Note: Descriptions are shown in the official language in which they were submitted.
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
SYNTHESIS OF METALLIC NANOPARTICLE DISPERSIONS
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Pat. App. No.
60/752,143, "Low Temperature Curing Inks Utilizing Metallic Nanoparticles As A
Sintering
Aid", filed December 20, 2005, U.S. Provisional Pat. App. No. 60/752,144, "Low
Temperature
Curing Inks Containing Metallic Nanoparticle Dispersions", filed December 20,
2005, U.S.
Provisional Pat. App. No. 60/752,628, "Capacitance Coupled Interactive
Electronics Using
Printed Conductors", filed December 21, 2005, and U.S. Non-Provisional Pat.
App. No.
11/613,136, "Synthesis of Metallic Nanoparticle Dispersions", filed December
19, 2006. The
entirety of each of these applications is incorporated by reference herein in
their entirety.
FIELD OF THE INVENTION
[0002] The present invention pertains to the field of nanoparticles. The
present
invention also pertains to the fields of conductive inks and of printable
conductive features.
BACKGROUND OF THE INVENTION
[0003] Various scientific and patent publications are referred to herein. Each
is
incorporated by reference in its entirety.
[0004] Thin, conductive metal films have a wide range of uses, and have
particular
application as connectors in microelectronic devices, e.g., U.S. Pat. No.
6,855,378, to Subhash,
N., and also as connectors in photovoltaic devices. Accordingly, the ability
to make highly
conductive traces and films at low temperatures and moderate cost is of
enormous commercial
interest to the electronics industry.
[0005] The economic feasibility of making devices such as radio frequency
identification ("RFID") tags, flexible displays, low cost consumer
electronics, and printed thin
film transistors (TFTs) depends on the ability to efficiently and quickly
manufacture such
materials. Thus, application of printing technologies, such as inkjet
printing, roll-to-roll,
printing, gravure printing, and the like, to forming conductive traces and
other structures is of
keen interest to the electronics, display, and energy industries. As is known,
e.g., Lee, et al.,
Nanotechnology, 2006, 17, 2424, forming conductive structures via printing
methods has clear
advantages over conventional photolithography and etching processes for
producing such
structures. First, printing is more environmentally friendly than more
traditional methods of
-1-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
manufacture. Second, printing involves relatively less material loss than
traditional processes.
Third, printing processes are customizable. Fourth, printing processes are
capable of high
productivity, and, in certain printing processes, furthermore, are also
capable of depositing
precise amount of material to a specified location via a non-contact and
directed deposition
process capable of being controlled by a computer.
[0006] Present methods of printing conductive compositions onto substrates
have
certain limitations. Commercial conductive ink systems, such as those used to
print membrane
touch switches, typically contain metal flake with a cross-sectional dimension
of about 1000 nm
as a conductive constituent and also contain polymers that function as
binders. These flake-
based inks, however, generally cannot be processed at temperatures lower than
120 C or at times
less than 1 minute, regardless of temperature. Furthermore, these materials
are only capable of
achieving conductivities of only 2 to 10% of bulk metal conductivity because
of the continuous
polymeric matrix and the manner with which the flakes pack together.
[0007] Furthermore, metallic flakes do not sinter together, so a continuous
metallic
network is not formed by such materials and the network conducts electricity
instead by
incidental contact of the metal flakes, resulting in conductivity that often
only 10% of the bulk
metal conductivity. Further, ink systems containing flake as the metal
constituent are limited in
the ultimate thickness that can be obtained; because the nominal flake size is
the range of 1-2
microns, the thinnest traces possible may be 3-4 microns.
[0008] Other conductive ink systems employ the use of metallo-organic
additives, U.S.
Pat. No. 5,071,826 to Anderson, J.T, et al, U.S. Pat. No. 5,338,507, to
Anderson, J.T, et al; U.S.
Pat. No. 4,599,277 to Brownlow, J.M., et al; U.S. Pat. No. 7,115,218, to Kydd,
P.H., et al.,
which, upon decomposition, result in atomic metal that acts as a chemical
binding agent for the
metal flakes. Conductivities as high as 20% of the bulk metal are possible
when using such
additives, but high temperatures are needed to decompose the metallo-organic
into a conductive
structure, and these high temperatures accordingly limit the range substrates
suitable for use in
conjunction with such ink systems.
[0009] Many metallic nanoparticle systems have been described in the
literature and
there are accordingly many methods for preparing and stabilizing metallic
nanoparticles. Haes,
A.J. and Van Duyne, R.P., J. Am. Chem. Soc., 2002,124, 10596; Chen, S., et al,
J. Phys. Chem.
B, 1992, 124, 9898; Li, Y., et al., Langmuir, 2002, 18, 4921; Taleb, A., et
al., Chem. Mat., 1997,
9, 950; Yi, K.C., et al., J. Phys. Chem. B., 1994, 98, 38721 Esumi, K., et al,
Langmuir, 2000, 16,
2604; Harfensit, S.A., et al, J. Phys. Chem. B, 1996, 100, 13904; Bunge, S.D.,
Boyle, T.J., et al.,
Nano. Letters, 2003, 3, 901; Wang, W., et al, Langmuir, 1998, 14, 602;
Yamamoto, M., et al.,
-2-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
Langmuir, 2006, 22, 8581; Yamamoto, M., Chem. Letters, 2003, 32, 452. Some of
these
methods include: condensation of metal vapor to create particles, thermal
decomposition of
atomized metal organic salts, and use of chemically reduced metal salt
solutions to produce
particles. All of these production or synthesis methods require that the
produced nanoparticles
be stabilized in order that the nanoparticles do not interact with each other
or aggregate to form
larger particles that become unsuitable for a given application. Thus,
preventing metallic
nanoparticle aggregation can be crucial to a ink composition having maximum
utility.
[0010] The mechanism of stabilization is important in that the manner by which
agents
that stabilize metallic nanoparticles against aggregation also affect the
ability of the metallic
particles to sinter at a low temperature. A low temperature sintering process
is critical to
application of conductive inks to substrates, such as paper, that can not
tolerate high
temperatures. Affecting the ability of the metallic nanoparticles to sinter in
turn defines the cure
characteristics of the ink; systems that form conductive structures only after
curing at
temperatures above 150 C and only after long cure times may possess such
suboptimal cure
behavior in part because of the method used to stabilize the particles. Hence,
an ideal stabilizing
species prevents permanent aggregation of the particles while not interfering
with the particle
sintering process when heat is applied. Other considerations present in
choosing a stabilizing
agent or agents also include the effect of the stabilizer on the
nanoparticles' shelf life and their
cost.
[00111 An additional consideration in crafting a system for stabilizing
metallic
nanoparticles against aggregation is the choice of the medium in which the
metallic nanoparticles
are dispersed. Certain conductive ink compositions spread out on a substrate
surface before
curing, thus adversely impacting the ability to form structures of a certain,
defined shape. In
addition, certain compositions incorporate organic solvents, which are
difficult to dispose of.
Furthermore, systems comprising metallic nanoparticles dispersed in an organic
solvent medium
suffer from poor sintering characteristics once deposited onto a substrate,
and may require
comparatively long exposure to comparatively high temperatures in order to
form conductive
traces after deposition. E.g., Lee, et al., J. of Colloid and Interface Sci,
2006, 304, 92-97.
[0012] One potential hindrance to metallic nanoparticles' use in conductive
inks is their
limited ability to form relatively thick conductive traces. While large
metallic particles -
typically characterized as having diameters of greater than about 500 nm - are
limited in
application because they do not sinter at low temperatures, large particles
are nonetheless
capable of forming thicker metal traces than metallic nanoparticles are
capable of forming.
Hence, because of large metallic particles' inability to sinter and form
conductive structures at
-3-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
low temperatures, manufacturers are limited in their ability to form thick
conductive structures
on substrates that are incapable of withstanding the temperatures necessary
for large particles to
cohere into conductive structures.
[0013] Accordingly, based on the current state of the art, there is a need for
an aqueous
metallic nanoparticle composition, that exhibits little to no particle
aggregation and is capable of
forming a cohesive, conductive structure on a broad range of substrates after
exposure to
moderate temperatures for short periods of time following deposition via a
printing process, and
method of making such a composition. There is also a related need for a method
of forming
conductive structures by using such a composition.
[0014] Furthermore, the difficulty in inducing large metallic particles to
form
conductive structures under moderate processing conditions also gives rise to
a need for a
method for forming thick conductive structures on substrates capable of
tolerating only low
temperatures. There is also a related need for any compositions suitable for
such a method.
SUMMARY OF THE INVENTION
[0015] In overcoming the challenges associated with providing an aqueous
metallic
nanoparticle composition capable of being deposited on a substrate and forming
a cohesive
conductive structure under moderate temperature conditions, the present
invention provides, inter,
alia, a composition comprising a population of metallic nanoparticles
dispersed in an aqueous
medium, wherein at least a portion of the population comprising individual
metallic
nanoparticles characterized as having an average cross-sectional dimension in
the range of from
about 1 nm to about 100 nm.
[0016] In another aspect, the present invention provides a composition,
comprising: a
metallic nanoparticle mixture capable of forming a cohesive structure of less
than about 10 m in
thickness following curing at a temperature of less than about 140 C for less
than about 90
seconds, wherein the cohesive structure has a resistivity in the range of from
about 2 times to
about 15 times the bulk resistivity of the corresponding metal.
[0017] Additionally, the present invention provides methods for synthesizing a
metallic
nanoparticle dispersion. As will be disclosed in further detail, the methods
include reacting in an
aqueous medium: at least one ligand, wherein the ligand comprises a heteroatom
head group
bonded to a tail comprising from 1 to about 20 carbon atoms; at least one
reducing agent; and, at
least one metallic salt in an aqueous dispersing solution, wherein the
metallic salt is present in
the dispersion at a concentration in the range of from about 10 grams/liter to
about 600
grams/liter based on volume of the dispersing solution, and wherein the
metallic salt comprises
-4-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
at least one cation comprising silver, copper, gold, zinc, cadmium, palladium,
iridium,
ruthenium, osmium, rhodium, platinum, iron, nickel, cobalt, indium, or any
combination thereof.
[0018] The present invention further provides methods for forming a conductive
structure on a substrate. As will be set forth in greater detail, the methods
comprise depositing a
composition onto the substrate, wherein the composition comprises at least one
population of
metallic nanoparticles, wherein at least a portion of the population
comprising individual
metallic nanoparticles characterized as having an average cross-sectional
dimension in the range
of from about 1 nm to about 100 nm; wherein each of the nanoparticles comprise
at least one
ligand bound to its surface, the ligand comprising a heteroatom head group
bound to the
nanoparticle surface and a tail bound to the heteroatom head group; and,
curing the deposited
composition.
[0019] In additional aspects, the present invention provides methods for
forming a
conductive structure. These methods, as will be set forth in further detail,
comprise depositing a
metallic nanoparticle composition onto the substrate, wherein the composition
is capable of
forming, after curing at a temperature of less than about 140 C for less than
about 90 seconds, a
cohesive and conductive structure having a resistivity in the range of from
about 2 times to about
15 times the bulk resistivity of the corresponding metal and having a
thickness of less than about
20 m, and curing the deposited composition.
[0020] The general description and the following detailed descri ption are
exemplary
and explanatory only and are not restrictive of the invention, as defined in
the appended claims.
Other aspects of the present invention will be apparent to those skilled in
the art in view of the
detailed description of the invention as provided herein.
BRIEF DESCRIPTION OF THE DRAWINGS
[0021] The summary, as well as the following detailed description, is further
understood when read in conjunction with the appended drawings. For the
purpose of
illustrating the invention, there are shown in the drawings exemplary
embodiments of the
invention; however, the invention is not limited to the specific methods,
compositions, and
devices disclosed. In addition, the drawings are not necessarily drawn to
scale. In the drawings:
[0022] FIG. 1(A) depicts a transmission electron microscope ("TEM") micrograph
of
silver nanoparticles synthesized by the present invention;
[0023] FIG. 1(B) illustrates a scanning electron microscope ("SEM") micrograph
of a
trace comprised of a composition of the present invention cured for 1 minute
at 100 C;
[0024] FIG. 1(C) depicts a SEM micrograph of a trace comprised of a
composition of
the present invention cured for 3 minutes at 85 C;
-5-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
[0025] FIG. 2 depicts a particle size distribution, on a weight basis, of a
composition of
the present invention; and,
[0026] FIG. 3 depicts the weight resistivity versus cure time for certain
compositions
provided by the present invention and for certain prior art compositions.
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
[0027] The present invention may be understood more readily by reference to
the
following detailed description taken in connection with the accompanying
figures and examples,
which form a part of this disclosure. It is to be understood that this
invention is not limited to the
specific devices, methods, applications, conditions or parameters described
and/or shown herein,
and that the terminology used herein is for the purpose of describing
particular embodiments by
way of example only and is not intended to be limiting of the claimed
invention. Also, as used in
the specification including the appended claims, the singular forms "a," "an,"
and "the" include
the plural, and reference to a particular numerical value includes at least
that particular value,
unless the context clearly dictates otherwise. The term "plurality", as used
herein, means more
than one. When a range of values is expressed, another embodiment includes
from the one
particular value and/or to the other particular value. Similarly, when values
are expressed as
approximations, by use of the antecedent "about," it will be understood that
the particular value
forms another embodiment. All ranges are inclusive and combinable.
[0028] It is to be appreciated that certain features of the invention which
are, for clarity,
described herein in the context of separate embodiments, may also be provided
in combination in.
a single embodiment. Conversely, various features of the invention that are,
for brevity,
described in the context of a single embodiment, may also be provided
separately or in any
subcombination. Further, reference to values stated in ranges include each and
every value
within that range.
Terms
[0029] As used herein, the term "aqueous" means containing water.
[0030] As used herein, the term "bonding" means covalently bonding, ionically
bonding, hydrogen bonding, coordinate bonding, and the like.
[0031] As used herein, the term "tail" means a straight, branched, or cyclic
chain of
carbon atoms, wherein the chain may be aliphatic, and wherein the chain may
have one or more
additional groups bound to one or more of its member carbon atoms. An example
would be a
chain of aliphatic carbon atoms with an alcohol group attached to one of the
chain members.
-6-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
[0032] As used herein, the term "heteroatomic head group" means a group
including at
least one atom wherein at least one atom within the group is atom other than
carbon. Examples
include nitrogen, sulfur, or oxygen.
[0033] As used herein, the term "cohesive" means united as a single entity and
resisting
separation.
[0034] As used herein, the term "complexing" means forming coordinating bonds
with
a metal atom or ion.
[0035] As used herein, the term "ligand" means a molecule or a molecular group
that
binds to another chemical entity to form a larger complex. Examples include a
molecular group
that becomes bound to a metal or metal ion by a coordinate covalent bond
through donating
electrons from a lone electron pair of the ligand into an empty metal electron
orbital.
[0036] As used herein, the term "agglomeration" means two or more particles
reversibly clustered together, wherein the surfaces of the particles do not
come into contact with
one another.
[0037] As used herein, the term "floc" means two or more particles reversibly
clustered
together, wherein the surfaces of the particles do not come into contact with
one another.
[0038] As used herein, the term "bulk resistivity" means the inherent
resistivity of a
material that makes up a specified object. For example, the bulk resistivity
of a ingot made of
silver would be the inherent conductivity of silver. As another example, the
bulk resistivity of an
ingot made of an alloy comprising silver and gold would be the inherent
conductivity of the
silver and gold alloy.
[0039] As used herein, the terms "aggregate", "aggregation", and similar forms
mean a
unified structure comprised of two or more particles irreversibly fused,
connected, or necked
together.
[0040] Compositions of the present invention typically include a population of
metallic
nanoparticles dispersed in an aqueous medium, wherein at least a portion of
the population
comprising individual metallic nanoparticles characterized as having an
average cross-sectional
dimension in the range of from about 1 nm to about 100 nm; and, wherein each
of the
nanoparticles comprise at least one ligand bound to its surface, the ligand
comprising a
heteroatom head group bound to the nanoparticle surface and a tail bound to
the heteroatom head
group.
[0041] Nanoparticle populations typically comprise particle agglomerate
comprised of
two or more individual nanoparticles, nanoparticle floc comprised of two or
more individual
nanoparticles, or any combination thereof. The ratio, by weight, of the
population of individual
-7-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
metallic nanoparticles to particle agglomerate is contemplated as being in the
range of from
about 1:99 to 99:1, and the ratio, by weight, of the population of individual
metallic
nanoparticles to particle floc is contemplated as being in the range of from
about 1:99 to 99:1.
[0042] In certain embodiments, a nanoparticle agglomerate has an average cross-
sectional dimension in the range of from about 100 nm to about 10000 nm; a
nanoparticle floc
has an average cross-sectional dimension in the range of from about 100 to
about 10000 nm.
[0043] An individual metallic nanoparticle may include silver, copper, gold,
zinc,
cadmium, palladium, iridium, ruthenium, osmium, rhodium, platinum, iron,
nickel, cobalt,
indium, silver oxide, copper oxide, gold oxide, zinc oxide, cadmium oxide,
palladium oxide,
iridium oxide, ruthenium oxide, osmium oxide, rhodium oxide, platinum oxide,
iron oxide,
nickel oxide, cobalt oxide, indium oxide, or any combination thereof.
[0044] It is contemplated that the ligand tail comprises from about 1 to about
20 carbon
atoms. The tail can comprise a straight-chain segment, a branched segment, a
cyclic segment, or
any combination thereof, and can further comprise an aliphatic chain, an acid
group, an alcohol
group, an amphiphillic group, an amine group, and the like, or any combination
thereof.
[0045] Suitable heteroatom head groups comprise oxygen, sulfur, nitrogen, and
the like.
100461 The aqueous medium comprises water, and it is envisioned that the
aqueous
medium can further comprise one or more polar organic solvents, one or more
non-polar organic
solvents, or any combination thereof. A suitable polar organic solvent
comprises an alcohol, a
polyol, a glycol ether, 1-methyl pyrolidinone, pyridine, methylethylketone, or
any combination
thereof. A suitable non-polar organic solvent comprises tetrahydrofuran,
toluene, xylene, a C5 to
C14 branched paraffin, a C5 to C14 unbranched paraffin, N,N - dimethyl
formamide, or any
combination thereof.
[0047] The aqueous medium is typically capable of solvating the metallic salt
in a
range of from about 10 grams/liter to about 600 grams/liter, or even 50 to
200, or even 80 to 120.
[0048] It is envisioned that the nanoparticles are present in the range of
from about 0.5
wt %to about 70 wt %, the ligand is present in the range of from about 0.5 wt
% to about 75 wt
%, and the medium is present in the range of from about 30 to about 98 wt %.
[0049] In some embodiments, the composition is capable of forming a cohesive
structure of less than about 10 m in thickness following curing at a
temperature of less than
about 110 C for less than about 90 seconds. The structure suitably has a
resistivity in the range
of from about 2 times to about 15 times the bulk resistivity of the
corresponding metals present
in the composition.
-8-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
[0050] Compositions also comprise a metallic nanoparticle mixture capable of
forming
a cohesive structure of less than about 10 m in thickness following curing at
a temperature of
less than about 110 C for less than about 60 seconds, or capable of forming a
cohesive structure
of less than about 5 m in thickness following curing at a temperature of less
than about 140 C
for less than about 15 seconds, or capable of forming a cohesive structure of
less than about 2
m in thickness following curing at a temperature of less than about 110 C for
less than about 10
seconds, or capable of forming a cohesive structure of less than about 2 m in
thickness
following curing at a temperature of less than about 140 C for less than about
5 seconds, wherein
the cohesive structure has a resistivity in the range of from about 2 times to
about 15 times the
bulk resistivity of the corresponding metal in the composition.
[0051] Suitable mixtures comprise a population of metallic nanoparticles, a
ligand, an
aqueous medium, or any combination thereof.
[0052] In some embodiments, the metallic nanoparticle populations can comprise
individual nanoparticles, particle agglomerate comprised of two or more
individual
nanoparticles, particle floc comprised of two or more individual
nanoparticles, or any
combination thereof. The ratio, by weight, of the population of individual
metallic nanoparticles
to particle agglomerate can be in the range of from about 1:99 to 99:1, and
the ratio, by weight,
of the population of individual metallic nanoparticles to particle floc is
typically in the range of
from about 1:99 to 99:1. In other embodiments, substantially all of the
nanoparticles are
agglomerated. In other embodiments, substantially all of the nanoparticles are
discrete
individual nanoparticles.
[0053] In some embodiments of the invention, individual metallic nanoparticles
have
an average cross-sectional dimension in the range of from about 1 nm to about
100 nm; or even
from about 5 nm to about 30 nm, or even from about 10 nm to about 20 nm.
Particle size can be
measured using an acoustic attenuation spectroscopy method substantiated by
transmission
electron microscopy. Particle agglomerates have an average cross-sectional
dimension of at least
about 2 nm, or even at least about 20 nm, or even at least about 200 nm, or in
the range of from
about 100 nm to about 10000 nm; and particle flocs have an average cross-
sectional dimension
in the range of from about 100 to about 10000 nm. Individual metallic
nanoparticles and ligands
are as described elsewhere herein; ligands are typically characterized as
bound to a surface of
one or more metallic nanoparticles by a heteroatom head group so as to give
rise to one or more
metallic nanoparticles stabilized against irreversible aggregation.
[0054] The aqueous medium of these compositions typically comprises water, and
can
further comprise one or more polar organic solvents, one or more non-polar
organic solvents, or
-9-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
any combination thereof. The aqueous medium is typically capable of solvating
the metallic salt
in a range of from about 10 grams/liter to about 600 grams/liter, and suitable
polar organic
solvents include an alcohol, a polyol, a glycol ether, 1-methyl pyrolidinone,
pyridine,
methylethylketone, or any combination thereof. Suitable non-polar organic
solvents comprises
tetrahydrofuran, toluene, xylene, a C5 to C14 branched paraffin, a C5 to C14
unbranched paraffin,
N,N - dimethyl formamide, or any combination thereof.
[0055] The nanoparticles can be present in the range of from about 0.5 to
about 70 wt
%, the ligand can be present in the range of from about 0.5 to about 75 wt %,
and the medium
can be present in the range of from about 30 to about 98 wt %. The
nanoparticles can be present
in the range of from about 10 to about 60 wt %, the ligand can be present in
the range of from
about 1 to about 30 wt %, and the medium is present in the range of from about
30 to about 98
wt %. The nanoparticles can be present in the range of from about 15 to about
55 wt %, the
ligand is present in the range of from about 2 to about 25 wt %, and the
medium is present in the
range of from about 30 to about 98 wt %. Also, the amount of ligand can be
about 10% based on
weight relative to the weight of the nanoparticles.
[0056] In one embodiment, the present invention involves the chemical
reduction of
metal salt in the presence of a ligand, which ligand is capable of complexing
or bonding to the
metal in a dispersing medium. The metal salt can be solvated by the solvent or
dispersed in the
solvent as a solid if the salt is insoluble in the solvent phase. Suitable
solvents include aqueous
solvents substantially free of organic solvents. Suitable solvents also
include some polar organic
solvents, e.g., if the metal salt can be solvated in a sufficiently high
concentration, e.g., about 0.3
to about 0.9 M, or about 0.45 to about 0.7 M, or about 0.55 to about 0.6 M.
The metal may
include silver, copper, gold, zinc, cadmium, palladium, iridium, ruthenium,
osmium, rhodium,
platinum, iron, nickel, cobalt, indium, or any combination thereof. The salt
anion may include
nitrates, carboxylates, sulfates, or chlorides. The reducing agent must be of
sufficient
electrochemical potential and concentration to effectively reduce the
respective metal salt.
Strong reducing agents such as hydrazine, hydrazine hydrate, or hydrogen, that
do not produce
undesirable ionic byproducts are suitable; other reducing agents such as
sodium borohydride may
be used.
[0057] Ligands can be chosen on their ability to complex with metal particles
and
stabilize the particles against aggregation; one consideration is the ability
of the ligand to allow
the particles to consolidate and sinter during drying and thermal treatment.
The temperature at
which the particles sinter is in some part controlled by the ligand adsorbed
to the metal. The
ligand can be characterized as bonding to the metal through a heteroatom such
as oxygen, sulfur,
-10-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
or nitrogen. In some embodiments the heteroatom portion of the ligand is
provided as a carboxyl,
sulfonyl, thiol, and the like. Without being bound to any particular theory of
operation, it is
believed that.based on the relative thermal stability of the complexing
portion and aliphatic
backbone of the ligand compound, an intermediate salt may result during
thermal treatment that
adversely affecting the sintering of the metal nanoparticles. Ligands having a
straight-chain
aliphatic backbone comprising from about 1 to about 20 carbon atoms are
particularly suitable.
Branched or cyclic backbones having up to about 20 carbon atoms may be used,
for example, if
the ligand is sufficiently stable in the solvent system. Suitable ligands can
preferably have from
about 5 to aboiit 12 carbon atoms in the aliphatic tail.
[0058] In the present invention, no post-synthesis treatment such as washing
or phase
transfer is needed in order to remove residual byproducts such as the metal
salt anion. Although
this step is not needed, additional washing and post-processing steps can be
used. The
byproducts of the reaction are left in the nanoparticle mixtures to catalyze
the decomposition of
the ligands on the nanoparticles surface. In particular, nitrate anions can
react with organic acid
ligands in self-propagating chemical decomposition or anionic oxidation-
reduction synthesis of
superconducting oxides to prevent intermediate metal salts. Alternatively, a
compound such as
an amine could be added to the reaction product or be part of the ligand
molecule which
similarly catalyzes the decomposition of the ligands and sintering of the
nanoparticles. The
particles are sometimes allowed to settle in order to concentrate them for
forming films.
[0059] Without being bound by a particular theory of operation, it is believed
that the
metallic nanoparticles are able to remain dispersed in the aqueous phase by
the formation of self-
assembled surfactant structures, e.g., an interdigitated bi-layer, of the
ligand or vesicle structures
around the metallic nanoparticles. In other cases, the nanoparticles can phase
separate from the
aqueous phase giving rise to an oily ligand-rich phase comprising concentrated
nanoparticles and
a second aqueous phase. The particles can be stabilized by ligands binding to
the surface of the
silver through nucleophilic head groups with the aliphatic portion extending
outward. The
aliphatic portion of ligands not bound to the nanoparticle surface can
associate with the aliphatic
portion of the bound ligands forming a vesicle around the nanoparticle. Also
without being
bound to any particular theory of operation, it is believed that if no bi-
layer formed, the metallic
nanoparticles may phase-separate into an oily phase. Accordingly, ligands can
form a bi-layer
around the particles. The bi-layer can be broken down causing the
nanoparticles to form a
hydrophobic phase by either modifying the pH or by adding a salt or to the
aqueous sol.
[0060] Accordingly, methods for synthesizing a metallic nanoparticle
dispersion
include reacting in an aqueous medium: at least one ligand, wherein the ligand
comprises a
-11-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
heteroatom head group bonded to a tail comprising from 1 to about 20 carbon
atoms; at least one
reducing agent; and, at least one metallic salt in an aqueous dispersing
solution, wherein the
metallic salt is present in the dispersion at a concentration in the range of
from about 10
grams/liter to about 600 grams/liter based on volume of the dispersing
solution, and wherein the
metallic salt comprises at least one cation comprising copper, gold, zinc,
cadmium, palladium,
iridium, ruthenium, osmium, rhodium, platinum, iron, nickel, cobalt, indium,
or any combination
thereof. Preferably the metallic salt comprises silver.
[0061] Typically, the tail is as described elsewhere herein; suitable
heteroatom head
groups comprise oxygen, sulfur, nitrogen, and the like. A suitable ligand is
characterized as
being capable of binding by its heteroatom head group to a surface of a
metallic nanoparticle so
as to give rise to a metallic nanoparticle stabilized at least in part against
aggregation.
[0062] Suitable reducing agents include strong reducing reagents that
typically are
capable of reducing metals in aqueous systems, e.g., hydrazine, hydrazine
hydrate, hydrogen,
sodium borohydride, lithium borohydride, ascorbic acid, a primary amine, a
secondary amine, a
secondary amine, a tertiary amine, and the like, or any combination thereof.
[0063] The metallic salt typically comprises at least one anion, wherein the
anion
comprises acetate, nitrate, carboxylate, sulfate, chloride, hydroxide, or any
combination thereof.
[0064] A suitable dispersing solution comprises an aqueous medium. Another
suitable
dispersing solution comprises an aqueous medium substantially free of organic
solvents, and can
comprise water. The dispersing solution can further comprise one or more polar
organic
solvents, one or more non-polar organic solvents, or any combination thereof.
Suitable polar and
non-polar solvents are as described elsewhere herein.
[0065] Reacting can comprise contacting, mixing, stirring, sonicating,
agitating, and the
like; after reacting, one or more ligand heteroatom head groups are
characterized as bound to a
surface of one or more metallic nanoparticles so as to give rise to one or
more metallic
nanoparticles stabilized against irreversible aggregation.
[0066] The method typically comprises combining the ligand and metallic salt
in a
respective molar ratio in the range of from about 0.1:1 to about 0.2:0.7, or
even in the range of
from about 0.1:1 to about 0.3:0.5; combining the metallic salt and reducing
agent in a respective
molar ratio in the range of from about 0.7:1 to about 1:2, in other cases the
metallic salt and
reducing agent in a respective molar ratio in the range of from about 4:1 to
about 1:2, in other
cases the metallic salt and reducing agent in a respective molar ratio in the
range of from about
0.6:1 to about 1.2:1. The method can, in some embodiments, include adjusting
the relative
amounts of ligand, reducing agent, metallic salt, aqueous dispersing solution,
adjusting the pH of
-12-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
the aqueous medium, or any combination thereof, so as to give rise to a pH in
the range of from
about 3 to about 12. In certain embodiments, the pH can vary between the basic
and acidic
regimes during the reaction.
[0067] In some configurations, the method includes heating the aqueous medium,
ligand, reducing agent, and metallic salt in aqueous dispersing solution, or
any combination
thereof, to a temperature of from about 5 C to about 200 C prior to reaction,
to a temperature of
from about 35 C to about 70 C prior to reaction, or to a temperature of from
about 40 C to about
60 C prior to reaction.
[0068] The method typically includes a recovery step following reaction. The
recovery
step can include allowing the passage of sufficient time such that the
concentration of
nanoparticles in any aqueous medium present after reaction can be in the range
of from about 0
wt % to about 70 wt%, or in the range of from about 0.5 wt % to about 30 wt.%,
or in the range
of from about 2 wt % to about 20 wt.%, or in the range of from about 3 wt % to
about 7 wt.%,
and then recovering the reaction products. In some cases, the recovery step
comprises allowing
the passage of sufficient time such that the concentration of nanoparticles in
any aqueous
medium present can be in the range of from about 0.5 wt % to about 70 wt.%, or
in the range of
from about 5 wt % to about 60 wt.%, decanting the aqueous medium, recovering
the reaction
products, and ultrafiltration of the decanted aqueous medium to recover any
nanoparticles
residing in the decanted medium. In some cases, a cake comprising
nanoparticles will be
formed. Such a cake can have from about 25 wt.% to about 70 wt.%. In other
embodiments, a
supernatant is formed, which can comprise from 0 wt.% up to about 30 wt.%
nanoparticles.
Distribution of the nanoparticles can be distributed between supematant and
cake. The recovery
step can include ultrafiltration of any aqueous medium present following
reaction when there are
no settled reaction products so as to recover nanoparticles present in the
medium.
[0069] , In some embodiments, the reacting comprises continuously introducing
the
aqueous medium, ligand, and reducing agent into a first stirred reactor
capable of fluid
communication with the contents of a second stirred reactor. Suitable medium,
ligand, and
reducing agent are described elsewhere herein, as are the suitable ratios of
these entities to one
another. The aqueous medium, ligand, reducing agent, and metallic salt in
aqueous dispersing
solution may be heated as set forth elsewhere herein. Typically, the residence
time of the first
reactor is sufficient to as to give rise to the reaction progressing to
substantial completion, and
the method can include continuously transporting the contents of the first
reactor to the second
reactor; the residence time in the second reactor is envisioned as sufficient
to allow the reaction
to progress to essentially total completion.
-13-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
[0070] The methods described herein can also include one or more recovery
steps.
[0071] Metallic nanoparticles synthesized according to the present invention
and the
structures formed by curing these nanoparticles are shown in FIGS 1(A), 1(B),
and 1(C). FIG.
1(A) depicts silver nanoparticles made according to the present invention. As
can be seen by
comparison of the particles to the scale bar in FIG. 1(A), typical
nanoparticles made in
accordance with the present invention have widths of well under 100 nm. FIG
1(B) depicts a
structure formed by metallic nanoparticles made according to the present
invention after curing
at about 100 C for about 1 minute. FIG. 1(C) depicts a structure formed by
metallic
nanoparticles made according to the present invention after curing at about 85
C for about 3
minutes.
[0072] The existence of individual particles along with nanoparticle
agglomerate in
certain embodiments of the present invention is shown in FIG. 2. That figure
depicts, on a
weight basis, the proportion of individual metallic nanoparticles synthesized
according to the
present invention relative to nanoparticle agglomerate comprised of the
individual nanoparticles.
[0073] Methods for forming a conductive structure on a substrate comprise
depositing a
composition onto the substrate, wherein the composition comprises at least one
population of
metallic nanoparticles, at least a portion of the population comprising
individual metallic
nanoparticles characterized as having an average cross-sectional dimension in
the range of from
about 1 mn to about 30 nm; wherein each of the nanoparticles comprise at least
one ligand bound
to its surface, the ligand comprising a heteroatom head group bound to the
nanoparticle surface
and a tail bound to the heteroatom head group; and, curing the deposited
composition.
[0074] The depositing can include a printing method; suitable printing methods
include
flexographic printing, rotogravure printing, lithographic printing, intaglio
printing, relief
printing, screen printing, inkjet printing, laser printing, or any combination
thereof.
[0075] As pertaining to the composition of these methods, typical populations
of
metallic nanoparticles are as described elsewhere herein, as are suitable
ligands, and acceptable
aqueous media.
[0076] A further consideration in formulating metallic nanoparticle-based inks
is
rheology. The ink rheology is influenced by the deformation behavior of the
solid components
and the flow behavior of the components. Mezger, T.G., The Rheology Handbook,
2002,
published by Vincentz Verlag, Hannover, Germany; Verstrat, D.W., Research
Report,
Formulating with Associative Rheology Modifiers, Alco Chemical website,
www.alcochemical.com, Alco Chemical Company, Division of National Starch and
Chemical
Company, Chattanooga, TN; Manshausen, P., Borchers GmbH, Monheim, Germany,
Presented
-14-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
at the 6'h Nurnberg Congress, April, 2001. These behaviors are responses to
shear forces
imparted on the ink during the process of depositing or printing the ink onto
the desired
substrate. Additives can modify the ink rheology such that the desired flow
properties are
achieved with minimal adverse affects on the electrical properties and
adhesion of the metallic
trace or film.
[0077] There are many choices for rheology modifiers, Manshausen, P., Borchers
GmbH, Monheim, Germany, Presented at the 6'h Nurnberg Congress, April, 2001;
Young, V.L.
and Hickman, A.D., Dow Latex Technotes, January 6, 1992, including associative
and non-
associative organic thickeners, as well as inorganic thickeners. Associative
thickeners generally
associate with ingredients in the inks such as the metal nanoparticles and the
polymeric binder
particles incorporated for adhesion. Non-associative thickeners interact with
the aqueous phase,
essentially thickening the water.
[0078] The composition of the method can include one or more rheology
modifiers.
Some such modifiers can include an associative thickener such as
hydrophobically modified
polyether polyurethane, hydrophobically modified polyether, hydrophobically
modified acrylic
thickener, hydrophobically modified cellulose ether, and the like.
Alternatively, the rheology
modifier can include a thickening agent such as an alkali-soluble emulsion,
such as a polymer
comprising units polymerized from (meth)acrylic acid, wherein a suitable
polymer comprises a
homopolymer of (meth)acrylic acid, a co-polymer of (meth)acrylic acid and
(meth)acrylate
esters, maleic acid, or any combination thereof. A thickening agent can also
include a cellulose
based material such as hydroxyethyl cellulose, hydroxypropyl cellulose,
arabinogalactin,
dextran, starches, an acid swellable emulsion, a polyvinyl alcohol, a
polyacrylamide,
polyethylene glycol, or any combination thereof. Typically, a rheology
modifier can be present
in the range of from about 0 wt% to about 15 wt%, or in the range of from
about 0 wt% to about
7 wt%, or even in the range of from about 0 wt% to about 3 wt%.
[0079] Preparation of a formulation that is viable as an ink to be printed on
commercial
printing equipment also typically requires the addition of agents to enable or
enhance adhesion of
the cured ink to the desired substrate, to enhance the wetting of the ink on
the substrate, and to
modify the rheological or flow characteristics of the ink.
[0080] Typically, metallic nanoparticles will not adhere to untreated
substrates that are
commonly used such as polyester, polypropylene, and paper. Thus, adhesives,
binders, or any
combination thereof, may be added to the metallic nanoparticle dispersion such
that additive
establishes a chemical or physical bond with the surface of the desired
substrate. Ideally, these
additives do not prevent or hamper the process of curing or sintering the
metallic nanoparticles
-15-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
into a continuous, conductive film or structure. In addition, the adhesion-
enhancing additive
should be chosen such that it does not affect the stability of the
nanoparticles. Adhesion-
promoting additives generally include surfactants that contribute to the ink
wetting the substrate
surface.
[0081] Accordingly, the composition of the disclosed method further comprises
a
binder, which can include a latex, any polymer soluble in the solvent medium
of the
nanoparticles, or compatible with the nanoparticles, a polymer latex, an
emulsion polymer,
polyimide, a silicone, a fluorocarbon, a polyamic acid, a polyurethane, a
polyester, an epoxy,
polyvinylalcohol, polyacrylamide, or any combination thereof. It is envisioned
that the binder is
present in the range of from about 0 wt% to about 20 wt%, or in the range of
from about 0 wt%
to about 7 wt%., or in the range of from about 0 wt% to about 5 wt%.
[0082] Substrates suitable for the method include a glass, a ceramic, a
polymer, a
silicon, a nitride, a carbides, a ceramic precursor, or any combination
thereof. Suitable polymers
include a polyester, a polyolefin, a polycarbonate, an acrylic polymer,
polyethylene naphthalate,
polyimide, polyamideimide, polyvinyl chloride, polypropylene, a liquid crystal
polymer,
polycarbonate, or any combination thereof. In some embodiments, the substrate
comprises
paper, synthetic engineered paper, cardboard, a coated corrugated cardboard,
uncoated
corrugated cardboard, a fabric, and the like.
[0083] In some instances, it is envisioned that at least a portion of a
surface of the
substrate is capable of being modified to give rise to a surface capable of
adhering to the
deposited composition.
[0084] In some embodiments of the invention, the composition further comprises
metallic particles. Such particles typically have a width in the range of from
about 200 nm to
about 20000 mn, in the range of from about 500 nm to about 10000 nm, or in the
range of from
about 800 nm to about 3000 nm. Suitable particles comprise silver, copper,
gold, zinc, cadmium,
palladium, iridium, ruthenium, osmium, rhodium, platinum, iron, nickel,
cobalt, indium, silver
oxide, copper oxide, gold oxide, zinc oxide, cadmium oxide, palladium oxide,
iridium oxide,
ruthenium oxide, osmium oxide, rhodium oxide, platinum oxide, iron oxide,
nickel oxide, cobalt
oxide, indium oxide, or any combination thereof.
[0085] The curing aspect of the method typically comprises exposing the
deposited
composition to a temperature of less than about 110 C for less than about 90
seconds; a structure
formed by the method typically has a thickness of less than about 20 m; or
exposing the
deposited composition to a temperature of less than about 110 C for less than
about 60 seconds;
a structure formed by the method typically has a thickness of less than about
15 m; or exposing
-16-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
the deposited composition to a temperature of less than about 140 C for less
than about 30
seconds; a structure formed by the method typically has a thickness of less
than about 15 m; or
exposing the deposited composition to a temperature of less than about 110 C
for less than about
30 seconds; a structure formed by the method typically has a thickness of less
than about 8 m;
or exposing the deposited composition to a temperature of less than about 140
C for less than
about 20 seconds; a structure formed by the method typically has a thickness
of less than about 8
m.
[0086] Methods for forming a conductive structure include depositing a
metallic
nanoparticle composition onto the substrate, wherein the composition is
capable of forming after,
curing at a temperature of less than about 110 C for less than about 90
seconds a cohesive and
conductive structure having a resistivity in the range of from about 2 times
to about 15 times the
bulk resistivity of the corresponding metal and having a thickness of less
than about 20 m; and,
curing the deposited composition.
[0087] Suitable deposition processes are described elsewhere herein.
Nanoparticle
compositions are envisioned as including a population of metallic
nanoparticles, a ligand, a
medium, or any combination thereof, all as discussed elsewhere herein.
[0088] Suitable compositions further can also include rheology modifiers as
described
elsewhere herein. The composition is envisioned as further comprising a
binder, as described
elsewhere herein. Suitable compositions may also include metallic particles,
as detailed
elsewhere.
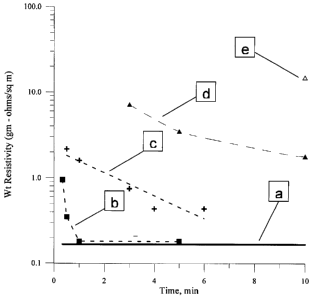
[0089] FIG. 3 depicts resistivity as a function of cure time for prior art
compositions
and compositions provided by the present invention. As shown, the resistivity
of a composition
comprising metallic silver nanoparticles synthesized by the present invention
(trace (b)) achieve
a resistivity comparable to that of bulk silver (trace (a)) after curing at a
temperature of about
85 C for about 1 minute. Trace (c) represents a composition comprising
metallic silver
nanoparticles synthesized by the present invention and certain additives such
as rheology
modifiers and binders; as shown, that composition also approaches the
resistivity of bulk silver
after curing at a temperature of about 100 C for about 6 minutes. Trace (d)
represents a
composition produced by Sumitomo Metal Mining Co (Japan),
http:www.smm.co.jp/b info E/b10 E.html which composition, when cured at 150 C,
achieved
resistivity higher than that of compositions made according to the present
invention at all cure
times. Trace (e) represents a composition produced by Sumitomo (Japan), which,
when cured at
100 C, and also is characterized as having a resistivity several orders of
magnitude greater than
that of compositions made according to the present invention at all cure
times.
-17-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
EXAMPLES
[0090] The following are non-limiting examples that are representative only
and that do
not necessarily restrict the scope of the present invention.
Example 1
[0091] An initial solution was prepared by adding 7.5 grams of ammonium
hydroxide
(30% ammonia by weight) to 275 grams of water; 13.5 grams of heptanoic acid
was added to this.
solution followed by 20.9 grams of 50 % hydrazine hydrate aqueous solution.
The ammonium
hydroxide is necessary to allow the acid to dissolve in the water. Separately,
36 grams of silver
nitrate was dissolved in 175 grams of water. The silver nitrate solution was
added to the initial
solution while stirring under nitrogen. The resultant product was flocculated
and allowed to
settle. Excess water was decanted off. The concentrated product was spread
onto 5 mil
polyester film with a 0.5 mil wire wound rod and then cured at 80 C and 100 C
for 1-2 minutes
resulting in cohesive and conductive silver films.
Example 2
[0092] An initial solution was prepared by adding 2.1 grams of ammonium
hydroxide
(30% ammonia by weight) to 50 grams of water; 7.8 grams of heptanoic acid was
added to this
solution followed by 3 grams of 50 % hydrazine hydrate aqueous solution.
Separately, 10 grams
of silver nitrate was dissolved in 50 grams of water. The silver nitrate
solution was added to the
initial solution while stirring under nitrogen. The resultant product was
allowed to settle and the
excess water decanted off.
[0093] The concentrated product was spread onto 5 mil polyester film with a
0.5 mil
wire wound rod and then cured at 80 C and 100 C for 1-2 minutes resulting in
cohesive and
conductive silver films. The weight resistivity of a sample cured at 100 C for
1 minute was
measured to be 0.39 gram-ohms/m2 (-2x bulk silver).
Example 3
[0094] An ink composition was prepared by adding 50 grams of spherical silver
powder
(1-2 um mean diameter) to 50 grams of 35 wt% nanoparticle dispersion of
Example 1 also
containing 3 wt% of an acrylic copolymer latex (55 wt% polymer), 2 wt% of
polyvinyl
alcohol (25 wt% in water, MW of 8,000-9,000), and 1 wt% ethylene glycol. The
materials were
mixed well together, and were milled in a mortar and pestle until a
homogeneous mixture was
obtained. A film of the resulting ink was deposited onto 0.005" thick
untreated polyester film
-18-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
with a 0.0015" Bird film applicator. The wet film was cured in a 100 C for 30
seconds followed
by 60 seconds at 140 C. The weight resistivity of the resulting silver films
was measured to be
1.3 gram-ohms/m2, approximately 8 times the resistivity of bulk silver. The
adhesion of the film
to the substrate was tested by applying a 4" long strip of Scotch brand tape
(3M Corporation) to
the film, insuring good adhesion to the film by applying pressure with the
index finger (not the
fingernail). The tape is then rapidly removed, pulling upward at a 90 angle,
perpendicular to the
substrate. This tape test method is derived from the ASTM D3359-02 Standard
Test Method for
Measuring Adhesion by Tape Test. Slight removal of the silver from the bulk of
the trace was
observed (4.215,with 5 being a clean tape), but none of the silver was removed
from the
substrate. The failure was observed to be a cohesive failure between the
silver particles.
Example 4
[0095] An ink composition was prepared by adding 50 grams of spherical silver
powder
(1 -2 micron mean diameter) to 50 grams of 35 wt% nanoparticle dispersion also
containing 10
wt% polyvinlacetate-polyethylene copolymer latex (50 wt% polymer), 2 wt% of
polyvinyl
alcohol (25 wt% in water, Mw of 8,000-9,000), and 1 wt% ethylene glycol. The
materials were
mixed well together, and were milled in a mortar and pestle until a
homogeneous mixture was
obtained. A film of the resulting ink was deposited onto 0.005" thick
untreated polyester film
with a#16 wire wound rod (0.016" wire diameter, 0.001" wet film thickness)..
The wet film was
cured in a 100 C for 30 seconds followed by 30 seconds at 140 C. The weight
resistivity of the
resulting silver films was measured to be 1.0 gram-ohm/m2, approximately 6.2
times the
resistivity of bulk silver. The adhesion of the film to the substrate was
evaluated by utilizing the
tape test method previously described in Example 2. The adhesion of the film
to the substrate
was very good (4.8/5), with only a trace of silver removed from the surface
(cohesive failure),
and no silver removal from the substrate was observed.
Example 5
[0096] An ink composition was prepared by adding 25 grams of spherical silver
powder
(1 -2 um mean diameter) to 50 grams of 35 wt% nanoparticle dispersion also
containing 3 wt%
acrylic copolymer (55 wt% polymer), and 4 wt% of polyacrylamide (50 wt% in
water). The
materials were mixed well together, and were milled in a mortar and pestle
until a homogeneous
mixture was obtained. A film of the resulting ink was deposited onto 0.005"
thick untreated
polyester film with a #16 wire wound rod (0.01 6" wire diameter, 0.00 1" wet
film thickness).
The wet film was cured in a 100 C for 30 seconds followed by 60 seconds at 130
C. The weight
-19-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
resistivity of the resulting silver films was measured to be 1.71 gram-
ohms/m2, approximately
10.7 times the resistivity of bulk silver. The adhesion of the film to the
substrate was evaluated
by utilizing the tape test method previously described in Example 2. The
adhesion of the film to
the substrate was very good (4.8/5), with only a trace of silver removed from
the surface
(cohesive failure), and no silver removal from the substrate was observed.
Example 6
[0097] An ink composition was prepared by adding 65 grams of spherical silver
powder
(1-2 pm mean diameter) to 80 grams of 35 wt% nanoparticle dispersion also
containing 3 wt%
acrylic copolymer (55 wt% polymer), 1.5 wt% of polyacrylamide (50 wt% in
water), and 1 wt%
propylene glycol. The materials were mixed well together, and were milled in a
mortar and
pestle until a homogeneous mixture was obtained. A film of the resulting ink
was deposited onto
0.005" thick untreated polyester film with a #16 wire wound rod (0.016" wire
diameter, 0.001"
wet film thickness). The wet film was cured in a 100 C for 30 seconds followed
by 60 seconds at
130 C. The weight resistivity of the resulting silver films was measured to be
1.3 1 gram-
ohms/m2, aproximately 8 times the resistivity of bulk silver. The adhesion of
the film to the
substrate was evaluated by utilizing the tape test method previously described
in Example 2. The
adhesion of the film to the substrate was good (4.215), with some removal
(cohesive failure) of
the silver from the bulk of the trace (4.215, with 5 being a clean tape), but
no silver removal from
the substrate.
Example 7
[0098] An ink composition was prepared by adding 52 grams of spherical silver
powder
(1 -2 pm mean diameter) to 64 grams of 35 wt% nanoparticle dispersion also
containing 3 wt%
acrylic copolymer (55 wt% polymer), 1.5 wt% of polyacrylamide (50 wt% in
water), and 1 wt%
propylene glycol. The materials were mixed well together, and were further
mixed in vortex
paint mixer for 5 minutes. A film of the resulting ink was deposited onto
0.005" thick untreated
polyester film with a #16 wire wound rod (0.016" wire diameter, 0.001" wet
film thickness). The
wet film was cured in a 60 C for 20 seconds followed by 40 seconds at 130 C.
The weight
resistivity of the resulting silver films was measured to be 1.00 gram-
ohms/m2, appproximately 6
times the resistivity of bulk silver. The adhesion of the film to the
substrate was evaluated by
utilizing the tape test method previously described in Example 2. The adhesion
of the film to the
substrate was very good (4.9/5), with only a slight trace of silver removed
from the surface
(cohesive failure), and no silver removal from the substrate was observed.
Further, resulting
-20-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
samples were folded in expansive mode (single crease) and than compressive
mode (single
crease), and a hard crease was made with the tip of the finger (not the finger
nail) on each
sample. Minimal loss of conductivity was observed for each sample.
Example 8
[0099] An ink composition was prepared by adding 10 grams of Floetrol (The
Flood
Company) to 40 grams of 35 wt% nanoparticle dispersion. The materials were
mixed well
together and a film of the resulting ink was deposited onto 0.005" thick
untreated polyester film
with a 0.0005" diameter wire wound rod and then cured at 130 C for 90 seconds
resulting in
cohesive and conductive silver films. The adhesion of the film to the
substrate was tested by
applying a 4" long strip of Scotch brand tape (3M Corporation) to the film,
insuring good
adhesion to the film by applying pressure with the index finger (not the
fingernail). The tape is
then rapidly removed, pulling upward at a 90' angle, perpendicular to the
substrate. This tape test
method is derived from the ASTM D3359-02, Standard Test Method for Measuring
Adhesion by
Tape Test. No material was removed from the substrate.
Example 9
[0100] An ink composition was prepared by adding 10 grams of a 25 wt% solution
ofpolyvinyl alcohol (9,000-10,000 Mw, 80% hydrolyzed) to 40 grams of 35 wt%
silver
nanoparticle dispersion. The materials were mixed well together and a film of
the resulting ink
was deposited onto 0.005" thick polyester film with a 0.0005" diameter wire
wound rod and then
cured at 130 C for 90 seconds resulting in cohesive and conductive silver
films. The adhesion of
the film to the substrate was evaluated by utilizing the tape test method
previously described.
Some material was removed from the substrate, however, most of the ink
remained on the
substrate.
Example 10
[0101] A film of the as-prepared, 35 wt% silver nanoparticle dispersion was
deposited
onto 5 mil polyester film with a 0.0005" diameter wire wound rod and then
cured at 85 C for 60
seconds resulting in cohesive and conductive silver films. The resulting film
had a weight
resistivity of 0.38 gram-ohms/m2 (IPC-TM-650, number 2.5.17.2). The adhesion
of the film to
the substrate was evaluated by utilizing the tape test method previously
described. All of the
material was removed from the substrate.
-21 -
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
Example 11
[0102] An ink composition was prepared by adding 2.6 grams of a 1 or 2 wt%
solution
of commercially available hydrophobically modified hydroxyethylcellulose to
19.2 grams of 40
wt% silver nanoparticle dispersion. The materials were mixed well together and
a film of the
resulting ink was deposited onto 0.005" thick polyester film with a 0.0003"
diameter wire wound
rod and then cured at 130 C for 90 seconds resulting in a cohesive and
conductive silver film.
The adhesion of the film to the substrate was evaluated by utilizing the tape
test method
previously described. All of the material was removed from the substrate.
Example 12
101031 An ink composition was prepared by adding 0.5 grams of a solution of
hydrophobically modified ethoxylated urethane rheology modifier to 10 grams of
34 wt% silver
nanoparticle dispersion. The materials were mixed well together and a film of
the resulting ink
was deposited onto 0.005" thick polyester film with a 0.0003" diameter wire
wound rod and then
cured at 100 C for 60 seconds resulting in a cohesive and conductive silver
film. The adhesion of
the film to the substrate was tested using the tape test method described
above. All of the
material was removed from the substrate.
Example 13
[0104] An ink composition was prepared by adding 0.36 grams of Arabinogalactan
wood gum (Larex Grade 100) to 18.2 grams of 35 wt% silver nanoparticle
dispersion. The
materials were mixed well together and a film of the resulting ink was
deposited onto 0.005"
thick polyester film with a 0.0003" diameter wire wound rod and then cured at
100 C for 60
seconds resulting in a cohesive and conductive silver film. The adhesion of
the film to the
substrate was tested using the tape test method described above. Some of the
material was
removed.
Example 14
[0105] An ink composition was prepared by adding 0.63 grams of a 50 wt. %
polyacrylamide solution (Aldrich 10,000 Mw) to 12.57 grams of 40 wt% silver
nanoparticle
dispersion. The materials were mixed well together and a film of the resulting
ink was deposited
onto 0.005" thick polyester film with a 0.0003" diameter wire wound rod and
then cured at
100 C for 60 seconds resulting in a cohesive and conductive silver film. The
adhesion of the film
-22-
CA 02634457 2008-06-19
WO 2008/048316 PCT/US2006/048699
to the substrate was evaluated by utilizing the tape test method previously
described in Example
1. None of the material was removed from the substrate.
Example 15
[0106] An ink composition was prepared by adding 0.44 grams of a 25 wt. %
polyvinyl
alcohol solution (Aldrich 9,000-10,000 Mw) and 1.14 grams of an acrylic
nanoparticle latex
dispersion to 22.2 grams of 35 wt% silver nanoparticle dispersion. The
materials were mixed
well together and a film of the resulting ink was deposited onto 0.005" thick
polyester film with
a 0.0003" diameter wire wound rod and then cured at 130 C for 30 seconds
resulting in a
cohesive and conductive silver film. The adhesion of the film to the substrate
was evaluated by
utilizing the tape test method previously described in Example 2. Some of the
material was
removed from the substrate was removed from the substrate
COMPARATIVE EXAMPLES
[0107] The material of Example 1 was transferred to hexane by sodium chloride
induction similar to the method of Hirai. Hirai, et al., Chemistry Letters,
1992, 1527-1530; Hirai,
et al., J. of Colloid and Interface Sci., 1993, 161, 471-474. Hexane and a
sodium chloride
solution was added to concentrated material from Example 1 and the two phases
mixed with a
magnetic stir bar for 10 minutes. The silver nanoparticles transferred phases
to the non-aqueous
phase presumably leaving all ionic species in the aqueous phase. The solvent
phase with the
suspended silver particles was separated from the water phase. When an attempt
was made to
cure the phase transferred material at 120 C, the silver did not cure and an
oily silver film
remained even after extended periods at this temperature.
-23-