Note: Descriptions are shown in the official language in which they were submitted.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
1
TISSUE ENGINEERING WITH HUMAN EMBRYONIC STEM CELLS
STATEMENT OF GOVERNMENT INTEREST
This disclosure was developed at least in part using funding from the National
Institutes of Health, Grant Number RO1 AR47839-2, and the National Science
Foundation-
Integrative Graduate Education and Research Traineeship Program, Grant Number
DGE-
0114264. The U.S. government may have certain rights in the invention.
CROSS-REFERENCE TO RELATED APPLICATIONS
This application a continuation-in-part of International Application No.
PCT/US2005/24269 filed July 8, 2005, which claims the benefit of U.S.
Provisional
Application Serial No. 60/586,862 filed on July 9, 2004; this application also
claims the
benefit of U.S. Provisional Application No. 60/789,851, filed April 5, 2006,
and also claims
the benefit of U.S. Provisional Application No. 60/789,853, filed April 5,
2006, and also
claims the benefit of U.S. Provisional Application No. 60/789,855, filed April
5, 2006 all of
which are incorporated herein by reference.
BACKGROUND
Tissue engineering is an area of intense effort today in the field of
biomedical
sciences. The development of methods of tissue engineering and replacement is
of particular
importance in tissues that are unable to heal or repair themselves, such as
articular cartilage.
Articular cartilage is a unique avascular, aneural and alymphatic load-bearing
live tissue,
which is supported by the underlying subchondral bone plate. Articular
cartilage damage is
common and does not normally self-repair. Challenges related to the cellular
component of
an engineered tissue include cell sourcing, as well as expansion and
differentiation. Findings
of recent well-designed studies suggest that autologous chondrocyte
implantation is the most
efficacious technique of repairing symptomatic full-thickness hyaline
articular cartilage
defects, which engender a demand for cell-based strategies for cartilage
repair. Further
studies have also attempted to engineer cartilage via the combination of
biodegradable or
biocompatible scaffolds with differentiated chondrocytes. According to these
studies, it is
unlikely that a sufficient supply of differentiated chondrocytes will be
available for clinical
applications.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
2
To overcome the deficiency in the supply of differentiated chondrocytes,
alternate
sources of cells from tissues other than cartilage have been researched. A
number of
researchers have investigated various adult tissues including bone marrow,
muscle, and
adipose tissue as alternative cell sources for cartilage tissue engineering.
However,
autologous procurement of these tissues has potential limitations. Stem cells
represent a
valuable source for this purpose.
A progenitor cell, also referred to as a stem cell, is generally considered an
undifferentiated cell that can give rise to other types of cells. A progenitor
cell has the
potential to develop into cells with a number of different phenotypes.
Differentiation usually
involves the selective expression of a subset of genes, which vary from cell
type to cell type,
without the loss of chromosomal material. Thus, the lineal descendants of a
progenitor cell
can differentiate along an appropriate pathway to produce a fully
differentiated phenotype.
All differentiated cells have, by definition, a progenitor cell type, for
example, neuroblasts for
neurons and germ cells for gamete cells.
Progenitor cells share the three following general characteristics: (1) the
ability to
differentiate into specialized cells, i.e., not terminally differentiated, (2)
the ability to
regenerate a finite number of times, and (3) the ability to relocate and
differentiate where
needed. Progenitor cells may give rise to one or more lineage-committed cells,
some of which
are also progenitor cells, that in turn give rise to various types of
differentiated cells and
tissues. Progenitor cells generally constitute a small percentage of the total
number of cells
present in the body and vary based on their relative level of commitment to a
particular
lineage. Because progenitor cells have the ability to produce differentiated
cell types, they
may be useful, among other things, for replacing the function of aging or
failing cells in many
tissues and organ systems.
There are three major classes of progenitor cells, based on what they have the
potential to become. The earliest cells, from the fertilized egg through the
first few division
cycles, are totipotent. A totipotent cell has the gentic potential to create
every cell of the
body, including the placenta and extra-embryonic tissues.
Next come the pluripotent, or multipotent, cells, which can become more than
one
kind of cell, but do not have the potential to become all cell types. A
pluripotent cell (i.e., an
embroyonic progenitor cell) has the potential to create every cell of the
body, but not the
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
3
necessary placenta and extra-embryonic tissues required to form a human being.
Pluripotent
cells can be isolated from embryos and the germ line cells of fetuses. A
multipotent cell, or a
multipotent adult progenitor cell ("MAPC"), can give rise to a limited number
of other
particular types of cells. Multipotent cells are found in both developing
fetuses and fully
developed human beings and have been observed to develop into a variety of
cell types such
as cardiomyocytes, hepatocytes, and epithelial cells. For example,
hematopoietic cells (blood
cells) in the bone marrow are multipotent and give rise to the various types
of blood cells,
including red blood cells, white blood cells, and platelets. Unlike
pluripotent cells,
multipotent cells are often present in a fully developed human being. But
multipotent cells
may only be present in minute quantities, and their numbers can decrease with
age.
Multipotent cells from a specific patient may take time to mature in culture
in order to
produce adequate amounts for treatment.
And finally there are unipotent cell types, such as the muscle-cell
progenitors. These
still have the quality of regenerating, but may be more differentiated or
committed to a
certain cell type.
DRAWINGS
Some specific example embodiments of the disclosure may be understood by
referring, in part, to the following description and the accompanying
drawings.
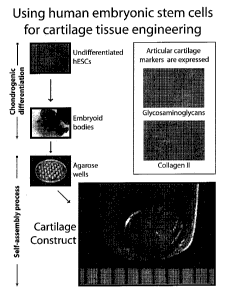
FIGURE 1 is a schematic diagram describing one example of a method of using
human embryonic stem cells to tissue engineer articular cartilage using a
process that does
not involve the use of exogenous scaffolds.
FIGURE 2 is an image of embryoid bodies after four weeks of culture, according
to
one embodiment of the present disclosure
FIGURE 3 is a photomicrograph image of embryoid body morphology after analysis
with A) immunohistochemistry for collagen type II, and B) alcian blue staining
for
glycosaminoglycans.
FIGURE 4 is an image of the gross morphology of constructs after 2 weeks of
tissue
engineering. Figure 4A shows a construct with a thickness of approximately 1
mm. Figure 4B
shows a construct with a diameter of 3 mm. Distance between each bar is 1 mm.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
4
FIGURE 5 is a photomicrograph image of constructs made with A) 0% serum and B)
20 % serum. Shown are collagen type II (left column) and glycosaminoglycan
(right column)
stained constructs.
FIGURE 6 shows chondrogenic differentiation of BGO1 V and H9. Embryoid Bodies
were treated with one of two differentiation regimens. Collagen type II and
Alcian blue
staining were observed in both cells lines with all serum levels tested.
Staining at t=4 wks is
shown for representative embryoid bodies. FIGURE 6A shows results for 20% FBS
BGOl V
Embryoid Bodies, and FIGURE 6B shows results for 0% FBS H9 Embryoid Bodies.
FIGURE 7 shows representative constructs for each cell line. A) Self-assembled
construct (t=2 wks of self-assembly) made from chondrogenically-differentiated
BGO1 V
Embryoid Bodies. For 2 wks of self-assembly, constructs received TGF-(31 + IGF-
I. This
particular construct received no serum. Constructs that received 1% and 20%
serum looked
similar to this construct. B) Self-assembled construct (t=4 wks of self-
assembly) made from
chondrogenically-differentiated H9 cells. Pictured is a construct that
received 20% FBS and
TGF- 01 + IGF-I throughout self-assembly. Constructs that received 0% and 1%
serum
looked similar to this construct. The markings are 1 mm apart.
FIGURE 8 shows the expression of collagen type II in self-assembled
constructs.
After 2 wks of self-assembly, these representative constructs exhibit collagen
type II, which
was seen after the differentiation phase of 4 wks, suggesting that the
chondrocytic phenotype
is maintained. A) Shown is collagen type II staining for the sample pictured
in FIGURE 7A
(0% FBS). B) This construct received 20% FBS but the same differentiation
agents and
growth factors as (A). Both were BGO l V constructs.
FIGURE 9 shows analysis for chondrogenic differentiation of hESCs at t=4 wks
FIGURE 9A shows collagens type I and II detected in all three differentiation
conditions with
immunohistochemistry at t=4 wks (l Ox). The EBs in all groups appeared highly
hydrated and
cellular with a loosely organized ECM. Due to this, obtaining good frozen
sections for these
structures was challenging. Calcified tissue (i.e., bone), muscle, adipose
were not detected
(data not shown). FIGURE 9B shows that SOX-9 transcription factor was detected
in all
three differentiation regimens at t=4 wks (green). The blue fluorescence is a
Hoechst stain for
the nucleus. While CM and D1 cells were approximately the same size and had a
similar
rounded shape as the positive control of native articular chondrocytes (bottom
row, left), D2
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
cells were larger and appeared fibroblastic. The negative control of MEFs
(bottom row, right)
did not stain for SOX-9. The white bar is 10 m (40x).
FIGURE 10 shows gross morphology and histology of self-assembled constructs at
t=8 wks. FIGURE l0A shows that dissociated cell (DC) constructs appeared more
uniform
5 than embryoid body (EB) constructs. The DC group also held their shape when
manipulated,
while the EB group did not. D1 constructs were generally smaller than
constructs from the
other two groups, as shown in the pictures and the morphological measurements.
EB
constructs were engineered larger (5-mm molds vs 3-mm molds for DC constructs)
because
the EBs at t=4 wks were too large for the 3-mm wells. FIGURE l OB shows that
collagens I
and II (top two rows) were detected in CM and D2 groups with
immunohistochemistry at t=8
wks, regardless of self-assembly mode (EB or DC) (lOx). D1 constructs had
collagen type II
but did not demonstrate much collagen type I staining. Intense picrosirius red
and spotty
Alcian blue stains (4x) are shown in the bottom row for each differentiation
condition.
Calcified tissues (i.e., bone), muscle, and adipose were not detected at t=8
wks (data not
shown).
FIGURE 11 shows biochemical analysis of total collagen and sulfated GAGs at
t=8
wks. FIGURE 11A illustrates that self-assembly with DCs caused an increase in
total
collagen content compared to self-assembly with EBs (p=0.002). Significant
differences were
also detected due to differentiation agent, with CM and D2 constructs being
higher than Dl
constructs (p=0.0007). Note: The convention used to show statistically
different results are
upper or lower case letters (one set for each experimental factor). Groups not
connected by
the same letter are significantly different (p<0.05). FIGURE 11 B shows that
sulfated GAG
content was higher in DC constructs compared to EB constructs (p=0.038).
Differentiation
condition was not a significant factor for GAG production.
FIGURE 12 shows ELISAs for collagens I and II. FIGURE 12A illustrates that the
picogreen results from the ELISA digest showed that CM constructs had higher
cell numbers
than D1 constructs at t=8 wks. Particularly notable is the fact that CM
dissociated cell (DC)
constructs had almost twice as many cells as the other two DC groups. All
constructs were
initially seeded with the same amount of cells. Additionally, D1 embryoid body
(EB)
constructs exhibited lower cell numbers than the other EB constructs. These
results generally
mirror the gross morphology of the constructs. FIGURE 12B shows that collagen
type I per
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
6
cell was undetectable in D1 constructs, while CM and D2 constructs exhibited
relatively high
amounts of collagen type I per cell. Overall, CM constructs had higher
collagen type I
content (p<0.0001). Also, DC constructs had more collagen type I per cell than
EB constructs
(p<0.0001). FIGURE 12C shows that collagen type II per cell demonstrated
differences
between EB and DC constructs (p=0.008). CM constructs had more collagen type
II per cell
than D2 constructs (p=0.001).
FIGURE 13 shows compressive properties of the constructs at t=8 wks.
Dissociated
cell (DC) constructs had a higher instantaneous modulus than embryoid body
(EB) constructs
(p=0.005). Differentiation condition had no effect.
FIGURE 14 shows the tensile properties of dissociated cell (DC) constructs at
t=8
wks. FIGURE 14A shows that DC constructs had enough mechanical integrity to be
tested
under tension, while embryoid body (EB) constructs did not have this degree of
mechanical
integrity and could not be tested. In terms of both tensile modulus and
ultimate tensile
strength, D2 constructs were significantly higher than CM and D1 constructs.
Also notable
was the fact that the values for these properties were on the order of
megapascals. FIGURE
14B shows that collagen alignment (demonstrated by picrosirius red and
polarized light) in
the specimens along the axis of tensile testing (double headed arrow) was seen
best in the D2
group, while the CM and D1 specimens demonstrated no preferred direction (top
row).
Pictured on the top row are one-half of the tensile specimens, with the broken
end (where
failure occurred) being on the left of each picture (white arrow, 10x).
Analyzing the untested
whole constructs (bottom row) also demonstrated a higher degree of collagen
alignment in
D2 constructs compared to the other groups (lOx).
The patent or application file contains at least one drawing executed in
color. Copies
of this patent or patent application publication with color drawing(s) will be
provided by the
Office upon request and payment of the necessary fee.
While the present disclosure is susceptible to various modifications and
alternative
forms, specific example embodiments have been shown in the figures and are
herein
described in more detail. It should be understood, however, that the
description of specific
example embodiments is not intended to limit the invention to the particular
forms disclosed,
but on the contrary, this disclosure is to cover all modifications and
equivalents as illustrated,
in part, by the appended claims.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
7
DESCRIPTION
The present disclosure is generally in the field of improved methods for
tissue
engineering. More particularly, the present disclosure relates to methods for
inducing
differentiation human embryonic stem cells to serve as a source of
chondrocytes and
associated methods of use in forming tissue engineered constructs.
The methods of the present disclosure generally comprise aggregating
undifferentiated human embryonic stem cells to form embryoid bodies; and
culturing the
embryoid bodies in culture medium in the presence of growth factors that
induce
chondrogenic differentiation of the embryoid bodies.
In certain other embodiments, the methods of the present disclosure comprise
aggregating undifferentiated human embryonic stem cells to form embryoid
bodies; culturing
the embryoid bodies in culture medium in the presence of growth factors that
induce
chondrogenic differentiation of the embryoid bodies; sedimenting the
differentiated embryoid
bodies onto a hydrogel coated culture vessel; and allowing the differentiated
embryoid bodies
to self-assemble to form a construct. The term "human embryonic stem cell" is
defined herein
to include cells that are self-replicating or can divide and to form cells
indistinguishable from
the original, derived from human embryos or human fetal tissue, and are known
to develop
into cells and tissues of the three primary germ layers, the ectoderm,
mesoderm, and
endoderm. Although human embryonic stem cells may be derived from embryos or
fetal
tissue, such stem cells are not themselves embryos. The term "embryoid bodies"
is defined
herein to include any cluster or aggregate of human embryonic stem cells. The
term
"chondrogenic differentiation" is defined herein to include any process that
would result in
cells that produce glycosaminoglycans and collagen type II.
The term "construct" or "tissue engineered construct" as used herein refers to
a three-
dimensional mass having length, width, and thickness, and which comprises
living
mammalian tissue produced in vitro. As used herein, "self-assemble" or "self-
assembly" as
used herein refers to a process in which specific local interactions and
constraints between a
set of components cause the components to autonomously assemble, without
external
assistance, into the final desired structure through exploration of
alternative configurations.
Among other things, the methods of the present disclosure may be used to
produce
human cartilage constructs. Another advantage of the methods of the present
disclosure is
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
8
that human embryonic stem cells can be easily expanded in culture, and human
embryonic
stem cells possess the ability to maintain their phenotype stably in culture
theoretically over
limitless numbers of passages (an immortal cell line), while native
chondrocytes and other
stem cells will lose their phenotype when expanded over just a few passages.
In addition to
their expansion capability, the pluripotency of human embryonic stem cells
makes them
attractive for various regenerative medicine approaches, including cartilage
tissue
engineering. These features are especially attractive for cartilage tissue
engineering, where
scarcity of chondrocytes is considered a major impediment. Establishing human
embryonic
stem cells for this purpose requires a protocol for chondrogenic
differentiation and a method
to harness the cells' synthetic potential. The methods of the present
disclosure may provide
for the specific formation of cartilage, at least until 6 weeks of total
culture, which is apparent
due to the lack of other tissues in our engineered constructs. In certain
embodiments, the
methods of the present disclosure do not involve the use of fetal bovine
serum, which is an
animal product. The ability to produce constructs without the use of fetal
bovine serum is a
milestone that may ease the translation of the present disclosure to
therapeutic applications.
The present disclosure also provides for a system for studying tissue
engineering with
human embryonic stem cells that can discern functional differences between
engineered
cartilages made from chondrogenically-differentiated human embryonic stem
cells that were
exposed to distinct differentiation conditions.
The modular design of this tissue engineering methodology accommodates
perturbations to each of the key components during each phase to study how
human
embryonic stem cells differentiate and how these differentiated cells can be
used to engineer
cartilage. With this system, a number of investigations into the effects of
different seeding
densities, different growth environments, and other biochemical and
biomechanical
differentiation agents can be imagined. The developed methodology can also be
used as a
model system for fundamental research.
Referring initially to FIGURE 1, a schematic diagram of the process of
utilizing
undifferentiated human embryonic stem cells to form tissue engineered
constructs, the
methods of the present disclosure generally comprise aggregating
undifferentiated human
embryonic stem cells to form embryoid bodies, culturing the embryoid bodies in
culture
medium in the presence of growth factors that induce chondrogenic
differentiation of the
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
9
embryoid bodies, sedimenting the differentiated embryoid bodies onto a
hydrogel coated
culture vessel, and allowing the differentiated embryoid bodies to self-
assemble to form a
construct.
Source of Undifferentiated Human Embryonic Stem Cell
The human embryonic stem cells suitable for use in conjunction with the
methods of
the present disclosure can be obtained from a variety of sources. For example,
two NIH-
approved human embryonic stem cell lines, BG01 V and H9 may be used in
conjunction with
the methods of the present disclosure. The human embryonic stem cells may be
cultured
according to standard embryonic cell culture protocols available to those of
ordinary skill in
the art.
Alternatively, the cells may be obtained from an embryonic stem cell bank or
from
the process of somatic cell nuclear transfer. An embryonic stem cell bank
containing 150
human embryonic stem cell lines could be used for HLA (antigen) matching a
human
embryonic stem cell line to about 85% of all possible recipients (published in
Lancet,
December 2005). The principles described herein could be applied to any of
these human
embryonic stem cell lines to produce tissue engineered constructs with minimal
possibility of
immune rejection.
Somatic cell nuclear transfer would involve the creation of a patient-specific
human
embryonic stem cell line by transferring genetic material from one of the
patient's adult cells
(i.e., a skin cell) to an unfertilized human ovum. After 5 days in culture,
human embryonic
stem cells can be derived from the inner cell mass and treated with the
methods described
herein to obtain patient-specific construct.
Culture Medium
One of ordinary skill in the art, with the benefit of this disclosure, will
recognize that
suitable culture medium should be used in conjunction with the methods of the
present
disclosure such that human embryonic stem cells may proliferate and preferably
such that
stem cells may aggregate to form embryoid bodies, and be induced to
differentiate. In certain
embodiments, the medium used may comprise fetal bovine serum. The fetal bovine
serum
may be present in the range of about 1% to about 20% of culture medium. In
certain
embodiments, the culture media may be substantially free of fetal bovine
serum. The ability
to produce constructs without the use of fetal bovine serum is an advantage of
the methods of
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
the disclosure that may ease the translation of the present present.disclosure
to therapeutic
applications. One example of suitable medium for use in conjunction with the
methods of
the present disclosure is medium comprising high glucose Dulbecco's Modified
Eagle
Medium (DMEM), 10-7 M dexamethasone, 50 g/ml ascorbic acid, 40 g/ml L-
proline, 100
5 g/mi sodium pyruvate, 1% FBS, and ITS+Premix (6.25 ng/ml insulin, 6.25
mg/ml
transferrin, 6.25 ng/ml selenious acid, 1.25 mg/ml bovine serum albumin, and
5.35 mg/ml
linoleic acid).
Another example of suitable medium for use in conjunction with the methods of
the
present disclosure is medium comprising DMEM with 4.5 g/L-glucose and L-
glutamine, 0.1
10 M dexamethasone, 50 g/ml ascorbate-2-phosphate, 40 g/ml proline, 100
g/mi sodium
pyruvate, 1% fungizone, 1% Penicillin/Streptomycin, and 1 x ITS+Premix (6.25
g/ml
insulin, 6.25 g/ml transferrin, 6.25 ng/ml selenious acid, 1.25 mg/ml BSA,
and 5.35 mg/ml
linoleic acid)
Chondrogenic Differentiation of Undifferentiated Embryoid Bodies
The human embryonic stem cells used in conjunction with the methods of the
present
disclosure may be aggregated to form embryoid bodies. The embryoid bodies may
be
differentiated using culture medium in the presence growth factors that induce
chondrogenic
diffentiation. A variety of growth factors can be used in conjunction with the
methods of the
present disclosure. Suitable examples of growth factors include, but are not
limited to, TGF-
01, IGF-I, BMP-2, and TGF-(33.
In certain embodiments, the chondrogenic potential of human embryonic stem
cells
can be altered with soluble growth factors. In certain embodiments, TGF-(33
may be
administered during the critical early period of embryoid body differentiation
when the
specification of inesodermal cells into precursors of different lineages may
occur. After this
initial stage, the combination of TGF-01 with IGF-I or BMP-2 alone may be
administered to
the embryoid bodies.
In certain embodiments, the embryoid bodies are cultured in medium
supplemented
by a combination of TGF-(31 and IGF-I. In certain embodiments, the TGF-(31 is
present at a
concentration of about 10 ng/mL of culture medium. In certain embodiments, the
IGF-I may
be present at a concentration of 100 ng/mL of culture medium. The embryoid
bodies may be
exposed to the combination of TGF-(31 and IGF-I for a period of about four
weeks.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
Il
In certain embodiments,. the embryoid bodies may be induced to differentiate
by
exposure to TGF-(31, IGF-I, and TGF-(33. The TGF-(33 may be exposed to the
embryoid
bodies in the culture prior to exposure of the embryoid bodies to TGF-01 and
IGF-I. In
certain embodiments, the TGF-03 is present at a concentration of about 10
ng/mL of culture
media and is present in the media for a period of about one week. Following
the removal of
the TGF-03 from culture, TGF-01 and IGF-I may be introduced into the medium at
a
concentration of about 10 ng/mL of culture media and 100 ng/mL of culture
media,
respectively, for a period of about four weeks.
In certain other embodiments, only TGF-(33 may present at a concentration of
about
10 ng/mL of culture media for a period of about one week followed by exposure
of the
embryoid bodies to BMP-2 at a concentration of about lOng/mL of culture medium
for a
period of about three weeks.
Hydrogel Coating of Culture Vessels
The culture vessels may be coated with a hydrogel in conjunction with the
methods of
present disclosure . In certain embodiments, the bottoms and sides of a
culture vessel may be
coated with 2% agarose (w/v). While 2% agarose is used in certain embodiments,
in other
embodiments, the agarose concentration may be in the range of about 0.5% to
about 4%
(w/v). The use of lower concentrations of agarose offers the advantage of
reduced costs;
however, at concentrations below about 1% the agarose does not stiffen enough
for optimal
ease of handling.
As an alternative to agarose, other types of suitable hydrogels may be used
(e.g.
aliginate). A "hydrogel" is a colloid in which the particles are in the
external or dispersion
phase and water is in the internal or dispersed phase. Suitable hydrogels are
non-toxic to the
cells, are non-adhesive, do not induce chondrocyte attachment, allow for the
diffusion of
nutrients, do not degrade significantly during culture, and are firm enough to
be handled.
Sedimentation and Self-Assembly of Embryoid Bodies to Form Tissue Engineered
Constructs
The chondrogenically differentiated embryoid bodies may be sedimented on
hydrogel
coated culture vessels. In certain embodiments, the embryoid bodies may be
seeded at a
concentration of 1x106 cells per well in 3 mm wells with culture medium. In
certain
embodiments, the culture medium may be supplemented with TGF-01 and IGF-I. In
certain
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
12
embodiments, the TGF-0 1 is present at a concentration of about 10 ng/mL of
culture medium.
In certain embodiments, the IGF-I may be present at a concentration of 100
ng/mL of culture
medium.
In certain embodiments, the amount of growth factor may be varied to provide
for
tissue engineered constructs with different ranges of collagen that are more
representative of
the range of collagen found in native tissues.
In certain embodiments, the embryoid bodies may be chemically dissociated
prior to
sedimentation on the hydrogel coated culture vessels. In certain embodiments,
the embryoid
bodies may be enzymatically dissociated during the transition from
differentiation to self-
assembly. This dissociation provides differentiated embryoid bodies that may
then be used to
produce the tissue engineered constructs of the present disclosure.
In certain embodiments, the embryoid bodies may be pressurized to 10 MPa at
1Hz
using a sinusoidal waveform function. In other embodiments, the embryoid
bodies may be
pressurized during self-assembly of the embryoid bodies. In particular
embodiments, a
loading regimen (e.g. compressive, tensile, shear forces) may be applied to
the embryoid
bodies during self-assembly based on physiological conditions of the native
tissue in vivo.
Loading of the embryoid bodies during self-assembly and/or construct
development may
cause enhanced gene expression and protein expression in the constructs.
In particular embodiments, the constructs may be treated with staurosporine, a
protein
kinase C inhibitor and actin disrupting agent, during the self-assembly
process to reduce
synthesis of aSMA, a contractile protein. Reducing aSMA in the constructs via
staurosporine
treatment may reduce construct contraction and may also upregulate ECM
synthesis.
Hydrogel Molds
In certain embodiments, the chondrogenically differentiated embryoid bodies
may be
sedimented on a hydrogel coated culture vessel, allowed to self-assemble into
a tissue
engineered construct, and molded into a desired shape. In certain embodiments,
the self-
assembly of the embryoid bodies into a construct may occur on hydrogel coated
culture
vessels before the construct is transferred to a shaped hydrogel negative mold
for molding the
construct into the desired shape.
Alternatively, rather than sedimenting the chondrogenically induced embryoid
bodies
on a hydrogel coated culture vessel, in certain embodiments, the cells may be
sedimented
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
13
directly onto a shaped hydrogel negative mold. The shaped hydrogel negative
mold may
comprise agarose. Other non-adhesive hydrogels, e.g. alignate, may be used in
conjunction
with the methods of the present disclosure. In other embodiments, the hydrogel
mold may be
a two piece structure comprising, a shaped hydrogel negative mold and a shaped
hydrogel
positive mold. The shaped hydrogel negative and positive molds may comprise
the same non-
adhesive hydrogel or may be a comprised of different non-adhesive hydrogels.
In certain embodiments, the chondrogenically differentiation embryoid bodies
may be
sedimented onto a hydrogel coated culture vessel and allowed to self-assemble
into a
construct. The construct may be transferred to a shaped hydrogel negative
mold. A shaped
hydrogel positive mold may be applied to the negative mold to form a mold-
construct
assembly. The mold-construct assembly may then further be cultured. As used
herein, the
term "mold-construct assembly" refers to a system comprising a construct or
cells within a
shaped positive and a shaped negative hydrogel mold.
In certain embodiments, the molds may be shaped from a 3-D scanning of a total
joint
to result in a mold fashioned in the shape of said joint. In other
embodiments, the molds may
be shaped from a 3-D scanning of the ear, nose, or other non-articular
cartilage to form molds
in the shapes of these cartilages. In certain embodiments, the mold may be
shaped to be the
same size as the final product. In other embodiments, the molds may be shaped
to be smaller
than the final product. In certain embodiments, the molds may be fashioned to
a portion of a
joint or cartilage so that it serves as a replacement for only a portion of
said joint or cartilage.
Other examples of shaped hydrogel molds and methods of developing scaffoldless
tissue engineered constructs that may be useful in conjunction with the
methods of the
present disclosure may be found in co-pending application entitled "A Shape-
Based
Approach for scaffoldless Tissue Engineering," the disclosure of which is
incorporated by
reference herein.
Analysis of the Constructs
The properties of constructs may be tested using any number of criteria
including, but
not limited to, morphological, biochemical, and biomechanical properties,
which also may be
compared to native tissue levels. In this context, morphological examination
includes
histology using safranin-O and fast green staining for proteoglycan and GAG
content, as well
as picro-sirius red staining for total collagen, immunohistochemistry for
collagens I and II,
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
14
and confocal and scanning electron microscopies for assessing cell-matrix
interactions.
Biochemical assessments includes picogreen for quantifying DNA content, DMMB
for
quantifying GAG content, hydroxyproline assay for quantifying total collagen
content, and
ELISA for quantifying amounts of specific collagens (I and II), and RT-PCR for
analysis of
mRNA expression of proteins associated with the extracellular matrix (e.g.
collagen and
aggrecan).
Constructs also may be evaluated using one or more of incremental tensile
stress
relaxation incremental compressive stress relaxation, and biphasic creep
indentation testing to
obtain moduli, strengths, and viscoelastic properties of the constructs.
Incremental
compressive testing under stress relaxation conditions may be used to measure
a construct's
compressive strength and stiffness. Incremental tensile stress relaxation
testing may be used
to measure a construct's tensile strength and stiffness. Additionally,
indentation testing under
creep conditions may be used to measure a construct's modulus, Poisson's
ratio, and
permeability.
Without wishing to be bound by theory or mechanism, although both collagen
type II
and glycosaminoglycans (GAGs) are excellent predictors of biomechanical
indices of
cartilage regeneration, typically only collagen type II exhibits a positive
correlation. Though
seemingly this hypothesis is counterintuitive for compressive properties, as
GAG content is
usually thought to correlate positively with compressive stiffness, our
results show that in
self-assembled constructs, GAG is negatively correlated with the aggregate
modulus
(R2=0.99), while collagen type II is positively correlated (RZ=1.00).
The constructs of the present disclosure may be assessed morphologically
and/or
quantitatively. Quantitatively, the constructs of the present disclosure may
be evaluated using
a functionality index (FI ) as described in Eq. 1. The functionality index is
an equally
weighted analysis of ECM production and biomechanical properties that includes
quantitative
results corresponding to the constructs' salient compositional characteristics
(i.e., amounts of
collagen type II and GAG) and biomechanical properties (compressive and
tensile moduli
and strengths).
FI=1 1 (Gta,-Ga) V 1 ~Cn`-CJ) I
1 + c~
4 Qat C,,ar 2 2 Ec,., 2 ~nr 2
Eq. (1)
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
In this equation, G represents the GAG content per wet weight, C represents
the
collagen type II content per wet weight, ET represents the tensile stiffness
modulus, Ec
represents the compressive stiffness modulus, ST represents the tensile
strength, and Sc
represents the compressive strength. Each term is weighted to give equal
contribution to
5 collagen, GAG, tension, and compression properties. The subscripts nat and
sac are used to
denote native and self-assembled construct values, respectively. The aggregate
modulus is
not used in Eq. 1, as it is expected to mirror the compressive modulus
obtained from
incremental compressive stress relaxation. Similarly, the amount of collagen
type I is not be
used in Eq. 1, as this type of collagen may not appear in a measurable
fashion; however, if
li 'ble, FI may be altered accordingly to account for
10 the amount of collagen type I is non-neg~
it.
Each term grouped in parentheses in Eq. 1 calculates how close each construct
property is with respect to native values, such that scores approaching 1
denote values close
to native tissue properties. Equal weight is given to GAG, collagen type II,
stiffness (equally
15 weighted between compression and tension), and strength (also equally
weighted between
compression and tension). This index, FI, will be used to assess the quality
of the construct
compared to native tissue values, with a lower limit of 0 and an unbounded
upper limit, with
a value of 1 being a construct possessing properties of native tissue.
However, the FI can
exceed 1 if optimization results in constructs of properties superior to
native tissue.
Methods of Using the Tissue Engineered Constructs
In certain embodiments, applications of the tissue engineered construct
include the
replacement of tissues, such as cartilaginous tissue, the knee meniscus, joint
linings, the
temporomandibular joint disc, tendons, or ligaments of mammals.
The constructs may be treated with collagenase, chondroitinase ABC, and BAPN
to
aid in the integration of the constructs with native, healthy tissue
surrounding the desired
location of implantation. The integration capacity of a construct with native
tissue is crucial
to regeneration. A wound is naturally anti-adhesive, but debridement with
chondroitinase
ABC and/or collagenase removes anti-adhesive GAGs and enhances cell migration
by
removing dense collagen at the wound edge. BAPN, a lysyl oxidase inhibitor,
may cause the
accumulations of matrix crosslinkers and may, thus, strengthen the interface
between the
construct and native tissue at the desired location of implantation.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
16
The tissue engineered constructs may be implanted into a subject and used to
treat a
subject in need of tissue replacement. In certain embodiments, the constructs
may be grown
in graded sizes (e.g. small, medium, and large) so as to provide a resource
for off-the-shelf
tissue replacement. In certain embodiments, the constructs may be formed to be
of custom
shape and thickness. In other embodiments, the constructs may be devitalized
prior to
implantation into a subject.
To facilitate a better understanding of the present disclosure, the following
examples
of specific embodiments are given. In no way should the following examples be
read to limit
or define the entire scope of the disclosure.
EXAMPLES
Example 1: Chondrogenic differentiation of human embryonic stem cells.
This study investigated the potential of two NIH-approved human embryonic stem
cell (hESC) lines, BG01 V and H9, to differentiate into cells that produce
collagen type II and
GAGs. The cell lines were cultured to passages 20-25 using established
protocols. To induce
the process of differentiation, embryoid bodies (EBs) were formed by exposing
undifferentiated hESC colonies to 0.1% (w/v) dispase. Two differentiation
agent regimens
were used: TGF-(33 (lOng/ml) for 1 wk followed by TGF-(31 (10 ng/ml) + IGF-I
(100 ng/ml)
for 3 wks was used with BGO1Vi cells, and TGF-(31 (10 ng/ml) + IGF-I (100
ng/ml) was used
with H9 cells for 4 wks. Controls received neither of these differentiation
agent regimens. H9
cells received no serum. The BGO1 V controls and groups exposed to the
differentiation
agents were tested at three levels of FBS: 0%, 1%, and 20%. EBs were cultured
in non-
adherent bacteriological petri dishes, and medium was changed every 48 hrs for
the duration
of the experiment. The medium was composed of DMEM with 4.5 g/L-glucose and L-
glutamine supplemented with 0.1 M dexamethasone, 50 ~tg/ml ascorbic acid, 40
g/ml
proline, 100 g/mi sodium pyruvate, and lx ITS+Premix.
After 4 wks, EBs were cryosectioned at 12 m, and Alcian blue staining for
GAGs
and immunohistochemistry for collagen type II were positive with both
differentiation agent
regimens with all the serum levels tested (FIGURE 6). Controls also showed
staining for
GAGs and collagen type II (data not shown), but the staining was not as
consistent as seen
with treatment groups. Encouragingly, staining for other tissues was negative,
1) including
Oil Red 0 for adipose tissue, 2) von Kossa for bone, and 3) Masson's Trichrome
for muscle
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
17
(data not shown). In summary, within 4 wks, the results demonstrate that the
differentiation
agent regimens were able to induce the expression of GAGs and collagen type II
in both
hESC lines.
Example 2 Self-assembly of chondro eng ically-differentiated hESCs
Self-assembly of the BG01 V and H9 EBs was initiated by placing enough EBs to
cover the bottom of agarose wells (approximately 3x105 cells). Media
components were the
same as those used for chondrogenic differentiation. This preliminary study
used the
combination of TGF-01 (10 ng/ml) + IGF-I (100 ng/ml) for both cell lines, and
the serum
level used in the differentiation phase stayed the same in the self-assembly
phase (0%, 1%,
and 20% FBS). The media and growth factors were changed every 48 hrs. After 4
days, the
constructs were transferred to 12-well agarose coated plates so that they
could grow without
confinement. After 2 wks in self-assembly, the BGO1 V constructs were easily
handled and
relatively uniform, as shown in FIGURE 7A. The H9 constructs were cultured to
4 wks, and
appeared similar to the BG01 V samples (FIGURE 7B). At these time points (t=2
wks for
BGOl V and t=4 wks for H9), the constructs were cryosectioned at 12 m and
stained using
Alcian blue for GAG (data not shown) and immunohistochemistry for collagen
type II
(FIGURE 8). Again, staining was negative for other mesodennal tissues,
indicating robust
chondrogenic differentiation. The self-assembled constructs were then tested
under biphasic
creep indentation conditions, yielding compressive modulus values in the same
range as those
obtained for self-assembled constructs using articular chondrocytes at their
respective time
points. It was remarkable to note that, using hESCs, tissue engineered
constructs of cartilage-
like characteristics could be produced with the self-assembly process.
Specifically, this study
shows that constructs of 1.5 mm thickness and 3 mm dia., with appropriate
chondrocytic
markers, can be formed using two different hESC lines.
Example 3: Morphological Assessment of the Embryoid Bodies
Undifferentiated human embryonic stem cells were incubated with 0.1 % (w/v)
dispase (Gibco) at 37 C and 5% CO2 for 15-30 min, removing colonies intact.
The colonies
were pelleted and resuspended in medium, consisting of Dulbecco's Modified
Eagle Medium
(DMEM) with 4.5 g/L-glucose and L-glutamine supplemented with 10-7 M
dexamethasone,
50 g/ml ascorbic acid, 40 g/ml proline, 100 g/mi sodium pyruvate, and 50
mg/ml
ITS+Premix (6.25 g/ml insulin, 6.25 g/ml transferrin, 6.25 ng/ml selenious
acid, 1.25
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
18
mg/ml BSA, and 5.35 mg/ml linoleic acid). Additionally, the differentiation
was performed at
three levels of fetal bovine serum (FBS): 0%, 1%, and 20%. The colonies were
placed in 100
mm bacteriological petri dishes (VWR) and formed cell aggregates called
embryoid bodies.
For directed differentiation, two differentiation regimens were used: 1)
Transforming growth
factor (TGF)-(31 (lOng/ml) with Insulin-like growth factor (IGF)-I (100 ng/ml)
for 4 wks, and
2) TGF-03 (10 ng/ml) for 1 wk followed by TGF-(31 (10 ng/ml) with IGF-I (100
ng/ml) for 3
wks. The medium and differentiation agents were replaced together every 48
hours.
The embryoid bodies (see FIGURE 2) were analyzed four weeks after seeding for
the
articular cartilage specific extracellular matrix proteins glycosaminoglycans
and collagen
type II using an Alcian blue stain and immunohistochemistry, respectively.
Stains for
unwanted differentiation in the form of bone (von Kossa), muscle (Masson's
Trichrome), and
adipose (Oil Red 0) were also performed on the constructs.
Immunohistochemistry showed
production of collagen type II, and histology at this time point demonstrated
the presence of
abundant g1Ycosaminog1Ycans for all three levels of FBS (FIGURE 3). Other
mesodermal
tissues were not detected by histology, including bone, muscle, and adipose
four weeks after
seeding.
Example 4: Morphological Assessment of the Tissue engineered Constructs
Undifferentiated human embryonic stem cells were incubated with 0.1 %(w/v)
dispase (Gibco) at 37 C and 5% COZ for 15-30 min, removing colonies intact.
The colonies
were pelleted and resuspended in medium, consisting of Dulbecco's Modified
Eagle Medium
(DMEM) with 4.5 g/L-glucose and L-glutamine supplemented with 10 7 M
dexamethasone,
50 [tg/ml ascorbic acid, 40 [tg/ml proline, 100 g/m1 sodium pyruvate, and 50
mg/ml
ITS+Premix (6.25 [tg/ml insulin, 6.25 [tg/ml transferrin, 6.25 ng/ml selenious
acid, 1.25
mg/ml BSA, and 5.35 mg/ml linoleic acid). Additionally, the differentiation
was performed at
three levels of fetal bovine serum (FBS): 0%, 1%, and 20%. The colonies were
placed in 100
mm bacteriological petri dishes (VWR) and formed cell aggregates called
embryoid bodies.
For directed differentiation, two differentiation regimens were used: 1)
Transforming growth
factor (TGF)-(31 (1 Ong/ml) with Insulin-like growth factor (IGF)-I (100
ng/ml) for 4 wks, and
2) TGF-03 (10 ng/ml) for 1 wk followed by TGF-(31 (10 ng/ml) with IGF-I (100
ng/ml) for 3
wks. The medium and differentiation agents were replaced together every 48
hours.
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
19
The bottoms and sides of 96-well plates were coated with 100 l 2% agarose
(w/v),
and the plates were shaken vigorously to remove excess agarose. The surface
area at the
bottom of the well in a 96-well plate is 0.2 cm2. Chilled plates were then
rinsed with culture
medium before the introduction of cells.
After 4 weeks of chondrogenic differentiation, embryoid bodies were placed
into
hydrogel-coated wells at 1x106 cells per well with 500 l of culture medium.
The medium
had the same composition as used during chondrogenic differentiation. The
growth factors
TGF-0 1 (lOng/ml) with IGF-I (100 ng/ml) were used to culture these
constructs.
After two weeks of culture on the hydrogel coated tissue culture wells (6
weeks after
initial seeding), the developing constructs were analyzed for the articular
cartilage specific
extracellular matrix proteins glycosaminoglycans and collagen type II using an
Alcian blue
stain and immunohistochemistry, respectively. Stains for unwanted
differentiation in the form
of bone (von Kossa), muscle (Masson's Trichrome), and adipose (Oil Red 0) were
also
performed on the constructs. At this time point, the embryoid body constructs
were 3 mm in
diameter and 1 mm thick (FIGURE 4). Glycosaminoglycans and collagen type II
are
expressed in these constructs at all three levels of FBS (FIGURE 5). Other
mesodermal
tissues were not detected by histology, including bone, muscle, and adipose at
this time
point..
Example 5: Determination of the Aggregate Modulus of the Constructs
After two weeks of culture (6 weeks after initial seeding) on the hydrogel
coated
wells, the aggregate modulus of the developing constructs was analyzed using
prior art
techniques. "Aggregate modulus" is a conventional measurement used in
characterizing
cartilage. Mechanical testing of the representative aggregate or construct
yielded a modulus
of 6 kPa at 6 weeks after seeding.
Example 6: Expansion of Human Embryonic Stem Cells
The NIH-approved hESC line BGOl V (American Type Culture Collection,
Manassas, VA, http://www.atcc.org) was cultured according to standard
protocols. Briefly, a
feeder layer of gamma-irradiated CF-1 (Charles River Laboratories, Wilmington,
MA,
http://www.criver.com) mouse embryonic fibroblasts (MEFs) at a density of
5x105 MEFs per
well of a Nunc 6-well dish (Fisher Scientific, Hampton, NH,
http://www.fishersci.com) was
used in the expansion of the hESCs. Frozen hESCs at passage 16 (p16) were
thawed
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
according to standard protocol and sub-cultured. A growth medium comprising
DMEM/F-12
(Gibco, Gaithersburg, MD, http://www.invitrogen.com), ES-qualified FBS (ATCC),
L-
glutamine (Gibco), knock out serum replacer (Gibco), and nonessential amino
acids (NEAA,
Gibco) was used. The hESCs were passaged with collagenase IV (Gibco) every 4-5
days, and
5 cells were utilized for the experiment at p21.
Example 7: Embryoid Body Formation, Differentiation Conditions, and Analysis
Dispase solution (0.1 % w/v in DMEM/F-12) was applied for 10-15 min to
colonies
of undifferentiated hESCs in monolayer when the colonies reached 70-80%
confluence. This
enzymatic treatment predominantly lifts the hESC colonies from the culture
dish, leaving
10 MEFs behind and forming embryoid bodies (EBs) from the hESC colonies as
described in
Zhang SC, Wernig M, Duncan ID, et al. In vitro differentiation of
transplantable neural
precursors from human embryonic stem cells. Nature Biotech 2001; 19:1129-1133.
After two
washes and centrifugations with DMEM/F-12, the EBs were suspended in a
chondrogenic
medium (CM) comprising high-glucose DMEM (Gibco), 10-7 M dexamethasone, ITS+
15 Premix (6.25 ng/ml insulin, 6.25 mg transferrin, 6.25 ng/mi selenious acid,
1.25 mg/ml
bovine serum albumin, and 5.35 mg/ml linoleic acid; Collaborative Biomedical,
San Jose,
CA, http://www.bdbiosciences.com), 40 g/ml L-proline, 50 g/ml ascorbic acid,
100 gg/ml
sodium pyruvate, and 1% FBS (Gemini Bio-Products, West Sacramento, CA,
http://www.gembio.com). The EBs were distributed into bacteriological petri
dishes (Fisher)
20 by placing EBs from two 6-well culture plates into each petri dish and
using 18 ml of
medium per dish. Three differentiation conditions were applied to the EBs in
this experiment:
1) CM alone for 28 days (designated CM), (2) CM with TGF-(33 (10 ng/ml) for 7
days
followed by the combination of TGF-(31 (10 ng/ml) and IGF-I (100 ng/ml) for 21
days
(designated Differentiation Condition 1(D1)), and (3) CM with TGF-P3 (10
ng/ml) for 7
days followed by BMP-2 (10 ng/ml) for 21 days (designated Differentiation
Condition 2
(D2)). For the entire experiment, medium, and, when applicable, growth factors
were
completely changed every 48 hrs. EBs were used for self-assembly or for
histological
analysis at t=4 wks.
EBs were also cryo-sectioned and stained for collagens using picrosirius red,
GAGs
using Alcian blue, and collagen type I and collagen type II using
immunohistochemistry
(IHC), as previously described in Hu JC and Athanasiou KA. A self-assembling
process in
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
21
articular cartilage tissue engineering. Tissue Eng 2006; 12:969-979.. Other
stains for
mesodermal tissue markers were used to detect unwanted differentiation. These
included von
Kossa (calcified tissues such as bone), Masson's trichrome (muscle), and Oil
red O(adipose).
Standard protocols were followed for each of these stains.
During the 4 wks of differentiation in EB form, EBs noticeably grew in size
with the
CM (chondrogenic medium without growth factors) and D2 (CM with additives of
TGF-03
followed by BMP-2) groups, while D 1(CM with additives of TGF-03 followed by
TGF-01
and IGF-I) EBs did not appear to change in size. The morphology and histology
of the EBs at
t=4 wks is shown in FIGURE 9A. The collagen type I and collagen type II IHC
illustrate that
the cartilaginous matrix in the EBs was loosely connected and unorganized,
with all three
differentiation conditions exhibiting collagen type I most prominently. Alcian
blue staining
for all groups at this time point was minimal (data not shown). Dissociation
of the EBs with
trypsin resulted in a cell suspension, though some cells were still connected
with extracellular
matrix (ECM) after the 1-hr digestion. Most of the cell suspension was used to
make
constructs, with at least 8 DC constructs being self-assembled from each
differentiation
regimen. Similarly, at least 8 EB constructs were self-assembled from each
group.
At t=4 wks, a small number of EBs from each differentiation condition were
collected
for analysis. For visualization of Sox-9, some of the cells obtained from the
trypsin digestion
at 4 wks of differentiation were plated at a density of 4.0x105 per ml onto a
glass slide and
allowed to attach overnight. The cells were then fixed with 3.7%
paraformaldehyde for 20
min, incubated with Triton-X 100 for 20 min at room temperature, blocked with
3% BSA for
min, incubated with Sox-9 primary antibody (Anaspec, Inc., San Jose, CA) for 2
hrs, and
then incubated with Alexa Fluor 546 conjugated goat anti-rabbit IgGI
secondary antibody
(Invitrogen, Carlsbad, CA, http://www.invitrogen.com) for 1 hr. PBS washes
were performed
25 between each of these steps.
A small portion of the cell suspension was used to analyze Sox-9 expression
and cell
morphology (FIGURE 9B). While the cells generated from each differentiation
regimen at
t=4 wks exhibited Sox-9 protein expression, they exhibited distinct cell
morphologies. CM
and D1 cells were rounded and approximately the same size as native articular
chondrocytes.
30 D2 cells appeared larger and fibroblastic. Histological analyses for
calcified tissue (von
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
22
Kossa), muscle (Masson's trichrome), and adipose (Oil red 0) were negative at
this time
point (data not shown).
Example 8: Self-assembly of chondrogenically-differentiated hESCs and Analysis
After 28 days of differentiation (t=4 wks), EBs in each of the three
differentiation
groups were separated into two equal subgroups. One subgroup of EBs from each
differentiation condition was digested in trypsin-EDTA (Gibco) for 1 hr. Cells
from each
digest were counted with a hemocytometer, washed with DMEM containing 1% FBS,
centrifuged at 200 x g, and resuspended at a concentration of 5.0x105 cells
per 20 l in CM.
Constructs were made by seeding the dissociated cell (DC) suspension into 3 mm
wells of
2% agarose (5.0x105 cells per well).
The other subgroup comprised the undigested EBs, which were centrifuged at 200
x g
and resuspended in 4 ml CM. EBs were seeded into 5 mm wells of 2% agarose
using an
equivalent of 1x106 cells per construct (based on the hemocytometer count).
The two self-
assembly modes (EB and DC) were carried out over the ensuing 4 wks, culturing
all
constructs made from the three differentiation conditions in CM without any
exogenous
growth factors or stimulation.
At the t=8 wks time point (after 4 wks of self-assembly), each construct was
measured
for wet weight after carefully blotting excess water. Diameter and thickness
measurements
were made using di 'tal cali ers with an accuracy of 0.01 mm Mituto o Aurora,
IL,
~ p ( Y > >
http://www.mitutoyo.com). Constructs were either used for histology,
biochemical assays, or
biomechanical testing. Histological assessments for self-assembled constructs
were exactly
the same as that for the EBs (above), except Sox-9 was not assessed at this
time point.
Additionally, picrosirius red samples were analyzed with a polarized
microscope (Nikon,
Melville, NY, http://www.nikonusa.com) to visualize collagen alignment.
Data were analyzed with a two factor ANOVA, using Tukey's post hoc test when
applicable and a significance value of p<0.05. At least four samples were
analyzed for
biochemical assays and biomechanical tests for all groups. All data are
reported as mean
standard deviation. Statistical differences between groups are denoted by a
standard
convention using letters. This convention illustrates significant differences
between groups
when the groups are not connected by the same letter. Since two experimental
factors were
assessed, upper and lower case letters were designated to each factor, with
differentiation
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
23
conditions (CM, D1, and D2) having lower case letters and self-assembly mode
(EB or DC)
having upper case letters.
After the initial seeding of the dissociated cells (DCs) into the 3-mm agarose
wells,
cells coalesced within 24 hrs into constructs that were slightly smaller than
the well. Over the
following weeks, the spacing between cells in each construct increased as they
produced
ECM, causing the constructs to appear smooth and cartilaginous (FIGURE l0A).
The amount
of EBs for each group seeded into the 5 mm wells was enough to cover the
entire bottom
surface initially. Over the ensuing weeks, CM and D2 constructs filled the
well, while D1
constructs appeared to shrink away from the outer edges. EB constructs never
achieved
homogeneity during the experiment. A clear matrix connected EBs in a
construct, and the
constructs appeared highly hydrated (FIGURE l0A).
Construct morphological measurements are shown in FIGURE 10 below the gross
morphological pictures. D1 constructs had significantly lower thickness and
wet weight
compared to CM constructs for both EB and DC groups (p<0.05), while D2
constructs were
not different from either of the differentiation conditions. At t=8 wks, CM
and D2 constructs
demonstrated uniform staining for collagens I and II, regardless of self-
assembly mode (EB
or DC, FIGURE lOB). Dl constructs also demonstrated uniform staining for
collagen type II
but no significant staining for collagen type I (FIGURE 10B), for both EB and
DC self-
assembled constructs. Intense picrosirius red staining in all self-assembled
constructs
illustrated the matrix-producing capacity of the differentiated cells (FIGURE
lOB).
Conversely, Alcian blue staining was minimal (FIGURE 10B). An interesting
finding with
histology was that a central pocket of fluid had formed within the DC
constructs (FIGURE
lOB). This was noted primarily in the CM and D2 constructs. At the end of the
8 wk
experiment, other mesodermal tissues (bone, muscle, adipose) were not detected
by histology
(data not shown).
Example 9: Biochemical analysis of the Constructs
Biochemical assays included dimethylmethylene blue (DMMB), hydroxyproline,
picogreen, and ELISAs for collagens I and II. Samples were lyophilized for 48
hrs, and dry
weights were measured. Previously described protocols were used for DMMB and
hydroxyproline tests, and one set of samples was used for these two assays.
For collagens I
and II, Chondrex reagents and protocols were used (Chondrex, Redmond, WA,
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
24
http://www.chondrex.com), with the exception that constructs were digested
with papain
(rather than pepsin) at 4 C for 4 days, followed by a 1 day elastase digest.
The picogreen
assay for DNA content was performed using this set of samples, and a multiple
of 7.7 pg
DNA per cell was used.
When comparing between EB and DC self-assembled groups for biochemical
content,
normalized by dry weight (dw), DC constructs demonstrated greater matrix
production (both
collagen and GAG) (p<0.05), as shown in FIGURE 11. The measurements for
hydroxyproline showed that the D1 DC group did not produce as much collagen
(5.2% by
dw) as the other two groups, with CM and D2 DC constructs producing 17.9% and
24.1% by
dw, respectively (FIGURE 11A). Although Alcian blue staining was not
substantial, the
DMMB assay demonstrated the presence of sulfated GAGs in all constructs
(FIGURE 11B).
The water content for engineered constructs in all groups was approximately
90%
(91.1 2.7% for CM DC, 85.5 5.8% for D1 DC, 89.7 5.1% for D2 DC, 92.8% 3.3% for
CM
EB, 94.2% 2.6% for Dl EB, and 91.7 2.3% for D2 EB).
Picogreen demonstrated that the number of cells per construct was
significantly
different between CM and D1 groups (p<0.05), while D2 constructs were not
different from
the other two groups (FIGURE 12A). ELISAs for collagens I and II demonstrated
that the
production of collagens I and II varied between each differentiation regimen
and between DC
and EB constructs (FIGURE 12B and FIGURE 12C). Specifically, collagen type I
production
per cell was significantly higher in CM constructs compared to the other two
differentiation
agents (for example, in g x 10-2/cell, 4.8 1.2 for CM DC, -0.5 0.5 for Dl DC,
and 3.8 0.9
for D2 DC, p<0.05). Dl constructs demonstrated undetectable collagen type I,
which echoed
the IHC results for this group. The ELISA data also demonstrated that DC
constructs had
higher collagen type I and lower collagen type II production per cell than EB
constructs
(p<0.05). Differentiation condition was a significant factor when analyzing
the collagen type
II ELISA, with CM constructs having higher collagen type II content compared
to D2
constructs. For example, CM DC samples had over 2-fold higher collagen type II
content per
cell than D2 DC samples (0.8 0.4 vs. 0.3 0.1 g x 10-5/cell, p<0.05). D1
constructs were not
significantly different compared to the other two differentiation agents in
terms of collagen
type II content per cell.
Example 10: Biomechanical Analysis of the Constructs
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
Biomechanical testing included tensile testing using an Instron 5565 (Instron,
Norwood, MA, http://www.instron.us) and unconfined compression using a
modified creep
indentation apparatus as described in Mow VC, Gibbs MC, Lai WM, et al.
Biphasic
indentation of articular cartilage--II. A numerical algorithm and an
experimental study. J
5 Biomech 1989; 22:853- 861.
For tensile testing, specimens were cut from the cylindrical constructs into
dog-bone
shapes and pulled at a strain rate of 1%/s until failure. Gauge length,
thickness and width of
the specimens were measured with digital calipers so that load and extension
measurements
could be converted to stress and strain. Similar to the whole constructs,
collagen alignment of
10 the tensile specimens was analyzed with picrosirius red staining and
polarized light. For
unconfined compression testing, constructs were allowed to equilibrate in PBS
for 10 min,
and then subjected to an instantaneous 1.96 mN test load. The creep test was
allowed to run
for at least 1 hr, which was long enough to achieve deformation equilibrium.
With the
unconfined compression creep data, intrinsic material properties of the
constructs were
15 obtained using a previously developed viscoelastic model as described in
Leipzig ND and
Athanasiou KA. Unconfined creep compression of chondrocytes. J Biomech 2005;
38:77-85.
Data were analyzed with a two factor ANOVA, using Tukey's post hoc test when
applicable and a significance value of p<0.05. At least four samples were
analyzed for
biochemical assays and biomechanical tests for all groups. All data are
reported as mean
20 standard deviation. Statistical differences between groups are denoted by a
standard
convention using letters. This convention illustrates significant differences
between groups
when the groups are not connected by the same letter. Since two experimental
factors were
assessed, upper and lower case letters were designated to each factor, with
differentiation
conditions (CM, D1, and D2) having lower case letters and self-assembly mode
(EB or DC)
25 having upper case letters.
Unconfined compression testing of the self-assembled constructs demonstrated
that
DC constructs had a significantly higher instantaneous modulus compared to EB
constructs
(p<0.05), while there was no significant difference between CM, D1, and D2
constructs
(FIGURE 13). There was no statistical difference between any treatments in
terms of their
relaxed modulus (2.2 1.5 kPa for CM DC, 1.7 0.8 kPa for Dl DC, 1.3 0.3 kPa for
D2 DC,
0.7 0.1 for CM EB, 1.8 0.7 kPa for D1 EB, and 0.8 0.2 kPa for D2 EB). The CM
and D2
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
26
DC constructs exhibited a higher apparent viscosity than all other treatments
(2778 817 kPa-
s for CM DC, 1489 857 kPa-s for D1 DC, 2487 980 kPa-s for D2 DC, 539 208 kPa-s
for
CM EB, 1445 572 kPa-s for D1 EB, and 693 356 kPa-s for D2 EB). Tensile testing
(FIGURE 14 A) showed that D2 DC constructs had an over 5.5-fold higher tensile
modulus
(3.3 0.7 vs. 0.6 0.5 MPa) and 2.8-fold higher ultimate tensile strength
compared to D 1
constructs (1.1 0.1 vs. 0.410.3 MPa). Comparing these tensile properties of D2
to CM
constructs yielded similar increases (6.6-fold and 2.8-fold, respectively).
Polarized light
microscopy performed directly on tensile tested specimens demonstrated
collagen alignment
in the direction of tensile testing for D2 DC tensile specimens while CM and D
1 tensile
specimens did not (FIGURE 14 B). Moreover, D2 constructs exhibited a higher
degree of
collagen alignment than CM and D1 constructs in the untested DC samples. EB
constructs
were not testable under tension.
Differences were observed at t=4 wks in terms of cell morphology and at t=8
wks in
terms of construct morphology (FIGURE 10), biochemistry (FIGURE 11 and FIGURE
12),
and tensile properties (FIGURE 14). Since cells from each differentiation
condition were
cultured in the basal chondrogenic medium without exogenous growth factors
during self-
assembly, these data collectively indicate that the cells generated after 4
wks of EB
differentiation had varying capacities to produce cartilage.
The constructs engineered according to the previous examples generally
exhibited
properties most similar to the fibrocartilages, particularly the TMJ disc and
the outer portion
of the knee meniscus. The constructs had relatively high total collagen
contents (up to 24%
by dw in this study vs. -80% by dw for native TMJ and outer meniscus), low
sulfated GAG
contents (about 4% by dw in this study vs. 0.6 to 10% for native TMJ and outer
meniscus),
and relatively high tensile properties (order of 1 MPa in this study vs. order
of 10-100 MPa
for the native fibrocartilages). These fibrocartilages are also notable for
their high collagen
type I content and low to absent collagen type II content. Both CM and D2
constructs
demonstrated this pattern, while Dl constructs did not contain detectable
collagen type I.
Compared to studies using biomaterials as scaffolds, as well as our original
work
describing self-assembly, the constructs produced by chondrogenically-
differentiated hESCs
have comparable collagen content (around 1 to 2% by wet weight), but lower
sulfated GAG.
Even though the current examples produced mostly fibrocartilage and these
previous tissue-
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
27
engineering studies produced hyaline-like cartilage with native chondrocytes,
this comparison
demonstrates the matrix-producing capacity of the differentiated hESCs. The
tensile
properties have been measured on the order of 1 MPa with native chondrocyte
self-assembled
constructs.
The most dramatic difference between differentiation conditions was revealed
by the
tensile testing. D2 tensile specimens exhibited the highest degree of collagen
alignment, and
this finding appears to account for the higher tensile modulus and ultimate
tensile strength of
this group (FIGURE 14). Whether this is a true functional difference needs
further
investigation. One explanation for the apparent differences in degree of
alignment and tensile
properties is that the D2 cells, which had a more fibroblastic morphology
(FIGURE 9B), had
a better ability to organize the collagen network. The link between cell shape
and function
has been well established in various types of cartilage. Additionally, in
native cartilages, the
resident cells, such as chondrocytes, remodel the matrix on a regular basis.
Another curious finding was the pocket of fluid inside of the CM and D2
constructs.
Our initial self-assembly study used bovine cells and bovine serum, and
encountered no fluid-
filled region . A possibility for the fluid-filled interior encountered in
this study is that a
different cell population (chondrogenic or non-chondrogenic) accumulated in
this space, but
the histological evidence did not offer support of this idea.
While characterization of the differentiation process was one major goal of
this study,
we also determined how the differentiated hESCs responded to the transition
from
differentiation in EB form to tissue engineering. While constructs made with
both self-
assembly modes, EB and DC, expressed cartilage proteins, the gross appearance
(FIGURE
10), total collagen and sulfated GAG contents (FIGURE 11), and biomechanical
properties
(compressive, FIGURE 13, and tensile, FIGURE 14) of the DC constructs were
better.
Additionally, the ELISA results (FIGURE 12) suggested that the process of
digesting the EBs
after 4 wks of differentiation and subsequently placing the cells into agarose
wells for self-
assembly increases collagen type I content and decreases collagen type II
content. In
comparing EB and DC constructs, it is important to note that the difference in
initial construct
size (3 mm wells for DC constructs and 5 mm wells for EB constructs) was
necessary due to
difficulty with seeding the EBs into 3 mm wells. This difference in construct
size between EB
and DC groups necessitated comparisons normalized by cell number and dry
weight. Given
CA 02648327 2008-10-03
WO 2007/115337 PCT/US2007/066092
28
the marked differences found between these two groups with this analysis, it
was postulated
that the ECM produced by the EBs during the first 4 wks hindered cell-cell
contacts and
lowered the concentration of cells when they were placed in agarose molds for
self-assembly.
On the other hand, enzymatic dissociation of the EBs and subsequent seeding of
the cells into
agarose molds promoted direct cell contacts and a higher cell density. Even in
normal
development of cartilaginous tissues, such as articular cartilage, mesenchymal
precursors
aggregate at high density with direct cell contacts as an early step of
chondrogenesis.
The preceding examples illustrate a new methodology to study cartilage tissue
engineering with hESCs. The use of self-assembly as a tissue engineering
strategy resulted in
quantitative data that addressed two hypotheses. First, we investigated
whether cells with
different chondrogenic potentials would be generated when hESCs were exposed
to distinct
growth factor regimens for 4 wks. We assessed this after the cells had formed
neocartilage (at
t=8 wks), showing differences in the chondrogenic potential of CM
(chondrogenic medium),
D1 (TGF-(33 followed by TGF-01 with IGF-I, added to CM), and D2 (TGF-(33
followed by
BMP-2, added to CM) cartilage constructs in terms of morphology, biochemistry,
and
biomechanics. These properties also illustrated that DC constructs outperform
EB constructs
and thereby highlighting the importance of enzymatic dissociation of EBs prior
to self-
assembly. These findings represent incremental steps toward functional
engineering of
different types of musculoskeletal cartilages with hESCs.
Notwithstanding that the numerical ranges and parameters setting forth the
broad
scope of the invention are approximations, the numerical values set forth in
the specific
examples are reported as precisely as possible. Any numerical value, however,
inherently
contain certain errors necessarily resulting from the standard deviation found
in their
respective testing measurements.
Therefore, the present invention is well adapted to attain the ends and
advantages
mentioned as well as those that are inherent therein. While numerous changes
may be made
by those skilled in the art, such changes are encompassed within the spirit of
this invention as
illustrated, in part, by the appended claims.