Note: Descriptions are shown in the official language in which they were submitted.
CA 02667414 2014-05-30
METHODS AND COMPOSITIONS FOR MODIFICATION OF THE HUMAN
GLUCOCORTICOID RECEPTOR LOCUS
TECHNICAL FIELD
[0003] The present disclosure is in the fields of immunology, immune
system
modulation and genome modification, including targeted mutagenesis, targeted
genomic
integration and targeted recombination.
BACKGROUND
[0004] The human glucocorticoid receptor (GR) is expressed in almost
all cells of the
body. Upon binding of glucocorticoid hormones such as Cortisol the receptor is
translocated
to the cell nucleus and activates a tissue-specific set of target genes. The
fact that GR target
genes vary from one tissue to another results in a pleiotropic pattern of GR
effects in different
tissues.
[0005] Many of the physiological actions of glucocorticoid hormones
are of medical
interest and present potential areas for clinical intervention. For example,
in
1
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
Cushing's syndrome, excess GR activity leads to high blood pressure. In the
brain,
abnormalities in the GR pathway have been linked to depression and mood
disorders;
and, in the lung, such abnormalities have been associated with asthma and
chronic
airway diseases.
[0006] One of the best-characterized clinical activities of glucocorticoid
hormones is their anti-inflammatory action, which is due to their immuno-
suppressive
effects. Exposure of T-cells to glucocorticoid hormones leads to T-cell anergy
and
interferes with T-cell activation. For a recent review, see Rhen, T. et al.
(2005). N.
Engl. I Med. 353(16):1711-23. Long-term treatment with glucocorticoids leads
to
serious side effects like diabetes and osteoporosis. See discussion in Rosen,
J. et al.
(2005) Endocr. Rev. 26(3):452-64. Moreover, suppression of the entire immune
system can lead to the reactivation of latent viruses (see Reinke, P. etal.
(1999)
Transpl Infect Dis 1(3):157-64) and interferes with immunotherapy approaches;
e.g.,
the delivery of a beneficial subset of immune cells to patients.
[0007] Many of the problems associated with the GR overactivation that
accompanies glucocorticoid treatment could be solved if a method was available
which allows selective disruption of GR function in a subset of cells; e.g., a
characterized population of T-cells. One such method would be to alter the
sequence
of the gene encoding the GR. Indeed, the ability to manipulate (i.e. edit) the
DNA
sequence at specific locations in the genome has been a major goal of human
genome
biology for some time. A variety of techniques have previously been tested for
this
purpose, but the frequencies of genome modification achieved with these
methods
have generally been too low for therapeutic applications. See, e.g., Yanez,
R.J. et al.
(1998) Gene Ther. 5(2): 149-159.
[0008] Another important application of genome editing is the insertion of
clinically useful transgenes into the genome. However, a crucial requirement
for any
genome editing method is that it allow for targeted insertion into a defined
location.
The importance of the requirement for precisely targeted integration of a
therapeutic
transgene was underscored by the recent observation, in a clinical trial for
treatment
of X-linked SCID that the random integration of transgenes used for human gene
therapy resulted, in certain cases, in insertional mutagenesis which led to
oncogenic
transformation of target cells. Hacein-Bey-Abina, S. et al. (2003). Science
302(5644):415-9.
2
CA 02667414 2014-05-30
[0009] Various methods and compositions for targeted cleavage of
genomic DNA
have been described. Such targeted cleavage events can be used, for example,
to induce
targeted mutagenesis, induce targeted deletions of cellular DNA sequences, and
facilitate
targeted recombination at a predetermined chromosomal locus. See, for example,
United
States patent application publications US 2003/0232410 (December 18, 2003), US
2005/0026157 (February 3, 2005), US 2005/0064474 (March 24, 2005), US
2005/0208489
(September 22, 2005) and US 2006/0188987 (August 24, 2006). Targeted
integration of
exogenous sequences can also be accomplished. See United States patent
publication
US 2007/0134796 (filed July 26, 2006). See also PCT WO 2005/084190 (September
15,
2005).
[0010] However, methods and compositions for specific cleavage of the
human
glucocorticoid receptor gene, and for modulation of immune function by
modification of the
GR gene, have not heretofore been described.
SUMMARY
[0011] Certain exemplary embodiments can provide a fusion protein
comprising: (i) a
zinc finger DNA-binding domain that has been engineered to bind a target
sequence in a
glucocorticoid receptor (GR) gene, wherein the target sequence bound by the
zinc finger
DNA-binding protein comprises a sequence of any one of SEQ ID Nos.: 1 to 7;
and (ii) a
nuclease domain.
[0011a] Disclosed herein are methods and compositions for alteration
of the nucleotide
sequence of the human gene encoding the glucocorticoid receptor (GR). In
certain
embodiments, alteration of the sequence of the human GR gene inactivates GR
function.
[0012] The methods include expression, in a cell, of a pair of zinc
finger nucleases
targeted to the human GR gene, which catalyze double stranded cleavage of
sequences in the
GR gene. Zinc finger nucleases are fusion proteins, comprising a zinc finger
DNA-binding
domain that has been engineered to bind to a target sequence and a cleavage
half-domain.
Expression of the zinc finger nucleases in a cell can be achieved by
introduction of the
nucleases themselves, RNA encoding the nucleases, or DNA encoding the
nucleases, into the
3
CA 02667414 2014-05-30
cell. GR-targeted zinc finger nucleases comprise zinc finger DNA-binding
domains that have
been engineered to bind to target sites in the GR gene. Engineering of a zinc
finger DNA-
binding domain includes determination of the amino acid sequence of the zinc
fingers
required for binding to the target nucleotide sequence (which may be achieved
by
3a
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
computational or empirical means) and construction of a polynucleotide or
polypeptide sequence corresponding to the desired amino acid sequence(s).
[0013] Exemplary engineered zinc finger DNA-binding domains targeted
to
the human GR gene are shown in Table 2 and their target sequences are shown in
Table 1. Thus, this disclosure provides zinc finger nucleases targeted to any
site in
the human GR gene, and polynucleotides encoding said zinc finger nucleases.
Cells
comprising the aforementioned zinc finger nucleases and polynucleotides are
also
provided, for example, isolated cells, either primary cells or cells in
culture.
[0014] In certain embodiments, targeted cleavage of the human GR
gene by
the zinc finger nucleases induces sequence alterations resulting from non-
homologous
end-joining (NHEJ). In additional embodiments, two zinc finger nucleases are
expressed in a cell, and a donor polynucleotide is introduced into the cell.
The donor
polynucleotide contains a first region of homology to sequences upstream of
the
double-strand break created by the zinc finger nucleases, and a second region
of
homology to sequences downstream of the double-strand break. The donor
polynucleotide optionally contains exogenous sequences that are non-homologous
to
the GR gene, which may comprise a transgene such as, for example, a chimeric T-
cell
receptor.
[0015] Inactivation of GR function by altering the primary
nucleotide
sequence of the GR gene, as described herein, can be used to prevent GR-
mediated
immune suppression in a variety of applications.
[0016] In one aspect, provided herein is a fusion protein
comprising: (i) a zinc
finger DNA-binding domain that has been engineered to bind a target sequence
in the
GR gene, and (ii) a cleavage half-domain. In certain embodiments, the zinc
finger
DNA-binding domain comprises a set of amino acid sequences in the order shown
in
a row of Table 2. Polynucleotides encoding any of the fusion proteins
described
herein are also provided.
[0017] In another aspect, the present disclosure provides a method
for
inactivating glucocorticoid receptor (GR) function in a cell, the method
comprising:
= 30 expressing in the cell a pair of fusion proteins, wherein each
fusion protein comprises:
(i) a zinc finger DNA-binding domain that has been engineered to bind a target
sequence in the GR gene, and (ii) a cleavage half-domain; such that the fusion
proteins catalyze a double-strand break in the GR gene. In certain
embodiments, the
zinc finger DNA-binding domain of a fusion protein comprises a set of amino
acid
4
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
sequences in the order shown in a row of Table 2. Any of the methods described
herein may further comprise the step introducing a polynucleotide into the
cell,
wherein the polynucleotide comprises a first region of homology to sequences
upstream of the double-strand break and a second region of homology to
sequences
downstream of the double-strand break. Optionally, the polynucleotide further
comprises exogenous sequences (e.g., a transgene such as a modified receptor)
that
are non-homologous to the GR gene. Furthermore, any of the methods may prevent
glucocorticoid-mediated immune suppression and/or T-cell anergy.
[0018] In yet another aspect, the disclosure provides methods of
selecting
cells into which an exogenous sequence has been introduced into a OR gene. The
method comprises expressing ZFNs as described herein to cause a double-
stranded
break in a GR gene and introducing a donor polynucleotide (comprising GR
homology arms and the exogenous sequence) into the cell. Cells in which the
donor
polynucleotide has been inserted into a OR gene are then selected for by
growing the
cells in the presence of a corticosteroid, which kills cells expressing normal
amounts
of GR. The term "corticosteroid" includes naturally occurring steroid hormones
such
as coritsol, corticosterone, cortisone and aldosterone. The term also includes
synthetic drugs with corticosteroid-like effect including, for example,
dexamethasone,
prednisone, Fludrocortisone (FlorineM) and the like. In certain embodiments,
the
corticosteroid is dexamethasone. In any of these methods, the exogenous
sequence
may comprise a transgene (a sequence encoding a polypeptide of interest).
Alternatively, the exogenous sequence may be a non-coding sequence.
[0019] In any of the methods described herein, the ZFNs are expressed
using a
viral delivery vector, for example, a replication-defective viral vector. In
certain
embodiments, the viral delivery vector is an adenovirus, a hybrid adenovirus
or a non-
integrating lentivirus.
[0020] Accordingly, the disclosure includes, but is not limited to,
the
following embodiments.
1. A method for inactivating glucocorticoid receptor (GR)
function in a
cell, the method comprising:
expressing in the cell a pair of fusion proteins, wherein each fusion protein
comprises:
(i) a zinc finger DNA-binding domain that has been engineered to bind
a target sequence in the OR gene, and
5
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
(ii) a cleavage half-domain;
such that the fusion proteins catalyze a double-strand break in the GR gene.
2. The method of 1, wherein the zinc finger DNA-binding domain of a
fusion protein comprises a set of amino acid sequences in the order shown in a
row of
Table 2.
3. The method of 1, further comprising introducing a polynucleotide into
the cell, wherein the polynucleotide comprises a first region of homology to
sequences upstream of the double-strand break and a second region of homology
to
sequences downstream of the double-strand break.
4. The method of 3, wherein the polynucleotide further comprises
exogenous sequences that are non-homologous to the GR gene.
5. The method of 4, wherein the exogenous sequences comprise a
transgene.
6. The method of 5, wherein the transgene encodes a modified receptor.
7. The method of 1, wherein inactivation of GR function prevents
glucocorticoid-mediated immune suppression.
8. The method of 1, wherein inactivation of GR function prevents T-cell
allergy.
9. A fusion protein comprising:
(i) a zinc finger DNA-binding domain that has been engineered to bind
a target sequence in the GR gene, and
(ii) a cleavage half-domain.
10. The fusion protein of 9, wherein the zinc finger DNA-binding domain
comprises a set of amino acid sequences in the order shown in a row of Table
2.
11. A polynucleotide encoding the fusion protein of 9.
12. The method of 1, wherein the expressing step comprises contacting the
cell with a viral delivery vector.
13. The method of 12, wherein the vector is replication-defective.
14. The method of 12, wherein the viral delivery vector is an adenovirus, a
hybrid adenovirus or a non-integrating lentivirus.
15. A method of selecting cells comprising an exogenous sequence in a GR
gene, the method comprising
expressing a pair of fusion proteins in the cell, wherein each fusion protein
comprises:
6
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
(i) a zinc finger DNA-binding domain that has been engineered to bind
a target sequence in a GR gene, and
(ii) a cleavage half-domain;
such that the fusion proteins catalyze a double-strand break in the GR
gene;
introducing a polynucleotide into the cell, wherein the polynucleotide
comprises a first region of homology to sequences upstream of the double-
strand
break, a second region of homology to sequences downstream of the double-
strand
break and the exogenous sequence; and
treating the cells with a natural or synthetic corticosteroid under conditions
such that cells not comprising the exogenous sequence in a GR gene are killed,
thereby selecting cells into which an exogenous sequence has been introduced
into a
GR gene.
16. The method of 15, wherein the corticosteroid is synthetic.
17. The method of 16, wherein the corticosteroid is dexamethasone.
18. The method of 15, wherein the exogenous sequence comprises a
transgene.
19. The method of 15, wherein the expressing step comprises contacting the
cell with a viral delivery vector.
20. The method of 19, wherein the vector is replication-defective.
21. The method of 19, wherein the viral delivery vector is an
adenovirus, a
hybrid adenovirus or a non-integrating lentivirus.
BRIEF DESCRIPTION OF THE DRAWINGS
[0021] Figure 1 is a schematic diagram of an exemplary plasmid construct
encoding a zinc finger nuclease. "CMV promoter" denotes the human
cytomegalovirus immediate early promoter, "ZFN" denotes sequences encoding a
zinc finger nuclease (e.g., a zinc finger DNA-binding domain fused to a
cleavage half-
domain), "BGH polyA" denotes the polyadenylation signal from the bovine growth
hormone gene, "SV40 promoter" denotes the major promoter from simian virus 40,
"NeoR" denotes an open reading frame encoding neomycin resistance, "SV40 pA"
denotes the polyadenlyation signal from the simian virus 40 major
transcription unit,
"ColE 1" denotes a replication origin from Colicin El and "AmpR" denotes the
(3-
lactamase gene encoding ampicillin resistance.
7
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0022] Figure 2 is a schematic of an exemplary plasmid construct
encoding
two different zinc finger nucleases. Abbreviations are the same as in Figure 1
with
the following additions. "2A" denotes the Foot-and-Mouth Disease virus (FMDV)
ribosome stuttering signal, "KanR" denotes an open reading frame which encodes
kanamycin resistance and "pUC on" denotes the origin of replication from the
pUC
19 plasmid.
[0023] Figure 3, panels A and B, show the results of Cell assays
demonstrating cleavage by ZFNs in the GR locus in hematopoietic cells (K562).
Fig.
3A shows analysis of cleavage in Exon 3, and Fig. 3B shows analysis of
cleavage in
exon 6. An ethidium bromide stain of a 10% acrylamide gel is shown.
[0024] Figure 4, panels A and B, show ZFN mediated targeted
integration of
a zetakine transgene into the GR locus. Fig 4A shows PCR analysis of CEM14
cells
transfected with GR-ZFNs and a zetakine-donor ZFN construct in the presence or
absence of dexamethasone. Lanes with a "+" indicate cells treated with
dexamethasone and lanes with a "-" indicate cells not treated with
dexamethasone.
"M" indicates the marker lane; "un" indicates untransfected cells; "zetakine"
indicates
cells transfected with the GR-ZFNs and the zetakine-donor construct; "p.c."
indicates
the positive control; and "n.c." indicates the negative control.
[0025] Fig. 4B shows Southern blot analysis of CEM14 genomic DNA
digested with SexAl. "M" indicates the marker lane; "un" indicates
untransfected
cells treated with dexamethasone; and "zetakine" indicates cells transfected
with the
GR-ZFNs and the zetakine-donor construct and treated with dexamethasone. Also
shown with arrows are a 1.6 kb marker band; wild-type 5.2 kb band; and 2.0 kb
band
representing integrated zetakine transgene (TI).
[0026] Figure 5 shows a chemiluminescent image of a protein blot that was
probed with an antibody to the human glucocorticoid receptor. "CEM14" denotes
untransfected cells; "ZFN" denotes CEM14 cells that had been transfected with
a
plasmid containing sequences encoding two ZFNs targeted to exon 3 of the GR
gene,
sequences containing a zetakine cassette, and sequences homologous to the GR
gene.
Bands corresponding to full-length and truncated GR protein are indicated to
the left
of the photograph.
[0027] Figure 6 shows the results of Cel I assays demonstrating
cleavage by
ZFNs in exon 3 of the GR locus in CD-8+ T-cells. An autoradiogram of a 10%
acrylamide gel is shown. Abbreviations are as follows: "un" denotes
untransfected
8
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
cells, "GFP" denotes cells transfected with a plasmid encoding green
fluorescent
protein. The identities of the zinc finger portion of the zinc finger
nucleases
expressed in the transfected cells are shown above the two rightmost lanes;
see Tables
1 and 2 for details.
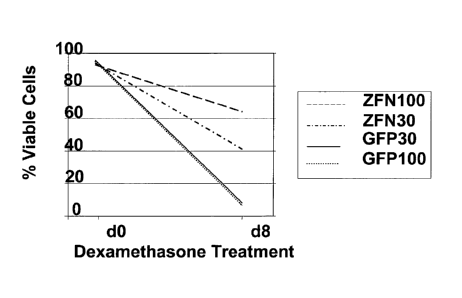
[0028] Figure 7, panels A and B, shows the results of Cel I assays
demonstrating GR cleavage by ZFNs in CD-8+ T-cells before (FIG. 7A) and after
(FIG. 7B) treatment with dexamethasone. "ZFN-10" denotes cells infected with
Ad5/F35 vector carrying ZFN pairs 9666 and 9674 at a multiplicity of infection
(moi)
of 10; "ZFN-30" denotes cells infected with Ad5/F35 vector carrying ZFN pairs
9666
and 9674 at moi of 30; "ZFN-100" denotes cells infected with Ad5/F35 vector
carrying ZFN pairs 9666 and 9674 at moi of 100; and "GFP" denotes cells
transfected
with a control Ad5/F35 virus encoding green fluorescent protein at an moi of
100.
Modification frequencies (percentages) are shown beneath various lanes.
[0029] Figure 8 is a graph depicting glucocorticoid resistance of
CD8+ cell
pools described in Example 7, as determined by comparing cell viability before
and
after a second treatment with 10-4M dexamethasone for 8 days.
[0030] Figure 9, panels A and B, show Western blot analysis for GR
protein
from two separate experiments. Panel A shows results of proteins extracted
from
CD8+ cell pools described in Example 7; from the zetakine expressing CD8+ cell
pool that was used for the virus transduction ('IL-13 ZK pool'); and from a
subclone
(`10A1') of the ZFN 100 pool. The antibody used for probing each panel is
listed to
the left of the blot. TFITB (Santa Cruz Antibodies) was used as a loading
control. The
GR antibody was obtained from BD Biosciences. Panel B shows GR protein levels
in
various subclones of CD8+ cells with specificity to CMV treated with the GR-
ZFN
expressing Ad5/F35 virus. Clone names are indicated above the lanes and "mock"
refers to the mock infected starting CMV-targeted CD8+ cell pool.
[0031] Figure 10, panels A through D, are graphs depicting RT-PCR
analysis
of ZFN treated CD8+ T-cells for expression of the indicated genes. Panel A
shows
expression of hcBa; panel B shows expression of GILZ; panel C shows expression
of
MKP-1; and panel E shows expression of IFNy. The samples tested are shown
below
the bars and were either untreated ("un") or treated with dexamethasone
("dex") for
20hrs.
9
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0032] Figure 11 is a graph depicting 1FN-y cytokine release by ZFN
Treated
CD8+ T-Cells upon stimulation with glioma cells. The CD8+ T-cell pools are
indicated below the bars. "Dex" refers to cells treated with 10-6 M
dexamethasone.
"U87MG" refers to cells cultured in the present of glioblastoma stimulator
cells.
[0033] Figure 12, panels A to E, are graphs depicting results of chromium
release assays using control and GR-ZFN-treated CD8+ T-cells. The cells used
are
noted above each graph. Samples were obtained using the target cell lines
indicated
on the right at various effector: target ratios. The percentage of chromium
release is
plotted against the effector: target ratio for each data point.
[0034] Figure 13, panels A to C, are graphs depicting photons emitted from
tumor cells in an orthotopic glioblastoma mouse model using luciferase labeled
U87MG cells. Panel A shows photon emission from PBS control injections into
the
indicated animals. Panel B shows photon emission from animals injected with
GFP100 controls. Panel C shows photon emission from animals injected with
ZFN100 (GR-targeted ZFN at moi 100).
[0035] Figure 14, panels A to D, are graphs depicting photons emitted
from
tumor cells in the orthotopic mouse glioblastoma model in the presence or
absence of
administered glucocorticoid hormone. Figs. 14A and 14B show photon emission
from PBS control injections in the absence (Fig. 14A) or presence (Fig. 14B)
of
dexamethasone. Figs. 14C and 14D show photon emission from tumor cells of the
mice following injecting of ZFN treated clone 10A1 into the tumor cells in the
absence (Fig. 14C) or presence of dexamethasone (Fig. 14D).
[0036] Figure 15 is a schematic depicting an experimental outline for
determining the DNA-binding specificity of an individual zinc finger DNA-
binding
domain by SELEX. Hemaglutinin-tagged ZFNs were incubated with a pool of
randomized DNA sequences in the presence of biotinylated anti-HA Fab antibody
fragments. The tagged ZFN-DNA complexes were captured with streptavidin-coated
magnetic beads, and the bound DNA was released and amplified by PCR. This
process was repeated three times using the previous eluted, amplified pool of
DNA as
a starting sequence. After four iterations, the eluted DNA fragments were
sequenced,
and the consensus sequence was determined.
CA 02667414 2014-05-30
DETAILED DESCRIPTION
[0037] Disclosed herein are compositions and methods useful for
altering the primary
sequence of the gene encoding the glucocorticoid receptor (GR), utilizing
fusion proteins
comprising an engineered zinc finger DNA-binding domain and a cleavage domain
(or
cleavage half-domain), referred to herein as "zinc finger nucleases." Such
sequence
alterations can result in inactivation of human GR function. As is known in
the art, zinc
finger DNA-binding domains can be engineered, by selection methods or using
techniques of
rational design, to bind any target DNA sequence of choice. Fusion of
engineered zinc finger
DNA-binding domains to various types of functional domain, including
transcriptional
activation domains, transcriptional repression domains and nuclease domains,
has also been
described. See, for example, U.S. Patents 6,534,261 and 6,933,113 and U.S.
Patent
Application Publication No.2005-0064474. Thus, by fusion of an engineered zinc
finger
binding domain to a nuclease domain, also known as a cleavage domain (i.e., a
polypeptide
domain with the ability to cleave double-stranded DNA), a custom endonuclease,
having
cleavage specificity for a sequence of choice, can be constructed. In certain
embodiments, an
engineered zinc finger DNA-binding domain is fused to a "cleavage half-domain"
(i.e., a
polypeptide domain which, when dimerized, possesses double-stranded DNA
cleavage
activity) and a pair of such fusion proteins is used for targeted DNA
cleavage.
[0038] Cleavage of genomic DNA can result in the induction of a
cellular repair
mechanism known as non-homologous end joining (NHEJ). In the process of
rejoining
broken DNA ends, NHEJ often introduces mutations into the sequence at or
around the site of
the DNA break. The error-prone nature of the repair process, coupled with the
ability of the
zinc finger nuclease(s) to continue to bind and cleave their target
sequence(s) until error-prone
repair causes an alteration of the target sequence(s), results in the
accumulation of mutations
at or near the site of cleavage at a high frequency. Accordingly, targeted
cleavage of
endogenous genomic DNA sequences with zinc finger nucleases can be used to
induce
sequence changes (i.e., mutations) at or around the site of targeted cleavage.
If such changes
in nucleotide sequence occur in a region of the genome that encodes a protein,
they usually
result in alterations of the amino acid sequence of the encoded protein. For
example,
alteration of reading frame can result in production of a truncated protein
due to premature
11
CA 02667414 2014-05-30
translation termination. Alternatively, incorrect amino acids may be encoded.
In either case, a
non- functional polypeptide is produced. An additional consequence of sequence
alteration
following NHEJ is nonsense-mediated decay of mRNA encoded by the altered
sequence.
Thus, targeted DNA cleavage using zinc finger nucleases can be used to
inactivate the
function of a gene of choice. Inactivation can be achieved either by
mutagenesis of both
alleles or by mutagenesis of a single allele to generate a dominant negative
mutant protein.
[0039] Targeted cleavage at a predetermined site in endogenous
chromosomal DNA
can also be used to facilitate integration of exogenous sequences at or near
the site of
cleavage, by both homology-directed and homo logy-independent mechanisms. For
homology-dependent integration, a "donor sequence," containing sequences
homologous to
genomic sequences on both sides of the targeted cleavage site, is provided to
cells in addition
to the zinc finger nuclease(s). Such a donor sequence can also contain
sequences that are
nonhomologous to genomic sequences in the vicinity of the targeted cleavage
site, optionally
disposed between two stretches of homologous sequence. See, for example,
United States
Patent Application Publication No. 2005-0064474 (March 24, 2005) and United
States Patent
Application No. 11/493,423 (July 26, 2006). If integration of exogenous
sequences occurs
within the transcribed region of a gene, at both alleles, inactivation of the
gene can result.
Finally, targeted cleavage at two or more sites in endogenous chromosomal DNA
can result in
deletion of genomic sequences between the cleavage sites. See U.S. Patent
Application
Publication No. 2006-0188987 (August 24, 2006). Thus, gene function can be
inactivated by
any of the foregoing mechanisms, all of which depend upon targeted cleavage of
endogenous
chromosomal DNA with one or more zinc finger nucleases.
[0040] The present disclosure provides methods and compositions for
mutating the
human glucocorticoid receptor (GR) gene. Such mutations can cause loss of GR
function and
result in modulation of immune function in a subject. In certain embodiments,
mutation of
the GR gene results from zinc finger nuclease-mediated integration of
exogenous sequences
into the human GR locus. In additional embodiments, the exogenous sequences
comprise
sequences encoding a modified receptor molecule.
12
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0041] The methods and compositions disclosed herein allow permanent
abolition of glucocorticoid receptor function in a specified population of
cells. This
makes it possible, for example, to treat patients with immunosuppressant
glucocorticoid hormones, while allowing those patients to retain a subset of
immune
cells able to effect specific immune responses.
[0042] Also provided are methods and compositions which facilitate
the use
of the GR locus as a defined integration site for therapeutic transgenes.
General
[0043] Practice of the methods, as well as preparation and use of the
compositions disclosed herein employ, unless otherwise indicated, conventional
techniques in molecular biology, biochemistry, chromatin structure analysis,
computational chemistry, cell culture, recombinant DNA and related fields as
are
within the skill of the art. These techniques are fully explained in the
literature. See,
for example, Sambrook et al. MOLECULAR CLONING: A LABORATORY MANUAL, Cold
Spring Harbor Laboratory Press, Second edition, 1989, Third edition, 2001;
Ausubel
et al., CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, New
York, 1987 and periodic updates; and the series METHODS IN ENZYMOLOGY,
Academic Press, San Diego.
Definitions
[0044] The terms "nucleic acid," "polynucleotide," and
"oligonucleotide" are
used interchangeably and refer to a deoxyribonucleotide or ribonucleotide
polymer, in
linear or circular conformation, and in either single- or double-stranded
form. For the
purposes of the present disclosure, these terms are not to be construed as
limiting with
respect to the length of a polymer. The terms can encompass known analogues of
natural nucleotides, as well as nucleotides that are modified in the base,
sugar and/or
phosphate moieties (e.g., phosphorothioate backbones). In general, an analogue
of a
particular nucleotide has the same base-pairing specificity; i.e., an analogue
of A will
base-pair with T.
[0045] The terms "polypeptide," "peptide" and "protein" are used
interchangeably to refer to a polymer of amino acid residues. The term also
applies to
amino acid polymers in which one or more amino acids are chemical analogues or
modified derivatives of a corresponding naturally-occurring amino acids.
13
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0046] "Binding" refers to a sequence-specific, non-covalent
interaction
between macromolecules (e.g., between a protein and a nucleic acid). Not all
components of a binding interaction need be sequence-specific (e.g., contacts
with
phosphate residues in a DNA backbone), as long as the interaction as a whole
is
sequence-specific. Such interactions are generally characterized by a
dissociation
constant (Kd) of 10-6 M-1 or lower. "Affinity" refers to the strength of
binding:
increased binding affinity being correlated with a lower Kd.
[0047] A "binding protein" is a protein that is able to bind non-
covalently to
another molecule. A binding protein can bind to, for example, a DNA molecule
(a
DNA-binding protein), an RNA molecule (an RNA-binding protein) and/or a
protein
molecule (a protein-binding protein). In the case of a protein-binding
protein, it can
bind to itself (to form homodimers, homotrimers, etc.) and/or it can bind to
one or
more molecules of a different protein or proteins. A binding protein can have
more
than one type of binding activity. For example, zinc finger proteins have DNA-
binding, RNA-binding and protein-binding activity.
[0048] A "zinc finger DNA binding protein" (or binding domain) is a
protein,
or a domain within a larger protein, that binds DNA in a sequence-specific
manner
through one or more zinc fingers, which are regions of amino acid sequence
within
the binding domain whose structure is stabilized through coordination of a
zinc ion.
The term zinc finger DNA binding protein is often abbreviated as zinc finger
protein
or ZFP.
[0049] Zinc finger binding domains can be "engineered" to bind to a
predetermined nucleotide sequence. Non-limiting examples of methods for
engineering zinc finger proteins are design and selection. A designed zinc
finger
protein is a protein not occurring in nature whose design/composition results
principally from rational criteria. Rational criteria for design include
application of
substitution rules and computerized algorithms for processing information in a
database storing information of existing ZFP designs and binding data. See,
for
example, US Patents 6,140,081; 6,453,242; and 6,534,261; see also WO 98/53058;
W098/53059; W098/53060; WO 02/016536 and W003/016496.
[0050] A "selected" zinc finger protein is a protein not found in
nature whose
production results primarily from an empirical process such as phage display,
interaction trap or hybrid selection. See e.g., US 5,789,538; US 5,925,523; US
6,007,988; US 6,013,453; US 6,200,759; WO 95/19431; WO 96/06166; WO
14
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
98/53057; WO 98/54311; WO 00/27878; WO 01/60970 WO 01/88197 and WO
02/099084.
[0051] The term "sequence" refers to a nucleotide sequence of any
length,
which can be DNA or RNA; can be linear, circular or branched and can be either
single-stranded or double stranded. The term "donor sequence" refers to a
nucleotide
sequence that is inserted into a genome. A donor sequence can be of any
length, for
example between 2 and 10,000 nucleotides in length (or any integer value
therebetween or thereabove), preferably between about 100 and 1,000
nucleotides in
length (or any integer therebetween), more preferably between about 200 and
500
nucleotides in length.
[0052] A "homologous, non-identical sequence" refers to a first
sequence
which shares a degree of sequence identity with a second sequence, but whose
sequence is not identical to that of the second sequence. For example, a
polynucleotide comprising the wild-type sequence of a mutant gene is
homologous
and non-identical to the sequence of the mutant gene. In certain embodiments,
the
degree of homology between the two sequences is sufficient to allow homologous
recombination therebetween, utilizing normal cellular mechanisms. Two
homologous
non-identical sequences can be any length and their degree of non-homology can
be
as small as a single nucleotide (e.g., for correction of a genomic point
mutation by
targeted homologous recombination) or as large as 10 or more kilobases (e.g.,
for
insertion of a gene at a predetermined ectopic site in a chromosome). Two
polynucleotides comprising the homologous non-identical sequences need not be
the
same length. For example, an exogenous polynucleotide (i.e., donor
polynucleotide)
of between 20 and 10,000 nucleotides or nucleotide pairs can be used.
[0053] Techniques for determining nucleic acid and amino acid sequence
identity are known in the art. Typically, such techniques include determining
the
nucleotide sequence of the mRNA for a gene and/or determining the amino acid
sequence encoded thereby, and comparing these sequences to a second nucleotide
or
amino acid sequence. Genomic sequences can also be determined and compared in
this fashion. In general, identity refers to an exact nucleotide-to-nucleotide
or amino
acid-to-amino acid correspondence of two polynucleotides or polypeptide
sequences,
respectively. Two or more sequences (polynucleotide or amino acid) can be
compared by determining their percent identity. The percent identity of two
sequences, whether nucleic acid or amino acid sequences, is the number of
exact
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
matches between two aligned sequences divided by the length of the shorter
sequences and multiplied by 100. An approximate alignment for nucleic acid
sequences is provided by the local homology algorithm of Smith and Waterman,
Advances in Applied Mathematics 2:482-489 (1981). This algorithm can be
applied
to amino acid sequences by using the scoring matrix developed by Dayhoff,
Atlas of
Protein Sequences and Structure, M.O. Dayhoff ed., 5 suppl. 3:353-358,
National
Biomedical Research Foundation, Washington, D.C., USA, and normalized by
Gribskov, Nucl. Acids Res. 14(6):6745-6763 (1986). An exemplary implementation
of this algorithm to determine percent identity of a sequence is provided by
the
Genetics Computer Group (Madison, WI) in the "BestFit" utility application.
The
default parameters for this method are described in the Wisconsin Sequence
Analysis
Package Program Manual, Version 8 (1995) (available from Genetics Computer
Group, Madison, WI). A preferred method of establishing percent identity in
the
context of the present disclosure is to use the MPSRCH package of programs
copyrighted by the University of Edinburgh, developed by John F. Collins and
Shane
S. Sturrok, and distributed by IntelliGenetics, Inc. (Mountain View, CA). From
this
suite of packages the Smith-Waterman algorithm can be employed where default
parameters are used for the scoring table (for example, gap open penalty of
12, gap
extension penalty of one, and a gap of six). From the data generated the
"Match"
value reflects sequence identity. Other suitable programs for calculating the
percent
identity or similarity between sequences are generally known in the art, for
example,
another alignment program is BLAST, used with default parameters. For example,
BLASTN and BLASTP can be used using the following default parameters: genetic
code = standard; filter = none; strand = both; cutoff= 60; expect = 10; Matrix
=
BLOSUM62; Descriptions = 50 sequences; sort by = HIGH SCORE; Databases =
non-redundant, GenBank + EMBL + DDBJ + PDB + GenBank CDS translations +
Swiss protein + Spupdate + PIR. Details of these programs can be found at the
following interne address: http://www.ncbi.nlm.gov/cgi-bin/BLAST. With respect
to
sequences described herein, the range of desired degrees of sequence identity
is
approximately 80% to 100% and any integer value therebetween. Typically the
percent identities between sequences are at least 70-75%, preferably 80-82%,
more
preferably 85-90%, even more preferably 92%, still more preferably 95%, and
most
preferably 98% sequence identity.
16
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0054] Alternatively, the degree of sequence similarity between
polynucleotides can be determined by hybridization of polynucleotides under
conditions that allow formation of stable duplexes between homologous regions,
followed by digestion with single-stranded-specific nuclease(s), and size
determination of the digested fragments. Two nucleic acid, or two polypeptide
sequences are substantially homologous to each other when the sequences
exhibit at
least about 70%-75%, preferably 80%-82%, more preferably 85%-90%, even more
preferably 92%, still more preferably 95%, and most preferably 98% sequence
identity over a defined length of the molecules, as determined using the
methods
above. As used herein, substantially homologous also refers to sequences
showing
complete identity to a specified DNA or polypeptide sequence. DNA sequences
that
are substantially homologous can be identified in a Southern hybridization
experiment
under, for example, stringent conditions, as defined for that particular
system.
Defining appropriate hybridization conditions is within the skill of the art.
See, e.g.,
Sambrook et al., supra; Nucleic Acid Hybridization: A Practical Approach,
editors
B.D. Hames and S.J. Higgins, (1985) Oxford; Washington, DC; IRL Press).
[0055] Selective hybridization of two nucleic acid fragments can be
determined as follows. The degree of sequence identity between two nucleic
acid
molecules affects the efficiency and strength of hybridization events between
such
molecules. A partially identical nucleic acid sequence will at least partially
inhibit the
hybridization of a completely identical sequence to a target molecule.
Inhibition of
hybridization of the completely identical sequence can be assessed using
hybridization assays that are well known in the art (e.g., Southern (DNA)
blot,
Northern (RNA) blot, solution hybridization, or the like, see Sambrook, et
al.,
Molecular Cloning: A Laboratory Manual, Second Edition, (1989) Cold Spring
Harbor, N.Y.). Such assays can be conducted using varying degrees of
selectivity, for
example, using conditions varying from low to high stringency. If conditions
of low
stringency are employed, the absence of non-specific binding can be assessed
using a
secondary probe that lacks even a partial degree of sequence identity (for
example, a
probe having less than about 30% sequence identity with the target molecule),
such
that, in the absence of non-specific binding events, the secondary probe will
not
hybridize to the target.
[0056] When utilizing a hybridization-based detection system, a
nucleic acid
probe is chosen that is complementary to a reference nucleic acid sequence,
and then
17
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
by selection of appropriate conditions the probe and the reference sequence
selectively hybridize, or bind, to each other to form a duplex molecule. A
nucleic
acid molecule that is capable of hybridizing selectively to a reference
sequence under
moderately stringent hybridization conditions typically hybridizes under
conditions
that allow detection of a target nucleic acid sequence of at least about 10-14
nucleotides in length having at least approximately 70% sequence identity with
the
sequence of the selected nucleic acid probe. Stringent hybridization
conditions
typically allow detection of target nucleic acid sequences of at least about
10-14
nucleotides in length having a sequence identity of greater than about 90-95%
with
the sequence of the selected nucleic acid probe. Hybridization conditions
useful for
probe/reference sequence hybridization, where the probe and reference sequence
have
a specific degree of sequence identity, can be determined as is known in the
art (see,
for example, Nucleic Acid Hybridization: A Practical Approach, editors B.D.
Hames
and S.J. Higgins, (1985) Oxford; Washington, DC; IRL Press).
[0057] Conditions for hybridization are well-known to those of skill in the
art.
Hybridization stringency refers to the degree to which hybridization
conditions
disfavor the formation of hybrids containing mismatched nucleotides, with
higher
stringency correlated with a lower tolerance for mismatched hybrids. Factors
that
affect the stringency of hybridization are well-known to those of skill in the
art and
include, but are not limited to, temperature, pH, ionic strength, and
concentration of
organic solvents such as, for example, formamide and dimethylsulfoxide. As is
known to those of skill in the art, hybridization stringency is increased by
higher
temperatures, lower ionic strength and lower solvent concentrations.
[0058] With respect to stringency conditions for hybridization, it is
well
known in the art that numerous equivalent conditions can be employed to
establish a
particular stringency by varying, for example, the following factors: the
length and
nature of the sequences, base composition of the various sequences,
concentrations of
salts and other hybridization solution components, the presence or absence of
blocking agents in the hybridization solutions (e.g., dextran sulfate, and
polyethylene
glycol), hybridization reaction temperature and time parameters, as well as,
varying
wash conditions. The selection of a particular set of hybridization conditions
is,
selected following standard methods in the art (see, for example, Sambrook, et
al.,
Molecular Cloning: A Laboratory Manual, Second Edition, (1989) Cold Spring
Harbor, N.Y.).
18
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0059] "Recombination" refers to a process of exchange of genetic
information between two polynucleotides. For the purposes of this disclosure,
"homologous recombination (HR)" refers to the specialized form of such
exchange
that takes place, for example, during repair of double-strand breaks in cells.
This
process requires nucleotide sequence homology, uses a "donor" molecule to
template
repair of a "target" molecule (i.e., the one that experienced the double-
strand break),
and is variously known as "non-crossover gene conversion" or "short tract gene
conversion," because it leads to the transfer of genetic information from the
donor to
the target. Without wishing to be bound by any particular theory, such
transfer can
involve mismatch correction of heteroduplex DNA that forms between the broken
target and the donor, and/or "synthesis-dependent strand annealing," in which
the
donor is used to resynthesize genetic information that will become part of the
target,
and/or related processes. Such specialized HR often results in an alteration
of the
sequence of the target molecule such that part or all of the sequence of the
donor
polynucleotide is incorporated into the target polynucleotide.
[0060] "Cleavage" refers to the breakage of the covalent backbone of
a DNA
molecule. Cleavage can be initiated by a variety of methods including, but not
limited
to, enzymatic or chemical hydrolysis of a phosphodiester bond. Both single-
stranded
cleavage and double-stranded cleavage are possible, and double-stranded
cleavage
can occur as a result of two distinct single-stranded cleavage events. DNA
cleavage
can result in the production of either blunt ends or staggered ends. In
certain
embodiments, fusion polypeptides are used for targeted double-stranded DNA
cleavage.
[0061] A "cleavage domain" comprises one or more polypeptide
sequences
which possesses catalytic activity for DNA cleavage. A cleavage domain can be
contained in a single polypeptide chain or cleavage activity can result from
the
association of two (or more) polypeptides. A" cleavage half-domain" is a
polypeptide sequence which, in conjunction with a second polypeptide (either
identical or different) forms a complex having cleavage activity (preferably
double-
strand cleavage activity). The terms "first and second cleavage half-domains;"
"+ and
¨ cleavage half-domains" and "right and left cleavage half-domains" are used
interchangeably to refer to pairs of cleavage half-domains that dimerize.
[0062] An "engineered cleavage half-domain" is a cleavage half-domain
that
has been modified so as to form obligate heterodimers with another cleavage
half-
19
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
domain (e.g., another engineered cleavage half-domain). See, also, U.S. Patent
Publication Nos. 20050064474 and 20060188987 and U.S. Provisional Application
No. 60/808,486 (filed May 25, 2006), incorporated herein by reference in their
entireties.
[0063] "Chromatin" is the nucleoprotein structure comprising the cellular
genome. Cellular chromatin comprises nucleic acid, primarily DNA, and protein,
including histones and non-histone chromosomal proteins. The majority of
eukaryotic cellular chromatin exists in the form of nucleosomes, wherein a
nucleosome core comprises approximately 150 base pairs of DNA associated with
an
octamer comprising two each of histones H2A, H2B, H3 and H4; and linker DNA
(of
variable length depending on the organism) extends between nucleosome cores. A
molecule of histone H1 is generally associated with the linker DNA. For the
purposes
of the present disclosure, the term "chromatin" is meant to encompass all
types of
cellular nucleoprotein, both prokaryotic and eukaryotic. Cellular chromatin
includes
both chromosomal and episomal chromatin.
[0064] A "chromosome," is a chromatin complex comprising all or a
portion
of the genome of a cell. The genome of a cell is often characterized by its
karyotype,
which is the collection of all the chromosomes that comprise the genome of the
cell.
The genome of a cell can comprise one or more chromosomes.
[0065] An "episome" is a replicating nucleic acid, nucleoprotein complex or
other structure comprising a nucleic acid that is not part of the chromosomal
karyotype of a cell. Examples of episomes include plasmids and certain viral
genomes.
[0066] An "accessible region" is a site in cellular chromatin in
which a target
site present in the nucleic acid can be bound by an exogenous molecule which
recognizes the target site. Without wishing to be bound by any particular
theory, it is
believed that an accessible region is one that is not packaged into a
nucleosomal
structure. The distinct structure of an accessible region can often be
detected by its
sensitivity to chemical and enzymatic probes, for example, nucleases.
[0067] A "target site" or "target sequence" is a nucleic acid sequence that
defines a portion of a nucleic acid to which a binding molecule will bind,
provided
sufficient conditions for binding exist. For example, the sequence 5' GAATTC
3' is a
target site for the Eco RI restriction endonuclease.
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0068] An "exogenous" molecule is a molecule that is not normally
present in
a cell, but can be introduced into a cell by one or more genetic, biochemical
or other
methods. "Normal presence in the cell" is determined with respect to the
particular
developmental stage and environmental conditions of the cell. Thus, for
example, a
molecule that is present only during embryonic development of muscle is an
exogenous molecule with respect to an adult muscle cell. Similarly, a molecule
induced by heat shock is an exogenous molecule with respect to a non-heat-
shocked
cell. An exogenous molecule can comprise, for example, a functioning version
of a
malfunctioning endogenous molecule or a malfunctioning version of a normally-
functioning endogenous molecule.
[0069] An exogenous molecule can be, among other things, a small
molecule,
such as is generated by a combinatorial chemistry process, or a macromolecule
such
as a protein, nucleic acid, carbohydrate, lipid, glycoprotein, lipoprotein,
polysaccharide, any modified derivative of the above molecules, or any complex
comprising one or more of the above molecules. Nucleic acids include DNA and
RNA, can be single- or double-stranded; can be linear, branched or circular;
and can
be of any length. Nucleic acids include those capable of forming duplexes, as
well as
triplex-forming nucleic acids. See, for example, U.S. Patent Nos. 5,176,996
and
5,422,251. Proteins include, but are not limited to, DNA-binding proteins,
transcription factors, chromatin remodeling factors, methylated DNA binding
proteins, polymerases, methylases, demethylases, acetylases, deacetylases,
kinases,
phosphatases, integrases, recombinases, ligases, topoisomerases, gyrases and
helicases.
[0070] An exogenous molecule can be the same type of molecule as an
endogenous molecule, e.g., an exogenous protein or nucleic acid. For example,
an
exogenous nucleic acid can comprise an infecting viral genome, a plasmid or
episome
introduced into a cell, or a chromosome that is not normally present in the
cell.
Methods for the introduction of exogenous molecules into cells are known to
those of
skill in the art and include, but are not limited to, lipid-mediated transfer
(i.e.,
liposomes, including neutral and cationic lipids), electroporation, direct
injection, cell
fusion, particle bombardment, calcium phosphate co-precipitation, DEAE-dextran-
mediated transfer and viral vector-mediated transfer.
[0071] By contrast, an "endogenous" molecule is one that is normally
present
in a particular cell at a particular developmental stage under particular
environmental
21
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
conditions. For example, an endogenous nucleic acid can comprise a chromosome,
the genome of a mitochondrion, chloroplast or other organelle, or a naturally-
occurring episomal nucleic acid. Additional endogenous molecules can include
proteins, for example, transcription factors and enzymes.
[0072] A "fusion" molecule is a molecule in which two or more subunit
molecules are linked, preferably covalently. The subunit molecules can be the
same
chemical type of molecule, or can be different chemical types of molecules.
Examples of the first type of fusion molecule include, but are not limited to,
fusion
proteins (for example, a fusion between a ZFP DNA-binding domain and a
cleavage
domain) and fusion nucleic acids (for example, a nucleic acid encoding the
fusion
protein described supra). Examples of the second type of fusion molecule
include,
but are not limited to, a fusion between a triplex-forming nucleic acid and a
polypeptide, and a fusion between a minor groove binder and a nucleic acid.
[0073] Expression of a fusion protein in a cell can result from
delivery of the
fusion protein to the cell or by delivery of a polynucleotide encoding the
fusion
protein to a cell, wherein the polynucleotide is transcribed, and the
transcript is
translated, to generate the fusion protein. Trans-splicing, polypeptide
cleavage and
polypeptide ligation can also be involved in expression of a protein in a
cell. Methods
for polynucleotide and polypeptide delivery to cells are presented elsewhere
in this
disclosure.
[0074] A "gene," for the purposes of the present disclosure, includes
a DNA
region encoding a gene product (see infra), as well as all DNA regions which
regulate
the production of the gene product, whether or not such regulatory sequences
are
adjacent to coding and/or transcribed sequences. Accordingly, a gene includes,
but is
not necessarily limited to, promoter sequences, terminators, translational
regulatory
sequences such as ribosome binding sites and internal ribosome entry sites,
enhancers,
silencers, insulators, boundary elements, replication origins, matrix
attachment sites
and locus control regions.
[0075] "Gene expression" refers to the conversion of the information,
contained in a gene, into a gene product. A gene product can be the direct
transcriptional product of a gene (e.g., mRNA, tRNA, rRNA, antisense RNA,
ribozyme, structural RNA or any other type of RNA) or a protein produced by
translation of a mRNA. Gene products also include RNAs which are modified, by
processes such as capping, polyadenylation, methylation, and editing, and
proteins
22
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
modified by, for example, methylation, acetylation, phosphorylation,
ubiquitination,
ADP-ribosylation, myristilation, and glycosylation.
[0076] "Modulation" of gene expression refers to a change in the
activity of a
gene. Modulation of expression can include, but is not limited to, gene
activation and
gene repression.
[0077] "Eucaryotic" cells include, but are not limited to, fungal
cells (such as
yeast), plant cells, animal cells, mammalian cells and human cells (e.g., T-
cells).
[0078] A "region of interest" is any region of cellular chromatin,
such as, for
example, a gene or a non-coding sequence within or adjacent to a gene, in
which it is
desirable to bind an exogenous molecule. Binding can be for the purposes of
targeted
DNA cleavage and/or targeted recombination. A region of interest can be
present in a
chromosome, an episome, an organellar genome (e.g., mitochondrial,
chloroplast), or
an infecting viral genome, for example. A region of interest can be within the
coding
region of a gene, within transcribed non-coding regions such as, for example,
leader
sequences, trailer sequences or introns, or within non-transcribed regions,
either
upstream or downstream of the coding region. A region of interest can be as
small as
a single nucleotide pair or up to 2,000 nucleotide pairs in length, or any
integral value
of nucleotide pairs.
[0079] The terms "operative linkage" and "operatively linked" (or
"operably
linked") are used interchangeably with reference to a juxtaposition of two or
more
components (such as sequence elements), in which the components are arranged
such
that both components function normally and allow the possibility that at least
one of
the components can mediate a function that is exerted upon at least one of the
other
components. By way of illustration, a transcriptional regulatory sequence,
such as a
promoter, is operatively linked to a coding sequence if the transcriptional
regulatory
sequence controls the level of transcription of the coding sequence in
response to the
presence or absence of one or more transcriptional regulatory factors. A
transcriptional regulatory sequence is generally operatively linked in cis
with a coding
sequence, but need not be directly adjacent to it. For example, an enhancer is
a
transcriptional regulatory sequence that is operatively linked to a coding
sequence,
even though they are not contiguous.
[0080] With respect to fusion polypeptides, the term "operatively
linked" can
refer to the fact that each of the components performs the same function in
linkage to
the other component as it would if it were not so linked. For example, with
respect to
23
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
a fusion polypeptide in which a ZFP DNA-binding domain is fused to a cleavage
domain, the ZFP DNA-binding domain and the cleavage domain are in operative
linkage if, in the fusion polypeptide, the ZFP DNA-binding domain portion is
able to
bind its target site and/or its binding site, while the cleavage domain is
able to cleave
DNA in the vicinity of the target site.
[0081] A "functional fragment" of a protein, polypeptide or nucleic
acid is a
protein, polypeptide or nucleic acid whose sequence is not identical to the
full-length
protein, polypeptide or nucleic acid, yet retains the same function as the
full-length
protein, polypeptide or nucleic acid. A functional fragment can possess more,
fewer,
or the same number of residues as the corresponding native molecule, and/or
can
contain one ore more amino acid or nucleotide substitutions. Methods for
determining the function of a nucleic acid (e.g., coding function, ability to
hybridize
to another nucleic acid) are well-known in the art. Similarly, methods for
determining
protein function are well-known. For example, the DNA-binding function of a
polypeptide can be determined, for example, by filter-binding, electrophoretic
mobility-shift, or immunoprecipitation assays. DNA cleavage can be assayed by
gel
electrophoresis. See Ausubel et al., supra. The ability of a protein to
interact with
another protein can be determined, for example, by co-immunoprecipitation, two-
hybrid assays or complementation, both genetic and biochemical. See, for
example,
Fields et al. (1989) Nature 340:245-246; U.S. Patent No. 5,585,245 and PCT WO
98/44350.
Design of zinc finger DNA-binding domains
[0082] Construction of zinc finger nucleases is described, for
example, in U.S.
Patent Application Publications 2003-0232410, 2005-0026157, 2005-0064474 and
2005-0208489, the disclosures of which are incorporated by reference in their
entireties. Briefly, a non-naturally-occurring zinc finger DNA-binding domain,
comprising 2, 3, 4, 5, 6 or more zinc fingers, is engineered to bind to a
predetermined
target nucleotide sequence. The engineered zinc finger binding domain is fused
to a
nuclease domain, a cleavage domain or a cleavage half-domain to form a zinc
finger
nuclease capable of DNA cleavage at or near the target nucleotide sequence. In
certain embodiments, following conceptual design of the zinc finger DNA-
binding
domain, a polynucleotide encoding the zinc finger nuclease is constructed,
using
standard molecular biological methods.
24
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0083] Zinc finger nucleases, for facilitating mutagenesis of the
human
glucocorticoid receptor gene, are designed and synthesized as follows. The
nucleotide sequences of relevant portions of the human glucocorticoid receptor
gene
are obtained. The sequences thus obtained are scanned, optionally using a
computer
program containing a listing of individual zinc fingers and their target sites
and/or a
listing of two-finger modules and their target sites, for a pair of target
sequences,
separated by 5-6 nucleotide pairs, wherein each target sequence can be bound
by a 3-,
4-, 5- or 6-finger zinc finger protein. See, for example, U.S. Patent No.
6,785,613;
WO 98/53057; WO 01/53480 and U.S. Patent Application Publication No.
2003/0092000. Additional methods for ZFP design are disclosed, for example, in
U.S. Patents 5,789,538; 6,013,453; 6,410,248; 6,733,970; 6,746,838; 6,785,613;
6,866,997; 7,030,215; WO 01/088197; WO 02/099084; and US Patent Application
Publications 2003/0044957; 2003/0108880; 2003/0134318 and 2004/0128717.
[0084] For each target sequence identified in the previous step, a
gene
encoding a fusion between a Fokl cleavage half-domain and a zinc finger
protein that
binds to the target sequence is synthesized. See, for example, U.S. Patent No.
5,436,150; WO 2005/084190 and U.S. Patent Application Publication No.
2005/0064474. Each fusion protein can be tested for the affinity with which it
binds
to its target sequence, using an ELISA assay as described, for example, by
Bartsevich
et al. (2003) Stem Cells 21:632-637. Proteins having target sequence binding
affinities which exceed a predetermined threshold value can subjected to
further
testing in a cell-based reporter assay.
[0085] Optionally, the binding specificity of one or more fusion
proteins as
described above can be assessed and, if necessary, improved, by alteration
(including
randomization) of one or more amino acid residues followed by a phage display
assay
against the target sequence (see, for example, WO 96/06166), and/or by methods
of
iterative optimization described in U.S. Patent No. 6,794,136.
[0086] Cell-based testing is conducted as described, for example, in
Urnov et
al. (2005) Nature 435:646-651 and U.S. Patent Application Publication No.
2005/0064474. Briefly, a target sequence pair, identified as described above,
is
inserted into a defective chromosomal green fluorescent protein (GFP) gene,
under
the transcriptional control of a doxycycline-inducible promoter, in an
appropriate cell
line. Cells are transfected with nucleic acids encoding two zinc finger/FokI
fusion
proteins (each of which binds to one of the target sequences) and with a
nucleic acid
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
containing sequences that, if they serve as template for homology-directed
repair of
the defective chromosomal GFP gene, will reconstitute a functional GFP gene.
Cells
in which homology-directed repair has occurred can be identified and
quantitated by
fluorescence-activated cell sorting, following induction with doxycycline.
Zinc finger binding domains targeted to the human glucocorticoid
receptor gene
[0087] Methods for inactivation of GR disclosed herein utilize zinc
finger
nucleases, comprising (1) a zinc finger DNA-binding domain which has been
engineered to bind a target sequence of choice and (2) a cleavage domain or
cleavage
half-domain. Any such zinc finger nuclease having a target site in a human GR
gene
can be used in the disclosed methods. Alternatively, any pair of zinc finger
nucleases,
each comprising a cleavage half-domain, whose target sequences are separated
by the
appropriate number of nucleotides, can also be used. See, for example, U.S.
Patent
Application Publication No. 2005-0064474; Smith et al. (2000) Nucleic Acids
Res.
28:3361-3369 and Bibikova et al. (2001) MoL Cell. Biol. 21:289-297.
[0088] Exemplary zinc finger binding domains having target sites in
the
human GR gene are disclosed in Tables 1 and 2. Table 1 provides the target
sequences of the exemplary binding domains and the location of those target
sites in
the GR gene. Table 2 shows the amino acid sequences of the engineered
recognition
regions (responsible for DNA-binding specificity) of these binding domains.
Zinc
finger sequences are shown in amino-to-carboxy order, with Fl denoting the
zinc
finger nearest the amino terminus of the protein.
26
CA 02667414 2009-04-23
WO 2008/060510 PCT/US2007/023745
Table 1: Target Sequences for GR-Targeted ZFNs
Namel Target Sequence2
Location3
8718, 9967 GACCTGt TGATAG nt 778-790 sense (exon 2)
(SEQ ID NO:1)
8893 GACCTGtTGATAGATG nt 778-793 sense (exon 2)
(SEQ ID NO: 2 )
8719, 10415, 10404 TCCAAGGACTCT nt 761-772 antisense (exon 2)
(SEQ ID NO:3)
8667,9666 CAACAGGACCAC nt 1370-1381 sense (exon 3)
(SEQ ID NO:4)
8668,8669,9671, GTTGAGGAGCTG nt 1353-1364 antisense (exon
9674, 10201, 10205 ( SEQ ID NO : 5 ) 3)
8531, 9737, 9846 AATGAGTAAGTTG nt 2020-2023 sense (exon 6) +
(SEQ ID NO: 6) first 9 nt in intron 6
8653 TCAGATCAGGAG nt 2003-2014 antisense (exon
( SEQ ID NO : 7 ) 6)
1 Each zinc finger binding domain is represented by a four- or five-digit
number. Relevant amino acid
sequences of these binding domains are shown in Table 2.
2 Nucleotides in uppercase represent those present in target subsites bound by
individual zinc fmgers;
nucleotides indicated in lowercase are not present in a subsite. See U.S.
Patent No. 6,453,242 and U.S.
Patent Application Publication No. 2005-0064474 (both incorporated by
reference) for a description of
target subsites.
3 Locations in the human glucocorticoid receptor locus are given with respect
to the published
sequence of the GRa mRNA. Hollenberg, S.M. et al. (1985). Nature 318(6047):635-
41; GenBank
accession number X03225.
27
CA 02667414 2009-04-23
WO 2008/060510 PCT/US2007/023745
Table 2: Amino Acid Sequences of Recognition Regions of GR-Targeted ZFNs
Name Fl F2 F3 F4 F5
Exon 2
8718 RSDYLST QNAHRKT RSDVLSA DRSNRIK
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:8) NO:9) NO:10) NO:11)
9967 RSDYLST QRSHRNT RSDVLSA DRSNRIK
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:8) NO:12) . NO:10) NO:11)
8893 RSDALTQ RSDYLST QNAHRKT RSDVLSE DRSNLTR
(SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:13) NO:8) NO:9) NO:14)
NO:15)
8719 DSDHLTE DRANLSR RSDNLSN TNSNRIK
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:16) NO:17) NO:18) NO:19)
10404 TSSDRKK DRANLSR RSDTLRC TNSNRIK
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:20) NO:17) NO:21) NO:19)
10415 TSSDRKK DRANLSR RSDNLSN ERRSLRY
(SEQ IL) (SEQ ID (SEQ ID (SEQ ID
NO:20) NO:17) NO:18) NO:22)
, Exon 3 _
8667 TSRALTA DRANLSR RSDNLSE QNANRKT
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:23) NO:17) NO:24) NO:25)
9666 TSRALTA DRANLSR RSDNLSE ERANRNS
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:23) NO:17) NO:24) NO:26) .
8668 RSDVLSE RSANLTR RSDNLST HSHARIK
(SEQ ID (SEQ ID (SEQ ID (SEQ ID
NO:14) NO:27) NO:28) NO:29)
28
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
Table 2 (continued)
Name Fl F2 F3 F4
8669 RSDVLSE RSANLTR TSGNLTR TSGSLTR
(SEQ ID NO:14) (SEQ ID NO:27) (SEQ ID NO:30) (SEQ ID NO:31)
9671 DGWNRDC RSANLTR TSGNLTR TSGSLTR
(SEQ ID NO:32) (SEQ ID NO:27) (SEQ ID NO:30) (SEQ ID NO:31)
9674 DSWNLQV RSANLTR TSGNLTR TSGSLTR
(SEQ ID NO:33) (SEQ ID NO:27) (SEQ ID NO:30) (SEQ ID NO:31)
10201 TNRDLND DRANLSR RSDNLSE ERANRNS
(SEQ ID NO:34) (SEQ ID NO:17) (SEQ ID NO:24) (SEQ ID NO:26)
10205 NRKNLRQ DRANLSR RSDNLSE ERANRNS
(SEQ ID NO:35) (SEQ rD NO:17) (SEQ NO:24) (SEQ ID NO:26)
Exon 6
8531 RSDSLSA RNDNRKT RSDNLSR TNQNRIT
(SEQ ID NO:36) (SEQ ID NO:37) (SEQ ID NO:38) (SEQ ID NO:39)
9737 RQDCLSL RNDNRKT RSDNLSR TNQNRIT
(SEQ ID NO:40) (SEQ ID NO:37) (SEQ ID NO:38) (SEQ ID NO:39)
9846 HKHVLDN RNDNRKT RSDNLSR TNQNRIT
(SEQ ID NO:41) (SEQ ID NO:37) (SEQ ID NO:38) (SEQ ID NO:39)
8653 RSANLAR RSDNLRE QSSNLAR QSADRTK
(SEQ ID NO:42) (SEQ ID NO:43) , (SEQ ID NO:44) (SEQ ID NO:45)
29
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
Cleavage Domains
[0089] Any zinc finger that binds to a target site in a GR gene can be
combined with a nuclease to form a zinc finger nuclease. As noted above, any
cleavage domain or cleavage half-domain can be used in the zinc finger
nucleases
described herein. See, U.S. Patent Publication 2005-0064474. Thus, the
cleavage
domain portion of the fusion proteins disclosed herein can be obtained from
any endo-
or exonuclease. Exemplary endonucleases from which a cleavage domain can be
derived include, but are not limited to, restriction endonucleases and homing
endonucleases. See, for example, 2002-2003 Catalogue, New England Biolabs,
Beverly, Mass.; and Belfort et al. (1997) Nucleic Acids Res. 25:3379-3388.
Additional enzymes which cleave DNA are known (e.g., Si Nuclease; mung bean
nuclease; pancreatic DNase I; micrococcal nuclease; yeast HO endonuclease; see
also
Linn et al. (eds.) Nucleases, Cold Spring Harbor Laboratory Press, 1993). One
or
more of these enzymes (or functional fragments thereof) can be used as a
source of
cleavage domains and cleavage half-domains. In certain embodiments, the
cleavage
domain is obtained from a nuclease that has separable binding and cleavage
domains,
for example a yeast HO endonuclease.
[0090] Exemplary cleavage half-domains can be obtained from any
endonuclease. In certain embodiments, the cleavage half-domain is obtained
from a
nuclease that has separable binding and cleavage domains, for example a Type
ITS
restriction endonuclease such as Fokl. In addition, engineered cleavage half-
domains
(also referred to as dimerization domain mutants) that minimize or prevent
homodimerization are described, for example, in U.S. Patent Application
Publication
Nos. 2005-0064474 and 2006-0188987, incorporated by reference in their
entireties
herein. Amino acid residues at positions 446, 447, 479, 483, 484, 486, 487,
490, 491,
496, 498, 499, 500, 531, 534, 537, and 538 of Fok I are all targets for
influencing
dimerization of the Fok I cleavage half-domains.
[0091] Described herein are additional engineered cleavage half-domains
of
Fok I that form an obligate heterodimer. The first cleavage half-domain
includes
mutations at amino acid residues at positions 490 (E in the wild-type
sequence,
underlined below) and 538 (I in the wild-type sequence, underlined below) of
Fok I
and the second cleavage half-domain includes mutations at amino acid residues
486
(Q in the wild-type sequence, underlined below) and 499 (Tin the wild-type
sequence,
underlined below).
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
Wild type FokI cleavage half domain
QLVKSELEEKKSELRHKLKYVPHEYIEL IEIARNSTQDRILEMKVMEFFMKVYGYRG
KHLGGSRKPDGAIYTVGSPIDYGVIVDTKAYSGGYNLPIGQADEMQRYVEENQTRNK
HINPNEWWKVYPSSVTEFKFLFVSGHFKGNYKAQLTRLNHITNCNGAVLEELLIG
GEMIKAGTLTLEEVRRKFNNGEINF (SEQ ID NO:46)
E490K:I538K dimerization mutant
QLVKSELEEKKSELRHKLKYVPHEYIEL IEIARNSTQDRILEMKVMEFFMKVYGYRG
KHLGGSRKPDGAIYTVGSPIDYGVIVDTKAYSGGYNLPIGQADEMQRYVKENQTRNK
HINPNEWWKVYPSSVTEFKFLFVSGHFKGNYKAQLTRLNHKTNCNGAVLEELLIG
GEMIKAGTLTLEEVRRKFNNGEINF (SEQ ID NO:47)
Q486E:I499L dimerization mutant
QLVKSELEEKKSELRHKLKYVPHEYIEL IEIARNSTQDRILEMKVMEFFMKVYGYRG
KHLGGSRKPDGAIYTVGSPIDYGVIVDTKAYSGGYNLPIGQADEMERYVEENQTRNK
HLNPNEWWKVYPSSVTEFKFLFVSGHFKGNYKAQLTRLNHITNCNaVLSVEELLIG
GEMIKAGTLTLEEVRRKFNNGEINF (SEQ ID NO:48)
[0092] As shown above, the mutation at 490 replaces Glu (E) with Lys (K);
the mutation at 538 replaces Ile (I) with Lys (K); the mutation at 486
replaces Gln (Q)
with Glu (E); and the mutation at position 499 replaces Ile (I) with Leu (L).
Specifically, the engineered cleavage half-domains described herein were
prepared by
mutating positions 490 (E¨>K) and 538 (I---4() in one cleavage half-domain to
produce an engineered cleavage half-domain designated "E490K:I538K" as shown
above, and by mutating positions 486 (Q¨>E) and 499 (I¨>L) in another cleavage
half-domain to produce an engineered cleavage half-domain designated
"Q486E:I499L" as shown above. These mutations result in a diminished ability
of
two cleavage half-domains containing the E490K:I538K mutations to form a
homodimer, (compared to wild-type Fold cleavage half-domains); similarly, two
cleavage half-domains containing the Q486E:I499L mutations are also unable to
form
a homodimer. However, a cleavage half-domain containing the E490K:I538K
mutation is capable of forming a heterodimer with a cleavage half-domain
containing
the Q486E:I499L mutations to reconstitute a functional cleavage domain capable
of
double-strand DNA cleavage. Furthermore, heterodimerization between
E490K:I538K- and Q486E:I499L-containing cleavage half-domains occurs with an
efficiency similar to that of dimerization between wild-type Fokl cleavage
half-
31
CA 02667414 2014-05-30
domains. Thus, the engineered cleavage half-domains described herein are
obligate
heterodimer mutants in which aberrant cleavage is minimized or abolished.
[0093] Engineered cleavage half-domains described herein can be
prepared using any
suitable method, for example, by site-directed mutagenesis of wild-type
cleavage half-
domains (e.g., Fok I) as described in U.S. Patent Application Publication No.
2005-0064474
(Example 5) and U.S. Patent Application Serial No. 11/493,423 (Example 38).
Methods of Treatment
[0094] In certain types of cancer immimotherapy, T-cells are
engineered to express
cell-surface proteins that recognize tumor cell-specific antigens, and these
engineered T-cells
are introduced into a subject. For example, glioblastoma patients, who have
tumor cells that
overexpress an IL- 13 receptor on their surface, can be treated, following
surgical resection of
the tumor(s), with cytolytic T-cells that express a "zetakine" tethered to
their cell surface.
Zetakines are chimeric transmembrane immunoreceptors, comprised of an
extracellular
domain comprising a soluble receptor ligand linked to a support region capable
of tethering
the extracellular domain to a cell surface, a transmembrane region and an
intracellular
signaling domain. When expressed on the surface of T lymphocytes, such
chimeric receptors
direct T cell activity to those specific cells expressing a receptor for which
the soluble
receptor ligand is specific.
[0095] For treatment of gliomas and glioblastomas, zetakines are targeted
to cells
expressing IL- 13 receptors. Thus, a zetakine can constitute a glioma-specific
immunoreceptor
comprising the extracellular targeting domain of the IL-13Ralpha.2-specific IL-
13 mutant IL-
13(E 13Y) linked to the Fe region of IgG, the transmembrane domain of human
CD4, and the
human CD3 zeta chain. T-cells expressing such a zetakine are able to detect
and kill IL-13-
overexpressing glioma and glioblastoma tumor cells remaining after surgical
tumor resection.
See, for example, Kahlon, K. S. et al. (2004) Cancer Res. 64:9160-9166 and US
patent
application publication numbers 2006-0067920, 2005-0129671, and 2003-0171546.
[0096] However, the clinical use of this approach is hampered by the
fact that brain
tumor patients must also be treated with glucocorticoid hormones following
tumor resection,
to prevent inflammation and swelling of the brain. This
32
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
glucocorticoid treatment inhibits activation of the zetakine-containing T-
cells, thus
preventing their cell-killing activity.
[0097] The methods and compositions disclosed herein make it possible to
eradicate GR function in the zetakine-containing T-cells, thereby making them
resistant to the inhibitory effect of glucocorticoids. Accordingly, by use of
the
disclosed methods and compositions, the post-surgical glioblastoma patient can
be
treated both with glucocorticoids (to prevent swelling and inflammation) and
with the
zetakine-containing 1-cells, to remove residual tumor cells. Indeed,
inactivation of
the GR gene in the T-cells can be accomplished by targeted integration of a
sequence
encoding the zetakine into the GR locus, accomplishing both objectives in a
single
step.
[0098] Accordingly, in certain embodiments, GR-targeted zinc finger
nucleases are expressed in T-cells and cleave at a site in the GR locus. Cells
containing GR-targeted zinc finger nucleases are optionally contacted with a
donor
DNA molecule which encodes a zetakine, such that the zetakine-encoding
sequences
are integrated into the GR locus, thereby inactivating GR function in those
cells.
Additional applications
[0099] Treatment of patients with engineered T-cells or isolated 1-cells
is
compromised if these patients are also treated with immune-suppressant drugs
such as
decadrone. However, modification of such therapeutic immune cells, using ZFNs
targeting the GR locus to inactivate GR function as disclosed herein, allows
generation of a population of immune cells that is not subject to
glucocorticoid-
mediated immune suppression. The high efficiency of the methods described
herein
allows simultaneous disruption of both alleles of the GR gene in the absence
of a
selection marker, which is not possible with any other technique. The speed
with
which mutations in both alleles of the GR gene (including homozygous
deletions) can
be obtained is also an important consideration since T-cells, like all primary
cells,
have a limited replication potential and therefore a finite lifespan.
[0100] The methods disclosed herein for modification of the sequence of
the
GR locus in human cells allow generation of pools of cells containing a
substantial
number of cells lacking GR function and also allow isolation of clonal cell
lines
lacking GR activity. Such cells include but are not limited to 1-cells and
other cells
of the immune system (e.g., B-cells, NK cells, memory cells, macrophages) all
of
33
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
which are normally prevented from undergoing proper activation in the presence
of
glucocorticoid hormones, due to the action of the glucocorticoid receptor.
[0101] As discussed above, one application of these methods is the use of
ZFNs to render zetalcine expressing T-cells, which target brain tumor cells,
non-
responsive to glucocorticoids. Other examples include ZFN-mediated
inactivation of
GR in T-cells used for treating opportunistic infections, e.g., in transplant
patients
receiving immunosuppressants, or in other immunocompromised patients.
[0102] Additional clinical complications of undesired GR activity can
also be
alleviated by inactivation of GR activity in a specific target cell
population. In this
context, it is noted that the pleiotropic activity of the GR is based, in
part, on the
existence of multiple receptor isoforms generated from the same gene. See, for
example, Zhou, J. et al. (2005) Steroids 70:407-417. The single nucleotide-
level
resolution of ZFN-mediated GR gene modifications described herein, in
combination
with tissue-specific ZFN expression, (see, for example, U.S. Patent No.
6,534,261 and
U.S. Patent Application Publication No. 2005-0064474, both incorporated by
reference) together enable disruption of specific receptor isoforms in
specific tissues:
an approach that is not possible with any other technology. In addition,
alteration of
the sequence of the GR gene using ZFNs can be used to replace the wild-type GR
with an isoform that is regulated exclusively by a specific ligand of choice.
[0103] Conversely, in patients with mutations in the GR locus, ZFN-
mediated
genome editing can be used to restore GR activity in a target tissue of
choice, either
through correction of the mutation itself or by targeted insertion of a
sequence (e.g.,
cDNA) encoding a functional GR.
[0104] The methods and compositions disclosed herein can also be used to
generate cell lines for research applications, drug screening and target
validation. For
instance, abolishing GR function in a cell line of choice, using ZFNs as
disclosed
herein, allows generation of a matched pair of isogenic cell lines that differ
only in the
presence or absence of GR function. As another example, insertion of a
reporter gene
into the GR locus, or fusion of a reporter to the GR protein, will facilitate
high-
resolution studies of the properties and regulation of the glucocorticoid
receptor.
Such lines can be used for research purposes, as well as for industrial
applications
such as target validation and drug screening. Similarly, ZFN-mediated gene
correction can be used to introduce specific changes into the GR locus to
generate cell
lines for studying the function of various receptor domains or isoforms.
34
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
[0105] Finally, in cases in which inactivation of the GR locus is not the
main
purpose of the intervention, but would have no negative effects on the
modified cells,
the GR locus can be used as a "safe harbor" integration site for any transgene
in any
gene therapy application. Thus, the disclosure also provides methods of
selection for
cells into which an exogenous sequence has been integrated into a GR gene. The
methods involve cleaving an endogenous GR gene in a cell with ZFNs as
described
herein and introducing an exogenous sequence (e.g., transgene), typically on a
donor
construct with GR-homology arms, into the cells under conditions such that the
exogenous sequence is integrated into the GR gene. Cells with the integrated
exogenous sequence can then be selected for by exposing the cells to a
naturally
occurring or synthetic corticosteroid (e.g., cortisol, dexamethasone, etc.),
which kills
cells without the integrated sequence (cells with normal GR expression). See,
also,
Example 4.
Vectors for delivery of zinc finger nucleases and donor DNA sequences
[0106] Any vector can be used for delivery, to a cell, of DNA sequences
encoding zinc finger nucleases and/or delivery of donor DNA. Exemplary viral
vectors include adenoviruses, adeno-associated viruses, poxviruses,
herpesviruses,
papovaviruses, retroviruses and lentiviruses. DNA can also be delivered to
cells by
transfection, electroporation, lipid-mediated methods, biolistics and calcium
phosphate-mediated transfer.
[0107] Because the methods and compositions disclosed herein utilize a
transient event (ZFN-mediated double-strand cleavage) to effect a permanent
genomic
alteration (e.g., targeted mutation or targeted integration of exogenous
sequences), it
is not necessary to use a delivery vector that persists in the cells.
Accordingly, non-
replicating viral vectors can be used as delivery vehicles. Thus, replication-
defective
adenoviruses, hybrid adenoviruses (e.g., Ad 5/35) and non-integrating
lentivirus
vectors are all suitable as delivery vehicles.
[0108] Non-limiting examples of adenovirus (Ad) vectors that can be used
in
the present application include recombinant (such as El deleted),
conditionally
replication competent (such as oncolytic) and/or replication competent Ad
vectors
derived from human or non-human serotypes (e.g., Ad5, Adll, Ad35, or porcine
adenovirus-3); chimeric Ad vectors (such as Ad5/35) or tropism-altered Ad
vectors
with engineered fiber knob proteins (such as peptide insertions within the HI
loop of
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
the knob protein); and/or "gutless" Ad vectors, e.g., an Ad vector in which
all
adenovirus genes from the Ad genome have been removed to reduce
inimunogenicity
and to increase the size of the DNA payload to allow simultaneous delivery of
both
ZFNs and donor molecule, especially large transgenes to be integrated via
targeted
integration.
[0109] Replication-deficient recombinant adenoviral vectors (Ad) can be
produced at high titer and readily infect a number of different cell types.
Most
adenovirus vectors are engineered such that a transgene replaces the Ad El a,
Elb,
and/or E3 genes; subsequently the replication defective vector is propagated
in human
293 cells that supply deleted gene function in trans. Ad vectors can transduce
multiple types of tissues in vivo, including nondividing, differentiated cells
such as
those found in liver, kidney and muscle. Conventional Ad vectors have a large
carrying capacity. An example of the use of an Ad vector in a clinical trial
involved
polynucleotide therapy for antitumor immunization with intramuscular
injection.
Sterman et al. (1998) Hum. Gene Ther. 7:1083-1089.
[0110] Additional examples of the use of adenovirus vectors for gene
transfer
in clinical trials include Welsh etal. (1995) Hum. Gene Ther. 2:205-218;
Rosenecker
et al. (1996) Infection 24:5-10; Alvarez et al. (1997) Hum. Gene Ther. 5:597-
613 and
Topf et al. (1998) Gene Ther. 5:507-513.
[0111] In certain embodiments, the Ad vector is chimeric adenovirus
vector,
containing sequences from two or more different adenovirus genomes. For
example,
the Ad vector can be an Ad5/35 vector. Ad5/35 is created by replacing the
fiber
protein of Ad5 with the fiber protein from B group Ad35. The Ad5/35 vector and
characteristics of this vector are described, for example, in Ni et al. (2005)
Hum. Gene
Ther. 16:664-677; Nilsson et al. (2004) MoL Ther. 9:377-388; Nilsson et al.
(2004)
Gene. Med. 6:631-641; Schroers etal. (2004) Exp. Hematol. 32:536-546;
Seshidhar etal. (2003) Virology 311:384-393; Shayalchmetov etal. (2000) J.
Virol.
74:2567-2583 and Soya et al. (2004) Mol. Ther. 9:496-509.
EXAMPLES
[0112] The following examples are presented as illustrative of, but not
limiting, the claimed subject matter.
36
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
Example 1: Construction of plasmid delivery vehicles encoding zinc
finger nucleases targeted to a human GR gene
[0113] Target sites for zinc finger DNA-binding domains were selected by
scanning the sequence of the human glucocorticoid receptor (GR) gene,
optionally
using a computer program containing a listing of individual zinc fingers and
their
target sites and/or a listing of two-finger modules and their target sites,
for a pair of
target sequences, separated by 5-6 nucleotide pairs, wherein each target
sequence can
be bound by a 3-, 4-, 5- or 6-finger zinc finger protein. See, for example,
U.S. Patent
No. 6,785,613; WO 98/53057; WO 01/53480 and U.S. Patent Application
Publication
No. 2003/0092000. Additional methods for ZFP design are disclosed, for
example, in
U.S. Patents 5,789,538; 6,013,453; 6,410,248; 6,733,970; 6,746,838; 6,785,613;
6,866,997; 7,030,215; WO 01/088197; WO 02/099084; and US Patent Application
Publications 2003/0044957; 2003/0108880; 2003/0134318 and 2004/0128717.
[0114] For certain of the target sequences identified in the previous
step, a
gene encoding a fusion between a Fold cleavage half-domain and a zinc finger
protein
that binds to the target sequence was synthesized. See, for example, U.S.
Patent No.
5,436,150; WO 2005/084190 and U.S. Patent Application Publication No.
2005/0064474.
[0115] Standard molecular biological methods were used to construct such
fusion genes and introduce them into plasmids. Exemplary expression constructs
are
shown in Figures 1 and 2.
Example 2: Targeted cleavage of a human GR gene using engineered zinc
finger nucleases
[0116] K562 cells (ATCC No. CCL243) were cultured in RPMI medium
(Invitrogen, Carlsbad, CA). At a density of 1 x 106 cells/ml, 2 x 106 cells
were
pelleted and transfected with 2.5 lag each of two ZFN expression vectors,
using an
Amaxa nucleofection device (Amaxa, Gaithersburg, MD). One set of cells was
transfected with plasmids encoding zinc finger nucleases comprising the exon 3-
targeted 9666 and 9674 binding domains (see Tables 1 and 2). A second set of
cells
was transfected with plasmids encoding zinc finger nucleases comprising the
exon 6-
targeted 8653 and 9737 binding domains (see Tables 1 and 2). Controls included
cells transfected with a plasmid encoding green fluorescent protein (GFP) and
untransfected cells.
37
CA 02667414 2009-04-23
WO 2008/060510 PCT/US2007/023745
[0117] Three days after transfection, DNA was isolated from the cells
using a
DNeasy kit (Qiagen, Valencia, CA). This DNA (100 ng) was used as template for
PCR amplification using primers specific for either exon 3 or exon 6 of the GR
locus
(see Table 3). The amplification products were denatured, then reannealed; and
the
reannealed products were exposed to the mismatch-specific nuclease Cel-I
(Transgenomic, Omaha, NE). Products of Cel-I treatment were analyzed on a 10%
polyacrylamide gel. If the population of amplification products is homogeneous
with
respect to nucleotide sequence, perfectly-matched duplexes, that are resistant
to Gel-I
cleavage, should be produced following denaturation and reannealing. If, on
the other
hand, the amplification products are heterogeneous due to the presence of
insertions,
deletions and/or mismatches in some of the amplification products, reannealing
will
generate some duplexes containing sequence mismatches, susceptible to Gel-1
cleavage. As a result, products smaller than the amplification product will be
detected
on the gel.
Table 3: PCR primers in the human GR locus
Exon 3 sense TCATAACACTGTTCTTCCCCTTCTTTAGCC (SEQ 1D
NO:49)
Exon 3 antisense TCAAAACACACACTACCTTCCACTGCTC (SEQ ID NO:50)
Exon 6 sense ACACCTGGATGACCAAATGACCCTAC (SEQ ID NO:51)
Exon 6 antisense CCTAGATACCTAGTAGGATTGTTTCAGTCCTG (SEQ ID
NO:52)
[0118] The results are shown in Figure 3. Amplification products of exon
3-
and exon 6-specific DNA from untransfected cells, and from cells transfected
with a
plasmid encoding GFP, yield a single band after denaturation, reannealing and
Gel I
treatment (left and middle lanes, respectively, of each panel), indicative of
a
homogeneous population of amplification products. By contrast, exon 3-specific
amplification products from cells transfected with plasmids encoding zinc
finger
nucleases having zinc finger DNA-binding domains with target sites in exon 3
(9666
and 9674) yield two smaller products after denaturation, reannealing and Gel I
treatment (left panel, rightmost lane). The presence of these smaller products
indicates the existence of a sequence mismatch at a unique location in the
reannealed
DNA. Similarly, exon 6-specific amplification products from cells transfected
with
plasmids encoding zinc finger nucleases having zinc finger DNA-binding domains
38
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
with target sites in exon 6 (8653 and 9737) yield two smaller products (of
similar
molecular weight), indicative of targeted cleavage in exon 6.
Example 3: Nature of mutations induced by targeted cleavage in a
human GR gene
[0119] CEM14 cells (a glucocorticoid-sensitive lymphoid cell line
obtained
from M. Jensen, City of Hope Medical Center, Duarte, CA) were cultured in RPMI
medium. At a density of 1 x 106 cells/ml, 2x 106 cells were pelleted and
transfected
with plasmids encoding ZFNs with the exon 3-targeted 8667 and 8668 DNA-binding
domains (see Tables 1 and 2), using an Amaxa nucleofection device. 2.5 g of
each
plasmid was used. Cells lacking GR function were selected by exposure to 1(15
M
dexamethasone for 14 days; and DNA isolated from dexamethasone-resistant cells
was amplified by PCR using GR exon 3-specific primers (see Table 3). The
amplification reaction mixture was fractionated on an agarose gel and a band
corresponding in size to the expected amplification product (based on the
location of
the primer sequences in the GR gene) was excised from the gel. DNA in this
band
was cloned using a Topo cloning kit (Invitrogen, Carlsbad, CA) and nucleotide
sequences of individual clones was determined.
[0120] In one clone, sequence analysis revealed the presence of a two-
nucleotide deletion near the site of targeted cleavage. In a second clone, an
11-
nucleotide deletion was detected. A third clone comprised a mixture of
sequences:
one of which was a duplication of four nucleotides, the other of which was a
19-
nucleotide deletion.
[0121] These results show that targeted cleavage of the GR gene, using
zinc
finger nucleases, induced both insertion and deletion mutations in the gene,
all of
which resulted in a change in the translational reading frame.
[0122] RNA analysis in these dexamethasone-resistant cells revealed lower
levels of GR mRNA, compared to untransfected cells; indicative of nonsense-
mediated decay of aberrant GR transcripts.
Example 4: Introduction of a transgene into the human GR locus
[0123] Glucocorticoid hormones trigger apoptosis or slow down cell growth
in many primary cells and cell lines. Glucocorticoid hormone treatment can
therefore
be used in combination with ZFNs targeting the GR locus to select or enrich
for a)
39
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
cells containing a ZFN mediated biallelic mutation of the GR locus, b) cells
containing ZFN mediated targeted integration of donor DNA sequences into both
alleles of the GR locus, resulting in the inactivation of GR function or c)
cells
containing a ZFN mediated GR mutation on one allele of the GR locus and ZFN
mediated targeted integration on the other allele.
[0124] Sequences that can be integrated into the GR locus in this manner
include but are not limited to expression cassettes with any transgene of
choice.
Potential benefits of using integration into the GR locus include but are not
limited to
a) avoiding the use of a selection marker on the donor molecule, b) long term
stability
of the expression of the transgene from a locus compatible with high level
gene
expression and not subjected to silencing, c) avoiding insertional mutagensis
events
that can occur upon random integration of transgenes. The following
experiments
were conducted using a zetakine transgene.
[0125] CEM14 cells were cultured in RPMI medium and transfected (as
described in previous examples), in separate experiments, with 25 lig of an
expression
construct encoding the GR-targeted ZFNs 9666 and 9674 and a zetakine
transgene.
The zetakine-donor ZFN construct was transfected into CEM14 cells and an
aliquot of
the transfected cells was incubated with dexamethasone (which kills cells
having
normal GR function) for 2 weeks, while the remaining cells were left
untreated.
Untransfected cells were used as controls and treated identically. After
dexamethasone treatment, targeted integration of the zetakine transgene into
the GR
locus was detected by PCR (Fig. 4A) and by Southern blotting (Fig. 4B).
Immunostaining shows high-level zetakine expression in the dexamethasone
selected
CEM14 cells transfected with both the GR ZFNs and the zetakine donor construct
(not shown).
[0126] These results demonstrated that targeted integration of a donor
sequence in the human GR gene was accomplished, that integration of the donor
sequence inactivated GR function, and that normal function of a transgene
contained
in the donor sequence was obtained.
Example 5: GR polypeptide production after targeted integration of an
exogenous sequence in the GR gene
[0127] CEM14 cells were cultured in RPMI medium and transfected as above
with 25 g of an expression vector containing a zetakine cassette and encoding
the
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
GR exon 3-targeted ZFN pair 8667 and 8669, flanked by regions of homology to
the
GR gene. Two days after transfection, 10-6M dexamethasone was added to the
growth medium, and glucocorticoid-resistant clones were obtained after two
weeks of
selection.
[0128] Protein extracts from transfected cells that survived the
dexamethasone
treatment were prepared by whole cell lysis, and Western Blot analysis was
performed using an antibody to the human GR (Catalog number 611226, BD
BioSciences Pharmingen, San Jose CA). Results are shown in Figure 5. A band
corresponding to the wild-type receptor was detected in untransfected CEM 14
cells
(left lane), while a clone of CEM14 cells treated with the ZFNs and the donor
sequence contained no immunoreactive material corresponding to the size of the
wild-
type receptor; instead, a band corresponding in size to a truncated receptor
form was
detected (right lane).
Example 6: Targeted cleavage of a human GR gene in CD8+ T-cells
[0129] Fresh human peripheral blood CD8+ T-Cells were obtained from
AllCells (Berkeley, CA). 2x106 cells were transfected, using an Amaxa
nucleofection
device and protocol, with 51.1g of an expression plasmid encoding two zinc
finger
nucleases targeting exon 3 of the human GR locus, in which the ZFN coding
sequences were separated by a 2A sequence. Two exon 3-targeted ZFN pairs (9666-
9671 and 9666-9674) were tested in separate transfections. Controls were
untransfected cells and cells transfected with a plasmid encoding green
fluorescent
protein (GFP).
[0130] Transfected CD8+ cells were cultured for 48 hours in X-VIV015
medium containing 5% human serum (both purchased from Cambrex, Walkersville,
MD). Thereafter, DNA was isolated and Cel I analysis was performed as
described in
Example 2 above, using exon 3-specific amplification primers, except that 5
ptCi each
of a-32P-dCTP and a-32P-dATP (Perkin-Elmer, Boston, MA) were added to the PCR
reaction.
[0131] Following Cel I treatment, digestion products were fractionated on
a
10% acrylamide gel. An autoradiographic image of the gel was developed using a
Storm PhosphorImager (GE Healthcare, Piscataway, NJ). Results are shown in
Figure 6. Frequencies of non-homologous end joining (NHEJ) were determined by
quantitation of the Cell cleavage products and are indicated in the rightmost
two
41
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
lanes of Figure 6. Thus, GR-targeted ZFNs efficiently modify the GR locus in
the
CD8+ T-cells.
Example 7: Analysis of GR-ZFN-treated CD-8+ T-cells
A. ZFNs Generate Mutations at GR locus
[0132] Fifty million human CD8+ T-cells cells expressing a zetakine
transgene ('IL-13 ZK Pool') were stimulated at day 0 and infected at day 7
with
Ad5/F35 expressing the ZFN pair 9666 and 9674 at the following multiplicity of
infections (mois): 10 ("ZFN10-"), 30 ("ZFN30-") and 100 ("ZFN100-"). As a
control, CD8+ cells were also infected with a GFP expressing Ad5/F35 virus at
the
same mois ("GFP10-", "GFP30-", "GFP100-").
[0133] Cells were grown as described above and 7 days post-infection
cells
were treated for 6 days with the glucocorticoid hormone dexamethasone at a
concentration of 104 M in the absence of cytokines. The resulting cell pools
(`ZFN10', `ZFN30', `ZFN100` and `GFP10', `GFP30', `GFP100`, respectively) were
restimulated and 12 days after restimulation cells were harvested. DNA was
isolated
and analyzed for modification of the GR locus by PCR of the ZFN target region
followed by the Cel I SurveyorTM endonuclease assay as described in Example 2
above. As shown in Fig. 7, panels A and B, transient ZFN expression generated
GC-
Resistant CD8+ T-cells with mutations at the GR locus.
[0134] The PCR products were also subcloned into the PCR4 TOPO vector
and the insert sequence analyzed using the T7 primer. Sequencing of the ZFN
target
region in exon 3 of the GR locus in cells pool ZFN100 demonstrated that 70% of
the
GR alleles contained mutations in the ZFN binding region. By contrast, no
mutations
were found in the GR alleles when cells had been infected with the GFP100
control
virus.
[0135] Glucocorticoid hormone resistance of the CD8+ cell pools was also
determined by comparing cell viability before and after a second treatment
with 10-
4M Dexamethasone for 8 days. Viability was measured using the Guava cell
analyzer.
As shown in Fig. 8, ZFN treated cells showed increased resistance to
glucocorticoid
hormone.
42
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
B. GR Protein Expression in ZFN-modified CD8+ T-cells
[0136] ZFN-treated CD8+ T-cells were also tested for the presence of full-
length GR protein.
[0137] In one experiment, protein extract from CD8+ cell pools described
above and from the zetakine expressing CD8+ cell pool that was used for the
virus
transduction ('IL-13 ZK pool') were analyzed by Western Blotting using GR (BD
BioSciences) and TFIlB (Santa Cruz Antibodies) antibodies. In addition a
subclone
(`10A1') of the ZFN 100 pool was analyzed alongside the cell pools. As shown
in
Fig. 9A, ZFN treated CD8+ T-cells showed a loss of GR protein as compared to
controls.
[0138] In a separate experiment, expression of GR was also assessed in
ZFN
treated CMV-targeted CD8+ subclones. In particular, GR-negative CD8+ cells
that
target CMV were generated using ZFNs as described herein. Single cell derived
subclones of these cells were isolated and protein extracts were obtained from
these
clones as described above. Genotyping of 26 subclones identified 2 wildtype
clones
and 24 clones with mutations in the GR locus comprising the ZFN binding
regions.
As above, protein extracts were analyzed by Western Blotting with a human GR
antibody (BD BioSciences). As shown in Fig. 9B, ZFN-treated CMV-targeted CD8+
clones showed loss of GR protein.
[0139] Thus, ZFNs as described herein can reduce or eliminate GR protein
expression in primary cells.
C. RT-PCR
[0140] In addition, GR-negative (ZFN-treated) CD8+ T-cells were analyzed
by RT-PCR to determine the effect of glucocorticoid addition on the expression
of
known GR target genes. Results are shown in Figs. 10A-10D. Cells were left
untreated ("un") or treated with 10-6M dexamethasone for 20 hrs ("dex") as
indicated.
RNA was isolated using standard protocols, and the mRNA levels of various
glucocorticoid regulated genes analyzed by RT-PCR using the TaqmanTm protocol
(Applied Biosystems). RNA values for GR target genes were corrected by the
values
for the GAPDH housekeeping gene obtained from the same RNA sample. Taqman
probes and primers for GR target genes were obtained from Applied Biosystems.
As
shown in Figs. 10A-D, glucocorticoid hormone treatment affected the target
genes
less in ZFN-treated CD8+ T-cells.
43
CA 02667414 2009-04-23
WO 2008/060510
PCT/US2007/023745
D. Cvtokine Release
[0141] Cytokine release from ZFN-treated CD8+ T-Cells stimulated with
glioma cells was also evaluated. GFP100, ZFN100 and untreated CD8+ T-cell
pools
were cultured alone or in the presence of U87MG glioblastoma stimulator cells.
Subsequently, 10-6M dexamethasone was added to the culture. After 20 hrs, cell
culture supernatants were harvested and IFN-y levels analyzed using a
commercially
available ELISA kit (R&D systems).
[0142] As shown in FIG. 11, cytokine release by ZFN treated CD8+ T-cells
upon stimulation with glioma cells was maintained and was rendered resistant
to
glucocorticoid hormones.
E. Chromium Release Assay For Cytolytic Activity
[0143] To analyze the cytolytic activity of ZFN treated CD8+ T-cells
chromium release assays were conducted at various ratios of effector (ZFN-
treated or
control CD8+ T-cells) to target cells (IL13Ra2 positive cell lines and control
cell
line).
[0144] As shown in Fig. 12, panels A to E, ZFN-treated CD8+ T-cells
maintain the ability to kill IL13Ra2+ target cells.
Example 8: In Vivo Administration of ZFN-Treated, GR-Negative CD8+
T-cells
[0145] ZFN-treated GR-negative CD8+ T-cells were also analyzed for tumor
cell killing activity in vivo. Tumor cell cyotoxicity was measured in an
orthotopic
glioblastoma mouse model using luciferase labeled U87MG cells injected into
the
brain at day 0. At day 5, controls or ZFN-treated CD8+ T-cells were injected
into the
brain and tumor volumes were determined by measuring luciferase activity up to
day
24.
[0146] As shown in Fig. 13, ZFN-treated GR-negative CD8+ T-cells (panel
C)
reduced tumor volumes as compared to controls. Furthermore, the experiment
shown
in Fig. 14 demonstrates that the anti-tumor activity of the ZFN treated GR-
negative
CD8+ T-cells in the mouse tumor model was not negatively affected by the
administration of glucocorticoid hormone. Accordingly, ZFN-treated GR-negative
44
CA 02667414 2014-05-30
CD8+ T-cells can be administered for treatment of glioblastoma in patients
receiving
glucocorticoid hormones.
Example 9: Analysis of GR ZFN Specificity
[0147] To analyze the specificity of the GR ZFNs 9666 and 9674 in cells, we
first
determined consensus DNA binding site for both the 9666 and the 9674 ZFNs in
vitro using
an affinity-based target site selection procedure (SELEX). See, also, U.S.
Serial
No. 11/805,707, filed May 23, 2007. An experimental overview is provided in
Figure 15.
Briefly, hemaglutinin-tagged ZFNs were incubated with a pool of randomized DNA
sequences in the presence of biotinylated anti-HA Fab antibody fragments. The
tagged ZFN-
DNA complexes were captured with streptavidin-coated magnetic beads, and the
bound DNA
was released and amplified by PCR. This process was repeated three times using
the previous
eluted, amplified pool of DNA as a starting sequence. After four iterations,
the eluted DNA
fragments were sequenced, and base frequencies at each position in the binding
sites of ZFN
9666 and ZFN 9674 were determined.
[0148] A consensus based on the site selection data was used to guide
a genome-wide
bioinformatic prediction of the most similar putative off-target sites in the
human genome.
The resulting list of potential cleavage sites was then ranked to give
priority to those sites
with the highest similarity to the experimentally derived binding site
preferences.
[0149] NR3C1 (the GR locus) contains the best match to the binding site
preferences
determined in the site selection experiments. Of the 15 potential off-target
sites, 10 fall within
annotated genes, and only 2 occur within exonic sequences. For none of these
ten genes, has
mutation or disruption been associated with any known pathology in CD8+ T
cells.
[0150] Genotyping of clone 10A1 (a T cell derived clone) confirmed
that none of the
15 sites with highest similarity to the consensus were modified under
conditions that resulted
in bi-allelic modification of the GR (on-target) locus.