Note: Descriptions are shown in the official language in which they were submitted.
CA 02613483 2010-01-18
HYDROGEN PRODUCTION USING ELECTROCHEMICAL REFORMING AND
ELECTROLYTE REGENERATION
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of the filing
date of United States Provisional Patent Application No.
60/693,316 filed June 23, 2005,
BACKGROUND OF THE INVENTION
[0002] This invention relates to processes for producing
hydrogen gas.
[0003] There is a need for new low cost routes to hydrogen
production. Hydrogen is widely used as an intermediate in the
petrochemical industry. The largest single use is in the
refining of crude oil into fuels. Hydrogen is also an
intermediate in the production of ammonia for fertilizers.
Many companies are working on hydrogen fuel cells as a
replacement for gasoline or diesel fueled vehicles. If this
fuel cell development is successful, large additional supplies
of hydrogen will be needed for hydrogen to power fuel cell
vehicles.
[0004] Gas phase steam reforming of natural gas is commonly
used to make low cost hydrogen. Methane is the majority
component in natural gas. However, the kinetics of methane
steam reforming are slow. As a result, this reaction is
conducted at high temperatures, typically 600 C-900 C. An
equation for the steam reforming of methane is given in
reaction (1).
CH4, + 2 H2O (vapor) CO2 + 4 H2 (1)
[0005] Other oxidizable fuels have also been steam reformed
to make hydrogen. Equations similar to (1) can be written for
the steam reforming of other hydrocarbons, oxygenates and
various other oxidizable fuels. For example, the steam
reforming of methanol is given by reaction (2). Methanol
1
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
steam reforming proceeds at a much faster rate than methane
steam reforming. Typical gas phase methanol steam reforming
temperatures are 250 C-350 C.
CH3OH (vapor) + H2O (vapor) C02 + 3 H2 (2)
[0006] The typical steam reforming product, known as
synthesis gas, is a mixture of hydrogen, carbon monoxide,
carbon dioxide and steam. In most applications, the synthesis
gas is passed sequentially through one or more "water gas
shift" reactors, where most of the carbon monoxide is reacted
with steam making additional hydrogen and carbon dioxide via
reaction (3).
CO + H2O (vapor) C02 + H2 (3)
[0007] Finally, purification steps are used to remove the
carbon dioxide, carbon monoxide, steam and other impurities.
Hydrogen product purity requirements vary widely depending on
the final hydrogen usage application.
[0008] Current energy markets are in a state of flux. In
recent years, the supply of domestic natural gas has been
limited and its price has increased several-fold over historic
levels. Offshore natural gas is viewed as a low cost
alternative to expensive domestic natural gas. Unfortunately,
most offshore natural gas cannot be transported via gas
pipelines. Many companies are considering the use of tankers
to transport liquefied natural gas (LNG). LNG tankers are
much more expensive than petroleum tankers. Furthermore,
transport of LNG poses serious safety and liability issues in
the event of a spill in a waterway. Others view the shipment
of liquids such as methanol or dimethyl ether as viable
alternatives to LNG. Both fuels can utilize less expensive
tankers and in the event of a spill, these materials are not
subject to the same safety issues presented by a LNG spill.
If these fuels become widely available, their prices may fall
below that of domestic natural gas and these liquid fuels can
2
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
be attractive alternative feedstocks for the production of
hydrogen. Both fuels can be processed in the gas phase by
steam reforming. The resulting synthesis gas can similarly be
processed sequentially, as described above, using well known
water gas shift reactors and hydrogen purification processes.
[0009] An early patent in this field, Canadian Patent No.
787831 (June 18, 1968), P. Grimes et al., teaches a liquid
phase process for making hydrogen by reforming various
oxidizable fuels. Liquid phase reforming can be conducted in
various aqueous electrolytes but the reforming kinetics are
more favorable in alkaline electrolytes, especially
hydroxides. Conductive catalysts are used to promote
reforming reactions by activating electrochemical pathways.
Preferred catalysts are from the Group VIIIA transition
metals. Reaction (4) describes the overall liquid phase
reforming of methanol to produce hydrogen.
CH3OH (liquid) + H2O (liquid) CO2 + 3 H2 (4)
[0010] The patent discloses a batch process using a mixture
of water, an ionic conductive electrolyte, and an organic
compound (fuel) which react in the presence of an electronic
conductive catalyst, oxidizing the fuel and producing
hydrogen. The reactions are said to occur in the liquid phase
and are believed to proceed via electrochemical pathways.
Thus for convenience herein, this type of liquid phase
reforming in alkaline electrolytes is referred to as
electrochemical reforming (ECR). Alcohol and a wide range of
organic fuels, including biomass, are disclosed.
High-pressure hydrogen production is disclosed and hydroxides
are described as preferred electrolytes.
[0011] Recent patents to Cortright et al., U.S. Patent Nos.
6,964,757, 6,699,457, and 6,964,758 and published U.S. Patent
Application 20050207971, and Reichman et al., U.S. Patent Nos.
6,890,419 and 6,994,839 and published U.S. Patent Application
3
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
20050163704 are similar in many respects to the disclosure in
Grimes. These include liquid phase reforming of alcohols,
sugars, biomass, hydrocarbons and various oxygenated
hydrocarbons to make hydrogen. These patents and published
applications disclose the use of various ionic conducting
electrolytes in the liquid phase and the use of conductive
metal catalysts from Group VIII and related catalysts. The
processes disclosed by Cortright et al., are conducted at
pH<10, where the by-product generally carbon dioxide leaves as
an impurity with the product hydrogen. U.S. Patent No.
6,994,839 and published U.S. Patent Application 20050163704
further disclose that alkali hydroxide electrolytes are
converted in a batch process to less active alkali carbonate
and bicarbonates and that the spent electrolyte can be
regenerated using a three step process: (1) make an alkali
carbonate solution via liquid phase reforming in alkali
hydroxide; (2) treat the alkali carbonate with a solid
alkaline earth oxide/hydroxide to regenerate the caustic while
precipitating an alkaline earth carbonate; and (3) use heat to
regenerate the alkaline earth carbonate to an oxide for re-use
in step (2). However, this approach is economically
unfavorable because significant heat is required to regenerate
alkaline earth oxide/hydroxide reactants resulting in
significant cost.
[0012] U.S. Patent No. 6,607,707 discloses that hydrogen
can be produced by combining an alcohol such as methanol with
a base and further in the presence of a catalyst such as a
transition metal and wherein the pH of the mixture is "at
least 10.3," but nothing specific is provided beyond that
limited disclosure.
[0013] U.S. Patent No. 6,890,419 discloses an
electrochemical cell consisting of anode and cathode
electrodes across which an external voltage is impressed and
4
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
employing acidic to strongly basic electrolyte solutions,
including the use of KOH up to 12M, in order to effect
production of hydrogen.
[0014] Liquid phase reforming provides desirable advantages
over conventional steam reforming because liquid phase
reforming offers electrochemical pathways that are not
accessible in conventional gas phase steam reforming.
Advantages include:
1. Higher efficiency than steam reforming;
2. Simplified product clean-up;
3. Eliminate the need for compressors; and
4. Provide a continuous, fully integrated process.
SUMMARY OF THE INVENTION
[0015] A process for producing hydrogen gas comprising
contacting in the liquid phase at least one oxidizable organic
substance in the presence of a mixture comprising at least one
conductive catalyst and an aqueous alkaline carbonate
electrolyte, wherein at least one bicarbonate composition
produced by reaction of the electrolyte is regenerated and the
at least one oxidizable organic substance comprises a
oxygenated hydrocarbon, for example methanol and/or dimethyl
ether. In a preferred embodiment the process for producing
hydrogen gas comprises contacting in the liquid phase at least
one oxidizable organic substance, such as methanol, in the
presence of a conductive catalyst and an alkaline electrolyte,
wherein: (A) hydrogen gas is generated in a reactor having a
top and bottom, wherein the at least one oxidizable organic
substance is introduced into said reactor at a point
substantially midway between said top and bottom; (B) an
alkaline electrolyte solution comprising at least one metal
carbonate is introduced into the reactor at a point
substantially at the top of the reactor such that the metal
carbonate solution and the hydrogen gas flow substantially
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
countercurrent to one another, thereby resulting in the
production of at least one metal bicarbonate composition; (C)
the at least one metal bicarbonate composition is regenerated.
In one regeneration process or step used in conjunction with
the hydrogen production sequence, the alkaline electrolyte is
regenerated using steam.
BRIEF DESCRIPTION OF THE DRAWINGS:
[0016] Figure 1 compares the energetics of steam reforming
with liquid phase reforming in various alkaline media.
[0017] Figure 2 shows a schematic of a batch reactor test
station used for liquid phase reforming catalyst evaluations.
[0018] Figure 3 shows a schematic of a continuous reactor
used for liquid phase reforming catalyst evaluations.
[0019] Figure 4 shows a schematic of a liquid phase
reformer integrated with a hot carbonate process for removing
carbon dioxide from product hydrogen and for regenerating
spent reformer electrolyte.
[0020] Figure 5 shows a schematic of a liquid phase
reformer in close-coupled co-current integration with a hot
carbonate process for removing carbon dioxide from product
hydrogen and for regenerating spent reformer electrolyte.
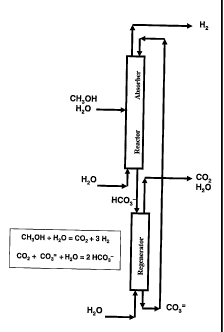
[0021] Figure 6 shows a schematic of a liquid phase
reformer in a close-coupled counter-current integration with a
hot carbonate process for removing carbon dioxide from product
hydrogen and for regenerating spent reformer electrolyte.
[0022] Figure 7 shows rate comparison data using a series
of catalysts for hydrogen generation from methanol in KOH
electrolyte.
[0023] Figure 8 compares hydrogen generation in fresh and
regenerated carbonate electrolytes using a Pt/C catalyst.
DETAILED DESCRIPTION
[0024] As described earlier, there is a need for a liquid
process that reforms oxidizable fuels to produce high-
6
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
pressure, high purity hydrogen without adding fresh alkaline
electrolyte. Unfortunately, prior art processes disclosing
liquid phase reforming are not economically feasible for at
least the following reasons:
(0025] The cost of making hydrogen in alkaline reforming is
effectively determined by the price of the caustic reagent
rather than by the price of the oxidizable fuel, when such
processes are conducted in alkaline electrolyte, for example
at pH>7, or pH>9, such as pH of about 7 to about 12. Although
strongly alkaline electrolytes (for example, pH>13) can
produce relatively pure hydrogen with low levels of carbon
dioxide and carbon monoxide, the product purity is achieved
because the carbon dioxide by-product reacts immediately with
the alkaline electrolyte, rapidly converting the electrolyte
into a bicarbonate-rich mixture thereby reducing the pH and
the reaction rate. In order to effectively and efficiently
conduct the process in an alkaline electrolyte, particularly a
continuous process at high pH, it thus becomes necessary to
add fresh caustic electrolyte along with fresh oxidizable
fuel. At current prices, the cost of makeup caustic, whether
in the form of a metal hydroxide or metal carbonate, far
exceeds the cost of the oxidizable fuel being reformed.
Consequently, the price of the caustic controls the product
price, which is unfavorable.
[0026] On the other hand, if the prior art processes are
conducted in acidic or neutral electrolytes (for example,
pH<7), the hydrogen product requires additional cleanup
processing. These cleanup processes add cost and reduce
process yield. In acidic or neutral electrolytes, the carbon
dioxide by-product does not react with the electrolyte. As a
result, a continuous process can be run in neutral or acidic
electrolytes without the need to add fresh electrolyte with
additional fuel. However, the reforming rates are lower than
7
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
in alkaline electrolytes, and, more importantly, the carbon
dioxide leaves the process in the product hydrogen, requiring
significant cleanup and the associated costs. In addition,
the carbon dioxide and hydrogen can react via the reverse of
reaction (3) above to produce carbon monoxide, and under some
conditions, hydrogen and carbon dioxide can further react to
form methane. All of these impurities must be removed in
subsequent water gas shift and/or purification processes. The
cost of cleanup processes and hydrogen yield losses can
thereby unfavorably affect the hydrogen production cost.
[0027] Alternatively, if the prior art processes are
conducted in mildly alkaline electrolytes (for example,
pH<10), the hydrogen product also requires additional cleanup
processing. In this case alkali carbonates are quickly
converted to bicarbonates, dropping the pH. As pH drops below
7, carbon dioxide again becomes a serious impurity in the
product hydrogen. A significantly higher pH can only be
maintained by the continuous addition of a caustic reagent.
In this case, the cost of making hydrogen is again controlled
by the more expensive caustic reagent rather than by the fuel.
[0028] As is described in detail below, the present
invention provides embodiments for continuous methods of
producing pressurized hydrogen from oxidizable materials via
liquid phase reforming in an alkaline carbonate electrolyte
and then regenerating the electrolyte so that the electrolyte
can be reused, thereby avoiding the negative impacts described
above. The oxidizable fuel is reacted with an aqueous
electrolyte in a high temperature liquid phase process in the
presence of a suitable catalyst. Furthermore, there are
several alternative and suitable ways of integrating the
reforming step with the electrolyte regeneration step.
[0029] The present invention relates to processes for
producing hydrogen gas and includes several alternative
8
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
embodiments. For example, one embodiment relates to batch or
continuous processes for the production of hydrogen gas from
oxidizable organic substances utilizing reactions with an
alkaline electrolyte and for the regeneration of that
electrolyte. In another embodiment, this invention relates to
the continuous production of high-pressure hydrogen gas
utilizing reactions with an oxygenated hydrocarbon such as a
lower alcohol, e.g., methanol, or an ether such as dimethyl
ether or mixtures of alcohols and ethers, with an alkaline
electrolyte and the regeneration of that electrolyte using
steam. For example, as will be described in greater detail
below, this invention reforms methanol and related fuels,
particularly in liquid systems, using liquid alkaline
electrolytes.
[0030] Oxidizable organic substances suitable for use in
the present invention include saccharides, celluloses,
starches, sugars, alcohols, ethers, carboxylic acids,
aldehydes, ketones, biomass and biomass derived materials and
mixtures of the foregoing. For example, suitable saccharides
include monosaccharides, disaccharides, oligosaccharides,
polysaccharides and mixtures thereof; suitable alcohols
include C1-C6 alcohols and mixtures thereof, particularly
methanol, ethanol and their mixtures; suitable ethers include
dimethyl ether, methylethyl ether, diethyl ether and mixtures
thereof. A particularly useful alcohol is methanol and a
particularly useful ether is dimethyl ether.
[0031] Alkaline electrolytes suitable for use in the
present invention include metal hydroxides, carbonates,
bicarbonates and mixtures thereof. Furthermore, suitable
metals of such electrolytes include alkali metals, alkaline
earth metals and mixtures thereof. Particularly suitable
metals of the alkaline electrolyte are selected from the group
consisting of sodium, lithium, potassium, cesium, rubidium and
9
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
mixtures thereof. Preferably, the electrolyte is present as
an aqueous solution. Typically the electrolyte concentration,
with reference to the reformer, is about 0.5 Normal (N) to
about 12 N (within solubility limits for the compound being
used); preferably about 1 N to about 8 N; more preferably
about 2 N to about 6 N; for example about 2 N to about 4 N;
such as about 3 N. Suitable concentrations of the electrolyte
or mixed electrolytes present in the process as a consequence
of regeneration may be less than the values expressed above,
but will be within the ranges expressed in order for the
process to operate efficiently. Fresh electrolyte may need to
be provided in order to replace gradual losses of electrolyte,
if any. In the absence of the physical loss of electrolyte
components, the process of the present invention can be
operated according to the disclosure provided herein,
particularly allowing for selection of a convenient and
suitable pH that can be achieved after regeneration. Thus,
even if, after regeneration, the process approaches but does
not return to the identical initial pH level selected at the
start of the process, the process can nevertheless be operated
so as to substantially approach the initial value and
thereafter operate on a continuing basis at close to the
initial value. Therefore, in the alternative, the pH level
achieved following regeneration can identified and such a
value can be selected as a convenient operation or target
value.
[0032] The aqueous metal carbonate electrolyte composition
suitable for use in the present invention optionally, but
preferably, includes a metal bicarbonate as well. The molar
ratio of metal carbonate to metal bicarbonate can be varied in
order to prepare a useful aqueous alkaline electrolyte
suitable for use in the present invention. Typically, the
feeds to the reactor comprise one or more streams of an
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
oxidizable organic substance or fuel and an aqueous
electrolyte, wherein the latter has a high molar ratio of
metal carbonate to metal bicarbonate. Useful ratios include
molar ratios of about 1 to about 1000; preferably about 2 to
about 100; more preferably about 3 to about 20. As the
reforming reaction proceeds, the carbonate /bicarbonate ratio
is reduced by the formation of reaction products, as discussed
below. Consequently, the mixture in the regeneration step
will have a lower ratio of carbonate/bicarbonate. Typically,
the feed in the regeneration step can have a molar ratio of
about 0.001 to about 5; preferably about 0.1 to about 3; most
preferably about 0.6 to about 1. The pH of the electrolyte
composition can be used as a convenient measure of the
bicarbonate ratio. However, since the presence of methanol in
the electrolyte can also affect the pH, the amount of methanol
that is present can also be compensated for in order to arrive
at an appropriate ratio in order to reach the target pH, or
vice versa. For convenience herein, suitable electrolyte
compositions of the present invention exhibiting a target pH
and comprising a high concentration of a metal carbonate
relative to bicarbonate, are referred to as carbonate-rich,
even if such compositions comprise less than about 50 mol%
metal carbonate. Alternatively, the pH of the reaction system
can be used as a gauge to establish that the electrolyte is at
a suitable concentration. Typically, the reforming reaction
is conducted at a pH of about 7 to about 14; preferably about
a pH about 8 to about 12; more preferably about 9 to about
11.5. As discussed further hereinbelow, other optional
additives and salts can be included for purposes other than
the reforming reaction, including corrosion control and
efficient bicarbonate regeneration. Furthermore, the scope of
the present invention includes optionally adding a minor
amount of a caustic reagent to the electrolyte, such as sodium
11
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
or potassium hydroxide, in order to achieve a specific pH
level, particularly higher levels of pH. In such instances,
it is expected that the amount of caustic will be quite small,
for example, from greater than zero to less than about 10 wt%
or less than 5 wt%, perhaps less than 3 wt% so that the
overall process will remain economically attractive even
though the caustic is not regenerated and a continuing
addition would be required to maintain the pH.
(0033] Catalysts suitable for use in the present invention
can be selected from the group consisting of compounds,
complexes, alloys and mixtures thereof, comprising at least
one metal selected from the Group VIII transition metals of
the Periodic Table of the Elements (the Groups of elements as
identified in the Periodic Table published in the CRC Handbook
of'Chemistry and Physics, 69th Ed., CRC Press, 1988). Suitable
catalysts can further comprise at least one metal. selected
from the metals of Group IB, Group IIB, Group VIIB, and
mixtures thereof. A particularly useful catalyst comprises
platinum alone or further comprising a metal selected from the
group consisting of copper, zinc and rhenium. Useful catalyst
concentrations in the reactor, expressed in volume%, are
typically about 0.1% to about 50%; preferably about 1% to
about 40%; more preferably about 2% to about 20%. In a
particularly useful embodiment, platinum is typically present
at a wt % concentration of about 0.5% to about 40%; preferably
about 1% to about 30%; more preferably about 5% to about 20%;
for example about 10% to about 20%. In another useful
embodiment, nickel is typically present at a wt% concentration
of about 2% to about 100%; preferably about 25% to about 100%;
more preferably about 40% to about 100%; for example about 60%
to about 80%. In this regard, wt% refers to the amount of'
catalytically active metal present in the catalyst component.
Thus, in the absence of a carrier, support or other inactive
12
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
component, the catalyst is present at 100 wt%. Additionally,
a useful form of the catalyst is where the catalyst is
supported on or in a conductive or non-conductive material
selected from the group consisting of metals, metal oxides,
silica, alumina, silica-alumina, zirconia, titania, ceria,
carbon, silicon carbide, silicon nitride, silicon boride and
mixtures thereof. Furthermore, useful forms of supports
include those selected from the group consisting of beads,
powders, coatings on extruded substrates or monoliths and
mixtures thereof.
[0034] The reforming reaction is typically conducted at a
temperature of about 150 C to about 350 C; preferably about
200 C to about 300 C; more preferably about 200 C to about
250 C; for example, the reforming process is suitably
conducted at about 200 C.
[0035] The reforming reaction is typically conducted such
that the hydrogen is generated at elevated pressure.
Typically the hydrogen pressure is about 1 atmosphere (atm) to
about 1000 atm; preferably about 5 atm to about' 500 atm; more
preferably about 10 atm. to about 100 atm. However, it can be
appreciated that useful pressures for operating the process of
the present invention can be determined by one skilled in the
art based on the use to which the hydrogen that is produced
will be put. Thus, useful pressures can be any pressure
including about 1 atm to about 1000 atm and all values and
ranges therebetween.
[0036] Typically, alkaline electrolytes react with the C02
in reformate by-product to yield carbonates and bicarbonates.
The preferred electrolytes are alkaline solutions of metal
carbonate/bicarbonate salts. An example of the overall
reaction is illustrated in equations (5) and (6) using
methanol and alkaline potassium compounds:
CH3OH (liquid) + 2 KOH 3 H2 + K2CO3 (5)
13
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
CH3OH (liquid) + 2 H2O + K2CO3 3 H2 + 2 KHCO3 (6)
[0037] Reforming methanol in alkaline electrolytes
increases the Free Energy driving force over conventional gas
phase or liquid phase reforming as shown in Figure 1. Heats
of reaction were calculated using standard Free Energy,
Enthalpy, and Entropy values as published in the CRC Handbook
of Chemistry and Physics (previously identified). (it is
understood that a negative value of Free Energy means that a
reaction is thermodynamically favorable and a positive value
of Free Energy is thermodynamically unfavorable.) Steam
reforming of methanol is favorable at room temperature but the
kinetics are sluggish. Methanol steam reforming is typically
conducted at about 250 C to about 350 C. The increased
temperature increases both the thermodynamic driving force and
the rate of reaction. Figure 1 shows that the Free Energy
driving force for methanol reforming in condensed water, in
KOH (at pH 14) or in K2CO3 (at pH 12) is much more favorable
than methanol steam reforming, even at much higher
temperatures. Without wishing to be bound by theory, it is
believed that this additional Free Energy advantage arises
from an acid-base reaction between carbon dioxide, a weak
acid, and KOH or K2CO3, strong bases. This supports the
experimental finding that methanol reforming in aqueous and
alkaline electrolytes can be run at lower temperatures than
those typically needed for methanol steam reforming.
[0038] Based on heating values of the fuel and product, the
theoretical conversion efficiency for methanol reforming under
these conditions is greater than 100%. This is a significant
advantage. Reacting water with methanol produces 3 H2
molecules. Lower Heating Values (LHV) of methanol and H2 are
638 kJ/mole and 242 kJ/mole, respectively. Comparing the
heating value of fuel and product gives a theoretical
conversion efficiency of 114%. The theoretical efficiency
14
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
using Higher Heating Values (HHV) is 119%. HHV and LHV are
defined in standard engineering and reference texts as
follows: LHV is the energy liberated by the complete
combustion of a substance where "the water produced is in the
gas phase." In contrast, HHV is "the corresponding value when
the water is in the liquid phase. . . . The difference
between the two corresponds to the heat of vaporization of the
water formed." (Smith et al., "Introduction to Chemical
Engineering Thermodynamics," McGraw Hill, Second Ed., p. 143,
1959) The additional energy, resulting in -efficiencies
greater than 100%, arises from endothermic heat that is
supplied to drive the reforming reaction. Methanol is not
unique regarding reforming efficiencies of greater than 100%,
the "theoretical" efficiency for methane steam reforming is
even higher. However, the slow reaction kinetics for methane
steam reforming requires supplying endothermic heat at
temperatures above 600 C. In contrast, alkaline reforming
according to the present invention can be conducted at
temperatures as low as about 150 C. In this temperature
range methanol and dimethyl ether are particularly preferred.
[0039] A significant advantage of the process of the
present invention is that the product obtained is nearly pure
hydrogen. In KOH, the gaseous carbon dioxide that is usually
produced by steam reforming remains in the liquid as K2CO3 and
in carbonates it remains as KHCO3. Thus the chemical activity
of the by-product carbon dioxide approaches zero, so that any
carbon monoxide production, via the reverse water gas shift
reaction (3), is greatly suppressed. Sequestration of the
carbon dioxide greatly simplifies the product clean up. In
contrast, carbon dioxide cleanup is a major expense in
conventional steam reforming of natural gas. In methane steam
reforming processes, carbon monoxide and carbon dioxide
cleanup often requires a two-stage water gas shift reactor
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
followed by a pressure swing adsorption unit, adding to cost
and increasing energy consumption.
[0040] Another advantage of present invention is that a
pressurized hydrogen product can be generated without the need
for gas phase compressors. The reactor operating pressure is
often dictated by the application to which the product
hydrogen will be put. Various applications can require
anywhere from near-atmospheric pressure hydrogen to hydrogen
at several hundred atmospheres or more. To operate above
ambient pressures, liquid feeds are pressurized up to reactor
operating pressure and then hydrogen is evolved at the
pressure of the reactor. In most applications, it is much
less expensive and more efficient to pressurize liquid feeds
than to use compressors to increase the pressure of the
hydrogen produced.
[0041] Figure 2 shows a schematic of a batch reactor for
conducting liquid-phase reforming. The reactor is a pressure
vessel which is initially charged with a mixture of an aqueous
liquid electrolyte, an oxidizable fuel, such as methanol, and,
typically, a catalyst. The pressure vessel is then sealed and
heated to the reaction temperature. Hydrogen (and carbon
dioxide when pH <7) is evolved as the reaction proceeds, as
described in reactions (5) and/or (6), and the pressure
gradually increases. Catalyst activity, for example, can be
evaluated as a function of pressure versus time (as well as
other process variables amenable to a batch experiment).
Eventually one reactant becomes exhausted, the reaction slows
to a halt and the pressure approaches a constant value. In
typical batch operations, even those on a commercial scale,
all reactants must be charged at the beginning of the process
and at the end of the reaction, the product and spent
reactants must be removed. During such start-up and shutdown
periods, no product is being produced and, consequently, such
16
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
processes are' not efficient, particularly compared to
continuous processes.
(0042] It is often preferable to conduct chemical processes
in a continuous rather than a batch processing mode and a
continuous process is a preferred embodiment of the present
invention. Typical advantages for continuous operation
include greater productivity and easier process control. For
example, batch processes can include frequent start-up and
shutdown conditions, and during these periods, the product
produced may not meet specifications or product may not be
produced at all. In addition, in a batch reactor, the
reaction mixture is constantly changing so that process
control is complicated requiring the need to monitor
conditions that necessarily change with time. In contrast, a
continuous process is productive essentially 100% of the time,
or at least until a decision is made to terminate the reaction
or an unforeseen complication arises. Consequently, less
off-specification product is produced. In addition, process
control variables in a continuous process typically do not
change with time, so that process control can be based on
fixed rather than changing control points. Since virtually
all large-scale hydrogen applications, such as the
petrochemical and fertilizer industries, require a continuous
feed of high-purity hydrogen, similarly such operations can
benefit greatly from a process that includes continuous
regeneration of a critical reaction component, the
electrolyte. In other words, if liquid phase reforming for
the production of hydrogen is to find its place in large-scale
industrial processes, then the improvements of the present
invention are an important aspect of that advance. Unlike the
processes of the prior art that are typically conducted in
batch operations, the present invention allows for continuous
operation and production of hydrogen with simultaneous
17
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
regeneration of the electrolyte in an efficient and
cost-effective process.
[0043] Figure 3 is a schematic illustration of an
experimental, continuous reactor for conducting liquid phase
reforming. In this arrangement, the reactor is filled with a
catalyst. It is continuously fed with a mixture of an
electrolyte and an oxidizable reactant or fuel, such as
methanol. Hydrogen is continuously evolved as the reactants
pass through the reactor bed. Typically the reactor is sized
so that the reactions shown in equations (5) and/or (6) reach
a high level of fuel conversion. Conversion is typically
about 20% to about 100%; preferably about 75% to about 100%;
more preferably about 95% to about 100%; most preferably about
98% to 100% conversion. In such a continuous process,
start-up and shutdown times are minimized and, as a result,
the product hydrogen can be produced at a constant rate almost
100% of the time. In the integrated, continuous process of
the present invention including regeneration, a regeneration
unit or section, including various configurations as further
described herein below, can be included after the
"accumulation tank" in Figure 3. Liquid from the accumulation
tank, comprising spent electrolyte, water and unreacted fuel,
for example, methanol is fed to a regenerator. Steam is fed
to the regenerator and by-product carbon dioxide leaves the
regenerator as well as a liquid stream comprising the
regenerated electrolyte, which is returned to the reformer
section of the process, for example, the feed tank as
illustrated in Figure 3. A continuous, integrated process is
not limited to such an arrangement of elements, as other
configurations will be apparent to those skilled in the art in
view of the teachings and disclosures herein.
[0044] It is known to remove carbon dioxide from methane
synthesis gas by using hot carbonate, promoted hot carbonate
18
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
or similar processes utilizing amines. For purposes of the
present invention, the phrase hot carbonate refers to an
aqueous composition comprising alkali carbonate and
bicarbonate salts at temperatures above ambient (ambient
typically considered to be about 20 C to about 25 C). Many
compounds have been used to increase the rate of carbon
dioxide absorption and desorption in hot carbonate processes,
such compounds sometimes referred to herein as promoting
agents. When a promoting agent is employed in the present
invention, at least one compound can be added to at least one
of the reaction mixture, the absorption step, the regeneration
step or a combination thereof. At least one compound can be
selected from the group consisting of various amines, such as
methyl ethyl amine, alcohol amines, hindered amines, as well
as borates, arsenates, glycines, piperazine and mixtures
thereof. In the present invention, one or more of such
promoting agents can be used to positively affect or promote
the absorption reaction, the regeneration reaction or both.
Promoters are typically added in concentrations of about 0.1
to about 3 molar; preferably about 0.5 to about 1 molar. The
use of a promoter ordinarily does not significantly affect the
amount of carbon dioxide that can be adsorbed in the hot
carbonate electrolyte, but a promoter increases the rate of
absorption of carbon dioxide as well as the rate of
regeneration of the hot carbonate electrolyte. Promoters can
be useful because they can facilitate processing of the same
amount of carbon dioxide in smaller processing vessels,
thereby reducing capital cost of the processing unit. A
typical hot carbonate process integrated with a methane steam
reforming/water gas shift process is comprised of two towers.
The first or absorber tower is located downstream of a water
gas shift reactor. An aqueous solution of Na2CO3 enters the
top of the absorber. Carbon dioxide from the shifted
19
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
synthesis gas reacts with the carbonate and water to form
dissolved bicarbonates via reaction (7).
CO2 + K2CO3 + H2O 2KHCO3 (7)
[0045] A stream containing synthesis gas and the dissolved
bicarbonates exits from the top of the absorber substantially
reduced in carbon dioxide. This bicarbonate-rich solution is
sent to the top of the second tower, the regenerator.
Pressure is typically reduced prior to entering the
regenerator in this as well as the other, alternative process
arrangements of the present invention described herein. For
example, whatever operating pressure is selected for the-
production of hydrogen, the pressure in the regenerator is
typically reduced to about 0.3 atm to about 2 atm; preferably
to about 0.5 atm to about 1.5 atm; for example about ambient
pressure. A combination of thermodynamics and economics
provides that hot carbonate processes should be regenerated
near or below atmospheric pressure in order to reduce stream
.consumption. Furthermore, the partial pressure of carbon
dioxide in the absorber liquid product is used to drive the
gas out of solution via the reverse of reaction (7). If the
regenerator pressure is reduced below the absorber product
partial pressure, large amounts of carbon dioxide tend to
flash out of solution, simplifying and reducing the work-load
on the regenerator. Steam is introduced at the bottom of the
regenerator and acts to reverse reaction (7); this reaction is
illustrated using the potassium salt, but any suitable alkali
metal or alkaline earth metal carbonate, or mixture, can be
used, for example, sodium carbonate. A sodium or potassium
carbonate-rich stream leaves the bottom of the regenerator and
is recycled back to the stripper. Carbon dioxide and steam
exit from the top of the regenerator. There are several
variations of this process including the use of solutions that
can be promoted or unpromoted. Hot carbonate and promoted hot
CA 02613483 2010-01-18
carbonate processes are described by Danckwerts et al., "The
Absorption of Carbon Dioxide Into Solutions Of Alkalis and
Amines", The Chemical Engineer, pp. 244-280, (1966); Kohl et
al., "Alkaline Salt Solution for Hydrogen Sulfide and Carbon
Dioxide Absorption" in Gas Purification 3rd Edition, Gulf
Publishing Company Houston, TX, 1979, 158-221; and
Pierantozzi, "Carbon Dioxide", Kirk-Othmer Encyclopedia of
Chemical Technology, 4th Ed. 1993, vol. 5, 42-53.
[00461 Other useful hot carbonate processes include various
heat recuperation and thermal integration schemes or staging
based on process particulars. For example, in one embodiment
the regenerator can be run at a lower (or higher) temperature
than the absorber. In such embodiment(s) the liquid effluents
from each tower are run into a recuperative heat exchanger
which pre-heats the liquid feed to the absorber and cools the
liquid feed to the regenerator. An example of recuperative
heat exchangers is illustrated in Perry's Chemical Engineers
Handbook, 4th edition, McGraw Hill, Fig. 14-19, p. 14-31, 1963.
In alternate embodiments the absorber and regenerator towers
can be staged so that liquid product from an upper absorber
will be cycled to an upper regenerator and the liquid product
from a lower absorber(s) will be cycled to a lower
regenerator. Staging alternatives are also described in the
above-noted reference by Kohl et al.
[0047] The regeneration step, preferably comprising the hot
carbonate regeneration method described herein, can be
conducted at suitable temperatures as described herein.
Useful temperatures typically are about 85 C to about 200 C;
alternatively, about 100 C to about 190 C; such as about
110 C to about 180 C; for example, about 130 C to about
175 C; or about 150 C to about 170 C. In those instances
where the temperature exceeds about 100 C, the regeneration
21
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
step is necessarily conducted in a pressurized vessel or
environment such that a liquid-phase process can be continued.
In preferred embodiments, regeneration is conducted at
temperatures of about 85 C to about 100 C; or about 90 C to
about 100 C; alternatively, greater than about 100 C and
less than about 200 C.
[0048] There are various alternative embodiments of the
present invention that usefully integrate the liquid phase,
electrochemical reforming (ECR) process with hot carbonate
regeneration processes for the production of hydrogen from a
fuel such as methanol.
[0049] Figure 4 is a schematic representation of a process
wherein a carbonate-rich solution from the regenerator flows
co-currently with the product hydrogen along a reactor, for
example a plug flow ECR-reactor. For purposes of the present
invention, plug flow is defined as follows:
[0050] In an ideal plug flow reactor or vessel every fluid
element entering the vessel follows the element that entered
before it without any intermixing and exits the reactor in
exactly the same order. At any instant then, the exit stream
is made up of fluid elements, all of which have been resident
in the reactor for exactly the same length of time. Stated
another way, idealized plug flow is a simple way of modeling
flow of a fluid in, for example, a pipe, reactor or any vessel
in general, wherein the structure of the vessel or the
conditions of flow (or both) are such that "plugs" or finite
volumetric elements of fluid pass through the vessel with
substantially no back mixing between the elements or plugs.
Under practical operating conditions, an efficient plug flow
reactor can substantially approach the above definition of an
ideal plug flow vessel or reactor, but it is expected that, in
typical operation of the present invention, at least some
22
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
hydrogen gas product will bypass the liquid feed and some
back-mixing can occur.
[0051] For reference purposes the hot carbonate process is
encircled in the figure. Most of the carbon dioxide produced
during the hydrogen forming reaction is absorbed into the
electrolyte to form the corresponding metal bicarbonate.
Typically about 50% to about 99% or more of the carbon dioxide
is removed; preferably about 70% to about 95% or more; for
example, about 90% to about 99%. If any gaseous carbon
dioxide leaves with the product hydrogen, it can be
substantially removed by sending the product into the
absorber. Typically, the absorber operates to remove about
50% to about 99% or more of the carbon dioxide that may be
present; preferably about 90% to about 99% or more; more
preferably about 95% to about 99% or more. The bicarbonate-
rich streams from the reactor and the absorber are combined
and sent to the regenerator. Again, steam is used to reverse
reaction (7) and to substantially regenerate the
carbonate-rich stream. The carbonate-rich stream is then
split to feed 'both the ECR reactor and the absorber.
Typically about 50 to about 99% or more of the regenerated
carbonate-rich stream is sent as feed to the reactor;
preferably, about 90% to about 99% or more of the regenerated
carbonate-rich stream. Substantially all of the carbon
dioxide formed in the process leaves the process with steam
from the top of the regenerator.
[0052] The process scheme illustrated in Figure 4 is an
improvement over sequentially integrating an ECR reformer,
typically including catalyst, with a hot carbonate process
because the reforming reactions occur in the liquid phase and
most (for example, greater than 80%) of the carbon dioxide is
converted directly to bicarbonate. As a result, the absorber
in Figure 4 is much smaller because it can be sized to remove
23
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
less than about 20% of the carbon dioxide from the gaseous
hydrogen product stream rather than about 100%.
[0053] The process embodiment of the present invention
illustrated in Figure 4 can be further simplified.
Substantially all carbon dioxide from a plug flow (or non-plug
flow) ECR reactor can be captured as bicarbonate. For
example, typically, about 90% to about 99% or more of the
carbon dioxide that may be present is removed; preferably
about 95% to about 99% or more; more preferably about 98% to
about 99% or more; for example about 99%. In this embodiment,
the absorber tower can be eliminated as shown in Figure 5.
Again, a carbonate-rich solution is mixed with a fuel, e.g.,
methanol, feed and flows co-currently along a plug flow ECR
reactor, also typically containing catalyst. In this
embodiment, substantially all of the carbon dioxide leaves the
reactor in the bicarbonate-rich stream. A substantially
carbon dioxide-free hydrogen product is produced as noted
above. The bicarbonate-rich stream is again sent to the
regenerator where steam is used to regenerate the
carbonate-rich stream. Substantially all of the
carbonate-rich stream is re-circulated as a feed to the ECR
reactor. This preferred embodiment is a simplification of the
process illustrated in Figure 4 because a separate absorption
tower is totally eliminated.
[0054] A further improvement over the embodiments
illustrated in Figures 4 and 5 is shown in Figure 6. The
process again uses two towers. In contrast to Figure 4, the
first tower is loaded with catalyst so that the tower or
reactor acts as both the ECR reactor and absorber. As
described hereinabove, a suitable catalyst is selected from
one or more of those useful in the reforming reaction. The
second tower continues to act as a regenerator. In this
embodiment, a feed of water and methanol enters at a feed
24
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
plate at an intermediate point along the first tower. The
specific feed location depends on such factors as the
operating temperature of the reactor and on the particular
catalyst employed. However, typically the feed will enter the
tower in the middle third of the tower; alternatively, in the
top third quarter or in the bottom second quarter; for
example, about midway in the tower. The product hydrogen and
the absorption carbonate solution run counter-current to each
other. The down flowing carbonate-rich stream acts to capture
and remove carbon dioxide as a bicarbonate. If any methanol
vapors or carbon dioxide are carried up the column with the
gaseous hydrogen product, the incoming carbonate stream
absorbs them from the gas and pulls them downward in the
liquid phase. The section of the first tower below the feed
ensures that substantially all of the methanol is reacted
before the bicarbonate-rich stream exits the first tower.
This bicarbonate-rich stream is again preferably reduced in
pressure and sent to the regenerator tower where it is purged
or reacted with steam in order to remove substantially all of
the carbon dioxide and to regenerate the carbonate-rich
stream. Stream flow rates and sizing will depend on the scale
of the reformer and such quantities are readily determinable
by an engineer using standard engineering knowledge.
(0055] The countercurrent flow of product hydrogen versus
the regenerated electrolyte flow in Figure 6 is superior to
the co-current integrations as shown in Figure 4 and Figure 5
because countercurrent flow enables a more complete separation
of the carbon dioxide and methanol from the product hydrogen
and a more complete reaction of methanol or other volatile
fuel. The down flowing electrolyte acts to absorb gas phase
methanol and/or carbon dioxide from the hydrogen product,
thereby reducing the impurity levels of these components in
the product by as much as about 20 times over the co-current
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
integrations. Furthermore, conversion of the fuel is higher
because substantially all of, for example, the methanol is
contained in the reactor column, thereby providing for
increased conversion as a result of a longer residence time.
While actual improvements are typically dependent on the
design specifics, countercurrent flow has the potential to
increase the residence time of the oxidizable fuel (e.g.,
methanol) by 2 to 3 times over a similarly sized co-current
process. Under such circumstances, if the residence time of a
first order reaction is increased by a factor of 2 to 3 fold,
a 90% conversion in a co-current design could be increased to
99% and 99.9% respectively. Even an improvement of 1.5 to 2
fold can be significant. For example, an improvement of 1.5
fold can increase conversion from 90% to 97%, with the
associated significant economic benefit.
[0056] A significant economic advantage of the present
invention in its various embodiments is that hydrogen can be
produced without the need to continuously add fresh caustic
reagents (hydroxides, carbonates and or their mixtures) using
instead an effective and economical electrolyte regeneration
method. Also, liquid phase reforming in alkaline electrolytes
produces a relatively pure hydrogen product that is
significantly lower in carbon monoxide and carbon dioxide than
typical steam reformed hydrogen products. Furthermore, the
hydrogen can be produced at pressures exceeding 1000 psi so
that expensive compressors are not needed downstream of the
reformer to obtain a high pressure product.
[0057] The following examples are provided as specific
illustrations of embodiments of the claimed invention. It
should be understood, however, that the invention is not
limited to the specific details set forth in the examples.
All parts and percentages in the examples, as well as in the
specification, are by weight unless otherwise specified.
26
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
Furthermore, any range of numbers recited in the specification
or claims, such as that representing a particular set of
properties, units of measure, conditions, physical states or
percentages, is intended to literally incorporate expressly
herein by reference or otherwise, any number falling within
such range, including any subset of numbers within any range
so recited. For example, whenever a numerical range with a
lower limit, RL, and an upper limit Ru, is disclosed, any
number R falling within the range is specifically disclosed.
In particular, the following numbers R within the range are
specifically disclosed: R = RL + k (Ru -RL), where k is a
variable ranging from 1% to 100% with a 1% increment, e.g., k
is 1%, 2%, 3%, 4%, 5%. ... 50%, 51%, 52%. ... 95%, 96%, 97%, 98%,
99%, or 100%. Moreover, any numerical range represented by
any two values of R, as calculated above is also specifically
disclosed.
[0058] For purposes of the present invention, unless
otherwise defined with respect to a specific property,
characteristic or variable, the term "substantially" as
applied to any criteria, such as a property, characteristic or
variable, means to meet the stated criteria in such measure
such that one skilled in the art would understand that the
benefit to be achieved, or the condition or property value
desired is met.
[0059] Throughout the entire specification, including the
claims, the word "comprise" and variations of the word, such
as "comprising" and "comprises," as well as "have," "having,"
"includes," "include" and "including," and variations thereof,
means that the named steps, elements or materials to which it
refers are essential, but other steps, elements or materials
may be added and still form a construct within the scope of
the claim or disclosure. When recited in describing the
invention and in a claim, it means that the invention and what
27
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
is claimed is considered to be what follows and potentially
more. These terms, particularly when applied to claims, are
inclusive or open-ended and do not exclude additional,
unrecited elements or methods steps.
[0060] As used throughout the specification, including the
described embodiments, the singular forms "a," an," and "the"
include plural referents unless the context clearly dictates
otherwise. Thus, for example, reference to "an oxidizable
fuel" includes a single fuel as well a two or more different
fuels in combination, reference to "a metal carbonate"
includes mixtures of two or more metal carbonates as well as a
single metal carbonate, and the like.
[0061] The term "about" encompasses greater and lesser
values than those specifically recited provided that the value
of the relevant property or condition facilitates reasonably
meeting the technologic objective(s) of the present invention
as described in detail in the specification and claims. More
specifically, the term "about" when used as a modifier for, or
in conjunction with, a variable, is intended to convey that
the numbers and ranges disclosed herein are flexible and that
practice of the present invention by those skilled in the art
using, for example, concentrations, amounts, contents, carbon
numbers, temperatures, pressures, properties such as density,
purity, etc., that are outside of a stated range or different
from a single value, will achieve the desired result, namely,
the efficient production of hydrogen including regeneration of
an alkaline reactant.
EXAMPLES
[0062] Example 1
[0063] Catalysts for liquid phase reforming in' caustic
electrolytes were experimentally evaluated in a test stand
shown schematically in Figure 2. Mixtures of methanol,
aqueous caustic electrolytes and catalysts were initially
28
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
charged into a reaction vessel and then sealed. The vessel
was then heated to reaction temperatures between 150 2C-
250 2C. Kinetics were measured as a pressure build-up caused
by evolved hydrogen. Gas chromatography (GC) analysis of the
product gases using potassium hydroxide typically showed high
purity hydrogen with only trace amounts (less than 1000 ppm)
of carbon monoxide, carbon dioxide or methane. A wet test
meter (WTM) was used to monitor the amount of hydrogen gas
evolved.
[0064] A series of conductive metallic catalysts were
evaluated using the test stand shown schematically in
Figure 2. Figure 7 compares the activity from these catalyst
evaluations. The results are presented as hydrogen pressure
increase after the reactor was heated to a steady state
temperature of 200 C. All tests utilized substantially the
same charge, 40 ml methanol and 250 ml 45wt% KOH, so that the
pressure curves are directly comparable as a function of
catalyst loading and catalyst type. The catalysts tested are
identified in the legend accompanying Figure 7. Although KOH
electrolyte was used in these tests, it is expected that
comparable results will be observed using an electrolyte
composition comprising carbonate, as disclosed herein.
[0065] These experiments demonstrated that platinum
catalysts exhibited the highest activity. Even so, there was
considerable overlap in activity between the lowest platinum
containing catalysts and the highest nickel containing
catalysts. Since platinum is about 1000 times more expensive
than nickel, a nickel catalyst can be more cost-effective in a
particular process embodiment. Amongst both of the catalyst
groups or samples including platinum or nickel, the higher
catalyst surface area generally correlated with higher
activity. However, higher activity per unit surface area was
generally observed with low surface area particles. This
29
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
observation is generally consistent with the understanding
that an internal diffusion, mass transfer limitation within
the catalyst particles can affect catalyst performance. This
suggests that high surface area catalysts are most active when
using small particle size catalysts and may also suggest that
a nickel slurry catalyst as the most cost-effective. On the
other hand, an experiment using Raney nickel exhibited an
exceptionally high activity per unit surface area. In one of
the experiments represented by curve 3 in Figure 7, the amount
of oxidizable reactant (methanol) relative to platinum
catalyst, was significantly decreased in order to demonstrate
that in the reforming reaction the reactant can be
substantially completely reformed. Complete reformation was
achieved in this experiment, but since the amount of methanol
present was less than in the other experiments, the total
amount of hydrogen produced (and consequently its pressure)
was less, resulting in the distinctly different pressure
curve.
[0066] Example 2
[0067] A series of experiments are described which
illustrate significant features or steps of the invention.
These steps are common to the process integrations described
in the application, particularly in Figures 4, 5 and 6. These
tests were conducted in the form of batch experiments for
demonstration purposes. Furthermore, in view of the
comprehensive teachings provided herein, they can be
integrated to form a continuous, multi-step process.
[0068] The steps are described as follows:
[0069] Step A: Generation of hydrogen from a fuel, in
this instance methanol was used, in a liquid composition at a
pH between 10.5 and <12, producing a bicarbonate-rich
electrolyte, in other words a solution in which the reforming
reaction proceeds via an electrochemical . pathway, an
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
ion-conducting electrolyte, thereby lowering its pH to a range
between 8 and 10.5. In this step, a substantial portion, but
not all of the carbonate is converted to bicarbonate;
[0070] Step B: Using steam to substantially regenerate
the bicarbonate-rich electrolyte to a carbonate-rich
electrolyte thereby increasing the ratio of carbonate to
bicarbonate to approach the original ratio and increasing the
electrolyte pH to approach the starting pH in Step A;
[0071] Step C: Using the regenerated electrolyte to
produce additional hydrogen from the fuel, e.g., methanol.
EXPERIMENT
[0072] Step A: Making H2 from fuel (methanol) at pH<12.
[0073] The batch reactor, schematically shown in Figure 2,
was charged with an aqueous solution of potassium carbonate
and potassium bicarbonate, as shown in Table 2 (starting
pH = 11.6), containing methanol and a reforming catalyst
(20 wt% Pt supported on carbon) provided by E-Tek, Inc.,
(Somerset, NJ). The reactor was run at 200 C using pressure to
follow the reaction progress. Reaction (6) generates hydrogen
and converts carbonates to bicarbonates. In the process, the
pH is gradually lowered as the level of bicarbonate increases.
[0074] Reactor pressure at the start of the 200 C hold was
300 psi. The reaction progressed for about 95 hours. At the
end of this period the pressure had increased to 495 psi. At
the end of this period, the reaction rate had declined to a
negligible rate even though 70% of the methanol and 70% of the
carbonate from the original reaction 'charge still remained in
the reactor. The final pH was 10. A pressure versus time
record for this experiment is plotted in Figure 8. Table 2
summarizes the initial and final compositions (based on
calculated pressure changes in the batch reactor), pH as
measured with and without methanol and the initial and final
reaction rates. (The presence of methanol has a small effect
31
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
on pH.) Carbonate composition was also measured via titration
before and after the experiment. Gas chromatograms documented
hydrogen and carbon dioxide content in the reaction product at
various points during Step A. The electrolyte was not
separated from the catalyst, but instead the whole mixture was
saved for Step B, except for small samples taken for
titration.
[0075] Step B: Regenerate electrolyte back toward original
pH
[0076] Spent electrolyte from Step A was placed into a
flask and heated to generate steam. Vapors from the flask
were refluxed using a condenser coil. The steam. effected a
reversal of reaction (7) and eventually stripped most of the
carbon dioxide that had been trapped in the electrolyte during
Step A. Carbon dioxide vapor was allowed to escape from the
condenser and its volume was measured using a wet test meter.
[0077] Boiling continued over 14 hours until the carbon
dioxide evolution rate had dropped substantially, thus
indicating that regeneration had approached completion.
During this refluxing the condenser returned substantially all
of the water and methanol to the boiling flask. Comparison of
the pressure buildup during Step A with the carbon dioxide
measured by the wet test meter showed that about 100% of the
bicarbonate created in Step A was removed in Step B. At the
end of this period the condenser was removed and boiling
continued for another 1.5 hours in order to remove
substantially all of the unreacted methanol. The pH measured
after this step was 11.7. Some water was lost as water vapor
during this distillation. Electrolyte carbonate composition
was again measured via titration after the steam stripping.
The regenerated electrolyte was saved for Step C. Table 2
summarizes the total carbon dioxide evolved, the initial and
final electrolyte compositions (based on calculated pressure
32
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
changes in the batch reactor and carbon dioxide evolution in
the Step B, and pH as measured with methanol.
[0078] Step C: Using regenerated electrolyte to make H2
from methanol.
[0079] The batch reactor was re-charged with the
regenerated electrolyte from Step B. Fresh water and methanol
were added to replace that removed by reaction in Step A and
by distillation in the final stages of Step B. A small
increment of catalyst was also added to make up estimated
losses from transfers and titrations that occurred in Steps A
and B. The reactor was again run at 200 sC using pressure
build-up to follow the reaction progress. After addition of
the makeup water, the electrolyte pH dropped to 11.2 prior to
methanol addition and increased to 11.4 after methanol
addition. The pH of the initial mixture charged for Step A
was 11.6. This close approach to the original pH in Step A is
further evidence that Step B converted substantially all of
the bicarbonate formed during Step A back to carbonate.
[0080] Reactor pressure at the start of the 200 C hold was
340 psi. This pressure is slightly higher than that in Step A
(+40 psi). In highly concentrated alkali carbonates, methanol
is known to form a second methanol-rich phase. The presence
of a methanol-rich phase exhibits a higher methanol vapor than
when methanol is fully dissolved in the aqueous carbonate
phase. Alternatively, the additional pressure could suggest
that regeneration of the original carbonate charge in Step A
was slightly less than 100%, causing a small incremental
increase in carbon dioxide partial pressure. The reaction
progressed for about 90 hours. At the end of this period
pressure had increased to 480 psi. Also at the end of this
period, the hydrogen generation rate had declined to a
negligible rate compared to the original, even though 69%
methanol and 69% of the carbonate in its starting reaction
33
CA 02613483 2007-12-21
WO 2007/002503 PCT/US2006/024645
charge remained in the reactor. Final pH was 10.6. The
pressure versus time record for Step C is also plotted in
Figure 8 for comparison with Step A. The improved initial
rate in Step C is consistent with Step A and the
interpretation that the electrolyte approached full
regeneration in Step B. Table 2 summarizes the initial and
final compositions (based on calculated pressure changes in
the batch reactor), and pH (as measured with and without
methanol). Carbonate composition was also measured via
titration before and after Step C. GC data again documented
hydrogen and carbon dioxide content in the reaction product at
various points during Step C.
[0081] Table 2
Summary of Electrolyte Regeneration Using Pt/C Catalyst
Variable* Begin End Begin End Begin End
Expt. 1 Expt. 1 Expt. 1 Expt. 1 Methanol Expt. 1 Expt. 1
Step A Step A Step B Step B Distillation Step C Step C
K2CO3 0.4995 0.3496 0.344 0.4912 0.4809 0.4679 0.3247
KHCO3 0.0512 0.351 - 0.0511 0.05 0.0486 0.3349
CH3OH 0.518 0.368 0.363 0.356 0 0.469 0.326
H2O 12.03 11.73 11.54 11.69 9.614 11.35 11.07
Catalyst 3.01 3.01 2.96 2.96 2.96 2.96 2.96
(gm)
Volume 250 245 241 237 185 235 230
(ML)
pH, no 11.45 na na na na 11.22 na
CH3OH
pH + 11.63 10.06 10.06 11.17 na 11.43 10.6$
CH3OH
CO2 - - - 0.1533 - - -
Evolution
Total H2 - 0.4498 - - - - 0.4294
Evolved
* Chemical components in moles except as indicated
$ Measured at 21.3 C
34
CA 02613483 2007-12-21
[0082] Example 3
[0083] The series of experiments described in Example 2
were repeated using a Ni catalyst (spherical nickel powder,
commercially available as Novamet Type 4SP-10, from Inco
Special Products, Inco Limited, Wyckoff, NJ) The results
were substantially similar to those reported in Example 2.
Hydrogen was again generated in Step A and Step C. Nickel is
a less active catalyst than platinum and as a result the
amount of catalyst was increased from 3g to 104g. In spite of
its slower kinetics, nickel is about 1000 times less expensive
than platinum per unit weight and thus nickel may be a more
cost-effective catalyst. In Step A the pH decreased from
10.95 to 10.54. In Step B, the electrolyte (which contained a
nickel slurry and was difficult to separate by decantation),
was steam stripped until the final pH increased from 10.54 to
11.14. In Step C, the regenerated electrolyte was again
recharged with fresh methanol. The hydrogen generation
returned to the initial rate observed in Step A, again
suggesting that regeneration of the electrolyte in Step B was
substantially 100%. The amounts of hydrogen produced in
Step A, the carbon dioxide observed in Step B and the
titrations were more consistent in Example 2 than they were in
Example 3, possibly due to experimental error.
[0084] Alternative embodiments of the invention are set
forth in the following numbered paragraphs:
[0085] A process for producing hydrogen gas comprising
contacting in the liquid phase at least one oxidizable organic
substance in the presence of a conductive catalyst and an
alkaline electrolyte, wherein:
(A) hydrogen gas is generated in a reactor having a top
and bottom, wherein said at least one oxidizable organic
substance is introduced into said reactor at a point
substantially midway between said top and bottom;
CA 02613483 2007-12-21
(B) an alkaline electrolyte comprising at least one
metal carbonate is introduced into said reactor at a point
substantially at the top of said reactor such that said metal
carbonate and said hydrogen gas flow substantially
countercurrent to one another, thereby resulting in the
production of at least one metal bicarbonate composition;
(C) said at least one metal bicarbonate composition is
regenerated and said at least one oxidizable organic substance
comprises an oxygenated hydrocarbon.
[0086] The process of paragraph 85 wherein a process stream
comprising said metal bicarbonate is reduced in pressure
compared to the pressure in said reactor and introduced into a
second reactor where it is contacted with steam, thereby
producing carbon dioxide and substantially regenerating said
alkaline electrolyte.
[0087] The process of paragraph 85 wherein said regenerated
alkaline electrolyte is introduced into said hydrogen
generating reactor.
[0088] The process of paragraph 85, wherein said oxygenated
hydrocarbon is selected from the group consisting of a
saccharides, celluloses, starches, sugars, alcohols, ethers,
carboxylic acids, aldehydes, ketones, biomass and mixtures
thereof.
[0089] The process of paragraph 88, wherein said saccharide
is selected from the group consisting of monosaccharides,
disaccharides, oligosaccharides, polysaccharides and mixtures
thereof.
[0090] The process of paragraph 88, wherein said alcohol is
selected from the group consisting of Cl-C6 alcohols and
mixtures thereof.
[0091] The process of paragraph 90, wherein said alcohol is
selected from methanol, ethanol and mixtures thereof.
36
CA 02613483 2007-12-21
[0092] The process of paragraph 91 wherein said alcohol is
methanol.
[0093] The process of paragraph 88, wherein said ether is
selected from dimethyl ether, methylethyl ether, diethyl ether
and mixtures thereof.
[0094] The process of paragraph 93, wherein said ether is
dimethyl ether.
[0095] The process of paragraph 88, wherein said oxygenated
hydrocarbon is a mixture of methanol and dimethyl ether.
[0096] The process of paragraph 85, wherein said alkaline
electrolyte is selected from the group consisting of metal
hydroxides, carbonates, bicarbonates and mixtures thereof.
[0097] The process of paragraph 85, wherein said catalyst
is selected from the group consisting of compounds, complexes,
alloys and mixtures thereof comprising a metal selected from
the Group VIII transition metals of the Periodic Table of the
Elements.
[0098] The process of paragraph 97, wherein said catalyst
further comprises at least one metal selected from the metals
of Group IB, Group IIB, Group VIIB, and mixtures thereof.
[0099] The process of paragraph 97, wherein said catalyst
is selected from the group consisting of platinum, nickel,
palladium, rhodium, iridium, cobalt, ruthenium, iron and
mixtures thereof.
[0100] The process of paragraph 98, wherein said catalyst
further comprises a metal selected from the group consisting
of copper, zinc, silver and rhenium.
[0101] The process of paragraph 97, wherein said catalyst
is supported on or in a conductive or non-conductive material
selected from the group consisting of metals, metal oxides,
silica, alumina, silica-alumina, zirconia, titania, ceria,
carbon, silicon carbide, silicon nitride, silicon boride and
mixtures thereof.
37
CA 02613483 2010-01-18
[0102] The process of paragraph 101, wherein said support
is in a form selected from the group consisting of beads,
powders, coatings extruded substrates, monoliths and mixtures
thereof.
[0103] The process of paragraph 85, wherein said oxidizable
organic substance and said alkaline electrolyte are reacted in
the presence of water.
[0104] The process of paragraph 96, wherein said alkaline
electrolyte is selected from the group consisting of alkali
metal or alkaline earth metal: hydroxides, carbonates,.
bicarbonates and mixtures thereof.
[0105] The process of paragraph 104, wherein the metal of
said alkaline electrolyte is selected from the group
consisting of sodium, lithium, potassium, cesium, rubidium and
mixtures thereof.
[0106] The process of paragraph 85 wherein regeneration is
conducted at a temperature of about 85 C to about 200 C, or
alternatively at a temperature of about 150 C to about 170 C
or about 85 C to about 100 C.
[0107] The process of paragraph 85 conducted continuously.
[0108]
The principles,
preferred embodiments, and modes of operation of the present
invention have been described in the foregoing specification.
Although the invention herein has been described with
reference to particular embodiments, it is to be understood
that these embodiments are merely illustrative of the
principles and applications of the present invention. It is
therefore to be understood that numerous modifications may be
made to the illustrative embodiments and that other
arrangements may be devised without departing from the
38
CA 02613483 2010-01-18
scope of the present invention as defined by the appended
claims.
39