Note: Descriptions are shown in the official language in which they were submitted.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
FUSED IMIDAZOLES FOR CANCER TREATMENT
Field of application of the invention
The invention relates to fused imidazole compounds, which are used in the
pharmaceutical industry for the manufacture of pharmaceutical compositions.
Known technical background
Cancer is the second most prevalent cause of death in the United States,
causing
450,000 deaths per year. While substantial progress has been made in
identifying
some of the likely environmental and hereditary causes of cancer, there is a
need
for additional therapeutic modalities that target cancer and related diseases.
In
particular there is a need for therapeutic methods for treating diseases
associated
with dysregulated growth / proliferation.
Cancer is a complex disease arising after a selection process for cells with
ac-
quired functional capabilities like enhanced survival / resistance towards
apop-
tosis and a limitless proliferative potential. Thus, it is preferred to
develop drugs
for cancer therapy addressing distinct features of established tumors.
One pathway that has been shown to mediate important survival signals for
mammalian cells comprises receptor tyrosine kinases like platelet-derived
growth
factor receptor (PDGF-R), human epidermal growth factor 2/3 receptor (HER2/3),
or the insulin-like growth factor 1 receptor (IGF-1R). After activation the
respec-
tives by ligand, these receptors activate the phoshatidylinositol 3-kinase
(Pi3K)/Akt pathway. The phoshatidylinositol 3-kinase (Pi3K)/Akt protein kinase
pathway is central to the control of cell growth, proliferation and survival,
driving
progression of tumors. Therefore within the class of serine-threonine specific
sig-
nalling kinases, Akt (protein kinase B; PKB) with the isoenzmyes Akti (PKBa),
Akt2 (PKB 11) and Akt3 (PKB y) is of high interest for therapeutic
intervention. Akt
is mainly activated in a Pi3-kinase dependent manner and the activation is
regu-
lated through the tumor suppressor PTEN (phosphatase and tensin homolog),
which works essentially as the functional antagonist of Pi3K.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
2
The Pi3K/Akt pathway regulates fundamental cellular functions (e.g.
transcription,
translation, growth and survival), and is implicated in human diseases
including
diabetes and cancer. The pathway is frequently overactivated in a wide range
of
tumor entities like breast and prostate carcinomas. Upregulation can be due to
overexpression or constitutively activation of receptor tyrosine kinases (e.g.
EGFR, HER2/3), which are upstream and involved in its direct activation, or
gain-
or loss-of-function mutants of some of the components like loss of PTEN. The
pathway is targeted by genomic alterations including mutation, amplification
and
rearrangement more frequently than any other pathway in human cancer, with the
possible exception of the p53 and retinoblastoma pathways. The alterations of
the Pi3K/Akt pathway trigger a cascade of biological events, that drive tumor
pro-
gression, survival, angiogenesis and metastasis.
Activation of Akt kinases promotes increased nutrient uptake, converting cells
to
a glucose-dependent metabolism that redirects lipid precursors and amino acids
to anabolic processes that support cell growth and proliferation. These
metabolic
phenotype with overactivated Akt lead to malignancies that display a metabolic
conversion to aerobic glycolysis (the Warburg effect). In that respect the
Pi3K/Akt
pathway is discussed to be central for survival despite unfavourable growth
con-
ditions such as glucose depletion or hypoxia.
A further aspect of the activated PI3K/Akt pathway is to protect cells from
pro-
grammed cell death ("apoptosis") and is hence considered to transduce a
survival
signal. By acting as a modulator of anti-apoptotic signalling in tumor cells,
the
Pi3K/Akt pathway, particular Akt itself is a target for cancer therapy.
Activated Akt
phosphorylates and regulates several targets, e.g. BAD, GSK3 or FKHRL1, that
affect different signalling pathways like cell survival, protein synthesis or
cell
movement. This Pi3K/Akt pathway also plays a major part in resistance of tumor
cells to conventional anti-cancer therapies. Blocking the Pi3K/Akt pathway
could
therefore simultaneously inhibit the proliferation of tumor cells (e.g. via
the inhibi-
tion of the metabolic effect) and sensitize towards pro-apoptotic agents.
Akt inhibition selectively sensitized tumor cells to apoptotic stimuli like
Trail,
Campthothecin and Doxorubicin. Dependent on the genetic background / molecu-
lar apperations of tumors, Akt inhibitors might induce apoptotic cell death in
monotherapy as well.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
3
In the European patent EP1268478 phenyl-substituted imidazopyridines are
disclosed as H3-antagonists for treating diseases in the central nervous
system.
In the International patent application W02005014598 substituted
imidazopyrimidines are disclosed for the treatment of cancer. In the
International
patent application W02007025090 substituted imidazopyridazines are disclosed
for the treatment of cancer. In the International patent applications
W02004096131, W02005100344, W02006036395, W02006065601,
W02006091395 and W02006135627 Akt inhibitors are described.
Description of the invention
It has now been found that the fused imidazole compounds, which are described
in detail below, have surprising and advantageous properties.
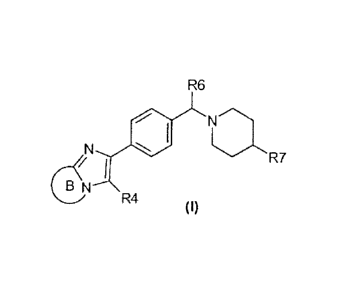
In accordance with a first aspect, the invention relates to compounds of
formula
(I)
R6
N
R7
I
B N
R4
(I)
wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N R1 N R1 R1
NN
N
R3 R3 R3
R4 R4 R3
R4
R4
R1 N R1N R1N R1 N
X ,N NN
R3 N R3 ¨ R3 R3
R4 R4 R4
R4
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
4
R1
1---5--: R14_71:.:
.....kg
N " N / N
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cyano, 3-
7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-7C-cycloalkoxy,
mono- or di-1-4C-alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -
C(NH)NH2, -C(0)NH2 or -C(0)0R1 0
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, thia-
zolyl or oxazolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or 1-4C-alkyl,
R7 is -W-Y,
W is a monocyclic 5-membered heteroarylene comprising 1 nitrogen atom
and optionally 1 or 2 further heteroatoms independently selected from
oxygen, nitrogen and sulphur,
and wherein the heteroarylene is optionally substituted by R8,
R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
Y is phenyl or a monocyclic 5- or 6-membered heteroaryl comprising 1
nitro-
gen atom and optionally 1 or 2 further heteroatoms independently selected
from oxygen, nitrogen, sulphur
and wherein the heteroaryl is optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
CA 02695251 2010-02-01
WO 2009/021990
PCT/EP2008/060686
53799AWO
2008 08 13
In accordance with a second aspect, the invention relates to compounds of for-
mula (I)
R6
0 N
/1\1 R7
IN I
R4
(I)
5 wherein ring B and the imidazole to which it is fused form a ring system
selected
from
R1N R1 N k ,2 R1 R1 s N
>,` )
NN
R3 R3 R3
',q
R4 R4 R3 R
R4
4
R1 N m R1N R1)._,>¨_N R1 NN
)___11 ) / 1\1---,.? N? N /
R3 N N R3 N R3 N R3 N
R4 R4 R4
R4
R1 Si.....>..-1
..... JR R1¨
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cya-
no, 3-7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-70-
cycloalkoxy, mono- or di-1-4C-alkylamino, mono- or di-1-4C-
alkylaminocarbonyl, -C(NH)NH2, -C(0)NH2 or -C(0)0R10
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl, pyridinyl,
thia-
zoly1 or oxazolyl,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
6
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or 1-4C-alkyl,
R7 is -W-Y,
W is a monocyclic 5-membered heteroarylene comprising 1 nitrogen atom
and optionally 1 or 2 further heteroatoms independently selected from
oxygen, nitrogen and sulphur,
and wherein the heteroarylene is optionally substituted by R8,
R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
Y is a monocyclic 5- or 6-membered heteroaryl comprising 1 nitrogen
atom
and optionally 1 or 2 further heteroatoms independently selected from oxy-
gen, nitrogen, sulphur
and wherein the heteroaryl is optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
1-4C-Alkyl is a straight-chain or branched alkyl group having 1 to 4 carbon
atoms.
Examples are methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, sec-
butyl and
tert-butyl.
Mono- or di-1-4C-alkylamino radicals contain in addition to the nitrogen atom,
one
or two of the abovementioned 1-4C-alkyl radicals. Examples are the methyl-
amino, the ethylamino, the isopropylamino, the dimethylamino, the diethylamino
and the diisopropylamino radical.
Mono- or di-1-4C-alkylaminocarbonyl radicals contain in addition to the
carbonyl
group one of the abovementioned mono- or di-1-4C-alkylamino radicals. Exam-
ples are the N-methylaminocarbonyl, the N,N-dimethylaminocarbonyl, the N-
ethylaminocarbonyl, the N-propylaminocarbonyl, the N,N-diethylaminocarbonyl
and the N-isopropylaminocarbonyl.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
7
Halogen within the meaning of the present invention is iodine, or particularly
bro-
mine, chlorine and fluorine.
1-4C-Alkoxy represents radicals, which in addition to the oxygen atom, contain
a
straight-chain or branched alkyl radical having 1 to 4 carbon atoms. Examples
which may be mentioned are the butoxy, isobutoxy, sec-butoxy, tert-butoxy, pro-
poxy, isopropoxy, ethoxy and methoxy radicals.
3-7C-Cycloalkyl stands for cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or
cycloheptyl.
3-7C-Cycloalkyloxy stands for cyclopropyloxy, cyclobutyloxy, cyclopentyloxy,
cyclohexyloxy or cycloheptyloxy.
2-4C-Alkenyl is a straight chain or branched alkenyl radical having 2 to 4
carbon
atoms. Examples are the but-2-enyl, but-3-enyl (homoallyl), prop-1-enyl, prop-
2-
enyl (ally1) and the ethenyl (vinyl) radicals.
2-4C-Alkynyl is a straight chain or branched alkynyl radical having 2 to 4
carbon
atoms. Examples are the but-2-ynyl, but-3-ynyl (homopropargyl), prop-1-ynyl, 1-
methylprop-2-ynyl (1-methylpropargy1), prop-2-ynyl (propargyl) and the ethinyl
radicals.
The term "monocyclic 5-or 6-membered heteroaryl" comprised without being re-
stricted thereto, the 5-membered heteroaryl radicals furyl, thienyl, pyrrolyl,
oxa-
zolyl, isoxazolyl, thiazolyl, isothiazolyl, imidazolyl, pyrazolyl, triazolyl
(1,2,4-
triazolyl, 1,3,4-triazoly1 or 1,2,3-triazoly1), thiadiazolyl (1,3,4-
thiadiazolyl, 1,2,5-
thiadiazolyl, 1,2,3-thiadiazoly1 or 1,2,4-thiadiazoly1) and oxadiazolyl (1,3,4-
oxadiazolyl, 1,2,5-oxadiazolyl, 1,2,3-oxadiazoly1 or 1,2,4-oxadiazoly1), as
well as
the 6-membered heteroaryl radicals pyridinyl, pyrimidinyl, pyrazinyl and
pyridaz-
inyl. Preferred 5- or 6-membered heteroaryl radicals are furanyl, thienyl,
pyrrolyl,
thiazolyl, oxazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyrimidinyl,
pyrimidinyl,
pyrazinyl or pyridazinyl. More preferred 5- or 6-membered heteroaryl radicals
are
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
8
furan-2-yl, thien-2-yl, pyrrol-2-yl, thiazolyl, oxazolyl, 1,3,4-thiadiazolyl,
1,3,4-
oxadiazolyl, pyridin-2-yl, pyridin-4-yl, pyrimidin-2-yl, pyrimidin-4-yl,
pyrazin-2-y1 or
pyridazin-3-yl.
The term "monocyclic 5-membered heteroarylene" is a divalent radical in which
arbitrary one hydrogen atom is eliminated from the above described
"heteroaryl"
and may include, without being restricted thereto, the 5-membered heteroaryl
radicals furylene, thienylene, pyrrolylene, oxazolylene, isoxazolylene, thia-
zolylene, isothiazolylene, imidazolylene, pyrazolylene, triazolylene (1,2,4-
triazolylene, 1,3,4-triazolylene or 1,2,3-triazolylene), thiadiazolylene
(1,3,4-
thiadiazolylene, 1,2,5-thiadiazolylene, 1,2,3-thiadiazolylene or 1,2,4-
thiadiazolylene) and oxadiazolylene (1,3,4-oxadiazolylene, 1,2,5-
oxadiazolylene,
1,2,3-oxadiazolylene or 1,2,4-oxadiazolylene). Prefered 5-membered heteroaryl
radicals are triazolylene, pyrazolylene, oxadiazolylene or imidazolylene. More
prefered 5-membered heteroaryl radicals are 1,2,4-triazolylene, pyrazolylene,
1,2,4-oxadiazolylene or imidazolylene.
In general and unless otherwise mentioned, the heteroarylic or heteroarylenic
radicals include all the possible isomeric forms thereof, e.g. the positional
isomers
thereof. Thus, for some illustrative non-restricting example, the term
pyridinyl or
pyridinylene includes pyridin-2-yl, pyridin-2-ylene, pyridin-3-yl, pyridin-3-
ylene,
pyridin-4-y1 and pyridin-4-ylene; or the term thienyl or thienylene includes
thien-2-
yl, thien-2-ylene, thien-3-y1 and thien-3-ylene.
Constituents which are optionally substituted as stated herein, may be substi-
tuted, unless otherwise noted, at any possible position. Analogously it is
being
understood that it is possible for any heteroaryl group if chemically suitable
that
said heteroaryl group may be attached to the rest of the molecule via any
suitable
atom.
The heteroarylic or heteroarylenic groups mentioned herein may be substituted
by their given substituents or parent molecular groups, unless otherwise
noted, at
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
9
any possible position, such as e.g. at any substitutable ring carbon or ring
nitro-
gen atom.
Unless otherwise noted, rings containing quaternizable amino- or imino-type
ring
nitrogen atoms (-N=) may be preferably not quaternized on these amino- or
imino-type ring nitrogen atoms by the mentioned substituents or parent
molecular
groups.
Unless otherwise noted, any heteroatom of a heteroarylic or heteroarylenic
ring
with unsatisfied valences mentioned herein is assumed to have the hydrogen
atom(s) to satisfy the valences.
When any variable occurs more than one time in any constituent, each
definition
is independent.
In another embodiment of the above-mentioned first or second aspect, the
invention relates to compounds of formula (I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1 N R1 N 11 R1 R1
r_.,.
. _ _T 1
NN
R3 R3 R3 N /
R4 R4 R3 R
R4
4
R1 ) KI R1 R1 R1 N
>1")..1\,1
>-.-........"----..-%N
,>---....."--.-.<-.N --.:--
---N
,N / ,N-,? NN--_,? NN--,?
R3 N R3 N R3 R3
R4 R4 R4 R4
Or.N R1 0 Sr.....-_-N
R1¨<\ -:::- R1¨<\
Ns'N N Ns'N
R4 R3 / R4 R4
wherein
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cyano, 3-
7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-7C-cycloalkoxy,
mono- or di-1-4C-alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -
C(NH)NH2, -C(0)NH2 or
5 R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, thia-
zoly1 or oxazolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
10 R6 is hydrogen or 1-4C-alkyl,
R7 is -W-Y,
W is a monocyclic 5-membered heteroarylene comprising 1 nitrogen atom
and optionally 1 or 2 further heteroatoms independently selected from
oxygen, nitrogen and sulphur,
and wherein the heteroarylene is optionally substituted by R8,
R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
Y is a monocyclic 5- or 6-membered heteroaryl comprising 1 nitrogen
atom
and optionally 1 or 2 further heteroatoms independently selected from oxy-
gen, nitrogen, sulphur
and wherein the heteroaryl is optionally substituted by R9,
R9 is 1-4C-alkyl or halogen,
or a salt, as well as the stereoisomer and salt of the stereoisomer thereof.
In a further embodiment, the invention relates to compounds of formula (I),
wherein
ring B and the imidazole to which it is fused form a ring system selected from
)
R1 N R1 N--N R1 R1 S.____ N
)---...."-..-....%,` )-.-=
NN
R3 R3 R3 N /
R
R4 R4 R3
R4
R4
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
11
R1 N R1N R1,N R1 N
___121 )* )7,121
/ ---.? N--.,? N /
R3 N N R3 N N N
R3 R3 N
R4 R4 R4
R4
R1 Ir_ R1 [-----R
N---11 N"--N /
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cyano, 3-
7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-7C-cycloalkoxy,
mono- or di-1-4C-alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -
C(NH)NH2 or -C(0)NH2,
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl or thia-
zolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is triazolylene, pyrazolylene, oxadiazolylene or imidazolylene,
each of which is optionally substituted by R8,
R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
Y is thiazolyl, oxazolyl, thiadiazolyl, oxadiazolyl, pyridinyl,
pyrimidinyl, pyraz-
inyl or pyridazinyl, each of which is optionally substituted by R9,
R9 is 1-40 alkyl or halogen,
or a salt, as well as the stereoisomer and salt of the stereoisomer thereof..
In another embodiment, the invention relates to compounds of formula (I),
wherein
ring B and the imidazole to which it is fused form a ring system selected from
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
RlN 12
R1 N 1\1 / R1 R1
R3 R3
)7,
N
R3 N
R4 R4 R3 R4
R4
R1 N R1N R1N R1 )7 NN ,.
X X 1\1-..,.? N? N
R3 N R3 N R3 N R3 N
R4 R4 R4 R4
R1
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cyano, 3-
7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-7C-cycloalkoxy,
mono- or di-1-4C-alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -
C(NH)NH2 or -C(0)NH2,
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl or thia-
zolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene, 1,2,4-oxadiazolylene or
imidazolylene,
Y is thiazol-2-yl, thiazol-4-yl, oxazol-2-yl, oxazol-4-yl, 1,3,4-thiadiazol-
2-yl,
1,3,4-oxadiazol-2-yl, pyridin-2-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-
y1
or pyridazin-3-yl,
or a salt as well as the stereoisomer and salt of the stereoisomer thereof. .
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
13
ring B and the imidazole to which it is fused form a ring system selected from
R1>---%N ) R1 N m R1 R1 s N
.."..
NN
R3 R3 R3
' , \ r,
R4 R4 R3 R
R4
4
)' R1¨<\
A /
R32N N R4
R4 R3 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, trifluoromethyl, cyano, 2-
40-
alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, mono- or di-1-4C-alkylamino, mono- or
di-1-4C-alkylaminocarbonyl, -C(NH)NH2 or -C(0)NH2,
R3 is hydrogen,
R4 is unsubstituted phenyl or thienyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene, 1,2,4-oxadiazolylene or
imidazolylene,
Y is pyridin-2-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-y1 or
pyridazin-3-yl,
or a salt thereof. .
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R11\1 )-7 R1 N m R1 R1 s N
,12
N%N
R3
' , \ r,
R3 R3
R4 R4 R3 R
R4
4
>,` ) R1
A /
R32 N N R4
R4 R3 R4
wherein
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
14
R1 is hydrogen, 1-4C-alkyl, halogen, amino, trifluoromethyl, cyano, 1-
40-
alkoxy, -C(NH)NH2 or -C(0)NH2,
R3 is hydrogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl or thia-
zolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, 1,2,4-oxadiazolylene or pyrazolylene,
Y is pyridin-2-y1 or pyrazin-2-yl,
and the salts, as well as the stereoisomers and salts of the stereoisomers
thereof
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N * R1 N m R1 R1 r-%..",
NN
RS----T:
R3 R3
R4 R4 R3 R
R4
4
R3
)'
..._,,R
N /
N2R4 R3 N R4 R1 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, trifluoromethyl, cyano, 1-
40-
alkoxy, -C(NH)NH2 or -C(0)NH2,
R3 is hydrogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl, pyridinyl or
thia-
zolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, 1,2,4-oxadiazolylene or pyrazolylene,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Y is pyridin-2-y1 or pyrazin-2-yl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
5
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N R1)Nr_ki R1 R1
. ..-1
N%N
R3 R3 R3 N /
R4 R4 R3 R
R4
4
S N R1N
R1¨ TR
N,N /
R4 R3 N
R4
wherein
10 R1 is hydrogen, methyl, halogen, trifluoromethyl, cyano, methoxy, -
C(NH)NH2
or -C(0)NH2,
R3 is hydrogen,
R4 is unsubstituted phenyl,
R6 is hydrogen,
15 R7 is -W-Y,
W is 1,2,4-triazolylene,
Y is pyridin-2-yl,
and the salts, as well as the stereoisomers and salts of the stereoisomers
thereof.
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
16
R1N R1 N m R1 R1
)%-
>N---.,? N / kq ...,-N /
R3 R3-___: IN = R3
R4 R4 R3
R4
R4
Sr___--N R1N
R1¨ _....kg
N IN
R4 R3 N
R4
wherein
R1 is hydrogen, methyl, halogen, trifluoromethyl, cyano, methoxy, -
C(NH)NH2
or -C(0)NH2,
R3 is hydrogen,
R4 is unsubstituted phenyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene,
Y is pyridin-2-yl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further embodiment, the invention relates to compounds of formula (I),
wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N R1 N m R1 R1
* )2
NN
R3 R3 RS----TI:N
R4 R4 R3 R
R4 4
R1 N m R1N R1N R1 N m
)2'
X N / X --.? N---__? N /
R3 N R3 N N N
R3 R3 N
R4 R4 R4 R4
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
17
R1 klq OR R1¨
N--N
N---"
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl,
cyano, 3-
7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-7C-cycloalkoxy,
mono- or di-1-4C-alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -
C(NH)NH2, -C(0)NH2 or -C(0)0R1 0
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, oxa-
zolyl or thiazolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is triazolylene, pyrazolylene, oxadiazolylene or imidazolylene,
each of which is optionally substituted by R8,
R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
Y is phenyl, furanyl, thienyl, pyrrolyl, thiazolyl, oxazolyl,
thiadiazolyl, oxadia-
zolyl, pyridinyl, pyrimidinyl, pyrazinyl or pyridazinyl, each of which is op-
tionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In another embodiment, the invention relates to compounds of formula (I),
wherein
ring B and the imidazole to which it is fused form a ring system selected from
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
RlN 18
)
R1 N 11 R1 R1
N
R3 R37,R3 N
R4 R4 R3
R4
R4
R1 N R1N R1N R1 )7 NN ,.
X X 1\1-..,.? N
R3 N R3 N R3 R3 N
R4 R4 R4
R4
R1 0 N S N
R1 I R1¨ TR/
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2,
trifluoromethyl, cya-
no, 3-7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-70-
cycloalkoxy, mono- or di-1-4C-alkylamino, mono- or di-1-4C-
alkylaminocarbonyl, -C(NH)NH2, -C(0)NH2 or -C(0)0R10
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, oxa-
zolyl or thiazolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene, 1,2,4-oxadiazolylene or
imidazolylene,
Y is phenyl, furan-2-yl, thien-2-yl, pyrrol-2-yl, pyridin-4-yl, thiazol-2-
yl, thiazol-
4-yl, oxazol-2-yl, oxazol-4-yl, 1,3,4-thiadiazol-2-yl, 1,3,4-oxadiazol-2-yl,
pyridin-2-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-y1 or pyridazin-3-yl,
each of which is optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
19
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N R1 N m R1 R1
NN
R3 R3
R3 N
R4 R4 R3 R
R4
4
R1 N N R1
R1
--N
N
R3 N R3 N R4
R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, -SR2, amino, trifluoromethyl,
cyano, 2-
4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, mono- or di-1-4C-alkylamino, mo-
no- or di-1-4C-alkylaminocarbonyl, -C(NH)NH2, -C(0)NH2 or -C(0)0R10
R2 is 1-4C-alkyl,
R3 is hydrogen or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, oxa-
zolyl or thiazolyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene or 1,2,4-oxadiazolylene,
Y is phenyl, furan-2-yl, thien-2-yl, pyrrol-2-yl, pyridin-4-yl,
thiazol-2-yl, pyridin-
2-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-y1 or pyridazin-3-yl, each of
which is
optionally substituted by R9,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
In a further preferred embodiment the invention relates to compounds of
formula
(I), wherein ring B and the imidazole to which it is fused form a ring system
selected from
Ri R1 N R1 R1
)7,11
N
R3 R3R3 N
R4 R4 R3
R4
R4
R1 N R1N R1N R1 N
XX 1\1-..,.? NN? NN
R3 N R3 N R3 R3
R4 R4 R4
R4
R1 0 N S N
R1 I R1
N"--N
R3
R4 R4 R4
5
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2,
trifluoromethyl, cya-
no, 3-7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-70-
cycloalkoxy, mono- or di-1-4C-alkylamino, mono- or di-1-4C-
10 alkylaminocarbonyl, -C(NH)NH2, -C(0)NH2 or -C(0)0R10
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl,
oxazolyl or thiazolyl,
15 R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is triazolylene, pyrazolylene, oxadiazolylene or
imidazolylene,
each of which is optionally substituted by R8,
20 R8 is 1-4C-alkyl or 3-7C-cycloalkyl,
CA 02695251 2010-02-01
WO 2009/021990
PCT/EP2008/060686
53799AWO
2008 08 13
21
Y is furanyl, thienyl, pyrrolyl, thiazolyl, oxazolyl,
thiadiazolyl, oxadia-
zolyl, pyridinyl, pyrimidinyl, pyrazinyl or pyridazinyl, each of which is
optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the second aspect, the invention relates
to
compounds of formula (I), wherein ring B and the imidazole to which it is
fused
form a ring system selected from
R1>\,.-_1\1 R1 N R1 R1
R3 R3 R3 N /
R4 R4 R3 R
R4 4
R1 N m R1N R1N R1 N
)2' )* )7,12.1
X / X 1\1-..,.? N--.? N /
R3 N N R3 N R3 N R3 N
R4 R4 R4 R4
0 N R1 0 N Sr..--N
R1 I-----R
N---N / ....:kR R1-1\1
R3
R4 R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, amino, -SR2, trifluoromethyl, cya-
no, 3-7C-cycloalkyl, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, 3-70-
cycloalkoxy, mono- or di-1-4C-alkylamino, mono- or di-1-4C-
alkylaminocarbonyl, -C(NH)NH2, -C(0)NH2 or -C(0)0R10
R2 is hydrogen, 1-4C-alkyl or 3-7C-cycloalkyl,
R3 is hydrogen, 1-4C-alkyl or halogen,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
22
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl, oxa-
zoly1 or thiazolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen or methyl,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene, 1,2,4-oxadiazolylene or
imidazolylene,
Y is furan-2-yl, thien-2-yl, pyrrol-2-yl, pyridin-4-yl, thiazol-2-yl,
thiazol-4-yl,
oxazol-2-yl, oxazol-4-yl, 1,3,4-thiadiazol-2-yl, 1,3,4-oxadiazol-2-yl, pyridin-
2-yl,
pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-y1 or pyridazin-3-yl, each of which
is op-
tionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the second aspect, the invention relates
to
compounds of formula (I), wherein ring B and the imidazole to which it is
fused
form a ring system selected from
R1N R1 N N R1 R1
. ..-
....-1
R3 R3 N R3 N
R4 R4 R3 R4
R4
R3
)12,
..._,,R
N /
N7_R4 R3 N R4 R1 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, -SR2, amino,
trifluoromethyl, cya-
no, 2-4C-alkenyl, 2-4C-alkynyl, 1-4C-alkoxy, mono- or di-1-4C-
alkylamino, mono- or di-1-4C-alkylaminocarbonyl, -C(NH)NH2, -
C(0)NH2 or -C(0)0R1 0
R2 is 1-4C-alkyl,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
23
R3 is hydrogen or halogen,
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl,
oxazolyl or thiazolyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, pyrazolylene or 1,2,4-oxadiazolylene,
Y is furan-2-yl, thien-2-yl, pyrrol-2-yl, pyridin-4-yl, thiazol-
2-yl, pyridin-
2-yl, pyrimidin-2-yl, pyrimidin-4-yl, pyrazin-2-y1 or pyridazin-3-yl,
each of which is optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1¨,=N )-7 R1 N m R1 R1 s N
,12
NN
R3 R3
R3 1
R4 R4 R3 R4
R4
)2R4 R1
N /
R3 N R3 N R4 R4
wherein
R1 is hydrogen, 1-4C-alkyl, halogen, -SR2, amino, trifluoromethyl, cya-
no, 1-4C-alkoxy,
-C(NH)NH2, -C(0)NH2 or -C(0)0R10,
R2 is 1-4C-alkyl,
R3 is hydrogen or halogen,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
24
R4 is phenyl substituted by R5, unsubstituted phenyl, thienyl,
pyridinyl or thia-
zolyl,
R5 is 1-4C-alkyl, halogen or 1-4C-alkoxy,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene, 1,2,4-oxadiazolylene or pyrazolylene,
Y is pyridin-2-y1 or pyrazin-2-yl, each of which is optionally
substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment, the invention relates to compounds of
formula
(I), wherein
ring B and the imidazole to which it is fused form a ring system selected from
R1N R1 N m R1 R1
NN (S1
..., /
IN =
R3 R3
R3 -N
R4 R4 R3
R4
R4
Sr..--N R1N
R1¨ _...,,R
R4 R3 N
R4
wherein
R1 is hydrogen, methyl, halogen, -SR2, trifluoromethyl, cyano,
metho-
xy, -C(NH)NH2, -C(0)NH2 or -C(0)0R10,
R2 is 1-4C-alkyl,
R3 is hydrogen or halogen,
R4 is unsubstituted phenyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Y is pyridin-2-y1 which is optionally substituted by R9,
R9 is 1-4C-alkyl, 1-4C-alkoxy or halogen,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein
10 ring B and the imidazole to which it is fused form a ring system
selected from
).
R1 N R1 N N R1 R1 N
NN
R3 R3 R3 N /
R4 R4 R3 R
R4
4
N
Srl_..--__N R1
R1¨ )---..---"--....---=-
N--N A-..?
R4 R3 N
R4
wherein
R1 is hydrogen, methyl, halogen, -SR2, trifluoromethyl, cyano, methoxy,
-
C(NH)NH2, -C(0)NH2 or -C(0)0R10,
R2 is 1-4C-alkyl,
15 R3 is hydrogen,
R4 is unsubstituted phenyl,
R6 is hydrogen,
R7 is -W-Y,
W is 1,2,4-triazolylene,
20 Y is pyridin-2-yl,
R10 is hydrogen or 1-4C-alkyl,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
26
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1>,,N
N--._
R3
R4, R3 is hydrogen, R6 is hydrogen and R1, R2, R4, R5, R7, R8,
R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1 N
)7.,...1
N
R3 /
R4, R3 is hydrogen, R6 is hydrogen and R1, R2, R4, R5, R7, R8,
R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1
NN
>1\1-._.
R3
R4, R3 is hydrogen, R6 is hydrogen and R1, R2, R4, R5, R7, R8,
R9, W and Y are as described above,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
27
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1
N/
R3
R4, R3 is hydrogen, R6 is hydrogen and R1, R2, R4, R5, R7, R8,
R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1
1\1-._.
R3 N
R4, R3 is hydrogen, R6 is hydrogen and R1, R2, R4, R5, R7, R8,
R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
28
S N
R1 .---R,
R4, R6 is hydrogen and R1, R2, R4, R5, R7, R8, R9 ,Wand Y
are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1N
N.--...,
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl and R1, R2, R7,
R8, R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1 N
).-=----N
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl and R1, R2, R7,
R8, R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
29
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1
NI\I
N--._
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl and R1, R2, R7,
R8, R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R1 S.........m,..._-A
N
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl and R1, R2, R7,
R8, R9, W and Y are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
R3 N kl---.?
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl and R1, R2, R7,
R8, R9, W and Y are as described above,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
5 In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused form the following ring system
Sr......-_-N
R1
N--N
R4, R6 is hydrogen, R4 is phenyl and R1, R2, R7, R8, R9, W
and Y are as described above,
10 or a salt, particularly a pharmaceutically acceptable salt, a tautomer,
or a stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
15 the invention relates to compounds of formula (I), wherein ring B and
the
imidazole to which it is fused is
R1
N
R3 --____
-
>N
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl, R7 is -W-Y, W
is 1,2,4-triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
20 somer of said compound, or a salt, particularly a pharmaceutically
acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
25 imidazole to which it is fused is
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
31
R1 N
).----:;-N
>1\1-._..
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl, R7 is ¨W-Y, W
is 1,2,4-triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused is
R1
NN
kl---.?
R3
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl, R7 is ¨W-Y, W
is 1,2,4-triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused is
RR3 N /
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl, R7 is ¨W-Y, W
is 1,2,4-triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
32
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused is
R1>,,N
N--._
R3 N
R4, R3 is hydrogen, R6 is hydrogen, R4 is phenyl, R7 is ¨W-Y, W
is 1,2,4-triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
In a further preferred embodiment of the above-mentioned first or second
aspect,
the invention relates to compounds of formula (I), wherein ring B and the
imidazole to which it is fused is
S N
R1 [-----R,
N,N /
R4, R6 is hydrogen, R4 is phenyl, R7 is ¨W-Y, W is 1,2,4-
triazolylene, Y is pyridin-2-y1 and R1 and R2 are as described above,
or a salt, particularly a pharmaceutically acceptable salt, a tautomer, or a
stereoi-
somer of said compound, or a salt, particularly a pharmaceutically acceptable
salt, of said tautomer or said stereoisomer.
Salts of the compounds according to the invention include all inorganic and
organic acid addition salts and salts with bases, especially all
pharmaceutically
acceptable inorganic and organic acid addition salts and salts with bases,
particularly all pharmaceutically acceptable inorganic and organic acid
addition
salts and salts with bases customarily used in pharmacy.
Examples of acid addition salts include, but are not limited to,
hydrochlorides,
hydrobromides, phosphates, nitrates, sulfates, salts of sulfamic acid,
formates,
acetates, propionates, citrates, D-gluconates, benzoates, 2-(4-hydroxybenzoyI)-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
33
benzoates, butyrates, salicylates, sulfosalicylates, lactates, maleates,
laurates,
malates, fumarates, succinates, oxalates, malonates, pyruvates, acetoacetates,
tartarates, stearates, benzensulfontes, toluenesulfonates, methanesulfonates,
trifluoromethansulfonates, 3-hydroxy-2-naphthoates, benzenesulfonates,
naphthalinedisulfonates, and trifluoroacetates.
Examples of salts with bases include, but are not limited to, lithium, sodium,
potassium, calcium, aluminum, magnesium, titanium, meglumine, ammonium
salts optionally derived from NH3 or organic amines having from 1 to 16 C-
atoms
such as e.g. ethylamine, diethylamine, triethylamine, ethyldiisopropylamine,
monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine,
dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine,
lysine, ethylendiamine, N-methylpiperindine and guanidinium salts.
The salts include water-insoluble and, particularly, water-soluble salts.
According to the person skilled in the art the compounds of formula (I)
according
to this invention as well as their salts may contain, e.g. when isolated in
crystalline form, varying amounts of solvents. Included within the scope of
the
invention are therefore all solvates and in particular all hydrates of the
compounds of formula (I) according to this invention as well as all solvates
and in
particular all hydrates of the salts of the compounds of formula (I) according
to
this invention.
The compounds according to the invention and their salts can exist in the form
of
tautomers. In particular, those compounds of the invention which contain a
pyrazole moiety for example can exist as a 1H tautomer, or a 2H tautomer, or
even a mixture in any amount of the two tautomers, or a triazole moiety for
example can exist as a 1H tautomer, a 2H tautomer, or a 4H tautomer, or even a
mixture in any amount of said 1H, 2H and 4H tautomers:
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
34
H
3 -,145
3 N
N=
H
I H-tautomer 2H-tautomer 4H-tautomer
The compounds according to the invention and the salts thereof include
stereoisomers. Each of the stereogenic centers present in said stereoisomers
may have the absolute configuration R or the absolute configuration S
(according
to the rules of Cahn, IngoId and Prelog). Accordingly, the stereoisomers (1S)
and
(1R) in case of a compound of formula (1a*)
R6
0 1 N
N R7
I\1 I
R4 (1a*)
and the salts thereof are part of the invention.
The invention further includes all mixtures of the stereoisomers mentioned
above
independent of the ratio, including the racemates.
Some of the compounds and salts according to the invention may exist in
different crystalline forms (polymorphs) which are within the scope of the
invention.
Furthermore, derivatives of the compounds of formula (I) and the salts thereof
which are converted into a compound of formula (I) or a salt thereof in a
biological
system (bioprecursors or pro-drugs) are covered by the invention. Said
biological
system is e.g. a mammalian organism, particularly a human subject. The
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
bioprecursor is, for example, converted into the compound of formula (I) or a
salt
thereof by metabolic processes.
The intermediates used for the synthesis of the compounds of claims 1-5 as
5 described below as well as their use for the synthesis of the compounds
of claims
1-5 are one further aspect of the present invention.
The compounds according to the invention can be prepared as follows:
xi As shown in reaction scheme 1 the compounds of formula (I), wherein ring
B and
the imidazole to which it is fused, R4 and R7 have the above mentioned
meanings and R6 is hydrogen or 1-4C-alkyl, can be obtained by a reductive
amination reaction of a corresponding compound of formula (III), wherein R has
the meaning ¨C(0)R6, with a piperidine derivative of formula (II), wherein R7
has
15 the above-mentioned meanings. The reductive amination can be carried out
according to standard procedures, for example by the use of NaBH(OAc)3 or
NaBH3CN in a suitable solvent exemplified by dimethylformamide (DMF) or
methanol or mixtures of methanol and DMF.
20 The piperidine derivatives of formula (II), wherein R7 has the above-
mentioned
meanings are known or can be prepared according to known procedures (they
may contain protecting group(s) in certain cases to protect other
functionalities
such as but not limited to NH functions).
25 The use of the compounds of formula (II) for the synthesis of the
compounds of
claims 1-5 is one aspect of the present invention.
Compounds of formula (III), wherein R has the meaning ¨C(0)H can be obtained
from corresponding compounds of formula (III), wherein R has the meaning -
30 C(0)0(1-4C-alkyl), in a one or two step procedure. The ester group is
selectively
reduced to the aldehyde group by methods known to the skilled person, for
example by the use of DIBALH under low temperature for example -80 to -60 C
in the one step procedure. Alternatively, the ester group is reduced to the
alcohol
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
36
group (-CH2OH) according to known procedures, for example by the use of
LiAIH4 or NaBH4, and then, the resulting alcohol is selectively oxidized to
the -
C(0)H group by methods known to the skilled person, for example with S03-
pyridine complex or Dess-Martin Periodinane, in the two step procedure.
Alternatively to the reaction sequence described above, the compounds of
formula (I), wherein ring B and the imidazole to which it is fused, R4 and R7
have
the above mentioned meanings and R6 is hydrogen or 1-4C-alkyl, can be
obtained by reaction of a corresponding compound of formula (111a), wherein X
is
a suitable leaving group, such as for example a halogen atom or a sulfonester,
with piperidine derivatives of formula (II), wherein R7 has the above-
mentioned
meanings. The reaction is preferably carried out in an inert solvent, such as
for
example DMF, at a temperature of from 60 to 100 C in presence of a base, such
as for example triethylamine.
Compounds of formula (111a), wherein X is a suitable leaving group, for
example a
halogen atom can be obtained from corresponding compounds of formula (III),
wherein R is ¨CH(R6)0H and R6 is hydrogen or 1-4C-alkyl, by a halogenation
reaction. Such a halogenation reaction can be accomplished, for example, by
the
use of PBr3 in dichloromethane.
Alternatively, compounds of formula (111a), wherein X is a suitable leaving
group,
for example a halogen atom can be obtained from corresponding compounds of
formula (III), wherein R is -CH2R6 and R6 is hydrogen or 1-4C-alkyl, by means
of
benzylic halogenation. Benzylic halogenation can, for example, be achieved by
the use of N-bromosuccinimide (NBS).
Compounds of formula (III), wherein R is ¨CH(R6)0H and R6 is hydrogen or 1-
4C-alkyl, can, for example, be obtained from corresponding compounds of
formula (III), wherein R is ¨C(0)R6, by methods known to the person skilled in
the art, for example by reduction with NaBH4 or LiAIH4.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
37
Alternatively, compounds of formula (III), wherein R is ¨CH(R6)0H and R6 is
hydrogen or 1-4C-alkyl, can be obtained from corresponding compounds of
formula (III), wherein R is ¨CH2R6, by means of benzylic oxidation, which can
be
achieved, for example, by the use of catalytic or equimolar amounts of Se02.
In a further alternative, compounds of formula (III), wherein R is ¨CH(1-4C-
alkyl)OH can be obtained from corresponding compounds of formula (III),
wherein
R is ¨C(0)H by the addition of a suitable metal organic reagent, such as, but
not
limited to Gringnard or Lithium reagents.
If necessary for the reactions in reaction scheme 1, for the synthesis of
compounds of formula (III), wherein ring B and the imidazole to which it is
fused
and R4 have the above mentioned meanings and R is ¨C(0)R6 or ¨CH(R6)0H,
these groups can be protected in some or all of the precursors by suitable
protecting groups known to the person skilled in the art. Compounds of formula
(III), wherein ring B and the imidazole to which it is fused and R4 have the
above
mentioned meanings and R is a protected ketone, aldehyde or alcohol group, can
be deprotected by art-known removal of the protecting groups to generate the
corresponding deprotected compounds.
Compounds of formula (III), wherein R has the meanings ¨C(0)0(1-4C-alkyl), ¨
C(0)R6,
¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl, can be obtained as
shown in reaction scheme 1 by cyclocondensation of compounds of formula (IV)
with compounds of formula (V), wherein R4 has the meanings given above, R
has the meanings ¨C(0)0(1-4C-alkyl),
¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl and X1 is a
suitable leaving group, such as for example a halogen or a sulfonate. This
reaction can be carried out for example in DMF at a temperature from 80 to
140 C.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
38
Reaction scheme 1:
R4 \_¨M R
(VIII) (VII)
0 =
R4 (VI) X1 R4 (V)
0
R4 R
(Xa) (IX)
R6
H,
N
H2 N
gs,N
(II) R7
I
(IV) I\1 I
R4 (III) R4
(I)
R6
X
\/R7
(II)
(111a)
R4
In the case that the substituent of ring B and the imidazole to which it is
fused,
which is described above as R1 or R3 is a halogen, preferably Cl, Br or I,
these
halogens can be transformed to other functionalities at this or a later stage
of the
overall synthesis. This transformation can be achieved for example by the
means
of catalyzed or uncatalyzed replacement of the halogen by certain reagents ex-
emplified but not limited to boronic acids, tin reagents, Gringnard reagents,
cya-
nide salts, alcohols or amines. Certain Cu or Pd complexes are examples of
cata-
lysts, which can be employed for these transformations.
As further shown in reaction scheme 1, compounds of formula (V), wherein R4
has the meanings given above, R has the meanings ¨C(0)0(1-4C-alkyl), ¨
C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl and X1 is a
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
39
suitable leaving group, such as for example a halogen or a sulfonate, can be
ob-
tained from the corresponding compounds of formula (VI) by procedures known
to the skilled person, for example by alpha-halogenation reaction of ketones
e.g.
using CuBr in suitable solvents such as a mixture of chloroform and
ethylacetate.
This can also lead to a concomitant cleavage of certain protecting groups,
which
are part of R, for example acetal protection groups.
Compounds of formula (VI), wherein R4 has the meanings given above and R
has the meanings ¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and
R6 is hydrogen or 1-4C-alkyl, can for example be synthesized starting from ni-
triles of formula (VII), wherein R has the meanings ¨C(0)0(1-4C-alkyl),
¨C(0)R6,
¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl, by means of addition
of a metal organic reagent of formula (VIII), wherein R4 has the above-
mentioned
meanings.
Alternatively, compounds of formula (VI), wherein R4 has the meanings given
above and R has the meanings ¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨
CH2R6 and R6 is hydrogen or 1-4C-alkyl, can be synthesized starting from com-
pounds of formula (Xa), wherein R4 has the above-mentioned meanings, by
means of addition of a metal organic reagent of formula (IX), wherein R has
the
meanings ¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hy-
drogen or 1-4C-alkyl. The metal organic reagent of formula (IX) can be a Grig-
nard or a lithium reagent; if necessary the addition of the metal organic
reagent is
followed by an oxidation reaction.
The oxidation reaction can be carried out by using reagents known to those
skilled in the art, for example the pyridine-S03 complex or Dess-Martin Perio-
dinane.
Compounds of formulae (VII), (VIII), (IX) and (Xa) are either commercially
avail-
able or can be prepared from commercially available compounds by methods
known to the person skilled in the art.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
An alternative synthesis route to compounds of formula (III) is described in
the
reaction scheme 2.
Compounds of formula (III), wherein ring B and the imidazole to which it is
fused
5 and R4 have the meanings mentioned above and R is ¨C(0)0(1-4C-alkyl), ¨
C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl, can be
obtained by reaction of a corresponding compound of formula (XI) with
compounds of formula (XIII), wherein X3 has the meaning of Cl, Br, I or -
OS(02)CF3. This reaction can, for example, be conducted with palladium
10 acetate, triphenyl phosphine and triethylamine in dioxan.
Compounds of formula (XIII), are either commercially available or can be
prepared from commercially available compounds by methods known to the
person skilled in the art.
Alternatively, compounds of formula (III), wherein ring B and the imidazole to
which it is fused and R4 have the meanings mentioned above and R is ¨
C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-
4C-alkyl, can be obtained by a transition metal catalysed C-C bond formation
of a
corresponding compound of formula (XII), wherein X2 is Cl, Br or I, with a
corresponding compound of formula (XIV), wherein R4-A is R4-B(OH)2, R4-B(0-
1-4C-alkly)2,
R4-BOCH(CH3) CH(CH3)0, R4ZnCI, R4ZnBr, R4ZnI or (1-4C-alky1)3SnR4
and R4 has the meaning mentioned above. This reaction can be carried out for
example by the use of tetrakis triphenylphosphine palladium in suitable
solvent
exemplified by dioxan or mixtures of ethanol in water at a temperature of from
60
to 100 C.
Compounds of formula (XIV) are either commercially available or can be
prepared from commercially available compounds by methods known to the
person skilled in the art.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
41
Compounds of the formula (XII), wherein ring B and the imidazole to which it
is
fused have the meaning mentioned above and R is ¨C(0)0(1-4C-alkyl), ¨
C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-4C-alkyl and X2 is Cl,
Br or I, can be obtained by a halogenation reaction of a corresponding
compound
of formula (XI) with, for example, N-bromosuccinimide (NBS) or N-
iodosuccinimide (NIS).
Compounds of formula (XI), wherein ring B and the imidazole to which it is
fused
and R is
¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or 1-
4C-alkyl, can be obtained by cyclocondensation of corresponding compounds of
formula (IV) with a corresponding compound of formula (XV), wherein X4 has the
meaning of a halogene or tosylate. This reaction can be carried out, for
example,
in DMF at temperature from 80-140 C.
Compounds of formula (XV), wherein R is ¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨
CH(R6)0H or
¨CH2R6 and R6 is hydrogen or 1-4C-alkyl and X4 is mesylate or toyslate, can be
prepared with a corresponding compound of formula (XVII) by treatment with
suitable reagents such as, but not limited, to sulfonylchlorides.
Compounds of the formula (XV), wherein R is ¨C(0)0(1-4C-alkyl), ¨C(0)R6, ¨
CH(R6)0H or
¨CH2R6 and R6 is hydrogen or 1-4C-alkyl and X4 is halogen, can be prepared
by a halogenation reaction of a corresponding compound of formula (XVI) with,
for example, Br2 or NBS in the case that the halogen is Br.
Compounds of formulae (XVI) or (XVII) are either commercially available or can
be prepared from commercially available compounds by methods known to the
person skilled in the art.
Compounds of the formula (III) in reaction scheme 2, wherein ring B and the
imi-
dazole to which it is fused and R4 have the meaning mentioned above and R is ¨
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
42
C(0)0(1-4C-alkyl), ¨C(0)R6, ¨CH(R6)0H or ¨CH2R6 and R6 is hydrogen or
1-4C-alkyl, can be transformed into compounds of formula (I) or into compounds
of formula (111a) and then to compounds of formula (I), as described above in
re-
action scheme 1.
If necessary for the reactions in reaction scheme 2, for the synthesis of com-
pounds of formula (III), wherein ring B and the imidazole to which it is fused
have
the meaning mentioned above and R is ¨C(0)R6 or ¨CH(R6)0H and R6 is hy-
drogen or 1-4C-alkyl, these groups can be protected in some or all of the
precur-
sors by suitable protecting groups known to the person skilled in the art. Com-
pounds of formula (III), in which R is a protected ketone, aldehyde or alcohol
group, can be deprotected by art-known removal of the protecting groups to gen-
erate the corresponding deprotected compounds.
Reaction scheme 2:
= R R R
0 0 0
__________________________ ).
(XVI) (XV) H--.0
X4 (XVII)
H2
(IV)
R
I R4-X3
(XII
1),1/4
(XI)
I R4 (III)
R R4(XIVA)
x2 (XU)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
43
One preferred aspect of the invention is the process for the preparation of
the
compounds of claims 1-5 according to the examples.
Optionally, compounds of the formula (I) can be converted into their salts,
or, op-
tionally, salts of the compounds of the formula (I) can be converted into the
free
compounds. Corresponding processes are customary for the skilled person.
It is known to the person skilled in the art that, if there are a number of
reactive
centers on a starting or intermediate compound, it may be necessary to block
one
or more reactive centers temporarily by protective groups in order to allow a
reaction to proceed specifically at the desired reaction center. A detailed
description for the use of a large number of proven protective groups is
found, for
example, in T. W. Greene, Protective Groups in Organic Synthesis, John Wiley &
Sons, 1999, 3rd Ed., or in P. Kocienski, Protecting Groups, Thieme Medical
Publishers, 2000.
The compounds according to the invention are isolated and purified in a manner
known per se, e.g. by distilling off the solvent in vacuo and recrystallizing
the
residue obtained from a suitable solvent or subjecting it to one of the
customary
purification methods, such as column chromatography on a suitable support ma-
terial.
Salts of the compounds of formula (I) according to the invention can be
obtained
by dissolving the free compound in a suitable solvent (for example a ketone
such
as acetone, methylethylketone or methylisobutylketone, an ether such as
diethyl
ether, tetrahydrofuran or dioxane, a chlorinated hydrocarbon such as methylene
chloride or chloroform, or a low molecular weight aliphatic alcohol such as
methanol, ethanol or isopropanol) which contains the desired acid or base, or
to
which the desired acid or base is then added. The acid or base can be employed
in salt preparation, depending on whether a mono- or polybasic acid or base is
concerned and depending on which salt is desired, in an equimolar quantitative
ratio or one differing therefrom. The salts are obtained by filtering,
reprecipitating,
precipitating with a non-solvent for the salt or by evaporating the solvent.
Salts
CA 02695251 2014-08-13
44
obtained can be converted into the free compounds which, in turn, can be con-
verted into salts. In this manner, pharmaceutically unacceptable salts, which
can
be obtained, for example, as process products in the manufacturing on an
industrial scale, can be converted into pharmaceutically acceptable salts by
processes known to the person skilled in the art.
Pure diastereomers and pure enantiomers of the compounds and salts according
to the invention can be obtained e.g. by asymmetric synthesis, by using chiral
starting compounds in synthesis and by splitting up enantiomeric and
diasteriomeric mixtures obtained in synthesis.
Enantiomeric and diastereomeric mixtures can be split up into the pure enantio-
mers and pure diastereomers by methods known to a person skilled in the art.
Preferably, diastereomeric mixtures are separated by crystallization, in
particular
fractional crystallization, or chromatography. Enantiomeric mixtures can be
separated e.g. by forming diastereomers with a chiral auxiliary agent,
resolving
the diastereomers obtained and removing the chiral auxiliary agent. As chiral
auxiliary agents, for example, chiral acids can be used to separate
enantiomeric
bases such as e.g. mandelic acid and chiral bases can be used to separate
enantiomeric acids via formation of diastereomeric salts. Furthermore,
diastereomeric derivatives such as diastereomeric esters can be formed from
enantiomeric mixtures of alcohols or enantiomeric mixtures of acids,
respectively,
using chiral acids or chiral alcohols, respectively, as chiral auxiliary
agents.
Additionally, diastereomeric complexes or diastereomeric clathrates may be
used
for separating enantiomeric mixtures. Alternatively, enantiomeric mixtures can
be
split up using chiral separating columns in chromatography. Another suitable
method for the isolation of enantiomers is the enzymatic separation.
As will be appreciated by persons skilled in the art, the invention is not
limited to the
examples and particular embodiments described herein, but covers all
modifications
of said embodiments as defined by the appended claims. Therefore, the scope of
the
claims should not be limited by the preferred embodiments set forth in the
examples,
but should be given the broadest interpretation consistent with the
description as a
whole.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
The following examples illustrate the invention in greater detail, without
restricting
it. Further compounds according to the invention, of which the preparation is
not
explicitly described, can be prepared in an analogous way.
5 The compounds, which are mentioned in the examples and the salts thereof
represent preferred embodiments of the invention as well as a claim covering
all
subcombinations of the residues of the compound of formula (I) as disclosed by
the specific examples.
10 The term "according to" within the experimental section is used in the
sense that
the procedure referred to is to be used "analogously to".
Examples
15 The following abbreviations are used: In the examples, m.p. stands for
melting
point, h or hrs for hour(s), min for minutes, conc. for concentrated, calc.
for calcu-
lated, fnd. for found, EF for elemental formula, MS for mass spectrometry, M
for
molecular ion in mass spectrometry, TLC: thin layer chromatography, HPLC for
high performance liquid chromatography, 1H-NMR for 1H nuclear magnetic reso-
20 nance spectroscopy (chemical shifts are reported as ppm against
tetramethylsi-
lane as internal standard, coupling constants J are reported in Hz), w/w for
weight
by weight, RT for room temperature (20-25 C), DCM for dichloromethane, THF
for tetrahydrofurane, DMSO for dimethylsulfoxide, DBU for 1,8-
diazabicyclo[5.4.0]undec-7-ene, Et0Ac for ethyl acetate, DIBAL for diisobuty-
25 laluminiumhydrid, DCM for dichloromethane, ACN for acetonitril and other
abbre-
viations have their meanings customary per se to the skilled person.
Intermediate A: 2-[3-(piperidin-4-yI)-1H-1, 2,4-triazol-5-yl] pyridine dihydro-
chloride (procedure described in U5401 1218 or W02005100344)
30 Step-I: pyridine-2-carbohydrazonamide
A solution of pyridine-2-carbonitrile 20g (192mM), hydrazine hydrate (3 eq.)
in
ethanol (50 ml) is stirred at room temperature for 18 hrs. Reaction mass is
then
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
46
diluted with water, extracted with ethyl acetate, dried over anhydrous sodium
sul-
phate and concentrated under vacuum to yield desired compound.
MS (M+1): 137.28
1H NMR (300MHZ, CDCI3): 6 8.53 (d, 1H, J=8 & 2.3 Hz), 8.02 (d, 1H, J=7.8&
2.1 Hz), 7.72 (t, 1H, J=8.2 & 2Hz ), 7.29 (t, 1H, J=8.4 & 2.1 Hz), 5.42 (bs,
2H),
4.60 (bs, 2H).
Step-II: tert-butyl 4-({(2Z)-2-[amino (pyridin-2-y1) methylidene] hydrazinyl}
carbonyl) piperidine-1-carboxylate
To as solution of 1-(tert-butoxycarbonyl) piperidine-4-carboxylic acid 37g
(167
mM) in dichloromethane (300m1) is added carbonyl diimidazole (1 eq.) in small
portion over a period of 30 min. pyridine-2-carbohydrazonamide is then added
to
reaction mixture and stirred at room temperature for 3 hrs. Dichloromethane is
evauporated and reaction mass is then stirred in water for 30 min. solid
precipi-
tated is filtered and dried to afford the desired compound.
MS (M+1): 348.07
1H NMR (300MHZ, CDCI3): 610.75 (s, 1H), 8.56 (d, 1H, J=4.5Hz), 8.10 (d, 1H,
J=8.3Hz), 7.75 (dt, 1H, J=8.2 & 1.3Hz), 7.34 (dt, 1H, J=7.9 & 1.5Hz), 4.18
(bs,
2H), 3.46 (s, 1H), 2.88 (t, 2H), 1.91 (m, 2H), 1.72 (m, 4H), 1.47 (s, 9H).
Step-Ill: tert-butyl 4-[5-(pyridin-2-yI)-1H-1, 2,4-triazol-3-yl] piperidine-1-
carboxylate
Tert-butyl 4-({(2Z)-2-[amino (pyridin-2-y1) methylidene] hydrazinyl} carbonyl)
piperidine-1-carboxylate 45g (129 mM) obtained in step II is melted at 220 C
under nitrogen atmosphere for 1 hr. Reaction is then cooled to 150 C and added
ethanol till solid get dissolved. Ethanol is then evapourated to get desired
compound.
MS (M+1): 330.5
1H NMR (300MHZ, DMS0): 6 9.11 (s, 1H), 8.74 (dd, 1H, J= 4.8 & 1.3 Hz), 8.17
(dt,2H, J=8.2 & 2.1 Hz), 7.66 (dt, 1H, J=8.0 & 1.3 Hz), 3.34 (m, 2H), 3.18 (m,
1H), 3.06 (m, 2H), 2.20 (m, 2H), 1.99 (m, 2H), 1.28 (s, 9H).
Step-IV: 2-[3-(piperidin-4-yI)-1H-1, 2,4-triazol-5-yl] pyridine
dihydrochloride
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
47
To a solution of tert-butyl 4-[5-(pyridin-2-yI)-1H-1, 2,4-triazol-3-yl]
piperidine-1-
carboxylate 39g (iii mM) in methanol 50 is added 100 ml solution of HCI in
dioxane and stirred at room temperature for 3 hrs. Solid precipitated is then
filtered and washed with cold acetonitrile to obtained 2-[3-(piperidin-4-yI)-
1H-1,
2,4-triazol-5-yl] pyridine dihydrochloride as white solid.
1H NMR (300MHZ, DMS0): 6 9.11 (s, 1H), 8.97 (s, 1H), 8.74 (dd, 1H, J= 4.8 &
1.3 Hz), 8.17 (dt,2H, J=8.2 & 2.1 Hz), 7.66 (dt, 1H, J=8.0 & 1.3 Hz), 3.34 (m,
2H),
3.18 (m, 1H), 3.06 (m, 2H), 2.20 (m, 2H), 1.99 (m, 2H)
Intermediate B: 4[3-(piperidin-4-y1)-1H-1,2,4-triazol-5-ylipyridine
Prepared according to intermediate A.
Intermediate C: 2-methyl-6- [3-(piperidin-4-yI)-1H-1, 2,4-triazol-5-
yl]pyridine
Prepared according to intermediate A.
Intermediate D: 5-methyl-2- [3-(piperidin-4-yI)-1H-1, 2,4-triazol-5-
yl]pyridine
Prepared according to intermediate A.
Intermediate E : 5-chloro-2[3-(piperidin-4-y1)-1H-1,2,4-triazol-5-ylipyridine
Prepared according to intermediate A.
Intermediate F: 2[3-(piperidin-4-y1)-1H-1, 2,4-triazol-5-ylipyrimidine
Prepared according to intermediate A.
Intermediate G: 445-(1,3-thiazol-2-y1)-1H-1, 2,4-triazol-3-ylipiperidine
Prepared according to intermediate A.
Intermediate H : 4[5-(furan-2-y1)-1H-1,2,4-triazol-3-ylipiperidine
Prepared according to intermediate A.
Intermediate I: 4[5-(thiophen-2-y1)-1H-1, 2,4-triazol-3-yl] piperidine
Prepared according to intermediate A.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
48
Intermediate J : 445-(1H-pyrrol-2-y1)-1H-1,2,4-triazol-3-ylipiperidine
Step-I : 1H-pyrrole-2-carbohydrazonamide
A solution of 1H-pyrrole-2-carbonitrile lOg (108 mM) and sodium methoxide (1
eq) in ethanol (20 ml) and stirred for 10 min. Hydrazine hydrate (3 eq.) is
then
added and resulting reaction mixture is stirred at room temperature for 18
hrs.
Reaction mass is then diluted with water, extracted with ethyl acetate, dried
over
anhydrous sodium sulphate and concentrated under vacuum to yield desired
compound.
Step II, III, IV are similar to intermediate A.
Intermediate K: 2[3-(piperidin-4-y1)-IH-pyrazol-5-ylipyridine
Prepared according to intermediate A.
Intermediate L: 2[3-(piperidin-4-y1)-I,2,4-oxadiazol-5-ylipyridine
Intermediate L is prepared according to intermediate A.
Intermediate M : 4-(5-phenyl-1H-1,2,4-triazol-3-yl)piperidine
Intermediate M is prepared according to intermediate J
Example 1: 3-phenyl-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
Step 1: 144-(dimethoxymethyl)pheny1]-2-phenylethanol
A mixture of Mg turnings 2.4g (0.1mol) and 2m11-bromo-4-
(dimethoxymethyl)benze (0.012mol) in THF (10m1) is heated under nitrogen at-
moshere until the reaction starts. Subsequently additional 1-bromo-4-
(dimethoxymethyl)benze 14.71m1 (0.088 mol) dissolved in 30m1 THF is added
slowly and the reaction refluxed for lh mixture to complete formation of the
Gringnard reagent. A solution of 11.70m1 phenylacetaldehyde (0.1mol) in 100m1
THF is added at to 0 C and the reaction refluxed for 2h upon completion of the
addition. The mixture is worked up by pouring into saturated aqueous NH4C1 and
extraction with ethyl acetate. The combined organic layers are washed with
brine,
CA 02695251 2014-08-13
49
dried over MgSO4 and the solvent is evaporated under reduced pressure. The
brown-black oily product is used for the next step without purification.
Step 2: 144-(dimethoxymethyl)pheny1]-2-phenylethanone
29.16g (0.183mol) sulfur trioxide pyridine complex is added in portions to a
solu-
tion of 33g 144-(dimethoxymethyl)pheny1]-2-phenylethanol in dichloromethane
(540m1), DMSO (140m1) and triethylamine (25.5m1) at 10 C. The mixture is
slowly
warmed to room temperature and stirred for 2h. Water is added and the organic
phase is separated, washed with lmol/IHCI, 3 times with 5% w/w sodium thiosul-
th fate solution and saturated NaCI. The combined organic phases are dried
over
sodium sulfate and the solvens is evaporated. The residue is purified on a
silica
gel column chromatography (n-Hexan/Et0Ac).
MS (M+1): 271
Characteristic 1H NMR (300MHz, dDMS0) signals: 8.1 ppm (d, 2H); 7.6 ppm (d,
2H); 5.4 ppm (s, 1H), 4.3 (s, 2H)
Step 3: 4-[bromo(phenyl)acetyl]benzaldehyde
A solution of 3.0g 114-(dimethoxymethyl)pheny11-2-phenylethanone (0.011mol) in
60mlethyl acetate and 60m1 chloroform is heated to reflux. Powdered cupric
bromide 4.96g (0.022mol) is added over 2h period in small portions under nitro-
gen atmosphere. Refluxing is continued for lh until the green color and dark
cu-
pric bromide disappears, the reaction is then mixture cooled to room
temperature
and filtered though celitem. The solvens is evaporated and the residue
purified on
silica gel.(n-Hexan/Et0Ac).
MS (M+ -Br): 223
Characteristic 1H NMR (300MHz, dDMS0) signals: 10.0ppm (s, 1H); 8.3 ppm (d,
2H); 8.0 ppm (d, 2H); 7.2 ppm (s, 1H)
Step 4: 4-(3-phenylimidazo[1,2-a]pyrimidin-2-yl)benzaldehyde
4.6g 44bromo(phenyl)acetyl]benzaldehyde and 1.64 g pyrimidin-2-amine are
stirred in 140 ml DMF at 90 C for 4h. The DMF is evaporated and the residue
suspended in ethylacetat over night, collected by filtration.
MS (M+1): 300
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Characteristic 1H NMR (400MHz, dDMS0) signals: 1Oppm (s,1H), 7.0 ppm (1H)
Step 5: 3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1H-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
5 0.14m1triethylamine is added to a solution of 156mg 4-(3-
phenylimidazo[1,2-
a]pyrimidin-2-yl)benzaldehyde in 10m1 methanol. To this solution a solution of
140mg 2-(5-Piperidin-1,2,4]triazol-3-y1)-pyridine2HC1 (Intermediate A prepared
from tert-butyl 4-(hydrazinocarbonyl)piperidine-1-carboxylate and pyridine-2-
carbonitrile according to a procedure described in US4011218 or
10 W02005100344) is added, followed by 0.07m1 glacial acetic acid and 198mg
NaBH(OAc)3. The resulting mixture is stirred at room temperature. Three addi-
tional portions of 198mg NaBH(OAc)3 are added after 2, 4 and 20 hours. The sol-
yens is removed by evaporation after 24h and the residue purified by chromatog-
raphy on silica gel (dichlormethan/ methanol).
15 MS (M+1): 513
Characteristic 1H NMR (400MHz, dDMS0) signals: 8.6ppm (1H); 7ppm (1H);
3.5ppm (s, 2H)
Example 2: 3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1H-1,2,4-triazol-3-yl)piperidin-1-
20 yl]methyl}phenyl)imidazo[1,2-a]pyridine
Example 2 is prepared in a manner according to example 1 by using pyridin-2-
amine in step 4.
MS (M+1): 512
Characteristic 1H NMR (400MHz, dDMS0) signals: 8.7 ppm (1H); 6.9 ppm (1H);
25 3.5ppm (s, 2H)
Example 3: 7-pheny1-6-(4-{[4-(5-pyridin-2-y1-1H-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-b][1,2,4]triazine
Example 3 is prepared in a manner according to example 1 by using 1,2,4-
triazin-
30 3-amine in step 4.
MS (M+1): 514
Characteristic 1H NMR (400MHz, dDMS0) signals: 8.0 ppm (1H); 7.9 ppm (1H);
3.5 ppm (s, 2H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
51
Example 4: 3-phenyl-2-(4-{[4-(3-pyrazin-2-y1-1,2,4-oxadiazol-5-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner similiar to 1 by using commercially avail-
able 2-(5-piperidin-4-y1-1,2,4-oxadiazol-3-yl)pyrazine in the last step.
Analytical data
MS (M+1): 515
Characteristic 1H NMR (400Hz, dDMS0) signals: 9.2 ppm (s, 1H); 7.1ppm (dd,
1H); 3.5 ppm (s, 2H).
Example 5: 3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyridine-8-carbonitrile
This example is prepared in a manner according to example 12 by using 2-
aminonicotinonitrile in the first step.
MS (M+1): 537.23
1H NMR (CD30D): 6 8.675(dd, J = 7.2 Hz, 1.2 Hz, 1H), 8.073(d, J = 7.8 Hz, 1H),
7.945-7.87(m, 2H), 7.65(d, J = 8.1 Hz, 2H), 7.60-7.56(m, 2H), 7.49-7.46(m,
3H),
7.444-7.419 (m, 3H), 7.345 (d, J= 8.4 Hz, 2 H), 7.010 (t, 1H), 3.75(s, 2H),
3.16-
3.12(m, 2H), 3.01-2.91(m, 1H), 2.48-2.41 (m, 2H), 2.15-1.96(m, 3H).
Example 6: 6-bromo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Step 1: 4-(6-bromo-3-phenylimidazo[1,2-a]pyridin-2-yl)benzaldehyde
3.3mMol (2-Bromo-2-phenyl-ethanoyI)-benzaldehyde and 1.2 equivalents of 5-
Bromo-pyridin-2-ylamine are dissolved in 10m1 DMF. The reaction mixture is
heated at 100 C for 18 hours. The reaction mixture is cooled and water is
added.
It is then extracted with ethyl acetate. The organic layer is dried and concen-
trated. The crude is purified by column chromatography to yield the desired
com-
pound.
MS (M+1): 377.27, 379.27.
1H NMR (CDCI3): 6 9.950(s, 1H), 8.1(dd, J= 1.8Hz, 0.9Hz, 1H), 7.86-7.76(m,
4H), 7.72(dd, J = 9.3Hz, 0.6Hz, 1H), 7.6-7.54(m, 5H), 7.5 (dd, J = 9.3Hz,
1.8Hz,
1H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
52
Step 2: [4-(6-bromo-3-phenylimidazo[1,2-a]pyridin-2-yl)phenylimethanol
To the stirred solution 4-(6-bromo-3-phenylimidazo[1,2-a]pyridin-2-
yl)benzaldehyde (0.795 mM) in 15mL of methanol is added NaBH4 (1.5 eq) at 0
C and the reaction is allowed to stir at room temperature for 1 hr. The
reaction is
concentrated and quenched with water. The precipitated solid is filtered and
dried
to yield the desired product.
MS (M+1): 379.27, 381.27.
1H NMR (DMSO-d6): 6 8.1(dd, J= 1.8Hz, 0.6Hz, 1H). 7.7-7.525(m, 8H), 7.5(dd, J
= 8.4Hz, 1.8Hz, 1H), 7.2 (d, J = 8.4Hz, 2H), 5.4 (bs, 1H), 4.5 (s, 2H).
Step 3: 6-bromo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
To the stirred solution of 0.52 mM of [4-(6-bromo-3-phenylimidazo[1,2-
a]pyridin-2-
yl)phenyl]methanol in 15mL of dichlormethane is added methanesulfonyl chloride
(1.1 eq) at 0 C followed by triethylamine (1.5 eq). The reaction mixture is
allowed
to stir at room temperature for 3h. The reaction is quenched with water and ex-
tracted with DCM. The organic layer is dried and concentrated. It is then
taken up
in the next reaction without further purification. The crude is dissolved in
5mL of
DMF. To this solution 2-(5-Piperidin-[1,2,4]triazol-3-y1)-pyridine2HCI (leg),
triethylamine (4 eq) are added. The reaction mixture is heated at 80 C for 3h.
The
reaction mixture is quenched with water and extracted with ethyl acetate. The
organic layer is dried and concentrated. The crude obtained is triturated with
pen-
tane to obtain the desired compound.
MS (M+1): 590.13, 592.07.
1H NMR (DMSO-d6): 6 14.2-14.0 (bs, 1H), 8.7(d, J= 4.2Hz 1H), 8.1-7.9 (m, 3H),
7.7-7.5 (m, 8H), 7.4(dd, J = 9.3Hz, 1.8Hz, 2H), 7.2 (d, J = 8.4Hz, 2H), 3.5(s,
2H),
2.8 (d, 2H) 2.8-2.7(m, 1H), 2.1(t, 2H), 2.0 (t, 2H), 1.8(t, 2H).
Example 7: 6-chloro-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
This compound is prepared according to example 6 by using 5-chloropyridin-2-
amine in step1.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
53
MS (M+1 ): 546.20,548.20.
1H NMR (CDCI3): 6 8.7(s, 1H). 8.2 (d, J = 7.5Hz, 1H), 8.0 (d, J = 1.2Hz, 1H),
7.8
(t, J= 7.5Hz, 1H), 7.6-7.5(m, 6H), 7.5(dd, J= 7.8Hz, 2.1Hz, 2H), 7.2 (d, J=
8.1 Hz, 2H), 7.3 (s, 1H), 7.2 (dd, J = 9.3Hz, 1.8Hz, 1H), 3.5 (s, 2H), 3.0-2.8
(m,
3H), 2.1-1.9 (m, 6H)
Example 8: 8-methyl-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
This compound is prepared according to example 6 by using 3-methylpyridin-2-
amine in step1.
MS (M+1): 526.27.
1H NMR (CDCI3): 6 8.7 (d, J = 3.3Hz, 1H), 8.2(d, J = 7.8Hz, 1H), 7.9-7.7(m,
2H),
2.2-1.9(m, 7H), 7.6(d, J = 8.1Hz, 1H), 7.5-7.5(m, 5H), 7.3(t, J = 5.7Hz, 1H),
7.2(d,
J = 8.1 Hz, 3H), 7.0(d, J = 6.9Hz, 1H), 6.7(t, J = 6.9Hz, 1H), 3.5(s, 2H), 3.0-
2.8 (m,
2H), 2.7(s, 3H)
Example 9: 3-(4-fluoropheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Stepl: 443-(4-fluorophenyl)imidazo[1,2-a]pyridin-2-ylibenzaldehyde
0.46g (1.5 mM) 4-(3-bromoimidazo[1,2-a]pyridin-2-yl)benzaldehyde (prepared as
described by Sundberg et al.; J. Heterocyclic Chem., 25, 129, 1988) is
dissolved
in 5mL of toluene. To this mixture is added 4-fluorphenyl boronic acid (1.5
eq)
followed by tetrakis triphenylphosphine palladium (0) 10% by wt, K2003 (3 eq)
and ethanol water mixture (4mL). The reaction mixture is heated at 90 C for 4-
6
h. The reaction is cooled to room temperature and 20mL of water is added and
the reaction mixture is extracted with ethyl acetate. The organic layer is
dried and
concentrated. The crude is purified by flash column chromatography to obtain
the
desired compound.
MS (M+1): 317.33.
1H NMR(DMSO-d6): 6 9.9 (s, 1H), 8.0(d, J= 6.9Hz,1H), 7.85(d, J= 6.6Hz, 2H),
7.8(d, J = 8.4Hz, 2H), 7.45(d, J = 9.3Hz, 1H), 7.6(m, 2H), 7.45(m, 2H),
7.35(m,
1H),6.9(dt, J = 6.9Hz, 1.2Hz, 1H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
54
Step 2: 3-(4-fluoropheny1)-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
To a solution of 0.1g (0.31mM) 443-(4-fluorophenyl)imidazo[1,2-a]pyridin-2-
yl]benzaldehyde in 5mL of THF is added triethylamine (2eq). The reaction
mixture
is stirred for 5 minutes. To this solution is added 2-(5-Piperidin-
[1,2,4]triazol-3-y1)-
pyridine*2HCI (1.5 eq) followed by acetic acid (2.5eq). The reaction mixture
is
stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq) over a
period
of 40 minutes. The reaction mixture is stirred overnight. The reaction mixture
is
quenched with methanol and concentrated. The residue obtained is taken up in
chloroform and washed with water, dried and concentrated. The crude is
purified
on a flash column chromatography to obtain the desired copound.
MS (M+1): 530.27.
1H NMR(CD30D): 6 8.7 (d, 1H), 8.1(m, 2H), 7.9(dt, J = 1.8Hz, 7.8Hz, 1H),7.6-
7.7(m, 3H), 7.2-7.5(m, 8H), 6.9(dt, J = 6.9Hz, 1.2Hz, 1H), 3.9(s, 2H), 3.0(m,
1H),
2.7(t, 2H), 2.1(m, 4H), 2.0(m,2H).
Example 10: 5-phenyl-6-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[2,1-b][1,3]thiazole
Step1: methyl 4-(phenylacetyl)benzoate:
33g methyl-4-(1-hydroxy-2-phenylethyl)benzoate (prepared as described by Berk
et al.; J. Org. Chem. 1988, 53, 5791) is dissolved in 200mL of
dichloromethane.
The solution is cooled to 0-10 C. To this is added DMSO (10 eq) and triethyl-
amine (1.5 eq) followed by addition of 1.5 eq of pyridine-sulphur trioxide-
com-
plex. The reaction is stirred overnight at RT 200m1 of water is added. Then
ethyl
acetate is added and the organic layer is separated, dried, and concentrated
to
obtain the crude compound. It is purified by column chromatography to obtain
the desired compound.
1H NMR (DMSO-d6) 6 8.15(d, J= 8.4Hz, 2H), 8.06(d, J= 8.4Hz, 2H), 7.40-
7.19(m, 5H), 4.44(s, 2H), 3.87(s, 3H).
Step 2: methyl 4-[bromo(phenyl)acetyl]benzoate: methyl 4-
(phenylacetyl)benzoate (3.8g, 14.9 mM) is dissolved in 300mL of ethyl
acetate/chloroform mixture. The reaction mixture is refluxed and cupric
bromide
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
(2 eq) is added in portions over a period of 2h while constantly purging with
nitro-
gen. The reaction mixture is further refluxed for 4h. The reaction mixture is
cooled, filtered and evaporated. The crude product is purified by column
chroma-
tography.
5 1H NMR (DMSO-d6) 6 8.18(d, J= 8.1Hz, 2H), 8.05(d, J= 7.5Hz, 2H), 7.6-
7.3(m,
5H), 7.21(s, 1H), 3.87(s, 3H).
Step 3: methyl 4-(5-phenylimidazo[2,1-13][1,3]thiazol-6-yl)benzoate
0.7g (2.1 mM) of methyl 4-[bromo(phenyl)acetyl]benzoate and 2-aminothiazole
10 (2.5 eq) are stirred in 20m1DMF at 90 C for 6h. The DMF is evaporated
and the
crude product is purified by column chromatography.
MS (M+1): 335.27.
1H NMR (DMSO-d6): 6 7.9(d, J = 8.4Hz, 2H), 7.7(d, J = 4.5Hz,1H), 7.6(d, J =
8.7Hz, 2H), 7.4-7.6(m, 5H), 7.3(d, J = 4.5Hz, 1H), 3.8 (s, 3H).
Step 4: [4-(5-phenylimidazo[2,1-13][1,3]thiazol-6-yl)phenylimethanol:
0.12g (0.36 mM) of methyl 4-(5-phenylimidazo[2,1-b][1,3]thiazol-6-yl)benzoate
is
dissolved in 8mL of dry THF. To this is added lithium alumium hydride (5 eq)
at
0 C. The reaction mixture is brought to RT and stirred for 2h. It is then
quenched
with sat. solution of sodium sulphate and filtered. The filtrate is evaporated
and
extracted in ethyl acetate. The organic layer is dried and concentrated to
obtain
the desired compound .
MS (M+1): 307.27.
1H NMR (DMSO-d6) 6 7.7(d, J = 4.5Hz, 1H), 7.5-7.4(m, 7H), 7.3(d, J = 4.5Hz,
1H), 7.2(d, J= 8.1Hz, 2H), 5.2(t, J= 5.7Hz, 1H), 4.5(d, J= 5.7Hz, 2H).
Step 5: 5-phenyl-6-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[2,1-b][1,3]thiazole
To the stirred solution of 0.12g (0.39mM) of [4-(5-phenylimidazo[2,1-
b][1,3]thiazol-6-yl)phenyl]methanol in 5mL of dichlormethane is added
methansul-
fonyl chloride (1.5 eq) at 0 C followed by triethylamine (2.5 eq). The
reaction mix-
ture is allowed to stir at room temperature for 3h. The reaction is quenched
with
water and extracted with DCM. The organic layer is dried and concentrated. It
is
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
56
then taken up in the next reaction without further purification. The crude
product
is dissolved in 4mL of DMF. To this is added 2-(5-Piperidin-[1,2,4]triazol-3-
y1)-
pyridine*2HCI (1.2eq), NaHCO3 (4 eq). The reaction mixture is stirred at RT
for
12h and then heated at 80 C for 3h. The reaction mixture is quenched with
water
and extracted with ethyl acetate. The organic layer is dried and concentrated.
The
crude obtained is converted to its HCI salt by addition of ethereal HCI. The
solid
obtained is washed with acetone/methanol to obtain the desired compound.
MS (M+1): 518.2.
1H NMR (D20): 6 8.7(d, J = 5.7Hz, 1H), 8.5(t, J = 8.1Hz, 1H), 8.4(d, J =
7.8Hz,
1H), 8.0-7.9(m, 1H), 7.8(d, J = 4.5Hz, 1H), 4.3(s, 2H), 3.6(br d, J = 12Hz,
2H),
7.6-7.40(m, 10H), 3.4-3.2(m, 1H), 3.2-3.1(m, 2H), 2.1-1.9(m, 2H), 2.3(br d, J=
12.6, 2H).
Example 11: 5-phenyl-6-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[2,1-b][1,3,4]thiadiazole
Step 1: methyl 4-(5-phenylimidazo[2,1-13][1,3,4]thiadiazol-6-yl)benzoate
0.7g (2.1mM) methyl 4-[bromo(phenyl)acetyl]benzoate (prepared as described
under example 10) and 2-aminothiadiazole (2 eq) are stirred in 15m1 DMF at
90 C for 6h. The DMF is evaporated and the residue suspended in ethylacetat
over night. The desired product is collected by filtration.
MS (M+1): 336.20.
1H NMR (DMSO-d6): 6 9.2(s, 1H), 7.91(d, 2H, J= 7.2Hz), 7.7(d, 2H, J= 6.9 Hz),
7.5-7.4(m, 5H), 3.8(s, 3H).
Step 2: [4-(5-phenylimidazo[2,1-13][1,3,4]thiadiazol-6-yl)phenylimethanol:
methyl 4-(5-phenylimidazo[2,1-b][1,3,4]thiadiazol-6-yl)benzoate (0.25g, 0.77
mM)
is dissolved in 25mL DCM. To this is added DIBAL (5 eq) at RT. The reaction
mixture is stirred for 20 min. The reaction mixture is diluted with DCM and
the
organic layer is washed with water. The organic layer is dried and
concentrated to
obtain the desired compound as a white solid.
MS (M+1): 308.20.
1HNMR(CD30D): 6 9.02(s, 1H), 7.58-7.50(m, 4H), 7.43 -7.38(m, 3H), 7.32(d, 2H,
J = 9Hz), 4.6(s, 2H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
57
Step 3: 5-phenyl-6-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[2,1-b][1,3,4]thiadiazole:
To the stirred solution of 0.21g (0.684 mM) of [4-(5-phenylimidazo[2,1-
b][1,3,4]thiadiazol-6-yl)phenyl]methanol in 15 mL of dichlormethane is added
methansulfonyl chloride (1.5 eq) at 0 C followed by triethylamine (2.0 eq).
The
reaction mixture is allowed to stir at room temperature for 3h. The reaction
is
concentrated. It is then taken up in the next reaction without further
purification.
The crude product is dissolved in 10mL of DMF. To this is added 2-(5-Piperidin-
[1,2,4]triazol-3-yl)pyridine*2HCI (2.5 eq), triethylamine (3 eq). The reaction
mix-
ture is heated at 80 C for 8h. The reaction mixture is quenched with water
and
extracted with ethyl acetate. The organic layer is dried and concentrated. The
crude product is purified by cc to obtain the desired compound.
MS (M+1): 519.27.
1HNMR(0D013): 6 8.67(d, 1H, J= 4.2Hz), 8.5(s, 1H), 8.1(d, 1H, J= 7.8Hz),
7.8(dt,
1H, J =7.8, 8.1Hz), 7.66-7.62(m, 4H), 7.4-7.2(m, 6H), 3.59 (s, 2H), 3.0(d,
2H),
2.9(t, 1H), 2.1-1.9(m, 6H).
Example 12: 6-bromo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
Step 1: 4-(6-bromo-3-phenylimidazo[1,2-a]pyrimidin-2-yl)benzaldehyde
0.15g (0.5mM) of 4-[bromo(phenyl)acetyl]benzaldehyde and 2.0 equivalents of 5-
bromopyrimidin-2-amine are dissolved in 6m1DMF. The reaction mixture is
heated at 90 C overnight. The reaction mixture is cooled and water is added.
It is
then extracted with ethyl acetate. The organic layer is dried and
concentrated.
The crude product is purified by column chromatography to obtain the desired
compound.
MS (M+1): 378.27, 380.27.
1H NMR(DMSO-d6): 6 10.0 (s, 1H), 8.7 (d, 1H, J= 2.4Hz), 8.6(d, 1H, J= 2.4Hz),
7.9(d, 2H, J = 8.7Hz), 7.8(d, 2H, J = 8.4Hz), 7.5-7.6 (m, 5H).
Step 2: 6-bromo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
58
To a solution of 0.044g (0.116mM) 4-(6-bromo-3-phenylimidazo[1,2-a]pyrimidin-
2-yl)benzaldehyde in 3mL of THF is added triethylamine (2eq). The reaction mix-
ture is stirred for 5 minutes. To this solution is added 2-(5-Piperidin-
[1,2,4]triazol-
3-y1)-pyridine*2HCI (1.5 eq) followed by acetic acid (2.5eq). The reaction
mixture
is stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq) over a
pe-
riod of 40 minutes. The reaction mixture is stirred overnight. The reaction
mixture
is quenched with methanol and concentrated. The residue obtained is taken up
in
chloroform and washed with water, dried and concentrated. The crude is
purified
on a flash column chromatography to obtain the desired compound.
MS (M+1): 591.27, 593.13.
1H NMR(CDCI3): 6 8.63(d, 1H, J = 4.8Hz), 8.61(d, 1H, J = 2.4Hz), 8.57(d, 1H, J
=
2.4Hz), 8.1(d, 1H, J= 7.8Hz), 7.9(t, 1H, J= 7.5Hz, 1.9 Hz), 7.59-7.61 (m, 5H),
7.49-7.52 (m, 2H), 7.4(t, 1H), 7.3(d, 2H, J= 8.4Hz), 3.7(s, 2H), 3.1(d, 2H),
2.9(t,
1H), 2.4(t, 2H), 2.1(d, 2H), 2.0(t, 2H).
Example 13: 8-methoxy-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Example13 is prepared according to example 12.
MS (M+1): 542.20.
1H NMR(CD30D): 6 8.64(d, 1H, J = 4.2 Hz), 8.06(d, 1H, J = 8.1Hz), 7.91(dt, 1H,
J
= 7.8, 7.5Hz), 7.5-7.7 (m, 6H), 7.42-7.44 (m, 3H), 7.33(d, 2H, J = 8.1 Hz),
6.82(t,
1H), 6.75 (d, 1H, J = 7.5Hz), 4.06(s, 3H), 3.83(s, 2H), 3.1(q, 2H), 3.03(m,
1H),
2.57(t, 2H), 2.12(d, 2H), 2.02(t, 2H).
Example 14: 3-(3-fluoropheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Example 14 is prepared according to example 9.
MS (M+1): 530.20.
1H NMR(DMSO-d6): 6 8.7(s, 1H), 8.07(d, J= 6.9Hz, 1H), 8.0(d, J= 7.2Hz, 1H),
8.0 (m, 1H) 7.7-7.6(m, 2H), 7.5(d, J = 8.1Hz,2H), 7.4-7.2(m, 7H), 6.9(t,1 H),
3.5(s,2H), 2.9(d, 2H), 2.6(m,1H), 2.1(t, 2H), 1.9(d, 2H), 1.8(t, 2H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
59
Example 15: 3-(4-methoxypheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Example 15 is prepared according to example 9 by using 4-methoxyphenyl bo-
ronic acid in step1.
MS (M+1): 542.27.
1H NMR(CD30D): 6 8.7 (d, 1H), 8.1(m, 2H), 7.9(dt, J = 1.8Hz, 7.8Hz, 1H), 7.6-
7.7(m, 3H), 7.2-7.5(m, 8H),6.9(dt, J= 6.9Hz, 1.2Hz, 1H), 4.1(s, 2H),3.9(s,
3H),
3(m, 1H), 2.7(t, 2H), 2.1(m, 4H), 2.0(m, 2H).
Example 16: 3-pyridin-4-y1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Step1: 4-(3-pyridin-4-ylimidazo[1,2-a]pyridin-2-yl)benzaldehyde
0.2g (0.9 mM) 4-imidazo[1,2-a]pyridin-2-ylbenzaldehyde (prepared as described
by Sundberg et al.; J. Heterocyclic Chem., 25, 129, 1988) is dissolved in 5mL
of
Dioxane. To this is added 05003 (1.1 eq), palladium acetate (8mol %),
triphenyl
phosphine (16mol %), triethylamine (2 eq) and 4-bromo-pyridine(1.4 eq). The re-
action mixture is heated (microwave) for 45 min at 100 C. The reaction
mixture is
cooled to RT, diluted with DCM. The organic layer is washed with water, dried
and concentrated. The crude product purified by column chromatography to ob-
tam n the desired compound.
MS (M+1): 300.33.
1H NMR(DMSO-d6): 6 10(s, 1H), 8.8(d, J= 4.2Hz, 2H), 8.3(d, J= 6.9Hz, 1H),
7.9(d, J= 8.4Hz, 2H), 7.7(d, J=8.1Hz, 2H), 7.6(d, J=5.7 Hz, 2H), 7.73(s, 1H),
7.4(m, 1H), 7(dt, J= 6.9Hz, 1.2Hz, 1H).
Step 2: 3-pyridin-4-y1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyridine
To a solution of 0.05g (0.16mM) 4-(3-pyridin-4-ylimidazo[1,2-a]pyridin-2-
yl)benzaldehyde in 5mL of THF is added triethylamine (2eq). The reaction mix-
ture is stirred for 5 minutes. To this solution is added 2-(5-Piperidin-
[1,2,4]triazol-
3-y1)-pyridine2HCI (1.5 eq) followed by acetic acid (2.5eq). The reaction
mixture
is stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq) over a
pe-
riod of 40 minutes. The reaction mixture is stirred overnight. The reaction
mixture
is quenched with methanol and concentrated. The residue obtained is taken up
in
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
chloroform and washed with water, dried and concentrated. The crude is
purified
on a flash column chromatography to obtain the desired compound.
MS (M+1): 513.33.
1H NMR(CD30D): 6 8.7 (d, J = 1.5Hz, 2H), 8.65 (d, J = 4.8Hz, 1H), 8.1(d, J =
5 6.9Hz, 1H),8.1(d , J= 7.8Hz, 1H), 7.93(dd, J= 1.8Hz, J= 7.8Hz, 1H),
7.9(s, 1H),
7.7(d, J = 9Hz, 1H), 7.3-7.7(m, 8H), 7.0(dt, J = 6.9Hz,1.2Hz, 1H), 3.9(s, 2H),
3(m,
1H), 2.7(t, 2H), 2.1(m, 4H), 2(m, 2H).
Example 17: 2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
10 yl]methyl}pheny1)-3-(2-thienyl)imidazo[1,2-a]pyridine
Example 17 is prepared according to example 9 by using 2-thienylboronic acid
in
step.
MS (M+1): 518.20.
1H NMR(CD30D): 6 8.65 (d, J = 4.2Hz, 1H), 8.1(t, J = 6.9Hz, 2H), 7.9(m,
15 1H),7.8(dd J = 2.1 Hz, 4.5Hz, 1H), 7.7(d, J = 8.1 Hz, 2H), 7.6(d, J =
9.3Hz, 1H),
7.4(m, 4H), 7.3(m, 2H),7(dt, J = 6.9Hz, 1.2Hz, 1H), 3.9(s, 2H),3(m,1H), 2.7(t,
2H),
2.1(m, 4H), 2(m,2H).
Example 18: 3-(4-fluoropheny1)-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-
20 yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
Step1: 1-[4-(1,3-dioxolan-2-yl)phenyl]ethanone
To the stirred solution of methyl magnesium chloride in ether is added 4-(1,3-
dioxolan-2-yl)benzonitrile (15g, 85.6mM, 0.2 eq) in dry ether over a period of
15
min. The reaction mixture is stirred over night. Reaction mixture is cooled to
0 C
25 and slowly quenched with sat. solution of ammonium chloride. Ether is
added and
the separated organic layer is dried and concentrated. The crude product is
puri-
fied by column chromatography to obtain the desired compound as a white solid.
1H NMR (CDCI3): 6 2.622(s, 3H), 4.165-4.04(m, 4H), 5.875(s, 1H), 7.588(d, J=
8.1 Hz, 2H), 7.987(d, J = 8.4 Hz, 2H).
Step 2: 4-(bromoacetyl)benzaldehyde
5.5g (28.6mM) of 1-[4-(1,3-dioxolan-2-yl)phenyl]ethanone is dissolved in
acetone
water mixture (200 mL). Catalytic amount of 4-methylbenzenesulfonic acid is
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
61
added and the reaction mixture is refluxed for 4h. The organic solvent is
evapo-
rated and the crude is extracted in ethylacetate. The organic layer is dried,
con-
centrated to obtain the crude product, which is dissolved in a mixture of
ethyl ace-
tate and chloroform (100 mL). The reaction mixture is refluxed. To this is
added
cupric bromide (2 eq) in portions while purging N2 gas. The reaction mixture
is
refluxed for 8h. It is then filtered, concentrated and the crude is purified
by cc to
obtain the desired compound.
1H NMR (DMSO-d6): 6 10.12(s, 1H), 8.18(d, J = 8.1Hz, 2H), 8.06(d, J = 6.6Hz,
2H),5.09(s, 2H).
Step 3: 4-imidazo[1,2-a]pyrimidin-2-ylbenzaldehyde.
4-(bromoacetyl)benzaldehyde is dissolved in acetone. To it is added pyrimidin-
2-
amine (leg). The reaction mixture is stirred overnight. The precipitated solid
is
filtered off and dried to obtain the desired compound as a white solid.
MS (M+1): 224.13.
1H NMR (DMSO-d6): 6 10(s, 1H), 9(dd, J= 6.6Hz, 1.5Hz, 1H), 8.57(dd, J =
2.1 Hz, 4.2Hz, 1H), 8.5(s, 1H), 8.3(d, J = 8.4Hz, 2H), 8.0(d, J = 8.4Hz, 2H),
7.1(dd, J= 6.6Hz, 3.9Hz, 1H).
Step 4: 4-(3-bromoimidazo[1,2-a]pyrimidin-2-yl)benzaldehyde:
This compound 13 is prepared according to the procedure reported by Sundberg
et al. (J. Heterocyclic Chem., 25, 129, 1988).
Step 5: 443-(4-fluorophenyl)imidazo[1,2-a]pyrimidin-2-ylibenzaldehyde
0.225g (0.74mM) 4-(3-bromoimidazo[1,2-a]pyrimidin-2-yl)benzaldehyde is dis-
solved in 5mL of toluene. To this mixture is added 4-fluorphenyl boronic acid
(1.5
eq) followed by tetrakis triphenylphosphine palladium (0) 10% by wt, K2003 (3
eq) and ethanol water mixture (4mL). The reaction mixture is heated at 90 C
for
4-6h. The reaction is cooled to room temperature and 20mL of water is added
and the reaction mixture is extracted with ethyl acetate. The organic layer is
dried
and concentrated. The crude is purified by flash column chromatography to
yield
the desired compound.
MS (M+1): 318.33.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
62
1H NMR (DMSO-d6): 6 10.07(s, 1H),8.89(dd, J= 2.1Hz, 6.9Hz, 1H), 8.68(dd, J =
1.8Hz, 4.2Hz,1H), 8.36(d, J= 8.1Hz, 2H), 8.08(d, J= 8.4Hz, 2H), 7.26(dd, J=
4.2Hz, 6.9Hz, 1H).
Step 6: 3-(4-fluoropheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
To a solution of 0.23g (0.72mM) 4-[3-(4-fluorophenyl)imidazo[1,2-a]pyrimidin-2-
yl]benzaldehyde in 5mL of THF is added triethylamine (2eq). The reaction
mixture
is stirred for 5 minutes. To this solution is added 2-(5-Piperidin-
[1,2,4]triazol-3-y1)-
pyridine*2HCI (1.5 eq) followed by acetic acid (2.5 eq). The reaction mixture
is
stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq) over a
period
of 40 minutes. The reaction mixture is stirred overnight. The reaction mixture
is
quenched with methanol and concentrated. The residue obtained is taken up in
chloroform and washed with water, dried and concentrated. The crude is
purified
on a flash column chromatography to obtain the desired compound.
MS (M+1): 531.27.
1H NMR(CD30D): 6 8.7 (m, 2H), 8.5(d, 1H), 8.1(d, 1H), 7.9(dt, J = 1.8Hz,
7.8Hz,
1H),7.7-7.6(m, 3H), 7.2-7.5(m, 8H), 6.9(dt, J = 6.9Hz, 1.2Hz, 1H), 3.9(s, 2H),
3(m,1H), 2.7(t, 2H), 2.1(m, 4H), 2.0(m, 2H).
Example 19: 7-methyl-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared according to compound 12 by using 4-methylpyrimidin-
2-amine in step1.
MS (M+1): 527.13.
1H NMR(CD30D): 6 8.7 (s,1 H), 8.35(d, J = 6.9Hz ,1H),8.1(d, J = 8.1Hz, 1H),
7.6-
7.5(m, 6H), 7.5-7.4(m, 3H), 7.3(d, J = 7.8Hz, 2H),6.9(d, J = 6.9Hz, 1H),
3.7(s,
2H), 3.1(m, 2H), 2.9(m, 1H), 2.6(s, 3H), 2.3(m, 2H), 2.1-1.9(m, 4H).
Example 20: 3-(4-methoxypheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 18 by using 4-
methoxyphenyl boronic acid in step5.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
63
MS (M+1): 543.27.
1H NMR(CD30D): 6 8.65 (d, J = 4.2Hz, 1H), 8.61(dd, J = 2.1 Hz, 4.2Hz, 1H),
8.4(dd, J = 1.9Hz, 6.9Hz, 1H), 8.05(d, J = 7.8Hz, 1H), 7.92(t, 1H), 7.73(d, J
=
8.1Hz, 2H).
Example 21: 6-chloro-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 12 by using 5-
chloropyrimidin-2-amine in step1.
MS (M+1): 547.20, 549.20.
1H NMR (CD30D): 6 8.65(d, J = 3.6Hz, 1H), 8.60(d, J = 2.4Hz, 1H), 8.54(d, J =
2.4Hz, 1H), 8.06(d, J = 7.8Hz 1H), 7.97-7.89(m, 1H), 7.70 (d, J = 8.4Hz, 2H)
7.65-7.57(m, 3H), 7.55-7.42(m, 5H), 4.20(s, 2H), 3.46(brd, J= 10.2Hz, 2H), 3.4-
3.35(m, 1H), 3.1-2.95(m, 2H), 2.28(brd, 2H), 2.2-2.0(m, 2H).
Example 22: 6-fluoro-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
This example is prepared in a manner according to example 12 by using 5-
fluoropyridin-2-amine in the first step.
MS (M+1): 530.27.
1H NMR (CD30D): 6 8.65(brd, J = 4.2Hz, 1H), 8.10-8.02(m, 2H), 7.96-7.88(m,
1H), 7.71-7.55(m, 5H), 7.49-7.40(m, 5H), 7.40-7.32(m, 1H), 7.24-7.12(m, 1H)
4.08(s, 2H), 3.44-3.32(m, 2H), 3.15-3.05(m, 1H), 3.00-2.8(m, 2H), 2.30-2.19(m,
2H), 2.19-2.00(m, 2H).
Example 23: 6-iodo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 12 by using 5-
iodopyrimidin-2-amine in the first step.
MS (M+1): 639.13.
1H NMR (CD30D): 6 8.67 (d, J = 2.4Hz, 1H), 8.64 (d, J = 4.5Hz, 1H), 8.58 (d, J
=
2.1 Hz, 1H), 8.07 (d, J = 7.8Hz, 1H), 7.91 (dt, J = 1.8,7.5Hz, 1H), 7.68-7.57
(m,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
64
5H), 7.51-7.47 (m, 2H), 7.44 (t, 1H), 7.34 (d, J = 8.1Hz, 2H), 3.8 (s, 2H),
3.11 (d,
2H), 2.90 (m, 1H), 2.42 (t, 2H), 2.15 (d, 2H), 1.96 (t, 2H).
Example 24: 7-methoxy-3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 12 by using 4-
methoxy-2-amine in the first step.MS (M+1): 543.13.
1H NMR (CD30D): 6 8.64 (d, J = 4.5Hz, 1H), 8.21 (d, J = 7.2Hz, 1H), 8.07 (d, J
=
8.1Hz, 1H), 7.89 (dt, J= 2.1,7.8Hz, 1H), 7.59-7.52 (m, 5H), 7.49-7.44 (m, 3H),
7.28 (d, J= 8.4Hz, 2H), 6.54 (d, J= 7.2Hz, 1H), 4.07 (s, 3H), 3.73 (s, 2H),
3.12
(d, 2H), 2.92 (m, 1H), 2.44 (t, 2H), 2.14 (d, 2H),. 2.0 (t, 2H).
Example 25: 8-bromo-3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
This example is prepared in a manner according to example 12 by using 3-
bromopyridin-2-amine in the first step.
MS (M+1): 590.07, 592.07.
1H NMR (CD30D): 6 8.64(d, J = 4.5Hz, 1H), 8.07 (d, J = 6.6Hz, 2H), 7.92 (dt, J
=
1.8Hz, 7.8Hz, 1H), 7.65-7.54 (m, 6H), 7.48-7.42 (m, 3H), 7.34 (d, J= 8.1Hz,
2H),
6.81 (t, 1H), 3.84 (s, 2H), 3.20 (d, 2H), 2.98 (m, 1H), 2.57 (t, 2H), 2.14 (d,
2H),
2.06 (t, 2H).
Example 26: 8-chloro-3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrazine
This example is prepared in a manner according to example 12 by using 3-
chloropyrazin-2-amine in the first step.
MS (M+1): 547.20, 549.20.
1H NMR (CD30D): 6 8.64 (d, J = 4.8Hz, 1H), 8.08 (t, 1H), 8.06 (s, 1H), 7.92
(dt, J
= 1.8, 7.8Hz, 1H), 7.68 (t, 3H), 7.62-7.59 (m, 3H), 7.56-7.46 (m, 2H), 7.44-
7.57
(m, 3H), 3.83 (s, 2H), 3.18 (d, 2H), 2.98 (m, 1H), 2.58 (t, 2H), 2.16 (d, 2H),
2.01 (t,
2H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Example 27: 8-methoxy-3-phenyl-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrazine
This example is prepared in a manner according to example 12 by using 3-
methoxypyrazin-2-amine in the first step.
5 MS (M+1): 543.07.
1H NMR (CD30D): 6 8.26 (d, J = 3.9Hz, 1H), 8.06 (d, J = 7.8Hz, 1H), 7.90 (t,
1H),
7.65 (d, J = 4.8Hz, 1H), 7.55-7.53 (m, 5H), 7.45-7.38 (m, 4H), 7.29 (d, J =
8.1 Hz,
2H), 4.16 (s, 3H), 3.59 (s, 2H), 3.0 (s, 2H), 2.85 (m, 1H), 2.28 (t, 2H), 2.08
(d,
2H), 1.90(d, 2H).
Example 28: 3-phenyl-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-7-(trifluoromethyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 12 by using 4-
(trifluoromethyl)pyrimidin-2-amine in the first step.
MS (M+1): 581.13.
1H NMR (CD30D): 6 8.73(d, J= 7.2Hz, 1H), 8.65(br d, J= 3.6Hz, 1H), 8.12-
8.05(m, 1H), 7.97-7.89(m, 2H), 7.73-7.53(m, 5H), 7.49-7.38(m, 4H), 7.37-7.30
(m,
1H) 3.86(s, 2H), 3.2-2.9(m, 3H), 2.7-2.5(m, 2H), 2.2-1.95(m, 4H).
Example 29: 3-phenyl-2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyridine-7-carbonitrile
This example is prepared in a manner according to example 12 by using 2-
aminoisonicotinonitrile in the first step.
MS (M+1): 537.23
1H NMR (CDCI3): 6 8.675(d, J = 6.5 Hz, 1H), 8.177 (d, J = 8.1 Hz, 1H), 8.16
(s,
1H), 8.019(d, J = 7.2 Hz, 1H) 7.858 (t, J = 7.2Hz, 1H) 7.664(d, J = 8.1 Hz,
2H)
7.608-7.457 (m, 5H) 7.36 (d, 8.1 Hz, 2 H) 7.285 (m, 3H), 6.907 (dd, J = 7.2
Hz,
1.5 Hz, 1H), 3.714 (s, 2H), 3.185-2.155 (m, 9H).
3-phenyl-244-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)phenyl]imidazo[1,2-a]pyridine-7-carbonitrile (2E)-but-2-enedioate
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
66
To 3.0g 3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyllphenyl)imidazo[1,2-a]pyridine-7-carbonitrile (prepared as described
un-
der example 29) in 50m1 acetone is added a solution of 0.714g fumaric acid in.
The reaction mixture is stirred at ambient temperature for 3d. The desired com-
pound is collected by filtration and dried.
Characteristic 1H NMR (dDMS0,300MHz) Signals: 8.66 (d, 1H); 8.43 (s, 1H),
8.13 (d, 1H), 8.01 (d, 1H),7.93 (t, 1H), 7.61 (m, 5H), 7.41 (t, 1H), 7.33 (d,
2H),
7.16 (d, 1H), 6.6 (2H)
Example 30: 3-pheny1-2-(4-{[4-(3-pyridin-2-yl-pyrazol-5-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner similiar to 1 by using 2-(5-piperidin-4-
yl-
pyrazol-3-yl)pyridine in the last step which was prepared as described in
Bioorg.
Med. Chem. Lett.; EN; 12; 3; 2002; 383 ¨ 386.
Analytical data
MS (M+1): 510
Characteristic 1H NMR (400Hz, dDMS0) signals: 8.4ppm (d, 1H); 7.1ppm (dd,
1H);
Example 31: 6-chloro-3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-b]pyridazine.
This example is prepared in a manner according to example 12 by using 6-
chloropyridazin-3-amine in the first step.
MS (M+1): 547.13.
1H NMR (300 MHz, DMSO-d6): 6 8.65 (d, 1H), 8.278 (d, J= 9.6Hz, 1H), 8.014 (d,
J = 7.8Hz, 1H), 7.92 (s, 1H), 7.59-7.51(m, 7H), 7.41(d, J = 9.3Hz, 3H), 7.3(d,
J =
7.5Hz, 2H), 3.49 (s, 1H), 2.86-2.73 (m, 3H), 2.07 (s, 2H) 1.97-1.76 (m, 5H).
Example 32: 6-methoxy-3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-b]pyridazine.
To a suspension of example 31 (0.2g, 0.366mM) in 15mL of methanol is added
NaOCH3 (10 eq.) and the reaction mixture is refluxed for 6h. The reaction
mixture
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
67
is concentrated and quenched with water. The resulting solid is filtered and
dried
to obtain the desired compound.
MS (M+1): 543.13.
1H NMR (300 MHz, CD30D) 6 8.65(d, J = 4.2Hz, 1H), 8.1(d, J = 8.1Hz, 1H),
7.94-7.9 (m, 2H), 7.6-7.5(m, 4H), 7.5-7.4(m, 4H), 7.32(d, J = 8.1Hz, 2H), 6.9
(d, J
= 9.6Hz, 1H), 3.9 (s, 3H), 3.6(s, 2H), 3.04-3.0 (m, 2H) 2.88-2.87 (m, 1H),
2.25-
2.18 (m, 2H), 2.07-1.88(m, 4H).
Example 33: 3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1 -
xi yl]methyl}phenyl)imidazo[1,2-a]pyridine-8-carboximidamide.
To a solution of hydroxylamine hydrochloride (10 eq) in 5mL of anhyd. DMSO, is
added, KtBuO (10 eq) at 5 C in portions. To this is added example 5 (0.15g,
0.28mM). The reaction is stirred overnight. The reaction mixture is quenched
with
water and the resulting ppt is filtered to obtain 170mg of crude
hydroxyamidine.
This is dissolved in 3mL of acetic acid and acetic anhydride is added (0.1mL).
The reaction mixture is stirred overnight at RT. The reaction mixture is
concen-
trated and triturated with ether to obtain acetylated hydroxyamidine. This
crude is
dissolved in methanol and to this is added Pd/C 10%, and the reaction mixture
is
stirred under hydrogen atmosphere for 2h. After the workup, the crude is
purified
by prep HPLC to obtain the desired compound.
MS (M+1): 554.2.
1H NMR (300 MHz, CD30D) 6: 8.75(d, J= 3.3Hz, 1H), 8.50 (s, 1H), 8.37(d, J =
6.9Hz, 1H), 8.22(d, J = 6.6Hz, 1H), 8.076(d, J = 7.8Hz, 1H), 7.95-7.89 (m,
2H),
7.71-7.50 (m, 5H), 7.44(t, 1H), 7.35(d, J = 7.8Hz, 2H), 7.14(t, 1H) 4.87(s,
2H),
3.24-3.23(d, 2H), 3.13-3.1(d, 2H), 2.9(m, 1H), 2.13-1.96(m, 2H).
Example 34: 2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-3-(3-thienyl)imidazo[1,2-a]pyrimidine
This example is prepared according to example 18 by using 3-thienyl boronic
acid
in step5.
MS (M+1): 519.13.
1H NMR(CD30D): 6 8.7 (d, J = 4.8Hz, 1H), 8.6 (m, 1H), 8.5 (dd, J = 1.8, 6.9Hz,
1H), 8.1 (d, J = 8.1Hz, 1H), 7.9 (m, 1H), 7.7 (m, 4H), 7.5 (m, 1H), 7.4 (d, J
=
CA 02695251 2014-08-13
,
68
8.1Hz, 1H), 7.2 (dd, J= 1.5, 5.1Hz, 1H), 7 (m, 1H), 3.8 (s, 2H), 3.2 (m, 2H),
3 (m,
1H), 2.5 (m,2H), 2.2 (m,2H), 2 (m,2H).
Example 35: 2-(4-([4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-3-(2-thienyl)imidazo[1,2-a]pyrimidine
This example is prepared according to example 18 by using 2-thienyl boronic
acid
in step5.
MS (M+1): 519.13.
1H NMR(CD30D): 8 8.7 (dd, J = 4.2, 2.1Hz, 2H), 8.5 (dd, J = 1.8, 6.6Hz, 1H),
8.1
(d, J= 8.1, 1H), 7.95 (m, 1H), 7.8 (m, 3H), 7.5 (m, 3H), 7.4 (m, 2H), 7.1 (m,
1H),
4.1 (s, 2H), 3.4 (m, 2H), 3.1 (m, 2H), 2.9 (m, 1H), 2.3 (m, 2H), 2.1 (m, 2H).
Example 36: 3-pyridin-4-y1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 18 (first 3 steps).
The final 2 steps are conducted in a manner according to step 1 & 2 from exam-
ple 16.
MS (M+1): 514.2.
1H NMR (CD30D): 6 8.8-8.6 (m, 5H), 8.1 (d, J = 7.8Hz, 1H), 7.9 (m, 1H), 7.7
(d, J
= 8.1Hz, 2H), 7.6 (m, 4H), 7.4 (m, 1H), 7.2 (m, 1H), 4.25 (s, 2H), 3.6 (m,
2H), 3.2
(m, 2H), 3.1 (m, 1H), 2.3 (m, 2H), 2.2 (m, 2H).
Example 37: 3-phenyl-2-(44[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyrazine
To a solution of example 26 (0.1g, 0.18nnM) in 10mL of methanol is added 40mg
Pd/C(10%). The reaction mixture is stirred at RT under hydrogen atmosphere
overnight. The reaction mixture is filtered through celiteTM and concentrated.
It is
then purified by column chromatography and washed with water to obtain the de-
sired compound.
MS (M+1): 513.20.
1H NMR (CD30D): 6 9.05 (d, J = 1.5Hz, 1H), 8.64 (d, J = 3.9Hz, 1H), 8.14 (dd,
J =
4.8 Hz, 1.5 Hz, 1H), 8.07 (d, J = 7.8Hz, 1H), 7.94-7.87(m, 2H), 7.68 (d, J =
8.4Hz,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
69
2H), 7.63 ¨ 7.56 (m, 3H), 7.52-7.46(m, 3H), 7.38(d, J = 8.1Hz, 2H), 3.81(s,
2H),
3.19(d, 2H), 2.98(m, 1H), 2.51(t, 2H), 2.21-1.92(m, 4H).
Example 38: 3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1 ,2,4-triazol-3-yl)piperidin-1 -
yl]methyl}phenyl)imidazo[1,2-a]pyridine-7-carboxamide.
Example 29 (0.1g, 0.018mM) is dissolved in 3mL of conc. H2SO4. It is stirred
overnight and quenched with water and basified with 20% w/w NaOH solution.
The precipitated out solid is filtered and dried. It is then purified by
column chro-
matography to obtain the desired compound.
MS (M+1): 555.13.
1H NMR (CDCI3): 6 8.64(d, J=4.5 Hz, 1H), 8.18 (s, 1H), 8.13 (d, J=7.2 Hz, 1H),
8.08(d, J=7.8 Hz, 1H) 7.94 (dd, J= 7.8Hz, 1.8Hz, 2H) 7.64-7.54(m, 5H) 7.50-
7.42
(m, 3H) 7.38-7.36 (m, 3 H) 7.33 (s, 2H), 3.30 (bs, 2H), 3.18(d, 2H), 2.95(m,
1H),
2.40(m, 2H), 2.15-1.93(m, 4H)
Example 39: 2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-3-(1,3-thiazol-2-y1)imidazo[1,2-a]pyrimidine
This example is prepared in a manner according to example 18 except step 5
where Stille coupling employing 2-(tributylstannyI)-1,3-thiazole is carried
out in-
stead of Suzuki coupling. Step 6 is according to example 18.
Step 5: 443-(1,3-thiazol-2-yl)imidazo[1,2-a]pyrimidin-2-ylibenzaldehyde
0.5g (1.65 mM) 4-(3-bromoimidazo[1,2-a]pyrimidin-2-yl)benzaldehyde is dis-
solved in 15 mL of 1,4-Dioxane. To this solution is added tetrakis triphenyl-
phosphine palladium (200 mg) 40%. The reaction mixture is heated at 100 C for
10 min. and 2-(tributylstannyI)-1,3-thiazole is added (681mg, 1.81mM). The
reac-
tion is heated (oil bath or microwave) at same temp for 5h. The reaction is
cooled
to room temperature and 50mL of water is added and the reaction mixture is ex-
tracted with ethyl acetate. The organic layer is dried and concentrated. The
crude is purified by flash column chromatography to yield the desired product.
MS (M+1): 307.20.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
1H NMR (DMSO-d6): 6 10.16 (s, 1H), 9.72 (dd, J= 1.8, 6.9 Hz, 1H), 8.7 (dd, J =
1.8, 4.2 Hz, 1H), 8.08 (d, J = 3.3 Hz, 1H), 8.06 (d, J = 8.1Hz, 2H), 7.94 (d,
J =
8.1Hz, 2H), 7.81 (d, J = 3.3 Hz, 1H), 7.35 (m, 1H).
5 Step 6: 2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-3-(1,3-thiazol-2-yl)imidazo[1,2-a]pyrimidine
MS (M+1): 520.13
1H NMR (CD30D): 6 9.93 (d, J = 5.1 Hz, 1H), 8.72 (dd, J = 4.2, 2.1 Hz, 1H),
8.63
(d, J = 4.2, 1H), 8.092 (d, J = 8.1, 1H), 7.98 (d, J = 3.3Hz, 1H), 7.92 (t,
1H), 7.71
10 (d, J = 8.1, 2H), 7.58 (d, J= 8.1 Hz, 2H), 7.54 (d, J= 3.3, 1H), 7.44
(t, 1H), 7.26
(dd, J= 4.5, 7.2 Hz, 1H) 3.92 (s, 2H), 3.21 (m, 2H), 3.18 (m, 1H), 2.55 (m,
2H),
2.15 (m, 2H), 2.14 (m, 2H).
15 Example 40: 3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-
yl]methyl}phenyl)imidazo[1,2-a]pyridine-8-carboxamide.
This example is prepared from example 5 in a manner according to example 38.
MS (M+1): 555.13.
1H NMR (CD30D): 6 8.63 (d, J = 5 Hz, 1H), 8.19-8.13 (m, 2H), 8.07 (d, J =
7.8Hz,
20 1H), 7.93-7.87 (m, 1H), 7.65 (d, J = 8.4Hz, 2H), 7.62-7.56 (m, 3H), 7.51-
7.48 (m,
2H), 7.44-7.40 (m, 1H), 7.29 (d, J = 8.4Hz, 2H), 7.01 (t, 1H) 3.56 (s, 2H)
3.05-
2.98 (br d, 1H), 2.91-2.78 (m, 1H), 2.23-2.11(m, 2H), 2.070-1.950 (m, 3H),
1.950-
1.911(m, 2H).
25 Example 41: 3-(2-fluoropheny1)-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyrimidine
This example is prepared according to example 9 using 2-fluorophenyl boronic
acid in step 1.
MS (M+1): 531.13.
30 1H NMR(CD30D): 6 8.64 (dd, J = 4.2, 1.2 Hz, 2H), 8.39 (dd, J =6.9, 2.1
Hz, 1H),
8.01 (d, J = 7.8 Hz, 1H), 7.93 (dt, J = 7.8, 1.8 Hz, 1H), 7.6-7.7 (m ,3H), 7.2-
7.5
(m, 6H ), 7.02 (dd, J = 6.9, 1.2 Hz, 1H), 3.65 (s, 2H), 3.18 (d, 2H), 2.9 (m,
1H),
2.06 (t, 2H), 1.9 (d, 2H), 1.8 (d, 2H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
71
Example 42: 6-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyl}pheny1)-5-(1,3-thiazol-2-yl)imidazo[2,1-b][1,3]thiazole
Stepl: 4-Imidazo [2,1-13] thiazol-6-yl-benzaldehyde
0.7g (2.1mM) 4-(2-Bromo-acetyl)-benzaldehyde and 2-aminothiazole (2 eq) are
stirred in 15m1DMF at 90 C for 6h. The DMF is evaporated and the residue
suspended in ethyl acetate over night. The desired product is collected by
filtration.
MS (M+1): 229.13
1H NMR (DMSO-d6): d 10.0 (s, 1H), 8.51 (s, 1H), 8.06 (d, J = 8.2 Hz, 2H), 8.04
(d, J = 4.5 Hz, 1H), 7.9 (d, J = 8.4 Hz, 2H), 7.40 (d, 1J = 4.5 Hz, 1H).
Step2: 4-(5-Bromo-imidazo [2,1-13] thiazol-6-y1)-benzaldehyde
4-Imidazo [2,1-13] thiazol-6-yl-benzaldehyde (3.8g, 14.9mM) is dissolved in
30mL
acetic acid. A solution of bromine (1.1 eq) in acetic acid (15mL) is added to
the
reaction mixture. The reaction mixture is stirred for 3 h at room temperature.
The
solid obtained is collected by filtration and re-dissolved in water (100m1)
and the
solution neutralized by aqueous ammonia. The solid obtained is collected by
filtration and dried.
MS (M+1): 307.13, 309.1
1H NMR (DMSO-d6): d 10.03(s, 1H), 8.2 (d, J = 8.4 Hz, 2H), 8.0 (d, J = 8.4Hz,
2H), 7.95 (d, J = 4.5 Hz, 1H), 7.48 (d, J = 4.5 Hz, 1H).
Step3: (5-Thiazol-2-yl-imidazo [2,1-13] thiazol-6-y1)-benzaldehyde
4-(5-Bromo-imidazo [2,1-13] thiazol-6-y1)-benzaldehyde 0.09g (0.37mM) is dis-
solved in 5mL of 1,4 dioxane and 0.5 eq of Pd(PPh3)4 is added followed by the
addition of 2-(tributylstannyI)-1,3-thiazole (1.1eq) and the reaction mixture
is
heated to reflux for 24h. The reaction mixture is cooled down to room tempera-
ture and diluted with 10mL of methylene chloride and 15mL of water. The
separated organic layer is washed with water and dried over Na2504. The crude
reaction mixture is used for next step.
MS (M+1): 312.1
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
72
Step4: 6-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyllpheny1)-5-
(1,3-thiazol-2-y1)imidazo[2,1-b][1,3]thiazole
To a solution of 0.1 g (0.31 mM) 4-(5-Thiazol-2-yl-imidazo [2,1-b] thiazol-6-
y1)-
benzaldehyde in 5mL of THF is added triethylamine (2 eq). The reaction mixture
is stirred for 5 minutes. To this solution is added 2-(5-Piperidin-
[1,2,4]triazol-3-y1)-
pyridine*2HCI (1.5 eq) followed by acetic acid (2.5eq). The reaction mixture
is
stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq) over a
period
of 40 minutes. The reaction mixture is stirred overnight. The reaction mixture
is
quenched with methanol and concentrated. The residue obtained is taken up in
chloroform and washed with water, dried and concentrated. The crude is
purified
on a flash column to obtain the desired compound.
Mass (M+1): 525.13
1H NMR(CD30D): d 8.64 (s, 1H), 8.55 (d, J = 4.5 Hz, 1H), 8.08 (d, J = 9.6 Hz,
1H), 9.92 (t, J = 9.3, 9.2 Hz, 1H), 7.84 (d, J = 3.3 Hz, 1H), 7.66 (d, J = 6.6
Hz,
2H), 7.53 (d, J = 7.8 Hz, 2H), 7.4 (d, J = 3.3Hz, 2H), 7.30 (d, J = 4.5 Hz,
1H), 3.8
(s, 2H), 3.2 (d, 2H), 3.0 (m, 1H), 2.5 (t, 2H), 2.14 (d, 2H), 2.00 (d, 2H).
Example 43: 5-(1,3-oxazol-2-y1)-6-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[2,1-b][1,3]thiazole
This example is prepared according to example 42 by using 2-(tributylstannyI)-
1,3-oxazole in step 3.
MS (M+1): 509.13.
1H NMR (CD30D): 6 8.62 (d, J = 4.5 Hz, 1H), 8.41 (d, J = 4.5 Hz, 1H), 8.07(d,
J =
7.8 Hz, 1H), 7.9 (m, 2H), 7.81(d, 2H, J= 8.1 Hz), 7.49-7.41 (m, 3H), 7.34 (d,
J=
4.2 Hz, 2H), 3.7 (s, 2H), 3.19( d, 2H), 3.0 (m, 1H), 2.4 (t, 2H), 2.1 (d, 2H),
2.0 (d,
2H).
Example 44: 6,8-Difluoro-3-phenyl-2- {4-[4-(5-pyridin-2-y1-4H- [1,2,4] triazol-
3-y1)-piperidin-1 -ylmethyli-phenyl}-imidazo [1,2-a] pyridine
Example 44 is prepared according to example 12
MS (M+1) : 548.20
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
73
1H NMR(300 MHz, CD30D): 6 8.64 (dd, J = 4.2, 1.2 Hz, 2H), 8.39 (dd, J =6.9,
2.1 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.93 (dt, J = 7.8, 1.8 Hz, 1H), 7.6-7.7
(m
,3H), 7.2-7.5 (m, 6H ), 7.02 (dd, J = 6.9, 1.2 Hz, 1H), 3.65 (s, 2H), 3.18 (d,
2H),
2.9 (m, 1H), 2.06 (t, 2H), 1.9 (d, 2H), 1.8 (d, 2H)
Example 45: 2-methyl-5-phenyl-644-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl)phenylpmidazo[2,1-13][1,3,4]thiadiazole
Example 45 is prepared according to example 12
MS (M+1): 533.13
1H NMR (300MHz, CDCI3) 6 8.67(d, J=3Hz, 1H), 8.17(d, J=6Hz, 1H), 7.9-7.2(m,
12H), 3.6(s, 2H), 3.1-2.8(m, 3H), 2.71(s, 3H), 2.4-1.9(m, 6H).
Example 46: 2-methyl-5-phenyl-644-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl)phenylpmidazo[2,1-13][1,3]thiazole
Example 46 is prepared according to example 12
MS (M+1) :532.13
1H NMR (300MHz, CDCI3) 6 8.651 (d, 1H), 8.089 (d, J=7.8,Hz 1H), 7.954 (t, J =
1.5 Hz 1H), 7.523-7.456 (m, 7H), 7.377-7.217 (m, 4H), 3.760 (s, 2H), 3.082 (d,
J=7.8Hz, 2H), 2.981-2.943 (m, 1H), 2.457 (s, 2H), 2.45 (s, 3H) 2.243-2.125 (m,
4H).
Example 47: 2-(methylsulfany1)-5-phenyl-644-({445-(pyridin-2-y1)-4H-1,2,4-
triazol-3-ylipiperidin-1-yl}methyl)phenylpmidazo[2,1-13][1,3,4]thiadiazole
Example 47 is prepared according to example 12
MS (M+1): 565.07
1H NMR (300 MHz,CDCI3):- 6 8.672 (d, J = 3.9 Hz, 1H), 8.163 (d, J =7.8Hz,1H),
7.828 (t, J = 1.2 Hz 1H), 7.637-7.561 (m, 4H), 7.448-7.338 (m, 4H), 7.288
(d,J=8.4Hz, 2H), 3.552 (s, 2H), 3.012 (d, J =11.1 Hz,2H), 2.873 (m, 1H), 2.73
(s, 3H), 2.195-1.970 (m, 6H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
74
Example 48: 5-phenyl-644-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-yl]piperidin-
1-y1) methyl) phenyl]-2-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazole
Example 48 is prepared according to example 12
MS (M+1): 587.05
1H NMR (300 MHz,CDCI3):- 6 8.683 (d, J = 4.5 Hz, 1H), 8.177 (d, J=7.8 Hz, 1H),
7.852 (t, J = 1.5 Hz 1H), 7.643 (s, 2H), 7.616 (s, 2H), 7.511-7.439 (m, 3H),
7.402-7.32 ( m, 3H), 3.637(s, 2H),3.065 (d, J =11.1Hz, 1H ), 2.928 (bs, 1H),
2.243-2.067 (m, 6H).
Example 49: 3-phenyl-244-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-yl]piperidin-
1-yl}methyl)phenyl]imidazo[1,2-a]pyridine-7-carbonitrile (2Z)-but-2-
enedioate
To 3.0g 3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-triazol-3-yl)piperidin-1-
yl]methyllphenyl)imidazo[1,2-a]pyridine-7-carbonitrile (prepared as described
un-
der example 29) in 50m1 acetone is added a solution of 0.714g malonic acid in
10m1 acetone dropwise. The reaction mixture is stirred at ambient temperature
for
18h. The desired compound is collected by filtration and dried.
Characteristic 1H NMR (dDMS0,300MHz) Signals: 8.68 (d, 1H); 8.44 (s, 1H),
8.15 (d, 1H), 8.01 (d, 1H),7.90 (t, 1H), 7.70 (m, 5H), 7.63 (t, 1H), 7.47 (d,
2H),
7.18 (d, 1H), 6.0 (2H)
Example 50: 7-methyl-3-phenyl-2- [4-({4[5-(pyridin-2-y1)-4H-1, 2,4-triazol-3-
yl] piperidin-1-y1) methyl) phenyl] imidazo [1,2-a] pyridine
Example 50 is prepared according to example 12
MS (M+1): 526.13
1H NMR (300 MHz,CDCI3):- 6 8.65(bs, 1H), 8.02 (d, J=7.5 Hz, 1H), 7.9 (d, J=7.2
Hz, 1H), 7.46-7.60 (m, 7H), 7.42 (s, 1H), 7.22 (d, J= 8.1 Hz, 2H), 3.45 (s,
2H),
2.9 (d, 2H), 2.37 (s, 3H), 1.71-2.07 (m, 6H)
Example 51: 2-ethyl-5-phenyl-6-(4-([4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl} phenyl)imidazo[2,1-b][1,3,4]thiadiazole
Example 41 is prepared according to example 12
MS (M+1): 547.07
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
1H NMR (300 MHz,CDCI3):- 6 8.67 (s, 1H), 8.18 (d, J=7.5 Hz, 1H), 7.9-7.25 (m,
12H), 3.56 (s, 2H), 3.1-2.8 (m, 3H), 2.3-1.9 (m, 6H), 1.43 (t, J=7.5 Hz, 3H),
0.9
(m, 2H)
5
Example 52: '6-bromo-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine-7-carbonitrile
Example 52 is prepared according to example 12
MS (M+1): 417.07
10 1H NMR (300 MHz,CDCI3):- 6 8.7(s, 1H), 8.0-8.2(m, 4H), 7.8(m, 1H), 7.5-
7.7 (m,
4H), 7.2-7.4(m, 5H), 3.9(s, 2H), 3 - 3.3(m, 3H), 2-2.6(m, 6H).
Example 53: '6-methyl-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl] methyl}phenyl)imidazo[1,2-b]pyridazine
15 Example 53 is prepared according to example 12
MS (M+1) :527.13
1H NMR (300 MHz,CDCI3):- 6 8.6 (m, 1H), 8.1(m,1H), 7.8-7.9(m,2H), 7.7(d, 2H),
7.6(d,2H), 7.3-7.5(m,6H), 6.9(d, 1H), 3.9(s, 2H), 3.0-3.3 (m,3H), 2.5(s,3H),
2.0-
2.3(m,6H).
Example 54: '7-chloro-3-phenyl-2-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[1,2-a]pyridine
Example 54 is prepared according to example 12
MS (M+1) :546.13
1H NMR (300 MHz,CDCI3):- 6 8.7 (d, J=4.5Hz, 1H), 8.1(d, J=8.1Hz, 1H), 7.8-7.9
(m, 2H), 7.6-7.7(m, 3H), 7.5-7.6(m, 3H), 7.3-7.5(m, 5H), 6.7(d, 1H), 3.9(s,
2H),
3.0-3.3(m, 3H), 2.1-2.8(m, 6H)
Example 55: '3-phenyl-2- (4-{[4-(5-pyridin-4-y1-1H-1, 2,4-triazol-3-y1)
piperidin-1-yl] methyl) phenyl) imidazo [1,2-a] pyrimidine.
To a solution of 0.094g (0.314mM) 4-(3-phenylimidazo [1,2-a] pyrimidin-2-y1)
benzaldehyde
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
76
in 3mL of THF is added triethylamine (2 eq). The reaction mixture is stirred
for 5
minutes. To this solution is added 4-[3-(piperidin-4-yI)-1H-1, 2,4-triazol-5-
yl] pyri-
dine 2HCI (1.5 eq) (Intermediate B) followed by acetic acid (2.5 eq). The
reaction
mixture is stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6 eq)
over a period of 40 minutes. The reaction mixture is stirred overnight. The
reac-
tion mixture is quenched with methanol and concentrated. The residue obtained
is taken up in chloroform and washed with water, dried and concentrated. The
crude is purified on a flash column chromatography to obtain the desired com-
pound.
MS (M+1) :513.2
1H NMR (300 MHz,CDCI3):- 6 8.66 (d, J = 5.7 Hz, 2H), 8.576 (s, 1H), 8.28 (dd,
J
= 6.6,1.5 Hz 1H), 7.98 (d, J = 6Hz, 2H), 7.65 (d, J=8.1 Hz 2H), 7.54
(d,J=6.3Hz,
3H), 7.457-7.431 ( m, 2H), 7.265-7.216 (m,2H),6.880-6.843 (m,1H ) 3.57 (s,
2H),
2.97 (d, J = 10.8Hz, 3H) 2.182-2.094 (m, 2H),1.992-1.910 (m,4H).
Example 56: '5-phenyl-6-(4-{[4-(5-pyridin-4-y1-4H-1,2,4-triazol-3-yl)piperidin-
1 -yl]methyl}phenyl)imidazo[2,1-b][1,3]thiazole
Example 56 is prepared according to example 55
MS (M+1) : 518.0
1H NMR (300MHz, DMSO-d6) 6 8.63(d, J=6Hz, 2H), 7.88(d, J=4.5Hz, 2H), 7.70(d,
J=4.5Hz, 1H), 7.55-7.15(m, 11H), 3.46(s, 2H), 3.0-2.7(m, 3H), 2.2-1.7(m, 6H).
Example 57: '3-phenyl-2-(4-{[4-(3-pyridin-4-y1-1 H-1,2,4-triazol-5-
yl)piperidin-
1 -yl]methyl}phenyl)imidazo[1,2-a]pyridine-7-carbonitrile
Example 57 is prepared according to example 55
MS (M+1) :537.13
1H NMR (300 MHz,CDCI3):- 6 8.69(dd, J=4.5 Hz, 1.2 Hz, 2H), 8.123 (s, 1H),
8.011(dd, J=7.2 Hz, 0.9 Hz, 1H), 7.95 (dd, J=4.5 Hz, 1.5Hz, 2H), 7.63(d, J=8.1
Hz, 2H), 7.60-7.52(m, 2H), 7.46-7.4 (m, 2H), 7.30 (d, J=8.4 Hz, 2H) 6.90 (dd,
J=6.9Hz, 1.5Hz, 1H), 3.60(m, 3H), 2.44-1.91 (m, 7H)
Example 58: '3-phenyl-2-(4-{[4-(5-pyridin-4-y1-4H-1,2,4-triazol-3-yl)piperidin-
1 -yl]methyl}phenyl)imidazo[1,2-a]pyridine
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
77
Example 58 is prepared according to example 55
MS (M+1) :512.07
1H NMR (300 MHz,CDCI3):- 6 8.7 (d, 2H),8(m, 3H), 7.7(d, 1H), 7.4to7.6 (m, 7H),
7.1to7.3(m, 3H), 6.8(t, 1H), 3.6(s,2H), 2.7to2.9(m,3H), 1.7to2.2(m,6H).
Example 59: '3-phenyl-2-(4-{[4-(5-pyridin-4-y1-4H-1,2,4-triazol-3-yl)piperidin-
1-yl]methyl}phenyl)imidazo[1,2-a]pyridine-8-carbonitrile
Example 59 is prepared according to example 55
MS (M+1) :537.13
1H NMR (300 MHz,CDCI3):- 6 8.7 (d, 2H), 8.2(d, 1H), 8(d, 1H), 7.2-7.7 (m,
10H), 6.8(t, 1H), 3.5(s,2H), 2.8-3.1(m,3H), 1.8-2.3(m,6H).
Example 60: '5-phenyl-6-(4-{[4-(5-pyridin-4-y1-4H-1,2,4-triazol-3-yl)piperidin-
1-yl]methyl}phenyl)imidazo[2,1-b][1,3,4]thiadiazole
Example 60 is prepared according to example 55
MS (M+1) : 519.0
1H NMR (300MHz, DMSO-d6) 6 9.22 (s, 1H), 8.65 (d, J=4.5Hz, 2H), 7.87 (d,
J=4.5Hz, 2H), 7.6-7.3 (m, 7H), 7.26 (d, J=8.1 Hz, 2H), 3.47 (s, 2H), 2.95-2.75
(m,
3H), 2.15-1.7 (m, 6H)
Example 61: 244-({4[5-(6-methylpyridin-2-y1)-4H-1, 2,4-triazol-3-yl]
piperidin-1-y1) methyl) phenyl]-3-phenylimidazo [1,2-a] pyridine-7-
carbonitrile
To a solution of 0.150g (0.464mM) 2-(4-formylphenyI)-3-phenylimidazo [1,2-a]
pyridine-7-carbonitrile in 3mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 2-methyl-4- [3-
(piperidin-
4-y1)-1H-1, 2,4-triazol-5-yl] pyridine dihydrochloride (Intermediate C) (1.5
eq) fol-
lowed by acetic acid (2.5eq). The reaction mixture is stirred for 10 minutes.
To
this mixture is added NaBH(OAc)3 (6 eq) over a period of 40 minutes. The reac-
tion mixture is stirred overnight. The reaction mixture is quenched with
methanol
and concentrated. The residue obtained is taken up in chloroform and washed
with water, dried and concentrated. The crude is purified on a flash column
chro-
matography to obtain the desired compound.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
78
MS (M+1) :551.2
1H NMR (300 MHz,CDCI3):- 6 8.15(s, 1H),7.9(m, 2H), 7.7(m, 3H), 7.55 (m,
3H),7.45(m,2H), 7.3(d, J=8.1 Hz, 2H), 7.2(d, J=7.5Hz, 1H),6.9(d, 1H),
3.6(s,2H),
3(d,2H), 2.9(m,1 H), 2.5(s, 3H), 2to2.6(m,6H).
Example 62: 244-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -yl}methyl) phenyl]-3-phenylimidazo[1,2-a]pyridine-8-carbonitrile
Example 62 is prepared according to example 61
MS (M+1) :551.2
1H NMR (300 MHz,CDCI3):- 6 8.15(d, J=7.2Hz, 1H),7.9(d, J=7.8Hz, 1H), 7.7(m,
4H), 7.55 (m, 3H),7.45(m,2H), 7.3(d, J=8.1Hz, 2H), 7.2(d, J=7.5Hz, 1H),6.8(t,
1H), 3.7(s,2H), 3.1(m,2H), 2.9(m,1H), 2.6(s, 3H), 2to2.6(m,6H).
Example 63: 244-({4[5-(6-methylpyridin-2-y1)-4H-1 ,2,4-triazol-3-ylipiperidin-
1-y1) methyl) phenyl]-3-phenylimidazo[1,2-a]pyrimidine
Example 63 is prepared according to example 61
MS (M+1) : 527.2
1H NMR (300 MHz,CDCI3):- 6 8.237 (dd, J = 6.6,1.8 Hz, 1H), 7.938 (d, J=7.5Hz,
1H), 7.751-7.668 (m, 3H), 7.598-7.525 (m, 3H), 7.47-7.439 (m, 2H), 7.30
(d,J=8.1 Hz, 2H), 7.193 ( d, J=7.5 Hz, 1H), 6.835-6.799 (m,1H) , 3.665 (s, 2H)
3.088-2.938 (m, 3H), 2.583 (s, 3H), 2.351 (t, J=7.8 Hz,2H) , 2.121-2.025
(m,4H).
Example 64: 244-({4[5-(6-methylpyridin-2-y1)-4H-1 ,2,4-triazol-3-ylipiperidin-
1 -y1) methyl) phenyl]-3-phenylimidazo[1,2-a]pyridine
Example 64 is prepared according to example 61
MS (M+1) : 526.1
1H NMR (300 MHz,CDCI3):- 6 8 (m, 2H),7.6to7.8(m, 4H), 7.4to7.6(m, 5H),
7.1to7.4 (m, 4H), 6.7(t, 1H), 3.6(s,2H), 2.8to3.1(m,3H), 2.5(s,3H),
1.8to2.4(m,6H).
Example 65: '8-methoxy-244-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1 -yl}methyl)phenyI]-3-phenylimidazo[1,2-a]pyrazine
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
79
Example 65 is prepared according to example 61
MS (M+1) :557.13
1H NMR (300 MHz,CDCI3):- 6 8 (d, J=7.8Hz, 1H), 7.65-7.75(m, 3H), 7.49-7.57
(m, 4H), 7.4-7.48(m, 2H), 7.27-7.35(m,2H), 7.2(d, J=7.8Hz, 1H
),4.2(s,2H),3.7(s,2H), 2.8-3.2(m,3H), 2.6(s,3H), 2-2.3(m,4H).
Example 66: '644-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -yl}methyl)phenyI]-5-phenylimidazo[2,1-b][1,3]thiazole
Example 66 is prepared according to example 61
MS (M+1) : 532.07
1H NMR (300 MHz,CDCI3):- 6 7.94(d, J=7.5Hz, 1H), 7.75-7.15(m, 13H), 6.81(d,
J=4.2Hz, 1H), 3.52(s, 2H), 3.05-2.75(m, 3H), 2.58(s, 3H), 2.2-1.8(m, 6H).
Example 67: '644-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -yl}methyl)phenyI]-5-phenylimidazo[2,1-b][1,3,4]thiadiazole
Example 67 is prepared according to example 61
MS (M+1) :533.13
1H NMR (300 MHz,CDCI3):- 6 8.54 (s, 1H), 7.94 (d, J=7.5Hz, 1H), 7.75-7.55 (m,
4H), 7.5-7.15 (m, 8H), 3.58 (s, 2H), 3.1-2.8 (m, 3H), 2.58 (s, 3H), 2.4-1.9
(m, 6H).
Example 68: '7-methoxy-244-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1}methyl)phenyl]-3-phenylimidazo[1,2-a]pyridine
Example 68 is prepared according to example 61
MS (M+1) : 557.3
1H NMR (300 MHz,CDCI3):- 6 8 (m, 2H), 7.65to7.75(m,3H), 7.4to7.6(m, 5H),
7.1to7.3(m,3H), 6.4(d, 1H), 4.1(s,3H), 3.6(s, 2H), 2.8to3.1 (m,3H), 2.5(s,
3H),
1.9to2.4(m,6H).
Example 69: 6-methyl-244-({445-(6-methylpyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1 -y1) methyl) phenyl]-3-phenylimidazo[1,2-b]pyridazine (467395)
Example 69 is prepared according to example 61
MS (M+1) :541.13
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
1H NMR (300 MHz,CDCI3):- 6 7.85to7.95 (m, 2H), 7.55to7.75(m,5H),
7.4to7.5(m,3H), 7.3to7.4(m, 2H), 7.2(d,1H), 6.9(d, 1H), 3.7(s, 2H), 2.9to3.1
(m,3H), 2.6(s,3H), 2.5(s,3H), 2to2.3(m,6H).
5 Example 70: '244-({445-(5-methylpyridin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-
1-yl}methyl) phenyl]-3-phenylimidazo[1,2-a]pyrimidine
To a solution of 0.110g (0.367mM) 4-(3-phenylimidazo [1,2-a] pyrimidin-2-y1)
benzaldehyde
in 3mL of THF is added triethylamine (2eq). The reaction mixture is stirred
for 5
10 minutes. To this solution is added 5-methyl-2- [3-(piperidin-4-yI)-1H-1,
2,4-triazol-
5-yl] pyridine
dihydrochloride(Intermediate D) (1.5 eq) followed by acetic acid (2.5eq). The
re-
action mixture is stirred for 10 minutes. To this mixture is added NaBH(OAc)3
(6
eq) over a period of 40 minutes. The reaction mixture is stirred overnight.
The
15 reaction mixture is quenched with methanol and concentrated. The residue
ob-
tained is taken up in chloroform and washed with water, dried and
concentrated.
The crude is purified on a flash column chromatography to obtain the desired
compound
MS (M+1): 527.13
20 1H NMR (300 MHz,CDCI3):- 6 8.57-8.55 (q,1H), 8.43 (bs, 1H), 8.23 (dd,
J=6.9,1.8Hz, 1H), 8.04 (d, J=8.1Hz, 2H), 7.64-7.59(m, 1 H), 7.58-7.52 (m, 3H),
7.49-7.47 ( m, 2H), 7.30 (d, J =8.4Hz 2H), 6.83-6.79(q,1 H), 3.63 (s,2H), 3.06
(d,2H), 2.91 (bs,1H), 2.39 (s, 3H), 2.72 (bs, 1H), 2.10-1.87 (m, 4H).
25 Example 71: '2-[4-({4-[5-(5-methylpyridin-2-yI)-4H-1, 2,4-triazol-3-yl]
piperidin-1 -y1) methyl) phenyl]-3-phenylimidazo [1,2-a] pyridine-8-
carbonitrile
Example 71 is prepared according to example 70
MS (M+1) :551.2
30 1H NMR (300 MHz,CDCI3):- 6 8.5 (s, 1H), 8.1(d, J=6.9Hz, 1H), 8 (d,
J=8.1Hz,
1H), 7.6to7.7(m, 4H), 7.5to7.6(m, 3H), 7.4to7.5(m, 2H), 7.3(m, 2H), 6.8(d,
1H),
3.6(s, 2H), 2.8to3.1(m, 3H), 2.4(s, 3H), 1.9to2.2(m, 6H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
81
Example 72: '244-({445-(5-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -yl}methyl) phenyl]-3-phenylimidazo[1,2-a]pyridine-7-carbonitrile
Example 72 is prepared according to example 70
MS (M+1) :551.27
1H NMR (300 MHz, DMSO-d6) 68.50(bs, 1H), 8.45 (s, 1H), 8.13 (dd, J=7.2Hz,
0.9Hz, 1H), 7.92 (d, J=8.1Hz,1H), 7.75 (bs, 1H), 7.67-7.53 (m, 7H), 7.28 (d,
J=8.4Hz, 2H), 7.17 (dd, J=7.2Hz, 1.8Hz, 1H) 3.47- (s, 2H), 2.87-2.72(m, 3H),
2.11-1.74(m, 6H)
Example 73: '644-({445-(5-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -yl}methyl)phenyI]-5-phenylimidazo[2,1-b][1,3]thiazole
Example 73 is prepared according to example 70
MS (M+1) : 532.07
1H NMR (300 MHz,CDCI3):- 6 8.47 (s, 1H), 8.04 (d, J=7.8Hz, 1H), 7.65-7.2(m,
12H), 6.81 (d, J=4.5Hz, 1H), 3.58 (s, 2H), 3.1-2.8 (m, 3H), 2.39 (s, 3H), 2.3-
1.9
(m, 6H)
Example 74: '644-({445-(5-methylpyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1 -y1) methyl) phenyl]-5-phenylimidazo[2,1-b][1,3,4]thiadiazole
Example 74 is prepared according to example 70
MS (M+1) : 533.07
1H NMR (300 MHz,CDCI3):- 6 8.53 (s, 1H), 8.47 (s, 1H), 8.04 (d, J=7.8 Hz, 1H),
7.68-7.25 (m, 11H), 3.57 (s, 2H), 3.1-2.8 (m, 3H), 2.39 (s, 3H), 2.3-1.9 (m,
6H)
Example 75: '244-({4[5-(5-chloropyridin-2-y1)-1 H-1 ,2,4-triazol-3-
ylipiperidin-
1 -yl}methyl) phenyl]-3-phenylimidazo[1,2-a]pyridine-8-carbonitrile
To a solution of 0.230g (0.712mM) of 2-(4-formylphenyI)-3-phenylimidazo [1,2-
a]
pyridine-8-carbonitrile in 5mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 5-chloro-2- [3-
(piperidin-
4-yI)-1H-1, 2,4-triazol-5-yl] pyridine dihydrochloride (Intermediate E) (1.5
eq) fol-
lowed by acetic acid (2.5eq). The reaction mixture is stirred for 10 minutes.
To
this mixture is added NaBH(OAc)3 (6 eq) over a period of 40 minutes. The reac-
tion mixture is stirred overnight. The reaction mixture is quenched with
methanol
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
82
and concentrated. The residue obtained is taken up in chloroform and washed
with water, dried and concentrated. The crude is purified on a flash column
chro-
matography to obtain the desired compound.
MS (M+1): 571.2
1H NMR (300 MHz,CDCI3):- 6 8.61 (bs,1H), 8.11 (t, 2H), 7.78-7.58 (m, 2H), 7.71-
7.7.43 (m, 6H), 7.28-7.17(m, 4 H), 6.85 (t, 1H), 4.88 ( bs, 1H), 3.55
(s,2H),2.97(,3H),2.21-1.7(m,5H).
Example 76: '244-({445-(5-chloropyridin-2-y1)-4H-1,2,4-triazol-3-ylipiperidin-
1-yl}methyl) phenyl]-3-phenylimidazo[1,2-a]pyrimidine
Example 76 is prepared according to example 75
MS (M+1): 547.07
1H NMR (300 MHz,CDCI3):- 6 8.612 (bs, 1H), 8.55 (q, 1H), 8.24 (dd, J= 2.1 Hz,
6.9Hz,1H), 8.10 (d, J= 8.4Hz, 1H), 7.75 (dd, J=2.1,8.4Hz, 1 H), 7.68 (d, J=8.1
Hz,2H), 7.58-7.52 (m,3H), 7.48-7.431 (m, 2H), 7.26-7.22 (m, 2 H),6.82 (q,
1H),3.52 (s, 2H),2.98-2.86(m,3H),2.157-1.95 (m,6H).
Example 77: 3-phenyl-244-({445-(pyrimidin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl) phenyliimidazo[1,2-a]pyridine-8-carbonitrile
To a solution of 0.130g (0.582mM) of 2-(4-formylphenyI)-3-phenylimidazo [1,2-
a]
pyridine-8-carbonitrile in 4mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 2-[3-(piperidin-4-
yI)-1H-
1, 2,4-triazol-5-yl] pyrimidine
Dihydrochloride (Intermediate F) (1.5 eq) followed by acetic acid (2.5eq). The
re-
action mixture is stirred for 10 minutes. To this mixture is added NaBH(OAc)3
(6
eq) over a period of 40 minutes. The reaction mixture is stirred overnight.
The
reaction mixture is quenched with methanol and concentrated. The residue ob-
tained is taken up in chloroform and washed with water, dried and
concentrated.
The crude is purified on a flash column chromatography to obtain the desired
compound.
MS (M+1) : 538.2
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
83
1H NMR (300 MHz,CDCI3):- 6 8.15(s, 1H),7.9(m, 2H), 7.7(m, 3H), 7.55 (m,
3H),7.45(m,2H), 7.3(d, J=8.1 Hz, 2H), 7.2(d, J=7.5Hz, 1H),6.9(d, 1H),
3.6(s,2H),
3(d,2H), 2.9(m,1H), 2.5(s, 3H), 2-2.6(m,6H).
Example 78: 3-pheny1-244-({445-(pyrimidin-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl) phenylpmidazo[1,2-a]pyridine-7-carbonitrile
Example 78 is prepared according to example 77
MS (M+1) : 538.2
1H NMR (300 MHz,CDCI3):- 6 8.92(d, J= 4.5 Hz, 1H), 8.45 (s, 1H), 8.13 (d,
J=7.2
Hz, 1H), 7.53-7.66 (m, 9H), 7.29 (d, J=7.8 Hz, 2H), 7.15-7.18 (dd, J= 1.5 Hz,
6.9
Hz, 2H), 3.49 (s, 2H), 2.85 (bs, 2H), 1.755-2.101 (m, 5H)
Example 79: '3-pheny1-2-(4-{[4-(5-pyrimidin-2-y1-1H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl} phenyl)imidazo[1,2-a]pyrimidine
Example 79 is prepared according to example 77
MS (M+1) :514.13
1H NMR (300 MHz,CDCI3):- 6 8.88 (d, J = 4.2 Hz, 2H), 8.564 (s, 1H), 8.24 (dd,
J
= 7.5,1.8 Hz 1H), 7.70 (d, J = 8.1Hz, 2H), 7.611-7.516 (m, 3H), 7.467-7.435
(m,
2H), 7.326 (m,1 H), 7.282-7.255 ( m, 2H), 6.835-6.799 (m,1 H), 3.604 (s, 2H),
3.01
(d, J = 11.1Hz, 3H) 2.356-2.063 (m, 6H).
Example 80: '3-pheny1-2-(4-{[4-(5-pyrimidin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl] methyl) phenyl)imidazo[1,2-a]pyridine
Example 80 is prepared according to example 77
MS (M+1) : 513.13
1H NMR (300 MHz, DMSO-d6) 6 9.0 (s, 2H), 8(d,1H), 7.4-7.7(m, 9H), 7.1to7.3(m,
3H), 6.9(t, 1H), 3.5(s,2H), 2.6-3.2(m,3H), 1.7-2.3(m,6H).
Example 81: '5-pheny1-6-(4-{[4-(5-pyrimidin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl} phenyl)imidazo[2,1-b][1,3]thiazole
Example 81 is prepared according to example 77
MS (M+1) :519.07
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
84
1H NMR (300 MHz, DMSO-d6) 68.91(d, J=4.2Hz, 2H), 7.7(d, J=4.5Hz, 1H), 7.6-
7.18(m, 12H), 3.46(s, 2H), 2.88-2.63(m, 3H), 2.22-1.74(m, 6H).
Example 82: '5-pheny1-6-(4-{[4-(5-pyrimidin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl} phenyl)imidazo[2,1-b][1,3,4]thiadiazole
Example 82 is prepared according to example 77
MS (M+1) :520.13
1H NMR (300 MHz,CDCI3):- 6 8.90 (d, J=4.8Hz, 2H), 8.54 (s, 1H), 7.65-7.25 (m,
11H), 3.58 (s, 2H), 3.1-2.9 (m, 3H), 2.25-1.95 (m, 6H)
Example 83: '6-methy1-3-pheny1-2-(4-{[4-(5-pyrimidin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl] methyl) phenyl)imidazo[1,2-b]pyridazine
Example 83 is prepared according to example 77
MS (M+1) :528.2
1H NMR (300 MHz,CDCI3):- 6 9.0 (s, 1H), 7.9(d, 1H), 7.7(d, 2H), 7.3to7.6(m,
8H),
6.9(d, 1H), 4.1(s, 2H), 3.1-3.4(m, 3H), 2.8(m, 3H), 2.5(s, 1H), 2.2-2.4(m,
3H).
Example 84: '3-pheny1-244-({445-(1,3-thiazol-2-y1)-1H-1,2,4-triazol-3-
ylipiperidin-1-y1) methyl)phenylpmidazo[1,2-a]pyrimidine
To a solution of 0.340g (1.137mM) of 4-(3-phenylimidazo [1,2-a] pyrimidin-2-
y1)
benzaldehyde
in 7 mL of THF is added triethylamine (2eq). The reaction mixture is stirred
for 5
minutes. To this solution is added 4-[5-(1,3-thiazol-2-y1)-1H-1, 2,4-triazol-3-
yl]
piperidine dihydrochloride (Intermediate G) (1.5 eq) followed by acetic acid
(2.5eq). The reaction mixture is stirred for 10 minutes. To this mixture is
added
NaBH(OAc)3 (6 eq) over a period of 60 minutes. The reaction mixture is stirred
overnight. The reaction mixture is quenched with methanol and concentrated.
The residue obtained is taken up in chloroform and washed with water, dried
and
concentrated. The crude is purified on a flash column chromatography to obtain
the desired compound.
MS (M+1) :519.07
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
1H NMR (300 MHz,CDCI3):- 6 8.567(s, 1H), 8.26 (d, J= 6Hz 1H), 7.92 (d, J = 3
Hz 1H), 7.67 (d, J = 8.1Hz, 2H), 7.549-7.364 (m, 6H), 7.227 (s, 2H), 6.852-
6.816(m,1H), 3.585 (s, 2H), 2.99 (d, 3H) 2.211-1.971 (m, 6H).
5 Example 85: '3-phenyl-244-({445-(1,3-thiazol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1}methyl) phenylpmidazo[1,2-a]pyridine
Example 85 is prepared according to example 84
MS (M+1) :518.07
1H NMR (300 MHz,CDCI3):- 6 7.9 - 8.1 (m, 2H), 7.7(d,1H), 7.4-7.7(m, 8H), 7.1-
10 7.3(m, 3H), 6.8(t, 1H), 3.6(s,2H), 2.7-3.1(m,3H), 1.8-2.3(m,6H).
Example 86: '3-phenyl-244-({445-(1,3-thiazol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1}methyl) phenylpmidazo[1,2-a]pyridine-8-carbonitrile
Example 86 is prepared according to example 84
15 MS (M+1) : 543.07
1H NMR (300 MHz,CDCI3):- 6 8.2 (d, 1H), 8(s,1H), 7.6-7.7(m, 3H), 7.4-7.6(m,
6H),7.2-7.4(m, 2H), 6.8(t, 1H), 3.6(s,2H), 2.8-3.1(m,3H), 1.9-2.3(m,6H).
Example 87: '5-phenyl-644-({445-(1 ,3-thiazol-2-y1)-4H-1 ,2,4-triazol-3-
20 ylipiperidin-1-yl}methyl) phenylpmidazo[2,1-13][1,3]thiazole
Example 87 is prepared according to example 84
MS (M+1) : 524.07
1H NMR (300 MHz,CDCI3):- 6 7.95 (d, J=3.3Hz, 1H), 7.6-7.2 (m, 12H), 6.84 (d,
J=4.8Hz, 1H), 3.59 (s, 2H), 3.05-2.85 (m, 3H), 2.3-1.9 (m, 6H).
Example 88: '5-phenyl-644-({445-(1,3-thiazol-2-y1)-1 H-1 ,2,4-triazol-3-
ylipiperidin-1 -yl}methyl) phenylpmidazo[2,1-b][1,3,4]thiadiazole
Example 88 is prepared according to example 84
MS (M+1): 525.07
1H NMR (300 MHz,CDCI3):- 6 8.55 (s, 1H), 7.94 (d, J=3Hz, 1H), 7.65-7.25 (m,
11H), 3.58(s, 2H), 3.1-2.8 (m, 3H), 2.3-1.8 (m, 6H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
86
Example 89: '3-phenyl-244-({445-(1,3-thiazol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl) phenylpmidazo[1,2-a]pyridine-7-carbonitrile
Example 89 is prepared according to example 84
MS (M+1) :543.13
1H NMR (300 MHz,CDCI3):- 6 8.101(s 1H), 8.023 (dd J=7.2 Hz, 0.9 Hz, 1H),
7.966 (d, J=3.3 Hz, 1H), 7.64 (d, J=8.1 Hz, 2H), 7.611-7.559 (m, 3H), 7.479-
7.448
(m, 3H), 7.31 (d, J=8.4 Hz, 2H), 6.908(dd, J=7.2Hz, J=1.8 Hz, 1H) 3.615 (s,
2H),
3.053-2.915(m, 4H), 2.24-1.993 (m, 7H)
Example 90: '244-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-3-phenylimidazo[1,2-a]pyridine-8-carbonitrile
To a solution of 0.180g (0.557mM) 2-(4-formylphenyI)-3-phenylimidazo [1,2-a]
pyridine-8-carbonitrile in 5 mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 4-[5-(furan-2-yI)-
1H-1,
2,4-triazol-3-yl] piperidine dihydrochloride (Intermediate H) (1.5 eq)
followed by
acetic acid (2.5eq). The reaction mixture is stirred for 10 minutes. To this
mixture
is added NaBH(OAc)3 (6 eq) over a period of 60 minutes. The reaction mixture
is
stirred overnight. The reaction mixture is quenched with methanol and concen-
trated. The residue obtained is taken up in chloroform and washed with water,
dried and concentrated. The crude is purified on a flash column chromatography
to obtain the desired compound.
MS (M+1) :526.13
1H NMR (300 MHz,CDCI3):- 6 8.2 (d, 1H), 7.6-7.7(m,3H), 7.5-7.6(m, 3H),
7.5(m,1H), 7.4-7.5(m, 2H), 7.2-7.3(m, 2H), 7(d, 1H), 6.8(t, 1H), 6.5(q, 1H),
3.6(s,2H), 2.8-3.1(m,3H), 1.8-2.3(m,6H).
Example 91: '244-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-3-phenylimidazo[1,2-a]pyridine
Example 91 is prepared according to example 90
MS (M+1) : 501.13
1H NMR (300 MHz,CDCI3):- 6 8 (d, 1H), 7.7(d,1H), 7.4-7.7(m, 9H), 7.2-
7.3(m,3H), 7 (d, 1H), 6.8(t, 1H), 6.5(q, 1H), 3.6(s,2H), 2.8-3 (m,3H), 1.8-
2.2(m,6H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
87
Example 92: '244-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-6-methyl-3-phenylimidazo[1,2-b]pyridazine
Example 92 is prepared according to example 90
MS (M+1) : 516.13
1H NMR (300 MHz,CDCI3):- 6 7.9 (d, 1H), 7.6(m,4H), 7.4to7.5(m,4H), 7.2to7.3(m,
2H), 6.9to7(m,2H), 6.5(q, 1H), 3.6(s, 2H), 2.8to3.1 (m,3H), 2.5(s,3H),
1.9to2.2(m,6H).
Example 93: '644-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-5-phenylimidazo[2,1-13][1,3]thiazole
Example 93 is prepared according to example 90
MS (M+1) : 507.07
1H NMR (300 MHz,CDCI3):- 6 8.55 (s, 1H), 7.65-7.2 (m, 12H), 6.98 (d, J=3.3Hz,
1H), 6.55-6.45 (m, 1H), 3.56 (s, 2H), 3.1-2.8 (m, 3H), 2.3-1.8 (m, 6H)
Example 94: '644-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-5-phenylimidazo[2,1-13][1,3,4]thiadiazole
Example 94 is prepared according to example 90
MS (M+1) : 508.07
1H NMR (300 MHz,CDCI3):- 6 7.6-7.15(m, 11H), 6.97(d, J=3Hz, 1H), 6.82(d,
J=4.5Hz, 1H), 6.55-6.45(m, 1H), 3.51(s, 2H), 3.0-2.8(m, 3H), 2.2-1.8(m, 6H).
Example 95: '244-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-3-phenylimidazo[1,2-a]pyrimidine
Example 95 is prepared according to example 90
MS (M+1) : 502.2
1H NMR (300 MHz,CDCI3):- 6 8.57-8.55 (m, 1H), 8.25 (dd, J=6.6,1.8Hz, 1H), 7.69
(d, J = 8.4 Hz 2H), 7.56-7.43 (m, 6H), 7.24 (s, 2H), 6.98 (d,J=3.3Hz, 1H),
6.84-
6.81 ( m, 1H), 6.50-6.48 (m, 1H), 3.581 (s, 2H), 3.02-2.91 (m, 3H) 2.18(t,
2H),2.085-1.93(m,4H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
88
Example 96: '244-({445-(2-fury1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl)pheny1]-3-phenylimidazo[1,2-a]pyridine-7-carbonitrile
Example 96 is prepared according to example 90
MS (M+1) :526.13
1H NMR (300 MHz,CDCI3):- 6 8.07(s, 1H), 7.51 (d, J=5.4 Hz, 1H), 7.64(d, J=8.4
Hz, 2H), 7.58-7.43 (m 6H), 7.29(d, J=8.1 Hz, 2H), 7.98(d, J=3.3Hz, 1H), 6.89
(dd,
J=6.9 Hz, 4.5 Hz, 1H), 6.51 (dd, J=3.3 Hz, 1.8Hz, 1H) 3.6 (s, 2H), 3.04-
2.91(m,
3H), 2.4-1.94 (m, 8H)
Example 97: '3-phenyl-244-({445-(2-thieny1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl) phenylpmidazo[1,2-a]pyridine
To a solution of 0.120g (0.402 mM) of 4-(3-phenylimidazo [1,2-a] pyridin-2-y1)
benzaldehyde
in 5 mL of THF is added triethylamine (2eq). The reaction mixture is stirred
for 5
minutes. To this solution is added 4-[5-(thiophen-2-yI)-1H-1, 2,4-triazol-3-
yl]
piperidine dihydrochloride (Intermediate I) (1.5 eq) followed by acetic acid
(2.5eq). The reaction mixture is stirred for 10 minutes. To this mixture is
added
NaBH(OAc)3 (6 eq) over a period of 40 minutes. The reaction mixture is stirred
overnight. The reaction mixture is quenched with methanol and concentrated.
The residue obtained is taken up in chloroform and washed with water, dried
and
concentrated. The crude is purified on a flash column chromatography to obtain
the desired compound.
MS (M+1) :517.13
1H NMR (300 MHz,CDCI3):- 6 8 (d, 1H), 7.4-7.7(m,9H), 7.2-7.4(m, 4H),
7.1(m,1H), 6.8(t, 1H), 3.6(s, 2H), 2.8-3.1 (m,3H), 1.8-2.2(m,6H).
Example 98: '3-phenyl-244-({445-(2-thieny1)-4H-1,2,4-triazol-3-ylipiperidin-1-
yl}methyl) phenylpmidazo[1,2-a]pyridine-8-carbonitrile
Example 98 is prepared according to example 97
MS (M+1) : 542.13
1H NMR (300 MHz,CDCI3):- 6 8.1 (d, 1H), 7.6-7.7(m,4H), 7.5-7.6(m, 3H), 7.4-
7.5(m,2H), 7.2-7.4(m,3H), 7.1(m,1H), 6.8(t, 1H), 3.6(s, 2H), 2.8-3.1 (m,3H),
1.8-
2.3(m,6H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
89
Example 99: '6-methyl-3-phenyl-244-({445-(2-thieny1)-4H-1,2,4-triazol-3-
ylipiperidin-1-yl}methyl)phenylpmidazo[1,2-1Apyridazine
Example 99 is prepared according to example 97
MS (M+1) : 532.13
1H NMR (300 MHz,CDCI3):- 6 7.9 (d, J=9.3Hz, 1H), 7.5-7.7(m,5H), 7.4-7.5(m,3H),
7.3-7.35(d, J=5.1 Hz ,1H), 7.2-7.3(m ,2H), 3.6(s,2H),2.8-3(m,3H),2.5(s,3H),1.8-
2.2(m,6H).
Example 100: '5-phenyl-644-({445-(2-thieny1)-4H-1,2,4-triazol-3-ylipiperidin-
1-yl}methyl) phenyliimidazo[2,1-13][1,3]thiazole
Example 100 is prepared according to example 97
MS (M+1) : 523.07
1H NMR (300 MHz,CDCI3):- 6 7.66-7.0(m, 14H), 6.825(d, J=4.5Hz, 1H), 3.61(s,
2H), 3.15-2.85(m, 3H), 2.3-1.9(m, 6H).
Example 101: '5-phenyl-644-({445-(2-thieny1)-4H-1,2,4-triazol-3-ylipiperidin-
1-yl}methyl) phenylpmidazo[2,1-13][1,3,4]thiadiazole
Example 101 is prepared according to example 97
MS (M+1): 524.07
1H NMR (300 MHz,CDCI3):- 6 8.57(s, 1H), 7.7-7.05(m, 13H), 3.61(s, 2H), 3.14-
2.85(m, 3H), 2.3-1.9(m, 6H).
Example 102: '3-phenyl-244-({445-(2-thieny1)-4H-1,2,4-triazol-3-ylipiperidin-
1-yl}methyl) phenyliimidazo[1,2-a]pyridine-7-carbonitrile
Example 102 is prepared according to example 97
MS (M+1) :542.13
1H NMR (300 MHz, DMSO-d6) 6 8.44(s, 1H), 8.13 (d, J=7.2 Hz, 1H), 7.64=7.5(m,
10H), 7.28 (d, J=5.4Hz, 2H), 7.72-7.12(m, 2H), 3.51(bs,3H), 2.89-2.78(m 3H),
2.1-1.74(m, 6H)
Example 103: '7-chloro-3-phenyl-244-({445-(2-thieny1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1} methyl) phenyliimidazo[1,2-a]pyridine
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Example 103 is prepared according to example 97
MS (M+1) :551.07
1H NMR (300 MHz,CDCI3):- 6 7.9 (d, J=7.5Hz,1H), 7.7(d, J=2.1Hz,1H), 7.5-7.67
(m, 5H), 7.4-7.5(m,2H), 7.18-7.35(m,3H), 7.09(q,1H), 6.7(q, 1H), 3.5(s,2H),
2.8-
5 3(m,3H), 1.7-2.2(m,6H)
Example 104: '6-methyl-3-phenyl-244-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-
3-ylipiperidin-1-yl}methyl)phenylpmidazo[1,2-1Apyridazine
To a solution of 0.150g (0.479 mM) of 4-(6-methyl-3-phenylimidazo [1,2-b] pyri-
10 dazin-2-y1) benzaldehyde in 6 mL of THF is added triethylamine (2eq).
The reac-
tion mixture is stirred for 5 minutes. To this solution is added 445-(1 H-
pyrrol-2-y1)-
1H-1, 2,4-triazol-3-yl] piperidine dihydrochloride (Intermediate J) (1.5 eq)
followed
by acetic acid (2.5eq). The reaction mixture is stirred for 10 minutes. To
this mix-
ture is added NaBH(OAc)3 (6 eq) over a period of 40 minutes. The reaction mix-
15 ture is stirred overnight. The reaction mixture is quenched with
methanol and
concentrated. The residue obtained is taken up in chloroform and washed with
water, dried and concentrated. The crude is purified on a flash column chroma-
tography to obtain the desired compound.
MS (M+1) :515.13
20 1H NMR (300 MHz,CDCI3):- 6 9.5(s, 1H), 7.9 (d, J=9Hz, 1H), 7.55-7.65(m,
4H),
7.4-7.5 (m, 3H), 7.2-7.3(m, 2H), 6.8-7(m, 2H), 6.7(s, 1H), 6.2-6.3(s, 1H),
3.5(s,
2H), 2.8-3(m, 3H), 2.5(s, 3H) 1.7-2.2(m, 6H)
Example 105: '3-phenyl-244-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-3-
25 ylipiperidin-1-yl}methyl)phenyliimidazo[1,2-a]pyridine-8-carbonitrile
Example 105 is prepared according to example 104
MS (M+1) :525.13
1H NMR (300 MHz,CDCI3):- 6 9.5(s,1 H),8.1 (d, J=6.9Hz,1H), 7.6-7.7(m, 3H), 7.5-
7.6 (m, 3H), 7.4-7.5(m,2H), 7.2-7.3(m,2H), 6.5-7(m,3H), 6.3(m, 1H), 3.5(s,2H),
30 2.8-3(m,3H), 1.7-2.2(m,6H).
Example 106: '3-phenyl-244-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1) methyl)phenylpmidazo[1,2-a]pyrimidine
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
91
Example 106 is prepared according to example 104
MS (M+1) :501.13
1H NMR (300 MHz,CDCI3):- 6 8.56 (q, J = 2.1 Hz, 1H), 8.25 (dd, J=6.9,2.1Hz,
1H), 7.68 (d, J =8.1Hz,2H), 7.60-7.51 (m, 3H), 7.45-7.428 (m, 2H), 7.24 (s,
2H),
6.91-6.76 ( m, 3H), 6.24 (q, J =3.3Hz,1H), 3.56(s,2H ), 2.98 (d, J=11.1Hz,
2H),
2.88-2.81 (m, 1H), 2.18-1.93 (m, 6H),
Example 107: '7-chloro-3-phenyl-244-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-
3-ylipiperidin-1-yl}methyl)phenylpmidazo[1,2-a]pyridine
Example 107 is prepared according to example 104
MS (M+1) :534.13
1H NMR (300 MHz,CDCI3):- 6 8.56 (q, J = 2.1 Hz, 1H), 8.25 (dd, J=6.9,2.1Hz,
1H), 7.68 (d, J 9.5(s,1H),7.9(d, J=7.5Hz,1H), 7.4-7.7(m, 8H), 7.2-7.3 (m, 2H),
6.9(m,1 H), 6.7-6.8(m,2H), 6.3(q,1 H), 3.5(s,2H), 2.8-3(m,3H), 1.8-2.2(m,6H).
Example 108: '5-phenyl-644-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1) methyl)phenyliimidazo[2,1-13][1,3,4]thiadiazole
Example 108 is prepared according to example 104
MS (M+1) :507.13
1H NMR (300 MHz,CDCI3):- 6 9.7(s, 1H), 8.54(s, 1H), 7.66-7.24(m, 10H), 6.87(br
s, 1H), 6.73(br s, 1H), 6.26(m, 1H), 3.57(s, 2H), 3.1-2.6(m, 3H), 2.25-1.85(m,
6H).
Example 109: '5-phenyl-644-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1) methyl)phenyliimidazo[2,1-13][1,3]thiazole
Example 109 is prepared according to example 104
MS (M+1) :506.13
1H NMR (300 MHz,CDCI3):- 6 9.93 (s, 1H), 7.6-7.2 (m, 10H), 6.9-6.7 (m, 3H),
6.3-6.2 (m, 1H), 3.59 (s, 2H), 3.0-2.8 (m, 3H), 2.3-1.8 (m, 7H)
Example 110: '3-phenyl-244-({445-(1H-pyrrol-2-y1)-4H-1,2,4-triazol-3-
ylipiperidin-1-y1) methyl)phenylpmidazo[1,2-a]pyridine-7-carbonitrile
Example 110 is prepared according to example 104
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
92
MS (M+1) : 525.2
1H NMR (300 MHz,CDCI3):- 6 8.06(s, 1H), 8.8 (d, J=7.2 Hz, 1H), 7.614(d, J=4.2
Hz, 2H), 7.59-7.57(m 3H), 7.46-7.43(m, 2H), 7.26(s, 2H), 6.91-6.87 (m, 2H),
6.725 (s, 1H) 6.28 (dd, J=6Hz, 2.7 Hz, 1H), 3.515 (s, 1H), 2.96 (d, 2H), 2.82
(m,
1H), 2.15-1.71(m, 7H)
Example 111: '3-pheny1-2-(4-{[4-(3-pyridin-2-y1-1H-pyrazol-5-yl)piperidin-1-
yl]methyl} phenyl)imidazo[1,2-a]pyridine
To a solution of 0.250g (0.838 mM) of 4-(3-phenylimidazo [1,2-a] pyridin-2-y1)
benzaldehyde
in 6 mL of THF is added triethylamine (2eq). The reaction mixture is stirred
for 5
minutes. To this solution is added 2[3-(piperidin-4-y1)-1H-pyrazol-5-yl]
pyridine
dihydrochloride (Intermediate K) (1.5 eq) followed by acetic acid (2.5eq). The
re-
action mixture is stirred for 10 minutes. To this mixture is added NaBH(OAc)3
(6
eq) over a period of 40 minutes. The reaction mixture is stirred overnight.
The
reaction mixture is quenched with methanol and concentrated. The residue ob-
tained is taken up in chloroform and washed with water, dried and
concentrated.
The crude is purified on a flash column chromatography to obtain the desired
compound.
MS (M+1) : 511.13
1H NMR (300 MHz,CDCI3):- 6 8.6 (d, 1H), 7.9(d, 1H), 7.6-7.8 (m, 5H), 7.4-
7.6(m,
5H), 7.15-7.4(m,4H), 6.8(t,1H), 6.6(s,1H), 3.7(s,2H),3.1(m,2H), 2.8(m, 1H),
1.8-
2.4(m,6H).
Example 112: '5-pheny1-6-(4-{[4-(5-pyridin-2-y1-1H-pyrazol-3-yl)piperidin-1-
yl]methyl} phenyl)imidazo[2,1-13][1,3,4]thiadiazole
Example 112 is prepared according to example 111
MS (M+1) :518.07
1H NMR (300 MHz,CDCI3):- 6 9.22 (s, 1H), 8.53 (s, 1H), 7.9-7.75 (m, 2H), 7.6-
7.2
(m, 10H), 6.61 (s, 1H), 3.49 (s, 2H), 3.0-2.5 (m, 3H), 2.2-1.6 (m, 6H)
Example 113: '5-pheny1-6-(4-{[4-(5-pyridin-2-y1-1H-pyrazol-3-yl)piperidin-1-
yl]methyl} phenyl)imidazo[2,1-13][1,3]thiazole
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
93
Example 113 is prepared according to example 111
MS (M+1) :517.07
1H NMR (300 MHz,CDCI3):- 6 12.75(s, 1H), 8.53(d, J=4.8Hz, 1H), 7.92-7.18(m,
14H), 6.6(s, 1H), 3.45(s, 2H), 2.95-2.55(m, 3H), 2.2-1.55(m, 5H).
Example 114: '3-phenyl-2-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-
1-yl]methyl} phenyl)imidazo[1,2-a]pyridine-7-carbonitrile
Example 114 is prepared according to example 111
MS (M+1) : 536.2
1H NMR (300 MHz,CDCI3):- 6 8.57(d, J=4.8 Hz, 1H), 8.06(s, 1H), 8.02(d, J=7.2
Hz, 1H), 7.73-7.44 (m, 9H), 7.30 (d, J=8.1 Hz, 2H), 7.23-7.19 (m, 1H),6.89
(dd,
J=7.2 Hz, 1.8 Hz, 1H), 6.59(s, 1H), 3.56 (s, 2H), 2.50(d, 2H), 2.74 (m, 1H),
2.15-
1.82(m, 14H)
Example 115: '3-phenyl-2-(4-{[4-(5-pyridin-2-y1-1,2,4-oxadiazol-3-yl)piperidin-
1-yl]methyl} phenyl) imidazo[1,2-a]pyridine-7-carbonitrile
To a solution of 0.250g (0.838 mM) of 2-(4-formylphenyI)-3-phenylimidazo [1,2-
a]
pyridine-7-carbonitrile in 6 mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 2-[3-(piperidin-4-
yI)-
1,2,4-oxadiazol-5-yl] pyridine
dihydrochloride (Intermediate L)(1.5 eq) followed by acetic acid (2.5eq). The
reac-
tion mixture is stirred for 10 minutes. To this mixture is added NaBH(OAc)3 (6
eq)
over a period of 40 minutes. The reaction mixture is stirred overnight. The
reac-
tion mixture is quenched with methanol and concentrated. The residue obtained
is taken up in chloroform and washed with water, dried and concentrated. The
crude is purified on a flash column chromatography to obtain the desired com-
pound.
MS (M+1) : 537.6
1H NMR (300 MHz, DMSO-d6) 6 8.74(d, J=4.8 Hz, 1H), 8.43 (s, 1H), 8.113 (d,
J=7.2Hz, 1H), 8.061-7.99 (m, 2H), 7.64-7.51 (m, 8H), 7.3 (d, J=8.1 Hz, 2H),
7.15
(dd, J=6.9 Hz 1.5 Hz, 1H), 3.47- (s, 2H), 2.84-2.80(m, 2H), 2.17-2.05(m, 4H),
1.83-1.79(m, 2H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
94
Example 116: '3-pheny1-2-(4-{[4-(5-pyridin-2-y1-1,2,4-oxadiazol-3-yl)piperidin-
1-yl]methyl} phenyl)imidazo[1,2-a]pyrimidine
Example 116 is prepared according to example 115
MS (M+1): 514.13
1H NMR (300 MHz, DMSO-d6) 68.75 (d, J=4.8Hz, 1H), 8.59 (q, 1H), 8.55-8.44
(m,1 H), 8.07-7.98 (m, 2H), 7.64-7.47 (m, 8 H), 7.03 (q, /H), 7.31 (m,2H),
3.49 (s,
2H), 3.19-3.14 (m, 1 H), 2.91-2.82 (m, 2H), 2.24-2.06 (m, 4H), 1.89-
1.81(m,2H).
Example 117: 3-phenyl-2- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1) piperidin-1-
yl] methyl} phenyl) imidazo [1,2-a] pyridine-8-carbonitrile
To a solution of 0.250g (0.838 mM) of 2-(4-formylphenyI)-3-phenylimidazo [1,2-
a]
pyridine-8-carbonitrile in 6 mL of THF is added triethylamine (2eq). The
reaction
mixture is stirred for 5 minutes. To this solution is added 4-(5-phenyl-1H-1,
2,4-
triazol-3-y1) piperidine (Intermediate M) (1.5 eq) followed by acetic acid
(2.5eq).
The reaction mixture is stirred for 10 minutes. To this mixture is added
NaBH(OAc)3 (6 eq) over a period of 40 minutes. The reaction mixture is stirred
overnight. The reaction mixture is quenched with methanol and concentrated.
The residue obtained is taken up in chloroform and washed with water, dried
and
concentrated. The crude is purified on a flash column chromatography to obtain
the desired compound.
MS (M+1): 536.2
1H NMR (300 MHz,CDCI3):- 6 8.1 (d, 1H), 8.0(m, 2H), 7.6-7.7(m, 3H), 7.5-7.6(m,
3H), 7.4-7.5(m, 5H), 7.2-7.3 (m, 2H), 6.8(t, 1H), 3.6(s, 2H), 2.8-3.1(m, 3H),
1.9-
2.3(m, 6H).
Example 118: 3-phenyl-2- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1) piperidin-1-
yl] methyl} phenyl) imidazo [1,2-a] pyridine-7-carbonitrile
Example 118 is prepared according to example 117
MS (M+1): 536.2
1H NMR (300 MHz,CDCI3):- 6 8.1(s, 1H), 8.03-7.9(m, 3H), 7.62(d, J=8.4Hz, 2H),
7.57-7.54 (m, 3H), 7.46-7.39 (m, 5H), 7.3 (d, J=9.6Hz, 2H), 6.9 (dd, J=4.2Hz,
0.6Hz, 1H), 3.54 (s, 2H), 3.02-2.91(m, 3H), 2.185-1.87(m, 6H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
Example 119: 6-methyl-3-phenyl-2- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1)
piperidin-1-yl] methyl) phenyl) imidazo [1,2-b] pyridazine
Example 119 is prepared according to example 117
5 MS (M+1) : 526.13
1H NMR (300 MHz,CDCI3):- 6 8.1 (d, J=8.1Hz, 2H), 7.9(d, J=9.3Hz, 1H), 7.6(m,
4H), 7.4to7.5(m, 6H), 7.2to7.3(m, 2H), 6.9(d, J=9Hz, 1H), 3.6(s, 2H),
2.8to3.1(m,
3H), 2.5(s, 3H), 1.8to2.2(m, 6H).
10 Example 120: 7-chloro-3-phenyl-2- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1)
piperidin-1-yl] methyl) phenyl) imidazo [1,2-a] pyridine
Example 120 is prepared according to example 117
MS (M+1): 445.2
1H NMR (300 MHz,CDCI3):- 6 8.1 (d, 2H), 7.9(d, J=7.5Hz, 1H), 7.7(d, 1H),
15 7.6to7.65(m, 2H), 7.4to7.6(m, 7H), 7.2to7.3 (m, 2H), 6.8(dd, J=7.2Hz,
1H), 3.6(s,
2H), 2.8to3.1(m, 3H), 1.6to2.2(m, 6H).
Example 121: 3-phenyl-2- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1) piperidin-1-
yl] methyl) phenyl) imidazo [1,2-a] pyrimidine.
20 Example 121 is prepared according to example 117
MS (M+1): 512.27
1H NMR (DMSO d6):- 6 8.59 (q, J =2.4Hz,1H), 8.476 (dd, J=6.9,1.8Hz, 1H),
7.975 (d, J = 7.2 Hz 2H), 7.646-7.529 (m, 7H), 7.437-7.411 (m, 3H), 7.281
(d,J=7.8Hz, 2H), 7.034 ( q, 1H), 3.485 (s, 2H), 2.873 (d J=9.9Hz, 2H), 2.729
(t,
25 1H) 2.087(t, 2H), 1.925(t,2H),1.799(t,2H).
Example 122: 5-phenyl-6- (4-{[4-(5-pheny1-4H-1, 2,4-triazol-3-y1) piperidin-1-
yl] methyl) phenyl) imidazo [2,1-b][1,3] thiazole
30 Example 122 is prepared according to example 117
MS (M+1): 517.07
1H NMR (300 MHz,CDCI3):- 6 8.05-8.00(m, 2H), 7.6-7.21(m, 13H), 6.82(d,
J=4.5Hz, 1H), 3.58(s, 2H), 3.1-2.85(m, 3H), 2.3-1.9(m,6H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
96
Example 123: 5-phenyl-6- (4-{[4-(5-phenyl-4H-1, 2,4-triazol-3-y1) piperidin-1-
yl] methyl} phenyl) imidazo [2,1-b][1,3,4] thiadiazole
Example 123 is prepared according to example 117
MS (M+1): 518.13
1H NMR (300 MHz,CDCI3):- 6 8.54 (s, 1H), 8.07-7.98 (m, 2H), 7.68-7.22 (m,
12H), 3.56 (s,2H), 3.1-2.8 (m,3H), 2.25-1.8 (m, 6H).
Example 124: methyl 3-phenyl-2- [4-({4[5-(pyridin-2-y1)-4H-1, 2,4-triazol-3-
yl]
piperidin-1-y1} methyl) phenyl] imidazo [1,2-a] pyridine-7-carboxylate
Example 124 is prepared according to example 12
MS (M+1); 570.1
1H NMR (300 MHz,CDCI3):- 6 8.67 (d, J = 4.2 Hz, 1H), 8.414(s, 1H), 8.161 (d, J
=
7.8 Hz 1H), 7.967 (d, J = 6.6Hz, 1H), 7.825 (t, J=1.5Hz 1H), 7.621 (d,J=8.1Hz,
2H), 7.564-7.517 ( m, 3H), 7.373-7.319 (m,2H), 7.295 (s,2H ) 3.97 (s, 3H),
3.535
(s, 2H), 2.98 (d, J=11.4Hz, 2H),2.175-1.948 (m,6H).
Example 125: 3-phenyl-244-({445-(pyridin-2-y1)-4H-1,2,4-triazol-3-
yl]piperidin-1-yl}methyl) phenyl]imidazo[1,2-a]pyridine-7-carboxylic acid
To as solution of example 124 (0.100 gm 0.175mM) in methanol (3 ml) is added
1N NaOH solution (1 ml). The resulting reaction mixture is stirred at room tem-
perature for overnight. Reaction mass is then concentrated to remove methanol,
diluted with 4 ml of water and neutralized (pH ¨ 7). Solid precipitated is
filtered
and dried to afford the desired compound.
MS (M+1) :556.13
1H NMR (DMSO-d6):- 6 8.653 (bs, 1H), 8.32(s,1H), 8.174 (s, 1H), 8.079-8.015
(m, 2H), 7.93 (bs, 1H), 7.65-7.532 (m, 7H), 7.462 (bs, 1H), 7.337-7.263 ( m,
3H),
3.5 (s, 3H), 2.877 (d, J = 10.8Hz, 2H) 2.729 (m, 21H), 2.118 (t, J
=11.1Hz,2H),
1.953 (d,2H), 1.80 (t, J =10.5Hz,2H).
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
97
Example 126 : methoxy[5-phenyl-6-(4-{[4-(5-pyridin-2-y1-1H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl}phenyl)imidazo[2,1-b][1,3]thiazol-2-ylimethanol
Example 126 is prepared according to example 12
MS (M+1): 573.13
1H NMR (300 MHz,CDCI3):- 6 8.675(d, J=4.5Hz, 1H), 8.175 (d, J=7.8 Hz, 1H),
8.09 (s, 1H), 7.844 (td, J=7.8 Hz, 1.8Hz, 1H), 7.60 (d, J=8.1Hz, 2H), 7.511-
7.46
(m, 5H), 7.395-7.349 (m,1 H), 7.292 (d, J=6.3Hz, 1H) 3.934 (s, 3H), 3.55 (s,
2H),
3.011 (d, J=11.4 Hz, 2H), 2.88 (m, 2H), 2.257-1.975 (m, 8H)
Example 127 : '5-phenyl-6-(4-{[4-(5-pyridin-2-y1-4H-1,2,4-triazol-3-
yl)piperidin-1-yl]methyl} phenyl)imidazo[2,1-13][1,3]thiazole-2-carboxylic
acid
To as solution of example 126 (0.125 gm 0.219mM) in methanol 3 ml and water 2
ml is added potassium carbonate (3 eq.). The resulting reaction mixture is
stirred
at room temperature for overnight. Reaction mass is then concentrated to
remove
methanol, diluted with 4 ml of water and neutralized (pH ¨ 7). Solid
precipitated is
filtered and dried to afford the desired compound.
MS (M+1) :562.07
1H NMR (300 MHz, DMSO-d6) 6 14.44(bs, 1H), 8.67 (d, J=3 Hz, 1H), 8.02 (d,
J=7.5Hz, 1H), 7.95 (s, 1H), 7.68 (s, 1H), 7.57-7.44 (m, 8H), 7.34 (d, J=7.5Hz,
2H), 3.83-3.75 (bs, 2H) 3.17-2.89 (m, 4H), 2.18-1.69(m, 4H)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
98
Commercial utility
The compounds of formula (I) and the stereoisomers of the compounds of
formula (I) according to the invention are hereinafter referred to as the
compounds of the invention. In particular, the compounds of the invention are
pharmaceutically acceptable. The compounds according to the invention have
valuable pharmaceutical properties, which make them commercially utilizable.
In
particular, they inhibit the Pi3K/Akt pathway and exhibit cellular activity.
They are
expected to be commercially applicable in the therapy of diseases (e.g.
diseases
dependent on overactivated Pi3K/Akt.
Cellular activity and analogous terms in the present invention is used as
known to
persons skilled in the art, as an example, induction of apoptosis or
chemosensiti-
zation.
Chemosensitization and analogous terms in the present invention is used as
known to persons skilled in the art. These stimuli include, for example,
effectors
of death receptor and survival pathways as well as cytotoxic /
chemotherapeutic
and targeted agents and finally radiation therapy. Induction of apoptosis and
analogous terms according to the present invention are used to identify a com-
pound which excecutes programmed cell death in cells contacted with that com-
pound or in combination with other compounds routinely used for therapy.
Apoptosis in the present invention is used as known to persons skilled in the
art.
Induction of apoptosis in cells contacted with the compound of this invention
might not necessarily be coupled with inhibition of cell proliferation.
Preferably,
the inhibition of proliferation and/or induction of apoptosis are specific to
cells with
aberrant cell growth.
Further on, the compounds according to the present invention inhibit protein
kinase activity in cells and tissues, causing a shift towards dephosphorylated
substrate proteins and as functional consequence, for example the induction of
apoptosis, cell cycle arrest and/or sensitization towards chemotherapeutic and
target-specific cancer drugs. In a preferred embodiment, inhibition of
Pi3K/Akt
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
99
pathway induces cellular effects as mentioned herein alone or in combination
with
standard cytotoxic or targeted cancer drugs.
Compounds according to the present invention exhibit anti-proliferative and/or
pro-apoptotic and/or chemosensitizing properties. Accordingly, the compounds
of
the present invention are useful for treatment of hyperproliferative
disorders, in
particular cancer. Therefore the compounds of the present invention are used
in
the production of an anti-proliferative and/or pro-apoptotic and/or
chemosensitiz-
ing effect in mammals such as human being suffering from a hyperproliferative
disorders, like cancer.
Compounds according to the present invention exhibit anti-proliferative and/or
pro-apoptotic properties in mammals such as humans due to inhibition of meta-
bolic activity of cancer cells which are able to survive despite of
unfavourable
growth conditions such as glucose depletion, hypoxia or other chemo stress.
Thus, the compounds according to the present invention are for treating,
amelio-
rating or preventing diseases of benign or malignant behaviour as described
herein, such as e.g. for inhibiting cellular neoplasia.
Neoplasia in the present invention is used as known to persons skilled in the
art.
A benign neoplasia is described by hyperproliferation of cells, incapable of
form-
ing an aggressive, metastasizing tumor in-vivo. In contrast, a malignant
neoplasia
is described by cells with multiple cellular and biochemical abnormalities,
capable
of forming a systemic disease, for example forming tumor metastasis in distant
organs.
The compounds according to the present invention can be preferably used for
the
treatment of malignant neoplasia. Examples of malignant neoplasia treatable
with
the compounds according to the present invention include solid and hematologi-
cal tumors. Solid tumors can be exemplified by tumors of the breast, bladder,
bone, brain, central and peripheral nervous system, colon, endocrine glands
(e.g.
thyroid and adrenal cortex), esophagus, endometrium, germ cells, head and
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
100
neck, kidney, liver, lung, larynx and hypopharynx, mesothelioma, ovary, pan-
creas, prostate, rectum, renal, small intestine, soft tissue, testis, stomach,
skin,
ureter, vagina and vulva. Malignant neoplasias include inherited cancers
exempli-
fied by Retinoblastoma and Wilms tumor. In addition, malignant neoplasias in-
dude primary tumors in said organs and corresponding secondary tumors in dis-
tant organs ("tumor metastases"). Hematological tumors can be exemplified by
aggressive and indolent forms of leukemia and lymphoma, namely non-Hodgkins
disease, chronic and acute myeloid leukemia (CML / AML), acute lymphoblastic
leukemia (ALL), Hodgkins disease, multiple myeloma and T-cell lymphoma. Also
included are myelodysplastic syndrome, plasma cell neoplasia, paraneoplastic
syndromes, and cancers of unknown primary site as well as AIDS related malig-
nancies.
It is noted that a malignant neoplasia does not necessarily require the
formation
of metastases in distant organs. Certain tumors exert devastating effects on
the
primary organ itself through their aggressive growth properties. These can
lead to
the destruction of the tissue and organ structure finally resulting in failure
of the
assigned organ function and death.
Drug resistance is of particular importance for the frequent failure of
standard
cancer therapeutics. This drug resistance is caused by various cellular and mo-
lecular mechanisms. One aspect of drug resistance is caused by constitutive ac-
tivation of anti-apoptotic survival signals with PKB/Akt as a key signalling
kinase.
Inhibition of the Pi3K/Akt pathway leads to a resensitization towards standard
chemotherapeutic or target specific cancer therapeutics. As a consequence, the
commercial applicability of the compounds according to the present invention
is
not limited to 1st line treatment of cancer patients. In a preferred
embodiment,
cancer patients with resistance to cancer chemotherapeutics or target specific
anti-cancer drugs are also amenable for treatment with these compounds for
e.g.
2nd or 3rd line treatment cycles. In particular, the compounds according to
the pre-
sent invention might be used in combination with standard chemotherapeutic or
targeted drugs to resensitize tumors towards these agents.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
101
In the context of their properties, functions and utilities mentioned herein,
the
compounds according to the present invention are distinguished by unexpected
valuable and desirable effects related therewith, such as e.g. superior
therapeutic
window, superior bioavailability (such as e.g. good oral absorption), low
toxicity
and/or further beneficial effects related with their therapeutic and
pharmaceutical
qualities.
Compounds according to the present invention are for treatment, prevention or
amelioration of the diseases of benign and malignant behavior as described [De-
lo fore, such as e.g. benign or malignant neoplasia, particularly cancer,
especially a
cancer that is sensitive to Pi3K/Akt pathway inhibition.
The present invention further includes a method for treating, prevention or
ame-
lioration mammals, including humans, which are suffering from one of the
abovementioned conditions, illnesses, disorders or diseases. The method is
characterized in that a pharmacologically active and therapeutically effective
and
tolerable amount of one or more of compounds according to the present
invention
is administered to the subject in need of such treatment.
The present invention further includes a method for treating, preventing or
ame-
liorating diseases responsive to inhibition of the Pi3K/Akt pathway, in a
mammal,
including human, comprising administering a pharmacologically active and thera-
peutically effective and tolerable amount of one or more of the compounds ac-
cording to the present invention to said mammal.
The present invention further includes a method for treating
hyperproliferative
diseases of benign or malignant behaviour and/or disorders responsive to induc-
tion of apoptosis, such as e.g. cancer, particularly any of those cancer
diseases
described above, in a mammal, comprising administering a pharmacologically
active and therapeutically effective and tolerable amount of one or more of
the
compounds according to the present invention to said mammal.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
102
The present invention further includes a method for inhibiting cellular
hyperprolif-
eration or arresting aberrant cell growth in a mammal, comprising
administering a
pharmacologically active and therapeutically effective and tolerable amount of
one or more of the compounds according to the present invention to said mam-
mal.
The present invention further includes a method for inducing apoptosis in the
therapy of beningn or malignant neoplasia, particularly cancer, comprising
admin-
istering a pharmacologically active and therapeutically effective and
tolerable
amount of one or more of the compounds according to the present invention to a
subject in need of such therapy.
The present invention further includes a method for inhibiting protein kinase
activ-
ity in cells comprising administering a pharmacologically active and
therapeuti-
cally effective and tolerable amount of one or more of the compounds according
to the present invention to a patient in need of such therapy.
The present invention further includes a method for sensitizing towards chemo-
therapeutic or target-specific anti-cancer agents in a mammal, comprising
admin-
istering a pharmacologically active and therapeutically effective and
tolerable
amount of one or more of the compounds according to the present invention to
said mammal.
The present invention further includes a method for treating benign and/or
malig-
nant neoplasia, particularly cancer, in a mammal, including human, comprising
administering a pharmacologically active and therapeutically effective and
toler-
able amount of one or more of the compounds according to the present invention
to said mammal.
The present invention further relates to the use of the compounds for the pro-
duction of pharmaceutical compositions, which are employed for the treatment,
prophylaxis, and/or amelioration of one or more of the illnesses mentioned.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
103
The present invention further relates to the use of the compounds for the manu-
facture of pharmaceutical compositions for treating, preventing or
ameliorating
hyperproliferative diseases and/or disorders responsive to the induction of
apop-
tosis, such as e.g. beningn or malignant neoplasia, in particular cancer.
The present invention further relates to the use of the compounds according to
this invention for the production of pharmaceutical compositions for treating,
pre-
venting or ameliorating benign or malignant neoplasia, particularly cancer,
such
as e.g. any of those cancer diseases described above.
The invention further relates to a compound according to the invention or a
phar-
maceutically acceptable salt thereof, for the treatment and/or prophylaxis of
(hy-
per)proliferative diseases and/or disorders responsive to induction of
apoptosis,
which include benign neoplasia and malignant neoplasia, including cancer.
The invention further relates to a pharmaceutical composition, comprising a
com-
pound according to the invention or a pharmaceutically acceptable salt
thereof,
for the treatment and/or prophylaxis of (hyper)proliferative diseases and/or
disor-
ders responsive to induction of apoptosis, which include benign neoplasia and
malignant neoplasia, including cancer.
The present invention further relates to the use of compounds and pharmaceuti-
cally acceptable salts according to the present invention for the manufacture
of
pharmaceutical compositions, which can be used for sensitizing towards chemo-
therapeutic and/or target specific anti-cancer agents.
The present invention further relates to the use of compounds according to the
present invention for the manufacture of pharmaceutical compositions, which
can
be used for sensitizing towards radiation therapy of those diseases mentioned
herein, particularly cancer.
The present invention further relates to the use of the compounds according to
the present invention for the manufacture of pharmaceutical compositions,
which
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
104
can be used in the treatment of diseases sensitive to protein kinase inhibitor
ther-
apy and different to cellular neoplasia. These non-malignant diseases include,
but
are not limited to benign prostate hyperplasia, neurofibromatosis, dermatoses,
and myelodysplastic syndromes.
The present invention further relates to pharmaceutical compositions
comprising
one or more of the compounds according to this invention and a
pharmaceutically
acceptable carrier or diluent.
The present invention further relates to pharmaceutical compositions
comprising
one or more of the compounds according to this invention and pharmaceutically
acceptable auxiliaries and/or excipients.
The pharmaceutical compositions according to this invention are prepared by
processes, which are known per se and familiar to the person skilled in the
art. As
pharmaceutical compositions, the compounds of the invention (= active com-
pounds) are either employed as such, or preferably in combination with
suitable
pharmaceutical auxiliaries and/or excipients, e.g. in the form of tablets,
coated
tablets, dragees, pills, cachets, granules, capsules, caplets, suppositories,
patches (e.g. as TTS), emulsions (such as e.g. micro-emulsions or lipid emul-
sions), suspensions (such as e.g. nano suspensions), gels, solubilisates or
solu-
tions (e.g. sterile solutions), or encapsuled in liposomes or as beta-
cyclodextrine
or beta-cyclodextrin derivative inclusion complexes or the like, the active
com-
pound content advantageously being between 0.1 and 95% and where, by the
appropriate choice of the auxiliaries and/or excipients, a pharmaceutical
admini-
stration form (e.g. a delayed release form or an enteric form) exactly suited
to the
active compound and/or to the desired onset of action can be achieved.
The person skilled in the art is familiar with auxiliaries, vehicles,
excipients, dilu-
ents, carriers or adjuvants which are suitable for the desired pharmaceutical
for-
mulations, preparations or compositions on account of his/her expert
knowledge.
In addition to solvents, gel formers, ointment bases and other active compound
excipients, for example antioxidants, dispersants, emulsifiers, preservatives,
CA 02695251 2015-04-28
=
105
solubilizers (such as e.g. polyoxyethylenglyceroltriricinoleat 35, PEG 400,
Tween TM
80, CaptisolTM, SolutolTM HS15 or the like), colorants, complexing agents,
permeation
promoters, stabilizers, fillers, binders, thickeners, disintegrating agents,
buffers,
pH regulators (e.g. to obtain neutral, alkaline or acidic formulations),
polymers,
lubricants, coating agents, propellants, tonicity adjusting agents,
surfactants, fla-
vorings, sweeteners or dyes, can be used.
In particular, auxiliaries and/or excipients of a type appropriate to the
desired for-
mulation and the desired mode of administration are used.
The administration of the compounds, pharmaceutical compositions or combina-
tions according to the invention may be performed in any of the generally ac-
cepted modes of administration available in the art. Illustrative examples of
suit-
able modes of administration include intravenous, oral, nasal, parenteral,
topical,
transdermal and rectal delivery. Oral and intravenous deliveries are
preferred.
Generally, the pharmaceutical compositions according to the invention can be
administered such that the dose of the active compound is in the range
customary for Pi3K/Akt pathway inhibitors. In particular, a dose in the range
of
from 0.01 to 4000 mg of the active compound per day is preferred for an
average
adult patient having a body weight of 70 kg. In this respect, it is to be
noted that
the dose is dependent, for example, on the specific compound used, the species
treated, age, body weight, general health, sex and diet of the subject
treated,
mode and time of administration, rate of excretion, severity of the disease to
be
treated and drug combination.
The pharmaceutical composition can be administered in a single dose per day or
in multiple subdoses, for example, 2 to 4 doses per day. A single dose unit of
the
pharmaceutical composition can contain e.g. from 0.01 mg to 4000 mg, prefera-
bly 0.1 mg to 2000 mg, more preferably 0.5 to 1000 mg, most preferably 1 to
500
mg, of the active compound. Furthermore, the pharmaceutical composition can
be adapted to weekly, monthly or even more infrequent administration, for exam-
ple by using an implant, e.g. a subcutaneous or intramuscular implant, by
using
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
106
the active compound in form of a sparingly soluble salt or by using the active
compound coupled to a polymer.
The choice of the optimal dosage regime and duration of medication,
particularly
the optimal dose and manner of administration of the active compounds neces-
sary in each case can be determined by a person skilled in the art.
The present invention further relates to combinations comprising one or more
first
active ingredients selected from the compounds of the invention and one or
more
second active ingredients selected from chemotherapeutic anti-cancer agents
and target-specific anti-cancer agents e.g. for treating, preventing or
ameliorating
diseases responsive or sensitive to inhibition of the Pi3K/Akt pathway, such
as
hyperproliferative diseases of benign or malignant behaviour and/or disorders
responsive to the induction of apoptosis, particularly cancer, such as e.g.
any of
those cancer diseases described above.
The invention further relates to the use of a pharmaceutical composition
compris-
ing one or more of the compounds according to this invention as sole active in-
gredient(s) and a pharmaceutically acceptable carrier or diluent in the
manufac-
ture of pharmaceutical products for the treatment and/or prophylaxis of the
ill-
nesses mentioned above.
Depending upon the particular disease, to be treated or prevented, additional
therapeutic active agents, which are normally administered to treat or prevent
that
disease, may optionally be coadministered with the compounds according to this
invention. As used herein, additional therapeutic agents that are normally
admin-
istered to treat or prevent a particular disease are known as appropriate for
the
disease being treated.
The above mentioned second active ingredient, which is a chemotherapeutic anti-
cancer agents, includes but is not limited to (i) alkylating/carbamylating
agents
such as Cyclophosphamid (Endoxan@), lfosfamid (Holoxan@), Thiotepa (Thio-
tepa Lederle@), Melphalan (Alkeran@), or chloroethylnitrosourea (BCNU); (ii)
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
107
platinum derivatives like cis-platin (Platinex@ BMS), oxaliplatin (Eloxatin@),
sa-
traplatin or carboplatin (Cabroplat@ BMS); (iii) antimitotic agents / tubulin
inhibi-
tors such as vinca alkaloids (vincristine, vinblastine, vinorelbine), taxanes
such as
Paclitaxel (Taxol@), Docetaxel (Taxotere@) and analogs as well as new formula-
tions and conjugates thereof (like the nanoparticle formulation Abraxane@ with
paclitaxel bound to albumin), epothilones such as Epothilone B (Patupilone@),
Azaepothilone (Ixabepilone ) or ZK-EPO, a fully synthetic epothilone B analog;
(iv) topoisomerase inhibitors such as anthracyclines (exemplified by
Doxorubicin /
Adriblastin@), epipodophyllotoxines (examplified by Etoposide / Etopophos@)
and
camptothecin and camptothecin analogs (exemplified by Irinotecan / Camptosar@
or Topotecan / Hycamtin@); (v) pyrimidine antagonists such as 5-fluorouracil
(5-
FU), Capecitabine (Xeloda@), Arabinosylcytosine / Cytarabin (Alexan@) or Gem-
citabine (Gemzar@); (vi) purin antagonists such as 6-mercaptopurine (Puri-
Nethol@), 6-thioguanine or fludarabine (Fludara@) and (vii) folic acid
antagonists
such as methotrexate (Farmitrexat@) or premetrexed (Alimta@).
The above mentioned second active ingredient, which is a target specific anti-
cancer agent, includes but is not limited to (i) kinase inhibitors such as
e.g.
Imatinib (Glivec@), ZD-1839 / Gefitinib (Iressa ), Bay43-9006 (Sorafenib,
Nexavar@), SU11248 / Sunitinib (Sutent@), OSI-774 / Erlotinib (Tarceva@),
Dasatinib (Sprycel@), Lapatinib (Tykerb@), or, see also below, Vatalanib,
Vandetanib (Zactima@) or Pazopanib; (ii) proteasome inhibitors such as PS-341
/
Bortezumib (Velcade@); (iii) histone deacetylase inhibitors like SAHA
(Zolinza@),
PXD101, MS275, MGCD0103, Depsipeptide / FK228, NVP-LBH589, Valproic
acid (VPA), GRA / PCI 24781, ITF2357, SB939 and butyrates (iv) heat shock pro-
tein 90 inhibitors like 17-allylaminogeldanamycin (17-AAG) or 17-
dimethylaminogeldanamycin (17-DMAG); (v) vascular targeting agents (VTAs)
like combretastin A4 phosphate or AVE8062 / AC7700 and anti-angiogenic drugs
like the VEGF antibodies, such as Bevacizumab (Avastin@), or KDR tyrosine
kinase inhibitors such as PTK787 / ZK222584 (Vatalanib@) or Vandetanib (Zac-
tima@) or Pazopanib; (vi) monoclonal antibodies such as Trastuzumab (Her-
ceptin@), Rituximab (MabThera / Rituxan@), Alemtuzumab (Campath@), Tositu-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
108
momab (Bexxar@), 0225/ Cetuximab (Erbitux@), Avastin (see above) or Panitu-
mumab (Vectibix@) as well as mutants and conjugates of monoclonal antibodies,
e.g. Gemtuzumab ozogamicin (Mylotarg@) or Ibritumomab tiuxetan (Zevalin@),
and antibody fragments; (vii) oligonucleotide based therapeutics like G-3139 /
Oblimersen (Genasense@) or the DNMT1 inhibitor MG98; (viii) Toll-like receptor
/
TLR 9 agonists like Promune@, TLR 7 agonists like Imiquimod (Aldara@) or Isa-
toribine and analogues thereof, or TLR 7/8 agonists like Resiquimod as well as
immunostimulatory RNA as TLR 7/8 agonists; (ix) protease inhibitors; (x) hormo-
nal therapeutics such as anti-estrogens (e.g. Tamoxifen or Raloxifen), anti-
androgens (e.g. Flutamide or Casodex), LHRH analogs (e.g. Leuprolide, Gosere-
lin or Triptorelin) and aromatase inhibitors (e.g. Femara, Arimedex or
Aromasin).
Other target specific anti-cancer agents includes bleomycin, retinoids such as
all-
trans retinoic acid (ATRA), DNA methyltransferase inhibitors such as 5-Aza-2'-
deoxycytidine (Decitabine, Dacogen@) and 5-azacytidine (Vidaza@), alanosine,
cytokines such as interleukin-2, interferons such as interferon a2 or
interferon-y,
bcI2 antagonists (e.g. ABT-737 or analogs), death receptor agonists, such as
TRAIL, DR4/5 agonistic antibodies, FasL and TNF-R agonists (e.g. TRAIL recep-
tor agonists like mapatumumab or lexatumumab).
Specific examples of the second active ingredient include, but is not limited
5 FU,
actinomycin D, ABARELIX, ABCIXIMAB, ACLARUBICIN, ADAPALENE, ALEM-
TUZUMAB, ALTRETAMINE, AMINOGLUTETHIMIDE, AMIPRILOSE, AMRUBI-
CIN, ANASTROZOLE, ANCITABINE, ARTEMISININ, AZATHIOPRINE,
BASILIXIMAB, BENDAMUSTINE, BEVACIZUMAB, BEXXAR, BICALUTAMIDE,
BLEOMYCIN, BORTEZOMIB, BROXURIDINE, BUSULFAN, CAMPATH, CAPE-
CITABINE, CARBOPLATIN, CARBOQUONE, CARMUSTINE, CETRORELIX,
CHLORAMBUCIL, CHLORMETHINE, CISPLATIN, CLADRIBINE, CLOMIFENE,
CYCLOPHOSPHAMIDE, DACARBAZINE, DACLIZUMAB, DACTINOMYCIN,
DASATINIB, DAUNORUBICIN, DECITABINE, DESLORELIN, DEXRAZOXANE,
DOCETAXEL, DOXIFLURIDINE, DOXORUBICIN, DROLOXIFENE, DRO-
STANOLONE, EDELFOSINE, EFLORNITHINE, EMITEFUR, EPIRUBICIN, EPI-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
109
TIOSTANOL, EPTAPLATIN, ERBITUX, ERLOTINIB, ESTRAMUSTINE, ETO-
POSIDE, EXEMESTANE, FADROZOLE, FINASTERIDE, FLOXURIDINE, FLU-
CYTOSINE, FLUDARABINE, FLUOROURACIL, FLUTAMIDE, FORMESTANE,
FOSCARNET, FOSFESTROL, FOTEMUSTINE, FULVESTRANT, GEFITINIB,
GENASENSE, GEMCITABINE, GLIVEC, GOSERELIN, GUSPERIMUS, HER-
CEPTIN, IDARUBICIN, IDOXURIDINE, IFOSFAMIDE, IMATINIB, IMPROSUL-
FAN, INFLIXIMAB, IRINOTECAN, IXABEPILONE, LANREOTIDE, LAPATINIB,
LETROZOLE, LEUPRORELIN, LOBAPLATIN, LOMUSTINE, LUPROLIDE,
MELPHALAN, MERCAPTOPURINE, METHOTREXATE, METUREDEPA, MI-
BOPLATIN, MIFEPRISTONE, MILTEFOSINE, MIRIMOSTIM, MITOGUAZONE,
MITOLACTOL, MITOMYCIN, MITOXANTRONE, MIZORIBINE, MOTEXAFIN,
MYLOTARG, NARTOGRASTIM, NEBAZUMAB, NEDAPLATIN, NILUTAMIDE,
NIMUSTINE, OCTREOTIDE, ORMELOXIFENE, OXALIPLATIN, PACLITAXEL,
PALIVIZUMAB, PAN ITUMUMAB, PATUPILONE, PAZOPANIB, PEGASPAR-
GASE, PEGFILGRASTIM, PEMETREXED, PENTETREOTIDE, PENTOSTATIN,
PERFOSFAMIDE, PIPOSULFAN, PIRARUBICIN, PLICAMYCIN, PREDNI-
MUSTINE, PROCARBAZINE, PROPAGERMANIUM, PROSPIDIUM CHLORIDE,
RALOXIFEN, RALTITREXED, RANIMUSTINE, RANPIRNASE, RASBURICASE,
RAZOXANE, RITUXIMAB, RIFAMPICIN, RITROSULFAN, ROMURTIDE,
RUBOXISTAURIN, SARGRAMOSTIM, SATRAPLATIN, SIROLIMUS, SOBU-
ZOXANE, SORAFENIB, SPIROMUSTINE, STREPTOZOCIN, SUNITINIB, TA-
MOXIFEN, TASONERMIN, TEGAFUR, TEMOPORFIN, TEMOZOLOMIDE,
TENIPOSIDE, TESTOLACTONE, THIOTEPA, THYMALFASIN, TIAMIPRINE,
TOPOTECAN, TOREMIFENE, TRAIL, TRASTUZUMAB, TREOSULFAN, TRI-
AZIQUONE, TRIMETREXATE, TRIPTORELIN, TROFOSFAMIDE, UREDEPA,
VALRUBICIN, VATALANIB, VANDETANIB, VERTEPORFIN, VINBLASTINE,
VINCRISTINE, VINDESINE, VINORELBINE, VOROZOLE, ZEVALIN and
ZOLINZA.
The anti-cancer agents mentioned herein above as combination partners of the
compounds according to this invention are meant to include pharmaceutically
acceptable derivatives thereof, such as e.g. their pharmaceutically acceptable
salts.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
110
The person skilled in the art is aware of the total daily dosage(s) and
administra-
tion form(s) of the additional therapeutic agent(s) coadministered. Said total
daily
dosage(s) can vary within a wide range.
In practicing the present invention, the compounds according to this invention
may be administered in combination therapy separately, sequentially, simultane-
ously, concurrently or chronologically staggered (such as e.g. as combined
unit
dosage forms, as separate unit dosage forms, as adjacent discrete unit dosage
forms, as fixed or non-fixed combinations, as kit-of-parts or as admixtures)
with
one or more standard therapeutics (chemotherapeutic and/or target specific
anti-
cancer agents), in particular art-known anti-cancer agents, such as any of
e.g.
those mentioned above.
In this context, the present invention further relates to a combination
comprising a
first active ingredient, which is at least one compound according to this
invention,
and a second active ingredient, which is at least one art-known anti-cancer
agent,
such as e.g. one or more of those mentioned herein above, for separate, sequen-
tial, simultaneous, concurrent or chronologically staggered use in therapy,
such
as e.g. in therapy of any of those diseases mentioned herein.
The term "combination" in the present invention is used as known to persons
skilled in the art and may be present as a fixed combination, a non-fixed
combi-
nation or kit-of-parts.
A "fixed combination" in the present invention is used as known to persons
skilled
in the art and is defined as a combination wherein the said first active
ingredient
and the said second active ingredient are present together in one unit dosage
or
in a single entity. One example of a "fixed combination" is a pharmaceutical
com-
position wherein the said first active ingredient and the said second active
ingre-
dient are present in admixture for simultaneous administration, such as in a
for-
mulation. Another example of a "fixed combination" is a pharmaceutical combina-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
111
tion wherein the said first active ingredient and the said second active
ingredient
are present in one unit without being in admixture.
A non-fixed combination or "kit-of-parts" in the present invention is used as
known
to persons skilled in the art and is defined as a combination wherein the said
first
active ingredient and the said second active ingredient are present in more
than
one unit. One example of a non-fixed combination or kit-of-parts is a
combination
wherein the said first active ingredient and the said second active ingredient
are
present separately. The components of the non-fixed combination or kit-of-
parts
may be administered separately, sequentially, simultaneously, concurrently or
chronologically staggered.
The present invention further relates to a pharmaceutical composition
comprising
a first active ingredient, which is at least one compound according to this
inven-
tion, and a second active ingredient, which is at least one art-known anti-
cancer
agent, such as e.g. one or more of those mentioned herein above, and, option-
ally, a pharmaceutically acceptable carrier or diluent, for separate,
sequential,
simultaneous, concurrent or chronologically staggered use in therapy.
The present invention further relates to a combination product comprising
a.) at least one compound according to this invention formulated with a pharma-
ceutically acceptable carrier or diluent, and
b.) at least one art-known anti-cancer agent, such as e.g. one or more of
those
mentioned herein above, formulated with a pharmaceutically acceptable carrier
or
diluent.
The present invention further relates to a kit-of-parts comprising a
preparation of
a first active ingredient, which is a compound according to this invention,
and a
pharmaceutically acceptable carrier or diluent; a preparation of a second
active
ingredient, which is an art-known anti-cancer agent, such as one of those men-
tioned above, and a pharmaceutically acceptable carrier or diluent; for simul-
taneous, concurrent, sequential, separate or chronologically staggered use in
therapy. Optionally, said kit comprises instructions for its use in therapy,
e.g. to
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
112
treat hyperproliferative diseases and diseases responsive or sensitive to
inhibition
of the Pi3K/Akt pathway, such as e.g. beningn or malignant neoplasia,
particularly
cancer, more precisely, any of those cancer diseases described above.
The present invention further relates to a combined preparation comprising at
least one compound according to this invention and at least one art-known anti-
cancer agent for simultaneous, concurrent, sequential or separate
administration.
The present invention further relates to combinations, compositions,
formulations,
preparations or kits according to the present invention having Pi3K/Akt
pathway
inhibitory activity.
In addition, the present invention further relates to a method for treating in
combi-
nation therapy hyperproliferative diseases and/or disorders responsive to the
in-
duction of apoptosis, such as e.g. cancer, in a patient comprising
administering a
combination, composition, formulation, preparation or kit as described herein
to
said patient in need thereof.
In addition, the present invention further relates to a method for treating
hyperpro-
liferative diseases of benign or malignant behaviour and/or disorders
responsive
to the induction of apoptosis, such as e.g. cancer, in a patient comprising
admin-
istering in combination therapy separately, simultaneously, concurrently,
sequen-
tially or chronologically staggered a pharmaceutically active and
therapeutically
effective and tolerable amount of a pharmaceutical composition, which
comprises
a compound according to this invention and a pharmaceutically acceptable
carrier
or diluent, and a pharmaceutically active and therapeutically effective and
toler-
able amount of one or more art-known anti-cancer agents, such as e.g. one or
more of those mentioned herein, to said patient in need thereof.
In further addition, the present invention relates to a method for treating,
prevent-
ing or ameliorating hyperproliferative diseases and/or disorders responsive to
induction of apoptosis, such as e.g. benign or malignant neoplasia, e.g.
cancer,
particularly any of those cancer diseases mentioned herein, in a patient
compris-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
113
ing administering separately, simultaneously, concurrently, sequentially or
chronologically staggered to said patient in need thereof an amount of a first
ac-
tive compound, which is a compound according to the present invention, and an
amount of at least one second active compound, said at least one second active
compound being a standard therapeutic agent, particularly at least one art-
known
anti-cancer agent, such as e.g. one or more of those chemotherapeutic and tar-
get-specific anti-cancer agents mentioned herein, wherein the amounts of the
first
active compound and said second active compound result in a therapeutic
effect.
In yet further addition, the present invention relates to a method for
treating, pre-
venting or ameliorating hyperproliferative diseases and/or disorders
responsive
to induction of apoptosis, such as e.g. benign or malignant neoplasia, e.g.
cancer,
particularly any of those cancer diseases mentioned herein, in a patient
compris-
ing administering a combination according to the present invention.
In addition, the present invention further relates to the use of a
composition, com-
bination, formulation, preparation or kit according to this invention in the
manufac-
ture of a pharmaceutical product, such as e.g. a commercial package or a me-
dicament, for treating, preventing or ameliorating hyperproliferative
diseases,
such as e.g. cancer, and/or disorders responsive to the induction of
apoptosis,
particularly those diseases mentioned herein, such as e.g. malignant or benign
neoplasia.
The present invention further relates to a commercial package comprising one
or
more compounds of the present invention together with instructions for simulta-
neous, concurrent, sequential or separate use with one or more chemotherapeu-
tic and/or target specific anti-cancer agents, such as e.g. any of those
mentioned
herein.
The present invention further relates to a commercial package consisting essen-
tially of one or more compounds of the present invention as sole active
ingredient
together with instructions for simultaneous, concurrent, sequential or
separate
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
114
use with one or more chemotherapeutic and/or target specific anti-cancer
agents,
such as e.g. any of those mentioned herein.
The present invention further relates to a commercial package comprising one
or
more chemotherapeutic and/or target specific anti-cancer agents, such as e.g.
any of those mentioned herein, together with instructions for simultaneous,
con-
current, sequential or separate use with one or more compounds according to
the
present invention.
The compositions, combinations, preparations, formulations, kits or packages
mentioned in the context of the combination therapy according to this
invention
may also include more than one of the compounds according to this invention
and/or more than one of the art-known anti-cancer agents mentioned.
The first and second active ingredient of a combination or kit-of-parts
according to
this invention may be provided as separate formulations (i.e. independently of
one another), which are subsequently brought together for simultaneous, concur-
rent, sequential, separate or chronologically staggered use in combination
ther-
apy; or packaged and presented together as separate components of a combi-
nation pack for simultaneous, concurrent, sequential, separate or
chronologically
staggered use in combination therapy.
The type of pharmaceutical formulation of the first and second active
ingredient of
a combination or kit-of-parts according to this invention can be according,
i.e.
both ingredients are formulated in separate tablets or capsules, or can be
differ-
ent, i.e. suited for different administration forms, such as e.g. one active
ingredi-
ent is formulated as tablet or capsule and the other is formulated for e.g.
intrave-
nous administration.
The amounts of the first and second active ingredients of the combinations,
com-
positions or kits according to this invention may together comprise a
therapeuti-
cally effective amount for the treatment, prophylaxis or amelioration of a
hyper-
proliferative diseases and/or a disorder responsive to the induction of
apoptosis,
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
115
particularly one of those diseases mentioned herein, such as e.g. malignant or
benign neoplasia, especially cancer, like any of those cancer diseases
mentioned
herein.
In addition, compounds according to the present invention can be used in the
pre-
or post-surgical treatment of cancer.
In further addition, compounds of the present invention can be used in combina-
tion with radiation therapy.
A combination according to this invention can refer to a composition
comprising
both the compound(s) according to this invention and the other active anti-
cancer
agent(s) in a fixed combination (fixed unit dosage form), or a medicament pack
comprising the two or more active ingredients as discrete separate dosage
forms
(non-fixed combination). In case of a medicament pack comprising the two or
more active ingredients, the active ingredients are preferably packed into
blister
cards, which are suited for improving compliance.
Each blister card preferably contains the medicaments to be taken on one day
of
treatment. If the medicaments are to be taken at different times of day, the
me-
dicaments can be disposed in different sections on the blister card according
to
the different ranges of times of day at which the medicaments are to be taken
(for
example morning and evening or morning, midday and evening). The blister cavi-
ties for the medicaments to be taken together at a particular time of day are
ac-
commodated in the respective range of times of day. The various times of day
are, of course, also put on the blister in a clearly visible way. It is also
possible, of
course, for example to indicate a period in which the medicaments are to be
taken, for example stating the times.
The daily sections may represent one line of the blister card, and the times
of day
are then identified in chronological sequence in this column.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
116
Medicaments which must be taken together at a particular time of day are
placed
together at the appropriate time on the blister card, preferably a narrow
distance
apart, allowing them to be pushed out of the blister easily, and having the
effect
that removal of the dosage form from the blister is not forgotten.
Biological investigations
Cellular PI3K / Akt pathway assay
In order to study the cellular activity of the compounds according to the
present
invention, an Enzyme Linked Immunosorbent Assay (ELISA)-based assay has
been used specific phospho-AKT. The assay is based on a Sandwich ELISA kit
(PathScanTM Phospho-Akt1 (5er473); Cell Signaling, USA; #7160).
The ELISA Kit detects endogenous levels of phosphorylated Akt protein. A phos-
pho-Akt (5er473) antibody (Cell Signaling, USA; #9271) has been coated onto
the microwells. After incubation with cell lysates, the coated antibody
captures the
phosphorylated Akt protein. Following extensive washing, Akt1 monoclonal anti-
body (Cell Signaling, USA; #2967) is added to detect the captured phospho-Akt1
protein. HRP-linked anti-mouse antibody (HRP: horseradish peroxidase; Cell
Signaling, USA; #7076) is then used to recognize the bound detection antibody.
HRP substrate (= 3,3',5,5'-tetramethylbenzidine (TMB); Cell Signaling, USA;
#7160) is added to develop colour. The magnitude of optical density for this
de-
veloped color is proportional to the quantity of phosphorylated Akt protein.
MCF7 cells (ATOG HTB-22) are seeded into 96 well fate bottom plates at a den-
sity of 10000 cells/well. 24 hours after seeding, the cells are serum starved
using
low-serum medium (IMEM media including 0,1% charcoal treated FCS (FCS: fe-
tal calf serum)). After 24 hours 1 pl each of the compound dilutions (test com-
pounds were dissolved as 10 mM solutions in dimethylsulfoxide (DMSO) and
subsequently diluted) are added into each well of the 96 well plates and incu-
bated for 48h at 37 C in a humidified athmosphere containing 5% CO2. To stimu-
late Akt phosphorylation, R-Heregulin (20ng/m1R-HRG) is added in parallel to
the
compounds. Wells containing unstimulated control cells (no R-Heregulin stimula-
tion) are incubated with or without the diluted compound. Wells containing un-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
117
treated control cells (no compound) are filled with medium containing 0.5% v:v
DMSO and are or are not stimulated with R-Heregulin.
Cells are harvested and lysed with brief sonification in lx cell lysis buffer
(20mM
Tris (pH7.5), 150 mM NaCI, 1 mM ethylene diaminetetraacetate (EDTA), 1 mM
ethylene glycolbis(2-aminoethyl)-N,N,N1,N1-tetraacetic acid (EGTA), 1vol%
Triton
X-100, 2.5mM sodium pyrophosphate, 1 mM 6-glycerolphosphate, 1 mM Na3VO4,
1pg/m1 leupeptin). The lysate is centrifuged for 10 min. at 4 C and the super-
natant is transferred to a new tube. 100 pl of sample diluent (0.1vol% Tween-
20,
0.1vol% sodium azide in phosphate buffered saline (PBS)) are added to a micro-
centrifuge tube and 100 pl of cell lysate are transferred into the tube and
vor-
texed. 100 pl of each diluted cell lysate are added to the appropriate ELISA
well,
and incubated overnight at 4 C. The plates are washed 4 times with lx wash
buffer (1vol% tween-20, 0.33vo1% thymol, in PBS). Next 100 pl of detection
anti-
body (Akt1 (2H10) monoclonal detection antibody; Cell Signaling, USA; #2967)
are added to each well and incubation continued for lh at 37 C. The washing
procedure is repeated between each step. 100plof secondary antibody (anti-
mouse IgG HRP-linked antibody; Cell Signaling, USA; #7076) are added to each
well and incubated for 30 min. at 37 C. Than, 100p1of TMB substrate (0.05%
3,3',5,5' tetramethylbenzidine, 0.1% hydrogen peroxide, complex polypeptides
in
a buffered solution; Cell Signaling, USA; #7160) are added to each well and
incu-
bated for 30 min. at 25 C. Finally 100 pl of STOP solution (0.05vol% a and [3
un-
saturated carbonyl compound) are added to each well and the plate are shaked
gently. The absorbance is measured at 0 0450 nm (Wallac Victor2; Perkin Elmer,
USA) within 30 min. after adding the STOP solution. The analysis of the data
is
performed using a statistical program (Excel; Microsoft, USA). Preferred com-
pounds show an inhibitory activity towards Akt phosphorylation below 10pM.
Cellular pGSK3 assay:
In order to study the cellular activity of the compounds according to the
present
invention, an ELISA-based assay has been established for the phosphorylated
protein glycogen synthetase kinase 3 (GSK3). The assay is based on a solid
phase sandwich ELISA that detects endogenous levels of phosphorylated GSK3
using a phospho-GSK3 (Ser9) specific antibody (BioSource International, Inc.;
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
118
Catalog #KH00461). After incubation with cell lysates, the coated antibody cap-
tures the phosphorylated GSK3 protein. Following extensive washing, GSK3
polyclonal antibody is added to detect the captured phospho-GSK3 protein. Sec-
ondary antibody (anti-rabbit IgG-HRP) is then used to recognize the bound
detec-
tion antibody. After the second incubation and washing to remove all the
excess
anti-rabbit IgG-HRP, a substrate solution is added, which is acted upon by the
bound enzyme to produce color. The intensity of this colored product is
directly
proportional to the concentration of GSK-313 [pS9] present in the original
speci-
men.
MCF7 cells (ATCC HTB-22) were seeded into 96 well fate bottom plates at a
density of 10000 cells/well. After 24 h 1 pl each of the compound dilutions
(test
compounds were dissolved as 10 mM solutions in dimethylsulfoxide (DMSO) and
subsequently diluted) were added into each well of the 96 well plates and incu-
bated for 48h at 37 C in a humidified athmosphere containing 5% CO2.
Cells were harvested and lysed in cell extraction buffer (10 mM Tris, pH 7.4,
100
mM NaCI, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P207, 2 mM
Na3VO4, 1% Triton X-100, 10vol% glycerol, 0.1vol% SDS, 0.5vol% deoxycholate,
1 mM phenylmethylsulfonylfluorid (PMSF)). The lysate were centrifuged for 10
min. at 4 C and the supernatant were transferred to a new tube. 50 pl of
sample
diluent (standard diluent buffer, Biosource) were added and 100 pl of cell
lysate
transferred into the tube and vortexed. 100 pl of each diluted cell lysate
were
added to the appropriate ELISA well plate and incubated for 3h at room tempera-
ture. The plates were washed 4 times with lx wash buffer (Biosource). 50 pl of
detection antibody (GSK3 (Ser9) detection antibody; BioSource) were added to
each well and incubated for 30 min. at room temperature. The washing procedure
was repeated between each step. 100p1 of HRP-linked secondary antibody (anti-
mouse IgG HRP-linked antibody) were added to each well and incubated for 30
min. at room temperature. 100p1 of TMB substrate (0.05vol% 3,3',5,5' tetrame-
thylbenzidine, 0.1vol% hydrogen peroxide, complex polypeptides in a buffered
solution; Biosource) were added to each well and incubated for 30 min. at room
temperature. Finally 100 pl of Stop solution (0.05vol% a and 13. unsaturated
car-
bonyl compound) were added to each well and the plate were shaked gently for a
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
119
few seconds. The absorbance was measured at A 450 nm (Wallac Victor2; Perkin
Elmer, USA) within 30 min. after adding the stop solution.
The analysis of the data was performed using a statistical program (Excel;
Micro-
soft, USA) and the 1050 of pGSK3 inhibition was determined.
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
120
Table:
Cellular PI3K / Akt pathway inhibition//Cellular pGSK3 assay
Cellular Cellular Cellular Cellular Cellular Cellular
PI3K 1 Pkt pGSK3 PI3K 1 Pkt pGSK3 PI3K / Pkt pGSK3
Example pathway pathway Example pathway pathway Example pathway pathway
No. assay assay No. assay assay No. assay assay
I +++ ++ 43 ++ 85
2 +++ ++ 44 +++ ++ 86
3 +++ +++ 45 87
4 ++ 46 +++ ++ 88
+++ +++ 47 89
6 +++ ++ 48 90
7 +++ ++ 49 91
8 +++ ++ 50 92
9 ++ ++ 51 93 +++
+++ +++ 52 94
11 +++ ++ 53 95
12 +++ +++ 54 96
13 +++ ++ 55 97
14 +++ ++ 56 98 +++
+ ++ 57 99
16 ++ ++ 58 100
17 +++ ++ 59 +++ 101
18 ++ ++ 60 102
19 +++ +++ 61 +++ 103
+ + 62 +++ 104
21 +++ ++ 63 105 +++
22 +++ ++ 64 106 +++
23 +++ ++ 65 +++ 107
24 +++ +++ 66 +++ 108
+++ +++ 67 109
26 ++ 68 +++ 110
27 +++ ++ 69 111
28 ++ 70 112
29 +++ +++ 71 +++ 113
++ +++ 72 114
31 +++ +++ 73 115
32 +++ ++ 74 116
33 ++ + 75 117 +++
34 +++ +++ 76 118
++ 77 +++ 119
36 ++ 78 120
37 +++ ++ 79 121
38 +++ +++ 80 122
39 +++ ++ 81 123
+++ +++ 82 125
41 +++ ++ 83 124 +++
42 +++ + 84 126
127
IC50 > 1 OpM +
5 10pM > IC50 > 1 pM ++
1 pM > IC50 +++
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
121
Cellular proliferation / Cytotoxicity assay:
The anti-proliferative activity of the compounds as described herein, is
evaluated
using the OvCAR3, HCT116 and A549 cell lines and the Alamar Blue (Resazurin)
cell viability assay (O'Brien et al. Eur J Biochem 267, 5421-5426, 2000).
Resazu-
rin is reduced to the fluorescent resorufin by cellular dehydrogenase
activity, cor-
relating with viable, proliferating cells. Test compounds are dissolved as 10
mM
solutions in DMSO and subsequently diluted. Cells like HCT116 or A549 cells
were seeded into 96 well flat bottom plates at a density of 10000 cells/well
(OvCAR3 cells), 1000 cells/well (HCT116 cells) or 2000 cells/well (A549 cells)
in
a volume of 200 p1/well. 24 hours after seeding, 1 pl each of the compound
dilu-
tions are added into each well of the 96 well plates. Each compound dilution
is
tested as at least as duplicates. Wells containing untreated control cells
were
filled with 200 pl DMEM (Dulbecco's Modified Eagle Medium) containing 0.5vol%
v:v DMSO. The cells are then incubated with the substances for 72h at 37 C in
a
humidified atmosphere containing 5vol% CO2. To determine the viability of the
cells, 20 pl of a Resazurin solution (90mg / I) are added. After 4h incubation
at
37 C, the fluorescence is measured by extinction at A= 544 nm and an emission
of A = 590 nm (Wallac Victor2; Perkin Elmer, USA). For the calculation of the
cell
viability, the emission value from untreated cells is set as 100% viability
and the
fluorescence intensity of treated cells are set in relation to the values of
untreated
cells. Viabilities are expressed as (:)/0 values. The corresponding IC50
values of
the compounds for cytotoxic activity are determined from the concentration-
effect
curves by means of non-linear regression. The analysis of the data is
performed
using a biostatistical program (GraphPad Prism, USA).
Representative IC50 values for anti-proliferative / cytotoxic potency
determined in
the aforementioned assay follow from the following table, in which the numbers
of
the compound correspond to the numbers of the examples.
CA 02 695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
122
Table:
Anti-proliferative / Cytotoxic activity (OvCAR3 cells and A549 cells)
Anti- Anti- Anti-
Anti- proliferative Anti- proliferative Anti-
proliferative
proliferative / Cytotoxic proliferative / Cytotoxic
proliferative / Cytotoxic
/ Cytotoxic activity / Cytotoxic activity /
Cytotoxic activity
Example activity (A549 (OvCAR3 Example activity (A549
(OvCAR3 Example activity (A549 (OvCAR3
No. cells) cells) No. cells) cells) No. cells)
cells)
1 + ++ 43 85 +
2 ++ ++ 44 + ++ 86 +
3 ++ 45 + 87 ++
4 + + 46 ++ 88 ++
++ ++ 47 + 89 ++
6 ++ ++ 48 + 90 +
7 ++ ++ 49 91 ++
8 ++ ++ 50 + 92 ++
9 ++ + 51 ++ 93 ++
++ ++ 52 + 94 ++
11 ++ ++ 53 ++ 95 +
12 ++ ++ 54 ++ 96 ++
13 ++ ++ 55 + 97 ++
14 ++ ++ 56 ++ 98 +
++ + 57 ++ 99 ++
16 + ++ 58 ++ 100 ++
17 ++ ++ 59 ++ 101 ++
18 + ++ 60 ++ 102 ++
19 ++ ++ 61 + 103 ++
+ + 62 + 104 ++
21 ++ ++ 63 + 105 ++
22 + ++ 64 ++ 106 +
23 + ++ 65 ++ 107 ++
24 ++ ++ 66 ++ 108 ++
++ ++ 67 ++ 109 ++
26 + ++ 68 ++ 110 ++
27 ++ ++ 69 ++ 111 ++
28 + ++ 70 ++ 112 ++
29 + ++ 71 ++ 113 ++
+ ++ 72 ++ 114 ++
31 + ++ 73 ++ 115 +
32 ++ ++ 74 ++ 116 ++
33 ++ ++ 75 + 117 +
34 + ++ 76 ++ 118 ++
+ ++ 77 + 119 ++
36 + + 78 ++ 120 ++
37 + ++ 79 + 121 ++
38 + ++ 80 + 122 ++
39 + ++ 81 + 123 ++
+ ++ 82 ++ 125 +
41 + ++ 83 + 124 +
42 ++ ++ 84 + 127 +
126 ++
IC50 > 1 OpM +
5 10pM > IC50 ++
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
123
Chemosensitization assay:
The herein disclosed compounds are evaluated for the ability to sensitize
cancer
cells towards apoptotic stimuli. Inhibitors of Akt are tested alone and in
combina-
tion with chemotherapeutic and targeted cancer therapeutics to determine the
effect on apoptosis induction.
Cancer cells are seeded in 96 well plates at concentrations ranging from 2x103
to
lx 104 cells per well in their respective growth media. 48-72 hours later, the
apop-
tosis assay are set up as follows:
For combination assays with a chemotherapeutic agent especially preferred to-
poisomerase inhibitors (such as doxorubicin, etoposide, camptothecin or
mitoxan-
trone) or antimitotic agents / tubulin inhibitors (such as vincristine),
compounds
are added at respective concentrations indicated and plates incubated at 37 C
in
a CO2 incubator for 18 hours. For standard combination assays utilizing
treatment
with chemotherapeutic agent are added at the same time at the respective con-
centrations indicated.
For combinations assays involving addition of targeted pro-apoptotic agents
like
the death receptor ligand TRAIL/Ap02L (Research Diagnostics) compounds are
added for 1,5 hours prior to addition of TRAIL and plates incubated an
additional
3 to 4 hours post TRAIL addition. In the case of the time course, plates are
incu-
bated for 2, 3, 4 and 6 hours with TRAIL ligand before ending the assay.
For both procedures, total final volumes do not exceed 250p1. At the end of
the
incubation time, the cells are pelleted by centrifugation (200 x g; 10 min. at
RT)
and the supernatant is discarded. The cells are resuspended and incubated
using
lysis buffer for 30 min. at RT (Cell Death Detection ELISAPLus, Roche, Cat.
No.11774425001). After the centrifugation is repeated (200 x g; 10 min. at RT)
an
aliquot of the supernatant is transferred to a streptavidin-coated well of a
mi-
croplate. Followed by the incubation (2h, RT) and binding of nucleosomes in
the
supernatant with, anti-histone antibody (biotin-labeled) and anti-DNA antibody
(peroxidase-conjugated; Cell Death Detection ELISAPLus, Roche, Cat.
No.11774425 001). The antibody-nucleosome complexes are bound to the mi-
croplate. The immobilized antibody-histone complexes are washed three times at
RT to remove cell components that are not immunoreactive. The substrate solu-
CA 02695251 2010-02-01
WO 2009/021990 PCT/EP2008/060686
53799AWO
2008 08 13
124
tion (2,2'-AZINO-bis [3-ethylbenziazoline-6-sulfonic acid (ABTS); Cell Death
De-
tection ELISAPLus, Roche, Cat. No. 11 774 425 001) is added and the samples
were incubated for 15 min., RT. The amount of colored product is determined
spectrophotometrically (absorbance at A = 405 nm). Data are expressed as per-
cent activity of control with cisplatin used as a positive control. Apoptosis
induc-
tion by 50 M cisplatin is arbitrarily defined as 100 cisplatin units (100
CPU).