Note: Descriptions are shown in the official language in which they were submitted.
CA 02626028 2013-03-21
INDOLE DERIVATIVES AS HISTAMINE 3 RECEPTOR INHIBITORS FOR THE
TREATMENT OF COGNITIVE AND SLEEP DISORDERS, OBESITY AND OTHER
CNS DISORDERS
, s
TECHNICAL FIELD
This invention relates to compounds having pharmacological activity, to
compositions containing these compounds, and to a method of treatment
employing the
compounds and compositions. More particularly, this invention concerns certain
indole
derivatives and their salts and solvates. These compounds alter H3 histamine
receptor activity.
This invention also relates to pharmaceutical compositions containing these
compounds and
to a method of treating disorders in which histamine H3 receptor blockade is
beneficial.
BACKGROUND OF THE INVENTION
=
Histamine is a chemical messenger involved in various complex biological
actions. When released, histamine interacts with specific macromolecular
receptors on the
cell surface or within a target cell to elicit changes in many different
bodily functions.
Various cell types including smooth muscle, blood cells, cells of the immune
system,
endocrine and exocrine cells as well as neurons respond to histamine by
modulating the
formation of intracellular signals, including of phosphatidylinositol, or
adenylate cyclase.
Evidence that histamin. e plays a role as a neurotransmitter was established
by the mid-to-late
1970's (Schwartz, 1975) Life Sci. 17:503-518. Immunohistochemical studies
identified
histaminergic cell bodies in the tuberomarmnillary nucleus of the posterior
hypothalamus
with widespread projections in the diceneephalon and telencephalon (Inaga)d et
al., 1998) .I.
Comp. Neurol. 273:283-300.
Two histamine receptors (H1 and H2) were reported to mediate the
biochemical actions of histamine oil neurons. =More recently, studies have
demonstrated the
existence of a third subtype of histamine receptor, the histamine H3 receptor
(Schwartz et al.,
=
_ _
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
1986) TIPS 8: 24-28. Various studies have now demonstrated that histamine H3
receptors are
found on the histaminergic nerve terminals in the brains of several species,
including man
(Arrang et al., 1983) Nature 302: 832-837. The H3 receptor found on the
histaminergic nerve
terminal was defined as an autoreceptor and could intimately control the
amount of histamine
released from the neurons. Histamine, the natural compound, was capable of
stimulating this
autoreceptor but testing of known H1 and H2 receptor agonists and antagonists
suggested that
the H3 receptor has a distinct pharmacological profile. Further, H3 receptors
have been
identified on cholinergic, serotoninergic and monoamine nerve terminals in the
peripheral
nervous system (PNS) and central nervous system including the cerebral cortex
and cerebral
vessels. These observations suggest that H3 receptors are uniquely located to
modulate
histamine as well as other neurotransmitter release, and compounds that bind
H3 receptors
could be important mediators of neuronal activity.
As stated, CNS histaminergic cell bodies are found in the magnocellular nuclei
of the hypothalamic,mammillary region and these neurons project diffusely to
large areas of
the forebrain. The presence of histaminergic cell bodies in the
tuberomammillary nucleus of
the posterior hypothalamus, a brain area involved in the maintenance of
wakefulness, and
their projections to the cerebral cortex suggest a role in modulating the
arousal state or sleep-
wake cycle. The histaminergic projection to many limbic structures such as the
hippocarnpal
formation and the amygdaloid complex suggest roles in functions such as
autonomic
regulation, control of emotions and motivated behaviors, and memory processes.
The concept that histamine is important for the state of arousal, as suggested
by the location of histaminergic pathways, is supported by other types of
evidence. Lesions
of the posterior hypothalamus are well known to produce sleep. Neurochemical
and
electrophysiological studies have also indicated that the activity of
histaminergic neurons is
maximal during periods of wakefulness and is suppressed by barbiturates and
other hypnotics.
Intraventricular histamine induces the appearances of an arousal EEG pattern
in rabbits and
increased spontaneous locomotor activity, grooming and exploratory behavior in
both saline
and pentobarbital-treated rats.
In contrast, a highly selective inhibitor of histidine decarboxylase, the sole
enzyme responsible for histamine synthesis, has been shown to impair waking in
rats. These
data support the hypothesis that histamine may function in modulating
behavioral arousal.
The role of the H3 receptor in sleep-waking parameters has been demonstrated
(Lin et al.,
1990) Brain Res. 592: 325-330. Oral administration of RAMHA, a H3 agonist,
caused a
significant increase in deep slow wave sleep in the cat. Conversely,
thioperamide, a H3
2
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
antagonist/inverse agonist, enhanced wakefulness in a dose-dependent fashion.
Thioperamide has also been shown to increase wakefulness and decrease slow-
wave and
REM sleep in rats. These findings are consistent with in vivo studies
demonstrating that
thioperamide caused an increase in synthesis and release of histamine.
Together, these data
demonstrate that selective H3 antagonists or inverse agonists may be useful in
the treatment
of arousal states and sleep disorders.
Serotonin, histamine, and acetylcholine have all been demonstrated to be
diminished in the Alzheimer's (AD) brain. The histamine H3 receptor has been
demonstrated
to regulate the release of each of these neurotransmitters. An H3 receptor
antagonist or
inverse agonist would therefore be expected to increase the release of these
neurotransmitters
in the brain. Since histamine has been demonstrated to be important in arousal
and vigilance,
H3 receptor antagonists or inverse agonists might enhance arousal and
vigilance via
increasing levels of neurotransmitter release and thereby improve cognition.
Thus, the use of
compounds that bind the use of H3receptor in AD, attention deficit disorders
(ADD), age-
related memory dysfunction and other cognitive disorders would be supported.
H3 receptor antagonists or inverse agonists may be useful in treating several
other CNS disorders. It has been suggested that histamine may be involved in
cerebral
circulation, energy metabolism, and hypothalmic hormone secretion. For
example, H3
receptor antagonists or inverse agonists have been demonstrated to affect food
intake and
body weight gain in rodents. Recent evidence has indicated the possible use of
H3
antagonists or inverse agonists in the treatment of epilepsy. Work has
demonstrated an
inverse correlation between the duration of clonic convulsions and brain
histamine levels.
Thioperamide was also shown to significantly and dose-dependently decrease the
durations
of every convulsive phase after electrically-induced convulsions and increase
the
electroconvulsive threshold.
In spite of their low density, H3 receptor binding sites can be detected
outside
the brain. Several studies have revealed the presence of H3 heteroreceptors in
the
gastrointestinal tract, as well as upon neurons of the respitory tract.
Accordingly, an H3
receptor binding compound may be useful in the treatment of diseases and
conditions such as
asthma, rhinitis, airway congestion, inflammation, hyper and hypo motility and
acid secretion
of the gastrointestinal tract. Peripheral or central blockage of H3 receptors
may also
contribute to changes in blood pressure, heart rate and cardiovascular output
and could be
used in the treatment of cardiovascular diseases, and in the treatment of
diseases or
conditions such as obesity, migraine, inflammation, motion sickness, pain,
ADHD, dementia,
3
CA 02626028 2008-04-14
WO 2007/047775 PCT/US2006/040744
depression, Parkinson's disease, schizophrenia, epilepsy, narcolepsy, acute
myocardial
infarction and asthma.
Various indole derivatives are disclosed in U.S. Patent Nos. 5,631,381 and
6,630,496 B1; WO 93/25524; WO 99/43672 and WO 2004/099192.
SUMMARY OF THE INVENTION
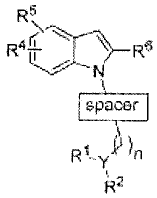
The present invention provides, in its principal aspect, compounds of the
general formula:
R5
>
R4.1- R6
\
spacer
R1-y n
R2
wherein
spacer is
R3¨E
R3-T R3--y
0,s5s5, I I
A
Y is CH or N, provided that if Y is CH then n is 0 ¨ 2; if Y is N then n is 2
¨ 4;
if Y is CH then RI and R2 taken together are ¨(CH2)a-NR"-(CH2)2- where a is 1-
2 which
when taken together with Y form a piperidine or pyrrolidine ring which is
optionally
substituted with 1-3 groups selected from fluoro, fluoroalkyl, (C1-C4)alkyl,
alkoxy, aryl, (C3-
C7)cycloalkyl, heterocycloalkyl containing 1-2 hetero atoms selected from (0,
S) and (CI-
C5)alky1-0-(C1-05)alkyl; and
if Y is N then RI and R2 independently are (CI-05)alkyl or (C3-C6)cycloalkyl,
or RI and R2
taken together with the nitrogen to which they are attached form a 5-7 member
heterocyclic
ring system with 0-1 additional hetero atoms selected from 0 and S which is
optionally
4
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
substituted with 1-3 (Ci-05)alkyl, fluoroalkyl or (C3-C6)cycloalkyl groups, or
R1 and R2 taken
together are ¨(CH2)a-NR11-(CH2)2- ,where a is 2-3, which when taken together
with Y form a
piperazine or homopiperazine ring which is optionally substituted with 1-3
groups selected
from fluoro, fluoroalkyl, (Ci-C4)alkyl, alkoxy, aryl, (C3-C7)cycloalkyl,
heterocycloalkyl
containing 1-2 hetero atoms selected from (0, S) and (Ci-05)alky1-0-(Ci-
05)alkyl;
R3 is 0-2 of groups selected from halogen, (Ci-C6)a1ky1, (C1-C6)alkoxy, (C3-
C7)cycloalkyl,
(C3-C7)cycloalkyl-(CI-C6)alkyl, heterocycloalkyl containing 1-3 hetero atoms
selected from
(0, S) and (CI-C3)allcy1-0-(C1-05)alkyl;
R4 and R5 are selected independently from H, (Ci-05)alkyl, (Ci-C8)alkoxy, (CI-
05)alky1-0-
(CI-05)alkyl, (C3-C6)cycloalkyl, aryl, CF3 and halogen;
R6 isCONR7R8, -(CH2)x-O-R9, alkyl, fluoroalkyl or SO2NR7R8;
x is 1 ¨ 4;
R7 and R8 independently are hydrogen, (C1-05)alkyl or (C3-C6)cycloalkyl, or R7
and R8
together with the nitrogen to which they are attached form a 5-7 member
heterocyclic ring
system with 0-1 additional hetero atoms selected from 0, S and N(R1 ), wherein
the resulting
ring is optionally substituted with 1-3 (C1-05)alkyl or (C3-C6)cycloalkyl
groups;
R9 is hydrogen, (Ci-05)alkyl, (C3-C7)cycloalkyl or aryl;
Rto is
(u C5)alkyl, (Ci-C8)alkoxy, (Ci-05)alky1-0-(CI-05)alkyl, (C3-C6)cycloalkyl or
aryl;
and
=
R11 is (Ci-05)alkyl, fluoroalkyl or (C3-C6)cycloalkyl and the pharmaceutically
acceptable
salts, and individual stereoisomers thereof.
This invention also provides pharmaceutical compositions comprising a
pharmaceutically acceptable carrier in combination with an effective amount of
at least one
compound of formula (I).
The present invention also provides a method of treating conditions in which
modulation of histamine H3 receptors may be of therapeutic importance such as
inflammation,
migraine, motion sickness, pain, Parkinson's Disease, epilepsy, cardiovascular
disease (i.e.
hyper or hypotension, acute myocardial infarction), gastrointestinal disorders
(acid secretion,
motility) and CNS disorders involving attention or cognitive disorders (i.e.,
Alzheimer's,
5
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Attention Deficit Disorder, age-related memory dysfunction, stroke, etc.),
psychiatric
disorders (i.e., depression, schizophrenia, obsessive-compulsive disorders,
etc.); sleep
disorders (i.e. narcolepsy, sleep apnea, insomnia, disturbed biological and
circadian rhythms,
hyper and hypsomnolence), and disorders such as obesity, anorexia/bulimia,
thermoregulation, hormone release) comprising administering an effective
amount of a
compound of formula (I) to a patient in need of such treatment.
DETAILED DESCRIPTION OF THE INVENTION
Preferably for compounds of formula (I), R1-Y-R2is-( ________________________
; R3 is H; R4 is H;
5-methoxy, 5-fluoro or methyl; R5 is H; and R6 is-CH2OCH3 or-(CH2)2 OCH3.
Presently preferred compounds include:
2:Methyl-I -[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
-Methyl-1-[4-(3-piperidin-l-ylpropoxy)pheny1]-1H-indole;
2-Methy1-1-{4-[3-(2R-methylpyrrolidin-1-yppropoxy]phenyll-1H-indole;
1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
5-Methoxy-2-methy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
5-Methyl-1 -[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
5-Bromo-144-(3-pyrrolidin-1-ylpropoxy)phenylj-1H-indole;
4-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
5-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
2,5-Dimethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
6-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
2-Methy1-5-fluoro-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
1 -[3 -Methoxy-4 -(3 -pyrrolidin-l-ylpropoxy)pheny1]-2-methy1-1H-indole;
1-[3-Chloro-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-methy1-1H-indole;
2-Propy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole;
6
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
5-Methoxy-2-methy1-1-[4-(4-pyrrolidin-l-ylbut-1-ynyl)phenyl]-1H-indole;
(5-Methoxy-1-{4-[3-(2R-methylpyrrolidin-1-y1)propoxy]pheny1}-1H-indo1-2-
yl)pyrrolidin-1-
ylmethanone;
1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclobutylamide;
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclopentylamide;
1-14-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclohexylamide;
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
cycloheptylamide;
1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indol-2-yOpyrrolidin-1-
ylmethanone;
2-(3-Morpholin-4-ylpropoxy)-6,7,8,9-tetrahydropyrido[1,2-a]indole;
(1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]pheny1}-1H-indo1-2-y1)morpholin-4-
ylmethanone;
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
butylamide;
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny1}-1H-indole-2-carboxylic acid
isobutylamide;
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclohexylmethylamide;
5-Methoxy-1-{4-[3-(2R-methylpyrrolidin-1-yppropoxy]phenyll-1H-indole-2-
carboxylic acid
cyclohexylamide;
1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole-2-carboxylic acid ethyl
ester;
{1-[4-(3-Pyrrolidin-l-ylpropoxy)pheny1]-1H-indo1-2-yllmethariol;
2-Methoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
2-Cyclohexyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
2-Isopropoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
2-Cyclopentyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
{5-Methoxy-1-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-1H-indo1-2-yllmethanol;
7
CA 02626028 2013-03-21
2-Cyclopropy1-144-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
2-Propy1-144-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole;
2-Cyclopropy1-144-(3-pyn-olidin-1-ylpropoxy)cyclohexyl]-1H-indole;
2-(2-Methoxyethyl)-144-(3-pyrrolidin-l-ylpropoxy)phenyll-1H-indole; and
2- {14443 -P yrroli din-l-ylprop oxy)pheny1]-1H-indo1-2-yll ethanol.
Certain compounds of the invention may exist in different isomeric (e.g.
enantiomers and distereoisomers) forms. The invention contemplates all such
isomers both
in pure form and in a mixture, including racemic mixtures. Enol and tautomeric
forms are
also included.
The compounds of the invention can exist in linsolvated as well as solvated
forms, including hydrated forms, e.g., hemi-hydrate. In general, the solvated
forms, with
pharmaceutically acceptable solvents such as water, ethanol, and the like are
equivalent to the
unsolvated forms for the purposes of the invention.
Certain compounds of the invention also form pharmaceutically acceptable
salts, e.g., acid addition salts. For example, the nitrogen atoms may form
salts with acids.
, Examples of suitable acids for salt formation are hydrochloric, sulfuric,
phosphoric, acetic,
citric, oxalic, malonic, salicylic, malic, fumaric, succinic, ascorbic,
maleic, methanesulfonic
and other mineral carboxylic acids well known to those in the art. The salts
are prepared by
contacting the free base form with a sufficient amount of the desired acid to
produce a salt in
the conventional manner. The free base forms may be regenerated by treating
the salt with a
suitable dilute aqueous base solution such as dilute aqueous hydroxide,
potassium carbonate,
ammonia, and sodium bicarbonate. The free base forms differ from their
respective salt
fonns somewhat in certain physical properties, such as solubility in polar
solvents, but the
acid salts are equivalent to their respective free base forms for purposes of
the invention.
(See, for example S. M. Berge, et al., "Pharmaceutical Salts," J. Pharni.
Sci., 66: 1-19 (1977),
As throughout this specification and appended claims, the following terms
- have the meanings ascribed to them:
The term "alkyl" as used herein refers to straight or branched chain radicals
derived from saturated hydrocarbons by the removal of one hydrogen atom.
Representative
8
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
examples of alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl,
sec-butyl, iso-
butyl, tert-butyl, and the like.
The term "cycloalkyl" as used herein refers to an aliphatic ring system having
3 to 10 carbon atoms and 1 to 3 rings, including, but not limited to
cyclopropyl, cyclopentyl,
cyclohexyl, norbornyl, and adamantly among others. Cycloalkyl groups can be
unsubstituted
or substituted with one, two or three substituents independently selected from
lower alkyl,
haloalkyl, alkoxy, thioalkoxy, amino, alkylamino, dialkylamino, hydroxyl,
halo, mercapto,
nitro, carboxaldehyde, carboxy, alkoxycarbonyl and carboximide.
"Cycloalkyl" includes cis or trans forms. Furthermore, the substituents may
either be in endo or exo positions in the bridged bicyclic systems.
The term "halo" or "halogen" as used herein refers to I, Br, Cl or F.
The tem' "heteroatom" as used herein refers to at least one N, 0 or S atom.
The teiiii "heterocycly1" as used herein, alone or in combination, refers to a
non-aromatic 3- to 10- membered ring containing at least one endocyclic N, 0,
or S atom.
The heterocycle may be optionally aryl-fused. The heterocycle may also
optionally be
substituted with at least one substituent which is independently selected from
the group
consisting of hydrogen, halogen, hydroxyl, amino, nitro, triflouromethyl,
trifluoromethoxy,
alkyl, aralkyl, alkenyl, alkynyl, aryl, cyano, carboxy, carboalkoxy,
carboxyalkyl, oxo,
arylsulfonyl and aralkylaminocarbonyl among others.
As used herein, the term "composition" is intended to encompass a product
,
comprising the specified ingredients in the specified amounts, as well as any
product which
results, directly or indirectly, from a combination of the specified
ingredients in the specified
amounts.
The compounds of the present invention can be used in the form of
pharmaceutically acceptable salts derived from inorganic or organic acids. The
phrase
"pharmaceutically acceptable salt" means those salts which are, within the
scope of sound
medical judgment, suitable for use in contact with the tissues of humans and
lower animals
without undue toxicity, irritation, allergic response and the like and are
commensurate with a
reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well-
known in the art. For
example, S. M. Berge et al. describe pharmaceutically acceptable salts in
detail in J.
Pharmaceutical Sciences,1977, 66: 1 et seq. The salts can be prepared in situ
during the final
isolation and purification of the compounds of the invention or separately by
reacting a free
base function with a suitable organic acid. Representative acid addition salts
include, but are
not limited to acetate, adipate, alginate, citrate, aspartate, benzoate,
benzenesulfonate,
9
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
bisulfate, butyrate, camphorate, camphorsulfonate, digluconate,
glycerophosphate,
hemisulfate, heptanoate, hexanoate, fumarate, hydrochloride, hydrobromide,
hydroiodide, 2-
hydroxyethansulfonate (isothionate), lactate, maleate, methanesulfonate,
nicotinate, 2-
naphthalenesulfonate, oxalate, palmitoate, pectinate, persulfate, 3-
phenylpropionate, picrate,
pivalate, propionate, succinate, tartrate, thiocyanate, phosphate, glutamate,
bicarbonate, p-
toluenesulfonate and undecanoate. Also, the basic nitrogen-containing groups
can be
quatemized with such agents as lower alkyl halides such as methyl, ethyl,
propyl, and butyl
chlorides, bromides and iodides; dialkyl sulfates like dimethyl, diethyl,
dibutyl and diamyl
sulfates; long chain halides such as decyl, lauryl, myristyl and stearyl
chlorides, bromides and
iodides; arylalkyl halides like benzyl and phenethyl bromides and others.
Water or oil-soluble
or dispersible products are thereby obtained.' Examples of acids which can be
employed to
form pharmaceutically acceptable acid addition salts include such inorganic
acids as
hydrochloric acid, hydrobromic acid, sulphuric acid and phosphoric acid and
such organic
acids as oxalic acid, maleic acid, succinic acid and citric acid.
Basic addition salts can be prepared in situ during the final isolation and
purification of compounds of this invention by reacting a carboxylic acid-
containing moiety
with a suitable base such as the hydroxide, carbonate or bicarbonate of a
pharmaceutically
acceptable metal cation or with ammonia or an organic primary, secondary or
tertiary amine.
Pharmaceutically acceptable salts include, but are not limited to, cations
based on alkali
metals or alkaline earth metals such as lithium, sodium, potassium, calcium,
magnesium and
aluminum salts and the like and nontoxic quaternary ammonia and amine cations
including
ammonium, tetramethylammonium, tetraethylammonium, methylarrunonium,
dimethylammonium, trimethylammonium, triethylammonium, diethylammonium, and
ethylammonium among others. Other representative organic amines useful for the
formation
of base addition salts include ethylenediamine, ethanolamine, diethanolamine,
piperidine,
piperazine and the like.
Dosage foans for topical administration of a compound of this invention
include powders, sprays, ointments and inhalants. The active compound is mixed
under
sterile conditions with a pharmaceutically acceptable carrier and any needed
preservatives,
buffers or propellants which can be required. Opthalmic formulations, eye
ointments,
powders and solutions are also contemplated as being within the scope of this
invention.
Actual dosage levels of active ingredients in the pharmaceutical compositions
of this invention can be varied so as to obtain an amount of the active
compound(s) which is
effective to achieve the desired therapeutic response for a particular
patient, compositions and
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
mode of administration. The selected dosage level will depend upon the
activity of the
particular compound, the route of administration, the severity of the
condition being treated
and the condition and prior medical history of the patient being treated.
However, it is within
the skill of the art to start doses of the compound at levels lower than
required to achieve the
desired therapeutic effect and to gradually increase the dosage until the
desired effect is
achieved.
When used in the above or other treatments, a therapeutically effective amount
of one of the compounds .of the present invention can be employed in pure form
or, where
such forms exist, in pharmaceutically acceptable salt, ester or prodrug form.
Alternatively,
the compound can be administered as a pharmaceutical composition containing
the
compound of interest in combination with one or more pharmaceutically
acceptable
excipients. The phrase "therapeutically effective amount" of the compound of
the invention
means a sufficient amount of the compound to treat disorders, at a reasonable
benefit/risk
ratio applicable to any medical treatment. It will be understood, however,
that the total daily
usage of the compounds and compositions of the present invention will be
decided by the
attending physician within the scope of sound medical judgment. The specific
therapeutically
effective dose level for any particular patient will depend upon a variety of
factors including
the disorder being treated and the severity of the disorder; activity of the
specific compound
employed; the specific composition employed; the age, body weight, general
health, sex and
diet of the patient; the time of administration, route of administration, and
rate of excretion of
the specific compound employed; the duration of the treatment; drugs used in
combination or
coincidental with the specific compound employed; and like factors well known
in the
medical arts. For example, it is well within the skill of the art to start
doses of the compound
at levels lower than required to achieve the desired therapeutic effect and to
gradually
increase the dosage until the desired effect is achieved.
The total daily dose of the compounds of this invention administered to a
human or lower animal may range from about 0.0001 to about 1000 mg/kg/day. For
purposes
of oral administration, more preferable doses can be in the range of from
about 0.001 to about
5 mg/kg/day. If desired, the effective daily dose can be divided into multiple
doses for
purposes of administration; consequently, single dose compositions may contain
such
amounts or submultiples thereof to make up the daily dose.
The present invention also provides pharmaceutical compositions that
comprise compounds of the present invention formulated together with one or
more non-toxic
pharmaceutically acceptable carriers. The pharmaceutical compositions can be
specially
11
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
formulated for oral administration in solid or liquid form, for parenteral
injection or for rectal
administration.
The pharmaceutical compositions of this invention can be administered to
humans and other mammals orally, rectally, parenterally, , intracisternally,
intravaginally,
intraperitoneally, topically (as by powders, ointments or drops), bucally or
as an oral or nasal
spray. The term "parenterally," as used herein, refers to modes of
administration which
include intravenous, intramuscular, intraperitoneal, intrasternal,
subcutaneous and
intraarticular injection and infusion.
In another aspect, the present invention provides a pharmaceutical
composition comprising a component of the present invention and a
physiologically tolerable
diluent. The present invention includes one or more compounds as described
above
formulated into compositions together with one or more non-toxic
physiologically tolerable
or acceptable diluents, carriers, adjuvants or vehicles that are collectively
referred to herein as
diluents, for parenteral injection, for intranasal delivery, for oral
administration in solid or
liquid form, for rectal or topical administration, among others.
The compositions can also be delivered through a catheter for local delivery
at
a target site, via an intracoronary stent (a tubular device composed of a fine
wire mesh), or
via a biodegradable polymer. The compounds may also be complexed to ligands,
such as
antibodies, for targeted delivery.
Compositions suitable for parenteral injection may comprise physiologically
acceptable, sterile aqueous or nonaqueous solutions, dispersions, suspensions
or emulsions
and sterile powders for reconstitution into sterile injectable solutions or
dispersions.
Examples of suitable aqueous and nonaqueous carriers, diluents, solvents or
vehicles include
water, ethanol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and
the like),
vegetable oils (such as olive oil), injectable organic esters such as ethyl
oleate, and suitable
mixtures thereof.
These compositions can also contain adjuvants such as preserving, wetting,
emulsifying, and dispensing agents. Prevention of the action of microorganisms
can be
ensured by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol,
phenol, sorbic acid, and the like. It may also be desirable to include
isotonic agents, for
example sugars, sodium chloride and the like. Prolonged absorption of the
injectable
pharmaceutical form can be brought about by the use of agents delaying
absorption, for
example, aluminum monostearate and gelatin.
12
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Suspensions, in addition to the active compounds, may contain suspending
agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene
sorbitol and sorbitan
esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-
agar and
tragacanth, or mixtures of these substances, and the like.
In some cases, in order to prolong the effect of the drug, it is desirable to
slow
the absorption of the drug from subcutaneous or intramuscular injection. This
can be
accomplished by the use of a liquid suspension of crystalline or amorphous
material with
poor water solubility. The rate of absorption of the drug then depends upon
its rate of
dissolution which, in turn, may depend upon crystal size and crystalline form.
Alternatively,
delayed absorption of a parenterally administered drug form is accomplished by
dissolving or
suspending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices of the
drug in biodegradable polymers such as polylactide-polyglycolide. Depending
upon the ratio
of drug to polymer and the nature of the particular polymer employed, the rate
of drug release
can be controlled. Examples of other biodegradable polymers include
poly(orthoesters) and
poly(anhydrides). Depot injectable foimulations are also prepared by
entrapping the drug in
liposomes or microemulsions which are compatible with body tissues.
The injectable formulations can be sterilized, for example, by filtration
through a bacterial-retaining filter or by incorporating sterilizing agents in
the form of sterile
solid compositions which can be dissolved or dispersed in sterile water or
other sterile
injectable medium just prior to use.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders and granules. In such solid dosage forms, the active compound may be
mixed with
at least one inert, pharmaceutically acceptable excipient or carrier, such as
sodium citrate or
dicalcium phosphate and/or a) fillers or extenders such as starches, lactose,
sucrose, glucose,
mannitol and silicic acid; b) binders such as carboxymethylcellulose,
alginates, gelatin,
polyvinylpyrrolidone, sucrose and acacia; c) humectants such as glycerol; d)
disintegrating
agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic
acid, certain
silicates and sodium carbonate; e) solution retarding agents such as paraffin;
(f) absorption
accelerators such as quaternary ammonium compounds; g) wetting agents such as
cetyl
alcohol and glycerol monostearate; h) absorbents such as kaolin and bentonite
clay and i)
lubricants such as talc, calcium stearate, magnesium stearate, solid
polyethylene glycols,
sodium lauryl sulfate and mixtures thereof. In the case of capsules, tablets
and pills, the
dosage form may also comprise buffering agents.
13
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Solid compositions of a similar type may also be employed as fillers in soft
and hard-filled gelatin capsules using such excipients as lactose or milk
sugar as well as high
molecular weight polyethylene glycols and the like.
The solid dosage forms of tablets, dragees, capsules, pills and granules can
be
prepared with coatings and shells such as enteric coatings and other coatings
well-known in
the pharmaceutical formulating art. They may optionally contain opacifying
agents and may
also be of a composition such that they release the active ingredient(s) only,
or preferentially,
in a certain part of the intestinal tract, optionally, in a delayed manner.
Examples of
embedding compositions which can be used include polymeric substances and
waxes.
The active compounds can also be in micro-encapsulated form, if appropriate,
with one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions, solutions, suspensions, syrups and elixirs. In addition
to the active
compounds, the liquid dosage forms may contain inert diluents commonly used in
the art
such as, for example, water or other solvents, solubilizing agents and
emulsifiers such as
ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl
alcohol, benzyl
benzoate, propylene glycol, 1,3-butylene glycol, dimethyl founamide, oils (in
particular,
cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol,
tetra.hydrofurfuryl
alcohol, polyethylene glycols and fatty acid esters of sorbitan and mixtures
thereof.
Besides inert diluents, the oral compositions may also include adjuvants such
as wetting agents, emulsifying and suspending agents, sweetening, flavoring
and perfuming
agents.
Compositions for rectal or vaginal administration are preferably suppositories
which can be prepared by mixing the compounds of this invention with suitable
non-irritating
excipients or carriers such as cocoa butter, polyethylene glycol or a
suppository wax which
are solid at room temperature but liquid at body temperature and therefore
melt in the rectum
or vaginal cavity and release the active compound.
Compounds of the present invention can also be administered in the form of
liposomes. As is known in the art, liposomes are generally derived from
phospholipids or
other lipid substances. Liposomes are formed by mono- or multi-lamellar
hydrated liquid
crystals which are dispersed in an aqueous medium. Any non-toxic,
physiologically
acceptable and metabolizable lipid capable of forming liposomes can be used.
The present
compositions in liposome form can contain, in addition to a compound of the
present
14
CA 02626028 2013-03-21
=
inventiOn, stabilizers, preservatives, excipients and the like. The preferred
lipids are natural
and synthetic phospholipids and phosphatidyl cholines (lecithins) used
separately or together.
Methods to form liposomes are known in the art. See, for example, Prescott,
Ed., Methods in Cell BiologyNolume XEV, Academic Press, New York, N.Y. (1976),
p. 33
et seq.
= The term "pharmaceutically acceptable prodrugs" as used herein represents
those prodrugs of the compounds of the present invention which are, within the
scope of
sound medical judgment, suitable for use in contact with the tissues of humans
and lower
animals without undue toxicity, irritation, allergic response, and the like,
commensurate with
a reasonable benefit/risk ratio, and effective for their intended use, as well
as the zwitterionic
forms, where possible, of the compounds of the invention. Prodrugs of the
present invention
may be rapidly transformed in vivo to the parent compound of the above
formula, for
example, by hydrolysis in blood. A thorough discussion is provided in T.
Higuchi and V.
Stella, Pro-drugs as Novel Delivery Systems,V. 14 of the A.C.S. Symposium
Series, and in
Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American
Pharmaceutical
Association and Pergamon Press (1987).
Compounds of the present invention that are formed by in vivo conversion of a
different compound that was administered to a mammal are intended to be
included within
the scope of the present invention.
= Compounds of the present invention may exist as stereoisomers wherein
asymmetric or chiral centers are present. These stereoisomers are "R" or "S"
depending on
the configuration of substituents around the chiral carbon atom. The present
invention
contemplates various stereoisomers and mixtures thereof. Stereoisomers include
enantiomers
and diastereomers, and mixtures of enantiomers or diastereomers. Individual
stereoisomers of
compounds of the present invention may be prepared synthetically from
commercially
available starting materials which contain asymmetric or chiral centers or by
preparation of
racemic mixtures followed by resolution well-known to those of ordinary skill
in the art.
These methods of resolution are exemplified by (1) attachment of a mixture of
enantiomers to
a chiral auxiliary, separation of the resulting mixture of diastereomers by
recrystallization or
chromatography and liberation of the optically pure product from the auxiliary
or (2) direct
separation of the mixture of optical enantiomers on chiral chromatographic
columns.
The compounds of the invention can exist in unsolvated as well as solvated
forms, including hydrated forms, such as hemi-hydrates. In general, the
solvated forms, with
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
pharmaceutically acceptable solvents such as water a.nd ethanol among others
are equivalent
to the unsolvated forms for the purposes of the invention.
The invention may be illustrated by the following representative scheme and
examples.
Scheme 1
criNH2
= N
2 BBr3
N OMe Cul, K3PO4 __ * 110
dioxane, 90 C
OMe
1110
Br-C1= ION pyrrolidine N N
K2CO3 K2CO3, KI, MEIZ
= MEK, 70 C
111 =
OH 0¨\ /CI
0¨\ /1\-\D
Example 1
2-Methyl-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny11-1H-indole
=
1101 N\
O\
1-(4-Methoxypheny1)-2-methyl-1H-indole. 1-(4-Methox3Theny1)-2-methyl-1H-indo
le was
synthesized according to Buchwald et al. J Am. Chem. Soc. 2001, 123, 7727.
2-
Methylindole (157 mg, 1.2 mmol), 4-iodoanisole (234 mg, 1 mmol), copper(I)
iodide (2 mg,
0.01 mmol), trans-1,2-diaminocyclohexane (11.4 mg, 0.1 mmol), and potassium
phosphate
tribasic (446 mg, 2.1 mmol) were stirred in dioxane (1 mL) at 90 C overnight.
The reaction
mixture was filtered through a pad of silica and washed with ethyl acetate.
Si02
16
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
chromatography with 5-20% ethyl acetate/hexanes gave 175 mg of the desired
product (74%
yield). LC-MS (C16H15N0 calc'd 237) 171/Z 238 (M+H).
4-(2-Methylindo1-1-yl)phenol. 1-(4-Methoxypheny1)-2-methy1-1H-indole (175 mg,
0.73
mmol) was dissolved in dichloromethane (2 mL) and cooled to 0 C. Boron
tribromide (2.19
mL, 1 M solution in dichloromethane, 2.19 mmol) was added dropwise, and the
reaction was
stirred for 2 hours. The reaction was quenched by addition of saturated sodium
bicarbonate
solution, then extracted with dichloromethane followed by ethyl acetate. The
organic extracts
were dried over MgSO4 and concentrated. The reaction was assumed to be
quantitative. LC-
MS (C15H13NO calc'd 223) m/z 224 (M+H).
144-(3-Chloropropoxy)pheny1]-2-methy1-1H-indole. 4-(2-Methylindo1-1-yl)phenol
(6.36
mmol) was heated at 70 C in 2-butanone (3 mL) with 1-bromo-3-chloropropane
(0.107 mL,
1.08 mmol) and potassium carbonate (0.15 g, 1.08 mmol) overnight. The solvent
was
evaporated. The resulting residue was diluted with ethyl acetate and washed
with saturated
ammonium chloride solution. The aqueous layer was back-extracted with ethyl
acetate. The
organic extracts were dried over MgSO4 and concentrated. Si02 chromatography
with 5-20%
ethyl acetate/hexanes gave the desired product as a colorless oil, 60 mg (56%
yield, 2 steps).
LC-MS (C181-118C1N0 calc'd 299) 171/Z 300, 302 (M+H).
2-Methyl-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole. 1-
[4-(3-
Chloropropoxy)pheny1]-2-methy1-1H-indole (20 mg, 0.067 mmol) was dissolved in
N-
methylpyrrolidinone (0.5 mL), and pyrrolidine (0.017 mL, 0.2 mmol), potassium
carbonate
(46 mg, 0.34 mmol), and a catalytic amount of potassium iodide were added. The
reaction
was heated to 70 C overnight. The reaction was diluted with saturated sodium=
bicarbonate
solution and extracted with ethyl acetate. The organic extracts were dried
over MgSO4 and
concentrated. The residue was purified by semi-prep LC-MS to give 9.6 mg of
the desired
product (43% yield). LC-MS (C22H26N20 calc'd 334) nilz 335 (M+H); 11-1 NMR
(300 MHz,
CDC13) 8 7.58 ¨ 7.53 (m, 1H), 7.26 ¨ 7.25 (m, 3H), 7.12 ¨ 6.99 (m, 4H), 6.37
(s, 1H), 4.10 (t,
J= 6 Hz, 2H), 2.99 ¨ 2.90 (m, 6H), 2.24 (s, 3H), 2.22 ¨ 2.14 (m, 2H), 2.00 ¨
1.90 (m, 6H).
17
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 2
2-Methyl-144-(3-piperidin-1-ylpropoxy)pheny11-1H-indole
N
110
0-\ ___________________________________________ /0
2-Methy1-1-[4-(3-piperidin-1-ylpropoxy)phenyl]-1H-indole was synthesized by a
method
analogous to that used for Example 1 using piperidine in place of pyrrolidine
in the final step.
LC-MS (C23H28N20 calc'd 348) m/z 349 (M+H); 1H NMR (300 MHz, CDC1a) 8 7.58 -
7.52
(m, 1H), 7.23 - 7.21 (m, 3H), 7.12 - 6.99 (m, 4H), 6.37 (s, 1H), 4.09 (t, J= 6
Hz, 2H), 2.79 -
2.68 (m, 6H), 2.27 (s, 311), 2.20 - 2.09 (m, 2H), 1.77 - 1.69 (m, 4H), 1.56 -
1.52 (m, 2H).
Example 3
2-Methyl-1-{443-(2R-methylpyrrolidin-1-yl)propoxylphenyl}-1H-indole
\
110
O\
2-Methy1-1-{443-(2R-methylpyrrolidin-1-yl)propoxy]phenyll-1H-indole was
synthesized by
a method analogous to that used for Example 1 using (R)-2-methylpyrrolidine in
place of
pyrrolidine in the final step. LC-MS (C23H28N20 calc'd 348) m/z 349 (M+H); 1H
NMR (300
MHz, CDC13) 8 7.58 - 7.52 (m, 1H), 7.26 - 7.25 (m, 3H), 7.12 - 7.00 (m, 411),
6.37 (s, 1H),
4.16 - 4.05 (m, 2H), 3.46 - 3.39 (m, 1H), 3.22 - 3.13 (m, 111), 2.75 - 2.63
(m, 111), 2.57 -
2.42 (m, 2H), 2.27 (s, 3H), 2.21 - 1.55 (m, 6H), 1.25 (d, J= 6.3 Hz, 3H).
18
CA 02626028 2013-03-21
=
Scheme 2
01'Cl 401
110
\ -
N
o,
OH NaH, Na l nANH2.
DMF, 70 C = -21NH2
= cul, K3PO4
dioxane, 90 C
Example 4
144-(3-Pyrrolidin-J.-ylpropoxy)pheny11-1H-indole
N\
00
O=
1-13-(4-Todophenoxy)propylipyrrolidine. 4-Todophenol (2.2 g, 10 mmol) was
dissolved in
N,N-dimethylformamide (30 mL) under N2, and sodium hydride (0.48 g, 60%
dispersion in
mineral oil, 12 mmol) was added in portions. 1-(3-Chloropropyl)pyrrolidine
(1.77 g, 12
mmol) and sodium iodide (1.8 g, 12 mrnol) were added, and the reaction mixture
was stirred
at 70 C overnight. The reaction mixture was diluted with ethyl acetate and
=washed with
water. The ethyl acetate solution was washed with 1 N HC1 (2 x). The acidic
extracts were
made basic with 2 N NaOH, then were extracted with ethyl acetate (2 x). All
ethyl acetate
extracts were combined, dried over MgSO4 and concentrated to give a yellow
oil, 2.98 g
(90% crude yield). LC-MS (C131-118INO calc'd 331) ni/z 332 (M+H).
144-(3-Pyrrolidin-1-ylp rop o xy)p henyl]
indole. 1-[3-(4-Iodophenoxy)propy1]-
= pyrrolidine (66 mg, 0.2 mmol), indole (28 mg, 0.24 mmol), copper(I) iodide
(0.4 mg, 0.002
mmol), trans-1,2-diaminocyclohexane (0.0024 mmol, 0.02 nunol), and potassium
phosphate
tribasic (89 mg, 0.42mmol) were stirred at 90 C in dioxane overnight. The
reaction mixture
was filtered through celiteTM and washed with dichloromethane. The filtrate
was concentrated
and the resulting residue was purified by semi-prep LC-MS = to. give 22.4 mg
of the desired
19
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
product (35% yield). LC-MS (C211-124N20 calc'd 320) in/z 321 (M+H); 11-1 NMR
(300 MHz,
CDC13) 5 7.67 (d, J¨ 6.9 Hz, 1H), 7.45 (d, J= 8.4 Hz, 1H), 7.40 ¨ 7.35 (m,
2H), 7.27 (d, J=
3 Hz, 1H), 7.22 ¨ 7.10 (m, 2H), 7.04 ¨ 6.99 (m, 2H), 6.64 (d, J = 3 Hz, 1H),
4.08 (t, J= 6.3
Hz, 2H), 2.66 (t, J= 7.2 Hz, 2H), 2.57 ¨ 2.52 (m, 4H), 2.09 ¨ 2.00 (m, 2H),
1.83 ¨ 1.78 (m,
4H).
=
Example 5
5-Methoxy-2-methyl-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
0
1\1\
5-Methoxy-2-methyl- I -[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was
synthesized by a
method analogous to that used for Example 4 using 5-methoxy-2-methylindole in
place of
indole in the final step. LC-MS (C23H28N202 calc'd 364) m/z 365 (M+H); 1H NMR
(300
MHz, CDC13) 5 7.23 ¨ 7.20 (m, 2H), 7.03 (d, J= 2.4 Hz, 1H), 7.02 ¨ 6.97 (m,
2H), 6.93 (d, J
= 8.7 Hz, 1H), 6.72 (dd, J= 9 Hz, 2.4 Hz, 1H), 6.30 (s, 1H), 4.10 (t, J= 6 Hz,
2H), 3.85 (s,
3H), 3.03 ¨ 2.96 (m, 4H), 2.25 (s, 3H), 2.24 ¨ 2.16 (m, 2H), 1.99 ¨ 1.94 (m,
4H).
Example 6
5-Methyl-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
111101 N\
o
5-Methy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by a
method
analogous to that used for Example 4 using 5-methylindole in place of indole
in the final step.
LC-MS (C22H26N20 calc'd 334) 711/Z 335 (M+H); 1H NMR (300 MHz, CDC13) 5 7.46
(s, 1H),
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
7.40 - 7.33 (m, 2H), 7.23 (d, J= 3.3 Hz, 1H), 7.23 (d, J= 3.3 Hz, 1H), 7.04 -
6.98 (m, 2H),
6.56 (d, J= 3.3 Hz, 1H), 4.08 (t, J= 6.3 Hz, 2H), 2.80 - 2.70 (m, 6H), 2.17 -
2.06 (m, 2H),
1.88 - 1.84 (m, 4H).
Example 7
5-Bromo-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
Br.
110
o
5-Bromo-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by a
method
analogous to that used for Example 4 using 5-bromoindole in place of indole in
the final step.
LC-MS (C211-123BrN20 calc'd 399) m/z 400, 402 (M+H); 1HNMR (300 MHz, CDC13) 6
7.79
(s, 1H), 7.37 (d, J= 4.8 Hz, 2H), 7.29 - 7.26 (m, 3H), 7.01 (d, J= 8.7 Hz,
2H), 6.58 (d, J=
3.3 Hz, 1H), 4.09 (t, J= 6.3 Hz, 2H), 2.92 - 2.82 (m, 6H), 2.20 - 2.11 (m,
2H), 1.94 - 1.89
(m, 411).
Example 8
4-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
CI
o
1101
4-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by a
method
analogous to that used for Example 4 using 4-chloroindole in place of indole
in the final step.
LC-MS (C211423C1N20 calc'd 354) m/z 355, 357 (M+H); 1H NMR (300 MHz, CDC13) 6
7.39
- 7.30 (m, 4H), 7.17 - 7.08 (m, 2H), 7.04 - 6.99 (m, 2H), 6.75 (d, J= 3.3 Hz,
1H), 4.09 (t, J
= 6 Hz, 2H), 2.97 - 2.89 (m, 6H), 2.22 - 2.13 (m, 2H), 1.96 - 1.92 (m, 4H).
21
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 9
5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
oO\
o
41,
5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by
a method
analogous to that used for Example 4 using 5-methoxyindole in place of indole
in the final
step. LC-MS (C22H26N202 calc'd 350) m/z 351 (M+H).
Example 10
5-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
ci
o
5-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was synthesized by a
method
analogous to that used for Example 4 using 5-chlorolindole in place of indole
in the final
step. LC-MS (C21H23C1N20 calc'd 354) m/z 355, 357 (M+H); NMR (300 MHz, CDC13)
6 7.63 (d, J= 2.1 Hz, 1H), 7.37 ¨ 7.32 (m, 3H), 7.28 (d, J= 3 Hz, 1H), 7.14
(dd, J= 8.7 Hz,
1.8 Hz, 1H), 7.04 ¨ 6.99 (m, 2H), 6.58 (d, J = 3 Hz, 1H), 4.09 (t, J= 6.3 Hz,
2H), 2.83 ¨ 2.73
(m, 6H), 2.16 ¨ 2.07 (m, 2H), 1.90¨ 1.85 (m, 4H).
Example 11
2,5-Dimethy1-144-(3-pyrrolidin-1-ylpropoxy)pheny1F1H-indole
1110 N\
22
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
2,5-Dimethy1-144-(3-pyrrolidin-1-ylpropoxy)phenyll-1H-indole was synthesized
by a
method analogous to that used for Example 4 using 2,5-dimethylindole in place
of indole in
the final step. LC-MS (C23H28N20 calc'd 348) m/z 349 (M+H); 1H NMR (300 MHz,
CDC13)
8 7.40 (s, 1H), 7.23 ¨ 7.19 (m, 2H), 7.04 ¨ 6.99 (m, 2H), 6.95 ¨ 6.90 (m, 2H),
6.28 (s, 1H),
4.10 (t, J= 6.3 Hz, 2H), 2.68 (t, J= 7.2 Hz, 2H), 2.57 (m, 4H), 2.43 (s, 3H),
2.25 (s, 3H),
2.12 ¨ 1.99 (m, 2H), 1.84 ¨ 1.80 (m, 4H).
Example 12
6-Chloro-144-(3-pyrrolidin-1-ylpropoxy)pheny1F1H-indole
N\
CI
6-Chloro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by a
method
analogous to that used for Example 4 using 6-chloroindole in place of indole
in the final step.
LC-MS (C211123C1N20 calc'd 354) m/z 355, 357 (M+H); 1H NMR (300 MHz, CDC13) 8
7.57
(d, J= 8.4 Hz, 1H), 7.38 (s, 1H), 7.37 ¨ 7.34 (m, 2H), 7.26 ¨ 7.25 (m, 1H),
7.11 (dd, J = 8.4
Hz, 1.8 Hz, 1H), 7.06 ¨7.01 (m, 2H), 6.62 (d, J = 3.3 Hz, 1H), 4.10 (t, J =
6.3 Hz, 2H), 2.69
(t, J 7.5 Hz, 2H), 2.59 (m, 4H), 2.12 ¨ 2.03 (m, 2H), 1.88 ¨ 1.78 (m, 4H).
Example 13
2-Methyl-5-fluoro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
F
2-Methyl-5-fluoro-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was
synthesized by a
method analogous to that used for Example 4 using 5-fluoro-2-methylindole in
place of
indole in the final step. LC-MS (C22H25FN20 calc'd 352) m/z 353 (M+H).
23
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 14
1[3-Methoxy-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-methyl-1H-indole
N
=
0ON
u
1-[3-Methoxy-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-methy1-1H-indole was
synthesized by a
method analogous to that used for Example 4, using 4-bromoguaiaeol in the
first step rather
than 4-iodophenol and 2-methylindole in the final step. LC-MS (C23H28N202
eale'd 364) m/z
365 (M+H); 1H NMR (300 MHz, CDC13) 5 7.58 ¨ 7.55 (m, 1H), 7.14 ¨ 7.07 (m, 3H),
6.99 (d,
J= 8.7 Hz, 1H), 6.88 (dd, J= 8.4 Hz, 2.4 Hz, 2H), 6.83 (d, J= 2.4 Hz, 1H),
6.38 (s, 1H), 4.17
(t, J¨ 6.3 Hz, 2H), 3.83 (s, 3H), 3.02 ¨ 2.94 (m, 6H), 2.29 (s, 3H), 2.27 ¨
2.20 (m, 2H), 1.97
¨ 1.93 (m, 4H).
Example 15
1[3-Chloro-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-methyl-1H-indole
N\
Cl
143-Chloro-4-(3-pyrrolidin-1-ylpropoxy)pheny11-2-methyl-1H-indole was
synthesized by a
method analogous to that used for Example 4, using 4-bromo-2-ehlorophenol in
the first step
rather than 4-iodophenol and 2-methylindole in the final step. LC-MS
(C22H25C1N20 cale'd
368) m/z 369, 371 (M+H); 1H NMR (300 MHz, CDC13) 5 7.58 ¨ 7.54 (m, 1H), 7.37
(d, J =
2.4 Hz, 1H), 7.20 (dd, J= 8.7 Hz, 2.7 Hz, 1H), 7.11 ¨ 7.02 (m, 4H), 6.38 (s,
1H), 4.19 (t, J=
6.3 Hz, 2H), 2.88 (t, J= 7.2 Hz, 2H), 2.77 (m, 4H), 2.24 ¨ 2.15 (m, 2H), 1.91
¨ 1.87 (m, 4H).
24
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 16
2-Propy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole
ON
o
2-Propy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole was synthesized by a
method
2-Propylindole was prepared according to Kuyper et al. (J. Med. Chem. 1996,
39, 892). LC-
MS (C24H30N20 calc'd 362) m/z 363 (M+H); 1H NMR (300 MHz, CDC13) 5 7.58 (m,
1H),
7.25 ¨ 7.21 (m, 2H), 7.12 ¨ 6.99 (m, 5H), 6.38 (s, 1H), 4.10 (t, J= 6 Hz, 2H),
3.01 ¨ 2.93 (m,
6H), 2.55 (t, J= 7.8 Hz, 2H), 2.25 ¨ 2.15 (m, 2H), 1.98 ¨ 1.91 (m, 4H), 1.66 ¨
1.54 (m, 2H),
Scheme 3
0 0
0 Br
=
\ = \
= Cul, K3PO4, PhMe
80 C
Br
0
0
Cul, Pd(PPI13)4 MsCI, DCM, TEA
TEA, 80 C 0 C-rt =
=
OH çN
25
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 17
5-Methoxy-2-methy1-144-(4-pyrrolidin-1-ylbut-1-ynyl)pheny11-1H-indole
,
\\
1-(4-Bromopheny1)-5-methoxy-2-methyl-1H-indole and 1-(4-Iodopheny1)-5-methoxy-
2-
methy1-1H-indole. 5-Methoxy-2-methylindole (500 mg, 3.1 mmol) and 1-bromo-4-
iodobenzene (877 mg, 3.1 mmol) were dissolved in toluene (6 mL). To the
resulting solution
were added copper(I) iodide (12 mg, 0.062 mmol), potassium phosphate tribasic
(1.32 g, 6.2
mmol), and N,/V'-dimethylethylenediamine (6.6 4, 0.062 mmol). The mixture was
heated at
80 C overnight, allowed to cool to room temperature and filtered through a
pad of silica. The
resulting solution was concentrated to give a mixture of both the bromo and
the iodo
halophenyl indoles, which were used without further purification (assumed
quantitative).
444-(5-Methoxy-2-methylindo1-1-yl)phenyl]but-3-yn-1-ol. To a solution of the
above
mixture of indoles (50 mg) in triethylamine (1 mL) was added copper(I) iodide
(6 mg, 0.03
mmol) and tetralcis(triphenylphosphine)palladium(0) (17 mg, 0.015 mmol). After
3-butyn-1-
ol (15pL, 0.20 mmol) was added, the resulting mixture was heated at 80 C
overnight. The
reaction was allowed to cool and was filtered through a pad of silica. The
silica was washed
with ethyl acetate. Concentration gave the desired alcohol, which was used
without further
purification (assumed quantitative).
5-Methoxy-2-methyl-144-(4-pyrrolidin-1-ylbut-1-ynyl)pheny11-1H-indole. To a
solution
of 444-(5-Methoxy-2-methylindo1-1-yl)phenyl]but-3-yn-1-ol (60 mg, 0.19 mmol)
in
methylene chloride (1 mL) at 0 C was added triethylamine (54 pL, 0.39 mmol)
and
methanesulfonylchloride (18 pL, 0.39 mmol). After the solution was stirred at
0 C for 2
hours, pyrrolidine (1634, 1.95 mmol) was added and the reaction was allowed to
warm to
room temperature overnight. After the reaction was quenched with water, the
organic layer
was dried over MgSO4, and concentrated. The residue was purified by HPLC to
give the
26
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
desired indole (2.8 mg). LC-MS (C24H26N20 calc'd 358) 177/Z 359 (M+H); NMR
(300
MHz, CDC13) 5 7.53 (d, J= 8.4 Hz, 2H), 7.26 (d, J= 8.4 Hz, 2H), 7.03 (d, J=
2.4 Hz, 1H),
6.99 (d, J= 9.0 Hz, 1H), 6.73 (dd, J= 2.7, 9.0 Hz, 1H), 6.32 (s, 1H), 3.85 (s,
3H), 2.92 ¨
2.83 (m, 4H), 2.76 (m, 4H), 2.28 (s, 3H), 1.88 (m, 4H).
Scheme 4
101 r1
ANH2 Me0
\
211\1H2 = N O_\Me0NaOH
co2Et (:)`=
110
1144 N Cul, K3PO4
dioxane, 90 C
Me0 rat 0
Me0 =
pyrrolidine
N OH N
PyBrOP
1114 DIEA
DMF =
O
Example 18
(5-Methoxy-1-{443-(2R-methylpyrrolidin-1-yl)propoxylphenyl}-1H-indo1-2-
yl)pyrrolidin-1-ylmethanone
0
0
N \].)
NC
1-{443-(2R-Methyl-pyrrolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
ethyl ester
was synthesized by a method analogous to that used for Example 4 starting from
ethyl 5-
methoxyindole-2-carboxylate. 1-{4-[3-(2R-Methyl-pyrrolidin-1-
yl)propoxy]phenyll-1H-
27
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
indole-2-carboxylic acid ethyl ester (0.43 mmol) was dissolved in THF (2.4
mL), methanol
(1.2 mL), and water (0.4 mL), and sodium hydroxide (103 mg, 2.58 mmol) was
added. The
reaction mixture was heated at 50 C overnight. A 1 N solution of HCl was
added until the
pH measured 7, and the solvents were evaporated. A portion of the residue (ca.
0.2 mmol)
was dissolved in N,N-dimethylformamide (1 mL), and pyrrolidine (0.017 mL, 0.2
mmol),
PyBrOP (0.14 g, 0.3 mmol), and diisopropylethylamine (0.104 mL, 0.6 mmol) were
added.
The reaction mixture was stirred overnight, and then the solvent was
evaporated. The residue
was purified by semi-prep LC-MS to give the desired product and a PyBrOP-
related side
product. The residue was further purified by Si02 chromotography with ethyl
acetate, then
10% methanol/ethyl acetate, then 2% triethylamine/10% methanol/ethyl acetate
to give the
desired product, 15.1 mg. LC-MS (C28H35N303 calc'd 461) m/z 462 (M+H); 114 NMR
(300
MHz, CDC13) 8 7.30 (d, J= 9 Hz, 2H), 7.17 (d, J= 9 Hz, 1H), 7.10 (d, J= 2.4
Hz, 1H), 6.97
(d, J= 8.7 Hz, 2H), 6.88 (dd, J= 9 Hz, 2.4 Hz, 1H), 6.76 (s, 111), 4.09 - 4.04
(m, 2H), 3.86
(s, 3H), 3.49 (t, J= 6.6 Hz, 2H), 3.36 (t, J= 6.2 Hz, 2H), 3.22 (dt, J= 2.7
Hz, 8.7 Hz, 1H),
3.07 - 2.98 (m, 1H), 2.36 - 1.65 (m, 12 H), 1.51 - 1.39 (m, 1H), 1.12 (d, J= 6
Hz, 3H).
Example 19
1-{443-(2R-Methylpyrrolidin-1-yl)propoxylpheny1}-1H-indole-2-carboxylic acid
cyclobutylamide
\0
N HN-0
=
1-14-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
cyclo-
butylamide was synthesized by a method analogous to that used for Example 18
using
Cyclobutylamine in place of pyrrolidine. LC-MS (C27H33N302 calc'd 431) 772/2
432 (M+H);
'FT NMR (300 MHz, CDC13) 8 7.68 (d, J= 7.8 Hz, 1H), 7.29 - 7.02 (m, 8H), 5.91
(d, J= 7.8
Hz, 1H), 4.51 - 4.37 (m, 1H), 4.10 (t, J= 6.3 Hz, 2H), 3.25 - 3.14 (m, 4H),
3.07 - 2.98 (m,
1H), 2.40- 1.60 (m, 11H), 1.51 - 1.38 (m, 1H), 1.12 (d, J= 6 Hz, 3H).
28
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 20
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl}-1H-indole-2-carboxylic acid
cyclopentylamide
\ o
N
1_{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclopentylamide was synthesized by a method analogous to that used for
Example 18 using
cyclopentylamine in place of pyrrolidine. LC-MS (C28H35N302 calc'd 445) m/z
446 (M+H).
Example 21
1-f4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
cyclohexylamide
\0
N HN-0
f\C'
o
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
cyclohexylamide was synthesized by a method analogous to that used for Example
18 using
cyclohexylamine in place of pyrrolidine. LC-MS (C29H37N302 calc'd 459) m/z 460
(M+H).
Example 22
1-44-[3-(2R-Methylpyrrolidin-1-yl)propoxylpheny1}-1H-indole-2-carboxylic acid
cycloheptylamide
0
HN-0
sit I\r1
= 29
CA 02626028 2013-03-21
1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]pheny1}-1H-indole-2-carboxylic acid
cycloheptylarnide was synthesized by a method analogous to that used for
Example 18 using
cycloheptylamine in place of pyrrolidine. LC-MS (C30H39N302 calc'd 473) rth
474 (M+H).
Example 23
(144-P-(2R-Methylpyrrolidin-1-yl)propoxy]pheny1}-11/-indol-2-yl)pyrrolidin-1-
ylmethanone
=\O=
N 14q3
* Nr1
(1-{413-(2R-Methylpyrrolidin-1-yl)propoxylpheny1}-1H-indol-2-y1)pyrrolidin-1-
ylmethanone was synthesized by a method analogous to that used for Example 18.
LC-MS
(C271133N302 calc'd 431) n2/z 432 (M+H).
Example 24
(R)-( 1-(4-(3-(2-methylpyrrolidin-1-yppropoxy)phenyl)-1H-indol-2-y1)(piperidin-
1-
yl)methadone
=0
\
N
(1- {4-[3 -(2R-Methylpyrrolidin-1-yl)propoxy]phenyl} -1H-indo1-2-yl)piperidin-
l-ylmethanone
was synthesized by a method analogous to that used for Example 18 using
piperidine in place
of pyrrolidine. LC-MS (C281135N302 calc'd 445) m/z 446 (M+H).
Example 25
(1-{4-[3-(2K-Methylpyrrolidin-1-y1)propoxy]phenyl}-1H-indol-2-yl)morpholin-4-
ylmethanone
=\O
N
410-01
=
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
(1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]phenyll-1H-indo1-2-y1)morpholin-4-
ylmethanone was synthesized by a method analogous to that used for Example 18
using
morpholine in place of pyrrolidine. LC-MS (C27H33N303 calc'd 447) m/z 448
(M+H).
Example 26
1-{443-(2R-Methylpyrrolidin-1-yl)propoxylphenyl}-1H-indole-2-carboxylic acid
butylamide
0
4110 N HN
=
1-{443-(2-Methylpyrrolidin-1-yl)propoxylphenyll-1H-indole-2-carboxylic acid
butylamide
was synthesized by a method analogous to that used for Example 18 using
butylamine in
place of pyrrolidine. LC-MS (C27H35N302 calc'd 433) m/z 434 (M+H).
Example 27
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl}-1H-indole-2-carboxylic acid
isobutylamide
40, \ 0 \
N HN
=
1-{4-[3-(2-Methylpynolidin-1-yl)propoxy]phenyll-1H-indole-2-carboxylic acid
isobutylamide was synthesized by a method analogous to that used for Example
18 using
isobutylamine in place of pyrrolidine. LC-MS (C271135N302 calc'd 433) m/z 434
(M+H).
31
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 28
1-{443-(2R-Methylpyrrolidin-1-yl)propoxylpheny1}-1H-indole-2-carboxylic acid
cyclohexylmethylamide
=\
N HN
1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]pheny11-1H-indole-2-carboxylic acid
cyclohexylmethylamide was synthesized by a method analogous to that used for
Example 18
using cyclohexylmethylamine in place of pyrrolidine. LC-MS (C30H39N302 calc'd
473) m/z
474 (M+H).
Example 29
5-Methoxy-1-{4-[3-(2R-methylpyrrolidin-1-yl)propoxylphenyl}-1H-indole-2-
carboxylic
acid cyclohexylamide
Me0
0
N HN-0
5-Methoxy-1-{443-(2R-methylpyrrolidin-1-y1)propoxy]pheny1}-1H-indole-2-
carboxylic acid
cyclohexylarnide was synthesized by a method analogous to that used for
Example 18 using
cyclohexylamine in place of pyrrolidine. LC-MS (C30H39N303 calc'd 489) m/z 490
(M+H).
Scheme 5
\0
LiAIH4 _______________________________________ ON OH Me0H
N
Ts0H N
32
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 30
144-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole-2-carboxylic acid ethyl ester
\0
N 0-\
1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole-2-carboxylic acid ethyl ester
was
synthesized by a method analogous to that used for Example 4 starting from
ethyl indole-2-
carboxylate. LC-MS (C24H28N203 calc'd 392) m/z 393 (M+H); 11-1NMR (300 MHz,
CDC13)
5 7.72 (d, J= 7.8 Hz, 1H), 7.43 (s, 1H), 7.29 - 7.15 (m, 4H), 7.06 (d, J= 8.4
Hz, 1H), 6.99
(d, J= 8.7 Hz, 2H), 4.23 (q, J= 7.2 Hz, 2H), 4.10 (t, J= 6 Hz, 2H), 3.06 -
3.01 (m, 4H), 2.27
-2.17 (m, 2H), 2.01 - 1.96 (m, 6H), 1.26 (t, J= 7.2 Hz, 3H).
Example 31
{1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indo1-2-yl}methanol
N OH
=
0
(1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole-2-carboxylic acid ethyl
ester (0.5 g,
1.27 mmol) was dissolved in THF (10 mL) and added dropwise to lithium aluminum
hydride
(1.53 mL, 1 M solution in THF, 1.53 mmol) in THF (10 mL). The reaction was
stirred at 60
C for 2 hours. Water (0.3 mL), 2 N NaOH (0.3 mL), and water (0.9 mL) =were
added, and
the solvent was evaporated. The resulting residue was diluted with water and
extracted with
dichloromethane. The dichloromethane extracts were dried over MgSO4 and
concentrated to
give a white solid, 0.36 g. A small amount of the product was purified by semi-
prep LC-MS
to give 3.5 mg of pure desired product. LC-MS (C22H26N202 calc'd 350) in/z 351
(M+H); 1H
NMR (300 MHz, CDC13) 5 7.65 - 7.61 (m, 1H), 7.35 (d, J= 8.7 Hz, 2H), 7.16 -
7.08 (m,
33
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
3H), 7.01 (d, J= 8.7 Hz, 2H), 6.65 (s, 1H), 4.64 (s, 2H), 4.10 (t, J= 6 Hz,
2H), 2.93 ¨ 2.86
(m, 6H), 2.21 ¨ 2.12 (m, 2H), 1.94 ¨ 1.90 (m, 4H).
Example 32
2-Methoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1F1H-indole
=
0-7-1
{1-[4-(3-Pyrrolidin-l-ylpropoxy)pheny1]-1H-indo1-2-yllmethanol (15 mg) was
dissolved in a
mixture of methanol, acetonitrile, and 1 N HC1. After standing at room
temperature for 2
hours, the solution was purified by semi-prep LC-MS to give 1.1 mg of the
desired product.
LC-MS (C23H2.8N202 calc'd 364) m/z 365 (M+H); 1H NMR (300 MHz, CDC13) 8 7.66 ¨
7.61
(m, 1H), 7.33 (d, J= 9 Hz, 2H), 7.20 ¨ 6.75 (m, 5H), 6.66 (s, 1H), 4.40 (s,
2H), 4.10 (t, J--
6.3 Hz, 2H), 3.28 (s, 3H), 2.82 ¨ 2.71 (m, 6H), 2.17 ¨ 2.08 (m, 2H), 1.87 (m,
4H).
Example 33
2-Cyclohexyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny11-1H-indole
o
N
2-Cyclohexyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was
synthesized
by a method analogous to that used for Example 32 using cyclohexanol in place
of methanol.
LC-MS (C28H36N202 calc'd 432) m/z 433 (M+H).
34
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 34
2-Isopropoxymethy1-1-[4-(3-pyrrolidin-1.-ylpropoxy)pheny1]-1H-indole
110
N 0¨(
Öc
o
2-Isopropoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyll-1H-indole was
synthesized by a
method analogous to that used for Example 32 using isopropanol in place of
methanol. LC-
MS (C25H32N202 cale'd 392) m/z 393 (M+H); 1H NMR (300 MHz, CDC13) 8 7.63 ¨
7.61 (m,
1H), 7.35 ¨ 7.32 (m, 2H), 7.14 ¨ 7.07 (m, 3H), 7.01 (d, J= 8.7 Hz, 2H), 6.64
(s, 1H), 4.42 (s,
2H), 4.10 (t, J= 6.3 Hz, 2H), 3.58 ¨ 3.48 (m, 1H), 2.76 ¨ 2.38 (m, 6H), 2.08 ¨
2.02 (m, 2H),
1.83 ¨ 1.71 (m, 4H), 1.08 (d, J= 6Hz, 6H).
Example 35
2-Cyclopentyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny11-1H-indole
110
N 0-0
o
2-Cyclopentyloxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was
synthesized
by a method analogous to that used for Example 32 using cyclopentanol in place
of methanol.
LC-MS (C27H34N202 ealc'd 418) m/z 419 (M+H).
Example 36
(5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indo1-2-y1}methanol
e.0
\
N OH
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
{5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indol-2-yllmethanol was
synthesized by a method analogous to that used for Example 31 starting with
ethyl 5-
methoxyindole-2-carboxylate. LC-MS (C23H28N203 calc'd 380) nilz 381 (M+H);
IHNMR
(300 MHz, CDC13) 5 7.33 (d, J= 8.1 Hz, 2H), 7.08 (s, 1H), 6.99 (d, J= 8.4 Hz,
3H), 6.80 (d,
J= 8.7 Hz, 1H), 6.56 (s, 1H), 4.61 (s, 2H), 4.07 (m, 2H), 3.85 (s, 3H), 2.83
(m, 6H), 2.14 (m,
2H), 1.91 (m, 4H).
Scheme 6
¨
40 1>= I CH3COCI ). = \ 1
pyridine pdc12(pPh3)2 HN
NH2
Cul, dioxane/TMG
90 C
Eel \ 4
Oo
cui, K3PO4, PhMe
/---\ 1000C
c/C
-NH HN-
N,
c--
Example 37
2-Cyclopropy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny11-1H-indole
\ 4
4104
O
N-(2-Iodophenyl)acetamide. 2-Iodoaniline (1.00 g, 4.56 mmol) was dissolved in
pyridine
(5 mL) and cooled to 0 C. After acetyl chloride (314 !IL, 5.94 mmol) was
added, the
reaction was stirred at 0 C for 1 hour and then at room temperature for 2
hours. The reaction
was diluted with 1 N HC1 and extracted with ether. The organic layer was dried
(MgS0.4) and
36
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
concentrated to give the desired acetamide (assumed quantitative), which was
used in the
next reaction without further purification.
2-Cyclopropy1-1H-indole. To a solution of N-(2-iodophenyl)acetamide (100 mg,
0.38
mmol) in dioxane (750 mL) and 1,1,3,3-tetramethylguanidine (750 mL) was added
cyclopropylacetylene (41 mL, 0.49 mmol), bis(triphenylphosphine)palladium(II)
chloride (35
mg, 0.05 mmol), and copper(I) iodide (10 mg, 0.05 mmol). The reaction was
stirred overnight
at 80 C. The solution was cooled and partitioned between water and methylene
chloride. The
organic layer was dried (MgSO4) and concentrated to give the uncyclized
Sonagashira
coupling product. Dioxane (750 mL) and 1,1,3,3-tetramethylguanidine (750 mL)
were added
and the reaction was stirred overnight at 90 C. The solution was again
partitioned between
water and methylene chloride. The organic layer was dried (MgSO4) and
concentrated to give
the desired indole, which was used without further purification.
2-Cyclopropy1-144-(3-pyrrolidin-1-ylpropoxy)pheny11-1H-indole. To a solution
of 2-
cyclopropy1-1H-indole (17 mg, 0.11 mmol) and 1-[3-(4-
iodophenoxy)propyl]pyrrolidine (36
mg. 0.11 mmol) in toluene (0.2 mL) was added copper iodide (0.2 mg), potassium
phosphate
(47 mg, 0.22 mmol), and N, N-dimethylethylenediamine (1.2 1.1.L, 0.11 mmol).
The reaction
was stirred at 100 C overnight. After cooling to room temperature, the
reaction was filtered
through a pad of silica. The reaction was concentrated and purified by
preparative HPLC to
give 1.6 mg of the desired indole. LC-MS (C24H28N20 calc'd 360) nilz 361
(M+H); 1H NMR
(300 MHz, CDC13) 5 7.55-7.52 (m, 1H), 7.35-7.32 (m, 2H), 7.10-7.02 (m, 5H),
6.16 (s, 1H),
4.11 (t, J= 6.3 Hz, 2H), 2.70 (t, J= 7.5 Hz, 2H), 2.59 (m, 4H), 2.08 (quint,
J= 7.0 Hz, 1H),
1.83 (m, 2H), 1.64 (m, 4H), 0.88-0.81 (m, 2H), 0.79-0.73 (m, 2H).
Example 38
2-Propy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole
N\
o
37
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
2-Propy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was synthesized by a
method
analogous to that used for Example 37 using 1-pentyne in the second step. LC-
MS
(C24H30N20 cale'd 362) m/z 363 (M+H); 11-1 NMR (300 MHz, CDC13) 5 7.61 - 7.54
(m, 1H),
7.25 - 7.21 (m, 2H), 7.09 - 6.99 (m, 5H), 6.38 (s, 1H), 4.10 (t, J= 6 Hz, 2H),
3.01 - 2.93 (m,
6H), 2.55 (t, J= 7.8 Hz, 2H), 2.25 - 2.15 (m, 2H), 1.98 - 1.93 (m, 4H), 1.60
(sextet, J=7.5
Hz, 2H), 0.91 (t, J= 7.5 Hz, 3H).
Scheme 7
A
O 1.1NH
, NH2
Cul
NaBH(OAc)3)- NH
Pd(PPh3)2Cl2*-
Et3N
DMF
reflux
0, DCM, AcOH
I*1
Cul
0, / 0, /
\1 1 O\1
TBAF N
THF
NaH, Nal
DMF
OH
Example 39
2-Cyclopropy1-1-[4-(3-pyrrolidin-1-ylpropoxy)eyelohexy1]-1H-indole
1
0,7-1N,
4-(tert-Butyldimethylsilanyloxy)eyelohexanone. Synthesized by literature
procedure.
Carrell , M. C.; Urbano, A.; Di Vitta C. J Org. Chem. 1998, 63, 8320.
38
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
[4-(tert-Butyldimethylsilanyloxy)cyclohexy1]-(2-iodophenyl)amine. To a
solution of 2-
iodoaniline (2.5 g, 10.9 mmol) in dichloromethane (160 mL) was added 4-(tert-
butyldimethylsilanyloxy)cyclohexanone (2.39 g, 10.9 mmol) and acetic acid (8
mL). After
the reaction was stirred for 1 hour at room temperature, sodium
triacetoxyborohydride (3.47
g, 16.4 mmol) was added and the reaction was stirred at room temperature
overnight. The
reaction was quenched with saturated sodium bicarbonate and extracted with
dichloromethane. The organic solution was dried (MgSO4) and concentrated to
give 4.47 g of
the desired amine, which was used without further purification. LC-MS
(C18H30INOSi calc'd
431) in/z432 (M+H).
[4-(tert-Butyldimethylsilanyloxy)cyclohexyl]-(2-
cyclopropylethynylphenyl)amine. To a
solution of [4-(tert-butyldimethylsilanyloxy)cyclohexyl]-(2-iodophenypamine
(4.47 g, 10.4
mmol) in triethylamine (70 mL) was added copper(I) iodide (198 mg, 1.04 mmol),
followed
by bis(triphenylphosphine)palladium(II) chloride (730 mg, 1.04 mmol) and
cyclopropylacetylene (1.73 mL, 20.8 mmol). The reaction was stirred under
nitrogen at room
temperature overnight. After the reaction mixture was concentrated, the
residue was
dissolved in ether and filtered through Celite. Concentration gave the crude
product, which
was used without further purification in quantitative yield. LC-MS (C23H35NOSi
calc'd 369)
m/z 370 (M+H).
144-(tert-Butyldimethylsilanyloxy)cyclohexy11-2-cyclopropyl4H-indole. To a
solution of
[4-(tert-butyldimethylsilanyloxy)cyclohexyl]-(2-cyclopropylethynylphenyl)amine
(3.84 g,
10.4mmol) in N,N-dimethylfoiniamide (60 mL) was added copper(I) iodide (100
mg, 0.525
mmol). After the reaction was refluxed for 48 hours, it was allowed to cool to
room
temperature, and the solvent was removed in vacuo. The residue was partitioned
between
water and dichloromethane. The dichloromethane was dried (MgSO4) and
concentrated to
give a dark residue that was used in the next step with no further
purification. LC-MS
(C23H35NOSi calc'd 369) m/z 370 (M+H).
4-(2-Cyclopropylindo1-1-Acyclohexanol. Crude 1-[4-(tert-
butyldimethylsilanyloxy)cyclohexyl]-2-cyclopropy1-1H-indole from above (10.4
mmol) was
dissolved in tetrahydrofuran (150 mL), and tetrabutylammonium fluoride (21 mL,
1 M in
THF, 21 mmol) was added. After the reaction was stirred for 72 hours it was
concentrated
39
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
and the residue partitioned between ethyl acetate and water. The organic layer
was dried
(MgSO4), concentrated and purified by Si02 chromatography (10-50% ethyl
acetate/hexanes)
to give two (cis/trans) isomers (355 mg of the more polar isomer, 681 mg of
the less polar
isomer) of the desired alcohol. LC-MS (C17H211\10 calc'd 255) 777/Z 256 (M+H).
2-Cyclopropy1-144-(3-pyrrolidin-l-ylpropoxy)cyclohexy1FIR-indole. To a
solution of 4-
(2-cyclopropylindo1-1-yl)cyclohexanol (25 mg, 0.098 mmol, more polar isomer)
in N,N'-
dimethylformamide (2 mL) was added sodium iodide (8 mg) and sodium hydride (6
mg, 60%
dispersion in mineral oil, 0.15 mmol). After the reaction was allowed to stir
at room
temperature for 5 minutes, 1-(3-chloropropyl)pyrrolidine (22 mg, 0.15 mmol,)
was added,
and the reaction was stirred at 85 C for 3. hours. The reaction was allowed
to cool to room
temperature and partitioned between water and dichloromethane. The organic
layer was dried
(MgSO4), and concentrated. The residue was purified by preparative LCMS to
give 8.0 mg of
the desired amine. 1H NMR (300 MHz, CDC13) 8 7.50 (d, J¨ 7.2 Hz, 1H), 7.42 (d,
J= 8.1
Hz, 1H), 7.12 - 6.99 (m, 2H), 6.14 (s, 1H), 4.64 - 4.53 (m, 3H), 3.58 (t, J=
6.2 Hz, 2H), 3.42
(tt, J= 3.9, 10.8 Hz, 1H), 2.91 - 2.82 (m, 4H), 2.42 (m, 2H), 2.24 (m, 2H),
2.00 - 1.79 (m,
9H), 1.45 (m, 2H), 0.95 (m, 2H), 0.74 (m, 2H); LC-MS (C24H34N20 calc'd 366)
in/z 367
(M+H).
Spectral data for the product founed from the less polar isomer of 4-(2-
cyclopropylindo1-1-
ypcyclohexanol:
1H NMR (300 MHz, CDC13) 8 7.56 - 7.48 (m, 2H), 7.10 - 6.96 (m, 2H), 6.12 (s,
1H), 4.58 (tt,
J= 4.2, 12.6 Hz, 1H), 3.65 (s, 1H), 3.55 (t, J= 6.0 Hz, 2H), 2.78 - 2.62 (m,
7H), 2.15 (d, J-
14.7 Hz, 2H), 2.00 - 1.83 (m, 8H), 1.69 - 1.25 (m, 4H), 0.984 - 0.873 (m, 2H),
0.764 - 0.713
(m, 2H); LC-MS (C24H34N20 calc'd 366) 177/Z 367 (M+H).
40
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Scheme 8
= l Si,/
/
____________________________________________ 401
Cul, DMF
reflux
NH2 no,i(Dok r.1
r- ukr 1312v12, Cul NH2
NEt3
I la le
O
/3\ =
OBn
TBAF
401 N
Cu!, K3PO4
OBn
toluene 100 C
o/
110 OH 1. NaH, Mel
2. H2, Pd/C
N
3- Cl^'NLD
0
OBn
Example 40
2-(2-Methoxyethyl)-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyll-1H-indole
o/
O
1.1 \
244-(tert-Butyldimethylsilanyloxy)but-1-ynyllphenylamine. 2-Iodoaniline (1.76
g, 8
mmol) was dissolved in triethylamine (50 mL) and placed under N2. tert-
Butylbut-3-
ynyloxydimethylsilane (2.58 g, 14 mmol) was added, followed by
bis(triphenylphosphine)palladium(II) chloride (30 mg, 0.042 mmol) and
copper(I) iodide (7
mg, 0.036 mmol), and the reaction was stirred overnight at room temperature.
Triethylamine
was evaporated, and the residue was diluted with ether and filtered through
Celite. The
filtrate was concentrated, and the residue was purified by Si02 chromatography
(5-20% ethyl
acetate/hexanes) to give the desired product. The reaction was assumed to be
quantitative.
41
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
2-[2-(tert-Butyldimethylsilanyloxy)ethy1]-1H-indole. 2- [4-(tert-
Butyldimethylsilanyloxy)but-1-ynyliphenylamine (8 mmol) was heated at reflux
in N,N-
dimethylformamide (30 mL) with copper(I) iodide (5 mg, 0.026 mmol) for 3
hours. The
solvent was evaporated, and the residue was diluted with ether and filtered
through Celite.
The filtrate was concentrated, and the residue was purified by Si02
chromatography (5-20%
ethyl acetate/hexanes) to give the desired product, 0.88 g. 1HNMR (300 MHz,
CDC13) 68.62
(br, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.13 - 7.03 (m,
2H), 6.22 (s, 1H),
3.92 (t, J = 5.7 Hz, 2H), 2.95 (t, J = 5.7 Hz, 2H), 0.95 (s, 9H), 0.08 (s,
6H).
1-(4-Benzyloxypheny1)-2[2-(tert-butyldimethylsilanyloxy)ethy1]-1H-indole. 242-
(tert-
Butyldimethylsilanyloxy)ethy1]-1H-indole (0.44 g, 1.6 mmol) and (4-
benzyloxy)iodobenzene
(0.6 g, 1.92 mmol) were dissolved in toluene (1.6 mL), and NN-
dimethylethylenediamine
(0.034 mL, 0.32 mmol), copper(I) iodide (16 mg, 0.08 mmol), and potassium
phosphate (0.72
g, 3.36 mmol) were added. The mixture was heated at 100 C overnight, then
filtered through
a plug of silica with ether. The filtrate was concentrated, and the residue
was purified by
Si02 chromatography (0-10% ethyl acetate/hexanes) to give the desired product,
0.62 g. LC-
MS (C29H35NO2Si calc'd 457) m/z 458 (M+H).
2-[1-(4-Benzyloxypheny1)-1H-indo1-2-yl]ethanol. 1-(4-Benzyloxypheny1)-2-[2-
(tert-
butyldimethylsilanyloxy)ethy1]-1H-indole (0.62 mmol, 1.35 mmol) was dissolved
in
tetrahydrofuran (6 mL) under N2, and tetrabutylammonium fluoride (1.49 mL, 1 M
in
tetrahydrofuran, 1.49 mmol) was added. The reaction was stirred for 2 hours,
then quenched
with saturated ammonium acetate. The mixture was extrated with ethyl acetate,
dried over
MgSO4, and concentrated. The residue was passed through a plug of silica with
ethyl acetate.
The filtrate was concentrated to give the desired product. The reaction was
assumed to be
quantitative. LC-MS (C23H2INO2 calc'd 343) m/z 344 (M+H).
1-(4-Benzyloxypheny1)-2-(2-methoxyethyl)-1H-indole. 2-[1-(4-Benzyloxypheny1)-
1H-
indol-2-yl]ethanol (0.675 mmol) was dissolved in tetrahydrofuran (5 mL) under
N2, and
sodium hydride (81 mg, 60% dispersion in mineral oil, 2.03 mmol) was added.
The reaction
was heated to reflux, at which time iodomethane (0.42 mL, 6.75 mmol) was
added. The
reaction was stirred at reflux for 3 hours, then carefully quenched with
water. The mixture
was extracted with ethyl acetate, dried over MgSO4, and concentrated. The
residue was
42
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
purified by Si02 chromatography (5-20% ethyl acetate/hexanes) to give the
desired product,
0.14 g. LC-MS (C24.H23NO2 calc'd 357) n2/z 358 (M+H).
4-[2-(2-Methoxyethyl)indo1-1-yllphenol. 1-(4-Benzyloxypheny1)-2-(2-
methoxyethyl)-1H-
indole (0.14 g, 0.39 mmol) was dissolved in tetrahydrofaran (2 mL) and
methanol (1 mL). A
catalytic amount of palladium on carbon (wet, 10% dry basis) was added, and
the flask was
purged with N2 and H2. The reaction was stirred under 1 atm of H2 overnight.
The mixture
was filtered through Celite, and the filtrate was concentrated to give the
desired product. The
reaction was assumed to be quantitative. LC-MS (C17E1171\102 calc'd 267) m/z
266 (M-H).
2-(2-Methoxyethyl)-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]--1H-indole. 4.4242-
Methoxyethyl)indo1-1-yl]phenol (0.39 mmol) was dissolved in N,N-
dimethylfoiniamide (4
mL), and 1-(3-chloropropyl)pyrrolidine (58 mg, 0.39 mmol), sodium hydride (19
mg, 60%
dispersion in mineral oil, 0.47 mmol), and sodium iodide (59 mg, 0.39 mmol)
were added.
The reaction was heated at 70 C for 1.5 hours, then carefully quenched with
saturated
sodium bicarbonate solution. The mixture was extracted with ethyl acetate,
dried over
MgSO4 and concentrated. The residue was purified by Si02 chromatography to
give 70 mg
of the desired product. LC-MS (C24H30N202 calc'd 378) m/z 379 (M+H); 1H NMR
(300
MHz, CDC13) 5 7.60 - 7.56 (m, 1H), 7.25 - 7.21 (m, 2H), 7.10 - 7.00 (m, 5H),
6.45 (s, 1H),
4.11 (t, J = 6.3 Hz, 2H), 3.57 (t, J = 7.2 Hz, 2H), 3.30 (s, 3H), 2.89 (t, J =
7.2 Hz, 2H), 2.74 (t,
J = 7.5 Hz, 2H), 2.65 (m, 4H), 2.16 -2.05 (m, 2H), 1.90.- 1.81 (m, 4H).
Scheme 9
401 N OTBS
Cul, K3PO4
N OTBS
OH
TBAF N
N
"
41,
I toluene 100 C =
43
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Example 41
2-{144-(3-Pyrrolidin-1-ylpropoxy)pheny11-1H-indo1-2-yllethanol
N OH
O
2-[2-(tert-Butyldimethylsilanyloxy)ethy1]-144-(3-pyrrolidin-1-
ylpropoxy)pheny11-1H-
indole. 2[2-(tert-Butyldimethylsilanyloxy)ethy1]-1H-indole (0.11 g, 0.4 mmol)
and 14344-
Iodophenoxy)propyl]pyrrolidine (0.16 g, 0.48 mmol) were dissolved in toluene
(0.4 mL), and
N,N-dimethylethylenediamine (0.017 mL, 0.16 mmol), copper(I) iodide (30 mg,
0.16 mmol),
and potassium phosphate (0.18 g, 0.84 mmol) were added. The reaction was
heated at 100 C
overnight. The mixture was filtered through Celite with dichloromethane. The
filtrate was
concentrated, and taken forward without purification (crude product contains
some starting
material). LC-MS (C291442N202Si calc'd 478) m/z 479 (M+H).
2-{1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indol-2-yl}ethanol. 2- [2-(tert-
Butyldimethylsilanyloxy)ethy1]-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole (0.4
mmol) was dissolved in tetrahydrofuran (2 mL) under N2, and
tetrabutylam_monium fluoride
(0.44 mL, 1 M in tetrahydrofuran, 0.44 mmol) was added. The reaction was
stirred at room
temperature for 3 hours, then quenched with saturated ammonium chloride. The
mixture was
diluted with saturated sodium bicarbonate solution and extracted with ethyl
acetate. The
organic extracts were dried over MgSO4 and concentrated. The residue was
purified by semi-
prep LC-MS to give the desired product, 15.8 mg. LC-MS (C23H28N202 calc'd 364)
m/z 365
(M+H); 1H NMR (300 MHz, CDC13) 6 7.61 - 7.59 (m, 1H), 7.24 - 7.20 (m, 2H),
7.13 - 7.07
(m, 2H), 7.03 - 7.00 (m, 3H), 6.48 (s, 1H), 4.09 (t, J = 6.3 Hz, 2H), 3.75 (t,
J = 6.6 Hz, 2H),
2.89 (t, J = 6.6 Hz, 2H), 2.66 (t, J = 7.5 Hz, 2H), 2.60 - 2.49 (m, 4H), 2.10 -
2.01 (m, 2H),
1.85 - 1.76 (m, 4H).
44
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Scheme 10
la Me0
4"-OBn 1101 \
Me0 LAH
\ N OEt
OEt Cul, K3PO4
H -N, toluene 100C
OBn
= Me0 0¨
Me0401 0¨
Me0 40 \ OH \
Mel, Ag20 =
H2, Pd/C
OBn OBn OH
Me0 0¨
CIN
=
Example 42
5-Methoxy-2-methoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole
0 0¨
=
0
1-(4-Senzyloxyphenyl)-5-methoxy-1H-indole-2-carboxylic acid ethyl ester. 5-
Methoxyindole-2-ethyl ester (0.59 g, 2.7 mmol), 1-benzyloxy-4-iodobenzene (1
g, 3.23
mmol), N,N-dimethylethylenediamine (0.057 mL, 0.54 mmol), copper(I) iodide
(0.1 g, 0.54
mmol), and potassium phosphate tribasic (1.2 g, 5.67 mmol) were heated in
toluene at 100 C
for 24 hours. The mixture was filtered through a plug of silica with ethyl
acetate, and the
filtrate was concentrated. Si02 chromatography with 5-20% ethyl
acetate/hexanes gave the
desired product (0.51 g, 57% yield), along with some mixed fractions (0.36 g)
that were
saved for future purification. LC-MS (C25H23N04 calc'd 401) miz 402 (M+H).
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
[1-(4-Benzyloxypheny1)-5-methoxy-1H-indo1-2-3711methanol. Lithium aluminum
hydride
(1.53 mL, 1 M in tetrahydrofuran, 1.53 mmol) was diluted with tetrahydrofuran
(5 mL) under
N2, and 1-(4-benzyloxypheny1)-5-methoxy-1H-indole-2-carboxylic acid ethyl
ester (0.51 g,
1.27 mmol) in tetrahydrofuran (5 mL) was added dropwise. The reaction was
stirred at reflux
for 2 hours, then cooled to room temperature. Water (0.3 mL) was added
carefully, followed
by 2 N NaOH (0.3 mL) and water (0.9 mL). The solvent was evaporated, and the
residue was
partitioned between water and ethyl acetate. The organic was separated, dried
over MgSO4,
and concentrated to give 0.41 g (88% yield) of crude product. LC-MS (C23H21NO3
calc'd
359) m/z 360 (M+H).
1-(4-Benzyloxypheny1)-5-methoxy-2-methoxymethyl1H-indole. [1-(4-
Benzyloxypheny1)-
5-methoxy-1H-indo1-2-yl]methanol (0.2 g, 0.56 mmol) was dissolved in
acetonitrile (2 mL),
and iodomethane (0.35 mL, 5.6 mmol) and silver(I) oxide (0.39 g, 1.68 mmol)
were added.
The mixture was stirred overnight at 40 'IC, then cooled to room temperature
and filtered
through a pad of Celite. The filtrate was concentrated. Si02 chromatography
with 3-50%
ethyl acetate/hexanes gave 0.16 g (77% yield) of the desired product. LC-MS
(C24H23NO3
calc'd 373) m/z 374 (M+H).
4-(5-Methoxy-2-methoxymethylindo1-1-yl)phenol. 1-(4-Benzyloxypheny1)-5-methoxy-
2-
methoxymethy1-1H-indole (0.16 g, 0.43 mmol) was dissolved in methanol (3 mL)
and
tetrahydrofuran (1 mL). Palladium on carbon (0.32 g, 10% wet) and ammonium
formate
(0.14 g, 2.14 mmol) were added, and the reaction was stirred at reflux for 2
hours. The
mixture was cooled to room temperature and filtered through a pad of Celite.
The filtrate was
concentrated, and the crude material taken on without purification. LC-MS
(C17H17NO3
calc'd 283) nilz 284 (M+H).
5-Methoxy-2-methoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-indole.
445-
Methoxy-2-methoxymethylindo1-1-yl)phenol (0.43 mmol) was dissolved in N,N-
dimethylformamide under N2. 1-(3-Chloropropyl)pyrrolidine (74 mg, 0.5 mmol),
sodium
hydride (20 mg, 60 % wt dispersion in mineral oil, 0.5 mmol) and sodium iodide
(75 mg, 0.5
mmol) Were added, and the mixture was heated at 70 C for 2 hours. The
reaction was cooled
to room termperature, diluted with water, and extracted with ethyl acetate.
The organic
extracts were dried over MgSO4 and concentrated. Purification by semi-prep LC-
MS gave
the desired product, 67.4 mg (40% yield, 2 steps). LC-MS (C24H30N203 calc'd
394) 772/Z 395
46
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
(M+H); 1H NMR (300 MHz, CDC13) 5 7.34 ¨ 7.29 (m, 2H), 7.09 (d, J = 2.4 Hz,
1H), 7.04 ¨
6.98 (m, 3H), 6.82 (d, J = 2.4 Hz, 1H), 6.58 (s, 1H), 4.37 (s, 2H), 4.10 (t, J
= 6.3 Hz, 2H),
2H), 3.86 (s, 3H), 3.28 (s, 3H), 2.69 ¨ 2.64 (m, 2H), 2.58 ¨2.54 (m, 4H), 2.11
¨2.01 (m, 2H),
1.83 ¨ 1.79 (m, 4H).
Example 43
5-Methyl-2-methoxymethy1-1-14-(3-pyrrolidin-l-ylpropoxy)phenylPH-indole
0¨
NI\
=
0
5-Methy1-2-methoxymethy1-1-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-1H-indole was
synthesized by a method analogous to that used for Example 42. LC-MS
(C24H30N202 calc'd
378) 117/Z 379 (M+H); 114 NMR (300 MHz, CDC13) 6 7.42 ¨ 7.41 (m, 1H), 7.34¨
7.29 (m,
2H), 7.04 ¨ 6.95 (m, 4H), 6.57 (s, 1H), 4.38 (s, 2H), 4.09 (t, J = 6.3 Hz,
2H), 3.27 (s, 3H),
2.69 ¨ 2.64 (m, 2H), 2.58 ¨ 2.53 (m, 4H), 2.44 (s, 3H), 2.10 ¨ 2.01 (m, 2H),
1.83 ¨ 1.79 (m,
4H).
Example 44
5-Fluoro-2-methoxymethy1-144-(3-pyrrolidin-l-ylpropoxy)phenyl]-1H-indole
F
0-
5-Fluoro-2-methoxymetby1-144-(3-pyrrolidin-l-ylpropoxy)phenyl]-1H-indole was
synthesized by a method analogous to that used for Example 42. LC-MS
(C23H27FN202
calc'd 382) m/z 383 (M+H); 1HNMR (300 MHz, CDC13) 5 7.34 ¨ 7.24 (m, 3H), 7.03
¨ 6.99
(m, 3H), 6.88 (td, J = 9 Hz, 2.4 Hz, 1H), 6.61 (s, 1H), 4.36 (s, 2H), 4.10 (t,
J = 6.3 Hz, 2H),
3.28 (s, 3H), 2.69 ¨ 2.64 (m, 2H), 2.57 ¨ 2.53 (m, 4H), 2.10 ¨ 2.01 (m, 2H),
1.83 ¨ 1.79 (m,
4H).
47
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Representative compounds of the present invention that were prepared by the
procedures of Examples 1-41 were evaluated in binding assays against cells
expressing
human H3 receptor by the following procedure.
Cell culture
Materials
[125I]iodoproxyfan (2000 Ci/mmol) was obtained from Amersham Bioscience
Piscataway, NJ). [3H]Na-methyhistamine (85 Ci/mmmol) was purchased from Perkin
Elmers Life Science (Boston, MA). Calcium 3 dye kit was from Molecular Devices
(Sunnyvale, CA). All other chemicals were either from Sigma-Aldrich (St.
Louis, MO) or
Tocris Cookson Inc. (Ellisville, MO).
RAGE methodology
The human histamine H3 receptor was stably expressed in HT1080 cells
containing
the chimeric G-protein, Gq i5 (Coward et al., Anal Biochein 1999; 270:242-8).
HT1080-
Gqcci5 cells were grown in alpha-modified MEM containing 10% fetal bovine
serum and 7
jug/mlblasticidin at 37 C in 5% CO2/95% atmosphere. Cells (4.8x109) were
irradiated with
50 rads from a 137Cs source and the pFG8-HH3 RAGE (Random Activation of Gene
Expression; see Harrington et al., Nature Biotechnology. 2001; 19:440-45)
vector was
subsequently integrated into the cells via electroporation (250V, 600g, 50S2).
The RAGE
vector pFG8-HH3 contained cDNA sequence coding for the first exon (83 amino
acids) of
human H3 receptor. After electroporation, cells were plated in T75 flasks and
grown in
alpha-modified MEM. The culture medium was replaced 48 hours after
electroporation with
alpha-modified MEM, 10% fetal bovine serum, 5001..tg/m1 hygromycin B and 3
jig/m1
puromycin. Medium was replaced every four days during cell expansion. To
identify RAGE
activated cells expressing the H3 receptor, pools of approximately 10,000
colonies (5x107-
1.5x108 cells total) were screened by PCR for the desired gene product (using
primers
specific to the RAGE vector and exon 2 of the H3 receptor). Pools that were
found to contain
the appropriate transcript, as confirmed by sequencing, were subcloned into
pools of 100
cells/well. Positive 100 cells/well pools were identified by PCR, confirmed by
sequencing,
and subsequently subcloned to 0.8 cells/well. Once clones expressing the H3
receptor were
identified by PCR analysis, assays (FLIPR or radioligand binding) were
performed to
48
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
confirm that the activated gene produced functional protein. The protein
expression in the
initial clones obtained from the RAGE library was increased by growth in the
presence of
methotrexate. Since the integrated RAGE vector contains the DHFR gene, such
treatment
selects for cells that have amplified the genetic locus containing the RAGE
insert. Subclones
obtained after methotrexate amplification were tested for functional activity
in FLIPR assays
to identify the clone that was most suitable for HTS. The final HT1080-Gqai5
RAGE clone
(RAGE-H3) expressing the human histamine H3 receptor was grown in alpha-
modified
MEM containing 10% fetal bovine serum, 3 jug/m1 puromycin, 500 ii.g/m1
hygromycin B, 3.2
[IM methotrexate at 37 C in 5% CO2/95% atmosphere.
Membrane preparation
RAGE-H3 cells (109) were washed twice with cold PBS, scraped off the plates,
and
centrifuged at 1000 x g for 5 minutes. Cells were resuspended in ice-cold 10
mM Tris HC1,
pH 7.4, containing 5 mM EDTA and protease inhibitor cocktail tablets (Roche
Molecular
Biochemicals). After incubating on ice for 10 minutes, the cells were
homogenized with a
dounce homogenizer or a polytron tissue grinder, and centrifuged at 1000 x g
for 10 minutes
at 4 C. The resulting supernatant was centrifuged at 32, 000 x g for 30
minutes at 4 C. The
membrane pellets were resuspended in 50 mM Tris HC1, pH 7.4, and stored at ¨
80 C until
use. Protein concentration was deteauined by the Bradford method (Bio-Rad
Laboratories,
CA).
Radioligand binding assays
Binding assays were carried out in 96-well polypropylene plates in 50 mM Tris
HC1,
pH 7.4, containing 1 mM EDTA. Reaction mixtures contained 100 ill of membrane
suspension, 50 .1 of 4% DMSO, and 50 .1 of increasing amounts of
[125I]iodoproxyfan (final
concentration 0.0005-1.8 nM for human H3 receptor saturation binding assay).
Nonspecific
binding was defined by adding 10 jtM clobenpropit to the reaction mixtures.
Competition
binding assays were performed in a reaction mixture containing 100 i.1.1 of
membrane
suspension (¨ 20 lig of protein/well), 50 ill of [125I]iodoproxyfan (final
concentration of ¨
0.15 nM) and 50 jul of test compound. Compounds were dissolved in DMSO and
then diluted
with 4% DMSO; the final maximal DMSO concentration in the binding assays was
1%.
Incubations were performed for 1.5 hours at room temperature and reactions
were terminated
49
CA 02626028 2011-10-11
by rapid filtration over glass fiber GF/C filters (Perkin Elmer, MA) using a
Brandel cell
harvester. The filters were presoaked in 0.3% polyethyleneimine for 30 minutes
and were
washed with 500 ml of ice-cold 50 mM Tris HC1, pH 7.4. The filters were dried,
impregnated
with Meltilex wax scintillate (Perkin Elmer, MA) and counted with a Betaplate
scintillation
counter (Perkin Elmer, MA).
Calcium mobilization assays
RAGE-H3 or HT1080-mH3 cells were seeded in black 384-well plates and incubated
overnight at 37 C in a 5% CO2/95% atmosphere. After removing medium, cells
were treated
with CsC1 Ringer's buffer (136 mM CsCl, 5.4 mM KC1, 5.5 In.M D-Glucose, 20 mM
Hepes,
pH 7.4, 2.1 mM MgC12, 1.2 mM CaC12) containing the Calcium 3 dye (Molecular
Device,
CA) and probenecid (3.75 uM) for 60 minutes, according to manufacture's
instruction.
Compounds were diluted in CsCI Ringer's buffer containing 0.2% bovine serum
albumin and
1.0% DMSO. The dose response of (R)-a-methylhistamine-stimulated Ca2+ flux was
measured on a Fluorometric Imaging Plate Reader (FLIPR, Molecular Devices, CA)
and the
concentration of (R)-a-methylhistarnine to stimulate 75% of maximum response
was used to
test the inhibitory effect of compounds.
Data analysis
All data were analyzed by nonlinear least squares curve fitting using Prism
4.0 software. The
KD and Bmax for [125I]iodoproxyfan were derived from the equation RL=RtL /(KD
+L), where
RL is concentration of receptor-bound ligand at equilibrium, L is the free
ligand
concentration, and Rt is the total receptor concentration (i.e., %tax). For
competition binding
experiments, IC50 values (the concentration of compound producing 50%
inhibition of
specific binding) and Hill Coefficients (nH) were derived from fitting the
data to a 4-
parameter logiStic equation. Apparent Ki values were calculated using the
Cheng-Prussof
equation of K = IC50/(1+(L/KD)), where L is the ligand concentration. Agonist
stimulation
and antagonist inhibition in FLIPR were fitted to sigmoidal dose response
curves using the
equation Y = Bottom + (Top-Bottom)/(1+10A(LogEC50-X)), where X is the
logarithm of
concentration of compounds and Y is the fluorescent response. Z' values [15]
were derived to
evaluate the quality of the assays. Figures are representative of two to three
separate
experiments performed in triplicates or quadruplicates.
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
The results of this assay are set forth in the following Table 1.
Table 1
Selected Examples
Human 113
Chemical Name
(uM)
2-Methy1-144-(3-pyn-olidin-1-ylpropoxy)pheny1]-1H-
indole K1<0.01
Example 1
2-Methy1-144-(3-piperidin-1-ylpropoxy)pheny1]-1H-
indole K, <0.01
Example 2
2-Methy1-1-{443-(2R-methylpyrrolidin-1-
yl)propoxy]phenyll -1H-indole K, <0.01
Example 3
114-(3-Pyn-olidin-1-ylpropoxy)pheny1]-1H-indole
1c50<0.1
Example 4
5-Methoxy-2-methy1-1-[4-(3-pyrrolidin-1-
ylpropoxy)pheny1]-1H-indole 1050 <0.1
Example 5
5-Methy1-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole 1050 <1
Example 6
5-Bromo-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole 1050 <1
Example 7
4-Chloro-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole 1050<1
Example 8
5-Methoxy-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole 1050 <1
Example 9
5-Chloro-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole 1050 <1
Example 10
51
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Human H3
Chemical Name
(uM)
2,5-Dimethy1-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-
1H-indole Ki <0.1
Example 11
6-Chloro-144-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indole Ki <0.1
Example 12
2-Methy1-5-fluoro-144-(3-pyrrolidin-l-
ylpropoxy)phenyl]-1H-indole K, <0.01
Example 13
143-Methoxy-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-
methy1-1H-indole 1050<1
Example 14
1-[3-Chloro-4-(3-pyrrolidin-1-ylpropoxy)pheny1]-2-
methy1-1H-indole 1050<1
Example 15
2-Propy1-144-(3-pyrrolidin-l-ylpropoxy)phenyl]-1H-
indole K, <0.01
Example 16
5-Methoxy-2-methy1-144-(4-pyrrolidin-1-ylbut-1-
ynyl)pheny1]-1H-indole IC50<0.1
Example 17
(5-Methoxy-1-{443-(2R-methylpyrrolidin-1-
yppropoxy]phenyll-1H-indol-2-yppyrrolidin-1-
ylmethanone K, <0.01
Example 18
1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl} -1 H-
indole-2-carboxylic acid cyclobutylamide Ki <0.01
Example 19
1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl} -1H-
indole-2-carboxylic acid cyclopentylamide K. <0.01
Example 20
1-{4-[3-(2R-Methylpyrrolidin-1-yppropoxy]phenyll -1H-
indole-2-carboxylic acid cyclohexylamide K, <0.1
Example 21
1-{443-(2R-Methylpyrrolidin-1-yppropoxy]phenyl} -1H-
indole-2-carboxylic acid cycloheptylamide K. <0.1
Example 22
52
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Human 1-13
Chemical Name
(uM)
(1-{4-[3-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl} -
1H-indo1-2-yl)pyrrolidin-1-ylmethanone K, <0.01
Example 23
2-(3-Morpholin-4-ylpropoxy)-6,7,8,9-
tetrahydropyrido[1,2-a]indole lc <0.01
Example 24
(1-{443-(2R-Methylpyrrolidin-1-yl)propoxy]phenyl}-
1H-indol-2-y1)morpholin-4-ylmethanone ic <0.01
Example 25
1-{443-(2R-Methylpyrrolidin-1-yppropoxy]phenyll-1H-
indole-2-carboxylic acid butylamide K, <0.01
Example 26
1-{443-(2R-Methylpyrrolidin-l-yppropoxy]phenyl}-1H-
indole-2-carboxylic acid isobutylamide K1<0.01
Example 27
1-{443-(2R-Methylpyrrolidin-1-yl)propoxylphenyll-1H-
indole-2-carboxylic acid cyclohexylmethylamide K, <0.01
Example 28
5-Methoxy-1-{443-(2R-methylpyrrolidin-1-
yppropoxylpheny1}-1H-indole-2-carboxylic acid
cyclohexylamide K, <0.01
Example 29
144-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indole-2-
carboxylic acid ethyl ester Ki <0.01
Example 30
{144-(3-Pyrrolidin-l-ylpropoxy)pheny11-1H-indol-2-
y1}methanol K, <0.01
Example 31
2-Methoxymethy1-144-(3-pyrrolidin-1-
ylpropoxy)phenyl]-1H-indole K, <0.01
Example 32
2-Cyclohexyloxymethy1-144-(3-pyrrolidin-1-
ylpropoxy)phenyl]-1H-indole Ki <0.01
Example 33
2-Isopropoxymethy1-144-(3-pyrrolidin-1-
ylpropoxy)pheny1]-1H-indole K, <0.01
Example 34
53
CA 02626028 2008-04-14
WO 2007/047775
PCT/US2006/040744
Human H3
Chemical Name
(uM)
2-Cyclopentyloxymethyl- 1 -[4-(3-pyrrolidin- 1-
ylpropoxy)pheny1]-1H-indole Ki <0.0 1
Example 35
{5-Methoxy-1-[4-(3-pyrrolidin-1-ylpropoxy)pheny1]-1H-
indo1-2-y1} methanol Ki <0.01
Example 36
2-CycloProw1-144-(3 -pyrrolidin- 1 -ylpropoxy)pheny1]-
1H-indole 1050 <0.1
Example 37
2-Propy1-1-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-1H-indole
Ki <0.01
Example 38
2-Cyclopropyl- 14443 -pyrrolidin-1-
ylpropoxy)cyclohexyl]-1H-indole K, <0.01
Example 39
2-(2-Methoxyethyl)- 1 4443 -pyrrolidin-1-
ylpropoxy)pheny1]-1H-indole K, <O. 0 1
Example 40
2-{1-[4-(3-Pyrrolidin-1-ylpropoxy)pheny1]-1H-indol-2-
y1} ethanol K, <O. 01
Example 41
5-Methoxy-2-methoxymethyl- 14443 -pyrrolidin-1-yl-
propoxy)-pheny1]-1H-indole K, <0.01
Example 42
5-fluoro-2-methoxymethyl- 1 4443 -pyrrolidin-1 -y1 -
propoxy)-phenyl]-IH-indole K. <0.01
Example 43
5-methyl-2methoxymethyl- 1.-14-(3-pyrrolidin- 1-y1 -
propoxy)-pheny1]-1H-indole K, <O. 0/
Example 44
aIC50 values were determined using FLIPR.
54