Note: Descriptions are shown in the official language in which they were submitted.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
NANOPARTICULATE MEGESTROL FORMULATIONS
INFORMATION ON RELATED APPLICATIONS
[0001] This application is a continuation-in-part of Application No.
11/093,149,
filed on March 30, 2005, which is a continuation-in-part of Application No.
10/412,669
filed on April, 14, 2003, which claims the priority benefit of U.S.
provisional patent
Application No. 60/371,680, filed on April 12, 2002, and U.S. provisional
Application
No. 60/430,348, filed on December 3, 2002. In addition, this application
claims the
priority benefit of U.S. provisional Application No. 60/693,127, filed on June
22, 2005.
FIELD OF THE INVENTION
[0002] The present invention relates to nanoparticulate compositions
comprising
megestrol and preferably at least one surface stabilizer associated with the
surface of the
drug. The nanoparticulate megestrol particles have an effective average
particle size of
less than about 2000 nm. Methods of making and using the compositions are also
encompassed by the invention. The invention also relates to methods of
increasing
appetite and/or effecting weight gin in a subject suffering from weight loss
and/or
decreased appetite as a result of anorexia and/or cachexia, including
anorexia/cachexia
due to HIV//AIDS, cancer, chemotherapy, or related conditions or treatments.
BACKGROUND OF THE INVENTION
A. Background Rmarding Nanoparticulate Active A2ent Compositions
[0003] Nanoparticulate active agent compositions, first described.in U.S.
Patent
No. 5,145,684 ("the '684 patent"), are particles consisting of a poorly
soluble therapeutic
or diagnostic agent having adsorbed onto or associated with the surface
thereof a non-
crosslinked surface stabilizer. The '684 patent does not describe
nanoparticulate
compositions of megestrol.
1
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0004] Methods of making nanoparticulate active agent compositions are
described, for example, in U.S. Patent Nos. 5,518,187 and 5,862,999, both for
"Method of
Grinding Pharmaceutical Substances;" U.S. Patent No. 5,718,388, for
"Continuous
Method of Grinding Phannaceutical Substances;" and U.S. Patent No. 5,510,118
for
"Process of Preparing Therapeutic Compositions Containing Nanoparticles."
Nanoparticulate active agent compositions are also described, for example, in
U.S. Patent
Nos. 5,298,262 for "Use of Ionic Cloud Point Modifiers to Prevent Particle
Aggregation
During Sterilization;" 5,302,401 for "Method to Reduce Particle Size Growth
During
Lyophilization;" 5,318,767 for "X-Ray Contrast Compositions Useful in Medical
Imaging;" 5,326,552 for "Novel Formulation For Nanoparticulate X-Ray Blood
Pool
Contrast Agents Using High Molecular Weight Non-ionic Surfactants;" 5,328,404
for
"Method of X-Ray Imaging Using Iodinated Aromatic Propanedioates;" 5,336,507
for
"Use of Charged Phospholipids to Reduce Nanoparticle Aggregation;" 5,340,564
for
"Formulations Comprising Olin 10-G to Prevent Particle Aggregation and
Increase
Stability;" 5,346,702 for "Use of Non-Ionic Cloud Point Modifiers to Minimize
Nanoparticulate Aggregation During Sterilization;" 5,349,957 for "Preparation
and
Magnetic Properties of Very Small Magnetic-Dextran Particles;" 5,352,459 for
"Use of
Purified Surface Modifiers to Prevent Particle Aggregation During
Sterilization;"
5,399,363 and 5,494,683, both for "Surface Modified Anticancer Nanoparticles;"
5,401,492 for "Water Insoluble Non-Magnetic Manganese Particles as Magnetic
Resonance Enhancement Agents;" 5,429,824 for "Use of Tyloxapol as a
Nanoparticulate
Stabilizer;" 5,447,710 for "Method for Making Nanoparticulate X-Ray Blood Pool
Contrast Agents Using High Molecular Weight Non-ionic Surfactants;" 5,451,393
for "X-
Ray Contrast Compositions Useful in Medical Imaging;" 5,466,440 for
"Formulations of
Oral Gastrointestinal Diagnostic X-Ray Contrast Agents in Combination with
Pharmaceutically Acceptable Clays;" 5,470,583 for "Method of Preparing
Nanoparticle
Compositions Containing Charged Phospholipids to Reduce Aggregation;"
5,472,683 for
"Nanoparticulate Diagnostic Mixed Carbamic Anhydrides as X-Ray Contrast Agents
for
Blood Pool and Lymphatic System Imaging;" 5,500,204 for "Nanoparticulate
Diagnostic
Dimers as X-Ray Contrast Agents for Blood Pool and Lymphatic System Imaging;"
2
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
5,518,738 for "Nanoparticulate NSAID Formulations;" 5,521,218 for
"Nanoparticulate
Iododipamide Derivatives for Use as X-Ray Contrast Agents;" 5,525,328 for
"Nanoparticulate Diagnostic Diatrizoxy Ester X-Ray Contrast Agents for Blood
Pool and
Lymphatic System Imaging;" 5,543,133 for "Process of Preparing X-Ray Contrast
Compositions Containing Nanoparticles;" 5,552,160 for "Surface Modified NSAID
Nanoparticles;" 5,560,931 for "Formulations of Compounds as Nanoparticulate
Dispersions in Digestible Oils or Fatty Acids;" 5,565,188 for "Polyalkylene
Block
Copolymers as Surface Modifiers for Nanoparticles;" 5,569,448 for "Sulfated
Non-ionic
Block Copolymer Surfactant as Stabilizer Coatings for Nanoparticle
Compositions;"
5,571,536 for "Formulations of Compounds as Nanoparticulate Dispersions in
Digestible
Oils or Fatty Acids;" 5,573,749 for "Nanoparticulate Diagnostic Mixed
Carboxylic
Anydrides as X-Ray Contrast Agents for Blood Pool and Lymphatic System
Imaging;"
5,573,750 for "Diagnostic Imaging X-Ray Contrast Agents;" 5,573,783 for
"Redispersible
Nanoparticulate Film Matrices With Protective Overcoats;" 5,580,579 for "Site-
specific
Adhesion Within the GI Tract Using Nanoparticles Stabilized by High Molecular
Weight,
Linear Poly(ethylene Oxide) Polymers;" 5,585,108 for "Formulations of Oral
Gastrointestinal Therapeutic Agents in Combination with Pharmaceutically
Acceptable
Clays;" 5,587,143 for "Butylene Oxide-Ethylene Oxide Block Copolymers
Surfactants as
Stabilizer Coatings for Nanoparticulate Compositions;" 5,591,456 for "Milled
Naproxen
with Hydroxypropyl Cellulose as Dispersion Stabilizer;" 5,593,657 for "Novel
Barium
Salt Formulations Stabilized by Non-ionic and Anionic Stabilizers;" 5,622,938
for "Sugar
Based Surfactant for Nanocrystals;" 5,628,981 for "Iinproved Formulations of
Oral
Gastrointestinal Diagnostic X-Ray Contrast Agents and Oral Gastrointestinal
Therapeutic
Agents;" 5,643,552 for "Nanoparticulate Diagnostic Mixed Carbonic Anhydrides
as X-
Ray Contrast Agents for Blood Pool and Lymphatic System Imaging;" 5,718,388
for
"Continuous Method of Grinding Pharmaceutical Substances;" 5,718,919 for
"Nanoparticles Containing the R(-)Enantiomer of Ibuprofen;" 5,747,001 for
"Aerosols
Containing Beclomethasone Nanoparticle Dispersions;" 5,834,025 for "Reduction
of
Intravenously Administered Nanoparticulate Formulation Induced Adverse
Physiological
Reactions;" 6,045,829 "Nanocrystalline Formulations of Human Immunodeficiency
Virus
3
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
(HIV) Protease Inhibitors Using Cellulosic Surface Stabilizers;" 6,068,858 for
"Methods
of Malcing Nanocrystalline Formulations of Human Immunodeficiency Virus (HIV)
Protease Inhibitors Using Cellulosic Surface Stabilizers;" 6,153,225 for
"Injectable
Formulations of Nanoparticulate Naproxen;" 6,165,506 for "New Solid Dose Form
of
Nanoparticulate Naproxen;" 6,221,400 for "Methods of Treating Manvnals Using
Nanocrystalline Formulations of Human hnmunodeficiency Virus (HN) Protease
Inhibitors;" 6,264,922 for "Nebulized Aerosols Containing Nanoparticle
Dispersions;"
6,267,989 for "Methods for Preventing Crystal Growth and Particle Aggregation
in
Nanoparticle Compositions;" 6,270,806 for "Use of PEG-Derivatized Lipids as
Surface
Stabilizers for Nanoparticulate Compositions;" 6,316,029 for "Rapidly
Disintegrating
Solid Oral Dosage Form," 6,375,986 for "Solid Dose Nanoparticulate
Compositions
Comprising a Synergistic Combination of a Polymeric Surface Stabilizer and
Dioctyl
Sodium Sulfosuccinate," 6,428,814 for "Bioadhesive Nanoparticulate
Compositions
Having Cationic Surface Stabilizers;" 6,431,478 for "Small Scale Mill;"
6,432,381 for
"Methods for Targeting Drug Delivery to the Upper and/or Lower
Gastrointestinal Tract,"
6,592,903 for "Nanoparticulate Dispersions Comprising a Synergistic
Combination of a
Polymeric Surface Stabilizer and Dioctyl Sodium Sulfosuccinate," 6,582,285 for
"Apparatus for sanitary wet milling;" 6,656,504 for "Nanoparticulate
Compositions
Comprising Amorphous Cyclosporine;" 6,742,734 for "System and Method for
Milling
Materials;" 6,745,962 for "Small Scale Mill and Method Thereof;" 6,811,767 for
"Liquid
droplet aerosols of nanoparticulate drugs;" 6,908,626 for "Compositions having
a
combination of immediate release and controlled release characteristics;"
6,969,529 for
"Nanoparticulate compositions comprising copolymers of vinyl pyrrolidone and
vinyl
acetate as surface stabilizers;" and 6,976,647 for "System and Method for
Milling
Materials," all of which are specifically incorporated by reference.
[0005] In addition, U.S. Patent Publication No. 20020012675 Al, for
"Controlled
Release Nanoparticulate Compositions;" U.S. Patent Publication No. 20050276974
for
"Nanoparticulate Fibrate Formulations;" U.S. Patent Publication No.
20050238725 for
"Nanoparticulate compositions having a peptide as a surface stabilizer;" U.S.
Patent
Publication No. 20050233001 for "Nanoparticulate megestrol formulations;" U.S.
Patent
4
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
Publication No. 20050147664 for "Compositions comprising antibodies and
methods of
using the same for targeting nanoparticulate active agent delivery;" U.S.
Patent
Publication No. 20050063913 for "Novel metaxalone compositions;" U.S. Patent
Publication No. 20050042177 for "Novel compositions of sildenafil free base;"
U.S.
Patent Publication No. 20050031691 for "Gel stabilized nanoparticulate active
agent
compositions;" U.S. Patent Publication No. 20050019412 for " Novel glipizide
compositions;" U.S. Patent Publication No. 20050004049 for "Novel griseofulvin
compositions;" U.S. Patent Publication No. 20040258758 for "Nanoparticulate
topiramate
formulations;" U.S. Patent Publication No. 20040258757 for " Liquid dosage
compositions of stable nanoparticulate active agents;" U.S. Patent Publication
No.
20040229038 for "Nanoparticulate meloxicam formulations;" U.S. Patent
Publication No.
20040208833 for "Novel fluticasone formulations;" U.S. Patent Publication No.
20040195413 for " Compositions and method for milling materials;" U.S. Patent
Publication No. 20040156895 for "Solid dosage forms comprising pullulan;" U.S.
Patent
Publication No. U.S. Patent Publication No. U.S. Patent Publication No.
20040156872 for
"Novel nimesulide compositions;" U.S. Patent Publication No. 20040141925 for
"Novel
triamcinolone compositions;" U.S. Patent Publication No. 20040115134 for
"Novel
nifedipine compositions;" U.S. Patent Publication No. 20040105889 for "Low
viscosity
liquid dosage forms;" U.S. Patent Publication No. 20040105778 for "Gamma
irradiation
of solid nanoparticulate active agents;" U.S. Patent Publication No.
20040101566 for
"Novel benzoyl peroxide compositions;" U.S. Patent Publication No. 20040057905
for
"Nanoparticulate beclomethasone dipropionate compositions;" U.S. Patent
Publication
No. 20040033267 for "Nanoparticulate compositions of angiogenesis inhibitors;"
U.S.
Patent Publication No. 20040033202 for "Nanoparticulate sterol formulations
and novel
sterol combinations;" U.S. Patent Publication No. 20040018242 for
"Nanoparticulate
nystatin formulations;" U.S. Patent Publication No. 20040015134 for "Drug
delivery
systems and methods;" U.S. Patent Publication No. 20030232796 for
"Nanoparticulate
polycosanol formulations & novel polycosanol combinations;" U.S. Patent
Publication
No. 20030215502 for "Fast dissolving dosage forms having reduced friability;"
U.S.
Patent Publication No. 20030185869 for "Nanoparticulate compositions having
lysozyme
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
as a surface stabilizer;" U.S. Patent Publication No. 20030181411 for
"Nanoparticulate
compositions of mitogen-activated protein (MAP) kinase inhibitors;" U.S.
Patent
Publication No. 20030137067 for "Compositions having a combination of
immediate
release and controlled release characteristics;" U.S. Patent Publication No.
20030108616
for "Nanoparticulate compositions comprising copolymers of vinyl pyrrolidone
and vinyl
acetate as surface stabilizers;" U.S. Patent Publication No. 20030095928 for
"Nanoparticulate insulin;" U.S. Patent Publication No. 20030087308 for "Method
for
high through put screening using a small scale mill or microfluidics;" U.S.
Patent
Publication No. 20030023203 for "Drug delivery systems & methods;" U.S. Patent
Publication No. 20020179758 for "System and method for milling materials; and
U.S.
Patent Publication No. 20010053664 for "Apparatus for sanitary wet milling,"
describe
nanoparticulate active agent compositions and are specifically incorporated by
reference.
[0006] Amorphous small particle compositions are described, for exanlple, in
U.S.
Patent Nos. 4,783,484 for "Particulate Composition and Use Thereof as
Antimicrobial
Agent;" 4,826,689 for "Method for Making Uniformly Sized Particles from Water-
Insoluble Organic Compounds;" 4,997,454 for "Method for Making Uniformly-Sized
Particles From Insoluble Compounds;" 5,741,522 for "Ultrasmall, Non-aggregated
Porous
Particles of Uniform Size for Entrapping Gas Bubbles Within and Methods;" and
5,776,496, for "Ultrasmall Porous Particles for Enhancing Ultrasound Back
Scatter."
B. Background Regarding Megestrol
[0007] Megestrol acetate, also known as 17a-acetyloxy-6-methylpregna-4,6-
diene-3,20-dione, is a synthetic progestin with progestational effects similar
to those of
progesterone. It is used in abortion, endometriosis, and menstrual disorders.
It is also
used in a variety of situations including treatment of breast cancer,
contraception, and
hormone replacement therapy in post-menopausal women. Megestrol acetate is
also
frequently prescribed as an appetite enhancer for patients in a wasting state,
such as HIV
wasting, cancer wasting, or anorexia. In combination with ethynyl estradiol it
acts as an
oral contraceptive. It is also administered to subjects after castration.
6
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0008] Megestrol acetate is marlceted by Par Pharmaceuticals, Inc. and under
the
brand name Megace by Bristol Myers Squibb Co. Typical commercial formulations
are
relatively large volume. For example, Par Pharmaceuticals, Inc. megestrol
acetate oral
suspension contains 40 mg of micronized megestrol acetate per ml, and the
package insert
recommends an initial adult dosage of megestrol acetate oral suspension of 800
mg/day
(20 mL/day). The commercial formulations of megestrol acetate are highly
viscous
suspensions, which have a relatively long residence time in the mouth and any
tubing.
Highly viscous substances are not well accepted by patient populations,
particularly
patients suffering wasting and those that are intubated.
[0009] U.S. Patent No. 6,028,065 for "Flocculated Suspension of Megestrol
Acetate," assigned to Pharmaceutical Resources, Inc. (Spring Valley, NY),
describes oral
pharmaceutical micronized megestrol acetate compositions in the form of a
stable
flocculated suspension in water. The compositions comprise at least one
compound
selected from the group consisting of polyethylene glycol, propylene glycol,
glycerol, and
sorbitol; and a surfactant, wherein polysorbate and polyethylene glycol are
not
simultaneously present. U.S. Patent No. 6,268,356, also for "Flocculated
Suspension of
Megestrol Acetate," and assigned to Pharmaceutical Resources, Inc., describes
methods of
treating a neoplastic condition comprising adininistering the composition of
U.S. Patent
No. 6,028,065.
[0010] Another company that has developed a megestrol formulation is Eurand
(Milan, Italy). Eurand's formulation is a modified form of megestrol acetate
having
increased bioavailability. Eurand structurally modifies poorly soluble drugs
to increase
their bioavailability. See www.eurand.com. For megestrol acetate, Eurand uses
its'
"Biorise" process, in which a New Physical Entity (NPE) is created by
physically
breaking down megestrol's crystal lattice. This results in drug nanocrystals
and/or
amorphous drug, which are then stabilized with biologically inert carriers.
Eurand uses
three types of carriers: swellable microparticles, composite swellable
microparticles, and
cyclodextrins. See e.g., http://www.eurand.com/page.php?id=39. Such a delivery
system
can be undesirable, as "breaking down" an active agent's crystalline structure
can modify
7
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
the activity of the active agent. A drug delivery system which does not alter
the structure
of the active agent is preferable.
[0011 ] Among the progestins, megestrol acetate is one of the few that can be
administered orally because of its reduced first-pass (hepatic) metabolism,
compared to
the parent hormone. In addition, it is claimed to be superior to 19-nor
compounds as an
antifertility agent because it has less effect on the endometrium and vagina.
See
Stedman's Medical Dictionary, 25th Ed., page 935 (Williams & Wilkins, MD
1990).
[0012] There is a need in the art for megestrol formulations which exhibit
increased bioavailability, less variability, and/or less viscosity as compared
to
conventional microparticulate megestrol formulations. The present invention
satisfies
these needs.
SUMMARY OF THE INVENTION
[0013] The invention relates to nanoparticulate megestrol compositions. The
compositions comprise megestrol and preferably at least one surface stabilizer
associated
with the surface of the megestrol particles. The nanoparticulate megestrol
particles have
an effective average particle size of less than about 2000 nm.
[0014] Another aspect of the invention is directed to pharmaceutical
compositions
comprising a nanoparticulate megestrol composition of the invention. The
pharmaceutical compositions preferably comprise megestrol, at least one
surface
stabilizer, and a pharmaceutically acceptable carrier, as well as any desired
excipients.
[0015] The invention encompasses megestrol acetate compositions witli improved
physical (viscosity) and pharmacokinetic profiles (such as less variability)
over traditional
forms of megestrol acetate.
[0016] This invention fu.rther discloses a method of making a nanoparticulate
megestrol composition according to the invention. Such a method comprises
contacting
megestrol particles and at least one surface stabilizer for a time and under
conditions
sufficient to provide a nanoparticulate megestrol composition. The one or more
surface
stabilizers can be contacted with megestrol either before, during, or after
size reduction of
the megestrol.
8
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0017] The present invention is also directed to methods of treatment using
the
nanoparticulate compositions of the invention for conditions such as
endometriosis,
dysmenorrhea, hirsutism, uterine bleeding, neoplastic diseases, methods of
appetite
enhancement, contraception, hormone replacement therapy, and treating patients
following castration. In particular, the invention relates to methods of
increasing appetite
and/or effecting weight gin in a subject suffering from weight loss and/or
decreased
appetite as a result of anorexia and/or cachexia, including anorexia/cachexia
due to
HIV//A.IDS, cancer, chemotherapy, or'related conditions or treatments. Such
methods
comprises administering to a subject a therapeutically effective amount of a
nanoparticulate megestrol composition according to the invention.
[0018] Both the foregoing general description and the following brief
description
of the drawings and detailed description of the invention are exemplary and
explanatory
and are intended to provide fitrther explanation of the invention as claimed.
Other
objects, advantages, and novel features will be readily apparent to those
skilled in the art
from the following detailed description of the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
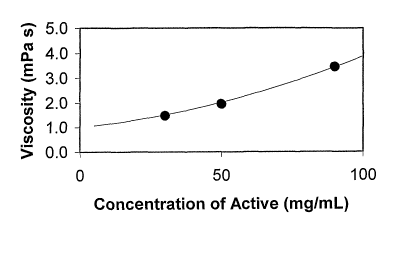
[0010] Figure. 1: Illustrates viscosity in units of mPa s as a function of
concentration.
Circles indicate the experimental values and the line illustrates the
expected trend;
[0011] Figure. 2: Illustrates viscosity in units of Pa s as a function of
shear rate for
two commercial samples, Bristol Myers Squibb and Par
Pharmaceuticals, both at an active concentration of 40 mg/mL; and
[0012] Figure 3: Shows a photograph of, from left to right, a nanoparticulate
dispersion of megestrol acetate, 'a commercial sample of megestrol
acetate marketed by Par Pharmaceuticals, and a commercial sample
of megestrol acetate marketed by Bristol Myers Squibb.
[0013] Figure 4: The figure graphically shows the comparative bioavailability
(via
plasma concentration (ng/mL)) of several nanoparticulate megestrol
9
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
compositions (575 mg/5m1, 625 mg/5m1 and 675 mg/5m1) versus a
conventional megestrol acetate marlceted by Bristol Myers Squibb.
[0014] Figure 5: The figure graphically shows on a natural log scale the
comparative
bioavailability (via plasma concentration (ng/mL)) of several
nanoparticulate megestrol compositions (575 mg/5m1, 625 mg/5m1
and 675 mg/5ml) versus a conventional megestrol acetate marketed
by Bristol Myers Squibb.
[0015] Figs 6A&B: Contain data showing weight in Kg for each subject receiving
MEGACEO OS megestrol acetate oral suspension (conventional
microcrystalline megestrol acetate) over the course of 12 weeks.
Also shown in the average data with standard deviations and
percent change. Data may contain imputed values.
[0016] Figure 7: Contains data showing weight in Kg for each subject receiving
an
oral dose of a dispersion of nanoparticulate megestrol acetate over
the course of 12 weeks. Also shown is the average data with
standard deviations and percent change. Data may contain imputed
values.
[0017] Figure 8: Contain two graphs. The first graph shows the percent change
in
weight from the initial baseline weight after the course of 12
weeks. The second graph depicts the average weight of the
subjects over the course of 12 weeks. Both graphs contain data
points for MEGACEO OS megestrol actetate oral suspension
(conventional microcrystalline megestrol acetate) and for an oral
dose of a dispersion of nanoparticulate megestrol acetate. Data
may contain imputed values.
[0018] Figs 9A&B: Contain data regarding subject's response to the fifth BACRI
question "To what extent has your appetite changed since the start
of treatment? [much worse - much better]" for those patients
receiving MEGACEO OS megestrol acetate oral suspension
(conventional microcrystalline megestrol acetate). Also shown is
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
the average data with standard deviations. Data may contain
imputed values.
[0019] Figs 10A&B: Contain data regarding subject's response to the fifth
BACRI
question "To what extent has your appetite changed since the start
of treatment? [much worse - much better]" for those patients
receiving an oral dose of a dispersion of nanoparticulate megestrol
acetate. Also shown is the average data with standard deviations.
Data may contain imputed values.
[0020] Figure 11: contains a graph depicting the average weight BACRI score to
the
fifth question "To what extent has your appetite changed since the
start of treatment? [much worse - much better]" for those patients
receiving an oral dose of a dispersion of nanoparticulate megestrol
acetate and those receiving MEGACE OS megestrol actetate oral
suspension (conventional microcrystalline megestrol acetate). Data
may contain imputed values.
[0021] Figs 12A&B: Contain data regarding subject's response to the 24 hour
recall
question "How would you describe the amount of food you ate
yesterday" on a scale where 1=typical, 2=considerably less, and
3=considerably more, for those patients receiving MEGACE OS
; megestrol acetate oral suspension (conventional microcrystalline
megestrol acetate). Also shown is the average data with standard
deviations. Data may contain imputed values.
[0022] Figs 13A&B: Contain data regarding subject's response to the 24 hour
recall
question "How would you describe the amount of food you ate
yesterday" on a scale where 1=typical, 2=considerably less, and
3=considerably more, for those patients receiving an oral dose of a
dispersion of nanoparticulate megestrol acetate. Also shown is the
average data with standard deviations. Data may contain imputed
values.
11
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0023] Figs 14A&B: Contain data showing the subjects' bioimpedance data at day
1
(baseline) and weelc 12, as well as the change in body fat and lean
muscle over the 12 weeks for those patients receiving MEGACEO
OS megestrol acetate oral suspension (conventional
microcrystalline megestrol acetate). Also shown is the average data
with standard deviations. Data may contain imputed values.
[0024] Figs 15A&B: Contain data showing the subjects' bioimpedance data at day
1
(baseline) and week 12, as well as the change in body fat and lean
muscle over the 12 weelcs for those patients receiving an oral dose
of a dispersion of nanoparticulate megestrol acetate. Also shown
is the average data with standard deviations. Data may contain
imputed values.
[0025] Figure 16: Contains a graph depicting the amounts of lean muscle and
body fat
the 12 weeks versus the amounts of lean muscle and body fat at day
1 for those patients receiving an oral dose of a dispersion of
nanoparticulate megestrol acetate and those receiving MEGACEO
OS megestrol actetate oral suspension (conventional
microcrystalline megestrol acetate). Data may contain imputed
values.
DETAILED DESCRIPTION OF THE INVENTION
[0015] The present invention is directed to nanoparticulate compositions
comprising megestrol particles having an effective average particle size of
less than about
2 microns. The compositions comprise megestrol and preferably at least one
surface
stabilizer associated with the surface of the drug.
[0016] As taught in the '684 patent, not every combination of surface
stabilizer
and active agent will result in a stable nanoparticulate composition. It was
surprisingly
discovered that stable nanoparticulate megestrol compositions can be made.
12
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0017] For example, nanoparticulate megestrol compositions with hydroxypropyl
methylcellulose (HPMC) and sodium lauryl sulfate (SLS) as surface stabilizers
remained
stable in an electrolyte solution mimicking the physiological pH of the
stomach.
Nanoparticulate megestrol compositions comprising HPMC and SLS are stable for
several weelcs at temperatures up to 40 C with only minimal particle size
growth. In
addition, nanoparticulate megestrol compositions with hydroxypropylcellulose
(HPC) and
dioctyl sodium sulfosuccinate (DOSS) as surface stabilizers, HPMC and DOSS as
surface
stabilizers, polyvinylpyrrolidone (PVP) and DOSS as surface stabilizers, and
Plasdone
S630 and DOSS as surface stabilizers were stable in electrolyte fluids and
exhibited
acceptable physical stability at 5 C for 4 weeks. (Plasdone" S630 (ISP) is a
random
copolymer of vinyl acetate and vinyl pyrrolidone.) Moreover, the
nanoparticulate
mege'stroUHPMC/SLS and nanoparticulate megestrol/HPMC/DOSS compositions also
exhibited acceptable physical stability at 25 C and 40 C for 4 weeks.
[0018] Advantages of the nanoparticulate megestrol compositions of the
invention
include, but are not limited to: (1) low viscosity liquid nanoparticulate
megestrol dosage
forms; (2) for liquid nanoparticulate megestrol compositions having a low
viscosity -
better subject compliance due to the perception of a lighter formulation which
is easier to
consume and digest; (3) for liquid nanoparticulate megestrol compositions
having a low
viscosity - ease of dispensing because one can use a cup or a syringe; (4)
faster onset of
action; (5) smaller doses of megestrol required to obtain the same
pharmacological effect
as compared to conventional microcrystalline forms of megestrol; (6) increased
bioavailability as compared to conventional microcrystalline forms of
megestrol;
(7) substantially similar pharmacokinetic profiles of the nanoparticulate
megestrol
compositions when administered in the fed versus the fasted state; (8)
bioequivalency of
the nanoparticulate megestrol compositions when administered in the fed versus
the
fasted state; (9) redispersibility of the nanoparticulate megestrol particles
present in the
compositions of the invention following administration; (10) bioadhesive
nanoparticulate
megestrol compositions; (11) improved pharmacokinetic profiles, such as more
rapid
megestrol absorption, greater megestrol absorption, and longer megestrol dose
retention in
the blood following administration; (12) the nanoparticulate megestrol
compositions can
13
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
be used in conjunction with other active agents; (13) the nanoparticulate
megestrol
compositions preferably exhibit an increased rate of dissolution as compared
to
conventional microcrystalline forms of megestrol; (14) improved performance
characteristics for oral, intravenous, subcutaneous, or intramuscular
injection, such as
higher dose loading and smaller tablet or liquid dose volumes; (15) the
nanoparticulate
megestrol compositions are suitable for parenteral administration; (16) the
nanoparticulate
megestrol compositions can be sterile filtered; and (17) the nanoparticulate
megestrol
compositions do not require organic solvents or pH extremes.
[0019] Moreover, it has now been surprisingly shown that administration of a
nanoparticulate megestrol formulation, such as nanoparticulate megestrol
acetate,
provides improved appetite, increased weight gain, and increased food intake
in
comparison to MEGACE megestrol acetate oral suspension, which is a
composition of
conventional, microparticulate megestrol actetate. In particular, a trial
performed in
accordance with the clinical study protocol provided herein resulted in the
data shown in
Figures 6-16, and is described in further detail in Example 11.
[0020] To summarize the results of the study described in Example 11, subjects
receiving a nanoparticulate megestrol acetate composition ("MEGACE ES")
gained an
average of 5.3 kg over the course of the 12 week study, and 38% of the
patients reported
an increase in food intake. In contrast, patients receiving the MEGACE OS
megestrol
acetate oral suspension (a conventional, microparticulate megestrol acetate
composition)
gained only 3.55 kg on average, and only 19% of the patients reported an
increase in food
intake.
[0021 ] The study described in Example 11 demonstrates weight gain in adult
HIV-positive subjects who have weiglit loss associated with AIDS-related
wasting
(anorexia/cachexia) in the first 12 weeks of treatment with a nanoparticulate
megestrol
acetetate composition. The study results are significant in that they
demonstrate that
weight gain upon administration of a nanoparticulate megestrol formulation is
not just
observed with healthy patients, but it is also observed with subjects having a
condition
that may affect their metabolism or other factors affecting weight gain.
14
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0022] Accordingly, the present invention relates to a method of increasing at
least
one of appetite, weight gain, and food intake comprising administering an
effective
amount of a nanoparticulate megestrol composition, such as nanoparticulate
megestrol
acetate, to a subject in need thereof.
[0023] The present invention is described herein using several definitions, as
set
forth below and throughout the application.
[0024] "About" will be understood by persons of ordinary skill in the art and
will
vary to some extent on the context in which the term is used. If there are
uses of the term
which are not clear to persons of ordinary skill in the art given the context
in which it is
used, "about" will mean up to plus or minus 10% of the particular term.
[0025] As used herein with reference to stable drug particles, "stable" means
that
the megestrol particles do not appreciably flocculate or agglomerate due to
interparticle
attractive forces or otherwise increase in particle size.
[0026] "Conventional active agents or drugs" refers to non-nanoparticulate
compositions of active agents or solubilized active agents or drugs. Non-
nanoparticulate
active agents have an effective average particle size of greater than about 2
microns.
A. Preferred Characteristics of the Nanoparticulate
Megestrol Compositions of the Invention
1. Low Viscosity
[0027] Typical commercial formulations of megestrol, such as Megace , are
relatively large volume, highly viscous substances that are not well accepted
by patient
populations, particularly subjects suffering from wasting. "Wasting" is a
condition in
which a subject finds it difficult to eat because, for example, food makes the
subject
nauseous. A highly viscous medicine is not compatible with treating such a
condition, as
frequently the highly viscous substance can cause additional nausea.
[0028] Moreover, viscous solutions can be problematic in parenteral
administration because these solutions require a slow syringe push and can
stick to tubing.
In addition, conventional formulations of poorly water-soluble active agents,
such as
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
megestrol, tend to be unsafe for intravenous administration techniques, which
are used
primarily in conjunction with highly water-soluble substances.
[0029] Liquid dosage forms of the nanoparticulate megestrol compositions of
the
invention provide significant advantages over conventional liquid megestrol
dosage
forms. The low viscosity and silky texture of liquid dosage forms of the
nanoparticulate
megestrol compositions of the invention results in advantages in both
preparation and use.
These advantages include, for example: (1) better subject compliance due to
the
perception of a lighter formulation which is easier to consume and digest; (2)
ease of
dispensing because one can use a cup or a syringe; (3) potential for
formulating a higher
concentration of megestrol resulting in a smaller dosage volume and thus less
volume for
the subject to consume; and (4) easier overall fonnulation concerns.
[0030] Liquid megestrol dosage forms which are easier to consume are
especially
important when considering juvenile patients, terminally ill patients, and
patients
suffering from gastrointestinal tract dysfunction or other conditions where
nausea and
vomiting are symptoms. For example, patients suffering from cancer or AIDS-
related
complications are commonly hypermetabolic and, at various stages of disease,
exhibit
gastrointestinal dysfunction. Additionally, drugs used to treat these
conditions often
cause nausea and vomiting. Viscous or gritty formulations, and those that
require a
relatively large dosage volume, are not well tolerated by patient populations
suffering
from wasting associated with these diseases because the formulations can
exacerbate
nausea and encourage vomiting.
[0031] The viscosities of liquid dosage forms of nanoparticulate megestrol
according to the invention are preferably less than about 1/200, less than
about 1/175, less
than about 1/150, less than about 1/125, less than about 1/100, less than
about 1/75, less
than about 1/50, or less than about 1/25 of existing commercial liquid oral
megestrol
acetate compositions, e.g. Megace , at about the same concentration per ml of
megestrol.
[0032] Typically the viscosity of liquid nanoparticulate megestrol dosage
forms of
the invention is from about 175 mPa s to about 1 mPa s, from about 150 mPa s
to about 1
mPa, from about 125 mPa s to about 1 mPa s, from about 100 mPa s to about 1
mPa s,
from about 75 mPa s to about 1 mPa s, from about 50 mPa s to about 1 mPa s,
from about
16
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
25 mPa s to about 1 mPa s, from about 15 mPa s to about 1 mPa s, or from about
5 mPa s
to about 1 mPa s. Such a viscosity is much more attractive for subject
consumption and
may lead to better overall subject compliance.
[0033] Viscosity is concentration and temperature dependent. Typically, a
higher
concentration results in a higher viscosity, while a higher temperature
results in a lower
viscosity. Viscosity as defined above refers to measurements taken at about 20
C. (The
viscosity of water at 20 C is 1 mPa s.) The invention eizcompasses equivalent
viscosities
measured at different temperatures.
[0034] A viscosity of 1.5 mPa s for a nanoparticulate megestrol dispersion
having
a concentration of 30 mg/mL, measured at 20 C, was obtained by the inventors.
An
equivalent viscosity at 4% active agent concentration would be 1.7 mPa s.
Higher and
lower viscosities can be obtained by varying the temperature and concentration
of
megestrol.
[0035] Another important aspect of the invention is that the nanoparticulate
megestrol compositions of the invention are not turbid. "Turbid," as used
herein refers to
the property of particulate matter that can be seen with the naked eye or that
which can be
felt as "gritty." The nanoparticulate megestrol compositions of the invention
can be
poured out of or extracted from a container as easily as water, whereas a
conventional
standard commercial (i.e., non-nanoparticulate or solubilized) megestrol
liquid dosage
form exhibits notably more "sluggish" characteristics.
[0036] The liquid formulations of this invention can be formulated for dosages
in
any volume but preferably equivalent or smaller volumes than existing
commercial
formulations.
2. Fast Onset of Activity
[0037] The use of conventional formulations of megestrol is not ideal due to
delayed onset of action. In contrast, the nanoparticulate megestrol
compositions of the
invention exhibit faster therapeutic effects.
[0038] Preferably, following administration the nanoparticulate megestrol
compositions of the invention have a T,,,ax of less than about 5 hours, less
than about 4.5
17
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
hours, less than about 4 hours, less than about 3.5 hours, less than about 3
hours, less than
about 2.75 hours, less than about 2.5 hours, less than about 2.25 hours, less
than about 2
hours, less than about 1.75 hours, less than about 1.5 hours, less than about
1.25 hours,
less than about 1.0 hours, less than about 50 minutes, less than about 40
minutes, less than
about 30 minutes, less than about 25 minutes, less than about 20 minutes, less
than about
15 minutes, or less than about 10 minutes.
3. Increased Bioavailability
[0039] The nanoparticulate megestrol compositions of the invention preferably
exhibit increased bioavailability and require smaller doses as compared to
prior
conventional megestrol compositions administered at the same dose.
[0040] Any drug, including megestrol, can have adverse side effects. Thus,
lower
doses of megestrol which can achieve the same or better therapeutic effects as
those
observed with larger doses of conventional megestrol compositions are desired.
Such
lower doses can be realized with the nanoparticulate megestrol compositions of
the
invention because the greater bioavailability observed with the
nanoparticulate megestrol
compositions as compared to conventional drug formulations means that smaller
doses of
drug are required to obtain the desired therapeutic effect. Specifically, a
once a day dose
of about 375 mg/5 mL (75 mg/mL) of a nanoparticulate megestrol acetate
composition is
considered equivalent to an 800 mg dose of Megace .
[0041] Administration of nanoparticulate megestrol formulations of the present
invention can exhibit bioavailability, as determined by AUCO-t, in an amount
of about
3000 ng hr/ml to about 15,000 ng hr/ml, wherein Cmax is about 300 ng/ml to
about 1400
ng/ml, 1500 ng/ml, 1600 ng/ml, 1645 ng/ml or 1700 ng/ml in a fed human subject
and
AUCO-t in an amount of about 2000 ng hr/ml to about 9000 ng hr/ml, wherein
Cmax is
about 300 ng/ml to about 2000 ng/ml in a fasted human subject. Preferably,
nanoparticulate megestrol formulations of the present invention exhibit
comparable
bioavailability in a range of between about 75 and about 130%, more preferably
between
about 80% and about 125%, of the specified therapeutic parameter (e.g., AUCO-t
or
Cmax).
18
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
4. The Pharmacokinetic Profiles of the Nanoparticulate
Megestrol Compositions of the Invention are not Substantially
Affected by the Fed or Fasted State of the Subject
Ingesting the Compositions
[0042] The invention encompasses nanoparticulate megestrol compositions
wherein the pharmacokinetic profile of the megestrol is not substantially
affected by the
fed or fasted state of a subject ingesting the composition. This means that
there is no
substantial difference in the quantity of megestrol absorbed or the rate of
megestrol
absorption when the nanoparticulate megestrol compositions are administered in
the fed
versus the fasted state. Thus, the invention encompasses nanoparticulate
megestrol
compositions that can substantially eliminate the effect of food on the
pharmacokinetics
of megestrol.
[0043] The difference in absorption of the nanoparticulate megestrol
composition
of the invention (Cmax or AUC), when administered in the fed versus the fasted
state, is
less than about 600%, less than about 575%, less than about 550%, less than
about 525%,
less than about 500%, less than about 475%, less than about 450%, less than
about 425%,
less than about 400%, less than about 375%, less than about 350%, less than
about 325%,
less than about 300%, less than about 275%, less than about 250%, less than
about 225%,
less than about 200%, less than about 175%, less than about 150%, less than
about 125%,
less than about 100%, less than about 95%, less than about 90%, less than
about 85%, less
than about 80%, less than about 75%, less than about 70%, less than about 65%,
less than
about 60%, less than about 55%, less than about 50%, less than about 45%, less
than
about 40%, less than about 35%, less than about 30%, less than about 25%, less
than
about 20%, less than about 15%, less than about 10%, less than about 5%, or
less than
about 3%. This is an especially important feature in treating patients with
difficulty in
maintaining a fed state.
[0044] In addition, preferably the difference in the rate of absorption (i.e.,
T,,,aX) of
the nanoparticulate megestrol compositions of the invention, when administered
in the fed
versus the fasted state, is less than about 100%, less than about 90%, less
than about 80%,
19
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
less than about 70%, less than about 60%, less than about 50%, less than about
40%, less
than about 30%, less than about 20%, less than about 15%, less than about 10%,
less than
about 5%, less than about 3%, or essentially no difference.
[0045] Benefits of a dosage form which substantially eliminates the effect of
food
include an increase in subject convenience, tliereby increasing subject
compliance, as the
subject does not need to ensure that they are taking a dose either with or
without food.
5. Bioequivalency of Megestrol Compositions of the
Invention When Administered in the Fed Versus the Fasted State
[0046] The invention also encompasses provides a nanoparticulate megestrol
composition in which administration of the composition to a subject in a
fasted state is
bioequivalent to administration of the composition to a subject in a fed
state.
[0047] In one embodiment of the invention, the invention encompasses
compositions comprising a nanoparticulate megestrol, wherein administration of
the
composition to a subject in a fasted state is bioequivalent to adininistration
of the
composition to a subject in a fed state, in particular as defined by CmaX and
AUC
guidelines given by the U.S. Food and Drug Administration and the
corresponding
European regulatory agency (EMEA). Under U.S. FDA guidelines, two products or
methods are bioequivalent if the 90% Confidence Intervals (CI) for AUC and
C,Y,aX are
between 0.80 to 1.25 (Tmax measurements are not relevant to bioequivalence for
regulatory purposes). To show bioequivalency between two compounds or
administration
conditions pursuant to Europe's EMEA guidelines, the 90% CI for AUC must be
between
0.80 to 1.25 and the 90% CI for Cma,{ must between 0.70 to 1.43.
6. Redispersibility Profiles of the Nanoparticulate
Megestrol Compositions of the Invention
[0048] An additional feature of the nanoparticulate megestrol compositions of
the
invention is that the compositions redisperse such that the effective average
particle size
of the redispersed megestrol particles is less than about 2 microns. This is
significant, as
if upon administration the nanoparticulate megestrol particles present in the
compositions
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
of the invention did not redisperse to a substantially nanoparticulate
particle size, then the
dosage form may lose the benefits afforded by formulating megestrol into a
nanoparticulate particle size.
[0049] This is because nanoparticulate megestrol coinpositions benefit from
the
small particle size of megestrol; if the nanoparticulate megestrol particles
do not
redisperse into the small particle sizes upon administration, then "clumps" or
agglomerated megestrol particles are formed. With the formation of such
agglomerated
particles, the bioavailability of the dosage form may fall.
[0050] Preferably, the redispersed megestrol particles of the invention have
an
effective average particle size, by weight, of less than about 2 microns, less
than about
1900 nm, less than about 1800 nm, less than about 1700 mn, less than about
1600 nm,
less than about 1500 nm, less than about 1400 nm, less than about 1300 nm,
less than
about 1200 nm, less than about 1100 nm, less than about 1000 nm, less than
about 900
nm, less than about 800 nm, less than about 700 nm, less than about 600 nm,
less than
about 500 nm, less tlian about 400 rim, less than about 300 nm, less than
about 250 nm,
fpss than about 200 nm, less than about 150 nm, less than about 100 mn, less
than about
75 nm, or less than about 50 nm, as measured by light-scattering methods,
microscopy, or
other appropriate methods.
[0051] Moreover, the nanoparticulate megestrol compositions of the invention
exhibit dramatic redispersion of the nanoparticulate megestrol particles upon
administration to a mammal, such as a human or animal, as demonstrated by
reconstitution in a biorelevant aqueous media. Such biorelevant aqueous media
can be
any aqueous media that exhibit the desired ionic strength and pH, which form
the basis
for the biorelevance of the media. The desired pH and ionic strength are those
that are
representative of physiological conditions found in the human body. Such
biorelevant
aqueous media can be, for example, aqueous electrolyte solutions or aqueous
solutions of
any salt, acid, or base, or a combination thereof, which exhibit the desired
pH and ionic
strength.
[0052] Biorelevant pH is well known in the art. For example, in the stomach,
the
pH ranges from slightly less than 2 (but typically greater than 1) up to 4 or
5. In the small
21
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
intestine the pH can range from 4 to 6, and in the colon it can range from 6
to 8.
Biorelevant ionic strength is also well lcnown in the art. Fasted state
gastric fluid has an
ionic strength of about 0.1M while fasted state intestinal fluid has an ionic
strength of
about 0.14. See e.g., Lindahl et al., "Characterization of Fluids from the
Stomach and
Proximal Jejunum in Men and Women," Pharm. Res., 14 (4): 497-502 (1997).
[0053] It is believed that the pH and ionic strength of the test solution is
more
critical than the specific chemical content. Accordingly, appropriate pH and
ionic
strength values can be obtained through numerous combinations of strong acids,
strong
bases, salts, single or multiple conjugate acid-base pairs (i.e., weak acids
and
corresponding salts of that acid), monoprotic and polyprotic electrolytes,
etc.
[0054] Representative electrolyte solutions can be, but are not limited to,
HCl
solutions, ranging in concentration from about 0.001 to about 0.1 M, and NaCl
solutions,
ranging in concentration from about 0.001 to about 0.1 M, and mixtures
thereof. For
example, electrolyte solutions can be, but are not limited to, about 0.1 M HC1
or less,
about 0.01 M HCl or less, about 0.001 M HCl or less, about 0.1 M NaCI or less,
about
0.01 M NaCI or less, about 0.001 M NaCl or less, and mixtures thereof. Of
these
electrolyte solutions, 0.01 M HCl and/or 0.1 M NaCI, are most representative
of fasted
human physiological conditions, owing to the pH and ionic strength conditions
of the
proximal gastrointestinal tract.
[0055] Electrolyte concentrations of 0.001 M HCI, 0.01 M HCI, and 0.1 M HCI
correspond to pH 3, pH 2, and pH 1, respectively. Thus, a 0.01 M HCl solution
simulates
typical acidic conditions found in the stomach. A solution of 0.1 M NaCI
provides a
reasonable approximation of the ionic strength conditions found throughout the
body,
including the gastrointestinal fluids, although concentrations higher than 0.1
M may be
employed to simulate fed conditions within the human GI tract.
[0056] Exemplary solutions of salts, acids, bases or combinations thereof,
which
exhibit the desired pH and ionic strength, include but are not limited to
phosphoric
acid/phosphate salts + sodium, potassium and calcium salts of chloride, acetic
acid/acetate
salts + sodium, potassium and calcium salts of chloride, carbonic
acid/bicarbonate salts +
22
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
sodium, potassium and calcium salts of chloride, and citric acid/citrate salts
+ sodium,
potassium and calcium salts of chloride.
7. Bioadhesive Nanoparticulate Megestrol Compositions
[0057] Bioadhesive nanoparticulate megestrol compositions of the invention
comprise at least one cationic surface stabilizer, which are described in more
detail below.
Bioadhesive formulations of megestrol exhibit exceptional bioadhesion to
biological
surfaces, such as mucous.
[0058] In the case of bioadhesive nanoparticulate megestrol compositions, the
term "bioadhesion" is used to describe the adhesion between the
nanoparticulate
megestrol compositions and a biological substrate (i.e. gastrointestinal
mucin, lung tissue,
nasal mucosa, etc.). See e.g., U.S. Patent No. 6,428,814 for "Bioadhesive
Nanoparticulate
Compositions Having Cationic Surface Stabilizers," which is specifically
incorporated by
reference.
[0059] The bioadhesive megestrol compositions of the invention are useful in
any
situation in which it is desirable to apply the compositions to a biological
surface. The
bioadhesive megestrol compositions coat the targeted surface in a continuous
and uniform
film which is invisible to the naked human eye.
[0060] A bioadhesive nanoparticulate megestrol composition slows the transit
of
the composition, and some megestrol particles would also most likely adhere to
tissue
other than the mucous cells and therefore give a prolonged exposure to
megestrol, thereby
increasing absorption and the bioavailability of the administered dosage.
8-. Pharmacokinetic Profiles of the Nanoparticulate
Megestrol Compositions of the Invention
[0061] The present invention also provides compositions of nanoparticulate
megestrol having a desirable pharmacokinetic profile when administered to
mammalian
subjects. The desirable phannacokinetic profile of the compositions comprising
megestrol includes but is not limited to: (1) a C,,,aX for megestrol, when
assayed in the
plasma of a mammalian subject following administration, that is preferably
greater than
23
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
the Cmax for a non-nanoparticulate formulation of the saine megestrol,
administered at the
same dosage; and/or (2) an AUC for megestrol, when assayed in the plasma of a
mammalian subject following administration, that is preferably greater than
the AUC for a
non-nanoparticulate formulation of the same megestrol, administered at the
same dosage;
and/or (3) a Tmax for megestrol, when assayed in the plasma of a mammalian
subject
following administration, that is preferably less than the T,,,ax for a non-
nanoparticulate
formulation of the same megestrol, administered at the same dosage. The
desirable
pharmacokinetic profile, as used herein, is the pharmacokinetic profile
measured after the
initial dose of megestrol or a salt or derivative thereof.
[0062] The desirable pharmacokinetic profile of the nanoparticulate megestrol
compositions preferably comprise the parameters: (1) that the T,,,ax of
megestrol, when
assayed in the plasma of the mammalian subject, is less than about 5 hours;
and (2) a Cmax
of megestrol is greater than about 30 nghnl. Preferably, the T,,,ax parameter
of the
pharmacokinetic profile is not greater than about 3 hours. Most preferably,
the Tmax
parameter of the pharmacokinetic profile is not greater than about 2 hours.
[0063] The desirable pharmacokinetic profile, as used herein, is the
pharmacokinetic profile measured after the initial dose of megestrol. For
example, in a
subject receiving 40 mg of megestrol four times a day, the T,,,ax and Cmax
after the initial
dose must be less than about 5 hours and greater than about 30 ng/ml,
respectively. The
compositions can be formulated in any way as described below.
[0064] Current formulations of megestrol include oral suspensions and tablets.
According to the package insert of Megace , the pharmacokinetic profile of the
oral
suspension contains parameters such that the median T,,,ax is 5 hours and the
mean Cmax is
753 ng/ml. Further, the Tax and Cmax for the Megace0 40 mg tablet, after the
initial
dose, is 2.2 hours and 27.6 ng/ml, respectively. Physicians Desk Reference,
55th Ed.,
2001. The nanoparticulate megestrol compositions of the invention
simultaneously
improve upon at least the T,,,ax and Cmax parameters of the pharmacokinetic
profile of
megestrol.
24
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0065] In one embodiment, a threshold blood plasma concentration of megestrol
of about 700 ng/ml is attained in less than about 5 hours after administration
of the
formulation, and preferably not greater than about 3 hours.
[0066] A preferred nanoparticulate megestrol composition of the invention
exhibits in comparative pharmacokinetic testing with a standard commercial
formulation
of megestrol, such as Megace oral suspension or tablet from Bristol Myers
Squibb, a
T,,,aX which is less than about 100%, less than about 90%, less than about
80%, less than
about 70%, less than about 60%, less than about 50%, less than about 40%, less
than
about 30%, less than about 25%, less than about 20%, less than about 15%, or
less than
about 10% of the T,,,a,t exhibited by the standard commercial formulation of
megestrol.
[0067] A preferred nanoparticulate megestrol coinposition of the invention
exhibits in comparative pharmacokinetic testing with a standard commercial
formulation
of megestrol, such as MegaceS oral suspension or tablet from Bristol Myers
Squibb, a
C,,,aX which is greater than about 5%, greater than about 10%, greater than
about 15%,
greater than about 20%, greater than about 30%, greater than about 40%,
greater than
about 50%, greater than about 60%, greater than about 70%, greater than about
80%,
greater than about 90%, greater than about 100%, greater than about 110%,
greater than
about 120%, greater than about 130%, greater than about 140%, greater than
about 150%,
greater than about 200%, greater than about 500% or greater than about 800%
than the
C,,,aX exhibited by the standard commercial formulation of megestrol.
[0068] A preferred nanoparticulate megestrol composition of the invention
exhibits in comparative pharmacokinetic testing with a standard commercial
formulation
of megestrol, such as Megace oral suspension or tablet from Bristol Myers
Squibb, an
AUC which is greater than about 5%, greater than about 10%, greater than about
15%,
greater than about 20%, greater than about 30%, greater than about 40%,
greater than
about 50%, greater than about 60%, greater than about 70%, greater than about
80%,
greater than about 90%, greater than about 100%, greater than about 110%,
greater than
about 120%, greater than about 130%, greater than about 140%, greater than
about 150%,
greater than about 200%, greater than about 500% or greater than about 800%
than the
AUC exhibited by the standard commercial formulation of megestrol.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0069] There is no critical upper limit of blood plasma concentration so long
as
the dosage amounts set out below are not significantly exceeded. A suitable
dose of
megestrol, adininistered according to the method of the invention, is
typically in the range
of about 1 mg/day to about 1000 mg/day, or from about 40 mg/day to about 800
mg/day.
In one embodiment, a nanoparticulate megestrol composition is administered at
a dose of
575 mg/day. In other embodiments, the nanoparticulate megestrol composition is
administered at doses of 625 mg/day or 675 mg/day. Preferably, the
therapeutically
effective amount of the nanoparticulate megestrol compositions of the
invention is about
1/6, 1/5, 1/4, 1/3, 1/2, 2/3, 3/4 or 5/6 of the therapeutically effective
amount of existing
commercial megestrol formulations.
[0070] Any standard pharmacokinetic protocol can be used to determine blood
plasma concentration profile in humans following administration of a
nanoparticulate
megestrol composition, and thereby establish whether that composition meets
the
pharmacokinetic criteria set out herein. For example, a randomized single-dose
crossover
study can be performed using a group of healthy adult human subjects. The
number of
subjects should be sufficient to provide adequate control of variation in a
statistical
analysis, and is typically about 10 or greater, although for certain purposes
a smaller
group can suffice. Each subject receives by oral administration at time zero a
single dose
(e.g., 300 mg) of a test formulation of megestrol, normally at around 8 am
following an
overnight fast. The subjects continue to fast and remain in an upright
position for about 4
hours after administration of the megestrol formulation. Blood samples are
collected
from each subject prior to administration (e.g., 15 minutes) and at several
intervals after
administration. For the present purpose it is preferred to take several
samples within the
first hour, and to sample less frequently thereafter. Illustratively, blood
samples could be
collected at 15, 30, 45, 60, and 90 minutes after administration, then every
hour from 2 to
hours after administration. Additional blood samples may also be taken later,
for
example at 12 and 24 hours after administration. If the same subjects are to
be used for
study of a second test formulation, a period of at least 7 days should elapse
before
administration of the second formulation. Plasma is separated from the blood
samples by
centrifugation and the separated plasma is analyzed for megestrol by a
validated high
26
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
performance liquid chromatography (HPLC) procedure, such as for example Garver
et al.,
J. Pharna. Sci. 74(6):664-667 (1985), the entirety of which is hereby
incorporated by
reference. Plasma concentrations of megestrol referenced herein are intended
to mean
total megestrol concentrations including both free and bound megestrol.
[0071] Any formulation giving the desired phannacokinetic profile is suitable
for
administration according to the present methods. Exemplary types of
formulations giving
such profiles are liquid dispersions and solid dose forms of nanoparticulate
megestrol.
Dispersions of megestrol have proven to be stable at teinperatures up to 50 C.
If the
liquid dispersion medium is one in which the nanoparticulate megestrol has
very low
solubility, the nanoparticulate megestrol particles are present as suspended
particles. The
smaller the megestrol particles, the higher the probability that the
formulation will exhibit
the desired pharmacokinetic profile.
9. Combination Pharmacokinetic Profile Compositions
[0072] In yet another embodiment of the invention, a first nanoparticulate
megestrol composition providing a desired pharmacokinetic profile is co-
administered,
sequentially administered, or combined with at least one other megestrol
composition that
generates a desired different pharmacokinetic profile. More than two megestrol
compositions can be co-administered, sequentially administered, or combined.
While the
first megestrol composition has a nanoparticulate particle size, the
additional one or more
megestrol compositions can be nanoparticulate, solubilized, or have a
conventional
microparticulate particle size.
[0073] For exanlple, a first megestrol composition can have a nanoparticulate
particle size, conferring a short T,,,ax and typically a higher Cmax= This
first megestrol
composition can be combined, co-administered, or sequentially administered
with a
second composition comprising: (1) megestrol having a larger (but still
nanoparticulate
as defined herein) particle size, and therefore exhibiting slower absorption,
a longer T,,,ax,
and typically a lower Cmax; or (2) a microparticulate or solubilized megestrol
composition,
exhibiting a longer Tmax, and typically a lower C,t,ax.
27
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0074] The second, third, fourth, etc., megestrol compositions can differ from
the
first, and from each other, for example: (1) in the effective average particle
sizes of
megestrol; or (2) in the dosage of megestrol. Such a combination composition
can reduce
the dose frequency required.
[0075] If the second megestrol composition has a nanoparticulate particle
size,
then preferably the megestrol particles of the second composition have at
least one surface
stabilizer associated with the surface of the drug particles. The one or more
surface
stabilizers can be the same as or different from the surface stabilizer(s)
present in the first
megestrol composition.
[0076] Preferably where co-administration of a "fast-acting" formulation and a
"longer-lasting" formulation is desired, the two formulations are combined
within a single
composition, for example a dual-release composition.
10. Combination Active Agent Compositions
[0077] The invention encompasses the nanoparticulate megestrol compositions of
the invention formulated or co-administered with one or more non-megestrol
active
agents, which are either conventional (solubilized or microparticulate) or
nanoparticulate.
Methods of using such combination compositions are also encompassed by the
invention.
The non-megestrol active agents can be present in a crystalline phase, an
amorphous
phase, a semi-crystalline phase, a semi-amorphous phase, or a mixture thereof.
[0078] The compound to be administered in combination with a nanoparticulate
megestrol composition of the invention can be formulated separately from the
nanoparticulate megestrol composition or co-formulated with the
nanoparticulate
megestrol composition. Where a nanoparticulate megestrol composition is co-
formulated
with a second active agent, the second active agent can be formulated in any
suitable
manner, such as immediate-release, rapid-onset, sustained-release, or dual-
release form.
[0079] If the non-megestrol active agent has a nanoparticulate particle size
i.e., a
particle size of less than about 2 microns, then preferably it will have one
or more surface
stabilizers associated with the surface of the active agent. In addition, if
the active agent
has a nanoparticulate particle size, then it is preferably poorly soluble and
dispersible in at
28
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
least one liquid dispersion media. By "poorly soluble" it is meant that the
active agent has
a solubility in a liquid dispersion media of less than about 30 mg/mL, less
than about 20
mg/mL, less than about 10 mg/mL, or less than about 1 mg/mL. Useful liquid
dispersion
medias include, but are not limited to, water, aqueous salt solutions,
safflower oil, and
solvents such as ethanol, t-butanol, hexane, and glycol.
[0080] Such non-megestrol active agents can be, for exaniple, a therapeutic
agent.
A therapeutic agent can be a pharmaceutical agent, including biologics. The
active agent
can be selected from a variety of known classes of drugs, including, for
example, amino
acids, proteins, peptides, nucleotides, anti-obesity drugs, central nervous
system
stimulants, carotenoids, corticosteroids, elastase inhibitors, anti-fungals,
oncology
therapies, anti-emetics, analgesics, cardiovascular agents, anti-inflammatory
agents, such
as NSAIDs and COX-2 inhibitors, anthelmintics, anti-arrhythmic agents,
antibiotics
(including penicillins), anticoagulants, antidepressants, antidiabetic agents,
antiepileptics,
antihistamines, antihypertensive agents, antimuscarinic agents,
antimycobacterial agents,
antineoplastic agents, immunosuppressants, antithyroid agents, antiviral
agents,
anxiolytics, sedatives (hypnotics and neuroleptics), astringents, alpha-
adrenergic receptor
blocking agents, beta-adrenoceptor blocking agents, blood products and
substitutes,
cardiac inotropic agents, contrast media, corticosteroids, cough suppressants
(expectorants
and mucolytics), diagnostic agents, diagnostic imaging agents, diuretics,
dopaminergics
(antiparkinsonian agents), haemostatics, immunological agents, lipid
regulating agents,
muscle relaxants, parasympathomimetics, parathyroid calcitonin and
biphosphonates,
prostaglandins, radio-pharmaceuticals, sex hormones (including steroids), anti-
allergic
agents, stimulants and anoretics, sympathomimetics, thyroid agents,
vasodilators, and
xanthines.
[0081] A description of these classes of active agents and a listing of
species
within each class can be found in Martindale's The Extra Pharmacopoeia, 31st
Edition
(The Pharmaceutical Press, London, 1996), specifically incorporated by
reference. The
active agents are commercially available and/or can be prepared by techniques
known in
the art.
29
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0082] Exemplary nutraceuticals and dietary supplements are disclosed, for
example, in Roberts et al., Nutraceuticals: The Complete Encyclopedia of
Supplements,
Herbs, Vitamins, and Healing Foods (American Nutraceutical Association, 2001),
which
is specifically incorporated by reference. Dietary suppleinents and
nutraceuticals are also
disclosed in Ph.ysicians' Desk Reference for Nutritional Supplements, 1st Ed.
(2001) and
The Playsicians' Desk Reference for Herbal Medicines, 1 st Ed. (2001), botli
of which are
also incorporated by reference. A nutraceutical or dietary supplement, also
known as a
phytochemical or functional food, is generally any one of a class of dietary
supplements,
vitamins, minerals, herbs, or healing foods that have medical or
pharmaceutical effects on
the body.
[0083] Exemplary nutraceuticals or dietary supplements include, but are not
limited to, lutein, folic acid, fatty acids (e.g., DHA and ARA), fruit and
vegetable
extracts, vitamin and mineral supplements, phosphatidylserine, lipoic acid,
melatonin,
glucosamine/chondroitin, Aloe Vera, Guggul, glutamine, amino acids (e.g.,
arginine, iso-
leucine, leucine, lysine, methionine, phenylanine, threonine, tryptophan, and
valine),
green tea, lycopene, whole foods, food additives, herbs, phytonutrients,
antioxidants,
flavonoid constituents of fruits, evening primrose oil, flax seeds, fish and
marine animal
oils, and probiotics. Nutraceuticals and dietary supplements also include bio-
engineered
foods genetically engineered to have a desired property, also known as
"pharmafoods."
11. Sterile Filtered Nanoparticulate Megestrol Compositions
[0084] The nanoparticulate megestrol compositions of the invention can be
sterile
filtered. This obviates the need for heat sterilization, which can harm or
degrade
megestrol, as well as result in crystal growth and particle aggregation.
[0085] Sterile filtration can be difficult because of the required small
particle size
of the composition. Filtration is an effective method for sterilizing
homogeneous
solutions when the membrane.filter pore size is less than or equal to about
0.2 microns
(200 mn) because a 0.2 micron filter is sufficient to remove essentially all
bacteria.
Sterile filtration is normally not used to sterilize conventional suspensions
of micron-
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
sized megestrol because the megestrol particles are too large to pass through
the
membrane pores.
[0086] A sterile nanoparticulate megestrol dosage form is particularly useful
in
treating immunocompromised patients, infants or juvenile patients, and the
elderly, as
these patient groups are the most susceptible to infection caused by a non-
sterile liquid
dosage form.
[0087] Because the nanoparticulate megestrol compositions of the invention can
be sterile filtered, and because the compositions can have a very small
megestrol effective
average particle size, the compositions are suitable for parenteral
administration.
12. Miscellaneous Benefits of the Nanoparticulate
Megestrol Compositions of the Invention
[0088] The nanoparticulate megestrol compositions preferably exhibit an
increased rate of dissolution as compared to conventional microcrystalline
forms of
megestrol. In addition, the compositions of the invention exliibit improved
performance
characteristics for oral, intravenous, subcutaneous, or intramuscular
injection, such as
higher dose loading and smaller tablet or liquid dose volumes. Moreover, the
nanoparticulate megestrol compositions of the invention do not require organic
solvents
or pH extremes.
[0089] Another benefit of the nanoparticulate megestrol compositions of the
invention is that is was surprisingly discovered that upon administration,
nanoparticulate
compositions of megestrol acetate reach therapeutic blood levels within one
dose. This is
in dramatic contrast to the current commercially available megestrol acetate
composition
(Megace by Bristol Myers Squibb Co.), which requires multiple doses,
administered
over several days to a week, to build up to a therapeutic level of drug in the
blood stream.
B. Compositions
[0090] The invention provides compositions comprising nanoparticulate
megestrol particles and preferably at least one surface stabilizer. The one or
more surface
stabilizers are preferably associated with the surface of the megestrol
particles. Surface
31
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
stabilizers useful herein preferably do not chemically react with the
megestrol particles or
itself. Individual molecules of the surface stabilizer are essentially free of
intermolecular
cross-linlcages.
[0091 ] The present invention also includes nanoparticulate megestrol
compositions together with one or more non-toxic physiologically acceptable
carriers,
adjuvants, or vehicles, collectively referred to as carriers. The compositions
can be
formulated for parenteral injection (e.g., intravenous, intramuscular, or
subcutaneous),
oral administration in solid, liquid, or aerosol form, vaginal, nasal, rectal,
ocular, local
(powders, ointments or drops), buccal, intracistemal, intraperitoneal, or
topical
administration, and the like.
1. Megestrol Particles
[0092] As used herein the term megestrol, which is the active ingredient in
the
composition, is used to mean megestrol, megestrol acetate (17a-acetyloxy-6-
methylpregna-4,6-diene-3,20-dione), or a salt thereof. The megestrol particles
can be
present in a crystalline phase, an amorphous phase, a semi-crystalline phase,
a semi-
amorphous phase, or a mixture thereof.
[0093] Megestrol acetate is well known in the art and is readily recognized by
one
of ordinary skill. Generally, megestrol is used for treating breast cancer,
endometrial
cancer and, less frequently, prostate cancer. Megestrol is also frequently
used as an
appetite stimulant for patients in a wasting state, such as HIV wasting,
cancer wasting,
and anorexia. Megestrol may be used for other indications where progestins are
typically
used, such as hormone replacement therapy in post-menopausal women and oral
contraception. Further, megestrol may be used for ovarian suppression in
several
conditions such as endometriosis, hirsutism, dysmenorrhea, and uterine
bleeding, as well
as uterine cancer, cervical cancer, and renal cancer. Megestrol is also used
in patients
following castration.
32
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
2. Surface Stabilizers
[0094] The choice of a surface stabilizer for megestrol is non-trivial.
Accordingly, the present invention is directed to the surprising discovery
that
nanoparticulate megestrol compositions can be made.
[0095] Combinations of more than one surface stabilizer can be used in the
invention. Preferred surface stabilizers include, but are not limited to,
hydroxypropyl
methylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, random
copolymers of
vinyl pyrrolidone and vinyl acetate, sodium lauryl sulfate,
dioctylsulfosuccinate or a
combination thereof. . Preferred primary surface stabilizers include, but are
not limited
to, hydroxypropyl methylcellulose, hydroxypropylcellulose,
polyvinylpyrrolidone, random
copolymers of vinyl pyrrolidone and vinyl acetate, or a combination thereof.
Preferred
secondary surface stabilizers include, but are not limited to, sodium lauryl
sulfate and
dioctylsulfosuccinate.
[0096] Other surface stabilizers which can be employed in the invention
include,
'but are not limited to, known organic and inorganic#armaceutical excipients.
Such
excipients include various polymers, low molecular weight oligomers, natural
products,
and surfactants. Surface stabilizers include nonionic, cationic, ionic, and
zwitterionic
surfactants.
[0097] Representative examples of surface stabilizers include hydroxypropyl
methylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, sodium lauryl
sulfate,
dioctylsulfosuccinate, gelatin, casein, lecithin (phosphatides), dextran, gum
acacia,
cholesterol, tragacanth, stearic acid, benzalkonium chloride, calcium
stearate, glycerol
monostearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan
esters,
polyoxyethylene alkyl ethers (e.g., macrogol ethers such as cetomacrogol
1000),
polyoxyethylene castor oil derivatives, polyoxyethylene sorbitan fatty acid
esters (e.g., the
commercially available Tweens such as e.g., Tween 20 and Tween 80 (ICI
Specialty
Chemicals)); polyethylene glycols (e.g., Carbowaxs 3550 and 934 (Union
Carbide)),
polyoxyethylene stearates, colloidal silicon dioxide, phosphates,
carboxymethylcellulose
calcium, carboxymethylcellulose sodium, methylcellulose,
hydroxyethylcellulose,
hydroxypropylmethylcellulose phthalate, noncrystalline cellulose, magnesium
aluminium
33
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
silicate, triethanolamine, polyvinyl alcohol (PVA), 4-(1,1,3,3-
tetramethylbutyl)-phenol
polymer with ethylene oxide and formaldehyde (also lciown as tyloxapol,
superione, and
triton), poloxamers (e.g., Pluronics F68 and F108 , which are block
copolymers of
ethylene oxide and propylene oxide); poloxamines (e.g., Tetronic 908 , also
lcnown as
Poloxamine 908 , which is a tetrafunctional block copolymer derived from
sequential
addition of propylene oxide and ethylene oxide to ethylenediamine (BASF
Wyandotte
Corporation, Parsippany, N.J.)); Tetronic 1508 (T-1508) (BASF Wyandotte
Corporation), Tritons X-200 , which is an alkyl aryl polyether sulfonate (Rohm
and
Haas); Crodestas F-110 , which is a mixture of sucrose stearate and sucrose
distearate
(Croda Inc.); p-isononylphenoxypoly-(glycidol), also known as Olin-lOG or
Surfactant
10-G (Olin Chemicals, Stamford, CT); Crodestas SL-40 (Croda, Inc.); and
SA9OHCO,
which is C18H37CH2(CON(CH3)-CH2(CHOH)4(CH2OH)2 (Eastman Kodak Co.);
decanoyl-N-methylglucamide; n-decyl (3-D-glucopyranoside; n-decyl (3-D-
maltopyranoside; n-dodecyl (3-D-glucopyranoside; n-dodecyl [i-D-maltoside;
heptanoyl-
N-methylglucamide; n-heptyl-(3-D-glucopyranoside; n-heptyl (3-D-thioglucoside;
n-hexyl
(3-D-glucopyranoside; nonanoyl-N-methylglucamide; n-noyl (3-D-glucopyranoside;
octanoyl-N-methylglucamide; n-octyl-(3-D-glucopyranoside; octyl (3-D-
thioglucopyranoside; PEG-phospholipid, PEG-cholesterol, PEG-cholesterol
derivative,
PEG-vitamin A, PEG-vitamin E, lysozyme, random copolymers of vinyl pyrrolidone
and
vinyl acetate, and the like.
[0098] Examples of useful cationic surface stabilizers include, but are not
limited
to, polymers, biopolymers, polysaccharides, cellulosics, alginates,
phospholipids, and
nonpolymeric compounds, such as zwitterionic stabilizers, poly-n-
methylpyridinium,
anthryul pyridinium chloride, cationic phospholipids, chitosan, polylysine,
polyvinylimidazole, polybrene, polymethylmethacrylate trimethylammoniumbromide
bromide (PMMTMABr), hexyldesyltrimethylammonium bromide (HDMAB), and
polyvinylpyrrolidone-2-dimethylaminoethyl methacrylate dimethyl sulfate.
[0099] Other useful cationic stabilizers include, but are not limited to,
cationic
lipids, sulfonium, phosphonium, and quarternary anunonium compounds, such as
stearyltrimethylammonium chloride, benzyl-di(2-chloroethyl)ethylammonium
bromide,
34
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
coconut trimethyl ammonium chloride or bromide, coconut methyl dihydroxyethyl
ammoniuin chloride or bromide, decyl triethyl ammonium chloride, decyl
dimethyl
hydroxyethyl ammonium chloride or bromide, C12_15dimethyl hydroxyethyl
ammonium
chloride or bromide, coconut dimethyl hydroxyethyl aminonium chloride or
bromide,
myristyl trimethyl ammonium methyl sulphate, lauryl dimethyl benzyl ammonium
chloride or bromide, lauryl dimethyl (ethenoxy)4 ammonium chloride or bromide,
N-alkyl
(C12_18)dimethylbenzyl ammonium chloride, N-alkyl (C14_18)dimethyl-benzyl
ammonium
chloride, N-tetradecylidmethylbenzyl ammonium chloride monohydrate, dimethyl
didecyl
ammonium chloride, N-alkyl and (C12_14) dimetllyl 1-napthylmethyl ammonium
chloride,
trimetliylammonium halide, alkyl-trimethylammonium salts and dialkyl-
dimethylammonium salts, lauryl trimethyl ammonium chloride, ethoxylated
alkyamidoalkyldialkylammonium salt and/or an ethoxylated trialkyl ammonium
salt,
dialkylbenzene dialkylammonium chloride, N-didecyldimethyl ammonium chloride,
N-
tetradecyldimethylbenzyl arnmonium, chloride monohydrate, N-alkyl(C12_14)
dimethyl 1-
naphthylmethyl ammonium chloride and dodecyldimethylbenzyl ammonium chloride,
dialkyl benzenealkyl ammonium chloride, lauryl trimethyl ammonium chloride,
alkylbenzyl methyl ammonium chloride, alkyl benzyl dimethyl ammonium bromide,
C12,
C15, C17 trimethyl ammonium bromides, dodecylbenzyl triethyl ammonium
chloride,
poly-diallyldimethylammonium chloride (DADMAC), dimethyl ammonium chlorides,
alkyldimethylammonium halogenides, tricetyl methyl ammonium chloride,
decyltrimethylammonium bromide, dodecyltriethylammonium bromide,
tetradecyltrimethylammonium bromide, methyl trioctylammonium chloride (ALIQUAT
336TM), POLYQUAT lOTM, tetrabutylammonium bromide, benzyl trimethylammonium
bromide, choline esters (such as choline esters of fatty acids), benzalkonium
chloride,
stearalkonium chloride conipounds (such as stearyltrimonium chloride and Di-
stearyldimonium chloride), cetyl pyridinium bromide or chloride, halide salts
of
quaternized polyoxyethylalkylamines, MIRAPOLTM and ALKAQUATTM (Alkaril
Chemical Company), alkyl pyridinium salts; amines, such as alkylamines,
dialkylamines,
alkanolamines, polyethylenepolyamines, N,N-dialkylaminoalkyl acrylates, and
vinyl
pyridine, amine salts, such as lauryl amine acetate, stearyl amine acetate,
alkylpyridinium
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
salt, and alkylimidazolium salt, and amine oxides; imide azolinium salts;
protonated
quaternary acrylamides; methylated quaternary polymers, such as poly[diallyl
dimethylammonium chloride] and poly-[N-methyl vinyl pyridinium chloride]; and
cationic guar.
[00100] Such exemplary cationic surface stabilizers and other useful
cationic surface stabilizers are described in J. Cross and E. Singer, Cationic
Suzfactants:
Analytical and Biological Evaluation (Marcel Dekker, 1994); P. and D. Rubingh
(Editor),
Cationic Surfactants: Physical Chemistzy (Marcel Deldcer, 1991); and J.
Richmond,
Cationic Suifactants: Organic Clzeznistzy, (Marcel Deldcer, 1990).
[00101] Particularly preferred nonpolymeric primary stabilizers are any
nonpolymeric compound, such benzalkonium chloride, a carbonium compound, a
phosphonium compound, an oxonium compound, a halonium compound, a cationic
organometallic compound, a quarternary phosphorous compound, a pyridinium
compound, an anilinium compound, an ammonium compound, a hydroxylammonium
compound, a primary ammonium compound, a secondary ammonium compound, a
tertiary ammonium compound, and quarternary ammonium compounds of the formula
NR1R2R3R4(+). For compounds of the formula NR1RZR3R4(+):
(i) none of RI-R4 are CH3;
(ii) one of Rl-R4 is CH3;
(iii) three of RI-R4 are CH3;
(iv) all of RI-R4 are CH3;
(v) two of RI-R4 are CH3, one of RI-R4 is C6H5CH2, and one of RI-R4 is an
alkyl chain of seven carbon atoms or less;
(vi) two of Rl-R4 are CH3, one of RI-R4 is C6H5CH2, and one of RI-R4 is an
alkyl chain of nineteen carbon atoms or more;
(vii) two of RI-R4 are CH3 and one of RI-R4 is the group C6H5(CH2),,, where
n>1;
(viii) two of RI-R4 are CH3, one of RI-R4 is C6H5CH2, and one of RI-R4
comprises at least one heteroatom;
36
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
(ix) two of R1-R4 are CH3, one of RI-R4 is C6H5CHZ, and one ofRl-R4
comprises at least one halogen;
(x) two of R1-R4 are CH3, one of RI-R4 is C6H5CH2, and one of RI-R4
comprises at least one cyclic fragment;
(xi) two of RI-R4 are CH3 and one of RI-R4 is a phenyl ring; or
(xii) two of RI-R4 are CH3 and two of RI-R4 are purely aliphatic fragments.
[00102] Such compounds include, but are not limited to, behenalkonium
chloride, benzethonium chloride, cetylpyridinium chloride, behentrimonium
chloride,
lauralkonium chloride, cetalkonium chloride, cetrimonium bromide, cetrimonium
chloride, cethylamine hydrofluoride, chlorallylmethenamine chloride
(Quaternium-15),
distearyldimonium chloride (Quaternium-5), dodecyl dimethyl ethylbenzyl
ammonium
chloride(Quaternium-14), Quaternium-22, Quaternium-26, Quaternium-18
hectorite,
dimethylaminoethylchloride hydrochloride, cysteine hydrochloride,
diethanolammonium
POE (10) oletyl ether phosphate, diethanolammonium POE (3)oleyl ether
phosphate,
tallow alkonium chloride, dimethyl dioctadecylarnmoniumbentonite,
stearalkonium
chloride, domiphen bromide, denatonium benzoate, myristalkonium chloride,
laurtrimonium chloride, ethylenediamine dihydrochloride, guanidine
hydrochloride,
pyridoxine HCI, iofetamine hydrochloride, meglumine hydrochloride,
methylbenzethonium chloride, myrtrimonium bromide, oleyltrimonium chloride,
polyquaternium-l, procainehydrochloride, cocobetaine, stearalkonium bentonite,
stearalkoniumhectonite, stearyl trihydroxyethyl propylenediamine
dihydrofluoride,
tallowtrimonium chloride, and hexadecyltrimethyl ammonium bromide.
[00103] Most of these surface stabilizers are known pharmaceutical
excipients and are described in detail in the Handbook of Pharmaceutical
Excipients,
published jointly by the American Pharmaceutical Association and The
Pharmaceutical
Society of Great Britain (The Pharmaceutical Press, 2000), specifically
incorporated by
reference. The surface stabilizers are commercially available and/or can be
prepared by
techniques known in the art.
37
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
3. Other Pharmaceutical Excipients
[00104] Pharmaceutical megestrol compositions according to the invention
may also comprise one or more binding agents, filling agents, lubricating
agents,
suspending agents, sweeteners, flavoring agents, preservatives, buffers,
wetting agents,
disintegrants, effervescent agents, and other excipients. Such excipients are
known in the
art.
[00105] Examples of filling agents are lactose monohydrate, lactose
anhydrous, and various starches; examples of binding agents are various
celluloses and
cross-linked polyvinylpyrrolidone, microcrystalline cellulose, such as Avicel
PH 101 and
Avicel PH102, microcrystalline cellulose, and silicified microcrystalline
cellulose
(ProSolv SMCCTM).
[00106] Suitable lubricants, including agents that act on the flowability of
the powder to be compressed, are colloidal silicon dioxide, such as Aerosil
200, talc,
stearic acid, magnesium stearate, calcium stearate, and silica gel.
[0100] Examples of sweeteners are any natural or artificial sweetener, such as
sucrose, xylitol, sodium saccharin, cyclamate, aspartame, and acsulfame.
Examples of
flavoring agents are Magnasweet (trademark of MAFCO), bubble gum flavor, and
fruit
flavors, and the like.
[0101] Examples of preservatives are potassium sorbate, methylparaben,
propylparaben, benzoic acid and its salts, other esters of parahydroxybenzoic
acid such as
butylparaben, alcohols such as ethyl or benzyl alcohol, phenolic compounds
such as
phenol, or quarternary compounds such as benzalkonium chloride.
[0102] Suitable diluents include pharmaceutically acceptable inert fillers,
such as
microcrystalline cellulose, lactose, dibasic calcium phosphate, saccharides,
and/or
mixtures of any of the foregoing. Examples of diluents include
microcrystalline cellulose,
such as Avicel PH101 and Avicel PH102; lactose such as lactose monohydrate,
lactose
anhydrous, and Pharmatose DCL21; dibasic calcium phosphate such as Emcompress
;
mannitol; starch; sorbitol; sucrose; and glucose.
38
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0103] Suitable disintegrants include lightly crosslinked polyvinyl
pyrrolidone,
corn starch, potato starch, maize starch, and modified starches,
croscarmellose sodium,
cross-povidone, sodium starch glycolate, and mixtures thereof.
[0104] Examples of effervescent agents are effervescent couples such as an
organic acid and a carbonate or bicarbonate. Suitable organic acids include,
for example,
citric, tartaric, malic, fumaric, adipic, succinic, and alginic acids and
anhydrides and acid
salts. Suitable carbonates and bicarbonates include, for example, sodium
carbonate,
sodium bicarbonate, potassium carbonate, potassium bicarbonate, magnesium
carbonate,
sodium glycine carbonate, L-lysine carbonate, and arginine carbonate.
Alternatively, only
the sodium bicarbonate component of the effervescent couple may be present.
4. Nanoparticulate Megestrol or Active Agent Particle Size
[0105] As used herein, particle size is determined on the basis of the weight
average particle size as measured by conventional particle size measuring
techniques well
known to those skilled in the art. Such techniques include, for example,
sedimentation
field flow fractionation, photon correlation spectroscopy, light scattering,
and disk
centrifugation.
[0106] The compositions of the invention comprise nanoparticulate megestrol
particles which have an effective average particle size of less than about
2000 nm (i.e., 2
microns). In other embodiments of the invention, the megestrol particles have
an
effective average particle size of less than about 1900 nm, less than about
1800 nm, less
than about 1700 nm, less than about 1600 nm, less than about 1500 nm, less
than about
1400 nm, less than about 1300 nm, less than about 1200 rim, less than about
1100 nm,
less than about 1000 nm, less than about 900 nm, less than about 800 nm, less
than about
700 nm, less than about 600 nm, less than about 500 nm, less than about 400
nm, less
than about 300 nm, less than about 250 nm, less than about 200 nm, less than
about 150
nm, less than about 100 nm, less than about 75 rim, or less than about 50 nm,
when
measured by the above techniques.
39
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0107] If the nanoparticulate megestrol composition additionally comprises one
or
more non-megestrol nanoparticulate active agents, then such active agents have
an
effective average particle size of less than about 2000 nm (i.e., 2 microns).
In other
embodiments of the invention, the nanoparticulate non-megestrol active agents
can have
an effective average particle size of less than about 1900 nm, less than about
1800 nm,
less than about 1700 nm, less than about 1600 nm, less than about 1500 nm,
less than
about 1400 nm, less than about 1300 nm, less than about 1200 nm, less than
about 1100
nm, less than about 1000 nm, less than about 900 nm, less than about 800 nm,
less than
about 700 nm, less than about 600 nm, less than about 500 nm, less than about
400 nm,
less than about 300 nm, less than about 250 nm, less than about 200 nm, less
than about
150 nm, less than about 100 nm, less than about 75 nm, or less than about 50
nm, as
measured by light-scattering methods, microscopy, or other appropriate
methods.
[0108] By "an effective average particle size of less than about 2000 nm" it
is
meant that at least 50% of the nanoparticulate megestrol or nanoparticulate
non-megestrol
active agent particles have a particle size of less than about 2000 nm, by
weight (or by
other suitable measurement technique, such as by number, volume, etc.), when
measured
by the above-noted techniques. Preferably, at least about 60%, at least about
70%, at least
about 80%, at least about 90%, at least about 95%, or at least about 99% of
the
nanoparticulate megestrol or nanoparticulate non-megestrol active agent
particles have a
particle size of less than the effective average, i.e., less than about 2000
nm, less than
about 1900 nm, less than about 1800 nm, etc.
[0109] If the nanoparticulate megestrol composition is combined with a
conventional or microparticulate megestrol composition or non-megestrol active
agent
composition, then such a composition is either solubilized or has an effective
average
particle size of greater than about 2 microns. By "an effective average
particle size of
greater than about 2 microns" it is meant that at least 50% of the
conventional megestrol
or non-megestrol active agent particles have a particle size of greater than
about 2
microns, by weight, when measured by the above-noted techniques. In other
embodiments of the invention, at least about 70%, about 90%, about 95%, or
about 99%
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
of the conventional megestrol or non-megestrol active agent particles have a
particle size
greater than about 2 microns.
[0110] In the present invention, the value for D50 of a nanoparticulate
megestrol
composition is the particle size below which 50% of the megestrol particles
fall, by
weight. Similarly, D90 is the particle size below which 90% of the megestrol
particles
fall, by weight.
5. Concentration of Nanoparticulate Megestrol and Surface Stabilizers
[0111] The relative amounts of nanoparticulate megestrol and one or more
surface
stabilizers can vary widely. The optimal amount of the individual components
can
depend, for example, the hydrophilic lipophilic balance (HLB), melting point,
and the
surface tension of water solutions of the stabilizer, etc.
[0112] The concentration of megestrol can vary from about 99.5% to about
0.001%, from about 95% to about 0.1%, or from about 90% to about 0.5%, by
weight,
based on the total combined dry weight of the megestrol and at least one
surface
stabilizer, not including other excipients.
[0113] The concentration of the at least one surface stabilizer can vary from
about
0.5% to about 99.999%, from about 5.0% to about 99.9%, or from about 10% to
about
99.5%, by weight, based on the total combined dry weight of the megestrol and
at least
one surface stabilizer, not including other excipients.
[0114] If a combination of two or more surface stabilizers is employed in the
composition, the concentration of the at least one primary surface stabilizer
can vary from
about 0.01% to about 99.5%, from about 0.1% to about 95%, or from about 0.5%
to about
90%, by weight, based on the total combined dry weight of the megestrol, at
least one
primary surface stabilizer, and at least one secondary surface stabilizer, not
including
other excipients. In addition, the concentration of the at least one secondary
surface
stabilizer can vary from about 0.01 % to about 99.5%, from about 0.1 % to
about 95%, or
from about 0.5% to about 90%, by weight, based on the total combined dry
weight of the
megestrol, at least one primary surface stabilizer, and at least one secondary
surface
stabilizer, not including other excipients.
41
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
C. Methods of Making Nanoparticulate Megestrol Compositions
[0115] The nanoparticulate megestrol compositions can be made using, for
example, milling, homogenization, precipitation, freezing, template emulsion
techniques,
or any combination thereof. Exemplary methods of malcing nanoparticulate
active agent
compositions are described in the '684 patent.
[0116] Methods of making nanoparticulate compositions are also described in
U.S. Patent No. 5,518,187 for "Method of Grinding Pharmaceutical Substances;"
U.S.
Patent No. 5,718,388 for "Continuous Method of Grinding Pharmaceutical
Substances;"
U.S. Patent No. 5,862,999 for "Method of Grinding Pharmaceutical Substances;"
U.S.
Patent No. 5,665,331 for "Co-Microprecipitation of Nanoparticulate
Pharmaceutical
Agents with Crystal Growth Modifiers;" U.S. Patent No. 5,662,883 for "Co-
Microprecipitation of Nanoparticulate Pharmaceutical Agents with Crystal
Growth
Modifiers;" U.S. Patent No. 5,560,932 for "Microprecipitation of
Nanoparticulate
Pharmaceutical Agents;" U.S. Patent No. 5,543,133 for "Process of Preparing X-
Ray
Contrast Compositions Containing Nanoparticles;" U.S. Patent No. 5,534,270 for
"Method of Preparing Stable Drug Nanoparticles;" U.S. Patent No. 5,510,118 for
"Process of Preparing Therapeutic Compositions Containing Nanoparticles;" and
U.S.
Patent No. 5,470,583 for "Method of Preparing Nanoparticle Compositions
Containing
Charged Phospholipids to Reduce Aggregation," all of which are specifically
incorporated
by reference.
[0117] The resultant nanoparticulate megestrol compositions can be utilized in
solid or liquid dosage formulations, such as controlled release formulations,
solid dose
fast melt formulations, aerosol formulations, lyophilized formulations,
tablets, capsules,
etc.
1. Milling to Obtain Nanoparticulate Megestrol Dispersions
[0118] Milling megestrol to obtain a nanoparticulate megestrol dispersion
comprises dispersing megestrol particles in a liquid dispersion medium in
which
megestrol is poorly soluble, followed by applying mechanical means in the
presence of
grinding media to reduce the particle size of megestrol to the desired
effective average
42
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
particle size. The dispersion medium can be, for example, water, safflower
oil, ethanol, t-
butanol, glycerin, polyethylene glycol (PEG), hexane, or glycol.
[0119] The megestrol particles can be reduced in size in the presence of at
least
one surface stabilizer. Alternatively, the megestrol particles can be
contacted with one or
more surface stabilizers after attrition. Other compounds, such as a diluent,
can be added
to the megestrol/surface stabilizer composition either before, during, or
after the size
reduction process. Dispersions can be manufactured continuously or in a batch
mode.
2. Precipitation to Obtain Nanoparticulate Megestrol Compositions
[0120] Another method of forming the desired nanoparticulate megestrol
composition is by microprecipitation. This is a method of preparing stable
dispersions of
poorly soluble active agents in the presence of one or more surface
stabilizers and one or
more colloid stability enhancing surface active agents free of any trace toxic
solvents or
solubilized heavy metal impurities. Such a method comprises, for example: (1)
dissolving megestrol in a suitable'solvent; (2) adding the formulation from
step (1) to a
solution corimprising at least one surface stabilizer; and (3) precipitating
the formulation
from step (2) using an appropriate non-solvent. The method can be followed by
removal
of any formed salt, if present, by dialysis or diafiltration and concentration
of the
dispersion by conventional means.
3. Homogenization to Obtain Nanoparticulate Megestrol Compositions
[0121 ] Exemplary homogenization methods of preparing nanoparticulate active
agent compositions are described in U.S. Patent No. 5,510,118, for "Process of
Preparing
Therapeutic Compositions Containing Nanoparticles."
[0122] Such a method comprises dispersing megestrol particles in a liquid
dispersion medium, followed by subjecting the dispersion to homogenization to
reduce
the particle size of the megestrol to the desired effective average particle
size. The
megestrol particles can be reduced in size in the presence of at least one
surface stabilizer.
Alternatively, the megestrol particles can be contacted with one or more
surface
stabilizers either before or after attrition. Other compounds, such as a
diluent, can be
43
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
added to the megestrol/surface stabilizer composition either before, during,
or after the
size reduction process. Dispersions can be manufactured continuously or in a
batch
mode.
4. Cryogenic Methodologies to Obtain Nanoparticulate Megestrol
Compositions
Another method of forming the desired nanoparticulate megestrol composition is
by spray freezing into liquid (SFL). This technology comprises an organic or
organoaqueous solution of megestrol with stabilizers, which is injected into a
cryogenic
liquid, such as liquid nitrogen. The droplets of the megestrol solution freeze
at a rate
sufficient to minimize crystallization and particle growth, thus formulating
nanostructured
megestrol particles. Depending on the choice of solvent system and processing
conditions, the nanoparticulate megestrol particles can have varying particle
morphology.
In the isolation step, the nitrogen and solvent are removed under conditions
that avoid
agglomeration or ripening of the megestrol particles.
As a complementary technology to SFL, ultra rapid freezing (URF) may also be
used to created equivalent nanostructured megestrol particles with greatly
enhanced
surface area. URF comprises an organic or organoaqueous solution of megestrol
with
stabilizers onto a cryogenic substrate.
5. Emulsion Methodologies to Obtain
Nanoparticulate Megestrol Compositions
Another method of forming the desired nanoparticulate megestrol composition is
by template emulsion. Template emulsion creates nanostructured megestrol
particles with
controlled particle size distribution and rapid dissolution performance. The
method
comprises an oil-in-water emulsion that is prepared, then swelled with a non-
aqueous
solution comprising the megestrol and stabilizers. The particle size
distribution of the
megestrol particles is a direct result of the size of the emulsion droplets
prior to loading
with the megestrol a property which can be controlled and optimized in this
process.
Furthermore, through selected use of solvents and stabilizers, emulsion
stability is
achieved with no or suppressed Ostwald ripening. Subsequently, the solvent and
water
44
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
are removed, and the stabilized nanostructured megestrol particles are
recovered. Various
megestrol particles morphologies can be achieved by appropriate control of
processing
conditions.
D. Methods of Using Nanoparticulate Megestrol Formulations of the Invention
1. Applications of the Nanoparticulate Megestrol
Compositions of the Invention
[0123] The nanoparticulate megestrol compositions of the invention may be used
as an appetite stimulant to treat wasting conditions or cachexia. As used
herein, the term
"wasting" is used to mean a condition where the patient is losing body mass as
a side
effect of a disease progression, a disease treatment, or other condition.
Examples of
conditions where wasting is prevalent include, but are not limited to, HIV or
AIDS,
cancer, cachexia and anorexia.
[0124] Additional conditions where the nanoparticulate megestrol compositions
of
the invention may be used include, but are not limited to, neoplastic diseases
where the
disease normally regresses or the patient's symptoms are normally reduced in
response to
megestrol, or any other progestin.
[0125] The nanoparticulate megestrol compositions of the invention may also be
used to treat conditions such as breast cancer, endometrial cancer, uterine
cancer, cervical
cancer, prostate cancer, and renal cancer. As used herein, the term "cancer"
is used as one
of ordinary skill in the art would recognize the term. Examples of cancers
include, but are
not limited to, neoplasias (or neoplasms), hyperplasias, dysplasias,
metaplasias, and
hypertrophies. The neoplasms may be benign or malignant, and they may
originate from
any cell type, including but not limited to epithelial cells of various
origin, muscle cells,
and endothelial cells.
[0126] The present invention also provides methods of hormone replacement
therapy in post-menopausal women, or in subjects after castration, comprising
administering a nanoparticulate megestrol composition of the invention.
Further, the
compositions of the present invention may be used for ovarian suppression in
several
situations such as endometriosis, hirsutism, dysmenorrhea, and uterine
bleeding.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0127] The present invention also provides methods of oral contraception
comprising administering a nanoparticulate megestrol composition of the
invention. In
one embodiment, the compositions of the invention are administered in
combination with
estrogen or a synthetic estrogen.
2. Dosage Forms of the Invention
[0128] The nanoparticulate megestrol compositions of the invention can be
administered to a subject via any conventional means including, but not
limited to, orally,
rectally, ocularly, parenterally (e.g., intravenous, intramuscular, or
subcutaneous),
intracistemally, pulmonary, intravaginally, intraperitoneally, locally (e.g.,
powders,
ointments or drops), or as a buccal or nasal spray. As used herein, the term
"subject" is
used to mean an animal, preferably a mammal, including a human or non-human.
The
terms patient and subject may be used interchangeably.
[0129] Moreover, the nanoparticulate megestrol compositions of the invention
can
be formulated into any suitable dosage form, including but not limited to
liquid
dispersions, gels, aerosols, ointments, creams, controlled release
formulations, fast melt
formulations, lyophilized formulations, tablets, capsules, delayed release
formulations,
extended release formulations, pulsatile release formulations, and mixed
immediate
release and controlled release formulations.
[0130] Nanoparticulate megestrol compositions suitable for parenteral
injection
may comprise physiologically acceptable sterile aqueous or nonaqueous
solutions,
dispersions, suspensions or emulsions, and sterile powders for reconstitution
into sterile
injectable solutions or dispersions. Examples of suitable aqueous and
nonaqueous
carriers, diluents, solvents, or vehicles including water, ethanol, polyols
(propyleneglycol,
polyethylene-glycol, glycerol, and the like), suitable mixtures thereof,
vegetable oils (such
as olive oil) and injectable organic esters such as ethyl oleate. Proper
fluidity can be
maintained, for example, by the use of a coating such as lecithin, by the
maintenance of
the required particle size in the case of dispersions, and by the use of
surfactants.
46
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0131] The nanoparticulate megestrol compositions may also contain adjuvants
such as preserving, wetting, emulsifying, and dispensing agents. Prevention of
the growth
of microorganisms can be ensured by various antibacterial and antifungal
agents, such as
parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be
desirable to
include isotonic agents, such as sugars, sodium chloride, and the like.
Prolonged
absorption of the injectable pharmaceutical form can be brought about by the
use of
agents delaying absorption, such as aluminum monostearate and gelatin.
[0132] Solid dosage forms for oral administration include, but are not limited
to,
capsules, tablets, pills, powders, and granules. In such solid dosage forms,
the active
agent is admixed with at least one of the following: (a) one or more inert
excipients (or
carriers), such as sodium citrate or dicalcium phosphate; (b) fillers or
extenders, such as
starches, lactose, sucrose, glucose, mannitol, and silicic acid; (c) binders,
such as
carboxymethylcellulose, alignates, gelatin, polyvinylpyrrolidone, sucrose, and
acacia; (d)
humectants, such as glycerol; (e) disintegrating agents, such as agar-agar,
calcium
carbonate, potato'~or tapioca starch, alginic acid, certain complex silicates,
and sodium
carbonate; (f) solution retarders, such as paraffin; (g) absorption
accelerators, such as
quaternary ammonium compounds; (h) wetting agents, such as cetyl alcohol and
glycerol
monostearate; (i) adsorbents, such as kaolin and bentonite; and (j)
lubricants, such as talc,
calcium stearate, magnesium stearate, solid polyethylene glycols, sodium
lauryl sulfate, or
mixtures thereof. For capsules, tablets, and pills, the dosage forms may also
comprise
buffering agents.
[0133] Liquid nanoparticulate megestrol dosage forms for oral administration
include pharmaceutically acceptable emulsions, solutions, suspensions, syrups,
and
elixirs. In addition to megestrol, the liquid dosage forms may comprise inert
diluents
commonly used in the art, such as water or other solvents, solubilizing
agents, and
emulsifiers. Exemplary emulsifiers are ethyl alcohol, isopropyl alcohol, ethyl
carbonate,
ethyl acetate, benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-
butyleneglycol,
dimethylformainide, oils, such as cottonseed oil, groundnut oil, corn germ
oil, olive oil,
castor oil, and sesame oil, glycerol, tetrahydrofurfuiyl alcohol,
polyethyleneglycols, fatty
acid esters of sorbitan, or mixtures of these substances, and the like.
47
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0134] Besides such inert diluents, the composition can also include
adjuvants,
such as wetting agents, emulsifying and suspending agents, sweetening,
flavoring, and
perfuming agents.
3. Dosage Quantities for the Nanoparticulate
Megestrol Compositions of the Invention
[0135] The present invention provides a method of achieving therapeutically
effective plasma levels of megestrol in a subject at a lower dose than the
standard
commercial formulations. This can permit smaller dosing volumes depending on
the
megestrol concentration chosen. Such a method comprises orally administering
to a
subject an effective amount of a nanoparticulate megestrol composition.
[0136] The nanoparticulate megestrol composition, when tested in fasting
subjects
in accordance with standard pharmacokinetic practice, produces a maximum blood
plasma concentration profile of megestrol of greater than about 30 ng/ml in
less than
about 5 hours after the initial dose of the coinposition.
[0137] As used herein, the phrase "maximum plasma concentration" is
interpreted
as the maximum plasma concentration that megestrol will reach in fasting
subjects.
[0138] A suitable dose of inegestrol, administered according to the method of
the
invention, is typically in the range of about 1 mg/day to about 1000 mg/day,
or from about
40 mg/day to about 800 mg/day. Preferably, the therapeutically effective
amount of the
megestrol of this invention is about 1/6, about 1/5, about 1/4, about 1/3ra'
or about 1/Z of the
therapeutically effective amount of existing commercial megestrol
formulations, e.g.,
Megace .
[0139] "Therapeutically effective amount" as used herein with respect to a
drug
dosage, shall mean that dosage that provides the specific pharmacological
response for
which the drug is administered in a significant number of subjects in need of
such
treatment. It is emphasized that "therapeutically effective amount,"
administered to a
particular subject in a particular instance will not always be effective in
treating the
diseases described herein, even though such dosage is deemed a
"therapeutically effective
amount" by those skilled in the art. It is to be further understood that drug
dosages are, in
48
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
particular instances, measured as oral dosages, or with reference to drug
levels as
measured in blood.
[0140] One of ordinary skill will appreciate that effective amounts of
megestrol
can be detennined empirically and can be employed in pure form or, where such
forms
exist, in pharmaceutically acceptable salt, ester, or prodrug form. Actual
dosage levels of
megestrol in the nanoparticulate compositions of the invention may be varied
to obtain an
amount of megestrol that is effective to obtain a desired therapeutic response
for a
particular composition and method of administration. The selected dosage level
therefore
depends upon the desired therapeutic effect, the route of administration, the
potency of the
administered megestrol, the desired duration of treatment, and other factors.
[0141] Dosage unit compositions may contain such amounts of such submultiples
thereof as may be used to make up the daily dose. It will be understood,
however, that the
specific dose level for any particular patient will depend upon a variety of
factors: the
type and degree of the cellular or physiological response to be achieved;
activity of the
specific agent or composition employed; the specific agents or composition
employed; the
age, body weight, general health, sex, and diet of the patient; the time of
administration,
route of administration, and rate of excretion of the agent; the duration of
the treatment;
drugs used in combination or coincidental with the specific agent; and like
factors well
known in the medical arts.
[0142] The following examples are given to illustrate the present invention.
It
should be understood, however, that the invention is not to be limited to the
specific
conditions or details described in these examples. Throughout the
specification, any and
all references to a publicly available document, including a U.S. patent, are
specifically
incorporated by reference.
[0143] In the examples that follow, the value for D50 is the particle size
below
which 50% of the megestrol particles fall. Similarly, D90 is the particle size
below which
90% of the megestrol particles fall.
49
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0144] The formulations in the examples that follow were also investigated
using
a light microscope. Here, "stable" nanoparticulate dispersions (unifonn
Brownian
motion) were readily distinguishable from "aggregated" dispersions (relatively
large,
nonuniform particles without motion). Stable, as known in the art and used
herein, means
the particles don't substantially aggregate or ripen (increase in f-undamental
particle size).
Example 1
[0145] The purpose of this example was to describe preparation of
nanoparticulate
dispersions of megestrol acetate.
[0146] Fonnulations 1, 2, 3, 4 and 5, shown in Table 1, were milled under high
energy milling conditions using a NanoMill (Elan Drug Delivery, Inc.) (see
e.g., WO
00/72973 for "Small-Scale Mill and Method Thereof') and a Dyno -Mill (Willy
Bachofen AG).
TABLE 1
Formulation Quantity Identity and Quantity Identity and Quantity Mean D90 (nm)
of of Primary Surface of Secondary Surface (nm)
Megestrol Stabilizer Stabilizer
1 5% 1% HPC-SL 0.05% DOSS 167 224
2 5% 1% HPMC 0.05% DOSS 156 215
3 5% 1% PVP 0.05% DOSS 167 226
4 5% 1% Plasdone S630* 0.05% DOSS 164 222
5% 1% HPMC 0.05% SLS 148 208
* Plasdone S630 (ISP) is a random copolymer of vinyl acetate and vinyl
pyrrolidone.
[0147] Formulations 1-5 showed small, well-dispersed particles using the
Horiba
La-910 Laser Scattering Particle Size Distribution Analyzer (Horiba
Instruments, Irvine,
CA) and light microscopy. Formulations 1-5 were stable in electrolyte fluids
and had
acceptable physical stability at 5 C for 4 weeks. Electrolyte fluids are
representative of
physiological conditions found in the human body. Formulations 1, 2, 3, and 4
also
exhibited acceptable stability at 25 C and 40 C for 4 weeks. Formulation 5
exhibited
acceptable stability at 40 C for at least 3 weelcs.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
Example 2
[0148] This example compares the pharmacokinetic parameters of nanoparticulate
megestrol acetate formulations of the present invention with conventional
microparticulate formulations of megestrol acetate.
[0149] Twelve male beagles, at least twelve months of age, were divided into 2
groups based on whether they were fasting or being fed. The dogs were
acclimated for
thirteen days prior to dosing. The animals weighed approximately 11.4 to 14.3
kg at the
time of dosing, and the dose was adjusted to 10 mg/kg. Water was available ad
libituna.
The animals were fasted (food only) for twelve to sixteen hours prior to
dosing on day 1.
On day 1, each dog was administered a formulation by gavage. Following dosing,
the
gavage tube was flushed with 18 ml of water. In the fed study, the animals
were fed a
high fat meal about 1 hour prior to dosing.
[0150] The dogs were subdivided into four groups, with each group receiving
eitlier Formulation A (nanoparticulate megestrol dispersion #1, comprising
4.0%
megestrol acetate, 0.8% HPMC,, and 0.4% DOSS), Formulation B (nanoparticulate
megestroLdispersion #2, comprising 4.0% megestrol acetate, 0.8% HPMC, and
0.04%
SLS), Formulation C (suspension of microparticulate megestrol acetate, Par
Pharmaceutical, Inc., New York) or Formulation D(Megace Oral Suspension,
which is
a suspension of microparticulate megestrol acetate). Each formulation was
adjusted to
administer a dose of 10 mg/kg ofinegestrol acetate to the subject.
[0151] Prior to dosing, blood samples were taken from each subject. Blood
samples were then collected from each subject at 15 and 30 minutes, as well as
1, 2, 3, 4,
6, 8, 24, 48, and 72 hours after dosing and centrifuged. Plasma was then
separated and
diluted when necessary, and subsequently analyzed for megestrol acetate by
HPLC.
[0152] Tables 2 and 3 summarize the pharmacokinetic data of the four
formulations administered to fasted dogs and fed dogs, respectively.
51
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
TABLE 2
Summary of Pharmacokinetic Data in Fasted Do s
Parameters Formulation A n=3 Formulation B n=3 Formulation C n=3 Formulation D
n=3
Mean ~ SD) (Mean -+ SD) (Mean -+ SD) (Mean SD)
AUCo-t 37774.23f 11648.60 21857.68f 10737.53 17395.95f 10428.73 10094.30
1990.89
AUCO-inf 49408.88f3392.80 27863.56~:15279.16 6948.48:L* 12007.13f1923.80
Cmax 2209.74 351.54 1563.02 787.37 484,98f321.70 339.92 175.86
Tmax 0.8310.29 0.50:W.00 18.67 9.24 2.67 0.58
tyz 42.01.+33.81 30.09-+19.37 26.57 * 25.59-~7.11
K.1 0.025 0.018 0.032 0.024 0.026 * 0.028 Ø007
AUCo_t (ng.hr/ml) = Area under the curve from time zero to the last measurable
concentration;
AUCo-ffif(ng.hr/ml)= Area under the curve from time zero to infinity;
Cm~ (ng/ml)= Maximum plasma concentration;
T. (hr)= Time to occurrence of Cmax;
ti~ (hr)= Apparent eliniination half-life;
Kel (1/hr)= elimination rate constant;
*n=1.
TABLE 3
Summary of Pharmacokinetic Data in Fed Dogs
Parameters Formulation A Formulation B Formulation C Formulation D
n=3 n=3 n=3 n=3
(Mean SD) (Mean f SD) (Mean SD) (Mean SD)
AUCo-t 48543.56 11608.55 36687.92f12016.26 27332.11 6488.79 31397.16 5823.79
AUC04nf 61734.90 4918.52 42787.74 14630.92 31720.98 5580.32 40218.66f8649.33*
Cmax 3777.34 2489.41 2875.8211334.32 2180.73f406.28 2577.83 665.31
Tmax 1.67th2.02 3.00 4.33 1.00 0.00 0.83 0.29
Z'y2 34.35 12.10 26.67~7.80 26.16 10.88 36.60~9.62*
K.1 0.02210.009 0.028 0.010 0.31 0.16 0.20-+0.005
AUCo_t (ng.hr/ml)= Area under the curve from time zero to the last measurable
concentration;
AUCo_~f(ng.hr/ml)= Area under the curve from time zero to infinity;
Cmax (ng/ml)= Maximum plasma concentration;
T,,,ax (hr)= Time to occurrence of Cmax;
ty, (lhr)= Apparent elimination half-life;
Kei (1/hr) = elimination rate constant;
*n=2.
[0153] The results in the fasted dogs show that the nanoparticulate megestrol
formulations (Formulations A and B) showed dramatically superior
bioavailability, as
52
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
evidenced by the superior AUC and C,,,ax results, as compared to the
conventional
microparticulate megestrol formulations (Formulations C and D). Formulation A,
with a
Cmax of 2210, had a maximum concentration more than 4%z times that of
Formulation C
(485), and a maximum concentration more than 6%a times that of Formulation D
(340).
Formulation B, with a Cmax of 1563, had a maximum concentration more than 3.2
times
that of Formulation C (485), and a maximum concentration more than 4.6 times
that of
Formulation D (340). Also, Formulation A, with an AUC of 49,409 ng hr/mL, had
an
oral bioavailability more than 7 times that of Formulation C (6948 ng hr/mL)
and an oral
bioavailability of more than 4 times that of Formulation D (12007 ng hr/mL).
Formulation B, with an AUC of 27,864 ng hr/mL, had an oral bioavailability
more than 4
times that of Formulation C (6949 ng hr/mL) and an oral bioavailability more
than 2 times
that of Formulation D (12,007 ng hr/mL).
[0154] In addition, in the fasted dogs the nanoparticulate megestrol
formulations
(Formulations A and B) showed dramatically superior faster onset of action, as
evidenced
by the superior T,,,ax results, as compared to the conventional
microparticulate megestrol
formulations (Formulations C and D). Formulation A, with a T,,,ax of 0.83 hr,
reached a
maximum concentration of megestrol in less than 1/20t" the time of Formulation
C (18.67
hr), and in less than 1/3ra the time of Formulation D (2.67 hr). Formulation
B, with a Tmax
of 0.50 hr, reached a maximum concentration in less than 1/37th the time of
Formulation
C (18.67 hr), and in less than 1/5th the time of Formulation D (2.67 hr).
[0155] Similarly, the results in the fed dogs show that the nanoparticulate
megestrol formulations (Formulations A and B) showed dramatically superior
bioavailability, as evidenced by the superior AUC and Cmax results, as
compared to the
conventional microparticulate megestrol formulations (Formulations C and D).
Formulation A, with a Cmax of 3777, had a maximum concentration of about more
than
1.7 times that of Formulation C(2181), and a maximum concentration of about
more than
1.5 times that of Formulation D (2578). Formulation B, with a Cmax of 2876,
had a
maximum concentration of about more tlian 1.3 times that of Formulation
C(2181), and a
maximum concentration of about more than 1.1 times that of Formulation D
(2578).
53
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
Formulation A, with an AUC of 61,735 ng hr/mL, had an oral bioavailability of
more than
1.9 times that of Formulation C (31721 ng hr/mL) and more than 1.5 times that
of
Formulation D (40219 ng hr/mL). Formulation B, with an AUC of 42788 ng hr/mL,
had
an oral bioavailability of more tha.n 1.3 times that of Fonnulation C (31721
ng hr/mL) and
an oral bioavailability of more than 1.1 times that of Formulation D (40218 ng
hr/mL).
Example 3
[0156] This example demonstrates the physical stability of megestrol acetate
dispersions at various concentrations and with the addition of sucrose,
flavoring, and
preservatives.
[0157] Megestrol acetate was milled under high energy milling conditions using
a
NanoMillTM2 System (Elan Drug Delivery, Inc.) in the presence of a
preservative / buffer
system consisting of sodium benzoate, citric acid monohydrate, and sodium
citrate
dihydrate. After milling, the resulting dispersion was diluted with water,
sucrose,
flavoring, and additional preservative / buffer to prepare dispersions
containing 3% (w/w),
5% (w/w), or 9% (w/w) megestrol acetate. The resulting formulations are shown
in Table
4. The physical stability of the formulations was then monitored at 25 C, 40
C, and 50 C.
TABLE 4
Formulation Summary
Concentrated Diluted, Flavored Dispersions
Nanoparticle
Dispersion
Formulation E Formulation F Formulation G
3% Dispersion 5% Dispersion 9% Dispersion
API and Excipients g/kg g/kg g/kg g/kg
Megestrol Acetate, USP 325.000 30.000 50.000 90.000
Hydroxypropyl Methylcellulose, USP 65.000 6.000 10.000 18.000
Docusate Sodium, USP 3.250 0.300 0.500 0.900
Sodium Benzoate, USP 1.214 1.826 1.777 1.681
Sodium Citrate Dihydrate, USP 0.910 0.091 0.089 0.084
Citric Acid Monohydrate, USP 0.061 1.369 1.333 1.260
Sucrose, USP 50.000 50.000 50.000
Natural and Artificial Lemon Flavor 0.400 0.400 0.400
Artificial Lime Flavor 0.400 0.400 0.400
Purified Water, USP 604.600 909.614 885.500 837.280
API = active pharmaceutical ingredient
54
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0158] Particle size measurements (Table 5) were used to assess the physical
stability. The results show almost no increase in the mean particle size at
either 25 C or
40 C, and only a slight increase in the mean particle size at 50 C. 126 days
of stability
measurements were obtained for the 5% and 9% dispersions and 33 days of
stability were
obtained for the 3% dispersion, which was prepared at a later date.
TABLE 5
Mean article size (nm)
3% Dis ersion 5% Dis ersion 9% Dispersion
25 C 40 C 50 C 25 C 40 C 50 C 25 C 40 C 50 C
0 days 148 148 148 169 169 169 169 169 169
30 days 172 171 187 172 170 179
33 days 141 144 173
126 days 171 174 188 168 175 182
Example 4
[0159] The purpose of this Example was to demonstrate the improved viscosity
characteristics of the dispersions of this invention.
[0160] The viscosities of three formulations of this invention (E, F, and G as
described in Example 3) and two conventional commercial formulations
(Formulations C
and D as described in Example 2) were determined using a rheometer (model CVO-
50,
Bohlin Instruments). The measurements were performed at a temperature of 20 C
using a
double gap (40/50) geometry.
[0161] The viscosities of the Formulations of this invention were found to be
nearly Newtonian (i. e., the viscosity being independent of shear rate), and
were 1.5, 2.0,
and 3.5 mPa s for the 30, 50, and 90 mg/mL concentrations, respectively.
[0162] The viscosity dependence on concentration is illustrated in Figure 1.
[0163] The commercial formulations C and D were shear thinning in nature. Such
samples cannot be characterized by a single viscosity but rather a series of
viscosities
measured at different shear rates. This is most conveniently illustrated as
viscosity - shear
rate curves as shown in FIG. 2.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0164] The comrnercial samples and the three formulations of this invention
are
compared in Table 6 below. Viscosities are in units of mPa s.
TABLE 6
Shear Rates of Comniercial Megestrol Formulations (D and C) and the
Nano articulate Megestrol Formulations of the Invention (E, F, & G)
Shear Rate Commercial Samples Formulations E, F, & G
s" Formulation D Formulation C(E) 30 mg/mL (F) 50 mg/mL (G) 90 mg/mL
(mPa s) (mPa s) (mPa s) (mPa s) (mPa s)
0.1 4010 2860 1.5 2.0 3.5
1 929 723 " " "
215 183
100 49.9 46.3
These samples were not measured at the 0.1 and 1 s"1 shear rates (the shear
range was ca 2 to 100 s"I) but
the assessment that these exhibit Newtonian flow properties justifies the
entries.
Example 5
[0165] The purpose of this Example was to visually demonstrate the difference
between the viscosity character'istics of liquid megestrol formulations of the
invention as
compared to conventional liquid megestrol formulations.
[0166] A sample of a 50 mg/mL nanoparticulate dispersion of megestrol acetate
and two conventional commercial formulations at 40 mg/mL (Formulations C and D
as
described in Example 2) were each placed in a vial, which was then shaken.
Attached as
Figure 3 is a photograph of the thee vials, which from left to right are the
nanoparticulate
megestrol acetate dispersion, Formulation C, and Formulation D.
[0167] The vial with the nanoparticulate dispersion shows a thin, silky,
almost
shear film coating the vial. In contrast, the vials containing the two
commercial
formulations show a gritty residue coating. Such a gritty residue is the same
residue
which coats a patient's mouth and throat upon administration. Such a coating
is highly
unpleasant, particularly for patients suffering from wasting (i.e., unable to
eat). Thus,
Figure 3 visually demonstrates the appeal of a liquid oral nanoparticulate
megestrol
formulation of the invention as compared to conventional commercial liquid
oral
megestrol formulations.
56
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
Example 6
[0168] The purpose of this example was to prepare nanoparticulate compositions
of megestrol acetate using various surface stabilizers.
[0169] 5% megestrol acetate (Par Pharmaceuticals, Inc.) was combined with
1.25% of various surface stabilizers: tyloxapol (Sterling Organics), Tween 80
(Spectrum
Quality Products), Pluronic F-108 (BASF), Plasdone S-630 (ISP),
hydroxypropylmethylcellulose (HPMC) (Shin Etsu), hydroxypropylcellulose (HPC-
SL)
(Nippon Soda Co., Ltd.), Kollidon K29/32 (polyvinylpyrrolidone) (ISP), or
lysozyme
(Fordras).
[0170] For each combination of megestrol acetate and surface stabilizer, the
surface stabilizer was first dissolved in 7.875 g water for injection (WFI)
(Abbott
Laboratories, Inc.), followed by the addition of the milling media, PolyMillTM-
500 (Dow
Chemical, Co.), and 0.42 g megestrol.
[0171] The slurries were charged into each of eight 18 cc NanoMill (Elan Drug
Delivery) chambers and milled for 30 min. Upon completion of milling the
dispersions
were harvested with a 26 gauge needle yielding the following particle sizes
shown in
Table 7.
[0172] All particle size distribution analyses were conducted on a Horiba LA-
910
Laser Light Scattering Particle Size Distribution Analyzer (Horiba
Instruments, Irvine,
CA). RO-water was utilized as the liquid dispersing medium and a flow-through
sample
cell was used for all measurements. All samples were assayed in 150 cc liquid
medium.
TABLE 7
Megestrol Surface Stabilizer/Conc. Mean Particle Size
Conc.
5% tyloxapol; 1.25% 214 nm
5% Tween 80; 1.25% 210 nm
5% Pluronic F-108; 1.25% 459 nm
5% Plasdone S-630; 1.25% 292 nm
5% HPMC; 1.25% 314 nm
5% HPC-SL; 1.25% 623 nm
5% PVP K29/32; 1.25% 24816 nm
5% lysozyme; 1.25% 179 nm
57
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0173] The results show that tyloxapol, Tween 80, and lysozyme produced small
particles without substantial aggregation. Pluronic F-108, Plasdone S-630,
HPMC, HPC-
SL, and K29/32 had larger particle sizes, indicating that aggregation was
occurring. Thus,
at the particular concentration of drug and surface stabilizer, using the
described milling
method, Pluronic F-108, Plasdone S-630, HPMC, HPC-SL, and K29/32 were not
preferable surface stabilizers. These surface stabilizers may be useful in
nanoparticulate
compositions of megestrol at different drug or surface stabilizer
concentrations, or when
used in conjunction with another surface stabilizer.
Example 7
[0174] The purpose of this example was to prepare nanoparticulate compositions
of megestrol acetate using various surface stabilizers.
[0175] Megestrol acetate (Par Pharmaceuticals, Inc.) and various surface
stabilizers, as shown in Table 8, were combined and milled, followed by
determination of
the particle size and stability of the resulting composition. Materials were
obtained as in
Example 6.
[0176] All of the samples were milled using a Dyno -Mill (Model KDL-Series,
Willy Bachofen AG, Basel, Switzerland) equipped with a 150 cc stainless steel
batch
chamber. Cooling water (approximate temperature 5 C) was circulated through
the mill
and chamber during operation.
[0177] All particle size distribution analyses were conducted on a Horiba LA-
910
Laser Light Scattering Particle Size Distribution Analyzer (Horiba
Instruments, Irvine,
CA), as described above in Example 6.
[0178] Qualitative microscopic assessments of the formulations were performed
using a Leica light microscope (Type 301-371.010). Sample preparation involved
diluting the product dispersions in RO-water and dispensing about 10 L onto a
glass
slide. Oil immersion was utilized in conjunction with 1000x magnification.
[0179] The physical stability was assessed by storing the dispersion is 20 ml
glass
scintillation vials in a temperature / humidity controlled chamber at either 5
C, (25 C /
58
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
60% RH), (40 C / 75% RH), (50 C /75% RH), or 55 C. Samples were taken at
varying
time intervals and the particle size was analyzed.
[0180] For all formulations, the surface stabilizer(s) was first dissolved in
WFI
(Abbott Laboratories, Inc.) (75.0 g for Exp. Nos. 1, 2, 3, 7, and 8; 75.2 g
for Exp. Nos. 4
and 9; 74.9 g for Exp. Nos. 5 and 6; 70.3 g for Exp. Nos. 10 and 11), followed
by
combining the surface stabilizer solution nlegestrol acetate and Po1yMi11TM-
500
polymeric grinding media. This mixture was then added to the appropriate
milling
chamber, milled for the time period shown in Table 8, followed by harvesting
and
vacuum filtering of the megestrol acetate dispersion.
59
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
M
~ O
00 o ~ o
0 0 _ Y
rn 'j 0 ~ N 'n 'n cn v N U
i o
~ ~ ~= O V c~
Cc3 N O-'J _" O~ F H cn
in 4 c~d -- Od O
ca b ~ 'd O O cri~ ca '+ cd cd a v N
Z rd ~ b r, r,
0 o cn
N o c~ cri
> o v o 75 U~a c~d V
U
'd M 0i o ~.
A c~i y, o o Z ('U' Z o U H a~i U '~i
~ o Y o ~' o o s~ x ,~ x s =~ o >,
9 o > '
~ ~ o 9 r:l
a~ y (1) U .(~~ u~,~
4' sHQ. t~ N~' .~-+~ Y t;'
b~0 dp' vWi dp' ~~ U~ 0~ U'd p p sN Yr.
Y m Y C/] i..;
V1 ~4 ~
P. ~ .~ Vp do = C N
Cd
>-, cn cn -j vi vi
+ 'd ~f ~ EG ~ b 0' N C d (1) 01~0 cq3 d v~ '3
w t~" V t tn ~ p r~+ p 4-4 ~o ~." 4 4 \o r n O~+ O O~q O~ y~0
ct, c~ co N p N 0 N
'C O U O o o y- v o r Nv o ~' v o o V~ o O N o O o O N,-~ O O N.-. ~~A W
C~ ~==+ (.) -F=> Y C~ Y C~ =--1 Y =~ Y =~ M 4-1 C*
v
~ cc3 ) cd cd p cd ,=p cd O N~ O N t-a O
en O N 0 N N ::3 Cd N'~' cd N =-+ Cd F-+ N Cd i-~ O
N~ bA V bA bA V P. bA -L bA P+ 4-
0 0 0 cn a)
En Y O
cq3 tYo cd rn p
[Ya ~ cn U Q) r~) U (n U U N
rn
OO p 3==~"' V -i~ S~"' ~"i ,-i .3=". ,D r
W O M P. ~Y+cn~ En P, cn Q. U) v f~. O
~ v v~
r ~ -+ r~ r~ 0 P. 00 N ~
~ ~ kn
~
~
~
A A
N kn (14 N Lo ~ d Lr) kn
d N
00 o ~ a a
y 6 U C,) rr~
~ ~
~., o 0 0 ,_, o~1 0 0 0 0 0
a , N
tõ V, kP, l(, ~.o V, Ln V, kf)
~ N N N \ N O N O N N
- .-i r-i .-~ .-+ p '=-i O =--i r-+ ,--r O
y o 0 0 0 0 0 0 0 0
y o \ ~ \ \ \ \ \ \
~ U tn ln V'1 tn kA tn tn tn
m V) \O l~ 00
W ~'
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
~
~
i
rn c'n
~
0 0
4-i
N bp ~, i-, p
O N ~ N ~ N
~~~b=~
Q cn cncd
'd
c cd cd ~
m
rz Cd
cn C~C M ~N cC{
cn
Cd
o ~ ~ ~ ~"tz) ~ ybb
rn
o o o
+~+ 4-~ f~ 4-a 4 a
~o o ~ U ~ o
0 0 0 0 >1
~ ~t d= ~ ~r
U U p
Cd N N N 0
bo
U U
bo O ~ O O
3 ~ C ~ O ~ Cd
00 o 0 0 ~
4.10
~~~~~~~~0 ~
~.
\10 00
o 0 0
U
,-,
N
U U cn p U
u (U
Q
N O N O'ci'
~ w
w
a o 0 o v~i
~ U O o o
~ C/O]
R" O o --~ N O
0
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0181] The results shown in Table 8 indicate that the use of lysozyme (Exp.
No.
1) as a surface stabilizer resulted in small well dispersed particles with a
mean particle
size of 209 mn, but the formulation showed aggregation when diluted into a
normal saline
solution. A megestrol acetate/tyloxapol sample was also stable at higher drug
and
stabilizer concentrations (Exp. No. 12).
[0182] Tween 80, tyloxapol, and Pluronic F127 (Exp. Nos. 2, 3, and 4) were
effective primary surface stabilizers and produced well-dispersed particles
without
significant aggregation. Stability measurements, however, revealed rapid
crystal growth
for all three stabilizers. 5% megestrol acetate/1.25% Tween 80 grew from 157
nm to 577
nm after 15 days at 5 C. 5% megestrol acetate/1.25% tyloxapol showed needle-
like
crystals when observed under optical microscopy. 5% megestrol acetate/1.25%
Pluronic
F127 grew from 228 nm to 308 nm after 5 days at 25 C. Because of the rapid
crystal
growth observed, Tween 80, tyloxapol, and Pluronic F127 were deemed not
suitable
surface stabilizers at the described drug/surface stabilizer concentrations
prepared under
the conditions described above.
[0183] The HPC-SL formulation (Exp. No. 8) showed substantial aggregation
indicating that a secondary charged stabilizer would be needed. SLS was added
(Exp. No.
6) and the new formulation grew from 167 to 194 nm after storage at 40 C for
15 days
and did not show any substantial aggregation upon incubation in either 0.O1N
HC1 or
normal saline. The SLS appeared effective at preventing the aggregation but
the sample
showed some particle size growth.
[0184] The HPMC formulation (Exp. No. 7) showed substantial aggregation
indicating that a secondary charged stabilizer would be needed. SLS was added
(Exp.
Nos. 5 and 11), and the new formulations showed only minimal growth from 161
nm to
171 nm (Exp. No. 5), and from 146 to 149 nm (Exp. No. 11), after storage at 40
C for 19
days. In addition, the composition of Exp. No. 5 did not show any substantial
aggregation
upon incubation in either 0.01N HCl or normal saline. The SLS was effective at
preventing the aggregation without causing significant crystal growth.
[0185] An attempt was made to reduce the concentration of the primary and
secondary stabilizers (Exp. No. 9) and resulted in a post-milling mean
diameter of 152
62
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
nm. Incubation for 30 minutes at 40 C in normal saline resulted in particle
sizes of 539
nm. Optical microscopy confirmed that aggregation was present in the sample
incubated
in saline.
[0186] Docusate sodium (DOSS) was tried as a secondary stabilizer (Exp. No.
10)
and resulted in well-dispersed particles with a mean diameter of 150 nm. Upon
storage at
40 C for 40 days, the sample had a mean diameter of 146 nm. Optical microscopy
revealed small, well-dispersed particles. DOSS seemed to result in even less
particle size
growth than SLS.
Example 8
[0187] The purpose of this example was to prepare nanoparticulate compositions
of megestrol acetate using various surface stabilizers and further including
preservatives
or excipients.
[0188] The materials and methods were the same as in Example 7, except that
for
several of the examples different sources of megestrol acetate were used (See
Table 9). In
addition, for Exp. Nos. 5 , a NanoMi11 milling system (Elan Drug Delivery)
was used.
Several different combinations of megestrol acetate, surface stabilizer(s),
and one or more
preservatives or excipients were prepared, following by testing the
compositions for
particle size and stability.
[0189] The surface stabilizer(s) and one or more preservatives were first
dissolved
in WFI, followed by combining the solution with megestrol acetate and the
grinding
media. This mixture was then added to the milling chamber and milled for the
time
period set forth in Table 9, below.
[0190] For several of the experiments, following milling the megestrol acetate
dispersion was combined with a flavored suspension. The stability of the
resultant
composition was then evaluated.
[0191] The formulation details and results are shown in Table 9, below.
63
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
M
~
bQ O b bA o9
.s~ o ~ ~ 9
0 V IRotIt
~ O +~ ~
;~o'b ~~~~~ a a
Ln ~
Z b y to =~ ~d ' U w ,~ ,n~ n rn
0
a o Q o~~
o
a~ ~' a~~'p~",=
oCIS 6D "0 c
.y,
(1)
U O~ b.i -p a O~
~O =~o0
to o~~~~o~ ~
y " 9 9 c+
~b
cn N v ~ d d
~~~r
g e'~ cN
o
~c . o o~
0
oo '~ o 0 0 0 +;
cd - U N 97 cu o +d '"
tw ,y o o 'a o 'u ao
0
>
w
~~"Z o~
v, oro~~
~i~ oua).o ~ A~ ti ti a a
cn ~oM U)~ a
'o
cl)
tn ~
00 ~ b~A ON
O M 00 N ~ 4 N bA N bA N ~~; +N+ bA ~'~'' ~
bA cd i ~.y ,..,~ Nyy 7~y
O ~ ~
a~ ~ O aa O N O a~ ~ O a~ ~ O
o A o 0 0 0 0 o A a o (R o
~
..b
d t~1 U U a1 U f~7 U W U ~ Pa U ~
0 00 o~ 0 0.5 0 0.~
M cnU voU uOU v) cnU m cnU
~ ~ U~ U ~
r i)
a~"i oUv~ 0 0 O O
U~A
vi in c c~ o c ~
O \.-+ V1 M h M
N O ~n O ~n O ~O O ~O O
ti
~ cd
~ v \ \ \ o
~ o 0 o tn
U O N N M ~.o
~y~~ N crl d vl
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0192] In Exp. No. 1 of Table 9, a sweetened, flavored dispersion was prepared
by
mimiclcing the current commercial formulation of megestrol acetate that
contains sucrose,
xanthan gum, glycerol, lemon and lime flavors, and is preserved and buffered
with
sodium benzoate and citric acid. Upon storage at 40 C for 24 days the sample
showed
aggregation with a mean diameter of 837 nm. Incubation for 30 minutes at 40 C
in 0.O1N
HCl or normal saline resulted in particle sizes of 206 nm and 3425 nm,
respectively.
Optical microscopy confinned that the sample incubated in saline had
aggregated. The
aggregation upon storage indicated that this particular combination of drug
and surface
stabilizer, at the concentrations used and methodology employed to make the
compositions, would not be an effective formulation.
[0193] For Exp. Nos. 4 and 5, the formulation was scaled-up in a NanoMillTM-2
system to determine if the scale-up would effect the physical stability. Two
different
sources of megestrol acetate were tested: Pharmacia and Pharmabios. The
product of
Exp. No. 4 had a mean diameter of 160 nm without ultrasound. Upon storage at
50 C for
44 days the mean diameter was 190 nm. The composition of Exp. No. 5 had a post-
milling mean diameter of 147 nm without ultrasound. Upon storage at 50 C for
44 days
the mean diameter was 178 nm. Both sources of active agent milled effectively
and
showed little particle size growth even at 50 C.
[0194] The results of Examples 6 and 7 showed that high energy milling with
polymeric attrition media could be used to produce stable nanoparticulate
colloidal
dispersions of megestrol acetate suitable for oral administration to animals
or humans.
The primary stabilizer HPMC required the presence of DOSS or SLS to prevent
aggregation at the concentrations of drug and stabilizer tested (other
combinations of drug
and HPMC concentrations may result in a stable composition without the
addition of a
second surface stabilizer). In general, average particle sizes of less than
about 160 nm
could be obtained. Tests conducted with two sources of megestrol acetate
revealed that
both sources milled effectively and exhibited excellent physical stability.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0195] Based on mean particle size, physical stability, and the pre-clinical
dog
study, the best nanoparticulate megestrol acetate formulation for commercial
development, based on the results of the data given in the examples, consisted
of 32.5%
megestrol acetate, 6.5% HPMC, and 0.325% DOSS (i.e., a drug:HPMC ratio of 1:5
and a
drug:DOSS ratio of 1:100. The formulation milled effectively in the presence
of
preserved water (0.2% sodium benzoate, 0.01% sodium citrate dihydrate, and
0.15% citric
acid monohydrate). Upon dilution with preserved water, flavors, and sucrose
none of the
dispersions showed severe aggregation, except for the dispersions containing
xanthan
gum (data not shown) or low levels of DOSS. The alcohol-based flavors did not
effect
the physical stability nor did several freeze-thaw cycles (data not shown).
Example 9
[0196] This example compares the pharmacokinetic parameters of nanoparticulate
megestrol acetate formulations of the invention with a conventional
microparticulate
formulation of megestrol acetate. Results were obtained from a fasted study
group
1. ~
consisting of 36 male subjects, 18 years of age or older. For a fed study
group, results
from 32 subjects were analyzed.
[0197] Subjects in the fasted study group and the fed study group were
administered study drugs in four successive periods. Treatment A(1 x 150 mg
drug as 5
ml of a 3% megestrol acetate nanoparticulate formulation) was administered in
the first
period. Reference Treatment B (1 x 800 mg drug as 20 ml of a 4% megestrol
acetate
Megace Oral Suspension) was administered in the second period. Treatment C (1
x 250
mg drug as 5 ml of a 5% megestrol acetate nanoparticulate formulation) was
administered
in the third period. Treatment D (1 x 450 mg drug as 5 ml of a 9% megestrol
acetate
nanoparticulate formulation) was administered in the fourth period. The
formulations of
Treatments A, C, and D are listed in Table 10 below, with particle size
information
(microns) provided in Table 11.
[0198] In each period, subjects were confined from at least 10 hours prior to
drug
administration to after the last sample collection. In the fasted study group,
no food was
66
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
consumed from at least 10 hours before dosing to at least 4 hours after
dosing. In the fed
study group, a high-calorie breakfast (containing about 800 to 1000 calories,
approximately 50% of which were from fat) was served within 30 minutes prior
to
dosing; dosing occurred within 5 minutes after the brealcfast was completed. A
controlled
meal was served to both groups after 4 hours after dosing, and standard meals
were served
at appropriate times thereafter. The meals in all four periods were identical.
Subjects in
the fasted study group were not allowed fluid intalce from 1 hour before
dosing to 1 hour
after. Subjects in the fed study group were also not allowed fluid intake
during this period
except for fluids provided with the high-calorie breakfast. Water was provided
ad libituin
to both study groups at all other times.
[0199] Blood samples were obtained before dosing, at half-hourly intervals in
the
6 hours following dosing, and at 7, 8, 12, 16, 20, 24, 36, 48, 72, and 96
hours after dosing.
Megestrol acetate in plasma samples was then determined.
[0200] Table 12 below summarizes pharmacokinetic data for the fasted study
group, and Table 13 below summarizes pharmacokinetic data for the fed study
group.
[0201] Treatments A, C, and D in fasting subjects produced dose-normalized
values for AUCo_t and AUCo_;,,f that were approximately twice those of
Reference
Treatment B. Maximum dose-normalized megestrol acetate concentrations in
Treatments
A, C, and D were approximately 9 to 12 times that of Reference Treatment B.
The
maximum megestrol acetate concentration for the 150 mg-dose of Treatment A was
approximately twice that of the 800 mg-dose of reference Treatment B.
Moreover,
comparable values of AUCo_t and AUCo_iõf were observed for the 450 mg-dose of
Treatment D and the 800 mg-dose of Reference Treatment B.
[0202] Treatments A, C, and D in fed subjects produced dose-normalized values
for AUCo_t and AUCo_iõf that were approximately 8 to 10% greater than those of
Reference
Treatment B. Maximum dose-normalized megestrol acetate concentrations in
Treatments
A, C, and D were approximately 38 to 46% greater than that of Reference
Treatment B.
Megestrol acetate onset for Treatments A, C, and D was comparable to Reference
Treatment B.
67
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0203] Nanoparticulate megestrol acetate formulations, therefore, exhibited
superior oral bioavailability, relative to the Megace Oral Suspension, in
fasting and fed
human subjects.
TABLE 10
Formulations for Megestrol Acetate Oral Suspension 3, 5% and 9%
Strengths
Ingredients 3% W/W 5% W/W 9% W/W
(30 mg/mL) (50 mg/mL) (90 mg/mL)
Megestrol Acetate 3.000 5.000 9.000
Hydrox ro yl Methylcellulose 0.600 1.000 1.800
Docusate Sodium 0.030 0.050 0.090
Sodium Benzoate 0.183 0.178 0.168
Sodium Citrate Dihydrate 0.009 0.009 0.008
Citric Acid Monohydrate 0.137 0.133 0.126
Sucrose 5.000 5.000 5.000
Natural and Artificial Lemon Flavor 0.040 0.040 0.040
Artificial Lime Flavor 0.040 0.040 0.040
Purified Water 90.961 88.550 83.727
TOTAL 100.000 100.000 100.000
TABLE 11
Particle Size Data for the Me estrol Acetate Oral Suspensions*
Stren th 30m g/g Stren th 50 m/ Stren h 90 g/g
d0.1 d0.5 d0.9 d0.1 d0.5 d0.9 d0.1 d0.5 d0.9
Initial 0.068 0.123 0.223 0.069 0.125 0.229 0.068 0.124 0.227
ACC/1 month 0.070 0.129 0.237 0.070 0.127 0.231 0.070 0.127 0.230
ACC/2 months 0.070 0.127 0.231 0.070 0.127 0.233 0.073 0.126 0.221
ACC/3 months 0.070 0.129 0.237 0.070 0.128 0.235 0.070 0.128 0.234
RT 3 months 0.070 0.128 0.237 0.073 0.128 0.224 0.067 0.121 0.223
* All particle sizes are given in microns. "d(0.1)" means distribution of
smallest 10% of the
particles, i.e., d(0.1) 10 m means 10% of the particles are less than 10%.
Similarly, "d(0.5)"
means distribution of the smallest 50% of the particles, and "d(O.9)" means
distribution of the
smallest 90% of the particles. Thus, d(0.9) means that 90% of the particles
are less than XX gm.
68
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
TABLE 12
Summar of Pharmacokinetic Data in Fasted Human Sub'ects*
Parameters Treatment A Ref. Treatment B Treatment C Treatment D
(Mean SD) (Mean SD) (Mean SD) (Mean tL SD)
AUC0 -t 2800 } 900 7000 5000 4700 1800 8500 3200
AUC0-inf 3100 1000 9000 9000 5200 2100 9000 4000
Cmax 410 120 190 110 650 200 950 270
Tmax 1.7 0.9 6 6 1.6 1.0 1.7 1.1
ty2 35 t 13 31 t 19 34 t 10 34 t 12
l~l 0.023 0.011 0.026 0.009 0.022 0.008 0.023 0.008
AUCo_t (ng.hr/ml) = Area under the curve from time zero to the last measurable
concentration;
AUCo_;,,f(ng.hr/ml)= Area under the curve from time zero to infinity;
C,õ,., (ng/ml)= Maximum plasma concentration;
T,,,aõ (hr)= Time to occurrence of C,,,.;
ty, (hr)= Apparent elimination half-life;
Kei (l/hr)= Elimination rate constant;
*n=36.
TABLE 13
Summar of Pharmacokinetic Data in Fed Human Sub'ects*
Parameters Treatment A Ref. Treatment B ! Treatment C Treatment D
Mean f SD) Mean ~ SD) (Mean SD) (Mean A: SD)
A[JC0 -t 3500 1100 17000 5000 5700 1600 10500 3000
AUCO-inf 3900 1300 19000 6000 6300 2000 12000 4000
Cmax 380 140 1400 400 590 170 1080 290
Tmax 3.8 3.5 3.9 0.9 3.4 1.7 3.2 1.7
t,2 35 12 33 9 35 10 38 } 12
'i 0.023 0.013 0.023 0.007 0.023 0.009 0.021 0.008
AUCo_t (ng.hr/ml) = Area under the curve from time zero to the last measurable
concentration;
AUCo_iõf(ng.hr/ml)= Area under the curve from time zero to infinity;
C,,,a,, (ng/ml)= Maximum plasma concentration;
T,,,,, (hr)= Time to occurrence of C,,,,_,;
t./, (hr)= Apparent elimination half-life;
Kei (1/hr)= Elimination rate constant;
*n=32.
Example 10
[0204] This example compares the pharmacokinetic parameters of a
nanoparticulate megestrol acetate formulations to a conventional
microparticulate
formulation of megestrol acetate (Megace by Bristol Myers Squibb Co.).
Results were
69
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
obtained from a fasted study group consisting of 33 male subjects, 18 years of
age or
older.
[0205] The nanoparticulate megestrol acetate compositions were prepared as
described in Example 10.
[0206] Subjects were administered study drugs in four successive periods.
Treatinent A (575 ing of nanoparticulate megestrol acetate fomiulation in 5 ml
oral
suspension) was administered in the first period. Reference Treatment B (800
mg of
megestrol acetate (Megace by Bristol Myers Squibb Co.) in 20 ml oral
suspension) was
administered in the second period. Treatment C (625 mg of nanoparticulate
megestrol
acetate formulation in 5 ml oral suspension) was administered in the third
period.
Treatment D (675 mg of nanoparticulate megestrol acetate formulation in 5 ml
oral
suspension) was administered in the fourth period.
[0207] Table 14 provides the formulations of Treatments A, C and D.
Table 14
Formulations of Nanoparticulate Megestrol Acetate Oral Suspensions
Dosage 115 mg/mL 125 mg/mL 135 mg/mL
FINAL AMOUNTS Weight (g) Conc. Weight (g) Conc. Weight (g) Conc.
m/mL m/mL m/mL
Megestrol Acetate 37,500.0 115.00 37,500.0 125.00 37,500.0 135.00
HPMC 7,500.0 23.00 7,500.0 25.00 7,500.0 27.00
Docusate Sodium 375.0 1.15 375.0 1.25 375.0 1.35
Sodium Benzoate 530.4 1.63 481.4 1.60 439.7 1.58
Sodium Citrate Dihydrate 26.5 0.08 24.0 0.08 22.0 0.08
Citric Acid Monohydrate 397.8 1.22 361.1 1.20 329.8 1.19
Sucrose 15,473.0 47.45 14,044.0 46.81 12,826.7 46.18
Lemon Flavor 123.8 0.38 112.4 0.37 102.6 0.37
Lime Flavor 123.8 0.38 112.4 0.37 102.6 0.37
Water 277,080.1 - 251,489.7 - 229,690.5 -
TOTAL (Weight, g) 339,130.4 312,000.0 - 288,888.9 -
TOTAIr (volume, L) 326.1 300.0 - 277.8 -
[0208] The nanoparticulate megestrol acetate formulations were prepared by
milling a concentrated dispersion of the drug substance followed by dilution
to yield the
final products. Hydroxypropyl methylcellulose and docusate sodium were used as
stabilizing agents. The formulations were processed in a NanoMill-10
horizontal media
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
mill (Netzsch USA) for 20 hours. The attrition media used was 500 m
crosslinlced
polystyrene (PolyMillTM-500). The dispersion further comprised 0.13% sodium
benzoate,
0.01% sodium citrate dihydrate, and 0.1% citric acid monohydrate. Milled
dispersion was
diluted to final megestrol acetate concentrations of 115 mg/mL (575 mg/5 mL),
125
mg/mL (625 mg/5 mL) and 135 mg/mL (675 mg/5 mL). The final compositions
additionally contained sweetening and flavoring agents.
[0209] Particle size determinations were performed on a Malvern Mastersizer
2000 instrument. The particle size distributions of the nanoparticulate
megestrol acetate
compositions are provided in Table 15.
Table 15
Concentration (mg/mL) Mean particle size nm 50% < nm 90% < nm
115 144 130 234
125 144 127 237
135 145 131 236
[0210] In each period, subjects were confined from at least 11 hours prior to
drug
administration until after the 24.0 hour post-dose sample collection. After a
supervised
fast of at least 10 hours, subjects were fed a high-calorie meal containing
about 800 to
1000 calories (approximately 150 calories from carbohydrates and 500-600
calories from
fat). The meal consisted of two eggs fried in butter, two slices of toast with
butter, two
strips of bacon, approximately 128 g of hash brown potatoes and 200 ml of
whole milk.
The meals in all four periods were identical. The meal was completed within 30
minutes,
and subjects were dosed 30 minutes after starting the meal.
[0211] The suspensions of Treatments A, B, C and D were administered via Slip
Tip syringe directly into the mouth and swallowed. The syringe was rinsed
three (3) times
with approximately 5 ml (Treatments A, C and D) or 20 ml (Treatment B) of
water.
Following drug administration, approximately 225 ml (Treatments A, C and D) or
180 ml
(Treatment B) of water was ingested.
[0212] For each period, a total of 24 blood samples were drawn from each
subject.
Blood samples were collected in EDTA blood tubes prior to drug administration
and
0.250, 0.500, 0.750, 1.00, 1.50, 2.00, 2.50, 3.00, 3.50, 4.00, 4.50, 5.00,
5.50, 6.00, 8.00,
71
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
12.0, 16.0, 20.0, 24.0, 36.0, 48.0, 72.0 and 96.0 hours post-dose (1 x 7 mL
for each
sampling time).
[0213] Table 16 below summarizes the pharmacokinetic data, while Table 17
provides the statistical comparisons of the treatments.
Table 16
Pharmacokinetic Parameters
Test-1 (Megtestrol Acetate 575 mg/5 niL Reference: (Megace 40 mg/mL (B))
(A))
Parameters Mean SD CV (%) Mean ~ SD Cv (%)
AUCo-t (ng-h/mL) 13657.52 ~ 3900.50 28.56 16896.21 ~ 4942.51 29.25
AUCo-inf (ng-h/mL) 14743.33 f 4451.31 30.19 18274.06 ~ 5623.07 30.77
Cmax (ng/mL) 1420.73 f 420.79 2962 1400.66 ZL 350.57 25.03
Tm. (h) 3.75 1.57 41.85 3.88 ~ 1.02 26.38
Tõ,.* (h) 4.50 f 1.00 - 4.50 ~ 1.00 -
KeI (0) 0.0224 ~ 0.0062 27.44 0.0238 ~ 0.0054 22.84
T1/2 e, (h) 32.78 f 7.47 22.80 30.53 ~ 6.66 21.80
Test-2 (Megtestrol Acetate 625 mg/5 mL Test-3 (Megestrol Acetate 675 mg/5 mL
(D))
(C))
Parameters Mean f SD CV (%) f ~ SD Cv (%)
AUCo-, (ng-h/mL) 14682.3 f 4844.60 33.00 15323.29 f 4525.94 29.54
7
AUCo-inf (ng-h/mL). 16081.7 f 5563.09 34.59 16738.88 f 5432.52 32.45
6
Cm, (ng/mL) 1516.79 ~ 389.01 25.65 1645.74 f 455.71 27.69
T. (h) 2.52 ~ 1.60 63.52 3.13 ~ 1.64 52.55
T,,m* (h) 2.50 ~ 3.50 - 3.50 3.00 -
Ke1 (h 1) 0.0211 f 0.0055 26.21 0.0211 2=
0.0054 25.64
T1/2e1 (h) 34.75 f 7.81 22.48 34.83 8.12 23.30
*Median and interquartile ranges are presented.
AUCo-t (ng.h/ml) = Area under the curve from time zero to the last measurable
concentration
AUCo-;,,f(ng.h/ml) = Area under the curve from time zero to infmity
Cm,,x (ng/ml) = Maximum plasma concentration
Tm,,
(h) = Time to occurrence of C,,,a,
ti eI (h) = elimination half-life
Kel (1/h) = elimination rate constant
Table 17
Treatment Comparisons
Statistical Treatment Ratio' 90 % Geometric CL2 Intra-
Analysis Subject
CV
(ANOVA) Comparisons Lower Upper
AUCo-, Megestrol Actate 575 mg/5 mL (A) vs 81.06% 78.20% 84.03%
Megace 40 mg/niL (B)
0
Megestrol Actate 625 mg/5 mL (C) vs 86.29% 83.24% 89.45% 8.82%
Megace 40 mg/mL (B)
72
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
Table 17
Treatment Comparisons
Statistical Treatment Ratio' 90 % Geometric CLZ Intra-
Analysis Subject
CV
(ANOVA) Comparisons Lower Upper
Megestrol Actate 675 mg/5 mL (D) vs 90.63% 87.43 l0 93.95%
Megace 40 mg/mL (B)
AUCo_i f Megestrol Actate 575 mg/5 mL (A) vs 80.92% 77.95% 84.00%
Megace 40 mg/mL (B)
Megestrol Actate 625 mg/5 mL (C) vs 87.33% 84.12% 90.65%
Megace 40 mg/mL (B) 9.16%
Megestrol Actate 675 mg/S mL (D) vs 91.31 % 87.96% 94.79%
Megace 40 mg/inL (B)
C,,,a., Megestrol Actate 575 mg/5 mL (A) vs 100.62% 94.10% 107.69%
Megace 40 mg/mL (B)
Megestrol Actate 625 mg/5 mL (C) vs 108.18% 101.17% 115.69%
Megace 40 mg/mL (B) 16.51 %
Megestrol Actate 675 mg/5 mL (D) vs 116.72% 109.15% 124.82%
Megace 40 mg/mL (B)
Calculated using least-squares means
290% Geometric Confidence Interval using In-transformed data
[0214] Tables 16 and 17 demonstrate that Treatments A, C, and D produced
similar pharmakinetics as Treatment B. Figures 4 and 5 show that Treatments A,
C and D
produce similar concentration-time curves as Treatment B.
Examule 11
[0215] This example describes a randomized, open-labeled, multicenter,
multinational, pilot study comparing the weight gain effect in adult HIV-
positive subjects
of (1) a nanoparticulate megestrol acetate composition as compared to (2)
MEGACE OS,
which is a conventional, microparticulate megestrol acetate formulation.
[0216] The nanoparticulate megestrol acetate formulation contained 115 mg of
nanoparticulate megestrol acetate per ml, docusate sodium and hydroxyproyl
methylcellulose as surface stabilizers, alcohol, artificial lime flavor,
citric acid
monohydrate, natural and artificial lemon flavor, purified water, sodium
benzoate, sodium
citrate dihydrate, and sucrose. The megestrol acetate particles in the
nanoparticulate
megestrol acetate formulation had a volume weighted mean of no greater than
180 mn
(this is roughly equivalent to a D50 particle size).
73
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PA=R Pharmaceufical CONFIDENTIAL -Page v of ix
'Finai Clinical Protocol PAR-002 v.4. Version: 01 Nov'04
'PROTOCOL SYNOPSIS
Name of S onsor/Com an. : Par Pharmaceutical
Name of Compound: =Megestrol Acetate Oral Name of Active Ingredient: megestrol
acetate
Suspension NanoCrystalT~+ Dispersion (NCD) (nanocrystalline formulation)
formulation
Title of Study: A Randomized, Open-labeled,,Pilot Study Comparing Weight Gain
in Adults with AIDS-
related Wasting (Anorexia/Cachexia) Given Either,Megestrol Acetate Oral
Suspension NCD
Formulation or Megestrol Acetate Oral Suspension (Megace ).
Investigators: -Multicenter (approximately 20 centers) in =India, South Africa
and United States
Study Centers: TBD
Study Period . ears. : Phase of Development:
Date of :ptanned :first enrollment November 2004 II
Date of.ptanned completion April 2005
Objectivesc'
Primary: To explore weight gain in adult HIV positive subjects who have weight
loss associated with
AIDS-related wasting (anorexia/cachexia) in the *st 12 weeks of treatment with
either megestrol
acetate oralsuspension NCD formulation or:Megace .
Secondary:1) To explore changes from :baseline in lean body =mass, fat-free
mass, appetite, and
Quality-of-t_ife assessments at multiple time points over a 12-week.period
among subjects who receive
megestrol acetate oral suspensionCD formufation and Nlegace(g, 2) -to assess
the safety and
tolerability of megestrol acetate.oral suspension NCD formulation and Megace
in adult HIV-positive
subjects who have weight loss associated with AIDS-related wasting and 3)
explore pharmacokinetic
variables in the.target population.
Methodology: This Js a-randomized, open-labeled,
multicenter,.rnultinational,:pilot study intended to
explore differences in.weight gain within the first-12-weeks of treatment with
megestrol acetate NCD
fiormulation or Megace in -adult men and women vuith AIDS-related wasting.
This pilot -study includes
a total'of 60 HIV-positive adults.who have weight loss associated with AIDS-
related wasting and meet
the inclusion/exclusion criteria. Subjects=will be centrally randomized In
equal proportions to receive
one of the two treatments given as oral suspensions: megestrol acetate NCD
formulation 575 mg or
Megace@) -800 mg-per day as single daily doses for 12-weeks. Subjects return
to the clinic weekly for
the 12 weeks on =treatment and then foilowed :by a brief clinic visit 30 days
later. Study-related
assessments for the study are summarized in Table 2.
Prepared by Quintiles, Inc.
74
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PAR'Pharmaceutical GONFIDENTIAI. Page vi of ix
Final Clinical Protocol PAR-002 v.4. Version: 01 Nov 04
=Methodoiogy: -(con't) Serial assessments of pharmacokinetic variables wiii be
assessed on the first
day of treatment and at the Week 6 visit. Blood samples will be obtained after
a standardized meal at
the following time points (hrs post dose):=Baseline (0), 0.5, 1.0, 1.5, 2.0,
2.5, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0,
12.0, and 24;0 =hours. Trough levels=of study drug will be collected at Day
3(t1 day) after beginning
tr.eatment=and at all other clinic visits prior =to the time of the daily
dose.
Number of Subjects '(Planned and Analyzed): Total of 60 subjects enrolled to
yield a total of 40
evaluable, HIV-positive adult subjects (30 enrolled per.treatment group).
Diagnosis =and'Main Criteria for=Inciusion: Adult men and women with HIV
infection between the
ages of 18 and 70 years of age who -have an unintentional weight loss -
resulting in 10%7ess than the
lower %limit of Ideal Body Weight for frame size (as defined in Metropolitan -
Height and Weight Tables or
other standardized tables appropriate for the locale) or a recent history of
unintentional weight loss of
10% from the subjeets baseline. Weight losses must=be clinically associated
with AIDS-related wasting.
Women of childbearing potential may -not be pregnant or nursing =and -must
agree to use effective
contraception for the duration of the study and -for two weeks -after the last
dose. =Subjects must =be
capable of and willing to return to the clinic regularly for study vi''sits;
=must be willing to abstain from any
illegal or recreational drug substances for the duration of the trial; must
abstain from taking any other
medications or substances known to affect appetite or weight gain (eg,
steroids [other than those
inhaled for:treatment =of asthmatic conditions], nutritional supplements,
dronabinol).
Subjects may have=none'of the following criteria: active AIDS-defining illness
or other uncontrolled or
: clinically significant medical problems or laboratory abnormalities;
evidence of or history of diabetes
mellitus, fiypoadrenalism or adrenal insufficiency (stimulated serum cortisol
of <18pgIdL) ; evidence of
clinical,depression identified -by GRID-HAMD-17'screening assessment; recent
history of significant
psychiatric =iliness that may compromise the subject's ability to comply with
the study requirements; or a
history *or evidence of thromboembolic events (or any first degree relative
w(ith a history of
thromboembolic events).
Test Product, Dose and Mode of Administration, Batch Number: Megestrol acetate
oral
suspension NCD formulation with 115 mg of nanocrystalline megestrol acetate
per mL for a daily dose
of 575 mg =per day (5 mL dose). Lot Number. =041787.
=Duration of Treatment: 12-weeks
Reference -Product, :Dose and .Mode of Administration, Batch"Number Megace
(megestrol
acetate) Oral Suspension (Bristol-Myers Squibb, Princeton, NJ) contains 40 mg
micronized megestrol
acetate per mL.'Dose: 800 mg daily. Lot Number: 4D80437.
Criteria -for Evaluation:
Primary,Endpoints: Weight gain will be assessed at baseline, then weekly x12
during treatment.
Serial weight measurements for each subject should be obtained using the same
scale for each-
assessment at approximately the same time of day. Subjects should be weighed
In street clothes and
without shoes.
Prepared by Quintites, Inc.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
~PAR Pharmaceutical CONFIDENTIAL Page vii of=ix
:Final Ciinical 'Protocoi PAR-002 v.4. Version:'01 Nov. 04
Criteria for Evaluation: con't
SecondaryZndpoints:'Lean body mass, body fat and fat-free,body mass will be
assessed by
bioimpedance analysis at baseline, Weeks 6 and 12. Total body muscle -mass
will be assessed b.y
anthropometric measurements (mid-arm, waist and hip circumferences and
ttriceps skinfold
measurement). Appetite:and Tood intake will be assessed by compietion -of a 3-
day food intake diary
completed .prior-to each clinic visit -and a 24-hour recall food diary =at the
time of the clinic visit. Quality-
of-Life Assessments will be made using a validated QOL -instrument, 'Bristol-
Myers Anorexia/Cachexia
Recovery Instrument (BACRI), administered weekly through week 12 and =at the
30-day post treatment
visit. Appetite will be assessed via a visual analogue scale included as part
of'the BACRI. Safety will be
assessed by coliection of adverse events and vital signs at each clinic visit.
Physical examinations and
routine clinical laboratory samples including hematology, seruni chemistry,
lipid profile and routine
urinalysis will be assessed at baseline, Weeks 3, 6, and 12. Additional
laboratory assessments may be =
made =at the investigator's discretion. Trough levels for study dnag
andconcomitant medication
information will'be collected at each clinic visit. Pharmacokinetic
=assessments will be obtained on the
first day and at the Week=6 visit and will include Cm,, (ng/mL), AUC at (h-
ng/mL), and TmOx (h).
Anaiytical Methodology: Changes and percentage changes from 'baseline for
continuous
measurements will be calculated at Weeks 1 through 6, 9, and 12. For weight
gain, an "area under the
cunre" analysis =will also be conducted. to assess the overall difference in
effect of the two therapies
over the first 12.weeks. Adve~se events will be coded using'MedDRA dictionary
and reported by
preferred term and treatment group.
An interim analysis reporting the results of all the endpoints for the first
40 subjects is planned.
Safety: Safety will be assessed -by adverse events, vital signs, .periodic
physical-examinations and
routine clinical laboratory testing. Samples for routine hematology (complete
blood count with platelet
count), serum chemistry (sodium, potassium, chloride, bicarbonate, BUN,
albumin, glucose, creatinine,
alkaline phosphatase, total biiirubin, liver function tests, and lipid panel)
and routine urinalyses
(dipstick) will be collected at baseline/screening, Weeks 3,.6. and 12.
Pregnancy testing will be
performed on all women of childbearing potential at screening/baseline (serum
j3-hCG) and at each
clinic visit (by urine pregnancy test) through Week 12. Adrenocorticotropin
(ACTH) stimulation testing,
including resting cortisol levels, and hemoglobin Al C will be assessed at
screening and at week 12 (or
last clinic visit).
Statistical Methods: The primary goal for this -pilot study Is to explore the
rate of weight gain during
the =first 12 weeks of treatment with megestrol acetate NCD formulation or
Megace oral suspensions.
No fonnal statistical analyses are planned; only exploratory analyses will be
performed. Results for
each variable will be =provided with appropriate summary statistics. Due to -
the exploratory nature of the
analyses, missing :individual observations will be interpolated based on.prior
and subsequent values.
Treatment differences will be estimated and 95% confidenceintervals will be
provided. Analyses will.be
performed on an Intent-to-Treat population that will include all randomized
subjects with at least one
post-randomization visit. The Per-Protocol population will include
all,subjects who completed the study
requirements with =no more than one missing visit and no major protocol
violations.
Safety population will include all subjects who received any study medication.
Version Date of Synopsis: 01 :November 2004
Prepared by Quintiles, Inc.
76
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PAR, Pharmaceutical CONFIDENTIAL Page viii of ix
=Final Clinical Protocol PAR-002 v.4. Version: 01 :Nov 04
,LIST'OF ABBREVIATIONS AND DEFINITIONS OF TERMS
= g/dL Micrograms per deciliter
ACTH A.drenocorticotropin
AE(s) Adverse event(s)
AIDS Acquired immunodeficiency syndrome
ALT =(SGPT) Alanine arninotransferase
AST (SGOT) Aspartate aminotransferase'
AUC Area under the ,plasma drug concentration-time curve
:(3-hCG Beta human chorionic gonadotropin
:BACRI Bristol-Myers Anorexia/Cachexia Recovery Instrument
BIA Bioimpedance analysis
BUN Blood -urea nitrogen
CBC Complete blood count
CD4+ Specific T-lymphocyte decreased in patients with HtV
infections
Cmax Peak drug concentration
CRF Case Report Forra
FFM Fat-free mass
GCP Good Clinical Practice
GGT Gamma-glutamyl transferase
GRID-FL41y1D Structure interview Guide to assess depression in multiple
functional areas
HAART :Highly-active antiretroviral therapy
HAMD Hamilton Rating Scale for Depression
HIV Human immunodeficiency virus
ICH International Conference on Harmonization
IEC Independent Ethics Committee
IRB Institutional Review Board
TTT Intent-To-Treat
1U 'International unit
L Liter
LDH Lactic dehydrogenase
LFT Liver function tests
MedDRA Medical Dictionary for Regulatory Activities
Prepared by Quintiles, Inc.
77
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceuticai= CONFIDENTIAL Page =ix of ix
Final Clinical Protocol PAR-002 v.4. Version: =01Nov 04
mg Milligram
NCD NanoCrystalTM Dispersion
PK Pharmacokinetics
PP Per-Protocol
'QOL ~Quality-of-Life
RBC Red blood cells
SAE(s) Serious adverse event(s)
t1/2 Apparent =terninal half-life
Tm.X Time of observed maximum=.concentration
VAS Visual Analogue Scale
WBC White blood cells
Prepared by Quintiles, Jnc.
78
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR =Pharmaceutical CONFIDENTIAL Page 1 of'50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
1.0 INTRODUCTION
The AIDS-related wasting :(HIV-wasting) syndrome as defined by Center for
Disease Control
and Prevention =(CD=C) is an :41DS-defming illness characterized by a profound
involuntary
=weight =loss of >10% of baseline body weight plus either chronic diarrhea or
chronic
weakness and documented fever in the absence of a concurrent illness or
condition other
than HIV infection that explain these findings! The nature of the weight loss
is characterized
by the loss of lean 'body mass, predominantly muscle.protein? Even
asymptomatic patients in
the early stages of the disease may have a reduction in body mass3 and
continuing losses in
weight, fat-free mass, body cell mass aiid fat:mass are significant indicators
of mortality in
AIDS-related wasting syndrome.4 S For the purposes of this study, AIDS-
related wasting will
be defined as the involuntary weight loss of>10% of baseline weight in the
absence of a
concurrent illness or condition=other than HIV infection. The additional
criteria of chronic
diarrhea, chronic weakness .or documented fever required -by the CDC defmition
of AIDS-
.related wasting need=not be present to qualify for the study.
yVhile the icausative agent is unclear, -the consequences of AIDS-related
wasting are well
=documented. Tang et al 6 reported in alongitudinal study of 678 H1V-positive
participants
receiving highly-active antiretroviral therapy (HAART) in the Nutrition for
Healthy Living
study that weight loss of>_10% either from =baseline or from the previous
visit was
significantly associated with a four- to six-fold increase in mortality
compared with
maintenance of or increase in weight. Even one episode of weight.loss _3 0o
from baseline or
>5 fo from the ;previous visit was predictive of mortality in this population.
In Tang's study,
weight loss emerged as the single strongest independent predictor of mortality
over changes
=in fat-free mass, body cell mass or fat mass.
Despite the success in improving overall survival with the advent of HAART,
A,IDS-related
wasting remains problematic. Wanke 7 et al reported on the results of
a.prospective cohort of
469 HIV-infected =adults to study the impact of H1V on nutrition in patients
taking HA.,A:RT.
In the ,population studied, 58% of the cohort lost >1.5 kg of weight within 6
months (between
2 study visits) despite the :prevalence of HAART therapy. While no defmitive
cause 'has been
,established for this condition, several possible theories'have been proposed
including
-increased energy expenditure, decreased energy intake, malabsorption,
inefficient use of
energy, 'hormonal factors and =cytokine effects.$ 9,10,11,12
Current=therapies for AIDS-related wasting include nutritional education and
support,
nutritional supplementation, hormonal therapies (testosterone and testosterone
analogues,
oxandrolone, nandrolone, other androgenic compounds), recombinant human growth
hormone,.exercise training and cytokine modulators.13
Prepared by Quintiles, Inc.
79
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL =,Page 2 of 50
Final Clinical Protocol =PAR-002 v.4 Version: 01 Nov 04
2:0 ;.BACKGROUN.D
Megestrol =acetate is a synthetic derivative of;progesterone. It has slight
=glucocorticoid
activity and =a=very slight degree of miiieralocorticoid activity. Megestrol
has no estrogenic,
androgenic or anabolic activity.14
The ,precise mechanism'by which megestirol acetate :produces effects in
'anorexia and
..cachexia is =unknown at this time. However, evidence from clinical studies
indicates that the
increase in'body weight observed during megestrol therap.y is related to the
drug's appetite-
=stimulant or:metabolic effects rather than its .glucocorticoid=like effects
or the ;production of
edema. It has been =suggested that megestrol and%r :its =rnetabolites may,
either directly or
:indir.ectly, stimulate appetite resulting in weight gain or may alter
metabolic pathways via
interference with the production or action of ~mediators such as cachectin (a
hormone that
-inhibits adipocyte lipogenic. enzymes). 14
*Megestrol acetate (Ivlegace , Bristol-Myers Squibb, Princeton, NJ) oral
suspension has been
'widely studied as a treatment for anorexia and cachexia in both cancer
patients .15, 16, i7 and
:patients with ATDS-related wasting syndrome.l 8,19, 20 ;~ile the exact
mechanism by which
the m egestrol acetate improves appetite and facilitates weight gain is
unclear, the results of
previous studies have demonstrated its efficacy in these populations.
Published studies have
=reported weight gain and improvement in appetite after 4 and 12 weeks of
treatment;
however, .reports of weight gain and appetite changes within the first few
weeks )lave not
been reported.
The.pivotal studies for the parent compound, Megace 1$-19 had significant
patient attrition
rates of approximately 25-29% of the enrolled subjects within the first 12
weeks of treatment.
Reasons for the relatively significant level of attrition were not evident in
the published
reports. Coincidentally, the same ]evel of=attrition was noted in both the
megestrol-treated
and pIacebo groups. The comparable attrition rates may indicate patient-
related factors
influencing motivation to continue treat.ment not measured in these studies
such as a lack of
-subjective ,improvement early in the lreatment,course. 'Oster-et al 18
reported a summary of
.reasons patients -discontinued treatment during the 12-week study; however,
no details of the
reasons for the attrition by interval were reported.
The scientific question to be explored in this study is whether or not the
onset of the
improved appetite, weight= gain and perception of:improved=quality-of-life
would begin
sooner with megestrol acetate NCD formulation than the reference product,
Megace . The
:clinical relevance of this hypothesis would be if patients noticed
improvement sooner after
beginning treatment, this could possibly influence patient compliance to
continue with.
:treatment.
Prepared by Quintiles, Inc.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical ~CONFIDENTIAL Page 3 of 50
Final Clinical Protocol'PAR-002 v.4 Version: 01 Nov 04
3.0 STUDY OBJECT=IVES
3.1 Pnirnary Objective
The primary objective of the study is to:
= explore weight gain in adult, HIV-positive subjects who have weight loss
.associated with AIDS-related wasting :(anorexia/cachexia) =in the 'first 12
weeks of
treatznent with either megestrol acetate oral suspension NCD formulation or
Megace .
=3.2 Secondar.y'Objectives
The secondary. objectives of this study are:
= To explore changes from baseline in lean body mass, fat-free mass, total
body
muscle mass, appetite, and Quality-of-Life assessments at multiple time.points
over a 12-week period among subjects who receive megestrol acetate oral
suspension NCD formulation or *Megace
To =assess the safety and tolerability of megestrol acetate oral suspension
NCD
-formulation and Megace in adult HIV-positive subjects who have weight loss
associated with AIDS-related wasting
= To explore pharmacokinetic variables in the target population.
4.0 .INVEST.IGAT.IONAL 'P.LAN
.4.1 Study Design and Rationale for Study Design
This is a randornized,.open-labeled, 'multicenter, xnultinational, pilot.study
intended to
explore -differences in weight =gain within the first 12-weeks of treatment
with megestrol
acetate NCD formulation or Iviegace in adult men and women with AIDS-related
wasting.
This pilot study includes a total of 60 HIV-positive adults who have weight
loss associated
with AIDS-related wasting and meet the inclusion/exclusion criteria. Subjects
will be
centrally randomized in equal proportions to receive one of the two
treatments: megestrol'
acetate NCD formulation 575 mg or 1Vlegace oral suspensions 800 mg per day as
single
doses for 12 weeks. Subjects will return to the clinic weekly for the 12 weeks
on treatment
and have a brief clinic visit 30 days after treatment stops.
Prepared by Quintiles, Inc.
81
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
-PAR =Phannaceutical CONFIDENTIAL =Page 4 of 50
finai Clinical'Protocoi PAR-002 v.4 Version: 01 Nov 04
Serial :assessments of pharmacokinetic variables will be assessed on the first
day of treatment
:and at the Week 6 visit. Blood samples will be obtained after a standardized
meal at the
=following time points (hours post dose): Baseline (0), 0.5, 1.0, 1.5, 2.0,
2.5, 3.0, 4.0, 5.0, 6.0,
7.0, -;80, 12:0, =and 24.0 hours. Trough levels of study drug will be
collected at all other clinic
visits prior to the time of the daily =dose.
Results from =this pilot study will be used in designing the sample size,
=natare and number of
-assessments.planned foi the next.pivotal study.
4:2 S.e'lection of Study Population.
Subjects will :be recruited from sites in the TJnited States, India and'South
Africa that provide
:care for this population. Subjects enrolled in the study must meet 411 of the
inclusion criteria
and >none of the =exclusion .criteria. Exceptions to these criteria may -only
:be made after
,agreement by Par and the Medical Monitor responsible for the conduct of the
trial.
4.2.1 'Inclusion 'Criteria
l.. Adult man or woman between the ages of 18 and 70 years of age;
2. 'Capable of and willing to .provide informed consent;
3. Evidence of HIV infection (either HN-seropositive, CD4" T-cell count of
<350/nun3 or other clinically accepted indicator);
4. An unintentional weight :loss resulting in -a weight 10 fo less than the
lower limit of
Ideal Body Weight for frame size (as defined =in Metropolitan Height and
Weight
Tables or other standardized tables appropriate for the locale) or a recent
history of
uninteintional weight loss of 10% from the subjects baseline;
5. Weight losses must be clinically associated with AIDS-related wasting and
not
related to any other-disease process;
6. Women of childbearing potential must agree to use effective contraception
for the
duration of the study and for two weeks after the last dose;
7. Clinical laboratory values must be within normal limits or out-of-range
limits must
be designated as =not clinically significant. The following out-of-range
laboratory
values may be permissible based -upon individual circumstances:
= Hemoglobin (Hgb) values should be > 9.0gm/dL; however, values
between 7.0 and 8.9 gm/dL may be admitted after consultation with the
study medical monitor, Hgb values < 7:0 .gm/dL are exclusionary
Prepared by Quintiles, Inc.
82
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR -P.harmaceutical CONFIDENTIAL Page 5 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
~ Liver function =tests (LFT) including AST, ALT, LDH) -should be < 3
times the upper limits of normal (ULN). Acceptance of =LFT values 3-5x
ULN =should be based .on clinical significance as determined'=by the
:investigator and requires notification of=the Medical 1Vionitor
:8. Normal adrenal function at baseline as evidenced=by basal cortisol levels
(of
> l0 g/dL) and -adrenocorticotropin (ACTH) stimulated cortisol levels (of
>18 g/dL);
-9. . Able to read and write in -the study related documents :translated :into
the ;primary
aocal ,langaage;
.10. Capable of and willing to return to the clinic =regularly for =study
visits;
11. 11~ust be taking a stable regimen of accepted ,HIV anti-retroviral
treatments for at
:least two weeks prior to study entry;
12. Capable of completing :a 3-day -food intake diary with =instruction;
13. Willing to abstain from any .illegal= or recreational drug substances for
the duration
=of the trial; and
14. Will.ing -to abstain from taking any .other medications or substances
known to affect
appetite. or weight :gain (eg, steroids [other than those inhaled for
treatment of
asthmatic conditions], -nutritional supplements =[bther than vitamins or
minerals],
=dronabinol, =recombinant human growth hormone, etc.).
4.2.2 Exclusion =Crite.ria
1. Age is less than 18 years and greater than 70 years of age;
2. Weight loss due to factors other than AIDS-related wasting;
3. Enrollment in= any other clinical trial;
.4. Lack of access to :regular meals;
5. Women of childbearing potential may not be :pregnant or nursing;
6. Clinically severe depression evidenced by a baseline score of 17 or -more
on the
=Hamilton Depression Rating Scale (GRID-HAMD-17);
7. Recent evidence of or history of significant psychiatric illness that may
compromise the subject's ability to comply with the study requirements;
Prepared by Quintiles, Inc.
83
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical =CONFIDENTIAL. Page'6 of 50
Final Clinical Protocol PAR-002 v.4 'Version: 01 =Nov 04
'8. Intractable or frequent vomiting that regularly interferes with eating;
9. =Clinically significant diarrhea that would interfere with absorption of
foods or
medications;
.10. Clinically -significant oral 'lesions or dental conditions that interfere
with eating a
.regular diet;
11. 'History or evidence ofthromboembolic=events or any first degree relative
with a
=history of thromboembolic events;
12. Active AIl7S-defining .illness or other clinically significant or
uncontrolled medical
:problems;
13. :Current evidence of or bistory of diabetes =mellitus or hypoadrenalism
or,
>14.- Systemic txeatment with glucocorticoids within the past 12 months.
4.2.3 'Rernoval, :Replacement, or Early Withdrawals of Patients from
' Therapy or Assessment
4.2.3.1 :INfthdrawaf of Subjects
A subject =is free to withdraw from the study at any time for any reason
without prejudice to
their future medical care by the physician or at the institution. The
Investigator or Sponsor
may also withdraw the subject at any time in the interest of subject safety or
study integrity:
Any subject who develops de novo diabetes mellitus or adrenal insufficiency
while on study
will be discontinued from taking additional study drug, followed up and
treated
appropriately. Please refer to Section 10.4 for additional guidance in this
circumstance.
Additionally, Par reserves the right to terminate the study at any time. The
primary reason
for withdrawal -of subjects -must be recorded in the subject's medical record
and on the
withdrawal form in the Case Report Form (CRF).
The withdrawal of a subject from the study should be discussed where possible
with the Medical Monitor before the subject stops medication. Final
evaluations will be performecl as
completely as possible at the time of the subject's withdrawal (refer to
Section 7.1.11). Any
comments (spontaneous or elicited) or coinplain.ts made by the subject and the
reason for
termination, .date of stopping the study medication and the total amount of
study medication
.must be =recorded =in the CRF and source documents. An attempt should be
made'to perform a
follow-up evaluation. If the site is unable to contact the subject after three
phone calls and a
certified letter, the subject should be considered lost to follow-up.
Prepared by Quintiles, Inc.
84
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 7 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
If a subject is withdrawn for more than one reason, each reason should be
documented in the
source document and the most medically significant reason should be eintered
on the CRF.
Subjects that =are removed or withdrawn early will not be -replaced.
5.0 STUDY TREATMENTS
5.1 - Identity of .Investigafiiona! Products
5.1.1 ;Me.gestrol Acetate Oral Suspension :NCD :Fon7neilation
1Vlegestrol :acetate"oral suspension NanoCrystalTM Dispersion (NCD)
formulation contains
imegestrol acetate, a synthetic derivative of the naturally occurring steroid
hormone
progesterone. Megestrol acetate is a white, crystalline solid and chemically
described as 17-
Hydroxy-6-methylpregna-4,6-diene-3,20-dione acetate. .
Ivlegestrol acetate oral suspension NCD is supplied as a suspension containing
115 mg of
<nanocrystalline megestrol acetate per mL. It also contains the following
inactive ingredients:
alcohol :(max 0.06% v/v from'flavor), artificial lime flavor, citric -acid
monohydrate, docusate
sodium, ~hydrox.y.propyl methylcellulose, natural and artificial lemon flavor,
purified water,
sodium benzoate, sodium citrate dihydrate, and sucrose.
.5.1.2 Megace (me.gestrol acetate orai suspension)
Megace (megestrol acetate oral suspension, Bristol-Myers Squibb, Princeton
NJ) is a
commercially available oral suspension with micronized megestrol acetate. It
contains the
following inactive ingredients: alcohol (max 0.06% v/v from flavor), citric
acid, lemon-lime
flavor, polyethylene glycol, polysorbate 80, purified water, sodium benzoate,
sodium citrate,
sucrose and xanthan. gum. Megace is supplied as an oral suspension containing
40 mg of
.the micronized megestrol acetate per mL.
Prepared by Quintiles, Inc.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR'Pharmaceutical CONFIDENTIAL :Page 8, of 50
'Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
5.1.3 Chemical Structure
Figure 1: Chemical Structure of Megestrol Acetate
,=:,i, ..~.f ..,....,:'=:.=o-;I:. ,..,:'::==id:'.I:.4
~ 1, ~~~~r~~;j,E{ C~~-~F~'l~~~C!J~~~~~~ rf'~il!~f! ~~k~~ ~~~~!i'i~~~!I~ XI
- r 1 i'h ly I 1 !'~ F
? ,, ry ~=~,9~~:,' (nr~~l,~~~ii~~
' e:,~'.', =, ~e!:i
~i,::i.::~.:::::.':'.f:',::'.;='.'.:I:::~,,J~';~:;!;,;,;a::G ;
l>::;:;i:a::';'::::'.::f.'. ~:;
:;~ =;;:h~;,;: ~:..,, ; .'~ :!: .C~.l.l.a;l. . ~, ] 1 ( 1 !I ~ ~~. (1.. ~~j~ r
''' i 1 t ;.i~.. ~..; . ~~.:~=:::::: ~;{;:!::r ....::.::::: :.=
4 = .:I .: h:..~.~ ., ( ~:~:': ~:
' ::: ,~==,:~.~;a''' '1 (;,~.:~~::=' ~:I.:::~: ,. ,
t.,=. ''r=. :;;~~=,}: :~;~i:r.L.:~P{ ti:f.?:.::::. ~: iiil~i~ ' ~i.. ~
.~:!::~il::=~i~ =~t~.~e
= ~ _=~ -a,~~.:~ =;kj;.= ~jf ,.: ..~~;~~:w..~ == ~k
a'i ~= : G==~.;r: F ~ ~.:.}:.~::. :.j: =~=::
::.;,=; -=1:.'.,.,, . ~i;::'~-. ~:: :~::::. i::::a:~::~ :::qih :~::~: ~ d..
~:~::.
:,~' = ,.:r:~3 ~:..~. ~1' 1:~ ~:a:':.: ~:'.
.i: ' ..~sk;;~:4'~: p ~~..~i, ~F~;=~i'ii:F~~~!:~b a+' , I::: .i(.~lii:''~
~'~~'~
~'=:=~;'~i:*P'i; ==~:~::r::l:{.a:f:. ~~t Ii:'I:~k Li::.'.:i ~ir~
.ar:.:k~~iFC:f{~:r;i~'i~;~:;:',..= Ii.: !i Li: ~a..l:
.,,. =..:,~.. i=~ ,.;:::::,: ! ~~ ~:.~ =,i,:i,:,.1~=.:: ,.:i ,1=
li:i =::!~I:;;i: ~=.. ~I:::x.;..l' r:.?1,. ~~ ~.F .1: i::-Ekf:~!';E;i:~E:f:
!::F::
=, = '~,i =:,,.. ..~f= _ ~'i:~:i;: il, a~ ~I i~~'i'li.;'i!!'.~.: ~''I ,, -
.....1;
~:~:c;==;' = " ~~) i.::.:aY.' ~:: =~ i;+:iii~i:.i:'.:r~..
.;;.,rt,:I .i.: J.....i~.:j'::'~ e: 1:.'=..(:::t.:~ ,~:, .,~tt.,;~.~. a:=
;:::..:.
,:.:,.;:. ~=,=N::,:~: '.f; = ' ~~a:~:::;.,..i ,;... r.a:.;:~::..~ ,
. :,, (;;~~~ ::..r,.,,i ..1..~.;:rC , . ,,.1 j ::.b,:=~:i: i i, ~ i ~ J E
:~,,,., ;~ ~.I{ ~ai~!~,:j ~! !:; ~;: =.; : ~~,~~ ~= f. :. .~. I~~; .
~:I.l~,' :.r~~?::i::::;=:::M'rr=,..a- J3:I~EE:Y=;'~::::::'.:~I:i~:;!:!:::,:,
~'. '; !
a~',1=:~a ,:~'~T:'.:~... :r:==:,.t~~.+~y,'''!;"Ã.~:.
tr..:,l:::::.,~:~:~....:.,...~a':~(=:õ
,
:_:,. i:.:h:~;; ~a'=I~ t i5, ~; i~' ~=:~"!~:,:~.{: ,.: ,.::;
':~" ;..~..,;:kl=:lit.'
='~~':i~yr:~~!C:;I~'::i;;:i:;l'fai:Ii'.;~ya;li~':i:'=.~a.~.{.= 'p .=::::..
r:r:1=,.. ':~+;:i~:: r?r.~ i,.i:!.,.~:A:+7'~.. . :=:=:lji:i~i
~~' q:<
i =;: ,.... >; ~,=t ,,:..~.... ~. a. :Gc:::.:: ~~~::~ ::~~~ ai} :...~: ~G::
; i::,=; . . .. 4.. :~..!~: =kf .i .i. =~=..:::: ~~~ i a: ~.i T:~~:::.:::6rL
:~t:!i ii. ~::i ~1= 1.~.,.9a...r..j:.:.:.: 91:?iC ~ i i =. .I~i~=iu4
Table l.: =Co.m,parison of Characteristics of Megestrol Acetate NCD and Megace
1Vlegestrol Acetate NCD Megace@
Molecular Formula C24H3204 C24H3204
Molecular=Weight 384.51 =384.51'
,Concentration 115 mg/mL 40 mg/mL
Bioequivalent doses 575 mg '800 mg
Batch/Lot number 041787 4D80437
Manufacturing Date 12/14/03 (see note below)
Note: No date of manufacture was available for the Megace product; however,
the expiry date provided by the
manufacturer is May 2006.
Both formulations are lemon-lime flavored solutions and identical in
appearance. Study
medications will be dispensed in identical containers labeled only .by:the
investigational label.
An example of the investigational label is displayed in Section 5.4.
.5.2 Method of Assigning Patients to Treatment Group
Subjects will be randomized -by site via sealed randomized treatment cards to
receive either
megestrol acetate NCD il'ormulation or Megace in a 1:1 ratio. Treatment will
be open-
labeled.
Prepared by Quintiles, ]nc.
86
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR ~Pharmaceutical CONFIDENTIAL Page 9 of 50
Final Ciinical=Protocoi PAR-002 v,4 Version: 01 Nov 04
5.3 Dose.,. Dose Schedule and 'Route of Administration
Megestrol acetate NCD :formulation will be adiministered as a single daily
dose of 575 .mg
:administered as -a 5 mL dose (115mg/mL -concentration).
MegaceV w.ill be given according to the standard dose specified in the product
labeling with
a=single' daily dose of .800 mg megestrol acetate administered as =a 20 mL
dose (40mg/mL
concentration).
In :both treatment groups, subjects will be in:structed fio take one dose by
mouth per day each
xnorning for -a total of 12 weeks.
S:4- :Packaging, ,Labeling and Retention .of 'Supplies
'Single lots of each of the study medications will be used for the study.
Megace will be obtained from a commercially available lot provided by Par
Pharmaceutical
and shipped to Quintiles, Inc. in its original packaging (240 mL bottles). Par
Pharmaceutical
wilYsupply megestrol acetate NCD formulation to Quintiles, Xnc in bottles of
150 mL.
Quintiles, Inc. will re-label study medication in their original bottles with
clinical labels and
distribute them to the investigational sites. The clinical label will be a 2-
part perforated label
-containing the following..information: Par Pharm4eeutical, protocol number,
patient number,
.patient initials, randomization number (treatment assignment nuniber), date
dispensed,
dosing instructions, cautionary statement required by Federal law, storage
requirements, and
lot number.
Prepared by Quintiles, Inc.
87
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
'PAR.Pharmaceutical CONFIDENTIAL. Page 10 of 50
=Finai Clinical =Protocol PAR-002 v.4 Version: 01 =Nov 04
Study medications will be dispensed in bottles of 240 mL (Megace -as is
commercially
-available) and ] 50 mL (NCD formulation) along with a reusable, plastic
medication cup of
20 :mL volume. Thedosing instructions for the megestrol acetate NCD
formulation will be
labeled as 5 m.L per dose. For the Ivlegace , dosing will be labeled =as 20 mL
per. dose.
Additional measuring cups will be available at the anvestigational sites..
The investigational product labeling will be cbmpliant with local regulatory
requirements. An
iexample of the investigational label is shown below:
Caution:=For Investigational Use Oniy
Study No: PAR-002 Randomization Number:
Subject-ID number. =Date Dispensed:
Shake containerwelt before use
Dosing Instructions: Take 5(or 20) mL by mouth every moming
Lot Number. =
Protect from heat and keep between 59' and 77 F (15 to 25-C)
Sponsorediby: PAR Pharmaceutical. inc:, Spring Valley, NY USA
5:5 Treatment Com pliance
,,
Subjects Will be asked to return the containers from the previous week at each
clinic visit to
.determine compliance. :In addition, trough blood levels for study medication
will be obtained
at each of the clinic visits.
&'6 %P.rior and Concomitant Treatments
Use of other appetite stimulating medications including any .of the following -
must be
= discontinued at 'least 1month prior to study entry. Tn addition, no other
appetite-stimulating
medications may be taken concurrently during the study.
This includes (but is not limited to) the following medications:
= megestrol acetate (Megace )
.= dronabinol (Marinol )
.= cyproheptadine .(Periactin )
= anabolic androgenic steroids including:
Prepared by Quintiles, Inc.
88
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PAR Pharmaceuticai CONFIDENTIAL Page 11 of 50
Final'Ciinical Protocol PAR-002 v.4 Version: 01 Nov 04
- testosterone, intramuscular=(Depo-Testosterone and others), transdermal,
topical gels
- testosterone analogues (dihydrotestosterone or DIIT),
> anabolic steroids including oxymetnolbne (A.nadrol ), oxandrolone
(Oxandrin ), methandrostenolone (Dianabol )
- = other androgenic compounds (dihydroepiandrosterone, androstenedione)
= recombinant human :growth hormone (Serostim )
= cytokine modulators (thalidomide, pentoxifylline)
..Trihaled steroids foir astthma and asthma-like conditions may be .given as
needed =as well as
,short term topical steroid treatments for'localized cutaneous conditions.(eg,
poison ivy or
,contact.dermatitis).
Current anti-retroviral medication regimen for treatment of HIV should be well-
established
,for at least two weeks prior to study entry. Subjects who require frequent
changes in
-medication should be deferred until a suitable regimen of inedication has
been established
and would be unlikely to vary considerably from baseline regimen. Medication
history
-should be carefully collected :at baseline and updated at each clinic visit.
No'other investigational agents may be used concurrently during this study.
Systemic -exposure to any glucocorticoids within the past 12 months prior to
screening is
exclusionary.
Any supplements (herbal, over-the-counter, or other) that may affect appetite
in any way are
specifically -excluded; however, multivitamin and mineral supplements are
allowed.
Nutritional products intended as caloric food supplements (eg, protein-
fortified drinks) are
: allowed.
Prepared by Quintiles, Inc.
89
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
-PAR Pharmaceutical CONFIDENTIAL Page 12 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
6.0 EFFICACY AND SAF-ETY ANALYSIS VARIA=BLES
6.1 !Efficacy
6.1.1 'Primary" ;Efficacy Endpoint
The primary efficacy endpoint is weight gain from baseline value. Baseline
weight will be
established -at screening then assessed weekly for the 12 weeks of treatment
and at the 30-day
follow-up. Subjects will'be weighed on the same scale, =in street clothes and
without shoes .for
each assessment.
6.1:2 Secondary Endpoints
iSecondary endpoints :include -changes from baseline in:
= lean body rnass, body fat and fat-free body assessed by bioimpedance
analysis at
Weeks 6 and 12 post treatment
= total body muscle mass assessed by anthropometric measures (mid-arm, waist
and hip
circumferenaes, :triceps skinfold measurements) weekly through Week 12
= food intake will be assessed by 1) diary record of the number and time of
meals
during a 3-day interval beginning at baseline and prior to each clinic visit
thereafter
:and 2) a 24-hour =recall food diary obtained at each clinic visit
= appetite assessed weekly by visual analog scale (included .in Quality of
Life
assessment described below)
= Quality-of-Life Assessments (Bristol-Myers Anorexia/Cachexia Recovery
Instrument
=or BACRI) completed weekly from Week 1 through Week .12 and at the 30-day
follow-up
= Pharmacokinetic studies =conducted on Day I and Week 6 visits. Trough levels
of
study drug will be collected at Day 3=( 1 day) after beginning treatment and
at each
=clinic visit during the treatment period thereafter
-6.2 Safety
Safety endpoints include weekly assessments of incidence and nature of adverse
events,
changes in vital signs, and pregnancy testing for women of childbearing
potential. Routine
clinical laboratory assessments (hematology, chemistry, and urinalyses) will
be assessed at
Prepared by Quintiles, Inc.
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical =CONFIDENTIAL Page 13 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
baseline and Weeks 3,.6 and 12 (end of study), and physical exaniinations
will'be performed
at baseline and Week 12. Hemoglobin A1C and ACTH stimulation testing will be
performed
at screeningand Week 12 (or last visit). Additional clinical laboratory
assessments may be
made at the discretion -o:f the Principal Investigator=if clinically
indicated.
Routine clinicdl laboratory sarnples for the sites :in the United States will
be =processed by a
centralized clinical lab and sent to the following address:
-Quintiles Laboratories, Ltd.
5500 Highlands Parkway
Suite -600
Smyrna, ;GA 30082
Routine clinical laboratory samples for the sites in South Africa will be
processed by a
centralized clinical lab and sent to =the following address:
Quintiles Laboratories South Africa
Pencardia 1 Ground Floor
:509 Pretorius Street
:Pretoria, RSA=
Routine clinical laboratory samples for the sites in India will be processed
by a centralized,
clinical lab and sent to the following address:
SRL Ranbaxy Ltd.
113, MIDC-15th Street
Andheri (East), Mumbai - 400 093
II\TDIA
-6.3 =P:harmacokinetics
Two pharmacokinetic:(PK) studies will be performed on each subject; the first
will be
:performed on the=first day of treatment and the second, during the Week 6
clinic visit. The
sampling times for the PK studies will be identical at each study and are
described in the
following sections. Pharmacokinetic assessments will include Cm", AUC o-t, and
T m~,..
Trougb -levels for study drug will also be assessed during each clinic visit.
=6.3.1 'Day I Pharmacokinetic Study
Afler eligibility has been established, subjects will be asked to come to the
clinic after at least
a 10-hour fast on Day 1. An indwelling venous access device will be placed to
allow for
i'repared by Quintiles, Inc.
91
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
'PAR'Pharmaceuticai CONFIDENTIAL Page 14 of 50
Finai Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
multiple blood samples and the baseline sample will be drawn. The daily dose
of the assigned
study =medication will be administered and the tinie recorded. The subject
will then be served
a standardized breakfast. The contents of the standardized meal will vary by
country and
=minimum requirements will be specified in the Study Piocedures Manual. The
schedule for
the subsequent PK samples is as :follows (time points refer to hours post
dose):
Baseline .(0), =0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 12.0,
and 24.0 hours.
Subjects will be served an additional meal at approximately 4 hours after the
dose of study
drug and the amount of.food consumed will be recorded according to the
standards defined in
ithe 3-day food intake diary. Additional=details =regarding these
diaries=are=presented in the
'Study Procedures Manual.
*6.3.2 Wee'k .6 ;Pharmacokinetic Study
The second PK study will follow the same procedures as the first study with
identical
=sampling times.
Baseline :(0), 0.5, 1.0, 1.5, 2:0, 2.5, 3.0, 4.0, 5.0, 6.0, 7:0, 8.0, 12.0,
and 24.0 hours.
During the PK study, =the subject will be asked to record his or her =food
intake on the 3-day
food intake diary.
6.3.3 Processing =of =Pharmacokinetic Samples
'Samples for pharmacokinetic testing will be labeled with the unique subject
identification,
date and time of sample. Each sample requires 51nL of blood collected in an
EDTA K3 tube.
'Samples should be stored on wet ice until centrifugation, then spun within 50
minutes and the
resulting plasma separated into 2 equal volumes and stored in 2 labeled
cryotubes of 5 mL
volume. Aliquots should be stored at approximately -20 C (nominally) or colder
in a
temperature-monitored freezer until shipment. Frozen samples should be stored
until Par (or
designee) :indicates the timing ofsample shipment. =Samples should be sent on
enough dry ice
=to keep samples frozen for approximately 72 hours. Samples should be sent via
overnight
courier to the following address:
Prepared by Quintiles, Inc.
92
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL 'Page 15 of 50
=Final Clinical Protocol.PAR-002 v.4 Version: 01 Nov 04
SFBC Anapharm Inc.
2050, blvd. Rene=L6vesque West
Saiinte-Foy (Qu6bec), Canada, GIV 2K8
Phone: .(418) 527=4000
-Fax: (418) 527-3456
Attention: Mr. Louis-Philippe Beauregard, Sample Controller Coordinator
.A.dditional -details regarding handling of the PK samples are provided in
Appendix D. -
7:0 STUDY PROCEDURES .;A'N'D 'SCHEDULE
Study-specific ;procedures including,protocols for the bioimpedance analyses,
and
-.anthropometric assessments (mid-ann, waist and hip circumferences and
triceps skinfold
=measurements) will be provided to each Investigator in an additional Study
Procedures
manual.
7.1 Study Schedule
The schedule of study -related assessments is sumzinarized by interval in
Table 2.
7.1.1 Screening Visit
Screening assessments may satisfy the baseline requirements if the screening
assessments are
completed within 7 days of study entry. Pregnancy testing, however, must be
completed
immediately before study drug is dispensed'regardless of timing of the
previous pregnancy
test.
The following assessments are made at screening:
= Review study procedures and- obtain informed consent
= Medical history with particular attention to the review of gastrointestinal
system
= Height and weight (in street clothes without shoes)
= Physical examination including vital signs (temperature, pulse, blood
pressure and
respirations)
= Routine clinical laboratories including:
- hematology including complete blood count with differential, platelet count
and
hemoglobin A1 C
Prepared by Quintiles, Inc.
93
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical 1CONFIDENTIAL 'Page 16 of 50
'Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
> serum claemistry including sodium, potassium, =chloride, bicarbonate, BUN,
albumin, glucose, creatinine, alkaline phosphatase, =total bilirubin, liver
function
-tests (AST/SGOT,..ALT/SGPT, LDH), and'lipid panel
> =routine -urinalysis =by dipsticlC (pH, .specific :gravity, =glucose,
protein, ketones,
nitrites, leukocyte= esterase and urobilinogen)
=- beta human chorionic gonadotropin ((3-hCG),for women of childbearing
: potential
= ACTH -stimulation =study with serum cor.tisol samples at baseline, 30 and 60
minutes
post-stimulation
= Hamilton Rating Scale .for Depression .(GRID-HAMD-17)
Prepared by Quintiles, lna
94
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
a3~,
.'a1 a X
Qo oMi
c
0 m S m
o
Q c~
~ XXXXXXXXX X =~
g
.
'n
> Na- '~
t- (1) (D y~otf , X X x xX mN m g~
(D ~ .b c ~
o
oo ~ o
c m ~ -E
dCD
x x x x x
y
'. .t0 .= C [+J =C
1 ~C ~co E.m
X X o =V
'
a ot{ >C ?( X "" "'
-3 C
;~ ~ :C p =O = N
'm ,G N ~ O C
;~C : 'c to u, v
eD 'XXx Xx XX X X 0=~
O C1 N U O V
'O C_.C O 'm p
x X y,N-~ =~ N .N C U~+
X X X N
:20 :a) O
~ O. N 1a
to;
Z c~vod mE
=lu =-= O U N =C ~
~~E
~ x xXx xx xx
z ~~~=T ~~,~ ~ A~,
O m m'>= E 3
U ~~~ E n
-J =- N O=wo N
N b~ ~_EI a
IVca X . x X X X
.~ r ~.~ C N C m t
! O.O N'C y m'~ Ol
O
f(1a,
U.p m c o E wiv ai c
L~ ~ = x X =N T~ C N C=O 7~
C 'C C N r O.N
'p =N E y y y N~ m cn (D a~Y .~-
f0 N na N
S ) E C m
==C~ . GI ~ y 7~
9 N~p v T'O p p)
~ C r a a a a ' a ' ' N
='~y :~ . X X X"X X X X ' X X~ X x .+ C+' 3 y U 7 0 C S O
m :Q =! .CN ~ a jq O
m
v m.:o d.o o -
~'+ = ,c~ '~ :n ~ ;c m ,fl ~= ~.
E
XlCXXXXX?<XXXx v"af6m
=m 'N .p ....
~m oooc~~oc3
~ ~ ~ ~ ma' ~ c=:c
o L - ~, ~. oWo
~ C ,C N p~'C C_N O
~ CFy C ~7 'O T, ~ ~ p)_ C m..0 O.p=*
pQ p : c c N. a~ ~
am Np Si "
y:~ N a~ e'o ~ v ~ 'Ã w M
N t0 Y. a1 ,o . T
3 ,d
o m 4: c~ c nIS
o y E _ r_ o U=N C~ ~ v o~ w a~ 3"2 2
'
a v m y E m.m.'. m E 'Qo
~ c o ,C = . m Q lt{ ~ o c . N O .lm .... y ~1 ' ) c N > >.
c
~ v< p ~ E o c c p a~ U' cc m o m v
N :U .y . ~ .y = ~ .~ ~ ~ ~ C =N O > N ~ ~ =S O Gyi E T N E ~
~ c N: Q a ~ 3 c-oo c~ v a~ 'E toa ~ w uoi r 3. vyi A m'n
y' >~= 3 o c c o.c'o a>o3 ~~,
C- V = . Ã ~ c =y =N E E .~ c 6 aD ~ ~i E uyi m
c CC aa a a~ 2i 'ai ~' a>i m f o ~n o ~
Q fi 2'~ = fp Q S fl' n t2 ~~ a F- [] ~x~p~xnr~-o
A .O tl V O 0 t.~
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
.1c X X
: O L~O ~.
O O
m O
XXiG xxX scp
tN V pC7
r_Y ' ~ = v t V
= =~ ~/ ~ X X X X X /l = m N
is
1- +"a
O
b 'N
O=
XXXX 'XX
r. m
= m
=oo
(D'otS XXX>C XX
' S =a ai
Y N v1 .m
~ -
tn X X X X XXX o
o
m~.=
.N m
aA, E
~ =~ = : . = .E 7 N
,J oa XXXX XX ~cM
.Q .,~ d' i, b=~Q :lO
7
z ~ w
=W m
-~ XX>L:X XX d
p .aE
$ o
a'S XXXX XX ~oNm
y tN
N A 7 ~
R7 =--= b
,~ . X
M
E ~~=~
mUN
= r w == m q
X X X XXX ETEc
'
N N
~
9 _>
v c> E
> f/) a=;~ .m
N LC =3
c~ Q d ~
O C
~, = C M m
.. Q .. tQ Y' a)
N'N U b
n a ccg N~E m~:
=N
C> O ,41 .r~. m CN/1
m:E
'a L e ia E
0' :y N=m g N ' S~ m
m o
af v m ._
E~ o vi EEM o c m.tS L~ E
m c t!~ ~' Om o Uo m' T
c' Q c -'mN3 -
n U m o~ vi g m ' ~ o'm cv
m r U ~ ami ;o
o. N m a E m a~~ - m E
96
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharinaceutical :CONFIDENTIAL Page 19 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
7.1.2 Baseline and Treatment Day(Day 1)
The following activities are to be completed after eligibility is determined
and the subject
agrees to enroll in the study:
= Review =eligibility criteria and screening assessments
+ Review informed consent is appropriately signed
= Update any screening assessments if initial screening was longer than 7 days
prior
;to Study Day '1
= Obtain =baseline weight
= Obtain urine pregnancy test for women of childbearing potential
: =Obtain randomized treatment =assigntnent.
If subject continues to be,eligible, then insert.indwelling venous access
device for the
pbarmacokinetic study.
7.1.2.1 Phannacokinetic Study
- Time 0:'Baseline sample of approximately 5 mL in an EDTA K3 tube will be
drawn, labeled with the unique subject identifier, date and time
- The daily dose of the assigned randomized study medication will be
administered, the time recorded, and then subject will be served a
standardized breakfast
- Samples for PK analysis require 5 mL in a tube containing EDTA K3 for each
=sample. The schedule for the subsequent PK samples are as follows (time
points refer to hours post dose):
:0.5, 1.0, 1.5, 2:0, 2.5, 3.0, 4.0, 5.0, 6:0,.7.0, 8.0, 12.0, and 24:0 hours..
- An additional meal will be served at approximately 4=hours post dose and the
time of the meal will be recorded. During this.meal, the subject will be
instructed in the proper completion of the 3-day food intake diary
- After the conclusion of the PK study, the subject will receive the remaining
study medication to take home. The 24-hour post dose sample should be
obtained prior to the next day's dose
The folIowing assessments may be collected during the conduct of the PK study:
Prepared by Quintiles, Inc.
97
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
MR Pharmaceutical CONFIDENTIAL =. Page 20 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
= Anthropometric assessments (mid-arm, bip and waist circumferences and
triceps
skinfold measurenaents)
= Bioimpedance analysis
=-Schedule next follow-up for Post-Treatment days -i and 7=(Week 1)
= Dispense study medication
='bispense wallet subject 'identification card and review its'use
Dispense 3-day food intake diar.y and review its=use
== 'Complete 24-hour recall food diary and record f ndings and
~p 'Subjects should be instructed uot to -take the =daily dose of study before
coming to
the =next clinic visit in order to obtain adequate trough levels. The daily
dose of
study medication may be taken anytime after -the trough level is drawn
7:1.3 ;Post-treatment Day 3
= A trough level of study drug should be obtained within the first week of
dosing,
preferably at Day 3(f1 day)
== Complete BACRI Quality-of-Life Assessment
.= Adverse events and concomitant medications should also be assessed at this
time
:7.1.4 'Post-treatment Weeks 1 !and 2
Follow-up appointments should try to be at approximately the same time of day
each week
(pr.eferably morning) if possible. All visits may vary by a 2-day window on
either side of the
expected date.
= Weight on same scale used for'baseline; subject in street clothes and no
shoes
= Vital signs (temperature, pulse, respirations and blood pressure)
~ Urine pregnancy testing for women of childbearing potential prior to
dispensing
study drug
= Trough levels of study -drug (5 mL in an EDTA K3 tube)
~ Review 3-day food intake diary for accuracy, provide blank diary for
following
week
Prepared by Quintiles, Inc:
98
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 21 Df 50
'Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
'= Complete 24-hour recall food diary and record findings
= Complete BACRI Quality-of-Life Assessment
= Anthropometric assessments (mid-arm, waist and hip circumferences and
triceps
skinfold =measurements)
= Return study drug dispensed from .prior week, dfspense study drug for
following
week
: Assess for adverse.events or changes in concom=itant=medications
= 'Schedule next week's appointment
Unscheduled laboratory'assessments -may'be obtaiined at any -time to ensure
the safety and
well being of the subject based upon the clinical judgment of.the 1'rincipal
Investigator.
Abnormal laboratory values obtained as part of the routine assessments may be
repeated if
:the Investigator judges that the results are suspect and repeat testing would
be clinically
-indicated. However, if abnormalities persist on a-subsecluent assessment, the
-abnormality will
be considered an -adverse event.
7-1:5 .Post-treatment'Week 3
Follow-up appointments should try to be at approximately the same time of day
each week
(morning) if possible.
V Weight on same scale, used for baseline; subject in street clothes and no
shoes
.= Vital signs (temperature, pulse, respirations and blood;pressure)
= Routine clinical laboratory samples should be obtained :in a fasting state.
Samples
for the following assessments should be obtained:
- hematology inclqding complete blood count with -differential and platelet
count
- serum chemistry including sodium, potassium,.chloride, bicarbonate, BUN,
albumin,.glucose, creatinine, alkaline phosphatase, total bilirubin, liver
function tests (AST/SGOT, ALT/SGPT, =LDH) and lipid,panel
- routine urinalysis by dipstick (pH, specific.gravity, glucose, protein,
ketones, nitrites, leukocyte esterase and urobilinogen)
.= Urine pregnancy testing for women of childbearing potential prior to
dispensing
study drug
Prepared by Quintiles, Inc.
99
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutioal CONFIDENTIAL Page 22 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
= Trough =levels of study drug (5 mL in EDTA K3 tube)
= Return study drug dispensed firom prior week, dispense study drug for
following
week
Review. 3-day food intake diary for accuracy, provide ~blank diary for
following
week
= Complete 24-hour recall 'food diary .and record =findings
= Complete BACRI Quality-of-Life Assessment,
= Anthropometric assessments (mid-arm, waist and hip circumferences and
triceps
skinfold measurements)
A Assess for adverse events or changes in concomitant medications
= Schedule next week's appointment
7.1.6 :Post-tr.eatment Weeks =4 and '5
Repeat =assessments under Weeks 1 and 2.
7.1.7 Post-treatment Week :6
Subjects should schedule-the Week 6 visit for a morning appointment in order
to obtain the
baseline blood sample for the PK study in =a fasting state and prior to that
morning's daily
:dose of study medication.
= Repeat assessments under Week 3 and include the following additional
assessments
= Physical examination
= Bioimpedance =analysis
= Repeat Pharmacokinetic Study (same -as Day 1):
- Time 0: Baseline sample of approximately 5 mL will be drawn and added to
an EDTA K3 tube, labeled with the unique subject identifier, date and time
- The daily dose of the assigned randomi,zed study medication will be
administered, the time recorded, and then subject will be served a
standardized breakfast
Prepared by Quintiles, Inc.
100
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
+PAR =Phanraceutical CONFIDENTIAL Page 23 of 50
Final Clinical'Protocol PAR-002 v.4 Version: 01 Nov 04
- Each subsequent sample for PK =analysis requires 5 mL =in an'EDTA K3 tube.
The schedule for the subsequent PK samples are as follows (time points refer
=to:hours;post dose)
0.5, 1:0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 12:0, and= 24:0 hours
-.A.n additional meal will =be served at =approxiumately 4 hours post dose
and= the
time of the meal will'be recorded. During this meal, the subject will record
the
food consumed sn the meal in the 3-day food intake diary
- After the conclusion of the PK study, the subject will receive the -
remaining
study medication to take home. The 24-hour post =dose 'sample should be
obtained prior to the next day's dose
7.1.8 Post-treatment Weeks 7 ;and B
= Repeat assessments under Weeks 1 and 2
7.1.9 'Post-tr.eatment'Week 9
= Repeat assessments unde=r Week 3
~ Clinical =laboratory samples may'be omitted unless clinically indicated as
judged
by the Principal Investigator
7.1.10- ftst-treatment Weeks 10 and 11
= Repeat assessments :under Weeks I and 2
7.1.11 =Post=treatment Week 12 (End-of-Study :Drug Treatment Visit)
= Repeat assessments under Weeks 1 and 2
= Routine clinical laboratory samples should'be obtainedin a fasting state.
Samples for the following assessments should be obtained.
- Hematology including complete blood count with differential, platelet count
and hemoglobin A1C
- Serum chemistry including sodium, potassium, chloride, bicarbonate, BUN,
albumin, glucose, creatinine, alkaline phosphatase, total bilirubin, liver
function tests (AST/SGOT, ALT/SGPT, LDH) and lipid panel
- Routine urinalysis by dipstick (pH, specific gravity, glucose,
protein,'ketones,
nitrites, leukocyte esterase and urobilinogen)
Prepared by Quintiles, Inc.
101
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
-PAR =Pharmaceuticai CONFIDENTIAL Page 24 of 50
'Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
= Repeat ACTH stimulation testing
= Review last 3-day food diary
Complete 24=hour recall food diary and recoid findings
= No dispensing ofadditional =food diary or study drug
= Schedule 30-day follow-up
7.1.12 30-day 'Follow-u,p (End of'Study Visit)
= Weight on same scale used for:baseline; subject in street clothes and no
shoes
= Complete BAC.RT Quality-of Life Assessment
= Review status of any adverse events ongoing at the last clinic visit (Week
12)
:and assess:for any.new serious adverse events
,B:0 -STATiSTICS
8:1 'Statistical:Plan
8.1.1 Primar.y. Endpoint Analysis
The primary endpoint is change in body weight from baseline. The primary goal
for this pilot
study is to explore the rate of weight gain over timed intervals; therefore,
only exploratory
analyses will be .performed. For weight gain, an "area under the curve"
analysis will also be
-conducted to assess the overall difference in effect of the two therapies
over the first 12
weeks.
Each .measurernent will be provided with appropriate summary statistics.
Treatment
"differences will be-estimated and 95%0' confidence intervals will be
provided. Tviissing
,individual observations will be interpolated based on prior and subsequent
values.
'8.1.2 Secondary Endpoint Analysis
For analysis of secondary endpoints, each variable will be ,provided with
appropriate
summary statistics. Changes from baseline for the secondary endpoints will be
explored by
treatment group. Treatment differences will be estimated and 95% confidence
intervals will
be provided. However,.because of the exploratory nature of the analyses,
missing individual
observations will be interpolated based on prior and subsequent values.
Prepared by Quintiles, Inc.
102
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PARPhannaceutical CONFIDENTIAL, Page 25 of=50
Final Clinical Protocol PAR=002 v.4 Version: 01 Nov 04
Safety analysis will include the incidence of adverse events coded using
Medical Dictionary
for Regulatory Activities =(MedDRA), version 6.0 dictionary and reported by
preferred term
and treatiuent group. Descriptive statistics will be used for clinical
laboratory data and vital
sign data. Abnormalities in non-numeric data (eg, physical examination
results) will be
presented in listings.
=8.9.3 Study Populations
Study populations intended for analysis will be defined as follows: Intent-to-
Treat,
Per-Protocol and Safety.
~8.1.3.1 :intent-to-Treat
The Intent-to-Treat population will consist of all randomized subjects who
were dispensed
medication and had at least one post-randomization visit. Subjects will be
analyzed by
treatment assigned. Analyses of the primary endpoint will be performed on the
Intent-to-
Treat and the Per-Protocol populations
:8.1:3.2 Per-Protocol
The Per-Protocol (evaluable) population will include all subjects who
completed .the study.
requirements with no more than one missing visit and no major protocol
violations.
8.1.3.3 Safety Population
The Safety population will consist of all subjects who received at least one
dose of study
medication and will be analyzed according to actual treatment received rather
than treatnaent
assigned.
8.1.4 Planned Analyses
8.1.4.1 Patient Disposition
A detailed description of patient disposition will be provided and will
include:
= A summary of=data on patient discontinuation
= A summary of data on overall qualification status of all patients
== An account of all identified .protocol violations
All randomized patients .entered in the study will be accounted for in the
summary. The
number of patients who do not qualify for analysis, who die, or who
discontinue before
treatment begins will be specified. Patients discontinuing due to lack of
treatment effect will
be considered as treatment failures. '
Prepared by Quintiles, Inc.
103
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
.PAR;Pharmaceutical CONFIDENTIAL Page 26 of 50
:Final Clinical'Protocol PAR-002 v.4 Version: 01 Nov 04
8.1.4.2 Patient .Characteristics
Patient characteristics will include a summary of the following:
= Patient demographics
io Baseline disease characteristics
= Medical history
,= Prior medications
'= Concomitant=drugs
Other patient characteristics will be summarized as deemed appropriate.
8.1.4.3 Safety Analysis
Adverse events will be coded using MedDRA, version 6:0. Frequency of AEs will
be
calculated for each system organ class and .preferred term by treatment group.
The number
of patients and proportion reporting each AE will be summarized. The severity
of the AE
and .relationship to study medication will be summarized for each system organ
class and
preferred term by treatment group.
Descriptive statistics (number of observations, mean, standard deviation,
minimum, 'median
and maximum values) will be calculated for clinical laboratory tests
(hematology, serum
=chemistry and urinalysis) at applicable visits.
Vital signs (systolic and diastolic blood pressure, and pulse) and physical
examination results
will be summarized by treatment group using appropriate descriptive
statistics. Continuous
variables will be summarized using number of observations, mean, standard
deviation,
minimum, median, and maximum values. Categorical values will be summarized
using
=number of observations and percentages.
Withdrawals from the.study w.ill :be summarized by treatment group.
Additional details regarding the intended analyses are provided in the
Statistical Analysis
Plan.
8.2 Determination of:Sarnple Size
'This is an exploratory study; therefore, sample size was not contingent upon
enrolhnent
=numbers required to achieve adequate statistical power.
Prepared by Quintiles, Inc.
104
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
VARPhanraceuticai CONFIDENTIAL Page 27 of 50
Finai Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
9:0 A=D'1/ERSE EVENTS
9.1 Adverse Event Definitions
An Adverse Event (AE) is any untoward medical occurrence reported in a subject
enrolled in
=clinical investigation which does not necessarily.have a causal relationship
with the study
treatment. An adverse event can therefore be any unfavorable and unintended
sign (including
an =abnormal -laboratory fnding), symptom, disease or exacerbation of a pre-
existing
condition temporally associated with the use of a medicinal
(investigational).product.) (ICH
Quidance E2A: Clinical Safety Data Management: Definitions and Standards for
Fxpedited
[t.eporting, pctober 1994)
Each AE .requires a complete and thorough description including date of onset
and corrective
actions taken. Additionally, the intensity of the AB and its relationship to
the investigational
product, as well as its outcome, must be reported.
Tn order to avoid bias =in eliciting AEs, subjects should be asked, a
non=leading question, such
as 'How are you feeling?' It is also important to question the subject in a
non-leading way
about changes in .their -health or concomitant med7ication usage since their
last visit. This
:information should be collected prior to completion of assessments at all
study visits. In
addition, any symptoms/conditions reported during assessments deemed to be
clinically
significant by the lnvestigator should be reported as AEs.
All AEs (related and unrelated, serious and non-serious) will be recorded for
the interval
-beginning from the time the informed consent is signed until 30 days after
the end of
treatment =exposure. All ABs are to be recorded on the appropriate AB ;pages
in the Case
IReport Form (CRF) and in source documents. Where possible, a diagnosis rather
-than a list
of symptoms should be recorded. If a diagnosis has not been made, then each
symptom
should be listed individually.
All AEs will be followed until one of the following milestones is reached: A)
the event is
resolved :(defined as the subject's health has retumed to his/her baseline
status or all variables
have returned to normal); 2) the event is stabilized or designated as a
chronic=condition (the
investigator does not expect any further improvement or worsening of the
event); or 3) the
event is .otherw=ise .explained regardless of whether the subject is still
participating in the
study. Where appropriate, medical tests and examinations will be performed to
document
=resolution of event(s).
Prepared by Quintiles, Inc.
105
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
,PAR'Pharmaceutical =CONFIUENTIAL Page 28 of'50
Final Clinical Protocol PAR-002 v.4 Version:.01 Nov 04
.9.2 Reporting Adverse =Event -Intensity
In general, the intensity of a particular AE is reported as the worst
intensity experienced by
the subject during the course of the event. However, worsening .of.pre-
treatment events after
initiation of investigational ,product or an increase :in severity of
a=previously reported, post-
treatment adverse event must be recorded as new .A Es. For exampl'e, if a
subject experiences
mild hypertension at study entry -(prior to dosing of investigational product)
and the
=hypertension =becomes severe and more frequent after the investigational
product has been
administered, a new AE of severe hypertension (with the appropriate date of
onset indicating
the change in severity) will be recorded on the appropriate CRF. Similarly, if
an adverse
eveint is first Identified.as mild and then increases in severity during the
study, an additional
adverse event should be recorded'to document the change in severity.
The medical assessment of intensity wxll be determined by =using the following
defmitions:
Mild: The A:E is easily tolerated and does not interfere with usual activity.
Moderate: The AE interferes with daily activity, but the subject is still able
to
function.
Severe; The AE is incapacitating and the- subject is unable to work or
complete usual
activity.
9.3 :Reporting .Relationships of AdverseEvents to Study Drug
The Investigator must make the determinationof relationship between the event
and the
investigational product for each AE. The Investigator should -decide whether,
in his or her -
medical judgment, there is a reasonable possibility that the event may have
been caused by
-the investigational product. If no valid reason exists for suggesting a
relationship, then the
AE should'be classified as 'unrelated'. Otherwise, if there is any valid
reason, even if
-undetermined or untested, for suspecting a:possible cause-and-
effect:relationship between the
investigational product and the .occurrence of the AE, then the AE should be
considered
"related".
Unrelated: The -event can be readily explained by other factors such as the
subject's
underlying medical condition, concomitant therapy or accident and no
obvious temporal relationship exists between the investigational product
and the event.
Possybly related: There may be some temporal relationship between the event
and the
administration of the investigational product but there remains some
ambiguity as to the cause.
Prepared by Quintiles, Inc.
106
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 29 of 50
Final Clinical Protocol PAR-602 v.4 Version: 01 Nov 04
Probably related: The temporal relationship between the event and the
administration of
the :investigational product is :compelling, and/or follows =a known or
suspected :response =pattern to that product, and the event -cannot be
explained by th=e subject's medical condition, other therapies or accident.
If the causal relationshxp between an AE and the investigational product is
determined to be
'.possible'=or 'probable' the event will be considered related to
investigational product for the
;purposes of expedited regulatory reporting.
.9.4 Notifcatio.n .about =Serious or *.Unexpected Adverse Events
A SeriousAdverse :Event (SA;E) is any untoward medical occurrence (whether
considered to
'be related to investigational product or not) that at =any dose:
= .results in =death
= is life-threatening
NOTE: The term "life-threatening" in the definition of "serious" refers to an
event in which
the patient was at risk of death at the time of the event; it does not refer
to an event which
hypothetically might have caused death if =it was more severe.
= requ'vres inpatient -hospitalization or -prolongation of
existing'hospitalization
= results in persistent or significant disability/incapacity
= is a congenital abnormality/birth defect
Medical and scientific judgment should be exercised in deciding whether
expedited reporting
is appropriate in other situations. Important medical events that may not be
immediately life-
threatening or result in death or hospitalization but may jeopardize the
patient or may require
=intervention to prevent one of the other outcomes listed in the definition
above should :be
considered for ex.pedited reporting. These should also usually be considered
serious.
All SAEs .(related and unrelated) will be .recorded from the time the informed
consent is
signed -until 30 days following the end of treatment exposure. Any SAEs
considered possibly
or.probably related to the investigational product and discovered by the
Investigator at my
interval after the study should be reported. All SAEs must be reported within
one business
day of the first awareness of the event. The Investigator must complete, sign
and date the
SAE pages, verify the accuracy of the information recorded on the SAE pages
with the
corresponding source documents, and send a copy (by fax) to the Quintiles
Pharmacovigilance office =using the toll-free contact numbers noted in the
following table.
Prepared by Quintiles, Inc.
107
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Phannaceutical CONFIDENTIAL Page 30 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 =Nov 04
Quintiles Pharmacovigilance staff pan be reached directly by telephone from
8:00 AM to
5:00 PM (GMT); however, faxed reports may be sent at any time.
Quintiles Pharmacovigilance Contact Information
Country Fax Number Telephone Number
India = AT&T Access Code 000117 AT&T Access Code -000117
+ Pause + Pause
+ 877-264-10 39 +877-264-10 40
South Africa 0 800 99 42 08 0 800 99 42 07
'United States 1 (800) 414-8460 1 (800) 414-8451
At a=minimum, the adverse event name, the name of the person making the
report, .the name
of the suspected investigational product, and p*atient identifiers, and a
description of the event
should be provided. The Investigator's preliminary assessment of causality
must be ,provided
at the time of the initial report. Additional follow-up information, if
required or available,
should'be faxed to Quintiles Pharmacovigilance within one'business day of
receipt. This
-should be completed on a follow up SAE form and placed with the original in
the appropriate
section of the CRF/study file.
The Investigator is encouraged to discuss with the Quintiles Medical Monitor
any AEs for
which the issue of level of reportability is unclear or questioned.
Par Pharmaceutical (or designee) .is responsible for notifying the relevant
regulatory
authorities of serious adverse events. Additionally, some events may require
immediate
reporting to relevant local regulatoryauthorities -in accordance with local
requirements.
It is the Principal Investigator's -responsibility to notify his or her
Institutional Review Board
=(IRB), Tndependent Ethics Committee :(IEC) or the-relevant local iegulatory
authority of all
SAEs that :occur at'his ,or her site. Investigators will also be notified of
all unexpected,
serious, drug-related events (7/15-Day Safety Reports) that occur at other
sites during the
.study. Each site is responsible for notifying their IRB, IEC or the relevant
local regulatory
authority of these additional SAEs.
9.5 Notification of Adverse Events of Interest
The sponsor has identified certain adverse events of interest that should be
reported to
Quintiles Pharmacovigilance in the same manner and timeframe as specified in
the previous
Prepared by Quin6les, Inc.
108
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical =CONFIDENTIAL 'Page 31 of 50
Final Clinical Protocol =PAR 002 v.4 Versfon: 01,Nov.04
section for serious adverse event reporting. As previously noted, there have
been rare
occurrences of de =novo diabetes mellitus and hy.poadrenalism in the stressed
and non-stressed
:states. in subjects who 'have received megestrol -acetate. Should the
investigator observe or
suspect =any of these events, reports should be made promptly to the sponsor
via Quintiles
Pharmacovigilance following the same reporting mechanisna as for SAEs.
In addition, any .pregnancy identified on -study should be followed to term
and any fetal
abnormality(s) detected reported by the same expedited reporting mechanism.
Any subject
who =becomes pregnant on study should be discontinued ~rom the study but
followed until
delivery or pregnancy termination.
1,0.0 'EM'E'RGE'NCY PROCE'DURES
10.1 Emergency Sponsor Contact
In case of an Emergency, the Medical Monitor responsible for the study should
be contacted.
The contact -xnformation for the responsible Medical Monitor is displayed
below:
Richard Levine, MD
Quintiles Medical Advisor
1801 Rockville Pike, Suite 300
Rockville, IvID 20852
Office.phone:= (301) 272-3224
Cell phone: (301) 266-0132 (24 hours)
Fax: ' =(301) 272-2153
:Email:Richard.Levine@qiuintiles.com
10.2 Emergency Identification of Study Medication
This is an open-labeled study; therefore,.emergency identification of study
medication is not
applicable.
10.3 .Emer.gency. Treatment
10.3.1 Overdosage
No serious and unexpected side effects resulted from studies involving
megestrol acetate oral
suspension (Megace ) administered .in dosages as high as 1200 mg/day.
Megestrol acetate
has not been tested for dialyzability; however, due to its low solubility, it
is postulated that
dialysis would not be an effective means of treating overdose. 21
Prepared by Quinliles, Inc.
109
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
.PAR Pharmaceutical =CONFIDENTIAL Page 32 of 50
Final Glinical Arotocol PAR-002 v.4 Version: 01 Nov 04
10:4 Guidance for the -investigator =Regarding the Possibility of
Adrenal Insufficiency
The glucocorticoid activity of Megace Oral Suspension has not been fully
evaluated.
=Clinical cases of de novo diabetes mellitus, exacerbation of.pre-existing
diabetes mellitus,
and overt =Cushing's= syndrome have been reported =in association with the
chronic use of
Megace . In addition, clinical cases of adrenal insufficiency in the stressed
and non-stressed
states have been observed in patients receiving or recently withdrawn from
chronic Megace
therapy. Furthermore, adrenocorticotropin.(ACTH) stimulation testing has
revealed the
frequent occurrence of asymptomatic suppression of the hypothalarnic-pituitary-
adrenal axis
'in such :patients al Finally, there have been reports in the literature of
these events occurring
:in subjects within the =frst few weeks of Ivlegace therapy 22
Therefore, 4he possibility of adrenal insuff ciency should be considered in
the differential
diagnosis whein patients.receiving or=recently withdrawn from any form of
inegestrcil acetate
therapy (IJCD or Megace(b) present with symptoms and/or signs suggestive of
hypoadrerialism (e.g., bypotension, nausea, vomiting, dizziness or weakness)
in either the
stressed or non-stressed states. Laboratory evaluation to rule out adrenal
insufficiency and
-consideration of treatment with -replacement =or stress doses of a rapidly
acting glucocorticoid
are strongly =recommended in such patients. Failure to .recognize suppression
of the
hypothalamic-pituitary-adrenal axis may, in certain circumstances, result in
death. Finally,
during periods of stress or serious intercurrent illness (e.g., surgery or
serious infection) in
patients receiving or recently withdrawn from any form of megestrol acetate
therapy,
,consideration should be given to the use of empiric therapy with stress doses
of a rapidly
acting glucocorticoid.
If, at any time during the study, a patient manifests symptoms suggestive of
adrenal
insu$iciency, and subsequent laboratory evaluation reveals a significantly low
basal serum
cortisol level (<l0ug/dL) and/or stimulated serum cortisol level (<1811g/dL)
30 minutes after
ACTH administration, the patient should-be withdrawn from the study and the
study
medication discontinued. These symptomaticpatients should be treated
with'apprropriate
.replacement or stress -doses of glucocorticoid therapy (as should symptomatic
patients who
manifest clinical adrenal insufficiency at study termination or following
withdrawal of
megestrol acetate therapy). In addition, at study termination (when .protocol-
directed ACTH
stimulation testing is scheduled to be performed), if an abnormal basal serum
cortisol
-(typically accompanied -by an abnormal stimulated serum cortisol) is observed
in an
asymptomatic patient, replacement glucocorticoid therapy should be instituted
as well. In
either instance described above, serial ACTH stimulation tests should then be
perfonned at
appropriate intervals. When the basal serum cortisol level exceeds l0ug/dL,
daily
maintenance therapy can be discontinued. However, until the stimulated serum
cortisol level
Prepared by Quintiles, Inc.
110
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
'PAR Pharmaceutical CONFIDENTIAL Page 33 of 50
Final Clinical Protocol PAR-002 v.4 Verslon: 01 Nov 04
exceeds 18 ug/dL, empiric thera.py with stress doses of a rapid acting
glucocorticoid should
be:provided during stress or serious intercurrent illness and the patient
should carry a wallet
-card identifying his/her potential for adrenal -insufticiency.
I''I :0 ETHICS
1-1.1 Institutional 'Review'Board or:lndependent Ethics Committee
The Investigator will submit the protocol and informed consent for the
Institutional Review
Board or Tnstitutional Ethics Conimittee (IRB/IEC) :responsible for the
conduct of'human
trials at his or her site. In addition, the Investigator agrees to .provide to
Par (or designee) the
documentation of ethical review board (1RI.i/IEC) approval of the protocol and
the infornaed
consent document before the study may begin -at the investigative site(s). Any
member of the
ethical review board who is directly affiliated with this study as an
investigator or as site
personnel must abstain from the ethical review board'=s vote on the approval
of the .protocol.
The ethical reviewboard(s) will review the.protocol and any subsecluent
amendments to the
study prior -to implementation.
1Vlinimally, the Investigator will supply the following documents to the
IRB/IEC for their
review and approval:
= the protocol approved by Par (or designee)
= any amendments made to the protocol after the original approval
= the current Investigator's Brochure, package labeling and any updates made.
to
these documents during the course of the study
= informed consent document
The Investigator will forward written documentation of the IRB/IEC approval to
Par (or
designee) prior to shipment of any study medications.
The Investigator is also responsible for notifying the IRB/IEC .in a timely
manner of any
serious adverse events -(SAEs) reported in subjects enrolled at his or her
site as well as
relaying =any communication from Par (or designee) to the Investigator
advising the
Tnvestigator of SAEs reported at other sites.
:Prepared by Quintiles, Inc.
111
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR ;Pharmaceutical CONFIDENTIAL =Page 34 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
1 1.2 -Ethical Conduct of the Study
The 1'rincipal Znvestigator is -expected to conduct the study'in accordance
with the ethical
.;principles that have their origin in the Declaration of Helsinki and in a
manner consistent
with Good Clinical Practice (GCP), as well as adhering =to'local and federal
regulatory
:guid'elines.
11.3 Subject -Information and Informed Consent
The Investigator is responsible for ensuring that the patient understands the
risks and benefits
of participating in the study. This, includes answering any questions the
.patient may :have
-throughout.the study and sharing any new information that maybe relevant-to
the patient's
willingness to continue his or her participation in the trial =in a-timely
manner.
The :informed consent document will 'be used to explain the risks and
benefitsof study
participation to the patient in simple terms before the patient is enrolled
into the study. In
.addition, the informed consent documents that the patient is satisfied with
his or her
:understanding of the risks and benefits of participating in the study and
desires to participate
in the study. The informed consent should also clarify the subject's right to
privacy .in
relation to the protection of personal health information as a research
subject.
'The investigator is responsible for ensuring that the informed consent given
to each patient or
legal representative is approved by the IRBIIEC and is specific to this study.
This
responsibility includes obtaining the appropriate signatures and dates on the
informed
:consent-document -prior to the performance of any protocol procedures,
including screening,
and prior to the administration of study drug.
11.4 Protocol Amendments
Any amendment to the protocol must be approved by Par (or designee) and the
xesponsible
IRB/IEC at investigational site prior to implementing any change to the
protocol.
Prepared by QuinCiles, Inc.
112
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL 'Page 35 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
1-2.0 STUDY ADMINiST:RATION
.12.1 :C.Iinical Monitoring
In order to assure the quality of the data, =the Principal Investigator agrees
to a11ow
representatives from Par (or.designee) to periodically review study documents,
audit clinical
data collected during the conduct of the trial, -and review source
documentation and drug
taccountability records according to .GCP .guidelines. Clinical monitoring may
also include
regulatory authorities .if indicated. Monitoring personnel, bqund by
professional secrecy, will
not disclose any ,protected health information or personal medication
:information outside of
fitlfilling their responsibilities to ensuring the integrity of the data.
12.2 Data 'Quality Assurance
:Quality assurance methods will .be used to ensure the quality and integrity
of the data. These
methods include the following activities associated with the conduct of the
study:
= provide instructional material to the study sites, as appropriate
= sponsor a start-up training session to instruct the investigators and study
coordinators This -session will give instruction on the protocol, the
completion of
the CRFs, and study procedures.
= make periodic visits to the study site
= be available for consultation and stay in contact with the study site
personnel by
:mail, telephone, and/or fax
In addition, -data:quality assurance practices will include standardized
practices according to
-the Standard bperating Procedures of the Data Management team at Quintiles,
Inc..(the
responsible contract research organization) including, but not limited to the
following:
periodic auditing of data at clinical site against source documents, double
data entry (or other
:duplicative :method of verification), periodic audits of the electronic
dataset of clinical data
against Case ReportForms, programmatic data checks for inconsistencies and
resolution of
.outstanding -data queries and clarifications prior to database lock.
Electronic centralized laboratory. data will be stored at the central
laboratory facility and
transferred to the Data management team at the appropriate time.
Prepared by Quintiles, Inc.
113
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR'Pharmaceutical CONFIDENTIAL -Page 36 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
12.3 Retention of Study Records
'12.3.1 Case Report,Forms
Case Report Forms (CRFs) will be supplied by Quintiles and should =be'handled
in
accordance with instructions from the Quintiles staff.
The Investigator is responsible for maintaining adequate and accurate source
documents and
CRFs. CRFs have been designed to fecord all observations and other data
pertinent to the
=clinical investigation and should be filled out completely by the
Investigator'(or designate
study site representative). All CRFs should be completed in a neat, legible
manner to ensure
=accurate interpretation of the data. Black ball-point pen should be used to
ensure.the clarity
of reproduced copies of all-CRFs.
Tncorrect entries should be crossed with a single 1ine. Corrections must be
made adjacent to
.the item to be altered, initialed and dated with the reason for the
correction if necessary, by
an authorized member of the investigational site team (eg, Investigator or
designee).
Overwriting of this information or use:of liquid correcting fluid is not
allowed.
The'CRFs are reviewed, signed and dated by the Investigator.
-Once the site monitor has verified the contents of the completed CRF pages
against the
source data, the duplicate pages will be collected and forwarded to Quintiles
for data entry.
Data queries may. be raised if the data is unclear or contradictory; these
queries must be
addressed by the Investigator.
12.3.2 Recording, And :Retention of Source Data
Source data collected during this study will include, but is not restricted
to: subject's medical
file, subject diaries, original laboratory reports, or any other medical
records generated
during the time of the study conduct.
All clinical data recorded in the CRF must also be recorded in the subject's
medical notes.
The monitor (auditors, IEC/IltB or regulatory inspectors) will check the CRF
entries against
the source documents. The consent form will include,a statement by which the
subjects
:allow the monitor/auditor/inspector from the IEC/IRB or regulatory authority
access to
source data (e.g., subject's medical file, appointment:books, original
laboratory xeports, X-
rays, etc.) which substantiate information recorded in the case report forms.
As described in the ICH GCP Guidelines, 'essential documents', include CRFs,
source
docuinents, consent forms, laboratory test results, and medication inventory
records. These
records should be retained by the Investigator until: 1) at least 2 years
after the last approval
Prepared by Quintiles, Inc.
114
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical =CONFIDENTIAL Page 37 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
of a marketing application in an ICH region and there are no pending or
contemplated
marketing applications in an ICH region or 2) at least 2 years have elapsed
since the formal
=discontinuation of clinical development of the investigational product. These
documents
should be retained for a longer period, however, if required by the applicable
regulatory
requirements or =by an agreement with Par. The investigator should obtain
written permission
fxom Par prior to= the destruction of any study document.
These,records will =be made available at reasonable times for inspection and
duplication, if
required, =by a properly authorized representative of the United States Food
and Drug
Admioistration in accordance with 21 CFR 312.68 or other regulatory
authorities.
12.3.3 ~Study 'Drug Accountability
All study =drug required for cornpletion of this study will be provided by Par
(or designee).
The recipient will acknowledge receipt of the .drug indicating shipment
content and
condition. Damaged supplies will be -replaced. Accurate records of all study
drugs
:dispensed, used and returned will be maintained.
12.4 Confidentiality
Data collected during this study may be used to support the development,
registration or
marketing of megestrol acetate oral suspension NCD formulation. All data
collected during
=the study will be controlled by Par (or designee) and will abide by all
relevant data protection
-laws and regulations according to the standards of the participating
countries. After subjects
have consented to take part in the study, their medical records and the data
collected during
the study will be reviewed by representatives of Par =(or designee) to confirm
that the data
collected are accurate for analyzing the results. These records and xesultant
data may
additionally be reviewed by auditors, interested commercial parties or by
regulatory
authorities. The subject's name, however, will not be disclosed outside the
study site.
Subject data, outside -of t-he investigational site source records, will only
be identified by a
unique subject.number.
The handling of confidential study data will be in compliance with the
guidelines established
by the standards of the participating countries =such as the Health Insurance
Portability and
Accountability Act of 1996 =(HIPAA), Final Rule, published August 17, 2000 for
sites in the
United States.
12.5 =Publication,Poiicy
All manuscripts, abstracts or other modes of presentation arising from the
results of this
study must be reviewed and approved in writing by Par, in advance of
submission. The
Prepared by Quintiles, Inc..
115
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical WNFIDENTIA! Page 38 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
review is aimed at protecting Par's pre-existing.propriety information and
commercial
interests. Further information regarding publications shall be governed in the
agreement
.signed between =each indidiiiual center and PAR.
The Investigator will submit =an.y. :proposed=publication relating to
or:referring to the results of
-this study to Par for review at least sixty (60) days =prior to'the proposed
date of submission
for publication. Par will complete =its review of the proposed publication
within sixty (60)
days of receipt and, upon Par's written,request, the proposed publication will
be delayed up
to an additional sixty (60) days to enable Par to secure adequate intellectual
property
:protection of confidential information that would be affected by the proposed
publication.
No publication of confidential =information shall be made without Par's prior
written consent.
No publication shall be made ;prior to completion .of the multi-site study.,
If no multi-site
publication has-been made six (6) months after completion of this study, the
Investigator may
publish in accordance with the terms of the signed Investigator Agreement
between the
Investigator and Par. Par's written consent for the Investigator to ;publish
data from this
study will not be unreasonably withheld. The content of the publication,
whether written or
oral, will be given to Par to allow sufficient time for considered comment.
fihe object of this
policy -to -ensure consistency between data submitted to.R.egulatory
Authorities and that
-appearing :in publications.
Prepared by Quintiles, Inc.
116
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 39 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
1.3Ø SIGNATURE(S) OF INVESTIGATOR(S)
Thave read this Par Pharmaceutical protocol No. PAR-002:
'Title: A Randomized, Open-labeled, Pilot Study Comparing'Weight Gain =in
Adults with AIDS-related Wasting Giv,en Either 1Vlegestrol Acetate NCD
Formulation Oral Suspension or Megestrol Acetate Oral Suspension
(Megace ).
I have full.y -discussed the objectives of this trial and the contents of this
protocol with the
Sponsor's (Par Pharmaceutical) representative.
I=understand that -the information in this protocol is confidential and should
not be disclosed,
other than to those directly involved in the execution or the ethical xeview
of the study,
without written authorization from Par. It is, however, permissible to provide
information to
a subject =in order to obtain consent once JRB/IEC approval is obtained.
I=agree to conduct this trial according to this .protocol and to comply with
its requirements,
subject to .ethical :and safety considerations and .guidelines, and to conduct
the trial in
accordance with ICH guidelines on GCP and=with the applicable regulatory
requirements.
I understand that Par may decide to suspend or prematurely terminate the trial
at any time for
whatever reason; such a decision will be communicated to me =in writing.
Conversely, should
I decide to withdraw from execution of the trial I will communicate my
intention
=immediately -in writing to Par.
Investigator Signature: Date:
Name and Title ofTnvestigator: Investigational Site:
Investigator Address:
Prepared by Quintiles, Inc.
117
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
-PAR Pharmaceutical CONFIDENTIAL Page 40 of'50
-Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
14.O.REFERENGES
1. Centers for Disease Control and'Prevention. 1993'Revised classification
system for HIV
infection and expanded surveillance case definition forAIDS among adolescents
and
-adults. MMWR 1992;41.(RR-17):1-19
2. Coodley GO, Loveless MO, 'Nlerrill TM. The HIV Wasting Syndrome; a review.
J Acquir
Immune Defic Syndr. 1994;7:681-694
3. Ott M, Lembcke'B, Fischer H, Jager R, Polat H, Geier H, -et la. :Early
changes of body
composition in human immunodeficiency virus-infected patients; tetrapolar body
.impedance analysis indicates significant mainutrition. Am J Clin IVutr
1992;15-19.
4. Tang AM. Weight 'loss, wasting, -and survival in HIV=positive patients:
current strategies.
AIDS Read. 2003;13(12 Suppl):S23-27.
5. Wheeler,DA. Weight loss and disease -progression in -HIV infection. AIDS
Read.
1999;9(5):347-353.
6. Tang AM, Forrester J, Spiegelman D, Knox TA, Tchetgen E, Gorbach S. Weight
loss
=and survival in.HIV-positive patients in the era of hi,ghly active
antiretroviral therapy. J
Acquir Immune Def Syndr. 2002:31(2):230-236.
7. Wanke CA, Silva M, Knox TA, Forrester J, Speigelman D, Gorbach SL. Weight
loss and
wasting remain common complications-in individuals infected with human
-immunodeficiency virus in the era of highly active antiretroviral therapy..
Clin Infect Dis.
2000;31-:803-805.
8. Grunfeld C, Pang .M, Shimizu L, et al. Resting energy expenditure, calorie
intake and
short term weight change in immunodeficiency virus infection and acquired
-immunodeficiency syndrome. Am J Clin Nutr. 1992;55:455460.
.9. .McCallan'DC, Nobel C, :Baldwin C et al. Calorie -expenditure and wasting
in human
immunodeficiency virus infection. New Engl J Med 1995;333:83=88.
10. Hellerstein MK, GruunfeldC, Wu K, et al. Increased de novo hepatic
lipogenesis-in
-human immunodeficiency virus infection. J Clin Endocrinol Metab.
1993;76:559=565:
11. Mulligan X, Grunfeld C, Hellerstein MK-et al. Anabolic effects
of,recombinant'human
-growth hormone in patients with wasting *associated human immunodeficiency
virus
-infection. J -Clin Endocrinol Metab.1993;77:956-962.
12. Dibbs AS, Dempsey MA, Ladenson PW, Polk .BF. Endocrine disorders in men
infected
with human immunodeficiency virus. Am J Med 1988'84:611-616.
13. Corcoran C, Grinspoon S. Treatments for wasting in patients with acquired
immunodeficiency syndrome. New Engi J Med 1999;340:1740-1750.
Prepared by Quintiles, Inc.
118
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
;PAR Pharmaceutioal CONFIDENTIAL 'Page 41 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
14. MaEvoy GK, ~ed. AHFS Drug Information 2001, American Society of'Health-
System
Pharmacists, Bethesda, MD. Published by the American Society of -Health-System
Pharmacists; 2001:1050-1052.
15. =Loprinzi C,'Kugler JW, Sloan JA, Malliard JA, Krook JE, V1lilwerding MB,
et al.
Randomized comparison of inegestrol acetate versus dexamethasone versus
-fluomesterone for the treatment of cancer anorexia/cachexia. J Clin Oncol
1999;17.(10):3299-3306.
16. Jatoi A, Windschitl HE, Loprinzi CL, Sloan JA, Dakhil SR, Mailliard JA, et
al. Dronabinol
versus megestrol acetate versus combination therapy for cancer-associated
anorexia: a
=North Central=Cancer Treatment Group Study. J Clin Onco12002;20(2):567-573.
17. Aisner J, Parnes H, Tait'N, Hickman M, 'Forrest A, Greco =FA,
Tchekmedy'ian NS.
Appetite stimulation and weight.gain with megestrol.acetate. Sem Oncol
1990:17(6):2-7.
18.,Oster:MH,'Enders'SR, Samuels SJ, Cone LA, flooton TM,'Browden HP, Flynn
NM.
Megestrol acetate =in =patients with AIDS and cachexia. Ann Intern Med
1994;121(6):400-
-408.
19. Von Roenn JH, Armstrong'D, =Kotler DP, Cohn DL, Klimas -NG, Tchekmedyian
NS, .et:al.
Megestrol acetate :in :patients with AIDS-related cachexia. Ann Intern Med
1994;121(6):393-399.
20. Tchekmedyian NS. -Hickman -M, tHeber D. Treatment of anorexia and weight
loss with
.megestrol acetate =in patients with cancer or acquired immunodeficiency
syndrome. Sem
-Oncol 1991;18(1 Su ppl 2):35-42.
21. =Megace Oral Suspension (megestrol acetate) Product Labeling. Bristol-
Myers Squibb
Company, Princeton, NJ, =Revised 2002.
~22. -Mann M, Koller E, ~Murgo =A, Malozowski S, =Bacsanyi J, -Leinung M.
Glucocorticoidlike
.activity of megestrol. A summary of Food-and Drug Administration -experience
and
review of the 'literature. Arch lntern Med 1997;157(15):1651-1656.
Prepared by Quintiles, Jno.
119
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutiaal' CONFlDENTIAL Page 42 of 50
Final Clinical Protocol =PAR-002 v.4 Version: 01 Nov 04
APPENDIX A: METROPOLITAN HEIGHT AND WEIGHT TABLES
TABLE 1
1999 -METROPOLITAN HEIGHT AND WEIGHT TABLES FOR
MEN AND WOMEN
According to Frame, Ages 25-59
WOMEN
Weight in Pounds (In Indoor Clothing)*
HEIGHT SMALL MEDIUM LARGE
(In Shoes)+ FRAME FRAME FRAME
Feet Inches
4 10 102-111 109-121 118-131
4 17 103-113 111-123 120-134
'5 0 104-115 113-126 122-137
'5 1 106-118 115-129 125-140
2 108-121 118-132 128-143
5 3 111-124 121-135 131-147
5 4 114-127 124-138 134-151
5 5 117-130 127-141 137-155
5 6 120-133 130-144 140-159
5 7 123-136 133-147 143-163
.5 '8 126-139 136-150 146-167
5 .9 129-142 139-153 149-17.0
5 10 132-145 142-156 152-173
5 11 135-148 145-159 155-176
6 0 138-151 148-162 158-179
Prepared by Quintites, Inc.
120
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 43 of 50
Final Clinical Protocoi'PAR-002 v.4 'Version, 01 Nov 04
TABLE I
1999:METROPOLITAN =HEiGHT AND WEIGHT TABLES FOR
MEN AND WOMEN
According to'Frame, Ages 25-59
MEN
Weight in Pounds (in Indoor Clothing)*
HEIGHT SMALL MEDIUM LARGE
(In =Shoes)+ FRAME FRAME ;FRAME
Feet Inches 5 2 128-134 131-141 138-150
3 130-136 133-143 140-153
5 4 132-138 135-145 142-156
- --------- -----
5 5 134-140 137-148 144-160
5 6 136-142 139-151 146-164
5 7 138-145 142-154 149-168
5 8 140-148 145-157 152-172
5 9 142-151 148-160 155-176
5 10 144-154 151-163 158-180
5 11 146-157 154-166 161-184
6 0 149-160 157-170 164-188
6 1 152-164 160-174 168-192
6 2 155-168 164-178 172-197
..6 3 158-172 167-182 176-202
6 4 162-176 171-187 181-207
Prepared by Quintiles, Inc.
121
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR =Pharmaceutical CONFIDENTIAL Page 44 of 50
=Final Clinical'Protocol PAR-002 v.4 Version: 01 Nov 04
APPENDIX B: .HAMILTOWDEP.RESSION Ri4TING SCALE
,These questions are representative of the questions asked on the GRYD-HAMD-17
Structured
Interview Guide. Please refer to the GRID-HAYvID-17 Structured Interview Guide
=in the
Study Procedures for specific infor.mation,regard'vng the assessment.
'Patient's Name (or Study :Identifier)
Date of Assessment
;For each item, write the Correct=number on the !line next to -the item. (Only
one
,response ;per item)
1. DEPRESSED:MOOD (Sadness, hopeless, =helpless, worthless)
0= Absent
1 = These feelings:states Indicated only on questioning
2= These feelings states spontaneously reported verbally
3=-Communicates=feeling states non-verbally-ie, through facial expression,
posture, voice
and tendency to vveep
4= Patient reports VIRTUALLY ONLY these feeling states in his spontaneous
verbal and
non-verbal communication
2. =FEELINGS OF GUILT
0= Absent
1= Self reproach, feels he has let people down
2= Ideas of guilt or rumination over past errors or sinful deeds
3= Present illness is a punishment. Delusions of guilt
4= Hears accusatory or denunciatory voices or experiences threatening visual
hallucinations
3. SUICIDE
.0= Absent
1= Feels life is not worth living
2= Wishes he were dead or any thoughts of possible death to self
3= Suicidal ideas or.gestures
4= Attempts at suicide (any serious attempt rates 4)
4.:INSOMNIA-E,ARLY
.0==No difficulty:falling asleep
1= Complains of occasional difficulty falling asleep- i.e., more thari hour
2= Complains ofnightly difficulty falling asleep
5. 4NSOMNIA MIDDLE
0=.No difficulty
1= Patient complains of being restless and disturbed during the night
2= Waking during the night-any.getting out of bed'rates 2 (except for voiding)
Prepared by Qc.intiles, Inc.
122
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAt, = Page 45 of 50
,Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
6. INSOMNIA LATE
0= No difficulty
1= Waking in early hours of the moming =but goes back to sleep
2= Unable to fall asleep again if he gets out of bed
7. WORK AND ACTIVITIES
0= No.difficulty =
1= Thoughts and feelings of incapacity, fatigue or weakness related to
activities; work or
hobbies
2= Loss of Interest in activity; hobbies orwork-either.directly reported
by.patient or indirect
by listlessness, indecision, and vacillation (feels he has to push self to do
work or
activities
=3= Decrease in actual time spent in activities or decreased productivity
4= Stopped working because of present illness
S. RETARDATION: =PSYCHOMOTOR.(Slowness of thought and speech; impaired ability
.to concentrate; decreased motor activity)
.0= =Normal speech and thought
1= Slight retardation at interview
2= Obvious retardation at interview
.3= Interview difficult
=4= Complete stupor
9. AGITATION
.0= None
1= Subjective tension and irritability
2= Worrying about minor matters '
3= Apprehensive attitude apparent in face or speech
4= Fears expressed without questioning
10. ANXIETY (PSYCHOLOGICAL)
0= No difficutty
1= Subjective tension and irritability
2= Worrying over minor matters
3= Apprehensive attitude apparent in face and speech
4= Fears expressed without questioning
11. ANXIETY SOMATIC: Physiological concomitants of anxiety (i.e. effects of
autonomic
overactivity, "butterflies', indigestion, stomach cramps, belching, diarrhea,
palpitations,
hyperventilation, paresthesia, sweating, flushing, tremor, headache, urinary
frequency). Avoid
asking about possible medication side effects (i.e., dry mouth, constipation)
0= Absent
1= Mild
2= Moderate
3= Severe
4=1nr,apacitating
Prepared by Quinti.les, Ina
123
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
'PAR Pharmaceutical CONFIDENTIAL 'Page 46 6f'50 =
'Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
12. SOMATIC SYMPTOMS (GASTROINTESTINAL):
0= None
1= =Loss of appetite but eating without encouragement from, others. Food
intake about
tnormal.
~2='Difficulty eating without urging from others,'Marked reduction of appetite
and food
;intake
13. SOMATIC SYMPTOMS GENERAL
=0= None
,1= Heaviness in limbs, back, or head. Backaches, headaches, muscle aches.
Loss of
energy and fatigability
2= Any clear cut symptom rates 2
14. =GENITAL =SYMPTOMS (symptoms such as loss of libido; impaired sexual
performance; menstrual disturbances)
=:0= Absent
1=,Mild
'2=,Severe
15. -HYPOCHONDRIASIS
.0=.Not present
=1='Self absorption (bodily)
2= Preoccupation with health
.3=~.Frequent complaints, requests for help, etc.
4==Hypochondriacal delusions
'16.:LOSS OF WEIGHT
(A. When rating by history)
'0= No weight loss
1==Probably weight associated weight loss with present illness
2= =Definite (according to patient) weight loss
3=Not assessed
1T. INSIGHT
0= Acknowledges being depressed and ill
1=Acknowledges illness but attributes cause to bad food, climate, overwork,
virus, need
for rest, =etc.
2= Denies being ill at all
18. -DIURNAL VARIATION
A. Note whether symptoms are worse in morning or evening. If NO diurnal
variation; mark
=none.
:O=.No variation
1= Worse in A.M.
2= Worse in P.M.
B. When present, mark severity of the variation. Mark "None" if NO variation
0='None
1= Mild
2= Severe
Prepared by Quintiles, bic.
124
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
iPAR Pharmaceutical CONFIDENTIAL Page 47 of 50
Final Clinical Protocol PAR-002 v.4 Version: 01 Nov 04
19. -DEPERSONALIZATION AND DtREALIZATION (Such as: Feelings of unreality;
+Nihilistic ideas)
0= Absent
1=,Mild
2= IVloderate
'3==Severe
4=lncapacitating
=20. !PARANOID SYMPTOMS
:0= None
1= Suspicious
2='Ideas =of reference
.3= Aelusions of reference =and,persecution
'21. =OBSESSIONAL'AND'COMPULSIVE SYMPTOMS
i0= Absent
1= Mild
2= Severe
Adapted from Hedlung and Viewig,The Hamilton rating scale for depression.
Journal.of
~Operational Psyclz iatry, 1979; 10(2): 149-165.
' 1997 Glaxo Wellcome Inc. All rights reserved-
Prepared by Quintiles, Inc.
125
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
.PAR'Pharmaceutical 'CONFIDENTIAL Page 48 of 50
'Finat Clinical Protocol PAR-002 v.4' 'Version: 01 Nov 04
APPENDIX C: THE BRISTOL-MYERS ANOREXIA/CACHEXIA.RECOVERY
'INSTRUMENT (BACRI)
:INSTRUCTIONS (TO BE FILLED OUT BY THE PATIENT)
Below are several -questions pertaining to your well-being. To ansWer a
question, place an "X" on the
'line below each question at the point which best shows us what is happening
to you at present. (The
-description "normal for me" in some cases means what was normal prior to
illness). =in some
questions, if no=change has occurred, mark the midpoint of the line to so
signify.
'Example:'HOW DO'YOU FEEL ABOUT YOUR.ABILITY TO CONCENTRATE?
Cannot
=Concentrate ~ Can=Concentrate
=At A!)
This "X" shows that you are able to =concentrate fairly well.
1. Since you have begun treatment with the test drug, do you feel that any
change in weight has had
a significant;impact on your health?
Health
Worsened Health Improved
2. Are you more or less concemed about your weight now than when you started
treatment?
Much Less
Concerned Much More
Concemed
3. To what extent has your appearance changed =since.treatment started?
Much Worse (
I- Much Better
Prepared by Quintiles, Inc.
126
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
PAR Pharmaceutical CONFIDENTIAL Page 49 of 50
=Final Clinical Protocol 'PAR-002 v.4 Version: 01 Nov 04
4. Based on comments from friends,,coworkers and loved ones, how do you fell
your appearance has
changed since the start of treatment?
Favorably Unfavorably
5.'To what extent has your appetite changed since the start of treatment?
tMuch Worse Much Better
,6. Do you enjoy eating -more -or'Iess'than'before treatment began?
Much Less ;I t I Much More
7. 'Since the beginning of treatment, .do you feel better or worse overall?
Auch Worse I --I Much B"etter
=I ~
-8. Do you think this treatmenthas been of benefit to you?
'Not at All Very Much
9. Since the beginning of treatment, has your quality of life become better or
worse?
:Much Worse Much Berier
Taken -from: Celia -DF, VonRoenn J, Lloyd S, =Browder HP. The Bristol-Myers
Anorexia/Cachexia
Recove .ry Instrument (BACRI): a brief assessment of patient's subjective
response to treatment for
anorexia/cachexia. .Qual of Life Res. 1995;4:221-231.
Prepared by Quintiles, Inc.
127
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
=PAR =Pharmeceutical CONFIDENTIAI. -Page 50 of 50
'Final Clinical Protocol PAR-002 v.4 Version: 04 Nov 04
APPENDIX D: =PHARMACOKINETIC'SAMPLE HANDLING
The following procedures will be followed for each pharmacokinetic assessment
(including
trough samples).
1) Samples of 5=mL of venous blood will be collected in a sterile manner into
an evacuated
EDTA K3 tube for each sample.
2) 'Sample will be placed immediately in a containei with a mixture of =ice-
water at a
=temperature of approximately 4 C until.separation.
3) Samples must'be,centrifuged within 50 :minutes of sampling time.
:4) Samples must be centrifuged =at 3000rpm for 10 minutes =in a refrigerated
centrifuge
;(4 C). There =may be no moie than 60 minutes from the beginning of
centrifitgation until
the aliquots are separated.
5) Resulting plasma as aliquoted into at'least two separate samples into
propylene tubes of at
least 5 mL volume with -no less than 1.2 mL.per aliquot. Each aliquot must be
labeled
-with the subject identifier, date, time of draw and sample time (eg, pre-,
0.5 h, 1.0 h, etc.).
The -samples should be stored in separate boxes as they will be shipped to the
pharmacokinetic analysis'lab in 2 separate shipments. Each sample must have an
identical label with the pertinent information clearly legible.
:6) Aliquots should be stored as soon as possible in an upright position in a
freezer of 20 C
or lower. The freezer should be maintained, and monitored in case of power
failure.
7) Samples for an individual subject.should be retained by the investigational
laboratory
until the second pharmacokinetic study for that :patient is completed. At that
time, the
investigator should send one set of samples from each of the two
pharmacokinetic studies
for a-given subject onenough dry ice to ensure the samples remain frozen for
at least 72
hours. More than -one subject's samples may be shipped ;in a single shipment;
bowever,
:ino shipment shoitld contaiii all of one patient's samples of any PK test
date. Once the
bioanalytic lab confirms receipt of the first set of samples for a given
patient, then the
:second =set ofsamples may be sent.
:=S) Samples should be sent via=overnight courier on Monday or Tuesday to:
SFBC Anapharm
2050, =boul Rend-Levesque Ouest
Quebec (Quebec) Gl V 2K8
CANADA
To the attention of M. Louis-Phillipe Beauregard, Sample Controller
Coordinator
Prepared by Quintiles, Inc.
128
CA 02613466 2007-12-20
WO 2007/002315 PCT/US2006/024349
[0217] It will be apparent to those skilled in the art that various
modifications and
variations can be made in the methods and compositions of the present
invention without
departing from the spirit or scope of the invention. Thus, it is intended that
the present
invention cover the modifications and variations of this invention provided
they come
within the scope of the appended claims and their equivalents.
129