Note: Descriptions are shown in the official language in which they were submitted.
CA 02676892 2010-06-14
KETAL AMIDE COMPOUNDS, METHODS OF MAKING, AND APPLICATIONS
This is a division of Canadian Patent Application No. 2,648,012 filed on
January 15,
2009.
FIELD OF THE INVENTION
The invention relates to ketal amide functional compounds. The compounds are
synthesized by the reaction of ketal acids or ketal esters with polyamines.
The
invention further relates methodology useful to make the ketal amide
functional
compounds. The invention further relates to applications of ketal amide
functional
compounds in various formulations and articles.
BACKGROUND
Many known chemical products such as surfactants, plasticizers, solvents, and
polymers are currently manufactured from non-renewable, expensive, petroleum-
derived or natural gas-derived feedstock compounds. High raw material costs
and
uncertainty of future supplies requires the discovery and development of
surfactants, plasticizers, solvents, and polymers that can be made from
inexpensive
renewable biomass-derived feedstocks and by simple chemical methods. Using
renewable resources as feedstocks for chemical processes will reduce the
demand
on non-renewable fossil fuels currently used in the chemical industry and
reduce
the overall production of carbon dioxide, the most notable greenhouse gas.
A potential source of materials that are useful as chemical building blocks
are cyclic ketals and acetals of oxocarboxylates with polyols. It is known,
for
1
CA 02676892 2010-06-14
example, that polyhydric alcohols, or polyols, having 1,2 and 1,3 hydroxy
conformations can react with a ketone or aldehyde to form a cyclic ketal or an
acetal (Carey, F.A. and Sundberg, R.J., "Advanced Organic Chemistry Part B:
Reactions and Synthesis" 2nd ed., 1983, Plenum Press, NY, NY, p. 544). The
1,2
and 1,3 configurations of hydroxyl groups on a hydrocarbon chain are shown
below
as (a) and (b), respectively.
HO\ H OH OH
õC-C~
(a) (b)
Diols such as 1,2-ethane diol (ethylene glycol) and 1,3 propanediol (propylene
glycol) are examples of such polyols. Diols having a 1,2 hydroxyl group
configuration will form dioxolanes when reacted with ketone or aldehyde
moieties,
while 1,3 diols will form dioxanes.
Ketal acids and esters are starting materials from which the compounds of
the invention are synthesized. Ketals of glycerol and levulinic acid or an
ester
thereof are described in U.S. Patent Publication No. 2008/0242721. The ketal
reaction product of glycerol with a levulinate results in a monoketal acid or
monoketal ester as shown below,
O OH O
H+ O
A--~y OR HO OH 0" HO-__ O
OR
O
wherein R is hydrogen or an alkyl group. Combining a levulinate ester with
glycerol
provides a levulinate-glycerol ketal that is bifunctional and available from
100%
renewable feedstocks. The levulinate-glycerol ketals are useful for synthesis
of a
wide variety of surfactants, plasticizers, polymers, and the like. Other
monoketals
synthesized from various oxocarboxylic acids or esters thereof such as
acetoacetates and pyruvates, with triols such as 1,1,1-trimethylolpropane and
2
CA 02676892 2010-06-14
1,1,1-trimethylolethane, are described in International Patent Publication WO
2009/032905. The monoketal acids and esters have the general structure
0 R3
H2
HO-C O-X
R4 bo
0
wherein a is 0 or 1, b is 0 or 1, R3 is hydrogen or an alkyl group having
between 1
and 12 carbons, R4 is an alkyl group having between 1 and 12 carbons, and X is
any substituent. Substituents R3 and R4 may further be substituted with one or
more functional groups, such as halogen, ether, cyano, and the like. These
materials are useful as, or for the synthesis of, a wide variety of
surfactants,
plasticizers, polymers, and the like. The compounds described in these
applications are capable of self condensation to provide oligomers or polymers
having hydroxyl endgroups, and further are capable of condensation with one or
more diols to give oligomeric or polymeric polyols. Monoketals are starting
materials for the synthesis of the compounds of the invention.
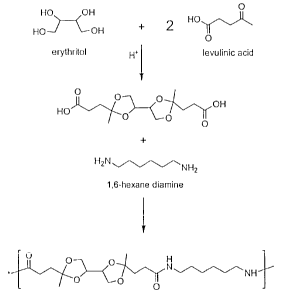
Polyketal acids and polyketal esters are compounds having at least two
contiguous or semi-contiguous ketal acid or ketal ester moieties per molecule.
Various oxocarboxylic acids and esters are useful in synthesizing the
disclosed
compounds, as well as various tetrols and higher polyols. In one nonlimiting
example, combining a levulinate ester with erythritol provides a bisketal
starting
material from renewable feedstocks. This reaction is shown below,
HO
HO O O 0
+ 2 RO H40.RO 0 0 OR
HO O
HO
wherein R is hydrogen or an alkyl group. Further, the polyketal acids and
esters
are useful as, or for the synthesis of, a wide variety of surfactants,
plasticizers,
polymers, and the like, as disclosed in the application.
3
CA 02676892 2009-08-19
The synthetic routes described therein are useful as a basis for efficient
reaction of a number
of oxocarboxylic acids and esters thereof with diols, triols, and higher
polyols and are useful
in making any of the above mentioned ketal moieties, all of which are starting
materials for
the compounds of the invention.
SUMMARY
Disclosed herein are ketal amide compounds, which are cyclic ketals and
acetals of
oxocarboxamides. The compounds of the invention are, in sonic embodiments,
synthesized
by the reaction of diamines and higher polyamines with cyclic ketal and acetal
acids, cyclic
ketal and acetal esters, cyclic ketal and acetal polyesters, cyclic polyketal
and polyacetal
acids, cyclic polyketal and polyacetal esters, cyclic polyketal polyesters,
and cyclic
polyacetal polyesters. Polymeric and nonpolymeric compounds, as well as
methods to make
these compounds, are aspects of the invention.
The compounds of the invention are useful in a number of applications.
Nonlimiting
examples of uses for the compounds of the invention include plasticizers,
surfactants,
coalescing solvents, interfacial modifiers, and phase transfer materials in
one or more
formulations. Some of the ketal amide compounds of the invention are employed,
in
embodiments, as monomers in the synthesis of various polymers such as
polyesters,
polyisocyanates, polyurethanes, poly(urethane urea)s, poly(ester urethane)s,
polycarbonates,
polyamides, and copolymers thereof. In other embodiments, the compounds of the
invention
are functionalized with acrylates, methacrylates, allyl or oxirane groups;
these groups are, in
embodiments, further reacted or polymerized. In some embodiments, the multiple
functionalities of the compounds of the invention serve as crosslinking
moieties for one or
more polymeric networks.
In embodiments, the compounds of the invention are reacted to form a polymer
having
a substantial degree of polymerization, that is, a degree of polymerization of
about 2 to 500,
for example in some embodiments about 10 to 200, or about 10 to 100. Glass
transition
temperature of some of polymers of the invention is about 0 C to 110 C, or
about 5 C to
80 C. In some embodiments, the polymers of the invention are ductile. The
polymers of the
invention are, in some embodiments, transparent to visible light and are light
amber to orange
in color. The polymers of the invention, in some embodiments, are hydrophilic
and therefore
4
CA 02676892 2010-06-14
compatible with damp surfaces, enabling certain applications of the polymers
requiring adhesion to a damp, water-coated, or water-saturated surface.
One aspect of the invention is a novel aminolysis methodology. The
methodology is based on use of an organic guanidine type catalyst that drives
the
polymerization of diesters and diamines to form polyamides at surprisingly low
temperatures. Thus, compounds of the invention are employed in conjunction
with
the method of the invention to result in polyamides under mild conditions.
High polymers and crosslinked polymer networks containing one or more
compounds of the invention are useful in a variety of applications. Due to
superior
properties such as high tensile strength and high glass transition
temperatures, the
polymeric compounds of the invention are well suited for many commercially
valuable applications such as use in fibers for nonwoven or woven fabrics;
formation of articles, such as structural members, having high strength; and
many
other applications that typically and advantageously employ polyamides such as
Nylon 6, Nylon 6,6, and other related structures.
Additional advantages and novel features of the invention will be set forth in
part in the description that follows, and in part will become apparent upon
examination of the following, or may be learned through routine
experimentation
upon practice of the invention.
Another aspect of the invention relates to a compound comprising one or
more fragments having structure V:
O Rs 0~0 Rs O
X ~-a 0 O a N-R'
R3R4 R4R3 I
R2 V
wherein
5
CA 02676892 2010-12-23
[3 is an integer of at least 1;
each X is 0 or NR2;
each a is independently 0 or an integer of I to 12;
each R' is independently a linear, branched, or cyclic alkyl, a linear,
branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl group, optionally
comprising one
or more heteroatoms;
each R2 is independently hydrogen or an alkyl group having between 1 and 6
carbon atoms, or when X is NR2, R2 optionally forms a piperazine ring with R';
and
each R3, R4, and R5 is independently hydrogen, a linear, branched, or cyclic
alkyl, a linear, branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl group,
optionally
comprising one or more heteroatoms.
Another aspect of the invention relates to a formulation comprising one or
more compounds as defined above and one or more additional polymeric
compounds, one or more additives, one or more solvents, or a combination
thereof.
Another aspect of the invention relates to an article comprising the
formulation as defined above.
Another aspect of the invention relates to a method of making a polyamide
as defined above, said method comprising:
a. contacting a diester with a diamine,
b. adding about 20-2000ppm, based on the mass of the combined
diester and diamine, of 1,5,7-triazabicyclo[4.4.0]dec-5-ene to form a reaction
mixture, and
c. affecting a reaction between the diester and the diamine to form the
polyamide.
BRIEF DESCRIPTION OF THE DRAWINGS
5a
CA 02676892 2010-06-14
FIG. 1 A - 1 D depict representative synthetic schemes and structures of the
invention.
FIG. 2 is a plot of the stress-strain measurements for a compound of the
invention.
FIG. 3 is a plot of the differential scanning calorimetry measurements for a
compound of the invention.
FIG. 4 is a plot of the differential scanning calorimetry measurements for a
compound of the invention.
FIG. 5 is a plot of the 'H NMR measurements for a compound of the
invention.
FIG. 6 is a plot of the differential scanning calorimetry measurements for a
compound of the invention.
FIG. 7 is a plot of the differential scanning calorimetry measurements for a
compound of the invention.
5b
CA 02676892 2009-08-19
DETAILED DESCRIPTION
Various embodiments will be described in detail. Reference to various
embodiments
does not limit the scope of the claims attached hereto. Additionally, any
examples set forth
in this specification are not intended to be limiting and merely set forth
some of the many
possible embodiments for the appended claims.
The compounds of the invention have, in embodiments, one or more isomers.
Where
an isomer can exist, it should be understood that the invention embodies all
isomers thereof,
including stereoisomers, conformational isomers, and cis, trans isomers;
isolated isomers
thereof; and mixtures thereof.
STRUCTURE I.
In some embodiments, the invention embodies compounds having Stricture I:
R9
R8 b
O R5 R
R7 [jR1
O a
R6 R3 R4 O a
I
wherein
R is a linear, branched, or cyclic alkyl, alkenyl, or alkynyl group, or an
aryl or
alkaryl group, or a polymeric group, wherein the alkyl, alkenyl, aryl,
alkaryl, or
polymeric groups can have one or more heteroatoms; or R' together with R2 can
form
a cyclic structure that is the residue of a cyclic diamine, such as
piperazine;
R2 is hydrogen or an alkyl group having 1 to 6 carbon atoms, wherein each R2
may be
the sane or different; or R2 together with R' can form a cyclic structure that
is the
residue of a cyclic diamine, such as piperazine;
R3 and R4 are independently hydrogen, halogen, amine, mercapto, phosphate,
phosphonooxy, silyl, siloxane, alkynyl, or a linear, branched, or cyclic alkyl
or
alkenyl groups having 1 to 18 carbon atoms, or an aryl or alkaryl group,
wherein the
6
CA 02676892 2009-08-19
alkyl, alkenyl, aryl, or alkaryl groups can have one or more heteroatoms, and
wherein
each R3 and R 4 may be the same or different;
R 5 is hydrogen, alkynyl, or a linear, branched, or cyclic alkyl or alkenyl
group having
I to 18 carbon atoms, or an aryl or alkaryl group, wherein the alkyl, alkenyl,
aryl, or
alkaryl groups can have one or more heteroatoms, and wherein each R 5 may be
the
same or different;
R6, R8, and R9 are independently hydrogen, halogen, or an alkyl group having
between I and 6 carbon atoms and optionally one or more heteroatoms, and
wherein
each R6, R8, and R9 may be the same or different;
R7 is -CH2OH, -CH2NH2, -NH2, -CH2SH, -CH2Br,
0
II H2 H2 H2
A-C X-C O=C=N-B-X-C - D-X-C - -
or
wherein A, B, and D are independently linear, branched, or cyclic alkyl,
alkenyl, or
alkynyl groups, or aryl or alkaryl groups, wherein the alkyl, alkenyl,
alkynyl, aryl, or
alkaryl groups have I to 36 carbon atoms and can further have one or more
heteroatoms; and X is 0, S, or NH; wherein each R7 may be the same or
different;
a is 0 or an integer of between 1 and 12, and each a is the same or different;
b is 0 or 1, wherein b = 0 indicates a five membered ring,
R8
R7 X0 R5
R6 O~
and b = 1 indicates a 6 membered ring,
R9
R8 0 0Ra
R7 O
R6
and b is the same or different for each occurrence; and
7
CA 02676892 2010-06-14
a is an integer of 1 or more.
"Heteroatoms" present in the one or more R1, R3, R4, R5, R6, R', R8, R9, or A
groups of Structure I can include, for example, halogen, nitrogen, oxygen,
sulfur,
silicon, phosphorus, and the like and can be embodied in a functional group
such
as amino, carbonate, imide, amide, sulfone, sulfonamide, urethane, mercapto,
disulfide, ether, ester, phosphate, phosphonooxy, silane, or silyl functional
groups,
or a combination thereof. The "polymeric group" of R1 is not particularly
limited and
can be derived from, for example, a polyethylene having multiple amino
residues, a
poly(ethyleneimine), a polyoxyalkyleneamine, a branched polyurea having
primary
amide groups, or some other polymer. The polymeric group can have, in
embodiments, a linear, branched, hyperbranched, or starburst morphology. The
polymer can be, in embodiments, present as a surface, as a layer on top of a
surface, or as a particle surface, a porous particle interior, and the like.
In some embodiments, the compounds of Structure I are made by the
reaction of ketal esters with diamines or higher polyamines. The term "ketal
ester"
means the cyclic ketal or acetal of a keto acid, semialdehyde, or ester
thereof. The
ketal esters are, in embodiments, any of those disclosed in International
Patent
Publication No. WO 2009/032905, or U.S. Patent Publication No. 2008/0242721.
In
other embodiments, the ketal esters are made from oxocarboxylates as disclosed
in
the incorporated references and diols capable of forming cyclic ketals with
ketone
or aldehyde moieties. In general, such diols are those having 1,2 and 1,3
hydroxy
conformations (Carey, F.A. and Sundberg, R.J., "Advanced Organic Chemistry
Part
B: Reactions and Synthesis" 2nd ed., 1983, Plenum Press, NY, NY, p. 544).
The
8
CA 02676892 2010-06-14
1,2 and 1,3 configurations of hydroxyl groups on a hydrocarbon chain are shown
below as (a) and (b), respectively.
HO\ , H iH iH
-IC
/ 1
S
(a) (b)
8a
CA 02676892 2009-08-19
Diols such as 1,2-ethane diol and 1,3 propanediol are examples of such diols.
Other
suitable diols include any of those having (a) or (b) type configurations,
such as for example
1,2-propanediol, 2,2-dimethyl-1,3-propanediol (neopentyl glycol), 3-
aminopropane-1,2-diol
(aminoglycerol), 3-sulfanylpropane-1,2-diol (thioglycerol), 1,4-bis-
sulfanylbutane-2,3-diol
(dithiothreitol), 1,2-butanediol, 1,3-butanediol, cyclohexane-1,2-diol, 1,4-
dioxane-2,3-diol,
3-butene-1,2-diol, benzene-1,2-diol (catechol), 3-chlorocatechol, indane-l,2-
diol, tartaric
acid, and 2,3-dihydroxyisovaleric acid.
Employing a triol to form a ketal ester imparts, in some embodiments, a
methylol
(e.g. -CH2OH) group to the resulting ketal ester. Trimethylolethane,
trimethylolpropane, and
glycerol are examples of triols that react with oxocarboxylates to form ketal
esters having a
methylol group adjacent to the ketal functionality. The methylol group remains
intact when a
diamine or higher polyamine is reacted with the ester group, providing two or
more hydroxyl
moieties in the resulting poly(ketal amide). In some embodiments, the triol
employed in the
reaction is glycerol. Glycerol is an inexpensive renewable compound that is
readily available
as a by-product of biodiesel production or via fermentation of carbohydrates.
Since glycerol
forms the backbone of triglycerides, it is produced upon saponification or
transesterification
of these compounds. Soap-making and biodiesel production are respective
examples.
Glycerol is a roughly 10% by-product of biodiesel manufacture, via
transesterification of
vegetable oils.
In some embodiments of Structure I, RI is -(CH2)3-. In other embodiments, RI
is 1,2-
cyclohexyl. In still other embodiments, R' is -(CH2)6-. In some embodiments, a
is 0, 1, or 2.
In other embodiments, the value of a is 2 and all R3 and R4 are hydrogen. In
embodiments,
R 5 is methyl. In embodiments, b is 0 and R6 and R8 are hydrogen.
In some embodiments, the compounds of Structure I are formed from the reaction
of
a diamine or higher polyamine with the ketal formed from pyruvic acid (a = 0),
acetoacetic
acid (a = 1, R3, R4 = H), or levulinic acid (a = 2, all R3, R4 = H) or an
ester thereof with
glycerol (b = 0, R7 = CH2OH, R6, R8 = H), 1,1,1-trimethylo]prop ane (b = 1, R7
= -CH2OH,
R8 = CH2CH3, R6, R' = H), or 1,1,1-trimethylolethane (b = 1, R7 = -CH2OH, R8 =
CH3, R6,
R9 = H). Levulinic acid is an abundant feedstock that is prepared on an
industrial scale by
acidic degradation of hexoses and hexose-containing polysaccharides such as
cellulose,
starch, sucrose, and the like.
9
CA 02676892 2009-08-19
Suitable diamines and higher polyamines that are, in embodiments, reacted with
a
ketal acid or ester to form the poly(ketal amide)s of Structure I include
hydrazine, ethane-1,2-
diamine, 1,6-hexanediamine, but-2-ene-l,4-diamine, Metformin, butane-1,4-
diamine,
propane-1,2- diamine, piperazine, 2,2,4-trimethyl-1,6-hexanediamine, 2,4,4-
trimethyl-1,6-
hexanediamine, benzene- 1,3-diamitie, 2-methylbenzene-1,3-diamine, 4-
chlorobenzene-1,3-
diamine, methanediamine, 1,3,5-triazine-2,4,6-tiiamine, N-(2-aminoethyl)ethane-
1,2-
diamine, N-(6-aminohexyl)hexane-1,6-diamine, N,N'-bis(2-aminoethyl)ethane-1,2-
diamine,
N-[2-(3-aminopropylamino)ethyl]propane-1,3-diamine, 4-(3,4-di
aminophenyl)benzene-1,2-
diamine, spennine (N,N'-bis(3-aminopropyl)butane-1,4-diamine), diethylene
triamine,
dipropylene triamine, dihexylene triamine, 1,2,4-triazole-3,4,5-triamine,
2,4,5-
triaminotoluene, melamine (1,3,5-triazine-2,4,6-triamine), benzene-1,3,5-
tiiamine,
triethylene tetramine, norspennine, N-[2-(3-aminopropylamino)ethyl] propane-
1,3-diamine,
4-(3,4-diaminophenyl)benzene-1,2-diamine, a polyethyleneimine, a
polyoxyalkyleneamine
having two or more amine groups, such as those sold under the trade name
JEFFAMINE R,
(available from the Huntsman Corp. of Salt Lake City, UT), or any diamine or
higher amine
compound such as those sold under the trade name ELASTAMINE R (available from
the
Huntsman Corporation).
The compounds of Structure I are not particularly limited as to the method
employed
to make them; in general, any of the known methods of forming an amide from a
carboxylic
acid or ester thereof may be employed to synthesize a compound having
Structure I from a
ketal acid or ester thereof and a diamine or higher polyamine. In some
embodiments, a ketal
acid or ketal ester and a polyamine are simply mixed together and heated to a
temperature of
about 150 C to 250 C, or about 180 C to 210 C to affect the reaction to form
the compounds
of Structure I. In some such embodiments, water or alcohol evolved during the
course of the
reaction is removed. In some embodiments, catalysts are employed in the
synthesis of the
compounds of Structure I. For example, titanium alkoxides such as titanium
(IV) butoxide or
titanium (IV) isopropoxide are employed in some embodiments to catalyze the
reaction. In
other embodiments, a Lewis acid catalyst such as antimony trichloride,
aluminum chloride,
antimony trifluoride, ferric chloride, antimony pentachloride, niobium
pentachloride,
tantalum tetrachloride, titanium tetrachloride, boron trifluoride, antimony
pentafluoride,
stannic fluoride, aluminum bromide, thallium trichloride, uranyl nitrate,
uranium
CA 02676892 2010-06-14
tetrachloride, uranyl acetate, uranium oxides such as U02, and the like are
employed in the synthesis of the compounds of Structure I. In embodiments
where
a catalyst is employed, the reaction proceeds at temperatures as low as about
200 C, or between about 80 C and 200 C, or even as low as about 80 C to 180 C.
In another method that is employed, in embodiments, to synthesize the
compounds of Structure I, an oxocarboxylate is first reacted with a diamine or
higher polyamine to form a bisamide, trisamide, or higher amide functional
intermediate compound. The amide functional intermediate compound is then
reacted with a diol such as those disclosed above, or a triol or higher polyol
such as
those disclosed in International Patent Publication No. WO 2009/032905, or
U.S.
Patent Publication No. 2008/0242721, to form the ketal moiety. For example,
where the diol used to form the ketal is 1-aminoglycerol, it is advantageous
in some
embodiments to first employ a polyamine to form an amide functional
intermediate,
then employ the aminoglycerol to form the ketal functionality, because it
avoids a
side reaction between aminoglycerol and the carboxyl moiety of the
oxocarboxylate.
Some representative compounds having Structure I and made from of ketal
esters of pyruvic acid, acetoacetic acid, or levulinic acid with glycerol,
aminoglycerol, or trimethylolpropane and further reacted with diamines or
triamines
are shown below as la - Ic; it will be understood that these representative
examples are not limiting to the overall body of compounds encompassed by
Structure I but rather are intended to illustrate the breadth of structures
available.
H
HOO Hz N- CH H OOH
~C -f ~ z)s_N~Cz~
O O
la
11
CA 02676892 2010-06-14
0
HzN~O ^N /v ~ ~ _o p NHz
lb
l la
CA 02676892 2009-08-19
HO OH
~O NNN O O
p O O O
HO O
Ic
Structure I, Embodiment 1.
Where R7 of Structure I is amino (-NH2), methylol (-CH2OH), methylamino (-
CH2NH2), or methylthio (-CH2SH), the compounds of Structure I are, in some
embodiments,
soluble in water and lower alcohols and hydrophilic coating formulations. In
other
embodiments, for example wherein R7 is amino, methylol, methylamino, or
methylthio; and
R' is a long chain alkyl group, for example dodecyl, the compounds of
Structure I are soluble
in hydrophobic formulations. In yet other embodiments where R7 is amino,
methylol,
methylamino, or methylthio, the various other R groups of Stricture I
determine solubility in
one or more formulations; in some such embodiments, the compounds of Structure
I are
surfactants, solubilizers, interfacial modifiers, and the like.
For the purposes of the invention, compounds having Structure I wherein R7 is
methylol are referred to as "poly(ketal amide)ols." In similar embodiments,
compounds of
Structure I are, where R7 is amino or methylamino are poly(ketal amide)
amines; and where
R7 is methylthio, poly(ketal amide) thiols. Where a is 2, the compounds of
Structure I are
bisketal amide diols (or diamines or dithiols); where a is 3, the compounds of
Structure I are
trisketal amide triols (or triamines or trithiols); and so on. Such poly(ketal
amide)s having
two or more reactive hydrogen atoms have synthetic applicability to a wide
variety of
polymeric structures. In embodiments where R7 is described as methylol, it
will be
understood for the purposes of the discussion that follows that in many
embodiments the
methylol group can be replaced with amino, methylamino, or methylthiol to
provide
compounds of similar reactivity in the corresponding structure formation
and/or utility in the
corresponding formulation.
The various compounds of Structure I, Embodiment I are useful for
incorporation
into various formulations. For example, where the compounds are hydrophilic,
they are
useful as incorporated into coating formulations such as concrete coatings,
floor coatings,
12
CA 02676892 2009-08-19
and other coatings designed for damp substrates. Due to the hydrophilicity of
some
compounds of Structure I, Embodiment 1, they are miscible in waterborne and
other
hydrophilic coating formulations, and provide a compound of substantial
molecular weight;
this in turn allows the compounds, in some embodiments, to act as a coalescing
solvent as the
coating dries; in other embodiments, these compounds provide a means for such
coatings to
gain better adhesion to the desired substrate by increasing compatibility
between the coating
and the substrate or by some other means. In other embodiments, the poly(ketal
amide)ols,
the poly(ketal amide)amines, and the poly(ketal amide)thiols are useful as
nucleating agents
for polymers in the solid state. For example, it is known that polylactide
polymers are
nucleated by bisamides to induce crystal formation; see, for example,
Nishimura et al., U.S.
Patent Publication No. 2005/0165142, Tweed et al., U.S. Patent Publication No.
2007/0116909, and McDaniel, U.S. Patent Publication No. 2007/0003774; poly-a-
olefins
such as poly(l-butene) are also nucleated by bisamide compounds; see, for
example,
Chatterjee, U.S. Patent Nos. 4,645,792 and 4,322,503. The compounds of
Structure I,
Embodiment I are, in some embodiments, similarly useful as nucleating agents
for one or
more polymers that may be polylactide polymers, poly-a-olefins, or various
other polymer
structures.
Structure 1, Embodiment IA.
Where a of Structure I is I and R7 is NH2 or -CH2NH2i homopolymerization of
the
compound of Structure I leads, in embodiments, to a compound having a
structure
R9 R9
Re b0 R5 R8 b0 R5
H H
N N
O a O a
R6 R3 R4 0 or R6 R3 R4 O
respectively. Such polymeric structures are obtained, in embodiments, by
reacting an
oxocarboxylic acid with 1-amino-2,3-prop anediol or 2-amino-1,3-propanediol
(collectively,
"aminoglycerols") to result in the ammonium salt, followed by heating to form
the amide;
13
CA 02676892 2010-06-14
heat or catalysis or both would then result in homopolymerization via
ketalization.
This reaction scheme is shown in FIG. 1A.
The method is carried out, in embodiments of the invention, by starting with
a precursor oxocarboxylate having free acid groups, for example pyruvic acid,
acetoacetic acid, or levulinic acid. A stoichiometric balance of a precursor
oxocarboxylic acid and an aminoglycerol is achieved by forming the 1:1
ammonium
salt in aqueous solution of about 10% to 80%, or about 50%, by weight of the
combined compounds in water. Stoichiometry is achieved by controlling the pH
of
the solution by addition of the oxocarboxylic acid to lower the pH, or
addition of the
aminoglycerol to raise the pH. Subsequent concentration of the salt to a
slurry of
about 60% by weight or greater is then achieved, in embodiments, by removing
some of the water at a temperature of about 100 C or greater. Concentration is
followed by amide formation by heating the concentrated slurry to a
temperature
greater than 100 C, in embodiments as high as 200 C or even 250 C to remove
water and form the amide bond. In some embodiments, a pressure of about
1.7MPa or greater is employed during part of all of the amide formation by
allowing
escape of water. In some embodiments, no additional catalyst is required to
form
the amide from the ammonium salt.
After the amide is formed, homopolymerization is carried out under relatively
mild conditions by employing a catalyst and heating the reaction to a
temperature
sufficient to cause ketalization and remove water. The catalyst employed may
be a
Lewis or Bronsted acid and the conditions and catalyst used to cause
polyketalization are the same or similar, in some embodiments, to the methods
employed to synthesize ketal esters as described in U.S. Patent Publication
No.
2008/0242721.
The compounds of Structure I, Embodiment 1A have unique and useful properties
that enable their use in a wide range of applications. In various embodiments,
the
compounds of Structure I, Embodiment 1A have good transparency, high levels of
14
CA 02676892 2010-06-14
stiffness, high levels of hardness, good creep resistance, good dimensional
stability, little processing shrinkage, good heat distortion properties, high
melt
viscosity, high melt strength, ability to alloy with other polyamides that are
amorphous or semicrystalline to achieve a wide additional range of properties,
low
water uptake, good surface properties, good barrier
14a
CA 02676892 2009-08-19
properties, resistance to nonpolar solvents, good impact strength, ductility
at moderate
temperatures, good weatherability, and stress-crack resistance to polar
solvents.
Structure I, Embodiment 2.
Where R7 of Structure I is
O
II HZ
A-C X- -
and A is a linear, branched, or cyclic alkyl, alkenyl, or alkynylgroup, or an
aryl or alkaryl
group, wherein the alkyl, alkenyl, alkynyl, aryl, or alkaryl group has I to 36
carbon atoms
and can further have one or more heteroatoms; and X is 0, S, or NH; the
compounds are
poly(ketal amide)esters, poly(ketal amide)amides, or poly(ketal
amide)thioesters. The
character of the A group is easily tailorable to impart the desired property,
such as degree of
hydrophobicity or hydrophilicity, melting point, and so forth to the compounds
of Structure I.
Poly(ketal amide)esters, poly(ketal amide)amides, and poly(ketal
amide)thioesters are
obtained, in embodiments, by the reaction of, respectively, a poly(ketal
amide)ol, poly(ketal
amide)amine, or poly(ketal amide)thiol of Structure I, Embodiment I with one
or more
carboxylate compounds. The methylol, methylamino, or methylthio moieties of
Structure I,
Embodiment 1 have two or more reactive hydrogen atom sites that are available
for reaction
with, in various embodiments, carboxylic acids, carboxylic acid esters,
carboxylic acid
anhydrides, or carboxylic acid halides such as a carboxylic acid chloride.
Such reactions are
well known in the literature and any of the commonly employed methods to form
carboxylates from hydroxyl groups, carboxamides from amine groups, or
thioesters from
mercapto groups are useful in one or more embodiments. Carboxylic acids,
carboxylic acid
esters, carboxylic acid anhydrides, or carboxylic acid halides useful in such
reactions are, in
various embodiments, any of the carboxylic acids, carboxylic acid esters,
carboxylic acid
anhydrides, or carboxylic acid halides found in the literature. Many
structural variations of
the poly(ketal amide)esters, poly(ketal amide)amides, or poly(ketal
amide)thioesters of the
invention are easily envisioned.
In one set of embodiments, a fatty acid ester is transesterified with a
poly(ketal
amide)ol, poly(ketal amide)amine, or poly(ketal amide)thiol of Structure I,
Embodiment I to
CA 02676892 2009-08-19
give the compounds of Structure I, Embodiment 2. In some such embodiments, the
fatty acid
ester is a mixture of unsaturated and saturated fatty acid esters. In some
such embodiments,
the mixture is predominantly unsaturated fatty acid ester. In some such
embodiments, the
mixture contains a triglyceride of a vegetable oil, such as soybean oil,
linseed oil, canola oil,
safflower oil, sunflower oil, corn oil, castor oil, or a blend thereof;
soybean oil or canola oil
are particularly useful. The mixture includes, in some embodiments, high oleic
canola oil, an
ester of 10-undecylenic acid, or a mixture of methyl esters of fatty acids
derived from
transesterification of a vegetable oil. In some embodiments, the fatty acid
ester is an
epoxidized unsaturated fatty acid ester; in some such embodiments the epoxide
an
unsaturated fatty acid ester is composed of a mixture of an unsaturated fatty
acid ester and
one or more saturated fatty acid esters. In some such embodiments, the epoxide
of an
unsaturated fatty acid ester contains at least one epoxidized double bond; in
other
embodiments, the epoxidized unsaturated fatty acid ester contains a majority
of epoxidized
double bonds. An unsaturated fatty acid ester is, in some embodiments,
partially
hydrogenated. In some embodiments, an unsaturated fatty acid ester is
isomerized to change
position or stereochemistry of the double bonds.
The poly(ketal amide)esters, poly(ketal amide)amides, or poly(ketal
amide)thioesters
of Structure I, Embodiment 2 are useful in a broad range of applications. For
example, in
some embodiments where A is an alkyl group having between I and 18 carbons,
the
compounds of the invention are useful as plasticizers, a coalescing solvents,
cosolvents,
phase transfer agents, compatibilizing agents, interfacial modifiers, or
surfactants in one or
more formulations. In some embodiments, the compounds are useful as
plasticizers in one or
more polymer formulations. In still other embodiments, the compounds are
nucleating
agents for one or more solid polymer formulations. A representative example of
one
embodiment of Structure I, Embodiment 2 wherein cx is 2 and R7 groups are
octanoate groups
is shown in FIG. I B.
Structure I, Embodiment 2A.
Where R7 of Structure I is
16
CA 02676892 2009-08-19
O
11 Hz
A-C-XC _
and A contains a secondary or tertiary nitrogen bonded to the C=O group and a
linear,
branched, or cyclic alkyl, alkenyl, or alkynylgroup, or an aryl or alkaryl
group, wherein the
alkyl, alkenyl, alkynyl, aryl, or alkaryl group has I to 36 carbon atoms and
can further have
one or more heteroatoms; and X is 0, S, or NH, the compounds of Structure I,
Embodiment
2A are poly(ketal amide) urethanes, poly(ketal amide)ureas, or poly(ketal
amide)
thiocarbamates. The A moiety is easily tailorable to impart the desired
property, such as
degree of hydrophobicity or hydrophilicity, melting point, and so forth to the
compounds of
Structure I.
The poly(ketal amide)urethanes, poly(ketal amide)ureas, or poly(ketal amide)
thiocarbamates are obtained, in embodiments, by the reaction of, respectively,
a poly(ketal
amide)ol, poly(ketal amide)amine, or poly(ketal amide)thiol of Structure I,
Embodiment I
with one or more monoisocyanate compounds. The methylol, methylamino, or
methylthio
moieties of Structure I, Embodiment I have two or more reactive hydrogen atom
sites that
are available for reaction with an isocyanate group. Such reactions are well
known in the
literature and any of the commonly employed methods to form urethanes from
hydroxyl
groups, ureas from amine groups, or thiocarbamates from mercapto groups are
useful in one
or more embodiments.
Monoisocyanates useful in such reactions are, in various embodiments, any of
the
monoisocyanates found in the literature. Examples of suitable monoisocyanates
include, in
embodiments, methyl isocyanate, ethyl isocyanate, 2-chloroethyl isocyanate,
propyl
isocyanate, isopropyl isocyanate, n-butyl isocyanate, isobutyl isocyanate, sec-
butyl
isocyanate, t-butyl isocyanate, hexyl isocyanate, heptyl isocyanate, octyl
isocyanate, nonyl
isocyanate, decyl isocyanate, 2-(perfluorooctyl)ethyl isocyanate, undecyl
isocyanate, dodecyl
isocyanate, tetradecyl isocyanate, pentadecyl isocyanate, hexadecyl
isocyanate, 8-hexadecyl
isocyanate, cyclopentyl isocyanate, cyclohexyl isocyanate, cyclooctyl
isocyanate,
eyelododeeyl isocyanate, p-tolyl isocyanate, o-tolyl isocyanate, benzyl
isocyanate, p-anisyl
isocyanate, m-fluorophenyl isocyanate, 2-ethoxyphenyl isocyanate,
perfluorophenyl
isocyanate, p-nitrophenyl isocyanate, 4-phenylbutyl isocyanate, chlorosulfonyl
isocyanate,
17
CA 02676892 2009-08-19
naphthyl isocyanate, ally] isocyanate, furfuryl isocyanate, and the like, as
well as mixtures
thereof.
The poly(ketal amide)urethanes, poly(ketal amide)ureas, or poly(ketal amide)
thiocarbamates are useful in a broad range of applications. For example, in
some
embodiments where A contains an alkyl group having between 1 and 18 carbons,
the
compounds of the invention are useful as plasticizers, a coalescing solvents,
cosolvents,
phase transfer agents, compatibilizing agents, interfacial modifiers, or
surfactants in one or
more formulations. In some embodiments, the compounds are useful as
plasticizers in one or
more polymer formulations. In still other embodiments, the compounds are
nucleating
agents for one or more solid polymer formulations.
Structure I, Embodiment 3.
Where R7 of Structure I is
H2
O=C=N-B-X-C--
the compounds of Structure I are poly(ketal amide)isocyanates. In such
embodiments, B is a
linking group that is the residue of the reaction between a poly(ketal
amide)ol, poly(ketal
amide)amine, or poly(ketal amide)thiol of Structure I, Embodiment 1 and a
diisocyanate.
The nmethylol, methylamino, or methylthio moieties of Structure I, Embodiment
1
have two or more reactive hydrogen atom sites that are available for reaction
with a
diisocyanate to form a urethane, urea, or thiocarbamate linkage and an
isocyanate endgroup.
The urethane, urea, or thiocarbamate linkage is thus, in some such
embodiments, part of
group B. The degree of isocyanate functionality imparted to the compounds of
the invention
depends, in embodiments, on the value of a. For example, where the trisketal
amide trio] is
formed from the reaction of product of levulinate ester and glycerol, followed
by amidation
by diethylene triamine (Structure Ic), a trisketal amide tiisocyanate is, in
some
embodiments, formed by reacting the compound of Structure Ic with 3
equivalents of a
diisocyanate. In other embodiments employing the poly(ketal amide)ols of the
invention, the
value of a is 2; in still other embodiments, the value of a is as high as 100
or even 1000 or
more. In general, the precursors to poly(ketal amide)isocyanates are any of
the compounds
of Structure I, Embodiment 1 described above.
18
CA 02676892 2009-08-19
In some embodiments, the degree of isocyanate functionality imparted to the
compounds of the invention depends on the isocyanate functionality of one or
more
compounds reacted with the poly(ketal amide)ols of the invention to form one
or more
poly(ketal amide)isocyanates. For example, where a = 2 of Structure I,
Embodiment 1, the
subsequent reaction with two molar equivalents of a triisocyanates results, in
embodiments,
in a poly(ketal amide)isocyanate having four isocyanate moieties.
The poly(ketal amide)ols are converted to poly(ketal amide)isocyanates by
forming
urethane, urea, or thiocarbamate linkages with diisocyanates or higher
polyisocyanates,
wherein one of the two available isocyanate groups react with an available
hydroxyl group of
a poly(ketal amide)ol, or the corresponding amino or thiol group of the
poly(ketal
amide)amine or poly(ketal amide)thiol. Suitable diisocyanates useful for
embodiments of the
urethane, urea, or thiocarbamate forming reactions include, without
limitation, those
represented by formula OCN-B'-NCO, in which B' represents a divalent aliphatic
hydrocarbon group having 4 to 18 carbon atoms, a divalent cycloaliphatic
hydrocarbon group
having 5 to 15 carbon atoms, a divalent aralkyl hydrocarbon group having 7 to
15 carbon
atoms, or a divalent aromatic hydrocarbon group having 6 to 15 carbon atoms.
Non-limiting examples of suitable organic diisocyanates include 1,4-
tetramethyl ene
diisocyanate, 1,6-hexamethylene diisocyanate, 2,2,4-trimethyl-1,6-
hexamethylene
diisocyanate, 1,12-dodecamethylene diisocyanate, cyclohexane-l,3-
diisocyanate,
cyclohexane -1,4-diisocyanate, I -isocyanato-2-isocyanatomethyl cyclopentane,
I -
isocyanato-3-isocyanatomethyl-3,5,5-trimethyl-cyclohexane (isophorone
diisocyanate or
IPDI), bis-(4-isocyanatocyclohexyl) methane, 2,4'-dicyclohexyl-methane
diisocyanate, 4,4'-
dicyclohexyl-methane diisocyanate, 1,3-bis-(isocyanatomethyl)-cyclohexane, 1,4-
bis-
(isocyanatomethyl)-cyclohexane, bis-(4-isocyanato-3-methyl-cyclohexyl)methane,
a, a,
a',a'-tetramethyl-l,3-xylylene diisocyanate, a, a, a',a'-tetramethyl-l,4-
xylylene
diisocyanate, 1-isocyanato-l-methyl-4(3)-isocyanatomethyl cyclohexane, 2,4-
hexahydrotolylene diisocyanate, 2,6-hexahydrotolylene diisocyanate, 1,3-
phenylene
diisocyanate, 1,4-phenylene diisocyanate, 2,4- tolylene diisocyanate, 2,6-
tolylene
diisocyanate, 2, 2'-diphenylmethane diisocyanate, 2,4'- diphenylmethane
diisocyanate , 4,4'-
diphenylmethane diisocyanate, 1,5-diisocyanato naphthalene; and mixtures
thereof.
19
CA 02676892 2010-06-14
Also suitable for making one or more poly(ketal amide) polyisocyanates of
the invention are polyisocyanates containing 3 or more isocyanate groups.
Nonlimiting examples of suitable polyisocyanates include 4-isocyanatomethyl-
1,8-
octamethylene diisocyanate, aromatic polyisocyanates such as 4,4',4"-
triphenyl methane diisocyanate, and polyphenyl polymethylene polyisocyanates
obtained by phosgenating aniline/formaldehyde condensates.
One or more poly(ketal amide) polyisocyanates of the invention are
synthesized, in some embodiments, in the form of a poly(ketal amide)
polyisocyanate adduct. Suitable poly(ketal amide) polyisocyanate adducts are
those containing isocyanurate, uretdione, biuret, urethane, allophanate,
carbodiimide and/or oxadiazinetrione groups.
In some embodiments, diisocyanates employed to make one or more
poly(ketal amide) polyisocyanates of the invention include the various isomers
of
diphenylmethane diisocyanate and mixtures thereof, IPDI, 4,4'-dicyclohexyl-
methane diisocyanate, and polymeric isocyanates based on diphenylmethane
diisocyanate, such as MondurTM MRS (available from Bayer MaterialScience LLC
of Pittsburgh, PA).
Methods used to make one or more poly(ketal amide)isocyanates of the
invention include conventional techniques known in the literature for the
synthesis
of polyisocyanates from polyols and diisocyanates. A representative technique
for
making one or more polyketal polyisocyanates of the invention from one or more
poly(ketal amide) polyols is that employed in U.S. Patent Publication No.
2008/0242721. The technique of the incorporated Application employs an excess
of diisocyanate, as determined by hydroxyl equivalents per mole of polyol, in
the
presence of dibutyltin dilaurate to give the corresponding polyisocyanate.
Poly(ketal amide)amines and poly(ketal amide)thiols are also reacted with
diisocyanates and higher polyisocyanates using standard literature techniques.
The reaction of poly(ketal amide)amines with isocyanate groups typically
requires
CA 02676892 2010-06-14
no catalyst; contacting the primary amine with a polyisocyanate is sufficient,
in
some embodiments, to bring about the reaction to form a urea group. In other
embodiments, the addition of heat to the reaction mixture is required.
Similarly,
thiocarbamates are formed in some embodiments without the additional of
catalyst,
wherein addition of heat is sufficient to bring about the reaction between a
poly(ketal amide) thiol and a polyisocyanate. Literature methods, such as
those
described by Iwakura et al., Can. J.
20a
CA 02676892 2009-08-19
Chem. 38, 1960, 2418-24; and Movassagh et al., Monatschefle fiir Chemie
139(2), 2007, 137-
140, for example, are useful in making poly(ketal amide)thiocarbamates from
poly(ketal
amide)thiols of the invention.
Structure I, Embodiment 4.
Where R7 of Structure I is
O
II HZ
A XC - -
such that A is an ethylene or propylene group and X is 0 or NH, R7 has the
structure
0
11 H2
R1'rC-X-C
wherein R10 is hydrogen or methyl. In such embodiments, Structure I,
Embodiment 4 is a
poly(ketal amide) acrylate. For example, in such embodiments, Structure I is a
bisketal
amide diacrylate where a is 2, X is 0, and R10 is hydrogen; a bisketal amide
dimethacrylate
where a is 2, X is 0, and R10 is methyl; an N-bisketal amide bisacrylamide
where a is 2, X is
NH, and R10 is hydrogen; and an N-bisketal amide dimethacrylamide where a is
2, X is NH,
and R10 is methyl. In general the following discussion relates to the
formation of bisketal
amide diacrylate structures. However, it will be understood that the
corresponding
methacrylate, acrylamide, and methacrylamide adducts, as well as higher
adducts, e.g.
poly(ketal amide)acrylates, poly(ketal amide)methacrylates, poly(ketal
amide)acrylamides
and poly(ketal amide)methacrylamides, are also generally available by
employing the
corresponding compounds and methodology, or methodology available in the
literature by
employing suitable compounds of Structure I wherein a is greater than 2, X is
NH, and/or R10
of group (b) of R7 in Structure I is methyl. As used herein, the terms
"acrylic", "acrylate",
and the like is also intended to include and incorporate the corresponding
methacrylate,
acrylamide, or methacrylamide moieties.
Bisketal amide diacrylates are synthesized by reacting compounds of Structure
I,
Embodiment 1 wherein a is 2 and R' is CH2OH or CH2NH2 with two equivalents of
a
suitable acrylate compound. Acrylic functionality is imparted, in embodiments,
by
21
CA 02676892 2009-08-19
employing conventional techniques for the reaction of alkanols to form
acrylates. In one
such embodiment a bisketal amide diol is employed in an esterification
reaction with acrylic
acid or to form a bisketal amide diacrylate. Another embodiment employs
acrylyl chloride in
a reaction with a bisketal amide diol to form the corresponding bisketal amide
diacrylate.
The reaction results in the formation of hydrochloric acid (HC1) that is
advantageously
scavenged by a base, for example ammonia.
In a related set of embodiments, a poly(ketal amide) isocyanate of Structure
I,
Embodiment 3 may be reacted with a hydroxyl-functional acrylate or
methacrylate to form a
urethane moiety linking the poly(ketal amide) to the acrylate or methacrylate
moiety. For
example, a bisketal amide diisocyanate is, in embodiments, reacted with a 3-
methacrylyl-2-
hydroxylpropyl ester to give the corresponding bisketal amide dimethacrylate.
In another
example, the bisketal amide diisocyanate is reacted with 2-hydroxypropyl
acrylate to give the
corresponding bisketal amide diacrylate. Urethane acrylates are known in the
literature and
are typically formed by synthesizing polyurethane from a diol and a
diisocyanate, followed
by endcapping the polyurethane isocyanate endgroup with a hydroxy functional
acrylate or
methacrylate as described herein above. Alternatively, the polyurethane is
hydroxy
endcapped and is esterified with acrylic acid or methacrylic acid. For
example, Barbeau, et
al., Journal of Polymer- Science Part B: Polymer Physics, 38(21), 2750 - 68
(2000) disclose
one reaction scheme for a prepolymer that is a polyurethane having isocyanate
endgroups,
endcapped with an acrylate group. In some embodiments, the poly(ketal amide)
isocyanates
of the invention are acrylate functionalized using this or a similar method.
The acrylate
functionality is then polymerized to give an acrylate polymer network. In yet
another
variation of this chemistry, an isocyanate endcapped material is crosslinked
with a hydroxy-
functional polymer, such as poly(2-hydroxypropyl acrylate) or poly(vinyl
alcohol); see, for
example, Decker et al., Macromol. Mater. Eng. 286, 5-16 (2001). In some
embodiments, the
poly(ketal amide)isocyanates of the invention are functionalized with a
previously
synthesized acrylate polymer using this or a similar method.
The various poly(ketal amide)acrylates and poly(ketal amide)methacrylates of
the
invention have, in various embodiments, two or more acrylic functionalities.
The a, f-
unsaturated portion of acrylic functionalities are capable of radical,
cationic, or anionic
polymerization to result in a polymer network. Such reactions are widely used
in the
22
CA 02676892 2009-08-19
industry and one or more acrylate functional poly(ketal amide)acrylates or
poly(ketal
amide)methacrylates of the invention may be reacted using any of the known
techniques of
polymerization or crosslinking of acrylate functionalities. Many references
are available that
discuss these techniques. Radical polymerization or crosslinking reactions
initiated by
thermal, redox, electromagnetic radiation such as ultraviolet (UV), or
electron beam (ebeam)
are the most common of these known techniques. Some useful references
discussing such
means of polymerization of acrylate functional materials are Decker et al.,
Macromol. Mater.
Eng. 286, 5-16 (2001); Burlant, W., U.S. Patent No. 3,437,514; Endruweit, et
al., Polymer
Composites 2006, 119-128; Decker, C., Pigment and Resin Technology 30(5), 278-
86 (2001);
and Jonsson et al., Progress in Organic Coatings 27, 107-22 (1996). Other
known and
useful methods are those taught by U.S. Patent Nos. 3,437,514; 3,528,844;
3,542,586;
3,542,587; 3,641,210. Such polymerization reactions are particularly
advantageous where
one or more poly(ketal amide)acrylates or poly(ketal amide)inethacrylates of
the invention
are polymerized or crosslinked, for example, in situ in a coated formulation,
in a syrup
preparation for coating, and the like. Any of the techniques employed in these
references
may be advantageously employed to react the acrylate functional poly(ketal
amide)s of the
invention, resulting in branched or crosslinked polymer networks.
Many useful extensions of the above embodiments of the invention are readily
envisioned wherein poly(ketal amide)acrylates and poly(ketal
amide)methacrylates are
employed. For example, in one embodiment, a bisketal amide diacrylate is
employed as a
crosslinker when blended with additional acrylate functional compounds,
typically
monoacrylate functional compounds, in a radical polymerization reaction. In
other
embodiments, any one or more of the polymers described above are provided with
acrylic
functionality by employing the reactions described above, to form polyketal
acrylate
prepolymers. In some such embodiments, the polyketal acrylate prepolymers are
processed,
for example by coating, extruding, mold filling, and so forth, with or without
additional
solvents, prior to reaction of the acrylate groups. The prepolymers may
further be blended
with one or more additional acrylate functional compounds and/or additional
vinyl functional
compounds. After processing, the prepolymers are reacted to form a branched
and/or
crosslinked network. The resulting networks are thermoset or thermoplastic. It
is readily
23
CA 02676892 2009-08-19
understood that the properties of the networks will vary greatly depending on
both the nature
of the compounds used and crosslink density.
Additional acrylate functional compounds include compounds having one or more
acrylate, alkylacrylate, acrylamide, or alkylacrylamide residues. Non-limiting
examples of
useful acrylate functional compounds include acrylic acid, methacrylic acid,
acrylamide,
methacrylamide, N-hydroxymethyl acrylamide, methacryloxyethyl phosphate,
acrylonitrile,
methacrylonitrile, 2-acrylamido-2-methylpropanesulfonie acid and salts
thereof; maleic acid,
its salt, its anhydride and esters thereof; monohydric and polyhydric alcohol
esters of acrylic
and alkylacrylic acid such as 1,6 hexane diol diacrylate, neopentyl glycol
diacrylate, 1,3
butylene dimethacrylate, ethylene glycol diacrylate, trimethylolpropane
triacrylate,
pentaerythritol triacrylate, pentaerythritol tetracrylate, etc.; other
oxygenated derivatives of
acrylic acid and alkylacrylie acids, e.g., glycidyl methacrylate, hydroxyethyl
methacrylate,
hydroxypropyl methacrylate, etc.; halogenated derivatives of the same, e.g.,
chloroacrylic
acid and esters thereof; and diacrylates and dimethacuylates, e.g., ethylene
glycol diacrylate.
In some embodiments, the additional acrylate functional compounds are present
in blends
with poly(ketal amide)acrylates of up to about 99.9 mole percent of the the
additional
acrylate compounds, such as between about 50 mole percent to 99.9 mole percent
of
additional acrylate functional compounds relative to the poly(ketal
amide)acrylates of the
invention.
Additional vinyl functional compounds include non-acrylate functional a, f3-
unsaturated compounds capable of copolymerizing with the acrylate functional
compounds
and/or poly(ketal amide)acrylates and poly(ketal amide)methacrylates. Non-
limiting
examples of additional vinyl compounds include aromatic polyvinyl compounds
such as
divinyl benzene, aromatic monovinyl compounds such as styrene, methyl
substituted styrenes
such as a-methyl styrene, vinyl toluene, t-butyl styrene, chlorostyrene, and
the like; aliphatic
monovinyl compounds such as a-olefins, e.g. propylene, 1-octene, and the like.
Other
additional vinyl functional compounds useful in blends with the poly(ketal
amide)acrylates
and poly(ketal amide)methacrylates are the divinyl and tetravinyl compounds
disclosed in
U.S. Patent Nos. 3,586,526; 3,586,527; 3,586,528; 3,586,529; 3,598,530;
3,586,531;
3,591,626; and 3,595,687.
24
CA 02676892 2010-06-14
Structure I, Embodiment 5.
In embodiments of Structure I where R7 is
HZ
D-X-C - -
such that X is 0 or NH and D is
[R11
wherein R11 is hydrogen, a linear, cyclic, or branched alkyl, alkenyl, or
alkynyl
group, an aralkyl group, or an aromatic group, the compounds of Structure I
are
poly(ketal amide)allyl ethers or poly(ketal amide)N-allylamines. For example,
in
such embodiments, Structure 1, Embodiment 5 is a bis(ketal amide)allyl ether
where
a is 2 and X is 0; or a tris(ketal amide)N-allylamine where a is 3 and X is
NH. As
used herein, the term "allyl functionality" means a -CH2-CH=CH2 moiety that is
capable of subsequent polymerization or crosslinking reactions utilizing a
free
radical or redox mechanism. Allylic (polyketal amide)s are, in general,
synthesized
from poly(ketal amide)ols or poly(ketal amide)amines with allyl halides.
Examples of suitable allyl halides for use in forming the poly(ketal
amide)allyl
ethers and poly(ketal amide)-N-allylamines of the invention include, without
limitation, allyl chloride, allyl bromide, methallyl chloride, 2-
(trimethylsilylmethyl)allyl
chloride, 1-halo-2-alkenes such as 1-bromo-2-butene, 1-chloro-2-pentene, 1-
bromo-2-cyclohexylethylene, cis/trans isomers thereof, and the like.
In general, any known technique may be employed to form the poly(ketal
amide)allyl ethers of the invention. In particular, the reaction of organic
alcohols
with allyl halides is known in the literature. Typically, sodium hydride (NaH)
is
employed to facilitate the reaction of allyl bromide and an alkanol to form
the
corresponding alkyl-allyl ether. For example, see, Green et al., Protective
Groups
in Organic Synthesis, Wiley-Interscience, New York, 1999, 67-74, 708-711. This
and other methods are used, in embodiments, to synthesize the poly(ketal
CA 02676892 2010-06-14
amide)allyl ethers of Structure I, Embodiment 5 of the invention. Similarly,
literature
methods are also known for synthesis of N-allyl alkylamines. For example, de
Jesus et al., React. Kinet. Catal. Lett. 84, 2, 255-62 (2005) disclose
palladium
catalyzed formation of allylamines; and Hachemaoui et al., Mendeleev Commun.
2005, 15(3), 124-125
25a
CA 02676892 2009-08-19
disclose a montmorillonite clay catalysis route to allylamines. These
techniques are
employed, in various embodiments, to synthesize the poly(ketal amide)-N-allyl
amines of the
invention.
The one or more allylic compounds of Structure I, Embodiment 5 are, in
embodiments, polymerized using any of the techniques known in the literature.
For example,
heating allyl monomers in the presence of thermal free-radical initiators
gives polymeric
products. Typically, allyl polymers are made by charging the allyl monomer and
a free-
radical initiator to a reactor, and heating the mixture at a temperature
effective to polymerize
the monomer (see, e.g. "Kirk-Othmer Encyclopedia of Chemical Technology," 411'
ed.,
Volume 2, pp. 161-179). Improved methods of polymerizing allyl compounds are
also
usefully employed with one or more allylic polyketals of the invention. For
example, Guo et
al., U.S. Patent No. 5,420,216 discloses that gradual addition of initiator is
key to high
conversion in ally] polymerization.
In some embodiments of the invention, the two or more reactive double bonds
per
poly(ketal amide)allyl ether or poly(ketal amide)-N-allyl amine yield solid,
high molecular
weight polymers by initiation with a suitable free-radical catalyst. Such
embodiments are
useful to provide, for example, heat-resistant cast sheets and thermoset
moldings. In some
such embodiments, the reactivity of compounds having two or more ally] group
permits
polymerization in two stages: a solid prepolyrner containing reactive double
bonds is molded
by heating; then completion of polymerization gives cross-linked articles of
superior heat
resistance. In embodiments, the relatively slow rate of polymerizations is
controlled more
readily than in the polymerization of polyfunctional vinyl compounds to give
soluble
prepolymers containing reactive double bonds.
One useful embodiment of one or more allylic compounds of Structure I,
Embodiment 5 employs minor proportions of one or more polyfunctional
poly(ketal amide)
allyl ethers or poly(ketal amide)-N-allyl amines for cross-linking or curing
preformed vinyl-
type polymers. Among the preformed polymers cured by minor additions of ally]
functional
monomers and catalysts followed by heat or irradiation are polyethylene, PVC,
and
acrylonitrile-butadiene-styrene (ABS) copolymers. These reactions are examples
of graft
copolymerization in which specific added peroxides or high energy radiation
achieves
optimum cross-linking. In other embodiments, small proportions of poly(ketal
amide)allyl
26
CA 02676892 2009-08-19
ethers or poly(ketal amide)-N-allyl amines are added as regulators or
modifiers of vinyl
polymerization for controlling molecular weight and polymer properties. In yet
other
embodiments, poly(ketal amide)allyl ethers or poly(ketal amide)-N-allyl amines
are of high
boiling point and compatibility are employed as stabilizers against oxidative
degradation and
heat discoloration of polymers.
One useful embodiment of one or more thermoset poly(ketal amide)allyl ethers
or
poly(ketal amide)-N-allyl amines are of the invention is in moldings and
coatings for
electronic devices requiring high reliability under long-term adverse
environmental
conditions. These devices include electrical connectors and insulators in
communication,
computer, and aerospace systems. Other embodiments are readily envisioned.
Structure I, Embodiment 6.
In embodiments of Structure I where R' is
H2
D-X-C--
such that X is O and D is
Structure I is a poly(ketal amide) glycidyl ether. Poly(ketal amide)ols are
useful, in
embodiments, for the synthesis of poly(ketal amide) glycidyl ethers. Such
embodiments
include bisketal amide diglycidyl compounds derived from Structure I,
Embodiment 1
wherein a is 2; or poly(ketal amide) glycidyl compounds wherein a is between
about 3 and
100.
In some embodiments, an epihalohydrin such as epichlorohydrin is used to
functionalize one or more poly(ketal amide)ols of Structure I, Embodiment 1.
The reaction
between an alcohol and epichlorohydrin to form a glycidyl ether is known in
the literature.
For example, the reaction of the alcohol Bisphenol A with epichlorohydrin is a
well known
reaction by which epoxy resins are formed; see, for example, Andrews et al.,
U.S. Patent No.
5,420,312 and Chanda, M. and Roy, S., eds., Plastics Technology Handbook, 4"'
ed., (D 2007
27
CA 02676892 2009-08-19
Taylor & Francis Group, LLC, pp. 4-114 to 4-1 16. These and other conventional
techniques
are employed, in embodiments, to react epichlorohydrin with one or more
poly(ketal
amide)ols of Structure I, Embodiment 1 to form poly(ketal amide) glycidyl
ethers of the
invention.
Another technique employed, in some embodiments, to provide compounds having
Structure 1, Embodiment 6 is to react a poly(ketal amide) ally] ether of
Structure I,
Embodiment 5 with a peroxide. For example, Au, U.S. Patent No. 5,036,154
discloses a
method whereby an ethylenically unsaturated ester group, such as an allyl
ester, is reacted
with hydrogen peroxide in the presence of an alkali metal or alkaline earth
metal salt of
tungstic acid, phosphoric acid, and a phase transfer catalyst to give the
epoxidized product of
the unsaturated moiety. Such a technique is used, in embodiments, to form a
poly(ketal
amide)glycidyl ether from the corresponding allyl ether. Other techniques
employed in the
literature are similarly useful to obtain one or more epoxidized products of
allyl esters of the
invention.
In a related embodiment that results in an oxirane functional compound, one or
more
unsaturated fatty acid esters of one or more poly(ketal amide)ols, e.g. the
poly(ketal amide)
esters described in Structure 1, Embodiment 2, are reacted with a peroxide to
form a
poly(ketal amide) oxirane adduct. The techniques that are, in embodiments,
employed to
carry out such reactions are the same as those described above for the
reaction of a peroxide
with an allyl ether. Additionally, Du et al., J. Am. Org. Chein. Soc. 81(4)
477-480 (2004)
describe the esterification of a carboxylate with one or more unsaturated
fatty acids,
followed by reaction of one or more unsaturated sites of the unsaturated fatty
acid ester with
hydrogen peroxide to form the corresponding oxiranyl adducts. Any of these
methods are
used, in various embodiments, to form the oxiranyl adducts of poly(ketal
amide) esters of
unsaturated fatty acids.
One or more poly(ketal amide) glycidyl ethers or poly(ketal amide) oxiranyl
adducts
of the invention are, in embodiments, subsequently polymerized using standard
techniques
such as those found in the literature. The polymerization of epoxy groups, for
example with
amines, amides, or anhydrides, is widely known. A useful summary of compounds
and
mechanisms of curing epoxy groups is found in Chanda, M. and Roy, S., eds.,
Plastics
Technology Handbook, 4 i ed., 2007 Taylor & Francis Group, LLC, pp. 4-116 to
4-122.
28
CA 02676892 2009-08-19
Any of the techniques employed or referenced therein are used, in various
embodiments, to
polymerize the epoxy groups present on one or more poly(ketal amide) glycidyl
ethers or
poly(ketal amide) oxiranyl esters of the invention to form the corresponding
linear or
crosslinked polymer.
Applications of polymerized oxiranyl and glycidyl compounds are numerous and
broad in scope. Due to their high strength, variable crosslink density, and
variable chemical
starting materials, such compounds have found broad applicability for numerous
applications. Many of the most common applications are set forth in Chanda, M.
and Roy,
S., eds., Plastics Technology Handbook, 4`" ed., 2007 Taylor & Francis
Group, LLC, pp.
2-80 to 2-81, 7-26, and 4-124 to 4-125. The resins formed by curing the
glycidyl and
oxiranyl functional poly(ketal amide) compounds of the invention are, in
various
embodiments, useful in one or more of these applications.
STRUCTURE II.
The invention embodies compounds having one or more fragments corresponding to
Structure II
R9 R9
R bO RS I Z Z RS O R8
8
-R'-N b
O X~~ a 0
X-H2C HZC-X E
R6 R3 R4 0 0 R4 R3 R6 Y
II
wherein R1, R2, R3, R4, R5, R6, R8, R9, X, a, and b are as defined for
Structure I;
yyis 0 or 1;
0 is an integer of at least 1; and
E is a linking group selected from (a) - (d) as follows,
0 O O
N-F-N8
(a), H H (b),
29
CA 02676892 2009-08-19
O 0 ~ O O O
N-F-N X'-G-X N-F-N AH8
H H H H (c), or (d),
wherein E is the same or different for each repeat unit represented by)3.
It will be recognized that where linking group E is (a), the compound of
Structure II
is the divalent residue of phosgene or an organic bicarbonate; where linking
group E is (b)
the compound of Structure It is the divalent residue of a diisocyanate; where
linking group E
is (c), the compound of Structure II is the divalent residue of a diol,
diamine, dithiol, or a
hybrid compound such as an aminoalcohol, thioalcohol, and the like reacted
with a
diisocyanate; and where linking group E is (d), the compound of Structure II
is the divalent
residue of a diacid, diester, anhydride, or diacid chloride.
Combinations of various linking groups E are available, in embodiments, in a
single
compound corresponding to Structure It. For example, a polymeric compound of
Structure II
having repeat unit a wherein one or more linking groups are (a) may have
terminal groups
that are hydroxyl; the hydroxy-terminated compound is then reacted with one or
more
polyisocyanates to provide linking groups (b). Many other embodiments wherein
more than
one linking group E is present in a single compound are easily envisioned. Any
of these
combinations are possible in the Structure II Embodiments 1 through 4
described below and
none of the Embodiments described are limited to a single linking group E, but
rather are
grouped according to discussion of each different linking group E.
In some embodiments of the compounds of Structure II, 'y is 1. In some such
embodiments, the starting materials for making the compounds of Structure II
include one or
more fragments that are based on the poly(ketal amide)ols, poly(ketal
amide)amines, and
poly(ketal amide)thiols of Structure I, Embodiment I as well as and poly(ketal
amide)isocyanates of Structure 1, Embodiment 3. Where the compounds of
Structure I are
employed as the starting materials for the synthesis of the compounds of
Structure II, the
compounds of Structure II have 'y = 1: that is, the bis(ketal amide) diols,
bis(ketal amide)
diamines, bis(ketal amide) dithiols, and diisocyanates thereof of Structure I,
embodied by a=
2, are the starting materials in forming the compounds of Structure II. Such
starting
materials are, for example, the compounds of Structure 1, Embodiment I wherein
a is 2 and
CA 02676892 2010-06-14
both R7 are methylol, methylamino, or methylthiol as well as compounds of
Structure I, Embodiment 3 wherein a is 2.
In other embodiments of Structure 11 where y is 1, the starting materials for
making the compounds of Structure II are ketal esters and diamines, which are
the
same starting materials as those described for the synthesis of compounds
having
Structure I, along with fragments attributable to any one of the linking
groups E.
The ketal esters are, in embodiments, any of those disclosed in International
Patent
Publication No. WO 2009/032905, or U.S. Patent Publication No. 2008/0242721.
The ketal esters and diamines are reacted, in various embodiments, with
phosgene
or an organic bicarbonate; a diisocyanate; a diol, dithiol, or a hybrid
compound such
as an aminoalcohol, thioalcohol, and the like; a diacid, diester, anhydride,
or diacid
chloride; or a mixture of one or more of these. When all starting materials
are
mixed and reacted in a single step, some diamines react, in embodiments, with
two
equivalents of ketal ester, to result in one or more fragments of Structure II
where y
is 1. For example, such fragments can arise where the ketal ester is more
reactive
toward the diamine than another chemical in the reaction mixture, such as a
diester
or a bicarbonate, with which the diamine is also capable of reacting.
In similar embodiments, where all starting materials are mixed and reacted in
a single step as described immediately above, the resulting compound of
Structure
II contains a mixture of fragments having y = 1 and y = 0. Such embodiments
arise
where a diamine reacts both with a ketal ester and with phosgene or an organic
bicarbonate; a diisocyanate; a diol, dithiol, or a hybrid compound such as an
aminoalcohol, thioalcohol, and the like; a diacid, diester, anhydride, or
diacid
chloride; or a mixture of one or more of these in a statistical fashion based
on
stoichiometry, relative reactivities of the various chemical compounds in the
reaction mixture, or both.
In some embodiments of the compounds of Structure II, y is 0. In some such
31
CA 02676892 2010-06-14
embodiments, the starting materials for making the compounds of Structure II
include one or more compounds that are based on the reaction of a diamine with
phosgene or an organic bicarbonate; a diisocyanate; a diacid, diester,
anhydride, or
diacid chloride; or a mixture of one or more of these. These starting
materials are
then reacted with a ketal ester or a poly(ketal ester) to give the compounds
of
Structure II. In other words, order of addition of
31a
CA 02676892 2009-08-19
the reagents in various embodiments will result in a Compound of Structure II
wherein y is 0,
1, or a mixture wherein fragments having both y= 0 and y= 1 are contained in a
single
compound.
In some embodiments of Structure II, RI is -(CH2)3-. In other embodiments, R1
is
1,2-cyclohexyl. In still other embodiments, R1 is -(CH2)6-. In some
embodiments of
Structure II, is 0, 1, or 2. In other embodiments, the value of a is 2 and all
R3 and R4 are
hydrogen. In embodiments, R 5 is methyl. In embodiments, b is 0 and R6 and R8
are
hydrogen.
In some embodiments, the compounds of Structure II are formed from starting
materials such as, for example, pyruvic acid (a = 0), acetoacetic acid (a = 1,
R3, R4 = H), or
levulinic acid (a = 2, all R3, R4 = H) or an ester thereof with glycerol (b =
0, R7 = CH2OH,
R6, R8 = H), 1,1,1-trimethylolpropane (b = 1, R7 = -CH2OH, R8 = CH2CH3, R6, R9
= H), or
1,1,1-trimethylolethane (b = 1, R' = -CH2OH, R8 = CH3, R6, R9 = H). Levulinic
acid is an
abundant feedstock that is prepared on an industrial scale by acidic
degradation of hexoses
and hexose-containing polysaccharides such as cellulose, starch, sucrose, and
the like.
Structure II, Embodiment 1
The various embodiments described for Embodiment 1 recite compounds and
methods used in the synthesis of the compounds of Structure II that result in
y= 1. However,
the discussion above regarding order of addition, stoichiometry, and relative
reactivity of the
compounds employed in the synthesis of the compounds of Structure II apply
with equal
force to the compounds of Embodiment 1. It will be understood that Embodiment
1 also
extends to compounds of Structure II wherein -y is 0, 1, or a mixture thereof.
In embodiments where linking group E of Stricture II is (a), the polymeric
structure
is a poly(bisketal amide carbonate) where X is 0, poly(bisketal amide urea)
where X is NH,
or poly(bisketal amide thiocarbonate) where X is S. Notably, thiocarbonate
compounds can
be further stabilized against degradation to form CS2 (carbon disulfide) using
the techniques
set forth in Green II et al., U.S. Patent No. 5,340,593. The following
discussion relates to the
formation of poly(bisketal amide carbonate)s. However, it will be understood
that the
corresponding urea and thiocarbonate functional polymers are also generally
available by
employing similar compounds and methodology, or methodology available in the
literature.
32
CA 02676892 2009-08-19
Also, it will be readily understood that similar polymeric structures
containing branched or
crosslinked morphologies are available by employing analogs of Structure I,
Embodiments I
and 3 wherein the value of a is more than 2; such analogs may be present as
part of a reactive
mixture to form one or more branched or crosslinked analogs of the polymers of
Structure II
where linking group E is structure (a).
In some embodiments, the poly(bisketal amide carbonate)s of the invention have
a
value of a that is 1. In other embodiments, a is between 2 and 12. In still
other
embodiments, the value of 0 is as high as 100. In still other embodiments, the
value of a is as
high as 1000.
Poly(bisketal amide carbonate) synthesis is carried out, in embodiments, by
employing any known and conventional technique for making polycarbonates. One
such
technique employs phosgene. For example, in one such embodiment, a bisketal
amide diol is
treated with sodium hydroxide, followed by an interfacial reaction between the
sodium
alkoxide of the bisketal amide diol and phosgene. Alternatively, one or more
poly(bisketal
amide carbonate)s of the invention are synthesized, in embodiments, by
transesterification of
a polyketal ester with a difunctional carbonate having the general structure
O
R12_O--L- O-R13
where R12 and R13 may be the same or different and are, in embodiments, a
linear, cyclic, or
branched alkyl, alkenyl, or alkynyl group; an aralkyl group, or an aromatic
group. In some
embodiments, R12 and R13 together with the carbonate bond forms a cyclic
carbonate; in these
embodiments, a poly(bisketal amide carbonate) is formed by a ring opening
reaction.
Poly(bisketal amide carbonate)s are also formed, in embodiments, by reacted a
dibromo
bisketal amide compound with potassium carbonate. Thus, in one such
embodiment, a
precursor bisketal amide of Structure I wherein a is 2 and R7 is -CH2Br is
reacted with
potassium carbonate to form a poly(bisketal amide carbonate) of the invention.
The poly(bisketal amide carbonate)s of the invention have a range of available
properties due to the broad range of bisketal amide diols of the invention
that are available as
starting materials. Polycarbonates are known to be tough, transparent,
thermally stable
materials suitable for a range of engineering plastics applications. Suitable
applications for
one or more poly(bisketal amide carbonate)s of the invention include, but are
not limited to,
33
CA 02676892 2009-08-19
fabrication of items requiring molding, laminating, thermoforming such as
extruding or
coextruding, or machining or other conventional means of working. Examples of
useful
items include compact discs, riot shields, baby bottles and other water/drink
bottles and food
containers, electrical components, automobile headlamps, as a component of a
safety glass
laminate, eyeglass lenses, safety helmets, and the like.
The poly(bisketal amide carbonate)s of the invention do not employ Bisphenol A
(4,4'- dihydroxy-2,2-d1phenylpropane), the most commonly employed
polycarbonate polyol
starting material. Bisphenol A has been the subject of toxicity concerns since
the 1930s,
particularly in food or drink contact applications (e.g., baby bottles,
water/drink bottles, food
containers). One or more polycarbonates of the invention are, in one or more
embodiments,
are useful for food or drink applications where it is desirable to eliminate
Bisphenol A.
Additionally, some aliphatic poly(bisketal amide carbonate)s of the invention
are, in
some embodiments, biodegradable. Biodegradable polycarbonates are useful for
one or more
applications, for example, in food or drink contact applications, to enable
disposable
embodiments of various containers. Other applications where biodegradability
is
advantageous include disposable medical supplies such as eye shields and the
like. In
various embodiments, the polyketal ester polycarbonates of the invention
advantageously
supply the desirable properties associated with polycarbonates and
additionally supply
biodegradability thereof.
In some embodiments, poly(bisketal amide carbonate)s of the invention, when
terminated by hydroxyl endgroups, are suitable as diols for use in
polyurethane synthesis.
Poly(bisketal amide carbonate urethane)s are synthesized, in some embodiments,
by
employing bisketal amide diols in the synthesis of a polycarbonate and
controlling
stoichiometry of the polymerization in order to provide hydroxyl functionality
at the ends of
the poly(bisketal amide carbonate). In other embodiments, a poly(bisketal
amide carbonate)
is transesterified at each end with a diol to provide hydroxyl endgroup.
Various poly(bisketal
amide carbonate)s having hydroxyl endgroups are reacted, in embodiments, with
one or more
diisocyanates to form a poly(bisketal amide carbonate urethane). Poly(bisketal
amide
carbonate urethane)s are synthesized using, in some embodiments, the
techniques described
below to make polyketal polyurethanes. In other embodiments, techniques used
to form the
polyketal poly(carbonate urethane)s of the invention are those outlined in
Moore et al., Novel
34
CA 02676892 2009-08-19
Co-Polymer Polycarbonate Diols for Polyurethane Elastomer Applications,
Proceedings of
the Polyurethanes Expo 2003, October 1-3, 2003 ((D 2003, American Chemistry
Council)
Structure II, Embodiment 2.
The various embodiments described for Embodiment 2 recite compounds and
methods used in the synthesis of the compounds of Structure II that result in
y = 1. However,
the discussion above regarding order of addition, stoichiometry, and relative
reactivity of the
compounds employed in the synthesis of the compounds of Structure II apply
with equal
force to the compounds of Embodiment 2. It will be understood that Embodiment
I also
extends to compounds of Stricture II wherein -y is 0, 1, or a mixture thereof.
In embodiments where linking group E of Structure II is (b), Structure II is a
poly(bisketal amide urethane) where X is 0, poly(bisketal amide urea) where X
is NH, or
poly(bisketal amide thiocarbamate) where X is S. The identity of F within
linking group D is
not particularly limited; it can be any of the groups B' listed above (in the
section Structure I,
Embodiment 3) for the diisocyanate structure OCN-B'-NCO. The diisocyanate that
results in
the residue (b), when incorporated into a poly(bisketal amide urethane), can
be any of the
diisocyanates and analogs thereof described above, in various nonlimiting
examples. In
general the following discussion relates to the formation of poly(bisketal
amide urethane)
structures. However, it will be understood that urea and thiocarbamate
functional polymers
are also generally available by employing the corresponding compounds and
methodology,
or methodology available in the literature, where X of Structure II is NH or
S, respectively.
Also, it will be readily understood that similar polymeric structures
containing branched or
crosslinked morphologies are available by employing analogs of Structure I
where the value
of a is more than 2; such analogs may be present as part of a reactive mixture
to form one or
more branched or crosslinked analogs of the polymers of Structure II where
linking group D
is structure (b).
Bisketal amide diols having Structure I, Embodiment 1 are reacted, in some
embodiments, with diisocyanates such as any of those described above to result
in the
compounds of Structure II wherein linking group E is (b). Whereas a
stoichiometric excess
of diisocyanate relative to bisketal amide diol results in a poly(ketal
amide)isocyanate as
described above, a 1:1 stoichiometric ratio of bisketal amide diol and
diisocyanate results in
CA 02676892 2010-06-14
the formation of a linear poly(bisketal amide urethane) having at least one
repeat unit corresponding to the repeat unit of Structure II such that /3 is
at least 1.
In other embodiments, ,8 is between 2 and 12. In still other embodiments, the
value
of ,G is as high as 100. In still other embodiments, the value of fl is as
high as 1000.
While diisocyanates are employed in various embodiments to form the
poly(bisketal amide urethane)s of the invention, additional polyisocyanates of
higher functionality may also be incorporated. Blends of diisocyanates with
polyisocyanates having three or more isocyanate moieties are employed, in
embodiments, to provide a tailored level of branching or crosslinking in the
resulting
poly(bisketal amide urethane) matrix. Additionally, other diols and higher
polyols
that are not bisketal amide diols are optionally blended with the bisketal
amide diols
of the invention in a urethane synthesis to provide variation in the
properties of the
poly(bisketal amide urethane)s of the invention by varying their structure.
Useful
polyols for such embodiments include any diols and higher polyols. Various
other
embodiments are easily envisioned. In forming polymers with values of B
greater
than about 2 to 12, it is important to control stoichiometry carefully so as
to maintain
a ratio of hydroxyl to isocyanate functionality of as close to 1:1 as
possible. Blends
of polyisocyanate functional and polyhydroxylated materials are used, in
embodiments, to form poly(bisketal amide urethane)s having a broad range of
ketal
content, branching and/or crosslink density and a wide range of available
physical
properties including glass transition temperature, tensile strength,
ductility, clarity,
rigidity, elasticity and the like.
In one set of embodiments, poly(bisketal amide urethane)s are present as
blocks in a copolymer with other polyurethane or poly(urethane urea), or
polyurethane thiocarbamate) blocks. Such block copolymers are easily achieved
by controlling stoichiometry of the reactions to reach the desired residual
endgroups, then employing those endgroups as initiation points for an
additional
polymerization reaction with a different monomer mixture. For example, a
bisketal
amide diol of
36
CA 02676892 2010-06-14
the invention may be reacted with a first diisocyanate to form a poly(bisketal
amide
urethane) oligomer; the stoichiometry of the reaction is adjusted, using
conventional
techniques, to result in hydroxyl endgroups. The hydroxyl terminated
poly(bisketal
amide urethane) oligomer is then reacted with a second diisocyanate in a blend
with a second diol to provide a diblock type polyurethane polymer. Various
36a
CA 02676892 2009-08-19
embodiments that are variations of this embodiment are easily envisioned, such
as providing
two different bisketal amide diols in two oligomerizations with the same or
two different
diisocyanates, then reacting the two oligomers in a final reaction to form
diblock
poly(bisketal amide urethane)s.
In another set of embodiments, a poly(bisketal amide ester) or copolyester
thereof is
synthesized according to the methods set forth below and employing a one or
more bisketal
amide diols, adjusting stoichiometry such that residual hydroxyl endgroups are
present, 0 is
about 2 to 12, or about 12 to 100, or even 100 to 1000, and linking group E is
(d)
(embodiments which are more fully described below). The hydroxyl endgroups are
useful in
one or more subsequent reactions to form a poly(bisketal amide urethane) by
reacting the
hydroxyl terminated polyester or copolyester with a polyisocyanate to produce
a
poly(bisketal amide ester urethane). Several additional variations of such
embodiments are
possible.
It will be appreciated that in various embodiments the ketal content of the
resulting
poly(bisketal amide urethane) polymers is widely variable, and a wide range of
physical
properties such as glass transition temperature, tensile strength, elasticity,
and ductility are
attainable in various embodiments of the invention.
The reactions and processes used to form various poly(bisketal amide
urethane)s of
the invention employ conventional techniques of polyurethane synthesis; such
techniques
typically involve blending the two reagents in a stoichiometry that will
result in oligomeric or
polymeric molecular weights. In embodiments, the reaction is catalyzed.
Catalysts useful in
poly(bisketal amide urethane) formation include, in embodiments, tertiary
amines.
Nonlimiting examples of suitable tertiary amines include
dimethylcyclohexylamine, 1,4-
diazabicyclo[2.2.2] octane (also called DABCO or TEDA), and bis-(2-
dimethylaminoethyl)ether. In other embodiments, organometallic compounds, such
as
dibutyltin dilaurate, potassium octanoate, or bismuth octanoate may be used to
catalyze
poly(bisketal amide urethane) formation. In some embodiments where X of
Structure II is
NH or S, no catalyst is required to drive the reaction. Addition of heat is,
in some
embodiments, sufficient to bring about the reaction of a bisketal amide diol,
bisketal amide
dithiol, or bisketal amide diamine with a diisocyanate. The processes
described above for the
reaction of poly(ketal amide)amines and poly(ketal amide)thiols with
polyisocyanates to
37
CA 02676892 2009-08-19
form poly(ketal amide)isoeyanates apply in general to the analogous reactions
of bisketal
amide diamines and bisketal amide dithiols with diisocyanates to form high
polymers having
urea and thiocarbamate linkages.
Processes employing the poly(bisketal amide urethane)s of the invention
include, in
embodiments, reaction injection molding, prepolymerization to a coatable syrup
followed by
coating and curing, and the like. The various poly(bisketal amide urethane)s
of the invention
are not particularly limited as to the methods employed in the processing
thereof.
Foamed formulations employing the various poly(bisketal amide urethane)s of
the
invention are useful embodiments of the invention. Foams are formed during the
polymerization reaction, typically by the addition of one or more blowing
agents. In some
embodiments, a blowing agent is added to the polymer during processing to
facilitate
foaming when the polymer is heated, for example in a thermoforming process.
Suitable
blowing agents include water, certain halocarbons such as HFC-245fa (1,1,1,3,3-
pentafluoropropane) and HFC-134a (1,1,1,2-tetrafluoroethane), and hydrocarbons
such as n-
pentane. In some embodiments, blowing agents are incorporated into e.g. the
polyketal
polyol prior to the polymerization; in other embodiments the blowing agent is
added as an
auxiliary stream. Halocarbons and hydrocarbons are chosen such that they have
boiling
points at or near room temperature; these blowing agents volatilize into a gas
during the
exothermic polymerization reaction. In addition, high density microcellular
foams are
formed, in embodiments, without the addition of blowing agents by mechanically
frothing or
nucleating the polyol component of the reaction mixture prior to
polymerization.
In some embodiments, surfactants are employed to modify the characteristics of
the
foam during the foaming process. In embodiments, they are used to emulsify the
liquid
components, regulate cell size, and stabilize the cell structure to prevent
collapse and surface
defects. Rigid foam surfactants produce, in embodiments, very fine cells and
very high
closed cell content. In other embodiments, flexible foam surfactants stabilize
the reaction
mass while maximizing open cell content to prevent the foam from shrinking.
The need for,
and choice of, surfactant is determined, in embodiments, by choice of reaction
components,
component compatibility, system reactivity, process conditions and equipment,
tooling, part
shape, and shot weight.
38
CA 02676892 2009-08-19
Various embodiments of the poly(bisketal amide urethane)s of the invention are
useful in a broad range of applications. Polyurethane polymers, in general,
are compounds of
exceptional industrial utility; they find numerous applications because the
final properties of
the resulting polymer can be influenced greatly through selection of active
hydrogen
monomers (typically, polyhydroxyl compounds) and isocyanates used, and by
selecting the
conditions used to prepare the finished polymer products. In various
embodiments, the
poly(bisketal amide urethane)s of the invention are lightweight, strong,
durable and resistant
to abrasion and corrosion. Depending on choice of monomers, poly(bisketal
amide
urethane)s range from stiff to flexible at ambient temperatures. The broad
range of bisketal
amide diol chemistry as well as the range of linkages available from ester,
urethane, urea, and
thiocarbamate in various embodiments provides extensive flexibility in choice
of structure
that leads to a broad range of properties and, in turn, applications.
Without providing any particular limitations, the various poly(bisketal amide
urethane)s of the invention are useful, in embodiments, as adhesives or
sealants, particularly
for exterior uses or building construction applications where extremely
challenging
conditions are encountered; as binders; as coating materials where durability
and/or
challenging environmental conditions exist; in reactive spray coatings of 100%
solids; as
elastomers for applications such as rollers and belts for carrying heavy
and/or abrasive
materials, roller blades, and other footwear parts such as shoe soles; as
vibration damping
materials; and in the fabrication of medical devices, for example for surface
modification, as
a protective coating, or within moving parts (e.g. for elastomeric materials).
In foamed form,
these materials also find utility as insulation materials; low density
vibration damping
materials; flexible foam for indoor furniture such a seat cushions and
mattresses, and other
similar applications such as automobile seat cushions.
Structure II, Embodiment 3.
The various embodiments described for Embodiment 3 recite compounds and
methods used in the synthesis of the compounds of Structure II that result in
'y = 1. However,
the discussion above regarding order of addition, stoichiometry, and relative
reactivity of the
compounds employed in the synthesis of the compounds of Structure II apply
with equal
39
CA 02676892 2009-08-19
force to the compounds of Embodiment 3. It will be understood that Embodiment
1 also
extends to compounds of Structure II wherein y is 0, 1, or a mixture thereof.
Similarly to polymeric compounds of Structure 11 where linking group E is (b),
in
embodiments where linking group E of Structure II is (c), Structure II is a
poly(bisketal
amide urethane), poly(bisketal amide urea), poly(bisketal amide
thiocarbamate), or any one
of a variety of hybrid heteroatomic structures depending on whether X, X', and
X" are 0,
NH, or S. It will be understood that the various linking groups E available
having
substructure (c) are the result of the reaction of compounds with two active
hydrogen atoms,
HX'-G-X"H, with the poly(ketal amide)isocyanate compounds of Structure I,
Embodiment 3
to result in linking group (c). Compounds HX'-G-X"H are dithiols, diols,
diamines, or
"hybrid" compounds such as amino alcohols (e.g. compounds such as
ethanolamine). Any
of the diols, dithiols, or diamines listed in the above sections are useful in
forming the linking
groups (c) in combination with the poly(ketal amide)isocyanates of Structure
I, Embodiment
3. Such diols, dithiols, or diamines include the bisketal amide diols,
bisketal amide diamines,
or bisketal amide dithiols described in Structure I, Embodiment 1.
Suitable "hybrid" compounds include, without limitation, 2-aminoethanol, 3-
aminopropan-l-ol, isopropanolamine, 2-aminopropan-l-ol, 2-aminobutan-l-ol, 2-
amino-3-
methylbutan-l-ol, 2-amino-4-methylpentan-I-ol, 6-aminohexan-l-ol, I-amino-3-
chloropropan-2-ol, 7-aminobicyclo[2.2.2]octan-8-ol, 2-aminopyridin-3-oI, 2-
amino-4-
phenylphenol, 5-aminonaphthalen-l-ol, 4-(4-aminophenyl)phenol, 2-
mercaptoethanol, 3-
methyl-3 -hydroxybutane- 1- thiol, pyridoxine-4-thiol, I1-mercapto-l-
undecanol, and the like.
It will be readily understood that similar polymeric structures containing
branched or
crosslinked morphologies are available by employing analogs of Structure I,
Embodiments I
and 3 where the value of a is more than 2; such analogs may be present as part
of a reactive
mixture to form one or more branched or crosslinked analogs of the polymers of
Structure II
where linking group E is structure (c). Similarly, multifunctional bisketal
amide
polyisocyanates, formed by the reaction of bisketal amide dithiols, bisketal
amide diols, or
bisketal amide diamines with polyisocyanates having more than two isocyanate
functionalities, are employed in some embodiments of the invention as
compounds of
Structure II wherein linking group E is (c) to provide branching and/or
crosslinking to the
polymer matrixes formed therefrom.
CA 02676892 2009-08-19
In general, the reagents employed, methodology, physical properties of the
resulting
polymer matrixes, and applications that are addressed using the above
described
poly(bisketal amide urethane)s wherein linking group E is (b) are the same or
similar to those
described where linking group E is (c). Some additional variations, however,
bear
discussion.
In some embodiments, poly(bisketal amide urethane)s, poly(bisketal amide
urea)s, or
poly(bisketal amide thiocarbamate)s are formed by the reaction of one or more
bisketal
amide diols, bisketal amide dithiols, or bisketal amide diamines of the
invention with one or
more bisketal amide diisocyanates or bisketal amide polyisocyanates of the
invention. In still
other embodiments, one or more bisketal amide diisocyanates or bisketal amide
polyisocyanates are reacted with one or more polyols that are not bisketal
amide diols, to
form a poly(bisketal amide urethane).
Additionally, in some embodiments, one or more isocyanate moieties of the
bisketal
amide diisocyanates or bisketal amide polyisocyanates of the invention are
partially reacted
with water to form the corresponding amine group and carbon dioxide. It is
known that an
isocyanate can be reacted with water to form a primary amine group and carbon
dioxide; the
primary amine is then available to react with another isocyanate group to form
a urea linkage.
Thus, in embodiments, one or more bisketal amide diisocyanates or bisketal
amide
polyisocyanates of the invention are reacted with water to form one or more
poly(bisketal
amide urethane urea)s via a this known pathway. In some such embodiments, the
evolution
of carbon dioxide acts as a foaming agent as the reaction progresses, thus
providing for a
foamed poly(ketal amide urethane urea)s matrix. Water reacts with isocyanate
groups to
create carbon dioxide gas, which fills and expands cells created during the
mixing process,
and causes the formation of urea groups.
In still other embodiments, one or more bisketal amide diisocyanates or
bisketal
amide polyisocyanates are reacted with one or more polyamines or polythiols to
form
poly(ketal amide urethane urea)s or poly(ketal amide urethane thiocarbamate)s,
respectively.
Suitable polyamines for forming one or more poly(bisketal amide urethane
urea)s of the
invention include, for example, hydrazine, ethane-l,2-diamine, 1,6-
hexanediamine, but-2-
ene-l,4-diamine, Metformin, butane-1,4- diamine, propane-1,2- diamine, benzene-
1,3-
diamine, 2-methylbenzene-1,3-diamine, 4-chlorobenzene-1,3- diamine,
methanediamine,
41
CA 02676892 2009-08-19
1,3,5-tiiazine-2,4,6-triarnine, N-(2-aminoethyl)ethane-l,2-diamine, N-(6-
aminohexyl)hexane-1,6-diamine, N,N'-bis(2-aminoethyl)ethane-1,2-diamine, N-[2-
(3-
aminopropylamino)ethyl]propane- l,3-diamine, 4-(3,4-diaminophenyl)benzene-1,2-
diamine,
spennine (N,N'-bis(3-aminopropyl)butane-1,4-diamine), a polyethyleneimilie, a
polyoxyalkyleneamine having two or more amine groups, such as those sold under
the trade
name JEFFAMINE 1z , (available from the Huntsman Corp. of Salt Lake City, UT),
or any
diamine or higher amine compound such as those sold under the trade name
ELASTAMINE (available from the Huntsman Corporation). Suitable polythiol
compounds include, for example, dithiols such as ethane-1,2-dithiol, propane-
1,3-dithiol,
propane-l,2-dithiol, propane-1,1-dithiol, 3-chlorobutane-1,2-dithiol, 1-
chlorobutane-2,3-
dithiol, 2-chloro-2-methylpropane-1,3-dithiol, butane-1,4-dithiol, butane-1,3-
dithiol, hexane-
1,6-dithiol, octane-l,8-dithiol, decane-1,10-dithiol, 3-ethoxypropane-1,2-
dithiol, 3,4-
dimethoxybutane-1,2-dithiol, 2-methyl-5-(1-sulfanylpropan-2-yl)cyclohexane-1-
thiol,
benzene-1,2-dithiol, benzene-1,3-dithiol, 4-methylbenzene-1,2-dithiol, 4-(4-
sulfanylphenyl)benzenethiol, butane-1,4-dithiol, 3-(phenoxy)propane-1,2-
dithiol; and
trithiols such as ethane-1,1,2-trithiol, propane-1,2,3-trithiol, pentane-1,3,5-
trithiol, octane-
1,3,8-trithiol, cyclohexane-1,2,4-trithiol, cyclododecane-1,4,8-trithiol,
1,3,5-trithiane-2,4,6-
trithiol, nonane-1,5,6-trithiol, benzene-1,2,3-trithiol, benzene-1,3,5-
trithiol, 2-
methylbenzene-1,3,5-trithiol, and naphthalene-1,2,3-trithiol.
Structure II, Embodiment 4.
The various embodiments described for Embodiment 4 recite compounds and
methods used in the synthesis of the compounds of Structure II that result in
y = 1. However,
the discussion above regarding order of addition, stoichiornetry, and relative
reactivity of the
compounds employed in the synthesis of the compounds of Structure II apply
with equal
force to the compounds of Embodiment 4. It will be understood that Embodiment
I also
extends to compounds of Structure II wherein -y is 0, 1, or a mixture thereof.
In embodiments where linking group E of Structure II is (d), Structure II is a
poly(ketal amide ester) where X is 0, poly(ketal amide amide) where X is NH,
or poly(ketal
amide thioester) where X is S. In general the following discussion relates to
the formation of
polyester structures. However, it will be understood that polyamide and
polythioester
42
CA 02676892 2009-08-19
adducts are also generally available by employing the corresponding compounds
and
methodology, or methodology available in the literature, where X of Structure
II is NH or S,
respectively. Also, it will be readily understood that similar polymeric
structures containing
branched or crosslinked morphologies are available by employing analogs of
Structure I
where the value of a is more than 2; such analogs may be present as part of a
reactive
mixture to form one or more branched or crosslinked analogs of the polymers of
Structure II
where linking group E is structure (d).
Poly(ketal amide ester)s are formed by reacting one or more bisketal amide
diol with
one or more diacids or diesters having structures incorporating moiety H in
linking group D
of structure (d). The identity of H within linking group D is not particularly
limited. For
example, bisketal amide diols as described in Structure I, Embodiment I are,
in
embodiments, polymerized with one or more diacids or diesters thereof, such as
adipic acid
or methyl isophthalate, to give a perfectly alternating copolyester. A
nonlimiting,
representative example of a bisketal amide diol and a diester is shown in FIG.
113. Other
structural variations are easily envisioned. The polyesters include, in some
embodiments,
one or more additional diols, or a mixture of diacids or diesters, in order to
provide structural
variability. Additionally, in some embodiments, triols such as a trisketal
amide triol (i.e.
where a is 3 in Structure I, Embodiment 1) or another trios, or a higher
polyol, a higher
poly(ketal amide)ol, a triacid, triester or higher poly(acid) or ester
thereof, are incorporated to
provide crosslinking or branching sites to the polyester. Other diols or
higher polyols, such
as any of the those described above, are also incorporated, in embodiments,
via one or more
copolymerization reactions in conjunction with diacids or diesters and one or
more bisketal
amide diols of the invention to result in one or more poly(ketal amide ester)
copolymers.
In various embodiments, the poly(ketal amide ester)s of the invention have a
value of
that is 1. In other embodiments, 0 is between 2 and 500. In still other
embodiments, the
value of 0 is between about 10 and 200. In still other embodiments, the value
of 0 is between
about 10 and 100. It will be appreciated that by varying the structure of a
diacids or diesters
employed in the copolymerization, a wide range of properties are available,
for example,
hydrophobicity, hydrophilicity, amphiphilic character, tensile strength,
solvent resistance,
crystallinity, optical transparency, glass transition temperature, and the
like.
43
CA 02676892 2010-06-14
Non-limiting examples of suitable diacids (or esters of diacids) for use in a
polymerization reaction to provide poly(ketal amide esters) and copolymers
thereof
include aliphatic, cycloaliphatic or aromatic dicarboxylic acids, for example,
succinic
acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid,
sebacic acid,
nonanedicarboxylic acid, decanedicarboxylic acid, terephthalic acid,
isophthalic
acid, o-phthalic acid, tetrahydrophthalic acid, hexahydrophthalic acid, maleic
acid,
fumaric acid, naphthalene dioic acid, dimerized fatty acids, or hydrogenated
dimerized fatty acids. The methyl, ethyl, propyl, butyl or phenyl esters of
the acids
listed above are suitable substitutes for the diacid component, as well as
acid
anhydrides such as o-phthalic, maleic or succinic acid anhydride or a mixture
thereof. Some examples of suitable triacids include 1,3,5-trimethylcyclohexane-
1,3,5-tricarboxylic acid, cis or trans aconitic acid, propane- 1,2,3-
tricarboxylic acid,
hemmellitic acid, isocitric acid, and the like.
Copolyesters are also formed where mixtures of diacids and/or diesters are
employed, as well as additional diols such as any of the diols described
above.
Additionally, mixtures of bisketal amide diols are used in some embodiments to
form poly(ketal amide ester ) copolymers of the invention.
In embodiments, the poly(ketal amide ester)s and copolymers thereof are
synthesized using conventional transesterification and/or polymerization
catalysts
and conditions. The catalyst may be any of the known esterification or
transesterification catalysts in general. For example, acidic catalysts such
as a
toluenesulfonic acid, sulfuric acid, sulfamic acid, or a sulfonic acid are
employed in
various embodiments. In a preferred embodiment an organometallic catalyst is
employed, for example a catalyst based on titanium or tin, such as titanium
(IV)
tetrabutoxide (Ti(OBu)4), or tin (I1) octanoate. The choice of catalyst is not
particularly limited within the scope of the invention. In embodiments,
reaction
conditions are optimized to reach high molecular weight. Such reaction
conditions
include, in embodiments, the techniques, conditions, and catalysts employed in
polyesterification reactions described in U.S. Patent Publication No.
2008/0242721.
44
CA 02676892 2010-06-14
In other embodiments, the bisketal amide diols or poly(ketal amide)ols
of the invention are employed in the ring opening reaction of one or more
lactones
to form the corresponding poly(ketal amide ester). In such embodiments, fl is
at
least 1 and in many such embodiments /3 equals 1. Ring opening polymerization
of
lactones is carried out using one or more catalysts and using reaction
conditions
suitable for ring opening polymerization. Catalysts and reaction conditions
employed in such reactions are any of those used in the art for ring opening
reactions of lactones. For example, some ring opening polymerization catalysts
are
based on transition metals such as zinc, tin, or titanium. Without limiting
the
species of catalysts or reaction conditions employed, any of the catalysts or
reaction conditions described in Hori et al., U. S. Patent No. 5,516,883 or
Schechtman et al., U. S. Patent No. 5,648,452 are useful. Activated carbon as
employed by Endo et al., EP1857484 or organic catalysts employed as described
in
a web-published article from IBM Company of Armonk, NY, may be used to affect
the ring opening polymerization of lactones using the poly(ketal amide)
polyols of
the invention as the initiating polyol. The above examples are not limiting as
to the
type of catalyst or set of reaction conditions that can be employed in a ring
opening
polymerization of lactones.
Suitable lactones for the ring opening polymerization initiated by one or more
poly(ketal amide)ols of the invention include, without limitation,
propiolactone,
pivalolactone, diketene, dimethyldiketene, /3-butyrolactone, 4-butyrolactone,
4-
valerolactone, 6-caprolactone, E-caprolactone, 5-ethenyl-5-methyloxolan-2-one,
gluconolactone, glucuronolactone, D-galactonolactone, coumarin, hydrocoumarin,
ascorbic acid lactone, a-angelicalactone, 2-acetylbutyrolactone, 6-propyloxan-
2-
one, 6-ethyloxan-2-one, ribonolactone, arabonolactone, A-nonalactone,
bicyclononalactone, 5-nonalactone, A-decalactone, pantolactone, 2-
dehydropantolactone, 5-butoxolan-2-one, isocrotonolactone, 6-hexyloxan-2-one 5-
heptyloxolan-2-one, 5-propyloxolan-2-one, 6-[(b)-pent-2-enyl]oxan-2-one,
cocolactone, isocitric lactone, 2-hydroxy-6-methylpyran-4-one, 1-
oxacyclododecan-
CA 02676892 2010-06-14
2-one, E-dodecalactone, 1-oxacyclopentadecan-2-one, 1-oxacycloheptadecan-2-
one, L-arabino-1,4-lactone, 4-hydroxy-4-methyloxan-2-one, homoserine lactone,
4-
methyl-7-propan-2-yloxepan-2-one, and the like.
In one embodiment of a lactone ring opening polymerization, one or more
poly(ketal amide)ols of the invention are employed in the ring opening
polymerization of SEGETOLIDETM (available from Segetis, Inc. of Golden Valley,
MN) or its dimer to form the corresponding ketal functional polyester. The
structure
of SEGETOLIDET"' and its dimer, as well as methods for the ring opening
polymerization of both compounds, are found in U.S. Patent Publication No.
2008/0242721. The methods disclosed therein are suitable, in embodiments, for
initiating the ring opening polymerization using the poly(ketal amide) polyols
of the
invention as initiators.
It will be understood that many additional variations are possible employing
the poly(ketal amide)ols of the invention, as well as their poly(ketal
amide)thiol and
poly(ketal amide)amine analogs. The thermal and environmental stability of one
or
more poly(ketal amide ester)s or copolymers thereof is excellent. The
poly(ketal
amide esters) and copolymers based on one or more poly(ketal amide)ols of the
invention are, in some embodiments, stable in air up to about 150 C. In other
embodiments, the copolyesters of the invention are stable in air up to 200 C.
Under an inert atmosphere or under conditions where oxygen is excluded, such
as
in one layer of a multilayer film, the copolyesters of the invention are
stable up
temperatures as high as 300 C. The poly(ketal amide esters) and copolyesters
of
the invention also have, in embodiments, excellent tensile properties that
make
them useful for a wide variety of commercial applications. Many other
embodiments will be readily envisioned; it will be appreciated that the ketal
content
of the resulting polymers are variable, and a wide range of physical
properties such
as glass transition temperature, tensile strength, elasticity, and ductility
are
attainable in various embodiments of the invention.
46
CA 02676892 2010-06-14
STRUCTURE III.
The invention embodies compounds having one or more fragments
corresponding to Structure III:
R9
b0 R5 R2
8
R
HO N-R'-OH
O a
R6 R3 R4 O
wherein:
46a
CA 02676892 2009-08-19
R' is a linear, branched, or cyclic alkyl, alkenyl, or alkynyl group, or an
aryl or
alkaryl group, wherein the alkyl, alkenyl, aryl, alkaryl groups can have one
or more
heteroatoms;
R2 is hydrogen or an alkyl group having 1 to 6 carbon atoms and can further
include a
hydroxyl group;
R3 and R4 are independently hydrogen, halogen, amine, mercapto, phosphate,
phosplionooxy, silyl, siloxane, alkynyl, or a linear, branched, or cyclic
alkyl or
alkenyl groups having 1 to 18 carbon atoms, or an aryl or alkaryl group,
wherein the
alkyl, alkenyl, aryl, or alkaryl groups can have one or more heteroatoms, and
each R3
and R4 may be the same or different;
R5 is hydrogen, alkynyl, or a linear, branched, or cyclic alkyl or alkenyl
group having
1 to 18 carbon atoms, or an aryl or alkaryl group, wherein the alkyl, alkenyl,
aryl, or
alkaryl groups can have one or more heteroatoms;
R6, R8, and R9 are independently hydrogen, halogen, or an alkyl group having
between 1 and 6 carbon atoms and optionally one or more heteroatoms;
a is 0 or an integer of between 1 and 12; and
b is 0 or 1, wherein b = 0 indicates a five membered ring,
R8
HO ORs
R6 O\
and b = 1 indicates a 6 membered ring,
R9
R8 0Ra
HO O S~
R6
"Heteroatoms" present in the one or more R', R3, R4, R5, R6, R8, or R9 groups
can
include, in various embodiments, halogen, nitrogen, oxygen, sulfur, silicon,
phosphorus, and
the like and can be embodied in a functional group such as amino, carbonate,
imide, amide,
47
CA 02676892 2009-08-19
sulfone, sulfonamide, urethane, mercapto, disulfide, ether, ester, phosphate,
phosphonooxy,
silane, or silyl functional groups, or a combination thereof.
The compounds of Structure III are ketal amide diols or, where R2 contains an
additional OH group, ketal amide triols. It will be understood that the
compounds of
Structure III are analogs to the compounds of Structure I wherein R7 is CH2OH.
In some
embodiments, the ketal amide diols and triols of Structure III are made by the
reaction of
ketal esters with aminoalcohols. The ketal esters are, in embodiments, those
described above
for Structures I and II. Examples of suitable aminoalcohols include, without
any particular
limitation, those described in Structure IT, Embodiment 3. The methods
employed to make
the compounds of Structure III include, in general, the same methods employed
to make the
compounds of Structure I, except that the stoichiometry of the ketal ester to
aminoalcohol is,
in embodiments, different from that of the ketal ester to diamine. This is
because compounds
having Structure I represent the reaction product of two moles of ketal ester
to one mole of
diamine, while compounds of Structure III represent the reaction product of
one mole of
ketal ester to one mole of aminoalcohol.
In some embodiments of Structure III, RI is -(CH2)2-. In other embodiments, RI
is -
(CH2)6-. In some embodiments, a is 0, 1, or 2. In other embodiments, the value
of a is 2 and
all R3 and R4 are hydrogen. In embodiments, R5 is methyl. In embodiments, b is
0 and R6
and R8 are hydrogen.
In some embodiments, the compounds of Structure III are formed from pyruvic
acid
(a = 0), acetoacetic acid (a = 1, R3, R4 = H), or levulinic acid (a = 2, all
R3, R4 = H) or an
ester thereof with glycerol (b = 0, R6 , R8 = H), 1,1,1-trimethylolpropane (b
= 1, R8 =
CH2CH3, R6, R' = H), or 1,1,1-trimethylolethane (b = 1, R8 = CH3, R6, R9 = H).
Levulinic
acid is an abundant feedstock that is prepared on an industrial scale by
acidic degradation of
hexoses and hexose-containing polysaccharides such as cellulose, starch,
sucrose, and the
like.
The compounds having Structure III are useful in various formulations. The
compounds of Structure III are, in some embodiments, soluble in water and
lower alcohols
and hydrophilic coating formulations. In other embodiments, for example
wherein R1 is a
long chain alkyl group, for example dodecyl, the compounds of Structure III
are soluble in
hydrophobic formulations. In yet other embodiments, the various other R groups
of
48
CA 02676892 2009-08-19
Structure III determine solubility in one or more formulations; in some such
embodiments,
the compounds of Structure III are coalescing solvents, surfactants,
solubilizers, interfacial
modifiers, and the like.
The compounds having Structure III are, in various embodiments, useful to make
a
variety of compounds via subsequent reaction pathways. In general, any of the
Embodiments
listed for Structure I arising from embodiments wherein R7 is CH2OH are
available as
analogs of Structure III by employing similar reagents and methodology. For
example, the
ketal amide diols of Structure III are useful in transesterification reactions
to provide diesters,
analogous to those of Structure I, Embodiment 2. In other embodiments the
ketal amide
diols of Structure III are useful in reactions with polyisocyanates to form
isocyanate capped
compounds, analogous to those of Structure I, Embodiment 3. In other
embodiments the
ketal amide diols of Structure III are useful in reactions with acrylate,
methacrylate,
acrylamide, or methacrylamide compounds to form the acrylate functional
compounds that
are analogs to the acrylate compounds of Structure I, Embodiment 4; subsequent
reactions to
form the radically or conically polymerized analogs thereof are also
available. In other
embodiments the ketal amide diols of Structure III are useful in reactions
with allylic
compounds to provide ally] capped materials analogous to those of Structure I,
Embodiment
5; subsequent reactions to form the polymerized analogs thereof are also
available. And in
other embodiments the ketal amide diols of Structure III are useful in
reactions with epoxy
functional compounds, such as epihalohydrins, to provide glycidyl and other
epoxy
functional compounds analogous to those of Structure 1, Embodiment 6;
subsequent reactions
to form the polymerized analogs thereof are also available.
STRUCTURE IV.
The invention embodies compounds having one or more fragments corresponding to
Structure IV
49
CA 02676892 2009-08-19
O R5 R2
1R9
R8 b
O
O a
R6 R3 R4 O
IV
wherein
R1, R2, R3, R4, R5, R', R8, R9, X, a, and b are as defined for Structure III;
0 is an integer of at least 1; and
E is a linking group as defined for Structure II.
The compounds of Structure IV encompass oligorners of the compounds of
Structure
III, wherein a is about 2 to 12, as well as polymers wherein a is about 12 to
500, or about 10
to 200, or about 10 to 100. It will be understood that the compounds of
Structure IV are
analogous to those compounds of Structure II wherein X is 0 and that arise, in
turn, from
compounds of Structure I wherein R7 is CH2OH. Thus, analogs of various
Embodiments
described for Structure II wherein X is 0 are also available as their
Structure IV counterparts
by employing similar reagents and methodology in conjunction with the ketal
amide diols
and triols of Structure III. For example, in embodiments, ketal amide diols of
Structure III
are useful in making polycarbonate compounds analogous to those of Structure
II,
Embodiment 1. Employing some ketal amide triol in such a reaction incurs
branching or
crosslinking. In other embodiments, the ketal amide diols of Structure III are
useful in
making polyurethane compounds analogous to those of Structure II, Embodiments
2 and 3.
And in other embodiments, the ketal amide diols of Structure III are useful in
making
polyester compounds analogous to those of Structure II, Embodiment 4.
By employing a ketal amide triol in replacement of or partial replacement of a
ketal
amide diol, branching and/or crosslinking may be incurred. The ketal amide
triol is available
in embodiments wherein the amine employed to react with the ketal ester is,
for example,
diethanolamine.
CA 02676892 2009-08-19
STRUCTURE V.
The invention embodies compounds having one or more fragments corresponding to
Structure V:
R6 R7 R6 R7
Rg b R8 b Rio
O O 0 O
O O
R5 R5
X a a N~R1
R3 R4 R4 R3
R2
V
wherein
a is an integer of at least 1;
0 is an integer of at least 1;
R1 is a linear, branched, or cyclic alkyl, a linear, branched, or cyclic
alkenyl, alkynyl,
aryl, alkaryl, or an oligomeric or polymeric moiety; and optionally contains
one or
more heteroatoms; and R' may be the same or different for each occurrence;
R2 is hydrogen or an alkyl group having between 1 and 6 carbon atoms;
R3, R4, R5, R6, and R7 are independently hydrogen, a linear, branched, or
cyclic alkyl,
a linear, branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl; and
optionally contains
one or more heteroatoms; and R3, R4, R5, R6, and R' may be the same or
different for
each occurrence;
R8 is a covalent bond, methylene, ethylene, hydroxymethylene, oxygen, or -CH2-
O-
CH2- and R7 is the same or different for each occurrence;
R9 and R10 are independently hydrogen, a linear, branched, or cyclic alkyl, a
linear,
branched, or cyclic alkenyl, alkynyl, aryl, alkaryl, or a polymeric moiety;
and
optionally contains one or more heteroatoms;
X is 0 or NR2, wherein R2 is as defined above;
51
CA 02676892 2010-06-14
a is 0 or an integer of 1 to 12; and
b is 0 or 1, wherein b = 0 indicates a five membered ring:
,~s 1
0 0
and b = 1 indicates a 6 membered ring:
R6 R7
0 RO
I5
and b may be the same or different for each occurrence.
The invention also embodies compounds having one or more fragments
corresponding to Structure V:
5 0 0 R5 0
a5 0 O a
R3 R4 R4 R3
R2
I R
V'
wherein R1, R2, R3, R4, R5, X, a, and a are as defined for Structure V.
Structure V
is an analog of Structure V, wherein the tetrol basis for the bisketal
functionality is
pentaerythritol, C(CH2OH)4.
The compounds of Structures V and V are polyketal polyamides. Polyketal
polyamides are derived from precursor polyketal compounds. The precursor
polyketal compounds to Structure V have the structure
52
CA 02676892 2009-08-19
R5 R6 l RR66 R5
R8-~~ b R7 R9
O O O IO
O O
~-~ R4 R4
X ~< a a O-R1
RJ-O
R2 R3 R3 R2
wherein
a is an integer of at least 1;
R' is hydrogen, a metal cation, an organic cation, a linear, branched, or
cyclic alkyl, a
linear, branched, or cyclic alkenyl, alkynyl, aryl, alkaryl, or an oligomeric
or
polymeric moiety; and optionally contains one or more heteroatoms; and R' may
be
the same or different for each occurrence;
R2, R3, R4, R5, and R6 are independently hydrogen, a linear, branched, or
cyclic alkyl,
a linear, branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl; and
optionally contains
one or more heteroatoms; and R2, R3, R4, R5, and R6 may be the same or
different for
each occurrence;
R7 is a covalent bond, methylene, ethylene, hydroxymethylene, oxygen, or -CH2-
O-
CH2- and R7 is the same or different for each occurrence;
R8 and R9 are independently hydrogen, a linear, branched, or cyclic alkyl, a
linear,
branched, or cyclic alkenyl, alkynyl, aryl, alkaryl, or a polymeric moiety;
and
optionally contains one or more heteroatoms;
a is 0 or an integer of 1 to 12; and
b is 0 or 1, wherein b = 0 indicates a five membered ring:
I~S
O O
and b = 1 indicates a 6 membered ring:
53
CA 02676892 2009-08-19
R5 R6
OKO
,2, R4
and b may be the same or different for each occurrence.
The precursor polyketal compounds to Structure V have the structure
O ?aOY:O)' 4 OO R4 O
R1-O >--~ a O-R1
R2 R3 R3 R2
wherein
each R' is independently hydrogen, a metal cation, an organic cation, a
linear,
branched, or cyclic alkyl, a linear, branched, or cyclic alkenyl, alkynyl,
aryl, alkaryl,
or an oligomeric or polymeric moiety; and optionally contains one or more
heteroatoms;
each R2 and R3 are independently hydrogen, a linear, branched, or cyclic
alkyl, a
linear, branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl; and optionally
contain
one or more heteroatoms;
each R4 is independently linear, branched, or cyclic alkyl; linear, branched,
or cyclic
alkenyl; alkynyl; aryl; or alkaryl; and optionally contains one or more
heteroatoms;
and
each a is independently 0 or an integer of 1 to 12.
The precursor polyketal compounds to compounds of Structure V are the reaction
product of at least two molar equivalents of an oxocarboxylate with one molar
equivalent of
a first polyol which is a tetrol or higher polyol. The precursor compounds to
compounds of
Structure V are bisketal adducts of pentaerythritol, C(CH2OH)4, with two molar
equivalents
of a keto acid or ester thereof. Precursor polyketal compounds for the
polyketal polyamides
having Structure V, and precursor bisketal compounds for polyketal polyamides
having
54
CA 02676892 2009-08-19
Structure V are collectively referred to herein as "precursor polyketal
compounds." One
example of a reaction wherein a precursor polyketal compound is formed is
shown in FIG.
1D.
In some embodiments of Structures V and V', R1 is -(CH2)3-. In other
embodiments
of Structures V and V', R' is 1,2-cyclohexyl. In still other embodiments of
Structures V and
V', R1 is -(CH2)6-. In still other embodiments of Structures V and V', RI and
R2 combine to
form a cyclic moiety, for example the residue of piperazine. In some
embodiments of
Structures V and V', a is 0, 1, or 2. In other embodiments of Structures V,
the value of a is 2
and all R3 and R4 are hydrogen; similarly, for Structure V', all R2 and R3 are
hydrogen. In
embodiments of Structures V and V', R4 is methyl. In embodiments of Structure
V, b is 0
and R6 and R8 are hydrogen. In embodiments of Structures V and V', 0 is 1. In
other
embodiments of Structures V and V', 0 is 2 or greater. In embodiments of
Structures V and
V', 0 is between about 2 and 500. In other embodiments of Structures V and V',
0 is about
10 to 200. In other embodiments of Structures V and V', 0 is about 10 to 100.
In some embodiments, the compounds of Structure I are formed from pyruvic acid
(a
= 0), acetoacetic acid (a = 1, R3, R4 = H), or levulinic acid (a = 2, all R3,
R4 = H) or an ester
thereof with glycerol (b = 0, R7 = CH2OH, R6, R8 = H), 1,1,1-
trimethylolpropane (b = 1, R7 _
-CH2OH, R8 = CH2CH3, R6, R9 = H), or 1,1,1-trimethylolethane (b = 1, R7 = -
CH2OH, R8 =
CH3, R6, R9 = H). Levulinic acid is an abundant feedstock that is prepared on
an industrial
scale by acidic degradation of hexoses and hexose-containing polysaccharides
such as
cellulose, starch, sucrose, and the like.
Some illustrative examples of polyketal precursor compounds that are suitable
for use
in the synthesis of polyketal polyamides having Structure V and V are, without
limiting the
full range of structures as described in the incorporated application:
O O
O RO~O
O O\~v 11 OR OR
O O
RO O
O O (a), OH (b),
CA 02676892 2009-08-19
HO
O
HO OR
0 0 0 ""~ 0 O O
O O
OR RO
RO O O
O
HO H
RO O O ~~ 11 OR
RO O p O p OR O O O O
O~ ~p RO
p
HCH2~CH-CHz~CH2-CHzH
O O 111111 ~~~~~~ OH O O
OR RO OR
~O DC O~
o (g), and O O (h)
wherein each R is independently hydrogen, a metal cation, an organic cation, a
linear,
branched, or cyclic alkyl, a linear, branched, or cyclic alkenyl, alkynyl,
aryl, or alkaryl, and
optionally contains one or more heteroatoms, and in, n, and o are integers of
1 or more. The
polyketal precursors to compounds of Structures V and V' are bisketals and
higher polyketals
of tetrols and higher polyols with two or more equivalents of an
oxocarboxylate. In some
embodiments, such as in polyketal precursor (a), the polyol employed is
erythritol. In other
embodiments, such as in polyketal precursors (d), (e), and (f), the polyol
employed is
sorbitol. In other embodiments, such as in polyketal precursor (c), the polyol
employed is
diglycerol (a tetrol that is a mixture of glycerol dimers). For compounds of
Structure V the
polyol employed is pentaerythritol and polyketal precursor compounds are those
such as (h).
As used herein, erythritol and threitol, which are diastereomers, are used
interchangeably in various embodiments of the reaction. Similarly, sorbitol
and its
stereoisomer mannitol are used interchangeably in various embodiments. Where
no
stereochemistry is indicated in a chemical structure, any stereoisomer may be
employed in
the embodiments of the invention. Further, any indication of stereochemistry
is not meant to
limit any particular embodiment to that stereochemistry only; any
stereochemical isomer may
be used in one or more embodiments of the compounds of the invention.
56
CA 02676892 2009-08-19
In some embodiments, a precursor polyketal compound is reacted with one or
more
diamines or higher polyamines to result in a polyketal polyamide of Structure
V or W. In
some such embodiments, X is NR2. In other embodiments, a precursor polyketal
compound
is polymerized to form the corresponding polyketal polyester by reaction with
diol, and then
the polyester so formed is subsequently reacted with one or more diamines or
higher
polyamines to result in a polyketal polyamide of Structure V or W. In some
such
embodiments, X is 0 or NR2 or a mixture of these. In still other embodiments,
a polyketal
polyamide is subjected to transamidation with a diamine or higher polyamine to
result in a
new polyketal polyamide structure. In such embodiments, X is NR2. It will be
appreciated
that copolymers of any of these structures are easily obtained. It will also
be appreciated that
certain methods of chain extension, such as reacting amino endgroups of a
polyketal
polyamide of the invention with a diisocyanate, are also available as an
extension of any of
the synthetic methodology and the unique polyketal polyamide structures of the
invention to
increase molecular weight, or otherwise effect the physical properties of the
polyketal
polyamides of the invention.
In forming the polyketal polyamides of the invention from the precursor
polyketal
compounds, any of the diamines or higher polyamines described for the
embodiments of
Structure I are suitable. One representative, nonlimiting example of a
polyketal polyamide
synthetic scheme, starting from a precursor polyketal compound, is shown in
FIG. 1D.
One useful method for making the polyketal polyamides of the invention is to
form a
"nylon salt" of the precursor polyketal compound and a diamine, followed by
heating to form
a polyketal polyamide of Structure V or W. The method is carried out, in
embodiments of
the invention, by starting with a precursor polyketal compound having free
acid groups; thus,
for example, compounds (a)-(h) wherein R is H. A stoichiometric balance of a
precursor
polyketal and a diamine or higher polyamine is achieved by forming the 1:1
ammonium salt
in aqueous solution of about 10% to 80%, or about 50%, by weight of the
combined
compounds in water. Stoichiometry is achieved by controlling the pH of the
solution by
addition of the polyketal precursor or the diamine. Subsequent concentration
of the salt to a
slurry of about 60% by weight or greater is then achieved by removing some of
the water at a
temperature of about 100 C or greater. Concentration is followed by
polymerization by
heating the concentrated slurry to about 200 C or greater, or between about
200 C and 250 C,
57
CA 02676892 2009-08-19
or to about 210 C. During the polymerization, the temperature is, in some
embodiments,
raised to about 260 C to 300 C, or to about 275 C. In some embodiments, a
pressure of
about 1.7MPa or greater is employed during part of all of the polymerization
reaction by
allowing escape of water. No additional catalyst is required using this
method. Notably, in
such embodiments, all X of Structure II fragments will be NR2 - only endgroups
of the
compounds formed using this method will have X as 0.
The polyketal polyamides having Structures V and V' are, in embodiments of the
invention, synthesized via amidolysis. In amidolysis, a precursor polyketal
ester, for
example any of compounds (a)-(h) wherein R is a linear, branched, or cyclic
alkyl, a linear,
branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl group, is reacted with
one or more
diamines or higher polyamines to form a polyketal polyamide of Structure V or
W. As with
the nylon salt method, in such embodiments, all X of Structure II fragments
will be NR2 -
only endgroups of the compounds formed using this method will have X as 0.
Aminolysis is carried out, in some embodiments, by employing one of the
techniques
known in the literature. For example, methods of reacting of diesters with
diamines to form
polyamides is described employed in Pryde et al., U.S. Patent No. US
3,223,683; Tashiro et
al., U.S. Patent No. 3,597,376; Brill, U.S. Patent No. 3,763,234. In
embodiments, a polyketal
precursor compound and a diamine or higher polyamine are contacted in a vessel
in amounts
that correspond to a 1:1 molar amount of ester to primary amine groups. The
contacted
compounds are simply heated to affect the reaction, by allowing for removal of
the product
alcohol that forms upon reaction of the amines with the ester groups. The
compounds are
heated to about 200 C, in embodiments between about 200 C and 250 C, in other
embodiments between 250 C and 300 C, and in still other embodiments to about
300 C. A
vacuum is applied, in some embodiments, in order to help drive the reaction to
form the
polyketal polyamide by facilitating removal of the alcohol byproduct of the
aminolysis
reaction, which corresponds to the ester group. In some embodiments, an inert
solvent is
employed to facilitate the reaction; for example, benzene, toluene, xylene,
hexane, octane,
chlorinated aliphatic hydrocarbons such as 1,1,2-trichloroethane, and the like
may be used in
various embodiments of the reaction. In some embodiments, for example where
the amine is
a liquid at room temperature, it is preferable to employ no solvent.
58
CA 02676892 2009-08-19
In some embodiments, a Lewis acid is employed as a catalyst in the aminolysis
reaction to form the polyketal polyamides of the invention. Examples of
suitable Lewis acids
include, in embodiments, antimony trichloride, aluminum chloride, antimony
trifluoride,
ferric chloride, antimony pentachloride, niobium pentachloride, tantalum
tetrachloride,
titanium tetrachloride, boron trifluoride, antimony pentafluoride, stannic
fluoride, aluminum
bromide, thallium trichloride, uranyl nitrate, uranium tetrachloride, uranyl
acetate, uranium
oxides such as U02, and the like. In embodiments where a Lewis acid catalyst
is employed,
the reaction proceeds at temperatures as low as about 250 C, or between about
100 C and
250 C, or even as low as about 80 C to 100 C.
In other embodiments, aminolysis can be carried out using mild conditions when
organic catalysts are employed. For example, Sabot et al., Tetrahedron Letters
48 (2007)
3863-6 disclose solvent-free aminolysis of monoesters with monoamines
catalyzed by 1,5,7-
triazabicyclo[4.4.0]dec-5-ene, or TBD, as low as room temperature. In the
reactions of the
invention, the addition of heat is required in order to reach appreciable
molecular weight,
because of the general tendency of polyamides to form high melting, very hard
solids even
with a low degree of polymerization such as 2-3; raising the temperature
allows a higher
degree of polymerization to be reached than the same reaction at ambient
temperatures.
However, we have found, surprisingly, that lower temperatures than those
required to
synthesize polyamides in any of the previously described techniques may be
employed and
similar degrees of polymerization are reached, in embodiments, to those
reached by
employing the high temperatures to synthesize polyketal polyamides described
above. For
example, a polyketal polyamide is formed, in embodiments, by contacting a
bisketal ester
with a diamine at molar ratios of about 2:1 to about 1:2, or in some
embodiments about 3:2,
in other embodiments about 1:1, and in still other embodiments about 1.1:1 to
1.2:1 [bisketal
ester]:[diamine]; and adding TBD in an amount of about 200-2000ppm, or in some
embodiments about 750-1000ppm, based on the mass the combined reagents, to
form a
reaction mixture. In some embodiments, one or more inert solvents such as
toluene, hexane,
and the like are added to the reaction mixture; in some embodiments, no
solvent is added to
the reaction mixture. In embodiments, no heat is added to the reaction
mixture; in other
embodiments, the reaction mixture is heated to a temperature of about 20 C to
200 C; in
other embodiments, the reaction mixture is heated to a temperature of about 70
C to 150 C;
59
CA 02676892 2009-08-19
in other embodiments, the reaction mixture is heated to a temperature of about
120 C to
140 C. The reaction of the bisketal ester with the diamine is carried out for
about 1 minute to
50 hours, in some embodiments about 1 hour to 45 hours, in other embodiments
about 10 to
40 hours, and in still other embodiments about 30 to 40 hours.
It will be understood that the method of the invention is not limited by the
nature of
either the diester or the diamine employed in the reaction. That is, the
method is employed in
various embodiments with non-ketal based diesters in addition to the polyketal
precursor
compounds that are employed in various embodiments in the synthesis of
polyketal
polyamides having Structure V and W. The mildness of the reaction conditions
and the
favorable polyamide products of narrow polydispersity are advantageous in many
polyamide
syntheses, some embodiments of which include the synthesis of the polyketal
polyamides of
the invention.
The result of the reaction carried out using the method of the invention is
the
formation of a polyamide having a degree of polymerization of about 2 to 500,
or about 10 to
200, or about 10 to 100 depending on reaction stoichiometry, temperature, and
reaction time.
The polymers formed by the method of the invention are characterized by a
narrow
polydispersity. For example, for polymers having a molecular weight of about
2000 to
10,000 g/mol, polydispersity index (the ratio of weight average molecular
weight to number
average molecular weight) is, in embodiments, about 1 to 3. In other
embodiments, the
polydispersity index is about 1.7 to 1.8.
The polymers formed by the method of the invention are, in embodiments,
subjected
to further reactions to increase molecular weight or add functionality to the
resulting
polymer. For example, where a stoichiometric excess of diamine is contacted
with bisketal
ester, such that amine endgroups are formed in the resulting polymer, the
polymer is, in
embodiments, additionally contacted with a diisocyanate to form polyurea
linkages between
chains initially formed by aminolysis. In another example, where a
stoichiometric excess of
bisketal ester is contacted with a diamine such that ester endgroups are
formed in the
resulting polymer, the polymer is, in embodiments, additionally contacted with
a diol to form
polyester linkages between chains initially formed by aminolysis.
In a related embodiment, aminolysis of a polyketal polyester is carried out to
form the
polyketal polyamides of the invention. In such embodiments, the starting ester
functionality
CA 02676892 2010-06-14
is a residue of a polyketal polyester instead of a precursor polyketal
compound.
Polyketal polyesters, copolyesters thereof, have at least one repeat unit
corresponding to the structure
R5 R6 R6 5
R8 b R7 b R9
O O O 0 O O
R4 R4
O a a O-R1
R2 R3 R3 R2
wherein
R' is a linear, branched, or cyclic alkyl, a linear, branched, or cyclic
alkenyl,
alkynyl, aryl, alkaryl, or an oligomeric or polymeric moiety; and optionally
contains one or more heteroatoms; and R1 is the same or different for each
occurrence;
R2, R3, R4, R5, and R6 are independently hydrogen, a linear, branched, or
cyclic alkyl, a linear, branched, or cyclic alkenyl, alkynyl, aryl, or
alkaryl; and
optionally contains one or more heteroatoms; and R2, R3, R4, R5, and R6 is
the same or different for each occurrence;
R7 is a covalent bond, methylene, ethylene, hydroxymethylene, oxygen, or -
CH2-O-CH2- and R7 is the same or different for each occurrence;
R8 and R9 are independently hydrogen, a linear, branched, or cyclic alkyl, a
linear, branched, or cyclic alkenyl, alkynyl, aryl, alkaryl, or a polymeric
moiety; and optionally contains one or more heteroatoms;
a is 0 or an integer of 1 to 12; and
b is 0 or I wherein b = 0 indicates a five membered ring,
61
CA 02676892 2010-06-14
S'/ 1
0
0~<W
b = 1 indicates a 6 membered ring,
R5 R6
O O
2 R4
and b may be the same or different for each occurrence;
a is an integer of at least 1; and
/3 is an integer of at least 1.
Other polyketal polyesters additionally have, in embodiments, the structure
O Ra O Ra
O a 0 O a O-Rl
R2 R3 R3 R2 R
wherein
R1 is a linear, branched, or cyclic alkyl, a linear, branched, or cyclic
alkenyl,
alkynyl, aryl, alkaryl, or an oligomeric or polymeric moiety; and optionally
contains one or more heteroatoms; and R1 is the same or different for each
occurrence;
R2 and R3 are independently hydrogen, a linear, branched, or cyclic alkyl, a
linear, branched, or cyclic alkenyl, alkynyl, aryl, or alkaryl; and optionally
contains one or more heteroatoms; and R2 and R3 are the same or different
for each occurrence;
62
CA 02676892 2010-06-14
R4 is a linear, branched, or cyclic alkyl, a linear, branched, or cyclic
alkenyl,
alkynyl, aryl, or alkaryl; and optionally contains one or more heteroatoms;
and R4 is the same or different for each occurrence;
a is 0 or an integer of 1 to 12; and
$ is an integer of at least 1.
The polyketal polyesters are the polyester analogs of the polyketal precursor
compounds described above. They are formed, in embodiments, by
polyesterification techniques, employing precursor polyketal compounds and
diols.
Aminolysis of the polyketal polyesters is generally carried out according to
the
techniques described above and employs the same catalysts, solvents or lack
thereof, and reaction conditions. In embodiments where no solvent is employed
to
affect the reaction, the polyester is heated to its melt temperature in the
presence of
a diamine or higher polyamine in order to affect the aminolysis. In such
embodiments, no catalyst is required to affect the reaction and the reaction
proceeds smoothly to high molecular weights using conventional methods. In
some
such embodiments, application of vacuum during the reaction is useful for
removing
diol molecules that are the byproduct of the aminolysis.
The aminolysis reaction between diamines or higher polyamines and polyketal
polyesters is, in some embodiments, only a partial aminolysis. In such
embodiments, the polyamine reacts with the polyketal polyester to form a
polyketal
poly(ester amide), or a compound of Structure V or V wherein X is 0 or a
mixture
of 0 and NR2. How far the aminolysis reaction proceeds to complete the removal
of diol and form a polyketal polyamide with very few or no fragments wherein X
is 0
is dependent, in embodiments, upon both reaction conditions and stoichiometry
of
amine to ester functionality. For example, where less than 100 mole% of amine
groups are added to an aminolysis reaction, compared to ester groups, the
reaction
proceeds to form a polyketal poly(ester amide). In some embodiments, about 99
63
CA 02676892 2010-06-14
mole% to 95 mole% of amine groups are added as compared to ester groups. In
other embodiments, about 95 mole% to 80 mole% of amine groups are added as
compared to ester groups. In still other embodiments, as low as 50% of amine
groups are added compared to ester groups. In general, as the mole% of amine
groups are lowered with respect to ester groups, the glass transition
temperature of
the resulting copolymer is observed to become lower. Thus, glass transition
temperatures of amide-ester copolymers of the invention are targeted based on
the
desired end use.
In other embodiments, the polyketal polyamides of the invention are
synthesized
using transamidation of an amide and a polyamine. For example, a polyketal
polyamide is, in some embodiments, synthesized employing one of the above-
described methods; the
63a
CA 02676892 2009-08-19
polyketal polyamide is then subjected to transamidation with a second diamine
or higher
polyamine using techniques described, for example, in Stahl et al., U.S.
Patent No. 7,154,004
to arrive at a polyketal polyamide having an polyamino fragment attributable
to the second
polyamine. In other embodiments, a precursor polyketal ester, for example any
of
compounds (a)-(h) wherein R is a linear, branched, or cyclic alkyl, a linear,
branched, or
cyclic alkenyl, alkynyl, aryl, or alkaryl group, is reacted with a monoamine
such as 1-
aminohexane, 1-propanamine, N-butylhexan-l-amine, or any other primary or
secondary
alkyl or alkenyl monoamine, to form a precursor polyketal amide; the polyketal
amide is then
subjected to transamidation with a diamine or higher polyamine to result in a
polyketal
polyamide of the invention. In embodiments, a metal catalyst based on Sc, Ti,
or Al is
employed to catalyze the transamidation reaction. In some embodiments, the
catalyst
employed is Sc(OTf)3; in other embodiments, Ti(NMe2)4 or A12(NMe2)6 are used.
The
reactions are preferably carried out at temperatures of about 250 C or less.
An inert solvent,
such as toluene, is employed in some embodiments; in other embodiments, no
solvent is
employed to affect the transamidation reaction.
By using any of the above methods described to make compounds having
Structures
V and V', a wide range of copolymers are easily formed. A mixture of precursor
polyketal
compounds are used, in embodiments, in a single reaction to form a polyketal
copolyamide.
Similarly, a mixture of diamines or higher polyamines are employed in other
embodiments.
In some embodiments, combining one or more precursor polyketal compounds with
one or
more diacids that are not precursor polyketal compounds in a "nylon salt"
reaction together
with one or more diamines or higher polyamines results in polyketal
copolyamides. Suitable
diacids that are employed in such reactions include, for example, aliphatic,
cycloaliphatic or
aromatic dicarboxylic acids such as succinic acid, glutaric acid, adipic acid,
pimelic acid,
suberic acid, azelaic acid, sebacic acid, nonanedicarboxylic acid,
decanedicarboxylic acid,
terephthalic acid, isophthalic acid, o-phthalic acid, tetrahydrophthalic acid,
hexahydrophthalic acid, maleic acid, fumaric acid, naphthalene dioc acid,
dimerized fatty
acids, or hydrogenated dimerized fatty acids.
Similarly, one or more precursor polyketal compounds are combined with one or
more diesters that are not precursor polyketal compounds in an aminolysis
reaction together
with one or more diamines or higher polyamines to result in various polyketal
copolyamides.
64
CA 02676892 2009-08-19
Any of the known ester moieties, such as the methyl, ethyl, propyl, butyl or
phenyl esters of
any of the diacids listed above, are suitable for copolymerization with
precursor polyketal
compounds and polyamines. In some embodiments, it is advantageous to provide
an ester
group that corresponds to an alcohol byproduct that is easily removed from a
reaction vessel
during the reaction, in order to help drive the reaction to completion. For
example, in
embodiments where vacuum and/or heat is employed in an aminolysis reaction, it
is
advantageous to employ an ester having an alcohol byproduct that boils at or
below the
temperature of the reaction to facilitate removal of the alcohol by
evaporation.
Combining one or more polyketal polyesters with a second polyester in an
aminolysis
reaction together with a diamine or higher polyamine also results in a
polyketal copolyamide.
Combining one or more polyketal polyesters with one or more diesters that are
not precursor
polyketal compounds in an aminolysis reaction together with one or more
diamines or higher
polyamines results in various polyketal copolyamides. And combining one or
more
precursor polyketal compounds with one or more polyesters that are not
polyketal polyesters
in an aminolysis reaction together with one or more diamines or higher
polyamines results in
various polyketal copolyamides. Transamidation similarly lends itself to a
wide range of
copolyamide structures, as will be easily envisioned. A wide range of
copolyamides is
available using the described techniques, resulting in materials with a wide
range of
properties including tensile strength, ductility, thermal stability, and the
like.
Crosslinked or branched analogs of the polyketal polyamides of the invention
are
readily formed by employing a major proportion of precursor polyketal
compounds or
polyesters thereof, diacids or esters thereof, and diamines with a minor
proportion of, in
embodiments, tricarboxylic acid or higher polyacid or ester thereof, or
triamine or higher
polyamine in any of the polyamide forming reactions described above. The
precursor
polyketal compounds are, in some embodiments, trisketal compounds or precursor
polyketal
compounds of higher functionality.
Additional functionality and increase in molecular weight of the polyketal
polyamides
are also realized, in embodiments, by providing additional reagents to the
polyketal
polyamides during or after the polymerization methods described above. For
example, by
including a diol or higher polyol into an amidation reaction between a
precursor polyketal
compound and a diamine or higher polyamine, residual ester endgroups are taken
up by
CA 02676892 2009-08-19
transesterification reactions and ultimate chain length is increased; a
polyketal poly(ester
amide) is thereby formed. Similarly, by adding diisocyanate at the end of a
reaction in which
a polyketal polyamide is formed having amino endgroups, chain extension by
formation of
urea groups results in a polyketal poly(amide urea). Many other variations are
possible, such
as reacting residual endgroups of a polyketal polyamide to provide reactive
acrylate, allyl, or
oxirane functionalities that in turn can be polymerized to provide chain
extension,
crosslinking, or branching. It will be recognized that many conventional
techniques can be
employed to provide further variations in structure and molecular weight of
the polyketal
polyamides of the invention.
The amide-functional polymers and copolymers of the invention have unique and
useful properties that enable their use in a wide range of applications. In
various
embodiments, the amide-functional polymers and copolymers of the invention
have good
transparency, high levels of stiffness, high levels of hardness, good creep
resistance, good
dimensional stability, little processing shrinkage, good heat distortion
properties, high melt
viscosity, high melt strength, ability to alloy with other polyamides that are
amorphous or
semicrystalline to achieve a wide additional range of properties, low water
uptake, good
surface properties, good barrier properties, resistance to nonpolar solvents,
good impact
strength, ductility at moderate temperatures, good weatherability, and stress-
crack resistance
to polar solvents.
Amide-functional polymers and copolymers incorporating one or more repeat
units
attributable to Structures I - V are useful in a wide variety of industrially
useful and
significant applications. The amide-functional polymers and copolymers of the
invention
are, in embodiments, used in blends, optionally obtained by reactive
extrusion. Blends
include blends of various species of the amide-functional polymers and
copolymers of the
invention as well as blends with such polymers as aliphatic/aromatic
copolyesters, as for
example polybutylene terephthalate adipate (PBTA), polybutylene terephthalate
succinate
(PBTS), and polybutylene terephthalate glutarate (PBTG); biodegradable
polyesters such as
polylactic acid, poly-E-caprolactone, polyhydroxybutyrates such as poly-3-
hydroxybutyrates,
poly-4-hydroxybutyrates and polyhydroxybutyrate-valerate, polyhydroxybutyrate-
propanoate, polyhydroxybutyrate-hexanoate, polyhydroxybutyrate-decano ate,
polyhydroxybutyrate-dodecano ate, polyhydroxy-butyrate-hexadecanoate,
66
CA 02676892 2009-08-19
polyhydroxybutyrate-octadecanoate, and polyalkylene succinates and their
copolymers with
adipic acid, lactic acid or lactide and caprolactone and their combinations,
and the like;
polystyrene and copolymers thereof; polyurethanes; polycarbonates; polyamides
such as
Nylon 6 and Nylon 6,6; polyolefins such as polyethylene, polypropylene, and
copolymers
thereof; or any other industrially useful polymeric compounds. Blends also
include, in some
embodiments, composites with gelatinized, destructed and/or complexed starch,
natural
starch, flours, and other materials of natural, vegetable or inorganic origin.
The amide-
functional polymers and copolymers of the invention are, in some embodiments,
blended
with polymers of natural origin, such as starch, cellulose, chitosan,
alginates, natural rubbers
or natural fibers (such as for example jute, kenaf, hemp). The starches and
celluloses can be
modified, such as starch or cellulose esters with a degree of substitution of
between 0.2 and
2.5, hydroxypropylated starches, or modified starches with fatty chains, among
others.
In some embodiments, alloys of two or more polyamide polymers or copolymers,
including at least one polymer having one or more repeat units attributable to
Structures I -V,
are formed in blends thereof. The term "alloy" means a blend wherein the two
or more
polymers have a chemical interaction evidenced by a shift in glass transition
temperatures as
described in M. Kohen, ed., "Nylon Plastics Handbook", 1995 by Carl Hanser
Verlag,
Munich, Germany, p. 380-1. The polyamide blended with the at least one
polyamide or
copolyamide of the invention is, in embodiments, a second polyamide or
copolyamide of the
invention, or some other polyamide that is amorphous or semicrystalline.
The amide-functional polymers and copolymers according to the invention, and
blends of thereof, possess properties and values of viscosity that render them
suitable for use,
by appropriately adjusting the molecular weight, in numerous practical
applications, such as
films, injection-molded products, extrusion coated products, fibers, foams,
thermoformed
products, extruded profiles and sheets, extrusion blow molding, injection blow
molding,
rotomolding, stretch blow molding and the like.
In the case of films, production technologies like film blowing, casting, and
coextrusion can be used. Moreover such films can be subject to monoaxial or
biaxial
orientation in line or after film production. It is also possible that the
stretching is obtained in
presence of an highly filled material with inorganic fillers. In such a case,
the stretching can
generate micropores and the so obtained film can be suitable for hygiene
applications.
67
CA 02676892 2009-08-19
The amide-functional polymers and copolymers according to the invention are
suitable for the production of films. A "film" is defined, for the purposes of
the invention, as
a sheet type material that is flexible to e.g. bending and is between about
1.im to 5mm thick.
Films employing the amide-functional polymers and copolymers of the invention
are, in
embodiments, one-directional or two-directional, single layer or multilayer,
and employ the
polyketal polymers of the invention as a single component or in a blend with
other materials,
as described above. The films are useful for various applications including
agricultural
mulching films; printable films for graphics or text; cling films (extensible
films) for
foodstuffs, films for bales in the agricultural sector and for wrapping of
refuse; shrink films
such as for example for pallets, mineral water, six pack rings, and so on;
bags and liners such
as for collection of refuse, holding foodstuffs, gathering mowed grass and
yard waste, and
the like; thermoformed single-layer and multilayer packaging for foodstuffs,
such as for
example containers for milk, yogurt, meat, beverages, etc.; and in multilayer
laminates with
layers of paper, plastic materials, aluminum, metalized films for a wide
variety of
applications.
The amide-functional polymers and copolymers of the invention are also useful
for
coatings that form a layer on top of a film, an article, and the like.
Coatings of the invention
are applied, in embodiments, by extrusion coating, die coating, slot coating,
brush coating,
spray coating, or any other generally known technique employed in the coating
industry.
Coatings employing the amide-functional polymers and copolymers of the
invention are
useful as protective coatings, paint components, adhesives or glues, barrier
layers, and the
like. The coatings of the invention are applied, in embodiments, with or
without additional
solvent(s), such as coalescing solvents, and with our without additives such
as UV blocking
agents, antibacterial agents, colorants, fillers, and the like. The coatings
of the invention are,
in some embodiments, crosslinked after application.
The amide-functional polymers and copolymers of the invention are also useful
in
forming articles. An "article", as defined for the purposes of the invention,
includes objects
that are be rigid or flexible; that exist as standalone objects or as part of
an assembly or
laminate; and that include one or more amide-functional polymers and
copolymers of the
invention or a blend thereof, optionally with one or more additional
materials. Some
examples of useful articles that include amide-functional polymers and
copolymers of the
68
CA 02676892 2009-08-19
invention are punnets for foodstuffs, I-beams for construction, casings for
e.g. pens,
computer screens, and the like; parts for automobile construction, table tops,
and the like;
decorative items such as lamp parts, jewelry, vases, architectural features,
and the like;
children's toys; drink bottles; and many other articles. The invention is not
particularly
limited in terms of what articles maybe formed employing the amide-functional
polymers
and copolymers of the invention.
Articles that can be formed using the amide-functional polymers and copolymers
of
the invention include, in various embodiments, loose covers for optical wave
guides, e.g.
loose jacketing; measuring and pressure reservoirs for liquids and gases, e.g.
filter bowls;
pump casings, styled articles such as toiletry, glasses frames, handles, and
the like; shaped
articles for medical and biological apparatus, barrier layers in foodstuff
packaging, and the
like. The transparent nature of many of the the amide-functional polymers and
copolymers
of the invention make them ideally suited for many useful applications where
tough
polyamide properties are required and transparency is also desirable or
required.
Other examples of articles that can be formed using the amide-functional
polymers
and copolymers of the invention are foamed articles. Foaming of polyurethanes
is discussed
above; these techniques and others generally known in the industry are used,
in
embodiments, to form foamed articles from the various amide-functional
polymers and
copolymers of the invention. Foamed articles include both rigid and flexible
foams. Some
examples of useful foamed materials include cushions for automobile seats,
interior or
exterior furniture, and the like; foamed or foamable beads for the production
of pieces
formed by sintering; foamed blocks made up of pre-foamed particles; foamed
sheets,
thermoformed foamed sheets, and containers obtained therefrom for the
packaging of
foodstuffs.
Articles also include fibrous articles. Examples of fibrous articles include
standard
scale fibers, microfibers, nanofibers, and composite fibers. Composite fibers
have, in
embodiments, a core constituted by a rigid polymer such as PLA, PET, PTT, etc.
and an
external shell made with one or more polyketal polymers of the invention;
other composite
fibers have various section configurations (from round to multilobed). Fibers
also include
flaked fibers, woven and non-woven fabrics or spun-bonded or thermobonded
fabrics for the
69
CA 02676892 2009-08-19
sanitary sector, the hygiene sector, the agricultural sector, georemediation,
landscaping and
the clothing sector.
EXPERIMENTAL SECTION
General Experimental Procedures
Gas Chromatography (GC) and GC-Mass Spectrometry (GC-MS) Analyses
GC and GC-MS analyses are carried out according to standard laboratory
techniques.
Standard GC analysis is carried out by flame ionization detector. The
integration peak areas
of all peaks in the chromatogram are automatically calculated by an Agilent
Technologies
ChemStation (Agilent Technologies of Santa Clara, CA). The calculated peak
areas are
reported as a weighted percent (expressed as abundance) relative to the area
of all of the
detected peaks in the chromatogram (total area). These calculations are used
elsewhere
herein to report all percent yield, percent yield "based on theoretical",
percent yield "as
determined by GC-MS", and any other percent reaction statements resulting from
GC or GC-
MS analyses.
Gel Permeation Chromatography (GPC
Molecular weight determination is carried out by GPC using a Waters Isocratic
HPLC
System (from Waters Corp. of Milford, Massachusetts) that includes a Waters
2414
Differential Refractometer, Waters 1515 Isocratic Pump, Waters 717
Autosampler, and
Waters Column Heater and Empower GPC Software for molecular weight analysis.
For
samples with an expected molecular weight of 20,000-400,000 Daltons a PLgel
Mixed D
5 m column, 300X7.5 mm, is used; for samples with an expected molecular weight
of less
than 20,000 a PLgel Mixed E 5 m column, 300X7.5 mm, is used; and for samples
with an
expected molecular weight between 20,000 and 2,000,000 a PLgel Mixed C 5 m
column,
300X7.5 mm is used. All columns were obtained from Polymer Labs, a division of
Varian
Inc. of Palo Alto, CA.
All samples are analyzed using either tetrahydrofuran (THF) or dimethyl
formamide
(DMF) mobile phase. The THE mobile phase is employed at 1 ml/min and weight
average
CA 02676892 2009-08-19
molecular weight (M,,,) is calculated against polystyrene narrow molecular
weight standards.
The DMF mobile phase with 0.05M lithium bromide is employed at I ml/min and
weight
average molecular weight (Mw) is calculated against polymethylmethacrylate
narrow
molecular weight standards.
Differential Scanning Calorimetry (DSC)
Glass transition temperature (Tg) is determined by following ASTM D-3418,
employing a TA Q200 instrument with refrigerated cooling and TA Thermal
Advantage
software (from TA Instruments of New Castle, DE). Homogeneous samples of
between
about 5 and 15mg are prepared, weighed, placed in a Tzero pan and crimped with
a Tzero lid,
(pan and lid both available from TA Instruments). The mass of the sample is
entered into the
Thermal Advantage software. The thermal analysis is carried out according to
one or the sets
of parameters below:
Cycle 0: Equilibrate at -40 C
Isotherm for 2.00 minutes
End of Cycle 0
Cycle 1: Ramp 10 C/min to 240 C
Isotherm for 2.00 minutes
End of Cycle 1
Cycle 2:Ramp 10 C/min to -40 C
Isotherm for 2.00 minutes
End of Cycle 2
Cycle 3: Ramp 10 C/min to 240 C
Isotherm for 2.00 minutes
End of Cycle 3
Repeat at Cycle 0
Cycle 0: Equilibrate at -80 C
Isotherm for 2.00 minutes
End of Cycle 0
Cycle 1: Ramp 10 C/min to 200 C
Isotherm for 2.00 minutes
End of Cycle 1
Cycle 2:Ramp 10 C/min to -80 C
Isotherm for 2.00 minutes
End of Cycle 2
Cycle 3: Ramp 10 C/min to 200 C
Isotherm for 2.00 minutes
End of Cycle 3
71
CA 02676892 2010-06-14
Repeat at Cycle 0
Synthesis of Certain Starting Materials
The following ketal compounds were synthesized for further reactions in the
Examples.
EtLGK
The glycerol ketal of ethyl levulinate, 1,3-dioxolane-2-propanoic acid, 4-
(hydroxymethyl)-2-methyl, ethyl ester, will be referred to as "EtLGK".
0
off/
HO O
0
EtLGK
EtBLEK
The erythritol bisketal of ethyl levulinate, [4,4'-bi-1,3-dioxolane]-2,2'-
dipropanoic acid, 2,2'-dimethyl-2,2'-diethyl ester, will be referred to as
"EtBLEK".
o
o J~ 'K,
EtBLEK
Et BLPK
The pentaerythritol bisketal of ethyl levulinate will be referred to as
"EtBLPK".
72
CA 02676892 2009-08-19
IOII 0
O O
EtBLPK
EtBPEK
The bisketal of erythritol and ethyl pyruvate was synthesized as follows. A
1000 mL,
three neck round bottom flask was charged with 122.12g (1.00 mol) erythritol
(obtained from
Cargill of Wayzata, MN), 348.36g (3.00 mol) ethyl pyruvate (obtained from the
Sigma-
Aldrich Company of St. Louis, MO), and 235g toluene (obtained from Fisher
Scientific of
Waltham, MA). The flask was equipped with a thermocouple, mechanical stirrer,
and Dean-
Stark trap with an attached condenser. A bubbler was attached to the top of
the condenser of
the Dean Stark trap to release positive pressure in the flask.
The reaction was stirred and heated to 110 C using a heating mantle. Upon
reaching
110 C, 29 L of concentrated sulfuric acid (obtained from the Sigma-Aldrich
Company of St.
Louis, MO) was quickly added by metered micropipette. A liquid was observed to
collect in
the Dean-Stark trap; the trapped liquid separated into two layers upon
cooling. The top layer
was presumed to be toluene, and the bottom layer presumed to be water. The top
layer was
allowed to return to the flask while the bottom layer continued to collect in
the Dean-Stark
trap. Heating and stirring were continued for approximately 5 hours, at which
time 2/3 the
theoretical amount of water had been collected in the Dean-Stark trap. The
contents of the
flask were allowed to cool to room temperature. The cooled contents of the
flask were
analyzed by GC-MS. The GC trace showed about 10% yield of the bisketal
structure of
erythritol and ethyl pyruvate, referred to as "EtBPEK". The toluene was
stripped from the
contents of the flask by rotary evaporation.
0
0
O 0 I___C O 0
O
O
EtBPEK
EtBLDK
73
CA 02676892 2010-06-14
The bisketal of diglycerol (3,3'-oxybis-l,2-propanediol) and ethyl levulinate
will be referred to as "EtBLDK".
0 0
0 --,~O :~- 0 -COK,--~O
0 0
EtBLDK
EtBAEK
The bisketal of erythritol and acetoacetate will be referred to as "EtBAEK".
0
0 j
0 0
0 0
0 0
EtBAEK
Example 1
5 $ 0
2 0 7 H 10 12 H
N
HO 3 p 6 Y N
9 11 H
1 4 0
Proton assignments
D E O
A O G H H J L OH
HO B O F I K H
C 0
74
CA 02676892 2010-06-14
Carbon assignments
Exact Mass: 460.28
Molecular Weight: 460.56
74a
CA 02676892 2009-08-19
A flame-dried 250mL four-necked flask was cooled under a stream of nitrogen
and
equipped with a Dean-Stark apparatus. To this flask was added 33.5 g (153
mmol) EtLGK
and 8.95 g (77 mmol) 1,6-hexamethylene diamine (obtained from Acros Organics
of Geel,
Belgium). The reaction mixture was heated to 200 C for 2.5 hours, after which
time 7 mL
of a liquid accumulated in the Dean Stark trap (theoretical yield of ethanol =
8.8 mL). The
liquid was removed and vacuum of about 5 torr was applied to the reaction
mixture for 3
hours. The reaction mixture was then allowed to cool to ambient temperature.
Upon
cooling, 0.132 g of a yellow viscous oil was isolated for characterization.
IR (cm) = 3306 (OH, NH amide), 2935 (CH aliphatic), 1647 (C=O amide). 'H
NMR (CDC13) 6 (ppm) = 6.15, 5.96 (2H, br s, 8); 4.20 (2H, in, 3); .88 (4H, in,
4); 3.86 (2H, t,
1); 3.64 (4H, m, 2); 3.21 (4H, m, 9); 2.29 (4H, in, 7); 2.11 (2H, m, CH2 12);
1.98 (4H, in, 6);
1.48 (4H, br t, 10); 1.36, 1.32 (6H, s, 5); 1.32 (4H, s, 11). 13C NMR (CDC13)
S (ppm) _
174.24, 173.26 (C=O amide, H); 110.36, 110.04 (D); 77.10, 76.49 (B); 66.26,
65.50 (C);
63.08, 62.11 (A); 39.17, 39.09 (G); 34.96, 34.38 (F); 31.38, 31.07 (E); 29.34,
29.15 (1);
25.96 (J); 24.91 (K); 23.90 (L).
Example 2
To the crude product of Example 1 was added 14.92 g (77 mmol) dimethyl
terephthalate (obtained from the Sigma Aldrich Company of St. Louis, MO) under
nitrogen
purge. The mixture was heated to 180 C and then 7.6 L (300 ppm) Ti(O-nBu)4
(obtained
from Acros Organics of Geel, Belgium) added. The reaction mixture was heated
to 200 C
for 2 hours, then 220 C for 5 hours, after which time 1.5 mL of a liquid
(theoretical yield of
methanol is 3.0 mL) was collected. The collected methanol was removed from the
Dean
Stark trap and a vacuum of about 5 torn was applied to the reaction flask and
maintained for 5
hours. The contents of the reaction flask were then allowed to cool to room
temperature and
the vacuum was released. Upon cooling, the reaction contents were transparent,
brittle, and
amber-colored.
Yield: 12.84 g, 22.0%. GPC: M,, = 1314 PDI = 2.31. DSC (-40 to 240 C): Tg =
54.08 C (OH = 0.45 J/(g* C)).
CA 02676892 2009-08-19
Example 3
A 250mL, 3-neck roundbottom flask was charged with 53.82g (0.247mol) of EtLGK,
15.97g(0.082mo1) of dimethyl terephthalate, "DMT" (obtained from the Sigma
Aldrich
Company of St. Louis, MO), and 10.30g(0.166mo1) ethylene glycol, "EG"
(obtained from
Fisher Scientific of Waltham, MA), for a mole ratio of 3:1:2 EtLGK:DMT:EG,
respectively.
The flask was equipped with a Dean Stark trap and condenser, mechanical
stirrer,
nitrogen/vacuum inlet, and nitrogen outlet. The system was degassed and
backfilled with N2
a total of five times, applying a vacuum of about 20 torr, and after the five
degassing cycles
the system was backfilled with nitrogen and a nitrogen sweep was commenced
from the inlet
through the outlet. Titanium(IV) n-butoxide (16.2 L, 200ppm) (obtained from
Acros
Organics of Geel, Belgium) was injected using a metered micropipette and the
mixture was
stirred. The flask was immersed in an oil bath heated to 190 C for
approximately 22.5 hours.
Liquid was observed to collect in the Dean Stark trap, and once the liquid
neared the
theoretical yield of ethanol based on observed volume in the trap, a vacuum of
approximately
15 torr was applied to the system. The vacuum was maintained for about 2
hours. The flask
was then filled with nitrogen and cooled, and a sample taken for GPC analysis.
Then, 9.13g (0.079 mol) of 1,6-hexamethylene diamine, "HD" (obtained from
Acros
Organics of Geel, Belgium) was added to the flask. The flask was then
degassed/backfilled
by alternating vacuum and nitrogen an additional three times, followed by
filling the flask
with nitrogen and applying a nitrogen sweep through the flask. The flask was
immersed in
an oil bath set to 180 C for 1 hour. The temperature of the oil bath was then
increased to
200 C and this was maintained for an additional 1 hour. The temperature of the
oil bath was
increased again to 210 C for an additional hour. At this point, vacuum was
applied using 2
Teflon pumps (approximately 10 torr) for 3 hours, and then high vacuum applied
using an oil
pump (500-750mtorr). The high vacuum was maintained for 1 hour, and then the
temperature of the oil bath was increased to 220 C. This temperature and
vacuum was then
maintained for an additional 48 hours. The flask was placed under nitrogen and
cooled to
ambient temperature, and the polymer removed from the flask for analysis by
GPC and DSC.
The results of the analysis are shown in Table 1.
76
CA 02676892 2009-08-19
Examples 4-7
Employing the methodology of Example 3, reactions were carried out with
varying
ratios of reagents EtLGK, DMT, EG, and HD and with varying time under high
vacuum.
Table 1 shows the reactions and the results of analysis for Examples 4-7.
Example Molar ratios Properties Observa- Time
No. Et- DMT EG HD Polyol Tg, tions at end Under
LGK MW, C of reaction High
g/mol Vacuum
3 3 1 2 0.96 Mn = 1708 36 viscous (but 48.33 hrs.
Mw = 2670 not climbing
stir rod)
4 1 1 2 0.9 Mn = 1296 62 not very 135 min.
Mw = 2369 viscous (but
still
crystalline)
5 2 1 2 0.84 Mn = 1248 30 not very 13 hrs.
Mw = 1969 viscous
6 2 1 2 0.9* Mn = 6664 49 very viscous 21.66 hrs.
Mw = (polymer
23204 climbing stir
rod)
7 5 1 2 0.87 Mn = 1494 25 viscous (but 31.25 hrs.
Mw = 2272 not climbing
stir rod)
* In this experiment, the diamine was added as two separate fractions. The
first fraction
accounted for 0.5mol equiv., and the second fraction (which was added
approximately 22 hrs.
later) accounted for 0.4mol equiv.
Table 1. Reaction parameters and analytical results obtained for syntheses of
Examples 3-7.
Example 8
A flame-dried 250 mL four-necked flask was cooled under a stream of nitrogen
and
equipped with a mechanical stirrer, Dean-Stark trap, nitrogen inlet, and
nitrogen
outlet/vacuum port. To the flask was added 7.79 g (67 mmol) 1,6-hexamethylene
diamine
(obtained from Acros Organics of Geel, Belgium), 24.98 g (67 mmol) of EtBLEK,
and
10mg (0.07 mmol) 1,5,7-triazabicyclo[4.4.0]dec-5-ene (obtained from Acros
Organics).
77
CA 02676892 2009-08-19
With mechanical stirring under a slow nitrogen sweep, the mixture was heated
to 50 C for
16 hours, then 200 C for 4 hours, during which a liquid was observed to
collect in the Dean-
Stark trap. At the end of the 20 hours reaction time, liquid collection
subsided. Heating for
an additional 16 hours at 200 C resulted in increased solution viscosity.
Subsequent
application of vacuum (8 torr) at 220 C for 2 hours was accompanied by
bubbling of the
reaction mixture and further increase in solution viscosity.
The contents of the reaction flask were cooled to ambient temperature,
yielding a
transparent, brittle, amber-colored material. Yield was 19.63 g, or 59.8 wgt%.
GPC analysis
revealed Mn = 2153, PDI = 1.33. DSC was carried out using temperature profile
3; Tg =
56.91 C (AH = 0.37 J/(g* C)).
Example 9
A 250ml 3 neck round bottom flask was charged with 50.4g (0.135mol) of EtBLEK,
17.Og (0.27mol) of ethylene glycol (obtained from the Fisher Scientific
Company of of
Waltham, MA) and 13.5 l (200ppm) of titanium tetrabutoxide (Ti(O-nBu)4,
obtained from
Acros Organics of Geel, Belgium). The flask was equipped with a mechanical
stirrer, a Dean
Stark trap and condenser, and a nitrogen inlet and nitrogen/vacuum outlet. The
contents of
the flask were degassed at room temperature by subjecting to 5 vacuum/nitrogen
purge
cycles. After the degassing cycles were completed the flask was backfilled
with nitrogen and
a slow nitrogen sweep was commenced. The flask was placed in an oil bath set
to a
temperature of 190 C. The flask was stirred in the oil bath for about 13
hours. At this point
a vacuum was applied to the contents until the pressure was between 2 and 3.5
torr. This was
maintained for about half an hour, after which time the temperature in the oil
bath was
increased to 210 C. The temperature and vacuum were maintained for about 1.5
hours.
The contents of the flask were then cooled to about 65 C, at which point a
sample
was removed for analysis. GPC showed that the number average molecular weight
(Mõ) was
3,600 g/mol and weight average molecular weight (Mw) was about 7,400 g/mol,
for a
polydispersity index of about 2.8.
The reaction flask was then additionally charged with 13.5g (0.1 l6mol) of 1,6-
hexamethylenediamine (obtained from Acros Organics of Geel, Belgium) and the
flask was
degassed with a 3 vacuum/nitrogen purge cycles. At the end of the degassing
cycles, the
78
CA 02676892 2009-08-19
flask was backfilled with nitrogen, and a slow nitrogen sweep was initiated
with stirring.
The flask was placed in an oil bath having a temperature preset to 180 C. This
temperature
was maintained for 1 hour and then the temperature of the oil bath was
increased to 200 C.
This temperature was maintained for another hour and then the temperature was
increased to
210 C. This temperature was maintained for about 1.5 hours, then a vacuum was
applied to
the flask until the pressure in the flask reached between about 3 and 5 torr.
Over the ensuing
3 hours, the vacuum in the flask was observed to decrease to about 0.5 torr
(500 millitorr).
The contents of the reaction flask were observed to undergo significant
increase in viscosity
during this time. The reaction was stopped when the contents of the reaction
flask could no
longer be stirred due to the contents climbing the stir rod.
Upon cooling the contents of the flask to ambient temperature, a transparent,
orange
solid was obtained. The solid was insoluble and could not be broken by hand.
Glass
transition temperature was measured by DSC to be about 60 C.
Tensile testing was carried out according to ASTM D638-90, specimen type IV.
The
specimens for testing were prepared by removing material from the flask and
placing it
between TEFLON coated aluminum foil sheets (BYTAC , obtained from Fisher
Scientific
of Waltham, MA) and pressing with a spacer of 1.5mm using a Carver Model 4122
pneumatic heated platen press (obtained from Carver, Inc. of Wabash, IN)
preheated to
180 C, at a pressure of about 2268kg, for about 10 minutes. The pressed sample
was
removed from the press and cut into pieces. The pieces were placed on top of
TEFLON
molds machined to specifications of ASTM D638-90, specimen IV, 1 mm deep. A
BYTAC sheet was placed on top of the samples and mold. The BYTAC sheet,
samples,
and mold were pressed at 180 C at 2268kg for about 10 minutes. The samples
were cooled
to room temperature and placed in a vacuum oven at 50 C for about 16 hours;
the samples
were removed from the oven immediately prior to tensile testing.
Testing was performed with nominal strain at outset of about 0.17. The result
of
tensile tests carried out on seven samples prepared as described above are
shown in Table 2.
The stress-strain curve for each of the seven samples is shown in FIG. 2. It
can be observed
in FIG. 2 that the samples are ductile, having a yield stress at about 15-20%
strain, but
ultimate strain between about 22-72%.
79
CA 02676892 2009-08-19
Examples 10-12
The procedure of Example 9 was repeated, except that the mole percent (mol%)
of
1,6-hexamethylenediamine ("HD") to EtBLEK was varied. Glass transition
temperature was
measured by DSC; tensile data were measured using the technique of Example 9.
The mol%
HD and thermal and tensile data are reported in Table 2.
Example HD, mol % Tensile Properties
No. based on Tg, oC Yield stress, Tensile Strain at
EtBLEK MPa strength, MPa break, %
9 86% 60 57 36 34
95% 65 --- --- ---
11 98% 66 67 45 29
12 100% 66 69 45 22
Table 2. HD content and resulting properties for polymers of Examples 9-12.
Example 13
10 A 250 mL, three-neck roundbottom flask was charged with 31.5 g (81.2 mmol)
EtBLPK and 10.1g (163 mmol) ethylene glycol (obtained from Fisher Scientific
of Waltham,
MA), and 8.3 L (200 ppm) Ti(O-nBu)4 (obtained from Acros Organics of Gee],
Belgium).
The flask was equipped with a mechanical stirrer, Dean Stark trap,
nitrogen/vacuum inlet,
and nitrogen outlet. The flask was degassed with three cycles of evacuating
the flask to
approximately 5 torn followed by backfilling with nitrogen. After the three
cycles, the flask
was backfilled with nitrogen and a nitrogen sweep through the flask was
commenced. The
mixture was stirred in an oil bath set to about 190-210 C under nitrogen
sweep for about
18.5 hours, and then under a vacuum of about 4-13 torn for about 4 hours, then
under a
vacuum of 0.4 torr for about 2.5 hrs.
The flask was then cooled to room temperature and 8.5g (73.3 mmol) 1,6-
hexamethylenediamine (obtained from Acros Organics of Geel, Belgium) was added
to the
flask contents. The flask was placed in an oil bath at a temperature of
between about 180-
210 C under nitrogen sweep for about 5 hours, then a vacuum of about 26 torr
was applied
for about 3.5 hours. The contents of the flask were cooled to room temperature
and a sample
CA 02676892 2009-08-19
was removed for analysis by DSC. The contents of the flask were found to have
a Tg of
about 7 C.
Example 14
A 250 mL three-neck roundbottom flask was equipped with a mechanical stirrer,
Dean Stark trap, nitrogen/vacuum inlet, and nitrogen outlet. The flask was
charged 39.4g
(124 mmol) EtBPEK and 14.5g (125 mmol) 1,6-hexamethylenediamine (obtained from
Acros Organics of Geel, Belgium). The flask was degassed with three cycles of
evacuating
the flask to approximately 20 tort followed by backfilling with nitrogen.
After the three
cycles, the flask was backfilled with nitrogen and a nitrogen sweep through
the flask was
commenced. The mixture was stirred in an oil bath set to a temperature of
about 190-200 C
for 20 hours, then the temperature of the oil bath was increased to about 200-
210 C and a
vacuum of 10-35 tort was applied to the flask for about 7 hours The contents
of the flask
were cooled to room temperature and a sample was removed for analysis by DSC.
The
contents of the flask were found to have a Tg of about 45 C.
Example 15
A 250 mL, three neck roundbottom flask was charged with 7.6g (0.0392 mol)
dimethyl terephthalate (obtained from the Sigma-Aldrich Company of St. Louis,
MO), 7.6g
(0.0392 mol) dimethyl isophthalate (obtained from the Sigma-Aldrich Company),
27.3g
(0.073 mol) EtBLEK, and 18.Og (0.155mo1) of 1,6-hexamethylene diamine
(obtained from
Acros Organics of Geel, Belgium). The flask was equipped with a Dean Stark
trap,
mechanical stirrer, nitrogen/vacuum inlet, and nitrogen outlet. The flask was
degassed 5
times by applying a vacuum of about 5.0 tort and then releasing the vacuum
with a nitrogen
flow. After the five degassing cycles vacuum was released by applying a
nitrogen sweep
through the flask. The flask was placed in an oil bath set to a temperature of
180 C with
stirring for about 30 minutes. The temperature in the oil bath was then
increased to 210 C
and this temperature was maintained for about 2 hours with stirring. After
that the vacuum
was applied to the flask. Over a period of about 2 hours the vacuum in the
flask was
observed to go from about 18.0 tort to about 1.0 tort. The temperature and
stirring were
maintained with vacuum for the next 11 hours during which time the vacuum was
observed
81
CA 02676892 2009-08-19
to fall to 0.5 torn. At the end of the 11 hours the contents of the reaction
flask were observed
to have sufficiently high viscosity that stirring could not be maintained and
the contents of
the flask were allowed to cool to room temperature.
A portion of the contents of the flask were removed for DSC analysis, which
revealed
that the glass transition temperature (Tg) was about 88 C. No crystalline
transition was
observed in the DSC trace. The DSC trace is shown in FIG. 3.
Example 16
The procedure of Example 15 was repeated except that 27.88g (0.0745mol) of
EtBLEK, 18.38g (0.158mo1) 1,6-hexamethylene diamine, 16.4g (0.0845mo1) of
dimethyl
terephthalate, and no dimethyl isophthalate was used. The contents of the
reaction flask at
the end of the procedure were determined by DSC to have a glass transition
temperature of
97 C and no crystalline transition. The DSC trace is shown in FIG. 4.
Example 17
A 3 neck 250m1 flask was charged with 50.50g (0.135mo1) EtBLEK and 12.Og
(0.103mol) of 1,6-hexamethylene diamine (obtained from Acros Organics of Geel,
Belgium).
The flask was equipped with a Dean Stark trap, mechanical stirrer, and
nitrogen/vacuum inlet
amd outlet. The flask was subjected to 5 degassing cycles by alternating
between a vacuum
of about 10 torr and nitrogen sweep. At the end of the 5 cycles the flask was
placed under a
nitrogen sweep, and with stirring was placed in an oil bath set to 180 C. The
temperature of
the oil bath was then ramped at about 2 C/hour until the final temperature of
200 C was
reached. The temperature of 200 C was then maintained for the next 4 hours. A
vacuum
was then applied to the flask and the vacuum in the flask was observed to go
from 10.7torr to
about 2.8torr over the period of about 1 hour. The oil bath was then shut off
and allowed to
slowly cool to 100 C.
After the oil bath had cooled to about 100 C the vacuum to the flask was
released by
backfilling with nitrogen, and the flask was quickly opened under nitrogen and
charged with
47.4g (0.244mo1) of dimethyl terephthalate (obtained from the Sigma-Aldrich
Company of
St. Louis, MO), 32.1 g (0.35mol) of 1,4-butane diol (obtained from the Sigma
Aldrich
Company), and 16.1 l (200ppm) of Ti(O-nBu)4 (obtained from Acros Organics).
The flask
82
CA 02676892 2009-08-19
was then closed and degassed with 5 cycles of a vacuum of about 10 torr
alternated with a
nitrogen sweep. After the degassing cycles the flask was placed under nitrogen
sweep and
the temperature of the oil bath was ramped up to 180 C with stirring. This
temperature was
maintained for about 2 hours and then the temperature in the flask was
increased to 200 C
and this temperature was maintained for about 2 hours. Then with the
temperature still at
200 C a vacuum was applied to the flask and the vacuum inside the flask was
observed to go
from 34.4 torr to 7.7 torr over a period of 2.5 hours. The temperature of the
oil bath was then
increased to 210 C while still under vacuum and the temperature was then
maintained for
another %2 hour and then the temperature of the oil bath was increased to 220
C for the next
hour. The contents of the reaction flask were then observed to be at
sufficiently high
viscosity that stirring could not be continued and stirring was stopped and
the flask was
allowed to cool to room temperature.
The contents of the reaction flask were subjected to DSC, which showed the
reaction
product had a glass transition temperature (Tg) of 39 C and a melt temperature
(Tm) of
130 C.
Example 18
A flame-dried 250 mL three-neck flask was charged with 41.82g (100 mmol)
EtBLDK and 11.84g (102 mmol) 1,6-hexamethylene diamine (obtained from Acros
Organics
of Geel, Belgium). The flask was equipped with a mechanical stirrer, nitrogen
inlet, nitrogen
outlet, and Dean Stark trap. Nitrogen sweep was applied through the flask. The
flask was
immersed in an oil bath set to 180 C, and the temperature was gradually raised
to 200 C at
the rate of about 2 C/min. Heating was continued at 200 C for an additional 14
hours,
resulting in collection of 15.5 mL of liquid in the Dean-Stark trap. After the
liquid in the
Dean-Stark trap was drained, a vacuum of 10 - 20 torn was applied to the flask
for 1 hour.
The vacuum level was then decreased to about 6 torr while the temperature of
the oil bath
was raised to 210 T. Heating was continued for 6 hours, and the vacuum level
was
gradually decreased from 6 Torr to 0.2 Torr. The crude polymer was isolated
after slight
cooling. The crude polymer was isolated as a red brittle solid and analyzed by
DSC and
GPC. Yield: 33.19 g. DSC (-80 to 200 C, 10 C/min, Tg = 34.9 C). GPC: Mn =
10700, PDI
= 10.1.
83
CA 02676892 2009-08-19
Example 19
A flame-dried, 250 mL four-neck roundbottom flask was charged with 38.31g (11
1
mmol) EtBAEK and 14.02g (226 mmol) ethylene glycol (obtained from Fisher
Scientific of
Waltham, MA). The flask was equipped with a mechanical stirrer, Dean-Stark
trap, and
nitrogen/vacuum inlet. The flask was degassed at room temperature with three
repetitions of
evacuating the flask to about 1.5 torr and back-filling with nitrogen. After
degassing, the
flask was back-filled with nitrogen and 10.5 L (200 ppm) Ti(OBu)4 (obtained
from Acres
Organics of Geel, Belgium) was added to the reaction mixture. The flask was
placed in an
oil bath with a temperature set to 190 C and was stirred under nitrogen for 14
hours. The
temperature of the oil bath was then increased to 200 C and was maintained at
that
temperature for 2.5 hours, then the temperature of the oil bath was increased
to 210 C and
was maintained at the temperature for 30 minutes. During this time about 4.Og
of a liquid
was observed to collect in the Dean Stark trap. The liquid was drained, and a
vacuum was
applied to the flask. The vacuum level started at about20 torr, and was
gradually lowered to 5
torn over the next 3 hours while the oil bath temperature was maintained at
210 C. After 3
hours, the flask was back-filled with nitrogen and was allowed to cool to room
temperature.
At this point, 11.68g (101 mmol) 1,6-hexamethylene diamine (obtained from
Acros
Organics of Geel, Belgium) was added to the flask. The flask was placed in an
oil bath set to
a temperature of 180 C with stirring under nitrogen, and held at this
temperature for 1 hour;
then the temperature of the oil bath was increased to 200 C and held at this
temperature for
minutes; then the temperature of the oil bath was increased to 220 C and held
at this
temperature for 1 hour. At this point a vacuum of about 4 - 10 torr was
applied to the flask
while the temperature of the oil bath was maintained at 220 C for 15 hours.
Then the flask
25 was back-filled with nitrogen and allowed to cool slightly before the crude
reaction product
was isolated.
The crude reaction product was isolated as a black brittle solid and was
analyzed by
DSC and GPC. Yield: 12.29 g. DSC: Tg = 36.9 C). GPC: Mn = 5500, PDI = 2.62.
84
CA 02676892 2009-08-19
Example 20
A 500 mL four-neck roundbottom flask was flame-dried and charged with 99.25g
(265 mmol) EtBLEK, 33.38g (538mmo1) ethylene glycol (obtained from Fisher
Scientific of
Waltham, MA), and 27 pL (200ppm) Ti(O-nBu)4 (obtained from Acros Organics of
Geel,
Belgium). The flask was equipped with a mechanical stirrer, Dean Stark
apparatus, and
nitrogen/vacuum inlet. The system was degassed while stirring in an oil bath
set to 40 C
with three repetitions of evacuating the flask to approximately 1 torr and
back-filling with
nitrogen. After degassing, the flask was backfilled with nitrogen. The
temperature of the oil
bath was increased to 190 C and the contents of the flask were stirred for 22
hours, resulting
in collection of 22 mL of liquid in the Dean Stark trap. At this point a
vacuum of about 10
torr was applied to the flask for 1 hour, then the temperature of the oil bath
was increased to
210 C and this temperature was maintained for 1.5 hours, resulting in 13 mL of
liquid
collected in the Dean Stark trap. The flask was back-filled with nitrogen, and
the contents of
the flask were allowed to cool before collecting the crude reaction product.
The reaction
crude product was analyzed by GPC to determine Mõ = 1117, PDI = 2Ø
A 250 mL three-neck round bottom flask was charged with the 31.58g of the
crude
reaction product. The flask was equipped with a mechanical stirrer, Dean Stark
apparatus,
and nitrogen/vacuum inlet. An addition funnel was installed and charged with
10.6g (102
mmol) bis(2-aminoethyl)amine (obtained from Acros Organics of Geel, Belgium).
The flask
was warmed to about 40 C using an oil bath before degassing with three
repetitions of
evacuating the flask to approximately 1 torr and back-filling with nitrogen.
After degassing,
a nitrogen sweep was applied through the flask. The temperature of the oil
bath was then
increased to 190 C and the contents of the flask were stirred under nitrogen
sweep while
bis(2-aminoethyl)amine was added dropwise over about 1 hour. After the
addition was
complete, stirring was continued for 1 additional hour at 190 C under
nitrogen, at which
point a vacuum was applied to the flask. The reaction mixture was then stirred
at 190 C
under 0.5 torr vacuum for 40 minutes, resulting in collection of 5.5 mL of
liquid in the Dean
Stark trap. The temperature was increased to 210 C and 0.5 torr applied for 1
additional
hour. At this point the flask was back-filled with nitrogen and allowed to
cool slightly before
the crude product was isolated as a brittle red solid and analyzed by DSC and
GPC. Yield:
CA 02676892 2009-08-19
23.28 g. DSC revealed Tg = 72.68 C (OH = 0.42 J/(g* C)). GPC (DMF mobile
phase: Mõ _
5293, PDI = 1.93.
Example 21
A 250 mL four-neck roundbottom flask was charged with 12.63g (32.5 mmol)
EtBLPK, 13.72g (32.8 mmol) EtBLDK, and and 7.31g (62.9 mmol) 1,6-hexamethylene
diamine (obtained from Acros Organics of Geel, Belgium). The flask was
equipped with a
mechanical stirrer, Dean Stark trap, and nitrogen/vacuum inlet. The flask was
degassed at
ambient temperature with three repetitions of evacuating the flask to
approximately 1 torr
and back-filling with nitrogen. The flask was placed in an oil bath with
temperature set to
150 C and was stirred under nitrogen for 2 hours, resulting in collection of 6
mL of liquid in
the Dean Stark trap. The temperature of the oil bath was increased to 190 C
and this
temperature was maintained for 1 hour; then the temperature of the oil bath
was increased to
200 C and this temperature was maintained for 16 hours, resulting in
collection of 1.2 mL of
liquid in the Dean Stark trap.
The contents of the flask were allowed to cool slightly before the crude
product was
collected. The crude product was isolated as a hard orange solid that was
insoluble in THE
and DMF and was analyzed by DSC to determine that Tg = 37.14 C (OH = 0.40
J/(g* C)).
Example 22
A flame-dried 250 mL four-neck roundbottom flask was flame dried, then cooled
to
ambient temperature under nitrogen. The flask was charged with 40.2 mg (0.29
mmol)
1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]-pyrimidine (TBD, obtained from the
Sigma-
Aldrich Company of St. Louis, MO), 12.78 g (110 mmol) 1,6-hexamethylene
diamine
(obtained from the Fluka Chemical Corporation of Milwaukee, WI) and 41.16g
(110 mmol)
EtBLEK. The flask was equipped with a mechanical stirrer, Dean Stark trap, and
nitrogen/vacuum inlet. The contents of the flask were degassed with three
repetitions of
evacuating the flask to approximately 1 torr and back-filling with nitrogen.
After degassing,
the flask was back-filled with nitrogen. The flask was placed in an oil bath
set to a
temperature of 120 C and was stirred for 29 hours, resulting in the
collection of 3 mL of
86
CA 02676892 2009-08-19
liquid in the Dean Stark trap. A sample of the contents of the flask was
removed for analysis
by GPC: Mn = 4903, PDI = 1.69.
Then the flask was placed in an oil bath set to a temperature of 140 C for 16
hours
under nitrogen, leading to collection of an additional 1 mL of liquid in the
Dean Stark trap.
A sample of the contents of the flask was removed for analysis by GPC: Mõ =
6627, PDI =
1.83.
Finally, the flask was placed in an oil bath set to a temperature of 180 C
for 4 hours,
then 200 C for 0.5 hours, under nitrogen. When the oil bath reached 200 C a
vacuum of 0.5
torr was applied to the flask for about 1 hour, resulting in a viscous amber
mixture. The
vacuum was released by back-filling with nitrogen and the contents of the
flask were isolated
after slight cooling. Yield: 26.25 g.
Example 23
A 500 mL four-neck flask was flame dried and cooled under nitrogen. The flask
was
charged with 99.25g (265 mmol) EtBLEK, 33.38g (538 mmol) ethylene glycol
(obtained
from Fisher Scientific of Waltham, MA), and 27 L (200 ppm) Ti(O-nBu)4
(obtained from
Acros Organics of Geel, Belgium). The flask was equipped with a mechanical
stirrer, Dean
Stark trap, and nitrogen/vacuum inlet. The flask was placed in an oil bath set
to 40 C and
was degassed with three repetitions of evacuating the flask to approximately 1
torn and back-
filling with nitrogen. After degassing, the flask was back-filled with
nitrogen. The
temperature of the oil bath was increased to 190 C and the flask was stirred
under nitrogen
for 22 hours, resulting in collection of 22 mL of liquid in the Dean Stark
trap. Then a 10 torr
vacuum was applied to the flask while maintaining the oil bath temperature at
190 C for 1
hour. The temperature of the oil bath was then increased to 210 C and this
temperature was
maintained for 1.5 hours, resulting in an additional 13 mL of liquid collected
in the Dean
Stark trap.
The crude product was analyzed by GPC: Mõ = 1117, PDI = 2Ø
A 250 mL four-neck roundbottom flask was flame-dried and cooled under
nitrogen.
The flask was charged with 30.63 g of the crude reaction product of EtBLEK and
ethylene
87
CA 02676892 2009-08-19
glycol, 8.34 g (72 mmol) 1,6-hexamethylene diamine (obtained from Acros
Organics of
Geel, Belgium), and 24.82 g (143 mmol) dimethyl adipate (obtained from the
Sigma-Aldrich
Company of St. Louis, MO). The flask was equipped with a mechanical stirrer,
Dean Stark
trap, and nitrogen/vacuum inlet. The flask was placed in an oil bath at a
temperature of about
40 C and was degassed with three repetitions of evacuating the flask to
approximately 1 torr
and back-filling with nitrogen. The oil bath temperature was then increased to
about 220 C
and the contents of the flask were stirred under nitrogen for 1.5 hours,
resulting in collection
of 2 mL of liquid in the Dean Stark trap. At this point a vacuum of 3 torn was
applied to the
flask for 45 minutes, resulting in collection of an additional 10 mL of
liquid. Then the
temperature of the oil bath was decreased to about 210 C and a vacuum of 0.5
torn was
applied to the flask for 22 hours, resulting in collection of 1.3 mL of liquid
in the Dean Stark
trap. The flask was back-filled with nitrogen and cooled to room temperature.
The flask was then additionally charged with 7.01g (113 mmol) ethylene glycol
(obtained from Fisher Scientific of Waltham, MA) and the contents of the flask
were stirred
in the reactor at 100 C under nitrogen over 16 hours. An additional 14 L
(200ppm) Ti(O-
nBu)4 was added and the flask, and the temperature of the oil bath was then
increased to
about 210 C. The contents of the flask were stirred under nitrogen for 17
hours, resulting in
collection of 1.3 mL of liquid in the Dean Stark trap. A vacuum of 0.5 torn
was then applied
to the flask for 16 hours, resulting in an additional 4.7 mL of liquid
collected and substantial
buildup in viscosity of the reaction mixture. The flask was then back-filled
with nitrogen and
removed from the oil bath, and allowed to cool slightly before the crude
product was
isolated. The crude product was a brown, rubbery solid and was analyzed by DSC
and GPC.
Yield: 34.56 g. DSC: Tg = 9.78 C, OH = 0.42 J/(g* C)). GPC: Mn = 13700, PDI =
4.95.
Example 24
A 250 mL four-neck round bottom flask was charged with 26.06g (69.6 mmol)
EtBLEK and 8.40g (72.3 mmol) 1,6-hexamethylene diamine (obtained from Acros
Organics
of Geel, Belgium). The flask was equipped with a mechanical stirrer, Dean
Stark apparatus,
and nitrogen/vacuum inlet. The contents of the flask were placed in an oil
bath set to a
temperature of 50 C and stirred under nitrogen purge for 16 hours. The
contents of the flask
were then degassed with three repetitions of evacuating the flask to
approximately 1 torr and
88
CA 02676892 2009-08-19
back-filling with nitrogen. After degassing, the flask was back-filled with
nitrogen. The
flask was additionally charged with 6.9 L (200 ppm) Ti(O-nBu)4 (obtained from
Acros
Organics of Geel, Belgium). The temperature of the oil bath was increased to
190 C and the
contents of the flask were stirred for 21 hours under nitrogen. The
temperature of the oil bath
was then increased to 210 C and a vacuum of about 0.5 tort was applied to the
flask for 7
hours. The flask was then back-filled with nitrogen and the contents of the
flask allowed to
cool slightly before a sample of the crude reaction product was removed for
analysis by
DSC: Tg = 55.11 C, AH = 0.37 J/(g* C)).
Dimethyl adipate (obtained from the the Sigma-Aldrich Company of St. Louis,
MO)
was dried by placing an aliquot of the compound in a flask, which was placed
in an oil bath
set to a temperature of 60 C under nitrogen sweep for 16 hours. The flask
containing the
crude reaction product of EtBLEK and 1,6-hexamethylene diamine was then
additionally
charged with 12.16g (70 mmol) of the dried dimethyl adipate. The flask was
placed in an oil
bath set to a temperature of 90 C and degassed with three repetitions of
evacuating the flask
to approximately 1 torn and back-filling with nitrogen. The temperature of the
oil bath was
then increased to about 210 C and he contents of the flask were stirred under
nitrogen for 1
hour, after which time the reactor was equipped with an addition funnel
charged with 7.23g
(62.2 mmol) 1,6-hexamethylene diamine. The contents of the addition funnel
were melted
with a heat gun and then added dropwise over about 7 hours to the reaction
mixture stirred at
210 C under nitrogen. A marked increase in solution viscosity was observed,
accompanied
by 4.0 mL of liquid collecting in the Dean Stark trap. The temperature of the
oil bath was
then increased to 220 C and a vacuum of 0.5 tort was applied to flask for 3.5
hours. The
temperature of the oil bath was then increased to 240 C under 0.5 tort vacuum
for 3.0 hours.
The flask was back-filled with nitrogen and allowed to cool slightly before
the contents of
the flask were isolated. Upon cooling to room temperature, the isolate was an
amber opaque
solid that was completely insoluble in THE and DMF. The isolate was analyzed
by DSC: Tg
= 45.08 C AH = 0.39 J/(g* C), T,,, = 170.9 C.
Example 25
A 250 mL three-neck roundbottom flask was charged with 25.35g (67.7 mmol)
EtBLEK and 7.83g (67.4 mmol) 1,6-hexamethylene diamine (obtained from Acros
Organics
89
CA 02676892 2009-08-19
of Geel, Belgium). The flask was equipped with a mechanical stirrer, Dean
Stark trap, and
nitrogen/vacuum inlet. The contents of the flask were degassed in an oil bath
set to a
temperature of 30 C by three repetitions of evacuating the flask to
approximately 1 torr and
back-filling with nitrogen. After degassing, the flask was back-filled with
nitrogen. The
temperature of the oil bath was increased to 190 C and the contents of the
flask were stirred
under nitrogen for 16 hours. The temperature of the oil bath was then
increased to 210 C for
2 hours, resulting in 3 mL of liquid collected in the Dean Stark trap and an
observed increase
in solution viscosity. A sample of the contents of the flask were removed for
analysis by
DSC: Tg = 50.76 C, DII = 0.39 J/(g* C) and GPC: Mn = 10967, PDI = 1.81.
Dimethyl adipate (obtained from the the Sigma-Aldrich Company of St. Louis,
MO)
was dried by placing an aliquot of the compound in a flask, which was placed
in an oil bath
set to a temperature of 60 C under nitrogen sweep for 16 hours. The flask
containing the
crude reaction product of EtBLEK and 1,6-hexamethylene diamine was then
additionally
charged with 11.94g (68.5 mmol) of the dried dimethyl adipate and 8.55g (127.7
mmol)
ethylene glycol (obtained from Fisher Scientific of Waltham, MA). The flask
was placed in
an oil bath set to a temperature of 60 C for 16 hours while a nitrogen sweep
was applied
across the flask. Then 16 L (300 ppm) Ti(O-nBu)4 (obtained from Acros Organics
of Geel,
Belgium) was added. The contents of the flask were degassed with three
repetitions of
evacuating the flask to approximately 1 torr and back-filling with nitrogen.
Once degassed,
the reaction mixture back-filled with nitrogen and stirred for 24 hours at in
an oil bath set to a
temperature of 190 C. Then a vacuum of 0.3 torr was applied to the flask for
1.5 hours. The
the temperature of the oil bath was increased to 210 C for 2 hours under a
vacuum of 0.4
torr. The flask was back-filled with nitrogen and allowed to cool slightly
before the contents
were isolated. The isolate was analyzed with DSC: Tg = 18.74 C, OH = 0.38
J/(g* C) and
GPC: Mõ = 14441, PDI = 2.47.
Example 26
A 250mL 3-neck roundbottom flask was charged with 38.07g (0.174mo1) EtLGK and
10.70g (0.175mol) 2-aminoethanol (obtained from TCI America of Portland, OR).
The flask
was equipped with a mechanical stirrer, a Dean-Stark trap, and a nitrogen
inlet/outlet. The
flask was degassed by three repetitions of applying vacuum of about 40 torr to
the flask,
CA 02676892 2009-08-19
followed by back-filling with nitrogen at room temperature. After degassing
was complete,
the flask was back-filled with nitrogen and 16.7 L (200 ppm) Ti(O-nBu)4
(obtained from
Acros Organics of Geel, Belgium) was added to the flask. The flask was
degassed an
additional three times and back-filled with nitrogen. Then the flask was
placed in an oil bath
at room temperature and the oil bath was then heated to 150 C, and the flask
was stirred
under nitrogen for about 20.5 hours. The flask was removed from the oil bath
and cooled to
about 100 C. A sample of the contents of the flask was removed for analysis by
1H NMR
(300MHz, DMF-d7 solvent); the carboxamide structure was confirmed. The 'H NMR
is
shown in FIG. 5.
Dimethyl terephthalate (obtained from the Sigma-Aldrich Company of St. Louis,
MO) was ground into a powder and 33.68g (173 mmol) of the powder was added to
the flask.
The flask was placed under nitrogen in the warm oil bath and the bath
temperature was set to
210 C. This temperature was maintained for about 6.5 hours with stirring, and
then a
vacuum of approximately 100 torr was applied to the flask. After 1 hour, the
pressure was
decreased to approximately 1 torr. After three more hours the flask was back-
filled with
nitrogen and removed from the oil bath.
The contents of the flask were analyzed by DSC and GPC. GPC: Mn = 8131, M,,,
19129, PDI = 2.35. DSC: Tg = 64 C.
Example 27
A 250mL 3-neck roundbottom flask was charged with 25.61g (132 mmol) dimethyl
terephthalate (obtained from the Sigma-Aldrich Company of St. Louis, MO) and
8.36g (137
mmol) 2-aminoethanol (obtained from TCI America of Portland, OR). The flask
was
equipped with a mechanical stirrer, Dean-Stark trap, and a nitrogen
inlet/outlet. The flask
was degassed by applying three vacuum/nitrogen cycles employing a vacuum of
about 15
torr followed by back-filling with nitrogen. After degassing, the flask was
back-filled with
nitrogen, then 12.7 L (200 ppm) Ti(O-nBu)4 (obtained from Acros Organics of
Geel,
Belgium) was added to the flask via microliter pipette. The flask was
degassed/backfilled an
additional 3 times, followed by back-filling with nitrogen. The flask was
placed in an oil
bath and the temperature of the oil bath was set to 150 C and left for 1 hour.
Then the flask
was cooled to 120 C, and 28.82g (132 mmol) EtLGK was added to the flask. The
oil bath
91
CA 02676892 2009-08-19
temperature was increased to 210 C and maintained for 7 hours. The pressure in
the flask
was then decreased to 20 torr over the ensuing 90 minutes and these conditions
maintained
for another 4 hours. The pressure was then further decreased to between 750
millitorr and
1000 millitorr and maintained for 8.5 hours. The flask was then backfilled
with nitrogen and
removed from the oil bath. The resulting polymer was analyzed by DSC and found
to have a
T9 of 51'C.
Example 28
A 250mL 3-neck roundbottom flask was charged with 43.27g (116 mmol) EtBLEK
and 7.06g (116 mmol) 2-aminoethanol (obtained from TCI America of Portland,
OR). The
flask was equipped with The flask was equipped with a mechanical stirrer, Dean-
Stark trap,
and a nitrogen inlet/outlet. The flask was degassed by applying three
vacuum/nitrogen cycles
employing a vacuum of about 30 torn followed by back-filling with nitrogen.
After
degassing, the flask was back-filled with nitrogen, then 10.2 L (200 ppm)
Ti(O-nBu)4
(obtained from Acros Organics of Geel, Belgium) was added to the flask via
microliter
pipette. The flask was degassed/backfilled an additional 3 times, followed by
back-filling
with nitrogen. The flask was placed in an oil bath and the temperature of the
oil bath was set
to 150 C and maintained for 14 hours. The temperature of the oil bath was then
increased to
180 C for 45 minutes, followed by 190 C for 1 hour, then 200 C for 40 minutes.
The
pressure in the flask was then decreased to about 15 torn over the next hour
and maintained
for an additional 22.5 hours. The resulting polymer was analyzed by DSC, and
the Tg was
found to be 24 C. A small sample was also dissolved in DMF and analyzed by
GPC. The
polymer was found to have Mõ = 7476, M,,, = 15986, for PDI = 2.14.
Example 29
A 250 mL four-neck round bottom flask was charged with 26.83g (71.7 mmol)
EtBLEK, 5.28g (6.0 mL, 71.2 mmol) 1,3-diaminopropane (obtained from Acros
Organics of
Geel, Belgium) and 30.3 mg (0.2 mmol, 1000 ppm) 1,3,4,6,7,8-hexahydro-2H-
pyrimido[1,2-
a]-pyrimidine (TBD, obtained from the Sigma-Aldrich Company of St. Louis, MO).
The
flask was equipped with a mechanical stirrer, Dean-Stark apparatus, and a
nitrogen/vacuum
inlet. The contents of the flask were degassed with three repetitions of
evacuating the flask
to approximately 1 torr and back-filling with nitrogen while stirring in an
oil bath having a
92
CA 02676892 2009-08-19
temperature of about 25 C. After degassing was complete, the reaction mixture
back-filled
with nitrogen and the oil bath was heated to 120 C. The flask was stirred in
the oil bath for
15 hours, at which point 1.0 mL of liquid had collected in the Dean-Stark trap
and solution
viscosity had increased noticeably. Then the temperature of the oil bath was
raised to 200 C
over the next about 6 hours and total of 4.2 mL of liquid was observed to
collect in the Dean-
Stark trap. The Dean Stark trap was emptied and 0.5 torr vacuum applied to the
flask at
200 C for 6 hours. The reaction product was a red transparent solid. The
amount of reaction
product collected was 16.15 g. The reaction product was analyzed by DSC (Tg =
76.7 C,
OH = 0.40 J/(g* C)).
Example 30
To a 250 mL three-neck flask was charged with 25.1 Og (67.0 mmol) EtBLEK and
8.03g (69.1 mmol) 1,6-hexamethylene diamine (obtained from Acros Organics of
Geel,
Belgium). THe flask was equipped with a mechanical stirrer, Dean Stark
apparatus, and
nitrogen/vacuum inlet. The flask was degassed at room temperature with three
repetitions of
evacuating the flask to approximately 1 torn and back-filling with nitrogen.
After degassing
was completed the flask was back-filled with nitrogen, and the reaction
mixture was placed
in an oil bath having a temperature set to 190 C, with stirring, and
maintained at this
temperature for 18 hours, resulting in collection of 7.0 mL of liquid in the
Dean Stark trap.
The oil bath was then heated to 200 C and 0.6 g (2.4 mmol) methylene-bis(4,4'-
diphenyl)
diisocyanate (obtained from the Sigma-Aldrich Company of St. Louis, MO) was
added
dropwise by syringe, resulting in the immediate increase in mixture viscosity
to the point
where stirring became completely ineffective. The contents of the flask were
analyzed by
DSC (Tg = 57.1 C MI = 0.38 J/(g* C)).
Example 31
A 1L, 3-neck roundbottom flask was warmed by placing it in an oil bath set to
50 C.
The flask was charged with 209.26g (1.20 mol) dimethyl adipate (obtained from
the Sigma-
Aldrich Company of St. Louis, MO) and 148.01g (1.27 mol) 1,6
hexamethylenediamine
(obtained from the Fluka Chemical Corporation of Milwaukee, WI) which was
melted prior
to its addition to the flask. The flask was equipped with a magnetic stir bar,
nitrogen inlet, a
93
CA 02676892 2009-08-19
Dean-Stark trap, and a nitrogen outlet running to a mineral oil bubbler.
Nitrogen flow was
commenced and 75 mg (210 ppm) 1,3,4,6,7,8-hex ahydro-2H-pyrimido[1,2-a]-
pyrimidine
(obtained from the Sigma-Aldrich Company of St. Louis, MO) was added to the
flask. The
oil bath in which the flask was immersed was maintained at 50 C for about 24
hours while
under a slow stream of nitrogen and with stirring. Then the flask was removed
from the oil
bath and approximately 300 mL of deionized water was added to the flask and
the contents
stirred. The water was then stripped off by placing the contents of the flask
into a 1 L single-
neck roundbottom flask and placing the flask onto a rotary evaporator at a
pressure of 10 torr.
The stripped residue was a solid. The residue was placed in a vacuum oven at a
temperature
of about 60 C and a pressure of about 10 torr for about 60 hours. After vacuum
dried, the
residue was not soluble in any common solvents and therefore could not be
analyzed by GC,
or GPC. An 'H-NMR spectrum run in CDC13/Hexafluoroisopropanol (HFIP) (95:5v/v)
confirmed the presence of a 1:1 adduct of dimethyl adipate and 1,6-
hexamethylene diamine,
but the presence of very broad peaks suggested that some polymerization may
have taken
place.
A 250mL, 3-neck roundbottom flask was charged with 21.159 (96.9 mmol) EtLGK
and 25.1 Og (97.1mmol based on theoretical structure from 1:1 condensation) of
the 1:1
adduct of dimethyl adipate and 1,6-hexamethylene diamine. The flask was
equipped with a
mechanical stirrer, nitrogen inlet, a Dean-Stark trap, and a nitrogen outlet
running to a
mineral oil bubbler. The flask was placed under a stream of nitrogen and
placed in an oil bath
set to a temperature of 160 C. The flask was stirred in the oil bath for about
2 hours, at
which point the temperature of the oil bath was changed to 180 C, and
maintained at this
temperature for about 25 minutes. Then the temperature of the oil bath was
increased to
200 C, and maintained at this temperature for about 90 minutes. Then the oil
bath
temperature was increased to 220 C and maintained at this temperature for
about 1 hour; then
the temperature of the oil bath was increased to 240 C. After about 30
minutes, 4.7 L
(100ppm) Ti(O-nBu)4 (obtained from Acros Organics of Geel, Belgium) was added
to the
flask via metered micropipette; stirring at 240 C was continued for about 8.5
hours. The heat
was then shut off and the reaction cooled to room temperature. The product was
an amber
solid which contained small, inhomogeneous pieces. The contents of the flask
were
insoluble in common solvents and were analyzed only by DSC, which showed a Tg
of 13 C.
94
CA 02676892 2009-08-19
Example 32
A 250 mL, four-neck round bottom flask was charged with 26.06 g (69.6 mmol)
EtBLEK and 8.40g (72.3 mmol) 1,6-hexamethylene diamine (obtained from Acros
Organics
of Geel, Belgium). The flask was equipped with a mechanical stirrer, Dean
Stark trap, and
nitrogen/ vacuum inlet. The contents of the flask were placed in an oil bath
set to a
temperature of 50 C under nitrogen purge for about16 hours. The contents of
the flask were
then degassed with three repetitions of evacuating the flask to approximately
1 torr and back-
filling with nitrogen. After degassing was complete, the flask was back-filled
with nitrogen.
Then 6.9 L (200ppm) Ti(O-nBu)4 (obtained from Acros Organics) was added to the
flask via
metered micropipette, and the temperature of the oil bath was increased to 190
C. The
contents of the flask were stirred under nitrogen in the oil bath for 21
hours, then the oil bath
temperature was increased to 210 C and a vacuum of 0.5 torr applied to the
flask for 7 hours.
The flask was then removed from the oil bath and back-filled with nitrogen. A
small sample
of the contents of the flask were removed and analyzed by DSC, which showed Tg
= 55.1 C,
AH = 0.37 J/(g* C).
Example 33
To the flask containing the reaction product of Example 33 was added 12.16g
(70
mmol) dimethyl adipate (obtained from the Sigma-Aldrich Company of St. Louis,
MO). The
contents of the flask were placed in an oil bath set to a temperature of 90 C
and degassed
with three repetitions of evacuating the flask to approximately 1 torr and
back-filling with
nitrogen. After the degassing was completed the flask was back-filled with
nitrogen. The
contents were stirred and the temperature of the oil bath was increased to 210
C for one
hour. Then the flask was equipped with an addition funnel charged with 7.23g
(62.2 mmol)
1,6-hexamethylene diamine (obtained from Acros Organics of Geel, Belgium). The
1,6-
hexamethylene diamine was melted with a heat gun and the contents added
dropwise over
about 7 hours to the flask. The addition of 1,6-hexamethylene diamine was
accompanied by
marked increase in solution viscosity and collection of 4.0 mL of liquid in
the Dean Stark
trap. The oil bath temperature was increased to about 220 C and a vacuum of
about 0.5 torr
was applied to the flask for about 3.5 hours, then the temperature of the oil
bath was
CA 02676892 2009-08-19
increased to about 240 C for about 3 hours. Then the flask was removed from
the oil bath
and was allowed to cool under nitrogen. An opaque amber solid was isolated.
The solid was
completely insoluble in THE and DMF. DSC analysis of the solid showed T. =
47.33 C
(AH = 0.35 J/(g* C)), T,,, = 187.95 C (AHf = 17.30 J/g), and T = 168.18 C
(OHf = 20.26
J/g).
Example 34
A 250 mL four-neck flask was charged with 25.52g (68.2 mmol) EtBLEK, 14.98g
(74.1 mmol) diethyl adipate (obtained from SAFC of Lenexa, KS), and 15.57g
(134 mmol)
1,6-hexamethylene diamine (obtained from Acros Organics of Geel, Belgium). The
flask
was equipped with a mechanical stirrer, Dean Stark trap, and nitrogen/vacuum
inlet. The
contents of the flask were degassed with three repetitions of evacuating the
flask to
approximately 1 torn and back-filling with nitrogen. After degassing, the
flask was back-
filled with nitrogen. The contents of the flask were stirred while immersed in
an oil bath
having a temperature set to 190 C for 16 hours, leading to the collection of
13.3 mL of liquid
in the Dean Stark trap. The temperature of the oil bath was raised to 220 C
and a vacuum of
about 1 torr was applied to the flask for about 1 hour, and which point a
yellow transparent
solid was isolated. DSC analysis revealed Tg = 34.88 C (AH = 0.29 J/(g* C)),
Trõ = 177.72
C (OHf = 36.74 J/g), and T = 126.15 C (AHf = 34.46 J/g).
Comparison of the DSC results from Example 34 and Example 35 reveal that
controlling the order of addition of reagents, which gives rise to a segmented
polymer
structure, results in a significant difference in the polymer's thermal
properties.
Example 35
A 250mL round bottom flask was charged with 50.1g (0.230mo1) of EtLGK and
40.Og (0.230mol) of dimethyl adipate (obtained from the Sigma-Aldrich Company
of St.
Louis, MO). The flask was equipped with a mechanical stirrer, nitrogen inlet,
and a Dean
Stark trap with a nitrogen inlet/outlet. The flask was immersed in an oil bath
set to a
temperature of 60 C and the contents of the flask were degassed with 5 cycles
of applying a
vacuum of about 0.4 torr followed by back-filling with nitrogen. After the
degassing was
completed the flask was backfilled with nitrogen and a nitrogen sweep through
the flask was
96
CA 02676892 2009-08-19
commenced with stirring, and this was maintained for about 12 hours. At the
end of the 12
hours the nitrogen sweep was stopped and the flask placed under a nitrogen
blanket (e.g.
nitrogen outlet from the flask was closed and the nitrogen flow directed to a
bubbler). At this
point, 23.4 l (200ppm) of titanium tetrabutoxide (obtained from Acros Organics
of Geel,
Belgium) was injected into the flask by a metered pipette and the flask was
stirred for about
10-20 minutes. Then 26.7g (0.230mo1) of 1,6-hexamethylenediamine (obtained
from Acros
Organics) was added all at once to the flask. The flask was then degassed
again with 3
cycles of vacuum of about 1 torr followed by back-filling with nitrogen.
After degassing was complete, the flask was back-filled with nitrogen and the
oil bath
temperature was set to 190 C. This temperature was maintained for about 2
hours and then
the temperature of the oil bath was raised to 200 C. This was maintained for
about 2 hours,
then the temperature of the oil bath was increased to 210 C. This temperature
was
maintained for about 30 minutes, then the temperature of the oil bath was
increased to 220 C.
After about 1 hour at 220 C a vacuum was applied to the flask. The vacuum was
observed to
go from about 45 torr to about 8 torr over about 15 hours. At this point a
high vacuum was
applied to the flask and the pressure in the flask was observed to be about
0.5 torr, this
vacuum was maintained for about 8 hours. At this point the flask was open
under a rapid
nitrogen flow and a small sample removed for analysis by DSC. The DSC showed
that the
product of the reaction had glass transition temperature (Tg) of about 4.3 C
and a melt
temperature of about 178.3 C. During the cooling cycle, recrystallization was
observed with
a strong peak at about 148.7 C. The DSC trace for the sample is shown in FIG.
6.
Example 36
A 250mL, 3-neck roundbottom flask was charged with 26.02g (119 mmol) EtLGK
and and 13.75g (118 mmol) 1,6-hexamethylene diamine (obtained from the Fluka
Chemical
Corporation of Milwaukee, WI). The flask was equipped with a mechanical
stirrer, nitrogen
inlet, a Dean-Stark trap, and a nitrogen outlet running to a mineral oil
bubbler. The flask was
placed under nitrogen and placed in an oil bath. The oil bath was set to 175
C, and
maintained for about 16.5 hours. A small sample of the contents of the flask
was removed
for analysis.
97
CA 02676892 2009-08-19
The oil bath was cooled to 75 C, and 20.54g (118 mmol) dimethyl adipate
(obtained
from the Sigma-Aldrich Company of St. Louis, MO) was added to the flask. After
1 hour,
the temperature was increased to 180 C over the next three hours and then
maintained for
another 3.5 hours. The oil bath was then allowed to cool to 120 C, and 12.2 L
(200ppm)
Ti(O-nBu)4 (obtained from Acros Organics of Geel, Belgium) was added to the
flask. The
temperature of the oil bath was then increased to 200 C, and maintained at
this temperature
for about 15 hours. The pressure in the flask was then reduced to about 10
torr, and
maintained for 90 minutes, at which point the pressure was further reduced to
about 5 torr,
and maintained for approximately 4 hours. The flask was backfilled with
nitrogen and
allowed to cool to room temperature. The reaction product was rubbery and
dark, and was
analyzed by GPC and DSC. The GPC showed that M, = 1761, M, = 2610, and PDI =
1.48.
The DSC result showed that the polymer had a Tg of approximately 7 C and no
crystalline
transition. The DSC plot is shown in FIG. 7. By comparing the DSC results of
Example 35
with this Example, it can be seen that affecting the degree of segmentation of
the polymer by
affecting the order of addition of reagents has a profound effect on the
ability of the resulting
polymer to form crystalline structure.
A sample of the reaction product was analyzed by 1H NMR (DMSO-d6). The NMR
was consistent with a proportion of monomer fragments corresponding to two
EtLGK
molecules condensed with one mole of diamine (e.g. the bis(ketal amide)).
Example 37
A 250 mL four-neck flask was charged with 23.3g (62.2 mmol) EtBLEK, 8.2g
(132.1
mmol) ethylene glycol (obtained from Fisher Scientific of Waltham, MA), 5.30g
(61.5
mmol) piperazine (obtained from Sigma-Aldrich Company of St. Louis, MO), and
35.7 mg
(0.26 mmol) 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]-pyrimidine (obtained from
the
Sigma-Aldrich Company). The flask was equipped with a mechanical stirrer, Dean
Stark
trap. The contents of the flask were degassed at room temperature with three
repetitions of
evacuating the flask to approximately 1 torr, then back-filling with nitrogen.
After degassing
was complete the flask was back-filled with nitrogen. The reaction mixture was
placed in an
oil bath having a temperature set to 120 C and stirred for 16 hours, leading
to collection of
1.0 mL of liquid in the Dean Stark trap. The temperature was raised to 200 C
over 7 hours,
98
CA 02676892 2009-08-19
leading to a total of 6.5 mL of liquid collected in the Dean Stark trap. The
trap was drained
and 1 torn vacuum was applied for 7 hours at 220 C, leading to collection of
an additional
6.9 mL of liquid. The flask was back-filled with nitrogen and allowed to cool,
resulting in
the collection of 15.60g of a dark red solid. DSC analysis of the solid
revealed Tg = 58.05 C
(OH = 0.41 J/(g* C)).
Example 38
A 250mL, 3-neck roundbottom flask was charged with 39.40g (181 mmol) EtLGK,
34.99g (180 mmol) dimethyl terephthalate (DMT, obtained from the Sigma-Aldrich
Company of St. Louis, MO) and 11.15g (183 mmol) 2-aminoethanol (obtained from
TCI
America of Portland, OR). The flask was equipped with a mechanical stirrer,
Dean-Stark
trap and condenser, and a nitrogen inlet/outlet. The contents of the flask
were degassed with
three repetitions of evacuating the flask to approximately 20 torn and back-
filling with
nitrogen. After degassing, the flask was back-filled with nitrogen. Then the
flask was
briefly opened and 17.3 L (about 200ppm) of Ti(O-nBu)4 (obtained from Acros
Organics of
Geel Belgium), was added to the flask, followed by degassing with three
repetitions of
evacuating the flask to approximately 20 torr and back-filling with nitrogen.
After
degassing, the flask was back-filled with nitrogen.
The flask was placed, with stirring, in an oil bath having a temperature set
to 150 C.
The flask was maintained at these conditions for approximately 2.5 hours, and
then the
temperature of the oil bath was increased to 190 C for 17 hours. The pressure
in the flask
was then reduced to approximately 35 torn, and the temperature of the oil bath
was increased
to 210 C. After 2.5 hours, the pressure was further reduced to approximately
ltorr, and these
conditions maintained for approximately 10 hours. The flask was then
backfilled with
nitrogen and removed from the oil bath and allowed to cool to room
temperature. The crude
reaction product was analyzed by GPC (DMF solvent) and DSC. The reaction
product was
not completely soluble in DMF and was filtered using a 0.45 m PTFE filter
prior to analysis
in order to remove the insoluble portions. The GPC data for the DMF-soluble
fraction
showed Mõ=6617, Mw 39226, PDI = 5.93. The DSC data showed that the reaction
product
had a T. of 62 C.
99
CA 02676892 2009-08-19
Comparison of this Example to the results obtained in Example 26 shows that a
narrower polydispersity index (PDI) is obtained, in this embodiment of the
invention, by
controlling the order of addition of the reagents and thereby the morphology
of the resulting
reaction product.
The present invention may suitably comprise, consist of, or consist
essentially of, any
of the disclosed or recited elements. The invention illustratively disclosed
herein can be
suitably practiced in the absence of any element which is not specifically
disclosed herein.
The various embodiments described above are provided by way of illustration
only and
should not be construed to limit the claims attached hereto. It will be
recognized that various
modifications and changes may be made without following the example
embodiments and
applications illustrated and described herein, and without departing from the
true spirit and
scope of the following claims.
100