Note: Descriptions are shown in the official language in which they were submitted.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
TREATMENT OF QT INTERVAL PROLONGATION AND DISEASES
ASSOCIATED THEREWITH
Field of the Invention
The present invention relates to cardiology and, in particular, methods for
treating
QT interval prolongation and diseases associated therewith, such as, but not
limited to,
congenital long QT syndrome, acquired long QT syndrome, rriyocardial ischemia,
heart
failure, diabetes or stroke.
Background of the Invention
Ion channels are macromolecular aqueous protein tunnels that span cell
membranes. A vast number of ion channels are known to exist. These channels
generate
and orchestrate a variety of electrical signals that pass through the brain,
heart and muscle
each second of life. Ion channels are classified based on the type of ion that
they allow to
pass - e.g. sodium, potassium, calcium or chloride and their gating
properties. Often
times there are different channels for each type of ion. The direction of
ionic movement
in an ion channel is governed by electrical and chemical concentration
gradients. In
many channels the movement of ions is controlled by gating structures that
form the basis
for a broad classification of gated ion channels into mechano-, voltage- and
ligand-gated
subtypes. Thus, ion channels are not opened continuously. Instead, they have
"gates"
which open briefly and then close again. The synchronized activity of gated
ion channels
within individual cells produces the complex and vital voltage waveforms
characteristic
of excitable tissues.
During the resting stage, cells maintain an electrical potential difference
(voltage)
across their cell membranes depending on whether the ion channels on their
cell
membrane are open or closed. Typically, the interior of cells (cytoplasm) is
electrically
negative relative to the extracellular fluid, so the cells are polarized
during the restiiig
stage. This electrical potential difference across the cell membrane is called
the resting
membrane potential. For cardiac muscle cells, the resting membrane potential
is about
-90 mV.
1
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Electrically excitable cells become excited when they are exposed to different
stimuli which can cause the ion channels to open or close. The main types of
stimuli that
are known to change (or gate) ion channel activity are a change in the voltage
across the
membrane (i.e., voltage-gated channels), a mechanical stress (i.e.,
mechanically gated
channels) or the binding of a ligand (i.e., ligand-gated channels). When a
cell is excited,
it undergoes a cycle of transmembrane potential change which is referred to as
the action
potential.
In the heart, the action potential in a heart ventricular cell comprises five
phases.
Phase 0 is the rapid depolarization phase when the cell membrane rapidly
transits from
the negative resting potential to a positive potential due to an almost
exclusive influx of
positively charged sodium ions into the cell. This influx causes the membrane
potential
to become positive. Phase 0 is also referred to as the "upstroke" of the
action potential
because it lasts less than one millisecond and is the fastest phase. During
depolarization,
the potential difference is actually reversed, so that the potential of the
cytoplasm exceeds
that of the extracellular fluid by about 20 mV. The upstroke is immediately
followed by a
brief period of partial, or early repolarization (Phase 1) which is mediated
mainly by the
transient efflux of potassium ions, which is then followed by a plateau phase
(Phase 2).
During the plateau phase, there is an influx of positively charged calcium
ions which is
counterbalanced by the efflux of positive charged potassium ions. Following
the plateau
phase, the membrane repolarizes (Phase 3) back to the resting state of
polarization (i.e,
the change in membrane potential back to an negative value after
depolarization). This
final repolarization occurs when the efflux of potassium ions begins to exceed
the influx
of calcium ions. The Phase 3 repolarization develops more slowly than does the
depolarization Phase 0. The potassium currents through the potassium channels
play a
major role in determining the duration of Phase 3 and thus, the duration of
the action
potential. The last phase of the action potential (Phase 4) is silent in terms
of the
membrane potential changes. Phase 4 is the phase during which the ionic
concentrations
are restored via the elimination of the sodium and calcium ions that entered
into the cell
in exchange with the potassium ions that exited the cell during the action
potential.
2
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Each action potential of a cardiac muscle cell causes a contraction of that
cell.
The contraction of all the cardiac muscle cells in concert forms a coordinated
heart
contraction or a heart beat. At the same time, an integrated electrical signal
(action
potentials) from all the cardiac muscle cells is emitted to the surface of the
body. This
signal can be recorded on an electrocardiogram (such as an ECG or EKG) which
produces a characteristic wave form. The different parts of the wave form are
designated
by the letters - P, Q, R, S and T, which represent sum of the action
potentials from
different regions of the heart. Certain intervals of time between the
different parts of the
wave provide valuable information about the condition of the heart. For
example, the
period of time from the beginning of the QRS complex of the wave to the end of
the T
wave (known as the "QT" interval) provides a measure of the duration of
ventricular
depolarization and repolarization. In other words, it is a measurement of the
duration of
cardiac ventricular action potential.
Unfortunately, in some individuals, the duration of the QT interval is
prolonged.
Prolongation of the QT interval, which is clinically referred to as "long QT
syndrome"
(hereinafter "LQTS"), has been associated with increasing the risk of certain
medical
conditions, such as ventricular tachyarrhythmia, particularly, torsade de
pointes, which
can lead to sudden cardiac death. QT interval prolongation or an increase in
action
potential duration can result from an increase of the inward (or influx)
sodium or calcium
currents, or inhibition of one or more of the outward (or efflux) potassium
currents. Two
of the potassium channels involved in the Phase 3 repolarization of the heart
are referred
to as the rapidly and slowly activating components of the delayed rectifier
potassium
channels, IK, and IKS. These channels have a significant role in determining
the duration
of the action potential and thus, the QT interval. ' Any defect or blockage of
either of
these channels slows the repolarization Phase 3, thereby prolonging the
duration of the
action potential and the QT interval. The rapidly delayed rectifier potassium
channel IKr
is encoded by the human ether-a-go-go-related gene (hereinafter referred to as
"hERG").
Thus, the channel is also known as a"hERG" channel. The prolongation of the QT
interval is generally believed to result from one or more genetic defects
(which are
referred to as "congenital LQTS") in these ion channels, or through the action
of one or
3
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
more drugs (which is referred to herein as "acquired LQTS"). Nonetheless,
despite this,
prolonged QT interval has been found in a number of cardiovascular or other
related
diseases, including myocardial ischemia (Puddu, et al., Journal of
Electrocardiology,
19(3):203-11 (1986)), heart failure (Brooksby et al., European Heart Journal,
20(18):1335-41 (1999)), diabetes (Veglio et al., Journal ofIrzternal Medicine,
251(4):317-24 (2002)) and stroke (Wong et al., Heart (British Cardiac
Society),
89(4):377-81 (2003)), all of which are associated with high rates of death.
The measurement and determination of baseline QT intervals has been shown,
albeit not consistently, to be a prognostic indicator of the mortality in
patients with
myocardial ischemia, heart failure and stroke. This suggests that QT
prolongation may
be a factor for mortality associated with the above mentioned diseases.
Indeed,
electrophysiological studies using isolated myocytes from heart failure
patients
(Beuckelmann, et al., Circulation Research, 73(2):379-85 (1993)) and from
animals with
myocardial infarction (Kaprrielian et al., American Journal of Physiology,
283:H1157-
Hl 168 (2002)) and experimentally induced heart failure (Despa et al.,
Circulation,
105:2543-2548 (2002); Rose et al., American Journal ofPhysiology, 288:H2077-
H2087
(2005)) have shown that currents through the transient outward (Ito), the
inward rectifier
(IKl) K+ channels, and delayed rectifier currents can be reduced as compared
to healthy
normal controls (Janse, Cardiovascular Research, 61: 208-217 (2004)).
Treatment options for congenital LQTS include reduction of the QT interval
directly and indirectly through (3-blocker therapy, cardiac pacing and
implantable
cardioverter defibrillators, (Ackerman, M.J., Mayo Clin. Proc., 73:250-269
(1998);
Wehrens et al., Ann. Intern. Med., 137:981-992 (2002); Khan, Am. Heart J.
143:7-14
(2002)). Pharmacological modulation of ion channels has had some success.
Sodium
channel blockers can reduce the QT interval directly in patients with LQT3
since QT
prolongation is due to a defect in sodium channel inactivation causing the
mutated
sodium channels to be overactive during the cardiac action potential plateau.
This "gain
of function" can be reversed pharmacologically with sodium channel blockers
such as
mexiletine and flecainide (Schwartz et al., Circulation, 92:3381-3386 (1995);
Wang et
4
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
al., J. Clin. Invest. 99:1714-1720 (1997); Windle et al., Electrocardiol.,
6:153-158
(2001); Liu et al., J. Pharrnacogenonaics, 3:173-179 (2003)). This approach is
mechanism based and effective but limited to the minority of congenital LQTS
patients
with LQT3. However, the potential for cardiac toxicity is well established for
sodium
channel blockers (which are referred to as Class I anti-arrhythmic drugs) due
to their
slowing of conduction in potential reentrant circuits and triggering
arrhythinias (Nattel,
Cardiovasc. Res., 37:567-577 (1998)). Methods based on increasing
extracellular
potassium concentration, such as intravenous (i.v.) infusion of potassium,
shorten the QT
interval by increasing the activity of repolarizing potassium channels. QT
intervals have
been significantly shortened in patients that received this treatment (Compton
et al.,
Circulation, 94:1018-1022 (1996)). However, this therapy is not widely used
because it
requires incontinent i.v. infusion and achieving sufficiently high long-term
potassium
levels is difficult (Etheridge et al., J. Am. Coll. Cardiol., 42:1777-1782
(2003)). The
ATP-sensitive potassium (KATP) channel opener, nicorandil, has been shown to
normalize
the congenitally prolonged QT interval in patients (Shimizu et al., Curr.
Pharna. Des.
11:1561-1572 (2005)). However, KATp channel openers are all associated with
unwanted
vasodilating activities (e.g. hypotension) due to the presence of KATP
channels not only in
the heart, but also in vascular smooth muscle (Quast et al., Cardiovasc. Res.,
28:805-810
(1994)). Therapies targeting ion channels directly can be successful, as is
the case for
LQT3, and are likely to be useful generally.
It is also known in the art that intracellular calcium overloads contribute to
myocardial ischemia injury (Farber J.L., Laboratory Investigation, 47(2):114-
23 (1982)).
Increased cytosolic calcium concentrations (calcium overload) result in
irreversible
increases in the resting tension of the cardiac muscle and thus prevent normal
relaxation
of the heart (Lowe et al., Jour nal of Molecular= & Cellular Cardiology,
11(10):1017-31
(1979)). The opening of potassium channels could provide cardioprotection via
shortening the action potential plateau and speeding repolarization, thus
leading to a
reduction of the influx of calcium through L-type calcium channels. Efforts
have been
made to identify potassium channel openers that are safe and effective for the
treatment
of myocardial ischemia (Gomma et al., Drugs, 61(12):1705-10 (2001)). So far
only KATP
openers have been pursued. As discussed above, the prevalence of KA-rp
channels in non-
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
target tissues increases the likelihood of unwanted side effects with these
agents.
Moreover, KATP openers can significantly shorten QT intervals and be
arrhythmogenic
under certain circumstances.
Reversing QT prolongation or calcium overload by increasing the activity of
Iic,
potassium channels may also be beneficial in treatment of the acquired and
inherited
LQTS, myocardial ischemia, heart failure, diabetes and stroke. Two recent
reports (Kang
et al., Mol. Pharmacol., 67:827-836 (2005); Zhou et al., Mol. Pharniacol.,
68:876-884
(2005)) describe drug molecules that act as hERG agonists and may permit
development
of a drug therapy for reversal of prolonged QT. However, micromolar
concentrations of
these compounds are required to increase hERG current (which may not be
achievable in
human after oral dosing) and they shorten normal action potential duration and
QT
intervals (which may translate into a proarrhythmic risk).
Thereupon, there is a need in the art for pharmaceutically acceptable
potassium
channel openers that can be used to safely shorten prolonged QT intervals and
reduce the
calcium overload. Such agents would be usefia.l in treating a variety of
cardiovascular or
other related diseases such as LQTS, heart failure, diabetes, stroke,
myocardial ischemia
etc.
Summary of the Present Invention
In one embodiment, the present invention relates to methods for shortening a
QT
interval in a patient suffering from QT prolongation. The methods involve the
following
steps:
administering to a patient suffering from QT prolongation a therapeutically
effective amount of at least one pharmaceutically acceptable human ether-a-go-
go-
related gene ("hERG") channel agonist wherein said at least one hERG channel
agonist
does not shorten the QT interval when administered to a patient that is not
suffering from
QT prolongation.
6
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
The patient suffering from QT prolongation to be treated by the above method
may be suffering from congenital long QT syndrome, acquired long QT syndrome,
myocardial ischemia, heart failure, diabetes or stroke.
The administration of the hERG channel agonist in the above-described method
increases the currents of the hERG channel in said patient. Specifically, the
increase in
the current of the hERG channel was found to be voltage dependent. More
specifically, it
was found that the at least one hERG channel agonist increases the current of
the hERG
channel at a positive transmembrane potential. This positive transmembrane
potential is
between about +0.1 mV and about +50 mV, preferably between about +5 mV and
about
+30 mV and most preferably from about +10 mV and about +20 mV.
hERG channel agonists that can be used in the above-described method include
those having the following formula:
R4
::x::
R1
wherein Rl is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
an unsubstituted or substituted Cl-Clo haloalkyl;
7
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
an unsubstituted or substituted Cl-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)nR, where n is an integer from 0 to 5; or
NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted C1-C10 alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-C10 alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted Cl-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7 -
membered
cyclic amino group.
wherein R2 is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
an unsubstituted or substituted C1-Clo haloalkyl;
an unsubstituted or substituted CI-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
T7n ~p' ,
iVi~t~ ,
where R or R' is each independently a hydrogen, a unsubstitated or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
8
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Cio alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R3 is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted Cl-Clo alkyl;
an unsubstituted or substituted Cl-Clo haloalkyl;
an unsubstituted or substituted C1-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
NRR' .
~
where R or R' is each independently a hydrogen, a unsubstituted or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R5 is
a hydrogen;
9
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
an unsubstituted or substituted Cl-Clo haloalkyl;
an unsubstituted or substituted C1-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein Rb is
S R7 R7
N R7
~ / \ I
N
R8 Rs or Ra
R7 is hydrogen, a C1-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a C1-
C5 alkoxycarbonyl, carbamoyl or C1-C4 alkyl aminocarbonyl group; and
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
R8 is hydrogen, a Ci-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a Cl-
C5 alkoxycarbonyl, carbamoyl or Cl-C4 alkyl aminocarbonyl group.
Examples of hERG channel agonists having the above fonnula are selected from
the group consisting of: 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-
methylthiazole-5-
carboxylic acid, 2-[3-cyano-4-(3-hydroxy-2-methylpropoxy)phenyl]-4-methyl-5-
thiazolecarboxylic acid, 2-[3-cyano-4-(2-hydroxy-2-methylpropoxy)phenyl]-4-
methyl-5-
thiazolecarboxylic acid, 2-(3-cyano-4-hydroxyphenyl)-4-methyl-5-
thiazolecarboxylic
acid, 2-[4-(2-carboxypropoxy)-3-cyanophenyl]-4-methyl-5-thiazolecarboxylic
acid and a
pharmaceutically acceptable salt thereof.
In another embodiment, the present invention relates to a method of treating a
patient suffering from myocardial ischemia, heart failure, diabetes or stroke.
The method
involves the steps of:
administering to a patient suffering from myocardial ischemia, heart failure,
diabetes or stroke a therapeutically effective amount of at least one
pharmaceutically
acceptable human ether-a-go-go-related gene ("hERG") channel agonist wherein
said at
least one hERG channel agonist does not shorten the QT interval when
administered to a
patient that is not suffering from QT prolongation.
The'administration of the hERG channel agonist in the above-described method
increases the currents of the hERG channel in said patient. Specifically, the
increase in
the current of the hERG channel was found to be voltage dependent. More
specifically, it
was found that the at least one hERG channel agonist increases the current of
the hERG
channel at a positive transmembrane potential. This positive transmembrane
potential is
between about +0.1 mV and about +50 mV, preferably between about +5 mV and
about
+30 mV and most preferably from about +10 mV and about +20 mV.
hERG channel agonists that can be used in the above-described method include
those having the following formula:
11
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Rq.
::x::
Ri
wherein Rl is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
an unsubstituted or substituted Ct-Clo haloalkyl;
an unsubstituted or substituted Cl-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)nR, where n is an integer from 0 to 5; or
NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted Cl-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted Cl-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted CI-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
12
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
wherein R2 is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
an unsubstituted or substituted CI-Clo haloalkyl;
an unsubstituted or substituted C1-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
NRR' =
~
where R or R' is each independently a hydrogen, a unsubstituted or
substituted Cl-C10 alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted Cl-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted Cl-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstitated aralkyl'amino group, or a 5- to 7 -
membered
cyclic amino group.
wherein R3 is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted C1-Clo alkyl;
13
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
an unsubstituted or substituted C1-Cro haloalkyl;
an unsubstituted or substituted C1-Clo alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
NRR';
where R or R' is each independently a hydrogen, a unsubstitated or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted Cj-Clo alkyl, aryl or aralkyl.group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted Ci-Cto alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R5 is
a hydrogen;
a carboxyl;
a halogen atom;
a nitro group;
a cyano group;
a formyl group;
an unsubstituted or substituted Cz-Clo alkyl;
an unsubstituted or substituted Cl-Clo haloalkyl;
an unsubstituted or substituted Cl-Cz0 alkoxy;
an unsubstituted or substituted hydroxyalkoxy;
OR;
S(O)õR, where n is an integer from 0 to 5; or
NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
14
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-C10 alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R6 is
S R7 R
N R7 7
R8 Rs or Re
R7 is hydrogen, a C1-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a C1-
CS alkoxycarbonyl, carbamoyl or C1-C4 alkyl aminocarbonyl group; and
R8 is hydrogen, a C1-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a C1-
C5 alkoxycarbonyl, carbamoyl or Cr-C4 alkyl aminocarbonyl group.
Examples of hERG channel agonists having the above formula are selected from
the group consisting of: 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-
methylthiazole-5-
carboxylic acid, 2-[3-cyano-4-(3-hydroxy-2-methylpropoxy)phenyl]-4-methyl-5-
thiazolecarboxylic acid, 2-[3-cyano-4-(2-hydroxy-2-methylpropoxy)phenyl]-4-
methyl-5-
thiazolecarboxylic acid, 2-(3-cyano-4-hydroxyphenyl)-4-methyl-5-
thiazolecarboxylic
acid, 2-[4-(2-carboxypropoxy)-3-cyanophenyl]-4-methyl-5-thiazolecarboxylic
acid and a
pharmaceutically acceptable salt thereof.
Brief Description of the Drawinjzs
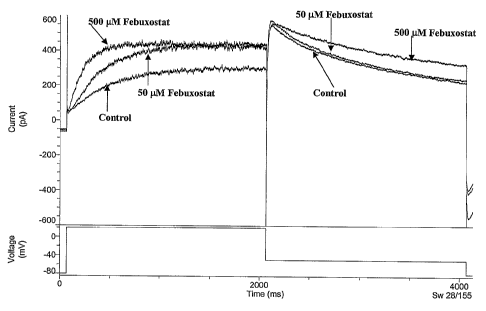
Figure 1 shows sample HEK/hERG current trace before and after 50 and 500 M
application of 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-
carboxylic
acid (hereinafter referred to as "febuxostat"). HEK/hERG currents [Current
(pA); Time
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
(ms)] were obtained using the voltage procedure [Voltage (mV)] described for
concentration-response (shown in the lower panel). Current records in the
presence of
febuxostat were obtained after three minutes of equilibration at the indicated
concentration.
Figure 2 shows a sample HEK/hERG current trace before and after 0.1 and 1 M
febuxostat application. HEK/hERG currents [Current (pA); Time (ms)] were
obtained
using the voltage procedure [Voltage (mV)] described for concentration-
response (shown
in the lower panel). The current records in the presence of 0.1 and 1 M
febuxostat was
obtained after at least three minutes of exposure to febuxostat.
Figure 3 shows the sample time course of HEK/hERG current measured before,
during and after 50 M febuxostat application at +20 mV.
Figure 4 shows the use- or frequency-dependence of febuxostat effect on
HEKIHERG peak tail current. Before and after 500 M febuxostat equilibration,
repetitive test pulses at frequencies 0.3 Hz (Top Panel) and 3.0 Hz (Bottom
Panel) were
applied. Current amplitudes were normalized to the first pulse and plotted
against time.
Data are the average of two cells.
Figure 5 shows sample CHO/hERG current trace before and during febuxostat
application. CHO/hERG currents [Current (pA); Time (ms)] were obtained using
the
voltage procedure described for concentration-response and is shown in the
lower panel.
The steady state effect record was obtained 7 minutes after the start of
febuxostat
application.
Figure 6 shows a sample time course of CHO/bERG current measured before and
after 1 M febuxostat application at +20 mV. The cell was superfused with
Tyrode's
solution from an array of three capillary tubes placed adjacent to the cell.
To control for
solution flow artifacts, control solution was switched between two capillary
tubes
(Control 1 and Control 2) before switching to febuxostat containing solution.
16
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Figure 7 shows a sample time course of CHO/hERG current measured before and
after 0.1 M febuxostat application at +20 mV. The cell was superfused with
Tyrode's
solution from an array of three capillary tubes placed adjacent to the cell.
To control for
solution flow artifacts, control solution was switched between two capillary
tubes
(Control 1 and Control 2) before switching to febuxostat containing solution.
Figure 8 shows the concentration-response of the initial maximum effect of
febuxostat (also known as "TMX-67") on hERG current at +20 mV. The mean
fractional
currents present after application of febuxostat (circles) S.E.M. were fit
to a simple
binding equation (Solid Line). The calculated EC50 was 0.003 M. Number of
observations is shown in parentheses.
Figure 9 shows the concentration-response of the steady state effect of
febuxostat
on hERG current at +20 mV. The mean fractional currents present after
application of
febuxostat (circles) S.E.M. were fit to a simple binding equation (Solid
Line). The
calculated EC50 was 0.070 M. Number of observations is shown in parentheses.
Figure 10 shows the use-dependence of febuxostat agonist effect measured at
+60
mV. Before and after 1 M febuxostat equilibration repetitive test pulses at
frequencies
of 0.3 Hz (Top Panel) and 3.0 Hz (Bottom Panel) were applied. The train of
pulses was
generated by repetition of this step waveform: depolarization +60 mV for 250
ms;
repolarization: -50 mV for 70 ms; followed by return to the holding potential
of -80 mV.
Peak current amplitudes were measured at the onset of the +60 mV step. Peak
currents
were normalized to the train second pulse amplitude in control and in
febuxostat solution
so that steady state drug effects before the start of the train did not
overlap the frequency
dependent effects. Normalized currents were plotted against time. Data are the
average
of three cells.
17
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Figure 11 shows the use-dependence of febuxostat agonist effect measured at -
50
mV. Before and after 1 M febuxostat equilibration repetitive test pulses at
frequencies
of 0.3 Hz (Top Panel) and 3.0 Hz (Bottom Panel) were applied. The train of
pulses was
generated by repetition of this step waveform: depolarization +60 mV for 250
ms;
repolarization: -50 mV for 70 ms; followed by return to the holding potential
of -80 mV.
Peak tail current amplitudes were measured at -50 mV following channel
activation and
inactivation at +60 mV. Peak tail currents were normalized to the train first
pulse
amplitude in control and in febuxostat solution so that steady state drug
effects before the
start of the train did not overlap the frequency dependent effects. Normalized
currents
were plotted against time. Data are the average of three cells.
Figure 12 shows the effect of febuxostat on voltage-dependence of activity and
steady state I-V relation. Current values (Mean S.E.M.) measured in 3 cells
at the end
of the 4-second activating voltage step in control and after equilibration
with 1 M
febuxostat are plotted for each voltage step. Data were normalized to the
maximum
value in control for each cell.
Figure 13 shows the effect of febuxostat on the steady state I-V current
records.
Each panel shows 16 superimposed current records from a CHO/hERG cell produced
by
the voltage protocol diagrammed below the currents in control (upper panel)
and 1 M
febuxostat (lower panel).
Figure 14 shovvs the steady state G-V relation in control and febuxostat
treated
cells. Normalized conductance measured from peak tail current amplitude values
(Mean:L S.E.M.) in 3 CHO/bERG cells during the -50 mV repolarizing voltage
step of
the steady-state I-V relation protocol. Measurements in control and after
equilibration
with 1 M febuxostat are plotted for each voltage during the preceding
variable voltage
step. Data in control and in febuxostat were fit to a Boltzmann equation of
the form
Normalized Current =1/(l+e-(V-Vli2)/Kv)
18
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
where V is the voltage of the 4 second activating voltage step of the steady-
state I-V
relation protocol preceding the -50 mV repolarizing step, V112 is the
potential at which
half maximal conductance occurs, and K, is the exponential slope factor
setting the
steepness of the curve. Values for Vli2 in control and febuxostat were 0.9 and
-2.1 mV,
respectively. Values for Kv in control and febuxostat were 9.9 and 9.8 mV,
respectively.
Figure 15 shows the fully activated I-V relation in control and febuxostat
treated
cells. Normalized peak current values (Mean S.E.M.) measured in 3 cells in
control
and after equilibration with 1 RM febuxostat are plotted for each voltage
step. Peak
current measurements were made during the second, 5-second duration variable
voltage
step of the voltage protocol. Data were normalized to the maximum value in
control for
each cell.
Figure 16 shows the effect of febuxostat on the fully activated I-V current
records.
Each panel shows 15 superimposed current records from a CHO/hERG cell produced
by
the voltage protocol diagrammed below the currents in control (upper panel)
and 1 M
febuxostat (lower panel).
Figure 17 shows the effect of febuxostat on voltage-dependence of
inactivation.
Normalized channel availability values (Mean zL S.E.M.) measured in 3 CHO/hERG
cells
in control and after equilibration with 1 M febuxostat are plotted for each
voltage step.
Peak current measurements were made during the second, 1 second duration,
variable
voltage step of the voltage protocol. Data were normalized to the maximum
value in
control for each cell. Data were fit to an equation of the form:
Channel Availability = 1/(l+e(V-Vli2)/K,))
where V is the voltage of the variable voltage step in the protocol, V1/2 is
the voltage for
half-maximal channel availability, Kv is the exponential slope factor setting
the steepness
of the curve. Values for V1i2 in control and febuxostat were -68 and -67 mV,
19
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
respectively. Values for K, in control and febuxostat were 28 and 30 mV per e-
fold
change, respectively. The fraction of channels inactivated is "1- channel
availability".
Figure 18 shows the effect of febuxostat on the alternate I-V relation. Each
panel
shows 17 superimposed current records from a CHO/hERG cell produced by the
voltage
protocol diagrammed below the currents in control (upper panel) and 1 M
febuxostat
(lower panel).
Figure 19 shows the effect of febuxostat on instantaneous I-V relation.
Current
values (Mean S.E.M.) measured in 3 cells at the beginning of the 1 second
variable
voltage step in the alternate I-V relationship voltage protocol, in control
and after
equilibration with 1 M febuxostat, are plotted for each voltage step. Data
were
normalized to the 0 mV value in control for each cell.
Figure 20 shows the effect of febuxostat on action potential. Superimposed
records before (control) and after equilibration with increasing
concentrations of
febuxostat (10, 100 and 1000 nM). Febuxostat did not cause significant changes
in any
of the action potential parameters. Temperature was maintained at 37 :L 1 C
and the BCL
was set to 2s.
Figure 21 shows the effect of 50 M sotalol on action potential. Superimposed
records before (control) and after equilibration of 50 M sotalol at 23 min
(1), 50 M
sotalol at 46 min (2) and 50 M sotalol at 69 min (3). Temperature = 37 1
C, BCL =
2s. Sotalol significantly prolonged APD.
Figure 22 shows the effect of 20 nM ATX II on action potential.. Superimposed
records before (control) and after equilibration of 20 nM ATX II at 23 min
(1), at 46 min
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
(2) and at 69 min (3). Temperature = 37 1 C, BCL = 2s. ATX II significantly
prolonged APD.
Figure 23 shows the effects of febuxostat (also known as TMX) and sotalol on
action potential duration. Percent change in APD90 (BCL = 2s) relative to
baseline was
plotted versus exposure period. In the febuxostat group (open diamonds, n = 4)
10, 100
and 1000 nM febuxostat concentrations were applied cumulatively during
exposure
periods 1, 2 and 3, respectively. In the sotalol group (filled squares, n =
4), 50 M sotalol
was applied continuously throughout exposure periods 1, 2 and 3. In the
coinbined
sotalol and febuxostat group (open triangles, n = 4) 50 M sotalol, 50 M
sotalol + 100
nM febuxostat, and 50 M sotalol + 1000 nM febuxostat were applied
cumulatively
during exposure periods 1, 2 and 3, respectively. The sotalol data was
overlaid on the
sotalol + febuxostat data by normalization of the sotalol data by the sotalol
+ febuxostat
exposure period 1 value. The normalized sotalol data (crosses) overlays the
period 2 and
3 sotalol + febuxostat data, indicating that febuxostat at 100 and 1000 nM
concentration
had no effect on the time course of sotalol APD90 prolongation.
Figure 24 shows the effect of febuxostat on ATX II-induced APD90 prolongation.
Measurements were made at BCLs of 2 s (A), 1 s (B) and 0.34 s (C). The percent
change
in APD90 (Mean SEM) at each BCL was plotted versus exposure period during
sequential applications of 20 nM ATX II + 100 nM febuxostat (exposure periods
1 and
2), 20 nM ATX II + 1000 nM febuxostat (exposure period 3). Data graphed with
diamond, triangle and square symbols were obtained at BCLs of 2, 1 and 0.34 s,
respectively. Filled and open symbols represented data obtained in ATX II
alone (n = 4
fibers) and ATX II + febuxostat (n= 7 fibers). *Statistically significant
difference
between ATX II and ATX II + febuxostat groups (P<0.05, Student's t-test).
21
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Detailed Description of the Invention
As used in this specification and the appended claims, the singular forms "a,"
"an"
and "the" include plural references unless the context clearly dictates
otherwise. Thus, for
example, reference to "an active agent" includes a single active agent as well
two or more
different active agents in combination.
Definitions
In describing and claiming the present invention, the following terminology
will
be used in accordance with the definitions set out below.
The term "acquired LQTS" refers to the prolongation of the QT interval in a
patient that is believed to be the result of the action of one or more drugs.
The terms "administer", "administering", "administered" or "administration"
refer
to any manner of providing a drug to a subject or patient. Routes of
administration can
be accomplished through any means known by those skilled in the art. Such
means
include, but are not limited to, oral, buccal, intravenous, subcutaneous,
intramuscular, by
inhalation and the like.
The term "congenital LQTS" refers to the prolongation of the QT interval in a
patient that is believed to be the result of one or more genetic defects.
The term "dosage form" refers to any solid object, semi-solid, or liquid
pharmaceutical composition designed to contain a specific pre-determined
amount (i.e.
dose) of a certain active ingredient (i.e, at least one hERG channel agonist).
Suitable
dosage forms may be pharmaceutical drug delivery systems, including those for
oral
administration, buccal administration, rectal administration, topical or
mucosal delivery
or subcutaneous implants, or other implanted drug delivery systems and the
like.
Preferably, the dosage form of the pharmaceutical composition of the present
invention is
considered to be solid, however, they may containing liquid or semi-solid
components.
22
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
More preferably, the dosage form is an orally administered system for
delivering an
active ingredient to a patient.
By an "effective amount" or a "therapeutically effective amount" of an active
ingredient (i.e., at least one hERG channel agonist) is meant a nontoxic but
sufficient
amount of the active ingredient to provide the desired effect. The amount of
active
ingredient that is "effective" will vary from subject to subject, depending on
the age and
general condition of the individual, the particular active ingredient or
active ingredient,
and the like. Thus, it is not always possible to specify an exact "effective
amount."
However, an appropriate "effective amount" in any individual case may be
determined by
one of ordinary skill in the art using routine experimentation.
The term "human ether-a-go-go-related gene ("hERG") channel agonist" refers to
a compound, peptide, active ingredient or drug that potentiates or increases
the current in
a hERG channel in the heart of a patient that is suffering from QT
prolongation thereby
reversing or shortening the QT interval in said patient. Additionally, the
hERG channel
agonist used in the methods of the present invention does not shorten the QT
interval
when administered to a patient that is not suffering from QT prolongation.
Examples of hERG channel agonists that can be used in the present invention
are
those compounds having the below formula I:
R4
::::
R1 Formula I
wherein Rl is
23
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
a hydrogen; a carboxyl; a halogen atom; a nitro group; a cyano group; a
formyl group; an unsubstituted or substituted C1-Clo alkyl; an unsubstituted
or
substituted Ct-Cto haloalkyl; an unsubstituted or substituted Cl-Cza alkoxy;
an
unsubstituted or substituted hydroxyalkoxy; OR; S(O)õR, where n is an integer
from 0 to
5; or NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted Cl-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted Cl-Cio alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted Cl-Cto alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted Cl-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R2 is
a hydrogen; a carboxyl; a halogen atom; a nitro group; a cyano group; a
formyl group; an unsubstituted or substituted Cl-Clo alkyl; an unsubstituted
or
substituted Cl-Clo haloalkyl; an unsubstituted or substituted C1-Clo alkoxy;
an
unsubstituted or substituted hydroxyalkoxy; OR; S(O)nR, where n is an integer
from 0 to
5; or NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted Cl-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted C1-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
24
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
wherein R3 is
a hydrogen; a carboxyl; a halogen atom; a nitro group; a cyano group; a
formyl group; an unsubstituted or substituted C1-Clo alkyl; an unsubstituted
or
substituted C1-Clo haloalkyl; an unsubstituted or substituted CI-Clo alkoxy;
an
unsubstituted or substituted hydroxyalkoxy; OR; S(O)nR, where n is an integer
from 0 to
5; or NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted Cl-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted C1-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted Cl-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted C1-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7-
membered
cyclic amino group.
wherein R5 is
a hydrogen; a carboxyl; a halogen atom; a nitro group; a cyano group; a
formyl group; an unsubstituted or substituted Cl-C10 alkyl; an unsubstituted
or
substituted C1-Clo haloalkyl; an unsubstituted or substituted C1-Clo alkoxy;
an
unsubstituted or substituted hydroxyalkoxy; OR; S(O)õR, where n is an integer
from 0 to
5; or NRR';
where R or R' is each independently a hydrogen, a unsubstituted or
substituted C1-Clo alkyl, aryl, aralkyl, alkylcarbonyl, arylcarbonyl or
aralkylcarbonyl
group or where R and R' taken together with a nitrogen atom bonded thereto
form an
unsubstituted or substituted 5- to 7- membered heterocyclic ring; or COR"
where R" is a
unsubstituted or substituted Cl-Clo alkyl, aryl or aralkyl group, a hydroxyl
group; a
unsubstituted or substituted Cl-Clo alkoxy, aryloxy, aralkyloxy, an amino
group, an
unsubstituted or substituted Cl-Clo alkyl amino, an unsubstituted or
substituted aryl
amino group, a substituted or unsubstituted aralkyl amino group, or a 5- to 7 -
membered
cyclic amino group.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
wherein R6 is
S R7 R7
'ii>'i:: Rs or Rs
R7 is hydrogen, a C1-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a Cl-
C5 alkoxycarbonyl, carbamoyl or C1-C4 alkyl aminocarbonyl group; and
R8 is hydrogen, a C1-C4 alkyl, carboxyl, COO-Glucoronide or COO-Sulfate, a C1-
C5 alkoxycarbonyl, carbamoyl or Cl-C4 alkyl aminocarbonyl group.
In formula I above, substituents which may have further substituent(s),
namely, a
pyridyl, thienyl, fia.ryl or naphthyl group; CI-Clo alkyl, aryl, aralkyl,
alkylcarbonyl,
arylcarbonyl or aralkylcarbonyl group; 5- to 7- membered heterocyclic ring; C1-
Clo
alkoxy, aryloxy or aralkyloxy group; a unsubstituted or substituted
hydroxyalkoxy; and
C1-CIo alkyl (mono- or di-substituted) amino, aryl (mono- or disubstituted)
amino group,
on chain or cyclic moiety thereof, can be substituted by one or more of: a Cl-
C4
halogenated alkyl, carboxyl, alkylcarbonyl, alkyloxy, alkylcarbonyloxy,
hydroxyl, mono-
or di-substituted alkylamino, amino, nitro, cyano or formyl group, or halogen
atom,
heterocyclic ring such as 5- to 7- membered cyclic-secondary amino group, etc.
Preferred substituents are a halogen atom, methyl group, ethyl group, methoxy
group and
ethoxy group.
As used herein, the teml "Cl-C4 alkyl" refers to a methyl group, ethyl group,
propyl (iso- or n-) group and butyl (iso-, n-, tert- or sec-) group.
As used herein, the term "C1-C4 alkyl aminocarbonyl" refers to a group
comprising an alkyl group of one to four carbon atoms and an aminocarbonyl
group.
26
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
As used herein, the term " unsubstituted or substituted C1-Cio alkyl" group
refers
to a C1-Clo straight-chain or branched aliphatic hydrocarbon residue, cyclic
aliphatic
hydrocarbon residue or chain-cyclic aliphatic hydrocarbon residue which can be
mono-
or di-substituted. Examples include, but are not limited to, methyl, ethyl, n-
propyl, iso-
propyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, n-pentyl, iso-pentyl, neo-
pentyl, n-hexyl,
n-octyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclopropylmethyl,
cyclohexylmethyl, cyclohexylpropyl, methoxyethyl, ethoxyethyl, and the like.
As used herein, the term "unsubstituted or substituted C1-Clo alkoxy" refers
to an
alkyl group (which can be mono- or di-substituted) in which one hydrogen atom
has been
replaced by an oxy group. Examples include, but are not limited to, methoxy,
ethoxy,
propoxy (n- or iso-), butoxy (n-, iso-, sec- or tert-), 3-methylbutoxy, 2-
ethylbutoxy,
pentyloxy, hexyloxyl, 3-methyl-2-butenyloxy, geranyloxy, cyclopentyloxy,
cyclohexyloxy, cyclohexyl- Cl-Clo -alkyloxy (e.g., cyclohexylmethyloxy), and
the like.
As used herein, the term "C1-C5 alkoxycarbonyl" refers to a group comprising
an
alkoxy group having one to five carbon atoms and a carbonyl group.
As used herein, the term "unsubstituted or substituted Cz-Clo alkyl amino"
refers
to a group comprising an alkyl (which can be mono- or di-substituted) group
and an
amino group. Examples include, but are not limited to, methylamino,
ethylamino,
dimethylamino, diethylamino groups, and the like.
As used herein, the term "aminocarbonyl" refers to a group comprising an amino
group and a carbonyl group.
As used herein, the term "aryl" group refers to aromatic hydrocarbon residues
or
aromatic heterocyclic ring groups comprising a 5- or 6-membered monocyclic or
fused
ring. Examples include, but are not limited to, phenyl, 1-naphthyl, 2-
naphthyl, 2-
pyrrolyl, 2-furyl, 2-thienyl, 2-pyridyl, and the like. If said aromatic
hydrocarbon resisdue
27
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
or aromatic heterocyclic ring or fused ring is mono- or di-substituted than
said aryl group
can be considered to be a "substituted" aryl group.
As used herein, the term "unsubstituted or substituted aryl amino" refers to a
group that comprises an aryl (which can be mono- or di-substituted) group and
an amino
group. Examples include, but are not limited to, phenylamino,
methylphenylamino, and
the like.
As used herein, the term "aryloxy" group refers to an aryl group and an oxy
group. Examples include, but are not limited to, phenoxy, 1-naphthoxy, and the
like.
As used herein, the term "aralkyl" refers to an alkyl group (such as any of a
C1-
Clo alkyl group) that is substituted by an aryl group. Examples include, but
are not
limited to, benzyl, 1-phenylethyl, 1-methyl-l-phenylethyl, 2-phenylethyl, 3-
phenylpropyl, cinnamyl, 2-pyrrolylmethyl, furfuryl, thenyl, and the like, and
a benzyl
group is preferred.
As used herein, the term "unsubstituted or substituted aralkyl amino" refers
to a
group that comprises an aralkyl (mono- or di-substituted) group and an amino
group.
Examples include, but are not limited to, benzylamino, methylbenzylamino, and
the like.
As used herein, the term "alkylcarbonyl" refers to a group comprising an alkyl
group and a carbonyl group. Examples include, but are not limited to, C2-C7
lower
aliphatic acyl groups such as acetyl, propanoyl, butanoyl, 2-methylpropanoyl,
pentanoyl,
2-methylbutanoyl, 3-methylbutanoyl, pivaloyl, hexanoyl, cyclopropylcarbonyl,
and the
like.
As used herein, the term "arylcarbonyl" refers to a group comprising an aryl
group and a carbonyl group. Examples include, but are not limited to, benzoyl,
toluoyl,
2-pyrrolcarbonyl, 2-fluoyl, 2-thiophenecarbonyl, and the like.
28
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
As used herein, the term aralkylcarbonyl" refers to a group comprising an
aralkyl
group and a carbonyl group. Examples include, but are not limited to, Cs-Cio
aralkylcarbonyl groups such as phenylacetyl, 3-phenylpropanoyl, 4-
phenylbutanoyl,
cinnamoyl, 2-pyrrolylacetyl, 2-furylacetyl, 2-thienylacetyl, and the like.
As used herein, the term "aralkyloxy" refers to a group comprising an aralkyl
group and an oxy group. Examples include, but are not limited to, benzyloxy, 1-
phenylethoxy, 1-methyl-l-phenylethoxy, and the like.
As used herein, the term "halo" or "halogen" refers to fluorine, chlorine,
bromine
and iodine atoms. Chlorine and fluorine are particularly preferred.
As used herein, the term "haloalkyl" refers to a group comprising a halogen
atom
and an alkyl group.
As used herein, the term "unsubstituted or substituted C1-Clo haloalkyl"
refers to
a haloalkyl group comprising from one to ten carbon atoms, in which the alkyl
group can
be mono- or di-substituted.
As used herein, the term "hydroxyalkoxy" refers to alkoxy group in which one
hydrogen atom has been replaced by a hydroxy group. Examples, include, but are
not
limited to, hydroxymethoxy and 2-hydroxyethoxy.
In formula I above, examples of OR include, but are not limited to, ethoxy,
propoxy (n- or iso-), butoxy (n-, iso-, sec- or tert-), pentyloxy, n-hexyloxy,
cyclopropylmethyloxy, cyclohexyloxy, phenyloxy, benzyloxy, phenetyloxy,
methoxethyloxy, ethoxyethyloxy, acetoxy, propanoyloxy, butanoyloxy,
benzoyloxy, and
the like.
In formula I above, examples of S(O)n R include, but are not limited to,
ethylthio,
isopropylthio, isopropylsulfinyl, isopropylsulfonyl, pentylsulfonyl,
phenylthio,
29
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
phenylsulfinyl, phenylsulfonyl, and the like.
In formula I above, examples of NRR' include, but are not limited to,
dimethylamino, diethylamino, benzylamino, phenethylamino, and the like.
In formula I above, where R and R' taken together with each other nitrogen
atom
bonded thereof, represent atoms can forrn an unsubstituted or substituted 5-
to 7-
membered heterocyclic ring. Examples of a heterocyclic ring include, but are
not limited
to, morpholino, 1-pyrrolyl, 1-pyrrolidinyl, piperidino, piperazino, and the
like.
In formula I above, examples of the 5- to 7- membered cyclic-secondary ainino
group include, but are not limited to, morphorino, l-pyrrolyl, 1-pyrrolidino,
piperidino,
and the like.
Examples of hERG channel agonists having the above-described formula I
include, but are not limited to, 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-
methylthiazole-
5-carboxylic acid (which shall also be referred to herein as "febuxostat"), 2-
[3-cyano-4-
(3-hydroxy-2-methylpropoxy)phenyl]-4-methyl-5-thiazolecarboxylic acid, 2-[3-
cyano-4-
(2-hydroxy-2-methylpropoxy)phenyl]-4-methyl-5-thiazolecarboxylic acid, 2-(3-
cyano-4-
hydroxyphenyl)-4-methyl-5-thiazolecarboxylic acid, 2-[4-(2-carboxypropoxy)-3-
cyanophenyl]-4-methyl-5-thiazolecarboxylic acid or pharmaceutically acceptable
salts
thereof. Methods for making these compounds are described in U.S. Patent No.
5,614,520, which is herein incorporated by reference. Additionally, it is
known in the art
that febuxostat does not prolong the QT interval in healthy subjects (See, Yu,
P., et al., J.
Clin. Phar7nacol., 44(10):1195 (2004)).
The term "long QT syndrome" or "LQTS" refers to prolongation of the QT
interval in a patient.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
The term "patient" refers to an animal, preferably a mammal, including a human
or non-human. The terms patient and subject may be used interchangeably
herein.
By "pharmaceutically'acceptable," such as in the recitation of
a"pharmaceutically
acceptable excipient," or a "pharmaceutically acceptable additive," is meant a
material
that is not biologically or otherwise undesirable, i.e., the material may be
incorporated
into a pharmaceutical composition administered to a patient without causing
any
undesirable biological effects.
The terms "treating" and "treatment" refer to reduction in severity and/or
frequency of symptoms, elimination of symptoms and/or underlying cause,
prevention of
the occurrence of symptoms and/or their underlying cause, and improvement or
remediation of damage. Thus, for example, "treating" a patient involves
prevention of a
particular disorder or adverse physiological event in a susceptible individual
as well as
treatment of a clinically symptomatic individual by inhibiting or causing
regression of a
disorder or disease.
The Invention
As mentioned briefly above, the present invention relates to methods for
reversing
or shortening the QT interval of a patient suffering from QT prolongation.
Specifically,
the methods of the present invention can be used to treat patients suffering
from
congenital or acquired LQTS, myocardial ischemia, heart failure, diabetes or
stroke
(methods for determining whether a patient is suffering from any of the
aforementioned
are well known to those skilled in the art). Additionally, the methods of the
present
invention can also be used to reduce intracellular calcium overload in
patients suffering
diseases with intracellular calcium overload such as myocardial ischemia and
in need of a
therapy.
The methods of the present invention will generally comprise administering to
a
patient in need of such therapy a therapeutically effective amount of at least
one
pharmaceutically acceptable hERG channel agonist. As will be discussed in more
detail
31
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
herein, the administration of at least one hERG channel agonist potentiates or
increases
the currents of the hERG channel in a heart of a patient suffering from QT
prolongation
(such as in a patient suffering from congenital or acquired LQTS, myocardial
ischemia,
heart failure, diabetes or stroke), thereby shortening the QT interval of said
patient.
However, the hERG channel agonists of the present invention are different from
other
hERG channel agonists known in the art in that they do not shorten the QT
interval when
administered to a patient that is not suffering from QT prolongation.
As mentioned briefly above, using the methods of the present invention, QT
prolongation can be reversed (i.e., shortened) in patients suffering from QT
prolongation
(congenital or acquired LQTS, myocardial ischemia, heart failure, diabetes or
stroke) by
increasing the activity of repolarizing potassium channels in particular, the
hERG channel
or IKr via the administration to said patients of a therapeutically effective
amount at least
one pharmaceutically acceptable hERG channel agonist. Specifically,
administration of a
therapeutically effective amount of at least one pharmaceutically acceptable
bERG
channel agonist to a patient suffering from QT prolongation potentiates or
increases the
outward potassium currents, particularly the currents in the hERG channel
(i.e., the I,.,),
thus reversing or shortening the QT interval in said patient. However, not
only do the
hERG channel agonists described for use in the methods herein increase the
current in the
bERG channel in patients suffering from QT prolongation, but, most
importantly, the
hERG channel agonists of the present invention do not shorten the QT interval
in normal
patients that do not suffer from a prolonged QT interval (such as, for
example, a normal,
healthy patient).
Additionally, the potentiation or increase in the current in the hERG channel
after
administration of at least one hERG channel agonist to a patient suffering
from QT
prolongation was found to be voltage dependent. More specifically, the
increase in the
current of a hERG channel was found to occur at positive transmembrane
potentials,
specifically, from about +0.1 mV to about +50 mv, more preferably at from
about +5 mV
to about +30 mV, and even more preferably, at about +10 mV to about +20 mV.
The
hERG channel agonists of the present invention potentiate or increase the
currents in the
32
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
hERG channel during the action potential plateau (in patients suffering from
QT
prolongation). Normally, hERG channels are mostly inactivated at plateau
potentials,
whereas the hERG channel agonists of the present invention potentiate or
increase the
currents during this period.
Methods for identifying hERG channel agonists that can be used in the methods
of the present invention can be readily achieved using routine techniques
known to those
skilled in the art. For example, as described in the examples herein, whole
cell patch
clamp measurements can be performed on cell lines (such as HEK293 and CHO
cells)
that have been transfected with hERG cDNA to screen for hERG channel agonists
that
increase the currents in the hERG channel as described herein. Once such hERG
channel
agonists have been identified, they can be further screened to determine
whether or not
these compounds reduce or reverse QT prolongation in patients that suffer from
QT
prolongation. This can be achieved by administering such hERG channel agonists
to a
patient that suffer from QT prolongation and then taking an ECG/EKG of said
patient
during the time when the at least one hERG channel agonist is circulating in
the blood.
One skilled in the art could easily determine, by reading the ECG/EKG, whether
the
administration of the hERG channel agonist to said patient has shortened the
patient's
prolonged QT interval.
For patients suffering from myocardial ischemia, heart failure, diabetes or
stroke,
one skilled in the art could easily monitor, using routine techniques, the
reduction of
mortality (death) or frequency of a disease event (i.e., meaning how often a
patient may
experience a stroke and/or heart attack) associated with any of the above
diseases, and/or
an improvement in the symptoms, biochemical markers (i.e., for a patient
suffering from
myocardial ischemia a reduction creatine phosphate kinase (CPK), a reduction
in C
reactive protein (CRP) in patient in suffering from myocardial ischemia or
stroke, etc.)
and/or ECG/EKG abnormality associated with these diseases, after
administration of the
hERG channel agonist to said patient.
33
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
In addition to the methods described above, some pharmaceutical compounds that
have shown utility in preventing a broad variety of disease states never
benefit the public
because these compounds are prone to prolonging the QT interval thus causing
acquired
LQTS. With the discovery of the methods described herein, these drugs may now
be
available to benefit the public. In particular, a hERG channel agonist that is
capable of
selectively shortening the QT interval (the hERG channel agonists described
herein are
selective in that these compounds shorten or reverse the QT interval only in
patients
suffering from QT prolongation and not in normal patients that do not suffer
from QT
prolongation) can be co-administered with compounds that would otherwise
benefit the
public but for the fact that these compounds prolong the QT interval. By co-
administration of a hERG channel agonist, the detrimental effects of these
compounds
can be assuaged to make them useful for their intended purpose. Such drugs may
come
from a wide variety of compound classes and include but are not limited to
antihistamines, antidepressants, neuroleptics, antimalaria drugs, macrolide
antibiotics,
serotonin antagonists and calcium antagonists.
Compositions containing at least one hERG channel agonist in combination with
another pharmaceutical compound are therefore part of the present invention.
Using the
excipients and dosage forms described below, formulations containing such
combinations
are a matter of choice for those skilled in the art. Further, those skilled in
the art will
recognize that various coatings or other separation techniques may be used in
cases
where the combination of compounds are incompatible.
The hERG channel agonists used in accordance with the methods of the present
invention can be provided in the form of pharmaceutically acceptable salts
derived from
inorganic or organic acids. Pharmaceutically acceptable salts are well-known
in the art.
For example, S. M. Berge et al. describe pharmaceutically acceptable salts in
detail in J.
Pharmaceutical Sciences, 66: 1 et seq. (1977). The salts can be prepared in
situ during
the final isolation and purification of the compounds or separately by
reacting a free base
function with a suitable organic acid. Representative acid addition salts
include, but are
not limited to, acetate, adipate, alginate, citrate, aspartate, benzoate,
benzenesulfonate,
34
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
bisulfate, butyrate, camphorate, camphor sulfonate, digluconate,
glycerophosphate,
hemisulfate, heptanoate, hexanoate, fumarate, hydrochloride, hydrobromide,
hydroiodide,
2-hydroxyethansulfonate (isothionate), lactate, maleate, methane sulfonate,
nicotinate, 2-
naphthalene sulfonate, oxalate, palmitoate, pectinate, persulfate, 3-
phenylpropionate,
picrate, pivalate, propionate, succinate, tartrate, thiocyanate, phosphate,
glutamate,
bicarbonate, p-toluenesulfonate and undecanoate. Also, basic nitrogen-
containing groups
can be quaternized with such agents as lower alkyl halides such as methyl,
ethyl, propyl,
and butyl chlorides, bromides and iodides; dialkyl sulfates like dimethyl,
diethyl, dibutyl
and diamyl sulfates; long chain halides such as decyl, lauryl, myristyl and
stearyl
chlorides, bromides and iodides; arylalkyl halides like benzyl and phenethyl
bromides
and others. Water or oil-soluble or dispersible products are thereby obtained.
Examples of
acids which can be employed to form pharmaceutically acceptable acid addition
salts
include such inorganic acids as hydrochloric acid, hydrobromic acid, sulphuric
acid and
phosphoric acid and such organic acids as oxalic acid, maleic acid, succinic
acid and
citric acid.
Basic addition salts can be prepared in situ during the final isolation and
purification of compounds by reacting a carboxylic acid-containing moiety with
a
suitable base such as the hydroxide, carbonate or bicarbonate of a
pharmaceutically
acceptable metal cation or with ammonia or an organic primary, secondary or
tertiary
amine. Pharmaceutically acceptable salts include, but are not limited to,
cations based on
alkali metals or alkaline earth metals such as lithium, sodium, potassium,
calcium,
magnesium and aluminum salts and-the like and nontoxic quaternary ammonia and
amine
cations including ammonium, tetramethylammonium, tetraethylammonium,
methylammonium, dimethylammonium, trimethylammonium, triethylammonium,
diethylammonium, and ethylammonium among others. Other representative organic
amines useful for the formation of base addition salts include
ethylenediamine,
ethanolamine, diethanolamine, piperidine, piperazine and the like.
The at least one hERG channel agonist may be formulated in a variety of ways
that is largely a matter of choice depending upon the delivery route desired.
For
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
example, solid dosage forms for oral administration include capsules, tablets,
pills,
powders and granules. In such solid dosage forms, the hERG channel agonist can
be
mixed with at least one inert, pharmaceutically acceptable excipient or
carrier, such as
sodium citrate or dicalcium phosphate and/or a) fillers or extenders, such as,
but not
limited to, starches, lactose, sucrose, glucose, mannitol and silicic acid; b)
binders, such
as, but not limited to, carboxymethylcellulose, alginates, gelatin,
polyvinylpyrrolidone,
sucrose and acacia; c) humectants, such as, but not limited to glycerol; d)
disintegrating
agents, such as, but not limited to, agar-agar, calcium carbonate, potato or
tapioca starch,
alginic acid, certain silicates and sodium carbonate; e) solution retarding
agents, such as,
but not limited to, paraffin; f) absorption accelerators, such as, but not
limited to,
quaternary ammonium compounds; g) wetting agents, such as, but not limited to,
cetyl
alcohol and glycerol monostearate; h) absorbents, such as, but not limited to,
kaolin and
bentonite clay; and i) lubricants, such as, but not limited to, talc, calcium
stearate,
magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate and
mixtures
thereof.
Solid compositions of a similar type may also be employed as fillers in soft
and
hard-filled gelatin capsules using such excipients as lactose or milk sugar as
well as high
molecular weight polyethylene glycols and the like.
Solid dosage forms comprising tablets, capsules, pills and granules can be
prepared with coatings and shells such as enteric coatings and other coatings
well-known
in the pharnlaceutical formulating art. They may optionally contain opacifying
agents and
may also be of a composition such that they release the active ingredient(s)
only, or
preferentially, in a certain part of the intestinal tract, optionally, in a
delayed manner.
Examples of embedding compositions that can be used include polymeric
substances and
waxes.
Liquid dosage forms for oral administration include pharmaceutically
acceptable
emulsions, solutions, suspensions, syrups and elixirs. In addition to the bERG
channel
agonist, the liquid dosage forms may contain inert diluents commonly used in
the art such
36
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
as, for example, water or other solvents, solubilizing agents and emulsifiers,
such as, but
not limited to, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl
acetate, benzyl
alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethyl
formamide,
oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and
sesame oils),
glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid
esters of sorbitan
and mixtures thereof.
The compositions can also be delivered through a catheter for local delivery
at a
target site, via an intracoronary stent (a tubular device coiuposed of a fine
wire mesh), or
via a biodegradable polymer.
Compositions suitable for parenteral injection may comprise physiologically
acceptable, sterile aqueous or nonaqueous solutions, dispersions, suspensions
or
emulsions and sterile powders for reconstitution into sterile injectable
solutions or
dispersions. Examples of suitable aqueous and nonaqueous carriers, diluents,
solvents or
vehicles include, but are not limited to, water, ethanol, polyols (propylene
glycol,
polyethylene glycol, glycerol, and the like), vegetable oils (such as olive
oil), injectable
organic esters such as ethyl oleate, and suitable mixtures thereof.
These compositions can also contain adjuvants such as preserving, wetting,
emulsifying, and dispensing agents. Prevention of the action of microorganisms
can be
ensured by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to
include
isotonic agents, for example, sugars, sodium chloride and the like. Prolonged
absorption
of the injectable pharmaceutical form can be brought about by the use of
agents delaying
absorption, for example, aluminum monostearate and gelatin.
Suspensions, in addition to the active agent (i.e., hERG channel agonist), may
contain suspending agents, as for example, ethoxylated isostearyl alcohols,
polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose,
aluminum
37
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these
substances, and
the like.
Proper fluidity can be maintained, for example, by the use of coating
materials
such as lecithin, by the maintenance of the required particle size in the case
of dispersions
and by the use of surfactants.
In some cases, in order to prolong the effect of the drug (i.e. hERG channel
agonist), it is desirable to slow the absorption of the drug from subcutaneous
or
intramuscular injection. This can be accomplished by the use of a liquid
suspension of
crystalline or amorphous material with poor water solubility. The rate of
absorption of
the drug then depends upon its rate of dissolution which, in turn, may depend
upon
crystal size and crystalline form. Alternatively, delayed absorption of a
parenterally
administered drug form is accomplished by dissolving or suspending the drug in
an oil
vehicle. Injectable depot forms are made by forming microencapsule matrices of
the
drug in biodegradable polymers such as polylactide-polyglycolide. Depending
upon the
ratio of drug to polymer and the nature of the particular polymer employed,
the rate of
drug release can be controlled. Examples of other biodegradable polymers
include
poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also
prepared
by entrapping the drug in liposomes or microemulsions which are compatible
with body
tissues.
The injectable formulations can be sterilized, for example, by filtration
through a
bacterial-retaining filter or by incorporating sterilizing agents in the form
of sterile solid
compositions which can be dissolved or dispersed in sterile water or other
sterile
injectable medium just prior to use.
Dosage forms for topical administration of the compounds of this present
invention include powders, sprays, ointments and inhalants. The active
compound(s) is
mixed under sterile conditions with a pharmaceutically acceptable carrier and
any needed
preservatives, buffers or propellants that can be required. Opthalmic
formulations, eye
38
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
ointments, powders and solutions are also contemplated as being within the
scope of this
invention.
It will be understood that formulations to be used in the methods of the
present
invention generally will comprise a therapeutically effective amount of one or
more
hERG channel agonists. The phrase "therapeutically effective amount" as used
herein
means a sufficient amount of, for example, the composition, hERG channel
agonist, or
formulation necessary to treat the desired disorder (i.e., the prolonged QT
interval), at a
reasonable benefit/risk ratio applicable to any medical treatment. As with
other
pharmaceuticals, it will be understood that the total daily usage of a
pharmaceutical
composition of the invention will be decided by a patient's attending
physician within the
scope of sound medical judgment. The specific therapeutically effective dose
level for
any particular patient will depend upon a variety of factors including the
disorder being
treated and the severity of the disorder; activity of the specific compound
employed; the
specific composition employed; the age, body weight, general health, sex and
diet of the
patient; the time administration, route of administration, and rate of
excretion of the
specific compound employed; the duration of the treatment; drugs used in
combination or
coincidental with the specific compound employed; and other factors known to
those of
ordinary skill in the medical arts. For example, it is well within the skill
of the art to start
doses of the compound at levels lower than required to achieve the desired
therapeutic
effect and to gradually increase the dosage until the desired effect is
achieved.
Formulations of the present invention are administered and dosed in accordance
with sound medical practice, taking into account the clinical condition of the
individual
patient, the site and method of administration, scheduling of administration,
and other
factors known to medical practitioners.
Therapeutically effective amounts for purposes herein thus can readily be
determined by such considerations as are known in the art. The daily
pharmaceutically
effective amount of the hERG channel agonist administered to a patient in
single or
divided doses range from about 0.01 to about 750 milligram per kilogram of
body weight
39
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
per day (mg/kg/day). More specifically, a patient may be administered from
about 5.0
mg to about 1000 mg daily, preferably from about 20 mg to about 500 mg daily
and most
preferably from about 40 mg to about 300 mg daily of a hERG channel agonist.
As mentioned previously, the present invention relates to methods for
reversing
or shortening a QT interval in a patient suffering from QT prolongation. The
methods of
the present invention involve increasing the currents of the hERG channel in a
heart of a
patient suffering from QT prolongation by administering to a patient a
therapeutically
effective amount of at least one pharmaceutically acceptable hERG channel
agonist.
Once a patient has been administered at least one hERG channel agonist as
described
herein, the effectiveness and progress of the treatment in reversing or
shortening the QT
interval can be monitored by performing an ECG/EKG on said patient and
determining
the QT interval of said patient using routine techniques known to those
skilled in the art.
An ECG/EKG can be repeated as many times as necessary until the QT interval
has been
reversed or shortened to the satisfaction of the treating physician.
For patients suffering from myocardial ischemia, heart failure, diabetes or
stroke,
one skilled in the art could easily monitor, using routine techniques, the
reduction of
mortality (death) or frequency of a disease event (i.e., meaning how often a
patient may
experience a stroke and/or heart attack) associated with any of the above
diseases, and/or
an improvement in the symptoms, biochemical markers (i.e., for a patient
suffering from
myocardial ischemia a reduction creatine phosphate kinase (CPK), a reduction
in C
reactive protein (CRP) in patient in suffering;from myocardial ischemia or
stroke, etc.)
and/or ECG/EKG abnormality associated with these diseases, after
administration of the
hERG channel agonist to said patient.
By way of example and not of limitation, examples of the present invention
shall
now be given.
EXAMPLE 1: Effect of febuxostat on cloned hERG channels expressed in Human
Embryo Kidney (HEK)293 cells: whole cell patch clamp measurements focusing on
peak tail current
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Materials and Methods
Solutions and Chemicals
All chemicals used in preparation of bath and electrode solutions were
purchased
from Sigma (St. Louis, MO) unless otherwise noted and were of ACS reagent
grade
purity or higher. Febuxostat was obtained from Teijin Limited (Yamaguchi,
Japan). All
solutions containing febuxostat were prepared in glass containers whenever
possible.
Test solutions of febuxostat and terfenadine (positive control) were prepared
daily using a
modified HEPES-buffered Tyrode's (HBT) solution (composition in mM): NaCI,
137;
KC1, 5.4; CaCla, 1.8; MgClz, 1; HEPES, 10; Glucose, 10; pH adjusted to 7.4
with NaOH.
The HBT solution was freshly prepared weekly. Terfenadine solution was
prepared in
HBT at a concentration of 60 nM. The HBT solution was warmed to room
temperature
before preparing febuxostat or terfenadine solutions. Fresh test and control
solutions were
prepared on each experimental day. Pipette solution for whole cell recordings
was
(composition in mM): K-aspartate, 130; MgC12, 5; EGTA, 5; ATP, 4; HEPES, 10;
pH
adjusted to 7.2 with KOH. The pipette solution was prepared in batches, stored
at -20 C,
and was freshly thawed each day of use.
Cell Culture
HEK293 cells were stably transfected with hERG cDNA. Stable transfectants
were selected by coexpression of the hERG cDNA and G418 gene incorporated into
the
expression plasmid. Selection pressure was maintained by including G418 in the
culture
medium. Cells were cultured in Dulbecco's Modified Eagle Medium / Nutrient
Mixture
F-12 (D-MEM/F-12) supplemented with 10% fetal bovine serum, 100 U/mL
penicillin G
sodium, 100 g/mL streptomycin sulfate and 500 g/mL G418. Cells were
maintained in
tissue culture incubators at 37 C in a humidified 5% C02 atmosphere, with
stocks
maintained in cryogenic storage. Cells used for electrophysiology were plated
on 35 mm
tissue culture dishes or glass coverslips. All experiments were performed at
room
temperature (22 C - 25 C) unless otherwise noted. Each cell acted as its own
control.
41
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Electrophysiology
Warner PC501A and Axon Instruments Axopatch 200B patch clamp amplifiers
were used for whole cell patch clamp recordings. Current records were analog
filtered at
0.2 of the sampling frequency for digital conversion by Axon Instruments
Digidata
1320A AD/DA converters attached to PC-compatible desktop computers. Axon
Instruments Clampex 8.2 software was used to acquire data and generate
stimulus voltage
waveforms. The suite of Axon Instruments pCLAMP8.2 applications (Molecular
Devices Corp., Sunnyvale, CA) and Microsoft Excel 2000 spreadsheet software
were
used to analyze the data.
Cells attached to glass coverslips or plastic 35mm Petri dishes were
transferred to
the recording chamber and superfused with HBT solution. Patch pipettes were
fabricated
from TW 150-F glass capillaries on a P 97 horizontal puller (Sutter Instrument
Co.,
Novato, CA) to generate pipettes with 1- 5 MSZ resistances after fire
polishing. The
concentration-response relationship for febuxostat modulation of hERG channel
function
was evaluated at concentrations of febuxostat ranging from 0.1 to 500 M.
These
concentrations were applied cumulatively to cells expressing hERG channels.
Each
concentration had an n> 3 where n = number of measurements. Only one or two
concentrations of febuxostat were applied to each cell. Terfenadine (60 nM)
was applied
to two cells as a positive control.
Patch Clamp Voltage Protocols
Concetitration-Response
Cells stably expressing hERG were held at -80 mV. Onset and steady state
modulation of hERG current due to febuxostat or terfenadine was measured using
a pulse
pattern with fixed amplitudes (depolarization: +20 mV for 2 s; repolarization:
-50 mV for
2 s) repeated at 10 s intervals. Peak tail current was measured during the 2 s
step to -50
mV. A steady state was maintained for at least 30 s before applying febuxostat
or
terfenadine. Peak tail currents after application of febuxostat or terfenadine
were
measured until a new steady state was achieved.
42
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Ft-equency-Dependence
Cells were held at -80 mV for at least 1 minute. A train of pulses
(depolarization:
+60 mV for 250 ms; repolarization: -50 mV for 70 ms) sufficient to reach a
steady state
value (typically in the range of 20 to 30 pulses) was then applied with pulses
in the train
repeated at frequencies of 0.3 Hz and 3 Hz. Frequency-dependence of
febuxostat's effect
on peak tail currents was measured before and after equilibration with 500 M
febuxostat. Peak tail current was measured during the step to -50 mV in each
pulse of the
train.
Data Analysis
Data acquisition and analyses were performed using the suite of pCLAMP8.2
applications (Molecular Devices Corp., Sunnyvale, CA). Steady state was defmed
as a
limiting constant rate of change with time (linear time dependence). The
steady states
before and after test article application were used to measure drug effects.
Analysis of frequency-dependence of febuxostat hERG modulation
Data for analysis of frequency-dependence (use-dependence) were normalized to
the peak current of the first pulse in each train of pulses and normalized
data from trains
at each frequency were pooled to construct average time courses.
Analysis of teinpeYature-dependence
Data for analysis of temperature-dependence compared the currents in the
presence of 500 M febuxostat at room temperature (22 C - 25 C) and at near
physiological temperature (35 2 C) in at least two cells at each temperature
Results
Febuxostat modulation of HEK/hERG peak tail currents.
These initial patch-clamp measurements were to evaluate potential hERG
blocking effect of febuxostat. In contrast, the steady state effect of
febuxostat on the time
43
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
course of normalized peak tail currents over the concentration range tested
(0.1 to 500
M) revealed a slight increase from 1.01 to 1.09 in mean current and no change
in tail
current kinetics (Table 1). Moreover, febuxostat produced a voltage-dependent
increase
in the hERG currents that was prominent during the +20 mV step. Individual
HEK/hERG voltage-clamp current-time (I-T) records acquired before and
following
equilibration with 500, 50, 1 and 0.1 M febuxostat are shown superimposed in
Figures 1
and 2. The magnitude of the effect at +20 mV was variable, but consistently
present.
The time course of the normalized peak current response to febuxostat
application at
+20 mV, measured in consecutive records repeated at 10 second intervals,
consisted of an
initial rapid increase that declined to a smaller steady state effect (Figure
3). These results
indicate that febuxostat is not a hERG blocker and will not cause QT
prolongation,
instead, it is a novel hERG agonist.
Evaluation of the use- or frequency-dependence of 500 M febuxostat modulation
of HEK/hERG peak tail current magnitude (measured at -50 mV) showed no
difference
in the time course of normalized peak tail current amplitudes in control and
after
equilibration with febuxostat when activating pulses were repeated at a
frequency of 0.3
Hz. At the higher activation frequency of 3 Hz, febuxostat produced a small,
insignificant reduction in the time course of normalized peak tail current
magnitude
relative to control (Figure 4). This suggests that difference in heart rate
will not
significantly affect the lack of blocking activity of febuxostat on hERG
channel.
Raising the temperature of the bath to 35 2 C did not significantly change
the
effect on peak tail current magnitude caused by application of 500 M
febuxostat from
that obtained at room temperatizre (22 C -25 C) (Table 2). The agonist effect
on the
hERG current at +20 mV was still present at 35 2 C and was qualitatively
similar.
These results demonstrate that the effects of febuxostat observed at room
temperature
mentioned above should also occur in the body at body temperature.
Terfenadine is an established and potent hERG blocker and was used as a
positive
control. The effect of application of 60 nM terfenadine on peak tail current
was
44
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
measured. As expected, 60 nM terfenadine blocked 77 ~: 3 % (n=2) of HEK/hERG
peak
tail current (Table 3).
In summary, these results indicate that febuxostat at concentrations up to 500
M
has no undesirable blocking effect on the HEK/hERG peak tail current. And the
lack of a
blocking effect of 500 M febuxostat on peak tail current was not use- or
temperature-
dependent. Instead, a voltage dependent increase in hERG currents during +20
mV was
observed, suggesting that febuxostat is a novel hERG agonist.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 1: Effect of febuxostat on HEK/HERG peak tail current
Febuxostat Concentration
Mean a SEM b N
0.1 1.01 0.03 3
1 1.03 0.05 3
50 1.07 0.04 4
500 1.09 0.04 4
a Mean fraction of current (ITest/Icontr I) at each febuxostat concentration,
standard error
of the mean (SEM), and 'number of observations (n) for each febuxostat
concentration.
Table 2: Comparison of fraction of HEK/hERG peak tail current after
application
of febuxostat at room temperature and 35 C
500 M TMX-67
35 2 C 22 C -25 C
Cell ID TMX-67/kontrol Ce11ID TMX-67/IControl
c 1 bw 020411 0001 1.12 c 1 bw 020403 0001 1.14
d 1 bw 0204110001 1.04 a 1 bw 0204050001 1.00
b I bw 020405 0001 0.94
a 1 bw, 0204080003 0.96
Mean a SEM 6 1.08 0.04 Mean SEM 1.09 0.04
a Mean fraction of current (ITeSt/k ntr l) with febuxostat at 500 M, b
standard error
of the mean (SEM).
Table 3: Fraction of HEK/hERG current after a lication of terfenadine
60 nM terfenadine
Cell ID ITerfenadine/lControl
f I bw 020401 0000 0.26
h 1 bw 0204010000 0.21
Mean a SEM b 0.23 0.03
a Mean fraction of current (ITest/IC ntr l) with terfenadine at 60 nM, b
standard
error of the mean (SEM).
46
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
EXAMPLE 2: Effect of febuxostat on cloned hERG channels expressed in Chinese
Hamster Ovary (CHO) cells: whole cell patch clamp measurements focusing on
agonist effects at positive potential.
Material and Methods
Solutions and Chemicals
All chemicals used in preparation of bath and electrode solutions were
purchased
from Sigma (St. Louis, MO) unless otherwise noted and were of ACS reagent
grade
purity or higher. Febuxostat was obtained from Teijin Limited (Yamaguchi,
Japan). All
solutions containing febuxostat were prepared in glass containers whenever
possible.
Test solutions of febuxostat and terfenadine (positive control) were prepared
daily using a
modified HEPES-buffered Tyrode's (HBT) solution (composition in mM): NaCl,
137;
KCl, 5.4; CaC12, 1.8; MgC12, 1; HEPES, 10; Glucose, 10; pH adjusted to 7.4
with NaOH.
The HBT solution was freshly prepared weekly. Terfenadine solution was
prepared in
HBT at a concentration of 60 nM. The HBT solution was warmed to room
temperature
before preparing febuxostat or terfenadine solutions. Fresh test and control
solutions were
prepared on each experimental day. Pipette solution for whole cell recordings
was
(composition in mM): K-aspartate, 130; MgC12, 5; EGTA, 5; ATP, 4; HEPES, 10;
pH
adjusted to 7.2 with KOH. The pipette solution was prepared in batches, stored
at -20 C,
and was freshly thawed each day of use.
Cell Culture
CHO cells were stably transfected with hERG cDNA. Stable transfectants were
selected by coexpression of the hERG cDNA and G418 gene incorporated into the
expression plasmid. Selection pressure was maintained by including G41 S in
the culture
medium. Cells were cultured in Dulbecco's Modified Eagle Medium / Nutrient
Mixture
F-12 (D-MEM/F-12) supplemented with 10% fetal bovine serum, 100 U/mL
penicillin G
sodium, 100 g/mL streptomycin sulfate and 500 g/mL G418. Cells were
maintained in
tissue culture incubators at 37 C in a humidified 5% C02 atmosphere, with
stocks
maintained in cryogenic storage. Cells used for electrophysiology were plated
on 35 mm
47
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
tissue culture dishes or glass coverslips. All experiments were performed at
room
temperature (22 C - 25 C) unless otherwise noted. Each cell acted as its own
control.
Electrophysiology
Warner PC501A and Axon Instruments Axopatch 200B patch clamp amplifiers
were used for whole cell patch clamp recordings. Current records were analog
filtered at
0.2 of the sampling frequency for digital conversion by Axon Instruments
Digidata
1320A AD/DA converters attached to PC-compatible desktop computers. Axon
Instruments Clampex 8.2 software was used to acquire data and generate
stimulus voltage
waveforms. The suite of Axon Instruments pCLAMP8.2 applications (Molecular
Devices Corp., Sunnyvale, CA) and Microsoft Excel 2000 spreadsheet software
were
used to analyze the data.
Cells attached to glass coverslips or plastic 35mm Petri dishes were
transferred to
the recording chamber and superfused with HBT solution. Patch pipettes were
fabricated
from TW150-F glass capillaries on a P 97 horizontal puller (Sutter Instrument
Co.,
Novato, CA) to generate pipettes with 1- 5 MS2 resistances after fire
polishing. The
concentration-response relationship for febuxost modulation of hERG channel
function
0.0001 to 10 M. These
was evaluated at concentrations of febuxostat ranging fr
concentrations were applied cumulatively to cells expressing hERG channels.
Each
concentration had an n> 3 where n = number of measurements. Only one or two
concentrations of febuxostat were applied to each cell. Terfenadine (60 nM)
was applied
to two cells as a positive control.
Patch Clamp Voltage Protocols
Concentration-Response
Cells stably expressing hERG were held at -80 mV. Onset and steady state
modulation of hERG current due to febuxostat (0.0001 to 10 M) or terfenadine
(60 nM)
was measured using a pulse pattern with fixed amplitudes (depolarization: +20
mV for 2
48
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
s; repolarization: -50 mV for 2 s) repeated at 10 s intervals. Peak tail
current was
measured during the 2 s step to -50 mV. For modulation of hERG current at
positive
potentials, peak current during the step to +20 mV was measured. A steady
state was
maintained for at least 30 s before applying febuxostat or terfenadine. Peak
currents after
application of febuxostat or terfenadine were measured until a new steady
state was
achieved.
Frequency-Dependence
Cells were held at -80 mV for at least 1 minute. A train of pulses
(depolarization:
+60 mV for 250 ms; repolarization: -50 mV for 70 ms) sufficient to reach a
steady state
value (typically in the range of 20 to 30 pulses) was then applied with pulses
in the train
repeated at frequencies of 0.3 Hz and 3 Hz. Frequency-dependence of febuxostat
modulation of hERG channel function was measured before and after
equilibration with 1
M febuxostat. Febuxostat frequency-dependent modulation of hERG channels was .
measured as the time course of peak current magnitude measured during the
steps to +60
mV and -50 mV in each pulse of the train.
Steady State I-VRelation
From the holding potential of -80 mV, 4 s depolarizing voltage steps to
voltages
from -70 to +80 mV in 10 mV increments, followed by repolarization to -50 mV
for 5 s,
were used to measure the steady-state I-V relation in control and in the
presence of 1 M
febuxostat. The voltage protocol was repeated at 15 s intervals. A normalized
steady-state I-V relation was generated using the current amplitude at the end
of the
depolarizing pulse for normalization.
Voltage-Dependence ofActivation (G-Vrelationship)
Peak tail currents were measured during the repolarization step (-50 mV) in
the
steady-state I-V protocol (above) in control and in the presence of 1 M
febuxostat.
49
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Fully activated hERG I-V Relationship
From the holding potential of -80 mV, cells were depolarized to +60 mV for 1
sec to fully activate and partially inactivate hERG currents, and then
repolarized for 5 sec
to voltages ranging from -100 to +40 mV in 10 mV increments. The interval
between
voltage protocol repetitions was 15 s. Peak currents were measured during the
repolarizing step and plotted as a function of voltage. The fully activated
hERG I-V
relation was measured in control and in the presence of 1 M febuxostat.
Alteriiate I- VRelationship (Conservative Protocol)
From a holding potential of 0 mV, a 25 ms hyperpolarizing pulse to -80 mV was
followed by a 1 s depolarizing step to potentials from -120 mV to +40 mV in 10
mV
increments. The voltage protocol was repeated at 10 s intervals. A normalized
peak I-V
relation was generated using the peak current amplitude during the variable
voltage step
plotted as a function of voltage. The alternate hERG I-V relation was measured
in
control and in the presence of 1 M febuxostat.
holtage Dependence of Inactivation ~
The steady state inactivation-voltage relation was measured by calculating the
ratio of the initial current to the steady state current in the 1 s variable
voltage step at each
step voltage from the Alternate I-V Relationship protocol.
Data Analysis
Data acquisition and analyses were performed using the suite of pCLAMP8.2
applications (Molecular Devices Corp., Sunnyvale, CA). Steady state was
defined as a
limiting constant rate of change with time (linear time dependence). The
steady states
before and after test article application were used to measure drug effects.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Analysis of EC50 data
To quantify febuxostat's agonist effects, a Hill equation of the following
form
was used:
ITest/Icontrol = Imax/{1+( EC50/[Test])N} + Io, (1)
where EC50 is the concentratioR of febuxostat that produces half-maximal
stimulation,
Imax is the maximum stimulation value, Io is the initial, control current,
[Test] is the
concentration of febuxostat, ITest/koõtrol is the ratio of steady state
channel current
amplitudes in test and control solutions and N, the Hill coefficient, is a
measure of
cooperativity. If N is fixed at 1, equation (1) becomes a simple one-to-one
binding model
for current stimulation.
Analysis of frequency-dependence offebuxostat hERG modulation
Data for analysis of frequency-dependence (use-dependence) were normalized to
the peak current at -50 mV of the first pulse and to the peak current at +60
mV of the
second pulse in each train of pulses and normalized data from trains at each
frequency
were pooled to construct average time courses.
Analysis of voltage dependerice of IzERG activation
Voltage dependence of activation was fit with a single Boltzmann distribution
of
the form:
ITai1(V)/hail M. = 1/{ l+e-(V-V 1/2)/K~} (2)
Where ITail(v) is the peak tail current elicited by the variable voltage V
activating
step in the steady state I-V relation protocol. ITail MaX was calculated as
the average of the
peak values for currents during voltage steps to 60, 70 and 80 mV. V1/2 and Kv
are the
midpoint potential and the exponential slope factor for this Boltzmann
distribution.
51
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Analysis of voltage dependence of hERG inactivation
An equation similar to equation (2) was used to fit the voltage dependence of
inactivation with a single Boltzmann distribution of the form:
(IPeakM-ISteady(V))/IPeak(V) = 1 / {l+e(V-Vii2)/I~ }, (3)
where Isteady(v) is the current at the end of the 1 s variable voltage step
when steady state
inactivation is attained for potentials greater than -80 mV in the alternate I-
V relationship
and IPeakM is the current at the beginning of the 1 s step at each voltage V.
For potentials
equal to or less than -80 mV, IsteadyM was the extrapolated value of the
current at the
beginning of the 1 s variable voltage step. The extrapolated value was
obtained by fitting
a single exponential function to the decaying phase of the current transient.
All initial
measures for inactivation were relative to the inactivation present at -80 mV
(value of
IPeak(v=-80mv))= This relative measure was renormalized so that the asymptotic
value for
channel availability at negative potentials was 1. The best fit value of V lia
and K, was
determined by nonlinear least squares fitting. Vli2 and K, are the midpoint
potential and
the exponential slope factor for this Boltzmann distribution. The term on the
left of the
equal sign is the channel availability and channel inactivation is defmed as
"1 - channel
availability".
Results
HEK293 cells have an endogenous delayed rectifier current that overlaps the
heterologously expressed hERG currents at positive potentials. In order to
characterize
the agonist effects of febuxostat at positive potentials, CHO cells were used
to
heterologously express hERG channels (CHO/hERG), since untransfected CHO cells
have only small time-independent background currents over the range of
potentials at
which febuxostat modulated hERG channel activity as observed in the above
HEK/hERG
example.
52
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Agonist effect of febuxostat on CHO/hERG current at +20 mV
When hERG was expressed in CHO cells, febuxostat again produced a voltage-
dependent increase in hERG currents that was prominent during the +20 mV step
but
much more reduced during the -50 mV step. Sample CHO/hERG voltage-clamp I-T
records acquired during control and after application of 1 M febuxostat are
shown in
Figure 5. The time course of the agonist action of febuxostat application,
measured as
the maximum current at +20 mV in consecutive records acquired at 0.1 Hz, was
comprised of an initial rapid increase in current that rose to a maximum
during the first 1-
2 minutes followed by a slow decline to a smaller steady state (maintained)
current that
was established after at least 3 minutes of febuxostat application. The time
course in two
cells of the peak current evoked by the +20 mV voltage step before (Control 1
and
Control 2) and following application of 1 M (Figure 6) and 0.1 M (Figure 7)
febuxostat both show the initial and steady state agonist responses and
washout of the
effect at 0.1 M. Summary statistics for the initial current increase (Table
4) and for the
steady-state component (Table 5) measured from the time course of normalized
peak
currents at +20 mV showed the agonist effect febuxostat was concentration
dependent.
The concentration-response relations for the initial and steady state response
components
measured from the time course of peak currents at +20 mV gave EC50 values of
0.003 for
the initial component (Figure 8) and 0.070 M for the steady-state component
(Figure 9).
Table 4: Initial current increase at +20 mV following febuxostat application
Concentration Mean f SEM a n
0.00010 0.98 + 0.01 2
0.0010 1.27 + 0.07 3
0.010 1.28+0.02 3
0.10 1.62 + 0.06 11
1.0 1.54 + 0.08 8
1.55 + 0.05 4
a Mean fraction of current (ITest/Icontrol) at each febuxostat concentration,
standard error of
the mean (SEM), and number of observations (n) for each febuxostat
concentration.
53
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 5: Steady-state current increase at +20 mV foRowing febuxostat
application
Concentration Mean + SEM a n
0.00010 0.98+0.01 2
0.0010 1.10 + 0.02 2
0.010 1.14 + 0.03 3
0.10 1.20 + 0.06 10
1.0 1.42+0.14 7
1.29 0.02 4
a Mean fraction of current (ITest/IControj) at each febuxostat concentration,
standard error of
the mean (SEM), and number of observations (n) for each febuxostat
concentration.
Effect of febuxostat on CHO/hERG current at -50 mV (tail current)
Summary statistics for the peak tail current (Table 6) and tail current at the
end of
the two-second step to -50 mV (Table 7) confirmed that the agonist effect of
febuxostat
retained the voltage sensitivity identified in HEK/hERG cells. The
measurements
presented in Tables 4- 7 were obtained from the same set of CHO/hERG cells.
The
effects of febuxostat on hERG peak tail currents during the -50 mV voltage
step were too
small to fit EC50 values. Application of the positive control (60 nM
terfenadine) blocked
CHO/hERG peak tail currents by 76 + 5% (Table 8), as expected and similar to
block of
HEK/hERG by terfenadine (see above in Example 1).
Table 6: Peak tail current increase at -50 mV following febuxostat application
Concentration Mean + SEM a n
0.00010 0.87+0.08 2
0.0010 1.04 + 0.03 3
0.010 1.12 + 0.04 3
0.10 1.11 + 0.03 11
1.0 1.06+0.03 8
10 1.06 + 0.02 4
Mean fraction of current (ITest/Icontrot) at each febuxostat concentration,
standard error of
the mean (SEM), and number of observations (n) for each febuxostat
concentration.
54
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table7: End tail current increase at -50 mV foRowing febuxostat a lication.
Concentration Mean ~ SEM a n
0.00010 0.83 ~ 0.12 2
0.0010 1.06 0.06 3
0.010 1.08 0.03 3
0.10 1.05 0.03 11
1.0 0.95 ~ 0.03 8
1.01 ~ 0.02 4
a Mean fraction of current (ITest/kontrol) at each febuxostat concentration,
standard error of
the mean (SEM), and number of observations (n) for each febuxostat
concentration. End
tail currents were measured at the end of a two second voltage step to -50 mV.
Table 8: Fraction of CHO/hERG current after a lication of terfenadine
60 nM terfenadine
Cell ID ITerfenadine/IControl
a 1 bw 020625 000 0.29
b 1 bw 0206250000 0.19
Mean SEM 0.24 0.05
Frequency- or use-dependence of the effect with febuxostat on CHO/HERG current
In experiments to measure frequency-dependence of the agonist effect with 1 M
febuxostat, enhancement of CHO/hERG peak current measured at +60 mV and -50 mV
was not observed at 0.3 Hz stimulus repetition frequency but was pronounced at
3 Hz
frequency (Figures 10 and 11). These results indicate that agonist effect of
febuxostat was
frequency or use-dependent. The effect may be more pronounced at higher
frequencies.
Febuxostat modulation of CHO/hERG voltage gating parameters
The families of current traces from one cell, as an example, analyzed to
produce
the steady state I-V relation in Figure 12 are shown superimposed in Figure
13. The
steady-state current-voltage (I-V) relation was measured in three cells before
and after
exposure to febuxostat (Figure 12). The maximum increase of CHO/hERG current
by 1
.M febuxostat occurs at +10 and +20 mV. This result is consistent with the
pronounced
effect on current at +20 mV and the relative lack of effect on current at -50
mV seen in
Figures 1, 2 and 5.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
The steady state conductance-voltage (G-V) relation (Figure 14) was
constructed
from measurements of the peak tail current at voltages less than +60 mV
normalized by
the average of peak tail current measured at +60, +70 and +80 mV. The values
for the
midpoint potential (Vlia) were 0.9 and -2.1 mV in the absence and presence of
febuxostat, respectively. The slope factors (K,) were 9.9 and 9.8 mV per e-
fold change
during equilibration with control and 1 M febuxostat, respectively. The
differences in
the absence and presence of febuxostat for the V zi2 and K,, values were
insignificant and
small, indicating that the agonist effect of febuxostat is not the result of a
simple shift in
the voltage dependence of hERG activation to more negative potentials.
The families of current traces from a CHO/hERG cell, as an example, analyzed
to
produce Figure 15 are shown superimposed in Figure 16. The fully-activated I-V
relation
(Figure 15) shows the agonist effect of febuxostat developed at +60 mV was
abolished by
repolarizing to potentials of -60 mV or less. The stimulation persists through
the potential
range where inactivation gating causes rectification of the fully activated I-
V relation
(positive to -50 mV). This is consistent with the reduced agonist effect seen
in peak tail
current measurement during negative potentials.
The families of current traces from a CHO/hERG cell, as an example, analyzed
to
produce Figure 17 are shown superimposed in Figure 18. The channel
availability-
voltage relationship (Figure 17), like the normalized G-V relationship,
changes little in
response to application of febuxostat. Channel inactivation is equal to "1 -
channel
availability". The gating parameter values for the midpoint potential Vl/a in
the absence
and presence of 1 M febuxostat were -67.6 and -67.3 mV, respectively. The
values for
K,, were 27.9 and 29.6 mV per e-fold change for control and 1 gM febuxostat,
respectively. These results suggest that a simple voltage shift in channel
availability to
more positive potentials does not explain the agonist effect of febuxostat.
56
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
'1'he instantaneous I-V relation (r'igure 19) measures the conductance
properties of
open hERG channels. This measurement is based on the ability of the voltage
clamp to
change membrane potential much faster than channels gate, so that the number
of
channels open at -80 mV remains unchanged for a short time after the change in
membrane potential specified in the voltage protocol. The current measured
immediately
after the change in voltage from -80 mV is free of channel gating and reflects
only the
conductance properties of the open hERG channel. Like many potassium channels,
the I-
V relation measured for the open channel is linear over the voltage range
measured and
all the rectification associated with the hERG channel is derived from voltage-
dependent
gating. There is a small increase in the slope of the instantaneous I-V for
hERG channels
indicating a small increase in the number of open channels in the presence of
febuxostat,
but the linearity of the I-V relation is unaffected, demonstrating that
febuxostat does not
alter the conductance properties of open hERG channels.
In summary, febuxostat had an agonist effect on hERG currents measured with
the whole cell patch clamp method in CHO cells stably expressing cloned hERG
channels. The agonist effect was voltage dependent and more pronounced at
positive
potentials with maximal effect occurring at +10 and +20 mV. The agonist
response was
biphasic with an initial maximum and a smaller steady state effect during
maintained
application of febuxostat to the cells. The concentration dependence of the
initial
maximum and steady state effects yielded EC50 values of 0.003 M and 0.070 M,
respectively.
The agonist effect of febuxostat is voltage dependent and occurs rapidly.
Closed
channels are much less stimulated by febuxostat. Open channels and depolarized
potentials are required for stimulation and the stimulatory effect
equilibrates with open
channels rapidly. The agonist effect of febuxostat is not the result of a
simple shift in the
voltage dependence of hERG activation to more negative potentials, nor is a
simple
voltage shift in channel inactivation to more positive potentials. Febuxostat
does not alter
the conductance properties of open hERG channels. While not wishing to be
bound any
57
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
theory, the inventors believe that one possible mechanism for the agonist
effect consistent
with these observations could be to increase burst duration of hERG channel
openings.
EXAMPLE 3: Effect of febuxostat on action potentials and prolongation of
action
potential duration induced by dl-sotalol and ATX II in isolated cardiac
Purkinje
fibers
Material and Methods
Solutions and Chemicals
Chemicals used in preparation of experimental solutions were obtained from
Sigma-Aldrich (St. Louis, MO) or Calbiochem (San Diego, CA) and were of ACS
reagent grade purity. All solutions containing febuxostat were prepared in
glass
containers whenever possible. Test solutions of febuxostat were prepared daily
by
diluting stock solutions into a modified Tyrode's solution prepared fresh
weekly and
refrigerated, (composition in mM): NaCI, 131; KC1, 4.0; CaCl2, 2.0; MgCl2,
0.5;
NaHCO3, 18.0; NaH2PO4, 1.8; Glucose, 5.5. Before use, the Tyrode's solution
was
aerated with a mixture of 95% 02 aind 5% CO2 (pH 7.2 at room temperature).
Febuxostat
concentrations were prepared by serially diluting a 1000 .M stock solution in
Tyrode's
solution. The Tyrode's solution was warmed to room temperature before
preparing
febuxostat or positive control solutions. Febuxostat solutions were prepared
freshly no
more than six hours before use and protected from light. Febuxostat was tested
at
concentrations of 10, 100, and 1000 nM in the Purkinje fiber assay.
dl-Sotalol (Sigma-Aldrich) is a potent (3-adrenergic receptor antagonist with
class
III antiarrhythmic properties. The drug prolongs the cardiac action potential
duration
(APD) by selectively blocking the rapid delayed rectifier potassium current,
IKr. dl-
Sotalol solutions were prepared fresh daily by directly dissolving the
chemical into
Tyrode's solution.
58
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
ATX II (toxin II, Anenionia sulcata) was obtained from Calbiochem and is a
toxic
polypeptide component of sea anemone venom. ATX II acts specifically on
voltage-
gated Na+ channels of excitable membranes to induce persistent non-
inactivating Na+
currents. These persistent Na currents cause APD prolongation. Test solutions
were
prepared by dilution with Tyrode's solution of a 1000-fold concentrated stock
prepared in
distilled water.
Purkinje Fiber Electrophysiology
Fiber Preparation
Purkinje fibers were excised from canine ventricles by standard methods
(Gintant
et al., 2001). Briefly, 5-7 purpose-bred Beagle dogs (young adult female,
Marshall Farms
USA Inc., NY) were housed in AAALAC accredited facilities. On each test day a
dog
was anesthetized with sodium pentobarbital (30 mg/kg i.v.). The heart was
rapidly
removed through a left lateral thoracotomy, placed in a container with
chilled,
oxygenated, storage Tyrode's solution (8 mM KCl), and transported to ChanTest
on wet
ice. All usable free-running Purkinje fibers from both ventricles were removed
along
with their muscle attachments. The fibers were stored at room temperature in
oxygenated
standard Tyrode's solution (4 mM KCl) until use.
Electroplzysiological Recording
Purkinje fibers were mounted in a glass-bottomed Plexiglas chamber
(approximate volume, 1 ml) affixed to a heated platform, and superfu.sed at
approximately 4 ml/min with standard Tyrode's solution. The bath temperature
was
maintained at 37 1 C using a combination of SH-27B in-line solution pre-
heater,
Series 20 chamber platform heater, and TC-344B dual channel feedback
temperature
controller (Warner Instruments, Inc., Hamden, CT). Bath temperature was
recorded
using a thermistor probe. Intracellular membrane potentials were recorded
using
conventional intracellular microelectrodes pulled from borosilicate glass
capillary tubing
on a Sutter Instruments P-97 horizontal puller (Sutter Instrument Co., Novato,
CA), filled
with 3 M KCl solution and connected via Ag-AgCI wire to a Wamer Instruments IE
210
59
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
intracellular electrometer amplifier (Wamer Instruments, Inc., Hamden, CT).
Membrane
potential was referenced to a Ag-AgCl wire electrode in contact with the
Tyrode's
solution via a 3 M KCl-agar bridge.
Action potentials were evoked by repetitive electrical stimuli (0.1-3 ms
duration,
approximately 1.5 times threshold amplitude). A bipolar, insulated (except at
the tip)
platinum wire electrode was used to deliver pulses generated by a Dagan Corp.
S-900
photo-isolated, electronic stimulator (Dagan Corp., Minneapolis, MN). Analog
signals
were low-pass filtered at 20 kHz before digitization at 50 kHz with a DT3010
AD/DA
board (Data Translation, Inc., Marlboro, MA), and stored on hard disk using a
PC-
compatible computer controlled by NOTOCORD-HEM 3.5 software (Notocord Systems
SA, Croissy sur Seine, France).
Concentration-response and rate-dependence were determined by the following
test procedure. Purkinje fibers were paced continuously at a BCL of 2 s
(equivalent to
stimulation frequency of 0.5 Hz) during a stabilization period of at least 25
minutes
before obtaining control AP responses. Only fibers with resting potentials
more negative
than -80 mV and normal AP morphology (APD90 = 250-450 ms) were used.
Acceptable
fibers were stimulated continuously at BCL of 2 s for 20 minutes. At the end
of this
period, baseline APD rate- or frequency- dependence under control conditions
was
measured using stimulus pulse trains consisting of approximately 50 pulses at
BCL of 2,
1 and 0.34 s (equivalent to stimulation frequency of 1 and 3Hz, respectively).
After
returning to BCL of 2 s, test solution at the lowest concentration was applied
for 20
minutes to allow equilibration, and the stimulus trains repeated. The entire
sequence (20
minutes of equilibration followed by three cycles of stimulus trains at
decreasing BCL, a
total of 23-minuts per cycle) was repeated at increased drug concentration
cumulatively.
The average responses from the last five recorded action potentials from each
stimulus
train were analyzed for each test condition.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Purkinje Fiber Electrophysiological Response to Febuxostat
Three concentrations of febuxostat (10, 100 and 1000 nM) were applied
cumulatively (e.g. three 23-minute exposure periods) to a group of four
Purkinje fibers as
outlined above to examine the effect of febuxostat on action potential
parameters and
rate-dependence of these effects.
Modulation of Purkinje Fiber Electrophysiological Response to Sotalol or ATX H
by Febuxostat
Modulation of Purkinje fiber response to sotalol by febuxostat was assayed by
measuring responses in fibers exposed to both compounds and comparing to the
response
to sotalol alone. In the sotalol alone group, sotalol at 50 M was applied to
four Purkinje
fibers with exposure times in each fiber approximately the same as in the
febuxostat test
group (three 23-minute exposure periods). In the sotalol plus febuxostat
group, sotalol at
50 M was applied throughout the measurement periods and febuxostat at 100 and
1000 nM was applied during the second and third, respectively, 23-minute
exposure
periods.
In a similar series of experiments with ATX II, the responses of four Purkinje
fibers to application of 20 nM ATX II alone for all three 23-minute exposure
periods
were measured. The experiment was repeated in seven fibers but with addition
of 100
(exposure periods 1 and 2) and 1000 nM (exposure period 3) febuxostat to ATX
R.
Data Analysis
Action Potential Analysis
Data were analyzed with the AP analysis module of Notocord-Hem version 3.5
and Microsoft Excel 2000. The following parameters were determined: RMP
(resting
membrane potential, mV), APA (action potential amplitude, mV), Vmax (maximum
rate
of rise V/s), APD60 and APD90 (action potential duration at 60 and 90%
repolarization,
respectively, ms). Concentration-response data are presented relative to
baseline before
test article application. APD60, APD90 and Vmax at each stimulus frequency are
61
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
presented as percent change (A%) from baseline at each concentration. RMP and
APA
data are presented as absolute change in membrane potential (OmV).
Statistical Analysis
Data were reported as mean I SEM. Pooled data were tabulated for each
condition: control baseline, drug concentration and stimulus frequency.
Changes in action
potential parameters induced by febuxostat, 50 M sotalol or 20 nM ATX II were
evaluated using a two-tailed Student's t-test for paired samples to determine
whether the
means obtained during the drug-free control period are significantly different
(P<0.05)
from those obtained after equilibration in each drug concentration. The
effects of
febuxostat on sotalol- or ATX II-induced changes in action potential
parameters were
evaluated by a Student's t-test comparing the data obtained in the presence of
20 nM
ATX-II or 50 M sotalol alone, and the data from time-matched experiments
performed
in the presence of one of these agents together with febuxostat. Statistical
analyses were
performed in Microsoft Excel 2000.
Results
Effect of febuxostat on action potential parameters
At BCL that simulates bradycardia (BCL=2s), the average change in APD90 was
-2.4 0.8%, -1.9 0.7% and -6.8 3.7%, respectively, at febuxostat
concentrations 10,
100 and 1000 nM. (Table 9, Figure 20). At shorter cycle lengths of ls and
0.34s
(simulating normocardia and tachycardia, respectively) the average change in
APD90 was
-2.4 1.0% and -2.0 1.0%, respectively, at 1000 nM febuxostat (Table 10 and
11).
None of these small effects were statistically significant (P<0.05), and also
they are not
considered to be biologically significant. As shown in Tables 12-14,
febuxostat did not
significantly change the maximum rate of rise (Vmax), action potential
amplitude or
resting potential amplitude at any concentrations or BCL regimens.
In summary, despite the fact that febuxostat is a bERG agonist, it had no
effect by
itself on action potential parameters.
62
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 9: Effect of febuxostat on action potential duration at 2 second BCL
Fiber ID Febuxostat APD60 APD90
(nM) (ms) (A%) (ms) (A%)
06 MR 0212100 0 269.3 NA 336.5 NA
07 GEK 0212120 0 232.5 NA 281.2 NA
307 GEK 0212120 0 261.4 NA 309.2 NA
07 GEK 0212100 0 277.0 NA 351.9 NA
Mean 260.1 319.7
SEM 9.7 15.6
06 MR 0212100 10 244.1 -9.4 329.0 -2.2
07 GEK 0212120 10 231.9 -0.2 274.9 -2.2
07 GEK 0212120 10 260.8 -0.2 307.7 -0.5
07 GEK 0212100 10 267.7 -3.4 335.9 -4.5
Mean 251.1 -3.3 311.9 -2.4
SEM 8.1 2.2 13.7 0.8
06 MR 0212100 100 241.9 -10.2 330.9 -1.6
07 GEK 0212120 100 233.7 0.6 276.7 -1.6
307 GEK 0212120 100 263.7 0.9 307.6 -0.5
07 GEK 0212100 100 273.9 -1.1 337.9 -4.0
Mean 253.3 -2.5 313.3 -1.9
SEM 9.3 2.6 13.8 0.7
06 MR 0212100 1000 255.0 -5.3 335.8 -0.2
07 GEK 0212120 1000 229.4 -1.3 270.2 -3.9
307 GEK 0212120 1000 246.6 -5.7 291.3 -5.8
07 GEK 0212100 1000 279.4 0.8 291.3 -17.2
Mean 252.6 -2.9 297.1 -6.8
SEM 10.4 1.6 13.8 3.7
BCL, basic cycle length. 0%, Percent change from control values. NA, not
applicable.
63
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 10: Effect of febuxostat on action potential duration at 1 second BCL
Fiber ID Febuxostat APD60 APD90
(nM) (ms) (A%) (ms) (A%
06 MR 0212100 0 246.1 NA 305.1 NA
07 GEK 0212120 0 207.6 NA 253.2 NA
307 GEK 0212120 0 233.8 NA 278.0 NA
07 GEK 0212100 0 248.6 NA 313.9 NA
Mean 234.0 287.6
SEM 9.4 13.8
06 MR 0212100 10 218.4 -11.3 295.3 -3.2
07 GEK 0212120 10 210.4 1.3 252.7 -0.2
307 GEK 0212120 10 226.9 -3.0 274.0 -1.5
07 GEK 0212100 10 241.7 -2.8 298.9 -4.8
Mean 224.3 -3.9 -280.2 -2.4
SEM 6.7 2.6 10.7 1.0
06 MR 0212100 100 220.4 -0.1 299.0 -2.0
07 GEK 0212120 100 215.2 3.7 258.1 1.9
307 GEK 0212120 100 242.1 3.5 284.3 2.2
07 GEK 0212100 100 250.0 0.6 307.3 -2.1
Mean 231.9 1.9 287.2 0.0
SEM 8.4 1.0 10.8 1.2
06 MR 0212100 1000 214.7 -12.7 295.1 -3.3
07 GEK 0212120 1000 211.1 1.7 254.0 0.3
07 GEK 0212120 1000 230.1 -1.6 275.2 -1.0
07 GEK 0212100 1000 246.3 -0.9 300.0 -4.4
Mean 225.6 -3.4 281.1 -2.1
SEM 8.0 3.2 10.5 1.1
BCL, basic cycle length. A%, Percent change from control values. NA, not
applicable.
64
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 11: Effect of febuxostat on action potential duration at 0.34 second BCL
Fiber ID Febuxostat APD60 APD90
(nM) (ms) (A%) (ms) (A%)
06 MR 0212100 0 147.1 NA 202.4 NA
07 GEK 0212120 0 153.7 NA 197.2 NA
307 GEK 0212120 0 166.2 NA 212.0' NA
07 GEK 0212100 0 170.8 NA 221.2 NA
Mean 159.4 208.2
SEM 5.5 5.3
06 MR 0212100 10 136.2 -7.4 205.1 1.3
07 GEK 0212120 10 156.6 1.9 199.4 1.1
307 GEK 0212120 10 159.9 -3.8 206.2 -2.7
07 GEK 0212100 10 166.7 -2.4 211.7 -4.3
Mean 154.8 -2.9 205.6 -1.1
SEM 6.6 1.9 2.5 1.4
06 MR 0212100 100 129.9 -11.7 201.7 -0.3
07 GEK 0212120 100 156.5 1.8 200.6 1.7
307 GEK 0212120 100 168.6 1.5 211.8 -0.1
07 GEK 0212100 100 163.2 -4.5 207.7 -6.1
Mean 154.6 -3.2 205.5 -1.2
SEM 8.6 3.2 2.6 1.7
06 MR 0212100 1000 118.8 -19.2 195.1 -3.6
07 GEK 0212120 1000 154.8 0.7 199.1 0.9
307 GEK 0212120 1000 160.8 -3.2 206.4 -2.6
07 GEK 0212100 1000 174.6 2.2 215.1 -2.7
Mean 152.3 -4.9 203.9 -2.0
SEM 11.9 4.9 4.4 1.0
A%, Percent change from control values. NA, not applicable.
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 12: Effect of febuxostat on resting and action potential amplitudes and
maximum action potential rate of rise at 2 second basic cycle length
Febuxostat RMP APA Vmax
(nM) (mV) (,&m (mV) (bm (V/s) (t~%)
0 -89.8 0.2 NA 109.8 2.7 NA 349.1 75.5 NA
-90.6 0.6 -0.8 0.6 109.2=L 3.1 -0.7 1.3 336.9-J, 79.1 -4.5 5.7
100 109.8 2.7 -0.7 0.4 108.4 3.4 1.510.8 311.1=L76.4 -12.3 4.0
1000 109.8 2.7 0.5 1.5 101.3=1: 4.4 -8.5 4.4 255.1+- 44.1 -20.5 5.8
Data are expressed as mean + SEM from n= 4 fibers. A%, Percent change from
baseline
values; AmV, absolute change from baseline in millivolts; NA, not applicable;
RMP,
resting membrane potential; APA, action potential amplitude; Vmax, maximum
action
potential rate of rise.
Table 13: Effect of febuxostat on resting and action potential amplitudes and
maximum action potential rate of rise at 1 second basic cycle length
Febuxostat RMP APA Vmax
(nM) (mV) (AmV) (mV) (AmV) (V/s) (A%)
0 -91.6 zL 0.8 NA 112.7=L 2.4 NA 363.8 zL 90.6 NA
10 -92.3 0.6 -0.7 1.3 112.0+ 2.7 -0.7 :1:1.2 357.7f 96.7 -3.0 5.3
100 -92.3 0.5 -0.7 :L 0.9 111.7~ 3.1 -1.0 ~ 0.8 340.6:~101.5 -9.2 5.5
1000 -91.3 1.2 0.3 :L 1.8 106.6:h 2.6 -6.1 ~z 2.7 268.&z 57.3 -24.8 4.8
Data are expressed as mean SEM from n= 4 fibers. A%, Percent change from
baseline
values; AmV, absolute change from baseline in millivolts; NA, not applicable;
RMP,
resting meinbrane potential; APA, action potential amplitude; Vmax, maximum
action
potential rate of rise.
Table 14: Effect of febuxostat on resting and action potential amplitudes and
maximum action potential rate of rise at 0.34 second basic cycle length
Febuxostat RMP APA Vmax
(nM) (mV) (AmV) (mV) . (AmV) (V/s) (A%)
0 -90.6 10.8 NA 113.7zL 1.7 NA 351.6 89.5 NA
10 -92.3~0.6 -1.7 0.6 114.2~: 2.1 0.4~0.8 360.11110.4 -1.2:L 5.9
100 -92.0 10.9 -1.4 1.1 113.1+ 2.5 -0.6 ~ 1.5 347.9 113.3 -5.5 + 5.9
1000 -91.2 1.0 -0.6 1.3 110.3 1.9 -3.4 2.0 272.2164.2 -22.0 ~ 3.2
Data are expressed as mean SEM from n= 4 fibers. A%, Percent change from
baseline
values; AmV, absolute change from baseline in millivolts; NA, not applicable;
RMP,
resting membrane potential; APA, action potential amplitude; Vmax, maximum
action
potential rate of rise.
66
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Febuxostat modulation of prolonged action potential duration induced by dl-
sotalol
and ATX II
In contract to febuxostat, under identical recording conditions, the positive
control
dl-sotalol at 50 M produced significant APD prolongation (Figure 21). The
effect of the
same concentration of dl-sotalol increased overtime during each 23-minute
exposure
period, reflecting a slow component of sotalol equilibration with Purkinje
fiber tissue. At
the end of the third 23-minute exposure period, there were APD90 prolongation
of 40.9
8.8%, 34.8 7.5% and 14.6 7.2% at BCL of 2s (Figure 21, Table 15), 1s
(Table 16),
and 0.34s (Table 17), respectively. Sotalol at 50 M did not significantly
change the
maximum rate of rise (Vmax), action potential amplitude or resting potential
amplitude at
BCL 2s, ls and 0.34s. Figure 23 shows addition of 100 and 1000 nM febuxostat
together
with 50 M dl-sotalol did not change the prolongation of action potential
duration at 2s,
(Table 18, and Figure 23) or at is and 0.34s BCL (Tables 19 and 20).
Febuxostat
together with sotalol did not change the maximum rate of rise (Vmax), action
potential
amplitude or resting potential amplitude.
67
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 15: Effect of sotalol on action potential duration at 2 s basic cycle
len th
50 M Sotalol APD60 APD90
(ms) (0%) (ms) 0%)
Baseline 239.1 27.6 NA 301.64: 27.2 NA
Period 1 323.8=L 44.5* 35.5 8.0 393.2=L 46.7* 29.9:L 6.3
Period 2 334.1Jz 47.8* 39.8 10.2 404.1 51.7* 33.3:~ 8.0
Period 3 356.7 54.6* 48.9~--11.4 428.7:L 59.1* 40.9 8.8
Data are expressed as mean SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between baseline control and sotalol mean values
(P<0.05, t-test
paired samples).
Table 16: Effect of sotalol on action otential duration at 1 s basic cycle
length
50 M Sotalol APD60 APD90
(ms) (A%) (ms) (A%)
Baseline 212.6 20.8 NA 272.5 19.7 NA
Period 1 279.8:L 35.3* 31.5 8.3 344.8 36.5* 26.0 6.3
Period 2 284.9~ 37.6* 33.6 8.3 351.4 -38.6* 28.4:L7.0
Period 3 303.6 39.6* 42.6:L- 10.0 369.9-+42.9* 34.8f 7.5
Data are expressed as mean + SEM from n= 4 fibers.
d%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and sotalol mean values (P<0.05, t-test
paired
samples).
Table 17: Effect of sotalol on action otential duration at 0.34 s basic cycle
length
50 M Sotalol APD60 APD90
(ms) (A%) (ms) (A%)
Baseline 154.0:L 15.0 NA 205.1 11.4 NA
Period 1 163.5111.4 8.3+ 9.2 224.0-+ 10.6 10.2- 7.6
Period 2 169.3 10.1 12.3+- 9.2 232.5 10.7 14.4:L 7.6
Period 3 171.2~ 7.8 14.1f 10.6 232.9 8.9 14.6=L 7.2
Data are expressed as mean SEM from n= 4 fibers.
0%, Percent change from baseline control values. NA, not applicable.
68
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 18: Effect of febuxostat on sotalol-induced action potential duration
prolongation at 2 s basic cycle length
Fiber ID Sotalol Febuxostat APD60 APD90
( h'j) (nN]) (ms) (A%) (ms) (A%)
A 06 AR 0212171 0 0 275.3 NA 328.8 NA
B 06 AR 0212170 0 0 274.2 NA 326.4 NA
07 GK 0212170 0 0 248.1 NA 324.2 NA
B 07 GK 0212170 0 0 230.7 NA 276.5 NA
Mean 257.1 314.0
SEM 10.8 12.5
A06AR0212171 50 0 417.4 51.6 485.6 47.7
B 06 AR 0212170 50 0 370.9 35.3 446.8 36.9
07 GK 0212170 50 0 357.0 43.9 467.0 44.1
B 07 GK 0212170 50 0 384.2 66.5 442.8 60.2
Mean 382.4* 49.3 460.6* 47.2
SEM 12.9 6.6 9.9 4.9
A 06 AR 0212171 50 100 444.1 61.4 519.0 57.8
B 06 AR 02121.70 50 100 383.8 40.0 460.6 41.1
07 GK 0212170 50 100 357.9 44.3 473.2 46.0
B 07 GK 0212170 50 100 357.9 55.1 473.2 71.2
Mean 385.9* 50.2 481.5* 54.0
SEM 20.3 4.9 12.9 6.7
A 06 AR 0212171 50 1000 423.9 54.0 529.4 61.0
B 06 AR 0212170 50 1000 404.6 47.6 480.5 47.2
07 GK 0212170 50 1000 406.6 63.9 480.5 48.2
B 07 GK 0212170 50 1000 446.0 93.3 480.5 73.8
Mean 420.3* 64.7 492.7* 57.6
SEM 9.6 10.1 12.2 6.3
Data are expressed as mean ~ SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and sotalol mean values (P<0.05, t-test
paired
samples).
69
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 19: Effect of febuxostat on sotalol-induced action potential duration
rolon ation at 1 s basic cycle length
Fiber ID Sotalol EBITXOSTAT APD60 APD90
( M) (nM) (ms) (A%) (ms) (A%
06 AR 0212171 0 0 244.8 NA 294.7 NA
306 AR 0212170 0 0 248.3 NA 297.9 NA
07 GK 0212170 0 0 231.3 NA 296.4 NA
307 GK 0212170 0 0 213.5 NA 258.5 NA
Mean 234.5 286.9
SEM 7.9 9.5
06 AR 0212171 50 0 362.9 48.2 425.5 44.4
306 AR 0212170 50 0 327.3 31.8 393.0 31.9
07 GK 0212170 50 0 323.1 39.7 414.2 39.8
307 GK 0212170 50 0 325.7 52.6 382.7 48.0
Mean 334.8* 43.1 403.9* 41.0
SEM 9.4 4.6 9.8 3.5
06 AR 0212171 50 100 390.7 59.6 457.9 55.4
306 AR 0212170 50 100 337.1 35.8 403.8 35.5
07 GK 0212170 50 100 323.0 39.6 419.6 41.6
307 GK 0212170 50 100 333.0 56.0 396.3 53.3
Mean 345.9* 47.7 419.4* 46.5
SEM 15.2 5.9 13.7 4.7
06 AR 0212171 50 1000 367.4 50.1 461.2 56.5
306 AR 0212170 50 1000 353.8 42.5 419.9 41.0
07 GK 0212170 50 1000 357.7 54.6 452.9 52.8
07 GK 0212170 50 1000 374.4 75.4 436.9 69.0
Mean 363.3* 55.7 442.7* 54.8
SEM 4.7 7.0 9.1 5.8
Data are expressed as mean ~ SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and sotalol mean values (P<0.05, t-test
paired
samples).
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 20: Effect of febuxostat on sotalol-induced action potential duration
rolon ation at 0.34 s basic cycle length
Fiber ID Sotalol FEBUXOSTAT APD60 APD90
( M) (nryl) (ms) (A%) (ms) (A%)
06 AR 0212171 0 0 158.5 NA 200.0 NA
306 AR 0212170 0 0 151.2 NA 193.7 NA
07 GK 0212170 0 0 168.6 NA 210.8 NA
307 GK 0212170 0 0 153.5 NA 194.0 NA
Mean 157.9 199.6
SEM 3.9 4.0
06 AR 0212171 50 0 192.5 21.5 251.1 25.6
306 AR 0212170 50 0 178.7 18.2 232.2 19.9
07 GK 0212170 50 0 207.2 22.9 266.5 26.4
307 GK 0212170 50 0 194.1 26.4 246.7 27.2
Mean 193.1 22.3 249.1 24.8
SEM 5.8 1.7 7.1 1.7
06 AR 0212171 50 100 196.6 24.1 260.1 30.1
306 AR 0212170 50 100 179.5 18.7 236.3 22.0
07 GK 0212170 50 100 209.5 24.2 269.3 27.7
307 GK 0212170 50 100 189.9 23.7 249.4 28.5
Mean 193.9 22.7 253.8 27.1
SEM 6.3 1.3 7.1 1.8
06 AR 0212171 50 1000 155.3 -2.0 241.5 20.7
306 AR 0212170 50 1000 173.6 14.8 230.8 19.2
07 GK 0212170 50 1000 204.1 21.1 261.4 24.0
307 GK 0212170 50 1000 202.1 31.7 261.3 34.7
Mean 183.8 16.4 248.7 24.6
SEM 11.8 7.1 7.6 3.5
A%, Percent change from control values. NA, not applicable.
Like sotalol, 20 nM ATX II induced significant APD prolongation, which was
increased during each 23-minute exposure period, reflecting a slow component
of ATX II
equilibration with Purkinje fiber tissue. In addition, it elevated the plateau
potential as
well (Figure 22). At the end of the third 23-minute exposure period, ATX II at
20 M
71
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
induced a maximum prolongation of APD90 to 75.1 8.1% at 2s BCL (Table 21,
Figure
22), 46.4 6.2% at ls BCL (Table 22), and 13.6 2.8% at 0.34s BCL (Table
23).
Febuxostat at 1000 nM reduced the maximum ATX II-induced prolongation of APD90
to
37.2 3.6% (A% = -50% vs ATX II alone) at 2s BCL (Figure 24, Table 24), 27.7
2.5%
(A% _-40% vs ATX II alone) at 1 s BCL (Table 25), and 9.0 1.2% (A% = -34% vs
ATX II alone) at 0.34s BCL (Table 26). The blunting effect of 1000 nM
febuxostat on
ATX II-induced prolongation was statistically significant at BCL 2s and ls
(Figure 24).
Febuxostat at 100 nM moderately shortened APD prolongation induced by ATX-II
at all
stimulus intervals tested. However, the effect was only statistically
significant at BCL
0.34s (Figure 24). ATX II by itself or in association with febuxostat did not
alter the
maximum rate of rise (Vmax), action potential amplitude or resting potential
amplitude.
Table 21: Effect of ATX II on action potential duration at 2 s basic cycle
length
20 nM ATX II APD60 APD90
(ms) (A%) (ms) (A%)
Baseline 308.7=L 25.0 NA 367.9=L 22.2 NA
Period 1 420.6::L 28.7* 37.1::L6.3 490.6125.9* 34.0-L 6.3
Period 2 510.0+ 38.2* 65.9 7.1 585.3 39.1 * 59.6 8.7
Period 3 569.3 33.6* 86.4+-11.8 641.2:L33.6* 75.1=L 8.1
Data are expressed as mean + SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and ATX II mean values (P<0.05, t-test
paired
samples).
Table 22: Effect of ATX II on action potential duration at 1 s basic cycle
length
20 nM ATX II APD60 APD90
(ms) (~%) (ms) (A%)
Baseline 273.7:L 18.0 NA 327.6 15.0 NA
Period 1 343.0+ 18.0* 25.9+4.4 401.3 15.2* 22.8-J: 4.3
Period 2 384.1 22.2* 40.8 4.6 445.1zL 22.3* 36.2- 5.8
Peri od 3 423.0 19.9* 56.0 9.7 478.0 18.5* 46.4L 6.2
Data are expressed as mean SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and ATX II mean values (P<0.05, t-test
paired
samples).
72
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 23: Effect of ATX II on action potential duration at 0.34 s basic cycle
length
20 nM ATX II APD60 APD90
(ms) (~%) (ms)
Baseline 164.9 6.9 NA 210.9 5.3 NA
Period 1 186.0+ 7.6 12.9 3.1 230.3 5.8* 9.2 0.3
Period 2 194.4L 8.6* 18.04:3.6 238.6~: 4.9* 13.2 0.9
Period 3 195.4:L6.7* 19.0:L5.9 239.2 3.5* 13.6:L 2.8
Data are expressed as mean SEM from n= 4 fibers.
A%, Percent change from baseline control values. NA, not applicable.
*Statistically
significant difference between control and ATX II mean values (P<0.05, t-test
paired
samples).
73
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Table 24: Effect of febuxostat on ATX II-induced action potential duration
rolon ation at 2 basic cycle length
Fiber ID ATX II Febuxostat APD60 APD90
(nM) (nM) (ms) d% (ms)
A09LG0302041 0 0 214.8 NA 292.3 NA
B09LG0302041 0 0 219.6 NA 323.6 NA
A09LG0302061 0 0 345.1 NA 405.4 NA
B09LG0302061 0 0 257.1 NA 309.0 NA
B07MR0302060 0 0 242.9 NA 280.4 NA
A06AR03032001 0 0 273.9 NA 321.3 NA
B06AR03032001 0 0 277.0 NA 327.6 NA
Mean 261.5 322.8
SEM 16.7 15.2
A09LG03 02041 20 100 252.2 17.4 333.0 13.9
B09LG0302041 20 100 312.8 42.5 419.6 29.7
A09LG0302061 20 100 416.6 20.7 485.2 19.7
B09LG0302061 20 100 340.6 32.5 386.2 37.7
B07MR0302060 20 100 279.6 15.1 315.0 12.3
A06AR03032001 20 100 334.1 22.0 380.9 18.5
B06AR03032001 20 100 335.2 21.0 386.2 17.9
Mean 324.4 24.5 386.6 21.4
SEM 19.7 3.6 21.2 3.4
A09LG0302041 20 100 360.6 67.9 476.5 63.0
B09LG0302041 20 100 445.3 102.8 544.1 68.1
A09L00302061 20 100 517.3 49.9 596.8 47.2
B09LG0302061 20 100 430.5 67.4 471.7 68.2
B07MR0302060 20 100 315.2 29.8 346.8 23.7
A06AR03032001 20 100 391.6 43.0 442.8 37.8
B06AR03032001 20 100 398.1 43.7 447.3 36.5
Mean 408.4 57.8 475.1 49.2
SEM 24.4 9.1 30.0 6.6
A09LG0302041 20 1000 331.6 54.4 424.7 45.3
B09LG0302041 20 1000 391.2 78.2 479.2 48.1
A09LG0302061 20 1000 492.6 42.8 558.3 37.7
B09LG0302061 20 1000 402.8 56.7 445.9 44.3
B07MR0302060 20 1000 308.5 27.0 340.7 21.5
A06AR03032001 20 1000 375.0 36.9 425.7 32.5
B06AR03032001 20 1000 380.0 37.2 429.4 31.1
74
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
Mean 383.1 47.6 443.4 37.2
SEM 22.2 6.4 24.8 3.6
A%, Percent change from control values. NA, not applicable.
Table 25: Effect of febuxostat on ATX II-induced action potential duration
rolon ation at 1 basic cycle len th
Fiber ID A IX Febuxostat APD60 APD90
nM nM (ms) (A%) (ms) (~%)
09LG0302041 0 0 191.0 NA 260.0 NA
309LG0302041 0 0 196.5 NA 277.6 NA
09LG0302061 0 0 293.1 NA 346.6 NA
309LG0302061 0 0 222.7 NA 269.9 NA
307MR0302060 0 0 224.2 NA 262.2 NA
06AR0303200 0 0 239.2 NA 285.8 NA
1
06AR0303200 0 0 237.8 NA 284.9 NA
1
Mean 229.2 283.9
SEM 12.8 11.1
09LG03 02041 20 100 221.2 15.8 294.6 13.3
309LG03 02041 20 100 248.1 26.2 332.8 19.9
09LG03 02061 20 100 343.4 17.2 397.4 14.7
309LG0302061 20 100 284.3 27.7 325.5 20.6
307MR0302060 20 100 256.1 14.2 289.0 10.2
06AR0303200 20 100 280.6 17.3 324.8 13.6
1
06AR0303200 20 100 268.1 12.7 317.5 11.4
1
Mean 271.7 18.7 325.9 14.8
SEM 14.4 2.2 13.4 1.5
09LG0302041 20 100 268.0 40.3 356.7 37.2
309LG0302041 20 100 335.7 70.8 411.0 48.0
09LG0302061 20 100 398.8 36.1 457.7 32.1
309LG0302061 20 100 320.0 43.7 357.2 32.3
307MR0302060 20 100 274.7 22.5 304.8 16.2
06AR0303200 20 100 313.8 31.2 359.5 25.8
1
06AR0303200 20 100 8.4 29.7 355.0 24.6
1
Mean 317.0 39.2 371.7 30.9
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
SEM 16.4 5.9 18.4 3.8
09LG0302041 20 1000 274.0 43.5 356.1 37.0
309LG0302041 20 1000 301.6 53.5 374.1 34.8
09LG0302061 20 1000 390.3 33.2 442.8 27.8
309LG0302061 20 1000 310.6 39.5 349.4 29.5
07MR0302060 20 1000 280.7 25.2 312.1 19.0
06AR0303200 20 1000 300.7 25.7 351.9 23.1
1
06AR0303200 20 1000 303.3 27.5 349.3 22.6
1
Mean 308.7 35.4 362.2 27.7
SEM 14.5 4.0 15.1 2.5
A%, Percent change from control values. NA, not applicable.
Table 26: Effect of febuxostat on ATX II-induced action potential duration
rolon ation at 0.34 basic cycle len th
Fiber ID ATX Febuxostat APD60 APD90
nM nM (ms) (A%) (ms) (A%)
A09LG0302041 0 0 135.6 NA 185.5 NA
B09LG0302041 0 0 135.4 NA 195.1 NA
A09LG0302061 0 0 172.2 NA 215.4 NA
B09LG0302061 0 0 153.0 NA 193.1 NA
307MR0302060 0 0 166.4 NA 205.3 NA
A06AR0303200 0 0 184.4 NA 227.7 NA
1
B06AR0303200 0 0 187.6 NA 229.7 NA
1
Mean 162.1 L 207.4
SEM 8.1 6.6
A09LG03 02041 20 100 142.5 5.1 193.7 4.4
B09L00302041 20 100 147.9 9.3 206.7 6.0
A09LG0302061 20 100 184.4 7.1 227.4 5.5
B09LG0302061 20 100 173.1 13.2 208.9 8.2
307MR0302060 20 100 172.4 3.7 208.9 1.8
A06AR0303200 20 100 200.2 8.6 242.2 6.4
1
B06AR0303200 20 100 192.4 2.6 236.5 3.0
1
Mean 173.3 7.1 217.8 5.0
SEM 8.2 1.4 6.7 0.8
76
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
A09LG03 02041 20 100 147.1 8.5 201.9 8.8
B09LG0302041 20 100 166.5 23.0 221.9 13.7
A09LG0302061 20 100 195.0 13.2 237.4 10.2
B09LG0302061 20 100 179.9 17.6 212.4 10.0
B07MR0302060 20 100 179.9 8.1 210.9 2.7
A06AR0303200 20 100 216.5 17.4 258.2 13.4
1
B06AR0303200 20 100 207.1 10.4 248.4 8.1
1
Mean 184.6 14.0 227.3 9.6
SEM 9.0 2.1 8.0 1.4
A09LG03 02041 20 1000 148.3 9.4 204.3 10.2
B 09LG03 02041 20 1000 162.4 20.0 220.4 13.0
A09LG03 02061 20 1000 196.6 14.1 235.9 9.5
B09LG03 02061 20 1000 174.9 14.4 209.2 8.3
B07MR0302060 20 1000 180.8 8.7 212.6 3.6
A06AR0303200 20 1000 212.5 15.2 254.2 11.6
1
B06AR0303200 20 1000 204.4 9.0 245.9 7.1
1
Mean 182.8 13.0 226.1 9.0
SEM 8.7 1.6 7.3 1.2
A%, Percent change from control values. NA, not applicable.
In conclusion, febuxostat at 10, 100 ancl. 1000 nM did not have any effects on
action potential parameters by itself. Febuxostat had no effect on sotalol-
induced action
potential prolongation. However, febuxostat at 100 and 1000 nM dose-
dependently
shortened the ATX II-induced action potential prolongation.
One skilled in the art would readily appreciate that the present invention is
well
adapted to carry out the objects and obtain the ends and advantages mentioned,
as well as
those inherent therein. The molecular complexes and the methods, procedures,
treatments, molecules, specific compounds described herein are presently
representative
of preferred embodiments, are exemplary, and are not intended as limitations
on the
scope of the invention. It will be readily apparent to one skilled in the art
that varying
substitutions and modifications may be made to the invention disclosed herein
without
departing from the scope and spirit of the invention.
77
CA 02630639 2008-05-21
WO 2007/062028 PCT/US2006/045042
All patents and publications mentioned in the specification are indicative of
the
levels of those skilled in the art to which the invention pertains. All
patents and
publications are herein incorporated by reference to the same extent as if
each individual
publication was specifically and individually indicated to be incorporated by
reference.
The invention illustratively described herein suitably may be practiced in the
absence of any element or elements, limitation or limitations which is not
specifically
disclosed herein. Thus, for example, in each instance herein any of the terms
"comprising," "consisting essentially of' and "consisting of' may be replaced
with either
of the other two terms. The terms and expressions which have been employed are
used as
terms of description and not of limitation, and there is no intention that in
the use of such
terms and expressions of excluding any equivalents of the features shown and
described
or portions thereof, but it is recognized that various modifications are
possible within the
scope of the invention claimed. Thus, it should be understood that although
the present
invention has been specifically disclosed by preferred embodiments and
optional
features, modification and variation of the concepts herein disclosed may be
resorted to
by those skilled in the art, and that such modifications and variations are
considered to be
within the scope of this invention as defmed by the appended claims.
78