Note: Descriptions are shown in the official language in which they were submitted.
CA 02689084 2009-12-18
20086-2271 F
MASS SPECTROMETRY METHOD AND APPARATUS
This application is a divisional of Canadian National Phase Patent
Application No. 2,441,589 filed March 20, 2002.
Field of Invention
This invention relates to a method and an
apparatus of mass spectrometry, and in particular to a
method and an apparatus for storage and injection of
ions into an electrostatic ion trap.
Background of the Invention
Mass spectrometers have been used to analyse a
wide range of materials, including organic substances
such as pharmaceutical compounds, environmental
compounds and biomolecules. They are particularly
useful, for example, for DNA and protein sequencing.
In such applications, there is an ever increasing
desire for high mass accuracy, as well as high
resolution of analysis of sample ions by the mass
spectrometer, notwithstanding the short time frame of
modern separation techniques such as gas
chromatography/mass spectrometry (GC/MS), liquid
chromatography/mass spectrometry (LC/MS) and so forth.
One of the new directions in the field of mass
spectrometry is the development of mass analysers
where ions are dynamically trapped in an electrostatic
field. Broadly, these may be divided into two classes:
those that employ frequency analysis by image current
detection, as disclosed in US-A-5,880,466 and
US-A-5,886,346, and those that employ time of flight
(TOF) separation and ion detection by secondary
electron conversion, as is disclosed, for example by
H. Wollnik, in J. Mass Spectrom. Ion Proc. (1994),
vol. 131, at pages 387-407, and by C. Piadyasa et al.,
in Rapid Commun. Mass Spectrom. (1999), vol. 13 at
pages 620-624. Although the trap fields may be ramped
at the beginning of the mass scan, they are typically
held very stable during the detection, or TOF
separation of ions, and so each of the foregoing mass
CA 02689084 2009-12-18
WO 02/078046 PCT/GB02/01373
2 -
analysers may be regarded as electrostatic traps
(ESTs).
Such EST mass analysers can achieve high and even
ultra-high mass resolutions (in excess of 100,000),
thus allowing more accurate determination of ion
masses. However, they all operate using an inherently
pulsed technique and as such the task of coupling to
any external continuous ion source is a serious
problem.
To improve duty cycle and sensitivity, it is
possible to use an external collision quadrupole ion
trap for ion cooling and storage between injections.
This technique has proved particularly successful when
combined with other inherently pulsed techniques such
as TOF mass analysis as is described by S. Michael
et al., in Rev. Sci., Instrum. (1992) vol. 63, pages
4277 to 4284. Here, ions are accumulated in the trap.
As suggested in US-A-5,572,022, it is possible to
control the number of ions in the trap to reduce
space-charge effects. Once ions have been stored in
the trap, they can be pulsed into the TOF mass
analyser by applying high voltages to the (normally
grounded) end caps of the trap. In US-A-5,569,917, the
ions are given a simultaneous "push" out of the trap
and a "suck" from the TOF mass analyser, so as to
improve the efficiency of ion injection into the
analyser. The spatially spread ion beam is focussed
into a tight pack in the "object" plane of the TOF
mass analyser.
Despite these improvements, quadrupole ion traps
are still currently a relatively inefficient technique
for injecting ions into a mass analyser (down to a few
percent), and they also suffer from low space charge
capacity due to the limited trap volume.
One approach that has been taken to address these
problems is to employ a different type of collisonal
storage device known as a linear trap (LT) or RF
CA 02689084 2009-12-18
WO 02/078046 PCT/G802/01373
3 -
multipole trap. US-A-5,179,278 shows such an
arrangement, wherein a two-dimensional multipole RF
field is generated. The-trap of US-A-5,179,278 is
limited by end lenses. Alternatively, the poles of the
trap may be split into sections as is shown in
US-A-5,420,425. Both split poles and end caps can be
employed together. The elevated voltages on the end
lenses or sections limit the ion movement along the
axis whilst the RF voltage provides a quasi-potential
well in the radial direction. If ions lose enough
energy during the first passes through the multipole,
then they may be trapped in it and squeezed towards
the axis during further collisions. The number of ions
in the trap can be controlled using a short pre-scan,
a technique disclosed in the above-referenced
US-A-5,572,022. Nevertheless, to inject ions from the
LT into the next stage of analysis, the voltage is
lowered on the exit lens and the ions in the LT are
allowed to flow out of the multipole. This flow
typically lasts up to hundreds or even thousands of
microseconds. These time scales are compatible with
the injection times for quadrupole ion traps (as
disclosed in the above-referenced US-A-5,179,278) or
for Fourier Transform Ion Cyclotron Resonance (FTICR)
as set out by M. Senko et al in JASMS, (1997), volume
8, pages 970-976. The time scales are also suitable
for orthogonal acceleration TOF mass spectrometry, see
for example US-A-6,011,259, US-A-6,020,586, and
W099/30350.
Segmented construction of the poles in the LT may
be employed, as set out by M. Belov et al, in
Analytical Chemistry (2001) volume 73, pages 253-261,
to reduce the injection time down to about
300-400 microseconds. The segmented construction of
the LT provides an axial field which causes ions to be
displaced towards the exit lens.
Even so, such injection times are too long for an
CA 02689084 2009-12-18
20086-2271 F
4 -
electrostatic trap. This is because ESTs require high
ion energies (typically 1-2 keV per charge) to achieve
dynamic trapping. If injection takes place over
hundreds of microseconds, at such energies the process
may last for hundreds of ion reflections. Without any
collisional cooling inside the electrostatic trap, ion
stability may be compromised.
Summary of the Invention
According to one aspect of the present invention, there is provided a
mass spectrometer comprising: an ion source for supplying sample ions for
analysis; an ion trap having an entrance through which the sample ions are
received, and an exit through which the sample ions are released; a voltage
source being configured to apply a first set of trapping voltages to the ion
trap to
confine the sample ions within the ion trap, the first set of trapping.
voltages being
selected to cause a center of an ion cloud defined by the sample ions
contained
within the ion trap to be positioned closer to the exit than to the entrance;
the
voltage source being further configured to apply a release voltage to the ion
trap
to release the sample ions from the ion trap through the exit.
According to another aspect of the present invention, there is
provided an ion trap for trapping sample ions in a mass spectrometer,
comprising:
an entrance through which the sample ions are received, and an exit through
which the sample ions are released; and a voltage source being configured to
apply a first set of trapping voltages to the ion trap to confine the sample
ions
within the ion trap, the first set of trapping voltages being selected to
cause a
center of an ion cloud defined by the sample ions contained within the ion
trap to
be positioned closer to the exit than to the entrance; the voltage source
being
further configured to apply a release voltage to the ion trap to release the
sample
ions from the ion trap through the exit.
CA 02689084 2009-12-18
20086-2271 F
-4a-
According to another aspect of the present invention, there is
provided a method for injecting sample ions into an electrostatic trap,
comprising
the steps of: receiving the sample ions through an entrance of an ion trap;
applying a first set of trapping voltages to the ion trap to confine the
sample ions
within the ion trap, the first set of trapping voltages being selected to
cause a
center of an ion cloud defined by the sample ions contained within the ion
trap to
be positioned closer to an exit of the ion trap than to the entrance; applying
a
release voltage to the ion trap to release the sample ions from the ion trap
through
the exit; and receiving the released sample ions through an entrance of the
electrostatic trap.
It is an object of some embodiments disclosed herein to provide a
method and an apparatus
which provides for adequate storage of ions prior to
injection of these ions into an electrostatic trap
over a timescale compatible with such a device.
According to an embodiment disclosed herein,a method of injection of
sample ions into an electrostatic trap, :comprises the
steps of: (a) generating a plurality of sample ions
to be analysed, each of which has a mass-to-charge
ratio m/z; (b) receiving the sample ions through a
storage device entrance in an ion storage device
having a plurality of storage device poles;
(c) supplying a trapping voltage to the storage device
so as to trap at least a proportion of the received
sample ions within a volume p in the storage device,
during at least a part of a. trapping period, the thus
trapped ions each having a kinetic energy Ek such that
there is an average kinetic energy Ek of the ions in
the volume p during the trapping period or the said
part thereof; (d) supplying a release voltage to the
storage device so as to controllably release at least
some of the said sample ions contained within the said
volume of the storage device from a storage device
exit, the release voltage being of a magnitude such
that the potential difference then experienced by the
CA 02689084 2009-12-18
20086-2271 F
-
ions across the volume p is greater than the said
average kinetic energy Ek during the trapping period
or said part thereof, and further wherein the release
voltage is such that the strength of the electric
5 field generated thereby at any first point across the
volume p, upon application of the said release
voltage, is no more than 50% greater.or smaller than
the strength of the electric field generated thereby
at any other second point across the volume p;
(e) receiving those sample ions released from the
storage device exit according to the criteria of step
(d) through an entrance of an electrostatic trap
having a plurality of trapping electrodes, the ions
arriving as a convolution of bunched time of flight
distributions for each m/z, each distribution having a
full width at half maximum (FWHM); and (f) trapping
the received sample ions within the electrostatic trap
by applying a potential to the electrodes such that
the sample ions describe movement having periodic
oscillations in at least one direction.
The method proposes
particular restrictions on the release potential
supplied to the storage device which ensures that
groups of ions of a given m/z arrive as a tightly
focussed `bunch' at or adjacent the electrostatic
trap. The two conditions are, respectively, that the
electric field experienced by the ions upon leaving
the storage device is relatively uniform and that the
potential drop experienced by the ions upon release is
larger than the average thermal (kinetic) energy of
the ions when trapped in the storage device, and
preferably much larger.
The first condition is a consequence of the
effective focussing of ions in time of flight from the
storage device and into the electrostatic trap. The
focal length, L, is given by L = aV/E, where V is the
final energy of the ions when released from the
NO02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 6 -
storage device, E is the electric field strength, and
a is a constant. If the electric field is non-
uniform, therefore, along the volume p, then ions of
the same m/z will be focussed at different lengths L
and will not arrive in the same tightly focussed
bunch. A variation in the electric field strength of
no more than about 30%-50% is preferred.
The second criterion ensures that the thermal
energy (i.e., the kinetic energy) of the trapped ions
is less than and preferably insignificant relative to
the gradient of the potential `slope' upon which the
ions find themselves when the release potential is
applied.
In an idealized storage device or ion trap, the
ions are all in the same place and leave with exactly
the same energy when ejected. This means that they
arrive at the same time at any chosen location
downstream of the ion trap. In reality, of course, the
ions have a range of kinetic energies and start off
from different locations within the trap. Hence the
ions of the same m/z arrive at different times
downstream of the ion trap. The purpose of TOF
focussing is therefore to cause ions further back in
the trap to `catch up' with ions ejected from the
front of the trap, by ensuring that the ions nearer
the front of the trap move more slowly than those
leaving later. The release potential is chosen so that
the ions are ejected without being affected
significantly by the random perturbations of thermal
energy spread. Although it is necessary that the
potential drop is at least as much as the average
kinetic energy, a multiple of two is preferred, of
five is more preferable, and at least one order of
magnitude is most preferable.
This second condition provides the further
advantage that the ions will be relatively energetic
upon arrival at or adjacent the electrostatic trap.
CA 02689084 2009-12-18
20086-2271 F
7 -
Electrostatic traps have an energy acceptance window,
which is centred at a relatively high energy, so that
the ions ejected in accordance with the conditions of
the present embodiment are within that energy
acceptance window.
Provided that the foregoing conditions are met,
the ions will arrive at the electrostatic trap as a
convolution of short, energetic packets of similar
m/z. Such packets are ideally suited to an
electrostatic trap (and particularly the preferred
embodiment of an orbitrap) because the FWHM of each
ion packet's TOF distribution for a given m/z ratio is
then less than the period of oscillation of those
sample ions having that m/z when in the electrostatic
trap. In other words, the packets are sufficiently
coherent for detection to take place.
In some 'embodiments, the ions are pre-
cooled, for example in the storage device. This
reduces the thermal energy of the ions and also their
energy spread, hence reducing the ratio of kinetic
energy to release voltage which is the second
prerequisite set out above. Pre-cooling may be
achieved by collisional cooling, for example.
Furthermore, the trapping voltage may be applied so as
2,5 to force the ions in the storage device towards the
exit thereof. This may either be carried out
throughout the trapping period or may, in preference,
be carried out only immediately prior to ejection.
To obtain.bunching of the sample ions at the
electrostatic trap, it is preferable to employ an
axially segmented linear trap (LT) as the storage
device. A differential, preferably DC, voltage is
applied between the two or more segments of the LT so
as to force the ion cloud within the LT towards the
exit thereof. This procedure may be carried out after
trapping and cooling of the sample ions received from
an ion source in the storage device, which may be
CA 02689084 2009-12-18
20086-2271 F
- 8 -
achieved by applying voltages to the pole segments so
as to create an axial potential well whose base is in
the middle of the LT. Alternatively, the bottom of the
potential well may be located from the outset at or
.5 . towards the exit from the storage device. It is
particularly preferable that the bottom of the
potential well is no more than twice the diameter
inscribed by the poles of the storage device away from
the storage device exit.
The ions in the storage device are preferably
gated out of the exit thereof by applying one or more
voltage pulses, to an end electrode of the storage
device for example.
The condition for correct focussing or bunching
of the sample ions at or adjacent the electrostatic
trap is met, in some embodiments, by the
requirement that the period of oscillation of those
ions when injected into the electrostatic trap is
shorter, and preferably much shorter, than the time of
20= flight of those sample ions between the-storage device
exit and arrival at the electrostatic trap entrance.
In alternative embodiments, the further condition
that the FWHM time of flight distribution of the ions
arriving at the electrostatic trap should be less than
the TOF along each detection electrode in the
electrostatic trap is*imposed as well.
In a particular embodiment, the.
storage device is arranged to receive ions along a
first direction and to release them from the storage
device along a second, orthogonal direction. This
permits much higher space charge capacity and better
ion beam parameters.
In that case, it is preferable that the storage
device should be curved along the first direction.
This improves geometric focussing. Optionally, lenses
may be added to convert a wide angle beam-into a
narrow beam.
CA 02689084 2009-12-18
20086-2271 F
9 -
The method may also include boosting the ion energies
prior to their arrival at the electrostatic trap
entrance.
In some embodiments, the method is employed
for the generation, storage and detection of sample
ions in MS-only mode. Alternatively, the method may be
employed for collision-induced dissociation of the
ions to produce daughter sample ion fragments. In
either case, it is preferable to select the release
voltage so as to focus the ions on the entrance to the
orbitrap. In an alternative embodiment, however, the
method may be employed to detect fragment ions using
surface-induced dissociation. In this case, the method
preferably further comprises focussing the sample ions
through the electrostatic trap and onto a collision
surface. The fragment ions which result are then
accelerated back towards the electrostatic trap.
It is particularly preferable that the ions
arrive at the electrostatic trap at an angle
tangential to a central plane of the electrostatic
trap. This is preferably achieved by lenses between
the ion trap-and the electrostatic trap. The benefit
of this is that no further excitation of the ions is
necessary once they enter the electrostatic trap. This
in turn reduces the amount of electronics necessary
for correct operation of the electrostatic trap, and
is to be compared with the arrangement of
US-A-5,886,346 referenced above.
The lenses between the ion trap and electrostatic
trap, where present, preferably offer no direct line
of sight between the inside of the ion trap and the
inside of the electrostatic trap. This arrangement
prevents streaming of ions and gas carryover from the
(relatively high pressure) ion trap into the
(relatively lower pressure) electrostatic trap.
Detection of the.sample ions in the electrostatic
trap may be achieved in a number of ways. Most
CA 02689084 2009-12-18
20086-2271 F
-
preferably, the electrostatic trap is of the orbitrap
type and ions are trapped in a hyper-logarithmic
field. As bunches of coherent ions of different m/z
pass by the outer electrodes of the orbitrap, an image
5 current is induced therein. This current may be
amplified and then processed to generate a TOE
spectrum, for example by Fourier transform analysis.
The field within the electrostatic trap may
preferably be compensated by applying a compensating
10 voltage (which may be time dependent) to a field
compensator during detection of the ions. This
procedure ensures minimum field perturbation within
the volume occupied by the ion trajectories.
Additionally or alternatively, during ion injection
into the electrostatic trap, the field compensator may
be arranged to act as a deflector to improve the
trapping efficiency of the electrostatic trip.
As with-previous ion.traps, and LTs in
particular, the ion trap may contain facilities for
- resonance or mass-selective instability scans to
provide for data-dependent excitation, fragmentation
or elimination of certain m/z ratios.
The optimum duration of ion trapping in the ion
trap may be determined prior to commencement of mass
analysis by carrying out a pre-scan. Preferably, a
-.secondary electron multiplier (SEM) or the like is
employed. The SEM may be located radially of the ion
trap and in that case mass-selective instability or a
resonance.excitation scan in the ion trap may be used.
Most preferably, however,., an axial SEM is employed
downstream of the electrostatic trap and on an ion
abeam axis. In this case, ions are preferably injected
into the electrostatic trap just as they would be for
subsequent mass analysis.
According to another embodiment disclosed herein, a mass spectrometer
comprises: (a) an ion source arranged to supply a
WO 02/078046 CA 02689084 2009-12-18 PCTIGB02/01373
- 11 -
plurality of sample ions to be analysed, each of which
has a mass-to-charge ratio m/z; (b) an ion storage
device comprising a plurality of storage device poles
and having a storage device entrance end through which
the said sample ions are received and a storage device
exit end through which the said sample ions may exit;
(c) a voltage source arranged to supply a trapping
voltage to the storage device poles so as to contain
at least a proportion of the sample ions received
through the storage device entrance end of the storage
device within a volume p of the storage device in a
trapping mode during at least a part of a trapping
period, the thus trapped ions each having a kinetic
energy Ek such that there is an average kinetic energy
Ek of the ions in the volume p during the trapping
period or the said part thereof, and to supply a
release voltage to the storage device in an ion
ejection mode. so as to controllably release at least
some of the said sample ions contained within the said
volume p of the storage device through the storage
device exit end, the-release voltage being of a
magnitude such that the potential difference then
experienced by the ions across the volume p is greater
than the said average kinetic energy Ek during the
trapping period or said part thereof, and further
wherein the release voltage is such that the electric
field generated thereby at any first point across the
volume p, upon application of the said release
voltage, is no more than 50% greater or smaller than
the electric field generated thereby at any other
second point across the volume p; and (d) an
electrostatic trap having an electrostatic trap
entrance arranged to. receive those ions released
through the storage device exit end and meeting the
criteria imposed by the applied trapping and release
potentials, as a convolution of bunched time of flight
distributions for each m/z, each distribution having a
CA 02689084 2009-12-18
20086-2271 F
12 -
full width at half maximum (FWHM); the electrostatic
trap further comprising a plurality of electrodes
arranged to trap ions received through the
electrostatic trap entrance therebetween so that the
said trapped ions describe movement having periodic
oscillations in at least one direction.
As with the embodiment of the method described above, a
segmented multipolar ion trap is preferable. In that
case, to maximise focussing or bunching of ion packets
at the entrance to the electrostatic trap, the length
of the segment of the pole pieces proximal the ion
trap is preferably shorter than twice the inscribed
diameter between the segments of the ion trap
(measured radially), and most preferably shorter than
the inscribed diameter. Also, the distance between
the ion trap exit and the centre of the segment
closest thereto is preferably greater than or equal to
the said inscribed diameter.
Other features and advantages of the invention
will become apparent with reference to the appended
claims and to the following specific description.
Brief description of the Drawings
The present invention may be put into practice in
a number of ways, and some specific embodiments will
now be described by way of example only and with
reference to the accompanying drawings in which:
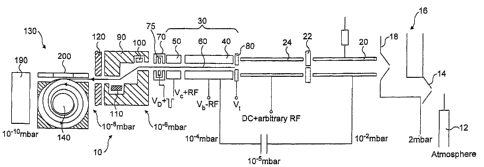
Figure .1 is a schematic side view of a mass
spectrometer- embodying the present invention and -
including an ion trap and electrostatic trap;
Figure 2 shows schematically and in perspective a
part of the ion trap of Figure. 1;
Figure 3 shows a front view of the electrostatic
trap of Figure 1;
35, Figure 4a shows, schematically, the potential
distribution in the ion trap of Figures-1, 2 and 3
when ions are trapped therein;
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 13 -
Figure 4b shows, schematically, the potential
distribution in the ion trap of Figures 1, 2 and 3 at
the point when trapped ions are ejected from the ion
trap;
Figure 5 shows a front view of an alternative
electrostatic trap for use in the mass spectrometer of
Figure 1;
Figure 6 shows an alternative configuration of
the mass spectrometer of Figure 1, again in schematic
side view and including a curved ion trap along with
an electrostatic trap;
Figure 7 shows a sectional view through the
curved ion trap of Figure 6; and
Figure 8 shows a side view of an electrostatic
trap for use with the mass spectrometer of Figure 1 OR
Figure 6, in combination with a collision surface
downstream of the electrostatic trap to allow
operation of-the mass spectrometer in surface-induced
dissociation mode.
Referring first to Figure 1, a mass spectrometer
10 is shown. The mass spectrometer comprises a
continuous or pulsed ion source 12, such as an
electron impact source, an electro-spray source (with
or without a collisional RF multipole), a matrix-
assisted laser desorption and ionisation (MALDI)
source, again with or without a collisional RF
multipole, and so forth. In Figure 1, an electrospray
ion source 12 is shown.
The nebulized ions from the ion source 12 enter
an ion source block 16 having an entrance.cone 14 and
an exit cone 18. As is described, for example, in
WO 98/49710, the exit cone 18 has an entrance at 90
to the ion flow in the block 16 so that it acts as a
skimmer to prevent streaming of ions into the
subsequent mass analysis components.
A first component downstream of the exit cone 18
is an ion cooler 20 which reduces the energy of the
WO02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 14 -
sample ions from the ion source 12. Cooled ions exit
the ion cooler 20 through an aperture 22 and arrive at
a quadrupole mass filter 24 which is supplied with a
D.C. voltage upon which is superimposed an arbitrary
r.f. signal. This mass filter extracts only those
ions within a window of m/z of interest and the chosen
ions are then released to a linear ion trap 30. The
ion trap 30 is segmented, in the embodiment of Figure
1, into an entrance segment 40 and an exit segment 50.
Although only two segments are shown in Figure 1, it
will be understood that three or more segments could
instead be employed. As better seen in Figure 2, the
segments 40, 50 are each formed from four rods which
are radially spaced so as to form a trapping volume 60
between them.
To trap ions within the trapping volume 60, a
voltage source (not shown) applies an RF voltage to
each of the segments 40, 50. The application of an RF
field generates a potential well in the axial
direction. Collisions between ions entering the linear
trap 30 rapidly cause these ions to sink towards the
bottom of the potential well.
The ends of the linear trap 30 are bounded by
exit and entrance electrodes 70, 80 respectively.
These electrodes are supplied with a DC voltage VD and
Va respectively. As will be familiar to those skilled
in the art, the linear trap 30 may also contain
facilities for resonance or mass-selective instability
scans, to provide data-dependent excitation,
fragmentation or elimination of selected mass-to-
charge ratios.
In preference, the length of the exit segments 50
is not in excess of the inscribed diameter D between
the rods (Figure 2). Also, the distance x between the
exit electrode 70 and the axial centre of the exit
segment 50 is preferably comparable to, or greater
than, the inscribed diameter D (again, see Figure 2).
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 15 -
The linear trap 30 may have a pressure gradient
therein. In this way, the conditions in one part of
the trap 30 are optimised for the best dissipation of
energy through ion collisions, whilst near the exit
electrode 70, the conditions may be optimised for the
best trapping, lowest fragmentation and so forth. The
pressure gradient may, for example, be created through
the introduction of additional gas inlets.
Downstream of the exit electrode is a deflection
lens arrangement 90 including deflectors 100, 110. The
deflection lens arrangement is arranged to deflect the
ions exiting the linear trap 30 in such a way that
there is no direct line of sight connecting the
interior of the linear trap 30 with the interior of an
electrostatic orbitrap 130 downstream of the
deflection lens arrangement 90. This prevents
streaming of energetic ions from the relatively high
pressure linear trap 30= into the relatively low
pressure orbitrap 130. The deflection lens arrangement
90 also acts as a differential pumping aperture.
Downstream.of the deflection lens arrangement 90
is a conductivity restrictor 120. This sustains a
pressure differential between the orbitrap 130 and the
lens arrangement 90.
Ions exiting the deflection lens arrangement 90
through the conductivity restrictor 120 arrive at the
orbitrap 130. The orbitrap 130 has a central electrode
140 as may better be seen with reference now to Figure
3. The central electrode 140 is connected to a high
voltage amplifier 150.
The orbitrap 130 also contains an outer electrode
split into two outer electrode parts 160, 170. Each of
the two outer electrode parts 160, 170 is connected to
a differential amplifier 180. Preferably, this
differential amplifier is maintained at virtual
ground.
Referring once more to Figure 1, downstream of
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 16 -
the orbitrap 130 is a secondary electron multiplier
190, on the optical axis of the ion beam. Although not
shown in Figure 1, the secondary electron multiplier
(SEM) 190 may also be located on the side of the
linear trap 130.
The system, and particularly the voltages applied
to the various parts of the system, is controlled by a
data acquisition system. This data acquisition system
is in itself known and does not form a part of the
present invention. Accordingly, it is not shown in the
Figures and will not be described further. The data
acquisition system may also carry out signal
processing as described below. Likewise, a vacuum
envelope is also provided, to allow differential
pumping of the system. Again, this is not shown in the
Figures, although the typical pressures are indicated
in Figure 1.
In operation, ions from the ion source 12 enter
the segmented linear trap 30 and are reflected by an
elevated potential VD on the exit electrode 70
thereof. AC voltages at RF frequencies are applied to
the segments of the trap to provide=a quasi-potential
well in the radial direction whilst DC voltages Va, Vb
and V,: provide a potential well along the axis of the
linear trap 30. The pressure inside the linear trap 30
is chosen in such a way that ions lose sufficient
kinetic energy during their first pass through the
trap that they accumulate near the bottom of the axial
potential well. Before ions are removed from the
linear trap 30, the DC voltages Va, Vb, Vd and VD may be
varied in such a way that the centre of ion cloud
within the linear trap 30 is shifted into the end
section of the linear trap, that is, into the volume
defined between the rods in the exit segment 50
adjacent to the exit electrode 70. As an alternative,
the bottom of the axial potential well may instead be
located in this exit segment 50 from.the start of ion
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 17 -
storage in the linear trap 30.
Figure 4a shows, not to scale, the potential
electrode 80 and the exit electrode 70 when ions are
trapped therein. It will be seen that the ions sit in
a potential well defined by the difference in
potentials between the exit electrode 70, the exit
segment 50 of the trap 30 and the entrance segment 40
thereof.
At the end of storage, the data acquisition
system starts to ramp the voltage applied to the
central electrode 140 in the orbitrap 130 and,
simultaneously, applies a voltage pulse to the exit
electrode 70 of the linear trap 30. In presently
preferred embodiments, a single pulse is applied to
empty the linear trap. However, multiple pulses may be
employed instead. In any event, the delay between
successive pulses is chosen in such a way that all of
the mass.range of interest arrives into the orbitrap
130 during the correct phase of the voltage applied to
the central electrode 140 thereof as it is ramped.
Although the ramping of the voltages on the central
electrode of the orbitrap 130 and the exit electrode
70 of the linear trap are timed to each other, they do
not however need to be synchronous. Thus, the voltage
applied to the central electrode 140 of the orbitrap
130 may start to ramp before the pulse is applied to
the exit electrode 70 on the linear trap, and may
continue to ramp for a period (e.g. tens of
microseconds) after the linear trap has been emptied.
Figure 4b shows the potential at the different
points along the ion trap 30 between the entrance 80
and the exit electrode 70 again not to scale, when
such a pulse is applied to the exit electrode 70.
Because the pulse is negative (in the convention
adapted to illustrate the specific embodiment
described, the ions previously trapped in the
potential well instantaneously find themselves on a
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 18 -
"slope" which accelerates them away from the ion trap
30. As will be explained below, the ejection
technique causes the ions leaving the ion trap 30 to
be time-of-flight focussed onto the entrance of the
electrostatic trap 130.
It is important, for correct trapping of ions in
the orbitrap 130, that they arrive at the entrance to
the orbitrap when the voltage on the central electrode
140 thereof is between approximately (D1/D2)' V, and V,
where V is the final, static voltage on the central
electrode 140, D1 is the outer diameter of the central
electrode 140, and D2 is the inner diameter of the
outer electrode formed from the outer electrode parts
160 and 170. The ion energy at the entrance to the
orbitrap 130 also needs to lie within a certain range.
Whilst the voltage applied to the central
electrode.140 of the orbitrap 130 is ramped, ions are
directed.and focussed by the linear trap 30 and the
deflection lens arrangement 90 to the entrance of the
orbitrap 130. The ions enter the field within the
orbitrap 130 tangentially to the outer electrodes
formed from the outer electrode parts 160, 170 and are
prevented from hitting this electrode again by a
monotonically-increasing electric field, which
squeezes the ions closer to the centre of the trap.
Tangential injection into the orbitrap 130 is achieved
by displacing the trap relative to the centre of the
beam of ions arriving from the ion trap 30. By way of
example only, the orbitrap 130 may be positioned so
that ions enter it at a radius of 17.4 mm with
z = 10 mm, the highest internal radius of the
electrodes being 20 mm in this specific example.
The rise time of the electric field depends on
the mass range to be trapped, ion parameters, and the
orbitrap 130, but is usually between 20 and
200 microseconds. Squeezing stops when there is no
more threat of losing ions onto the electrodes.
CA 02689084 2009-12-18
20086-2271 F
19 -
The orbitrap 130 is shaped so as to generate a
hyper-logarithmic field between the central electrode
140 and the outer electrode formed from the outer
electrode parts 160, 170. The potential distribution
of this hyper-logarithmic field may be described in
cylindrical coordinates (r, z) by the following
equation:
r2 k 2 r
V(r,z) = k 2 z 2 2 + 2(,) +C
where z = 0 is the plane of symmetry of the field, C,
k, R,(>0) are constants, and k >0 for positive ions.
Such a field creates a potential well along the z axis
direction which causes ion trapping in that potential
well provided that the incident energy is not too
great for the ion to escape. As the voltage applied to
the centre of electrode-140 increases, the field
intensity increases and therefore the force- on the
ions towards the longitudinal axis increases, thus-
decreasing the radius of spiral of the ions as may be
seen from Figures 1 and 2. Thus, the ions are forced
to rotate in spirals of smaller. radius as the sides of
the potential well increase in gradient.
There are three characteristic frequencies of
oscillation within the hyper-logarithmic field. The
first is the harmonic motion of the ions in the axial
direction where they oscillate in the potential well
with a frequency independent of energy in this
direction. The second characteristic frequency is
oscillation in the radial direction since not all of
the trajectories will be perfectly circular. The third
frequency characteristic of the trapped ions is the
frequency of angular rotation.
Further details of the preferred electrode
arrangement of the orbitrap 130 may be found in
US-A-5,886,346, referenced above.
CA 02689084 2009-12-18
20086-2271 F
- 20 -
It will, however, be-noted that, in the present case,
the ions enter the field tangentially and do not
,require a separate injection of radial force which in
turn reduces the amount of electronic control of the
orbitrap 130 that is necessary.
The ion packets arriving at the entrance to the
orbitrap 130 are bunched together due to the time of
flight focussing created by the ion ejection technique
from the linear trap 130. The ion packets are
sufficiently coherent that coherent axial oscillation
within the orbitrap 130 takes place without addition
excitation. The required degree of coherency depends
on the type of detection.
If all ions of the same mass--to-charge ratio were
to have the same initial kinetic energy, begin flight
at the same time, and from the same position. within
the trap, then. they would-a-11 leave the trap. together
and travel together to arrive at exactly the same
moment at any point downstream of the trap. This
idealized situation cannot of course be realized in
practice, primarily due to three factors which `smear'
the initial peak from a delta function. Firstly, the
starting position of different ions of the same mass-
to-charge ratio will be different, secondly, the time
at which flight begins, and thirdly the initial
kinetic energies of the different ions of the same
mass-to-charge ratio will be different.
The present invention addresses the non-ideal
30- nature of the ions in the trap by firstly minimizing
the spread of initial kinetic energies and by
`bunching' the ions together at one end of the trap
(so that they tend to leave from roughly the same
point in the trap) and secondly by focussing the ions
during flight so that any temporal, spatial or
energetic spread which still remains is reduced.
Figure 4b shows why this should be so.
WO02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 21 -
When the exit electrode 70 receives the negative
going pulse to eject the ions, those ions of the same
m/z, which at the moment of pulsing are closest to the
exit electrode 70, experience a smaller voltage drop
than those further away from the exit electrode 70. As
the ions of similar m/z which are closest to the exit
aperture have a smaller distance to travel to that
exit aperture, they pass through it earlier than the
ions `behind' them but at a lower velocity. In other
words, ions further away from the exit aperture 70 on
application of the negative pulse take longer to be
emptied from the tap but leave it at a higher
velocity. In this manner, the ions bunch up downstream
of the ion trap 30. By carefully selecting the
ejection parameters, the ions in the ion trap 30 may
be focussed onto the entrance of the orbitrap 130.
In the preferred embodiment shown in Figures 1
and.3., a. mass: spectrum is generated using image
current detection, which technique is again described
in US-A-5,886,346. An interference pattern of
frequencies of different mass-to-charge ratios
produces an image current on the outer electrode parts
160, 170. This current is amplified by the
differential amplifier 180 and then processed by the
data acquisition system by application of a Fourier
transform. For this type of ion detection, coherency
of the ion packets is achieved as soon as the duration
dT (m/z) of a given ion packet of a specific m/z is
smaller than the period of axial oscillations within
the orbitrap 130. This period of axial oscillation may
in turn be significantly less than the time of flight
between the linear trap 30 and the entrance to the
orbitrap 130. To achieve this level of coherence, time
of flight focussing or bunching of the ions as they
enter the orbitrap 130 is necessary. It is important
to distinguish this bunching from first or second
order focussing typical in time of flight (TOF) mass
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 22 -
analysers. The time of flight between the linear trap
30 and the orbitrap 130 is normally significantly
greater than the period of axial oscillations within
the orbit trap 130 because of the differential pumping
between the linear trap 30 and the orbitrap 130 which
necessarily requires a significant spatial separation
between the linear trap and the orbitrap 130.
The electric field strength and the ion trap
dimensions are chosen in such a way that the ions
catch up, that is, focus in time of flight, just at
the entrance to the EST 130. For example, for a
uniform field, the focal point is located 3.d away
from the starting point where d is the length of this
uniform field as measured from the starting point to
the field boundary. Non-uniform fields have focussing
properties but the mathematical relations are very
complex. The random energy distributions of the ions
trapped-in-the ion-trap 30 adversely affect the
quality of the TOF focussing; for example, the pulse
width becomes comparable with the total time of flight
as soon as the energy spread of the ions becomes
comparable with the acceleration voltage. In order to
address these problems, therefor, the bunching of the
ions is achieved, in practice, through a combination
of the following requirements.
Firstly, the relative variation of the electric
field strength along the ion cloud within the ion trap
30, or at least the portion to be injected into the
orbitrap 130, should be smaller than unity, and
preferably much smaller than unity (such as 10% or
even less). Such uniformity of the field along the
beam may be achieved by ion squeezing prior to pulsing
of the linear trap 30 which in turn is achieved by
moving the base of the potential well in the linear
trap 30 towards the exit electrode 70 as described
above. Collisional cooling of ions within the linear
trap also assists in this squeezing. Secondly, the
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
23 -
voltage drop across the ion cloud in the linear trap
should at least exceed the average kinetic energy
during storage, and preferably exceed it by an order
of magnitude or more. Such considerable field strength
reduces the time of flight spread caused by the
initial energies.
The optimum duration of trapping of ions within
the linear trap 30 may be determined using a pre-scan
after a short storage duration. The secondary electron
multiplier 190 allows detection. Where the SEM 190 is
located radially of the linear trap 30, mass-selective
instability or a resonance excitation scan in the
linear trap 30 may be used. It is preferable, however,
to use an axial SEM 190, after the orbitrap 130, so
that ions are injected in the same way as for the
analysis in the orbitrap 130. It will be understood
by those skilled in the art that any other known way
of ion detection could be used instead of SEM 190,,
such as a collector-with a charge-sensitive amplifier,
a photo-multiplier, semiconductor detectors and so
forth.
During detection of ions in the orbitrap 130, an
appropriate voltage, which may be time-dependent, may
be supplied to a field compensator 200 adjacent the
entrance to the orbitrap 130. This field compensator
ensures minimum field perturbation within the volume
occupied by the ion trajectories. During ion
injection, this field compensator 200 acts also as a
deflector to improve trapping efficiency within the
orbitrap 130.
Although a preferred embodiment has been
described, it will be understood that various
modifications are contemplated. For example, although
a linear ion trap 30 has been described for storage of
the sample ions from the ion source 12, it is to be
understood that a quadrupole ion trap could equally be
employed for ion storage, cooling and so forth.
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 24 -
Quadrupole ion traps are known as such and one example
is shown in EP-A-0,113,207.
Furthermore, a second form of orbitrap 130, seen
in front view in Figure 5, may be employed instead of
the one shown in Figure 3. Here, instead of split
outer electrode parts, the outer electrode is not
split. Instead; the central electrode 140' is axially
segmented with a centre part 220 connected to a pre-
amplifier 210. In this arrangement, pulses of image
current from passing ion packets of different m/z are
detected by this central, disc-shaped electrode part
220, amplified by the pre-amplifier 210, and then
processed to yield a time of flight spectrum. A
deconvolution method such as is described by M. May et
al, in Analytical Chemistry (1992) vol. 64, pages
1601-1605 could be used, or any other signal
reconstruction method. Two or more detection
electrodes could also be used. The pre-amplifier could
also be a differential pre-amplifier with a second
input connected to another detection electrode or
simply open-ended to improve common mode rejection.
For this type of detection, the best results are
achieved when the duration dT (m/z) of each ion packet
of a particular m/z is not only smaller than the
period of axial oscillations in the orbitrap 130, but
also does not exceed the time of flight along each of
the detection electrodes.
Furthermore, a mass selective instability scan,
such as is described in the above-referenced
US-A-5,886,346 could also be used. In this case, ion
injection could be performed also along the central
plane of the trap.
Figure 6 shows another mass spectrometer 10'
which embodies the present invention and currently
represents the preferred implementation. Features
common to Figures 1 and 6 are labelled with like
reference numerals.
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 25 -
As with the arrangement of Figure 1, the mass
spectrometer 10' comprises an electrospray ion source
12 which provides nebulized ions into an ion source
block 16 through an entrance cone 14. The ions exit
the block 16 via an exit cone 18 and pass into an ion
cooler 20 at around 10-1 mbar. The ions then arrive in
a quadrupole mass filter 24 via an aperture 22, and a
range of m/z of the incident ions is selected as
described previously.
Ions exiting the mass filter 24 enter an ion trap
300 in a first direction generally parallel with a `y'
axis (see Figure 6). The ions are however ejected from
the ion trap 300 in a second direction generally
orthogonal to their entrance direction, that is, in an
`x' direction as indicated in Figure 6. As
previously, the ions are focussed in time of flight
downstream of the linear trap. Orthogonal ejection of
trapped-.ions additionally allows a much higher space.
charge capacity than is provided by the arrangement of
Figure 1, and also provides better ion beam
parameters.
Although, to achieve orthogonal ejection, a
quadrupole assembly such as is shown in Figure 1 could
be used, this requires the RF voltages applied thereto
to be switched off instantaneously. This requires very
complex associated electronics, however, so, in
preference, the ion trap 300 is elongate in the `y'
direction but is curved in the x-y plane (such that
the longitudinal axis is likewise curved in the x-y
plane). Curvature of the ion trap 300 improves
geometrical focussing. By way of distinguishing from
the truly linear trap 130 of Figure 1, the ion trap
300 will henceforth be termed a `curved trap'.
The curved trap 300 includes lenses 310 extending
from the exit of the trap which together convert the
wide angle incident beam into a narrow beam.
The narrow ion beam exiting the curved trap 300
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 26 -
passes through a beam deflector 320 and into an
electrostatic trap 130. The beam deflector may take
one of a variety of forms. A plate in front of the
focal point of the focussing lenses could be used to
block both ions and gas along the line of sight
towards the entrance to the electrostatic trap 130
(for example, around 5 of arc). Gas arriving from
larger angles will not be along the line of sight
whereas corresponding ions can be diverted into the
electrostatic trap entrance by lenses. The problem
with this approach is that it requires the blocking
plate to be located in a field-free region which can
be difficult to arrange. As an alternative, a
toroidal deflector can be employed to achieve the
required beam deflection although this introduces
extra complexity.
The preferred beam deflector 320 is shown in
Figure:6 and.contains-a right-angled bend which
prevents gas carryover along the line of sight between
the curved trap 300 and the electrostatic trap 130. An
`S'-shaped beam deflector could of course be employed
instead (as is shown in Figure 1). If the
electrostatic trap is arranged with its entrance
parallel to the direction of exit of ions from the
curved trap 300. As with the arrangement of Figure 1,
the ions are usually focussed in time of flight onto
the electrostatic trap entrance which then detects in
the manner described previously. However, a different
focal point can be chosen, as for example when
operating in surface-induced dissociation mode (see
Figure 8 below).
Referring now to Figure 7, a sectional view of
the curved trap 300 of Figure 6 is shown. The curved
trap 300 is preferably RF only and comprises an outer
electrode plate 330, at a D.C. voltage preferably near
ground, along with an inner electrode plate 340 at the
same D.C. voltage. Sandwiched between the inner and
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 27 -
outer electrode plates 330, 340 are upper and lower
centre plate pairs 360a, 360b, to which is applied an
RF voltage having a phase indicated on the drawings as
RF-. The upper and lower centre plate pairs 360a,
360b in turn sandwich an axis plate pair 350 to which
is applied an RF voltage (labelled RF+ in Figure 7)
which is in antiphase to the voltage applied to the
upper and lower centre plate pairs 360a, 360b.
The geometry of the curved trap 300 is chosen in
such a way that the minimum of the effective potential
(that is, the minimum of the RF field) is located
exactly in the middle of the trap. This is the axis of
symmetry in the plane XZ and is labelled point Q in
Figure 7.
The electrodes 330, 340, 350, 360a and 360b are
curved in the XY plane. A DC offset, which is the same
as that applied to the inner and outer electrode
plates 330, 340=, is applied to the upper, lower and
axis plates. This-causes all masses to be stored in
the curved trap 300 and. cooled in.collisions with gas
at 0.1-1 mtorr. At the end of the storage, a positive
pulse is applied to the outer electrode plate 330, and
a negative pulse of the same amplitude is applied to
the inner electrode plate 340 (for positive ions).
Ions are extracted by the resulting electric field
through a trap exit 370. The RF voltages do not need
to be removed as they have little effect on the beam
parameters due to their symmetry. In addition, the RF
field strength near the X axis is relatively weak.
However, it is preferable to time the pulses applied
to the inner and outer electrode plates 330, 340 so
that they are applied in synchronism with the phase of
the RF voltages applied to the upper, lower and axis
plates 350, 360a, 360b.
After sufficient ions have been stored in the
trap, they are ejected towards the centre of curvature
of the curved trap 300 (see below), and also focussed,
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 28 -
both geometrically and in time-of-flight, into a
narrow beam that is then introduced into the
electrostatic trap 130.
Still referring to Figure 7, a liner 380 may be
provided between the trap exit 370 and the lenses 310.
The liner 380 is located in a substantially field-free
region of the curved trap. Another pulse, equal to the
required ion energy on the entrance to the
electrostatic trap 130, may be applied to the liner.
380 at the same time as the pulses are applied to the
inner and outer electrode plates 330, 340. The pulse
is applied to the liner so as to create an "energy
lift", that is, to produce a high ion energy in
conditions where both the DC offset applied to the
curved trap 300 and the potential applied to the outer
electrode parts 160, 170 of the orbitrap 130 (Figure
3) are maintained near ground voltage. If the curved
trap--300 floats at-the acceleration voltage then no
energy lift will be required. Nevertheless, it is
important that any ion source should have the
capability to float as well.
The length of the liner 380 is defined by the
required relative mass range: the ratio of-the maximum
to minimum masses is given by mmax/mmin= (Ll/L2) 2, where Ll
is the effective distance from the axis to the exit
from liner and L2 is the effective distance to the
entrance to the liner. The duration of the pulse
applied to the liner 380 is determined by calculating
the time-of-flight, to the liner exit, for the
lightest mass of interest, so that this mass emerges
from the liner 380 at its full energy whilst the
voltage on the liner 380 is already back to its normal
value (near ground). The duration of the pulses
applied to the inner and outer electrode plates 330,
340 should be longer than this.
It is to be understood that the liner 380 in the
curved trap 300 of Figures 6 and 7 is equally suitable
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02101373
- 29 -
for the linear trap 30 of Figure 1.
The foregoing description of some preferred
embodiments has also explained the principles involved
with reference to sample ions that are derived
directly from an atmospheric pressure ionization
source or the like, and are simply stored as such in
the ion trap. However, structural analysis of sample
ions may also be carried out using TOF focussing and
any of the ms/ms modes available. Three modes in
particular are contemplated.
In collision-induced dissociation (CID),
precursor ions may be selected either by the
quadrupole mass filter 24 (Fig. 1) or inside the ion
trap 30. Ejection of unwanted ions in each of these
cases may be performed, for example, by resonant
excitation between the opposite rods of each device or
by a mass selective instability scan (see, for
example, the.abo.ve-referenced US-A-5,886,346). This
may be achieved by DC biassing one set of rods
relative to the other, for example. Fragmentation may
be caused by collisions with collision gas at an
elevated pressure in a dedicated RF-only multipole or,
preferably, in the linear ion trap 30. The resulting
fragments are stored and collisionally cooled in the
ion trap 30 and then injected into the orbitrap 130 in
the same way as described previously. CID in
collisional multipoles is a technique which is well
known as such to those skilled in the art and the
technique of selecting only the required m/z is
likewise a known part of tandem mass spectrometry.
Further description of these techniques may be found
in "Protein Sequencing and Identification Using Tandem
Mass Spectrometry", by M. Kinter, N.E. Sherman, John
Wiley and Sons, 2000, and in "Mass Spectrometry/Mass
Spectrometry : Techniques and Applications of Tandem
Mass Spectrometry", by K. L. Busch, G. L. Glish, and
S. A. McLuckey, John Wiley and Sons, 1989.
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 30 -
In surface-induced dissociation (SID), precursor
ion selection is typically carried out in the same way
as in CID. Precursor ions are stored in the linear
trap 30 and then pulsed towards the orbitrap 130.
However, in this case the TOF focus is shifted behind
the plane of the entrance of the orbitrap 130, to a
separate plane where a collision surface is located.
This is shown schematically in Figure 8, which
illustrates in side view the orbitrap 130 along with
the electrode 140, the compensator 200 and the
secondary electron multiplier 190. The collision
surface 250 is located downstream of the secondary
.electron multiplier 190 and may be formed from a metal
or may instead be metal- or polymer-coated. A
fluorocarbon or hydrocarbon self-assembled monolayer
plate such as is disclosed in Science, Vol. 275, pages
1447-1450, by S. A. Miller, H. Luo, S. J. Pachuta and
R. G.. Cooks, (1997) could for example be used.
In this case, precursor ions pass tangentially
through the orbitrap 130, which has electric fields
that are low enough to prevent ion losses, and out
past the compensator 200 which in this part of the
process is switched off to allow passage of the
precursor ions. These precursor ions then decelerate
in front of the collision surface 250 in a
deceleration gap created by a grid 255 and collide
with the collision surface 250 at a collision energy
determined by a voltage difference between the
collision surface and the offset applied to the exit
segment 50 of the linear trap. Collision results in
the formation of fragment ions at low energies
(several electronvolts) which are accelerated by the
same electric field towards the orbitrap 130. Due to
the TOF focussing of these precursor ions and the
instantaneous nature of SID, fragment ions separate
(at least partially) on their way to the orbitrap 130
according to their mass/charge ratio and ions of each
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 31 -
mass/charge ratio enter the orbitrap 130 as a short
packet. The compensator 200 is switched on during this
part of the process so that the fragment ions are then
captured by the orbitrap. This allows ions to be
captured in the same way as in the MS-only mode
described in connection with Figures 1 to 5 above. If
low resolution of parent selection is sufficient for a
given application, then an ion gate (not shown) may be
installed between the orbitrap 130 and the collision
surface 250 to provide an alternative way of selecting
precursor ions. It is in fact possible to use the
deflection electrode 200 of the orbitrap 130 as an ion
gate.
Finally, metastable dissociation (MSD) mode may
be employed with the arrangement and principles
outlined previously. Precursor ions may either be
selected as described above in connection with the CID
and. SID modes,. or may instead be injected into the
orbitrap 130 without prior selection. The only
difference from the MS-only mode described in
connection with Figures 1 to 5 is the activation of
ions in the ion trap 30. Activation may be achieved
by pulsed ion extraction in the presence of gas at an
elevated pressure (either static or pulsed), wherein
the increase of ion internal energy is controlled by
the gas pressure, by pulsed electromagnetic radiation
(e.g. infrared radiation which can be used to excite
ions inside the. ion trap 30, or by resonant or
broadband dipolar excitation using at least two pairs
of quadrupole rod electrodes in the ion trap 30. In
that case, the increase of internal energy is
controlled by the amplitude of the excitation signal
and the gas pressure.
Although pulsed ion extraction with a high
pressure gas is preferable due to its simplicity, each
of the foregoing results in the excitation ("heating")
of ions and the consequent formation of metastable
WO 02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 32 -
ions with a controllable decay constant. The
magnitude of the decay constant can be controlled by
variation of the intensity of excitation. Before
fragmentation becomes noticeable, ions may be injected
into the orbitrap 130 and precursor ion selection may
even be achieved. On the other hand, excessively long
decay times lead to a decrease in the speed of
analysis. Therefore, optimum decay times range from
several milliseconds to tens of milliseconds.
Precursor ion selection is achieved by applying a
radio frequency voltage in resonance with the axial
oscillation of precursor ions at a correct phase. A
waveform generator (not shown), under the control of
the data processing system referred to in connection
with Figure 1, supplies this RF voltage either to the
central electrode 140 (parametric resonance de-
excitation at doubled ion frequency, set out in
Analytical.Chemistry'Volume 72, No. 6, p.1156-1162, by
Makarov, and in the above-referenced US-A-5,886,346),
or between the two outer electrode parts 160, 170
(resonance de-excitation at ion frequency) of the
orbitrap 130. Application of an RF voltage decreases
the amplitude of axial oscillation of ions so that
only precursor ions are brought into the plane of
symmetry of the orbitrap 130. Precursor ions are left
in this state long enough to allow metastable decay to
occur. The remaining precursor ions are then excited,
together with their fragment ions, by a broadband
excitation. Typically, a radio frequency voltage is
.30 applied to the two outer electrode parts 160, 170 by
the waveform generator. Coherent oscillations of ions
of each mass/charge ratio are detected by detecting an
image detection current via the differential amplifier
180 in the same way as described for MS-only mode.
Metastable decay of ions other than precursor ions
also results in the formation of fragment ions.
However, these are uniformly spaced along the orbitrap
WO02/078046 CA 02689084 2009-12-18 PCT/GB02/01373
- 33 -
130 and thus do not move coherently. No image current
is produced for detection in that case. Alternatively,
unwanted precursor or fragment ions may be removed by
an additional broadband excitation.