Note: Descriptions are shown in the official language in which they were submitted.
CA 02630755 2008-05-06
PROCESS FOR THE PRODUCTION OF LOW SULFUR
DIESEL AND HIGH OCTANE NAPHTHA
BACKGROUND OF THE INVENTION
[0001] This invention relates to a process for the production of low sulfur
diesel and high
octane naphtha. More specifically, the invention is an integrated process for
the
hydrodesulfurization of middle distillate hydrocarbon streams and the
hydrocracking of highly
aromatic hydrocarbon streams.
[0002] Petroleum refiners produce desirable products such as turbine fuel,
diesel fuel and
middle distillates, as well as naphtha and gasoline, by hydrocracking a
hydrocarbon feedstock
derived from crude oil, for example. Feedstocks most often subjected to
hydrocracking are gas
oils and heavy gas oils recovered from crude oil by distillation. Refiners
also subject middle
distillate hydrocarbon streams to hydrodesulfurization. Although a wide
variety of process flow
schemes, operating conditions and catalysts have been used in commercial
activities, there is
always a demand for new hydroprocessing methods which provide lower costs,
more valuable
product yields and improved operability.
INFORMATION DISCLOSURE
[0003] US 4,943,366 (Fischer et al.) discloses a hydrocracking process for
converting highly
aromatic, substantially dealkylated feedstock into high octane gasoline.
BRIEF SUMMARY OF THE INVENTION
[0004] The present invention is an integrated process for the production of
low sulfur diesel
and high octane naphtha. The process of the present invention utilizes a
middle distillate
hydrocarbon stream and a highly aromatic hydrocarbon stream. The middle
distillate
hydrocarbon feedstock is reacted with fresh make-up hydrogen in a
hydrodesulfurization reaction
zone and the highly aromatic hydrocarbon stream is reacted with recycle
hydrogen in a
hydrocracking zone. The resulting effluents from the two zones are introduced
into a common
high pressure vapor liquid separator to produce a hydrogen-rich recycle gas
and a liquid stream
containing desulfurized diesel and high octane naphtha which are then
subsequently separated.
CA 02630755 2008-05-06
[0005] The use of make-up hydrogen as the source of hydrogen for the
hydrodesulfurization
reaction zone minimizes the recycle compressor duty and the use of the common
separation and
fractionation zones result in the advantages of lower capital and operating
expenses.
100061 Other embodiments of the present invention encompass further details
such as
detailed descriptions of feedstocks, hydrodesulfurization catalysts,
hydrocracking catalysts, and
preferred operating conditions, all of which are hereinafter disclosed in the
following discussion
of each of these facets of the invention.
BRIEF DESCRIPTION OF THE DRAWING
[0007] The drawing is a simplified process flow diagram of a preferred
embodiment of the
present invention. The drawing is intended to be schematically illustrative of
the present
invention and not be a limitation thereof.
DETAILED DESCRIPTION OF THE INVENTION
[00081 The present invention is an integrated process for the
hydrodesulfurization of middle
distillate hydrocarbon streams and the hydrocracking of highly aromatic
hydrocarbon streams.
Preferred feedstocks to the hydrodesulfurization reaction zone include
distillate hydrocarbons
boiling at a temperature greater than 149 C (300 F) and more preferably
boiling in the range
from 149 C (300 F) to 399 C (750 F). Distillate hydrocarbon feedstocks are
most often
recovered from crude oil by distillation. However, distillate hydrocarbons may
be utilized from
any convenient source such as tar sand extract and gas to liquids for example.
Furthermore, the
distillate hydrocarbon feedstocks may contain from 0.1 to 4 wt-% sulfur.
100091 The preferred highly aromatic hydrocarbon feedstocks boil in the range
from 149 C
(300 F) to 343 C (650 F). Highly aromatic, substantially dealkylated
hydrocarbons are produced
during the fluid catalytic cracking (FCC) of vacuum gas oils to produce high
octane gasoline
boiling range hydrocarbons. FCC is a thermally severe process which is
operated without the
presence of added hydrogen to reject carbon to coke and to produce residual
fractions. During
catalytic cracking, the high molecular weight feedstock disproportionates into
relatively
hydrogen-rich light liquids and aromatic, hydrogen-deficient heavier
distillates and residues. The
catalytic cracking in the absence of hydrogen does not provide significant
desulfurization nor is
-2-
CA 02630755 2008-05-06
the nitrogen content of the feed selectively rejected with the coke. The
sulfur and nitrogen
therefore concentrate in heavier cracking products aild produces significant
quantities of highly
aromatic, hydrogen-deficient middle and heavy distillates with high sulfur and
nitrogen levels.
Recycling these liquids to the catalytic cracker is not an attractive option.
A typical light cycle oil
(LCO) from an FCC contains 3 wt-% sulfur, 700 wppm nitrogen and greater than
80 vol-%
aromatics. Present market requirements make refractory product streams such as
light cycle oil
particularly difficult to dispose of as commercially valuable products.
[0010] In one embodiment of the present invention, a highly aromatic, hydrogen
deficient
and substantially dealkylated hydrocarbon feedstock is introduced into a
hydrocracking zone. The
hydrocracking zone may contain one or more beds of the same or different
catalyst. In one
embodiment the preferred hydrocracking catalysts utilize amorphous bases or
low-level zeolite
bases combined with one or more Group VIII or Group VIB metal hydrogenating
components. In
another embodiment the hydrocracking zone contains a catalyst which comprises,
in general, any
crystalline zeolite cracking base upon which is deposited a minor proportion
of a Group VIII
metal hydrogenating component. Additional hydrogenating components may be
selected from
Group VIB for incorporation with the zeolite base. The zeolite cracking bases
are sometimes
referred to in the art as molecular sieves and are usually composed of silica,
alumina and one or
more exchangeable cations such as sodium, magnesium, calcium, rare earth
metals, etc. They are
further characterized by crystal pores of relatively uniform diameter between
4 and 14 angstroms
(10-10 meters). It s preferred to employ zeolites having a silica/alumina mole
ratio between 3 and
12. Suitable zeolites found in nature include, for example, mordenite,
stillbite, heulandite,
ferrierite, dachiardite, chabazite, erionite and faujasite. Suitable synthetic
zeolites include, for
example, the B, X, Y and L crystal types, e.g., synthetic faujasite and
mordenite. The preferred
zeolites are those having crystal pore diameters between 8 and 12 angstroms
(10-10 meters),
wherein the silica/alumina mole ratio is 4 to 6. A prime example of a zeolite
falling in the
preferred group is synthetic Y molecular sieve.
100111 The natural occurring zeolites are normally found in a sodium form, an
alkaline earth
metal form, or mixed forms. The synthetic zeolites are nearly always prepared
first in the sodium
form. In any case, for use as a cracking base it is preferred that most or all
of the original zeolitic
monovalent metals be ion-exchanged with a polyvalent metal and/or with an
ammonium salt
-3-
CA 02630755 2008-05-06
followed by heating to decompose the ammonium ions associated with the
zeolite, leaving in
their place hydrogen ions and/or exchange sites which have actually been
decationized by further
removal of water. Hydrogen or "decationized" Y zeolites of this nature are
more particularly
described in US 3,130,006.
100121 Mixed polyvalent metal-hydrogen zeolites may be prepared by ion-
exchanging first
with ammonium salt, then partially back exchanging with a polyvalent metal
salt and then
calcining. In some cases, as in the case of synthetic mordenite, the hydrogen
forms can be
prepared by direct acid treatment of the alkali metal zeolites. The preferred
cracking bases are
those which are at least 10 percent, and preferably at least 20 percent, metal-
cation-deficient,
based on the initial ion-exchange capacity. A specifically desirable and
stable class of zeolites are
those wherein at least 20 percent of the ion exchange capacity is satisfied by
hydrogen ions.
[00131 The active metals employed in the preferred hydrocracking catalysts of
the present
invention as hydrogenation components are those of Group VIII, i.e., iron,
cobalt, nickel,
ruthenium, rhodium, palladium, osmium, iridium and platinum. In addition to
these metals, other
promoters may also be employed in conjunction therewith, including the metals
of Group VIB,
e.g., molybdenum and tungsten. The amount of hydrogenating metal in the
catalyst can vary
within wide ranges. Broadly speaking, any amount between 0.05 percent and 30
percent by
weight may be used. In the case of the noble metals, it is normally preferred
to use 0.05 to 2 wt-
%. The preferred method for incorporating the hydrogenating metal is to
contact the zeolite base
material with an aqueous solution of a suitable compound of the desired metal
wherein the metal
is present in a cationic form. Following addition of the selected
hydrogenating metal or metals,
the resulting catalyst powder is then filtered, dried, pelleted with added
lubricants, binders or the
like if desired, and calcined in air at temperatures of, e.g., 371 -648 C (700
-1200 F) in order to
activate the catalyst and decompose ammonium ions. Alternatively, the zeolite
component may
first be pelleted, followed by the addition of the hydrogenating component and
activation by
calcining. The foregoing catalysts may be employed in undiluted form, or the
powdered zeolite
catalyst may be mixed and copelleted with other relatively less active
catalysts, diluents or
binders such as alumina, silica gel, silica-alumina cogels, activated clays
and the like in
proportions ranging between 5 and 90 wt-%. These diluents may be employed as
such or they
-4-
CA 02630755 2008-05-06
may contain a minor proportion of' an added hydrogenating metal such as a
Group VIB and/or
Group VIII metal.
[00141 Additional metal promoted hydrocracking catalysts may also be utilized
in the process
of the present invention which comprises, for example, aluminophosphate
molecular sieves,
crystalline chromosilicates and other crystalline silicates. Crystalline
chromosilicates are more
fully described in US 4,363,718 (Klotz).
[0015] The hydrocracking of the hydrocarbonaceous feedstock in contact with a
hydrocracking catalyst is conducted in the presence of hydrogen and preferably
at hydrocracking
reactor conditions which include a temperature from 260 C (500 F) to 426 C
(800 F), a pressure
from 7.0 MPa (1000 psig) to 10.5 MPa (1500 psig), a liquid hourly space
velocity (LHSV) from
0.1 to 30 hr 1, and a hydrogen circulation rate from 2000 (337 normal m3/m3)
to 25,000 (4200
normal m3/m3) standard cubic feet per barrel.
[0016] The resulting effluent from the hydrocracking zone is preferably
contacted with an
aqueous stream to dissolve any ammonium salts and partially condensed, and
then introduced
into a high pressure vapor-liquid separator operated at a pressure
substantially equal to the
hydrocracking zone and a temperature in the range from 38 C (100 F) to 71 C
(160 F). An
aqueous stream is recovered from the vapor-liquid separator. A hydrogen-rich
gaseous stream is
removed from the vapor-liquid separator to provide at least a majority and
preferably all of the
hydrogen introduced into the hydrocracking zone.
[0017] In one embodiment of the present invention, a distillate hydrocarbon
boiling at a
temperature greater than 149 C (300 F) is introduced into a desulfurization
reaction zone
together with a hydrogen-rich make-up stream at desulfurization reaction
conditions. Preferred
desulfurization reaction conditions include a temperature from 260 C (500 F)
to 426 C (800 F),
a pressure from 7.0 MPa (1000 psig) to 10.5 MPa (1500 psig), and a liquid
hourly space velocity
from 0.1 hr-I to 10 hr"1.
[00181 Suitable desulfurization catalysts for use in the present invention are
any known
convention desulfurization catalysts and include those which are comprised of
at least one Group
VIII metal, preferably iron, cobalt and nickel, more preferably cobalt and/or
nickel and at least
one Group VI metal, preferably molybdenum and tungsten, on a high surface area
support
material, preferably alumina. Other suitable desulfurization catalysts include
zeolitic catalysts, as
-5-
CA 02630755 2008-05-06
well as noble metal catalysts where the noble metal is selected from palladium
and platinum. It is
within the scope of the present invention that more than one type of
desulfurization catalyst be
used in the same reaction vessel. Two or more catalyst beds and one or more
quench points may
be utilized in the reaction vessel or vessels. The Group VIII metal is
typically present in an
amount ranging from 2 to 20 wt- ro, preferably from 4 to 12 wt-%. The Group VI
metal will
typically be present in an amount ranging from I to 25 wt-%, preferably from 2
to 25 wt-%.
[0019] The resulting effluent from the desulfurization reaction zone is
contacted with an
aqueous stream and partially condensed, and then introduced into the
previously described high
pressure vapor-liquid separator.
[0020] Fresh make-up hydrogen is directly introduced into the desulfurization
reaction zone
in order to supply high purity hydrogen to maximize the activity of the
desulfurization catalyst. In
a preferred embodiment, at least a majority of the hydrogen-rich recycle gas
which is recovered
in the high pressure vapor-liquid separator is recycled to the hydrocracking
zone and more
preferably essentially all of the hydrogen-rich recycle gas is recycled to the
hydrocracking zone.
DETAILED DESCRIPTION OF THE DRAWING
[0021] In the drawing, the process of the present invention is illustrated by
means of a
simplified schematic flow diagram in which such details as pumps,
instrumentation, heat-
exchange and heat-recovery circuits, compressors and similar hardware have
been deleted as
being non-essential to an understanding of the techniques involved. The use of
such
miscellaneous equipment is well within the purview of one skilled in the art.
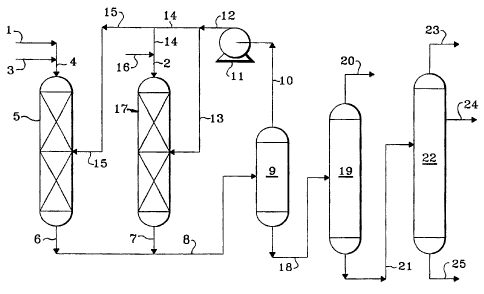
[0022] Referring now to the drawing, a distillate hydrocarbon feedstock is
introduced into the
process via line 3 and is admixed with a fresh make-up hydrogen stream which
is introduced via
line 1. The resulting admixture is transported via line 4 and introduced into
hydrodesulfurization
reaction zone 5. A resulting effluent from hydrodesulfurization reaction zone
5 is carried via
lines 6 and 8 and introduced into cold high pressure separator 9. A hydrogen-
rich gaseous stream
is removed from cold high pressure separator 9 via line 10 and introduced into
compressor 11. A
resulting compressed hydrogen-rich gaseous stream is transported via lines 12
and 14 and
admixed with a light cycle oil feedstock provided via line 16 and the
resulting admixture is
transported via line 2 and introduced into hydrocracking zone 17. A resulting
effluent from
-6-
CA 02630755 2008-05-06
hydrocracking zone 17 is transported via line 7 and 8 and introduced into cold
high pressure
separator 9. Another hydrogen-rich gaseous stream is introduced via line 15
into
hydrodesulfurization reaction zone 5. Yet another hydrogen-rich gaseous stream
is introduced via
line 13 into hydrocracking zorre 17 to provide quench. A hydrocarbonaceous
liquid stream is
removed from cold high pressure separator 9 via line 18 and introduced into
low pressure flash
drum 19. A gaseous stream contaiiiing hydrogen and low boiling gaseous
hydrocarbons are
removed from low pressure flash drum 19 via line 20 and recovered. A liquid
hydrocarbonaceous
stream is removed from low pressure flash drum 19 via line 21 and introduced
into fractionation
zone 22. A normally gaseous hydrocarbonaceous stream is removed from
fractionation zone 22
via line 23 and recovered. A high octane naphtha stream is removed from
fractionation zone 22
via line 24 and recovered. The high octane naphtha stream may be recovered
from fractionation
zone 22 in any convenient manner and is preferably a sidecut as shown. A low
sulfur diesel
stream is removed from fractionation zone 22 via line 25 and recovered.
[0023] The process of the present invention is further demonstrated by the
following
illustrative embodiment. This illustrative embodiment is, however, not
presented to unduly limit
the process of this invention, but to further illustrate the advantage of the
hereinabove-described
embodiment. The following data were not obtained by the actual performance of
the present
invention but are considered prospective and reasonably illustrative of the
expected performance
of the invention.
ILLUSTRATIVE EMBODIMENT
[0024] A blend of straight run diesel and light coker gas oil (LCGO) in an
amount of 4372
m'/day (27,500 barrels per day) and having the characteristics presented in
Table 1 is introduced
along with a high purity make-up hydrogen stream into a hydrodesulfurization
reaction zone
operated at hydrodesulfurization reaction conditions summarized and presented
in Table 2.
[0025] A stream of FCC light cycle oil (LCO) in an amount of 3657 m'/day
(23,000 barrels
per day) and having the characteristics presented in Table 1 is introduced
into a hydrocracking
zone operated at hydrocracking conditions summarized and presented in Table 2.
[0026] The resulting effluent from the hydrodesulfurization reaction zone and
'the
hydrocracking zone is cooled, partially condensed and introduced into a cold,
high pressure vapor
-7-
CA 02630755 2008-05-06
liquid separator operated at a pressure of 9.7 MPa (1400 psig) and a
temperature of 43 C (110 F)
to produce a hydrogen-rich recycle gas stream and a liquid hydrocarbonaceous
stream. The
hydrogen-rich recycle gas stream is recycled to the hydrocracking zone and the
liquid
hydrocarbonaceous stream is fractionated to produce a high octane naphtha
stream (gasoline)
boiling in the range of 88 C (185 F) to 193 C (380 F), in the amount of 2145
m3/day (13,500
barrels per day) and having an octane number of 87, and a diesel stream
boiling at a temperature
greater than 193 C (380 F) in an amount of 5500 m3/day (34,500 barrels per
day) and having a
sulfur content of <10 ppm and a cetane Index of 46.
TABLE 1 -- FEEDSTOCK ANALYSIS
Diesel/LCGO Light Cycle Oil
Specific gravity 0.87 0.96
Total sulfur, wt-% 2 1
Total nitrogen, weight ppm 725 900
Distillation, C ( F)
IBP 146 (295) 215 (420)
10% 222 (432) 263 (506)
50% 278 (543) 304 (580)
90% 336 (638) 349 (660)
EP 390 (735) 371 (700)
TABLE 2- OPERATING CONDITIONS
Hydrodesulfurization Hydrocracking
Pressure, MPa (psig) 9.7 (1400) 9.7 (1400)
Temperature, C ( F) 371 (700) 371 (700)
[00271 The foregoing description, drawing and illustrative embodiment clearly
illustrate the
advantages encompassed by the process of the present invention and the
benefits to be afforded
with the use thereof.
-8-