Note: Descriptions are shown in the official language in which they were submitted.
CA 02642563 2008-10-31
IRON(H) CATALYSTS CONTAINING DIIMINO-DIPHOSPHINE
TETRADENTATE LIGANDS AND THEIR SYNTHESIS
TECHNICAL FIELD
The present invention relates to catalytic materials for hydrogenation or
asymmetric hydrogenation. In particular, the invention relates to iron (II)
complexes
containing tetradentate diimino-diphosphine (P2N2) ligands for the catalytic
hydrogenation or asymmetric hydrogenation of carbonyl groups for use in
preparing
alcohols or non racemic alcohols, respectively. Imine groups can similarly be
hydrogenated or asymmetrically hydrogenated to provide amines, or non-racemic
amines,
respectively. These alcohols and amine products are important raw materials in
the
manufacturing of chemical products, pharmaceuticals, fragrance and flavours.
BACKGROUND
Asymmetric hydrogenation is an important method for generating single
enantiomer molecules that include intermediates and fine chemicals with
applications in
the pharmaceuticals, biotechnology, agrochemical, food, flavours, essential
oils, personal
care and advanced materials industries. Each enantiomer may have quite
different
properties and effectiveness. The use of a drug molecule as a single
enantiomer reduces
the risk of negative effects of a racemate, increases efficacy and accuracy of
dosage,
reduces the dosage compared to racemates by one half, with a subsequent
reduction in
cost and waste, environmental burden including agricultural and human waste
run-off.
This is particularly true since the US Food and Drug Administration, the
European
Committee for Proprietary Medicinal Products and other regulatory authorities
have
required characterization of enantiomers in proposed marketable drug products.
Examples of some of the top selling drug products that are chiral are:
LipitorTM, ZocorTM,
ZyprexaTM, NorvascTM, ProcritTM, PrevacidTM, NexiumTM, PlavixTM, AdvairTM and
ZoloftTm. In 2003 the total global sales for these products amounted to 48.3
billion dollars.
In the biotechnology sector the ability to synthesize enantiomerically pure
amino
acids, peptides and proteins is of great value. In the agrochemical business
about 25% of
CA 02642563 2008-10-31
the members of several classes of pesticides and herbicides exist as
enantiomers.
Currently the largest scale asymmetric hydrogenation process is the production
of the S
enantiomer of MetalochlorTM.
Volatile, enantiomerically pure alcohols are particularly valuable in the
flavours
and fragrances industries where each enantiomer provides a distinctive
olfactory
sensation. They are playing an increasingly important role in aromatherapy.
Single enantiomer helical molecules impart important optical, electronic and
magnetic properties to materials and nanomaterials with applications in
switches, motors,
sensors, polarizers and displays.
In the hydrogenation of complex molecules, the selectivity and activity of the
process is dependent on the catalyst structure. This structure must interact
with the
substrate to provide the diastereomeric transition state of lower energy that
leads to the
required enantiomer.
Conventional asymmetric hydrogenation catalysts utilize platinum group metals
(PGM) ruthenium, osmium, rhodium, iridium, palladium or platinum (De Vries et
al.,
"Handbook of Homogeneous Hydrogenation" Wiley-VCH, volumes 1-3, 2007). Their
ability to activate hydrogen gas toward addition to organic compounds is well
known.
However, these metals present potential toxicity problems and prolonged usage
of
pharmaceuticals containing traces of these metals might lead to harmful bio-
accumulation.
PGM are expensive and thereby add to the cost of the final product. In
addition, they are
in limited supply and will decrease in availability over time.
The direct hydrogenation of carbonyl and/or imine groups in an organic
molecule
using hydrogen gas is now becoming the preferred "green" method because no
waste is
produced and the separation of product is easier. Hydrogen is expected to be
an even
more abundant feedstock as it is used more as a green fuel. In a complimentary
way, the
catalytic hydrogenation or asymmetric hydrogenation of carbonyl and/or imine
groups in
an organic molecule by transfer from a hydrogen-donating molecule or mixture
has the
advantage of operational simplicity by avoiding the use of pressurized
hydrogen (Gladiali
2
CA 02642563 2008-10-31
et al., "Asymmetric transfer hydrogenation: chiral ligands and applications,"
Chem. Soc.
Rev. 35 (2006) pp 226-236).
The reduction of ketones is one of the fundamental reactions in the chemistry
field
and is used in many chemical transformations towards various products.
Asymmetric
reduction of the carbonyl group was achieved in the past using chiral
catalysts that are
based on platinum group metals (PGM) such as ruthenium, rhodium, iridium,
palladium
or platinum. Usually 'PrOH or H2 are used as a reducing agent in those
transformations
when they are activated by the metal-catalysts. The activation is normally
produced via
the in situ formation of the catalyst from pre-catalyst by the addition of a
strong base.
Reduction catalysis utilizing molecular hydrogen is more attractive compared
to
the reduction with iPrOH because of the low price of hydrogen gas, product
purification
simplicity and waste elimination. Reduction catalysis by hydrogen transfer
from 1PrOH is
preferred when pressurized hydrogen gas is not available or convenient.
Chiral alcohols and amines that are produced by the asymmetric hydrogenation
or
asymmetric transfer hydrogenation of ketones and imines, respectively, are
extensively
used in the synthesis of pharmaceuticals, agricultural chemicals, fragrances
and materials.
A non-limiting list of the examples of such compounds is presented below:
HO OH OH
400 *
1 2 3
,Ph
OH SD HN
*
*
4 5
Product 1 can be used in preparation of the (+)-compactin, an HMG-CoA-
reductase inhibitor. Product 2 can be used in the synthesis of 2,4-
diaminoquinazoline
derivatives which are possible SMN2 promoter activators which can be used in
the
treatment of spinal muscular atrophy. Product 3 may be used as a synthetic
building block
3
CA 02642563 2008-10-31
of the highest selling drug Fluoxetine (prozae). Product 4 may be used as a
chiral
synthetic intermediate in preparation of the benzazepine dopamine antagonist
Sch 39 166.
Although some PGM catalytic systems have enzyme-like enantioselectivities and
activities, their toxicity and high price make them unattractive for some
industrial
synthetic transformations.
Attempts have been made to solve this problem. For example, Gao et al. in 1996
in the journal Polyhedron (Gao et al. "Synthesis and characterization of
iron(2+) and
ruthenium(2+) diimino-diphosphine, diamino-diphosphine and diamido-diphosphine
complexes,"Polyhedron 1 (1996), pp. 1241-1251) reported the synthesis of iron
complexes with tetradentate ligands. The use and application of their iron
complexes
towards hydrogenation was not disclosed. They reported the synthesis of two
iron
complexes with diphosphinediimine ligands 6 and 7: trans-[Fe(NCMe)2(6)](C104)2
and
trans-[Fe(NCMe)2(7)](C104)2.
¨N n
PPh2 Ph2P
6 n = 2
7 n = 6
They also reported the iron complex with the diphosphinediamine ligand 8.
f¨N
NH HN
111PPPh2 Ph2P
8
Further, Gao et al. in 1996 in the journal Organometallics (Gao et al., "A
ruthenium(ii) complex with a c-2-symmetrical diphosphine/diamine tetradentate
ligand
for asymmetric transfer hydrogenation of aromatic ketones, "Organometallics 15
(1996),
pp. 1087-1089) disclosed that ruthenium complexes with the enantiopure ligands
9
4
CA 02642563 2008-10-31
((R,R)-cYP2N2) and 10 are catalysts for the asymmetric transfer hydrogenation
of ketones
with the latter displaying superior activity and selectivity. Rautenstrauch et
al.
(Rautenstrauch et al., "Hydrogenation versus Transfer Hydrogenation of
Ketones: Two
Established Ruthenium Systems Catalyze Both," Chem. Eur. J. 9 (2003), pp. 4954-
4967;
6,878,852 B2 5/2005 to Rautenstrauch et al.) showed that similar ruthenium
complexes
are active for the hydrogenation and asymmetric hydrogenation of ketones.
¨N N¨ NH HN
=
ip P13112 Ph2P 41, PPh2 Ph2P
9 10
Boaz etal. (6,690,115 B2 7/2003 to Boaz et al.; 2006/0135805 Al to Boaz et
al.)
made ketone hydrogenation catalysts based on PG metals such as Ru and Rh in
complexes of PNNP ligands of the type 11. Here the iron is part of the
ferrocenyl
substituent on the ligand which is known in the art to provide selectivity and
sometimes
activity to a PG metal catalyst.
/D\ R
NH HN---
Rn ph2p(Rn
Fe Fe
Rn Rn
11
Chen et al. (Chen et al., "Asymmetric transfer hydrogenation of ketones
catalyzed
by chiral carbonyl iron systems," Huaxue Xuebao 62 (2004), pp. 1745-1750)
reported an
asymmetric transfer hydrogenation system where one of the compounds 10, 12 or
13 of
the type P-NH-NH-P are added to [HFe3(CO)1 r to generate in situ catalysts for
the
transfer of hydrogen from isopropanol to ketones but the activity was low and
the nature
of the active catalyst was thought to be a cluster containing the three irons.
The structure
5
CA 02642563 2008-10-31
of this catalyst remains unknown. Other iron precursors Fe(C0)5 and
[Fe(C5H5)(CO2]2
did not lead to active catalyst mixtures.
Ph Ph
NH HN NH HN
111 PPh2 Ph2P 110 PPh2 Ph2P *
12 13
Bianchini et al. (Bianchini et al., "Chemoselective Hydrogen-Transfer
Reduction
of alpha,beta-Unsaturated Ketones Catalyzed by Isostnictural Iron(II),
Ruthenium(II),
and Osmium(II) cis Hydride eta(2)-Dihydrogen Complexes," Organometallics 12
(1993),
pp. 3753-3761) reported that iron complexes with a tetradentate PP3 ligand
were active
for the non-asymmetric hydrogenation of olefins under mild conditions.
Enthaler et al. (Enthaler et al., "Biomimetic transfer hydrogenation of
ketones
with iron porphyrin catalysts," Tet. Lett. 47 (2006), pp. 8095-8099) reported
that in situ-
generated iron complexes of achiral porphyrin ligands are somewhat active for
the
hydrogenation of ketones but no asymmetric hydrogenation reaction was possible
because of the lack of a chiral ligand.
Casey's group (Casey et al., "An efficient and chemoselective iron catalyst
for the
hydrogenation of ketones," J. Am. Chem. Soc. 129 (2007), pp. 5816-5817)
reported that
an achiral complex of the type Fe(arene-OH)H(C0)2 is a hydrogenation catalyst
but not
an asymmetric hydrogenation catalyst for ketones and imines at room
temperature. It also
catalyzes the hydrogenation of acetophenone by transfer from isopropanol. The
complex
[NMed [Fe3H(CO)i i] catalyzes the complete conversion of ketones to alcohols
at 80-100
C within 1-24 h by using alcohols as the reductant (Jothimony et al.
"Mechanism for
transfer hydrogenation of ketones to alcohols catalyzed by hydridotriiron
undecacarbonylate anion under phase transfer conditions," 52 J. Molec. Cat.
(1989), pp.
301-304) but this is not an asymmetric reduction. Bart et al. (Bart et al.,
"Preparation and
molecular and electronic structures of iron(0) dinitrogen and silane complexes
and their
application to catalytic hydrogenation and hydrosilation," J. Am. Chem. Soc.
126 (2004),
6
CA 02642563 2008-10-31
'
,
pp. 13794-13795) have reported achiral iron catalysts that hydrogenate olefins
under mild
conditions.
Thus, there is a need for new catalysts for hydrogenation, asymmetric
hydrogenation, transfer hydrogenation, and asymmetric transfer hydrogenation
which do
not require the use of PGMs.
SUMMARY OF THE INVENTION
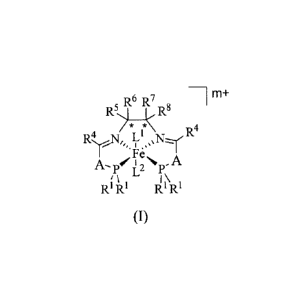
In one aspect, there is a provided a hexa-coordinate iron (II) complex
comprising
a compound of formula (I):
, R6 R7
R-)lc R8 -1m+
* *
R4 ...,1=1, Li ,/=1::_-.1, R4
Y
A.---piL2p-A
R`; µ 1,\ ,
R, I RI R`
(I)
wherein
each R1 is independently selected from the group consisting of aryl,
heteroaryl, C1-C8
alkyl, C2-C8 alkenyl, C1-C8 alkoxy, aryloxy, and cycloalkyl, all of which may
be
optionally substituted; two geminal RI groups may combine to form a C2-C4
linear alkyl
diradical or C3-C8 branched alkyl diradical, each of which may be optionally
substituted,
to form a ring together with the phosphorus atom to which they are attached;
or two R'
groups, each of which is located on a different phosphorus atom, may combine
to form a
linker M, wherein M is selected from the group consisting of C2-C4 linear
alkyl diradical
and C3-C8 branched alkyl diradical, each of which may be optionally
substituted, or M
may be a diradical ligand with a wide bite angle;
A is selected from:
7
CA 02642563 2008-10-31
(i)
/;and
a\n/V
R_23--7,Lcs
(ii)
wherein each R2 and R3 are independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl, and each n
is an integer independently selected from 1, 2, and 3;
each R4 is independently selected from the group consisting of H, substituted
or
unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8 alkenyl,
substituted or
unsubstituted aryl, and substituted or unsubstituted cycloalkyl;
each R5, R6, R7 and R8 is independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl; R5 and R6,
together with the carbon atom to which they are attached, may combine to form
a
substituted or unsubstituted cycloalkyl ring of size from 5-8 carbons; R7 and
R8, together
with the carbon atom to which they are attached, may combine to form a
substituted or
unsubstituted cycloalkyl ring of size from 5-8 carbons; or R5, R6, R7 and R8,
together with
the carbon atoms to which they are attached, may combine to form a group
selected from
, and 41
each of which may be optionally substituted with one or more substituents
selected from
the group consisting of C1-C8 alkyl, C1-C8 alkoxy, and halogen atoms;
LI and L2 are independently selected from the group consisting of CO; hydride;
pyridine
and derivatives thereof; imidazole and derivatives thereof; halide ion; NCR,
CNR and
OR, wherein R is independently selected from the group consisting of aryl,
heteroaryl,
8
CA 02642563 2008-10-31
C1-C8 alkyl, C2-C8 alkenyl and cycloalkyl, all of which may be optionally
substituted;
RaRbRN wherein Ra, Rb, and 11c are independently selected from the group
consisting of
H and Ci-C2 alkyl; and Re(CO)Rd wherein Rc and Rd are independently selected
from the
group consisting of C1-C8 alkyl, aryl, and heteroaryl;
m represents the charge of the compound of formula (I) and is 0, +1, or +2;
and when m
is +1 or +2, the iron (II) complex comprises at least one counter ion to
counterbalance the
charge of the compound of formula (I);
õ
with the proviso that when A is ,
then at least one of LI and L2 must be selected
from the group consisting of CO and CNR, wherein R is as defined above.
In another aspect, there is provided a process for the preparation of a hexa-
coordinate iron (II) complex of formula (I), the process comprising reacting a
phosphinaldehyde precursor of formula (V):
_00H 2+
R1213" A '4=0
R4--AõPRI2
HO
(V)
wherein
each R' is independently selected from the group consisting of aryl,
heteroaryl, C1-C8 alkyl, C2-C8 alkenyl, C1-C8 alkoxy, aryloxy, and cycloalkyl,
all of which may be optionally substituted; two geminal RI groups may
combine to form a C2-C4 linear alkyl diradical or C3-C8 branched alkyl
diradical, each of which may be optionally substituted, to form a ring
together with the phosphorus atom to which they are attached; or two RI
groups, each of which is located on a different phosphorus atom, may
combine to form a linker M, wherein M is selected from the group consisting
of C2-C4 linear alkyl diradical and C3-C8 branched alkyl diradical, each of
9
CA 02642563 2008-10-31
which may be optionally substituted, or M may be a diradical ligand with a
wide bite angle;
A is selected from:
el
(i) 4 ; and
RR23>.`,L,,nrsg,
(ii)
wherein each R2 and R3 are independently selected from the group consisting
of H, substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted
C8 alkenyl, substituted or unsubstituted aryl, and substituted or
unsubstituted
cycloalkyl, and each n is an integer independently selected from 1, 2, and 3;
each R4 is independently selected from the group consisting of H, substituted
or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8 alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl;
with a diamine of formula (VI):
.R5 R7
H2N NH2
(VI)
wherein
each R5, R6, R7 and R8 is independently selected from the group consisting of
H, substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-
C8
alkenyl, substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl; R5 and R6, together with the carbon atom to which they are
attached, may combine to form a substituted or unsubstituted cycloalkyl ring
of size from 5-8 carbons; R7 and R8, together with the carbon atom to which
they are attached, may combine to form a substituted or unsubstituted
CA 02642563 2008-10-31
=
cycloalkyl ring of size from 5-8 carbons; or R5, R6, R7 and R8, together with
the carbon atoms to which they are attached, may combine to form a group
selected from
,and
each of which may be optionally substituted with one or more substituents
selected from the group consisting of C1-C8 alkyl, C1-C8 alkoxy, and halogen
atoms;
in the presence of:
an iron (II) salt;
a ligand selected from the group consisting of CH3CN; pyridine and derivatives
thereof;
and imidazole and derivatives thereof; and
a strong base;
to form the compound of formula (I)
, R6 R7
R5 R8
R4 N, LI R4
A- pv vpA
-
RI RI R' R'
(I)
wherein A, R' -R8, and n are as defined above,
m is +2,
LI and L2 are both CH3CN; pyridine or a derivative thereof; or imidazole or a
derivative thereof;
and adding at least one counter ion to counterbalance the charge of the
compound of
formula (I).
11
CA 02642563 2008-10-31
In another aspect, there is provided, a process for preparing an alcoholic
compound wherein said process comprises a step of preparing the alcoholic
compound by
reducing a ketone or aldehyde with the reaction of hydrogen or a compound
donating
hydrogen in the presence of a hexa-coordinate iron (II) complex of formula
(I), with the
proviso that the ketone is not an unsubstituted cycloalkanone.
In still another aspect, there is provided a process for preparing an amine
compound wherein said process comprises a step of preparing the amine compound
by
reducing an imine with the reaction of hydrogen or a compound donating
hydrogen in
the presence of a hexa-coordinate iron (II) complex of formula (I).
In yet another aspect, there is provided a hydrogenation catalyst comprising a
hexa-coordinate iron(II) complex of formula (I)
R6 R7
RJ __ R8
R4 ...)\1, Li \N---1, R4
A
A- pv L2
I\
RI RI R`, R`,
(I)
wherein a trans coordination geometry is achieved at iron through nitrogen and
phosphorus donor bonds of a tetradentate diimino-diphosphine templated ligand
of the
formula (II):
R12P-A-C(R4)=N-C*(R5R6)-C*(R7R8)-N=C(R4)-A-PRI2
(II)
and LI and L2 are in an axial coordination above and below the templated
ligand,
respectively,
wherein the tetradentate diimino-diphosphine templated ligand is the reaction
product of
a phosphinaldehyde precursor of formula (V)
12
CA 02642563 2008-10-31
A OH
R4-)A'PRI2
HO
(V)
and a diamine precursor of formula (VI)
,R5 R7
H2N NH2
(VI)
wherein
each R' is independently selected from the group consisting of aryl,
heteroaryl, C1-C8
alkyl, C2-C8 alkenyl, C1-C8 alkoxy, aryloxy, and cycloalkyl, all of which may
be
optionally substituted; two geminal R1 groups may combine to form a C2-C4
linear alkyl
diradical or C3-C8 branched alkyl diradical, each of which may be optionally
substituted,
to form a ring together with the phosphorus atom to which they are attached;
or two RI
groups, each of which is located on a different phosphorus atom, may combine
to form a
linker M, wherein M is selected from the group consisting of C2-C4 linear
alkyl diradical
and C3-C8 branched alkyl diradical, each of which may be optionally
substituted, or M
may be a diradical ligand with a wide bite angle;
A is selected from:
cs
(i) Ts' ; and
R2>L.,
(ii) \ R3 /ncs'
wherein each R2 and R3 are independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
13
CA 02642563 2008-10-31
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl, and each n
is an integer independently selected from 1, 2, and 3;
each R4 is independently selected from the group consisting of H, substituted
or
unsubstituted Ci-C8 alkyl, substituted or unsubstituted C2-C8 alkenyl,
substituted or
unsubstituted aryl, and substituted or unsubstituted cycloalkyl;
each R5, R6, R7 and R8 is independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl; R5 and R6,
together with the carbon atom to which they are attached, may combine to form
a
substituted or unsubstituted cycloalkyl ring of size from 5-8 carbons; R7 and
R8, together
with the carbon atom to which they are attached, may combine to form a
substituted or
unsubstituted cycloalkyl ring of size from 5-8 carbons; or R5, R6, R7 and R8,
together with
the carbon atoms to which they are attached, may combine to form a group
selected from
, and 4110
each of which may be optionally substituted with one or more substituents
selected from
the group consisting of CI-Cs alkyl, Ci-C8 alkoxy, and halogen atoms;
LI and L2 are independently selected from the group consisting of CO; hydride;
pyridine
and derivatives thereof; imidazole and derivatives thereof; halide ion; NCR,
CNR and
OR, wherein R is independently selected from the group consisting of aryl,
heteroaryl,
Ci-C8 alkyl, C2-C8 alkenyl and cycloalkyl, all of which may be optionally
substituted;
RaRbRN wherein Ra, Rb, and Re are independently selected from the group
consisting of
H and C1-C2 alkyl; and Re(CO)Rd wherein Re and Rd are independently selected
from the
group consisting of C1-C8 alkyl, aryl, and heteroaryl;
m represents the charge of the compound of formula (I) and is 0, +1, or +2;
and when m
is +1 or +2, the iron (II) complex comprises at least one counter ion to
counterbalance the
charge of the compound of formula (I);
14
CA 02642563 2008-10-31
'111.
with the proviso that when A is S, then at least one of LI and L2 must be
selected
from the group consisting of CO and CNR, wherein R is as defined above.
DETAILED DESCRIPTION
Iron (II) complexes with PNNP donor ligands as catalytic materials for the
hydrogenation, asymmetric hydrogenation, transfer hydrogenation, and/or
asymmetric
transfer hydrogenation of ketones and imines are disclosed.
The asymmetric hydrogenation technology described herein that provides a
specified enantiomer enables a more economical, safer, efficient, and greener
chemical
way to generate compounds that are significantly enriched in the required
enantiomer.
As noted above, conventional asymmetric hydrogenation catalysts utilize
platinum group metals (PGM) ruthenium, osmium, rhodium, iridium, palladium or
platinum (De Vries et al., "Handbook of Homogeneous Hydrogenation" Wiley-VCH,
volumes 1-3, 2007). PGM are expensive and thereby add to the cost of the final
product.
In addition, they are in limited supply and not readily available. By
contrast, iron is
inexpensive, abundant and biocompatible. An unexpected feature of the
disclosed
catalysts is the high activity that they display in the activation of hydrogen
gas toward the
hydrogenation of ketones and in the activation of hydrogen-donor molecules
such as
isopropanol toward the transfer hydrogenation of ketones and imines.
In one embodiment, there is provided a hexa-coordinate iron (II) complex
comprising a compound of formula (I):
, R6 R7
RJ __ R8 7 m+
R4 Ir 1,õNzir R4
'Fe
A_ .A
P L2 P
I \
R1 RI R' R'
(I)
CA 02642563 2008-10-31
wherein
each RI is independently selected from the group consisting of aryl,
heteroaryl, Ci-C8
alkyl, C2-C8 alkenyl, C1-C8 alkoxy, aryloxy, and cycloalkyl, all of which may
be
optionally substituted; two geminal RI groups may combine to form a C2-C4
linear alkyl
diradical or C3-C8 branched alkyl diradical, each of which may be optionally
substituted,
to form a ring together with the phosphorus atom to which they are attached;
or two RI
groups, each of which is located on a different phosphorus atom, may combine
to form a
linker M, wherein M is selected from the group consisting of C2-C4 linear
alkyl diradical
and C3-C8 branched alkyl diradical, each of which may be optionally
substituted, or M
may be a diradical ligand with a wide bite angle;
A is selected from:
elcr.cs ; and
VINV
ç 3>4S
(ii) R ncs.
wherein each R2 and R3 are independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl, and each n
is an integer independently selected from 1, 2, and 3;
each R4 is independently selected from the group consisting of H, substituted
or
unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8 alkenyl,
substituted or
unsubstituted aryl, and substituted or unsubstituted cycloalkyl;
each R5, R6, R7 and R8 is independently selected from the group consisting of
H,
substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8
alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl; R5 and R6,
together with the carbon atom to which they are attached, may combine to form
a
16
CA 02642563 2008-10-31
substituted or unsubstituted cycloalkyl ring of size from 5-8 carbons; R7 and
R8, together
with the carbon atom to which they are attached, may combine to form a
substituted or
unsubstituted cycloalkyl ring of size from 5-8 carbons; or R5, R6, R7 and R8,
together with
the carbon atoms to which they are attached, may combine to form a group
selected from
, and 441
each of which may be optionally substituted with one or more substituents
selected from
the group consisting of C1-C8 alkyl, C1-C8 alkoxy, and halogen atoms;
LI and L2 are independently selected from the group consisting of CO; hydride;
pyridine
and derivatives thereof; imidazole and derivatives thereoff, halide ion; NCR,
CNR and
OR, wherein R is independently selected from the group consisting of aryl,
heteroaryl,
C1-C8 alkyl, C2-C8 alkenyl and cycloalkyl, all of which may be optionally
substituted;
RaRbReN wherein Ra, RI', and Re are independently selected from the group
consisting of
H and C1-C2 alkyl; and Re(CO)Rd wherein Re and Rd are independently selected
from the
group consisting of CI-Cs alkyl, aryl, and heteroaryl;
m represents the charge of the compound of formula (I) and is 0, +1, or +2;
and when m
is +1 or +2, the iron (II) complex comprises at least one counter ion to
counterbalance the
charge of the compound of formula (I);
rs
with the proviso that when A is , then at least one of LI and L2 must be
selected
from the group consisting of CO and CNR, wherein R is as defined above.
In another embodiment, a trans coordination geometry is achieved at iron
through
nitrogen and phosphorus donor bonds of a tetradentate diimino-diphosphine
templated
ligand of the formula (II):
R I 2P-A-C(R4)=N-C.(RsR6)_0R7R8)_N=c(R4)_A_pRi2
(II)
17
CA 02642563 2008-10-31
and L' and L2 are in an axial coordination above and below the templated
ligand,
respectively.
In one embodiment, the at least one counter ion is selected from BF4-; PFC;
SbF6-;
C104"; CH3S03-; CF3S03-; C6H5S03-; p-CH3C6H4S03-; FeC142-; FeBr42-; B(R*)4-,
wherein
R* is selected from phenyl, C6H3(CF3)2 and C6F5; halides; pseudohalides; C1-C8
alkoxides; and aryloxides. In another embodiment, the at least one counter ion
is BF4-.
In another embodiment, the at least one counter ion is BPh4".
In another embodiment, R1 is substituted or unsubstituted aryl. In other
embodiments, R1 is phenyl.
In another embodiment, A is /. In
another embodiment, R4 is H. In yet
another embodiment, R5, R6, R7 and R8, together with the carbon atoms to which
they are
attached, combine to form . .. In certain embodiments, the chiral carbon atoms
denoted by asterisks both have an R configuration. In other embodiments, the
chiral
carbon atoms denoted by asterisks both have an S configuration.
R23¨;*s_ss
In still another embodiment, A is \ R in `v . In another embodiment, R4 is H.
In
another embodiment, R2=R3=H. In yet another embodiment, n=1.
In another embodiment, R5=R8=substituted or unsubstituted aryl and R6=R7=H.
In another embodiment, R5=R8=phenyl. In still another embodiment, the chiral
carbon
atoms bearing the substituents R5 and R6, and R7 and R8, respectively, both
have an R
configuration. In
another embodiment, these chiral carbon atoms have an S
configuration.
In another embodiment, R4¨R5¨R6¨R7¨R8¨H.
18
CA 02642563 2008-10-31
In another embodiment, LI and L2 are CH3CN. In still another embodiment, LI is
CH3CN and L2 is selected from CO or CNR, wherein R is C1-C8 alkyl. In another
embodiment, L2 is CNtBu.
In another embodiment, the hexa-coordinate iron (II) complex comprises a
compound having the structure:
Ph
NC0i3 --12+
Ph
>7.¨k
C"'-Fe.sµ"
I N
Ph2 Ph2
NCCH3
(III).
In another embodiment, the chiral carbon atoms denoted by asterisks both have
an
R configuration. In another embodiment, the chiral carbon atoms denoted by
asterisks
both have an S configuration.
As noted above, the A symbol represents the bridging group ¨(CR2R3).. In one
embodiment, n is 1, R3 is H and R2 is H. In other embodiments, R3 is H and R2
may be
selected from aryl or C1-C8 alkyl, each of which may be optionally
substituted. When R2
the carbon bearing these substituents is chiral and may be enantiopure.
In other embodiments, n may be 2, and A is then ¨CR2R3CR2R3-. In further
embodiments, n may be 3 and A is then ¨CR2R3CR2R3CR2R3-. In one embodiment,
all R3
may be H. In another embodiment, each R3 may be different. Likewise, the R2
groups
may be the same or different.
In another embodiment, R5, R6, R7 and R8 can be selected to produce
enantiopure
structures. For instance, the cyclohexyldiyl structure noted above may be
present as the
(R,R) or (S,S) enantiopure isomer (having regard to the chiral carbon atoms
denoted by
asterisks),
19
CA 02642563 2008-10-31
=
The various chemical terms used herein are to be given their ordinary meaning
as
would be understood by persons skilled in the art, unless provided otherwise.
The following chemical terms presently described apply to all compounds and
processes disclosed herein, unless provided otherwise.
A "templated ligand" is a molecule that forms from precursor parts that
coordinate to a metal ion at geometrically defined positions such as
octahedral or square
planar, for example, and bond together. The metal ion acts as template for the
formation
of this ligand. Given the same reaction conditions, but in the absence of the
metal
template, the precursor parts usually either do not react, or do react but
form a mixture of
products, none of which have the structure of the templated ligand.
The compounds of formula (I) disclosed herein are referred to herein as
"catalysts". However, it will be understood by a person of skill in the art
that further
study may reveal that these compounds are in theory "pre-catalysts" and are
converted to
an active form during the hydrogenation reactions.
The term "C1-C8 alkyl" as used herein either alone or in combination with
another
substituent means acyclic, linear or branched chain alkyl substituent
containing from one
to eight carbons and includes for example, methyl, ethyl, 1-methylethyl, 1-
methylpropyl,
2-methylpropyl, butyl and the like.
The term "C2-C8 alkenyl", as used herein, either alone or in combination with
another radical, is intended to mean an unsaturated, acyclic linear chain
radical
containing from two to eight carbon atoms, at least two of which are bonded to
each other
by a double bond. Examples of such radicals include, but are not limited to,
ethenyl
(vinyl), 1-propenyl, 2-propenyl, and 1-butenyl. The alkenyl groups may contain
any
number of double bonds.
The term "aryl" as used herein, either alone or in combination with another
substituent, means an aromatic monocyclic system containing 6 carbon atoms or
an
aromatic bicyclic system containing 10 carbon atoms. The rings may have
substituents
including alkyl groups or alkoxy groups. For instance, a phenyl ring may have
CA 02642563 2015-06-11
substituents such as in the 3 and 5 positions, or 2 and 6 positions, or in the
4 position. The
term "aryl" includes but is not limited to a phenyl, tolyl (substituted aryl)
or naphthyl
group.
The term "heteroaryl" as used herein, either alone or in combination with
another
substituent means a 5, 6, 7, or 8-membered unsaturated heterocycle containing
one
oxygen or sulfur or from one to 4 nitrogen heteroatoms and which form an
aromatic
system. For example, the term "heteroaryl" includes a furyl, pyridyl, or
quinolinyl group.
The term "cycloalkyl" as used herein, either alone or in combination with
another
substituent, means a cycloalkyl substituent that includes for example, but is
not limited to,
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
The term "alkoxy" as used herein, either alone or in combination with another
radical, means the radical -0-(C1) alkyl wherein the alkyl group contains 1 or
more
carbon atoms, and includes for example methoxy, ethoxy, propoxy, 1-
methylethoxy,
butoxy, cyclohexyloxy and 1,1-dimethylethoxy. "Alkoxide" refers to the radial -
0-(C14
alkyl bearing a negative charge.
The term "aryloxy" as used herein, either alone or in combination with another
radical, means the radical -0-aryl wherein aryl is defined as above, such as
phenyl.
The term "aromatic diradical" includes groups such as benzo, as well as
naphthyl
diradical, binaphthyl diradical, and bisoxynaphthyl diradical as derived from
BINOL.
The term "branched alkyl diradical" includes groups such as 1,4-
dimethylbutanediyl. In
one aspect, the branched alkyl diradical may have between 3 and 8 carbon
atoms. Such
diradicals may be enantiopure. The term "linear alkyl diradical" includes C2-
C4 linear
alkyl diradicals such as 1,2-ethylene, 1,3-propylene, and 1,4-butylene.
The term "diradical that spans a wide bite angle" refers to aromatic
diradicals
such as naphthyl diradicals or tricyclic groups such as the 4,5-diradical of
9,9-
dimethylxanthene and other groups described in the article by Kramer et al.
Acc. Chem.
Res. 2001, 34, 895-904.
21
CA 02642563 2008-10-31
The term "halogen" refers to F, Cl, Br, and I. The term "halide ion" refers to
a
halogen atom bearing a negative charge.
The term "pseudohalide" refers to anions that behave chemically like halides.
These include OCN-, SCN-, CN- and NNN-.
As noted above, certain of the RI-R8 groups may be optionally substituted.
Those
of skill in the art will understand that a suitable substituent includes, for
example, methyl
substituents on aryl groups to generate tolyl or xylyl groups and the like.
Suitable
substituents for aryl, heteroaryl, and cycloalkyl functionalities include C1-
C8 alkyl,
branched or linear, alkoxy or halogen atoms. Suitable substituents for each
"R" group
mentioned in the claims include methyl, isopropyl, tertiary-butyl and phenyl.
It is to be understood that a suitable substituent is a substituent that does
not
interfere with the formation of the desired product by the claimed processes
and methods
disclosed herein. It is understood, of course, that the R groups defined
herein (RI-R8, etc.)
will not contain any substitution or substitution patterns which are
sterically impractical
and/or synthetically non-feasible.
As noted above, the LI, L2 symbols, taken separately, represent simultaneously
or
independently CO; hydride; pyridine and derivatives thereof, including but not
limited to
4-picoline or 3-picoline; imidazole and derivatives thereof, including but not
limited to
N-methyl imidazole; halide ion; NCR, CNR and -OR, wherein R is independently
selected from aryl, heteroaryl, C1-C8 alkyl, C2-C8 alkenyl and cycloalkyl, all
of which
may be optionally substituted; RaRbReN wherein Ra, RID, and Re are
independently selected
from H and C1-C2 alkyl; and Re(CO)Rd wherein Re and Rd are independently
selected
from C1-C8 alkyl, aryl, and heteroaryl.
The charge on the complex (m) depends on the nature of the P-N-N-P ligand and
the ligands LI and L2 and can vary from 0 to +2. The charge m+ on the metal is
2+ when
the ligands LI and L2 are neutral, 1+ when one of LI or L2 is anionic, 0 when
both LI and
L2 are anionic.
22
CA 02642563 2008-10-31
To counterbalance this charge in the metal complex salt, at least one counter
ion is
present. The term "counter ion" refers to an ion that is associated with the
compounds of
formula (I) disclosed herein in order to counterbalance the charge of the
compound of
formula (I) in the iron (II) complex. Such counter ions may include for
example anions
selected from the group comprising BF4-; PF6-; ShF6-; C104-; CH3503-; CF3503-;
C6H5503-;p-CH3C6H4S03"; FeC142-; FeBr42-; B(R*)4-, wherein R* is selected from
phenyl,
C6H3(CF3)2 and C6F5; halides; pseudohalides; alkoxides such as C1-C8 alkoxides
and
aryloxides such as phenoxide.
Compounds donating hydrogen include lower alcohols such as methanol, ethanol,
propanol, 2-propanol or butanol, and formic acid.
In particular the enantiopure complex (i) is useful for hydrogenation of
ketones
and imines, asymmetric hydrogenation of prochiral ketones and imines, and is
useful as a
precursor for the complex (ii). Complex (i) has been crystallized as the BF4-
and the
BPh4- salt (see Example 1) and characterized by elemental analyses, NMR, IR,
MS and
single crystal X-ray diffraction. The (S,5)-enantiomer of complex (i) has also
been
prepared.
NecH3
Ph Ph I 2+
Nõ N
N
Ph2 Ph2
NCC H3
(i)
The performance of the catalyst (i) was tested on 10 different aromatic
ketones
according to the reaction
Scheme 1
0 Catalyst (i)/H2(10atm) /KOIBu/iPrOH HO H
R p' R" SIC 200/1/15; T=35 C. *
R"
(S)
23
CA 02642563 2008-10-31
where S:C:B refers to the substrate to catalyst to base ratio. The procedure
of the catalytic
runs was performed as follows: (a)
Table 1. The hydrogenation of ketones catalyzed by (i) and base KO'Bu (S/C/B=
200/1/15) in 9 mL isopropanol at 35 C under 10 atm H2.
Entry Substrate Time Cony. e.e. (S)
(min) (%) (%)
1 Ph-CO-Me 30 40-90 81
2 Ph-CO-Et 25 35-80 92
3 Ph-CO-iPr 30 5 99
45-90/56-
4 Ph-CH2-CH2-CO-Me 25/50 98 1
(4'-C1C6H4)-CO-Me 20 55-91 91
6 (4'-Me0C6H4)-CO-Me 20 60-94 88
7 (3 ' -C1C6H4)-CO-Me 180 10-45 82
8 (3 '-BrC6H4)-CO-Me 30 5-30 86
9 (2'-C1C6H4)-CO-Me 30 35-58 75
1-Acetonaphthone 360 55-96 95
5 (a)
In the N2 glovebox, the iron complex (10 mg, 0.007 mmol), KCY8u (12.3 mg,
0.107 mmol) and the
substrate were separately dissolved in the 3 mL of 2-propanol, each. The
resulting solutions in the order
substrate, then base, and then catalyst were injected into a 50 cm3 Parr
hydrogenator reactor at the desired
pressure and temperature, maintained by use of a Fischer Scientific Isotemp
1016D water bath under a
hydrogen atmosphere..
10 Complex (ii), shown below, has been crystallized as the BPh4- salt (see
Example 2)
and characterized by elemental analyses, NMR, IR, MS and single crystal X-ray
diffraction. The (S,S)-enantiomer has also been prepared and completely
characterized.
Enantiopure complex (ii) is useful for the transfer hydrogenation of ketones
and imines
and asymmetric transfer hydrogenation of prochiral ketones and imines.
Nccn3
Ph_ Ph 7 2+
Nõ
I/ I NP
Ph2 C Ph2
0
(ii)
24
CA 02642563 2008-10-31
Table 2. The transfer hydrogenation of ketones to the (5) alcohols catalyzed
by (ii) and
base KO'Bu (S/C/B= 1600/1/8 unless specified) isopropanol at 22 C.(a)
Entry Substrate Time (min.) Cony. % ee %
1 Ph-CO-Me) 30 90 83
2 Ph-CO-Et(c) 50 84 93
3 (4-C1C61-14)-CO-Me(c) 50 93 70
4 (4-Me0-C6H4)-CO-MeM 50 78 81
1 -acetonaphthone(d) 50 93 95
6 Ph-CO-'Pr (e) 50 89 91
(a)In the N2 glovebox, the iron complex (ii) (2.0 mg, 0.0014 mmol), KOtBu (1.3
mg, 0.0114 mmol) and
ketone (2.2 mmol) were separately dissolved in the 5 mL of 2-propanol, each.
The resulting solutions were
5 added to a vial charged with a stirring bar in the order: substrate,
catalyst followed by base. The samples of
the reaction mixture were analyzed by GC. (b) in 15 mL isopropanol with S/C/B
2000/1/8. (c) in 12 mL
isopropanol. (d) in 14 mL isopropanol.
The enantiopure complex trans-[Fe(NCMe)(C0)(9)](BEI)2 (iii), wherein 9 is as
defined above:
_N N-
=PPh2 Ph2P
9
has also been prepared. This complex is inactive for catalytic hydrogenation
directly
from H2 gas but is useful for the asymmetric transfer hydrogenation of pro
chiral ketones
and is useful for the transfer hydrogenation of ketones and imines. Complex
(iii) has been
crystallized as the BFI (see Example 4) and the BPh4- salt and characterized
by elemental
analyses, NMR, IR, MS and single crystal X-ray diffraction. The (S,5)-
enantiomer of
complex (iii) has also been prepared and characterized. The enantiopure
complex trans-
[Fe(NCMe)(CN`Bu)(9)](BF4)2 (iv) has also been prepared and characterized (see
Example 5).
CA 02642563 2008-10-31
Me
(BF4)2
--N1
Fe
I \DP, *
Ph2 L2
(iii) L2 = CO, * = R configuration
(iv) L2 = tBuNC, * = R configuration
Table 3. Transfer hydrogenation of ketones and imines from 2-propanol
catalyzed by (iii)
and KOtBu (S/C/B = 200/1/8) at 22 C.Ial
Time Cony. e.e. TOFIei
Entry Substrate
(h) (%) (%) (11-1)
11b1 Ph-CO-Me 0.4 95 29 (S) 907
2M Ph-CO-Me 0.7 33 39 (S) 93
3 Ph-CO-Me 0.4 95 33 (S) 454
4 (T-C1-C6H4)-CO-Me 0.2 >99 18 (S) 995
5 (3'-C1-C6H4)-CO-Me 0.4 99 24 (S) 495
6 (4'-C1-C6H4)-CO-Me 0.2 94 26 (S) 938
7 (4'-Br-C6R4)-CO-Me 0.2 93 33 (S) 930
8 (4'-Me-C6H4)-CO-Me 0.6 86 33 (S) 279
9 (4'-0Me-C6H4)-CO-Me 0.5 69 23 (S) 260
Ph-CO-Et 3.6 95 61(5) 26
11 C 10H7-CO-Me[di 0.3 94 25 (S) 564
12 Ph-CO-Ph 0.4 94 470
13 Ph-(CH2)2-CO-Me 0.6 100 29 (S) 315
14 Ph-CHO 2.4 94 77
Ph-CH=N-Ph 17 100 - 12
16 Ph-CMe=N-Ph 17 <5
17 Cyclohexanone 17 0
[a]In an Ar or N2 glovebox at 22 C, the iron complex (5 mg, 0.005 mmol, [Cat]
= 1.04 mM), KOtBu (5mg,
0.045 mmol) and the substrate (200 equiv) were stirred in 5 mL of 2-propanol.
The conversion and
enantiomeric excess of the products were determined by NMR spectroscopy and
GC. [bi S:C:B = 400:1:8,
[Cat] = 0.1 mM, 10 mL iPrOH. lc] S:C:B = 200:1:2, [Cat] = 0.1 mM, 5 mL iPrOH.
[d] C10H7-CO-Me = 2-
10 acetonaphthone. [e] TOF = turn over frequencies.
As can be seen from Table 3, the electronic properties of the substituents on
the
phenyl ring of the ketone changed the reduction rate but had less effect on
the
26
CA 02642563 2008-10-31
enantioselectivity (18-33%). An acetophenone substituted in the para position
by an
electron releasing group, such as 4'-methyl and 4'-methoxy, is reduced more
slowly than
acetophenone (entries 3, 8 and 9). The chloro substituted acetophenones are
all reduced
faster, especially for the ortho position (entries 3-7). This trend is
opposite to the
generally observed trend for Noyori's transfer hydrogenation catalysts in
which an ortho-
Cl substitution decreases the rate of the reduction (S. Hashiguchi, A. Fujii,
J. Takehara, T.
Ikariya, R. Noyori, J. Am. Chem. Soc. 1995, 117, 7562). The catalyst (iii)
with KOtBu is
also efficient for the transfer hydrogenation of propiophenone, 2-
acetonaphthone,
benzophenone, benzylacetone, benzaldehyde and N-benzylideneaniline (entries 11-
15).
The hydrogenation of propiophenone gave 1-phenylpropanol in 61% e.e (S) (entry
10).
The more difficult ketimine N-phenyl-(1-phenylethylidene)amine (Ph-CMe=N-Ph)
was
only partially reduced (< 5%) after 18 h under the same conditions (entry 16),
while
cyclohexanone was not hydrogenated (entry 17). Transfer hydrogenation of
unsaturated
ketones was complicated by some reduction of the C=C double bond (Scheme 2).
Scheme 2. Transfer hydrogenation of unsaturated ketones.
0 OH OH
3/KOtBuhPrOH
Me
Ph Me - __________ Ph me + Ph
S:C:B = 200:1:8,22 C, 23 h 18% 82%
e.e. = 45% (S) e.e. = 27% (S)
Complex (iv) is useful for the asymmetric transfer hydrogenation of ketones.
Complex (iv) was used in the transfer hydrogenation of acetophenone, using the
same
reaction conditions as noted for complex (iii) (see [a] in Table 3 above).
After 2.6 hours
the conversion was 34% and the e.e. was 76% (S).
The mechanism of the catalysis is uncertain. The tetradentate ligand complex
may
be hydrogenated in the reaction medium to produce the amine intermediate
[FeH(C0){(R,R)-cyP2(NH)2}]+; however, such a hydride has not yet been
synthesized or
observed in the catalytic solution. Such a complex might be expected to
transfer a hydride
from iron and a proton from nitrogen to polar bonds in an outer sphere
hydrogenation, the
mechanism postulated for the related complexes [RuH2{(S,S)-cyP2(NH)21][15] and
[RuH2{PPh2(o-C6H4)CH2NHCMe2CMe2NHCH2(o-C6H4)PPh21] (T. Li, R. Churlaud, A. J.
27
CA 02642563 2008-10-31
Lough, K. Abdur-Rashid, R. H. Morris, Organometallics 2004, 23, 6239). Since
there is
poor chemoselectivity for the reduction of the C=0 bond versus the C=C during
the
hydrogenation of trans-4-phenyl-3-buten-2-one, another mechanism might be
involved.
During the transfer hydrogenation of acetophenone catalyzed by (iii) (entry 3,
Table 3), the 31P {1H} NMR shows an AB pattern at 56 and 74 ppm (d, 2Jp_p = 28
Hz) due
to an, as yet, unidentified intermediate. There is also a singlet for the free
ligand 9 (R,R)-
cyP2N2, and some other minor, unassigned peaks at 29 and -12.3 ppm. For the
reaction
catalyzed by (iv), the AB pattern for the intermediate is observed at 54 and
58 ppm (d,
2Jp_p = 31 Hz). This intermediate decomposes upon attempt to isolate it from
the catalytic
mixture. Without being bound by theory, it is thought that it might be a
complex such as
[Fe(C0)(X){(R,R)-cyP2N2}](BF4), X = alkoxide or hydride, but further study is
required.
The observation of free PNNP ligand in the catalytic solution may suggest the
formation of colloidal iron; however, there is evidence that the active
catalyst is
homogeneous instead of heterogeneous in that the reaction solutions are clear.
The e.e. of
the product alcohols are reproducible. There is no poisoning of catalysis by
mercury
when it is added during the reaction (C. A. Jaska, I. Manners, J. Am. Chem.
Soc. 2004,
126, 9776).
As it follows from Table 2, TOF (turn over frequencies), TON (turn over
numbers) and enantioselectivity of the catalyst (ii) are much higher compared
to the
catalysts (iii) and (iv). At a certain moment of the reaction when equilibrium
between
product and a substrate is established, catalytic racemization of the product
starts taking
place. It is hard to propose a reliable mechanistic explanation for such
behavior of the
catalyst at this point of investigation, but the conditions of the reduction
can be
optimized, so the product can be obtained in high yields and enantiopurity.
When a
smaller amount of the base is used the rate of the reaction is lower and thus
the time at
which racemization is taking place can be defined. If the reaction is quenched
by simple
exposure to air at this point of the process, high enantioselectivity and
yields of the
reaction can be achieved. Those conditions have a disadvantage: the overall
rate of the
reaction and TOF are reduced. In order to reach high enantioselectivity and
conversion
28
CA 02642563 2008-10-31
of the process the substrate concentration was increased. That increased the
time of the
reaction enough to determine when the equilibrium is established without
reduction of
the TOF and product was obtained in good ee, conversion and excellent TOF and
TON.
Yellow solutions of complex (iii) are stable to oxidation in air for at least
one day.
The 1I-1 NMR spectrum of (iii) showed the presence of a singlet for the imine
protons at
9.11 ppm while the 13C {1H} NMR spectrum displayed a pseudo-triplet for the
carbonyl
carbon. The 11-INMR spectrum of complex (iv) has two distinct resonances for
the imines
protons. The 31P NMR spectra consist of AB patterns at ca. 51 and 48 ppm
(2Jp_p
40 Hz) for (iii) and ca. 58 and 48 ppm (2Jp_p = 51 Hz) for (iv). The IR
spectra of (iii) and
(iv) proved valuable. The carbonyl ligand of (iii) absorbs at 2000 cm-I.
Complex (iv) has
absorptions at 2151 and 2173 cm-1 for the tBuNC and MeCN ligands.
Similarly, the enantiopure complex (v) is useful for asymmetric transfer
hydrogenation of prochiral ketones and imines and is useful for the transfer
hydrogenation of ketones and imines. Complex (v) has been crystallized as the
BF4- salt
(see Example 7) and characterized by elemental analyses, NMR, IR, MS. The
(S,S)-
enantiomer of complex (v) has also been prepared and characterized including a
single
crystal X-ray diffraction study.
0
Ph 8 pi-j1 BF4)2
(11'
1\11.`, I N¨
Fe
4.0
1 i
Ph "h 111 PNh Ph
(v)
Table 4. Transfer hydrogenation of ketones from 2-propanol (6 mL) catalyzed by
(v) and
KOtBu (S/C/B = 600/1/8 unless specified) at 24 C under N2.(a)
Time Cony. e.e.
Entry Substrate
(min) (%) (%)
1 Ph-CO-Me 30 71 63 (S)
29
CA 02642563 2008-10-31
2 Ph-CO-Et 30 75 70 (S)
3 Ph-CO-4)r 30 58 94 (S)
4 Ph-00-'13u b 15 93 96 (S)
(2'-C1-C6F14)-CO-Me 30 93 29 (S)
6 (31-C1-C6E)-00-Me 30 68 45 (S)
7 (4'-CI-C6H4)-CO-Me 30 81 38 (S)
8 3-C101-17-CO-Me 30 61 52 (S)
9 2- C10H7-CO-Me 30 73 61(S)
Ph-(CH2)2-CO-Me 15 91 57 (S)
11 Me-CO-Pr 15 63 12 (S)
(a)To a mixture of (v) (0.005 mmol) and KOtBu (0.04 mmol) was added a solution
of ketone in 6 ml of
iPrOH; (b) S/C/B = 200/1/8
Other A groups of formula (I) can be envisaged such as the ferrocenyl
substituent
shown as part of compound 11.
5 The above complexes can be prepared using an efficient, economical,
template
synthesis utilizing air stable phosphinoaldehyde precursor. The synthesis of
(I) is shown
schematically as follows:
1) Me0Hm+
R6 R7
OH 2+ 2) Fe2' R5..)*
A 1
RI2p ________ Ra 3) Strong base
R4 ___________ PRI2
4) Li, L2 R4 N, 1-1 ,Nz-_¨( R4
jr,L.
HO A R5 R7 A¨ / leN ,A
/P\ L2 P
(V) 5) R6¨) __
RI RI R' RI
H2N NH2
(VI) (I)
In one embodiment, there is provided a process for the preparation of a hexa-
10
coordinate iron (II) complex of formula (I), the process comprising reacting a
phosphinaldehyde precursor of formula (V):
2+
, A 1211
R'2P R
R47.L,
A
HO
(V)
wherein
CA 02642563 2008-10-31
each RI is independently selected from the group consisting of aryl,
heteroaryl, C1-C8 alkyl, C2-C8 alkenyl, C1-C8 alkoxy, aryloxy, and cycloalkyl,
all of which may be optionally substituted; two geminal RI groups may
combine to form a C2-C4 linear alkyl diradical or C3-C8 branched alkyl
diradical, each of which may be optionally substituted, to form a ring
together with the phosphorus atom to which they are attached; or two RI
groups, each of which is located on a different phosphorus atom, may
combine to form a linker M, wherein M is selected from the group consisting
of C2-C4 linear alkyl diradical and C3-C8 branched alkyl diradical, each of
which may be optionally substituted, or M may be a diradical ligand with a
wide bite angle;
A is selected from:
/;and
R23>.`_4õ,
R /nc?
wherein each R2 and R3 are independently selected from the group consisting
of H, substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted
Ca-
C8 alkenyl, substituted or unsubstituted aryl, and substituted or
unsubstituted
cycloalkyl, and each n is an integer independently selected from 1, 2, and 3;
each R4 is independently selected from the group consisting of H, substituted
or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-C8 alkenyl,
substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl;
with a diamine of formula (VI):
6R5 R7
R
H2N NH2
31
CA 02642563 2008-10-31
(VI)
wherein
each R5, R6, R7 and R8 is independently selected from the group consisting of
H, substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C2-
C8
alkenyl, substituted or unsubstituted aryl, and substituted or unsubstituted
cycloalkyl; R5 and R6, together with the carbon atom to which they are
attached, may combine to form a substituted or unsubstituted cycloalkyl ring
of size from 5-8 carbons; R7 and R8, together with the carbon atom to which
they are attached, may combine to form a substituted or unsubstituted
cycloalkyl ring of size from 5-8 carbons; or R5, R6, R7 and R8, together with
the carbon atoms to which they are attached, may combine to form a group
selected from
, and it
each of which may be optionally substituted with one or more substituents
selected from the group consisting of C1-C8 alkyl, C1-C8 alkoxy, and halogen
atoms;
in the presence of:
an iron (II) salt;
a ligand selected from the group consisting of CH3CN; pyridine and derivatives
thereof;
and imidazole and derivatives thereof; and
a strong base;
to form the compound of formula (I)
R6 R7
R8
*
Feµõ
A--p/ 2 121-1-1
=-= I \
RI RI RI RI
(I)
32
CA 02642563 2008-10-31
wherein A, RI-R8, and n are as defined above,
m is +2,
LI and L2 are both CH3CN; pyridine or a derivative thereof; or imidazole or a
derivative thereof;
and adding at least one counter ion to counterbalance the charge of the
compound of
formula (I).
In one embodiment, the at least one counter ion is selected from BEt-; FF6-;
SbF6-;
C104-; CH3S03-; CF3S03-; C6H5S03-; p-CH3C6114S03-; FeC142-, FeBr42-, B(R*)4-,
wherein
R* is selected from phenyl, C6H3(CF3)2 and C6F5; halides; pseudohalides; C i-
C8
alkoxides; and aryloxides. In another embodiment, the at least one counter ion
is BF4-.
In another embodiment, the at least one counter ion is BP4.
In another embodiment, the compound of formula (I), wherein LI and L2 are both
CH3CN, pyridine or a derivative thereof, or imidazole or a derivative thereof,
is further
reacted with CO; hydride; halide ion; NCR, CNR or -OR, wherein R is
independently
selected from the group consisting of aryl, heteroaryl, C1-C8 alkyl, C2-C8
alkenyl and
cycloalkyl, all of which may be optionally substituted; RaRbReN wherein Ra,
RI), and Re
are independently selected from the group consisting of H and C1-C2 alkyl; or
Re(CO)Rd
wherein Re and Rd are independently selected from the group consisting of CI-
C8 alkyl,
aryl, and heteroaryl, to produce a compound of formula (Villa):
, R6 R7
______________________________________ R8
RN
m+
* *
124 N, õNz...---r R4
Fe'
A- pl A
I
RI RI R' R'
(VIIIa)
wherein A, RI-R8, and n are as defined for formula (I),
L1 is CH3CN; pyridine or a derivative thereoff, or imidazole or a derivative
thereoff, and
33
CA 02642563 2008-10-31
1,2' is selected from the group consisting of CO; hydride; halide ion; NCR,
CNR or OR,
wherein R is independently selected from the group consisting of aryl,
heteroaryl, Ci-C8
alkyl, C2-C8 alkenyl and cycloalkyl, all of which may be optionally
substituted; RaRbReN
wherein Ra, Rb, and Rc are independently selected from the group consisting of
H and C1-
C2 alkyl; or Re(CO)Rd wherein Rc and Rd are independently selected from the
group
consisting of C1-C8 alkyl, aryl, and heteroaryl,
and m is +1 or +2.
The synthesis is conducted in an atmosphere of N2 (1 atm) or Ar (1 atm) or
another suitable gas to prevent reaction with atmospheric oxygen.
The concentration of the dimer V for use in forming the complexes disclosed
herein can range from 0.5 M to 0.0005 M with a preferred concentration of
0.03M. The
concentration of the diamine for use in forming the complexes disclosed herein
can range
from 0.5 M to 0.0005 M with a preferred concentration of 0.03M.
Suitable iron (II) salts for use in forming the complexes disclosed herein
include
[Fe(H20)6]2+ with counterions as noted herein, namely, Fe(BF4)2, Fe(PF6)2;
Fe(SbF6)2;
Fe(C104)2; Fe(MeS03)2; Fe(CF3S03)2; Fe(C6H5S03)2; Fe(p-CH3C6H4S03)2; FeC142-;
FeBr42-; Fe[B(R*)4]2, wherein R* is selected from phenyl, C6H3(CF3)2 and C6F5;
FeX2
wherein X is a halide or pseudohalide; Fe[0(Ci-C8 alkyl)]2; FeSO4; Fe(NO3)2;
and
Fe[R**C(0)012, wherein R** is C1-C3 alkyl, CF3, or phenyl, and hydrates
thereof. The
preferred range of iron concentrations for the template synthesis of complex
II is 1 M to
0.001 M with a preferred concentration of 0.05 M
Suitable strong bases for use in forming the complexes disclosed herein
include
alkoxides, such as Na0Me, DBU, a phosphazene, or an alkaline or alkaline-earth
metal
carbonate salt, carboxylate salt, alkoxide salt or hydroxide salt. In one
embodiment, the
strong base may be MOR***, wherein M is an alkaline metal selected from Na and
K, and
R*** is C1-C4 alkyl. The preferred base to iron ratio is 1.3:1.
Suitable solvents for forming the complexes disclosed herein include Me0H,
Et0H, PrOH, iPrOH, BuOH, CH3CN, EtCN, pyridine, picoline, imidazole,
34
CA 02642563 2008-10-31
methylimidazole. The preferred solvents are alcoholic solvents, such as Me0H.
The
preferred total volume of the solvent in the synthesis ranges from 1 mL to
20,000 mL
with a preferred volume of 10 mL.
The temperature for the template synthesis can range between 0 and 120 C with
the preferred temperature being between 20 C and 40 C.
The catalysts disclosed herein comprising a compound of formula (I) and with
LI
L2 = MeCN are surprisingly active and selective for the hydrogenation, by use
of
hydrogen gas, of ketones to produce valuable chiral and non-chiral alcohols in
the
presence of a base and an appropriate solvent. The use of complex (i) provides
a
particularly active and usefully enantioselective catalyst system.
The hydrogenation reaction involving a catalyst disclosed herein may or may
not
require solvent. When the use of the solvent is preferred for practical
reasons, any solvent
can be utilized for better performance of the catalyst. Non-limiting examples
include
primary, secondary and tertiary alcohols with hydrocarbon skeleton containing
2-15
carbons or aromatic solvents or ethers or hydrocarbon solvents.
In the solvent, the catalyst can be used at concentrations of 0.001 mM to 0.1
mM
while the substrate ketone or imine can be used in concentrations of 2 mM to
10 M. The
pressure of hydrogen gas can range from 0.5 atm to 100 atm with a preferred
pressure of
10 atm. Preferred concentrations of catalyst and substrate are 0.8 mM and 0.16
M,
respectively, with a ketone to catalyst ratio of 200:1.
In another embodiment, the phosphinaldehyde precursor is:
ph2p0H
HOr PPh2
PIL
the diamine is:
Ph Ph
H2N *NH2
and the product is a compound of formula (I) having the structure:
CA 02642563 2008-10-31
. .
. .
Ph
NCCH3 --1 2+
Ph
>;"" 4-,
Fe''''µI\I
I
P P
Ph2 Ph2
NCCH3
(III).
In one embodiment, the chiral carbon atoms denoted by asterisks in (III) above
both have an R configuration. In another embodiment, these chiral carbon atoms
both
have an S configuration.
The processes outlined herein generates a catalyst with sections derived from
the
precursor diamine (VI) and phosphine (V) precursor building blocks with the
iron ion
acting as a template to orient the precursors to ensure a high yield of the
compound of
formula (I). The sections are shown in structure (VII) below:
diamine
R6 R7 R8 ¨1 wri
RL) \<
/ LI
R4 "j...i =,"
..-
A..__p t, 4R
(
RI' 1 \pl
RI IV -
phosphine osphine
(VII)
This is an advantage since different catalysts can be rapidly synthesized from
a
phosphine precursor and a diamine precursor with a variety of substitutents,
providing
flexibility to appropriately optimize manufacturing costs and end product
quality
specifications, such as a high enantiomeric excess. The methods disclosed
herein allow
for tuning of the coordinating ligand to obtain the easy introduction of
chiral elements
such as enantiomerically pure diamines into the catalyst because of the
modular nature of
the coordinating ligand. As a consequence, the iron (II) complex with PNNP
ligand is
easily modified by introducing substituents to produce a catalyst structure
capable of
interaction with a substrate and ensuring selectivity. Where both enantiomers
of these
36
CA 02642563 2008-10-31
diamines are available, both enantiomers of an iron catalyst can be easily
prepared to
hydrogenate a substrate to either enantiomer of the target molecule.
The phosphine-aldehyde precursors (V) are prepared by methods known in the art
from commercially available or readily prepared phosphine starting materials
PHRI2 or
PC1R12 and compounds XCR2R3Y where X is a halide or tosylate or other good
leaving
group known in the art and Y is a formyl group ¨CHO or a protected formyl
group ¨
CH(OR)2. The diamines NH2CR5R6CR7R8NH2 are available from commercial sources.
A most interesting catalyst has the discrete structure (i), shown above (also
see
Example 1). The chiral ligand can have an (R, R) or (S, S) configuration. To
counterbalance the 2+ charge in the metal complex salt, anions such as 13F4-,
PF6-, SbF6-,
FeC142-, FeBr42-, tetraarylborates where the aryl is Ph, C6H3(CF3)2 or C6F5,
or halides or
pseudohalides or alkoxides and others noted above may be used.
Catalysts of structure (i), for example the tetraphenylborate salt, are
prepared in a
similar fashion to that of other iron complexes reported by Mikhailine et al.
(Mikhailine
et al. "Template Syntheses of Iron(II) Complexes Containing Chiral P¨N¨N¨P and
P¨N¨N Ligands," Inorg. Chem. 47 (2008), pp 6587 ¨ 6589) by the template
reaction of
the phosphonium salt shown below with (R,R)-dpen as described in Example 1:
(S,S)-
dpen can alternatively be used to generate the other enantiomer of (i).
Scheme 3
37
CA 02642563 2008-10-31
1) Me0H Me
OH 2) [Fe(1120)6]2+ Ph NPh
Ph2P
HO h2 (Br)2 3) Na0Me
4) MeCNP
P
5) (R,R)- or Ph2 Ph2
(S,S)- dpen (S,S)-enantiomer
Me
or
Me
Ph, N Ph
Ni I N
C (BP114)2
PP
Ph2 Ph2
(R,R)-enantiomer = (i)
Me
The complexes are precipitated as the BPh4- salts in high yield and
characterized
by NMR, electrospray ionization mass spectrometry, and elemental analysis. The
detailed
procedure of the complex (i) preparation is described as Example 1.
The reaction of complex I where Li = L2 = acetonitrile or another nitrogen
donor
ligand such as imidazole or pyridine with carbon monoxide yields the
monocarbonyl
catalysts of formula (VIII).
R6 R7 m+
R5 R8
-
rR4 N, Li ,Ni
Fe
A-- /1 rA
P CO P
RI RI RI RI
(VIII)
For example when complex (i) in acetone is treated with 5 atm CO, the
monocarbonyl
complex (ii) is formed (see Example 2). When the complex trans-
[Fe(NCM02(9)liBF4)2
(Sui-Seng et al., "Highly Efficient Catalyst Systems Using Iron Complexes with
a
Tetradentate PNNP Ligand for the Asymmetric Hydrogenation of Polar Bonds."
Angew.
Chem. Int. Ed. EngL 47 (2008), pp. 940-943) in acetone is reacted with 1 atm
CO, the
carbonyl complex trans-[Fe(NCMe)(C0)(9)](BF4)2 (iii) is formed (see Example
4).
38
CA 02642563 2008-10-31
Similarly when [Fe(NCMe)2(9)](BF4)2in acetone is reacted with tertiary-
butylisocyanide,
the complex [Fe(NCMe)(CNtl3u)(9)](BF4)2 (iv) is formed (see Example 5). The
reaction
of complex (vi) (Mikhailine et al. "Template Syntheses of Iron(II) Complexes
Containing
Chiral P-N-N-P and P-N-N Ligands," Inorg. Chem. 47 (2008), pp 6587 - 6589)
(Example 8) with CO produces the complex (vii; Example 9).
Scheme 4
- Me - - CO -
C
12 _____..CO(g)
C Fe_ -3 BPh4i
aceton CFep [ BPh41 2
Ph2N Ph2 Ph2 r:4 Ph2
¨ Me _ ¨ Me _
(vi) (vii)
Complex vi has less than optimum activity (< 5% conversion) for the
hydrogenation of acetophenone at 35 C, 25 atm H2 with KOtBu in iPrOH, and is
inactive
for the transfer hydrogenation of ketones in basic isopropanol.
Catalyst (vii) can be used for transfer hydrogenation. In the N2 glovebox, the
iron complex (vii) (8.7 mg, 0.007 mmol), KO'Bu (6.3 mg, 0.056 mmol) and
acetophenone (168 mg, 1.4 mmol) were separately dissolved in the 3 mL of 2-
propanol,
each. The resulting solutions were added to a vial charged with a stirring bar
in the order:
substrate, catalyst followed by base and stirred at room temperature. The
samples of the
reaction mixture were analyzed by GC. The conversion was 92% after 75 minutes.
Complex trans-[Fe(MeCN)2(6)RBF4)2(Example 10) wherein 6 is
k`k
¨N n
IIPPPh2 Ph2P
, n = 2, was prepared and tested. For the hydrogenation of acetophenone with
H2 (25 atm)
with a catalyst to base to substrate ratio of 1:15:225 in isopropanol the
conversion was
4% after 18 h. It was found to be inactive for the transfer hydrogenation of
acetophenone
in basic isopropanol under the standard conditions.
39
CA 02642563 2008-10-31
=
The iron (II) complex trans-[Fe(MeCN)2(6)1(BF4)2, can be reacted with CO to
produce
sc? ¨i(BF4)2
41.0,
_______________________________________ Ph I N
Ph in Ph INPh
(viii) (Example 11).
Catalyst (viii) can be used for transfer hydrogenation. To a mixture of (viii)
(0.005
mmol) and KOtBu (0.04 mmol) was added a solution of ketone in 6 ml of iPrOH.
Catalyst (viii) was found to be highly active for the transformation of
acetophenone to 1-
phenylethanol at room temperature using a catalyst:base:substrate ratio of
1:8:600 (85%
conversion after 60 min).
The bis-acetonitrile complexes
trans-[Fe(NCMe)2 {9)1] [BF4]2, trans-
[Fe(MeCN)2(6)](BF4)2, wherein 6 is
--NA1.1
PPh2 Ph2P where n = 2 and trans-[Fe(NCMe)2{(R,R)-
PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2}1[BF4]2were prepared by reaction of the
known PNNP ligands 6 (Jeffery, J. C.; Rauchfuss, T. B.; Tucker, P. A. Inorg.
Chem. 1980,
19, 3306-3316) 9, and PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2 (J.-X. Gao et al.
Chirality 2000, 12, 383) with iron salts such as [Fe(0H2)6](BF4)2 in
acetonitrile as
described in the examples below.
The iron complexes trans-[Fe(NCMe)(C0)(6)][BF4], (R,R)- or (S,S)-trans-
[Fe(NCMe)(C0){9)}i[BF4]2 and (R,R)- or (S,S)- trans-
[Fe(NCMe)(C0)(PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2)][BF4]2 were obtained as
orange solids in good yields when the corresponding bis-acetonitrile compounds
just
mentioned were stirred under a CO atmosphere in acetone. The new compounds are
fairly
air stable, both as a solid and in solution. They are soluble in acetonitrile
and
CA 02642563 2008-10-31
methylenechloride, poorly soluble in acetone, chloroform, 2-propanol and
insoluble in
tetrahydrofuran, ether and hydrocarbons. The new compounds were characterized
by 1H
and 13C and 31P NMR techniques, elemental analysis, mass spectroscopy, IR and
the
solid state structures were confirmed by X-ray crystallography. The 31P {1H}
NMR
spectrum of trans-[Fe(NCMe)(C0)(6)][BE4]2 shows a singlet while those for
(R,R)- or
(S,S)-trans-[Fe(NCMe)(C0){9)}][BE4J2 and (R,R)- or (S,S)- trans-
[Fe(NCMe)(CO)(PPh2C61-14CHNCHPhCHPhNCHC6H4PPh2)][BE]2 show two doublets.
Mass spectra (ES!) show the cationic fragment without the acetonitrile and
carbonyl
ligands.
In one embodiment, there is provided a process for preparing an alcoholic
compound wherein said process comprises a step of preparing the alcoholic
compound by
reducing a ketone or aldehyde with the reaction of hydrogen or a compound
donating
hydrogen in the presence of a hexa-coordinate iron (II) complex of formula
(I), with the
proviso that the ketone is not an unsubstituted cycloalkanone.
As can be seen from Table 3, no conversion was observed for the transfer
hydrogenation of cyclohexanone catalyzed by (iii). However, it is envisioned
that cyclic
ketones having substituents such as aromatic groups may be better substrates.
In another embodiment, the hexa-coordinate iron (II) complex comprises a
compound of formula (I) having the structure:
Ph
NCCH3 2+
Ph
>;--;c
Nõ ,N
r- "-Fe,"
Ph2 Ph2
NCCH3
(III).
In another embodiment, the chiral carbons atoms denoted by asterisks both have
an R
configuration. In another embodiment, the chiral carbons atoms denoted by
asterisks
both have an S configuration, In another embodiment, the reaction uses
hydrogen.
41
CA 02642563 2008-10-31
In yet another embodiment, the substrate is a ketone. In another embodiment,
the
ketone is an aromatic ketone. In yet another embodiment, the ketone is
prochiral.
In another embodiment, there is provided a process for preparing an amine
compound wherein said process comprises a step of preparing the amine compound
by
reducing an imine with the reaction of hydrogen or a compound donating
hydrogen in the
presence of a hexa-coordinate iron (II) complex of formula (I). In another
embodiment,
the hexa-coordinate iron (II) complex comprises a compound of formula (I)
having the
structure:
Me
(BF4)2
PFe
= p
ph2 co ph2
,o In another embodiment, the chiral carbons atoms denoted by asterisks
both have an R
configuration. In another embodiment, the chiral carbons atoms denoted by
asterisks
both have an S configuration. In another embodiment, the reaction uses a
compound
donating hydrogen. In another embodiment, the imine is not prochiral.
The catalysts I disclosed herein can reduce aldehydes, ketones and imines with
general structure (IX):
R"
(IX)
where the R', R" symbols, taken separately, represent simultaneously or
independently
a hydrogen atom, a linear or branched alkyl or alkenyl chain containing 1-8
carbon atoms,
possibly substituted, a cycloalkyl radical or an aryl group, possibly
substituted. The
symbol Q represents simultaneously or independently an oxygen atom or NR"
group,
where R" ' symbol represent simultaneously or independently a hydrogen atom, a
linear
42
CA 02642563 2008-10-31
or branched alkyl or alkenyl chain containing 1-8 carbon atoms, possibly
substituted, a
cycloalkyl radical or an aryl group, possibly substituted. Possible
substituents include
alkyl groups (such as Ci-C8 alkyl), aryl groups, halogens, and alkoxy groups.
The reduction of ketones and imines with general structure (IX) produce
products,
namely alcohols and amines, respectively with general structure (X). When the
correct
asymmetric hydrogenation or transfer hydrogenation catalyst I is applied, the
products are
obtained in one enantiomeric form. For example the use of complex (i) in
asymmetric
hydrogenation gives the S-alcohol in high e.e. while the use of complex (ii)
or (v) in
asymmetric transfer hydrogenation gives the S-alcohol in high e.e. The correct
catalyst I
might also be used for other catalytic asymmetric reactions such as the
transfer of
hydrogen from a hydrogen donor such as isopropanol or ethanol to a ketone or
imine.
The use of the catalysts disclosed herein for addition of a hydrosilane to a
ketone or imine,
an asymmetric Michael addition of donor to an acceptor, an asymmetric Diels-
Alder
reaction of an olefin to a diene or an asymmetric cyclopropanation reaction
may also be
possible.
QH
)<-H
R' R"
(X)
The catalysts disclosed herein comprising a compound of formula (I) and with
LI
= 1,2 = MeCN are surprisingly active and selective for the hydrogenation, by
use of
hydrogen gas, of ketones to produce valuable chiral and non-chiral alcohols in
the
presence of a base and an appropriate solvent. The use of complex (i) provides
a
particularly active and usefully enantioselective catalyst system.
The hydrogenation reaction involving a catalyst disclosed herein may or may
not
require solvent. When the use of the solvent is preferred for practical
reasons, any solvent
can be utilized for better performance of the catalyst. Non-limiting examples
include
primary, secondary and tertiary alcohols with hydrocarbon skeleton containing
2-15
carbons or aromatic solvents or ethers or hydrocarbon solvents.
43
CA 02642563 2008-10-31
In the solvent, the catalyst can be used at concentrations of 0.001 mM to 0.1
mM
while the substrate ketone or imine can be used in concentrations of 2 mM to
10 M. The
pressure of hydrogen gas can range from 0.5 atm to 100 atm with a preferred
pressure of
atm. Preferred concentrations of catalyst and substrate are 0.8 mM and 0.16 M,
5 respectively, with a ketone to catalyst ratio of 200:1.
The base in the hydrogenation process using H2 gas can be substrate (if it has
a
basic functionality) or a strong neutral base such as DBU or a phosphazene, or
an
alkaline or alkaline-earth metal carbonate salt, carboxylate salt, alkoxide
salt or hydroxide
salt. The base in the process can be used in a concentration of between one
and fifty times
10 the concentration of the catalyst concentration. The preferred base to
catalyst ratio is 15.
The temperature of the direct hydrogenation with hydrogen gas catalyzed by
complexes comprising a compound of formula (I) and with LI = L2 = MeCN can
range
between 0 and 120 C with the preferred temperature being 35 C.
Catalysts such as (ii) are particularly active and selective for the
asymmetric
transfer hydrogenation of ketones to non racemic alcohols in basic isopropanol
solvent or
other alcohols or mixtures such as formic acid/triethylamine known in the art
to transfer
hydrogen. Similarly, complexes (iii), (iv) and (v) can also be used as
catalysts for the
asymmetric transfer hydrogenation of ketones and the transfer hydrogenation of
certain
imines. The catalysts (VIII) with the ligand LI = MeCN or another nitrile
donor ligand
and L2 = CO are surprisingly active and selective for the reduction of ketones
to non-
racemic alcohols by transfer of hydrogen from basic isopropanol or other
alcohols or
mixtures such as formic acid/triethylamine known in the art to transfer
hydrogen.
The conditions for the transfer hydrogenation catalyzed by catalysts (ii),
(iii), (iv),
(v) and of the type (VIII) are surprising mild. The preferred temperature is
room
temperature but a range of temperatures is possible from 0 and 150 C. The
turnover
numbers reported in the examples are unprecedented for non-PGM catalysts that
operate
at room temperature.
44
CA 02642563 2008-10-31
The transfer hydrogenation catalysts (ii), (iii), (iv), (v) and of the type
(VIII) can be used
at concentrations of 0.001 mM to 1 mM while the substrate ketone or imine can
be used
in concentrations of 2 mM to 5 M. Preferred concentrations of catalyst and
substrate are
0.1 mM and 0.2 M, respectively, with a ketone to catalyst ratio of 1600:1 for
catalyst (ii)
and 200:1 for catalyst (iii) and 600:1 for catalyst (v) or in general a
substrate to catalyst
ratio of 500:1.
The base in the transfer hydrogenation process can be substrate (if it has a
basic
functionality) or a strong neutral base such as DBU or a phosphazene, or an
alkaline or
alkaline-earth metal carbonate salt, carboxylate salt, alkoxide salt or
hydroxide salt. The
base in the process can be used in a concentration of between one and fifty
times the
concentration of the catalyst concentration. The preferred base to catalyst
ratio is 8.
The invention will now be described in further detail by way of the following
examples, wherein the temperatures are indicated in degrees centigrade and the
abbreviations have the usual meaning in the art.
Examples
General Considerations.
All preparations and manipulations were carried out under an argon or nitrogen
atmosphere using standard Schlenk, vacuum-line, and glove-box techniques. Dry,
oxygen-free solvents were prepared by distillation from appropriate drying
agents and
employed throughout. The synthesis of the ligands (R,R)-cyP2N2 (9) and (R,R)-
PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2 have been reported previously (J.-X. Gao, H.
Zhang, X.-D. Yi, P.-P. Xu, C.-L. Tang, H.-L. Wan, K.-R. Tsai, T. Ikariya,
Chirality 2000,
12, 383). All other reagents used in the experiments were obtained from
commercial
sources and used as received. The mass spectroscopy (ESI+, Me0H) and elemental
analyses were performed at the University of Toronto, on sample handled under
argon for
the EA. Varian Gemini 400 MHz and 300 MHz spectrometers were employed for
recording 1H (400 MHz and 300 MHz), 13C { IH} (100 MHz and 75 MHz), and 31P
{1H}
CA 02642563 2008-10-31
(161 MHz and 121 MHz) NMR spectra at ambient temperature. The 111 and 13C NMR
spectra were referenced to solvent resonances, as follows: 7.26 and 77.16 ppm
for CHC13
and CDC13, 1.94 and 1.24 ppm for CH3CN and CD3CN). The 3113 NMR spectra were
referenced to 85% H3PO4 (0 ppm). All infrared spectra were recorded on a
Nicolet 550
Magna-IR spectrometer.
The samples of hydrogenation reaction mixtures were analyzed by 11-1 NMR
spectroscopy and GC using a Perkin Elmer Autosystem XL chromatograph with a
chiral
column (CP chirasil-Dex CB 25 m x 2.5 mm). Hydrogen was used as a mobile phase
at a
column pressure of 6 psi. The injector temperature was 250 C, and a FID
temperature
was 275 C. The retention times of the substrates are listed in Table 5.
trans-4-phenyl-3-buten-2-one: the GC analysis were conducted as above, except
that for the GC conditions the oven temperature was 140 C. The retention
times were
trans-4-phenyl-3-buten-2-one 7.8 min, I-4-phenyl-2-butanol 11.1 min, (S)-4-
pheny1-2-
butanol 11.4 min, trans-4-phenyl-3-buten-2-one 13.4 min, trans-(R)-4-pheny1-3-
buten-2-
ol 15.9 min, trans-(S)-4-phenyl-3-buten-2-ol 16.2 min. The product was also
identified by
1H NMR spectroscopy and the data obtained matches literature values.
N-Benzylideneaniline and benzophenone: the conversion of the product was
determined by 1H NMR spectroscopy and the data matches those of the commercial
samples.
General procedure for the iron catalyzed H2-hydrogenation of polar bonds: In
an
Ar or N2 glovebox, the iron complex (8 mg, 0.008 mmol) was suspended in 2 mL
of 2-
propanol and acetophenone (225 equiv) in 1 mL of 2-propanol. The solution of
base was
prepared by dissolution of KOtBu (15 equiv) in 2 mL of 2-propanol. The
solution
containing the substrate and then the one with base, followed by the
suspension of
catalyst were injected into a 50 cm3 Parr hydrogenator reactor filled with
hydrogen at the
desired pressure and temperature, maintained by use of a Fischer Scientific
Isotemp
1016D water bath.
46
CA 02642563 2008-10-31
,
The procedures for the iron catalyzed transfer hydrogenation of polar bonds
are
found in the footnotes of the Tables above.
Table 5.
GC analytical data for the reduced substrates (ts = retention time of
substrate; t1, t2 =
retention times of the products)
Substrate Oven Temp. ts (min) t1 (min) t2 (min)
( C)
Ph-CO-Me 130 5.0 8.5 9.1
(2'-C1-C6H4)-CO-Me 145 4.7 10.0 11.7
(3'-C1-C6H4)-CO-Me 130 7.8 16.6 17.7
(4'-C1-C6H4)-CO-Me 145 5.9 11.1 12.0
(4'-Br-C6H4)-CO-Me 155 6.5 11.4 12.1
(4'-Me-C6H4)-CO-Me 125 6.5 9.6 10.4
(4'-0Me-C6H4)-CO-Me 130 15.6 21.8 23.2
Ph-CO-Et 105 18.4 48.5 51.5
C oH7-CO-Meral 150 21.8 35.7 37.5
10H7-CO-Merbl 140 24.1 63.6 73.9
Ph-CHO 130 3.9 5.9
Ph-CO-iPr 114 11.0 37.2 37.8
Ph-CH2-CH2-CO-Me 135 7.9 11.8 12.6
Ph-CO-tBu 140 5.9 11.6 12.2
CH3-CO-CH-(CH3)2 60 2.9 8.2 8.5
[a] 2-acetonaphthone. [b] 1-acetonaphthone
EXAMPLE 1
47
CA 02642563 2008-10-31
, )
,
Preparation of the catalyst (R, R)-
[Fe(Ph2PCH2CH=NCH(Ph)CH(Ph)N¨CHCH2PPh2)(CH3CN)2][BP114]2, (i)
¨
Me
Ph C N Ph
./----
(BPh4)2
1"--P ' 1 -µ.1)--i
Ph2 N Ph2
C
¨ Me ¨
Synthesis of the diphenylphosphino-acetaldehyde hydrobromide dimer:
The procedure for the synthesis of the diphenylphosphino-acetaldehyde
hydrobromide dimer has been previously reported by Matt et al. (Matt, D.;
Ziessel, R.; De
Cian, A.; Fischer, J. New J. Chem. 1996, 20, 1257-1263) and was used in this
study with
modifications. Potassium hydride (413 mg, 10.3 mmol) was partially dissolved
in 10 mL
of dry THF. Diphenylphosphine (1.60 g, 8.58 mmol) was added to the resulting
mixture
to give a purple solution. After 30 min the solution was cooled to -78 C and
bromoacetaldehyde diethyl acetal (1.691 g, 8.58 mmol) was added over the
course of 15
min. The mixture was brought to room temperature to give a yellow solution. A
diluted
hydrobromic acid (10 mL, 1.17 mol_L-1) were added and the mixture was heated
at 40 C
overnight. The solvent volume was reduced by one half The white precipitate
was
recovered by filtration and washed with 20 mL of water and 20 mL
eyelohexane:ethyl
acetate (1:1 by volume). Drying in vacuo yielded 4 (2.51 g, 4.06 mmol) as a
white
powder. Analytical data were the same as those that have been reported by Matt
et al.
Synthesis of the catalyst (R,
R)-
[Fe(Ph2PCH2CH=NCH(Ph)CH(Ph)N=CHCH2PPh2)(CH3CN)2][BPh4]2, (i)
The diphenylphosphino-acetaldehyde hydrobromide dimer (200 mg, 0.324 mmol) was
completely dissolved in Me0H (6 mL). [Fe(H20)6][BE4]2 (164 mg, 0.485 mmol) was
added to the reaction mixture. Na0Me (34.9 mg, 0.647 mmol) was added as a Me0H
(1
mL) solution and the color of the solution changed from colorless to clear
yellow. After
48
CA 02642563 2008-10-31
=
min of stirring, 1 mL of acetonitrile was added. To this solution was added,
over the
course of 20 min, a solution of (1R,2R)-(+)-1,2-diphenylethylenediamine (R,R-
dpen, 69
mg, 0.323 mmol) in 0.5 mL of acetonitrile. The solution changed color to
purple after the
addition. After 20 h the resulting solution was added to a solution of NaBPh4
(250 mg,
5 0.658 mmol) in 1 mL of Me0H to cause the formation of the precipitate. A
pink solid
was recovered by filtration and dried under vacuum. Yield of (i): 83% (380
mg); 11-1
NMR (400 MHz, CD3CN) (5: 1.54 (s, 6H, CH3CN), 3.95-4.15 (m, 2H, HCP), 4.26-
4.38
(m, 2H, HCP), 5.43 (m, 2H, HC-N), 6.80-7.75 (m, 70H, ArH), 8.10-8.27 (m, 2H,
HC=N).
31P {H} NMR (121 MHz; CD3CN): 72.63 ppm (s). Anal. Calcd for C94H84N4P2FeB2:
C,
10 80.14; H, 6.01; N, 3.98. Found: C, 79.20; H, 6.08; N, 4.65. MS (EST)
Calcd for
[C46H44N4P2Fe-2(CH3CN)]2 : 344.3 m/z. Found: 344.1 m/z. MS (EST-) Calcd for
[B(Ph)4]-: 319.2 m/z. Found: 319.2 m/z.
EXAMPLE 2
Preparation of the catalyst (R, R)-
[Fe(Ph2PCH2CH=NCH(Ph)CH(Ph)N=CHCH2PPh2)(CH3CN)(C0)][13Ph412, (ii)
CO -
Ph, Ph
BPh41 2
Ph2 N Ph2
Me
The tetraphenylborate salt of the bisacetonitrile complex (i) (200 mg, 0.142
mmol)
was dissolved in (10 mL) of degassed acetone under inert atmosphere. Resulting
solution
was placed in the CO high pressure reactor and was stirred under 5 atmosphere
of CO for
12 hours at room temperature. Solvent was evaporated under reduced pressure
and
resulting solid was washed with diethyl ether (5 mL) three times. Yellow solid
was dried
under vacuum. Yield of (ii): 75 % (149 mg); Ili NMR (400 MHz, acetone-d6) (3:
1.54 (s,
3H, CH3CN), 4.42-4.57 (m, 2H, HC-N), 5.58-5.76 (m, 4H, HCP), 6.80-7.75 (m,
70H,
ArH), 8.14-8.23 (m, 2H, HC=N); 31P {111} NMR (121 MHz; acetone-d6): 69.3 ppm
(d,
49
CA 02642563 2008-10-31
Jp-p=30Hz); 65.7 ppm (d, Jp..p=30Hz); MS (ESI+) Calcd for [C46H4.4N4P2Fe-
(CO+CH3CN)]2+: 344.3 m/z. Found: 344.1 m/z. MS (ESI-) Calcd for [B(Ph)4I:
319.2 m/z.
Found: 319.2 m/z , IR (KBr) 2294 cm- I (vC 4%1, MeCN), 2001 cm-I (vC0).
EXAMPLE 3
Preparation of the complex [Fe(NCMe)2{9)}][BF4]2
Me
N (BF4)2
z
¨ Ns y
PpP
Ph2 h2
III
Me
*= R configuration
A suspension of (R,R)-cyP2N2 (9) (317 mg, 0.48 mmol) in 7 mL of MeCN was
added dropwise to a solution of [Fe(H20)6][BR]2 (162 mg, 0.48 mmol) in MeCN
(12
mL). After stirring for 20 mm at room temperature, the red solution was
concentrated to 1
mL and 10 mL of Et20 were added. A red-orange powder precipitated and was
isolated
by filtration and washed with Et20. Recrystallization of [Fe(NCMe)2{9}][BF42
from a
CHCb/ether solution gave the product (435 mg, 92% yield). A CDC13 solution in
a NMR
tube yielded red crystals suitable for X-ray diffraction studies and elemental
analysis.
1H NMR (400 MHz, CDC13) 6= 9.26 (s, HC=N), 8.06-6.63 (m, ArH), 3.68 (s, CH),
2.70-
2.13 (m, CH2), 1.75 (s, CH3CN). '3C {'H} NMR (100 MHz, CDCb) 6: 172.45 (s,
HC=1\1),
138.66-124.84 (m, C aromatic and CaN), 71.52, 66.05 (s, CH), 31.54, 29.26,
24.16, 22.82 (s,
CH2), 1.22 (s, CH3CN). {1H} NMR (161 MHz, CDC13) 6: 53.4 (s) ppm.
311){1H} NMR (161 MHz, CD3CN) 6: 52.6 (s) ppm. Anal. Calcd. for
C48H46N4B2F8P2Fe1Ø5CHC13: C, 56.55; H, 4.55; N, 5.44. Found: C, 56.45; H,
4.91; N,
5.04. IR (KBr) 2284 crn-' (vC 4\1, MeCN). MS (ESI+, Me0H) for [Fe(9)]2+ (m/z =
357.1).
CA 02642563 2008-10-31
EXAMPLE 4
Preparation of the catalyst [Fe(NCMe)(C0){9}][BEt]2,
Me
(BF)2
1711
y _.
p Fie p
Ph2 I CO Ph2
* = R configuration
Method A. A solution of [Fe(MeCN)2{9}][BF42 (Example 3, 200 mg, 0.21 mmol)
in acetone (10 mL) was stirred under 2 atm CO overnight at room temperature.
The
resulting orange-yellow solution was evaporated to dryness to give an orange
powder
(quantitative yield).
Method B. A solution of [Fe(MeCN)2{9}][BE4]2 (Example 3, 160 mg, 0.17 mmol)
in CHC13 (3 mL) was refluxed under 2 atm CO for 48 hours. The resulting orange-
yellow
solution was evaporated to dryness to give an orange powder (iii)
(quantitative yield).
1H NMR (400 MHz, CDC13) 6: 9.11 (s, CH=N), 8.21-6.35 (m, ArH), 3.53-3.32 (m,
CH),
2.77-1.21 (m, CH2), 1.75 (s, CH3CN). 13C{11-1} NMR (100 MHz, CDC13) 6: 213.75
(t, 2Jc_
p = 27.2 Hz, CO), 171.87 (d, 3Jc_p = 25.3 Hz, HC=N), 139.42-123.85 (m,
Caromatic and
70.56, 65.99 (s, CH), 32.22, 30.89, 24.36, 23.71 (s, CH2), 1.03 (s, CH3CN).
31P {'H} NMR (161 MHz, CDC13) 5: 51.82 (d, 2Jp_p = 40.6 Hz), 48.03 (d, 2Jp_p =
40.6 Hz).
Anal. Calcd. for C47H43N30B2F8P2FeØ25CHC13: C, 57.49; H, 4.42; N, 4.26.
Found: C,
57.36; H, 4.99; N, 4.10. IR (KBr) 2294 cm-1 (vC 44, MeCN), 1999 cm-1 (vCO). MS
(ESI+, Me0H) for [Fe {9 }]2+ (m/z = 357.1).
EXAMPLE 5
51
CA 02642563 2008-10-31
=
Preparation of the catalyst [Fe(NCMe)(C1\143u){9}][BF4]2, (iv).
Me
(BF)2
?NJ
¨N, y
põ,pies,p 41,
ph2, ph2
rJ
ii3u
* = R configuration
A solution of [Fe(MeCN)2{9}][BN2 (Example 3; 95 mg, 0.098 mmol) and
tBuNC (22 AL, 0.196 mmol) in acetone (3 mL) was stirred for 2 h at room
temperature.
The resulting orange-yellow solution was evaporated to dryness to give an
orange powder
of (iv). (quantitative yield). 'H NMR (400 MHz, CDC13) 6: 9.27, 8.87 (s,
CH=N), 8.30-
6.55 (m, ArH), 3.71-1.58 (m, CH and CH2), 2.17 (s, CH3CN), 1.21 (s,
(CH3)3CNC).
13C{1H} NMR (100 MHz, CDC13) (5: 173.47, 171.62 (s, HC=N), 139.60-125.38 (m,
Cammatic, C1\1 and N=C), 75.8, 73.61 (s, CH), 32.38, 31.76, 24.76, 23.90 (s,
CH2), 29.45
(s, (CH3)3CNC), 1.03 (s, CH3CN). 31P {111} NMR (161 MHz, CDC13) 6: 58.22 (d,
2Jp_p =
51 Hz), 48.48 (d, 2Jp_p = 51 Hz). IR (KBr) 2151, 2173 cm-1 (vC 4\1, MeCN and
tBuNC).
EXAMPLE 6
Preparation of transtFe(NCMe)2{(R,R)-
PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2}1[13F4]2
A solution
of
(R,R)-PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2 (510 mg, 0.78 mmol) and [Fe(H2
0)61[BF4]2 (260 mg, 0.78 mmol) in MeCN (10 mL) was stirred for 1 h at ambient
temperature. The solution was evaporated and the remaining red residue was
washed with
pentane. The analytically pure product was obtained after crystallization from
MeCN/Et20 as dark red crystals (510 mg, 64%). Recrystallization from a
52
CA 02642563 2008-10-31
MeCN/Me0H/Et20 solution yielded crystals suitable for X-ray diffraction
studies. 1 H
NMR (400 MHz, CD3CN): 9.32 (s, CH=N), 7.82-7.21 (m, Ar-H), 6.94 (m, Ar-H),
6.85
(m, Ar-H, 5.97 (s, N-CH), 1.96 (s, CH3CN). 31P{111} NMR (161 MHz, CDC13 ):
51.8
(s). Anal. Calcd for C56 H48 N4 B2 F8 P2 Fe : C, 62.95; H, 4.53; N, 5.24.
Found: C,
62.69; H, 4.79; N, 5.81.
EXAMPLE 7
Preparation of catalyst (v): trans-[Fe(NCMe)(C0){(R,R)-
PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2}i[BE]2
0
III (BF
Ph 4)2, c
NJ, I
AM' /Pi Fieljix
Ph Ph Ph Ph
(V)
A solution of
trans-[Fe(NCMe)2 {(R,R)-
PPh2C6H4CHNCHPhCHPhNCHC6H4PPh2}RBF4]2 (0.51 g, 0.5 mmol) in acetone was
stirred under a 5 atm of CO at room temperature for 6 h. The solvents were
evaporated, to
obtain an orange powder. The powder was again dissolved in acetone and stirred
under
two atm of CO atmosphere for 12 h at room temperature. The solvents were
evaporated
and the remaining orange residue was washed with toluene and ether.
Crystallization
from acetone/CH2C12/Et20 gave the analytical pure compound as an orange solid.
Yield:
0.47 g (0.4 mmol, 80 %). 111 NMR (d3-MeCN, 300 MHz, 25 C): 6.05 (br s, 2 H,
CH),
6.69-8.06 (several m, 30 H, Ph), 9.43 (br s, 2 H, CH=N). 31P NMR (CD2C12, 121
MHz):
49.9 (d, Jr,, = 39 Hz), 53.0 (d, Jpy = 39 Hz). Anal. Calcd. for
C55H45B2F8N3P2Fe1: C,
62.59; H, 4.30; N, 3.98; Found: C, 61.93; H, 4.96; N, 3.67.
53
CA 02642563 2008-10-31
EXAMPLE 8
Preparation of catalyst (vi): Fe(Ph2PCH2CH=NC2H4N=CHCH2PPh2)(CH3CN)21(BPh4)21
¨ Me ¨
C
[BPh412
Ph2ril Ph2
¨ Me _
Preparation of precursor solution (A): The reaction was performed in the glove-
box under N2 atmosphere at room temperature. The diphenylphosphino-
acetaldehyde
hydrobromide dimer from Example 1 (200 mg, 0.324 mmol) was partially dissolved
in
CH3CN (6 mL). After 5 min of stirring [Fe(H20)6][BF4]2(164 mg, 0.485 mmol) was
added to the reaction mixture. t-BuOK (74.0 mg, 0.645 mmol) was added to the
reaction
mixture and the color of the solution changed from white to yellow. The
mixture was
stirred at room temperature for 30 min without any observable changes.
A stock solution of the diamine was prepared by dissolving 85.5 mg of 1, 2-
ethylenediamine in 1.1 mL of acetonitrile. A portion (0.250 mL) of stock
solution was
added to the precursor solution (A) over the course of 20 min at room
temperature. The
solution changed color to red-orange after the addition. After 3 h the
solution became
deep orange. The solution was added to a solution of NaBP1i4 (250 mg, 0.658
mmol) in
1.5 mL of Me0H to cause the formation of the precipitate. The orange-pink
solid was
filtered and washed with 0.35 mL of Me0H three times and dried under vacuum.
Yield:
82% (0.33 mg); IHNMR (400 MHz, CD3CN) J: 1.36 (s, 6H, CH3CN ), 4.10-4.25 (m,
4H,
HCP), 4.10-4.25 (m, 4H, HC-N), 6.80-7.55 (m, 60H, ArH), 8.65-8.80 (m, 2H,
HC=N).
31P {H} NMR (121 MHz; CD3CN): 74.01 ppm (s). Anal. Calcd for
C82H761\14132FeB2: C,
78.38; H, 6.08; N, 4.46. Found: C, 77.58; H, 6.03; N, 4.26. MS (ESI+) Calcd.
for
[C34H36N4P2Fe-2(CH3CN)]2+: 268.2 m/z. Found: 268.1 m/z. MS (ESI-) Calcd for
[B(Ph)4]-: 319.2 m/z. Found: 319.2 m/z. The crystals were obtained by
diffusion of Et20
(1.0 mL) into the deep orange solution (1 mL) obtained as above but before the
addition
of NaBPh4.
54
CA 02642563 2008-10-31
EXAMPLE 9
Preparation of catalyst (vii):
[Fe(Ph2PCH2CH=NC2H4N=CHCH2PPh2)(CH3CN)(C0)](BPh4)2
- CO
1- BPh41
Ph2 N Ph2
_ Me
The complex [Fe(Ph2PCH2CH=NC2H4N=CH-CH2PPh2)(CH3CN)21(BP1142 (vi)
(200 mg, 0.159 mmol) was dissolved in (10 mL) of degassed acetone under inert
atmosphere. Resulting solution was placed in the CO high pressure reactor and
was
stirred under 5 atmosphere of CO for 12 hours at room temperature. Solvent was
evaporated under reduced pressure and resulting solid was washed with diethyl
ether (5
mL) three times. Yellow solid was dried under vacuum. Yield of (vii): 80 %
(158 mg); 1H
NMR (400 MHz, acetone-do) (5: 1.72 (s, 3H, CH3CN), 3.95-4.50 (m, 4H, HC-N),
3.95-
4.50 (m, 4H, HCP), 6.55-7.89 (m, 60H, ArH), 8.18-8.46 (m, 2H, HC=N); 31P {H}
NMR
(121 MHz; acetone-do): 69.1 ppm (s).
EXAMPLE 10
Preparation of transtFe(MeCN)2(6)1(BF4)2, wherein 6 is
PPh2 Ph2P
where n = 2
This ligand 6 was prepared as described in Jeffery, J. C.; Rauchfuss, T. B.;
Tucker, P. A.
Inorg. Chem. 1980, 19, 3306-3316. The complex transtFe(MeCN)2(6)](BF4)2 was
prepared as follows. A suspension of 6 (149 mg, 0.25 mmol) in 5 mL of MeCN was
added to a solution of [Fe(H20)6][BF42 (84 mg, 0.25 mmol) in MeCN (10 mL).
After
CA 02642563 2008-10-31
,
stirring for 1 h, the red solution was concentrated to 1 mL and 10 mL of Et20
was added.
A purple powder precipitated. The powder was isolated and washed with hexane.
(200
mg, 87%). Crystals suitable for X-ray diffraction studies were obtained from a
MeCN/Et20 solution. 11-1 NMR (400 MHz, CDC13): 9.46 (s, CH=1\1), 8.07-6.71 (m,
An]),
4.35 (s, CH2), 2.00 (s, CH3CN); 31P {11-1} NMR (161 MHz, CDC13) 54.4 (s).
Anal. Calcd
for C44H401\14B2F8P2Fe: C, 57.68; H, 4.40; N, 6.12%. Found: C, 57.16; H, 4.40;
N, 5.86%.
EXAMPLE 11
Preparation of catalyst (viii): trans-[Fe(MeCN)(CO) (6)](BF4)2
--103F4)2
Ph Ph !;,1 Ph Ph
(viii)
Complex trans-[Fe(MeCN)2(6)KBN2 was reacted with CO (2 atm) in acetone at
room temperature to produce complex (viii). Yield: 1.14 g (1.3 mmol, 87 %.).
1H NMR
(d3-MeCN, 300 MHz): 1H NMR 4.01 (br s, 4 H, CH2), 7.20-7.98 (several m, 20 H,
Ph),
9.21 (br s, 2 H, CH-1\1). 31P NMR (CD2C12, 121 MHz): 50.8 (s). Anal. Calcd.
for
C43H37B2F8N3P2Fe1: C, 57.18; H, 4.13; N, 4.65; Found: C, 56.12; H, 4.15; N,
4.83.
EXAMPLE 12
Preparation of complex: trans-(R,R)-
[Fe(MeCN)2(PPh2CH2CHNC6H10NCHCH2PP112)KBF4)2
56
CA 02642563 2008-10-31
, =
CCH3
C
F N
[BP114]2
Ph2 Ph2
CCH3
The diphenylphosphino-acetaldehyde hydrobromide dimer from Example 1 (200 mg,
0.324 mmol) was completely dissolved in Me0H (6 mL). [Fe(H20)6][BF4]2 (164 mg,
0.485 mmol) was added to the reaction mixture. Na0Me (34.9 mg, 0.647 mmol) was
added as a Me0H (1 mL) solution and the color of the solution changed from
colorless to
clear yellow. After 10 min of stirring, 1 mL of acetonitrile was added to give
precursor
solution B.
(1R,2R)-(-)-1,2-diaminocyclohexane (37 mg, 0.32 mmol) was dissolved in 0.5 mL
of
acetonitrile and was added to the precursor solution over the course of 20
min. The
solution changed color to purple after addition. The resulting solution was
heated at 40
C for 20 h to give an deep orange solution. The solvent volume was reduced by
one half
and the resulting solution was added to a solution of NaBPh4 (250 mg, 0.658
mmol) in
1.5 mL of Me0H to cause the formation of a precipitate. An orange-red solid
was
recovered by filtration and washed with 0.15 mL of Me0H three times and dried
with
vacuum. Yield: 54% (0.23 mg); 11-1 NMR (400 MHz, CD3CN) (3: 1.33 (s, 6H,
CH3CN),
1.29-1.39 (m, 2H, H of C6H10), 1.68-1.76 (m, 2H, H of C6H10), 1.98-2.28 (m,
2H, H of
C6H10), 2.70-2.78 (m, 2H, H of C6Hio), 3.54-3.58 (m, 2H, HC-N), 3.88-4.01 (m,
2H,
HCP), 4.34-4.49 (m, 2H, HCP), 6.8-7.5 (m, 60H, ArH), 8.60-8.74 (m, 2H, HC=N).
3IP
{H} NMR (121 MHz; CD3CN): 73.96 ppm (s). Anal. Calcd for C86H82N4P2FeB2: C,
78.78; H, 6.31; N, 4.27. Found: C, 77.00; H, 5.99; N, 4.34. MS (EST+) Calcd
for
[C38H42N4P2Fe-2(CH3CN)]2+: 268.1 m/z. Found: 268.1 m/z. MS (ESI-) Calcd for
[B(Ph)4]-: 319.2 m/z. Found: 319.2 m/z.
EXAMPLE 13
Preparation of trans-(R,R)-[Fe(C0)(NCMe)(PPh2CH2CHNC6HIONCHCH2PPh2)i(BF4)2
57
CA 02642563 2008-10-31
NCCH3 _
[BPha]2
I P
Ph2 t Ph2
0
The complex was prepared according to the method of example 9. 31P NMR(1H) d
(66.78, 67.05) and d(70.52, 70. 79) J=81 Hz.
EXAMPLE 14
Preparation of complex: trans-{Fe(MeCN)2(PPh2CH2CHNC6H4NCHCH2PPh2)] (BF4)2
NCCH3 _
[BPh4]2
Ph2 Ph2
NCCH3
Ortho-phenylenediamine (35 mg, 0.32 mmol) was dissolved in 0.5 mL of
acetonitrile
and was added to the precursor solution (A) of Example 8 over the course of 20
minutes
at 22 C. The solution changed color to orange after the addition. The
resulting residue
was added to the solution of NaBPh4 (250 mg, 0.658 mmol) in 1 mL of Me0H to
cause
the formation of the precipitate. The red-orange solid was isolated by
filtration and
washed with 0.15 mL of Me0H three times and dried under vacuum. Yield: 86%
(0.36
mg); 1H NMR (400 MHz, CD3CN) (5: 2.10 (s, 6H, CH3CN), 4.52-4.60 (m, 4H, HCP),
6.80-8.20 (m, 64H, HAr), 9.32-9.44 (m, 2H, HC=N). 31P {H} NMR (121 MHz;
CD3CN):
68.33 ppm (s). Anal. Calcd for C38H36N4P2FeB2: C, 79.19; H, 5.87; N, 4.29.
Found: C,
58
CA 02642563 2015-06-11
76.83; H, 5.80; N, 4.15. MS (ES1 ) Calcd for [C86H76N4P2Fe-2(CH3CN)]2+: 292.2
m/z.
Found: 292.1 m/z. MS (ESIs) Calcd for [B(Ph)4]": 319.2 m/z. Found: 319.2 m/z.
While the present invention has been described with reference to examples, it
is to be
understood that the invention is not limited to the disclosed examples. To the
contrary,
the invention is intended to cover various modifications and equivalent
arrangements as
would be apparent to a person of skill in the art.
59