Note: Descriptions are shown in the official language in which they were submitted.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
1,1-DIOXO-THIOMORPHOLINYL INDOLYL METHANONE DERIVATIVES FOR USE AS H3
MODULATORS
The present invention is concerned with novel 1,1-dioxo-thiomorpholinyl
indolyl
methanone derivatives, their manufacture, pharmaceutical compositions
containing them
and their use as medicaments. The active compounds of the present invention
are useful in
treating obesity and other disorders.
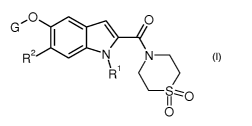
In particular, the present invention relates to compounds of the general
formula
G .O O
I ~ I
N
R2 N'
~
R
~
S~O
O
wherein
Ri is selected from the group consisting of hydrogen, lower alkyl, lower
hydroxyalkyl,
lower alkoxyalkyl, lower halogenoalkyl, lower cycloalkylalkyl,
lower alkanoyl, lower alkoxycarbonyl, lower cyanoalkyl, lower alkylsulfonyl,
phenylsulfonyl wherein the phenyl ring may be unsubstituted or substituted
with one
or two groups independently selected from lower alkyl, halogen, cyano, lower
halogenoalkyl, lower alkoxy, lower halogenoalkoxy and lower hydroxyalkyl;
phenyl unsubstituted or substituted with one or two groups independently
selected
from lower alkyl, halogen, cyano, lower halogenoalkyl, lower alkoxy, lower
halogenoalkoxy and lower hydroxyalkyl;
lower phenylalkyl, wherein the phenyl ring may be unsubstituted or substituted
with
one or two groups independently selected from lower alkyl, halogen, cyano,
lower
halogenoalkyl, lower alkoxy, lower halogenoalkoxy and lower hydroxyalkyl; and
heteroaryl unsubstituted or substituted with one or two groups independently
selected from lower alkyl, lower alkoxy, halogen, lower halogenoalkyl, lower
halogenoalkoxy and cyano;
R~ is hydrogen or halogen;
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-2-
G is a group selected from
R 3 R5 R5,
6
N In R~
Rs N(CH2)q
m N
R4 A R'
J-~tp R
G1 G2 G3
RN11~(CH2)q
and 19
R
G4
wherein
m is 0, l or 2;
R is selected from lower alkyl, lower halogenoalkyl, cycloalkyl,
halogenocycloalkyl, lower cycloalkylalkyl and lower phenylalkyl;
n is 0, l or 2;
R4 is lower alkyl;
p is 0, l or 2;
q is 0, l or 2;
A is selected from CR R ' 0 and S;
R, R', R, R', R, R', R and R 'independently from each other are selected from
the group consisting of hydrogen, lower alkyl, hydoxy, halogen and
dialkylamino, or
R and R together form a double bond;
R8 is lower alkyl;
R is C3-C6-alkyl;
and pharmaceutically acceptable salts thereof.
The compounds of formula I are antagonists and/or inverse agonists at the
histamine
3 receptor (H3 receptor).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-3-
Histamine (2-(4-imidazolyl) ethylamine) is one of the aminergic
neurotransmitters
which is widely distributed throughout the body, e. g. the gastrointestinal
tract (Burks 1994
in Johnson L.R. ed., Physiology of the Gastrointestinal Tract, Raven Press,
NY, pp. 211 -
242). Histamine regulates a variety of digestive pathophysiological events
like gastric acid
secretion, intestinal motility (Leurs et al., Br J. Pharmacol. 1991, 102, pp
179-185),
vasomotor responses, intestinal inflammatory responses and allergic reactions
(Raithel et
al., Int. Arch. Allergy Immunol. 1995, 108, 127-133). In the mammalian brain,
histamine is
synthesized in histaminergic cell bodies which are found centrally in the
tuberomammillary nucleus of the posterior basal hypothalamus. From there, the
histaminergic cell bodies project to various brain regions (Panula et al.,
Proc. Natl. Acad.
Sci. USA 1984, 81, 2572-2576; Inagaki et al., J. Comp. Neuro11988, 273, 283 -
300).
According to current knowledge, histamine mediates all its actions in both the
CNS
and the periphery through four distinct histamine receptors, the histamine H
1, H2, H3 and
H4 receptors.
H3 receptors are predominantly localized in the central nervous system (CNS).
As an
autoreceptor H3 receptors constitutively inhibit the synthesis and secretion
of histamine
from histaminergic neurons (Arrang et al., Nature 1983, 302, 832-837; Arrang
et al.,
Neuroscience 1987, 23, 149-157). As heteroreceptors, H3 receptors also
modulate the
release of other neurotransmitters such as acetylcholine, dopamine, serotonin
and
norepinephrine among others in both the central nervous system and in
peripheral organs,
such as lungs, cardiovascular system and gastrointestinal tract (Clapham &
Kilpatrik, Br. J.
Pharmacol. 1982, 107, 919- 923; Blandina et al. in The Histamine H3 Receptor
(Leurs RL
and Timmermann H eds, 1998, pp 27-40, Elsevier, Amsterdam, The Netherlands).
H3
receptors are constitutively active, meaning that even without exogenous
histamine, the
receptor is tonically activated. In the case of an inhibitory receptor such as
the H3 receptor,
this inherent activity causes tonic inhibition of neurotransmitter release.
Therefore it may
be important that a H3R antagonist would also have inverse agonist activity to
both block
exogenous histamine effects and to shift the receptor from its constitutively
active
(inhibitory) form to a neutral state.
The wide distribution of H3 receptors in the mammalian CNS indicates the
physiological role of this receptor. Therefore the therapeutic potential as a
novel drug
development target in various indications has been proposed.
The administration of H3R ligands - as antagonists, inverse agonists, agonists
or
partial agonists - may influence the histamine levels or the secretion of
neurotransmitters
in the brain and the periphery and thus may be useful in the treatment of
several disorders.
Such disorders include obesity, (Masaki et al; Endocrinol. 2003, 144, 2741-
2748; Hancock
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-4-
et al., European J. of Pharmacol. 2004, 487, 183-197), cardiovascular
disorders such as
acute myocardial infarction, dementia and cognitive disorders such as
attention deficit
hyperactivity disorder (ADHD) and Alzheimer's disease, neurological disorders
such as
schizophrenia, depression, epilepsy, Parkinson's disease and seizures or
convulsions, sleep
disorders, narcolepsy, pain, gastrointestinal disorders, vestibular
dysfunction such as
Morbus Meniere, drug abuse and motion sickness (Timmermann, J. Med. Chem.
1990, 33,
4-11).
It is therefore an object of the present invention to provide selective,
directly acting
H3 receptor antagonists respectively inverse agonists. Such antagonists /
inverse agonists
are useful as therapeutically active substances, particularly in the treatment
and/or
prevention of diseases which are associated with the modulation of H3
receptors.
In the present description the term "alkyl", alone or in combination with
other
groups, refers to a branched or straight-chain monovalent saturated aliphatic
hydrocarbon
radical of one to twenty carbon atoms, preferably one to sixteen carbon atoms,
more
preferably one to ten carbon atoms.
The term "lower alkyl" or "Ci-Cg-alkyl", alone or in combination, signifies a
straight-
chain or branched-chain alkyl group with 1 to 8 carbon atoms, preferably a
straight or
branched-chain alkyl group with 1 to 6 carbon atoms and particularly preferred
a straight
or branched-chain alkyl group with 1 to 4 carbon atoms Examples of straight-
chain and
branched Ci-Cg alkyl groups are methyl, ethyl, propyl, isopropyl, butyl,
isobutyl, tert.-
butyl, the isomeric pentyls, the isomeric hexyls, the isomeric heptyls and the
isomeric
octyls, preferably methyl and ethyl and most preferred methyl.
The term "lower alkenyl" or "C2-g-alkenyl", alone or in combination, signifies
a
straight-chain or branched hydrocarbon radical comprising an olefinic bond and
up to 8,
preferably up to 6, particularly preferred up to 4 carbon atoms. Examples of
alkenyl groups
are ethenyl, 1-propenyl, 2-propenyl, isopropenyl, 1-butenyl, 2-butenyl, 3-
butenyl and
isobutenyl. A preferred example is 2-propenyl.
The term "lower alkynyl" or "C2-g-alkynyl", alone or in combination, signifies
a
straight-chain or branched hydrocarbon residue comprising a triple bond and up
to 8,
preferably up to 6, particularly preferred up to 4 carbon atoms. Examples of
alkynyl groups
are ethinyl, 1-propinyl, or 2-propinyl. A preferred example is 2-propinyl.
The term "cycloalkyl" or "C3_7-cycloalkyl" denotes a saturated carbocyclic
group
containing from 3 to 7 carbon atoms, such as cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl or cycloheptyl. Especially preferred are cyclopropyl, cyclopentyl
and cyclohexyl.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-5-
The term "lower cycloalkylalkyl" or "C3_7-cycloalkyl-C1_g-alkyl" refers to
lower alkyl
groups as defined above wherein at least one of the hydrogen atoms of the
lower alkyl
group is replaced by cycloalkyl. A preferred example is cyclopropylmethyl.
The term "alkoxy" refers to the group R'-O-, wherein R' is lower alkyl and the
term
"lower alkyl" has the previously given significance. Examples of lower alkoxy
groups are
e.g. methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec. butoxy
and tert.-
butoxy, preferably methoxy and ethoxy and most preferred methoxy.
The term "lower alkoxyalkyl" or "Ci_g-alkoxy-Ci_g-alkyl" refers to lower alkyl
groups
as defined above wherein at least one of the hydrogen atoms of the lower alkyl
groups is
replaced by an alkoxy group, preferably methoxy or ethoxy. Among the preferred
lower
alkoxyalkyl groups are 2-methoxyethyl or 3-methoxypropyl.
The term "alkylsulfanyl" or "Ci_g-alkylsulfanyl" refers to the group R'-S-,
wherein R'
is lower alkyl and the term "lower alkyl" has the previously given
significance. Examples of
alkylsulfanyl groups are e.g. methylsulfanyl or ethylsulfanyl.
The term "lower alkylsulfanylalkyl" or "C1_g-alkylsulfanyl-C1_g-alkyl" refers
to lower
alkyl groups as defined above wherein at least one of the hydrogen atoms of
the lower alkyl
groups is replaced by an alkylsulfanyl group, preferably methylsulfanyl. An
example for a
preferred lower alkylsulfanylalkyl group is 2-methylsulfanylethyl.
The term "alkylsulfonyl" or "lower alkylsulfonyl" refers to the group R'-S(O)z-
,
wherein R' is lower alkyl and the term "lower alkyl" has the previously given
significance.
Examples of alkylsulfonyl groups are e.g. methylsulfonyl or ethylsulfonyl.
The term "halogen" refers to fluorine, chlorine, bromine and iodine, with
fluorine,
chlorine and bromine being preferred.
The term "lower halogenoalkyl" or "halogen-Ci_g-alkyl" refers to lower alkyl
groups
as defined above wherein at least one of the hydrogen atoms of the lower alkyl
group is
replaced by a halogen atom, preferably fluoro or chloro, most preferably
fluoro. Among
the preferred halogenated lower alkyl groups are trifluoromethyl,
difluoromethyl,
fluoromethyl and chloromethyl, with trifluoromethyl being especially
preferred.
The term "lower halogenoalkoxy" or "halogen-C1_g-alkoxy" refers to lower
alkoxy
groups as defined above wherein at least one of the hydrogen atoms of the
lower alkoxy
group is replaced by a halogen atom, preferably fluoro or chloro, most
preferably fluoro.
Among the preferred halogenated lower alkyl groups are trifluoromethoxy,
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-6-
difluoromethoxy, fluormethoxy and chloromethoxy, with trifluoromethoxy being
especially preferred.
The term "lower hydroxyalkyl" or "hydroxy-Ci_g-alkyl" refers to lower alkyl
groups
as defined above wherein at least one of the hydrogen atoms of the lower alkyl
group is
replaced by a hydroxy group. Examples of lower hydroxyalkyl groups are
hydroxymethyl
or hydroxyethyl.
The term "dialkylamino" refers to the group -NR'R", wherein R' and R" are
lower
alkyl and the term "lower alkyl" has the previously given significance. A
preferred
dialkylamino group is dimethylamino.
The term "lower dialkylaminoalkyl" or "Ci_g-dialkylamino-Ci_g-alkyl" refers to
lower
alkyl groups as defined above wherein at least one of the hydrogen atoms of
the lower alkyl
group is replaced by a dialkylamino group, preferably dimethylamino. A
preferred lower
dialkylaminoalkyl group is 3-dimethylaminopropyl.
The term "lower alkanoyl" refers to the group -CO-R', wherein R' is lower
alkyl and
the term "lower alkyl" has the previously given significance. Preferred is a
group -CO-R',
wherein R' is methyl, meaning an acetyl group.
The term "lower alkoxycarbonyl" or "Ci_g-alkoxycarbonyl" refers to the group
-COOR', wherein R' is lower alkyl and the term "lower alkyl" has the
previously given
significance. Preferred is a group -COOR', wherein R' is methyl.
The term "carbamoyl" refers to the group -CO-NHz.
The term "dialkylcarbamoyl" or "Ci_g-dialkylcarbamoyl" refers to the group
-CO-NR'R" wherein R' and R" are lower alkyl and the term "lower alkyl" has the
previously given significance. A preferred dialkylcarbamoyl group is
dimethylcarbamoyl.
The term "lower dialkylcarbamoylalkyl" or "C1_g-dialkylcarbamoyl-C1_g-alkyl"
refers
to lower alkyl groups as defined above wherein at least one of the hydrogen
atoms of the
lower alkyl group is replaced by a dialkylcarbamoyl group as defined herein
before. A
preferred lower dialkylcarbamoylalkyl groups is dimethylcarbamoylmethyl.
The term "lower phenylalkyl" or "phenyl-Ci_g-alkyl" to lower alkyl groups as
defined
above wherein at least one of the hydrogen atoms of the lower alkyl group is
replaced by a
phenyl group. Preferred lower phenylalkyl groups are benzyl or phenethyl.
The term "heteroaryl" refers to an aromatic 5- or 6-membered ring which can
comprise one, two or three atoms selected from nitrogen, oxygen and/or
sulphur.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-7-
Examples of heteroaryl groups are e.g. furyl, pyridyl, pyrazinyl, pyrimidinyl,
pyridazinyl,
thienyl, isoxazolyl, thiazolyl, isothiazolyl, oxazolyl, imidazolyl, or
pyrrolyl. Especially
preferred are furyl and pyridyl.
The term "lower heteroarylalkyl" or "heteroaryl-C1_g-alkyl" refers to lower
alkyl
groups as defined above wherein at least one of the hydrogen atoms of the
lower alkyl
group is replaced by a heteroaryl group as defined above.
The term "heterocyclyl" refers to a saturated or partly unsaturated 5- or 6-
membered
ring which can comprise one, two or three atoms selected from nitrogen, oxygen
and/or
sulphur. Examples of heterocyclyl rings include piperidinyl, piperazinyl,
azepinyl,
pyrrolidinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, pyridinyl,
pyridazinyl,
pyrimidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl,
isothiazolidinyl,
thiadiazolylidinyl, dihydrofuryl, tetrahydrofuryl, dihydropyranyl,
tetrahydropyranyl and
thiomorpholinyl. A preferred heterocyclyl group is piperidinyl.
The term "lower heterocyclylalkyl" or "heterocyclyl-C1_g-alkyl" refers to
lower alkyl
groups as defined above wherein at least one of the hydrogen atoms of the
lower alkyl
group is replaced by a heterocyclyl group as defined above.
The term "pharmaceutically acceptable salts" refers to those salts which
retain the
biological effectiveness and properties of the free bases or free acids, which
are not
biologically or otherwise undesirable. The salts are formed with inorganic
acids such as
hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric
acid and the
like, preferably hydrochloric acid and organic acids such as acetic acid,
propionic acid,
glycolic acid, pyruvic acid, oxylic acid, maleic acid, malonic acid, salicylic
acid, succinic
acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid,
mandelic acid,
methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic
acid, N-
acetylcystein and the like. In addition these salts may be prepared form
addition of an
inorganic base or an organic base to the free acid. Salts derived from an
inorganic base
include, but are not limited to, the sodium, potassium, lithium, ammonium,
calcium,
magnesium salts and the like. Salts derived from organic bases include, but
are not limited
to salts of primary, secondary and tertiary amines, substituted amines
including naturally
occurring substituted amines, cyclic amines and basic ion exchange resins,
such as
isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine,
ethanolamine, lysine, arginine, N-ethylpiperidine, piperidine, polymine resins
and the like.
The compound of formula I can also be present in the form of zwitterions.
Particularly
preferred pharmaceutically acceptable salts of compounds of formula I are the
hydrochloride salts.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
8-
The compounds of formula I can also be solvated, e.g. hydrated. The solvation
can
be effected in the course of the manufacturing process or can take place e.g.
as a
consequence of hygroscopic properties of an initially anhydrous compound of
formula I
(hydration). The term "pharmaceutically acceptable salts" also includes
physiologically
acceptable solvates.
"Isomers" are compounds that have identical molecular formulae but that differ
in
the nature or the sequence of bonding of their atoms or in the arrangement of
their atoms
in space. Isomers that differ in the arrangement of their atoms in space are
termed
"stereoisomers". Stereoisomers that are not mirror images of one another are
termed
"diastereoisomers" and stereoisomers that are non-superimposable mirror images
are
termed "enantiomers", or sometimes optical isomers. A carbon atom bonded to
four
nonidentical substituents is termed a "chiral center".
In detail, the present invention relates to compounds of the general formula
G .O O
I ~ I
N
R2 N '
~
R
~
S~O
O
wherein
Ri is selected from the group consisting of hydrogen, lower alkyl, lower
hydroxyalkyl,
lower alkoxyalkyl, lower halogenoalkyl, lower cycloalkylalkyl,
lower alkanoyl, lower alkoxycarbonyl, lower cyanoalkyl, lower alkylsulfonyl,
phenylsulfonyl wherein the phenyl ring may be unsubstituted or substituted
with one
or two groups independently selected from lower alkyl, halogen, cyano, lower
halogenoalkyl, lower alkoxy, lower halogenoalkoxy and lower hydroxyalkyl;
phenyl unsubstituted or substituted with one or two groups independently
selected
from lower alkyl, halogen, cyano, lower halogenoalkyl, lower alkoxy, lower
halogenoalkoxy and lower hydroxyalkyl;
lower phenylalkyl, wherein the phenyl ring may be unsubstituted or substituted
with
one or two groups independently selected from lower alkyl, halogen, cyano,
lower
halogenoalkyl, lower alkoxy, lower halogenoalkoxy and lower hydroxyalkyl; and
heteroaryl unsubstituted or substituted with one or two groups independently
selected from lower alkyl, lower alkoxy, halogen, lower halogenoalkyl, lower
halogenoalkoxy and cyano;
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-9-
R is hydrogen or halogen;
G is a group selected from
R 3 R5 R5,
6
N In R~
Rs N(CH2)q
m N
R4 A R'
J-~tp R
G1 G2 G3
RN11~(CH2)q
and 19
R
G4
wherein
m is 0, l or 2;
R is selected from lower alkyl, lower halogenoalkyl, cycloalkyl,
halogenocycloalkyl, lower cycloalkylalkyl and lower phenylalkyl;
n is 0, l or 2;
R4 is lower alkyl;
p is 0, l or 2;
q is 0, l or 2;
A is selected from CR R ' 0 and S;
R, R', R, R', R, R', R and R 'independently from each other are selected from
the group consisting of hydrogen, lower alkyl, hydoxy, halogen and
dialkylamino, or
R and R together form a double bond;
R8 is lower alkyl;
R is C3-C6-alkyl;
and pharmaceutically acceptable salts thereof.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-10-
Furthermore, ccompounds of formula I according to the present invention are
preferred, wherein Ri is selected from the group consisting of
hydrogen, lower alkyl, lower hydroxyalkyl, lower alkoxyalkyl, lower
halogenoalkyl,
lower cycloalkylalkyl, lower cyanoalkyl,
lower alkylsulfonyl and
phenyl unsubstituted or substituted with one or two groups independently
selected
from lower alkyl, halogen, cyano, lower halogenoalkyl, lower alkoxy, lower
halogenoalkoxy and lower hydroxyalkyl.
More preferred are those compounds of formula I, wherein Ri is selected from
the
group consisting of hydrogen, lower alkyl and lower halogenoalkyl, with those
compounds, wherein Ri is hydrogen, or those compounds, wherein Ri is lower
halogenoalkyl, being especially preferred. Most preferably, Ri is
trifluoroethyl.
Further preferred compounds of formula I are those, wherein Ri is lower
cyanoalkyl.
Especially preferred is 1-cyanoethyl (propionitrile).
Also preferred are compounds of formula I of the present invention, wherein Rl
is
lower hydroxyalkyl or lower alkoxyalkyl. More preferably, Rl is selected from
the group
consisting of 2-hydroxyethyl, 3-hydroxypropyl, 2-methoxyethyl and 3-
methoxypropyl.
Furthermore, compounds of formula I of the present invention are preferred,
wherein Ri is heteroaryl unsubstituted or substituted with one or two groups
independently selected from lower alkyl, lower alkoxy, halogen, lower
halogenoalkyl, lower
halogenoalkoxy and cyano. Most preferably, heteroaryl is pyridyl or
pyrimidinyl.
W is hydrogen or halogen. Compounds of formula I, wherein W is selected from
the
group consisting of hydrogen, chloro and bromo, are preferred.
Especially preferred compounds of formula I according to the invention are
those,
wherein W is hydrogen.
Furthermore, compounds of formula I according to the present invention are
preferred, wherein G signifies
R3
N
G1
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-11-
wherein m is 0, 1 or 2 and R3 is selected from lower alkyl, lower
halogenoalkyl, cycloalkyl,
halogenocycloalkyl, lower cycloalkylalkyl and lower phenylalkyl.
Within this group, those compounds of formula I are preferred, wherein R3 is
lower
alkyl. Most preferably, R3 is isopropyl.
Preferred are those compounds of formula I, wherein m is 1, thus meaning
compounds, wherein G1 is represented by a piperidinyl group.
Also preferred are compounds of formula I, wherein m is 0, thus meaning
compounds, wherein G1 is represented by a pyrrolidinyl group.
Furthermore, compounds of formula I according to the present invention are
preferred, wherein G signifies
In
N
R4
G2
~
wherein n is 0, 1 or 2 and R4 is lower alkyl.
Also preferred are compounds of formula I according to the invention, wherein
G
signifies
R5 R5,
R6,
Rs N11~(CH2)q
A R'
R
Hp~
G3
wherein p is 0, 1 or 2, q is 0, 1 or 2; A is selected from CR10Ri0' 0 and S;
and
R5, RS', R6, R6', W, W', R10 and RiO' independently from each other are
selected from the
group consisting of hydrogen, lower alkyl, hydoxy, halogen and dialkylamino,
or
R6 and R10 together form a double bond.
In addition, compounds of formula I according to the present invention are
preferred, wherein G signifies
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 12-
R8
N11~ (CH2)q
19
R
G4
wherein q is 0, 1 or 2, R8 is lower alkyl and R9 is lower alkyl.
Particularly preferred compounds of formula I of the present invention are the
following:
(1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-yloxy)-1-(2,2,2-
trifluoro-
ethyl)-1H-indol-2-yl] -methanone,
(1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-
yl] -
methanone,
(1, 1-dioxo-thiomorpholin-4-yl)- [ 1-isopropyl-5-(1-isopropyl-piperidin-4-
yloxy)-1H-indol-
2-yl] -methanone,
(1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-pyrrolidin-3S-yloxy)-1H-indol-2-
yl] -
methanone,
5-[2-(1,1-dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isopropyl-piperidin-4-
yloxy)-
indol-1-yl] -pyridine-2-carbonitrile,
[6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl]-(1,1-dioxo-l~6-
thiomorpholin-4-yl)-methanone,
[6-bromo-l-isopropyl-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl] -(1,1-
dioxo-l',~ 6-
thiomorpholin-4-yl)-methanone,
[6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-1H-indol-
2-yl] -(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone,
[6-bromo-l-(2-chloro-pyridin-4-yl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-
2-yl] -
(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone,
(1,1-dioxo-1,~6-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-yloxy)-1-(3-
trifluoromethyl-phenyl)-1H-indol-2-yl] -methanone,
[1-(2-chloro-pyridin-4-yl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl]-
(1,1-dioxo-
1',~6-thiomorpholin-4-yl)-methanone,
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 13-
2-[2-(1,1-dioxo-l',~6-thiomorpholine-4-carbonyl)-5-(1-isopropyl-piperidin-4-
yloxy)-
indol-l-yl] -propionitrile,
(1,1-dioxo-1,~6-thiomorpholin-4-yl) - [ 5- (1-isopropyl-piperidin-4-yloxy) -1-
pyrimidin-5-yl-
1H-indol-2-yl]-methanone,
[6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1-(2-methoxy-ethyl)-1H-indol-2-yl]-
(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone,
[6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1-(3-methoxy-propyl)-1H-indol-2-yl]
-(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone,
[6-bromo-1-(2-hydroxy-ethyl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl] -
(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone,
[6-bromo-1-(3-hydroxy-propyl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl]
-(1,1-
Dioxo-1,~6-thiomorpholin-4-yl)-methanone,
(S)-2-[2-(1,1-dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isopropyl-piperidin-4-
yloxy)-
indol-l-yl] -propionitrile,
(R)-2-[2-(1,1-dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isopropyl-piperidin-4-
yloxy)-
indol-l-yl] -propionitrile,
[ 5- (1-cyclobutyl-piperidin-4-yloxy) -1-isopropyl-lH-indol-2-yl] - (1,1-dioxo-
1~ 6-
thiomorpholin-4-yl)-methanone,
and pharmaceutically acceptable salts thereof.
Furthermore, the pharmaceutically acceptable salts of the compounds of formula
I
and the pharmaceutically acceptable esters of the compounds of formula I
individually
constitute preferred embodiments of the present invention.
Compounds of formula I may form acid addition salts with acids, such as
conventional pharmaceutically acceptable acids, for example hydrochloride,
hydrobromide, phosphate, acetate, fumarate, maleate, salicylate, sulphate,
pyruvate, citrate,
lactate, mandelate, tartarate and methanesulphonate. Preferred are the
hydrochloride salts.
Also solvates and hydrates of compounds of formula I and their salts form part
of the
present invention.
Compounds of formula I can have one or more asymmetric carbon atoms and can
exist in the form of optically pure enantiomers, mixtures of enantiomers such
as, for
example, racemates, optically pure diastereoisomers, mixtures of
diastereoisomers,
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-14-
diastereoisomeric racemates or mixtures of diastereoisomeric racemates. The
optically
active forms can be obtained for example by resolution of the racemates, by
asymmetric
synthesis or asymmetric chromatography (chromatography with a chiral adsorbens
or
eluant). The invention embraces all of these forms.
It will be appreciated, that the compounds of general formula I in this
invention may
be derivatised at functional groups to provide derivatives which are capable
of conversion
back to the parent compound in vivo. Physiologically acceptable and
metabolically labile
derivatives, which are capable of producing the parent compounds of general
formula I in
vivo are also within the scope of this invention.
A further aspect of the present invention is the process for the manufacture
of
compounds of formula I as defined above, which process comprises
b) reacting a compound of formula II
.O ~
G O
I ~ II
R2 \ OH
R'
or a salt thereof,
wherein Ri and W are as defined herein before,
with the amine of the formula III
H N S~~o III
O
to obtain a compound of the formula I
G .O O
I ~ I
N
R2 N '
~
R
~
S~O
O
wherein R1, W and G are as defined herein before,
and if desired,
converting the compound obtained into a pharmaceutically acceptable acid
addition salt.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 15-
The term a salt of a compound of formula II embraces all acid addition salts
with
acids, for example hydrochloride, hydrobromide, phosphate, acetate, fumarate,
maleate,
salicylate, sulphate, pyruvate, citrate, lactate, mandelate, tartrate and
methanesulphonate.
Preferred are the hydrochloride salts. In addition, these hydrochloric salts
may contain an
equivalent of an alkali chloride salt, such as lithium chloride, sodium
chloride or
potassium chloride.
In more detail, the compounds of formula I can be manufactured by the methods
given below, by the methods given in the examples or by analogous methods. The
preparation of compounds of formula I of the present invention may be carried
out in
sequential or convergent synthetic routes. Syntheses of the invention are
shown in the
following schemes. The skills required for carrying out the reaction and
purification of the
resulting products are known to those skilled in the art. The substituents and
indices used
in the following description of the processes have the significance given
herein before
unless indicated to the contrary.
Scheme 1
0
11;0
G I ~ ~
' O C
Rz ~ H O
lb x HO,, B" OH
d) R or
R
H O o) VIII X
~S~\\ O
o 1 1~o
v ~ III g~
X OH
HO ~ or ~
OEt G G G0 OEt Gi0 \ N
Rz H O a) Rz H O Rz I ~ \ O
R
IV ViI Ia
X o)
VIII 1 b)
R H~ -O
G0 OEt III O
Rz
N O
R
IX
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-16-
Compounds of general formula I can be prepared according to scheme 1 as
follows:
a) The syntheses of ethers are widely described in literature and the
procedures are
known to those in the art (For reaction conditions described in literature
affecting such
reactions see for example: Comprehensive Organic Transformations: A Guide to
Functional Group Preparations, 2nd Edition, Richard C. Larock. John Wiley &
Sons, New
York, NY. 1999). The transformation can be affected by employing reaction
conditions
which are commonly utilised in the so called "Mitsunobu reaction" which is
known to
those in the art and widely described (Hughes, David L. The Mitsunobu
reaction. Organic
Reactions, New York, 1992 42 335-656.) We find it convenient to couple a
phenolic
alcohol IV with alcohols HO-G VI (either commercially available or accessible
by methods
described in references or by methods known in the art, as appropriate) under
conditions
employing a phosphine such as tributylphosphine or triphenylphosphine and the
like and
a diazo-compound like diethyl-azodicarboxylate, diisopropyl-azodicarboxylate
(optionally
polymer bound), di-tert-butylazodicarboxylate, tetramethyl azodicarboxamide
and the like
in a solvent commonly used in such transformations like tetrahydrofuran,
toluene,
dichloromethane and the like. There is no particular restriction on the nature
of the
solvent to be employed, provided that it has no adverse effect on the reaction
or the
reagents involved and that it can dissolve the reagents, at least to some
extent. The reaction
can take place over a wide range of temperatures and the precise reaction
temperature is
not critical to the invention. We find it convenient to carry out the reaction
with heating
from ambient temperature to reflux. The time required for the reaction may
also vary
widely, depending on many factors, notably the reaction temperature and the
nature of the
reagents. However, a period of from 0.5 h to several days will usually suffice
to yield the
compounds of formula VII.
Alternatively, compounds of formula IV can be subjected to a reaction in which
the
phenolic OH will substituted by G-X V (either commercially available or
accessible by
methods described in references or by methods known in the art, as
appropriate).
Conditions commonly used in such types of transformation are widely described
in
literature and known to those in the art. The leaving group X can be any
halogen group or
pseudo halogen (e.g. trifluoromethylmethanesulfonyl, para-toluenesulfonyl,
methanesulfonyl and the like). The reaction might be carried out in the
presence or
absence of a solvent and preferably in the presence of a base. Solvents like
N,N-
dimethylformamide, N,N-dimethyl acetamide, tetrahydrofuran, diethyl ether,
dioxane,
acetonitrile, butanone and the like are conveniently used. There is no
particular restriction
on the nature of the solvent to be employed, provided that it has no adverse
effect on the
reaction or the reagents involved and that it can dissolve the reagents, at
least to some
extent. Usually the reaction is carried out in the presence of a base.
Suitable bases include
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 17-
sodium hydride, N-ethyldiisopropylamine, sodium carbonate and cesium carbonate
and
the like. The reaction can take place over a wide range of temperatures and
the precise
reaction temperature is not critical to the invention. We find it convenient
to carry out the
reaction with heating from ambient temperature to reflux. The time required
for the
reaction may also vary widely, depending on many factors, notably the reaction
temperature and the nature of the reagents. However, a period of from 0.5 h to
several days
will usually suffice to yield the title compounds VII.
b) Compounds VII can be subjected to a reaction in which the indole NH will be
substituted by lower alkyl substituents, benzyl substituents, alkyl, alkanoyl
and arylsulfonyl
substituent, e.g. through a reaction with an alkylating, acylating or
sulfonylating agent
(either commercially available or accessible by methods described in
references or by
methods known in the art, as appropriate). Typical examples of an alkylating
or acylating
agent VIII are methyl iodide, 2-bromopropane, 2,2,2-trifluoroethyl-
methanesulfonate,
methanesulfonylchloride or phenylsulfonylchloride. Conditions commonly used in
such
types of transformation are widely described in literature and known to those
in the art. X
signifies a leaving group such as any halogen group (chlorine, bromine,
iodine) or pseudo
halogens group (e.g. trifluoromethylmethanesulfonyl, paratoluensulfonyl,
methanesulfonyl
and the like). The reaction might be carried out in the presence or absence of
a solvent and
preferably in the presence of a base. Solvents like N,N-dimethylformamide, N,N-
dimethyl
acetamide, tetrahydrofuran, diethyl ether, dioxane, acetonitrile, butanone and
the like are
conveniently used. There is no particular restriction on the nature of the
solvent to be
employed, provided that it has no adverse effect on the reaction or the
reagents involved
and that it can dissolve the reagents, at least to some extent. Usually the
reaction is carried
out in the presence of a base. Suitable bases include sodium hydride, N-
ethyldiisopropylamine, sodium carbonate and cesium carbonate and the like. The
reaction
can take place over a wide range of temperatures and the precise reaction
temperature is
not critical to the invention. We find it convenient to carry out the reaction
with heating
from ambient temperature to reflux. The time required for the reaction may
also vary
widely, depending on many factors, notably the reaction temperature and the
nature of the
reagents. However, a period of from 0.5 h to several days will usually suffice
to yield the
title compounds IX.
c) The compounds of formula IX are transformed into the free acids under basic
conditions, for example by using lithium hydroxide monohydrate as a base. The
free acid
or any of its suitable salt is coupled to thiomorpholine-1,1-dioxide
(purchased at Syntec,
ref M1201) by the procedures known to those in the art (for reaction
conditions described
in literature affecting such reactions see for example: Comprehensive Organic
Transformations: A Guide to Functional Group Preparations, 2nd Edition,
Richard C.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 18-
Larock. John Wiley & Sons, New York, NY. 1999). We found it convenient to use
2-(1H-
benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and
diisopropylethylamine in dimethylformamide, yielding a compound of formula Ia.
d) The indoles Ib might be the desired products, however, they might
optionally be
subjected to a subsequent alkylating reaction as described above under point
b) to furnish
the desired compounds Ia.
Alternatively, compound Ib can be alkylated or arylated by a boronic acid or a
boronic ester of formula X (either commercially available or accessible by
methods
described in references or by methods known in the art, as appropriate).
Conditions
commonly used in such types of transformation are described in literature and
known to
those in the art (e.g. Mederski, W. W. K. R.; Lefort, M.; Germann, M. Kux, D.
Tetrahedron
1999 55 12757). Ri can be any aryl, cycloalkyl or heteroaryl compounds.
Alternatively, compound Ib can be arylated by compound of general formula Ri-X
(either commercially available or accessible by methods described in
references or by
methods known in the art, as appropriate). The transformation can be affected
by
employing reaction conditions which are known to those in the art and widely
described
(e.g. Watanabe, M; Nishiyama, M.; Yamamoto, T.; Koie, Y, Tetrahedron Letters
2000, 41,
481; Old, D. W.; Harris, M. C.; Buchwald, S. L 2000 2 10 1403; Klapars, A.;
Antilla, J. C.;
Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2001 123 7727). The leaving group
X can be
any halogen group (chlorine, bromine, iodine) or pseudo halogen group (e.g.
trifluoromethylmethanesulfonyl, paratoluensulfonyl, methanesulfonyl and the
like) and Ri
can be any aryl or heteroaryl groups.
As described above, the compounds of formula I of the present invention can be
used as medicaments for the treatment and/or prevention of diseases which are
associated
with the modulation of H3 receptors.
In this context, the expression 'diseases associated with the modulation of H3
receptors' means diseases which can be treated and/or prevented by modulation
of H3
receptors. Such diseases encompass, but are not limited to, obesity, metabolic
syndrome
(syndrome X), neurological diseases including Alzheimer's disease, dementia,
age-related
memory dysfunction, mild cognitive impairment, cognitive deficit, attention
deficit
hyperactivity disorder, epilepsy, neuropathic pain, inflammatory pain,
migraine,
Parkinson's disease, multiple sclerosis, stroke, dizziness, schizophrenia,
depression,
addiction, motion sickness and sleep disorders including narcolepsy and other
diseases
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-19-
including asthma, allergy, allergy-induced airway responses, congestion,
chronic
obstructive pulmonary disease and gastro-intestinal disorders.
In a preferable aspect, the expression 'diseases associated with modulation of
H3
receptors' relates to obesity, metabolic syndrome (syndrome X) and other
eating disorders,
with obesity being especially preferred.
The invention therefore also relates to pharmaceutical compositions comprising
a
compound as defined above and a pharmaceutically acceptable carrier and/or
adjuvant.
Further, the invention relates to compounds as defined above for use as
therapeutically active substances, particularly as therapeutic active
substances for the
treatment and/or prevention of diseases which are associated with the
modulation of H3
receptors.
In another embodiment, the invention relates to a method for the treatment
and/or
prevention of diseases which are associated with the modulation of H3
receptors, which
method comprises administering a therapeutically active amount of a compound
of
formula I to a human being or animal. A method for the treatment and/or
prevention of
obesity is preferred.
The invention further relates to the use of compounds of formula I as defined
above
for the treatment and/or prevention of diseases which are associated with the
modulation
of H3 receptors.
In addition, the invention relates to the use of compounds of formula I as
defined
above for the preparation of medicaments for the treatment and/or prevention
of diseases
which are associated with the modulation of H3 receptors. The use of compounds
of
formula I as defined above for the preparation of medicaments for the
treatment and/or
prevention of obesity is preferred.
Furthermore, the present invention relates to the use of a compound of formula
I for
the manufacture of a medicament for the treatment and prevention of obesity in
a patient
who is also receiving treatment with a lipase inhibitor and particularly,
wherein the lipase
inhibitor is orlistat.
It is a further preferred object of the present invention to provide a method
for the
treatment or prevention of obesity and obesity related disorders which
comprises
administration of a therapeutically effective amount of a compound according
to formula I
in combination or association with a therapeutically effective amount of other
drugs for
the treatment of obesity or eating disorders so that together they give
effective relief.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-20-
Suitable other drugs include, but are not limited to, anorectic agents, lipase
inhibitors,
selective serotonin reuptake inhibitors (SSRI) and agents that stimulate
metabolism of
body fat. Combinations or associations of the above agents may be encompassing
separate,
sequential or simultaneous administration.
The term "lipase inhibitor" refers to compounds which are capable of
inhibiting the
action of lipases, for example gastric and pancreatic lipases. For example
orlistat and
lipstatin as described in U.S. Patent No. 4,598,089 are potent inhibitor of
lipases. Lipstatin
is a natural product of microbial origin and orlistat is the result of a
hydrogenation of
lipstatin. Other lipase inhibitors include a class of compound commonly
referred to as
panclicins. Panclicins are analogues of orlistat (Mutoh et al, 1994). The term
"lipase
inhibitor" refers also to polymer bound lipase inhibitors for example
described in
International Patent Application WO 99/34786 (Geltex Pharmaceuticals Inc.).
These
polymers are characterized in that they have been substituted with one or more
groups that
inhibit lipases. The term "lipase inhibitor" also comprises pharmaceutically
acceptable salts
of these compounds. The term "lipase inhibitor" preferably refers to
tetrahydrolipstatin.
Administration of a therapeutically effective amount of a compound according
to formula
I in combination or association with a therapeutically effective amount of
tetrahydrolipstatin is especially preferred.
Tetrahydrolipstatin (orlistat) is a known compound useful for the control or
prevention of obesity and hyperlipidemia. See, U.S. Patent No. 4,598,089,
issued July 1,
1986, which also discloses processes for making orlistat and U.S. Patent No.
6,004,996,
which discloses appropriate pharmaceutical compositions. Further suitable
pharmaceutical
compositions are described for example in International Patent Applications WO
00/09122
and WO 00/09123. Additional processes for the preparation of orlistat are
disclosed in
European Patent Applications Publication Nos. 0 185 359, 0 189 577, 0 443 449
and 0 524
495.
Suitable anorectic agents of use in combination with a compound of the present
invention include, but are not limited to, APD356, aminorex, amphechloral,
amphetamine,
axokine, benzphetamine, bupropion, chlorphentermine, clobenzorex, cloforex,
clominorex, clortermine, CP945598, cyclexedrine, CYT009-GhrQb,
dexfenfluramine,
dextroamphetamine, diethylpropion, diphemethoxidine, N-ethylamphetamine,
fenbutrazate, fenfluramine, fenisorex, fenproporex, fludorex, fluminorex,
furfurylmethylamphetamine, levamfetamine, levophacetoperane, mazindol,
mefenorex,
metamfepramone, methamphetamine, metreleptin, norpseudoephedrine, pentorex,
phendimetrazine, phenmetrazine, phentermine, phenylpropanolamine, picilorex,
rimonabant, sibutramine, SLU319, SNAP 7941, SR147778 (Surinabant), steroidal
plant
extract (e.g. P57) and TM30338 and pharmaceutically acceptable salts thereof.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-21-
Most preferable anorectic agents are sibutramine, rimonabant and phentermine.
Suitable selective serotonin reuptake inhibitors of use in combination with a
compound of the present invention include: fluoxetine, fluvoxamine, paroxetine
and
sertraline and pharmaceutically acceptable salts thereof.
Suitable agents that stimulate metabolism of body fat include, but are not
limited to,
growth hormone agonist (e.g. AOD-9604).
The use of a compound of formula I in the manufacture of a medicament for the
treatment and prevention of obesity in a patient who is also receiving
treatment with a
compound selected from the group consisting of a lipase inhibitor, an
anorectic agent, a
selective serotonin reuptake inhibitor and an agent that stimulates metabolism
of body fat,
is also an object of the present invention.
The use of a compound of formula I in the manufacture of a medicament for the
treatment and prevention of obesity in a patient who is also receiving
treatment with a a
lipase inhibitor, preferably with tetrahydrolipstatin, is also an object of
the present
invention.
It is a further preferred object to provide a method of treatment or
prevention of
Type II diabetes (non-insulin dependent diabetes mellitus (NIDDM)) in a human
which
comprises administration of a therapeutically effective amount of a compound
according
to formula I in combination or association with a therapeutically effective
amount of a
lipase inhibitor, particularly, wherein the lipase inhibitor is
tetrahydrolipstatin. Also an
object of the invention is the method as described above for the simultaneous,
separate or
sequential administration of a compound according to formula I and a lipase
inhibitor,
particularly tetrahydrolipstatin.
It is a further preferred object to provide a method of treatment or
prevention of
Type II diabetes (non-insulin dependent diabetes mellitus (NIDDM)) in a human
which
comprises administration of a therapeutically effective amount of a compound
according
to formula I in combination or association with a therapeutically effective
amount of an
anti-diabetic agent.
The term "anti-diabetic agent" refers to compounds selected from the group
consisting of 1) PPARy agonists such as pioglitazone (actos) or rosiglitazone
(avandia) and
the like; 2) biguanides such as metformin (glucophage) and the like; 3)
sulfonylureas such
as glibenclamide, glimepiride (amaryl), glipizide (glucotrol), glyburide
(DiaBeta) and the
like; 4) nonsulfonylureas such as nateglinide (starlix), repaglimide (prandin)
and the like;
5) PPAR(r,/y agonists such as GW-2331 and the like 6) DPP-IV- inhibitors such
as LAF-237
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 22 -
(vildagliptin), MK-0431, BMS-477118 (saxagliptin) or GSK23A and the like; 7)
Glucokinase activators such as the compounds disclosed in e.g. WO 00/58293 Al
and the
like; 8) a-Glucosidase inhibitors such as acarbose (precose) or miglitol
(glyset) and the
like.
Also an object of the invention is the method as described above for the
simultaneous, separate or sequential administration of a compound according to
formula I
and a therapeutically effective amount of an anti-diabetic agent.
The use of a compound of formula I in the manufacture of a medicament for the
treatment and prevention of Type II diabetes in a patient who is also
receiving treatment
with an anti-diabetic agent is also an object of the present invention.
It is a further preferred object to provide a method of treatment or
prevention of
dyslipidemias in a human which comprises administration of a therapeutically
effective
amount of a compound according to formula I in combination or association with
a
therapeutically effective amount of a lipid lowering agent.
The term "lipid lowering agent" refers to compounds selected from the group
consisting of 1) bile acid sequestrants such as cholestyramine (questran),
colestipol
(colestid) and the like; 2) HMG-CoAreductase inhibitors such as atorvastatin
(lipitor),
cerivastatin (baycol), fluvastatin (lescol), pravastatin (pravachol),
simvastatin (zocor) and
the like; 3) cholesterol absorption inhibitors such as ezetimibe and the like;
4) CETP
inhibitors such as torcetrapib, JTT 705 and the like; 5) PPARa-agonists such
as
beclofibrate, gemfibrozil (lopid), fenofibrate (lipidil), bezafibrate
(bezalip) and the like; 6)
lipoprotein synthesis inhibitors such as niacin and the like; and 7) niacin
receptor agonists
such as nicotinic acid and the like.
Also an object of the invention is the method as described above for the
simultaneous, separate or sequential administration of a compound according to
formula I
and a therapeutically effective amount of a lipid lowering agent.
The use of a compound of formula I in the manufacture of a medicament for the
treatment and prevention of dyslipidemias in a patient who is also receiving
treatment with
a lipid lowering agent, is also an object of the present invention.
It is a further preferred object to provide a method of treatment or
prevention of
hypertension in a human which comprises administration of a therapeutically
effective
amount of a compound according to formula I in combination or association with
a
therapeutically effective amount of an anti-hypertensive agent.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-23-
The term "anti-hypertensive agent" or "blood-pressure lowering agent" refers
to
compounds selected from the group consisting of 1) Angiotensin-converting
Enzyme
(ACE) Inhibitors including benazepril (lotensin), captopril (capoten),
enalapril (vasotec),
fosinopril (monopril), lisinopril (prinivil, zestril), moexipril (univasc),
perindopril
(coversum), quinapril (accupril), ramipril (altace), trandolapril (mavik) and
the like; 2)
Angiotensin II Receptor Antagonists including candesartan (atacand),
eprosartan
(teveten), irbesartan (avapro), losartan (cozaar), telmisartan (micadisc),
valsartan (diovan)
and the like; 3) Adrenergic Blockers (peripheral or central) such as the beta-
adrenergic
blockers including acebutolol (sectrol), atenolol (tenormin), betaxolol
(kerlone),
bisoprolol (zebeta), carteolol (cartrol), metoprolol (lopressor; toprol-XL),
nadolol
(corgard), penbutolol (levatol), pindolol (visken), propranolol (inderal),
timolol
(blockadren) and the like; alpha/beta adrenergic blockers including carvedilol
(coreg),
labetalol (normodyne) and the like; alpha-1 adrenergic blockers including
prazosin
(minipress), doxazosin (cardura), terazosin (hytrin), phenoxybenzamine
(dibenzyline) and
the like; peripheral adrenergic-neuronal blockers including guanadrel
(hylorel),
guanethidine (ismelin), reserpine (serpasil) and the like; alpha-2 adrenergic
blockers
including a-methyldopa (aldomet), clonidine (catapres), guanabenz (wytensin),
guanfacine (tenex) and the like; 4) Blood Vessel Dilators (Vasodilators)
including
hydralazine (apresoline), minoxidil (lonitren), clonidine (catapres) and the
like; 5)
Calcium Channel Blockers including amlodipine (norvasc), felodipine (plendil),
isradipine
(dynacirc), nicardipine (cardine sr), nifedipine (procardia, adalat),
nisoldipine (sular),
diltiazem (cardizem), verapamil (isoptil) and the like; 6) Diuretics such as
thiazides and
thiazides-like agents, including hydrochlorothiazide (hydrodiuril, microzide),
chlorothiazide (diuril), chlorthalidone (hygroton), indapamide (lozol),
metolazone
(mykrox) and the like; loop diuretics, such as bumetanide (bumex) and
furosemide (lasix),
ethacrynic acid (edecrin), torsemide (demadex) and the like; potassium-sparing
diuretics
including amiloride (midamor), triamterene (dyrenium), spironolactone
(aldactone) and
the tiamenidine (symcor) and the like; 7) Tyrosine Hydroxylase Inhibitors,
including
metyrosine (demser) and the like; 8) Neutral Endopeptidase Inhibitors,
including BMS-
186716 (omapatrilat), UK-79300 (candoxatril), ecadotril (sinorphan), BP-1137
(fasidotril),
UK-79300 (sampatrilat) and the like; and 9) Endothelin Antagonists including
tezosentan
(R00610612), A308165 and the like.
Also an object of the invention is the method as described above for the
simultaneous, separate or sequential administration of a compound according to
formula I
and a therapeutically effective amount of a anti-hypertensive agent.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 24 -
The use of a compound of formula I in the manufacture of a medicament for the
treatment and prevention of hypertension in a patient who is also receiving
treatment with
an anti-hypertensive agent, is also an object of the present invention.
As described above, the compounds of formula I and their pharmaceutically
acceptable salts possess valuable pharmacological properties. Specifically, it
has been found
that the compounds of the present invention are good histamine 3 receptor
(H3R)
antagonists and/or inverse agonists.
The following test was carried out in order to determine the activity of the
compounds of formula (I).
Binding assay with 3H-(R)a-methylhistamine
Saturation binding experiments were performed using HR3-CHO membranes
prepared as described in Takahashi, K, Tokita, S., Kotani, H. (2003) J.
Pharmacol. Exp.
Therapeutics 307, 213-218.
An appropriate amount of membrane (60 to 80 g protein/well) was incubated
with
increasing concentrations of 3H(R)a-Methylhistamine di-hydrochloride (0.10 to
10 nM).
Non specific binding was determined using a 200 fold excess of cold (R)a-
Methylhistamine dihydrobromide (500 nM final concentration). The incubation
was
carried out at room temperature (in deep-well plates shaking for three hours).
The final
volume in each well was 250 l. The incubation was followed by rapid
filtration on GF/B
filters (pre-soaked with 100 l of 0.5% PEI in Tris 50 mM shaking at 200 rpm
for two
hours). The filtration was made using a cell-harvester and the filter plates
were then
washed five times with ice cold washing buffer containing 0.5 M NaC1. After
harvesting,
the plates were dried at 55 C for 60 min, then we added scintillation fluid
(Microscint 40,
40 microl in each well) and the amount of radioactivity on the filter was
determined in
Packard top-counter after shaking the plates for two hours at 200 rpm at room
temperature.
Binding Buffer: 50 mM Tris-HC1 pH 7.4 and 5 mM MgCl2x 6H20 pH 7.4. Washing
Buffer: 50 mM Tris-HC1 pH 7.4 and 5 mM MgC12x6HzO and 0.5 M NaC1 pH 7.4.
Indirect measurement of affinity of H3R inverse agonists: twelve increasing
concentrations (ranging from 10 M to 0.3 nM) of the selected compounds were
always
tested in competition binding experiments using membrane of the human HR3-CHO
cell
line. An appropriate amount of protein, e.g. approximately 500cpm binding of
RAMH at
Kd, were incubated for 1 hour at room temperature in 250 l final volume in 96-
well
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-25-
plates in presence of 3H(R)a-Methylhistamine (1 nM final concentration = Kd).
Non-
specific binding was determined using a 200 fold excess of cold (R)a -
Methylhistamine
dihydrobromide.
All compoundswere tested at a single concentration in duplicates. Compounds
that
showed an inhibition of [3H]-RAMH by more than 50% were tested again to
determine
IC50 in a serial dilution experiment. Ki's were calculated from IC50 based on
Cheng-Prusoff
equation ( Cheng, Y, Prusoff, WH (1973) Biochem Pharmaco122, 3099-3108).
The compounds of the present invention exhibit K. values within the range of
about
0.1 nM to about 1000 nM, preferably of about 0.1 nM to about 300 nM and more
preferably of about 0.1 nM to about 100 nM. The following table shows measured
values
for some selected compounds of the present invention.
The following table shows measured values for some selected compounds of the
present invention.
K; (nM)
Example 1 3
Example 4 49
Demonstration of additional biological activities of the compounds of the
present
invention may be accomplished through in vitro, ex vivo and in vivo assays
that are well
known in the art. For example, to demonstrate the efficacy of a pharmaceutical
agent for
the treatment of obesity-related disorders such as diabetes, Syndrome X, or
atherosclerotic
disease and related disorders such as hypertriglyceridemia and
hypercholesteremia, the
following assays may be used.
Method for Measuring Blood Glucose Levels
db/db mice (obtained from Jackson Laboratories, Bar Harbor, ME) are bled (by
either eye or tail vein) and grouped according to equivalent mean blood
glucose levels.
They are dosed orally (by gavage in a pharmaceutically acceptable vehicle)
with the test
compound once daily for 7 to 14 days. At this point, the animals are bled
again by eye or
tail vein and blood glucose levels are determined.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-26-
Method for Measuring Triglyceride Levels
hApoAl mice (obtained from Jackson Laboratories, Bar Harbor, ME) are bled (by
either eye or tail vein) and grouped according to equivalent mean serum
triglyceride levels.
They are dosed orally (by gavage in a pharmaceutically acceptable vehicle)
with the test
compound once daily for 7 to 14 days. The animals are then bled again by eye
or tail vein
and serum triglyceride levels are determined.
Method for Measuring HDL-Cholesterol Levels
To determine plasma HDL-cholesterol levels, hApoAl mice are bled and grouped
with equivalent mean plasma HDL-cholesterol levels. The mice are orally dosed
once daily
with vehicle or test compound for 7 to 14 days and then bled on the following
day. Plasma
is analyzed for HDL-cholesterol.
The compounds of formula (I) and their pharmaceutically acceptable salts and
esters
can be used as medicaments, e.g. in the form of pharmaceutical preparations
for enteral,
parenteral or topical administration. They can be administered, for example,
perorally, e.g.
in the form of tablets, coated tablets, drag6es, hard and soft gelatine
capsules, solutions,
emulsions or suspensions, rectally, e.g. in the form of suppositories,
parenterally, e.g. in the
form of injection solutions or infusion solutions, or topically, e.g. in the
form of
ointments, creams or oils.
The production of the pharmaceutical preparations can be effected in a manner
which will be familiar to any person skilled in the art by bringing the
described compounds
of formula (I) and their pharmaceutically acceptable, into a galenical
administration form
together with suitable, non-toxic, inert, therapeutically compatible solid or
liquid carrier
materials and, if desired, usual pharmaceutical adjuvants.
Suitable carrier materials are not only inorganic carrier materials, but also
organic
carrier materials. Thus, for example, lactose, corn starch or derivatives
thereof, talc, stearic
acid or its salts can be used as carrier materials for tablets, coated
tablets, drag6es and hard
gelatine capsules. Suitable carrier materials for soft gelatine capsules are,
for example,
vegetable oils, waxes, fats and semi-solid and liquid polyols (depending on
the nature of
the active ingredient no carriers are, however, required in the case of soft
gelatine
capsules). Suitable carrier materials for the production of solutions and
syrups are, for
example, water, polyols, sucrose, invert sugar and the like. Suitable carrier
materials for
injection solutions are, for example, water, alcohols, polyols, glycerol and
vegetable oils.
Suitable carrier materials for suppositories are, for example, natural or
hardened oils,
waxes, fats and semi-liquid or liquid polyols. Suitable carrier materials for
topical
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-27-
preparations are glycerides, semi-synthetic and synthetic glycerides,
hydrogenated oils,
liquid waxes, liquid paraffins, liquid fatty alcohols, sterols, polyethylene
glycols and
cellulose derivatives.
Usual stabilizers, preservatives, wetting and emulsifying agents, consistency-
improving agents, flavour-improving agents, salts for varying the osmotic
pressure, buffer
substances, solubilizers, colorants and masking agents and antioxidants come
into
consideration as pharmaceutical adjuvants.
The dosage of the compounds of formula (I) can vary within wide limits
depending
on the disease to be controlled, the age and the individual condition of the
patient and the
mode of administration and will, of course, be fitted to the individual
requirements in each
particular case. For adult patients a daily dosage of about 1 mg to about 1000
mg,
especially about 1 mg to about 100 mg, comes into consideration. Depending on
the
dosage it is convenient to administer the daily dosage in several dosage
units.
The pharmaceutical preparations conveniently contain about 0.1-500 mg,
preferably
0.5-100 mg, of a compound of formula (I).
The following examples serve to illustrate the present invention in more
detail. They
are, however, not intended to limit its scope in any manner.
Examples
Intermediate 1
5-(1-Isoproprl-piperidin-4-yloxy)-1H-indole-2-carboxylic acid ethyl ester
a) Step 1: 1-Isoproprl-piperidin-4-ol
To a cold (0 C) solution of 1-isopropylpiperidone (purchased at Chemie
Brunschwig AG, 100 g, 1.0 eq.) in ethanol (500 mL) was added sodium
borohydride (19.3
g, 0.7 eq.) in small portions. The reaction mixture was allowed to warm up to
room
temperature and was stirred overnight. After concentration in vacuo, ice water
(1 kg),
sodium hydroxide aqueous solution (28% in mass, 0.5 L) and dichloromethane (1
L) were
added. The mixture was stirred vigorously for 4 h and the aqueous layer was
extracted with
dichloromethane. Combined organic layers were washed with brine, dried over
sodium
sulfate, filtered and purified by fractionated vacuum distillation (20 mBar).
One fraction
(95 C at 20 mBar) was isolated to yield 61.3 g(60 Io) of the title product as
colorless oil.
MS (m/e): 144.5 (MH+, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-28-
b) Step 2: 5-(1-Isoproprl-p3rrolidin-3-yloxy)-1H-indole-2-carboxylic acid
ethyl ester
To a cold (0 C) mixture of 5-hydroxy-lH-indole-2-carboxylic acid ethyl ester
(10 g,
1.0 eq.), 1-isopropyl-piperidin-4-ol (intermediate 1, step 1, 7.32 g, 1.05
eq.) and
triphenylphosphine (15.3 g, 1.2 eq.) in tetrahydrofuran (280 ml) was slowly
added a
solution of diisopropylazodicarboxylate (11.8 g, 1.2 eq.) in tetrahydrofuran
(20 mL). The
mixture was stirred 30 min at 0 C and overnight at room temperature, was
concentrated
in vacuo, dissolved in methyltertiobutylether (310 mL), washed with sodium
hydroxide
aqueous solution (0.5N), brine, dried over NazSO4, filtered and evaporated.
The residue
was purified on silica eluting with dichloromethane/methanoUammoniac. One
fraction
was isolated and dried in vacuo, to yield 7.0 g(43 Io) of the desired product
as white solid.
MS (m/e): 331.5 (MH+, 100%)
Example 1
(1,1-Dioxo-thiomorpholin-4-yl)-f 5-(1-isoproprl-piperidin-4-yloxy)-1-(2,2,2-
trifluoro-
ethyl)-1H-indol-2-yll-methanone
a) Step 1: 5-(1-Isoprop,rl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-1H-
indole-2-
carboxylic acid ethyl ester
To a mixture of 5-(1-isopropyl-pyrrolidin-3-yloxy)-1H-indole-2-carboxylic acid
ethyl ester (intermediate, 4 g, 1.0 eq.) in dimethylformamide (40 mL) was
added sodium
hydride (dispersion in oil, 60% in mass, 533 mg, 1.1 eq.) in several portions.
The solution
was stirred 30 min at 70 C. Then 2,2,2-
trifluoroethyltrifluoromethanesulfonate (3.37 g,
1.2 eq.) was added and the reaction mixture was stirred at 70 C overnight.
The reaction
mixture was partitioned between an aqueous solution of sodium
hydrogenocarbonate and
ethyl acetate. The organic layer was washed with water and brine, evaporated
in vacuo and
then purified on silica eluting with dichloromethane/methanoUammoniac. One
fraction
was isolated and dried in vacuo, to yield 3.9 g(78 Io) of the desired product
as off-white
solid. MS (m/e): 413.5 (MH+, 100%)
b) Step 2: 5-(1-Isoprop,rl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-1H-
indole-2-
carboxylic acid, hydrochloric salt with one equivalent of lithium chloride
To a solution of 5-(1-isopropyl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-
1H-
indole-2-carboxylic acid ethyl ester (3.9g, 1.0 eq.) in tetrahydrofuran (30
mL), water (15
mL) and methanol (7 mL) was added lithium hydroxide monohydrate (460 mg, 1.16
eq.).
The reaction mixture was refluxed overnight. After concentration in vacuo the
residue was
acidified (pH:2) with hydrochloric acid (2N). The resulting mixture was dried
in vacuo to
yield 4.4 g(99 Io) of the desired product as off-white solid. MS (m/e): 462.5
(M-H-, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-29-
c) Step 3: (1,1-Dioxo-thiomorpholin-4-yl)-f5-(1-isoproprl-piperidin-4-yloxy)-1-
(2,2,2-
trifluoro-ethyl)-1H-indol-2-yll-methanone
A mixture of 5-(1-isopropyl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-1H-
indole-
2-carboxylic acid, hydrochloric salt with one equivalent of lithium chloride
(650 mg, 1.0
eq.), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylurunium tetrafluoroborate
(563 mg, 1.2
eq.), thiomorpholine-1,1-dioxide (purchased at Syntec, ref. M1201) and
diisopropylethylamine (1.22 mL, 5 eq.) in dimethylformamide was stirred at
room
temperature for 24h and then partitioned between an aqueous solution of sodium
hydrogenocarbonate and ethyl acetate. The aqueous layer was extracted with
ethyl acetate
and the combined organic layers were washed with water and brine, dried over
sodium
sulfate, filtered, evaporated in vacuo and purified on silica eluting with
dichloromethane/methanoUammoniac. One fraction was isolated and dried in
vacuo, to
yield 468 mg (66%) of the desired product as off-white solid. MS (m/e): 502.5
(MH+,
100%).
Example 2
(1,1-Dioxo-thiomorpholin-4-yl)-f 5-(1-isoproprl-piperidin-4-yloxy)-1H-indol-2-
yll -
methanone
a) Step 1: 5-(1-Isoproprl-piperidin-4-yloxy)-1H-indole-2-carboxylic acid
hydrochloric salt
with one equivalent of lithium chloride
A mixture of 5-(1-isopropyl-pyrrolidin-3-yloxy)-1H-indole-2-carboxylic acid
ethyl
ester (intermediate 1, 1.98 g, 1.0 eq.) and lithium hydroxide monohydrate (300
mg, 1.15
mmol) in tetrahydrofuran (30 mL), methanol (30 mL) and water (15 mL) was
heated to
100 C for 2 h. The organic solvents were removed and aq. 1N HC1 was added to
adjust the
pH of the solution in a range of 2 to 3. Subsequently, the mixture was
evaporated to
dryness and the mixture was used without further purification in the next
step. MS (m/e):
301.5 (M-H-, 100%).
b) Step 2: (1,1-Dioxo-thiomorpholin-4-yl)-f5-(1-isoproprl-piperidin-4-yloxy)-
1H-indol-
2-yll-methanone
In analogy to the procedure described for the synthesis of example 1, step 3,
the title
compound was synthesized from 5-(1-isopropyl-piperidin-4-yloxy)-1H-indole-2-
carboxylic acid hydrochloric salt with one equivalent of lithium chloride
(example 2, step
1) and thiomorpholine-1,1-dioxide (purchased at Syntec, ref. M1201). The title
product
was obtained in 75% yield as white solid. MS (m/e): 420.5 (MH+, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 30 -
Example 3
(1,1-Dioxo-thiomorpholin-4-yl)-[ 1-isopropyl-5-(1-isoproprl-piperidin-4-yloxy~-
IH-
indol-2-yll-methanone
To a mixture of (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-
yloxy)-
1H-indol-2-yl]-methanone (Example 2, 100 mg, 1.0 eq.) and isopropyl-
methanesulfonate
(67 mg, 2.0 eq.) in dimethylformamide (4 mL) was added cesium carbonate (156
mg, 2.0
eq.). The solution was stirred 22h at 95 C. The reaction mixture was
concentrated in vacuo
and the residue partitioned between water and methyl-tert-butylether. The
aqueous layer
was extracted with methyl-tert-butylether. The combined organic layer was
washed with
brine, dried over sodium sulfate, filtered, concentrated in vacuo and then
purified on silica,
eluting with cyclohexane/ethyl acetate. One fraction was isolated and dried in
vacuo, to
yield 52 mg (47%) of the desired product as off-white solid. MS (m/e): 462.5
(MH+, 100%)
Example 4
(1,1-Dioxo-thiomorpholin-4-yl)-[5-(1-isoprop,rl-p3rrolidin-3S-yloxy)-1H-indol-
2-yll-
methanone
a) Step 1: 5-((S)-1-Benzyl-p3rrolidin-3-yloxy)-1H-indole-2-carboxylic acid
ethyl ester
To a cold (0 C) mixture of ethyl-5-hydroxyindole-2-carboxylate (purchased at
Biosynth, H-6350, 20.5 g, 1.0 eq.), (R)-1-benzyl-3-pyrrolidinol (23 g, 1.3
eq.) and tri-n-
butylphosphine (58 mL, 2.0 eq.) was slowly added 1,1'-
(azodicarbonyl)dipiperidine (50.4
g, 2.0 eq.) in several portions. The reaction mixture was stirred at room
temperature
overnight and then filtered off. The filtrate was concentrated in vacuo and
diethylether was
added. The precipitate was filtered off and the filtrate was concentrated in
vacuo and
purified on silica eluting with dichloromethane/methanoUammoniac. One fraction
was
isolated and dried in vacuo, to yield 18 mg (49%) of the desired product as
light yellow
foam. MS (m/e): 365.5 (MH+, 100%).
b) Step 2: 5-((S)-1-Isoprop,rl-p3rrolidin-3-yloxy)-1H-indole-2-carboxylic acid
ethyl ester
To a mixture of 5-((S)-1-benzyl-pyrrolidin-3-yloxy)-1H-indole-2-carboxylic
acid
ethyl ester (18.0 g, 1.0 eq.) and acetic acid (28 mL, 10 eq.) in ethanol (500
mL) was added
palladium on activated charchoal (10% in mass, 2.0 g, 0.04 eq.) and the
reaction vessel was
flushed with hydrogen (1 Atm). The reaction mixture was stirred 18h at room
temperature
and then filtered off and concentrated in vacuo. The residue (28.4 g) was
dissolved in
dimethylformamide (500 mL) and potassium carbonate was added. The mixture was
stirred 15 min at room temperature. Then 2-iodopropane (42 g, 5.0 eq.) was
added and the
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-31-
mixture was stirred 4h at 50 C. The reaction mixture was filtered off and the
filtrate was
evaporated in vacuo. The residue was dissolved in acetone, filtered and the
filtrate was
evaporated in vacuo then purified on silica eluting with dichloromethane/
methanoU
ammoniac. One fraction was isolated and dried in vacuo, to yield 9.3 g(59 Io)
of the desired
product as light brown solid. MS (m/e): 317.4 (MH+, 100%).
c) Step 3: 5-((S)-1-Isoproprl-p3rrolidin-3-yloxy)-1H-indole-2-carboxylic acid
hydrochloric salt with one equivalent of lithium chloride
To a solution of 5-((S)-1-isopropyl-pyrrolidin-3-yloxy)-1H-indole-2-carboxylic
acid
ethyl ester (8.9 g, 1.0 eq.) in tetrahydrofuran (100 mL), water (50 mL) and
methanol (10
mL) lithium hydroxide monohydrate (1.3 g, 1.10 eq.) was added. The reaction
mixture was
refluxed overnight and then concentrated in vacuo. The residue was acidified
(pH: 2) with
hydrochloric acid (2N). The resulting mixture was dried in vacuo yielding 10.5
g(100 Io) of
the desired product as brown foam. MS (m/e): 287.0 (M-H-, 100%).
d) Step 4: (1,1-Dioxo-thiomorpholin-4-yl)-[5-(1-isoprop,rl-p3rrolidin-3S-
Yloxy~-IH-
indol-2-yll -methanone
In analogy to the procedure described for the synthesis of example 1, step 3,
the title
compound was synthesized from 5-((S)-1-isopropyl-pyrrolidin-3-yloxy)-1H-indole-
2-
carboxylic acid hydrochloric salt with one equivalent of lithium chloride
(example 4, step
3) and thiomorpholine-1,1-dioxide (purchased at Syntec, ref. M1201). The
desired product
was obtained in a yield of 67 % as white solid. MS (m/e): 406.5 (MH+, 100%).
Example 5
5-[2-(1,1-Dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isoproprl-piperidin-4-
yloxy
indol-l-yll -p3ridine-2-carbonitrile
To a mixture of (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-
yloxy)-
1H-indol-2-yl]-methanone (Example 2, 150 mg, 1.0 eq.), anhydrous copper(II)
acetate
(131 mg, 2 eq.), pyridine (120 microL, 4 eq.) in dichloromethane (3.5 mL), 2-
cyanopyridine-5-boronic acid pinacol ester (247 mg, 3 eq.) was added. The
reaction
mixture was stirred for 4 days and then concentrated in vacuo. The residue was
then
purified on silica eluting with dichloromethane/ methano198:2 v:v. One
fraction was
isolated and dried in vacuo, to yield 57 mg (23%) of the desired product as
light yellow oil.
MS (m/e): 522.5 (MH+, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 32 -
Example 6
f6-Bromo-5-(1-isoproprl-piperidin-4-yloxy)-1H-indol-2-yll-(1,1-dioxo-l~6-
thiomorpholin-4-yl)-methanone
a) Step 1: 6-Bromo-5-hydroxy-lH-indole-2-carboxylic acid ethyl ester
The solution of 8.30 g (27.8 mmol) 6-bromo-5-methoxy-lH-indole-2-carboxylic
acid ethyl ester (prepared according to J. Org. Chem. 1974, 39, 3580) in 160
mL
dichloromethane was cooled to -78 C. At this temperature, 55.7 mL boron
tribromide
(55.7 mmol; 1M solution in dichloromethane) were added. The solution was
allowed to
warm to room temperature and after 30 min. The solution was poured on 10%
aqueous
sodium bicarbonate solution, the phases were separated and the aqueous phase
was
extracted three times with ethyl acetate. The combined organic layers were
washed with
water followed by brine, dried over magnesium sulfate, filtered and
evaporated. The
residue was flash-chromatographed on silica gel with n-hexane : ethyl acetate
(2: 1 v/v) as
eluant to give 5.7 g(72 Io) of the product as a light yellow solid. MS (m/e):
282.2 (M-H+,
100%).
b) Step 2: 6-Bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-indole-2-carboxylic
acid ethyl
ester
To the suspension of 0.25 g (0.88 mmol) 6-bromo-5-hydroxy-lH-indole-2-
carboxylic acid ethyl ester in 5 mL tetrahydrofuran, 0.15 g (1.05 mmol) 1-
isopropyl-
piperidin-4-ol (commercially available) and 0.28 g (1.07 mmol)
tributylphosphine were
added. The suspension was cooled to 0 C, 0.244 g (1.06 mmol) di-tert-butyl
azodicarboxylate was added and the reaction was allowed to reach room
temperature. After
48 hours the suspension was filtered and the filtrate was evaporated. The
residue was flash-
chromatographed on silica gel with a gradient of dichloromethane : methanol
(100 : 0 to
60 : 40 v/v) to give 0.20 g(55 Io) of the product as a light yellow foam. MS
(m/e): 409.0
(M-H+, 100%).
c) Step 3: 6-Bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-indole-2-carboxylic
acid,
hydrochloric salt in mixture with lithium chloride
A mixture of 6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-indole-2-carboxylic
acid ethyl ester (3.4 g, 1.0 eq.) and lithium hydroxide (249 mg 1.25 eq.) in a
mixture of
tetrahydrofuran (170 mL) and water (85 mL) was refluxed for 2h then the
volatiles where
removed in vacuo and the pH was adjusted to ca. 2. The suspension was dried by
azeotropic removal of water (toluol) to yield 3.95 g of the desired product as
light yellow
solid which was used without further purification. MS (m/e): 416.5.0 (M-H+,
100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-33-
d) Step 4: [6-Bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl]-( 1,1-
Dioxo-1',~6-
thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 1, step 3
the title
compound was synthesized from 6-Bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indole-
2-carboxylic acid, hydrochloric salt in mixture with lithium chloride (Example
6 step 3)
and thiomorpholine-1,1-dioxide (purchased at Syntec, ref. M1201). The desired
product
was obtained in a yield of 60 % as light yellow solid. MS (m/e): 499.5 (MH+,
100%).
Example 7
[6-Bromo-l-isopropyl-5-(1-isoproprl-piperidin-4-yloxy)-1H-indol-2-yll -(1,1-
dioxo-1',~6-
thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 3, the
title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and
isopropylmethanesulfonate. The desired product was obtained in a yield of 69 %
as light
yellow solid. MS (m/e): 540.3 (M+H, 100%).
Example 8
[6-Bromo-5-(1-isoproprl-piperidin-4-yloxy)-1-(2,2,2-trifluoro-ethyl)-1H-indol-
2-yll-
(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and
2,2,2-
trifluoroethyltrifluoromethanesulfonate. The desired product was obtained in a
yield of 15
Io as white solid. MS (m/e): 580.1 (M+H, 100%).
Example 9
[6-Bromo-l-(2-chloro-p3ridin-4-yl)-5-(1-isoprop rl-piperidin-4-,~y)-1H-indol-2-
,~
(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 5, the
title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and 2-
chloropyridine-4-boronic acid. The desired product was obtained in a yield of
7 Io as white
solid. MS (m/e): 609.0 (M+H, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 34 -
Example 10
(1,1-Dioxo-l~,6-thiomorpholin-4-yl)-[5-(1-isoproprl-piperidin-4-yloxy)-1-(3-
trifluoromethyl-phenyl)-1H-indol-2-yll-methanone
In analogy to the procedure described for the synthesis of example 5, the
title
compound was synthesized from (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-
piperidin-4-yloxy)-1H-indol-2-yl]-methanone (Example 2) and (3-
trifluoromethyl) -
phenylboronic acid. The desired product was obtained in a yield of 77 % as
light yellow
solid. MS (m/e): 564.5 (MH+, 100%).
Example 11
[1-(2-Chloro-p3ridin-4-yl)-5-(1-isoprop,rl-piperidin-4-yloxy)-1H-indol-2-yll-
(1,1-dioxo-
1',~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 5, the
title
compound was synthesized from (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-
piperidin-4-yloxy)-1H-indol-2-yl]-methanone (Example 2) and 2-chloropyridine-4-
boronic acid. The desired product was obtained in a yield of 8 Io as an off-
white solid. MS
(m/e): 531.5 (M+, 100%).
Example 12
rac-2-[2-(1,1-Dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isoproprl-piperidin-4-
yloxy
indol-l-yll-propionitrile
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-
piperidin-4-yloxy)-1H-indol-2-yl]-methanone (Example 2) and 2-
bromopropionitrile. The
desired product was obtained in a yield of 20 % as a light yellow foam. MS
(m/e): 473.6
(MH+, 100%).
Example 13
(1,1-Dioxo-l~,6-thiomorpholin-4-yl) - [ 5- (1-isoproprl-piperidin-4-ylox,
rp3rimidin-5-yl-
1H-indol-2-yll-methanone
To a mixture of (1,1-dioxo-thiomorpholin-4-yl)-[5-(1-isopropyl-piperidin-4-
yloxy)-
1H-indol-2-yl]-methanone (Example 2, 1 g, 1.0 eq.), trans-1,2-
diaminocyclohexane (186
microL, 0.65 eq.), copper(I) iodide (54 mg, 0.12 eq.), potassium phosphate
(1.06 g, 2.1 eq.)
and potassium carbonate (692 mg, 2.1 eq.) in dioxane (20 mL) 5-bromopyrimidine
(417
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-35-
mg, 1.1 eq.) was added. The reaction mixture was stirred for 5 days and then
concentrated
in vacuo. The residue was then purified on silica eluting with
dichloromethane/ methanol
98:2 v:v. One fraction was isolated and dried in vacuo, to yield 158 mg (13%)
of the desired
product as light yellow oil. MS (m/e): 498.6 (MH+, 100%).
Example 14
[6-Bromo-5-(1-isoproprl-piperidin-4-yloxy)-1-(2-methox, -r~yl)-1H-indol-2-yll-
(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and (2-
bromoethyl)-methylether. The desired product was obtained in a yield of 70 %
as white
foam. MS (m/e): 556.3 (M+H, 100%).
Example 15
[6-Bromo-5-(1-isoprop,rl-piperidin-4-yloxy)-1-(3-methox, -propyl)-1H-indol-2-
yll-(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and 1-
bromo-3-
methoxypropane. The desired product was obtained in a yield of 65 % as light
yellow
foam. MS (m/e): 570.4 (M+H, 70%).
Example 16
f6-Bromo-1-(2-h, d, -r~yl)-5-(1-isoprop,rl-piperidin-4-yloxy)-1H-indol-2-yll-
(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone
a) step 1: [6-Bromo- 1- [2- (tert-butyl-dimethyl-silanyloxy) -ethyl] -5- (1-
isopropyl-piperidin-
4-yloxy)-1H-indol-2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and (2-
bromoethoxy)-tert-butyldimethylsilane. The desired product was obtained in a
yield of 50
% as colorless oil. MS (m/e): 656.4 (M+H, 50%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 36 -
b) step 2: [6-Bromo-1-(2-hydroxy-ethyl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-2-
yl] -(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone
Amixture of [6-bromo-1-[2-(tert-butyl-dimethyl-silanyloxy)-ethyl]-5-(1-
isopropyl-
piperidin-4-yloxy)-1H-indol-2-yl]-(1,1-dioxo-l,,~6-thiomorpholin-4-yl)-
methanone (130
mg, 1.0 eq.) and trifluoroacetic acid in dichloromethane was stirred for lh at
room
temperature and concentrated in vacuo. The crude mixture was partitioned
between an
aqueous solution of sodium hydroxide and dichloromethane. The aqueous lyer was
extracted with dichloromethane. Combined organic layers were washed with
brine, dried
over sodium sulfate, filtered and evaporated in vacuo, to yield 106 mg (99%)
of the desired
product as white foam. MS (m/e): 542.1 (M+H, 100%).
Example 17
[6-Bromo-l-(3-h, d, -propyl)-5-(1-isoprop,rl-piperidin-4-yloxy)-1H-indol-2-yll-
(1,1-
dioxo-1,~6-thiomorpholin-4-yl)-methanone
a) Step 1: [6-Bromo-l-[3-(tert-butyl-dimethyl-silanyloxy)-propyl]-5-(1-
isopropyl-
piperidin-4-yloxy)-1H-indol-2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-
methanone
In analogy to the procedure described for the synthesis of example 1, step 1,
the title
compound was synthesized from [6-bromo-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-
2-yl]-(1,1-dioxo-1,~6-thiomorpholin-4-yl)-methanone (Example 6, step 4) and (3-
bromopropoxy)-tert-butyldimethylsilane. The desired product was obtained in a
yield of
65 % as colorless oil. MS (m/e): 670.5 (M+H, 100%).
b) Step 2: [6-Bromo-1-(3-hydroxy-propyl)-5-(1-isopropyl-piperidin-4-yloxy)-1H-
indol-2-
yl] -(1,1-Dioxo-l,~6-thiomorpholin-4-yl)-methanone
In analogy to the procedure described for the synthesis of example 16, step 1,
the title
compound was synthesized from [6-bromo-l-[3-(tert-butyl-dimethyl-silanyloxy)-
propyl]-
5-(1-isopropyl-piperidin-4-yloxy)-1H-indol-2-yl]-(1,1-dioxo-1,~6-thiomorpholin-
4-yl)-
methanone (Example 17, step 1). The desired product was obtained in a yield of
99 % as a
white foam. MS (m/e): 556.5 (M+H, 70%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-37-
Example 18 and 19
(S)-2-[2-(1,1-Dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-isoproprl-piperidin-4-
yloxy~-
indol-1-yll-propionitrile and (R)-2-[2-(1,1-dioxo-1,~6-thiomorpholine-4-
carbonyl)-5-(1-
isoproprl-piperidin-4-yloxy)-indol-l-yll-propionitrile
Aracemic mixture ofrac-2-[2-(1,1-dioxo-1,~6-thiomorpholine-4-carbonyl)-5-(1-
isopropyl-piperidin-4-yloxy)-indol-1-yl]-propionitrile (Example 12, 190 mg) in
ethanol (8
mL) was resolved by chiral chromatography using a DAICEL Chiralcel OD with
EthanoUheptane 25:75 v:v. 2 fractions of opposite optical rotation were
isolated and dried
in vacuo.
Fraction 1, negative rotation at 220 nm, 60 mg of a yellow solid, (32%) MS
(m/e):
473.5 (MH+, 100%).
Fraction 2, positive rotation at 220 nm, 84 mg of a yellow solid, (44%) MS
(m/e):
473.5 (MH+, 100%).
Example 20
[5-(1-C, clyl-piperidin-4-yloxy)-1-isopropyl-lH-indol-2-yll-(1,1-dioxo-1',~6-
thiomorpholin-4-yl)-methanone
a) Step 1: 5-Benzyloxy-1-isopropyl-lH-indole-2-carboxylic acid ethyl ester
To a mixture of 5-benzyloxyindole-2-carboxylic acid ethyl ester (30 g, 1.0
eq.) and
cesium carbonate (58. g, 1.75 eq.) in acetonitrile (200 mL), was added
isopropylmethane
sulfonate (24.8 g, 1.75 eq.). The reaction mixture was refluxed for 18h and
then
concentrated in vacuo. The residue was partitioned between water and methyl-
tert-
butylether. The aqueous layer was extracted with methyl-tert-butylether and
the combined
organic layers were washed with brine, dried over sodium sulfate, filtered and
dried in
vacuo. then purified on silica eluting with dichloromethane/ methano198:2 v:v.
The crude
material was purified by crystallization in ethanol and dried in vacuo to
yield 31 g(90 Io) of
the desired product as an off-white oil. MS (m/e): 338.4 (MH+, 100%).
b) Step 2: 5-Benzyloxy-1-isopropyl-lH-indole-2-carboxylic acid
5-Benzyloxy-1-isopropyl-lH-indole-2-carboxylic acid ethyl ester (47.5 g, 1.0
eq.) and
lithium hydroxide (6.56 g, 1.1 eq.) in a mixture of tetrahydofuran (360 mL),
water (180
mL) and methanol (120mL) was refluxed for 2h then the reaction mixture was
concentrated in vacuo. The residue was stirred vigorously in aqueous
hydrochloric acid
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-38-
(2N, final pH: 2). The white precipitate was filtered, washed with water then
dried in vacuo
to yield 41.6 g(96 Io) of the desired product as white oil. MS (m/e): 308.5 (M-
H, 100%).
c) Step 3: 5-Hydroxy-1-isopropyl-lH-indole-2-carboxylic acid
A mixture of 5-benzyloxy-1-isopropyl-lH-indole-2-carboxylic acid (41.6 g, 1.0
eq.)
and palladium on activated charchoal (10% m:m, 4.3 g, 0.03 eq.) in ethyl
acetate (330 mL)
and ethanol (235 mL) was flushed with hydrogen, then vigorously stirred for
2h30 at room
temperature. The resulting back suspension was filtered through a dicalite
pad. The pad
was washed with a mixture of ethyl acetate and ethanol then the liquor was
evaporated in
vacuo to yield 26.3 g (quant.) of the desired product as off-white oil. MS
(m/e): 218.2 (M-
H, 100%).
d) Step 4: (1,1-Dioxo-1,~6-thiomorpholin-4-yl)-(5-hydroxy-1-isopropyl-lH-indol-
2-yl)-
methanone
In analogy to the procedure described for the synthesis of example 1, step 3,
the title
compound was synthesized from 5-Hydroxy-1-isopropyl-lH-indole-2-carboxylic
acid
(Example 20, step 3) and thiomorpholine-1,1-dioxide (purchased at Syntec, ref.
M1201).
The desired product was obtained in a yield of 75 % as white solid. MS (m/e):
335.5 (M-H,
100%).
e) Step 5: 4-[2-(1,1-Dioxo-1,~6-thiomorpholine-4-carbonyl)-1-isopropyl-lH-
indol-5-
yloxy] -piperidine- 1-carboxylic acid tert-butyl ester
To a cold (0 C) mixture of (1,1-dioxo-1,~6-thiomorpholin-4-yl)-(5-hydroxy-1-
isopropyl-lH-indol-2-yl)-methanone (890 mg, 1.0 eq.), 1-tert-butyloxycarbonyl-
4-
hydroxy-piperidine (659 mg, 1.2 eq.) and triphenylphosphine (858 mg, 1.2 eq.)
in
tetrahydrofuran (6 mL) was added dropwise a solution of di-tert-butyl-
azodicarboxylate
(746 mg, 1.0 eq.) in tetrahydrofuran (4 mL). The reaction mixture was stirred
for 15h at
room temperature, concentrated in vacuo, then purified on silica eluting with
a gradient of
cyclohexane / ethyl acetate. One fraction was isolated to yield 434 mg (31.6
Io) of the
desired product as white oil. MS (m/e): 520.7 (MH+, 100%).
f) Step 6: (1,1-Dioxo-l,~6-thiomorpholin-4-yl)-[1-isopropyl-5-(piperidin-4-
yloxy)-1H-
indol-2-yl] -methanone
To a cold (0 C) mixture of 4-[2-(1,1-dioxo-1,~6-thiomorpholine-4-carbonyl)-1-
isopropyl-lH-indol-5-yloxy]-piperidine-1-carboxylic acid tert-butyl ester (410
mg, 1.0 eq.)
in dichloromethane (8 mL) was added dropwise trifluoroacetic acid (920 mg, 10
eq.). The
mixture was stirred overnight at room temperature then concentrated in vacuo.
The
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 39 -
residue was partitioned between a potassium carbonate aqueous solution and
ethyl acetate.
The aqueous layer was extracted with ethyl acetate and the combined organic
layers were
dried over sodium sulfate, filtered and dried in vacuo. The residue was then
purified on
silica eluting with dichloromethane/ methanol / ammoniac 95:5:0.25 v:v:v. to
yield 296 mg
(86%) of the desired product as off-white oil. MS (m/e): 420.4 (MH+, 100%).
g) Step 7: [5-(1-Cyclobutyl-piperidin-4-yloxy)-1-isopropyl-lH-indol-2-yl]-(1,1-
dioxo-1',~6-
thiomorpholin-4-yl)-methanone
To a mixture of (1,1-dioxo-1,~6-thiomorpholin-4-yl)-[1-isopropyl-5-(piperidin-
4-
yloxy)-1H-indol-2-yl]-methanone (268 mg, 1.0 eq.) in acetic acid (115 mg, 3.0
eq.) was
added a solution of cyclobutane (90 mg, 2.0 eq.) in tetrahydrofuran (8 mL).
The mixture
was stirred for 2h at 55 C. Then the mixture was cooled to room temperature
and sodium
triacetoxyborohydride (279 mg, 2.0 eq.) was added. The reaction mixture was
stirred
overnight at 65 C. The residue was partitioned between water and ethyl
acetate. The
organic layer was washed with a sodium hydrogenocarbonate aqueous solution,
dried over
sodium sulfate, filtered and dried in vacuo. Then the residue was purified on
silica eluting
with a gradient of dichloromethane/ methanol to yield 97 mg (32%) of the
desired product
as off-white oil. MS (m/e): 474.5 (MH+, 100%).
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-40-
Example A
Film coated tablets containing the following ingredients can be manufactured
in a
conventional manner:
Inuedients Per tablet
Kernel:
Compound of formula (I) 10.0 mg 200.0 mg
Microcrystalline cellulose 23.5 mg 43.5 mg
Lactose hydrous 60.0 mg 70.0 mg
Povidone K30 12.5 mg 15.0 mg
Sodium starch glycolate 12.5 mg 17.0 mg
Magnesium stearate 1.5 mg 4.5 mg
(Kernel Weight) 120.0 mg 350.0 mg
Film Coat:
Hydroxypropyl methyl cellulose 3.5 mg 7.0 mg
Polyethylene glyco16000 0.8 mg 1.6 mg
Talc 1.3 mg 2.6 mg
Iron oxide (yellow) 0.8 mg 1.6 mg
Titanium dioxide 0.8 mg 1.6 mg
The active ingredient is sieved and mixed with microcrystalline cellulose and
the
mixture is granulated with a solution of polyvinylpyrrolidone in water. The
granulate is
mixed with sodium starch glycolate and magnesiumstearate and compressed to
yield
kernels of 120 or 350 mg respectively. The kernels are lacquered with an
aqueous solution /
suspension of the above mentioned film coat.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-41-
Example B
Capsules containing the following ingredients can be manufactured in a
conventional manner:
Inuedients Per capsule
Compound of formula (I) 25.0 mg
Lactose 150.0 mg
Maize starch 20.0 mg
Talc 5.0 mg
The components are sieved and mixed and filled into capsules of size 2.
Example C
Injection solutions can have the following composition:
Compound of formula (I) 3.0 mg
Gelatine 150.0 mg
Phenol 4.7 mg
Sodium carbonate to obtain a final pH of 7
Water for injection solutions ad 1.0 ml
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
- 42 -
Example D
Soft gelatin capsules containing the following ingredients can be manufactured
in a
conventional manner:
Capsule contents
Compound of formula (I) 5.0 mg
Yellow wax 8.0 mg
Hydrogenated Soya bean oil 8.0 mg
Partially hydrogenated plant oils 34.0 mg
Soya bean oil 110.0 mg
Weight of capsule contents 165.0 mg
Gelatin capsule
Gelatin 75.0 mg
Glycero185 % 32.0 mg
Karion 83 8.0 mg (dry matter)
Titanium dioxide 0.4 mg
Iron oxide yellow 1.1 mg
The active ingredient is dissolved in a warm melting of the other ingredients
and the
mixture is filled into soft gelatin capsules of appropriate size. The filled
soft gelatin capsules
are treated according to the usual procedures.
CA 02630307 2008-05-16
WO 2007/062997 PCT/EP2006/068649
-43-
Example E
Sachets containing the following ingredients can be manufactured in a
conventional
manner:
Compound of formula (I) 50.0 mg
Lactose, fine powder 1015.0 mg
Microcrystalline cellulose (AVICEL PH 102) 1400.0 mg
Sodium carboxymethyl cellulose 14.0 mg
Polyvinylpyrrolidone K 30 10.0 mg
Magnesium stearate 10.0 mg
Flavoring additives 1.0 mg
The active ingredient is mixed with lactose, microcrystalline cellulose and
sodium
carboxymethyl cellulose and granulated with a mixture of polyvinylpyrrolidone
in water.
The granulate is mixed with magnesium stearate and the flavoring additives and
filled into
sachets.