Note: Descriptions are shown in the official language in which they were submitted.
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
7-SUBSTITUTED PURINE DERIVATIVES FOR IIVIlVIUNOSUPPRESSION
FIELD OF THE INVENTION
[0001] The invention relates to purinone derivatives useful as
immunosuppressants.
BACKGROUND OF THE INVENTION
[0002] Immunosuppression is an important clinical approach in treating
autoimmune
disease and in preventing organ and tissue rejection. The clinically available
immunosuppressants, including azathioprine, cyclosporine and tacrolimus,
although
effective, often cause undesirable side effects including nephrotoxicity,
hypertension,
gastrointestinal disturbances and gum inflammation. Inhibitors ofthe tyrosine
kinase Jak3
are known to be useful as immunosuppressants (see US patent 6,313,129).
[0003] The members of the Janus kinase (Jak) family of non-receptor
intracellular
tyrosine kinases are components of cytokine signal transduction. Four family
members
have been identified to date: Jakl, Jak2, Jak3 and Tyk2. The Jaks play a key
role in the
intracellular signaling mediated through cytokine receptors. Upon binding of
cytokines to
their receptors, Jaks are activated and phosphorylate the receptors, creating
docking sites
for other signaling molecules, in particular members of the signaltransducer
and activator
of transcription (STAT) family. Whi1e expression of Jakl, Jak2 and Tyk2 is
relatively
ubiquitous, Jak3 expression is temporally and spatially regulated. Jak3 is
predominantly
expressed in cells of hematopoietic lineage; it is constitutively expressed in
natural killer
(NK) cells and thymacytes and is inducible in T cells, B cells and myeloid
cells (reviewed
in Ortmann, et al., 1999 and Yamaoka, et al., 2004). Jak3 is also is expressed
in mast cells,
and its enzymatic activity is enhanced by IgE receptor/FceRI cross-linking
(Malaviya and
Uckun, 1999).
1
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0004] A specific, orally active Jak3 inhibitor, CP-690,550, has been shown to
act as an
effective immunosuppressant and prolong animal survival in a murine model of
heart
transplantation and a primate model of kidney transplantation (Changelian, et
al., 2003).
[0005] Furthermore, aberrant Jak3 activity has been linked to a leukemic form
of
cutaneous T-cell lymphoma (Sezary's syndrome) and acute lymphoblastic leukemia
(ALL), the most common form of childhood cancer. The identification of Jak3
inhibitors
has provided the basis for new clinical approaches in treating leukemias and
lymphomas
(reviewed in Uckun, et al, 2005). Two dimethox-yquinazoline derivatives, WHI-
P131
(JANEX-1) and WHI-P154 (JANEX-2), have been reported to be selective
iinhibitors of
Jak3 in leukemia cells (Sudbeck et al..: 1999).
[0006] Jak3 has also been shown to play a role in mast-cell mediated allergic
reactions
and inflammatory diseases and serves as a target in indications such as asthma
and
anaphylaxis.
[0007] Therefore, compounds that inhibit Jak3 are useful for indications such
as
leukemias and lymphomas, organ and bone marrow transplant rejection, mast cell-
mediated allergic reactions and inflammatory diseases and disorders.
SUMMARY OF THE INVENTION
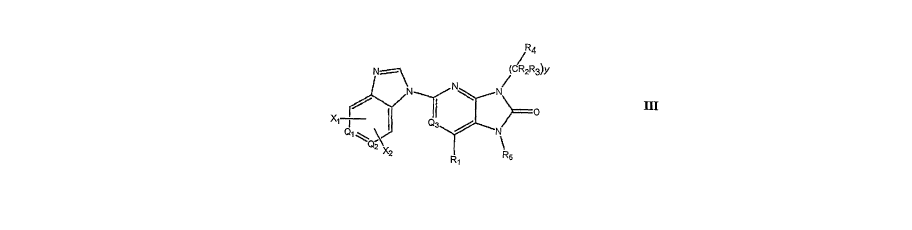
[0008] It has now been found that compounds of general formula III are potent
and
selective inhibitors of Jak3:
Rq,
N1.-- (CR2R3)y
~ N
-~ ~ o
Q3 N X=
QZ
R6
III
2
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
In these compounds,
Ql and Q2 are independently selected from the group consisting of CX1, CX2 and
nitrogen
wherem Ql and Q2 are not both nitrogen;
Q3 is N or CH;
Xr and X2 are independently selected from the group consisting of hydrogen,
(Cl-C6)alkyl,
cyano, halo, halo(Cl-C6)alkyl, hydroxyl, (Cl-C6)alkoxy; halo(Cl-C6)alkoxy, and
nitro;
Rl is selected from the group consisting of hydrogen and (Cl-C6)alkyl;
y is zero or aninteger selected from 1, 2 and 3;
R2 and R3 are selected independently for each occurrence of (CR2R3) from the
group
consisting of hydrogen and (Ci-C6)alkyl;
R4 is selected from a group consisting of alkyl, heterocyclyl, aryl,
heteroaryl, substituted
alkyl, substituted heterocyclyl, substituted aryl, substituted heteroaryl; and
R5 is selected from the group consisting of alkyl, heterocyclyl, heterocyclyl,
and Cl-Cs
alkyl wherein
(a) one or two CHa is replaced by a group chosen from NH and N(alkyl);
(b) one or two CH2 is replaced by 0;
(c) one or two CH2 is replaced by (C=O);
(d) two CH2 are replaced by CH=CH or C=C; or
(e) any chemically stable combination of (a), (b) (c) and (d);
and wherein from zero to three hydrogens is replaced by a substituent chosen
from:
(a) halogen, hydroxy, cyano, loweralkylsulfonyl, loweralkylsulfonyloxy, amino,
loweralkylamino, diloweralkylamino, alkoxyamino, sulfonylamino, acylamino,
arylamino,
loweralkoxry;
3
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
(b) heterocyclyl and heterocyclyl substituted with from one to three
substituents chosen
from halogen, hydroxy, alkoxy, alkyl and alkoxycarbonyl;
(c) phenyl and phenyl substituted with from one to three substituents chosen
from halogen,
hydroxy, alkoxy, alkyl, acylamino, cyano, carboxy, allcoxycarbonyl, haloalkyl
and
heterocyclyl; and
(d) heteroaryl and heteroaryl substituted with from one to three substituents
chosen from
halogen, hydroxy, alkoxy, alkyl and alkoxycarbonyl.
[0009] The members of these genera are useful in inhibiting Jak3 activity and
as such are
useful in indications where clinical irnmunosuppression is desired and in the
treatment of
hematological cancers. The compounds are more selective for Jak3 than for
Aurora A
kinases than are the corresponding compounds in which R5 is hydrogen.
[0010] In another aspect, the invention relates to pharmaceutical compositions
comprising a therapeutically effective amount of at least one compound of
general formula
III, or a phannaceutically acceptable salt thereof; and a pharmaceutically
acceptable
carrier.
[0011] In another aspect, the invention relates to a method for treating a
disease by
altering a response mediated by Jak3 tyrosine kinase. The method comprises
bringing into
contact with Jak3 at least one compound of general formula III.
[0012] In yet another aspect the present invention relates to a method of
suppressing the
immune system in a subject in need thereof comprising administering to the
subject a
therapeutically effective amount of at least one compound of general formula
III.
[0013] In a fiu-ther aspect of the present invention relates to a method for
treating a
disease or disorder selected from an autounmune disease, an inflammatory
disease, a mast
cell mediated disease, hematological malignancy and organ transplant rejection
in a subject
in need thereof comprising administering to a subject a therapeutically
effective amount of
at least one compound of general formula III.
4
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0014] Suppression of immune system activity is desirable for preventing or
treating
tissue or organ rejection following transplant surgery and for preventing and
treating
diseases and disorders arising from aberrant activity of the immune system, in
particular
autoimmune disorders and diseases. Exemplary autoimmune disorders include
grafft versus
host disease (GVHD), insulin-dependent diabetes (Type I), Hashimoto's
thyroiditis and
Graves' disease, pernicious anemia, Addison's disease, chronic active
hepatitis, Crohn's
disease, ulcerative colitis, rheumatoid arthritis, multiple sclerosis,
systemic lupus
erythematosus, psoriasis, scleroderma and myasthenia gravis.
[0015] The compounds of the present invention are useful in preventing and
treating
diseases and disorders related to mast cell-mediated allergic reactions and
inflammation,
such as keratoconjuctivitis sicca.
[0016] Other indications in which the Jak3 inhibitors are useful include
leukemias and
lymphonias.
DETAILED DESCRIPTION OF THE INVENTION
[0017] Throughout this specification the substituents are defmed when
introduced and
retain their defmitions.
[0018] In a first aspect the invention relates to purinones and
imidazopyridinones having
general formula III:
R4
N~ (CR2Rg)y
~ N N
O
I Q3
Q, "-~
Q2 R$
Rl
III
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0019J The members of the genus III may be conveniently divided into subgenera
based
on the values of Q. When Ql is nitrogen and Qa is carbon, a subgenus of
purinones and
iinidazo[4,5-b]pyridinones having an attached imidazo[4,5-c]pyridiine arises.
When Ql is
carbon and Q2 is nitrogen, a subgenus of purinones and imidazo[4,5-
b]pyridinones having
an attached imidazo[5,4-c]pyridine arises. When Qland Q2 are both carbon, a
subgenus of
purinones and imidazo[4,5-b]pyridinones having an attached benzimidazole
arises. The
genus could similarly be divided on the basis of Q3. When Q3 is nitrogen, a
subgenus of
purinones having an attached iunidazo[4,5-c]pyridine, imidazo[5,4-c]pyridine
or
benzimidazole arises. When Q3 is carbon, a subgenus of imidazo[4,5-
b]pyridinones
having an attached imidazo[4,5-c]pyridine, imidazo[5,4-c]pyridine or
benzimidazole
arises. The structures of these subgenera are shown below:
R4 ~4
N (C/RzR3)Y N (dR2R3)Y
N N N1,,\ N
I N O N N 0
N ~ N
Rl RS Ri R5
Xz Xz
R4 /R4
N (C/'R2R3)Y N~ (CR2R3)y
N N N N`'~ N N
N >==o N ~~ N ~
~
X, N X, N
1 R5 Ri R$
6
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
/ R4 / R4
N~--~ (CR2R3)Y N GR2R3)Y
N I o
~N NVR6 N N
N>~O `
X~ Xi Ri R5
X2 X2
[0020] In certain embodiments, X1 and X2 are selected from hydrogen, cyano,
chloro,
fluoro, trifluoromethyl, trifluoromethoxy and methyl; in other embodiments Rl
is H. In
one subdivision, y is zero; in another y is 1 or 2 and R2 and R3 are hydrogen
or methyl.
Examples of R4 include: cyclopentyl, cyclohexyl, piperidine, oxepane,
benzoxepane,
dihydrocyclopentapyridine, phenyl, benzyl, tetralin, indane, tetrahydropyran,
tetrahydrofuran, tetrahydroindole, isoquinoline, tetrahydroisoquinoline,
quinoline,
tetrahydroquinoline, chroman, pyridine, pyrimidine, pyrazine, dihydropyran,
dihydrobenzofuran, tetrahydrobenzofuran, tetrahydrobenzothiophene, furan,
dihydropyrano[2,3-b]pyridine (see example below), tetrahydroquinoxaline,
tetrahydrothiopyran (thiane), thiochroman (dihydrobenzothiin) or any of the
foregoing
rings carrying from 1-3 additional substituents, such as halogen, methyl,
methoxy,
trifluoromethyl, cyano, hydroxy, oxo, oxide and acetyl. Additional examples in
which R4
is alkyl or substituted alkyl include subgenera in which R4 is oxaalkyl
(alkoxyalkyl).
[0021] In certain embodiments of genus III, y is 1 or 2; R2 and R3 are
hydrogen or
methyl and R4 is phenyl, quinoline, pyridine, pyrazine or substituted phenyl,
quinoline,
pyridine or pyrazine. In other embodiments of genus III, y is zero and R4 is
cyclopentyl,
cyclohexyl, phenyl, indane, piperidine, oxepane, benzoxepane,
dihydrocyclopentapyridine,
tetralin, tetrahydropyran, tetrahydrofuran, tetrahydroindole, isoquinoline,
tetrahydroisoquinoline, quinoline, tetrahydroquinoline, chroman, pyridine,
pyrimidine,
dihydropyran, dihydrobenzofuran, tetrahydrobenzofuran,
tetrahydrobenzothiophene, furan,
dihydropyrano[2,3-b]pyridine, tetrahydroquinoxaline, tetrahydrothiopyran
(thiane),
7
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
thiochroman (dihydrobenzothiin) or a substituted ring from the foregoing list.
In further
embodiments, (a) y is zero and R4 is selected from cyclohexyl, oxepane,
tetralin, indane,
dihydrocyclopentapyridine, tetrahydropyran, tetrahydroquinoline, chroman,
dihydrobenzofuran, tetrahydrobenzof-uran, dihydropyrano[2,3-b]pyridine and
tetrahydroquinoxaline, each optionally substituted with hydroxy, oxo, or
halogen; or (b) y
is 1 or 2, R2 and R3 are hydrogen or methyl and R4 is selected from phenyl,
pyridine and
pyrazine, each optionally substituted with halogen. When y is zero, R4 may be
tetrahydropyran-4-yl, 4-hydroxycyclohexyl, 4-oxocyclohexyl, oxepan-4-yl,
chroman-4-yl;
3,4-dihydronaphthalen-1(2H)-on-4-yl; 2,3-dihydroinden- 1 -on-4-yl and their
fluoro
substituted counterparts. It appears that, although both enantiomers are
active, conipounds
in which the carbon at 4 ofthe chroman is of the (R) configuration have higher
potency.
Certain of the foregoing subgenera in which y is zero may also be described by
a
representation in which R41s
W
~ J (CH2)p ~
~rtir~rv~
[4022] According to this representation, W is CH2, C=O or O; p zs 1, 2 or 3;
and A is a
six-membered heteroaromatic ring containing 1 or 2 nitrogens or a benzene ring
optionally
substituted with one or two fluorines. The wavy line denotes the point of
attachment to the
purinone. Compounds in which the carbon marked with an asterisk
--Z
~
(CH2)p J A
V'tF.J%I1
8
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
is of the (R) configuration appear to be more potent than their corresponding
(S)
enantiomers. Other embodiments include compounds in which y is 1 and R4is
selected
from difluorophenyl, fluorophenyl, chlorophenyl, chlorofluorophenyl, pyridin-3-
yl and
pyrazin-3-yl.
[0023] In certain embodiments of genus III, Xi and XZ are selected from
hydrogen,
cyano, chloro, fluoro, trifluoromethyl, trifluoromethoxy and methyl. In
narrower
embodiments, Xl is selected from hydrogen, cyano and fluoro; Ql and Q2 are
CXI; Q3 is N
and Rl is H. In some embodiments, y is zero and R4 is selected from
cyclohexyl, tetralin,
indane, oxepane, dihydrocyclopentapyridine, tetrahydropyran, tetrahydroquino
line,
chroman, dihydrobenzofuran, tetrahydrobenzofuran, dihydropyrano[2,3-b]pyridine
and
tetrahydroquinoxaline, each optionally substituted with hydroxy, oxo, or
halogen. In other
embodiments y is 1 or 2, R2 and R3 are hydrogen or methyl and R4 is selected
from
phenyl, pyridine and pyrazine, each optionally substituted with halogen.
[0024] In certain embodiments, R5 is Cl-C6 allcyl wherein certain replacements
and
substitutions have been made. In one embodiment, (a), one or two CHZ may be
replaced
by NH or N(alkyl). These residues are also referred to as azaalkyl. An example
of such an
R5 is -CH2CH2CH2N(CH3)CH2CN, which may be considered 5-cyanopentane in which
one CH2has been replaced by N(CH3). In one embodiment, (b), one or two C112
rnay be
replaced by O. These residues are also referred to as oxaalkyl. In one
embodiment, (c),
one or two C112 may be replaced by (C=O). An example of such an R5 is
0 \
O N
, which may be considered a 2-(4-morpholinyl)ethyl in which
one CH2has been replaced by C=fl. In one embodiment,(d), two CHa are replaced
by
CH=CH or C=C. An example of such an R5 is 3-methylbut-2-ene-1-yl
[-CH2CH=C(CH3)2]. A fiu-ther embodiment includes (e), any chemically stable
9
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
combination of (a), (b) (c) and (d). An example of such an R5 is
-CH2CH2CH2NHC(=O)CH2CN, which may be considered 6-cyanohexane in which one
CH2 has been replaced by NH and one CH2has been replaced by C=O. The term
"chemically stable" is readily understood by persons of skill in the chemical
art. It
encompasses compounds that may be isolated and purified and that either do not
measurably decompose at physiological pH in aqueous solution or do so at a
rate such that
>99% remains after one hour at ambient temperature. In all of these compounds,
from
zero to three hydrogens may be replaced by a substituent chosen from:
(a) halogen, hydroxy, cyano, loweralkylsulfonyl, loweralkylsulfonyloxy, amino,
loweralkylamino, diloweralkylamino, alkoxyamino, sulfonylamino, acylamino,
arylamino,
loweralkoxy;
(b) heterocyclyl and heterocyclyl substituted with from one to three
substituents chosen
from halogen, hydroxy, alkoxy, alkyl and alkoxycarbonyl;
(c) phenyl and phenyl substituted with from one to three substituents chosen
from halogen,
hydroxy, alkoxy, alkyl, acylamino, cyano, carboxy, alkoxycarbonyl, haloalkyl
and
heterocyclyl.
[0025] Some examples of the aforementioned substituents for Cl-C6 alkyl
include but are
not limited to:
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0
II
F CI 0 CH3 /NO
O H
> . > >
0
II O N O IH~
N ~ 3 H H iNO
> > >
O
N
> ! > >
0 0 0
`2, N `'Z~`2. N
ll~) ~ N
`Z. NH Q
> > >
0 O OMe 0
N
NH "i.
OH
> > >
11
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0 OH
O O
N N
OH
[0026] In certain embodiments of genus III, Ql is CXI, Q2 is CX2, Xl is
hydrogen, X2 is
a substituent at the 6 position of the benzimidazole, and X2 is chosen from
hydrogen,
fluoro and cyano.
[0027] All of the compounds falling within the foregoing parent genera and
their
subgenera are useful as Jak3 inhibitors.
Definitions
[0028] For convenience and clarity certain terms employed in the
specification,
examples and claims are described herein.
[0029] Alkyl is intended to include linear, branched, or cyclic hydrocarbon
structures
and combinations thereof. Lower alkyl refers to alkyl groups of from 1 to 6
carbon atoms.
Examples of lower alkyl groups include methyl, ethyl, propyl, isopropyl,
butyl, s-and t-
butyl and the like. Preferred alkyl groups are those of C20 or below; more
preferred are Cl-
C$ alkyl. Cycloalkyl is a subset of alkyl and includes cyclic hydrocarbon
groups of from 3
to 8 carbon atoms. Examples of cycloalkyl groups include cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl, norbornyl and the like.
[0030] Cl to C20 hydrocarbon includes alkyl, cycloalkyl, alkenyl, alkynyl,
aryl and
combinations thereof. Examples include phenethyl, cyclohexylmethyl, camphoryl
and
naphthylethyl.
12
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0031] Alkoxy or alkoxyl refers to groups of from 1 to 8 carbon atoms of a
straight,
branched, cyclic configuration and combinations thereof attached to the parent
structure
through an oxygen. Examples include methoxy, ethoxy, propoxy, -isopropoxy,
cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to groups
containing
one to four carbons. The term oxaalkyl is intended as it is understood in the
art [see
Naming and Indexing of Chemical Substances for Chemical Abstracts, published
by the
American Chemical Society, 196, but without the restriction of 127(a)], i.e.
it refers to
compounds in which the oxygen is bonded via a single bond to its adjacent
atoms (forming
ether bonds); it does not refer to doubly bonded oxygen, as would be found in
carbonyl
groups.
[0032] Acyl refers to groups of from 1 to 8 carbon atoms of a straight,
branched, cyclic
configuration, saturated, unsaturated and aromatic and combinations thereof,
attached to
the parent structure through a carbonyl functionality. One or more carbons in
the acyl
residue may be replaced by nitrogen, oxygen or sulfur as long as the point of
attachment to
the parent remains at the carbonyl. Examples include acetyl, benzoyl,
propionyl,
isobutyryl, t-butoxycarbonyl, benzyloxycarbonyl and the like. Lower-acyl
refers to groups
containing one to four carbons.
[0033] Aryl and heteroaryl mean a 5- or 6-membered aromatic or heteroaromatic
ring
containuig 0-3 heteroatoms selected from 0, N, or S; a bicyclic 9- or 10-
membered
aromatic or heteroaromatic ring system containiiig 0-3 heteroatoms selected
from 0, N, or
S; or a tricyclic 13- or 14-membered aromatic or heteroaromatic ring system
containing 0-
3 heteroatoms selected from 0, N, or S. The aromatic 6- to 14membered
carbocyclic
rings include, e.g., benzene and naphthalene, and for the purposes of the
present invention,
fused moieties such as tetrahydronaphthalene (tetralin), indane and fluoruie,
in which one
or more rings are aromatic, but not all need be. The 5- to 10-membered
aromatic
heterocyclic rings include, e.g., imidazole, pyridine, indole, thiophene,
benzopyranone,
thiazole, furan, benzimidazole, quinoline, isoquinoline, quinoxaline,
pyrimidine, pyrazine,
tetrazole and pyrazole.
13
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0034] Arylalkyl refers to a substituent in which an aryl residue is attached
to the parent
structure through alkyl. Examples are benzyl, phenethyl and the like.
Heteroarylalkyl
refers to a substituent in which a heteroaryl residue is attached to the
parent structure
through alkyl. Examples include, e.g., pyridinyhnethyl, pyrimidinylethyl and
the like.
[0035] Heterocycle means a cycloalkyl :iri which from one to three carbons is
replaced by
a heteroatom selected from the group consisting of N, 0 and S. The nitrogen
and sulfur
heteroatoms may optionally be oxidized. In some contexts (other than the
present) the
term heterocycle may be interpreted to include heteroaryl; for the purpose of
this
application, heterocycle is a saturated heterocycle and does not include
heteroaryl. When
heteroaryl is intended, it is expressly named. Examples of heterocycles
include
pyrrolidine, morpholine, dioxane, tetrahydrofuran, and the like. Examples of
heterocyclyl
residues additionally include piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl,
2-oxo-
pyrrolidinyl, 4-piperidinyl, pyrazolidinyl, oxazolidinyl, isoxazolidinyl,
thiazolidinyl,
quinuclidinyl, tetrahydrofuryl, tetrahydropyranyl, thiamorpholinyl,
thiamorpholinylsulfoxide, and thiamorpholinylsulfone. A nitrogenous
heterocycle is a
heterocycle containing at least one nitrogen in the ring; it ma.y contain
additional nitrogens,
as well as other heteroatoms.
[0036] Substituted alkyl, aryl, cycloalkyl, heterocyclyl, heteroaryl etc.
refer to alkyl, aryl,
cycloalkyl, heterocyclyl, or heteroaryl wherein up to three H atoms in each
residue are
replaced with halogen, haloalkyl, hydroxy, loweralkoxy, hydroxyloweralkyl,
carboxy,
carboalkoxy (also referred to as alkoxycarbonyl), carboxamido (also referred
to as
alkylaminocarbonyl), heterocyclylcarbonyl, cyano, carbonyl, nitro, amino,
alkylamino,
dialkylamino, loweralkoxyamino, arylaminocarbonyl, mercapto, alkylthio,
sulfoxide,
sulfoxide amino, sulfone, acylamino, aniidino, alkenyl, cycloalkyl, phenyl,
substituted
phenyl, benzyl, substituted benzyl, heteroaryl, phenoxy, benzenesulfonyl,
benzyloxy, or
heteroaryloxy. When the parent is a heterocycle that allows such substitution,
the term
also includes oxides, for example pyridine-N-oxide, thiopyran sulfoxide and
thiopyran-
S,S-dioxide. As mentioned above, two hydrogens on a single carbon may be
replaced by a
14
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
carbonyl to form an oxo derivative. Noteworthy oxo-substituted aryl residues
include
tetralone (3,4-dihydronaphthalen-.1(2H)-one) and indanone (2,3-dihydroinden-l-
one).
[0037] The terms "halogen" and "halo" refer to fluorine, chlorine, bromine or
iodine.
[0038] Some of the conzpounds described herein may contain one or more
asymmetric
centers and may thus give rise to enantiomers, diastereomers, and other
stereoisomeric
forms that may be defined, in terrns of absolute stereochemistry, as (R)- or
(S)-. The
present invention is meant to include all such possible isomers, as well as,
their racemic
and optically pure forms. Optically active (R)- and (S)- isomers may be
prepared using
chiral synthons or chiral reagents, or resolved using conventional techniques.
When the
compounds described herein contain olefmic double bonds or other centers of
geometric
asymmetry, and unless specified otherwise, it is intended that the compounds
include both
E and Z geometric isomers. Likewise, all tautomeric forms are also intended to
be
included. The configuration of any carbon-carbon double bond appearing herein
is selected
for convenience only and is not intended to designate a particular
configuration; thus a
carbon-carbon double bond depicted arbitrarily herein as trans may be Z, E or
a mixture of
the two in any proportion.
[0039] The graphic representations of racemic, ambiscalemic and scalemic or
enantiomerically pure compounds used herein are taken from Maehr J. Chem. Ed.
62, 114-
120 (1985): solid and broken wedges are used to denote the absolute
configuration of a
chiral element; wavy lines indicate disavowal of any stereochemical
implication which the
bond it represents could generate; solid and broken bold lines are geometric
descriptors
indicating the relative configuration shown but denoting racemic character;
and wedge
outlines and dotted or broken lines denote enantiomerically pure compounds of
indeterminate absolute configuration.
[00440] It will be recognized that the compounds ofthis invention can exist in
radiolabeled form, i.e., the compounds may contain one or more atoms
containiing an
atomic mass or mass number different from the atomic mass or mass number
usually found
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
in nature. Radioisotopes of hydrogen, carbon, phosphorous, fluorine, chlorine
and iodine
include 3H, 14C, 35S,18F, 36C1 and 125I, respectively. Compounds that contain
those
radioisotopes and/or other radioisotopes of other atoms are within the scope
ofthis
invention. Tritiated, i.e. 3H, and carbon-14, i.e., 14C, radioisotopes are
particularly
preferred for their ease in preparation and detectability. Radiolabeled
compounds of this
invention can generally be prepared by methods well known to those skilled in
the art.
Conveniently, such radiolabeled compounds can be prepared by carrying out the
procedures disclosed in the Examples by substituting a readily available
radiolabeled
reagent for a non-radiolabeled reagent. Because of the high affmity for the
JAK3 enzyme
active site, radiolabeled compounds ofthe invention are useful for JAK3
assays.
[0041] In one embodiment, R4 is a heterocycle. Heterocycles that appear in the
examples are monocyclic and bicyclic heterocycles or monocyclic and bicyclic
heterocycles substituted with one or two substitutions. When y is not zero,
heteroaryl is a
preferred subset of heterocyclyl for R4. Exemplary nitrogenous heterocycles
include
piperidine, pyridine, pyrazine, pyrimidine, pyridine, quinoline, isoquinoline,
tetrahydroquinoline, tetrahydroisoquinoline, and their variously substituted
derivatives,
such as
N
N N H3C
~ ~ 1 I
N
N CI N` ` C H 3 I N+
N ~-)~ ~ " '/ N O-
16
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
CN I ~
N s~ ,~ N
N
and H
[0042] An oxygenous heterocycle is a heterocycle containing at least one
oxygen in the
ring; it may contain additional oxygens, as well as other heteroatoms.
Exemplary
oxygenous heterocycles include tetrahydropyran, chroman and their variously
substituted
derivatives, such as:
S, C3
Q .,~~, Q
I I I
F F
17
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
Chemical Synthesis
[0043] Terminology related to "protecting", "deprotecting" and "protected"
functionalities occurs throughout this application. Such terminology is well
understood by
persons of skill in the art and is used in the context of processes that
involve sequential
treatment with a series of reagents. In that context, a protecting group
refers to a group
which is used to mask a functionality during a process step in which it would
otherwise
react, but in which reaction is undesirable. The protecting group prevents
reaction at that
step, but may be subsequently removed to expose the original functionality.
The removal
or "deprotection" occurs after the completion of the reaction or reactions in
which the
functionality would interfere. Thus, when a sequence of reagents is specified,
as it is in the
processes of the invention, the person of ordinary skill can readily envision
those groups
that would be suitable as "protecting groups". Suitable groups for that
purpose are
discussed in standard textbooks in the field of chemistry, such as Protective
Groups in
Organic Synthesis by T.W. Greene [John Wiley & Sons, New York, 1991], which is
incorporated herein by reference.
[0044] A comprehensive list of abbreviations utilized by organic chemists
appears in the
first issue of each volume of the Journal of Organic Chemistry. The list,
which is typically
presented in a table entitled "Standard List of Abbreviations", is
incorporated herein by
reference.
[0045] In general, the compounds of the present invention may be prepared by
the
methods illustrated in the general reaction schemes as, for example, described
below, or by
modifications thereof, using readily available starting materials, reagents
and conventional
synthesis procedures. In these reactions, it is also possible to make use of
variants that are
in themselves known, but are not mentioned here. The starting materials, for
example in
the case of suitably substituted benzimidazole ring compounds, are either
commercially
available, synthesized as described in the examples or may be obtained by the
methods
well known to persons of skill in the art
18
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0046] The present invention firther provides pharmaceutical compositions
comprising
as active agents, the compounds described herein.
[0047] As used herein a "pharmaceutical composition" refers to a preparation
of one or
more of the compounds described herein, or physiologically acceptable salts or
solvents
thereof, with other chemical components such as physiologically suitable
carriers and
excipients.
[0048] Pharmaceutical compositions for use in accordance with the present
invention
thus may be formulated in conventional manner using one or more
physiologically
acceptable carriers comprising excipients and auxiliaries, which facilitate
processing of the
active compounds into preparations which, can be used pharmaceutically. Proper
formulation is dependent upon the route of administration chosen.
[0049] Compounds that inhibit Jak-3 can be formulated as pharmaceutical
compositions
and administered to a mammalian subject, such as a human patient in a variety
of forms
adapted to the chosen route of administration, i.e., orally or parenterally,
by intravenous,
intramuscular, topical, transdermal or subcutaneous routes.
[0050] For oral administration, the compounds can be formulated readily by
combining
the active compounds with pharmaceutically acceptable carriers well known in
the art.
Such carriers enable the compounds of the invention to be formulated as
tablets, pills,
dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like,
for oral
ingestion by a patient. Pharmacological preparations for oral use can be made
using a solid
excipient, optionally grinding the resulting mixture, and processing the
mixture of
granules, after adding suitable auxiliaries if desired, to obtain tablets or
dragee cores.
Suitable excipients are, in particular, fillers such as sugars, including
lactose, sucrose,
mannitol, or sorbitol; cellulose preparations such as, for example, maize
starch, wheat
starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose,
hydroxypropylmethyl-cellulose, sodium carbomethylcellulose; and/or
physiologically
acceptable polymers such as polyvinylpyrrolidone (PVP). If desired,
disintegrating agents
19
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
may be added, such as cross-linked polyvinyl pyrrolidone, agar or alginic acid
or a salt
thereof such as sodium alginate.
[0051] In addition, enteric coating may be useful as it is may be desirable to
prevent
exposure of the compounds of the invention to the gastric environment.
[0052] Pharmaceutical compositions, which can be used orally, include push-fit
capsules
made of gelatin as well as soft, sealed capsules made of gelatin and a
plasticizer, such as
glycerol or sorbitol. The push-fit capsules may contain the active ingredients
in admixture
with filler such as lactose, binders such as starches, lubricants such as talc
or magnesium
stearate and, optionally, stabilizers.
[0053] In soft capsules, the active compounds may be dissolved or suspended in
suitable
liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
In addition,
stabilizers may be added. All formulations for oral administration should be
in dosages
suitable for the chosen route of administration.
[0054] For injection, the compounds of the invention may be formulated in
aqueous
solutions, preferably in physiologically compatible buffers such as Hank's or
Ringer's
solution or physiological saline buffer. For transmucosal and transdermal
adrninistration,
penetrants appropriate to the barrier to be permeated may be used ui the
composition. Such
penetrants, including for example DMSO or polyethylene glycol, are known in
the art.
[0055] For administration by inhalation, the compounds for use according to
the present
invention are conveniently delivered in the form of an aerosol spray
presentation from a
pressurized pack or a nebulizer with the use of a suitable propellant, e. g.,
dichlorodifluoromethane, trichlorofluoromethane, dichloro-tetrafluoroethane or
carbon
dioxide. In the case of a pressurized aerosol, the dosage unit may be
determined by
providing a valve to deliver a metered amount. Capsules and cartridges of, e.
g., gelatin for
use in an inhaler or insufflator may be formulated containing a powder mix of
the
compound and a suitable powder base such as lactose or starch.
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0056] Pharmaceutical compositions for parenteral administration include
aqueous
solutions of the active ingredients in water-soluble form. Additionally,
suspensions of the
active compounds may be prepared as appropriate oily iiiiection suspensions.
Suitable
lipophilic solvents or vehicles include fatty oils such as sesame oil, or
synthetic fatty acids
esters such as ethyl oleate, triglycerides or liposomes. Aqueous injection
suspensions may
contain substances, which increase the viscosity of the suspension, such as
sodium
carboxymethyl cellulose, sorbitol or dextran. Optionally, the suspension may
also contain
suitable stabilizers or agents, which increase the solubility of the
compounds, to allow for
the preparation of highly concentrated solutions.
[0057] The compounds of the present invention may also be formulated in rectal
compositions such as suppositories or retention enemas, using, e.g.,
conventional
suppository bases such as cocoa butter or other glycerides..
[0058] Depending on the severity and responsiveness of the condition to be
treated,
dosing can also be a single administration of a slow release composition, with
course of
treatment lasting from several days to several weeks or until cure is effected
or diminution
of the disease state is achieved. The amount of a composition to be
administered will, of
course, be dependent on many factors including the subject being treated, the
severity of
the affliction, the manner of administration, the judgment of the prescribing
physician. The
compounds of the invention may be administered orally or via inj"ection at a
dose from
0.001 to 2500 mg/kg per day. The dose range for adult humans is generally from
0.005 mg
to 10 g/day. Tablets or other forms of presentation provided in discrete units
may
conveniently contain an amount of compound of the invention which is effective
at such
dosage or as a multiple of the same, for instance, units containing 5 mg to
500 mg, usually
around 10 mg to 200 mg. The precise amount of compound administered to a
patient will
be the responsibility of the attendant physician. However, the dose employed
will depend
on a number of factors, including the age and sex of the patient, the precise
disorder being
treated, and its severity. Also, the route of administration may vary
depending on the
condition and its severity.
21
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0059] As used herein, and as would be understood by the person of skill in
the art, the
recitation of "a compound" is intended to include salts, solvates and
inclusion complexes
of that compound. The temi "solvate" refers to a compound of Formula I or II
in the solid
state, wherein molecules of a suitable solvent are incorporated in the crystal
lattice. A
suitable solvent for therapeutic administration is physiologically tolerable
at the dosage
administered. Examples of suitable solvents for therapeutic administration are
ethanol and
water. When water is the solvent, the solvate is referred to as a hydrate. In
general,
solvates are formed by dissolving the compound in the appropriate solvent and
isolating
the solvate by cooling or using an antisolvent. The solvate is typically dried
or azeotroped
under ambient conditions. Inclusion complexes are described in Remington: The
Science
and Practice of Pharmacy 19th Ed. (1995) volume 1, page 176-177, which is
incorporated
herein by reference. The most commonly employed inclusion complexes are those
with
cyclodextrins, and all cyclodextrin complexes, natural and synthetic, are
specifically
encompassed within the claims.
[0060] The term "pharmaceutically acceptable salt" refers to salts prepared
from
pharmaceutically acceptable non-toxic acids or bases including inorganic acids
and bases
and organic acids and bases. When the compounds of the present invention are
basic, salts
may be prepared from pharmaceutically acceptable non-toxic acids includiing
inorganic and
organic acids. Suitable pharmaceutically acceptable acid addition salts for
the compounds
of the present invention include acetic, benzenesulfonic (besylate), benzoic,
camphorsulfonic, citric, ethenesulfonic, fumaric, gluconic, glutamic,
hydrobromic,
hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic,
mucic, nitric,
pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric acid, p-
toluenesulfonic, and the
like. When the compounds contain an acidic side chain, suitable
pharmaceutically
acceptable base addition salts for the compounds ofthe present invention
include metallic
salts made from aluminum, calcium, lithium, ma.gnesium, potassium, sodium and
zinc or
organic salts made from lysine, N,N'-dibenzylethylenediamine, chloroprocaine,
choline,
diethanolamine, ethylenediamme, meglumine (N-methylglucamine) and procaine.
22
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0061] The term "preventing" as used herein refers to administering a
medicament
beforehand to forestall or obtund an attack. The person of ordinary skill in
the medical art
(to which the present method claims are directed) recognizes that the term
"prevent" is not
an absolute term. In the medical art it is understood to refer to the
prophylactic
administration of a drug to substantially diminish the likelihood or
seriousness of a
condition, and this is the sense intended herein.
[0062] It should be understood that in addition to the ingredients
particularly mentioned
above, the formulations of this invention may include other agents
conventional in the art
having regard to the type of formulation in question, for example those
suitable for oral
administration may include flavoring agents.
[0063] The compositions may be presented in a packaging device or dispenser,
which
may contain one or more unit dosage forms containing the active ingredient.
Examples of a
packaging device include metal or plastic foil, such as a blister pack and a
nebulizer for
inhalation. The packaging device or dispenser may be accompanied by
instructions for
administration. Compositions coniprising a compound of the present iilvention
formulated
in a compatible pharmaceutical carrier may also be placed in an appropriate
container and
labeled for treatment of an indicated condition.
Indications
[0064] The compounds of the present invention are useful in inhibiting the
activity if
Jak3 or in inhibiting Jak3 mediated activity and are useful as
immunosuppressive agents
for tissue and organ transplants, including bone marrow transplant and in the
treatment of
autoimmune and inflammatory diseases and of complications arising therefrom.
[0065] Hyperacute, acute and chronic organ transplant rejection may be
treated.
Hyperacute rejection occurs within minutes of transplantation. Acute rejection
generally
occurs within six to twelve months of the transplant. Hyperacute and acute
rejections are
typically reversible where treated with iTnmunosuppressant agents. Chronic
rejection,
23
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
characterized by gradual loss of organ function, is an ongoing concern tor
transplant
recipients because it can occur anytime after transplantation.
[0066] There are about 75 different autoimmune disorders known that may be
classified
into two types, organ-specific (directed mainly at one organ) and non-organ-
specific
(affecting multiple organs).
[0067] Examples of organ-specific autoimmune disorders are insulin-dependent
diabetes
(Type I) which affects the pancreas, Hashimoto's thyroiditis and Graves'
disease which
affect the thyroid gland, pernicious anemia which affects the stomach,
Cushing's disease
and Addison's disease which affect the adrenal glands, chronic active
hepatitis which
affects the liver; polycystic ovary syndrome (PCOS), celiac disease,
psoriasis,
inflammatory bowel disease (IBD) and ankylosing spondylitis.
[0068] Examples of non-organ-specific autoimmune disorders are rheumatoid
arthritis,
multiple sclerosis, systemic lupus and myasthenia gravis.
[0069] Type I diabetes ensues from the selective aggression of autoreactive T-
cells
against insulin secreting (3 cells of the islets of Langerhans. Targeting Jak3
in this disease
is based on the observation that multiple cytokines that signal through the
Jak pathway are
known to parYicipate iii the T-cell mediated autoimmune destruction of 0
cells. Indeed, a
Jak3 inhibitor, JANEX- 1 was shown to prevent spontaneous autoimmune diabetes
development in the NOD mouse model of type I diabetes.
[0070] Graft-versus-host disease (GVHD) is a donor T-cell initiated
pathological
condition that frequently follows allogeneic bone marrow transplantation
(BMT).
Substantial experimental and clinical research have demonstrated that donor T-
cells are the
principal mediators and effectors of GVHD. Jak3 plays a key role in the
induction of
GVHD and treatment with a Jak3 inhibitor, JANEX-1, was shown to attenuate the
severity
of GVHD (reviewed in Cetkovic-Cvrlje and Ucken, 2004).
24
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0071] Mast cells express Jak3 and Jak3 is a key regulator of the IgE mediated
mast cell
responses including the release of inflammatory mediators. Jak3 was shown to
be a valid
target in the treatment of mast cell mediated allergic reaction.
[0072] Allergic disorders associated with mast cell activation include Type I
immediate
hypersensitivity reactions such as allergic rhinitis (hay fever), allergic
urticaria (hives),
angioedema, allergic asthma and anaphylaxis, i.e., "anaphylatic shock." These
disorders
are treated or prevented by inhibition of Jak3 activity, for example, by
administration of a
Jak3 inhibitor according to the present invention.
[0073] According to the present invention, the Jak3 inhibitors may be
administered
prophylactically, i.e., prior to onset of acute allergic reaction, or they may
be administered
after onset of the reaction, or at both times.
[0074] Inflammation of tissues and organs occurs in a wide range of disorders
and
diseases and in certain variations, results from activation of the cytokine
family of
receptors. Exemplary inflanunatory disorders associated with activation of
Jak3 include, in
a non-limiting manner, skin inflammation due radiation exposure, asthma,
allergic
inflammation and chronic iinflammation.
[0075] The Jak3 inhibitors of the present invention are also useful in
treating certain
malignancies, including skin cancer and hematological malignancy such as
lymphomas
and leukemias.
[0076] The following examples will further describe the invention, and are
used for the
purposes of illustration only, and should not be considered as limiting the
invention being
disclosed.
EXAMPLES
[0077] The following abbreviations and terms have the indicated meaning
throughout:
Ac = acetyl
Bu = butyl
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
DCM = dichloromethane = methylene chloride = CH2UI2
DEAD = diethyl azodicarboxylate
DIC = diisopropylcarbodiimide
DIEA = N,N-diisopropylethyl amine
DMF = N,N-dirnethylformamide
DMSO = dimethyl sulfoxide
EA (EtOAc)= Ethyl Acetate
GC = gas chromatography
h= hours
HOAc = acetic acid
HOBt = hydroxybenzotriazole
Me = methyl
Pd(dppf)2C12 = dichloro[1,1'-bis(diphenylphosphinoferrocene]palladiurn
Ph= phenyl
PhOH = phenol
RT = room temperature
sat'd = saturated
s- = secondary
t- = tertiary
TBDMS = t-butyldimethylsilyl
TFA = trifluoroacetic acid
THF = tetrahydrofuran
TMOF = trimethyl orthoformate
TMS = trimethylsilyl
tosyl = p-toluenesulfonyl
Trt = triphenylmethyl
Examples 1-15 describe syntheses of certain precursors and intermediates of
the invention.
[0078] Example 1. Synthesis of 3,4-Diaminobenzonitrile
.~ NHz H2, Pd/C ~ NH2 EtOCH=C(CN)2 ,~ ~ N
~ --" ~ ~
~
NC ~ NO~ EtOH NC NHZ iPrOH NC H
1 2 3
[0079] A solution of 4-amino-3-nitrobenzonitrile (1) (3.0 g) in ethanol (80
mL) was
sparged for 5 minutes with nitrogen. Palladium on carbon (10%, 300 mg) was
added and
the mixture was saturated with hydrogen. The mixture was stirred under a
hydrogen
balloon for seven hours. The mixture was sparged with nitrogen and filtered
through celite.
26
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
The filtrate was concentrated in vacuo to provide the title compound, 3,4-
diaminobenzonitrile (2).
[00801 Example 2: Synthesis of 3H-Benzo[d]imidazole-5-carbonitrile
[0081] A mixture of 3,4-diaminobenzonitrile (2) (1.0 g) and
(ethoxymethylene)malononitrile (1.4 g) was refluxed in 50 mL of isopropyl
alcohol for 16
h. The mixture was concentrated in vacuo to provide the title compound, 3H-
benzo[d]imidazole-5-carbonitrile (3).
[0082] Example 3: Synthesis of 6-(Trifluoromethoxy)-1H-benzo[d]imidazole
N
F3C0 \ !N
H
4
[0083] 6-(trifluoromethoxy)-1H-benzo[d]imidazole (4) was prepared in two steps
from
2-nitro-4-(trifluoromethoxy)aniline (5) using procedures identical to those
used to make
3H-benzo[d]imidazole-5-carbonitrile (3) from 4-amino-3-nitrobenzonitrile (1,
examples 1,
2).
[0084] Example 4: Synthesis of 5,6-Difluoro-lH-benzo[d]imfdazole
F~\ NH2 Na2S2 04 F NH2 EtOCH=C(CN)2 F,i ~ N
-~ ~ \
~\
~~ iPrOH F N
F NOa NaHCO3 F NH~ H
THF / H20 !
6 MeOH 7 8
[0085] A solution of 4,5-difluoro-2-nitroaniline (6)(1.0 g) in 30 mL of THF
was treated
with a solution comprised of 6 g of Na2S2O4 and 3 g NaHCO3. in 30 mL of water.
Methanol
(10 mL) was added after the addition of the aqueous solution so that the
mixture remained
homogeneous. The mixture was stirred for two hours and then diluted with 100
mL of
27
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
ethyl acetate and 100 mL of water. The organic layer was separated and the
aqueous layer
was extracted again with 100 mL of methylene chloride. The combined organic
layers
were dried over sodium sulfate, filtered, and concentrated to provide the
crude intermediate
4,5-difluorobenzene-1,2-diamine (7). The intermediate was refluxed with
(ethoxymethylene)malononitrile (1.1 g) in 25 mL of isopropyl alcohol for 16 h.
The
mixture was concentrated in vacuo and the resulting crude product was
suspended in water
and filtered. The precipitate was washed with water and air-dried to provide
380 mg of 5,6-
difluoro-lH-benzo[d]imidazole (8).
[0086] Example 5: Synthesis of 5,6-Dimethoxy-lH-benzo[d]imidazole
MeO NHZ HCOOH Me~a)aNH2 N>
Me O N
C H
MeQ MW a~ 220
9 10
[0087] The title compound 5,6-dimethoxy-lH-benzo[d]imidazole (10) was made by
heating 4,5-dimethoxy-1,2-phenylenediamine dihydrochloride (9) in formic acid
at 220 C
in a microwave followed by concentration in vacuo.
[0088] Example 6: Synthesis of 6-Fluoro-lH-benzo[d]imidazole (11) and 6-
(trifluoromethyl)-1H-benzo[d]imidazole (12).
[0089] The title compounds were by made as described in US Application
Publication
No. 2004/0087601, of some ofthe present inventors.
\ I ~ N~
JaN
F H F3C H
11 12
28
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0090] Example 7: Benzimidazole (13), 5-azabenzimidazole (14), 6-chloro-5-
fluorobenzimidazole (15), and 5-methylbenzimidazole (16).
[0091] The title compounds were commercially available.
OtT> \ I ~ N
)OCN>
H H F H
16
13 14 1
[0092] Example S. Synthesis of the primary amine, pyrazin-2-yhnethanamine
1. Raney Ni / H2
N C
N NH3 / MeOH iN NH
2
U
N 2. (3ocaO / ' ~
CH2CIZ N
ch ro matography
17 3. TFA / CH2CI2 18
[00931 Raney nickel catalyst was carefully washed with THF and methanol making
sure
that the catalyst remained moist. The weight of the moist catalyst was 2.5 g
after washing.
This material was added to a solution of pyrazinecarbonitrile (17) (3.0 g) in
7N methanolic
ammonia (120 mL). The mixture was shaken under a 50 p.s.i. atmosphere of
hydrogen for
1.5 hours. The mixture was filtered and the filtrate was concentrated in vacuo
to provide
the crude title compound. Purification was accomplished by conversion of the
crude amine
to the tert-butyl carbamate with excess di-tert-butyl dicarbonate in methylene
chloride.
Column chromatography (70:27:3 hexanes:ethyl acetate:methanol) provided 0.50 g
of pure
tert-butyl pyrazin-2-ylrnethylcarbamate. Pure pyra2in-2-yhnethanamine (18) was
obtained
as the TFA salt from deprotection of the carbamate with P,.1 TFA / CH2C12.
29
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0094] Example 9: Synthesis of 3-Aininomethy~2-fluoropyridine.
I.~ CN H2, Pd/C .~ I NH2
~
N F EtOH N F
19 20
[0095] A round bottom flask was charged with 0.3g (2.46mM) of 3-cyano-2-
fluoropyridine (19), which was then diluted in 20mL EtOH. The solution was
flushed with
argon, and then while under a blanket of argon, 60mg of 10% Pd/C (20% by
weight), was
added. The system was then sealed by septum and put under vacuum. A hydrogen
balloon
was then added, and the reaction was stirred for three hours (followed by
TLC). The
reaction was then put under vacuum again, then exposed to air, and filtered
(keeping
catalyst wet). The resulting solution was dried and evaporated to give 0.28g
(90%) of the
title compound, 3-aminomethyl-2-fluoropyridine (20).
[0096] Example 10: Synthesis of 3-Aminomethyl-6-lnethogypyridine (21), 3-
Aminomethyl-6-methylpyridine (22), and 3-Aminomethylquinoline (23).
[0097] The title amines were obtained from the corresponding nitriles using
the same
procedure that was used to obtain 3-aminomethyl-2-fluoropyridine (20) from 3-
cyano-2-
fluoropyridine (see Example 9).
rN OMe N N
H2N I H2N H2N
21 22 23
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[0098] Example 11: Synthesis of 3-aminomethyl-2-methoxypyridine.
N
HZ
I ~ CHO NH4OAc CC
N OMe NaBH3CN OMe
MeOH
24 25
[0099] Around bottom flask was charged with 0.44g (3.23mM) of 2-methoxy-3-
pyridine
carboxaldehyde (24), 1.24g (16.15mM) of ammonium acetate, and 0.61 g (19.69mM)
of
sodium cyanoborohydride. The flask was then flushed with argon, and then 50mL
of dry
MeOH was added by syringe. The reaction was stirred for 2 days, at which point
the
MeOH was evaporated off. 25mL of water was added, and the mixture was brought
to pH
2 with conc. HC1. This was extracted twice with EtOAc to remove the alcohol
side
product. The mixture was brought to pH 10 using sodium hydroxide pellets,
saturated with
NaCI, and extracted twice with DCM and once with EtOAc. The combined organics
were
dried and evaporated to give 0.31g (69%) of 3-aninomethyl-2-methoxypyridine
(25).
[00100] Example 12: Synthesis of 3-(a-aminoethyl)-2-chloropyridine (26)
N
CI :P~)'
H2N 26
[00101] The title amine was obtained from the corresponding ketone using the
same
procedure that was used to obtain 3-aminomethyl-2-methoxypyridine from 2-
methoxy-3-
pyridine carboxaldehyde (24; Example 11).
31
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00102] Example 13: Synthesis of 3-aminomethyl-4-methylpyrictine
I~ CONH2 BH3 - Me2S NH2
THF N
N
27 28
[00103] A round bottom flask was charged with 0.45g (3.30mM) of 4-
methylnicotinamide (27). The flask was flushed with argon, and 50mL of dry THF
was
added by syringe. The resulting solution was cooled to 0 dg C, and 2. 5mL
(4.96mM) of a
2M solution of borane-dimethylsulfide complex (in THF) was added. A bubbler
was
attached, and the solution was allowed to wann to RT overnight. The solution
was
quenched with MeOH, and dried and evaporated to give 0.38g (95%) of 3-
aminomethyl-4-
methylpyridine (28).
[00104] Example 14: Synthesis of 5-fluoro-1,2,3,4-tetrahydronaphthalen-l-amine
F F
~O 1. 9-BBN /THF \ NaOH
% v 'OMe Me0 ~ / MeOH HO
2. 1-Fluo ro-2
29 iodobenzene, 0 30 O 31
NaOMe,
Pd(dppf)2CI2
1. (CICO)2 F
cat DMF F F
CHZCIz HONHZ-HCI \ H2, Pd/C \
-- -'- ~ -`- ~
2. AICI3 NaOAc / EtOH 1 EtOH ~
O HO-N NH2
32 33 34
[00105] Methyl 4-(2-fluorophenyl)butanoate (2-(4-
methylbutanoate)fluorobenzene, 30).
A round bottom flask was sealed with a rubber septum, flushed with argon, then
charged
with 5.32mL of methyl 3-buteneoate (29) and 100mL of a 0.5M solution of 9-BBN
in
THF. The solution was stirred at RT for three hours. A 2-necked round bottom
flask was
32
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
equipped with a condenser and flushed with argon, then charged wtth `/.36g ot
socaium
methoxide and 1.11g of Pd(dppf)2Clz. To this mixture were added 20mL of dry
THF and
5.22mL of 1-fluoro-2-iodobenzene. The hydroboration solution was added via
canula and
the resulting mixture was refluxed for 16 hours. The solution was cooled to
RT, diluted
with 150mL of water, and extracted three times with ether. The combined
organic layers
were washed with brine, dried, and evaporated. Column chromatography (5%
EtOAc/hexanes) gave 1.:79 g methyl 4-(2-fluorophenyl)butanoate (30).
[00106] 4-(2-Fluorophenyl)butanoic acid (31). A round bottom flask was charged
with
1;79g of 2-(4-methylbutanoate)fluorobenzene, which was dissolved in 17mL of
MeOH.
To this solution was added a solution of 1 g of sodium hydroxide. The
resulting mixture
was stirred 20 hours at RT. The solvent was evaporated and the crude material
diluted
with l5mL of 0. 5M HCI. Extraction with DCM three times gave 1.17g (92%) of 4-
(2-
fluorophenyl)butanoic acid (31).
[00107] 5-Fluoro-3,4-dihydronaphthalen-1(2H)-one (32). A round bottom flask
was
charged with 0.15 g of 4-(2-fluorophenyl)butanoic acid, which was dissolved
ii120mL
DCM and cooled to 0 C. Oxalyl chloride (0.15mL) was added, followed by 1 drop
of
DMF. A drying tube was attached, and the solution was stirred at 0 C for two
hours.
Aluminum chloride (0.121g) was added and the solution was allowed to slowly
warm to
RT overnight.. The mixture was poured onto ice water, and extracted three
times with
DCM. The combined organic layers were washed with 0.5 M NaOH and briiie. The
organic phase was dried, evaporated, and purified by column chromatography
(eluting with
20% EtOAc/Hexanes), to give 0.07g (53%) of 5-fluoro-3,4-dihydronaphthalen-
1(2H)-one
(32).
[00108] 5-Fluoro-1,2,3,4 tetrahydronaphthalen-l-amine (34). A round bottom
flask
was charged with 0.5g of 5-fluoro-3,4-dihydronaphthalen-1(2H)-one, 0.28g of
hydroxylamine hydrochloride, and 0.34g of sodium acetate. A condenser was
attached,
and the flask was purged with argon. 20inL of dry EtOH was added, and the
mixture was
33
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
stirred at reflux for 18 hours. The solution was cooled to K"l', diluted wrtri
EtVAc, and
washed with water. The organic phase was dried with sodium sulfate and
evaporated to
give 0.5g of the intermediate 5-fluoro-3,4-dihydronaphthalen-1(2H)-one oxiine
(33), which
was reduced with Pd/C in EtOH with hydrogen (50psi), to give 0.43g (86%) of 5-
fluoro-
1,2,3,4-tetrahydronaphthalen-l-amine (34).
[00109] Example 15: Synthesis of 8-fluorochroman-4-amine.
1. (CICO)2
F 3-Bromo F cat. DMF F
H O ~ ::': I~' HO I'~ 2 AICb
35 O 36 O 37
F F
HONHz-HCI O ~ H2, RaneyNi O ~
y I~
NaOA EI tc OH I~ EtOH
HO'N NH2
38 39
[00110] 3-(2-fluorophenogy)propanoic acid (36). A mixture of 2-fluorophenol
(35) (15
g), 3-bromopropanoic acid (20 g) and NaOH (11 g) was refluxed in 50mL of
water. The
solution was cooled to RT and acidified to pH 2 with 3 M HCI. The resulting
precipitate
was isolated by filtration to yield 9.27 g of title compound as a white solid.
The filtrate was
extracted three times with EtOAc to yield 2.5g of less pure compound (36).
[00111] 8-fluorochroman-4-one (37). Oxalyl chloride (8.79 mL) and one drop of
DMF
were added to an ice cold solution of 3-(2-fluorophenoxy)propanoic acid (9.27
g) in DCM
(50 mL). The solution was stirred at 0 C for two hours, aluminum chloride
(7.39g,
55.42mM) was added and the solution was stirred for 16 hours at RT. The
mixture was
poured onto ice water, and extracted three times with DCM. The combined
organics were
washed with 0.5M NaOH and brine, dried, evaporated, and purified by column
34
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
chromatography (eluting with 20% EtOAc/Hex) to give 8-fluorochroman-4-one (37)
(8.20g, 98%),
[001121 8-fluorochroman-4-amine (39). A round bottom flask was charged with 8-
fluorochroman-4-one (8.2g), hydroxylamine hydrochloride (3.78g) and sodium
acetate
(4.46g). A reflux condenser was added, the flask was purged with argon, dry
EtOH (20
mL) was added, and the mixture was stirred at reflux for 18 hours. The
solution was
cooled to RT, diluted with EtOAc, and washed with water. The organic phase was
dried,
and evaporated to give the intermediate 8-fluorochroman-4-one oxime (38),
which was
reduced with Raney Nickel in EtOH at 50 PSI to yield the titled amine (39)
(4.69g, 57%)..
[001131 Resolution of 8-fluorochroman-4-amine.
F F F
c 0 Novnzyme 435 1O O
I / MeOCH2COOMe "= ~
t-BuOMe
NH2 0 NH NH2
~
O 8 M H CI 39a
EtOH
F
O jkz
NH2
39b
[001141 In short, a mixture of 8-fluorochroman-4-amine (3.40 g), methyl 2-
methoxyacetate (2.44 g) and Novozyme 435 (Aldrich, 0.68 g) in anhydrous tert-
butyl
methyl ether (75mL) was heated at reflux under argon for two hours (at which
time the
ratio of acylated to unacylated pmduct was 1:1 by HPLC). The solid that formed
upon
cooling was collected via filtration and dissolved in EtOAc. The mixture was
filtered to
remove the biocatalyst and washed once with 0. 5M HCl to remove any lingering
(S)-
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
amine. The solvent was evaporated and the product was recrystallizea rrom fert-
nutyl
methyl ether to yield (R)-N-(8-fluorochroman-4-yl)-2-methoxyacetamide (0.78
g). The
reaction solvent and recrystallization mother liquor was washed three times
with 0.5 M
HCl and concentrated to yield additional (R)-N-(8-fluorochroman-4-yl)-2-
methoxyacetamide (0.83 g). The combined acidic aqueous layers were made basic
by
NaOH and extracted with DCM to yield (,S)-8-fluorochroman-4-amine (39a) (1.6
g). A
solution of (R)-N-(8-fluorochroman-4-yl)-2-methoxyacetamide (0.78 g) in 8M HC1
in
EtOH (50 mL) was heated at reflux for four hours. The solvents were removed
from the
cooled reaction mixture, the resulting solid was taken up in 50 mL of 0.5M
NaOH, salted
out with NaCl(s), and extracted four times with DCM to yield (R)-8-
fluorochroman-4-
amine (0.48 g(87%))(39b). The % ee was checked via chiral HPLC: Chiralcel OD-H
(0.46
x 25 cm analytical column, Daicel Chemical Industries) method: isocratic 5 %
(0.05%
TFA/EtOH) 95 0/0 (0.05 % TFA/Hex), Rt = 7.2 min (S)-enantiomer, Rt = 9.2 min
(R)-
enantiomer.
[00115] Example 16: Synthesis of 2-(1H-Benzo[d]imidazol-1-yl)-9-((R)-8-
fluorochroman-4-yl)-7H-purin-8(9H)-one.
H N F N~ N ~ F
~ Na `~ N~
CI N CI
N CIN` N ~` F~ N/ NOZ
N02 DIEA, THF TN.~NO O
2
Na25zO4
THF1H20
F
N r O~
N,,.N~ N CD~THF
N `~i_ ~.
N N N N &NH2 ~~ ~
H Nv 'F
36
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00116] 2-(1H-Benzo[d]imidazol-1-yl)-N-((1t)-x-fluorochroman-4-yt)-t)-
nitropyrimidin-4-amine. (R)-8-fluorochroman-4-amine (60 mg, Example 15) was
added
to a solution of 2,4-dichloro-5-nitropyrimidine (70 mg) and DIEA (0.14 mL) in
THF (5
mL) at -78 C. The reaction mixture was stirred for a further 15 min at -78 C
then
removed from the cold bath and allowed to warm to RT. A one molar solution of
the
sodium salt of benzimidazole (0.7 ml, stock solution prepared via the addition
of sodium
hydride to a benzimidazole solution in THF) was added to the reaction
intermediate ((R)-2-
chloro-N-(8-fluorochroman-4-yl)-5-nitropyrimidin-4-amine) and the resulting
mixture was
stirred at RT overnight. Purification via column chromatography (elution with
1
MeOH/DCM) gave the titled compound (120 mg), MH+ = 407.
[00117] 2-(lH-Benzo[d]imidazoll-yl)-9-((R)-8-fluorochroman-4-yl)-7.H-purin-
S(9H)-
one. A freshly prepared solution of sodium hydrosulfrte (tech, 0.5 g) and
sodium
bicarbonate (0.25 g) in H20 (5 mL) was added to a solution of the above nitro
compound
(120 mg) in THF (10 mL). The mixture was stirred vigorously for 30 min then
extracted
with EtOAc (2 x) and DCM (2 x), the combined organics were washed with brine,
dried,
filtered and concentrated to yield the intermediate 2-(1H benzo[d]imidazol-1-
yl)-.N4-((R)-
8-fluorochroman-4-yl)pyrimidine-4,5-diamine that was used as such in the next
step.
[00118] Carbonyldiimidazole (0.2 g) was added to a solution of the above amine
in THF
(10 mL). The resulting mixture was stirred at RT overnight, silica gel was
added, then the
solvents were removed under reduced pressure and purified via column
chromatography,
elution with 5 1o MeOH/DCM, to yield the titled product (28 mg), MH+ = 403,1H
NMR
(CDC13) S 10.6 (s, 1H), 8.9 (s, 1H), 8.3 (s, 111), 7.8 (m, 2H), 7.3 (m, 2H),
7.0 (m, 1H), 6.7
(m, 1 H), 5. 9(dd, 1 H), 4.7 (m, 1 H), 4.4 (t, 1 H), 2.7 (m, 1 H), 2.3 (m, 1
H) ppm,19F NMR S-
135.7 (m). Chiral HPLC - no evidence of other enantiomer, Method; Chiralcel OD-
H (0.46
x 25 cm analytical column, Daicel Chemical Industries) isocratic 15 % (0.05%
TFA/EtOH)
85 % (0.05 % TFA/Hex), Rt = 19.5 min (R)-enantiomer, Rt = 22.4 min (S)-
enantiomer.
37
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00119] Example 17: Non-regiospecific synthesis of benzimidazole purinone
derivatives: Synthesis of 5-Nitro-N-(pyridin-3-ylmethyp-2-(6-
(trifluoromethozy)-1H-
benzo[d]imidazol 1-yl)pyrimidin-4-amine (42) AND 5-nitro-N-(pgyridin-3-
ylmethyl)-
2-(5-(trifluoromethogy)-1H-benzo[d]imidazol 1-yl)pyrimidin-4-amine (44).
N
HZN \ N H ~ ~ I/' N
F3COH
CI N CI CI N N \ N
KZC03
N NOZ TEA/ CH3CN N NOZ
40 41
N~
N
~ N N N N 4
~ N~ NN~ N~O 3
~
NOZ N e N
F 42 F3CO H
-NN N N- N
F3C0 ~~ N~.,.~N` N 45 `
NO II O
2 FsCO ...r N _,.L N~
`~ ~H
[00120] 2-Chloro-5-nitro-N-(pyridin-3-ylmethynpyrimidin-4-amine (41). A
solution
of 2,4-dichloro-5-nitropyrimidine (40) (5 g) in methylene chloride (60 mL) was
cooled to -
78 C and treated with 3-(aminomethyl)pyridine (2.8 g). The mixture was
stirred at -78 C
for six hours, and concentrated in vacuo at RT ta provide crude 2-chloro-5-
nitro-N-
(pyridin-3-ylmethyl)pyrirnidin-4-amine (41), which was used without further
purification.
[00121] 5-Nitro-N-(pyridin-3-ylmethyl)-2-(6-(trifluoromethoxy)-1H-
benzo[d]imidazol-1-
yl)pyrlmidin-4-arnine (42) and 5-nitro-N-(pyridin-3-ylmethyl)-2-(5-
(trifluoromethoxy)-
1H-benzo[d]imidazol-1-yl)pyrimidin-4-amine (44). A suspension of crude 2-
chloro-5-
nitro-N-(pyridin-3-ylmethyl)pyrimidin-4-amine (52 mg) in acetonitrile (10 mL)
was
treated with 6-(trifluoromethoxy)- 1H-benzo [d]imidazole (40 mg), potassium
carbonate
38
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
(0.5 g), and heated at 80 C for four hours. The mixture was diluted witn
water ana
extracted with methylene chloride. The organic layer was separated, dried with
sodium
sulfate, filtered, and concentrated in vacuo. Column chromatography (70:22:8
methylene
chloride:ethyl acetate:methanol) provided 12 mg of 5-nitro-N-(pyridin-3-
ylmethyl)-2-(5-
(trifluoromethoxy)-1H-benzo[d]imidazol-1-yl)pyrimidin-4-amine as the first
eluting
isomer and 15 mg of 5-nitro-N-(pyridin-3-ylmethyl)-2-(6-(trifluoromethoxy)-1H-
benzo[d]imidazol 1-yl)pyrimidin-4-amine as the second eluting isomer.
[00122] High Rf isomer: 1H-NMR (CDC13) S 9.2 (s, 1H), 8.9 (s, 1H), 8.8 (m,
1H), 8.6 (s,
1H), 8.5 (d, 1H), 8.2 (d, 1H, assign: H-7 of benzimidazole ring), 7.6 (d, 1H),
7.6 (s, 1H,
assign: H-4 ofbenzimidazole ring), 7.2 (dd, 1H), 4.9 (d, 2H).
[00123] Low Rf isomer: 'H-NMR (CDCl3) S 9.2 (s, IH), 8.9 (s, 1H), 8.8 (rn,
1H), 8.6 (s,
1H), 8.5 (d, 1H), 8.2 (s, 1H, assign: H-7 of benzimidazole ring), 7.7 (d, 11-L
assign: H-4 of
benzimidazole ring), 7.6 (d, 1H), 7.2 (dd, 1H), 7.1 (d, 1H), 4.9 (d, 2H).
[00124] Example 18: Non-regiospecific synthesis of benzimidazole purinone
derivatives: Synthesis of 9-(Pyridin-3-ylmethyl)-2-(6-(trifluoromethoxy)-1H-
benzo[d]imidazol 1-yl)-7H-purin-8(9H) one (43) AND 9-(pyridin-3-yhnethyl)-2-(5-
(trifluoromethoxy)-1H-benzo[d]imidazol-1-yl)-7H-purin-8(9H)-one (45).
[00125] The title compounds were synthesized from 5-nitro-N-(pyridin-3-
ylmethyl)-2-(6-
(trifluoromethoxy)-lH-benzo[d]imidazol-l-yl)pyrimidin-4-arnine (42) and 5-
nitro-N-
(pyridin-3-ylmethyl)-2-(5-(trifluoromethoxy)-1 H-benzo [d] imidazo 1-1-
yl)pyrimidin-4-
amine (44) using the same procedures that were used to convert (R) tert-
butyl2õ4-
dimethoxybenzyl(5-nitro-6-(1-(pyridin 3-y1)ethylarnino)pyridin-2-yl)carbarnate
to (R)-tert-
buty12,4-dimethoxybenzyl(2-oxo-3-(1-(pyridin-3-y1)ethyl)-2,3-dihydro-lH-
imidazo [4,5-
b]pyridin-5-yl)carbarnate (67; Example 22 below).
39
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00126] 6-Trifluoromethoxy isomer (non-salt): 'H-NMR (C.D3OD) a 9.3 (s, 1ri),
s.N (ur s,
1H), 8.6 (s, 1H, assign: H-7 of benzimdazole ring), 8.6 (m, 1H), 8.4 (s, 1H),
8.1 (d, 1H),
7.9 (d, 1K assign: H-4 of benzimidazole ring), 7.5 (dd, 1H), 7.4 (dd, 1H), 5.4
(s, 2H).
[00127] 5-Trifluoromethoxy isomer (non-salt): 1H-NMR (CD3OD) S 9.3 (s, 1H),
8.8 (s,
1H), 8.7 (d, 1H, assign: H-7 of benzimdazole ring), 8.5 (d, 1H), 8.3 (s, 1H),
7.9 (d, 1H), 7.6
(s, 1H, assign: H-4 of benzimidazole ring), 7.4 (dd, 1H), 7.3 (dd, 1H), 5.3
(s, 211).
[00128] Example 19: Non-regiospecific synthesis of an ogoinudazopyridine and
an
imidazopyridine derivative: Synthesis of 5-(1H-Benzo[d]imidazol-1-yl)-3-
(pyridin-3-
yhnethyl)-1H-imidazo [4,5-b] pyridin-2(3H)-one (50).
CI N\ CI CI N N N
TEA
I ~,, H2N I N 91e
Ctr
N02 NOZ
46 47
N
I / } K2C03
N
H
Nn
XXL0
NH NOZ
2
49 48
CDI
CH$CI2
4 `
N~
N
>=
NI
O
N
H
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00129] 6-(1H-Senzo[d]imidazol-1-yl)-3-nitro-N-(pyridin-3-ylmethyl)pyridin-2-
amine (48). A solution of 2,6-dichloro-3-nitropyridine (46) (0.5 g) in
acetonitrile (20 mL)
was cooled to 0 C and treated with triethylaznine (0.36 mL) followed by 3-
(aminomethyl)pyridine (0.26 mL). The mixture was stirred for 30 minutes at 0 C
and eight
hours at RT. The resulting solution, which contained the intermediate 6-chloro-
3-nitro-N-
(pyridin-3-ylmethyl)pyridin-2-arnine (47), was transferred to a sealed tube
containing
benzimidazole (0.84 g) and potassium carbonate (3 g) and heated at 70 C for
16 h. The
mixture was cooled and filtered. The precipitated was washed with water and
air-dried to
provide 239 mg ofthe title conipound (48).
[00130] 5-(1H-Benzo[d]imidazol-1-yl)-3-(pyridin-3-ylmethyl)-1H-imidazo[4,5-
b]pyridin-
2(3H)-one (50) and 5-(1H-benzo[d]imida2o1-1-yl)-3-(pyridin-3-ylmethyl)-3H-
imidazo[4,5-
b]pyridine (51). A solution of 6-(1H-benzo[d]imidazol-1-yl)-3-nitro-N-(pyridin-
3-
ylmethyl)pyridin-2-amine (48) (50 mg) in 1 mL of DMSO was treated with a
solution of
NaZS2O4 (300 mg) in 1 mL of water. The mixture was stirred for two hours and
diluted
with 50 rnL of ethyl acetate. The mixture was washed three times with 50 niL
aliquots of
saturated sodium chloride solution, dried over sodium sulfate, filtered, and
concentrated in
vacuo to provide the intermediate 6-(1 H-benzo [d]imidazol- 1-yl)-N2-(pyridin-
3-
ylmethyl)pyridine-2,3-diamine (49). Half of the intermediate was dissolved in
methylene
chloride (2 mL) and treated with 1,1'-carbonyldiimidazole (46 mg) at RT for 16
h. The
resulting crude mixture was purified by preparative TLC (1000 microns, 5% MeOH
/
CH2Cl2) to provide 7.1 mg of 5-(1H-benzo[d]imidazol-1-yl)-3-(pyridin-3-
ylmethyl)-1H-
imidazo[4, 5-b]pyridin-2(3H)-one (50): 1H NMR (CDC13) S 10.0 (br s, 1H), 8.9
(s, 1H), 8.6
(d, 1H), 8. 5(s, 1H), 7.9 (m, 2H), 7.8 (m, 1H), 7.6 (d, 1H), 7.4 (ni, 2H), 7.4
(in, 1H), 7.3 (d,
1 H), 5.2 (s, 2H)..
[00131] Example 20: Regiospecific synthesis: Synthesis of 3-(9-(2,6-
Difluorobenzyl)-
8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile.
41
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F / MeO ~, OMe
H2N \ I HzN i
r
CI CI FCI:x7
-------------- --- -
Y~N DIEA1 THF NNO2 F DIEA ! THF
NOZ
40 52
MeO /
MeO F H
N~,!~ N N ~,. ~ Raney Ni NYN` NF~. I
II \ THF OMe 54 N./NH F
OMe NNO F Z
a
53 F `
Me0 I r''
CDI ~ H TFA lEt3SiH
=~ N N N F
THF ~j ~tz >==O CHZCI2
OMe N,N
55 H
F F PF: ` N02
Boc2O ,
TEA NCJ~ F
H2N N N F H2N N` N
O CHOCN N~N O IiH I DMF
~ 1
N DCM Boc
56 57
F
F
Np~ NH2 (MeO)3CH
N H IN NF NazS~a N N
pTsOH
N~~ THF / H20
Y I/ ~/ ~p M
eOH
=
CN 58 Boc CN 59 Boc
F PFF F \
Nz FA ! C H 1 /
N N ~ Zt ~$ix N N F
1~ NC H
NC
60 Boc 61
42
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00132] 1V2-(2,4-Dimethoxybenzyl) 1V¾-(2,6-difluorobenzyl)-5-nitropyriniidine-
2,4-
diamine (53). 2,6-Difluorobenzylamine (0.24 mL) was added dropwise over one
min. to a
solution of 2,4-dichloro-5-nitropyrimidine(40) (0.388 g) and DIEA (0.77 mL) in
THF in a
cold bath set to -78 C. The reaction mixture was stirred for a further 15 min
at -78 C then
removed from the cold bath and allowed to warm to RT. Additional DIEA (0.77
mL) was
added to the reaction intermediate (N-(2,6-difluorobenzyl)-2-chloro-5-
nitropyrimidin-4-
amine) (52) followed by the addition of 2,4-dimethoxybenzylamine (0.30 mL) and
the
resulting mixture was stirred at RT overnight. Purification via column
chromatography
(eluted with 1 and 2.5 % MeOH/DCM) gave N2-(2,4-dimetho~.~ybenzyl),1V4-(2,6-
difluorobenzyl)-5-nitropyrimidine-2,4-diamine (53) (0.80 g), MH+ = 432.
[00133] 2-(2,4-Dimethoxybenzylamino)-9-(2,Crdifluorobenzyl)-7H-purin-8(9H)-one
(55). Raney Ni was added to a solution of Na-(2,4-dimethoxybenzyl)-N4-(2,6-
difluorobenzyl)-5-nitropyrimidine-2,4-diamine (0.80 g) in THF (50 mL) under
argon flush.
The suspension was evacuated, charged with hydrogen (balloon) and stirred for
16 hr. The
resulting mixture was filtered through a celite plug that was thoroughly
rinsed with THF
and MeOH to yield N2-(2,4-dimethoxybenzyl)-N4-(2,6-difluorobenzyl)pyrimidine-
2,4,5-
triamine (54) that was used as such in the next reaction.
[00134] Carbonyldiimidazole (0.93 g) was added to a solution of N2-(2,4-
dimethoxybenzyl)1V4-(2,6-difluorobenzyl)pyrimidine-2,4,5-triamine (54) in THF
(20 mL)
and the resultant mixture stirred at RT overnight, then the solvents were
removed under
reduced pressure and the taken up in EtOAc and washed trice with water. The
organics
were dried, filtered and evaporated and purified via column chromatography,
elution with
2.5 and 4 % MeOH/DCM, to yield 2-(2,4-Dimethoxybenzylamino)-9-(2,6-
difluorobenzyl)-
7H-purin-8(9H)-one (55) (0.58 g), MH+ = 428.
[00135] tert-Butyl9-(2,6-difluorobenzyl)-2-amino-8-oxo-8,9-dihydropurine-7-
carboxylate (57). A 1:1 solution of TFA/DCM (10 mL) was added to 2-(2,4-
dimethoxybenzylamino)-9-(2,6-difluorobenzyl)-7H-purin-8(9H)-one (55) (0.58 g)
and
43
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
stirred for 30 min, after which triethylsilane (2 mL) was added and the
mixture was stirred
an additional 4 hr. The solvents were removed under in vacuo, the residue was
taken up in
minimal MeOH and triturated with Et20, to yield the TFA salt of 9-(2,6-
difluorobenzyl)-2-
amino-7H-purin-8(9H)-one (56) (0.55 g), MH+ = 278, as a salmon colored solid.
[00136] 9-(2,6-Difluorobenzyl)-2-amino-7H-purin-8(9H)-one (0.55 g) was
dissolved in a
mixture of MeOH/ACN/DCM (40 mL), Et3N (2 mL) and di-ter7-butyl dicarbonate
(0.61 g)
were added and the mixture was stirred at RT overnight. The reaction solvents
were
removed and the crude material was taken up in DCM and washed with H20,
evaporated
and purified via column chromatography, elution with 2 and 3 % MeOH/DCM gave
the
titled product (57) (0.36 g), MH+ = 378, MH+-Boc = 278 (major), (M +Na)+ = 400
and
(2M + Na){ = 777 were also observed.
[00137] tert-Butyl 9-(2,6-difluorobenzyl)-2-(5-cyano-2 nitrophenylamino)-8-ogo-
8,9-
dihydropurine-7-carboxylate (58). Sodium hydride (88 mg, 95%) was added, under
argon flush, to a solution of tert-butyl9-(2,6-difluorobenzyl)-2-amino-8-oxo-
8,9-
dihydropurine-7-carboxylate (57) (191 mg) and 3-fluoro-4-nitrobenzonitrile
(415 mg) in
DMF (5 mL) at -40 C. The reaction mixture was allowed to warm to -20 C over
3 hr
then quenched by the addition of sat. aq. NH4C1, once at RT the mixture was
diluted with
EtOAc and separated. The organics were washed with brine (3 x), dried,
filtered and
evaporated, purified via column chromatography, (eluted with DCM and 1 and 2.5
%
MeOH/DCM) to yield tert-Butyl 9-(2,6-difluorobenzyl)-2-(5-cyano-2-
nitrophenylamino)-
8-oxo-8,9-dihydropurine-7-carboxylate (58) (288 mg), MH+ = 524.
[00138] tert-Buty19-(2,6-difluorobenzy~-2-(6-cyano-1H=benzo[d]imidazol-1-yl)-8-
ogo-8,9-dihydropurine-7-carbogylate (60). A freshly prepared solution of
sodium
hydrosulfite (tech, 1 g) and sodium bicarbonate (0.5 g) in H20 (10 mL) was
added to a
solution ofthe above nitro compound (58) (288 mg) in THF (10 mL). The mixture
was
stirred vigorously for 5 min., extracted with DCM (3 x), the combined organics
were
washed with brine, dried, filtered and concentrated to yield the intermediate
tert-butyl 9-
44
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
(2,6-difluorobenzyl)-2-(2-amino- 5-cyanophenylamino)- 8-oxo-8,9-dihydropurine-
7-
carboxylate (59) that was used as such in the next step.
[00139] A catalytic amount ofpara-toluene sulfonic acid monohydrate was added
to a
solution ofthe above amine intermediate and trimethyl orthoformate (3 mL) in
MeOH (10
mL). After 1 hr the crude material was adsorbed onto silica gel and purified
by column
chromatography (eluted with 1 and 2 lo MeOH/DCM) to yield tert-Buty19-(2,6-
difluorobenzyl)-2-(6-cyano-lH-benzo [dJ imidazol-1-yl)-8-oxo-8,9-dihydropurine-
7-
carboxylate (60) (164 mg), MH+ = 504 and MH+-BOC = 404.
[00140] 3-(9-(2,6-Difluorobenzyl)-8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-
benzo[d]itnidazole-5-carbonitrile (61). A 1;1 solution of TFA/DCM (10 mL) was
added
to tert-Buty19-(2,6-difluorobenzyl)-2-(6-cyano-lH-benzo[d]imidazol-1-yl)-8-oxo-
8,9-
dihydropurine-7-carboxylate (60) and stirred for 1 hr. The solvents were
removed en
vacuo and the resulting solid was triturated with Et2(?, and suspended in 6N
HCI. Removal
of solvents and trituration with Et20 of the resulting solid gave the titled
compound (61)
(68 mg) as a HCI salt, MH+ = 404,1H NMR (d6-DMSO) S 11.8 (s, 1H), 9.2 (s, 1H),
8.8 (s,
1H), 8.0 (s, 1H), 7.9 (d, 1H), 7.8 (broad s, 1H), 7.4 (d, 1H), 7.4 (quintet,
1H), 7.1 (m, 2H),
5.2 (s, 2H) ppm, 19F NMR 8 -114.3 (m).
[00141] Example 21: Synthesis of 3-(8-Oxo-9-(tetrahydro-2H-pyran-4-yl)-8,9-
dihydro-7H-purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile (62).
0
N~A
N N,~
Y~N~O
v N
NC -'_H
62
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00142] The title compound can be synthesized using the same procedures as
described
for the synthesis of 3-(9-(2,6-difluorobenzyl)-8-oxo-8,9-dihydro-7H-purin-2-
yl)-3H-
benzo[dlimidazole-5-carbonitrile (61, Example 20).
[00143] 1H NMR (ds-DMSO)11.71(s, 1H), 9.34 (s, 1H), 8.94 (d, J= 1.5 Hz, 1H),
8.38 (s,
111), 8.00 (d, J = 8.1 Hz, 1H), 7.79 (dd, J = 8.1, 1.5 Hz, 1H), 4.57 (m, 1H),
4.04 (m, 2H),
3.50 (m, 2H), 2.59 (m, 2H), 1.79 (m, 2H); Mass (MH+) 362.1.
[00144] Example 22: Regiospecffic synthesis of an oxoimidazopyridine
derivative:
Synthesis of 3-(2-oxo-3-((R)-1-(pyridin-3 yl)ethyl)-2,3-dihydro-lH-imidazo[4,5-
b]pyridin-5-yl)-3H-benz,o[d]imidazole-5-carbonitril.e.
46
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
~ MeO ~õ OMe
H2N N
H2N I
CI I N CI CI t.NO2 N N ~., N \
NOa TEA/THF 63 TEA/THF
46
Me0 MeO / Boc H / ~
~. ~ N N N Boc2O / DMAP \( N N N \ N
OMe CH2CI2 OMe N
Oz
64 NO2 65
Me
Boc N N \ N CDI H Na2S20d ~
THF / /H MOZ eOH OMe ~~ THF / 50 C
NHZ
66
C Meo N~ ~ f
~c N TFA/Et3SiH HzN (~ N ~~O
N N>.. K2CO3
OMe ~~ N~O CH2CI2 O CH3CN
67 H 68
~
N!! NOz N~ /
NOZ H
H N N N NC F N N N Na2S2O4
2 >=0 IAH 1 DMF = I~ *~~!/~` ~O THF / H20 / MeOH
~N N %
Boc CN 70 Boc
69
l / l
N\ N N NH2 N (MeO)3CH N $(N)N TFA/ CHZCpTsOH ,N
v~~THF NC Boc NC ' H
CN 71
72 73
[00145] (R)-N6-(2,4-Dimethoxybenzyl)-3-nitro-N2-(1-(pyridin-3-ynethyl)pyridine-
2,6-diamine (64). A solution of 2,6-dichloro-5-nitropyridme (46) (0.5 g) in
THF (20 mL)
was cooled to 0 C and treated with 1.6 mL triethylamine followed by (R)-1-
pyridin-3-yl-
47
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
ethylamine (300 L). The mixture was stirred for 1.5 h, then warmed to RT and
stirred
another 20 h. 2,4-Dimethoxybenzylamine (0.8 mL) was added and the m`ixture was
heated
at 50 C for four hours. The mixture was diluted with ethyl acetate and washed
twice with
saturated sodium chloride solution. The organic layer was separated, dried
over sodium
sulfate, filtered, and concentrated. Column chromatography (50 4 100% ethyl
acetate in
hexanes) provided 761 mg of (R)-N6-(2,4-Dimethoxybenzyl)-3-nitro-N2-(1-
(pyridin-3-
yl)ethyl)pyridine-2,6-diamine (64).
[00146] (R)-tert-Butyl2,4-dimethoaybenzyl(5-nitro-6-(1-(pyridin-3-
ynethylamino)pyridin-2 yl)carbanrnate (65). A solution of (R)-N6-(2,4-
dimethoxybenzyl)-3-nitro-N2-(1-(pyridin-3-yl)ethyl)pyridine-2,6-diamine (64)
(367 mg) in
methylene chloride (20 mL) was treated with di-tert-butyl dicarbonate (1.0 g)
and 4-
dirnethylaminopyridlne (22 mg). The mixture was stirred for 16 h and
concentrated in
vacuo. Column chromatography (50 4 100% ethyl acetate iin hexanes) provided
500 mg of
(R) tert-Buty12,4-dimethoxybenzyl(5-nitro-6-(1-(pyridin-3-
yl)ethylamino)pyridin-2-
yl)carbamate (65).
[00147] (R)-tert-Buty12,4-dimethoxybenzyl(2-ogo-3-(1-(pyridin-3-yl)ethyl)-2,3-
dihydro-lH-imidazo[4,5-b]pyridin-5-yl)carbamate (67). A solution of (R)-tert-
butyl
2,4-dimethhoxybenzyl(5-nitro-6-(1-(pyridin-3-yl)ethylamino)pyridin-2-
yl)carbamate (500
mg) in THF (25 mL) was treated with an aqueous solution comprised of 2 g of
Na2S2O4
and 1 g Na.HCO3 in 20 mL of water followed by 1 mL of methanol. The mixture
was
stirred for 30 minutes, then diluted with ethyl acetate and washed with
saturated sodium
chloride solution. The organic layer was separated, dried over sodium sulfate,
filtered, and
concentrated to provide the intermediate (R)-tert-buty12,4-dimethoxybenzyl(5-
amino-6-(1-
(pyridin-3-yl)ethylamino)pyridin-2-yl)carbamate (66). The intermediate was
dissolved in
THF (50 mL) and treated with 1,1'-carbonyldiirnidazole (0.5 g) at 50 C for 20
h. The
mixture was concentrated and purified by column chromatography (2 4 5% MeOH in
methylene chloride) to provide 413 mg of(R)-tert-Butyl 2,4-dimethoxybenzyl(2-
oxo-3-(1-
(pyridin-3-yl)ethyl)-2,3-dihydro-lH-imidazo[4,5-b]pyridin-5-yl)carbamate (67).
48
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00148] (R)-tert-Butyl5-amino-2-oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-
dihydroimidazo[4,5-b]pyridi.ne-l-carboxylate (69). A solution of (R)-tert-
butyl 2,4-
dimethoxybenzyl(2-oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-dihydro-1 H=imidazo [4,5-
b]pyridin-
5-yl)carbamate in methylene chloride (15 mL) was treated with TFA (15 mL) and
triethylsilane (1:0 mL) for one hour. The mixture was concentrated to provide
the
intermediate (R)-5-amino-3-(1-(pyridin-3-yl)ethyl)-1H-imidazo[4,5-b]pyridin-
2(3H)-one
(68), which was dissolved in acetonitrile (50 mL) and stirred vigorously with
di-ter7-butyl
dicarbonate (1.0 g) and potassium carbonate (3.0 g) for 2 h. Methylene
chloride (200 mL)
and water (100 mL) was added and the organic layer was separated. The aqueous
layer was
extracted with another 100 mL of methylene chloride. The combined organic
layers were
separated, dried over sodium sulfate, filtered, and concentrated. Column
chromatography
(2 4 3-> 4% MeOH in methylene chloride) provided 235 mg (R)-tert-Butyl 5-amino-
2-
oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-dihydroimidazo[4,5-b]pyridine-1-carboxylate
(69).
[00149] (R)-tert-Buty15-(5-cyano-2-nitrophenylamino)-2-oxo-3-(1-(pyridin-3-
y~ethyl)-2,3-dihydronnidazo[4,5-b]pyridine-l-carboarylate (70). A solution of
(R)-tert-
butyl 5-amino-2-oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-dihydroimidazo [4,5-
b]pyridine-l-
carboxylate (94 mg) and 3-fluoro-4-nitrobenzonitrile (225 mg) in DMF (6 mL)
was cooled
to -25 C and treated with NaH (60% w/w in mineral oil, 75 mg) and slowly
allowed to
warm to -15 C. The mixture was stirred for four hours between -20 C and -15
C, then
diluted with EtOAc and quenched with saturated ammonium chloride solution. The
organic phase was washed three times with brine, separated, dried over sodium
sulfate,
filtered, and concentrated. Column chromatography (2% MeOH in methylene
chloride)
provided 100 mg (R)-tert-Butyl 5-(5-cyano-2-nitrophenylamino)-2-oxo-3-(1-
(pyridin-3-
yl)ethyl)-2,3-dihydroimidazo[4,5-b]pyridine-1-carboxylate (70).
[00150] tert-Butyl5-(6-cyano-lH-benzo[d]imidazol-1-yl)-2-oxo-3-((R)-1{pyridin-
3-
yl)ethyl)-2,3-dihydroimidazo[4,5-b]pyridine-l-carboxylate (72). A solution of
(R)-tert-
butyl 5-(5-cyano-2-nitrophenylamino)-2-oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-
dihydroimidazo[4,5-b]pyridine-1-carboxylate (70) (100 mg) in THF (5 mL) was
treated
49
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
with an aqueous solution comprised of 0.5 g of Na2S2O4 and 0.25 g NaHCO3 in 5
mL of
water. The mixture went quickly from a red color to a slightly yellow color,
which
indicated reduction of the nitro group. The mixture was diluted with ethyl
acetate and
washed with saturated sodium chloride solution. The organic layer was
separated, dried
over sodium sulfate, filtered, and concentrated to provide the intermediate
(R) tert-butyl5-
(2-amino-5-cyanophenylamino)-2-oxo-3-(1-(pyridin-3-yl)ethyl)-2,3-
dihydroimidazo [4,5-
b]pyridine-l-carboxylate (71). The intermediate was dissolved in THF (5 mL),
DMF (1
mL), and trimethylorthoforma.te (2 mL). The mixture was treated with 10 mg of
p-
toluenesulfonic acid and stirred for 20 h. The mixture was diluted with ethyl
acetate and
washed once with saturated sodium bicarbonate and twice with saturated NaCI
solution.
The organic layer was separated, dried over sodium sulfate, filtered, and
concentrated.
Column chromatography (2% MeOH in methylene chloride) provided 57 mg tert-
Buty15-
(6-cyano-lH-benzo [d]imidazol-l-yl)-2-oxo-3-((R)-1-(pyridin-3-yl)ethyl)-2,3-
dihydroimidazo[4,5-b]pyridine-l-carboxylate (72).
[00151] 3-(2-ogo-3-((R)-1-(pyridin-3-yl)ethyl)-2,3-dihydro-lH-imidazo[4,5-
b]pyridin-5-yl)-3H-benzo[d]imidazole-5-carbonitrile (73). A solution oftert-
butyl5-(6-
cyano-lH-benzo[d]imidazol-1-yl)-2-oxo-3-((R)-1-(pyridin-3-yl)ethyl)-2,3-
dihydroimidazo[4,5-b]pyridine-l-carboxylate (72) (57 mg) in methylene chloride
(1 mL)
was treated with TFA (1 mL) for one hour. The mixture was concentrated and the
resulting
TFA salt was converted to the HCl salt by dissolving it in 5 mL EtOH and
adding 0.5 mL
of conc. HCl, then concentrating the solution in vaciso. The process was
repeated and the
resulting residue was dissolved in a minimum amount of methanol and triturated
with the
addition of ethyl ether. After 3 triturations, 3-(2-oxo-3-((R)-1-(pyridin-3-
yl)ethyl)-2,3-
dihydro-lH-imidazo[4,5-b]pyridin-5-yl)-3H-benzo[d]imidazole-5-carbonitrile
(73) HCl
salt (39 mg) was isolated as a tan colored solid: 'H-NMR (CD3OD) S 9.9 (br s,
1H), 9.2 (s,
1 H), 9.0 (m, 2H), 8. 5(s, 1 H), 8.3 (m, 1 H), 8.2 (m, 1H), 8.1 (d, 1 H), 7.9
(d, 1 H), 7. 8(d,
1H), 6.3 (q, 1H), 2.3 (d, 3H).
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[001521 Example 24. Synthesis of 2-(1H-Benzo[d]nnidazol 1-yl)-9-(cis-3-methyl-
tetrahydro-2H-pyran-4-yl)-7H-purin-8(9H)-one.
0
NH2 c
CI N CI DIEA CI N NH BenzImld=
N,,N -- `r ~ --~
N02 THF N NO K2C03
2 THF
CIS
O
N C 1. Na2S204
NaHCO3 N
~ (rc~
N N[i NH THF / H20 / MeOH N N
.-- ~ ~ ~
NO 2. CDI / THF N J~ N O
z v 'H
[00153] 2-Chloro-N-(cis-3--methyl-tetrahydro-2H-pyran-4-yl)-5-nitropyrimidin-4-
amine. To a suspension of 0.24 g of the hydrochloride salt of czs-3-methyl-
tetrahydro-2H-
pyran-4-aniine (WO 2004/041161) and DIEA (1.5 mL) in THF (10 mL) at -78 C was
added 2,4-dichloro-5-nitropyrimidine (0.72 g). The rnix~ture was allowed to
slowly reach
room temperature and stirred for 16 hours. The mixture was diluted with EtOAc
and
washed 3 tirnes with brine. The organic layer was separated, dried over sodium
sulfate, and
concentrated in vacuo. Column chromatography (20 4 40% EtOAc / hexanes)
provided
289 mg ofthe title conipound.
[001541 2-(1H-Benzo[d]imidazol 1-yl)-N-(cis-3 methyl-tetrahydro-2H-pyran-4-yl)-
5-
nitropyrimidin-4-amine. To a solution of 2-chloro-N-(cis-3-methyl-tetrahydro-
2H-pyran-
4-yl)-5-nitropyrimidin-4-amine (115 mg) in acetonitrile (5 mL) was added
potassium
carbonate (300 rng) and benzimidazole (150 mg). The mixture was stirred at 70
C for 2.5
hours. After diluting with 70 mL EtOAc, the mixture was washed with brine,
dried over
sodium sulfate, and concentrated in vacuo. Column chromatography (50 4 100%
EtOAc /
hexanes) provided 99 mg of the title compound.
51
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00155] 2-(1H-Benzo[d]imidazol 1-yl)-9-(cis-3-methyl tetrahydro-2H-pyran-4-yl)-
7H-purin-8(9H)-one. To a solution of 2-(1H-benzo[d]imidazol-1-yl)-N-(cis-3-m(,-
thyl-
tetrahydro-2H-pyran.-4-y1)-5-nitropyrunidin-4-amine (51 mg) in THF (10 mL) was
added a
solution of sodium hydrosulfite (300 mg) and sodium bicarbonate (150 mg) in
water (10
mL). The mixture briefly became blue followed by colorless. Methanol (1 mL)
was added
to maintain the homogeneity of the solution. The mixture was diluted with 70
mL EtOAc
and washed twice with brine. The aqueous washes were extracted with another 50
mL of
EtOAc and then the combined organic layers were dried over sodium sulfate and
concentrated invacuoto provide 2-(1H-benzo[d]imidazol-1-yl)-N4-(cis-3-methyl-
tetrahydro-2H-pyran-4-yl)pyrimidine-4,5-diamine. The diamine intermediate was
dissolved in THF (5 mL) and treated with 1,1'-carbonyldiimidazole (80 mg) at
50 C for
16 hours. The mixture was diluted with 50 mL EtOAc and washed 3 times with
brine. The
organic layer was separated, dried over sodium sulfate, and concentrated in
vacuo. Column
chromatography (2 4 4% MeOH / DCM) provided 19.3 mg of the title compound. 'H-
NMR (300 MHz, 5% CD3DD in CDC13) S 8.9 (s, 1H), 8.5 (d, 1H), 8.2 (s, 1H), 7.8
(d, 1H),
7.4 (t, 1H), 7.3 (t, 1H), 4.7 (m, 1H), 4.2 (d(br), 1H), 3.9 (d, 11-1), 3.7 (d,
1H), 3.5 (n-4 2H),
2.3 (t(br), 1H), 1.8 (d(br), 1H), 1.2 (d, 3H).
[00156] Example 25.
O F
N-.~ `:
~
CI NNN N ~O
.,.,. ~
N
CI H
[00157] 2-(5,6-Dichloro-lH-benzo[d]imidazol-1-yl)-9-((R)-S-fluorochroman-4-yn-
7H-
purin-S(9H)-one. A solution of (R)-2-chloro-N-(8-fluorochroman-4-yl)-5-
nitropyrimidin-4-amine in acetonitrile was treated with 5,6-
dichlorobenzimidazole and
52
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
potassium carbonate. The mixture was stirred at reflux for 6 hours, cooled to
room
temperature, diluted with 150 mL of EtOAc, and washed twice with 30 mL
portions of
water. The organic layer was separated, dried with magnesium sulfate,
filtered, and
concentrated in vacuo. Purification by column chromatography (2% MeOH/DCM)
gave
the intermediate nitropyrimidinamine. The title compound was synthesized from
the
intermediate nitropyrimidinamine via the procedures described in Example 24.
1H-NMR
(300 MHz, CDC13) 6 8.7 (s, 1H), 8.3 (s, 1H), 8.2 (s, 1H), 7.8 (t, 1H), 7.0 (t,
1H), 6.6 (m,
2H), 5.9 (t, 1H), 4.6 (m, 1H), 4.4 (m, 1H), 3.0 (m, 1H), 2.3 (m, 1H).
O F
N` ';
~---
! NN NO
J" _ f
H
[00158] 2-(5,6-Dimethyl-lH-benzo[d]imidazol-1-yl)-9-((R)-8-fluorochroman-4-yn-
7H-purin-8(9H)-one. The title compound was synthesized from 5,6-
dichlorobenzimidazole via the procedures described in Example 25;1H-NMR (300
MHz,
CDC13) S 9.6 (s, 1H), 8.8 (s, 11-1), 8.3 (s, 1H), 8.0 (s, 1H), 7.6 (s, 1H),
7.0 (t, 1H), 6.7 (m,
1H), 5.9 (t, 1H), 4.7 (m, 1H), 4.5 (m, 1H), 3.2 (m, 2H), 2.4 (d, 6H).
r0 F F
N , f ~ N_ ', / ~
N N\N f ~ N N
N N~O F3C 6 ~ N YN~O
F3C ~H =.~^"H
53
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00159] 9-((R)-B-Fluorochroman-4-yl)-2-(6-(trifluoromethyl)-1H-
benzo[d]imidazol-l-
yl)-7H-purin-8(9H)-one and 9-((R)-8-fluorochroman-4-yl)-2-(5-(trifluoromethyl)-
1H-
benzo[d]imidazol-1-y1)-7H-purin-8(9H)-one. The title compound was synthesized
from
5-trifluoromethylbenzimidazole (US 2004/0087601) via the procedures described
in
Example 25 Purification by column chromatography (2% MeCH/DCM) eluted the 6-
trifluoromethyl isomer first ('H-NMR (300 MHz, CDC13) & 8.8 (d, 2H), 8.4 (s,
1H), 7.9 (d,
1H), 7.6 (d, 2H), 7.0 (t, 1H), 6.7 (ni, 2H), 5.9 (t, 1H), 4.7 (m, 1H), 4.4
(ni, 1H), 3.0 (m,
1H), 2.4 (m, 1H).) followed by the 5-trifluoromethyl isomer (1H-NMR (300 MHz,
CDC13)
S 9.0 (s, 1H), 8.4 (s, 1H), 8.1 (s, 1H), 8.0 (d, 1H), 7.6 (d, 2H), 7.0 (m,
1H), 6.8 (ni, 2H),
5.9 (t, 1H), 4.7 (m, 1H), 4.4 (m, 1H), 2.9 (m, 1H), 2.4 (in, 1H).
F
Q F
N ~ I ~ ~O 1
N Jc02 .,~ Nf \ `ll ~
r "~
N02
[00160] N-((R)-B-Fluorochroman-4-y1)-2-(3H-imidazo[4,5-c]pyridin-3-yl)-5-
nitropyrimidin-4-a.mine and N-((R)-8-fluorochroman-4-yl)-2-(1H-imidazo[4,5-
c]pyridin-1-yl)-5-nitropyrimidin-4-amine. The title compound was syntheszzed
from 5-
azabenzimidazole via the procedure described in Example 25. Purification by
column
chromatography (1% MeOH / DCM) provided N-((R)-8-fluorochroman-4-yl)-2-(3H-
imida2o[4,5-c]pyridin-3-yl)-5-nitropyrimidin-4-amine as the first eluting
isomer: (1H-
NMR (300 MHz, CDC13) S 9.8 (s, 11-1), 9.4 (s, 1H), 9.2 (s, 1H), 8.9 (d, 1H),
8.6 (d, 1H),
7.8 (d, 111), 7.1(n4 2H), 6.9 (ni, 1H), 5. 8(q, 1 H), 4.6 (m, 1 H), 4.4 (m, 1
H), 2.6 (m, 1 H),
2.4 (m, 1H).). N-((R)-8-Fluorochroman-4-yl)-2-(1H-imidazo[4,5-c]pyridin-1-yl)-
5-
nitropyrimidin-4-amine eluted second: ('H-NMR (300 MHz, CDC13) S 9.4 (s, 1H),
9.2 (s,
-54
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
1H), 9.1 (s, 1H), 8.9 (d, 1H), 8.6 (d, 1H), 8.4 (d, 1H), 7.1 (m, 2H), 6.9 (m,
1H), 5.7 (q, 1H),
4.5 (m, 1 H), 4.4 (m, 1H), 2.6 (m, 1H), 2.4 (xn, 1 H). ).
N_
0 F
~ ~--
~ \ N N N
Y~ ~o
Nr N ..~ N
H
[00161] 9-((R)-8-fluorochroman-4-yl)-2-(3H-imidazo[4,5-c]pyrid.in-3-yl)-7H-
purin-
8(9Ii)-one. The title compound was synthesized from N-((R)-8-fluorochroman-4-
yl)-2-
(3H-imida.zo[4,5-c]pyridin-3-yl)-5-nitropyrimidin-4-amine via the procedures
described in
Example 24. 'H-NMR (300 MHz, CDC13) S 9.8 (s, 1H), 9.4 (s, 1H), 8.6 (d, 1H),
8.3 (m,
2H), 7.0 (t, 1H), 6.7 (m, 2H), 5.9 (t, 1H), 4.6 (m, 1H), 4.4 (m, 1H), 2.8 (m,
1H), 2.4 (m,
1 H).
0 F
N_
~
Nf N Y, N~ N~
N~ O
N
H
[00162) 9-((R)-S-fluorochroman-4-yl)-2-(1H-imidazo[4,5-c]pyridin-l-yl)-7H-
purin-
8(91i)-one. The title compound was synthesized from N-((R)-8-fluorochroman-4-
yl)-2-
(1H-imida2o[4,5-c]pyridin-1-yl)-5-nitropyrimidin-4-atnine via the procedures
described in
Example 24. 1H-NMR (300 MHz, CDC13) S 9.3 (d, 2H), 8.4 (d, 1H), 8.3 (d, 21-1),
7.0 (t,
1H), 6.7 (rn, 2H), 5.9 (t, 1H), 4.6 (m, 1H), 4.4 (in, 1H), 2.8 (rn, 1H), 2.4
(m, 1H).
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00163] Example 26. Synthesis of 3-(9-((R)-6,8-clifluorochroman-4-y1)-8-ogo-
8,9-
dihydro-7H-purin 2-yl)-3H-benzo[d]imidazole-5-carbonitrile:
OMe .' OMe
I~ F H2N DIEA \ N ~. ~
141
NC NO2 THF # ~ OMe
OMe NC NO2
[00164] 4-(2,4-dimethoxybenzylamino)-3-nitrobenzonitrile. A solution of 4-
fluoro-3-
nitrobenzonitrile (5.0 g) in THF (100 mL) was treated with DIEA (6.3 mL) and
2,4-
dimethoxybenzylamine (5.0 mL), and then stirred for 24 h. The solvent was
evaporated
and the crude mixture was dissolved in EtOAc (100 mL). The solution was washed
once
with 1 M HCI and twice with saturated aqueous NaCI (100 mL each). The organic
layer
was separated, dried over Na2SO4, filtered, and concentrated in vacuo. Column
chromatography (20% EtOAc / DCM) provided 9.25 g of the title conipound.
pMe OMe
~ I
Na2S2O4 H
~ N ~., NaHCO3 ~ N ~
~~ OMe THF / Hz I,~ OMe
NC N02 MeOH NC NH2
[00165] 4-(2,4-dimethoxybenzylamino)-3-amuiobenzonitrile. A solution of 4-(2,4-
dimethoxybenzylamino)-3-nitrobenzonitrile (4.54 g) in THF (400 mL) was treated
with a
solution of sodium hydrosulfite (20 g) and sodium bicarbonate (10 g) in
distilled water
(350 mL). Enough methanol was immediately added (50 mL) to maintain a
homogeneous
solution. After 15 minutes, EtOAc (500 mL) and saturated aqueous NaCI (500 mL)
were
added and the organic layer was separated, The aqueous layer was extracted
again with
400 mL EtOAc. The combined organic layers were washed with saturated aqueous
NaCI
(500 mL) and separated. The organic phase was dried over Na2SO4, filtered, and
concentrated in vacuo to provide 4.33 g of the title compound.
56
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
r OMe
OMe CI ~ N ~ ~
H
NN I~2CO3 ~ / OMe
NC NH
NC NH OMe SCN CH3CN
2 NO2 N ~ N
~SCN
NOZ
[00166] 4-(2,4-dimethoxybenzylainino)-3-(5-nitro-4-thiocyanatopyximidin-2-
ylamino)benzonitrile. A solution of 4-(2,4-dimethoxybenzylaniino)-3-
aminobenzonitrile
(3.9 g) in acetonitrile (100 mL) was cooled to 0 C and treated with potassium
carbonate
(6.3 g) followed by a solution containing 3 g of 2-chloro-5-nitro-4
thiocyanatopyrimidine
(WO 2003/032994) in acetonitrile (50 mL). The mixture was stirred for 30
minutes at 0 C
and 30 minutes at room temperature resulting in the formation of a
precipitate. The mixture
was quenched at 0 C by the addition of 4 o acetic acid (150 mL) and filtered.
The
precipitate was swirled in 100 niL acetonitrile and filtered again. The
precipitate was
washed with acetonitrile, which resulted in the slow dissolution of product
into the filtrate.
After air-drying, 1.5 g ofthe title compound remained as the precipitate cake.
The filtrate
was extracted with EtOAc, dried over Na2SO4, filtered, and concentrated in
vacuo. Column
chromatography (0320% EtOAc / DCM) and recrystallization from acetonitrile
provided
0.415 g of additional title compound.
OMe OMe
H ~ N F ~ H
,~ ~
`/ OMe KZC03 I~ OMe H N C (0, N H
N~ N F CH3CN / NJN -'O
Y S~ NH2 DMSO Yl- N F
NO2 NOZ H
F
[001671 (R)-4-(2,4-dimethoxybenzylamino)-3-(4-(6,8-difluorochroman-4-ylamino)-
5-
nitropyrimidin-2-ylamino)benzonitrile. A partial suspension of 4-(2,4-
57
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
dimethoxybenzylamino)-3-(5-nitro-4 thiocyanatopyrimidin-2-ylamino)benzonitrile
(415
mg) in 40 mL of acetonitrile was treated with a solution 9f (R)-6,8-
difluorochroman-4-
amine HC1 salt (320 mg) in DMSO (10 mL) followed by potassium carbonate (1.0
g). The
mixture was stirred for 24 hours, and then diluted with EtOAc (200 mL). The
mixture was
washed once with saturated aqueous ammonium chloride (200 mL) and 3 times with
saturated aqueous NaCI (200 mL each). The organic layer was separated, dried
over
Na2SO4, filtered, and concentrated in vacuo. Column chromatography (20440%
EtOAc /
hexanes) provided 358 mg of the title compound.
OMe
OMe OMe H
N ` ~ N \ I , ~.~N
\ Na2Sa04 ~ (~~j` OMe
NC I f NH OMe NaHC03 NC '~ NH OMe CDI NC N~H
THF/H20 THF N~'N
N N l~O MeOH NN -"0
N F H~~[N F
Y N F I
2 H \\
NOZ H NH
O
F F F
[00168] (R)-4-(2,4-dimethoxybenzylamino)-3-(9-(6,8-difluorochroman-4-yl)-8-ogo-
8,9-dihydro-7H-purin-2-ylamino)benzonitrile. A solution of (R)-4-(2,4-
dimethoxybenzylamino)-3-(4-(6, 8-difluorochroman-4-ylamino)-5-nitropyrimidin-2-
ylamino)benzonitrile (358 mg) in THF (25 mL) was treated with a solution of
sodium
hydrosulfite (1.5 g) and sodium bicarbonate (1<5 g) in 20 mL of distilled
water. Methanol
(5 mL) was added to maintain a homogeneous solution. After 15 minutes, the
mixture was
diluted with EtOAc (100 mL) and washed with saturated aqueous NaCI (2 X 100
mL). The
organic layer was separated, dried over Na2SO4, filtered, and concentrated in
vacuo to
provide the intermediate (R)-4-(2,4-dimethoxybenzylamino)-3-(5-amino-4-(6,8-
difluorochroman-4-ylamino)pyrirnidin-2-ylamino)benzonitrile. The intermediate
was
dissolved in THF (5 mL) and treated with carbonyldiimidazole (0.55 g) for 16
hours. The
58
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
mixture was diluted with EtOAc (100 mL) and washed twice with saturated
aqueous NaCI
(2 X 100 mL). The organic layer was separated, dried over Na2SO4, filtered,
and
concentrated in vacuo. Column chromatography (24 3% MeOH / DCM) provided 230
mg
of the title compound.
OMe
N 1 ~ NH2 -- ~ - N
a TFANC ~/ NH (Me )3CH NC `-NNC" NH OMe Et3SiH p-TsOH
~ JN~N CHZCl~ N~ ~ THF N N
N
` I
N F HN-~ F
HN-o ~ 0-F O o F F
F
[00169] 3-(9-((R)-6,8-difluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-yl}-
3H-
benzo[d]imidazole-5-carbonitrile. A solution of (R)-4-(2,4-
dimethoxybenzylamino)-3-
(9-(6,8-difluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-
ylarnino)benzonitrile (230
mg) in DCM (5 mL) was treated with TFA (5 mL) and triethylsilane (1 mL) for 16
h. The
mixture was concentrated in vacuo to provide the intermediate (R)-4-amino-3-(9-
(6,8-
difluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-ylamino)benzonitrile. The
intermediate was dissolved in 5 niL THF and treated with 3 mL
trimethylorthoformate
followed by p-toluenesulfonic acid (3 mg). After 1 hour, the mixture was
diluted with
EtOAc (100 mL) and washed once with saturated aqueous sodium bicarbonate (100
mL).
The organic layer was separated, dried over Na2SO4, filtered, and concentrated
in vaeuo.
Column chromatography (504 100 1o EtOAc / hexanes) provided 78 mg of the title
compound.1H-NMR (300 MHz, 5% CD3OD in CDC13) S 8.8 (s, 1H), 8.7 (s, 1H), 8.2
(s,
1H), 7.8 (d, 1H), 7.6 (dd, 1H), 6.8 (td, 1H), 6.4 (dd, IH), 5.8 (dd, 1H), 4.6
(m, 1H), 4.4 (td,
1H), 2.9 (ni, 1H), 2.3 (m, 1H).
59
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
N`
---
~ N N N
N f N~U
NC H
[00170] 3-(9-((R)-chroman-4-yl)-8-ogo-8,9-dihydro-7H-purin-2 yl)-3H-
benzo[d]imidazole-5-carbonitrile. The title compound was synthesized from (R)-
chroman-4-amine via the procedures described in Example 26. 'H-NMR (300 MHz,
5%
CD30D in CDC13) S 8.8 (s, 1H), 8.5 (s, 1H), 8.2 (s, 1H), 7.8 (d, 1H), 7.5 (dd,
1H), 7.1 (m,
2H), 6.8 (d, 1H), 6.7 (td, 1H), 5.8 (dd, 1H), 4.5 (m, 1H), 4.3 (td, 1H), 2.8
(m, 1H), 2.3 (m,
1 H).
Q F
N 'r \
~
N N N
N ~, ~O
N
NC H
[00171] 3-[9-(8-Fluoro-chroman-4-yl)-8-ogo-8,9-dihydro-7H-purin-2-yl]-3H-
benzoimidazole-5-carbonitrile. The title compound was synthesized from (R)-8-
fluorochroman-4-amine via the procedures described in Example 26. 'H-NMR (300
MHz,
CDC13) 6 8.8 (s, 1H), 8.6 (s, 1H), 8.2 (s, IH), 7.8 (d, 1H), 7.6 (d, 1H), 7.0
(t, 1H), 6.6 (m,
2H), 5.8 (1, 1H), 4.6 (rn, 1H), 4.4 (m, 1H), 2.8 (nn, 1H), 2.4 (m, 1H). , ~
1 \
~ F
NNI N>--
N
NC H
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00172] 3-(9-((R)-6-fluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-yp-3H-
benzo[d]imidazole-5-carbonitrile. The title compound was synthesized from (R)-
6-
fluorochroman-4amine via the procedures described in Example 26. 'H NMR (300
MHz,
CDCl3+5%CD3OD): S 8.86 (s, 1H), 8.41 (s, 1H), 8.18 (s, 1H), 7.72 (d, 1H), 7.51
(d, 1H),
7.0-7.1(m, 1H), 6.8-6.9 (m, IH), 6.49 (dd, 1H), 5.76 (br t, 1H), 4.4-4.5 (m,
1H), 4.24 (br t,
1H), 2.7-2.9 (m, 111), 2.2-2.3 (m, 1H). Conditions for introduction of the
chromanyl amine
were improved as described below:
OMe ~Q OMe O
NH H F =k NH '~~ ~ =
H F
N N SCN NH2.HCi ~
Y N Y N NH
MeO i .
~ ~ - Meo
N/ N02 DIEA, DMSO N~XNO2
CN CN
[00173] (R)-4-(2,4-dimethogybenzylamino)-3-(4-(6-fluorochroman-4-ylamino)-5-
nitropyrimidin-2-ylamino)benzonitrile. A solution of 4-(2,4-
dimethoxybenzylamino)-3-
(5-nitro-4-thiocyanatopyrimidin-2-ylamino)benzonitrile (139 mg) in anhydrous
DMSO (3
mL) was added to solution of (R)-6-fluorochroman-4-amine hydrochloride (79 mg)
in
anhydrous DMSO (3 mL) and DIEA (0.21 mL), the resulting dark red solution was
stirred
at RT under an atmosphere of Ar over which time the solution lighten to
yellow. Upon
completion of the reaction, the mixture was cooled to 0 C with an ice bath,
and water (25
mL) was added (exotherm). The resulting yellow solid was collected via
filtration, washed
with additional water, air dried, then dissolved in CH2C12, the organic
solution was dried
(MgSO4), filtered and evaporated to yield the titled compound (quant.), NMR
CDCl3 1H S
9.0 (s, 1H), 8.6(d, 1H), 7.7 (br s, 1H), 7.4 (dd, 1H), 7.1 (d, 1H), 7.0-6.8
(m, 4H), 6.5-6.4
(m, 2H), 5.2 (br s, 1H), 4.3 (s, 2H), 4.2 (br s, 2H), 3.8 (s, 6H), 2.2 (br s,
111), 1.8 (br s, 1H);
19FS-123ppm;MH+=572.
[00174] This material was taken on using the same procedures outlined in
Example 26 to
give 3-(9-((R)-6-fluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-
benzo [ei`J imidazole-5-carbonitrile.
61
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
"`
I
~ `
N C F
~ ( N~O
N
NC H
[00175] 3-(9-((R)-7-fluorochroman-4-yiJ-B-oxo-8,9-dihydro-7H-purin-2 yl)-3H-
benzo[d]imidazole-5-carbonitrrle. The title compound was synthesized from (R)-
7-
fluorochroman-4-amine via the procedures described in Example 26. 'H NMR (300
MHz,
CDC13+5%CD3OD): S 8.86 (s, 1H), 8.58 (s, 1H), 8.19 (s, 1H), 7.79 (d, 1H), 7.56
(d, 1H),
6.7-6.9 (m, 2H), 6.4-6.5 (m, 1H), 5.78 (br t, IH), 4.5-4.6 (m, 1H), 4.32 (br
t, 1H), 2.7-2.9
(m, 1H), 2.2-2.4 (ni, 1H).
F
N~
N N
N i ~F
ON
NC H
[00176] 3-[9-(5,8-Difluoro-chroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-y1]-3H-
benzoimidazole-5-carbonitrile. The title compound was synthesized from (R)-5,8-
difluorochroman-4-amine via the procedures described in Example 26. 'H-NMR
(300
MHz, CDC13) S 8.8 (s, 11-1), 8.7 (s, 1H), 8.2 (s, 1H), 7.8 (d, 1H), 7.6 (d,
1H), 7.0 (m, 1H),
6.4 (m, 1H), 5.9 (t, 1H), 4.6 (in, 1H), 4.4(m, 1H), 2.5(m, 2H).
[00177] Example 27. Synthesis of 2-(6-fluoro-lH-benzo[d]imidazol-1-yl)-9-((R)-
S-
fluoro-chroman-4-yl)-7H-purin-8(9hI)-one:
62
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
BOC
N Hz I
~ (BOC)ZO 1, N'BOC TFA
~
F I'~ N02 DMAP, DCM F 1`~ NO DCM
2
BOC BOC
~ NH NazS2O4 / NaHCO3 ` NH
FI''~ NOz THF I H20 / MeOH F 'XNH2
[00178] 4-Fluoro-2-nitro-phenyl di-tert-butyl imidodicarbonate. A catalytic
amount of
DMAP was added to a mixture of 4-fluoro-2-nitrobenzenamine (0.78 g) and di-
terl-butyl
dicarbonate (2.18 g) in DCM (20 mL) and stirred at room temperature for 15 hr.
The
mixture was diluted with H20 and twice extracted with DCM, the combined
organics were
dried, filtered and evaporated to yield the bis-BOC material (quant). 'H-NMR
(300 MHz,
CDC13) S 7.8 (dd, 1H), 7.3 (in, 2H), 1.4 (s, 18H).
[00179] tert-Butyl4-fluoro-2-nitrophenylcarbamate. (procedure; Connell, R. D.;
Rein,
T.; Akermark, B.; Helquist, P. J. J. Org. Chem. 1988, 53, 3845) To a stirred
solution ofthe
Bis-BOC material in DCM (20 mL) was added TFA (0. 58 mL). After 3 hr the
reaction
was quenched with aq. NaHCO3 (5 mL), brine was added, the mixture separated
and
extracted with additional DCM. The combined organics were evaporated, purified
via
column chromatography (eluted with 7.5% EtOAc/Hex) to give the titled product
(1.12 g).
'H-NMR (300 MHz, CDC13) S 9.5 (br 1H), 8.5 (dd, 1H), 7.9 (dd, 1H), 7.3 (m,
1H), 15 (s,
9H).
[00180] tert-Butyl2-amino-4-fluorophenylcarbamate. To a solution of terl-butyl
4-
fluoro-2-nitrophenylcarbamate (0.34 g) in THF (30 mL) was added a premixed
solution of
sodium hydrosulfite (2 g) and sodium bicarbonate (1g) in water (50 mL). MeOH
(10 mL)
was also added to aid solution of the mixture, which was stirred at room
temperature for 30
min, when sodium chloride was added to saturate the solution. The resultant
mixture was
extracted with EtOAc (2x). The combined organics were dried, filtered and
evaporated to
63
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
yield the titled compound (quant) that was used as such for the next step. 1H-
NMR (300
MHz, CDC13) S 7.5 (dd, 1H), 6.6 (dd, 1H), 6.5 (m, 1H), 6.4 (br 1H), 4.7 (br
2H), 1.5 (s,
9H); MH+ = 227 (minor) 127 (-BOC), 171 (-tBu).
BOC
I ~ NH BOC
F"v'NH NH H
CI YN` CI KSCN~ CI Y N~ SCN _} , N N SCN
NV 'NOz EtOH N/~NOa K2COs, ACN NXN02
F
[00181] 2-Chloro-5-nitro-4-thiocyanatopyrimidine. (compound known, e.g. WO
2003/032994) Potassium thiocyanate (0.97 g, 10 mM) was added to a solution of
2,4-
dichloro-5-nitropyrimidine (1.94 g 10, mM) in EtOH (40 mL) cooled to 0 C via
an ice
bath. The solution was stirred at 0 C for 30 min, then the bath was removed
and the
resulting suspension allowed to come to RT over 60 min, when water (100 mL)
was added.
The precipitate was collected via filtration, washed with ice cold water,
dissolved with
DCM, dried (MgSO4), filtered and evaporated to yield the titled compound (1.,7
g).1H-
NMR (300 MHz, CDC13) S 9.4 (s, 1H).
[00182] tert-Butyl4-fluoro-2-(5-nitro-4-thiocyanatopyrimidin-2-
ylamino)phenylcarbamate. Potassium carbonate (207 mg) was added to a stirred
solution
of 2-chloro-5-nitro-4-thiocyanatopyrimidine (108 mg) and tert-butyl4-fluoro-2-
nitxophenylcaa-bamate (113 nig) in ACN (5 mL) and stirred for 15 hr. The
solution was
diluted with brine and extracted with EtOAc (2x). The combined organics were
evaporated arnd purified via column chromatography, elution with 30 %
EtOAc/Hex gave
the titled compound (144 mg, 71 % yield). 1H-NMR (300 MHz, DMSO-d6) S 10.5 (br
s,
1H), 9.3 (br s, 1H), 8.9 (br s, 1H), 7.7-7.4 (m, 2H), 7.1 (br s, 111), 1.5 (s,
911), 1.5 (s, 9H);
MH+ = 407, 307 (-BOC), 351 (-tBu).
64
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
F F
BOC.NH P )/0
~N SCN NHZ BOC.NH INapSpO4 /NaHCO3 : BOC.NH ', ~ I~ CDI
~ - H
~I N~NOZ K2C03,ACN . ~ .,~N NH THF/HpO/MeOH E I NN NH THF
F ~ N~OZ ~ NNHz
F F
F F F
BOC,NH H TFA NHZ H p-TsOH Njz,
\~~N N~N~O DCM 0 N N~N~O HC(Me) I~ N N~N~O
F H F H N
F H
[00183] (R)-tert-Butyl4-fluoro-2-(4-(8-fluorochroman-4-ylamino)-5-
nitropyrimidin-
2-ylamino)phenylcarbamate. A solution of the (R)-8-fluorochroman-4-amine
hydrochloride (104 mg) in DMSO (2 mL) and potassium carbonate (141 mg) were
added
to a stirred solution of tert-butyl 4-fluoro-2-(5-nitxo-4-thiocyanatopyrimidin-
2-
ylamino)phenylcarbamate (140 mg) in ACN (10 mL). The mixture was stirred for
15 hr at
room temperature then partitioned between brine and EtOAc and separated. The
aq. layer
was washed with additional EtOAc, the combined organics were evaporated and
purified
via column chromatography, elution with 20-30 % EtOAc/H gave the titled
product in 83
% yield. 'H-NMR (300 MHz, CDC13) S 9.1 (s, 1H), 8.7(m, 1H), 8.2 (br s, 1H),
7.7 (m,
1 H), 7.3 (m, 1 H), 7.3-6.8 (m, 4H), 6. 5(s, 1H), 5. 5(br s,1 H), 4.4 (m 2H),
2.4 (m, 1H), 2.2
(m, 1H), 1.5 (s, 9H); MW = 515, 459 (-tBu).
[00184] (R)-tert-Butyl4-fluoro-2-(9-(8-fluorochroman-4-yl)-8-ogo-8,9-dihydro-
7H-
purin-2-ylamino)phenylcarbamate. To a solution of (R)-tert-butyl 4-fluoro-2-(4-
(8-
fluorochroman-4-ylamino)-5-nitropyrimidin-2-ylarnino)phenylcarbamate (141 mg)
in THF
(20 mL) was added a premixed solution of sodium hydrosulfite (0.6 g) and
sodium
bicarbonate (0.3g) in water (50 mL). MeOH (5 mL) was also added to aid
solution of the
mixture, which was stirred at room temperature for 30 min, when sodium
chloride was
added to saturate the solution. The resultant mixture was extracted with EtOAc
(2x), the
combined organics were dried, filtered and evaporated to yield (R)-ter7-butyl2-
(5-amino-
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
4-(8-fluorochroman-4-ylamino)pyrimidin-2-ylamino)-4-fluorophenylcarbamate that
was
used as such for the next step, MH} = 485.
[00185] To a stirred solution of the above material in THF (5 mL) was added
CDI (131
mg). After 15 hr brine and EtOAc were added and the mixture was separated. The
aq.
layer was washed with additional EtOAc and the combined organics were
evaporated and
purified by column chromatography (eluted 3 % MeOH/DCM) to yield titled
product (86
mg, 62 % yield for two steps). 'H NMR (300 MHz, 5% CD3OD in CDC13) S 7.9 (s,
1H),
7.4 (dd, 1H), 7.3 (m, 1H), 6.9 (dd, 1H), 6.7-6.5 (m, 3H), 5.7 (dd,1H), 4.6 (m
1H), 4.3(td,
1H), 2.9 (m, 1H), 2.2 (m, 1H), 1.5 (s, 9H); MH+ = 511, 411 (-BOC), 455 (-tBu).
[00186] 2-(6-Flaoro-iH-benzo[d]imidazol-l-yl)-9-((R)-8-fluorochroman-4-yl)-7.H-
purin-8(9R)-one. A freshly prepared solution of 30 % TFA/DCM (5 mL) was added
to
(R)-teri-butyl4-fluoro-2-(9-( 8-fluorochroman-4-yl)-8-oxo- 8,9-dihydro-7H-
purin-2-
ylamino)phenylcarbamate and the solution was stirred at room teniperature for
60 min then
the solvents were removed in vacuo to yield (R)-2-(2-amino-5-
fluorophenylamino)-9-(8-
fluorochroman-4-yl)-7H-purin-8(9.H)-one that was used as such MH* = 411.
[00187] To the above di-amine was added MeOH (2 mL), trimethylorthoformate (2
mL)
and p-TsOH (cat). The mixture was stirred at RT for 60 min then the solvents
were
reduced and the resultant material partitioned between DCM and brine and
separated. The
crude product was purified via column chromatography (eluted with 4 1o
MeOH/DCM) to
yield the titled cornpound (46 mg). 1H-NMR (300 MHz, 5% CD3OD in CDC13) S 8.7
(s,
1 H), 8.1 (s, 1 H), 7.5 (m, 2H), 6.9 (nn, 2H), 6.6 (m, 2H), 5. 8(dd, 1 H), 4.
6(m 1 H), 4.3 (td,
1H), 2.8 (m, 1H), 2.3 (m, 1H); MH+ = 421:
r 0 .-
N ': ~
~
NNNI N~O F
- ~
N
F H
66
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00188] 2-(6-Fluoro-lH-benzo[d]imidazol-1-yl)-9-((R)-6-fluorochroman-4-yl)-7H-
purin-8(9H)-one. The title compound was synthesized from (R)-6-fluorochroman-4-
amine
via the procedures described in Example 27. 'H NMR (300 MHz, CDC13+5%CD3(?D):
S
8.72 (s, 1H), 8.16 (s, 1H), 7.5-7.7 (m, 2H), 6.9-7.0 (m, 2H), 6.8-6.9 (m, 1H),
6.54 (dd, 1H),
5.78 (brt, 1H), 4.4-4.5 (m, 1H), 4.26 (m, 1H), 2.7-2.8 (rn, 1H), 2.2-2.3 (m,
1H).
N::~~I
NY N N
`,. N ~ N~O
F H
[00189] 2-(6-Fluoro-lH-benzo[d]imidazol-1-yl)-9-(tetrahydro-2H-pyran-4-yl)-7H-
purin-8(9H)-one. The title compound was synthesized from 4-
aminotetrahydropyran via
the procedures described in Example 27. 'H NMR (d6-DMSO) 8 11.65 (s, 1H), 9.13
(s,
1H), 8.34 (s, IH), 8.28 (m, 1H), 7.82 (n-4 1H), 7.25 (td, J = 9.0, 2.4 Hz,
1H), 4.56 (m, 1H),
4.03 (dd, J= 11.1, 3.9 Hz, 2H), 3.50 (t, J=11.1 Hz, 2H), 2.59 (m, 2H), 1.78
(m, 2H).
O F
r
N
N,~N H
NI` IN~O
C! H
[00190] 2-(6-Chloro-lH-benzo[d]imidazol-1-yn-9-((R)-8-fluorqchroman-4-yl)-7Ff
purin-8(9H)-one. The title compound was synthesized from (R)-8-fluorochroman-4-
amine
and 4-chloro-2-nitrobenzenamine via the procedures described in Example 27. 1H
NMR
(300 MHz, CDC13+50/oCD34D): S 8.7 (s, 1H), 8.2 (s, 1H), 8.1(s, 1H), 7.6 (d,
1H), 7.2 (dd,
1H), 6.9 (td, 1H), 6.7-6.5 (m, 2H). 5.8 (dd, 1H), 4.6 (m 1H), 4.4 (td, 1H),
2.9 (in, 1H), 2.3
(m, 1 H).
67
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00191] Example 28: Synthesis of 2-(5-Fluoro-lH-benzo[cl]imidazol-1-yl)-9-((R)-
8-
fluorochroman-4-yl)-7.lY-purin-S(9I1)-one:
OMe
MeO OMe MeDa
Me0 F N H
~ _ F ~ N ~~ ~e DIF~1_ ~~ Raney Ni ~
Np2 H2N ~~ THF N02 THF, Hs NH2
2
[00192] N-(2,4-Dimethogybenzyl)-5-fluoro-2-nitrobenzenamine. A solution of 2,4-
difluoro-1-nitrobenzene (1.1 mL), 2,4-dimethoxy benzylamine (1.5 mL) and DIEA
(5.2
mL), in THF (40 mL) was heated at 60 C for 60 min, allowed to cool to RT,
partitioned
between EtOAc and H20, separated, dried (MgSO4), filtered and evaporated to
yield the
titled product as a yellow solid (3.14 g). 1H-NMR (300 MHz, CDC13) S 8.5 (br
s, 1H), 7.2
(dd, 1H), 7.2 (d, 1H), 6.6-6.4 (m, 3H), 6.3 (m, 1H), 4.3(d, 2H), 3.9 (s, 3H),
3.8 (s, 3H).
[00193] M-(2,4-Dimethoxybenzyl)-5-fluorobenzene-1,2-diamine. Under a flush of
Ar,
a catalytic amount of a Raney Ni solution in water was added to a solution
of.N-(2,4-
dimethoxybenzyl)-5-fluoro-2-nitrobenzenamine (0.5 g) in THF (20 mL). The flask
was
closed with a septunn, evacuated under house vacuum and hydrogen added via
balloon.
The resulting suspension was stirred at RT for 16 hr, when the Ha balloon was
removed,
mixture evacuated and filtered through a plug of celite, that was thoroughly
rinsed with
THF and MeOH, to yield the titled diamine that was used as such.
68
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F F H2N ~ F
r0 ~ rQ ~ I XH N\{ F OMe O N H CI~N~ CI NH2.HCI CI N NH Me0 Me M~ NYN NH
~ Y .~ ~
N.
N"~ NO2 DIEA, THF N~ NO2 IC2C03, ACN F N NOz
F
OMe rO OMe rO
Ir~ I CDI ~ NH '~1
Ni NH -~.- ~ H N
H2, THF~ Me0 N~N NH THF Aryep ~ N ~O
N F~ N~N
F N HZ H
~--O F O F
f~f ~~ ~ ,
~ N =-
TFA NH2 Id N~ N ~ (MeO)3CH N
_ N N
E St3 ~H F~. N O p-TsOH F,' N~ ~O
H H
[00194] (R)-2-Chloro-N-(8-fluorochroman-4-y1)-5-nitropyrimidin-4-amine. A
solution
of (R)-8-fluorochroman-4-amine hydrochloride (1.02 g) and DIEA (2.6 mL) in DCM
(10
mL) was slowly added to a solution of 2,4-dichloro-5-nitropyrimidine (0.97 g)
THF (25
mL) at -78 C. The reaction mixture was stured for 30 min at -78 C then
allowed to
warm to RT overnight. The reaction was quenched with the addition of sat.
NH4Cl (1 mL),
the solvent volume was reduced in vacuo, and the resulting mixture partition
between
EtOAc and water then separated. The crude material was purified via column
chromatography, elution with 30 % EtOAc/Hex gave the titled product (1.43 g).
'H-NMR
(300 MHz, CDC13) S 9.1 (s, 1H), 8.6 (br d, 1H), 7.1-6.8 (m, 3H), 5.6 (dd,1H),
4.4 (m 1H),
4.3(m, 1H), 2.4 (ni, 1H), 2.2 (in, 1H).
[00195] (R)1V2-(2-(2,4-Dimethozybenzylamino)-4-fluorophenyl)-1V4-(8-
fluorochroman-4-yl)-5-nitropyrimidine-2,4-diamine. A mixture of (R)-2-chloro
1V-(8-
fluorochroman-4-yl)-5-nitropyrimidin-4-amine (32 mg), N1-(2,4-dimethoxybenzyl)-
5-
fluorobenzene-1,2-diamine (28 mg)and KCO3 (41 mg) in ACN was heated at 65 C
for 3
hr, cooled to RT, diluted with brine and extracted with EtOAc (2x). The
combined
organics were evaporated, and purified by column chromatography (eluted with
30 %
EtOAc/Hex) to yield the titled product (21 mg). 'H-NMR (300 MHz, CDC13.) S 9.0
(s, 1H),
69
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
8.6 (br d, 1H), 7.2-6.8 (in, 6H), 6.5-6.3 (m,4H), 4.4-4.2 (m, 4H), 3.8 (s,
6H), 2.3-2.2 (m,
2H).
[00196] (R)-2-(2-(2,4-Dimethoa-ybenzylamino)-4-fluorophenylamino)-9-(8-
fluorochroman-4-yl)-7H-purin-8(9H)-one. Under an Ar atmosphere, a catalytic
amount
of a Raney Ni solution in water was added to a solution of (R)-N2-(2-(2,4-
dimethoxybenzylamino)-4-fluorophenyl)-N4-(8-fluoro chroman-4-yl)- 5-
nitropyrimidine-
2,4-diamine (21 mg) in THF. The flask was closed with a septum, evacuated
under house
vacuum and hydrogen added via balloon. The resulting suspension was stirred at
RT for 2
hr, when the H2 balloon was removed, the nuxture evacuated and filtered
through a plug of
celite, that was thoroughly rinsed with THF and MeOH, to yield (R)-1V`-(2-(2,4-
dimethoxybenzylamino)-4-fluorophenyl)-.N4-(8-fluorochroman-4-yl)pyrimidine-
2,4, 5-
triamine that was used directly.
[00197] To a stirred solution of the above material in THF (5 mL) was added
CDI (12
mg). After 18 hr brine and EtOAc were added and the mixture was separated. The
organic
layer was evaporated and purified by column chromatography (eluted 4 %
MeOH/DCM)
to yield titled product (14 mg). 'H NMR (300 MHz, CDC13+5 loCD3OD): 8 7.8 (s,
1H), 7.3
(s, 1H), 7.1 (d, 1H), 6.9 (m, 2H), 6.7-6.2 (m, 6H), 5.7 (dd, 11-1), 4.5 (m
1H), 4.2 (m, 1H),
4.1 (s, 2H), 3.8 (s, 3H), 3.7 (s, 3H), 2.8 (m, 1H), 2.2 (m, 1H).
[00198] 2-(5-Fluoro-lH-benzo[d]imidazol-1-yl)-9-((R)-8-fluorochroman-4-yl)-7H-
purin-
8(9H)-one.
[00199] A mixture of (R)-2-(2-(2,4-dimethoxybenzylamino)-4-fluorophenylamino)-
9-(8-
fluorochroman-4-yl)-7H-purin-8(9.H)-one (14 mg) and TFA (1 mL) was stirred for
60 min
when triethyl silane (0.5 mL) was added. The resulting solution was stirred at
RT for 16
hr, then the solvents were reduced in vacuo to yield (R)-2-(2-amino-4-
fluorophenylamino)-
9-(8-fluorochroman-4-yl)-7H purin-8(9H)-one that was used as such.
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00200] A catalytic amount ofp-TsOH was added to a solution of the above amine
in
trimethylorthoformate (2 mL). The mixture was stirred at RT for 15 hr then the
solvents
were reduced and the resultant material partitioned between DCM and brine and
separated.. The crude product was purified via column chromatography (eluted
with 5 %
MeOH/DCM) to yield the titled compound (9 mg). 'H-NMR (300 MHz, CDC13) S 10.0
(s,
1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.8 (dd, 1H), 7.4 (d, 1H), 7.1(m, 211), 6.8 (m,
2H), 5.9 (dd,
1H), 4.7 (rn. 1H), 4.4 (td, 1H), 2.9 (m, 1H), 2.4 (m, 1H).
[00201] Example 29. Synthesis and Resolution of 8-Fluorochroman-4-amine:
1 .; (CiCC~)z
F 3-Bromo F cat. DMF F
HO ,~ propionic acid Q .~ CHZCI2
I/ NaOH I Ha0 HO I~ 2. AI 3CI I/
O 0
F F
HONHZ-HCI O b HZ, Raney Ni NaOAc ! EtOH EtOH
jy NH2
HO'
[00202] 3-(2-Fluorophenogy)propanoic acid. A mixture of 2-fluorophenol (15 g),
3-
bromopropanoic acid (20 g) and NaOH (11 g) was refluxed in 50mL of water. The
solution was cooled to room temperature and acidified to pH 2 with 3 M HCI.
The
resulting precipitate was isolated by filtration to yield 9.27 g of title
compound as a white
solid. The filtrate was extracted 3 times with EtOAc to yield 2.5g of less
pure compound.
[00203] 8-Fluorochroman-4-one. Oxalyl chloride (8.79 mL) and 1 dmp of DMF were
added to an ice cold solution of 3-(2-fluorophenoxy)propanoic acid (9.27 g) in
DCM (50
mL). The solution was stirred at 0 C for 2 hours, then aluminum chloride
(7.39g,
55.42mM) was added and the solution was stirred for 16 hours at room
temperature. The
mixture was poured onto ice water, and extracted three times with DCM. The
combined
71
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
organics were washed with 0.5M NaOH and brine, then dried, evaporated, and
purified by
column chromatography (eluting with 20% EtOAc/Hex) to give of the title
compound
(8.20g, 98%).
[00204] 8-Fluorochroman-4-amine. A round bottom flask was charged with 8-
fluorochroman-4-one (8.2g), hydroxylamine hydrochloride (3.78g) and sodium
acetate
(4.46g). A reflux condenser was added, the flask was purged with argon, dry
EtOH (20
mL) was added, and the mixture was stirred at reflux for 18 hours. The
solution was
cooled to room temperature, diluted with EtOAc, and washed with water. The
organic
phase was dried, and evaporated to give the intermediate 8-fluorochroman-4-one
oxime,
which was reduced with Raney Nickel in EtOH at 50 PSI to yield the titled
amine (4.69g,
57%).
F F F
O ` Novozyme 435
MeOCH2COOMe NH2 t-BuOMe 0 T NH NH2
O 8 M HC1
EtOH
F
O .~
NH2
[00205] Resolution of 8-flnorochroman-4-atnine. (Procedure based on
US0157739). A
mixture of 8-fluorochroman-4-amine (3.40 g), methyl 2-methoxyacetate (2.44 g)
and
Novozyme 435 (Aldrich, 0.68 g) in anhydrous tert-butyl methyl ether (75mL) was
heated
at reflux under argon for 2 hours (at which time the ratio of acylated to
unacylated product
was 1:1 by HPLC). The solid that formed upon cooling was collected via
filtration and
dissolved in EtOAc. The mixture was filtered to remove the biocatalyst and
washed once
with 0. 5M HCl to remove any lingering (S)-amine. The solvent was evaporated
and the
72
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
product was recrystallized from tert-butyl methyl ether to yield (R)-1V-(8-
fluorochroman-4-
yl)-2-methoxyacetamide (0.78 g). The reaction solvent and recrystallization
mother liquor
was washed 3 times with 0. 5 M HCl and concentrated to yield additional (R)-N-
(8-
fluorochroman-4-yl)-2-methoxyacetamide (0.83 g). The combined acidic aqueous
layers
were made basic by NaOH and extracted with DCM to yield (S)-8-fluorochroman-4-
amine
(1.6g). A solution of (R)-N-(8-fluorochroman-4-yl)-2-methoxyacetamide (0.78g)
in 8M
HCl in EtOH (50mL) was heated at reflux for 4 hours. The solvents were removed
from
the cooled reaction mixture, the resulting solid was taken up in 50 mL of 0.5M
NaOH,
salted out with NaCl(s), and extracted 4 times with DCM to yield (R)-8-
fluorochroman-4-
amine (0.48 g(87%)). The % ee was checked via chiral HPLC: Chiralcel OD-H
(0.46 x 25
cm analytical column, Daicel Chemical Industries) method: isocratic 5 % (0.05%
TFA/EtOH) 95 % (0.05 % TFA/Hex), Rt = 7.2 min (S)-enantiomer, Rt = 9.2 min (R)-
enantiomer.
[00206] Example 30. Chroman-4-amine, 5-fluorochroman-4-amine, 6-
fluorochroman-4-amine, 6-chlorochroman-4-amine, 6-methylchroman-4-amine, 6-
methoxychroman-4-amine, 7=fluorochroman-4-amine, 5,8-difluorochroman-4-amine,
and 6,8-difluorochroman-4-amine.
~
F
a ~~
~
NHz F NH2 NH2 NH2 NH2
F F
~ O F 0
Me ~ I
NH2 NHz F NH2 NH2
[00207] These amines were prepared via procedures described in Example 29 for
the
synthesis of 8-fluorochroman-4-amine. The corresponding chroman-4-ones were
commercially available as advanced intermediates for the synthesis of chroman-
4-amine,
6-fluorochroman-4-anune, 6-chlorochroman-4-amine, 6-methylchroman-4-amira.e,
and 6-
methoxychroman-4-amine. For the synthesis of 5-fluorochroman-4-amine, the
intermediate
73
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
5-fluorochroman-4-one was obtained using procedures from GB 2355264, which
also
provided 7-fluorochroman-4-one. 7-Fluorochroman-4-one could be used in the
synthesis of
7-fluorochroman-4-amine. Chroman-4-am'ine, 5-fluorochroman-4-amrne, 6-
fluorochroman-4-amine, 7-fluorochroman-4-amine, 5,8-difluorochroman-4-amine,
and
6,8-difluorochroman-4-amine were resolved via the procedure described in
Example 29 for
the resolution of 8-fluorochroman-4-amine.
[00208] Example 31. 1-Methyl-4,5,6,7-tetrahydro-lH-indol4-amine.
NH2
16N
The title compound was obtained from 1-methyl-6,7-dihydro-lH-indol-4(5H)-one
(Heterocycles (1984), 22, 2313) via the procedure described in Example 29 that
were used
to obtain 8-fluorochroman-4-amine from 8-fluorochroman-4-one.
[002091 Example 32. Synthesis of 5,6-Uifluorochroman-4-amine:
Br Br Br
~
~ OH BrCH2CH2COQH O'I~COOH 1) (COC02
~
F NaOH F 2) AfCl3 F /
F F O
Br
NHzOH-HCI, NaOAc ~ O 1.. H2 / Raney-Ni `~ O
EtOH, reflux F ` ~ 2. H2 ! Pd/C F ~
F NH2
'O Y H ~
74
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00210] 3-(2-Bromo-4,5-difluorophenoxy)propanoic acid. A solution of 1.68 g of
NaOH (42 mmol) in 5 mL of water was added slowly to the suspension of 2.29 mL
(20
mmol) 2-bromo-4,5-difluorophenol and 3.07 g (20 mmol) 3-bromopropionic acid.
The
mixture was heated at 100 C in an oil bath for 5 hours, and then allowed to
cool to room
temperature. Water was added to completely dissolve any solid material and the
reaction
mixture was made acidic with concentrated HCI. The product was extracted into
ether (3
times), and the combined organic layers was dried over Na2SO4 and evaporated
to give 3.7
g (66%) ofthe title compound as a light brown solid. 'H NMR (300 MHz, CDC13):
8 7.4
(t, 1H), 6.8 (q, 1H), 4.3 (t, 2H), 2.9 (t, 2H).
[00211] 8-Bromo-5,6-difluoro-2,3-dihydrochromen-4-one. Oxalyl chloride (1.7
rnL, 20
mmol) was added to the solution of 2.8 g (10 mmol) of 3-(2-bromo-4,5-
difluorophenoxy)-
propanoic acid in 40 mL of anhydrous DCM followed by a drop of DMF. After 1.5
hours,
a drying tube was attached and the solution was cooled in an ice-water bath.
AIC13 (1.5 g,
11 mmol) was added and the dark red solution was allowed to slowly reach room
temperature while being stirred for 16 hours. The mixture was poured into ice
and the
organic layer was separated. The aqueous layer was extracted with DCM twice.
The
combined organic layers were washed with 0.5 N NaOH and brine, then dried over
Na2SO4
and concentrated. Column chromatography of this residue with hexane and EtOAc
provided 1.9 g of the title compound as an off-white solid (73%). 1H NMR (300
MHz,
CDC13): S 7.6 (t, 1H), 4.65 (t, 2H), 2.85 (t, 2H).
[00212] 8-Bromo-5,6-difluoro-2,3-dihydrochromen-4-one oxime. To a solution of
8-
bromo-5,6-difluoro-2,3-dihydrochromen-4-one (7.2 mmol) in 40 mL of ethanol was
added
hydroxylamine hydrochloride (0.55 g, 7.9 mmol) and sodium acetate (0.65 g, 7.9
mmol).
This mixture heated at reflux for 20 hrs. The mixture was cooled, diluted with
EtOAc,
washed with water and brine, and then dried over Na2SO4. Concentration of the
solvent
provided the title compound as a white solid (1.9 g).1H NMR (300 MHz, 10%
CD30D
CDC13): S 7.3 (t, 1H), 4.2 (t, 2H), 2.9 (t, 2H).
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00213] 5,6-Difluoro-3,4-dihydro-2H-chromen-4-amine. Raney-Ni (5 mL slurry in
water) was added to a solution of 8-bromo- 5,6-difluoro-2,3-dihydrochromen-4-
one oxime
(1.9 g) in 200 mL MeOH. The mixture was hydrogenated at 50 psi for 24 hrs to
provide 8-
bromo-5,6-difluoro-3,4-dihydro-2H-chromen-4-amine. Pd/C (0.3 g) was added to
the
mixture and hydrogenation was resumed at 50 psi for 4 hours. The title
compound was
obtained after filtration and concentration in vacuo. 1H NMR (300 MHz, 10%
CD30D in
CDC13): S 7.15 (q, 1H), 6.6 (in, 1H), 4.6 (bm, 1H), 4.25 (bm, 2H), 2.2-2.4 (m,
2H).
[00214] Example 33. Synthesis of 4-Amino-3,4-dihydro-2H-chromene-8-
carbonitrile:
CN CN
0 O~ I NH4OAc / NaCNBH3 O,~
~ MeOH / 3A sieves
0 NH2
[00215] 4-Amfno-3,4-d&ydro-2H-chromene-8-carbonitrile. A mixture of 260 mg of
4-
oxo-3,4-dihydro-2H-chromene-8-carbonitrile (made from 2-hydroxybenzonitrile
via the
procedure described in Example 29), ammonium acetate (1.2 g), and 3A molecular
sieves
(1.5 g) in 10 mL of methanol was stirred for 5 days. The mixture was filtered
through
celite and the filtrate concentrated in vacuo. The crude residue was treated
with 100 mL of
1 M HCl and extracted with ethyl ether (3 X 100 mL). The aqueous layer was
made basic
to pH 10 with saturated NaOH and extracted with DCM (3 X 100 mL). The combined
DCM layers were washed with brine, dried over magnesium sulfate, and
concentrated in
vacuo to provide 150 mg of the title compound.
NC
NH2
76
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00216] 4-Amino-3,4-dihydro-2H-chromene-6-carbonitrile. The title compound was
made from 6-cyano-4-chromanone (Syntech) via the same procedure that was
described in
Example 33.,
O
NH2
[00217] 4-Amino-1,2,3,4-tetrahydronaphthalen-1-yl acetate. The title compound
was
made from 4-oxo- 1,2,3,4tetrahydronaphthalen-l-yl acetate (Tetrahedron:
Asymmetry
2001, 12, 2283) via the same procedure that was described in Example 33.
H2N
[00218] 6,7-Dihydro-5H-cyclopenta[b]pyridin-5-amine. The title compound was
made
as described in WO 03/045924.
[00219] Example 34. Synthesis of (R)-5,6,7,8-Tetrahydroquinogalin-5-amine:
N Boc2O N I H2NNH2 N I TFA N
'N~ 'N~ N~ i ', ' N
DMAP MeOH CH2CI2
HN,~r CH3CN Boc'N,,r Boc' NH NH2
O O
[00220] (R)-tert-Butyl acetyl(5,6,7,8-tetrahydroquinogalin-5-yl)carbamate. A
solution
containing 483 mg of (R)-N-(5,6,7,8-tetrahydroquinoxalin-5-yl)acetamide (J.
Org. Chem.
(2003), 68, 3546) in acetonitrile (20 mL) was treated with Boc2O (3 g) and
DMAP (5 mg).
The mixture was heated at 60 C for 1.5 h and then concentrated in vacuo.
Column
chromatography (50% EtOAc / hexanes) provided 293 mg of the title compound.
77
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00221] (R)-tert-Butyl 5,6,7,8-tetrahydroquinozalin-5-ylcarbamate. A solution
of (R)-
tert-butyl acetyl(5,6,7,8-tetrahydroquinoxalin-5-yl)carbarnate (293 mg) in
methanol (10
mL) was treated with hydrazine hydrate (0.5 mL) for 1:;5 h. The mixture was
diluted with
EtOAc and washed twice with saturated aqueous sodium chloride. The organic
layer was
separated, dried with sodium sulfate, and concentrated in vacuo to provide 238
mg of the
title compound.
[00222] (R)-5,6,7,8-Tetrahydroquinogalin-5-amine. A solution of (R)-tert-
buty15,6,7,8-
tetrahydroquinoxalin-5-ylcarbamate (238 mg) in 10 ml of 1:1 TFA / DCM was
stirred for
30 minutes. The mixture was concentrated in vacuo to provide the title
compound as the
TFA salt.
[00223] Example 35. Synthesis of 2-(1H-Senzo[d]imidazol-1-yl)-9-(4,5,6,7-
tetrahydro-lH-indol-4-yl)-7H-purin-8(9H)-one:
O O NH2
N ~ N
::r:::e
Fi SOZPh SO2Ph
,
~1502Pt1
/ NH
N
N~ J~ 4 N NaOH N~ ~
N~N I NO MeOH /\
~ NYN N
~
NN N~,
H H
[00224] 1-(Phenylsulfonyl)-4-oxo-4,5,6,7-tetrahydroindole. To a suspension of
NaOH
(4.44 g) in 1,2-dichloroethane (250 mL) was added 4-oxo-4,5,6,7-
tetrahydroindole (5.0 g).
The mixture was then cooled to 0 C and stirred for 30 min, following which a
solution of
phenylsulfonyl chloride (5.7 mL) in 1,2-dichloroethane (50 mL) was added
dropwise over
78
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
a period of 30 min. After 30 min of stirring, the reaction mixture was allowed
to come to
room temperature and stirred overnight. The reaction was quenched by pouring
onto
distilled water (100 mL). The organic layer was separated, and the aqueous
layer was
extracted with dichloromethane (3 x 50 mL). The combined organic extract was
washed
with distilled water to neutrality, dried over MgSO4, and concentrated in
vacuo to afford
7.0 g of the title compound.
[00225] 1-(Phenylsulfonyl)-4,5,6,7-tetrahydro-lH-indol-4-anune. The title
compound
was obtained from 1-(phenylsulfonyl)-4-oxo-4,5,6,7-tetrahydroindole via the
procedure
described in Example 29 that was used to obtain 8-fluorochroman-4-amine from 8-
fluorochroman-4-one.
[00226] 2-(1H-Benzo[d]imidazol-1-yl)-9-(1-(phenylsulfonyl)-4,5,6,7-tetrahydro-
lH-
indol-4-yl)-7H-purin-8(9H)-one. The title compound was obtained from 1-
(phenylsulfonyl)-4,5,6,7-tetrahydro- 1H-indol-4-amine via the procedures
described in
Example 24.
[00227] 2-(lH-benzo[d]imidazol-l-yl)-9-(4,5,6,7-tetrahydro-lH-indol-4y1)-7H-
purin-
8(9li)-one. To a solution of 2-(1H-benzo[d]imidazol-1-yl)-9-(1-
(phenylsulfonyl)-4,5,6,7-
tetrahydro-lH-indol-4-yl)-7H-purin-8(9H)-one (50 mg) in MeOH (1 mL) was added
4 N
NaOH (1 mL), and the mixture was refluxed overnight and cooled.. Volatiles
were removed
under reduced pressure, and the resultant neutralized with 4 N HCI. The white
precipitate
was filtered, washed with a small amount of water, and dried in vacuo to
afford 36 mg of
the title compound. 'H NMR (d&-DMSO) 8 11.6 (s, 1H), 10.7 (s, 1H), 8.86 (s,
1H), 8.27 (s,
1H), 7.69 (d, J = 7.8 Hz, 111), 7.58 (d, J = 8.1 Hz, 1H), 7.33 (m, 2H), 6.53
(t, J= 2.4 Hz,
1H), 5.64 (t, J= 2.4 Hz, 1H), 5.54 (m, 1H), 2.72 (m, 2H), 2.30 (m, 1H), 2.07
(m, 2H), 1.84
(m, 1H).
79
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00228] Examples 36 and 37'(Dihydrobenzofurans): 2-(1H-benzo[d]imidazol-1-yl)-
9-
(2,3-dihydrobenzofuran-3-yl)-7H-purin-8(9H)-one and 2-(1H-benzo[d]imidazol-1-
yn-
9-(4-fluoro-2,3-dihydrobenzofuran-3-yn-7H-purin-S(9H)-one.
e F O F O
~ QH {COCI}Z O
i Ci 1) 2.5 eq TM5CHN2
I' O DCM Q
~ I 2) HOAc O
OH
F N Hg {q~ ~ F NH2
NH2OH-HCI
NaOAc ~ ba THF ~~ Q
[00229] 2-fluoro-6-methogybenzoyl chloride. Oxalyl chloride (0.56 mL, 6.4
mmol) was
added to the solution of 1.0 g (5.9 mmol) 2-fluoro-6-methoxybenzoic acid in 5
mL
anhydrous CH2C12. Then a drop of DMF was added. After one hour, when the slow
bubbling was ceased, volatiles were removed under reduced pressure to afford
1.1 g (95%)
acid chloride as pale-yellow liquid.
[00230] 1H NMR (300 MHz, CDC13): 6 7.45 (q, 1H), 6.7-6.8 (m, 2H), 3.9 (s, 3H).
[00231] 4-fluorobenzofuran-3(2H)-one. The yellow (trimethylsilyl)diazomethane
ether
solution (2.0 M, 3.7 mL) was added to 0.57 g (3.0 mmol) above acid chloride
with stirring.
After 3 hours, solvent was evaporated. The yellow residue was dissolved in 3
mL acetic
acid (strong gas and heat evolution, used a water bath to cool the flask for a
minute), and
stirred for 15 min at room temperature. The solvents were removed under
vacuum, and the
red residue was taken into 2 mL CH2Cl2, washed with water twice, then brine,
and dried
over Na2SO4. This crude product was purified by column chromatography (eluting
with
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
10% EtOAc in hexanes) to give 0.24 g (53%) 4-fluorobenzofuran-3(2H)-one as
white
solid.
[00232] 'H NMR (300 MHz, CDC13): S 7.58 (m, 1H), 6.92 (br d, 1H), 6.71 (t,
1H), 4.65
(s, 2H).
[00233] (Z)-4-fluorobenzofuran-3(2H)-one oxhne. The above ketone (0.70g, 4.6
nimol)
was dissolved in 5 ml ethyl alcohol, and then added 0.64 g (9.2 nunol)
hydroxylamine
hydrochloride and 0.75 g (9.2 mmol) sodium acetate. This suspension was
brought to
reflux for 1 hr. The mixture was cooled to room temperature and added 4 mL
water to
dissolve the excess reagents. Suction filtration, and then wash the solid cake
with small
amount of cold water provided 0.48 g (63%) desired oxime as white needle
crystals.
[00234] 'H NMR (300 MHz, CDC13): S 8.38 (s, 1H), 7.38 (q, 1H), 6.6-6.8 (m,
2H), 5.21
(s, 2H).
[002351 4-fluoro-2,3-dihydrobenzofuran-3-anine. The above oxime (0.48 g) was
dissolved in 40 mL anhydrous THF under Argon. Fresh prepared aluminum amalgam
(by
dippnzg 1 g polished aluminum foil sequentially in 2% HgC12 aqueous solution,
water, and
fmally THF) was added quickly and the rnixture was refluxed for 24 hours under
Argon.
Shinny mercury beads appeared at the bottom of the flask. The mixture was
allowed to
coolto room temperature and filtered through a pad of celite. Flask and solid
cake were
washed with THF three times, then methanol three times. The combined filtrate
was rotary
evaporated to give 0.41g yellow solid as a mixture of approximately
(determined by NMR)
20% desired amine with 80% starting oxime. This mixture was used for next step
without
purification.
81
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00236] 1H NMR (300 MHz, CDCl3): S 7.18 (q, 1H), 6.5-6.7 (m, 2H), 4.8-4.9 (1n,
1H),
4.69 (t, 1H), 4.2-4.3 (m, 1H).
N~
--~
N ~1_ N~O
N
H
[00237] The above racemic 2-(1H-benzo[d]imidazol-l-yl)-9-(2,3-
dihydrobenzofuran-3-
yl)-7H-purin-8(9H)-one can be separated on chiral CHIRALCEL OD-H column
(Cellulose
tris(3,5-dimethylphenylcarbamate) on a 5 M silica-gel substrate) eluting with
85:15
hexanes:ethanol (both with 0.1 Jo diethylamine). One enantiomer has a
retention time of 25
min, the other one 33.5 min.
[00238] Procedures and NMR Data for 7-Alkylated Purinone Analogs.
_ _
F F
N~ "= ~ lodomethane N~ ~`' \ j
NZ~N~ N N PS-BEMP YO
N ~,,
F H F
[00239] Example 38: 2-(6-fluoro-iFl-benzo[dJimidazol 1-yl)-9-((R)-8-
fluorochroman-
4-yl)-7-methyl-7H-purin-8(9H)-one. A suspension of 2-(6-fluoro-1H-
benzo[dJinudazol-
1-yl)-9-((R)-8-fluorochroman-4-yl)-7H-purin-8(9H)-one (5 mg), 2-tert-
butylimino-2-
diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorin.e on polystyrene (30
mg, 2.2
mM/g) and iodomethane (5 L) in ACN (1 mL) was stirred for 1 hr at RT, then
filtered
through a short plug of celite, which was thoroughly rinsed with MeOH.
Evaporation of
the solvents and silica gel chromatography (elution with 4 Ao MeOH/DCM) gave
the titled
compound (4 mg). 1H-NMR (300 MHz, CDC13) S 83 (s, 1H), 8.2 (s, 1H), 7.7 (m,
2H), 7.0
82
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
(m, 2H), 6.7 (m, 2H), 5.9 (dd, 1 H), 4.7 (m 1H), 4.4 (td, 1 H), 3.6 (s, 3H),
2.9 (m, 1 H), 2.4
(m, 1 H).
0 coo
' j N N N \ N N N
...- N ~ ~O N>=O
N
NC H PS-BEMP NC
/0
[00240] Example 39: 3-[9-Chroman-4-yl-7-(2-methoxy-ethyl)-8-oxo-8,9-dihydro-7H-
pnrin-2-yl]-3H-benzoiunidazole-5-carbonitrile. A solution of 3-(9-((R)-chroman-
4-yl)-8-
oxo-8,9-dihydro-7H-purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile (10 mg) in
acetonitrile (10 mL). was treated with 2-bromoethyhnethyl ether (23 p,L) and 2-
tert-
butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine on
polystyrene
(500 mg, 2.2 mmol base/g). The mixture was stirred at room temperature 16
hours, and
then filtered through celite. The celite was washed with acetonitrile and
methanol, and then
the collected filtrate was dried with magnesium sulfate and concentrated in
vacuo.
Purification by column chromatography (1% MeOH/DCM) gave 4 mg afthe title
compound. 'H-NMR (300 MHz, CDC13) S 8.8 (s, 1H), 8.6 (s, 1H), 8.4 (s, 1H), 7.9
(d,
1H), 7.6 (d, 1H), 7.2 (m, 2H), 6.9 (d, 1H), 6.8 (d, 1H), 5.9 (t, 1H), 4.6 (m,
1H), 4.4 (m,
1H), 4.2 (rn, 2H), 3.8 (n4 2H), 3.4 (s, 3H), 2.8 (m, 1H), 2.4 (rn, 1H).
r O ~
se \ ~
N N N ~:~ N~N N O F
, O F - N~ X N~O
N~ PS BEMP F
F H
0H
83
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00241] Example 40: 2-(6-fluoro-lH-benzo [d] imidazol-l-yl)-9-((R)-6-fluoro
chroman-4
yl)-7-(2-hydroxyethyl)-7H-purin-8(9H)-one. To the solution of 5 mg of 2-(6-
fluoro-lH-
benzo[d]imidazol 1-yl)-9-((R)-6-fluorochroman-4-yl)-7H-purin-8(9H)-one in 1 mL
anhydrous acetonitrile was added 50 mg (excess) of 2-tert-butylimino-2-
diethylamino-1,3-
dimethyl-perhydro-1,3,2-dia.zaphosphorine on polystyrene (2.2 mmol base/g,
Fluka), then
L (excess) of 2-iodoethanol. The mixture was stirred at room temperature for
24
hours. Column chromatography with 3% MeOH in CH2Cl2and then preparative HPLC
provided 3 mg of the desired product as TFA salt. 'H NMR (300 MHz, CDC13): S
9.14 (s,
1H), 8.40 (s, 1H), 7.7-7.9 (m, 2H), 7.1-7.2 (m, 1H), 7.0-7.1(ni, 1H), 6.9-7.0
(m, 1H), 6.59
(dd, 1H), 5.88 (brt, 1H), 4.5-4.6 (m, 1H), 4.32 (t, 1H), 4.0-4.2 (m, 4H), 2.7-
2.9 (bs+m,
2H), 2.2-2.3 (m, IH).
O c
~ -- N
N~ ,~ ~ HCI ~ _ .
NYN N p N Il N- N~O
~ ~ NN
N ` N
NC H PS-BEMP NC ~
N1
A
[00242] Example 41: 3-[9-Chroman-4-yl-7-(2-dimethylamino-ethyl)-8-0$0-8,9-
dihydra-7H-purin-2-yl]-3H-benzoimidazole-5-carbonitrile. A solution of 3-(9-
((R)-
chroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-benzo[d]imidazole-5-
carbonitrile (65
mg) in acetonitrile (10 mL). was treated with 2-chloro-N,N-dimethylethylamine
hydrochloride (25 mg) and 2-tert-butylimino-2-diethylamino-l,3-dimethyl-
perhydro-1,3,2-
diazaphosphorine on polystyrene (325 mg, 2.2 mmol base/g). The mixture was
stirred at
room temperature for 16 hours, and then filtered through celite. The celite
was washed
with acetonitrile and methanol, and then the collected filtrate was dried with
magnesium
sulfate and concentrated in vacuo. Purification by column chromatography (2%
84
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
MeOH/DCM) gave 21 mg of the title conipound. 'H-NMR (300 MHz, CDC13) S 8.9 (s,
1H), 8.5 (s, 1H), 8.4 (s, 1H), 7.8 (d, 1H), 7.60 (d, 1H), 7.2 (m, 2H), 6.9 (d,
111), 6.8 (d,
1 H), 5. 9(t, 1 H), 4.4 (m, 4H), 3. 6(m, 3H), 3.0 (s, 6H), 2.2 (m, 1H).
.!" ~
N~ '1
BrCN ~NyjN ~O NN
NC H PS-BEMP NC
NC-i
[00243] Example 42: 3-(9-Chroman-4-y1-7-cyanomethyl-S-oxo-S,9-dihydro-7H-
purin-2-yl)-3H-benzoimidazole-5-carbonitrile. A solution of 3-(9-((R)-chroman-
4-yl)-
8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile (10 mg)
in
acetonitrile (10 mL). was treated with bromoacetonitrile (17 L) and 2-tert-
butylimino-2-
diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine on polystyrene (500
mg, 2.2
mmol baselg). The mixture was stirred at room temperature 16 hours, and then
filtered
through celite. The celite was washed with acetonitrile and methanol, and then
the
collected filtrate was dried with magnesium sulfate and concentrated in vacuo.
Purification by column chromatography (1 lo MeOH/DCM) gave 11 mg of the title
compound.1H-NMR (300 MHz, CDC13) S 8.8 (s, 1H), 8.5 (s, 1H), 8.4 (s, 1H), 7.8
(d, 1H),
7.6 (d, 111), 7.2 (in, 2H), 6.8 (m, 2H), 5.9 (t, 1H), 5.0 (s, 2H), 4.6 (m,
1H), 4.4 (m, 1H), 2.8
(m, 1H), 2.4 (m, 1H).
F
F `. , \
' ..~-
`'= N`I
N,~N I N O F BrCOOMe N N N~. N~ F
(
N :/`~N~ N
NC H PS-BEMP NC
MeOOC
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00244] Example 43: Methyl 2-(2-(6-cyano-lH-benzo[d]imidazol-1-yl)-9-((R)-6,8-
difluorochroman-4-yl)-8-oxo-8,9-dihydropurin-7-yl)acetate. A solution of 3-(9-
((R)-
6,8-difluorochroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-benzo
[d]imidazole-5-
carbonitrile (10 mg) in acetonitrile (2 mL). was treated with methyl
bromoacetate (90 mg)
and 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-
diazaphosphorine on
polystyrene (100 mg, 2.2 mmol base/g). The mixture was stirred at room
temperature 48
hours, and then filtered. The collected filtrate was concentrated in vacuo.
Purification by
preparative TLC (5% MeOH/DCM) followed by column chromatography to give 6 mg
of
the title compound. 'H-NMR (300 MHz, CDC13) 58.94 (s, 1H), 8.78 (m, 1H), 8.21
(s, 1H),
7.91 (d, J= 8.1 Hz, 1 H), 7.63 (dd, J= 8.1, 1.2 Hz, 1 H), 6.83 (m, 1 H), 6.42
(m, 1 H), 5.89
(m, 1H), 4.77 (s, 211), 4.68 (m, 1H), 4.41 (m, 1H), 3.87 (s, 3H), 2.98 (m,
1H), 2.40 (m,
1 H).
F F
NL ~ N_ '%
LiOH
N N N F ~\ N N N F
N ~ N ~O MeOH(THFM20 ~ N I ~p
N
NC NC
MeO4 HO
0 0
[00245] Example 44: 2-(2-(6-cyano-lH-benzo[aijimidazol-1-yl)-9-((R)-6,8-
difluorochroman-4-yl)-8-ogo-8,9-dihydropurin-7-yl)acetic acid. To a solution
of methyl
2-(2(6-cyano-lH-benzo[d]imidazol-1-yl)-9-((R)-6,8-difluorochroman-4-yl)-8-oxo-
8,9-
dihydropurin-7-yl)acetate (4 mg) in MeOH (0.75 mL) and THF (0.25 mL) at 0 C
was
added a solution of lithium hydroxide (2 mg) iiz water (0.25 rnL). The
reaction was allowed
to warm to room temperature and stirred for 2 h. After concentration under
reduced
pressure, the residue was diluted with water and acidified with 2 N HC1 to pH
4. The
precipitated solid was filtered, washed with a small amount of cold water,
treated with 1 N
HCl in MeOH (0.5 mL), and concentrated -in vacuo to afford 2 mg of the title
compound.
86
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
1H-NMR (300 MHz, CDC13) cS9.04 (s, 1H), 8.75 (m, 1H), 8.46 (s, 1H), 7.87 (d,
J= 8.4 Hz,
1H), 7.68 (dd, J= 8.4, 1 5 Hz, 1H), 6.92 (m, 1H), 6.70 (m, 1H), 5.97 (m, 1H),
4.77 (s, 2H),
4.63 (m, 1H), 4.44 (in, 1 H), 2.91 (m, 1 H), 2.42 (ni, 1 H).
[00246] Example 45. Synthesis of 2-(6-Fluoro-lH-benzo[djimidaxol 1-yl)-7-
(pip eridin-4-yn-9-(tetrahydro-2H-pyran-4-yl)-7H-purin-8(9.II)-one.
0 0
J~\
N~ HO-( N-Boc
N
N N N ~/ N N OLN
O
N / ~ DIAD, DCE I ~
N F
H Q--PPh3
2) H* F NH
[00247] Resin bound triphenylphosphine (264 mg, 2.15 mM/g, Argonaut) and tert-
butyl
4-hydroxypiperidine-l-carboxylate 6(24 mg) were added to 2-(6-fluoro-lH-
benzo[dlimidazol-1-yl)-9-(tetrahydro-2H-pyran-4-yl)-7H-purin-8(9H)-one in
dichloroethane (6 mL), the mixture was stirred for 10 min when diisopropyl
azodicarboxylate (64 L) was added. After 18 hr, the material was filtered,
the resins were
thoroughly rinsed with ACN and MeOH, the combined organics were evaporated and
purified via RP-HPLC (TFA removed Boc upon evaporation) to yield the titled
compound
as a TFA salt (16 mg). 1H-NMR (300 MHz, CDC13 +5% CD3OD) S 9.0 (s, 1H), 8.6
(s,
1H), 8.3 (dd, 1H), 7.7 (dd, 1H), 7.1 (td, 1H), 4.7-4.4 (m, 2H), 4.2 (dd, 2H),
3.6 (t,2H), 3.3
(br d, 2H), 2.9-2.7 (m, 4H), 2.4-2.2 (m, 4H), 2.9 (m, 1H), 1.9 (d, 2H), 1.8
(d, 2H);19F 8 -
76, -117 ppm; MH+ = 438; IR (CDC13) 1726 (C=O stretch).
87
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
Cr76
N~ ~ \ N N N
,~ N ~ ~O
N
N
[00248] 2-(1H-benzo[d]imidazol-1-y1)-9-((R)-S-fluorochroman-4-y1)-7-methyl-7H-
purin-8(9H)-one. This compound was synthesized by the method described zn
example
38. 'H-NMR (300 MHz, CDC13) S 8.8 (s, 1H), 8.2 (s, 1H), 8.1 (m, 1H), 7.8 (m,
2H), 7.3
(m, 2H), 7.0 (m, 1H), 6.7 (m, 2H), 5.9 (t, 1H), 4.7 (m iH), 4.4 (td, 1H), 3.6
(s, 3H), 2.9 (m,
1 H), 2.4 (m, 1 H).
O
I ~ N ~ N F
~ N
N
[00249] 2-(1H-benzo[d]imidazol-1-yl)-9-(6-fluorochroman-4-yl)-7-methyl7H-purin-
8(9H)-one. This compound was synthesized by the method described in example
38. 1H
NMR (300 MHz, CDC13): S 8.80 (s, 1H), 8.19 (s, 1H), 7.96 (d, 1H), 7.78 (d,
1H), 7.2-7.4
(m, 2H), 6.8-7,1(m, 2H), 6.61 (dd, 1H), 5.88 (brt, 1H), 4.54 (m, 1H), 4.32
(rn, 1H), 3.56
(s, 3H), 2.84 (m, 1H), 2.30 (m, 1H).
F
O &NicoF N~
N
N
88
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00250] 2-(1H-benzo[d]imidazol1-yl)-9-((R)-5,8-difluorochroman-4-yl)-7-methyl-
7H-purin-8(9H)-one. This compound was synthesized by the method described in
example 38. 1H NMR (300 MHz, CDC13): S 8.7 (s, 1H), 8.2 (s, 1H), 8.0 (d, 1H),
7.9 (d,
1H), 7.4 (m, 2H), 7.0 (t-d, 1H), 6.5 (t-d, 1H), 6.0 (t, 1H), 4.6 (m, 1H),4.4
(m, 1H), 3.5 (s,
3H), 2.6 (m, 1H), 2.5 (rn, 1H).
F
~O
., 1
N
~~ N N N N~OF
~` ` /-
,'N
[00251] 2-(1H-benzo[d]imidazol-1-yl)-9-((R)-5,8-difluorochroman-4-y1)-7-ethyl-
7H-
purin-8(9H)-one. This compound was synthesized by the method described in
example 38
using iodoethane. 'H NMR (300 MHz, CDC13): S 8.8 (s, 1H), 8.2 (s, 1H), 8.0 (d,
1H), 7.9
(d, 1H), 7.4 (m, 2H), 7.0 (t-d, 1H), 6.5 (t-d, 1H), 6.0 (t, 1H), 4.6 (m,
1H),4.4 (m, 1H), 4.0
(m, 2H), 2.6 (m, 1H), 2.5 (m, 1H), 1.5 (t, 3H).
F
N~ ,..-
O 6
~ N N
\ Yi !- N ~OF
N 89
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00252] 2-(1H-benzo[d]imidazol-1-y1)-9-((R)-5,8-difluorochroman-4-yl)-7-gropyl-
7H-
purin-8(9H)-one. This compound was synthesized by the method described in
example 38
using iodopropane. 'H NMR (300 MHz, CDC13): S 9.8 (s, 111), 8.2 (s, 1H), 8.0
(d, 1H), 7.9
(d, 1H), 7.4 (m, 2H), 7.0 (t-d, 1H), 6.5 (t-d, 1H), 6.0 (t, 1H), 4.6 (m,
1H),4.4 (m, 1H), 4.0
(m, 2H), 2.6 (m, 1H), 2.5 (m, 1H), 1.9 (m, 2H), 1.0 (t, 3H).
F
~0
N~ ''=j~
N N
N f ~OF
N
F
[00253] 9-((R)-5,8-difluorochroman-4-yp-2-(6-fluoro-lH-benzo[d]imidazol-1-yl)-
7-
methyl7H-purin-8(9H)-one. This compound was synthesized by the method
described in
example 38. 1H-NMR (300 MHz, CDC13) S 8.7 (s, 1H), 8.0 (rn, 1H), 7.7 (dd, 1H),
7.1 (m,
2H), 6.5 (td, 1H), 5.9 (dd, 1H), 4.6 (m 1H), 4.5 (m, 1H), 3.5 (s, 3H), 2.6 (m,
1H), 2.5 (m,
1 H).
F
0
NN f TN F
r-- /--
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00254] 9-((R)-5,8-difluorochroman-4-y4-2-(6-fluoro-lH-benzo[dJimidazol-1-yl)-
7-(2-
methoxyethyl)-7H-purin-8(9H)-one. This compound was synthesized by the method
described in example 39. 'H-NMR (300 MHz, CDC13) S 8.7 (s, 1H), 8.4 (s, 1H),
8.0 (rn,
1H), 7. 8(dd, 1 H), 7.2-7.0 (m, 2H), 6. 5(td, 1 H), 6.0 (t, 1H), 4.6 (m 1H),
4.5 (m, 1 H), 4.1(t,
2H), 3.7 (t, 2H), 3.4 (s, 3H), 2.7 (m, 1H), 2.4 (m, 1H).
F
/ N N
~ N ' ~ F
N
F ~
N--
[00255] 9-((R)-5,8-difluorochroman-4-yl)-7-(2-(dimethylamino)ethyl)-2-(6-
fluoro-lH-
benzo[d]imidazol-1-y1)-7H-purin-8(9B)-one. This compound was synthesized by
the
method described in example 41.;.1H-NMR (300 MHz, CDC13) S 9.2 (s, 1H), 8.5
(s, 1H),
7.9 (m, 1H), 7.8 (dd, 1H), 7.2 (td, 1H), 7.1 (td, 1H), 6.5 (td, 1H), 5.9 (t,
1H), 4.6 (m 1H),
4.5 (m, 3H), 3.6 (nz, 2H), 3.0 (s, 6H), 2.7 (m, 1H), 2.5 (m, 1H).
O F
N_ '~ I ~`
---
N N f N~O
r-- ~
CI
[00256] 2-(6-Chloro-lH-benzo[d]imidazol-1-yl)-9-((R)-S-fluorochroman-4-yl)-7-
methyl-7H purin-8(91Y)-one. This compound was synthesized by the method
described in
example 38. 1H-NMR (300 MHz, CDC13) 8 8.7 (s, 1H), 8.3 (rn, 1H), 8.2 (s, 1H),
7.7 (d,
91
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
1H), 7.3 (dd, 1H), 7.0 (td, 1H), 6.8-6.6 (m, 2H), 5.9 (dd, 1H), 4.7 (m 1H),
4.5 (td, 1H), 3.6
(s, 3H), 3.0 (m, 1H), 2.4 (m, 1H).
N~
N N N
N I N O
NC ~
[00257] 3-(7-methyl-8-ozo-9-(tetrahydro-2H-pyran-4 yl)-8,9-dihydro-7H-purin-2-
yl)-
3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38.1H-NMR (300 MHz, CDC13) S 9.37 (s, 1H), 8.90 (m, 111),
8.58
(s, 1 H), 8.00 (d, J= 8.4 Hz, 1H), 7.79 (dd, J= 8.4, 1. 5 Hz, 1H), 4.63 (m,
1H), 4.03 (m,
211), 3.51(m, 2H), 3.44 (s, 311), 2.58 (in, 2H), 1.80 (m, 2H).
O ~
N ~ ~~
N N
N iF-N >==O
NC ~
92
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00258] 3-(9-Chroman-4-yl-7-methyl-8-oxo-8,9-dihydro-7H-purin-2-yl)-3H-
benzoimidazole-5-carbonitrile. This compound was synthesized by the method
described in example 38. 'H-NMR (300 MHz, CDC13) S 8.8 (s, 1H), 8.5 (s, 1H),
8.2 (s,
IH), 7.8 (d, 1H), 7.6 (d, IH), 7.2 (m, 2H), 6.9 (d, 1H), 6.8 (d, 1H), 5.9 (t,
11-1), 4.6 (m, 1H),
4.4(m, 1H), 3.6 (s, 3H), 2.8 (m, 1H), 2.4 (m, 1H).
F
( \ N N N >=
~ O
N
N~ 1
[00259] 3-[9-(8-Fluoro-chroman-4-yl)-7-methyl-8-oxo-8,9-dihydro-7H-purin-2-yl]-
3H-benzoimfdazole-5-carbonitrile. This compound was synthesized by the method
described in exaniple 38. 1H-NMR (300 MHz, CDC13) f> 8.8 (d, 2H), 8.2 (s, 1H),
7.8 (d,
1H), 7.6 (d, 1H), 7.0 (t, 1H), 6.6 (m, 2H), 5.9 (t, 1H), 4.6 (m, 1H), 4.4 (m,
1H), 3.6 (s,
3H),2.8(m, 1H),2.4(m, 1H).
ro
N
N~Q F
N~NXN
NC
[00260] 3-(9-((R)-6-flnorochroman-4-yl)-7-methyl8-oga-8,9-dihydro-7H-purin-2-
yl)-
3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38. 1H NMR (300 MHz, CDC13): S 8.90 (s, 1H), 8.52 (s,
1H), 8.21 (s,
93
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
1H), 7.84 (d, 1H), 7.59 (d, 11-1), 7.1-7.2 (m, 1H), 6.9-7.0 (m, 1H), 6.59 (dd,
1H), 5.89 (br t,
1H), 4.5-4.6 (m, 1H), 4.34 (br t, 1H), 3.59 (s, 311), 2.8-2.9 (m, 1H), 2.3-2.4
(m, 1H).
flyF
N~N~ N~O
N
NC
[00261] 3-(9-((R)-'7-fluorochroman-4-yl)-7-methyl8-oxo-8,9-dihydro-7H-purj.n.-
2-yl)-
3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38. 'H NMR (300 MHz, CDC13): S 8.90 (s, 1H), 8.62 (s,
1H), 8.19 (s,
1H), 7.84 (d, 1H), 7.59 (d, 1H), 6.8-7.0 (m, 2H), 6.4-6.6 (m, 1H), 5.86 (brt,
1H), 4.5-4.6
(m, 1H), 4.36 (brt, 1H), 3.59 (s, 3H), 2.8-2.9 (m, 1H), 2.3-2.4 (m, 1H).
O F
N~ '`,'
~--
N ix= F
N
NC
[00262] 3-(9-((R)-6,8--difluorochroman-4-yl)-7-methyl-8-oxo-8,9-dihydro-7H-
purin-2-
yl)-3H-benzo[d]iunidazole-5-carbonitrile. This compaund was synthesized by the
method
described in example 38. 'H-NMR (300 MHz, CDC13) 6 8.9 (s, 1H), 8.8 (s, 1H),
8.2 (s,
1H), 7.9 (d, 1H), 7.6 (dd, 1H), 6.8 (td, 1H), 6.4 (dd, 1H), 5.9 (dd, 1H), 4.7
(m, 1H), 4.4 (td,
1H), 3.6 (s, 3H), 3.0 (m, 1H), 2.4 (m, 1H).
94
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O ~ ~
N Cf
N N
N' N
rr ~F
-
NC 1
[00263] 3-[9-(5,8-Difluoro-chroman-4-yl)-7-methyl-8-ogo-8,9-dihydro-7H-purin-2-
yl]-3H-benzoimidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38. 'H-NMR (300 MHz, CDC13) S 8.8 (s, 1H), 8.7 (s, 1H),
8.2 (s,
1H), 7.8 (d, IH), 7.6 (d, 1H), 7.0 (m, 1H), 6.5 (m, 1H), 5.9 (t, 1H), 4.5 (in,
2H), 3.5 (s, 3H),
2.5(m, 2H).
F
N N
~ ~ =o
N
NC J
[00264] 3-[7-Ethyl-9-(8-fluoro-chroman-4-yl)-8-oxo-8,9-dihydro-7H-purin-2-ylJ-
3H-
benzoimidazole-5-carbonitrile. This compound was synthesized by the method
described in example 38 using iodoethane. 1H-NMR (300 MHz, CDC13) fi 8.8 (s,
2H), 8.2
(s, 1H), 7.9 (d, 1H), 7.6 (d, 1H), 7.0 (t, 1H), 6.7 (m, 2H), 5.9 (t, 1H), 4.6
(m, 1H), 4.4(m,
1H), 4.0 (q, 2H), 2.9 (m, 1H), 2.4 (m, 1H), 1.4 (t, 3H).
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N~ = f F
A
N r'N N
;~C
NC
[00265] 3-(7-ethyl-9-((R)-7-fluorochroman 4y1)-8-oxo-8,9-dihydro-7H-purin-2-
yl)-
3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38 using iodoethane. 'H NMR (300 MHz, CDC13)= S 8.88 (s,
1H),
8.62 (s, 1H), 8.21 (s, 1H), 7.84 (d, 1H), 7.59 (d, 1H), 6.8-7.0 (m, 2H), 6.4-
6.6 (m, 1H), 5.86
(br t, 111), 4.5-4.6 (m, 111), 4.36 (br t, 1H), 4.0-4.1(m, 211), 2.8-2.9 (m,
111), 2.3-2.4 (m,
1H), 1.48 (t, 3H).
C F
N_ ; ! \
-
f \ NLr>= F
~NC ~
[00266] 3-(9-((R)-6,8-difl.uorochroman-4-yl)-7-ethyl-8-oxo-8,9-dihydro-7H-
purin-2-
yl)-3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38 using iodoethane. 'H-NMR (300 MHz, CDC13) S 8.9 (s,
1H), 8.8
(s, 1H), 8.2 (s, 1H), 7.9 (d, 1H), 7.6 (dd, 1H), 6.8 (td, 1H), 6.4 (dd, 1H),
5.9 (dd, 1H), 4.7
(m, 1H), 4.4 (td, 1H), 4.1 (q, 2H), 3.0 (m, 1H), 2.4 (rn, 1H), 1.5 (t, 3H).
96
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
N N
N $i1_JN
~ i N _OF[00267] 3-[9-(5,S-Difluoro-chrornan-4-yl)-7-ethyl-S-oxo-S,9-dihydro-
7H-puriu-2-yl]-
3H-benzoimidazole-5-carbonitrile. This compound was synthesized by the method
described in example 38 using iodoethane. 1H-NMR (300 MHz, CDC13) 6 8.8 (s,
1H), 8.7
(s, 1H), 8.2 (s, 1H), 7.9 (d, 1H), 7.6 (d, 1H), 7.0 (m, 1H), 6.4 (m, 1H), 5.9
(t, 1H), 4.6 (m,
1H), 4.4(m, 1H), 4.0 (q, 21-1), 2.5(ni, 2H), 1.4 (t, 3H).
F
cO
N N
oF
~' N ~_-
N
NC
[00265] 3-[9-(5,8-Difluoro-chroman-4-yn-7-isobutyl-S-oxo-8,9-dihydro-7H-purin-
2-
yl]-3H-benzoimidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38 usmg iso-butyl 'iodide. 'H-NMR (300 MHz, CDCl3) S 8.9
(s, 1H),
8.8 (s, 1H), 8.2 (s, 1H), 7.9 (d, 1H), 7.6 (d, 1H), 7.1 (m, 1H), 6.5 (m, 1H),
5.9 (t, 1H), 4.6
(m, IH), 4.4(rn, 1H), 3.8 (q, 2H), 2.7 (m, 1H), 2.5 (m, 1H), 2.3 (q, 1H), 1.0
(s, 61-1).
97
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N~
N r N N
NC
[00269] 3-(7-benzyl-8-ozo-9-(tetrahydro-2H-pyran-4-yl)-8,9-dihydro-7H-purin-2-
yl)-
3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized by the
method
described in example 38 using benzyl bromide. 'H NMR (300 MHz, CDC13): S 9.30
(s,
1H), 9.01(m, 1H), 8.23 (s, 1H), 7.89 (m, 1H), 7.66 (dd, J= 8.1, 1.5 Hz, 1H),
7.40-7.32 (in,
5H), 5.16 (s, 2H), 4.75 (m, 1H), 4.18 (m, 2H), 3.63 (m, 2H), 2.81(m, 2H), 1.90
(m, 2H).
O F
Nrj;o N F
NC
OH
[00270] 3-(9-(6,8-difluorochroman-4-yl)-7-(3-hydrogypropyl)-8-oxo-8,9-dihydro-
7H-
purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile. This compound was synthesized
by
the method described in example 40 using 3-iodopropanol. 1H NMR (300 MHz,
CDC13): S
8.92 (s, 1 H), 8.77 (m, 1 H), 8.38 (s, 1H), 7.90 (d, J= 8.4 Hz, 1H), 7.62 (dd,
J= 8.4, 1.5 Hz,
1H), 6.83 (m, 1H), 6.38 (m, 1H), 5.88 (m, 1H), 4.67 (m, 1H), 4.41 (m, 1H),
4.19 (t, J= 6.5
Hz, 2H), 3.74 (t, J= 5.6 Hz, 2H), 2.93 (m, 1H), 2.40 (m, 1H), 2.08 (m, 2H).
98
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0
1 ~ F
N ' l
~ ~
N'r N N
N` N >=0
NC
[00271] 3-(7-(2-(diethylamino)ethyl)-9-((R)-7-fluorochroman-4-yl)-8-oxo-8,9-
dihydro-
7H-purin-2-yl)-3H-benzo[d]imidazole-5-carbonitrile. This compound was
synthesized by
the method described in example 41 using 2-chloro-N,N-diethylethylamine
hydrochloride.
1H NMR (300 MHz, CDC13): 6 8.91(s, 1H), 8.64 (s, 1H), 8.28 (s, 1H), 7.84 (d,
1H), 7.59
(d, 1H), 6.8-7.0 (m, 2H), 6.4-6.6 (m, 1H), 5.86 (br t, 1H), 4.5-4.7 (m, 1H),
4.3-4.4 (m, 1H),
4.0-4.1(m, 2H), 2.8-3.0 (m, 3H), 2.58 (q, 4H), 2.3-2.4 (m, 11-1), 0.96 (t,
6H).
Jak3 kinase assay
[00272] Human Jak3 cDNA was amplified by PCR. A fragment encoding the
catalytic
domain of Jak3 (508aa to 1 124aa) was ligated with GST at 5' end. This fused
GST-Jak3
DNA fragment was cloned into the EcoRl site of the donor plasmid pFastBac 1
(Life
Technologies #10359-016). The transformation, transposition, and transfection
of insect
cells (SD) were performed according to the manufacture's instructions. The
cell lysate
containing recombinant GST-Jak3 was used in the kinase assay. Anti-GST
antibody (10
g/rnl, Sigma #G1417) was coated onto a 384-well plate at 4 C overnight. Cell
lysate
containing GST-Jak3 (1;100 dilution) was added to the anti-GST coated plates,
and GST-
Jak3 was captured by immobilized anti-GST antibody. Testing compounds and
substrate
mix (50 mM HEPES, pH 7, 0.5 mM Na3VO4, 25 mM MgC12, 1 mM DTT, 0.005% BSA, 1
99
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
pM ATP, and 4.5 g/ml liiotinyl poly-G1u,Ala,Tyr) were added to the plate to
initiate the
reaction. After a 60-min incubation, the reaction was stopped by 4 mM EDTA,
and
phosphorylation of biotinyl poly-G1u,Ala,Tyr was detected using 17 g/ml Cy5-
streptavidin (Amersharn, #PA92005) and 2.7 g/m1 Europium-conjugated anti-
phosphotyrosine antibody (PerkinEhner #AD0069) using homogeneous time-resolved
fluorescence (HTRF) technology.
Jak3 ceIlular assay
[00273] The mouse F7 pre-B lymphocyte cell line was used for the cellular Jak3
assay.
Human IL-2R(3c cDNA is stably expressed in F7 cells (Kawahara et al., 1995).
F7 cells
were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum
plus
IL-3. Cells (30,000 cells/well) in serum-free medium were seeded in 96-well
plates for the
cell proliferation assay. Testing compounds were added to cells, followed by
the addition
of IL-2 (fina120 ng/ml). After a 24-h incubation, the number of viable cells
was
determined by the Ce1lTiter-Glo Luminescent Cell Viability Assay kit (Promega,
#G7573)
according to the manufacturer's instructions.
[00274] The results of testing of representative species are shown below. The
compounds in Table 1 exhibited IC50 less than 100nM. The compounds in Table 2
exhibited IC50 between 101 nM and 1 M.
100
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
Synthesis Reference
Example No. TABLE 1
F
O
N_ )
501
f\N N N
F
y O
N ~
N
NC ,
CH3
F
O
N ~, I \
_
N N N
F
502 I o
N
N
I
HO
101
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N
~ N N
Example 42 503 N
O
.r---
N N >==
NC
NC
O
rr
N~` ~'=r,,~,~~ I
504 NIN
N F
O
N N
NC
CH3
F
O
N
N n
505 N N N
Y O
N N
CH3
CN
102
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
N~ N
506 N O
N
N
N "~
OH
CN
F
Q
~ F
507 NIN N
N 0
N
NC 0
1
H3Gf II~'O
0
103
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N
508
' ~ N N
N F
O
N
NC
CH3
0
F
N N
509 ` N o
N
, N
N~
\-NOH
CN
104
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
I
510 NIN~ O F
N
NC
NC
F
O
N~_
N
511 cJ(N F
o N N{CH3}2
NC
105
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
ro
N co
512 \ N `N
)
N F
I O
.--r"
N N
NC Et
F
a
N-- N
Example 44 513 N N
N >=O
N
NC
COOH
O
N
514 "`'~~ N N
~- N
~ ~ I O
N
NC
CH3
106
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N Fr~ir
-
Example 43 515 N Ny N
F
~
N N
NC
\-,COOMe
F
O
N ,r 1
516
F
N ~N N
>~o
~- N
NC Et
107
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N~~
N
517 >=--O
N
NC
N
1 ~
O
/ I
'fff.f` F
N~ N
518 N 0
N
, N
N`~
CH3
H3C
CN OH
108
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
`
F
N~ N
519 N o
N
N "--~
NC
F
O F
N~ N
520 N O
N
N
I N
f
CN OH
109
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
N O
521 N N N F
y C
N
N
NC CH3
O
N's
N
.~--'
~ `+=
Example 40 522 F
F
OH
F
co
~
N ~
"J~
523 C
N
NC ~
Et
110
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N,`
~ '.
~ ~ N N N
Example 39 524
I C
N~
NC
OCH3
r O N F
-
525 N 'N r
N
N o
NC ~
CH3
0
N
1
Example 41 526 I ~ N ~ N N
N >==o
N
NC \,,,N(CH3)2
111
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
,f I
r 1\
N ,,
527 '~~
/ ~ N 'fN N
I O
~-,-"
N ~ N
F
CH3
O
F
528 N /,.N
NC N(Et}z
F
O
N~'
529
A
N >==O F
N
F \,,N(CH3)2
112
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
rf~ ~
530 I ~ N ~N N F
f_---
N >=O
N
F
OCH3
O
N s~f~ J 531
N
N
>=O
F 1
CH3
113
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
0
C
N
_~~'~
532
N F
r----'
C
N~ N
F
CH3
0
533 N
N
C
N \-,CN
O
rF
N
534 N ~,N
N
J I O
N
~'`~ N
NC ,
Et
114
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0
N
N
535
l
_----
N
NC
F
O
I
N_
536 N N~ N F
~.r I O
N / N
NC CH3 --< CH3
115
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
N,~
537 QNyN
O
NN
CI
CH3
O
Nõ~
~--"
,,
538 N N N
~ F
F \,,,N(CH3)2
O
N~~-
539 N N N
F
I O
N >=--
N
F \~`" CN
116
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
Example 540 N N N F
l o
N
N
F \H3
O
N~
541 N
>=O
N ~ N
NC CH3
117
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
4
Nõ`
Example 542
\\ N N N F
I C
N
N
~H3
F
N
. ~~.
543
~ N N N
f~ (
>=-- O
N
CH3
118
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
N
N_
r---'
NIN N
544 f N >~o
N
F ,
CH3
O
N_
545 NIN N
F
.-- J N
N
,
CH3
F
0
N c
Ir
546
N N
N F
N
N O
Et
119
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
0 701 N N
N
~
N N OH
N `
F
O
N~
702 N ~/N N F
1 ~
N N
If
O
120
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N_
N N N
703 y
F
N
N
NHZ
F
O
'~.
'~.
N II N N F
704 N N
N
HN
O
121
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
0
N F
705
N
N
N
0
N
~ \N-p
~~
N
F
c,o 706
F
N N N
0
N~` 1i N "
O
N
122
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N
F
707 N N
'/.
N :D N
N
N
N
0
9
N N
~
N
708 N (
N
N ~-.
N
123
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
F
N~
N O
709 N
N
N
F
F
O
CI
N~
N N N
F
, O
710 ff N N
If
N
HN
` H3
124
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
N~
711 '
F
N N N
~
z
N N
N
O
N~\
9
N N
712 N j ~~ O
N
NH
Nf
125
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
F
713 N N N
N
NC
NH
F
0
cd/
N
N N N
714 _-- ~ ~
N N
N
N
'CL,
0
126
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
N ~
i
X
.
N N N
O
715 --"^ N
F
O O
-71
N OMe
N
716 \ N N
~ ` N
( O
~-~ N
M HN
NC
127
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N
N
~--~--
~ N N N
O
N 717 N
F
O
N
OH
F
O
NN N N
Co
718 I o
N
N
N
H2N
128
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N ~'.. !
>==o
N II N N
719 N -/~ N
NC
N
0
F
O
~
.
`
N
~
~---'
N N N
y ~
710 N N
F
O
N
OH
129
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00275] The results oftesting of representative species are shown below. The
compounds in Table 1 exhibited JC$o less than lOOnM. The compounds in Table 2
exhibited ICso between 101 nM and 1 M.
Synthesis Reference
Example No. TABLE 1
F
O
~
N_
501
I N(N N
F
N >==o
NC
CH3
F
0
r
N'`" Ef'`s
N N N
F
502 I >==o
N
X,
HO
130
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N
`t = ~.~
~ N N
Example 42 503 N
~~ N
N
NC
NC
O
.~~
r''~
504 N N
N F
I O
N
NC
CH3
F
O
l N
N n
505 N N N
y O
CH3
CN
131
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O \
F
N~ N
506 N O
N
N'1-
OH
CN
F
N
00
N N
0 N
507 yN
N
NC 0
I I
H3C'I I~C
0
132
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
~
~ t
N,'r~~i '/ ~
508 N ~,- N
F
__--
N ~* N
NC CH3
0
F
N~ N
509 N
N
N
N
~ H
CN
133
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
~ixi
510 N N N F
y ~
NC
NC
F
O
N~4
511 =
N N F
N ~ N
NC \,,N(CH3)2
134
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
r0
~
N,`~'
512 \ N N
N F
N >=--O
N
NC
Et
F
0
N
Example 44 513 ~ ~ N N
N N
O F
N
NC
COOH
"+.~~ >==
r
514 '"~~ N
~ f O
N
N
NC
CH3
135
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N
Example 43 515 N N N
F
~
N
N
NC
\-,COOMe
F
O
~
~
N'" "fi /"~
516
N
F
o
.~--`
N N >==
NC Et
136
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
N /jf~ef I ~
N N
517 I >=--:o
N
NC
d
N
O a
F
N~ N N
518 O
,
N
N
N~
CH3
H3C
CN OH
137
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
F
N~ N
C
519
N
N
"~r
N
N"-~
NC
F
O F
520 N
r==\
N
N
N ".,--
CN OH
138
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
521 N N N F
~ O
N
N
NC , H3
_---^'"
N ~ N
Example 40 522 F
N ~ N
F
OH
F
0
r ~ `~`~y~
523
O
N ,``~=.
N
NC Et
139
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
s ' \
N~,,,` ~s=~.
N N
Example 39 524
N~ N
NC
OCH3
O
= I ~
N.~..~ f='=.
""-~ f=
`y = ~...-
525 N fN N
N >=:=o
N
NC
CH3
O
N~~
-
Example 41 526 N
N O
N
NC N(CHs)z
140
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
r f \
N
527
\ N ~N
~ \ N
>==o
N~. N
F , H3
O
F
N
528
I O
N~. N
1 N{Et}2
NG
F
O
N~'
529
N N F
o
N N >==
N{CH~Z
F
141
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
0
r,,
N.,,~
530 N N F
_--~ (
N >==o
N
F
OCH3
~
N
531 N
N >=O
N
F , H3
142
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
ro
532
N F
N
F CH3
O
I
F
533 N C 0
N N
~
N
\\---CN
~ \\ F
! f~
534 N
0
N N
NC Et
143
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0
N
N~IN N
535
l o
N
NC
F
O
Nõ~
536 , N N F
~
~_ y o
N
NC CH3
CH3
144
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
\
N I~ !
d/
_r
537 \ N N
N
~.... ~ >==O
N
CI
CH3
O
N,~~
538 ~ N f N F
..~ I
N >=O
N
\,,N(cH3)2
F
O
N'Z
539 NIN N C F
N
F V"CN
145
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
Example 540 N N. N F
I C
''rf N
F CH3
O
N
541
>=-::o
N~ N
NC CH3
146
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N=,,,rf~!! I
Example 542
NiN N F
O
~-~ N !
N
CH3
F
..~---'
O
N
~`.
543
~ N N N
O
N
CH3
147
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
N
N
~
~_
N
NIN N
544
0
N
N
F CH3
d
N
1 `
~ ...~
545 N N F
Yi o N N
,
C:H3
F
0
N
~1 ~'~ ~='/
546 N
N
O F
N
N
,t
148
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
I
F
701 N
N
N N O H
N
N `
F
O
r4) 702 N f N F
1 >==O
N N
N
0
149
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
0
N
N
N N\ N
703 F
, o
N
N
II
N
NHz
F
O
'~.
'~.
~ ~õ~...
N~N N F
O
'-- ~
704 N N
N
HN
/----o
150
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
705 F
N N
N N
~ `N-0
~_
N
F
0
N:
706 F
N N
o
~~-- N N 1'
N
151
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N~ F
N N
707 ~ N ~ D N~ N
11
O
N.~
N
708 N
N _--~ /
N
I I
N
152
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
0
1
F
N:===\ N p,
N
709
N
N '--~
F %
N
F
co
NyN N
710 N N
N
HN
CHs
153
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
I
yf~~jr"
711 Nn F
N N
o
N N Nx
O
N7---
9
N
~
71Z N ~ `o 0
f N N
NH
N
154
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
~
~f'~~.,,
N~--~
713 F
N N
N N
NC
NH
F
O
N~~
N N N F
714 X o
N =~
ll
N
O
155
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O
N ~. /
4 ~J
N N
715 N
F
O O
N OMe
O
c
N.~~~ ~. /
...r"'
716 N IN\ N
HN
NC
156
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
Q
N_ --, / N
NIN N
717 N
F
N
OH
F
rc,
N
N N N
718 + F
~--f N 0
-~ N
N
H2N
157
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
O
r
~--~'
N N N
II
719 ff N
NC
N
0
F
O
N++~+~~~..~~~~
~ M N N
`~ I O
710 N N
F
O Z:: ~- ~,
N
<:]-",OH
IL-2-induced IFN-y production in the mouse
158
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00276] Administration of IL-2 leads to an increase in serum IFN-y in the
mouse due to
NK secretion of the cytokine (Thornton S, Kuhn KA, Finkelman FD and Hirsch R.
NK
cells secrete high levels of IFN-7 in response to in vivo administration of
ILr2. Eur J
Immunol 2001 31:3355-3360). The experiment was carried out essentially
according to
the protocol in Thornton et al. and the test compounds were administered in
order to
determine the level of inhibition attained. In summary, female BALB/c mice
were fasted
for 12-18 hours before a study but had free access to water at all times. Test
compounds
were administered by gavage one hour before intraperitoneal injection of IL-2
and capture
antibody. At termination of the studies, the mice were sacrificed by carbon
dioxide
inhalation, terminal blood samples were collected by cardiac puncture and
serum was
generated. Serum was stored frozen until it was assayed for IFN-y, as
described by the kit
manufacturer (BD PharmingenTM, San Diego, CA).
[00277] Using the above method, the compounds of experimental examples 16, 28,
and
Table 1 examples 501, 504 and 505, at 30 mg/kg, were shown to inhibit IL-2-
induced IFN-
gamma production by >40% in vivo in the mouse. A reference compound, CP690550,
exhibited 96% inhibition at 30 mg/kg in this screen.
[00278] The 7-substituted purinones exhibit increased selectivity for Jak3
compared to
their 7-unsubstituted congeners.
[00279] Aurora A kinase assay was performed using a fluorescence polarization
format.
A 100 nM solution of fluorescien-labeled FAM PKAtide (Molecular Devices), the
substrate for Aurora A (Upstate Biotechnology), was incubated with Aurora A(S0
ng/ml)
and 30 mM ATP at room temperature for 1 hour in the presence of an appropriate
concentration oftest inhibitor. The reaction was terminated by adding IMAP
Progressive
Binding Reagent mix according to the manufacturer's instructions (Molecular
Devices).
The polarization signal was detected using Aquest (Molecular Devices).
159
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
[00280] Some comparative examples are shown below. On the left is the 7-
unsubstituted
compound and its ratio of Jak3 to Aurora A inhibition given as "x-fold"
selectivity. On the
right are shown the 7-substituted congeners along with theiT selectivity. All
of the ICso's
for Jak3 are below 100 nM.
160
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
7-UNSUBSTITUTED 7-SUBSTITUTED
---C
a
~
N 1
N N F N~-NJN _ ~o ~~~ ~~0
N NC CH
NC 19-fold H 112~-fold 3
N //--C - F N /--o F
N %~
N
N N ~ /
F N ~ X ~a F N
N N
14-fold H 58 fold CH3
O N.,, ,~-- C
N ~ .____~
N
~" N
N
NC N~ _p NG NN` ~C
N
15-fold H 4-1-fold CH3
`N.
N \,-N
NG NN ~C
56-fold ~CN
161
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F
O F
r
N N.,r-~N N >_O F
I-' .N N
N`
~~
~O F
N
N~ NC
or
3
NC H 192-fold CH3
13-fold
,- O ,~
~~
~~
NN,~--'N F
O
N
NC Et
'~ 40-fold O F
f-
N~ f ~ ,1
N
,,N,~~ N F
-
~~
~_~ ~ ~
:1 -O
NC `'-COOMe
'l 73-fold F
~ O
N~ `=: ~..,t~~'1
I~ ;-1 N N , N\_ F
N~---0
N
NC 0
11 -fold
OH
F
N -,
.~-
~ J N N N F
~
.~ ~
~ ~;0
NC ~~OH
200-fold
162
CA 02669686 2009-05-12
WO 2008/060301 PCT/US2006/061004
F F
0 f /1 O\~
c
~ j ~N.~ 3
N ~ .
(NN N F N~ F
l >==o 0
~
~
NC H N NC
73-fold 236-fold CH3
F
N;
N
NC
Et
1 23-fold
F
;N-=~
N N
N, F
N O
NC ~ ~,"- = ~
'_,N(CH3)Z
724-fold
[00281] Although the foregoing invention has been described in some detail for
purposes
of illustration, it will be readily apparent to one skilled in the art that
changes and
modifications may be made without departing from the scope of the invention
described
herein.
163