Note: Descriptions are shown in the official language in which they were submitted.
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
PHYTOESTROGENIC FORMULATIONS AND USES THEREOF
Background of the Invention
This work was supported by National Institute of Mental Health
Intramural Research Program (P.J.S.) and Grant MH67159 (R.D.B.),
National Institute of Aging Grants AG06647 (J.H.M.), AG16765 (J.H.M.,
A.C.G.), and AG14751 and AG026572 (R.D.B.), and the Kenneth T. and
Eileen L. Norris Foundation (R.D.B.).
This application claims priority to U.S.S.N. 60/819,849 mailed by
express mail label number ER455959795US on August 1, 2006; U.S.S.N.
60/889,920 filed February 14, 2007, and U.S.S.N. 60/943,190 filed June 11,
2007.
The demographics suggest that we face a devastating increase in the
prevalence of AD, reinforcing the immediate need for basic and translational
neuroscience to develop safe and efficacious ET and HT regimens for the
brain. Of those affected with AD, 68% are female and 32% are male
(Brookmeyer et al., 1998 Am J Public Health 88:13372). Because women
have a longer life expectancy than men, the absolute number of women with
AD exceeds that of men. However, a double danger exists for women.
Results of a meta-analysis of seven sex-specific studies concluded that
women are 1.5 times more likely to develop AD than age-matched men (Gao
et al., 1998 Arch Gen Psychiatry 55:809), which was supported by the Cache
County analysis that showed a clear female gender increase in the incidence
of AD (Zandi et al., 2002 JAMA 288:21239).
At the tum of the new millennium in the United States, there were
nearly 42 million women over the age of 50 years and, of these, more than 31
million women were over the age of 55 years (North American Menopause
Society, 2004). Worldwide, there are currently more than 470 million women
aged 50 years or older, and 30% of those are projected to live into their 80s
(North American Menopause Society, 2004). These women can anticipate
spending one-third to one-half of their lifetime in the menopausal state.
Reports on prevalence of AD vary, but of the 18 million American women in
1
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
their mid to late 70s, as many as 5 million may suffer from AD, and this
figure increases dramatically at older ages (Brookmeyer et al., 1998). The
projected exponential increase in the prevalence of AD, along with the
anticipated impact on families and society, highlights the imperative for
developing strategies to prevent or delay the onset of AD sooner rather than
later.
The profound disparities between the largely positive basic science
findings of gonadal steroidal action in brain and the adverse outcomes of
recent estrogen or hormone therapy ("ET/HT") clinical trials in women who
are either aged postmenopausal or postmenopausal with Alzheimer's disease
(AD), has led to an intense reassessment of gonadal hormone action and the
model systems used in basic and clinical science. One key factor that could
contribute to the negative results of the Women's Health Initiative Memory
Study ("WHIMS") trial was the advanced age, more than ten years following
menopause, at which ET/HT was initiated in women. Data from both basic
science analyses and clinical studies indicate a "healthy cell bias" of
estrogen
action in the neurons / brains, suggesting that ET/HT acts as an effective
preventative therapeutic strategy for age-related cognitive decline and
neurodegenerative disorders, such as Alzheimer's disease ("AD"), while it is
not an effective treatment strategy. The current widely prescribed ET,
conjugated equine estrogens ("CEE"), is a highly complex ET with over 200
different components. Whether CEE provides the optimal therapeutic
efficacy has been questioned. Another key issue challenging HT is the
optimal composition. The progestin and its timing of administration in
conjunction with ET, remains to be determined. Moreover, while ET/HT has
long been used in postmenopausal women to delay or reverse some of the
problems associated with menopause, epidemiological and clinical studies
have uncovered potential long-term risks related to this therapy. The
recently revealed risks associated with ET/HT have greatly increased interest
in the development of estrogen alternatives that promote beneficial effects of
estrogen in brain, bone and the cardiovascular system, while not eliciting
deleterious effects in other organs, particularly in breast and uterine
tissues.
2
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Two nuclear receptors for estrogen (ERs), ERa and ERP, have been
identified. In the central nervous system, both ERa and ERP are expressed in
the hippocampus and cortex of rodent and human brains. Previous studies
have demonstrated that both ERa and ERP can equivalently promote
neuronal survival by activating estrogen mechanisms of action in rat
hippocampal neurons. Increasing evidence indicates that ER(3 is a key
requirement for activation of mechanisms that underlie estrogen-inducible
neuronal morphological plasticity, brain development, and cognition. ERa,
on the other hand, is more predominant in mediating the sexual
characteristics of estrogen effects in the reproductive organs such as breast
and uterus. Taken together, these data establish a potential therapeutic
application for ERP as a pharmacological target to promote memory function
and neuronal defense mechanisms against age-related neurodegeneration
such as Alzheimer's disease (AD), while avoiding activating untoward
estrogenic proliferative effects in the breast and uterus, although this might
be at the cost of lower efficacy due to the lack of activation of ERP in the
brain. Other potential therapeutic advantages associated with ER(3 include
regulation of estrogen vasculoprotective action and development of
interventions targeting diseases such as depression, colon cancer, prostate
cancer, obesity, leukemia, and infertility. However, a potential disadvantage
of an ERP-selective ligand is the lack of activation of ERa in bone, as ERa
has been demonstrated to mediate estrogen regulation of bone density.
Although there is still controversy regarding the differential roles of
two estrogen receptor ("ER ') subtypes, ERa and/or ER(3, in mediating
estrogen actions in the brain and/or neurons, it has been widely demonstrated
that ER(3 plays a key role in regulating brain development, neurogenesis and
estrogen-induced improved neuronal plasticity and survival. In addition, as
compared with ERa, ERP is less effective in mediating the sexual
characteristics of estrogen action in reproductive tissues, avoiding
activating
untoward estrogenic proliferative effects in the breast and uterus. Therefore,
ERP represents a potentially safer therapeutic target for promoting memory
function and neuroprotection. However, this safety may be at the cost of
3
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
lower efficacy, due to the lack of activation of ERa in the brain. Other
potential advantages for ERP-target therapeutics arise from its regulation of
estrogen's cardioprotective effects. ERO-selective ligands may also provide
effective therapeutics for preventing or treating inflammation, depression,
S anxiety, colon cancer, prostate cancer, obesity, leukaemia, and infertility.
In searching for an effective ER(3-selective estrogen alternative
replacement therapy for promoting neurological function and preventing age-
related neurodegeneration, such as AD, in postmenopausal women, it is of
particular interest to identify and develop naturally occurring molecules or
analogues that potentially have a less toxic profile for long-term
administration. It is known that several plant-derived estrogenic molecules
(referred to as "phytoestrogens") bind to ERa and to ERP subtypes, and
some of these molecules possess moderate binding selectivity for ERP and
exert estrogenic effects in multiple tissues.
The therapeutic efficacy of phytoestrogens in the brain remains
controversial. On the one hand, when administered singly, phytoestrogens
appeared to be moderately neuroprotective. On the other hand, a recent
clinical trial revealed that a soy protein supplement that contains a mixture
of
phytoestrogens did not show improved cognitive function in postmenopausal
women, when treatment was initiated at the age of 60 years or older. The
clinical trial of phytoestrogens reported that a soy protein supplement
containing a complex formulation of isoflavones did not improve cognitive
function in postmenopausal women when treated at the age of 60 years or
older, Kreijkamp-Kaspers, et al. JAMA 2004, 292, 65-74, also indicating that
when started 10 or more years following menopause in postmenopausal
women when age-related neuronal reorganization has taken place, ET/HT
has no benefit on neural function. Age and hormonal "history" may be
important factors that were responsible for these negative results, as was the
case for the WH1MS trials.
Another issue that can substantially impact the efficacy of a mixture
of phytoestrogens action in the brain is the formulation of phytoestrogens,
since when administered alone, a number of phytoestrogens were protective
4
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
to neurons from neurodegenerative insults. Zhao, et al. Exp. Biol. Med. 2002,
227, 509-519. Soy extracts or soy protein supplements generally contain
multiple phytoestrogenic molecules, some of which may be ERa-selective
agonists, while others may be ER(3-selective agonists, and others may be
ineffective in activating either ERa or ERP but may function as inhibitors of
ER binding of those ERa and/or ERP phytoestrogenic agonists. The
ineffectiveness of a complex form.ulation of phytoestrogens in promoting
beneficial effects of estrogen in brain, such as a soy-derived preparation,
may
also arise from antagonizing actions among the different phytoestrogens, in
addition to the possible ER antagonism, likely from the activation of both
ERa and ERP in the same context. Co-administration of an ERa-selective
agonist and an ER(3-seiective agonist is less effective than treatment with
either agonist alone in various neuroprotective measurements.
ERa and ERP have a yin/yang relationship in many contexts where
one receptor may antagonize the actions of the other. Weihua, et al. FEBS
Lett. 2003, 546, 17-24; Gustafsson, J. A. Trends Pharmacol. Sci. 2003, 24,
479-485. Studies confirmed this observation, showing that coadministration
of ERa-selective agonist PPT and ERR-selective agonist DPN was less
efficacious than either PPT or DPN alone in protecting hippocampal neurons
against excitotoxic insults. Based on this analysis, a presumption can be
made that the ineffectiveness of administering a mixture of phytoestrogens
(i.e. a soy protein supplement) may partly come from the antagonizing
actions among different phytoestrogens, which may be ERa selective or ERP
selective. These findings indicate that although both ERa and ERP
contribute to estrogen promotion of neuronal survival, simultaneous
activation of both ER subtypes, ERa and ER(3, in the same context may
diminish the efficacy. In addition, the different ratio and distinct function
of
homodimer and heterodimer induced by co-administration of an ERa-
selective agonist and an ER(3-selective agonist may also account for the
reduced efficacy exerted by the combination of both agonists.
Development of an ER(3-selective phytoestrogen formulation could
maximize the therapeutic benefits associated with activation of ERP in the
5
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
brain while minimizing the adverse effects associated with the activation of
ERa in reproductive tissues. Moreover, selective targeting of ER(3
potentially reduces antagonistic actions that may occur in a complex soy-
derived preparation. This naturally occurring ideal formulation would have
tremendous therapeutic value in promoting neurological function and
preventing AD in a population at risk for losing neurological capacity and
losing memory function, i.e., postmenopausal women. To date, no such
phytoestrogen formulation exists. Thus, there is a need to discover and
develop a novel select phytoestrogen formulation, generally, and particularly,
a formulation that functions in the brain.
It is therefore an object of the present invention to provide an ERP-
selective phytoestrogen formulation maximizing the therapeutic benefits
associated with activation of ERP in the brain while minimizing the adverse
effects associated with the activation of ERa in reproductive tissues.
It is a further object of the invention to provide such a composition
wherein the active ingredients are isolated from natural substances.
Summary of the Invention
Select phytoestrogen pharmaceutical compositions and methods of
use for promoting neurological health and prevention of age-related
neurodegeneration, such as AD, have been developed. These select
phytoestrogen formulations are composed of a number of plant-derived
estrogenic molecules andlor their structural analogues and exhibit binding
preference to ERP over ERa and agonist activity in the brain. These ERP-
selective phytoestrogen formulations cross the blood-brain-barrier and
promote estrogen-associated neurotrophism and neuroprotection mechanisms
in the brain, without activating proliferative mechanisms in the reproductive
tissues and are therefore devoid of other estrogen-associated problematic
aspects. The select phytoestrogen formulations are therapeutically useful to
both women and men for sustaining neurological health and preventing age-
related cognitive decline and neurodegenerative disorders, such as AD.
These are administered enterally, transdermally, transmucosally,
intranasally or parenterally, in a dosage effective to prevent or alleviate
6
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
neuronal damage, effect neuronal regeneration or sustain viability, increase
expression of anti-apoptotic proteins, and/or decrease indicators of
Alzheimer's Disease. The formulations preferably contain combinations of
compounds, and can be formulated for daily, sustained, delayed or
weekly/monthly administration. In a preferred embodiment, these are
administered to women who are in menopause or post menopausal, most
preferably early in menopausal.
Brief Description of the Drawings
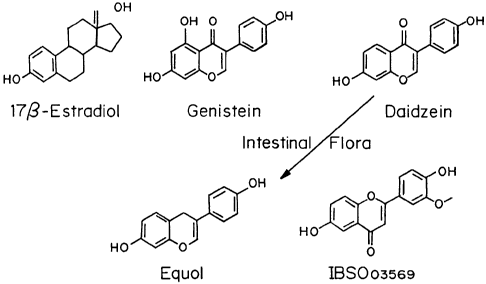
Figure 1 shows the chemical Structures of 17p-estradiol and the
phytoSERMs genistein, daidzein, equol, and IBS003569.
Figures 2A and 2B show the competition binding curves for ERa
and ER(3 (molar concentration versus fluorescence polarization (mP)) of G,
D, E, I or combinations: G+D, G+D+E, or G+D+E+I.
Figure 3 is a graph showing the neuroprotective efficacy of four
ER(3-selective phytoestrogenic molecules when administered alone at
concentrations that elicited the maximal neuroprotective effects as revealed
from the dose-response analyses (100 nM for all four molecules Genistein
(G), Daidzein (D), Equol (E) and IBS003569 (I)), or co-administered: G+D,
G+D+E, or G+D+E+I, against supraphysiological glutamate (100 M)-
induced neurotoxicity in primary hippocampal neurons by measurement of
calcein AM staining.
Figures 4A and 4B are graphs showing the effect of four ER(3-
selective phytoestrogenic molecules when co-administered (100 nM for all
four molecules) as G+D, G+D+E, or G+D+E+I, on the expression of the
anti-apoptotic proteins, Bcl-2 and Bcl-xL, in primary hippocampal neurons.
Figure 5 is a graph illustrating the effect of four ER(3-selective
phytoestrogenic molecules when co-administered (100 nM for all four
molecules), G+D, G+D+E, or G+D+E+I, on the expression of the anti-p-
amyloid protein, insulin-degrading enzyme ("IDE"), in primary hippocampal
neurons.
Figure 6 is a graph illustrating the effect of four ER(3-selective
phytoestrogenic molecules when co-administered (100 nM for all four
7
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
molecules): G+D, G+D+E, or G+D+E+I, on the expression of the spine
marker, spinophilin, in primary hippocampal neurons.
Figures 7A-7D are graphs shows the neuroprotective efficacy of G,
D, E, and I, alone and in combination: G+D, G+D+E, or G+D+E+I, against
(7A) glutamate- and (7B) P-amyloidl-42-induced neurotoxicity in rat
primary hippocampal neurons, controls live/dead cells (7C); dead cells (7D).
Figures 8A-8C are graphs showing the effects of G, D, E, and I,
alone and in combination: G+D, G+D+E, and G+D+E+I, on insulin-
degrading enzyme (IDE) expression on neprilysin (NEP) expression in
hippocampal tissues derived from adult ovariectomized rats.
Figures 9A-9E are graphs showing the effects of G, G+D+E, and
G+D+E+l on forebrain mitochondrial cytochrome c oxidase (COX) activity
in adult ovariectomized rats.
Figures 10A-10E are graphs showing the effects of G, G+D+E, and
G+D+E+I on percent increase forebrain mitochondrial respiratory activity in
adult ovariectomized rats.
Figures 11A-11C are schematics showing estrogen mechanisms of
action that lead to neurotrophic and neuroprotective outcomes. 11A, 17-0-
Estradiol (E2) acting via a membrane-associated site (mER) activates a
cascade required for multiple responses that lead to enhanced neural
plasticity, morphogenesis, neurogenesis, and neural survival. The signaling
sequence induced by E2 at the membrane site is as follows: (1) E2 binding to
mER, (2) E2-mER complexes with p85 to activate PI3K, (3) activating
calcium-independent PKC, (4) phosphorylating the L-type calcium channel,
(5) inducing calcium influx, (6) activating calcium-dependent PKCs, (7)
activating Src kinase, (8) activating the MEK/ERKI/2 pathway, (9) ERK
translocates to the nucleus, (10) activating and phosphorylating CREB, (11)
enhancing transcription of antiapoptotic genes Bcl-2 and Bcl-xl, which
enhance mitochondrial vitality, and spinophilin, which encourages synaptic
growth, (12) simultaneously, estrogen activation of P13 K leads to activation
of Akt, which phosphorylates and inhibits the proapoptotic protein BAD.
I 1B, Estrogen-induced neuroprotective mechanisms converge on
8
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
mitochondria. Estrogen-activated cellular signaling cascade promotes
enhanced mitochondrial function, leading to increased calcium load
tolerance, enhanced electron transport chain efficiency, and promotion of
antioxidant defense mechanisms. These actions are mediated by the
regulation of both nuclear and mitochondrial encoded genes initiated by the
activation of second-messenger signaling cascades. 11 C, Conceptual
schematic of NeuroSERM design and therapeutic use. Consistent with the
healthy cell bias of estrogen benefit hypothesis, selective molecules would be
administered before neurodegenerative insult while neurons are still healthy.
NeuroSERM exposure would lead to enhanced neural survival mechanisms,
represented as mitochondria with Bcl-2 additions, that promote neural
defense against neurodegenerative insults associated with age-associated
diseases such as Alzheimer's and Parkinson's. Designer NeuroSERM
molecules target the membrane site of estrogen action, whereas PhytoSERM
molecules preferentially target estrogen receptorp. Abbreviations: AMPAR,
AMPA receptor; C, cytochrome oxidase; F' , F'1, ATPase subunits; LTD,
long-term depression; LTP, long-term potentiation; NAD, nicotinamide
adenine dinucleotide; NADH, nicotinamide adenine dinucleotide; VDCC,
voltage-dependent calcium channel.
Detailed Description of the Invention
I. Definitions
"Estrogen Receptor", as used herein, refers to any protein in the
nuclear receptor gene family that binds estrogen, including, but not limited
to, any isoforms and variants thereof. Human estrogen receptors include the
alpha- and beta-isoforms (referred to herein as "ERa" and "ERP").
"Estrogen Receptor Modulator", as used herein, refers to a compound
that can act as an estrogen receptor agonist or antagonist of an estrogen
receptor or estrogen receptor isoform having an IC50 or ECS with respect to
ERa, ERP and/or other estrogen receptor isoforms of no more than about 50
p.M as determined using the ERa, and/or ERP transactivation assay described
herein. More typically, estrogen receptor modulators have IC50 or EC50
values (as agonists or antagonists) of not more than about 10 M.
9
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Representative compounds are predicted to exhibit agonist or antagonist
activity via an estrogen receptor. Compounds preferably exhibit an
antagonist or agonist TC5o or EC5o with respect to ERa and/or ERP of about
M, more preferably, about 500 nM, even more preferably about 1 nM,
5 and most preferably, about 500 pM, as measured in the ERa and/or ER(3
transactivation assays. "ICSO" is that concentration of compound which
reduces or inhibits the activity of a target (e.g., ERa or ERP) to half-
maximal
level. "EC50" is that concentration of compound which provides half-
maximum effect.
10 "Selective Estrogen Receptor Modulator" (or "SERM"), as used
herein, refers to a compound that exhibits activity as an agonist or
antagonist
of an estrogen receptor (e.g., ERa, ERP or other estrogen receptor isoform)
in a tissue-dependent or receptor dependent manner. Thus, as will be
apparent to those of skill in the biochemistry, molecular biology and
endocrinology arts, compounds that function as SERMs can act as estrogen
receptor agonists in some tissues, e.g., bone, brain, and/or cardiovascular,
and as antagonists in other tissue types, e.g., the breast and/or uterine
tissue.
"Phytoestrogen" refers to a naturally occurring compound of plants,
such as soybeans, or plant products, such as whole grain cereals, that acts
like estrogen or binds to an estrogen receptor.
As used herein, the terrrm "NeuroSERM" refers to compounds that
target the membrane site of estrogen action.
As used herein, the term "PhytoSERM" refers to natural source
compounds that preferentially target estrogen receptor beta.
As used herein, the term "analogue" refers to a chemical compound
with a structure similar to that of another (reference compound) but differing
from it in respect to a particular component, functional group, atom, etc.
As used herein, the term "derivative" refers to compounds which are
formed from a parent compound by chemical reaction(s).
Ix. Compositions
Compositions containing one or more phytoestrogens are described
herein. A number of phytoestrogens have been isolated and identified and
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
additional analogs created, all of which have estrogen receptor binding
selectivity. In one embodiment, of the composition contains two or more
plant-derived estrogenic molecules and/or structural analogues, which
possess ERP-binding selectivity and exhibit neuroprotective activity when
administered individually. These compositions are useful for preventing
estrogen-defzciency associated symptoms and disorders, particularly age-
related cognitive decline and neurodegenerative diseases, such as
Alzheimer's disease ("AD").
A. PhytoSERMs
The compositions described herein contain one or more
phytoestrogens or natural source selective estrogen receptor modulators
(SERMs) exhibiting a binding preference for ERj3. PhytoSERMs can be
identified as described in Example 1. Suitable phytoSERMs include, but are
not limited to, genistein, daidzein, equol, IBS003569 and combinations
thereof The structures of genistein, daidzein, equol, and IBS003569 are
shown in Figure 1. Others are listed in Table 1. Preferred compounds cross
the blood brain barrier.
As demonstrated by Example 2, combinations of two or more
PhytoSERMS are more effective than administration of one PhytoSERM.
The compounds can be used in the form of salts derived from
inorganic or organic acids. These salts include, but are not limited to, the
following: acetate, adipate, alginate, citrate, aspart ate, benzoate,
benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate,
digluconate, cyclopentanepro-pionate, dodecylsulfate, ethanesulfonate,
glucoheptanoate, glycerophosphate, hemi-sulfate, heptanoate, hexamate,
fumarate, hydrochloride, hydrobromide, hydroiodide, 2-
hydroxyethanesulfonate, lactate, maleate, methanesulfonate, nicotinate, 2-
napthalenesulfanate, oxalate, parnoate, pectinate, sulfate, 3-
phenylpropionate, picrate, pivalate, propionate, succinate, tartrate,
thiocyanate, p-toluenesulfonate and undecanoate. Also, any basic nitrogen-
containing groups can be quaternized with agents such as lower alkyl
halides, such as methyl, ethyl, propyl, and butyl chloride, bromides, and
11
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl, and diamyl
sulfates,
long chain halides such as decyl, lauryl, myristyl and stearyl chlorides,
bromides and iodides, aralkyl halides like benzyl and phenethyl bromides,
and others. Wafer or oil-soluble or dispersible products are thereby obtained.
Examples of acids which may be employed to form pharmaceutically
acceptable acid addition salts include such inorganic acids as hydrochloric
acid, sulfuric acid, and phosphoric acid, and organic acids such as oxalic
acid, maleic acid, succinic acid and citric acid. Basic addition salts can be
prepared in situ during the final isolation and purification of the compounds,
or separately by reacting carboxylic acid moieties with a suitable base such
as the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable
metal cation or with ammonia, or an organic primary, secondary or tertiary
amine. Pharmaceutically acceptable salts include, but are not limited to,
cations based on the alkali and alkaline earth metals, such as sodium,
lithiurn,
potassium, calcium, magnesium, and aluminum salts, as well as n.on-toxic
ammonium, quatemary ammonium, and mine cations, including, but not
limited to ammonium, tetramethylammonium, tetraethylammonium,
methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine,
and the like. Other representative organic amines useful for the formation of
base addition salts include diethylamine, ethylenediamine, ethanolamine,
diethanolamine, and piperazine.
Appropriate carriers can be added that assist the compounds to cross
the blood-brain-barrier.
B. Additional Active Agents
While the compounds can be administered as the sole active
pharmaceutical agent, they can also be used in combination with one or more
other compound as described herein, and/or in combination with other agents
used in the treatment and/or prevention of estrogen receptor-mediated
disorders. Alternatively, the compounds can be administered sequentially
with one or more such agents to provide sustained therapeutic and
prophylactic effects. Suitable agents include, but are not limited to, other
SERMs as well as traditional estrogen agonists and antagonists.
12
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Representative agents useful in combination with the compounds for
the treatment of estrogen receptor-mediated disorders include, for example,
tamoxifen, 4-hydroxytamoxifen, raloxifene, toremifene, droloxifene, TAT-
59, idoxifene, RU 58,688, EM 139,1CT 164,384, ICI 182,780, clomiphene,
MER-25, DES, nafoxidene, CP-336,156, GW5638, LY 139481, LY353581,
zuclomiphene, enclomiphene, ethamoxytriphetol, delmadinone acetate,
bisphosphonate. Other agents that can be combined with one or more of the
compounds include aromatase inhibitors such as, but not 1 imited to, 4-
hydroxymdrostenedione, plomestane, exemestane, aminogluethimide,
rogletimide, fadrozole, vorozole, letrozole, and anastrozole .
Still other agents useful in combination with the compounds
described herein include, but are not limited to antineoplastic agents, such
as
alkylating agents, antibiotics, hormonal antineoplastics and antimetablites.
An example includes the compounds used to treat or prevent osteoporosis.
Other ingredients include vitamins, nutritional supplements, anti-oxidant
agents, coenzymes, etc.
The additional active agents may generally be employed in
therapeutic amounts as indicated in the PHYSICIANS' DESK REFERENCE
(PDR) 53rd Edition (2003), or such therapeutically useful amounts as would
be known to one of ordinary skill in the art. The compounds and the other
therapeutically active agents can be administered at the recommended
maximum clinical dosage or at lower doses. Dosage levels of the active
compounds in the compositions may be varied to obtain a desired therapeutic
response depending on the route of admi:nistration, severity of the disease
and the response of the patient. The combination can be administered as
separate compositions or as a single dosage form containing both agents.
When administered as a combination, the therapeutic agents can be
formulated as separate compositions that are given at the same time or
different times, or the therapeutic agents can be given as a single
composition.
13
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
C. Pharmaceutical Compositions
The compounds can be administered enterally, transdermally,
transmucisally, intranasally or parenterally. Excipients for oral formulation
are known to those skilled in the art, as discussed briefly below, and can be
used to provide immediate, sustained, delayed, or pulsed release. The
compounds can also be administered via a transdermal patch, a depo,
vaginally or rectally using a topical carrier such as a gel, lotion, ointment,
liposomal formulation, suspension, foam, spray or supposity, via the
pulmonary or nasal route, buccally or sublingual via the mucosal membranes
of the mouth. The appropriate excipients for all of these formulations are
known. The compounds may be dissolved or suspended in saline, sterile
water or phosphate buffered saline, or a suitable oil for injection iv, im,
subcu, or ip.
Suitable pharmaceutically acceptable excipients include processing
agents and drug delivery modifiers and enhancers, such as, for example,
calcium phosphate, magnesium stearate, talc, monosaccharides,
disaccharides, starch, gelatin, cellulose, methyl cellulose, sodium
carboxymethyl cellulose, dextrose, hydroxypropyl-.beta.-cyclodextrin,
polyvinylpyrrollidone, low melting waxes, and ion exchange resins, as well
as combinations of any two or more thereof. Other suitable pharmaceutically
acceptable excipients are described in Remington's Pharmaceutical Sciences,
Mack Pub. Co., New Jersey (1991).
Pharmaceutical compositions containing estrogen receptor
modulating compounds may be in any form suitable for the intended method
of administration, including, for example, a solution, a suspension, or an
emulsion. Liquid carriers are typically used in preparing solutions,
suspensions, and emulsions. Liquid carriers contemplated for use include, for
example, water, saline, pharmaceutically acceptable organic solvent(s),
pharmaceutically acceptable oils or fats, as well as mixtures of two or more
thereof. The liquid carrier may contain other suitable phaxma.ceutically
acceptable additives such as solubilizers, emulsifiers, nutrients, buffers,
preservatives, suspending agents, thickening agents, viscosity regulators, or
14
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
stabilizers. Suitable organic solvents include, for example, monohydric
alcohols, such as ethanol, and polyhydric alcohols, such as glycols. Suitable
oils include, for example, soybean oil, coconut oil, olive oil, safflower oil,
cottonseed oil. For parenteral administration, the carrier can also be an oily
ester such as ethyl oleate, isopropyl myristate. Compositions may also be in
the form of microparticles, microcapsules, liposomal encapsulates, as well as
combinations of any two or more thereof.
The compounds may be administered orally, parenterally,
sublingually, by inhalation spray, rectally, vaginally, or topically in dosage
unit forxnulations containing conventional nontoxic pharmaceutically
acceptable carriers, adjuvants, and vehicles as desired. Topical
administration may also involve the use of transdermal administration such
as transderrrial patches or ionophoresis devices. The term parenteral as used
herein includes subcutaneous injections, intravenous, intramuscular,
intrastemal injection, or infusion techniques.
Injectable preparations, for example, sterile injectable aqueous or
oleaginous suspensions may be formulated according to the known art using
suitable dispersing or wetting agents and suspending agents. The sterile
injectable preparation may also be a sterile injectable solution or suspension
in a nontoxic parenterally acceptable diluent or solvent, for example, as a
solution io. 1,3-propanediol. Among the acceptable vehicles and solvents that
may be employed are water; Ringer's solution, and isotonic sodium chloride
solution. In addition, sterile, fixed oils are conventionally employed as a
solvent or suspending medium. For this purpose, any bland fixed oil may be
employed including synthetic mono- or diglycerides. In addition, fatty acids
such as oleic acid can be useful in the preparation of injectables.
Suppositories for rectal or vaginal administration of the drug can be
prepared by mixing the drug with a suitable nonirritating excipient such as
cocoa butter and polyethylene glycols that are solid at ordinary temperatures
but liquid at the rectal temperature and will therefore melt in the rectum and
release the drug.
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Solid dosage forms for oral administration may include capsules,
tablets, pills, powders, and granules. In such solid dosage forms, the active
compound may be admixed with at least one inert diluent such as sucrose
lactose or starch. Such dosage forms may also comprise, as is normal
practice, additional substances other than inert diluents, e.g., lubricating
agents such as magnesium stearate. In the case of capsules, tablets, and
pills,
the dosage forms may also comprise buffering agents. Tablets and pills can
additionally be prepared with enteric eoa.tings.
Liquid dosage forms for oral administration may include
pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and
elixirs containing inert diluents commonly used in the art, such as water.
Such compositions may also comprise adjuvants, such as wetting agents,
emulsifing and suspending agents, cyclodextrins, and sweetening, flavoring,
and perfuming agents.
The compounds can also be administered in the form of lipsomes. As
is known in the art, liposomes are generally derived from phospholipids or
other lipid substances. Liposomes are formed by mono- or multilamellar
hydrated liquid crystals that are dispersed in an aqueous medium. Any non-
toxic, physiologically acceptable and metabolizable lipid capable of forming
liposomes can be used. The present compositions in liposome form can
contain, in addition to a compound, stabilizers, preservatives, excipients.
The
preferred lipids are the phospholipids and phosphatidyl eholines (lecithins),
both natural and synthetic. Methods to form liposomes are known in the art
(Prescott 1976).
Transdermal patches are well known for delivery of nicotine,
nitroglycerin and birth control. These can be utilized with these formulations
as well. Depos that are implanted under the skin or ip can also be used,
similarly to the manner of delivering birth control.
III. Methods of Administration
Compounds can be administered in a variety of ways including
enteral, parenteral, pulmonary, nasal, mucosal and other topical or local
routes of administration. For example, suitable modes of administration
16
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
include oral, subcutaneous, transdermal, transmucosal, iontophotetic,
intravenous, intramuscular, intraperitoneal, intranasal, subdural, rectal,
vaginal and inhalation.
An effective amount of the compound or composition is administered
to treat andJor prevent an estrogen receptor-mediated disorder in a human or
animal subject. Modulation of estrogen receptor activity results in a
detectable suppression or up-regulation of estrogen receptor activity either
as
compared to a control or as compared to expected estrogen receptor activity.
Effective amounts of the compounds generally include any amount sufficient
to detectably modulate estrogen receptor activity by any of the assays
described herein, by other activity assays known to those having ordinary
skill in the art, or by detecting prevention andlor alleviation of symptoms in
a
subject afflicted with an estrogen receptor-mediated disorder.
The effective amount will also be determined based on when the
compounds are administered. Estrogen/hormone therapy (ET/HT) has been
associated with the reduced risk of developing AD when treated at the
menopausal transition in women Brinton, R. D. Impact of estrogen therapy
on Alzheimer's disease: a fork in the road? CNS Drugs 2004, 18, 405-422.
For example, results of the Cache County Study indicate that women who
receive ET/HT at the time of menopause and continue for 10 years have a 3-
fold lower risk of developing AD, Zandi, et al. JAMA 2002, 288, 2123-2129,
whereas the recent data from the Wornen's Health Initiative Memory Study
indicate that women who begin the therapy late in menopause have a greater
risk of developing AD, Espeland, et al. Women's Health Initiative Memory
Study. JAMA 2004, 291, 2959-2968; Shumaker, et al., JAAIA 2004, 291,
2947-2958. These clinical observations are consistent with basic science
analyses of estrogen-inducible molecular mechanisms in the brain, indicating
a healthy cell bias of estrogen action.
Estrogen receptor-mediated disorders that may be treated include any
biological or medical disorder in which estrogen receptor activity is
implicated or in which the inhibition of estrogen receptor potentiates or
retards signaling through a pathway that is characteristically defective in
the
17
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
disease to be treated. The condition or disorder may either be caused or
characterized by abnormal estrogen rcceptor activity. Representative
estrogen receptor-mediated disorders include, for example, osteoporosis,
atherosclerosis, estrogen-mediated cancers (e.g., breast and endometrial
cancer), Turner's syndrome, benign prostate hyperplasia (i.e., prostate
enlargement), prostate cancer, elevated cholesterol, restenosis,
endometriosis, uterine fribroid disease, hot flashes, and skin and/or vagina
atrophy. Other estrogen receptor-mediated conditions that may be treated
include neurological diseases and disorders including memory loss and
dementia, and neurodegenerative disease, including Alzheimer's disease.
In addition to the potential beneficial effects of estrogen on episodic
memory, some evidence suggests that HT reduced the risks of both dementia
(including AD) and mild cognitive impairment (MCI). MCI is a condition
thought to represent a transitional state between normal cognition and
dementia in some individuals, with a 12% conversion rate from MCI to
dementia each year. Observational studies repeatedly document that women
taking HT enjoy an 30% reduced risk for dementia compared with women
not taking HT [odds ratio range, 0.306 (Yaffe et a1.,1998 JAMA 279:688;
Hogervorst et al., 2003 Cochrane Database Syst Rev CD003122)]. Thus,
observational studies suggest that declining reproductive function could be a
modifiable risk factor for dementia or that HT/ET could serve a protective
role against some of the risks for developing dementia.
Several recent observational studies have identified that the stage of
reproductive aging at which HT/ET is started modifies the risk of dementia.
In these studies, women who take HT/ET during the late menopause
transition or early postmenopause have a lower risk of dementia than those
starting HT/ET later (Zandi et al., 2002 JAMA 288:21239; Henderson et al.,
J Neurol Neurosurg Psychiatry 76:103 2005). Thus, the timing of starting
HT/ET relative to the menopause has been proposed to be one factor
explaining the otherwise discordant observations between the observational
studies and the RCTs (Resnick and Henderson, 2002 JAMA 288:21702;
18
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Manson et al., 2006 Menopause 13:139). Recent preclinical studies reviewed
below highlight the importance of timing of ET in this report.
Successful treatment of a subject may result in the prevention,
inducement of a reduction in, or alleviation of symptoms in a subject
afflicted with an estrogen receptor-mediated medical or biological disorder.
Thus, for example, treatment can result in a reduction in breast or
endometrial tumors and/or various clinical markers associated with such
cancers. Treatment of Alzheimer's disease can result in a reduction in rate of
disease progression, detected, for example, by measuring a reduction in the
rate of increase of dementia.
Historically, there has been a presumption that declining reproductive
function plays no role in the onset of mood disorders that occur during
midlife in women. The symptoms of depression during the menopause
transition also were assumed to be transient and of such minor severity that
they were dismissed to be of little clinical consequence. Recent studies,
however, suggest that these presumptions are incorrect. First, several
community-based longitudinal studies have reported the relative
independence of depressions during the menopause transition and hot
flushes: both occur at this stage of life, but depression is not simply caused
by hot flushes (Avis et al., 2001 Soc Sci Med 52:345). Second, recent
longitudinal studies that followed women with no past history of depression
demonstrated an increased risk of first-onset depressions during the late
menopause transition (Schmidt et al., 2004 Am J Psychiatry 161:22384;
Cohen et al., 2006 Arch Gen Psychiatry 63:385; Freeman et al., 2006 Arch
Gen Psychiatry 61:62). Finally, both major and minor depressions are
clinically significant to women at midlife, because both are associated with
an increased risk for several other medical conditions (Wassertheil-Smoller et
al., 2004 Arch Intern Med 164:289) relevant to the health of women at
midlife (e.g., cardiovascular disease, dementia, and the metabolic syndrome).
The majority of women do not develop depression during the
menopause transition, and, therefore, reproductive aging is not uniformly
associated with either depressive symptoms or the syndrome of depression.
19
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Nonetheless, despite numerous studies concluding that the menopause is not
associated with an increased risk for developing depression in women,
several other longitudinal, community-based studies reported an association
between the menopause transition and an increased risk for depression
(Schmidt, 2005 Am J Med 118:54). Indeed, five recent longitudinal studies
all have documented an increased risk for depression during the menopause
transition, with odds ratios ranging from 1.8 to 2.9 compared with the
premenopause (Bromberger et al., 2001 Am J Public Health 91:14352;
Freeman et al., 2004 Arch Gen Psychiatry 61:62, 2006 Arch Gen Psychiatry
63:375; Schmidt et al., 2004 Am J Psychiatry 161:223 84; Cohen et al., 2006
Arch Gen Psychiatry 63:385). These data suggest that events surrounding the
final menstrual period may predispose some women to develop clinically
significant depressive illness. Although several factors could precipitate
depression in these women, endocrine events are suggested by the stage of
the menopause transition (i.e., late) during which depressions appeared. The
late transition is characterized by more prolonged hypogonadism than the
early perimenopause, during which estradiol secretion may be increased.
Thus, the timing of appearance of the depressions observed suggest an
endocrine mechanism related to the perimenopause (estradiol withdrawal
and/or recent-onset of prolonged hypogonadism) in the pathophysiology of
perimenopausal depression.
Efforts to investigate the potential role of declining ovarian honnone
secretion in the onset of depression have examined the effects on mood of
administering HT/ET in women with perimenopausal and postmenopausal
depression. The antidepressant efficacy ofestradiol has been examined in
three relatively recent RCTs of women meeting standardized diagnostic
criteria for major and minor depression, who were randomly assigned to
enter double-blind, placebo-controlled trials (Schmidt et al., 2000; Soares et
al., 2001 Arch Gen Psychiatry 58:529 ; Morrison et al., 2004 Biol Psychiatry
55:406). In perimenopausal women, short-term administration (3 weeks) of
estradiol significantly decreased depression scores compared with both
baseline and placebo conditions. In one study, a full or partial therapeutic
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
response was seen in 80% of perimenopausal women on estradiol compared
with 22% of those on placebo (Schmidt et al., 2000). The efficacy of ET in
perimenopausal depression is consistent with the observed effect size (0.69)
in a recent meta-analysis of studies examining the effects of estrogen on
mood (Zweifel and O'Brien, 1997 Psychoneuroendocrinology 22:189). The
therapeutic response to estradiol was observed in both major and minor
depression as well as in women with and without hot flushes. Thus, the
efficacy of ET in perimenopausal depression is not solely a product of its
ability to reduce the distress of hot flushes. In contrast to these studies in
perimenopausal depression, the administration of estradiol under similar
conditions failed to improve mood in depressed women who were 5 years
postmenopause (Morrison et al., 2004). Thus, the effects of estradiol on
depression may be limited to perimenopausal women. Additionally, as with
the potential effects of estrogen on the course of dementia, the stage of
reproductive aging at which women present and/or commence ET might
modify the observed outcomes.
In summary, the majority of women do not develop depression during
or after the menopause transition. Nevertheless, recent prospective studies
monitoring both reproductive status and mood have documented that, for
some women, perimenopause-related events increase the risk for the onset of
depression. The role of ovarian function in these episodes of depression is
suggested by both the timing of their onset relative to the last menstrual
period and the antidepressant efficacy of short-term ET.
The amount of active ingredient that may be combined with the
carrier materials to produce a single dosage form will vary depending upon
the estrogen-mediated disease, the host treated and the particular mode of
administration. It will be understood, however, that the specific dose level
for any particular patient will depend upon a variety of factors including the
activity of the specific compound employed, the age, body weight, general
health, sex, diet, time of administration, route of administration, rate of
excretion, drug combination, and the severity of the particular disease
undergoing therapy. The prophylactically or therapeutically effective amount
21
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
for a given situation can be readily determined by routine experimentation
and is within the skill and judgment of the ordinary clinician.
For exemplary purposes, a prophylactically or therapeutically
effective dose will generally be from about 0.01 mg/kg/day to about 100
mg/kg/day, preferably from about 0.1 mg/kg/day to about 2 0 mg/kg/day,
and most preferably from about 1 mg/kg/day to about 10 mg/kg/day of a
estrogen receptor modulating compound , which may be administered in one
or multiple doses.
IV. Kits
Kits may be provided which contain the formulation to be
administered. The formulation may be administered once a day or more than
once a day. The formulation can be administered enterally, parenterally, or
topically. The kits typically contain the active agent(s) to be administered,
excipients and carriers, and instructions for administration of the
formulation. The kits may also contain equipment/devices used to
administer the formulation, such as syringes.
The present invention will be further understood by reference to the
following non-limiting examples.
Examples
Example 1: Identification of PhytoSERMS
ERP has been associated with estrogen-induced promotion of
memory function and neuronal survival. Based on the optimized complex
structure of human ERP LBD bound with genistein, computer-aided
structure-based virtual screening against a natural source chemical database
was conducted to determine the occurrence of plant-based ERO-selective
ligands. Twelve representative hits derived from database screening were
assessed for their binding profiles to both ERs, three of which displayed over
100-fold binding selectivity to ERP over ERa.
22
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Materials and Methods
Identification of Molecules
Identification of Compounds in Database
All computational work was performed on a SGI Octane workstation
equipped with the IRIX 6.5 operating system (Silicon Graphic Inc.). First,
the 3D crystallographic structure of human ERP LBD complexed with
genistein was downloaded from the Protein Data Bank (PDB ID: 1QKM).
The complex structure was fixed and energy minimized with the Accelrys
molecular modeling software package lnsightll 2000 (Accelrys Inc.). An in-
house 2D natural source chemical collection containing approximately 25
000 plant-based natural molecules or derivatives was converted to a 3D
multiconformational database with the Accelrys modeling software package
Catalyst 9.8 (Accelrys Inc.).
The receptor-docking site was defined based on the binding position
of genistein in the receptor and specified as all atoms within 10 A of the
center carbon of genistein. GOLD 2.0 (Genetic Optimization for Ligand
Docking), an automated ligand docking program distributed by CCDC
(Cambridge Crystallographic Data Center), was applied to calculate and rank
the molecules based on their complementarities with the receptor binding
site, on both geometrical and chemical features.
Prior to the database screening, initial validation using genistein as
the test ligand was conducted. The aim of the validation test was to evaluate
the effectiveness of the algorithm of the docking program in identifying the
experimentally observed binding mode of the ligand in the receptor, to
determine whether the program is applicable to the specific target system in
this study. Tn addition, the validation test was used to determine the optimal
parameter settings for the later database screening. Twenty docking runs
were carried out on the test complex, using the fastest default generic
algorithm parameters optimized for virtual library screening, and the
GoldScore fitness function was applied. The validation test demonstrated
that, based on the specified parameter settings, GOLD was effective in
capturing the contributive hydrogen bond donor (NDI in His 475) crucial to
23
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
the binding and reproducing the nearly coincident solution in terms of both
the binding orientation and conformation of genistein as observed in the
experimental measurement (see Figure 1). The root-mean-square (RMS)
deviations were computed between the observed experimental position and
the GOLD solutions, with RMSD 0.3299 and 0.4483 compared to top-ranked
and worst solutions, respectively. The average RMSD of all solutions was
0.3566, which is regarded as a good prediction based on the subjective
classifications defined by the program developer (refer to the program
manual), suggesting that this program is reliable and applicable to the
database screening toward ERP.
Using the parameter settings determined in the validation test, the 3D
natural source chemical database was input and docked into the prepared
ERP binding site in a flexible docking manner (full ligand and partial
protein) and scored based on the GoldScore fitness function. Five hundred
resultant top-scoring molecules were filtered via visual screening in the
context of the receptor in Insightli. Based on visual analysis, 100 molecules
underwent further analysis by Affinity, a more complex and predictive ligand
docking program to refine the binding modes predicted by GOLD. The
criteria used for the selection of candidate molecules for investigation
included the following (a) formation of hydrogen bond with donor atom
ND1 in His 475; (b) hydrophobic and hydrophilic balance appearing in the
structure (e.g., the molecule should potentially have two relatively
hydrophilic sides and a hydrophobic center to enhance both the steric and
electrostatic complementarity with the receptor); (c) bound pose of the
molecule in the receptor; and d) structural diversity. Finally, molecules that
met the above criteria were computationally predicted for their drug-likeness
(Lipinski's Rule of Five) and blood-brain barrier (BBB) penetration
properties.
The ligand binding domains of the human ERa and ERP are
approximately 60% homologous. Structural modeling and mutational
analyses indicate that two variant amino acid residues along the ligand
binding pocket, Leu 384 and Met 421 in ERa, which are replaced with Met
24
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
336 and Ile 373, respectively, in ERf3, are the key molecular constituents
underlying discriminative binding of selective ligands to either receptor
subtypes. Sun, et al. Mol. Endocrinol. 2003, 17, 247-258. This slight
structural variance serves as the foundation for both design and discovery of
ER specific ligands. The similarities in the chemical features of both pairs
of
residues presents a substantial challenge to discover a selective ligand based
on this difference. Of the known natural source ERj3-selective ligands,
genistein remains the most selective. However, an increasing number of
synthetic compounds are emerging showing greater selectivity than genistein
for ERP, as evidenced by the compound DPN developed in
Katzellenebogen's laboratory. Computer-aided structure-based virtual
database screening provides an efficient approach to rationally highlight a
small group of lead candidates from a large number of compounds for
investigation at the bench.
Determination of BindingAfflnity and Selectivity
The binding affinity and selectivity of candidate molecules yielded
from database screening were determined by a fluorescent polarization
competitive binding assay using purified baculovirus-expressed human ERP
or ERa and a fluorescent estrogen ligand EL Red (PanVera Corp.). Test
molecules were serially diluted to a 2x concentration in assay buffer (200
[.M to 200 pM). Fifty microliters of preincubated 2x complex of ER(3 (30
nM) or ERP (60 nM) and EL Red (2 nM) was added to each well in a 96-
well Non-binding Surface black microplate (Coming Life Sciences) for a
final volume of 100 VL. Negative controls containing ER and EL Red
(equivalent to 0% inhibition) and positive controls containing only free EL
Red (equivalent to 100% inhibition) were included. After a 2-h incubation
period at room temperature, the polarization values were measured using a
Tecan GENios Pro reader at 535 nm/590 nm excitation/emission and plotted
against the logarithm of the test molecule concentration. IC50 values
(concentration of test molecule that displaces half of the EL Red from ER)
were determined from the plot using a nonlinear least-squares analysis.
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Results
31 molecules that can form a hydrogen bond with NDI in His 475
were selected and grouped into three categories based upon the chemical
features that favored both the van der Waals (VDW) contact (the number of
the rings in the structure) and electrostatic interactions (the number of the
hydrogen bonds) with the receptor. 10 molecules that have strong VDW
interactions with the receptor, but without contributive hydrogen bonding,
were grouped in Category IV. These molecules contain three or four five- or
six-membered rings in their structures that could promote the hydrophobic
interactions with the center of the receptor binding site as observed in
endogenous esttogen 170-estradiol that consists of four rings in its structure
and binds to the estrogen receptor with a high affinity.
Table 1 summarizes the 1C5n binding results of test molecules to both
ERa and ERP as well as the binding selectivity of representative molecules
selected from four categories.
26
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
T"aisle 1BfiaEnp~ Affinit~ (1~) and SeteaLi~ty ef
Representative ~~~~~ulas forEstrogen RerePta~ Ct an'd A
C~mpd S1~~~~ctuae E Ra , eo(tt vity
NC*
-AYM
4,66 N,7 ~7,2
~~
~
751 M~ 4,07
nM nu
2 N~' 0,68 > 100
NO ;?,;:};:E
4
rQ
~~~ ~~~ ~ 100
6 NC NC
7 r1h 85.7 4,10 IOR
~ ~C, 4r46 gm
.:= ~~
KC, NC
NC, NC
õN`= ~y~yu
4 r~ ~,7,P^~ <ri~.+~Y1 py.l~-f
2ni~6 .k.'v'l,r
.El. .
c'`~, ~'~r~ =,y , e
Y.e 5~
*NC: Nmoanvergence within the dose r:aiigea predicfin~ that
ei~lier the- motsc~~ ~oes not l.hia ta th+e r~~eptor or that tie
binding ~.inity io ven- lnva, with an ICDo ~~ater iT-An I mlvL
~
As expected, the negative control steroid, progesterone, does not bind
to either ER. As a positive naturai source estrogen control, genistein was
found to bind to ER(3 with a 47.2-fold greater binding selectivity over ERa,
27
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
but at an affinity one-fourth of 17~-estradiol. Among 12 molecules tested,
five molecules, 1, 2, 5, 7, and 8, showed binding selectivity to ERP over
ERa, 3 of which, 2, 5, and 8, displayed the selectivity over 100-fold.
Preliminary structure and binding activity relationship analyses revealed that
both the central hydrophobic skeletal structure and the connected two polar
`arms' contribute to the binding affinity of ligands to both ERs. The
enhanced VDW contact derives mainly from the central hydrophobic feature
of the molecule. For example, the number of rings increases the binding
affinity of molecules to the receptor, as indicated by the VDW value of 17p-
estradiol (-67.98) versus that of genistein (-60.75) and molecule 9 (-58.04),
which are well correlated with their order-different binding affinities.
Meanwhile, the hydrogen bonds derived from the two polar "arms" of the
molecule are essential for the binding as we11. The lack of one "arm" of the
hydrogen bond, as represented by molecule 4 and 6, or two `arms', as
represented by 10 and 12, even though the latter two molecules can elicit
strong VDW interactions with the receptor, with the VDW value of -72.58
and -69.19, respectively, leads to either very weak or no binding. With
respect to the binding selectivity, as demonstrated in the modeling complex
structures of a synthetic ERP-selective agonist, PPT, Stauffer, et al.. J.
Med.
Chem. 2000, 43, 4934-4947 and a synthetic ERP-selective agonist, DPN,
Meyers, et al., J. Med. Chem. 2001, 44, 4230-425I, with both ERs, Zhao, et
al. 2004 Abstract Book; The Keystone Symposia: Nuclear Receptors: Steroid
Sisters, Keystone, CO; February 2004, relatively larger molecular size favors
the binding selectivity for ERD over ERa, as represented by molecule 3 and
11.
These analyses shed light on the future search and design of more
active and selective ER subtype-selective ligands. Further, 3 out of 12
representative molecules yielded from database searching displayed over
100-fold selectivity toward ERP over ERa, demonstrating the effectiveness
of this computer-aided virtual screening approach applied in the present
study in the discovery of potential molecules that preferentially interact
with
ERP.
28
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Example 2: Preclinical Identification of ERji-Selective PhytoSERM
Combinations for Prevention of Neurodegeneration
The impact of ERb-selective PhytoSERMs when administered singly
or in combination on neuronal survival and molecular/functional markers
associated with prevention of neurodegeneration and Alzheimer's disease
(AD) was investigated.
Materials and Methods
1713-Estradiol was purchased from Steraloids (Newport, RI).
Genistein, daidzein and equol were purchased from Indofine Chemical
(Hillsborough, NJ). IBS003569 was purchased from InterBioScreen
(Moscow, Russia). The structures of these compounds are shown in Figure
1.
In Vitro Treatments: Test compounds (or combinations) were first
dissolved in analytically pure DMSO (10 mM) and diluted in Neurobasal
medium to the working concentrations right before treatments.
In Vivo Treatments: Test compounds (or combinations) were first
dissolved in analytically pure DMSO and diluted in corn oil (50 ml of
DMSO in 950 ml of corn oil) to the working concentrations at 100 mg/ml for
17p-estradiol and 10 mg/mI for phytoSERMs.
In vitro Assays
ERa Binding Assays
ERa receptor (aboutØ2 mg/ml, Affinity Bioreagents) is diluted to
about 2 x 103 mg/ml in phosphate-buffered saline ("PBS") at a pH of 7.4.
Fifty microliters of the EPa -PBS solution is then added to each of the wells
of a flashplate. The plates are sealed and stored in the dark at 4 C for 16-18
hours. The buffered receptor solution is removed just prior to use, and the
plates are washed 3 times with 200 microliters per well of PBS. The washing
is typically performed using a slow dispense of reagent into the wells to
avoid stripping the receptor from the well surface.
For library screening, 150 microliters of 1 nM 3 H-estradiol (New
England Nuclear, Boston, Mass.) in 20 mM Tris-HCI, 1 mM EDTA, 10%
29
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
glycerol, 6 mM monothioglycerol, 5 mM KCI, pH 7.8 is mixed with 50
microliters of the test compound (in same buffer) in a 96 well mictrotiter
plate, resulting in a final estradiol concentration of 0.6 nM. In addition,
several dilutions of estradiol, centered on the IC50 of 1-2 nM, are also added
to individual wells to generate a standard curve. The plates are gently shaken
to mix the reagents. A total of 150 microliters from each of the wells is
added to the corresponding wells of the pre-coated ERoc plates. The plates are
sealed and the components in the wells are incubated either at room
temperature for 4 hours or at 4 C overnight. The receptor bound ligand is
read directly after incubation using a scintillation counter. The amount of
receptor bound ligand is determined directly, i.e., without separation of
bound from free ligand. If estimates of both bound and free ligand are
required, the supernatant is removed from the wells, liquid scintillant is
added, and the wells are counted separately in a liquid scintillation counter.
ERfl Binding Assays
ERA receptor (.aboutØ2 mg/ml, Affinity Bioreagents) is diluted to
about 2 xI03 rng/ml in phosphate-buffered saline ("PBS") at a pH of 7.4.
Fifty microliters of the ERA -PBS solution is then added to each the wells of
a flashplate. The plates are sealed and are stored in the dark at 4 C for 16-
18
hours. The buffered receptor solution is removed just prior to use, and the
plates are washed 3 times with 200 microliters per well of PBS. The washing
is typically performed using a slow dispense of reagent into the wells to
avoid stripping the receptor from the well surface.
For library screening, 150 microliters of 1 nM 3H-estradiol (New
England Nuclear, Boston, Mass.) in 20 mM Tris-HC1, 1 mM EDTA, 10%
glycerol, 6 mM monothioglycerol, 5 mM KCI, pH 7.8 was mixed with 50
microliters af the test compound (in same buffer) in a 96 well microtiter
plate, resulting in a final estradiol concentration of 0.6 nM. In addition,
several dilutions of estradiol, centered on the IC50 of 1-2 nM is also added
to
individual wells to generate a standard curve. The plates are then gently
shaken to mix the reagents. A total of 150 microliters from each of the wells
is added to the corresponding wells of the pre-coated ERfi plates. The plates
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
are sealed and the components in the wells are incubated at room
temperature either for 4 hours or at 4 C overnight. The receptor bound ligand
is read directly after incubation using a scintillation counter. The atnount
of
receptor bound ligand is determined directly, i.e., without separation of
bound from free ligand. If estimates of both bound and free ligand are
required, the supernatant is removed from the wells, liquid scintillant is
added, and the wells are counted separately in a liquid scintillation counter.
ERo/ERfl Transactivation Assays
Construction of Transfected CHO Cells
Transfected CHO cells were derived from CHO KI cells obtained
from the American Type Culture Collection ("ATCC", Rockville, Md.). The
transfected cells were modified to contain the following four plasmid
vectors: (1) pKCRE with DNA for the human estrogen receptor, (2) pAG-60-
neo with DNA for the protein leading to neomycin resistance, (3) pRO-LUC
with DNA for the rat oxytocin promoter and for firefly luciferase protein,
and (4) pDR2 with DNA for the protein leading to hygromycine resistance.
All transformations with these genetically modified CHO cells are performed
under rec-VMT containment according to the guidelines of the COGEM
(Commzssie Genetische Modificatie). Screening is performed either in the
absence of estradiol (estrogenicity) or in the presence of estradiol (anti-
estrogenieity).
Assays to Assess Neuronal Function
Neuronal culture pret2aration
Primary cultures of hippocampal neurons were obtained from
Embryonic Day 18 (El 8d) rat fetuses. Briefly, after dissected from the brains
of the rat fetuses, the hippocampi were treated with 0.02% trypsin in Hank's
balanced salt solution (137 mM NaCl, 5.4 mM KC1, 0.4 mM KH2PO4, 0.34
mM Na2HPO4.7H20, 10 mM glucose, and 10 mM HEPES) for 5 min at
37 C and dissociated by repeated passage through a series of fire-polished
constricted Pasteur pipettes. Between 2 x 104 and 4 x 104 cells were plated
onto poly-D-lysine (10 g/ml) -coated 22 mm coverslips in covered 35 mm
petri dishes for morphological analysis, and 1 x 105 cells/ml were plated
31
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
onto poly-D-lysine-coated 24-well, 96-well culture plates or 3-5 x 105
cells/ml onto 0. 1% polyethylenimine-coated 60mm petri dishes for
biochemical analyses. Nerve cells were grown in phenol-red free Neurobasal
medium (NBM, Invitrogen Corporation, Carlsbad, CA) supplemented with
B27, 5 U/ml penicillin, 5 gg/mi streptomycin, 0.5 mM glutamine and 25 M
glutamate at 3 7 C in a humidified 10% CO2 atmosphere at 37 C for the first
3 days and NBM without glutamate afterwards. Cultures grown in serum-
free Neurobasal medium yield approximately 99.5% neurons and 0.5% glial
cells.
Neuroprotection measurements
Glutamate exposure
Primary hippocampal neurons were pretreated with compounds for
48 hr followed by exposure to 100 gM glutamate for 5 min at room
temperature in HEPES buffer containing 100 mM NaCI, 2.0 mM KCI, 2.5
mM CaC12, 1.0 mM MgSOq., 1.0 mM NaH2PO4, 4.2 mnM NaHCO3, 10.0
mM glucose and 12.5 mnM T-LEPES. Immediately following glutamate
exposure, cultures were washed once with HEPES buffer and replaced with
fresh Neurobasal medium containing the test compounds. Cultures were
returned to the culture incubator and allowed to incubate for 24 hr prior to
cell viability measurements on the following day.
Western immunoGlotting
CREB phosphorylation
Nuclear lysates were prepared as following: Briefly, hippocampal
neurons grown on poly-D-lysine coated culture dishes were treated with
compounds for appropriate periods, washed with cold PBS once and scraped
into I ml PBS. Cells were then centrifuged at 5,000 rpm for 5 min, and the
pellet was dissolved in Cytoplasm Extraction buffer (10 mM HEPES, 1 mM
EDTA, 60 mM KC1, 0.075% Igepal and protease and phosphatase inhibitor
cock-tail) and suspended by passage through a 200 ~L1 pipette tip. After 30-45
RPM of incubation at 4 C, the samples were centrifuged at 5,000 rpm for 5
min to generate the cytoplasmic extract in the supernatant. The supernatant
cytoplasmic extract was removed, and Nuclear Extraction buffer (20mM Tris
32
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
HCI, 1.5mM MgC12, 420mM NaCI, 0.2mM EDTA, 25% glycerol, 0.5%
Igepal and protease and phosphatase inhibitor cocktail) was added to the
pellet followed by 5M NaCI to break the nuclear membrane. Following 30-
45 RPM of incubation at 4 C, the samples were centrifuged at 12,000 rpm
for 10 min to generate a supernatant containing the nuclear extract.
Protein concentration was determined by the BCA method. An
appropriate volume of 2X sample buffer was added to the protein samples,
and samples were boiled at 95 C for 5 rnin. Samples (25 g of proteins per
well) were loaded on a 10% SDSPAGE gel and resolved by standard
electrophoresis at 90V. Proteins were then electrophoretically transferred to
Immobilon-P PVDF membranes overnight at 32 V at 4 C. Membranes were
blocked for 1 hr at room temperature in 10% non-fat dried milk in PBS
containing 0.0 5% Tween 20 (PBS-T), incubated with appropriate primary
antibodies against phospho-CREB (pSER133, mouse monoclonal, 1:2000;
Cell Signaling Technology, Beverly, MA), CREB (rabbit polyclonal, 1:1000;
Cell Signaling Technology, Beverly, MA), spinophilin (rabbit polyclonal,
1:1000; Upstate Biotecholagy, Lake Placid, NY), actio. (mouse monoclonal,
1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or histone H1
(mouse monoclonol, 1:250; Santa Cruz Biotechology, Inc., Santa Cruz, CA)
at temperatures and times specified by the antibody providers. All primary
antibodies were dissolved in PBS-T with 1 % horse serum (for mouse
monoclonal antibody) or goat serum (for rabbit polyclonol). After washing in
PBS-T, the membranes were incubated with horseradish peroxidase-
conjugated anti-mouse IgG (1:5000; Vector Laboratories, Inc., Burlingame,
CA) in PBS-T with 1% horse serum or anti-rabbit IgG (1:5000; Vector
Laboratories, Inc., Burlingame, CA) in PBS-T with 1% goat serum for 1 hr.
Immunoreactive bands were visualized by TMI3 detection kit (Vector
Lahoratories. Inc., Burlingame, CA) and quantified using Un-Scan-It gel
image software (Silk Scientific, Inc., Orem, UT). Following transfer, gels
were stained with Coomassie blue (Bio-rad Laboratories, Hercules, CA) to
ensure equal protein loading.
33
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Bc1-2 and Bcl-xl expression
Primary hippocampal neurons were pretreated with compounds for
48 hr before the cells were lysed by incubation in ice-cold lysis buffer
containing: 0.005% SDS, 0.1% Igepal, 0.2 mM sodium orthovanadate, 0.2
mM phenylmethylsulfonylfluoride and protease inhibitor mixture in PBS at
4 C for 45 min. Cell lysates were centrifuged at 10,000 rpm at 4 C for 10
min, and the concentration of protein in the supematant was detennined
using the BCA Protein Assay (Pierce Biotechnslogy, Inc., Rockford, IL). 25
g of total protein were diluted in 15 l 2X SDS containing sample buffer
and the final volume was made 30 p1 with water. After denaturalization on a
hot plate at 95-100 C for 5 min, 25 g1 ol'the mixture were loaded per lane on
10% SDS-polyacrylamide mini-gels followed by electrophoresis at 90V. The
proteins were then electro-transferred to polyvinylidene difluoride
membranes (Millipore Corp., Bedford, MA) from the ge1s. Nonspecific
binding sites were blocked with 5% nonfat dry milk in PBS containing
0.05% TweenTm-20 (PBS-TweenTM). Membranes were incubated with the
primary monoclonal antibody against Bc1-2 (Zymed Laboratories, Inc., S.
San Francisco, CA) diluted 1:250 in PBS-TweenTM with 1% horse serum
(Vector Laborataries, Inc., Burlingame, CA) overnight at 4"C, then
incubated with the secondary horseradish peroxidase (HRP)-conjugated
horse anti-mouse IgG (Vector Laboratories, Inc, Burlingame, CA) diluted
1:5,000 in PBS-TweenTM with 1% horse serum for 2 hr at room temperature,
and Bcl-2 proteins were visualized by developing the membranes with TMB
substrate for peroxidase (Vector Laboratories, Inc., Burlingame, CA). (3-
Actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) level was
determined to ensure equal protein loading, and high-range Precision Protein
Standards (Bio-Rad Laboratories, Hercules, CA) was used to determine
protein sizes. Relative intensities of bands were quantified by optical
density
analysis using an image digitizing software Un-Scarn-Tt version 5.1 (Silk
Scientific, Inc., Orem, UT).
34
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Statistics
Statistically significant differences between groups were determined
by a one way analysis of variance (ANOVA) followed by a Newman-Keuls
post hoc analysis.
In vivo Assa'ys
Immature Rat Uterotrophic Bioassay for Estrogenicity Anti-Estrogenicity
Antiestrogenic activity was determined by the ability of a test
compound to suppress the increase in uterine wet weight resulting from the
administration of 0.2 gg 17-(3-estradiol ("E2") per day. Any statistically
significant decreases in uterine weight in a particular dose group as
compared with the E2 control group are indicative of anti-estrogenicity .
One hundred forty (140) female pups (19 day s old) in the 35-50 g
body weight range are selected for the study. On day 19 of age, when the
pups weigh approximately 35-50 g, they are body weight-order randomized
into treatment pups. Observations for mortality, morbidity, availability of
food and water, general appearance and signs of toxicity are made twice
daily. Pups not used in the study are euthanized along with the foster dams.
Initial body weights are taken just prior to the start of treatment at day 19
of
age. The final body weights are taken at necropsy on day 22 of age.
Treatment commences on day 19 of age and continues until day 20
and. 21 of age. Each animal is given three subcutaneous ("sc") injections
daily for 3 consecutive days. Three rats in each of the control and mid- to
high-level dose test groups are anesthetized with a ketamine/xylazine
mixture. Their blood is collected by exsanguination using a 22 gauge needle
and 5 ml syringe flushed with 10 USP with sodium heparin/mi through the
descending vena cava; and then transferred into a 5 ml green top plasma tube
(sodium heparin (freeze-dried), 72 USP units). Plasma samples are collected
by centrifugation, frozen at -70 C, and analyzed using mass spectrographic
to determine the presence and amount of test compound in the serum . Blood
chemistry is also analyzed to determine other blood parameters. The uteri
from the rats are excised and weighed. The remaining rats are sacrificed by
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
asphyxiation under CO2. The uteri from these rats are excised, nicked,
blotted to remove fluid, and weighed to the nearest 0.1 mg.
In order to determine whether the test compound significantly
affected final body weight, a parametric one-way analysis of variance
(ANOVA) is performed (SIGMASTAT version 2.0, available commercially
from Jandel Scientific, San Rafael, Calif.). Estrogen agonist and antagonist
activity is assessed comparing uterine wet weights across treatment groups
using a parametric ANOVA on loglo transformed data. The data are
transformed to meet assumptions of normality and homogeneity of variance
of the parametric AWQVA. The F value is determined and a Student-
Newman-Kuel s multiple range test is performed to determine the presence
of significant differences among the treatment groups. The test compound is
determined to act as a mixed estrogen agonist/antagonist if the test
compound does not completely inhibit the 17-P-estradiol stimulated
uterotrophic response.
The use of animals was approved by the Institutional Animal Care
and Use Committee at the University of Southern California (Protocol
Number: 10780). Embryonic day 18 Sprague-Dawley rat (Harlan,
Indianapolis, IN) fetuses were used to obtain primary hippocampal neuronal
cultures for in vitro experiments. Young adult (14 to 16-week-old, weighing
from 270-290 g) female ovariectomized Sprague-Dawley rats (Harlan) were
used for in vivo experiments.
In vitro neuroprotection and associated mechanistic studies were
conducted in primary hippocarnpai neurons obtained from embryonic day 18
rat fetuses. Adult female ovariectomized rats were used to relate the in vitro
findings to in vivo environment, along with the assessment of the impact of
PhytoSERMs on brain mitochondrial functions and uterine weight.
During a 2-week surgery recovery following ovariectomy, but before
treatment, rats were placed on a phytoSERM-reduced diet, TD.96155
(Harlan Teklad). Rats were given, once daily, 2 subcutaneous injections of
vehicle (control), 17 P -estradiol (70 pg/kg BW), genistein (6 mg/kg BW), or
36
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
phytoSERM combinations (6 mg/kg BW). Dosages used here are
commensurate with those used in humans.
Following the second injection, animals fasted for 24 hours prior to
sacrifice and brain dissection. Hippocampal and cortical tissues were
collected from one hemisphere and stored for biochemical analyses. The
remaining brain tissues minus cerebellum, pineal gland, and brainstem were
utilized for mitochondrial isolations, followed immediately by mitochondrial
respiratory activity measurements. The rest of mitochondrial samples were
stored for cytochrome c oxidase activity measurements. Uteri were excised,
trimmed of fat and connective tissue, and both a wet weight and a dry weight
were recorded.
Results
The PhytoSERMs tested are shown in Figure 1.
Selective Binding for both ER and ERa
Figure 2 presents the competition binding curves of four known ER
ligands for both ERP and ERa. The IC50 determined for these ligands from
the binding curves are consistent with the previously reported values using
alternative methods such as radioligand assay, demonstrating the reliability
of this assay in determining the binding profiles of small molecules to both
ERs.
Figures 2A and 2B show the competition binding curves for ERa and
ER(3. Data were generated with a fluorescence polarization-based
competitive binding assay using full-length human ERa and ER 0, and
plotted against the logarithm of serially diluted concentrations of the test
compounds (or combinations). Progesterone served as a negative control. 17
P -Estradiol served as a positive control. Combined formulations were
composed of equivalent molar of individual phytoSERMs included. G:
genistein; D: daidzein; E: equol; I: IBS403569. 17p-estradiol has no
binding preference to ERcx or to ER(3. The concentration of a test molecule
resulting in the half-maximum shift in polarization value equals its ICSo.
Non-convergence within the dose range, predicts that either the molecule
does not bind to the receptor or that the binding affinity is very low.
37
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Table 2 shows the binding data for ERa and ERP.
Table 2. Binding data for ERa and ERD
COmAPOatn,& ~Ra
Icst RHA Icso RSA
0~) (W Oa~ ow"
Pa-ogesterone Nsra-t3im~ 1,T=-Bindiag.
179-E~trzdio1 4.0253 1.001 4.9391 0.0325 100.0 0.9611 0.78
Gemzstein 4_735 0.5343 0.9911 0.47799 41.12 0.9909 60.0
.Daidaek 26.65 0.949 0.7V6 1.738 1.967 0.9583 14,27
Equdl 5,876 0.4306 0.9948 0.5825 5571 U986 30.09
13800350 1695 40015 0,9917 7.819 0.435 0.9959 >100
G+-D 9_896 00.2557 0.9365 0.1574 20.62 0.9970 62.87
G+D+E 85.71 0.1610 0.9M 0_1902 1746 0.9m
G+Y}.ÃE-rI 15.85 0.1595 0.9532 01615 12A1 0.9891 60_61
AR13A (%) refers to the relative binding affinity of the test compound
(combination) that is
expressed as the percent of the binding affinity of 17 P -estradiol (RBA =
100%).
IIRZ refers to goodness of fit of nonlinear regression between the binding
curve and the data.
Between 0,0 and 1.0, higher values indicate that the curve fits the data
better. A fit with a R2
at 1.0 indicates that all points lie exactly on the curve with no scatter.
Neuropxotective Effect
Table 3 and Figure 3 show the dose-dependent neuroprotective
effects of four ERR-selective phytoestrogenic molecules against
supraphysiological glutamate (100 VÃM)-induced neurotoxicity in primary
hippocampal neurons by measurement of LDH release. ## P< 0.01
compared to vehicle alone-treated cultures; * P < 0.05 and ** P < 0.01
compared to glutamate alone-treated cultures.
38
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Table 3. Dose-dependent effects of individual phytoSERMs against
glutamate-induced neurotoxicity in primary hippocampal neurons by
LDH
measurementsA
Treatment Genistein LDH Release (% of Control)
Control 100.00 3.09
Glutamate alone 410.99 8.27
1nM 361.03 7.71 **
10nM 350.02 8.21 **
100 nM 347.24 16.96 **
l M 356.79 11.15
377.84 8.45 * *
5
Treatment Daidzein LDH Release (% of Control)
Control 100.00 4.28
Glutamate alone 378.26 11.95
1 nM 338.39 16.49
10 nM 333.98 9.10 *
100 nM 3 01.42 7.70 **
I M 318.49 15.92 * *
10 M 325.41 26.12 *
Treatment Equol LDH Release (% of Control)
Control 100.00 14.95
Glutamate alone 460.27 12.20
1 nM 453.50 23.37
10 nM 403.78 17.02 *
100 n1VI 331.59 9.67 * *
1 M 381.80 12.01 **
10 M 390.21 9.40**
Treatment IBS003569 LDH Release (% of Control)
Control 100.00 2.05
Glutamate alone 281.17 6.77
1 nM 262.41 10.60
lO nM 270.86 12.94
100 nM 220.56 6.80 * *
1 M 246.30 7.70**
10 M 307.53 2.62
39
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
A1'rimary hippocampal neurons grown for 7 DIV were pretreated with the test
phytoSERMs
at serially diluted concentrations for 48 h, followed by a 5-min exposure to
100 mM
glutamate. The arnount of LDH released into the culture media was measured 24
h later.
BData are derived from a single experiment and are representative of at lease
three
independent experiments. Results are presented as the percent of LDH release
from
vehicletreated control cultures and expressed as means ~: S.E.M., n 6. 4#I' <
0.01 compared
to vehicle-treated control cultures, * P < 0.05 and **P < 0.01 compared to
glutamate alone-
treated cultures; [tP < 0.05 and IIP < 0.01 compared to cultures treated with
10 nM
phytoSERMs; P < 0.05 and pP < 0.01 compared to cultures treated with 1[M
phytoSERMs; pP < 0.05 and pfP <
0.01 compared to cultures treated with 10 mM phytoSERMs.
Figure 3 shows the neuroprotective efficacy of four ER(3-selective
phytoestragenic molecules when administered alone at concentrations that
elicited the maximal neuroprotective effects as revealed from the dose-
response analyses (100 nM for all.four molecules), or co-administered,
against supraphysiologicai glutamate (100 M)-induced neurotoxicity in
primary hippocampal neurons by measurement of calcein AM staining.
Results are presented in terms of neuroprotective efficacy
NE =(Vtreatment - vgintamate) / (vcoatrol - Vglutamate) * 100%,
(1)
where V,eatmC1t is the individual value from phytoestrogen-treated cultures,
Vglõtamaw is a mean value from glutamate alone-treated cultures, and Vwõtrot
is
a mean value from vehicle-treated control cultures. 00 P < 0.01 compared to
vehicle alone-treated cultures; * P < 0.05 and **P < 0.01 compared to
glutamate alone-treated cultures.
Data presented in Figure 3 and Table 3 demonstrate that although the
four ERj3-selective phytoestrogenic molecules, when administered
individually, are concentration-dependent and are protective against
excitotoxic glutamate-induced neurotoxicity in primary neurons, these
effects are moderate and arise from the weaker binding to the estrogen
receptor compared to the endogenous estrogen 17p-estradiol (E2). Figure 3
demonstrates that co-administration of 3 or 4 of these phytoestrogens
afforded much greater neuroprotective efficacy compared to administration
of single phytoestrogens or a combination of 2 phytoestrogens.
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Expression of Anti-Apoptotic Proteins Bcl-2 and Bcl-xL
These outcomes are paralleled by the results derived from the vvestern
analyses of the expression of anti-apoptotic proteins, Bcl-2 and Bcl-xL, in
primary neurons. Figures 4A-4B shows the effects on Bcl-2 and Bcl-XL
expression in rat primary hippocampal neurons and hippocampal tissues
derived from adult ovariectomized rats. Primary hippocampal neurons
grown for 7 divisions were treated with the test compounds (or
combinations) for 48 hr followed by Western blot anaiyses. Adult
ovariectomized rats were given, once daily, 2 subcutaneous injections of the
test compounds (or combinations). Rats were sacrificed 24 h later following
the 2nd injection. Hippocampal tissues were homogenized followed by
Western blot analyses. Combined formulations were composed of equivalent
molar in (A) and equivalent weight in (B) of individual phytoSERMs
including G: genistein; D: daidzein; E: equol; and L= IBS003569.
Incubation of neurons with a combination of four phytoestrogens for
48 hours induces a significantly increased expression of both proteins
comparable to those induced by E2. This is illustrated in Figure 4, which
shows the effect of four ERP-selective phytoestrogenic molecules when co-
administered (100 nM for all four molecules) on the expression of the anti-
apoptotic proteins, Bcl-2 and Bcl-xL, in primary hippocampal neurons. **P
< 0.01 compared to vehicle alone-treated cultures. By comparison, a
combination of two phytoestrogens is not sufficient to induce a significant
increase in the expression of both proteins, as also illustrated in Figures 4A
and 4B.
Up-regulation of the Bcl-2 family anti-apoptotic proteins have been
associated with the neuroprotective mechanism elicited by E2. These data
indicate that a combined used of multiple ERP-selective phytoestrogens is
effective to activate the neuroprotective mechanism leading to improved
neuronal survival against neurodegenerative insults. Estrogen receptor
interaction with p85/PI3K also enhances pAkt, which phosphorylates the
proapoptotic protein Bcl-2-associated death protein (BAD) to prevent
heterodimerization with, and inactivation of, Bcl-2. In cortical neurons,
41
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
estradiol induced pAkt translocation to the nucleus. Recent analyses indicate
that estradiol, via the P13K signaling pathway, activates both the Akt and the
ERK1/2 cascades in the same population of cortical and hippocam.pal
neurons. Simultaneous activation of two pathways that prevent mitochondria
from activating cell-death cascades is likely to promote neuron survival.
Increased Expression of the anti-i3-amyloicl- proteinLIDE
Figure 5 illustrates the effect of four ERR-selective phytoestrogenic
molecules when co-administered (100 nM for all four molecules) on the
expression of the anti-o-amyloid protein, insulin-degrading enzyme ("IDE")
in primary hippocampal neurons. **P < 0.01 compared to vehicle alone-
treated cultures. Figure 5 demonstrates the effects of these various
combinations of phytoestrogens along with E2 on the expression of the anti-
(3-amyloid (anti-AP) protein, insulin-degrading enzyme (IDE) in primary
neurons. Data showed that all three combinations composed of two, three or
four phytoestrogens significantly increases IDE expression in neurons.
Among them, a combination of three phytoestrogens induced the greatest
neuronal response, with an efficacy greater than E2 as well as a combination
of two phytoestrogens.
It is clear that one neuropathological hallmark of AD is a significant
deposition of extracellular Ap peptide, as referred to A(3 plaque. Impaired
Ap clearance and/or degradation has been demonstrated to contribute in part
to AD plaque formation in AD brain. Besides degrading insulin and several
regulatory peptides, IDE, a metalloprotease enzyme, has been demonstrated
to play a key role in degrading A(3 peptide monomer in the brain. Choronic
upregulation of IDE represents a novel efficacious therapeutic approach to
lowering the steady-state Aj3 level in the brain and eventually preventing the
occurrence of Alzheimer-type pathology. Therefore, these data indicate that
coadministration of multiple ERD-selective phytoestrogens have the potential
to activate the anti-Ap mechanism, and as a result, maintain the brain in a
long-term healthy status.
42
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
Upregulation of Spinophilin
Figure 6 illustrates the effect of four ERP-selective phytoestrogenic
molecules when co-administered (100 nM for all four molecules) on the
expression of the spine marker, spinaphilin, in primary hippocampal
neurons. **P < 0.01 compared to vehicle alone-treated cultures.
Spinophilin, a protein that is enriched in the heads of neuronal dendritic
spines, has been demonstrated to play a significant role in modulating both
dendritic morphology and glutamatergic synaptic activity. Upregulation of
spinophilin has been correlated with estrogen regulation of neuronal synaptic
plasticity. Therefore, theses results indicate that these phytoestrogen
combinations are effective to promote neurotrophism, thereby sustaining the
brain staying in a synaptically active status, and prevent cognitive decline
and memory loss.
Neuroprotection against Glutamate
Figures 7A-7D shows the neuroprotective efficacy of the compounds
against glutamate (Figure 7A) and amyloidl..42-induced neurotoxicity in rat
primary hippocampal neurons. Primary hippocampal neurons grown for 7
divisions were pretreated with the test compounds (or combinations) for 48
h, followed by a 5-min exposure to 100 mM glutamate. Neurons were
incubated for an additiona124 h prior to neuronal viability analyses by
calcein AM staining. Following pretreatment with the compounds (or
combinations)1'or 48 hr, neurons were exposed to 3 mM 0-amyloidi-42 for 2
d. Neuronal viability was analyzed by fluorometric measurements of
activities of the LDH and dead-cell protease released in the culture media,
and the live-cell protease exclusively entering intact viable neurons.
Results are presented as neuroprotective efficacy (NE), which is
defined as the percentage of neurotoxin-induced toxicity prevented by the
test compounds (or combinations) and quantitated by the equation:
NE _ (Vtreatment -Vneurotoxin) / (Vcontrol- Vneurotoxin) * 100%
where Vt,e,,tn,ent is the individual value from the test compounds (or
combinations)-treated cultures, VneurotoX;n is a mean value from glutamate or
P-amyloidi-4z alone-treated cultures, and VCO,It,ol is a mean value from
43
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
vehicle-treated control cultures. ##P < 0.01 compared to vehicle-treated
control cultures; *P < 0.05 and **P < 0.01 compared to glutamate or ~3-
amyloid1-42 alone treated cultures; SP < 0.05 compared to E2-treated cultures;
~P < 0.05 compared to genistein-treated cultures; IP < 0.05 and "P < 0.01
compared to combination (G+D)-treated cultures; `''P < 0.05 compared to
combination (G+D+E+I) -treated cultures. Combined formulations were
composed of equivalent molar of individual phytoSERMs included. G:
genistein; D: daidzein; E: equol; I: IBS003569.
Effect on IDE/NEP Expression
Figures 8A-8C show the effects on insulin-degrading enzyme
(IDE)/neprilysin (NEP) expression in (A) rat primary hippocampal neurons
and (B) hippocampal tissues derived from adult ovariectomized rats. (A)
Primary hippocampal neurons grown for 7 DIV were treated with the test
compounds (or combinations) for 48 hr followed by Western blot analyses.
(B) Adult ovariectomized rats were given, once daily, 2 subcutaneous
injections of the test compounds (or combinations). Rats were sacrificed 24 h
later following the second injection. Hippocarnpal tissues were homogenized
followed by western blot analyses. Results are presented as the fold increase
in protein expression and expressed as the percent of control, n>_ 4. *P <
0.05 and **P < 0.01 compared to vehicle-treated control cultures or animals.
SP < 0.05 and "P < 0.01 compared to E2-treated cultures; ~"P < 0.01
compared to combination (G+D) or genisteintreated cultures; IP < 0.05
compared to combination (G+D+E+I) -treated cultures. Combined
formulations were composed of equivalent molar in (A) and equivalent
weight in (B) of individual phytoSERMs including G: genistein; D: daidzein;
E: equol; and I: IBS003569.
Effect on Forebrain Mitochondria
Figures 9A-9E show the effects on forebrain mitochondrial
cytochrome c oxidase (COX) activity in adult ovariectomized rats. Rats
were given, once daily, 2 subcutaneous injections of the test compounds (or
combinations). Rats were sacrificed 24 h later following the 2nd injection.
Forebrain mitochondria were isolated followed by a spectrophotometric
44
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
measurement of COX activity using an immunocapture method. Colorimetric
absorbance at 550 nm was recorded every 5 min for 115 min. COX activity
is presented as the initial rate of oxidation of reduced cytochrome c, and
determined by calculating the initial slope between two time points (< 20
min) within the linear region. (Upper Panel) Time-lapse change in
absorbance; (Lower Panel) % increase in mitochondrial COX activity, n _ 4;
*P < 0.05 and **P < 0.01 compared to vehicle-treated control animals; 'VP <
0.05 compared to genistein-treated animals. Combined formulations were
composed of equivalent weight of individual phytoSERMs including E2:
17bestradiol; G: genistein; D: daidzein; E: equol; I: 1BS003569.
Figures 10A-10E show the effects on forebrain mitochondrial
respiratory activity in adult ovariectomized rats. Rats were treated as above.
Forebrain mitochondria were isolated followed immediately by a
polygraphical measurement of respiratory activity using an oxygen electrode.
Following a basal recording, mitochondrial state 4 respiration was measured
following the addition of substrates, malate/glutamate. State 3 respiration
was measured following the addition of ADP. Respiratory control ratio
(RCR) was calculated as the ratio between the rate of oxygen uptake at state
3 and the rate of oxygen uptake at state 4. (Figures 12A-12D) Time-lapse
oxygen uptake; (Figure 12E) % increase in mitochondrial respiratory
activity, n _ 4; *P < 0.05 and **P < 0.01 compared to vehicle-treated control
animals; IP < 0.05 compared to genistein-treated animals. Combined
formulations were composed of equivalent weight of individual
phytoSERMs including E2: 17b-estradiol; G: genistein; D: daidzein; E:
equol; and I: IBS003569; Mito: mitochondria; Mal/Glut: malate/glutamate.
Effect on Uterine Weight
Table 4 shows the effects on uterine weight in adult ovariectomized
rats. Changes in uterine weight in response to estrogenic stimulation can be
used to evaluate the estrogenic characteristics of test compounds on uterine
tissues. In one example, described below, immature female rats having low
endogenous levels of estrogen are dosed with test compound
(subcutaneously) daily for 3 days. Compounds are formulated as appropriate
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
for subcutaneous injection. As a control, I7-P estradiol is administered alone
to one dose group. Vehicle control dose groups are also included in the
study. Twenty-four hours after the last treatment, the animals are necropsied,
and their uteri excised, nicked, blotted and weighed. Any statistically
significant increases in uterine weight in a particular dose group as compared
to the vehicle control group demonstrate evidence of estrogenicity.
Table 4. Effects on uterine weight in adult ovariectomized ratsA
Treatment 1Jferine-Weigltt
Nvet Zi'eight (mg) Inc ase (%) ~ Dry VVeight (mg) Increase (%)
Controi 127.62 10.75 0.00 t 8.42 26.42 ~ 2.45 0.40 ~ 9.27
(veilictg}
17 PEstradiol. 281.06 32.04 * "`D 120.23 25.07 -"-' 46.70 ~ 4.13 76_74 +
15.63
(70 itg/icg BW)
Genistein 144.11 10:18 12.92 7,97 28.14 { 2.ÃB4 6.49 t 7_71
(6 mg(kg liW)
G+D+E 119.84 1 _15 -6_ 1Q {}.93 23 _71 0.+34 40.26 0.13
(6 M&9 Bwf
G+Ui-E 1 146 .99 18.45 15.17 t 14.46 28_73 3.67 8_73 t 13.90
~i M]lc B f
AAdult ovariectomized rats were given, daily once, 2 subcutaneous injections
of the test
compounds (or combinations) (n ?4 for each group). Rats were sacrificed 24 h
later
following
the 2nd injection. Uteri were immediately excised and a wet weight was
recorded. Uteri
were
then air dried for 1 hour followed by at 70 C overnight, and the dry weight
was recorded.
BIncrease in uterine weight compared with vehicle-treated control animals and
expressed as
the percent of control (set as 0).
eCombined formulations were composed of equivalent weight of individual
phytoSERMs
included for a total amount of 6 mg/kg BW given to animals. G: genistein; D:
daidzein; E:
equol; I: IBS003569.
3 **t' < 0.01 compared to any other treatment groups.
Summary
Both in vitro and in vivo analyses demonstrated that combined use of
select test phytoSERMs provided significantly increased efficacy in
sustaining neuronal survival when challenged with neurotoxins, promoting
expression of proteins as key players in neuroprotection and
metabolism/clearance of j3-amyloid in neurons/brain, and enhancing brain
mitochondrial functions. In particular, combined use of genistein, daidzein
and equol at an equivalent weight afforded the maximal efficacy comparable
46
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
or greater than 17b-estradiol in neuronal/brain assays. In contrast, such a
combination showed no impact on uterine weight, which however was
markedly increased by 17b-estradiol.
The present study indicates that combined use of select ER(3-selective
PhytoSERMs can be more therapeutically effective than single
administrations and alternative combined formulations. In particular, the
present study suggests the potential of the combination of genistein, daidzein
and equol, at an equivalent weight, for prevention of neurodegeneration and
AD, along with management of climacteric symptoms in postmenopausal
women.
Figures 11A-11C are schematics showing estrogen mechanisms of
action that lead to neurotrophic and neuroprotective outcomes. 17-0-
Estradiol (E2) acting via a membrane-associated site (mER) activates a
cascade required for multiple responses that lead to enhanced neural
plasticity, morphogenesis, neurogenesis, and neural survival. The signaling
sequence induced by E2 at the membrane site is as follows: (1) E2 binding to
mER, (2) E2-mER complexes with p85 to activate PI3K, (3) activating
calcium-independent PKC, (4) phosphorylating the L-type calcium channel,
(5) inducing calcium influx, (6) activating calcium-dependent PKCs, (7)
activating Src kinase, (8) activating the MEK/ERK1/2 pathway, (9) ERK
translocates to the nucleus, (10) activating and phosphorylating CREB, (11)
enhancing transcription of antiapoptotic genes Bcl-2 and Bcl-xl, which
enhance mitochondrial vitality, and spinophilin, which encourages synaptic
growth, (12) simultaneously, estrogen activation of P13K leads to activation
of Akt, which phosphorylates and inhibits the proapoptotic protein BAD.
Estrogen-induced neuroprotective mechanisms converge on
mitochondria. Estrogen-activated cellular signaling cascade promotes
enhanced mitochondrial function, leading to increased calcium load
tolerance, enhanced electron transport chain efficiency, and promotion of
antioxidant defense mechanisms. These actions are mediated by the
regulation of both nuclear and mitochondrial encoded genes initiated by the
activation of second-messenger signaling cascades.
47
CA 02659905 2009-02-02
WO 2008/016768 PCT/US2007/073505
These mechanisms and the data herein demonstrate that, consistent
with the healthy cell bias of estrogen benefit hypothesis, selective molecules
can be administered before neurodegenerative insult while neurons are still
healthy and that phytoSERM exposure will lead to enhanced neural survival
mechanisms, represented as mitochondria with Bcl-2 additions, that promote
neural defense against neurodegenerative insults associated with age-
associated diseases such as Alzheimer's and Parkinson's.
These studies exemplify the therapeutic promise of select ERP-
selective phytoestrogens when used in combination for sustaining memory
function and preventing age-related neurodegenerative insults and AD.
These ER(3-selective phytoestrogen formulations, which optimize activation
of ERP while minimizing or avoiding activating ERa, should serve as an
effective estrogen alternative replacement therapy for sustaining neurological
health, function and prevention of AD without induction of proliferative
responses in the reproductive tissues as seen with the current ET/HT.
Moreover, in light of the most recent data indicating that activation of ERj3
significantly reduces both ApoE mRNA and protein expression in neurons,
ERP-selective phtoestrogen formulations may serve as a particular viable
strategy for reducing a major risk factor of AD in ApoE4 carriers.
48