Note: Descriptions are shown in the official language in which they were submitted.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-1-
METHODS OF FORMING NANOPARTICLES
FIELD OF THE INVENTION
The present invention relates to methods for preparing nanoparticles of group
IV elements. It
relates particularly to the preparation of nanoparticles of Si, Ge and Sn, and
binary and ternary
alloys of these elements.
BACKGROUND TO THE INVENTION
The invention relates to quantum dots, also known as nanoparticles or
nanocrystals.
The term "nanoparticle" is generally invoked to refer to particles that have
an average diameter
between about 1 nm and about 100 nm. Nanoparticles have a size intermediate
between
individual atoms and macroscopic bulk solids. Nanoparticles that have
dianeters smaller or
comparable to the Bohr exciton radius of the material can exhibit quantum
confinement effects.
Such effects can alter the optical, electronic, catalytic, optoelectronic,
thermal and magnetic
properties of the material.
Many nanoparticles exhibit photoluminescence effects that are significantly
greater than the
photoluminescence effects observed for macroscopic crystals having the same
composition.
Additionally, these quantum confinement effects may vary as the size and
surface chemistry of
the nanoparticle is varied. For example, size-dependent discrete optical and
electronic transitions
exist for nanoparticles of group II-VI semiconductors (e.g., CdSe) and group
III-V
semiconductors (e.g., InP).
The gas phase synthesis of group IV nanoparticles tlirough processes such as
metal organic
chemical vapour deposition (MOCVD) is well-known. However, such approaches
produce low
yields and are highly capital intensive. The solution-phase synthesis
techniques used to
synthesise group II-VI and III-V semiconductors have not been readily applied
to group IV
materials, largely due to the high temperatures required to produce highly
crystalline
nanopar-ticles in a high yield. The strong covalent bonding of amorphous Si
and Ge means that
synthesis temperatures significantly higher than those used for the group II-
VI materials are
required in order to achieve highly crystalline cores at commercially viable
production rates. In
addition, the temperature required to thermally degrade many liquid phase
group IV precursors
exceeds the boiling points of many typical solvents at atmospheric pressure.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-2-
There have been a few reports of the solution-phase reduction of group IV
salts and of aerosol
methods. However, the nanoparticles produced by these methods often have
extremely broad
size distributions and poor visible luminescence efficiencies.
In this specification, where reference has been made to external sources of
information, including
patent specifications and other documents, this is generally for the purpose
of providing a context
for discussing the features of the present invention. Unless stated otherwise,
reference to such
sources of information is not to be construed, in any jurisdiction, as an
admission that such
sources of information are prior art or form part of the common general
knowledge in the art.
OSJECT OF THE INVENTION
It is an object of the present invention to provide a method of making
nanoparticles that provides
an alternative to those currently available; and/or to provide a method of
making nanoparticles
that produces nanoparticles of good mono-dispersity and/or of high
luminescence.
SUMMARY OF THE INVENTION
In a first aspect, the present invention provides a method of preparing
nanoparticles of one or
more group IV metals or alloys thereof comprising the steps of: reacting,
under an inert
atmosphere, at atmospheric pressure and with heating, one or more group IV
metal precursors
with a decomposition-promoting reagent in a liquid reaction medium comprising
a high
temperature surfactant; adding a surface-bonding agent; and recovering the
nanoparticles':
In one embodiment, the liquid reaction medium may comprise a high temperature
solvent and a
high temperature surfactant.
In a preferred embodiment, the group IV metal precursor comprises a compound
of the general
formula: G(Ar)XY4,; wherein G is the group IV metal, Ar is aryl, Y is halo and
x takes a value
that is at least 0 and no greater than 4.
In an alternative embodiment, the group IV metal precursor comprises a
compound of the general
formula: G(Ar)yY2_y wherein G is the group IV metal, Ar is aryl, Y is halo and
y takes a value
that is at least 0 and no greater than 2.
Preferably, the decomposition-promoting reagent is selected from one of:
a) a strong reducing agent; or
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-3-
b) S, Se, Te, P or As or a compound comprising one or more of these elements
in a
zero valence state.
In one embodiment, the method of the invention includes a further step of
adding =a quenching
agent prior to adding the surface-bonding agent. Preferably, the step of
adding a quenching agent
is prior to adding the surface-bonding agent but after adding the
decomposition-promoting
reagent.
In a preferred embodiment, the decomposition-promoting agent is selected from
the group
consisting of: S; Se; Te; P; As; and compounds comprising one or more of these
elements in a
zero valence state, and the me`thod includes the step of adding a quenching
agent.
In one embodiment, the surface-bonding agent may also act as the quenching
agent.
Preferably, the step of adding the surface-bonding agent is effective to
prevent aggregation of the
nanoparticles. Preferably, the surface-bonding agent interacts with the
nanoparticles to provide
an organic layer surrounding the nanoparticles.
In a preferred embodiment, the surface-bonding agent is a carboxylic acid,
aldehyde, amide or
alcohol. More preferably, the surface-bonding agent is a carboxylic acid and,
therefore, the
resulting nanoparticles are "acid-terminated".
In another preferred embodiment, the surface-bonding agent comprises an
alkenyl or=alkynyl
moiety.
Accordingly, one preferred embodiment of the invention comprises preparing
acid-terminated
nanoparticles of one or more group IV metals or alloys thereof; preferably
acid-terminated
nanoparticles of Ge, Si or Sn, or a binary or ternary alloy thereof.
Preferably, the step of reacting includes heating to a temperature between
about 100 C and about
400 C; more preferably to between about 200 C and about 400 C; more preferably
to about
300 C.
Preferably, the method of the invention is complete in less than about 30
minutes. More
preferably, the method is complete in less than about 20 minutes.
Preferably, the method includes a further step of purifying the nanoparticles.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-4-
Preferably, the method produces nanoparticles with size in the range about 1
nm to about 20 nm,
more preferably about 1 nm to about 10 nm.
Preferably, the method produces a monodisperse nanoparticle size distribution
such that the
nanoparticle diameter has a standard deviation of less than 20% of the mean
diameter. More
preferably, the method produces a monodisperse nanoparticle size distribution
such that the
nanoparticle diameter has a standard deviation of less than 5% of the mean
diameter.
Preferably, the method produces a solution of nanoparticles having a
concentration >1 gl-1; more
preferably > 10 gl'1.
Preferably, the method produces nanoparticles with a chemical reaction yield
>50%; more
preferably >60%.
Preferably, the method produces nanoparticles that produce luminescence in
response to optical
excitation with a quantum efficiency in excess of 1%. More preferably, the
nanoparticles
produce luminescence in response to optical excitation with a quantum
efficiency in excess of
20%.
Preferably, the method produces nanoparticles with a high degree of
crystallinity. In a preferred
embodiment, wherein G is germanium, the crystal structure is substantially
that of diamond.
In a further aspect, the present invention provides nanoparticles of a group
IV metal or a group;
IV metal alloy prepared substantially according to the method of the
invention.
In a yet further aspect, the present invention provides a method of preparing
chemically
functionalised nanoparticles of one or more group IV metals or alloys thereof
comprising the
steps of: reacting hydrogen-terminated nanoparticles of the group IV metals or
alloys thereof
with a compound of the formula L-R-N; wherein R is alkyl, alkenyl or aryl, L
is a group having
the desired functionality and N is a functional group capable of bonding to
the hydrogen-
terminated nanoparticle surface; and recovering the chemically functionalised
nanoparticles.
Suitable N groups include, but are not limited to: -NH2; -COOH; -CONH2; -
CONH2; -OH; -
CHO; -SO3H; -PO3H2; -PH2; -SH; -CH=CH2; -C-CH; -Cl; -F; -Br; and -I.
Preferred chemically functionalised nanoparticles include water soluble
nanoparticles prepared
by reacting hydrogen-terminated nanoparticles with a compound of the formula L-
R-N; wherein
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-5-
L is a polar functional group. An alternative embodiment provides
biochemically functionalised
nanoparticles reacting hydrogen-terminated nanoparticles with a compound of
the formula
L-R-N; wherein L is a functional group capable of binding to a biological
antibody and/or
biologically active molecule. Suitable L groups include, but are not limited
to: -NH2; -COOH;
-CONH2; -OH; -CHO; -SO3H; -P03H2; -PH2; -SH; -CH=CH2,= -C-CH; -Cl; -F; -Br;
and -I.
In another aspect, the present invention provides chemically functionalised
nanoparticles of one
or more group IV metals or alloys thereof prepared according to the method of
the invention.
In another aspect, the present invention provides a method of preparing
hydrogen-terminated
nanoparticles of one or more group IV metals or alloys thereof; preferably
hydrogen-terminated
nanoparticles of Ge, Si or Sn, or a binary or ternary alloy thereof;
comprising reacting acid-,
aldehyde-, alcohol- or amide-terminated nanoparticles of the invention with a
hydride reducing
agent in the absence of water and oxygen; and recovering the hydrogen-
terminated nanoparticles.
In a preferred embodiment, the hydrogen-terminated nanoparticles are prepared
from
acid-terminated nanoparticles of the invention.
In another aspect, the present invention provides hydrogen-terminated
nanoparticles of one or
more group IV metals or alloys thereof prepared substantially according to the
method f the
invention.
Nanoparticles of the invention are suitable for incorporation into a matrix of
asecond material
such as a polymeric, ceramic, metallic or other material. They are also
suitable for the
preparation of devices such as optoelectronic devices, including photovoltaic
devices, and
biochemical imaging devices.
Other aspects of the invention may become apparent from the following
description which is
given by way of example only and with reference to the accompanying drawings.
As used herein, the term "aryl" is intended to include optionally substituted
aromatic radicals
including, but not limited to: phenyl; naphthyl; indanyl; biphenyl; and the
like; and optionally,
substituted heteroaromatic radicals including, but not limited to:
pyrimidinyl; pyridyl; pyrrolyl;
furyl; oxazolyl; thiophenyl; and the like.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-6-
As used herein, the term "alkyl" is intended to include optionally substituted
straight chain,
branched chain and cyclic saturated hydrocarbon groups.
As used herein, the term "alkenyl" is intended to include optionally
substituted straight chain,
branched chain and cyclic mono-unsaturated hydrocarbon groups.
As used herein, the term "alkynyl" is intended to include optionally
substituted straight chain and
branched chain hydrocarbon groups that include a-C=C- moiety.
As used herein, the term "halo" refers to an iodo, bromo, chloro or fluoro
group.
Where a particular compound includes more than one alkyl, alkenyl, alkynyl,
aryl or halo group,
each of such groups may be independently selected.
As used herein, the term "optionally substituted" is intended to mean that one
or more hydrogen
atoms in the group is replaced with one or more independently selected
suitable substituents,
provided that the normal valency of each atom to which the optional
substituent/s are attached is
not exceeded.
As used herein, the terms "nanoparticle", "nanocrystal" and "quantum dot"
refer to any particle
less than 100 nanometres in size.
Although a "nanocrystal" may have a higher degree of crystallinity than a
nanoparticle,
references to nanoparticles in this specification should be understoodby one
skilled in the art to
also include nanocrystals and quantuxn dots.
As used herein, the "size" of a nanoparticle refers to the diameter of the
core of the nanoparticle.
A nanoparticle will typically comprise a core of one or more first materials
and can optionally be
surrounded by a shell of a second material.
A nanoparticle of the invention will typically comprise a "core" of a group IV
metal (such as
silicon, germanium or tin) or an alloy thereof and can be optionally
surrounded by a "shell" of a
second material. The term "core" refers to the central region of the
nanoparticle. A core can "
substantially include a single homogeneous material. A core may be
crystalline, polyatomic or
amorphous. While a core may be referred to as crystalline, it is understood
that the surface of the
core may be amorphous or polycrystalline and that this non-crystalline surface
layer may extend
to a finite depth into the core. The "shell" of a nanoparticle may coinprise a
layer of either
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-7-
organic or inorganic material or a bi-layer comprising both an inner inorganic
layer and an outer
organic layer, or vice versa. The shell material may be selected to minimise
the number of
"dangling bonds" at the surface of the nanoparticle core. The shell material
in the nanoparticles
of the present invention is generally dictated by the surface-bonding agent
employed in the
method of the inventiorz.
As used herein, the term "photoluminescence" of nanoparticles refers to the
emission of light of a
first wavelength (or range of wavelengths) by the nanoparticles following
irradiation with light of
a second wavelength (or range of wavelengths). The first wavelength (or range
of wavelengths)
is longer than the second wavelength (or range of wavelengths).
As used herein, the term "quantum efficiency" of the nanoparticles refers to
the ratio of the
number of photons emitted by the nanoparticles to the number of photons
absorbed by the
nanoparticles.
As used herein, the term "monodisperse", with respect to nanoparticles, refers
to a population of
nanoparticles wherein at least 75% and preferably 100% of the population, (or
an integer or non-
integer there between) falls within a specified particle size range. A
population of
`monodispersed' particles has a standard deviation of less than 20% of the
mean diameter and a
`highly monodispersed' population has a standard deviation of less than 5% of
the mean
diameter.
As used herein, the term "surfactant molecule" refers to a molecule containing
a non-polar end
comprising an alkyl, alkenyl or aryl group or combination thereof and a polar
end containing one
or more groups selected from: various acids, such as carboxylic, sulfinic,
sulfonic, phosphinic
and phosphonic acids, and their salts; primary, secondary, ternary or
quaternary amines; halides;
oxides; sulfides; thiols; phosphines; phosphides; phosphates; and glycols.
As used herein, the term "decomposition-promoting reagent" refers to a
compound or material
that accelerates a chemical reaction involving a group IV metal precursor
compound at a given
temperature, such as to yield one or more group IV metals.
As used herein, the term "and/or" means "and" or "or", or both.
As used herein, "(s)" following a noun means the plural and/or singular forms
of the noun.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-8-
The term "comprising" as used in this specification means "consisting at least
in part of'. When
interpreting statements in this specification and claims which include that
term, the features,
prefaced by that term in each statement or claim, all need to be present, but
other features can
also be present. Related terms such as "comprise" and "comprised" are to be
interpreted in the
same manner.
It is intended that reference to a range of numbers disclosed herein (by way
of example only, with
respect to the size of nanoparticles, 1 nm to 10 nm) also incorporates
reference to all integers and
non-integers within that range (for example, 1, 1.1, 2, 3, 3.9, 4, 5, 6, 6.5,
7, 8, 9 and 10) and also
any range of integers and non-integers within that range (for example, 2 to 8,
1.5 to 5.5 and 3.1 to
4.7).
This invention may also be said broadly to consist in the parts, elements and
features referred to
or indicated in the specification of the application, individually or
collectively, and any or all
combinations of any two or more of said parts, elements or features, and where
specific integers
are mentioned herein wllich have known equivalents in the art to which this
invention relates,
such known equivalents are deemed to be incorporated herein as if individually
set forth.
BRIEF DESCRIPTION OF THE DRAWINGS
The invention will now be described by way of example only and with reference
to the drawings
in which:
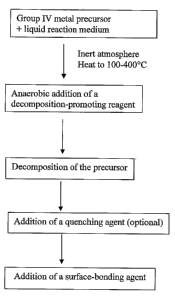
Figure 1: is a generalised flow diagram of the method of the invention;
Figure 2: is a flow diagram of one embodiment of the method of the invention;
Figure 3: is a flow diagram of an alternative embodiment of the method of the
invention;
Figure 4a: is a transmission electron micrograph of germanium nanoparticles
prepared
according to the invention;
Figure 4b: is another transmission electron micrograph of germanium
nanoparticles prepared
according to the invention;
Figure 5: is the X-ray diffraction data for germanium nanoparticles prepared
according to the
invention;
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-9-
Figure 6: shows electron diffraction by germanium nanoparticles prepared
according to the
invention;
Figure 7: shows the luminescence spectra of germanium nanoparticles prepared
according to
the invention;
Figure 8: is a diagram of typical glassware apparatus useful in a method of
the invention;
Figure 9a: represents a chemically functionalised nanoparticle, having a
surface organic layer,
prepared according to the invention;
Figure 9b: represents a hydrogen-terminated nanoparticle prepared according to
the invention;
and
Figure 9c: shows a carboxylic acid group bound to the surface of an acid-
terminated
nanoparticle prepared according to the invention.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to a method of forming nanoparticles comprising
one or more
group IV elements. In a preferred embodiment, the method comprises forming
nanoparticles that
include a semiconductor material consisting of one or more of the elements Si,
Ge and Sn.
The method comprises reacting, in a liquid reaction medium at atmospheric
pressure and under
an inert atmosphere, one or more group IV metal precursors, with a
decomposition-promoting
reagent. The reaction is carried out by heating the reaction mixture. In a
preferred embodiment,
the group IV metal precursors are sources of semiconductor material consisting
of one or more of
Si, Ge and Sn.
In general terms, the decomposition-promoting reagent may be a reducing agent,
a polymerising
agent, or other suitable substance. The method also includes the use of a high
temperature
surfactant in the reaction mixture. The-method includes the addition of a
strongly-interacting
surface-bonding agent to the reaction mixture. This surface-bonding agent
forms an organic
coating on the nanoparticle, preventing aggregation of the nanoparticles
during subsequent
crystal growth.
The general method of preparing nanoparticles is illustrated in Figure 1.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-10-
As shown in Figure 1, the method of the invention comprises four main steps:
(a) heating, under an inert gas at atmospheric pressure, a solution comprising
a liquid
reaction medium and one or more group IV metal precursors;
(b) introducing a decomposition-promoting reagent to accelerate the
decoinposition
of the group IV metal precursors;
(c) optionally introducing a quenching agent to remove excess decomposition-
promoting reagent. This optional step is not necessary if, for example, there
is no
excess decomposition-promoting reagent, and may not be necessary if the
decomposition-promoting reagent is a reductant (as discussed below); and
(d) introducing a surface-bonding agent to prevent aggregation of the
resultant
nanoparticles.
Each of these steps will be discussed in detail below.
Group IV metal precursor
A number of group IV metal precursors are suitable for use in a method of the
invention. These
include organometallic compounds containing a silicon, germanium or tin atom.
Preferably,
these organometallic compounds containing the silicon, germanium or tin atoms
are tetravalently
bonded to a combination of aryl groups and halides following the molecular
formula: G(Ar)XY4_X
where G is" the group IV metal, Ar is aryl, Y is halo and x takes a value that
is at least 0 and no
greater than 4.
Such compounds include, but are not limited to: tetraphenylgermane;
triphenylchlorogermane;
diphenyldichlorogermane; phenyltrichlorogermane; tetraphenylsilane;
triphenylchlorosilane;
diphenyldichlorosilane; phenyltrichlorosilane; tetraphenylstannane;
triphenylchlorostannane;
diphenyldichlorostannane; phenyltrichlorostannane; as well as their bromo-,
iodo- and fluoro-
analogues.
Alternatively, the group IV metal precursor species includes a group IV atom
divalently bonded
to a combination of aryl groups and halides following the molecular formula:
G(Ar)yY2_y wheare
G is the group IV metal, Ar is aryl, Y is halo and y takes a value that is at
least 0 and no greater
than 2.
In preferred embodiments, Ar is optionally substituted phenyl; more preferably
phenyl.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-11-
In a preferred embodiment, the group'IV precursor is the phenyl-substituted
precursor
G(Ph)XY4_x. This precursor is particularly preferred when the decomposition-
promoting reagent
is selected from the group consisting of: S; Se; Te; P; As; and compounds
comprising one or
more of these elements in a zero valence state (see below).
The group IV precursor may be a mixture of different group IV metal
precursors, including those
having different group IV metals. This can result in alloy or interphase
formation in the final
nanoparticles.
Liquid reaction ynedium
The primary role of the liquid reaction medium is to solvate the group IV
metal precursors and
the various reagents to ensure a homogenous liquid phase reaction. The liquid
reaction medium
also provides a barrier to nanoparticle aggregation through the interaction of
a high temperature
surfactant with the nanoparticle surface. These measures can be effective in
producing a high
degree of monodispersity of the size of the nanoparticles.
Those persons skilled in the art will appreciate that the liquid reaction
medium should be
thermally stable at the reaction temperature.
In one embodiment, the liquid reaction medium comprises a high temperature
surfactant and a
high temperature solvent. Alternatively, the high temperature surfactant also
acts as the high
temperature solvent. I
As used herein, the term "high temperature" means that the surfactant or
solvent should be
thermally stable under an inert atmosphere at the reaction temperature and
have a boiling point in
excess of the reaction temperature.
The reaction temperature is preferably between about 100 C and about 400 C;
more preferably
between about 200 C and about 400 C; more preferably about 300 C.
The surfactant possesses a molecular structure containing a functional group
capable of
interacting with the nanoparticle surface. This acts to form a thin layer of
solvent material that is
weakly bound to the nanoparticle surface. In such a way aggregation of the
nanoparticles can be
inhibited. Preferred surfactants do not take part in parasitic side-reactions
with the reagents or
precursors.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-12-
Suitable surfactants that meet the above criteria may include, but are not
limited to compounds of
the formulae: R-NH2; R-PH2; R3N; R3P; R2NH; R2PH; R4N+; and REY; wherein R is
alkyl,
alkenyl or aryl, E is an ethylene glycol group and N, P and H take their
common IUPAC
meaning.
Preferred high temperature surfactants include oleylamine, hexadecylamine,
trioctylamine and
trioctylphosphine. These surfactants act as co-ordinating solvents and so need
no additional
solvent.
In those embodiments wherein the liquid reaction medium comprises a high
temperature
surfactant and a high temperature solvent, suitable high boiling point organic
solvents include,
but are not limited to: alkanes, alkenes, aromatic hydrocarbons and other
paraffins; mono- or
poly-ethylene glycol ethers; crown ethers; and phenyl ethers.
The group IV metal precursor can be introduced into the liquid reaction medium
either prior to or
subsequent to heating of the liquid reaction medium to the reaction
temperature. In a preferred
embodiment, the liquid reaction medium is heated to the desired reaction
temperature under a
flow of a suitable inert gas to purge gaseous contaminants from the reaction
vessel prior to the
introduction of the group IV metal precursor and other reagents.
Suitable inert gases are known to those persons skilled in the art. Such gases
include, but are not
limited to: nitrogen; argon; and helium. Preferably.the inert gas is nitrogen.
The relative chemical stability of the precursors precludes the need for the
reaction to be carried
out in a controlled atmosphere glovebox, and simple Schlenk line or similar
gas-purged
glassware apparatus can be used. Figure 8 shows a typical benchtop glassware
apparatus suitable
for this synthesis. The liquid reaction medium 1 is contained in a three-
necked round bottomed
flask 2. The temperature is monitored using a thermometer probe 3 housed in a
protective glass
enclosure 4. The solution is stirred with a magnetic stir bar 5 and heat is
applied 6, by a heating
mantle, heat bath or similar apparatus. Inert gas is admitted into the flask 2
via the gas inlet 7
and leaves via the outlet 10. Reagents can be added to the flask 2 using a
syringe temporarily.
admitted at the gas inlet 7. A condenser 8 is also used to cool and condense
vapour above the
liquid reaction medium 1. The condenser can be water cooled through the water
inlet 9 and
outlet 9 to an outer glass sheath.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-13-
This reaction mixture is usually heated to the reaction temperature during
step a), although it is
possible to heat the mixture to the reaction temperature during step b).
Decomposition-promoting reagent
After degassing, the decomposition-promoting reagent is added to the solution
of the group N
metal precursor in the liquid reaction medium, typically under conditions of
heat and an inert
atmosphere.
The decomposition-promoting reagent may be injected into the reaction vessel
as a solution or
suspension. In a preferred embodiment, the decomposition-promoting reagent is
added as a
solution in a carrier liquid, which may be the same as that comprising the
liquid reaction
medium.
The decomposition-promoting reagent accelerates the decomposition of the group
IV metal
precursor at the reaction temperature to the elemental form of the group IV
metal - typically Ge
or Si or Sn (or alloys of these) as nanoparticles. The generalised reaction
scheme is as follows:
Ge, Si and/or Sn precursor/s + decoinposition-promoting reagent - group IV
metals or alloy (np) + by-products
wherein np indicates nanoparticle formation.
In one embodiment of the invention, illustrated in Figure 2, the decomposition-
promoting agent
is a strong reducing agent. The decompositiori-promoting agent must be able to
cleave the
substituent-Ge, substituent-Si or substituent-Sn bond.
In those embodiments wherein the group IV metal precursor is a phenylmetallic
compound, the
decomposition-promoting agent must be able to cleave one or more Si-Ph, Sn-Ph
or Ge-Ph
bonds. This strong covalent bond has previously been assumed to be stable to
reductive attack,
but the liquid phase method disclosed herein utilises a combination of high
reaction temperatures
and strong reducing agents to obtain elemental group IV metals from precursors
containing Ge-
Ph, Si-Ph or Sn-Ph bonds.
Example strong reducing,agents include, but are not limited to, one or more
of: sodium
borohydride; lithium borohydride; potassium borohydride; sodium naphthalide;
sodium
anthracide; lithium naphthalide; lithiuin anthracide; potassium naphthalide;
potassium
anthracide; lithium aluminium hydride; sodium aluminium hydride; potassium
aluminium
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-14-
hydride; sodium hydride; lithium hydride; potassium hydride; sodium; lithium;
potassium;
sodium sulfide; lithium sulfide; potassium sulfide; tin dichloride; tin
dioleate; tin dibromide; tin
di-iodide; sodium amide; sodium azide; and triphenyl phosphine.
In an alternative embodiment of the invention, illustrated in Figure 3, the
decomposition-
promoting reagent comprises one or more elements from group V or VI, or a
compound
comprising one or more of these elements in a zero valence state, wherein the
group V or VI
elements are selected from the group comprising: sulfur; selenium; tellurium;
phosphorus; and
arsenic.
In a preferred embodiment, the decomposition-promoting reagent comprises S or
Se (or a
compound comprising one of these elements in a zero valence state). A
generalised reaction
scheme for an example of this embodiment,'4herein the group IV metal precursor
is (Ph)3GeCl,
is shown below:
260-360 C, 5-30 mins
(Ph)3GeC1 + S or Se 10 Ge(õp) + by-products
wherein np indicates nanoparticle formation.
In those embodiments wherein the decomposition-promoting reagent comprises S
(or a
compound-comprising S in a zero valence state), the by-products may include:
Ph-S-Ph, Ph-Cl,
Ph, Ph-Ph, C12, S and GeS2.
Quenching agent
A quenching agent is typically used in the embodiment of the invention
illustrated in Figure 3.
However, a quenching agent may also be used in those embodiments wherein the
decomposition-
promoting reagent is a strong reducing agent.
The quenching agent acts to remove excess decomposition-promoting reagent from
the reaction
mixture, thus eliminating any inhibitory effects this may have on nanoparticle
growth. In
addition, the use of a quenching agent prevents the reaction of any excess
decomposition-
promoting reagent with the surface-bonding agent which, in some embodiments,
is added to the
reaction mixture after the quenching agent.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-15-
When the decomposition-promoting reagent is selected from the group consisting
of: S; Se; Te;
P; As; and compounds comprising one or more of these elements in a zero
valence state, a
quenching agent is preferably added subsequent to precursor decomposition.
In a preferred embodiment, wherein the decomposition-promoting reagent is
selected from one
of:
a) a strong reducing agent, or
b) S, Se, Te, P or As or a compound comprising one or more of these elements
in a zero
valence state;
the quenching reagent is a non-aqueous higli boiling point acid. Suitable non-
aqueous high
boiling point acids include, but are not limited to: carboxylic acids; and
compounds containing
one or more carboxylic acid groups.
In an alternative embodiment, wherein the decomposition-promoting reagent is
selected from the
group consisting of: S; Se; Te; P; As; and compounds comprising one or more of
these elements
in a zero valence state, the quenching agent may. be a hydride reducing agent
such as sodium
borohydride, lithium borohydride, potassium borohydride, lithium aluminium
hydride, sodium
aluminium hydride, potassium aluminium hydride, sodium hydride, lithium
hydride or potassium
hydride.
Surface-bonding agent
The surface-bonding agent is added to the reaction mixture to prevent
aggregation of the
nanoparticles. A surface-bonding agent can provide stronger interactions with
the nanoparticle
surface than those obtained from the high temperature surfactant within the
liquid reaction
medium. Typically, the surface-bonding agent is added in excess.
Advantageously, it is possible to avoid parasitic side-reactions of the
surface-bonding agent with
the decomposition-promoting reagent by adding the surface-bonding agent after
the initial
decomposition of the precursor. A second motivation for adding the strongly
binding surface-
bonding agent after the initial decomposition of the precursor is that very
strong bonds between
the nanoparticle and the surface-bonding agent may inhibit nanoparticle
growth.
Notwithstanding this point, it is possible to add the surface-bonding agent to
the reaction mixture
prior to, or at the same time as, the decomposition-promoting agent.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-16-
Suitable surface-bonding agents that may be susceptible to reduction as a
parasitic side-reaction
in those embodiments wherein the decomposition-promoting reagent is a strong
reducing agent
include, but are not limited to: trioctylphosphine oxide; carboxylic acids;
sulfonic acids;
phosphonic acids; alcohols; thiols; and other proton-donating compounds.
Suitable surface-bonding agents that may be susceptible to parasitic side-
reactions in those
embodiments wherein the decomposition-promoting reagent is selected from the
group
consisting of S; Se; Te; P; As; and compounds comprising one or more of these
elements in a
zero valence state, include, but are not limited to: primary, secondary and
ternary phosphines.
Advantageously, in those embodiments wherein the surface-bonding agent
comprises an alkenyl
or alkynyl moiety, the surface-bonding agent may form stable bonds between the
group IV metal
and carbon at the nanoparticle surface~:
In an alternative embodiment, the surface-bonding agent comprises a compound
of the formula
R-N, wherein R is alkyl, alkenyl or aryl and N is a functional group capable
of bonding to the
surface of the group IV metal nanoparticle.
Alternatively, the surface-bonding agent may comprise a bi-functional compound
of the formula
L-R-N wherein R is alkyl, alkenyl or aryl, L is a group having the desired
functionality and N is a
functional group capable of bonding to the surface of the group IV metal
nanoparticle.
In a preferred embodiment, L and N are each independently selected from the
group comprising:
-NH2; -COOH; -CONH2; -OH; -CHO; -SO3H; -PO3H2; -PH2; -SH; -CH=CH2; -C=CH; -Cl;
-F;
-Br; and -I.
In a preferred embodiment, the surface-bonding agent is the same as the
quenching agent. In this
embodiment, the excess surface-bonding agent acts to remove excess
decomposition-promoting
reagent from the reaction mixture.
The nanoparticles remain in suspension following the addition of the surface-
bonding agent. The
particles are typically subject to a purification/recovery step:
Nanoparticle recovery and purification
The nanoparticles are recovered and optionally purified following their
preparation according to
the methods disclosed above. Recovery and purification may proceed by a number
of techniques
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-17-
known to those skilled in the art. Such techniques include, but are not
limited to: nanoparticle
flocculation and centrifugation; or two phase separation in an appropriate
choice of immiscible
solvents.
An example purification procedure, which is illustrative only, is:
(a) addition of a flocculent to the reaction mixture;
(b) centrifugation of the flocculated mixture at a rotational acceleration
above 1000G;
(c) removal and disposal of the supernatant;
(d) re-suspension in an organic solvent with a boiling point below about 120 C
(a low
boiling point solvent); and, optionally
(e) repetition of steps (a) to (d) until the required purity is obtained.
Suitable flocculents include, but~are not limited to: distilled or suitably
purified water; alcohols
including methanol, ethanol, propanol and butanol; methyl formamide; and
acetone.
Suitable low boiling point solvents include, but are not limited to: toluene;
tetrahydrofuran
(THF); chloroform; dichloromethane; and liquid alkanes having fewer than 10
carbon atoms.
Characterisation of nanoparticles
The methods of the present invention lead to the formation of nanoparticles
and/or nanocrystals
of group IV elements or alloys thereof. The particles may be robust,
chemically stable,
crystalline, and may be coated by an organic or inorganic passivating layer.
Although the product of the method of the invention is described herein as a
nanoparticle or
nanocrystal, those persons skilled in the art will appreciate that the group
IV metal nanoparticle
has a surrounding layer, the composition of which is dependent on the surface-
bonding agent.
For example, when the surface-bonding agent is oleic acid, the oleic acid will
be bound to the
surface as illustrated in Figures 9a and 9c.
Figure 9a shows the group IV metal nanoparticle core 70 with the surface-
bonding agent 71,
which may be a carboxylic acid such as oleic acid, forming a surface organic
layer. Figure 9b :is
a diagram of a hydrogen-terminated group IV metal nanoparticle produced
according to one
aspect of the present invention. Figure 9c shows a carboxylic acid group, such
as that present in
oleic acid, binding to the surface of the group IV metal nanoparticle.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-18-
Other common examples of functional groups that may bind to the surface of the
group IV metal
nanoparticle include -NH2, -COOH, -CONH2, -OH, -CHO, -SO3H, -P03H2, -PH2, -SH,
-CH=CH2, -C=CH, -Cl, -F, -Br, -I and -H.
Further steps
Hydrogen- terminated nanoparticles
In many potential applications of group IV metal nanoparticles it is
advantageous to be able to
chemically functionalise the nanoparticles by the addition of specific
molecules to the
nanoparticle surface. The ability to obtain clean unfunctionalised crystal
surfaces from which the
functionalisation reaction can proceed is, therefore, important. Of particular
interest is the
hydrogen-terminated crystal surface illustrated in Figure 9b.
A fu.rther aspect of this invention is the preparation of hydrogen-terminated
group IV metal
nanoparticles from acid-, aldehyde-, alcohol- or amide-terminated group IV
metal nanoparticles
grown outside of a glovebox environment.
In a preferred embodiment, the hydrogen-terminated nanoparticles are prepared
from acid-
terminated nanoparticles.
This is achieved according to the following method, which is illustrative
only:
(a) adding a hydride reducing agent, under an inert gas at atmospheric
pressure, to a
purified mixture of acid-, aldeliyde-, alcohol- or amide-terminated group IV
metal
nanoparticles in a solvent at a temperature at or below the boiling point of
the
solvent;
(b) reacting the resulting mixture for a period of up to 48 hrs in the absence
of water
and oxygen; and
(c) quenching the reaction.
Suitable hydride reducing agents include, but are not limited to: lithium
aluminium hydride;
sodium aluminium hydride; potassium aluminium hydride; lithium
triethylborohydride; sodium
triethylborohydride; sodium borohydride; lithium borohydride; potassium
borohydride; hydrogen
gas; sodium hydride; potassium hydride; lithium hydride; borane-
tetrahydrofuran complex;
lithium tri-teNt-butoxyaluminium hydride; sodium cyanoborohydride; and di-
isobutylaluminum
hydride.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
- T9 -
Suitable inert gases are known to those persons skilled in the art. Such gases
include, but are not
limited to: nitrogen; argon; and helium. Preferably, the inert gas is
nitrogen.
In a preferred embodiment, the reaction is quenched with an alcohol. Suitable
alcohols include,
but are not limited to: methanol; ethanol; butanol; and propanol.
Chenaical functionalisation
A further aspect of this invention is the preparation of chemically
functionalised group IV metal
nanoparticles by the reaction of a specific chemically active species with the
surface of the
hydrogen-terminated nanoparticles of this invention.
The chemically active species may be a compound of the formula L-R-N wherein R
is alkyl,
alkenyl or aryl, L is a groiap having the desired functionality and N is a
functional group capable
of bonding to the hydrogen-terminated nanoparticle surface. Such groups
include, but are not
limited to: -NH2; -COOH; -CONH2; -OH; -CHO; -Cl; -F; -Br; -I; -PO2H; -PH2; -
SH; -SO3H;
-CH=CH2; and -C-CH.
In one specific embodiment of this invention, water soluble nanoparticles may
be produced by
reacting the hydrogen-terminated nanoparticles with a compound of the formula
L-R-N, wherein
L is a polar functional group. In an alternative embodiment, biochemically
functionalised
nanoparticles may be prepared a compound of the formula L-R-N, wherein L is a
functional
group capable of binding to a biological antibody and/or biologically active
molecule. Suitable L
groups include, but are not limited to: -NH2; -COOH; -CONH2; -OH; -CHO; -SO3H;
-P03H2;
-PH2; -SH; -CH=CH2; -C=CH; -Cl; -F; -Br; and -I.
Favourable Characteristics
In preferred embodiments, the methods of the present invention, and the
resultant nanoparticles,
incorporate a number of favourable characteristics.
Relative ease ofpreparation
Advantageously, the method of the present invention may be carried out in a
fumehood or
benchtop location. In contrast, many of the prior art methods require high
pressures and/or the
use of controlled atmosphere glove-box techniques or other protective
environments. This is
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
- 20 -
because the method of the present invention is carried out at atmospheric
pressure and uses
comparatively benign or non-toxic reactants when compared with many of the
prior art methods.
Accordingly, preferred embodiments of the method of the present invention are
carried out under
atmospheric pressure utilising standard glassware apparatus purged with an
inert gas. Such
apparatus is illustrated in Figure 8.
High yields
The methods described herein are capable of achieving high reaction yields of
nanoparticles
exceeding 60%. Such yields are achieved through the use of non-hydride
decomposition agents,
thus obviating the formation of the volatile by-products silane, germane,
stannane and derivatives
thereof. In this way, the group IV elements in the precursor species remain
within the reaction
vessel until coinpletion of the reaction. The reaction terminates at solid
phase nanoparticles, and
the crystallisation of elemental particles from the reaction solution ensures
that the
decomposition reaction drives to completion.
Those persons skilled in the art will appreciate that the presence of species
surface-bonded to the
nanoparticles gives rise to nanoparticle yields that are difficult to
ascertain with any accuracy.
The yield figures given herein refer to the yields from syntheses in the
absence of quenching
agents, surfactants or surface active material.
Relatively high throughput
Preferred embodiments of the invention require short reaction times, typically
less than one hour
and more preferably less than 30 minutes. In addition, the nanoparticles can
be synthesised with
solution concentrations in excess of 2 gl-1 and, in preferred embodiments,
with concentrations in
excess of 10 gl-1. Such production rates represent a significant improvement
over many of the
prior art processes.
Good monodispersity
Figures 4a and 4b show transmission electron microscope (TEM) images of
germaniuin
nanoparticles prepared according to the methods of the present invention. The
scale bar in both
photographs is 20 nm. High monodispersity is observed. This is achievable
through the
homogeneous solution-phase nature of the reaction.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-21-
Figure 5 shows the X-ray diffraction data of germanium nanoparticles prepared
according to the
methods of the present invention and demonstrates the high crystallinity of
the nanoparticles.
The copper peaks observed in the X-ray diffraction data are due to sampling of
the TEM grid
bars. Preferred embodiments of the invention produce nanoparticles with a
diameter standard
deviation of less than 20% of mean diameter; more preferably less than 5% of
mean diameter.
Figure 6 shows the electron diffraction pattern from 8 nm germanium
nanoparticles prepared
according to the methods of the present invention.
Spectral properties
In one embodiment, under optical excitation the nanoparticles of the present
invention emit
coloured light at wavelengths within the visible spectrum. In other
embodiments, the
nanoparticles emit light under optical excitation at near-infrared and
infrared wavelengths less
than the bandgap of the bulk crystalline material. The wavelength of the light
emitted by the
nanoparticles may be tuned through manipulation of the nanoparticle size, the
chemical make-up
of the passivating layer and the chemical composition of the group IV metal or
alloy comprising
the nanoparticle. In one embodiment, the method disclosed herein provides a
passivated
germanium nanoparticle displaying discrete optical transitions and
photoluminescence. Figure 7
shows the typical luminescence (as a function of excitation wavelength) of a
solution of
germaniuni nanoparticles. Preferred embodiments of the method of the present
invention
produce nanoparticles with luminescence having a quantum efficiency > 1%, more
preferably up
to and in excess of 20%.
APPLICATIONS
The nanoparticles of the present invention have many possible applications, as
would be
understood by one skilled in the art.
The nanoparticles are observed to luminesce in the visible spectrum and may be
utilised in the
field of biomedical imaging or as a starting material for novel optoelectronic
devices. Group IV
nanoparticles are of particular inteirest in the field of biomedical imaging
where the long lifetime,
high stability and low toxicity of germanium, silicon and tin is highly
attractive compared to
current alternatives.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-22-
The nanoparticles of the present invention may be incorporated into
biochemical imaging
markers. For such applications, the nanoparticles may have an average particle
diameter of
between about 1 and about 200 angstroms and will produce nearly monochromatic
luminescence
within the visible spectrum in response to optical excitation. The biochemical
imaging system
may include a linker attaching:a biochemically active molecule to the
nanoparticle, enabling the
tagging of specific chemical compounds. The system may contain more than one
type of linker
type, with each type of linker being attached to nanoparticles that luminesce
at a different and
nearly monochromatic, visible wavelength.
The nanoparticles of the present invention may be incorporated into
optoelectronic materials by a
variety of methods, including the deposition of a film of the nanoparticles
upon a substrate. The
nanoparticles may include, for example, silicon or germanium or an alloy
thereof. For such
applications, the nanoparticles may have an average particle diameter of
between about 1 and
about 200 angstroms. The deposited nanoparticles may be sintered at a
temperature between
about 300 C and 900 C to produce a film of material exhibiting optoelectronic
properties. In
certain embodiments, the film may consist of a plurality of nanoparticles
resulting in a
polycrystalline material. This material may be further used to produce a
variety of electronic
devices, including photovoltaic devices, infrared emitting devices and light
emitting devices.
Another embodiment of an optoelectronic material may include the incorporation
of one or more
nanoparticles into a matrix of one or more polymeric species whereby electrons
can be
transferred to and from the nanoparticles by means of external electronic
contacts. The
nanoparticles may include silicon or germanium or an alloy thereof. At least
one of the
nanoparticles may have an average particle diaineter of between about 1 nm to
about 20 nm. The
said nanoparticle-incorporating matrix may be deposited upon a substrate or
flexible film. This
material may be further used to produce a variety of electronic devices
including photovoltaic
devices, infrared emitting devices and light emitting devices.
The nanoparticles of the present invention may be incorporated into
photovoltaic devices for the
generation of electrical power from optical and near-infrared radiation. Such
devices may
include a plurality of nanoparticles. The photovoltaic devices may include
electrical contacts at
their anode and cathode. The photovoltaic devices may include nanoparticles of
a size optimised
to provide maximum absorption of an incident solar spectrum.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
- 23 -
EXAMPLES
Various aspects of the in-vention will now be illustrated in non-limiting ways
by reference to the
following examples.
Example 1
1) Place 4.5g of hexadecylamine (HAD) or 7.5m1 of oleylamine in a three necked
round
bottom flask
2) Add 0.05g triphenylchlorogermane to flask
3) Place flask in a stirring heating mantle under a cold water condenser and
nitrogen purge
flow
4) Heat to 285-300 C whilst stirring
5) Inject a solution of 0.005g sulfur in 1.5m1 trioctylamine
6) Wait approximately 5 minutes
7) Observe swift (<30 seconds) reaction as solution turns dark
8) Imfnediately add 0.2-5m1 oleic acid. The solution will rapidly clear
9) Heat to a defined temperature between 285 C and 360 C for up to one hour
10) Cool to 150 C and then quench with a 1:1 mixture of ethanol and toluene,
added
dropwise
11) Remove from flask. Add ethanol (for hexadecylamine) or methanol (for
oleylamine)
drop-wise until flocculation is observed
12) Centrifuge out flocculated sediment and remove and keep supernatant
13) Further dilute precipitate with flocculent and centrifuge. Repeat steps 9-
11 until required
purity is reached. Steps 9-11 may also be repeated on the supeinatant to yield
size
selective precipitation of nanoparticles
14) Resuspend final precipitated material in toluene, hexane or ethanol
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-24-
Example 2
An alternative procedure omits steps (2) and (3) from the procedure described
in Example 1 and
instead utilises solutions of sulfur in oleylamine and triphenylchlorogermane
in oleylamine
which are injected into the heated solvent at a temperature between 260 C and
360 C. The
procedure is then followed as from step (5) of Example 1.
Example 3
1) Place 4.5g of hexadecylamine or 7.5m1 of oleylainine in a three-necked
round bottom
flask
2) Add 0.012g selenium to flask
3) Place flask in stirring heating mantle under a cold water condenser and
nitrogen purge
flow
4) Heat to 285-300 C whilst stirring
5) Inject solution of 0.05g triphenylchiorogermane in 1.5ml trioctylamine
6) React for 30 minutes
7) Cool to 150 C and then quench with a 1:1 mixture of ethanol and toluene,
added
dropwise
8) Remove from flask. Add ethanol (for hexadecylamine) or methanol (for
oleylamine)
drop-wise until flocculation is observed
9) Centrifuge out flocculated sediment and remove a.nd keep supernatant
10) Further dilute precipitate with flocculent and centrifuge. Repeat steps 9-
11 until required
purity is reached. Steps 9-11 may also be repeated on the supematant to yield
size
selective precipitation of nanoparticles
11) Resuspend the final precipitated material in toluene, hexane or ethanol
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
- 25 -
Example 4
1) Place 6g of hexadecylamine or l Oml of oleylamine in a three-necked round
bottom flask
2) Add 0.05g triphenylchlorogermane to flask
3) If tetraglyme is used in step 6 a defined quantity of trioctylphosphine may
also be added
to the flask at this point
4) Place flask in stirring heating mantle under a cold water condenser and
nitrogen purge
flow
5) Heat to 285 C (boiling point of triphenylchlorogermane) whilst stirring
6) Inject 1 ml of 1 M NaBH4 solubilised in either tetraglyme or
trioctylphosphine. The
solution will rapidly clear
7) Heat to a defined temperature between 285 C and 360 C for up to one hour
8) Cool to 150 C and then quench with methanol, added dropwise
9) Remove from flask. Add ethanol (for hexadecylamine) or methanol (for
oleylamine)
drop-wise until flocculation is observed
10) Centrifuge out- flocculated sediment and remove and keep supematant
11) Further dilute precipitate with flocculent and centrifuge. Repeat steps 9-
11 until required
purity is reached. Steps 9-11 may also be repeated on the supernatant to yield
size
selective precipitation of nanoparticles
12) Resuspend final precipitated material in toluene, hexane or ethanol
Example 5
An alternative procedure omits steps (2) and (3) from the procedure described
in Example 4 and
instead utilises a solution of triphenylchlorogermane in oleylamine and
trioctylphosphine, which
is injected into the heated solvent at a temperature between 260 C and 360 C
at the same time as
step (6). The procedure is then followed as from step (6) of Example 4.
CA 02662006 2009-02-25
WO 2008/030110 PCT/NZ2007/000246
-26-
It is apparent that a wide variety of reducing agents are capable of
accelerating the decomposition
of aryl-containing group IV metal precursors at temperatures in excess of 240
C, even though a
number of such reducing agents are not stable at these temperatures. For
example, sodium
napthalide breaks down at a temperature close to the boiling point of
naphthalene (-220 C), but
addition of naphthalene to a mixture containing triphenylchlorogermane and
sodium metal at
275 C exhibits decomposition of the group IV metal precursor in the time
required for the
naphthalene to enter the solution, attain thermal equilibrium with its
surroundings and then re-
volatilise.
It is important that the reducing agent be homogeneously distributed within
the solution.
Preferably, the reducing agent should be fully soluble in the utilised solvent
system such that
crystallisation occurs from a completely homogeneous distribution of
decomposed precursor
molecules. However, this requirement may be relaxed, provided the
heterogeneity of the
distribution of the reducing agent is on a length scale that is comparable
with the average
diffusion length within the solution for a reduced group IV atom undergoing
crystallisation. For
example, the thermal breakdown of lithium aluminium hydride to LiH and A1H3 at
temperatures
above 200 C is not a barrier to its utilisation in the methods of the present
invention because the
resulting highly disperse emulsion of LiH reacts completely with the precursor
prior to
aggregation of the insoluble hydride salt to form a homogeneous distribution
of precursor
decomposition products within the solution. Vigorous stirring can assist the
production of a
homogeneous distribution of reduced or partially-reduced precursor molecules.
Although the invention has been described by way of example and with reference
to particular
embodiments, it is to be understood that modifications and/or improvements may
be made
without departing from the scope of the invention as set out in the
accompanying claims.
In addition, where features or aspects of the invention are described in terms
of Markush groups,
those skilled in the art will recognise that the invention is also thereby
described in terms of any
individual member or subgroup of members of the Markush group.