Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE I)E CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME DE _2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.
CA 02640416 2008-10-03
79783-69
BIFUNCTIONAL FUSION MOLECULES FOR THE DELIVERY OF ANTIGENS
TO PROFESSIONAL ANTIGEN-PRESENTING CELLS
FIELD OF THE INVENTION
The present invention relates generally to
bifunctional fusion molecules for the delivery of antigens
to professional antigen-presenting cells.
BACKGROUND OF THE INVENTION
In vivo targeting of the dendritic cell DEC-205
receptor has been shown to be effective in generating immune
responses to protect the host against cancer, viral
infection and autoimmune disease. Four types of DEC-205
targeting systems have been reported to date, including: a)
HB290 scFv coated liposome [van Broekhoven et al. Cancer
Res, 64:4357-65, 2004]; b) chemical cross-linking of HB290
with antigen [Mahnke et al. Cancer Res, 65:7007-12, 2005;
Bonifaz et al. J Exp Med, 199:815-24, 2004; Bruder et al.
Diabetes, 54:3395-401, 2005; Bonifaz et al. J Exp Med,
196:1627-38, 2002]; c) HB290 hybrid antibody [Trumpfheller
et al. J Exp Med. 203:607-17, 2006; Bozzacco et al. Proc
Natl Acad Sci USA, 104:1289-94, 2007; Hawiger et al. J Exp
Med, 194:769-79, 2001]; and d) bsmAb targeting system
[Wang et al. J Immunol Methods, 306:80-92, 2005]. There
are several limitations with these systems. Currently, the
above strategies have only demonstrated delivery of protein
or peptide to dendritic cells. However, these delivery
systems may not be versatile and flexible for clinical
applications, for example, in the delivery of DNA,
glycolipids and multiple, different antigens.
Limitations in the scFv coated liposome targeting
strategy includes instability of the scFv on the liposome
1
CA 02640416 2008-10-03
79783-69
surface (scFv could be lost during circulation); liposome
instability; scale-up issues; relative complexity in
encapsulation process; and only certain class of antigens
may be encapsulated efficiently (hydrophilic antigens
constantly leach from liposome). Issues including batch to
batch variation (cross-linking and purification) and
Fc domain mediated effects are some of the limitations in
chemical cross-linking of the whole mAb with antigen.
Hybrid antibody or recombinant antigen fusion protein
generation using mammalian expression system requires
careful monitoring of in cell productivity, and
consideration of issues such as post-translational chemical
modifications, degradation and aggregation of the final
product [Filpula. Biomol Eng, 24:201-15, 2007]. Moreover,
a new antibody fusion protein is required for each antigen
(protein or peptide) and the process is often time consuming
and costly. A bsmAb produced from a quadroma is also not
suitable for clinical applications and the limitations
include the yields and purity [Wang et al. J Immunol
Methods, 306:80-92, 2005]. Drawbacks for these systems may
also include dose-limiting toxicity (prolonged circulation
time) and immunogenicity against the targeting system that
can alter pharmacokinetics (biodistribution and clearance),
block receptor-antigen interactions, induce hypersensitivity
reactions and injection site reactions [Filpula. Biomol Eng,
24:201-15, 2007].
Accordingly, there remains a need for molecules
useful for delivering antigens to antigen presenting cells.
2
CA 02640416 2008-10-03
79783-69
SUMMARY OF THE INVENTION
In one aspect, the invention relates to a
bifunctional fusion molecule comprising: a first functional
domain comprising a first immunoglobulin variable region, a
second immunoglobulin variable region and a linker for
connecting the first and second variable regions; a second
functional domain comprising a moiety for binding to an
antigenic agent; wherein the first and second functional
domains are linked; and wherein the first functional domain
specifically binds to a surface molecule of a professional
antigen-presenting cell.
In another aspect, the invention relates to an
antigen delivery system comprising: a bifunctional fusion
molecule comprising: a first functional domain comprising an
immunoglobulin heavy chain variable region, an
immunoglobulin light chain variable region and a linker for
connecting the heavy chain variable region and the light
chain variable region; a second functional domain comprising
a moiety for binding to an antigenic agent; wherein the
first and second functional domains are linked; and wherein
the first functional domain binds to a surface molecule of a
professional antigen-presenting cell; and an antigenic agent
comprising an antigen and a particle; wherein the antigen is
conjugated to the particle, and wherein the particle binds
to the moiety of the second domain of the bifunctional
fusion molecule.
In another aspect, the invention relates to a
method of delivering an antigen to a professional
antigen-presenting cell, said method comprising: contacting
the professional antigen-presenting cell with the
bifunctional fusion molecule as described herein; and
3
CA 02640416 2008-10-03
79783-69
contacting the bifunctional fusion molecule with a plurality
of the antigenic agents as described herein.
In another aspect, the invention relates to a
method of modulating an immune response of a subject to an
antigen, said method comprising: administering to the
subject the bifunctional fusion molecule as described
herein, a plurality of the antigenic agents as described
herein and optionally, a co-stimulatory molecule.
In one exemplary embodiment, the invention
provides single-chain antibody-core-streptavidin
bifunctional fusion molecules (bfFp) that can form a complex
with any biotinylated antigen with one arm and targets the
antigen to the dendritic cells via the DEC-205 receptor with
its second paratope. Such a system has several unique
properties over the other targeting systems including
dendritic cell targeting with a variety of antigens. Almost
any antigen may be biotinylated by chemical conjugation (for
instance, NHS-LC-Biotin), photoactivation (photobiotin
acetate) or incorporated by synthetic strategies. This
simple strategy avoids the need of encapsulation, chemical
cross-linking and construction of a new hybrid fusion
antibody. The bfFp in addition lacks an Fc domain and the
E.coli based production is consistent and economical.
Faster clearance rate (kidney glomerular filtration cut-off
is 70 kDa) and lower immunogenicities are expected due to
smaller molecular weight (-46 kDa) of the vector. If
required, bfFp stability and half-life may be increased by
polyethylene glycol linkage [Holliger and Hudson. Nat
Biotechnol, 23:1126-36, 20051 or by isolation of the
tetrameric form of bfFp. Moreover, such a targeting vehicle
may be translated into clinical applications; several bfFp
(tetrameric form) have been applied in clinical studies for
4
CA 02640416 2008-10-03
79783-69
pretargeting radioimmunotherapy [Zhang et al. Proc Natl
Acad Sci USA, 100:1891-5, 2003; Graves et al. Clin Cancer
Res, 9:3712-21, 2003].
Other aspects and features of the present
invention will become apparent to those of ordinary skill in
the art upon review of the following description of specific
embodiments of the invention in conjunction with the
accompanying tables and figures.
BRIEF DESCRIPTION OF DRAWINGS
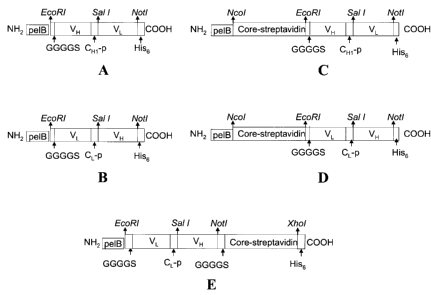
Figure 1. The construct of the HB290 scFv (A)
VH-VL, (B) VL-VH; and bfFp (C) WET5, (D) WET6 and (E) WET7.
The genes were cloned in different orientation to maximize
the production in E.coli system. Abbreviations: pelB,
bacterial leader sequence; VL, variable domain light chain;
VH, variable domain heavy chain; CH1_P, partial constant heavy
chain region 1, 15 amino acids linker; CL_P, partial constant
light chain region, 15 amino acids linker; G, glycine; S,
serine; His6, six histidine amino acid tag.
Figure 2. Expression and purification of
recombinant proteins. The scFv and bfFp genes were
chemically transformed, cultured, induced and the whole-cell
bacterial pellets were analyzed by Western blot. (A)
Western blot probed with anti-His6 mAb for analysis of scFv
and bfFp expression. Lane 1: HB290 scFv VH-VL; lane 2: scFv
VL-VH; lane 3: bfFp WET5, core-streptavidin-VH-VL; lane 4:
bfFp WET6, core-streptavidin-VL_VH; lane 5: bfFp WET7,
VL_VH_core-streptavidin. WET7 has the highest protein
expression level. (B) SDS-PAGE of WET7 before and after
IMAC purification. Lane 1: WET7 periplasmic protein;
lane 2: IMAC-purified WET7. M: molecular weight marker.
5
CA 02640416 2008-10-03
79783-69
The values at the left of each figure denote molecular
weights.
Figure 3. Demonstration of IMAC-purified bfFp
bifunctional activity and thermal stability. (A) IMAC
purified bfFp was incubated either at 60 C (lane 3) or 95 C
(lane 4) for 10 min and probed with B-BSA followed by
streptavidin-HRPO. Lanes 1 (HB290 scFv VH-VL) and 2 (HB290
scFv VL_VH) are controls. The values at the left of the
figure denote molecular weights. The IMAC-purified bfFp
bifunctional activity was tested against DC 2.4 cells and
B-OVA via ELISA method (B). Competition binding studies of
HB290 mAb and bfFp for binding to dendritic cells. The
binding of bfFp is confirmed by B-OVA and streptavidin-HRPO.
The error bars are the standard deviations.
Figure 4. Analysis of humoral immune responses to
biotinylated antigens in vivo. Groups of five mice were
immunized with different antigen combinations along with a
PBS control. The specific components, amounts, and the
schedule of immunizations are outlined in Table 2. The mice
were analyzed individually and the data was pooled. The
humoral response, as quantified by serum antibody titres on
day 21 post immunization, was measured by ELISA method
against the respective antigens listed in Table 1B. The
method involved coating 10 g of antigen in microtiter
plates followed by addition of 1:1000 diluted serum
antibody, and detection using GAM-HRPO. The ELISA
measurements were done in quadruplicate for each mouse. The
mean ELISA values obtained for each individual mouse were
further averaged. The error bars represent the standard
deviation in a group of 5 mice.
6
CA 02640416 2008-10-03
79783-69
Figure 5. Analysis of cell-mediated immune
responses based on IFN-y estimation. Spleen T cells
(responder cells) from the groups of mice immunized with
different antigens (shown on X-axis and for details see
Tables lB and 2) were isolated and purified using a nylon
wool column. The five spleens in each group were pooled,
mixed in DMEM prior to nylon wool purification. Stimulator
cells prepared from the spleen cells of naive mice were
isolated and treated with 50 g/ml mitomycin C. Purified
responder cells from both immunized and non-immunized mice
were aliquoted to a tissue culture plate in quadruplicate
with or without stimulator cells. The cells were then
incubated with 10 g of antigen for 3 days at 37 C in a CO2
atmosphere. After incubation, the IFN-y concentration in
the supernatant was determined using mouse IFN-T ELISA
Ready-SET-Go kit using the protocol from eBioscience. Each
data set is shown following subtraction of the corresponding
ELISA values obtained without stimulator cells. The error
bars are the standard deviations.
Figure 6. Analysis of serum reactivity towards
biotin, bfFp and core-streptavidin. The mice (5 mice per
group) were analyzed individually and the data was pooled.
The mouse group is listed on the X-axis. The serum
reactivity, as quantified by serum antibody titres on day 21
post immunization, was measured by ELISA method against
B-BSA, bfFp and core-streptavidin. The method involved
coating of 10 g antigen in microtiter plates followed by
addition of 1:1000 diluted serum antibody, and detection
using GAM-HRPO. The ELISA measurements were done in
quadruplicate for each mouse. The mean ELISA values
obtained for each individual mouse were further averaged.
7
CA 02640416 2008-10-03
79783-69
The error bars represent the standard deviation in a group
of 5 mice.
Figure 7. Amino acid sequence of the VH region of
HB290 (SEQ ID NO: 17).
Figure 8. Nucleotide sequence of the VH region of
HB290 (SEQ ID NO: 18).
Figure 9. Amino acid sequence of the VL region of
HB290 (SEQ ID NO: 19).
Figure 10. Nucleotide sequence of the VL region of
HB290 (SEQ ID NO: 20).
Figure 11. Amino acid (SEQ ID NO: 22), coding
(SEQ ID NO: 21) and non-coding (SEQ ID NO: 23) nucleotide
sequences of the VH-CH1 region of HB290.
Figure 12. Amino acid (SEQ ID NO: 25), coding
(SEQ ID NO: 24) and non-coding (SEQ ID NO: 26) nucleotide
acid sequences of the VL-CL region of HB290.
DETAILED DESCRIPTION
The bifunctional fusion molecules of the present
invention may be used to deliver one or more antigens, such
as an infectious disease antigen, to a professional
antigen-presenting cell, such as a dendritic cell.
The in vivo targeting strategy of the present
invention is effective in generating an immune response that
protects the host against viral infection, autoimmune
disease and transplant rejection. The presentation of
antigen may result in immunostimulation or immunoregulation
depending on the presence or absence, respectively, of
co-stimulation of the professional antigen-presenting cell,
8
CA 02640416 2008-10-03
79783-69
for example, with a CD40 agonist (such as an anti-CD40
antibody, CpG, polyI:C and MF59). As a result, the
bifunctional fusion molecule of the present invention may be
used to target one or more antigens to professional
antigen-presenting cells, such as dendritic cells, to
stimulate an immune response against a viral or bacterial
pathogen; or to induce tolerance by delivery antigen in the
absence of a co-stimulatory molecule.
Other co-stimulatory molecules that may mature
and/or activate professional antigen presenting cells
include, but are not limited to, whole bacteria or
bacterial-derived antigens, inflammatory cytokines, or other
molecules that can cross-link to a a select cell surface
receptor such as CD40, and viral products
The bifunctional fusion molecules, antigens,
methods and uses described herein relate to a bifunctional
fusion molecule that has a first functional domain specific
for a cell surface molecule of a professional
antigen-presenting cell and a second functional domain
comprising a moiety that is specific for a particle, where
the particle is not a target of the first functional domain,
in combination with an antigenic agent comprising an antigen
and a particle that associates with the moiety of the second
functional domain of the antibody, to direct antigens to a
professional antigen-presenting cell. In one embodiment,
the bifunctional fusion molecule may be in monomeric form.
As used herein, a "moiety" may comprise any
molecule that can associate specifically with the particle
of the antigenic agent.
To develop the bifunctional fusion molecule,
general molecular approaches may be used to clone the
9
CA 02640416 2008-10-03
79783-69
expressed VH and VL fragments from a hybridoma expressing an
antibody of known specificity. Theses fragments are then
joined by a linker and combined with a molecule, such as a
core streptavidin fragment, resulting in a bifunctional
fusion molecule.
Antibodies (also termed immunoglobulin) are
protein molecules produced by lymphocytes that bind with
high specificity for its cognate antigen. Typically,
antibodies consist of two heavy and two light chains that
are covalently linked to each other via disulfide bonds.
Each chain comprises variable domains and constant domains.
A variable domain comprising three complementary regions
(CDRs) is located at the N-terminus of each chain.
Together, variable regions of the heavy and light chains
determine antigen specificity of the antibody. Single chain
variable fragment (scFv) antibodies consists of variable
heavy (VH) and variable light (VL) domains linked by a
flexible amino acid linker (Bird et al. Science,
242:423-426, 1988; Huston et al. Proc Natl Acad Sci USA,
85:5879-5883, 1988).
scFvs are encoded by a single gene and the
resulting scFv may be expressed in microbial systems such as
yeast and prokaryotic systems (reviewed in Verma et al.
J. Immunol. Methods, 216:165-181, 1998), allowing for rapid
selection of specific high affinity molecules, using
techniques such as phage display or ribosome display. Due
to the absence of a Fc portion, scFv antibodies do not exert
toxic effects via antibody-dependent or complement-dependent
cell-mediated cytotoxicity. Further, scFv antibodies show
good tissue penetration abilities. Due to these advantages,
scFv fragments have found broad applications in medicine
(reviewed by Huston et al. Int Rev Immunol, 10:195-217,
CA 02640416 2008-10-03
79783-69
1993) and have potential in biotechnology (Harris. Trends
Biotechnol, 17:290-296, 1999).
scFvs have been successfully used in intracellular
applications (Worn et al. Curr Opin Rheumatol, 16:38-42,
2000; Auf der Maur et al. J Biol Chem, 277:45075-85, 2002;
Stocks. Drug Discov Today, 15:960-966, 2004). In general,
intracellular expression of functional scFvs is limited by
their instability, insolubility, and tendency to form
aggregates. Thus, in vivo screening systems for scFv
antibodies have been developed using a "Quality Control"
screen (W00148017; Auf der Maur et al. FEBS Lett
508:407-412, 2001; Auf der Maur et al. Methods, 34:215-224,
2004), leading to the identification of particularly stable
and soluble scFv framework sequences (W003097697). These
frameworks are highly expressed, stabile and soluble under
natural, oxidizing conditions in the extracellular
environment. Accordingly, scFvs are suitable for
therapeutic applications.
The immunoglobulins of the present invention may
be adapted for use in a recipient. For example, murine
antibody fragments may be adapted for use in humans by
humanizing the fragments. Humanized antibodies may be
produced, without limitation, by merging the DNA that
encodes the binding portion of a non-human (e.g. mouse)
antibody with human antibody-producing DNA. The extent of
the CDRs, the human frameworks to use and the substitution
of residues from the rodent mAb into the human framework
regions (back mutations) may also be considered in the
design of the humanized antibody.
As used herein, "linker" refers, without
limitation, to a molecule or a chemical bond that links two
11
CA 02640416 2008-10-03
79783-69
domains. In one embodiment, the VH and VL domains are linked
by chemical cross-linking. In another embodiment, the
linker is a peptide linking VH and VL that permits folding of
the antibody fragments, i.e. an appropriate folding of the VH
and VL domains and their capacity to be brought together. In
addition, the linker permits folding into a monomeric
functional unit. When the antibody fragments are assembled
in the VH to VL orientation (Vx-linker-VL) , a linker of 3 to
12 residues may not fold into a functional Fv domain and
instead may associate with a second molecule to form a
bivalent dimer. Reducing below 3 residues may lead to
trimers. Direct ligation of VL to Vi., may lead to the
formation of tetramers. A typical linker of the present
invention may have at least 12 and preferably less than
25 amino acids, preferably between 14-18, 14-16, or 15 amino
acids.
In an embodiment the VH and VL regions may be
linked through the dimerisation of two monomers (diabodies).
The first functional domain and the second
functional domain of the bifunctional fusion molecule may be
linked by a linker as described above.
The first functional domain of the bifunctional
fusion molecule comprises, consists or consists essentially
of a Vx region and a VL region joined by a linker. In one
embodiment, the first functional domain comprises an scFv
antibody.
The first and second functional domains may be
joined in any number of configurations. One configuration
of the bifunctional fusion molecule is: (NH2) VH-linker-VL-
second functional domain (COOH). Another configuration of
the bifunctional fusion molecule is: (NH2) VL-linker-VH-
12
CA 02640416 2008-10-03
79783-69
second functional domain (COOH). Another configuration of
the bifunctional fusion molecule is: (COOH) VH-linker-VL-
second functional domain (NH2). A further configuration of
the bifunctional fusion molecule is: (COOH) VL-linker-VH-
second functional domain (NH2). The preferred configuration
of the bifunctional fusion molecule is: (NH2) VL-linker-VH-
second functional domain (COOH).
The second functional domain comprises a moiety
that interacts with a cognate particle on an antigenic
agent. The second functional domain may comprise any moiety
as long as it is not reactive with the immunoglobulin
fragments of the first functional domain. In one
embodiment, the moiety of the second functional domain is a
protein, for instance an enzyme such as streptavidin, core
streptavidin or avidin. In another embodiment, the moiety
of the second functional domain is an antibody that binds
biotin.
Streptavidin is a 53 kDa tetrameric protein
purified from the bacterium Streptomyces avidinii. It finds
wide use in molecular biology through its extraordinarily
strong affinity for the vitamin biotin; the dissociation
constant (Kd) of the biotin-streptavidin complex is on the
order of -10-15 mol/L, ranking among one of the strongest
known non-covalent interactions. Streptavidin mutants may
also be engineered in the form of a stable, single-chain
dimer, (Aslan et al., Proc Natl Acad Sci USA, 102:8507-8512,
2005). Further, streptavidin may be modified, for example,
by substitution of charged, aromatic, or large hydrophobic
residues on the surface of streptavidin with smaller neutral
residues to reduce the molecule's immunogenicity
(Meyer et al. Protein Science, 10:491-503, 2001). As used
herein, "core streptavidin" refers to a streptavidin product
13
CA 02640416 2008-10-03
79783-69
that had been reduced to a minimal size that still retains
full biotin-binding activity.
The bifunctional fusion molecule may further
comprise a tag, for example, a protein tag. Protein tags
may find use in protein purification, specific enzymatic
modification and chemical modification. Protein tags are
known in the art and include, but are not limited to,
affinity tags (e.g. poly-Histidine), solubilization tags
(e.g. thioredoxin), chromatography tags (e.g. FLAG), epitope
tags (e.g. c-myc-tag) and fluorescent tags (e.g. GFP). In
some instances, these tags are removable by chemical agents
or by enzymatic means.
The first functional domain of the bifunctional
fusion molecule targets a cell surface molecule of a
professional antigen-presenting cell.
As used herein, a "surface molecule" refers to any
molecule that associated with the plasma membrane of a
professional antigen-presenting cell. Preferably, the
surface molecule is present on the extracellular side of the
plasma membrane. Exemplary surface molecules include, but
are not limited to, receptors, cell adhesion molecules,
cellular transporters and other molecules displayed on the
cell surface.
The term "cell" or "cells" refers to a single
cell, as well as a plurality of cells, a culture of cells, a
growth of cells, a population of cells or a cell line, and
may be in vitro or in vivo, where context permits, unless
otherwise specified. In vitro cells include ex vivo cells
explanted from a subject. Similarly, reference to "cells"
also includes reference to a single cell where context
permits, unless otherwise specified.
14
CA 02640416 2008-10-03
79783-69
The adaptive immune system has evolved to
recognize and respond to a vast number of diverse antigens
based on a repertoire of lymphocytes of unique antigen
binding specificities. Lymphocytes generally work in tandem
with professional antigen-presenting cells which capture
antigen and present them to the lymphocytes, resulting in
lymphocyte activation or inactivation, depending on the
presence or absence of co-stimulatory signals, respectively.
Most cells in the body can present antigen to CD8+ T cells
via MHC class I molecules however, as used herein,
"professional antigen-presenting cells" (or "APCs") refers
generally to those specialized cells that can prime T cells.
Professional antigen-presenting cells, in general, express
MHC class II as well as MHC class I molecules, and can
stimulate CD4+ ("helper") cells as well as CD8+
("cytotoxic") T cells.
There are three main types of professional
antigen-presenting cell: 1) Dendritic cells; 2) Macrophages;
and 3) B-cells.
Dendritic cells are the most specialized and
potent professional antigen-presenting cells in the immune
system and play a critical role in innate and adaptive
immune responses, especially in priming and activating
T cell and B cell immunity. Dendritic cells may originate
from the myeloid (including Langerhan cells) or lymphoid (or
plasmacytoid) lineage. The term includes dendritic cells at
any stage of development, including, but not limited to,
immature dendritic cells and activated dendritic cells.
Dendritic cells are motile cells that ingest
antigens and present them to B and T lymphocytes. Dendritic
cells also play critical roles in the induction of central
CA 02640416 2008-10-03
79783-69
and peripheral immunological tolerance, the regulation of
the types of T cell immune responses, and functions in
innate immunity against microbes (reviewed in Sato and
Fujita. Allergology International, 56:183-191, 2007;
Banchereau and Steinman. Nature, 392:245-252, 1998;
Diebold. Immunology and Cell Biology, 86:389-397, 2008).
During maturation, dendritic cells change the
uptake, processing and presentation of material from their
environment (Mellman and Steinman. Cell, 106:255-258,
2001). Immature dendritic cells constantly ingest material
from their environment, but are not efficient in antigen
presentation. Once the cells are activated and undergo
maturation, processing and presentation of antigens from the
ingested material is induced, resulting in increase levels
of major histocompatibility complex (MHC) class II molecules
at the cell surface (Inaba et al. J Exp Med, 191:927-936,
2000).
Dendritic cells utilizes surface receptors
(Gb3/CD77, CD40, 02 integrins, Fc receptors, and C-type
lectin receptors) to internalize, process and present
antigens to MHC class I and MHC class II pathways
(Tacken et al. Immunobiology, 211:599-608, 2006).
Exemplary dendritic cell surface receptors that may be used
for the targeting of antigens to dendritic cells are listed
in Table 1.1.
Table 1.1 also shows the expression pattern of the
receptors and also indicates whether antigen targeting to
the receptor required co-stimulation for induction of immune
responses. In summary, antigen targeting to Gb3, LOX-I and
CD40 receptors does not require co-stimulation for induction
of immune responses. DC-SIGN expression is restricted to
16
CA 02640416 2008-10-03
79783-69
dendritic cells and macrophages; whereas other receptors are
distributed on a variety of cells. DEC-205 targeting
strategies appear to be extensively studied and applied for
a variety of applications.
DEC-205 (CD205) is a C-type lectin receptor that
is present in both immature and mature dendritic cells in
lymphoid tissues, lymph nodes and spleen (Inaba et al. Cell
Immunol, 163:148-56, 1995; den Haan et al. J Exp Med,
192:1685-96, 2000; Witmer-Pack et al. Cell Immunol,
163:157-62, 1995; Granelli-Piperno et al. J Immunol,
175:4265-73, 2005; Pack et al. Immunology, 123:438-46,
2008). DEC-205 is also expressed at low levels on
macrophages and at moderate levels by B cells and is up-
regulated during the pre-B cell to B cell transition
(Inaba et al. Cell Immunol, 163:145, 1995). DEC-205
behaves as an antigen uptake/processing receptor for
dendritic cells (Kato et al. International Immunology,
18:857-869, 2006). In the presence of co-stimulatory
molecules, DEC-205 targeting of antigens by an antibody is
an efficient strategy to protect the host against tumor
growth (van Broekhoven et al. Cancer Res, 64:4357-65,
2004), rejection of existing tumor (Mahnke et al. Cancer
Res, 65:7007-12, 2005), protection against airway challenge
of virus (Trumpfheller et al. J Exp Med. 203:607-17, 2006),
and enhance resistance to an established rapidly growing
tumor, as well as viral infection (Bonifaz et al. J Exp
Med, 199:815-24, 2004).
DEC-205 targeting of a single protein enables
cross-presentation of several peptides more efficiently than
CD206 and CD209 (Bozzacco et al. Proc Natl Acad Sci U S A,
104:1289-94, 2007) and induces stronger T cell immunity at
much lower doses of protein antigen, plasmid DNA or
17
CA 02640416 2008-10-03
79783-69
recombinant adenovirus (Trumpfheller et al. J Exp Med.
203:607-17, 2006). In the absence of co-stimulation, DEC-205
targeting of antigen can be used to suppress the development
of autoimmunity, such as type 1 diabetes (Bruder et al.
Diabetes, 54:3395-401, 2005). Similarly, other studies have
demonstrated that DEC-205 targeting, depending on the
presence or absence of an additional activation signal
(i.e. CD40 agonist, such as an anti-CD40 antibody), may
result in either immunostimulatory or immunoregulatory
effects, respectively (Bonifaz et al. J Exp Med, 196:1627,
2002; Hawiger et al. J Exp Med, 194:769, 2001; Hawiger et
al. Immunity, 20:695, 2004; Bonifaz et al. J Exp Med,
199:815, 2004).
Other dendritic cell surface molecules that may be
used in the context of the invention may be identified by
methods known in the art. For example, a typical method for
identifying other suitable dendritic cells surface molecules
as a target comprise first directing a molecule, such as a
ligand or an antigen, towards the professional
antigen-presenting cell surface molecule. Flow cytometry
may be performed on those cells to identify upregulation or
downregulation of MHC, CD and/or other cell surface
molecules. DNA microarray, 2D SDS-PAGE, cytokine
monitoring, cells proliferation study, in vivo fluorescence
imaging, in vivo immune system study and animal challenge
study may be subsequently used to identify the cell surface
molecule.
In one embodiment, the first functional domain of
the bifunctional fusion molecule is directed towards
DEC-205.
In this regard, the VH region of the bifunctional
18
CA 02640416 2008-10-03
79783-69
fusion molecule may comprise, consist of, or consist
essentially of an amino acid sequence that has at least 70%,
at least 75%, at least 80%, at least 83%, at least 85%, at
least 87%, at least 89%, at least 91%, at least 93%, at
least 95%, at least 97%, at least 99% or has 100% sequence
identity to SEQ ID NO: 17 (amino acid sequence of VH),
wherein the VH in association with VL have specificity
towards DEC-205.
In one embodiment, the VH region of the
bifunctional fusion molecule is encoded by a nucleic acid
sequence that has at least 70%, at least 75%, at least 80%,
at least 83%, at least 85%, at least 87%, at least 89%, at
least 91%, at least 93%, at least 95%, at least 97%, at
least 99% or has 100% sequence identity to SEQ ID NO: 18
(nucleic acid sequence of VH), wherein the VH in association
with VL have specificity towards DEC-205.
The VL region of the bifunctional fusion molecule
may comprise, consist of, or consist essentially of an amino
acid sequence that has at least 70%, at least 75%, at least
80%, at least 83%, at least 85%, at least 87%, at least 89%,
at least 91%, at least 93%, at least 95%, at least 97%, at
least 99% or has 100% sequence identity to SEQ ID NO: 19
(amino acid sequence of VL), wherein the VL in association
with VH have specificity towards DEC-205.
In one embodiment, the VL region of the
bifunctional fusion molecule is encoded by a nucleic acid
sequence that has at least 70%, at least 75%, at least 80%,
at least 83%, at least 85%, at least 87%, at least 89%, at
least 91%, at least 93%, at least 95%, at least 97%, at
least 99% or has 100% sequence identity to SEQ ID NO: 20
19
CA 02640416 2008-10-03
79783-69
(nucleic acid sequence of VL) , wherein the VL in association
with VH have specificity towards DEC-205.
Fragments of VH and VL regions that retain the
binding specificity of the full length VH and VL regions may
be used. Such fragments may be e.g. at least 10, 20, 30,
40, 50, 100 or 200 amino acids in length.
As used herein, "% sequence identity" is
determined by comparing two optimally aligned sequences over
a comparison window, where the fragment of the polypeptide
or polynucleotide sequence in the comparison window may
comprise additions or deletions (e.g., gaps or overhangs) as
compared to the reference sequence (which does not comprise
additions or deletions) for optimal alignment of the two
sequences. The percentage is calculated by determining the
number of positions at which the identical amino acid
residue or nucleic acid residue occurs in both sequences to
yield the number of matched positions, dividing the number
of matched positions by the total number of positions in the
comparison window and multiplying the result by 100 to
provide the percentage of sequence identity. Algorithms to
align sequences are known in the art. Exemplary algorithms
include, but are not limited to, the local homology
algorithm of Smith and Waterman (Add APL Math, 2: 482,
1981); the homology alignment algorithm of Needleman and
Wunsch (J Mol Biol, 48: 443, 1970); the search for
similarity method of Pearson and Lipman (Proc Natl Acad Sci
USA, 85: 2444, 1988); and computerized implementations of
these algorithms (GAP, BESTFIT, BLAST, PASTA, and TFASTA in
the Wisconsin Genetics Software Package, Genetics Computer
Group (GCG), 575 Science Dr., Madison, Wis.). In one
embodiment, two sequences may be aligned using the "Blast 2
Sequences" tool at the NCBI website at default settings
CA 02640416 2008-10-03
79783-69
(Tatusova and Madden. FEMS Microbiol Lett, 174: 247-250,
1999). Alternatively, amino acid sequences or nucleic acids
sequences may be aligned by human inspection.
As used herein, "consists essentially of" or
"consisting essentially of" means that the bifunctional
fusion molecule may include amino acid residues, including
within the amino acid sequence or at one or both ends of the
amino acid sequence, but that the additional residues do not
materially affect the function of the bifunctional fusion
molecule.
In this regard, the VH region may include residues
from the constant region of the heavy chain (see SEQ ID
NO: 21 and SEQ ID NO: 22) and/or the VL regions may include
residues from the constant region of the light chain (see
SEQ ID NO: 24 and SEQ ID NO: 25).
The bifunctional fusion molecule associates with
an antigenic agent through interaction at the second
functional domain. An antigenic agent comprises an antigen
and a particle, where the particle binds the moiety of the
second functional domain of the bifunctional fusion
molecule. In one embodiment the antigen is linked to the
particle. Further, the antigen and particle of the
antigenic agent may be linked by chemical cross-linking or
by a linker.
Antigens may comprise, without limitation,
proteins, peptides, nucleic acids and/or glycolipids. As
used herein, an "antigen" refers to molecules that react
with antibodies, B-cell receptors and/or T-cell receptors.
Some antigens do not by themselves elicit an immune
response.
21
CA 02640416 2008-10-03
79783-69
As used herein, the terms "peptide",
"oligopeptide", "polypeptide" and "protein" may be used
interchangeably. Polypeptides may comprise non-natural
amino acids and may be joined to linker elements known to
the skilled person. Polypeptides may also comprise
modifications to the amino acid side chains or the backbone
structure, such modifications may occur during polypeptide
synthesis or processing or following treatment with isolated
modifying enzymes. Further, polypeptides may be monomeric
or multimeric, and may include derivatives, variants,
fragments, analogs or homologs thereof.
Polypeptides may comprise a contiguous span of at
least 5, at least 10, at least 25, at least 50, at
least 100, at least 250, at least 500, at least 1000, at
least 1500, or at least 2500 consecutive amino acids and may
retain the desired activity and/or structure of the full
length polypeptide.
The term "protein" may also refer to a full length
or essentially the full length product encoded by a gene.
As used herein, "essentially" means that a protein may
include or lack one or more amino acid residues, but that
the additional or missing amino acid residues do not
materially affect the function and/or structure of the
protein.
As used herein, "nucleotide sequence", "nucleic
acid" or "nucleic acid molecule" refers to a polymer of DNA
or RNA which can be single or double stranded and optionally
containing synthetic, non-natural or altered nucleotide
bases capable of incorporation into DNA or RNA polymers.
"Nucleic acids", "Nucleic acid sequences" or "Nucleic acid
molecules" may encompass genes, cDNA, DNA and RNA encoded by
22
CA 02640416 2008-10-03
79783-69
a gene. Nucleic acids or nucleic acid sequences may
comprise at least 3, at least 10, at least 100, at least
1000, at least 5000, or at least 10000 nucleotides or base
pairs.
As used herein, "glycolipid" refers, without
limitation, to any compound containing one or more
monosaccharide residues bound by a glycosidic linkage to a
hydrophobic moiety including, but not limited to, an
acylglycerol, a sphingoid, a ceramide (N-acylsphingoid) or a
prenyl phosphate.
The antigens of the invention include, without
limitation, those derived from cancer, autoimmune diseases
and infectious disease agents such as viruses and bacteria.
Exemplary infectious viral and/or bacterial
antigens include proteins derived from, and nucleic acids
encoding proteins derived from bacteria and viruses
including, but not limited to, avian influenza, ebola,
Bacillus anthracis (anthrax), Severe acute respiratory
syndrome-coronavirus (SARS-CoV), Western Equine Encephalitis
Virus (WEEV), poliovirus, human rhinovirus, hepatitis A
virus, human immunodeficiency virus, human influenza, human
papillomavirus, herpes simplex virus, picornaviruses such as
foot-and-mouth disease virus, Dengue and West Nile viruses,
Yersinia pestis, and respiratory syncytial virus.
Exemplary cancer disease associated and/or
specific antigens include, but are not limited to, MUC-1 of
breast cancer, GM2 and GM3 of myeloma cancer.
Exemplary antigens associated with autoimmune
disease include, but are not limited to, transglutaminase in
celiac disase, muscle actin in autoimmune hepatitis, Bullous
23
CA 02640416 2008-10-03
79783-69
Pemphigoid antigen 1 and 2 in bullous pemphigoid, basement
membrane collagen Type IV protein in Goodpasture's syndrome,
ganglioside in Guillain-Barre syndrome, myelin basic protein
in multiple sclerosis, desmogein 3 in Pemphigus Vulgaris,
p62/splOO/mitochondrial(M2) in primary biliary cirrhosis,
rheumatoid factor in rheumatoid arthritis, and topoisomerase
in Scleroderma.
The antigenic agent further comprises a particle.
The particle may be, without limitation, a vitamin, a
peptide, a protein, a glycoconjugate or any natural or
synthethic conjugate, so long as it associates with the
moiety of the second functional domain of the bifunctional
fusion molecule but not with the immunoglobulin fragments of
the first functional domain. In one embodiment, the antigen
may have high affinity specifically for the moiety of the
second functional domain of the bifunctional fusion
molecule. Pairs of molecules with strong affinity for one
another that may be suitable for use in the context of the
invention are known in the art and include, but not limited
to, streptavidin and biotin, and an antibody and its cognate
antigen. The skilled person would appreciate that methods
to attach a particle to an antigen are known in the art and
that the choice of a suitable method would depend, among
other factors, on the nature and/or composition of the
antigen and the particle. Typical methods of attaching a
particle to an antigen include, but are not limited to,
covalent, ionic or electrostatic interactions. In one
embodiment, an antigen is attached to a particle by a
linker.
As noted above, various configurations of the
first and second functional domains are possible. For the
24
CA 02640416 2008-10-03
79783-69
purpose of illustration, the following are a number of non-
limiting examples of particular combinations of particular
bifunctional fusion molecules according to the invention:
1. VL-VH-Avidin
2. VH-VL-Avidin
3. Avidin-VL-VH
4. Avidin-VH-VL
5. VH*-VL*-VH-VL
6. VL*-VH*-VH-VL
7. VH*-VL*-VL-VH
8. VL*-VH*-VL-VH
9. VH-VL-VH*-VL*
10. VL-VH-VH*-VL*
11. VH-VL-VL*-VH*
12. VL-VH-VL*-VH*
13. VH*-VL-VH-VL* (bispecific diabody)
14. VL*-VH-VL-VH* (bispecific diabody)
In Examples 1 to 4, the second functional domain
comprises avidin as the moiety which specifically binds a
particle (in this instance biotin) attached to the antigen.
Alternative suitable moiety/particle pairs may of course be
used.
CA 02640416 2008-10-03
79783-69
Examples 5 to 12 are bispecific scFv wherein the
second functional domain comprises immunoglobulin heavy and
light chain variable regions (identified by the *) as the
moiety which binds to a particle attached to the antigen.
Again, the particle could be e.g. biotin, but may be any
particle to which the immunoglobulin moiety is specific.
Examples 13 and 14 are variants on the strategy
shown in examples 5 to 12, and depict bispecific diabody
configurations, in which the second functional domain again
comprises immunoglobulin heavy and light chain variable
regions (identified by the *), but are separated, one at
either end of the first functional domain.
In one embodiment, the moiety of the second
functional domain of the bifunctional fusion molecule is
streptavidin or avidin and the particle of the antigenic
agent is biotin. Alternatively, the moiety of the second
functional domain may be biotin, and the particle of the
antigen agent may be streptavidin or avidin. Biotinylation
may be effected by methods known in the art, including, but
not limited to, chemical conjugation (for instance,
NHS-LC-Biotin), photoactivation (photobiotin acetate) or
incorporated by synthetic strategies.
The construction, expression, purification and
analysis of the bifunctional fusion molecule and the
antigenic agent may be achieved by methods known in the art.
With respect to construction, a skilled person can
construct the bifunctional fusion molecule based on known
molecular techniques. In one embodiment, the bifunctional
fusion molecule may be expressed from a vector encoding VH
and VL fragments separated by a linker and fused to a second
functional domain. In another embodiment, the first
26
CA 02640416 2008-10-03
79783-69
functional domain may comprise variable regions and a linker
that were each encoded by separate vectors, expressed and
subsequently joined together, for example, by chemical
conjugation. In a further embodiment, the VH and VL
fragments may be cloned from a hybridoma expressing an
antibody of desired specificity. Alternatively, VH, VL and
linker fragments may be synthesized chemically separately
and then joined together by methods known in the art, or the
fragments may be synthesized as a single molecule.
Similarly, the skilled person would know how to
construct an antigenic agent comprising an antigen and a
particle, depending on the nature of the antigen and that of
the particle. The antigen may be chemically synthesized or
cloned from a cell expressing the antigen. The antigen and
the particle may be expressed and/or produced in tandem or
separately.
Methods of expressing the bifunctional fusion
molecule and the antigenic agent will depend on their
composition. Exemplary methods include microbial,
mammalian, plant cell cultures and cell free culture systems
known to the skilled person.
The expressed bifunctional fusion molecule and the
antigenic agent may be purified with methods and techniques
known in the art. Exemplary purification methods include,
but are not limited to, affinity chromatography, size
exclusion chromatography, Immobilized Metal Chelating
Chromatography (IMAC), and agarose / acrylamide gel
electrophoresis.
Following purification, the bifunctional fusion
molecule and the antigenic agent may be detected and/or
assessed for biological activity. Exemplary methods of
27
CA 02640416 2008-10-03
79783-69
detecting the bifunctional fusion molecule and the antigenic
agent may include, but are not limited to, Western blot
analysis, ELISA and gas chromatography. Exemplary methods
of assessing the biological function of the bifunctional
fusion molecule and the antigenic agent may include, but are
not limited to, serum neutralization inhibition assays, and
in vivo assays to assess the antibody production as
indication of a humoral immune response and IFN-y as
indication of cell mediated immune response.
The bifunctional fusion molecule and the antigenic
agent may in used serially, in either order, simultaneously
or as part of a treatment strategy. In one embodiment,
there is provided a pharmaceutical composition comprising a
bifunctional fusion molecule and a pharmaceutically
acceptable carrier. In another embodiment, there is
provided a pharmaceutical composition comprising a
bifunctional fusion molecule, an antigenic agent and a
pharmaceutically acceptable carrier. In a further
embodiment, there is provided a bifunctional fusion
molecule, an antigenic agent and a kit. The pharmaceutical
composition, as described herein, may be used to immunize a
subj ect .
Kits and commercial packages containing the
bifunctional fusion molecules and/or the antigenic agents
described herein or kits and commercial packages containing
a pharmaceutical composition as described herein, are
contemplated. Such a kit or commercial package will also
contain instructions regarding use of the included
bifunctional fusion molecules, antigenic agents and/or
pharmaceutical compositions, for example, to treat
infectious diseases in accordance with the methods described
herein.
28
CA 02640416 2008-10-03
79783-69
In one aspect, a plurality of bifunctional fusion
molecules directed to distinct cell surface antigens may be
administered to a subject in combination with a plurality of
distinct antigenic agents, wherein the antigenic agents
associate with the moiety of the second functional domain of
the bifunctional fusion molecule.
In another aspect, a plurality of bifunctional
fusion molecules directed to distinct cell surface antigens
may be administered to a subject in combination with a
plurality of identical antigenic agents, wherein the
antigenic agents associate with the moiety of the second
functional domain of the bifunctional fusion molecule.
In another aspect, a plurality of bifunctional
fusion molecules directed to the same cell surface antigen
may be administered to a subject in combination with a
plurality of distinct antigenic agents, wherein the
antigenic agent associates with the moiety of the second
functional domain of the bifunctional fusion molecule.
In a further aspect, a plurality of bifunctional
fusion molecules directed to the same cell surface antigen
may be administered to a subject in combination with a
plurality of identical antigenic agents, wherein the
antigenic agent associates with the moiety of the second
functional domain of the bifunctional fusion molecule.
As used herein, "immunize" or "immunization" and
"vaccinate" or "vaccination" are used interchangeably and
refer to a means for providing protection against a pathogen
by inoculating a host with an immunogenic preparation
containing a bifunctional fusion molecule and an antigenic
agent, in combination with an APC co-stimulatory molecule,
such as a CD40 agonist, such that the host immune system is
29
CA 02640416 2008-10-03
79783-69
stimulated and prevents or attenuates subsequent pathology
associated with the host reactions to subsequent exposures
of the pathogen. Alternatively, a host may be inoculated
solely with an immunogenic preparation containing a
bifunctional fusion molecule and an antigenic agent to
induce tolerance in treating an autoimmune disease.
A person skilled in the art would know how to
prepare suitable vaccine formulations. Conventional
procedures and ingredients for the selection and preparation
of suitable formulations are described, for example, in
Remington's Pharmaceutical Sciences (16th edition, Osol, A.
Ed. (1980) Mack Printing Company, Easton, PA) and in The
United States Pharmacopeia: The National Formulary (USP 24
NF19) published in 1999.
To aid in administration of such an antibody
and/or antigenic agent to a subject, such as a subject in
need of treatment of an infectious disease, an antibody
and/or antigenic agent may be formulated as an ingredient in
a pharmaceutical composition.
The pharmaceutical composition may further include
a pharmaceutically acceptable diluent or carrier. The
invention in one aspect therefore also includes such
pharmaceutical compositions for use in treating an
infectious disease. The compositions may routinely contain
pharmaceutically acceptable concentrations of salt,
buffering agents, preservatives and various compatible
carriers. Pharmaceutically acceptable carriers, diluents
and excipients are known in the art and are described, for
example, in Remington's Pharmaceutical Sciences 16th
edition, Osol, A. Ed. (1980) Mack Printing Company,
Easton, PA.
CA 02640416 2008-10-03
79783-69
The forms of the pharmaceutical compositions
suitable for injectable use include sterile aqueous
solutions or dispersion and sterile powders for the
extemporaneous preparation of sterile injectable solutions
or dispersions, wherein the term sterile does not extend to
any cell that may comprise the pharmaceutical product of
interest that is to be administered. In all cases the form
must be sterile and must be fluid to the extent that easy
syringability exists.
The dose of the pharmaceutical composition that is
to be used depends on the particular condition being
treated, the severity of the condition, individual patient
parameters including age, physical condition, size and
weight, the duration of the treatment, the nature of
concurrent therapy (if any), the specific route of
administration and other similar factors that are within the
knowledge and expertise of the health practitioner. These
factors are known to those of skill in the art and can be
addressed with minimal routine experimentation. Typical
dosages of the bifunctional fusion molecule and/or the
antigenic agent is about 1 mg, about 750 g, about 500 g,
about 250 g, about 100 g, about 10 g, about 1 g, about
750 ng, about 500 ng, about 250 ng, about 100 ng, about
50 ng, about 10 ng, about 1 ng, about 750 pg, about 500 pg,
about 250 pg or about 100 pg.
The pharmaceutical composition may be administered
to a subject in a variety of forms depending on the selected
route of administration, as will be understood by those
skilled in the art. The composition of the invention may be
administered orally, by injection (intramuscular,
intradermal, subcutaneous, intraperitoneal), by puncture,
transdermally or intranasally.
31
CA 02640416 2008-10-03
79783-69
The bifunctional fusion molecule may be conjugated
to a moiety to reduce its immunogenicity or to increase its
circulating half-life. In one embodiment, the bifunctional
fusion molecule is conjugated to polyethylene glycol (PEG).
PEG refers to an oligomer or polymer of ethylene oxide.
PEGs are prepared by polymerization of ethylene oxide and
are commercially available over a wide range of molecular
weights from 300 g/mol to 10,000,000 g/mol. Polyethylene
glycol has a low toxicity. PEG attached drugs, such as
IFN-a show reduced immunogenicity and decreased clearance
resulting in longer circulating half-life in vivo.
In another embodiment, the immunogenicity and/or
half-life of the bifunctional fusion molecule and/or the
antigenic agent may be modified using a nanoparticle or
liposome.
In a further embodiment, the immunogenicity and/or
half-life of the bifunctional fusion molecule and/or
antigenic agent may be modified by lipidization of the
bifunctional fusion molecule and/or the antigenic agent
(Yuan et al., J. Controlled Release, 129: 11-17, 2008).
The pharmaceutical compositions of the present
invention may be useful in the therapeutic and/or
prophylactic treatment of infectious diseases and/or in
modulating the immune response to an antigen. In one
embodiment the pharmaceutical composition, in the absence of
a co-stimulatory molecule, may be used for the treatment of
an autoimmune disease. In another embodiment, the
pharmaceutical composition, in the absence of a co-
stimulatory molecule, may be used for the treatment of organ
transplant.
32
CA 02640416 2008-10-03
79783-69
As used herein, "treat" or "treatment" refers to
an approach for obtaining beneficial or desired results,
including clinical results. Beneficial or desired clinical
results can include, but are not limited to, alleviation or
amelioration of one or more symptoms or conditions,
diminishment of extent of disease, stabilization of the
state of disease, prevention of development of disease,
prevention of spread of disease, delay or slowing of disease
progression, delay or slowing of disease onset, amelioration
or palliation of the disease state, and remission (whether
partial or total). "Treating" can also mean prolonging
survival of a subject beyond that expected in the absence of
treatment. "Treating" can also mean inhibiting the
progression of disease, slowing the progression of disease
temporarily, although more preferably, it involves halting
the progression of the disease permanently.
The pharmaceutical composition of the present
invention may be useful for modulating the immune response
of a subject to an antigen. As used herein, "modulate" or
"modulating" may refer to increasing the strength, magnitude
and/or duration of the immune response, or to suppressing
the immune response resulting in unresponsiveness to the
antigen.
In one embodiment, the subject is administered a
bifunctional fusion molecule, an antigenic agent and a CD40
agonist such as an anti-CD40 antibody or another
co-stimulatory molecule, inducing the immune response of the
subject to the antigen. In another embodiment, the subject
is administered a bifunctional fusion molecule and an
antigenic agent but without a CD40 agonist such as an
anti-CD40 antibody or a co-stimulatory molecule, thus
33
CA 02640416 2008-10-03
79783-69
suppressing, anergizing or inactivating the immune response
of the subject to the antigen.
The invention is further illustrated by the
following, non-limiting examples.
EXAMPLES
MATERIALS AND METHODS
Materials
DC 2.4 is a DEC-205 expressing mouse bone marrow
dendritic cell-line transduced with GM-CSF, myc and raf
oncogenes [Shen et al. J Immunol, 158:2723-30, 1997].
HB290, a rat anti-mouse DEC-205 hybridoma, was obtained from
ATCC. BSA (bovine serum albumin), streptavidin-HRPO
(horseradish peroxidase), anti-His6 mAb (monoclonal
antibody), NHS-LC-Biotin (biotinamidohexanoic acid
3-sulfo-N-hydroxysuccinimide ester), photobiotin acetate,
OVA and goat-anti-mouse-HRPO (GAM-HRPO) were from Sigma
(Oakville, Canada). Biotinylated MUC-1 peptide with amino
sequence of B-GVTSAPDTRGVTSAPDTR (N-terminal biotinylated)
was kindly provided by Biomira, Inc. (Edmonton, Alberta,
Canada). The streptavidin gene was kindly provided by
Dr. T. Sano, Center for Molecular Imaging, Diagnosis and
Therapy and Basic Science Laboratory, Boston, MA, USA. HSF
(hybridoma serum free media), DMEM, PSG (penicillin,
streptomycin and L-glutamine) and FBS (fetal bovine serum)
were purchased from Gibco BRL (Burlington, Canada). B-BSA
[(biotin)n labeled BSA] was prepared by biotinylation of BSA
with NHS-LC-Biotin as per vendor's protocol. TMB
(3, 3', 5, 5'-tetramethylbenzidine) peroxidase substrate was
purchased from Kirkegaard & Perry Laboratory Inc
(Gaithersburg, USA). Hybond ECL (enhanced chemiluminiscent)
34
CA 02640416 2008-10-03
79783-69
nitrocellulose membrane and the ECL Western blotting kit
were from Amersham Pharmacia Biotech (Baie dUrfe, Canada).
The E. coli strain BL21-CodonPlus (DE3)-RIPL and
pBlueScript II KS-(PKS) were purchased from Stratagene
5(Cedar Creek, USA). T7 promoter and terminator primers and
the expression vector pET-22b (+) were from Novagen
(Madison, USA). pVAX1 mammalian expression vector and
molecular cloning materials (TOP10 cells, FastTrack mRNA
isolation kit, modifying and restriction enzymes) were from
Invitrogen (Burlington, Canada). Protein assay reagent was
purchased from Bio-Rad (Mississauga, Canada). Ni-NTA
agarose was purchased from Qiagen (Mississauga, Canada).
Mouse IFN-T (Interferon gamma) ELISA Ready-SET-Go was
purchased from eBioscience (San Diego, USA). LAL (limulus
amebocyte lysate) PYROGENT'~' Plus Single Test Vials was
purchased from Cambrex (Walkersville, USA).
Rat anti-mouse CD40 mAb was prepared from the
hybridoma IC10, kindly provided by Dr. M. Gold (University
of British Columbia, Canada). pVHX-6, WEEV DNA encoding El
and E2 proteins was provided by Dr. L. Nagata (Chemical
Biological Defense Section, Defence R&D Canada)
[Nagata et al. Vaccine, 23:2280-3, 20051. SARS-CoV
membrane codon optimized DNA was purchased from GENEART.
EBOV GP1,2 DNA [Wahl-Jensen et al. J Virol, 79:10442-50,
2005], EBOV GP1,2 mammalian expressed protein and SARS-CoV
spike DNA were from Dr. S. Jones and Dr. T. Booth (Special
Pathogens Program, National Microbiology Laboratory,
Canada). Gangliosides and ganglioside conjugates were from
Dr. D. Bundle (University of Alberta, Canada). Gangliosides
GM2 and GM3 antigens were biotinylated (B-GM2, B-GM3) and
conjugated with BSA (B-BSA-GM2, B-BSA-GM3). Anthrax
Protective Antigen (PA), extracted from the S-layer of
CA 02640416 2008-10-03
79783-69
recombinant Caulobacter crescentus, was provided by
Dr. J. Smit (University of British Columbia, Canada).
Cells and Antigen Preparation
DC 2.4 was cultured at 37 C with 5% CO2 in DMEM-10
medium [10% (v/v) FBS and 1% (v/v) PSG]. HB290 hybridoma
was cultured under the same conditions in HSF medium
[1% (v/v) FBS and 1% (v/v) PSG]. The various primers and
antigens used for all the experiments are listed in
Tables 1A and 1B. SARS-CoV spike DNA (encoding Sl, S2, RBD
and transmembrane domain), SARS-CoV membrane DNA, EBOV GP1,2
DNA were PCR (polymerase chain reaction) amplified using
primers WPOll [SEQ ID NO: 11] and WPO12 [SEQ ID NO: 121,
WPO13 [SEQ ID NO: 13] and WPO14 [SEQ ID NO: 14], or WP015
[SEQ ID NO: 15] and WPO16 [SEQ ID NO: 16] respectively
(Table 1A). These primers inserted Kozak translation
initiation sequence and initiation codon (ATG) into the
restriction sites BamHI, EcoRI. The PCR fragments were
gel-purified, double digested with BamHI and EcoRI, ligated
to pVAX1 mammalian expression vector and transformed into
TOP10 cells. The positive clones were screened by
restriction digestion fragment mapping (BamHI and EcoRI) and
then biotinylated using photobiotin acetate [McInnes et al.
Methods Enzymol. 184:588-600, 1990]. Core-streptavidin,
WEEV El, WEEV E2 and EBOV GP1 (Subfragment D) were prepared
based on previous work [Das et al. Protein Expr Purif,
54:117-25, 2007; Wang et al. Mol Biotechnol, 31:29-40,
2005; Das et al. Virus Res, 128:26-33, 2007; Das et al.
Antiviral Res, 64:85-92, 20041. EBOV GP2, SARS-CoV spike
S1, S2, RBD and membrane proteins were produced from E. coli
expression system. All antigens for immunization were
labelled with biotin. DNA vectors were biotin labelled
using photobiotin acetate (B-pVHX-6, B-pEBOV GP1,2,
36
CA 02640416 2008-10-03
79783-69
B-pSARS-CoV spike, B-pSARS-CoV membrane), all proteins were
labelled with NHS-LC-Biotin (B-OVA, B-SARS-CoV spike RBD,
B-EBOV GPl, B-Anthrax PA), glycolipids (B-GM2, B-GM3,
B-BSA-GM2, B-BSA-GM3) and peptide (B-MUC-1) were
synthetically labelled with biotin. Biotinylation of all
antigens was confirmed by dot blot assay probed with
streptavidin-HRPO.
Sequencing and Cloning of HB290 Gene
Sequencing of HB290 Fab
1 x 109 HB290 cells were harvested and the mRNA was
isolated using FastTrack mRNA isolation kit according to
vendor's protocol. PCR primers were designed based on
protocols in Methods in Molecular Biology volume 178
[O'Brien and Aitken. Methods in molecular biology,
vol. 178: antibody phage display: methods and protocols,
Humana Press, 2002]. cDNA of mAb from the variable regions
(VL and VH) to the constant light chain region (CL) and heavy
chain region (CH2) were generated by RT-PCR (reverse
transcription polymerase chain reaction) and TD-PCR (touch
down polymerase chain reaction) using rat specific PCR
primers and inserted in PKS plasmid. Positive clones were
selected by restriction digestion mapping and DNA
sequencing. The positive cloned fragment was sequenced
using M13 primers by CEQTM2000 (Beckman Coulter, USA). DNA
alignment between the forward sequence and backward sequence
was done using DS Gene 1.1 software (Discovery Studio). The
sequence generated after DNA alignment was compared with the
known mAb sequences in NCBI BLAST databank. The consensus
sequence of 2 individual clones was selected from two
independent PCR resulted in 100% match in both nucleotide
sequence and amino acid sequence.
37
CA 02640416 2008-10-03
79783-69
Cloning of HB290 scFv and bfFp
The various cloning constructs of HB290 scFv and
bfFp are listed in Figure 1. The genes were cloned in
different orientations to determine the best expression in
E.coli. The heavy and light chain variable regions
with/without partial constant regions of anti-DEC205
antibody were amplified from PKS plasmid containing HB290
Fab DNA by PCR and cloned into pET-22b (+) plasmid. The
heavy chain variable region gene of HB290 was fused to the
3' end of light chain variable region gene with a linker of
amino acids from constant heavy chain region 1 sequence
using the PCR (PCR primers: WP005-WP008). Subsequently
restriction digest/ligation methods were employed to
generate VH-VL scFv gene (Fig. 1A). The PCR primers WP005
15 [SEQ ID NO: 5] and WP006 [SEQ ID NO: 6] were inserted into
the restriction sites EcoRI and SalI; PCR primers WP007 [SEQ
ID NO: 7] and WP008 [SEQ ID NO: 8] were inserted into the
SalI and NotI. The PCR fragments were gel-purified, double
digested to the respective inserted restriction sites, and
ligated to pET-22b (+) plasmid. VL-VH scFv gene was
generated also by the same methods utilizing PCR primers
WP001-WP004 (WP001 [SEQ ID NO: 1] and WP002 [SEQ ID NO: 2]
were inserted into the EcoRI and SalI; WP003 [SEQ ID NO: 3]
and WP004 [SEQ ID NO: 4]were inserted into the SalI and
NotI). The light chain variable region gene of HB290 was
fused to the 3' end of heavy chain variable region gene via
15 amino acids of constant light chain sequence (Fig. 1B).
Both scFv orientations were inserted into pET-22b (+)
containing the core-streptavidin gene [Wang et al. Eur J
Pharm Biopharm, 65:398-405, 2007] in 3' orientation fusion
with the core-streptavidin gene (Figs. 1C and 1D). VL-VH
scFv gene was fused in 5' terminus with core-streptavidin
38
CA 02640416 2008-10-03
79783-69
gene by the same method described above (Fig. 1E). Briefly,
core-streptavidin gene was PCR amplified using WP009 [SEQ ID
NO: 9] and WPO10 [SEQ ID NO: 101 primers. These primers
were inserted into restriction sites NotI and XhoI. The PCR
fragment was gel-purified, double digested with NotI and
XhoI, and ligated to pET-22b (+) containing VL-VH scFv gene
(Fig. 1E). All clones were screened and characterized by
both PCR and restriction digestion fragment mapping. The
positive cloned fragment was sequenced using T7 promoter and
terminator primers by CEQTM2000. The positive clones were
named as follows: WET5 encoding core-streptavidin-VH-VL, WET6
encoding core-streptavidin-VL-VH and WET7 encoding
VL-VH-core-streptavidin.
Expression, Purification and Characterization of HB290
Recombinant Proteins
Expression of HB290 scFv and bfFp
Expression of scFv and bfFp methods and conditions
were previously described [Wang et al. Eur J Pharm
Biopharm, 65:398-405, 20071. Briefly, the pET-22b (+)-scFv
or bfFp genes (WET5-7) were chemically transformed into
BL21-CodonPlus (DE3)-RIPL. E.coli transformants were
cultured and induced and the whole-cell bacterial pellets
were analyzed by Western blot. The pellets were resuspended
in reducing SDS dye (50 mM Tris-HC1, pH 6.8, 2% SDS,
0.1% bromophenol blue, 10% glycerol, 5 mM 2-mercaptoethanol)
and heated at 95 C for 10 min prior to SDS-PAGE. The pellets
were electrophoresed and transferred to a Hybond ECL
nitrocellulose membrane using the Trans blot apparatus
(Bio-Rad). The membrane was then blocked with 5% skim milk,
probed with mouse anti-His6 mAb and GAM-HRPO, and revealed by
ECL according to the manufacturer's protocol. The bfFp gene
39
CA 02640416 2008-10-03
79783-69
with the highest protein expression level was selected for
medium scale expression in culture flask. Affinity
purification from E.coli periplasm using IMAC purification
protocol was performed as described previously [Wang et al.
Eur J Pharm Biopharm, 65:398-405, 2007]. The induced
periplasm and IMAC purified fractions were analyzed by
SDS-PAGE using 10% polyacrylamide gels under reducing
conditions following staining with Coomassie brilliant blue.
The fractions were heated at 95 C for 10 min prior to loading
on the polyacrylamide gel.
Western Blot: bfFp Biotin Binding and Heat Stability
IMAC purified bfFp was heated at either 60 C or
95 C for 10 min under reducing conditions and resolved in
10% SDS-PAGE. The resolved proteins were
electrophoretically transferred onto a nitrocellulose
membrane and probed with B-BSA followed by
streptavidin-HRPO.
ELISA: bfFp Bispecificity and Binding to DC 2.4 Cells
DC 2.4 cells were seeded on a 96-well V-bottomed
plate (Nunc, Denmark) in quadruplicate (1.0 x 105
cells/well). The plates were washed with PBS (phosphate
buffer saline) and blocked with 1% PBS dialyzed BSA (to
remove traces of biotin) for 3 h at 4 C. After incubation,
the plates were washed with PBS, and bfFp (40 g/ml in
100 l volume) was added to bind DEC-205 receptors. The
plates were incubated at 4 C for 3 h, then washed with PBS
and B-OVA (20 g/ml in 100 l volume) was added to each well
and incubated for 1 h at 4 C. After the incubation, the
plates were washed and then incubated with streptavidin-HRPO
(10 g/ml in 100 pl volume) for 1 h at 4 C. Finally, the
plates were washed with PBS, and TMB substrate was then
CA 02640416 2008-10-03
79783-69
added. The OD650 nm was taken after 15 min using an ELISA Vmax
kinetic microplate reader (Molecular Devices Corp,
California, USA) and subtracted with control (dendritic
cells with only streptavidin-HRPO incubated at 4 C for 1 h).
The bfFp DEC-205 receptor binding specificity was confirmed
by competition study with full-length HB290 mAb. Various
concentrations of H3290 mAb (50, 100, 200, 300 g/ml in
100 l volume) was added after bfFp binding to DEC-205. The
mAb was incubated for 2 h at 4 C then washed with PBS and the
bfFp was detected using B-OVA, streptavidin-HRPO and TMB as
mentioned above.
In Vivo Targeting of Dendritic cells in Mice
Mice and Immunization Protocol
groups of female BALB/c mice (5 mice per group,
15 average 6-8 weeks old) were obtained from Health Sciences
Laboratory Animals Services of the University of Alberta,
Edmonton, Canada. Animal treatment, care and euthanasia
were carried out according to the Canadian Council of Animal
Care guidelines. Mice were injected subcutaneously near the
inguinal lymph node area with 0.1 ml of various formulations
of antigens and bfFp in saline. The immunization protocol
is listed in Table 2. Three separate experiments were
conducted and each design has its own control group. All
groups had n=5 mice. The first set of experiments
demonstrated the versatility of bfFp based delivery of
protein, peptide, glycolipids and DNA to dendritic cells in
generating immune responses. B-pVHX-6, B-OVA, B-MUC-1,
B-GM2 & GM3 were studied (Groups 2-7 mice). Group 2-4 mice
focused on the DNA delivery to dendritic cells and to verify
the essential requirement of dendritic cell targeting
vehicle and co-stimulatory molecule. Group 2 mice were
41
CA 02640416 2008-10-03
79783-69
immunized with B-Pvhx-6 and bfFp without the presence of
anti-CD40 mAb to verify the essential role of anti-CD40 mAb
co-stimulation on dendritic cells. Group 3 mice were
co-immunized with anti-CD40 mAb without the bfFp to
determine the non-targeted immune responses. Group 4 mice
were co-immunized with both bfFp and anti-CD40 mAb. Protein
antigen delivery is studied in Group 5 mice, peptide
delivery is focused in Group 6 mice, and glycolipids were
investigated in Group 7 mice. The second group of
experiments was designed to confirm the DNA delivery
strategy. A variety of infectious disease viral DNA were
targeted to dendritic cells using bfFp mediated delivery
system (Group 9, EBOV GP1,2 DNA; Group 10, SARS-CoV spike
DNA; Group 11, SARS-CoV membrane DNA). Multivalent immune
responses against a mixture of antigens were studied in the
third set of experiments. Groups 13-15 were designed to
evaluate multivalent immune responses against proteins and
peptide. B-OVA, B-EBOV GPl, B-SARS-CoV spike RBD, B-MUC-l,
B-Anthrax PA were immunized as a mixture in saline in
Groups 13-15 mice. Group 13 mice were co-immunized with
anti-CD40 mAb and core-streptavidin without the bfFp to
determine the non-targeted immune responses. Group 14 mice
were co-immunized with bfFp without the presence of
anti-CD40 mAb to verify the essential role of anti-CD40 mAb
co-stimulation. Group 15 mice were immunized with antigens,
bfFp and anti-CD40 mAb. Groups 1, 8 and 12 are the control
groups for the three separate animal experiments. All mice
were boosted with the same concentration of antigen(s) in
PBS 12 days following primary immunization. Prior to
immunization, every reagent (antigens, bfFp and mAb) was
checked using LAL PYROGENT Plus Single Test Vials kits to
identify lipopolysaccharide (LPS) contamination (endotoxin
sensitivity at 0.125 EU/ml). The mice were sacrificed after
42
CA 02640416 2008-10-03
79783-69
9 days of boost. The blood and the spleen were collected.
The serum was isolated using standard procedure
[Coligan et al. (Eds.) Current Protocols in Immunology
1.7.1-1.7.8. Wiley, 1995] and used to evaluate humoral
immune responses. Spleens were used to study IFN-y immune
responses. The spleens from the 15 different groups of
immunized mice were aseptically removed and each group was
pooled. The responder cells were isolated using nylon wool
columns and the stimulator cells from the naive mouse
spleens were treated with mitomycin C as previously
described [Wang et al. J Immunol Methods, 306:80-92, 2005].
Evaluation of Humoral Immune Responses and IFN-y
Antibody titres were measured individually in each
mouse by ELISA following immunization of the respective
antigen (Table 1B). The ELISA method was done by overnight
coating of the specific antigen in the Nunc 96-well ELISA
microplates (10 g/ml in 100 l volume). After overnight
coating, the plates were washed with PBST (0.1% Tween 20 in
PBS, pH 7.3) and the plates were blocked with BSA for 3 h at
RT. After incubation, the plates were washed with PBST and
the 1:1000 serially diluted serum from each mouse in
quadruplicate was added and incubated for 2 h at RT. Plates
were then washed with PBST and incubated with GAM-HRPO for
1 h at RT. After 1 h, the plates were washed again with
PBST and TMB was then added to each well and OD650nm was taken
after 10 min using microplate reader. Statistical analyses
were performed as described [Wang et al. J Immunol Methods,
306:80-92, 2005] to determine the significance between
antibody responses in different groups. IFN-y activity was
determined by the IFN-T concentration generated after 3 days
of incubation of responder cells (2.5 x 105 cells) and/or
stimulator cells (3 x 105 cells) with the respective
43
CA 02640416 2008-10-03
79783-69
antigens: OVA, SARS-CoV spike RBD, EBOV GP1, EBOV GP2, EBOV
GP1,2, SARS-CoV spike Sl, SARS-CoV spike S2, SARS-CoV spike
RBD, SARS-CoV membrane, Anthrax PA, B-MUC-1, B-GM2, B-GM3,
WEEV El and WEEV E2 (10 g per antigen, each antigen is
separately incubated). IFN-y ELISA Ready-SET-Go kit was
used to determine the IFN-y concentration.
Immune responses towards biotin, bfFp and core-streptavidin
All the 15 groups of mice serum were tested
against B-BSA, bfFp and core-streptavidin using ELISA
method. The serum reactivity was determined by the same
method as described above. The plates were coated with
B-BSA, bfFp or core-streptavidin (10 g/well). The rest of
procedures and statistical analyses were as enumerated in
the above section.
Results
Construction, Expression and Purification of HB290 scFv
and bfFp
The genes encoding HB290 Fab were generated by
RT-PCR and TD-PCR. The sequence generated was compared with
the known mAb sequences in NCBI BLAST database indicating
the sequence is antibody related. HB290 VL amino acid
sequence from the recombinant clone was 100% identical to a
previous publication on cloning of NLDC-145 (HB290) scFv
[Demangel et al. Mol Immunol, 42:979-85, 20051. However,
HB290 VH deduced amino acid sequence
(EVKLVESGGGLVQPGGSLRLSCAASGFTFNDFYMNWIRQPPGQAPEWLGVIRNKGNGYT
TEVNTSVKGRFTISRDNTQNILYLQMNSLRAEDTAIYYCARGGPYYYSGDDAPYWGQGVM
VTVSS - SEQ ID NO: 17) shared only 46% homology. The VH
amino acid sequence appears to be unique from other
published amino acid sequences in the NCBI protein database;
44
CA 02640416 2008-10-03
79783-69
whereas, the amino acid sequence published by Demangel and
co-workers is 99% identical (only 2 amino acid difference in
the 5' terminal) with single chain antibody against rice
stripe virus protein P20 (Accession: AAG28706). HB290 VL-VH
and Vg-VI, genes were successfully cloned into separate
pET-22b (+) plasmid by PCR, restriction digest and DNA
ligation methods. The plasmid vectors WET5 and WET6 were
successfully constructed by inserting the sequence encoding
the HB290 single-chain anti-DEC205 antibody (VL-VH or VH- VL
respectively) into a core-streptavidin containing pET22b (+)
plasmid next to the pelB leader sequence (Figs. 1A-D). The
plasmid vector WET7 was constructed by inserting the
core-streptavidin sequence into HB290 VL-VH scFv containing
pET22b (+) plasmid (Figs. 1B and 1E). The core-streptavidin
sequence is at the N-terminus of the scFv in WET5 and WET6
constructs; whereas, in WET7 the core-streptavidin is in the
C-terminal. The scFv and bfFp vectors were transformed into
E.coli, cultured, induced and the whole-cell bacterial
pellets were analyzed by Western blot using the anti-His6 mAb
(Fig. 2A). The optimal scFv and bfFp orientation for
expression were determined by the Western blot. HB290 VL-VH
scFv, WET6 and WET7 were successfully expressed and the
proteins are shown at the desired MW (molecular weight) band
either at -30 kDa or -46 kDa (Fig. 2A). WET7 had higher
level of protein expression compare to WET6. WET7 was
subjected to medium scale expression, and the bfFp in the
periplasmic space was extracted and affinity purified by
IMAC column. Both periplasmic fraction and the affinity
purified fraction were analyzed on SDS-PAGE (Fig. 2B). IMAC
purification of the WET7 bfFp was successful; a clear band
is shown at 46 kDa in affinity purified fraction (Fig. 2B).
Approximately 1.0 mg of WET7 bfFp was affinity purified from
a 2 L culture.
CA 02640416 2008-10-03
79783-69
Characterization of bfFp
E. coli expressed bfFp appears in 3 isoforms:
monomeric, dimeric and tetrameric forms similar to a
published report [Kipriyanov et al. Protein Eng, 9:203-11,
1996]. Following affinity purification by either IMAC or
iminobiotin column the fusion protein was subjected to size
exclusion chromatography for isolation of the tetrameric
fusion protein [Kipriyanov et al. Protein Eng, 9:203-11,
1996; Schultz et al. Cancer Res, 60:6663-9, 20001. Fusion
protein is known to be heat sensitive and heating at 95 C
dissociates the tetramer completely into monomeric forms.
The tetrameric form remains stable at temperature below 60 C
[Kipriyanov et al. Protein Eng, 9:203-11, 19961. The bfFp
isoforms and the biotin binding activity of the bfFp were
analyzed by Western blot probed with B-BSA (Fig. 3A). WET7
bfFp was detected using B-BSA and streptavidin-HRPO in
Western blot. The bfFp appears predominantly in monomeric
form after heating at either 60 C or 95 C (Fig. 3A). The
predominant monomeric form may be due to the difference in
the linker between core-streptavidin and scFv [Wang et al.
Eur J Pharm Biopharm, 65:398-405, 20071.
The bispecificity of the bfFp was confirmed by
cell ELISA. The anti-DEC-205 activity was demonstrated on
DC 2.4 cells employing B-OVA with streptavidin-HRPO for
detection. In addition, specific DEC-205 receptor binding
activity was also confirmed by competitive displacement of
bfFp with increasing concentrations of HB290 mAb (Fig. 3B).
bfFp Mediated Immune Responses: Humoral and IFN-y
In vivo studies were carried out to investigate
the ability and efficacy of bfFp targeting of antigens to
dendritic cells. Four classes of antigens were chosen to
46
CA 02640416 2008-10-03
79783-69
demonstrate the versatility and diversity of antigen
delivery. These antigens were either made by recombinant
DNA technology or obtained from several sources. LPS is
identified in E.coli expressed proteins (SARS-CoV proteins,
EBOV proteins, WEEV proteins, bfFp), OVA and Anthrax PA.
DNA vectors, MUC-1 peptide, anti-CD40 mAb and mammalian
expressed GP1,2 were LPS free. Both humoral and
cell-mediated responses were investigated using a variety of
antigens listed in Table 1B. The immunization protocol is
described in detail in Table 2 and the results of humoral
and cell-mediated responses are shown in Figures 4 and 5.
Humoral responses were measured by the antibody titres
against the immunized antigens or its respective proteins
from DNA vectors individually in each mouse by ELISA. The
magnitude of cell-mediated immune response is determined by
the amount of IFN-T secreted from spleen T cells in response
to the antigens. Individual mouse spleen was not studied
since the humoral response data was reproducible from five
individual mice. The spleen cells were pooled to average
the quality of data. Responder cells from both immunized
and non-immunized mice without stimulator cells had minimal
IFN-T secretion. 15 groups of mice were divided into three
different experiments to demonstrate the diversity and
efficacy of dendritic cell targeting strategies. Groups 1,
8 and 12 are the control groups for the three separate
animal experiments, being only immunized and boosted with
PBS. Both humoral and cell-mediated immune responses are
minimal (Figs. 4 and 5).
bfFp mediated protein, peptide, gangliosides and DNA
targeting
Groups 2-7 were designed to demonstrate the
versatility of bfFp based delivery of protein, peptide,
47
CA 02640416 2008-10-03
79783-69
glycolipids and DNA to dendritic cells in generating immune
responses. Group 2-4 mice focused on the DNA delivery to
dendritic cells and to verify the essential requirement of
bfFp and anti-CD40 mAb. Group 2 mice were immunized with
B-pVHX-6 and bfFp without the presence of anti-CD40 mAb.
Group 3 mice were co-immunized with anti-CD40 mAb without
the bfFp. Group 4 mice were co-immunized with both bfFp and
anti-CD40 mAb. The results indicate that Group 4 has
highest antibody titre (statistically significant) and
augmented IFN-T secretion against WEEV El and WEEV E2
proteins compare to Groups 1-3 (Figs. 4A and 5A). Immune
responses against WEEV E2 antigen appear to be higher than
WEEV El in Group 4 (Figs. 4A and 5A). Immune responses in
Groups 2 and 3 are minimal towards WEEV El and E2 protein
antigens, probably due to LPS, anti-CD40 mAb or bfFp effect
(Figs. 4A and 5A). Anti-CD40 mAb and bfFp appeared to be
essential for DNA targeting strategy. The strategy was
applied to protein (OVA), peptide (MUC-1) and glycolipids
(GM2 and GM3). Essentially similar humoral responses and
cell-mediated immune responses are shown in mice immunized
with protein (Group 5), peptide (Group 6) and glycolipids
(Group 7) in the presence of bfFp and anti-CD40 mAb
(Figs. 4A and 5A). In summary, anti-CD40 mAb and bfFp are
required to achieve immune responses in dendritic cell
delivery of DNA.
The efficiency of targeting different classes of
antigens such as protein, peptide, glycolipid and DNA to
dendritic cells has been demonstrated. However, one DNA
targeting model may not be sufficient to document the
successful DNA targeting to dendritic cells. Thus, the same
targeting strategy for a variety of infectious disease DNAs
were tested. Groups 9-11 were immunized with different DNA
48
CA 02640416 2008-10-03
79783-69
vectors encoding genes for different viral proteins:
(Group 9, EBOV GP1,2 DNA; Group 10, SARS-CoV spike DNA;
Group il, SARS-CoV membrane DNA). pEBOV GP1,2 encodes EBOV
GP1 and GP2 protein, pSARS-CoV spike encodes SARS-CoV S1, S2,
RBD and transmembrane domain, and pSARS-CoV membrane encodes
SARS-CoV membrane protein. Strong humoral and cell-mediated
responses were achieved against the encoded viral proteins
by targeting viral DNA to dendritic cells (Figs. 4B and 5B).
EBOV GP1 generates highest immune responses compare to GP2
and GP1,2. The immune responses against mammalian expressed
GP1,2 protein are lower than E.coli expressed fragments (GP1
and GP2) (Figs. 4B and 5B). Immune response against EBOV
GP1,2 is relatively lower than its fragment may be due to
the glycosylation masking of the epitopes for serum or T
cell reactivity [Dowling et al. J Virol, 81:1821-37, 2007].
There is no significant difference in serum titer activity
between SARS-CoV spike proteins, but higher IFN-y
concentration is found in SARS-CoV RBD. Thus the naked DNA
delivery strategy was successful in several infectious
diseases models.
bfFp mediated multiple antigen targeting strategy
Groups 13-15 were designed to evaluate multivalent
immune responses by targeting multiple biotinylated proteins
and peptide antigen at same time in nanogram concentrations.
Group 13 reflects the possible involvement of
core-streptavidin targeting of a mixture of biotinylated
proteins and peptide to dendritic cells. Groups 14 and 15
demonstrated the efficacy of the bfFp in the absence and
presence of the anti-CD40 mAb co-stimulator respectively.
The results show that Group 15 has the highest antibody
titre (statistically significant) and augmented IFN-7
secretion against OVA, SARS-CoV RBD, Anthrax PA and MUC-1
49
CA 02640416 2008-10-03
79783-69
compare to Groups 12-14 (Figs. 4C and 5C). Groups 13-15
have shown minimal immune responses against EBOV GP1
(Figs. 4C and 5C). In Group 15, highest antibody titre was
found against OVA and lowest against Anthrax PA (Fig. 4C);
whereas, highest IFN-T secretion was found against Anthrax
PA (Fig. 5C). Minimal humoral or cell-mediated immune
responses generated from Groups 12-14 may be due to the
anti-CD40 mAb, LPS and/or core-streptavidin effects
(Figs. 4C and 5C). MUC-1 and OVA immune responses were
compared between single antigen and multiple antigen
targeting strategies. Higher IFN-ry secretion and serum
titre against MUC-1 were achieved in single antigen
targeting strategy of MUC-1 peptide antigen (Group 6) in
comparison to multiple antigens targeting strategy
(Group 15) (Figs. 4A, 4C, 5A, 5C). The serum titre against
OVA is not different between Groups 5 and 15 (Figs. 4A and
4C). However, Group 5 has higher IFN-T secretion compare to
Group 15 (Figs. 5A and 5C). To summarize, both bfFp and
anti-CD40 mAb are required to achieve strong immune
responses against protein and peptide delivered to dendritic
cells. Single antigen targeting appears to achieve strong
humoral and cell-mediated immune responses in comparison to
multi-antigen targeting strategy. Immune responses are
variably shifted in multi-antigen targeting method, possibly
towards immunodominant antigens.
Immunogenicity of bfFp
Serum titre against biotin, bfFp and
core-streptavidin were analyzed from every one of the
15 groups to evaluate the immunogenicity of the bfFp
targeting vehicle. The bfFp, B-BSA and core-streptavidin
proteins were coated on the ELISA plate. Serum reactivity
was found to a minor extent against core-streptavidin
CA 02640416 2008-10-03
79783-69
(OD -0.25) and bfFp (OD -0.15). This level of serum
reactivity is not significant compare to the control groups
(Fig. 6). The serum reactivity towards B-BSA was minimal
and not statistically significant between all groups
(Fig. 6).
Table 1
A
Primer Function Primer Sequence (5'-3') SEQ ID NO:
WP001 5'PCR primer, H8290 VL-C~P (strep-V VH) ATC AGT GAA TTC GGG AGG TGG CGG
ATC AGA CAT CCA GAT GAC ACA GTC T 1
WPOO2 3'PCR primer, HB290 VL-C, (strep-VL-VH) TGG TTT CGC TCA TGC TAG GTC GAC
CGT GGA TGG TGG GAA GAT AGA 2
WP003 5'PCR primer, HB290 VH (strep-V VH) GTT AAT GTC GAC GAA GTG AAG CTG GTG
GAA TCT 3
WPOO4 3'PCR primer, HB290 VH (strep-VL-VH) TAC TAA GCG GCC GCA AGC TGA GGA GAC
TGT GAC 4
WP005 5'PCR pnmer, HB290 VH CHI_P (strep-VH VL) ATC AGT GAA TTC GGG AGG TGG
CGG ATC AGA AGT GAA GCT GGT GGA ATC T 5
WP006 3'PCR primer, HB290 VH CH1. (strep-VH-VL) TGG TTT CGG TCA TGA TAG GTC
GAC AGC AGT TCC AGG AGC CAG T 6
WP007 5'PCR primer, H8290 VL (strep-VH Vi) GTT AGG GTC GAC GAC ATC CAG ATG ACA
CAG TCT 7
WP008 3'PCR primer, HB290 VL (strep-VH VL) TAC TAA GCG GCC GCA AGC CCG TTT CAA
TTC CAG C 8
WP009 5'PCR primer, core-streptavidin (VL-VH strep)
TACTAATGCGGCCGCGGAGGTGGCGGATCAGAGGCCGGCATCACCGGCA 9
WP010 3'PCR primer, core-streptavidin (VCVH strep)
ATTACTCTCGAGGGAGGCGGCGGACGGCTTC 10
WP011 5'PCR primer, pSARS-CoV spike
AAGAGGGGGATCCTACCATGGGTAGTGACCTTGACCGGTGCACCACT 11
WP012 3'PCR primer, pSARS-CoV spike
CTCGCTCGAGAGAATiCTATTATGTGTAATGTAATTTGACACCCTTGAG 12
WPO13 5'PCR primer, pSARS-CoV membrane
AAGAGGGTCTTCATATGGGGGATCCTACCATGGCAGACAACGGTACTATTACCGTTGAG 13
WPO14 3'PCRprimer,pSARS-CoV membrane
CTCGCTCGAGAGAATTCTAGTGATGATGGTGGTGATGCTGTACTAGCAAAGCAATATTGT 14
CGTT
WPO15 5'PCR pdmer, pEBOV GP1,2 AAGAGGGGGATCCTACCATGGGCGTTACAGGAATATTGCAGTTACCT
15
WPO16 3'PCR pnmer, pEBOV GP1,2
CTCGCTCGAGAGAATTCTAAAAGACAAATTTGCATATACAGAATAAAGC 16
B
Classification Immunized antigens Testing antigens
Protein B-OVA OVA
B-SARS-CoV spike RBD SARS-CoV spike RBD
B-EBOV GP1 EBOV GP1
B-Anthrax PA Anthrax PA
Peptide B-MUC-1 B-MUC-1
Glycolipid B-GM2 B-GM2 (IFN-yAssay)
B-GM3 B-GM3 (IFN-yAssay)
B-BSA-GM2 (Humoral study)
B-BSA-GM3 (Humoral study)
DNA B-pVHX-6 WEEV El
B-pEBOV GP1,2 WEEV E2
B-pSARS-CoV spike EBOV GP1
B-pSARS-CoV membr-ane EBOV GP2
EBOV GP1,2
SARS-CoV spike S1
SARS-CoV spike S2
SARS-CoV spike RBD
SARS-CoV membrane
Table 1. (A) PCR primers for cloning of HB290 scFv (VL-VH and
VH-VL) and bfFp (core-streptavidin-VH-VL, core-streptavidin-
51
CA 02640416 2008-10-03
~79783-69
VL-VH and VL-VH-core-streptavidin orientations) into pET-22b
(+) E.coli perplasmic expression vector. These primers
contain the following restriction inserts: EcoRI and SalI
for WPOO1, 002, 005 and 006; SalI and NotI for WP003, 004,
007 and 008; NotI and XhoI for WP009 and WPO10. WPOll-16 are
the PCR primers for cloning of viral DNA into pVAX1
mammalian expression vector. These primers contain Kozak
translation initiation sequence, initiation codon, and BamHI
and EcoRI restriction sites. (B) Summary table for the
antigen categories used for testing and targeted for in vivo
study.
Table 1.1
Surface Rece tors Li and Distribution Co-Stimulation
p g Re uirement Appfications
Gb3 (glycosphingolipid) Shiga toxin Monocytes, dendritic, endothelial,
epithelial, and B No Vaccine vector
cells
fl2 integrins (CDI1GCD18) CyaA Myeloid dendritic cells, macrophages,
monocytes, Yes Vaccine vector
activated B cells, natural killer cells, granulocytes Tumor protection
CD40 receptors CD40 ligand Dendritic cells, B cells, macrophages, endothelial
No Vaccine vector
cells, keratinocytes, fibroblasts, thymic epithelial Tumor protection
cells, CD34 hematopoietic cell progenitors Tumor therapy
Adenovirus delivery
C-type lectin receptors: Unknown DendriGc cells, Langerhans cells, monocytes,
B Yes Vaccine vector
DEC-205 (CD205) cells, natural killer cells, T cells, respiratory tracts,
Liposome delivery
thymic and gut epithelial cells Tumor protection
Tumor therapy
lnfection protection
Suppress autoimmunity
C-type lectin receptors: Hsp70 Immature dendritic cells, macrophages,
fibroblasts, No Vaccine vector
LOX-I (lectin-like oxidized smooth muscle cells and endothelial cells Tumor
therapy
low-density lipoprotein
receptor-I Myocardial ischemia
Atherosclerosis
C-type lectin receptors: Mannose Immature dendrific cells, macrophages and
Unknown Vaccine vector
DC-SIGN (CD209) rnegakaryocytes Liposome delivery
Adenovirus delivery
C-type lectin receptors: Mannose Immature dendritic cells, macrophaqes,
interstitial Yes Vaccine vector
mannose receptor (CD206) Mannan dendritic cells, dermal denddtic cells,
lymphatic Immunosuppression
endothelium, tracheal smooth muscle cefis, kidney Immunoactivation
mesangial cells
Cancer therapy
Fc receptors: FcyRl (CD64) Fc Dendritic cells, monocytes, macrophages, and
Unknown Vacoine vector
activated neutrophils Tumor protection
Tumor therapy
Table 1.1. Summary of dendritic cell surface receptors for
antigen targeting. This table shows the expression pattern
of the receptors and also indicates whether antigen
52
CA 02640416 2008-10-03
79783-69
targeting to the receptor requires co-stimulation for
induction of immune responses.
Table 2
Groups Day 0 Day 12 Day 21 Day 24
1 Control PBS PBS Spleen (T cell) IFN-yAssay
Serum (Humoral)
B-pVHX-6 500 ng 500 ng Spleen (T cell) IFN--yAssay
2 bfFp 20 N g 0 Serum (Humoral)
B-pVHX-6 500 ng 500 ng Spleen (T cell) IFN-yAssay
3 Anti-CD40 mAb 25 N g 0 Serum (Humoral)
B-pVHX-6 500 ng 500 ng Spleen (T cell) IFN-yAssay
4 bfFp 20 N g 0 Serum (Humoral)
Anti-CD40 mAb 25 U g 0
B-OVA 200 ng 200 ng Spleen (T cell) IFN-yAssay
bfFp 20 N g 0 Serum (Humoral)
Anti-CD40 mAb 25 N g 0
B-MUC-1 200 ng 200 ng Spleen (T cell) IFN-yAssay
6 bfFp 20 p g 0 Serum (Humoral)
Anti-CD40 mAb 25 p g 0
B-GM3 1 p g 1 p g Spleen (T cell) IFN-yAssay
B-GM2 1 p g 1 p g Serum (Humoral)
7
bfFp 20 N g 0
Anti-CD40 mAb 25 N g 0
Groups Day 0 Day 12 Day 21 Day 24
8 Control PBS PBS Spleen (T cell) IFN-yAssay
Serum (Humoral)
B-pEBOV GP1,2 500 ng 500 ng Spleen (T cell) IFN-yAssay
g bfFp 20 p g 0 Serum (Humoral)
Anti-CD40 mAb 25 u g 0
B-pSARS-CoV spike 500 ng 500 ng Spleen (T cell) IFN-yAssay
bfFp 20 p g 0 Serum (Humoral)
Anti-CD40 mAb 25 p g 0
B-pSARS-CoV membrane 500 ng 500 ng Spleen (T cell) IFN-yAssay
11 bfFp 20 N g 0 Serum (Humoral)
Anti-CD40 mAb 25 N g 0
53
CA 02640416 2008-10-03
79783-69
Groups Day 0 Day 12 Day 21 Day 24
12 Control PBS PBS Spleen (T cell) IFN-yAssay
Serum (Humoral)
B-OVA 200 ng 200 ng Spleen (T cell) IFN-7Assay
B-EBOV GP1 200 ng 200 ng Serum (Humoral)
13 B-SARS-CoV spike RBD 200 ng 200 ng
B-MUC-1 200 ng 200 ng
B-Anthrax PA 200 ng 200 ng
Anti-CD40 mAb 25 p g 0
Core-streptavidin 10 N g 0
B-OVA 200 ng 200 ng Spleen (T cell) IFN--yAssay
B-EBOV GP1 200 ng 200 ng Serum (Humoral)
14 B-SARS-CoV spike RBD 200 ng 200 ng
B-MUC-1 200 ng 200 ng
B-Anthrax PA 200 ng 200 ng
bfFp 20 N g 0
B-OVA 200 ng 200 ng Spleen (T cell) IFN--yAssay
B-EBOV GP1 200 ng 200 ng Serum (Humoral)
15 B-SARS-CoV spike RBD 200 ng 200 ng
B-MUC-1 200 ng 200 ng
B-Anthrax PA 200 ng 200 ng
bfFp 20 N g 0
Anti-CD40 mAb 25 u g 0
Table 2. Immunization protocol for evaluating humoral and
cell-mediated immune responses to biotinylated antigens in
mice (n=5 for each group). The amounts of antigens,
antibodies and bfFp are either in g or ng per mouse. All
mice were injected subcutaneously near inguinal lymph node.
Groups 1, 8 and 12 are the control groups and each group of
the control mice were immunized and boosted subcutaneously
with PBS. The first experiment demonstrated the versatility
of bfFp based delivery of protein, peptide, glycolipids and
DNA to dendritic cells in generating immune responses
(Groups 2-7 mice). Groups 2-4 mice focused on the DNA
delivery to dendritic cells and to verify the essential
requirement of DC targeting vehicle and co-stimulatory
molecule. Protein antigen delivery is studied in Group 5
mice, peptide delivery is focused in Group 6 mice, and
glycolipids were investigated in Group 7 mice. A variety of
infectious diseases viral DNA were targeted to dendritic
cells using bfFp mediated delivery system (Group 9, EBOV
GP1,2 DNA; Group 10, SARS-CoV spike DNA; Group 11, SARS-CoV
54
CA 02640416 2008-10-03
79783-69
membrane DNA). Multivalent immune responses against a
mixture of antigens were studied in the third set of
experiments. Groups 13-15 were designed to evaluate
multivalent immune responses against proteins and peptide.
All mice were boosted with the same concentration of
antigen(s) in PBS 12 days following primary immunization.
The mice were sacrificed after 9 days of boost.
The citation of any publication is for its
disclosure prior to the filing date and should not be
construed as an admission that the present invention is not
entitled to antedate such publication by virtue of prior
invention.
As used in this specification and the appended
claims, the singular forms "a," "an," and "the" include
plural reference unless the context clearly dictates
otherwise. Unless defined otherwise all technical and
scientific terms used herein have the same meaning as
commonly understood to one of ordinary skill in the art to
which this invention belongs.
Although the foregoing invention has been
described in some detail by way of illustration and example
for purposes of clarity of understanding, it is readily
apparent to those of ordinary skill in the art in light of
the teachings of this invention that certain changes and
modifications may be made thereto without departing from the
spirit or scope of the appended claims.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.