Note: Descriptions are shown in the official language in which they were submitted.
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
COMPRESSED SOLID DOSAGE FORMS WITH DRUGS OF LOW SOLUBILITY
AND PROCESS FOR MAHING THE SAME
The present invention concerns a process to promote dissolution of poorly
water
soluble drugs in order to achieve rates of dissolution and absorption
comparably similar to, or
even faster than, the recent generation of technologies and products in the
prior art.
BACKGROUND OF THE INVENTION
When solid dosage forms are taken orally, in many cases, the drug must
dissolve in
aqueous gastrointestinal fluids, for example in the patient's stomach, before
the drug can exert
a therapeutic effect. A recurring problem with compressed solid oral dosage
forms, such as
tablets, capsules and caplets (i.e., capsule-shaped tablets), is that the
dissolution rate of some
drugs from the dosage form limits their biological availability. This problem
arises from the
fact that many drugs are small organic molecules with low solubility in
aqueous fluids. There
are several ways to address the solubility problem of poorly soluble drugs
For example, the drug itself can be modified. The physical form of the drug
can be
manipulated by various techniques to optimize the rate at which the drug
dissolves. Of these
techniques, the one most relevant to the present invention is particle size
reduction. The rate
of dissolution of a solid may often depend upon the surface area that is
exposed to the
dissolving medium and since the surface area of a given mass of a substance is
generally
inversely proportional to the substance's particle size, reducing the particle
size of a powder
or granular substance may increase its dissolution rate.
Where it is effective, particle size reduction often increases the dissolution
rate of a
particulate solid by increasing the surface area that is exposed to the
dissolving medium.
However, particle size reduction is not always effective at increasing the
dissolution rate of a
drug from a compressed solid dosage form. Many hydrophobic drugs have a strong
tendency
to agglomerate during the dosage form manufacturing process into larger
particles with an
overall decrease in effective surface area. Remington: Tlze Science and
Practice of
Pharmacy, 20t1a ed. 656, 657 (A.R. Gennaro, ed., Lippincott Williams &
Wilkins:
Philadelphia 2000), incorporated by reference herein, contains a more thorough
discussion of
the concept of "effective surface area" and the effect of particle size on
dissolution. A drug
1
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
that has ostensibly been milled to a fine particle size will sometimes display
dissolution
characteristics of a larger particle due to agglomeration or similar effect.
Recently in the field of the pharmaceutical industry pharmaceutical companies
have
been trying to develop a new generation of products with increased surface
area of the drug
particles with concomitant increased dissolution rates and absorption. This is
achieved by a
variety of methods including production of a formulation with nano particles
(i.e., with
diameters less thanl gm), creating stable nano-particles of amorphous material
stabilized by
hydrophilic polymer, and stabilizing small particles by making a complex with
surface active
agent or agents via melting of the two substances. In general the new
approaches of the
pharmaceutical industry face two major barriers. The first barrier is the
technical /mechanical
limitation of breaking the drug particles into the size of nano particles. The
second barrier is
the stabilization of these small particles of the drug substance in the dosage
form, whether
they are in crystalline or amorphous form.
SUMMARY OF THE INVENTION
The present invention provides a process for making a pharmaceutical
formulation of
an active pharmaceutical ingredient (API), i.e., drug, having low aqueous
solubility, the
process comprising
(A) fixing the drug in a strong matrix comprising at least one at least
partially
amorphous sugar, which sugar is preferably substantially amorphous or entirely
amorphous,
to obtain a sugar-drug matrix; and
(B) milling, preferably intensely, the sugar-drug matrix to obtain a milled
sugar-drug
matrix as the pharmaceutical formulation.
The fixing step, i.e., step (A), of the above process of the invention is
performed by
heating a mixture of the drug, optionally pre-mixed or pre-granulated with at
least one
inactive excipient, and the at least one at least partially amorphous sugar
followed by cooling.
Alternatively, the fixing step of the above process of the invention is
performed by heating a
mixture of the drug, optionally pre-mixed or pre-granulated with inactive
excipient, and at
least one sugar followed by cooling, wherein the at least one sugar is
converted to the at least
one at least partially amorphous sugar. Preferably, the heating is accompanied
with mixing
of the drug and the sugar. The "sugar-drug matrix" obtained in the fixing
step, i.e., step (A),
of the above process preferably is a homogeneous dispersion of particles of
the drug in the
strong matrix. However, the sugar-drug matrix may also be only partially
homogeneous
without departing from the spirit of the invention but preferably the sugar-
drug matrix
2
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
obtained in the fixing step will be substantially homogeneous which is
understood to be of an
even consistency by visual inspectioiz. Step (B) involves milling, preferably
intensely, the
"sugar-drug matrix" obtained in step (A), e.g., by using a comininuting mill
exemplified by a
FitzmillTM Communitor (Fitzpatrick "Knives" forward).
In a first embodiment of the process of the invention for making the
pharmaceutical
formulation of the drug having low aqueous solubility, step (A) is performed
as step (a)
below and step (B) is performed as step (b) below, such that the embodiment
comprises
(a) mixing while heating the drug, having low aqueous solubility, e.g.,
fenofibrate
145 mg, with the following excipient(s) to form a sugar-drug matrix (wherein
the drug can be
a formulated particulate composition which has been previously compounded):
(i) at least one at least partially amorphous sugar, which sugar is preferably
substantially amorphous or entirely amorphous, in a Sugar-Drug ratio of from
about 1.5:1 to about 10:1, preferably from about 2:1 to about 8:1 and more
preferably from about 3:1 to about 6:1, wherein the amorphous sugar was
prepared for example, by heating sucrose, glucose and water to a high
temperature to form a mix and then cooling the mix,
(ii) optionally a surface active agent, e.g., polysorbate, glycerol
monostearate
and, preferably, sodium lauryl sulfate (SLS), in an amount of from about 1 to
about 5 weight % of the final phannaceutical formulation, and
(iii) optionally, a dispersing agent, e.g., poloxamer, polysorbate, and
preferably polyvinylpyrrolidone (PVP), in an amount of from about 1 to
about 10 weight % of the final pharmaceutical formulation; and
(b) milling, preferably intensely, the sugar-drug matrix to obtain a milled
sugar-drug
matrix as the pharmaceutical forinulation, wherein the milling is optionally
repeated until the
"drug-sugar matrix" achieves the desired dissolution property, and wherein the
sugar-drug
matrix is about 10% to about 99 weight%, preferably from about 30% to about 95
weight%,
and more preferably from about 65% to about 90 weight%, of the final
pharmaceutical
formulation.
In a second einbodiment of the process of the invention for making the
pharmaceutical formulation of the drug having low aqueous solubility, step (A)
is performed
by conducting steps (a')-(c') described below and step (B) is performed by
conducting step
(d') below, such that the second embodiment comprises:
(a') blending the drug having low aqueous solubility in a particulate form,
the at least
one at least partially amorphous sugar in powder form, the optional surface
active agent (e.g.,
3
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
polysorbate, glycerol monostearate and, preferably, sodium lauryl sulfate) and
the optional
dispersing agent (e.g., poloxamer, polysorbate and, preferably,
polyvinylpyrrolidone),
wherein the drug can be a formulated particulate composition which has been
previously
compounded;
(b') heating the blend with mixing, preferably to a temperature ranging from
about
50 C to about 200 C and, more preferably from about 70 C to about 120 C,
until a
"smooth" mixture is obtained, wherein the temperature, time and intensity of
mixing should
be controlled, based on the mixing instrumentation used, to obtain the
"smooth" mixture;
(c') cooling the "smooth" mixture to room temperature or below room
temperature to
obtain a sugar-drug matrix; and
(d') milling, preferably intensely, the sugar-drug matrix obtained in step
(c')
optionally with a glidant, e.g., by using a comminution inill exemplified by a
FitzmillTM
Communitor ("Knives" forward) from The Fitzpatrick Company, or any similarly
intensive
milling machinery, to obtain a milled sugar-drug matrix as the pharmaceutical
formulation,
wherein the milled sugar-drug matrix comprises a powder.
The second embodiment of the process of the invention can optionally further
comprise re-working the milled sugar-drug matrix as shown below:
(e') making a blend with a procedure comprising the following steps:
(e' 1) heating the milled sugar-drug matrix with mixing, preferably to a
temperature ranging from about 50 C to about 200 C and, more preferably
from about 70 C to about 120 C, until a "smooth" mixture is obtained;
(e'2) cooling the "smooth" mixture to room teinperature or below room
temperature to obtain a cooled matrix;
(e' 3) milling, preferably intensely, the cooled matrix obtained in step
(e'2),
e.g., by using a comminuting mill exemplified by a FitzmillTM Communitor
("knives" forward) froin The Fitzpatrick Coinpany, or any similarly intensive
milling machinery, to obtain a milled matrix as a blend containing a powder;
and
(e'4) optionally repeating steps (e' 1) to (e'3) until the powder in the
milled
matrix reaches a desired dissolution profile;
(f) optionally adding other pharmaceutically acceptable excipients, such as
diluents, disintegrants, lubricants and glidants, to the blend obtained from
step
(d') or (e') in order to improve the handlin.g and/or compression of the
blend;
and
4
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
(g') tabletting the blend or filling the blend into capsules or sachets.
The present invention also provides a pharmaceutical fonnulation of the drug
having
low aqueous solubility prepared by anyone of the embodiments of the process of
the
invention.
BRIEF DESCRIPTION OF THE DRAWINGS
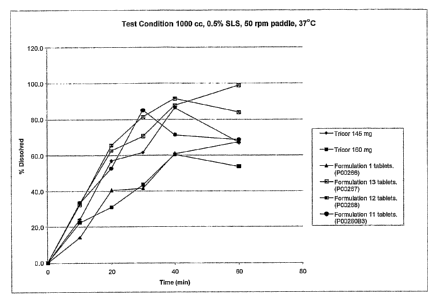
Fig. 1 shows the dissolution data of fenofibrate tablets having a medium
content of at
least partially amorphous sugar (Fonnulation 11) and fenofibrate tablets
having a high
content of a different type of at least partially amorphous sugar (Formulation
12) of the
invention compared with that of commercially available fenofibrate tablets
(Tricor 145 mg
and Tricor 160 mg), fenofibrate tablets made by conventional wet granulation
(Formulaion
1), and fenofibrate tablets made by using fenofibrate and crystalline sugar
(Formulation 13).
Fig. 2 shows the dissolution data of two fenofibrate blends having a high
content of at
least partially amorphous sugar (Formulations 4 and 7) according to the
invention compared
with that of commercially available fenofibrate formulations (Tricor 145 mg
and Tricor 160
mg).
Fig. 3 shows the dissolution data of fenofibrate tablets 145 mg made with
blends
having a high ratio of sugar to drug in the "sugar-drug matrix" (Formulations
4 and 7)
prepared according to the invention compared with that of the commercially
available
fenofibrate tablets (Tricor 145 mg and Tricor 160 mg).
Fig. 4 shows the dissolution data of fenofibrate tablets 145 mg having a high
ratio of
sugar to drug in the "sugar-drug matrix" using a"formulated granulate of
fenofibrate" as the
source of the "drug" in the process of preparing the "sugar-drug matrix"
according to the
invention coinpared with the dissolution data of the commercially available
Tricor 145 mg
and Formulation K-29740 ( bioequivalent to Tricor 160 mg, K-29740 being not a
formulation of the invention).
DETAILED DESCRIPTION OF THE INVENTION
A drug has "low aqueous solubility" or is "poorly soluble in water" if the
water
solubility of the un-ionized form of the drug is less than about 1% by weight,
and typically
less than about 0.1% or 0.01% by weight. Such drugs include fenofibrate,
bicalutamide,,
atorvastatin, fluvastatin, , simvastatin, , paclitaxel, aripiprazole,
glyburide, ezetimibe,
oxcarbazepine, meloxicam, celecoxib, rofecoxib, valdecoxib and raloxifene.
5
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
The term "at least partially amorphous sugar" means that the sugar is partly
or purely
amorphous. The term "at least partially amorphous sugar" encompasses
situations where part
of the sugar used is crystalline, either due to incomplete conversion from
crystalline to
amorphous form or due to re-crystallization of a portion of an amorphous
sugar. A sugar is
"substantially amorphous" if the sugar is about 60-100 weight % such as about
60-95 weight
%, more preferably about 80-99 weight %, and even more preferably about 90-99
weight %,
amorphous. Formulations of the invention can be made from amorphous sugar that
has been
prepared without using glucose.
The tenn "strong matrix comprising at least one at least partially amorphous
sugar"
refers to a dense and strong material coinprising at least one at least
partially amorphous
sugar and having physical characteristics, such as rigidity, hardness and
denseness, very
similar to that of boiled sugar candy or hard sugar candy.
The ternn "milling" is used generically to reflect methods of particle size
reduction
such as grinding of many types.
In the process of the invention, the term "'smooth" mixture' means that drug
particles
are evenly distributed in the sugar matrix and that the drug particles do not
form lumps or
agglomerates in the sugar-drug matrix.
In the pharmaceutical formulation of the drug of low aqueous solubility
according to
the invention, it is important to control and define the weight ratio between
the at least one at
least partially amorphous sugar and the drug in the "sugar-drug matrix" well.
Preferably, it
is also important to control and define the weight % of the "sugar-drug
matrix" in the final
formulation.
Generally, a high ratio of the at least one partially amorphous sugar to the
drug in the
"sugar-drug matrix" would be preferred, while the weight % of the "sugar-drug
matrix" in the
final formulation should be as small as possible. The extent to which the two
parameters can
be adjusted is dependent on the maximum allowed size of the dosage form, e.g.,
2 grams for
capsules or tablets, that must be swallowed, and the dose to be administered.
For example,
the ratio of the at least one at least partially amorphous sugar, and drug of
low aqueous
solubility can range from about 1:10 to about 1000:1 such as from about 1:1 to
about 600:1,
or from about 5:1 to about 300:1, or from about 10:1 to about 100:1, and the
weight % of the
"sugar-drug matrix" in the final formulation can range from about 0.5% to
about 99.5% such
as from about 5% to about 99%. For instance, for drugs generally used at a
relatively high
dose ( e.g. fenofibrate 145 mg), the weight ratio of the at least one at least
partially
amorphous sugar : drug can range from about 1.5:1 to about 10:1, preferably
from about 2:1
6
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
to about 8:1 and more preferably from about 3:1 to ab'out 6:1, and the weight
% of the
"sugar-drug matrix" in the final formulation can range from about 10 to about
99 weight %,
preferably, from about 30 to about 95 weight % and, more preferably, from
about 65 to about
90 weight %. On the other hand, for drugs generally used at a relatively low
dose (e.g.
glyboride 1 mg), the weight ratio of the at least one at least partially
amorphous sugar:drug
can be from about 1.5:1 to about 500:1, preferably from about 5:1 to about
50:1 and more
preferably from about 10:1 to about 25:1, and the weight % of the "sugar-drug
matrix" in the
final formulation can range from about 10 to about 90 weight %, preferably,
from about 15 to
about 60 weight % and, more preferably, from about 20 to about 40 weight %.
In preparing formulations of the present invention with a specified dose of
145 mg, an
example of using a relatively high weight ratio of the at least one at least
partially amorphous
sugar to drug in the final formulation is about 6:1 to about 4:1; an example
of a medium
weight ratio of the at least one at least partially amorphous sugar to drug in
the final
formulation is about 3:1 to about 4:1, e.g., about 3.4:1; and an example of a
relatively low
weight ratio of the at least one at least partially amorphous sugar to drug in
the final
formulation is about 0.5:1 to less than about 3:1, e.g., about 0.66:1, about
1.5:1, and about
2.6:1. In the formulation of the present invention with a specified dose of
145 mg, an
example of a relatively high weight % of the "drug-sugar matrix" in the final
formulation is
about 65 weight% to about 95 weight%, e.g., about 78 weight%; and an example
of a
relatively low weight% of the "drug-sugar matrix" in the final formulation is
about 10
weight% to less than about 65 weight%, e.g., about 20 weight%, about 23.4
weight%, about
40 weight%, about 42.5 weight%, about 58 weight% and about 60 weight%.
The solubility of the drug obtained by this process can be controlled by
controlling the
temperature, mixing intensity and/or mixing time of the heated mass. All these
parameters
enable controlling the geometry and "architecture" of the resultant sugar-drug
matrix, and
consequently may control the extent to which the milling of the sugar-drug
matrix effects the
solubility of the drug particles.
The process of the invention can involve two main operations: 1) the formation
of a
homogeneous dispersion of the drug particles in a strong sugar matrix, and 2)
strong milling
of the sugar-drug matrix. These two operations result in fine particles, which
yield good
dissolution and consequently good absorption.
Two main parameters are involved in the first operation of the process of the
invention: the mixing temperature and mixing time of the "hot mixing stage",
e.g., step (a) of
the first embodiment or step (b') of the second embodiment, of the drug and
the at least one
7
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
at least partially amorphous sugar. When the mixing temperature is below the
melting point
of the drug, no melting of the drug particles should occur and the mixing of
the drug and
sugar results in good dispersion of the drug particles in the sugar-drug
matrix. When the
mixing temperature is above the melting point of the drug, at least a portion
of the drug
particles in the heated mass should melt or be deformed (depending on the
mixing
temperature) resulting in even better dispersion of the drug in the sugar-drug
matrix. For any
given mixing temperature, longer mixing time of the heated mass should improve
the
dispersion of the drug in the final sugar-drug matrix. A third parameter which
may have an
effect on the final dispersion of the drug in the drug-sugar matrix is the
mixing of the drug
with the at least one at least partially amorphous sugar before the heating
stage, e.g., step (a')
of the second embodiment, or any optional mixing of the drug and the at least
one at least
partially amorphous sugar before step (a) of the first embodiment. It is
assumed that good
dispersion of the drug particles in the mixture of the drug and the at least
one at least partially
amorphous sugar before heating can further improve the distribution of the
drug particles in
the sugar-drug matrix at the heating stage and consequently in the final
"sugar-drug matrix".
An example for the procedure for pre-mixing the drug and sugar comprises: I)
direct mixing
of the drug with the at least one at least partially amorphous sugar (with or
without the
addition of the surface active agent and dispersing agent, II) applying any
other
conventional or non-conventional granulation process which can improve the
dispersion of
drug particles to the product of step I) to form a pregranulated powder, and
followed by III)
using the pregranulated powder as a "drug source" in step (A) of the general
process of the
invention, step (a) of the first embodiment, or step (b') of the second
embodiment.
The second operation of the above process involves strong milling of the final
rigid
sugar-drug matrix resulting in a break down of the rigid sugar-drug matrix
into fine small
particles. It is believed that the milling breaks down potential drug
agglomerates, which are
fixated across the breaking surfaces of the rigid sugar-drug matrix, an event
which can further
increase the solubility of the drug in the final blend. It is further believed
that higher milling
intensity would increase the efficiency of the milling and, consequently,
would also increase
the dissolution and absorption.
The desired dissolution rate is not always defined as "as fast as possible."
In certain
circumstances, it is desired to control the rate at which the drug having low
aqueous
solubility, e.g., fenofibrate, is released by adjusting the parameters of the
process of
manufacture as above, or adjusting the content of the at least one at least
partially amorphous
sugar.
8
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
ExMIe 1
(CONTROL FOR COMPARISON)
Formulation 1 (P00266)
A fenofibrate tablet was made by conventional wet granulation based on the
four
steps described below. The ingredients in Table 1 were wet granulated and then
compressed
into tablets weighing 1368 mg. The dissolution profile of Formulation 1 in
1000 mL of
0.5% aqueous SLS solution, paddle (Apparatus II) at 50 rpm, was determined and
compared
with commercial versions of fenofibrate tablets ("old version" Tricor 160 mg
and "newer
version" Tricor 145 mg with supposedly increased dissolution and
bioavailability).
Table 1
(Formulation 1; about 56.5% sugar)
Ingredient Weight Approx.Weight Percent
(mg/tablet)
Part I
Fenofibrate 145 10.6
Sodium Lauryl Sulfate (SLS) 50 3.7
Polyvinylpyrrolidone 100 7.3
(PVP K-30)
Sucrose (SUGAT - commercially available 644 47.1
in food industry)
Glucose monohydrate 128.8 9.4
Part II
Pregelatinized Starch 217 15.9
(Starch 1500)
Croscarmellose Sodium 50 3.7
(Ac-di-soITM)
Colloidal Si 02 (AerosilTM 200) 13 1
Part III
Magnesium stearate 20 1.5
1. The Part I ingredients were thoroughly blended.
2. The blend of Part I was granulated by adding water (approx. 0.5 cc per unit
dose),
granules were dried at about 65 C and milled with a small laboratory scale
mill
(IKAO Werke GmbH & Co.) fitted with a 0.5mm aperture screen.
3. The Part II ingredients were then blended with the granules of step 2 for
about 2
minutes.
9
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
4. The Part III ingredient was then blended with the blend of step 3 for about
2 minutes.
The final blend was compressed into capsule shaped tablets of 10mm/2lmm
dimensions.
Examples 2-7
Examples 2-7 display the effects of manipulating the parameters discussed
above
including, a high weight ratio of the at least one at least partially
amorphous sugar to drug; a
high weight % of the "drug-sugar matrix" in the final formulation; low or high
mixing
temperature and short or long mixing time.
A fenofibrate tablet weighing 1368 mg was made from the ingredients listed in
Table
2 by the -process of the invention. Formulations 2-7 are examples of using a
relatively high
weight ratio (for the specified dose of 145 mg) of the at least one at least
partially amorphous
sugar to drug, e.g., about 6:1 to about 4:1; relatively high weight % of the
"sugar-drug
matrix" in the final forinulation, e.g., about 65 to about 95 weight percent;
low or high
mixing temperatures; and short or long mixing time of the heated mass.
The dissolution profiles of Formulations 4 and 7 (before and after tabletting)
in 1000
mL of 0.5% aqueous SLS solution, paddle (Apparatus II) at 50 rpm, were
determined and
compared with the dissolution profiles of commercial versions of fenofibrate
tablets ("old
version" Tricor 160 mg and "newer version" Tricor 145 mg) (for results see
dissolution rate
study below).
If the dissolution profiles of Formulations 1-3, 5 and 6 are determined, a
trend of
improved dissolution rate with increases in mixing time and/or mixing
temperature should be
observed.
30
10
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 2
Formulations 2-7
Ratio of the at least one at least partially amorphous sugar to drug = 5.3:1
Weight % of the "sugar-drug matrix" in the final formulation = 78%
Ingredient Weight Approx.
(mg/tablet) Weight Percent
Part I
SACTOSe (SUGAT- commercially 644 47.1
available in food industry)
Glucose 128.8 9.4
Part II
Fenofibrate 145 10.6
Sodium Lauryl Sulfate (SLS) 50 3.7
Polyvinylpyrrolidone 100 7.3
(PVP K-30)
Part III
Pregelatinized Starch 217 15.9
(Starch 1500)
Croscarmellose Sodium 50 3.7
(Ac-dl-SOITM)
Colloidal Si 02 (AerosilTM 13 1
200)
Part IV
Magnesium stearate 20 1.5
20
11
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 3
Parameter Table
Formulation Xl X2 X3
(vessel tem .) (blend tem . (extra time)
Formulation 2 154 60-80 0 min
P00255A1
Formulation 3 154 60-80 1.5 min
P00255A2
Formulation 4 154 60-80 3 min
P00255A3
Formulation 5 170 90 0 min
P00255B1
Formulation 6 1700 90 1.5 min
P00255B2
Formulation 7 170 90 3 m1n
P00255B3
1. To the sucrose of Part 1, 322 gL of water were added. The mixture was
heated until the temperature reached 125 C. At this stage the glucose of Part
1 was
added to the mixture and the heating was continued until temperature reached
156 C.
At this stage the solution was allowed to cool to room temperature. The
amorphous
sugar obtained was milled by using a FitzmillTM Communitor (1000 rpm, knives
forward, 0.5 mm aperture screen).
2. Powder of step 1 and ingredients of Part II were thoroughly blended. The
blend was transferred into a pot heated externally to a vessel wall
temperature of X1,
and the blend was mixed while heated ( the blend temperature, about X2, was
measured by sticking a thermometer into the blended mass). The mixing of the
blend
proceeded until the powder collapsed into a sticky mass in approximately 3
min, and
then the sticky mass was mixed for an extra period of X3 ininutes. After
mixing, the
mass was allowed to cool to room temperature (at approximately 20 C), and
stored
under dry conditions (<30% Relative Humidity) for about 1 hour. See the values
of
X1, X2 and X3 in the Parameter Table.
3 The rigid mass of step 2 was milled using a FitzmillTM Communitor (2000
rpm, knives forward, 0.5 mm aperture screen).
4. The Part III ingredients were then mixed and then blended with the powder
of
step 3 for about 15 minutes.
12
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
5. The Part IV ingredient was then blended with the powder of step 4 for about
5
minutes.
The final blend was compressed into capsule shaped tablets having dimensions
of 10
mm/21 mm. Tablet hardness was found to be approximately 12-20 Strong-Cobb
units (SCU).
Examples 8, 9 and 10
Examples 8-10 exemplify a low weight ratio of the at least one at least
partially
amorphous sugar to total drug content in the finished dosage form - high
temperature; short
or long mixing time.
Fenofibrate tablets comprising a relatively low weight ratio of the at least
one at least
partially amorphous sugar to drug in the sugar-drug matrix (for a fenofibrate
dose of 145 mg),
i.e., from about 0.5:1 to about 1.5:1, can be prepared as per the process of
the invention as
described in Examples 2 through 7 using less sugar and thus the ratio of sugar
to fenofibrate
is decreased. It is believed, however, that if a low ratio of the at least one
at least partially
amorphous sugar to drug is desired whilst wishing to maintain, at least
partially the
advantages of the invention, the preferred method would be to produce the
Milled Rigid
Mass, i.e. the milled sugar-drug matrix, at the same or similar ratio of
fenofibrate to the at
least one at least partially amorphous sugar as produced by step 3 of the
above process and
then to add additional fenofibrate in the blending of step 4 to the step 3
materials, and
continuing the process so as to achieve a total composition with ratio of the
at least one at
least partially amorphous sugar to total drug between about 0.5:1 to about 3:1
amorphous
sugar in the finished dosage form.
These formulations will similarly be influenced by the process conditions of
temperature and time as described above when producing the hot blended mass.
But overall
it will be expected that those formulations comprising less amorphous sugar
will have lower
dissolution rates as compared to the formulations where all the fenofibrate is
fixed within the
sugar-drug matrix, i.e., all the fenofibrate is homogeneously dispersed in the
sugar-drug
matrix.
Formulations 8-10 are examples of employing low total contents of amorphous
sugar.
The dissolution profiles of Formulation 8-10 can be detennined in 1000 mL of
0.5 lo aqueous
SLS solution, paddle (Apparatus II) at 50 rpm,
Table 4 (Not Performed Yet)
13
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Formulations 8, 9 and 10
Ratio of amorphous sugar to total drug = 0.66:1 (Formulation 8); 1.5:1
(Fonnulation 9);
2.6:1 (formulation 10), and weight % of the "sugar-drug-matrix" in the final
formulation =
23.4% (Formulation 8); 42.5% (Formulation 9); and 58% (formulation 10)
Approx.
Weight (Weight %)
Ingredient Formulation 8 Formulation 9 Formulation 10
Part I
Sucrose 80 (-14%) 180 (-25.6%) 310 (-35 l0)
Glucose 16 (2.9%) 36 (5.1%) 62 (7.0%)
Part II
Fenofibrate 18 (3.2%) 40.5 (5.8%) 70 (7.9%)
Sodium Lauryl Sulfate (SLS) 6.2 (l.1%) 14 (2.0%) 24 (2.7%)
Polyvinylpyrrolidone 12.4 (2.2%) 28 (4.0%) 48 (5.4%)
(PVP K-30)
Part III
Fenofibrate 127 (22.6%) 104.5 (14.9%) 75 (8.4%)
Pregelatinized Starch 217 (38.8%) 217 (30.9%) 217 (24%)
(Starch 1500)
Croscarmellose Sodium 50 (8.9%) 50 (7.1%) 50 (5.6%)
(Ac-dl-SOITM)
Colloidal Si 02 (AerosilTM 13 (2.3%) 13 (1.8%) 13 (1.5%)
200)
Part IV
Magnesium stearate 20 (3.5%) 20 (2.8%) 20 (2.2%)
1. To the sucrose of part 1 water is added (40, 90 and 155 [t1 for
formulations 8,
9 and 10, respectively). The mixture is heated until the temperature reaches
125 C.
At this stage the glucose of Part 1 is added to the inixture and the heating
is continued
until temperature reaches 156 C. At this stage the solution is allowed to
cool to
room temperature. The amorphous sugar obtained is milled by FitzmillTM
Communitor (1000 rpm, knives forward, 0.5 mm aperture screen).
2. Powder of step 1 and ingredients of Part II are thoroughly blended. The
blend
is transferred into a pot heated externally to the set temperature, the blend
is heated
while mixing, the blend temperature is measured by sticking a thermometer into
the
blended mass. The mixing proceeds until the powder collapses into a sticky
mass,
followed by extra predefined time of mixing the mass. After mixing, the mass
is
14
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
allowed to cool to room temp (-20 C), and stored under dry conditions (<30%
Relative Humidity) for about 1 hour.
3 The rigid mass of step 2 is milled by using a FitzmillTM Conimunitor (2000
rpm, knives forward, 0.5 mm aperture screen).
4. The Part III ingredients, including the remaining fenofibrate are then
blended
with the powder of step 3 for about 15 minutes.
5. The Part IV ingredient is then blended with the powder of step 4 for about
five
minutes.
6. The final blend is compressed into tablets.
Example 11
Formulation 11 (P-00260B3; medium ratio of amorphous sugar to drug;
high temperature; long mixing time).
Fenofibrate tablets of the invention can also be prepared where the ratio of
the
amorphous sugar to fenofibrate in the sugar-drug matrix is lower than that in
Examples 2
through 7 while ALL of the fenofibrate is fixed in the sugar-drug matrix. The
formulation
exemplified in Example 11 is described as having a"inedium ratio" (for the
specified dose of
145 mg) of the amorphous sugar to drug, i.e., having a lower weight ratio of
the at least one
at least partially amorphous sugar to the drug than that used in formulations
exemplified by
Examples 2 through 7 which are examples of high weight ratio of the at least
one at least
partially amorphous sugar to the drug.
Fenofibrate tablets weighing 1095 mg were made from the ingredients listed in
Table
5 by the process of the invention. Formulation 11 is an examples in which a
medium ratio
(for the specified dose of 145 mg) of amorphous sugar to drug, i.e., about 3:1
to about 4:1;
relatively high weight % of the "drug-sugar matrix" in the final formulation,
e.g., about 65 to
about 95% weight percent; high mixing temperatures and high mixing time of the
heated
mass were used. The dissolution profile of Formulation 11 in 1000 mL of 0.5%
aqueous
SLS solution, paddle (USP Apparatus II) at 50 rpm, was determined and compared
with
commercial versions of Fenofibrate tablets ("Old version" Tricor 160 mg and
"Newer
version" Tricor 145 mg ) (see dissolution rate study below).
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 5
Formulation 11
Ratio of amorphous sugar to drug = 3.4 :1
Weight % of the "sugar-drug-matrix" in the final formulation = 72.5%
Formulation 11
Ingredient Weight in mg/tablet
(Weight %)
Part I
SUCTOSe (SUGAT - connnercial for 416.6 (-38%)
food industry)
Glucose 83 (7.6%)
Part II
Fenofibrate 145 (13.2%)
Sodium Lauryl Sulfate (SLS) 50 (4.6%)
Polyvinylpyrrolidone 100 (9.1%)
(PVP K-30)
Part III
Pregelatinized Starch 217 (-20%)
(Starch 1500)
Croscannellose Sodium 50 (4.6%)
(Ac-dl-solTM)Ol
Colloidal Si 02 (AerosilTM 13 (1.2%)
200)
Part IV
Magnesium stearate 20 (1.8%)
Formulation 11 was prepared using the following procedure.
1. To the sucrose of part I, 208 L (per tablet) of water were added. The
mixture
was heated until the temperature reached 125 C. At this stage, the glucose of
Part I
was added to the mixture and the heating was continued until the temperature
reached
156 C. At this stage the solution was allowed to cool to room temperature.
The
amorphous sugar obtained was milled by using a FitzmillTM Communitor (1000
rpm,
knives forward, 0.5 mm aperture screen).
2. Powder of step 1 and ingredients of Part II were tlioroughly blended. The
blend was transferred into a pot heated externally to 170 C, and the blend
was heated
while mixing the blend (the temperature, which was measured by inserting the
thermometer directly into the blended mass, was about 90 C). The mixing
proceeded
16
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
until the powder collapsed into a sticky mass (-3 minutes), followed by extra
3
minutes of mixing the mass. After mixing, the mass was allowed to cool to room
temperature (about 16 C), and stored under dry conditions (<30% relative
humidity
for about 1 hour).
3 The rigid mass of step 2 was milled by using a FitzmillTM Communitor (2000
rpm, knives forward, 0.5 mm aperture screen).
4. The Part III ingredients were then mixed and then blended with the powder
of
step 3 for about 15 minutes.
5. The Part IV ingredient was then blended with the powder of step 4 for about
five minutes.
6. The final blend was compressed into capsule shaped tablets having
dimensions
of 10 mm/21mm.
Tablet hardness was found to be - 12-20 Strong-Cobb units (SCU).
Example 12 P-00268)
Fonnulations 12 - Different type of amorphous su ar high ratio of amorphous
sugar to drug=
high weight % of the "drug-sugar matrix" in the final formulation
Formulations of the invention can also be made from amorphous sugar that has
been
prepared without the use of glucose.
Fenofibrate tablets weighing 1239 mg were made from the ingredients listed in
Table
6 by the process of the invention. Formulation 12 is an example of using a
relatively high ratio (for the specified dose of 145 mg) of amorphous sugar to
drug, e.g.,
about 6:1 to about 4:1; a relatively high weight % of the "drug-sugar matrix"
in the final
formulation, e.g., about 65 to about 95% weight percent; and a mixing time of
2 minutes of
the heated mass. A major difference between Fonnulation 12 and Formulations 2-
7 is that
Formulation 12 used only sucrose as a source for the preparation of the
amorphous sugar,
while Formulations 2-7 used sucrose and glucose. The dissolution profile of
Formulation 12
in 1000 mL of 0.5% aqueous SLS solution, paddle (Apparatus II) at 50 rpm, was
determined
and compared with commercial versions of Fenofibrate tablets ("Old version"
Tricor 160 mg
and "Newer version" Tricor 145 mg) (see dissolution rate study below).
17
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 6
Formulation 12
Ratio of a.inorphous sugar to drug = 4.4 : 1
Weight % of the "sugar-drug matrix" in the final formulation = 75.7%
Ingredient Weight (mg/tablet) Weight %
Part I
Sucrose (SUGAT - comerical for food 644 -52%
industry)
Part II
Fenofibrate 145 11.7%
Sodium Lauryl Sulfate (SLS) 50 4.0%
Polyvinylpyrrolidone 100 8.0%
(PVP K-30)
Part III
Pregelatinized Starch 217 17.5%
(Starch 1500)
Croscarmellose Sodium 50 4.0%
(Ac-di-so1TM)
Colloidal Si 02 (AerosilTM 13 1.0%
200)
Part IV
Magnesium stearate 20 1.5%
1. To the sucrose of part I, 322gL of water were added. The mixture was heated
until the temperature reached 156 C. At this stage the solution was allowed
to cool
to room temperature. The amorphous sugar obtained was milled by using a
FitzmillTM
Communitor (1000 rpm, knives forward, 0.5 mm aperture screen).
2. Powder of step 1 and ingredients of Part II were thoroughly blended. The
blend was transferred into a pot heated externally to 170 C, and the blend
was heated
while mixing (the blend temperature was measured by sticking a thermometer
into the
blended mass and was about 90 C). The mixing proceeded until the powder
collapsed into a sticky mass (-3 min), followed by extra 2 minutes of mixing
the
mass. After mixing the mass was allowed to cool to room temp (about 20 C),
and
stored under dry conditions (<30% RH for about 1 hour).
3 The rigid mass of step 2 was milled by a small laboratory scale mill (IK.AO
Werke GmbH & Co.) fitted with a 0.5 mm aperture screen.
18
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
4. The Part III ingredients were then mixed and then blended with the powder
of
step 3 for about 15 minutes.
5. The Part IV ingredient was then blended with the powder of step 4 for about
five minutes.
6. The lubricated granules were compressed into capsule shaped tablets having
dimensions of 10 mm/21 mm.
Tablet hardness was found to be - 12-20 Strong-Cobb units (SCU).
Example 13 P00267)Formulations 13 (comparative; using entirely crystalline
sugar instead
of amorphous su ag-r -- higLi temperature; long lnixing time)
Whereas the formulations of the invention are manufactured using a process
that
involves heating fenofibrate and amorphous sugar, a comparative example was
performed
wherein this same process was performed where the sugar used was in
crystalline form and
thus the preparatory step of producing the amorphous sugar was absent.
Fenofibrate tablets weighing 1368 mg were made from the ingredients listed in
Table 7.
Formulations 13 is an example of using a relatively high content of
crystalline sugar from
about 50 weight % to about 99 weight %, employing high mixing temperatures and
long
mixing time (of the heated mass). The dissolution profile of Formulation 13 in
1000 mL of
0.5% aqueous SLS solution, paddle (Apparatus II) at 50 rpm was tested and
compared with
comrnercial versions of Fenofibrate tablets ("Old version" Tricor 160 mg and
"Newer
version" Tricor 145 mg) (see dissolution rate study, below).
30
19
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 7
(Formulation 13)
Ingredient Weight (mg/tablet) Weight Percent
Part I
Sucrose (SUGAT - comeiical for food 644 47.1
industry)
Glucose 128.8 9.4
Fenofibrate 145 10.6
Sodium Lauryl Sulfate (SLS) 50 3.7
Polyvinylpyrrolidone 100 7.3
PVP k-30
Part II
Pregelatinized Starch 217 15.9
(Starch 1500)
Croscarmellose Sodium 50 3.7
(Ac-di-so1TM)
Colloidal Si 02 (AerosilTM 13 1
200)
Part III
Magnesium stearate 20 1.5
1. Ingredients of Part I were thoroughly blended. The blend was transferred
into
a pot heated externally to 170 C, and the blend was heated while mixing. The
mixing was preceded for 5 minutes. After mixing the mass was allowed to cool
to
room temperature (about 20 C).
2 The mass of step 1 was milled by using a small laboratory scale mill (IKA
Werke GmbH & Co.), fitted with a 0.5 mm aperture screen.
3. The Part II ingredients were then blended with the powder of step 2 for
about
fifteen minutes.
4. The Part III ingredient was then blended with the powder of step 3 for
about
five minutes.
5. The lubricated blend was compressed into capsule shaped tablets of
dimensions 12.0
mm x 5.5 mm.
Tablet hardness was found to be 9 Strong-Cobb units.
Example 14 (K-35211)
Formulation 14 - Use of formulated granulate as a drug source in the "drug-
sugar matrix"
relatively high ratio of amorphous sugar to drug(for the specified dose of 145
mg), high
weight % of the "su as r-drug- matrix" in the final formulation
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
A fenofibrate tablet weighing 1444 mg was made from the ingredients listed in
Table
8 by the process of the invention. Formulation 14 is example of using a
formulated granulate
of fenofibrate as a drug source in the process for preparing the sugar-drug
matrix. Also this
example used a relatively high ratio (for the specified dose of 145 mg) of the
amorphous
sugar to drug, e.g., about 6:1 to about 4:1; and a relatively high weight % of
the "sugar-
drug-matrix" in the final formulation, e.g., about 65 to about 95 weight %.
Also this example used high mixing temperature and 1 inin mixing time of the
heated
mass. The dissolution profiles of Formulation 14 in 1000 mL of 0.5% aqueous
SLS solution,
paddle (Apparatus II) at 50 rpm, were determined and compared with that of the
commercially available "newer version" Tricor 145 and Teva's formulation k-
29740
(bioequivalent to Tricor 160 mg ) (for results see dissolution rate study
below).
Table 8
Ingredient Weight Approx.W
(mg/tablet) eight
Percent
Part I
Sucrose 453.0 31.4
Glucose 90.6 6.3
Part II
"Granulated Fenofibrate" see below 682 47.2
Part III
Colloidal Si 02 (AerosilTM 200) 50 3.5
Part IV
Sodium Starch Glycolate 42 2.9
Primojeld')
Croscarinellose Sodium 42 2.9
Ac-dl-SO1TM)
Croscarmellose Sodium 42 2,9
(Ac-di-solTM)
Part V
Magnesium stearate NF 42.4 2.9
1. To the Sucrose of part 1, 227 L of water were added. The mixture was
heated until
the temperature reached 125 C. At this stage the glucose of part 1 was added
to the
mixture and the heating was continued until temperature reached 156 C. At
this stage the
solution was allowed to cool to room temperature. The amorphous sugar obtained
was
milled by FitzmillTM Communitor (medium speed, knives forward, 1.65 mm
aperture
screen).
21
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
2. Powder of stage 1 and ingredients of Part II were thoroughly blended. The
blend
was transferred into a pot heated externally to 160 C vessel wall
temperature, and the blend
was mixed while heated (the blend teinperature was measured by sticking
thermometer into
the blended mass and was about 90 C. The mixing proceeded until the powder
collapsed
into a sticky mass (approx. tmin), followed by an extra 2-3minute of mixing
the mass. After
mixing, the mass was allowed to cool to room temp (approximately 20 C), and
stored under
dry conditions (<30% Relative Humidity) for about 1 hour.
3. The Part III ingredient was added to the rigid mass of stage 2 and together
were
milled with a FitzinillTM Communitor (medium speed, knives forward, 2 to 0.5
mm aperture
screen ).
4. The Part IV ingredients were then mixed and then blended with the powder of
stage
3 for about 15 minutes.
5. The Part V ingredient was then blended with the powder of stage 4 for about
15
minutes.
6. The lubricated final blend was compressed into capsule shaped tablets.
Preparation of the "granulated fenofibrate" used in Part II of Table 8
The "granulated fenofibrate" was prepared according to the following
procedures
using the ingredients of Table 9.
Table 9
Ingredient Weight Approx.Weight
(mg/tablet) Percent
Part I
Fenofibrate 145 21
Polyvinylpyrrolidone 16 2.3
(PVP K-30)
Polyvinylpyrrolidone 38 5.6
(PVP K-25)
Sodium Starch Glycolate 44 6.5
(Primoj el)
Croscarmellose Sodium 44 6.5
(Ac-di-so1TM)
Crospovidone NF 44 6.5
Microcrystaline Cellulose 127 18.6
(Avicel)
Part II
Sodium Lauryl Sulfate (SLS) 14 2.1
Lactose 192 28.2
22
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Part III
Colloidal Si 02 7 1.0
Aerosil 200
Part IV
Pruv (Sodium Stearyl Fumarate) 11 1.6
1. Part I ingredients were thoroughly blended.
2. Lactose of Part II was dissolved in 192 mg of water heated to about 70 C.
3. SLS of Part II was dissolved in about 10 mg of water.
4. The blend of stage 1 was granulated by adding the lactose and SLS solutions
of stage
2 and 3.
5. The granules were dried in a fluidized bed dryer (inlet air 55 C, outlet
air not more
than40 C).
6. Aerosil of Part III was blended with the granules of stage 5 and then
milled by
FitzmillTM ( fitted with a 0.5 mm aperture screen).
7. The Part IV ingredients were then blended with the granules of stage 6 for
about 2
minutes.
8. The final blend was compressed into tablets.
9. The tablets were milled with a FitzmillTM Communitor (knives forward, 0.5
mm
aperture screen).
Dissolulation Rate Study
The dissolution properties of the blends or tablets prepared according to
Formulation
1(control), Formulations 4, 7, 11 and 12 (invention), Formulation 13
(comparative;
crystalline sugar), Formulation 14 (invention), K-29740 (control - used as an
alternative to
Tricor 160 mg), Tricor 145 mg and Tricor 160 mg were determined using 1000 cc
of 0.5%
aqueous SLS solution, 50 rpm paddle, 37 C for up to 60 minutes. The results
are presented
in Tables 10-13 and illustrated in Fig. 1-3.
Table 10
Time (min) Tricor 145mg Tricor 160mg Formulation I Formulation 13 Formulation
Formulation
tablets. tablets. 12 tablets. 11 tablets.
(P00266) (P00267) (P00268) (P00260B3)
0 0.0 0.0 0.0 0.0 0.0 0.0
10 24.3 22.5 14.1 33.2 32.5 33.4
20 56.8 31.0 40.5 62.6 65.6 52.7
61.5 43.6 41.6 70.6 81.3 85.0
86.4 60.2 60.8 87.6 91.5 71.3
60 67.0 53.7 67.6 98.9 83.9 68.6
23
CA 02626234 2008-04-16
WO 2007/073389 PCT/US2005/047260
Table 11
Formulation 4 Formulation
Time final Blend 7 final Blend
min Tricor 145m Tricor 160mg (P00255A3) (P00255B3)
0 0.0 0.0 0.0 0.0
32.3 30.9 47.5 55.7
70.3 40.9 62.2 68.5
67.8 48.2 68.4 68.0
87.7 61.1 80.6 81.6
60 94.1 73.3 81.6 75.4
Table 12
5
Formulation 4 Formulation
Time tablets. 7 tablets.
min Tricor 145mg Tricor 160mg (P00255A3) (P00255B3)
0 0.0 0.0 0.0 0.0
10 52.4 30.4 39.8 42.9
20 88.8 58.9 67.0 73.8
30 92.7 68.8 77.9 87.8
60 83.3 72.1 81.3 88.3
Table 13
Time (min) Tricor 145mg K-29740 Formulation 14
tablets.
(K-35211)
0 0.0 0.0 0.0
10 48.5 37.2 39.6
20 64.0 47.7 55.2
30 66.6 55.5 58.4
40 67.8 51.6 52.9
60 66.1 56.9 52.4
24