Note: Descriptions are shown in the official language in which they were submitted.
CA 02626142 2008-03-18
Stabilized Aqueous Solutions of Ergoline Compounds
The present invention relates to stabilized aqueous
solutions of ergoline compounds or their physiologically
tolerable salts or derivatives in which the aqueous
solution contains 0.05 to 90.00% of at least one oxygen-
containing cosolvent. The present invention likewise
relates to the use of an aqueous solution stabilized in
this way for production of an agent for parenteral
treatment of neurodegenerative diseases or brain trauma.
Ergoline compounds such as lisuride or its physiological
tolerable salts are used today for treatment of patients
suffering from Parkinson's disease (F. Stocchi et al.
(2002) Brain 125: 2058-2066), Meige syndrome (G. Ransmayr
et al. (1988) Clinical Neuropharmacology 11: 68-76),
Dystonias (N. P. Quinn et al. (1984) Neurology 34: 223)
or other serious diseases. The required active ingredient
solutions are administered intravenously, intramuscularly,
transdermally or subcutaneously.
However, many ergoline compounds have proven to be unstable
in dissolved form (water as the solvent). Especially when
there are 8a-amino groups on the ring system, e.g., a
diethylurea group in the case of lisuride, these compounds
are extremely unstable in solution and tend to decompose
rapidly (hydrolysis). Another disadvantage is the poor
CA 02626142 2008-03-18
2
solubility of natural ergot alkaloids as well as their
salts and derivatives in water as the solvent.
Therefore, the corresponding ergoline compounds, in
particular lisuride, are currently being provided only in
lyophilized form. These must be reconstituted, i.e.,
returned to a liquid form by the patient, by adding a
corresponding salt solution before being administered to
the patient.
Only after this reconstitution can the substance be
administered to the patient. In particular, when using a
programmable minipump or micropump, the solution must first
be transferred to a special syringe. Only then can the
final filling of the minipump or micropump take place.
It is therefore easy to see that the type and number of
steps to be performed when using a lyophilizate make this
procedure extremely complex and difficult. This procedure
has proven to be extremely difficult especially for
patients who suffer from movement disorders (as in
Parkinson's disease) or dystonia because of their primary
illness.
The lisuride solution reconstituted from the lyophilizate
currently has only a limited stability so that
administration of this solution by a minipump or micropump,
for example, over a long period of time must be ruled out.
In addition, the multistep procedure described above often
entails the risk that the solutions are not sterile because
they must be prepared by the user. In such cases, the
CA 02626142 2008-03-18
3
safety of the patient is at risk because administration of
unsterile solutions entails unforeseeable risks.
Because of the aforementioned disadvantages of the
multistep procedure, it would be desirable if an active
ingredient solution could be made available directly.
Earlier publications especially with regard to the
stability of solutions and the solubility of ergot
alkaloids have taught us that the disadvantages can be
overcome to a great extent by using mixtures of water and
alcohols as the solvent. To do so, the use of linear
monofunctional alcohols and polyfunctional alcohols in
amounts of 60.00% to 100.00% in particular has been
described. Within the context of monofunctional alcohols,
ethanol is used in particular. Representatives of
polyfunctional alcohols that may be used include propylene
glycol, glycerol or polyethylene glycol (GB 2 062 568 A,
DE 27 35 587 Al, BE 881967 A, GB 1 539 083 A).
The following publications expand this information to
mixtures consisting of water and exclusively polyfunctional
alcohols with an alcohol content of 10.00% to 90.00%. In
particular the use of 25.00% to 80.00% has proven suitable
here (DD 43 402 A; EP 0 101 879 A2). This use is based on
strictly oral therapy in these publications.
With regard to tolerability for the patient, there are in
general problems with large amounts of cosolvents such as
the alcohols mentioned above. Especially the use of high
percentages of these compounds results in the solutions no
longer meeting the requirements of the corresponding
guidelines.
CA 02626142 2008-03-18
4
Furthermore, at higher concentrations of cosolvent, the
viscosity of the solutions also increases. This may lead to
problems in administration, e.g., when using a minipump or
a micropump, because the solution is more difficult to
pump.
Addition of corresponding alcohols in the literature has
also been limited to use with naturally occurring ergot
alkaloids and their 9,10-dihydro derivatives. Since these
compounds do not have an 8a-amino group, they are
significantly more stable than the ergot alkaloids with a
corresponding amino group in a position in position 8 on
the ring system. An amino group, optionally with additional
substituents, has a greater tendency to instability. This
pertains in particular to the diethylurea group
-NHCON (C2H5) 2 as in the case of the lisuride, terguride,
bromerguride or proterguride or their pharmaceutically
tolerable salts and esters, for example. On the basis of
this increased instability, the stabilization methods
mentioned above are regarded as completely unsuitable for
these compounds.
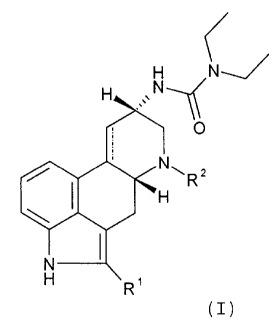
Therefore, the object of the present invention is to
provide aqueous stabilized solutions of ergoline compounds
of general formula I
CA 02626142 2008-03-18
H N
H N__~
0
NRs
I H
N I
H R,
(I)
This object is achieved by a stabilized aqueous solution of
an ergoline compound according to Claim 1, which contains
0.05% to 90.00% of at least one oxygen-containing
cosolvent. The percentage amounts in the following table
are to be understood as mass per volume (m/v).
Additional preferred embodiments are derived from the
dependent claims.
In other words, the object is achieved by a stabilized
aqueous solution which contains an ergoline compound of
formula I
~
H N~
H N__~
0
NR2
H
N
H R
(I)
CA 02626142 2008-03-18
6
or its physiologically tolerable salt or derivative in
which R' is a hydrogen atom or a halogen atom and R2 as an
alkyl group or an alkenyl group with 1 to 4 carbon atoms
and ----- denotes a single bond or double bond and also
0.05% to 90.00% of at least one oxygen-containing
cosolvent.
The at least one oxygen-containing cosolvent is preferably
a polyvalent alcohol. It may preferably have 2 to 6 carbon
atoms and at least two hydroxyl groups.
In a preferred embodiment, the polyvalent alcohol is
selected from the group of 1,2-ethanediol (glycol), 1,2-
propanediol (propylene glycol), 1,3-propanediol, 1,2,3-
propanetriol (glycerol) or 1,3-butanediol.
The at least one oxygen-containing cosolvent is preferably
also a polyethylene glycol with a molecular weight of 200-
35,000 g/mol. It is especially preferable here for the at
least one oxygen-containing cosolvent to be a polyethylene
glycol with a molecular weight of 200-4,000 g/mol. The at
least one oxygen-containing cosolvent is especially
preferably a polyethylene glycol with a molecular weight of
400 g/mol.
The concentration of the at least one oxygen-containing
cosolvent is preferably 0.05% to 20.00%. A concentration of
the at least one oxygen-containing cosolvent of 0.50% to
9.50% is especially preferred.
In a preferred embodiment, the at least one oxygen-
containing cosolvent is propylene glycol, and its
concentration amounts to 0.05% to 9.50%.
CA 02626142 2008-03-18
7
In another preferred variant, the at least one oxygen-
containing cosolvent is a nonionic detergent. It is
preferably selected from the group of reaction products of
polyethylene glycol, ethylene oxide or polyglycerol with
fatty alcohols, alcohols, hydrogenated castor oil, fatty
acids, hydroxy fatty acids or alkylphenols such as
nonylphenol or derivatives thereof.
It is preferable here for the nonionic detergent to be
selected from the group of reaction products of ethylene
oxide and castor oil, the reaction products of hydrogenated
castor oil and ethylene oxide or the polyethylene glycol 15
hydroxystearates, such as preferably Macrogol 15
hydroxystearate (Ph. Eur.) [European Pharmacopeia].
It is especially preferable for nonionic detergents from
the group of reaction products of ethylene oxide and castor
oil with a molar ratio of 20-60:1 or reaction products of
hydrogenated castor oil and ethylene oxide with a molar
ratio of 20-60:1 to be selected. The nonionic detergent is
preferably selected from the group of reaction products of
ethylene oxide and castor oil with a molar ratio of 30-60:1
or the reaction products of hydrogenated castor oil and
ethylene oxide with a molar ratio of 30-60:1. Most highly
preferably, the nonionic detergent is selected from the
group of reaction products of ethylene oxide and castor oil
with a molar ratio of 35:1 or the reaction products of
hydrogenated castor oil and ethylene oxide with a molar
ratio of 40:1 or 60:1.
In addition, it is preferable for the nonionic detergent to
be selected from the group of polyoxysorbitan fatty acid
CA 02626142 2008-03-18
8
esters, sorbitan fatty acid esters or polyoxyethylene-
polyoxypropylenes.
The nonionic detergent is preferably present in a
concentration of 0.05% to 90.00%. It is especially
preferably present in a concentration of 0.20% to 20.00%.
The nonionic detergent is most preferably present in a
concentration of 0.20% to 10.00%.
The ergoline compound is preferably selected from the group
of lisuride, terguride, proterguride and bromerguride. The
ergoline compound is most preferably lisuride.
In a preferred embodiment, the ergoline compound is present
in the form of its salt with sulfuric acid, sulfurous acid,
phosphoric acid, phosphorous acid, nitric acid, nitrous
acid, perchloric acid, hydrobromic acid, hydrochloric acid,
formic acid, acetic acid, propionic acid, succinic acid,
oxalic acid, gluconic acid (glyconic acid, dextronic acid),
lactic acid, malic acid, tartaric acid, tartric acid
(hydromalonic acid, hydroxypropane diacid), fumaric acid,
citric acid, ascorbic acid, maleic acid, malonic acid,
hydroxymaleic acid, pyruvic acid, phenylacetic acid, ortho-
toluic acid, meta-toluic acid, para-toluic acid, benzoic
acid, para-aminobenzoic acid, para-hydroxybenzoic acid,
salicylic acid, para-aminosalicylic acid, methanesulfonic
acid, ethanesulfonic acid, hydroxyethanesulfonic acid,
ethylenesulfonic acid, para-toluenesulfonic acid,
naphthylsulfonic acid, naphthylaminesulfonic acid,
sulfanilic acid, camphorsulfonic acid, china acid (quinic
acid), ortho-methylmandelic acid, hydrogenbenzenesulfonic
acid, picric acid (2,4,6-trinitrophenol), adipic acid,
D-(ortho-tolyl)tartaric acid or an amino acid.
CA 02626142 2008-03-18
9
Another preferred variant is implemented by the ergoline
compound being present in the form of its salt with an
amino acid from the group of methionine, tryptophan and
arginine.
In addition, it is preferable for the ergoline compound to
be present in the form of its salt with an acid-containing
amino acid from the group of glutamic acid and aspartic
acid.
An extremely preferred embodiment is implemented by the
ergoline compound being present in the form of its salt
with maleic acid.
The ergoline compound or its physiologically tolerable salt
or derivative is preferably present in a concentration of
0.01 to 25.00 mg/mL. A concentration of the ergoline
compound or its physiologically tolerable salt or
derivative of 0.25 to 10.00 mg/mL is especially preferred.
It is highly preferable for the ergoline compound or its
physiologically tolerable salt or derivative to be present
in a concentration of 0.50 to 3.00 mg/mL.
In addition, the stabilized aqueous solution may contain
organic and/or inorganic compounds for adjusting the
osmolarity in the case of a hypotonic solution and/or for
adjusting the pH.
The stabilized solution is preferably stored in a prefilled
syringe or in a carpule.
The invention also relates to the use of the stabilized
aqueous solution for preparing an agent for parenteral
CA 02626142 2008-03-18
treatment of neurodegenerative diseases or brain trauma.
The stabilized aqueous solution includes an ergoline
compound of formula I
H N
H N--~
O
NR2
H
N
H R
(I)
or its physiologically tolerable salt or derivative in
which R1 denotes a hydrogen atom or a halogen atom and R2
denotes an alkyl group or an alkenyl group with 1 to 4
carbon atoms and ----- denotes a single bond or double bond
which also contains 0.05% to 90.00% of at least one oxygen-
containing cosolvent.
The parenteral treatment is preferably administered
subcutaneously, intramuscularly, intravenously,
transdermally or through a pump implanted in the
bloodstream or in the tissue. With regard to pumps in
general, many different pumps or micropumps may be used.
In one embodiment, the neurodegenerative diseases are
Parkinson's disease or dystonias.
The brain trauma may be caused by a stroke or a traumatic
brain injury.
CA 02626142 2008-03-18
11
It has surprisingly been found that the inventive ergoline
compounds or their physiologically tolerable salts or
derivatives may be dissolved in mixtures of water and at
least one oxygen-containing cosolvent with a concentration
of the cosolvent of 0.05% to 90.00%. It is also possible to
use definitely lower concentrations than those described
for stabilized ergot alkaloids in the state of the art. In
particular, concentrations of 0.05% to 20.00%, especially
preferably from 0.50% to 9.50% are possible here.
Since the at least one cosolvent surprisingly improves the
solubility of the active ingredient, the volume to be
administered to a patient each day in a parenteral
dopaminergic therapy may be reduced significantly. The
solubility of an inventive model compound has been improved
by a factor of 1.5 by using an inventive cosolvent, in
particular even by a factor of 2, where the concentration
of the inventive cosolvent was less than 10% and the pH of
the solution was neutral.
The resulting solutions have, contrary to all expectations,
a stability of at least 6 months, in particular when using
the alcohols listed above. The stabilization is even
significantly improved with regard to increases in
temperature. On the whole, in comparison with the
unstabilized solution, an improved stability is observed
even when using the nonionic detergents. When using the
inventive alcohols, especially when using propylene glycol,
the positive effect on solubility and stability of the
invention ergoline compound is most noticeable. Especially
with regard to the alcohols listed above, in particular
propylene glycol, the result is a dual vantage on the
whole: first the solubility of the respective active
CA 02626142 2008-03-18
12
ingredient is increased and secondly the chemical stability
of the respective active ingredient is surprisingly
improved significantly.
In testing samples, it has been found, for example, that
storage at 25 C with light protection results in
decomposition of 5% within a period of 180 days when not
using a cosolvent according to this invention. With storage
at 6-8 C, the decomposition amounted to 0.5% to 0.8%. In
the case of storage at 40 C, up to 30% of the active
ingredient had decomposed. In contrast with that, addition
of an inventive cosolvent led to less than 0.5%
decomposition with storage under refrigeration (6-8 C) for
180 days in the absence of light. At room temperature
(25 C) an average decomposition of less than 0.5% was
observed. In the case of storage at 40 C in the presence of
the inventive cosolvent, 89.8% of the active ingredient
could still be detected, i.e., decomposition amounted to
only approximately 10%.
Owing to the solutions stabilized according to this
invention, it is now possible to supply patients directly
with solutions. This is a significant improvement in
comparison with the lyophilizates used in the past. At the
same time, the problems with regard to sterility are also
eliminated. Since the use of extremely low concentrations
of the cosolvent was also surprisingly possible, parenteral
administration is greatly facilitated. At low
concentrations, the viscosity of the solutions can be kept
in a range that is very suitable for handling. In addition,
with the solutions stabilized according to this invention,
it is possible to avoid exposing the patient to
CA 02626142 2008-03-18
13
unnecessarily high doses of the alcohols over a long period
of time.
For example, the estimated amount that is acceptable as a
daily oral dose of propylene glycol is 25 mg/kg body weight
(17th Report of the Joint FAO/WHO Expert Committee on Food
Additives, 1974). Likewise, values for which daily amount
is not acceptable for subcutaneous administration have also
been published. Therefore, to reduce the safety risks for
the patient, it is considered necessary to expose the
patient to preferably only small quantities of the
respective alcohol.
With the assumption of a daily dose of 2 mg lisuride
hydrogen maleate, this yields a burden on the patient of
240 mg propylene glycol when using 8.00% propylene glycol
and an assumed active ingredient concentration of 2 mg
active ingredient per 3 mL solution. In the case of an
average body weight of 70 kg, this dose thus amounts to
less than 15% of the acceptable amount for oral
administration. This can be optimized according to this
invention by further increasing the active ingredient
concentration and/or reducing the propylene glycol
concentration.
The inventive stabilized aqueous solution may comprise
organic and/or inorganic compounds, as mentioned above, to
adjust the osmolarity in the case of a hypotonic solution
and/or to adjust the pH.
By adjusting the pH, the solubility of the inventive
ergoline compounds or their physiologically tolerable salts
or derivatives can be increased. A reduction in pH leads to
CA 02626142 2008-03-18
14
an increase in the amount of the ionized form of the
inventive ergoline compound and thus to an improved
solubility. For example, a reduction in pH from pH 7 to
pH 6.2 with a representative inventive ergoline compound
yields an improvement in solubility by a factor of
approximately 5, and with a reduction in pH from 7 to 5.5
the solubility is even improved by a factor of
approximately 20. However, the stability of the inventive
ergoline compounds has an inverse relationship to the
solubility. It has been observed that the stability becomes
worse when the pH is reduced into the acidic range. On the
whole, in adjusting the pH it has proven to be preferable
to adjust a pH in the range of 4.00 to 8.00. A pH range
from 4.50 to 7.50 is especially preferred, and a range of
5.00 to 7.00 is most especially preferred.
To adjust the pH, the stabilized aqueous solution may
include a buffer system from the group of citrate buffer,
carbonate buffer, phosphate buffer or maleate buffer. A
citrate buffer is preferred as the buffer system.
The in-vivo tolerability of the inventive stabilized
aqueous solution of an inventive ergoline compound or its
physiologically tolerable salt or derivative can be
optimized by adjusting the osmolarity. This is the case
with an inventive solution in comparison with a solution
that is hypotonic with blood. The term "hypotonic" as used
here is understood to refer to a solution having a lower
osmotic pressure than human blood. On the average, blood
has an osmolarity (= osmotic pressure) of 290 mosmol/L
(milliosmol per liter). Then by adding physiologically
tolerable excipients, the solution is preferably adjusted
to an osmolarity of 250 to 350 mosmol/L, most preferably to
CA 02626142 2008-03-18
an osmolarity of 270 to 320 mosmol/L. To adjust the
osmolarity, sodium chloride is preferred. Sodium chloride,
which is a physiologically tolerable excipient is
preferably used to adjust a hypotonic solution that is
isotonic with blood.
For structurally related active ingredients which are based
on the same basic structure, similar physicochemical
activities may be expected in vitro, in vivo and in
clinical studies. The lisuride derivatives terguride and
proterguride have alkyl substituents on the nitrogen (N6)
of the basic structure and have a single bond between
positions 9 and 10. In fact, these compounds yield a
solubility in water comparable to that of lisuride. The
solubility of the free proterguride amounts to
2.6 mg/100 mL, for example, in a phosphate-buffered
solution (pH 7.4). The solubility of free lisuride may then
be estimated at 2.2 mg/100 mL accordingly, but this value
was calculated based on the solubility data of the
respective active ingredients/active ingredient salts
(I. Zimmermann (1983) International Journal of
Pharmaceutics 13: 57-65).
The invention is illustrated below in greater detail on the
basis of examples.
CA 02626142 2008-03-18
16
Examples
Example 1
Stability of lisuride in buffered aqueous solutions
Solutions of lisuride hydrogen maleate with a concentration
of 2 mg active ingredient per 3 mL solution were
investigated with regard to their temperature-dependent
stability. These solutions were buffered using a citrate
buffer system, so that the pH value of the pure medium was
adjusted to 5.1, 4.5 and 3.5. Aqueous solutions of citric
acid monohydrate and trisodium citrate dihydrate (0.53 mM
each) were prepared.
Suitable mixing of these solutions yielded aqueous buffer
systems with a pH of 4.5 and/or 3.5. After 33.3 mg lisuride
hydrogen maleate had been weighed out in a scaled 50 mL
flask, 40 mL of the corresponding buffer medium was added.
The flasks were agitated for several minutes and then
exposed to ultrasound for several minutes until the active
ingredient was completely dissolved. The solutions were
cooled to room temperature and topped off to the mark with
buffer medium. In conclusion, the flasks were again
agitated for one minute.
In the case of the more strongly basic medium (pH 5.1), a
supply solution was prepared, containing citric acid
monohydrate (0.38 mM) and trisodium citrate dihydrate
(0.68 mM). This was used to dissolve the active ingredient.
In principle, double-distilled water was used to prepare
the solutions.
The resulting solutions were stored under three different
conditions: under refrigeration at 6 to 8 C, at room
temperature (25 C) and at an elevated temperature of 40 C.
CA 02626142 2008-03-18
17
The solutions were stored in sealed glass ampoules which
were wrapped with aluminum foil for protection from light.
After suitable intervals of time, samples were analyzed by
means of a specific reverse phase HPLC method with UV
detection (running time 30 min, retention time of lisuride
-13 min). It was observed that the decomposition of the
active ingredient depends on the temperature and pH. The
decomposition proceeded to a greater extent at high
temperatures and at high concentrations of oxonium ions.
In each of the cases, a decomposition of 5% was already
discernible in storage at room temperature within a period
of 180 days. In storage under refrigeration, the
decomposition amounted to only approximately 0.7%. In the
case of storage at 40 C, up to 30% of the active ingredient
had decomposed.
All solutions that had been stored at 40 C showed a
significant yellow discoloration after only 30 days,
turning brown after another 60 days.
CA 02626142 2008-03-18
18
Table 1: Purity data for lisuride hydrogen maleate
dissolved in aqueous citrate-buffered solutions.
The data are given in percent of the lisuride peak area
relative to the total peak area of the chromatogram. "SD"
stands for standard deviation.
Room temperature Elevated temperature
Refrigeration (6-8 C)
(25 C) (40 C)
pH pH pH
5.1 5.1 5.1
Average Area t Average Area t Average Area
t [d]
area SD [d] area SD [d] area SD
0 100.00 0 100.00 0 100.00
7 99.90 0.05 7 99.79 0.04 7 98.86 0.06
30 99.72 0.03 30 99.36 0.03 30 94.71 0.26
90 99.59 0.02 90 98.64 0.14 90 90.99 0.37
180 99.29 0.04 180 95.45 1.17 180 87.98 0.61
pH pH pH
4.5 4.5 4.5
Average Area t Average Area t Average Area
t [d]
area SD [d] area SD [d] area SD
0 99.79 0 99.79 0 99.79
7 99.88 0.03 7 99.84 0.01 7 98.82 0.12
30 99.73 0.01 30 99.26 0.06 30 94.96 0.09
90 99.75 0.11 90 98.14 0.28 90 90.39 0.30
180 99.11 0.13 180 95.90 1.19 180 87.71 0.19
pH pH pH
3.5 3.5 3.5
Average Area t Average Area t Average Area
t [d]
area SD [d] area SD [d] area SD
0 99.87 0 99.87 0 99.87
7 99.92 0.01 7 99.89 0.01 7 98.89 0.02
30 99.80 0.02 30 99.37 0.07 30 90.41 0.12
90 99.57 0.07 90 98.70 0.13 90 73.37 1.15
180 99.22 0.15 180 94.14 1.90 180 69.91 0.89
CA 02626142 2008-03-18
19
Example 2
Addition of 9% propylene glycol
An aqueous solution of lisuride hydrogen maleate which
additionally contained 9% propylene glycol was
investigated. The active ingredient concentration was
adjusted to 2 mg per 3 mL. No buffer system was added. The
remaining procedure included weighing 33.3 mg of the active
ingredient into a scaled 50 mL flask, then adding the
propylene glycol (4.5 g) and also adding 40 mL water. The
flask was agitated for several minutes and then exposed to
ultrasound until the active ingredient was completely
dissolved. The flask was cooled to room temperature and
topped off with double-distilled water up to the mark. In
conclusion, to ensure homogeneity, agitation was continued
for another minute.
The solution was poured into transparent glass ampoules
with a volume of 1 mL. These ampoules were sealed airtight
and wrapped with aluminum foil to protect them from light.
The ampoules were each stored at 6-8 C, 25 C and 40 C. HPLC
analysis revealed that these solutions were significantly
more stable than the solutions prepared without the
addition of propylene glycol (see Example 1).
After storage under refrigeration for 180 days,
approximately 0.4% of the active ingredient had decomposed.
The samples stored at room temperature also revealed a
remaining active ingredient content of 98.4% in comparison
with the initial concentration. In the case of storage at
40 C, only approximately 10% of the active ingredient had
decomposed.
In addition, the discoloration of the solutions occurred
with a time lag in comparison with the solutions from
CA 02626142 2008-03-18
Example 1. A slight yellow discoloration was observed only
after a storage time of 90 days at an elevated temperature.
Another advantage of using propylene glycol as the low-dose
additive (less than 10%) was the improved solubility of the
active ingredient (lisuride hydrogen maleate). Accordingly,
the volume to be administered to a patient each day can be
greatly reduced after optimizing the drug concentration in
a parenteral dopaminergic treatment of neurodegenerative
diseases.
Table 2: Purity data on lisuride hydrogen maleate dissolved
in double-distilled water with the addition of 9% propylene
glycol.
The data are given in percentage of the lisuride peak area
to the total peak area of the chromatogram. "SD" stands for
standard deviation.
Room temperature Elevated temperature
Refrigeration (6-8 C)
(25 C) (40 C)
Average Area t Average Area t Average Area
t [d]
area SD (d] area SD [d] area SD
0 99.80 0 99.80 0 99.80
7 99.79 0.09 7 99.79 0.01 7 99.33 0.06
99.67 0.01 30 99.46 0.02 30 97.59 0.18
90 99.52 0.05 90 99.15 0.02 90 94.06 0.93
180 99.36 0.08 180 98.35 0.34 180 89.76 0.50
CA 02626142 2008-03-18
21
Example 3
Addition of Cremophor ELP
An aqueous lisuride hydrogen maleate solution containing
various additives was investigated with regard to its
improved stability. The additives used were propylene
glycol and Cremophor ELP in concentrations between 1.00%
and 10.00%.
The solutions were prepared by analogy with the procedure
used in examples 1 and 2.
The solutions were stored in the dark in glass containers
at 60 C for one week.
It was found that propylene glycol would lead to a
significantly improved stability of the active ingredient
in comparison with Cremophor ELP. The remaining active
ingredient content when using propylene glycol was 92.5%.
In contrast with that, under the same conditions when using
Cremophor ELP, then degradation of the active ingredient
amounted to 14.3-21.3% (1% and 10% Cremophor ELP,
respectively).
This also points to the better suitability of propylene
glycol in particular in comparison with other additives
because on the whole this yields a dual advantage. First,
the solubility of the respective active ingredient is
increased and secondly, the chemical stability of the
respective active ingredient is surprisingly also improved.