Note: Descriptions are shown in the official language in which they were submitted.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
BENZIMIDAZOLE FORMULATION
FIELD OF INVENTION
The present invention relates to the field of pharmaceutical formulation
science. In
particular, the present invention relates to pharmaceutical formulations
comprising acid
labile benzimidazoles. The invention provides cost-effective production
methods providing
stable formulations.
BACKGROUND
Due to the acid lablie nature of the benzimidazoles it is necessary to protect
the drug
substance from exposure to acids, especially in the presence of humidity. In
the first
stage, the benzimidazole has to be protected from acid attack during storage
of the drug.
In the prior art, this is provided by contacting the benzimidazole with an
alkaline substance
present in the pharmaceutical formulation. In the next stage the benzimidazole
must be
protected from acid attack in the stomach. The person skilled in the art will
realise that this -
can be achieved by applying an enteric coating. It:is however acknowledged in
the art that
an enteric coating will introduce a stability problem in that commonly used
enteric coatings
are acidic by nature. This stability problem is described in EP 0244380
(Hassle). Applying a
neutral subcoating for protection of the acid labile benzimidazole was
suggested by EP 244
380 for solving the stability problem.
Wet granulation techniques are traditionally employed in such formulation
approaches. EP
0244380 e.g. discloses use of conventional granulation process, wherein a wet
binder is
used as well as an aqueous granulation liquid. EP 0589981 e.g. discloses use
of
polyvinylpyrrolidone as a wet binder.
WO 05009410 (Dr. Reddy's) discloses an enteric coated benzimidazole
formulation,
wherein the tablets are arrived at via different combinations of wet mixing
and dry mixing.
Example 7 e.g. discloses dry mixing and compression of esomeprazole and
magnesium
oxide.
WO 9850019 (Sage) discloses an enteric coated formulation comprising
omeprazole or
lanzoprazole. In Example 2C, ten grams of omeprazole were mixed with
pharmaceutical
excipient lactose anhydrous USP/NF and then passed through a screen to obtain
a
homogenous granule size. In Example 4, it is stated that "The individual core
granulation
was mixed with lactose and talc or magnesium stearate and compressed into
tablets by
known pharmaceutical techniques." No stability assays are disclosed with
tablets according
to Example 2C+4.
WO 04075881 (Ranbaxy) discloses an enteric coated formulation comprising
rabeprazole
and a low viscosity hyd roxypropyice flu lose optionally in combination with
antioxidants
produced by a method comprising dry granulation.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
2
Several approaches of providing a stable pharmaceutical formulation of
benzimidazoles are
thus suggested in the art. The suggested manufacturing processes are however
relatively
laborious. There is thus a need in the art for simple and cost efficient
manufacturing
methods for producing benzimidazole formulations with good stability
properties and good
dissoiution profiles. There is furthermore a need in the art for .obtaining
such tablets in a
relatively small size for efficient passage and drug delivery in the
intestinal tract.
SUMMARY OF INVENTION "
The object of the present invention thus to provide a stable and cost-
efficient
pharmaceutical formuiation intended for oral administration and subsequent
efficient
delivery of the active benzidimazole in the intestinal tract, wherein said
formulation has a
good shelf life stability and release profile.
In particular, the present invention relates to a method for producing a
pharmaceutical
tablet formulation comprising a benzimidazole as the biologically active
component,
wherein:
- said formulation comprises an enteric coating for protection of the active
component
from acid attack in the stomach,
- said benzimidazole is further stabilized by an alkaline substance in the
tablet,
- said method comprising dry granulating steps and dry compressing of tablets,
wherein
said formulation is further chaaracterized by one or more of the following
features:
(i) the alkaline substance is an alkali metal carbonate with high water
solubility
and a BET area of at least about 1 m2/g prior to any dry granulation and/or
dry
compression steps,
(li) the alkaline substance is an alkaline earth metal carbonate with low
water
solubility and a BET area of at least about 1 mZ/g prior to any dry
granulation
and/or dry compression steps,
(iii) the benzimidazole and the alkaline substance have been mixed and dry
granulated together prior to dry compression,
(iv) the weight ratio of benzimidazole and alkaline substance is from about
1:0.2 -
1:5,
(v) the alkaline substance has a pKa of at least about 10 and a BET area of at
least
about .1 m2/g prior to any dry granulation and/or dry compression steps,
(vi) if the alkaline substance is polyvalent, said alkaline substance has a
pKa1-value
of 6 or more and a BET area of at least about 1 m2/g prior to any dry
granulation and/or dry compression steps,
(vii) the alkaline substance has a BET-area of at least about 1 mZJg prior to
any dry
granulation and/or dry compression steps,
(viii) the tablet formulation further comprises a disintegrant in an amount of
about
1-30% by weight.
The invention furthermore relates to a pharmaceutical tablet formulation
comprising a
benzimidazole as the biologically active component, wherein:
- said formulation comprises an enteric coating for protection of the active
component
from acid attack in the stomach,
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
3
- said benzimidazole is further stabilized by an alkaline substance in the
tablet, wherein
said formulation is further characterized by one or more of the following
features:
(P) the alkaline substance raw material is an alkali metal carbonate with high
water solubility and a BET area of at least about 1 n-i2/g,
(ii) the alkaline substance raw material is an alkaline earth metal carbonate
with
low water solubility and a BET area of at least about 1 mz/g,
(iii) the benzimidazole and the alkaline substance raw material have been
mixed
and dry granulated together prior to dry compression,
(iv) the weight ratio of benzimidazole and alkaline substance is from about
1:0.2
- 1:5,
(v) the alkaline substance raw material has a pKa of at least about 10 and a
BET
area of at least about 1 mZ/g,
(vi) if the alkaline substance is polyvalent, said alkaline substance has a
pKal-
value of 6 or more and a BET area of at least about I mZ/g,
(vii) the alkaline substance raw material has a BET-area of at least about 1
m2/g,
(viii) the tablet formulation further comprises a disintegrant in an amount of
about
1-30 lo by weight.
The manufacturing process involves only few production steps and the use of
any liquid is
avoided rendering an expensive drying step superFluous. A liquid can, however,
be
applicable in the subsequent coating steps providing an enteric coat and
optionally a
subcoat.
DETAILED DESCRIPTION OF THE INVENTION
In a ffrst aspect, the present invention relates to a pharmaceutical tablet
formulation
comprising a benzimidazole as the bioiogically active component, wherein:
said formulation comprises an enteric coating for protection of the active
component
from acid attack in the stomach,
- said benzimidazole is further stabilized by an alkaline substance in the
tablet,
wherein said formulation is 'further characterized by one or more of the
following features:
(i:) the alkaline substance raw material is an alkali metal carbonate with
high
water solubility and a BET area of at least about 1 m2/g,
(if) the alkaline substance raw material is an alkaline earth metal carbonate
with
low water solubility and a BET area of at least about 1 m2/g,
(iii) the benzimidazole and the alkaline substance raw material have been
mixed
and dry granulated together prior to dry compression,
(iv) the weight ratio of benzimidazole and alkaline substance is from about
1:0.2
- 1:5,
(v) the alkaline substance raw material has a pKa of at least about 10 and a
BET
area of at least about 1 ma/g,
(vi) if the alkaline substance is polyvalent, said alkaline substance has a
pKal-
value of 6 or more and a BET area of at least about 1 mz/g,
(vii) the alkaline substance raw material has a BET-area of at least about 1
m2/g,
(viii) the tablet formulation further comprises a disintegrant in an amount of
about
1-30% by weight.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
4
In a second aspect, the present invention relates to a method for producing a
pharmaceutical tablet formulation comprising a benzimidazole as the
biologically active
component, wherein:
- said formulation comprises an enteric coating for protection of the active
component
from acid attack in the stomach,
- said benzimidazole is further stabilized by an alkaline substance in the
tablet,
- said method comprising dry granutating steps and dry compressing of tabiets
wherein said formulation is further characterized by one or more of the
following features:
(i) the alkaline substance is an alkali metal carbonate with high water
solubility
and a BET area of at least about i m2/g prior to any dry granulation and/or
dry
compression steps,
(ii) the alkaline subStance is an alkaline earth metal carbonate with low
water
solubility and a BET area of at least about i m2/g prior to any dry
granulation
and/or dry compression steps,
(iii) the benzimidazole and the alkaline substance have been mixed and dry
granulated together prior to dry compression,
(iv) the weight ratio of benzimidazole and alkaline substance is from about
1:0.2 -
1:5,
(v) the alkaline substance has a pKa of at least about 10 and a BET area of at
least
about 1 mz/g prior to any dry granulation and/or dry compression steps,
(vi) if the alkaline substance is polyvalent, said alkaline substance has a
pKal-value
of 6 or more and a BET area of at least about 1 mZ/g prior to any dry
granulation and/or dry compression steps,
(vii) the alkaline substance has a BET-area of at least about 1 mZ/g prior to
any dry
granulation and/or dry compression steps,
(viii) the tablet formufiation further comprises a disintegrant in an amount
of about
1-30% by weight.
In a preferred embodiment the benzimidazole is pantoprazole such as
pantoprazole sodium
hydrate or pantoprazole sodium sesquihydrate. In other preferred embodiments
the
benzimidazole may be omeprazole or a salt and/or a hydrate thereof,
lansoprazole or a
salt and/or a hydrate thereof, esomeprazol or a salt and/or a hydrate thereof,
aripiprazole
or a salt and/or a hydrate thereof, rabeprazol or a salt and/or a hydrate
thereof, or
timoprazole or a salt and/or a hydrate thereof.
In another preferred embodiment, said formulation further comprises other
pharmaceutically acceptable excipients such as fillers, dry binders, glidants
and lubricants.
In a particularly preferred embodiment, said formulation comprises
crospovidone as a
disintegrant in an amount of from about 5-20%, preferably 7,5-15%, most
preferably 10-
13% by weight.
In yet another embodiment, said formulation comprises a subcoat. In a
particularly
preferred embodiment however, said formulation does not comprise a subcoat. in
the
Examples it is shown that it is possible according to the present invention to
obtain tablet
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
formulations with good stability properties and good dissolution profiles
without the
laborious steps of applying a subcoat.
In yet another preferred embodiment, the alkaline substance is a salt of an
organic or an
5 inorganic acid where the anion of the salt is carbonate (C032"),
hydrogenphosphate (HP042"
) or phosphate (P043"). The alkaline substance may also be a salt of an
organic or an
inorganic acid where the kation is sodium (Na}), caicium (CaZ+) or magnesium
(Mg2+).
Preferably, the salt of the organic and/or inorganic acid according is
sodiumcarbonate
(NaZCO3), or Calciumcarbonate (CaCO3) trisodiumphosphate (Na3PO4),
disodiumhydrogenphosphate (NazHPO4), hydrazine or derivatives thereof, lysine
or a
derivative thereof, arginine or a derivative thereof, or histidine or a
derivative thereof.
In yet another preferred embodiment, dry granulation is provided by means of a
roller
compactor.
In a final preferred embodiment, the mixture has been subject to sieving prior
to tablet
compression with a sieve size (Roller compactor) of 1,25 mm or less. It is
shown in the
examples that relatively small and, relatively homogenous particles result in
a more
accurate dosing of the active compound. Doze accuracy is of particular
importance in
production of relatively small tablets.
In a final aspect, the present invention relates to products obtainable
or.obtained by the
methods disclosed herein.
Definitions
Pharmaceutical tablet formulation:
A pharmaceutical tablet formulation according to the present invention is
equivalent to a
solid dosis form.
Druas:
According to the present invention, the drug substance belongs to the group of
benzimidazoles or salts and/or hydrates thereof. The benzimidazole is
preferably
pantoprazole, omeprazole, lansoprazole, timoprazol, aripiprazole, rabeprazol
or
esomeprazole, as well as pharmaceutically acceptable salts, hydrates and
mixtures
thereof. Preferably, the benzimidazole is pantoprazole sodium sesquihydrate.
Any
pharmaceutically acceptable salt can be used. Examples of conventionally used
salts are
sodium or potassium salts of the drug substance.
A pharmaceutical formulation according to the present invention comprises
about 1 to 500
mg drug pr. dose; such as 1 to 200 mg; or 1 to 100 mg. Preferably, the unit
dose
comprises 10-120 mg; 15-100 mg; 15-80 mg, 15-70 mg; 15-60 mg; 15-50 mg; 15-45
mg; such as 20, 30, or 40 mg of benzimidazole, preferably pantoprazole.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
6
Unit dose is a pharmaceutically formulated unit comprising the dosage of drug
substance
intended for administration. The dosage unit can be a tablet.
Alkaline substance:
Due to the acid labile nature of the drug substance, the unit dose comprises
an alkaline
substance, or a mixture of two or more different alkaline substances, in the
core to confer
shelf life stability of the pharmaceutical formulation,
The alkaline substance according to the present invention may be soluble in
water or even
practically insoluble in water. E.g., I part of water soluble alkaline
substance might be
dissolved in about e.g. 100, 50, 30, or 10 parts of water or less. 1 part of
alkaline
substance with low water solubility may be dissolved in at least about 100,
300, 500,
1000, 10,000 or even more than 10,000 parts of water. This is in contrast tc)
conventional
production methods employing wet granulation wherein water soluble alkaline
substances
are preferred. The rationale behind using water soluble alkaline substances in
conventional
methods is that water soluble alkaline substances are thought to generate a
humid
environment with an alkaline pH protecting the active drug substance during
disintegration
of the tabiet in the gastric system. In the present invention it is
surprisingly demonstrated
in the Examples 6, 8 og 15 that alkaline substances such as calcium carbonate
which are
practically insoluble in water (1 part in more than 10,000 parts of water
according to
handbook of Pharmaceutical Excipients, 5" ed.) may result in stable
formulations with
good dissolution profiles. It however appears that alkaiine substances with
low water
solubility should preferably have a relatively large BET area (about 1 mZ/g or
more).
Generally, alkaline earth metal salts (such as e.g. calcium carbonate,
magnesium oxide,
magnesium carbonate) tend to have low water solubility. On the other hand,
alkali metal
,. -
salts (such as e.g. sodium carbonate and potassium carbonate) tend to be more
water
soluble.
According to the prior art such as e.g. W005009410 (Dr. Reddy's), ratios
between
benzimidazole and alkaline substance of about 1:0.17 are disclosed (Examples 1
and 2).
On basis of the existing knowledge in the art, the skilled man would thus not
expect that it
would be possible to use ratios of 1:0.2 and above and definitely not ratios
of about 1:0.5
or 1:1 or more since increasing amounts of base would be expected to result in
slow '
dissolution profiles of the active compound. According to the present
invention however,
the weight ratio between the drug substance and the alkaline substance ranges
between
1:0.2 to 1:10, preferably between 1:0.5 and 1:5, and most preferably between
1:1 to 1:5
while surprisingly still providing formulations with a combination of good
shelf life
stabilities and good dissolution profiles (examples 6, 8, and 15). The weight
ratio between
the drug and the alkaline substance may thus be about 1:0.2, or 1:0.3, or
1:0.4, or 1:0.5,
or 1:0.6, or 1: 0.7, or 1:
0.8,or1:0.9or1:1,or1:1.5,or1:2,or1:2.5,or1:3,or1:3.5,
or1:4,or1:4.5,or1:5,or1:6,or1:7,or1:8,or1:9,or1:10.
It is preferred that the pKa of the chosen alkaline material is at least 10.
However, this
alone is not sufficient. If the alkaiine material is polyvalent the pKal,
(where pKaI is the
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
7
most acidic pKa value) should be above 6. As shown in ex. 6 the use of
tricalcium
phosphate which has a pKal of 2.2 results in a poorer stability than if
disodium carbonate
(which has a pKal of 6.4) is used.
Alkaline substance preferably having a pKa value of 6 or above. The alkaline
substance will
typically provide an alkaline pH in the range of 7-12, when being dissolved
and/or
dispersed in water at room temperature in an amount of about 10-100 mg/mi.
Accordingly, the term alkaline substance includes the corresponding base of an
organic or
an inorganic acid, such as provided in the form of a pharmaceutically
acceptable salt of an
organic or inorganic acid and/or a mixture thereof, and some amino acids.
According to the
present invention, it is understood that a pharmaceutical formulation may very
well
comprise more than one aikaline substance, if appropriate.
The alkaline substance raw material is understood to be the alkaline substance
prior to any
formulation processing steps.
Examples of alkaline substances are listed in the following table.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
8
Table 1: Examples of alkaline substances
Substance Examples Structure pKa*
Saft of carbonic acid DiSodium carbonate Na2CO3 10.3
(carbonate) and
phosphoric acid Sodium hydrogen NaHCO3 6.4
(phosphate). Soluble carbonate
salts with pKa-values of 9
and above Trisodium phosphate Na3PO4 12.4
Disodium hydrogen Na2HPO4 7.2
phosphate
Sodium dihydrogen NaH2PO4 2.2
phosphate
Salt of carbonic acid Calcium carbonate CaCO3 10.3
(carbonate). Practically
insoluble with pKa-value
of 9 or above
Amino acids with pKa3- Lysine C6H1402N2 pKa2: 8.9
Values of 9 or above pKa3: 10.3
Arginine C6H14N402 pKa2:9.1
pKa3: 13.2
pHz11.4
(100 g/L Hz0)
Histidine - C6HgOZN3 pKa3:9.0
pH~z;7.7
(10 g/L HZO)
* The pKa-values in this table are approximate values and refer to the pKa of
the acid.
Only relevant pKa values are included.
A suitable disintegration time means that the pharmaceutical formulation must
comply
with the standards set up in the European Pharmacopoeia. Those skilled in the
art will
appreciate that it is desirable for compressed tablets to disintegrate within
30 minutes,
most desirable within 15 minutes upon contact with an aqueous solution,
provided that the
enteric coating is absent or bursted. Disintegration is preferably performed
in a dissolution
apparatus such as the Ph.Eur. Basket method as disclosed in e.g. example 11.
Furthermore, it should be understood that the alkaline substance should be
provided in
solid form, such as in the form of a powder, granulate or the like.
In connection with the present invention (example 16) it has been demonstrated
that
different alkaline substances may have different surface areas (BET areas) and
that the
same compound purchased under different trade names (e.g. calcium carbonate -
"Sturcal
L" and "Scoralite") may have different BET areas (see the SEM pictures in the
figures). It is
furthermore demonstrated that alkaline substances with reiativeiy large BET
areas (at least
about 0.5, 0.6, 0.7, 0.8, 0.9, preferably at least about 1.0, 1.1, 1.2, 1.3,
1.4, and most
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
9
preferably 1.5 mZ/g or more) tend to result in tablets with improved stability
properties
while at the same time retaining good dissolution properties (example 15). A
plausible
explanation for this finding is that porous alkaline substances with
relatively large BET
areas used as a raw material tend to be crushed into fine particles upon
mechanical
pressure such.as e.g. dry granulation and/or dry compression. In contrast,
substances with
relatively small BET areas (such as e.g. calcium carbonate purchased under the
trade
name "Scora{ite") tends to either not being affected by mechanical pressure
and/or to be
crushed into relatively large particles and/or to show a slightly improved
distribution of the
particles around the drug substance. It is thus preferred to use porous and/or
polycrystallic
alkaline substances with relative large BET area having a tendency to be
crushed into very
fine particles upon mechanical pressure. Such alkaline substances in the form
of very fine
particles in the resulting tablet most likely provide a better "alkaline
shield" against acid
and humidity attacks of the active compound by providing a close physical
contact
between the drug and the protective alkaline substance. It is shown in Example
10 that a
particularly preferred way of providing a close physical contact and thus a
stable tablet
with good dissolution profiles is to include a step wherein the two substances
are mixed
and subsequently dry granulated together prior to compression into a tablet.
Pharmaceutical excipients:
It is shown in the Examples that it is possible to compress tablets with good
stability and
dissolution profiles where the only pharmaceutical exciplent, apart from the
alkaline
substance, is minute amounts of MgStearate (example 5). When manufacturing
tablets
according to the invention in larger scale, such as in production scale, it
can however be
advantageous to add at least one of the following ingredients: a l~g_dant, a
lubricant, a
filler, a dry binder, a color, and a disinte rq ant to the formulation
comprising alkaline
substance and a benzimidazole. Preferably, the only additional pharmaceutical
excipient is
a glidant or a lubricant, preferably along with at least one disintegrant,
thus providing a
simple and cost efficient production method.
Examples of lig dant and lubricants are stearic acid, metallic stearates,
talc, colloidal silica,
sodium stearyl fumarate and alkyl sulphates.
In the present invention, a dry binder such as e.g. sorbitol, isomalt, or
mixtures thereof
may be used. The dry binder provides the effect of binding a material and
thereby
providing a powder that can be compressed into a tablet.
A ftller substance is any pharmaceutically acceptable substance that does not
interact with
the drug substance or with other excipients. Commonly used filler substances
are:
mannitol, Dextrins, maltodextrins (e.g. Lodex 5 and Lodex 10), inositol,
erythritol,
isomalt, lactitol, maltitol, mannitol, xylitol, low-substituted
hydroxypropylcellulose (e.g LH
11, LH 20, LH 21, LH 22, LH 30, LH 31, LH 32 available from Shin-Etsu Chemical
Co.),
starches or modified starches (e.g potato starch, maize starch, rice starch,
pre-gelatinised
starch), polyvinylpyrrolidone, polyvinylpyrrolidone/vinyl acetate copolymer,
agar (e.g.
sodium alginate), , carboxyalkylcellulose, dextrates, gelatine, gummi
arabicum,
hydroxypropyl cellulose, hydroxypropyimethylcellulose, methylceliulose,
polyethylene
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
glycol, polyethylene oxide, polysaccharides e.g. dextran, soy polysaccharide,
sodium
carbonate, and sodium chloride.
A wet binder is an excipient that in combination with water facilitates a
powder to be =
5 compressed into coherent bodies such as tablets or facilitates a powder to
be granulated
into a paiticulate matter. A wet binder must, at least to some extent, be
soluble in water.
Examples of wet binders are PVP (polyvinylpyrrolidone), HPMC
(hydroxymethylpropylcellulose) or getatine. If a wet binder is used according
to the
present invention, the wet binder will merely act as a filler and will not
exhibit the binding
10 properties normally associated with such wet binders. It will therefore be
understood that
excipients conventionally regarded as wet binders might be used as mere
fillers in the
context of the present invention.
A disintegrant is a pharmaceutically acceptable substance that improves the
disintegration
of tablets without interacting with the drug substance or with any other
exciplents. The
disintegrant has the capability of swelling upon contact with water, causing
the tablet to
swell/disintegrate and thus releasing the active compound. This effect is
shown in the
Examples (example 18), where dissolution profiles are improved upon addition
of
disintegrant in the tablet. Traditional wet granulated benzimidazole
formulations normally
comprise large amounts of disintegrants (at least about 30%) since "wet"
production steps
cause a significant proportion of the disintegrant to swell and thus
irreversibly reducing its
swelling capacity. However, in connection with the present invention it has
surprisingly
been shown that it is possible to obtain a good dissolution profile using
relatively small
amounts of disintegrant thus enabling production of smaller tablets using more
cost-
efficient methods (disintegrants are often relatively expensive ingredients).
Small tablets
have the advantage of being easier to swallow, pack, store, transport, etc.
Small tablets
furthermore improve movement through the gastric system and are less dependent
on the
gastric emptying. This effect is probably achieved because dry granulatian
techniques do
not result in unwanted preswelling of the disintegrant. It is furthermore
shown that it is
possible to use water in connection with the subcoating and/or the enteric
coating process
without causing unwanted preswelling and/or disintegration of the tablets
(example 12,
figure 8).
Examples of commonly used disintegrants are: Alginic acid - alginates,
carboxymethylcellulose calcium, carboxymethylcellulose sodium, crospovidone,
hydroxypropylcellulose, hydroxypropylmethylcel lu lose (HPMC), cellulose
derivatives such
as low-substituted hydroxypropylcellulose (e.g LH 11, LH 20, LH 21, LH 22, LH
30, LH 31,
LH 32 available from Shin-Etsu Chemical Co.) and microcrystalline cellulose,
polacrilin ,
potassium or sodium, polyacrylic acid, polycarbofif, polyethylene glycol,
polyvinylacetate,
crosslinked polyvinylpyrrolidone (e.g. Polyvidon(D CL, Polyvidon CL-M,
Kollidon CL,
Polyplasdone XL, Poiyplasdone XL-10); sodium carboxymethyl starch (e.g.
Primogel(D
and Explotabo), sodium croscarmellose (i.e. cross-linked
carboxymethylcellulose sodium
salt; e.g. Ac-Di-SalO), sodium starch glycolate, starches (e.g potato starch,
maize starch,
rice starch), and pre-gelatinised starch.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
11
The disintegrant may be present in the tablet in an amount of about 1-30%,
preferably 3-
25%, more preferably 5-20% and most preferably 10-15 !0, and even most
preferably
about 8-14%.
Granulation and compression:
Powders comprising either the drug in question, the alkaline substance, the
pharmaceutical
excipient(-s), or any combination thereof are subjected to a dry granulation
process. The
dry granulation process causes the powder to agglomerate into larger particles
having a
size suitable for further processing. Dry granulation can 'thus be said to
improve the
flowability of a mixture in order to be able to produce tablets that comply
with the demand
of mass variation or content uniformity set out in the European Pharmacopoeia.
Formulations according to the invention may be produced using one or more
mixing and
dry granulations steps. The order and the number of the mixing and granulation
steps do
not seem to be critical. However, it seems to be of importance that at least
one of the
alkaline substance and the drug has been subject to dry granulation before
compression
into tablets. Dry granulation of drug and alkaline substance together prior to
tablet
compression seem, surprisingly, to be a simple, inexpensive and efficient way
of providing
close physical contact between the alkaline substance and the drug and thus a
tablet
formulation with good stability properties. Relatively large BET areas of the
alkaline raw
material do also have a beneficial effect on the stability properties.
Dry granulation is carried out by a mechanical process, which transfers energy
to the
mixture without any use of any liquid substances (neither in the form of
aqueous
solutions, solutions based on organic solutes, or mixtures thereof) in
contrast to
conventional wet granulation processes. Generally, the mechanical process
requires
compaction such as the one provided by roller compaction. An example of an
alternative
method for dry granulation is slugging.
Roller compaction is a process comprising highly intensive mechanical
compacting of one
or more substances. The powder is pressed, that is roller compacted, between 2
counter
rotating rollers to make a solid sheet which is subsequently crushed in a
sieve to form a
particulate matter. In this particulate matter a close mechanical contact
between the
substance(-s) has been obtained. An example of equipment is Minipactoro or a
Gerteis
3W-Polygran from Gerteis Maschinen + Processengineering AG.
Tablet comaression according to the present invention takes place without any
use of any
liquid substances (neither in the form of aqueous solutions, solutions based
on organic
solutes, or mixtures thereof). In a typical embodiment the resulting core or
tablet must
have a crushing strength in the range of 10 to 150 N; such as 15 to 125 N,
preferably in
the range of 20 to 100 N.
A core is thus provided by compression of a powder or a particulate matter.
Typically the
core has a weight in the range of 75 mg to 2.5 g; such as 80 mg to 1 g; such
as 80 mg to
500 mg; such as 100 mg to 300 mg. Preferably, the core is a tablet with a
weight in the
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
12
range of 75 mg to 2.5 g; such as 80 mg to 1 g; such as 80 mg to 500 mg; such
as 100 mg
to 300 mg. In the present invention, the core is further coated with an
enteric coat and
optionally a subcoat to obtain the desired tablet formulation.
Tablets according to the present invention may be smaller than conventional
tablets, i.e.
having a diameter of about 7, preferably about 6 and most preferably about 5
mm or
below. In contrast to tablets with a diameter of e.g. 7.S mm or above, tablets
having a
smaller diameter will be able to move freely through the pylorus sphincter
into the small
intestine and thus be less dependent on the gastric emptying.
Subcoatinci:
Any conventionally used water soluble film forming excipient can be used for
subcoating
such as a sugar, polyethylene glycol, polyvinylpyrrolidone, polyvinyl alcohol,
hydroxypropyl
cellulose, methyl cellulose, hydroxymethyl cellulose, hydroxypropyl
methylcellulose,
polyvinyl acetal diethylenaminoacetate, "Kollicoat IR" (polyvinyl alcohol -
polyethylene
grycol graft copolymer), etc. Water or any conventionally used organic solvent
or a
mixture thereof is suitable as a subcoating solvent.
It is a general teaching within the field that the subcoat is critical to the
stability of the
entire formulation. In Sage (WO 9850019) it is e.g. disclosed that the enteric
coating and
the drug must be separated by a subcoat in order to avoid acid degradation of
the drug
(page 8, lines 16-30). Even though absence of a subcoat may be suggested in
various
patent documents (e.g. as a non-enabling statement in WO 04075881 on page 3,
line 21)
the existing knowledge leads the skilled person to the conclusion that tablets
produced
without subcoating have a poor drug stability. In contrast to the general
teaching, the
inventors of the present invention have surprisingly shown that is possible to
produce
tablets without subcoating having a drug stability compared with tablets with
conventional subcoating (table 18 and figure 9). As the subcoating process is
laborious and
time consuming, a production process according to the present invention
without laborious
subcoating steps is thus far more cost-efficient while still obtaining a
product with the
desired stability and dissolution properties.
Enteric coating
Any conventionally used enteric coating polymer can be used such as celiulose
acetate
phthalate such as Aquacoat CPD (FMC) or C-A-P NF (Eastman Chemical),
polyvinyl
acetate phthalate such as Sureteric (Colorcon), carboxymethylethylcellulose,
co-
polymerized methacrylic acid/methacrylic acid methyl esters such as Eudragit
L 30 D, or
Eudragit L 12.5 or Eudragit L 100 (Degussa - Rbhm Pharma Polymers) or
Kollicoat MAE
30 DP or Kollicoat 1OOP (BASF) or Acryl-Eze (Colorcon) or Eastacryl 30 D
(Eastman
Chemical) etc.
Preferred plasticizers include cetanol, triacetin, citric acid esters such as
Citroflexo (Pfizer),
phthalic acid esters, dibutyl succinate, acetylated monoglyceride,
acetyltributyl,
acetyltributyl citrate, acetyltriethyl citrate, benzyl benzoate, calcium
stearate, castor oil,
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
13
cetanot, chlorebutanol, colloidal silica dioxide, dibutyl phthalate, dibutyl
sebacate, diethyl
oxalate, diethyl malate, diethyl maleate, diethyl malonate, diethyl fumarate,
diethyl
phthalate, diethyl sebacate, diethyl succinate, dimethylphthalate, dioctyl
phthalate,
glycerin, glyceroltributyrate, glyceroltriacetate, glyceryl behanate, glyceryl
monostearate,
hydrogenated vegetable oil, lecithin, leucine, magnesium silicate, magnesium
stearate,
polyethylene glycol, propylene, glycol, polysorbate, silicone, stearic acid,
talc, titanium
dioxide, triacetin, tributyl citrate, triethyl citrate, zinc stearate, PEG
(polyethylene glycol),
etc. Methods for enteric coating are well known in the art such as described
in e.g. (Stuart
C. Porter in Remmington 215t Ed. 2005, pp 929), hereby incorporated by
reference.
Stability
Preferably, at least 95% (w/w) of the declared content of drug substance
remains in the
tablet formulation according to the present invention after storage at 25C /60
%RH
(relative humidity) of a period of 2, 3, 4, or 5 years. Alternatively, the
stability can be
determined after storage at other conditions according to appropriate ICH
guidelines.
Methods of assessing stability are described in the Examples.
Brief description of drawings
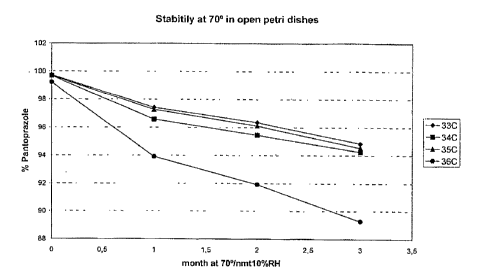
Flgure 1: Stability test of coated tablets containing Sodium Carbonate,
Calcium Carbonate
(Sturcal L), or Tri calcium phosphate. Test performed in open petri dishes at
70 and
not more than (nmt) 10 % relative humidity. Example 6.
Figure 2: Stability test of coated tablets containing Sodium Carbonate or
Calcium
Carbonate (Sturcal L) in the mixing ratio's of 1:0.2 or 1:0.8. Test performed
in open petri
dishes at 700 and not more than (nmt) 10 % relative humidity. Example 8.
Figure 3: Stability test of coated tablets containing Sodium Carbonate
(Sturcal L) 1:0.8
based on different sequential order of mixing and roller compaction. Test
performed in
open petri dishes at 70 and not more than (nmt) 10 %-relative humidity.
Example 10.
Figure 4: Particle size distribution using two different sieve sizes (1.25 mm
and 1.0 mm)
during roller compaction. Example 11.
Figure 5: Impact on dose variation of Roller compactor sieve size illustrated
by tablet core
dissolution. Example 11.
Figure 6: Impact on dissolution of amount of subcoat, HPMC E15, applied.
Example 12.
Figure 7: Impact on dissolution of type of subcoat, HPMC E 5 and HPMC E 15.
Evaluated on
subcoated tablets. Example,12.
Figure 8: Impact on dissolution of type of subcoat, HPMC E 5 and HPMC E 15.
Evaluated
for enteric coated tablets. Example 12.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
14
Figure 9: Stability of batches 13030634 and 31030634 (without the application
of a sub
coat) at 70 C in open petri dishes. Example 14.
Figure 10: Pantoprazole Sodium Sesquihydrate (SEM picture).
Figure 11: Calcium Carbonate (Sturcal L) (SEM picture).
Figure 12: Calcum Carbonate (Scoralite) (SEM picture).
Figure 13: Sodium Carbonate (SEM picture).
Figure 14: Sodium Carbonate (SEM picture).
Figure 15: Pantoprazole Sodium Sesquihydrate and Calcium Carbonate (Sturcal
L);
Mixing followed by slugging (SEM picture).
Figure 16: Pantoprazole Sodium Sesquihydrate and Calcium Carbonate (Scoralite)
Mixing followed by slugging (SEM picture).
Figure 17: Pantoprazole Sodium Sesquihydrate and Sodium Carbonate;
Mixing followed by slugging (SEM picture).
Figure 18: Pantoprazole Sodium Sesquihydrate and Sodium Carbonate
Pre rollercompaction of Pantoprazole, mixing with sodium carbonate, and
slugging (SEM
picture).
Figure 19: Impact of disintegrant on tablet core dissolution. Example 18.
It should be noted that, according to the present invention, embodiments and
features
described in the context of one of the aspects of the present invention also
apply to the
other aspects of the invention.
The following non-limiting examples are meant to illustrate the present
invention.
EXAMPLES
Example 1
Dry manufacture of tablets containing pantoprazole or omeprazole. Dry
granulation is
followed by compressing the resulting particulate matter into tablets.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
Table 2
Raw Manufactured bathes, no.
Materials * 1 2 3 4 5 6 L 8 9 10 11 12
a+b a+b
1 Pantoprazole 20,0 20.0 20.0 20.0 20.0 20.0
Na 1.51iz0
1 Omeprazole 20.0 20.0 20.0 20.0 20.0 20.0
Na
2 CaCO3 56.4 56.4 56.4 56.4
2 Na3PO4 56.4 56.4 56.4 56.4
2 NaZCO3 56.4 56.4 56.4 56.4
3 Sorbitok 17.6 17.6 17.6 17.6 17.6 17.6
3 Mannitol 17.6 17.6 17.6 17.6 17.6 17.6
4 MCC 5.25 5.25 5.25 5.25 5.25 5.25 5.25 5.25 5.25 5.25 5.25 5.25
5 Mg-stearate 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75
*: Amounts in % wjw
Table 3: List of ingredients
1 PantoprazoEe sodium sesquihydrate
1 Omeprazole sodium
2 CaCO3 Sturcal L
2 Na3PO4 trisodium phosphate
2 Na2CO3 sodium carbonate
3 Sorbitol
3 Mannitol
4 MCC; Cellulose Microcrystalline type 1010
5 Mg-stearate; magnesium stearate
5
The drug substance 1) (having a mean particle size of about 7 pm) was mixed by
hand
with the alkaline substance 2) and with 3).
10 The resulting mixture of the ingredients 1) to 3) was subjected to roller
compaction by use
of the following set of parameters:
Rpm: 2.0
Gab size 2.5 mm
15 Sieve size 1.25 mm
Force 10 kN/cm
The resulting particulate matter of the dry granulated ingredients 1) to 3)
was admixed
with 4) and 5). Thereafter, tablets were compressed of the mixture of
ingredients 1) to 5)
using a Diaf TM 20 press and 7.5 mm standard concave punch design. Unless
otherwise
stated, a relatively low compression force was used. '
Crushing strength and time of disintegration of the obtained tablets were
measured by use
of a Schleuniger E2 hardness tester (n=10) and a Sotax DT 2 (n=6). Both tests
were
performed according to the European Pharmacopoeia.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
16
Table 4: Crushing strength and disintegration time
Batch no. Crushing strength Disintegration time in water
[ N ] [sec. ]
1 39.5 291.0
2 39.5 151.0
3 41.3 520.0
4 39.9 457.8
41.1 496.5
6 42.1 523.0
7 32.4 281.8
8 30.9 477.5
9 36.0 300
38.0 284.7
11a 45.3 269.7
Low Comp.
Force
lib 90.6 301.8
High Comp.
Force
12a 40.7 256.8
Low Comp.
Force
12b 61.7 271.2
High Comp.
Force
The results from table 4 show that tablets containing pantoprazole or
omeprazole having a
5 satisfactory crushing strength and disintegration time can be manufactured
from a dry
manufacturing process based on a particulate matter provided by dry
granulation resulting
from roller compaction.
Furthermore, batches 11a+b and 12 a+b illustrate that the crushing strength
can be
10 increased without any significant influence on the disintegration time.
This means that the
tablets are of a quality that allows the application of a standard enteric
coating using
standard coating equipment and parameters. Optionally a sub coat comprising a
standard
water-soluble film like HPMC (Hydroxypropyl methylcellulose) can be used to
protect the
enteric coat from the alkaline reacting core.
Example 2
Crushing strength and disintegration time resulting from compression of
particulate matter
based on roller compaction
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
17
Table 5
Raw Materials 1 Manufactured batches, no.
13 14
1 Pantoprazole sodium 21.3 21.3
sesquihydrate
(Mean particle size around 7pm)
2 Na3PO4 trisodium phosphate 60.0 60.0
3 Sorbitol 18.7 -
3 Mannitol - 18.7
Amounts in % w/w
1) was mixed by hand with 2) and 3).
The resulting mixture of the ingredients 1) to 3) was roller compacted by use
of the
following set of parameters:
Rpm: 2.0
Gab size 2.5 mm
Sieve size 1.25 mm
Force 10 kN/cm
The particulate matter resulting from roller compaction of the ingredients 1)
to 3) was
compressed into tablets using a Diaf TM 20 press and 7.5 mm standard concave
punch
design. Crushing strength and time of disintegration of the obtained tablets
were
measured by use of a Schleuniger E2 hardness tester (n=10) and a Sotax DT 2
disintegrations tester (n=6).
Table 6: Crushing strength and disintegration time
Batch no. Crushing strength [N] Disintegration time [sec.]
13 42.6 443.0
14 52.6 486.7
The results from table 6 show that tablets containing pantoprazole having a
satisfactory
crushing strength and disintegration time can be manufactured by compression
of a
particulate matter provided by a dry granulation process based on roller
compacted
granulates, without the addition of further excipients (apart from a dry
binder). Addition of
magnesium stearate could, however, be advantageous in production scale.
The tablets are thus of a quality that allows the application of a standard
enteric coating
using standard coating equipment and parameters. Optionally a sub coat
comprising a
standard water-soluble film like HPMC can be used to protect the enteric coat
from the
alkaline reacting core.
Example 3
Crushing strength and disintegration time of tablets resulting from direct
compression of
mixtures of pantoprazole and compactable alkaline excipient
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
18
Table 7
Raw material D(v;0.5) 10060531 10060532
[pm]
1 Pantoprazole sodium 7 20.00 20.00
sesquihydrate
2 Trisodium phosphate, 203 79.26 71.34
coarse
2 Trisodium phosphate, 40 7.92
fine
3 Magnesium stearate - 0.74 0.74
*: Amounts in % w/w
The drug substance 1) and the ingredient 2) were mixed by hand followed by
admixing of
3). The mixture of ingredients 1) to 3) was compressed into tablets using a
Diaf TM 20
press and 7.5 mm standard concave punch design.
Crushing strength and time of disintegration of the obtained tablets were
measured by use
of a Schleuniger E2 hardness tester (n=10) and a Sotax DT 2 disintegration
tester (n=6).
Table 8: Crushing strength and disintegration time
Batch no. Crushing strength [N] Disintegration time Mass variation
[sec.] [ s.rel - roj
10060531 37.4 657 0.96
10060532 41.2 631
The results shown in table 8 show that tablets containing pantoprazole having
a
satisfactory crushing strength and disintegration time result from direct
compression of the
compactable alkaline substance. The mass variation illustrates that the
flowability of the
mixture of ingredients 1) to 3) is acceptable. It is conceivable that the
coarse alkaline
substance functions both as a filler substance and an alkaline substance in
this
formulation.
The resulting tablets are of a quality that allows the application of a
standard enteric
coating using standard coating equipment and parameters. Optionally a sub coat
comprising a standard water soluble film like HPMC can be used to protect the
enteric coat
from the alkaline reacting core.
Example 4
Manufacture of tablets having a diameter of 5 mm based on a particulate matter
resulting
from dry granulation in a roller compactor
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
19
Table 9
Raw Materials * 1 2 3 4 5 6 7 8
1 Pantoprazole 40 40 40 40
sodium 1.5H20
1 Omeprazole 40 40 40 40
sodium
2 Na3PO4 40 40 40 40
2 Na2CO3 40 40 40 40
3 Sorbitol 18.25 18.25 18.25 18.25
3 Mannitol 18.25 18.25 18.25 18.25
4 Mg-stearate 0.75 0.75 0.75 0.75 0.75 0.75 0.75 0.75
Amounts in %w/w
Table 10: List of ingredients
1 Pantoprazole sodium sesquihydrate
1 Omeprazole sodium
2 Na3PO4 trisodium phosphate
2 NaZCO3 sodium carbonate
3 Sorbitol
3 Mannitol
4 Mg-stearate; magnesium stearate
The drug substance 1) was mixed by hand with 2) and 3).
The resulting mixture of the ingredients 1) to 3) was dry granulated by roller
compaction
by use of the following set of parameters:
Rpm: 2.0
Gab size 2.5 mm
Sieve size 1.25 mm
Force 10 kN/cm
The resulting particulate matter of the dry granulated ingredients 1) to 3)
was admixed
with 4). The resulting mixture of the ingredients 1) to 4) was compressed into
tablets
using a 5.0 mm standard concave punch design. The resulting tablets are of a
quality that
allows applying a standard enteric coat using standard coating equipment and
parameters.
Optionally a sub coat consisting of a standard water soluble film like FIPMC
can be used to
protect the enteric coat from the alkaline reacting core.
Example 5:
Dry manufacture of tablets containing pantoprazole and alkaline excipients
followed by
application of a sub coat and an enteric coating.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
Table 11: Composition of tablet core % w/w:
Batch no. 07030631 10030631 13030634 13030636
"33C" "34C" "35C" "36C"
Granulate by roller compaction
Pantoprazo{e, 11/z H20 13.3 11.7 14.1 12.9
Sodium carbonate 13.3 55.2 -
Calcium carbonate - 54.9 - -
(Sturcal L)
TriCalciumPhosphate 60.6
Pantoprazole:alkaline excipient 1:1 1:4.7 1:3.9 1:4.7
Mannitol - 13.5 - 14.9
Crospovidone 0.4 1.1 1.1 1.2
Tablet core excipients
DiCafos A 62.7 8.5 19.2 -
Crospovidone 9.7 9.7 10.0 9.8
Mg Stearate 0.5 0.4 0.5 0.5
Table 12: Composition of sub coat [% w/w]:
Raw materials Coating liquid Dry film
composition composition
Hypromellose 15 5 55,60lo
Propylenglycol 1 11,1%
Talc 3 33,3%
Water, purified 91
% Plasticizer of polymer 20,0%
5 Table 13: Composition of enteric coat [% w/w]::
Raw materials Coating -iquid Dry film
composition composition
Methacryl acid-acryl 41,76 62,49
Copolymer disp. 30%
Triethylcitrate 1,25 6,24
Talc 6,27 31,28
Water, purified 50,92
% Plasticizer of Polymer 10,0
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
21
Table 17: Approximately amount of film dry matter applied [% w/w]:
Coat 07030631 10030631 13030634 13030636
"33C" "34C" "35C" "36C"
Theoretical applied 9,7 10,3 10,9 9,0
amount of sub coat
[mg polymer/cmz]
Theoretical applied 5,5 5,4 5,3 + 2 5,7
amount of enteric coat
[mg polymer/cmZ]
Pantoprazole was mixed with the alkaline excipient in a tumble mixer together
with
Crosspovidone and mannitol followed by roller compaction as described in
example 1.
The remaining tablet core excipients were admixed and tablets were compressed
by use of
a Korsch PH106 tablet press and 6 mm concave punches aiming at a mean weight
of 160
mg and a crushing strength of 50 N. Thereafter the tablet cores were coated
with the sub
coat followed by the enteric coat by use of a lab-scale Combi Coata. The
obtained coated
tablets were used for stability testing as described in example 6.
Example 6 (stability testing)
A stability testing program with batches obtained in example 5 was performed.
The
batches were stored at accelerated stability testing conditions (open petri
dishes at 70 C
and not more than 10% relative humidity for three months). Such conditions
probably
correspond to shelf life stability testing of at least two years.
The analytical method is as follows: 10 tablets are transferred to a 200 ml
volumetric
flask. 150 ml mobile phase (the initial composition) is added and sample is
shaken for 90
minutes. After the solutions pH-values have been adjusted to 8.0, mobile phase
is added
to the mark. The sample solution is filtered through 0.45 m filter and
analysed by reverse
phase HPLC in order to quantify the amount of Pantoprazole as well as
degradation
products thereof. The amount is given in % of total area, see table 19.
Furthermore, in
figure 1 is shown the amount of pantoprazole as a function of time.
Table 18: HPLC method parameters
Mobile phase ammonium phosphate buffer: methanoi: acetonitril
Time % Buffer % Methanol % Acetonitril
0 min 65 10 25
Gradient profile 25 min 30 10 60
min 30 10 60
31 min 65 10 25
Flow 1,0 mI/min
Column Waters Spherisorb, 250X4,6 mm, 5 pm particle size
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
22
Column temperature 30 C
Auto sampler temperature 4 C
Detection UV-290 nm
Injection volume 10 ial
Run time 40 min.
Pantoprazole 10,7 min
Approx. Retention time Impurity B 6,6 min
Impurity A 13,4 min
Table 19: Results of stability study
Batch 07030631 10030631 13030634 13030636
"33C" "34C" "35C" "36C"
Pantoprazole:sodium carbonate 1:1 1:3,9
Pantoprazole:calcium carbonate 1:4,7
Pantoprazole:calcium phosphate 1:4,7
Pantoprazole:DiCafos A 1:4,7 1:0;7 1:1,4
9 25160 C lmonth 99,7 99,7 99,7 99,2
' 70110 OP 1 month 97,4 96,6 97,3 93,9
o ca
Q2
70/10 OP 2 months 96,4 95,5 96,1 91,9
70/10 OP 3 months 94,9 94,3 94,6 89,3
cu
40/75 3 months 99,4 99,4 99,5 97,2
25/60 C 1month 0,18 0,15 0,20 0,56
0- 70/10 OP lmonth 2,2 3,2 2,3 6,0
M
a)
~~~ 70/10 OP 2 months 3,3 4,0 3,6 7,6
70/10 OP 3 months 4,3 4,8 4,4 9,3
a
40175 3 months 0,5 0,5 0,5 2,6
70/10 OP: 70 C / 10 % RH in Open petri dishes
25/60: 25 C / 60% RH in closed containers
40/75: 40 C / 75% RH in closed containers
According to table 19 and figure 1, use of Ca$(P04)3OH as alkaline substance
result in a
relatively larger degree of pantoprazole degradation compared with use of
Na2CO3, CaCO3i
and DiCafos A as alkaline excipients. The stability data for the stressed
stability testing
conditions (70 C open Petri dishes, 70/10 OP) probably corresponds to a shelf
life of at
least two years.
Example 7:
Dry manufacture of tablets containing pantoprazole and Sodium carbonate or
calcium
carbonate and a disintegrant followed by application of a sub coat and an
enteric coating.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
23
Table 20: Composition of tablet core % w/w:
01050633 3F-C 05050632 29050636 29050640
2F-C 7F-C 8F-C
Granulate by roller
compaction
Pantoprazole, 11/2 H20 14.1 14.1 14.1 14.1
Sodium carbonate 2.82 11.3
Calcium carbonate
2.82 11.3
(Sturcal L)
Pantoprazole: alkaline exciplent 1:0.2 1:0.8 1:0.2 1:0,8
Tabiet core excipients
DlCafos A 69.6 61.1 69.6 61.1
Crosspovidone 13.0 13 13.0 13
Mg Stearate 0.5 0.5 0.5 0.5
Based on the tablet core compositions listed above coated tablets were
manufactured as
described in example 5 with the following exception:
The pantoprazole was pre-rollercompacted prior to mixing with the alkaline
excipient by
use of the following parameters
Rpm: 2.0
Gap size: 1.0 mm
Sieve size: 1.25 mm
Force: 4 kfV/cm
The pre-roller compaction leads to formation of pantoprazole granules.
The obtained coated tablets were used for stability testing with the purpose
of
investigating the impact of lower amounts of alkaline material than used in
example 5 and
the use of pre-roffercompaction of the pantoprazole. The stability testing is
disclosed in
example B.
ExampEe 8: stability testing
A stability program, including batches obtained in example 7 was performed.
The batches
were stored in open petri dishes at 70 C and not more than 10 % RH for up to
six weeks.
The anaEytical method is as described in example 6
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
24
Table 21: Result of stability study
01050633 05050632 29050636 29050640
batch 3F-C 2F-C 7F-C 8F-C
Pantoprazole:sodium carbonate 1:0.8 1:0.2
Pantoprazole:calcium carbonate 1:0.2 1:0.8
Pantoprazole:DiCafos A 1:5 1:4,3 1:5 1:4,3
Pantoprazole 25/60 C 2 weeks 99,7 99,8 99,8 99,1
in % area 70/10 OP 2weeks 98,3 97,9 98,3 98,1
70/10 OP 6weeks 97,5 97,2
Degradation 25/60 C 2 weeks 0,20 0,15 0,10 0,52
products in 70/10 OP 2weeks 1,2 1,6 1,4 1,2
% area 70/10 OP 6weeks 2,0 2,4
It appears in table 21 and figure 2 that use of sodium carbonate and calcium
carbonate as
alkaline excip9ent at the mixing ratios of 1:0.2 and 1:0.8 result in nearly
identical and
acceptable degradation ratios when tested at 70 C and not more than 10 % RH
for up to
six weeks. It is surprising that the solubility of the alkaline material
apparently does not
affect stability of the benzimidazole composition (calcium carbonate is poorly
soluble in
water).
Example 9:
Dry manufacture of tablets containfng pantoprazole and alkaline excipients
focusing on
order of mixing and roller compaction followed by application of a sub coat
and an enteric
coating.
Table 22: Composition of tablet core % w/w:
Granulate by roller compaction
Pantoprazole, 1l/z H20 14.1
Sodium carbonate 11.3
Pantoprazole:alkafine excipient 1:0.8
Tablet core excipients
DiCafos A 61.1
Crospovidone 13
Mg Stearate 0.5
The composition shown in table 22 was mixed and roller compacted in different
sequential
order as shown in table 23
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
Table 23: Sequential order of mixing and roller compaction:
Batch no. 1 2 3 4
(Of coated Mixing with Pre-roller Mixing with Roller
tablets) alkaline compaction alkaline compaction
excipient (solely excipient
pantoprazole)
01050633 3F-C Not used X X X
05050635 X Not used Not used X
05050638 12F-C Not used X X Not used
Mixing and roller compaction were carried out as described in example 5.
5 The remaining tablet core excipients (Dicafos A, Crosspovidone and Mg-
stearate) were
admixed and tablets were compressed and coated in accordance with example 5.
The batches 01050633 and 05050638 were used for stability testing in example
10 and
05050635 was used for comparison with batches of example 16 with the purpose
of
10 evaluating excipient homogeneity of granules.
Example 10: stability testing
A stability program, including batches mentioned in example 9 was performed.
The
15 batches were stored in open petri dishes at 70 C for 2 weeks.
The analytical method is as described in example 6
Table 24: Results of stability testing:
01050633 3F-C 05050638 12F-C
batch
Pantoprazole:sodium carbonate 1:0.8 1:0.8
Pantoprazole:DiCafos A 1:5 1:4,3
Pre-
rollercompacting, Pre-
mixing, rollercompacting,
- rollercompacting mixing
Pantoprazole 25160 C 2 weeks 99,7 99,5
in % area 70/10 OP 2weeks 98,3 97,2
70/10 OP 6weeks 97,5
Degradation 25/60 C 2 weeks 0,20 0,33
products in 70/10 OP 2weeks 1,2 2,9
% area 70/10 OP 6weeks 2,0
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
26
It appears in table 24 and figure 3 that panoprazole degradation is relatively
small when
pantoprazole is pre-roller compacted, mixed with the alkaline excipient and
then roller
compacted again. The pantoprazole degradation is relatively high when
pantoprazole is
pre-roller compacted and mixed with the alkaline excipient without including a
step of
roller compacting pantoprazole and alkaline substance together. The impact on
stability is
seen already after two weeks.
Example 11:
Dry manufacture of tablets containing pantoprazole and alkaline excipients.
Roller
compaction has been carried out using two different sieve sizes. Tableting was
followed by
application of a sub coat and an enteric coating.
Table 25: Composition of tablet core % w/w:
Granulate by roller compaction
Pantoprazole, 11/2 HZ0 14.1
Sodium carbonate 2.82
Pantoprazole:alkaline excipient 1:0.2
Tablet core excipients
DiCaPos A 69.6
Crospovidone 13.0
Mg Stearate 0.5
Based on the tablet core compositions listed above coated tablets were
manufactured as
described in example 5 with the following exception:
The pantoprazole was pre-rollercompacted prior to mixing with the alkaline
excipient by
use of the following parameters
Rpm: 2.0
Gap size: 1.0 mm
Sieve size: 1.25 mm (used for batch 29050641) or 1.0 mm (used for batch
29050645)
Force: 4 kN/cm
The pre-roller compacted pantoprazole and the sodium carbonate were mixed by
use of a
tumble-mixer and roller compacted using the following parameters:
Rpm: 2.0
Gap size: 2.5 mm
Sieve size: 1.25 mm (used for batch 29050641) or 1.0 mm (used for batch
29050645)
Force: 10 kN/cm
The remaining tablet core excipients were admixed and tablets were compressed
and
coated in accordance with example 5.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
27
Evaluation of the impact of the sieve size was based on sieve analysis and
dissolution
testing. Dissolution was carried out by use of the following method:
Table 26: Dissolution method.
USP/Ph.Eur Dissolution apparatus 1
Spindle Basket
Rotation 150 rpm
Temperature 37 C 0,5 C
Filter Whatman GF/F (0.7 pm)
Dissolution medium 0- 120 minutes 600 ml 0.1 N HC!
After 120 minutes the dissolution medium 200 ml 0.20 M Na3PO4 added to the
is changed to pH 6.8 vessel (method A, USP)
Detection, UV 288 nm
Sampling time The absorbance is measured by each
minutes at 288 nm
5
From figure 4 it can be seen that the use of a sieve size of 1.0 mm results in
a relatively
narrow particle size distribution compared to sieve size 1.25. The narrow size
distribution
results in a better dose variation of pantoprazole as can be seen form figure
S.
Example 12:
Dry manufacture of tablets containing pantoprazole and alkaline excipients
followed by
application of different types and amounts of sub coat.
Table 27: Composition of tablet core % w/w:
Granulate by roller compaction
Pantoprazole, 11/2 H20 14,1
Sodium carbonate 55,2
Pantoprazole: allcaline excipient 1:3,9
Crospovidone 1,1
Tablet core excipients
DlCafos A 19,2
Crospovidone 10,0
Mg Stearate 0,5
Tablet cores according to the above mentioned composition were manufactured as
described in example S. Sub coat was applied as laid out in example 5 with the
exception
of variation in applied amount and type of polymer as shown En table 28:
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
28
Table 28: Amount (approximately) and type of sub coat applied:
Batch no. HPMC type Theoretical amount of HPMC
Cm9/cm2]
13030634 E15 10.9 (1/1 amount)
31030633 (C) E15 5.3 (1/2 amount)
21040634 E5 9.9 (1/1 amount)
31030631 - -
Enteric coat was applied as described in example 5.
Evaluation of the impact of amount and type of sub coat was based following
dissolution
results obtained obtained in example 11. However, tablets only coated with a
sub coat
were analysed solely in a dissolution medium with pH 6.8.
Figures 6, 7 and 8 disclose the impact of type of sub coat on the dissolution
rate form both
sub coated tabiets and sub + enteric coated tablets. A HPMC E5 based sub coat
results in a
relatively quick dissolution rate.
Example 13:
Dry manufacture of tablets containing pantoprazole and alkaline excipients
followed by
application of an enteric coating.
Tablet cores of batch 13030634 and 31030634 were manufactured as described in
example 12. The tablet cores of batch 13030634 were enteric coated as
described in
example S. Batch 31030634 was produced with an enteric coating but without the
application of a sub coat. The enteric coated tablets were used for stability
testing in
example 14.
Example 14: stability testing
A stability program, including batches mentioned in example 13 was performed.
The
batches were stored in open petri dishes at 70 C for respectively 2 and 3
months.
The analytical method is as described in example 6
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
29
Table 29: Results of stability testing
Batch 13030634 31030634
Without sub coat
25/60 C
1 month 99,7 99,7
Pantoprazole 70/10 OP 97,3 97,9
in % of area I month
70/10 OP
2 months 96,1 97,4
25/60 C
1 month 0,20 0,22
Degradation products. 70/10 OP
in % of area 1 month 2,3 1,7
70/10 OP
2 months 3,6 2,2
70/10 OP: 70 C / 10 % RH in Open petri dishes
25/60: 25 C / 60% RH in closed containers
It appears from table 29 and figure 9 that the stability (70 C / 10 % RH in
Open petri
dishes and 25 C / 60% RH in closed containers) of sub-coated tablets is fully
comparable
with tablets which has not been sub-coated.
Example 15:
Impact of type and amount of alkaline excipient on deg'radation of
pantoprazole in a stress
test by the addition of a weakly acidic component (ibuprofen)
Table 30: Composition of mixtures [gram]
Raw materials Amounts in gram
Sodium carbonate 4 16 20 25
Calcium carbonate 4 16 20 25
(Sturcal L)
Ibuprofen 0.8 0.8 0.8 0.8 0.8 0.8 0.8 0.8
Pantoprazole 11/2 H20 20 20 10 5 20 20 10 5
Pantoprazole:alkaline 10.2 1:0.8 1:2 1:5 1:0.2 1:0.8 1:2 1:5
excipient
Table 31: Composition of mixtures [gram]
Raw materials Amounts in gram
Calcium carbonate 4 16 20 25
(Scoralite)
Tri calcium phosphate 4 16 20 25
Ibuprofen 0.8 0.8 0.8 0.8 0.8 0.8 0.8 0.8
Pantoprazole 11/2 HZO 20 20 10 5 20 20 10 5
Pantoprazole:alkaline 1:0.2 1:0.8 1:2 1:5 1:0.2 1:0.8 1:2 1:5
excipient
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
Mixtures were made by grinding the raw materials in a steel bowl and
subsequently placing
the mixture in petri dishes in sealed alu-bags for two weeks at ambient
conditions.
5 The evaluation of this stability test is shown in table 32 (pantoprazole
degradation
products are coloured). Discoloration is evaluated based on a scale ranging
from 1 - 10
where "1" indicates no discoloration and "10" indicates a severe
discoloration.
Table 32. Discoloration scale of powder mixtures of table 30 and 31:
Alkaline material Mixing ratio Mixing ratio Mixing ratio Mixing ratio
1:0.2 1:0.8 1:2 1:5
Sodium carbonate 9 8 4 1
Calcium carbonate 10 8 4 1
(Sturcal L)
Calcium carbonate 10 10 7 7
(Scoralite)
Tri calcium 9 6 - 1
phosphate
The results in table 32 are discussed in example 16.
Example 16:
Characterisation of alkaline materials.
Alkaline materials:
* Calcium carbonate, Sturcal L
* Calcium carbonate, Scoralite
* Sodium carbonate anhydrate
Alkaline substance raw material particle sizes have. been measured by laser
light scattering
(Malvem) and BET area has been measured by use of a Micromeritics Gemini 2375
at
relative target pressures (P/Po) of 0.1 and 0.2 and 0.3. Samples have been
dried for
minimum 12 hours at 40 C prior to the measurements.
Table 33: BET-areas
Raw materials BET-area [mz/g] Particle size D(v 0,5) [pm]
Calcium carbonate, Sturcal L 3.0 9
Calcium carbonate, Scoralite 0.2-0.5 29
Sodium carbonate anhydrate 2.1 99
The SEM pictures (figures 10-18) illustrate considerable differences in size
and morphology
on the alkaline raw materials. It should be noted that even though Sodium
carbonate
anhydrate has a larger particle size than the Calcium carbonate (Scoralite),
sodium
carbonate anhydrate has the largest BET area. This difference is further
illustrated in
example 17 by use of Scanning Electron Microscope pictures. The impact of the
BET area
and particle size on stability was illustrated in example 15.
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
31
The discoloration shown in table 32 demonstrates the impact of BET area (see
example 16,
table 33) and mixing ratio on pantoprazole stability. A high BET area favours
good stability
results. It appears from example 16 that a relatively small particle size may
not suffice to
ensure a satisfactory stability. Relatively big particles can be useful,
provided that their
porosity leads to a sufficiently high BET area.
Furthermore, it is illustrated that a high amount of alkaline material is
preferred.
However, the discoloration test cannot reveal smaller amount of degradation
product as
can be seen by comparison of the addition of tri calcium phosphate in example
6. This
means that tri calclum phosphate is not as efficient as e.g. sodium carbonate
and calcium
carbonate (Sturcal L).
Example 17
Impact of type and amount of alkaline excipient on homogeneity of a mixture
with
pantoprazole with alkaline materials.
Composition of powder mixtures
* Pantoprazole, 11/2 H20 : Calcium carbonate (Sturcal L), 1:0.8
* Pantoprazole, 11/z H20 : Calcium carbonate (Scoralite), 1:0.8
= Pantoprazole, 11I2 H20 (pre-roller compacted) : sodium carbonate, 1:0.8
Pantoprazole, 11/2 H20 : Calcium carbonate (Sturcal L) and Pantoprazole, 11/2
H20 :
Calcium carbonate (Scoralite) batches have been mixed in a lab. scale high
shear mixer for
1 minute. The Pantoprazole, 11/7. H20 (pre-roller compacted) batch was
manufactured as
described in example 7 prior to mixing with sodium carbonate. The mixing was
done as
described above.
All the mixtures were slugged on a single punch tableting machine using 11.3
mm flat
faced punches. The slugs were evaluated for mixture homogeneity by use of
scanning
electronic microscopy (SEM). For reference, SEM pictures of the individual raw
materials
were obtained.
The SEM pictures are shown in the figures 10 - 18. Figure 11 and 12 disclose a
considerable difference between Calcium carbonate (Sturcal L) and Calcium
carbonate
(Scoralite). SEM appearances of Sturcal L and Scoralite are in full accordance
with the BET
areas measured in example 16.
Figure 13 is a magnification of the particles shown in figure 14. The
magnification showing
the porosity of the particles clearly supports the finding of a high BET area
of Sodium
Carbonate is illustrated.
Figures 15 and 16 show that the use of Calcium carbonate (Sturcal L) leads to
a much
more homogeneous mixture than Calcium carbonate (Scoralite). The impact on
stability of
this difference was illustrated in example 15. Figure 17 shows that the sodium
carbonate
CA 02614526 2008-01-09
WO 2006/105798 PCT/DK2006/000409
32
particles are crushed during manufacturing. The physical structure of Sturcal
L results in
an improved distribution of the particles during mechanical pressure. An
acceptable
stability is obtained as illustrated in examples 15 and 6, 8 and 10.
Figure 18 iliustrates the effect of pre-roller compacting pantoprazole prior
to mixing with
alkaline substance. Slugging (or roller compaction) of the mixture results in
"coating" of
the surface of the relative large pantoprazole granules with calcium carbonate
particles
(Sturcal L). This "coating" also leads to an acceptable stability as shown in
example 6 and
8. The need for using roller compaction to apply this coat is indicated in
example 9, which
was based on the use of sodium carbonate.
Example 18.
Impact of disintegrant on dissolution rate.
Table 34: Composition of tablet core % w/w:
Pantoprazole, 1112 H20 14.1 14.1
Calcium carbonate
11.3 11.3
(Sturcal L)
Pantoprazole:alkaiine 1,0 8 1=0,8
excipient ~ '
DiCafos A 74.1 61.1
Crospovidone - 13
Mg Stearate 0.5 0.5
Based on the tablet core compositions listed above tablets were manufactured
as described
in example 7 with the following exception that no coat has been applied.
The cores were tested with respect to dissolution rate as described in example
11 with the
exception that tablet cores were analysed solely in a dissolution medium with
pH 6.8.
The results shown in figure 19 illustrate the advantages of the incorporation
a disintegra,nt
as the presence of a disintegrant in the formulation significantly increases
the dissolution
rate.