, as such, have very long reaction times
when mixed with aqueous solutions of synthetic polymers containing multiple
nucleophilic groups.
Certain polymers, such as polyacids, can be derivatized to contain
two or more functional groups, such as succinimidyl groups. Polyacids for use
105
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
in the present invention include, without limitation, trimethylolpropane-based
tricarboxylic acid, di(trimethylol propane)-based tetracarboxylic acid,
heptanedioic acid, octanedioic acid (suberic acid), and hexadecanedioic acid
(thapsic acid). Many of these polyacids are commercially available from
DuPont Chemical Company (Wilmington, DE). According to a general method,
polyacids can be chemically derivatized to contain two or more succinimidyl
groups by reaction with an appropriate molar amount of N-hydroxysuccinimide
(NHS) in the presence of N,N1-dicyclohexylcarbodiimide (DCC).
Polyalcohols such as trimethylolpropane and di(trimethylol
propane) can be converted to carboxylic acid form using various methods, then
further derivatized by reaction with NHS in the presence of DCC to produce
trifunctionally and tetrafunctionally activated polymers, respectively, as
described in U.S. application Ser. No. 08/403,358. Polyacids such as
heptanedioic acid (HOOC-(CH2)5-COOH), octanedioic acid (HOOC-(C1-12)6-
COOH), and hexadecanedioic acid (HOOC-(CH2)14-COOH) are derivatized by
the addition of succinimidyl groups to produce difunctionally activated
polymers.
Polyamines such as ethylenediamine, tetramethylenediamine,
pentamethylenediamine (cadaverine), hexamethylenediamine, bis (2-
aminoethyparnine, and tris(2-aminoethyl)amine can be chemically derivatized to
polyacids, which can then be derivatized to contain two or more succinimidyl
groups by reacting with the appropriate molar amounts of N-
hydroxysuccinimide in the presence of DCC, as described in U.S. application
Ser. No. 08/403,358. Many of these polyamines are commercially available
from DuPont Chemical Company.
In a preferred embodiment, the first synthetic polymer will contain
multiple nucleophilic groups (represented below as "X") and it will react with
the
second synthetic polymer containing multiple electrophilic groups (represented
below as "Y"), resulting in a covalently bound polymer network, as follows:
Polymer-Xm + Polymer-Y ¨4 Polymer-Z-Polymer
wherein m n and m + n
106
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
where exemplary X groups include -NH2, -SH, -OH, -PH2, CO-NH-
NH2, etc., where the X groups may be the same or different in polymer-Xm;
where exemplary Y groups include -0O2-N(COCH2)2, -CO2H, -
CHO, -CHOCH2 (epoxide), -N=C=O, -S02-CH=CH2, -N(COCH)2 (La, a five-
membered heterocyclic ring with a double bond present between the two CH
groups), -S-S-(C5H4N), etc., where the Y groups may be the same or different
in
polymer-Y; and
where Z is the functional group resulting from the union of a
nucleophilic group (X) and an electrophilic group (Y).
As noted above, it is also contemplated by the present invention
that X and Y may be the same or different, Le., a synthetic polymer may have
two different electrophilic groups, or two different nucleophilic groups, such
as
with glutathione.
In one embodiment, the backbone of at least one of the synthetic
polymers comprises alkylene oxide residues, e.g., residues from ethylene
oxide, propylene oxide, and mixtures thereof. The term 'backbone' refers to a
significant portion of the polymer.
For example, the synthetic polymer containing alkylene oxide
residues may be described by the formula X-polymer-X or Y-polymer-Y,
wherein X and Y are as defined above, and the term "polymer" represents -
(CH2CH2 0)n- or -(CH(CH3)CH2 0)n- or -(CH2-CH2-0)n-(CH(CH3)CH2-0)n-. In
these cases the synthetic polymer would be difunctional.
The required functional group X or Y is commonly coupled to the
polymer backbone by a linking group (represented below as "0"), many of
which are known or possible. There are many ways to prepare the various
functionalized polymers, some of which are listed below:
Polymer-Q1-X + Polymer-Q2-Y --> Polymer-Q1-Z-Q2-Polymer
Exemplary Q groups include ¨0-(CH2)n-; -S-(CH2)n-, -NH-(CH2)n-;
-02C-NH-(CH2)n-; -02C-(CH2)n-; -02C-(CR1H)n-; and -0-R2-CO-NH-, which
provide synthetic polymers of the partial structures: polymer-0-(CH2)n-(X or
Y);
107
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
polymer-S-(CH2)-(X or Y); polymer-NH-(CH2),;(X or Y); polymer-02C-NH-
(CH2)-(X or Y); polymer-02C-(CH2)-(X or Y); polymer-02C-(CR1H)n-(X or Y);
and polymer-O-R2-CO-NH-(X or Y), respectively. In these structures, n = 1-10,
R1 = H or alkyl (i.e., CH3, 02H5, etc.); R2 = CH2, or CO-NH-CH2CH2; and Q1 and
Q2 may be the same or different.
For example, when Q2 = OCH2CH2 (there is no Q1 in this case); Y
= -0O2-N(COCH2)2, and X = -NH2, -SH, or -OH, the resulting reactions and Z
groups would be as follows:
Polymer-NH2 + Polymer-O-CH2-CH2-0O2-N(COCH2)2
Polymer-NH-CO-CH2-CH2-0-Polymer;
Polymer-SH + Polymer-O-CH2-CH2-0O2-N(COCH2)2
Polymer-S-COCH2CH2-0-Polymer; and
Polymer-OH + Polymer-O-CH2-CH2-0O2-N(COCH2)2
Polymer-O-COCH2CH2-0-Polymer.
An additional group, represented below as "D", can be inserted
between the polymer and the linking group, if present. One purpose of such a
D group is to affect the degradation rate of the crosslinked polymer
composition
In vivo, for example, to increase the degradation rate, or to decrease the
degradation rate. This may be useful in many instances, for example, when
drug has been incorporated into the matrix, and it is desired to increase or
decrease polymer degradation rate so as to influence a drug delivery profile
in
the desired direction. An illustration of a crosslinking reaction involving
first and
second synthetic polymers each having D and Q groups is shown below.
Polymer-D-Q-X + Polymer-D-Q-Y Polymer-D-Q-Z-Q-D-Polymer
Some useful biodegradable groups "D" include polymers formed
from one or more a-hydroxy acids, e.g., lactic acid, glycolic acid, and the
cyclization products thereof (e.g., lactide, glycolide), E-caprolactone, and
amino
acids. The polymers may be referred to as polylactide, polyglycolide, poly(co-
lactide-glycolide); poly-e-caprolactone, polypeptide (also known as poly amino
acid, for example, various di- or tri-peptides),and poly(anhydride)s.
108
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In a general method for preparing the crosslinked polymer
compositions used in the context of the present invention, a first synthetic
polymer containing multiple nucleophilic groups is mixed with a second
synthetic polymer containing multiple electrophilic groups. Formation of a
three-dimensional crosslinked network occurs as a result of the reaction
between the nucleophilic groups on the first synthetic polymer and the
electrophilic groups on the second synthetic polymer.
The concentrations of the first synthetic polymer and the second
synthetic polymer used to prepare the compositions of the present invention
will
vary depending upon a number of factors, including the types and molecular
weights of the particular synthetic polymers used and the desired end use
application. In general, when using multi-amino PEG as the first synthetic
polymer, it is preferably used at a concentration in the range of about 0.5 to
about 20 percent by weight of the final composition, while the second
synthetic
polymer is used at a concentration in the range of about 0.5 to about 20
percent
by weight of the final composition. For example, a final composition having a
total weight of 1 gram (1000 milligrams) would contain between about 5 to
about 200 milligrams of multi-amino PEG, and between about 5 to about 200
milligrams of the second synthetic polymer.
Use of higher concentrations of both first and second synthetic
polymers will result in the formation of a more tightly crosslinked network,
producing a stiffer, more robust gel. Compositions intended for use in tissue
augmentation will generally employ concentrations of first and second
synthetic
polymer that fall toward the higher end of the preferred concentration range.
Compositions intended for use as bioadhesives or in adhesion prevention do
not need to be as firm and may therefore contain lower polymer concentrations.
Because polymers containing multiple electrophilic groups will
also react with water, the second synthetic polymer is generally stored and
used in sterile, dry form to prevent the loss of crosslinking ability due to
hydrolysis which typically occurs upon exposure of such electrophilic groups
to
109
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
aqueous media. Processes for preparing synthetic hydrophilic polymers
containing multiple electrophylic groups in sterile, dry form are set forth in
U.S.
Patent 5,643,464. For example, the dry synthetic polymer may be compression
molded into a thin sheet or membrane, which can then be sterilized using
gamma or, preferably, e-beam irradiation. The resulting dry membrane or
sheet can be cut to the desired size or chopped into smaller size
particulates.
In contrast, polymers containing multiple nucleophilic groups are generally
not
water-reactive and can therefore be stored in aqueous solution.
In certain embodiments, one or both of the electrophilic- or
nucleophilic-terminated polymers described above can be combined with a
synthetic or naturally occurring polymer. The presence of the synthetic or
naturally occurring polymer may enhance the mechanical and/or adhesive
properties of the in situ forming compositions. Naturally occurring polymers,
and polymers derived from naturally occurring polymer that may be included in
in situ forming materials include naturally occurring proteins, such as
collagen,
collagen derivatives (such as methylated collagen), fibrinogen, thrombin,
albumin, fibrin, and derivatives of and naturally occurring polysaccharides,
such
as glycosaminoglycans, including deacetylated and desulfated
glycosaminoglycan derivatives.
In one aspect, a composition comprising naturally-occurring
protein and both of the first and second synthetic polymer as described above
is used to form the crosslinked matrix according to the present invention. In
one aspect, a composition comprising collagen and both of the first and second
synthetic polymer as described above is used to form the crosslinked matrix
according to the present invention. In one aspect, a composition comprising
methylated collagen and both of the first and second synthetic polymer as
described above is used to form the crosslinked matrix according to the
present
invention. In one aspect, a composition comprising fibrinogen and both of the
first and second synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
110
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
composition comprising thrombin and both of the first and second synthetic
polymer as described above is used to form the crosslinked matrix according to
the present invention. In one aspect, a composition comprising albumin and
both of the first and second synthetic polymer as described above is used to
form the crosslinked matrix according to the present invention. In one aspect,
a
composition comprising fibrin and both of the first and second synthetic
polymer
as described above is used to form the crosslinked matrix according to the
present invention. In one aspect, a composition comprising naturally occurring
polysaccharide and both of the first and second synthetic polymer as described
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising glycosaminoglycan and both of the
first and second synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
composition comprising deacetylated glycosaminoglycan and both of the first
and second synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
composition comprising desulfated glycosaminoglycan and both of the first and
second synthetic polymer as described above is used-to form the crosslinked
matrix according to the present invention.
In one aspect, a composition comprising naturally-occurring
protein and the first synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
composition comprising collagen and the first synthetic polymer as described
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising methylated collagen and the first
synthetic polymer as described above is used to form the crosslinked matrix
according to the present invention. In one aspect, a composition comprising
fibrinogen and the first synthetic polymer as described above is used to form
the crosslinked matrix according to the present invention. In one aspect, a
composition comprising thrombin and the first synthetic polymer as described
111
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising albumin and the first synthetic
polymer
as described above is used to form the crosslinked matrix according to the
present invention. In one aspect, a composition comprising fibrin and the
first
synthetic polymer as described above is used to form the crosslinked matrix
according to the present invention. In one aspect, a composition comprising
naturally occurring polysaccharide and the first synthetic polymer as
described
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising glycosaminoglycan and the first
synthetic polymer as described above is used to form the crosslinked matrix
according to the present invention. In one aspect, a composition comprising
deacetylated glycosaminoglycan and the first synthetic polymer as described
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising desulfated glycosaminoglycan and the
first synthetic polymer as described above is used to form the crosslinked
matrix according to the present invention.
In one aspect, a composition comprising naturally-occurring
protein and the second synthetic polymer as described above is used to form
the crosslinked matrix according to the present invention. In one aspect, a
composition comprising collagen and the second synthetic polymer as
described above is used to form the crosslinked matrix according to the
present
invention. In one aspect, a composition comprising methylated collagen and
the second synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
composition comprising fibrinogen and the second synthetic polymer as
described above is used to form the crosslinked matrix according to the
present
invention. In one aspect, a composition comprising thrombin and the second
synthetic polymer as described above is used to form the crosslinked matrix
according to the present invention. In one aspect, a composition comprising
albumin and the second synthetic polymer as described above is used to form
112
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
the crosslinked matrix according to the present invention. In one aspect, a
composition comprising fibrin and the second synthetic polymer as described
above is used to form the crosslinked matrix according to the present
invention.
In one aspect, a composition comprising naturally occurring polysaccharide and
the second synthetic polymer as described above is used to form the
crosslinked matrix according to the present invention. In one aspect, a
composition comprising glycosaminoglycan and the second synthetic polymer
as described above is used to form the crosslinked matrix according to the
present invention. In one aspect, a composition comprising deacetylated
glycosaminoglycan and the second synthetic polymer as described above is
used to form the crosslinked matrix according to the present invention. In one
aspect, a composition comprising desulfated glycosaminoglycan and the
second synthetic polymer as described above is used to form the crosslinked
matrix according to the present invention.
The presence of protein or polysaccharide components which
contain functional groups that can react with the functional groups on
multiple
activated synthetic polymers can result in formation of a crosslinked
synthetic
polymer-naturally occurring polymer matrix upon mixing and/or crosslinking of
the synthetic polymer(s). In particular, when the naturally occurring polymer
(protein or polysaccharide) also contains nucleophilic groups such as primary
amino groups, the electrophilic groups on the second synthetic polymer will
react with the primary amino groups on these components, as well as the
nucleophilic groups on the first synthetic polymer, to cause these other
components to become part of the polymer matrix. For example, lysine-rich
proteins such as collagen may be especially reactive with electrophilic groups
on synthetic polymers.
In one aspect, the naturally occurring protein is polymer may be
collagen. As used herein, the term "collagen" or "collagen material" refers to
all
forms of collagen, including those which have been processed or otherwise
modified and is intended to encompass collagen of any type, from any source,
113
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
including, but not limited to, collagen extracted from tissue or produced
recombinantly, collagen analogues, collagen derivatives, modified collagens,
and denatured collagens, such as gelatin.
In general, collagen from any source may be included in the
compositions of the invention; for example, collagen may be extracted and
purified from human or other mammalian source, such as bovine or porcine
corium and human placenta, or may be recombinantly or otherwise produced.
The preparation of purified, substantially non-antigenic collagen in solution
from
bovine skin is well known in the art. U.S. Patent No. 5,428,022 discloses
methods of extracting and purifying collagen from the human placenta. U.S.
Patent No. 5,667,839, discloses methods of producing recombinant human
collagen in the milk of transgenic animals, including transgenic cows.
Collagen
of any type, including, but not limited to, types I, II, Ill, IV, or any
combination
thereof, may be used in the compositions of the invention, although type I is
generally preferred. Either atelopeptide or telopeptide-containing collagen
may
be used; however, when collagen from a xenogeneic source, such as bovine
collagen, is used, atelopeptide collagen is generally preferred, because of
its
reduced immunogenicity compared to telopeptide-containing collagen.
Collagen that has not been previously crosslinked by methods
such as heat, irradiation, or chemical crosslinking agents is preferred for
use in
the compositions of the invention, although previously crosslinked collagen
may
be used. Non-crosslinked atelopeptide fibrillar collagen is commercially
available from !named Aesthetics (Santa Barbara, CA) at collagen
concentrations of 35 mg/ml and 65 mg/ml under the trademarks ZYDERM I
Collagen and ZYDERM II Collagen, respectively. Glutaraldehyde crosslinked
atelopeptide fibrillar collagen is commercially available from !named
Corporation (Santa Barbara, CA) at a collagen concentration of 35 mg/ml under
the trademark ZYPLAST Collagen.
114
= CA 02610948 2012-12-14
Collagens for use in the present invention are generally in
aqueous suspension at a concentration between about 20 mg/ml to about 120
mg/ml; preferably, between about 30 mg/ml to about 90 mg/ml.
Because of its tacky consistency, nonfibrillar collagen may be
preferred for use in compositions that are intended for use as bioadhesives.
The term "nonfibrillar collagen" refers to any modified or unmodified collagen
material that is in substantially nonfibrillar form at pH 7, as indicated by
optical
clarity of an aqueoue suspension of the collagen.
Collagen that is already in nonfibrillar form may be used in the
compositions of the invention. As used herein, the term "nonfibrillar
collagen" is
intended to encompass collagen types that are nonfibrillar in native form, as
well as collagens that have been chemically modified such that they are in
nonfibrillar form at or around neutral pH. Collagen types that are
nonfibrillar (or
microfibrillar) in native form include types IV, VI, and VII.
Chemically modified collagens that are in nonfibrillar form at
neutral pH include succinylated collagen and methylated collagen, both of
which can be prepared according to the methods described in U.S. Pat. No.
4,164,559, issued Aug. 14, 1979, to Miyata et al.
Due to its inherent tackiness, methylated collagen is
particularly preferred for use in bioadhesive compositions..
Collagens for use in the crosslinked polymer compositions of the
present invention may start out in fibrillar form, then be rendered
nonfibrillar by
the addition of one or more fiber disassembly agent. The fiber disassembly
agent must be present in an amount sufficient to render the collagen
substantially nonfibrillar at pH 7, as described above. Fiber disassembly
agents
= for use in the present invention include, without limitation, various
biocompatible
alcohols, amino acids (e.g., arginine), inorganic salts (e.g., sodium chloride
and
potassium chloride), and carbohydrates (e.g., various sugars including
sucrose).
115
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
In one aspect, the polymer may be collagen or a collagen
derivative, for example methylated collagen. An example of an in situ forming
composition uses pentaerythritol poly(ethylene glycol)ether tetra-sulfhydryl)
(4-
armed thiol PEG), pentaerythritol poly(ethylene glycol)ether tetra-
succinimidyl
glutaratel (4-armed NHS PEG) and methylated collagen as the reactive
reagents. This composition, when mixed with the appropriate buffers can
produce a crosslinked hydrogel. (See, e.g., U.S. Patent Nos. 5,874,500;
6,051,648; 6,166,130; 5,565,519 and 6,312,725).
In another aspect, the naturally occurring polymer may be a
glycosaminoglycan. Glycosaminoglycans, e.g., hyaluronic acid, contain both
anionic and cationic functional groups along each polymeric chain, which can
form intramolecular and/or intermolecular ionic crosslinks, and are
responsible
for the thixotropic (or shear thinning) nature of hyaluronic acid.
In certain aspects, the glycosaminoglycan may be derivatized.
For example, glycosaminoglycans can be chemically derivatized by, e.g.,
deacetylation, desulfation, or both in order to contain primary amino groups
available for reaction with electrophilic groups on synthetic polymer
molecules.
Glycosaminoglycans that can be derivatized according to either or both of the
aforementioned methods include the following: hyaluronic acid, chondroitin
sulfate A, chondroitin sulfate B (dermatan sulfate), chondroitin sulfate C,
chitin
(can be derivatized to chitosan), keratan sulfate, keratosulfate, and heparin.
Derivatization of glycosaminoglycans by deacetylation and/or desulfation and
covalent binding of the resulting glycosaminoglycan derivatives with synthetic
hydrophilic polymers is described in further detail in commonly assigned,
allowed U.S. patent application Ser. No.,08/146,843, filed Nov. 3, 1993.
In general, the collagen is added to the first synthetic polymer,
then the collagen and first synthetic polymer are mixed thoroughly to achieve
a
homogeneous composition. The second synthetic polymer is then added and
mixed into the collagen/first synthetic polymer mixture, where it will
covalently
bind to primary amino groups or thiol groups on the first synthetic polymer
and
116
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
primary amino groups on the collagen, resulting in the formation of a
homogeneous crosslinked network. Various deacetylated and/or desulfated
glycosaminoglycan derivatives can be incorporated into the composition in a
similar manner as that described above for collagen. In addition, the
introduction of hydrocolloids such as carboxymethylcellulose may promote
tissue adhesion and/or swellability.
Administration of the Crosslinked Synthetic Polymer Compositions
The compositions of the present invention having two synthetic
polymers may be administered before, during or after crosslinking of the first
and second synthetic polymer. Certain uses, which are discussed in greater
detail below, such as tissue augmentation, may require the compositions to be
crosslinked before administration, whereas other applications, such as tissue
adhesion, require the compositions to be administered before crosslinking has
reached "equilibrium." The point at which crosslinking has reached equilibrium
is defined herein as the point at which the composition no longer feels tacky
or
sticky to the touch.
In order to administer the composition prior to crosslinking, the
first synthetic polymer and second synthetic polymer may be contained within
separate barrels of a dual-compartment syringe. In this case, the two
synthetic
polymers do not actually mix until the point at which the two polymers are
extruded from the tip of the syringe needle into the patient's tissue. This
allows
the vast majority of the crosslinking reaction to occur in situ, avoiding the
problem of needle blockage which commonly occurs if the two synthetic
polymers are mixed too early and crosslinking between the two components is
already too advanced prior to delivery from the syringe needle. The use of a
dual-compartment syringe, as described above, allows for the use of smaller
diameter needles, which is advantageous when performing soft tissue
augmentation in delicate facial tissue, such as that surrounding the eyes.
117
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Alternatively, the first synthetic polymer and second synthetic
polymer may be mixed according to the methods described above prior to
delivery to the tissue site, then injected to the desired tissue site
immediately
(preferably, within about 60 seconds) following mixing.
In another embodiment of the invention, the first synthetic polymer
and second synthetic polymer are mixed, then extruded and allowed to
crosslink into a sheet or other solid form. The crosslinked solid is then
dehydrated to remove substantially all unbound water. The resulting dried
solid
may be ground or comminuted into particulates, then suspended in a
nonaqueous fluid carrier, including, without limitation, hyaluronic acid,
dextran
sulfate, dextran, succinylated noncrosslinked collagen, methylated
noncrosslinked collagen, glycogen, glycerol, dextrose, maltose, triglycerides
of
fatty acids (such as corn oil, soybean oil, and sesame oil), and egg yolk
phospholipid. The suspension of particulates can be injected through a small-
gauge needle to a tissue site. Once inside the tissue, the crosslinked polymer
particulates will rehydrate and swell in size at least five-fold.
Hydrophilic Polymer + Plurality of Crosslinkable Components
As mentioned above, the first and/or second synthetic polymers
may be combined with a hydrophilic polymer, e.g., collagen or methylated
collagen, to form a composition useful in the present invention. In one
general
embodiment, the compositions useful in the present invention include a
hydrophilic polymer in combination with two or more crosslinkable components.
This embodiment is described in further detail in this section.
The Hydrophilic Polymer Component:
The hydrophilic polymer component may be a synthetic or
naturally occurring hydrophilic polymer. Naturally occurring hydrophilic
polymers include, but are not limited to: proteins such as collagen and
derivatives thereof, fibronectin, albumins, globulins, fibrinogen, and fibrin,
with
118
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
collagen particularly preferred; carboxylated polysaccharides such as
polymannuronic acid and polygalacturonic acid; aminated polysaccharides,
particularly the glycosaminoglycans, e.g., hyaluronic acid, chitin,
chondroitin
sulfate A, B, or C, keratin sulfate, keratosulfate and heparin; and activated
polysaccharides such as dextran and starch derivatives. Collagen (e.g.,
methylated collagen) and glycosaminoglycans are preferred naturally occurring
hydrophilic polymers for use herein.
In general, collagen from any source may be used in the
composition of the method; for example, collagen may be extracted and purified
from human or other mammalian source, such as bovine or porcine corium and
human placenta, or may be recombinantly or otherwise produced. The
preparation of purified, substantially non-antigenic collagen in solution from
bovine skin is well known in the art. See, e.g., U.S. Pat. No. 5,428,022, to
Palefsky et al., which discloses methods of extracting and purifying collagen
from the human placenta. See also U.S. Patent No. 5,667,839, to Berg, which
discloses methods of producing recombinant human collagen in the milk of
transgenic animals, including transgenic cows. Unless otherwise specified, the
term "collagen" or "collagen material" as used herein refers to all forms of
collagen, including those that have been processed or otherwise modified.
Collagen of any type, including, but not limited to, types I, II, Ill, IV,
or any combination thereof, may be used in the compositions of the invention,
although type I is generally preferred. Either atelopeptide or telopeptide-
containing collagen may be used; however, when collagen from a source, such
as bovine collagen, is used, atelopeptide collagen is generally preferred,
because of its reduced immunogenicity compared to telopeptide-containing
collagen.
Collagen that has not been previously crosslinked by methods
such as heat, irradiation, or chemical crosslinking agents is preferred for
use in
the compositions of the invention, although previously crosslinked collagen
may
be used. Non-crosslinked atelopeptide fibrillar collagen is commercially
119
= CA 02610948 2012-12-14
available from McGhan Medical Corporation (Santa Barbara, Calif.) at collagen
concentrations of 35 mg/ml and 65 mg/ml under the trademarks ZYDERM I
Collagen and ZYDERM II Collagen, respectively. Glutaraldehyde-crosslinked
atelopeptide fibrillar collagen is commercially available from McGhan Medical
Corporation at a collagen concentration of 35 mg/ml under the trademark
ZYPLAST .
Collagens for use in the present invention are generally, although
not necessarily, in aqueous suspension at a concentration between about 20
mg/ml to about 120 mg/ml, preferably between about 30 mg/ml to about 90
mg/ml.
Although intact collagen is preferred, denatured collagen,
commonly known as gelatin, can also be used in the compositions of the
invention. Gelatin may have the added benefit of being degradable faster than
collagen.
Because of its greater surface area and greater concentration of
reactive groups, nonfibrillar collagen is generally preferred. The term
"nonfibrillar collagen" refers to any modified or unmodified collagen material
that
is in substantially nonfibrillar form at pH 7, as indicated by optical clarity
of an
aqueous suspension of the collagen.
Collagen that is already in nonfibrillar form may be used in the
compositions of the invention. As used herein, the term "nonfibrillar
collagen" is
intended to encompass collagen types that are nonfibrillar in native form, as
well as collagens that have been chemically modified such that they are in
nonfibrillar form at or around neutral pH. Collagen types that are
nonfibrillar (or
microfibrillar) in native form include types IV, VI, and VII.
Chemically modified collagens that are in nonfibrillar form at
neutral pH include succinylated collagen, propylated collagen, ethylated
collagen, methylated collagen, and the like, both of which can be prepared
according to the methods described in U.S. Pat. No. 4,164,559, to Miyata et
al.
Due to its inherent
120
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
tackiness, methylated collagen is particularly preferred, as disclosed in U.S.
Patent No. 5,614,587 to Rhee et al.
Collagens for use in the crosslinkable compositions of the present
invention may start out in fibrillar form, then be rendered nonfibrillar by
the
addition of one or more fiber disassembly agents. The fiber disassembly agent
must be present in an amount sufficient to render the collagen substantially
nonfibrillar at pH 7, as described above. Fiber disassembly agents for use in
the present invention include, without limitation, various biocompatible
alcohols,
amino acids, inorganic salts, and carbohydrates, with biocompatible alcohols
being particularly preferred. Preferred biocompatible alcohols include
glycerol
and propylene glycol. Non-biocompatible alcohols, such as ethanol, methanol,
and isopropanol, are not preferred for use in the present invention, due to
their
potentially deleterious effects on the body of the patient receiving them.
Preferred amino acids include arginine. Preferred inorganic salts include
sodium chloride and potassium chloride. Although carbohydrates, such as
various sugars including sucrose, may be used in the practice of the present
invention, they are not as preferred as other types of fiber disassembly
agents
because they can have cytotoxic effects in vivo.
As fibrillar collagen has less surface area and a lower
concentration of reactive groups than nonfibrillar, fibrillar collagen is less
preferred. However, as disclosed in U.S. Patent 5,614,587, fibrillar collagen,
or
mixtures of nonfibrillar and fibrillar collagen, may be preferred for use in
compositions intended for long-term persistence in vivo, if optical clarity is
not a
requirement.
Synthetic hydrophilic polymers may also be used in the present
invention. Useful synthetic hydrophilic polymers include, but are not limited
to:
polyalkylene oxides, particularly polyethylene glycol and poly(ethylene oxide)-
poly(propylene oxide) copolymers, including block and random copolymers;
polyols such as glycerol, polyglycerol (particularly highly branched
polyglycerol), propylene glycol and trimethylene glycol substituted with one
or
121
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
more polyalkylene oxides, e.g., mono-, di- and tri-polyoxyethylated glycerol,
mono- and di-polyoxyethylated propylene glycol, and mono- and di-
polyoxyethylated trimethylene glycol; polyoxyethylated sorbitol,
polyoxyethylated glucose; acrylic acid polymers and analogs and copolymers
thereof, such as polyacrylic acid per se, polymethacrylic acid,
poly(hydroxyethyl-methacrylate), poly(hydroxyethylacrylate),
poly(methylalkylsulfoxide methacrylate), poly(methylalkylsulfoxide acrylate)
and
copolymers of any of the foregoing, and/or with additional acrylate species
such
as aminoethyl acrylate and mono-2-(acryloxy)-ethyl succinate; polymaleic acid;
poly(acrylamides) such as polyacrylamide per se, poly(methacrylamide),
poly(dimethylacrylamide), and poly(N-isopropyl-acrylamide); poly(olefinic
alcohol)s such as poly(vinyl alcohol); poly(N-vinyl lactams) such as
poly(vinyl
pyrrolidone), poly(N-vinyl caprolactam), and copolymers thereof;
polyoxazolines, including poly(methyloxazoline) and poly(ethyloxazoline); and
polyvinylamines. It must be emphasized that the aforementioned list of
polymers is not exhaustive, and a variety of other synthetic hydrophilic
polymers may be used, as will be appreciated by those skilled in the art.
The Crosslinkable Components:
The compositions of the invention also comprise a plurality of
crosslinkable components. Each of the crosslinkable components participates
in a reaction that results in a crosslinked matrix. Prior to completion of the
crosslinking reaction, the crosslinkable components provide the necessary
adhesive qualities that enable the methods of the invention.
The crosslinkable components are selected so that crosslinking
gives rise to a biocompatible, nonimmunogenic matrix useful in a variety of
contexts including adhesion prevention, biologically active agent delivery,
tissue
augmentation, and other applications. The crosslinkable components of the
invention comprise: a component A, which has m nucleophilic groups, wherein
m > 2 and a component B, which has n electrophilic groups capable of reaction
122
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
with the m nucleophilic groups, wherein n > 2 and m + n > 4. An optional third
component, optional component C, which has at least one functional group that
is either electrophilic and capable of reaction with the nucleophilic groups
of
component A, or nucleophilic and capable of reaction with the electrophilic
groups of component B may also be present. Thus, the total number of
functional groups present on components A, B and C, when present, in
combination is > 5; that is, the total functional groups given by m + n + p
must
be > 5, where p is the number of functional groups on component C and, as
indicated, is > 1. Each of the components is biocompatible and
nonimmunogenic, and at least one component is comprised of a hydrophilic
polymer. Also, as will be appreciated, the composition may contain additional
crosslinkable components D, E, F, etc., having one or more reactive
nucleophilic or electrophilic groups and thereby participate in formation of
the
crosslinked biomaterial via covalent bonding to other components.
The m nucleophilic groups on component A may all be the same,
or, alternatively, A may contain two or more different nucleophilic groups.
Similarly, the n electrophilic groups on component B may all be the same, or
two or more different electrophilic groups may be present. The functional
group(s) on optional component C, if nucleophilic, may or may not be the same
as the nucleophilic groups on component A, and, conversely, if electrophilic,
the
functional group(s) on optional component C may or may not be the same as
the electrophilic groups on component B.
Accordingly, the components may be represented by the
structural formulae
(I) R1(4Q1hrx)n, (component A),
(II) R2(4Q2ky)n (component B), and
(III) R3(-[Q3}5-Fn)p (optional component C),
wherein:
R1, R2 and R3 are independently selected from the group
consisting of C2 to C14 hydrocarbyl, heteroatom-containing C2 to C14
123
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
hydrocarbyl, hydrophilic polymers, and hydrophobic polymers, providing that at
least one of R1, R2 and R3 is a hydrophilic polymer, preferably a synthetic
hydrophilic polymer;
X represents one of the m nucleophilic groups of component A,
Y represents one of the n electrophilic groups of component B,
and the various Y moieties on A may be the same or different;
Fn represents a functional group on optional component C;
Q1, Q2 and Q3 are linking groups;
m n m + n is q, and r are independently zero or 1,
and when optional component C is present, p and s
is independently zero
or 1.
Reactive Groups:
X may be virtually any nucleophilic group, so long as reaction can
25 Examples of nucleophilic groups suitable as X include, but are not
limited to, -NH2, -NHR4, -N(R4)2, -SH, -OH, -COON, -C6H4-0H, -PH2, -PHR5, -
P(R5)2, -NH-NH2, -CO-NH-NH2, -05H4N, etc. wherein R4 and R5 are
hydrocarbyl, typically alkyl or monocyclic aryl, preferably alkyl, and most
preferably lower alkyl. Organometallic moieties are also useful nucleophilic
124
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
groups for the purposes of the invention, particularly those that act as
carbanion
donors. Organometallic nucleophiles are not, however, preferred. Examples of
organometallic moieties include: Grignard functionalities -R6MgHal wherein R6
is a carbon atom (substituted or unsubstituted), and Hal is halo, typically
bromo,
iodo or chloro, preferably bromo; and lithium-containing functionalities,
typically
alkyllithium groups; sodium-containing functionalities.
It will be appreciated by those of ordinary skill in the art that
certain nucleophilic groups must be activated with a base so as to be capable
of reaction with an electrophile. For example, when there are nucleophilic
sulfhydryl and hydroxyl groups in the crosslinkable composition, the
composition must be admixed with an aqueous base in order to remove a
proton and provide an or -0- species to enable reaction with an
electrophile.
Unless it is desirable for the base to participate in the crosslinking
reaction, a
nonnucleophilic base is preferred. In some embodiments, the base may be
present as a component of a buffer solution. Suitable bases and corresponding
crosslinking reactions are described infra in Section E.
The selection of electrophilic groups provided within the
crosslinkable composition, i.e., on component B, must be made so that reaction
is possible with the specific nucleophilic groups. Thus, when the X moieties
are
amino groups, the Y groups are selected so as to react with amino groups.
Analogously, when the X moieties are sulfhydryl moieties, the corresponding
electrophilic groups are sulfhydryl-reactive groups, and the like.
By way of example, when X is amino (generally although not
necessarily primary amino), the electrophilic groups present on Y are amino
reactive groups such as, but not limited to: (1) carboxylic acid esters,
including
cyclic esters and "activated" esters; (2) acid chloride groups (-CO-CI); (3)
anhydrides (-(C0)-0-(C0)-R); (4) ketones and aldehydes, including a,13-
unsaturated aldehydes and ketones such as -CH=CH-CH=0 and -CH=CH-
C(CH3)=0; (5) halides; (6) isocyanate (-N=C=0); (7) isothiocyanate (-N=C=S);
(8) epoxides; (9) activated hydroxyl groups (e.g., activated with conventional
125
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
activating agents such as carbonyldiimidazole or sulfonyl chloride); and (10)
olefins, including conjugated olefins, such as ethenesulfonyl (-S02CH=CH2)
and analogous functional groups, including acrylate (-0O2-C=CH2),
methacrylate (-0O2-C(CH3)=CF12)), ethyl acrylate (-0O2-C(CH2CH3)=CH2), and
ethyleneimino (-CH=CH-C=NH). Since a carboxylic acid group per se is not
susceptible to reaction with a nucleophilic amine, components containing
carboxylic acid groups must be activated so as to be amine-reactive.
Activation
may be accomplished in a variety of ways, but often involves reaction with a
suitable hydroxyl-containing compound in the presence of a dehydrating agent
such as dicyclohexylcarbodiimide (DCC) or dicyclohexylurea (DHU). For
example, a carboxylic acid can be reacted with an alkoxy-substituted N-
hydroxy-succinimide or N-hydroxysulfosuccinimide in the presence of DCC to
form reactive electrophilic groups, the N-hydroxysuccinimide ester and the N-
hydroxysulfosuccinimide ester, respectively. Carboxylic acids may also be
activated by reaction with an acyl halide such as an acyl chloride (e.g.,
acetyl
chloride), to provide a reactive anhydride group. In a further example, a
carboxylic acid may be converted to an acid chloride group using, e.g.,
thionyl
chloride or an acyl chloride capable of an exchange reaction. Specific
reagents
and procedures used to carry out such activation reactions will be known to
those of ordinary skill in the art and are described in the pertinent texts
and
literature.
Analogously, when X is sulfhydryl, the electrophilic groups present
on Y are groups that react with a sulfhydryl moiety. Such reactive groups
include those that form thioester linkages upon reaction with a sulfhydryl
group,
such as those described in PCT Publication No. WO 00/62827 to Wallace et al.
As explained in detail therein, such "sulfhydryl reactive" groups include, but
are
not limited to: mixed anhydrides; ester derivatives of phosphorus; ester
derivatives of p-nitrophenol, p-nitrothiophenol and pentafluorophenol; esters
of
substituted hydroxylamines, including N-hydroxyphthalimide esters, N-
hydroxysuccinimide esters, N-hydroxysulfosuccinimide esters, and N-
126
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
hydroxyglutarinnide esters; esters of 1-hydroxybenzotriazole; 3-hydroxy-3,4-
dihydro-benzotriazin-4-one; 3-hydroxy-3,4-dihydro-quinazoline-4-one;
carbonylimidazole derivatives; acid chlorides; ketenes; and isocyanates. With
these sulfhydryl reactive groups, auxiliary reagents can also be used to
facilitate bond formation, e.g., 1-ethyl-343-dimethylaminopropyllcarbodiimide
can be used to facilitate coupling of sulfhydryl groups to carboxyl-containing
groups.
In addition to the sulfhydryl reactive groups that form thioester
linkages, various other sulfhydryl reactive functionalities can be utilized
that
form other types of linkages. For example, compounds that contain methyl
imidate derivatives form imido-thioester linkages with sulfhydryl groups.
Alternatively, sulfhydryl reactive groups can be employed that form disulfide
bonds with sulfhydryl groups; such groups generally have the structure -S-S-Ar
where Ar is a substituted or unsubstituted nitrogen-containing heteroaromatic
moiety or a non-heterocyclic aromatic group substituted with an electron-
withdrawing moiety, such that Ar may be, for example, 4-pyridinyl, o-
nitrophenyl, m-nitrophenyl, p-nitrophenyl, 2,4-dinitrophenyl, 2-nitro-4-
benzoic
acid, 2-nitro-4-pyridinyl, etc. In such instances, auxiliary reagents, i.e.,
mild
oxidizing agents such as hydrogen peroxide, can be used to facilitate
disulfide
bond formation.
Yet another class of sulfhydryl reactive groups forms thioether
bonds with sulfhydryl groups. Such groups include, inter alia, maleinnido,
substituted maleimido, haloalkyl, epoxy, imino, and aziridino, as well as
olefins
(including conjugated olefins) such as ethenesulfonyl, etheneimino, acrylate,
methacrylate, and a,13-unsaturated aldehydes and ketones. This class of
sulfhydryl reactive groups are particularly preferred as the thioether bonds
may
provide faster crosslinking and longer in vivo stability.
When X is -OH, the electrophilic functional groups on the
remaining component(s) must react with hydroxyl groups. The hydroxyl group
may be activated as described above with respect to carboxylic acid groups, or
127
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
it may react directly in the presence of base with a sufficiently reactive
electrophile such as an epoxide group, an aziridine group, an acyl halide, or
an
anhydride.
When X is an organometallic nucleophile such as a Grignard
functionality or an alkyllithium group, suitable electrophilic functional
groups for
reaction therewith are those containing carbonyl groups, including, by way of
example, ketones and aldehydes.
It will also be appreciated that certain functional groups can react
as nucleophiles or as electrophiles, depending on the selected reaction
partner
and/or the reaction conditions. For example, a carboxylic acid group can act
as
a nucleophile in the presence of a fairly strong base, but generally acts as
an
electrophile allowing nucleophilic attack at the carbonyl carbon and
concomitant
replacement of the hydroxyl group with the incoming nucleophile.
The covalent linkages in the crosslinked structure that result upon
covalent binding of specific nucleophilic components to specific electrophilic
components in the crosslinkable composition include, solely by way of example,
the following (the optional linking groups Q1 and Q2 are omitted for clarity):
TABLE
REPRESENTATIVE
NUCLEOPHILIC REPRESENTATIVE
COMPONENT ELECTROPHILIC
COMPONENT RESULTING LINKAGE
(A, optional
component C (B, FNEL)
element FNNu)
R1-NH2 R2-0-(C0)-0-N(COCH2) R1-NH-
(C0)-0-R2
(succinimidyl carbonate
terminus)
R1-SH R2-0-(CO)-0-N(COCH2) R1-S-
(C0)-0-R2
R1-0H R2-0-(C0)-0-N(COCH2) R1-0-
(C0)-R2
128
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
REPRESENTATIVE
NUCLEOPHILIC REPRESENTATIVE
COMPONENT ELECTROPHILIC
(A, optional COMPONENT RESULTING LINKAGE
component C (B, FNEL)
element FNNu)
R1-NH2 R2-0(CO)-CH=CH2 R1-NH-CH2CH2-(C0)-
(acrylate terminus) 0-R2
R1-SH R2-0-(C0)-CH=CH2 R1-S-CH2CH2-(C0)-0-
R2
R1-0H R2-0-(C0)-CH=CH2 R1-0-CH2CH2-(C0)-0-
R2
R1-NH2 R2-0(C0)-(CH2)3-0O2- R1-NH-(C0)-(C1-12)3-
N(COCH2) (C0)-0R2
(succinimidyl glutarate
terminus)
R1-SH R2-0(C0)-(CH2)3-0O2- RI-S-(C0)-(CH2)3-
N(COCH2) (C0)-0R2
R1-0H R2-0(C0)-(CH2)3-0O2- R1-0-(CC!)-(CH2)3-
N(COCH2) (C0)-0R4
R1-NH2 R2-0-CH2-0O2-N(COCH2) R1-NH-(C0)-CH2-0R2
(succinimidyl acetate
terminus)
R1-SH R2-0-CH2-0O2-N(COCH2) R1-S-(C0)-CH2-0R2
R2-0-CH2-CO2-N(COCH2) R1-0-(C0)-CH2-0R2
R1-NH2 R2-0-NH(C0)-(CH2)2-CO2- R1-NH-(CO)-(CH2)2-
N(COCH2) (C0)-NH-OR`
(succinimidyl succinamide
terminus)
R1-SH R2-0-NH(C0)-(CH2)2-0O2- R1-S-(C0)-(CH2)2-
N(COCH2) (C0)-NH-0R2
R1-0H R2-0-NH(C0)-(CH2)2-0O2- R1-0-(C0)-(CH2)2-
N(COCH2) (C0)-NH-0R2
a1-NH2 R2-0- (CH2)2-CHO R1-NH-(C0)-(CH2)2-
0R2
(propionaldehyde terminus)
129
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
REPRESENTATIVE
NUCLEOPHILIC REPRESENTATIVE
COMPONENT ELECTROPHILIC
COMPONENT RESULTING LINKAGE
(A, optional
component C (B, FNEL)
element FNNu)
R1-NFI2 0
/ \ R1-NH-CH2-CH(OH)-
R2-0¨CH2¨CH¨CH2 CH2-0R2 and
R1-N[CH2-CH(OH)-
(glycidyl ether terminus) CH2-0R212
R1-NH2 R2-0-(CH2)2-N=C=0 R1-NH-(C0)-NH-CH2-
OR2
(isocyanate terminus)
R1-NH2 R1-NH-CH2CH2-S02-R2
R2-S02-CH=CH2
(vinyl sulfone terminus)
R1-SH R2-S02-CH=CH2 R1-S-CH2CH2-S02-R2
Linking Groups:
The functional groups X and Y and FN on optional component C
may be directly attached to the compound core (R1, R2 or R3 on optional
component C, respectively), or they may be indirectly attached through a
linking
group, with longer linking groups also termed "chain extenders." In structural
formulae (I), (II) and (III), the optional linking groups are represented by
Q1, Q2
and Q3, wherein the linking groups are present when q, r and s are equal to 1
(with R, X, Y, Fn, m n and p as defined previously).
Suitable linking groups are well known in the art. See, for
example, International Patent Publication No. WO 97/22371. Linking groups
are useful to avoid steric hindrance problems that are sometimes associated
with the formation of direct linkages between molecules. Linking groups may
additionally be used to link several multifunctionally activated compounds
together to make larger molecules. In a preferred embodiment, 'a linking group
can be used to alter the degradative properties of the compositions after
130
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
administration and resultant gel formation. For example, linking groups can be
incorporated into components A, B, or optional component C to promote
hydrolysis, to discourage hydrolysis, or to provide a site for enzymatic
degradation.
Examples of linking groups that provide hydrolyzable sites,
include, inter alia: ester linkages; anhydride linkages, such as obtained by
incorporation of glutarate and succinate; ortho ester linkages; ortho
carbonate
linkages such as trimethylene carbonate; amide linkages; phosphoester
linkages; a-hydroxy acid linkages, such as may be obtained by incorporation of
lactic acid and glycolic acid; lactone-based linkages, such as may be obtained
by incorporation of caprolactone, valerolactone, y-butyrolactone and p-
dioxanone; and amide linkages such as in a dimeric, oligonneric, or poly(amino
acid) segment. Examples of non-degradable linking groups include
succinimide, propionic acid and carboxymethylate linkages. See, for example,
PCT WO 99/07417. Examples of enzymatically degradable linkages include
Leu-Gly-Pro-Ala, which is degraded by collagenase; and Gly-Pro-Lys, which is
degraded by plasmin.
Linking groups can also enhance or suppress the reactivity of the
various nucleophilic and electrophilic groups. For example, electron-
withdrawing groups within one or two carbons of a sulfhydryl group would be
expected to diminish its effectiveness in coupling, due to a lowering of
nucleophilicity. Carbon-carbon double bonds and carbonyl groups will also
have such an effect. Conversely, electron-withdrawing groups adjacent to a
carbonyl group (e.g., the reactive carbonyl of glutaryl-N-hydroxysuccinimidyl)
would increase the reactivity of the carbonyl carbon with respect to an
incoming
nucleophile. By contrast, sterically bulky groups in the vicinity of a
functional
group can be used to diminish reactivity and thus coupling rate as a result of
steric hindrance.
By way of example, particular linking groups and corresponding
component structure are indicated in the following Table:
131
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
TABLE
LINKING GROUP COMPONENT STRUCTURE
-0-(CH2)n- Component A: R1-0-(CH2)n-X
Component B: R2-0-(CH2)n-Y
Optional Component C: R3-0-(CH2)-Z
Component A: R1-S-(CH2)n-X
Component B: R2-S-(CH2)n-Y
Optional Component C: R3-S-(CH2)n-Z
-NH-(CF12)n- Component A: R1-NH-(CH2)n-X
Component B: R2-NH-(CH2)n-Y
Optional Component C: R3-NH-(CH2)n-Z
-0-(CO)-NH-(CH2)n- Component A: R1-0-(C0)-NH-(CH2)n-X
Component B: R2-0-(C0)-NH-(CH2)n-Y
Optional Component C: R3-0-(CO)-NH-
(CH2)n-Z
-NH-(C0)-0-(CH2)- Component A: R1-NH-(CO)-0-(CH2)n-X
Component B: R2-NH-(C0)-0-(CH2)n-Y
Optional Component C: R3-NH-(C0)-0-
(CH2)n-Z
-0-(C0)-(CH2)- Component A: R1-0-(C0)-(CH2)n-X
Component B: R2-0-(CO)-(CF12)n-Y
Optional Component C: R3-0-(C0)-(CH2)n-
Z
Component A: R1-(C0)-0-(CH2)n-X
Component B: R2-(CO)-0-(CH2)-Y
Optional Component C: R3-(C0)-0-(CH2)n-
Z
-0-(C0)-0-(CH2)n- Component A: R1-0-(C0)-0-(CH2)n-X
Component B: R2-0-(C0)-0-(CH2)-Y
Optional Component C: R3-0-(CO)-0-
(CH2)õ-Z
-0-(C0)-CHR7- Component A: R1-0-(C0)-CHR7-X
Component B: R2-0-(C0)-CHR7-Y
Optional Component C: R3-0-(C0)-CHR7-Z
132
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
LINKING GROUP COMPONENT STRUCTURE
-0-R8-(CO)-NH- Component A: R1-0-R8-(C0)-NH-X
Component B: R2- 0-R8-(C0)-NH-Y
Optional Component C: R3- O-R8-(C0)-NH-
In the above Table, n is generally in the range of 1 to about 10, R7
is generally hydrocarbyl, typically alkyl or aryl, preferably alkyl, and most
preferably lower alkyl, and R8 is hydrocarbylene, heteroatom-containing
hydrocarbylene, substituted hydrocarbylene, or substituted heteroatom-
containing hydrocarbylene) typically alkylene or arylene (again, optionally
substituted and/or containing a heteroatom), preferably lower alkylene (e.g.,
methylene, ethylene, n-propylene, n-butylene, etc.), phenylene, or
amidoalkylene (e.g., -(C0)-NH-CF12)-
Other general principles that should be considered with respect to
linking groups are as follows: If higher molecular weight components are to be
used, they preferably have biodegradable linkages as described above, so that
fragments larger than 20,000 mol. wt. are not generated during resorption in
the
body. In addition, to promote water miscibility and/or solubility, it may be
desired to add sufficient electric charge or hydrophilicity. Hydrophilic
groups
can be easily introduced using known chemical synthesis, so long as they do
not give rise to unwanted swelling or an undesirable decrease in compressive
strength. In particular, polyalkoxy segments may weaken gel strength.
The Component Core:
The "core" of each crosslinkable component is comprised of the
molecular structure to which the nucleophilic or electrophilic groups are
bound.
Using the formulae (I) R1-[Q1]1-X),õ for component A, (II) R2(4Q21r-Y),, for
component B, and (Ill)
R3(4P31s-Fn)p for optional component C, the "core" groups are R1,
R2 and R3. Each molecular core of the reactive components of the
=
133
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
crosslinkable composition is generally selected from synthetic and naturally
occurring hydrophilic polymers, hydrophobic polymers, and C2-C14 hydrocarbyl
groups zero to 2 heteroatoms selected from N, 0 and S, with the proviso that
at
least one of the crosslinkable components A, B, and optionally C, comprises a
molecular core of a synthetic hydrophilic polymer. In a preferred embodiment,
at least one of A and B comprises a molecular core of a synthetic hydrophilic
polymer.
Hydrophilic Crosslinkable Components
In one aspect, the crosslinkable component(s) is (are) hydrophilic
polymers. The term "hydrophilic polymer" as used herein refers to a synthetic
polymer having an average molecular weight and composition effective to
render the polymer "hydrophilic" as defined above. As discussed above,
synthetic crosslinkable hydrophilic polymers useful herein include, but are
not
limited to: polyalkylene oxides, particularly polyethylene glycol and
poly(ethylene oxide)-poly(propylene oxide) copolymers, including block and
random copolymers; polyols such as glycerol, polyglycerol (particularly highly
branched polyglycerol), propylene glycol and trimethylene glycol substituted
with one or more polyalkylene oxides, e.g., mono-, di- and tri-
polyoxyethylated
glycerol, mono- and di-polyoxyethylated propylene glycol, and mono- and di-
polyoxyethylated trimethylene glycol; polyoxyethylated sorbitol,
polyoxyethylated glucose; acrylic acid polymers and analogs and copolymers
thereof, such as polyacrylic acid per se, polymethacrylic acid,
poly(hydroxyethyl-methacrylate), poly(hydroxyethylacrylate),
poly(methylalkylsulfoxide methacrylate), poly(methylalkylsulfoxide acrylate)
and
copolymers of any of the foregoing, and/or with additional acrylate species
such
as aminoethyl acrylate and mono-2-(acryloxy)-ethyl succinate; polynnaleic
acid;
poly(acrylamides) such as polyacrylamide per se, poly(methacrylamide),
poly(dimethylacrylamide), and poly(N-isopropyl-acrylamide); poly(olefinic
alcohol)s such as poly(vinyl alcohol); poly(N-vinyl lactams) such as
poly(vinyl
134
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
pyrrolidone), poly(N-vinyl caprolactam), and copolymers thereof;
polyoxazolines, including poly(methyloxazoline) and poly(ethyloxazoline), and
polyvinylamines. It must be emphasized that the aforementioned list of
polymers is not exhaustive, and a variety of other synthetic hydrophilic
polymers may be used, as will be appreciated by those skilled in the art.
The synthetic crosslinkable hydrophilic polymer may be a
homopolymer, a block copolymer, a random copolymer, or a graft copolymer.
In addition, the polymer may be linear or branched, and if branched, may be
minimally to highly branched, dendrimeric, hyperbranched, or a star polymer.
The polymer may include biodegradable segments and blocks, either
distributed throughout the polymer's molecular structure or present as a
single
block, as in a block copolymer. Biodegradable segments are those that
degrade so as to break covalent bonds. Typically, biodegradable segments are
segments that are hydrolyzed in the presence of water and/or enzymatically
cleaved in situ. Biodegradable segments may be composed of small molecular
segments such as ester linkages, anhydride linkages, ortho ester linkages,
ortho carbonate linkages, amide linkages, phosphonate linkages, etc. Larger
biodegradable "blocks" will generally be composed of oligomeric or polymeric
segments incorporated within the hydrophilic polymer. Illustrative oligomeric
and polymeric segments that are biodegradable include, by way of example,
poly(amino acid) segments, poly(orthoester) segments, poly(orthocarbonate)
segments, and the like.
Other suitable synthetic crosslinkable hydrophilic polymers
include chemically synthesized polypeptides, particularly polynucleophilic
polypeptides that have been synthesized to incorporate amino acids containing
primary amino groups (such as lysine) and/or amino acids containing thiol
groups (such as cysteine). Poly(lysine), a synthetically produced polymer of
the
amino acid lysine (145 MW), is particularly preferred. Poly(lysine)s have been
prepared having anywhere from 6 to about 4,000 primary amino groups,
'corresponding to molecular weights of about 870 to about 580,000.
135
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Poly(lysine)s for use in the present invention preferably have a molecular
weight within the range of about 1,000 to about 300,000, more preferably
within
the range of about 5,000 to about 100,000, and most preferably, within the
range of about 8,000 to about 15,000. Poly(lysine)s of varying molecular
weights are commercially available from Peninsula Laboratories, Inc. (Belmont,
Calif.).
The synthetic crosslinkable hydrophilic polymer may be a
homopolymer, a block copolymer, a random copolymer, or a graft copolymer.
In addition, the polymer may be linear or branched, and if branched, may be
minimally to highly branched, dendrimeric, hyperbranched, or a star polymer.
The polymer may include biodegradable segments and blocks, either
distributed throughout the polymer's molecular structure or present as a
single
block, as in a block copolymer. Biodegradable segments are those that
degrade so as to break covalent bonds. Typically, biodegradable segments are
segments that are hydrolyzed in the presence of water and/or enzymatically
cleaved in situ. Biodegradable segments may be composed of small molecular
segments such as ester linkages, anhydride linkages, ortho ester linkages,
ortho carbonate linkages, amide linkages, phosphonate linkages, etc. Larger
biodegradable "blocks" will generally be composed of oligomeric or polymeric
segments incorporated within the hydrophilic polymer. Illustrative oligomeric
and polymeric segments that are biodegradable include, by way of example,
poly(amino acid) segments, poly(orthoester) segments, poly(orthocarbonate)
segments, and the like.
Although a variety of different synthetic crosslinkable hydrophilic
polymers can be used in the present compositions, as indicated above,
preferred synthetic crosslinkable hydrophilic polymers are polyethylene glycol
(PEG) and polyglycerol (PG), particularly highly branched polyglycerol.
Various
forms of PEG are extensively used in the modification of biologically active
molecules because PEG lacks toxicity, antigenicity, and immunogenicity (i.e.,
is
biocompatible), can be formulated so as to have a wide range of solubilities,
136
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
and do not typically interfere with the enzymatic activities and/or
conformations
of peptides. A particularly preferred synthetic crosslinkable hydrophilic
polymer
for certain applications is a polyethylene glycol (PEG) having a molecular
weight within the range of about 100 to about 100,000 mol. wt., although for
highly branched PEG, far higher molecular weight polymers can be employed --
up to 1,000,000 or more -- providing that biodegradable sites are incorporated
ensuring that all degradation products will have a molecular weight of less
than
about 30,000. For most PEGs, however, the preferred molecular weight is
about 1,000 to about 20,000 mol. wt., more preferably within the range of
about
7,500 to about 20,000 mol. wt. Most preferably, the polyethylene glycol has a
molecular weight of approximately 10,000 mol. wt.
Naturally occurring crosslinkable hydrophilic polymers include, but
are not limited to: proteins such as collagen, fibronectin, albumins,
globulins,
fibrinogen, and fibrin, with collagen particularly preferred; carboxylated
polysaccharides such as polymannuronic acid and polygalacturonic acid;
aminated polysaccharides, particularly the glycosaminoglycans, e.g.,
hyaluronic
acid, chitin, chondroitin sulfate A, B, or C, keratin sulfate, keratosulfate
and
heparin; and activated polysaccharides such as dextran and starch derivatives.
Collagen and glycosaminoglycans are examples of naturally occurring
hydrophilic polymers for use herein, with methylated collagen being a
preferred
hydrophilic polymer.
Any of the hydrophilic polymers herein must contain, or be
activated to contain, functional groups, i.e., nucleophilic or electrophilic
groups,
which enable crosslinking. Activation of PEG is discussed below; it is to be
understood, however, that the following discussion is for purposes of
illustration
and analogous techniques may be employed with other polymers.
With respect to PEG, first of all, various functionalized
polyethylene glycols have been used effectively in fields such as protein
modification (see Abuchowski et at., Enzymes as Drugs, John Wiley & Sons:
New York, N.Y. (1981) pp. 367-383; and Dreborg et at., Crit. Rev. Therap. Drug
137
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Carrier Syst. (1990) 6:315), peptide chemistry (see Mutter et at., The
Peptides,
Academic: New York, N.Y. 2:285-332; and Zalipsky et at., Int. J. Peptide
Protein
Res. (1987) 30:740), and the synthesis of polymeric drugs (see Zalipsky et
at.,
Eur. Polym. J. (1983) 19:1177; and Ouchi et al., J. Macromol. Sci. Chem.
(1987) A24:1011).
Activated forms of PEG, including multifunctionally activated PEG,
are commercially available, and are also easily prepared using known methods.
For example, see Chapter 22 of Poly(ethylene Glycol) Chemistry: Biotechnical
and Biomedical Applications, J. Milton Harris, ed., Plenum Press, NY (1992);
and Shearwater Polymers, Inc. Catalog, Polyethylene Glycol Derivatives,
Huntsville, Alabama (1997-1998).
Structures for some specific, tetrafunctionally activated forms of
PEG are shown in FIGS. 1 to 10 of U.S. Patent 5,874,500, as are generalized
reaction products obtained by reacting the activated PEGs with multi-amino
PEGs, i.e., a PEG with two or more primary amino groups. The activated PEGs
illustrated have a pentaerythritol (2,2-bis(hydroxymethyl)-1,3-propanediol)
core.
Such activated PEGs, as will be appreciated by those in the art, are readily
prepared by conversion of the exposed hydroxyl groups in the PEGylated polyol
(i.e., the terminal hydroxyl groups on the PEG chains) to carboxylic acid
groups
(typically by reaction with an anhydride in the presence of a nitrogenous
base),
followed by esterification with N-hydroxysuccinimide, N-
hydroxysulfosuccinimide, or the like, to give the polyfunctionally activated
PEG.
Hydrophobic Polymers:
The crosslinkable compositions of the invention can also include
hydrophobic polymers, although for most uses hydrophilic polymers are
preferred. Polylactic acid and polyglycolic acid are examples of two
hydrophobic polymers that can be used. With other hydrophobic polymers, only
short-chain oligomers should be used, containing at most about 14 carbon
atoms, to avoid solubility-related problems during reaction.
138
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Low Molecular Weight Components:
As indicated above, the molecular core of one or more of the
crosslinkable components can also be a low molecular weight compound, i.e., a
C2-C14 hydrocarbyl group containing zero to 2 heteroatoms selected from N, 0,
S and combinations thereof. Such a molecular core can be substituted with
nucleophilic groups or with electrophilic groups.
When the low molecular weight molecular core is substituted with
primary amino groups, the component may be, for example, ethylenediamine
(H2N-CH2CH2-NH2), tetramethylenediamine (H2N-(CH4)-NH2),
pentamethylenediamine (cadaverine) (H2N-(CH5)-NH2), hexamethylenediamine
(H2N-(CH6)-NH2), bis(2-aminoethyl)amine (HN-[CH2CH2-NH2}2), or tris(2-
aminoethyl)amine (N4CH2CH2-NH2l3).
Low molecular weight diols and polyols include
trimethylolpropane, di(trimethylol propane), pentaerythritol, and diglycerol,
all of
which require activation with a base in order to facilitate their reaction as
nucleophiles. Such diols and polyols may also be functionalized to provide di-
and poly-carboxylic acids, functional groups that are, as noted earlier
herein,
also useful as nucleophiles under certain conditions. Polyacids for use in the
present compositions include, without limitation, trimethylolpropane-based
tricarboxylic acid, di(trimethylol propane)-based tetracarboxylic acid,
heptanedioic acid, octanedioic acid (suberic acid), and hexadecanedioic acid
(thapsic acid), all of which are commercially available and/or readily
synthesized using known techniques.
Low molecular weight di- and poly-electrophiles include, for
example, disuccinimidyl suberate (DSS), bis(sulfosuccinimidyl) suberate (BS3),
dithiobis(succinimidylpropionate) (DSP), bis(2-succinimidooxycarbonyloxy)
ethyl sulfone (BSOCOES), and 3,3'-dithiobis(sulfosuccinimidylpropionate
(DTSPP), and their analogs and derivatives. The aforementioned compounds
are commercially available from Pierce (Rockford, Ill.). Such di- and poly-
electrophiles can also be synthesized from di- and polyacids, for example by
139
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
reaction with an appropriate molar amount of N-hydroxysuccinimide in the
presence of DCC. Polyols such as trimethylolpropane and di(trimethylol
propane) can be converted to carboxylic acid form using various known
techniques, then further derivatized by reaction with NHS in the presence of
DCC to produce trifunctionally and tetrafunctionally activated polymers.
Delivery Systems:
Suitable delivery systems for the homogeneous dry powder
composition (containing at least two crosslinkable polymers) and the two
buffer
solutions may involve a multi-compartment spray device, where one or more
compartments contains the powder and one or more compartments contain the
buffer solutions needed to provide for the aqueous environment, so that the
composition is exposed to the aqueous environment as it leaves the
compartment. Many devices that are adapted for delivery of multi-component
tissue sealants/hemostatic agents are well known in the art and can also be
used in the practice of the present invention. Alternatively, the composition
can
be delivered using any type of controllable extrusion system, or it can be
delivered manually in the form of a dry powder, and exposed to the aqueous
environment at the site of administration.
The homogeneous dry powder composition and the two buffer
solutions may be conveniently formed under aseptic conditions by placing each
of the three ingredients (dry powder, acidic buffer solution and basic buffer
solution) into separate syringe barrels. For example, the composition, first
buffer solution and second buffer solution can be housed separately in a
multiple-compartment syringe system having a multiple barrels, a mixing head,
and an exit orifice. The first buffer solution can be added to the barrel
housing
the composition to dissolve the composition and form a homogeneous solution,
which is then extruded into the mixing head. The second buffer solution can be
simultaneously extruded into the mixing head. Finally, the resulting
composition can then be extruded through the orifice onto a surface.
140
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
For example, the syringe barrels holding the dry powder and the
basic buffer may be part of a dual-syringe system, e.g., a double barrel
syringe
as described in U.S. Patent 4,359,049 to Redl et al. In this embodiment, the
acid buffer can be added to the syringe barrel that also holds the dry powder,
so as to produce the homogeneous solution. In other words, the acid buffer
may be added (e.g., injected) into the syringe barrel holding the dry powder
to
thereby produce a homogeneous solution of the first and second components.
This homogeneous solution can then be extruded into a mixing head, while the
basic buffer is simultaneously extruded into the mixing head. Within the
mixing
head, the homogeneous solution and the basic buffer are mixed together to
thereby form a reactive mixture. Thereafter, the reactive mixture is extruded
through an orifice and onto a surface (e.g., tissue), where a film is formed,
which can function as a sealant or a barrier, or the like. The reactive
mixture
begins forming a three-dimensional matrix immediately upon being formed by
the mixing of the homogeneous solution and the basic buffer in the mixing
head. Accordingly, the reactive mixture is preferably extruded from the mixing
head onto the tissue very quickly after it is formed so that the three-
dimensional
matrix forms on, and is able to adhere to, the tissue.
Other systems for combining two reactive liquids are well known
in the art, and include the systems described in U.S. Patent Nos. 6,454,786 to
Holm et al.; 6,461,325 to Delmotte et al.; 5,585,007 to Antanavich et al.;
5,116,315 to Capozzi et al.; and 4,631,055 to Redl et al.
Storage and Handling:
Because crosslinkable components containing electrophilic
groups react with water, the electrophilic component or components are
generally stored and used in sterile, dry form to prevent hydrolysis.
Processes
for preparing synthetic hydrophilic polymers containing multiple electrophilic
groups in sterile, dry form are set forth in commonly assigned U.S. Patent No.
5,643,464 to Rhee et al. For example, the dry synthetic polymer may be
141
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
compression molded into a thin sheet or membrane, which can then be
sterilized using gamma or, preferably, e-beam irradiation. The resulting dry
membrane or sheet can be cut to the desired size or chopped into smaller size
particulates.
Components containing multiple nucleophilic groups are generally
not water-reactive and can therefore be stored either dry or in aqueous
solution.
If stored as a dry, particulate, solid, the various components of the
crosslinkable
composition may be blended and stored in a single container. Admixture of all
components with water, saline, or other aqueous media should not occur until
immediately prior to use.
In an alternative embodiment, the crosslinking components can
be mixed together in a single aqueous medium in which they are both
unreactive, i.e., such as in a low pH buffer. Thereafter, they can be sprayed
onto the targeted tissue site along with a high pH buffer, after which they
will
rapidly react and form a gel.
Suitable liquid media for storage of crosslinkable compositions
include aqueous buffer solutions such as monobasic sodium phosphate/dibasic
sodium phosphate, sodium carbonate/sodium bicarbonate, glutamate or
acetate, at a concentration of 0.5 to 300 mM. In general, a sulfhydryl-
reactive
component such as PEG substituted with maleimido groups or succinimidyl
esters is prepared in water or a dilute buffer, with a pH of between around 5
to
6. Buffers with pks between about 8 and 10.5 for preparing a polysulfhydryl
component such as sulfhydryl-PEG are useful to achieve fast gelation time of
compositions containing mixtures of sulfhydryl-PEG and SG-PEG. These
include carbonate, borate and AMPSO (3-[(1,1-dimethy1-2-
hydroxyethyDamino]2-hydroxy-propane-sulfonic acid). In contrast, using a
combination of maleimidyl PEG and sulfhydryl-PEG, a pH of around 5 to 9 is
preferred for the liquid medium used to prepare the sulfhydryl PEG.
142
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Collagen + Fibrinogen and/or Thrombin (e.g., Costasis)
In yet another aspect, the polymer composition may include
collagen in combination with fibrinogen and/or thrombin. (See, e.g., U.S.
Patent
Nos. 5,290,552; 6,096,309; 5,997,811; 5,968,018, and 6,123,687). For
example, an aqueous composition may include a fibrinogen and FXIII,
particularly plasma, collagen in an amount sufficient to thicken the
composition,
thrombin in an amount sufficient to catalyze polymerization of fibrinogen
present in the composition, and Ca2+ and, optionally, an antifibrinolytic
agent in
amount sufficient to retard degradation of the resulting adhesive clot. The
composition may be formulated as a two-part composition that may be mixed
together just prior to use, in which fibrinogen/FXIII and collagen constitute
the
first component, and thrombin together with an antifibrinolytic agent, and
Ca2+
constitute the second component.
Plasma, which provides a source of fibrinogen, may be obtained
from the patient for which the composition is to be delivered. The plasma can
be used "as is" after standard preparation which includes centrifuging out
cellular components of blood. Alternatively, the plasma can be further
processed to concentrate the fibrinogen to prepare a plasma cryoprecipitate.
The plasma cryoprecipitate can be prepared by freezing the plasma for at least
about an hour at about -20 C, and then storing the frozen plasma overnight at
about 4 C to slowly thaw. The thawed plasma is centrifuged and the plasma
cryoprecipitate is harvested by removing approximately four-fifths of the
plasma
to provide a cryoprecipitate comprising the remaining one-fifth of the plasma.
Other fibrinogen/FXIII preparations may be used, such as cryoprecipitate,
patient autologous fibrin sealant, fibrinogen analogs or other single donor or
commercial fibrin sealant materials. Approximately 0.5 ml to about 1.0 ml of
either the plasma or the plasma-cryoprecipitate provides about 1 to 2 ml of
adhesive composition which is sufficient for use in middle ear surgery. Other
plasma proteins (e.g., albumin, plasminogen, von Willebrands factor, Factor
143
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
VIII, etc.) may or may not be present in the fibrinogen/FXII separation due to
wide variations in the formulations and methods to derive them.
Collagen, preferably hypoallergenic collagen, is present in the
composition in an amount sufficient to thicken the composition and augment the
cohesive properties of the preparation. The collagen may be atelopeptide
collagen or telopeptide collagen, e.g., native collagen. In addition to
thickening
the composition, the collagen augments the fibrin by acting as a
macromolecular lattice work or scaffold to which the fibrin network adsorbs.
This gives more strength and durability to the resulting glue clot with a
relatively
low concentration of fibrinogen in comparison to the various concentrated
autogenous fibrinogen glue formulations (L e., AFGs).
The form of collagen which is employed may be described as at
least "near native" in its structural characteristics. It may be further
characterized as resulting in insoluble fibers at a pH above 5; unless
crosslinked or as part of a complex composition, e.g., bone, it will generally
consist of a minor amount by weight of fibers with diameters greater than 50
nm, usually from about 1 to 25 volume A) and there will be substantially
little, if
any, change in the helical structure of the fibrils. In addition, the collagen
composition must be able to enhance gelation in the surgical adhesion
composition.
A number of commercially available collagen preparations may be
used. ZYDERM Collagen Implant (ZCI) has a fibrillar diameter distribution
consisting of 5 to 10 nm diameter fibers at 90% volume content and the
remaining 10% with greater than about 50 nm diameter fibers. ZCI is available
as a fibrillar slurry and solution in phosphate buffered isotonic saline, pH
7.2,
and is injectable with fine gauge needles. As distinct from ZCI, cross-linked
collagen available as ZYPLAST may be employed. ZYPLAST is essentially an
exogenously crosslinked (glutaraldehyde) version of ZCI. The material has a
somewhat higher content of greater than about 50 nm diameter fibrils and
144
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
remains insoluble over a wide pH range. Crosslinking has the effect of
mimicking in vivo endogenous crosslin king found in many tissues.
Thrombin acts as a catalyst for fibrinogen to provide fibrin, an
insoluble polymer and is present in the composition in an amount sufficient to
catalyze polymerization of fibrinogen present in the patient plasma. Thrombin
also activates FXIII, a plasma protein that catalyzes covalent crosslinks in
fibrin,
rendering the resultant clot insoluble. Usually the thrombin is present in the
adhesive composition in concentration of from about 0.01 to about 1000 or
greater N1H units (NIHu) of activity, usually about ito about 500 NIHu, most
usually about 200 to about 500 NIHu. The thrombin can be from a variety of
host animal sources, conveniently bovine. Thrombin is commercially available
from a variety of sources including Parke-Davis, usually lyophilized with
buffer
salts and stabilizers in vials which provide thrombin activity ranging from
about
1000 NIHu to 10,000 NIHu. The thrombin is usually prepared by reconstituting
the powder by the addition of either sterile distilled water or isotonic
saline.
Alternately, thrombin analogs or reptile-sourced coagulants may be used.
The composition may additionally comprise an effective amount of
an antifibrinolytic agent to enhance the integrity of the glue clot as the
healing
processes occur. A number of antifibrinolytic agents are well known and
include aprotinin, C1-esterase inhibitor and 6-amino-n-caproic acid (EACA). E-
amino-n-caproic acid, the only antifibrinolytic agent approved by the FDA, is
effective at a concentration of from about 5 mg/ml to about 40 mg/ml of the
final
adhesive composition, more usually from about 20 to about 30 mg/ml. EACA is
commercially available as a solution having a concentration of about 250
mg/ml. Conveniently, the commercial solution is diluted with distilled water
to
provide a solution of the desired concentration. That solution is desirably
used
to reconstitute lyophilized thrombin to the desired thrombin concentration.
Other examples of in situ forming materials based on the
crosslinking of proteins are described, e.g., in U.S. Patent Nos. RE38158;
4,839,345; 5,514,379, 5,583,114; 6,458,147; 6,371,975; 5,290,552; 6,096,309;
145
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
U.S. Patent Application Publication Nos. 2002/0161399; 2001/0018598 and
PCT Publication Nos. WO 03/090683; WO 01/45761; WO 99/66964 and WO
96/03159).
Self-Reactive Compounds
In one aspect, the therapeutic agent is released from a
crosslinked matrix formed, at least in part, from a self-reactive compound. As
used herein, a self-reactive compound comprises a core substituted with a
minimum of three reactive groups. The reactive groups may be directed
attached to the core of the compound, or the reactive groups may be indirectly
attached to the compound's core, e.g., the reactive groups are joined to the
core through one or more linking groups.
Each of the three reactive groups that are necessarily present in a
self-reactive compound can undergo a bond-forming reaction with at least one
of the remaining two reactive groups. For clarity it is mentioned that when
these compounds react to form a crosslinked matrix, it will most often happen
that reactive groups on one compound will reactive with reactive groups on
another compound. That is, the term "self-reactive" is not intended to mean
that each self-reactive compound necessarily reacts with itself, but rather
that
when a plurality of identical self-reactive compounds are in combination and
undergo a crosslinking reaction, then these compounds will react with one
another to form the matrix. The compounds are "self-reactive" in the sense
that
they can react with other compounds having the identical chemical structure as
themselves.
The self-reactive compound comprises at least four components:
a core and three reactive groups. In one embodiment, the self-reactive
compound can be characterized by the formula (I), where R is the core, the
reactive groups are represented by Xl, X2 and X3, and a linker (L) is
optionally
present between the core and a functional group.
146
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
X2
I
(2
L )
1 a
I (1)
)(1¨(L1
)¨R¨(L3)---X3
The core R is a polyvalent moiety having attachment to at least
three groups (i.e., it is at least trivalent) and may be, or may contain, for
example, a hydrophilic polymer, a hydrophobic polymer, an amphiphilic
polymer, a C2-14 hydrocarbyl, or a C2-14 hydrocarbyl which is heteroatom-
containing. The linking groups Ll, L2, and L3 may be the same or different.
The
designators p, q and r are either 0 (when no linker is present) or 1 (when a
linker is present). The reactive groups X1, X2 and X3 may be the same or
different. Each of these reactive groups reacts with at least one other
reactive
group to form a three-dimensional matrix. Therefore X1 can react with X2
and/or X3, X2 can react with X1 and/or X3, X3 can react with X1 and/or X2 and
so
forth. A trivalent core will be directly or indirectly bonded to three
functional
groups, a tetravalent core will be directly or indirectly bonded to four
functional
groups, etc.
Each side chain typically has one reactive group. However, the
invention also encompasses self-reactive compounds where the side chains
contain more than one reactive group. Thus, in another embodiment of the
invention, the self-reactive compound has the formula (II):
[ X' - (-4)a - Y' - (L5)13 i c¨R'
where: a and b are integers from 0-1; c is an integer from 3-12; R' is
selected
from hydrophilic polymers, hydrophobic polymers, amphiphilic polymers, C2-14
hydrocarbyls, and heteroatom-containing C2-14 hydrocarbyls; X' and Y' are
reactive groups and can be the same or different; and L4 and L5 are linking
groups. Each reactive group inter-reacts with the other reactive group to form
a
three-dimensional matrix. The compound is essentially non-reactive in an
initial
environment but is rendered reactive upon exposure to a modification in the
initial environment that provides a modified environment such that a plurality
of
147 '
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
the self-reactive compounds inter-react in the modified environment to form a
three-dimensional matrix. In one preferred embodiment, R is a hydrophilic
polymer. In another preferred embodiment, X' is a nucleophilic group and Y' is
an electrophilic group.
The following self-reactive compound is one example of a
compound of formula (II):
Rzto
oR4
R4ok
oR4
where R4 has the formula:
z ______________________ o/ 0
H)_ x
0
Thus, in formula (II), a and bare 1; c is 4; the core R' is the
hydrophilic polymer, tetrafunctionally activated polyethylene glycol, (C(CH2-0-
)4; Xis the electrophilic reactive group, succinimidyl; Y' is the nucleophilic
reactive group -CH-NH2; L4 is -C(0)-0-; and L5 is -(CH2- CH2-0-CH2)x-CH2-0-
C(0)-(CF12)2--
The self-reactive compounds of the invention are readily
synthesized by techniques that are well known in the art. An exemplary
synthesis is set forth below:
148
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
OH 0
....
-
__________________________ / / HO __________ -(\
R40 0
0 ______________________ / (,_ x +
- HN 0
R40/-1./ \I __ 0
OOW 0
eill
Mitsunobo
or
DCC
p
_ o __
R4o
o/
o ______________________ / H110
µ) __________________________________________________ 0
fik
R40/-1-/ 0
OW
=
1 H2, Pd/C
149
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
0 ________________________________________________ KD
R40
0- __________________________________________________ H2N OH
X
R40/+
OR4
0
MitSournob0
HO DCC
0
0
o/ 0
R40
0 H)_ X
R40* 0
OR4
The reactive groups are selected so that the compound is
essentially non-reactive in an initial environment. Upon exposure to a
specific
modification in the initial environment, providing a modified environment, the
compound is rendered reactive and a plurality of self-reactive compounds are
then able to inter-react in the modified environment to form a three-
dimensional
matrix. Examples of modification in the initial environment are detailed
below,
but include the addition of an aqueous medium, a change in pH, exposure to
ultraviolet radiation, a change in temperature, or contact with a redox
initiator.
The core and reactive groups can also be selected so as to
provide a compound that has one of more of the following features: are
biocompatible, are non-immunogenic, and do not leave any toxic, inflammatory
or immunogenic reaction products at the site of administration. Similarly, the
150
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
core and reactive groups can also be selected so as to provide a resulting
matrix that has one or more of these features.
In one embodiment of the invention, substantially immediately or
immediately upon exposure to the modified environment, the self-reactive
compounds inter-react form a three-dimensional matrix. The term "substantially
immediately" is intended to mean within less than five minutes, preferably
within
less than two minutes, and the term "immediately" is intended to mean within
less than one minute, preferably within less than 30 seconds.
In one embodiment, the self-reactive compound and resulting
matrix are not subject to enzymatic cleavage by matrix metalloproteinases such
as collagenase, and are therefore not readily degradable in vivo. Further, the
self-reactive compound may be readily tailored, in terms of the selection and
quantity of each component, to enhance certain properties, e.g., compression
strength, swellability, tack, hydrophilicity, optical clarity, and the like.
In one preferred embodiment, R is a hydrophilic polymer. In
another preferred embodiment, X is a nucleophilic group, Y is an electrophilic
group and Z is either an electrophilic or a nucleophilic group. Additional
embodiments are detailed below.
A higher degree of inter-reaction, e.g., crosslinking, may be useful
when a less swellable matrix is desired or increased compressive strength is
desired. In those embodiments, it may be desirable to have n be an integer
from 2-12. In addition, when a plurality of self-reactive compounds are
utilized,
the compounds may be the same or different.
A. Reactive Groups
Prior to use, the self-reactive compound is stored in an initial
environment that insures that the compound remain essentially non-reactive
until use. Upon modification of this environment, the compound is rendered
reactive and a plurality of compounds will then inter-react to form the
desired
151
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
matrix. The initial environment, as well as the modified environment, is thus
determined by the nature of the reactive groups involved.
The number of reactive groups can be the same or different.
However, in one embodiment of the invention, the number of reactive groups
are approximately equal. As used in this context, the term "approximately"
refers to a 2:1 to 1:2 ratio of moles of one reactive group to moles of a
different
reactive groups. A 1:1:1 molar ratio of reactive groups is generally
preferred.
In general, the concentration of the self-reactive compounds in the
modified environment, when liquid in nature, will be in the range of about 1
to
50 wt%, generally about 2 to 40 wt%. The preferred concentration of the
compound in the liquid will depend on a number of factors, including the type
of
compound (i.e., type of molecular core and reactive groups), its molecular
weight, and the end use of the resulting three-dimensional matrix. For
example, use of higher concentrations of the compounds, or using highly
functionalized compounds, will result in the formation of a more tightly
crosslinked network, producing a stiffer, more robust gel. As such,
compositions intended for use in tissue augmentation will generally employ
concentrations of self-reactive compounds that fall toward the higher end of
the
preferred concentration range. Compositions intended for use as bioadhesives
or in adhesion prevention do not need to be as firm and may therefore contain
lower concentrations of the self-reactive compounds.
1) Electrophilic and Nucleophilic Reactive Groups
In one embodiment of the invention, the reactive groups are
electrophilic and nucleophilic groups, which undergo a nucleophilic
substitution
reaction, a nucleophilic addition reaction, or both. The term "electrophilic"
refers to a reactive group that is susceptible to nucleophilic attack, i.e.,
susceptible to reaction with an incoming nucleophilic group. Electrophilic
groups herein are positively charged or electron-deficient, typically electron-
deficient. The term "nucleophilic" refers to a reactive group that is electron
rich,
152
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
has an unshared pair of electrons acting as a reactive site, and reacts with a
positively charged or electron-deficient site. For such reactive groups, the
modification in the initial environment comprises the addition of an aqueous
medium and/or a change in pH.
In one embodiment of the invention, X1 (also referred to herein as
X) can be a nucleophilic group and X2 (also referred to herein as Y) can be an
electrophilic group or vice versa, and X3 (also referred to herein as Z) can
be
either an electrophilic or a nucleophilic group.
X may be virtually any nucleophilic group, so long as reaction can
occur with the electrophilic group Y and also with Z, when Z is electrophilic
(ZEL). Analogously, Y may be virtually any electrophilic group, so long as
reaction can take place with X and also with Z when Z is nucleophilic (ZNu).
The only limitation is a practical one, in that reaction between X and Y, and
X
and ZEL, or Y and ZNu should be fairly rapid and take place automatically upon
admixture with an aqueous medium, without need for heat or potentially toxic
or
non-biodegradable reaction catalysts or other chemical reagents. It is also
preferred although not essential that reaction occur without need for
ultraviolet
or other radiation. In one embodiment, the reactions between X and Y, and
between either X and ZEL or Y and ZNu, are complete in under 60 minutes,
preferably under 30 minutes. Most preferably, the reaction occurs in about 5
to
15 minutes or less.
Examples of nucleophilic groups suitable as X or FnNu include, but
are not limited to: -NH2, -NHR1, -N(R1)2, -SH, -OH, -COOH, -C6H4-0H, -H,
-PH2,
-PHR1, -P(R1)2, -NH-NH2, -CO-NH-NH2, -05H4N, etc. wherein R1 is a
hydrocarbyl group and each R1 may be the same or different. R1 is typically
alkyl or monocyclic aryl, preferably alkyl, and most preferably lower alkyl.
Organometallic moieties are also useful nucleophilic groups for the purposes
of
the invention, particularly those that act as carbanion donors. Examples of
organometallic moieties include: Grignard functionalities -R2MgHal wherein R2
153
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
is a carbon atom (substituted or unsubstituted), and Hal is halo, typically
bromo,
iodo or chloro, preferably bromo; and lithium-containing functionalities,
typically
alkyllithium groups; sodium-containing functionalities.
It will be appreciated by those of ordinary skill in the art that
certain nucleophilic groups must be activated with a base so as to be capable
of reaction with an electrophilic group. For example, when there are
nucleophilic sulfhydryl and hydroxyl groups in the self-reactive compound, the
compound must be admixed with an aqueous base in order to remove a proton
and provide an -S- or -0- species to enable reaction with the electrophilic
group.
Unless it is desirable for the base to participate in the reaction, a non-
nucleophilic base is preferred. In some embodiments, the base may be present
as a component of a buffer solution. Suitable bases and corresponding
crosslinking reactions are described herein.
The selection of electrophilic groups provided on the self-reactive
compound, must be made so that reaction is possible with the specific
nucleophilic groups. Thus, when the X reactive groups are amino groups, the Y
and any ZEL groups are selected so as to react with amino groups.
Analogously, when the X reactive groups are sulfhydryl moieties, the
corresponding electrophilic groups are sulfhydryl-reactive groups, and the
like.
In general, examples of electrophilic groups suitable as Y or ZEL include, but
are
not limited to, -CO-CI, -(C0)-0-(C0)-R (where R is an alkyl group),
-CH=CH-CH=0 and -CH=CH-C(CH3)=0, halo, -N=C=O, -N=C=S,
-S02CH=CH2, -0(C0)-C=CH2, -0(C0)-C(CH3)=CH2, -S-S-(C5H4N),
-0(C0)-C(CH2CH3)=CH2, -CH=CH-C=NH, -COOH, -(CO)O-N(COCH2)2, -CHO,
-(CO)O-N(COCH2)2-S(0)20H, and -N(COCH)2.
When X is amino (generally although not necessarily primary
amino), the electrophilic groups present on Y and ZEL are amine-reactive
groups. Exemplary amine-reactive groups include, by way of example and not
limitation, the following groups, or radicals thereof: (1) carboxylic acid
esters,
including cyclic esters and "activated" esters; (2) acid chloride groups (-CO-
CI);
154
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
(3) anhydrides (-(C0)-0-(C0)-R, where R is an alkyl group); (4) ketones and
aldehydes, including a,13-unsaturated aldehydes and ketones such as
-CH=CH-C1-1=0 and -CH=CH-C(CH3)=0; (5) halo groups; (6) isocyanate group
(-N=C=0); (7) thioisocyanato group (-N.--C=S); (8) epoxides; (9) activated
hydroxyl groups (e.g., activated with conventional activating agents such as
carbonyldiimidazole or sulfonyl chloride); and (10) olefins, including
conjugated
olefins, such as ethenesulfonyl (-S02CH=CH2) and analogous functional
groups, including acrylate (-0(C0)-C=CH2), methacrylate
(-0(C0)-C(CH3)=CH2), ethyl acryiate (-0(C0)-C(CH2CH3)=CH2), and
ethyleneimino (-CH=CH-C=NH).
In one embodiment the amine-reactive groups contain an
electrophilically reactive carbonyl group susceptible to nucleophilic attack
by a
primary or secondary amine, for example the carboxylic acid esters and
aldehydes noted above, as well as carboxyl groups (-COOH).
Since a carboxylic acid group per se is not susceptible to reaction
with a nucleophilic amine, components containing carboxylic acid groups must
be activated so as to be amine-reactive. Activation may be accomplished in a
variety of ways, but often involves reaction with a suitable hydroxyl-
containing
compound in the presence of a dehydrating agent such as
dicyclohexylcarbodiimide (DCC) or dicyclohexylurea (DHU). For example, a
carboxylic acid can be reacted with an alkoxy-substituted N-hydroxy-
succinimide or N-hydroxysulfosuccinimide in the presence of DCC to form
reactive electrophilic groups, the N-hydroxysuccinimide ester and the N-
hydroxysulfosuccinimide ester, respectively. Carboxylic acids may also be
activated by reaction with an acyl halide such as an acyl chloride (e.g.,
acetyl
chloride), to provide a reactive anhydride group. In a further example, a
carboxylic acid may be converted to an acid chloride group using, e.g.,
thionyl
chloride or an acyl chloride capable of an exchange reaction. Specific
reagents
and procedures used to carry out such activation reactions will be known to
155
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
those of ordinary skill in the art and are described in the pertinent texts
and
literature.
Accordingly, in one embodiment, the amine-reactive groups are
selected from succinimidyl ester (-0(C0)-N(COCH2)2), sulfosuccinimidyl ester
(-0(C0)-N(COCH2)2-S(0)20H), maleimido (-N(COCH)2), epoxy, isocyanato,
thioisocyanato, and ethenesulfonyl.
Analogously, when X is sulfhydryl, the electrophilic groups present
on Y and ZEL are groups that react with a sulfhydryl moiety. Such reactive
groups include those that form thioester linkages upon reaction with a
sulfhydryl
group, such as those described in WO 00/62827 to Wallace et al. As explained
in detail therein, sulfhydryl reactive groups include, but are not limited to:
mixed
anhydrides; ester derivatives of phosphorus; ester derivatives of p-
nitrophenol,
p-nitrothiophenol and pentafluorophenol; esters of substituted hydroxylamines,
including N-hydroxyphthalimide esters, N-hydroxysuccinimide esters, N-
hydroxysulfosuccinimide esters, and N-hydroxyglutarimide esters; esters of 1-
hydroxybenzotriazole; 3-hydroxy-3,4-dihydro-benzotriazin-4-one; 3-hydroxy-
3,4-dihydro-quinazoline-4-one; carbonylimidazole derivatives; acid chlorides;
ketenes; and isocyanates. With these sulfhydryl reactive groups, auxiliary
reagents can also be used to facilitate bond formation, e.g., 1-ethyl-3-[3-
dimethylaminopropyl]carbodiimide can be used to facilitate coupling of
sulfhydryl groups to carboxyl-containing groups.
In addition to the sulfhydryl reactive groups that form thioester
linkages, various other sulfhydryl reactive functionalities can be utilized
that
form other types of linkages. For example, compounds that contain methyl
imidate derivatives form imido-thioester linkages with sulfhydryl groups.
Alternatively, sulfhydryl reactive groups can be employed that form disulfide
bonds with sulfhydryl groups; such groups generally have the structure -S-S-Ar
where Ar is a substituted or unsubstituted nitrogen-containing heteroaromatic
moiety or a non-heterocyclic aromatic group substituted with an electron-
withdrawing moiety, such that Ar may be, for example, 4-pyridinyl, o-
156
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
nitrophenyl, m-nitrophenyl, p-nitrophenyl, 2,4-dinitrophenyl, 2-nitro-4-
benzoic
acid, 2-nitro-4-pyridinyl, etc. In such instances, auxiliary reagents, i.e.,
mild
oxidizing agents such as hydrogen peroxide, can be used to facilitate
disulfide
bond formation.
Yet another class of sulfhydryl reactive groups forms thioether
bonds with sulfhydryl groups. Such groups include, inter alia, maleimido,
substituted maleimido, haloalkyl, epoxy, imino, and aziridino, as well as
olefins
(including conjugated olefins) such as ethenesulfonyl, etheneimino, acrylate,
methacrylate, and a,13-unsaturated aldehydes and ketones.
When X is -OH, the electrophilic functional groups on the
remaining component(s) must react with hydroxyl groups. The hydroxyl group
may be activated as described above with respect to carboxylic acid groups, or
it may react directly in the presence of base with a sufficiently reactive
electrophilic group such as an epoxide group, an aziridine group, an acyl
halide,
an anhydride, and so forth.
When X is an organometallic nucleophilic group such as a
Grignard functionality or an alkyllithium group, suitable electrophilic
functional
groups for reaction therewith are those containing carbonyl groups, including,
by way of example, ketones and aldehydes.
It will also be appreciated that certain functional groups can react
as nucleophilic or as electrophilic groups, depending on the selected reaction
partner and/or the reaction conditions. For example, a carboxylic acid group
can act as a nucleophilic group in the presence of a fairly strong base, but
generally acts as an electrophilic group allowing nucleophilic attack at the
carbonyl carbon and concomitant replacement of the hydroxyl group with the
incoming nucleophilic group.
These, as well as other embodiments are illustrated below, where
the covalent linkages in the matrix that result upon covalent binding of
specific
nucleophilic reactive groups to specific electrophilic reactive groups on the
self-
reactive compound include, solely by way of example, the following Table:
157
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Table
Representative
Nucleophilic Representative Electrophilic Resulting
Group (X, ZNL)) Group (Y, ZEL) Linkage
-NH2 -0-(C0)-0-N(COCH2)2 -NH-(C0)-0-
succinimidyl carbonate terminus
-SH -0-(C0)-0-N(COCH2)2 -S-(C0)-0-
-OH -0-(C0)-0-N(COCH2)2
-0-(C0)-
-NH2 -0(C0)-CH=CH2 -NH-CH2CH2-
acrylate terminus (C0)-0-
-SH -0-(C0)-CH=CH2 -S-
CH2CH2-
(C0)-0-
-OH -0-(C0)-CH=CH2 -0-
CH2CH2-
(C0)-0-
-NH2 -0(C0)-(CH2)3-0O2-
N(COCH2)2 -NH-(C0)-
succinimidyl glutarate terminus (CH2)3-(C0)-
0-
-SH -0(C0)-(C1-12)3-0O2-
N(COCH2)2 -S-(C0)-(CH2)3-
(C0)-0-
-OH -0(C0)-
(CH2)3-0O2-N(COCH2)2 -0-(C0)-(CH2)3-
(C0)-0-
-NH2 -0-CH2-0O2-N(COCH2)2 -NH-
(C0)-CH2-
succinimidyl acetate terminus 0-
-SH -0-CH2-0O2-N(COCH2)2 -S-(C0)-
CH2-0-
-OH -0-CH2-0O2-N(COCH2)2 -0-(C0)-
CH2-0-
-NH2 -0-NH(C0)-(CH2)2-0O2- -NH-
(C0)-
N(000I-12)2 (CH2)2-(C0)-NH-
succinimidyl succinamide 0-
terminus
-SH -0-NH(C0)-(CH2)2-0O2-
N(COCH2)2 (C0)-NH-0-
-OH -0-NH(C0)-(CH2)2-0O2- -0-
(CO)-(CF12)2-
N(COCH2)2 (C0)-NH-0-
-NH2 -0- (CH2)2-CHO -NH-
(C0)-
propionaldehyde terminus (CH2)2-0-
158
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Representative
Nucleophilic Representative Electrophilic Resulting
Group (X, ZNu) Group (Y, ZEL) Linkage
-NH2 /0\ -NH-CH2-
-0¨CH2¨CH¨CH2 CH(OH)-CH2-0-
glycidyl ether terminus and
-N[CH2-CH(OH)-
CH2-0-]2 _
-NH2 -0-(CH2)2-N=C=0 -NH-(C0)-NH-
(isocyanate terminus) CH2-0-
-
-NH2 -S02-CH=CH2 -NH-CH2CH2-
vinyl sulfone terminus SO2-
_
-SH -S02-CH=CH2 -S-CH2CH2-S02-
For self-reactive compounds containing electrophilic and
nucleophilic reactive groups, the initial environment typically can be dry and
sterile. Since electrophilic groups react with water, storage in sterile, dry
form
will prevent hydrolysis. The dry synthetic polymer may be compression molded
into a thin sheet or membrane, which can then be sterilized using gamma or e-
beam irradiation. The resulting dry membrane or sheet can be cut to the
desired size or chopped into smaller size particulates. The modification of a
dry
initial environment will typically comprise the addition of an aqueous medium.
In one embodiment, the initial environment can be an aqueous
medium such as in a low pH buffer, i.e., having a pH less than about 6.0, in
which both electrophilic and nucleophilic groups are non-reactive. Suitable
liquid media for storage of such compounds include aqueous buffer solutions
such as monobasic sodium phosphate/dibasic sodium phosphate, sodium
carbonate/sodium bicarbonate, glutamate or acetate, at a concentration of 0.5
to 300 mM. Modification of an initial low pH aqueous environment will
typically
comprise increasing the pH to at least pH 7.0, more preferably increasing the
pH to at least pH 9.5.
In another embodiment the modification of a dry initial
environment comprises dissolving the self-reactive compound in a first buffer
solution having a pH within the range of about 1.0 to 5.5 to form a
159
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
homogeneous solution, and (ii) adding a second buffer solution having a pH
within the range of about 6.0 to 11.0 to the homogeneous solution. The buffer
solutions are aqueous and can be any pharmaceutically acceptable basic or
acid composition. The term "buffer" is used in a general sense to refer to an
acidic or basic aqueous solution, where the solution may or may not be
functioning to provide a buffering effect (Le., resistance to change in pH
upon
addition of acid or base) in the compositions of the present invention. For
example, the self-reactive compound can be in the form of a homogeneous dry
powder. This powder is then combined with a buffer solution having a pH within
the range of about 1.0 to 5.5 to form a homogeneous acidic aqueous solution,
and this solution is then combined with a buffer solution having a pH within
the
range of about 6.0 to 11.0 to form a reactive solution. For example, 0.375
grams of the dry powder can be combined with 0.75 grams of the acid buffer to
provide, after mixing, a homogeneous solution, where this solution is combined
with 1.1 grams of the basic buffer to provide a reactive mixture that
substantially
immediately forms a three-dimensional matrix.
Acidic buffer solutions having a pH within the range of about 1.0
to 5.5, include by way of illustration and not limitation, solutions of:
citric acid,
hydrochloric acid, phosphoric acid, sulfuric acid, AMPSO (3-[(1,1-dimethy1-2-
hydroxyethypaminop-hydroxy-propane-sulfonic acid), acetic acid, lactic acid,
and combinations thereof. In a preferred embodiment, the acidic buffer
solution, is a solution of citric acid, hydrochloric acid, phosphoric acid,
sulfuric
acid, and combinations thereof. Regardless of the precise acidifying agent,
the
acidic buffer preferably has a pH such that it retards the reactivity of the
nucleophilic groups on the core. For example, a pH of 2.1 is generally
sufficient
to retard the nucleophilicity of thiol groups. A lower pH is typically
preferred
when the core contains amine groups as the nucleophilic groups. In general,
the acidic buffer is an acidic solution that, when contacted with nucleophilic
groups, renders those nucleophilic groups relatively non-nucleophilic.
160
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
An exemplary acidic buffer is a solution of hydrochloric acid,
having a concentration of about 6.3 mM and a pH in the range of 2.1 to 2.3.
This buffer may be prepared by combining concentrated hydrochloric acid with
water, i.e., by diluting concentrated hydrochloric acid with water. Similarly,
this
buffer A may also be conveniently prepared by diluting 1.23 grams of
concentrated hydrochloric acid to a volume of 2 liters, or diluting 1.84 grams
of
concentrated hydrochloric acid to a volume to 3 liters, or diluting 2.45 grams
of
concentrated hydrochloric acid to a volume of 4 liters, or diluting 3.07 grams
concentrated hydrochloric acid to a volume of 5 liters, or diluting 3.68 grams
of
concentrated hydrochloric acid to a volume to 6 liters. For safety reasons,
the
concentrated acid is preferably added to water.
Basic buffer solutions having a pH within the range of about 6.0 to
11.0, include by way of illustration and not limitation, solutions of:
glutamate,
acetate, carbonate and carbonate salts (e.g., sodium carbonate, sodium
carbonate monohydrate and sodium bicarbonate), borate, phosphate and
phosphate salts (e.g., monobasic sodium phosphate monohydrate and dibasic
sodium phosphate), and combinations thereof. In a preferred embodiment, the
basic buffer solution is a solution of carbonate salts, phosphate salts, and
combinations thereof.
In general, the basic buffer is an aqueous solution that neutralizes
the effect of the acidic buffer, when it is added to the homogeneous solution
of
the compound and first buffer, so that the nucleophilic groups on the core
regain their nucleophilic character (that has been masked by the action of the
acidic buffer), thus allowing the nucleophilic groups to inter-react with the
electrophilic groups on the core.
An exemplary basic buffer is an aqueous solution of carbonate
and phosphate salts. This buffer may be prepared by combining a base
solution with a salt solution. The salt solution may be prepared by combining
34.7 g of monobasic sodium phosphate monohydrate, 49.3 g of sodium
carbonate monohydrate, and sufficient water to provide a solution volume of 2
161
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
liter. Similarly, a 6 liter solution may be prepared by combining 104.0 g of
monobasic sodium phosphate monohydrate, 147.94 g of sodium carbonate
monohydrate, and sufficient water to provide 6 liter of the salt solution. The
basic buffer may be prepared by combining 7.2 g of sodium hydroxide with
180.0 g of water. The basic buffer is typically prepared by adding the base
solution as needed to the salt solution, ultimately to provide a mixture
having
the desired pH, e.g., a pH of 9.65 to 9.75.
In general, the basic species present in the basic buffer should be
sufficiently basic to neutralize the acidity provided by the acidic buffer,
but
should not be so nucleophilic itself that it will react substantially with the
electrophilic groups on the core. For this reason, relatively "soft" bases
such as
carbonate and phosphate are preferred in this embodiment of the invention.
To illustrate the preparation of a three-dimensional matrix of the
present invention, one may combine an admixture of the self-reactive
compound with a first, acidic, buffer (e.g., an acid solution, e.g., a dilute
hydrochloric acid solution) to form a homogeneous solution. This
homogeneous solution is mixed with a second, basic, buffer (e.g., a basic
solution, e.g., an aqueous solution containing phosphate and carbonate salts)
whereupon the reactive groups on the core of the self-reactive compound
substantially immediately inter-react with one another to form a three-
dimensional matrix.
2) Redox Reactive Groups
In one embodiment of the invention, the reactive groups are vinyl
groups such as styrene derivatives, which undergo a radical polymerization
upon initiation with a redox initiator. The term "redox" refers to a reactive
group
that is susceptible to oxidation-reduction activation. The term "vinyl" refers
to a
reactive group that is activated by a redox initiator, and forms a radical
upon
reaction. X, Y and Z can be the same or different vinyl groups, for example,
methacrylic groups.
162
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
For self-reactive compounds containing vinyl reactive groups, the
initial environment typically will be an aqueous environment. The modification
of the initial environment involves the addition of a redox initiator.
3) Oxidative Coupling Reactive Groups
In one embodiment of the invention, the reactive groups undergo
an oxidative coupling reaction. For example, X, Y and Z can be a halo group
such as chloro, with an adjacent electron-withdrawing group on the halogen-
bearing carbon (e.g., on the "L" linking group). Exemplary electron-
withdrawing
groups include nitro, aryl, and so forth.
For such reactive groups, the modification in the initial
environment comprises a change in pH. For example, in the presence of a
base such as KOH, the self-reactive compounds then undergo a de-hydro,
chloro coupling reaction, forming a double bond between the carbon atoms, as
illustrated below:
Cl
C-Ar CI
I 6-Ar
H
Ar-9-R-C-Ar
Cl Ar-C-R-C-Ar
KOH
CI
a C-Ar
e-Ar
Ar-C-R-CH-Ar
Ar-C-R-C-Ar Cl CI
61
For self-reactive compounds containing oxidative coupling
reactive groups, the initial environment typically can be can be dry and
sterile,
or a non-basic medium. The modification of the initial environment will
typically
comprise the addition of a base.
4) Photoinitiated Reactive Groups
In one embodiment of the invention, the reactive groups are
photoinitiated groups. For such reactive groups, the modification in the
initial
environment comprises exposure to ultraviolet radiation.
163
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In one embodiment of the invention, X can be an azide (-N3)
group and Y can be an alkyl group such as -CH(CH3)2 or vice versa. Exposure
to ultraviolet radiation will then form a bond between the groups to provide
for
the following linkage: -NH-C(CH3)2-CH2-. In another embodiment of the
invention, X can be a benzophenone (-(C6H4)-C(0)-(C6H5)) group and Y can be
an alkyl group such as -CH(CH3)2 or vice versa. Exposure to ultraviolet
radiation will then form a bond between the groups to provide for the
following
linkage:
OH
4.--(--)H3C ax CH3(-
/
i
For self-reactive compounds containing photoinitiated reactive
groups, the initial environment typically will be in an ultraviolet radiation-
shielded environment. This can be for example, storage within a container that
is impermeable to ultraviolet radiation.
The modification of the initial environment will typically comprise
exposure to ultraviolet radiation.
5) Temperature-sensitive Reactive Groups
In one embodiment of the invention, the reactive groups are
temperature-sensitive groups, which undergo a thermochemical reaction. For
such reactive groups, the modification in the initial environment thus
comprises
a change in temperature. The term "temperature-sensitive" refers to a reactive
group that is chemically inert at one temperature or temperature range and
reactive at a different temperature or temperature range.
In one embodiment of the invention, X, Y, and Z are the same or
different vinyl groups.
For self-reactive compounds containing reactive groups that are
temperature-sensitive, the initial environment typically will be within the
range of
about 10 to 30 C.
164
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
The modification of the initial environment will typically comprise
changing the temperature to within the range of about 20 to 40 C.
B. Linking Groups
The reactive groups may be directly attached to the core, or they
may be indirectly attached through a linking group, with longer linking groups
also termed "chain extenders." In the formula (I) shown above, the optional
linker groups are represented by L1, L2, and L3, wherein the linking groups
are
present when p, q and r are equal to 1.
Suitable linking groups are well known in the art. See, for
example, WO 97/22371 to Rhee et al. Linking groups are useful to avoid steric
hindrance problems that can sometimes associated with the formation of direct
linkages between molecules. Linking groups may additionally be used to link
several self-reactive compounds together to make larger molecules. In one
embodiment, a linking group can be used to alter the degradative properties of
the compositions after administration and resultant gel formation. For
example,
linking groups can be used to promote hydrolysis, to discourage hydrolysis, or
to provide a site for enzymatic degradation.
Examples of linking groups that provide hydrolyzable sites,
include, inter alia: ester linkages; anhydride linkages, such as those
obtained by
incorporation of glutarate and succinate; ortho ester linkages; ortho
carbonate
linkages such as trimethylene carbonate; amide linkages; phosphoester
linkages; a-hydroxy acid linkages, such as those obtained by incorporation of
lactic acid and glycolic acid; lactone-based linkages, such as those obtained
by
incorporation of caprolactone, valerolactone, y-butyrolactone and p-dioxanone;
and amide linkages such as in a dimeric, oligomeric, or poly(amino acid)
segment. Examples of non-degradable linking groups include succinimide,
propionic acid and carboxyrnethylate linkages. See, for example, WO 99/07417
to Coury et al. Examples of enzymatically degradable linkages include Leu-
165
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Gly-Pro-Ala, which is degraded by collagenase, and Gly-Pro-Lys, which is
degraded by plasmin.
Linking groups can also be included to enhance or suppress the
reactivity of the various reactive groups. For example, electron-withdrawing
groups within one or two carbons of a sulfhydryl group would be expected to
diminish its effectiveness in coupling, due to a lowering of nucleophilicity.
Carbon-carbon double bonds and carbonyl groups will also have such an effect.
Conversely, electron-withdrawing groups adjacent to a carbonyl group (e.g.,
the
reactive carbonyl of glutaryl-N-hydroxysuccinimidyl) would increase the
reactivity of the carbonyl carbon with respect to an incoming nucleophilic
group.
By contrast, sterically bulky groups in the vicinity of a reactive group can
be
used to diminish reactivity and thus reduce the coupling rate as a result of
steric
hindrance.
By way of example, particular linking groups and corresponding
formulas are indicated in the following Table:
Table
Linking group Component structure
-0-(CH2)x- -0-(CH2)x-X
-0-(CH2)x-Y
-0-(CH2)x-Z
-S-(CH2)x- -S-(CH2)x-X
-S-(CH2)x-Y
-S-(CH2)x-Z
-NH-(CF12)x- -NH-(CH2)x-X
-NH-(CF12)x-Y
-NH-(CH2)x-Z
166
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Linking group Component structure
-0-(C0)-NH-(CF12)x- -0-(C0)-NH-(CH2)x-X
-0-(C0)-NH-(CF12)x-Y
-0-(C0)-NH-(CH2)x-Z
-NH-(C0)-0-(CF12)x- -NH-(C0)-0-(CH2)x-X
-NH-(C0)-0-(CF12)x-Y
-NH-(CO)-0-(CH2)x-Z
-0-(CO)CF12)x- -0-(CO)-(CH2)x-X
-0-(C0)-(CF12)x-Y
-0-(C0)-(CH2)x-Z
-(C0)-0-(CF12)x- -(C0)-0-(CH2)n-X
-(C0)-0-(C1-12)n-Y
-(C0)-0-(CH2)n-Z
-0-(C0)-0-(CH2)x-X
-0-(CO)-0-(CH2)x-Y
-0-(CO)-0-(CH2)x-Z
-0-(C0)-CHR2- -0-(C0)-CHR2-X
-0-(C0)-CHR2-Y
-0-(CO)-CHR2-Z
-0-R3-(CO)-NH- -O.-R3-(CO)-NH-.X
- 0-R3-(C0)-NH-Y
- 0-R3-(C0)-NH-Z
In the above Table, x is generally in the range of 1 to about 10; R2
is generally hydrocarbyl, typically alkyl or aryl, preferably alkyl, and most
preferably lower alkyl; and R3 is hydrocarbylene, heteroatom-containing
hydrocarbylene, substituted hydrocarbylene, or substituted heteroatom-
containing hydrocarbylene) typically alkylene or arylene (again, optionally
substituted and/or containing a heteroatom), preferably lower alkylene (e.g.,
167
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
methylene, ethylene, n-propylene, n-butylene, etc.), phenylene, or
amidoalkylene (e.g., -(C0)-NH-CH2).
Other general principles that should be considered with respect to
linking groups are as follows. If a higher molecular weight self-reactive
compound is to be used, it will preferably have biodegradable linkages as
described above, so that fragments larger than 20,000 mol. wt. are not
generated during resorption in the body. In addition, to promote water
miscibility and/or solubility, it may be desired to add sufficient electric
charge or
hydrophilicity. Hydrophilic groups can be easily introduced using known
chemical synthesis, so long as they do not give rise to unwanted swelling or
an
undesirable decrease in compressive strength. In particular, polyalkoxy
segments may weaken gel strength.
C. The Core
The "core" of each self-reactive compound is comprised of the
molecular structure to which the reactive groups are bound. The molecular
core can a polymer, which includes synthetic polymers and naturally occurring
polymers. In one embodiment, the core is a polymer containing repeating
monomer units. The polymers can be hydrophilic, hydrophobic, or amphiphilic.
The molecular core can also be a low molecular weight components such as a
C2-14 hydrocarbyl or a heteroatom-containing C2-14 hydrocarbyl. The
heteroatom-containing C2_14 hydrocarbyl can have 1 or 2 heteroatoms selected
from N, 0 and S. In a preferred embodiment, the self-reactive compound
comprises a molecular core of a synthetic hydrophilic polymer.
1) Hydrophilic Polymers
As mentioned above, the term "hydrophilic polymer" as used
herein refers to a polymer having an average molecular weight and composition
that naturally renders, or is selected to render the polymer as a whole
"hydrophilic." Preferred polymers are highly pure or are purified to a highly
pure
168
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
state such that the polymer is or is treated to become pharmaceutically pure.
Most hydrophilic polymers can be rendered water soluble by incorporating a
sufficient number of oxygen (or less frequently nitrogen) atoms available for
forming hydrogen bonds in aqueous solutions.
Synthetic hydrophilic polymers may be homopolymers, block
copolymers including di-block and tri-block copolymers, random copolymers, or
graft copolymers. In addition, the polymer may be linear or branched, and if
branched, may be minimally to highly branched, dendrimeric, hyperbranched,
or a star polymer. The polymer may include biodegradable segments and
blocks, either distributed throughout the polymer's molecular Structure or
present as a single block, as in a block copolymer. Biodegradable segments
preferably degrade so as to break covalent bonds. Typically, biodegradable
segments are segments that are hydrolyzed in the presence of water and/or
enzymatically cleaved in situ. Biodegradable segments may be composed of
small molecular segments such as ester linkages, anhydride linkages, ortho
ester linkages, ortho carbonate linkages, amide linkages, phosphonate
linkages, etc. Larger biodegradable "blocks" will generally be composed of
oligomeric or polymeric segments incorporated within the hydrophilic polymer.
Illustrative oligomeric and polymeric segments that are biodegradable include,
by way of example, poly(amino acid) segments, poly(orthoester) segments,
poly(orthocarbonate) segments, and the like. Other biodegradable segments
that may form part of the hydrophilic polymer core include polyesters such as
polylactide, polyethers such as polyalkylene oxide, polyamides such as a
protein, and polyurethanes. For example, the core of the self-reactive
compound can be a diblock copolymer of tetrafunctionally activated
polyethylene glycol and polylactide.
Synthetic hydrophilic polymers that are useful herein include, but
are not limited to: polyalkylene oxides, particularly polyethylene glycol
(PEG)
and poly(ethylene oxide)-poly(propylene oxide) copolymers, including block and
random copolymers; polyols such as glycerol, polyglycerol (PG) and
particularly
169
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
highly branched polyglycerol, propylene glycol; poly(oxyalkylene)-substituted
diols, and poly(oxyalkylene)-substituted polyols such as mono-, di- and tri-
polyoxyethylated glycerol, mono- and di-polyoxyethylated propylene glycol, and
mono- and di-polyoxyethylated trimethylene glycol; polyoxyethylated sorbitol,
polyoxyethylated glucose; poly(acrylic acids) and analogs and copolymers
thereof, such as polyacrylic acid per se, polymethacrylic acid,
poly(hydroxyethylmethacrylate), poly(hydroxyethylacrylate),
poly(methylalkylsulfoxide methacrylates), poly(methylalkylsulfoxide acrylates)
and copolymers of any of the foregoing, and/or with additional acrylate
species
such as aminoethyl acrylate and mono-2-(acryloxy)-ethyl succinate; polymaleic
acid; poly(acrylamides) such as polyacrylamide per se, poly(methacrylamide),
poly(dimethylacrylamide), poly(N-isopropyl-acrylamide), and copolymers
thereof; poly(olefinic alcohols) such as poly(vinyl alcohols) and copolymers
thereof; poly(N-vinyl lactams) such as poly(vinyl pyrrolidones), poly(N-vinyl
caprolactams), and copolymers thereof; polyoxazolines, including
poly(methyloxazoline) and poly(ethyloxazoline); and polyvinylamines; as well
as
copolymers of any of the foregoing. It must be emphasized that the
aforementioned list of polymers is not exhaustive, and a variety of other
synthetic hydrophilic polymers may be used, as will be appreciated by those
skilled in the art.
Those of ordinary skill in the art will appreciate that synthetic
polymers such as polyethylene glycol cannot be prepared practically to have
exact molecular weights, and that the term "molecular weight" as used herein
refers to the weight average molecular weight of a number of molecules in any
given sample, as commonly used in the art. Thus, a sample of PEG 2,000
might contain a statistical mixture of polymer molecules ranging in weight
from,
for example, 1,500 to 2,500 daltons with one molecule differing slightly from
the
next over a range. Specification of a range of molecular weights indicates
that
the average molecular weight may be any value between the limits specified,
and may include molecules outside those limits. Thus, a molecular weight
170
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
range of about 800 to about 20,000 indicates an average molecular weight of at
least about 800, ranging up to about 20 kDa.
Other suitable synthetic hydrophilic polymers include chemically
synthesized polypeptides, particularly polynucleophilic polypeptides that have
been synthesized to incorporate amino acids containing primary amino groups
(such as lysine) and/or amino acids containing thiol groups (such as
cysteine).
Poly(lysine), a synthetically produced polymer of the amino acid lysine (145
MW), is particularly preferred. Poly(lysine)s have been prepared having
anywhere from 6 to about 4,000 primary amino groups, corresponding to
molecular weights of about 870 to about 580,000. Poly(lysine)s for use in the
present invention preferably have a molecular weight within the range of about
1,000 to about 300,000, more preferably within the range of about 5,000 to
about 100,000, and most preferably, within the range of about 8,000 to about
15,000. Poly(lysine)s of varying molecular weights are commercially available
from Peninsula Laboratories, Inc. (Belmont, Calif.).
Although a variety of different synthetic hydrophilic polymers can
be used in the present compounds, preferred synthetic hydrophilic polymers are
PEG and PG, particularly highly branched PG. Various forms of PEG are
extensively used in the modification of biologically active molecules because
PEG lacks toxicity, antigenicity, and immunogenicity (La, is biocompatible),
can
be formulated so as to have a wide range of solubilities, and does not
typically
interfere with the enzymatic activities and/or conformations of peptides. A
particularly preferred synthetic hydrophilic polymer for certain applications
is a
PEG having a molecular weight within the range of about 100 to about 100,000,
although for highly branched PEG, far higher molecular weight polymers can be
employed, up to 1,000,000 or more, providing that biodegradable sites are
incorporated ensuring that all degradation products will have a molecular
weight
of less than about 30,000. For most PEGs, however, the preferred molecular
weight is about 1,000 to about 20,000, more preferably within the range of
171
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
about 7,500 to about 20,000. Most preferably, the polyethylene glycol has a
molecular weight of approximately 10,000.
Naturally occurring hydrophilic polymers include, but are not
limited to: proteins such as collagen, fibronectin, albumins, globulins,
fibrinogen,
fibrin and thrombin, with collagen particularly preferred; carboxylated
polysaccharides such as polymannuronic acid and polygalacturonic acid;
aminated polysaccharides, particularly the glycosaminoglycans, e.g.,
hyaluronic
acid, chitin, chondroitin sulfate A, B, or C, keratin sulfate, keratosulfate
and
heparin; and activated polysaccharides such as dextran and starch derivatives.
Collagen and glycosaminoglycans are preferred naturally occurring hydrophilic
polymers for use herein.
Unless otherwise specified, the term "collagen" as used herein
refers to all forms of collagen, including those, which have been processed or
otherwise modified. Thus, collagen from any source may be used in the
compounds of the invention; for example, collagen may be extracted and
purified from human or other mammalian source, such as bovine or porcine
corium and human placenta, or may be recombinantly or otherwise produced.
The preparation of purified, substantially non-antigenic collagen in solution
from
bovine skin is well known in the art. For example, U.S. Patent No. 5,428,022
to
Palefsky et al. discloses methods of extracting and purifying collagen from
the
human placenta, and U.S. Patent No. 5,667,839 to Berg discloses methods of
producing recombinant human collagen in the milk of transgenic animals,
including transgenic cows. Non-transgenic, recombinant collagen expression in
yeast and other cell lines) is described in U.S. Patent No. 6,413,742 to Olsen
et
al., 6,428,978 to Olsen et al., and 6,653,450 to Berg et al.
Collagen of any type, including, but not limited to, types I, II, III, IV,
or any combination thereof, may be used in the compounds of the invention,
although type I is generally preferred. Either atelopeptide or telopeptide-
containing collagen may be used; however, when collagen from a natural
source, such as bovine collagen, is used, atelopeptide collagen is generally
172
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
preferred, because of its reduced imrnunogenicity compared to telopeptide-
containing collagen.
Collagen that has not been previously crosslinked by methods
such as heat, irradiation, or chemical crosslinking agents is preferred for
use in
the invention, although previously crosslinked collagen may be used.
Collagens for use in the present invention are generally, although
not necessarily, in aqueous suspension at a concentration between about 20
mg/ml to about 120 mg/ml, preferably between about 30 mg/ml to about 90
mg/ml. Although intact collagen is preferred, denatured collagen, commonly
known as gelatin, can also be used. Gelatin may have the added benefit of
being degradable faster than collagen.
Nonfibrillar collagen is generally preferred for use in compounds
of the invention, although fibrillar collagens may also be used. The term
"nonfibrillar collagen" refers to any modified or unmodified collagen material
that
is in substantially nonfibrillar form, i.e., molecular collagen that is not
tightly
associated with other collagen molecules so as to form fibers. Typically, a
solution of nonfibrillar collagen is more transparent than is a solution of
fibrillar
collagen. Collagen types that are nonfibrillar (or microfibrillar) in native
form
include types IV, VI, and VII.
Chemically modified collagens that are in nonfibrillar form at
neutral pH include succinylated collagen and methylated collagen, both of
which can be prepared according to the methods described in U.S. Patent No.
4,164,559 to Miyata et al. Methylated collagen, which contains reactive amine
groups, is a preferred nucleophile-containing component in the compositions of
the present invention. In another aspect, methylated collagen is a component
that is present in addition to first and second components in the matrix-
forming
reaction of the present invention. Methylated collagen is described in, for
example, in U.S. Patent No. 5,614,587 to Rhee et al.
Collagens for use in the compositions of the present invention
may start out in fibrillar form, then can be rendered nonfibrillar by the
addition of
173
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
one or more fiber disassembly agent. The fiber disassembly agent must be
present in an amount sufficient to render the collagen substantially
nonfibrillar
at pH 7, as described above. Fiber disassembly agents for use in the present
invention include, without limitation, various biocompatible alcohols, amino
acids, inorganic salts, and carbohydrates, with biocompatible alcohols being
particularly preferred. Preferred biocompatible alcohols include glycerol and
propylene glycol. Non-biocompatible alcohols, such as ethanol, methanol, and
isopropanol, are not preferred for use in the present invention, due to their
potentially deleterious effects on the body of the patient receiving them.
Preferred amino acids include arginine. Preferred inorganic salts include
sodium chloride and potassium chloride. Although carbohydrates, such as
various sugars including sucrose, may be used in the practice of the present
invention, they are not as preferred as other types of fiber disassembly
agents
because they can have cytotoxic effects in vivo.
Fibrillar collagen is less preferred for use in the compounds of the
invention. However, as disclosed in U.S. Patent No. 5,614,587 to Rhee et al.,
fibrillar collagen, or mixtures of nonfibrillar and fibrillar collagen, may be
preferred for use in compounds intended for long-term persistence in vivo.
2) Hydrophobic Polymers
The core of the self-reactive compound may also comprise a
hydrophobic polymer, including low molecular weight polyfunctional species,
although for most uses hydrophilic polymers are preferred. Generally,
"hydrophobic polymers" herein contain a relatively small proportion of oxygen
and/or nitrogen atoms. Preferred hydrophobic polymers for use in the invention
generally have a carbon chain that is no longer than about 14 carbons.
Polymers having carbon chains substantially longer than 14 carbons generally
have very poor solubility in aqueous solutions and, as such, have very long
reaction times when mixed with aqueous solutions of synthetic polymers
containing, for example, multiple nucleophilic groups. Thus, use of short-
chain
174
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
oligomers can avoid solubility-related problems during reaction. Polylactic
acid
and polyglycolic acid are examples of two particularly suitable hydrophobic
polymers.
3) Amphiphilic Polymers
Generally, amphiphilic polymers have a hydrophilic portion and a
hydrophobic (or lipophilic) portion. The hydrophilic portion can be at one end
of
the core and the hydrophobic portion at the opposite end, or the hydrophilic
and
hydrophobic portions may be distributed randomly (random copolymer) or in the
form of sequences or grafts (block copolymer) to form the amphiphilic polymer
core of the self-reactive compound. The hydrophilic and hydrophobic portions
may include any of the aforementioned hydrophilic and hydrophobic polymers.
Alternately, the amphiphilic polymer core can be a hydrophilic
polymer that has been modified with hydrophobic moieties (e.g., alkylated PEG
or a hydrophilic polymer modified with one or more fatty chains ), or a
hydrophobic polymer that has been modified with hydrophilic moieties (e.g.,
"PEGylated" phospholipids such as polyethylene glycolated phospholipids).
4) Low Molecular Weight Components
As indicated above, the molecular core of the self-reactive
compound can also be a low molecular weight compound, defined herein as
being a C2_14 hydrocarbyl or a heteroatom-containing C2..14 hydrocarbyl, which
contains 1 to 2 heteroatoms selected from N, 0, S and combinations thereof.
Such a molecular core can be substituted with any of the reactive groups
described herein.
Alkanes are suitable C2-14 hydrocarbyl molecular cores.
Exemplary alkanes, for substituted with a nucleophilic primary amino group and
a Y electrophilic group, include, ethyleneamine (H2N-C1-12CF12-Y),
tetramethyleneamine (H2N-(CH4)-Y), pentamethyleneamine (H2N-(CH5)-Y), and
hexamethyleneamine (H2N-(CF16)-Y).
175
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Low Molecular weight diols and polyols are also suitable C2-14
hydrocarbyls and include trimethylolpropane, di(trimethylol propane),
pentaerythritol, and diglycerol. Polyacids are also suitable C2-14
hydrocarbyls,
and include trimethylolpropane-based tricarboxylic acid, di(trimethylol
propane)-
based tetracarboxylic acid, heptanedioic acid, octanedioic acid (suberic
acid),
and hexadecanedioic acid (thapsic acid).
Low molecular weight di- and poly-electrophiles are suitable
heteroatom-containing C2-14 hydrocarbyl molecular cores. These include, for
example, disuccinimidyl suberate (DSS), bis(sulfosuccinimidyl) suberate (BS3),
dithiobis(succinimidylpropionate) (DSP), bis(2-succinimidooxycarbonyloxy)
ethyl sulfone (BSOCOES), and 3,3'-dithiobis(sulfosuccinimidylpropionate
(DTSPP), and their analogs and derivatives.
In one embodiment of the invention, the self-reactive compound of
the invention comprises a low-molecular weight material core, with a plurality
of
acrylate moieties and a plurality of thiol groups.
D. Preparation
The self-reactive compounds are readily synthesized to contain a
hydrophilic, hydrophobic or amphiphilic polymer core or a low molecular weight
core, functionalized with the desired functional groups, i.e., nucleophilic
and
electrophilic groups, which enable crosslinking. For example, preparation of a
self-reactive compound having a polyethylene glycol (PEG) core is discussed
below. However, it is to be understood that the following discussion is for
purposes of illustration and analogous techniques may be employed with other
polymers.
With respect to PEG, first of all, various functionalized PEGs have
been used effectively in fields such as protein modification (see Abuchowski
et
al., Enzymes as Drugs, John Wiley & Sons: New York, N.Y. (1981) pp. 367-
383; and Dreborg et al. (1990) Crit. Rev. Therap. Drug Carrier Syst. 6:315),
peptide chemistry (see Mutter et al., The Peptides, Academic: New York, N.Y.
176
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
2:285-332; and Zalipsky et al. (1987) Int J. Peptide Protein Res. 30:740), and
the synthesis of polymeric drugs (see Zalipsky et al. (1983) Eur. Polym. J.
19:1177; and Ouchi et al. (1987) J. MacromoL ScL Chem. A24:1011).
Functionalized forms of PEG, including multi-functionalized PEG,
are commercially available, and are also easily prepared using known methods.
For example, see Chapter 22 of Poly(ethylene Glycol) Chemistry: Biotechnical
and Biomedical Applications, J. Milton Harris, ed., Plenum Press, NY (1992).
Multi-functionalized forms of PEG are of particular interest and
include, PEG succinimidyl glutarate, PEG succinimidyl propionate, succinimidyl
butylate, PEG succinimidyl acetate, PEG succinimidyl succinamide, PEG
succinimidyl carbonate, PEG propionaldehyde, PEG glycidyl ether, PEG-
isocyanate, and PEG-vinylsulfone. Many such forms of PEG are described in
U.S. Patent No. 5,328,955 and 6,534,591, both to Rhee et al. Similarly,
various
forms of multi-amino PEG are commercially available from sources such as
PEG Shop, a division of SunBio of South Korea (www.sunbio.com), Nippon Oil
and Fats (Yebisu Garden Place Tower, 20-3 Ebisu 4-chome, Shibuya-ku,
Tokyo), Nektar Therapeutics (San Carlos, California, formerly Shearwater
Polymers, Huntsville, Alabama) and from Huntsman's Performance Chemicals
Group (Houston, Texas) under the name Jeffamine polyoxyalkyleneamines.
Multi-amino PEGs useful in the present invention include the Jeffamine
diamines ("D" series) and triamines ("T" series), which contain two and three
primary amino groups per molecule. Analogous poly(sulfhydryl) PEGs are also
available from Nektar Therapeutics, e.g., in the form of pentaerythritol
poly(ethylene glycol) ether tetra-sulfhydryl (molecular weight 10,000). These
multi-functionalized forms of PEG can then be modified to include the other
desired reactive groups.
Reaction with succinimidyl groups to convert terminal hydroxyl
groups to reactive esters is one technique for preparing a core with
electrophilic
groups. This core can then be modified include nucleophilic groups such as
primary amines, thiols, and hydroxyl groups. Other agents to convert hydroxyl
177
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
groups include carbonyldiimidazole and sulfonyl chloride. However, as
discussed herein, a wide variety of electrophilic groups may be advantageously
employed for reaction with corresponding nucleophilic groups. Examples of
such electrophilic groups include acid chloride groups; anhydrides, ketones,
aldehydes, isocyanate, isothiocyanate, epoxides, and olefins, including
conjugated olefins such as ethenesulfonyl (-S02CH=CH2) and analogous
functional groups.
Other in situ Crosslinking Materials
Numerous other types of in situ forming materials may be used in
combination with an anti-scarring agent in accordance with the invention. The
in situ forming material may be a biocompatible crosslinked polymer that is
formed from water soluble precursors having electrophilic and nucleophilic
groups capable of reacting and crosslinking in situ (see, e.g., U.S. Patent
No.
6,566,406). The in situ forming material may be hydrogel that may be formed
through a combination of physical and chemical crosslinking processes, where
physical crosslinking is mediated by one or more natural or synthetic
components that stabilize the hydrogel-forming precursor solution at a
deposition site for a period of time sufficient for more resilient chemical
crosslinks to form (see, e.g., U.S. Patent No. 6,818,018). The in situ forming
material may be formed upon exposure to an aqueous fluid from a physiological
environment from dry hydrogel precursors (see, e.g., U.S. Patent No.
6,703,047). The in situ forming material may be a hydrogel matrix that
provides
controlled release of relatively low molecular weight therapeutic species by
first '
dispersing or dissolving the therapeutic species within relatively hydrophobic
rate modifying agents to form a mixture; the mixture is formed into
rnicroparticles that are dispersed within bioabsorbable hydrogels, so as to
release the water soluble therapeutic agents in a controlled fashion (see,
e.g.,
6,632,457). The in situ forming material may be a multi-component hydrogel
system (see, e.g., U.S. Patent No. 6,379, 373). The in situ forming material
178
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
may be a multi-arm block copolymer that includes a central core molecule, such
as a residue of a polyol, and at least three copolymer arms covalently
attached
to the central core molecule, each copolymer arm comprising an inner
hydrophobic polymer segment covalently attached to the central core molecule
and an outer hydrophilic polymer segment covalently attached to the
hydrophobic polymer segment, wherein the central core molecule and the
hydrophobic polymer segment define a hydrophobic core region (see, e.g.,
6,730,334). The in situ forming material may include a gel-forming macromer
that includes at least four polymeric blocks, at least two of which are
hydrophobic and at least one of which is hydrophilic, and including a
crosslinkable group (see, e.g., 6,639,014). The in situ forming material may
be
a water-soluble macromer that includes at least one hydrolysable linkage
formed from carbonate or dioxanone groups, at least one water-soluble
polymeric block, and at least one polymerizable group (see, e.g., U.S. Patent
No. 6,177,095). The in situ forming material may comprise polyoxyalkylene
block copolymers that form weak physical crosslinks to provide gels having a
paste-like consistency at physiological temperatures. (See, e.g., U.S. Patent
No. 4,911,926). The in situ forming material may be a thermo-irreversible gel
made from polyoxyalkylene polymers and ionic polysaccharides (see, e.g., U.S.
Patent No. 5,126,141). The in situ forming material may be a gel forming
composition that includes chitin derivatives (see, e.g., U.S. Patent No.
5,093,319), chitosan-coagulum (see, e.g., U.S. Patent No. 4,532,134), or
hyaluronic acid (see, e.g., U.S. Patent No. 4,141,973). The in situ forming
material may be an in situ modification of alginate (see, e.g., U.S. Patent
No.
5,266,326). The in situ forming material may be formed from ethylenically
unsaturated water soluble macromers that can be crosslinked in contact with
tissues, cells, and bioactive molecules to form gels (see, e.g., U.S. Patent
No.
5,573,934). The in situ forming material may include urethane prepolymers
used in combination with an unsaturated cyano compound containing a cyano
group attached to a carbon atom, such as cyano(meth)acrylic acids and esters
179
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
thereof (see, e.g., U.S. Patent No. 4,740,534). The in situ forming material
may
be a biodegradable hydrogel that polymerizes by a photoinitiated free radical
polymerization from water soluble macromers (see, e.g., U.S. Patent No.
5,410,016). The in situ forming material may be formed from a two component
mixture including a first part comprising a serum albumin protein in an
aqueous
buffer having a pH in a range of about 8.0-11.0, and a second part comprising
a
water-compatible or water-soluble bifunctional crosslinking agent. (see, e.g.,
U.S. Patent No. 5,583,114).
In another aspect, in situ forming materials that can be used
include those based on the crosslinking of proteins. For example, the in situ
forming material may be a biodegradable hydrogel composed of a recombinant
or natural human serum albumin and poly(ethylene) glycol polymer solution
whereby upon mixing the solution cross-links to form a mechanical non-liquid
covering structure which acts as a sealant. See e.g., U.S. Patent No.
6,458,147
and 6,371,975. The in situ forming material may be composed of two separate
mixtures based on fibrinogen and thrombin that are dispensed together to form
a biological adhesive when intermixed either prior to or on the application
site to
form a fibrin sealant. See e.g., U.S. Patent No. 6,764,467. The in situ
forming
material may be composed of ultrasonically treated collagen and albumin which
form a viscous material that develops adhesive properties when crosslinked
chemically with glutaraldehyde and amino acids or peptides. See e.g., U.S.
Patent No. 6,310,036. The in situ forming material may be a hydrated adhesive
gel composed of an aqueous solution consisting essentially of a protein having
amino groups at the side chains (e.g., gelatin, albumin) which is crosslinked
with an N-hydroxyimidoester compound. See e.g., U.S. Patent No. 4,839,345.
The in situ forming material may be a hydrogel prepared from a protein or
polysaccharide backbone (e.g., albumin or polymannuronic acid) bonded to a
cross-linking agent (e.g., polyvalent derivatives of polyethylene or
polyalkylene
glycol). See e.g., U.S. Patent No. 5,514,379. The in situ forming material may
be composed of a polymerizable collagen composition that is applied to the
180
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
tissue and then exposed to an initiator to polymerize the collagen to form a
seal
over a wound opening in the tissue. See e.g., U.S. Patent No. 5,874,537. The
in situ forming material may be a two component mixture composed of a protein
(e.g., serum albumin) in an aqueous buffer having a pH in the range of about
8.0-11.0 and a water-soluble bifunctional polyethylene oxide type crosslinking
agent, which transforms from a liquid to a strong, flexible bonding
composition
to seal tissue in situ. See e.g., U.S. Patents 5,583,114 and RE38158 and PCT
Publication No. WO 96/03159. The in situ forming material may be composed
of a protein, a surfactant, and a lipid in a liquid carrier, which is
crosslinked by
adding a crosslinker and used as a sealant or bonding agent in situ. See e.g.,
U.S. Patent Application No. 2004/0063613A1 and PCT Publication Nos. WO
01/45761 and WO 03/090683. The in situ forming material may be composed
of two enzyme-free liquid components that are mixed by dispensing the
components into a catheter tube deployed at the vascular puncture site,
wherein, upon mixing, the two liquid components chemically cross-link to form
a
mechanical non-liquid matrix that seals a vascular puncture site. See e.g.,
U.S.
Patent Application Nos. 2002/0161399A1 and 2001/0018598A1. The in situ
forming material may be a cross-linked albumin composition composed of an
albumin preparation and a carbodiimide preparation which are mixed under
conditions that permit crosslinking of the albumin for use as a bioadhesive or
sealant. See e.g., PCT Publication No. WO 99/66964. The in situ forming
material may be composed of collagen and a peroxidase and hydrogen
peroxide, such that the collagen is crosslinked to from a semi-solid gel that
seals a wound. See e.g., PCT Publication No. WO 01/35882.
In another aspect, in situ forming materials that can be used
include those based on isocyanate or isothiocyanate capped polymers. For
example, the in situ forming material may be composed of isocyanate-capped
polymers that are liquid compositions which form into a solid adhesive coating
by in situ polymerization and crosslinking upon contact with body fluid or
tissue.
See e.g., PCT Publication No. WO 04/021983. The in situ forming material
181
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
may be a moisture-curing sealant composition composed of an active
isocyanato-terminated isocyanate prepolymer containing a polyol component
with a molecular weight of 2,000 to 20,000 and an isocyanurating catalyst
agent. See e.g., U.S. Patent No. 5,206,331.
In another embodiment, the reagents can undergo an
electrophilic-nucleophilic reaction to produce a crosslinked matrix. Polymers
containing and/or terminated with nucleophilic groups such as amine,
sulfhydryl, hydroxyl, -PH2 or CO-NH-NH2 can be used as the nucleophilic
reagents and polymers containing and/or terminated with electrophilic groups
such as succinimidyl, carboxylic acid, aldehyde, epoxide, isocyanate, vinyl,
vinyl sulfone, maleimide, -S-S-(C5H4N) or activated esters, such as are used
in
peptide synthesis can be used as the electrophilic reagents. For example, a 4-
armed thiol derivatized poly(ethylene glycol) (e.g., pentaerythritol
poly(ethylene
glycol)ether tetra-sulfhydryl) can be reacted with a 4 armed NHS-derivatized
polyethylene glycol (e.g., pentaerythritol poly(ethylene glycol)ether tetra-
succinimidyl glutarate) under basic conditions (pH > about 8). Representative
examples of compositions that undergo such electrophilic-nucleophilic
crosslinking reactions are described, for example, in U.S. Patent. Nos.
5,752,974; 5,807,581; 5,874,500; 5,936,035; 6,051,648; 6,165,489; 6,312,725;
6,458,889; 6,495,127; 6,534,591; 6,624,245; 6,566,406; 6,610,033; 6,632,457;
and PCT Application Publication Nos. WO 04/060405 and WO 04/060346.
In another embodiment, the electrophilic- or nucleophilic-
terminated polymers can further comprise a polymer that can enhance the
mechanical and/or adhesive properties of the in situ forming compositions.
This
polymer can be a degradable or non-degradable polymer. For example, the
polymer may be collagen or a collagen derivative, for example methylated
collagen. An example of an in situ forming composition uses pentaerythritol
poly(ethylene glycol)ether tetra-sulfhydryl) (4-armed thiol PEG),
pentaerythritol
poly(ethylene glycol)ether tetra-succinimidyl glutarate) (4-armed NHS PEG) and
methylated collagen as the reactive reagents. This composition, when mixed
182
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
with the appropriate buffers can produce a crosslinked hydrogel. (See, e.g.,
U.S. Patent Nos. 5,874,500; 6,051,648; 6,166,130; 5,565,519 and 6,312,725).
In another embodiment, the reagents that can form a covalent
bond with the tissue to which it is applied may be used. Polymers containing
and/or terminated with electrophilic groups such as succinimidyl, aldehyde,
epoxide, isocyanate, vinyl, vinyl sulfone, maleimide, -S-S-(C5H4N) or
activated
esters, such as are used in peptide synthesis may be used as the reagents.
For example, a 4 armed NHS-derivatized polyethylene glycol (e.g.,
pentaerythritol poly(ethylene glycol)ether tetra-succinimidyl glutarate) may
be
applied to the tissue in the solid form or in a solution form. In the
preferred
embodiment, the 4 armed NHS-derivatized polyethylene glycol is applied to the
tissue under basic conditions (pH > about 8). Other representative examples of
compositions of this nature that may be used are disclosed in PCT Application
Publication No. WO 04/060405 and WO 04/060346, and U.S. Patent
Application No. 10/749,123.
In another embodiment, the in situ forming material polymer can
be a polyester. Polyesters that can be used in in situ forming compositions
include poly(hydroxyesters). In another embodiment, the polyester can
comprise the residues of one or more of the monomers selected from lactide,
lactic acid, glycolide, glycolic acid, e-caprolactone, gamma-caprolactone,
hydroxyvaleric acid, hydroxybutyric acid, beta-butyrolactone, gamma-
butyrolactone, gamma-valerolactone, y-decanolactone, 6-decanolactone,
trimethylene carbonate, 1,4-dioxane-2-one or 1,5-dioxepan-2one.
Representative examples of these types of compositions are described in U.S.
Patent. Nos. 5,874,500; 5,936,035; 6,312,725; 6,495,127 and PCT Publication
Nos. WO 2004/028547.
In another embodiment, the electrophilic-terminated polymer can
be partially or completely replaced by a small molecule or oligomer that
comprises an electrophilic group (e.g., disuccininnidyl glutarate).
183
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In another embodiment, the nucleophilic¨terminated polymer can
be partially or completely replaced by a small molecule or oligomer that
comprises a nucleophilic group (e.g., dicysteine, dilysine, trilysine, etc.).
Other examples of in situ forming materials that can be used
include those based on the crosslinking of proteins (described in, for
example,
U.S. Patent Nos. RE38158; 4,839,345; 5,514,379, 5,583,114; 6,310,036;
6,458,147; 6,371,975; US Patent Application Publication Nos.
2004/0063613A1, 2002/0161399A1, and 2001/0018598A1, and PCT
Publication Nos. WO 03/090683, WO 01/45761, WO 99/66964, and WO
96/03159) and those based on isocyanate or isothiocyanate capped polymers
(see, e.g., PCT Publication No. WO 04/021983).
Other examples of in situ forming materials that are of particular
interest in the treatment of diverticula, both alone and in combination with
the
therapeutic agents described previously, can include reagents that comprise
one or more cyanoacrylate groups. These reagents can be used to prepare a
poly(a I kylcyanoacrylate) or poly(carboxyalkylcyanoacrylate)
(e.g.,
poly(methylcyanoacrylate) poly(ethylcyanoacrylate), poly(butylcyanoacrylate),
poly(isobutylcyanoacrylate),
poly(hexylcyanoacrylate),
poly(methoxypropylcyanoacrylate), and poly(octylcyanoacrylate)) as well as
copolymers and mixtures of these. Specific examples of blends include blends
of ethyl cyanoacrylate and methoxypropyl acrylate, methoxyproypl
cyanoacrylate and octyl cyanoacrylate and methoxybutyl cyanoacrylate and
butyl cyanoacrylate. Examples of commercially available cyanoacrylates that
can be used include DERMABOND, INDERMIL, GLUSTITCH, VETBOND,
HISTOACRYL, TISSUEMEND, TISSUMEND II, HISTOACRYL BLUE and
ORABASE SOOTHE-N-SEAL LIQUID PROTECTANT.
In another embodiment, the cyanoacrylate compositions can
further comprise additives to stabilize the reagents or alter the rate of
reaction
of the cyanoacrylate, alter the flexibility of the finally cured polymer, or
alter the
viscosity of the product. For example, a trimethylene carbonate based polymer
184
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
or an oxalate polymer of poly(ethylene glycol) or a c-caprolactone based
copolymer (linear, branched, triaxial, terta-axial) can be mixed with a 2-
alkoxyalkylcyanoacrylate (e.g., 2-methoxypropylcyanoacrylate). Examples of
stabilizers include sulfur dioxide (SO2) or polyphosphoric acid.
Representative
examples of these compositions are described in U.S. Patent Nos. 5,350,798
and 6,299,631.
In another embodiment, the cyanoacrylate composition can be
prepared by capping heterochain polymers with a cyanoacrylate group. The
cyanoacrylate-capped heterochain polymer preferably has at least two
cyanoacrylate ester groups per chain. The heterochain polymer can comprise
an absorbable poly(ester), poly(ester-carbonate), poly(ether-carbonate) and
poly(ether-ester). The poly(ether-ester)s described in U.S. Patent Nos.
5,653,992 and 5,714,159 can also be used as the heterochain polymers. A
triaxial poly(8-caprolactone-co-trimethylene carbonate) is an example of a
poly(ester-carbonate) that can be used. The heterochain polymer may be a
polyether. Examples of polyethers that can be used include poly(ethylene
glycol), poly(propylene glycol) and block copolymers of poly(ethylene glycol)
and poly(propylene glycol) (e.g., PLURONICS group of polymers including but
not limited to PLURONIC F127 or F68). Representative examples of these
compositions are described in U.S. Patent No. 6,699,940.
Within another aspect of the invention, the biologically active
fibrosis-inducing agent, anti-infective, and/or hemostatic agent can be
delivered
with a non-polymeric compound (e.g., a carrier). These non-polymeric carriers
can include sucrose derivatives (e.g., sucrose acetate isobutyrate, sucrose
oleate), sterols such as cholesterol, stigmasterol, 13-sitosterol, and
estradiol;
cholesteryl esters such as cholesteryl stearate; C12 -C24 fatty acids such as
lauric acid, myristic acid, palmitic acid, stearic acid, arachidic acid,
behenic
acid, and lignoceric acid; C18 -C38 mono-, di- and triacylglycerides such as
glyceryl monooleate, glyceryl monolinoleate, glyceryl monolaurate, glyceryl
mdnodocosanoate, glyceryl monomyristate, glyceryl monodicenoate, glyceryl
185
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
dipalmitate, glyceryl didocosanoate, glyceryl dimyristate, glyceryl
didecenoate,
glyceryl tridocosanoate, glyceryl trimyristate, glyceryl tridecenoate,
glycerol
tristearate and mixtures thereof; sucrose fatty acid esters such as sucrose
distearate and sucrose palmitate; sorbitan fatty acid esters such as sorbitan
monostearate, sorbitan monopalmitate and sorbitan tristearate; C16 -C18 fatty
alcohols such as cetyl alcohol, myristyl alcohol, stearyl alcohol, and
cetostearyl
alcohol; esters of fatty alcohols and fatty acids such as cetyl palmitate and
cetearyl palmitate; anhydrides of fatty acids such as stearic anhydride;
phospholipids including phosphatidylcholine (lecithin), phosphatidylserine,
phosphatidylethanolamine, phosphatidylinositol, and lysoderivatives thereof;
sphingosine and derivatives thereof; spingomyelins such as stearyl, palmitoyl,
and tricosanyl spingomyelins; ceramides such as stearyl and palmitoyl
ceramides; glycosphingolipids; lanolin and lanolin alcohols, calcium
phosphate,
sintered and unscintered hydoxyapatite, zeolites; and combinations and
mixtures thereof.
Representative examples of patents relating to non-polymeric
delivery systems and the preparation include U.S. Patent Nos. 5,736,152;
5,888,533; 6,120,789; 5,968,542; and 5,747,058.
Within certain embodiments of the invention, the therapeutic
compositions are provided that include (i) a fibrosis-inducing agent and/or
(ii) an
anti-infective agent. The therapeutic compositions may include one or more
additional therapeutic agents (such as described above), for example, a
hemostatic agent. Other agents that may be combined with the therapeutic
compositions include, e.g., anti-inflammation agents, matrix metalloproteinase
inhibitors, cytokine inhibitors, IMPDH inhibitors, immunomodulatory agents,
tyrosine inhibitors, p38 MAP kinase inhibitors, NFKO inhibitors, HMGCoA
reductase inhibitors, apoptosis antagonist, caspase inhibitors, and JNK
inhibitors.
In one aspect, the present invention provides compositions
comprising i) a fibrosing agent and ii) a polymer or a compound that forms a
186
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
crosslinked polymer in situ. The following are some, but by no means all, of
the
preferred fibrosis-inducing agents and classes of fibrosis-inducing agents
that
may be included in the inventive compositions:
la. A fibrosing agent that promotes cell regeneration.
2a. A fibrosing agent that promotes angiogenesis.
3a. A fibrosing agent that promotes fibroblast migration.
4a. A fibrosing agent that promotes fibroblast
proliferation.
5a. A fibrosing agent that promotes deposition of
extracellular
matrix.
6a. A fibrosing agent that promotes tissue remodeling.
7a. A fibrosing agent that is a diverticular wall irritant.
8a. A fibrosing agent that is silk (such as silkworm silk,
spider
silk, recombinant silk, raw silk, hydrolyzed silk, acid-treated silk,
and acylated silk)
9a. A fibrosing agent that is talc.
10a. A fibrosing agent that is chitosan.
11a. A fibrosing agent that is polylysine.
12a. A fibrosing agent that is fibronectin.
13a. A fibrosing agent that is bleomycin or an analogue or
derivative thereof.
14a. A fibrosing agent that is connective tissue growth factor
(CTGF). .
15a. A fibrosing agent that is metallic beryllium or an oxide
thereof.
16a. A fibrosing agent that is copper.
187
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
17a. A fibrosing agent that is saracin.
18a. A fibrosing agent that is silica.
19a. A fibrosing agent that is crystalline silicates.
20a. A fibrosing agent that is quartz dust.
21a. A fibrosing agent that is talcum powder.
22a. A fibrosing agent that is ethanol.
23a. A fibrosing agent that is a component of extracellular
matrix.
24a. A fibrosing agent that is collagen.
25a. A fibrosing agent that is fibrin.
26a. A fibrosing agent that is fibrinogen.
27a. A fibrosing agent that is poly(ethylene terephthalate).
28a. A fibrosing agent that is poly(ethylene-co-vinylacetate).
29a. A fibrosing agent that is N-carboxybutylchitosan.
30a. A fibrosing agent that is an RGD protein.
31a. A fibrosing agent that is a polymer of vinyl chloride.
32a. A fibrosing agent that is cyanoacrylate.
33a. A fibrosing agent that is crosslinked poly(ethylene glycol)-
methylated collagen.
34a. A fibrosing agent that is an inflammatory cytokine.
35a. A fibrosing agent that is TGF[3.
36a. A fibrosing agent that is PDGF.
37a. A fibrosing agent that is VEGF.
38a. A fibrosing agent that is TNFa.
188
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
39a. A fibrosing agent that is NGF.
40a. A fibrosing agent that is GM-CSF.
41a. A fibrosing agent that is IGF-a.
42a. A fibrosing agent that is IL-1
43a. A fibrosing agent is IL-8.
44a. A fibrosing agent is IL-6.
45a. A fibrosing agent that is a growth hormone.
46a. A fibrosing agent that is a bone morphogenic protein.
47a. A fibrosing agent that is a cell proliferative agent.
48a. A fibrosing agent that is dexamethasone.
49a. A fibrosing agent that is isotretinoin.
50a. A fibrosing agent that is 17-p-estradiol.
51a. A fibrosing agent that is estradiol.
52a. A fibrosing agent that is diethylstibesterol.
53a. A fibrosing agent that is cyclosporine A.
54a. A fibrosing agent that is a//-trans retinoic acid or an
analogue or derivative thereof.
55a. A fibrosing agent that is wool (including animal wool, wood
wool, and mineral wool).
56a. A fibrosing agent that is cotton.
57a. A fibrosing agent that is bFGF.
58a. A fibrosing agent that is polyurethane.
59a. A fibrosing agent that is polytetrafluoroethylene.
60a. A fibrosing agent that is poly(alkylcyanoacrylate).
189
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
61a. A fibrosing agent that is activin.
62a. A fibrosing agent that is angiopoietin.
63a. A fibrosing agent that is insulin-like growth factor (IGF).
64a. A fibrosing agent that is hepatocyte growth factor (HG F).
65a. A fibrosing agent that is a colony-stimulating factor (CSF).
66a. A fibrosing agent that is erythropoietin.
67a. A fibrosing agent that is an interferon.
68a. A fibrosing agent that is endothelin-1.
69a. A fibrosing agent that is angiotensin II.
70a. A fibrosing agent that is bromocriptine.
71a. A fibrosing agent that is methylsergide.
72a. A fibrosing agent that is fibrosin.
73a. A fibrosing agent that is fibrin.
74a. A fibrosing agent that is an adhesive glycoprotein.
75a. A fibrosing agent that is a proteoglycan.
76a. A fibrosing agent that is hyaluronan.
77a. A fibrosing agent that is secreted protein acidic and rich in
cysteine (SPARC).
78a. A fibrosing agent that is a thrombospondin.
79a. A fibrosing agent that is tenacin. =
80a. A fibrosing agent that is a cell adhesion molecule.
81a. A fibrosing agent that is an inhibitor of matrix
metalloproteinase.
190
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
82a. A fibrosing agent that is a tissue inhibitor of matrix
metalloproteinase.
83a. A fibrosing agent that is methotrexate.
84a. A fibrosing agent that is carbon tetrachloride.
85a. A fibrosing agent that is thioacetamide.
1. As mentioned above, the present invention provides
compositions comprising each of the foregoing 86 (i.e., 1 a
through 85a) listed fibrosing agents or classes of fibrosing
agents, with each of the following 98 (i.e., lb through 97b)
polymers and compounds:
2. lb. A crosslinked polymer.
3. 2b. A polymer that reacts with mammalian tissue.
3b. A polymer that is a naturally occurring polymer.
4b. A polymer that is a protein.
5b. A polymer that is a carbohydrate.
6b. A polymer that is biodegradable.
7b. A polymer that is crosslinked and biodegradable.
8b. A polymer that nonbiodegradable.
9b. Collagen.
10b. Methylated collagen.
11b. Fibrinogen.
12b. Thrombin.
13b. Albumin.
14b. Plasminogen.
15b. von Willebrands factor.
16b. Factor VIII.
191
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
17b. Hypoallergenic collagen.
18b. Atelopeptidic collagen.
19b. Telopeptide collagen.
20b. Crosslinked collagen.
21b. Aprotinin.
22b. Gelatin.
23b. A protein conjugate.
24b. A gelatin conjugate.
25b. Hyaluronic acid.
26b. A hyaluronic acid derivative.
27b. A synthetic polymer.
28b. A polymer formed from reactants comprising a synthetic
isocyanate-containing compound.
29b. A synthetic isocyanate-containing compound.
30b. A polymer formed from reactants comprising a synthetic
thiol-containing compound.
31b. A synthetic thiol-containing compound.
32b. A polymer formed from reactants comprising a synthetic
compound containing at least two thiol groups.
33b. A synthetic compound containing at least two thiol groups.
34b. A polymer formed from reactants comprising a synthetic
compound containing at least three thiol groups.
35b. A synthetic compound containing at least three thiol
groups.
36b. A polymer formed from reactants comprising a synthetic
compound containing at least four thiol groups.
37b. A synthetic compound containing at least four thiol groups.
192
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
38b. A polymer formed from reactants comprising a synthetic
amino-containing compound.
39b. A synthetic amino-containing compound.
40b. A polymer formed from reactants comprising a synthetic
compound containing at least two amino groups.
41b. A synthetic compound containing at least two amino
groups.
42b. A polymer formed from reactants comprising a synthetic
compound containing at least three amino groups.
43b. A synthetic compound containing at least three amino
groups.
44b. A polymer formed from reactants comprising a synthetic
compound containing at least four amino groups.
45b. A synthetic compound containing at least four amino
groups.
46b. A polymer formed from reactants comprising a synthetic
compound comprising a carbonyl-oxygen-succinimidyl group.
47b. A synthetic compound comprising a carbonyl-oxygen-
succinimidyl group.
48b. A polymer formed from reactants comprising a synthetic
compound comprising at least two carbonyl-oxygen-succinimidyl groups.
49b. A synthetic compound comprising at least two carbonyl-
oxygen-succinimidyl groups.
50b. A polymer formed from reactants comprising a synthetic
compound comprising at least three carbonyl-oxygen-succinimidyl groups.
51b. A synthetic compound comprising at least three carbonyl-
oxygen-succinimidyl groups.
52b. A polymer formed from reactants comprising a synthetic
compound comprising at least four carbonyl-oxygen-succinimidyl groups.
193
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
53b. A synthetic compound comprising at least four carbonyl-
oxygen-succinimidyl groups.
54b. A polymer formed from from reactants comprising a
synthetic polyalkylene oxide-containing compound.
55b. A synthetic polyalkylene oxide-containing compound.
56b. A polymer formed from reactants comprising a synthetic
compound comprising both polyalkylene oxide and biodegradable polyester
blocks.
57b. A synthetic compound comprising both polyalkylene oxide
and biodegradable polyester blocks.
58b. A polymer formed from reactants comprising a synthetic
polyalkylene oxide-containing compound having reactive amino groups.
59b. A synthetic polyalkylene oxide-containing compound
having reactive amino groups.
60b. A polymer formed from reactants comprising a synthetic
polyalkylene oxide-containing compound having reactive thiol groups.
61b. A synthetic polyalkylene oxide-containing compound
having reactive thiol groups.
62b. A polymer formed from reactants comprising a synthetic
polyalkylene oxide-containing compound having reactive carbonyl-oxygen-
succinimidyl groups.
63b. A synthetic polyalkylene oxide-containing compound
having reactive carbonyl-oxygen-succinimidyl groups.
64b. A polymer formed from reactants comprising a synthetic
compound comprising a biodegradable polyester block.
65b. A synthetic compound comprising a biodegradable
polyester block.
66b. A polymer formed from reactants comprising a synthetic
polymer formed in whole or part from lactic acid or lactide.
194
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
67b. A synthetic polymer formed in whole or part from lactic acid
or lactide.
68b. A polymer formed from reactants comprising a synthetic
polymer formed in whole or part from glycolic acid or glycolide.
69b. A synthetic polymer formed in whole or part from glycolic
acid or glycolide.
70b. A polymer formed from reactants comprising polylysine.
71b. Polylysine.
72b. A polymer formed from reactants comprising (a) protein
and (b) a compound comprising a polyalkylene oxide portion.
73b. A polymer formed from reactants comprising (a) protein
and (b) polylysine.
74b. A polymer formed from reactants comprising (a) protein
and (b) a compound having at least four thiol groups.
75b. A polymer formed from reactants comprising (a) protein
and (b) a compound having at least four amino groups.
76b. A polymer formed from reactants comprising (a) protein
and (b) a compound having at least four carbonyl-oxygen-succinimide groups.
77b. A polymer formed from reactants comprising (a) protein
and (b) a compound having a biodegradable region formed from reactants
selected from lactic acid, lactide, glycolic acid, glycolide, and epsilon-
caprolactone.
78b. A polymer formed from reactants comprising (a) collagen
and (b) a compound comprising a polyalkylene oxide portion.
79b. A polymer formed from reactants comprising (a) collagen
and (b) polylysine.
80b. A polymer formed from reactants comprising (a) collagen
and (b) a compound having at least four thiol groups.
195
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
81b. A polymer formed from reactants comprising (a) collagen
and (b) a compound having at least four amino groups.
82b. A polymer formed from reactants comprising (a) collagen
and (b) a compound having at least four carbonyl-oxygen-succinimide groups.
83b. A polymer formed from reactants comprising (a) collagen
and (b) a compound having a biodegradable region formed from reactants
selected from lactic acid, lactide, glycolic acid, glycolide, and epsilon-
caprolactone.
84b. A polymer formed from reactants comprising (a)
methylated collagen and (b) a compound comprising a polyalkylene oxide
portion.
85b. A polymer formed from reactants comprising (a)
methylated collagen and (b) polylysine.
86b. A polymer formed from reactants comprising (a)
methylated collagen and (b) a compound having at least four thiol groups.
87b. A polymer formed from reactants comprising (a)
methylated collagen and (b) a compound having at least four amino groups.
88b. A polymer formed from reactants comprising (a)
methylated collagen and (b) a compound having at least four carbonyl-oxygen-
succinimide groups.
89b. A polymer formed from reactants comprising (a)
methylated collagen and (b) a compound having a biodegradable region formed
from reactants selected from lactic acid, lactide, glycolic acid, glycolide,
and
epsilon-caprolactone.
90b. A polymer formed from reactants comprising hyaluronic
acid.
91b. A polymer formed from reactants comprising a hyaluronic
acid derivative.
196
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
92b. A polymer formed from reactants comprising pentaerythritol
poly(ethylene glycol)ether tetra-sulfhydryl of number average molecular weight
between 3,000 and 30,000.
93b. Pentaerythritol poly(ethylene glycol)ether tetra-sulfhydryl of
number average molecular weight between 3,000 and 30,000.
94b. A polymer formed from reactants comprising pentaerythritol
poly(ethylene glycol)ether tetra-amino of number average molecular weight
between 3,000 and 30,000.
95b. Pentaerythritol poly(ethylene glycol)ether tetra-amino of
number average molecular weight between 3,000 and 30,000.
96b. A polymer formed from reactants comprising (a) a synthetic
compound having a number average molecular weight between 3,000 and
30,000 and comprising a polyalkylene oxide region and multiple nucleophilic
groups, and (b) a synthetic compound having a number average molecular
weight between 3,000 and 30,000 and comprising a polyalkylene oxide region
and multiple electrophilic groups.
97b. A mixture of (a) a synthetic compound having a number
average molecular weight between 3,000 and 30,000 and comprising a
polyalkylene oxide region and multiple nucleophilic groups, and (b) a
synthetic
compound having a number average molecular weight between 3,000 and
30,000 and comprising a polyalkylene oxide region and multiple electrophilic
groups.
As mentioned above, the present invention provides compositions
comprising each of the foregoing 86 (1a through 85a) listed fibrosing agents
or
classes of fibrosing agents, with each of the foregoing 98 (lb through 97b)
polymers and compounds: Thus, in separate aspects, the invention provides
86 times 98 = 8,428 described compositions. In other words, each of the
following is a distinct aspect of the present invention:
197
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
1 a+1 b; 1 a + 213; 1 a + 3b; 1 a+4b, la+5b; 1 a+6b; 1 a+7b; 1 a+8b; 1 a+9b;
la+10b;
la+11b; 1 a+12b; 1 a+13b, 1 a+14b, 1 a+15b, la+16b; la+17b; la+18b; la+19b;
1 a+20b; la+21b; 1 a+22b, la+23b; 1 a+24b; 1 a+25b; la+26b; 1 a+27b; 1 a+28b;
la+29b; 1 a+30b; la+31b, la+32b; 1 a+33b; 1 a+34b, la+35b; 1 a+36b; 1 a+37b,
la+38b; 1 a+39b; 1 a+40b; 1 a+41b; 1 a+42b; 1 a+43b, 1 a+44b; la+45b; 1 a+46b;
1 a+47b; 1 a+48b; 1 a+49b, la+50b; 1 a+51b; 1 a+52b; la+53b; 1 a+54b; 1 a+55b;
la+55b; 1 a+57b; 1 a+58b; 1 a+59b; 1 a+60b; la+61b; 1 a+62b; la-'-63b; 1
a+64b;
1 a+65b; 1 a+66b; 1 a+67b; 1 a+68b; 1 a+69b; 1 a+70b; 1 a+71b, la+72b; 1
a+73b,
1 a+74b; 1 a+75b; 1 a+76b; la+77b; 1 a+78b; 1 a+79b, la+80b; la+81b; 1 a+82b;
1 a+83b; la+84b; 1 a+85b; 1 a+86b; 1 a+87b; 1 a+88b; 1 a+89b, 1 a+90b; 1
a+91b,
1 a+92b; 1 a+93b; la+94b; la+95b; 1 a+96b; la+97b; 2a+lb; 2a + 213; 2a + 3b;
2a+4b; 2a+5b; 2a+6b; 2a+7b, 2a+8b; 2a+9b; 2a+10b; 2a+11 b; 2a+12b;
2a+13b; 2a+14b; 2a+15b; 2a+16b; 2a+17b, 2a+18b; 2a+19b, 2a+20b, 2a+21b;
2a+22b; 2a+23b, 2a+24b; 2a+25b, 2a+26b; 2a+27b; 2a+28b; 2a+29b; 2a+30b;
2a+31b; 2a+32b; 2a+33b; 2a+34b; 2a+35b; 2a+36b; 2a+37b, 2a+38b; 2a+39b,
2a+40b; 2a+41b; 2a+42b; 2a+43b; 2a+44b; 2a+45b; 2a+46b; 2a+47b; 2a+48b;
2a+49b; 2a+50b; 2a+51b; 2a+52b; 2a+53b; 2a+54b; 2a+55b; 2a+55b; 2a+57b;
2a+58b; 2a+59b, 2a+60b; 2a+61b; 2a+62b; 2a+63b; 2a+64b; 2a+65b, 2a+66b;
2a+67b; 2a+68b, 2a+69b; 2a+70b; 2a+71b; 2a+72b; 2a+73b, 2a+74b; 2a+75b;
2a+76b; 2a+77b; 2a+78b, 2a+79b, 2a+80b, 2a+81b; 2a+82b; 2a+83b; 2a+84b;
2a+85b; 2a+86b; 2a+87b; 2a+88b; 2a+89b, 2a+90b; 2a+91b; 2a+92b; 2a+93b;
2a+94b; 2a+95b; 2a+96b; 2a+97b; 3a+22b; 3a+23b, 3a+24b; 3a+25b, 3a+26b;
3a+27b; 3a+28b; 3a+29b; 3a+30b, 3a+31b; 3a+32b; 3a.+33b; 3a+34b; 3a+35b,
3a+36b; 3a+37b; 3a+38b; 3a+39b; 3a+40b; 3a+41b, 3a+42b; 3a+43b, 3a+44b;
3a+45b; 3a+46b; 3a+47b; 3a+48b; 3a+49b; 3a+50b; 3a+51b; 3a+52b; 3a+53b,
3a+54b, 3a+55b; 3a+55b; 3a+57b; 3a+58b; 3a+59b; 3a+60b; 3a+61b; 3a+62b;
3a+63b; 3a+64b; 3a+65b; 3a+66b; 3a+67b; 3a+68b; 3a+69b; 3a+70b; 3a+71b;
3a+72b, 3a+73b; 3a+74b; 3a+75b; 3a+76b; 3a+77b; 3a+78b; 3a+79b, 3a+80b;
3a+81b; 3a+82b; 3a+83b; 3a+84b; 3a+85b; 3a+86b; 3a+87b; 3a+88b; 3a+89b,
3a+90b; 3a+91b; 3a+92b; 3a+93b, 3a+94b; 3a+95b; 3a+96b; 3a+97b; 4a+12b;
198
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
4a+13b, 4a+14b; 4a+15b, 4a+16b; 4a+17b; 4a+18b; 4a+19b; 4a+20b; 4a+21b;
4a+22b, 4a+23b; 4a+24b; 4a+25b; 4a+26b, 4a+27b; 4a+28b; 4a+29,b; 4a+30b;
4a+31b; 4a+32b; 4a+33b; 4a+34b; 4a+35b; 4a+36b; 4a+37b; 4a+38b; 4a+39b;
4a+40b; 4a+41b; 4a+42b; 4a+43b; 4a+44b; 4a+45b; 4a+46b; 4a+47b; 4a+48b;
4a+49b; 4a+50b; 4a+51b; 4a+52b; 4a+53b; 4a+54b; 4a+55b; 4a+55b; 4a+57b,
4a+58b; 4a+59b; 4a+60b; 4a+6113; 4a+62b; 4a+63b; 4a+64b; 4a+65b; 4a+66b;
4a+67b; 4a+68b; 4a+69b; 4a+70b; 4a+71b, 4a+72b; 4a+73b; 4a+74b, 4a+75b;
4a+76b, 4a+77b; 4a+78b; 4a+79b; 4a+80b; 4a+81b; 4a+82b; 4a+83b; 4a+84b;
4a+85b; 4a+86b; 4a+87b; 4a+88b; 4a+89b; 4a+90b; 4a+91 b; 4a+92b; 4a+93b;
4a+94b; 4a+95b; 4a+96b; 4a+97b; 5a+12b; 5a+13b; 5a+14b; 5a+15b, 5a+16b;
5a+17b; 5a+18b; 5a+19b; 5a+20b; 5a+21b; 5a+22b, 5a+23b; 5a+24b; 5a+25b;
5a+26b; 5a+27b; 5a+28b; 5a+29b; 5a+30b; 5a+31b; 5a+32b; 5a+33b; 5a+34b;
5a+35b; 5a+36b; 5a+37b; 5a+38b; 5a+39b; 5a+40b; 5a+41b; 5a+42b; 5a+43b;
5a+44b; 5a+45b, 5a+46b; 5a+47b; 5a+48b; 5a+49b; 5a+50b; 5a+51b, 5a+52b;
5a+53b; 5a+54b; 5a+55b, 5a+55b; 5a+57b, 5a+58b, 5a+59b; 5a+60b; 5a+61b;
5a+62b; 5a+63b; 5a+64b; 5a+65b; 5a+66b, 5a+67b; 5a+68b; 5a+69b; 5a+70b;
5a+71b, 5a+72b; 5a+73b; 5a+74b; 5a+75b; 5a+76b; 5a+77b, 5a+78b, 5a+79b;
5a+80b; 5a+81b; 5a+82b; 5a+83b; 5a+84b; 5a+85b; 5a+86b; 5a+87b, 5a+88b;
5a+89b; 5a+90b; 5a+91b; 5a+92b; 5a+93b; 5a+94b; 5a+95b; 5a+96b; 5a+97b;
6a+lb; 6a + 213; 6a + 3b; 6a+4b; 6a+5b; 6a+6b; 6a+7b; 6a+8b; 6a+9b, 6a+10b;
6a+11 b; 6a+12b; 6a+13b; 6a+14b; 6a+15b, 6a+16b; 6a+17b, 6a+18b; 6a+19b;
6a+20b; 6a+21b, 6a+22b; 6a+23b; 6a+24b; 6a+25b; 6a+26b, 6a+27b; 6a+28b,
6a+29b; 6a+30b; 6a+31b, 6a+32b; 6a+33b; 6a+34b; 6a+35b; 6a+36b; 6a+37b;
6a+38b; 6a+39b; 6a+40b, 6a+41b; 6a+42b, 6a+43b, 6a+44b; 6a+45b; 6a+46b;
6a+47b; 6a+48b; 6a+49b; 6a+50b, 6a+51b; 6a+52b; 6a+53b; 6a+54b; 6a+55b,
6a+55b; 6a+57b; 6a+58b; 6a+59b; 6a+60b; 6a+61b; 6a+62b; 6a+63b; 6a+64b;
6a+65b; 6a+66b; 6a+67b, 6a+68b; 6a+69b; 6a+70b; 6a+71b; 6a+72b; 6a+73b;
6a+74b; 6a+75b; 6a+76b; 6a+77b; 6a+78b; 6a+79b; 6a+80b; 6a+81b; 6a+82b,
6a+83b; 6a+84b; 6a+85b; 6a+86b; 6a+87b; 6a+88b; 6a+89b; 6a+90b; 6a+91b;
6a+92b; 6a+93b; 6a+94b; 6a+95b; 6a+96b; 6a+97b, 7a+lb; 7a + 2b; 7a + 3b;
199
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
7a+4b; 7a+5b; 7a+6b; 7a+7b, 7a+8b; 7a+9b, 7a+10b, 7a+11b; 7a+12b,
7a+13b, 7a+14b; 7a+15b, 7a+16b; 7a+17b; 7a+18b, 7a+19b; 7a+20b; 7a+21b;
7a+22b; 7a+23b; 7a+24b; 7a+25b; 7a+26b; 7a+27b; 7a+28b; 7a+29b; 7a+30b,
7a+31b; 7a+32b; 7a+33b; 7a+34b; 7a+35b; 7a+36b; 7a+37b, 7a+38b; 7a+39b;
7a+40b; 7a+41b; 7a+42b; 7a+43b; 7a+44b; 7a+45b; 7a+46b; 7a+47b; 7a+48b;
7a+49b; 7a+50b; 7a+51b, 7a+52b; 7a+53b; 7a+54b; 7a+55b; 7a+55b; 7a+57b,
7a+58b; 7a+59b; 7a+60b; 7a+61b; 7a+62b; 7a+63b; 7a+64b; 7a+65b; 7a+66b;
7a+67b; 7a+68b, 7a+69b; 7a+70b; 7a+71b; 7a+72b; 7a+73b, 7a+74b; 7a+75b,
7a+76b, 7a+77b; 7a+78b; 7a+79b, 7a+80b; 7a+81b; 7a+82b; 7a+83b, 7a+84b;
7a+85b; 7a+86b; 7a+87b; 7a+88b; 7a+89b; 7a+90b, 7a+91b; 7a+92b; 7a+93b;
7a+94b; 7a+95b; 7a+96b, 7a+97b; 8a+12b; 8a+13b; 8a+14b; 8a+15b; 8a+16b,
8a+17b, 8a+18b; 8a+19b; 8a+20b; 8a+21b; 8a+22b, 8a+23b; 8a+24b; 8a+25b,
8a1-26b; 8a+27b; 8a+28b; 8a+29b; 8a+30b; 8a+31b; 8a+32b; 8a+33b; 8a+34b;
8a+35b; 8a+36b; 8a+37b; 8a+38b; 8a+39b; 8a+40b; 8a+41b; 8a+42b; 8a+43b;
8a+44b; 8a+45b; 8a+46b; 8a+47b, 8a+48b; 8a+49b; 8a+50b; 8a+51b, 8a+52b;
8a+53b; 8a+54b; 8a+55b, 8a+55b; 8a+57b; 8a+58b, 8a+59b; 8a+60b; 8a+61b;
8a+62b; 8a+63b; 8a+64b; 8a+65b; 8a+66b; 8a+67b; 8a+68b; 8a+69b; 8a+70b;
8a+71b; 8a+72b, 8a+73b, 8a+74b; 8a+75b; 8a+76b; 8a+77b; 8a+78b; 8a+79b;
8a+80b; 8a+81b; 8a+82b; 8a+83b; 8a+84b; 8a+85b; 8a+86b; 8a+87b; 8a+88b;
8a+89b; 8a+90b; 8a+91b, 8a+92b; 8a+93b; 8a+94b; 8a+95b; 8a+96b; 8a+97b;
9a+1b; 9a + 2b; 9a + 3b; 9a+4b; 9a+5b; 9a+6b; 9a+7b; 9a-'-8b; 9a+9b; 9a+1 0b;
9a+11 b; 9a+12b, 9a+13b; 9a+14b; 9a+15b; 9a+16b; 9a+17b; 9a+18b; 9a+19b;
9a+20b; 9a+21b; 9a+22b; 9a+23b; 9a+24b; 9a+25b; 9a+26b; 9a+27b; 9a+28b;
9a+29b, 9a+30b, 9a+31b, 9a+32b; 9a+33b; 9a+34b; 9a+35b; 9a+36b; 9a+37b;
9a+38b; 9a+39b; 9a+40b; 9a+41b; 9a+42b; 9a+43b; 9a+44b; 9a+45b; 9a+46b;
9a+47b; 9a+48b; 9a+49b, 9a+50b, 9a+51b, 9a+52b, 9a+53b, 9a+54b; 9a+55b;
9a+55b; 9a+57b; 9a+58b, 9a+59b; 9a+60b; 9a+61b; 9a+62b; 9a+63b; 9a+64b;
9a+65b; 9a+66b; 9a+67b; 9a+68b; 9a+69b; 9a+70b; 9a+71b; 9a+72b, 9a+73b;
9a+74b, 9a+75b, 9a+76b, 9a+77b; 9a+78b; 9a+79b; 9a+80b; 9a+81b; 9a+82b;
9a+83b; 9a+84b; 9a+85b; 9a+86b; 9a+87b; 9a+88b; 9a+89b; 9a+90b; 9a+91b;
200
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
9a+92b; 9a+93b; 9a+94b; 9a+95b; 9a+96b, 9a+97b, 10a+lb; 10a + 2b, 10a +
3b; 10a+4b; 10a+5b, 10a+6b; 10a+7b; 10a+8b; 10a+9b, 10a+10b; 10a+11 b;
10a+12b; 10a+13b, 10a+14b, 10a+15b; 10a+16b; 10a-'-17b; 10a+18b;
10a+19b; 10a+20b; 10a+21b, 10a-'-22b, 10a+23b; 10a+24b; 10a+25b;
10a+26b, 10a+27b; 10a+28b; 10a+29b; 10a+30b; 10a+31b; 10a+32b;
10a+33b; 10a+34b; 10a+35b; 10a+36b; 10a+37b; 10a+38b; 10a+39b;
10a+40b; 10a+41b, 10a+42b, 10a+43b; 10a+44b; 10a+45b; 10a+46b,
10a+47b; 10a+48b; 10a+49b; 10a-'-50b, 10a+51b; 10a+52b; 10a+53b,
10a+54b; 10a+55b; 10a+55b; 10a+57b, 10a+58b; 10a+59b; 10a+60b,
10a+61b; 10a+62b, 10a+63b; 10a-'-64b; 10a+65b; 10a+66b; 10a+67b,
10a+68b; 10a+69b; 10a+70b, 10a+71b, 10a+72b; 10a+73b; 10a+74b,
10a+75b; 10a+76b; 10a+77b; 10a+78b; 10a+79b; 10a+80b; 10a+81b;
10a+82b; 10a+83b; 10a+84b; 10a+85b; 10a+86b; 10a+87b; 10a+88b;
10a+89b, 10a+90b; 10a+91b; 10a+92b; 10a+93b; 10a+94b; 10a+95b;
10a+96b; 10a+97b; 11 a+1 b; 11 a + 2b; 11 a + 3b; lla+4b; 11 a+5b; 1 1 a+6b;
lla+7b; 1 la+8b; lla+9b; lla+10b, lla+11b; lla+12b; 1 la+13b; 1 la+14b;
1 1 a+15b; 11 a+16b; 1 la+17b; 11 a+18b; 11 a+19b, 11 a+20b, lla+21b;
lla+22b, 11 a+23b; 11 a+4b; lla+25b; lla+26b, lla+27b; Ila+28b;
1 1 a+29b; 1 1 a+30b; lla+31b; 1 1 a+32b; 1 1 a+33b; 1 1 a+34b; 1 1 a+35b;
lla+36b; 11 a+37b, 11 a+38b; lla+39b; 11 a+40b; 11 a+41b; lla+42b;
11 a+43b, lla+44b; 11 a+45b; lla+46b; 11 a+47b; 1 1 a+48b; 1 1 a+49b;
11 a+50b; lla+51b; 11 a+52b; lla+53b; 11 a+54b; lla+55b; lla+55b;
lla+57b; Ila+58b; lla+59b; I la+60b; lla+61b; lla+62b; lla+63b;
lla+64b; lla+65b; 1 1 a+66b; lla+67b; 11 a+68b; 1 la+69b; 1 1 a+70b;
lla+71b; 11 a+72b; 1 1 a+73b; lla+74b; 1 la+75b; 1 la+76b; 1 1 a+77b;
1 1 a+78b; 1 1 a+79b; 1 1 a+80b; 1 la+81b; I 1 a+82b, 11 a+83b, lla+84b,
11 a+85b, lla+86b; 1 1 a+87b; 11 a+88b, 11 a+89b; I 1 a+90b, I la+91b;
lla+92b; lla+93b; lla+94b; lla+95b; lla+96b; lla+97b; 12a+1 b; 12a +2b;
12a + 3b; 12a+4b; 12a+5b; 12a+6b; 12a+7b; 12a+8b; 12a+9b; 12a+10b;
12a+11b; 12a+12b; 12a+13b; 12a+14b; 12a+15b; 12a+16b, 12a+17b,
201
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
12a+18b; 12a+19b; 12a+20b; 12a+21b, 12a+22b, 12a+23b; 12a+24b,
12a+25b, 12a+26b; 12a+27b, 12a+28b; 12a+29b; 12a+30b; 12a+31b,
12a+32b; 12a+33b; 12a+34b, 12a+35b, 12a+36b; 12a+37b; 12a+38b;
12a+39b; 12a+40b; 12a+41b; 12a+42b; 12a+43b; 12a+44b; 12a+45b;
12a+46b, 12a+47b; 12a+48b, 12a+49b; 12a+50b; 12a+51b; 12a+52b;
12a+53b; 12a+54b; 12a+55b; 12a+55b; 12a+57b; 12a+58b; 12a+59b;
12a+60b; 12a+61b; 12a+62b, 12a+63b; 12a+64b; 12a+65b, 12a+66b;
12a+67b; 12a+68b; 12a+69b; 12a+70b, 12a+71b, 12a+72b; 12a+73b;
12a+74b, 12a+75b; 12a+76b, 12a+77b; 12a+78b; 12a+79b; 12a+80b;
12a+81b; 12a+82b; 12a+83b, 12a+84b; 12a+85b; 12a+86b; 12a+87b,
12a+88b; 12a+89b; 12a+90b, 12a+91b; 12a+92b, 12a+93b; 12a+94b,
12a+95b; 12a+96b; 12a+97b; 13a+lb; 13a + 2b; 13a + 3h; 13a+4b, 13a+5b,
13a+6b; 13a+7b; 13a+8b, 13a+9b; 13a+10b, 13a+11b; 13a+12b; 13a+13b;
13a+14b; 13a+15b, 13a+16b; 13a+17b; 13a+18b; 13a+19b; 13a+20b;
13a+21b, 13a+22b; 13a+23b; 13a+24b; 13a+25b; 13a+26b; 13a+27b;
13a+28b, 13a+29b; 13a+30b; 13a+31b; 13a+32b; 13a+33b; 13a+34b;
13a+35b; 13a+36b; 13a+37b; 13a+38b; 13a+39b, 13a+40b, 13a+41b,
13a+42b, 13a+43b; 13a+44b; 13a+45b; 13a+46b; 13a+47b, 13a+48b;
13a+49b; 13a+50b; 13a+51b; 13a+52b; 13a+53b; 13a+54b; 13a+55b;
13a+55b; 13a+57b; 13a+58b; 13a+59b; 13a+60b; 13a+61b; 13a+62b;
13a+63b; 13a+64b; 13a+65b; 13a+66b; 13a+67b; 13a+68b; 13a+69b;
13a+70b; 13a+71b; 13a+72b; 13a+73b; 13a+74b; 13a+75b; 13a+76b;
13a+77b; 13a+78b; 13a+79b; 13a+80b; 13a+81b; 13a+82b; 13a+83b;
13a+84b; 13a+85b; 13a+86b; 13a+87b; 13a+88b; 13a+89b; 13a+90b,
13a+91b; 13a+92b; 13a+93b; 13a+94b; 13a+95b; 13a+96b; 13a+97b; 14a+lb;
14a +2b; 14a + 3h; 14a+4b; 14a+5b, 14a+6b; 14a+7b, 14a+8b, 14a+9b,
14a+10b; 14a+11b; 14a+12b; 14a+13b, 14a+14b; 14a+15b; 14a+16b,
14a+17b; 14a+18b; 14a+19b; 14a+20b; 14a+21b; 14a+22b, 14a+23b;
14a+24b; 14a+25b; 14a+26b; 14a+27b, 14a+28b; 14a+29b; 14a+30b,
14a+31b; 14a+32b; 14a+33b; 14a+34b, 14a+35b; 14a+36b; 14a+37b;
202
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
14a+38b; 14a+39b; 14a+40b; 14a+41b; 14a+42b; 14a+43b; 14a+44b;
14a+45b; 14a+46b; 14a+47b; 14a+48b; 14a+49b; 14a+50b; 14a+51b;
14a+52b; 14a+53b; 14a+54b; 14a+55b; 14a+55b, 14a+57b; 14a+58b;
14a+59b; 14a+60b, 14a+61b; 14a+62b; 14a+63b; 14a+64b; 14a+65b;
14a+66b; 14a+67b; 14a+68b; 14a+69b; 14a+70b; 14a+71b; 14a+72b;
14a+73b; 14a+74b; 14a+75b; 14a+76b; 14a+77b; 14a+78b; 14a+79b;
14a+80b; 14a-F81b; 14a+82b, 14a+83b; 14a+84b, 14a+85b; 14a+86b;
14a+87b; 14a+88b; 14a+89b; 14a+90b, 14a+91b; 14a+92b; 14a+93b;
14a+94b; 14a+95b; 14a+96b, 14a+97b, 15a+1b; 15a 1- 2b; 15a + 3b; 15a+4b;
15a-F5b; 15a+6b; 15a+7b; 15a+8b; 15a+9b; 15a+10b; 15a+11b; 15a+12b;
15a+13b; 15a+14b; 15a+15b; 15a+16b; 15a+17b; 15a+18b; 15a+19b;
15a+20b; 15a+21b; 15a+22b; 15a+23b; 15a+24b; 15a+25b; 15a+26b;
15a+27b; 15a+28b, 15a+29b, 15a+30b, 15a+31b, 15a+32b; 15a+33b;
15a+34b; 15a+35b, 15a+36b; 15a+37b; 15a+38b; 15a+39b; 15a+40b;
15a+41b; 15a+42b; 15a+43b; 15a+44b; 15a+45b; 15a+46b; 15a+47b;
15a+48b, 15a+49b; 15a+50b, 15a+51b; 15a+52b, 15a+53b, 15a+54b,
15a+55b; 15a+55b; 15a+57b; 15a+58b; 15a+59b; 15a+60b, 15a+61b;
15a+62b; 15a+63b; 15a+64b; 15a+65b, 15a+66b; 15a+67b; 15a+68b;
15a+69b; 15a+70b; 15a+71b; 15a+72b; 15a+73b; 15a+74b; 15a+75b;
15a+76b; 15a+77b, 15a+78b; 15a+79b; 15a+80b; 15a+81b; 15a+82b;
15a+83b; 15a+84b; 15a+85b; 15a+86b; 15a+87b; 15a+88b; 15a+89b;
15a+90b; 15a+91b; 15a+92b; 15a+93b, 15a+94b; 15a-F95b; 15a+96b;
15a+97b; 16a+1b; 16a + 2b; 16a + 3b; 16a+4b; 16a+5b; 16a+6b; 16a+7b;
16a+8b; 16a+9b; 16a+10b, 16a+11b; 16a+12b;,16a+13b; 16a+14b; 16a+15b;
16a+16b; 16a+17b; 16a+18b; 16a+19b; 16a+20b; 16a+21b; 16a+22b;
16a+23b; 16a+24b; 16a+25b; 16a+26b; 16a+27b; 16a+28b; 16a+29b;
16a+30b, 16a+31b, 16a+32b; 16a+33b; 16a+34b; 16a+35b; 16a+36b;
16a+37b; 16a+38b; 16a+39b; 16a+40b; 16a+41b; 16a+42b; 16a+43b;
16a+44b; 16a+45b, 16a+46b; 16a+47b; 16a+48b; 16a+49b; 16a+50b;
16a+51b; 16a+52b; 16a+53b; 16a+54b; 16a+55b; 16a+55b; 16a+57b;
203
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
16a+58b; 16a+59b; 16a+60b; 16a+61b, 16a+62b; 16a+63b; 16a+64b;
16a+65b; 16a+66b; 16a+67b; 16a+68b; 16a+69b, 16a+70b; 16a+71b;
16a+72b; 16a+73b; 16a+74b; 16a+75b; 16a+76b; 16a+77b; 16a+78b;
16a+79b; 16a+80b; 16a+81b; 16a+82b; 16a+83b; 16a+84b; 16a+85b;
16a+86b; 16a+87b; 16a+88b; 16a+89b; 16a+90b; 16a+91b; 16a+92b;
16a+93b; 16a+94b; 16a+95b; 16a+96b, 16a+97b; 17a+lb; 17a + 2b, 17a + 3b;
17a+4b; 17a+5b; 17a+6b, 17a+7b; 17a+8b; 17a+9b; 17a+10b; 17a+11 b;
17a+12b; 17a+13b; 17a+14b; 17a+15b, 17a+16b; 17a+17b; 17a+18b;
17a+19b; 17a+20b; 17a+21b; 17a+22b, 17a+23b; 17a+24b; 17a+25b;
17a+26b; 17a+27b; 17a+28b; 17a+29b; 17a+30b; 17a+31b; 17a+32b;
17a+33b; 17a+34b; 17a+35b; 17a+36b; 17a+37b; 17a+38b; 17a+39b;
17a+40b; 17a+41b; 17a+42b; 17a+43b; 17a+44b; 17a+45b; 17a+46b;
17a+47b; 17a+48b; 17a+49b, 17a+50b; 17a+51b; 17a+52b; 17a+53b;
17a+54b; 17a+55b; 17a+55b; 17a+57b; 17a+58b; 17a+59b; 17a+60b;
17a+61b; 17a+62b; 17a+63b; 17a+64b; 17a+65b; 17a+66b; 17a+67b,
17a+68b, 17a+69b; 17a+70b; 17a+71b; 17a+72b; 17a+73b; 17a+74b;
17a+75b, 17a+76b; 17a+77b, 17a+78b, 17a+79b; 17a+80b; 17a+81b;
17a+82b; 17a+83b; 17a+84b; 17a+85b; 17a+86b; 17a+87b, 17a+88b,
17a+89b; 17a+90b; 17a+91b; 17a+92b; 17a+93b, 17a+94b; 17a+95b;
17a+96b; 17a+97b; 18a+lb; 18a + 2b; 18a + 3b; 18a+4b; 18a+5b; 18a+6b;
18a+7b; 18a+8b; 18a+9b; 18a+10b; 18a+11b; 18a+12b; 18a+13b; 18a+14b;
18a+15b; 18a+16b; 18a+17b; 18a+18b; 18a+19b; 18a+20b, 18a+21b;
18a+22b; 18a+23b; 18a+24b; 18a+25b; 18a+26b; 18a+27b; 18a+28b;
18a+29b; 18a+30b; 18a+31b; 18a+32b, 18a+33b; 18a+34b, 18a+35b;
18a+36b; 18a+37b; 18a+38b; 18a+39b; 18a+40b; 18a+41b; 18a+42b;
18a+43b; 18a+44b; 18a+45b; 18a+46b; 18a+47b; 18a+48b; 18a+49b;
18a+50b; 18a+51b; 18a+52b; 18a+53b; 18a+54b; 18a+55b; 18a+55b;
18a+57b; 18a+58b; 18a+59b, 18a+60b; 18a+61b; 18a+62b; 18a+63b;
18a+64b, 18a+65b; 18a+66b; 18a+67b, 18a+68b; 18a+69b; 18a+70b;
18a+71b; 18a+72b; 18a+73b; 18a+74b; 18a+75b; 18a+76b; 18a+77b;
204
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
18a+78b; 18a+79b; 18a+80b; 18a+81b; 18a+82b; 18a+83b; 18a+84b,
18a+85b; 18a+86b; 18a+87b; 18a+88b; 18a+89b; 18a+90b; 18a+91b;
18a+92b; 18a+93b; 18a+94b; 18a+95b; 18a+96b; 18a+97b; 19a+1 b; 19a + 2b;
19a + 3b; 19a+4b, 19a+5b, 19a+6b; 19a+7b; 19a+8b; 19a+9b; 19a+10b;
19a+11b; 19a+12b; 19a+13b; 19a+14b; 19a+15b; 19a+16b; 19a+17b;
19a+18b; 19a+19b, 19a+20b; 19a+21b; 19a+22b; 19a+23b; 19a+24b;
19a+25b; 19a+26b, 19a+27b, 19a+28b; 19a+29b; 19a+30b, 19a+31b;
19a+32b; 19a+33b; 19a+34b; 19a+35b; 19a+36b; 19a+37b; 19a+38b;
19a+39b; 19a+40b; 19a+41b; 19a+42b, 19a+43b, 19a+44b; 19a+45b;
19a+46b; 19a+47b; 19a+48b, 19a+49b; 19a+50b, 19a+51b; 19a+52b;
19a+53b; 19a+54b; 19a+55b; 19a+55b; 19a+57b; 19a+58b; 19a+59b,
19a+60b; 19a+61b; 19a+62b; 19a+63b; 19a+64b; 19a+65b; 19a+66b;
19a+67b, 19a+68b; 19a+69b; 19a+70b; 19a+71b; 19a+72b; 19a+73b,
19a+74b, 19a+75b; 19a+76b; 19a+77b; 19a+78b, 19a+79b; 19a+80b,
19a+81b; 19a+82b; 19a+83b; 19a+84b; 19a+85b; 19a+86b; 19a+87b;
19a+88b; 19a+89b; 19a+90b; 19a+91b; 19a+92b; 19a+93b; 19a+94b;
19a+95b; 19a+96b; 19a+97b; 20a+lb, 20a + 213; 20a + 3b; 20a+4b, 20a+5b;
20a+6b; 20a+7b; 20a+8b; 20a+9b; 20a+10b; 20a+11b; 20a+12b; 20a+13b;
20a+14b; 20a+15b, 20a+16b; 20a+17b; 20a+18b; 20a+19b; 20a+20b,
20a+21b; 20a+22b; 20a+23b; 20a+24b; 20a+25b; 20a+26b; 20a+27b;
20a+28b; 20a+29b; 20a+30b, 20a+31 b; 20a+32b; 20a+33b; 20a+34b;
20a+35b; 20a+36b; 20a+37b; 20a+38b; 20a+39b; 20a+40b; 20a+41b;
20a+42b; 20a+43b; 20a+44b; 20a+45b; 20a+46b; 20a+47b; 20a+48b;
20a+49b; 20a+50b; 20a+51b; 20a+52b; 20a+53b; 20a+54b; 20a+55b,
20a+55b; 20a+57b; 20a+58b, 20a+59b; 20a+60b; 20a+61b; 20a+62b;
20a+63b; 20a+64b; 20a+65b, 20a+66b, 20a+67b; 20a+68b; 20a+69b;
20a+70b; 20a+71b; 20a+72b; 20a+73b; 20a+74b; 20a+75b; 20a+76b;
20a+77b; 20a+78b, 20a+79b; 20a+80b; 20a+81b; 20a+82b; 20a+83b;
20a+84b; 20a+85b; 20a+86b, 20a+87b, 20a+88b; 20a+89b; 20a+90b;
20a+91b; 20a+92b; 20a+93b; 20a+94b; 20a+95b; 20a+96b; 20a+97b; 21a+1b;
205
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
21a + 2b; 21a + 3b; 21a+4b; 21a+5b; 21a+6b; 21a+7b; 21a+8b; 21a+9b;
21a+10b, 21a+11 b; 21a+12b; 21a+13b; 21a+14b; 21a+15b; 21a+16b;
21a+17b; 21a+18b; 21a+19b; 21a+20b; 21a+21b, 21a+22b, 21a+23b;
21a+24b; 21a+25b; 21a+26b, 21a+27b; 21a+28b; 21a+29b; 21a+30b;
21a+31b; 21a+32b; 21a+33b; 21a+34b; 21a+35b; 21a+36b; 21a+37b;
21a+38b; 21a+39b; 21a+40b; 21a+41b; 21a+42b; 21a+43b; 21a+44b;
21a+45b; 21a+46b; 21a+47b; 21a+48b; 21a+49b; 21a+50b; 21a+51b;
21a+52b; 21a+53b; 21a+54b; 21a+55b; 21a+55b, 21a+57b; 21a+58b,
21a+59b; 21a+60b; 21a+61b; 21a+62b, 21a+63b; 21a+64b; 21a+65b,
21a+66b; 21a+67b; 21a+68b; 21a+69b; 21a+70b; 21a+71b; 21a-F72b;
21a+73b; 21a+74b; 21a+75b; 21a+76b; 21a+77b; 21a+78b; 21a+79b;
21a+80b; 21a+81b; 21a+82b; 21a+83b; 21a+84b; 21a+85b, 21a+86b,
21a+87b; 21a+88b; 21a+89b; 21a+90b; 21a+91b; 21a+92b; 21a+93b;
21a+94b; 21a+95b; 21a+96b; 21a+97b; 22a+1 b; 22a + 2b; 22a + 3b; 22a+4b;
22a+5b, 22a+6b, 22a+7b; 22a+8b, 22a+9b; 22a+10b; 22a+11 b; 22a+12b;
22a+13b; 22a+14b; 22a+15b; 22a+16b; 22a+17b; 22a+18b; 22a+19b;
22a+20b; 22a+21b; 22a+22b; 22a+23b; 22a+24b; 22a+25b; 22a+26b;
22a+27b; 22a+28b; 22a+29b; 22a+30b; 22a+31b; 22a+32b; 22a+33b;
22a+34b; 22a+35b; 22a+36b; 22a+37b, 22a+38b; 22a+39b; 22a+40b;
22a+41b; 22a+42b; 22a+43b; 22a+44b; 22a+45b, 22a+46b; 22a+47b;
22a+48b; 22a+49b; 22a+50b; 22a+51b; 22a+52b; 22a+53b; 22a+54b,
22a+55b, 22a+55b; 22a+57b; 22a+58b; 22a+59b; 22a+60b; 22a+61b;
22a+62b; 22a+63b; 22a+64b; 22a+65b, 22a+66b; 22a+67b; 22a+68b,
22a+69b; 22a+70b; 22a+71b, 22a+72b; 22a+73b; 22a+74b, 22a+75b;
22a+76b; 22a+77b; 22a+78b; 22a+79b; 22a+80b; 22a+81b; 22a+82b;
22a+83b; 22a+84b; 22a+85b; 22a+86b; 22a+87b; 22a+88b; 22a+89b,
22a+90b; 22a+91b; 22a+92b; 22a+93b; 22a+94b, 22a+95b; 22a+96b;
22a+97b; 23a+1 b; 23a + 2b; 23a + 3b; 23a-F4b; 23a+5b; 23a+6b; 23a+7b,
23a+8b; 23a+9b; 23a+10b; 23a+1 1 b; 23a+12b; 23a+13b; 23a+14b; 23a+15b;
23a+16b, 23a+17b; 23a+18b; 23a+19b, 23a+20b; 23a+21b; 23a+22b,
206
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
23a+23b; 23a+24b; 23a+25b; 23a+26b, 23a+27b; 23a+28b; 23a+29b;
23a+30b; 23a+31b, 23a+32b; 23a+33b; 23a+34b; 23a+35b; 23a+36b;
23a+37b; 23a+38b; 23a+39b; 23a+40b; 23a+41b; 23a+42b; 23a+43b;
23a+44b, 23a+45b; 23a+46b; 23a+47b; 23a+48b; 23a+49b; 23a+50b;
23a+51b, 23a+52b; 23a+53b, 23a+54b; 23a+55b; 23a+55b; 23a+57b;
23a+58b; 23a+59b; 23a+60b; 23a+61b; 23a+62b; 23a+63b; 23a+64b;
23a+65b, 23a+66b; 23a+67b; 23a+68b; 23a+69b, 23a+70b; 23a+71b;
23a+72b; 23a+73b; 23a+74b; 23a+75b; 23a+76b; 23a+77b; 23a+78b;
23a+79b; 23a+80b; 23a+81b; 23a+82b; 23a+83b; 23a+84b; 23a+85b;
23a+86b; 23a+87b; 23a+88b; 23a+89b; 23a+90b; 23a+91b; 23a+92b;
23a+93b; 23a+94b; 23a+95b, 23a+96b; 23a+97b; 24a+lb; 24a + 2b; 24a + 3b;
24a+4b; 24a+5b; 24a+6b; 24a+7b; 24a+8b; 24a+9b; 24a+10b; 24a+11b,
24a+12b; 24a+13b; 24a+14b, 24a+15b; 24a+16b; 24a+17b; 24a+18b;
24a+19b; 24a+20b; 24a+21b; 24a+22b; 24a+23b; 24a+24b; 24a+25b;
24a+26b, 24a+27b; 24a+28b; 24a+29b; 24a+30b; 24a+31b; 24a+32b;
24a+33b; 24a+34b; 24a+35b; 24a+36b; 24a+37b; 24a+38b; 24a+39b;
24a+40b; 24a+41b; 24a+42b; 24a+43b; 24a+44b; 24a+45b; 24a+46b;
24a+47b; 24a+48b; 24a+49b; 24a+50b; 24a+51b; 24a+52b; 24a+53b,
24a+54b; 24a+55b; 24a+55b; 24a+57b; 24a+58b; 24a+59b, 24a+60b;
24a+61b; 24a+62b; 24a+63b; 24a+64b; 24a+65b; 24a+66b; 24a+67b;
24a+68b; 24a+69b; 24a+70b; 24a+71b; 24a+72b; 24a+73b; 24a+74b;
24a+75b; 24a+76b; 24a+77b; 24a+78b; 24a+79b; 24a+80b; 24a+81b;
24a+82b; 24a+83b; 24a+84b; 24a+85b; 24a+86b; 24a+87b; 24a+88b;
24a+89b; 24a+90b; 24a+91b; 24a+92b; 24a+93b; 24a+94b; 24a+95b;
24a+96b, 24a+97b, 25a+lb; 25a + 2b; 25a + 3b; 25a+4b; 25a+5b; 25a+6b;
25a+7b; 25a+8b; 25a+9b; 25a+10b; 25a+11 b; 25a+12b; 25a+13b; 25a+14b;
25a+15b; 25a+16b; 25a+17b; 25a+18b; 25a+19b, 25a+20b; 25a+21b;
25a+22b; 25a+23b; 25a+24b; 25a+25b; 25a+26b; 25a+27b; 25a+28b;
25a+29b; 25a+30b; 25a+31b; 25a+32b; 25a+33b; 25a+34b; 25a+35b;
25a+36b; 25a+37b; 25a+38b; 25a+39b; 25a+40b; 25a+41b; 25a+42b;
207
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
25a+43b; 25a+44b; 25a+45b; 25a+46b, 25a+47b; 25a+48b; 25a+49b;
25a+50b, 25a+51b; 25a+52b; 25a+53b; 25a+54b; 25a+55b, 25a+55b;
25a+57b, 25a+58b; 25a+59b; 25a+60b; 25a+61b; 25a+62b; 25a+63b;
25a+64b; 25a+65b; 25a+66b; 25a+67b; 25a+68b; 25a+69b; 25a+70b,
25a+71b; 25a+72b; 25a+73b, 25a+74b; 25a+75b; 25a+76b; 25a+77b;
25a+78b; 25a+79b, 25a+80b; 25a+81b; 25a+82b; 25a+83b; 25a+84b;
25a+85b, 25a+86b; 25a+87b; 25a+88b; 25a+89b; 25a+90b; 25a+91b;
25a+92b; 25a+93b, 25a+94b; 25a+95b; 25a+96b; 25a+97b; 26a+1 b; 26a + 2b;
26a + 3b, 26a+4b; 26a+5b, 26a+6b; 26a+7b; 26a+8b; 26a+9b; 26a+10b;
26a+11 b; 26a+12b; 26a+13b; 26a+14b; 26a+15b; 26a+16b; 26a+17b,
26a+18b; 26a+19b, 26a+20b; 26a+21b; 26a+22b; 26a+23b; 26a+24b;
26a+25b; 26a+26b; 26a+27b; 26a+28b; 26a+29b, 26a+30b; 26a+31b;
26a+32b; 26a+33b; 26a+34b; 26a+35b; 26a+36b; 26a+37b; 26a+38b;
26a+39b; 26a+40b; 26a+41b; 26a+42b; 26a+43b; 26a+44b; 26a+45b;
26a+46b, 26a+47b; 26a+48b; 26a+49b; 26a+50b; 26a+51b; 26a+52b;
26a+53b; 26a+54b, 26a+55b; 26a+55b; 26a+57b; 26a+58b; 26a+59b;
26a+60b; 26a+61b; 26a+62b; 26a+63b; 26a+64b, 26a+65b; 26a+66b,
26a+67b; 26a+68b, 26a+69b, 26a+70b; 26a+71b; 26a+72b; 26a+73b;
26a+74b; 26a+75b; 26a+76b; 26a+77b; 26a+78b; 26a+79b; 26a+80b,
26a+81b, 26a+82b; 26a+83b; 26a+84b; 26a+85b; 26a+86b; 26a+87b;
26a+88b; 26a+89b; 26a+90b; 26a+91b; 26a+92b; 26a+93b; 26a+94b;
26a+95b; 26a+96b; 26a+97b; 27a+1 b; 27a + 2b; 27a + 3b; 27a+4b, 27a+5b;
27a+6b; 27a+7b; 27a+8b; 27a+9b; 27a+10b; 27a+11 b; 27a+12b, 27a+13b;
27a+14b; 27a+15b; 27a+16b; 27a+17b; 27a+18b; 27a+19b; 27a+20b;
27a+21b; 27a+22b; 27a+23b; 27a+24b; 27a+25b; 27a+26b; 27a+27b;
27a+28b; 27a+29b; 27a+30b; 27a+31b, 27a+32b; 27a+33b; 27a+34b;
27a+35b; 27a+36b; 27a+37b; 27a+38b; 27a+39b; 27a+40b; 27a+41b;
27a+42b; 27a+43b; 27a+44b; 27a+45b; 27a+46b; 27a+47b; 27a+48b;
27a+49b; 27a+50b; 27a+51b; 27a+52b; 27a+53b; 27a+54b; 27a+55b,
27a+55b, 27a+57b; 27a+58b; 27a+59b, 27a+60b; 27a+61b; 27a+62b;
208
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
27a+63b; 27a+64b; 27a+65b; 27a+66b, 27a+67b; 27a+68b, 27a+69b;
27a+70b; 27a+71b; 27a+72b; 27a+73b; 27a+74b; 27a+75b; 27a+76b;
27a+77b; 27a+78b; 27a+79b; 27a+80b; 27a+81b; 27a+82b; 27a+83b;
27a+84b; 27a+85b; 27a+86b, 27a+87b; 27a+88b; 27a+89b; 27a+90b;
27a+91b; 27a+92b; 27a+93b; 27a+94b; 27a+95b; 27a+96b; 27a+97b; 28a+1 b;
28a + 2b; 28a + 3b; 28a+4b; 28a+5b; 28a+6b; 28a+7b; 28a+8b; 28a+9b;
28a+10b; 28a+11b; 28a+12b; 28a+13b; 28a+14b; 28a+15b; 28a+16b;
28a+17b; 28a+18b; 28a+19b, 28a+20b; 28a+21b; 28a+22b; 28a+23b;
28a+24b; 28a+25b; 28a+26b, 28a+27b; 28a+28b; 28a+29b; 28a+30b;
28a+31b; 28a+32b; 28a+33b; 28a+34b; 28a+35b; 28a+36b, 28a+37b,
28a+38b; 28a+39b; 28a+40b; 28a+41b; 28a+42b; 28a+43b; 28a+44b;
28a+45b; 28a+46b; 28a+47b; 28a+48b; 28a+49b; 28a+50b; 28a+51b;
28a+52b; 28a+53b; 28a+54b; 28a+55b; 28a+55b; 28a+57b; 28a+58b;
28a+59b, 28a+60b; 28a+61b; 28a+62b; 28a+63b; 28a+64b; 28a+65b;
28a+66b, 28a+67b; 28a+68b; 28a+69b; 28a+70b; 28a+71b; 28a+72b;
28a+73b; 28a+74b; 28a+75b; 28a+76b, 28a+77b; 28a+78b; 28a+79b;
28a+80b; 28a+81b; 28a+82b, 28a+83b, 28a+84b; 28a+85b; 28a+86b;
28a+87b; 28a+88b; 28a+89b; 28a+90b; 28a+91b; 28a+92b, 28a+93b;
28a+94b; 28a+95b; 28a+96b; 28a+97b; 29a+1b; 29a + 2b; 29a + 3h; 29a+4b;
29a+5b; 29a+6b; 29a+7b; 29a+8b; 29a+9b; 29a+10b; 29a+11 b; 29a+12b,
29a+13b; 29a+14b; 29a+15b; 29a+16b; 29a+17b; 29a+18b; 29a+19b;
29a+20b; 29a+21b; 29a+22b; 29a+23b; 29a+24b; 29a+25b; 29a+26b;
29a+27b, 29a+28b; 29a+29b; 29a+30b; 29a+31b; 29a+32b; 29a+33b;
29a+34b, 29a+35b; 29a+36b; 29a+37b; 29a+38b; 29a+39b; 29a+40b;
29a+41b; 29a+42b; 29a+43b; 29a+44b; 29a+45b; 29a+46b; 29a+47b;
29a+48b; 29a+49b, 29a+50b; 29a+51b; 29a+52b; 29a+53b; 29a+54b;
29a+55b; 29a+55b; 29a+57b; 29a+58b, 29a+59b; 29a+60b; 29a+61b;
29a+62b; 29a+63b; 29a+64b; 29a+65b; 29a+66b; 29a+67b; 29a+68b;
29a+69b; 29a+70b; 29a+71b; 29a+72b; 29a+73b; 29a+74b; 29a+75b;
29a+76b; 29a+77b; 29a+78b; 29a+79b; 29a+80b; 29a+81b; 29a+82b;
209
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
29a+83b; 29a+84b; 29a+85b; 29a+86b, 29a+87b; 29a+88b; 29a+89b,
29a+90b; 29a+91b; 29a+92b; 29a+93b; 29a+94b; 29a+95b; 29a+96b;
29a+97b; 30a+1 b; 30a + 2b; 30a + 3b; 30a+4b; 30a+5b, 30a+6b, 30a+7b,
30a+8b; 30a+9b; 30a+1 0b, 30a+1 1 b; 30a+12b; 30a+13b; 30a+14b; 30a+1 5b;
30a+16b, 30a+17b; 30a+18b, 30a+19b; 30a+20b, 30a+21b; 30a+22b;
30a+23b; 30a+24b; 30a+25b, 30a+26b; 30a+27b; 30a+28b; 30a+29b;
30a+30b; 30a+31b; 30a+32b; 30a+33b, 30a+34b; 30a+35b; 30a+36b;
30a+37b; 30a+38b; 30a+39b, 30a+40b; 30a+41b; 30a+42b; 30a+43b;
30a+44b; 30a+45b; 30a+46b; 30a+47b, 30a+48b; 30a+49b; 30a+50b;
30a+51b; 30a+52b; 30a+53b; 30a+54b, 30a+55b, 30a+55b; 30a+57b;
30a+58b; 30a+59b; 30a+60b; 30a+61b, 30a+62b; 30a+63b; 30a+64b;
30a+65b; 30a+66b; 30a+67b; 30a+68b, 30a+69b, 30a+70b; 30a+71b,
30a+72b; 30a+73b; 30a+74b, 30a+75b; 30a+76b; 30a+77b; 30a+78b,
30a+79b; 30a+80b; 30a+81b; 30a+82b; 30a+83b; 30a+84b; 30a+85b;
30a+86b; 30a+87b; 30a+88b; 30a+89b; 30a+90b; 30a+91b; 30a+92b;
30a+93b; 30a+94b; 30a+95b; 30a+96b; 30a+97b; 31a+lb; 31a + 2b; 31a + 3b;
31a+4b; 31a+5b; 31a+6b; 31a+7b; 31a+8b; 31a-'-9b; 31a+10b; 31a+1 1 b;
31a+12b; 31a+13b; 31a+14b; 31a+15b; 31a+16b; 31a+17b; 31a+18b;
31a+19b; 31a+20b; 31a+21b; 31a+22b; 31a+23b; 31a+24b, 31a+25b,
31a+26b; 31a+27b; 31a+28b; 31a+29b; 31a+30b, 31a+31b; 31a+32b;
31a+33b; 31a+34b; 31a+35b; 31a+36b, 31a+37b, 31a+38b, 31a+39b,
31a+40b; 31a+41b; 31a+42b; 31a+43b; 31a+44b; 31a+45b; 31a+46b;
31a+47b; 31a+48b; 31a+49b; 31a+50b; 31a+51b; 31a+52b; 31a+53b;
31a+54b; 31a+55b; 31a+55b; 31a+57b; 31a+58b, 31a+59b; 31a+60b;
31a+61b, 31a+62b; 31a+63b; 31a+64b; 31a+65b; 31a+66b; 31a+67b;
31a+68b; 31a+69b; 31a+70b; 31a+71b; 31a+72b; 31a+73b; 31a+74b;
31a+75b; 31a+76b; 31a+77b; 31a+78b; 31a+79b; 31a+80b; 31a+81b;
31a+82b; 31a+83b; 31a+84b; 31a+85b, 31a+86b, 31a+87b, 31a+88b,
31a+89b, 31a+90b; 31a+91b; 31a+92b; 31a+93b; 31a+94b, 31a+95b;
31a+96b; 31a+97b; 32a+1 b; 32a + 21); 32a + 313; 32a+4b; 32a+5b; 32a+6b;
210
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
32a+7b; 32a+8b; 32a+9b; 32a+10b, 32a+11 b; 32a+12b; 32a+13b; 32a+14b;
32a+15b; 32a+16b, 32a+17b; 32a+18b, 32a+19b; 32a+20b; 32a+21b,
32a+22b, 32a+23b, 32a+24b; 32a+25b, 32a+26b; 32a+27b; 32a+28b;
32a+29b; 32a+30b; 32a+31 b; 32a+32b; 32a+33b; 32a+34b; 32a+35b;
32a+36b; 32a+37b; 32a+38b, 32a+39b; 32a+40b; 32a+41b, 32a+42b;
32a+43b; 32a+44b; 32a+45b, 32a+46b, 32a+47b; 32a+48b; 32a+49b;
32a+50b; 32a+51b, 32a+52b, 32a+53b, 32a+54b; 32a+55b; 32a+55b;
32a+57b; 32a+58b; 32a+59b; 32a+60b, 32a+61b; 32a+62b; 32a+63b;
32a+64b; 32a+65b; 32a+66b; 32a+67b, 32a+68b; 32a+69b; 32a+70b;
32a+71b; 32a+72b; 32a+73b; 32a+74b, 32a+75b; 32a+76b; 32a+77b;
32a+78b; 32a+79b; 32a+80b; 32a+81b; 32a+82b; 32a+83b; 32a+84b;
32a+85b, 32a+86b; 32a+87b; 32a+88b; 32a+89b, 32a+90b; 32a+91b;
32a+92b; 32a+93b; 32a+94b; 32a+95b; 32a+96b; 32a+97b; 33a+1 b; 33a + 2b;
33a + 3h; 33a+4b; 33a+5b; 33a+6b; 33a+7b; 33a+8b; 33a+9b, 33a+10b;
33a+11b, 33a+12b, 33a+13b; 33a+14b; 33a+15b; 33a+16b; 33a+17b;
33a+18b; 33a+19b; 33a+20b; 33a+21b; 33a+22b; 33a+23b, 33a+24b;
33a+25b; 33a+26b; 33a+27b; 33a+28b; 33a+29b; 33a+30b; 33a+31b;
33a+32b; 33a+33b; 33a+34b; 33a+35b; 33a+36b, 33a+37b, 33a+38b;
33a+39b; 33a+40b; 33a+41b; 33a+42b, 33a+43b; 33a+44b; 33a+45b;
33a+46b; 33a+47b, 33a+48b; 33a+49b, 33a+50b; 33a+51b; 33a+52b;
33a+53b, 33a+54b; 33a+55b; 33a+55b; 33a+57b; 33a+58b, 33a+59b;
33a+60b; 33a+61b; 33a+62b, 33a+63b; 33a+64b; 33a+65b; 33a+66b;
33a+67b; 33a+68b, 33a+69b; 33a+70b; 33a+71b; 33a+72b; 33a+73b,
33a+74b; 33a+75b; 33a+76b; 33a+77b; 33a+78b, 33a+79b; 33a+80b;
33a+81b; 33a+82b; 33a+83b; 33a+84b; 33a+85b; 33a+86b; 33a+87b;
33a+88b; 33a+89b; 33a+90b; 33a+91b; 33a+92b; 33a+93b; 33a+94b;
33a+95b, 33a+96b; 33a+97b; 34a+1 b; 34a + 2b; 34a + 3b; 34a+4b; 34a+5b;
34a+6b; 34a+7b; 34a+8b; 34a+9b; 34a+10b, 34a+11 b; 34a+12b; 34a+13b;
34a+14b; 34a+15b; 34a+16b; 34a+17b; 34a+18b, 34a+19b; 34a+20b;
34a+21b; 34a+22b; 34a+23b; 34a+24b; 34a+25b; 34a+26b; 34a+27b;
211
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
34a+28b; 34a+29b, 34a+30b; 34a+31b; 34a+32b; 34a+33b; 34a+34b,
34a+35b; 34a+36b; 34a+37b, 34a+38b; 34a+39b; 34a+40b; 34a+41b;
34a+42b; 34a+43b; 34a+44b; 34a+45b; 34a+46b; 34a+47b; 34a+48b;
34a+49b; 34a+50b, 34a+51b; 34a+52b; 34a+53b; 34a+54b; 34a+55b;
34a+55b; 34a+57b; 34a+58b; 34a+59b; 34a+60b, 34a+61b; 34a+62b;
34a+63b; 34a+64b; 34a+65b, 34a+66b; 34a+67b; 34a+68b; 34a+69b;
34a+70b; 34a+71b; 34a+72b; 34a+73b, 34a+74b; 34a+75b; 34a+76b;
34a+77b; 34a+78b; 34a+79b; 34a+80b; 34a+81b; 34a+82b; 34a+83b;
34a+84b, 34a+85b; 34a+86b; 34a+87b; 34a+88b; 34a+89b; 34a+90b;
34a+91b; 34a+92b; 34a+93b; 34a+94b; 34a+95b; 34a+96b; 34a+97b; 35a+1 b;
35a + 2b; 35a + 313; 35a+4b; 35a+5b; 35a+6b; 35a+7b; 35a+8b; 35a+9b;
35a+10b; 35a-11 b; 35a+12b; 35a+13b, 35a+14b; 35a+15b; 35a+16b;
35a+17b; 35a+18b, 35a+19b, 35a+20b; 35a+21b; 35a+22b; 35a+23b;
35a+24b; 35a+25b; 35a+26b; 35a+27b; 35a+28b, 35a+29b; 35a+30b;
35a+31b, 35a+32b; 35a+33b; 35a+34b, 35a+35b; 35a+36b; 35a+37b;
35a+38b, 35a+39b; 35a+40b; 35a+41b; 35a+42b; 35a+43b, 35a+44b;
35a+45b; 35a+46b; 35a+47b; 35a+48b; 35a+49b; 35a+50b; 35a+51b;
35a+52b; 35a+53b; 35a+54b; 35a+55b; 35a+55b; 35a+57b; 35a+58b;
35a+59b; 35a+60b; 35a+61b; 35a+62b; 35a+63b; 35a+64b; 35a+65b;
35a+66b; 35a+67b; 35a+68b; 35a+69b; 35a+70b; 35a+71b; 35a+72b;
35a+73b; 35a+74b; 35a+75b, 35a+76b; 35a+77b; 35a+78b; 35a+79b;
35a+80b; 35a+81b; 35a+82b; 35a+83b; 35a+84b, 35a+85b; 35a+86b;
35a+87b; 35a+88b; 35a+89b; 35a+90b; 35a+91b; 35a+92b; 35a+93b;
35a+94b; 35a+95b, 35a+96b; 35a+97b; 36a+1 b; 36a + 2b; 36a + 3b; 36a+4b;
36a+5b; 36a+6b; 36a+7b; 36a+8b; 36a+9b; 36a+10b; 36a+11 b; 36a+12b;
36a+13b, 36a+14b; 36a+15b; 36a+16b; 36a+1,7b; 36a+18b; 36a+19b;
36a+20b, 36a+21b; 36a+22b; 36a+23b; 36a+24b; 36a+25b; 36a+26b,
36a+27b; 36a+28b, 36a+29b; 36a+30b; 36a+31b; 36a+32b; 36a+33b;
36a+34b; 36a+35b; 36a+36b; 36a+37b; 36a+38b, 36a+39b; 36a+40b;
36a+41b; 36a+42b, 36a+43b, 36a+44b; 36a+45b; 36a+46b; 36a+47b;
212
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
36a+48b; 36a+49b, 36a+50b, 36a+51b, 36a+52b, 36a+53b; 36a+54b;
36a+55b; 36a-F55b; 36a+57b; 36a+58b; 36a+59b; 36a+60b; 36a+61b;
36a+62b; 36a+63b; 36a+64b; 36a+65b; 36a+66b; 36a+67b; 36a+68b;
36a+69b; 36a+70b; 36a+71b; 36a+72b, 36a+73b; 36a+74b; 36a+75b;
36a+76b; 36a+77b; 36a+78b; 36a+79b; 36a+80b; 36a+81b; 36a+82b;
36a+83b; 36a+84b; 36a+85b; 36a+86b, 36a+87b; 36a+88b; 36a+89b,
36a+90b; 36a+91b; 36a+92b; 36a+93b; 36a+94b; 36a+95b; 36a+96b;
36a+97b; 37a+1 b; 37a + 2b; 37a + 3b; 37a+4b; 37a+5b; 37a+6b; 37a+7b;
37a+8b; 37a+9b; 37a+10b; 37a+11 b; 37a+12b; 37a+13b; 37a+14b, 37a+15b;
37a+16b; 37a+17b; 37a+18b; 37a+19b; 37a+20b, 37a+21b; 37a+22b;
37a+23b; 37a+24b; 37a+25b; 37a+26b; 37a+27b; 37a+28b, 37a+29b;
37a+30b; 37a+31b; 37a+32b; 37a+33b; 37a+34b; 37a+35b; 37a+36b;
37a+37b; 37a+38b, 37a+39b; 37a+40b; 37a+41b; 37a+42b; 37a+43b;
37a+44b; 37a+45b; 37a+46b; 37a+47b; 37a+48b; 37a+49b; 37a+50b;
37a+51b; 37a+52b; 37a+53b; 37a+54b; 37a+55b; 37a+55b; 37a+57b;
37a+58b; 37a+59b; 37a+60b; 37a+61b; 37a+62b; 37a+63b; 37a+64b;
37a+65b, 37a+66b; 37a+67b, 37a+68b; 37a+69b; 37a+70b; 37a+71b;
37a+72b, 37a+73b; 37a+74b; 37a+75b; 37a+76b; 37a+77b; 37a+78b;
37a+79b; 37a+80b; 37a+81b; 37a+82b; 37a+83b; 37a+84b; 37a+85b;
37a+86b; 37a+87b; 37a+88b; 37a+89b; 37a+90b; 37a+91b; 37a+92b;
37a+93b; 37a+94b; 37a+95b; 37a+96b; 37a+97b; 38a+1b; 38a + 2b; 38a + 3b;
38a+4b; 38a+5b; 38a+6b; 38a+7b; 38a+8b; 38a+9b; 38a+10b; 38a+11 b;
38a+12b, 38a+13b; 38a+14b; 38a+15b; 38a+16b; 38a+17b; 38a+18b;
38a+19b; 38a+20b; 38a+21b; 38a+22b; 38a+23b, 38a+24b; 38a+25b;
38a+26b; 38a+27b; 38a+28b, 38a+29b; 38a+30b; 38a+31b; 38a+32b;
38a+33b; 38a+34b; 38a+35b; 38a+36b; 38a+37b; 38a+38b; 38a+39b;
38a+40b, 38a+41b; 38a+42b; 38a+43b; 38a+44b, 38a+45b, 38a+46b;
38a+47b; 38a+48b; 38a+49b; 38a+50b; 38a+51b; 38a+52b; 38a+53b;
38a+54b; 38a+55b; 38a+55b; 38a+57b; 38a+58b; 38a+59b; 38a+60b,
38a+61b; 38a+62b; 38a+63b; 38a+64b; 38a+65b; 38a+66b; 38a+67b;
213
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
38a+68b; 38a+69b; 38a+70b; 38a+71b; 38a+72b; 38a+73b, 38a+74b;
38a+75b; 38a+76b; 38a+77b, 38a+78b; 38a+79b; 38a+80b; 38a+81b,
38a+82b; 38a+83b; 38a+84b; 38a+85b; 38a+86b; 38a+87b; 38a+88b;
38a+89b; 38a+90b; 38a+91b; 38a+92b; 38a+93b, 38a+94b; 38a+95b,
38a+96b; 38a+97b; 39a+lb, 39a + 2b; 39a + 3b; 39a+4b; 39a+5b; 39a+6b;
39a+7b; 39a+8b, 39a+9b; 39a+10b; 39a+11 b; 39a+12b; 39a+13b; 39a+14b;
39a+15b, 39a+16b; 39a+17b; 39a+18b; 39a+19b; 39a+20b; 39a+21b;
39a+22b; 39a+23b; 39a+24b; 39a+25b; 39a+26b; 39a+27b; 39a+28b;
39a+29b; 39a+30b; 39a+31 b; 39a+32 b; 39a+33b; 39a+34b; 39a+35b;
39a+36b; 39a+37b; 39a+38b; 39a+39b; 39a+40b, 39a+41b; 39a+42b;
39a+43b; 39a+44b; 39a+45b; 39a+46b; 39a+47b, 39a+48b, 39a+49b;
39a+50b; 39a+51b; 39a+52b; 39a+53b; 39a+54b; 39a+55b; 39a+55b;
39a+57b, 39a+58b; 39a+59b; 39a+60b, 39a+61b; 39a+62b; 39a+63b;
39a+64b; 39a+65b; 39a+66b; 39a+67b; 39a+68b; 39a+69b; 39a+70b;
39a+71b; 39a+72b; 39a+73b, 39a+74b; 39a+75b; 39a+76b; 39a+77b;
39a+78b; 39a+79b; 39a+80b; 39a+81b; 39a+82b; 39a+83b; 39a+84b;
39a+85b; 39a+86b; 39a+87b, 39a+88b; 39a+89b, 39a+90b; 39a+91b;
39a+92b; 39a+93b; 39a+94b; 39a+95b, 39a+96b; 39a+97b, 40a+lb; 40a + 2b;
40a + 3b; 40a+4b; 40a+5b; 40a+6b; 40a+7b; 40a+8b; 40a+9b; 40a+10b;
40a+11 b; 40a+12b; 40a+13b, 40a+14b; 40a+15b; 40a+16b; 40a+17b;
40a+18b; 40a+19b; 40a+20b; 40a+21b; 40a+22b; 40a+23b; 40a+24b;
40a+25b; 40a+26b; 40a+27b; 40a+28b, 40a+29b; 40a+30b; 40a+31b;
40a+32b; 40a+33b; 40a+34b; 40a+35b.; 40a+36b; 40a+37b; 40a+38b;
40a+39b; 40a+40b; 40a+41b; 40a+42b; 40a+43b; 40a+44b; 40a+45b;
40a+46b, 40a+47b; 40a+48b, 40a+49b; 40a+50b; 40a+51b; 40a+52b;
40a+53b; 40a+54b; 40a+55b; 40a+55b; 40a+57b; 40a+58b; 40a+59b,
40a+60b; 40a+61b; 40a+62b; 40a+63b; 40a+64b; 40a+65b; 40a+66b,
40a+67b, 40a+68b; 40a+69b; 40a+70b; 40a+71b; 40a+72b; 40a+73b;
40a+74b, 40a+75b; 40a+76b; 40a+77b, 40a+78b; 40a+79b; 40a+80b;
40a+81b; 40a+82b; 40a+83b; 40a+84b; 40a+85b; 40a+86b; 40a+87b;
214
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
40a+88b, 40a+89b, 40a+90b, 40a+91b; 40a+92b; 40a+93b; 40a+94b;
40a+95b; 40a+96b; 40a+97b, 41a+lb; 41a + 2b, 41a + 3b; 41a+4b; 41a+5b,
41a+6b, 41a+7b; 41a+8b; 41a+9b; 41a+10b; 41a+1 1 b; 41a+12b; 41a+13b;
41a+14b; 41a+15b; 41a+16b; 41a+17b; 41a+18b; 41a+19b, 41a+20b,
41a+21b; 41a+22b; 41a+23b; 41a+24b; 41a+25b; 41a+26b; 41a+27b;
41a+28b; 41a+29b; 41a+30b; 41a+31b; 41a+32b; 41a+33b; 41a+34b;
41a+35b, 41a+36b; 41a+37b; 41a+38b; 41a+39b; 41a+40b; 41a+41b;
41a+42b; 41a+43b; 41a+44b; 41a+45b; 41a+46b; 41a+47b, 41a+48b;
41a+49b, 41a+50b; 41a+51b; 41a+52b; 41a+53b; 41a+54b; 41a+55b;
41a+55b; 41a+57b; 41a+58b; 41a+59b, 41a+60b, 41a+61b; 41a+62b;
41a+63b; 41a+64b, 41a+65b; 41a+66b; 41a+67b; 41a+68b; 41a+69b;
41a+70b; 41a+71b; 41a+72b, 41a+73b; 41a+74b; 41a+75b; 41a+76b;
41a+77b, 41a+78b; 41a+79b; 41a+80b, 41a+81b; 41a+82b; 41a+83b;
41a+84b, 41a+85b; 41a+86b; 41a+87b; 41a+88b; 41a+89b; 41a+90b;
41a+91b; 41a+92b; 41a+93b; 41a+94b; 41a+95b, 41a+96b, 41a+97b; 42a+lb;
42a + 2b; 42a + 3b; 42a+4b; 42a+5b; 42a+6b; 42a+7b; 42a+8b, 42a+9b;
42a+10b; 42a+11 b; 42a+12b; 42a+13b; 42a+14b; 42a+15b; 42a+16b,
42a+17b; 42a+18b; 42a+19b; 42a+20b; 42a+21b; 42a+22b; 42a+23b;
42a+24b; 42a+25b; 42a+26b; 42a+27b; 42a+28b; 42a+29b; 42a+30b;
42a+31b; 42a+32b; 42a+33b; 42a+34b; 42a+35b; 42a+36b; 42a+37b;
42a+38b; 42a+39b; 42a+40b; 42a+41b; 42a+42b; 42a+43b, 42a+44b;
42a+45b; 42a+46b; 42a+47b; 42a+48b; 42a+49b; 42a+50b; 42a+51b;
42a+52b; 42a+53b; 42a+54b; 42a+55b; 42a+55b; 42a+57b; 42a+58b,
42a+59b; 42a+60b; 42a+61b; 42a+62b; 42a+63b; 42a+64b; 42a+65b;
42a+66b, 42a+67b; 42a+68b; 42a+69b; 42a+70b; 42a+71b, 42a+72b;
42a+73b, 42a+74b; 42a+75b; 42a+76b, 42a+77b; 42a+78b; 42a+79b;
42a+80b; 42a+81b; 42a+82b; 42a+83b; 42a+84b; 42a+85b; 42a+86b;
42a+87b; 42a+88b; 42a+89b, 42a+90b; 42a+91b; 42a+92b; 42a+93b;
42a+94b; 42a+95b; 42a+96b, 42a+97b; 43a+1 b; 43a + 2b; 43a + 3b; 43a+4b;
43a+5b, 43a+6b; 43a+7b; 43a+8b; 43a+9b, 43a+10b; 43a+1 1 b; 43a+12b;
215
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
43a+13b; 43a+14b; 43a+15b; 43a+16b; 43a+17b; 43a+18b; 43a+19b;
43a+20b, 43a+21b; 43a+22b; 43a+23b; 43a+24b; 43a+25b; 43a+26b;
43a+27b; 43a+28b; 43a+29b; 43a+30b; 43a+31b; 43a+32b; 43a+33b;
43a+34b; 43a+35b; 43a+36b; 43a+37b; 43a+38b; 43a+39b; 43a+40b;
43a+41b; 43a+42b, 43a+43b; 43a+44b; 43a+45b; 43a+46b; 43a+47b;
43a+48b; 43a+49b; 43a+50b, 43a+51b; 43a+52b; 43a+53b, 43a+54b,
43a+55b, 43a+55b; 43a+57b; 43a+58b; 43a+59b; 43a+60b; 43a+61b;
43a+62b, 43a+63b, 43a+64b; 43a+65b; 43a+66b; 43a+67b; 43a+68b;
43a+69b; 43a+70b; 43a+71b; 43a+72b; 43a+73b; 43a+74b; 43a+75b,
43a+76b; 43a+77b; 43a+78b; 43a+79b; 43a+80b; 43a+81b; 43a+82b;
43a+83b, 43a+84b; 43a+85b; 43a+86b; 43a+87b; 43a+88b; 43a+89b;
43a+90b; 43a+91b; 43a+92b; 43a+93b; 43a+94b; 43a+95b; 43a+96b;
43a+97b, 44a+lb; 44a + 213; 44a + 3b; 44a+4b; 44a+5b; 44a+6b; 44a+7b;
44a+8b; 44a+9b; 44a+10b; 44a+11b; 44a+12b; 44a+13b; 44a+14b; 44a+15b;
44a+16b; 44a+17b; 44a+18b; 44a+19b; 44a+20b; 44a+2113; 44a+22b;
44a+23b; 44a+24b; 44a+25b, 44a+26b; 44a+27b; 44a+28b, 44a+29b,
44a+30b; 44a+31b; 44a+32b; 44a+33b; 44a+34b; 44a+35b; 44a+36b;
44a+37b; 44a+38b; 44a+39b; 44a+40b; 44a+41b; 44a+42b; 44a+43b,
44a+44b; 44a+45b, 44a+46b; 44a+47b; 44a+48b; 44a+49b, 44a+50b;
44a+51b; 44a+52b; 44a+53b; 44a+54b; 44a+55b, 44a+55b; 44a+57b;
44a+58b; 44a+59b; 44a+60b; 44a+61b; 44a+62b; 44a+63b; 44a+64b;
44a+65b; 44a+66b; 44a+67b; 44a+68b; 44a+69b; 44a+70b; 44a+71b;
44a+72b, 44a+73b; 44a+74b; 44a+75b; 44a+76b; 44a+77b; 44a+78b;
44a+79b; 44a+80b; 44a+81b; 44a+82b, 44a+83b; 44a+84b, 44a+85b;
44a+86b; 44a+87b, 44a+88b, 44a+89b; 44a+90b; 44a+91b; 44a+92b;
44a+93b; 44a+94b; 44a+95b; 44a+96b; 44a+97b, 45a+1 b; 45a + 2b; 45a + 3b;
45a+4b, 45a+5b; 45a+6b; 45a+7b; 45a+8b; 45a+9b; 45a+10b; 45a+11 b;
45a+12b; 45a+13b; 45a+14b; 45a+15b; 45a+16b; 45a+17b; 45a+18b;
45a+19b; 45a+20b; 45a+21b; 45a+22b; 45a+23b; 45a+24b; 45a+25b;
45a+26b; 45a+27b; 45a+28b; 45a+29b; 45a+30b; 45a+31b; 45a+32b;
216
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
45a+33b; 45a+34b; 45a+35b, 45a+36b; 45a+37b; 45a+38b; 45a+39b;
45a+40b, 45a+41b, 45a+42b; 45a+43b; 45a+44b; 45a+45b; 45a+46b;
45a+47b; 45a+48b; 45a+49b; 45a+50b; 45a+51b; 45a+52b, 45a+53b;
45a+54b; 45a+55b; 45a+55b; 45a+57b; 45a+58b; 45a+59b; 45a+60b;
45a+61b; 45a+62b; 45a+63b; 45a+64b; 45a+65b; 45a+66b; 45a+67b;
45a+68b, 45a+69b, 45a+70b; 45a+71b; 45a+72b; 45a+73b, 45a+74b;
45a+75b; 45a+76b; 45a+77b; 45a+78b; 45a+79b; 45a+80b; 45a+81b;
45a+82b; 45a+83b; 45a+84b; 45a+85b; 45a+86b; 45a+87b; 45a+88b;
45a+89b; 45a+90b; 45a+91b; 45a+92b; 45a+93b; 45a+94b; 45a+95b;
45a+96b; 45a+97b; 46a+lb; 46a + 2b; 46a + 3b; 46a+4b; 46a+5b; 46a+6b,
46a+7b; 46a+8b; 46a+9b; 46a+10b, 46a+11 b; 46a+12b; 46a+13b; 46a+14b;
46a+15b; 46a+16b; 46a+17b; 46a+18b; 46a+19b; 46a+20b, 46a+21b;
46a+22b; 46a+23b, 46a+24b, 46a+25b; 46a+26b, 46a+27b, 46a+28b;
46a+29b; 46a+30b; 46a+31b; 46a+32b; 46a+33b; 46a+34b; 46a+35b;
46a+36b; 46a+37b, 46a+38b; 46a+39b; 46a+40b; 46a+41b; 46a+42b;
46a+43b; 46a+44b; 46a+45b; 46a+46b; 46a+47b, 46a+48b, 46a+49b;
46a+50b; 46a+51b, 46a+52b; 46a+53b; 46a+54b; 46a+55b; 46a+55b;
46a+57b; 46a+58b; 46a+59b, 46a+60b; 46a+61b; 46a+62b; 46a+63b;
46a+64b; 46a+65b; 46a+66b; 46a+67b; 46a+68b; 46a+69b, 46a+70b;
46a+71b; 46a+72b; 46a+73b; 46a+74b; 46a+75b; 46a+76b; 46a+77b;
46a+78b; 46a+79b; 46a+80b; 46a+81b; 46a+82b; 46a+83b; 46a+84b;
46a+85b; 46a+86b; 46a+87b; 46a+88b; 46a+89b; 46a+90b; 46a+91b;
46a+92b, 46a+93b; 46a+94b; 46a+95b; 46a+96b, 46a+97b, 47a+lb; 47a + 2b;
47a + 3b; 47a+4b; 47a+5b, 47a+6b; 47a+7b; 47a+8b; 47a+9b; 47a+10b;
47a+11b; 47a+12b; 47a+13b, 47a+14b; 47a+15b; 47a+16b; 47a+17b;
47a+18b; 47a+19b; 47a+20b; 47a+21b; 47a+22b; 47a+23b; 47a+24b;
47a+25b; 47a+26b, 47a+27b; 47a+28b; 47a+29b; 47a+30b; 47a+31b;
47a+32b; 47a+33b; 47a+34b, 47a+35b; 47a+36b; 47a+37b; 47a+38b,
47a+39b, 47a+40b; 47a+41b; 47a+42b; 47a+43b; 47a+44b, 47a+45b;
47a+46b; 47a+47b, 47a+48b; 47a+49b; 47a+50b; 47a+51b; 47a+52b,
217
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
47a+53b; 47a+54b; 47a+55b; 47a+55b; 47a+57b; 47a+58b; 47a+59b;
47a+60b; 47a+61b, 47a+62b; 47a+63b, 47a+64b; 47a+65b; 47a+66b,
47a+67b; 47a+68b; 47a+69b; 47a+70b; 47a+71b; 47a+72b; 47a+73b;
47a+74b; 47a+75b; 47a+76b; 47a+77b; 47a+78b, 47a+79b; 47a+80b;
47a+81b; 47a+82b; 47a+83b; 47a+84b; 47a+85b; 47a+86b; 47a+87b;
47a+88b; 47a+89b; 47a+90b, 47a+91b; 47a+92b; 47a+93b; 47a+94b;
47a+95b, 47a+96b; 47a+97b; 48a+1b; 48a + 2b; 48a + 3b; 48a+4b; 48a+5b;
48a+6b; 48a+7b; 48a+8b; 48a-F9b; 48a+10b; 48a+11b; 48a+12b; 48a+13b;
48a+14b; 48a+15b; 48a+16b; 48a+17b; 48a+18b; 48a+19b; 48a+20b;
48a+21b; 48a+22b; 48a+23b; 48a+24b; 48a+25b, 48a+26b; 48a+27b;
48a+28b; 48a+29b; 48a+30b; 48a+31b; 48a+32b; 48a+33b; 48a+34b,
48a+35b; 48a+36b; 48a+37b; 48a+38b; 48a+39b; 48a+40b; 48a+41b;
48a+42b; 48a+43b; 48a+44b; 48a+45b; 48a+46b; 48a+47b; 48a+48b,
48a+49b; 48a+50b; 48a+51b; 48a+52b; 48a+53b; 48a+54b; 48a+55b;
48a+55b; 48a+57b; 48a+58b; 48a+59b; 48a+60b; 48a+61b; 48a+62b;
48a+63b, 48a+64b; 48a+65b; 48a+66b; 48a+67b; 48a+68b; 48a+69b,
48a+70b; 48a+71b; 48a+72b, 48a+73b; 48a+74b; 48a+75b; 48a+76b;
48a+77b; 48a+78b; 48a+79b; 48a+80b; 48a+81b; 48a+82b; 48a+83b;
48a+84b; 48a+85b; 48a+86b; 48a+87b; 48a+88b; 48a+89b, 48a+90b,
48a+91b, 48a+92b; 48a+93b; 48a+94b; 48a+95b; 48a+96b; 48a+97b, 49a+1b;
49a + 2b; 49a + 3b; 49a+4b; 49a+5b; 49a+6b; 49a+7b; 49a+8b; 49a+9b;
49a+10b; 49a+11b; 49a+12b, 49a+13b; 49a+14b; 49a+15b; 49a+16b,
49a+17b; 49a+18b; 49a+19b; 49a+20b; 49a+21b; 49a+22b; 49a+23b;
49a+24b; 49a+25b; 49a+26b; 49a+27b; 49a+28b, 49a+29b; 49a+30b,
49a+31b; 49a+32b, 49a+33b; 49a+34b; 49a+35b; 49a+36b; 49a+37b;
49a+38b; 49a+39b; 49a+40b, 49a+41b; 49a+42b, 49a+43b; 49a+44b;
49a+45b; 49a+46b; 49a+47b; 49a+48b; 49a+49b, 49a+50b; 49a+51b;
49a+52b; 49a+53b; 49a+54b; 49a+55b; 49a+55b; 49a+57b; 49a+58b,
49a+59b; 49a+60b; 49a+61b; 49a+62b; 49a+63b; 49a+64b; 49a+65b;
49a+66b; 49a+67b; 49a+68b; 49a+69b; 49a+70b; 49a+71b; 49a+72b;
218
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
49a+73b; 49a+74b, 49a+75b; 49a+76b, 49a+77b; 49a+78b, 49a+79b,
49a+80b, 49a+81b; 49a+82b; 49a+83b, 49a+84b; 49a+85b; 49a+86b;
49a+87b; 49a+88b; 49a+89b; 49a+90b; 49a+91b; 49a+92b, 49a+93b;
49a+94b; 49a+95b; 49a+96b; 49a+97b; 50a+1 b; 50a + 2b; 50a + 3b; 50a+4b;
50a+5b; 50a+6b; 50a+7b; 50a+8b; 50a+9b; 50a+10b; 50a+11b; 50a+12b;
50a+13b; 50a+14b; 50a+15b; 50a+16b; 50a+17b, 50a+18b, 50a+19b,
50a+20b; 50a+21b; 50a+22b; 50a+23b; 50a+24b; 50a+25b; 50a+26b;
50a+27b; 50a+28b; 50a+29b; 50a+30b; 50a+31b; 50a+32b; 50a+33b,
50a+34b, 50a+35b, 50a+36b; 50a+37b; 50a+38b; 50a+39b; 50a+40b;
50a+41b, 50a+42b; 50a+43b; 50a+44b; 50a+45b; 50a+46b, 50a+47b,
50a+48b; 50a+49b; 50a+50b; 50a+51b; 50a+52b; 50a+53b; 50a+54b;
50a+55b; 50a+55b; 50a+57b; 50a+58b; 50a+59b; 50a+60b, 50a+61b;
50a+62b; 50a+63b; 50a+64b; 50a+65b; 50a+66b; 50a+67b; 50a+68b,
50a+69b, 50a+70b; 50a+71b; 50a+72b; 50a+73b; 50a+74b; 50a+75b;
50a+76b, 50a+77b; 50a+78b; 50a+79b; 50a+80b; 50a+81b; 50a+82b,
50a+83b; 50a+84b; 50a+85b; 50a+86b; 50a+87b; 50a+88b; 50a+89b;
50a+90b; 50a+91b, 50a+92b; 50a+93b; 50a+94b; 50a+95b; 50a+96b;
50a+97b; 51a+1 b; 51a + 2b; 51a + 3b; 51a+4b; 51a+5b; 51a+6b; 51a+7b;
51a+8b; 51a+9b; 51a+10b; 51a+1 1 b; 51a+12b; 51a+13b; 51a+14b; 51a+15b;
51a+16b; 51a+17b; 51a+18b; 51a+19b; 51a+20b; 51a+21b, 51a+22b;
51a+23b; 51a+24b; 51a+25b; 51a+26b; 51a+27b; 51a+28b, 51a+29b;
51a+30b, 51a+31b; 51a+32b; 51a+33b; 51a+34b; 51a+35b; 51a+36b;
51a+37b; 51a+38b; 51a+39b; 51a+40b; 51a+41b, 51a+42b; 51a+43b;
51a+44b; 51a+45b; 51a+46b; 51a+47b; 51a+48b; 51a+49b; 51a+50b;
51a+51b; 51a+52b; 51a+53b; 51a+54b; 51a+55b; 51a+55b; 51a+57b;
51a+58b, 51a+59b; 51a+60b; 51a+61b; 51a+62b; 51a+63b; 51a+64b;
51a+65b; 51a+66b; 51a+67b; 51a+68b, 51a+69b; 51a+70b; 51a+71b;
51a+72b; 51a+73b; 51a+74b; 51a+75b; 51a+76b; 51a+77b; 51a+78b;
51a+79b; 51a+80b; 51a+81b, 51a+82b; 51a+83b; 51a+84b; 51a+85b;
51a+86b; 51a+87b; 51a+88b; 51a+89b; 51a+90b; 51a+91b; 51a+92b,
219
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
51a+93b; 51a+94b; 51a+95b; 51a+96b; 51a+97b; 52a+1 b; 52a + 2h; 52a + 3b;
52a+4b; 52a+5b; 52a+6b; 52a+7b; 52a+8b; 52a+9b; 52a+10b; 52a+11b;
52a+12b; 52a+13b; 52a+14b; 52a+15b; 52a+16b; 52a+17b; 52a+18b;
52a+19b, 52a+20b; 52a+21b; 52a+22b; 52a+23b; 52a+24b; 52a+25b;
52a+26b; 52a+27b; 52a+28b; 52a+29b; 52a+30b; 52a+31b; 52a+32b,
52a+33b; 52a+34b; 52a+35b; 52a+36b; 52a+37b; 52a+38b; 52a+39b;
52a+40b; 52a+41b, 52a+42b; 52a+43b; 52a+44b; 52a+45b; 52a+46b;
52a+47b; 52a+48b, 52a+49b, 52a+50b; 52a+51b; 52a+52b; 52a+53b;
52a+54b; 52a+55b; 52a+55b; 52a+57b, 52a+58b; 52a+59b; 52a+60b;
52a+61b; 52a+62b; 52a+63b; 52a+64b; 52a+65b; 52a+66b; 52a+67b;
52a+68b; 52a+69b; 52a+70b; 52a+71b; 52a+72b; 52a+73b; 52a+74b;
52a+75b; 52a+76b; 52a+77b; 52a+78b; 52a+79b; 52a+80b; 52a+81b;
52a+82b; 52a+83b; 52a+84b; 52a+85b; 52a+86b, 52a+87b; 52a+88b;
52a+89b; 52a+90b; 52a+91b; 52a+92b; 52a+93b; 52a+94b; 52a+95b,
52a+96b; 52a+97b; 53a+1 b; 53a + 2b; 53a + 3b; 53a+4b; 53a+5b; 53a+6b;
53a+7b; 53a+8b; 53a+9b; 53a+10b; 53a+11 b; 53a+12b; 53a+13b; 53a+14b;
53a+15b, 53a+16b, 53a+17b; 53a+18b, 53a+19b; 53a+20b; 53a+21b;
53a+22b; 53a+23b, 53a+24b; 53a+25b; 53a+26b; 53a+27b, 53a+28b,
53a+29b; 53a+30b; 53a+31b; 53a+32b; 53a+33b; 53a+34b; 53a+35b;
53a+36b; 53a+37b; 53a+38b; 53a+39b; 53a+40b; 53a+41b; 53a+42b;
53a+43b; 53a+44b; 53a+45b; 53a+46b; 53a+47b; 53a+48b; 53a+49b;
53a+50b; 53a+51b; 53a+52b; 53a+53b; 53a+54b; 53a+55b; 53a+55b;
53a+57b, 53a+58b; 53a+59b; 53a+60b; 53a+61b; 53a+62b; 53a+63b;
53a+64b, 53a+65b; 53a+66b; 53a+67b; 53a+68b; 53a+69b; 53a+70b;
53a+71b; 53a+72b; 53a+73b; 53a+74b; 53a+75b, 53a+76b, 53a+77b;
53a+78b; 53a+79b; 53a+80b; 53a+81b; 53a+82b; 53a+83b; 53a+84b;
53a+85b; 53a+86b, 53a+87b; 53a+88b; 53a+89b; 53a+90b; 53a+91b;
53a+92b; 53a+93b; 53a+94b; 53a+95b; 53a+96b; 53a+97b; 54a+1 b; 54a + 2b;
54a + 313; 54a+4b; 54a+5b; 54a+6b; 54a+7b, 54a+8b, 54a+9b; 54a+10b;
54a+11 b; 54a+12b; 54a+13b; 54a+14b; 54a+15b; 54a+16b; 54a+17b;
220
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
54a+18b; 54a+19b; 54a+20b; 54a+21b; 54a+22b, 54a+23b, 54a+24b;
54a+25b; 54a+26b; 54a+27b; 54a+28b, 54a+29b; 54a+30b; 54a+31b;
54a+32b; 54a+33b; 54a+34b; 54a+35b; 54a+36b; 54a+37b; 54a+38b,
54a+39b; 54a+40b; 54a+41b; 54a+42b, 54a+43b; 54a+44b; 54a+45b;
54a+46b; 54a+47b; 54a+48b; 54a+49b; 54a+50b; 54a+51b; 54a+52b;
54a+53b; 54a+54b; 54a+55b; 54a+55b, 54a+57b; 54a+58b; 54a+59b;
54a+60b; 54a+61b; 54a+62b; 54a+63b, 54a+64b; 54a+65b; 54a+66b;
54a+67b; 54a+68b; 54a+69b; 54a+70b; 54a+71b; 54a+72b, 54a+73b;
54a+74b; 54a+75b; 54a+76b; 54a+77b; 54a+78b; 54a+79b; 54a+80b;
54a+81b; 54a+82b; 54a+83b; 54a+84b, 54a+85b; 54a+86b; 54a+87b;
54a+88b; 54a+89b; 54a+90b; 54a+91b; 54a+92b; 54a+93b; 54a+94b;
54a+95b; 54a+96b; 54a+97b; 55a+1b; 55a + 2b; 55a + 3b; 55a+4b; 55a+5b;
55a+6b; 55a+7b; 55a+8b, 55a+9b; 55a+10b; 55a+11 b; 55a+12b; 55a+13b,
55a+14b; 55a+15b, 55a+16b; 55a+17b; 55a+18b; 55a+19b; 55a+20b;
55a+21b; 55a+22b; 55a+23b; 55a+24b; 55a+25b; 55a+26b; 55a+27b;
55a+28b; 55a+29b; 55a+30b; 55a+31b; 55a+32b; 55a+33b, 55a+34b;
55a+35b; 55a+36b; 55a+37b; 55a+38b; 55a+39b; 55a+40b; 55a+41b;
55a+42b; 55a+43b; 55a+44b; 55a+45b; 55a+46b, 55a+47b; 55a+48b;
55a+49b; 55a+50b; 55a+51b; 55a+52b; 55a+53b; 55a+54b; 55a+55b;
55a+55b, 55a+57b; 55a+58b; 55a+59b; 55a+60b; 55a+61b; 55a+62b;
55a+63b; 55a+64b; 55a+65b; 55a+66b; 55a+67b, 55a+68b; 55a+69b;
55a+70b; 55a+71b; 55a+72b; 55a+73b; 55a+74b; 55a+75b; 55a+76b;
55a+77b; 55a+78b; 55a+79b; 55a+80b; 55a+81b; 55a+82b; 55a+83b,
55a+84b; 55a+85b, 55a+86b, 55a+87b; 55a+88b; 55a+89b; 55a+90b;
55a+91b; 55a+92b; 55a+93b; 55a+94b; 55a+95b; 55a+96b; 55a+97b; 56a+lb;
56a + 2b; 56a + 3b, 56a+4b; 56a+5b; 56a+6b; 56a+7b; 56a+8b; 56a+9b;
56a+10b; 56a+11 b; 56a+12b; 56a+13b; 56a+14b; 56a+15b; 56a+16b;
56a+17b; 56a+18b; 56a+19b; 56a+20b; 56a+21b; 56a+22b; 56a+23b;
56a+24b, 56a+25b; 56a+26b; 56a+27b, 56a+28b; 56a+29b; 56a+30b,
56a+31b; 56a+32b; 56a+33b; 56a+34b, 56a+35b; 56a+36b; 56a+37b;
221
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
56a+38b, 56a+39b; 56a+40b, 56a+41b; 56a+42b; 56a+43b; 56a+44b;
56a+45b; 56a+46b; 56a+47b; 56a+48b; 56a+49b, 56a+50b; 56a+51b,
56a+52b; 56a+53b, 56a+54b; 56a+55b; 56a+55b; 56a+57b; 56a+58b,
56a+59b, 56a+60b; 56a+61b; 56a+62b; 56a+63b; 56a+64b; 56a+65b;
56a+66b; 56a+67b; 56a+68b; 56a+69b; 56a+70b; 56a+71b; 56a+72b;
56a+73b; 56a+74b, 56a+75b, 56a+76b; 56a+77b; 56a+78b; 56a+79b;
56a+80b; 56a+81b; 56a+82b; 56a+83b; 56a+84b; 56a+85b; 56a+86b;
56a+87b; 56a+88b; 56a+89b; 56a+90b, 56a+91b; 56a+92b; 56a+93b;
56a+94b; 56a+95b; 56a+96b; 56a+97b; 57a+1 b; 57a + 2b; 57a + 3h; 57a+4b;
57a+5b; 57a+6b, 57a+7b; 57a+8b, 57a+9b; 57a+10b; 57a+11b; 57a+12b;
57a+13b; 57a+14b; 57a+15b, 57a+16b; 57a+17b; 57a+18b, 57a+19b;
57a+20b; 57a+21b; 57a+22b; 57a+23b; 57a+24b; 57a+25b; 57a+26b;
57a+27b; 57a+28b, 57a+29b; 57a+30b; 57a+31b; 57a+32b; 57a+33b;
57a+34b; 57a+35b; 57a+36b; 57a+37b, 57a+38b; 57a+39b; 57a+40b,
57a+41b; 57a+42b; 57a+43b; 57a+44b; 57a+45b, 57a+46b, 57a+47b,
57a+48b, 57a+49b; 57a+50b; 57a+51b; 57a+52b; 57a+53b; 57a+54b;
57a+55b, 57a+55b, 57a+57b; 57a+58b; 57a+59b; 57a+60b; 57a+61b;
57a+62b; 57a+63b; 57a+64b; 57a+65b; 57a+66b; 57a+67b; 57a+68b;
57a+69b; 57a+70b, 57a+71b; 57a+72b, 57a+73b; 57a+74b, 57a+75b;
57a+76b; 57a+77b; 57a+78b; 57a+79b; 57a+80b; 57a+81b; 57a+82b;
57a+83b; 57a+84b, 57a+85b; 57a+86b; 57a+87b; 57a+88b; 57a+89b;
57a+90b, 57a+91b; 57a+92b; 57a+93b; 57a+94b, 57a+95b; 57a+96b;
57a+97b; 58a+1 b; 58a + 2b; 58a + 3b; 58a+4b, 58a+5b, 58a+6b; 58a+7b;
58a+8b; 58a+9b; 58a+10b; 58a+11 b; 58a+12b; 58a+13b; 58a+14b; 58a+15b;
58a+16b; 58a+17b, 58a+18b; 58a+19b; 58a+20b; 58a+21b, 58a+22b;
58a+23b, 58a+24b; 58a+25b; 58a+26b; 58a+27b; 58a+28b; 58a+29b;
58a+30b; 58a+31b; 58a+32b; 58a+33b; 58a+34b, 58a+35b; 58a+36b;
58a+37b; 58a+38b; 58a+39b; 58a+40b; 58a+41b; 58a+42b; 58a+43b;
58a+44b; 58a+45b; 58a+46b, 58a+47b; 58a+48b, 58a+49b; 58a+50b,
58a+51b; 58a+52b; 58a+53b; 58a+54b; 58a+55b; 58a+55b; 58a+57b,
222
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
58a+58b, 58a+59b, 58a+60b; 58a+61b; 58a+62b; 58a+63b; 58a+64b;
58a+65b; 58a+66b; 58a+67b; 58a+68b; 58a+69b; 58a+70b; 58a+71b;
58a+72b; 58a+73b; 58a+74b; 58a+75b; 58a+76b; 58a+77b; 58a+78b;
58a+79b; 58a+80b; 58a+81b; 58a+82b; 58a+83b; 58a+84b; 58a+85b;
58a+86b; 58a+87b; 58a+88b; 58a+89b; 58a+90b; 58a+91b, 58a+92b;
58a+93b; 58a+94b; 58a+95b, 58a+96b; 58a+97b; 59a-F1b; 59a + 2b; 59a + 3b;
59a+4b; 59a+5b; 59a+6b, 59a+7b; 59a+8b; 59a+9b; 59a+10b; 59a+11 b;
59a+12b; 59a+13b; 59a+14b, 59a+15b; 59a+16b; 59a+17b; 59a+18b;
59a+19b; 59a+20b; 59a+21b; 59a+22b; 59a+23b; 59a+24b; 59a+25b,
59a+26b; 59a+27b; 59a+28b, 59a+29b, 59a+30b; 59a+31b, 59a+32b;
59a+33b; 59a+34b; 59a+35b; 59a+36b; 59a+37b; 59a+38b; 59a+39b;
59a+40b; 59a+41b; 59a+42b; 59a+43b; 59a+44b; 59a+45b, 59a+46b;
59a+47b; 59a+48b, 59a+49b; 59a+50b; 59a+51b; 59a+52b; 59a+53b;
59a+54b; 59a+55b; 59a+55b; 59a+57b; 59a+58b; 59a+59b; 59a+60b;
59a+61b; 59a+62b; 59a+63b; 59a+64b; 59a+65b; 59a+66b; 59a+67b;
59a+68b; 59a+69b; 59a+70b; 59a+71b; 59a+72b; 59a+73b; 59a+74b;
59a+75b, 59a+76b; 59a+77b; 59a+78b; 59a+79b; 59a+80b; 59a+81b;
59a+82b, 59a+83b; 59a+84b; 59a+85b; 59a+86b; 59a+87b, 59a+88b;
59a+89b; 59a+90b; 59a+91b; 59a+92b; 59a+93b; 59a+94b; 59a+95b;
59a+96b; 59a+97b; 60a+1 b; 60a + 2b; 60a + 3b; 60a+4b; 60a+5b; 60a+6b;
60a+7b; 60a+8b; 60a+9b; 60a+10b; 60a+11 b; 60a+12b; 60a+13b; 60a+14b;
60a+15b; 60a+16b; 60a+17b; 60a+18b; 60a+19b, 606+20b; 60a+21b;
60a+22b; 60a+23b; 60a+24b; 60a+25b; 60a+26b; 60a+27,b; 60a+28b;
60a+29b; 60a+30b; 60a+31b; 60a+32b; 60a+33b; 60a+34b; 60a+35b;
60a+36b, 60a+37b; 60a+38b; 60a+39b; 60a+40b; 60a+41b; 60a+42b;
60a+43b; 60a+44b; 60a+45b, 60a+46b; 60a+47b; 60a+48b; 60a+49b;
60a+50b; 60a+51b; 60a+52b, 60a+53b; 60a+54b; 60a+55b; 60a+55b;
60a+57b; 60a+58b; 60a+59b, 60a+60b; 60a+61b; 60a+62b; 60a+63b;
60a+64b, 60a+65b, 60a+66b; 60a+67b; 60a+68b; 60a+69b; 60a+70b;
60a+71b; 60a+72b; 60a+73b; 60a+74b; 60a+75b; 60a+76b, 60a+77b;
223
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
60a+78b, 60a+79b; 60a+80b; 60a+81b; 60a+82b; 60a+83b; 60a+84b,
60a+85b; 60a+86b; 60a+87b; 60a+88b; 60a+89b; 60a+90b; 60a+91b;
60a+92b; 60a+93b; 60a+94b; 60a+95b; 60a+96b; 60a+97b; 61a+lb; 61a + 2b;
61a + 3b; 61a+4b, 61a+5b; 61a+6b; 61a+7b; 61a+8b; 61a+9b; 61a+10b;
61a+11 b; 61a+12b; 61a+13b; 61a+14b; 61a+15b; 61a+16b; 61a+17b;
61a+18b; 61a+19b; 61a+20b; 61a+21b; 61a+22b; 61a+23b; 61a+24b;
61a+25b; 61a+26b; 61a+27b; 61a+28b; 61a+29b; 61a+30b; 61a+31b;
61a+32b; 61a+33b, 61a+34b; 61a+35b; 61a+36b, 61a+37b; 61a+38b,
61a+39b; 61a+40b; 61a+41b; 61a+42b; 61a+43b; 61a+44b; 61a+45b;
61a+46b; 61a+47b; 61a+48b, 61a+49b; 61a+50b; 61a+51b; 61a+52b;
61a+53b; 61a+54b; 61a+55b; 61a+55b; 61a+57b; 61a+58b; 61a+59b;
61a+60b; 61a+61b; 61a+62b; 61a+63b, 61a+64b, 61a+65b, 61a+66b;
61a+67b; 61a+68b, 61a+69b; 61a+70b; 61a+71b; 61a+72b; 61a+73b;
61a+74b; 61a+75b; 61a+76b; 61a+77b; 61a+78b; 61a+79b; 61a+80b;
61a+81b, 61a+82b; 61a+83b; 61a+84b; 61a+85b; 61a+86b; 61a+87b;
61a+88b; 61a+89b; 61a+90b; 61a+91b; 61a+92b; 61a+93b; 61a+94b;
61a+95b; 61a+96b; 61a+97b; 62a+1 b; 62a + 2b; 62a + 3b; 62a+4b; 62a+5b;
62a+6b; 62a+7b; 62a+8b; 62a+9b; 62a+10b; 62a+1 1 b; 62a+12b; 62a+13b;
62a+14b; 62a+15b; 62a+16b; 62a+17b; 62a+18b; 62a+19b, 62a+20b;
62a+21b, 62a+22b; 62a+23b; 62a+24b; 62a+25b; 62a+26b, 62a+27b;
62a+28b; 62a+29b; 62a+30b; 62a+31b, 62a+32b, 62a+33b; 62a+34b;
62a+35b; 62a+36b; 62a+37b; 62a+38b; 62a+39b; 62a+40b; 62a+41b;
62a+42b; 62a+43b; 62a+44b; 62a+45b; 62a+46b; 62a+47b; 62a+48b;
62a+49b; 62a+50b, 62a+51b; 62a+52b; 62a+53b; 62a+54b; 62a+55b;
62a+55b, 62a+57b; 62a+58b; 62a+59b; 62a+60b; 62a+61b; 62a+62b;
62a+63b; 62a+64b; 62a+65b; 62a+66b; 62a+67b; 62a+68b, 62a+69b;
62a+70b; 62a+71b; 62a+72b; 62a+73b; 62a+74b; 62a+75b; 62a+76b;
62a+77b; 62a+78b, 62a+79b; 62a+80b; 62a+81b; 62a+82b; 62a+83b;
62a+84b, 62a+85b; 62a+86b; 62a+87b; 62a+88b; 62a+89b; 62a+90b;
62a+91b; 62a+92b; 62a+93b; 62a+94b; 62a+95b; 62a+96b; 62a+97b; 63a+1 b;
224
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
63a + 2b, 63a + 3b; 63a+4b; 63a+5b; 63a+6b; 63a+7b; 63a+8b; 63a+9b,
63a+10b; 63a+11 b; 63a+12b; 63a+13b; 63a+14b; 63a+15b, 63a+16b;
63a+17b; 63a+18b; 63a+19b; 63a+20b; 63a+21b; 63a+22b; 63a+23b;
63a+24b, 63a+25b; 63a+26b; 63a+27b, 63a+28b; 63a+29b; 63a+30b;
63a+31b; 63a+32b, 63a+33b, 63a+34b; 63a+35b; 63a+36b; 63a+37b;
63a+38b, 63a+39b; 63a+40b, 63a+41b; 63a+42b; 63a+43b; 63a+44b;
63a+45b; 63a+46b; 63a+47b, 63a+48b; 63a+49b; 63a+50b; 63a+51b;
63a+52b; 63a+53b; 63a+54b; 63a+55b; 63a+55b; 63a+57b; 63a+58b;
63a+59b; 63a+60b; 63a+61b; 63a+62b, 63a+63b; 63a+64b; 63a+65b;
63a+66b; 63a+67b; 63a+68b; 63a+69b; 63a+70b; 63a+71b; 63a+72b,
63a+73b, 63a+74b; 63a+75b; 63a+76b; 63a+77b; 63a+78b; 63a+79b,
63a+80b; 63a+81b; 63a+82b; 63a+83b; 63a+84b; 63a+85b; 63a+86b;
63a+87b; 63a+88b, 63a+89b; 63a+90b; 63a+91b; 63a+92b; 63a+93b;
63a+94b; 63a+95b; 63a+96b; 63a+97b; 64a+1 b; 64a + 2b, 64a + 3b; 64a+4b;
64a+5b; 64a+6b; 64a+7b; 64a+8b; 64a+9b; 64a+10b, 64a+11 b; 64a+12b;
64a+13b; 64a+14b, 64a+15b; 64a+16b; 64a+17b; 64a+18b; 64a+19b; ,
64a+20b, 64a+21b; 64a+22b; 64a+23b; 64a+24b; 64a+25b; 64a+26b;
64a+27b; 64a+28b; 64a+29b, 64a+30b; 64a+31b; 64a+32b; 64a+33b;
64a+34b; 64a+35b; 64a+36b; 64a+37b, 64a+38b, 64a+39b; 64a+40b;
64a+41b, 64a+42b; 64a+43b; 64a+44b, 64a+45b; 64a+46b; 64a+47b;
64a+48b; 64a+49b; 64a+50b; 64a+51b, 64a+52b; 64a+53b; 64a+54b,
64a+55b; 64a+55b; 64a+57b; 64a+58b, 64a+59b; 64a+60b; 64a+61b;
64a+62b; 64a+63b; 64a+64b; 64a+65b; 64a+66b; 64a+67b; 64a+68b;
64a+69b; 64a+70b; 64a+71b; 64a+72b, 64a+73b, 64a+74b; 64a+75b;
64a+76b; 64a+77b; 64a+78b; 64a+79b; 64a+80b; 64a+81b; 64a+82b;
64a+83b, 64a+84b; 64a+85b; 64a+86b; 64a+87b; 64a+88b; 64a+89b;
64a+90b; 64a+91b; 64a+92b; 64a+93b, 64a+94b; 64a+95b; 64a+96b;
64a+97b; 65a+lb; 65a + 2b; 65a + 3b; 65a+4b, 65a+5b, 65a+6b; 65a+7b;
65a+8b, 65a+9b; 65a-'-10b; 65a+11b, 65a+12b; 65a+13b; 65a+14b, 65a+15b;
65a+16b; 65a+17b; 65a+18b; 65a+19b; 65a+20b, 65a+21b; 65a+22b;
225
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
65a+23b, 65a+24b; 65a+25b, 65a+26b; 65a+27b; 65a+28b; 65a+29b;
65a+30b; 65a+31b; 65a+32b; 65a+33b; 65a+34b; 65a+35b; 65a+36b;
65a+37b; 65a+38b; 65a+39b, 65a+40b; 65a+41b; 65a+42b, 65a+43b;
65a+44b; 65a+45b; 65a+46b; 65a+47b; 65a+48b; 65a+49b; 65a+50b;
,65a+51b, 65a+52b; 65a+53b, 65a+54b; 65a+55b; 65a+55b; 65a+57b,
65a+58b, 65a+59b; 65a+60b; 65a+61b; 65a+62b; 65a+63b; 65a+64b,
65a+65b, 65a+66b; 65a+67b, 65a+68b; 65a+69b; 65a+70b; 65a+71b;
65a+72b; 65a+73b; 65a+74b; 65a+75b; 65a+76b; 65a+77b, 65a+78b;
65a+79b; 65a+80b, 65a+81b; 65a+82b; 65a+83b; 65a+84b; 65a+85b;
65a+86b; 65a+87b; 65a+88b, 65a+89b; 65a+90b; 65a+91b; 65a+92b;
65a+93b; 65a+94b; 65a+95b; 65a+96b, 65a+97b, 66a+lb; 66a + 2b; 66a + 3b;
66a+4b, 66a+5b, 66a+6b; 66a+7b, 66a+8b; 66a+9b; 66a+1 0b; 66a+11b;
66a+12b; 66a+13b, 66a+14b, 66a+15b; 66a+16b; 66a+17b; 66a+18b,
66a+19b; 66a+20b; 66a+21b; 66a+22b; 66a+23b; 66a+24b; 66a+25b;
66a+26b; 66a+27b, 66a+28b; 66a+29b; 66a+30b; 66a+31b, 66a+32b;
66a+33b; 66a+34b; 66a+35b, 66a+36b; 66a+37b; 66a+38b; 66a+39b;
66a+40b; 66a+41b; 66a+42b; 66a+43b; 66a+44b; 66a+45b; 66a+46b;
66a+47b, 66a+48b; 66a+49b, 66a+50b; 66a+51b; 66a+52b, 66a+53b;
66a+54b; 66a+55b, 66a+55b; 66a+57b; 66a+58b; 66a+59b; 66a+60b;
66a+61b; 66a+62b; 66a+63b; 66a+64b; 66a+65b; 66a+66b; 66a+67b;
66a+68b; 66a+69b; 66a+70b; 66a+71b, 66a+72b; 66a+73b; 66a+74b;
66a+75b; 66a+76b; 66a+77b; 66a+78b; 66a+79b; 66a+80b; 66a+81b;
66a+82b, 66a+83b; 66a+84b; 66a+85b; 66a+86b; 66a+87b; 66a+88b;
66a+89b; 66a+90b; 66a+91b; 66a+92b; 66a+93b; 66a+94b; 66a+95b;
66a+96b; 66a+97b; 67a+1 b; 67a + 213; 67a + 313; 67a+4b; 67a+5b; 67a+6b;
67a+7b; 67a+8b; 67a+9b; 67a+10b; 67a+11 b; 67a+12b, 67a+13b; 67a+14b;
67a+15b, 67a+16b; 67a+17b; 67a+18b; 67a+19b; 67a+20b, 67a+21b;
67a+22b; 67a+23b; 67a+24b; 67a+25b; 67a+26b; 67a+27b; 67a+28b,
67a+29b; 67a+30b; 67a+31b; 67a+32b; 67a+33b; 67a+34b; 67a+35b;
67a+36b; 67a+37b; 67a+38b; 67a+39b; 67a+40b; 67a+41b; 67a+42b,
226
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
67a+43b; 67a+44b; 67a+45b; 67a+46b, 67a+47b; 67a+48b; 67a+49b;
67a+50b; 67a+51b, 67a+52b; 67a+53b; 67a+54b; 67a+55b; 67a+55b;
67a+57b; 67a+58b; 67a+59b; 67a+60b; 67a+61b; 67a+62b; 67a+63b;
67a+64b; 67a+65b; 67a+66b; 67a+67b; 67a+68b; 67a+69b, 67a+70b;
67a+71b; 67a+72b; 67a+73b; 67a+74b, 67a+75b; 67a+76b; 67a+77b;
67a+78b; 67a+79b; 67a+80b; 67a+81 b; 67a+82b; 67a+83b, 67a+84b;
67a+85b; 67a+86b; 67a+87b; 67a+88b; 67a+89b; 67a+90b; 67a+91b;
67a+92b; 67a+93b, 67a+94b; 67a+95b; 67a+96b; 67a+97b; 68a+1 b; 68a + 2b;
68a + 3b; 68a+4b; 68a+5b; 68a+6b; 68a+7b, 68a+8b; 68a+9b; 68a+10b;
68a+11 b; 68a+12b; 68a+13b; 68a+14b; 68a+15b; 68a+16b, 68a+17b;
68a+18b; 68a+19b; 68a+20b; 68a+21b; 68a+22b; 68a+23b, 68a+24b,
68a+25b; 68a+26b; 68a+27b; 68a+28b; 68a+29b; 68a+30b; 68a+31b;
68a+32b; 68a+33b, 68a+34b, 68a+35b; 68a+36b; 68a+37b; 68a+38b;
68a+39b; 68a+40b; 68a+41b, 68a+42b; 68a+43b; 68a+44b; 68a+45b;
68a+46b, 68a+47b; 68a+48b; 68a+49b, 68a+50b, 68a+51b; 68a+52b;
68a+53b; 68a+54b; 68a+55b; 68a+55b; 68a+57b; 68a+58b; 68a+59b;
68a+60b; 68a+61b; 68a+62b; 68a+63b; 68a+64b; 68a+65b; 68a+66b;
68a+67b; 68a+68b, 68a+69b; 68a+70b; 68a+71b, 68a+72b; 68a+73b;
68a+74b; 68a+75b, 68a+76b; 68a+77b; 68a+78b; 68a+79b; 68a+80b;
68a+81b; 68a+82b; 68a+83b; 68a+84b; 68a+85b; 68a+86b; 68a+87b,
68a+88b; 68a+89b; 68a+90b; 68a+91b; 68a+92b; 68a+93b; 68a+94b;
68a+95b; 68a+96b; 68a+97b; 69a+1 b; 69a + 2b; 69a + 3b; 69a+4b; 69a+5b,
69a+6b; 69a+7b; 69a+8b; 69a+9b; 69a+10b; 69a+11 b; 69a+12b; 69a+13b;
69a+14b, 69a+15b; 69a+16b, 69a+17b, 69a+18b; 69a+19b; 69a+20b;
69a+21b, 69a+22b; 69a+23b; 69a+24b; 69a+25b; 69a+26b; 69a+27b;
69a+28b; 69a+29b; 69a+30b; 69a+31b; 69a+32b; 69a+33b; 69a+34b;
69a+35b; 69a+36b; 69a+37b; 69a+38b; 69a+39b; 69a+40b; 69a+41b;
69a+42b; 69a+43b; 69a+44b; 69a+45b; 69a+46b, 69a+47b; 69a+48b;
69a+49b; 69a+50b; 69a+51b; 69a+52b; 69a+53b; 69a+54b; 69a+55b,
69a+55b, 69a+57b; 69a+58b; 69a+59b; 69a+60b; 69a+61b; 69a+62b;
227
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
69a+63b; 69a+64b; 69a+65b; 69a+66b; 69a+67b; 69a+68b; 69a+69b;
69a+70b; 69a+71b, 69a+72b; 69a+73b, 69a+74b; 69a+75b; 69a+76b;
69a+77b; 69a+78b, 69a+79b; 69a+80b; 69a+81b; 69a+82b; 69a+83b;
69a+84b; 69a+85b; 69a+86b; 69a+87b; 69a+88b; 69a+89b; 69a+90b;
69a+91b, 69a+92b; 69a+93b; 69a+94b; 69a+95b, 69a+96b; 69a+97b; 70a+1 b;
70a + 2b; 70a + 3b; 70a+4b; 70a+5b; 70a+6b; 70a+7b; 70a+8b; 70a+9b,
70a+10b; 70a+11b; 70a+12b; 70a+13b; 70a+14b; 70a+15b; 70a+16b;
70a+17b; 70a+18b; 70a+19b; 70a+20b; 70a+21b; 70a+22b; 70a+23b;
70a+24b; 70a+25b; 70a+26b; 70a+27b; 70a+28b; 70a+29b; 70a+30b;
70a+31b; 70a+32b; 70a+33b; 70a+34b; 70a+35b; 70a+36b; 70a+37b,
70a+38b; 70a+39b; 70a+40b, 70a+41b; 70a+42b; 70a+43b; 70a+44b,
70a+45b; 70a+46b; 70a+47b; 70a+48b; 70a+49b; 70a+50b; 70a+51b;
70a+52b; 70a+53b; 70a+54b, 70a+55b; 70a+55b, 70a+57b; 70a+58b;
70a+59b; 70a+60b; 70a+61b, 70a+62b; 70a+63b; 70a+64b; 70a+65b;
70a+66b; 70a+67b; 70a+68b; 70a+69b; 70a+70b, 70a+71b; 70a+72b;
70a+73b, 70a+74b; 70a+75b; 70a+76b; 70a+77b; 70a+78b; 70a+79b;
70a+80b; 70a+81b; 70a+82b; 70a+83b; 70a+84b; 70a+85b; 70a+86b;
70a+87b; 70a+88b; 70a+89b; 70a+90b; 70a+91b; 70a+92b; 70a+93b;
70a+94b; 70a+95b; 70a+96b; 70a+97b; 71a+lb; 71a + 2b; 71a + 3b; 71a+4b;
71a+5b, 71a+6b; 71a+7b, 71a+8b; 71a+9b; 71a+10b; 71a+1 1 b; 71a+12b;
71a+13b; 71a+14b; 71a+15b; 71a+16b; 71a+17b; 71a+18b; 71a+19b;
71a+20b; 71a+21b; 71a+22b; 71a+23b; 71a+24b; 71a+25b; 71a+26b;
71a+27b; 71a+28b; 71a+29b; 71a+30b; 71a+31b; 71a+32b; 71a+33b;
71a+34b, 71a+35b; 71a+36b; 71a+37b; 71a+38b, 71a+39b; 71a+40b,
71a+41b; 71a+42b, 71a+43b, 71a+44b, 71a+45b, 71a+46b; 71a+47b;
71a+48b, 71a+49b; 71a+50b; 71a+51b; 71a+52b; 71a+53b; 71a+54b;
71a+55b; 71a+55b; 71a+57b; 71a+58b; 71a+59b; 71a+60b; 71a+61b,
71a+62b; 71a+63b; 71a+64b; 71a+65b; 71a+66b; 71a+67b; 71a+68b;
71a+69b; 71a+70b; 71a+71b; 71a+72b; 71a+73b, 71a+74b; 71a+75b,
71a+76b, 71a+77b; 71a+78b; 71a+79b; 71a+80b; 71a+81b; 71a+82b;
228
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
71a+83b; 71a+84b; 71a+85b; 71a+86b; 71a+87b; 71a+88b; 71a+89b;
71a+90b; 71a+91b; 71a+92b, 71a+93b, 71a+94b; 71a+95b; 71a+96b;
71a+97b; 72a+1b; 72a + 2b; 72a + 3b; 72a+4b; 72a+5b; 72a+6b; 72a+7b;
72a+8b; 72a+9b; 72a+10b; 72a+11b, 72a+12b; 72a+13b; 72a+14b; 72a+15b;
72a+16b; 72a+17b, 72a+18b, 72a+19b, 72a+20b; 72a+21b; 72a+22b,
72a+23b; 72a+24b; 72a+25b, 72a+26b; 72a+27b; 72a+28b; 72a+29b;
72a+30b; 72a+31b; 72a+32b; 72a+33b; 72a+34b; 72a+35b; 72a+36b;
72a+37b; 72a+38b; 72a+39b; 72a+40b; 72a+41b; 72a+42b; 72a+43b;
72a+44b, 72a+45b; 72a+46b; 72a+47b; 72a+48b; 72a+49b, 72a+50b,
72a+51b; 72a+52b; 72a+53b; 72a+54b; 72a+55b; 72a+55b, 72a+57b;
72a+58b; 72a+59b; 72a+60b; 72a+61b; 72a+62b; 72a+63b; 72a+64b;
72a+65b; 72a+66b; 72a+67b; 72a+68b; 72a+69b; 72a+70b; 72a+71b;
72a+72b, 72a+73b; 72a+74b; 72a+75b; 72a+76b; 72a+77b; 72a+78b,
72a+79b; 72a+80b; 72a+81b; 72a+82b; 72a+83b; 72a+84b; 72a+85b,
72a+86b; 72a+87b; 72a+88b; 72a+89b; 72a+90b; 72a+91b; 72a+92b;
72a+93b; 72a+94b; 72a+95b; 72a+96b; 72a+97b; 73a+1b, 73a + 2b; 73a + 3b;
73a+4b; 73a+5b; 73a+6b; 73a+7b; 73a+8b; 73a+9b; 73a+10b; 73a+11b;
73a+12b; 73a+13b; 73a+14b; 73a+15b; 73a+16b; 73a+17b, 73a+18b;
73a+19b; 73a+20b; 73a+21b; 73a+22b; 73a+23b; 73a+24b; 73a+25b,
73a+26b, 73a+27b; 73a+28b; 73a+29b; 73a+30b; 73a+31b; 73a+32b,
73a+33b; 73a+34b; 73a+35b; 73a+36b; 73a+37b; 73a+38b; 73a+39b;
73a+40b; 73a+41b, 73a+42b; 73a+43b; 73a+44b, 73a+45b; 73a+46b,
73a+47b, 73a+48b; 73a+49b; 73a+50b; 73a+51b; 73a+52b; 73a+53b;
73a+54b; 73a+55b, 73a+55b; 73a+57b; 73a+58b; 73a+59b; 73a+60b;
73a+61b; 73a+62b; 73a+63b; 73a+64b; 73a+65b; 73a+66b; 73a+67b;
73a+68b; 73a+69b, 73a+70b; 73a+71b; 73a+72b; 73a+73b; 73a+74b;
73a+75b; 73a+76b; 73a+77b; 73a+78b; 73a+79b; 73a+80b, 73a+81b;
73a+82b; 73a+83b; 73a+84b; 73a+85b; 73a+86b; 73a+87b; 73a+88b;
73a+89b; 73a+90b; 73a+91b; 73a+92b; 73a+93b, 73a+94b; 73a+95b;
73a+96b; 73a+97b; 74a+1b; 74a + 2h; 74a + 3b; 74a+4b, 74a+5b; 74a+6b;
229
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
74a+7b; 74a+8b; 74a+9b; 74a+10b; 74a+11b, 74a+12b, 74a+13b; 74a+14b,
74a+15b, 74a+16b, 74a+17b, 74a+18b; 74a+19b, 74a+20b; 74a+21b;
74a+22b; 74a+23b; 74a+24b; 74a+25b; 74a+26b; 74a+27b; 74a+28b;
74a+29b; 74a+30b; 74a+31b; 74a+32b; 74a+33b; 74a+34b; 74a+35b;
74a+36b, 74a+37b; 74a+38b; 74a+39b, 74a+40b; 74a+41b; 74a+42b;
74a+43b; 74a+44b; 74a+45b; 74a+46b, 74a+47b; 74a+48b; 74a+49b;
74a+50b; 74a+51b; 74a+52b, 74a+53b; 74a+54b; 74a+55b; 74a+55b;
74a+57b; 74a+58b; 74a+59b; 74a+60b; 74a+61b; 74a+62b, 74a+63b,
74a+64b; 74a+65b; 74a+66b; 74a+67b, 74a+68b; 74a+69b; 74a+70b;
74a+71b; 74a+72b; 74a+73b; 74a+74b; 74a+75b, 74a+76b; 74a+77b;
74a+78b; 74a+79b; 74a+80b; 74a+81b; 74a+82b; 74a+83b; 74a+84b;
74a+85b; 74a+86b; 74a+87b, 74a+88b, 74a+89b; 74a+90b; 74a+91b;
74a+92b; 74a+93b; 74a+94b; 74a+95b; 74a+96b, 74a+97b; 75a+1b; 75a + 213;
75a + 3b; 75a+4b; 75a+5b; 75a+6b; 75a+7b; 75a+8b, 75a+9b; 75a+10b;
75a+11b; 75a+12b; 75a+13b; 75a+14b, 75a+15b, 75a+16b; 75a+17b;
75a+18b; 75a+19b; 75a+20b, 75a+21b; 75a+22b, 75a+23b, 75a+24b;
75a+25b; 75a+26b; 75a+27b; 75a+28b, 75a+29b; 75a+30b, 75a+31b;
75a+32b; 75a+33b; 75a+34b; 75a+35b; 75a+36b; 75a+37b; 75a+38b;
75a+39b; 75a+40b; 75a+41b; 75a+42b; 75a+43b; 75a+44b; 75a+45b;
75a+46b; 75a+47b, 75a+48b; 75a+49b; 75a+50b; 75a+51b, 75a+52b;
75a+53b, 75a+54b; 75a+55b, 75a+55b; 75a+57b; 75a+58b; 75a+59b;
75a+60b; 75a+61b; 75a+62b; 75a+63b; 75a+64b; 75a+65b, 75a+66b;
75a+67b; 75a+68b, 75a+69b; 75a+70b; 75a+71b; 75a+72b, 75a+73b;
75a+74b; 75a+75b; 75a+76b; 75a+77b; 75a+78b, 75a+79b, 75a+80b,
75a+81b; 75a+82b; 75a+83b; 75a+84b; 75a+85b; 75a+86b; 75a+87b;
75a+88b; 75a+89b; 75a+90b; 75a+91b; 75a+92b; 75a+93b, 75a+94b,
75a+95b; 75a+96b; 75a+97b; 76a+1b; 76a + 2b, 76a + 313; 76a+4b, 76a+5b;
76a+6b,_76a+7b; 76a+8b; 76a+9b; 76a+10b, 76a+11b; 76a+12b; 76a+13b;
\ 76a+14b; 76a+15b; 76a+16b; 76a+17b; 76a+18b; 76a+19b; 76a+20b;
76a+21b; 76a+22b; 76a+23b, 76a+24b; 76a+25b; 76a+26b; 76a+27b;
230
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
76a+28b, 76a+29b; 76a+30b; 76a+31b; 76a+32b, 76a+33b; 76a+34b,
76a+35b; 76a+36b; 76a+37b; 76a+38b; 76a+39b; 76a+40b, 76a+41b;
76a+42b, 76a+43b; 76a+44b; 76a+45b; 76a+46b; 76a+47b; 76a+48b;
76a+49b; 76a+50b; 76a+51b; 76a+52b; 76a+53b; 76a+54b; 76a+55b;
76a+55b; 76a+57b; 76a+58b; 76a+59b; 76a+60b; 76a+61b; 76a+62b;
76a+63b; 76a+64b; 76a+65b; 76a+66b; 76a+67b; 76a+68b; 76a+69b;
76a+70b; 76a+71b; 76a+72b; 76a+73b; 76a+74b; 76a+75b; 76a+76b;
76a+77b; 76a+78b; 76a+79b; 76a+80b; 76a+81b; 76a+82b; 76a+83b;
76a+84b; 76a+85b; 76a+86b; 76a+87b; 76a+88b, 76a+89b; 76a+90b;
76a+91b; 76a+92b; 76a+93b; 76a+94b; 76a+95b; 76a+96b, 76a+97b; 77a+1 b;
77a + 2b; 77a + 3h; 77a+4b; 77a+5b; 77a+6b; 77a+7b; 77a+8b; 77a+9b;
77a+10b, 77a+11 b; 77a+12b; 77a+13b; 77a+14b; 77a+15b; 77a+16b;
77a+17b; 77a+18b; 77a+19b; 77a+20b, 77a+21b; 77a+22b; 77a+23b,
77a+24b; 77a+25b; 77a+26b; 77a+27b; 77a+28b; 77a+29b; 77a+30b;
77a+31b; 77a+32b, 77a+33b; 77a+34b; 77a+35b; 77a+36b, 77a+37b;
77a+38b; 77a+39b; 77a+40b; 77a+41b; 77a+42b; 77a+43b; 77a+44b,
77a+45b; 77a+46b; 77a+47b; 77a+48b; 77a+49b; 77a+50b; 77a+51b;
77a+52b; 77a+53b; 77a+54b; 77a+55b; 77a+55b; 77a+57b; 77a+58b,
77a+59b; 77a+60b; 77a+61b, 77a+62b; 77a+63b, 77a+64b; 77a+65b;
77a+66b, 77a+67b; 77a+68b; 77a+69b; 77a+70b; 77a+71b; 77a+72b;
77a+73b; 77a+74b; 77a+75b; 77a+76b; 77a+77b; 77a+78b; 77a+79b;
77a+80b; 77a+81b; 77a+82b; 77a+83b; 77a+84b; 77a+85b; 77a+86b;
77a+87b; 77a+88b, 77a+89b; 77a+90b; 77a+91b; 77a+92b; 77a+93b;
77a+94b; 77a+95b; 77a+96b; 77a+97b; 78a+1 b; 78a + 2b; 78a + 313; 78a+4b;
78a+5b; 78a+6b; 78a+7b; 78a+8b; 78a+9b; 78a+10b; 78a+11 b; 78a+12b;
78a+13b; 78a+14b; 78a+15b; 78a+16b; 78a+17b, 78a+18b; 78a+19b;
78a+20b; 78a+21b; 78a+22b; 78a+23b; 78a+24b; 78a+25b; 78a+26b,
78a+27b, 78a+28b; 78a+29b; 78a+30b; 78a+31b; 78a+32b; 78a+33b;
78a+34b; 78a+35b; 78a+36b; 78a+37b; 78a+38b; 78a+39b; 78a+40b;
78a+41b; 78a+42b; 78a+43b; 78a+44b; 78a+45b; 78a+46b; 78a+47b,
231
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
78a+48b; 78a+49b; 78a+50b; 78a+51b; 78a+52b, 78a+53b; 78a+54b,
78a+55b; 78a+55b; 78a+57b; 78a+58b; 78a+59b; 78a+60b; 78a+61b;
78a+62b, 78a+63b; 78a+64b; 78a+65b; 78a+66b; 78a+67b; 78a+68b,
78a+69b; 78a+70b; 78a+71b; 78a+72b; 78a+73b; 78a+74b; 78a+75b;
78a+76b, 78a+77b, 78a+78b; 78a+79b; 78a+80b, 78a+81b; 78a+82b,
78a+83b; 78a+84b, 78a+85b; 78a+86b, 78a+87b; 78a+88b; 78a+89b;
78a+90b; 78a+91b; 78a+92b, 78a+93b; 78a+94b; 78a+95b; 78a+96b,
78a+97b, 79a+1 b; 79a + 2b; 79a + 3b; 79a+4b; 79a+513; 79a+6b; 79a+7b,
79a+8b; 79a+9b; 79a+10b; 79a+11 b; 79a+12b; 79a+13b; 79a+14b; 79a+15b;
79a+16b; 79a+17b; 79a+18b; 79a+19b; 79a+20b; 79a+21b, 79a+22b;
79a+23b; 79a+24b; 79a+25b; 79a+26b; 79a+27b; 79a+28b; 79a+29b;
79a+30b; 79a+31b; 79a+32b; 79a+33b; 79a+34b; 79a+35b; 79a+36b;
79a+37b; 79a+38b; 79a+39b; 79a+40b; 79a+41b; 79a+42b; 79a+43b;
79a+44b, 79a+45b; 79a+46b, 79a+47b; 79a+48b; 79a+49b; 79a+50b;
79a+51b; 79a+52b; 79a+53b; 79a+54b; 79a+55b; 79a+55b; 79a+57b;
79a+58b; 79a+59b; 79a+60b; 79a+61b; 79a+62b; 79a+63b; 79a+64b;
79a+65b; 79a+66b; 79a+67b, 79a+68b; 79a+69b; 79a+70b; 79a+71b;
79a+72b; 79a+73b; 79a+74b; 79a+75b; 79a+76b; 79a+77b; 79a+78b,
79a+79b; 79a+80b, 79a+81b, 79a+82b, 79a+83b; 79a+84b; 79a+85b,
79a+86b; 79a+87b; 79a+88b; 79a+89b, 79a+90b; 79a+91b; 79a+92b;
79a+93b, 79a+94b; 79a+95b; 79a+96b; 79a+97b; 80a+1 b; 80a + 2b; 80a + 313;
80a+4b; 80a+5b; 80a+6b; 80a+7b; 80a+8b, 80a+9b; 80a+10b; 80a+11 b;
80a+12b; 80a+13b, 80a+14b; 80a+15b; 80a+16b; 80a+17b; 80a+18b;
80a+19b; 80a+20b; 80a+2113; 80a+22b; 80a+23b; 80a+24b; 80a+25b;
80a+26b; 80a+27b; 80a+28b; 80a+29b; 80a+30b; 80a+31b; 80a+32b;
80a+33b; 80a+34b; 80a+35b; 80a+36b; 80a+37b; 80a+38b; 80a+39b;
80a+40b; 80a+41b; 80a+42b; 80a+43b; 80a+44b; 80a+45b, 80a+46b;
80a+47b; 80a+48b; 80a+49b; 80a+50b; 80a+51 b; 80a+52b; 80a+53b,
80a+54b; 80a+55b; 80a+55b; 80a+57b; 80a+58b; 80a+59b; 80a+60b,
80a+61b, 80a+62b, 80a+63b; 80a+64b; 80a+65b; 80a+66b; 80a+67b;
232
CA 02610948 2007-12-05
WO 2006/124021
PCT/US2005/016871
80a+68b; 80a+69b; 80a+70b; 80a+71b; 80a+72b; 80a+73b; 80a+74b;
80a+75b, 80a+76b, 80a+77b; 80a+78b; 80a+79b; 80a+80b; 80a+81b;
80a+82b; 80a+83b; 80a+84b; 80a+85b; 80a+86b, 80a+87b, 80a+88b;
80a+89b; 80a+90b; 80a+91b; 80a+92b; 80a+93b; 80a+94b; 80a+95b,
80a+96b; 80a+97b; 81a+lb; 81a + 2b; 81a + 3h; 81a+4b; 81a+5b; 81a-'-6b,
81a+7b; 81a+8b; 81a+9b; 81a+10b; 81a+11b; 81a+12b; 81a+13b; 81a+14b;
81a+15b, 81a+16b; 81a+17b; 81a+18b; 81a+19b; 81a+20b; 81a+21b;
81a+22b; 81a+23b, 81a+24b; 81a+25b; 81a+26b; 81a+27b; 81a+28b;
81a+29b; 81a+30b; 81a+31b; 81a+32b; 81a+33b; 81a+34b; 81a+35b;
81a+36b; 81a+37b; 81a+38b; 81a+39b; 81a+40b; 81a+41b; 81a+42b;
81a+43b; 81a+44b, 81a+45b; 81a+46b; 81a+47b; 81a+48b; 81a+49b;
81a+50b; 81a+51b; 81a+52b; 81a+53b; 81a+54b; 81a+55b; 81a+55b,
81a+57b; 81a+58b, 81a+59b; 81a-F60b, 81a+61b, 81a+62b; 81a+63b;
81a+64b; 81a+65b; 81a+66b; 81a+67b, 81a+68b; 81a+69b; 81a+70b;
81a+71b; 81a+72b; 81a+73b; 81a+74b; 81a+75b; 81a+76b; 81a+77b;
81a+78b; 81a+79b; 81a+80b; 81a+81b, 81a+82b; 81a+83b; 81a+84b;
81a+85b; 81a+86b; 81a+87b; 81a+88b; 81a+89b; 81a+90b; 81a+91b;
81a+92b; 81a+93b; 81a+94b; 81a+95b; 81a+96b; 81a+97b; 82a+1 b; 82a + 2b;
82a + 3b; 82a+4b; 82a+5b; 82a+6b; 82a+7b, 82a+8b; 82a+9b; 82a+10b;
82a+1 1 b; 82a+12b; 82a+13b; 82a+14b; 82a+15b; 82a+16b; 82a+17b;
82a+18b; 82a+19b; 82a+20b; 82a+21b, 82a+22b; 82a+23b, 82a+24b,
82a+25b; 82a+26b; 82a+27b; 82a+28b; 82a+29b; 82a+30b; 82a+31b;
82a+32b; 82a+33b; 82a+34b; 82a+35b; 82a+36b, 82a+37b; 82a+38b;
82a+39b, 82a+40b; 82a+41b; 82a+42b; 82a+43b, 82a+44b; 82a+45b;
82a+46b; 82a+47b; 82a+48b; 82a+49b; 82a+50b; 82a+51b; 82a+52b;
82a+53b; 82a+54b; 82a+55b; 82a+55b; 82a+57b; 82a+58b; 82a+59b;
82a+60b; 82a+61b; 82a+62b; 82a+63b; 82a+64b; 82a+65b; 82a+66b;
82a+67b; 82a+68b; 82a+69b; 82a+70b; 82a+71b; 82a+72b; 82a+73b;
82a+74b; 82a+75b; 82a+76b; 82a+77b; 82a+78b; 82a+79b, 82a+80b;
82a+81b; 82a+82b; 82a+83b; 82a+84b; 82a+85b; 82a+86b, 82a+87b,
233
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
82a+88b; 82a+89b; 82a+90b; 82a+91b; 82a+92b; 82a+93b; 82a+94b,
82a+95b; 82a+96b; 82a+97b; 83a+1 b; 83a + 2b; 83a + 3b; 83a+4b; 83a+5b;
83a+6b; 83a+7b, 83a+8b; 83a+9b; 83a+10b; 83a+11 b; 83a+12b; 83a+13b;
83a+14b, 83a+15b; 83a+16b; 83a+17b; 83a+18b; 83a+19b; 83a+20b;
83a+21b; 83a+22b; 83a+23b; 83a+24b; 83a+25b, 83a+26b; 83a+27b,
83a+28b; 83a+29b; 83a+30b; 83a+31b; 83a+32b; 83a+33b, 83a+34b,
83a+35b; 83a+36b; 83a+37b; 83a+38b; 83a+39b; 83a+40b; 83a+41b;
83a+42b; 83a+43b; 83a+44b; 83a+45b; 83a+46b, 83a+47b; 83a+48b;
83a+49b, 83a+50b; 83a+51b; 83a+52b; 83a+53b; 83a+54b; 83a+55b,
83a+55b; 83a+57b; 83a+58b; 83a+59b, 83a+60b, 83a+61b; 83a+62b;
83a+63b; 83a+64b; 83a+65b; 83a+66b; 83a+67b; 83a+68b; 83a+69b;
83a+70b; 83a+71b, 83a+72b; 83a+73b, 83a+74b, 83a+75b; 83a+76b;
83a+77b; 83a+78b; 83a+79b; 83a+80b; 83a+81b; 83a+82b; 83a+83b;
83a+84b; 83a+85b; 83a+86b; 83a+87b; 83a+88b; 83a+89b; 83a+90b;
83a+91b; 83a+92b; 83a+93b; 83a+94b; 83a+95b, 83a+96b; 83a+97b, 84a+1 b;
84a + 2b; 84a + 3b; 84a+4b; 84a+5b; 84a+6b, 84a+7b; 84a+8b; 84a+9b;
84a+10b; 84a+11 b; 84a+12b; 84a+13b; 84a+14b; 84a+15b; 84a+16b,
84a+17b; 84a+18b; 84a+19b; 84a+20b; 84a+21b, 84a+22b, 84a+23b;
84a+24b; 84a+25b, 84a+26b; 84a+27b; 84a+28b; 84a+29b; 84a+30b;
84a+31b; 84a+32b; 84a+33b; 84a+34b; 84a+35b; 84a+36b; 84a+37b;
84a+38b; 84a+39b; 84a+40b; 84a+41b; 84a+42b; 84a+43b; 84a+44b;
84a+45b; 84a+46b; 84a+47b; 84a+48b; 84a+49b; 84a+50b; 84a+51b;
84a+52b; 84a+53b; 84a+54b; 84a+55b; 84a+55b; 84a+57b; 84a+58b;
84a+59b; 84a+60b; 84a+61b, 84a+62b; 84a+63b; 84a+64b; 84a+65b;
84a+66b; 84a+67b; 84a+68b; 84a+69b; 84a+70b; 84a+71b; 84a+72b;
84a+73b; 84a+74b, 84a+75b; 84a+76b; 84a+77b; 84a+78b; 84a+79b;
84a+80b; 84a+81b; 84a+82b; 84a+83b; 84a+84b; 84a+85b; 84a+86b;
84a+87b; 84a+88b; 84a+89b; 84a+90b; 84a+91b; 84a+92b; 84a+93b;
84a+94b; 84a+95b; 84a+96b; 84a+97b, 85a+1 b; 85a + 2b; 85a + 3b; 85a+4b;
85a+5b; 85a+6b; 85a+7b; 85a+8b; 85a+9b; 85a+10b; 85a+11 b; 85a+12b;
234
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
85a+13b; 85a+14b; 85a+15b; 85a+16b; 85a+17b; 85a+18b; 85a+19b;
85a+20b; 85a+21b; 85a+22b; 85a+23b; 85a+24b; 85a+25b; 85a+26b;
85a+27b; 85a+28b; 85a+29b; 85a+30b; 85a+31b, 85a+32b; 85a+33b;
85a+34b; 85a+35b; 85a+36b; 85a+37b; 85a+38b; 85a+39b; 85a+40b;
85a+41b; 85a+42b; 85a+43b; 85a+44b; 85a+45b; 85a+46b; 85a+47b,
85a+48b; 85a+49b; 85a+50b; 85a+51b; 85a+52b; 85a+53b; 85a+54b;
85a+55b, 85a+55b; 85a+57b; 85a+58b; 85a+59b; 85a+60b; 85a+61b;
85a+62b; 85a+63b; 85a+64b; 85a+65b; 85a+66b; 85a+67b; 85a+68b;
85a+69b; 85a+70b; 85a+71b, 85a+72b; 85a+73b; 85a+74b; 85a+75b;
85a+76b, 85a+77b; 85a+78b; 85a+79b; 85a+80b; 85a+81b, 85a+82b;
85a+83b; 85a+84b; 85a+85b, 85a+86b; 85a+87b; 85a+88b; 85a+89b;
85a+90b; 85a+91b; 85a+92b; 85a+93b; 85a+94b, 85a+95b; 85a+96b; and
85a+97b.
As described above, the fibrosing agent can be coated onto an
implant or a portion of an implant using the polymeric coatings described
above. This can be accomplished, for example, by dipping, spraying,
electrospinning, painting or by vacuum deposition. In addition to the coating
compositions and methods described above, there are various other coating
compositions and methods that are known in the art. Representative examples
of these coating compositions and methods are described in U.S. Patent. Nos.
6,610,016; 6,358,557; 6,306,176; 6,110,483; 6,106,473; 5,997,517; 5,800,412;
5,525,348; 5,331,027; 5,001,009; 6,562,136; 6,406,754; 6,344,035; 6,254,921;
6,214,901; 6,077,698; 6,603,040; 6,278,018; 6,238,799; 6,096,726; 5,766,158;
5,599,576; 4,119,094; 4,100,309; 6,599,558; 6,369,168; 6,521,283; 6,497,916;
6251964; 6,225,431; 6,087,462; 6,083,257; 5,739,237; 5,739,236; 5,705,583;
5648442; 5645883; 5,556,710; 5,496,581; 4,689,386; 6,214,115; 6,090,901;
6,599,448; 6,054,504; 4,987,182; 4,847,324; and 4,642,267, U.S. Patent
Application Publication Nos. 2003/0129130; 2001/0026834; 2003/0190420;
2001/0000785; 2003/0059631; 2003/0190405; 2002/0146581; 2003/020399;
235
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
2003/0129130, 2001/0026834; 2003/0190420; 2001/0000785; 2003/0059631;
2003/0190405; 2002/0146581; and 2003/020399, and PCT Publication Nos.
WO 02/055121; WO 01/57048; WO 01/52915; and WO 01/01957.
Within another aspect of the invention, the biologically active
fibrosing, hemostatic and/or anti-infective agent(s) can be delivered with a
non-
polymeric agent into the diverticula. Examples of non-polymeric agents include
sucrose derivatives (e.g., sucrose acetate isobutyrate, sucrose oleate),
sterols
such as cholesterol, stigmasterol, beta-sitosterol, and estradiol; cholesteryl
esters such as cholesteryl stearate; C12 -C24 fatty acids such as lauric acid,
myristic acid, palmitic acid, stearic acid, arachidic acid, behenic acid, and
lignoceric acid; C18 -C36 mono-, di- and triacylglycerides such as glyceryl
monooleate, glyceryl monolinoleate, glyceryl monolaurate, glyceryl
monodocosanoate, glyceryl monomyristate, glyceryl monodicenoate, glyceryl
dipalmitate, glyceryl didocosanoate, glyceryl dimyristate, glyceryl
didecenoate,
glyceryl tridocosanoate, glyceryl trimyristate, glyceryl tridecenoate,
glycerol
tristearate and mixtures thereof; sucrose fatty acid esters such as sucrose
distearate and sucrose palmitate; sorbitan fatty acid esters such as sorbitan
monostearate, sorbitan monopalmitate and sorbitan tristearate; C16 -C18 fatty
alcohols such as cetyl alcohol, myristyl alcohol, stearyl alcohol, and
cetostearyl
alcohol; esters of fatty alcohols and fatty acids such as cetyl palmitate and
cetearyl palmitate; anhydrides of fatty acids such as stearic anhydride;
phospholipids including phosphatidylcholine (lecithin), phosphatidylserine,
phosphatidylethanolamine, phosphatidylinositol, and lysoderivatives thereof;
sphingosine and derivatives thereof; spingomyelins such as stearyl, palmitoyl,
and tricosanyl spingomyelins; ceramides such as stearyl and palmitoyl
ceramides; glycosphingolipids; lanolin and lanolin alcohols, calcium
phosphate,
sintered and unscintered hydoxyapatite, zeolites; and combinations and
mixtures thereof.
236
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Representative examples of patents relating to non-polymeric
delivery systems and their preparation include U.S. Patent Nos. 5,736,152;
5,888,533; 6,120,789; 5,968,542; and 5,747,058.
Other carriers that may likewise be utilized to contain and deliver
fibrosis-inducing agents, hemostatic agents, and/or anti-infective agents
described herein include: hydroxypropyl cyclodextrin (Cserhati and Hollo, Int.
J.
Pharm. /08:69-75, 1994), liposomes (see, e.g., Sharma et al., Cancer Res.
53:5877-5881, 1993; Sharma and Straubinger, Pharm. Res. /1(60):889-896,
1994; WO 93/18751; U.S. Patent No. 5,242,073), liposome/gel (WO 94/26254),
nanocapsules (Bartoli et al., J. Microencapsulation 7(2):191-197, 1990),
micelles (Alkan-Onyuksel et al., Pharm. Res. /1(2):206-212, 1994),
nanoparticles (Violante and Lanzafame PAACR), nanoparticles - modified (U.S.
Patent No. 5,145,684), nanoparticles (surface modified) (U.S. Patent No.
5,399,363), micelle (surfactant) (U.S. Patent No. 5,403,858), synthetic
phospholipid compounds (U.S. Patent No. 4,534,899), gas borne dispersion
(U.S. Patent No. 5,301,664), liquid emulsions, foam, spray, gel, lotion,
cream,
ointment, dispersed vesicles, particles or droplets solid- or liquid-
aerosols,
microemulsions (U.S. Patent No. 5,330,756), polymeric shell (nano- and micro-
capsule) (U.S. Patent No. 5,439,686), emulsions (Tarr et al., Pharm Res. 4: 62-
165, 1987), nanospheres (Hagan et al., Proc. Intern. Symp. Control Rel.
Bioact.
Mater. 22, 1995; Kwon et al., Pharm Res. /2(2):192-195; Kwon et al., Pharm
Res. /0(7):970-974; Yokoyama et al., J. Contr. Rel. 32:269-277, 1994; Gref et
al., Science 263:1600-1603, 1994; Bazile et al., J. Pharm. ScL 84:493-498,
1994) and implants (U.S. Patent No. 4,882,168, Jampel et al., Invest.
Ophthalm. Vis. Science 34(11):3076-3083, 1993; Walter et al., Cancer Res.
54:22017-2212, 1994).
Within another embodiment, the fibrosis-inducing agent,
hemostatic agent, and/or anti-infective agent can further comprise a secondary
carrier. The secondary carrier can be in the form of microspheres (e.g., PLGA,
PLLA, PDLLA, PCL, gelatin, polydioxanone, poly(alkylcyanoacrylate)),
237
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
nanospheres (e.g., PLGA, PLLA, PDLLA, PCL, gelatin, polydioxanone,
poly(alkylcyanoacrylate)), liposomes, emulsions, microemulsions, micelles
(e.g.,SDS, block copolymers of the form X-Y, X-Y-X or Y-X-Y, where X is a
poly(alkylene oxide) or an alkyl ether thereof and Y is a polyester (e.g.,
PLGA,
PLLA, PDLLA, PCL, polydioxanone)), zeolites or cyclodextrins.
Within another embodiment, these therapeutic agent/secondary
carrier compositions can be a) incorporated directly into or onto a
biomaterial
implant, b) incorporated into an injectable solution, or c) incorporated into
a gel
or viscous solution, d) incorporated into the composition used for coating the
implant, or e) incorporated into or onto the implant following coating of the
implant with a coating composition.
For example, therapeutic agent-loaded PLGA microspheres may
be incorporated into a polyurethane coating solution, which is then
incorporated
into an implant. In yet another example, the implant can be coated with a
polyurethane and then allowed to partially dry such that the surface is still
tacky.
A particulate form of the fibrosis-inducing agent or fibrosis-inducing
agent/secondary carrier can then be applied to all or a portion of the tacky
coating after which the implant is dried. In addition to coating the implant
with
one or more of the therapeutic agents described herein, the agent can be mixed
with the materials that are used to make the implant such that the agent is
incorporated into the implant.
In yet another example, an implant can be coated with any one of
the coatings described above. A thermal treatment process can then be used
to soften the coating, after which the fibrosis-inducing agent or the fibrosis-
inducing agent/secondary carrier is applied to the entire implant or to a
portion
of the implant (e.g., outer surface).
In one embodiment, fibrosis-inducing agents, hemostatic agents,
and/or anti-infective agents can be incorporated directly into the formulation
to
produce a suspension or a solution (e.g., silk powder, bleomycin) or it can be
incorporated into a secondary carrier (e.g., micelles, liposomes,
micropsheres,
238
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
microparticles, nanospheres, micropaticulates, emulsions
and/or
microemulations) that is then incorporated into a bulking composition (e.g.,
fibrin, collagen, PEG, cyanoacrylate, or a mixture thereof). In another
embodiment, the therapeutic agent can be electrostatically or covalently bound
to one or more of the polymeric components of the in-situ forming composition.
In another embodiment, the therapuetic agent can be
incorporated into a bulking agent during the manufacture of the agent. For
example, silk powder can be added as a reagent during the manufacture of
microspheres that are used as bulking agents.
Within another aspect of the invention, a coated implant that
promotes an in vivo fibrotic reaction is further coated with a compound or
composition which delays the release of and/or activity of the fibrosis-
inducing
agent. Representative examples of such agents include biologically inert
materials such as gelatin, PLGA/MePEG film, PLA, polyurethanes, silicone
rubbers, surfactants, lipids, or polyethylene glycol, as well as biologically
active
materials such as heparin (e.g., to induce coagulation).
For example, in one embodiment of the invention, the active agent
on the implant is top-coated with a physical barrier. Such barriers can
include
non-degradable materials or biodegradable materials such as gelatin,
PLGA/MePEG film, PLA, polyethylene glycol, or the like. In one embodiment,
the rate of diffusion of the therapeutic agent in the barrier coat is slower
that the
rate of diffusion of the therapeutic agent in the coating layer. In the case
of
PLGA/ MePEG, once the PLGA/ MePEG becomes exposed to bodily fluids, the
MePEG can dissolve out of the PLGA, leaving channels through the PLGA to
an underlying layer containing the fibrosis-inducing agent (e.g., silk or
cyclosporine A), which can then diffuse into the vessel wall and initiate its
biological activity.
In another embodiment of the invention, for example, a particulate
form of the active agent (e.g., silk or cyclosporine A) may be coated onto the
implant using a polymer (e.g., PLG, PLA, or polyurethane). A second polymer,
239
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
that dissolves slowly or degrades (e.g., MePEG-PLGA or PLG) and that does
not contain the active agent, may be coated over the first layer. Once the top
layer dissolves or degrades, it exposes the under coating, which allows the
active agent to be exposed to the treatment site or to be released from the
coating.
Within another aspect of the invention, the outer layer of the
coated implant, which induces an in vivo fibrotic, hemostatic and/or anti-
infective response, is further treated to crosslink the outer layer of the
coating.
This can be accomplished by subjecting the coated implant to a plasma
treatment process. The degree of crosslinking and nature of the surface
modification can be altered by changing the RF power setting, the location
with
respect to the plasma, the duration of treatment as well as the gas
composition
introduced into the plasma chamber.
Protection of a biologically active surface can also be utilized by
coating the implant surface with an inert molecule that prevents access to the
active site through steric hindrance, or by coating the surface with an
inactive
form of the therapeutic agent, which is later activated. For example, the
implant
can be coated with an enzyme, which causes either release of the fibrosis-
inducing agent or activates the fibrosis-inducing agent.
In another strategy, the implant can be coated with an inactive
form of the fibrosis-inducing agent, hemostatic agent and/or anti-infective
agent,
which is then activated once the implant is deployed. Such activation may be
achieved by injecting another material into the diverticula after the implant
(as
described below) is deployed or after the fibrosis-inducing agent has been
administered to the treatment area (via, e.g., injections, spray, wash, drug
delivery catheters or balloons). For example, the implant may be coated with
an inactive form of the fibrosis-inducing agent. Once the implant is deployed,
the activating substance is injected or applied into or onto the treatment
site
where the inactive form of the fibrosis-inducing agent has been applied.
240
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
For example, a implant may be coated with a biologically active
fibrosis-inducing agent, hemostatic agent, and/or anti-infective agent in the
usual manner. The outer surface containing the active therapeutic agents(s)
may then be covered (e.g., coated) with polyethylene glycol. The PEG and the
therapeutic agent containing coating may be bonded through the formulation of
a bond between reactive groups on the two layers. For example, an ester bond
may be formed using a condensation reaction. Prior to the deployment of the
implant, an esterase is injected into the treatment site around the outside of
the
implant. The esterase can cleave the bond between the ester and the
therapeutic agent, thereby allowing the agent to initiate fibrosis (fibrosis-
inducing agent), hemostasis (hemostatic agent), and/or inhibition of bacterial
growth (anti-infective agent).
Within certain embodiments of the invention, the therapeutic
compositions may also comprise additional ingredients such as surfactants
(e.g., PLURONICS, such as F-127, L-122, L-101, L-92, L-81, and L-61), anti-
inflammatory agents, preservatives, and/or anti-oxidants.
Within certain embodiments of the invention, the therapeutic
agent or carrier can also comprise radio-opaque, echogenic materials and
magnetic resonance imaging (MRI) responsive materials (i.e., MRI contrast
agents) to aid in visualization of the implant under ultrasound, fluoroscopy
and/or MRI. For example, a therapeutic implant may be made with or coated
with a composition which is echogenic or radiopaque (e.g., made with
echogenic or radiopaque with materials such as powdered tantalum, tungsten,
barium carbonate, bismuth oxide, barium sulfate, metrazimide, iopamidol,
iohexol, iopromide, iobitridol, iomeprol, iopentol, ioversol, ioxilan,
iodixanol,
iotrolan, acetrizoic acid derivatives, diatrizoic acid derivatives, iothalamic
acid
derivatives, ioxithalamic acid derivatives, metrizoic acid derivatives,
iodamide,
lypophylic agents, iodipamide and ioglycamic acid or, by the addition of
microspheres or bubbles which present an acoustic interface). Visualization of
an implant by ultrasonic imaging may be achieved using an echogenic coating.
241
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Echogenic coatings are described in, e.g., U.S. Patent Nos. 6,106,473 and
6,610,016. For visualization under MRI, contrast agents (e.g,, gadolinium
(III)
chelates or iron oxide compounds) may be incorporated into or onto the
implant, such as, as a component in a coating or within the void volume of the
implant (e.g., within a lumen, reservoir, or within the structural material
used to
form theimplant). In some embodiments, a diverticular implant may include
radio-opaque or MRI visible markers (e.g., bands) that may be used to orient
and guide the implant during the implantation procedure.
Medical implants may, alternatively, or in addition, be visualized
under visible light, using fluorescence, or by other spectroscopic means.
Visualization agents that can be included for this purpose include dyes,
pigments, and other colored agents that can improve visualization during
endoscopic deployment. In one aspect, the medical implant may further include
a colorant to improve visualization of the implant in vivo and/or ex vivo.
Frequently, implants can be difficult to visualize upon insertion, especially
at the
margins of implant. A coloring agent can be incorporated into a diverticular
implant to reduce or eliminate the incidence or severity of this problem. The
coloring agent provides a unique color, increased contrast, or unique
fluorescence characteristics to the implant. In one aspect, a solid implant is
provided that includes a colorant such that it is readily visible (under
visible light
or using a fluorescence technique) and easily differentiated from its implant
site.
In another aspect, a colorant can be included in a liquid or semi-solid
composition. For example, a single component of a two component mixture
may be colored, such that when combined ex-vivo or in-vivo, the mixture is
sufficiently colored.
The coloring agent may be, for example, an endogenous
compound (e.g., an amino acid or vitamin) or a nutrient or food material and
may be a hydrophobic or a hydrophilic compound. Preferably, the colorant has
a very low or no toxicity at the concentration used. Also preferred are
colorants
that are safe and normally enter the body through absorption such as p-
242
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
carotene. Representative examples of colored nutrients (under visible light)
include fat soluble vitamins such as Vitamin A (yellow); water soluble
vitamins
such as Vitamin B12 (pink-red) and folic acid (yellow-orange); carotenoids
such
as p-carotene (yellow-purple) and lycopene (red). Other examples of coloring
agents include natural product (berry and fruit) extracts such as anthrocyanin
(purple) and saffron extract (dark red). The coloring agent may be a
fluorescent
or phosphorescent compound such as a-tocopherolquinol (a Vitamin E
derivative) or L-tryptophan. Derivatives, analogues, and isomers of any of the
above colored compounds also may be used. The method for incorporating a
colorant into an implant or therapeutic composition may be varied depending on
the properties of and the desired location for the colorant. For example, a
hydrophobic colorant may be selected for hydrophobic matrices. The colorant
may be incorporated into a carrier matrix, such as micelles. Further, the pH
of
the environment may be controlled to further control the color and intensity.
In one aspect, the composition of the present invention include
one or more coloring agents, also referred to as dyestuffs, which will be
present
in an effective amount to impart observable coloration to the composition,
e.g.,
the gel. Examples of coloring agents include dyes suitable for food such as
those known as F. D. & C. dyes and natural coloring agents such as grape skin
extract, beet red powder, beta carotene, annato, carmine, turmeric, paprika,
and so forth. Derivatives, analogues, and isomers of any of the above colored
compound also may be used. The method for incorporating a colorant into an
implant or therapeutic composition may be varied depending on the properties
of and the desired location for the colorant. For example, a hydrophobic
colorant may be selected for hydrophobic matrices. The colorant may be
incorporated into a carrier matrix, such as micelles. Further, the pH of the
environment may be controlled to further control the color and intensity.
In one aspect, the compositions of the present invention include
one or more preservatives or bacteriostatic agents, present in an effective
amount to preserve the composition and/or inhibit bacterial growth in the
243
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
composition, for example, bismuth tribromophenate, methyl hydroxybenzoate,
bacitracin, ethyl hydroxybenzoate, propyl hydroxybenzoate, erythromycin,
chlorocresol, benzalkonium chlorides, and the like. Additional examples of the
preservative include paraoxybenzoic acid esters, chlorobutanol, benzyl
alcohol,
phenethyl alcohol, dehydroacetic acid, sorbic acid, and the like. In one
aspect,
the compositions of the present invention include one or more bactericidal
(also
known as bacteriacidal) agents.
In one aspect, the compositions of the present invention include
one or more antioxidants, present in an effective amount. Examples of the
antioxidant include sulfites, alpha-tocopherol and ascorbic acid.
Within related aspects of the present invention, implants and
compositions are provided, which release an agent which induces fibrosis,
hemostasis and/or anti-infective activity in vivo upon deployment of the
implant
or administration of the composition. In certain aspects, the therapeutic
agent
or composition that comprises the therapeutic agent is delivered locally or
regionally to the treatment site from the composition.
Within certain aspects of the present invention, the therapeutic
composition are preferably biocompatible, and release one or more fibrosing
agents, hemostatic agents and/or anti-infective agents over a period of
several
hours, days, or months.
The implants of the present invention may be configured to
release the fibrosis-inducing agent, hemostatic agent and/or anti-infective
agent
at one or more phases, the one or more phases having similar or different
performance (e.g., release) profiles. The therapeutic agent may be made
available to the tissue at amounts which may be sustainable, intermittent, or
continuous; in one or more phases; and/or rates of delivery; effective to
increase or promote any one or more components of fibrosis (or scarring),
hemostasis or infection control.
Thus, the release rate may be programmed to impact fibrosis (or
scarring), hemostasis (clotting) and/or infection by releasing the fibrosis
244
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
inducing (scarring), hemostatic and/or anti-infective agent at a time such
that at
least one of the components of fibrosis, hemostasis and/or infection control
is
promoted or increased. Moreover, the predetermined release rate may reduce
agent loading and/or concentration as well as potentially providing minimal
drug
washout and thus, increases efficiency of drug effect. In one embodiment, the
rate of release may provide a sustainable level of the therapeutic agent to
the
treatment site. In another embodiment, the rate of release is substantially
constant. The rate may decrease and/or increase over time, and it may
optionally include a substantially non-release period. The release rate may
comprise a plurality of rates. In an embodiment, the plurality of release
rates
may include rates selected from the group consisting of substantially
constant,
decreasing, increasing, and substantially non-releasing.
The total amount of fibrosis-inducing agent, hemostatic agent,
and/or anti-infective agent made available on, in or near the diverticula may
be
in an amount ranging from about 0.01 pg (micrograms) to about 2500 mg
(milligrams). Generally, the fibrosis-inducing, hemostatic, and/or anti-
infective
agent may be in the amount ranging from 0.01 pg to about 10 pg; or from 10 pg
to about 1 mg; or from 1 mg to about 10 mg; or from 10 mg to about 100 mg; or
from 100 mg to about 500 mg; or from 500 mg to about 2500 mg.
The surface amount of fibrosis-inducing agent, hemostatic agent,
and/or anti-infective agent on, in or near the diverticula may be in an amount
ranging from less than 0.01 pg to about 250 pg per mm2 of surface area.
Generally, the therapeutic agent may be in the amount ranging from less than
0.01 pg/mm2; or from 0.01 pg to about 10 pg/mm2; or from 10 pg to about 25
pg/mm2; or from 25 pg to about 250 pg/mm2.
The fibrosis-inducing agent, hemostatic agent, and/or anti-
infective agent that is on, in or near the diverticula may be released from
the
composition and/or implant in a time period that may be measured from the
time of implantation, which ranges from about less than 1 day to about 180
days. Generally, the release time may also be from about less than 1 day to
245
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
about 7 days; from 7 days to about 14 days; from 14 days to about 28 days;
from 28 days to about 56 days; from 56 days to about 90 days; from 90 days to
about 180 days.
In one aspect, "quick release" or "burst" therapeutic compositions
are provided that release greater than 10%, 20%, or 25% (w/v) of a fibrosis-
inducing agent, hemostatic agent, and/or anti-infective agent over a period of
7
to 10 days. Such "quick release" compositions should, within certain
embodiments, be capable of releasing therapeutic levels (where applicable) of
a desired fibrosing, hemostatic, and/or anti-infective agent. Within other
embodiments, "slow release" therapeutic compositions are provided that
release less than 1% (w/v) of a fibrosis-inducing agent, hemostatic agent,
and/or anti-infective agent over a period of 7 to 10 days. Within other
embodiments therapeutic compositions are provided that release either less
than 1% (w/v) of a therapeutic agent over a period longer than 10 days or do
not release the therapeutic composition at all, but maintain the composition
for
a very long period of time such as for the entire duration of the diverticular
healing process in the body.
The amount of therapeutic agent released from the composition
and/or implant as a function of time may be determined based on the in vitro
release characteristics of the agent from the composition. The in vitro
release
rate may be determined by placing the fibrosis-inducing agent (scarring),
hemostatic agent, and/or anti-infective agent within the composition or
implant
in an appropriate buffer such as 0.1M phosphate buffer (pH 7.4)) at 37 C.
Samples of the buffer solution are then periodically removed for analysis by
either HPLC or by gravimetric means, and the buffer is replaced to avoid any
saturation effects.
Based on the in vitro release rates, the release of fibrosis-inducing
agent, hemostatic agent, and/or anti-infective agent per day may range from
about 0.0001 pg (micrograms) to about 2500 mg (milligrams). Generally, the
therapeutic agent that may be released in a day may be in the amount ranging
246
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
from 0.0001 to 0.01 pg; 0.01 pg to about 10 pg; or from 10 pg to about 1 mg;
or
from 1 mg to about 10 mg; or from 10 mg to about 100 mg; or from 100 mg to
about 500 mg; or from 500 mg to about 2500 mg. In one embodiment, the
fibrosis-inducing agent, hemostatic agent, and/or anti-infective agent is made
available to the diverticular tissue in a constant but substantially
unchanging
manner so that the agent remains at the tissue essentially permanently. In
another embodiment, the therapeutic agent is made available to the susceptible
diverticular tissue in a sustained and/or controlled manner which results in
increased efficiency and/or efficacy. Further, the release rates may vary
during
either or both of the initial and subsequent release phases. There may also be
additional phase(s) for release of the same substance(s) and/or different
substance(s).
In one embodiment, the release rate of the fibrosis-inducing
agent, hemostatic agent, and/or anti-infective agent may be classed as zero
order, first order, or a combination of zero and first order.
Further, therapeutic compositions of the present invention
preferably have a stable shelf-life for at least several months and capable of
being produced and maintained under sterile conditions. The composition may
be sterile either by preparing them under aseptic environment and/or they may
be terminally sterilized using methods available in the art. Many
pharmaceuticals are manufactured to be sterile and this criterion is defined
by
the USP XXII <1211>. The term "USP" refers to U.S. Pharmacopeia (see
www.usp.org, Rockville, MD). Sterilization may be accomplished by a number
of means accepted in the industry and listed in the USP XXII <1211>, including
gas sterilization, ionizing radiation or, when appropriate, filtration.
Sterilization
may be maintained by what is termed asceptic processing, defined also in USP
XXII <1211>. Acceptable gases used for gas sterilization include ethylene
oxide. Acceptable radiation types used for ionizing radiation methods include
gamma, for instance from a cobalt 60 source and electron beam. A typical
dose of gamma radiation is 2.5 MRad. Sterilization may also occur by
247
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
terminally using gamma radiation or electron beam sterilization methods.
Filtration may be accomplished using a filter with suitable pore size, for
example 0.22 jim and of a suitable material, for instance
polytetrafluoroethylene
(e.g., TEFLON). A combination of these methods may also be used to prepare
the composition in the sterile form.
In another aspect, the agents, compositions, and implants of the
present invention are contained in a container that allows them to be used for
their intended purpose. Properties of the container that are important are a
volume of empty space to allow for the addition of a constitution medium, such
as water or other aqueous medium, e.g., saline, acceptable light transmission
characteristics in order to prevent light energy from damaging the composition
in the container (refer to USP XXII <661>), an acceptable limit of
extractables
within the container material (refer to USP X)(II), an acceptable barrier
capacity
for moisture (refer to USP XXII <671>) or oxygen. In the case of oxygen
penetration, this may be controlled by including in the container, a positive
pressure of an inert gas, such as high purity nitrogen, or a noble gas, such
as
argon.
Typical materials used to make containers for pharmaceuticals
include USP Type I through III and Type NP glass (refer to USP XXII <661>),
polyethylene, TEFLON, silicone, and gray-butyl rubber. For parenterals, USP
Types Ito III glass and polyethylene are preferred.
In certain embodiments fibrosis inducing agents, hemostatic
agents, anti-infective agents or derivatives and analogues thereof as
described
herein, can be used to create variations of the above compositions. In
addition,
the therapeutic agent(s) may be used in a composition with a polymyer carrier
as described herein or without polymer carrier.
Other agents which may be incorporated into or onto the implant
or released from the implant include extracellular matrix components such as
fibrous structural proteins (e.g., fibrillar collagens, nonfibrillar collagen
and
elastins), adhesive glycoproteins (e.g., laminin and fibronectin),
proteoglycans
248
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
(e.g., heparin sulphate, chondroitin sulphate, dermatan sulphate), hyaluronan
(e.g., hyaluronic acid), secreted protein acidic and rich in cysteine (SPARC),
thrombospondins, tenacin, inhibitors of matrix metalloproteinases (e.g., TIMPs
and synthetic TIMPs such as marimistat, batimistat, doxycycline, tetracycline,
minocycline, TROCADE, Ro-1130830, CGS 27023A, BMS-275291) and
polylysine. Growth factors and inflammatory cytokines involved in
angiogenesis, fibroblast migration, fibroblast proliferation, ECM synthesis
and
tissue remodeling such as epidermal growth factor (EGF) family, transforming
growth factor-a (TGF- (X), transforming growth factor-3 (TGF-13-1, TGF-(3-2,
TGF-p-3), platelet-derived growth factor (PDGF), fibroblast growth factor
(acidic
¨ aFGF; and basic - bFGF), bone morphogenic proteins, activins, vascular
endothelial growth factor (VEGF, VEGF-B, VEGF-C, placental growth factor -
PIGF), angiopoietins, insulin-like growth factors (IGF), hepatocyte hrowth
factor
(HGF), connective tissue growth factor (CTGF), myeloid colony-stimulating
factors (CSFs), granulocyte-macrophage colony-stimulating factors (GM-CSF),
granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating
factor (M-CSF), erythropoietin, interleukins (particularly IL-1, IL-8, IL-6),
tumor
necrosis factor-a (TNFa), nerve growth factor (NGF), interferon-a, interferon-
13,
and growth hormone (GH) are also suitable for incorporation and release from
specific diverticular implants. Other agents which may be coated onto or
released by the diverticular implant include adhesives such as cyanoacrylate
or
materials made from 4-armed thiol PEG (10K), a 4-armed NHS PEG(10K) and
methylated collagen.
Devices for Treating or Preventing Diverticular Disease
In certain embodiments, medical implants are provided that
comprise at least one of (i) a fibrosis-inducing agent and (ii) a composition
that
comprises a fibrosis-inducing agent. Within another embodiment, a fibrosis-
inducing pharmacologic agent, an implant adapted to include or to release an
agent that induces fibrosis (through one or more of the mechanisms described
249
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
herein), an anti-infective agent, and/or a haemostatic agent are administered
onto or into diverticula. When placed within diverticula, the fibrosing agent
is
present to induce fibrosis formation that would otherwise not occur. The
fibrosis-inducing, pharmacologic agent and/or an implant/composition that
comprises a fibrosis-inducing agent (and/or an anti-infective agent and/or a
haemostatic agent) are introduced into a into a host (human or non-human
mammal) with diverticula according to methods described herein, such that the
agent causes the formation of fibrosis within the diverticula, which fibrosis
would otherwise not occur in the absence of the agent.
Also provided herein are devices for gaining access to the lumen
of the diverticula and for administering fibrosis-inducing therapy. The lumen
of
the body passageway can be accessed via an endoscope (e.g., colonoscope,
gastroscope, ERCP, bronchoscope, cystoscope etc.) or under radiographic (x-
ray, fluoroscopy, ultrasound, CT scanning, MRI scanning, PET scanning or
other imaging modality) guidance. Upon visulalizing the opening of the
diverticula into the lumen of the passageway, a variety of specialized
catheters
may be used for the delivery of the previously therapeutic agents and
compositions described herein. These devices all have lumens that enable the
localized delivery of fibrosis-inducing agents, hemostatic agents, and/or anti-
infective agents into the lumen of diverticula, which may be effected either
under direct vision (endoscopy) or radiographic guidance.
Catheters
Numerous intravascular catheters (catheter containing one or
more lumens suitable for the intravascular delivery of aqueous,
microparticulate, fluid, or gel formulations into the bloodstream, the
vascular
wall, plaque, or an aneurysm sac) may be used for direct, site-specific drug
delivery (e.g., microinjector catheters, catheters placed within or
immediately
adjacent to the target tissue), regional drug delivery (i.e., catheters placed
in an
artery that supplies the target organ or tissue), or systemic drug delivery
(i.e.,
250
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
intra-arterial and intravenous catheters placed in the peripheral
circulation). For
example, catheters and balloon catheters suitable for use can deliver
fibrosing
agents from an end orifice, through one or more side ports, through a
microporous outer structure, or through direct injection into the desired
tissue or
vascular location.
A variety of catheters are available for regional or localized drug-
delivery. Intravascular balloon and non-balloon catheters for delivering drugs
are described, for example, in U.S. Patent Nos. 5,180,366; 5,171,217;
5,049,132; 5,021,044; 6,592,568; 5,304,121; 5,295,962; 5,286,254; 5,254,089;
5,112,305; PCT Publication Nos. WO 93/08866, WO 92/11890, and WO
92/11895; and Riessen et al., JACC 23: 1234-1244 (1994); Kandarpa K. J.
Vasc. Interv. Radio. 11 (suppl.):419-423 (2000); and Yang, X. Imaging of
Vascular Gene Therapy 228:36-49 (2003).
In addition, numerous physical interventions described herein
such as the application of thermal energy (heat or cryotherapy), cautery,
electricity, RF energy, laser, radioactivity, ultrasound, balloon inflation,
physical
abrasion, etc., will have the added benefit of wounding the luminal surface of
the diverticula to initiate the healing/scarring cascade that may or may not
be
further amplified by the concurrent administration of a fibrosis-inducing
agent.
Although virtually any delivery catheter of the correct length, flexibility,
and
lumen size could be used in the delivery of therapeutic agents in and around
diverticula, the following devices are non-limiting examples of catheters that
may be used for treating diverticular disease.
Drug Delivery Catheters. A variety of drug delivery catheters may
be used, or that may be adapted for use, for delivery of an agent or
composition
comprising the agent into a host. A catheter is advanced through the side port
of the endoscope (or under radiographic guidance) until is enters the lumen of
the diverticula. The fibrosis-inducing agent or implant can then be
administered
into the lumen of the diverticula via the catheter. Examples of drug delivery
catheters include diffusion catheters, infusion catheters, and direct
injection
251
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
catheters. The drug delivery catheter may be an energy-enabled drug delivery
system, such as an electroporation, ultrasound, laser light, or other energy-
based delivery system that activates a drug and/or accelerates the delivery of
the drug into a tissue (e.g., a diverticular surface).
Representative examples of drug delivery catheters include
balloon catheters, such as the CHANNEL and TRANSPORT balloon catheters
from Boston Scientific Corporation (Natick, MA) and Stack Perfusion Coronary
Dilitation catheters from Advanced Cardiovascular Systems, Inc. (Santa Clara,
CA). Other examples of drug delivery catheters include infusion catheters,
such as the CRESCENDO coronary infusion catheter available from Cord is
Corporation (Miami Lakes, FL), the Cragg-McNamara Valved Infusion Catheter
available from Microtherapeutics, Inc. (San Clemente, CA), the DISPATCH
catheter from Boston Scientific Corporation, the GALILEO Centering Catheter
from Guidant Corporation (Houston, TX), and infusion sleeve catheters, such as
the INFUSASLEEVE catheter from LocalMed, Inc. (Sunnyvale, CA). Infusion
sleeve catheters are described in, e.g., U.S. Patent Nos. 5,318,531;
5,336,178;
5,279,565; 5,364,356; 5,772,629; 5,810,767; and 5,941,868. Catheters that
mechanically or electrically enhance drug delivery include, for example,
pressure driven catheters (e.g., needle injection catheters having injector
ports,
such as the INFILTRATOR catheter available from InterVentional Technologies,
Inc. (San Diego, CA)) (see, e.g., U.S. Patent No. 5,354,279) and
ultrasonically
assisted (phonophoresis) and iontophoresis catheters (see, e.g., Singh, J., et
al. (1989) Drug Des. DOW. 4:1-12 and U.S. Patent Nos. 5,362,309; 5,318,014;
5,315,998; 5,304,120; 5,282,785; and 5,267,985).
RE (radiofrequency) Ablation Catheters. A variety of catheters
that deliver energy may be used, or may be adapted for use, in for treating
diverticular disease. Radiofrequency ablation is a procedure by which high
frequence energy is delivered into tissue. Either of two types of RF ablation
may be used: temperature control or fluid-cooled ablation. A carefully
controlled amount of energy is delivered to the surface of the diverticulum
such
252
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
that the surface is disrupted (but not enough to cause perforation of the
diverticula) or injured enough to initiate (or stimulate) the healing
response.
Such a device may be used alone to induce fibrosis or may be used in
combination with administration of a fibrosis-inducing agent or implant into
the
lumen of the diverticula. Exemplary RF ablation catheters include catheters
manufactured by Medtronic, Minneapolis, MN (e.g., RF CONTACTR (Dual-
Curve Series; RFENHANCR (SC) Series; RF CONDUCTR (MC) Series; RF
MARINR (MC) Series; and 5F RF MARINR (SC) Series) and Boston Scientific
Corp., Natick, MA (e.g., BLAZER II XP; BLAZER II HTD; POLARIS T
temperature ablation catheter; CHILLI Cooled ablation catheter; CHILLI RPM
cooled ablation catheter; STEEROCATH-T temperature ablation catheter; and
EPT-1000 XP).
Thermal Energy Catheters. A variety of catheters that deliver
thermal energy (heat, microwaves, cryotherapy) (see, e.g., UROLOGIX
Transurethral Thermal Therapy (T3), Urologix, Inc., Minneapolis, MN; Neya et
al., Circulation 91:2445-53 (1995)) may be used, or that may be adapted for
use, for treating diverticular disease as described herein. A carefully
controlled
amount of thermal energy is delivered to the surface of the diverticulum such
that the surface is disrupted (but not enough to cause perforation of the
diverticula) or injured sufficiently to initiate (or stimulate) the healing
response.
A thermal energy catheter may be used alone to induce fibrosis or may be used
in combination with administration of a fibrosis-inducing agent or implant
into
the lumen of the diverticula. Examples of thermal energy catheters include
SPINECATH Intradiscal Catheter and AUTHERM Decompression Catheter
(Smith & Nephew, Inc., Andover, MA) and include cryoablation devices (those
that apply extremely low temperatures to tissues) such as CRYOCOR
(CryoCor, Inc., San Diego, CA); CLOSURE (VNUS Medical Technologies, Inc.,
San Jose, CA); and FREEZOR and CRYOTHERAPY (CryoCath Technologies,
Inc., Kirkland, Quebec).
253
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Laser Devices. A variety of catheters that deliver laser light
energy may be used, or may be adapted for use, for treatment of diverticular
disease. A carefully controlled amount of light energy is delivered to the
surface of the diverticulum such that the surface is disrupted, but not enough
to
cause perforation of the diverticula, or injured sufficiently to initiate
(stimulate or
promote) the healing response (see, e.g., Capon et al., Am. J. Clin. Dermatol.
4:1-12 (2003); Reddy, J. Clin. Laser Med. Surg. 22:141-50 (2004); Vladimirov
et
al., Biochemistry (Moscow) 69:81-90 (2004); Schindl, et al., J. Investig. Med.
48:312-26 (2000)). This device may be used alone to induce fibrosis, or may
be used in combination with administration of a fibrosis-inducing agent or
implant into the lumen of the diverticula. Examples of catheters that deliver
laser energy include CLIRPATH (Spectranetics, Corp., Colorado Springs, CO)
and Trimedyne laser catheter (Trimedyne, Inc., Irvine, CA; see, e.g., U.S.
Patent No. 5,496,309); an argon or Nd:YAG laser (see, e.g., Hunter, Surg.
Clin.
North Am. 69(6):1147-66 (1989).
Radioactivity and Brachytherapy Devices. A variety of catheters
and implants that deliver radioactive energy may be used, or that may be
adapted for use, for treating diverticular disease. A carefully controlled
amount
of radioactivity energy is delivered to the surface of the diverticulum such
that
the surface is disrupted (but not enough to cause perforation of the
diverticula)
or injured sufficiently to initiate (or stimulate or promote) the healing
response.
This intervention can be used alone to induce fibrosis, or in combination with
administration of a fibrosis-inducing agent or implant into the lumen of the
diverticula. Radioactivity at low doses stimulates the healing and fibrosis
response (see, e.g., Vladimirov et al., supra; Hill et al., mt. J. Radiat
Oncol.
Biol. Phys. 49:353-65 (2001); Rodemann et al., Radiother. Oncol. 35:83-90
(1995); O'Sullivan et al., Semin. Radiat. Oncoi. 13:274-89 (2003); Schindl, et
al., supra); Conlon et al., J. Clin. Periodontot 23:492-96 (1996), making
permanent and degradable local sources of radiation particularly useful for
the
practice of this embodiment.
254
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Any one of several radionuclides used for intravascular
brachytherapy may be applied to diverticula (see, e.g., Nath et al.,
Cardiovasc.
Radiat. Med. 5:88-96 (2004); von Neumann-Cosel, Phys. Med. Biol. 48:1855-62
(2003); Lehmann et al., J. App!. Clin. Med. Phys. 4:58-63 (2003); Yue et al.,
Cardiovasc. Radiat. Med. 5:142-50 (2004)). Systems and catheters for
delivering radioactivity to a tissue are available to persons skilled in the
art (see,
e.g., Guglielmi detachable coils (Qureshi et al., Neurosurg. Focus 10: Preview
of J. Neurosurg. 94:880-85 (2001)); wire source, balloon source (see, e.g.,
Lehmann et al., supra); balloon catheter as described herein). Commercially
available products that may be used to deliver therapeutic compositions to a
diverticulum include beta radiation catheters such as the BETA-CATH System
from Novoste Corporation (Atlanta, GA); the GALILEO lntravascular
Radiotherapy System from Guidant Corporation (Indianapolis, IN); and gamma
radiation catheters such as the CHECKMATE System from Johnson &Johnson
Corporation (New Brunswick, NJ). Other radioactivity and brachytherapy
devices that may be used or adapted for use according to the methods
described herein include the GammaMedplus from Varian Medical Systems
(Charlottesville, VA), which is used in the MAMMOSITE RTS device from North
Shore Medical Accelerator, PC, Smithtown, NY) and brachytherapy seeds for
implantation such as the I-PLANT High Activity 125Iodine Seeds (Implant
Sciences Corporation, Wakefield, MA).
Balloon Catheters. A variety of balloon catheters may be used for
treatment of diverticular disease. A balloon catheter is delivered into the
lumen
of the diverticulum and carefully expanded such that the surface of the
diverticulum is disrupted (but not enough to cause perforation of the
diverticula)
or injured enough to initiate (or stimulate) the healing response. This
intervention can be used alone to induce fibrosis, or in combination with
administration of a fibrosis-inducing agent or implant into the lumen of the
diverticula. Representative examples of drug delivery catheters include
balloon
catheters, such as the CHANNEL and TRANSPORT balloon catheters from
255
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Boston Scientific Corporation (Natick, MA) and Stack Perfusion Coronary
Dilitation catheters from Advanced Cardiovascular Systems, Inc. (Santa Clara,
CA).
The balloon catheter systems that can be used include systems in
which the balloon can be inflated at the desired location where the desired
fibrosis-inducing agents can be delivered through holes that are located in
the
balloon wall. Other balloon catheters that can be used include systems that
have a plurality of holes that are located between two balloons. The system
can be guided into the desired location such that the inflatable balloon
components are located on either side of the specific site that is to be
treated.
The balloons can then be inflated to isolate the treatment area. The
compositions containing the fibrosing agent are then injected into the
isolated
area through the plurality of holes between the two balloons. Representative
examples of these types of drug delivery balloons are described in U.S.
Patent.
Nos. 5,087,244; 6,623,452; 5,397,307; 4,636,195; and 4,994,033.
Ultrasonic Catheters. A variety of catheters that deliver ultrasonic
energy may be used for treatment of diverticular disease. A carefully
controlled
amount of sound energy is delivered to the surface of the diverticulum such
that
the surface is disrupted (but not enough to cause perforation of the
diverticula)
or injured sufficiently to initiate (or stimulate) the healing response. This
intervention can be used alone to induce fibrosis, or in combination with
administration of a fibrosis-inducing agent or implant into the lumen of the
diverticula. Examples of ultrasonic catheters and systems that may be used
include but are not limited to those described by Martin et al., IEEE
Transactions on Sonics and Ultrasonics Supp.27:277 (1980); Gage Applied
Technologies, Lachine, Quebec (e.g., COMPUSCOPE (CS) 8500); U.S. Patent
No. 4,602,633; U.S. Patent No. 4,692,139; Atar et al., Echocardiography¨ J.
Cardiovasc. Ultrasound Allied Tech. 18:233-37 (2001); Horiuchi et al., J.
Endourology 19:130-32 (2005); Dick et al., Investigative Radiology 33:85-90
(19985); (see also, e.g., Rasor Consulting Group, Los Gatos, CA.). See also,
256
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
e.g., U.S. Patent Nos. 6,723,064; 6,689,086; 6,623,444; 6,592,520; 6,296,619;
and 6,527,759. Representative examples of ultrasonic catheters include IVUS
catheters, diagnostic catheters, and delivery catheters, and commercially
available ultrasonic catheters such as the ACUNAV catheter from Siemens, the
ViewMate Catheter from EPMedSystems, and the EAGLE EYE GOLD catheter
from Volcano Therapeutics.
Physical Abrasion/Denudation of the Diverticular Surface. A
variety of catheters that abraid the surface of the tissues they contact may
be
used for treatment of diverticular disease. The surface of the diverticulum is
mechanically disrupted such that the surface is abraded (but not enough to
cause perforation of the diverticula) or injured sufficiently to initiate (or
stimulate
or promote) the healing response. This intervention can be used on its own to
induce fibrosis, or in combination with administration of a fibrosis-inducing
agent or implant into the lumen of the diverticula. An example of such a
device
is a rotoblade, which, is a high-speed spinning device. Atherectomy is a
procedure that removes plaque from the arteries supplying blood to the heart
muscle. Devices used for atherectory include a laser catheter or a rotating
shaver ("burr" device on the end of a catheter), which may be used or adapted
for use for treatment of a diverticular disease (see also, e.g., dissectional
catheterectomy catheters that shave off tissue).
Representative examples of devices for abrading diverticular
surfaces include those used in atherectomy procedures. An atherectomy
device typically has a small mechanically driven tool that either shaves,
cuts, or
breaks a tissue (typically a fatty deposit such as a plaque) into small
particles to
remove the blockage from the inside of a blood vessel (e.g. an artery). After
being shaved from the artery wall, this plaque is stored safely in the tip of
the
catheter and removed from the body. Atherectomy devices for vascular
application may be used as is or be adapted or modified for use in the present
invention.
257
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
In another embodiment, the atherectomy device may be a
rotational or directional atherectomy device. Rotational atherectomy is a
minimally invasive treatment that is sometimes used to pulverize hardened
plaque within a coronary artery. During rotational atherectomy, a high-speed
rotating device (referred to as a a "rotoblade" or "rotablator") that connects
to
the end of a catheter is used to grind away material at the treatment site. In
some embodiments, an atherectomy catheter may include a rotating cutter and
a collecting chamber for debris.
Several rotational and directional atherectomy devices are
commercially available. For example, rotational atherectomy devices include
the ROTOBLATOR Rotational Atherectomy System from Boston Scientific
Corporation (Natick MA) and the directional atherectomy devices such as the
FLEXI-CUT and FX miniRAIL cutting balloon devices from Guidant Corporation.
In one aspect, the atherectomy device may be an atherectomy
catheter. The atherectomy catheter may be, for example, a 9-F Simpson
directional atherectomy catheter (see, e.g., Zeller et al., Endovasc. Thor.
11(6):676-85 (2004) or Schechter et al., J. Vasc. Interv. Radio!. (1993)
4(6):819-24.
Representative example of atherectomy catheters and devices
which may be used or adapted for use in the practice of the methods described
herein include, without limitation, those described in U.S. Patent Nos. 5,
087,265; 5,423,846; 6,001,112; 5,865,844; 5,156,610; 5,356,418; 6,482,216;
5,312,427; 6,428õ551; 5,030,201; 6,503,261, and 6,596,005.
Exemplary devices that may be used or adapted for use to abrade
and/or denude a diverticular surface include commercially available products
such as the SILVERHAWK plaque excision device from FoxHollow
Technologies, Inc. (Redwood City, CA), laser catheters such as the CLIRPATH
line of excinrier laser catheters from Spectranetics (Colorado Springs, CO)
and
the atherectomy devices from ev3 Inc., Johnson & Johnson/Cordis Corporation,
Pathway Medical Technologies, Possis Medical Inc., Lumend (Redwood City,
258
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
CA), Intraluminal Therapeutics (Carlsbad, CA), Flowcardia (Sunnyvale, CA),
Corazon Technologies (Menlo Park, CA), and Medtronic Inc.(Minneapolis, MN).
Another method for abrading a diverticular surface is via an
"endoscopic scalpel." Endoscopic instrumentation and methods are described
in, e.g., International Application Publication Nos. WO 96/25107A1, WO
04/064623A2, WO 01/06943A1, WO 03/096871A2, and U.S. Patent Nos.
6,482,219; 6,277,135; 6,165,184; and 4,539,976.
Additional examples of devices that may be used to abrade the
diverticular tissue surface are described in International Application
Publication
Nos. WO 01/89370A2, and WO 01/166018A1.
In another embodiment, an endoluminal paving device may be
used to physically abrade or denude a diverticular surface. Endoluminal paving
devices may be used, for example, to inject into a diverticulum via a catheter
a
polymer which is in the form of a liquid, but hardens at body temperature to
seal
the diverticular cavity. The polymer may be, for example, a gel, such as a
hydrogel, or another type of polymer adapted for delivering a fibrosing agent
to
a diverticulum. Various endoluminal paving compositions and procedures are
described (see, e.g., Slepian M.J., Semin. Intent. CardioL 1996 Mar,1(1):103-
16; Slepian MJ., Cardiol. Clin. 1994 Nov;12(4):715-37; International
Application
Publication Nos. WO 90/01969A1, WO 04/087065A2, WO 91/12846A1; and
U.S. Patent Nos. 5,749,922; 5,328,471; and 5,749,915. Although versions of
the above catheters have been describe for the practice of other therapeutic
modalities, modifying these devices for use in gaining access to, and
delivering
treatment to, diverticula is an embodiment contemplated herein. In many cases
these interventions alone may be sufficient or can be used in combination with
a fibrosis-inducing agent, hemostatic agent, and/or anti-infective agent.
In additional embodiments, for each of the aforementioned
devices combined with each of the aforementioned agents, it is, for each
combination, independently disclosed that the agent may be present in a
composition along with a polymer. In one embodiment, the polymer is
259
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
biodegradable. In another embodiment, the polymer is non-biodegradable.
Other features and characteristics of the polymer, which may be included with
every combination of device and agent described above, are set forth in
greater
detail herein.
In addition to implants and fibrosis-inducing compositions for the
treatment of diverticular disease, methods are also provided. For example, for
each of the aforementioned implants, and for each possible combinations of an
implant with a fibrosis-inducing agent, methods are provided wherein a
specified implant is introduced into an animal (a human (patient or subject)
or
non-human mammal, which includes but is not limited to a rabbit, rat, mouse,
hamster, dog, non-human primate, cat, goat, pig, sheep, goat, horse, bovine),
advanced to the site of a diverticula, and a specified physical intervention
(physical disruption of the luminal surface of the diverticula as described
previously), and/or administration of a fibrosis-inducing agent or composition
induces fibrosis within the diverticula that would otherwise not occur. Each
of
the implants identified herein may be a "specified implant."
In certain embodiments, a medical implant may be modified by
attaching fibers (threads) to the surface of the implant. The fibers may be
polymeric and/or may be formed of or coated with a fibrosing material, such as
silk. For example, the threads may be formed from a silk suture material. The
presence of the threads can result in an enhanced cellular and/or
extracellular
matrix response to the exterior of the implant. The threads can be attached to
the implant by using any one or a combination of the following methods,
including use of an adhesive, thermal welding, stitching, wrapping, weaving,
knotting, and the like. The threads can be coated with a material that delays
the time it takes for the thread material to come into contact with the
surrounding tissue and blood, thus allowing placement of the implant without
concern of thrombotic events due to the presence of the polymeric threads.
Examples of materials that can be used to prepare coatings capable of
degrading or dissolving upon implantation include gelatin, polyesters (e.g.,
260
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
PLGA, PLA, MePEG-PLGA, PLGA-PEG-PLGA, and blends thereof), lipids, fatty
acids, sugar esters, nucleic acid esters, polyanhydrides, polyorthoesters, and
PVA. The coating may further contain a fibrosing agent and/or a biologically
active agent that may, for example, reduce the probability of an immediate
thrombotic event (e.g., heparin, hydrophobic quaternary amine heparin
complexes, and the like). In addition to the polymeric threads, all or a
portion of
the implant may be coated with a polymeric carrier that contains a fibrosis-
inducing agent.
The fibers (threads) may further comprise a coating or
composition that is affected by an applied magnetic field. For example, an
implant may be coated with polymeric threads that are coated, contain, or are
formed from a fibrosing agent (e.g., silk suture). A magnetic field can be
applied to the coated implant to orient and align the polymeric fibers
relative to
each other and the surface of the implant to increase the surface area of the
fibers exposed to biological mediators that would stimulate a fibrotic
reaction.
The magnetically active component can be associated with the polymeric fiber
using a variety of methods. The magnetically active component may be
incorporated during manufacture of the fiber, for example, by incorporating a
magnetically active material such as magnetite into a polymer feed prior to
extrusion of the polymeric fiber. The magnetically active component can be
coated onto the entire fiber or a portion of the fiber using, for example, an
adhesive or a polymeric coating. The polymeric fiber (or a portion thereof)
can
be heated or plasticized with a solvent and then rolled in the magnetically
active
component, such that the magnetic material protrudes above the surface of the
fiber or is embedded into the surface of the fiber.
The threads (either with or without a magnetic component) may
be attached to the implant in various configurations that can result in either
partial or complete coverage of the exterior of the implant. The polymeric
threads may be affixed to the ends of an implant or to the central portion of
an
261
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
implant, and the attachment may be in a vertical, horizontal, or diagonal
manner.
Systemic, Regional and Local Delivery of Fibrosis-Inducing Agents
A variety of drug-delivery technologies are available for systemic,
regional, and local delivery of therapeutic agents. Several of these
techniques
may be suitable to achieve preferentially elevated levels of fibrosis-inducing
agents in the vicinity of the medical implant, including (a) using drug-
delivery
catheters for local, regional, or systemic delivery of fibrosing agents to the
tissue surrounding the implant (typically, drug delivery catheters are
advanced
through the circulation or inserted directly into tissues under endoscopic or
radiographic guidance until they reach the desired anatomical location; the
fibrosing agent can then be released from the catheter lumen in high local
concentrations in order to deliver therapeutic doses of the drug to the tissue
surrounding the implant); (b) drug localization techniques such as magnetic,
ultrasonic, or MRI-guided drug delivery; (c) chemical modification of the
fibrosis-
inducing drug or formulation designed to increase uptake of the agent into
damaged tissues (e.g., modification of the drug or formulation to include
antibodies directed against damaged or healing tissue components such as
macrophages, neutrophils, smooth muscle cells, fibroblasts, extracellular
matrix
components, neovascular tissue); (d) chemical modification of the fibrosis-
inducing drug or formulation designed to localize the drug to areas of
bleeding
or disrupted vasculature such as encapsulation of the drug into site directed
liposomes; and/or (e) direct injection of the fibrosis-inducing agent, for
example
under endoscopic vision.
Infiltration of Fibrosis-Inducing Agents into the Tissue Surrounding an
Implant
Alternatively, the tissue cavity into which the implant is placed can
be treated with a fibrosis-inducing agent prior to, during, or after the
implantation procedure. This can be accomplished in several ways including
262
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
(a) topical application of the fibrosing agent into the anatomical space where
the implant can be placed (particularly useful for this embodiment is the use
of
polymeric carriers which release the fibrosing agent over a period ranging
from
several hours to several weeks; fluids, suspensions, emulsions,
microemulsions, microspheres, pastes, gels, microparticulates, sprays,
aerosols, solid implants and other formulations which release a fibrosing
agent
can be delivered into the region where the implant can be inserted via
specialized delivery catheters or other applicators); (b) microparticulate
silk
and/or silk strands (e.g., linear, branched, and/or coiled) are also useful
for
directed delivery into the implantation site; (c) sprayable collagen-
containing
formulations such as COSTASIS (Angiotech Pharmaceuticals, Inc., Vancouver,
BC) or materials made from 4-armed thiol PEG (10K), a 4-armed NHS
PEG(10K) and methylated collagen (described herein), or materials made from
4-armed thiol PEG (10K), a 4-armed NHS PEG(10K) and collagen or gelatin,
either alone, or loaded with a fibrosis-inducing agent, applied to the
implantation site (or the implant surface); (d) sprayable in situ forming PEG-
containing formulations such as COSEAL (Angiotech Pharmaceuticals, Inc.,
Canada), FOCALSEAL (Genzyme Corporation, Cambridge, MA), SPRAYGEL
or DURASEAL (both from Confluent Surgical, Inc., Waltham, MA), either alone,
or loaded with a fibrosis-inducing agent, applied to the implantation site (or
the
implant surface); (e) fibrinogen-containing formulations such as FLOSEAL or
TISSEAL (both from Baxter Healthcare Corporation; Fremont, CA), either
alone, or loaded with a fibrosis-inducing agent, applied to the implantation
site
(or the implant surface); (f) hyaluronic acid-containing formulations (either
non-
crosslinked, crosslinked or chemically modified) such as PERLANE or
RESTYLANE (both from Q-Med AB, Sweden), HYLAFORM (Inamed
Corporation; Santa Barbara, CA), SYNVISC (Biomatrix, Inc.; Ridgefied, NJ),
SEPRAFILM or SEPRACOAT (both from Genzyme Corporation; Cambridge,
MA) loaded with a fibrosis with a fibrosis-inducing agent applied to the
implantation site (or the implant surface); (g) polymeric gels for surgical
263
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
implantation such as REPEL (Life Medical Sciences, Inc.; Princeton, NJ) or
FLOWGEL (Baxter Healthcare Corporation, Deerfield, IL) loaded with a fibrosis-
inducing agent applied to the implantation site (or the implant surface); (h)
orthopedic "cements" used to hold prostheses and tissues in place loaded with
a fibrosis-inducing agent applied to the implantation site (or the implant
surface), such as OSTEOBOND (Zimmer, Inc., Warsaw, IN), LVC (Wright
Medical Technology, Inc., Arlington, TN), SIMPLEX P (Stryker Corporation,
Kalamazoo, MI), PALACOS (Smith & Nephew PLC Corporation, United
Kingdom), and ENDURANCE (Johnson & Johnson, Inc., New Brunswick, NJ);
(i) surgical adhesives containing one or more cyanoacrylate monomers (e.g.,
methyl cyanoacrylate, ethyl cyanoacrylate, butyl cyanoacrylate, octyl
cyanoacrylate, methoxypropyl cyanoacrylate) such as DERMABOND (Johnson
& Johnson, Inc.), INDERMIL (United States Surgical; Norwalk, CT),
GLUSTITCH (Blacklock Medical Products, Inc., Canada) or TISSUMEND II
(Veterinary Products Laboratories; Phoenix, AZ), VETBOND (3M Company; St.
Paul, MN), TISSUEMEND (TEI Biosciences, Inc.; Boston, MA), HISTOACRYL
or HISTOACRYL BLUE (Davis & Geck; St. Louis, MO) and ORABASE
SOOTHE-N-SEAL LIQUID PROTECTANT (Colgate-Palmolive Company; New
York; NY), either alone, or loaded with a fibrosis-inducing agent, applied to
the
implantation site (or the implant surface); (j) implants containing
hydroxyapatite
(or synthetic bone material such as calcium sulfate, VITOSS and CORTOSS
(both from Orthovita, Inc., Malvern, PA)) loaded with a fibrosis-inducing
agent
applied to the implantation site (or the implant surface); (k) other
biocompatible
tissue fillers loaded with a fibrosis-inducing agent, such as those made by
BioCure, Inc. (Norcross, GA), 3M Company and Neomend, Inc. (Sunnyvale,
CA), loaded with a fibrosis-inducing agent applied to the implantation site
(or
the implant surface); (I) polysaccharide gels such as the ADCON series of gels
(Gliatech, Inc.; Cleveland, OH); (m) films, sponges or meshes such as
INTERCEED or VICRYL mesh (Ethicon, Inc., a Johnson & Johnson Company,
Somerville, NJ), and GELFOAM (Pharmacia & Upjohn Company; Kalamazoo,
264
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
MI) loaded with a fibrosis-inducing agent applied to the implantation site (or
the
implant surface); (n) a hydrogel that is formed from an amino-functionalized
polyethylene glycol (e.g., 4-armed tetra-amino PEG [10k]) and a 4-armed NHS
functionalized PEG (e.g., pentaerythritol poly(ethylene glycol)ether tetra-
succinimidyl glutarate [101q). This hydrogel may further contain collagen,
methylated collagen and/or gelatin. This hydrogel can further comprise the
fibrosis-inducing agents described above (e.g., silk powder or silk threads),
and
(o) compositions that enhance osteointegration and/or osteogenesis, including
materials composed of beta -tricalcium phosphate (e.g., VITOSS,
PROOSTEON 500R made by E-Interpore-Cross International), hydroxyapatite
or Caio(PO4)60H (e.g., OSTEOGRAF made by Ceramed Denta, Inc.,
Lakewood, CO), calcium carbonate or CaCO3, calcium sulfate (e.g.,
OSTEOSET and ALLOMATRIX made by Wright Medical Technology, Inc.),
calcium phosphate (e.g., CALCIBON made by Merck & Co., Inc., Whitehouse
Station, NJ, NORIAN SRS made by Synthes-Strates, Switzerland), as well as
synthetic bone fillers (e.g., CORTOSS and processed bone fillers, e.g.,
BIOOSS made by Geistlich Biomaterials, Inc., Switzerland). Representative
examples of these materials are described in U.S. Patent Nos. 3,929,971,
4,861,733; 6,527,810; 4,772,468; 4,882,149; 5,167,961; 6,576,015; 4,839,215;
5,614,206; 5,807,567; 6,030,636; 6,652,887; 6,206,957; 6,485,754; 4,347,234;
4,291,013; 5,129,905; 5,336,264; 5,569,442; 5,571,493; 5,683,667; 5,709,742;
5,820,632; 5,658,332; 5,681,872; 5,914,356; 5,939,039; 6,325,987; 6,383,519;
6,458,162; 6,736,799; 6,521,246; and 6,709,744.
In one embodiment, the fibrosis-inducing agent may be delivered
as a solution. The fibrosis-inducing agent can be incorporated directly into
the
solution to provide a homogeneous solution or dispersion. In certain
embodiments, the solution is an aqueous solution. The aqueous solution may
further include buffer salts, as well as viscosity modifying agents (e.g.,
hyaluronic acid, alginates, CMC, and the like). In another aspect of the
265
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
invention, the solution can include a biocompatible solvent, such as ethanol,
DMSO, glycerol, PEG-200, PEG-300 or NMP.
In certain embodiments, a method for inducing fibrosis within a
diverticulm for treating a diverticular disease comprises irrigation (lavage,
cleansing) of the diverticulum and surrounding tissues. Prior to introducing a
fibrosing agent, composition, or implant, the diverticulum may be irrigated to
cleanse or remove particulate matter, cells, tissues, fecal matter (for GI
diverticulum), microorganisms, and the like, which are undesirable to retain
in a
diverticulum that is being sealed or closed. Such methods for irrigating,
cleansing, and rinsing tissue or organs are routinely practiced by persons
skilled in the medical art in preparation of a site for a medical procedure.
Any
one or all solutions used for irrigating a diverticulum may contain one or
more
anti-infective agents, such as an antibiotic, antiseptic, or other anti-
infective
agent described herein and used by a person skilled in the art. Cleansing or
irrigating a diverticulum with an anti-infective agent acts to prevent or
treat an
infection and/or to minimize dissemination of microorganisms. One or more
solutions may be applied in one or more irrigation steps, for example, which
steps may include washing to remove undesired matter, disinfecting the site,
and rinsing with a solution, the composition of which is the same as the
composition for delivering the fibrosing agent, composition, or implant.
In certain embodiments, a haemostatic agent or composition
comprising a haemostatic agent (e.g., COSTASIS) or sealant, may also be
included in an irrigating solution. A polymer or carrier polymer may be
combined with a haemostatic agent. A haemostatic agent or a composition
comprising a haemostatic agent, which may also include a polymer or carrier
polymer, may be applied to the diverticulum or tissue surrounding the
diverticulum to control bleeding at the site of the diverticulum and to induce
scarring.
Also as described in greater detail herein, methods for inducing
fibrosis in the diverticulum and/or treating a host who has a diverticular
disease
266
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
may also include a diagnostic step. To determine or confirm the presence of a
diverticulum in a host, the site within the body is examined by methods such
as
endoscopy or radiographic analysis. Endoscopy permits direct visualization of
the target tissue to confirm the presence of a diverticum and to guide
accurate
placement of an implant or composition. Endoscopes are used to allow direct
visualization in a minimally invasive fashion (i.e., not requiring open
surgery) by
inserting a small camera into the body via an orifice (mouth, anus) or a small
incision. Any endoscopic technology may be used but a particular endoscope
may be better suited for a particular tissue or site within the body. Examples
of
endoscopes include flexible endoscopes, rigid endoscopes, gastroscopes,
ERCP, bronchoscopes, proctoscopes, angioscopes, and colonoscopes.
Alternatively, diagnosis may include radiographic analysis.
Imaging technology is used to allow manipulation and intervention in a
minimally invasive fashion (i.e., not requiring open surgery). Several imaging
technologies are available for such use in the methods described herein. The
choice of technology may be determined in part on the tissue being treated.
Representative examples of imaging technology include x-ray, angiography,
MRI, CT scanning, ultrasound, PET scanning, and nuclear medicine scanning.
Methods for Treating or Preventing Diverticular Disease
Methods are also provided herein for treatment of diverticular
disease. Accordingly, for each of the aforementioned implants and for each of
the aforementioned combinations of the implants, compositions, or implants
with the fibrosis-inducing agents, methods are provided whereby a specified
implant is introduced into a mammal, and advanced to the site of a
diverticula.
A specified physical intervention (physical disruption of the luminal surface
of
the diverticula as described herein) and/or administration of a fibrosis-
inducing
agent or composition within the diverticula may induce fibrosis that would
otherwise not occur.
267
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Also provided herein are methods for treating patients undergoing
surgical, endoscopic or minimally invasive therapies where a medical implant
is
placed as part of the procedure. As described herein, it should be understood
that "induces fibrosis" refers to a statistically significant increase (or
clinically
significant increase) in the amount of scar tissue around the implant or an
improvement in the incorporation of the implant into the surrounding tissue
and
not to a permanent prohibition of any complications or failures of the
implant.
Diagnosis of diverticular disease may be made by barium enema,
which can outline radiographically the extent and severity of the diverticula.
Typical radiological findings are sacculations with retained contrast and
colonic
spasm. Often the entire colon is affected, but the left and the sigmoid colon
is
most often affected. However, most bleeding diverticula occur on the right
side
of the body. Endoscopic examination can rule out concomitant lesions, and
rigid sigmoidoscopy usually does not advance past the rectosigmoid junction.
Colonoscopy allows for the differentiation from diverticular diseaseas,
angiodysplasia or carcinoma. It also allows for direct visualization of the
active
bleeding site, which is ultimately found in up to 85% of patients with this
technique. Colonoscopic interventions allow for direct visualization of the
bleeding site and electrocoagulation of the site or irrigation with
epinepherine. It
also allows for the identification of other concominant disease.
The compositions and implants provided herein may be delivered
to the site of a diverticulum (e.g., via a catheter inserted through the
sideport of
a colonoscope for placement in the diverticulum) to treat infection, induce
hemostasis, and/or to permanently to scar the diverticulum shut. The
compositions may be delivered into the diverticulum directly or into tissue
surrounding the diverticulum (e.g., by injection or via a catheter). In one
embodiment, a fibrosing agent and/or an anti-infective agent may be combined
with a haemostatic agent or loaded into a haemostatic composition to arrest
bleeding or scar the diverticula shut. Representative examples of haemostatic
agents and haemostatic compositions are described herein and include
268
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
polymers (carrier polymers) e.g., CT3 (a composition comprising electrophilic
and nucleophilic PEG derivatives and methylated collagen from Angiotech
BioMaterials Corp., Palo Alto, CA) and commercially available surgical
sealants, such as sprayable collagen-containing formulations, which include
COSTASIS (Angiotech BioMaterials Corp.); sprayable PEG-containing
formulations such as COSEAL or ADHIBIT (Angiotech BioMaterials Corp.); and
fibrinogen-containing formulations such as FLOSEAL or TISSEAL (both from
Baxter Healthcare Corporation, Fremont, CA); GELFOAM (Pharmacia & Upjohn
Company, Kalamazoo, MI); and AVITINE (CR Bard, Inc., Plano, TX).
In one embodiment, the compositions may be in the form of
sustained-release preparations (e.g., injectable compositions, sprays, and
gels). Applicators and kits including the instant compositions for use in the
treatment of diverticulitis also are provided. In another embodiment, the
fibrosis-inducing agent and/or infective agent can be added to a cyanoacrylate
adhesive that is then delivered to the diverticuli (such as described herein
via a
catheter using a colonoscope.
In certain embodiments, a fibrosis-inducing (scarring) agent,
haemostatic agent, and/or infective agent (and compositions comprising one or
more of these agents) can be included in a polymeric sealant spray, which
solidifies into a film or coating to promote fibrosis and seal diverticuli.
The
spray, which includes a tissue adherent polymer containing a fibrosis-inducing
agent, haemostatic agent, and/or infective agent may be prepared from
microspheres as described above.
Numerous polymeric and non-polymeric carrier systems that can
be used are described herein. These compositions can further comprise one or
more fibrosis-inducing agents to promote the formation of granulation tissue.
These compositions can further comprise one or more hemostatic agents to
promote clotting and/or one or more anti-infective agents to prevent
infection.
For the in situ forming compositions, one or more fibrosis-inducing agent,
haemostatic agent, and/or infective agent can be incorporated directly into
the
269
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
formulation to produced a suspension or a solution (e.g., silk powder,
bleomycin) or the agent(s) can be incorporated into a secondary carrier (e.g.,
micelles, liposomes, micropsheres, microparticles, nanospheres,
micropaticulates, emulsions and/or microemulations) that are then incorporated
into the in situ-forming compositions. In another embodiment, the fibrosis-
inducing agent, haemostatic agent, and/or infective agent can be
electrostatically or covalently bound to one or more of the polymeric
components of the in situ-forming composition.
In another embodiment, the fibrosis-inducing agent, haemostatic
agent, and/or infective agent can be incorporated directly or via a secondary
carrier into a gel or thermogel (e.g,, hyaluronic acid, PLURONIC F127,
polyester-PEG-polyester [PLGA-PEG-PLGA}. These gels can be applied to the
treatment site prior or after the application of a sealant.
In another embodiment, the fibrosis-inducing agent, haemostatic
agent, and/or infective agent can be incorporated into a biodegradable or
dissolvable film or mesh that is then applied to the treatment site. An in
situ-
forming composition can then be sprayed over the film, thereby sealing the
diverticulum and the film or mesh to the treatment site.
In another embodiment, the in situ sealant can be applied to the
diverticuli after which a biodegradable or dissolvable film or mesh that
comprises a fibrosis-inducing agent, haemostatic agent, and/or infective agent
is applied to the treatment area. Exemplary materials for the manufacture of
these films or meshes are hyaluronic acid (crosslinked or non-crosslinked),
cellulose derivatives (e.g., hydroxypropyl cellulose), and crosslinked
poly(ethylene glycol).
In another embodiment, the fibrosis-inducing agent, haemostatic
agent, and/or infective agent can be an injectable form that can be injected
directly into the diverticulum (treatment site) or tissue surrounding the
treatment
site. The fibrosis-inducing agent, haemostatic agent, and/or infective agent
can
be incorporated directly into the formulation to produce a suspension or a
270
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
solution (e.g., silk powder, or bleomycin) or it can be incorporated into a
secondary carrier (e.g., micelles, liposomes, micropsheres, microparticles,
nanospheres, micropaticulates, emulsions and/or microemulations) that is then
incorporated into the in situ-forming compositions. In another embodiment, the
fibrosis-inducing agent, haemostatic agent, and/or infective agent can be
electrostatically or covalently bound to one or more of the polymeric
components of the in-situ forming composition. These injectable compositions
can further comprise a polymer to enhance the viscosity of the solution.
Polymers that can be used include hyaluronic acid, CMC, PLURONICs F127 as
well as gels (normal and therm gels) of the form X-Y, X-Y-X, or Y-X-Y where X
is a degradable polyester and Y is a polyalkylene oxide, prefereably
polyethylene glycol or the mono-methyl ether thereof. In another embodiment,
the injectable formulation can further comprise a biocompatible solvent. These
can include ethanol, DMSO, NMP, poly(ethylene glycol)-200, and/or
poly(ethylene glycol)-300.
In another embodiment, the fibrosis-inducing agent may be in the
form of a fiber, braid, coil, thread, monofilament fiber, or multifilament
fiber that
is introduced directly into the treatment site via a catheter or an endoscopic
delivery device. For example, a silk braid can be delivered directly into the
diverticulum.
In another embodiment, the fibrosis-inducing agent can be coated
onto or incorporated into a polymeric fiber, braid, coil, thread, monofilament
fiber, or multifilament fiber that is introduced directly into the treatment
site via a
catheter or an endoscopic delivery device. For example, a silk fiber can be
incorporated into a Dacron braid that is then delivered directly into the
diverticulum.
As described herein, potentially any adhesion or fibrosis-inducing
agent described herein may be used alone, or in combination, in the practice
of
this embodiment. Exemplary fibrosing agents for use in compositions for
treating diverticultis include talc, silk, chitosan, polylysine, fibroncectin,
271
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
bleomycin, and CTFG, as well as analogues and derivatives of the
aforementioned. Also as described herein, any fibrosis-inducing agent may be
used with any of the numerous anti-infective agents known in the art and
described herein, and any fibrosis-inducing agent may be used with any of the
numerous haemostatic agents known in the art and described in detail herein.
Any haemostatic agent may also be used with any anti-infective agent.
A variety of compositions and implants are described for the
treatment of diverticulitis. As implants (e.g., compositions and devices) are
made in a variety of configurations, forms, and sizes, the exact dose
administered will vary with implant size, surface area, and design. However,
certain principles can be applied in the application of this art. Drug dose
can be
calculated as a function of dose per unit area (of the portion of the implant
being coated), total drug dose administered can be measured and appropriate
surface concentrations of active drug can be determined. Regardless of the
method of application or incorporation of the drug into implant or
composition,
the exemplary fibrosing agents, used alone or in combination with an anti-
infective agent or haemostatic agent, may be administered under the following
dosing guidelines.
Utilizing talc as an exemplary fibrosis-inducing agent, whether it is
applied as a polymer coating, incorporated into the polymers which make up an
implant, or applied with or without a polymeric carrier, the total dose of
talc
delivered from an implant or composition, or coated onto the surface of an
implant, preferably does not exceed 100 mg (range of 1 lug to 100 mg). In one
embodiment, the total amount of talc released from the composition or implant
is in the range of 10 lig to 50 mg. The dose per unit area of the implant (L
e.,
the dosage of talc as a function of the surface area of the portion of the
implant
to which drug is applied and/or incorporated) falls within the range of 0.05
p,g -
10 vig per mm2 of surface area coated. In another embodiment, talc is applied
to an implant surface at a dose (amount) of 0.05 lig/mm2 ¨10 fig/mm2 of
surface
area coated. As specific (polymeric and non-polymeric) drug delivery vehicles
272
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
and specific implants release talc at differing rates, the above dosing
parameters may be utilized in combination with the release rate of the drug
from the composition or implant such that a minimum concentration of 0.01 nM -
1000 pM of talc is delivered to the tissue. In another embodiment, talc is
released from the surface of an implant such that fibrosis in the diverticulum
is
promoted for a period ranging from several hours to several months. For
example, talc may be released in effective concentrations for a period ranging
from 1 hour¨ 30 days. Analogues and derivatives of talc (as described
previously) with similar functional activity may also be utilized in the
compositions and methods described herein; the above dosing parameters are
then adjusted according to the relative potency of the analogue or derivative
as
compared to the parent compound (e.g., a compound twice as potent as talc is
administered at half the above parameters, a compound half as potent as talc
is
administered at twice the above parameters, etc.).
Utilizing silk as an exemplary fibrosis-inducing agent, whether it is
applied as a polymer coating, incorporated into the polymers which make up an
implant, or applied with or without a polymeric carrier, the total dose of
silk
delivered from an implant or composition, or coated onto the surface of an
implant, preferably not exceed 100 mg (range of 1 lig to 100 mg). In one
embodiment, the total amount of silk released from the composition or implant
is in the range of 10 j.ig to 50 mg. The dose per unit area of the implant
(i.e.,
the dosage of silk as a function of the surface area of the portion of the
implant
to which drug is applied and/or incorporated) falls within the range of 0.05
lig -
10 pg per mm2 of surface area coated. In another embodiment, silk is applied
to an implant surface at a dose of 0.05 .g/mm2 ¨10 pg/mm2 of surface area
coated. As specific (polymeric and non-polymeric) drug delivery vehicles and
specific implants release silk at differing rates, the above dosing parameters
may be utilized in combination with the release rate of the drug from the
composition or implant such that a minimum concentration of 0.01 nM 1000 pM
of silk is delivered to the tissue. In one embodiment, silk is released from
the
273
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. For example, silk may be
released in effective concentrations for a period ranging from 1 hour¨ 30
days.
Analogues and derivatives of silk (as described previously) with similar
functional activity can be utilized in the compositions and methods described
herein; the above dosing parameters are then adjusted according to the
relative
potency of the analogue or derivative as compared to the parent compound
(e.g., a compound twice as potent as silk is administered at half the above
parameters, a compound half as potent as silk is administered at twice the
above parameters, etc.).
Utilizing chitosan as an exemplary fibrosis-inducing agent,
whether it is applied as a polymer coating, incorporated into the polymers
which
make up an implant, or applied with or without a polymeric carrier, the total
dose of chitosan delivered from an implantor composition, or coated onto the
surface of an implant, preferably does not exceed 100 mg (range of 1 g to 100
mg). In one embodiment, the total amount of chitosan released from the
composition or implant is in the range of 10 g to 50 mg. The dose per unit
area of the implant (i.e., the dosage of chitosan as a function of the surface
area of the portion of the implant to which drug is applied and/or
incorporated)
is then within the range of 0.05 jig - 10 jig per mm2 of surface area coated.
In
another embodiment, chitosan is applied to an implant surface at a dose of
0.05
g/mm2 ¨10 g/mm2 of surface area coated. As specific (polymeric and non-
polymeric) drug delivery vehicles and specific implants will release chitosan
at
differing rates, the above dosing parameters may be utilized in combination
with
the release rate of the drug from the composition or implant such that a
minimum concentration of 0.01 nM - 1000 pM of chitosan is delivered to the
tissue. In one embodiment, chitosan is released from the surface of an implant
such that fibrosis in the diverticulum is promoted for a period ranging from
several hours to several months. For example, chitosan may be released in
effective concentrations for a period ranging from 1 hour¨ 30 days. Analogues
274
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
and derivatives of chitosan (as described previously) with similar functional
activity can be utilized in the compositions and methods described herein; the
above dosing parameters are then adjusted according to the relative potency of
the analogue or derivative as compared to the parent compound (e.g., a
compound twice as potent as chitosan is administered at half the above
parameters, a compound half as potent as chitosan is administered at twice the
above parameters, etc.).
Utilizing polylysine as an exemplary fibrosis-inducing agent,
whether it is applied as a polymer coating, incorporated into the polymers
which
make up an implant, or applied with or without a polymeric carrier, the total
dose of polylysine delivered from an implant or composition, or coated onto
the
surface of an implant, preferably does not exceed 100 mg (range of 1 jig to
100
mg). In one embodiment, the total amount of polylysine released from the
composition or implant is in the range of 10 jig to 50 mg. The dose per unit
area of the implant (i.e., the dosage of polylysine as a function of the
surface
area of the portion of the implant to which drug is applied and/or
incorporated)
then falls within the range of 0.05 jig - 10 jig per mm2 of surface area
coated.
In another embodiment, polylysine is applied to an implant surface at a dose
of
0.05 p.g/mm2 ¨10 vtg/mm2 of surface area coated. As specific (polymeric and
non-polymeric) drug delivery vehicles and specific implants release polylysine
at differing rates, the above dosing parameters may be utilized in combination
with the release rate of the drug from the composition or implant such that a
minimum concentration of 0.01 nM to 1000 pM of polylysine is delivered to the
tissue. In one embodiment, polylysine is released from the surface of an
implant such that fibrosis in the diverticulum is promoted for a period
ranging
from several hours to several months. For example, polylysine may be
released in effective concentrations for a period ranging from 1 hour¨ 30
days.
Analogues and derivatives of polylysine (as described previously) with similar
functional activity can be utilized for the compositions and methods described
herein; the above dosing parameters are then adjusted according to the
relative
275
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
potency of the analogue or derivative as compared to the parent compound
(e.g., a compound twice as potent as polylysine is administered at half the
above parameters, a compound half as potent as polylysine is administered at
twice the above parameters, etc.).
Utilizing fibronectin as an exemplary fibrosis-inducing agent,
whether it is applied as a polymer coating, incorporated into the polymers
which
make up an implant, or applied with or without a polymeric carrier, the total
dose of fibronectin delivered from an implant or composition, or coated onto
the
surface of an implant, preferably does not exceed 100 mg (range of 1 g to 100
mg). In one embodiment, the total amount of fibronectin released from the
composition or implant is in the range of 10 p.g to 50 mg. The dose per unit
area of the implant (i.e., the dosage of fibronectin as a function of the
surface
area of the portion of the implant to which drug is applied and/or
incorporated)
then falls within the range of 0.05 [tg - 10 lag per mm2 of surface area
coated.
In another embodiment, fibronectin is applied to an implant surface at a dose
of
0.05 g/mm2 ¨10 lAg/mm2 of surface area coated. As specific (polymeric and
non-polymeric) drug delivery vehicles and specific implants will release
fibronectin at differing rates, the above dosing parameters may be utilized in
combination with the release rate of the drug from the composition or implant
such that a minimum concentration of 0.01 nM 1000 pM of fibronectin is
delivered to the tissue. In one embodiment, fibronectin is released from the
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. For example, fibronectin
may be released in effective concentrations for a period ranging from 1 hour-
30 days. Analogues and derivatives of fibronectin (as described previously)
with similar functional activity may also be utilized for the compositions and
methods described herein; the above dosing parameters are then adjusted
according to the relative potency of the analogue or derivative as compared to
the parent compound (e.g., a compound twice as potent as fibronectin is
276
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
administered at half the above parameters, a compound half as potent as
fibronectin is administered at twice the above parameters, etc.).
Utilizing bleomycin as an exemplary fibrosis-inducing agent,
whether it is applied as a polymer coating, incorporated into the polymers
which
make up an implant, or applied with or without a polymeric carrier, the total
dose of bleomycin delivered from an implant or composition, or coated onto the
surface of an implant, preferably does not exceed 100 mg (range of 0.01 jig to
100 mg). In one embodiment, the total amount of bleomycin released from the
composition or implant is in the range of 0.10 pg to 50 mg. The dose per unit
area of the implant (i.e., the dosage of bleomycin as a function of the
surface
area of the portion of the implant to which drug is applied and/or
incorporated)
then falls within the range of 0.005 jig - 10 fig per mm2 of surface area
coated.
In another embodiment, bleomycin is preferably applied to an implant surface
at
a dose of 0.005 g/mm2 ¨10 g/mm2 of surface area coated. As specific
(polymeric and non-polymeric) drug delivery vehicles and specific implants
will
release bleomycin at differing rates, the above dosing parameters is utilized
in
combination with the release rate of the drug from the composition or implant
such that a minimum concentration of 0.001 nM 1000 pM of bleomycin is
delivered to the tissue. In one embodiment, bleomycin is released from the
surface of an implant such that fibrosis in the diverticulum is promoted for a
period ranging from several hours to several months. For example, bleomycin
may be released in effective concentrations for a period ranging from 1 hour-
days. Analogues and derivatives of bleomycin (as described previously)
with similar functional activity can be utilized for the compositions and
methods
25 described herein; the above dosing parameters are then adjusted
according to
the relative potency of the analogue or derivative as compared to the parent
compound (e.g., a compound twice as potent as bleomycin is administered at
half the above parameters, a compound half as potent as bleomycin is
administered at twice the above parameters, etc.).
277
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
Utilizing CTFG as an exemplary fibrosis-inducing agent, whether it
is applied as a polymer coating, incorporated into the polymers which make up
an implant, or applied with or without a polymeric carrier, the total dose of
CTFG delivered from an implant or composition, or coated onto the surface of
an implant, preferably does not exceed 100 mg (range of 0.01 g to 100 mg).
In one embodiment, the total amount of CTFG released from the composition or
implant is in the range of 0.101_ig to 50 mg. The dose per unit area of the
implant (i.e., the dosage of CTFG as a function of the surface area of the
portion of the implant to which drug is applied and/or incorporated) then
falls
within the range of 0.005 fig - 10 jig per mm2 of surface area coated. In
another
embodiment, CTFG is applied to an implant surface at a dose of 0.005 g/mm2
¨10 g/mm2 of surface area coated. As specific (polymeric and non-polymeric)
drug delivery vehicles and specific implants will release CTFG at differing
rates,
the above dosing parameters may be utilized in combination with the release
rate of the drug from the composition or implant such that a minimum
concentration of 0.001 nM 1000 pM of CTFG is delivered to the tissue. In one
embodiment, CTFG is released from the surface of an implant such that fibrosis
in the diverticulum is promoted for a period ranging from several hours to
several months. For example, CTFG may be released in effective
concentrations for a period ranging from 1 hour¨ 30 days.
Analogues and derivatives of CTFG (as described previously) with
similar functional activity can be utilized for compositions and methods
described herein; the above dosing parameters are then adjusted according to
the relative potency of the analogue or derivative as compared to the parent
compound (e.g., a compound twice as potent as CTFG is administered at half
the above parameters, a compound half as potent as CTFG is administered at
twice the above parameters, etc.).
Agents which are tissue irritants such tissue as silk, silica,
bleomycin, neomycin, talcum powder, metallic beryllium, and copper are
particularly suitable for the practice of this invention. Other agents which
may
278
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
be incorporated into or onto the implant (e.g., a device) or released from the
implant include extracellular matrix components such as fibrous structural
proteins (e.g., fibrillar collagens, nonfibrillar collagen and elastins),
adhesive
glycoproteins (e.g., laminin and fibronectin), proteoglycans (e.g., heparin
sulphate, chondroitin sulphate, dermatan sulphate), hyaluronan (e.g.,
hyaluronic acid), Secreted Protein Acidic and Rich in Cysteine (SPARC),
Thrombospondins, Tenacin, Inhibitors of Matrix Metalloproteinases (e.g., TIMPs
and synthetic TIMPs such as MARIMISTAT, BATIMISTAT, Doxycycline,
Tetracycline, Minocycline, TROCADE, Ro-1130830, CGS 27023A, BMS-
275291) and polylysine. Growth factors and inflammatory cytokines involved in
angiogenesis, fibroblast migration, fibroblast proliferation, ECM synthesis
and
tissue remodeling such as Epidermal Growth Factor (EGF) Family,
Transforming Growth Factor-a (TGF- a), Transforming Growth Factor-13 (TGF-
3-1, TGF-13-2, TGF-f3-3), Platelet-derived Growth Factor (PDGF), Fibroblast
Growth Factor (acidic ¨ aFGF, and basic - bFGF), Bone Morphogenic Proteins,
Activins, Vascular Endothelial Growth Factor (VEGF, VEGF-B, VEGF-C,
Placental Growth Factor - PIGF), Angiopoietins, Insulin-like Growth Factors
(IGF), Hepatocyte Growth Factor (HGF), Connective Tissue Growth Factor
(CTGF), Myeloid Colony-stimulating Factors (CSFs), Granulocyte-Macrophage
Colony-stimulating Factors (GM-CSF), Granulocyte Colony-stimulating Factor
(G-CSF), Macrophage Colony-stimulating Factor (M-CSF), Erythropoietin,
Interleukins (particularly IL-1, IL-8, IL-6), Tumor Necrosis Factor-a (TNFa),
Nerve Growth Factor (NGF), Interferon-a, Interferon-13, and growth hormone
(GH) are also suitable for release from specific implants described herein.
Other agents that may be coated onto or released by the implant include
adhesives such as cyanoacrylate or CT3.
Within related aspects of the present invention, implants,
implantable tissue bulking agents, and other compositions are provided,
wherein the implant releases an agent that induces fibrosis in vivo. "Release
of
an agent" refers to any statistically significant presence of the agent, or a
279
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
subcomponent thereof, which has disassociated from the implant and/or
remains active on the surface of (or within) the implant. Within yet other
aspects of the present invention, methods are provided for manufacturing an
implant, comprising the step of coating (e.g., spraying, dipping, wrapping, or
administering drug through) an implant. Additionally, the implant can be
constructed so that the implant itself is comprised of materialsthat induce
fibrosis in or around the implant. A wide variety of implants may be utilized
for
the methods and compositions described herein, depending on the site and
nature of treatment desired.
Within various embodiments, the implant is further coated with a
composition or compound, which delays the onset of activity of the fibrosis-
inducing agent for a period of time after implantation. Representative
examples
of such agents include heparin, PLGA/MePEG, PLA, and polyethylene glycol.
Within further embodiments the fibrosis-inducing implant is activated before,
during, or after deployment (e.g., an inactive agent on the implant is first
activated to one that induces or accelerates an in vivo fibrotic reaction).
Any of the previously described fibrosis inducing agents, or
derivatives and analogues thereof, can be utilized to create variations of the
above compositions. The agent may also be used in a composition with or
without a polymer carrier and that any one or more of the carriers described
herein may be used in a composition with a fibrosing agent, anti-infective
agent,
and/or haemostatic agent.
Methods for Treating Diverticular Disease
Also provided by the present invention are methods for treating
patients undergoing surgical, endoscopic, or minimally invasive therapies
wherein a therapeutic agent or implant is delivered into a diverticula.
Diverticular disease is a condition whereby there is herniation of the mucosa
and submucosa of a hollow organ, such as the gastrointestinal (GI) tract,
urinary tract or repiratory tract, which produces outpouchings through the
280
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
muscular wall of the body passageway. Although diverticula can occur in any
tubular organ, diverticular disease is of greatest clinical relevance in the
lower
GI tract (large bowel or colon) where it can cause life threatening
inflammation
and infection (diverticulitis) or bleeding (lower GI hemorrhage). Several
specific
clinical conditions, for example, diverticular disease including
diverticulosis and
diverticulitis, colonic diverticulosis, acute diverticulitis, GI hemorrhage,
diverticular hemorrhage, appendicitis, Zenker's diverticulum (esophageal),
Meckel's diverticulitis (small bowel abnormality), small bowel diverticulum,
gastric diverticulum, and urinary diverticulum for which diverticula can be
treated using the agents, formulations, and implants, described herein are
described in greater detail below.
By way of example, a method to treat diverticular bleeding and
thus to treat or prevent a diverticular disease in a host, such as a patient,
may
include the following. A patient exhibiting symptoms of a diverticular disease
(e.g., abdominal pain and lower GI bleeding) may undergo either a physical or
radiographic examination (e.g., X-ray, ultrasound, or a CT scan) or both to
determine the extent of the diverticular disease. Once the patient is
diagnosed
with diverticular disease, a patient may be treated using the methods and
compositions described herein immediately to induce fibrosis within the
diverticulum to stop the bleeding or, alternatively, after the acute episode
has
subsided (e.g., any time up to about 6 weeks) to prevent the recurrence of a
new bleed. To treat or prevent diverticular bleeding, a patient may be
administered intravenous sedation (e.g., midazolam with either demerol or
fentanyl) and then placed on the patient's left side. For example, when a
colonic diverticulum is diagnosed and treated an end viewing colonoscope is
advanced through the colon to inspect the entire bowel visually, identifying
sites
of diverticular disease and bleeding. Once the bleeding site(s) and pertinent
diverticula are identified, a spray tip delivery catheter is introduced
through the
biopsy portal of the colonoscope. A variety of delivery catheters are
described
herein and used in the medical art.
281
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
The implant or composition(s) may be delivered to the
diverticulum to induce fibrosis and to stop bleeding. For example, a spray tip
catheter includes a syringe portion configured to contain the components of a
multi-component therapy. The multi-component therapy (see description of kits
herein) includes an in situ forming hemostatic composition (e.g., VITAGEL from
Orthovita, Inc. (Malvern, PA), COSTASIS from Angiotech Pharmaceuticals, Inc.
(Vancouver, BC)) and a mixing portion. The syringe portion is configured to
combine and mix the components of the composition just prior to delivery into
the diverticulum. Examples of mixing and dispensing devices that can be used
in this procedure are described in a co-pending application
PCT/US2004/037450. One or more components of the composition may also
include a vasoconstrictive agent (e.g. epinephrine). The spray tip catheter is
delivered to the treatment site through the portal of the colonoscope and
manipulated to position the syringe tip inside the diverticulum or at the
orifice of
the diverticulum. The spray tip catheter plunger is pushed, activating the
syringe, and delivering the composition to the diverticulum. Subsequent cross-
linking of the composition occludes the lumen or orifice of the diverticulum,
resulting in control of the bleeding.
In another embodiment, the method for treating a diverticular
disease in a patient diagnosed with a diverticular disease, the following
exemplary method may be performed. A patient is administered intravenous
sedation (e.g., midazolam with either demerol or fentanyl) and then placed on
their left side. When, for example, when treating colonic diverticular
disease,
an end viewing colonoscope is advanced through the colon to inspect the entire
bowel visually and to locate diverticular openings in the bowel lumen. Once
the
opening(s) is identified, a sclerosing needle is advanced through the portal
of
the colonoscope. The needle is attached to a syringe that is loaded with an
irrigation solution (e.g., saline), which can include an anti-septic agent
(e.g.,
chlorhexadine), an antibiotic agent (e.g., gentamicin sulfate or doxycyline),
and/or any other anti-infective agent (which are described herein), such as a
282
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
chemotherapeutic agent with anti-microbial activity (e.g., 5-fluorouracil,
doxorubicin, mitoxantrone, methotrexate, or etoposide), to eliminate bowel
flora,
including anaerobes and aerobes present in the diverticulum. The needle is
advanced through the biopsy portal to the opening of the affected
diverticulum.
The diverticulum is gently irrigated with the irrigation solution. Once the
diverticulum is cleared of debris (cells, fecal matter) the needle in the
biopsy
portal is replaced with a spray tip delivery catheter. The spray tip delivery
catheter is used to deliver a multi-component in situ forming composition to
the
diverticulum, as described herein. Subsequent cross-linking of the composition
partially or completely occludes the lumen or orifice of the diverticulum,
thereby
decreasing the probability of bleeding, infection, or inflammation.
Colonic Diverticulosis
Diverticulosis of the large bowel is an acquired condition that is
extrememly common in the Western World. The incidence of diverticulosis
increases with age ¨ affecting 30% of people over the age of 60 and over 50%
of people over the age of 80. Since the incidence of diverticulosis continues
to
increase, it is a significant healthcare problem for which very few new
treatments have been developed.
Colonic diverticula are pouches of mucosa that protrude through
the colonic musculature. Typically, the mucosa escapes through defects in the
muscle where the vasa recta (the arteries that supply blood to the mucosa)
penetrate through the colonic musculature. Diagnosis of diverticular disease
can often be made by CT scan or by barium enema, which can outline
radiographically the extent and severity of the diverticula. Typical
radiological
findings are sacculations with retained contrast and colonic spasm. Often the
entire colon is affected, but the majority of diverticula occur in the sigmoid
colon
(95%), while right-sided diverticula (cecum and acending colon) are less
common (6-7% of cases). Most patients with diverticulosis remain symptom-
free, but 10-20% will develop complications such as infection
(diverticulitits),
283
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
fistulas (abnormal communication between 2 organs ¨ most often between the
colon and the bladder), bowel obstruction or hemorrhage. If symptomatic, the
current treatment of diverticular disease involves open surgical resection of
the
affected segment of the colon; the methods described herein provide minimally-
invasive treatments that can be performed as an alternative to surgery.
Acute Diverticulitis
In acute diverticulitis, without wishing to be bound by theory, a
small fragment of stool (a fecalith) may become trapped within a diverticulum,
which causes thinning of the diverticular wall and produces a localized (and
contained) microperforation with infection and inflammation at the site of
perforation. In some cases, the infection can progress to peridiverticular
abscess formation, phlegmon, abdominal or pelvic abscess formation, and/or
generalized peritonitis if free perforation occurs. Patients are
characteristically
treated with broad spectrum oral or intravenous antibiotics, but the localized
nature of the disease lends itself well to the administration of the
therapeutic
interventions described herein, in conjunction with or alternatively to
antibiotic
treatment alone.
Colonoscopy (or in some cases CT scan, ultrasound, or other
imaging technology) is used to identify the diverticula that has become
infected.
A catheter (several examples are described herein) is advanced through the
side port of the endoscope (or under radiographic guidance) until is enters
the
lumen of the diverticula. Depending upon the catheter used, the luminal
surface of the diverticulum may be disrupted by abrasion or the application of
energy (RF, thermal, laser, ultrasound etc. as described herein) to facilitate
healing and fibrosis. In cases in which the diverticulum is extremely inflamed
and friable, these catheters are preferably not used because they will
increase
the risk of perforation; under these circumstances the therapeutic agent(s)
may
be administered via a single or multi-lumen drug-delivery catheter. The
diverticulum is then infiltrated with an anti-infective agent and/or a
fibrosis-
284
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
inducing agent to treat the infection and/or encourage fibrosis and permanent
filling/closure of the diverticulum. As described previously, it may also be
beneficial to deliver the therapeutic agents in a sustained release
preparation
(or implant delivered into the lumen of the diverticulum) to fully treat the
infection and promote complete healing of the diverticululm. Formulations that
release the therapeutic agents for a minimum of 7 days, and preferably for 30-
60 days and beyond, are particularly effective for this embodiment. In
instances in which hemorrhage or risk or of hemorrhage is evident, a
hemostatic agent may also be used in addition to the anti-infective or
fibrosis-
inducing agent(s).
In one embodiment, the fibrosis-inducing agent, anti-infective
agent, and/or hemostatic may be delivered to the diverticulum through an
abdominal endoscopic approach. In this embodiment, an endoscope is
inserted into the abdomen (typically via the umbilicus) as part of a standard
abdominal endoscopic procedure. The diverticulum is located, and an injection
device that is delivered via the sideport of the endoscope pierces the outer
wall
of diverticulum such that it enters the lumen. The therapeutic agent(s) or
composition is then injected via a catheter into the lumen of the
diverticulum.
Due to the risk of rupture, this method may be reserved for cases for which
colonoscopy cannot be conducted.
Diverticular Hemorrhage
Diverticular bleeding occurs in 5-15% of patients who have
diverticulosis, and in many (up to one third), the bleeding is substantial
enough
to cause cardiovascular instablility. The
precise cause of diverticular
hemorrhage is unknown, but the close anatomic association of the diverticulum
to the vasa recta is thought to be important. According to non-limiting
theory,
the site of the herniations may be the same as the site of penetration of a
nutrient artery, and the local inflammation of diverticulitis causes erosion
of the
artery wall. This approximation of the neck of the sack of the diverticulum
and
285
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
the arterial supply may be the reason for the massive hemorrhages observed in
patients with diverticulosis. The average age of a patient at the time of
diverticular hemorrhage is 65. As a result, mortality and morbidity is high
(10%
to 20%), in part due to patient comorbidity with other conditions such as
cardiac, pulmonary, renal disease.
Diagnosis is made by barium enema, which can radiographically
outline the extent and severity of the diverticula present. The right colon is
the
most common source of bleeding and is the cause in 48% to 90% of patients.
Emergency angiography is appropriate for patients with brisk bleeding (0.5ml -
1.0m1/min). Evaluation begins with examination of the mesenteric artery
(because right colon bleeds are most common) and followed by examination of
the inferior mesenteric artery and the celiac artery. Angiography not only has
a
high predictive value, but it also enables the detection of other causes of GI
hemorrhage such as angiodysplasia, tumors or diffuse mucosal bleeds.
Angiography can also be used therapeutically for the introduction of
vasospastic substances or selective embolization. Risks associated with
vasospastic substances include a re-bleeding rate of 50% in patients after
withdrawal of the therapy, decreased coronary perfusion, hypertension and
cardiac arrythmias; embolization can sometimes stop the bleeding but it is
associated with colon infarctions. Nuclear scanning with technetium is used
diagnostically in the detection of the source of slow bleeds (i.e. when the
rate of
bleeding is less than 0.1m1/min).
Generally diverticular bleeds are massive, painless, and self
limiting. Patients are treated supportively with volume resuscitation,
correction
of coagulation abnormalities, and transfusion if required. Patients frequently
require multiple blood transfusions and the average patient receives 7.6 Units
of packed cells per episode of bleeding. Fortunately, diverticular bleeding
stops
spontaneously in 70 to 95% of cases by using supportive measures only.
Generally, bleeding stops in 99% of patients if they require less than 4 Units
of
blood within the first 24 hours. Approximately 15% of patients will continue
to
286
CA 02610948 2007-12-05
WO 2006/124021 PCT/US2005/016871
be unstable despite aggressive resuscitation and will require surgery in an to
attempt to stop the bleeding, but this is associated with a very high
mortality
rate (14% to 38%). Unfortunately, recurrent episodes of hemorrhage requiring
a second hospital admission are also common and occur in 25% of patients
who have had a previous diverticular hemorrhage. After a second bleeding
episode, the risk of recurrence increases further to the point where the
chance
of a having a third hemorrhage increases to over 50%. Thus, a significant need
exists for a therapy that can be used acutely to control bleeding, as well as
a
need for a treatment that reduces the risk that a patient will suffer a
subsequent
hemorrhage.
Endoscopic examination can determine the presence of
concomitant lesions such as malignancy and angiodysplasia. Colonoscopy
allows for direct visualization of the active bleeding site, which is
ultimately
found in up to 85% of patients with this technique. Colonoscopic interventions
allow for direct visualization of the bleeding site, electrocoagulation of the
bleeding site, irrigation of the area with epinephrine and identification of
other
concominant disease. For the purposes of the methods described herein,
colonoscopy (or in some cases CT scan, ultrasound, or other imaging
technology) is used to identify the diverticula that is the source of the
bleeding.
A specialized catheter (described in a previous section) is advanced through
the side port of the endoscope (or under radiographic guidance) until it
enters
the lumen of the diverticula wherethe hemorrhage originates. Depending upon
the catheter used, the luminal surface of the diverticulum may be disrupted by
abrasion or the application of energy (RF, thermal, laser, ultrasound etc. as
described herein) to facilitate thrombosis and initiate healing and fibrosis.
In
instances in which the diverticulum is extremely inflamed and friable, these
catheters are preferably not used because they will increase the risk of -
perforation; under these circumstances the therapeutic agent(s) may be be
administered via a single or multi-lumen drug-delivery catheter. The
diverticulum is then infiltrated with a hemostatic agent alone or in
combination
287
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 287
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 287
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: