Note: Descriptions are shown in the official language in which they were submitted.
CA 02647655 2016-11-03
1
TITLE:
NOVEL C-1 ANALOGS OF PANCRATISTATIN AND 7-
DEOXYPANCRATISTATIN AND PROCESSES FOR THEIR PREPARATION.
FIELD
The present application relates to novel C-1 analogs of pancratistatin and 7-
deoxypancratistatin and to processes for their preparation. The application
further relates to pharmaceutical compositions containing the novel analogs
and
to uses of the analogs.
BACKGROUND
Pacratistatin (1) and narciclasine (2) are natural products that are highly
active
against many cancer cell lines including murine P388 and lymphocytic leukemia;
human cancer cells pancreas BXPC-3, breast MCF-7, CNS SF-268, lung NCI-
H460, colon KM20L2 and prostate DU145. Although the exact mode of action for
pancratistatin remains unknown, narciclasine is believed to inhibit peptide
bond
formation in eukaryotic ribosomes. Lycoricidine (3) and 7-deoxypancratistatin
(4)
are significantly less active, than the corresponding C-7 hydroxylated
compound.
The reduced activity maybe due to the absence of the hydrogen bonded donor
acceptor pair in the phenanthridone functionality.
OH OH OH OH
HO 40 OH OH fib OH HO OH
<0 OH 0
7 OH <0 7 OH 0< w OH
0 NHo NH 0 NH 0 NH
OHO OHO 0 0
1 2 3 4
CA 02647655 2016-02-26
2
SUMMARY
The present application describes novel C-1 substitution analogs of
pancratistatin
and 7-deoxypancratistatin.
Accordingly, one aspect of the present application includes a compound of the
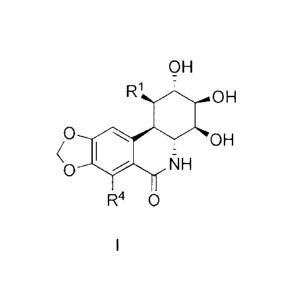
Formula I:
OH
R1 OH
O wOH
Co NH
R4 0
Formula I
wherein:
R1 is selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, CH=CR2R3, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3,
CH20C(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH200(0)R2; and
R2 and R3 are independently selected from H, C16alkyl, C2_6alkenyl, C3_
iocycloalkyl and C6_10ary1 said latter four groups being unsubstituted or
substituted with one to 5 groups independently selected from halo, OH, OCi_
4alkyl, OC(0)C1_6alkyl and nitro; and
R4 is selected from H and OH;
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
In another aspect of the application there is provided a pharmaceutical
composition comprising one or more compounds of Formula I as defined above,
or a pharmaceutically acceptable salt, solvate or prodrug thereof, and a
pharmaceutically acceptable carrier.
CA 02647655 2016-02-26
3
In a further aspect of the application there is provided a use of one or more
compounds of Formula I as defined above, or a pharmaceutically acceptable
salt,
solvate or prodrug thereof, to treat cancer.
In still a further aspect of the application there is provided a method of
treating
cancer comprising administering an effective amount of one or more compounds
of Formula I as defined above, or a pharmaceutically acceptable salt, solvate
or
prodrug thereof, to a subject in need thereof.
In another aspect of the present application is a use of one or more compounds
of Formula I, as defined above, or a pharmaceutically acceptable salt, solvate
or
prodrug thereof, as a medicament.
A further aspect of the present application is a process for the preparation
of an
intermediate of the Formula II:
0Pg
H(0)C 0Pg
0 vip
org
040 N- Pg
R4 0
wherein R4 is selected from H and 0Pg and each Pg may be the same or
different and represent suitable protecting groups or any two adjacent Pg are
joined to form a suitable cyclic protecting group;
the process comprising:
CA 02647655 2016-02-26
4
(i) reacting a compound of the Formula III with an aluminum acetylide derived
from a compound of the Formula IV, followed by protection to form a compound
of the Formula V, wherein R4 and each Pg is as defined above:
r el
+ < <0 0 . 0Pg
_________________________________________ lir _
0 R4 0 0Pg
-K/L`'OPg
PgKP'' R40P9
PgN
-0,
III IV V .
,
(ii) reducing the compound of Formula V to form a cis-alkene of the Formula
VI,
wherein R4 and each Pg is as defined above:
0 0 0Pg
<o =
-- 0Pg
0Pg
_
R4 0P9 --70 R4 el .iWo,
0Pg
-0
0P9 0 Pg
Pg1\i''' \-0
VI -
V ,
(iii) reacting the compound of the Formula VI under solid-state, silica gel
catalysis
conditions to form a compound of the Formula VII, wherein R4 and each Pg is as
defined above:
0Pg 0Pg
-- dh 0Pg AbAh 0P9
R4
40 õ..= 0Pg R4, l/1W 0Pg
Nµµ
1 NPg
0 Pg -00- 0
\--0
\-0
VI VII .
,
CA 02647655 2016-02-26
(iv) oxidatively cleaving the double bond in the compound of the Formula VII
to
form an intermediate diketone of the Formula VIII which cyclizes to form a
compound of the Formula IX, wherein R4 and each Pg is as defined above:
0Pg 0 0Pg 0Pg
H(0)C i& opg 0Pg 0_ 0Pg
R4 deW OP g _ip,, R4 si W
_ 0Pg -bi- (0 401 W_ 0Pg
0 0
VP NPg NPg 0 Mpg
R4 OH
VII ¨ ¨ VIII IX
5 ,
(v) oxidizing the compound of the Formula IX to form a compound of Formula II,
wherein R4 and each Pg is as defined above:
0Pg 0Pg
OHC 0Pg H(0)C 0Pg
( 0Pg _________
0 lei 0 is W
el 30. 0Pg
0 NPg 0 NPg
R4 OH R4 0
Ix II .
CA 02647655 2016-02-26
6
In a further aspect of the application there is included a process for
preparing a
compound of Formula I
OH
R1 OH
si 7 OH
0 NH
R4 0
wherein
R1 is selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3, CH=CR2R3,
CH200(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH200(0)R2 ; and
R2 and R3 are independently selected from H, C1_6alkyl, C2_6alkenyl, C3_
iocycloalkyl and C6_10aryl said latter four groups being unsubstituted or
substituted with one to 5 groups independently selected from halo, OH, OCi-
aalkyl, OC(0)C1_6alkyl and nitro, with the exception that R2 is not H when R1
is
C(0)R2; and
R4 is selected from H and OH,
comprising:
(i) reacting a compound of the Formula II, wherein R4 is selected from H and
0Pg
and each Pg may be the same or different and represent suitable protecting
groups or any two adjacent Pg are joined to form a suitable cyclic protecting
group under conditions to convert the aldehyde moiety to a group, other than
C(0)H, selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3, CH=CR2R3,
CA 02647655 2016-02-26
7
CH200(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH200(0)R2, wherein R2 and R3 are as defined above to form a compound of
the Formula X wherein R1, R4 and each Pg are as defined above:
0Pg 0Pg
oF
H(0)C di 0Pg R1 -g
0 40 v õ.g __ i,.
a i. 401 7 0Pg
< ov
0 NPg 0 NPg
R4 0 R4 0
II X .
,
(ii) removing the Pg groups to form a compound of the Formula I wherein R1 and
R4 are as defined above:
0Pg OH
R1 ib 0Pg R1 ib OH
si 7 ,. ____ vi.
<0 org 0 NH OH
40 7
0 .1Pg 0
R4 0 R4 0
X I .
Other features and advantages of the present application will become apparent
from the following detailed description. It should be understood, however,
that the
scope of the claims should not be limited by the preferred embodiments and
examples, but should be given the broadest interpretation consistent with the
description as a whole.
CA 02647655 2016-02-26
8
BRIEF DESCRIPTION OF THE DRAWINGS
The application will now be described in greater detail with reference to the
drawings in which:
Figure 1 is a scheme showing a process for the preparation of C-1 substituted
analogues of 7-deoxypancratistatin analogues according to an embodiment of
the present application;
Figure 2 is a graph showing the activity of the compounds 1(a), 1(c) and 1(d)
in
Jurkat cells.
Figure 3 is a graph showing the activity of the compound 1(b) at two
concentrations (0.5 uM and 1.0 uM) in Jurkat cells (human leukemia cells).
DETAILED DESCRIPTION
(I) DEFINITIONS
The term "Ci_salkyl" as used herein means straight and/or branched chain,
saturated alkyl groups containing from one to six carbon atoms and includes
methyl, ethyl, propyl, isopropyl, n-butyl, s-butyl, isobutyl, t-butyl, 2,2-
dimethylbutyl, n-pentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, n-
hexyl
and the like.
The term "C2_6alkenyl" as used herein means straight and/or branched chain,
unsaturated alkyl groups containing from two to six carbon atoms and one to
three double bonds, and includes (depending on the identity of n) vinyl,
allyl, 2-
methylprop-1-enyl, but-1-enyl, but-2-enyl, but-3-enyl, 2-methylbut-1-enyl, 2-
methylpent-1-enyl, 4-methylpent-1-enyl, 4-methylpent-2-enyl, 2-methylpent-2-
enyl, 4-methylpenta-1,3-dienyl, hexen-1-y1 and the like.
The term "C3_10cycloalkyl" as used herein a saturated carbocylic group
containing
from three to 10 carbon atoms and one or more rings and includes cyclopropyl,
cyclobutyl, cyclopentyl, cyclodecyl, bicyclo[2.2.21octane,
bicyclo[3.1.1]heptane,
octahydro-1H-indene and the like.
CA 02647655 2016-02-26
9
The term "halo" as used herein refers to a halogen atom and includes F, Cl, Br
and I.
In some cases the chemistries outlined herein may have to be modified, for
instance by use of protecting groups, to prevent side reactions of reactive
groups
attached as substituents. This may be achieved by means of conventional
protecting groups, for example as described in "Protective Groups in Organic
Chemistry" McOmie, J.F.W. Ed., Plenum Press, 1973 and in Greene, T.W. and
Wuts, P.G.M., "Protective Groups in Organic Synthesis", John Wiley & Sons, 3rd
Edition, 1999.
The terms "protective group" or "protecting group" or "Pg" or the like as used
herein refer to a chemical moiety which protects or masks a reactive portion
of a
molecule to prevent side reactions in those reactive portions of the molecule,
while manipulating or reacting a different portion of the molecule. After the
manipulation or reaction is complete, the protection group is removed under
conditions that do not destroy or decompose the molecule. Many conventional
protecting groups are known in the art for example as described in "Protective
Groups in Organic Chemistry" McOmie, J.F.W. Ed., Plenum Press, 1973 and in
Greene, T.W. and Wuts, P.G.M., "Protective Groups in Organic Synthesis", John
Wiley & Sons, 3rd Edition, 1999. These may include but are not limited to Boc,
Ts, Ms, TBDMS, TBDPS, Tf, Bn, allyl, Fmoc, C116acyl, silyl, acetal and the
like.
The term "leaving group" as used herein refers to a group that is readily
displaceable by a nucleophile, for example, under nucleophilic substitution
reaction conditions. Examples of suitable leaving groups include, halo, Ms,
Ts,
Ns, Tf, Bn, C16acy1, C1_16alkyl, alkylsulphonyl and the like.
CA 02647655 2016-02-26
The term "suitable", as in for example, "suitable protecting group", "suitable
leaving group" or "suitable reaction conditions" means that the selection of
the
particular group or conditions would depend on the specific synthetic
manipulation to be performed and the identity of the molecule but the
selection
5 would be well within the skill of a person trained in the art.
Boc as used herein refers to the group t-butyloxycarbonyl.
Ac as used herein refers to the group acetal.
Ts (tosyl) as used herein refers to the group p-toluenesulfonyl
10 Ms as used herein refers to the group methanesulfonyl
TBDMS as used herein refers to the group t-butyldimethylsilyl.
TBDPS as used herein refers to the group t-butyldiphenylsilyl.
Tf as used herein refers to the group trifluoromethanesulfonyl.
Ns as used herein refers to the group naphthalene sulphonyl.
In all of the compounds disclosed herein, that is compounds of the Formulae I-
X,
one or more, including all, of the hydrogen atoms may be replaced with F. A
person skilled in the art would appreciate that only those hydrogens available
for
substitution by fluorine would be replaceable by fluorine.
The term "compound(s) of the application" or "intermediate compounds" used
herein means compound(s) of Formulae I and ll as defined above, or any other
novel compounds or intermediates defined above, stereoisomers thereof or
pharmaceutically acceptable salts, solvates or prodrugs thereof, including
mixtures thereof.
The compounds of the application all have at least one asymmetric centre.
Where the compounds according to the application possess more than one
asymmetric centre, they may exist as diastereomers. It is to be understood
that
CA 02647655 2016-02-26
11
all such isomers and mixtures thereof in any proportion are encompassed within
the scope of the present application. It is to be understood that while the
stereochemistry of the compounds of the application may be as provided for in
any given compound listed herein, such compounds of the application may also
contain certain amounts (e.g. less than 20%, preferably less than 10%, more
preferably less than 5%) of compounds of the application having alternate
stereochemistry.
The term "pharmaceutically acceptable" means compatible with the treatment of
animals, in particular, humans.
The term "pharmaceutically acceptable salt" means an acid addition salt or
base
addition salt, which is suitable for or compatible with the treatment of
patients.
The term "pharmaceutically acceptable acid addition salt" as used herein means
any non-toxic organic or inorganic salt of any base compound of the
application,
or any of its intermediates. Illustrative inorganic acids which form suitable
salts
include hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as
metal salts such as sodium monohydrogen orthophosphate and potassium
hydrogen sulfate. Illustrative organic acids that form suitable salts include
mono,
di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic,
succinic,
glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic,
phenylacetic,
cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene
sulfonic
and methanesulfonic acids. Either the mono or di-acid salts can be formed, and
such salts may exist in either a hydrated, solvated or substantially anhydrous
form. In general, the acid addition salts of the compounds of the application
are
more soluble in water and various hydrophilic organic solvents, and generally
demonstrate higher melting points in comparison to their free base forms. The
selection of the appropriate salt will be known to one skilled in the art.
Other
CA 02647655 2016-02-26
12
non-pharmaceutically acceptable salts, e.g. oxalates, may be used, for
example,
in the isolation of the compounds of the application, for laboratory use, or
for
subsequent conversion to a pharmaceutically acceptable acid addition salt. In
embodiments of the application, the pharmaceutically acceptable acid addition
salt is the hydrochloride salt, or the H3PO4 salt. The formation of a desired
compound salt is achieved using standard techniques. For example, the neutral
compound is treated with an acid or base in a suitable solvent and the formed
salt is isolated by filtration, extraction or any other suitable method.
The term "pharmaceutically acceptable basic addition salt" as used herein
means
any non-toxic organic or inorganic base addition salt of any acid compound of
the
application, or any of its intermediates. Acidic compounds of the invention
that
may form a basic addition salt include, for example, where the group of R1 has
an acidic hydrogen, for example when R1 is C(0)0H. Illustrative inorganic
bases
which form suitable salts include lithium, sodium, potassium, calcium,
magnesium or barium hydroxide. Illustrative organic bases which form suitable
salts include aliphatic, alicyclic or aromatic organic amines such as
methylamine,
trimethylamine and picoline or ammonia. The selection of the appropriate salt
will be known to a person skilled in the art. Other non-pharmaceutically
acceptable basic addition salts, may be used, for example, in the isolation of
the
compounds of the invention, for laboratory use, or for subsequent conversion
to
a pharmaceutically acceptable acid addition salt.
The term "solvate" as used herein means a compound of the application or a
pharmaceutically acceptable salt of a compound of the application, wherein
molecules of a suitable solvent are incorporated in the crystal lattice. A
suitable
solvent is physiologically tolerable at the dosage administered. Examples of
suitable solvents are ethanol, water and the like. When water is the solvent,
the
molecule is referred to as a "hydrate". The formation of solvates of the
CA 02647655 2016-02-26
13
compounds of the application will vary depending on the compound and the
solvate. In general, solvates are formed by dissolving the compound in the
appropriate solvent and isolating the solvate by cooling or using an
antisolvent.
The solvate is typically dried or azeotroped under ambient conditions.
Compounds of the application includes prodrugs. In general, such prodrugs will
be functional derivatives of a compound of the application, which are readily
convertible in vivo into the compound from which it is notionally derived.
Prodrugs of the compounds of the application may be conventional esters formed
with available hydroxy, or amino groups. For example, an available OH or
nitrogen in a compound of the application may be acylated using an activated
acid in the presence of a base, and optionally, in inert solvent (e.g. an acid
chloride in pyridine). Some common esters which have been utilized as prodrugs
are phenyl esters, aliphatic (C5-C24) esters, acyloxymethyl esters, carbamates
and amino acid esters. In certain instances, the prodrugs of the compounds of
the application are those in which one or more of the hydroxy groups in the
compounds is masked as groups which can be converted to hydroxy groups in
vivo. Conventional procedures for the selection and preparation of suitable
prodrugs are described, for example, in "Design of Prodrugs" ed. H. Bundgaard,
Elsevier, 1985.
Compounds of the application includes radiolabeled forms, for example,
compounds of the application labeled by incorporation within the structure 3H
or
140 or a radioactive halogen such as 1251. A radiolabeled compound of the
application may be prepared using standard methods known in the art. For
example, tritium may be incorporated into a compound of the formula I using
standard techniques, for example by hydrogenation of a suitable precursor to a
compound of the application using tritium gas and a catalyst. Alternatively, a
compound of the formula I containing radioactive iodo may be prepared from the
CA 02647655 2016-02-26
14
corresponding trialkyltin (suitably trimethyltin) derivative using standard
iodination
conditions, such as [1251] sodium iodide in the presence of chloramine-T in a
suitable solvent, such as dimethylformamide. The trialkyltin compound may be
prepared from the corresponding non-radioactive halo, suitably iodo, compound
using standard palladium-catalyzed stannylation conditions, for example
hexamethylditin in the presence of tetrakis(triphenylphosphine) palladium (0)
in
an inert solvent, such as dioxane, and at elevated temperatures, suitably 50-
100 C.
The term "subject" as used herein includes all members of the animal kingdom
including human. The subject is preferably a human.
The term "cancer" as used herein refers to a class of diseases or disorders
characterized by uncontrolled division of cells and the ability of these cells
to
invade other tissues, either by direct growth into adjacent tissue through
invasion
or by implantation into distant sites by metastasis. Metastasis is defined as
the
stage in which cancer cells are transported through the bloodstream or
lymphatic
system. Examples of cancer that may be treated using the compounds of the
invention include but are not limited to, prostate cancer, colon cancer,
breast
cancer, bladder cancer, lung cancer, ovarian cancer, endometrial cancer renal
cancer and pancreatic cancer.
The term a "therapeutically effective amount", "effective amount" or a
"sufficient
amount" of a compound of the present invention is a quantity sufficient to,
when
administered to the subject, including a mammal, for example a human, effect
beneficial or desired results, including clinical results, and, as such, an
"effective
amount" or synonym thereto depends upon the context in which it is being
applied. In the context of disease, therapeutically effective amounts of the
compounds of the present invention are used to treat, modulate, attenuate,
CA 02647655 2016-02-26
reverse, or affect a disease or conditions for example, cancer in a subject.
An
"effective amount" is intended to mean that amount of a compound that is
sufficient to treat, prevent or inhibit such diseases or conditions. The
amount of a
given compound of the present invention that will correspond to such an amount
5 will vary depending upon various factors, such as the given drug or
compound,
the pharmaceutical formulation, the route of administration, the type of
disease or
disorder, the identity of the subject or host being treated, and the like, but
can
nevertheless be routinely determined by one skilled in the art. Also, as used
herein, a "therapeutically effective amount" of a compound of the present
10 invention is an amount which prevents, inhibits, suppresses or
reduces a disease
or conditions for example, cancer as determined by clinical symptoms or the
amount of cancer cells, in a subject as compared to a control. As defined
herein,
a therapeutically effective amount of a compound of the present invention may
be readily determined by one of ordinary skill by routine methods known in the
15 art.
In an embodiment, a therapeutically effective amount of a compound of the
present invention ranges from about 0.1 to about 40 mg/kg body weight,
suitably
about 1 to about 10 mg/kg body weight, and more suitably, from about 2 to
about
5 mg/kg body weight. The skilled artisan will appreciate that certain factors
may
influence the dosage required to effectively treat a subject, or prevent a
subject,
suffering from a disease or condition for example cancer, and these factors
include, but are not limited to, the severity of the disease or disorder,
previous
treatments, the general health and/or age of the subject and other diseases
present.
As used herein, and as well understood in the art, "treatment" is an approach
for
obtaining beneficial or desired results, including clinical results.
Beneficial or
desired clinical results can include, but are not limited to, alleviation or
CA 02647655 2016-02-26
16
amelioration of one or more symptoms or conditions, diminishment of extent of
disease, stabilized (i.e. not worsening) state of disease, preventing spread
of
disease, delay or slowing of disease progression, amelioration or palliation
of the
disease state, and remission (whether partial or total), whether detectable or
undetectable. "Treatment" can also mean prolonging survival as compared to
expected survival if not receiving treatment.
Moreover, a "treatment" or "prevention" regime of a subject with a
therapeutically
effective amount of the compound of the present invention may consist of a
single administration, or alternatively comprise a series of applications. For
example, the compound of the present invention may be administered at least
once a week. However, in another embodiment, the compound may be
administered to the subject from about one time per week to about once daily
for
a given treatment. In yet another embodiment the compound may be
administered more than once daily up to 5 times per day. The length of the
treatment period depends on a variety of factors, such as the severity of the
disease, the age of the patient, the concentration and the activity of the
compounds of the present invention, or a combination thereof. It will also be
appreciated that the effective dosage of the compound used for the treatment
or
prophylaxis may increase or decrease over the course of a particular treatment
or prophylaxis regime. Changes in dosage may result and become apparent by
standard diagnostic assays known in the art. In some instances, chronic
administration may be required.
As used herein, "administered contemporaneously" means that two substances
are administered to a subject such that they are both biologically active in
the
subject at the same time. The exact details of the administration will depend
on
the pharmacokinetics of the two substances in the presence of each other, and
can include administering one substance within 24 hours of administration of
the
CA 02647655 2016-02-26
17
other, if the pharmacokinetics are suitable. Designs of suitable dosing
regimens
are routine for one skilled in the art. In particular embodiments, two
substances
will be administered substantially simultaneously, i.e. within minutes of each
other, or in a single composition that comprises both substances.
"Palliating" a disease or disorder means that the extent and/or undesirable
clinical manifestations of a disorder or a disease state are lessened and/or
time
course of the progression is slowed or lengthened, as compared to not treating
the disorder.
The term "prevention" or "prophylaxis", or synonym thereto, as used herein
refers
to a reduction in the risk or probability of a patient becoming afflicted with
cancer
or manifesting a symptom associated with cancer.
To "inhibit" or "suppress" or "reduce" a function or activity, is to reduce
the
function or activity when compared to otherwise same conditions except for a
condition or parameter of interest, or alternatively, as compared to another
condition. The terms "inhibitor" and "inhibition", in the context of the
present
application, are intended to have a broad meaning and encompass compounds
of the application which directly or indirectly (e.g., via reactive
intermediates,
metabolites and the like) act on cancer.
The term "a cell" as used herein includes a plurality of cells. Administering
a
compound to a cell includes in vivo, ex vivo and in vitro treatment.
In understanding the scope of the present disclosure, the term "comprising"
and
its derivatives, as used herein, are intended to be open ended terms that
specify
the presence of the stated features, elements, components, groups, integers,
and/or steps, but do not exclude the presence of other unstated features,
CA 02647655 2016-02-26
18
elements, components, groups, integers and/or steps. The foregoing also
applies
to words having similar meanings such as the terms, "including", "having" and
their derivatives. Finally, terms of degree such as "substantially", "about"
and
"approximately" as used herein mean a reasonable amount of deviation of the
modified term such that the end result is not significantly changed. These
terms
of degree should be construed as including a deviation of at least 5% of the
modified term if this deviation would not negate the meaning of the word it
modifies.
(II) COMPOUNDS
The present application includes compounds Formula I:
OH
OH
O 7 OH
tC1H
R4 0
wherein
R1 is selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3, CH=CR2R3,
CH200(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH20C(0)R2; and
R2 and R3 are independently selected from H, C1_6a1ky1, C2_6alkenyl, C3.
iocycloalkyl and C6_10aryl
said latter four groups being unsubstituted or substituted with one to 5
groups
independently selected from halo, OH, OCiAalkyl, OC(0)Ci_6alkyl and nitro; and
R4 is selected from H and OH;
CA 02647655 2016-02-26
19
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
In an embodiment of the application R1 is selected from C(0)0H, C(0)0Me,
C(0)H, CH=NH and CH2NH2, CH2OH and CH200(0)CH3. In
another
embodiment of the application R4 is H.
(III) COMPOSITIONS AND USES/METHODS
As hereinbefore mentioned, novel compounds of the Formula I, and
intermediates of the Formula II have been prepared. Accordingly, the present
application includes all uses of the compounds of Formula I and the
intermediates of Formula II including their use in therapeutic methods and
compositions for treatment of cancer, their use in diagnostic assays and their
use
as research tools. In particular, the present application includes the use of
a
compound of Formula I as a medicament.
Another aspect of the application is a use of a compound of the application
for
treating cancer.
Another aspect of the application is a use of a compound of the application
for
the preparation of a medicament for treating cancer.
Another aspect of the application is a compound of the disclosure for use to
treat
cancer.
Also within the scope of the present application is a method of treating
cancer
comprising administering an effective amount of a compound of the application
to
a subject in need thereof.
CA 02647655 2016-02-26
The compounds of the application are suitably formulated into pharmaceutical
compositions for administration to human subjects in a biologically compatible
form suitable for administration in vivo. Accordingly, the present application
further includes a pharmaceutical composition comprising a compound of the
5 application and a pharmaceutically acceptable carrier and/or diluent.
The compositions containing the compounds of the application can be prepared
by known methods for the preparation of pharmaceutically acceptable
compositions, which can be administered to subjects, such that an effective
10 quantity of the active substance is combined in a mixture with a
pharmaceutically
acceptable vehicle. Suitable vehicles are described, for example, in
Remington's
Pharmaceutical Sciences (2000 - 20th edition) and in The United States
Pharmacopeia: The National Formulary (USP 24 NF19) published in 1999). On
this basis, the compositions include, albeit not exclusively, solutions of the
15 substances in association with one or more pharmaceutically acceptable
vehicles or diluents, and contained in buffered solutions with a suitable pH
and
iso-osmotic with the physiological fluids.
The compounds of the application may be used in the form of the free base, in
20 the form of salts and/or solvates. All forms are within the scope of the
application.
In accordance with the methods of the application, the described compounds,
salts or solvates thereof may be administered to a patient in a variety of
forms
depending on the selected route of administration, as will be understood by
those
skilled in the art. The compositions of the application may be administered,
for
example, by oral, parenteral, buccal, sublingual, nasal, rectal, patch, pump
or
transdermal (topical) administration and the pharmaceutical compositions
formulated accordingly.
Parenteral administration includes intravenous,
intraperitoneal, subcutaneous, intramuscular, transepithelial,
nasal,
CA 02647655 2016-02-26
21
intrapulmonary, intrathecal, rectal and topical modes of administration.
Parenteral administration may be by continuous infusion over a selected period
of time.
A compound of the application may be orally administered, for example, with an
inert diluent or with an assimilable edible carrier, or it may be enclosed in
hard or
soft shell gelatin capsules, or it may be compressed into tablets, or it may
be
incorporated directly with the food of the diet. For oral therapeutic
administration,
the compound of the application may be incorporated with excipient and used in
the form of ingestible tablets, buccal tablets, troches, capsules, elixirs,
suspensions, syrups, wafers, and the like.
A compound of the application may also be administered parenterally. Solutions
of a compound of the application can be prepared in water suitably mixed with
a
surfactant such as hydroxypropylcellulose. Dispersions can also be prepared in
glycerol, liquid polyethylene glycols, DMSO and mixtures thereof with or
without
alcohol, and in oils. Under ordinary conditions of storage and use, these
preparations contain a preservative to prevent the growth of microorganisms. A
person skilled in the art would know how to prepare suitable formulations.
Conventional procedures and ingredients for the selection and preparation of
suitable formulations are described, for example, in Remington's
Pharmaceutical
Sciences (2000 - 20th edition) and in The United States Pharmacopeia: The
National Formulary (USP 24 NF19) published in 1999.
The pharmaceutical forms suitable for injectable use include sterile aqueous
solutions or dispersion and sterile powders for the extemporaneous preparation
of sterile injectable solutions or dispersions. In all cases the form must be
sterile
and must be fluid to the extent that easy syringability exists. Ampoules are
convenient unit dosages.
CA 02647655 2016-02-26
22
Compositions for nasal administration may conveniently be formulated as
aerosols, drops, gels and powders. Aerosol formulations typically comprise a
solution or fine suspension of the active substance in a physiologically
acceptable aqueous or non-aqueous solvent and are usually presented in single
or multidose quantities in sterile form in a sealed container, which can take
the
form of a cartridge or refill for use with an atomizing device. Alternatively,
the
sealed container may be a unitary dispensing device such as a single dose
nasal
inhaler or an aerosol dispenser fitted with a metering valve, which is
intended for
disposal after use. Where the dosage form comprises an aerosol dispenser, it
will contain a propellant, which can be a compressed gas such as compressed
air or an organic propellant such as fluorochlorohydrocarbon. The aerosol
dosage forms can also take the form of a pump-atomizer.
Compositions suitable for buccal or sublingual administration include tablets,
lozenges, and pastilles, wherein the active ingredient is formulated with a
carrier
such as sugar, acacia, tragacanth, or gelatin and glycerine. Compositions for
rectal administration are conveniently in the form of suppositories containing
a
conventional suppository base such as cocoa butter.
Compositions for topical administration may include, for example, propylene
glycol, isopropyl alcohol, mineral oil and glycerin. Preparations suitable for
topical
administration include liquid or semi-liquid preparations such as liniments,
lotions, applicants, oil-in-water or water-in-oil emulsions such as creams,
ointments or pastes; or solutions or suspensions such as drops. In addition to
the
aforementioned ingredients, the topical preparations may include one or more
additional ingredients such as diluents, buffers, flavouring agents, binders,
surface active agents, thickeners, lubricants, preservatives, e.g. methyl
hydroxybenzoate (including anti-oxidants), emulsifying agents and the like.
CA 02647655 2016-02-26
23
Sustained or direct release compositions can be formulated, e.g. liposomes or
those wherein the active compound is protected with differentially degradable
coatings, such as by microencapsulation, multiple coatings, etc. It is also
possible to freeze-dry the compounds of the formula I and use the lypolizates
obtained, for example, for the preparation of products for injection.
The dosage administered will vary depending on the use and known factors such
as the pharmacodynamic characteristics of the particular substance, and its
mode and route of administration; age, health, and weight of the individual
recipient; nature and extent of symptoms, kind of concurrent treatment,
frequency of treatment, and the effect desired.
(IV) PROCESSES
The present application also includes a process for preparing a compound of
Formula II
0Pg
H(0)C 0Pg
0 4110
ut-'9
0
NPg
R4 0
wherein R4 is selected from H and 0Pg and each Pg may be the same or
different and represent suitable protecting groups or any two adjacent Pg are
joined to form a suitable cyclic protecting group;
the process comprising:
CA 02647655 2016-02-26
24
(i) reacting a compound of the Formula III with an aluminum acetylide derived
from a compound of the Formula IV, followed by protection to form a compound
of the Formula V, wherein R4 and each Pg is as defined above:
<0
0,, r
--'71."'OPg 0 el 0Pg \r,..0Pg + <00
_________________________________________ IP-
0 R4 0 0Pg
-
Pgr\i''' R4 0Pg
PgN'
III IV V =
,
(ii) reducing the compound of Formula V to form a cis-alkene of the Formula
VI,
wherein R4 and each Pg is as defined above:
<o0 40 0Pg 0Pg
0Pg--
_
R4
R4
.0::
N'
0 Rg
PgN''' \-0
V VI =
,
(iii) reacting the compound of the Formula VI under solid-state, silica gel
catalysis
conditions to form a compound of the Formula VII, wherein R4 and each Pg is as
defined above:
0Pg 0Pg
-- di 0P9 righdh 0P9
R4
0Pg R4 APIM P 0Pg
VP N Pg
\-0
L---0
VI VII =
,
CA 02647655 2016-02-26
(iv) oxidatively cleaving the double bond in the compound of the Formula VII
to
form an intermediate diketone of the Formula VIII which cyclizes to form a
compound of the Formula IX, wherein R4 and each Pg is as defined above:
0Pg 0 0Pg 0Pg
0Pg
/ - 0Pg H(0)C opg
ilh
R4 An" 0Pg R4 le WI
0Pg 0Pg
0
NPg NPg 0 NPg
0
R4 OH
VII VIII IX
5
(v) oxidizing the compound of the Formula IX to form a compound of Formula II,
wherein R4 and each Pg is as defined above:
0Pg 0Pg
OHC opg H(0)C 0Pg
0Pg 0 0Pg
0 NPg NPg
R4 OH R4 0
10 IX II
In an embodiment of the application, the Pg group on the aziridine nitrogen in
the
15 compound of Formula III is Ts. In another embodiment, the Pg groups
on the
oxygen atoms of the compound of Formula III are linked to form a cyclic acetal
group in particular dimethyl acetal. In a further embodiment the protecting
group
CA 02647655 2016-02-26
26
(Pg) added in step (i) to the oxygen following ring opening of the epoxide is
t-
butyldimethylsilyl(TBDMS).
According to a specific embodiment, the process of the present disclosure is
directed to the synthesis of a compound according to Formula II using the
reaction conditions shown in Figure 1. According to this embodiment the
aluminum acetylide of the compound of Formula IV described in (i) above is
formed by the addition of nBuLi and dimethylaluminum chloride in toluene at
reduced temperature, for example about -78 C. Reaction of the compound of
Formula III with the aluminum acetylide of the compound of Formula IV is
followed by protection of the hydroxyl product of the ring opening. In a
particular
embodiment the hydroxyl is protected by reaction with TBDMSOTf in the
presence of base, for example Et3N.
In another embodiment of the process, in (ii), reduction of the alkyne to the
cis
alkene is achieved using BH3 in a suitable solvent at reduced temperature, for
example about 0 C.
In another embodiment of the process, in (iii), the R2-OH ring opening of the
compound of the Formula IV is mediated by copper trifluoromethanesulfonate
(Cu (01-02).
In another embodiment of the process, in (iii), the cis alkene compound of
Formula VI is adsorbed onto silica gel and heated without solvent. In a
particular
embodiment the reaction mixture is heated to 120 C for approximately 24 hours
to provide a compound of Formula VII.
In a further embodiment of the process, in (iv), the compound of Formula VII
is
converted to the intermediate VIII by ozonolysis under suitable reaction
CA 02647655 2016-02-26
27
conditions, for example in Me2S and Me0H at reduced temperature, for example
at about -78 C. In another embodiment, in (iv), the intermediate of Formula
VIII is
formed by a two step process comprising oxidation with osmium tetroxide (0s04)
in the presence of N-methylmorpholine N-oxide (NMO), in CH2Cl2 to give a keto
alcohol intermediate of the Formula XI, followed by reduction, for example
with
sodium borohydride, then periodate cleavage under suitable conditions to give
the intermediate of Formula VIII.
OH 0Pg
0 dim& 0Pg
R4 IRPW 0Pg
NPg
0
XI
In another embodiment of the process, in (v), oxidation of the compound of
Formula IX to the compound of Formula ll is carried out using 2-iodoxybenzoic
acid (IBX) under suitable conditions.
Compounds of the Formulae III and IV are prepared using methods known in the
art [Schilling et al. Can J. Chem. 79:1659 (2001); Endoma, et al. Org. Process
Res. Dev. 6:525 (2002)]
CA 02647655 2016-02-26
28
In another aspect of the application there is include a process for preparing
a
compound of Formula I
OH
R1 OH
O 1110 2 OH
0 NH
R4 0
wherein
R1 is selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3, CH=CR2R3,
CH200(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH200(0)R2 ; and
R2 and R3 are independently selected from H, C1_6alkyl, C2_6alkenyl, C3_
iocycloalkyl and C6_10aryl said latter four groups being unsubstituted or
substituted with one to 5 groups independently selected from halo, OH, OCi-
4alkyl, OC(0)C1_6alkyl and nitro, with the exception that R2 is not H when R1
is
C(0)R2; and
R4 isselected from H and OH,
comprising:
(i) reacting a compound of the Formula II, wherein R4 is selected from H and
0Pg
and each Pg may be the same or different and represent suitable protecting
groups or any two adjacent Pg are joined to form a suitable cyclic protecting
group under conditions to convert the aldehyde moiety to a group, other than
C(0)H, selected from C(0)0R2, C(0)R2, C(0)NR2R3, CH=NR2, CH2NR2R3,
CH2OR2, CH2R2, NR2R3, NHC(0)R2, NHC(0)0R2, NHC(0)NR2R3, CH=CR2R3,
CA 02647655 2016-02-26
29
CH200(0)NR2R3, CH2NHC(0)R2, CH2NHC(0)0R2, CH2CHC(0)NR2R3 and
CH200(0)R2, wherein R2 and R3 are as defined above to form a compound of
the Formula X wherein R1, R4 and each Pg are as defined above:
0Pg 0Pg
H(0)C 0Pg opg
0 40
or r,
g 0Pg
0 NPg 0 NPg
R4 0 R4 0
II X
(ii) removing the Pg groups to form a compound of the Formula I wherein R1 and
R4 are as defined above:
0Pg OH
R1 di OH
0
01-9 40 r"`
g <0 40 7 OH
0 NPg 0 NH
R4 0 R4 0
X
In another aspect of the application there is provided a process for preparing
a
compound of Formula I wherein R1 is COOH, wherein in (i) above the conditions
comprise oxidizing the compound of Formula ll to form, after removal of the Pg
groups in (ii), the compound of the Formula I. In a particular embodiment the
conditions in step (i) comprise mCPBA Na2HPO4 in a suitable solvent at
elevated
temperature, for example, 40 C.
In another aspect of the application there is provided a process for preparing
a
compound of Formula I wherein R1 is C(0)0R2, wherein in (i) above the
CA 02647655 2016-02-26
conditions comprise oxidizing the compound of the Formula II followed by
alkylation with a compound of the Formula R2-LG, wherein LG is a suitable
leaving group, to form, after removal of the Pg groups in (ii), the compound
of the
Formula I.
5
In another aspect of the application there is provided a process for preparing
a
compound of Formula I wherein R1 is CH=NR2 wherein in (i) above the
conditions comprise reacting the compound of the Formula II with an amine of
the Formula R2-NH2 to form, after removal of the Pg groups in (ii), the
compound
10 of the Formula I.
In another aspect of the application there is provided a process for preparing
a
compound of Formula I wherein R1 is CH2NR2R3 wherein in step (i) above the
conditions comprise reacting the compound of the Formula II with an amine,
15 followed by reduction and optional alkylation with a compound of the
Formula R3-
LG, wherein LG is a suitable leaving group, to form, after removal of the Pg
groups in (ii), the compound of the Formula I.
In another aspect of the application there is provided a process for preparing
a
20 compound of Formula I wherein R1 is CH=CR2R3 wherein in step (i) above the
conditions comprise reacting the compound of the Formula II with a
phosphonium ylide of the formula R2R3CH=PPh3 under Wittig reaction conditions
to form, after removal of the Pg groups in (ii), the compound of Formula I.
25 In another aspect of the application there is provided a process for
preparing a
compound of Formula I wherein R1 is C(0)NR2R3, wherein in (i) above the
conditions comprise oxidizing the compound of Formula II as described above
followed by reaction with an amine of the formula NHR2R3 under amide bond
CA 02647655 2016-02-26
31
forming conditions to form, after removal of the Pg groups in (ii), the
compound of
the Formula I.
In another aspect of the application there is provided a process for preparing
a
compound of Formula I wherein R1 is NR2R3, wherein in (i) above the conditions
comprise oxidizing the compound of Formula ll as described above followed by
subjecting the C(0)0H group to Curtius rearrangement to form a compound of
Formula X, wherein R1 is NH2. This amine can be alkylated with various R2
and/or R3 groups. Alternatively, the C(0)0H groups can be converted to an
amide (CONH2) and the amide subjected to Hoffman degradation to produce,
after hydrolysis C-1 amine. These methods are known to those skilled in the
art.
Also included in the present application is a process for preparing a compound
of
Formula I
OH
R1 al OH
O 7 OH
0 fC11-1
R4 0
wherein R1 is C(0)H and R4 is H or OH, comprising removing the Pg groups from
a compound of Formula ll as defined above to form the compound of the
Formula I.
(V) EXAMPLES
The following non-limiting examples are illustrative of the present
application:
Examples 1-10 refer to compounds as shown in Figure 1.
CA 02647655 2016-02-26
32
Example 1: Preparation of (1S,2R,3R,4R,5S,6R)-3,4-(lsopropylidenedioxy)-5-
[(tert-butyldimethylsily0oxy]-6-2-Benzo[1.3]dioxol-5-ylethynyl-(4'-
methylphenylsulfonyl)-7-azabicyclo[4.1.0]heptane V(a)
<o0
9TBS
Cc/
WI 0/N
Tsl\r'
V(a)
To a solution of acetylene 4 (2.74 g, 18.75 mmol) in 18 mL dry toluene was
added at -78 C 8.33 mL of a solution of nBuLi in hexanes (2.25 M, 18.75
mmol).
The solution was stirred for 10 minutes before 18.75 mL of a solution of
Me2AICI
(1.0M in CH2Cl2, 18.75 mmol) was added dropwise. The reaction flask was
allowed to warm to room temperature and stir for 1h. The reaction flask was
then
cooled to -20 C and 18 mL of a solution of epoxide 3 (3.16 g, 9.38 mmol) in
toluene was added dropwise over 20 min. The reaction was stirred at -20 C for
3.5 h before being place in an ice bath and allowed to slowly warm to room
temperature and stir for 12h. The reaction was cooled in an ice bath and
quenched with 1 M HCI. Ethyl acetate (200 mL) was added and the layers where
separated. The aqueous phase was extracted 3 x 100 mL Et0Ac and the
combined organic layers dried over Na2SO4. Concentration under reduced
pressure and purification by flash column chromatography (hexanes:ethyl
acetate, 7:1 to 4:1) afforded alcohol intermediate which was immediately
subjected to protection protocol (2.01 g, 44%); [a]22D -113.05 (c 0.5, CHCI3);
Rf
0.30 (hexanes:ethyl acetate 2:1); IR (film) v3491, 2988, 1163 cm-1; 1H NMR
(300
MHz, CDCI3) 6: 7.78 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 6.91 (dd, J
=
8.2 Hz, 1.8 Hz 1H), 6.83 (d, J = 1.5 Hz, 1H), 6.73 (d, J = 7.9 Hz, 1H), 5.97
(s,
2H), 4.47 (d, J = 6.4 Hz, 1H), 4.22 (dd, J = 6.1, 4.4 Hz, 1H), 3.98 (m, 1H),
3.40
CA 02647655 2016-02-26
33
(d, J = 6.4 Hz, 1H), 3.24 (m, 2H), 3.06 (d, J = 9.6 Hz, 1H), 2.47 (s, 3H),
1.49 (s,
3H), 1.32 (s, 3H) ppm; 13C NMR (75 MHz, CDCI3) 6: 148.1, 147.5, 145.7, 134.2,
130.4, 128.1, 126.4, 116.2, 111.8, 110.3, 108.6, 101.5, 84.2, 83.8, 75.4,
70.1,
68.7, 42.3, 40.5, 31.1, 27.4, 25.2, 21.9 ppm; HRMS (FAB M+) calcd for
025H25N07S 484.1430, found 484.1428.
Alcohol intermediate (240mg, 0.49 mmol) was dissolved in 5 mL of CH2Cl2 and
triethylamine (0.14 mL, 1.04 mmol) was added. The reaction flask was cooled to
-78 C and t-butyldimethylsilytriflate (0.12 mL, 0.546 mmol) was added
dropwise
to the stirring solution. After stirring for 30 minutes at -78 0 the reaction
was
quenched with water and the two phases separated. The aqueous phase was
extracted with CH2012 (2 x 15 mL) and the combined organic solution was
washed sequentially with 5% citric acid (2 mL) and brine (2 mL) before drying
over sodium sulfate. The solvent was removed under reduced pressure and the
residue was purified by flash column chromatography (hexane:ethyl acetate, 9:1
to 2:1) affording V(a) (0.276g, 93%) as a colorless oil.; [a]24D +57.7 (c 0.5,
CHC13); Rf 0.49 (hexanes:ethyl acetate, 2:1); IR (film) v 2953, 2929, 2892,
2856,
1599,1490 cm-1; 1H NMR (300 MHz, CDCI3) 6: 7.83 (d, J = 8.1 Hz, 2H), 7.38 (d,
J
= 8.1 Hz, 2H), 6.94 (d, J = 8.1 Hz, 1H), 6.84 (s, 1H), 6.77 (d, J = 8.1 Hz,
1H),
5.99 (s, 2H), 4.45 (d, J = 5.1 Hz, 1H), 3.83 (m, 2H), 3.26 (m, 2H), 2.84 (d, J
= 7.5
Hz), 2.47 (s, 3H), 1.52 (s, 3H), 1.35 (s, 3H), 0.87 (s, 9H), 0.11 (s, 6H) ppm;
130
NMR (75 MHz, CDC13) 6: 147.8, 147.3, 134.7, 129.8, 127.9, 126.1, 111.6, 109.7,
108.4, 101.3, 86.3, 83.5, 71.7, 43.2, 39.53, 34.58, 27.9, 25.8, 25.79, 25.7,
21.7,
18.12, -4.4, -4.7 ppm; HRMS-E1 Calcd for 0301-136NO7SSi: 540.1481; Found,
540.1487; Anal. calcd for 0311-139NO7SSi C, 62.28; H, 6.58; found C, 62.22; H,
6.73
Example 2: 1S,2R,3R,4R,5S,6R)-3,4-(lsopropylidenedioxy)-5-gtert-
butyldimethylsily0oxyl-6-2-Benzo[1,3]dioxol-5-ylethenyl-(4'-
methylphenylsulfonyl)-7-azabicyclo[4.1.0]heptane VI(a).
CA 02647655 2016-02-26
34
OTBS
OZX
TsNrµ
0
\ ¨0
VI(a)
To a 1.0 M solution of BH3.THF complex (2.5 mL, 2.5 mmol) was added
cyclohexene (0.484 mL, 4.77 mmol) at 0 C. After 10 minutes a heavy
precipitate was formed. The reaction mixture was kept at 0 C for 1 h before
acetylene derivative V(a) (0.356 mg, 0.596 mmol) in 4.5 mL of THF was added.
The reaction mixture was stirred at 0 C until total consumption of starting
material (2 h, TLC) before being quenched with 1 mL HOAc. 60 mL Et0Ac were
added and the reaction mixture was washed with saturated aq. NaHCO3 (2 x 15
mL), H20 (2 x 15 mL), and brine (10 mL) before drying over Na2SO4 The solvent
was removed under reduced pressure and the residue was purified by flash
column chromatography (hexanes:ethyl acetate, 8:1) affording 0.271 g of VI(a)
(76%).; [a123D -26.14 (c 1.0, CHCI3; Rf 0.35 (hexanes:ethyl acetate, 4:1); IR
(film)
v 2986, 2930, 2894, 2856, 1598,1489 cm-1; 1H NMR (300 MHz, CDCI3) 6: 7.78
(d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 6.65 (m, 3H), 6.51 (d, J = 11.7
Hz,
1H), 5.97 (s, 2H), 5.54 (t, J = 11.3 Hz, 1H), 4.43 (d, J = 6, 1H), 3.85 (t, J
= 6.3,
1H), 3.61 (t, J = 7.2 Hz), 3.18 (d, J = 6.6, 1H), 2.91 (m, 2H), 2.44 (s, 3H),
1.52 (s,
3H), 1.33 (s, 3H), 0.79 (s, 9H), 0.02 (s, 3H), -0.04 (s, 3H) ppm; 13C NMR (150
MHz, CDCI3) 6: 147.5, 146.6, 144.6, 134.7, 132.0, 130.3, 129.8, 129.7, 129.5,
128.5, 127.9, 122.5, 109.35, 109.0, 108.1, 100.9, 83.2, 78.0, 72.6, 71.8,
43.7,
39.9, 39.5, 30.1, 27.8, 25.8, 25.79. 25.75, 25.72, 25.51, 23.7, 21.7, 18.1, -
4.3, -
4.7 ppm; HRMS-El Calcd for C311-141NO7SSi: 599.2373; Found, 599.2376; Anal.
calcd for 031 FI41 NO7SSi 0, 62.28; H, 6.58; found 0,61.30; H, 6.63
CA 02647655 2016-02-26
Example 3: N-[(1R,2aS,4aS,5S,5aR,12bR)-5-(tert-Butyl-dimethyl-silanyloxy)-3,3-
dimethy1-1,2a,4a,5,5a,12b-hexahydro-phenanthro[2,3-d][1,3]dioxo1-1-yl]4-methyl-
benzenesulfonamide VII(a)
5
OTBS
sip 0,
0/N
NHTs
0
VII(a)
A flame-dried 25-mL flask was charged with olefin 13 (336 mg, 0.561 mmol) and
10 silica gel which has been activate by heating under vacuum at 120 C
overnight
(1.5 g). The starting materials were suspended in 10 mL freshly distilled
methylene chloride and the solvent removed under reduced pressure. The silica
gel supporting the absorbed reactants was heated externally at 120 C under
nitrogen atmosphere for 24 h, after which time the silica gel was loaded onto
15 flash silica gel column and eluted with hexanes:ethyl acetate, 8:1 ¨ 5:1 to
give
175 mg (52%) of olefin VII(a) as a clear and colorless oil.; [a]23D -123.7 (c
1.0,
CHCI3); Rf 0.35 (hexanes:ethyl acetate, 2:1); IR (film) v3268, 2929, 2887,
2857,
1598,1503, 1485 cm-1; 1H NMR (600 MHz, CDCI3) 5: 7.43 (d, J = 7 Hz, 2H), 7.13
(d, J = 7 Hz, 2H), 6.49 (s, 2H), 6.34 (d, J = 8 Hz, 1H), 5.95 (s, 1H), 5.86
(s, 1H),
20 5.76 (d, J = 8 Hz, 1H), 4.51 (d, J = 7 Hz, 1H), 4.28 (m, 1H), 4.11 (m, 1H),
3.99
(m, 1H), 3.79 (m, 1H), 2.82 (m, 1H), 2.62 (dd, J= 11.1 Hz, J= 5.4 Hz, 1H),
2.40
(s, 3H), 1.43 (s, 3H), 1.33 (s, 3H), 0.89 (s, 9H), 0.11 (s, 3H), 0.07 (s, 3H)
ppm;
130 NMR (150 MHz, CDC13) 6: 146.7, 145.9, 142.1, 138.9, 128.9, 128.6, 127.7,
126.8, 126.3, 126.2, 110.4, 109.2, 107.0, 79.0, 78.3, 70.3, 54.1, 42.5, 41.5,
38.9,
25 27.8, 26.3, 25.7, 25.3, 22.7, 21.5, 18.0, -5.0, -5.0 ppm; HRMS-El Calcd for
CA 02647655 2016-02-26
36
C311-141N07SSi: 599.2373; Found, 599.2370; Anal. calcd for C311-141NO7SSi C,
62.07; H, 6.89; found C, 62.16; H, 6.94
Example 4: N-[(1 R,2aS,4aS,5S,5aS,12bR)-5-(tert-Butyl-dimethyl-silanyloxy)-6-
hydroxy-3,3-dimethy1-7-oxo-1,2a,4a,5,5a,6,7,12b-octahydro-phenanthro[2,3-
d][1,3]dioxo1-1-yl[4-methyl-benzenesulfonamide Xl(a)
OH OTBS
0 0\/
07N
NHTs
0 ,
\-0
To a solution of olefin VII(a) (0.240 mg, 0.4 mmol) in methylene chloride (10
mL)
was added 4-methylmorpholine N-oxide (58 mg, 0.48 mmol). The reaction
mixture was allowed to stir for 10 minutes before the introduction of a single
crystal of osmium tetroxide and two drops of water. The reaction was stirred
until
total consumption of starting material (10 h) before being quenched with a
saturated solution of saturated sodium bisulfite (6 mL). The two layers were
separated and the aqueous phase was extracted with ethyl acetate (3 x 30 mL).
The organic phase was dried over sodium sulfate, filtered, and concentrated in
vacuo to provide hydroxyketone Xl(a) as a white crystalline solid (0.227 g,
89%)
that was used without further purification; Rf 0.42 (hexanes:ethyl acetate,
1:1);
mp >200 C; IR (film) v3478, 3263, 2929, 2857, 1670, 1614, 1504, 1482, 1444,
1386, 1330, 1252, 1218, 1156, 1075, 1039 cm-1; 1H NMR (600 MHz, CDCI3) 6:
7.54 (d, J= 7.8 Hz, 2H), 7.49, (s, 1H), 7.18 (d, J= 7.8 Hz, 2H), 6.70 (s, 1H),
6.07
(s, 1H), 6.00 (s, 1H), 4.79 (d, J = 8.7 Hz, 1H), 4.71 (m, 2H), 4.19 (m, 1H),
4.08
(m, 1H), 3.74 (m, 2H), 3.08 (dd, J = 10.2 Hz, J = 1.8 Hz, 1H), 2.45 (m, 1H),
2.41
(s, 3H), 1.36 (s, 3H), 1.31 (s, 3H), 0.87 (s, 9H), 0.12 (s, 3H), 0.07 (s, 3H)
ppm;
130 NMR (150 MHz, CDCI3) 6: 196.6, 152.5, 147.9, 142.6, 140.5, 138.9, 129.1,
CA 02647655 2016-02-26
37
126.9, 124.7, 111.2, 109.6, 106.9, 102.1, 78.9, 78.7, 70.3, 65.9, 57.9, 49.4,
39.7,
27.9, 25.7, 21.5, 17.95, -5.1 ppm; HRMS-El Calcd for C27H32NO9SSi (M+-57):
574.1567, Found: 574.1572
Example 5: (3aS,3bR,10bR,11 R,12S,12aS)-12-(tert-Butyl-dimethyl-silanyloxy)-
2, 2-dimethy1-5-oxo-4-(toluene-4-sulfony1)-3a,3b, 4,5,10b,11,12, 12a-octahydro-
1, 3,7,9-tetraoxa-4-aza-dicyclopenta[a, h]phenanthrene-11-carbaldehyde II(a).
0 OTBS
H V
(0 010 NIPIF \
0 N,
Ts
0
II(a)
To a 10 mL round bottomed flask was added hydroxyl ketone Xl(a) (0.4 g, 0.63
mmol) and 6 mL of a 1:1 mixture of ethanol:dioxane. The reaction flask was
cooled externally in an ice bath and NaBH4 (24 mg, 0.63 mmol) was added in
one portion. The reaction was removed from the bath and allowed to warm to
room temperature over 1 h. The reaction was quenched with 1 N HCI (4 mL) and
separated. The aqueous phase was extracted with CH2Cl2 (3 x 20 mL) and the
organic phase combined before drying over sodium sulfate. The crude mixture
was concentrated in a 25 mL round bottomed flash and taken up in dioxane (8
mL). A stirring bar was added and the reaction was stirred while sodium
periodate (0.332, 1.5 mmol) was added. The flask was covered to exclude light
and H20 (15 drops) added. The reaction was stirred until total consumption of
starting material (23h) as monitored by TLC. The reaction was quenched with
H20 (10 mL) and separated. The aqueous phase was extracted with CH2Cl2 (3 x
50 mL) and the combined organic phases dried over sodium sulfate.
Concentration provided IX(a).
CA 02647655 2016-02-26
38
To a solution of hemi-aminal IX(a) (394 mg, 0.62 mmol) in N,N-
Dimethylformamide (3 mL) was added 2-lodoxybenzoic acid (520 mg, 1.86
mmol). After total consumption of starting material (by TLC), the reaction
mixture
was diluted with diethyl ether (200 mL) and washed sequentially with saturated
aqueous sodium bisulfite (10 mL), sodium bicarbonate (3 x 10 mL), H20 (10 x
1mL), and brine (10 mL). The organic phase was dried over magnesium sulfate,
filtered and concentrated. The final product was isolated by column
chromatography (hexanes:ethyl acetate, 4:1). Yield: 225 mg, 61%, white solid;
Rf 0.31 (hexanes:ethyl acetate, 4:1); mp >200 C, recrystallized from
hexanes/ethyl acetate 4:1; [a]D21 + 31.67 (c 0.5, CHC13); IR (film) v 2929,
2857,
1725, 1689, 1619, 1505, 1484, 1386, 1361, 1287, 1255, 1220, 1172, 1077, 1036
cm-1; 1H NMR (600 MHz, CDCI3) 6: 9.49 (s, 1H), 8.3 (d, J = 8.2 Hz, 2H), 7.58
(s,
1H), 7.33 (d, J = 8.2 Hz, 2H), 7.28 (s, 1H), 6.55 (s, 1H), 6.04 (d, J = 5 Hz,
2H),
5.81 (dd, J = 8.4 Hz, J = 5.2 Hz, 1H), 4.79 (m, 1H), 4.50 (dd, J = 12.7 Hz, J
= 8.4
Hz, 1H), 4.27 (dd, J = 5.2 Hz, J = 2.7 Hz, 1H), 3.83 (dd, J = 12.6, J = 4.0
Hz, 1H),
3.31 (m, 1H), 2.45 (s, 3H), 1.42 (s, 3H), 1.32 (s, 1H), 0.99 (s, 9H), 0.26 (s,
3H),
0.25 (s, 3H) ppm; 130 NMR (150 MHz, CD013) 6: 196.2, 166.0, 153.0, 147.1,
143.9, 138.8, 137.0, 128.9, 128.8, 110.1, 109.4, 104.2, 102.2, 72.4, 66.6,
65.5,
55.6, 35.4, 31.0, 27.9, 26.9, 25.7, 22.7, 21.7, 18.1, 14.2, -4.7, -4.9 ppm;
HRMS-
El Calcd for C301-136NO9SSi (M+-15): 614.1879, Found: 614.1870; Anal. calcd
for
C31 F139NO9SS C, 59.12; H, 6.24; found C, 59.31; H, 6.29.
Example 6: (3aS,3bR,10bR,11R,12S,12aS)-12-(tert-Butyl-dimethyl-silanyloxy)-
2,2-dimethy1-5-oxo-4-(toluene-4-sulfony1)-3a,3b,4,5,10b,11,12,12a-octahydro-
1,3,7,9-tetraoxa-4-aza-dicyclopenta[a,h]phenanthrene-11-carboxylic acid X(a)
CA 02647655 2016-02-26
39
0 OTBS
HO V
<0
0
N'Ts
WI
0
X(a)
To a solution of aldehyde IX(a) (144 mg, 0.229 mmol) in dry methylene chloride
(5 mL) was added sodium phosphate dibasic (81 mg, 0.57 mmol). The
suspension was stirred while 3-chloroperbenzoic acid (130 mg, 0.57 mmol) was
added. The reaction flask was sealed and heated at 40 C overnight. The
reaction mixture was diluted with methylene chloride (80 mL) and washed
sequentially with saturated aqueous sodium bisulfite (10 mL), sodium
bicarbonate (10 mL), and dried over sodium sulfate. The organic phase was
filtered and concentrated in vacuo to provide carboxylic acid X(a) as a white
crystalline solid (0.125g, 85%) that was used without further purification; ;
Rf 0.1
(hexanes/ethyl acetate, 1:1); mp >200 00; [a]p22 -35.09 (c 1.25, CHC13); IR
(KBr)
v 3246, 2930, 2891, 2857, 1710, 1688, 1619, 1505, 1484, 1361, 1240, 1220,
1172, 1078, 1033 cm-1; 1H NMR (300 MHz, CDCI3) 6: 8.29 (d, J = 8.3 Hz, 2H),
7.53 (s, 1H), 7.32 (d, J = 8.3 Hz, 2H), 7.28 (s, 1H), 6.56 (s, 1H), 6.02 (d, J
= 3
Hz, 2H), 5.77 (dd, J = 8.30 Hz, J = 5.3 Hz, 1H), 4.85 (dd, J = 12.5 Hz, J =
8.4 Hz,
1H)), 4.84 (t, J = 4.7 Hz, 1H), 4.22 (dd J = 5.22, J = 2.8 Hz, 1H), 3.76 (dd,
J =
12.4 Hz, J = 4.1 Hz, 1H), 3.38 (t, J = 3.5 Hz, 1H), 2.45 (s, 3H), 1.40 (s,
3H), 1.27
(s, 1H), 0.96 (s, 9H), 0.21 (s, 6H) ppm; 130 NMR (150 MHz, CDCI3) 6: 174.3,
166.2, 152.8, 146.9, 143.8, 138.9, 137.7, 129.0, 128.8, 122.4, 109.8, 109.2,
103.4, 102.1, 72.8, 68.2, 64.9, 48.0, 35.5, 27.4, 26.9, 25.7, 21.7, 18.0, -
4.9, -5.0
ppm; HRMS-E1 Calcd for C271-130N010SSi (M+-57): 588.1359, Found: 588.1354;
Anal. calcd for 031H39NOloSSi C, 57.65; H, 6.09; found C, 58.01; H, 6.37
CA 02647655 2016-02-26
Example 7: (3aS, 3bR, 1 ObR,11 R,12S,12aS)-12-(tert-Butyl-d imethyl-
silanyloxy)-
2, 2-dimethy1-5-oxo-4-(toluene-4-sulfony1)-3a, 3b,4, 5,10b,11, 12, 12a-
octahydro-
1, 3,7, 9-tetraoxa-4-aza-dicyclopenta [a, h]phenanthrene-11-carboxylic acid
methyl
ester X(b)
0 OTBS
0
(0 0/\
0
,Ts
WI
5 0
X(b)
To a solution of carboxylic acid X(a) (45 mg, 0.069 mmol) in diethyl ether (3
mL)
was added freshly prepared diazomethane solution in diethyl ether until the
10 persistence of yellow color and total consumption of starting material
(by TLC).
The reaction was quenched with one drop of acetic acid followed by saturated
sodium bicarbonate solution (1 mL), diluted with diethyl ether (30mL) and
washed with saturated sodium bicarbonate solution (2 x 1 mL), dried over
magnesium sulfate, filtered and concentrated. The crude reaction mixture was
15 passed through short silica plug using hexane/ethyl acetate 1:1 as
eluent and
concentrated to provide methyl ester X(b) that was used without further
purification. Yield: 38mg, 83%, white crystalline solid; Rf 0.45
(hexanes:ethyl
acetate, 1:1); mp >200 C; [a]D22 - 25.6809 (c 0.75, 0H013); IR (KBr) v 2986,
2953, 2931, 2896, 2858, 1739, 1692, 1620, 1598, 1505, 1485, 1361, 1289, 1264,
20 1173 cm-1; 1H NMR (300 MHz, CDCI3) 6: 8.30 (d, J = 8.4 Hz, 2H), 7.55 (s,
1H),
7.32 (d, J = 8.3 Hz, 2H), 6.58 (s, 1H), 6.02 (s, 2H), 5.78 (dd, J = 8.30 Hz, J
= 5.4
Hz, 1H), 4.9 (dd, J = 12.5 Hz, J = 8.3 Hz, 1H), 4.78 (t, J = 3.0 Hz, 1H), 4.24
(dd J
= 5.36 Hz, J = 2.9 Hz, 1H), 3.79 (dd, J = 12.4, J = 4.2 Hz, 1H), 3.56 (s, 3H),
3.40
(t, J = 3.7 Hz, 1H), 2.45 (s, 3H), 1.41 (s, 3H), 1.35 (s, 1H), 0.98 (s, 9H),
0.24 (s,
25 3H), 0.23 (s, 3H) ppm; 13C NMR (75 MHz, CDCI3) 6:169.4, 166.3, 152.8,
146.8,
143.7, 139.0, 138.2, 128.9, 128.8, 122.4, 109.8, 109.2, 103.5, 102.0, 72.9,
68.2,
CA 02647655 2016-02-26
41
65.2, 51.9, 48.1, 35.9, 27.5, 26.8, 25.7, 21.6, 18.0, -4.8, -4.9 ppm; HRMS-EI
Calcd for C28H32N010SS1 (M+-57): 602.1516, Found: 602.1516
Example 8: (3aS,3bR,10bR,11 R,12S,12aS)-12-(tert-Butyl-dimethyl-silanyloxy)-
2,2-dimethy1-5-oxo-3a,3b,4,5,10b,11,12,12a-octahydro-1,3,7,9-tetraoxa-4-aza
dicyclopenta[a,h]phenanthrene-11-carboxylic acid methyl ester X(c)
0 OTBS
Alk Ov
(0 a/ \
NH
0 WI
0
X(C)
To a solution of X(b) (52 mg, 0.079 mmol) in dry THF (1 mL) at -50 C was
added
a 0.5 M solution of Na/naphthalene in DME until a green color persisted and
total
consumption of starting material was observed (by TLC). The solution was
stirred for 10 minutes before the reaction was quenched with saturated aqueous
ammonium chloride solution (1 mL). The reaction was warmed to room
temperature and extracted with CH2Cl2 (6 x 15 mL). The combined organic
phase was dried over sodium sulfate, filtered, and concentrated. The final
product was isolated by column chromatography (hexanes:ethyl acetate, 5:1 to
2:1). Yield: 23 mg, 58%, clear oil; R10.28 (hexanes:ethyl acetate, 1:1);
[a]D22 ¨
14.51 (c 0.50, CHCI3); IR (film) v3320, 2952, 2930, 2895, 2857, 1743, 1669,
1619, 1504, 1484, 14601385, 1369, 1321, 1288, 1260, 1222 cm-1; 1H NMR (600
MHz, CDCI3) 6: 7.62 (s, 1H), 6.56 (s, 1H), 6.02 (s, 2H), 5.96 (s, 1H), 4.86
(t, J =
2.6, 1H), 4.41 (dd, J= 13.6 Hz, J= 8.2 Hz, 1H), 4.18 (dd, J= 8.25 Hz, J= 4.8
Hz,
1H), 4.11 (m, 1H), 3.66 (s, 3H), 3.40 (dd, J = 13.6 Hz, J = 3.7 Hz, 1H), 3.33
(m,
1H), 2.06 (s, 1H), 1.40 (s, 3H), 1.37 (s, 3H), 0.92 (s, 9H), 0.21 (s, 3H),
0.20 (s,
3H) ppm; 130 NMR (150 MHz, CDCI3) 6:169.6, 165.4, 151.4, 146.6, 135.4,
CA 02647655 2016-02-26
42
122.6, 110.5, 108.6, 103.3, 101.7, 69.2, 53.1, 51.9, 45.9, 33.4, 27.6, 26.5,
25.7,
17.9, -4.9, -5.0 ppm; HRMS-El Calcd for 025H35NO8Si (M+): 505.2132, Found:
505.2131
Example 9: (1 R, 2S, 3R,4 S, 4aR, 1 1 bR)-2, 3, 4-Trihydroxy-6-oxo-1 , 2, 3,4,
4a, 5,6,1 1 b-
octahydro-f1,3]dioxolo[4,5-j]phenanthridine-1-carboxylic acid methyl ester
1(a)
0 OH
OH
(0 OH
NH
0
0
1(a)
To a solution of the detosylated methyl ester X(c) (23 mg, 0.046 mmol) in
methanol (2 mL) was added 3% HCI in methanol (0.5 mL). The reaction mixture
was stirred until total consumption of starting material (3 days). The solvent
was
removed under reduced pressure and the residue was purified by flash column
chromatography using a 30:1 to 20:1 gradient of methylene chloride:methanol as
eluent to provide methyl ester 19 (11 mg, 69%) as a white crystalline solid.;
mp
>200 C (methylene chloride: methanol); Rf 0.06 (methylene chloride/methanol,
20:1); [a]D22 + 24.53 (c 0.25,Me0H); IR (KBr) v3311, 2913, 1732, 1648, 1609,
1497, 1462, 1349, 1259, 1037 cm-1; 1H NMR (300 MHz, Me0D) 6: 7.33 (s, 1H),
6.59 (s, 1H), 5.93 (d, J = 3.7, 2H), 4.50 (t, J = 3.12, 1H), 4.21 (dd, J =
13.1 Hz, J
= 10.1 Hz, 1H), 3.86 (m, 1H), 3.79, (dd, J = 10.1, J = 3.0, 1H), 3.51 (s, 3H),
3.39
(m, 1H), 3.29 (dd, J = 13.1, J = 4.1, 1H) ppm; 130 NMR (75 MHz, Me0D) 6:
170.8, 166.4, 151.7, 146.4, 137.3, 121.7, 106.9, 103.7, 101.8, 72.2, 71.9,
70.9,
51.4, 50.6, 44.8, 35.4 ppm, HRMS-FAB: (m/z) (M + H): Calcd for 016F117N08:
352.1032, Found: 352.0941
CA 02647655 2016-02-26
43
Example 10: (1 R, 2S, 3R, 4S, 4aR, 11bR)-2, 3,4-Trihydroxy-6-oxo-
1,2,3,4,4a,5,6,11b-octahydro-[1 ,3]dioxolo[4,5-llphenanthridine-1-carboxylic
acid
1(b)
0 OH
HO gib OH
<0 OH
0 WI NH
0
1(b)
To a solution of 1(a) (6 mg, 0.017 mmol) in methanol (0.5 mL) was added LiOH
(1
mg, 1.5 mmol). The reaction mixture was heated at 45 C and stirred until
total
consumption of starting material (2 days) as monitored by TLC. The reaction
was made slightly acidic with the addition of HCI (5 drops, 1M) and
concentrated
to provide acid 20 (5 mg, 95%) as a white crystalline solid.; mp >200 C; Rf
0.06
(methylene chloride:methanol, 4:1); IR (KBr) v 3412, 2920, 2115, 1641, 1505,
1471, 1409, 1462, 1363, 1267 cm-1; 1H NMR (300 MHz, Me0D) 6: 7.41 (s, 1H),
6.72 (s, 1H), 6.02 (d, J = 3.7, 2H), 4.64 (t, J = 3.12, 1H), 4.35 (dd, J =
13.1 Hz, J
= 10.1 Hz, 1H), 3.99 (m, 1H), 3.89, (dd, J = 10.1, J = 3.0, 1H), 3.45 (m, 1H),
3.38
(m, 1H) ppm; 130 NMR (75 MHz, Me0D) 6: 172.1, 166.4, 151.7, 146.4, 137.6,
121.7, 106.8, 103.8, 101.8, 72.4, 71.9, 71.1, 51.34, 45.03, 35.4 ppm.
Example 11: (3aS,3bR,10bR,11S,12S,12aS)-12-(tert-Butyl-dimethyl-silanyloxy)-
11-hydroxymethy1-2,2-dimethy1-4-(toluene-4-sulfony1)-3b,4,10b,11,12,12a-
hexahydro-3aH-1,3,7,9-tetraoxa-4-aza-dicyclopenta[a,h]phenanthren-5-one X(d)
CA 02647655 2016-02-26
44
OH OTBS
Ov
(0 o/N
0 Ts
X(d)
To a solution of II(a) (175 mg, 0.278 mmol) in Et0H/dioxane (1:1, 5 mL) at 0 C
was added NaBH4 (3 mg, 0.08 mmol). The reaction was allowed to warm to room
temperature over 1.5 hours before being quenched with a solution of saturated
NH4CI (1 mL). The Et0H/dioxane mixture was removed under reduced pressure
and the aqueous residue was extracted with CH2Cl2 (3 x 25 mL). The organic
phases were combined, dried over sodium sulfate, filtered, and concentrated to
provide alcohol X(d) which was used without further purification. Yield: 150
mg,
85%, clear oil; ; Rf 0.44 (hexanes:ethyl acetate, 1:1); [a]D22 ¨ 47.72 (c
1.50,
CHCI3); IR (film) v3547, 2986, 2932, 2586, 1692, 1616, 1594, 1508, 1481, 1360
cm-1; 1H NMR (300 MHz, CDCI3) 6: 8.28 (d, J = 8.3 Hz, 2H), 7.54 (s, 1H), 7.30
(d, J = 8.2 Hz, 2H), 6.77 (s, 1H), 6.04 (d, J = 1.6 Hz, 2H), 5.65 (dd, J = 8.8
Hz, J
= 5.6 Hz, 1H), 4.57 (d, J = 1.8 Hz, 1H), 4.32 (d, J = 4.6 Hz, 1H), 4.16 (dd J
=
12.8, J = 8.9 Hz, 1H), 3.78 (m 2H), 3.38 (dd, J = 11.3 Hz, J = 3.6 Hz, 1H),
2.55
(bs, 1H), 2.43 (s, 3H), 1.96 (bs, 1H), 1.43 (s, 3H), 1.35 (s, 3H), 0.96 (s,
9H), 0.20
(s, 6H) ppm; 13C NMR (75 MHz, 000I3) 6: 166.4, 153.1, 147.1, 143.7, 138.9,
137.0, 129.0, 128.7, 123.2, 109.1, 108.7, 104.9, 102.1, 73.1, 67.3, 64.8,
60.0,
46.9, 37.4, 28.1, 26.3, 25.8, 21.6, 18.0, -4.8, -4.9 ppm; HRMS-El Calcd for
031H41NO9SSi (M+-15): 616.2032, Found: 616.2032.
Example 12: Acetic acid (3aS, 3 bR,10bR,11 S, I 2S,12aS)-12-(tert-butyl-
dimethyl-
silanyloxy)-2, 2-d imethy1-5-oxo-4-(toluene-4-sulfony1)-3a, 3b, 4,5, 1 Ob, 11
, 12,12a-
octahydro-1,3,7,9-tetraoxa-4-aza-dicyclopenta[a,h]phenanthren-11-ylmethyl
ester
X(e)
CA 02647655 2016-02-26
OAc OTBS
Ov
<0 0/\
0 Ts
0
X(e)
To a solution of X(d) (150 mg, 0.237 mmol) in dry CH2Cl2 (10 mL) was added
5 DMAP (1.5 mg, 0.012 mmol), followed by pyridine (0.1 mL, 1.187 mmol).
Ac20
(45 ,uL, 0.475 mmol) was added and the reaction was stirred for 1 hour before
being quenched with saturated sodium bicarbonate (5 mL). The reaction was
diluted with Et20 (75 mL) and separated. The aqueous layer was extracted with
Et20 (2 x 75 mL) and the combined organic phases were washed with H20 (10
10 mL), brine (10 mL), dried over magnesium sulfate, filtered, and
concentrated.
The final product was isolated by column chromatography using 5:1 mixture of
hexanes: ethyl acetate as eluent. Yield: 128 mg, 81%, clear oil; Rf 0.51
(hexanes/ethyl acetate, 1:1); [a]p22 ¨ 41.081 (c 3.0, CHCI3); IR (film) v2988,
2952, 2930, 2858, 1742, 1694, 1619, 1598, 1505, 1485, 1395, 1362, 1254; 1H
15 NMR (600 MHz, CDCI3) 6: 8.29 (d, J = 8.3 Hz, 2H), 7.54 (s, 1H), 7.31 (d,
J = 8.2
Hz, 2H), 6.84 (s, 1H), 6.03 (d, J = 12.6 Hz, 2H), 5.62 (dd, J = 8.7 Hz, J =
5.6 Hz,
1H), 4.50 (s, 1H), 4.31 (d, J= 5.3 Hz, 1H), 4.18 (t, J = 11.1 Hz, 1H), 3.97
(dd, J=
13.0 Hz, J = 8.8 Hz, 1H), 3.85 (dd, J = 11.0 Hz, J = 3.6 Hz, 1H), 3.80 (dd, J
=
13.0, J = 4.2, 1H), 2.7 (d, J = 5.2, 1H), 2.44 (s, 3H), 2.03 (s, 3H), 1.42 (s,
3H),
20 1.36 (s, 3H), 0.96 (s, 9H), 0.19 (s, 1H); 130 NMR (150 MHz, CDCI3)
6:170.7,
166.2, 153.2, 147.3, 143.8, 138.8, 136.2, 129.1, 128.7, 123.2, 108.9, 108.8,
105.0, 102.2, 78.4, 73.0, 66.3, 64.4, 60.8, 44.0, 37.0, 28.3, 26.2, 25.8,
25.78,
25.75, 25.6, 21.6, 20.8, 18.1, -4.8, -5.0; HRMS-El Calcd for 032H40NO10SSi (M-
15): 658.2142, Found: 658.2152
CA 02647655 2016-02-26
46
Example 13: ((3aS,3bR,10bR,11 S,12S,12aR)-12-hydroxy-2,2-dimethy1-5-oxo-
3a,3b,4, 5,10b,11,12,12a-octahydrobis[1, 3]dioxolo[4, 5-c:4', 5'-
j]phenanthridin-11-
yl)methyl acetate
0
OH
OK
(OOH
0
To a solution of X(e) (137 mg, 0.203 mmol) in dry DME (5 mL) at -78 C was
added a 0.5 M solution of Na/naphthalene in DME until a green color persisted
and total consumption of starting material was observed (by TLC). The solution
was stirred for 10 minutes before the reaction was quenched with saturated
aqueous ammonium chloride solution (2 mL). The reaction was warmed to room
temperature, concentrated to remove DME, and extracted with CH2Cl2 (3 x 40
mL). The combined organic phase was dried over sodium sulfate, filtered, and
concentrated. The resulting crude acetate was taken up in THF (2.5 mL) and
cooled to 000. TBAF (0.1 mL, 1M in THE) was added dropwise over 2 minutes.
The reaction was stirred until total consumption of starting material was
observed
(TLC) before the stirring bar was removed, silica (200 mg added), and the
reaction concentrated to dryness. The final product was isolated by column
chromatography using 1:1 mixture of hexanes: ethyl acetate as eluent. Yield:
61
mg, 74%, white solid; mp >200 C Rf 0.059 (hexanes/ethyl acetate, 1:1); [a]D22
¨
38.301 (c 1.35, DMS0); IR (film) v3303, 2982, 2922, 2901, 2853, 1734, 1655,
1652, 1612, 1483, 1459, 1364, 1246, 1235, 1215; 1H NMR (300 MHz, DMSO) 6:
7.76 (s, 1H), 7.35 (s, 1H), 7.03 (s, 1H) 6.09 (d, J = 1.8, 2H), 5.48 (d, J =
4.2, 1H),
4.35, (s, 1H), 4.24(d, J= 5.3, 1H), 4.19 ¨ 4.10 (m, 3H), 3.46 (dd, J= 14.0 Hz,
J=
8.2 Hz, 1H), 3.21 (dd, J = 13.9 Hz, J = 3.8 Hz, 1H), 2.80 (bs, 1H), 2.02 (s,
1H),
CA 02647655 2016-02-26
47
1.39 (s, 3H), 1.31 (s, 3H); 130 NMR (75 MHz, DMSO) 6:170.9, 163.9, 151.3,
146.7, 134.5, 124.2, 108.9, 107.6, 105.4, 102.2, 77.9, 77.2, 65.3, 61.2, 53.5,
34.7, 28.3, 26.4, 21.2; HRMS-E1 Calcd for 020H23N08 (Mt): 405.1424, Found:
405.1431
Example 14: ((1S,2S,3R,4S,4aR,11bR)-2 ,3,4-trihydroxy-6-oxo-
1,2,3,4,4a ,5,6,11b-octahyd ro41,3]dioxolo[4,5-j]phenanthridin-1-yl)methyl
acetate
1(c)
0
)LO OH
Ai OH
(0 40/ OH
NH
0
0
1(c)
To a solution of the acetate of Example 13 (21 mg, 0.052 mmol) in Me0H (1 mL)
was added an HCI solution (3 % in Me0H, 3 mL). The reaction was stirred until
total consumption of starting material as monitored by TLC (3 h) before being
quenched to basic pH with saturated sodium bicarbonate solution. The crude
reaction mixture was concentrated to dryness. The final product was isolated
by
column chromatography (methlene chloride: methanol, 5:1). Yield: 6 mg, 45%,
white solid; mp >200 00; Rf 0.41 (methlene chloride: methanol, 5:1); [a]D22
97.32
(c 0.3, DMSO); 1H NMR (600 MHz, DMSO) 6:7.36 (s, 1H), 7.01 (s, 1H), 6.76, (s,
1H), 6.10, (s, 2H), 5.14, (bs, 3H), 4.38 (t, J = 10.7 Hz, 1H), 4.15 ¨ 4.10 (m,
2H),
3.84 (s, 1H), 3.70 (dd J = 9.8 Hz, J = 2.9 Hz, 1H), 3.50 (dd J = 13.2 Hz, J =
9.9
Hz, 1H), 3.27 (dd J = 13.3 Hz, J = 4.0 Hz, 1H), 2.69 (bs, 1H), 2.03 (s, 3H)
ppm;
130 NMR (150 MHz, DMSO) 6: 171.0, 164.1, 151.3, 146.6, 135.3, 123.8, 107.5,
CA 02647655 2016-02-26
48
105.5, 102.2, 73.1, 71.3, 69.1, 61.9, 51.6, 36.9, 21.3 ppm; HRMS-FAB Calcd for
017H20N08 (M + 1): 366.0988, Found: 366.1088.
Example 15: (1 S,2S,3R,4S,4aR,1 1 b R)-2,3,4-trihydroxy-1-
(hydroxymethyl)-
1 ,2,3,4,4a,5-hexahydro-[1 ,3]dioxolo[4,5-ilphenanthridin-6(1 1 bH)-one 1(d)
OH OH
OH
(0 Si OH
NH
0
0
1(d)
To a solution of acetate 1(c) (25 mg, 0.062 mmol) at 0 C, in Me0H (5 mL) was
added K2003 (40 mg, 0.62 mmol) and H20 (1 mL). The suspension was stirred
until total consumption of starting material (TLC) before being quenched with
HCI
(4 drops, 6N). The reaction was allowed to warm to room temperature and stir
(4
h). The pH of the reaction was made basic with the addition of saturated
sodium
bicarbonate solution and the methanol removed under reduced pressure. The
resulting aqueous phase was concentrated overnight on a freeze-dryer. The
salts were triturated with Me0H (5 x 5 mL) and the Me0H washes collected and
concentrated. The final product was isolated by column chromatography
(methlene chloride: methanol, 5:1). Yield: 15 mg, 75%, white solid; mp >200
C;
Rf 0.20 (methlene chloride: methanol, 5:1); [a]D22 90.91 (c 0.25, DMSO); IR
(film) v3361, 2916, 1646, 1608, 1503, 1460, 1385, 1361, 1252; 1H NMR (600
MHz, DMSO) 6: 7.34 (s, 1H), 6.97 (s, 1H), 6.66, (s, 1H), 6.09, (d, J = 0.78,
2H),
5.04 ¨4.97, (m, 3H), 4.47 (dd J = 6.6 Hz, J = 3.8 Hz, 1H), 4.19 (s, 1H), 3.89
(q, J
= 7.86 Hz, 1H), 3.82 (s, 1H), 3.69 ¨ 3.64 (m, 1H), 3.42 (dd J = 13.2 Hz, J =
9.9
Hz, 1H), 3.39 ¨ 3.32 (m, 1H), 3.15, (dd J = 13.3 Hz, J = 4.5 Hz, 1H), 2.41 (s,
1H)
ppm; 130 NMR (150 MHz, DMSO) 6: 164.2, 151.2, 146.3, 136.3, 123.7, 107.4,
CA 02647655 2016-02-26
49
105.6, 102.1, 73.3, 71.6, 69.7, 57.8, 51.8, 44.4, 37.3 ppm; HRMS-FAB Calcd for
C15H18N07 (M + 1): 324.1085, Found: 324.1084.
Example 16: Anti-cancer activity
The following cell lines and previously published research methods
[Siedlakowski, et al. Cancer Biology and Therapy, 7:376-384 (2008); Kekre, et
al.
Cancer Chemotherapy and Pharmacology, 56:29 (2005)] were used:
Cancer cell lines:
Human leukemia cells (Jurkat cells)
Human neuroblastoma cells (shsy5y cells)
Human Melanoma cells.
All these cells were obtained from ATCC, USA.
Normal non-cancerous cells:
Human peripheral nuclear blood cells (prepared from the blood donated by a
helthy volunteer)
Normal human fibroblasts (obtained from Corriel Cell repository, USA)
Methods and assays:
Apoptosis characterization:
1. Morphology: Nuclear condensation as observed by bright Hoechst staining.
2. Annexin-V binding assay.
Results:
After correction with solvent control, it was observed that compound 1(c)
induced
apoptosis selectively in human cancer cell lines. Compound 1(d) also showed
the
apoptosis-inducing activity, but to a lower extent. The ED50 for 1(c) was 500
nM.
These compounds did not induce apoptosis in normal non-cancerous cells. (See
Figures 2 and 3 for further results in Jurkat cells).