Note: Descriptions are shown in the official language in which they were submitted.
CA 02613125 2007-11-29
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME DE _2
NOTE: Pour les tomes additionels, veiliez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLLIME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
COMPOSITIONS AND METHODS FOR THERAPY AND
DIAGNOSIS OF PROSTATE CANCER
TECHNICAL FIELD
The present invention relates generally to therdpy and diagnosis of cancer,
such as''prostate cancer. The invention is more specifically related to
polypeptides
comprising at least a portion of a prostate tumor protein, and to
polynucleotides encoding
such polypeptides. Such polypeptides and polynucleotides may be used in
vaccines and
pharmaceutical compositions for prevention and treatment of prostate cancer,
and for the
diagnosis and monitoring of such cancers.
BACKGROUND OF THE INVENTION
Prostate cancer is the most common form of cancer among males, with an
estimated incidence of 30% in men over the age of 50. Overwhelming clinical
zv:.acnce
shows that human prostate cancer has the propensity to metastasize to bone,
and the disease
appears to progress inevitably from androgen dependent to androgen refractory
status, leading
to increased patient mortality. This prevalent disease is currently the second
leading cause of
cancer death among men in the U.S.
In spite of considerable research into therapies for the disease, prostate
cancer
remains difficult to treat. Commonly, treatment is based on surgery and/or
radiation therapy,
but these methods are ineffective in a significant percentage of cases. Two
previously
identified prostate specific proteins - prostate specific antigen (PSA) and
prostatic acid
phosphatase (PAP) - have limited therapeutic and diagnostic potential. For
example, PSA
levels do not always correlate well with the presence of prostate cancer,
being positive in a
percentage of non-prostate cancer cases, including benign prostatic
hyperplasia (BPH).
Furthermore, PSA measurements correlate with prostate volume, and do not
indicate the level
of metastasis.
In spite of considerable research into therapies for these and other cancers,
prostate cancer remains difficult to diagnose and treat effectively.
Accordingly, there is a
need in the art for improved methods for detecting and treating such cancers.
The present
invention fulfills these needs and further provides other related advantages.
SUMMARY OF THE INVENTION
Briefly stated, the present invention provides compositions and, methods for
the diagnosis and therapy of cancer, such as prostate cancer. In one aspect,
the present
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
2 invention provides polypeptides comprising at least a portion of a prostate
tumor protein, or a
variant thereof. Certain portions and other variants are immunogenic, such
that the ability of
the variant to react with antigen-specific antisera is not substantially
diminished. Within certain embodiments, the polypeptide comprises at least an
immunogenic portion of a
prostate tumor protein, or a variant thereof, wherein the tumor protein
comprises an amino acid sequence that is encoded by a polynucleotide sequence
selected from the group
consisting-of: (a) sequences recited in any one of SEQ ID NOs:I-111, 115-171,
173-175, 177,
179-305, 307-315, 326, 328, 330, 332-335, 340-375, 381, 382 or 384-472; (b)
sequences that
hybridize to any of the foregoing sequences under moderately stringent
conditions; and (c)
complements of any of the sequence of (a) or (b). In certain specific
embodiments, such a
polypeptide comprises at least a portion, or variant thereof, of a tumor
protein that includes an
amino acid sequence selected from the group consisting of sequences recited in
any one of
SEQ ID NO: 112-114, 172, 176, 178, 327, 329, 331, 336, 339, 376-380 and 383.
The present invention further provides polynucleotides that encode a
polypeptide as described above, or a portion thereof (such as a portion
encoding at least 15
amino acid residues of a prostate tumor protein), expression vectors
comprising such
polynucleotides and host cells transformed or transfected with such expression
vectors.
Within other aspects, the present invention provides pharmaceutical
compositions comprising a polypeptide or polynucleotide as described above and
a
physiologically acceptable carrier.
Within a related aspect of the present invention, vaccines are provided. Such
vaccines comprise a polypeptide or polynucleotide as described above and a non-
specific
immune response enhancer.
The present invention further provides pharmaceutical compositions that
comprise: (a) an antibody or antigen-binding fragment thereof that
specifically binds to a
prostate tumor protein; and (b) a physiologically acceptable carrier.
Within further aspects, the present invention provides pharmaceutical
compositions comprising: (a) an antigen presenting cell that expresses a
polypeptide as
described above and (b) a pharmaceutically acceptable carrier or excipient.
Antigen
presenting cells include dendritic cells, macrophages, monocytes, fibroblasts
and B cells.
Within related aspects, vaccines are provided that comprise: (a) an antigen
presenting cell that expresses a polypeptide as described above and (b) a non-
specific immune
response enhancer.
The present invention further provides, in other aspects, fusion proteins that
comprise at least one polypeptide as described above, as well as
polynucleotides encoding
such fusion proteins.
CA 02613125 2007-11-29
WO 00/04149 PCT/L3S99/15838 -
3
Within related aspects, pharmaceutical compositions comprising a fusion
protein, or a polynucleotide encoding a fusion protein, in combination with a
physiologically
acceptable carrier are provided.
Vaccines are further provided, within other aspects, that comprise a fusion
protein, or a polynucleotide encoding a fusion protein, in combination with a
non-specific
immune response enhancer.
Within further aspects, the present invention provides methods for inhibiting
the development of a cancer in a patient, comprising administering to a
patient a
pharmaceutical composition or vaccine as recited above.
The present invention fi.nther provides, within other aspects, methods for
removing tumor cells from a biological sample, comprising contacting a
biological sample
with T cells that specifically react with a prostate tumor protein, wherein
the step of
contacting is performed under conditions and for a time sufficient to permit
the removal of
cells expressing the protein from the sample.
Within related aspects, methods are provided for inhibiting the development of
a cancer in a patient, comprising administering to a patient a biological
sample treated as
described above.
Methods are further provided, within other aspects, for stimulating and/or
expanding T cells specific for a prostate tumor protein, comprising contacting
T cells with
one or more of: (i) a polypeptide as described above; (ii) a polynucleotide
encoding such a
polypeptide; and/or (iii) an antigen presenting cell that expresses such a
polypeptide; under
conditions and for a time sufficient to permit the stimulation and/or
expansion of T cells.
Isolated T cell populations comprising T cells prepared as described above are
also provided.
Within further aspects, the present invention provides methods for inhibiting
the development of a cancer in a patient, comprising administering to a
patient an effective
amount of a T cell population as described above.
The present invention further provides methods for inhibiting the development
of a cancer in a patient, comprising the steps of: (a) incubating CD4+ and/or
CD8+ T cells
isolated from a patient with one or more of: (i) a polypeptide comprising at
least an
immunogenic portion of a prostate tumor protein; (ii) a polynucleotide
encoding such a
polypeptide; and (iii) an antigen-presenting cell that expressed such a
polypeptide; and (b)
administering to the patient an effective amount of the proliferated T cells,
and thereby
inhibiting the development of a cancer in the patient. Proliferated cells may,
but need not, be
cloned prior to administration to the patient.
Within further aspects, the present invention provides methods for determining
the presence or absence of a cancer in a patient, comprising: (a) contacting a
biological
sample obtained from a patient with a binding agent that binds to a
polypeptide as recited
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
4
above; (b) detecting in the sample an amount of polypeptide that binds to the
binding agent;
and (c) comparing the amount of polypeptide with a predetermined cut-off
value, and
therefrom determining the presence or absence of a cancer in the patient.
Within preferred
embodiments, the binding agent is an antibody, more preferably a monoclonal
antibody. The
cancer may be prostate cancer.
The present invention also provides, within other aspects, methods for
monitoririg the progression of a cancer in a patient. Such methods comprise
the steps of: (a)
contacting a biological sample obtained from a patient at a first point in
time with a binding
agent that binds to a polypeptide as recited above; (b) detecting in the
sample an amount of
polypeptide that binds to the binding agent; (c) repeating steps (a) and (b)
using a biological
sample obtained from the patient at a subsequent point in time; and (d)
comparing the amount
of polypeptide detected in step (c) with the amount detected in step (b) and
therefrom
monitoring the progression of the cancer in the patient.
The present invention further provides, within other aspects, methods for
determining the presence or absence of a cancer in a patient, comprising the
steps of: (a)
contacting a biological sample obtained from a patient with an oligonucleotide
that hybridizes
to a polynucleotide that encodes a prostate tumor protein; (b) detecting in
the sample a level
of a polynucleotide, preferably mRNA, that hybridizes to the oligonucleotide;
and (c)
comparing the level of polynucleotide that hybridizes to the oligonucleotide
with a
predetermined cut-off value, and therefrom determining the presence or absence
of a cancer
in the patient. Within certain embodiments, the amount of mRNA is detected via
polymerase
chain reaction using, for example, at least one oligonucleotide primer that
hybridizes to a
polynucleotide encoding a polypeptide as recited above, or a complement of
such a
polynucleotide. Within other embodiments, the amount of mRNA is detected using
a
hybridization technique, employing an oligonucleotide probe that hybridizes to
a
polynucleotide that encodes a polypeptide as recited above, or a complement of
such a
polynucleotide.
In related aspects, methods are provided for monitoring the progression of a
cancer in a patient, comprising the steps of: (a) contacting a biological
sample obtained from
a patient with an oligonucleotide that hybridizes to a polynucleotide that
encodes a prostate
tumor protein; (b) detecting in the sample an amount of a polynucleotide that
hybridizes to
the oligonucleotide; (c) repeating steps (a) and (b) using a biological sample
obtained from
the patient at a subsequent point in time; and (d) comparing the amount of
polynucleotide
detected in step (c) with the amount detected in step (b) and therefrom
monitoring the
progression of the cancer in the patient.
Within further aspects, the present invention provides antibodies, such as
monoclonal antibodies, that bind to a polypeptide as described above, as well
as diagnostic
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
kits comprising such antibodies. Diagnostic kits comprising one or more
oligonucleotide
probes or primers as described above are also provided.
These and other aspects of the present invention will become apparent upon
reference to the following detailed description and attached drawings. All
references
disclosed herein are hereby incorporated by reference in their entirety as if
each was
incorporated individually.
BRIEF DESCRIPTION OF THE DRAWINGS AND SEQUENCE IDENTIFIERS
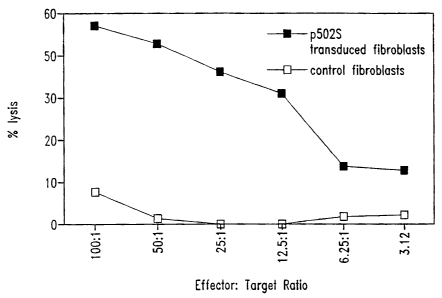
Figure 1 illustrates the ability of T cells to kill fibroblasts expressing the
representative prostate tumor polypeptide P502S, as compared to control
fibroblasts. The
percentage lysis is shown as a series of effector:target ratios, as indicated.
Figures 2A and 2B illustrate the ability of T cells to recognize cells
expressing
the representative prostate tumor polypeptide P502S. In each case, the number
of y-interferon
spots is shown for different numbers of responders. In Figure 2A, data is
presented for
fibroblasts pulsed with the P2S-12 peptide, as compared to fibroblasts pulsed
with a control
E75 peptide. In Figure 2B, data is presented for fibroblasts expressing P502S,
as compared to
fibroblasts expressing HER-21neu.
Figure 3 represents a peptide competition binding assay showing that the
P I S# 10 peptide, derived from P501 S, binds HLA-A2. Peptide P 1 S# 10
inhibits HLA-A2
restricted presentation of fluM58 peptide to CTL clone D150M58 in TNF release
bioassay.
D150M58 CTL is specific for the HLA-A2 binding influenza matrix peptide
fluM58.
Figure 4 illustrates the ability of T cell lines generated from P 1 S# 10
immunized mice to specifically lyse PIS#10-pulsed Jurkat A2Kb targets and
P501S-
transduced Jurkat A2Kb targets, as compared to EGFP-transduced Jurkat A2Kb.
The percent
lysis is shown as a series of effector to target ratios, as indicated.
Figure 5 illustrates the ability of a T cell clone to recognize and
specifically
lyse Jurkat A2Kb cells expressing the representative prostate tumor
polypeptide P501 S,
thereby demonstrating that the PIS#10 peptide may be a naturally processed
epitope of the
P501 S polypeptide.
Figures 6A and 6B are graphs illustrating the specificity of a CD8+ cell line
(3A-1) for a representative prostate tumor antigen (P501 S). Figure 6A shows
the results of a
5'Cr release assay. The percent specific lysis is shown as a series of
effector:target ratios, as
indicated. Figure 6B shows the production of interferon-gamma by 3A-1 cells
stimulated
with autologous B-LCL transduced with P501 S, at varying effector:target
rations as
indicated.
SEQ ID NO: I is the determined cDNA sequence for F 1-13
SEQ ID NO: 2 is the determined 3' cDNA sequence for F1-12
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838 6
SEQ ID NO: 3 is the determined 5' cDNA sequence for F I-12
SEQ ID NO: 4 is the determined 3' cDNA sequence for FI-16
SEQ ID NO: 5 is the determined 3' cDNA sequence for H 1-1
SEQ ID NO: 6 is the determined 3' eDNA sequence for H 1-9
SEQ ID NO: 7 is the determined 3' cDNA sequence for H1-4
SEQ ID NO: 8 is the determined 3' cDNA sequence for J1-17
SEQ ID NO: 9 is the determined 5' cDNA sequence for J I-17
SEQ ID NO: 10 is the determined 3' cDNA sequence for L1-12
SEQ ID NO: I 1 is the determined 5' cDNA sequence for L1-12
SEQ ID NO: 12 is the determined 3' cDNA sequence for N1-1862
SEQ ID NO: 13 is the determined 5' cDNA sequence for N1-1862
SEQ ID NO: 14 is the determined 3' cDNA sequence for J1-13
SEQ ID NO: 15 is the determined 5' cDNA sequence for J 1-13
SEQ ID NO: 16 is the determined 3' cDNA sequence for J1-19
SEQ ID NO: 17 is the determined 5' cDNA sequence for J1-19
SEQ ID NO: 18 is the determined 3' cDNA sequence for J1-25
SEQ ID NO: 19 is the determined 5' cDNA sequence for J 1-25
SEQ ID NO: 20 is the determined 5' cDNA sequence for J1.-24
SEQ ID NO: 21 is the determined 3' cDNA sequence for J 1-24
SEQ ID NO: 22 is the determined 5' cDNA sequence for K1-58
SEQ ID NO: 23 is the determined 3' cDNA sequence for K1-58
SEQ ID NO: 24 is the determined 5' cDNA sequence for K1.-63
SEQ ID NO: 25 is the determined 3' cDNA sequence for K1-63
SEQ ID NO: 26 is the determined 5' cDNA sequence for L1-4
SEQ ID NO: 27 is the determined 3' cDNA sequence for L 1-4
SEQ ID NO: 28 is the determined 5' cDNA sequence for L1-14
SEQ ID NO: 29 is the determined 3' cDNA sequence for L1-14
SEQ ID NO: 30 is the determined 3' cDNA sequence for J 1-12
SEQ ID NO: 31 is the determined 3' cDNA sequence for J1-16
SEQ ID NO: 32 is the determined 3' cDNA sequence for J1-21
SEQ ID NO: 33 is the determined 3' eDNA sequence for KI-48
SEQ ID NO: 34 is the determined 3' cDNA sequence for KI-55
SEQ ID NO: 35 is the determined 3' cDNA sequence for L1-2
SEQ ID NO: 36 is the determined 3' cDNA sequence for L1-6
SEQ ID NO: 37 is the determined 3' cDNA sequence for N1-1858
SEQ ID NO: 38 is the determined 3' cDNA sequence forNl-1860
SEQ ID NO: 39 is the determined 3' cDNA sequence for N1-1861
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
7
SEQ ID NO: 40 is the determined 3' cDNA sequence for N1-1864
SEQ ID NO: 41 is the determined cDNA sequence for P5
SEQ ID NO: 42 is the determined cDNA sequence for P8
SEQ ID NO: 43 is the determined cDNA sequence for P9
SEQ ID NO: 44 is the determined cDNA sequence for P18
SEQ ID NO: 45 is the determined eDNA sequence for P20
SEQ ID ~10: 46 is the determined cDNA sequence for P29
SEQ ID NO: 47 is the determined cDNA sequence for P30
SEQ ID NO: 48 is the determined cDNA sequence for P34
SEQ ID NO: 49 is the determined cDNA sequence for P36
SEQ ID NO: 50 is the determined cDNA sequence for P38
SEQ ID NO: 51 is the determined cDNA sequence for P39
SEQ ID NO: 52 is the determined cDNA sequence for P42
SEQ ID NO: 53 is the determined cDNA sequence for P47
SEQ ID NO: 54 is the determined cDNA sequence for P49
SEQ ID NO: 55 is the determined cDNA sequence for P50
SEQ ID NO: 56 is the determined cDNA sequence for P53
SEQ ID NO: 57 is the determined cDNA sequence for P55
SEQ ID NO: 58 is the determined cDNA sequence for P60
SEQ ID NO: 59 is the determined cDNA sequence for P64
SEQ ID NO: 60 is the determined cDNA sequence for P65
SEQ ID NO: 61 is the determined cDNA sequence for P73
SEQ ID NO: 62 is the determined eDNA sequence for P75
SEQ ID NO: 63 is the determined cDNA sequence for P76
SEQ ID NO: 64 is the determined cDNA sequence for P79
SEQ ID NO: 65 is the determined cDNA sequence for P84
SEQ ID NO: 66 is the determined cDNA sequence for P68
SEQ ID NO: 67 is the determined cDNA sequence for P80
SEQ ID NO: 68 is the determined cDNA sequence for P82
SEQ ID NO: 69 is the determined eDNA sequence for U1-3064
SEQ ID NO: 70 is the determined cDNA sequence for U1-3065
SEQ ID NO: 71 is the determined cDNA sequence for V 1-3692
SEQ ID NO: 72 is the determined cDNA sequence for 1A-3905
SEQ ID NO: 73 is the determined cDNA sequence for V1-3686
SEQ ID NO: 74 is the determined cDNA sequence for R1-2330
SEQ ID NO: 75 is the determined cDNA sequence for 1 B-3976
SEQ ID NO: 76 is the determined cDNA sequence for V 1-3679
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
8
SEQ ID NO: 77 is the determined cDNA sequence forl G-4736
SEQ ID NO: 78 is the determined cDNA sequence for 1G-4738
SEQ ID NO: 79 is the determined cDNA sequence for 1 G-4741
SEQ ID NO: 80 is the determined cDNA sequence for 1 G-4744
SEQ ID NO: 81 is the determined cDNA sequence for 1 G-4734
SEQ ID NO: 82 is the determined cDNA sequence for 1 H-4774
SEQ ID NO: 83 is the determined eDNA sequence for 1H-4781
SEQ ID NO: 84 is the determined cDNA sequence for 1 H-4785
SEQ ID NO: 85 is the determined cDNA sequence for 1 H-4787
SEQ ID NO: 86 is the determined cDNA sequence for 1H-4796
SEQ ID NO: 87 is the determined cDNA sequence for 11-4807
SEQ ID NO: 88 is the determined cDNA sequence for 1I-4810
SEQ ID NO: 89 is the determined cDNA sequence for 11-4811
SEQ ID NO: 90 is the determined cDNA sequence for 1 J-4876
SEQ ID NO: 91 is the determined cDNA sequence for 1 K-4884
SEQ ID NO: 92 is the determined cDNA sequence for 1 K-4896
SEQ ID NO: 93 is the determined cDNA sequence for 1 G-4761
SEQ ID NO: 94 is the determined cDNA sequence for I G-4762
SEQ ID NO: 95 is the determined cDNA sequence for 1 H-4766
SEQ ID NO: 96 is the determined cDNA sequence for 1H-4770
SEQ ID NO: 97 is the determined cDNA sequence for 1H-4771
SEQ ID NO: 98 is the determined cDNA sequence for IH-4772
SEQ ID NO: 99 is the determined cDNA sequence for 1 D-4297
SEQ ID NO: 100 is the determined cDNA sequence for 1D-4309
SEQ ID NO: 101 is the determined cDNA sequence for 1 D.1-4278
SEQ ID NO: 102 is the determined cDNA sequence for 1 D-4288
SEQ ID NO: 103 is the determined cDNA sequence for 1 D-4283
SEQ ID NO: 104 is the determined cDNA sequence for 1 D-4304
SEQ ID NO: 105 is the determined cDNA sequence for 1 D-4296
SEQ ID NO: 106 is the determined cDNA sequence for 1 D-4280
SEQ ID NO: 107 is the determined full length cDNA sequence for F 1-12 (also
referred to as
P504S)
SEQ ID NO: 108 is the predicted amino acid sequence for F1-12
SEQ ID NO: 109 is the determined full length cDNA sequence for J1-17
SEQ ID NO: I 10 is the determined full length eDNA sequence for L1-12
SEQ ID NO: 111 is the determined full length cDNA sequence for N 1-1862
SEQ ID NO: 112 is the predicted amino acid sequence for J 1-17
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
9
SEQ ID NO: 113 is the predicted amino acid sequence for L1-12
SEQ ID NO: 114 is the predicted amino acid sequence for N 1-1862
SEQ ID NO: 115 is the determined cDNA sequence for P89
SEQ ID NO: 116 is the determined cDNA sequence for P90
SEQ ID NO: 117 is the determined cDNA sequence for P92
SEQ ID NO: 118 is the determined eDNA sequence for P95
SEQ ID N0: 119 is the determined cDNA sequence for P98
SEQ ID NO: 120 is the determined cDNA sequence for P102
SEQ ID NO: 121 is the determined cDNA sequence for P110
SEQ ID NO: 122 is the determined cDNA sequence for P 111
SEQ ID NO: 123 is the determined cDNA sequence for P114
SEQ ID NO: 124 is the determined cDNA sequence for Pl 15
SEQ ID NO: 125 is the determined cDNA sequence for P116
SEQ ID NO: 126 is the determined cDNA sequence for P124
SEQ ID NO: 127 is the determined cDNA sequence for P126
SEQ ID NO: 128 is the detennined cDNA sequence for P130
SEQ ID NO: 129 is the deternlined cDNA sequence for P133
SEQ ID NO: 130 is the determined cDNA sequence for P138
SEQ ID NO: 131 is the determined cDNA sequence for P143
SEQ ID NO: 132 is the determined cDNA sequence for P151
SEQ ID NO: 133 is the determined cDNA sequence for P156
SEQ ID NO: 134 is the determined cDNA sequence for P157
SEQ ID NO: 135 is the determined cDNA sequence for P166
SEQ ID NO: 136 is the determined cDN.A sequence for P176
SEQ ID NO: 137 is the determined cDNA sequence for P178
SEQ ID NO: 138 is the determined cDNA sequence for P179
SEQ ID NO: 139 is the determined cDNA sequence for P185
SEQ ID NO: 140 is the determined cDNA sequence for P192
SEQ ID NO: 141 is the determined cDNA sequence for P201
SEQ ID NO: 142 is the determined cDNA sequence for P204
SEQ ID NO: 143 is the determined cDNA sequence for P208
SEQ ID NO: 144 is the deternined cDNA sequence for P211
SEQ ID NO: 145 is the determined cDNA sequence for P213
SEQ ID NO: 146 is the determined cDNA sequence for P219
SEQ ID NO: 147 is the determined cDNA sequeiice for P237
SEQ ID NO: 148 is the determined cDNA sequence for P239
SEQ ID NO: 149 is the determined cDNA sequence for P248
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
SEQ ID NO: 150 is the determined cDNA sequence for P251
SEQ ID NO: 151 is the determined cDNA sequence for P255
SEQ ID NO: 152 is the determined cDNA sequence for P256
SEQ ID NO: 153 is the determined cDNA sequence for P259
SEQ ID NO: 154 is the determined cDNA sequence for P260
SEQ ID NO: 155 is the determined cDNA sequence for P263
SEQ ID 140: 156 is the determined cDNA sequence for P264
SEQ ID NO: 157 is the determined cDNA sequence for P266
SEQ ID NO: 158 is the determined cDNA sequence for P270
SEQ ID NO: 159 is the determined cDNA sequence for P272
SEQ ID NO: 160 is the determined cDNA sequence for P278
SEQ ID NO: 161 is the determined cDNA sequence for P105
SEQ ID NO: 162 is the determined cDNA sequence for P107
SEQ ID NO: 163 is the determined cDNA sequence for P137
SEQ ID NO: 164 is the determined cDNA sequence for P194
SEQ ID NO: 165 is the determined cDNA sequence for P195
SEQ ID NO: 166 is the determined cDNA sequence for P196
SEQ ID NO: 167 is the determined cDNA sequence for P220
SEQ ID NO: 168 is the determined cDNA sequence for P234
SEQ ID NO: 169 is the determined cDNA sequence for P235
SEQ ID NO: 170 is the determined cDNA sequence for P243
SEQ ID NO: 171 is the determined cDNA sequence for P703P-DEI
SEQ ID NO: 172 is the predicted amino acid sequence for P703P-DEI
SEQ ID NO: 173 is the determined cDNA sequence for P703P-DE2
SEQ ID NO: 174 is the determined cDNA sequence for P703P-DE6
SEQ ID NO: 175 is the determined cDNA sequence for P703P-DE13
SEQ ID NO: 176 is the predicted amino acid sequence for P703P-DE13
SEQ ID NO: 177 is the determined cDNA sequence for P703P-DE14
SEQ ID NO: 178 is the predicted amino acid sequence for P703P-DE14
SEQ ID NO: 179 is the determined extended cDNA sequence for 1 G-4736
SEQ ID NO: 180 is the determined extended cDNA sequence for 1G-4738
SEQ ID NO: 181 is the determined extended cDNA sequence for 1 G-4741
SEQ ID NO: 182 is the determined extended cDNA sequence for 1 G-4744
SEQ ID NO: 183 is the determined extended cDNA sequence for 1H-4774
SEQ ID NO: 184 is the determined extended cDNA sequence for I H-4781
SEQ ID NO: 185 is the determined extended cDNA sequence for I H-4785
SEQ ID NO: 186 is the determined extended cDNA sequence for I H-4787
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
11
SEQ ID NO: 187 is the determined extended cDNA sequence for 1H-4796
SEQ ID NO: 188 is the determined extended cDNA sequence for 11-4807
SEQ ID NO: 189 is the determined 3' cDNA sequence for 1I-4810
SEQ ID NO: 190 is the determined 3' cDNA sequence for 11-4811
SEQ ID NO: 191 is the determined extended cDNA sequence for 1J-4876
SEQ ID NO: 192 is the determined extended cDNA sequence for 1 K-4884
SEQ ID N,O: 193 is the determined extended cDNA sequence for 1 K-4896
SEQ ID NO: 194 is the determined extended cDNA sequence for 1 G-4761
SEQ ID NO: 195 is the determined extended cDNA sequence for I G-4762
SEQ ID NO: 196 is the determined extended cDNA sequence for 1 H-4766
SEQ ID NO: 197 is the determined 3' cDNA sequence for 1 H-4770
SEQ ID NO: 198 is the determined 3' cDNA sequence for 1 H-4771
SEQ ID NO: 199 is the determined extended cDNA sequence for 1 H-4772
SEQ ID NO: 200 is the determined extended cDNA sequence for I D-4309
SEQ ID NO: 201 is the determined extended cDNA sequence for ID.1-4278
SEQ ID NO: 202 is the determined extended cDNA sequence for 1 D-4288
SEQ ID NO: 203 is the determined extended cDNA sequence for I D-4283
SEQ ID NO: 204 is the determined extended cDNA sequence for 1 D-4304
SEQ ID NO: 205 is the determined extended cDNA sequence for I D-4296
SEQ ID NO: 206 is the determined extended cDNA sequence for 1 D-4280
SEQ ID NO: 207 is the determined cDNA sequence for 10-d8fwd
SEQ ID NO: 208 is the determined cDNA sequence for 10-H10con
SEQ ID NO: 209 is the determined cDNA sequence for 11-C8rev
SEQ ID NO: 210 is the determined cDNA sequence for 7.g6fwd
SEQ ID NO: 211 is the determined cDNA sequence for 7.g6rev
SEQ ID NO: 212 is the determined eDNA sequence for 8-b5fwd
SEQ ID NO: 213 is the determined cDNA sequence for 8-b5rev
SEQ ID NO: 214 is the determined cDNA, sequence for 8-b6fwd
SEQ ID NO: 215 is the determined cDNA sequence for 8-b6 rev
SEQ ID NO: 216 is the determined cDNA sequence for 8-d4fwd
SEQ ID NO: 217 is the determined cDNA sequence for 8-d9rev
SEQ ID NO: 218 is the determined eDNA sequence for 8-g3fwd
SEQ ID NO: 219 is the determined cDNA sequence for 8-g3rev
SEQ ID NO: 220 is the determined cDNA sequence for 8-hl Irev
SEQ ID NO: 221 is the determined cDNA sequence for g-fl 2fwd
SEQ ID NO: 222 is the determined cDNA sequence for g-f3rev
SEQ ID NO: 223 is the determined cDNA sequence for P509S
CA 02613125 2007-11-29
WO 00/04149 PCT/US99115838
12
SEQ ID NO: 224 is the determined cDNA sequence for P510S
SEQ ID NO: 225 is the determined cDNA sequence for P703DE5
SEQ ID NO: 226 is the determined cDNA sequence for 9-A 11
SEQ ID NO: 227 is the determined cDNA sequence for 8-C6
SEQ ID NO: 228 is the determined cDNA sequence for 8-H7
SEQ ID NO: 229 is the determined cDNA sequence for JPTPNI3
SEQ ID 1N.O: 230 is the determined cDNA sequence for JPTPN14
SEQ ID NO: 231 is the determined eDNA sequence for JPTPN23
SEQ ID NO: 232 is the determined cDNA sequence for JPTPN24
SEQ ID NO: 233 is the determined cDNA sequence for JPTPN25
SEQ ID NO: 234 is the determined cDNA sequence for JPTPN30
SEQ ID NO: 235 is the determined cDNA sequence for JPTPN34
SEQ ID NO: 236 is the determined cDNA sequence for PTPN35
SEQ ID NO: 237 is the determined cDNA sequence for JPTPN36
SEQ ID NO: 238 is the determined cDNA sequence for JPTPN38
SEQ ID NO: 239 is the determined cDNA sequence for JPTPN39
SEQ ID NO: 240 is the determined cDNA sequence for JPTPN40
SEQ ID NO: 241 is the determined cDNA sequence for JPTPN41
SEQ ID NO: 242 is the determined cDNA sequence for JPTPN42
SEQ ID NO: 243 is the determined cDNA sequence for JPTPN45
SEQ ID NO: 244 is the determined cDNA sequence for JPTPN46
SEQ ID NO: 245 is the determined cDNA sequence for JPTPN5I
SEQ ID NO: 246 is the determined cDNA sequence for JPTPN56
SEQ ID NO: 247 is the determined eDNA sequence for PTPN64
SEQ ID NO: 248 is the determined eDNA sequence for JPTPN65
SEQ ID NO: 249 is the determined cDNA sequence for JPTPN67
SEQ ID NO: 250 is the determined cDNA sequence for JPTPN76
SEQ ID NO: 251 is the deternnined cDNA sequence for JPTPN84
SEQ ID NO: 252 is the determined cDNA sequence for JPTPN85
SEQ ID NO: 253 is the determined cDNA sequence for JPTPN86
SEQ ID NO: 254 is the determined cDNA sequence for JPTPN87
SEQ ID NO: 255 is the determined cDNA sequence for JPTPN88
SEQ ID NO: 256 is the determined cDNA sequence for JP1F1
SEQ ID NO: 257 is the determined cDNA sequence for JPIF2
SEQ ID NO: 258 is the determined cDNA sequence for JP 1 C2
SEQ ID NO: 259 is the determined cDNA sequence for JP1B1
SEQ ID NO: 260 is the determined cDNA sequence for JP1B2
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
13
SEQ ID NO: 261 is the determined cDNA sequence for JPID3
SEQ ID NO: 262 is the determined eDNA sequence for JP1A4
SEQ ID NO: 263 is the determined cDNA sequence for JP1 F5
SEQ ID NO: 264 is the determined cDNA sequence for JPIE6
SEQ ID NO: 265 is the determined cDNA sequence for JPID6
SEQ ID NO: 266 is the determined cDNA sequence for JPIB5
SEQ ID W: 267 is the determined cDNA sequence for JP1A6
SEQ ID NO: 268 is the determined cDNA sequence for JPIE8
SEQ ID NO: 269 is the determined cDNA sequence for JP 1 D7
SEQ ID NO: 270 is the determined cDNA sequence for JPI D9
SEQ ID NO: 271 is the determined cDNA sequence for JPIC10
SEQ ID NO: 272 is the determined cDNA sequence for JP1A9
SEQ ID NO: 273 is the determined cDNA sequence for JP1F12
SEQ ID NO: 274 is the determined cDNA sequence for JP1E12
SEQ ID NO: 275 is the determined cDNA sequence for JPID11
SEQ ID NO: 276 is the determined cDNA sequence for JP 1 C 11
SEQ ID NO: 277 is the determined cDNA sequence for JP1C12
SEQ ID NO: 278 is the determined cDNA sequence for JP I B 12
SEQ ID NO: 279 is the determined cDNA sequence for JP1A12
SEQ ID NO: 280 is the determined cDNA sequence for JP8G2
SEQ ID NO: 281 is the determined cDNA sequence for JP8HI
SEQ ID NO: 282 is the determined cDNA sequence for JP8H2
SEQ ID NO: 283 is the determined eDNA sequence for JP8A3
SEQ ID NO: 284 is the determined cDNA sequence for JP8A4
SEQ ID NO: 285 is the determined cDNA sequence for JP8C3
SEQ ID NO: 286 is the determined cDNA sequence for JP8G4
SEQ ID NO: 287 is the determined cDNA sequence for JP8B6
SEQ ID NO: 288 is the determined cDNA sequence for JP8D6
SEQ ID NO: 289 is the determined cDNA sequence for JP8F5
SEQ ID NO: 290 is the determined eDNA sequence for JP8A8
SEQ ID NO: 291 is the determined cDNA sequence for JP8C7
SEQ ID NO: 292 is the determined cDNA sequence for JP8D7
SEQ ID NO: 293 is the determined cDNA sequence for P8D8
SEQ ID NO: 294 is the determined cDNA sequence for JP8E7
SEQ ID NO: 295 is the determined cDNA sequence for JP8F8
SEQ ID NO: 296 is the determined cDNA sequence for JP8G8
SEQ ID NO: 297 is the determined cDNA sequence for JP8B10
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
14
SEQ ID NO: 298 is the determined cDNA sequence for JP8C10
SEQ ID NO: 299 is the determined cDNA sequence for JP8E9
SEQ ID NO: 300 is the determined cDNA sequence for JP8E10
SEQ ID NO: 301 is the determined cDNA sequence for JP8F9
SEQ ID NO: 302 is the determined eDNA sequence for JP8H9
SEQ ID NO: 303 is the determined cDNA sequence for JP8C12
SEQ ID I~D: 304 is the determined cDNA sequence for JP8E11
SEQ ID NO: 305 is the determined cDNA sequence for JP8E12
SEQ ID NO: 306 is the amino acid sequence for the peptide PS2412
SEQ ID NO: 307 is the determined cDNA sequence for P711 P
SEQ ID NO: 308 is the determined cDNA sequence for P712P
SEQ ID NO: 309 is the determined cDNA sequence for CLONE23
SEQ ID NO: 310 is the determined cDNA sequence for P774P
SEQ ID NO: 311 is the determined cDNA sequence for P775P
SEQ ID NO: 312 is the determined cDNA sequence for P715P
SEQ ID NO: 313 is the determined cDNA sequence for P710P
SEQ ID NO: 314 is the determined cDNA sequence for P767P
SEQ ID NO: 315 is the determined cDNA sequence for P768P
SEQ ID NO: 316-325 are the determined cDNA sequences of previously isolated
genes
SEQ ID NO: 326 is the determined cDNA sequence for P703PDE5
SEQ ID NO: 327 is the predicted amino acid sequence for P703PDE5
SEQ ID NO: 328 is the determined cDNA sequence for P703P6.26
SEQ ID NO: 329 is the predicted amino acid sequence for P703P6.26
SEQ ID NO: 330 is the determined cDNA sequence for P703PX-23
SEQ ID NO: 331 is the predicted amino acid sequence for P703PX-23
SEQ ID NO: 332 is the determined full length cDNA sequence for P509S
SEQ ID NO: 333 is the determined extended cDNA sequence for P707P (also
referred to as
11-C9)
SEQ ID NO: 334 is the determined eDNA sequence for P714P
SEQ ID NO: 335 is the determined cDNA sequence for P705P (also referred to as
9-F3)
SEQ ID NO: 336 is the predicted amino acid sequence for P705P
SEQ ID NO: 337 is the amino acid sequence of the peptide P1 S#10
SEQ ID NO: 338 is the amino acid sequence of the peptide p5
SEQ ID NO: 339 is the predicted amino acid sequence of P509S
SEQ ID NO: 340 is the determined cDNA sequence for P778P
SEQ ID NO: 341 is the determined cDNA sequence for P786P
SEQ ID NO: 342 is the determined cDNA sequence for P789P
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
15 SEQ ID NO: 343 is the determined cDNA sequence for a clone showing homology
to Homo
sapiens MM46 mRNA
SEQ ID NO: 344 is the determined cDNA sequence for a clone showing homology to
Homo
sapiens TNF-alpha stimulated ABC protein (ABC50) mRNA
SEQ ID NO: 345 is the determined cDNA sequence for a clone showing homology to
Homo
sapiens mRNA for E-cadherin
SEQ ID NO: 346 is the deten-nined cDNA sequence for a clone showing homology
to Human
nuclear-encoded mitochondrial serine hydroxymethyltransferase (SHMT)
SEQ ID NO: 347 is the determined cDNA sequence for a clone showing homology to
Homo
sapiens natural resistance-associated macrophage protein2 (NRAMP2)
SEQ ID NO: 348 is the determined cDNA sequence for a clone showing homology to
Homo
sapiens phosphoglucomutase-related protein (PGMRP)
SEQ ID NO: 349 is the determined cDNA sequence for a clone showing homology to
Human
mRNA for proteosome subunit p40
SEQ ID NO: 350 is the determined cDNA sequence for P777P
SEQ ID NO: 351 is the determined cDNA sequence for P779P
SEQ ID NO: 352 is the determined cDNA sequence for P790P
SEQ ID NO: 353 is the determined cDNA sequence for P784P
SEQ ID NO: 354 is the determined cDNA sequence for P776P
SEQ ID NO: 355 is the determined cDNA sequence for P780P
SEQ ID NO: 356 is the determined cDNA sequence for P544S
SEQ ID NO: 357 is the determined cDNA sequence for P745S
SEQ ID NO: 358 is the determined cDNA sequence for P782P
SEQ ID NO: 359 is the determined cDNA sequence for P783P
SEQ ID NO: 360 is the determined cDNA sequence for unknown 17984
SEQ ID NO: 361 is the determined cDNA sequence for P787P
SEQ ID NO: 362 is the determined cDNA sequence for P788P
SEQ ID NO: 363 is the determined cDNA sequence for unknown 17994
SEQ ID NO: 364 is the determined cDNA sequence for P781 P
SEQ ID NO: 365 is the determined cDNA sequence for P785P
SEQ ID NO: 366-375 are the determined cDNA sequences for splice variants of
B305D.
SEQ ID NO: 376 is the predicted amino acid sequence encoded by the sequence of
SEQ ID
NO: 366.
SEQ ID NO: 377 is the predicted amino acid sequence encoded by the sequence of
SEQ ID
NO: 372.
SEQ ID NO: 378 is the predicted amino acid sequence encoded by the sequence of
SEQ ID
NO: 373.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
16
SEQ ID NO: 379 is the predicted amino acid sequence encoded by the sequence of
SEQ ID
NO: 374.
SEQ ID NO: 380 is the predicted amino acid sequence encoded by the sequence of
SEQ ID
NO: 375.
SEQ ID NO: 381 is the determined cDNA sequence for B716P.
SEQ ID NO: 382 is the determined full-length cDNA sequence for P711P.
SEQ ID N'O: 383 is the predicted amino acid sequence for P711P.
SEQ ID NO: 384 is the cDNA sequence for P1000C.
SEQ ID NO: 385 is the cDNA sequence for CGI-82.
SEQ ID NO:386 is the eDNA sequence for 23320.
SEQ ID NO:387 is the cDNA sequence for CGI-69.
SEQ ID NO:388 is the cDNA sequence for L-iditol-2-dehydrogenase.
SEQ ID NO:389 is the cDNA sequence for 23379.
SEQ ID NO:390 is the cDNA sequence for 23381.
SEQ ID NO:391 is the cDNA sequence for KIAA0122.
SEQ ID NO:392 is the cDNA sequence for 23399.
SEQ ID NO:393 is the cDNA sequence for a previously identified gene.
SEQ ID NO:394 is the cDNA sequence for HCLBP.
SEQ ID NO:395 is the cDNA sequence for transglutaminase.
SEQ ID NO:396 is the cDNA sequence for a previously identified gene.
SEQ ID NO:397 is the cDNA sequence for PAP.
SEQ ID NO:398 is the cDNA sequence for Ets transcription factor PDEF.
SEQ ID NO:399 is the cDNA sequence for hTGR.
SEQ ID NO:400 is the cDNA sequence for KIAA0295.
SEQ ID NO:401 is the cDNA sequence for 22545.
SEQ ID NO:402 is the cDNA sequence for 22547.
SEQ ID NO:403 is the cDNA sequence for 22548.
SEQ ID NO:404 is the cDNA sequence for 22550.
SEQ ID NO:405 is the cDNA sequence for 22551.
SEQ ID NO:406 is the cDNA sequence for 22552.
SEQ ID NO:407 is the cDNA sequence for 22553.
SEQ ID NO:408 is the cDNA sequence for 22558.
SEQ ID NO:409 is the cDNA sequence for 22562.
SEQ ID NO:410 is the cDNA sequence for 22565.
SEQ ID NO:411 is the cDNA sequence for 22567.
SEQ ID NO:412 is the cDNA sequence for 22568.
SEQ ID NO:413 is the cDNA sequence for 22570.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
17
SEQ ID NO:414 is the cDNA sequence for 22571.
SEQ ID NO:415 is the cDNA sequence for 22572.
SEQ ID NO:416 is the cDNA sequence for 22573.
SEQ ID N0:417 is the cDNA sequence for 22573.
SEQ ID N0:418 is the cDNA sequence for 22575.
SEQ ID N0:419 is the cDNA sequence for 22580.
SEQ ID 1V0:420 is the cDNA sequence for 22581.
SEQ ID N0:421 is the cDNA sequence for 22582.
SEQ ID N0:422 is the cDNA sequence for 22583.
SEQ ID N0:423 is the cDNA sequence for 22584.
SEQ ID N0:424 is the cDNA sequence for 22585.
SEQ ID N0:425 is the cDNA sequence for 22586.
SEQ ID N0:426 is the cDNA sequence for 22587.
SEQ ID N0:427 is the cDNA sequence for 22588.
SEQ ID N0:428 is the cDNA sequence for 22589.
SEQ ID N0:429 is the cDNA sequence for 22590.
SEQ ID N0:430 is the cDNA sequence for 22591.
SEQ ID N0:431 is the cDNA sequence for 22592.
SEQ ID N0:432 is the cDNA sequence for 22593.
SEQ ID N0:433 is the cDNA sequence for 22594.
SEQ ID N0:434 is the cDNA sequence for 22595.
SEQ ID N0:435 is the eDNA sequence for 22596.
SEQ ID N0:436 is the cDNA sequence for 22847.
SEQ ID N0:437 is the cDNA sequence for 22848.
SEQ ID N0:438 is the cDNA sequence for 22849.
SEQ ID N0:439 is the cDNA sequence for 22851.
SEQ ID N0:440 is the cDNA sequence for 22852.
SEQ ID N0:441 is the cDNA sequence for 22853.
SEQ ID N0:442 is the cDNA sequence for 22854.
SEQ ID N0:443 is the cDNA sequence for 22855.
SEQ ID N0:444 is the cDNA sequence for 22856.
SEQ ID N0:445 is the cDNA sequence for 22857.
SEQ ID N0:446 is the cDNA sequence for 23601.
SEQ ID N0:447 is the cDNA sequence for 23602.
SEQ ID N0:448 is the eDNA sequence for 23605.
SEQ ID N0:449 is the cDNA sequence for 23606.
SEQ ID N0:450 is the cDNA sequence for 23612.
CA 02613125 2007-11-29
WO 00/04149 PCTlUS99/15838
18
SEQ ID NO:451 is the cDNA sequence for 23614.
SEQ ID NO:452 is the cDNA sequence for 23618.
SEQ ID NO:453 is the cDNA sequence for 23622.
SEQ ID NO:454 is the cDNA sequence for folate hydrolase.
SEQ ID NO:455 is the cDNA sequence for LIM protein.
SEQ ID NO:456 is the cDNA sequence for a known gene.
SEQ ID I~O:457 is the cDNA sequence for a known gene.
SEQ ID NO:458 is the cDNA sequence for a previously identified gene.
SEQ ID NO:459 is the cDNA sequence for 23045.
SEQ ID NO:460 is the cDNA sequence for 23032.
SEQ ID NO:461 is the cDNA sequence for 23054.
SEQ ID NOs:462-467 are cDNA sequences for known genes.
SEQ ID NOs:468-471 are cDNA sequences for P7lOP.
SEQ ID NO:472 is a cDNA sequence for P1001C.
DETAILED DESCRIPTION OF THE INVENTION
As noted above, the present invention is generally directed to compositions
and methods for the therapy and diagnosis of cancer, such as prostate cancer.
The
compositions described herein may include prostate tumor polypeptides,
polynucleotides
encoding such polypeptides, binding agents such as antibodies, antigen
presenting cells
(APCs) and/or immune system cells (e.g., T cells). Polypeptides of the present
invention
generally comprise at least a portion (such as an immunogenic portion) of a
prostate tumor
protein or a variant thereof. A "prostate tumor protein" is a protein that is
expressed in
prostate tumor cells at a level that is at least two fold, and preferably at
least five fold, greater
than the level of expression in a normal tissue, as determined using a
representative assay
provided herein. Certain prostate tumor proteins are tumor proteins that react
detectably
(within an immunoassay, such as an ELISA or Western blot) with antisera of a
patient
afflicted with prostate cancer. Polynucleotides of the subject invention
generally comprise a
DNA or RNA sequence that encodes all or a portion of such a polypeptide, or
that is
complementary to such a sequence. Antibodies are generally immune system
proteins, or
antigen-binding fragments thereof, that are capable of binding to a
polypeptide as described
above. Antigen presenting cells include dendritic cells, macrophages,
monocytes. fibroblasts
and B-cells that express a polypeptide as described above. T cells that may be
employed
within such compositions are generally T cells that are specific for a
polypeptide as described
above.
CA 02613125 2007-11-29
WO 00/04149 PCT/(JS99/15838
19
The present invention is based on the discovery of human prostate tumor
proteins. Sequences of polynucleotides encoding certain tumor proteins, or
portions thereof,
are provided in SEQ ID NOs:1-111, 115-171, 173-175, 177, 179-305, 307-315,
326, 328,
330, 332-335, 340-375, 381, 382 or 384-472. Sequences of polypeptides
comprising at least
a portion of a tumor protein are provided in SEQ ID NOs:112-114, 172, 176,
178, 327, 329,
331, 336, 339, 376-380 and 383.
PROSTATE TUMOR PROTEIN POLYNUCLEOTIDES
Any polynucleotide that encodes a prostate tumor protein or a portion or other
variant thereof as described herein is encompassed by the present invention.
Preferred
polynucleotides comprise at least 15 consecutive nucleotides, preferably at
least 30
consecutive nucleotides and more preferably at least 45 consecutive
nucleotides, that encode
a portion of a prostate tumor protein. More preferably, a polynucleotide
encodes an
immunogenic portion of a prostate tumor protein. Polynucleotides complementary
to any
such sequences are also encompassed by the present invention. Polynucleotides
may be
single-stranded (coding or antisense) or double-stranded, and may be DNA
(genomic, cDNA
or synthetic) or RNA molecules. RNA molecules include HnRNA molecules, which
contain
introns and correspond to a DNA molecule in a one-to-one manner, and mRNA
molecules,
which do not contain introns. Additional coding or non-coding sequences may,
but need not,
be present within a polynucleotide of the present invention, and a
polynucleotide may, but
need not, be linked to other molecules and/or support materials.
Polynucleotides may comprise a native sequence (i.e., an endogenous
sequence that encodes a prostate tumor protein or a portion thereof) or may
comprise a
variant of such a sequence. Polynucleotide variants may contain one or more
substitutions,
additions, deletions and/or insertions such that the immunogenicity of the
encoded
polypeptide is not diminished, relative to a native tumor protein. The effect
on the
immunogenicity of the encoded polypeptide may generally be assessed as
described herein.
Variants preferably exhibit at least about 70% identity, more preferably at
least about 80%
identity and most preferably at least about 90% identity to a polynucleotide
sequence that
encodes a native prostate tumor protein or a portion thereof.
Two polynucleotide or polypeptide sequences are said to be "identical" if the
sequence of nucleotides or amino acids in the two sequences is the same when
aligned for
maximum correspondence as described below. Comparisons between two sequences
are
typically performed by comparing the sequences over a comparison window to
identify and
compare local regions of sequence similarity. A "comparison window" as used
herein, refers
to a segment of at least about 20 contiguous positions, usually 30 to about
75, 40 to about 50,
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
in which a sequence may be compared to a reference sequence of the same number
of
contiguous positions after the two sequences are optimally aligned.
Optimal alignment of sequences for comparison may be conducted using the
Megalign program in the Lasergene suite of bioinformatics software (DNASTAR,
Inc.,
Madison, WI), using default parameters. This program embodies several
alignment schemes
described in the following references: Dayhoff, M.O. (1978) A model of
evolutionary change
in protein- - - Matrices for detecting distant relationships. In Dayhoff, M.O.
(ed.) Atlas of
Protein Sequence and Structure, National Biomedical Research Foundation,
Washington DC
Vol. 5, Suppl. 3, pp. 345-358; Hein J. (1990) Unified Approach to Alignment
and Phylogenes
pp. 626-645 Methods in Enzymology vol. 183, Academic Press, Inc., San Diego,
CA;
Higgins, D.G. and Sharp, P.M. (1989) CABIOS 5:151-153; Myers, E.W. and Muller
W.
(1988) CABIOS 4:11-17; Robinson, E.D. (1971) Comb. Theor 11:105; Santou, N.
Nes, M.
(1987) Mol. Biol. Evol. 4:406-425; Sneath, P.H.A. and Sokal, R.R. (1973)
Numerical
Taxonomy - the Principles and Practice of Numerical Taxonomy, Freeman Press,
San
Francisco, CA; Wilbur, W.J. and Lipman, D.J. (1983) Proc. Natl. Acad., Sci.
USA 80:726-
730.
Preferably, the "percentage of sequence identity" is determined by comparing
two optimally aligned sequences over a window of comparison of at least 20
positions,
wherein the portion of the polynucleotide or polypeptide sequence in the
comparison window
may comprise additions or deletions (i.e., gaps) of 20 percent or less,
usually 5 to 15 percent,
or 10 to 12 percent, as compared to the reference sequences (which does not
comprise
additions or deletions) for optimal alignment of the two sequences. The
percentage is
calculated by determining the number of positions at which the identical
nucleic acid bases or
amino acid residue occurs in both sequences to yield the number of matched
positions,
dividing the number of matched positions by the total number of positions in
the reference
sequence (i.e., the window size) and multiplying the results by 100 to yield
the percentage of
sequence identity.
Variants may also, or alternatively, be substantially homologous to a native
gene, or a portion or complement thereof. Such polynucleotide variants are
capable of
hybridizing under moderately stringent conditions to a naturally occurring DNA
sequence
encoding a native prostate tumor protein (or a complementary sequence).
Suitable
moderately stringent conditions include prewashing in a solution of 5 X SSC,
0.5% SDS, 1.0
mM EDTA (pH 8.0); hybridizing at 50 C-65 C, 5 X SSC, overnight; followed by
washing
twice at 65 C for 20 minutes with each of 2X, 0.5X and 0.2X SSC containing
0.1% SDS.
It will be appreciated by those of ordinary skill in the art that, as a result
of the
degeneracy of the genetic code, there are many nucleotide sequences that
encode a
polypeptide as described herein. Some of these polynucleotides bear minimal
homology to
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
21
the nucleotide sequence of any native gene. Nonetheless, polynucleotides that
vary due to
differences in codon usage are specifically contemplated by the present
invention. Further,
alleles of the genes comprising the polynucleotide sequences provided herein
are within the
scope of the present invention. Alleles are endogenous genes that are altered
as a result of
one or more mutations, such as deletions, additions and/or substitutions of
nucleotides. The
resulting mRNA and protein may, but need not, have an altered structure or
function. Alleles
may be Identified using standard techniques (such as hybridization,
amplification and/or
database sequence comparison).
Polynucleotides may be prepared using any of a variety of techniques. For
example, a polynucleotide may be identified, as described in more detail
below, by screening
a microarray of cDNAs for tumor-associated expression (i.e., expression that
is at least five
fold greater in a prostate tumor than in nonnal tissue, as determined using a
representative
assay provided herein). Such screens may be performed using a Synteni
microarray (Palo
Alto, CA) according to the manufacturer's instructions (and essentially as
described by
Schena et al., Proc. Natl. Acad. Sci. USA 93:10614-10619, 1996 and Heller et
al., Proc. Natl.
Acad. Sci. USA 94:2150-2155, 1997). Alternatively, polypeptides may be
amplified from
cDNA prepared from cells expressing the proteins described herein, such as
prostate tumor
cells. Such polynucleotides may be amplified via polymerase chain reaction
(PCR). For this
approach, sequence-specific primers may be designed based on the sequences
provided
herein, and may be purchased or synthesized.
An amplified portion may be used to isolate a full length gene from a suitable
library (e.g., a prostate tumor cDNA library) using well known techniques.
Within such
techniques, a library (cDNA or genomic) is screened using one or more
polynucleotide
probes or primers suitable for amplification. Preferably, a library is size-
selected to include
larger molecules. Random primed libraries may also be preferred for
identifying 5' and
upstream regions of genes. Genomic libraries are preferred for obtaining
introns and
extending 5' sequences.
For hybridization techniques, a partial sequence may be labeled (e.g., by nick-
translation or end-labeling with 32P) using well known techniques. A bacterial
or
bacteriophage library is then screened by hybridizing filters containing
denatured bacterial
colonies (or lawns containing phage plaques) with the labeled probe (see
Sambrook et al.,
Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratories, Cold
Spring
Harbor, NY, 1989). Hybridizing colonies or plaques are selected and expanded,
and the
DNA is isolated for further analysis. cDNA clones may be analyzed to determine
the amount
of additional sequence by, for example, PCR using a primer from the partial
sequence and a
primer from the vector. Restriction maps and partial sequences may be
generated to identify
one or more overlapping clones. The complete sequence may then be determined
using
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
22
standard techniques, which may involve generating a series of deletion clones.
The resulting
overlapping sequences are then assembled into a single contiguous sequence. A
full length
cDNA molecule can be generated by ligating suitable fragments, using well
known
techniques.
Alternatively, there are numerous amplification techniques for obtaining a
full
length coding sequence from a partial cDNA sequence. Within such techniques,
amplific'ation is generally performed via PCR. Any of a variety of
commercially available
kits may be used to perform the amplification step. Primers may be designed
using, for
example, software well lulown in the art. Primers are preferably 22-30
nucleotides in length,
have a GC content of at least 50% and anneal to the target sequence at
temperatures of about
68 C to 72 C. The amplified region may be sequenced as described above, and
overlapping
sequences assembled into a contiguous sequence.
One such amplification technique is inverse PCR (see Triglia et al., Nucl.
Acids Res. 16:8186, 1988), which uses restriction enzymes to generate a
fragment in the
known region of the gene. The fragment is then circularized by intramolecular
ligation and
used as a template for PCR with divergent primers derived from the known
region. Within an
alternative approach, sequences adjacent to a partial sequence may be
retrieved by
amplification with a primer to a linker sequence and a primer specific to a
known region. The
amplified sequences are typically subjected to a second round of amplification
with the same
linker primer and a second primer specific to the known region. A variation on
this
procedure, which employs two primers that initiate extension in opposite
directions from the
known sequence, is described in WO 96/38591. Another such technique is known
as "rapid
amplification of cDNA ends" or RACE. This technique involves the use of an
internal primer
and an external primer, which hybridizes to a polyA region or vector sequence,
to identify
sequences that are 5' and 3' of a known sequence. Additional techniques
include capture PCR
(Lagerstrom et al., PCR Methods Applic. 1:111-19, 1991) and walking PCR
(Parker et al.,
Nucl. Acids. Res. 19:3055-60, 1991). Other methods employing amplification may
also be
employed to obtain a full length cDNA sequence.
In certain instances, it is possible to obtain a full length cDNA sequence by
analysis of sequences provided in an expressed sequence tag (EST) database,
such as that
available from GenBank. Searches for overlapping ESTs may generally be
performed using
well known programs (e.g., NCBI BLAST searches), and such ESTs may be used to
generate
a contiguous full length sequence.
Certain nucleic acid sequences of cDNA molecules encoding at least a portion
of a prostate tumor protein are provided in SEQ ID NOs:1-111, 115-171, 173-
175, 177, 179-
305, 307-315, 326, 328, 330, 332-335, 340-375, 381, 382 or 384-472. Isolation
of these
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
23
polynucleotides is described below. Each of these prostate tumor proteins was
overexpressed
in prostate tumor tissue.
Polynucleotide variants may generally be prepared by any method known in
the art, including chemical synthesis by, for example, solid phase
phosphoramidite chemical
synthesis. Modifications in a polynucleotide sequence may also be introduced
using standard
mutagenesis techniques, such as oligonucleotide-directed site-specific
mutagenesis (see
Adelmari'Et al., DNA 2:183, 1983). Alternatively, RNA molecules may be
generated by in
vitro or in vivo transcription of DNA sequences encoding a prostate tumor
protein, or portion
thereof, provided that the DNA is incorporated into a vector with a suitable
RNA polymerase
promoter (such as T7 or SP6). Certain portions may be used to prepare an
encoded
polypeptide, as described herein. In addition, or alternatively, a portion may
be administered
to a patient such that the encoded polypeptide is generated in vivo (e.g., by
transfecting
antigen-presenting cells, such as dendritic cells, with a cDNA construct
encoding a prostate
tumor polypeptide, and administering the transfected cells to the patient).
A portion of a sequence complementary to a coding sequence (i.e., an
antisense polynucleotide) may also be used as a probe or to modulate gene
expression.
cDNA constructs that can be transcribed into antisense RNA may also be
introduced into
cells of tissues to facilitate the production of antisense RNA. An antisense
polynucleotide
may be used, as described herein, to inhibit expression of a tumor protein.
Antisense
technology can be used to control gene expression through triple-helix
formation, which
compromises the ability of the double helix to open sufficiently for the
binding of
polymerases, transcription factors or regulatory molecules (see Gee et al., In
Huber and Carr,
Molecular and Immunologic Approaches, Futura Publishing Co. (Mt. Kisco, NY;
1994)).
Alternatively, an antisense molecule may be designed to hybridize with a
control region of a
gene (e.g., promoter, enhancer or transcription initiation site), and block
transcription of the
gene; or to block translation by inhibiting binding of a transcript to
ribosomes.
A portion of a coding sequence, or of a complementary sequence, may also be
designed as a probe or primer to detect gene expression. Probes may be labeled
with a
variety of reporter groups, such as radionuclides and enzymes, and are
preferably at least 10
nucleotides in length, more preferably at least 20 nucleotides in length and
still more
preferably at least 30 nucleotides in length. Primers, as noted above, are
preferably 22-30
nucleotides in length.
Any polynucleotide may be further modified to increase stability in vivo.
Possible modifications include, but are not limited to, the addition of
flanking sequences at
the 5' and/or 3' ends; the use of phosphorothioate or 2' 0-methyl rather than
phosphodiesterase linkages in the backbone; and/or the inclusion of
nontraditional bases such
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
24
as inosine, queosine and wybutosine, as well as acetyl- methyl-, thio- and
other modified
forms of adenine, cytidine, guanine, thymine and uridine.
Nucleotide sequences as described herein may be joined to a variety of other
nucleotide sequences using established recombinant DNA techniques. For
example, a
polynucleotide may be cloned into any of a variety of cloning vectors,
including plasmids,
phagemids, lambda phage derivatives and cosmids. Vectors of particular
interest include
expressiori'-vectors, replication vectors, probe generation vectors and
sequencing vectors. In
general, a vector will contain an origin of replication functional in at least
one organism,
convenient restriction endonuclease sites and one or more selectable markers.
Other elements
will depend upon the desired use, and will be apparent to those of ordinary
skill in the art.
Within certain embodiments, polynucleotides may be formulated so as to
permit entry into a cell of a mammal, and expression therein. Such
formulations are
particularly useful for therapeutic purposes, as described below. Those of
ordinary skill in
the art will appreciate that there are many ways to achieve expression of a
polynucleotide in a
target cell, and any suitable method may be employed. For example, a
polynucleotide may be
incorporated into a viral vector such as, but not limited to, adenovirus,
adeno-associated
virus, retrovirus, or vaccinia or other pox virus (e.g., avian pox virus).
Techniques for
incorporating DNA into such vectors are well known to those of ordinary skill
in the art. A
retroviral vector may additionally transfer or incorporate a gene for a
selectable marker (to aid
in the identification or selection of transduced cells) and/or a targeting
moiety, such as a gene
that encodes a ligand for a receptor on a specific target cell, to render the
vector target
specific. Targeting may also be accomplished using an antibody, by methods
known to those
of ordinary skill in the art.
Other formulations for therapeutic purposes include colloidal dispersion
systems, such as macromolecule complexes, nanocapsules, microspheres, beads,
and lipid-
based systems including oil-in-water emulsions, micelles, mixed micelles, and
liposomes. A
preferred colloidal system for use as a delivery vehicle in vitro and in vivo
is a liposome (i.e.,
an artificial membrane vesicle). The preparation and use of such systems is
well known in
the art.
PROSTATE TUMOR POLYPEPTIDES
Within the context of the present invention, polypeptides may comprise at
least an immunogenic portion of a prostate tumor protein or a variant thereof,
as described
herein. As noted above, a "prostate tumor protein" is a protein that is
expressed by prostate
tumor cells. Proteins that are prostate tumor proteins also react detectably
within an
immunoassay (such as an ELISA) with antisera from a patient with prostate
cancer.
Polypeptides as described herein may be of any length. Additional sequences
derived from
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
the native protein and/or heterologous sequences may be present, and such
sequences may
(but need not) possess further immunogenic or antigenic properties.
An "immunogenic portion," as used herein is a portion of a protein that is
recognized (i.e., specifically bound) by a B-cell and/or T-cell surface
antigen receptor. Such
immunogenic portions generally comprise at least 5 amino acid residues, more
preferably at
least 10, and still more preferably at least 20 amino acid residues of a
prostate tumor protein
or a varialnt thereof. Certain preferred immunogenic portions include peptides
in which an N-
terminal leader sequence and/or transmembrane domain have been deleted. Other
preferred
immunogenic portions may contain a small N- and/or C-terminal deletion (e.g.,
1-30 amino
acids, preferably 5-15 amino acids), relative to the mature protein.
Immunogenic portions may generally be identified using well known
techniques, such as those summarized in Paul, Fundamental Immunology, 3rd ed.,
243-247
(Raven Press, 1993) and references cited therein. Such techniques include
screening
polypeptides for the ability to react with antigen-specific antibodies,
antisera and/or T-cell
lines or clones. As used herein, antisera and antibodies are "antigen-
specific" if they
specifically bind to an antigen (i.e., they react with the protein in an ELISA
or other
immunoassay, and do not react detectably with unrelated proteins). Such
antisera and
antibodies may be prepared as described herein, and using well known
techniques. An
immunogenic portion of a native prostate tumor protein is a portion that
reacts with such
antisera and/or T-cells at a level that is not substantially less than the
reactivity of the full
length polypeptide (e.g., in an ELISA and/or T-cell reactivity assay). Such
immunogenic
portions may react within such assays at a level that is similar to or greater
than the reactivity
of the full length polypeptide. Such screens may generally be performed using
methods well
known to those of ordinary skill in the art, such as those described in Harlow
and Lane,
Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988. For
example, a
polypeptide may be immobilized on a solid support and contacted with patient
sera to allow
binding of antibodies within the sera to the invnobilized polypeptide. Unbound
sera may
then be removed and bound antibodies detected using, for example,'ZSI-labeled
Protein A.
As noted above, a composition may comprise a variant of a native prostate
tumor protein. A polypeptide "variant," as used herein, is a polypeptide that
differs from a
native prostate tumor protein in one or more substitutions, deletions,
additions and/or
insertions, such that the immunogenicity of the polypeptide is not
substantially diminished.
In other words, the ability of a variant to react with antigen-specific
antisera may be enhanced
or unchanged, relative to the native protein, or may be diminished by less
than 50%, and
preferably less than 20%, relative to the native protein. Such variants may
generally be
identified by modifying one of the above polypeptide sequences and evaluating
the reactivity
of the modified polypeptide with antigen-specific antibodies or antisera as
described herein.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
26
Preferred variants include those in which one or more portions, such as an N-
terminal leader
sequence or transmembrane domain, have been removed. Other preferred variants
include
variants in which a small portion (e.g_, 1-30 amino acids, preferably 5-15
amino acids) has
been removed from the N- and/or C-terminal of the mature protein. Polypeptide
variants
preferably exhibit at least about 70%, more preferably at least about 90% and
most preferably
at least about 95% identity (determined as described above) to the identified
polypeptides.
Preferably, a variant contains conservative substitutions. A "conservative
substitution" is one in which an amino acid is substituted for another amino
acid that has
similar properties, such that one skilled in the art of peptide chemistry
would expect the
secondary structure and hydropathic nature of the polypeptide to be
substantially unchanged.
Amino acid substitutions may generally be made on the basis of similarity in
polarity, charge,
solubility, hydrophobicity, hydrophilicity and/or the amphipathic nature of
the residues. For
example, negatively charged amino acids include aspartic acid and glutamic
acid; positively
charged amino acids include lysine and arginine; and amino acids with
uncharged polar head
groups having similar hydrophilicity values include leucine, isoleucine and
valine; glycine
and alanine; asparagine and glutamine; and serine, threonine, phenylalanine
and tyrosine.
Other groups of amino acids that may represent conservative changes include:
(1) ala, pro,
gly, glu, asp, gln, asn, ser, thr; (2) cys, ser, tyr, thr; (3) val, ile, leu,
met, ala, phe; (4) lys, arg,
his; and (5) phe, tyr, trp, his. A variant may also, or alternatively, contain
nonconservative
changes. In a preferred embodiment, variant polypeptides differ from a native
sequence by
substitution, deletion or addition of five amino acids or fewer. Variants may
also (or
alternatively) be modified by, for example, the deletion or addition of amino
acids that have
minimal influence on the immunogenicity, secondary structure and hydropathic
nature of the
polypeptide.
As noted above, polypeptides may comprise a signal (or leader) sequence at
the N-terminal end of the protein which co-translationally or post-
translationally directs
transfer of the protein. The polypeptide may also be conjugated to a linker or
other sequence
for ease of synthesis, purification or identification of the polypeptide
(e.g., poly-His), or to
enhance binding of the polypeptide to a solid support. For example, a
polypeptide may be
conjugated to an immunoglobulin Fc region.
Polypeptides may be prepared using any of a variety of well known
techniques. Recombinant polypeptides encoded by DNA sequences as described
above may
be readily prepared from the DNA sequences using any of a variety of
expression vectors
known to those of ordinary skill in the art. Expression may be achieved in any
appropriate
host cell that has been transformed or transfected with an expression vector
containing a
DNA molecule that encodes a recombinant polypeptide. Suitable host cells
include
prokaryotes, yeast and higher eukaryotic cells. Preferably, the host cells
employed are
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
27
E. coli, yeast or a mammalian cell line such as COS or CHO. Supernatants from
suitable
hostlvector systems which secrete recombinant protein or polypeptide into
culture media may
be first concentrated using a commercially available filter. Following
concentration, the
concentrate may be applied to a suitable purification matrix such as an
affinity matrix or an
ion exchange resin. Finally, one or more reverse phase HPLC steps can be
employed to
further purify a recombinant polypeptide.
Portions and other variants having fewer than about 100 amino acids, and
generally fewer than about 50 amino acids, may also be generated by synthetic
means, using
techniques well known to those of ordinary skill in the art. For example, such
polypeptides
may be synthesized using any of the commercially available solid-phase
techniques, such as
the Merrifield solid-phase synthesis method, where amino acids are
sequentially added to a
growing amino acid chain. See Merrifield, J. Am. Chem. Soc. 85:2149-2146,
1963.
Equipment for automated synthesis of polypeptides is commercially available
from suppliers
such as Perkin Elmer/Applied BioSystems Division (Foster City, CA), and may be
operated
according to the manufacturer's instructions.
Within certain specific embodiments, a polypeptide may be a fusion protein
that comprises multiple polypeptides as described herein, or that comprises at
least one
polypeptide as described herein and an unrelated sequence, such as a known
tumor protein. A
fusion partner may, for example, assist in providing T helper epitopes (an
immunological
fusion partner), preferably T helper epitopes recognized by humans, or may
assist in
expressing the protein (an expression enhancer) at higher yields than the
native recombinant
protein. Certain preferred fusion partners are both immunological and
expression enhancing
fusion partners. Other fusion partners may be selected so as to increase the
solubility of the
protein or to enable the protein to be targeted to desired intracellular
compartments. Still
further fusion partners include affinity tags, which facilitate purification
of the protein.
Fusion proteins may generally be prepared using standard techniques,
including chemical conjugation. Preferably, a fusion protein is expressed as a
recombinant
protein, allowing the production of increased levels, relative to a non-fused
protein, in an
expression system. Briefly, DNA sequences encoding the polypeptide components
may be
assembled separately, and ligated into an appropriate expression vector. The
3' end of the
DNA sequence encoding one polypeptide component is ligated, with or without a
peptide
linker, to the 5' end of a DNA sequence encoding the second polypeptide
component so that
the reading frames of the sequences are in phase. This permits translation
into a single fusion
protein that retains the biological activity of both component polypeptides.
A peptide linker sequence may be employed to separate the first and the
second polypeptide components by a distance sufficient to ensure that each
polypeptide folds
into its secondary and tertiary structures. Such a peptide linker sequence is
incorporated into
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
28
the fusion protein using standard techniques well known in the art. Suitable
peptide linker
sequences may be chosen based on the following factors: (1) their ability to
adopt a flexible
extended conformation; (2) their inability to adopt a secondary structure that
could interact
with functional epitopes on the first and second polypeptides; and (3) the
lack of hydrophobic
or charged residues that might react with the polypeptide functional epitopes.
Preferred
peptide linker sequences contain Gly, Asn and Ser residues. Other near neutral
amino acids,
such as thr and Ala may also be used in the linker sequence. Amino acid
sequences which
may be usefully employed as linkers include those disclosed in Maratea et al.,
Gene 40:39-46,
1985; Murphy et al., Proc. Natl. Acad. Sci. USA 83:8258-8262, 1986; U.S.
Patent
No. 4,935,233 and U.S. Patent No. 4,751,180. The linker sequence may generally
be from I
to about 50 amino acids in length. Linker sequences are not required when the
first and
second polypeptides have non-essential N-terminal amino acid regions that can
be used to
separate the functional domains and prevent steric interference.
The ligated DNA sequences are operably linked to suitable transcriptional or
translational regulatory elements. The regulatory elements responsible for
expression of
DNA are located only 5' to the DNA sequence encoding the first polypeptides.
Similarly,
stop codons required to end translation and transcription termination signals
are only present
3' to the DNA sequence encoding the second polypeptide.
Fusion proteins are also provided that comprise a polypeptide of the present
invention together with an unrelated immunogenic protein. Preferably the
immunogenic
protein is capable of eliciting a recall response. Examples of such proteins
include tetanus,
tuberculosis and hepatitis proteins (see, for example, Stoute et al. New Engl.
J. Med., 336:86-
91, 1997).
Within preferred embodiments, an immunological fusion partner is derived
from protein D, a surface protein of the gram-negative bacterium Haemophilus
influenza B
(WO 91/18926). Preferably, a protein D derivative comprises approximately the
first third of
the protein (e.g., the first N-terminal 100-110 amino acids), and a protein D
derivative may be
lipidated. Within certain preferred embodiments, the first 109 residues of a
Lipoprotein D
fusion partner is included on the N-terminus to provide the polypeptide with
additional
exogenous T-cell epitopes and to increase the expression level in E. coli
(thus functioning as
an expression enhancer). The lipid tail ensures optimal presentation of the
antigen to antigen
presenting cells. Other fusion partners include the non-structural protein
from influenzae
virus, NS 1(hemaglutinin). Typically, the N-tenninal 81 amino acids are used,
although
different fragments that include T-helper epitopes may be used.
In another embodiment, the immunological fusion partner is the protein known
as LYTA, or a portion thereof (preferably a C-terminal portion). LYTA is
derived from
Streptococcus pneumoniae, which synthesizes an N-acetyl-L-alanine amidase
known as
CA 02613125 2007-11-29
WO 00/04149 PCT/[3S99/15838
29
amidase LYTA (encoded by the LytA gene; Gene 43:265-292, 1986). LYTA is an
autolysin
that specifically degrades certain bonds in the peptidoglycan backbone. The C-
terminal
domain of the LYTA protein is responsible for the affinity to the choline or
to some choline
analogues such as DEAE. This property has been exploited for the development
of E. coli C-
LYTA expressing plasmids useful for expression of fusion proteins.
Purification of hybrid
proteins containing the C-LYTA fragment at the amino terminus has been
described (see
Biotechriology 10:795-798, 1992). Within a preferred embodiment, a repeat
portion of LYTA
may be incorporated into a fusion protein. A repeat portion is found in the C-
terminal region
starting at residue 178. A particularly preferred repeat portion incorporates
residues 188-305.
In general, polypeptides (including fusion proteins) and polynucleotides as
described herein are isolated. An "isolated" polypeptide or polynucleotide is
one that is
removed from its original environment. For example, a naturally-occurring
protein is isolated
if it is separated from some or all of the coexisting materials in the natural
system.
Preferably, such polypeptides are at least about 90% pure, more preferably at
least about 95%
pure and most preferably at least about 99% pure. A polynucleotide is
considered to be
isolated if, for example, it is cloned into a vector that is not a part of the
natural environment.
BINDING AGENTS
The present invention further provides agents, such as antibodies and antigen-
binding fragments thereof, that specifically bind to a prostate tumor protein.
As used herein,
an antibody, or antigen-binding fragment thereof, is said to "specifically
bind" to a prostate
tumor protein if it reacts at a detectable level (within, for example, an
ELISA) with a prostate
tumor protein, and does not react detectably with unrelated proteins under
similar conditions.
As used herein, "binding" refers to a noncovalent association between two
separate molecules
such that a complex is formed. The ability to bind may be evaluated by, for
example,
determining a binding constant for the formation of the complex. The binding
constant is the
value obtained when the concentration of the complex is divided by the product
of the
component concentrations. In general, two compounds are said to "bind," in the
context of
the present invention, when the binding constant for complex formation exceeds
about 103
L/mol. The binding constant may be determined using methods well known in the
art.
Binding agents may be further capable of differentiating between patients with
and without a cancer, such as prostate cancer, using the representative assays
provided herein.
In other words, antibodies or other binding agents that bind to a prostate
tumor protein will
generate a signal indicating the presence of a cancer in at least about 20% of
patients with the
disease, and will generate a negative signal indicating the absence of the
disease in at least
about 90% of individuals without the cancer. To determine whether a binding
agent satisfies
this requirement, biological samples (e.g., blood, sera, urine and/or tumor
biopsies) from
CA 02613125 2007-11-29
WO 00/04149 PCT1US99/15838
patients with and without a cancer (as determined using standard clinical
tests) may be
assayed as described herein for the presence of polypeptides that bind to the
binding agent. It
will be apparent that a statistically significant number of samples with and
without the
disease should be assayed. Each binding agent should satisfy the above
criteria; however,
those of ordinary skill in the art will recognize that binding agents may be
used in
combination to improve sensitivity.
Any agent that satisfies the above requirements may be a binding agent. For
example, a binding agent may be a ribosome, with or without a peptide
component, an RNA
molecule or a polypeptide. In a preferred embodiment, a binding agent is an
antibody or an
antigen-binding fragment thereof. Antibodies may be prepared by any of a
variety of
techniques known to those of ordinary skill in the art. See, e.g., Harlow and
Lane,
Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988. In
general,
antibodies can be produced by cell culture techniques, including the
generation of
monoclonal antibodies as described herein, or via transfection of antibody
genes into suitable
bacterial or mammalian cell hosts, in order to allow for the production of
recombinant
antibodies. In one technique, an immunogen comprising the polypeptide is
initially injected
into any of a wide variety of mammals (e.g., mice, rats, rabbits, sheep or
goats). In this step,
the polypeptides of this invention may serve as the immunogen without
modification.
Alternatively, particularly for relatively short polypeptides, a superior
immune response may
be elicited if the polypeptide is joined to a carrier protein, such as bovine
serum albumin or
keyhole limpet hemocyanin. The immunogen is injected into the animal host,
preferably
according to a predetermined schedule incorporating one or more booster
immunizations, and
the animals are bled periodically. Polyclonal antibodies specific for the
polypeptide may then
be purified from such antisera by, for example, affinity chromatography using
the polypeptide
coupled to a suitable solid support.
Monoclonal antibodies specific for an antigenic polypeptide of interest may be
prepared, for example, using the technique of Kohler and Milstein, Eur. J.
Immunol. 6:511 -
519, 1976, and improvements thereto. Briefly, these methods involve the
preparation of
immortal cell lines capable of producing antibodies having the desired
specificity (i.e.,
reactivity with the polypeptide of interest). Such cell lines may be produced,
for example,
from spleen cells obtained from an animal immunized as described above. The
spleen cells
are then immortalized by, for example, fusion with a myeloma cell fusion
partner, preferably
one that is syngeneic with the immunized animal. A variety of fusion
techniques may be
employed. For example, the spleen cells and myeloma cells may be combined with
a
nonionic detergent for a few minutes and then plated at low density on a
selective medium
that supports the growth of hybrid cells, but not myeloma cells. A preferred
selection
technique uses HAT (hypoxanthine, aminopterin, thymidine) selection. After a
sufficient
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
31
time, usually about I to 2 weeks, colonies of hybrids are observed. Single
colonies are
selected and their culture supernatants tested for binding activity against
the polypeptide.
Hybridomas having high reactivity and specificity are preferred.
Monoclonal antibodies may be isolated from the supernatants of growing
hybridoma colonies. In addition, various techniques may be employed to enhance
the yield,
such as injection of the hybridoma cell line into the peritoneal cavity of a
suitable vertebrate
host, such.as a mouse. Monoclonal antibodies may then be harvested from the
ascites fluid or
the blood. Contaminants may be removed from the antibodies by conventional
techniques,
such as chromatography, gel filtration, precipitation, and extraction. The
polypeptides of this
invention may be used in the purification process in, for example, an affinity
chromatography
step.
Within certain embodiments, the use of antigen-binding fragments of
antibodies may be preferred. Such fragments include Fab fragments, which may
be prepared
using standard techniques. Briefly, immunoglobulins may be purified from
rabbit serum by
affinity chromatography on Protein A bead columns (Harlow and Lane,
Antibodies: A
Laboratory iLlanual, Cold Spring Harbor Laboratory, 1988) and digested by
papain to yield
Fab and Fc fragments. The Fab and Fc fragments may be separated by affinity
chromatography on protein A bead columns.
Monoclonal antibodies of the present invention may be coupled to one or more
therapeutic agents. Suitable agents in this regard include radionuclides,
differentiation
inducers, drugs, toxins, and derivatives thereof. Preferred radionuclides
include 9DY,1z3I, 125I,
131I, '86Re, 'ggRe, 21 'At, and 212 Bi. Preferred drugs include methotrexate,
and pyrimidine and
purine analogs. Preferred differentiation inducers include phorbol esters and
butyric acid.
Preferred toxins include ricin, abrin, diptheria toxin, cholera toxin,
gelonin, Pseudomonas
exotoxin, Shigella toxin, and pokeweed antiviral protein.
A therapeutic agent may be coupled (e.g., covalently bonded) to a suitable
monoclonal antibody either directly or indirectly (e.g., via a linker group).
A direct reaction
between an agent and an antibody is possible when each possesses a substituent
capable of
reacting with the other. For example, a nucleophilic group, such as an amino
or sulfhydryl
group, on one may be capable of reacting with a carbonyl-containing group,
such as an
anhydride or an acid halide, or with an alkyl group containing a good leaving
group (e.g., a
halide) on the other.
Alternatively, it may be desirable to couple a therapeutic agent and an
antibody via a linker group. A linker group can function as a spacer to
distance an antibody
from an agent in order to avoid interference with binding capabilities. A
linker group can
also serve to increase the chemical reactivity of a substituent on an agent or
an antibody, and
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
32
thus increase the coupling efficiency. An increase in chemical reactivity may
also facilitate
the use of agents, or functional groups on agents, which otherwise would not
be possible.
It will be evident to those skilled in the art that a variety of bifunctional
or
polyfunctional reagents, both homo- and hetero-functional (such as those
described in the
catalog of the Pierce Chemical Co., Rockford, IL), may be employed as the
linker group.
Coupling may be effected, for example, through amino groups, carboxyl groups,
sulfhydryl
groups or' oxidized carbohydrate residues. There are numerous references
describing such
methodology, e.g., U.S. Patent No. 4,671,958, to Rodwell et al.
Where a therapeutic agent is more potent when free from the antibody portion
of the immunoconjugates of the present invention, it may be desirable to use a
linker group
which is cleavable during or upon internalization into a cell. A number of
different cleavable
linker groups have been described. The mechanisms for the intracellular
release of an agent
from these linker groups include cleavage by reduction of a disulfide bond
(e.g., U.S. Patent
No. 4,489,710, to Spitler), by irradiation of a photolabile bond (e.g., U.S.
Patent
No. 4,625,014, to Senter et al.), by hydrolysis of derivatized amino acid side
chains (e.g., U.S.
Patent No. 4,638,045, to Kohn et al.), by serum complement-mediated hydrolysis
(e.g., U.S.
Patent No. 4,671,958, to Rodwell et al.), and acid-catalyzed hydrolysis (e.g.,
U.S. Patent
No. 4,569,789, to Blattler et al.).
It may be desirable to couple more than one agent to an antibody. In one
embodiment, multiple molecules of an agent are coupled to one antibody
molecule. In
another embodiment, more than one type of agent may be coupled to one
antibody.
Regardless of the particular embodiment, immunoconjugates with more than one
agent may
be prepared in a variety of ways. For example, more than one agent may be
coupled directly
to an antibody molecule, or linkers which provide multiple sites for
attachment can be used.
Alternatively, a carrier can be used.
A carrier may bear the agents in a variety of ways, including covalent bonding
either directly or via a linker group. Suitable carriers include proteins such
as albumins (e.g.,
U.S. Patent No. 4,507,234, to Kato et al.), peptides and polysaccharides such
as aminodextran
(e.g., U.S. Patent No. 4,699,784, to Shih et al.). A carrier may also bear an
agent by
noncovalent bonding or by encapsulation, such as within a liposome vesicle
(e.g., U.S. Patent
Nos. 4,429,008 and 4,873,088). Carriers specific for radionuclide agents
include
radiohalogenated small molecules and chelating compounds. For example, U.S.
Patent No.
4,735,792 discloses representative radiohalogenated small molecules and their
synthesis. A
radionuclide chelate may be formed from chelating compounds that include those
containing
nitrogen and sulfur atoms as the donor atoms for binding the metal, or metal
oxide,
radionuclide. For example, U.S. Patent No. 4,673,562, to Davison et al.
discloses
representative chelating compounds and their synthesis.
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
33
A variety of routes of administration for the antibodies and immunoconjugates
may be used. Typically, administration will be intravenous, intramuscular,
subcutaneous or
in the bed of a resected tumor. It will be evident that the precise dose of
the
antibody/immunoconjugate will vary depending upon the antibody used, the
antigen density
on the tumor, and the rate of clearance of the antibody.
T CELLS'
Immunotherapeutic compositions may also, or alternatively, comprise T cells
specific for a prostate tumor protein. Such cells may generally be prepared in
vitro or ex vivo,
using standard procedures. For example, T cells may be isolated from bone
marrow,
peripheral blood, or a fraction of bone marrow or peripheral blood of a
patient, using a
commercially available cell separation system, such as the CEPRATET"" system,
available
from Ce11Pro Inc., Bothell WA (see also U.S. Patent No. 5,240,856; U.S. Patent
No.
5,215,926; WO 89/06280; WO 91/16116 and WO 92/07243). Alternatively, T cells
may be
derived from related or unrelated humans, non-human mammals, cell lines or
cultures.
T cells may be stimulated with a prostate tumor polypeptide, polynucleotide
encoding a prostate tumor polypeptide and/or an antigen presenting cell (APC)
that expresses
such a polypeptide. Such stimulation is performed under conditions and for a
time sufficient
to permit the generation of T cells that are specific for the polypeptide.
Preferably, a prostate
tumor polypeptide or polynucleotide is present within a delivery vehicle, such
as a
microsphere, to facilitate the generation of specific T cells.
T cells are considered to be specific for a prostate tumor polypeptide if the
T
cells kill target cells coated with the polypeptide or expressing a gene
encoding the
polypeptide. T cell specificity may be evaluated using any of a variety of
standard
techniques. For example, within a chromium release assay or proliferation
assay, a
stimulation index of more than two fold increase in lysis and/or
proliferation, compared to
negative controls, indicates T cell specificity. Such assays may be performed,
for example, as
described in Chen et al., Cancer Res. 54:1065-1070, 1994. Alternatively,
detection of the
proliferation of T cells may be accomplished by a variety of known techniques.
For example,
T cell proliferation can be detected by measuring an increased rate of DNA
synthesis (e.g., by
pulse-labeling cultures of T cells with tritiated thymidine and measuring the
amount of
tritiated thymidine incorporated into DNA). Contact with a prostate tumor
polypeptide (100
ng/ml - 100 g/ml, preferably 200 ng/ml - 25 g/ml) for 3 - 7 days should
result in at least a
two fold increase in proliferation of the T cells. Contact as described above
for 2-3 hours
should result in activation of the T cells, as measured using standard
cytokine assays in which
a two fold increase in the level of cytokine release (e.g., TNF or IFN-y) is
indicative of T cell
activation (see Coligan et al., Current Protocols in Immunology, vol. 1, Wiley
Interscience
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
34
(Greene 1998)). T cells that have been activated in response to a prostate
tumor polypeptide,
polynucleotide or polypeptide-expressing APC may be CD4' and/or CD8'. Prostate
tumor
protein-specific T cells may be expanded using standard techniques. Within
preferred
embodiments, the T cells are derived from either a patient or a related, or
unrelated, donor
and are administered to the patient following stimulation and expansion.
For therapeutic purposes, CD4+ or CD8+ T cells that proliferate in response to
a prostattY tumor polypeptide, polynucleotide or APC can be expanded in number
either
in vitro or in vivo. Proliferation of such T cells in vitro may be
accomplished in a variety of
ways. For example, the T cells can be re-exposed to a prostate tumor
polypeptide, or a short
peptide corresponding to an immunogenic portion of such a polypeptide, with or
without the
addition of T cell growth factors, such as interleukin-2, and/or stimulator
cells that synthesize
a prostate tumor polypeptide. Alternatively, one or more T cells that
proliferate in the
presence of a prostate tumor protein can be expanded in number by cloning.
Methods for
cloning cells are well known in the art, and include limiting dilution.
PHARMACEUTICAL COMPOSITIONS AND VACCINES
Within certain aspects, polypeptides, polynucleotides, T cells and/or binding
agents disclosed herein may be incorporated into pharmaceutical compositions
or
immunogenic compositions (i.e., vaccines). Pharmaceutical compositions
comprise one or
more such compounds and a physiologically acceptable carrier. Vaccines may
comprise one
or more such compounds and a non-specific immune response enhancer. A non-
specific
immune response enhancer may be any substance that enhances an immune response
to an
exogenous antigen. Examples of non-specific immune response enhancers include
adjuvants,
biodegradable microspheres (e.g., polylactic galactide) and liposomes (into
which the
compound is incorporated; see e.g., Fullerton, U.S. Patent No. 4,235,877).
Vaccine
preparation is generally described in, for example, M.F. Powell and M.J.
Newman, eds.,
"Vaccine Design (the subunit and adjuvant approach)," Plenum Press (NY, 1995).
Pharmaceutical compositions and vaccines within the scope of the present
invention may also
contain other compounds, which may be biologically active or inactive. For
example, one or
more immunogenic portions of other tumor antigens may be present, either
incorporated into
a fusion polypeptide or as a separate compound, within the composition or
vaccine.
A pharmaceutical composition or vaccine may contain DNA encoding one or
more of the polypeptides as described above, such that the polypeptide is
generated in situ.
As noted above, the DNA may be present within any of a variety of delivery
systems known
to those of ordinary skill in the art, including nucleic acid expression
systems, bacteria and
viral expression systems. Numerous gene delivery techniques are well known in
the art, such
as those described by Rolland, Crit. Rev. Therap. Drug Carrier Systems 15:143-
198, 1998,
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
and references cited therein. Appropriate nucleic acid expression systems
contain the
necessary DNA sequences for expression in the patient (such as a suitable
promoter and
terminating signal). Bacterial delivery systems involve the administration of
a bacterium
(such as Bacillus-Calmette-Guerrin) that expresses an inununogenic portion of
the
polypeptide on its cell surface or secretes such an epitope. In a preferred
embodiment, the
DNA may be introduced using a viral expression system (e.g., vaccinia or other
pox virus,
retrovirus; or adenovirus), which may involve the use of a non-pathogenic
(defective),
replication competent virus. Suitable systems are disclosed, for example, in
Fisher-Hoch et
al., Proc. Natl. Acad. Sci. USA 86:317-321, 1989; Flexner et al., Ann. N. Y.
Acad. Sci.
569:86-103, 1989; Flexner et al., Vaccine 8:17-21, 1990; U.S. Patent Nos.
4,603,112,
4,769,330, and 5,017,487; WO 89/01973; U.S. Patent No. 4,777,127; GB
2,200,651;
EP 0,345,242; WO 91/02805; Berkner, Biotechniques 6:616-627, 1988; Rosenfeld
et al.,
Science 252:431-434, 1991; Kolls et al., Proc. Natl. Aead. Sci. USA 91:215-
219, 1994;
Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90:11498-11502, 1993; Guzman et
al.,
Circulation 88:2838-2848, 1993; and Guzman et al., Cir. Res. 73:1202-1207,
1993.
Techniques for incorporating DNA into such expression systems are well known
to those of
ordinary skill in the art. The DNA may also be "naked," as described, for
example, in Ulmer
et al., Science 259:1745-1749, 1993 and reviewed by Cohen, Science 259:1691-
1692, 1993.
The uptake of naked DNA may be increased by coating the DNA onto biodegradable
beads,
which are efficiently transported into the cells.
While any suitable carrier known to those of ordinary skill in the art may be
employed in the pharmaceutical compositions of this invention, the type of
carrier will vary
depending on the mode of administration. Compositions of the present invention
may be
formulated for any appropriate manner of administration, including for
example, topical, oral,
nasal, intravenous, intracranial, intraperitoneal, subcutaneous or
intramuscular administration.
For parenteral administration, such as subcutaneous injection, the carrier
preferably
comprises water, saline, alcohol, a fat, a wax or a buffer. For oral
administration, any of the
above carriers or a solid carrier, such as mannitol, lactose, starch,
magnesium stearate,
sodium saccharine, talcum, cellulose, glucose, sucrose, and magnesium
carbonate, may be
employed. Biodegradable microspheres (e.g., polylactate polyglycolate) may
also be
employed as carriers for the pharmaceutical compositions of this invention.
Suitable
biodegradable microspheres are disclosed, for example, in U.S. Patent Nos.
4,897,268 and
5,075,109.
Such compositions may also comprise buffers (e.g., neutral buffered saline or
phosphate buffered saline), carbohydrates (e.g., glucose, mannose, sucrose or
dextrans),
mannitol, proteins, polypeptides or amino acids such as glycine, antioxidants,
chelating
agents such as EDTA or glutathione, adjuvants (e.g., aluminum hydroxide)
and/or
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
36
preservatives. Alternatively, compositions of the present invention may be
formulated as a
lyophilizate. Compounds may also be encapsulated within liposomes using well
known
technology.
Any of a variety of non-specific immune response enhancers may be employed
in the vaccines of this invention. For example, an adjuvant may be included.
Most adjuvants
contain a, substance designed to protect the antigen from rapid catabolism,
such as aluminum
hydroxide-or mineral oil, and a stimulator of immune responses, such as lipid
A, Bortadella
pertussis or Mycobacterium tuberculosis derived proteins. Suitable adjuvants
are
commercially available as, for example, Freund's Incomplete Adjuvant and
Complete
Adjuvant (Difco Laboratories, Detroit, MI); Merck Adjuvant 65 (Merck and
Company, Inc.,
Rahway, NJ); aluminum salts such as aluminum hydroxide gel (alum) or aluminum
phosphate; salts of calcium, iron or zinc; an insoluble suspension of acylated
tyrosine;
acylated sugars; cationically or anionically derivatized polysaccharides;
polyphosphazenes;
biodegradable microspheres; monophosphoryl lipid A and quil A. Cytokines, such
as GM-
CSF or interleukin-2, -7, or -12, may also be used as adjuvants.
Within the vaccines provided herein, the adjuvant composition is preferably
designed to induce an immune response predominantly of the Thl type. High
levels of Th 1-
type cytokines (e.g., IFN-y, IL-2 and IL- 12) tend to favor the induction of
cell mediated
immune responses to an administered antigen. In contrast, high levels of Th2-
type cytokines
(e.g., IL-4, IL-5, IL-6, IL-10 and TNF-(3) tend to favor the induction of
humoral immune
responses. Following application of a vaccine as provided herein, a patient
will support an
immune response that includes Thl- and Th2-type responses. Within a preferred
embodiment, in which a response is predominantly Th1-type, the level of Thl-
type cytokines
will increase to a greater extent than the level of Th2-type cytokines. The
levels of these
cytokines may be readily assessed using standard assays. For a review of the
families of
cytokines, see Mosmann and Coffman, Ann. Rev. Immunol. 7:145-173, 1989.
Preferred adjuvants for use in eliciting a predominantly Thl-type response
include, for example, a combination of monophosphoryl lipid A, preferably 3-de-
O-acylated
monophosphoryl lipid A (3D-MPL), together with an aluminum salt. MPL adjuvants
are
available from Ribi ImmunoChem Research Inc. (Hamilton, MT; see US Patent Nos.
4,436,727; 4,877,611; 4,866,034 and 4,912,094). CpG-containing
oligonucleotides (in which
the CpG dinucleotide is unmethylated) also induce a predominantly Thl
response. Such
oligonucleotides are well known and are described, for example, in WO
96/02555. Another
preferred adjuvant is a saponin, preferably QS21, which may be used alone or
in combination
with other adjuvants. For example, an enhanced system involves the combination
of a
monophosphoryl lipid A and saponin derivative, such as the combination of QS21
and 3D-
MPL as described in WO 94/00153, or a less reactogenic composition where the
QS21 is
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
37
quenched with cholesterol, as described in WO 96/33739. Other preferred
formulations
comprises an oil-in-water emulsion and tocopherol. A particularly potent
adjuvant
formulation involving QS21, 3D-MPL and tocopherol in an oil-in-water emulsion
is
described in WO 95/17210. Any vaccine provided herein may be prepared using
well known
methods that result in a combination of antigen, immune response enhancer and
a suitable
carrier or excipient.
The compositions described herein may be administered as part of a sustained
release formulation (i.e., a formulation such as a capsule or sponge that
effects a slow release
of compound following administration). Such formulations may generally be
prepared using
well known technology and administered by, for example, oral, rectal or
subcutaneous
implantation, or by implantation at the desired target site. Sustained-release
formulations
may contain a polypeptide, polynucleotide or antibody dispersed in a carrier
matrix and/or
contained within a reservoir surrounded by a rate controlling membrane. Camers
for use
within such formulations are biocompatible, and may also be biodegradable;
preferably the
formulation provides a relatively constant level of active component release.
The amount of
active compound contained within a sustained release formulation depends upon
the site of
implantation, the rate and expected duration of release and the nature of the
condition to be
treated or prevented.
Any of a variety of delivery vehicles may be employed within pharmaceutical
compositions and vaccines to facilitate production of an antigen-specific
immune response
that targets tumor cells. Delivery vehicles include antigen presenting cells
(APCs), such as
dendritic cells, macrophages, B cells, monocytes and other cells that may be
engineered to be
efficient APCs. Such cells may, but need not, be genetically modified to
increase the
capacity for presenting the antigen, to improve activation and/or maintenance
of the T cell
response, to have anti-tumor effects per se and/or to be immunologically
compatible with the
receiver (i.e., matched HLA haplotype). APCs may generally be isolated from
any of a
variety of biological fluids and organs, including tumor and peritumoral
tissues, and may be
autologous, allogeneic, syngeneic or xenogeneic cells.
Certain preferred embodiments of the present invention use dendritic cells or
progenitors thereof as antigen-presenting cells. Dendritic cells are highly
potent APCs
(Banchereau and Steinman, Nature 392:245-251, 1998) and have been shown to be
effective
as a physiological adjuvant for eliciting prophylactic or therapeutic
antitumor immunity (see
Timmerman and Levy, Ann. Rev. Med. 50:507-529, 1999). In general, dendritic
cells may be
identified based on their typical shape (stellate in situ, with marked
cytoplasmic processes
(dendrites) visible in vitro) and based on the lack of differentiation markers
of B cells (CD19
and CD20), T cells (CD3), monocytes (CD14) and natural killer cells (CD56), as
determined
using standard assays. Dendritic cells may, of course, be engineered to
express specific cell-
CA 02613125 2007-11-29
WO 00/04149 PCT/[7S99/15838
38
surface receptors or ligands that are not commonly found on dendritic cells in
vivo or ex vivo,
and such modified dendritic cells are contemplated by the present invention.
As an
alternative to dendritic cells, secreted vesicles antigen-loaded dendritic
cells (called
exosomes) may be used within a vaccine (see Zitvogel et al., Nature Med. 4:594-
600, 1998).
Dendritic cells and progenitors may be obtained from peripheral blood, bone
marrow, tumor-infiltrating cells, peritumoral tissues-infiltrating cells,
lymph nodes, spleen,
skin, umbilical cord blood or any other suitable tissue or fluid. For example,
dendritic cells
may be differentiated ex vivo by adding a combination of cytokines such as GM-
CSF, IL-4,
IL- 13 and/or TNFa to cultures of monocytes harvested from peripheral blood.
Alternatively,
CD34 positive cells harvested from peripheral blood, umbilical cord blood or
bone marrow
may be differentiated into dendritic cells by adding to the culture medium
combinations of
GM-CSF, IL-3, TNFa, CD40 ligand, LPS, flt3 ligand and/or other compound(s)
that induce
maturation and proliferation of dendritic cells.
Dendritic cells are conveniently categorized as "immature" and "mature" cells,
which allows a simple way to discriminate between two well characterized
phenotypes.
However, this nomenclature should not be construed to exclude all possible
intermediate
stages of differentiation. Immature dendritic cells are characterized as APC
with a high
capacity for antigen uptake and processing, which correlates with the high
expression of Fcy
receptor, mannose receptor and DEC-205 marker. The mature phenotype is
typically
characterized by a lower expression of these markers, but a high expression of
cell surface
molecules responsible for T cell activation such as class I and class II MHC,
adhesion
molecules (e.g., CD54 and CD11) and costimulatory molecules (e.g., CD40, CD80
and
CD86).
APCs may generally be transfected with a polynucleotide encoding a prostate
tumor protein (or portion or other variant thereof) such that the prostate
tumor polypeptide, or
an immunogenic portion thereof, is expressed on the cell surface. Such
transfection may take
place ex vivo, and a composition or vaccine comprising such transfected cells
may then be
used for therapeutic purposes, as described herein. Alternatively, a gene
delivery vehicle that
targets a dendritic or other antigen presenting cell may be administered to a
patient, resulting
in transfection that occurs in vivo. In vivo and ex vivo transfection of
dendritic cells, for
example, may generally be performed using any methods known in the art, such
as those
described in WO 97/24447, or the gene gun approach described by Mahvi et al.,
Immunology
and cell Biology 75:456-460, 1997. Antigen loading of dendritic cells may be
achieved by
incubating dendritic cells or progenitor cells with the prostate tumor
polypeptide, DNA
(naked or within a plasmid vector) or RNA; or with antigen-expressing
recombinant
bacterium or viruses (e.g., vaccinia, fowlpox, adenovirus or lentivirus
vectors). Prior to
loading, the polypeptide may be covalently conjugated to an immunological
partner that
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
39
provides T cell help (e.g., a carrier molecule). Alternatively, a dendritic
cell may be pulsed
with a non-conjugated immunological partner, separately or in the presence of
the
polypeptide.
CANCER THERAPY
In further aspects of the present invention, the compositions described herein
may be u'sed for immunotherapy of cancer, such as prostate cancer. Within such
methods,
pharmaceutical compositions and vaccines are typically administered to a
patient. As used
herein, a "patient" refers to any warm-blooded animal, preferably a human. A
patient may or
may not be afflicted with cancer. Accordingly, the above pharmaceutical
compositions and
vaccines may be used to prevent the development of a cancer or to treat a
patient afflicted
with a cancer. A cancer may be diagnosed using criteria generally accepted in
the art,
including the presence of a malignant tumor. Pharmaceutical compositions and
vaccines may
be administered either prior to or following surgical removal of primary
tumors and/or
treatment such as administration of radiotherapy or conventional
chemotherapeutic drugs.
Within certain embodiments, immunotherapy may be active immunotherapy,
in which treatment relies on the in vivo stimulation of the endogenous host
immune system to
react against tumors with the administration of immune response-modifying
agents (such as
polypeptides and polynucleotides disclosed herein).
Within other embodiments, immunotherapy may be passive immunotherapy,
in which treatment involves the delivery of agents with established tumor-
immune reactivity
(such as effector cells or antibodies) that can directly or indirectly mediate
antitumor effects
and does not necessarily depend on an intact host immune system. Examples of
effector cells
include T cells as discussed above, T lymphocytes (such as CD8+ cytotoxic T
lymphocytes
and CD4' T-helper tumor-infiltrating lymphocytes), killer cells (such as
Natural Killer cells
and lymphokine-activated killer cells), B cells and antigen-presenting cells
(such as dendritic
cells and macrophages) expressing a polypeptide provided herein. T cell
receptors and
antibody receptors specific for the polypeptides recited herein may be cloned,
expressed and
transferred into other vectors or effector cells for adoptive immunotherapy.
The polypeptides
provided herein may also be used to generate antibodies or anti-idiotypic
antibodies (as
described above and in U.S. Patent No. 4,918,164) for passive immunotherapy.
Effector cells may generally be obtained in sufficient quantities for adoptive
immunotherapy by growth in vitro, as described herein. Culture conditions for
expanding
single antigen-specific effector cells to several billion in number with
retention of antigen
recognition in vivo are well known in the art. Such in vitro culture
conditions typically use
intermittent stimulation with antigen, often in the presence of cytokines
(such as IL-2) and
non-dividing feeder cells. As noted above, immunoreactive polypeptides as
provided herein
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
may be used to rapidly expand antigen-specific T cell cultures in order to
generate a sufficient
number of cells for immunotherapy. In particular, antigen-presenting cells,
such as dendritic,
macrophage, monocyte, fibroblast or B cells, may be pulsed with immunoreactive
polypeptides or transfected with one or more polynucleotides using standard
techniques well
known in the art. For example, antigen-presenting cells can be transfected
with a
polynucleotide having a promoter appropriate for increasing expression in a
recombinant
virus or dther expression system. Cultured effector cells for use in therapy
must be able to
grow and distribute widely, and to survive long term in vivo. Studies have
shown that
cultured effector cells can be induced to grow in vivo and to survive long
term in substantial
numbers by repeated stimulation with antigen supplemented with IL-2 (see, for
example,
Cheever et al., Immunological Reviews 157:177, 1997).
Alternatively, a vector expressing a polypeptide recited herein may be
introduced into antigen presenting cells taken from a patient and clonally
propagated ex vivo
for transplant back into the same patient. Transfected cells may be
reintroduced into the
patient using any means known in the art, preferably in sterile form by
intravenous,
intracavitary, intraperitoneal or intratumor administration.
Routes and frequency of administration of the therapeutic compositions
disclosed herein, as well as dosage, will vary from individual to individual,
and may be
readily established using standard techniques. In general, the pharmaceutical
compositions
and vaccines may be administered by injection (e.g., intracutaneous,
intramuscular,
intravenous or subcutaneous), intranasally (e.g., by aspiration) or orally.
Preferably, between
1 and 10 doses may be administered over a 52 week period. Preferably, 6 doses
are
administered, at intervals of 1 month, and booster vaccinations may be given
periodically
thereafter. Alternate protocols may be appropriate for individual patients. A
suitable dose is
an amount of a compound that, when administered as described above, is capable
of
promoting an anti-tumor immune response, and is at least 10-50% above the
basal (i.e.,
untreated) level. Such response can be monitored by measuring the anti-tumor
antibodies in a
patient or by vaccine-dependent generation of cytolytic effector cells capable
of killing the
patient's tumor cells in vitro. Such vaccines should also be capable of
causing an immune
response that leads to an improved clinical outcome (e.g., more frequent
remissions, complete
or partial or longer disease-free survival) in vaccinated patients as compared
to non-
vaccinated patients. In general, for pharmaceutical compositions and vaccines
comprising
one or more polypeptides, the amount of each polypeptide present in a dose
ranges from
about 100 g to 5 mg per kg of host. Suitable dose sizes will vary with the
size of the patient,
but will typically range from about 0.1 mL to about 5 mL.
In general, an appropriate dosage and treatment regimen provides the active
compound(s) in an amount sufficient to provide therapeutic andlor prophylactic
benefit. Such
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
41
a response can be monitored by establishing an improved clinical outcome
(e.g., more
frequent remissions, complete or partial, or longer disease-free survival) in
treated patients as
compared to non-treated patients. Increases in preexisting immune responses to
a prostate
tumor protein generally correlate with an improved clinical outcome. Such
immune
responses may generally be evaluated using standard proliferation,
cytotoxicity or cytokine
assays, which may be performed using samples obtained from a patient before
and after
treatment.'-
1VIETHODS FOR DETECTING CANCER
In general, a cancer may be detected in a patient based on the presence of one
or more prostate tumor proteins and/or polynucleotides encoding such proteins
in a biological
sample (for example, blood, sera, urine and/or tumor biopsies) obtained from
the patient. In
other words, such proteins may be used as markers to indicate the presence or
absence of a
cancer such as prostate cancer. In addition, such proteins may be useful for
the detection of
other cancers. The binding agents provided herein generally permit detection
of the level of
antigen that binds to the agent in the biological sample. Polynucleotide
primers and probes
may be used to detect the level of mRNA encoding a tumor protein, which is
also indicative
of the presence or absence of a cancer. In general, a prostate tumor sequence
should be
present at a level that is at least three fold higher in tumor tissue than in
normal tissue
There are a variety of assay formats known to those of ordinary skill in the
art
for using a binding agent to detect polypeptide markers in a sample. See,
e.g., Harlow and
Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988. In
general,
the presence or absence of a cancer in a patient may be determined by (a)
contacting a
biological sample obtained from a patient with a binding agent; (b) detecting
in the sample a
level of polypeptide that binds to the binding agent; and (c) comparing the
level of
polypeptide with a predetermined cut-off value.
In a preferred embodiment, the assay involves the use of binding agent
immobilized on a solid support to bind to and remove the polypeptide from the
remainder of
the sample. The bound polypeptide may then be detected using a detection
reagent that
contains a reporter group and specifically binds to the binding
agent/polypeptide complex.
Such detection reagents may comprise, for example, a binding agent that
specifically binds to
the polypeptide or an antibody or other agent that specifically binds to the
binding agent, such
as an anti-immunoglobulin, protein G, protein A or a lectin. Alternatively, a
competitive
assay may be utilized, in which a polypeptide is labeled with a reporter group
and allowed to
bind to the immobilized binding agent after incubation of the binding agent
with the sample.
The extent to which components of the sample inhibit the binding of the
labeled polypeptide
to the binding agent is indicative of the reactivity of the sample with the
immobilized binding
CA 02613125 2007-11-29
WO 00/04149 PCTlUS99/15838
42
agent. Suitable polypeptides for use within such assays include full length
prostate tumor
proteins and portions thereof to which the binding agent binds, as described
above.
The solid support may be any material known to those of ordinary skill in the
art to which the tumor protein may be attached. For example, the solid support
may be a test
well in a microtiter plate or a nitrocellulose or other suitable membrane.
Alternatively, the
support may be a bead or disc, such as glass, fiberglass, latex or a plastic
material such as
polystyrene or polyvinylchloride. The support may also be a magnetic particle
or a fiber
optic sensor, such as those disclosed, for example, in U.S. Patent No.
5,359,681. The binding
agent may be immobilized on the solid support using a variety of techniques
known to those
of skill in the art, which are amply described in the patent and scientific
literature. In the
context of the present invention, the term "immobilization" refers to both
noncovalent
association, such as adsorption, and covalent attachment (which may be a
direct linkage
between the agent and functional groups on the support or may be a linkage by
way of a
cross-linking agent). Inunobilization by adsorption to a well in a microtiter
plate or to a
membrane is preferred. In such cases, adsorption may be achieved by contacting
the binding
agent, in a suitable buffer, with the solid support for a suitable amount of
time. The contact
time varies with temperature, but is typically between about 1 hour and about
1 day. In
general, contacting a well of a plastic microtiter plate (such as polystyrene
or
polyvinylchloride) with an amount of binding agent ranging from about 10 ng to
about 10 g,
and preferably about 100 ng to about 1 g, is sufficient to immobilize an
adequate amount of
binding agent.
Covalent attachment of binding agent to a solid support may generally be
achieved by first reacting the support with a bifunctional reagent that will
react with both the
support and a functional group, such as a hydroxyl or amino group, on the
binding agent. For
example, the binding agent may be covalently attached to supports having an
appropriate
polymer coating using benzoquinone or by condensation of an aldehyde group on
the support
with an amine and an active hydrogen on the binding partner (see, e.g., Pierce
Immunotechnology Catalog and Handbook, 1991, at A12-A13).
In certain embodiments, the assay is a two-antibody sandwich assay. This
assay may be performed by first contacting an antibody that has been
immobilized on a solid
support, conunonly the well of a microtiter plate, with the sample, such that
polypeptides
within the sample are allowed to bind to the immobilized antibody. Unbound
sample is then
removed from the immobilized polypeptide-antibody complexes and a detection
reagent
(preferably a second antibody capable of binding to a different site on the
polypeptide)
containing a reporter group is added. The amount of detection reagent that
remains bound to
the solid support is then determined using a method appropriate for the
specific reporter
group.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
43
More specifically, once the antibody is immobilized on the support as
described above, the remaining protein binding sites on the support are
typically blocked.
Any suitable blocking agent known to those of ordinary skill in the art, such
as bovine serum
albumin or Tween 20TM (Sigma Chemical Co., St. Louis, MO). The immobilized
antibody is
then incubated with the sample, and polypeptide is allowed to bind to the
antibody. The
sample may be diluted with a suitable diluent, such as phosphate-buffered
saline (PBS) prior
to incub~mion. In general, an appropriate contact time (i.e., incubation time)
is a period of
time that is sufficient to detect the presence of polypeptide within a sample
obtained from an
individual with prostate cancer. Preferably, the contact time is sufficient to
achieve a level of
binding that is at least about 95% of that achieved at equilibrium between
bound and unbound
polypeptide. Those of ordinary skill in the art will recognize that the time
necessary to
achieve equilibrium may be readily determined by assaying the level of binding
that occurs
over a period of time. At room temperature, an incubation time of about 30
minutes is
generally sufficient.
Unbound sample may then be removed by washing the solid support with an
appropriate buffer, such as PBS containing 0.1% Tween 20T"'. The second
antibody, which
contains a reporter group, may then be added to the solid support. Preferred
reporter groups
include those groups recited above.
The detection reagent is then incubated with the immobilized antibody-
polypeptide complex for an amount of time sufficient to detect the bound
polypeptide. An
appropriate amount of time may generally be determined by assaying the level
of binding that
occurs over a period of time. Unbound detection reagent is then removed and
bound
detection reagent is detected using the reporter group. The method employed
for detecting
the reporter group depends upon the nature of the reporter group. For
radioactive groups,
scintillation counting or autoradiographic methods are generally appropriate.
Spectroscopic
methods may be used to detect dyes, luminescent groups and fluorescent groups.
Biotin may
be detected using avidin, coupled to a different reporter group (commonly a
radioactive or
fluorescent group or an enzyme). Enzyme reporter groups may generally be
detected by the
addition of substrate (generally for a specific period of time), followed by
spectroscopic or
other analysis of the reaction products.
To determine the presence or absence of a cancer, such as prostate cancer, the
signal detected from the reporter group that remains bound to the solid
support is generally
compared to a signal that corresponds to a predetermined cut-off value. In one
preferred
embodiment, the cut-off value for the detection of a cancer is the average
mean signal
obtained when the immobilized antibody is incubated with samples from patients
without the
cancer. In general, a sample generating a signal that is three standard
deviations above the
predetermined cut-off value is considered positive for the cancer. In an
altemate preferred
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
44
embodiment, the cut-off value is determined using a Receiver Operator Curve,
according to
the method of Sackett et al., Clinical Epidemiology: A Basic Science for
Clinical Medicine,
Little Brown and Co., 1985, p. 106-7. Briefly, in this embodiment, the cut-off
value may be
determined from a plot of pairs of true positive rates (i.e., sensitivity) and
false positive rates
(100%-specificity) that correspond to each possible cut-off value for the
diagnostic test result.
The cut-off.value on the plot that is the closest to the upper left-hand
corner (i.e., the value
that encldses the largest area) is the most accurate cut-off value, and a
sample generating a
signal that is higher than the cut-off value determined by this method may be
considered
positive. Alternatively, the cut-off value may be shifted to the left along
the plot, to minimize
the false positive rate, or to the right, to minimize the false negative rate.
In general, a sample
generating a signal that is higher than the cut-off value determined by this
method is
considered positive for a cancer.
In a related embodiment, the assay is performed in a flow-through or strip
test
format, wherein the binding agent is immobilized on a membrane, such as
nitrocellulose. In
the flow-through test, polypeptides within the sample bind to the immobilized
binding agent
as the sample passes through the membrane. A second, labeled binding agent
then binds to
the binding agent-polypeptide complex as a solution containing the second
binding agent
flows through the membrane. The detection of bound second binding agent may
then be
performed as described above. In the strip test format, one end of the
membrane to which
binding agent is bound is immersed in a solution containing the sample. The
sample migrates
along the membrane through a region containing second binding agent and to the
area of
immobilized binding agent. Concentration of second binding agent at the area
of
immobilized antibody indicates the presence of a cancer. Typically, the
concentration of
second binding agent at that site generates a pattern, such as a line, that
can be read visually.
The absence of such a pattern indicates a negative result. In general, the
amount of binding
agent immobilized on the membrane is selected to generate a visually
discernible pattern
when the biological sample contains a level of polypeptide that would be
sufficient to
generate a positive signal in the two-antibody sandwich assay, in the format
discussed above.
Preferred binding agents for use in such assays are antibodies and antigen-
binding fragments
thereof. Preferably, the amount of antibody immobilized on the membrane ranges
from about
25 ng to about I g, and more preferably from about 50 ng to about 500 ng.
Such tests can
typically be performed with a very small amount of biological sample.
Of course, numerous other assay protocols exist that are suitable for use with
the tumor proteins or binding agents of the present invention. The above
descriptions are
intended to be exemplary only. For example, it will be apparent to those of
ordinary skill in
the art that the above protocols may be readily modified to use prostate tumor
polypeptides to
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
detect antibodies that bind to such polypeptides in a biological sample. The
detection of such
prostate tumor protein specific antibodies may correlate with the presence of
a cancer.
A cancer may also, or alternatively, be detected based on the presence of T
cells that specifically react with a prostate tumor protein in a biological
sample. Within
certain methods, a biological sample comprising CD4+ and/or CD8' T cells
isolated from a
patient is incubated with a prostate tumor polypeptide, a polynucleotide
encoding such a
polypeptiYde and/or an APC that expresses at least an immunogenic portion of
such a
polypeptide, and the presence or absence of specific activation of the T cells
is detected.
Suitable biological samples include, but are not limited to, isolated T cells.
For example, T
cells may be isolated from a patient by routine techniques (such as by
Ficoll/Hypaque density
gradient centrifugation of peripheral blood lymphocytes). T cells may be
incubated in vitro
for 2-9 days (typically 4 days) at 37 C with prostate tumor polypeptide (e.g.,
5 - 25 g/ml). It
may be desirable to incubate another aliquot of a T cell sample in the absence
of prostate
tumor polypeptide to serve as a control. For CD4+ T cells, activation is
preferably detected
by evaluating proliferation of the T cells. For CD8+ T cells, activation is
preferably detected
by evaluating cytolytic activity. A level of proliferation that is at least
two fold greater and/or
a level of cytolytic activity that is at least 20% greater than in disease-
free patients indicates
the presence of a cancer in the patient.
As noted above, a cancer may also, or alternatively, be detected based on the
level of mRNA encoding a prostate tumor protein in a biological sample. For
example, at
least two oligonucleotide primers may be employed in a polymerase chain
reaction (PCR)
based assay to amplify a portion of a prostate tumor cDNA derived from a
biological sample,
wherein at least one of the oligonucleotide primers is specific for (i.e.,
hybridizes to) a
polynucleotide encoding the prostate tumor protein. The amplified eDNA is then
separated
and detected using techniques well known in the art, such as gel
electrophoresis. Similarly,
oligonucleotide probes that specifically hybridize to a polynucleotide
encoding a prostate
tumor protein may be used in a hybridization assay to detect the presence of
polynucleotide
encoding the tumor protein in a biological sample.
To permit hybridization under assay conditions, oligonucleotide primers and
probes should comprise an oligonucleotide sequence that has at least about
60%, preferably at
least about 75% and more preferably at least about 90%, identity to a portion
of a
polynucleotide encoding a prostate tumor protein that is at least 10
nucleotides, and
preferably at least 20 nucleotides, in length. Preferably, oligonucleotide
primers and/or
probes will hybridize to a polynucleotide encoding a polypeptide disclosed
herein under
moderately stringent conditions, as defined above. Oligonucleotide primers
and/or probes
which may be usefully employed in the diagnostic methods described herein
preferably are at
least 10-40 nucleotides in length. In a preferred embodiment, the
oligonucleotide primers
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
46
comprise at least 10 contiguous nucleotides, more preferably at least 15
contiguous
nucleotides, of a DNA molecule having a sequence recited in SEQ ID NO: 1-111,
115-171,
173-175, 177, 179-305, 307-315, 326, 328, 330, 332-335, 340-375 and 381.
Techniques for
both PCR based assays and hybridization assays are well known in the art (see,
for example,
Mullis et al., Cold Spring Harbor Symp. Quant. Biol., 51:263, 1987; Erlich
ed., PCR
Technology, Stockton Press, NY, 1989).
One preferred assay employs RT-PCR, in which PCR is applied in conjunction
with reverse transcription. Typically, RNA is extracted from a biological
sample, such as
biopsy tissue, and is reverse transcribed to produce cDNA molecules. PCR
amplification
using at least one specific primer generates a cDNA molecule, which may be
separated and
visualized using, for example, gel electrophoresis. Amplification may be
performed on
biological samples taken from a test patient and from an individual who is not
afflicted with a
cancer. The amplification reaction may be performed on several dilutions of
cDNA spanning
two orders of magnitude. A two-fold or greater increase in expression in
several dilutions of
the test patient sample as compared to the same dilutions of the non-cancerous
sample is
typically considered positive.
In another embodiment, the disclosed compositions may be used as markers
for the progression of cancer. In this embodiment, assays as described above
for the
diagnosis of a cancer may be performed over time, and the change in the level
of reactive
polypeptide(s) or polynucleotide evaluated. For example, the assays may be
performed every
24-72 hours for a period of 6 months to 1 year, and thereafter performed as
needed. In
general, a cancer is progressing in those patients in whom the level of
polypeptide or
polynucleotide detected increases over time. In contrast, the cancer is not
progressing when
the level of reactive polypeptide or polynucleotide either remains constant or
decreases with
time.
Certain in vivo diagnostic assays may be perfonned directly on a tumor. One
such assay involves contacting tumor cells with a binding agent. The bound
binding agent
may then be detected directly or indirectly via a reporter group. Such binding
agents may
also be used in histological applications. AlternativeIy, polynucleotide
probes may be used
within such applications.
As noted above, to improve sensitivity, multiple prostate tumor protein
markers may be assayed within a given sample. It will be apparent that binding
agents
specific for different proteins provided herein may be combined within a
single assay.
Further, multiple primers or probes may be used concurrently. The selection of
tumor protein
markers may be based on routine experiments to determine combinations that
results in
optimal sensitivity. In addition, or alternatively, assays for tumor proteins
provided herein
may be combined with assays for other known tumor antigens.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
47
DIAGNOSTIC KITS
The present invention further provides kits for use within any of the above
diagnostic methods. Such kits typically comprise two or more components
necessary for
performing a diagnostic assay. Components may be compounds, reagents,
containers and/or
equipment. For example, one container within a kit may contain a monoclonal
antibody or
fragmenf thereof that specifically binds to a prostate tumor protein. Such
antibodies or
fragments may be provided attached to a support material, as described above.
One or more
additional containers may enclose elements, such as reagents or buffers, to be
used in the
assay. Such kits may also, or alternatively, contain a detection reagent as
described above
that contains a reporter group suitable for direct or indirect detection of
antibody binding.
Alternatively, a kit may be designed to detect the level of mRNA encoding a
prostate tumor protein in a biological sample. Such kits generally comprise at
least one
oligonucleotide probe or primer, as described above, that hybridizes to a
polynucleotide
encoding a prostate tumor protein. Such an oligonucleotide may be used, for
example, within
a PCR or hybridization assay. Additional components that may be present within
such kits
include a second oligonucleotide and/or a diagnostic reagent or container to
facilitate the
detection of a polynucleotide encoding a prostate tumor protein.
The following Examples are offered by way of illustration and not by way of
limitation.
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838 48
EXAMPLES
EXAMPLE I
ISOLATION AND CHARACTERIZATION OF PROSTATE TUMOR POLYPEPTIDES
This Example describes the isolation of certain prostate tumor polypeptides
from a prostate tumor cDNA library.
A human prostate tumor cDNA expression library was constructed from
prostate tumor poly A' RNA using a Superscript Plasmid System for cDNA
Synthesis and
Plasmid Cloning kit (BRL Life Technologies, Gaithersburg, MD 20897) following
the
manufacturer's protocol. Specifically, prostate tumor tissues were homogenized
with
polytron (Kinematica, Switzerland) and total RNA was extracted using Trizol
reagent (BRL
Life Technologies) as directed by the manufacturer. The poly A' RNA was then
purified
using a Qiagen oligotex spin column mRNA purification kit (Qiagen, Santa
Clarita, CA
91355) according to the manufacturer's protocol. First-strand cDNA was
synthesized using
the NotI/Oligo-dT 18 primer. Double-stranded cDNA was synthesized. ligated
with
EcoRl/BAXI adaptors (Invitrogen, San Diego, CA) and digested with NotI.
Following size
fractionation with Chroma Spin-1000 columns (Clontech, Palo Alto, CA), the
cDNA was
ligated into the EcoRI/NotI site of pCDNA3.l (Invitrogen) and transformed into
ElectroMax
E. coli DHIOB cells (BRL Life Technologies) by electroporation.
Using the same procedure, a normal human pancreas cDNA expression library
was prepared from a pool of six tissue specimens (Clontech). The cDNA
libraries were
characterized by determining the number of independent colonies, the
percentage of clones
that carried insert, the average insert size and by sequence analysis. The
prostate tumor
library contained 1.64 x 10' independent colonies, with 70% of clones having
an insert and
the average insert size being 1745 base pairs. The normal pancreas eDNA
library contained
3.3 x 106 independent colonies, with 69% of clones having inserts and the
average insert size
being 1120 base pairs. For both libraries, sequence analysis showed that the
majority of
clones had a full length cDNA sequence and were synthesized from mRNA, with
minimal
rRNA and mitochondrial DNA contamination.
cDNA library subtraction was performed using the above prostate tumor and
normal pancreas cDNA libraries, as described by Hara et al. (Blood, 84:189-
199, 1994) with
some modifications. Specifically, a prostate tumor-specific subtracted cDNA
library was
generated as follows. Normal pancreas cDNA library (70 jig) was digested with
EcoRl, Notl,
and Sful, followed by a filling-in reaction with DNA polymerase Kienow
fragment. After
phenol-chloroform extraction and ethanol precipitation, the DNA was dissolved
in 100 l of
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
49
H2O, heat-denatured and mixed with 100 1 (100 g) of Photoprobe biotin
(Vector
Laboratories, Burlingame, CA). As recommended by the manufacturer, the
resulting mixture
was irradiated with a 270 W sunlamp on ice for 20 minutes. Additional
Photoprobe biotin
(50 gl) was added and the biotinylation reaction was repeated. After
extraction with butanol
five times, the DNA was ethanol-precipitated and dissolved in 23 pl H,O to
form the driver
DNA.
To form the tracer DNA, 10 g prostate tumor cDNA library was digested
with BamHI and XhoI, phenol chloroform extracted and passed through Chroma
spin-400
columns (Clontech). Following ethanol precipitation, the tracer DNA was
dissolved in 5 111
H20. Tracer DNA was mixed with 15 l driver DNA and 20 i of 2 x hvbridization
buffer
(1.5 M NaCI/10 mM EDTA/50 mM HEPES pH 7.5/0.2% sodium dodecyl sulfate),
overlaid
with mineral oil, and heat-denatured completely. The sample was immediately
transferred
into a 68 C water bath and incubated for 20 hours (long hybridization [LH]).
The reaction
mixture was then subjected to a streptavidin treatment followed by
phenol/chloroform
extraction. This process was repeated three more times. Subtracted DNA was
precipitated,
dissolved in 12 l H,O, mixed with 8 l driver DNA and 20 l of 2 x
hybridization buffer,
and subjected to a hybridization at 68 C for 2 hours (short hybridization
[SH]). After
removal of biotinylated double-stranded DNA, subtracted cDNA was ligated into
BamHI/XhoI site of chloramphenicol resistant pBCSK' (Stratagene, La Jolla, CA
92037) and
transformed into ElectroMax E. coli DH l OB cells by electroporation to
generate a prostate
tumor specific subtracted cDNA library (referred to as "prostate subtraction
1").
To analyze the subtracted cDNA library, plasmid DNA was prepared from 100
independent clones, randomly picked from the subtracted prostate tumor
specific library and
grouped based on insert size. Representative cDNA clones were further
characterized by
DNA sequencing with a Perkin Elmer/Applied Biosystems Division Automated
Sequencer
Model 373A (Foster City, CA). Six cDNA clones, hereinafter referred to as F1-
13, F1-12,
F1-16, Hl-1, H1-9 and H1-4, were shown to be abundant in the subtracted
prostate-specific
cDNA library. The determined 3' and 5' cDNA sequences for F 1-12 are provided
in SEQ ID
NO: 2 and 3, respectively, with determined 3' cDNA sequences for F 1-13, F 1-
16, H 1-1, H 1-9
and H1-4 being provided in SEQ ID NO: 1 and 4-7, respectively.
The eDNA sequences for the isolated clones were compared to known
sequences in the gene bank using the EMBL and GenBank databases (release 96).
Four of
the prostate tumor cDNA clones, F 1-13, F 1-16, H 1-1, and H 1-4, were
determined to encode
the following previously identified proteins: prostate specific antigen (PSA),
human
glandular kallikrein, human tumor expression enhanced gene, and mitochondria
cytochrome
C oxidase subunit 11. H1-9 was found to be identical to a previously
identified human
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
autonomously replicating sequence. No significant homologies to the cDNA
sequence for
F1-12 were found.
Subsequent studies led to the isolation of a full-length cDNA sequence for F 1-
12. This sequence is provided in SEQ ID NO: 107, with the corresponding
predicted amino
acid sequence being provided in SEQ ID NO: 108.
To clone less abundant prostate tumor specific genes, cDNA library
subtraction was performed by subtracting the prostate tumor cDNA library
described above
with the normal pancreas cDNA library and with the three most abundant genes
in the
previously subtracted prostate tumor specific cDNA library: human glandular
kallikrein,
prostate specific antigen (PSA), and mitochondria cytochrome C oxidase subunit
11.
Specifically, 1 g each of human glandular kallikrein, PSA and mitochondria
cytochrome C
oxidase subunit II cDNAs in pCDNA3.l were added to the driver DNA and
subtraction was
performed as described above to provide a second subtracted cDNA library
hereinafter
referred to as the "subtracted prostate tumor specific cDNA library with
spike".
Twenty-two cDNA clones were isolated from the subtracted prostate tumor
specific cDNA library with spike. The determined 3' and 5' cDNA sequences for
the clones
referred to as J1-17, L1-12, NI-1862, J1-13, J1-19, JI-25. J1-24, KI-58, KI-
63, I.l-4 and LI-
14 are provided in SEQ ID NOS: 8-9, 10-11, 12-13, 14-15, 16-17, 18-19, 20-21,
22-23, 24-
25, 26-27 and 28-29, respectively. The determined 3' cDNA sequences for the
clones referred
to as J1-12, JI-16, JI-21, K1-48, KI-55, LI-2, L1-6, N1-1858, N1-1860, N1-
1861, NI-1864
are provided in SEQ ID NOS: 30-40, respectively. Comparison of these sequences
with those
in the gene bank as described above, revealed no significant homologies to
three of the five
most abundant DNA species, (J1-17, LI-12 and N1-1862; SEQ ID NOS: 8-9, 10-11
and 12-
13, respectively). Of the remaining two most abundant species, one (J 1-12;
SEQ ID NO:30)
was found to be identical to the previously identified human pulmonary
surfactant-associated
protein, and the other (KI-48; SEQ ID NO:33) was determined to have some
homology to R.
norvegicus mRNA for 2-arylpropionyl-CoA epimerase. Of the 17 less abundant
cDNA
clones isolated from the subtracted prostate tumor specific cDNA library with
spike, four (J 1-
16, K1-55, L1-6 and N1-1864; SEQ ID NOS:31, 34, 36 and 40, respectively) were
found to
be identical to previously identified sequences, two (J1-21 and N1-1860; SEQ
ID NOS: 32
and 38, respectively) were found to show some homology to non-human sequences,
and two
(Ll-2 and N1-1861; SEQ ID NOS: 35 and 39, respectively) were found to show
some
homology to known human sequences. No significant homologies were found to the
polypeptides J1-13, JI-19, J1-24, J1-25, K1-58, K1-63, LI-4, L1-14 (SEQ ID
NOS: 14-15,
16-17, 20-21, 18-19, 22-23, 24-25, 26-27, 28-29, respectively).
Subsequent studies led to the isolation of full length cDNA sequences for J 1-
17, LI-12 and N1-1862 (SEQ ID NOS: 109-111, respectively). The corresponding
predicted
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
51
amino acid sequences are provided in SEQ ID NOS: 112-114. L1-12 is also
referred to as
P501 S.
In a further experiment, four additional clones were identified by subtracting
a
prostate tumor cDNA library with normal prostate cDNA prepared from a pool of
three
normal prostate poly A+ RNA (referred to as "prostate subtraction 2"). The
determined
cDNA se ; quences for these clones, hereinafter referred to as U I-3064, U l-
3065, V 1-3692 and
1A-3905,'are provided in SEQ ID NO: 69-72, respectively. Comparison of the
determined
sequences with those in the gene bank revealed no significant homologies to U
1-3065.
A second subtraction with spike (referred to as "prostate subtraction spike
2")
was performed by subtracting a prostate tumor specific cDNA library with spike
with normal
pancreas cDNA library and further spiked with PSA, J1-17, pulmonary surfactant-
associated
protein, mitochondrial DNA, cytochrome c oxidase subunit II, N 1-1862,
autonomously
replicating sequence, L 1-12 and tumor expression enhanced gene. Four
additional clones,
hereinafter referred to as V 1-3686, R 1-23 30, IB-3976 and V 1-3679, were
isolated. The
determined cDNA sequences for these clones are provided in SEQ ID NO:73-76,
respectively. Comparison of these sequences with those in the gene bank
revealed no
significant honiologies to VI-3686 and RI-2330.
Further analysis of the three prostate subtractions described above (prostate
subtraction 2, subtracted prostate tumor specific cDNA library with spike, and
prostate
subtraction spike 2) resulted in the identification of sixteen additional
clones, referred to as
1 G-4736, IG-4738, IG-4741, IG-4744, 1 G-4734, IH-4774, 1 H-4781, 1 H-4785, 1
H-4787,
1 H-4796, 11-4810, 11-4811, 1 J-4876, 1 K-4884 and 1 K-4896. The determined
cDNA
sequences for these clones are provided in SEQ ID NOS: 77-92, respectively.
Comparison of
these sequences with those in the gene bank as described above, revealed no
significant
homologies to IG-4741, 1G-4734, 11-4807, 1J-4876 and IK-4896 (SEQ ID NOS: 79,
81, 87,
90 and 92, respectively). Further analysis of the isolated clones led to the
determination of
extended cDNA sequences for I G-4736, 1 G-473 8, 1 G-4741, I G-4744, 1 H-4774,
I H-4781,
IH-4785, 1 H-4787, 1 H-4796, 11-4807, IJ-4876, IK-4884 and IK-4896, provided
in SEQ ID
NOS: 179-188 and 191-193, respectively, and to the determination of additional
partial
cDNA sequences for 11-4810 and 11-4811, provided in SEQ ID NOS: 189 and
190, respectively.
Additional studies with prostate subtraction spike 2 resulted in the isolation
of
three more clones. Their sequences were determined as described above and
compared to the
most recent GenBank. All three clones were found to have homology to known
genes, which
are Cysteine-rich protein, KIAA0242, and KIAA0280 (SEQ ID NO: 317, 319, and
320,
respectively). Further analysis of these clones by Synteni microarray
(Synteni, Palo Alto,
CA) demonstrated that all three clones were over-expressed in most prostate
tumors and
CA 02613125 2007-11-29
WO 00/04149 PCT/I3S99/15838
52
prostate BPH, as well as in the majority of normal prostate tissues tested,
but low expression
in all other normal tissues.
An additional subtraction was performed by subtracting a normal prostate
cDNA library witli normal pancreas cDNA (referred to as "prostate subtraction
3"). This led
to the identification of six additional clones referred to as I G-4761, 1 G-
4762, 1 H-4766, I H-
4770, 1H-4771 and 1H-4772 (SEQ ID NOS: 93-98). Comparison of these sequences
with
those in"the gene bank revealed no significant homologies to I G-4761 and I H-
4771 (SEQ ID
NOS: 93 and 97, respectively). Further analysis of the isolated clones led to
the determination
of extended cDNA sequences for 1 G-476 1, 1 G-4762, 1 H-4766 and 1 H-4772
provided in SEQ
ID NOS: 194-196 and 199, respectively, and to the determination of additional
partial cDNA
sequences for 1 H-4770 and IH-4771, provided in SEQ ID NOS: 197 and 198,
respectively.
Subtraction of a prostate tumor cDNA library, prepared from a pool of polyA+
RNA from three prostate cancer patients, with a normal pancreas eDNA library
(prostate
subtraction 4) led to the identification of eight clones, referred to as 1 D-
4297, 1 D-4309, 1 D.1-
4278, ID-4288, 1 D-4283, 1 D-4304, ID-4296 and ID-4280 (SEQ ID NOS: 99-107).
'rhese
sequences were compared to those in the gene bank as described above. No
significant
homologies were found to I D-4283 and ID-4304 (SEQ ID NOS: 103 and 104,
respectively).
Further analysis of the isolated clones led to the determination of extended
eDNA sequences
for ID-4309, 1 D.1-4278, ID-4288, 1 D-4283, 1 D-4304, 1 D-4296 and 1 D-4280,
provided in
SEQ ID NOS: 200-206, respectively.
cDNA clones isolated in prostate subtraction I and prostate subtraction 2,
described above, were colony PCR amplified and their mRNA expression levels in
prostate
tumor, normal prostate and in various other normal tissues were determined
using microarray
technology (Synteni, Palo Alto, CA). Briefly, the PCR amplification products
were dotted
onto slides in an array format, with each product occupying a unique location
in the array.
mRNA was extracted from the tissue sample to be tested, reverse transcribed,
and
fluorescent-labeled cDNA probes were generated. The microarrays were probed
with the
labeled cDNA probes, the slides scanned and fluorescence intensity was
measured. This
intensity correlates with the hybridization intensity. Two clones (referred to
as P509S and
P510S) were found to be over-expressed in prostate tumor and normal prostate
and expressed
at low levels in all other normal tissues tested (liver, pancreas, skin, bone
marrow, brain,
breast, adrenal gland, bladder, testes, salivary gland, large intestine,
kidney, ovary, lung,
spinal cord, skeletal muscle and colon). The determined eDNA sequences for
P509S and
P510S are provided in SEQ ID NO: 223 and 224, respectively. Comparison of
these
sequences with those in the gene bank as described above, revealed some
homology to
previously identified ESTs.
CA 02613125 2007-11-29
WO 00/04149 PCTlUS99/15838
53
Additional, studies led to the isolation of the full-length cDNA sequence for
P509S. This sequence is provided in SEQ ID NO: 332, with the corresponding
predicted
amino acid sequence being provided in SEQ ID NO: 339.
EXAMPLE 2
DETERMINATION OF TISSUE SPECIFICITY OF PROSTATE TUMOR
POLYPEPTIDES
Using gene specific primers, mRNA expression levels for the representative
prostate tumor polypeptides F 1-15, H 1-1, J 1-17 (also referred to as P502S),
L 1-12 (also
referred to -as P501S), F1-12 (also referred to as P504S) and N1-1862 (also
referred to as
P503S) were examined in a variety of normal and tumor tissues using RT-PCR.
Briefly, total RNA was extracted from a variety of normal and tumor tissues
using Trizol reagent as described above. First strand synthesis was carried
out using 1-2 g
of total RNA with SuperScript II reverse transcriptase (BRL Life Technologies)
at 42 C for
one hour. The cDNA was then amplified by PCR with gene-specific primers. To
ensure the
semi-quantitative nature of the RT-PCR, P-actin was used as an internal
control for each of
the tissues examined. First, serial dilutions of the first strand cDNAs were
prepared and RT-
PCR assays were performed using (3-actin specific primers. A dilution was then
chosen that
enabled the linear range amplification of the (3-actin template and which was
sensitive enough
to reflect the differences in the initial copy numbers. Using these
conditions, the P-actin
levels were determined for each reverse transcription reaction from each
tissue. DNA
contamination was minimized by DNase treatment and by assuring a negative PCR
result
when using first strand cDNA that was prepared without adding reverse
transcriptase.
mRNA Expression levels were examined in four different types of tumor
tissue (prostate tumor from 2 patients, breast tumor from 3 patients, colon
tumor, lung
tumor), and sixteen different normal tissues, including prostate, colon,
kidney, liver, lung,
ovary, pancreas, skeletal muscle, skin, stomach, testes, bone marrow and
brain. F 1-16 was
found to be expressed at high levels in prostate tumor tissue, colon tumor and
normal
prostate, and at lower levels in normai liver, skin and testes, with
expression being
undetectable in the other tissues examined. H1-1 was found to be expressed at
high levels in
prostate tumor, lung tumor, breast tumor, normal prostate, normal colon and
normal brain, at
much lower levels in normal lung, pancreas, skeletal muscle, skin, small
intestine, bone
marrow, and was not detected in the other tissues tested. J1-17 (P502S) and L1-
12 (P501S)
appear to be specifically over-expressed in prostate, with both genes being
expressed at high
levels in prostate tumor and normal prostate but at low to undetectable levels
in all the other
tissues examined. N1-1862 (P503S) was found to be over-expressed in 60% of
prostate
tumors and detectable in normal colon and kidney. The RT-PCR results thus
indicate that
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
54
F1-16, H1-1, J1-17 (P502S), N1-1862 (P503S) and L1-12 (P5O1S) are either
prostate specific
or are expressed at significantly elevated levels in prostate.
Further RT-PCR studies showed that F1-12 (P504S) is over-expressed in 60%
of prostate tumors, detectable in normal kidney but not detectable in all
other tissues tested.
Similarly, RI-2330 was shown to be over-expressed in 40% of prostate tumors,
detectable in
normal kidney and liver, but not detectable in all other tissues tested. U1-
3064 was found to
be over-expressed in 60% of prostate tumors, and also expressed in breast and
colon tumors,
but was not detectable in normal tissues.
RT-PCR characterization of R 1-2330, Ul-3064 and 1 D-4279 showed that
these three antigens are over-expressed in prostate and/or prostate tumors.
Northern analysis with four prostate tumors, two normal prostate samples, two
BPH prostates, and normal colon, kidney, liver, lung, pancrease, skeletal
muscle, brain,
stomach, testes, small intestine and bone marrow, showed that L1-12 (P501S) is
over-
expressed in prostate tumors and normal prostate, while being undetectable in
other normal
tissues tested. J1-17 (P502S) was detected in two prostate tumors and not in
the other tissues
tested. N1-1862 (P503S) was found to be over-expressed in three prostate
tumors and to be
expressed in normal prostate, colon and kidney, but not in other tissues
tested. F1-12
(P504S) was found to be highly expressed in two prostate tumors and to be
undetectable in all
other tissues tested.
The microarray technology described above was used to determine the
expression levels of representative antigens described herein in prostate
tumor, breast tumor
and the following normal tissues: prostate, liver, pancreas, skin, bone
marrow, brain, breast,
adrenal gland, bladder, testes, salivary gland, large intestine, kidney,
ovary, lung, spinal cord,
skeletal muscle and colon. L1-12 (P501S) was found to be over-expressed in
normal prostate
and prostate tumor, with some expression being detected in normal skeletal
muscle. Both J 1-
12 and F1-12 (P504S) were found to be over-expressed in prostate tumor, with
expression
being lower or undetectable in all other tissues tested. N 1-1862 (P503S) was
found to be
expressed at high levels in prostate tumor and normal prostate, and at low
levels in normal
large intestine and normal colon, with expression being undetectable in all
other tissues
tested. R1-2330 was found to be over-expressed in prostate tumor and normal
prostate, and
to be expressed at lower levels in all other tissues tested. ID-4279 was found
to be over-
expressed in prostate tumor and normal prostate, expressed at lower levels in
normal spinal
cord, and to be undetectable in all other tissues tested.
Further microarray analysis to specifically address the extent to which P501 S
(SEQ ID NO: 110) was expressed in breast tumor revealed moderate over-
expression not only
in breast tumor, but also in nietastatic breast tumor (2/31), with negligible
to low expression
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
in normal tissues. This data suggests that P501 S may be over-expressed in
various breast
tumors as well as in prostate tumors.
The expression levels of 32 ESTs (expressed sequence tags) described by
Vasmatzis et al. (Proc. Natl. Acad Sci. USA 95:300-304, 1998) in a variety of
tumor and
normal tissues were examined by microarray technology as described above. Two
of these
clones ~referred to as P1000C and PIOOIC) were found to be over-expressed in
prostate
tumor and normal prostate, and expressed at low to undetectable levels in all
other tissues
tested (normal aorta, thymus, resting and activated PBMC, epithelial cells,
spinal cord,
adrenal gland, fetal tissues, skin, salivary gland, large intestine, bone
marrow, liver, lung,
dendritic cells, stomach, lymph nodes, brain, heart, small intestine, skeletal
muscle, colon and
kidney. The determined cDNA sequences for P 1000C and P 1001 C are provided in
SEQ ID
NO: 384 and 472, respectively. The sequence of P1001C was found to show some
homology
to the previously isolated Human mRNA for JM27 protein. No significant
homologies were
found to the sequence of P1000C.
The expression of the polypeptide encoded by the full length cDNA sequence
for F1-12 (also referred to as P504S; SEQ ID NO: 108) was investigated by
immunohistochemical analysis. Rabbit-anti-P504S polyclonal antibodies were
generated
against the full length P504S protein by standard techniques. Subsequent
isolation and
characterization of the polyclonal antibodies were also performed by
techniques well known
in the art. Immunohistochemical analysis showed that the P504S polypeptide was
expressed
in 100% of prostate carcinoma samples tested (n=5).
The rabbit-anti-P504S polyclonal antibody did not appear to label benign
prostate cells with the same cytoplasmic granular staining, but rather with
light nuclear
staining. Analysis of normal tissues revealed that the encoded polypeptide was
found to be
expressed in some, but not all normal human tissues. Positive cytoplasmic
staining with
rabbit-anti-P504S polyclonal antibody was found in normal human kidney, liver,
brain,
colon and lung-associated macrophages, whereas heart and bone marrow were
negative.
This data indicates that the P504S polypeptide is present in prostate cancer
tissues, and that there are qualitative and quantitative differences in the
staining between
benign prostatic hyperplasia tissues and prostate cancer tissues, suggesting
that this
polypeptide may be detected selectively in prostate tumors and therefore be
useful in the
diagnosis of prostate cancer.
EXAMPLE 3
ISOLATION AND CHARACTERIZATION OF PROSTATE TUMOR POLYPEPTIDES
BY PCR-BASED SUBTRACTION
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
56
A cDNA subtraction library, containing cDNA from normal prostate
subtracted with ten other normal tissue cDNAs (brain, heart, kidney, liver,
lung, ovary,
placenta, skeletal muscle, spleen and thymus) and then submitted to a first
round of PCR
amplification, was purchased from Clontech. This library was subjected to a
second round of
PCR amplification, following the manufacturer's protocol. The resulting cDNA
fragments
were sub-cloned into the vector pT7 Blue T-vector (Novagen, Madison, WI) and
transformed
into XL-I Blue MRF' E. coli (Stratagene). DNA was isolated from independent
clones and
sequenced using a Perkin Elmer/Applied Biosystems Division Automated Sequencer
Model
373A.
Fifty-nine positive clones were sequenced. Comparison of the DNA
sequences of these clones with those in the gene bank, as described above,
revealed no
significant homologies to 25 of these clones, hereinafter referred to as P5,
P8, P9, P18, P20,
P30, P34, P36, P38, P39, P42, P49, P50, P53, P55, P60, P64, P65, P73, P75,
P76, P79 and
P84. The determined cDNA sequences for these clones are provided in SEQ ID NO:
41-45,
47-52 and 54-65, respectively. P29, P47, P68, P80 and P82 (SEQ ID NO: 46, 53
and 66-68,
respectively) were found to show some degree of homology to previously
identified DNA
sequences. To the best of the inventors' knowledge, none of these sequences
have been
previously shown to be present in prostate.
Further studies using the PCR-based methodology described above resulted in
the isolation of more than 180 additional clones, of which 23 clones were
found to show no
significant homologies to known sequences. The determined cDNA sequences for
these
clones are provided in SEQ ID NO: 115-123, 127, 131, 137, 145, 147-151, 153,
156-158 and
160. Twenty-three clones (SEQ ID NO: 124-126, 128-130, 132-136, 138-144. 146,
152,
154, 155 and 159) were found to show some homology to previously identified
ESTs. An
additional ten clones (SEQ ID NO: 161-170) were found to have some degree of
homology to
known genes. Larger cDNA clones containing the P20 sequence represent splice
variants of a
gene referred to as P703P. The determined DNA sequence for the variants
referred to as
DE1, DE13 and DE14 are provided in SEQ ID NOS: 171, 175 and 177, respectively,
with
the corresponding predicted amino acid sequences being provided in SEQ ID NO:
172. 176
and 178, respectively. The determined cDNA sequence for an extended spliced
form of P703
is provided in SEQ ID NO: 225. The DNA sequences for the splice variants
referred to as
DE2 and DE6 are provided in SEQ ID NOS: 173 and 174, respectively.
mRNA Expression levels for representative clones in tumor tissues (prostate
(n=5), breast (n=2), colon and lung) normal tissues (prostate (n=5), colon,
kidney, liver, lung
(n=2), ovary (n=2), skeletal muscle, skin, stomach, small intestine and
brain), and activated
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
57
and non-activated PBMC was determined by RT-PCR as described above. Expression
was
examined in one sample of each tissue type unless otherwise indicated.
P9 was found to be highly expressed in normal prostate and prostate tumor
compared to all normal tissues tested except for normal colon which showed
comparable
expression. P20, a portion of the P703P gene, was found to be highly expressed
in normal
prostate and prostate tumor, compared to all twelve normal tissues tested. A
modest increase
in expre~sion of P20 in breast tumor (n=2), colon tumor and lung tumor was
seen compared
to all normal tissues except lung (l of 2). Increased expression of P18 was
found in normal
prostate, prostate tumor and breast tumor conlpared to other normal tissues
except lung and
stomach. A modest increase in expression of P5 was observed in normal prostate
compared
to most other normal tissues. However, some elevated expression was seen in
normal lung
and PBMC. Elevated expression of P5 was also observed in prostate tumors (2 of
5), breast
tumor and one lung tumor sample. For P30, similar expression levels were seen
in normal
prostate and prostate tumor, compared to six of twelve other normal tissues
tested. Increased
expression was seen in breast tumors, one lung tumor sample and one colon
tumor sample,
and also in normal PBMC. P29 was found to be over-expressed in prostate tumor
(5 of 5)
and normal prostate (5 of 5) compared to the majority of normal tissues.
However,
substantial expression of P29 was observed in normal colon and normal lung (2
of 2). P80
was found to be over-expressed in prostate tumor (5 of 5) and normal prostate
(5 of 5)
compared to all other normal tissues tested, with increased expression also
being seen in
colon tumor.
Further studies resulted in the isolation of twelve additional clones,
hereinafter
referred to as 10-d8, 10-h10, 11-c8, 7-g6, 8-b5, 8-b6, 8-d4, 8-d9, 8-g3, 8-
h11, 9-fl 2 and 9-0.
The determined DNA sequences for 10-d8, 10-h10, 11-c8, 8-d4, 8-d9, 8-h11, 9-
f12 and 9-f3
are provided in SEQ ID NO: 207, 208, 209, 216, 217, 220, 221 and 222,
respectively. The
determined forward and reverse DNA sequences for 7-g6, 8-b5, 8-b6 and 8-g3 are
provided in
SEQ ID NO: 210 and 211; 212 and 213; 214 and 215; and 218 and 219,
respectively.
Comparison of these sequences with those in the gene bank revealed no
significant
homologies to the sequence of 9-f3. The clones 10-d8, 11-c8 and 8-hi I were
found to show
some homology to previously isolated ESTs, while 10-h10, 8-b5, 8-b6, 8-d4, 8-
d9, 8-g3 and
9-fl2 were found to show some homology to previously identified genes. Further
characterization of 7-G6 and 8-G3 showed identity to the known genes PAP and
PSA,
respectively.
mRNA expression levels for these clones were determined using the micro-
array technology described above. The clones 7-G6, 8-G3, 8-B5, 8-B6, 8-D4, 8-
D9, 9-F3, 9-
F 12, 9-H3, 10-A2, 10-A4, 11-C9 and 11-F2 were found to be over-expressed in
prostate
tumor and normal prostate, with expression in other tissues tested being low
or undetectabie.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
58
Increased expression of 8-F11 was seen in prostate tumor and normal prostate,
bladder,
skeletal muscle and colon. Increased expression of 10-H10 was seen in prostate
tumor and
normal prostate, bladder, lung, colon, brain and large intestine. Increased
expression of 9-B 1
was seen in prostate tumor, breast tumor, and normal prostate, salivary gland,
large intestine
and skin, with increased expression of 11-C8 being seen in prostate tumor, and
normal
prostate and large intestine.
An additional cDNA fragment derived from the PCR-based normal prostate
subtraction, described above, was found to be prostate specific by both micro-
array
technology and RT-PCR. The determined cDNA sequence of this clone (referred to
as 9-
A11) is provided in SEQ ID NO: 226. Comparison of this sequence with those in
the public
databases revealed 99% identity to the known gene HOXB 13.
Further studies led to the isolation of the clones 8-C6 and 8-H7. The
determined cDNA sequences for these clones are provided in SEQ ID NO: 227 and
228,
respectively. These sequences were found to show some homology to previously
isolated
ESTs.
PCR and hybridization-based methodologies were employed to obtain longer
cDNA sequences for clone P20 (also referred to as P703P), yielding three
additional cDNA
fragments that progressively extend the 5' end of the gene. These fragments,
referred to as
P703PDE5, P703P6.26, and P703PX-23 (SEQ ID NO: 326, 328 and 330, with the
predicted
corresponding amino acid sequences being provided in SEQ ID NO: 327, 329 and
331,
respectively) contain additional 5' sequence. P703PDE5 was recovered by
screening of a
cDNA library (#141-26) with a portion of P703P as a probe. P703P6.26 was
recovered from
a mixture of three prostate tumor cDNAs and P703PX 23 was recovered from cDNA
library
(#438-48). Together, the additional sequences include all of the putative
mature serine
protease along with part of the putative signal sequence. Further studies
using a PCR-based
subtraction library of a prostate tumor pool subtracted against a pool of
normal tissues
(referred to as JP: PCR subtraction) resulted in the isolation of thirteen
additional clones,
seven of which did not share any significant homology to known GenBank
sequences. The
determined cDNA sequences for these seven clones (P71 IP, P712P, novel 23,
P774P, P775P,
P710P and P768P) are provided in SEQ ID NO: 307-311, 313 and 315,
respectively. The
remaining six clones (SEQ ID NO: 316 and 321-325) were shown to share some
homology to
known genes. By microarray analysis, all thirteen clones showed three or more
fold over-
expression in prostate tissues, including prostate tumors, BPH and normal
prostate as
compared to normal non-prostate tissues. Clones P711P, P712P, novel 23 and
P768P showed
over-expression in most prostate tumors and BPH tissues tested (n=29), and in
the majority of
normal prostate tissues (n=4), but background to low expression levels in all
normal tissues.
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
59
Clones P774P, P775P and P710P showed comparatively lower expression and
expression in
fewer prostate tumors and BPH samples, with negative to low expression in
normal prostate.
The full-length cDNA for P711 P was obtained by employing the partial
sequence of SEQ ID NO: 307 to screen a prostate cDNA library. Specifically, a
directionally
cloned prostate cDNA library was prepared using standard techniques. One
million colonies
of this library were plated onto LB/Amp plates. Nylon membrane filters were
used to lift
these colonies, and the cDNAs which were picked up by these filters were
denatured and
cross-linked to the filters by UV light. The P71 1P cDNA fragment of SEQ ID
NO: 307 was
radio-labeled and used to hybridize with these filters. Positive clones were
selected, and
cDNAs were prepared and sequenced using an automatic Perkin Elmer/Applied
Biosystems
sequencer. The determined full-length sequence of P711P is provided in SEQ ID
NO: 382,
with the corresponding predicted amino acid sequence being provided in SEQ ID
NO: 383.
Using PCR and hybridization-based methodologies, additional cDNA
sequence information was derived for two clones described above, 11-C9 and 9-
F3, herein
after referred to as P707P and P714P, respectively (SEQ ID NO: 333 and 334).
After
comparison with the most recent GenBank, P707P was found to be a splice
variant of the
known gene HoxB 13. In contrast, no significant homologies to P714P were
found.
Clones 8-B3, P89, P98, P130 and P201 (as disclosed in U.S. Patent
Application No. 09/020,956, filed February 9, 1998) were found to be contained
within one
contiguous sequence, referred to as P705P (SEQ ID NO: 335, with the predicted
amino acid
sequence provided in SEQ ID NO: 336), which was determined to be a splice
variant of the
known gene NKX 3.1.
EXAMPLE 4
SYNTHESIS OF POLYPEPTIDES
Polypeptides may be synthesized on a Perkin Elmer/Applied Biosystems 430A
peptide synthesizer using FMOC chemistry with HPTU (O-Benzotriazole-N,N,N',N'-
tetramethyluronium hexafluorophosphate) activation. A Gly-Cys-Gly sequence may
be
attached to the amino terminus of the peptide to provide a method of
conjugation, binding to
an immobilized surface, or labeling of the peptide. Cleavage of the peptides
from the solid
support may be carried out using the following cleavage mixture:
trifluoroacetic
acid:ethanedithiol:thioanisole:water:phenol (40:1:2:2:3). After cleaving for 2
hours, the
peptides may be precipitated in cold methyl-t-butyl-ether. The peptide pellets
may then be
dissolved in water containing 0.1% trifluoroacetic acid (TFA) and lyophilized
prior to
purification by C18 reverse phase HPLC. A gradient of 0%-60% acetonitrile
(containing
0.1% TFA) in water (containing 0.1% TFA) may be used to elute the peptides.
Following
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
lyophilization of the pure fractions, the peptides may be characterized using
electrospray or
other types of mass spectrometry and by amino acid analysis.
EXAMPLE 5
FURTHER ISOLATION AND CHARACTERIZATION OF PROSTATE TUMOR
POLYPEPTIDES BY PCR-BASED SUBTRACTION
A cDNA library generated from prostate primary tumor mRNA as described
above was subtracted with cDNA from normal prostate. The subtraction was
performed
using a PCR-based protocol (Clontech), which was modified to generate larger
fragments.
Within this protocol, tester and driver double stranded cDNA were separately
digested with
five restriction enzymes that recognize six-nucleotide restriction sites
(MIuI, MscI, Pvull,
Sall and Stul). This digestion resulted in an average cDNA size of 600 bp,
rather than the
average size of 300 bp that results from digestion with Rsal according to the
Clontech
protocol. This modification did not affect the subtraction efficiency. Two
tester populations
were then created with different adapters, and the driver library remained
without adapters.
The tester and driver libraries were then hybridized using excess driver cDNA.
In the first hybridization step, driver was separately hybridized with each of
the two tester
cDNA populations. This resulted in populations of (a) unhybridized tester
cDNAs, (b) tester
cDNAs hybridized to other tester cDNAs, (c) tester cDNAs hybridized to driver
cDNAs and
(d) unhybridized driver cDNAs. The two separate hybridization reactions were
then
combined, and rehybridized in the presence of additional denatured driver
cDNA. Following
this second hybridization, in addition to populations (a) through (d), a fifth
population (e) was
generated in which tester cDNA with one adapter hybridized to tester cDNA with
the second
adapter. Accordingly, the second hybridization step resulted in enrichment of
differentially
expressed sequences which could be used as templates for PCR amplification
with adaptor-
specific primers.
The ends were then filled in, and PCR amplification was performed using
adaptor-specific primers. Only population (e), which contained tester cDNA
that did not
hybridize to driver cDNA, was amplified exponentially. A second PCR
amplification step
was then performed, to reduce background and further enrich differentially
expressed
sequences.
This PCR-based subtraction technique normalizes differentially expressed
cDNAs so that rare transcripts that are overexpressed in prostate tumor tissue
may be
recoverable. Such transcripts would be difficult to recover by traditional
subtraction
methods.
CA 02613125 2007-11-29
WO 00/04149 PCT/LJS99/15838
61
In addition to genes known to be overexpressed in prostate tumor, seventy-
seven further clones were identified. Sequences of these partial cDNAs are
provided in SEQ
ID NO: 29 to 305. Most of these clones had no significant homology to database
sequences.
Exceptions were JPTPN23 (SEQ ID NO: 231; similarity to pig valosin-containing
protein),
JPTPN30 (SEQ ID NO: 234; similarity to rat mRNA for proteasome subunit),
JPTPN45
(SEQ ID NO: 243; similarity to rat norvegicus cytosolic NADP-dependent
isocitrate
dehydrogenase), JPTPN46 (SEQ ID NO: 244; similarity to human subclone H8 4 d4
DNA
sequence), JP1D6 (SEQ ID NO: 265; similarity to G. gallus dynein light chain-
A), JP8D6
(SEQ ID NO: 288; similarity to human BAC clone RG016J04), JP8F5 (SEQ ID NO:
289;
similarity to human subclone H8 3 b5 DNA sequence), and JP8E9 (SEQ ID NO: 299;
similarity to human Alu sequence).
Additional studies using the PCR-based subtraction library consisting of a
prostate tumor pool subtracted against a normal prostate pool (referred to as
PT-PN PCR
subtraction) yielded three additional clones. Comparison of the cDNA sequences
of these
clones with the most recent release of GenBank revealed no significant
homologies to the two
clones referred to as P715P and P767P (SEQ ID NO: 312 and 314). The remaining
clone was
found to show some homology to the known gene KIAA0056 (SEQ ID NO: 318). Using
microarray analysis to measure mRNA expression levels in various tissues, all
three clones
were found to be over-expressed in prostate tumors and BPH tissues.
Specifically, clone
P715P was over-expressed in most prostate tumors and BPH tissues by a factor
of three or
greater, with elevated expression seen in the majority of nonnal prostate
samples and in fetal
tissue, but negative to low expression in all other normal tissues. Clone
P767P was over-
expressed in several prostate tumors and BPH tissues, with moderate expression
levels in half
of the normal prostate samples, and background to low expression in all other
normal tissues
tested.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
62
Further analysis, by microarray as described above, of the PT-PN PCR
subtraction library and of a DNA subtraction library containing cDNA from
prostate tumor
subtracted with a pool of normal tissue cDNAs, led to the isolation of 27
additional clones
(SEQ ID NO: 340-365 and 381) which were determined to be over-expressed in
prostate
tumor. The clones of SEQ ID NO: 341, 342, 345, 347, 348, 349, 351, 355-359,
361, 362 and
364 were also found to be expressed in normal prostate. Expression of all 26
clones in a
variety of normal tissues was found to be low or undetectable, with the
exception of P544S
(SEQ ID NO: 356) which was found to be. expressed in small intestine. Of the
26 clones, 10
(SEQ ID NO: 340-349) were found to show some homology to previously identified
sequences. No significant homologies were found to the clones of SEQ ID NO:
350-365.
EXAMPLE 6
PEPTIDE PRIMING OF MICE AND PROPAGATION OF CTL LINES
6.1. This Example illustrates the preparation of a CTL cell line specific for
cells expressing the P502S gene.
Mice expressing the transgene for human HLA A2.1 (provided by Dr L.
Sherman, The Scripps Research Institute, La Jolla, CA) were immunized with
P2S#12
peptide (VLGWVAEL; SEQ ID NO: 306), which is derived from the P502S gene (also
referred to herein as J1-17, SEQ ID NO: 8), as described by Theobald et al.,
Proc. Natl. Acad.
Sci. USA 92:11993-11997, 1995 with the following modifications. Mice were
immunized
with 100 g of P2S#12 and 120 g of an I-Ab binding peptide derived from
hepatitis B Virus
protein emulsified in incomplete Freund's adjuvant. Three weeks later these
mice were
sacrificed and using a nylon mesh single cell suspensions prepared. Cells were
then
resuspended at 6 x 10b cells/ml in complete media (RPMI-1640; Gibco BRL,
Gaithersburg,
MD) containing 10% FCS, 2mM Glutamine (Gibco BRL), sodium pyruvate (Gibco
BRL),
non-essential amino acids (Gibco BRL), 2 x 10"5 M 2-mercaptoethanol, 50U/ml
penicillin and
streptomycin, and cultured in the presence of inadiated (3000 rads) P2S# 12-
pulsed (5mg/mi
P2S#12 and 10mg/mi (32-microglobulin) LPS blasts (A2 transgenic spleens cells
cultured in
the presence of 7 g/ml dextran sulfate and 25 g/ml LPS for 3 days). Six days
later, cells (5 x
I0S/ml) were restimulated with 2.5 x 106/ml peptide pulsed irradiated (20,000
rads) EL4A2Kb
cells (Sherman et al, Science 258:815-818, 1992) and 3 x 106/ml A2 transgenic
spleen feeder
cells. Cells were cultured in the presence of 20U/ml IL-2. Cells continued to
be restimulated
on a weekly basis as described, in preparation for cloning the line.
P2S#12 line was cloned by limiting dilution analysis with peptide pulsed EL4
A2Kb tumor cells (1 x 10' cells/ well) as stimulators and A2 transgenic spleen
cells as feeders
( 5 x 105 cells/ well) grown in the presence of 30U/ml IL-2. On day 14, cells
were
CA 02613125 2007-11-29
WO 00/04149 PCT/[JS99/15838
63
restimulated as before. On day 21, clones that were growing were isolated and
maintained in
culture. Several of these clones demonstrated significantly higher reactivity
(lysis) against
human fibroblasts (HLA A2.1 expressing) transduced with P502S than against
control
fibroblasts. An example is presented in Figure 1.
This data indicates that P2S #12 represents a naturally processed epitope of
the
P502S protein that is expressed in the context of the human HLA A2.1 molecule.
6.2. This Example illustrates the preparation of murine CTL lines and CTL
clones specific for cells expressing the P501 S gene.
This series of experiments were performed similarly to that described above.
Mice were immunized with the PIS#10 peptide (SEQ ID NO: 337), which is derived
from the
P501 S gene (also referred to herein as L 1-12, SEQ ID NO: 110). The P 1 S# 10
peptide was
derived by analysis of the predicted polypeptide sequence for P501 S for
potential HLA-A2
binding sequences as defined by published HL,A-A2 binding motifs (Parker, KC,
et al, J.
Immunol., 152:163, 1994). P 1 S# 10 peptide was synthesized as described in
Example 4, and
empirically tested for HLA-A2 binding using a T cell based competition assay.
Predicted A2
binding peptides were tested for their ability to compete HLA-A2 specific
peptide
presentation to an HLA-A2 restricted CTL clone (D150M58), which is specific
for the HLA-
A2 binding influenza matrix peptide fluM58. D150M58 CTL secretes TNF in
response to
self-presentation of peptide fluM58. In the competition assay, test peptides
at 100-200 gg/ml
were added to cultures of D150M58 CTL in order to bind HLA-A2 on the CTL.
After thirty
minutes, CTL cultured with test peptides, or control peptides, were tested for
their antigen
dose response to the fluM58 peptide in a standard TNF bioassay. As shown in
Figure 3,
peptide PIS#10 competes HLA-A2 restricted presentation of fluM58,
demonstrating that
peptide P 1 S# 10 binds HLA-A2.
Mice expressing the transgene for human HLA A2.1 were immunized as
described by Theobald et al. (Proc. Natl. Acad. Sci. USA 92:11993-11997, 1995)
with the
following modifications. Mice were immunized with 62.5 g of P 1 S# 10 and 120
g of an I-
Ab binding peptide derived from Hepatitis B Virus protein emulsified in
incomplete Freund's
adjuvant. Three weeks later these mice were sacrificed and single cell
suspensions prepared
using a nylon mesh. Cells were then resuspended at 6 x 106 cells/ml in
complete media (as
described above) and cultured in the presence of irradiated (3000 rads) P I S#
10-pulsed (2
g/ml PIS#10 and 10mg/ml [32-microglobulin) LPS blasts (A2 transgenic spleens
cells
cultured in the presence of 7 g/ml dextran sulfate and 25 g/ml LPS for 3
days). Six days
later cells (5 x 195/ml) were restimulated with 2.5 x 106/mi peptide-pulsed
irradiated (20,000
rads) EL4A2Kb cells, as described above, and 3 x 106/ml A2 transgenic spleen
feeder cells.
Cells were cultured in the presence of 20 U/ml IL-2. Cells were restimulated
on a weekly
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
64
basis in preparation for cloning. After three rounds of in vitro stimulations,
one line was
generated that recognized P 1 S# 10-pulsed Jurkat A2Kb targets and P501 S-
transduced Jurkat
targets as shown in Figure 4.
A P 1 S# 10-specific CTL line was cloned by limiting dilution analysis with
peptide pulsed EL4 A2Kb tumor cells (1 x 104 cells/ well) as stimulators and
A2 transgenic
spleen cglls as feeders ( 5 x 105 cells/ well) grown in the presence of 30U/ml
IL-2. On day 14,
cells were restimulated as before. On day 21, viable clones were isolated and
maintained in
culture. As shown in Figure 5, five of these clones demonstrated specific
cytolytic reactivity
against P501 S-transduced Jurkat A2Kb targets. This data indicates that P I S#
10 represents a
naturally processed epitope of the P501 S protein that is expressed in the
context of the human
HLA-A2.1 molecule.
EXAMPLE 7
ABILITY OF HUMAN T CELLS TO RECOGNIZE PROSTATE TUMOR
POLYPEPTIDES
This Example illustrates the ability of T cells specific for a prostate tumor
polypeptide to recognize human tumor.
Human CD8+ T cells were primed in vitro to the P2S-12 peptide (SEQ ID NO:
306) derived from P502S (also referred to as J1-17) using dendritic cells
according to the
protocol of Van Tsai et al. (Critical Reviews in Immunology 18:65-75, 1998).
The resulting
CD8+ T cell microcultures were tested for their ability to recognize the P2S-
12 peptide
presented by autologous fibroblasts or fibroblasts which were transduced to
express the
P502S gene in a y-interferon ELISPOT assay (see. Lalvani et al., J. Exp. Med.
186:859-865,
1997). Briefly, titrating numbers of T cells were assayed in duplicate on 10'
fibroblasts in the
presence of 3 g/ml human p,-microglobulin and 1 ug/ml P2S-12 peptide or
control E75
peptide. In addition, T cells were simultaneously assayed on autologous
fibroblasts
transduced with the P502S gene or as a control, fibroblasts transduced with
HER-2/neu. Prior
to the assay, the fibroblasts were treated with 10 ng/ml y-interferon for 48
hours to upregulate
class I MHC expression. One of the microcultures (#5) demonstrated strong
recognition of
both peptide pulsed fibroblasts as well as transduced fibroblasts in a y-
interferon ELISPOT
assay. Figure 2A demonstrates that there was a strong increase in the number
of y-interferon
spots with increasing numbers of T cells on fibroblasts pulsed with the P2S-
12 peptide (solid
bars) but not with the control E75 peptide (open bars). This shows the ability
of these T cells
to specifically recognize the P2S-12 peptide. As shown in Figure 2B, this
microculture also
demonstrated an increase in the number of y-interferon spots with increasing
numbers of T
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
cells on fibroblasts transduced to express the P502S gene but not the HER-
2/neu gene. These
results provide additional confirmatory evidence that the P2S-12 peptide is a
naturally
processed epitope of the P502S protein. Furthermore, this also demonstrates
that there exists
in the human T cell repertoire, high affinity T cells which are capable of
recognizing this
epitope. These T cells should also be capable of recognizing human tumors
which express
the P502S gene.
EXAMPLE 8
PRIMING OF CTL IN VIVO USING NAKED DNA IMMUNIZATION WITH A
PROSTATE ANTIGEN
The prostate tumor antigen Li-12, as described above, is also referred to as
P501 S. HLA A2Kb Tg mice (provided by Dr L. Sherman, The Scripps Research
Institute, La
Jolla, CA) were immunized with 100 g VR10132-P501 S either intramuscularly or
intradermally. The mice were immunized three times, with a two week interval
between
immunizations. Two weeks after the last immunization, immune spleen cells were
cultured
with Jurkat A2Kb-P501 S transduced stimulator cells. CTL lines were stimulated
weekly.
After two weeks of in vitro stimulation, CTL activity was assessed against
P501 S transduced
targets. Two out of 8 mice developed strong anti-P501 S CTL responses. These
results
demonstrate that P501 S contains at least one naturally processed A2-
restricted CTL epitope.
EXAMPLE 9
GENERATION OF HUMAN CTL IN VITRO USING WHOLE GENE PRIMING AND
STIMULATION TECHNIQUES WITH PROSTATE TUMOR ANTIGEN
Using in vitro whole-gene priming with P501 S-retrovirally transduced
autologous fibroblasts (see, for example, Yee et al, The Journal of
Immunology, 157(9):4079-
86, 1996), human CTL lines were derived that specifically recognize autologous
fibroblasts
transduced with P501 S(also known as L 1-12), as determined by interferon-y
ELISPOT
analysis as described above. Using a panel of HLA-mismatched fibroblast lines
transduced
with P501 S, these CTL lines were shown to be restricted HLA-A2 class I
allele. Specifically,
dendritic cells (DC) were differentiated from monocyte cultures derived from
PBMC of
normal human donors by growing. for five days in RPMI medium containing 10%
human
serum, 50 ng/ml human GM-CSF and 30 ng/ml human IL-4. Following culture, DC
were
infected overnight with recombinant P501 S vaccinia virus at a multiplicity of
infection
(M.O.I) of five, and matured overnight by the addition of 3 g/ml CD40 ligand.
Virus was
inactivated by UV irradiation. CD8+ T cells were isolated using a magnetic
bead system, and
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
66
priming cultures were initiated using standard culture techniques. Cultures
were restimulated
every 7-10 days using autologous primary fibroblasts retrovirally transduced
with P501S.
Following four stimulation cycles, CD8+ T cell lines were identified that
specifically
produced interferon-y when stimulated with P501 S-transduced autologous
fibroblasts. The
P501 S-specific activity could be sustained by the continued stimulation of
the cultures with
P501 S-transduced fibroblasts in the presence of IL-15. A panel of HLA-
mismatched
fibroblast lines transduced with P501 S were generated to define the
restriction allele of the
response. By measuring interferon-y in an ELISPOT assay, the P501 S specific
response was
shown to be restricted by HLA-A2. These results demonstrate that a CD8+ CTL
response to
P501 S can be elicited.
EXAMPLE 10
IDENTIFICATION OF A NATURALLY PROCESSED CTL EPITOPE CONTAINED
WITHIN A PROSTATE TUMOR ANTIGEN
The 9-mer peptide p5 (SEQ ID NO: 338) was derived from the P703P antigen
(also referred to as P20). The p5 peptide is immunogenic in human HLA-A2
donors and is a
naturally processed epitope. Antigen specific CD8+ T cells can be primed
following repeated
in vitro stimulations with monocytes pulsed with p5 peptide. These CTL
specifically
recognize p5-pulsed target cells in both ELISPOT (as described above) and
chromium release
assays. Additionally, immunization of HLA-A2 transgenic mice with p5 leads to
the
generation of CTL lines which recognize a variety of P703P transduced target
cells
expressing either HLA-A2Kb or HLA-A2. Specifically, HLA-A2 transgenic mice
were
immunized subcutaneously in the footpad with 100 g of p5 peptide together
with 140 g of
hepatitis B virus core peptide (a Th peptide) in Freund's incomplete adjuvant.
Three weeks
post immunization, spleen cells from immunized mice were stimulated in vitro
with peptide-
pulsed LPS blasts. CTL activity was assessed by chromium release assay five
days after
primary in vitro stimulation. Retrovirally transduced cells expressing the
control antigen
P703P and HLA-A2Kb were used as targets. CTL lines that specifically
recognized both p5-
pulsed targets as well as P703P-expressing targets were identified.
Human in vitro priming experiments demonstrated that the p5 peptide is
immunogenic in humans. Dendritic cells (DC) were differentiated from monocyte
cultures
derived from PBMC of normal human donors by culturing for five days in RPMI
medium
containing 10% human serum, 50 ng/ml human GM-CSF and 30 ng/mi human IL-4.
Following culture, the DC were pulsed with p5 peptide and cultured with GM-CSF
and IL-4
together with CD8+ T cell enriched PBMC. CTL lines were restimulated on a
weekly basis
CA 02613125 2007-11-29
-
WO 00/04149 PCTIUS99/15838
67
with p5-pulsed monocytes. Five to six weeks after initiation of the CTL
cultures, CTL
recognition of p5-pulsed target cells was demonstrated.
EXAMPLE 11
EXPRESSION OF A BREAST TUMOR-DERIVED ANTIGEN
IN PROSTATE
Isolation of the antigen B305D from breast tumor by differential display is
described in US Patent Application No. 08/700,014, filed August 20, 1996.
Several different
splice forms of this antigen were isolated. The determined eDNA sequences for
these splice
forms are provided in SEQ ID NO: 366-375, with the predicted amino acid
sequences
corresponding to the sequences of SEQ ID NO: 292, 298 and 301-303 being
provided in SEQ
ID NO: 299-306, respectively.
The expression levels of B305D in a variety of tumor and normal tissues were
examined by real time PCR and by Northern analysis. The results indicated that
B305D is
highly expressed in breast tumor, prostate tumor, normal prostate tumor and
normal testes,
with expression being low or undetectable in all other tissues examined (colon
tumor, lung
tumor, ovary tumor, and normal bone marrow, colon, kidney, liver, lung, ovary,
skin, small
intestine, stomach).
EXAMPLE 12
ELICITATION OF PROSTATE TUMOR ANTIGEN-SPECIFIC CTL RESPONSES IN
HUMAN BLOOD
I
This Example illustrates the ability of a prostate tumor antigen to elicit a
CTL
response in blood of normal humans.
Autologous dendritic cells (DC) were differentiated from monocyte cultures
derived from PBMC of normal donors by growth for five days in RPMI medium
containing
10% human serum, 50 ng/ml GMCSF and 30 ng/ml IL-4. Following culture, DC were
infected ovemight with recombinant P501 S-expressing vaccinia virus at an
M.O.I. of 5 and
matured for 8 hours by the addition of 2 micrograms/mi CD40 ligand. Virus was
inactivated
by UV irradiation, CD8+ cells were isolated by positive selection using
magnetic beads, and
priming cultures were initiated in 24-well plates. Following five stimulation
cycles, CD8+
lines were identified that specifically produced interferon-gamma when
stimulated with
autologous P501 S-transduced fibroblasts. The P501 S-specific activity of cell
line 3A-1 could
be maintained following additional stimulation cycles on autologous B-LCL
transduced with
P501S. Line 3A-I was shown to specifically recognize autologous B-LCL
transduced to
CA 02613125 2007-11-29
WO 00/04149 PCT/US99115838
68
express P501 S, but not EGFP-transduced autologous B-LCL, as measured by
cytotoxity
assays (S'Cr release) and interferon-gamma production (Interferon-gamma
Elispot; see above
and Lalvani et al., J Exp. Med. 186:859-865, 1997). The results of these
assays are presented
in Figures 6A and 6B.
EXAMPLE 13
IDENTIFICATION OF PROSTATE TUMOR ANTIGENS
BY MICROARRAY ANALYSIS
This Example describes the isolation of certain prostate tumor polypeptides
from a prostate tumor cDNA library.
A human prostate tumor cDNA expression library as described above was
screened using microarray analysis to identify clones that display at least a
three fold over-
expression in prostate tumor and/or normal prostate tissue, as compared to non-
prostate
normal tissues (not including testis). 372 clones were identified, and 319
were successfully
sequenced. Table I presents a summary of these clones, which are shown in SEQ
ID
NOs:385-400. Of these sequences SEQ ID NOs:386, 389, 390 and 392 correspond to
novel
genes, and SEQ ID NOs: 393 and 396 correspond to previously identified
sequences. The
others (SEQ ID NOs:385, 387, 388, 391, 394, 395 and 397-400) correspond to
known
sequences, as shown in Table I.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
69
Table I
Sununary of Prostate Tumor Antigens
Known Genes Previously identified Genes Novel
Genes
T-cell gamma chain P504S 23379 (SEQ
ID NO:389)
Kallikrein P1000C 23399 (SEQ
ID NO:392)
Vector P501 S 23320 (SEQ
ID NO:386)
CGI-82 protein mRNA (23319; SEQ ID P503S 23381 (SEQ
NO:385) ID NO:390)
PSA P510S
Ald. 6 Dehyd. P784P
L-iditol-2 dehydrogenase (23376; SEQ ID P502S
NO:3 88 )
Ets transcription factor PDEF (22672; SEQ P706P
ID NO:398)
hTGR (22678; SEQ ID NO:399) 19142.2, bangur.seq (22621; SEQ
ID NO:396)
KIAA0295(22685; SEQ ID NO:400) 5566.1 Wang(23404; SEQ ID
NO:393)
Prostatic Acid Phosphatase(22655; SEQ ID P712P
NO:397)
SUBSTITUTE SHEET (RULE 26)
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838 70
transglutaminase (22611; SEQ ID NO:395) P778P
HDLBP (23508; SEQ ID NO:394)
CGI-69 Protein(23367; SEQ ID NO:387)
KIAA0122(23383; SEQ ID NO:391)
TEEG
CGI-82 showed 4.06 fold over-expression in prostate tissues as compared to
other normal tissues tested. It was over-expressed in 43% of prostate tumors,
25% normal
prostate, not detected in other normal tissues tested. L-iditol-2
dehydrogenase showed 4.94
fold over-expression in prostate tissues as compared to other normal tissues
tested. It was
over-expressed in 90% of prostate tumors, 100% of normal prostate, and not
detected in other
normal tissues tested. Ets transcription factor PDEF showed 5.55 fold over-
expression in
prostate tissues as compared to other normal tissues tested. It was over-
expressed in 47%
prostate tumors, 25% normal prostate and not detected in other normal tissues
tested. hTGR1
showed 9.11 fold over-expression in prostate tissues as compared to other
normal tissues
tested. It was over-expressed in 63% of prostate tumors and is not detected in
normal tissues
tested including normal prostate. KIAA0295 showed 5.59 fold over-expression in
prostate
tissues as compared to other normal tissues tested. It was over-expressed in
47% of prostate
tumors, low to undetectable in normal tissues tested including normal prostate
tissues.
Prostatic acid phosphatase showed 9.14 fold over-expression in prostate
tissues as compared
to other normal tissues tested. It was over-expressed in 67% of prostate
tumors, 50% of
normal prostate, and not detected in other normal tissues tested.
Transglutaminase showed
14.84 fold over-expression in prostate tissues as compared to other normal
tissues tested. It
was over-expressed in 30% of prostate tumors, 50% of normal prostate, and is
not detected in
other normal tissues tested. High density lipoprotein binding protein (HDLBP)
showed 28.06
fold over-expression in prostate tissues as compared to other normal tissues
tested. It was
over-expressed in 97% of prostate tumors, 75% of normal prostate, and is
undetectable in all
other normal tissues tested. CGI-69 showed 3.56 fold over-expression in
prostate tissues as
compared to other normal tissues tested. It is a low abundant gene, detected
in more than
90% of prostate tumors, and in 75% normal prostate tissues. The expression of
this gene in
normal tissues was very low. KIAA0122 showed 4.24 fold over-expression in
prostate
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
71
tissues as compared to other normal tissues tested. It was over-expressed in
57% of prostate
tumors, it was undetectable in all normal tissues tested including normal
prostate tissues.
19142.2 bangur showed 23.25 fold over-expression in prostate tissues as
compared to other
normal tissues tested. It was over-expressed in 97% of prostate tumors and
100% of normal
prostate. It was undetectable in other normal tissues tested. 5566.1 Wang
showed 3.31 fold
over-expression in prostate tissues as compared to other normal tissues
tested. It was over-
expressed in 97% of prostate tumors, 75% normal prostate and was also over-
expressed in
normal bone marrow, pancreas, and activated PBMC. Novel clone 23379 showed
4.86 fold
over-expression in prostate tissues as compared to other normal tissues
tested. It was
detectable in 97% of prostate tumors and 75% normal prostate and is
undetectable in all other
normal tissues tested. Novel clone 23399 showed 4.09 fold over-expression in
prostate
tissues as compared to other normal tissues tested. It was over-expressed in
27% of prostate
tumors and was undetectable in all normal tissues tested including normal
prostate tissues.
Novel clone 23320 showed 3.15 fold over-expression in prostate tissues as
compared to other
normal tissues tested. It was detectable in all prostate tumors and 50% of
normal prostate
tissues. It was also expressed in normal colon and trachea. Other normal
tissues do not
express this gene at high level.
EXAMPLE 14
IDENTIFICATION OF PROSTATE TUMOR ANTIGENS
BY ELECTRONIC SUBTRACTION
This Example describes the use of an electronic subtraction technique to
identify prostate tumor antigens.
Potential prostate-specific genes present in the GenBank human EST database
were identified by electronic subtraction (similar to that described by
Vasmatizis et al., Proc.
1Vat1. Acad. Sci. USA 95:300-304, 1998). The sequences of EST clones (43,482)
derived from
various prostate libraries were obtained from the GenBank public human EST
database. Each
prostate EST sequence was used as a query sequence in a BLASTN (National
Center for
Biotechnology Information) search against the human EST database. All matches
considered
identical (length of matching sequence >100 base pairs, density of identical
matches over this
region > 70%) were grouped (aligned) together in a cluster. Clusters
containing more than
200 ESTs were discarded since they probably represented repetitive elements or
highly
expressed genes such as those for ribosomal proteins. If two or more clusters
shared common
ESTs, those clusters were grouped together into a"supercluster," resulting in
4,345 prostate
superclusters.
CA 02613125 2007-11-29
WO 00/04149 PCTlUS99115838
72
Records for the 479 human cDNA libraries represented in the GenBank release
were downloaded to create a database of these cDNA library records. These 479
cDNA
libraries were grouped into three groups, Plus (normal prostate and prostate
tumor libraries,
and breast cell lines, in which expression was desired), Minus (libraries from
other normal
adult tissues, in which expression was not desirable), and Other (fetal
tissue, infant tissue,
tissue found only in women, non-prostate tumors and cell lines other than
prostate cell lines,
in which expression was considered to be irrelevant). A summary of these
library groups is
presented in Table II.
Table II
Prostate cDNA Libraries and ESTs
Library # of Libraries # of ESTs
Plus 25 43,482
Normal 11 18,875
Tumor 11 21,769
Cell lines 3 2,838
Minus 166
Other 287
Each supercluster was analyzed in terms of the ESTs within the supercluster.
The tissue source of each EST clone was noted and used to classify the
superclusters into four
groups: Type 1- EST clones found in the Plus group libraries only; no
expression detected in
Minus or Other group libraries; Type 2- EST clones found in the Plus and Other
group
libraries only; no expression detected in the Minus group; Type 3- EST clones
found in the
Plus, Minus and Other group libraries, but the expression in the Plus group is
higher than in
either the Minus or Other groups; and Type 4- EST clones found in Plus, Minus
and Other
group libraries, but the expression in the Plus group is higher than the
expression in the
Minus group. This analysis identified 4,345 breast clusters (see Table III).
From these
clusters, 3,172 EST clones were ordered from Research Genetics, Inc., and were
received as
frozen glycerol stocks in 96-well plates.
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838
73
Table III
Prostate Cluster Summarv
# of # of ESTs
Type Superclusters Ordered
1 688 677
2 2899 2484
3 85 11
4 673 0
Total 4345 3172
The inserts were PCR-amplified using amino-linked PCR primers for Synteni
microarray analysis. When more than one PCR product was obtained for a
particular clone, that
PCR product was not used for expression analysis. In total, 2,528 clones from
the electronic
subtraction method were analyzed by microarray analysis to identify electronic
subtraction breast
clones that had high tumor vs. normal tissue mRNA. Such screens were performed
using a
Synteni (Palo Alto, CA) microarray, according to the manufacturer's
instructions (and essentially
as described by Schena et al., Proc. Natl. Acad. Sci. USA 93:10614-10619, 1996
and Heller et al.,
Proc. Natl. Acad. Sci. USA 94:2150-2155, 1997). Within these analyses, the
clones were arrayed
on the chip, which was then probed with fluorescent probes generated from
normal and tumor
prostate cDNA, as well as various other normal tissues. The slides were
scanned and the
fluorescence intensity was measured.
Clones with an expression ratio greater than 3 (i.e., the level in prostate
tumor
cDNA was at least three times the level in normal prostate cDNA) were
identified as prostate
tumor-specific sequences (Table IV). The sequences of these clones are
provided in SEQ ID
NOs:401-453, with certain novel sequences shown in SEQ ID NOs:407, 413, 416-
419, 422, 426,
427 and 450.
Table IV
Prostate-tumor Specific Clones
SEQ ID NO. Sequence Comments
Designation
401 22545 previously identified P 1000C
402 22547 previouslv identified P704P
SUBSTITUTE SHEET (RULE 26)
CA 02613125 2007-11-29
WO 00/04149 PCTIUS99/15838
74
403 22548 known
404 22550 known
405 22551 PSA
406 22552 prostate secretory protein 94
407 22553 novel
408 22558 previously identified P509S
409 22562 glandular kallikrein
410 22565 previously identified P1000C
411 22567 PAP
412 22568 B 1006C (breast tumor antigen)
413 22570 novel
414 22571 PSA
415 22572 previously identified P706P
416 22573 novel
417 22574 novel
418 22575 novel
419 22580 novel
420 22581 PAP
421 22582 prostatic secretory protein 94
422 22583 novel
423 22584 prostatic secretory protein 94
424 22585 prostatic secretory protein 94
425 22586 known
426 22587 novel
427 22588 novel
428 22589 PAP
429 22590 known
430 22591 PSA
431 22592 known
432 22593 Previously identified P777P
433 22594 T cell receptor gamma chain
434 22595 Previously identified P705P
435 22596 Previously identified P707P
436 22847 PAP
43 7 22848 known
438 22849 prostatic secretory protein 57
SUBSTITUTE SHEET (RULE 26)
CA 02613125 2007-11-29
WO 00104149 PCT/[IS99/15838
439 22851 PAP
440 22852 PAP
441 22853 PAP
442 22854 previously identified P509S
443 22855 previously identified P705P
444 22856 previously identified P774P
445 22857 PSA
446 23601 previously identified P777P
447 23602 PSA
448 23605 PSA
449 23606 PSA
450 23612 novel
451 23614 PSA
452 23618 previously identified P1000C
453 23622 previously identified P705P
EXAMPLE 15
FURTHER IDENTIFICATION OF PROSTATE TUMOR ANTIGENS
BY MICROARRAY ANALYSIS
This Example describes the isolation of additional prostate tumor polypeptides
from a prostate tumor cDNA library.
A human prostate tumor cDNA expression library as described above was
screened using microarray analysis to identify clones that display at least a
three fold over-
expression in prostate tumor and/or normal prostate tissue, as compared to non-
prostate normal
tissues (not including testis). 142 clones were identified and sequenced.
Certain of these clones
are shown in SEQ ID NOs:454-467. Of these sequences SEQ ID NOs:459-461
correspond to
novel genes. The others (SEQ ID NOs:454-458 and 461-467) correspond to known
sequences.
EXAMPLE 16
FURTHER CHARACTERIZATION OF PROSTATE TUMOR ANTIGEN P710P
This Example describes the full length cloning of P71 OP.
SUBSTITUTE SHEET (RULE 26)
CA 02613125 2007-11-29
WO 00/04149 PCT/US99/15838 - 76
The prostate cDNA library described above was screened with the P710P
fragment described above. One million colonies were plated on LB/Ampicillin
plates. Nylon
membrane filters were used to lift these colonies, and the cDNAs picked up by
these filters
were then denatured and cross-linked to the filters by UV light. The P710P
fragment was
radiolabeled and used to hybridize with the filters. Positive cDNA clones were
selected and
their ciDNAs recovered and sequenced by an automatic ABI Sequencer. Four
sequences were
obtained, and are presented in SEQ ID NOs:468-471.
From the foregoing, it will be appreciated that, although specific embodiments
of the invention have been described herein for the purposes of illustration,
various
modifications may be made without deviating from the spirit and scope of the
invention.
Accordingly, the present invention is not limited except as by the appended
claims.
CA 02613125 2007-11-29
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME DE _2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME I OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.